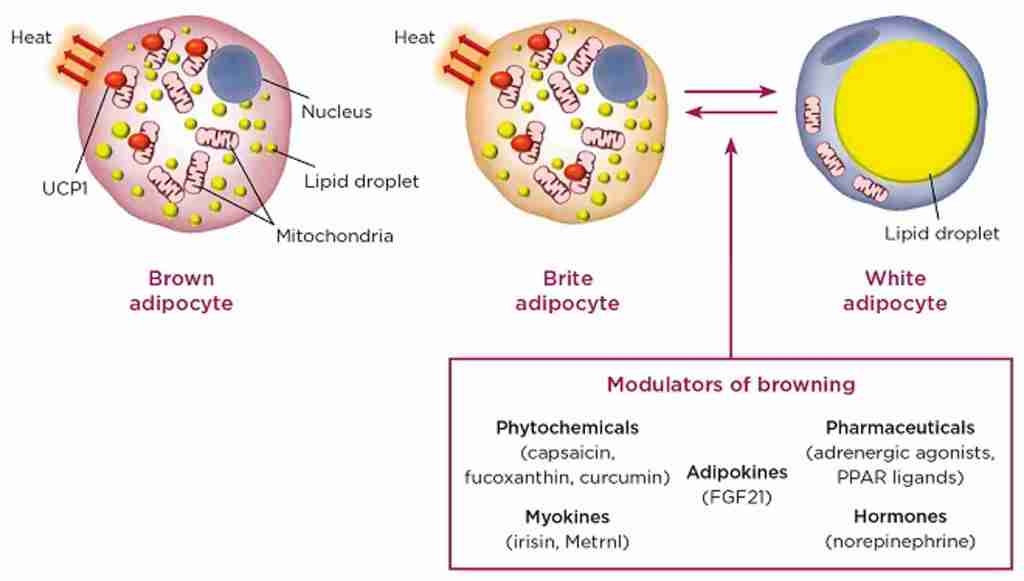
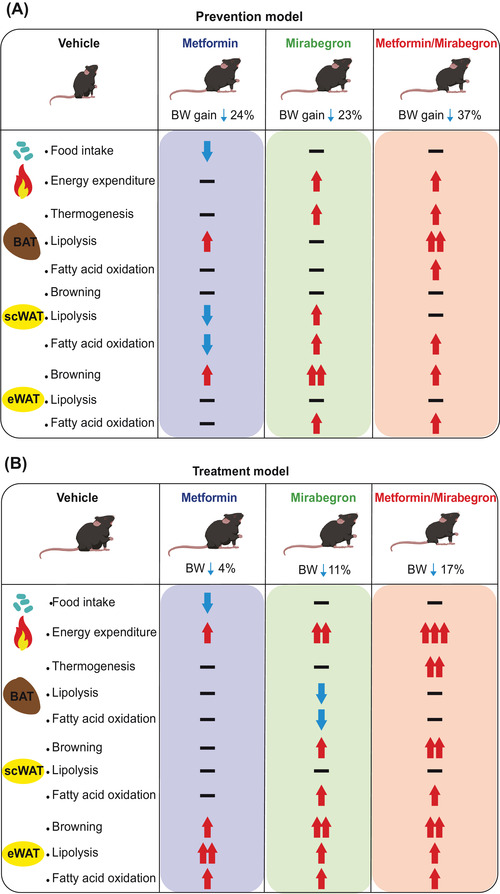
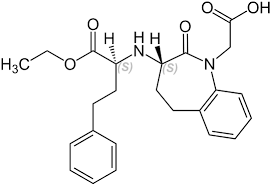
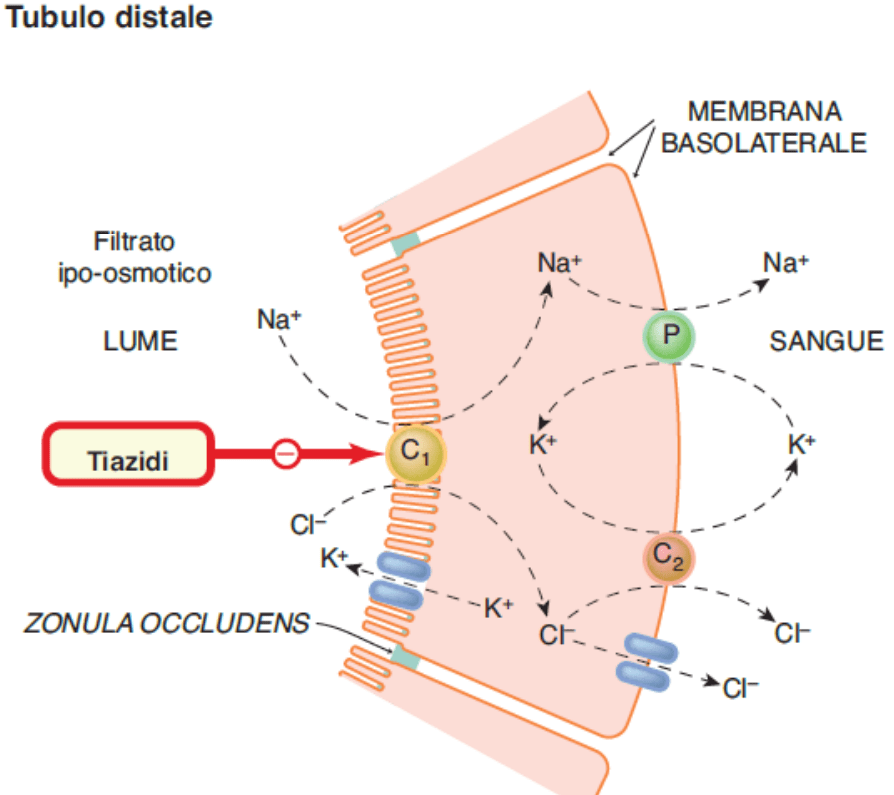
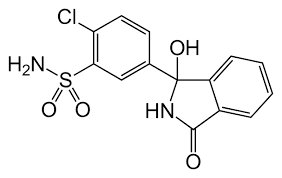
Introduzione:
La ricerca spasmodica di nuove molecole da aggiungere al già ricco corollario a disposizione dell’atleta enhanced rischia di farci trascurare la piena conoscenza di molecole “datate” ma lungi dall’essere completamente comprese nel loro pieno potenziale applicativo. Se ci pensiamo, molecole come la Metformina, dopo più di cento anni, stanno mostrando nuove possibilità applicative anche per ciò che concerne la composizione corporea. E vogliamo parlare del Boldenone e delle recenti scoperte effettuate direttamente sul campo riguardanti le sue potenzialità come “ormone esca” per l’Aromatasi? Non sto ovviamente affermando che non si debba proseguire con una ricerca volta alla identificazione di nuove molecole, ma se dobbiamo parlare di priorità in campo enhanced, appunto, il discorso cambia. Cambia essenzialmente per due cose: 1) presenza di numerose molecole che rendono obsoleto e/o non necessario l’inserimento di altre che, nonostante alcuni vantaggi più o meno degni di nota, non offrono cambiamenti tali e conoscenza gestionali pari o superiori alle “vecchie molecole” e 2) la mancanza di una universale, o quasi, conoscenza di molecole in uso da decenni.
Fatte queste doverose premesse, in questo articolo parlerò dei Sodium-dependent glucose cotransporters (o sodium-glucose linked transporter, SGLT; in italiano Cotrasportatori del Glucosio Sodio-Dipendenti), e dei loro inibitori i quali si stanno dimostrando dei promettenti farmaci ancillari soprattutto, ma non solo, per l’Harm Reduction cardio-renale.
Introduzione ai SGLT e inibitori SGLT:
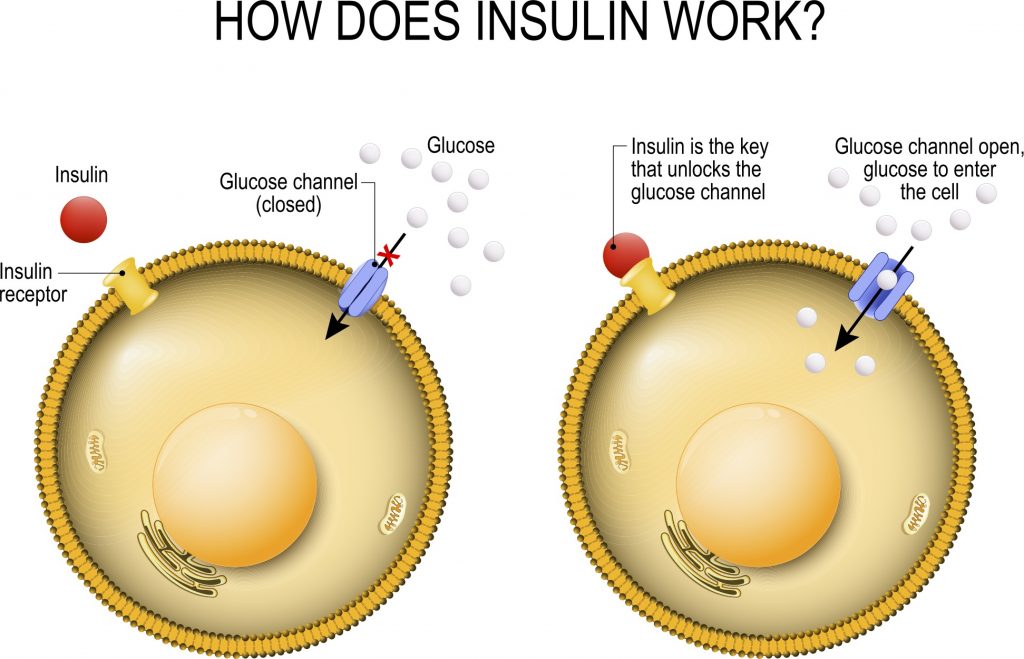
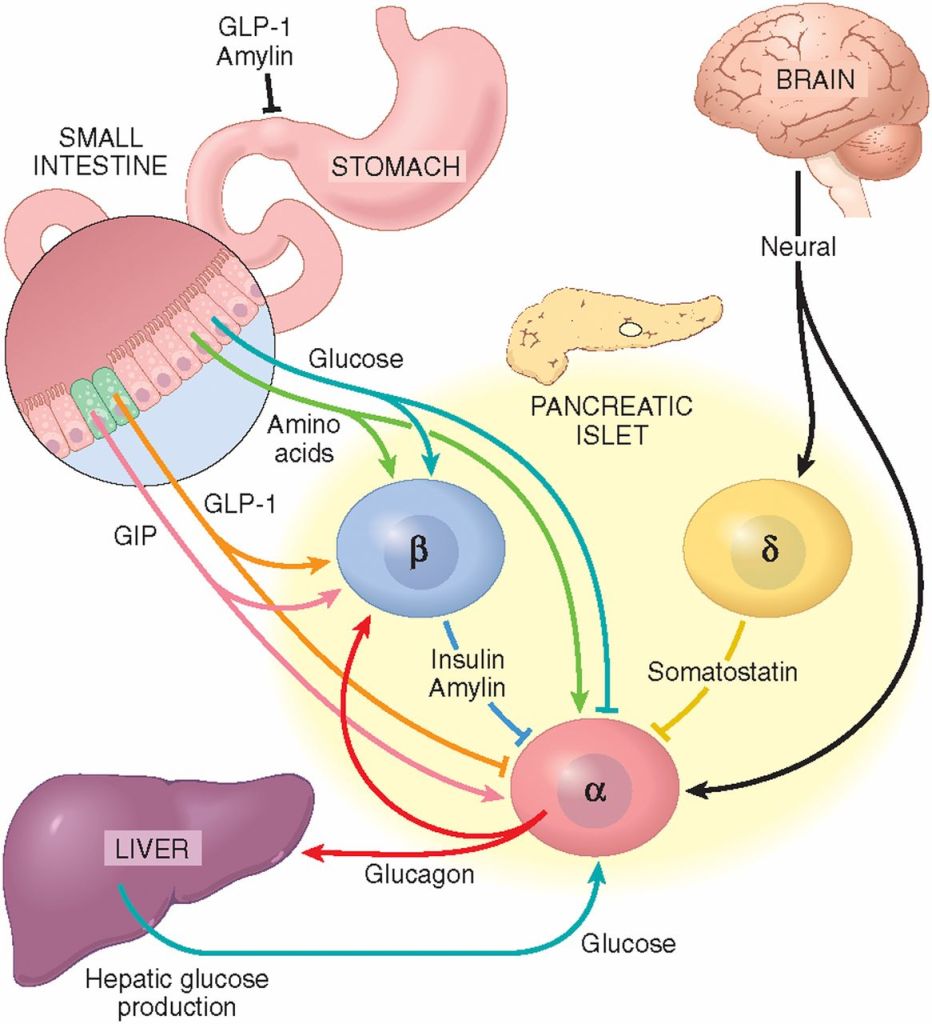
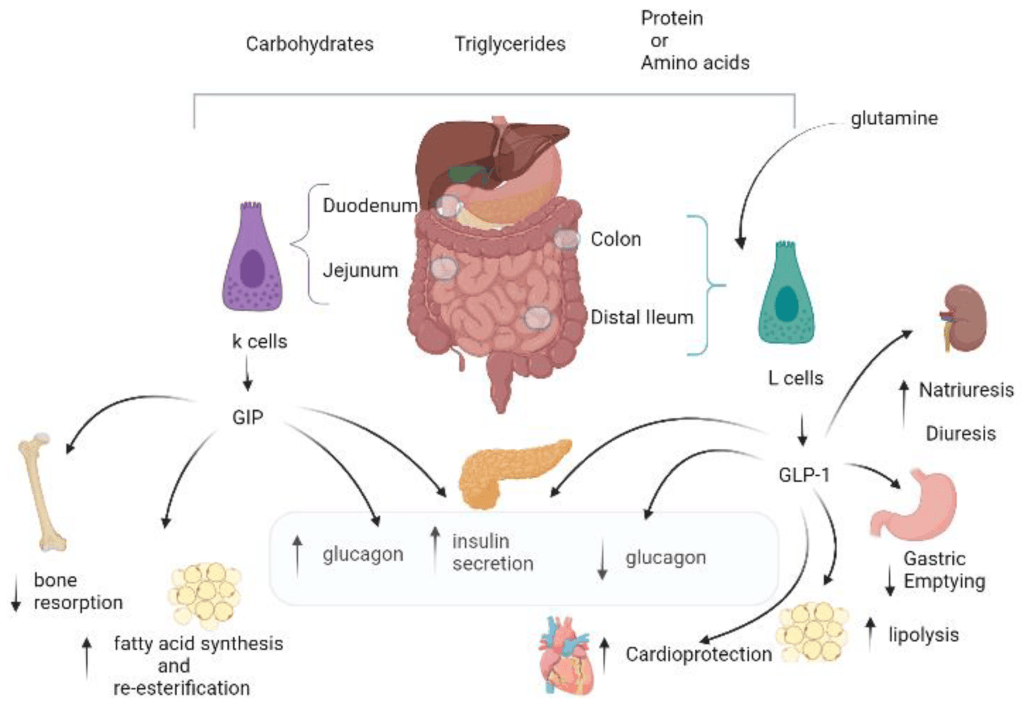
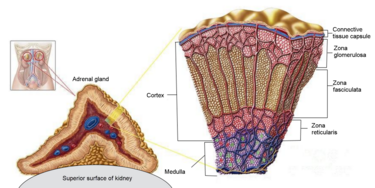
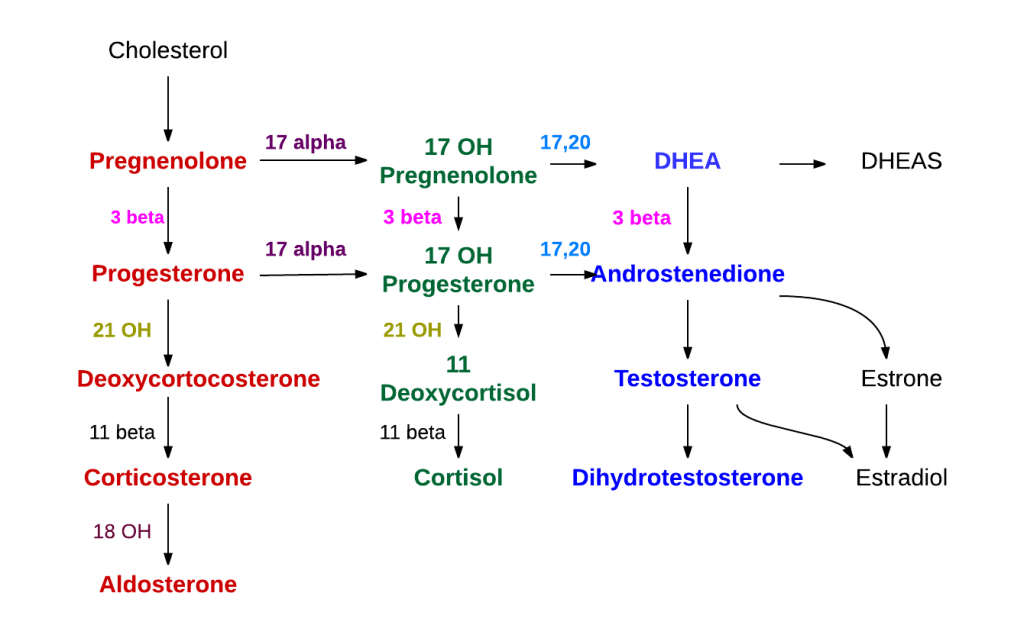
I Cotrasportatori di Glucosio Sodio-Dipendenti (o trasportatori sodio-glucosio, SGLT) sono una famiglia di trasportatori di glucosio presenti nella mucosa intestinale (enterociti) dell’intestino tenue (SGLT1) e nel tubulo prossimale del nefrone (SGLT2 nel tubulo contorto prossimale [PCT] e SGLT1 nel tubulo retto prossimale [PST]). Contribuiscono al riassorbimento renale del glucosio. Nei reni, il 100% del glucosio filtrato nel glomerulo deve essere riassorbito lungo il nefrone (98% nel PCT, tramite SGLT2). Se la concentrazione plasmatica di glucosio è troppo alta (iperglicemia), il glucosio passa nelle urine (glucosuria) perché gli SGLT sono saturi con il glucosio filtrato.
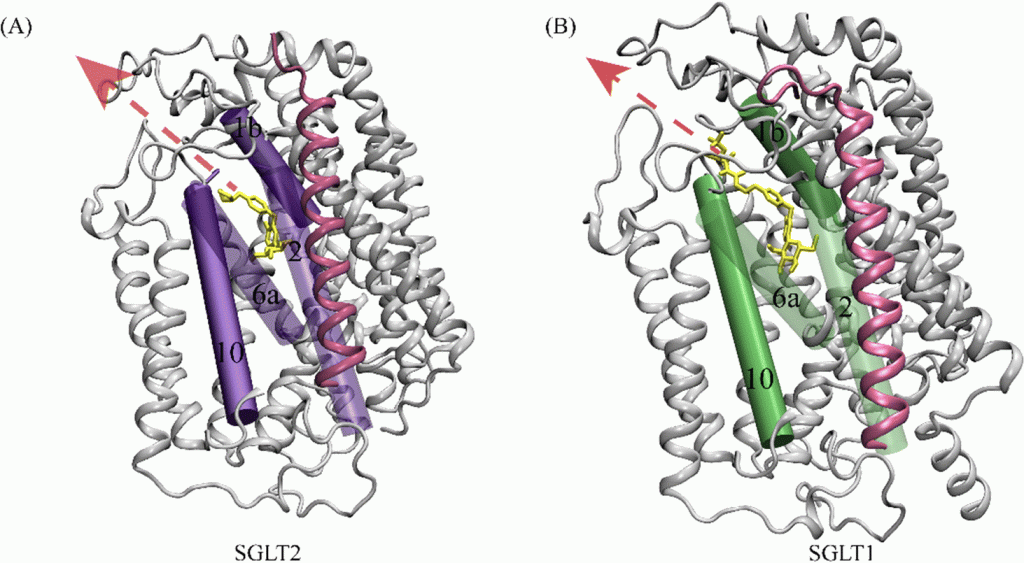
Nei mammiferi, il movimento del glucosio dentro e fuori le cellule è ottenuto dai trasportatori del glucosio (GLUT) sulla membrana cellulare. I GLUT si dividono in due tipi strutturalmente e funzionalmente distinti: (1 GLUT, che operano per diffusione facilitata (1, 2); e (2 SGLT, che trasportano attivamente il glucosio contro il gradiente di concentrazione accoppiandosi con il sodio (3, 4). I GLUT sono presenti in tutte le cellule del corpo per facilitare il trasporto del glucosio nelle cellule e le concentrazioni di glucosio dentro e fuori le cellule diventano uguali con l’operazione dei GLUT 1. Negli SGLT, che comprendono una famiglia di almeno sei diverse isoforme nell’uomo, glucosio e sodio vengono simultaneamente cotrasportati nelle cellule utilizzando il gradiente di concentrazione del sodio (5). Tra questi SGLT, SGLT1 e SGLT2 sono stati frequentemente studiati, perché svolgono un ruolo chiave nel trasporto di glucosio e sodio attraverso la membrana dell’orletto a spazzola delle cellule intestinali e renali (3 , 6) .
Nell’epitelio intestinale, l’afflusso di glucosio nelle cellule epiteliali è catalizzato da SGLT1 situato nella membrana apicale, e il glucosio fluisce nella circolazione attraverso GLUT2 situato nella membrana basolaterale (5, 7). Inoltre, i due tipi di trasportatori, GLUT e SGLT, lavorano insieme nelle cellule tubulari renali, con gli SGLT (SGLT2 e SGLT1) che trasportano il glucosio nelle cellule tubulari attraverso la membrana apicale, e i GLUT (GLUT2 e GLUT1) che trasportano il glucosio attraverso la membrana basolaterale nella circolazione sanguigna (7, 8).

Nell’agosto del 1960, a Praga, Robert K. Crane presentò per la prima volta la sua scoperta del cotrasporto sodio-glucosio come meccanismo di assorbimento intestinale del glucosio.[9]
La scoperta del cotrasporto da parte di Crane fu la prima proposta in assoluto di accoppiamento di flusso in biologia.[10][11]
Recentemente, sono stati sviluppati inibitori di SGLT2, basati su un nuovo concetto di azione antidiabetica mediante l’inibizione del riassorbimento renale del glucosio e l’aumento dell’escrezione di glucosio nelle urine. Gli inibitori di SGLT2 riducono la glicotossicità abbassando la glicemia, e studi clinici su larga scala hanno riportato una diminuzione della mortalità cardiovascolare e gli effetti protettivi renali (12) . Inoltre, SGLT1 è responsabile dell’assorbimento del glucosio nell’intestino tenue e del riassorbimento di parte del carico di glucosio filtrato nel rene (13) , e potrebbe essere un bersaglio interessante per il mantenimento di un buon controllo glicemico e il miglioramento della disfunzione renale (7 , 14) .

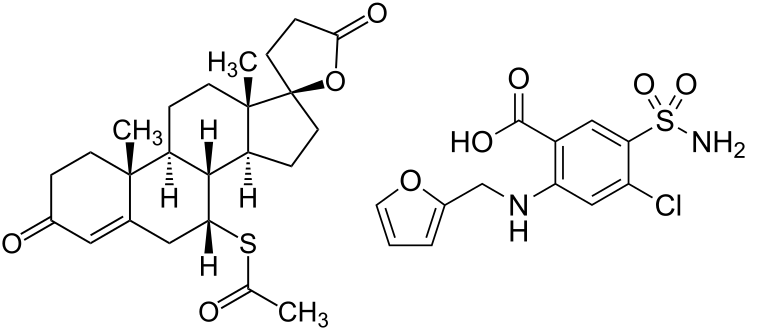
La Florizina, un diidrocalcone isolato dalla corteccia dei meli nel 1835, è nota per essere la prima sostanza naturale con attività inibitoria SGLT [15]. A causa delle sue somiglianze con gli estratti di china e salice, la florizina era precedentemente considerata un candidato per il trattamento di febbri, malattie infettive e malaria [16]. Circa 50 anni dopo, Chasis et al. [17] hanno riferito che la Florizina inibisce il riassorbimento renale del glucosio e aumenta l’escrezione urinaria di glucosio. L’associazione tra la Florizina e il sistema di trasporto attivo del glucosio dell’orletto a spazzola del tubulo prossimale è stata rivelata all’inizio degli anni ’70. Inoltre, diversi studi in vivo su modelli animali diabetici hanno dimostrato che la somministrazione di Florizina ha ridotto i livelli di glicemia a digiuno e/o postprandiali e aumentato la sensibilità all’Insulina [18,19,20,21]. Più recentemente, Katsuno et al. [22] hanno riportato che la Florizina ha inibito sia l’SGLT1 che l’SGLT2 umano, con valori di costante inibitoria (Ki) rispettivamente di 151 e 18,6 nM. Tuttavia, nonostante i suoi sufficienti effetti inibitori contro gli SGLT, la Florizina è stata infine considerata inappropriata per un ulteriore sviluppo come farmaco antiperglicemico a causa di alcuni svantaggi critici. In primo luogo, la Florizina inibisce sia SGLT1 che SGLT2 con bassa selettività terapeutica. L’inibizione di SGLT1, che è localizzata principalmente nell’intestino tenue, può causare diversi effetti collaterali gastrointestinali, come diarrea, disidratazione e malassorbimento. In secondo luogo, la Florizina è scarsamente assorbita nell’intestino tenue, a causa della sua bassa biodisponibilità orale. In terzo luogo, la Floretina, un metabolita idrolitico della Florizina catalizzato dalle β-glicosidasi, inibisce fortemente il trasportatore ubiquitario del glucosio 1 (GLUT1), che può quindi ostacolare l’assorbimento del glucosio in vari tessuti [23,24].
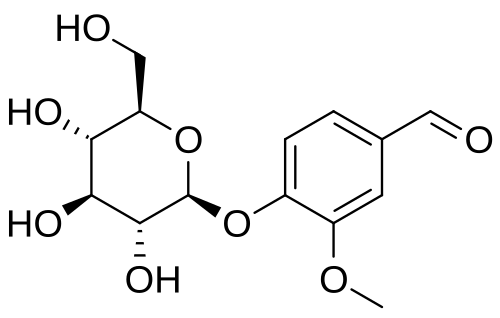
Per superare queste limitazioni, diverse aziende farmaceutiche hanno avviato ricerche approfondite per sviluppare nuovi analoghi a base di florizina con biodisponibilità e stabilità migliorate, nonché selettività per SGLT2. Nelle fasi iniziali, si sono concentrate sugli analoghi degli O-glicosidi ed è stato sviluppato un inibitore selettivo di SGLT2 disponibile per via orale, il T-1095 [25]. Il T-1095 ha subito un ampio metabolismo epatico nella sua forma attiva, il T-1095A, con conseguente diminuzione dose-dipendente del riassorbimento urinario del glucosio e soppressione dell’aumento della glicemia, insieme a un aumento dell’escrezione urinaria di glucosio. I valori di IC50 calcolati del T-1095A per SGLT1 e SGLT2 umani erano rispettivamente di circa 200 nM e 50 nM, il che rifletteva attività inibitorie più selettive e potenti rispetto al florizina [26]. Sono stati sviluppati altri derivati dell’O-glucoside sergliflozin [26], remogliflozin [27] e AVE2268 [28,29] e le loro proprietà farmacocinetiche e/o farmacodinamiche sono state valutate in vari contesti in vivo e clinici [30,31,32,33,34,35,36,37,38,39,40]. Sebbene questi inibitori dell’O-glicoside abbiano mostrato una degradazione mediata dalla glucosidasi minimizzata e una maggiore esposizione sistemica, la loro scarsa stabilità farmacocinetica e la selettività farmacologica incompleta per SGLT2 hanno rivolto l’interesse di molti scienziati e aziende farmaceutiche verso altri derivati della florizina, i C-glucosidi.
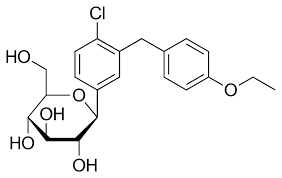
Da quando è stata eseguita la prima sintesi di analoghi del C-glicoside della florizina nel 2000 [41], sono stati effettuati numerosi tentativi di trovare sostituenti ottimali con sufficiente potenza e selettività contro SGLT2. Di conseguenza, nel 2008, Meng et al. [42] hanno sviluppato dapagliflozin, con sostituenti etossilici lipofili in posizione 4 sull’anello B della florizina. Dapagliflozin ha mostrato una potenza oltre 1200 volte superiore per SGLT2 umano [IC50 (nM): 1,12] rispetto a SGLT1 [IC50 (nM): 1391]. Una risposta glicosurica dose-dipendente e una diminuzione dei livelli di glucosio nel sangue a digiuno e postprandiale sono state osservate anche dopo somministrazione orale di dapagliflozin ai ratti [43,44]. In quanto candidato antidiabetico innovativo, l’efficacia del dapagliflozin è stata valutata in molti studi clinici e questo agente ha mostrato una significativa riduzione dei livelli di glucosio plasmatico e di emoglobina glicata (HbA1c), un migliore controllo glicemico e una riduzione del peso corporeo [45,46,47,48,49,50,51,52]. Il dapagliflozin è stato approvato e commercializzato per la prima volta in Europa nel 2012 e anche il comitato della Food and Drug Administration (FDA) degli Stati Uniti ha approvato questo farmaco per il trattamento del diabete di tipo 2 nel gennaio 2014.

A partire dalla comparsa di dapagliflozin, sono stati successivamente sviluppati diversi inibitori del C-glucoside. Canagliflozin, caratterizzato da un derivato tiofenico del C-glucoside [53], è stato approvato dalla FDA nel 2013. Insieme a una differenza di oltre 400 volte nelle attività inibitorie tra SGLT1 e SGLT2 umani [IC50 (nM) SGLT1: 910; SGLT2: 2,2] [53], canagliflozin ha mostrato buone proprietà antiperglicemiche paragonabili a dapagliflozin in molte pratiche cliniche [54,55,56,57]. Empagliflozin è stato il terzo agente nella classe gliflozin approvato sia dall’Agenzia europea per i medicinali (EMA) che dalla FDA nel 2014, avendo la più alta selettività per SGLT2 rispetto a SGLT1 (circa 2700 volte) tra gli inibitori SGLT2 sul mercato [57]. Negli ultimi anni, molte industrie farmaceutiche giapponesi hanno guidato lo sviluppo di inibitori SGLT2 di nuova generazione, tra cui ipragliflozin, tofogliflozin e luseogliflozin. Altri composti, ertugliflozin e LX-4211 (sotagliflozin, un doppio inibitore SGLT1/2), sono ora in fase avanzata di sperimentazione clinica [57].
- Riepilogo
Ricapitolando, gli SGLT costituiscono una vasta famiglia di proteine di membrana correlate a vari trasporti di glucosio, amminoacidi, vitamine e alcuni ioni attraverso la membrana apicale del lume, inclusi l’intestino tenue e i tubuli renali. Nell’uomo, sono state segnalate sei diverse isoforme e due trasportatori, le proteine SGLT1 (soluto-trasportatore [SLC]5A1) e SGLT2 (SLC5A2), sono state ampiamente studiate (58) . SGLT1 è stato scoperto tramite clonazione di espressione nel 1987 (59) , e SGLT2 è stato identificato tramite screening per omologia nel 1994 (60) . I GLUT equilibrano i livelli di glucosio su entrambi i lati della membrana plasmatica, poiché il gradiente di glucosio attraverso la membrana è la forza motrice, mentre SGLT2 può esercitare differenze nella concentrazione di glucosio, poiché il gradiente di sodio transmembrana è la forza motrice per l’assorbimento del glucosio.
SGLT1 è responsabile dell’assorbimento del glucosio nell’intestino tenue, mentre SGLT2 è responsabile del riassorbimento del glucosio nel rene (61, 62). Considerando le funzioni fisiologiche di SGLT1 e SGLT2, la ricerca sul recupero di farmaci mirati ai trasportatori è ragionevole. Nel 1987, è stato riportato che la florizina, un inibitore di SGLT1 e SGLT2, inverte il diabete sperimentale in ratti parzialmente pancreatectomizzati (63). Gli inibitori di SGLT sono stati sviluppati utilizzando la florizina come composto guida (64, 65), con conseguente sviluppo di inibitori di SGLT2, che sono stati lanciati con successo sul mercato (66, 67).

Per quanto riguarda gli altri SGLT, SGLT3 (nome del gene: SLC5A4), che è espresso nell’intestino, nella milza, nel fegato, nei reni, nel muscolo scheletrico e nei neuroni colinergici, non è un SGLT funzionale e sembra agire come un sensore del glucosio nella membrana plasmatica dei neuroni colinergici (67). Ci sono solo pochi rapporti sugli altri SGLT: SGLT4, SGLT5 e SGLT6. SGLT4 (nome del gene: SLC5A9) è espresso nell’intestino tenue, nei reni, nel fegato, nei polmoni, nel cervello, nella trachea, nell’utero e nel pancreas; SGLT5 (nome del gene: SLC5A10) è espresso solo nei reni; e SGLT6 (nome del gene: SLC5A11) è considerato un trasportatore di d-glucosio a bassa affinità nell’intestino tenue (67). I ruoli fisiologici di questi SGLT rimangono sconosciuti.
Proprietà basali del SGLT1:

La proteina SGLT1, codificata dal gene SLC5A sul cromosoma 22q13.1, è composta da 664 amminoacidi, comprendenti 14 domini α-elicoidali transmembrana, un singolo sito di glicosilazione tra le eliche transmembrana 5 e 6 e due siti di fosforilazione, tra le eliche transmembrana 6 e 7 e tra 8 e (68) . I terminali NH2 e COOH si trovano rispettivamente nelle membrane extracellulari e intracellulari e si suppone che il dominio di legame al glucosio includa i residui amminoacidici 457–460 (69). SGLT1 è un trasportatore ad alta affinità per il glucosio (costante di Michaelis-Menten [K m] = 0,4 mmol/L) e il galattosio, mentre il fruttosio non viene trasportato (70). Due ioni sodio vengono trasportati attraverso SGLT1 per ogni molecola di glucosio e questo cotrasportatore è autorizzato a trasportare il glucosio nelle cellule contro il suo gradiente di concentrazione.
L’espressione dell’mRNA SGLT1 è stata rilevata mediante reazione a catena della polimerasi con trascrizione inversa nei seguenti tessuti nell’uomo: intestino tenue, rene, muscolo scheletrico, fegato, polmone, cuore, trachea, prostata, testicolo, cervice uterina, stomaco, tessuto adiposo mesenterico, cellule α pancreatiche, colon e cervello (71). L’espressione della proteina SGLT1 è stata localizzata nell’orletto a spazzola apicale dell’intestino tenue e nei tubuli prossimali tardivi, ed è stata rilevata anche nei seguenti tessuti nell’uomo: ghiandole salivari, fegato, polmone, muscolo scheletrico, cuore e cellule α pancreatiche (72).
SGLT1 esercita l’attività di trasporto tramite numerose regolazioni molecolari, tra cui le protein chinasi. SGLT1 contiene siti di regolazione specifici per ceppo da parte della protein chinasi A (PKA) e della protein chinasi C (PKC): un sito PKA nell’uomo e nel coniglio, nessuno nel ratto; cinque siti PKC di consenso nell’uomo e nei ratti e quattro siti nei conigli (73). L’attivazione di PKA ha portato a un aumento del numero di proteine SGLT1 nella membrana dell’intestino tenue nei ratti (74) e l’attivatore di PKA, 8-bromo-adenosina monofosfato ciclico, o forskolina, ha aumentato la capacità di SGLT e l’attività di SGLT1 nella membrana plasmatica (75). L’espressione e l’attività di SGLT1 sono regolate positivamente dall’attività di PKA e gli effetti sull’attivazione di SGLT1 sono stati inibiti dall’inibitore di PKA, H-89 (76). Sono stati segnalati anche effetti mediati da PKC su SGLT1, ma sono ammesse evidenti differenze tra le specie e gli effetti sono controversi. L’attivazione di PKC ha ridotto la capacità di trasporto di SGLT1 nei ratti e nei conigli, ma ha aumentato la capacità negli esseri umani (74) .
In altri studi, l’attivazione della proteina chinasi attivata dall’adenosina monofosfato ha aumentato il trasporto massimo di glucosio sodio-dipendente (77) l’eliminazione della chinasi 3 inducibile dal siero e dai glucocorticoidi ha causato una diminuzione dell’attività intestinale di SGLT1 (78) e la chinasi ricca di prolina alanina correlata a Ste20p ha causato una diminuzione dell’abbondanza di SGLT1 nella membrana plasmatica (79).
L’attività e l’espressione intestinale di SGLT1 sono regolate dal contenuto di carboidrati nella dieta. L’attività e l’espressione di SGLT1 sono aumentate in topi, ratti e pecore alimentati con una dieta ricca di zuccheri (80) e sono mantenute dalla presenza di nutrienti luminali nell’intestino umano (81). Inoltre, l’attività e l’espressione di SGLT1 sono correlate a un ritmo diurno che correla le ore di veglia con la massima espressione di SGLT1 (82).
Proprietà basali del SGLT2:
La proteina SGLT2, codificata da SLC5A2, è composta da 672 amminoacidi e i suoi terminali NH2 e COOH sono extracellulari (83) . I valori di K m nell’SGLT2 umano per glucosio e sodio sono rispettivamente 2 e 25 mmol/L e, a differenza di SGLT1, SGLT2 è un trasportatore di glucosio a bassa affinità e alta capacità (84). SGLT2 è espresso prevalentemente nel rene di roditori e umani e basse espressioni di mRNA sono state rilevate nelle ghiandole mammarie, testicoli, fegato, polmoni, intestino, muscolo scheletrico, milza e cervelletto (85) . Inoltre, SGLT2 è segnalato come espresso nelle cellule α pancreatiche e correlato alla secrezione di glucagone (86). SGLT2 è localizzato nella membrana luminale dei segmenti (S)1 e S2 dei tubuli prossimali renali negli esseri umani e nei roditori, mentre SGLT1 è localizzato nella membrana luminale del segmento S3 (87) . SGLT2 è principalmente responsabile del riassorbimento del glucosio nel nefrone e ≥80% del glucosio filtrato viene riassorbito nei segmenti S1 e S2 dei tubuli prossimali attraverso SGLT2 (88) .
L’attivazione della proteina chinasi A e della PKC ha aumentato l’assorbimento del glucosio rispettivamente del 225 e del 150% nelle cellule renali embrionali umane che esprimono SGLT2 (89). Per quanto riguarda i meccanismi, l’effetto mediato dalla PKA potrebbe essere correlato a un aumento del tasso di fusione delle vescicole con la membrana; tuttavia, non è stato trovato alcun meccanismo simile sull’effetto mediato dalla PKC. Inoltre, l’espressione di SGLT2 è aumentata attraverso l’attivazione della proteina di scambio attivata direttamente dall’adenosina monofosfato ciclico/PKA attraverso la chinasi regolata dal segnale extracellulare/p38 e la proteina chinasi attivata dal mitogeno (90). Nella linea cellulare renale suina, l’interleuchina-6 e il fattore di necrosi tumorale-α hanno aumentato l’espressione dell’mRNA e della proteina SGLT2 (91) e, analogamente, la fosforilazione del fattore di crescita trasformante-β1 e del fattore di trascrizione a valle, smad3, hanno aumentato il livello della proteina SGLT2 nelle cellule tubulari prossimali renali umane (92).
Proprietà funzionali degli SGLT:
- Intestino tenue
L’SGLT1 nell’intestino tenue è localizzato nella membrana cellulare apicale che compone l’orletto a spazzola . SGLT1 è responsabile del trasporto di glucosio o galattosio dal lume alle cellule epiteliali, mentre il trasportatore facilitatore, GLUT2, è successivamente responsabile del trasporto di glucosio dalla membrana basolaterale alla circolazione sanguigna .
Il livello di espressione di SGLT1 determina la capacità di assorbimento del glucosio e subisce regolazioni a breve e lungo termine a seconda dei nutrienti luminali. Una dieta ricca di glucosio o una dieta ricca di sodio aumentano il livello di espressione di SGLT1 nell’intestino tenue. Inoltre, un aumento delle concentrazioni luminali di glucosio induce la traslocazione di GLUT2 alla membrana dell’orletto a spazzola.
L’espressione di SGLT1 nell’intestino tenue è aumentata nel diabete, che è considerato correlato alla risposta a un maggiore apporto di glucosio nella dieta. L’espressione intestinale dell’mRNA di SGLT1 è aumentata nei modelli animali diabetici, come i modelli diabetici indotti da streptozotocina e i ratti Otsuka Long-Evans Tokushima Fatty. Nei pazienti con diabete di tipo 2, l’espressione dell’mRNA e della proteina SGLT1 intestinale nella membrana dell’orletto a spazzola era più elevata e anche l’assorbimento intestinale del glucosio era elevato. Si ritiene che la regolazione positiva dell’assorbimento del glucosio mediato da SGLT1 nell’intestino tenue induca la rapida iperglicemia postprandiale nel diabete (93).
- Rene
Nel rene, il glucosio viene trasportato attraverso la membrana apicale del tubulo contorto prossimale da SGLT2 e SGLT1, ed esce attraverso la membrana basolaterale del tubulo prossimale dai trasportatori facilitatori GLUT2 e GLUT1 . SGLT2 è espresso nella parte superiore del tubulo prossimale, segmenti S1 e S2, mentre SGLT1 è espresso nella parte inferiore del tubulo prossimale, il segmento S3 negli esseri umani e nei roditori.
Nella capacità di riassorbimento del glucosio filtrato nell’euglicemia, SGLT2 esercita la funzione principale, mostrando il suddetto riassorbimento del glucosio ≥80%, mentre SGLT1 riassorbe il glucosio rimanente o circa il 5% del glucosio filtrato. Come punto da notare, il rapporto di accoppiamento di glucosio e sodio è diverso tra i due cotrasportatori: SGLT2 trasporta glucosio e sodio in un rapporto 1:1, mentre SGLT1 trasporta glucosio e sodio in un rapporto 1:2 . La proprietà di trasporto di SGLT2 aumenta il potere di concentrazione per riassorbire il glucosio consegnato alla parte distale del segmento S3 del tubulo prossimale 49. Inoltre, è stato riportato che SGLT1 prepara la capacità altamente riservata di riassorbimento del glucosio. Quando l’inibizione farmacologica di SGLT2 induce il flusso di glucosio a valle nel tubulo prossimale distale, SGLT1 può compensare il riassorbimento del glucosio. Di conseguenza, gli esseri umani euglicemici trattati con inibitori di SGLT2 hanno mantenuto un riassorbimento frazionario del glucosio del 40-50% , e il valore medio del riassorbimento frazionario del glucosio nei topi knockout (KO) SGLT2 euglicemici era del 36% . Nei topi selvatici, l’inibitore di SGLT2, empagliflozin, ha aumentato in modo dose-dipendente l’escrezione urinaria di glucosio, mentre la curva dose-risposta è stata spostata verso sinistra e la risposta massima è raddoppiata nei topi KO per SGLT1. L’effetto compensatorio di SGLT1 è supportato anche da studi su topi SGLT1/SGLT2 doppio KO e topi SGLT1 KO trattati con inibitore di SGLT2 87. L’iperglicemia sostenuta, che induce il superamento della capacità di trasporto dell’SGLT2 prossimale, ha aumentato il flusso di glucosio al tubulo prossimale distale e ha migliorato il riassorbimento del glucosio mediato da SGLT1. La capacità riservata di riassorbimento del glucosio e l’effetto compensatorio di SGLT1 sono proprietà notevoli in considerazione della funzione fisiologica.
Nei modelli animali di diabete di tipo 1 e di tipo 2, il livello della proteina renale SGLT2 è risultato aumentato, mentre i risultati riportati per i livelli renali di SGLT1 sono controversi. I ratti trattati con streptozotocina hanno mostrato un aumento dell’espressione di mRNA e proteine di SGLT1 nella corteccia renale (92, 93). Inoltre, l’espressione di mRNA renale di SGLT1 nei ratti Zucker grassi è risultata aumentata (94). Nei topi ob/ob, il livello della proteina SGLT1 della membrana renale è risultato aumentato, ma l’espressione di mRNA è risultata diminuita. Al contrario, è stato riportato che il livello della proteina SGLT1 della membrana renale è risultato diminuito nei topi Akita diabetici. Le proprietà di SGLT2 e SGLT1 nel riassorbimento renale del glucosio in condizioni euglicemiche sono ben comprese; tuttavia, tali proprietà nello stato diabetico rimangono poco note e, in particolare, una migliore comprensione del significato fisiologico nella regolazione renale di SGLT1 è un argomento fondamentale per il futuro.
- Cuore

La localizzazione della proteina SGLT1 è stata riscontrata nei capillari cardiaci nell’uomo e nei ratti, mentre l’espressione non è stata riscontrata nei capillari dell’intestino tenue . Inoltre, SGLT1 è stato segnalato come espresso nella membrana cellulare dei cardiomiociti nell’uomo e nei topi. Pertanto, SGLT1 cardiaco potrebbe essere coinvolto nel trasporto del glucosio dai capillari ai cardiomiociti. Al contrario, SGLT2 non è espresso nel cuore. Nel cuore, due trasportatori di glucosio facilitati, GLUT1 e GLUT4, svolgono un ruolo primario nell’assorbimento del glucosio: GLUT1 per l’assorbimento basale del glucosio e GLUT4 per l’assorbimento insulino-dipendente del glucosio. Considerati i ruoli fisiologici di SGLT1 nel cuore, il coinvolgimento con i trasportatori di glucosio facilitati è essenziale e non può essere ignorato.
L’espressione dell’mRNA cardiaco di SGLT1 è stata segnalata come aumentata nei pazienti con diabete di tipo 2 e cardiomiopatia diabetica. Nei ratti diabetici trattati con streptozotocina, l’espressione dell’mRNA e della proteina GLUT4 è diminuita, mentre l’espressione dell’mRNA di GLUT1 non è cambiata significativamente. La riduzione dell’attività cardiaca di GLUT4 ha portato a una diminuzione dell’assorbimento del glucosio e allo sviluppo di cardiomiopatia diabetica, mentre i ruoli fisiologici di GLUT1 nel cuore rimangono poco chiari.
Uno studio recente ha riportato che la sovraespressione cardiaca cronica di SGLT1 nei topi ha portato a ipertrofia cardiaca patologica e insufficienza ventricolare sinistra, e l’inibizione cardiaca di SGLT1 ha attenuato il fenotipo della malattia. Al contrario, uno studio recente ha anche riportato che il doppio inibitore di SGLT1/SGLT2 ha esacerbato la disfunzione cardiaca dopo infarto miocardico sperimentale nei ratti (95) . Considerando che SGLT2 non è espresso nel cuore, questo effetto potrebbe essere collegato all’inibizione di SGLT1. Non è ancora chiaro se l’inibizione cardiaca di SGLT1 eserciti effetti protettivi sulle malattie cardiovascolari. Sono necessarie ulteriori ricerche.
- Cervello
L’espressione dell’mRNA di SGLT1 è stata riscontrata nel cervello di esseri umani, conigli, maiali e roditori. Nei conigli e nei maiali, l’espressione dell’mRNA di SGLT1 è stata riscontrata nei neuroni della corteccia frontale, nelle cellule di Purkinje del cervelletto e nei neuroni dell’ippocampo 50. Nei roditori, l’espressione dell’mRNA di SGLT1 è stata riscontrata nei neuroni della corteccia cerebrale, dell’ippocampo, dell’ipotalamo, del corpo striato e del cervelletto. La proteina SGLT1 è stata espressa nei piccoli vasi del cervello dei roditori. Inoltre, un analogo del glucosio selettivo per SGLT1 marcato radioattivamente non è riuscito a superare la barriera emato-encefalica, suggerendo che SGLT1 è localizzato solo nella membrana luminale delle cellule endoteliali. Considerando la localizzazione e la funzione dell’SGLT1, quest’ultimo nel cervello potrebbe svolgere un ruolo chiave come fonte di approvvigionamento energetico per i neuroni in caso di aumento della richiesta di glucosio, come in caso di ipossiemia e ipoglicemia (96).
- Altri organi

Sono stati segnalati alcuni casi di SGLT1 nel polmone, nel fegato, nel pancreas e nei linfociti T. L’mRNA di SGLT1 è stato rilevato nella trachea, nei bronchi e nel tessuto polmonare negli esseri umani, e la proteina SGLT1 è stata rilevata nelle cellule alveolari di tipo 2 e nella membrana luminale delle cellule di Clara nei bronchioli negli esseri umani e nei ratti. L’assorbimento di glucosio mediato da SGLT1 potrebbe essere responsabile dell’assorbimento dei liquidi e fornisce energia per la produzione di tensioattivi nelle cellule alveolari di tipo 2 e di mucina e tensioattivi nelle cellule di Clara.
L’mRNA di SGLT1 è stato rilevato nel fegato e nella cistifellea negli esseri umani , e la proteina SGLT1 è stata rilevata nella membrana apicale delle cellule epiteliali dei dotti biliari negli esseri umani e nei ratti.
Piccole quantità di mRNA di SGLT1 sono state rilevate nel pancreas degli esseri umani, e l’espressione di mRNA e proteine di SGLT1 è stata trovata nelle cellule α pancreatiche di esseri umani e topi (97). Inoltre, l’espressione di mRNA di SGLT1 è stata trovata nei linfociti T attivati dei topi. I ruoli fisiologici di SGLT1 nel fegato, nel pancreas e nei linfociti T non sono ben compresi.
Potenziale terapeutico dell’inibizione di SGLT1 e SGLT2:
Il potenziale terapeutico degli inibitori selettivi di SGLT2 come strategia antiperglicemica è stato ampiamente dimostrato. Al contrario, il potenziale terapeutico, in termini di efficacia e sicurezza, dell’inibitore duale di SGLT2/SGLT1 o dell’inibitore selettivo di SGLT1 rimane meno chiaro.
- Inibitori SGLT1
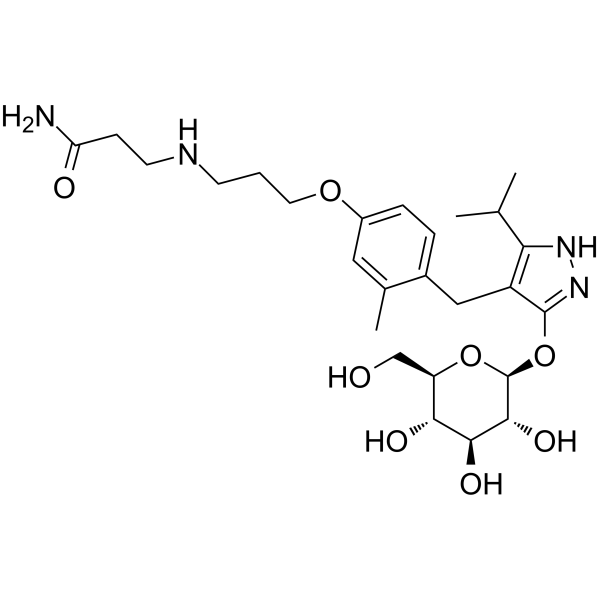
L’iperglicemia postprandiale è un fattore di rischio per l’insufficienza cardiovascolare e la microangiopatia diabetica, inclusa la retinopatia. Poiché l’assorbimento del glucosio dall’intestino tenue è principalmente mediato da SGLT1, un miglioramento dell’iperglicemia postprandiale con un inibitore di SGLT1 sarebbe sicuramente una terapia utile. Nei ratti diabetici, una singola dose di KGA-2727, un inibitore selettivo di SGLT1, ha migliorato l’iperglicemia postprandiale e la sua somministrazione cronica ha ridotto i livelli di emoglobina A1c, suggerendo che l’inibizione di SGLT1 potrebbe mantenere un buon controllo glicemico a lungo termine. In un test di tolleranza al glucosio orale con KGA-2727, i livelli plasmatici di insulina, così come i livelli di glucosio plasmatico, sono stati ridotti e sono attesi anche effetti protettivi sul pancreas.

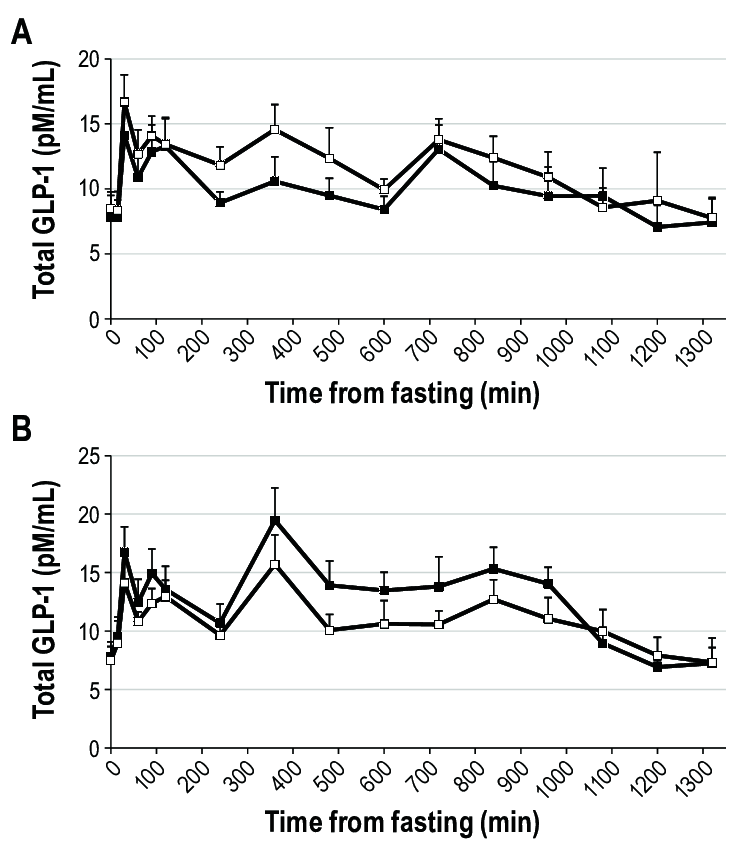
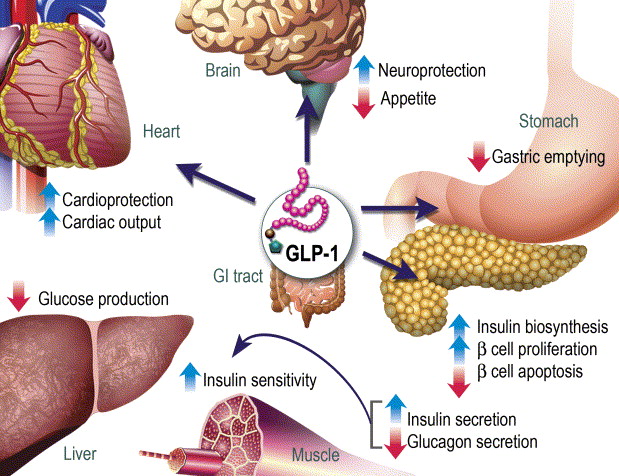
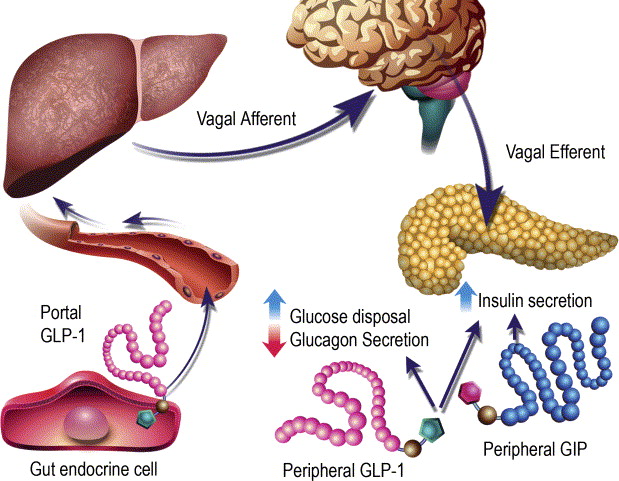
Studi recenti hanno riportato che i topi SGLT1 KO e i topi trattati con floridzina avevano livelli plasmatici di GLP-1 totale inferiori 5 minuti dopo la stimolazione con glucosio rispetto ai topi wild-type e ai topi di controllo, rispettivamente (98). Questo risultato suggerisce che SGLT1 è necessario per innescare la secrezione di GLP-1 nella fase precoce dopo la stimolazione con glucosio. Al contrario, un altro studio ha riportato che i topi SGLT1 KO avevano elevati livelli plasmatici di GLP-1 totale da 30 minuti a 6 ore dopo un pasto contenente glucosio. L’aumento del GLP-1 totale plasmatico nella fase tardiva dopo un pasto è stato osservato anche in esseri umani sani trattati con inibitore di SGLT1 e pazienti con diabete di tipo 2 trattati con inibitore di SGLT1/2. Un possibile meccanismo di rilascio ritardato di GLP-1 è la fermentazione del glucosio in acidi grassi a catena corta (SCFA). L’inibizione di SGLT1 nella fase iniziale dell’intestino riduce l’assorbimento del glucosio e quindi aumenta il suo apporto alle parti più distali dell’intestino tenue, dove il glucosio viene utilizzato dal microbioma per formare SCFA. Gli SCFA inducono la secrezione del peptide-1 simile al glucagone attraverso i recettori accoppiati alle proteine G, tra cui il recettore accoppiato alle proteine G 41 e il recettore accoppiato alle proteine G (98) . Da quanto sopra, sebbene l’inibizione di SGLT1 riduca il rilascio di GLP-1 stimolato dal glucosio nella fase iniziale, gli SCFA generati dalla fermentazione del glucosio inducono il rilascio di GLP-1 nella fase tardiva, suggerendo che l’inibitore di SGLT1 aumenta i livelli netti di GLP-1 circolante.
Un aspetto preoccupante è che, nell’intestino tenue, si ritiene che l’inibitore di SGLT1 induca effetti collaterali gastrointestinali, inclusa la diarrea, ma non sono stati osservati gravi effetti collaterali gastrointestinali nel trattamento degli inibitori selettivi di SGLT1, GSK-1614235 e KGA-2727, o di un inibitore duale di SGLT1/SGLT2, la sotagliflozin (99).
Gli inibitori di SGLT1 inducono un ritardo nell’assorbimento dei monosaccaridi e quindi nella loro ritenzione, e potrebbero migliorare le condizioni intestinali nei pazienti diabetici attraverso cambiamenti nel microbiota intestinale. Un aumento della produzione di propionato nel microbiota del colon con una maggiore esposizione al glucosio avrebbe contribuito a effetti metabolici intestinali positivi.
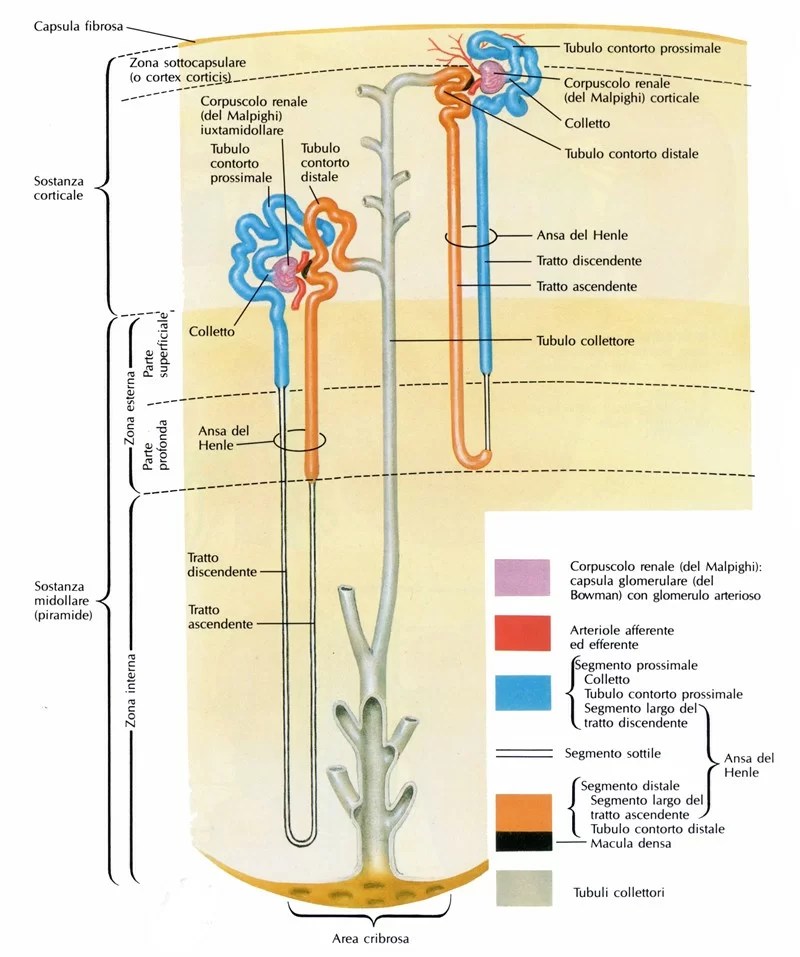
SGLT1 è espresso nella membrana dell’orletto a spazzola del segmento S3 del tubulo prossimale nel rene e riassorbe il glucosio che sfugge al riassorbimento mediato da SGLT2 nei segmenti S1 e S2. Studi su topi SGLT2 KO e inibitori selettivi di SGLT2 hanno descritto la capacità di trasporto renale di SGLT1, mostrando che il riassorbimento del glucosio mediato da SGLT1 viene mantenuto al 40-50% con l’inibizione di SGLT2 in condizioni euglicemiche . L’inibizione di SGLT2 in condizioni di iperglicemia prolungata e grave che supera la capacità di trasporto di SGLT2 attiva la piena capacità di trasporto renale di SGLT1 e SGLT1 esercita una funzione compensatoria nel riassorbimento renale del glucosio. Pertanto, si prevede che la terapia combinata di un inibitore SGLT1 e un inibitore SGLT2 o di un doppio inibitore SGLT1/SGLT2 induca una glicosuria e un controllo glicemico significativamente maggiori rispetto a un singolo inibitore SGLT1 o SGLT2 (100). Inoltre, è stato osservato un effetto più forte della doppia inibizione SGLT1/SGLT2 sui livelli di glucosio nel sangue nei topi con iperglicemia modesta, così come in quelli con euglicemia. Pertanto, gli effetti combinati della doppia inibizione SGLT1/SGLT2 potrebbero indurre effetti sinergici sui tubuli prossimali precoci e distali.
Sebbene gli inibitori selettivi dell’SGLT1 non siano ancora sul mercato, alcuni composti (ad esempio, LX2761 e JTT-662) sono in fase di sviluppo per il trattamento del diabete.
- Inibitori SGLT2
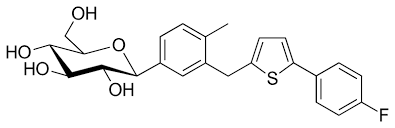
Gli inibitori selettivi dell’SGLT2 – dapagliflozin, canagliflozin, empagliflozin, ipragliflozin, luseogliflozin e tofogliflozin – sono stati approvati per il trattamento del diabete di tipo 2 (101) . Questi inibitori dell’SGLT2 riducono i livelli plasmatici di glucosio con un meccanismo diverso rispetto ad altri farmaci antidiabetici, che comporta un aumento dell’escrezione renale di glucosio attraverso l’SGLT2 nel tubulo prossimale, con conseguente riduzione della tossicità del glucosio. Al contrario, i meccanismi di altri farmaci sono come per la metformina: inibizione della gluconeogenesi nel fegato; derivati della sulfonilurea, analoghi del peptide glucagone-simile (GLP)-1 e inibitori del dipeptidil peptide-4: aumento della secrezione di insulina nel pancreas; e tiazolidinedioni: miglioramento della sensibilità all’insulina. Questi inibitori dell’SGLT2 hanno una diversa selettività per l’inibizione di SGLT2 rispetto a SGLT1. La selettività SGLT2/SGLT1 è ≥1.000 volte maggiore in dapagliflozin, empagliflozin, luseogliflozin e tofogliflozin, mentre la selettività di canagliflozin e ipragliflozin è inferiore, rispettivamente 190 e 250 volte (101) .
In studi preclinici su modelli animali diabetici, gli inibitori di SGLT2 hanno ridotto i livelli di glucosio a digiuno e non a digiuno, i livelli di emoglobina A1c e la pressione sanguigna, e migliorato l’intolleranza al glucosio (102). Inoltre, gli inibitori di SGLT2 hanno un meccanismo d’azione diverso dagli altri farmaci antidiabetici, come descritto sopra, e possono essere utilizzati in combinazione con questi farmaci, così come in monoterapia per il trattamento del diabete di tipo 2 (103).
Studi recenti hanno riportato che gli inibitori di SGLT2 hanno avuto un effetto protettivo renale in modelli animali di nefropatia diabetica (104). Gli effetti protettivi renali degli inibitori di SGLT2 sono stati dimostrati anche in studi clinici (105). Il meccanismo d’azione è ipotizzato come segue: l’inibitore SGLT2 aumenta la quantità di sodio trasportata al tubulo distale sopprimendo l’assorbimento di sodio nel tubulo prossimale. Di conseguenza, viene attivato il feedback tubuloglomerulare attraverso la macula densa, che consente la contrazione arteriolare afferente e normalizza la velocità di filtrazione glomerulare (106) .
Inibitori del SGLT2 nella perdita di peso e miglioramento della composizione corporea:
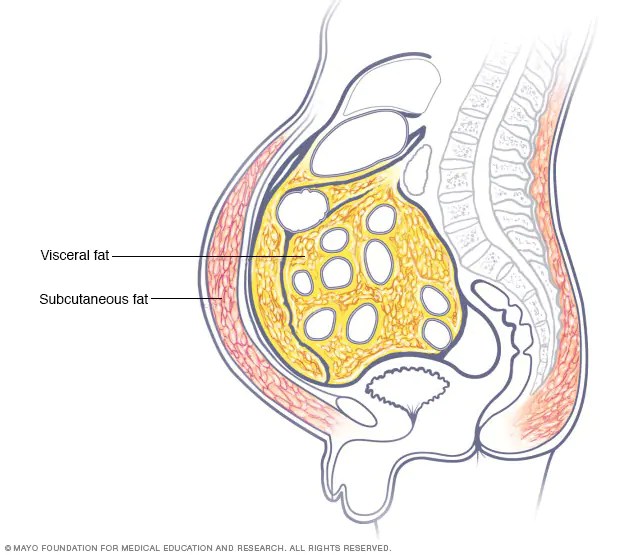
Sappiamo che gli inibitori del SGLT2 offrono un meccanismo insulino-indipendente per migliorare i livelli di glucosio nel sangue e sono approvati per il trattamento del diabete di tipo 2. Promuovono l’escrezione urinaria di glucosio inibendone il riassorbimento dall’urina nel tubulo prossimale del rene (fino a circa il 50%). L’entità della glicosuria risultante è proporzionale alla glicemia al di sopra della soglia [107].
Gli inibitori del SGLT2 (ad esempio, dapagliflozin, canagliflozin ed empagliflozin) e i GLP1-RA; ad esempio, exenatide, liraglutide e semaglutide) sono entrambi utilizzati per il trattamento del diabete di tipo 2, ma portano anche a una perdita di peso corporeo, in gran parte dovuta alla riduzione del grasso corporeo. Inoltre, gli effetti sulla glicemia e sul peso corporeo sono sostenuti per diversi anni con queste classi di farmaci [108]. Tuttavia, l’entità della perdita di peso è modesta sia nel diabete di tipo 2 che nell’obesità senza diabete. Per gli inibitori SGLT2 approvati si registra in media una perdita di peso di circa 1,5-2 kg (aggiustata per placebo), per gli inibitori del GLP1-RA di 2-4 kg e per la combinazione di 3-5 kg [109]. Pertanto, sono necessarie terapie dimagranti più efficaci per questo tipo specifico di pazienti.
- Controllo sulla glicemia
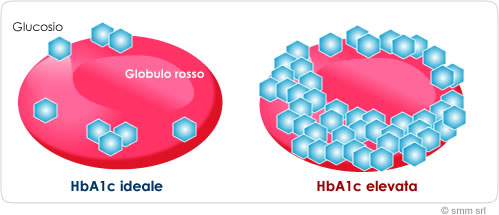
Gli inibitori SGLT2 hanno mostrato riduzioni costanti dei livelli di HbA1c rispetto al basale nei pazienti con diabete di tipo 2 in tutti i punti temporali. Le meta-analisi mostrano differenze medie nelle riduzioni di HbA1c rispetto al placebo da -1,4% a -0,5% [110]; queste riduzioni sono simili a quelle di altri agenti ipoglicemizzanti [111]. Questi risultati non sono sorprendenti per un farmaco sviluppato per il trattamento del diabete di tipo 2. Gli studi clinici che esaminano gli effetti degli inibitori SGLT2 sull’HbA1c in soggetti sovrappeso o obesi senza diabete di tipo 2 sono limitati. In due studi della durata di 12 e 24 settimane, gli inibitori SGLT2 da soli (rispettivamente canagliflozin e dapagliflozin) non hanno influenzato i livelli di HbA1c rispetto al placebo nei soggetti sovrappeso e obesi [112]. Tuttavia, gli inibitori SGLT2 in combinazione con un GLP1-RA hanno ridotto significativamente l’HbA1c nei soggetti obesi senza diabete, rispetto al placebo [113].
La meta-analisi mostra che gli inibitori SGLT2 riducono significativamente i valori di glicemia a digiuno da -2,0 a -1,1 mmol/L [114]. La capacità di abbassare la glicemia degli inibitori SGLT2 è dipendente dalla glicemia [115], il che riduce al minimo gli eventi ipoglicemici. La concentrazione plasmatica di glucosio è determinata principalmente da fattori ormonali e neurali (come insulina, glucagone e catecolamine), che regolano la produzione endogena di glucosio [116]. Gli inibitori SGLT2 stimolano la produzione epatica di glucosio e aumentano anche la secrezione di glucagone, che promuove la produzione endogena di glucosio e limita la loro capacità di abbassare la glicemia [117].
- Effetti sul peso corporeo e sull’adiposità:
Gli inibitori di SGLT2 causano direttamente la perdita di peso corporeo attraverso l’escrezione di glucosio (perdita di calorie) nei reni. L’inibizione di SGLT2 agisce in modo glucosio-dipendente e può comportare l’eliminazione di circa 60-100 g di glucosio al giorno nelle urine. La perdita di peso con la terapia con inibitori di SGLT2 è stata costantemente osservata in diversi studi sul diabete di tipo 2, indipendentemente dal fatto che i pazienti assumano inibitori di SGLT2 in monoterapia o in combinazione con ulteriori terapie ipoglicemizzanti. I risultati delle meta-analisi di rete mostrano riduzioni del peso corporeo rispetto al placebo per tutti i trattamenti con inibitori di SGLT2 di circa 1,5-2 kg [118] e questi effetti sono dose-dipendenti [119]. I dati clinici fino a 4 anni mostrano che la riduzione del peso corporeo con inibitori di SGLT2 viene mantenuta [120]. Tuttavia, gli inibitori SGLT2 causano una perdita di peso sostanzialmente inferiore a quella prevista dall’energia escreta tramite glicosuria, perché provocano un aumento adattativo dell’assunzione di energia, inclusi aumenti compensatori dell’appetito/apporto calorico [121]. Pertanto, la combinazione di inibitori SGLT2 con farmaci che agiscono attraverso meccanismi diversi potrebbe essere l’approccio più efficace per una perdita di peso significativa e affrontare i meccanismi controregolatori che mantengono il peso corporeo [122]. Studi recenti che valutano la co-somministrazione di inibitori SGLT2 con altre classi di farmaci hanno mostrato risultati promettenti. Ad esempio, lo studio DURATION-8 ha dimostrato che la perdita di peso corporeo media con la combinazione di exenatide (GLP1-RA, che sopprime l’appetito) una volta alla settimana e dapagliflozin (inibitore SGLT2) una volta al giorno nei pazienti con diabete di tipo 2 era maggiore di quella con le sole monoterapie [123].
Solo pochi studi hanno esaminato gli effetti degli inibitori SGLT2 sulla perdita di peso in soggetti obesi senza diabete. Bays et al. hanno dimostrato che canagliflozin 100 mg da solo riduce il peso corporeo di 2,8 kg [124]. La co-somministrazione di inibitori SGLT2 con GLP1-RA riduce il peso corporeo di 4,5 kg a 24 settimane di trattamento e questa perdita di peso viene mantenuta fino a 1 anno (-5,7 kg) in individui obesi senza diabete [125]. Ancora più importante, la perdita di peso è dovuta principalmente a una riduzione del tessuto adiposo sottocutaneo e viscerale, piuttosto che del tessuto magro. Un altro studio che esplora la terapia di combinazione di canagliflozin con fentermina, un farmaco simile all’anfetamina utilizzato per sopprimere l’appetito e approvato per la gestione del peso, ha dimostrato una perdita di peso superiore rispetto al placebo (-7,3 kg contro -0,6 kg) in un periodo di 26 settimane [126]. Tuttavia, gli studi che esplorano gli effetti della terapia combinata sono stati pochi e con dosi limitate. Pertanto, il pieno potenziale di riduzione del peso deve essere esplorato in più studi, tra cui l’ottimizzazione delle dosi, la sicurezza e l’inclusione di interventi sullo stile di vita. Sono attualmente in corso altri studi clinici che valutano gli effetti degli inibitori SGLT2 in soggetti obesi senza diabete, utilizzati da soli o in combinazione con altri farmaci (identificativo ClinicalTrial.gov: NCT03093103, NCT03710460, NCT02635386, NCT02695810).
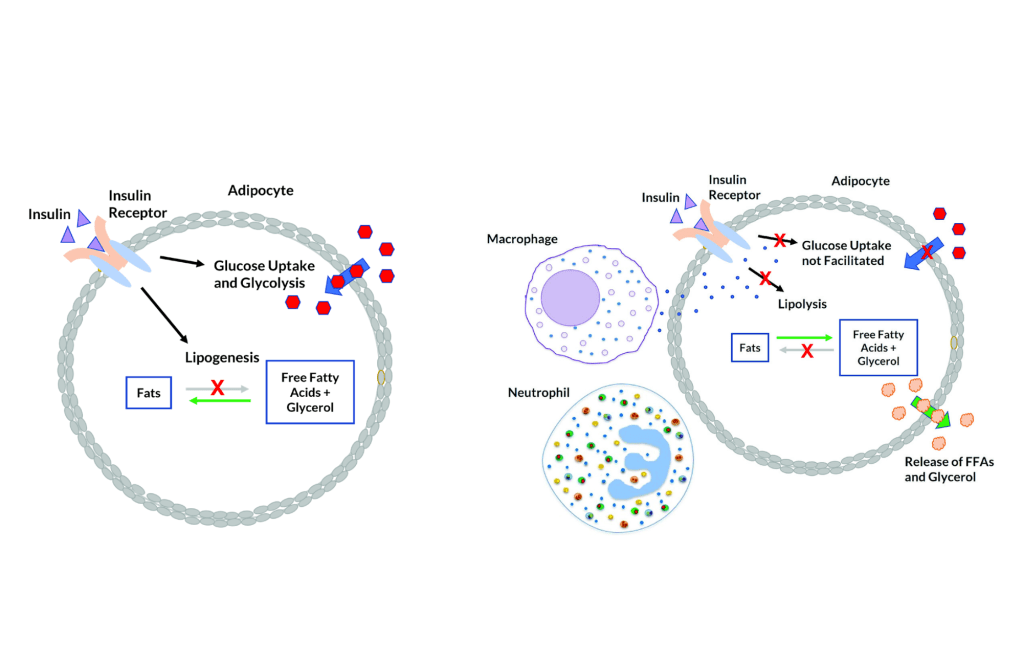
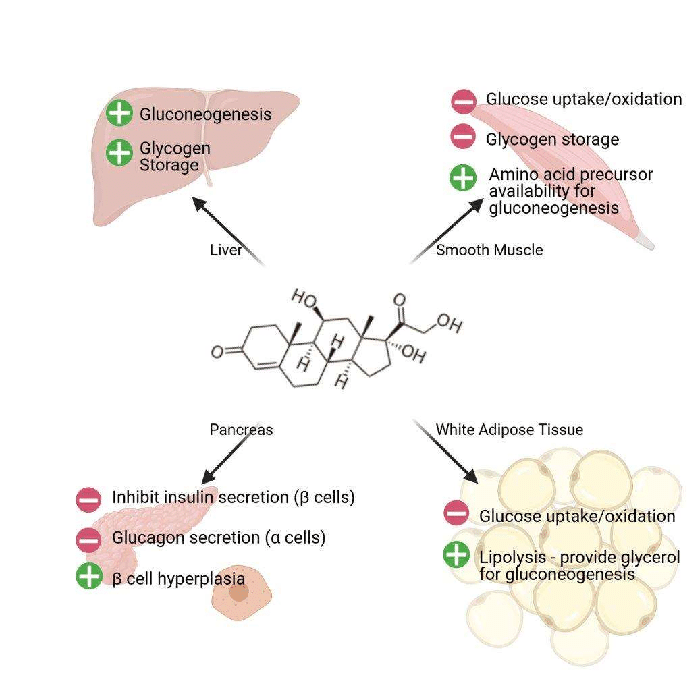
Nei ratti obesi indotti dalla dieta e trattati con inibitori dell’SGLT2, la lipolisi e i livelli circolanti di corpi chetonici risultano aumentati [127, 128]. Nei pazienti con diabete di tipo 2 o con obesità senza diabete, la glicosuria indotta dagli inibitori dell’SGLT2 riduce i livelli plasmatici di glucosio e insulina e aumenta le concentrazioni di glucagone a digiuno e post-prandiali. La riduzione della concentrazione circolante di glucosio, insieme ai cambiamenti ormonali, determina la mobilizzazione delle riserve lipidiche [129]. Ciò porta a cambiamenti nell’utilizzo dei substrati energetici, favorendo l’utilizzo dei lipidi per la produzione di energia [130]. In condizioni di ridotto rapporto portale insulina/glucagone, la lipolisi aumenta nel tessuto adiposo e rilascia acidi grassi non esterificati che vengono convertiti in corpi chetonici nel fegato attraverso la beta-ossidazione mitocondriale e la chetogenesi [131], determinando una condizione metabolica simile a un digiuno prolungato [132].
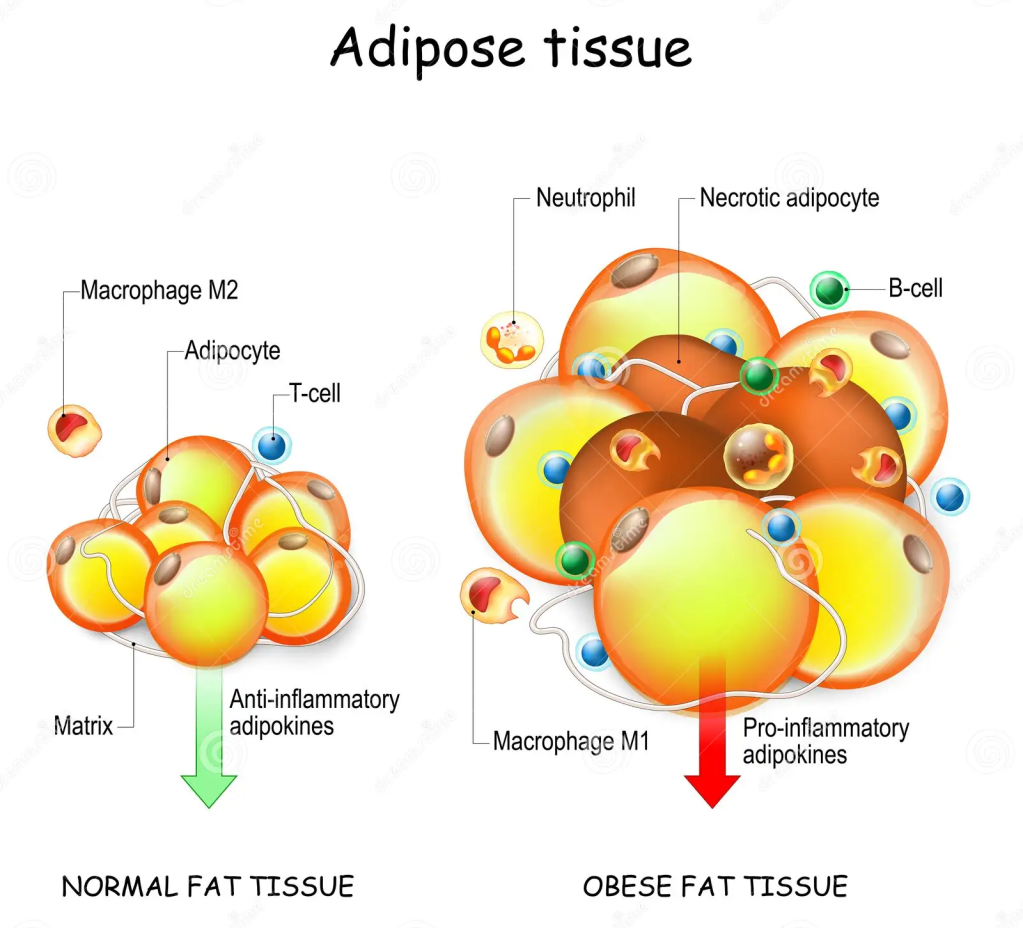
Inoltre, è stato dimostrato che gli inibitori SGLT2 riducono l’infiammazione del tessuto adiposo e aumentano il tessuto adiposo bruno nei modelli di roditori [133, 134]. La riduzione dell’infiammazione nel tessuto adiposo sarebbe particolarmente importante nell’obesità, poiché l’infiammazione cronica di basso grado nel tessuto adiposo è un importante mediatore nello sviluppo di complicazioni legate all’obesità, come la resistenza all’insulina e il diabete di tipo 2 [135].
- Lipidi ematici
In generale, il trattamento con inibitori SGLT2 ha effetti minori sul profilo lipidico. Sebbene alcuni studi abbiano dimostrato che gli inibitori SGLT2 aumentano modestamente i livelli di colesterolo HDL rispetto al placebo, sono disponibili anche dati che suggeriscono un piccolo aumento del colesterolo LDL [136, 137]. Pertanto, attualmente non vi sono prove chiare che le variazioni delle lipoproteine nel sangue siano importanti per i risultati clinici complessivi dopo il trattamento con inibitori SGLT2.
Recenti progressi nella scoperta di nuovi inibitori SGLT2 da fitocomposti:
Prima di giungere alle conclusioni sull’applicazione ed eventuali vantaggi d’uso degli inibitori selettivi SGLT2, è interessante ed utile riportare nuovi promettenti inibitori di questa categoria di origine naturale (fitocomposti).
Sebbene molti inibitori dei SGLT2 siano ora disponibili e ampiamente utilizzati, sono ancora in corso numerosi sforzi per trovare nuovi composti attivi da prodotti naturali/fitocomposti come candidati validi per nuovi inibitori dei SGLT2, come la Florizina. Le strutture chimiche di diversi importanti fitocomposti sono riportati di seguito:
- Sophora flavescens (Fabaceae)
La Sophora flavescens (S. flavescens) è una delle medicine tradizionali cinesi più popolari e importanti. La radice di questa specie (nota come “Kushen”) è ampiamente utilizzata per il trattamento di molte malattie, tra cui febbre, dissenteria, ittero, leucorrea, infezioni cutanee piogeniche, scabbia, gonfiore e dolore. Ad oggi, oltre 200 costituenti sono stati isolati da S. flavescens e i principali componenti di questa pianta sono alcaloidi e flavonoidi [138]. Oltre ai suoi usi tradizionali, sono stati condotti numerosi studi per scoprire altri effetti terapeutici di S. flavescens, come effetti antitumorali, antinfiammatori, antinocicettivi, antianafilattici, antiasmatici, antimicrobici, cardioprotettivi e immunoregolatori, utilizzando i suoi estratti grezzi e i principali composti attivi [138].
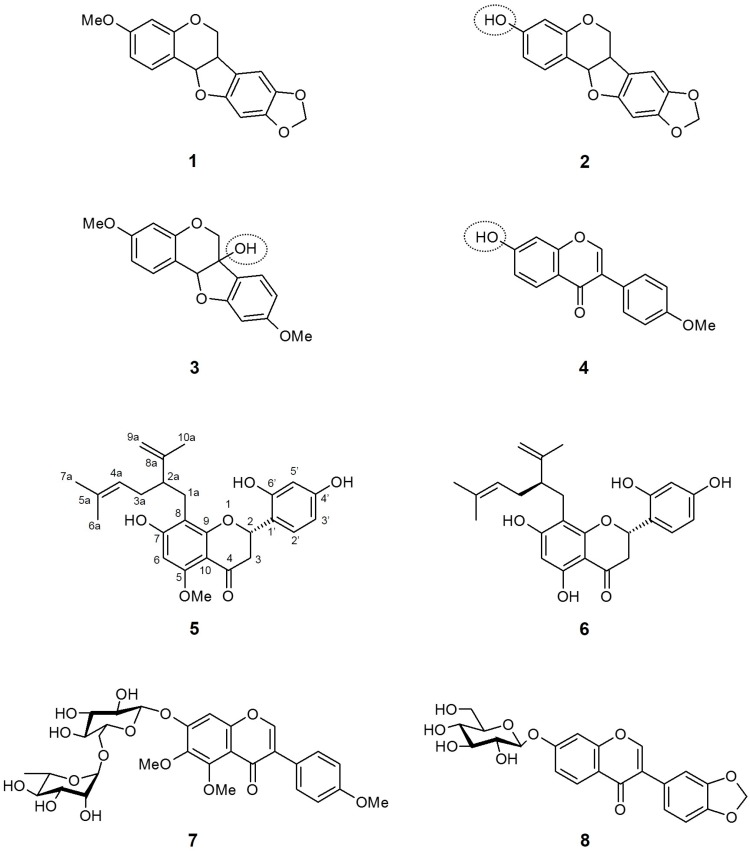
Sato et al. [139], come ricerca di follow-up sulla scoperta che l’estratto metanolo di S. flavescens ha una potente attività inibitoria di SGLT, hanno analizzato nove composti isolati dalla radice essiccata di S. flavescens per i loro effetti sull’inibizione di SGLT. È interessante notare che, ad eccezione della pterocarpina (1), tre composti con strutture a base di isoflavonoidi, vale a dire maackiaina (2), variabilina (3) e formononetina (4), hanno mostrato un’attività inibitoria esclusiva solo contro SGLT2, ma non contro SGLT1. Pertanto, si suggerisce che la presenza del gruppo funzionale idrossilico nell’isoflavonoide sia cruciale per l’acquisizione dell’attività inibitoria di SGLT2. Nel frattempo, la maggior parte dei composti flavanonici ha inibito ampiamente entrambi gli SGLT, con selettività contro SGLT2. I due composti più potenti erano (−)–kurarinone (5) e soforaflavanone G (6), con valori IC50 rispettivamente di 10,4 e 18,7 μM per SGLT1 e 1,7 e 4,1 μM per SGLT2. Si presumeva che l’aumento dell’inibizione di SGLT1 fosse attribuibile al gruppo funzionale lavandulile comune in posizione C-8.
Più recentemente, Yang et al. [140] hanno riportato gli effetti dei glicosidi isoflavonoidi dalla radice di S. flavescens sull’inibizione di SGLT2. Tutti e nove i composti isolati nella ricerca hanno mostrato attività inibitoria di SGLT2, con la più forte inibizione di SGLT2 nel composto 7 [IC50 (μM): 2,6 ± 0,18]. Inoltre, anche il composto 8 ha mostrato un’inibizione moderata per SGLT2 [IC50 (μM): 15,3 ± 1,44]. Poiché lo studio è stato progettato come una semplice valutazione di screening e l’attività inibitoria è stata stabilita solo per SGLT2, l’importanza conformazionale dell’attività inibitoria e la selettività contro SGLT2 non sono state discusse.
- Acer nikoense (Aceraceae)
Acer nikoense (A. nikoense) è originario e ampiamente distribuito in Giappone, e gli estratti della corteccia del suo fusto sono utilizzati nella medicina popolare giapponese per il trattamento di disturbi epatici e malattie oculari [141]. Diversi costituenti attivi di A. nikoense, tra cui specifiche acerogenine diarileptanoidi cicliche, sono stati valutati per i loro effetti antitumorali, antinfiammatori, antimicotici e antibatterici [142].
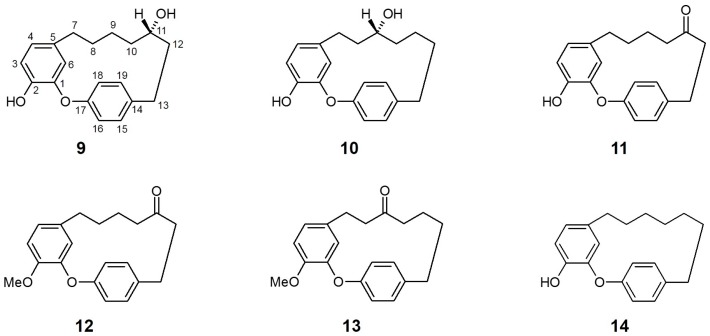
Morita et al. [143] hanno valutato gli effetti di quattro composti isolati dalla corteccia di A. nikoense e sedici derivati correlati sull’attività inibitoria degli SGLT. Due diarileptanoidi ciclici, acerogenina A (9) e B (10), hanno presentato una marcata inibizione sia per SGLT1 [IC50 (μM) 9: 20,0; 10: 26,0] che per SGLT2 [IC50 (μM) 9: 94,0; 10: 43.0], mentre altri composti isolati (incluso un diarileptanoide aciclico) non hanno mostrato un’inibizione sufficiente di nessuno dei due SGLT.
In particolare, i derivati chetonici in posizione C-11 (con o senza ulteriore sostituzione del gruppo idrossilico per l’estere metilico in posizione C-2) dell’acerogenina A/B, composti 11 (acerogenina C), 12 e 13, hanno acquisito un’attività inibitoria selettiva aumentata contro SGLT2 rispetto a SGLT1 rispetto ai loro precursori alla stessa concentrazione testata. Inoltre, un altro derivato dell’acerogenina A, caratterizzato da diidrossilazione in posizione C-11 (14), ha mostrato le attività inibitorie più potenti contro entrambi gli SGLT, senza selettività. Questi risultati suggeriscono che la posizione e/o la presenza di un gruppo idrossilico in posizione C-9 o C-11 e la loro stereochimica potrebbero non essere una struttura chiave per l’inibizione degli SGLT. La modifica del sistema ad anello diarileptanoide (come la sostituzione dell’ammide o la formazione di legami C-C insaturi) ha determinato una diminuzione dell’inibizione degli SGLT, indicando che la conformazione ad anello delle acerogenine e dei suoi derivati può influenzare l’effetto inibitorio di ciascun costituente contro gli SGLT.
- Alstonia macrophylla (Apocynaceae)
L’Alstonia macrophylla (A. macrophylla), chiamata anche alstonia dura o legno di latte duro, è originaria delle regioni del Sud-est asiatico come Indonesia, Malesia, Filippine, Thailandia e Vietnam. Questa pianta è stata tradizionalmente utilizzata come tonico generale, afrodisiaco, anticolerico, antidissenteriale, antipiretico, emmenagogo e agente vulnerario in Thailandia [144]. Essendo una ricca fonte di diversi fitochimici, sono state dimostrate diverse attività biologiche, tra cui effetti antimalarici, antimicrobici, antiossidanti, antidiabetici, antinfiammatori, antipiretici, antipsicotici, antifertilizzanti e antiprotozoari, utilizzando vari estratti e composti attivi di A. macrophylla [145].
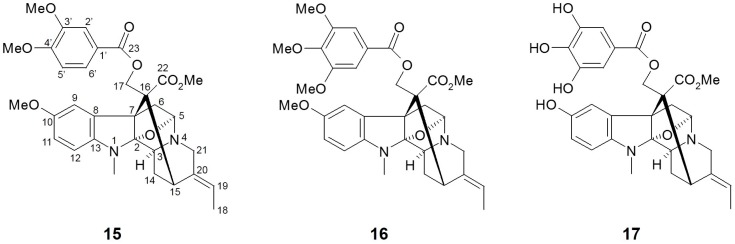
Arai et al. [146] hanno isolato venti composti alcaloidi dalle foglie di A. macrophylla e hanno valutato il potenziale inibitorio di SGLT di questi costituenti. Dei venti composti, cinque alcaloidi di tipo picralina hanno mostrato una buona inibizione di SGLT1 e SGLT2, con la più alta in 10-metossi-N(1)-metilburnammina-17-O-veratrato [15, IC50 (μM) SGLT1: 4,0; SGLT2: 0,5] e alstifillanina D [16, IC50 (μM) SGLT1: 5,0; SGLT2: 2,0]. Nel frattempo, altri composti alcaloidi di tipo ajmalina e macrolina non hanno mostrato attività inibitoria contro SGLT1 e/o SGLT2. I risultati dell’approccio della relazione struttura-attività (SAR) hanno suggerito che la presenza di una catena laterale estere in posizione C-17 può svolgere un ruolo fondamentale nell’attività inibitoria di SGLT. Quando è stato valutato un ulteriore studio SAR utilizzando otto derivati, il derivato idrossilico nell’alstifillina D (17) ha migliorato la selettività per SGLT2 più degli altri, sebbene il valore IC50 assoluto sia stato leggermente aumentato [IC50 (μM) SGLT1: 50,0; SGLT2: 7,0].
- Gnetum gnemonoides (Gnetaceae)
Gnetum gnemonoides (G. gnemonoides) è una specie di liane tropicali, ampiamente distribuita nella regione del Sud-est asiatico-Pacifico, tra cui Malesia, Indonesia, Filippine, Nuova Guinea e Arcipelago di Bismarck. Sebbene non esista ancora alcun rapporto scientifico sull’efficacia medicinale di G. gnemonoides, è noto che gli stilbeni isolati dalla specie Gnetum possiedono proprietà biologiche come attività epatoprotettiva, antiossidante, antimicrobica e inibitoria enzimatica [147].
Shimokawa et al. [148] hanno isolato due trimeri di stilbeni, gneulina A (18) e B (19), costituiti da unità di ossiresveratrolo, dalla corteccia essiccata di G. gnemonoides, e ne hanno semplicemente analizzato l’effetto sull’inibizione di SGLT1 e SGLT2. Questi due composti hanno entrambi mostrato un’attività inibitoria moderata e non selettiva per ciascun SGLT [IC50 (μM) 18 SGLT1: 27,0, SGLT2: 25,0; 19 SGLT1: 37,0, SGLT2: 18,0]. Hanno anche scoperto di recente due diidroflavonolo-C-glucosidi, il noidesol A e B, ma questi composti non hanno mostrato alcun potenziale inibitorio per gli SGLT.
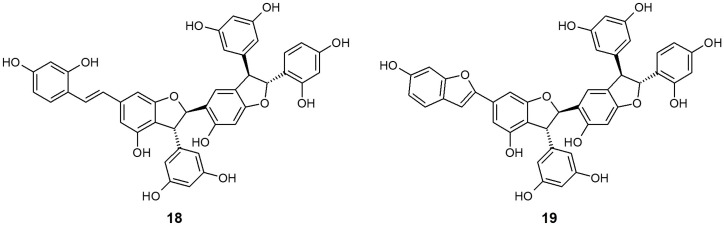
- Schisandra chinensis (Schisandraceae)
La Schisandra chinensis (S. chinensis, comunemente chiamata “bacca dai cinque sapori”) è originaria della Cina settentrionale e dell’Estremo Oriente russo, e i suoi frutti sono tradizionalmente utilizzati come agente anti-invecchiamento, antitussivo, sedativo e tonico [149]. La S. chinensis contiene vari fitochimici, tra cui polifenoli, lignani e triterpenoidi, e i suoi effetti farmacologici su vari sistemi organici sono stati ampiamente valutati [150].
Qu et al. [151] hanno recentemente valutato le attività inibitorie degli SGLT di Schisandrae Chinensis Fructus (SCF), allo scopo di identificare specifici composti inibitori dell’SGLT2. Nello screening iniziale con estratti acquosi ed etanolici di SCF a una concentrazione priva di citotossicità (1 mg/mL), sono stati osservati tassi di inibizione più potenti per entrambi gli SGLT con l’estratto etanolico. Dopo il frazionamento dell’estratto etanolico di SCF, un totale di nove frazioni (F1–F9) sono state sottoposte a un’ulteriore valutazione dell’inibizione di SGLT. Delle sei frazioni che hanno mostrato attività inibitoria contro SGLT1 e/o SGLT2, solo due frazioni (F8 e F9) hanno mostrato significativi pattern selettivi per SGLT2, con un tasso di inibizione rispettivamente del 41,9% e del 36,7% rispetto al controllo.
Sono stati infine studiati gli effetti di tre comuni composti lignanici isolati da F8, deossischisandrina, schisandrina B (γ-schisandrina) e schisandrina sull’inibizione di SGLT. Tuttavia, nessuno di essi ha mostrato attività inibitoria contro nessuno dei due SGLT, suggerendo che questi lignani principali non siano i componenti principali dell’inibizione di SGLT in SCF.
Conclusioni sugli inibitori dei SGLT1 e 2:
Da quando sono stati chiariti i ruoli importanti dell’SGLT2 nel riassorbimento renale del glucosio e nell’omeostasi sistemica del glucosio nell’organismo umano, l’inibizione dell’SGLT2 è stata considerata un promettente bersaglio terapeutico per il trattamento del diabete mellito di tipo 2. Dall’inizio del XXI secolo, diversi inibitori dell’SGLT2 derivati da un composto attivo naturale, la Florizina, hanno iniziato a essere commercializzati e ampiamente utilizzati come monoterapia o in combinazione con altri agenti ipoglicemizzanti orali.
Un vantaggio degli inibitori SGLT2, rispetto a diverse altre terapie ipoglicemizzanti, è il basso potenziale di indurre ipoglicemia, a meno che non siano combinati con insulina o secretagoghi dell’insulina. Questo perché l’escrezione urinaria di glucosio per impostazione predefinita diminuisce o cessa quando il glucosio plasmatico diminuisce, ma possono esserci anche contributi attraverso l’attivazione del sistema nervoso simpatico durante l’ipoglicemia che riduce la velocità di filtrazione glomerulare e quindi la glicosuria, nonché attraverso l’aumento della gluconeogenesi epatica, sebbene la cosomministrazione di Metformina può alterare questo aspetto.
In generale, gli inibitori SGLT2 sono ben tollerati e l’effetto avverso più comune è un aumento del rischio di infezioni genitali micotiche di circa quattro-sei volte rispetto al placebo o al comparatore attivo, e questo è osservato sia nelle donne che negli uomini. Questo è il risultato di una maggiore concentrazione di glucosio nelle urine che può facilitare l’insorgenza di infezioni nelle regioni urogenitali inferiori. Si prevede che lo stesso meccanismo promuova anche le infezioni del tratto urinario, ma nelle meta-analisi si riscontra una tendenza a un aumento minore fino a 1,5-piegare, e questo non è coerente tra gli studi. I rischi per tali effetti collaterali sono simili tra i diversi inibitori SGLT2.
Prove recenti suggeriscono che possono verificarsi anche episodi di chetoacidosi, e ciò potrebbe essere motivo di particolare preoccupazione tra gli individui con deficit di Insulina, compresi quelli con diabete di tipo 2, diabete di tipo 1 o diabete autoimmune latente negli adulti (LADA) di lunga data.
Infine, il programma CANVAS ha segnalato un aumento del rischio di fratture ossee e amputazioni degli arti inferiori con canagliflozin. Né fratture ossee né amputazioni degli arti inferiori sono state segnalate con gli altri inibitori SGLT2, né in un altro studio di analisi del mondo reale, pertanto sono necessarie ulteriori valutazioni prima di trarre conclusioni definitive.
Sebbene il bodybuilder nella media sia più propenso a sentirsi attirato all’uso off-label di questa classe di farmaci per il taglio calorico che possono contribuire a creare senza modifiche alimentari [rimando ai 60-100g di glucosio al giorno escreto con le urine corrispondente ad un range di perdita in Kcal pari a 240-400Kcal/die], gli effetti più interessanti sono da ricercarsi nel potenziale “harm reduction” nefro-cardiaco. Infatti, Gli inibitori del SGLT2 hanno dimostrato significativi effetti cardioprotettivi, che vanno oltre la semplice riduzione della glicemia. Questi benefici sono osservati nei pazienti con e senza diabete e includono una riduzione dei ricoveri ospedalieri per insufficienza cardiaca e un miglioramento della funzionalità cardiaca. I meccanismi sono molteplici e possono coinvolgere alterazioni del metabolismo cardiaco e vie antinfiammatorie e antifibrotiche.
Gli inibitori dell’SGLT2 possono alterare il modo in cui il cuore utilizza l’energia, migliorandone potenzialmente l’efficienza. Possono ridurre l’infiammazione cardiaca, un fattore chiave nelle malattie cardiache.
Gli inibitori dell’SGLT2 possono aiutare a prevenire o ridurre la formazione di tessuto cicatriziale (fibrosi) nel cuore, che può comprometterne la funzionalità.
Possono contribuire a smorzare l’iperattività del sistema nervoso simpatico, che può sovraccaricare il cuore.
Gli inibitori dell’SGLT2 possono migliorare la salute dei vasi sanguigni, importante per il trasporto di ossigeno e nutrienti al cuore.
Sebbene non sia il meccanismo primario, un certo grado di diuresi (aumento della minzione) e natriuresi (aumento dell’escrezione di sodio) può contribuire a ridurre la pressione e il volume sanguigno, il che può essere benefico per la salute del cuore.
Gli inibitori dell’SGLT2 possono aumentare i livelli di eritropoietina, determinando un aumento della produzione di globuli rossi e potenzialmente migliorando l’apporto di ossigeno al cuore. Questo ultimo punto potrebbe risultare problematico per gli atleti enhanced particolarmente sensibili allo stimolo della eritropoiesi.
Come già accennato, gli inibitori del SGLT2 hanno mostrato benefici significativi per la salute renale, anche in soggetti non diabetici. Sono sempre più riconosciuti per la loro capacità di rallentare la progressione della malattia renale cronica (CKD) e ridurre il rischio di insufficienza renale.
Gli inibitori dell’SGLT2 agiscono principalmente bloccando il riassorbimento di glucosio e sodio nei tubuli prossimali dei reni. Questo porta a una maggiore escrezione di glucosio nelle urine, contribuendo ad abbassare i livelli di glicemia. Riducendo il riassorbimento di sodio e glucosio, gli inibitori dell’SGLT2 possono ridurre il carico di lavoro sui reni e abbassare la pressione all’interno dei glomeruli (le unità filtranti dei reni).
È stato dimostrato che gli inibitori dell’SGLT2 riducono i livelli di albumina nelle urine (albuminuria), un marcatore di danno renale.
La velocità di filtrazione glomerulare stimata (eGFR) è una misura della funzionalità renale. È stato dimostrato che gli inibitori dell’SGLT2 rallentano il declino dell’eGFR, indicando una progressione più lenta della malattia renale cronica.
Di conseguenza, è stato dimostrato che gli inibitori dell’SGLT2 possono ridurre significativamente il rischio di insufficienza renale nei soggetti con e senza diabete.
Gli inibitori dell’SGLT2 offrono anche protezione cardiovascolare, riducendo il rischio di insufficienza cardiaca, infarto e ictus.
I benefici degli inibitori dell’SGLT2 nel rallentare la progressione della malattia renale cronica sono osservati in diversi stadi della malattia, compresi quelli precoci e avanzati.
Ciò nonostante, Gli inibitori dell’SGLT2 sono generalmente sconsigliati nei soggetti con eGFR molto basso (tipicamente inferiore a 20mL/min/1,73 m²).
La decisione di utilizzare gli inibitori SGLT2 deve essere presa consultando un medico, tenendo conto delle caratteristiche individuali del paziente e della sua storia clinica.
In conclusione, gli inibitori SGLT2 si sono rivelati una valida opzione terapeutica per la gestione della malattia renale, offrendo protezione contro l’insufficienza renale e altre complicanze associate alla malattia renale cronica e complicazioni cardiovascolari. Tali caratteristiche, hanno fatto si che gli inibitori degli SGLT2 venissero inseriti come ancillari nelle preparazioni di bodybuilding al fine di offrire una “riduzione del danno” a carico cardio-renale.
Gabriel Bellizzi [CEO BioGenTech]
Riferimenti:
- Mueckler M. Facilitative glucose transporters. Eur J Biochem 1994; 219: 713–725. [DOI] [PubMed] [Google Scholar]
- Joost HG, Thorens B. The extended GLUT‐family of sugar/polyol transport facilitators: nomenclature, sequence characteristics, and potential function of its novel members (review). Mol Membr Biol 2001; 18: 247–256. [DOI] [PubMed] [Google Scholar]
- Wright EM. Renal Na(+)‐glucose cotransporters. Am J Physiol Renal Physio 2001; 280: F10–F18. [DOI] [PubMed] [Google Scholar]
- Wood IS, Trayhurn P. Glucose transporters (GLUT and SGLT): expanded families of sugar transport proteins. Br J Nutr 2003; 89: 3–9. [DOI] [PubMed] [Google Scholar]
- Scheepers A, Joost HG, Schurmann A. The glucose transporter families SGLT and GLUT: molecular basis of normal and aberrant function. JPEN J Parenter Enteral Nutr 2004; 28: 364–371. [DOI] [PubMed] [Google Scholar]
- Hirayama BA, Wong HC, Smith CD, et al Intestinal and renal Na+/glucose cotransporters share common structures. Am J Physiol 1991; 261: C296–C304. [DOI] [PubMed] [Google Scholar]
- Song P, Onishi A, Koepsell H, et al Sodium glucose cotransporter SGLT1 as a therapeutic target in diabetes mellitus. Expert Opin Ther Targets 2016; 20: 1109–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santer R, Calado J. Familial renal glucosuria and SGLT2: from a mendelian trait to a therapeutic target. Clin J Am Soc Nephrol 2010; 5: 133–141. [DOI] [PubMed] [Google Scholar]
- Crane RK, Miller D, Bihler I (1961). “The restrictions on possible mechanisms of intestinal transport of sugars”. In Kleinzeller A, Kotyk A (eds.). Membrane Transport and Metabolism. Proceedings of a Symposium held in Prague, August 22–27, 1960. Czech Academy of Sciences & Academic Press. pp. 439–449.
- Wright EM, Turk E (February 2004). “The sodium/glucose cotransport family SLC5”. Pflügers Archiv. 447 (5): 510–8. doi:10.1007/s00424-003-1063-6. PMID 12748858. S2CID 41985805.
Crane in 1961 was the first to formulate the cotransport concept to explain active transport [7]. Specifically, he proposed that the accumulation of glucose in the intestinal epithelium across the brush border membrane was [is] coupled to downhill Na+ transport cross the brush border. This hypothesis was rapidly tested, refined, and extended [to] encompass the active transport of a diverse range of molecules and ions into virtually every cell type.
- Boyd CA (March 2008). “Facts, fantasies and fun in epithelial physiology”. Experimental Physiology. 93 (3): 303–14. doi:10.1113/expphysiol.2007.037523. PMID 18192340. S2CID 41086034.
p. 304. “the insight from this time that remains in all current text books is the notion of Robert Crane published originally as an appendix to a symposium paper published in 1960 (Crane et al. 1960). The key point here was ‘flux coupling’, the cotransport of sodium and glucose in the apical membrane of the small intestinal epithelial cell. Half a century later this idea has turned into one of the most studied of all transporter proteins (SGLT1), the sodium–glucose cotransporter.
- Wanner C, Inzucchi SE, Lachin JM, et al Empagliflozin and progression of kidney disease in type 2 diabetes. N Engl J Med 2016; 375: 323–334. [DOI] [PubMed] [Google Scholar]
- Lehmann A, Hornby PJ. Intestinal SGLT1 in metabolic health and disease. Am J Physiol Gastrointest Liver Physiol 2016; 310: G887–G898. [DOI] [PubMed] [Google Scholar]
- Tahrani AA, Barnett AH, Bailey CJ. SGLT inhibitors in management of diabetes. Lancet Diabetes Endocrinol 2013; 1: 140–151. [DOI] [PubMed] [Google Scholar]
- Ehrenkranz R.R.L., Lewis N.G., Kahn C.R., Roth J. Phlorizin: A review. Diabetes Metab. Res. Rev. 2005;21:31–38. doi: 10.1002/dmrr.532. [DOI] [PubMed] [Google Scholar]
- 18.White J.R., Jr. Apple trees to sodium glucose co-transporter inhibitors: A review of SGLT2 inhibition. Clin. Diabetes. 2010;28:5–10. doi: 10.2337/diaclin.28.1.5. [DOI] [Google Scholar]
- 19.Chasis H., Jolliffe N., Smith H.W. The action of phlorizin on the excretion of glucose, xylose, sucrose, creatinine and urea by man. J. Clin. Investig. 1933;12:1083–1090. doi: 10.1172/JCI100559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rossetti L., Smith D., Shulman G.I., Papachristou D., DeFronzo R.A. Correction of hyperglycemia with phlorizin normalizes tissue sensitivity to insulin in diabetic rats. J. Clin. Investig. 1987;79:1510–1515. doi: 10.1172/JCI112981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dimitrakoudis D., Vranic M., Klip A. Effects of hyperglycemia on glucose transporters of the muscle: Use of the renal glucose reabsorption inhibitor phlorizin to control glycemia. J. Am. Soc. Nephrol. 1992;3:1078–1091. doi: 10.1681/ASN.V351078. [DOI] [PubMed] [Google Scholar]
- Jonas J.C., Sharma A., Hasenkamp W., Ilkova H., Patane G., Laybutt R., Bonner-Weir S., Weir G.C. Chronic hyperglycemia triggers loss of pancreatic β cell differentiation in an animal model of diabetes. J. Biol. Chem. 1999;274:14112–14121. doi: 10.1074/jbc.274.20.14112. [DOI] [PubMed] [Google Scholar]
- Abdul-Ghani M.A., DeFronzo R.A. Inhibition of renal glucose absorption: A novel strategy for achieving glucose control in type 2 diabetes mellitus. Endocr. Pract. 2008;14:782–790. doi: 10.4158/EP.14.6.782. [DOI] [PubMed] [Google Scholar]
- Thorens B., Mueckler M. Glucose transporters in the 21st century. Am. J. Physiol. Endocrinol. Metab. 2010;298:E141–E145. doi: 10.1152/ajpendo.00712.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bays H. Sodium glucose co-transporter type 2 (SGLT2) inhibitors: Targeting the kidney to improve glycemic control in diabetes mellitus. Diabetes Ther. 2013;4:195–220. doi: 10.1007/s13300-013-0042-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oku A., Ueta K., Arakawa K., Ishihara T., Nawano M., Kuronuma Y., Matsumoto M., Saito A., Tsujihara K., Anai M., et al. T-1095, an inhibitor of renal Na+-glucose cotransporters, may provide a novel approach to treating diabetes. Diabetes. 1999;48:1794–1800. doi: 10.2337/diabetes.48.9.1794. [DOI] [PubMed] [Google Scholar]
- Katsuno K., Fujimori Y., Takemura Y., Hiratochi M., Itoh F., Komatsu Y., Fujikura H., Isaji M. Sergliflozin, a novel selective inhibitor of low-affinity sodium glucose cotransporter (SGLT2), validates the critical role of SGLT2 in renal glucose reabsorption and modulates plasma glucose level. J. Pharmacol. Exp. Ther. 2007;320:323–330. doi: 10.1124/jpet.106.110296. [DOI] [PubMed] [Google Scholar]
- Fujimori Y., Katsuno K., Nakashima I., Ishikawa-Takemura Y., Fujikura H., Isaji M. Remogliflozin etabonate, in a novel category of selective low-affinity sodium glucose cotransporter (SGLT2) inhibitors, exhibits antidiabetic efficacy in rodent models. J. Pharmacol. Exp. Ther. 2008;327:268–276. doi: 10.1124/jpet.108.140210. [DOI] [PubMed] [Google Scholar]
- Bickel M., Brummerhop H., Frick W., Glombik H., Herling A.W., Heuer H.O., Plettenburg O., Theis S., Werner U., Kramer W. Effects of AVE2268, a substituted glycopyranoside, on urinary glucose excretion and blood glucose in mice and rats. Arzneimittelforschung. 2008;58:574–580. doi: 10.1055/s-0031-1296559. [DOI] [PubMed] [Google Scholar]
- Derdau V., Fey T., Atzrodt J. Synthesis of isotopically labelled SGLT inhibitors and their metabolites. Tetrahedron. 2010;66:1472–1482. doi: 10.1016/j.tet.2009.12.003. [DOI] [Google Scholar]
- Fujimori Y., Katsuno K., Ojima K., Nakashima I., Nakano S., Ishikawa-Takemura Y., Kusama H., Isaji M. Sergliflozin etabonate, a selective SGLT2 inhibitor, improves glycemic control in streptozotocin-induced diabetic rats and Zucker fatty rats. Eur. J. Pharmacol. 2009;609:148–154. doi: 10.1016/j.ejphar.2009.03.007. [DOI] [PubMed] [Google Scholar]
- Katsuno K., Fujimori Y., Ishikawa-Takemura Y., Isaji M. Long-term treatment with sergliflozin etabonate improves disturbed glucose metabolism in KK-A(y) mice. Eur. J. Pharmacol. 2009;618:98–104. doi: 10.1016/j.ejphar.2009.07.001. [DOI] [PubMed] [Google Scholar]
- Hussey E.K., Clark R.V., Amin D.M., Kipnes M.S., O’Connor-Semmes R.L., O’Driscoll E.C., Leong J., Murray S.C., Dobbins R.L., Layko D., et al. Single-dose pharmacokinetics and pharmacodynamics of sergliflozin etabonate, a novel inhibitor of glucose reabsorption, in healthy volunteers and patients with type 2 diabetes mellitus. J. Clin. Pharmacol. 2010;50:623–635. doi: 10.1177/0091270009351879. [DOI] [PubMed] [Google Scholar]
- Hussey E.K., Dobbins R.L., Stoltz R.R., Stockman N.L., O’Connor-Semmes R.L., Kapur A., Murray S.C., Layko D., Nunez D.J. Multiple-dose pharmacokinetics and pharmacodynamics of sergliflozin etabonate, a novel inhibitor of glucose reabsorption, in healthy overweight and obese subjects: A randomized double-blind study. J. Clin. Pharmacol. 2010;50:636–646. doi: 10.1177/0091270009352185. [DOI] [PubMed] [Google Scholar]
- Dobbins R.L., O’Connor-Semmes R., Kapur A., Kapitza C., Golor G., Mikoshiba I., Tao W., Hussey E.K. Remogliflozin etabonate, a selective inhibitor of the sodium-dependent transporter 2 reduces serum glucose in type 2 diabetes mellitus patients. Diabetes Obes. Metab. 2012;14:15–22. doi: 10.1111/j.1463-1326.2011.01462.x. [DOI] [PubMed] [Google Scholar]
- Mudaliar S., Armstrong D.A., Mavian A.A., O’Connor-Semmes R., Mydlow P.K., Ye J., Hussey E.K., Nunez D.J., Henry R.R., Dobbins R.L. Remogliflozin etabonate, a selective inhibitor of the sodium-glucose transporter 2, improves serum glucose profiles in type 1 diabetes. Diabetes Care. 2012;35:2198–2200. doi: 10.2337/dc12-0508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussey E.K., Kapur A., O’Connor-Semmes R., Tao W., Rafferty B., Polli J.W., James C.D., Jr., Dobbins R.L. Safety, pharmacokinetics and pharmacodynamics of remogliflozin etabonate, a novel SGLT2 inhibitor, and metformin when co-administered in subjects with type 2 diabetes mellitus. BMC Pharmacol. Toxicol. 2013;14:25. doi: 10.1186/2050-6511-14-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur A., O’Connor-Semmes R., Hussey E.K., Dobbins R.L., Tao W., Hompesch M., Smith G.A., Polli J.W., James C.D., Jr., Mikoshiba I., et al. First human dose-escalation study with remogliflozin etabonate, a selective inhibitor of the sodium-glucose transporter 2 (SGLT2), in healthy subjects and in subjects with type 2 diabetes mellitus. BMC Pharmacol. Toxicol. 2013;14:26. doi: 10.1186/2050-6511-14-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykes A.P., O’Connor-Semmes R., Dobbins R., Dorey D.J., Lorimer J.D., Walker S., Wilkison W.O., Kler L. Randomized trial showing efficacy and safety of twice-daily remogliflozin etabonate for the treatment of type 2 diabetes. Diabetes Obes. Metab. 2015;17:94–97. doi: 10.1111/dom.12391. [DOI] [PubMed] [Google Scholar]
- Sykes A.P., Kemp G.L., Dobbins R., O’Connor-Semmes R., Almond S.R., Wilkison W.O., Walker S., Kler L. Randomized efficacy and safety trial of once-daily remogliflozin etabonate for the treatment of type 2 diabetes. Diabetes Obes. Metab. 2015;17:98–101. doi: 10.1111/dom.12393. [DOI] [PubMed] [Google Scholar]
- O’Connor-Semmes R., Walker S., Kapur A., Hussey E.K., Ye J., Wang-Smith L., Tao W., Dobbins R.L., Cheatham B., Wilkison W.O. Pharmacokinetics and pharmacodynamics of the SGLT2 inhibitor remogliflozin etabonate in subjects with mild and moderate renal impairment. Drug Metab. Dispos. 2015;43:1077–1083. doi: 10.1124/dmd.114.062828. [DOI] [PubMed] [Google Scholar]
- Link J.T., Sorensen B.K. A method for preparing C-glycosides related to phlorizin. Tetrahedron Lett. 2000;41:9213–9217. doi: 10.1016/S0040-4039(00)01709-3. [DOI] [Google Scholar]
- Meng W., Ellsworth B.A., Nirschl A.A., McCann P.J., Patel M., Girotra R.N., Wu G., Sher P.M., Morrison E.P., Biller S.A., et al. Discovery of dapagliflozin: A potent, selective renal sodium-dependent glucose cotransporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes. J. Med. Chem. 2008;51:1145–1149. doi: 10.1021/jm701272q. [DOI] [PubMed] [Google Scholar]
- .Han S., Hagan D.L., Taylor J.R., Xin L., Meng W., Biller S.A., Wetterau J.R., Washburn W.N., Whaley J.M. Dapagliflozin, a selective SGLT2 inhibitor, improves glucose homeostasis in normal and diabetic rats. Diabetes. 2008;57:1723–1729. doi: 10.2337/db07-1472. [DOI] [PubMed] [Google Scholar]
- Komoroski B., Vachharajani N., Feng Y., Li L., Kornhauser D., Pfister M. Dapagliflozin, a novel, selective SGLT2 inhibitor, improved glycemic control over 2 weeks in patients with type 2 diabetes mellitus. Clin. Pharmacol. Ther. 2009;85:513–519. doi: 10.1038/clpt.2008.250. [DOI] [PubMed] [Google Scholar]
- List J.F., Woo V., Morales E., Tang W., Fiedorek F.T. Sodium-glucose cotransport inhibition with dapagliflozin in type 2 diabetes. Diabetes Care. 2009;32:650–657. doi: 10.2337/dc08-1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilding J.P., Norwood P., T’joen C., Bastien A., List J.F., Fiedorek F.T. A study of dapagliflozin in patients with type 2 diabetes receiving high doses of insulin plus insulin sensitizers: Applicability of a novel insulin-independent treatment. Diabetes Care. 2009;32:1656–1662. doi: 10.2337/dc09-0517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrannini E., Ramos S.J., Salsali A., Tang W., List J.F. Dapagliflozin monotherapy in type 2 diabetic patients with inadequate glycemic control by diet and exercise: A randomized, double-blind, placebo-controlled, phase III trial. Diabetes Care. 2010;33:2217–2224. doi: 10.2337/dc10-0612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey C.J., Gross J.L., Pieters A., Bastien A., List J.F. Effect of dapagliflozin in patients with type 2 diabetes who have inadequate glycaemic control with metformin: A randomised, double-blind, placebo-controlled trial. Lancet. 2010;375:2223–2233. doi: 10.1016/S0140-6736(10)60407-2. [DOI] [PubMed] [Google Scholar]
- Strojek K., Yoon K.H., Hruba V., Elze M., Langkilde A.M., Parikh S. Effect of dapagliflozin in patients with type 2 diabetes who have inadequate glycaemic control with glimepiride: A randomized, 24-week, double-blind, placebo-controlled trial. Diabetes Obes. Metab. 2011;13:928–938. doi: 10.1111/j.1463-1326.2011.01434.x. [DOI] [PubMed] [Google Scholar]
- Nauck M.A., Del Prato S., Meier J.J., Durán-García S., Rohwedder K., Elze M., Parikh S.J. Dapagliflozin versus glipizide as add-on therapy in patients with type 2 diabetes who have inadequate glycemic control with metformin: A randomized, 52-week, double-blind, active controlled noninferiority trial. Diabetes Care. 2011;34:2015–2022. doi: 10.2337/dc11-0606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenstock J., Vico M., Wei L., Salsali A., List J.F. Effects of dapagliflozin, an SGLT2 inhibitor, on HbA1c, body weight, and hypoglycemia risk in patients with type 2 diabetes inadequately controlled on pioglitazone monotherapy. Diabetes Care. 2012;35:1473–1478. doi: 10.2337/dc11-1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura S., Sasamaki S., Hongu M., Kawanishi E., Koga Y., Sakamoto T., Yamamoto Y., Ueta K., Kimata H., Nakayama K., et al. Discovery of canagliflozin, a novel C-glucoside with thiophene ring, as sodium-dependent glucose cotransporter 2 inhibitor for the treatment of type 2 diabetes mellitus. J. Med. Chem. 2010;53:6355–6360. doi: 10.1021/jm100332n. [DOI] [PubMed] [Google Scholar]
- Rosenstock J., Aggarwal N., Polidori D., Zhao Y., Arbit D., Usiskin K., Capuano G., Canovatchel W., Canagliflozin DIA 2001 Study Group Dose-ranging effects of canagliflozin, a sodium-glucose cotransporter 2 inhibitor, as add-on to metformin in subjects with type 2 diabetes. Diabetes Care. 2012;35:1232–1238. doi: 10.2337/dc11-1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devineni D., Morrow L., Hompesch M., Skee D., Vandebosch A., Murphy J., Ways K., Schwartz S. Canagliflozin improves glycaemic control over 28 days in subjects with type 2 diabetes not optimally controlled on insulin. Diabetes Obes. Metab. 2012;14:539–545. doi: 10.1111/j.1463-1326.2012.01558.x. [DOI] [PubMed] [Google Scholar]
- Stenlöf K., Cefalu W.T., Kim K.A., Alba M., Usiskin K., Tong C., Canovatchel W., Meininger G. Efficacy and safety of canagliflozin monotherapy in subjects with type 2 diabetes mellitus inadequately controlled with diet and exercise. Diabetes Obes. Metab. 2013;15:372–382. doi: 10.1111/dom.12054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yale J.F., Bakris G., Cariou B., Yue D., David-Neto E., Xi L., Figueroa K., Wajs E., Usiskin K., Meininger G. Efficacy and safety of canagliflozin in subjects with type 2 diabetes and chronic kidney disease. Diabetes Obes. Metab. 2013;15:463–473. doi: 10.1111/dom.12090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grampler R., Thomas L., Eckhardt M., Himmelsbach F., Sauer A., Sharp D.E., Bakker R.A., Mark M., Klein T., Eickelmann P. Empagliflozin, a novel selective sodium glucose cotransporter-2 (SGLT-2) inhibitor: Characterisation and comparison with other SGLT-2 inhibitors. Diabetes Obes. Metab. 2012;14:83–90. doi: 10.1111/j.1463-1326.2011.01517.x. [DOI] [PubMed] [Google Scholar]
- Mudaliar S., Polidori D., Zambrowicz B., Henry R.R. Sodium-glucose cotransporter inhibitors: Effects on renal and intestinal glucose transport: From bench to bedside. Diabetes Care. 2015;38:2344–2353. doi: 10.2337/dc15-0642. [DOI] [PubMed] [Google Scholar]
- Poulsen SB, Fenton RA, Rieg T. Sodium‐glucose cotransport. Curr Opin Nephrol Hypertens 2015; 24: 463–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hediger MA, Coady MJ, Ikeda TS, et al Expression cloning and cDNA sequencing of the Na+/glucose co‐transporter. Nature 1987; 330: 379–381. [DOI] [PubMed] [Google Scholar]
- Kanai Y, Lee WS, You G, et al The human kidney low affinity Na+/glucose cotransporter SGLT2. Delineation of the major renal reabsorptive mechanism for D‐glucose. J Clin Investig 1994; 93: 397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright EM, Loo DD, Hirayama BA. Biology of human sodium glucose transporters. Physiol Rev 2011; 91: 733–794. [DOI] [PubMed] [Google Scholar]
- Rossetti L, Smith D, Shulman GI, et al Correction of hyperglycemia with phlorizin normalizes tissue sensitivity to insulin in diabetic rats. J Clin Investig 1987; 79: 1510–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFronzo RA, Davidson JA, Del Prato S. The role of the kidneys in glucose homeostasis: a new path towards normalizing glycaemia. Diabetes Obes Metab 2012; 14: 5–14. [DOI] [PubMed] [Google Scholar]
- Vallon V. The mechanisms and therapeutic potential of SGLT2 inhibitors in diabetes mellitus. Annu Rev Med 2015; 66: 255–270. [DOI] [PubMed] [Google Scholar]
- Abdul‐Ghani MA, Norton L, DeFronzo RA. Renal sodium‐glucose cotransporter inhibition in the management of type 2 diabetes mellitus. Am J Physiol Renal Physiol 2015; 309: F889–F900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diez‐Sampedro A, Hirayama BA, Osswald C, et al A glucose sensor hiding in a family of transporters. Proc Natl Acad Sci USA 2003; 100: 11753–11758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright EM, Loo DD, Hirayama BA, et al Surprising versatility of Na+‐glucose cotransporters: SLC5. Physiology 2004; 19: 370–376. [DOI] [PubMed] [Google Scholar]
- Turk E, Wright EM. Membrane topology motifs in the SGLT cotransporter family. J Membr Biol 1997; 159: 1–20. [DOI] [PubMed] [Google Scholar]
- Tyagi NK, Kumar A, Goyal P, et al D‐Glucose‐recognition and phlorizin‐binding sites in human sodium/D‐glucose cotransporter 1 (hSGLT1): a tryptophan scanning study. Biochemistry 2007; 46: 13616–13628. [DOI] [PubMed] [Google Scholar]
- Hediger MA, Rhoads DB. Molecular physiology of sodium‐glucose cotransporters. Physiol Rev 1994; 74: 993–1026. [DOI] [PubMed] [Google Scholar]
- Bonner C, Kerr‐Conte J, Gmyr V, et al Inhibition of the glucose transporter SGLT2 with dapagliflozin in pancreatic alpha cells triggers glucagon secretion. Nat Med 2015; 21: 512–517. [DOI] [PubMed] [Google Scholar]
- Lee WS, Kanai Y, Wells RG, et al The high affinity Na+/glucose cotransporter. Re‐evaluation of function and distribution of expression. J Biol Chem 1994; 269: 12032–12039. [PubMed] [Google Scholar]
- Hirsch JR, Loo DD, Wright EM. Regulation of Na+/glucose cotransporter expression by protein kinases in Xenopus laevis oocytes. J Biol Chem 1996; 271: 14740–14746. [DOI] [PubMed] [Google Scholar]
- Wright EM, Hirsch JR, Loo DD, et al Regulation of Na+/glucose cotransporters. J Exp Biol 1997; 200: 287–293. [DOI] [PubMed] [Google Scholar]
- Subramanian S, Glitz P, Kipp H, et al Protein kinase‐A affects sorting and conformation of the sodium‐dependent glucose co‐transporter SGLT1. J Cell Biochem 2009; 106: 444–452. [DOI] [PubMed] [Google Scholar]
- Ishikawa Y, Eguchi T, Ishida H. Mechanism of beta‐adrenergic agonist‐induced transmural transport of glucose in rat small intestine. Regulation of phosphorylation of SGLT1 controls the function. Biochim Biophys Acta 1997; 1357: 306–318. [DOI] [PubMed] [Google Scholar]
- Sopjani M, Bhavsar SK, Fraser S, et al Regulation of Na+‐coupled glucose carrier SGLT1 by AMP‐activated protein kinase. Mol Membr Biol 2010; 27: 137–144. [DOI] [PubMed] [Google Scholar]
- Banerjee SK, Wang DW, Alzamora R, et al SGLT1, a novel cardiac glucose transporter, mediates increased glucose uptake in PRKAG2 cardiomyopathy. J Mol Cell Cardiol 2010; 49: 683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandu C, Rexhepaj R, Grahammer F, et al Decreased intestinal glucose transport in the sgk3‐knockout mouse. Pflugers Archiv 2005; 451: 437–444. [DOI] [PubMed] [Google Scholar]
- Elvira B, Blecua M, Luo D, et al SPAK‐sensitive regulation of glucose transporter SGLT1. J Membr Biol 2014; 247: 1191–1197. [DOI] [PubMed] [Google Scholar]
- 65. Ferraris RP, Diamond J. Regulation of intestinal sugar transport. Physiol Rev 1997; 77: 257–302. [DOI] [PubMed] [Google Scholar]
- Dyer J, Hosie KB, Shirazi‐Beechey SP. Nutrient regulation of human intestinal sugar transporter (SGLT1) expression. Gut 1997; 41: 56–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turk E, Wright EM. Membrane topology motifs in the SGLT cotransporter family. J Membr Biol 1997; 159: 1–20.
[DOI] [PubMed] [Google Scholar] - Chen J, Williams S, Ho S, et al Quantitative PCR tissue expression profiling of the human SGLT2 gene and related family members. Diabetes Ther 2010; 1: 57–92.
[DOI] [PMC free article] [PubMed] [Google Scholar] - Sabolic I, Vrhovac I, Eror DB, et al Expression of Na+‐D‐glucose cotransporter SGLT2 in rodents is kidney‐specific and exhibits sex and species differences. Am J Physiol Cell Physiol 2012; 302: C1174–C1188.
[DOI] [PMC free article] [PubMed] [Google Scholar] - Bonner C, Kerr‐Conte J, Gmyr V, et al Inhibition of the glucose transporter SGLT2 with dapagliflozin in pancreatic alpha cells triggers glucagon secretion. Nat Med 2015; 21: 512–517.
[DOI] [PubMed] [Google Scholar] - Balen D, Ljubojevic M, Breljak D, et al Revised immunolocalization of the Na+‐D‐glucose cotransporter SGLT1 in rat organs with an improved antibody. Am J Physiol Cell Physiol 2008; 295: C475–C489. [DOI] [PubMed] [Google Scholar]
- Abdul‐Ghani MA, DeFronzo RA, Norton L. Novel hypothesis to explain why SGLT2 inhibitors inhibit only 30–50% of filtered glucose load in humans. Diabetes 2013; 62: 3324–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghezzi C, Wright EM. Regulation of the human Na+‐dependent glucose cotransporter hSGLT2. Am J Physiol Cell Physiol 2012; 303: C348–C354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunilkumar S, Ford SM. Elevated glucose concentration in culture media decreases membrane trafficking of SGLT2 in LLC‐PK1 cells via a cAMP/PKA‐dependent pathway. Am J Physiol Cell Physiol 2019; 316: C913–C924. [DOI] [PubMed] [Google Scholar]
- Maldonado‐Cervantes MI, Galicia OG, Moreno‐Jaime B, et al Autocrine modulation of glucose transporter SGLT2 by IL‐6 and TNF‐alpha in LLC‐PK(1) cells. J Physiol Biochem 2012; 68: 411–420. [DOI] [PubMed] [Google Scholar]
- Panchapakesan U, Pegg K, Gross S, et al Effects of SGLT2 inhibition in human kidney proximal tubular cells–renoprotection in diabetic nephropathy? PLoS ONE 2013; 8: e54442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E. Turk, MG. Martín; EM. Wright, Structure of the human Na+/glucose cotransporter gene SGLT1., in J Biol Chem, vol. 269, n. 21, Mag 1994, pp. 15204-9, PMID 8195156.
- Vallon V, Gerasimova M, Rose MA, et al SGLT2 inhibitor empagliflozin reduces renal growth and albuminuria in proportion to hyperglycemia and prevents glomerular hyperfiltration in diabetic Akita mice. Am J Physiol Renal Physiol 2014; 306: F194–F204.
[DOI] [PMC free article] [PubMed] [Google Scholar] - Connelly KA, Zhang Y, Desjardins JF, et al Dual inhibition of sodium‐glucose linked cotransporters 1 and 2 exacerbates cardiac dysfunction following experimental myocardial infarction. Cardiovasc Diabetol 2018; 17: 99.
[DOI] [PMC free article] [PubMed] [Google Scholar] - Poppe R, Karbach U, Gambaryan S, et al Expression of the Na+‐D‐glucose cotransporter SGLT1 in neurons. J Neurochem 1997; 69: 84–94.
[DOI] [PubMed] [Google Scholar] - Bonner C, Kerr‐Conte J, Gmyr V, et al Inhibition of the glucose transporter SGLT2 with dapagliflozin in pancreatic alpha cells triggers glucagon secretion. Nat Med 2015; 21: 512–517.
[DOI] [PubMed] [Google Scholar] - Moriya R, Shirakura T, Ito J, et al Activation of sodium‐glucose cotransporter 1 ameliorates hyperglycemia by mediating incretin secretion in mice. Am J Physiol Endocrinol Metab 2009; 297: E1358–E1365. [DOI] [PubMed] [Google Scholar]
- Tolhurst G, Heffron H, Lam YS, et al Short‐chain fatty acids stimulate glucagon‐like peptide‐1 secretion via the G‐protein‐coupled receptor FFAR2. Diabetes 2012; 61: 364–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cefalo CMA, Cinti F, Moffa S, et al Sotagliflozin, the first dual SGLT inhibitor: current outlook and perspectives. Cardiovascu Diabetol 2019; 18: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koepsell H. The Na(+)‐D‐glucose cotransporters SGLT1 and SGLT2 are targets for the treatment of diabetes and cancer. Pharmacol Therap 2017; 170: 148–165. [DOI] [PubMed] [Google Scholar]
- Tahara A, Kurosaki E, Yokono M, et al Effects of SGLT2 selective inhibitor ipragliflozin on hyperglycemia, hyperlipidemia, hepatic steatosis, oxidative stress, inflammation, and obesity in type 2 diabetic mice. Eur J Pharmacol 2013; 715: 246–255. [DOI] [PubMed] [Google Scholar]
- Abdel‐Wahab AF, Bamagous GA, Al‐Harizy RM, et al Renal protective effect of SGLT2 inhibitor dapagliflozin alone and in combination with irbesartan in a rat model of diabetic nephropathy. Biomed Pharmacother 2018; 103: 59–66. [DOI] [PubMed] [Google Scholar]
- Terami N, Ogawa D, Tachibana H, et al Long‐term treatment with the sodium glucose cotransporter 2 inhibitor, dapagliflozin, ameliorates glucose homeostasis and diabetic nephropathy in db/db mice. PLoS ONE 2014; 9: e100777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiviott SD, Raz I, Bonaca MP, et al Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl j Med 2019; 380: 347–357. [DOI] [PubMed] [Google Scholar]
- Perkovic V, Jardine MJ, Neal B, et al Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N Engl J Med 2019; 380: 2295–2306. [DOI] [PubMed] [Google Scholar]
- DeFronzo RA, Hompesch M, Kasichayanula S, Liu X, Hong Y, Pfister M, et al. Characterization of renal glucose reabsorption in response to dapagliflozin in healthy subjects and subjects with type 2 diabetes. Diabetes Care. 2013;36(10):3169–3176. doi: 10.2337/dc13-0387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- .Henry RR, Klein EJ, Han J, Iqbal N. Efficacy and Tolerability of Exenatide Once Weekly Over 6 Years in Patients with Type 2 Diabetes: an Uncontrolled Open-Label Extension of the DURATION-1 Study. Diabetes Technol Ther. 2016;18(11):677–686. doi: 10.1089/dia.2016.0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- .Frias JP, Guja C, Hardy E, Ahmed A, Dong F, Ohman P, et al. Exenatide once weekly plus dapagliflozin once daily versus exenatide or dapagliflozin alone in patients with type 2 diabetes inadequately controlled with metformin monotherapy (DURATION-8): a 28 week, multicentre, double-blind, phase 3, randomised controlled trial. The lancet Diabetes & endocrinology. 2016;4(12):1004–1016. doi: 10.1016/S2213-8587(16)30267-4. [DOI] [PubMed] [Google Scholar]
- Liu XY, Zhang N, Chen R, Zhao JG, Yu P. Efficacy and safety of sodium-glucose cotransporter 2 inhibitors in type 2 diabetes: a meta-analysis of randomized controlled trials for 1 to 2years. J Diabetes Complications. 2015;29(8):1295–1303. doi: 10.1016/j.jdiacomp.2015.07.011. [DOI] [PubMed] [Google Scholar]
- .Maruthur NM, Tseng E, Hutfless S, Wilson LM, Suarez-Cuervo C, Berger Z, et al. Diabetes Medications as Monotherapy or Metformin-Based Combination Therapy for Type 2 Diabetes: a Systematic Review and Meta-analysis. Ann Intern Med. 2016;164(11):740–751. doi: 10.7326/M15-2650. [DOI] [PubMed] [Google Scholar]
- .Bays HE, Weinstein R, Law G, Canovatchel W. Canagliflozin: effects in overweight and obese subjects without diabetes mellitus. Obesity (Silver Spring). 2014;22(4):1042–1049. doi: 10.1002/oby.20663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- .Lundkvist P, Sjostrom CD, Amini S, Pereira MJ, Johnsson E, Eriksson JW. Dapagliflozin once-daily and exenatide once-weekly dual therapy: a 24-week randomized, placebo-controlled, phase II study examining effects on body weight and prediabetes in obese adults without diabetes. Diabetes Obes Metab. 2017;19(1):49–60. doi: 10.1111/dom.12779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccardi F, Webb DR, Htike ZZ, Youssef D, Khunti K, Davies MJ. Efficacy and safety of sodium-glucose co-transporter-2 inhibitors in type 2 diabetes mellitus: systematic review and network meta-analysis. Diabetes Obes Metab. 2016;18(8):783–794. doi: 10.1111/dom.12670.
[DOI] [PubMed] [Google Scholar] - Monami M, Nardini C, Mannucci E. Efficacy and safety of sodium glucose co-transport-2 inhibitors in type 2 diabetes: a meta-analysis of randomized clinical trials. Diabetes Obes Metab. 2014;16(5):457–466. doi: 10.1111/dom.12244. [DOI] [PubMed] [Google Scholar]
- Gerich JE. Physiology of glucose homeostasis. Diabetes Obes Metab. 2000;2(6):345–350. doi: 10.1046/j.1463-1326.2000.00085.x. [DOI] [PubMed] [Google Scholar]
- Merovci A, Solis-Herrera C, Daniele G, Eldor R, Fiorentino TV, Tripathy D, et al. Dapagliflozin improves muscle insulin sensitivity but enhances endogenous glucose production. J Clin Invest. 2014;124(2):509–514. doi: 10.1172/JCI70704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mearns ES, Sobieraj DM, White CM, Saulsberry WJ, Kohn CG, Doleh Y, et al. Comparative efficacy and safety of antidiabetic drug regimens added to metformin monotherapy in patients with type 2 diabetes: a network meta-analysis. PLoS ONE. 2015;10(4):e0125879. doi: 10.1371/journal.pone.0125879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Yang W, Gao X, Chen Y, Zhou L, Zhang S, et al. The Association Between the Dosage of SGLT2 Inhibitor and Weight Reduction in Type 2 Diabetes Patients: a Meta-Analysis. Obesity. 2018;26(1):70–80. doi: 10.1002/oby.22066. [DOI] [PubMed] [Google Scholar]
- Bolinder J, Ljunggren O, Johansson L, Wilding J, Langkilde AM, Sjostrom CD, et al. Dapagliflozin maintains glycaemic control while reducing weight and body fat mass over 2 years in patients with type 2 diabetes mellitus inadequately controlled on metformin. Diabetes Obes Metab. 2014;16(2):159–169. doi: 10.1111/dom.12189. [DOI] [PubMed] [Google Scholar]
- Ferrannini G, Hach T, Crowe S, Sanghvi A, Hall KD, Ferrannini E. Energy Balance After Sodium-Glucose Cotransporter 2 Inhibition. Diabetes Care. 2015;38(9):1730–1735. doi: 10.2337/dc15-0355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibel RL, Rosenbaum M, Hirsch J. Changes in energy expenditure resulting from altered body weight. N Engl J Med. 1995;332(10):621–628. doi: 10.1056/NEJM199503093321001. [DOI] [PubMed] [Google Scholar]
- Frias JP, Guja C, Hardy E, Ahmed A, Dong F, Ohman P, et al. Exenatide once weekly plus dapagliflozin once daily versus exenatide or dapagliflozin alone in patients with type 2 diabetes inadequately controlled with metformin monotherapy (DURATION-8): a 28 week, multicentre, double-blind, phase 3, randomised controlled trial. The lancet Diabetes & endocrinology. 2016;4(12):1004–1016. doi: 10.1016/S2213-8587(16)30267-4.
[DOI] [PubMed] [Google Scholar] - Bays HE, Weinstein R, Law G, Canovatchel W. Canagliflozin: effects in overweight and obese subjects without diabetes mellitus. Obesity (Silver Spring). 2014;22(4):1042–1049. doi: 10.1002/oby.20663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundkvist P, Sjostrom CD, Amini S, Pereira MJ, Johnsson E, Eriksson JW. Dapagliflozin once-daily and exenatide once-weekly dual therapy: a 24-week randomized, placebo-controlled, phase II study examining effects on body weight and prediabetes in obese adults without diabetes. Diabetes Obes Metab. 2017;19(1):49–60. doi: 10.1111/dom.12779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollander P, Bays HE, Rosenstock J, Frustaci ME, Fung A, Vercruysse F, et al. Coadministration of Canagliflozin and Phentermine for Weight Management in Overweight and Obese Individuals Without Diabetes: a Randomized Clinical Trial. Diabetes Care. 2017;40(5):632–639. doi: 10.2337/dc16-2427. [DOI] [PubMed] [Google Scholar]
- Devenny JJ, Godonis HE, Harvey SJ, Rooney S, Cullen MJ, Pelleymounter MA. Weight loss induced by chronic dapagliflozin treatment is attenuated by compensatory hyperphagia in diet-induced obese (DIO) rats. Obesity. 2012;20(8):1645–1652. doi: 10.1038/oby.2012.59. [DOI] [PubMed] [Google Scholar]
- Yokono M, Takasu T, Hayashizaki Y, Mitsuoka K, Kihara R, Muramatsu Y, et al. SGLT2 selective inhibitor ipragliflozin reduces body fat mass by increasing fatty acid oxidation in high-fat diet-induced obese rats. Eur J Pharmacol. 2014;727:66–74. doi: 10.1016/j.ejphar.2014.01.040. [DOI] [PubMed] [Google Scholar]
- Busch RS, Kane MP. Combination SGLT2 inhibitor and GLP-1 receptor agonist therapy: a complementary approach to the treatment of type 2 diabetes. Postgrad Med. 2017;129(7):686–697. doi: 10.1080/00325481.2017.1342509.
[DOI] [PubMed] [Google Scholar] - Ferrannini E, Baldi S, Frascerra S, Astiarraga B, Heise T, Bizzotto R, et al. Shift to Fatty Substrate Utilization in Response to Sodium-Glucose Cotransporter 2 Inhibition in Subjects Without Diabetes and Patients With Type 2 Diabetes. Diabetes. 2016;65(5):1190–1195. doi: 10.2337/db15-1356.
[DOI] [PubMed] [Google Scholar] - Vasilakou D, Karagiannis T, Athanasiadou E, Mainou M, Liakos A, Bekiari E, et al. Sodium-glucose cotransporter 2 inhibitors for type 2 diabetes: a systematic review and meta-analysis. Ann Intern Med. 2013;159(4):262–274. doi: 10.7326/0003-4819-159-4-201308200-00007. [DOI] [PubMed] [Google Scholar]
- Zaccardi F, Webb DR, Htike ZZ, Youssef D, Khunti K, Davies MJ. Efficacy and safety of sodium-glucose co-transporter-2 inhibitors in type 2 diabetes mellitus: systematic review and network meta-analysis. Diabetes Obes Metab. 2016;18(8):783–794. doi: 10.1111/dom.12670. [DOI] [PubMed] [Google Scholar]
- Xu L, Nagata N, Nagashimada M, Zhuge F, Ni Y, Chen G, et al. SGLT2 Inhibition by Empagliflozin Promotes Fat Utilization and Browning and Attenuates Inflammation and Insulin Resistance by Polarizing M2 Macrophages in Diet-induced Obese Mice. EBioMedicine. 2017;20:137–149. doi: 10.1016/j.ebiom.2017.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugizaki T, Zhu S, Guo G, Matsumoto A, Zhao J, Endo M, et al. Treatment of diabetic mice with the SGLT2 inhibitor TA-1887 antagonizes diabetic cachexia and decreases mortality. NPJ Aging Mech Dis. 2017;3:12. doi: 10.1038/s41514-017-0012-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg AS, Obin MS. Obesity and the role of adipose tissue in inflammation and metabolism. Am J Clin Nutr. 2006;83(2):461S–465S. doi: 10.1093/ajcn/83.2.461S. [DOI] [PubMed] [Google Scholar]
- Zaccardi F, Webb DR, Htike ZZ, Youssef D, Khunti K, Davies MJ. Efficacy and safety of sodium-glucose co-transporter-2 inhibitors in type 2 diabetes mellitus: systematic review and network meta-analysis. Diabetes Obes Metab. 2016;18(8):783–794. doi: 10.1111/dom.12670. [DOI] [PubMed] [Google Scholar]
- Bays HE, Weinstein R, Law G, Canovatchel W. Canagliflozin: effects in overweight and obese subjects without diabetes mellitus. Obesity (Silver Spring). 2014;22(4):1042–1049. doi: 10.1002/oby.20663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X., Fang J., Huang L., Wang J., Huang X. Sophora flavescens Ait.: Traditional usage, phytochemistry and pharmacology of an important traditional Chinese medicine. J. Ethnopharmacol. 2015;172:10–29. doi: 10.1016/j.jep.2015.06.010.
[DOI] [PubMed] [Google Scholar] - Sato S., Takeo J., Aoyama C., Kawahara H. Na+-glucose cotransporter (SGLT) inhibitory flavonoids from the roots of Sophora flavescens. Bioorg. Med. Chem. 2007;15:3445–3449. doi: 10.1016/j.bmc.2007.03.011.
[DOI] [PubMed] [Google Scholar] - Yang J., Yang X., Wang C., Lin Q., Mei Z., Zhao P. Sodium-glucose-linked transporter 2 inhibitors from Sophora flavescens. Med. Chem. Res. 2015;24:1265–1271. doi: 10.1007/s00044-014-1200-0.
[DOI] [Google Scholar] - Nagai M., Kubo M., Fujita M., Inoue T., Matsuo M. Studies on the constituents of Aceraceae plants. II. Structure of aceroside I, a glucose of a novel cyclic diarylheptanoids from Acer nikoense Maxim. Chem. Pharm. Bull. (Tokyo) 1978;26:2805–2810. doi: 10.1248/cpb.26.2805.
[DOI] [Google Scholar] - Morita H., Deguchi J., Motegi Y., Sato S., Aoyama C., Takeo J., Shiro M., Hirasawa Y. Cyclic diarylheptanoids as Na+-glucose cotransporter (SGLT) inhibitors from Acer nikoense. Bioorg. Med. Chem. Letter. 2010;20:1070–1074. doi: 10.1016/j.bmcl.2009.12.036.
[DOI] [PubMed] [Google Scholar] - Shimokawa Y., Akao Y., Hirasawa Y., Awang K., Hadi A.H., Sato S., Aoyama C., Takeo J., Shiro M., Morita H. Gneyulins A and B, stilbene trimers, and noidesols A and B, dihydroflavonol-C-glucosides, from the bark of Gnetum gnemonoides. J. Nat. Prod. 2010;73:763–767. doi: 10.1021/np9007987.
[DOI] [PubMed] [Google Scholar] - Changwichit K., Khorana N., Suwanborirux K., Waranuch N., Limpeanchob N., Wisuitiprot W., Suphrom N., Ingkaninan K. Bisindole alkaloids and secoiridoids from Alstonia macrophylla Wall. ex G. Don. Fitoterapia. 2011;82:798–804. doi: 10.1016/j.fitote.2011.04.013. [DOI] [PubMed] [Google Scholar]
- Khyade M.S., Kasote D.M., Vaikos N.P. Alstonia scholaris (L.) R. Br. and Alstonia macrophylla Wall. ex G. Don: A comparative review on traditional uses, phytochemistry and pharmacology. J. Ethnopharmacol. 2014;153:1–18. doi: 10.1016/j.jep.2014.01.025. [DOI] [PubMed] [Google Scholar]
- Arai H., Hirasawa Y., Rahman A., Kusumawati I., Zaini N.C., Sato S., Aoyama C., Takeo J., Morita H. Alstiphyllanines E-H, picraline and ajmaline-type alkaloids from Alstonia macrophylla inhibiting sodium glucose cotransporter. Bioorg. Med. Chem. 2010;18:2152–2158. doi: 10.1016/j.bmc.2010.01.077.
[DOI] [PubMed] [Google Scholar] - Khyade M.S., Kasote D.M., Vaikos N.P. Alstonia scholaris (L.) R. Br. and Alstonia macrophylla Wall. ex G. Don: A comparative review on traditional uses, phytochemistry and pharmacology. J. Ethnopharmacol. 2014;153:1–18. doi: 10.1016/j.jep.2014.01.025.
[DOI] [PubMed] [Google Scholar] - Shimokawa Y., Akao Y., Hirasawa Y., Awang K., Hadi A.H., Sato S., Aoyama C., Takeo J., Shiro M., Morita H. Gneyulins A and B, stilbene trimers, and noidesols A and B, dihydroflavonol-C-glucosides, from the bark of Gnetum gnemonoides. J. Nat. Prod. 2010;73:763–767. doi: 10.1021/np9007987.
[DOI] [PubMed] [Google Scholar] - Huang T., Shen P., Shen Y. Preparative separation and purification of deoxyschisandrin and gamma-schisandrin from Schisandra chinensis (Turcz.) Baill by high-seed counter-current chromatography. J. Chromatogr. A. 2005;1066:239–242. doi: 10.1016/j.chroma.2005.01.025. [DOI] [PubMed] [Google Scholar]
- Chan S.W. Panax ginseng, Rhodiola rosea and Schisandra chinensis. Int. J. Food Sci. Nutr. 2012;63:75–81. doi: 10.3109/09637486.2011.627840. [DOI] [PubMed] [Google Scholar]
- Qu Y., Chan J.Y., Wong C.W., Cheng L., Xu C., Leung A.W., Lau C.B. Antidiabetic effect of Schisandrae Chinensis Fructus involves inhibition of the sodium glucose cotransporter. Drug Dev. Res. 2015;76:1–8. doi: 10.1002/ddr.21233. [DOI] [PubMed] [Google Scholar]