Introduzione:
Il concetto di “ricomposizione corporea” nel Fitness e nel BodyBuilding è senza dubbio considerabile come il “fattore dominante” ricercato dal momento che si tratta, molto semplicemente, del miglioramento quantitativo e qualitativo della massa contrattile (muscolo-scheletrico) a discapito della massa grassa e della ritenzione idrica extacellulare. Che si parli di “Natursl” o “Enhanced”, oltre alle variabili alimentari e allenanti vi sono quelle supplementative rappresentate, dipendentemente dalla “filosofia” scelta, da supplementi OTC e da farmaci utilizzati in ambito off-label.
Caffeina e p-Sinefrina rappresentano i lipolitici/termogenici OTC più utilizzati con un discreto margine di efficacia. Nel contesto “Enhanced”, invece, le classi di farmaci utilizzate al fine di accentuare la riduzione (direttamente o indirettamente) della massa grassa sono diverse e comprendono comunemente:
- i β2-agonisti (non selettivi e selettivi) come Efedrina, Clenbuterolo e Salbutamolo;
- i β3-agonisti selettivi come il Mirabegron;
- gli agenti anoressizzanti con azione sui neurotrasmettitori come la Sibutramina, la Lorcaserina, l’Amfepramone e il Benfluorex;
- gli anoressizzanti analoghi incretinici come la Semaglutide, il Liraglutide e il Tirzepatide;
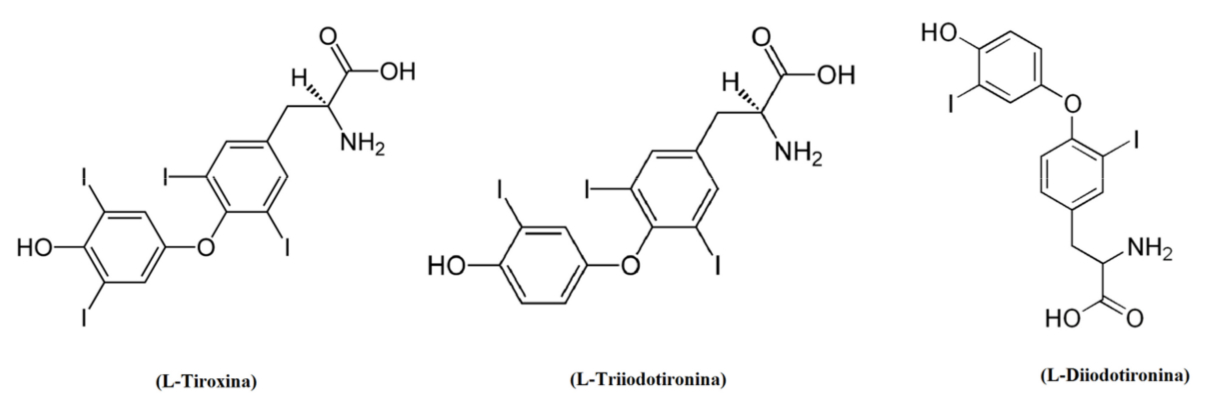
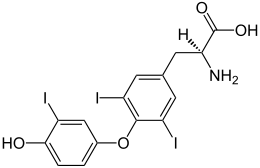
- i tiroidei Tiroxina (T4), Triiodotironina (T3) e Diiodotironina (T2);
- i tireomimetici come l’Eprotirome, il Sobetirome, il Resmetirome e il profarmaco VK2809;
- i disaccoppianti della fosforilazione ossidativa come il 2,4-dinitrofenolo (DNP);
- stimolanti il Il Peptide Natriuretico Atriale (ANP) – vedi, ad esempio, i β-bloccanti – ;
- α2-antagonisti come la Yohimbina e l’α-yohimbina [Rauwolscine];
- trattamenti mesoterapici a base di Fosfatidilcolina e/o Acidi Biliari.
A questo elenco, però, andrebbe aggiunta una classe di farmaci che molto poco intuitivamente ci fa pensare alla riduzione della massa grassa. Tale classe di farmaci è rappresentata dagli ACE II inibitori.
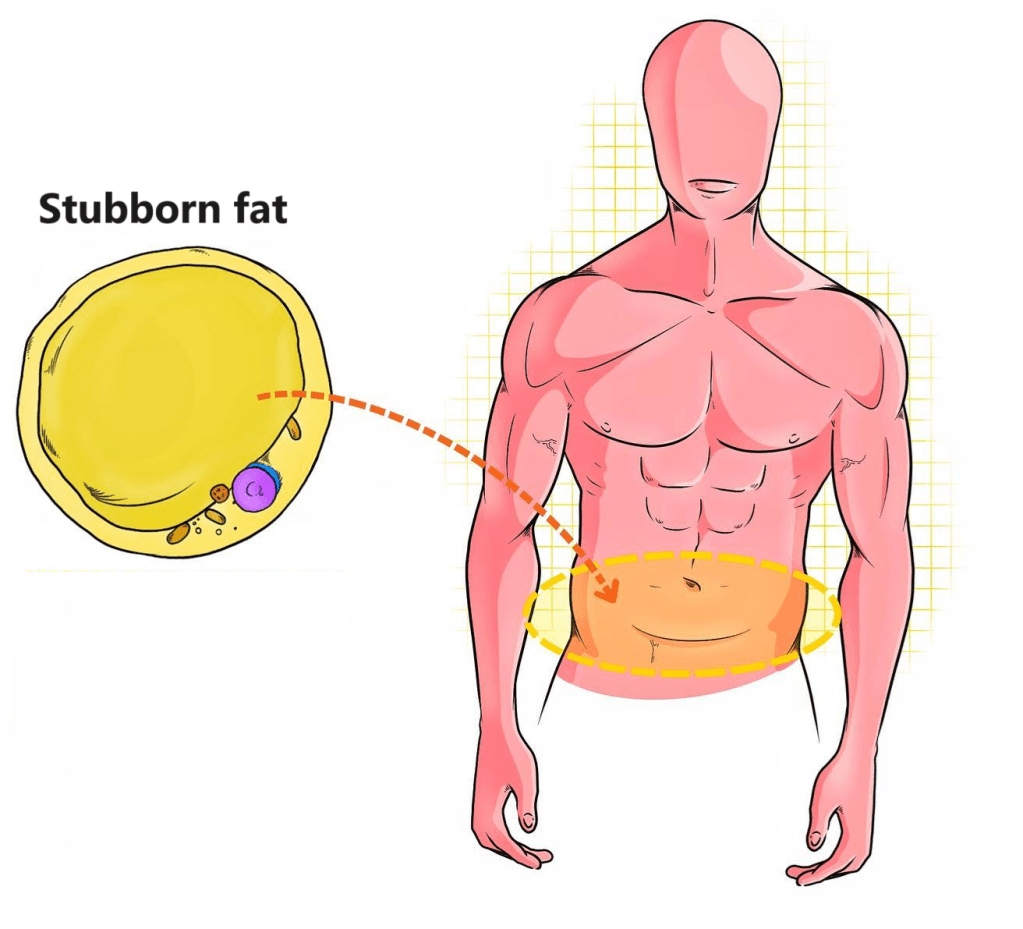
Per iniziare a comprendere del perchè questi farmaci possono rappresentare una componente funzionale nel miglioramento della composizione corporea, bisogna parlare di “Stubborn Fat” [“Grasso Testardo”]. Perchè è proprio in questa specifica e caratteristica area del tessuto adiposo che l’ACE II inibitore può contribuire alla riduzione della massa grassa.
Al fine di avere una visione di insieme più completa, è necessario trattare in modo adeguato tutte le componenti dell'”equazione”…
Tessuto adiposo e sue caratteristiche:
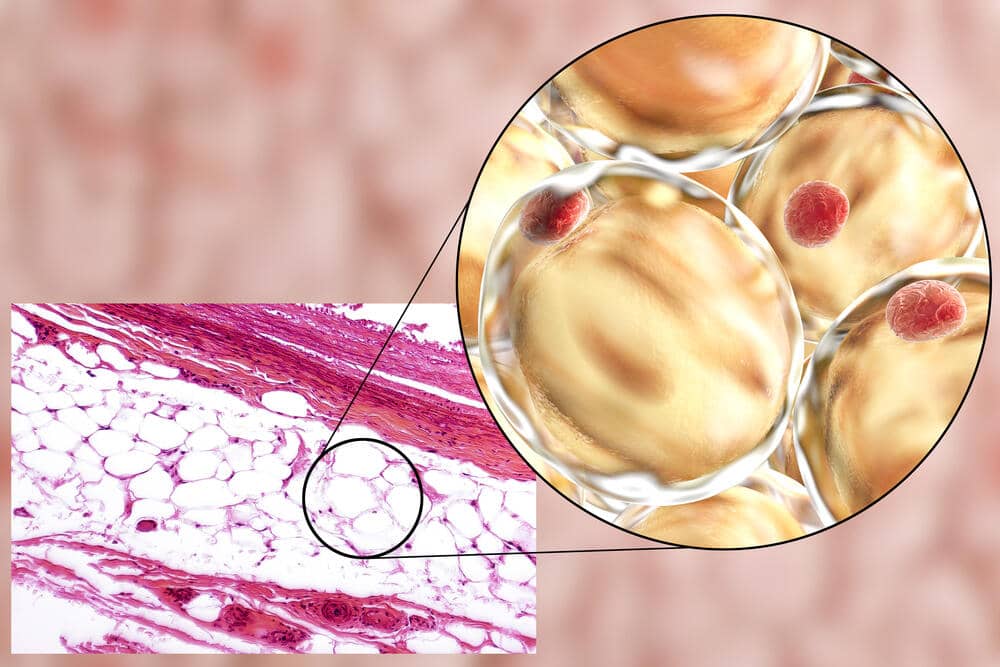
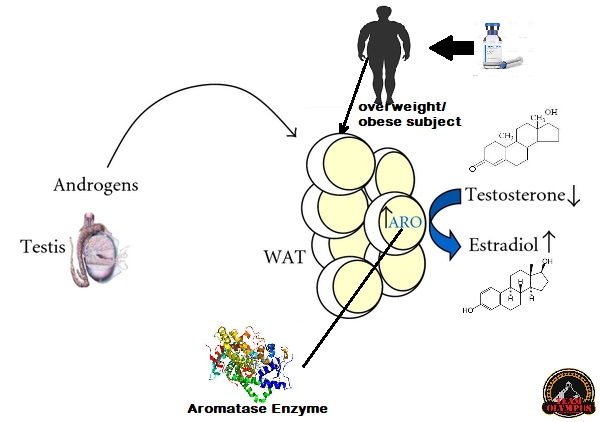
Il tessuto adiposo (noto anche come grasso corporeo o semplicemente grasso) è un tessuto connettivo lasso composto principalmente da adipociti.[1][2] Contiene anche la frazione vascolare stromale (SVF) di cellule tra cui preadipociti, fibroblasti, cellule endoteliali vascolari e una varietà di cellule immunitarie come i macrofagi del tessuto adiposo. Il suo ruolo non è semplicemente e solo quello di immagazzinare energia sotto forma di lipidi, ma anche di ammortizzare e isolare il corpo e rappresenta un vero e proprio organo endocrino.
Infatti, il tessuto adiposo veniva considerato inerte dal punto di vista ormonale, ma negli ultimi anni è stato riconosciuto come un importante organo endocrino,[3] in quanto produce ormoni come Leptina, Estrogeni, Resistina e Citochine (in particolare il TNFα). Nell’obesità, il tessuto adiposo è coinvolto nel rilascio cronico di marcatori pro-infiammatori noti come adipochine, che sono responsabili dello sviluppo della sindrome metabolica, una costellazione di malattie tra cui il diabete di tipo II, le malattie cardiovascolari e l’aterosclerosi.[2][4]
Il tessuto adiposo deriva dai preadipociti e la sua formazione sembra essere controllata in parte dal gene dell’adipe. Sappiamo ormai bene che vi sono due principali tipi di tessuto adiposo, il tessuto adiposo bianco (WAT), che immagazzina energia, e il tessuto adiposo bruno (BAT), che genera calore corporeo. Il tessuto adiposo, più precisamente il tessuto adiposo bruno, è stato identificato per la prima volta dal naturalista svizzero Conrad Gessner nel 1551.[5]
- Grasso Viscerale e Sottocutaneo:
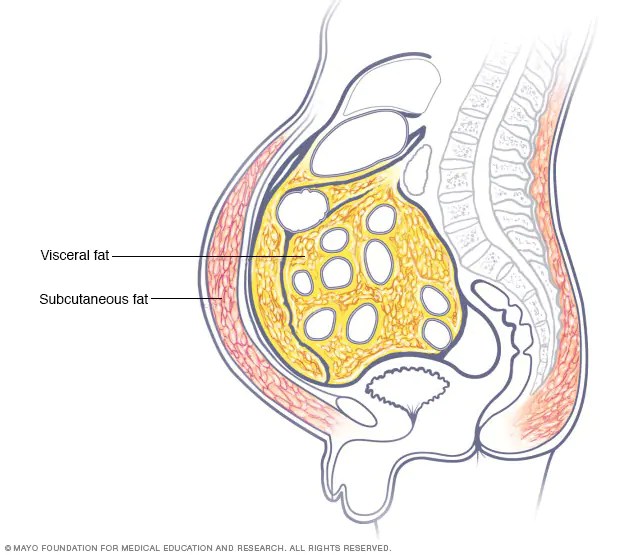
Grasso Viscerale: Il grasso viscerale o addominale[6] (noto anche come grasso d’organo o grasso intra-addominale) si trova all’interno della cavità addominale, stipato tra gli organi (stomaco, fegato, intestino, reni, ecc.). Il grasso viscerale è diverso dal grasso sottocutaneo e dal grasso intramuscolare presente nei muscoli scheletrici. Il grasso nella parte inferiore del corpo, come nelle cosce e nei glutei, è sottocutaneo e non è un tessuto omogeneo, mentre il grasso nell’addome è per lo più viscerale e semi-fluido.[7] Il grasso viscerale è composto da diversi depositi adiposi, tra cui il tessuto adiposo mesenterico, il tessuto adiposo bianco epididimale (EWAT) e i depositi perirenali. Il grasso viscerale viene spesso espresso in termini di area in cm2 (VFA, visceral fat area).[8]
Un eccesso di grasso viscerale è noto come obesità addominale, o “grasso della pancia”, in cui l’addome sporge eccessivamente. Nuovi sviluppi, come il Body Volume Index (BVI), sono specificamente progettati per misurare il volume addominale e il grasso addominale. L’eccesso di grasso viscerale è anche legato al diabete di tipo II,[9] all’insulino-resistenza,[10] alle malattie infiammatorie,[11] e ad altre patologie correlate all’obesità.[12] Allo stesso modo, è stato dimostrato che l’accumulo di grasso del collo (o tessuto adiposo cervicale) è associato alla mortalità.[13] Diversi studi hanno suggerito che il grasso viscerale può essere previsto da semplici misure antropometriche,[14] e predice la mortalità in modo più accurato dell’indice di massa corporea o della circonferenza vita.[15]
Gli uomini hanno maggiori probabilità di accumulare grasso nell’addome a causa delle differenze tra gli ormoni sessuali. L’estrogeno causa l’accumulo di grasso nei glutei, nelle cosce e nei fianchi delle donne.[16][17] Quando le donne raggiungono la menopausa e gli estrogeni prodotti dalle ovaie diminuiscono, il grasso migra dai glutei, dai fianchi e dalle cosce alla vita;[18] in seguito il grasso viene accumulato nell’addome.[7]
Il grasso viscerale può essere causato da un eccesso di livelli di cortisolo.[19] Almeno 10 ore MET a settimana di esercizio aerobico portano a una riduzione del grasso viscerale in chi non ha disturbi legati al metabolismo.[20] Anche l’allenamento contro-resistenza e la restrizione calorica riducono il grasso viscerale, anche se il loro effetto può non essere cumulativo.[21] Sia l’esercizio che la dieta ipocalorica causano la perdita di grasso viscerale, ma l’esercizio ha un effetto maggiore sul grasso viscerale rispetto al grasso totale. [22] L’esercizio fisico ad alta intensità è un modo per ridurre efficacemente il grasso addominale totale.[23][24] Una dieta ipocalorica combinata con l’esercizio fisico riduce il grasso corporeo totale e il rapporto tra tessuto adiposo viscerale e tessuto adiposo sottocutaneo, suggerendo una mobilitazione preferenziale del grasso viscerale rispetto al grasso sottocutaneo.[25] Il grasso addominale è fortemente soggetto alle variabili dell’Insulino-resistenza/sensibilità.
Grasso Sottocutaneo: La maggior parte del grasso non viscerale rimanente si trova appena sotto la pelle, in una regione chiamata ipoderma.[26] Questo grasso sottocutaneo non è correlato a molte delle classiche patologie legate all’obesità, come le malattie cardiache, il cancro e l’ictus, e alcune prove suggeriscono addirittura che potrebbe essere protettivo.[27] Il modello tipicamente femminile (o ginecoide) di distribuzione del grasso corporeo intorno ai fianchi, alle cosce e ai glutei è costituito da grasso sottocutaneo, e quindi rappresenta un rischio minore per la salute rispetto al grasso viscerale.[28][29]
Come tutti gli altri organi adiposi, il grasso sottocutaneo è parte attiva del sistema endocrino e secerne gli ormoni Leptina e Resistina.[26]
La relazione tra lo strato adiposo sottocutaneo e il grasso corporeo totale di una persona viene spesso modellata utilizzando equazioni di regressione. La più popolare di queste equazioni è stata creata da Durnin e Wormersley, che hanno testato in modo rigoroso molti tipi di dermoprotezione e, di conseguenza, hanno creato due formule per calcolare la densità corporea di uomini e donne. Queste equazioni presentano una correlazione inversa tra le pieghe cutanee e la densità corporea: all’aumentare della somma delle pieghe cutanee, la densità corporea diminuisce.[30]
Fattori come il sesso, l’età, le dimensioni della popolazione o altre variabili possono rendere le equazioni non valide e inutilizzabili e, a partire dal 2012, le equazioni di Durnin e Wormersley rimangono solo stime del reale livello di grassezza di una persona. Nuove formule sono ancora in fase di creazione.[30]
Gli adipociti del grasso sottocutaneo sono il target degli sforzi di manipolazione dietetica, allenante e supplementativa per ridurre al massimo la percentuale di grasso corporeo. Vi sono comunque aree di distribuzione del grasso sottocutaneo con tassi di mobilitazione lipidica differenti tra gli individui. Ed è proprio in riferimento alle aree di più difficile mobilitazione che ci si riferisce con il termina “grasso ostinato” .
- Fisiologia del tessuto adiposo:
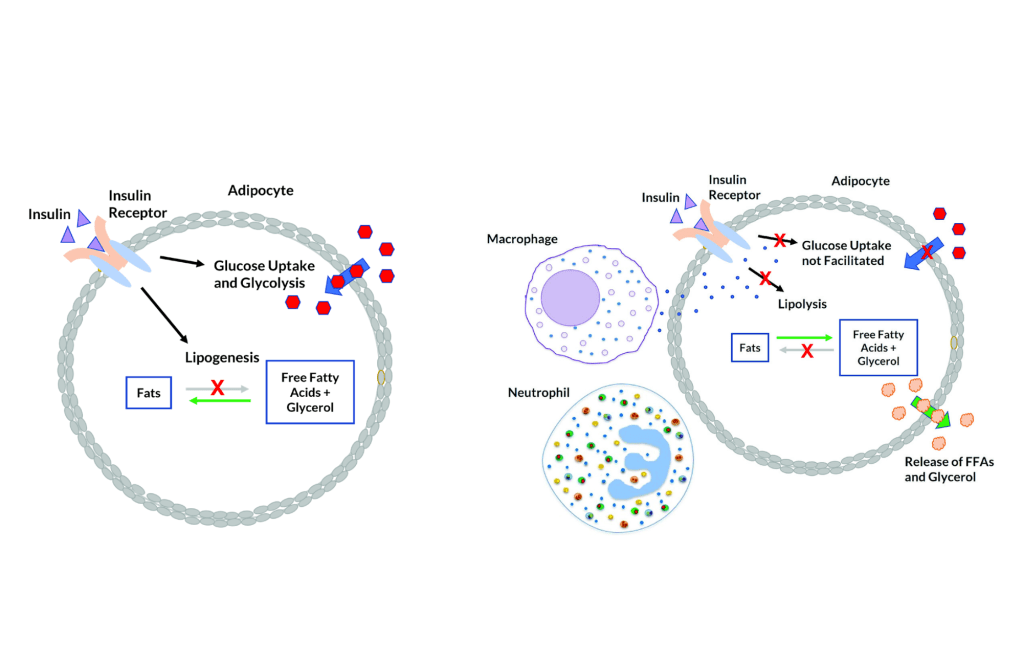
Gli acidi grassi liberi (FFA) vengono rilasciati dalla lipoproteina lipasi (LPL) ed entrano nell’adipocita, dove vengono riassemblati in trigliceridi mediante esterificazione con il glicerolo.[2] Il tessuto adiposo umano contiene circa l’87% di lipidi.[31]
Esiste un flusso costante di FFA che entrano ed escono dal tessuto adiposo.[2] La direzione netta di questo flusso è controllata dall’insulina e dalla leptina: se l’insulina è elevata, c’è un flusso netto di FFA verso l’interno e solo quando l’insulina è bassa gli FFA possono lasciare il tessuto adiposo. La secrezione di Insulina è stimolata dall’aumento della glicemia, dagli AA insulinogenici e in piccola parte dai grassi.[32]
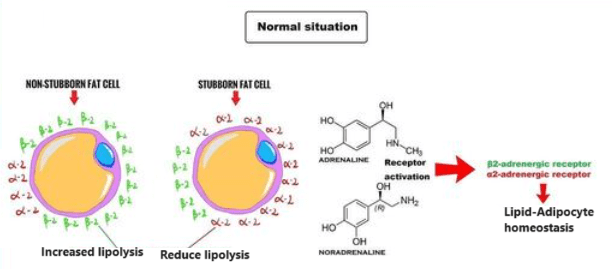
Nell’uomo, la lipolisi (idrolisi dei trigliceridi in acidi grassi liberi) è controllata attraverso il settaggio equilibrato dei recettori β-adrenergici lipolitici e dell’antilipolisi mediata dai recettori α2A-adrenergici.
L’equilibrio tra β2 e α2A-AR determina le caratteristiche peculiari dell’adipocita in termini di lisi dei trigliceridi di deposito (perdita di grasso). Infatti, se l’equilibrio tende a perdersi in favore dei α2A-AR a discapito dei β2-AR ci troviamo di fronte al già prima citato “grasso testardo”.
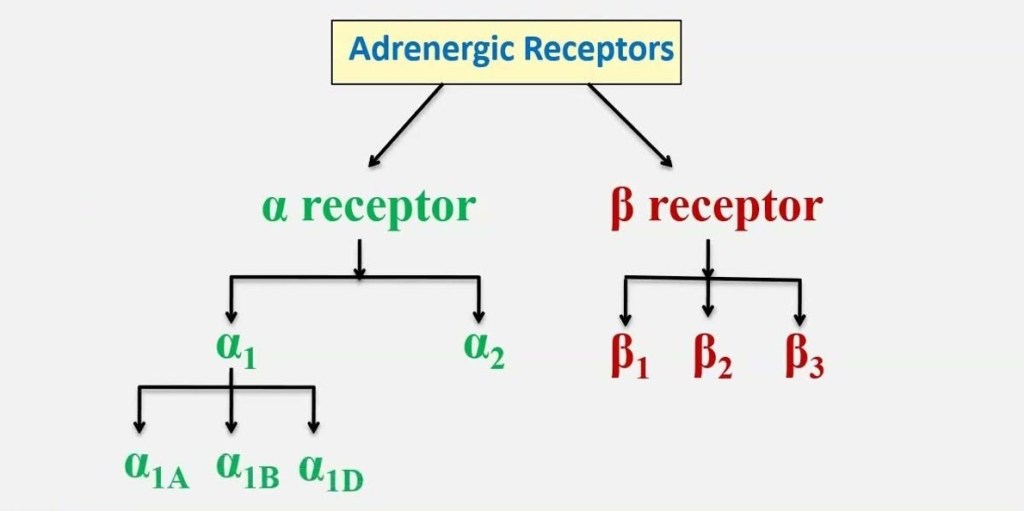
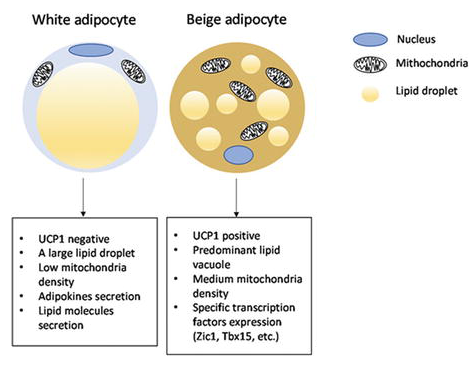
- Distribuzione degli Adrenocettori negli adipociti bianchi, bruni e beige
Gli adipociti bianchi sono il tipo di adipocita predominante nell’organismo e sono localizzati in depositi WAT distinti, caratterizzati da grasso intra-addominale (grasso viscerale che circonda gli organi interni, ovvero grasso mesenterico, perirenale e gonadico) o sottocutaneo (come il grasso inguinale). Gli adipociti bianchi immagazzinano energia (glucosio e acidi grassi) sotto forma di trigliceridi all’interno di un’unica goccia lipidica e il WAT agisce anche come organo endocrino per il rilascio di adipochine come la leptina e l’adiponectina che regolano l’omeostasi energetica dell’intero corpo (Galic, Oakhill, & Steinberg, 2010).
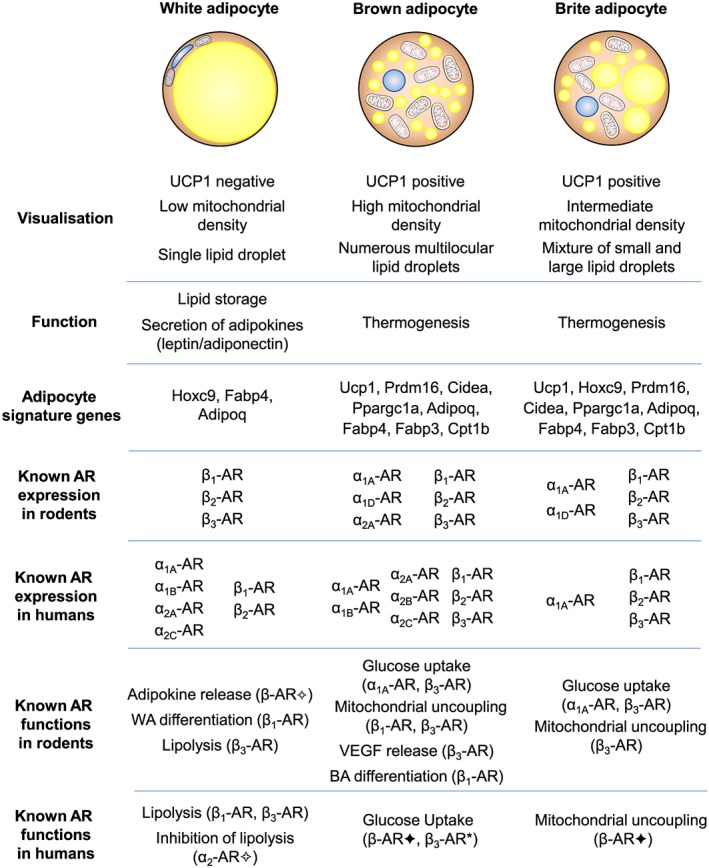
Nei roditori, tutti e tre i sottotipi di β-adrenocettori sono espressi in una serie di depositi sottocutanei e viscerali (Collins et al., 1994; Collins, Daniel, & Rohlfs, 1999; Germack, Starzec, Vassy, & Perret, 1997; Granneman, 1992; Hollenga & Zaagsma, 1989; Komai et al, 2016; Llado et al., 2002; Susulic et al., 1995), con il β3-adrenocettore che è il principale recettore responsabile della lipolisi mediata dal β-adrenocettore negli adipociti bianchi maturi. L’espressione del β-adrenocettore è influenzata anche dallo stato di differenziazione dell’adipocita bianco. L’agonista generale dei β-adrenocettori, l’Isoprenalina, ma non l’agonista altamente selettivo dei β3-adrenocettori, il CL316243, aumenta la proliferazione dei preadipociti, suggerendo un ruolo mediato dai β1-adrenocettori, mentre sia i β1-adrenocettori che i β3-adrenocettori mediano la lipolisi negli adipociti maturi (Germack et al., 1997; Klaus, Seivert, & Boeuf, 2001; Louis, Jackman, Nero, Iakovidis, & Louis, 2000; Susulic et al., 1995). È stato escluso un ruolo del β2-adrenocettore utilizzando antagonisti e agonisti selettivi del recettore.

Questi studi dimostrano collettivamente che i β-adrenocettori sono essenziali per la funzione del WAT, ma che esistono meccanismi di compensazione quando manca il β3-adrenocettore. Non ci sono prove convincenti di un contributo funzionale da parte degli α1- o α2-adrenocettori negli adipociti bianchi autentici dei roditori (Merlin, Sato, Nowell, et al., 2018). Le conoscenze sulla regolazione dell’adiponectina da parte degli adrenocettori sono meno numerose. L’adiponectina, una seconda adipochina secreta dagli adipociti bianchi e bruni, regola l’assorbimento del glucosio, la lipogenesi, la lipolisi e l’ossidazione degli acidi grassi in diversi tessuti, compreso il WAT, in modo autocrino.
Negli esseri umani, l’α1A-adrenocettore mostra una forte espressione in tutti i campioni adulti nativi, ma un’espressione trascurabile negli adipociti coltivati. Al contrario, l’mRNA per l’α1B-adrenocettore è osservato nei tessuti nativi ma anche negli adipociti differenziati di tutti i depositi, mentre l’espressione dell’α1D-adrenocettore è estremamente bassa sia nei tessuti che nelle colture primarie. L’α2A-adrenocettore mostra una forte espressione nei depositi di WAT adulto, un’espressione molto più bassa nel BAT e un’espressione bassa ma significativa nelle colture di adipociti umani maturi. L’espressione dell’α2B-adrenocettore è massima nel BAT fetale, mentre quella dell’α2C-adrenocettore è elevata nel WAT adulto e nel BAT fetale. Livelli significativi di mRNA di α2C-adrenocettori sono osservati anche negli adipociti bruni interscapolari fetali in coltura. Come accennato in precedenza, esiste un’ampia letteratura sul ruolo dei β-adrenocettori nel tessuto adiposo animale; è quindi interessante che tutti e tre i recettori siano espressi nei depositi adiposi umani nativi. Gli mRNA dei β1- e β2-adrenocettori sono presenti in tutti i depositi del BAT e del WAT, mentre l’mRNA del β3-adrenocettore è espresso principalmente nel BAT sopraclaveare adulto. Come altri marcatori termogenici, il numero di β3-adrenocettori è aumentato nel BAT sovraclaveare di un soggetto esposto al freddo (Chondronikola et al., 2016).
Rapporti precedenti hanno utilizzato la RT-PCR per dimostrare l’espressione dei β3-adrenocettori nel WAT, sebbene i segnali fossero costantemente più elevati nel BAT infantile o nel BAT perirenale (Krief et al., 1993; Lonnqvist et al., 1993; Tavernier et al., 1996). Il riscontro costante di una bassissima espressione di β3-adrenocettori nel WAT, sia da RT-PCR che da RNA-Seq, suggerisce che potrebbero esistere sottopopolazioni minori di cellule positive ai β3-adrenocettori nei depositi di WAT umano.
Le colture di adipociti derivate dalla SVF di depositi adiposi umani mostrano un’espressione trascurabile dei β3-adrenocettori, anche dopo il differenziamento in presenza di cocktail altamente adipogenici (Ding et al., 2018; Shinoda et al., 2015). L’mRNA del β1-adrenocettore è trascurabile anche negli adipociti umani primari, mentre il β2-adrenocettore è espresso nelle colture differenziate con valori medi di frammenti per kilobase per milione di reads di 1,8 (adipociti bruni sopraclavicolari) e 2,2 (adipociti bianchi sottocutanei). La mancanza di espressione dei β3-adrenocettori si verifica parallelamente a bassi livelli di mRNA per PPARGC1A, CPT1B e UCP1, tutti elementi centrali per il controllo cellulare della termogenesi. Ciò suggerisce che la differenziazione di adipociti bruni o beige termogenici è difficile da ottenere sperimentalmente negli adipociti umani primari derivati dalla SVF. Shinoda et al. (2015) hanno osservato che la differenziazione di colture clonali di adipociti bruni sopraclavicolari in presenza di 1 μM di Rosiglitazone e/o il trattamento degli adipociti maturi con 10 μM di Forskolina per 4 ore era sufficiente a indurre livelli di espressione di UCP1 simili a quelli osservati nelle biopsie scBAT native, come osservato nelle colture di adipociti bruni e beige di topo (Merlin, Sato, Chia, et al., 2018). Questo tipo di induzione potrebbe essere necessaria per promuovere l’espressione dei β3-adrenocettori, di PPARGC1A e di CPT1B.

L’espressione a basso livello dei sottotipi di β-adrenocettori è stata rilevata mediante qPCR nelle cellule staminali umane multipotenti di derivazione adiposa, con un rapporto di 3:12:1 per i β1:β2:β3-adrenocettori (Mattsson et al., 2011), ma solo gli agonisti dei β1- e β3-adrenocettori aumentano i livelli di mRNA e di proteina di UCP1 in queste cellule (Mattsson et al., 2011). Le cellule differenziate SGBS e PAZ6 sono state analizzate mediante RNA-Seq (Guennoun et al., 2015). L’espressione del β3-adrenocettore non è rilevabile nelle cellule SGBS, ma è significativa nelle cellule PAZ6 differenziate (2,5 RPKM (reads per kilobase per million mapped reads); Guennoun et al., 2015). È quindi evidente che i livelli di espressione degli adrenocettori e dei marcatori termogenici devono essere considerati in diversi sistemi modello quando si studiano potenziali agenti di “inbrunenti”.
Il WAT umano e gli adipociti bianchi dei roditori differiscono significativamente nell’espressione degli α2-adrenocettori, con un’alta espressione degli α2-adrenocettori nel WAT umano (Galitzky, Larrouy, Berlan, & Lafontan, 1990; Mauriege et al, 1991; Mauriege, Marette, et al, 1995; Mauriege, Prud’homme, Lemieux, Tremblay, & Despres, 1995), ma bassa espressione negli adipociti bianchi dei roditori (Merlin, Sato, Nowell, et al. , 2018; Valet et al., 2000). Ormai sappiamo che l’attivazione di α2-adrenocettori accoppiati a Gαi/o negli adipociti bianchi umani inibisce gli aumenti della lipolisi stimolati dalle catecolamine, contrastando così la lipolisi mediata dai β-adrenocettori (Stich et al, 1999), e gli adipociti bianchi degli esseri umani obesi presentano livelli aumentati di α2-adrenocettori, aumento di α2: β-adrenocettori e un aumento delle risposte mediate dagli α2-adrenocettori (Galitzky et al, 1990; Mauriege et al. , 1991; Mauriege, Marette, et al., 1995; Mauriege, Prud’homme, et al., 1995). Quando l’α2-adrenocettore umano è sovraespresso nel tessuto adiposo di topi KO con β3-adrenocettore, la lipolisi mediata dalla catecolamina negli adipociti bianchi è attenuata e i topi sviluppano una maggiore obesità con una dieta ad alto contenuto di grassi (Valet et al., 2000). Nonostante l’espressione significativa degli α1A- e α1B-adrenocettori nel tessuto adiposo umano nativo, non vi sono prove funzionali convincenti di un’attività diretta delle catecolamine.
- “Stubborn Fat”
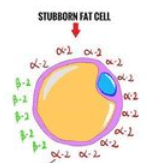
I due tipi di adrenocettori sopra citati, non controllano solo il metabolismo delle cellule grasse, ma anche il flusso sanguigno in entrata e in uscita da queste ultime. Di conseguenza, i β2-AR aumentano la lipolisi e il flusso sanguigno del tessuto adiposo mentre i α2A-AR inibiscono la lipolisi e il flusso sanguigno del tessuto adiposo.
Quindi, le diverse aree del grasso corporeo hanno una diversa distribuzione degli adrenorecettori β2 e α2A e questo influisce profondamente sulla capacità o meno di mobilitare e trasportare il grasso al di fuori di esse.
L’esempio più estremo è quello del grasso corporeo inferiore (fianchi e cosce), in cui è stato riscontrato un numero di recettori α2A circa 9 volte maggiore rispetto ai recettori β2. Alcune ricerche suggeriscono che il grasso addominale degli uomini ha una maggiore densità di recettori α2A (rispetto, ad esempio, al grasso viscerale), anche se non è così accentuato come per il grasso corporeo inferiore. Sebbene non sia stato studiato, è probabile che anche il grasso della parte inferiore della schiena sia relativamente resistente agli stimoli lipolitici, a causa di un numero maggiore di recettori α2A.
I dismorfismi sessuali sulla ripartizione calorica sembrano mostrare che nelle donne, dopo un pasto, può verificarsi una distribuzione calorica preferenziale nel grasso dell’area inferiore del corpo, oltre ad una ridistribuzione del grasso dalla parte superiore a quella inferiore del corpo.
Non è raro, infatti, che le donne lamentino una perdita sensibile nella parte superiore del corpo con una concomitante ed apparente peggioramento dei depositi adiposi nella parte inferiore. Una donna potrebbe mobilitare bene il grasso della parte superiore del corpo, ma immagazzinare parte di quel grasso nei depositi della parte inferiore del corpo. La parte superiore del corpo diventa più magra, quella inferiore più grassa. Questa possibilità può interessare a diverso grado anche gli uomini.
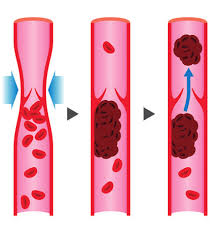
Come accennato in precedenza, oltre alle differenze nella reattività agli stimoli lipolitici, i depositi di “grasso testardo” hanno un flusso sanguigno significativamente più scarso rispetto ad altri depositi.
Alcuni studi hanno dimostrato che il flusso sanguigno nella parte inferiore del corpo può essere inferiore del 67% rispetto ad altri depositi. Il grasso viscerale ha un flusso sanguigno estremamente buono e viene mobilitato molto rapidamente.
La scarsa circolazione sanguigna ha due conseguenze importanti. In primo luogo, significa che gli ormoni trasportati dal sangue non possono raggiungere a concentrazioni ottimali le cellule adipose. In secondo luogo, un flusso sanguigno insufficiente rende più difficile far uscire il grasso mobilitato dalla cellula grassa per ossidarlo altrove.
Il motivo per cui il flusso sanguigno è così scarso non è ben definito. In parte potrebbe trattarsi semplicemente di un minor numero di vasi sanguigni, visto che gli studi di imaging ne mostrano pochi in quell’area. Inoltre, sembra che i vasi sanguigni della parte inferiore del corpo abbiano più recettori α2A che β2; ciò ha la stessa conseguenza della lipolisi. Più recettori α2A significano più vasocostrizione e meno vasodilatazione, il che si traduce in un minor flusso sanguigno.
Un fattore da tenere in considerazione è che, l’Estradiolo aumenta direttamente il numero di recettori α2A-adrenergici antilipolitici negli adipociti sottocutanei. L’aumento del numero di recettori α2A-adrenergici causa una risposta lipolitica attenuata delle Catecolamine o delle ammine simpaticomimentiche negli adipociti sottocutanei; al contrario, non è stato osservato alcun effetto degli estrogeni sull’espressione dell’mRNA dei recettori α2A-adrenergici negli adipociti del deposito di grasso intra-addominale.
Questi risultati mostrano che una cattiva gestione degli estrogeni abbassa la risposta lipolitica nel deposito di grasso sottocutaneo aumentando il numero di recettori α2A-adrenergici antilipolitici, mentre gli estrogeni non sembrano influenzare la lipolisi negli adipociti del deposito di grasso intra-addominale. Si è scoperto che questo effetto degli estrogeni è causato dal sottotipo α del recettore degli estrogeni (ERα).
Questi risultati dimostrano che una sovraespressione estrogenica attenua la risposta lipolitica attraverso la sovra-regolazione del numero di recettori α2A-adrenergici antilipolitici solo nel sottocutaneo e non nei depositi di grasso viscerale. Ciò rappresenta una spiegazione del modo in cui gli estrogeni mantengono la tipica distribuzione del grasso femminile nel sottocute, poiché gli estrogeni sembrano inibire la lipolisi solo nei depositi sottocutanei, spostando così l’assimilazione del grasso dai depositi intra-addominali a quelli sottocutanei peggiorando la situazione dei depositi di “grasso testardo” pre-esistenti e “generandone” di nuovi.
Antagonisti degli α2-AR:
Gli α2 bloccanti sono un sottoinsieme della classe dei farmaci α-bloccanti e sono antagonisti del recettore adrenergico α2. Sono utilizzati principalmente nella ricerca, avendo trovato un’applicazione clinica limitata nella medicina umana. Gli α2 bloccanti aumentano il rilascio di Noradrenalina e bloccano, per l’appunto, l’attività recettoriale degli α2-AR.
La Yohimbina, storicamente utilizzata come afrodisiaco, è talvolta impiegata in medicina veterinaria (anche se ora è stata ampiamente sostituita dall’atipamezolo) per invertire gli effetti degli α2-AR, come la Medetomidina, utilizzati come sedativi durante gli interventi chirurgici.[33]
Gli antidepressivi tetraciclici Mianserina e Mirtazapina sono α2-bloccanti , anche se la loro efficacia come antidepressivi può derivare dalla loro attività su altri siti recettoriali.
Meccanicamente, i α2-bloccanti aumentano i neurotrasmettitori adrenergici, dopaminergici e serotoninergici e inducono la secrezione di Insulina, riducendo i livelli di zucchero nel sangue.
La sospensione repentina degli α2-bloccanti può essere difficile o pericolosa, poiché la sottoregolazione globale dei neurotrasmettitori può causare sintomi di depressione e altri problemi neurologici, e l’aumento dei livelli di zucchero nel sangue insieme alla diminuzione della sensibilità all’insulina può causare in alcuni casi stati diabetici. Inoltre, può verificarsi una riduzione della microcircolazione insieme alla supersensibilità all’adrenalina in organi come il fegato.
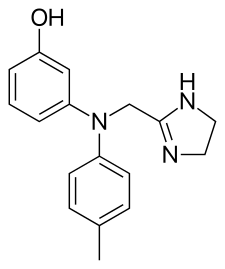
- Yohimbina e α-yohinbina
Non vi è dubbio che la Yohimbina rappresenti l’α2-antagonista più usato per ridurre il grasso corporeo e, nello specifico, le zone del “grasso testardo”.
Se assunta alla dose raccomandata (≤0,2mg per kg di peso corporeo), la Yohimbina può causare nausea, dolore addominale, vertigini, nervosismo e ansia.[34]
Dosi più elevate di Yohimbina possono essere pericolose; un rapporto del 2005 ha rilevato che la Yohimbina ha il più alto tasso di effetti tossici di qualsiasi prodotto botanico.[35] Casi di ingestione di Yohimbina in eccesso hanno suggerito che l’ansia, l’ipertensione (pressione alta), la tachicardia (frequenza cardiaca elevata), le aritmie e l’agitazione sono tra gli effetti collaterali più gravi di questo composto.[35]
La Yohimbina è un α2-antagonista adrenergico selettivo. In altre parole, ha come bersaglio e inattiva una classe di recettori del sistema nervoso che risponde al neurotrasmettitore Noradrenalina.[36] L’antagonismo dei recettori α2 aumenta il rilascio di Noradrenalina da parte del sistema nervoso simpatico, causando gli effetti stimolanti e “iperadrenergici” della Yohimbina.
La Yohimbina inibisce anche l’attività dei recettori α2 sulle cellule adipose, dove la Noradrenalina agisce normalmente per sopprimere il rilascio di grasso. L’inibizione dell’effetto antilipolitico della Noradrenalina consente una maggiore lipolisi (e conseguente ossidazione lipidica).[37]
Dosi giornaliere totali di 0,2mg/kg di peso corporeo sono state utilizzate con successo per aumentare la mobilitazione lipidica dai depositi di “grasso testardo” e la successiva ossidazione dei grassi senza implicazioni significative sui parametri cardiovascolari come la frequenza cardiaca e la pressione sanguigna. Ciò si traduce in un dosaggio giornaliero totale di:
- 14 mg per una persona di 68 kg
- 18 mg per una persona di 91 kg
- 22 mg per una persona di 113 kg.
Queste dosi totali giornaliere si riferiscono all’uso di Yohimbina come unico agente con azione riduttiva sulla attività dei recettori α2. Tali dosaggi vengono spesso suddivise e assunte in due o quattro dosi nel corso della giornata. Ad esempio, una persona di 68 kg potrebbe assumere 7mg due volte al giorno (lontano dai pasti) per raggiungere una dose totale di 14mg.
Nota: non tutti i soggetti sono in grado di tollerare la “dose piena” ricavata dalla sopra citata formula. In quel caso, l’utilizzatore mantiene la tose tollerabile raggiunta.
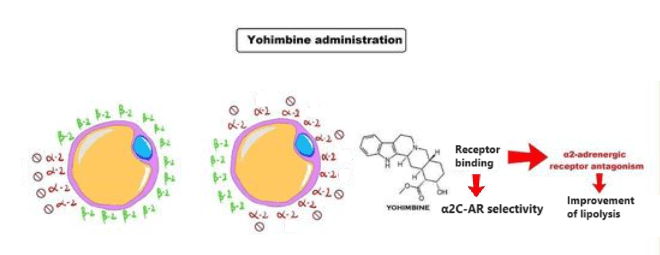
Se si considera lo stesso recettore α2, la Yohimbina sembra avere una selettività per la subunità α2C piuttosto che per la A o la B; la selettività è compresa tra 4 e 15 volte,[38] mentre la Rauwolscina [α-yohimbina] sembra non essere selettiva tra queste tre subunità.[39][38] La Rauwlscina sembra essere efficace a livello del recettore quanto la Yohimbina ma con una emivita di circa 5h contro i 30 minuti della prima emivita della Yohimbina.[40]
Il fatto che la Yohimbina è selettiva per la subunità α2C più che per altre subunità, compresa l’importante A, se parliamo di α2-AR adipocitari, la sua efficacia risulta moderatamente ridotta per la riduzione del “grasso testardo”, sebbene la subunità α2C sia ad un certo grado espressa anche nel WAT; o per lo meno lo è se utilizzata come unico agente interferente l’attività adipocitaria dei α2-AR.
Introduzione agli ACE II inibitori:
Leonard T. Skeggs e i suoi colleghi (tra cui Norman Shumway) scoprirono l’ACE [Inibitori dell’enzima di conversione dell’angiotensina] nel plasma nel 1956.[41] Le scoperte avvenute nel corso di un annosa ricerca hanno portato allo sviluppo del Captopril, il primo ACE-inibitore attivo per via orale, nel 1975.[42]
Gli ACE inibitori inibiscono l’attività dell’enzima di conversione dell’angiotensina, un componente importante del sistema renina-angiotensina che converte l’angiotensina I in angiotensina II e idrolizza la bradichinina.[43] Pertanto, gli ACE inibitori diminuiscono la formazione di angiotensina II, un vasocostrittore, e aumentano il livello di bradichinina, un vasodilatatore peptidico.[43] Questa combinazione è sinergica nell’abbassare la pressione sanguigna.
Gli ACE-inibitori riducono l’attività del sistema Renina-Angiotensina-Aldosterone (RAAS) come evento eziologico (causale) primario nello sviluppo dell’ipertensione nelle persone con diabete mellito, come parte della sindrome da insulino-resistenza o come manifestazione di una malattia renale.[44][45]

Il sistema renina-angiotensina-aldosterone è un importante meccanismo di regolazione della pressione sanguigna. I marcatori di squilibrio elettrolitico e idrico nell’organismo, come l’ipotensione, la bassa concentrazione di sodio nel tubulo distale, la diminuzione del volume sanguigno e l’elevato tono simpatico, innescano il rilascio dell’enzima renina dalle cellule dell’apparato juxtaglomerulare del rene.

La renina attiva un proormone circolante derivato dal fegato, l’angiotensinogeno, mediante scissione proteolitica di tutti i suoi residui aminoacidici, tranne i primi dieci, noti come angiotensina I. L’ACE (enzima di conversione dell’angiotensina) rimuove quindi altri due residui, convertendo l’angiotensina I in angiotensina II. L’ACE si trova nella circolazione polmonare e nell’endotelio di molti vasi sanguigni.[46] Il sistema aumenta la pressione sanguigna aumentando la quantità di sale e acqua trattenuta dal corpo, sebbene l’angiotensina II sia anche un potente vasocostrittore.[47]

Gli ACE-inibitori sono stati inizialmente approvati per il trattamento dell’ipertensione e possono essere utilizzati da soli o in combinazione con altri farmaci antipertensivi. In seguito, si sono rivelati utili per altre malattie cardiovascolari e renali[48], tra cui:
- Infarto miocardico acuto (attacco cardiaco)[49]
- Insufficienza cardiaca (disfunzione sistolica ventricolare sinistra)[50]
- Complicanze renali del diabete mellito (nefropatia diabetica), grazie alla riduzione della pressione arteriosa e alla prevenzione del danno da iperfiltrazione glomerulare[51].
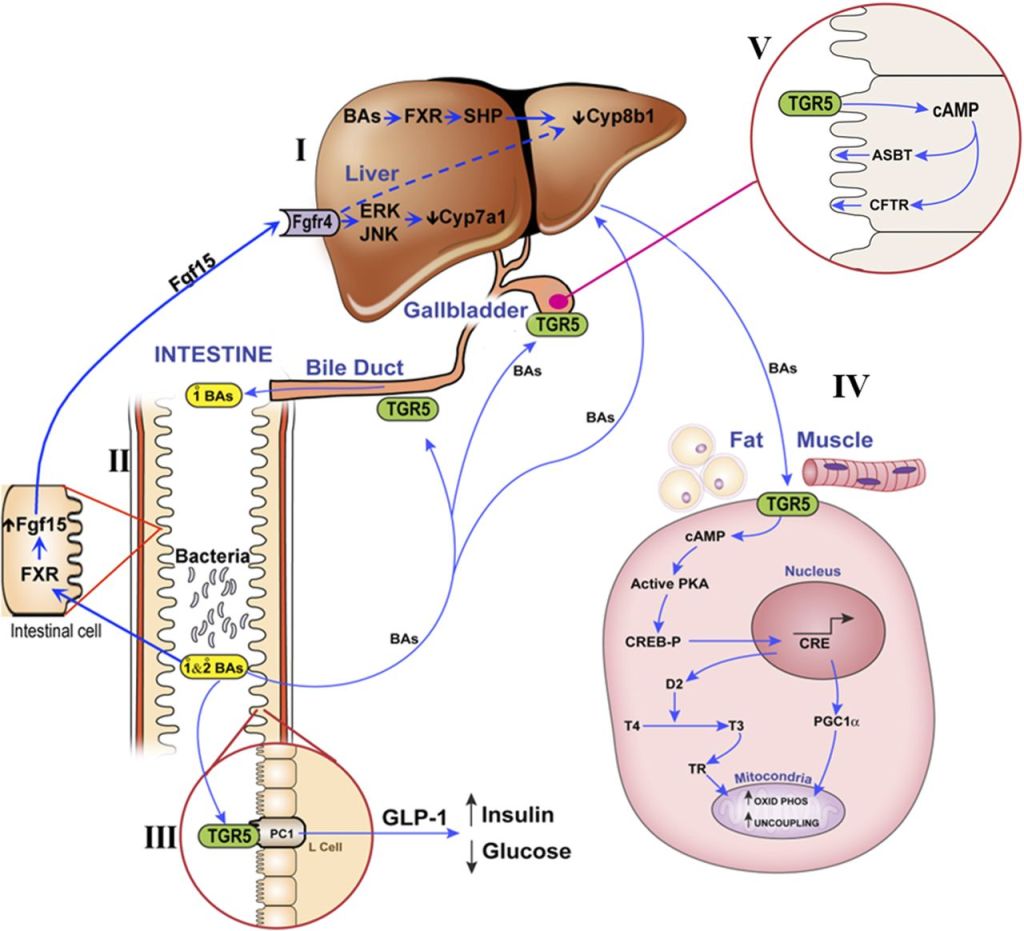
Angiotesina II e tessuto adiposo:
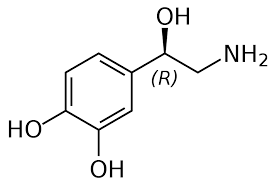
L’angiotensina II determina, tra le atre cose, un aumento del rilascio di catecolamine (Noradrenalina), della sensibilità alle catecolamine e della loro attività.[52]
L’angiotensina II può essere prodotta dal tessuto adiposo umano; a questo proposito, l’angiotensinogeno e gli enzimi coinvolti nella sua conversione in Ang II, nonché le vie RAS (renina, enzima di conversione dell’angiotensina: ACE) e non RAS (catepsina D, catepsina G) sono espressi nel tessuto adiposo umano. Inoltre, anche i recettori dell’Ang II sono espressi nel tessuto adiposo, il che suggerisce un ruolo locale di questo ormone nella regolazione dell’adipogenesi, del metabolismo lipidico e nella patogenesi dell’obesità28,48. L’influenza dell’Ang II sugli adipociti è mediata dall’attivazione dei recettori АТ1 e АТ2, coinvolgendo diversi sistemi di trasduzione del segnale, tra cui le risposte Са 2+, la proliferazione e la differenziazione cellulare, l’accumulo di trigliceridi, l’espressione dei geni delle adipochine e la secrezione di queste ultime [53]. L’angiotensina II ha anche un effetto anti-adipogenico, riducendo la differenziazione delle cellule pre-adipose umane [54]. Pertanto, questo ormone potrebbe rappresentare un fattore protettivo contro l’espansione incontrollata del tessuto adiposo [55].Questo effetto anti-adipogenico dell’Ang II è stato osservato anche nel grasso omentale di esseri umani affetti da obesità, con la partecipazione della via della chinasi regolata dal segnale extracellulare/1,2 (ERK/1,2) e la fosforilazione del recettore gamma attivato dal proliferatore del perossisoma (pPARG) [56]. Durante questo processo, l’origine dell’Ang II può essere sia da RAS che da vie non RAS; queste ultime potrebbero essere più importanti in questo processo [57]. Tuttavia, oltre a questo effetto, l’Ang II può aumentare il contenuto di trigliceridi e l’attività di due enzimi lipogenici (FAS: sintasi degli acidi grassi e GPDH: glicerolo-3-fosfato deidrogenasi) in colture primarie di cellule adipose umane, suggerendo un controllo dell’adiposità attraverso la regolazione della sintesi e dell’immagazzinamento dei lipidi negli adipociti [58]. L’Ang II regola anche il flusso sanguigno regionale verso il tessuto adiposo e le dimensioni e il numero delle cellule grasse [59]. Queste scoperte sono state confermate dal blocco sperimentale dell’Ang II, che influenza direttamente il peso corporeo e l’adiposità [60].

È stata documentata anche la regolazione autocrina dell’Ang II durante l’adipogenesi. L’angiotensina II può essere catabolizzata nei tessuti adiposi dall’enzima adiposo di conversione dell’angiotensina 2 (ACE2) per formare l’Ang 1-7. La regolazione autocrina del sistema angiotensinico locale implica la coespressione dei recettori dell’Ang II (AT1 e AT2) e dei recettori dell’Ang 1-7 (Mas) sugli adipociti. L’attivazione del recettore Mas da parte dell’Ang 1-7 ha un effetto contrario all’effetto anti-adipogenico dell’Ang II, inducendo l’adipogenesi attraverso l’attivazione delle vie PI3K/Akt e l’inibizione delle vie MAPK chinasi/ERK [61] . In questo contesto, la regolazione autocrina dell’asse Ang II/AT1-ACE2-Ang 1-7/Mas durante l’adipogenesi è in grado di produrre ormoni e citochine che promuovono l’infiammazione, l’accumulo di lipidi, l’IR e le componenti del RAS, che si attivano in presenza di obesità come meccanismi chiave correlati all’obesità dell’ipertensione e di altre componenti della sindrome cardiometabolica [62].
- Angiotesina II e α2A-AR
Una caratteristica di particolare interesse in riferimento all’Angiotesina II è il fatto che sia un polipeptide necessario per l’espressione di alcuni recettori α2 (ma non di tutti). Ciò significa che senza l’Angiotensina II i recettori α2 non possono essere sviluppati in alcune cellule. Di conseguenza, se sottoregoliamo l’Angiotensina II, prodotta naturalmente dall’organismo, il normale rinnovamento dei recettori α2 non avverrà. Bisogna capire che in ogni cellula c’è un costante rinnovamento recettoriale. Bloccando la formazione di un tipo specifico di recettore in una cellula (ad esempio i recettori α2), dopo un po’ di tempo non ci saranno più recettori α2 in questa cellula. I vecchi recettori saranno completamente degradati e avremo impedito alla nuova generazione di recettori di sostituire quelli vecchi.

Quindi, sotto-regolazione marcata dei α2 recettori . Il problema principale è se questa azione dell’Angiotensina II avviene nelle cellule adipose. L’Angiotensina II agisce solo sui recettori α2 che rispondono a due condizioni:
- Sembra avere il massimo effetto sui recettori α2 del sottotipo “A”. Ciò è positivo, poiché sono proprio questi recettori a trovarsi nelle cellule adipose. Quindi, la prima condizione è soddisfatta.
- L’Angiotensione II agisce solo sulle cellule ricche di recettori α2 e di recettori dell’Angiotensina II. Sappiamo già che le cellule adipose sono molto ricche di recettori α2. Da tempo i ricercatori sanno anche che le cellule adipose sono ricche di recettori dell’Angiotensina II.
Il punto chiave da ricordare è che nelle cellule grasse l’Angiotensina II è necessaria perché i recettori α2 si rinnovino normalmente. Se impediamo in qualche modo la formazione di Angiotensina II, causeremo grossi problemi nel rinnovo dei recettori α2A nelle cellule adipose.
Quindi, tutto ciò che occorre fare è alterare la produzione di Angiotensina II attraverso l’uso principale di ACE II inibitori. Nel giro di poche settimane il numero di recettori α2 diminuirà sensibilmente.
L’uso di ACE II inibitori ha quindi il potenziale di attenuare la sensibilità agli α2-adrenocettori negli adipociti umani. L’effetto del Quinapril, un ACE II inibitore lipofilo, è stato maggiore di quello dell’Enalapril [www.ncbi.nlm.nih.gov/pmc/articles], un ACE II inibitore idrofilo. Gli ACE II inibitori lipofili possono avere un effetto vasodilatatore più potente rispetto agli ACE II inibitori idrofili. La concentrazione di Angiotensina II nei tessuti piuttosto che nel plasma può contribuire alla sensibilità e il numero degli α2-adrenocettori.

È stato riportato che l’ACE inibitore lipofilo, Quinapril, riduce la concentrazione tissutale di Angiotensina II in misura maggiore rispetto all’ACE inibitore idrofilo, Enalapril, da 5 a 24 ore dopo una singola somministrazione orale nei ratti. I tempi di raggiungimento della concentrazione plasmatica massima del Quinapril e del suo metabolita attivo sono stati di 2-3 ore [63, 64]. Pertanto, per esaminare più chiaramente la cosa, ciascun farmaco è stato somministrato 22 e 3 ore prima dell’esame. Entrambi gli ACE inibitori hanno soppresso le attività plasmatiche dell’ACE per oltre il 90%. Questo risultato conferma i precedenti risultati ottenuti in soggetti giapponesi [65]. Sebbene la soppressione dell’attività dell’enzima convertitore dell’Angiotensina nel plasma e la pressione arteriosa sistemica non differissero tra i due farmaci, l’attenuazione della sensibilità degli α-adrenocettori alla Fenilefrina era maggiore nei soggetti trattati con Quinapril rispetto a quelli trattati con Enalapril. Le osservazioni e i rapporti precedenti [66] suggeriscono che la concentrazione di Angiotensina II nei tessuti piuttosto che quella nel plasma può contribuire alla sensibilità dei recettori α-adrenergici nei vasi ed in altri tessuti come quello adiposo. Inoltre, l’ACE inibitore lipofilo può essere più potente dell’ACE inibitore idrofilo. Infatti, il Quinapril ha attenuato la risposta vasopressore della Fenilefrina più dell’Enalapril e l’intervallo di confidenza del 95% per le differenze di ED50 tra Enalapril e Qinapril è stato di 31,1-397,5. Sebbene l’entità dell’attenuazione della sensibilità dei recettori α-adrenergici indotta dalla soppressione dell’ACE tissutale con Quinapril fosse varia, ciò è coerente con un altro esperimento in vitro [67].
Quando osservata, la concentrazione di Noradrenalina nel siero durante il riposo a letto non è cambiata prima e dopo la somministrazione del farmaco ACE inibitore. Rapporti precedenti hanno dimostrato che gli ACE inibitori attenuano il deflusso del nervo simpatico negli animali e nell’uomo [68, 69]. Negli studi in cui non è stato applicato alcun carico al sistema nervoso simpatico, non è stato possibile rilevare alcun cambiamento nel flusso simpatico indotto dagli ACE inibitori.
Applicazione degli ACE II inibitori nel trattamento del “grasso testardo”:
- La genesi dell’uso degli ACEI come PEDs

Nonostante il potenziale maggiore nella sotto-regolazione degli α2A-AR attribuita agli ACE inibitori con caratteristiche prettamente lipolifiche, la molecola appartenente a questa classe di farmaci maggiormente utilizzata per tale scopo e da più tempo è il Captopril. Questo storico ACE II inibitore mostra però caratteristiche idrofile. Certo, la sua maggiore diffusione è legata senza dubbio agli anni dalla sintesi e immissione nel circuito farmaceutico della molecola, ma anche, e soprattutto, al suo lancio come PEDs da parte, tra i primi, di Dan Duchaine (1952-2000).
Le proprietà potenziali sulla composizione corporea del Captopril vennero individuare per la prima volta in alcune atlete interessate ad assumere un farmaco che le desse un miglioramento della composizione corporea ma senza virilizzazione. Così quella divenne l’occasione giusta per testare il Captopril. La dieta delle atlete non venne cambiata. Le atlete hanno continuato per un paio di mesi ad assumere il Captopril come unico farmaco. Avevano migliorato leggermente il trofismo, ma non molto. Ciò che però colpì i “pionieri della preparazione” fu il fatto che avevano perso grasso in aree in cui prima non erano riuscite a perderlo in modo significativo.
Approfondendo le caratteristiche della molecola attraverso la consultazione di testi accademici reperiti alla biblioteca medica, scoprirono che la relazione tra il Captropril e i recettori α2.
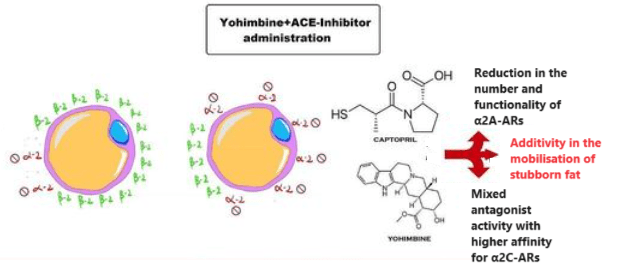
Con il procedere del tempo e le sperimentazione dose-tempo nell’applicazione del Captopril (ma non solo), si è notato che il farmaco poteva rendere possibile la riduzione totale della dose di Yohimbina migliorando notevolmente la compliance dell’utilizzatore.
Sappiamo, infatti, che la Yohimbina presenta una selettività maggiore per i recettori α2C piuttosto che ai sottogruppi “A” e “B”. Questa caratteristica risulta limitativa nell’azione ricercata nella Yohimbina come α2-antagonista adipocitario. L’inserimento del Captopril [o di altro ACE II inibitore] permette di 1) ridurre sensibilmente il numero di α2A-AR nell’adipocita e 2) di permettere, a dosaggio di 1/2 fino a 1/3, un legame antagonista da parte della Yohimbina nei confronti degli α2-AR rimasti. L’uso della α-yohimbina, non presentando tale affinità selettiva, migliora sensibilmente questo effetto sinergico.
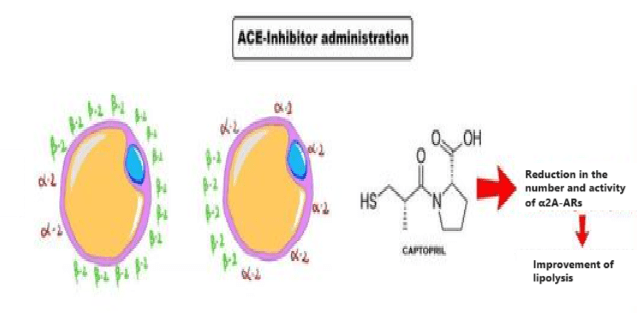
- Le limitazioni degli ACE II inibitori
- Il Captopril [e in generale gli ACE II inibitori] non è un farmaco che manifesta rapidamente i suoi effetti dal punto di vista estetico. Bisogna ricordare che la regolazione degli α2-AR richiede almeno due mesi prima di diventare significativa.
- È necessario seguire una dieta ipocalorica per vedere ottimi risultati in termini di perdita di grasso ostinato. Abbiamo detto, infatti, che i recettori α2-AR impediscono la normale perdita di grasso la dose si presentano in maggiori concentrazioni. Questo non significa, però, che si perderà automaticamente grasso di deposito solo perché si è ridotto il numero di recettori α2. Significa solo che la perdita di grasso ostinato indotta dalla dieta ipocalorica sarà più “facile”. Avrà un effetto permissivo sulla perdita di grasso ostinato, consentendo di ridurre i depositi adiposi con un rapporto di α2-AR più elevato.
- L’ultima limitazione è che esiste ancora una linea di difesa per le cellule adipose e la conservazione delle riserve lipiche. Eliminando parzialmente la linea di difesa rappresentata dagli α2-AR, se ne attiva una nuova costituita da recettori antilipolitici chiamati peptide YY, anch’essi localizzati sulle cellule adipose. Ciò significa che la riduzione del livello dei recettori α2-AR permetterà di perdere più grasso ostinato di quanto sarebbe stato normalmente possibile, ma le limitazioni genetiche saranno sempre presenti.
Ma l’uso di Captopril [o altro ACE II inibitore] può permettere di fare un grande passo avanti nella giusta direzione se l’obbiettivo è una marcata riduzione della body fat, soprattutto le aree ostinate.
- Esempi applicativi degli ACE II inibitori per il trattamento del “Stubborn Fat”
Nell’approccio protocollare di base, e se prendiamo come esempio di ACE II inibitore il Captopril:
- Captopril = 50mg/die [da raggiungere con gradualità e aumenti giornalieri di 6,25mg];
- Yohimbina = 5-10mg/die [dose da raggiungere con aumenti giornalieri (pari a 2,5mg) e test della sensibilità ];
- α-yohimbina = 3/5mg die [dose da raggiungere con aumenti giornalieri (pari a 0,5mg) e test della sensibilità ].
Nell’approccio protocollare intermedio:
- Captorpil = 50-75mg/die [da raggiungere con gradualità e aumenti giornalieri di 6,25mg];
- Yohimbina = 10mg/die [dose da raggiungere con aumenti giornalieri (pari a 2,5mg) e test della sensibilità ];
- α-yohimbina = 5-6mg die [dose da raggiungere con aumenti giornalieri (pari a 0,5mg) e test della sensibilità ];
- T3 = 25mcg/die [dose da raggiungere con aumenti giornalieri (pari a 12,5mcg) e controllo ematico del FT3].
Nell’approccio protocollare avanzato:
- Captorpil = 100mg/die [da raggiungere con gradualità e aumenti giornalieri di 6,25mg];
- Yohimbina = 0.2mg/Kg/die [dose da raggiungere con aumenti giornalieri (pari a 2,5mg) e test della sensibilità ];
- α-yohimbina = 0.1mg/Kg/die [dose da raggiungere con aumenti giornalieri (pari a 0,5mg) e test della sensibilità ];
- T3 = 50mcg/die [dose da raggiungere con aumenti giornalieri (pari a 12,5mcg) e controllo ematico del FT3];
- Salbutamolo = 8-12mg/die [dose da raggiungere con aumenti ogni 1-2 giorni (pari a 2mg)];
- Alternativa: Clenbuterolo = 1mcg/Kg/die (range 40-80mcg) [dose da raggiungere con aumenti ogni 2 giorni (pari a 10-20mcg) e test della sensibilità/tolleranza];
- Nedbivololo = 5mg/die [dose di partenza 2,5mg/die e valutazione della tolleranza].
*Nota bene: Nessuno dei protocolli sopra esposti rappresenta un indicazione d’uso o una prescrizione medica di applicazione. Tali informazioni SONO AD ESCLUSIVO SCOPO ESEMPLIFICATIVO!
- Effetti collaterali degli ACE II inibitori
- pressione bassa;
- tosse. Un altro possibile effetto avverso specifico degli ACE-inibitori, ma non di altri bloccanti del RAAS, è l’aumento del livello di bradichinina. La tosse secca persistente è un effetto avverso relativamente comune che si ritiene sia associato all’aumento dei livelli di bradichinina prodotto dagli ACE inibitori, anche se il ruolo della bradichinina nella produzione di questi sintomi è stato contestato. Tuttavia, molti casi di tosse in persone che assumono ACE inibitori potrebbero non essere dovuti al farmaco stesso. Alcuni (0,7%) sviluppano angioedema a causa dell’aumento dei livelli di bradichinina. Può esistere una predisposizione genetica. ;
- iperkaliemia. Il potassio elevato nel sangue è un’altra possibile complicazione del trattamento con un ACE-inibitore, dovuta al suo effetto sull’aldosterone. La soppressione dell’angiotensina II porta a una diminuzione dei livelli di aldosterone. Poiché l’aldosterone è responsabile dell’aumento dell’escrezione di potassio, gli ACE-inibitori possono causare una ritenzione di potassio. Alcune persone, tuttavia, possono continuare a perdere potassio durante l’assunzione di un ACE-inibitore. È necessario un attento monitoraggio dei livelli di potassio nei soggetti in trattamento con ACE-inibitori che sono a rischio di iperkaliemia.;
- cefalea;
- vertigini;
- affaticamento;
- nausea e compromissione renale. I soggetti che iniziano la terapia con un ACE-inibitore presentano di solito una modesta riduzione della velocità di filtrazione glomerulare (eGFR). Tuttavia, la riduzione può essere significativa in condizioni di preesistente ridotta perfusione renale, come stenosi dell’arteria renale, insufficienza cardiaca, malattia renale policistica o deplezione di volume. Una moderata riduzione della funzione renale, non superiore al 30% di aumento della creatinina sierica, che si stabilizza dopo una settimana di trattamento. La riduzione del eGFR è un problema soprattutto se il paziente assume contemporaneamente un FANS e un diuretico. Quando i tre farmaci vengono assunti insieme, il rischio di sviluppare un’insufficienza renale aumenta notevolmente.
- Una rara reazione allergica grave può colpire la parete intestinale e causare secondariamente dolore addominale.
Ma gli ARB/Sartani possono essere un sostituto agli ACE II inibitori per lo scopo qui discusso?
Sono circa vent’anni che si è scoperto che il Telmisartan, un Bloccante del Recettore dell’Angiotensina II (ARB) approvato per il trattamento dell’ipertensione, è anche un agonista parziale di PPARγ.[70-71] Mentre gli agonisti completi di PPARγ, come il Rosiglitazone e il Pioglitazone, promuovono l’aumento di peso alterando la distribuzione del grasso e la differenziazione degli adipociti, gli agonisti parziali (agonisti/antagonisti misti) di PPARγ possono avere la capacità di ritardare l’aumento di peso promuovendo al contempo la differenziazione degli adipociti.[72] Ad esempio, è stato scoperto che il Telmisartan può promuovere la differenziazione degli adipociti ma anche attenuare l’aumento di peso, migliorando al contempo il metabolismo del glucosio e dei lipidi nei ratti alimentati con una dieta ad alto contenuto di grassi e carboidrati.[70] Sharma et al[73] hanno riportato che il blocco del recettore dell’angiotensina II di tipo 1, di per sé, può promuovere la differenziazione degli adipociti e hanno proposto che questo possa contribuire agli effetti antidiabetici degli antagonisti del recettore dell’angiotensina II. Non è noto se molecole bifunzionali come il Telmisartan, che attivano PPARγ e bloccano il recettore dell’angiotensina II, esercitino effetti diversi sulle dimensioni degli adipociti e sui determinanti primari del peso corporeo rispetto ai normali bloccanti del recettore dell’angiotensina, come il Valsartan, che non hanno la capacità di attivare PPARγ.

Negli studi si è scoperto che il Telmisartan, ma non il Valsartan, aumenta l’espressione dei geni di un fattore di trascrizione nucleare (TFAM) che regola la funzione mitocondriale e di una proteina mitocondriale (MTCO1) coinvolta nella fosforilazione ossidativa. Rispetto agli agonisti totali convenzionali di PPARγ, come i Tiazolidinedioni, gli agonisti parziali del PPARγ, come il Telmisartan, possono avere la capacità di reclutare in modo preferenziale alcuni coattivatori trascrizionali che sono particolarmente importanti nella regolazione dei geni che controllano la funzione mitocondriale e il metabolismo energetico.[74-75] Ad esempio, gli agonisti parziali sembrano reclutare preferenzialmente il coattivatore 1-α di PPARγ, un coattivatore trascrizionale noto per stimolare l’espressione di TFAM, che, a sua volta, può aumentare l’espressione dei geni mitocondriali (ad esempio, MTCO1) e, in ultima analisi, la biogenesi mitocondriale.[75-76] Sebbene i precisi meccanismi cellulari e molecolari che mediano i robusti effetti del Telmisartan sul peso corporeo, sul dispendio energetico e sul metabolismo dei grassi rimangano da chiarire, gli studi sul reclutamento del coattivatore PPARγ e sull’espressione dei geni target, nonché sul numero, la struttura e la funzione dei mitocondri, potrebbero rappresentare aree di indagine potenzialmente fruttuose in futuro.
Ciò che si è anche notato con gli ARB, ma soprattutto con il Telmisartan, è che ha una azione sulla distribuzione del grasso più che sulla sua riduzione sistemica. Infatti, il Telmisartan ha mostrato di indurre la riduzione del grasso viscerale ma senza cambiamenti statistici sui deposito sottocutanei. Le più recenti review che hanno esaminato l’effetto del Telmisartan sulla condizione metabolica e composizione corporea dei pazienti trattati, hanno evidenziato che i risultati suggeriscono che questo sartano influisce sulla distribuzione del grasso, inducendo una riduzione del grasso viscerale, e quindi potrebbe essere utile nei pazienti ipertesi con obesità/sovrappeso, sindrome metabolica o intolleranza al glucosio.
Anche i dati aneddotici di un certo valore e design suggeriscono uno “spostamento” nell’equilibrio di mobilitazione delle riserve di grasso verso la perdita dei depositi viscerali invece di quelli sottocutanei. Ed è per tale motivo che diversi preparatori ne evitino l’uso sotto gara.
Questo “effetto shift” sul bilancio della mobilitazione delle riserve di grasso dal grasso sottocutaneo ad una prevalenza del viscerale si manifesta in modo significativo nel range di dosaggio di 80-160mg/die.
L’attività come agonista parziale del PPARγ è il motivo principale per il quale in Telmisartan agisce sul metabolismo lipidico adipocitario. Si è affermato che coloro i quali vogliono bypassare il problema dello shift della mobilitazione adiposa possono farlo assumendo l’Oleuropeina. Ora, non vi è nulla di certo e poco che superi la sottile linea tra ipotesi e dato realmente misurato, ma alcuni, soprattutto coloro i quali mal tollerano gli aumenti di bradichinina dati dagli ACE II inibitori, inseriscono questo supplemento erboristico nel tentativo di risolvere la sopra citata limitazione.
Peccato, però, che grazie a questa attività di agonista parziale del PPARγ, il Telmisartan può ridurre lo stoccaggio dei trigliceridi negli adipociti durante una dieta ipercalorica. In topi trattati per 28 giorni con ARB e ACE I, si è osservato un inferiore accumulo adiposo, minor peso corporeo, miglior controllo sull’assunzione di cibo rispetto ai topi non trattati con una dieta ad alto contenuto lipidico.
Nonostante, in teoria, l’effetto sul “grasso testardo” possa essere trattato anche attraverso il blocca del recettore dell’Angiotesina II, i dati a nostra disposizione ci mostrano una superiorità di azione e versatilità legata agli ACE II inibitori. L’uso di ARB, in particolar modo del Telmisartan, potrebbe avere un applicazione logica (se non si parla di soggetti obesi o in sovrappeso) nel gestione del grasso corporeo durante le fasi di ipercalorica, ad un dosaggio ipotetico di 40-80mg/die, al fine di ridurre l’accumulo adiposo e migliorare la qualità complessiva del peso raggiunto in Bulk.
- Effetti collaterali degli ARB:
- tachicardia e bradicardia (battito cardiaco accelerato o lento);
- ipotensione (pressione sanguigna bassa);
- edema (gonfiore di braccia, gambe, labbra, lingua o gola, quest’ultimo con conseguenti problemi di respirazione);
- potenziale manifestazione di reazioni allergiche;
- infezioni del tratto respiratorio superiore;
- diarrea;
- mal di schiena;
- problemi renali;
- iperkalemia.
Conclusioni:
Abbiamo visto come gli adrenocettori svolgono un ruolo importante nella biologia e nella fisiologia del tessuto adiposo, che comprende la regolazione della sintesi e dell’immagazzinamento dei trigliceridi (lipogenesi), la degradazione dei trigliceridi immagazzinati (lipolisi), la termogenesi (produzione di calore), il metabolismo del glucosio e la secrezione di ormoni derivati dagli adipociti che possono controllare l’omeostasi energetica dell’intero corpo. Questi processi sono regolati dal sistema nervoso simpatico attraverso l’azione di diversi sottotipi di adrenocettori espressi nei depositi di tessuto adiposo. In questa disamina, abbiamo evidenziato il ruolo dei sottotipi di adrenocettori negli adipociti bianchi, bruni e beige, e nel tessuto adiposo “testardo” ed abbiamo approfondito il ruolo potenziale degli ACE II inibitori nella modulazione sottoregolativa dell’attività degli α2-AR e l’impatto che questo può avere sul miglioramento della composizione corporea. Sono stati anche descritti gli effetti riscontrabili, nel medesimo contesto e fine, dei Sartani con le differenze tra l’applicabilità di questi confronto a quella degli ACE II inibitori.
Mentre il potenziale degli ACE II inibitori di migliorare la perdita di massa grassa in specie a carico dei depositi con una ratio sfavorevole tra α2:β2-AR, permettendo un importante sgravio sui dosaggi di α2-antagonisti, risulta un dato importante per la pianificazioni della preparazione alla gara, il potenziale effetto di riduzione del accumulo lipidico per attività di agonista parziale del PPARγ dato dal Telmisartan amplifica le applicazioni potenziali dei Sartani per il miglioramento della qualità del peso guadagnato in fase Bulk.
Ricordo, in fine, che tutto ciò che è stato detto è informazioni prettamente scientifica e non rappresenta in nessun modo un incitamento all’uso di farmaci fuori dalle linee di prescrizioni.
Gabriel Bellizzi [CEO BioGenTech]
Riferimenti:
- Birbrair A, Zhang T, Wang ZM, Messi ML, Enikolopov GN, Mintz A, Delbono O (August 2013). “Role of pericytes in skeletal muscle regeneration and fat accumulation”. Stem Cells and Development. 22 (16): 2298–2314. doi:10.1089/scd.2012.0647. PMC 3730538. PMID 23517218.
- Ye RZ, Richard G, Gévry N, Tchernof A, Carpentier AC (January 2022). “Fat Cell Size: Measurement Methods, Pathophysiological Origins, and Relationships With Metabolic Dysregulations”. Endocrine Reviews. 43 (1): 35–60. doi:10.1210/endrev/bnab018. PMC 8755996. PMID 34100954.
- Kershaw EE, Flier JS (June 2004). “Adipose tissue as an endocrine organ”. The Journal of Clinical Endocrinology and Metabolism. 89 (6): 2548–2556. doi:10.1210/jc.2004-0395. PMID 15181022.
- Mancuso P (May 2016). “The role of adipokines in chronic inflammation”. ImmunoTargets and Therapy. 5 (2016): 47–56. doi:10.2147/ITT.S73223. PMC 4970637. PMID 27529061.
- Cannon B, Nedergaard J (August 2008). “Developmental biology: Neither fat nor flesh”. Nature. 454 (7207): 947–948. Bibcode:2008Natur.454..947C. doi:10.1038/454947a. PMID 18719573. S2CID 205040511.
- Fat on the Inside: Looking Thin is Not Enough Archived 2016-11-17 at the Wayback Machine, By Fiona Haynes, About.com
- Jump up to:a b “Abdominal fat and what to do about it”. President & Fellows of Harvard College. September 2005.
Visceral fat more of a health concern than subcutaneous fat
- Nagai M, Komiya H, Mori Y, Ohta T, Kasahara Y, Ikeda Y (May 2010). “Estimating visceral fat area by multifrequency bioelectrical impedance”. Diabetes Care. 33 (5): 1077–1079. doi:10.2337/dc09-1099. PMC 2858179. PMID 20150289.
- Montague CT, O’Rahilly S (June 2000). “The perils of portliness: causes and consequences of visceral adiposity”. Diabetes. 49 (6): 883–888. doi:10.2337/diabetes.49.6.883. PMID 10866038.
- Kern PA, Ranganathan S, Li C, Wood L, Ranganathan G (May 2001). “Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance”. American Journal of Physiology. Endocrinology and Metabolism. 280 (5): E745–E751. doi:10.1152/ajpendo.2001.280.5.e745. PMID 11287357. S2CID 24306481
- Marette A (December 2003). “Molecular mechanisms of inflammation in obesity-linked insulin resistance”. International Journal of Obesity and Related Metabolic Disorders. 27 (Suppl 3): S46–S48. doi:10.1038/sj.ijo.0802500. PMID 14704744. S2CID 30693649.
- Mokdad AH, Ford ES, Bowman BA, Dietz WH, Vinicor F, Bales VS, Marks JS (January 2003). “Prevalence of obesity, diabetes, and obesity-related health risk factors, 2001”. JAMA. 289 (1): 76–79. doi:10.1001/jama.289.1.76. PMID 12503980.
- Maresky HS, Sharfman Z, Ziv-Baran T, Gomori JM, Copel L, Tal S (November 2015). “Anthropometric Assessment of Neck Adipose Tissue and Airway Volume Using Multidetector Computed Tomography: An Imaging Approach and Association With Overall Mortality”. Medicine. 94 (45): e1991. doi:10.1097/MD.0000000000001991. PMC 4912280. PMID 26559286.
- Brown JC, Harhay MO, Harhay MN (February 2018). “Anthropometrically predicted visceral adipose tissue and blood-based biomarkers: a cross-sectional analysis”. European Journal of Nutrition. 57 (1): 191–198. doi:10.1007/s00394-016-1308-8. PMC 5513780. PMID 27614626.
- Brown JC, Harhay MO, Harhay MN (January 2017). “Anthropometrically-predicted visceral adipose tissue and mortality among men and women in the third national health and nutrition examination survey (NHANES III)”. American Journal of Human Biology. 29 (1): e22898. doi:10.1002/ajhb.22898. PMC 5241265. PMID 27427402.
- “Reduce Abdominal Fat”. Archived from the original on 2011-09-28. Retrieved 2009-04-10.
Estrogen causes fat to be stored around the pelvic region, hips, butt and thighs (pelvic region)
- “Waistline Worries: Turning Apples Back Into Pears”. healthywomen.org. Archived from the original on 2009-06-09.
- Researchers think that the lack of estrogen at menopause plays a role in driving our fat northward. See: Andrews M (2006-12-01). “A Matter of Fat”. Yahoo Health. Women’s Health. Archived from the original on 2007-03-15.
- Singh AK, Loscalzo J, eds. (2014). The Brigham Intensive Review of Internal Medicine (2nd ed.). New York, NY: Oxford University Press. p. 483. ISBN 978-0-19-935827-4. Retrieved August 3, 2021.
- Ohkawara K, Tanaka S, Miyachi M, Ishikawa-Takata K, Tabata I (December 2007). “A dose-response relation between aerobic exercise and visceral fat reduction: systematic review of clinical trials”. International Journal of Obesity. 31 (12): 1786–1797. doi:10.1038/sj.ijo.0803683. PMID 17637702.
- Hoehn K, Marieb EN (2008). Anatomy & Physiology (3rd ed.). San Francisco, Calif.: Pearson/Benjamin Cummings. ISBN 978-0-8053-0094-9.
- Porter SA, Massaro JM, Hoffmann U, Vasan RS, O’Donnel CJ, Fox CS (June 2009). “Abdominal subcutaneous adipose tissue: a protective fat depot?”. Diabetes Care. 32 (6): 1068–1075. doi:10.2337/dc08-2280. PMC 2681034. PMID 19244087.
- “Belly fat in women: Taking – and keeping – it off”. MayoClinic.com. 2013-06-08. Retrieved 2013-12-02.
- Manolopoulos KN, Karpe F, Frayn KN (June 2010). “Gluteofemoral body fat as a determinant of metabolic health”. International Journal of Obesity. 34 (6): 949–959. doi:10.1038/ijo.2009.286. PMID 20065965. S2CID 21052919.
- Brodie D, Moscrip V, Hutcheon R (March 1998). “Body composition measurement: a review of hydrodensitometry, anthropometry, and impedance methods”. Nutrition. 14 (3): 296–310. doi:10.1016/S0899-9007(97)00474-7. PMID 9583375.
- Thomas LW (April 1962). “The chemical composition of adipose tissue of man and mice”. Quarterly Journal of Experimental Physiology and Cognate Medical Sciences. 47 (2): 179–188. doi:10.1113/expphysiol.1962.sp001589. PMID 13920823.
- Amitani M, Asakawa A, Amitani H, Inui A (2013). “The role of leptin in the control of insulin-glucose axis”. Frontiers in Neuroscience. 7: 51. doi:10.3389/fnins.2013.00051. PMC 3619125. PMID 23579596.
- Dhaliwal SS, Welborn TA (May 2009). “Central obesity and multivariable cardiovascular risk as assessed by the Framingham prediction scores”. The American Journal of Cardiology. 103 (10): 1403–1407. doi:10.1016/j.amjcard.2008.12.048. PMID 19427436.
- Park A (2009-08-08). “Fat-Bellied Monkeys Suggest Why Stress Sucks”. Time. Archived from the original on December 20, 2013. Retrieved 2013-12-19.
- Sugii S, Kida Y, Kawamura T, Suzuki J, Vassena R, Yin YQ, et al. (February 2010). “Human and mouse adipose-derived cells support feeder-independent induction of pluripotent stem cells”. Proceedings of the National Academy of Sciences of the United States of America. 107 (8): 3558–3563. Bibcode:2010PNAS..107.3558S. doi:10.1073/pnas.0910172106. PMC 2840462. PMID 20133714.
- Atzmon G, Yang XM, Muzumdar R, Ma XH, Gabriely I, Barzilai N (November 2002). “Differential gene expression between visceral and subcutaneous fat depots”. Hormone and Metabolic Research. 34 (11–12): 622–628. doi:10.1055/s-2002-38250. PMID 12660871. S2CID 33960130.
- Baglioni S, Cantini G, Poli G, Francalanci M, Squecco R, Di Franco A, et al. (4 May 2012). “Functional differences in visceral and subcutaneous fat pads originate from differences in the adipose stem cell”. PLOS ONE. 7 (5): e36569. Bibcode:2012PLoSO…736569B. doi:10.1371/journal.pone.0036569. PMC 3344924. PMID 22574183.
- Lemke, KA (June 2004). “Perioperative use of selective alpha-2 agonists and antagonists in small animals”. The Canadian Veterinary Journal. 45 (6): 475–80. PMC 548630. PMID 15283516.
- Hedner T, Edgar B, Edvinsson L, Hedner J, Persson B, Pettersson AYohimbine pharmacokinetics and interaction with the sympathetic nervous system in normal volunteersEur J Clin Pharmacol.(1992)
- Grossman E, Rosenthal T, Peleg E, Holmes C, Goldstein DSOral yohimbine increases blood pressure and sympathetic nervous outflow in hypertensive patientsJ Cardiovasc Pharmacol.(1993 Jul)
- Berlan M, Galitzky J, Riviere D, Foureau M, Tran MA, Flores R, Louvet JP, Houin G, Lafontan MPlasma catecholamine levels and lipid mobilization induced by yohimbine in obese and non-obese womenInt J Obes.(1991 May)
- Cimolai N, Cimolai TYohimbine use for physical enhancement and its potential toxicityJ Diet Suppl.(2011 Dec)
- Lalchandani SG, Lei L, Zheng W, Suni MM, Moore BM, Liggett SB, Miller DD, Feller DRYohimbine dimers exhibiting selectivity for the human alpha 2C-adrenoceptor subtypeJ Pharmacol Exp Ther.(2002 Dec)
- MacDonald E, Kobilka BK, Scheinin MGene targeting–homing in on alpha 2-adrenoceptor-subtype functionTrends Pharmacol Sci.(1997 Jun)
- Tan S, Curtis-Prior PBComparative effects of RX 781094, mianserin, yohimbine, rauwolscine and prazosin in reversing clonidine inhibition of MIX-stimulated lipolysis in hamster isolated white fat cellsPharmacol Res Commun.(1984 May)
- Bernstein KE, Ong FS, Blackwell WL, Shah KH, Giani JF, Gonzalez-Villalobos RA, et al. (January 2013). “A Modern Understanding of the Traditional and Nontraditional Biological Functions of Angiotensin-Converting Enzyme”. Pharmacological Reviews. 65 (1): 1–46. doi:10.1124/pr.112.006809. ISSN 0031-6997. PMC 3565918. PMID 23257181.
- Cushman DW, Ondetti MA (1991). “History of the design of captopril and related inhibitors of angiotensin converting enzyme”. Hypertension. 17 (4): 589–592. doi:10.1161/01.HYP.17.4.589. PMID 2013486. S2CID 30766421.
- Kaplan’s Essentials of Cardiac Anesthesia. Elsevier. 2018. doi:10.1016/c2012-0-06151-0. ISBN 978-0-323-49798-5.
Mechanisms of Action:ACE inhibitors act by inhibiting one of several proteases responsible for cleaving the decapeptide Ang I to form the octapeptide Ang II. Because ACE is also the enzyme that degrades bradykinin, ACE inhibitors increase circulating and tissue levels of bradykinin (Fig. 8.4).
- Jandeleit-Dahm K, Cooper ME (Sep 2006). “Hypertension and diabetes: role of the renin–angiotensin system”. Endocrinol Metab Clin North Am. 35 (3): 469–90, vii. doi:10.1016/j.ecl.2006.06.007. PMID 16959581.
- Wang W, McKinnie SM, Farhan M, Paul M, McDonald T, McLean B, et al. (May 2016). “Angiotensin Converting Enzyme 2 Metabolizes and Partially Inactivates Pyrapelin-13 and Apelin-17: Physiological Effects in the Cardiovascular System”. Hypertension. 68 (2): 365–77. doi:10.1161/HYPERTENSIONAHA.115.06892. PMID 27217402. S2CID 829514.
- Human Physiology, Silverthorn (Pearson Benjamin Cummings 2004)[page needed]
- Weir M (1999). “The renin-angiotensin-aldosterone system: a specific target for hypertension management”. American Journal of Hypertension. 12 (4). Oxford University Press (OUP): 205–213. doi:10.1016/s0895-7061(99)00103-x. ISSN 0895-7061. PMID 10619573.
- Jackson EK (2006). “Chapter 30. Renin and Angiotensin”. In Brunton LL, Lazo JS, Parker K (eds.). Goodman & Gilman’s The Pharmacological Basis of Therapeutics (11th ed.). New York: McGraw-Hill. ISBN 978-0-07-142280-2.
- “Myocardial Infarction”. The Lecturio Medical Concept Library. Retrieved 27 August 2021.
- “Congestive Heart Failure”. The Lecturio Medical Concept Library. 7 August 2020. Retrieved 27 August 2021.
- Kester M, Karpa KD, Vrana KE (2012). “Cardiovascular System”. Elsevier’s Integrated Review Pharmacology. Elsevier. pp. 125–151. doi:10.1016/b978-0-323-07445-2.00008-2. ISBN 978-0-323-07445-2.
ACE inhibitors also slow progression of kidney disease in patients with diabetic nephropathies. Renal benefits are probably a result of improved renal hemodynamics from decreased glomerular arteriolar resistance.
- Long AN, Dagogo-Jack S. Comorbidities of diabetes and hypertension: mechanisms and approach to target organ protection. J Clin Hypertens (Greenwich) 2011; 13:244-251. http://doi: 10.1111/j.1751-7176.2011.00434.x.
- Dolgacheva LP, Turovskaya MV, Dynnik VV, Zinchenko VP, Goncharov NV, Davletov B, Turovsky EA. Angiotensin II activates different calcium signaling pathways in adipocytes. Arch Biochem Biophys 2016; 593:38-49. http://doi: 10.1016/j.abb.2016.02.001. [ Links ]
- 50. Palominos MM, Dünner DH, Wabitsch M, Rojas CV. 2015. Angiotensin II directly impairs adipogenic differentiation of human preadipose cells. Mol Cell Biochem 2015; 408: 115-122. http://doi: 10.1007/s11010-015-2487-y.
- 51. Schling MM, Dünner NH, Wabitsch M, Rojas CV. Angiotensin II directly impairs adipogenic differentiation of human preadipose cells. Mol Cell Biochem 2015; 408:115-122. http://doi: 10.1007/s11010-015-2487-y.
- 52. Brücher R, Cifuentes M, Acuña MJ, Albala C, Rojas CV. Larger anti-adipogenic effect of angiotensin II on omental preadipose cells of obese humans. Obesity 2007; 15:1643-1646. http://doi: 10.1038/oby.2007.196.
- 53. Fuentes P, Acuña MJ, Cifuentes M, Rojas CV. The anti-adipogenic effect of angiotensin II on human preadipose cells involves ERK1,2 activation and PPARG phosphorylation. J Endocrinol 2010; 206:75-83. http:// doi: 10.1677/JOE-10-0049.
- Jones BH, Standridge MK, Moustaid N. Angiotensin II increases lipogenesis in 3T3-L1 and human adipose cells. Endocrinology 1997; 138:1512-1519. http://doi: 10.1210/endo.138.4.5038.
- Townsend RR. The effects of angiotensin-II on lipolysis in humans. Metabolism 2001; 50:468-472. http://doi: 10.1053/meta.2001.21021.
- Weisinger RS, Begg DP, Jois M. Antagonists of the renin-angiotensin system and the prevention of obesity. Curr Opin Investig Drugs 2009; 10: 1069-1077.
- Than A, Leow MK, Chen P. Control of adipogenesis by the autocrine interplays between angiotensin 1-7/Mas receptor and angiotensin II/AT1 receptor signaling pathways. J Biol Chem 2013; 288:15520-15531. http://doi: 10.1074/jbc.M113.459792.
- Sharma AM, Engeli S. The role of renin-angiotensin system blockade in the management of hypertension associated with the cardiometabolic syndrome. J Cardiometab Syndr 2006; 1:29-35. http://doi: 10.1111/j.0197-3118.2006.05422.x.
- Vertes V, Haynie R. Comparative pharmacokinetics of captopril, enalapril, and quinapril. Am J Cardiol. 1992;69:8C–16C. [PubMed] [Google Scholar]
- Nakajima T, Yamada T, Setoguchi M. Prolonged inhibition of local angiotensin-converting enzyme after single or repeated treatment with quinapril in spontaneously hypertensive rats. J Cardiovasc Pharmacol. 1992;19:102–107. [PubMed] [Google Scholar]
- Fukiyama F, Azuma J, Yoshida H, et al. A pharmacokinetic study of quinapril in normal Japanese men. J Clin Ther Med. 1993;9(Suppl 7):3–16. [Google Scholar]
- Weishaar RE, Panek RL, Major TC, Simmerman J, Rapundalo ST, Taylor J, DG DG. Evidence for a functional tissue renin-angiotensin system in the rat mesentric vasculature and its involvement in regulation blood pressure. J Pharmacol Exp Ther. 1991;256:568–574. [PubMed] [Google Scholar]
- Major TC, Overhiser RW, Taylor DG, Panek RL. Effect of quinapril, a new angiotensin-converting enzyme inhibitor, on vasoconstrictor activity in the isolated, perfused mesenteric vasculature of hypertensive rats. J Pharmacol Exp Ther. 1993;265:187–193. [PubMed] [Google Scholar]
- Willenbrock R, Ozcelik C, Osterziel KJ, Dietz R. Angiotensin-converting enzyme inhibition, autonomic activity, and hemodynamics in patients with heart failure who perform isometric exercise. Am Heart J. 1996;131:999–1006. [PubMed] [Google Scholar]
- Saitoh M, Miyakoda H, Kitamura H, Kinugawa T, Kotake H, Mashiba H. Effects of an angiotensin-converting enzyme inhibitor, alacepril, on cardiovascular and sympathetic nervous responses to mental stress in patients with essential hypertension. Intern Med. 1993;32:691–694. [PubMed] [Google Scholar]
- Schling P, Mallow H, Trindl A, Loffler G. Evidence for a Local Renin Angiotensin System in Primary Cultured Human Preadipocytes. Int J Obes Relat Metab Disord (1999) 23(4):336–41. doi: 10.1038/sj.ijo.080082 PubMed Abstract | CrossRef Full Text | Google Scholar
- Weisinger RS, Begg DP, Chen N, Jois M, Mathai ML, Sinclair AJ. The Problem of Obesity: Is There a Role for Antagonists of the Renin-Angiotensin System? Asia Pac J Clin Nutr (2007) 16 S1:359–67.Google Scholar
- Takahashi N, Li F, Hua K, Deng J, Wang CH, Bowers RR, et al. Increased Energy Expenditure, Dietary Fat Wasting, and Resistance to Diet-Induced Obesity in Mice Lacking Renin. Cell Metab (2007) 6(6):506–12. doi: 10.1016/j.cmet.2007.10.01 PubMed Abstract | CrossRef Full Text | Google Scholar
- Massiera F, Seydoux J, Geloen A, Quignard-Boulange A, Turban S, Saint-Marc P, et al. Angiotensinogen-Deficient Mice Exhibit Impairment of Diet-Induced Weight Gain With Alteration in Adipose Tissue Development and Increased Locomotor Activity. Endocrinology (2001) 142(12):5220–5. doi: 10.1210/endo.142.12.8556 PubMed Abstract | CrossRef Full Text | Google Scholar
- Jayasooriya AP, Begg DP, Chen N, Mathai ML, Sinclair AJ, Wilkinson-Berka J, et al. Omega-3 Polyunsaturated Fatty Acid Supplementation Reduces Hypertension in TGR(mRen-2)27 Rats. Prostaglandins Leukot Essent Fatty Acids (2008) 78(1):67–72. doi: 10.1016/j.plefa.2007.11.001 PubMed Abstract | CrossRef Full Text | Google Scholar
- De Blasi A, Cortellaro M, Costantini C. Enalapril in Essential Hypertension: A Comparative Study With Propranolol. Enalapril in Hypertension Study Group (Uk). Br J Clin Pharmacol (1984) 18(1):51–6. doi: 10.1111/j.1365-2125.1984.tb05021. PubMed Abstract | CrossRef Full Text | Google Scholar
- Zorad S, Dou JT, Benicky J, Hutanu D, Tybitanclova K, Zhou J, et al. Long-Term Angiotensin II AT1 Receptor Inhibition Produces Adipose Tissue Hypotrophy Accompanied by Increased Expression of Adiponectin and Ppargamma. Eur J Pharmacol (2006) 552(1-3):112–22. doi: 10.1016/j.ejphar.2006.08.062 PubMed Abstract | CrossRef Full Text | Google Scholar


















































![{\displaystyle K={\frac {[TBGT3]}{[T3][TBG]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/9ddd39492d87e75780e8491e2fc2f3c4e3cffb9c)
![{\displaystyle T3={\frac {[TBGT3]}{[K][TBG]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/ed67ca8c9784b9e5c530a91638f2d764c02d5e05)