
Introduzione:
Gli aminoacidi sono una classe di molecole biologiche costituenti le unità che formano le proteine, e svolgono numerose funzioni fondamentali per la corretta funzione del corpo umano. Rappresentano un elemento conosciuto a grandi linee da tutti gli assidui frequentatori di sala pesi, ma la maggior parte di loro è all’oscuro delle loro caratteristiche e reali richieste, quando una loro supplementazione risulta funzionale e quando, invece, si traduce in una pratica pressoché sterile. Questo primo articolo è finalizzato ad iniziare una approfondita disamina sugli aminoacidi facendo chiarezza sul significato biochimico e sulla loro più aggiornata applicazione in ambito sportivo soprattutto per quanto concerne gli Aminoacidi Essenziali.
Cosa sono gli Aminoacidi?
Gli aminoacidi sono composti organici che contengono sia gruppi funzionali amminici che carbossilici.[1] Sebbene in natura esistano oltre 500 amminoacidi, i più importanti sono i 22 α-amminoacidi incorporati nelle proteine.[2] Solo questi 22 compaiono nel codice genetico della vita.[3][4]
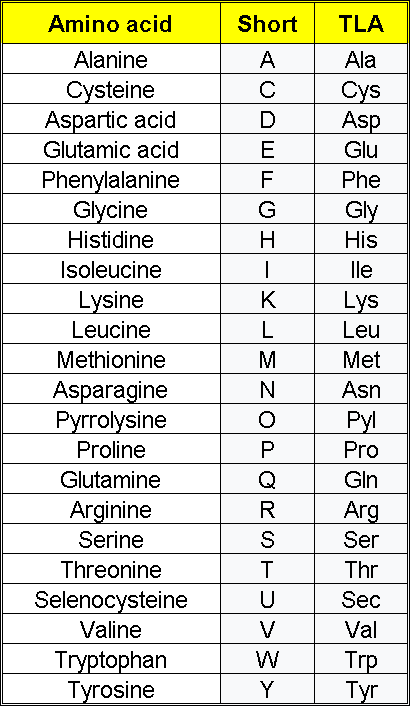
Gli aminoacidi possono essere classificati in base alla posizione dei gruppi funzionali strutturali principali (amminoacidi alfa (α-), beta (β-), gamma (γ-), ecc.); altre categorie riguardano la polarità, la ionizzazione e il tipo di catena laterale (alifatica, aciclica, aromatica, polare, ecc.). Sotto forma di proteine, i residui di aminoacidi costituiscono la seconda componente (l’acqua è la più grande) dei muscoli e degli altri tessuti umani.[5] Oltre al ruolo di residui nelle proteine, gli aminoacidi partecipano a una serie di processi come il trasporto e la biosintesi dei neurotrasmettitori.[6]
- Storia:

I primi amminoacidi furono scoperti all’inizio del 1800.[7][8] Nel 1806, i chimici francesi Louis-Nicolas Vauquelin e Pierre Jean Robiquet isolarono dagli asparagi un composto che fu poi chiamato asparagina, il primo amminoacido ad essere scoperto.[9][10] La cistina fu scoperta nel 1810,[11] anche se il suo monomero, la cisteina, rimase sconosciuto fino al 1884. [La glicina e la leucina furono scoperte nel 1820.[12[13] L’ultimo dei 20 aminoacidi comuni ad essere scoperto fu la treonina nel 1935 da William Cumming Rose, che determinò anche gli aminoacidi essenziali e stabilì il fabbisogno minimo giornaliero di tutti gli aminoacidi per una crescita ottimale.[14][15]
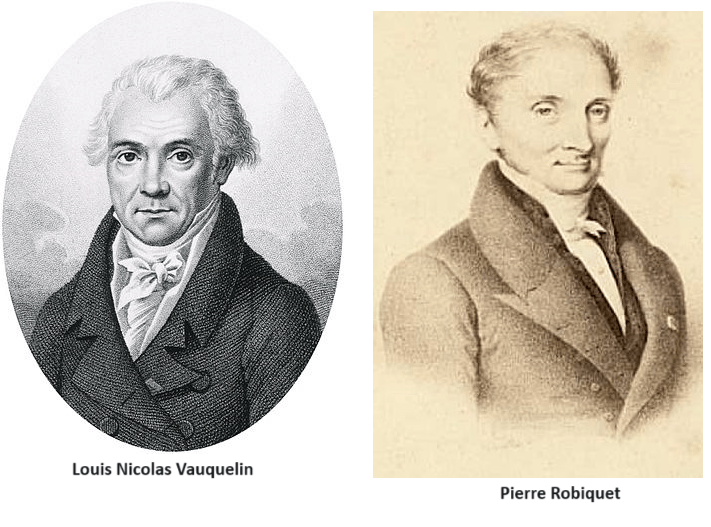
L’unità della categoria chimica fu riconosciuta da Wurtz nel 1865, ma non le diede un nome particolare.[16] Il primo uso del termine “aminoacido” in lingua inglese risale al 1898,[17] mentre il termine tedesco, Aminosäure, era già stato usato in precedenza.[18] Si è scoperto che le proteine producono aminoacidi in seguito a digestione enzimatica o idrolisi acida. Nel 1902, Emil Fischer e Franz Hofmeister proposero indipendentemente che le proteine sono formate da molti amminoacidi, per cui si formano legami tra il gruppo amminico di un amminoacido e il gruppo carbossilico di un altro, dando luogo a una struttura lineare che Fischer definì “peptide”.[19]

- Struttura generale
I 2-, alfa- o α-amminoacidi[20] hanno nella maggior parte dei casi la formula generica H2NCHRCOOH, dove R è un sostituente organico noto come “catena laterale”.[21]
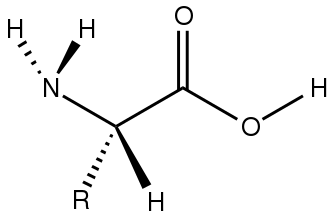
Delle centinaia di amminoacidi descritti, 22 sono proteinogenici (“costruiscono proteine”).[22][23][24] Sono questi 22 composti che si combinano per dare una vasta gamma di peptidi e proteine assemblate dai ribosomi.[25] Gli amminoacidi non proteinogenici o modificati possono derivare da modificazioni post-traslazionali o durante la sintesi di peptidi nonribosomiali.

L’atomo di carbonio vicino al gruppo carbossilico è chiamato carbonio α. Negli amminoacidi proteinogenici, porta l’ammina e il gruppo R o la catena laterale specifica di ciascun amminoacido. Con quattro sostituenti distinti, il carbonio α è stereogenico in tutti gli α-amminoacidi tranne la glicina. Tutti gli amminoacidi proteogenici chirali hanno la configurazione L. Sono “sinistrorsi”. Si tratta di enantiomeri “sinistrorsi”, che si riferiscono agli stereoisomeri del carbonio alfa.

Alcuni amminoacidi D (“destrorsi”) sono stati trovati in natura, ad esempio negli involucri batterici, come neuromodulatore (D-serina) e in alcuni antibiotici.[26][27] Raramente, i residui di amminoacidi D si trovano nelle proteine e vengono convertiti dall’amminoacido L come modifica post-traduzionale.[28]
Cinque amminoacidi possiedono una carica a pH neutro. Spesso queste catene laterali appaiono sulla superficie delle proteine per consentirne la solubilità in acqua, e le catene laterali con cariche opposte formano importanti contatti elettrostatici chiamati ponti salini che mantengono le strutture all’interno di una singola proteina o tra proteine interfacciate.[29] Molte proteine legano il metallo nelle loro strutture in modo specifico, e queste interazioni sono comunemente mediate da catene laterali cariche come l’aspartato, il glutammato e l’istidina. In determinate condizioni, ogni gruppo che forma ioni può essere carico, formando sali doppi.[30]

I due aminoacidi carichi negativamente a pH neutro sono l’aspartato (Asp, D) e il glutammato (Glu, E). I gruppi carbossilati anionici si comportano come basi di Brønsted nella maggior parte dei casi.[29] Gli enzimi in ambienti a pH molto basso, come la pepsina, proteasi aspartica nello stomaco dei mammiferi, possono avere residui catalitici di aspartato o glutammato che agiscono come acidi di Brønsted.

Ci sono tre amminoacidi con catene laterali che sono cationi a pH neutro: l’arginina (Arg, R), la lisina (Lys, K) e l’istidina (His, H). L’arginina ha un gruppo guanidino carico e la lisina un gruppo alchilico amminico carico e sono completamente protonati a pH 7. Il gruppo imidazolico dell’istidina ha un pKa di 6,0 ed è protonato solo per il 10% circa a pH neutro. Poiché l’istidina si trova facilmente nelle sue forme basiche e acide coniugate, partecipa spesso ai trasferimenti catalitici di protoni nelle reazioni enzimatiche.[29]
Gli aminoacidi polari e privi di carica serina (Ser, S), treonina (Thr, T), asparagina (Asn, N) e glutammina (Gln, Q) formano prontamente legami a idrogeno con l’acqua e con altri aminoacidi.[29] Non si ionizzano in condizioni normali; un’eccezione importante è rappresentata dalla serina catalitica nelle serina-proteasi. Questo è un esempio di grave perturbazione e non è caratteristico dei residui di serina in generale. La treonina ha due centri chirali, non solo il centro chirale L (2S) sul carbonio α condiviso da tutti gli amminoacidi, a parte la glicina achirale, ma anche (3R) sul carbonio β. La specifica stereochimica completa è (2S,3R)-L-treonina.

Le interazioni tra gli amminoacidi non polari sono la forza motrice principale dei processi di ripiegamento delle proteine nelle loro strutture tridimensionali funzionali.[29] Nessuna delle catene laterali di questi amminoacidi si ionizza facilmente e quindi non hanno pKas, ad eccezione della tirosina (Tyr, Y). L’idrossile della tirosina può deprotonarsi ad alto pH formando un fenolato carico negativamente. Per questo motivo si potrebbe collocare la tirosina nella categoria degli aminoacidi polari e privi di carica, ma la sua bassissima solubilità in acqua corrisponde bene alle caratteristiche degli aminoacidi idrofobici.

Diverse catene laterali non sono ben descritte dalle categorie cariche, polari e idrofobiche. La glicina (Gly, G) potrebbe essere considerata un amminoacido polare, poiché le sue piccole dimensioni fanno sì che la sua solubilità sia determinata in gran parte dai gruppi amminici e carbossilici. Tuttavia, la mancanza di catene laterali conferisce alla glicina una flessibilità unica tra gli amminoacidi, con ampie ramificazioni nel ripiegamento delle proteine.[29] Anche la cisteina (Cys, C) può formare facilmente legami idrogeno, il che la collocherebbe nella categoria degli amminoacidi polari, anche se spesso si trova nelle strutture proteiche a formare legami covalenti, detti legami disolfuro, con altre cisteine. Questi legami influenzano il ripiegamento e la stabilità delle proteine e sono essenziali nella formazione degli anticorpi. La prolina (Pro, P) ha una catena laterale alchilica e potrebbe essere considerata idrofobica, ma poiché la catena laterale si unisce di nuovo al gruppo alfa-amminico, diventa particolarmente inflessibile quando viene incorporata nelle proteine. Come la glicina, influenza la struttura delle proteine in un modo unico tra gli aminoacidi. La selenocisteina (Sec, U) è un raro amminoacido non codificato direttamente dal DNA, ma incorporato nelle proteine attraverso il ribosoma. La selenocisteina ha un potenziale redox più basso rispetto alla cisteina simile e partecipa a diverse reazioni enzimatiche uniche.[31] La pirrolisina (Pyl, O) è un altro aminoacido non codificato nel DNA, ma sintetizzato nelle proteine dai ribosomi.[34] Si trova in specie arcaiche dove partecipa all’attività catalitica di diverse metiltransferasi.
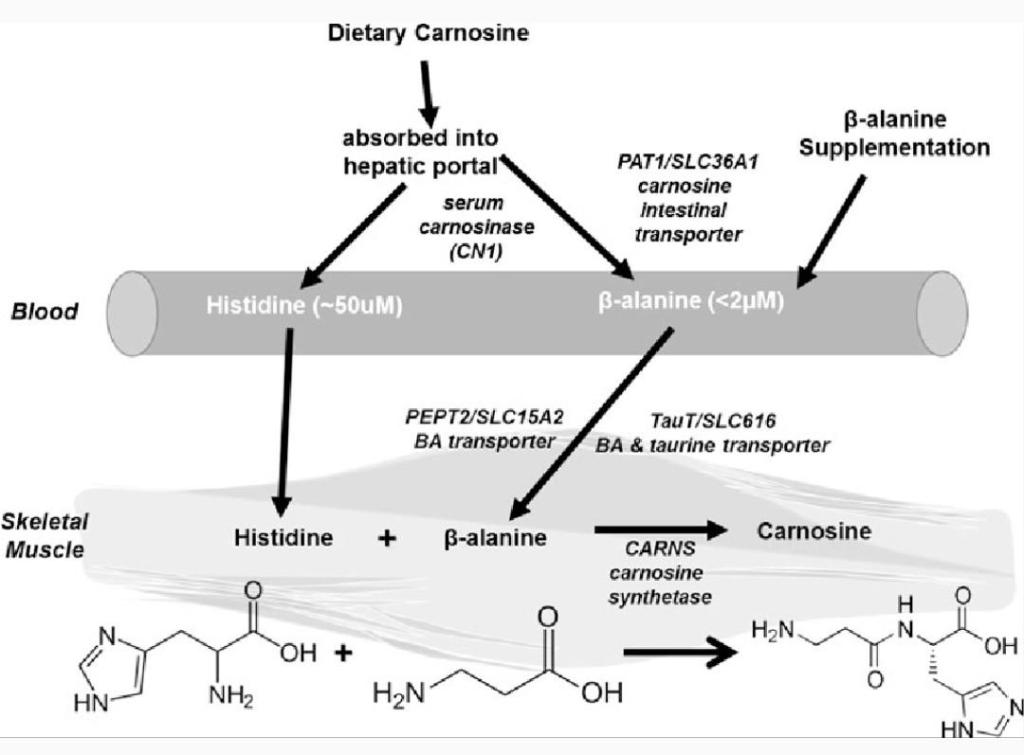
Gli amminoacidi con struttura NH+3-CXY-CXY-CO-2, come la β-alanina, componente della carnosina e di alcuni altri peptidi, sono β-amminoacidi. Quelli con la struttura NH+3-CXY-CXY-CXY-CO-2 sono γ-amminoacidi, e così via, dove X e Y sono due sostituenti (uno dei quali è normalmente H).[6]
Le forme naturali comuni di amminoacidi hanno una struttura zwitterionica, con gruppi funzionali -NH+3 (-NH+2- nel caso della prolina) e -CO-2 attaccati allo stesso atomo di C; sono quindi α-amminoacidi e sono gli unici che si trovano nelle proteine durante la traduzione nel ribosoma. In soluzione acquosa, a pH prossimo alla neutralità, gli amminoacidi esistono come zwitterioni, cioè come ioni dipolari con entrambi i gruppi NH+3 e CO-2 in stati carichi, per cui la struttura complessiva è NH+3-CHR-CO-2. A pH fisiologico le cosiddette “forme neutre” -Sebbene le due cariche della struttura zwitterionica si sommino a zero, è fuorviante definire “scarica” una specie con carica netta pari a zero.
In condizioni di forte acidità (pH inferiore a 3), il gruppo carbossilato viene protonato e la struttura diventa un acido carbossilico ammonio, NH+3
-CHR-CO2H. Ciò è rilevante per gli enzimi come la pepsina che sono attivi in ambienti acidi come lo stomaco e i lisosomi dei mammiferi, ma non si applica in modo significativo agli enzimi intracellulari. In condizioni altamente basiche (pH superiore a 10, normalmente non riscontrabile in condizioni fisiologiche), il gruppo ammonio viene deprotonato per dare NH2-CHR-CO-2.

Sebbene in chimica si utilizzino varie definizioni di acidi e basi, l’unica utile per la chimica in soluzione acquosa è quella di Brønsted:[32][33] un acido è una specie che può donare un protone a un’altra specie, mentre una base è una specie che può accettare un protone. Questo criterio viene utilizzato per etichettare i gruppi nell’illustrazione precedente. Le catene laterali carbossilate dei residui di aspartato e glutammato sono le principali basi di Brønsted nelle proteine. Allo stesso modo, la lisina, la tirosina e la cisteina agiscono tipicamente come acidi Brønsted. L’istidina, in queste condizioni, può agire sia come acido che come base di Brønsted.
Per gli amminoacidi con catene laterali non cariche, lo zwitterione predomina a valori di pH compresi tra i due valori di pKa, ma coesiste in equilibrio con piccole quantità di ioni netti negativi e positivi. Nel punto intermedio tra i due valori di pKa, la traccia di ioni negativi netti e la traccia di ioni positivi netti si bilanciano, in modo che la carica netta media di tutte le forme presenti sia pari a zero.[34] Questo pH è noto come punto isoelettrico pI, per cui pI = 1/2 (pKa1 + pKa2).

Per gli amminoacidi con catene laterali cariche, è coinvolto il pKa della catena laterale. Così per l’aspartato o il glutammato con catene laterali negative, il gruppo amminico terminale è essenzialmente interamente nella forma carica -NH+3, ma questa carica positiva deve essere bilanciata dallo stato con un solo gruppo carbossilato C-terminale carico negativamente. Questo si verifica a metà strada tra i due valori di pKa del carbossilato: pI = 1/2 (pKa1 + pKa(R)), dove pKa(R) è il pKa della catena laterale.[33]
Considerazioni simili valgono per altri amminoacidi con catene laterali ionizzabili, tra cui non solo il glutammato (simile all’aspartato), ma anche la cisteina, l’istidina, la lisina, la tirosina e l’arginina con catene laterali positive.
Gli amminoacidi hanno mobilità nulla nell’elettroforesi al loro punto isoelettrico, anche se questo comportamento è più sfruttato per i peptidi e le proteine che per i singoli amminoacidi. Gli zwitterioni hanno una solubilità minima al loro punto isoelettrico e alcuni amminoacidi (in particolare quelli con catene laterali non polari) possono essere isolati per precipitazione dall’acqua regolando il pH al punto isoelettrico richiesto.
- Proprietà fisico-chimiche
I 20 amminoacidi canonici possono essere classificati in base alle loro proprietà. Fattori importanti sono la carica, l’idrofilia o l’idrofobicità, la dimensione e i gruppi funzionali.[27] Queste proprietà influenzano la struttura delle proteine e le interazioni proteina-proteina. Le proteine idrosolubili tendono ad avere i loro residui idrofobici (Leu, Ile, Val, Phe e Trp) sepolti al centro della proteina, mentre le catene laterali idrofile sono esposte al solvente acquoso. (In biochimica, un residuo si riferisce a uno specifico monomero all’interno della catena polimerica di un polisaccaride, di una proteina o di un acido nucleico). Le proteine integrali di membrana tendono ad avere anelli esterni di aminoacidi idrofobici esposti che le ancorano al bilayer lipidico. Alcune proteine di membrana periferiche hanno una zona di aminoacidi idrofobici sulla loro superficie che si attacca alla membrana. In modo simile, le proteine che devono legarsi a molecole cariche positivamente hanno superfici ricche di aminoacidi carichi negativamente, come il glutammato e l’aspartato, mentre le proteine che si legano a molecole cariche negativamente hanno superfici ricche di aminoacidi carichi positivamente, come la lisina e l’arginina. Ad esempio, la lisina e l’arginina sono presenti in grandi quantità nelle regioni a bassa complessità delle proteine che legano gli acidi nucleici.[35] Esistono varie scale di idrofobicità dei residui di amminoacidi.[36]

Alcuni amminoacidi hanno proprietà speciali. La cisteina può formare legami disolfuro covalenti con altri residui di cisteina. La prolina forma un ciclo alla spina dorsale polipeptidica e la glicina è più flessibile di altri aminoacidi.
La glicina e la prolina sono fortemente presenti all’interno delle regioni a bassa complessità delle proteine sia eucariotiche che procariotiche, mentre l’opposto avviene con la cisteina, la fenilalanina, il triptofano, la metionina, la valina, la leucina, l’isoleucina, che sono altamente reattivi, o complessi, o idrofobici.[35][37][38]
Molte proteine subiscono una serie di modifiche post-traslazionali, in base alle quali gruppi chimici aggiuntivi vengono attaccati alle catene laterali dei residui aminoacidici, producendo talvolta lipoproteine (che sono idrofobiche),[39] o glicoproteine (che sono idrofile)[40] che consentono alla proteina di attaccarsi temporaneamente a una membrana. Ad esempio, una proteina di segnalazione può attaccarsi e poi staccarsi dalla membrana cellulare, perché contiene residui di cisteina a cui può essere aggiunto e successivamente rimosso l’acido grasso palmitico.[41]
- Sintesi
Nelle piante, l’azoto viene prima assimilato in composti organici sotto forma di glutammato, formato da alfa-chetoglutarato e ammoniaca nel mitocondrio. Per gli altri amminoacidi, le piante utilizzano le transaminasi per spostare il gruppo amminico dal glutammato ad un altro alfa-chetoacido. Ad esempio, l’aspartato aminotransferasi converte il glutammato e l’ossalacetato in alfa-chetoglutarato e aspartato.[42] Anche altri organismi utilizzano le transaminasi per la sintesi degli aminoacidi.

Gli amminoacidi non standard si formano solitamente attraverso modifiche agli amminoacidi standard. Ad esempio, l’omocisteina si forma attraverso la via della transulfurazione o mediante la demetilazione della metionina tramite il metabolita intermedio S-adenosilmetionina,[43] mentre l’idrossiprolina viene prodotta mediante una modifica post-traduzionale della prolina.[44]

I microrganismi e le piante sintetizzano molti amminoacidi non comuni. Ad esempio, alcuni microbi producono acido 2-amminoisobutirrico e lantionina, che è un derivato con ponti solforati dell’alanina. Entrambi questi amminoacidi si trovano nei lantibiotici peptidici come l’alameticina.[45] Tuttavia, nelle piante, l’acido 1-amminociclopropan-1-carbossilico è un piccolo amminoacido ciclico disostituito che è un intermedio nella produzione dell’ormone vegetale etilene.[46]
La produzione commerciale di amminoacidi si basa solitamente su batteri mutanti che avviano una sovrapproduzione di singoli amminoacidi utilizzando il glucosio come fonte di carbonio. Alcuni amminoacidi sono prodotti mediante conversioni enzimatiche di intermedi sintetici. L’acido 2-amminotiazolin-4-carbossilico è un intermedio in una sintesi industriale della L-cisteina, ad esempio. L’acido aspartico viene prodotto mediante l’aggiunta di ammoniaca al fumarato utilizzando una liasi.[47]
- Presenza e funzioni in biochimica
Gli amminoacidi sono i precursori delle proteine[25] e si uniscono tramite reazioni di condensazione per formare catene polimeriche corte chiamate peptidi o catene più lunghe chiamate polipeptidi o proteine. Queste catene sono lineari e non ramificate, con ogni residuo di amminoacido all’interno della catena attaccato a due amminoacidi vicini. In natura, il processo di creazione delle proteine codificate dal materiale genetico DNA/RNA è chiamato traduzione e comporta l’aggiunta graduale di aminoacidi a una catena proteica in crescita da parte di un ribozima chiamato ribosoma.[48] L’ordine di aggiunta degli aminoacidi viene letto attraverso il codice genetico da un modello di mRNA, che è una copia di RNA di uno dei geni dell’organismo.

Ventidue amminoacidi sono naturalmente incorporati nei polipeptidi e sono chiamati amminoacidi proteinogenici o naturali.[27] Di questi, 20 sono codificati dal codice genetico universale. Gli altri due, la selenocisteina e la pirrolisina, sono incorporati nelle proteine mediante meccanismi sintetici unici. La selenocisteina viene incorporata quando l’mRNA da tradurre include un elemento SECIS, che fa sì che il codone UGA codifichi la selenocisteina invece di un codone di stop.[49] La pirrolisina è utilizzata da alcuni archei metanogeni negli enzimi che usano per produrre metano. È codificata con il codone UAG, che in altri organismi è normalmente un codone di stop.[50] Questo codone UAG è seguito da una sequenza a valle PYLIS.[51]

I 20 aminoacidi codificati direttamente dai codoni del codice genetico universale sono chiamati aminoacidi standard o canonici. Una forma modificata di metionina (N-formilmetionina) è spesso incorporata al posto della metionina come aminoacido iniziale delle proteine nei batteri, nei mitocondri e nei cloroplasti. Altri aminoacidi sono chiamati non standard o non canonici. La maggior parte degli amminoacidi non standard sono anche non proteinogenici (cioè non possono essere incorporati nelle proteine durante la traduzione), ma due di essi sono proteinogenici, in quanto possono essere incorporati a livello di traduzione nelle proteine sfruttando informazioni non codificate nel codice genetico universale.

I due aminoacidi non standard proteinogenici sono la selenocisteina (presente in molti non eucarioti e nella maggior parte degli eucarioti, ma non codificata direttamente dal DNA) e la pirrolisina (presente solo in alcuni archei e in almeno un batterio). L’incorporazione di questi aminoacidi non standard è rara. Ad esempio, 25 proteine umane includono la selenocisteina nella loro struttura primaria,[52] e gli enzimi strutturalmente caratterizzati (selenoenzimi) impiegano la selenocisteina come moiety catalitica nei loro siti attivi.[53] La pirrolisina e la selenocisteina sono codificate tramite codoni varianti. Ad esempio, la selenocisteina è codificata dal codone di stop e dall’elemento SECIS.[54][55][56]
La N-formilmetionina (che è spesso l’amminoacido iniziale delle proteine nei batteri, nei mitocondri e nei cloroplasti) è generalmente considerata una forma di metionina piuttosto che un amminoacido proteinogenico separato. Le combinazioni codone-tRNA non presenti in natura possono anche essere utilizzate per “espandere” il codice genetico e formare nuove proteine note come alloproteine che incorporano aminoacidi non proteinogenici.[57][58][59]
Oltre ai 22 aminoacidi proteinogenici, sono noti molti aminoacidi non proteinogenici. Questi non si trovano nelle proteine (ad esempio la carnitina, il GABA, la levotiroxina) o non sono prodotti direttamente e isolatamente dai macchinari cellulari standard. Ad esempio, l’idrossiprolina viene sintetizzata dalla prolina. Un altro esempio è la selenometionina).
Gli aminoacidi non proteici che si trovano nelle proteine si formano tramite modificazioni post-traslazionali. Tali modifiche possono anche determinare la localizzazione della proteina, ad esempio l’aggiunta di lunghi gruppi idrofobici può far sì che una proteina si leghi a una membrana fosfolipidica.[60] Esempi:
la carbossilazione del glutammato permette di legare meglio i cationi di calcio,[61]
L’idrossiprolina, generata dall’idrossilazione della prolina, è uno dei principali componenti del collagene del tessuto connettivo[62].
L’ipusina nel fattore di iniziazione della traduzione EIF5A contiene una modifica della lisina.[63]
Alcuni aminoacidi non proteici non si trovano nelle proteine. Ne sono un esempio l’acido 2-amminoisobutirrico e il neurotrasmettitore acido gamma-amminobutirrico. Gli aminoacidi non proteinogenici sono spesso presenti come intermedi nelle vie metaboliche degli aminoacidi standard – ad esempio, l’ornitina e la citrullina sono presenti nel ciclo dell’urea, parte del catabolismo degli aminoacidi (vedi sotto).[64] Una rara eccezione alla predominanza degli α-amminoacidi in biologia è rappresentata dal β-amminoacido beta alanina (acido 3-amminopropanoico), utilizzato nelle piante e nei microrganismi nella sintesi dell’acido pantotenico (vitamina B5), un componente del coenzima A.[65]
Gli aminoacidi non sono una componente tipica del cibo: gli animali mangiano proteine. La proteina viene scomposta in aminoacidi durante il processo di digestione. Vengono quindi utilizzati per sintetizzare nuove proteine, altre biomolecole o vengono ossidati in urea e anidride carbonica come fonte di energia.[66] La via dell’ossidazione inizia con la rimozione del gruppo amminico da parte di una transaminasi; il gruppo amminico viene quindi immesso nel ciclo dell’urea. L’altro prodotto della transamidazione è un chetoacido che entra nel ciclo dell’acido citrico.[67] Gli amminoacidi glucogeni possono anche essere convertiti in glucosio, attraverso la gluconeogenesi.[68]
Dei 20 aminoacidi standard, nove (His, Ile, Leu, Lys, Met, Phe, Thr, Trp e Val) sono chiamati aminoacidi essenziali perché il corpo umano non è in grado di sintetizzarli da altri composti al livello necessario per la normale crescita. quindi devono essere ottenuti dal cibo.[69][70][71]
Inoltre, la cisteina, la tirosina e l’arginina sono considerati aminoacidi semiessenziali e la taurina un acido aminosolfonico semiessenziale nei bambini. Alcuni aminoacidi sono condizionatamente essenziali per determinate età o condizioni mediche. Le vie metaboliche che sintetizzano questi monomeri non sono completamente sviluppate.[72][73]
Molti amminoacidi proteinogenici e non proteogenici hanno funzioni biologiche oltre ad essere precursori di proteine e peptidi. Negli esseri umani, gli amminoacidi hanno anche ruoli importanti in diverse vie biosintetiche. Le difese contro gli erbivori nelle piante a volte impiegano gli aminoacidi.[74] Esempi:
Amminoacidi standard
– Il triptofano è un precursore del neurotrasmettitore serotonina.[75]
– La tirosina (e il suo precursore fenilalanina) sono precursori dei neurotrasmettitori catecolaminici dopamina, epinefrina e norepinefrina e di varie ammine in traccia.
– La fenilalanina è un precursore della fenetilammina e della tirosina nell’uomo. – Nelle piante è un precursore di vari fenilpropanoidi, importanti nel metabolismo vegetale.
– La glicina è un precursore delle porfirine come l’eme.[76]
– L’arginina è un precursore dell’ossido nitrico.[77]
– L’ornitina e la S-adenosilmetionina sono precursori delle poliammine.[78]
– Aspartato, glicina e glutammina sono precursori dei nucleotidi.[79] Tuttavia, non tutte le funzioni di altri abbondanti aminoacidi non standard sono note.
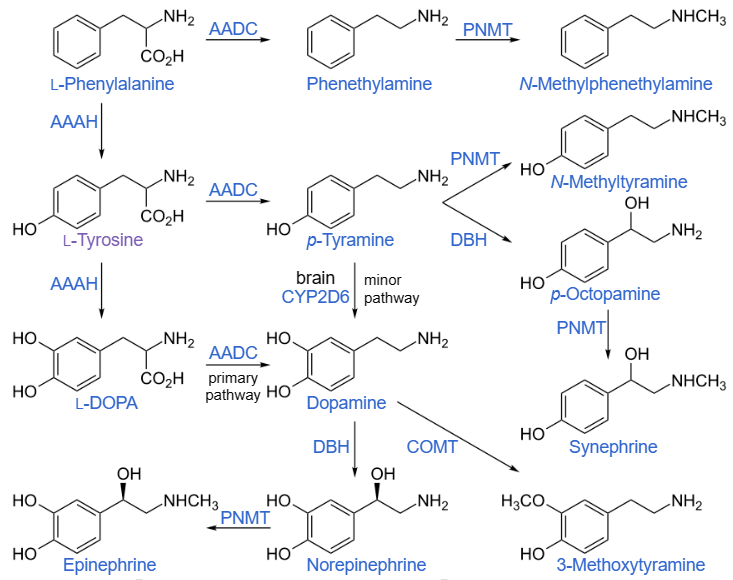
Ruoli degli amminoacidi non standard
– La carnitina è utilizzata nel trasporto dei lipidi.
– L’acido gamma-amminobutirrico è un neurotrasmettitore.[80]
– Il 5-HTP (5-idrossitriptofano) è utilizzato per il trattamento sperimentale della depressione.[81]
– L-DOPA (L-diidrossifenilalanina) per il trattamento del morbo di Parkinson,[82]
– L’eflornitina inibisce l’ornitina decarbossilasi e viene utilizzata nel trattamento della malattia del sonno.[83]
– La canavanina, un analogo dell’arginina presente in molti legumi, è un antifeedant, che protegge la pianta dai predatori.[84]
– La mimosina, presente in alcuni legumi, è un altro possibile antifeedant.[85] Questo composto è un analogo della tirosina e può avvelenare gli animali che pascolano su queste piante.
- Formazione del legame peptidico
Poiché sia i gruppi amminici che quelli carbossilici degli amminoacidi possono reagire per formare legami ammidici, una molecola di amminoacido può reagire con un’altra e unirsi attraverso un legame ammidico. Questa polimerizzazione degli amminoacidi è ciò che crea le proteine. Questa reazione di condensazione produce il legame peptidico appena formato e una molecola di acqua. Nelle cellule questa reazione non avviene direttamente; invece, l’amminoacido viene prima attivato mediante l’attaccamento a una molecola di RNA di trasferimento attraverso un legame estere. Questo amminoacil-tRNA viene prodotto in una reazione ATP-dipendente effettuata da un’amminoacil tRNA sintetasi.[86] Questo amminoacil-tRNA è quindi un substrato per il ribosoma, che catalizza l’attacco del gruppo amminico della catena proteica allungata sul legame estere.[87] Come risultato di questo meccanismo, tutte le proteine prodotte dai ribosomi vengono sintetizzate a partire dal loro terminale N e spostandosi verso il loro terminale C.
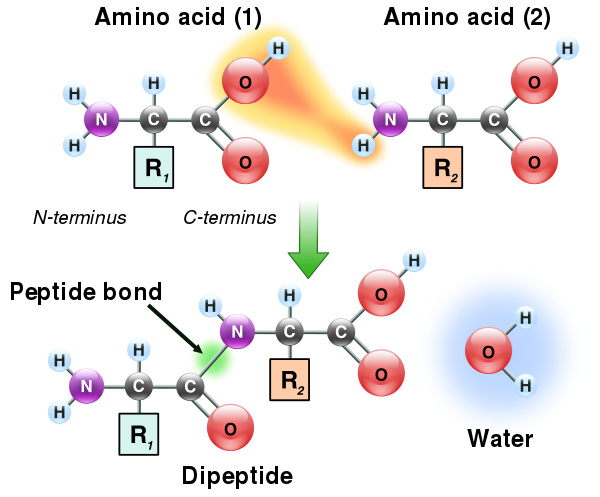
Tuttavia non tutti i legami peptidici si formano in questo modo. In alcuni casi, i peptidi vengono sintetizzati da enzimi specifici. Ad esempio, il tripeptide glutatione è una parte essenziale delle difese delle cellule contro lo stress ossidativo. Questo peptide viene sintetizzato in due fasi da amminoacidi liberi.[88] Nella prima fase, la gamma-glutamilcisteina sintetasi condensa la cisteina e il glutammato attraverso un legame peptidico formato tra il carbossile della catena laterale del glutammato (il carbonio gamma di questa catena laterale) e il gruppo amminico della cisteina. Questo dipeptide viene quindi condensato con la glicina dalla glutatione sintetasi per formare glutatione.[89]
In chimica, i peptidi vengono sintetizzati mediante una varietà di reazioni. Uno dei metodi più utilizzati nella sintesi peptidica in fase solida utilizza i derivati ossimici aromatici degli amminoacidi come unità attivate. Questi vengono aggiunti in sequenza sulla catena peptidica in crescita, che è attaccata a un supporto di resina solida.[90] Le librerie di peptidi vengono utilizzate nella scoperta di farmaci attraverso lo screening ad alto rendimento.[91]

Glucogenico, con prodotti che hanno la capacità di formare glucosio mediante gluconeogenesi
Chetogenico, poiché i prodotti non hanno la capacità di formare glucosio. Questi prodotti possono ancora essere utilizzati per la chetogenesi o la sintesi lipidica.
*Amminoacidi catabolizzati sia in prodotti glucogeni che chetogenici.
La combinazione di gruppi funzionali consente agli amminoacidi di essere efficaci ligandi polidentati per chelati metallo-amminoacidi.[92] Le molteplici catene laterali degli amminoacidi possono anche subire reazioni chimiche.
La degradazione di un amminoacido spesso comporta la deaminazione spostando il suo gruppo amminico nell’α-chetoglutarato, formando glutammato. Questo processo coinvolge le transaminasi, spesso le stesse utilizzate nell’amminazione durante la sintesi. In molti vertebrati il gruppo amminico viene poi eliminato attraverso il ciclo dell’urea ed escreto sotto forma di urea. Tuttavia, la degradazione degli aminoacidi può invece produrre acido urico o ammoniaca. Ad esempio, la serina deidratasi converte la serina in piruvato e ammoniaca.[56] Dopo la rimozione di uno o più gruppi amminici, il resto della molecola può talvolta essere utilizzato per sintetizzare nuovi amminoacidi, oppure può essere utilizzato per produrre energia entrando nella glicolisi o nel ciclo dell’acido citrico, come dettagliato nell’immagine a destra.
- Valutazione del contenuto di Azoto nella materia organica
Il contenuto totale di azoto della materia organica è formato principalmente dai gruppi amminici delle proteine. L’Azoto Totale Kjeldahl (TKN) è una misura dell’azoto ampiamente utilizzata nell’analisi di acque (reflue), suolo, alimenti, mangimi e materia organica in generale. Come suggerisce il nome, viene applicato il metodo Kjeldahl. Sono disponibili metodi più sensibili.[93][94]

Continua…
Gabriel Bellizzi [CEO BioGenTech]
Riferimenti:
- Nelson DL, Cox MM (2005). Principles of Biochemistry (4th ed.). New York: W. H. Freeman. ISBN 0-7167-4339-6.
- Flissi, Areski; Ricart, Emma; Campart, Clémentine; Chevalier, Mickael; Dufresne, Yoann; Michalik, Juraj; Jacques, Philippe; Flahaut, Christophe; Lisacek, Frédérique; Leclère, Valérie; Pupin, Maude (2020). “Norine: update of the nonribosomal peptide resource”. Nucleic Acids Research. 48 (D1): D465–D469. doi:10.1093/nar/gkz1000. PMC 7145658. PMID 31691799.
- Richard Cammack, ed. (2009). “Newsletter 2009”. Biochemical Nomenclature Committee of IUPAC and NC-IUBMB. Pyrrolysine. Archived from the original on 12 September 2017. Retrieved 16 April 2012.
- Rother, Michael; Krzycki, Joseph A. (1 January 2010). “Selenocysteine, Pyrrolysine, and the Unique Energy Metabolism of Methanogenic Archaea”. Archaea. 2010: 1–14. doi:10.1155/2010/453642. ISSN 1472-3646. PMC 2933860. PMID 20847933.
- Latham MC (1997). “Chapter 8. Body composition, the functions of food, metabolism and energy”. Human nutrition in the developing world. Food and Nutrition Series – No. 29. Rome: Food and Agriculture Organization of the United Nations. Archived from the original on 8 October 2012. Retrieved 9 September 2012.
- “Nomenclature and Symbolism for Amino Acids and Peptides”. IUPAC-IUB Joint Commission on Biochemical Nomenclature. 1983. Archived from the original on 9 October 2008. Retrieved 17 November 2008.
- Vickery HB, Schmidt CL (1931). “The history of the discovery of the amino acids”. Chem. Rev. 9 (2): 169–318. doi:10.1021/cr60033a001.
- Hansen S (May 2015). “Die Entdeckung der proteinogenen Aminosäuren von 1805 in Paris bis 1935 in Illinois” (PDF) (in German). Berlin. Archived from the original (PDF) on 1 December 2017.
- Vauquelin LN, Robiquet PJ (1806). “The discovery of a new plant principle in Asparagus sativus”. Annales de Chimie. 57: 88–93.
- Jump up to:a b Anfinsen CB, Edsall JT, Richards FM (1972). Advances in Protein Chemistry. New York: Academic Press. pp. 99, 103. ISBN 978-0-12-034226-6.
- Wollaston WH (1810). “On cystic oxide, a new species of urinary calculus”. Philosophical Transactions of the Royal Society. 100: 223–230. doi:10.1098/rstl.1810.0015. S2CID 110151163.
- Baumann E (1884). “Über cystin und cystein”. Z Physiol Chem. 8 (4): 299–305. Archived from the original on 14 March 2011. Retrieved 28 March 2011.
- Braconnot HM (1820). “Sur la conversion des matières animales en nouvelles substances par le moyen de l’acide sulfurique”. Annales de Chimie et de Physique. 2nd Series. 13: 113–125.
- Simoni RD, Hill RL, Vaughan M (September 2002). “The discovery of the amino acid threonine: the work of William C. Rose [classical article]”. The Journal of Biological Chemistry. 277 (37): E25. doi:10.1016/S0021-9258(20)74369-3. PMID 12218068. Archived from the original on 10 June 2019. Retrieved 4 July 2015.
- McCoy RH, Meyer CE, Rose WC (1935). “Feeding Experiments with Mixtures of Highly Purified Amino Acids. VIII. Isolation and Identification of a New Essential Amino Acid”. Journal of Biological Chemistry. 112: 283–302. doi:10.1016/S0021-9258(18)74986-7.
- Menten, P. Dictionnaire de chimie: Une approche étymologique et historique. De Boeck, Bruxelles. link Archived 28 December 2019 at the Wayback Machine.
- Harper D. “amino-“. Online Etymology Dictionary. Archived from the original on 2 December 2017. Retrieved 19 July 2010.
- Paal C (1894). “Ueber die Einwirkung von Phenyl-i-cyanat auf organische Aminosäuren”. Berichte der Deutschen Chemischen Gesellschaft. 27: 974–979. doi:10.1002/cber.189402701205. Archived from the original on 25 July 2020.
- Fruton JS (1990). “Chapter 5- Emil Fischer and Franz Hofmeister”. Contrasts in Scientific Style: Research Groups in the Chemical and Biochemical Sciences. Vol. 191. American Philosophical Society. pp. 163–165. ISBN 978-0-87169-191-0.
- “Alpha amino acid”. Merriam-Webster Medical. Archived from the original on 3 January 2015. Retrieved 3 January 2015..
- Clark, Jim (August 2007). “An introduction to amino acids”. chemguide. Archived from the original on 30 April 2015. Retrieved 4 July 2015.
- Jakubke HD, Sewald N (2008). “Amino acids”. Peptides from A to Z: A Concise Encyclopedia. Germany: Wiley-VCH. p. 20. ISBN 9783527621170. Archived from the original on 17 May 2016. Retrieved 5 January 2016 – via Google Books.
- Pollegioni L, Servi S, eds. (2012). Unnatural Amino Acids: Methods and Protocols. Methods in Molecular Biology. Vol. 794. Humana Press. p. v. doi:10.1007/978-1-61779-331-8. ISBN 978-1-61779-331-8. OCLC 756512314. S2CID 3705304.
- Hertweck C (October 2011). “Biosynthesis and Charging of Pyrrolysine, the 22nd Genetically Encoded Amino Acid”. Angewandte Chemie International Edition. 50 (41): 9540–9541. doi:10.1002/anie.201103769. PMID 21796749. S2CID 5359077.
- Jump up to:a b “Chapter 1: Proteins are the Body’s Worker Molecules”. The Structures of Life. National Institute of General Medical Sciences. 27 October 2011. Archived from the original on 7 June 2014. Retrieved 20 May 2008.
- Michal G, Schomburg D, eds. (2012). Biochemical Pathways: An Atlas of Biochemistry and Molecular Biology (2nd ed.). Oxford: Wiley-Blackwell. p. 5. ISBN 978-0-470-14684-2.
- Jump up to:a b c Creighton TH (1993). “Chapter 1”. Proteins: structures and molecular properties. San Francisco: W. H. Freeman. ISBN 978-0-7167-7030-5.
- Genchi, Giuseppe (1 September 2017). “An overview on d-amino acids”. Amino Acids. 49 (9): 1521–1533. doi:10.1007/s00726-017-2459-5. ISSN 1438-2199. PMID 28681245. S2CID 254088816.
- Garrett, Reginald H.; Grisham, Charles M. (2010). Biochemistry (4th ed.). Belmont, CA: Brooks/Cole, Cengage Learning. pp. 74, 134–176, 430–442. ISBN 978-0-495-10935-8. OCLC 297392560.
- Novikov, Anton P.; Safonov, Alexey V.; German, Konstantin E.; Grigoriev, Mikhail S. (1 December 2023). “What kind of interactions we may get moving from zwitter to “dritter” ions: C–O⋯Re(O4) and Re–O⋯Re(O4) anion⋯anion interactions make structural difference between L-histidinium perrhenate and pertechnetate”. CrystEngComm. 26: 61–69. doi:10.1039/D3CE01164J. ISSN 1466-8033. S2CID 265572280.
- Papp, Laura Vanda; Lu, Jun; Holmgren, Arne; Khanna, Kum Kum (1 July 2007). “From Selenium to Selenoproteins: Synthesis, Identity, and Their Role in Human Health”. Antioxidants & Redox Signaling. 9 (7): 775–806.
- Brønsted, J. N. (1923). “Einige Bemerkungen über den Begriff der Säuren und Basen” [Remarks on the concept of acids and bases]. Recueil des Travaux Chimiques des Pays-Bas. 42 (8): 718–728. doi:10.1002/recl.19230420815.
- Jump up to:a b Vollhardt, K. Peter C. (2007). Organic chemistry : structure and function. Neil Eric Schore (5th ed.). New York: W.H. Freeman. pp. 58–66. ISBN 978-0-7167-9949-8. OCLC 61448218.
- Fennema OR (19 June 1996). Food Chemistry 3rd Ed. CRC Press. pp. 327–328. ISBN 978-0-8247-9691-4.
- Ntountoumi C, Vlastaridis P, Mossialos D, Stathopoulos C, Iliopoulos I, Promponas V, et al. (November 2019). “Low complexity regions in the proteins of prokaryotes perform important functional roles and are highly conserved”. Nucleic Acids Research. 47 (19): 9998–10009. doi:10.1093/nar/gkz730. PMC 6821194. PMID 31504783.
- Urry DW (2004). “The change in Gibbs free energy for hydrophobic association: Derivation and evaluation by means of inverse temperature transitions”. Chemical Physics Letters. 399 (1–3): 177–183. Bibcode:2004CPL…399..177U. doi:10.1016/S0009-2614(04)01565-9.
- Marcotte EM, Pellegrini M, Yeates TO, Eisenberg D (October 1999). “A census of protein repeats”. Journal of Molecular Biology. 293 (1): 151–60. doi:10.1006/jmbi.1999.3136. PMID 10512723.
- Haerty W, Golding GB (October 2010). Bonen L (ed.). “Low-complexity sequences and single amino acid repeats: not just “junk” peptide sequences”. Genome. 53 (10): 753–62. doi:10.1139/G10-063. PMID 20962881.
- Magee T, Seabra MC (April 2005). “Fatty acylation and prenylation of proteins: what’s hot in fat”. Current Opinion in Cell Biology. 17 (2): 190–196. doi:10.1016/j.ceb.2005.02.003. PMID 15780596.
- Pilobello KT, Mahal LK (June 2007). “Deciphering the glycocode: the complexity and analytical challenge of glycomics”. Current Opinion in Chemical Biology. 11 (3): 300–305. doi:10.1016/j.cbpa.2007.05.002. PMID 17500024.
- Smotrys JE, Linder ME (2004). “Palmitoylation of intracellular signaling proteins: regulation and function”. Annual Review of Biochemistry. 73 (1): 559–587.
- Jones RC, Buchanan BB, Gruissem W (2000). Biochemistry & molecular biology of plants. Rockville, Md: American Society of Plant Physiologists. pp. 371–372. ISBN 978-0-943088-39-6.
- Brosnan JT, Brosnan ME (June 2006). “The sulfur-containing amino acids: an overview”. The Journal of Nutrition. 136 (6 Suppl): 1636S–1640S. doi:10.1093/jn/136.6.1636S. PMID 16702333.
- Kivirikko KI, Pihlajaniemi T (1998). “Collagen Hydroxylases and the Protein Disulfide Isomerase Subunit of Prolyl 4-Hydroxylases”. Advances in Enzymology and Related Areas of Molecular Biology. Advances in Enzymology – and Related Areas of Molecular Biology. Vol. 72. pp. 325–398. doi:10.1002/9780470123188.ch9. ISBN 9780470123188. PMID 9559057.
- Whitmore L, Wallace BA (May 2004). “Analysis of peptaibol sequence composition: implications for in vivo synthesis and channel formation”. European Biophysics Journal. 33 (3): 233–237. doi:10.1007/s00249-003-0348-1. PMID 14534753. S2CID 24638475.
- Alexander L, Grierson D (October 2002). “Ethylene biosynthesis and action in tomato: a model for climacteric fruit ripening”. Journal of Experimental Botany. 53 (377): 2039–2055. doi:10.1093/jxb/erf072. PMID 12324528.
- Drauz K, Grayson I, Kleemann A, Krimmer HP, Leuchtenberger W, Weckbecker C (2007). “Amino Acids”. Ullmann’s Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH.
- Rodnina MV, Beringer M, Wintermeyer W (January 2007). “How ribosomes make peptide bonds”. Trends in Biochemical Sciences. 32 (1): 20–26. doi:10.1016/j.tibs.2006.11.007. PMID 17157507.
- Driscoll DM, Copeland PR (2003). “Mechanism and regulation of selenoprotein synthesis”. Annual Review of Nutrition. 23 (1): 17–40. doi:10.1146/annurev.nutr.23.011702.073318. PMID 12524431.
- Krzycki JA (December 2005). “The direct genetic encoding of pyrrolysine”. Current Opinion in Microbiology. 8 (6): 706–712. doi:10.1016/j.mib.2005.10.009. PMID 16256420.
- Théobald-Dietrich A, Giegé R, Rudinger-Thirion J (2005). “Evidence for the existence in mRNAs of a hairpin element responsible for ribosome dependent pyrrolysine insertion into proteins”. Biochimie. 87 (9–10): 813–817.
- Kryukov GV, Castellano S, Novoselov SV, Lobanov AV, Zehtab O, Guigó R, Gladyshev VN (May 2003). “Characterization of mammalian selenoproteomes”. Science. 300 (5624): 1439–1443. Bibcode:2003Sci…300.1439K. doi:10.1126/science.1083516. PMID 12775843. S2CID 10363908. Archived from the original on 23 July 2018. Retrieved 12 June 2019.
- Gromer S, Urig S, Becker K (January 2004). “The thioredoxin system—from science to clinic”. Medicinal Research Reviews. 24 (1): 40–89. doi:10.1002/med.10051. PMID 14595672. S2CID 1944741.
- Tjong H (2008). Modeling Electrostatic Contributions to Protein Folding and Binding (PhD thesis). Florida State University. p. 1 footnote. Archived from the original on 28 January 2020. Retrieved 28 January 2020.
- Stewart L, Burgin AB (2005). “Whole Gene Synthesis: A Gene-O-Matic Future”. Frontiers in Drug Design & Discovery. 1. Bentham Science Publishers: 299. doi:10.2174/1574088054583318. ISBN 978-1-60805-199-1. ISSN 1574-0889. Archived from the original on 14 April 2021. Retrieved 5 January 2016.
- Elzanowski A, Ostell J (7 April 2008). “The Genetic Codes”. National Center for Biotechnology Information (NCBI). Archived from the original on 20 August 2016. Retrieved 10 March 2010.
- Xie J, Schultz PG (December 2005). “Adding amino acids to the genetic repertoire”. Current Opinion in Chemical Biology. 9 (6): 548–554. doi:10.1016/j.cbpa.2005.10.011. PMID 16260173.
- Wang Q, Parrish AR, Wang L (March 2009). “Expanding the genetic code for biological studies”. Chemistry & Biology. 16 (3): 323–336. doi:10.1016/j.chembiol.2009.03.001. PMC 2696486. PMID 19318213.
- Simon M (2005). Emergent computation: emphasizing bioinformatics. New York: AIP Press/Springer Science+Business Media. pp. 105–106. ISBN 978-0-387-22046-8.
- Blenis J, Resh MD (December 1993). “Subcellular localization specified by protein acylation and phosphorylation”. Current Opinion in Cell Biology. 5 (6): 984–989. doi:10.1016/0955-0674(93)90081-Z. PMID 8129952.
- Vermeer C (March 1990). “Gamma-carboxyglutamate-containing proteins and the vitamin K-dependent carboxylase”. The Biochemical Journal. 266 (3): 625–636. doi:10.1042/bj2660625. PMC 1131186. PMID 2183788.
- Bhattacharjee A, Bansal M (March 2005). “Collagen Structure: the Madras triple helix and the current scenario”. IUBMB Life. 57 (3): 161–172. doi:10.1080/15216540500090710. PMID 16036578. S2CID 7211864.
- Park MH (February 2006). “The post-translational synthesis of a polyamine-derived amino acid, hypusine, in the eukaryotic translation initiation factor 5A (eIF5A)”. Journal of Biochemistry. 139 (2): 161–169. doi:10.1093/jb/mvj034. PMC 2494880. PMID 16452303.
- Curis E, Nicolis I, Moinard C, Osowska S, Zerrouk N, Bénazeth S, Cynober L (November 2005). “Almost all about citrulline in mammals”. Amino Acids. 29 (3): 177–205. doi:10.1007/s00726-005-0235-4. PMID 16082501. S2CID 23877884.
- Coxon KM, Chakauya E, Ottenhof HH, Whitney HM, Blundell TL, Abell C, Smith AG (August 2005). “Pantothenate biosynthesis in higher plants”. Biochemical Society Transactions. 33 (Pt 4): 743–746. doi:10.1042/BST0330743. PMID 16042590.
- Sakami W, Harrington H (1963). “Amino acid metabolism”. Annual Review of Biochemistry. 32 (1): 355–398. doi:10.1146/annurev.bi.32.070163.002035. PMID 14144484.
- Brosnan JT (April 2000). “Glutamate, at the interface between amino acid and carbohydrate metabolism”. The Journal of Nutrition. 130 (4S Suppl): 988S–990S. doi:10.1093/jn/130.4.988S. PMID 10736367.
- Young VR, Ajami AM (September 2001). “Glutamine: the emperor or his clothes?”. The Journal of Nutrition. 131 (9 Suppl): 2449S–2459S, 2486S–2487S. doi:10.1093/jn/131.9.2449S. PMID 11533293.
- Young VR (August 1994). “Adult amino acid requirements: the case for a major revision in current recommendations”. The Journal of Nutrition. 124 (8 Suppl): 1517S–1523S. doi:10.1093/jn/124.suppl_8.1517S. PMID 8064412.
- Fürst P, Stehle P (June 2004). “What are the essential elements needed for the determination of amino acid requirements in humans?”. The Journal of Nutrition. 134 (6 Suppl): 1558S–1565S. doi:10.1093/jn/134.6.1558S. PMID 15173430.
- Reeds PJ (July 2000). “Dispensable and indispensable amino acids for humans”. The Journal of Nutrition. 130 (7): 1835S–1840S. doi:10.1093/jn/130.7.1835S. PMID 10867060.
- Imura K, Okada A (January 1998). “Amino acid metabolism in pediatric patients”. Nutrition. 14 (1): 143–148. doi:10.1016/S0899-9007(97)00230-X. PMID 9437700.
- Lourenço R, Camilo ME (2002). “Taurine: a conditionally essential amino acid in humans? An overview in health and disease”. Nutricion Hospitalaria. 17 (6): 262–270.
- Savelieva KV, Zhao S, Pogorelov VM, Rajan I, Yang Q, Cullinan E, Lanthorn TH (2008). Bartolomucci A (ed.). “Genetic disruption of both tryptophan hydroxylase genes dramatically reduces serotonin and affects behavior in models sensitive to antidepressants”. PLOS ONE. 3 (10): e3301. Bibcode:2008PLoSO…3.3301S. doi:10.1371/journal.pone.0003301. PMC 2565062. PMID 18923670.
- Shemin D, Rittenberg D (December 1946). “The biological utilization of glycine for the synthesis of the protoporphyrin of hemoglobin”. The Journal of Biological Chemistry. 166 (2): 621–625. doi:10.1016/S0021-9258(17)35200-6. PMID 20276176. Archived from the original on 7 May 2022. Retrieved 3 November 2008.
- Tejero J, Biswas A, Wang ZQ, Page RC, Haque MM, Hemann C, Zweier JL, Misra S, Stuehr DJ (November 2008). “Stabilization and characterization of a heme-oxy reaction intermediate in inducible nitric-oxide synthase”. The Journal of Biological Chemistry. 283 (48): 33498–33507. doi:10.1074/jbc.M806122200. PMC 2586280. PMID 18815130.
- Rodríguez-Caso C, Montañez R, Cascante M, Sánchez-Jiménez F, Medina MA (August 2006). “Mathematical modeling of polyamine metabolism in mammals”. The Journal of Biological Chemistry. 281 (31): 21799–21812. doi:10.1074/jbc.M602756200. PMID 16709566.
- Jump up to:a b Stryer L, Berg JM, Tymoczko JL (2002). Biochemistry (5th ed.). New York: W.H. Freeman. pp. 693–698. ISBN 978-0-7167-4684-3.
- Petroff OA (December 2002). “GABA and glutamate in the human brain”. The Neuroscientist. 8 (6): 562–573. doi:10.1177/1073858402238515. PMID 12467378. S2CID 84891972.
- Turner EH, Loftis JM, Blackwell AD (March 2006). “Serotonin a la carte: supplementation with the serotonin precursor 5-hydroxytryptophan”. Pharmacology & Therapeutics. 109 (3): 325–338. doi:10.1016/j.pharmthera.2005.06.004. PMID 16023217. S2CID 2563606. Archived from the original on 13 April 2020. Retrieved 12 June 2019.
- Kostrzewa RM, Nowak P, Kostrzewa JP, Kostrzewa RA, Brus R (March 2005). “Peculiarities of L-DOPA treatment of Parkinson’s disease”. Amino Acids. 28 (2): 157–164. doi:10.1007/s00726-005-0162-4. PMID 15750845. S2CID 33603501.
- Heby O, Persson L, Rentala M (August 2007). “Targeting the polyamine biosynthetic enzymes: a promising approach to therapy of African sleeping sickness, Chagas’ disease, and leishmaniasis”. Amino Acids. 33 (2): 359–366. doi:10.1007/s00726-007-0537-9. PMID 17610127. S2CID 26273053.
- Rosenthal GA (2001). “L-Canavanine: a higher plant insecticidal allelochemical”. Amino Acids. 21 (3): 319–330. doi:10.1007/s007260170017. PMID 11764412. S2CID 3144019.
- Hammond, Andrew C. (1 May 1995). “Leucaena toxicosis and its control in ruminants”. Journal of Animal Science. 73 (5): 1487–1492. doi:10.2527/1995.7351487x. PMID 7665380. Archived from the original on 7 May 2022. Retrieved 7 May 2022.
- Jump up to:a b
- Ibba M, Söll D (May 2001). “The renaissance of aminoacyl-tRNA synthesis”. EMBO Reports. 2 (5): 382–387. doi:10.1093/embo-reports/kve095. PMC 1083889. PMID 11375928.
- Lengyel P, Söll D (June 1969). “Mechanism of protein biosynthesis”. Bacteriological Reviews. 33 (2): 264–301. doi:10.1128/MMBR.33.2.264-301.1969. PMC 378322. PMID 4896351.
- Wu G, Fang YZ, Yang S, Lupton JR, Turner ND (March 2004). “Glutathione metabolism and its implications for health”. The Journal of Nutrition. 134 (3): 489–492. doi:10.1093/jn/134.3.489. PMID 14988435.
- Meister A (November 1988). “Glutathione metabolism and its selective modification”. The Journal of Biological Chemistry. 263 (33): 17205–17208. doi:10.1016/S0021-9258(19)77815-6. PMID 3053703.
- Carpino LA (1992). “1-Hydroxy-7-azabenzotriazole. An efficient peptide coupling additive”. Journal of the American Chemical Society. 115 (10): 4397–4398. doi:10.1021/ja00063a082.
- Marasco D, Perretta G, Sabatella M, Ruvo M (October 2008). “Past and future perspectives of synthetic peptide libraries”. Current Protein & Peptide Science. 9 (5): 447–467. doi:10.2174/138920308785915209. PMID 18855697.
- Konara S, Gagnona K, Clearfield A, Thompson C, Hartle J, Ericson C, Nelson C (2010). “Structural determination and characterization of copper and zinc bis-glycinates with X-ray crystallography and mass spectrometry”. Journal of Coordination Chemistry. 63 (19): 3335–3347.
- Muñoz-Huerta RF, Guevara-Gonzalez RG, Contreras-Medina LM, Torres-Pacheco I, Prado-Olivarez J, Ocampo-Velazquez RV (August 2013). “A review of methods for sensing the nitrogen status in plants: advantages, disadvantages and recent advances”. Sensors. 13 (8). Basel, Switzerland: 10823–43. Bibcode:2013Senso..1310823M. doi:10.3390/s130810823. PMC 3812630. PMID 23959242.
- Martin PD, Malley DF, Manning G, Fuller L (2002). “Determination of soil organic carbon and nitrogen at thefield level using near-infrared spectroscopy”. Canadian Journal of Soil Science. 82 (4): 413–422.
