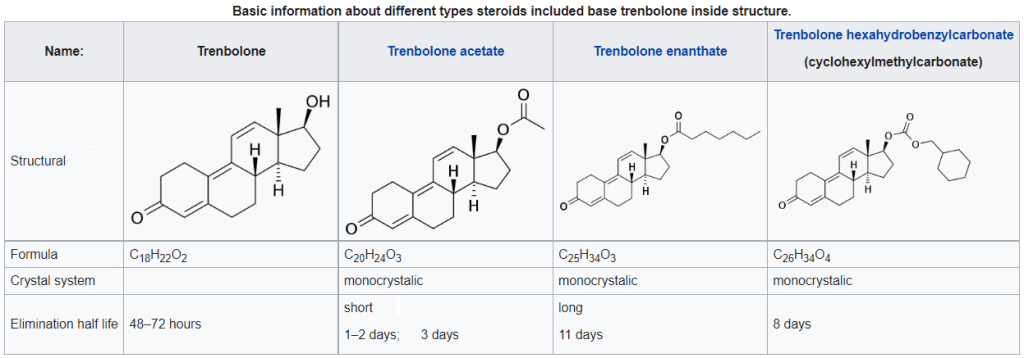
Introduzione alla molecola:
L’ACE-031 può rientrare a pieno titolo nel “club” delle molecole PEDs semisconosciute. Un peptide praticamente unico nel panorama “doped”, sicuramente promettente, specie nel BodyBuilding, ma del quale se ne parla poco.
Nel 2013 sembrava che la ricerca sul ACE-031 fosse stata definitivamente interrotta, nonostante funzionasse piuttosto bene.
Le aziende farmaceutiche Acceleron Pharma e Shire misero in pausa la ricerca sull’inibitore della Miostatina ACE-031 [Acceleronpharma.com 2 maggio 2013]. E questo evento risultò piuttosto strano. In un comunicato stampa congiunto rilasciato qualche tempo dopo il sopra citato annuncio, Muscle & Nerve aveva pubblicato uno studio che dimostrava che l’ACE-031 è un composto che un culturista supplementato farmacologicamente aggiungerebbe volentieri al suo “arsenale”.
L’ACE-031 iniettabile è un recettore sintetico dell’Attivina di Tipo IIB. Anche le cellule muscolari hanno questo recettore. È destinato a proteine come la Miostatina, il GDF11 e l’Attivina A e B. Se la Miostatina si lega al recettore dell’Attivina di Tipo IIB, la crescita delle fibre muscolari si riduce. Nelle circostanze “giuste” la Miostatina arriva addirittura a degradare il muscolo-scheletrico.

Se si somministra l’ACE-031, questo non accade o, comunque, l’effetto viene marcatamente ridotto. Il recettore sintetico dell’Attivina di Tipo IIB si lega con il tristemente noto peptide Miostatina impedendo a quest’ultimo di legarsi al sito recettore della cellula e compiere la sua attività di riduzione ipertrofica e degradazione del tessuto muscolo-scheletrico.
ACE-031 e “recettori esca”:
Come accennato pocanzi, l’ACE-031 non è altro che un “recettore esca”. Un recettore esca è un recettore in grado di riconoscere e legare in modo efficiente specifici fattori di crescita o citochine, ma non è strutturalmente in grado di segnalare o attivare il complesso recettoriale previsto. Agisce come un inibitore, legando un ligando e impedendogli di legarsi al suo recettore abituale. I recettori esca partecipano a un metodo comune di inibizione del segnale e sono anche abbondanti nei tessuti maligni, costituendo un argomento significativo nella ricerca sul cancro.[1]

IL1R2 è stato uno dei primi recettori esca identificati.[2] [3] Lega IL1A e IL1B e inibisce il loro legame con IL1R1, impedendo la risposta infiammatoria che è generalmente promossa dal legame delle interleuchine di tipo 1 con il recettore 1 dell’interleuchina di tipo I.[4]
Un altro membro di questa categoria è il recettore DcR3, conosciuto anche come TNFRSF6, che si trova principalmente nei tessuti maligni umani.[5] Agisce come recettore esca per i membri delle citochine TNF: FasL, LIGHT e TL1A, inibendo la capacità delle citochine di segnalare la morte cellulare o l’apoptosi.

Il VEGFR-1 è una tirosin-chinasi recettoriale che modula negativamente l’angiogenesi agendo come recettore esca.[6] La caratteristica di “esca” del VEGFR-1 è necessaria per lo sviluppo e l’angiogenesi normali. Il VEGFR-1 inibisce l’attività del VEGFR-2 sequestrando il VEGF, impedendo così al VEGFR-2 di legarsi al VEGF.
Quindi eccoci di nuovo con ACE-031. Esso è stato studiato in quanto è un recettore esca ingegnerizzato con attività inibitoria della Miostatina potenzialmente utile nel tentativo di trattare i bambini affetti da distrofia muscolare di Duchenne (DMD). Il recettore ACE-031 circola al di fuori della membrana della fibra muscolare. Poiché questo recettore si lega alla Miostatina, riduce la quantità di questo peptide che può legarsi al recettore nativo nella membrana (ActRIIB), impedendo alla Miostatina di fornire il segnale che limita la crescita muscolare e ne promuove il catabolismo.[7]
I principali studi su ACE-031:
Nel 2007 Acceleron Pharma aveva grandi aspettative su ACE-031. All’epoca l’azienda aveva condotto solo studi sugli animali. Tuttavia, nel marzo 2013 AP ha pubblicato uno studio sull’uomo in cui 48 donne sane di età compresa tra 45 e 75 anni hanno ricevuto una singola iniezione con 0.02, 0.05, 0.1, 0.3, 1 o 3 mg di ACE-031 per kg di peso corporeo. Il composto ha circolato per alcune settimane nell’organismo dei soggetti trattati. L’emivita è stata stimata essere di 10-15 giorni.
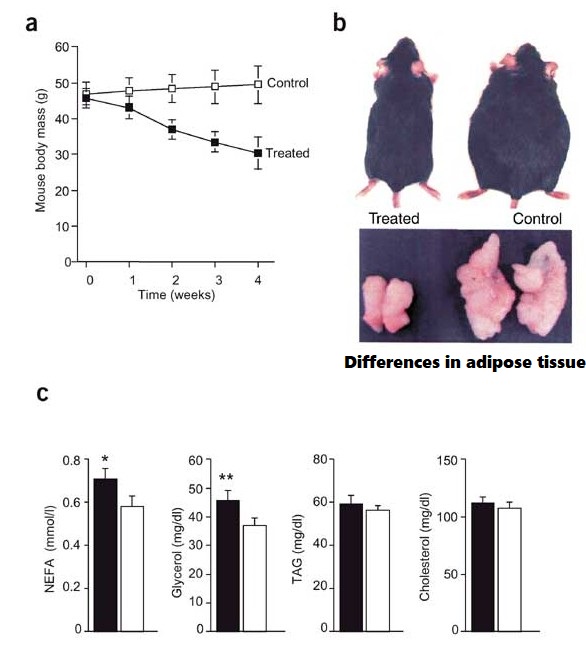
Tuttavia, questa singola iniezione ha prodotto una crescita muscolare. La dose di 3mg/kg ha mostrato un aumento del volume muscolare del 5%. La massa magra è aumentata del 3% [poco più di un chilo] e sembra anche diminuire la massa grassa.

L’iniezione ha ridotto la Leptina e aumentato la concentrazione di Adiponectina. Ciò suggerisce che l’ACE-031 riduce la massa grassa.
Inoltre, è aumentato l’inibitore della Miostatina, i livelli di fosfatasi alcalina specifica per le ossa [BSAP] nel sangue e si è ridotto quello del telopeptide C-terminale del collagene di tipo 1 [CTX]. Ciò suggerisce che l’ACE-031 rende le ossa più forti. Negli studi sugli animali con RAP-031, la versione per topi di ACE-031, Acceleron è riuscita a dimostrare questi effetti. [Endocrinology. 2010 Sep; 151 (9) :4289-300].
Se si legge lo studio su Muscle & Nerve, ci si chiede perché mai la Acceleron abbia interrotto lo sviluppo di ACE-031. E perché non agisce legalmente contro tutti gli store online che si puliscono le terga con i brevetti di Acceleron e vendono l’ACE-031 a un prezzo al quale una normale azienda farmaceutica non può trarre alcun profitto.[Muscle Nerve. 2013 Mar; 47 (3) :416-23.]
La risposta si trova in un messaggio sul sito web dell’Associazione per la Distrofia Muscolare. [Quest.mda.org 2 maggio 2013] In esso si legge che nel 2011, durante uno studio [NCT01099761] in cui i ricercatori somministravano l’ACE 031 a bambini affetti da malattie muscolari, sono emersi effetti collaterali che hanno costretto i ricercatori a interrompere lo studio.

“Gli eventi avversi che i partecipanti alla sperimentazione hanno subito – piccoli sanguinamenti del naso e delle gengive e dilatazione dei vasi sanguigni della pelle – non sono stati considerati di per sé pericolosi. Tuttavia, le aziende e le agenzie regolatorie coinvolte affermano di aver bisogno di comprendere appieno questi eventi prima di continuare gli studi clinici sull’ACE-031”. “
Un altro strano effetto collaterale è stato rivelato nello studio pubblicato su Muscle & Nerve. È emerso che la somministrazione di ACE-031 abbia ridotto fortemente la concentrazione di FSH nelle donne partecipanti. I ricercatori non ne conoscono la causa e le possibili conseguenze.
Sembrava che ACE-031 fosse stato definitivamente accantonato dalla ricerca fino alla pubblicazione nel 2017 di uno studio sul recettore esca , sempre su Muscle Nerve [Myostatin inhibitor ACE-031 treatment of ambulatory boys with Duchenne muscular dystrophy: Results of a randomized, placebo-controlled clinical trial]. L’ACE-031 è stato somministrato per via sottocutanea ogni 2-4 settimane a ragazzi affetti da DMD [distrofia muscolare di Duchenne] in uno studio randomizzato, in doppio cieco, controllato con placebo, a dose crescente. L’obiettivo primario era la valutazione della sicurezza. Gli obiettivi secondari comprendevano la caratterizzazione della farmacocinetica e della farmacodinamica.
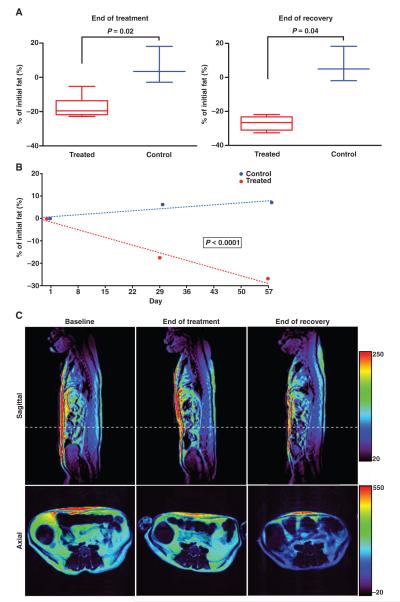
L’ACE-031, durante lo studio, non è stato associato a eventi avversi gravi o molto gravi. Lo studio è stato interrotto dopo il secondo regime di dosaggio a causa di potenziali problemi di sicurezza legati a epistassi e teleangectasie. È stata rilevata una tendenza al mantenimento della distanza del test del cammino di 6 minuti (6MWT) nei gruppi ACE-031 rispetto al calo osservato nel gruppo placebo (non statisticamente significativo), nonché una tendenza all’aumento della massa magra e della densità minerale ossea (BMD) e alla riduzione della massa grassa.
Anche in questo studio, l’uso dell’ACE-031 ha dimostrato tendenze per gli effetti farmacodinamici sulla massa magra, sulla massa grassa, sulla BMD e sul 6MWT (6-minute walk test). Ma, come successo in precedenza, gli eventi avversi non correlati ai muscoli hanno contribuito alla decisione di interrompere lo studio. Nonostante l’inibizione della Miostatina è un approccio terapeutico promettente per la DMD.

Neanche lo studio su MYO-029, il miostatinblokker della Wyeth, ha avuto successo. Nel 2008 uno studio deludente ha dimostrato che gli adulti con distrofia muscolare, dopo la somministrazione di MYO-029, non sono diventati più forti. [Ann Neurol. 2008 May, 63 (5) :561-71] e la Wyeth ha interrotto lo sviluppo del MYO-029.
Uso nel BodyBuilding e conclusioni:
Ora sappiamo che questo “recettore esca” può favorire lo sviluppo del muscolo-scheletrico legandosi alla Miostatina ed impedendo a questa di esercitare la sua azione di controllo e catabolismo muscolare. Sappiamo inoltre che gli studi effettuati su esseri umani sono stati promettenti ma non sufficientemente sicuri da permetterne uno sviluppo completo. I casi di epistassi e teleangectasie hanno spinto i ricercatori ad interrompere la ricerca. Ma come spesso accade, ogni qualvolta nel panorama scientifico si affaccia una molecola potenzialmente vantaggiosa per lo sportivo, e per il BodyBuilder in particolare, anche se la ricerca si interrompe non si può dire lo stesso per quella svolta illegalmente da improvvisate cavie umane. E questo evento si è verificato anche per l’ACE-031.
Partendo dalle prove emerse durante gli studi, sappiamo che una dose di 3mg/Kg ha comportato un aumento del volume muscolare del 5%, un aumento della massa muscolare del 3% e sembra portare anche a una riduzione della massa grassa. La molecola sembra ridurre la concentrazione di Leptina, condizione che potrebbe portare ad uno scompenso nella regolazione fame/sazietà, ed un aumento dell’Adiponectina, la quale è correlata ad un miglioramento della sensibilità all’Insulina.
Prove sul campo raccolte negli ultimi anni, hanno permesso di quantificare i dosaggi mediamente efficaci per un Bodybuilder e i tempi di somministrazione: 1-3mg per chilogrammo di peso corporeo ogni 15 giorni è risultato essere il range standard per ottenere i migliori risultati possibili. Per quanto concerne la lunghezza del trattamento, si presume che l’uso debba essere circoscritto in un arco temporale di circa 5-6 settimane, limite di conservazione che non dovrebbe essere superato.
Ricordo che il principale effetto collaterale di ACE-031 è la dilatazione dei vasi sanguigni. Tuttavia, questo effetto collaterale, se contenuto, non sembra avere svantaggi. Inoltre, l’uso di ACE-031 può causare epistassi e gengive sanguinanti. Non sono noti altri effetti collaterali. I soggetti emofiliaci sono a forte rischio emorragico potenziale con l’uso di ACE-031.
Anche se dovrebbe essere scontato, ribadisco il fatto che nessuno sta invitando all’uso sperimentale ed illegale di una molecola della quale, oltretutto, si sa poco. Le informazioni ivi presenti sono a puro scopo divulgativo e non rappresentano in alcun modo prescrizioni mediche e affini.
Gabriel Bellizzi
Riferimenti:
- Decoy Receptor”. Encyclopedia of Cancer. Springer Berlin Heidelberg. 2012. p. 1070.
- McMahan, CJ; Slack, JL; Mosley, B (1991). “A novel IL-1 receptor, cloned from B cells by mammalian expression, is expressed in many cell types”. The EMBO Journal. 10 (10): 2821–2832.
- Re, F; Muzio, M; De Rossi, M; et al. (1994). “The type II “receptor” as a decoy target for interleukin 1 in polymorphonuclear leukocytes: characterization of induction by dexamethasone and ligand binding properties of the released decoy receptor”. The Journal of Experimental Medicine. 179 (2): 739–743.
- “IL1R2 interleukin 1 receptor, type II [ Homo sapiens (human) ]”. ncbi.nlm.nih.gov. National Center for Biotechnology Information. 2015.
- Ashkenazi, Avi (1 June 2002). “Targeting death and decoy receptors of the tumour-necrosis factor superfamily”. Nature Reviews Cancer. 2 (6): 420–430.
- Meyer, Rosana D.; Mohammadi, Moosi; Rahimi, Nader (13 January 2006). “A Single Amino Acid Substitution in the Activation Loop Defines the Decoy Characteristic of VEGFR-1/FLT-1*”. The Journal of Biological Chemistry.
- Attie, Kenneth M (21 November 2012). “A single ascending-dose study of muscle regulator ace-031 in healthy volunteers”. Muscle and Nerve.