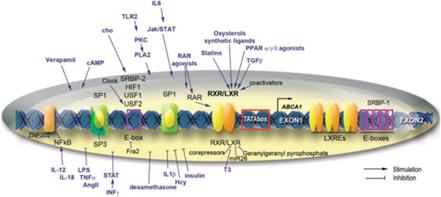
Nella seconda parte di questa serie di articoli dedicati alla alopecia androgenetica (e, quindi, anche AAS-correlata) ho discusso le modalità di trattamento più convenzionali per tale condizione , ovvero le versioni orali e topiche di Finasteride e Minoxidil con accenni alla Dutasteride. In questo articolo ne illustrerò alcune di più nuove o sperimentali, come gli Antagonisti topici del Recettore degli Androgeni, la terapia con plasma ricco di piastrine (PRP), i modulatori del segnale di Wnt e le Prostaglandine.
Antagonisti del Recettore degli Androgeni:
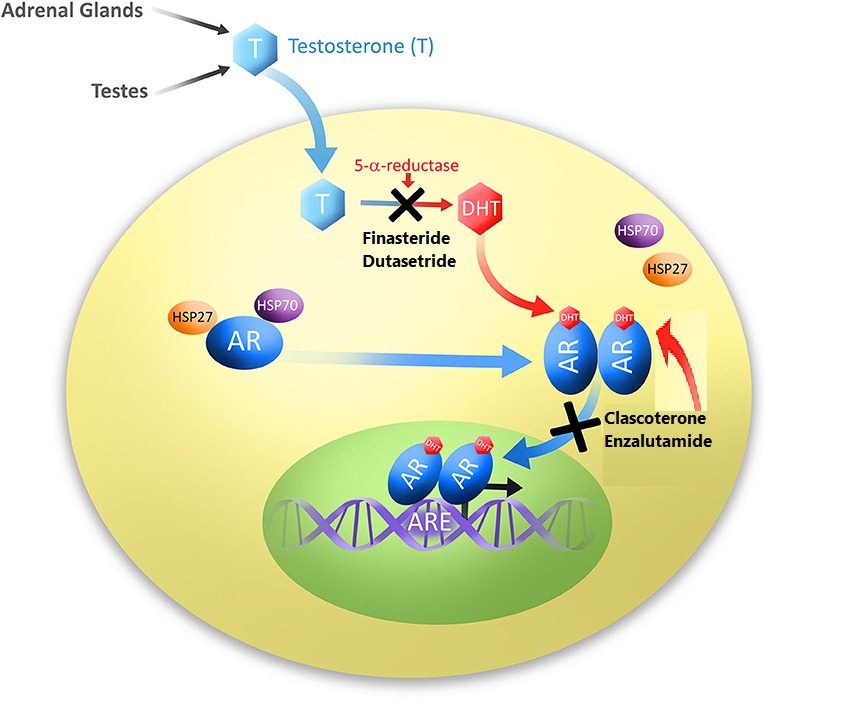
L’obbiettivo ottenuto con gli Antagonisti del Recettore degli Androgeni è simile a quello degli inibitori della 5α-reduttasi, come la Finasteride e la Futasteride: ridurre l’azione androgenica. Il meccanismo è tuttavia diverso. Gli inibitori della 5α-reduttasi bloccano la conversione del Testosterone nel più potente androgeno Diidrotestosterone (DHT). In questo modo, l’effetto androgeno del Testosterone non viene amplificato nel tessuto del cuoio capelluto. Gli Antagonisti del Recettore degli Androgeni bloccano l’azione androgena impedendo agli androgeni di legarsi al loro recettore. In questo modo, la loro azione viene bloccata a livello del Recettore degli Androgeni stesso, e quindi si rivolge praticamente a tutti gli androgeni piuttosto che a quello specifico, come nel caso degli inibitori della 5α-reduttasi. Il problema è che i suoi effetti devono rimanere localizzati al cuoio capelluto. Bloccare l’azione complessiva degli androgeni in altri tessuti, come quello muscolare, è decisamente indesiderato.
Rappresentazione grafica semplificata dell’attività degli inibitori della 5α-reduttasi e degli Antagonisti del Recettore degli Androgeni.
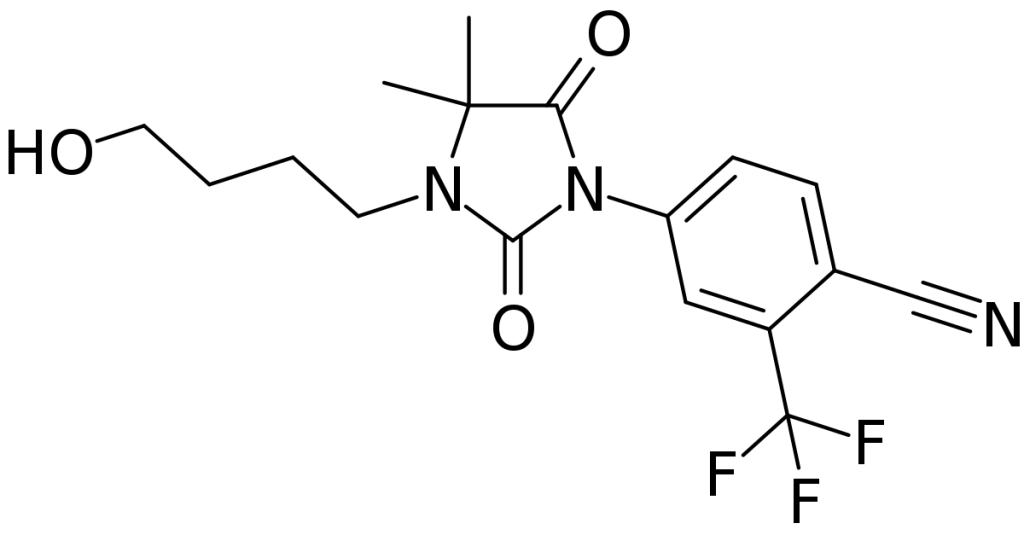
Uno di questi farmaci, attualmente in fase di sperimentazione clinica, è il Clascoterone (Breezula). La ricerca è condotta dall’azienda farmaceutica Cassiopea S.p.A. . Ricerche in vitro su cellule di papilla dermica umana hanno dimostrato che il composto è efficace nell’inibire l’azione degli androgeni [1]. Lo fa in misura maggiore rispetto all’Enzalutamide, un altro antagonista del recettore degli androgeni utilizzato nel trattamento del cancro alla prostata, e in misura paragonabile alla Finasteride. L’affinità per il AR è relativamente bassa, circa 100 volte inferiore all’affinità del DHT per il AR [2]. Questo non è un vero problema, si può rimediare semplicemente assicurandosi che le cellule del follicolo pilifero siano esposte a una concentrazione sufficientemente alta della molecola. Tuttavia, ci si chiede quale sia la sua affinità per altri recettori steroidei, come quello dei glucocorticoidi. Se non ha una specificità sufficientemente elevata per il Recettore degli Androgeni, si possono ottenere effetti fuori bersaglio legandosi a questi altri recettori. A sua volta, questo può portare a effetti collaterali. Anche questo non è necessariamente un problema se l’esposizione sistemica è minima o inesistente.
Nell’agosto 2020, la FDA ha approvato il Clascoterone crema 1 % (Winlevi) per il trattamento dell’acne vulgaris in pazienti di età pari o superiore a 12 anni [3]. È quindi in linea con le aspettative utili al fine di ottenere l’approvazione anche per l’alopecia androgenetica. Infatti, nel 2019 è stato completato uno studio di fase 2 su 404 uomini per il trattamento dell’alopecia androgenetica (EudraCT #2016-003733-23).
I soggetti sono stati trattati con una soluzione da 1mL di Clascoterone al 2,5, 5,0 o 7,5% da applicare due volte al giorno, oppure 0,0 (veicolo) e 7,5% una volta al giorno, o veicolo due volte al giorno, per un anno. Sebbene i risultati non siano stati pubblicati nella letteratura scientifica, possono essere consultati online nel registro degli studi clinici dell’UE. Il numero totale di peli nell’area è aumentato in modo significativo rispetto al gruppo con soluzione veicolante in tutti i gruppi di trattamento. (Soprattutto perché nel gruppo con soluzione veicolante si è verificata una diminuzione significativa del numero totale di capelli, che riflette la progressione dell’alopecia androgenetica). È interessante notare che le valutazioni della crescita dei capelli sono state simili tra tutti i gruppi, anche se un aumento è stato riportato con una frequenza leggermente maggiore nei gruppi di trattamento (dal 56,1 al 61,8% dei soggetti rispetto al 50,0% del gruppo con il solo veicolo). Gli eventi avversi sono stati simili tra i gruppi.
L’esposizione sistemica della crema all’1% utilizzata per il trattamento dell’acne è minima [4]. I dati relativi alla soluzione topica non sono purtroppo disponibili nella letteratura pubblicata. Gli effetti collaterali sessuali non sono stati monitorati nel loro studio, quindi è difficile ricavare una potenziale esposizione sistemica sulla base di questi risultati.
Come nota finale: è interessante vedere che nel 2016 è stato completato uno studio in cui una soluzione di clascoterone è stata confrontata con una soluzione di minoxidil al 5% o un placebo per il trattamento dell’alopecia androgenetica (NCT02279823). I risultati non sono mai stati pubblicati nella letteratura scientifica. Le ragioni possono essere molteplici, ma forse la più ovvia, dal punto di vista di un’azienda farmaceutica, è: risultati deludenti. Ho l’impressione che non abbia fatto molto bene rispetto al minoxidil.
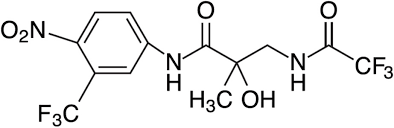
Un altro antagonista del recettore degli androgeni che sta facendo il giro di internet è RU58841 (noto anche come PSK-3841 o HMR-3841). Nel 2004 era in fase di sperimentazione II, ma da allora lo sviluppo del farmaco è stato interrotto. All’epoca era oggetto di ricerca da parte di Proskelia, l’unità francese del gruppo ProStrakan. Proskelia è stata poi acquisita da Galapagos nel 2006. È importante notare che i risultati degli studi clinici non sono mai stati riportati in letteratura. Si dice che ciò sia dovuto a motivi finanziari. Questo sembra plausibile, Proskelia era un’azienda relativamente piccola (visto che è stata acquisita per 16,5 milioni di dollari nel 2006). I costi degli studi clinici di fase 3 sono molto elevati. Si parla di almeno qualche migliaio di dollari per soggetto (in media costano diverse decine di migliaia di dollari per soggetto). Se si moltiplica questa cifra per i 1000-2000 soggetti necessari per una sperimentazione di questo tipo, diventa subito evidente che molto probabilmente hanno dovuto fare affidamento sugli investitori per realizzarla. Ciononostante, se Galapagos fosse stata interessata a questo composto, avrebbe potuto facilmente finanziare uno studio di fase 3. Va ricordato che la ragione principale per cui i farmaci non entrano nella fase 3 è la mancanza di efficacia o di sicurezza.
Mostra un’elevata affinità per il recettore degli androgeni umani, leggermente inferiore a quella del testosterone (il che è notevole, dato che la maggior parte degli antagonisti ha un’affinità sostanzialmente inferiore) [5]. Sono stati pubblicati alcuni dati di studi su animali. Mostra un’efficacia simile a quella della finasteride nei macachi dalla coda monca [6]. In topi nudi femmina condizionati con testosterone, lo xenotrapianto di tessuto del cuoio capelluto di uomini calvi ha mostrato risultati più favorevoli rispetto ai controlli [7]. Onestamente, questi studi sono preclinici per un motivo: forniscono solo un’indicazione sul fatto che potrebbe essere interessante o meno proseguire con gli studi clinici. Non forniscono altre informazioni, quindi li cito solo per completezza. Senza dati di sperimentazione clinica non si può dire molto su questo composto.
RU58841
Un’ultima osservazione che vorrei fare è che è stato suggerito che RU58841 può influenzare il recettore degli androgeni in modo allosterico [8]. Ciò significa che influisce sulla sua funzione legandosi a un sito diverso da quello di legame con il ligando (dove si legherebbero gli androgeni). Questo ha un’implicazione pratica molto importante. Se c’è un legame competitivo, la sua efficacia dipende dalla concentrazione di altri ligandi (come il DHT). In caso di legame allosterico, ciò non avviene, per cui il suo effetto è indipendente dalle concentrazioni di ligandi, il che sarebbe ideale per i consumatori di steroidi anabolizzanti, in quanto le dosi sovrafisiologiche utilizzate non influirebbero sulla sua efficacia. Purtroppo non sono in grado di accedere allo studio originale che ipotizza questa caratteristica.
Fluridil
Un altro antagonista topico dei recettori degli androgeni è il Fluridil, noto anche come topilutamide e venduto con il nome commerciale di Eucapil. È approvato per uso cosmetico nella Repubblica Ceca. È stato pubblicato uno studio clinico su piccola scala, ma i risultati non sembrano promettenti [9]. 43 soggetti con alopecia androgenetica sono stati randomizzati a ricevere una soluzione topica di Fluridil al 2% o un placebo per 9 mesi. Il conteggio dei capelli in fase anagen o telogen è stato effettuato a 0, 3, 6 e 9 mesi. Mentre nel gruppo del Fluridil si è registrato un aumento maggiore dei peli in fase anagen e una diminuzione maggiore dei peli in fase telogen rispetto al placebo a 3 mesi, non c’è stata alcuna differenza significativa a 9 mesi. (È piuttosto deludente. Studi futuri (che a questo punto non mi aspetto) potrebbero chiarire se si tratta di una peculiarità dello studio o meno. Si potrebbe ipotizzare che sia necessaria una maggiore concentrazione di soluzione di fluridil perché sia efficace.
Terapia con Plasma Ricco di Piastrine (PRP):
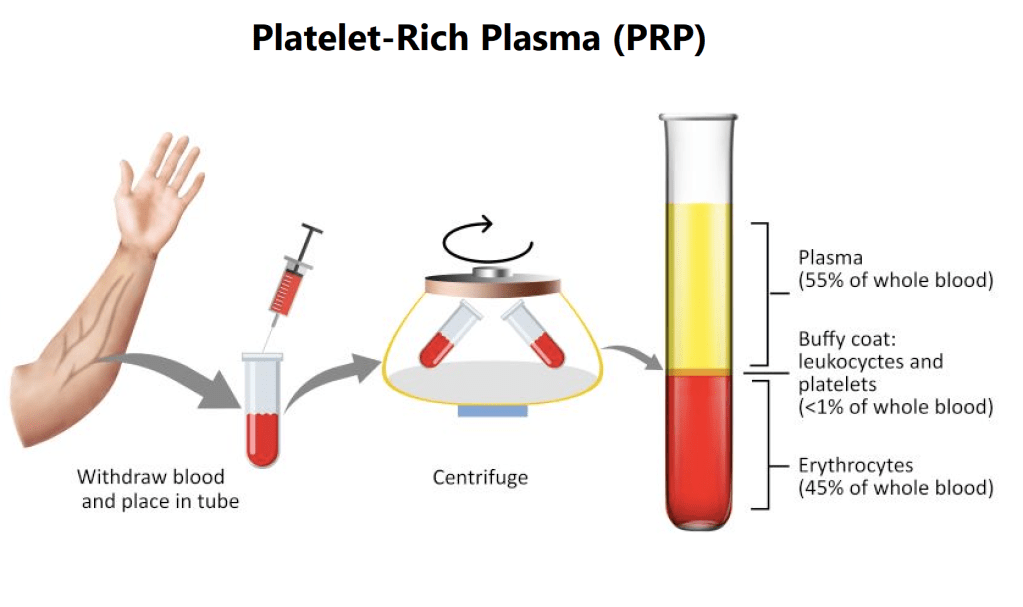
Credo che una piccola introduzione sul Plasma Ricco di Piastrine (PRP) sia necessaria. Che cos’è in realtà? In sostanza, è un concentrato di sangue con un’alta presenza di piastrine e la rimozione dei globuli rossi. Viene prodotto mediante un processo chiamato centrifugazione differenziale [9]. Il PRP prodotto contiene una concentrazione di piastrine da 2 a 8 volte superiore a quella del sangue intero. La concentrazione ottenuta dipende dal dispositivo e dal metodo utilizzato. Di solito, per preparare il PRP si prelevano circa 30ml di sangue.
Le piastrine sono importanti per la coagulazione, ma contengono anche una serie di fattori di crescita e citochine [10]. Queste molecole di segnalazione sono il motivo per cui vengono impiegate in diversi campi medici, tra cui la dermatologia, ad esempio nel trattamento dell’alopecia androgenetica. Le piastrine rilasciano questi fattori di crescita e citochine al momento dell’attivazione, che può avvenire dopo l’iniezione nel cuoio capelluto da parte dell’organismo oppure aggiungendo sali di calcio o trombina alle piastrine prima dell’iniezione. Si ritiene che questi fattori di crescita agiscano sulle cellule del follicolo pilifero, esercitando così il loro effetto benefico nel trattamento dell’alopecia (androgenetica).
Poiché non esiste una procedura standardizzata per l’applicazione del PRP, gli studi possono dimostrare risultati diversi a seguito di procedure PRP differenti. Sebbene manchino prove valide, si ritiene che la pre-attivazione delle piastrine prima dell’iniezione e la preparazione del PRP mediante il cosiddetto protocollo a doppia centrifuga portino a risultati migliori.
Una meta-analisi del 2020 ha valutato gli effetti della terapia con PRP come trattamento dell’alopecia androgenetica [11]. Ha incluso 30 studi randomizzati e controllati per l’analisi qualitativa e 5 di questi hanno potuto essere utilizzati per l’analisi quantitativa. La terapia con PRP è risultata efficace nell’aumentare la densità e lo spessore dei capelli. Di fatto, è apparsa più efficace del Minoxidil e della Finasteride. Mentre una meta-analisi del 2017 ha rilevato che la Finasteride e il Minoxidil topico al 5% aumentano la densità dei capelli rispettivamente di 18 e 15 capelli per cm quadrato, la terapia con PRP ha portato a un aumento medio di 33 capelli per cm quadrato.
Gli eventi avversi sono stati riportati nella metà degli studi e si sono limitati a dolore, eritema (rossore) ed edema locale, sanguinamento puntuale, mal di testa transitorio, sonnolenza, ematomi e sensibilità del cuoio capelluto. Non sono stati segnalati eventi avversi gravi. Nel complesso, la terapia con PRP è molto promettente.
Modulatori della via Wnt/β-catenina:
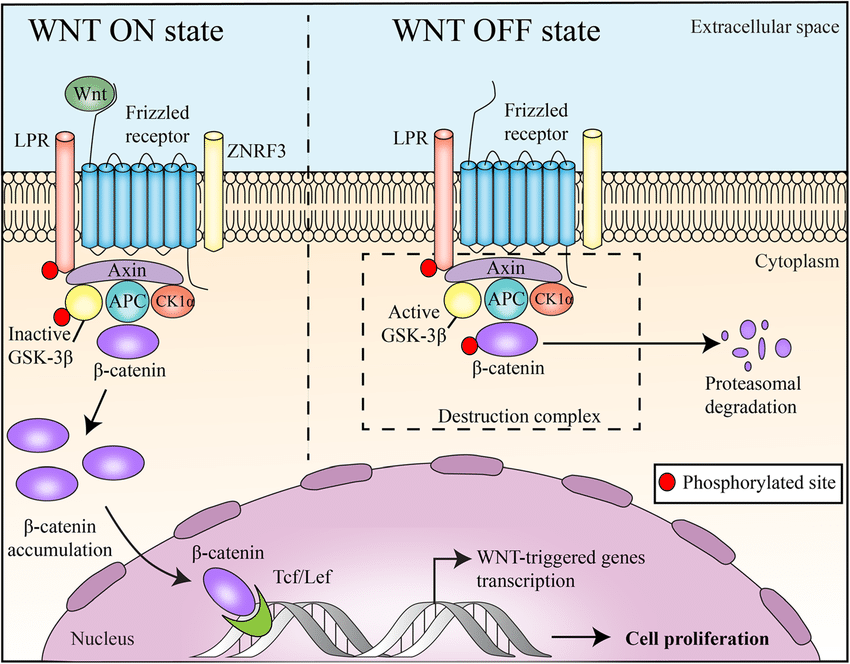
La via Wnt/β-catenina è coinvolta in numerosi processi cellulari. E, come si è scoperto, la via è anche coinvolta nella crescita e nello sviluppo del follicolo pilifero [12, 13]. La via canonica prevede il legame di una proteina Wnt a Frizzled, il suo recettore sulla superficie cellulare, e al suo co-recettore Proteina legata al recettore LDL (LRP) [14]. In assenza di segnalazione Wnt, la β-catenina viene continuamente degradata, mentre con l’attivazione da parte di una proteina Wnt, la β-catenina inizia ad accumularsi nel citosol. La β-catenina trasloca poi nel nucleo dove stimolerà la trascrizione dei geni bersaglio di Wnt.
Via canonica Wnt/β-catenina: Stato “WNT ON”: le proteine WNT, legandosi ai recettori frizzled e al co-recettore LRP, agiscono per sopprimere l’attività della glicogeno sintasi chinasi-3β (GSK-3β). ZNRF3 promuove la degradazione dei recettori WNT che funzionano come soppressori tumorali. Ciò impedisce la fosforilazione delle molecole a valle, consentendo l’associazione della β-catenina con Tcf/Lef nel nucleo e il conseguente aumento della proliferazione cellulare. Stato “WNT OFF”: in assenza del ligando WNT, il complesso di distruzione della β-catenina (contrassegnato dal riquadro tratteggiato), un complesso terziario formato da axina, APC, CK1α e GSK 3β, fosforila la β-catenina, che successivamente va incontro a degradazione proteasomica.
Un attivatore di Wnt attualmente in fase di studio per il trattamento dell’alopecia androgenetica è il SM04554 (noto anche come Dalosirvat). È stato sviluppato da Biosplice Therapeutics (precedentemente nota come Samumed) e sono stati registrati e completati 3 studi clinici: NCT02275351, NCT02503137, NCT03742518. Questi numeri NCT possono essere consultati sul sito www.clinicaltrials.gov per visualizzarne i dettagli. Lo studio registrato con il numero NCT03742518 è uno studio di fase II/III con 675 partecipanti che sono stati randomizzati in tre gruppi. Un gruppo ha utilizzato una soluzione di SM04554 allo 0,15% una volta al giorno, un altro ha utilizzato una soluzione allo 0,25% una volta al giorno e il terzo gruppo ha ricevuto una soluzione veicolo. Lo studio è durato 48 settimane e si è concluso il 31 dicembre 2020. Purtroppo non sono ancora stati pubblicati i risultati dello studio clinico nella letteratura scientifica. Tuttavia, facendo qualche ricerca su Google, è possibile trovare alcune diapositive utilizzate durante una presentazione al Congresso Internazionale di Dermatologia e Cosmetologia (INDERCOS) nel marzo 2019. In esse vengono presentati alcuni risultati di uno studio di fase II, tra cui questa diapositiva:
I partecipanti hanno ricevuto l’intervento per 90 giorni, dopodiché è stato effettuato un follow-up 45 giorni dopo. Ciò che mi colpisce è che la soluzione allo 0,25% ha fatto molto peggio della soluzione allo 0,15% e che la soluzione allo 0,15% ha iniziato a funzionare solo dopo aver terminato la somministrazione. (In ogni caso, i risultati non mi entusiasmano. E sospetto che anche la sperimentazione di fase III non sia andata molto bene. Se si utilizza la Wayback Machine per dare un’occhiata al sito web di Biosplice Therapeutics, si può vedere che SM04554 è ancora elencato nell’agosto 2021. Se si guarda oggi, il farmaco è scomparso e non è più presente nemmeno nella pagina della pipeline. Hanno rinunciato al farmaco?
Quindi, dov’è finito il SM04554?
Nel complesso, i modulatori della via di segnalazione Wnt sono promettenti, ma forse dovremo aspettare ancora un po’ prima di vedere il primo di questa classe di farmaci approvato dalla FDA.
Prostaglandine:
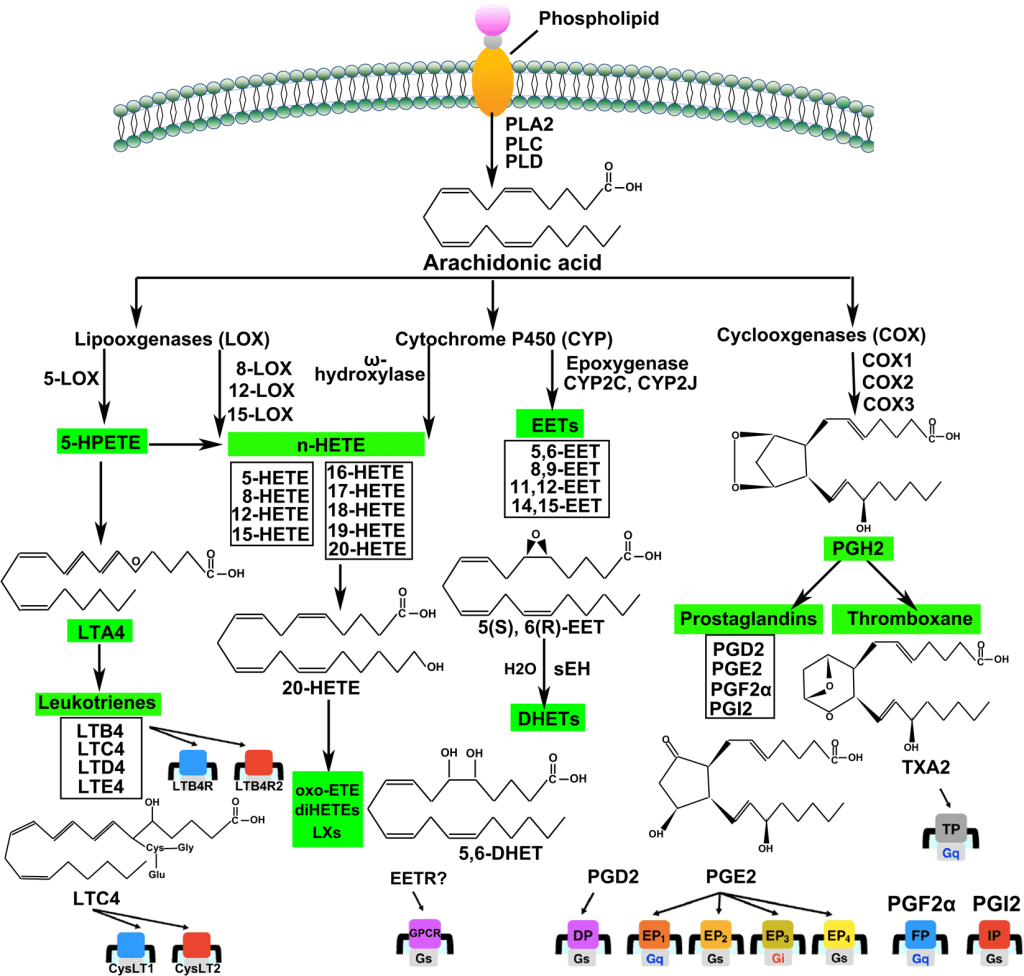
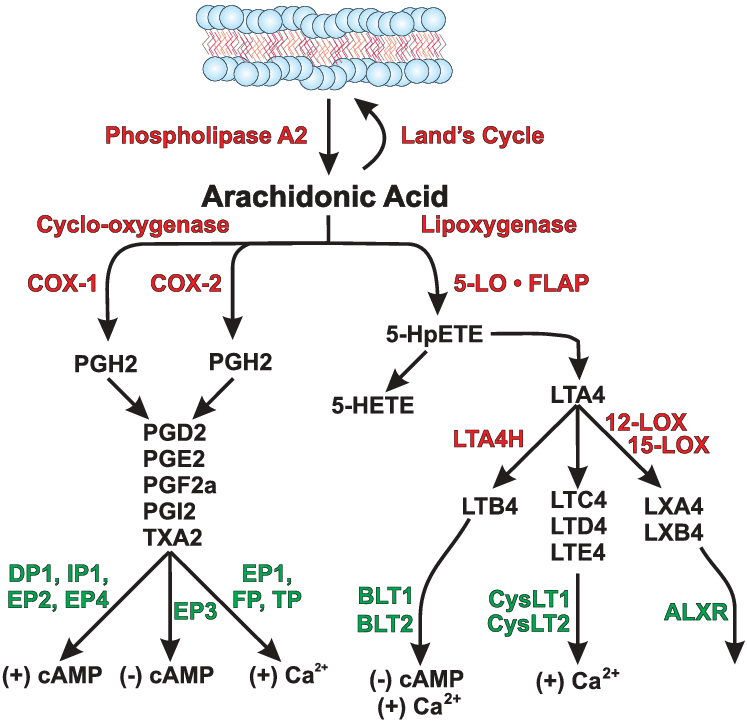
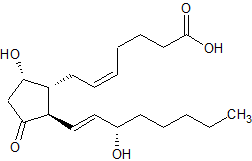
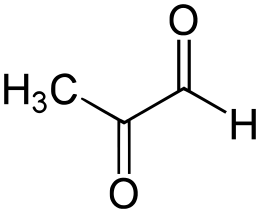
Le Prostaglandine sono emerse come importanti regolatori del ciclo del follicolo pilifero (poiché alcuni farmaci basati su di esse, di cui parlerò più avanti, si sono rivelati in grado di provocare una crescita localizzata dei capelli/ipertricosi). Sono sintetizzate dall’acido grasso arachidonico. In particolare, la Prostaglandina D2 (PGD2) è ritenuta responsabile dell’inibizione della crescita dei capelli nell’alopecia androgenetica [15]. La PGD2 è il prodotto di una reazione catalizzata dall’enzima Prostaglandina D2 Sintasi (PTGDS), il cui substrato è la Prostaglandina H2 (PGH2). La PGH2 è sintetizzata direttamente dall’Acido Arachidonico, una reazione catalizzata da un enzima ciclossigenasi (COX). Quindi, in poche parole:
Acido Arachidonico (COX)-> PGH2 (PTGDS)-> PGD2
Al contrario, la Prostaglandina F2α (PGF2α) e la Prostaglandina E2 (PGE2) stimolano la crescita dei capelli [16]. Sia la PGF2α che la PGE2 derivano anche dalla PGH2. Il primo sintetizzato dalla PGF2α Sintasi e il secondo dalla PGE2 Sintasi.
Bimatoprost
Anche l’industria farmaceutica ha esplorato quest’area di ricerca per il trattamento dell’alopecia androgenetica. Alcuni farmaci che sono stati sviluppati sono una soluzione topica di Bimatoprost (un analogo della PGE2) e una soluzione topica di Latanoprost (un analogo della PGF2α). Entrambi i farmaci sono stati originariamente utilizzati per trattare l’ipertensione oculare o il glaucoma, in quanto abbassano la pressione oculare. Tuttavia, si è scoperto per caso che provocano la crescita dei peli delle ciglia (ipertricosi). Alcuni studi clinici su piccola scala hanno valutato i loro effetti e sembrano promettenti [17, 18].
Setipiprant
Un altro farmaco è il Setipiprant, che agisce come Antagonista Selettivo del Recettore della Prostaglandina D2. Il farmaco è attualmente oggetto di studio da parte di Allergan Aesthetics e nell’ottobre 2021 sono stati pubblicati i risultati di uno studio di fase 2 [19]. I partecipanti hanno ricevuto il Setipiprant orale due volte al giorno (1g x 2 volte), 1mg di Finasteride una volta al giorno o un placebo, per 24 settimane. Sfortunatamente, però, non è stato possibile ottenere risultati migliori rispetto al placebo.
Penso che in futuro sentiremo ancora parlare di Prostaglandine (topiche), o di farmaci che potrebbero inibire la produzione di PGD2 inibendo l’enzima PTGDS (o di farmaci che stimolano la produzione di PGF2α o PGE2 stimolando i rispettivi enzimi che li sintetizzano).
Conclusione:
In questi tre articoli abbiamo imparato a conoscere una condizione (alopecia androgenetica) che interessa a diverso grado circa il 70% degli uomini ed il 40% delle donne. Abbiamo visto quali sono i farmaci approvati per il suo trattamento e quelli sperimentali più promettenti. Ora, sappiamo anche che con l’uso di dosi sovrafisiologiche di AAS riducono fortemente l’impatto apportato dagli inibitori della 5α-reduttasi i quali, comunque, interessano per lo più il Testosterone essendo il substrato principale per le 5α-reduttasi. Discorso diverso potrebbe essere fatto per ciò che concerne l’uso di soluzioni topiche contenenti Antagonisti del Recettore degli Androgeni. Ma, ad oggi, su questo punto non possiamo fare altro che analizzare la letteratura e ipotizzare.
Avrei sicuramente potuto parlare del potenziale effetto del TB-500 sulla sostanziale crescita di nuovi peli nella barba. La letteratura scientifica indica anche che la Timosina beta-4 attiva le cellule staminali nei follicoli (Questo è potenzialmente rilevante, poiché il TB-500 è, approssimativamente, un frammento della Timosina beta-4). È stato osservato che le applicazioni topiche giornaliere di TB-500 accelerano la crescita dei capelli. È stato osservato anche che i capelli risultanti sono più spessi, più scuri e più densi. Nel complesso, i risultati ottenuti su animali [topi e ratti] suggeriscono che, oltre ai suoi noti effetti angiogenici e di guarigione delle ferite, la Timosina β4 ( e potenzialmente il TB500) possa essere un modulatore naturale della crescita dei capelli che agisce stimolando la migrazione delle cellule staminali, la produzione di proteasi e la differenziazione. Attenzione però! Qui non si parla di una “resurrezione” del bulbo miniaturizzato e morto tipico della alopecia adrogenetica. Si parla di follicoli derivanti da sviluppo di cellule staminali, indi “nuovi”. Ma tutto questo rappresenta, ad oggi, poco più di una pura ipotesi nell’uomo.
L’unica soluzione, se la “rasata a 0” non è contemplata, è ovviamente il trapianto. Ma ciò non toglie che esso possa risultare migliorato da una combinazione di fattori iatrogeni, come quelli descritti in questo lavoro.
Gabriel Bellizzi [CEO BioGenTech]
Riferimenti:
Rosette, Caridad, et al. “Cortexolone 17α-propionate (clascoterone) is an androgen receptor antagonist in dermal papilla cells in vitro.” Journal of drugs in dermatology: JDD 18.2 (2019): 197-201.
Celasco, Giuseppe, et al. “Biological profile of cortexolone 17a-propionate (CB-03-01), a new topical and peripherally selective androgen antagonist.” Arzneimittelforschung 54.12 (2004): 881-886.
Dhillon, Sohita. “Clascoterone: first approval.” Drugs (2020): 1-6.
Mazzetti, Alessandro, et al. “Pharmacokinetic profile, safety, and tolerability of clascoterone (cortexolone 17-alpha propionate, CB-03-01) topical cream, 1% in subjects with acne vulgaris: an open-label phase 2a study.” Journal of drugs in dermatology: JDD 18.6 (2019): 563-563.
Battmann, T., et al. “RU 58841, a new specific topical antiandrogen: a candidate of choice for the treatment of acne, androgenetic alopecia and hirsutism.” The Journal of Steroid Biochemistry and Molecular Biology 48.1 (1994): 55-60.
Uno, H., et al. “Follicular regrowth with 5 α-reductase inhibitor (finasteride) or androgen receptor blocker (RU58841) in the bald scalp of the stumptailed macaque.” Journal of Investigative Dermatology 4.104 (1995): 658.
De Brouwer, B., et al. “A controlled study of the effects of RU58841, a non‐steroidal antiandrogen, on human hair production by balding scalp grafts maintained on testosterone‐conditioned nude mice.” British Journal of Dermatology 137.5 (1997): 699-702.
Poulos, Georgann A., and Paradi Mirmirani. “Investigational medications in the treatment of alopecia.” Expert opinion on investigational drugs 14.2 (2005): 177-184.
Dhurat, Rachita, and M. S. Sukesh. “Principles and methods of preparation of platelet-rich plasma: a review and author’s perspective.” Journal of cutaneous and aesthetic surgery 7.4 (2014): 189.
Alves, Rubina, and Ramon Grimalt. “A review of platelet-rich plasma: history, biology, mechanism of action, and classification.” Skin appendage disorders 4.1 (2018): 18-24.
Evans, Adam G., et al. “Platelet-rich plasma as a therapy for androgenic alopecia: a systematic review and meta-analysis.” Journal of Dermatological Treatment (2020): 1-14.
Beaudoin, Gerard MJ, et al. “Hairless triggers reactivation of hair growth by promoting Wnt signaling.” Proceedings of the National Academy of Sciences 102.41 (2005): 14653-14658.
Lei, Ming-Xing, Cheng-Ming Chuong, and Randall B. Widelitz. “Tuning Wnt signals for more or fewer hairs.” Journal of Investigative Dermatology 133.1 (2013): 7-9.
Clevers, Hans, and Roel Nusse. “Wnt/β-catenin signaling and disease.” Cell 149.6 (2012): 1192-1205.
Garza, Luis A., et al. “Prostaglandin D2 inhibits hair growth and is elevated in bald scalp of men with androgenetic alopecia.” Science translational medicine 4.126 (2012): 126ra34-126ra34.
Johnstone, Murray A., and Daniel M. Albert. “Prostaglandin-induced hair growth.” Survey of ophthalmology 47 (2002): S185-S202.
Blume-Peytavi, Ulrike, et al. “A randomized double-blind placebo-controlled pilot study to assess the efficacy of a 24-week topical treatment by latanoprost 0.1% on hair growth and pigmentation in healthy volunteers with androgenetic alopecia.” Journal of the American Academy of Dermatology 66.5 (2012): 794-800.
Barrón-Hernández, Yevher Lorena, and Antonella Tosti. “Bimatoprost for the treatment of eyelash, eyebrow and scalp alopecia.” Expert opinion on investigational drugs 26.4 (2017): 515-522.
DuBois, Janet, et al. “Setipiprant for Androgenetic Alopecia in Males: Results from a Randomized, Double-Blind, Placebo-Controlled Phase 2a Trial.” Clinical, Cosmetic and Investigational Dermatology 14 (2021): 1507.
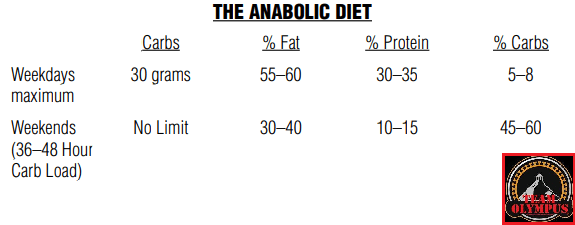
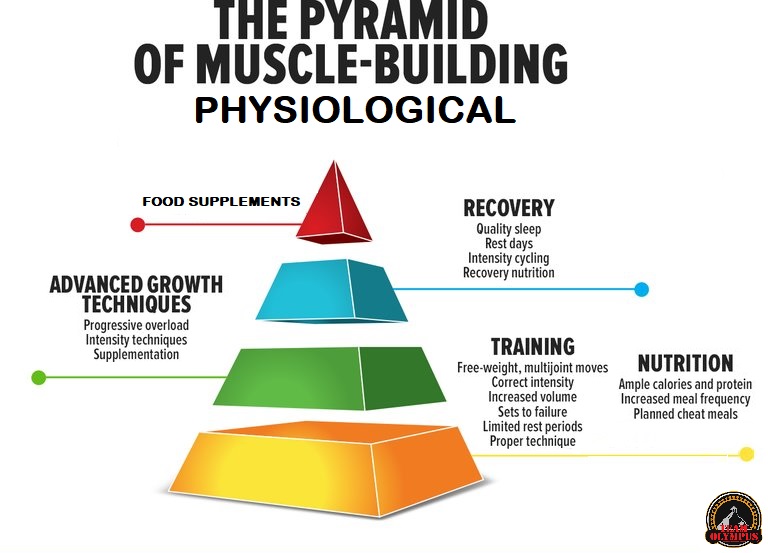
Il BodyBuilding si differenzia dagli sport di prestazione perché il giorno della gara gli atleti vengono giudicati in base all’aspetto piuttosto che alle capacità atletiche. I bodybuilder posano sul palco dove vengono giudicati per la muscolatura, la definizione e la simmetria. Nel corso di una stagione, i bodybuilder attraversano tre fasi diverse: la fase di crescita muscolare (Off-Season), la dieta per la competizione (preparazione alla gara) e la gara stessa. La maggior parte della letteratura riguarda la fase di dieta pre-gara e la peak week.[1]
Tuttavia, la letteratura scientifica sulle raccomandazioni alimentari per i bodybuilder durante la Off-Season è carente. Si tratta di una lacuna importante, poiché la maggior parte della carriera di un bodybuilder si svolge in questa fase, in cui l’obiettivo è aumentare la massa muscolare riducendo al minimo l’aumento eccessivo della massa grassa. I bodybuilder sono noti per avere atteggiamenti rigidi nei confronti della selezione degli alimenti, della frequenza dei pasti, dei tempi di alimentazione e dell’integrazione [2]. Storicamente, le informazioni sull’alimentazione e l’integrazione sono state trasmesse dalle riviste di bodybuilding e dai concorrenti di successo, ma recentemente sono emerse più informazioni attraverso Internet e i forum [3,4]. Di conseguenza, molte delle strategie alimentari utilizzate dai bodybuilder non hanno un solido supporto scientifico e la letteratura scientifica dimostra che alcune di queste strategie, tra cui l’uso massiccio di farmaci, ma anche di integratori più in generale, possono essere ovviamente dannosi per la salute [5,6,7].
Poiché i bodybuilder trascorrono la maggior parte del loro tempo in Off-Season, è evidente la necessità di raccomandazioni nutrizionali e di supplementazione, sia OTC che PEDs, il più possibile “sicure” e basate sull’evidenza per questa popolazione. È stato inoltre dimostrato che alcuni bodybuilder, e non soltanto i concorrenti di alto livello nel bodybuilding “Natural”, potrebbero essere interessati a informazioni basate sull’evidenza [8]. Con il supporto della review realizzata e pubblicata da Juma Iraki et al. che tratta del Off-Season a livello alimentare e integrativo, lo scopo di questo articolo sarà quello di riportare quanto evidenziato dalla letteratura scientifica sugli argomenti relativi all’alimentazione e all’integrazione alimentare e supplementazione PEDs rilevanti per i bodybuilder nella Off-Season e di fornire raccomandazioni pratiche sull’assunzione di energia, macronutrienti, frequenza dei pasti, tempistica dei nutrienti, integratori alimentari e PEDs .
Transizione dalla dieta pre-gara/peak week alla dieta in Off-Season – Reverse Diet Vs. Recovery Diet:
Il primo step che il bodybuilder si trova davanti è la gestione del passaggio da una dieta ipocalorica ad una ipercalorica. Ed è in questo frangente che emergono due strategie simili all’apparenza ma in realtà diverse: la “Recovery Diet” e la “Reverse Diet”.
Ora, molto semplicemente, la “Recovery Diet” consiste in un graduale aumento calorico ma di consistenza tale che l’atleta esca dalla condizione di ipocalorica nel giro di due settimane circa. Con la “Reverse Diet”, invece, abbiamo sempre un graduale aumento calorico ma caratterizzato da una ridotta consistenza dello stesso (si parla di circa 100Kcal/die a settimana). In questo caso specifico, il bodybuilder rimarrebbe in ipocalorica per diverse settimane con possibile emersione di problemi psicofisici legati al protrarsi dello stato stressorio.
Quindi, con il termine “Recovery Diet” ci riferiamo ad uno schema alimentare avente l’obiettivo generale di RECUPERARE da un periodo di dieta cronica sperimentato durante la preparazione alla gara. La “Recovery Diet” incoraggia i bodybuilder a guadagnare il 5-10% del loro peso di gara nelle prime 4-8 settimane successive all’evento. Questo con l’intento di accelerare l’aumento di grasso corporeo e far rientrare il soggetto in un range di grasso corporeo “sano”, fisiologico, il prima possibile. In seguito, si consiglia agli atleti di rallentare il ritmo di aumento del peso e di mantenere un surplus controllato, con un aumento medio dello 0,5-1% del peso corporeo al mese passando pienamente nella Off-Season. Questo fino a quando non raggiungono un punto in cui un ulteriore aumento di peso è considerato improduttivo. Con il termine “Reverse Diet” ci si riferisce ad una strategia la quale può ancora essere attuata con discreti vantaggi per aiutare un agonista a recuperare dopo il contest. Tuttavia, se rispettata e seguita correttamente, piccoli aumenti di cibo di ~100 Kcal/die a settimana potrebbero comunque protrarre il deficit calorico del soggetto, prolungando così il periodo di dieta ipocalorica. Sebbene questa possa essere una strategia utile in alcune circostanze, ad esempio durante l’avvicinamento alla competizione, le modalità di applicazione non permettono un recupero di una bf salubre in tempi ottimali. È risaputo che un bodybuilder in condizioni di picco non è necessariamente al massimo della salute, e questo è in gran parte correlato al livello di grasso corporeo. Accettare un certo aumento di grasso avrà effetti positivi su tutti gli aspetti della Off-Season come le prestazioni in allenamento, i marcatori ormonali, la disponibilità di energia, la qualità del sonno e, inoltre, sarà vantaggioso sulla longevità complessiva dello sport praticato.
In definitiva, se si parte da body fat estremamente basse, tipiche da gara, allora la “Recovery Diet” è la scelta migliore per shiftare dal regime ipocalorico che ha caratterizzato il periodo di preparazione alla gara a quello ipercalorico del Off-Season. Discorso diverso se ci troviamo di fronte ad un soggetto amatoriale, con una body fat del 8-10% arrivato al termine del percorso di “Cut”. In questo caso la “Reverse Diet” è la scelta più funzionale permettendo un controllo migliore degli incrementi calorici evitando che la massa grassa sfori eccessivamente e che il lavoro precedentemente svolto in “Cut” venga facilmente e totalmente compromesso. Anche “ibridazioni” con aumenti settimanali di 45-50g di CHO die possono essere applicati con buoni risultati.
Energia:
Durante la Off-Season, l’obiettivo principale di un bodybuilder è quello di aumentare la massa muscolare riducendo al minimo l’aumento della massa grassa attraverso l’uso di allenamenti contro-resistenza e il mantenimento di un bilancio energetico positivo. Per valutare con precisione il fabbisogno energetico dei bodybuilder durante la bassa stagione, è necessario considerare il volume, la frequenza e l’intensità dell’allenamento. Durante la fase off-season, è stato riportato che i bodybuilder si allenano alla resistenza 5-6 volte a settimana, esercitando ogni gruppo muscolare 1-2 volte a settimana [9]. È stato inoltre riferito che seguono una routine di allenamento ad alto volume con 4-5 esercizi per gruppo muscolare, eseguendo 3-6 serie per esercizio, 7-12 ripetizioni massime (RM) per ogni serie con 1-2 minuti di riposo tra le serie. La durata della sessione di allenamento è stata indicata in ~40-90 minuti. Tuttavia, i piani di allenamento possono variare notevolmente da atleta ad atleta. È necessario valutare anche l’apporto calorico medio dei bodybuilder. Nella fase off-season, l’apporto energetico è di solito sostanzialmente più elevato rispetto alla fase di dieta: tra i bodybuilder maschi è stato riportato un apporto medio di ~3800 kcal/giorno durante la fase off-season e di ~2400 kcal/giorno durante la fase di dieta [2].
Bilancio energetico positivo:
È stato dimostrato che un bilancio energetico positivo ha un importante effetto anabolico, anche in assenza di allenamento contro-resistenza [10]. Tuttavia, la combinazione di un bilancio energetico positivo con l’allenamento contro-resistenza rappresenta il metodo più efficace per garantire che gli effetti anabolici siano diretti all’aumento della massa muscolo-scheletrica [11,12]. L’entità del surplus energetico ideale per guadagnare massa muscolare limitando l’accumulo di tessuto adiposo può variare in base allo stato di allenamento. Nei soggetti non allenati, è stato dimostrato che un surplus energetico sostanziale di circa 2.000 kcal, combinato con l’allenamento contro-resistenza, fornisce un robusto aumento di peso, in cui il contributo della massa magra (LBM) può raggiungere il 100% [12]. Tuttavia, nei soggetti allenati, un surplus energetico sostanziale potrebbe non essere necessario o vantaggioso. Uno studio condotto su atleti d’élite ha esaminato l’effetto delle indicazioni dietetiche sui cambiamenti della composizione corporea tra gli atleti d’élite quando l’allenamento contro-resistenza è stato combinato con diverse entità di surplus energetico. Un gruppo con un peso corporeo medio di 75kg ha consumato energia ad libitum (2964 kcal) per raggiungere un surplus molto ridotto, mentre un secondo gruppo con un peso corporeo medio di 71kg ha ricevuto una consulenza dietetica e ha consumato ~600 kcal in più rispetto al gruppo ad libitum [13].
Entrambi i gruppi hanno seguito lo stesso programma di allenamento contro-resistenza di 4 giorni alla settimana per un periodo di 8-12 settimane. I ricercatori hanno ipotizzato che il gruppo ipercalorico avrebbe avuto un aumento maggiore del peso corporeo e della LBM. Sebbene il gruppo ipercalorico abbia ottenuto un aumento maggiore della LBM rispetto a quelli che mangiavano ad libitum, questo non ha raggiunto la significatività statistica (1,7kg contro 1,2kg, rispettivamente). Inoltre, rispetto al gruppo che mangiava a sazietà, hanno registrato un aumento significativamente maggiore della massa grassa (1,1kg contro 0,2kg, rispettivamente). I ricercatori hanno concluso che un surplus di 200-300 kcal al giorno negli atleti altamente allenati potrebbe essere più appropriato di 500 kcal per minimizzare il rischio di inutili aumenti di grasso corporeo. I soggetti non allenati, più lontani dal loro tetto genetico di massa muscolare, possono essere in grado di aumentare i muscoli a un ritmo più veloce rispetto agli individui allenati.
Il tasso di crescita muscolare può rallentare con l’avanzare dell’età [14]. Pertanto, un maggiore surplus energetico può essere più vantaggioso per i bodybuilder alle prime armi, mentre i bodybuilder avanzati potrebbero trarre maggiore beneficio da diete ipercaloriche conservative per limitare inutili aumenti di grasso corporeo. Studi precedenti hanno raccomandato ai bodybuilder di consumare una dieta leggermente ipercalorica, con un aumento dell’apporto energetico di circa il 15% rispetto al mantenimento nella Off-Season [15]. Tuttavia, ciò non tiene conto della storia di allenamento e del livello di esperienza del singolo bodybuilder. Poiché la capacità di aumentare la massa muscolare è limitata, un surplus aggressivo può portare a un inutile aumento del grasso corporeo, che aumenterebbe la durata o la gravità dei successivi periodi di preparazione alle gare, aumentando di conseguenza la durata o la gravità della scarsa disponibilità energetica. Pertanto, il numero di calorie che un bodybuilder consuma al di sopra del livello di mantenimento può essere stabilito in base al livello di esperienza e poi regolato in base al tasso di aumento di peso e ai cambiamenti nella composizione corporea. Dato che i bodybuilder spesso aumentano rapidamente di peso dopo una gara, potrebbe essere utile avere un obiettivo di aumento di peso per settimana e regolarsi di conseguenza [16,17].
Tuttavia, come detto precedentemente, inizialmente, dopo la gara, potrebbe essere utile un aumento di peso più rapido per aiutare a riportare il concorrente a uno stato di salute sia psicologico che fisiologico, prima che il tasso di aumento di peso venga rallentato per limitare l’accumulo eccessivo di tessuto adiposo. Nella letteratura scientifica si raccomanda di puntare a un aumento di peso di circa 0,25-0,5 kg a settimana per cercare di aumentare la LBM e ridurre al minimo l’aumento della massa grassa [14,18]. Per un bodybuilder avanzato, un potenziale aumento di 2kg di peso corporeo su base mensile potrebbe essere eccessivo e comportare un’inutile accumulazione di grasso corporeo; pertanto, questo tasso dovrebbe essere considerato con cautela. Sulla base delle prove attuali, potrebbe essere opportuno raccomandare ai bodybuilder di consumare una dieta leggermente ipercalorica (~10-20% sopra le calorie di mantenimento) nella Off-Season e raccomandare ai bodybuilder avanzati di puntare all’estremità inferiore di questa raccomandazione, o addirittura di essere più conservativi se si verificano aumenti sostanziali della massa grassa. Dato che i bodybuilder consumano in media 45 kcal/kg durante la bassa stagione, il surplus raccomandato equivale a circa 42-48 kcal/kg [2]. Potrebbe essere utile puntare a un aumento di peso di circa 0,25-0,5% del peso corporeo a settimana, regolando al contempo l’apporto energetico in base alle variazioni della composizione corporea. Inoltre, potrebbe essere più appropriato considerare le variazioni di peso medie settimanali basate su pesate giornaliere (o più volte alla settimana) per limitare gli errori delle fluttuazioni giornaliere del peso che possono verificarsi durante la settimana. Una volta determinato il surplus calorico, il passo successivo sarà quello di distribuire le calorie tra proteine, grassi e carboidrati.
Proteine:
Il turnover proteico del muscolo scheletrico è il rapporto tra la sintesi proteica muscolare (MPS) e la degradazione proteica muscolare (MPB). L’ipertrofia del muscolo scheletrico richiede un equilibrio netto in cui la MPS supera la MPB. L’esercizio contro-resistenza fornisce lo stimolo di tensione iniziale che induce l’ipertrofia risultante dall’aumento cumulativo della MPS dopo l’esercizio cronico [19]; tuttavia, l’aumento della massa grassa (FFM) può essere limitato se l’apporto proteico giornaliero è insufficiente [20]. Oltre alla quantità totale consumata al giorno, i ricercatori hanno ipotizzato che la qualità delle proteine possa aumentare il guadagno muscolare indotto dall’allenamento contro-resistenza [21]. Pertanto, entrambi questi argomenti saranno discussi nelle sezioni seguenti.
Introito proteico giornaliero:
Mentre l’attuale RDA per le proteine negli individui sani sedentari è di 0,8 g/kg, in una meta-analisi del 2018 di Morton e colleghi [22] è stato osservato che il doppio di questa quantità massimizza l’ipertrofia indotta dall’allenamento contro-resistenza. Inoltre, gli autori hanno osservato che “potrebbe essere prudente raccomandare ~2,2g di proteine/kg/die per coloro che cercano di massimizzare i guadagni di FFM indotti dall’allenamento contro-resistenza”, poiché 2,2g/kg era l’estremità superiore del limite di confidenza [22] e le differenze individuali impongono che alcuni atleti abbiano un fabbisogno proteico più elevato di altri [23]. Inoltre, la raccomandazione “meglio prevenire che curare” è probabilmente sicura, vista l’assenza di danni apparenti in studi di 1-2 anni tra i sollevatori che consumavano apporti proteici di almeno 2,2 g/kg [24,25]. Infine, la media e il limite superiore di confidenza del 95% per il fabbisogno proteico utilizzando la tecnica di ossidazione degli aminoacidi con indicatore tra i bodybuilder maschi nei giorni di non allenamento sono stati riportati rispettivamente come 1,7 e 2,2g/kg [26], che è simile al fabbisogno tra le donne quando è normalizzato alla FFM [27].
Tuttavia, è stato riportato che i bodybuilder consumano fino a 4,3g/kg di proteine al giorno tra i soggetti di sesso maschile e 2,8g/kg tra quelli di sesso femminile, superando di gran lunga queste raccomandazioni [2]. Le linee guida precedentemente fornite per i bodybuilder nella Off-Season erano di consumare il 25-30% del loro apporto energetico dalle proteine [15]. Potrebbe essere ragionevole opporsi all’indicazione di raccomandazioni basate su percentuali dell’apporto energetico totale, poiché un individuo con un peso non particolarmente elevato ma con un alto fabbisogno energetico potrebbe finire per consumare proteine che superano di gran lunga quelle necessarie e quindi richieste. Inoltre, questo può portare a un’assunzione insufficiente di carboidrati e grassi se l’atleta mira a un apporto calorico specifico. Pertanto, potrebbe essere più appropriato raccomandare un fabbisogno proteico basato sul peso corporeo. Pertanto, i bodybuilder dovrebbero consumare un minimo di 1,6g/kg di proteine nella Off-Season, anche se un obiettivo più vicino a 2,2 g/kg potrebbe garantire una risposta ottimizzata in modo più coerente in una maggiore percentuale di atleti.
E per i “Doped”? Dovremo ormai sapere che la fisiologia di base è la medesima per ogni individuo con le consuete variabili. Detto ciò, l’uso di PEDs va si ad alterare la fisiologia ma in questo specifico ambito, ossia introito proteico per massimizzare lo stimolo ipertrofico, hanno una azione di perfezionamento dell'”economia proteica cellulare”: in parole più semplici, sembra che l’uso di AAS porti ad una migliore resa nell’utilizzo degli amminoacidi scissi e assorbiti dalle proteine alimentari. Di conseguenza, a parità di apporto proteico, la veicolazione degli amminoacidi a scopo plastico è maggiore come minore è l’attività catabolica. Ciò significa che abusare delle proteine, in special modo durante una fase ipercalorica, perchè si è sotto AAS potrebbe risultare più inutile di quanto non lo sia in contesto “Natural”.
Infine, ed è necessario sottolinearlo, tra i bodybuilder che lottano con la fame in Off-Season e che di conseguenza assumono quantità caloriche che portano a un aumento di peso più rapido e all’accumulo di grasso in eccesso, un apporto proteico più elevato può essere utile (se non controindicato per motivi clinici). In uno studio condotto da Antonio e colleghi, i partecipanti ad allenamenti contro-resistenza che consumavano più proteine (4,4g/kg al giorno) e più calorie hanno guadagnato una quantità simile di FFM, ma non hanno guadagnato ulteriore grasso corporeo rispetto al gruppo che consumava meno proteine e meno calorie [28]. Allo stesso modo, in uno studio di follow-up, un gruppo che consumava 3,4g/kg di proteine al giorno ha guadagnato una quantità simile di FFM, ma ha perso una percentuale maggiore di grasso corporeo rispetto a un gruppo a basso contenuto proteico, ancora una volta, nonostante un apporto energetico più elevato [29]. Gli autori di questi studi sulla “vita libera” hanno ipotizzato che i loro risultati fossero dovuti a un aumento della termogenesi indotta dalla dieta attraverso protocolli alimentari ad alto contenuto proteico. Tuttavia, ciò è in contrasto con uno studio di Bray e colleghi del 2012 sul reparto metabolico, più strettamente controllato, in cui il contenuto proteico della dieta influenzava la percentuale di massa corporea acquisita, mentre la massa corporea totale era dettata dal solo contenuto energetico della dieta [30].
Pertanto, mentre la termogenesi indotta dalla dieta potrebbe essere significativamente più elevata con assunzioni di proteine nell’intervallo di 3 g/kg o superiore, la perdita di grasso o la mancanza di aumento di peso osservata da Antonio e colleghi, nonostante un apporto energetico più elevato, potrebbe con più probabilità riflettere l’effetto saziante di assunzioni proteiche molto elevate che diminuiscono l’assunzione calorica effettiva, piuttosto che un aumento della sola termogenesi.
Qualità delle Proteine:
Gli aminoacidi essenziali (EAA) sono gli unici aminoacidi necessari per stimolare il processo di MPS [31]. Sebbene tutti gli aminoacidi forniscano i “mattoni” necessari per la sintesi di nuovi tessuti, l’aminoacido Leucina in particolare sembra essere particolarmente importante come “innesco metabolico” della MPS [32]. È stato suggerito che una concentrazione sufficiente di Leucina è necessaria per raggiungere una “soglia di Leucina” che è richiesta per stimolare al massimo la MPS [33]. In breve, dal punto di vista della costruzione muscolare, le fonti proteiche che innescano una consistente risposta della MPS (quantità sufficiente di Leucina) e forniscono i mattoni essenziali per la costruzione di nuovo tessuto muscolare (contengono l’intero spettro di aminoacidi essenziali in abbondanza) possono essere considerate di “qualità superiore”.
Sebbene l’effetto meccanicistico della Leucina sulle MPS esuli dallo scopo di questo articolo, si invitano i lettori a leggere una rassegna che tratta questo argomento in dettaglio [34]. In generale, su una base di grammo per grammo, le fonti proteiche di origine animale contengono in genere più Leucina ed EAA, anche se ci sono eccezioni degne di nota. Le proteine della soia, uno dei più comuni integratori proteici di origine vegetale, contengono tutti gli EAA, ma in una quantità inferiore per grammo rispetto alle proteine del latte e quindi, in uno studio, hanno prodotto un aumento minore delle MPS rispetto al siero di latte dopo un’ingestione acuta [35]. È interessante notare che in questo stesso studio la soia ha prodotto un aumento maggiore delle MPS rispetto alla caseina, anch’essa una proteina casearia di “alta qualità”, presumibilmente a causa della più lenta velocità di digestione della caseina [35]. Rammentate sempre la differenza tra risposta “acuta” e “cronica”. Per l’appunto, ciò significa che, sebbene il contenuto di Leucina e di EAA di una fonte proteica debba essere preso in considerazione, la risposta acuta alla MPS non è l’unica variabile legata all’ipertrofia a lungo termine. Infatti, una proteina di alta qualità ma “lenta” come la caseina produce inizialmente una risposta MPS di minore ampiezza. Tuttavia, la caseina (e altre proteine a lenta digestione) può produrre un’area MPS sotto la curva simile o maggiore se osservata longitudinalmente rispetto a una fonte proteica “veloce” come il siero di latte, che determina un aumento iniziale maggiore e poi una brusca riduzione [36].
Inoltre, la risposta acuta della MPS a un determinato tipo di proteina non deve essere vista in una prospettiva riduzionista. Nel mondo reale si consumano quotidianamente più porzioni di varie fonti proteiche, rendendo probabilmente superflue alcune di queste distinzioni nel profilo aminoacidico e nella cinetica di digestione. Infatti, in una meta-analisi che ha confrontato i cambiamenti longitudinali della composizione corporea con diversi tipi di integratori proteici, non sono state riscontrate differenze significative tra i partecipanti che consumavano soia rispetto al siero di latte, ad altre proteine del latte o alle proteine isolate del manzo [37].
Come dimostrato in uno studio che ha messo a confronto gruppi che consumavano proteine dopo l’allenamento (in aggiunta a una dieta già composta dal 25% di proteine), sia che venissero forniti 48g di proteine del siero del latte (contenenti 5,5g di Leucina), sia che venissero forniti 48g di proteine del riso (contenenti 3,8g di Leucina), non è stato osservato alcun impatto sui cambiamenti della composizione corporea tra i gruppi dopo otto settimane [38]. Pertanto, se consumate in quantità sufficienti (soprattutto se si considera l’apporto proteico totale giornaliero), la qualità delle proteine di un singolo pasto è meno preoccupante. Tuttavia, se si volesse consumare una dieta dominata da fonti proteiche di origine vegetale, esistono alternative alla soia e al riso. Ad esempio, le proteine isolate del pisello sono ricche di EAA e di Leucina. In uno studio di 12 settimane, un gruppo che consumava 50g di proteine isolate di pisello al giorno ha registrato un aumento maggiore dello spessore muscolare indotto dall’allenamento di resistenza rispetto al placebo, non significativamente diverso da un gruppo che consumava 50g di siero di latte [39].
Pertanto, nel contesto delle indicazioni di questo articolo, la qualità delle proteine può essere un problema solo se si utilizza la fascia bassa delle linee guida sulle proteine (1,6g/kg) o se si consuma una dieta a base prevalentemente vegetale. In entrambi i casi, potrebbe essere utile integrare con fonti proteiche ricche di Leucina e di EAA, a seconda delle preferenze alimentari (ad esempio, proteine del latte o del pisello se si è vegani), per garantire la risposta attesa della MPS all’assunzione di proteine.
Grassi:
Il grasso è un nutriente fondamentale per molte funzioni dell’organismo. Tuttavia, non si sa molto dell’effetto dei grassi alimentari sull’ipertrofia del muscolo scheletrico. È stato riportato che l’assunzione di grassi alimentari tra i bodybuilder varia dall’8 al 33% delle calorie totali [2]. Sebbene i trigliceridi intramuscolari possano fungere da substrato energetico durante l’allenamento di resistenza, non sono un fattore limitante poiché i substrati derivano principalmente da processi anaerobici [40]. Di interesse per il bodybuilder, è dimostrato che negli atleti allenati contro-resistenza [41] e nei giocatori di hockey [42] le diete a basso contenuto di carboidrati (30-45% dell’energia o meno) possono influire sul rapporto Testosterone libero/Cortisolo (fTC), il che potrebbe avere un impatto negativo sul recupero. D’altra parte, la riduzione dei grassi alimentari nelle diete isocaloriche da ~30-40% a ~15-25% ha portato a riduzioni significative ma modeste dei livelli di Testosterone [43,44,45,46].
Tuttavia, non è chiaro se le variazioni di Testosterone all’interno di intervalli normali influenzino in modo significativo l’aumento della massa muscolare [47]. Nonostante la possibilità che i livelli di testosterone possano essere più elevati quando si consuma una percentuale maggiore di energia proveniente dai grassi alimentari, i cambiamenti effettivi nella massa muscolare durante gli studi longitudinali di individui allenati alla resistenza che seguono diete “chetogeniche” ad alto contenuto di grassi sono stati costantemente inferiori rispetto ad approcci moderati o a basso contenuto di grassi con ampi carboidrati [48,49,50,51]. Non è ancora stato chiarito se ciò sia dovuto a cambiamenti nella capacità di esercizio, ad alterazioni del rapporto fTC o a qualche altro meccanismo legato alla componente ad alto contenuto di grassi o a basso contenuto di carboidrati della dieta.
Tuttavia, ciò indica che forse si dovrebbe consumare una proporzione più moderata di grassi nella dieta, piuttosto che un apporto basso o alto. In letteratura sono state proposte raccomandazioni del 15-20% e del 20-30% delle calorie provenienti dai grassi alimentari [15,52]. Tuttavia, sono necessarie ulteriori ricerche per stabilire l’effetto e la quantità ottimale di grassi alimentari per favorire l’ipertrofia muscolare.
Sulla base delle evidenze attuali, può essere prudente raccomandare che i grassi alimentari rappresentino il 20-35% delle calorie, in linea con le raccomandazioni dell’American College of Sports Medicine per gli atleti [53], che nella maggior parte dei casi corrispondono a circa 0,5-1,5 g/kg/giorno. Inoltre, va notato che un apporto sufficiente di proteine e carboidrati non deve essere compromesso da un’elevata assunzione di grassi nella dieta.
Anche la qualità dei grassi, come gli essenziali omega 3 e gli omega 6, potrebbe essere importante per i bodybuilder. Se l’apporto di questi acidi grassi è sufficiente, non è necessario integrarli con una dieta di alta qualità contenente buone fonti di acidi grassi. Tuttavia, per alcuni potrebbe essere difficile assumere le quantità ottimali. Per questo motivo, l’argomento verrà trattato in modo più approfondito nella sezione dedicata agli integratori alimentari.
Carboidrati:
A differenza delle proteine e dei grassi, i carboidrati sono considerati non essenziali per la dieta umana perché l’organismo è in grado di produrre il glucosio necessario ai tessuti attraverso la gluconeogenesi [54]. Tuttavia, l’assunzione di carboidrati ha un ruolo importante nella dieta del bodybuilder come regolatore degli ormoni tiroidei e come contributo al fabbisogno di micronutrienti [55,56]. Inoltre, una dieta a basso contenuto di carboidrati potrebbe limitare la rigenerazione dell’adenosina trifosfato (ATP) e limitare la capacità dei muscoli di contrarsi con una forza elevata [57,58]. Durante l’esercizio ad alta intensità, il glicogeno muscolare è il principale contributore di substrato energetico ed è stato dimostrato che la glicolisi fornisce circa l’80% del fabbisogno di ATP di una serie di flessioni del gomito se portata al cedimento muscolare [59]. Nonostante ciò, parte del glicogeno utilizzato durante questo tipo di esercizio può essere risintetizzato dal lattato, il che potrebbe ridurre il fabbisogno di carboidrati. È stato inoltre dimostrato che l’allenamento contro-resistenza riduce il glicogeno muscolare del 24-40% in una singola sessione [59,60].
La quantità esaurita può variare in base alla durata, all’intensità e al lavoro svolto, ma l’allenamento tipico del bodybuilding con ripetizioni più elevate e carichi moderati sembra causare la maggiore riduzione delle scorte di glicogeno muscolare [61]. Inoltre, è stato suggerito che quando le scorte di glicogeno sono troppo basse (~70 mmol/kg), ciò può inibire il rilascio di calcio e accelerare l’insorgenza della fatica muscolare [62]. Un basso livello di glicogeno muscolare riduce significativamente il numero di ripetizioni eseguite quando si eseguono tre serie di Squat all’80% di 1RM [57].
Tuttavia, è stato dimostrato che il consumo di una dieta contenente 7,7 g/kg/die di carboidrati per 48 ore prima di una sessione di allenamento non ha un effetto maggiore sulle prestazioni rispetto a 0,37g/kg/die quando si eseguono 15 serie a 15RM di esercizi per la parte inferiore del corpo [63]. Analogamente, un altro studio ha rilevato che una dieta con il 70% di carboidrati rispetto a una dieta con il 50% di carboidrati non ha un effetto maggiore sulle prestazioni durante l’esercizio sopramassimale; tuttavia, una dieta composta dal 25% di carboidrati ha ridotto significativamente le prestazioni [64].
Inoltre, visti gli effetti negativi a lungo termine sulla massa muscolare osservati di recente in studi su popolazioni allenate alla resistenza che seguono diete chetogeniche [49,51], potrebbe essere prudente per i bodybuilder assicurarsi semplicemente un apporto sufficiente di carboidrati, visti questi risultati disparati. Pertanto, mentre le diete a moderato e alto contenuto di carboidrati sono probabilmente appropriate per il bodybuilding, le diete a bassissimo contenuto di carboidrati possono essere dannose per l’allenamento.
Nei bodybuilder maschi, sono stati riportati apporti medi di carboidrati pari a 5,3g/kg/giorno durante la Off-Season [2]. Tuttavia, non sono state stabilite le quantità ottimali di carboidrati per i bodybuilder. In letteratura sono state proposte raccomandazioni per gli sport di forza, tra cui il bodybuilding, con assunzioni di 4-7g/kg/giorno e 5-6g/kg [15,65]. I carboidrati sembrano essere importanti per il bodybuilder, ma per ottenere benefici possono essere necessarie solo quantità moderate. Pertanto, dopo aver destinato le calorie alle proteine (1,6-2,2g/kg/die) e ai grassi (0,5-1,5g/kg/die), le restanti calorie dovrebbero essere destinate ai carboidrati. Tuttavia, sulla base delle prove attuali, potrebbe essere ragionevole consumare quantità sufficienti di carboidrati nell’intervallo ≥3-5g/kg/giorno, se possibile.
Sono necessarie ulteriori ricerche tra i bodybuilder per stabilire se l’assunzione abituale di carboidrati, superiore o inferiore a quella osservata, possa produrre ulteriori benefici. La Tabella sottostante riassume le raccomandazioni per le calorie e i macronutrienti.
Raccomandazioni dietetiche per i bodybuilder in Off-Season.
Distribuzione e timing dei nutrienti:
Si dice che i bodybuilder consumino in media sei pasti al giorno [66]; tuttavia, non esistono studi che esaminino specificamente quale possa essere la frequenza ottimale dei pasti per questa popolazione [65]. Questa elevata frequenza dei pasti si basa sulla convinzione di un maggiore stato di anabolismo e persino di un migliore utilizzo dei nutrienti durante il giorno, che potrebbe tradursi in un miglioramento della composizione corporea.
Il concetto di temporizzazione dell’assunzione di proteine per massimizzare l’ipertrofia comprende diverse strategie di dosaggio. La prima a comparire in letteratura è stata il consumo di proteine in prossimità dell’allenamento contro-resistenza. I picchi di MPS sono più elevati in questo periodo quando si consumano proteine; pertanto, questa strategia è stata proposta per migliorare l’efficienza della riparazione e del rimodellamento del muscolo scheletrico [31]. Inoltre, a causa dell'”effetto muscolo pieno”, per cui un ulteriore apporto di proteine non aumenta la MPS finché non è trascorso un tempo sufficiente, distribuire uniformemente l’assunzione di proteine tra più pasti è un’altra strategia studiata per massimizzare la MPS totale giornaliera [67]. Infine, il consumo prima di andare a letto di proteine a lenta digestione (come la caseina) per evitare periodi catabolici prolungati durante il sonno è la strategia proposta più di recente per migliorare il bilancio proteico netto giornaliero [68], sebbene si sia dimostrata inutile nel perseguire il fine o, per lo meno, non molto diversa dalla risultante di una assunzione di isolate in un contesto alimentare con parità nel totale proteico giornaliero. Ciascuna di queste tre strategie sarà discussa in seguito.
Dosaggio proteico:
Il periodo post-allenamento consente un picco della MPS più elevato quando si consumano proteine [31] e per raggiungere il picco di MPS può essere necessaria un’adeguata dose di Leucina “soglia” [32]. Diversi studi hanno esaminato il dosaggio proteico necessario per massimizzare la MPS dopo l’allenamento [69,70,71]. In uno studio sono stati consumati 0, 5, 10, 20 o 40g di proteine d’uovo intere dopo l’esercizio contro-resistenza della parte inferiore del corpo, con 20g che stimolavano al massimo la MPS [69]. Risultati simili sono stati riscontrati anche in un altro studio, in cui 20 g di siero di latte sono stati sufficienti a stimolare al massimo i tassi post-assorbitivi di MPS sia a riposo che dopo un lavoro unilaterale delle gambe all’80% del 1RM [70]. Inoltre, 40g di siero di latte non hanno prodotto ulteriori aumenti di MPS in questo studio e hanno portato all’ossidazione amminoacidica e alla produzione di urea.
Tuttavia, uno studio recente ha rilevato che, durante l’esecuzione di esercizi contro-resistenza per tutto il corpo al 75% del 1RM, 40g di siero di latte hanno prodotto una risposta MPS significativamente più elevata rispetto a 20g [71]. Esiste quindi una relazione tra il volume di tessuto muscolare danneggiato e stimolato e l’assunzione adeguata di proteine. È interessante notare che gli autori di una meta-analisi del 2013 hanno osservato che, nonostante gli studi con traccianti a breve termine mostrassero risposte nella MPS maggiori quando le proteine venivano consumate nella “finestra anabolica” post-allenamento, negli studi longitudinali sull’allenamento non è stato riscontrato alcun effetto significativo sull’ipertrofia quando si controllava l’apporto proteico totale giornaliero, indipendentemente dal fatto che le proteine fossero consumate all’interno della “finestra anabolica” o al di fuori di essa [72].
Nutrient Timing:
Analogamente, i ricercatori di uno studio tracciante a breve termine che ha esaminato il dosaggio delle proteine nel corso di 12 ore hanno riportato una maggiore area sotto la curva della MPS quando sono state consumate quattro dosi di proteine del siero di latte da 20g ogni tre ore rispetto a due dosi da 40g a distanza di sei ore e otto dosi da 10g ogni ora e mezza [73]. In teoria, data la soglia oltre la quale le proteine supplementari consumate in una singola seduta non contribuiscono ulteriormente alla MPS [69] e a causa del “periodo refrattario” postprandiale durante il quale la MPS non può essere nuovamente stimolata al massimo [67], si potrebbe concludere che un bodybuilder dovrebbe raggiungere, ma non superare, questa dose soglia ogni poche ore per massimizzare l’ipertrofia a lungo termine. Tuttavia, gli autori di una review sistematica del 2018 sugli integratori proteici, comprendente 34 studi randomizzati e controllati, hanno riportato guadagni di massa magra simili tra i gruppi che utilizzavano un programma di dosaggio con i pasti (che comportava un minor numero di dosi di proteine di entità elevata) e tra i pasti (che comportava un maggior numero di dosi di proteine di entità moderata) [74].
È interessante notare che i dati che esaminano l’alimentazione proteica notturna mostrano uno distacco simile tra gli studi meccanicistici a breve termine e gli interventi di allenamento a lungo termine. Nel 2012 è stata condotta la prima ricerca che esaminava la risposta acuta all’alimentazione notturna con caseina [68]. Gli autori hanno riportato che 40g di caseina consumati prima di andare a letto sono stati digeriti, assorbiti e hanno stimolato la MPS e migliorato l’equilibrio proteico dell’intero corpo durante il periodo notturno in misura maggiore rispetto al placebo. Negli anni successivi sono stati pubblicati altri studi in acuto che hanno confermato [75] e riconfermato questi risultati in una popolazione più anziana [76]. Nel 2015, gli autori del primo studio longitudinale hanno riportato un aumento della forza e dell’ipertrofia in un gruppo a cui era stato somministrato un supplemento proteico notturno rispetto a un gruppo placebo [77].
Tuttavia, la quantità totale di proteine giornaliere non è stata equiparata, in quanto il gruppo con proteine notturne ha consumato 1,9g/kg/giorno, mentre il gruppo placebo ha consumato solo 1,3g/kg. È importante notare che in entrambi gli unici studi longitudinali con corrispondenza proteica che hanno confrontato l’integrazione notturna di caseina con i gruppi che hanno assunto l’integrazione prima, non sono state riportate differenze significative nell’aumento della FFM tra i gruppi [78,79]. Pertanto, la domanda è la stessa per ogni strategia di distribuzione: perché ci sono ripetuti distacchi tra gli studi meccanicistici a breve termine sulle MPS e le ricerche a lungo termine che esaminano l’effettiva ipertrofia? La risposta potrebbe risiedere nei metodi utilizzati negli studi sulla MPS, in quanto i partecipanti sono a digiuno, ricevono solo proteine in polvere in isolamento, spesso viene loro somministrato del siero di latte (che viene digerito molto rapidamente) e vengono osservati per brevi periodi. Questi contesti di laboratorio determinano tempi di digestione e cinetiche degli aminoacidi diversi da quelli che si verificano nel “mondo reale”. In particolare, in queste condizioni di laboratorio i livelli di base degli aminoacidi nel corpo sono più bassi del normale e la digestione e il successivo apporto di aminoacidi al muscolo sono più rapidi.
In condizioni di vita libera, le proteine vengono consumate principalmente da fonti alimentari intere, più volte al giorno e insieme ad altri alimenti, il che ritarda lo svuotamento gastrico. Per questi motivi, gli aminoacidi vengono titolati nel flusso sanguigno in modo più lento e costante; pertanto, in condizioni normali, le scorte sono quasi sempre prontamente disponibili [80]. Pertanto, l’efficacia della “finestra anabolica” e persino delle strategie di distribuzione delle proteine potrebbe non tradursi nella pratica. Inoltre, le limitazioni specifiche del laboratorio si estendono anche agli studi sull’alimentazione notturna. Si consideri, ad esempio, che 26g di proteine provenienti da una bistecca magra determinano un aumento sostenuto della MPS che dura almeno sei ore (l’intero periodo di tempo studiato) [81].
Inoltre, 26g sono solo il ~37% della dose di proteine contenuta in media in una cena americana [82], che richiederebbe più tempo per essere digerita a causa della maggiore porzione di proteine e dell’aggiunta di fibre, lipidi e altri nutrienti che ritarderebbero ulteriormente la digestione [80]. Pertanto, il tipico pasto finale potrebbe già soddisfare lo scopo di un frullato di caseina. Detto questo, nonostante queste discrepanze tra MPS e risultati della composizione corporea, non c’è nulla di male nel tentare queste strategie, soprattutto se attuate in modo pragmatico e senza introdurre ulteriori oneri logistici nel proprio programma quotidiano.
Pertanto, potrebbe essere prudente consigliare ai bodybuilder di suddividere l’assunzione giornaliera di 1,6-2,2 g/kg di proteine in più pasti contenenti ciascuno ~0,40-0,55g/kg [80] e di fare in modo che uno di questi pasti avvenga entro 1-2 ore prima o dopo l’allenamento, mentre un’alimentazione costituita da una fonte proteica e non proteica venga consumata 1-2 ore prima di dormire. Ad esempio, un bodybuilder di 90 kg potrebbe consumare 40-50g di proteine alle 8-9 del mattino per la colazione, allenarsi alle 11, consumare 40-50g di proteine alle 12-13 per il pranzo/post-allenamento, 40-50g di proteine a cena tra le 17-18, e poi un pasto finale di 40-50g di proteine non contenenti fonti proteiche grasse alle 21-10 prima di andare a letto entro le 23.
I carboidrati consumati prima dell’allenamento sono spesso una strategia utilizzata dagli atleti per migliorare le prestazioni negli esercizi ad alta intensità. La completa risintesi del glicogeno può essere raggiunta entro 24 ore da un allenamento che depaupera il glicogeno se si consumano quantità sufficienti di carboidrati [83]. Tuttavia, solo il 24-40% del glicogeno muscolare viene esaurito dopo un allenamento contro-resistenza [59,60]. Pertanto, una quantità di ≥3-5g/kg di carboidrati al giorno sarebbe probabilmente sufficiente per la risintesi del glicogeno. Questo elevato apporto giornaliero di carboidrati probabilmente riduce anche l’impatto della tempistica dei carboidrati pre-allenamento sulle prestazioni dell’esercizio.
Inoltre, per i bodybuilder che non hanno bisogno di enfatizzare il rifornimento di glicogeno, le proteine aumentano la MPS post-allenamento a livelli massimi anche senza l’aggiunta di carboidrati [86,87]. Anche se il consumo di carboidrati nel post-allenamento non è certo dannoso, è improbabile che questo favorisca l’ipertrofia a lungo termine, come discusso in precedenti review [1,88]. Pertanto, è meglio concentrarsi sul consumo di un’adeguata quantità di carboidrati giornalieri e basare la distribuzione dei carboidrati intorno all’allenamento sulle preferenze personali.
Supplementazione OTC:
In un recente sondaggio condotto tra i bodybuilder, è stato riportato che tutti i partecipanti assumevano integratori alimentari [9]. Gli integratori alimentari più comuni erano: integratori di proteine (86%), creatina (68%), aminoacidi a catena ramificata (67%), glutammina (42%), vitamine (40%), olio di pesce (37%) e prodotti contenenti caffeina/efedrina (24%).
Sebbene gli integratori proteici siano molto popolari tra i bodybuilder, vengono utilizzati prevalentemente come gli alimenti interi per raggiungere gli obiettivi proteici. Pertanto, non verranno discussi in dettaglio. I lettori sono invitati a leggere la posizione dell’ISSN su questo argomento [89]. Inoltre, la trattazione di tutti gli integratori comunemente utilizzati dai bodybuilder esula dallo scopo di questo articolo. L’attenzione si concentrerà piuttosto sugli integratori alimentari che potrebbero potenzialmente produrre un effetto ergogenico e sugli integratori che possono garantire un apporto sufficiente di micronutrienti e acidi grassi essenziali.
Creatina Monoidrato:
La Creatin-fosfato si trova in alte concentrazioni nel muscolo scheletrico e cardiaco, dove agisce come fonte di energia [90]. La Creatina può essere ottenuta anche attraverso la dieta nei soggetti che consumano carne; tuttavia, le concentrazioni di Creatina nella carne si riducono con la cottura [91].
Numerosi studi hanno osservato un aumento della massa e della forza muscolare in seguito a fasi di carico di Creatina, in genere di 20g al giorno per circa una settimana, spesso seguite da fasi di mantenimento di 2-3g di Creatina al giorno [92]. Tuttavia, la fase di carico potrebbe non essere necessaria. È stato dimostrato che la saturazione della Creatina muscolare dopo un’integrazione di 3g di Creatina Monoidrato per 28 giorni è simile al consumo di Creatina Monoidrato dopo la tipica fase di carico [93].
La maggior parte degli individui non raggiunge i 3g giornalieri con la dieta e può essere necessaria un’integrazione. Esistono numerose forme di Creatina negli integratori in commercio, tra le quali la Creatina Monoidrato è la più studiata. Le versioni più recenti di Creatina, come la kre-alkalyn [94] e la Creatina etil-estere [95], non si sono dimostrate superiori alla Creatina Monoidrato, nonostante abbiano in genere un prezzo più elevato. Pertanto, si raccomanda il consumo di 3-5g di Creatina Monoidrato al giorno. La tempistica di assunzione della Creatina non sembra avere importanza, poiché la saturazione delle riserve di Creatin-fosfato richiede circa 28 giorni per raggiungere le concentrazioni massime quando si consumano 3g al giorno e non ha un effetto in acuto [93].
Caffeina:
Uno degli integratori alimentari più utilizzati dai bodybuilder sono gli stimolanti, in particolare la Caffeina [9]. Oltre ad aumentare l’eccitazione [96], la Caffeina può ridurre il dolore e lo sforzo percepito durante l’esercizio [97] e migliora la gestione del Calcio, aumentando la potenza [98]. Studi sull’allenamento contro-resistenza hanno rilevato che la Caffeina riduce la fatica e aumenta la forza [99,100]. Tuttavia, non tutti gli studi hanno dimostrato un effetto ergogenico sull’allenamento contro-resistenza [101]. Gli studi che hanno dimostrato un effetto ergogenico hanno utilizzato dosaggi elevati di caffeina (5-6 mg/kg), che sono al limite superiore di quello che è considerato un dosaggio sicuro [99,100]. Tuttavia, può essere consigliabile consumare il dosaggio minimo efficace per individuo, poiché l’assunzione regolare può generare tolleranza [102]. A causa dell’effetto acuto della Caffeina, è consigliabile assumerla circa 1 ora prima dell’esercizio fisico [99]. Tuttavia, l’emivita della Caffeina è di circa 3-9 ore; pertanto, può essere consigliabile consumare la Caffeina all’inizio della giornata per favorire un sonno sano se l’esercizio fisico viene svolto più tardi nel corso della giornata [103]. Sono necessarie ulteriori ricerche per trovare un consenso sull’uso della Caffeina nell’allenamento contro-resistenza, ma sulla base delle prove attuali un dosaggio di 5-6 mg/kg consumato prima dell’esercizio potrebbe produrre un effetto ergogenico sulle prestazioni nell’allenamento contro-resistenza.
Beta-Alanina:
È stato dimostrato che l’ingestione di 4-6 g di beta-alanina aumenta i livelli di carnosina muscolare [104]. La carnosina agisce come tampone del pH nel muscolo scheletrico e può ritardare l’inizio dell’affaticamento muscolare durante l’esercizio ad alta intensità [105]. Una meta-analisi ha concluso che la beta-alanina potrebbe produrre effetti ergogenici durante l’esercizio ad alta intensità della durata di 60-240 secondi [104]. Inoltre, non sono stati riscontrati effetti benefici negli esercizi di durata inferiore a 60 secondi. La maggior parte degli studi inclusi nella meta-analisi riguardava l’esercizio di resistenza.
Tuttavia, è dimostrato che l’integrazione di beta-alanina può migliorare la resistenza muscolare negli atleti allenati alla resistenza [105] e può migliorare la composizione corporea [106]. Sono necessari ulteriori studi per esaminare l’effetto ergogenico della beta-alanina sulla composizione corporea e sulle prestazioni. Tuttavia, dato che i bodybuilder si allenano spesso con più di 10 ripetizioni per serie e spesso includono tecniche di intensità come drop set, pause di riposo, myo reps e altre, la beta-alanina potrebbe apportare un beneficio alla resistenza di queste serie [9].
Pertanto, potrebbe essere ragionevole per un bodybuilder consumare 3-5 g di beta alanina al giorno durante le fasi di allenamento ad alte ripetizioni o nelle fasi di allenamento in cui si incorporano diverse tecniche di intensità che prolungano la durata di un set. Come la creatina monoidrato, la beta-alanina non ha un effetto acuto, in quanto le concentrazioni di carnosina muscolare richiedono circa 4 settimane per raggiungere concentrazioni tali da produrre un effetto ergogenico, a condizione che se ne consumi una quantità sufficiente al giorno [104].
Citrullina Malato:
Recentemente, la Citrullina Malato ha guadagnato popolarità tra i bodybuilder. Il potenziale effetto ergogenico è dovuto all’aumento del flusso ematico al muscolo, alla produzione di ATP e alla potenziale capacità della Citrullina Malato di agire come agente tampone [107]. È stato dimostrato che il consumo di 8g di Citrullina Malato aumenta le ripetizioni fino al cedimento del 50% [107,108,109,110], riduce l’indolenzimento muscolare del 40% [107] e migliora la forza massimale e la potenza anaerobica [111].
Tuttavia, non tutti gli studi hanno osservato effetti ergogenici del consumo di Citrullina Malato. Due studi recenti non hanno mostrato un miglioramento delle prestazioni, un aumento della risposta del gonfiore muscolare dovuto all’allenamento, un’attenuazione della fatica o un aumento dell’attenzione e dell’energia in seguito all’integrazione di Citrullina Malato in uomini allenati contro-resistenza a livello amatoriale [112,113].
Una recente meta-analisi di Trexler et al. ha analizzato 12 studi sullla CM per le prestazioni di forza e potenza [114]. Sebbene abbiano riscontrato solo una piccola dimensione dell’effetto (0,20), hanno concluso che questo potrebbe essere rilevante per gli atleti di alto livello in cui i risultati delle competizioni si decidono su margini ridotti, come i culturisti agonisti di alto livello. Si consiglia di assumere la Citrullina Malato circa 60 minuti prima dell’esercizio fisico per consentire un assorbimento sufficiente.
Sono necessarie ulteriori ricerche per determinare l’efficacia della Citrullina Malato nell’esercizio contro-resistenza. Allo stato attuale, i dati indicano un effetto benefico o neutro sulle prestazioni. Pertanto, sulla base delle prove attuali, 8g al giorno di Citrullina Malato consumati prima dell’esercizio potrebbero avere dei benefici interessanti per i bodybuilder.
Alfa-GPC:
L’Alfa-GPC (alfa-glicerofosfocolina o colina alfoscerato) è un fosfolipide contenente colina. Quando viene ingerita, l’Alfa-GPC viene metabolizzata in colina e glicerolo-1-fosfato. La colina è un precursore dell’acetilcolina, un neurotrasmettitore coinvolto nella memoria, nell’attenzione e nella contrazione dei muscoli scheletrici. Il glicerolo-1-fosfato serve a sostenere le membrane cellulari.[https://pubmed.ncbi.nlm.]
L’Alfa-GPC sembra attraversare facilmente la barriera emato-encefalica e viene assorbito rapidamente. Attualmente è il miglior colinergico per aumentare i livelli plasmatici e cerebrali di colina.[https://pubmed.ncbi.nlm.]
L’integrazione orale di Alfa-GPC è interessante soprattutto per scopi nootropici o di potenziamento cognitivo. Esistono numerosi studi sui roditori che supportano questo effetto, ma non è ancora stato dimostrato negli esseri umani altrimenti sani. Negli anziani affetti da demenza lieve o moderata – che comporta un’alterazione della neurotrasmissione colinergica – l’Alfa-GPC migliora i sintomi cognitivi (ad esempio, disturbi della memoria e dell’attenzione).[https://pubmed.ncbi.nlm] L’Alfa-GPC può anche migliorare l’efficacia degli inibitori dell’acetilcolinesterasi (cioè i farmaci che aumentano la disponibilità di acetilcolina rallentandone la degradazione), utilizzati per il trattamento della malattia di Alzheimer.[https://pubmed.ncbi.nlm.]
Gli atleti sono un’altra popolazione che può trarre beneficio dall’integrazione di Alfa-GPC. Prove preliminari suggeriscono che l’alfa-GPC aumenta la potenza del salto verticale.[https://jissn.biomedcentral.com][https://pubmed.ncbi.nlm.] Inoltre, uno studio pilota ha riportato che l’Alfa-GPC ha aumentato il picco di forza nella panca, ma non la potenza di picco o il tasso di sviluppo della forza.[Ziegenfuss T, Landis J, Hofheins JJ Int Soc Sports Nutr.] Attualmente non è chiaro se l’Alfa-GPC aumenti la forza isometrica, ma i dati empirici e aneddotici sono incoraggianti [https://pubmed.ncbi.nlm.]
L’integrazione di un dosaggio pari a 600mg di Alpha-GPC prima di un test di potenza (spinte su panca) ha riportato un miglioramento della potenza del 14% rispetto al placebo quando assunta 45 minuti prima dell’attività; si trattava di uno studio pilota.[http://www.jissn.com] In media si è notato che il dosaggio di Alfa-GPC efficacie per trarre miglioramenti nella forza è nel range dei 300-600mg 45-30 minuti prima della seduta allenante.
Multi Vitaminico-Multi Minerale:
Storicamente, i bodybuilder hanno utilizzato diete restrittive che eliminano alimenti o interi gruppi di alimenti. Di conseguenza, sono comuni numerose carenze di vitamine e minerali. Nei bodybuilder a dieta sono state osservate carenze di Calcio, vitamina D, Zinco, Ferro e altre ancora [115,116,117]. Tuttavia, la maggior parte della letteratura sulle pratiche alimentari dei bodybuilder risale agli anni ’80 e ’90; pertanto, sono necessari dati più recenti [2].
Più di recente, le pratiche alimentari dei bodybuilder che seguono una dieta tradizionale restrittiva sono state confrontate con quelle degli agonisti che utilizzano un approccio dietetico basato sui macronutrienti, in cui nessun alimento o gruppo alimentare è off limits [118]. Non sorprende che i concorrenti che utilizzano un approccio dietetico più flessibile presentino meno carenze di micronutrienti. In particolare, la vitamina E, la vitamina K e le proteine sono risultate significativamente inferiori nelle donne che utilizzavano approcci dietetici rigidi rispetto a quelle che utilizzavano approcci più flessibili. Nel presente articolo, specie se si parla di Off-Season, si raccomanda di utilizzare un approccio dietetico flessibile, in cui nessun alimento o gruppo viene eliminato dalla dieta.
In questo modo, è meno probabile che si verifichino carenze di micronutrienti, soprattutto se si considera che le atlete in Off-Season hanno a disposizione una maggiore quantità di calorie rispetto a quelle a dieta per un contest, il che dovrebbe consentire loro di incorporare una maggiore varietà di alimenti.
Ciononostante, può essere consigliabile raccomandare un integratore multivitaminico/minerale a basso dosaggio (≤100% RDA) come misura di sicurezza per prevenire eventuali carenze di micronutrienti, sottolineando al contempo il consumo di una buona varietà di alimenti al giorno per soddisfare il fabbisogno di micronutrienti.
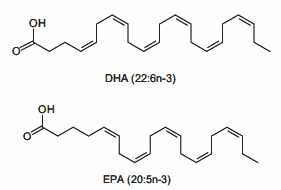
Omega 3 (EPA-DHA):
Gli acidi grassi polinsaturi con un doppio legame a tre atomi di distanza dal gruppo metilico terminale sono noti come ω-3 o acidi grassi omega-3 (O3). Un basso apporto di O3 nelle diete occidentali rispetto ad altre fonti di grassi alimentari (come gli acidi grassi omega-6) è associato a un peggioramento della salute multispettrale negli studi epidemiologici [119]. Pertanto, è interessante concentrarsi specificamente sulle modifiche della dieta per fornire acidi eicosapentaenoici e docosaesaenoici (EPA e DHA) – la carenza alimentare più comune nel mondo occidentale; ma vale la pena notare che la misurazione, l’interazione e l’effetto di O3 e acidi grassi omega-6 in relazione alla salute non sono chiari e vanno oltre lo scopo di questo articolo. Per una rassegna si rimanda ad altra pubblicazione [120].
Oltre alla salute, c’è interesse per i potenziali effetti anabolici degli integratori di EPA e DHA [121], che di solito vengono forniti attraverso l’olio di pesce o, in alcuni casi, l’olio di alghe. Tuttavia, ci sono dati contrastanti sulla capacità dell’olio di pesce di aumentare la risposta della sintesi proteica muscolare all’ingestione di proteine. Mentre un articolo di revisione del 2014 ha evidenziato una serie di studi secondo cui l’olio di pesce può aumentare la risposta [122], uno studio recente non ha rilevato alcun effetto sulla risposta della MPS a una sessione di allenamento contro-resistenza e all’ingestione di proteine dopo l’allenamento [123]. Inoltre, i dati sull’ipertrofia longitudinale sono pochi [124] e gli studi sulle prestazioni dell’allenamento contro-resistenza sono contrastanti [125] e in gran parte non applicabili o difficili da valutare a causa dell’uso di partecipanti non allenati o di allenamenti non standardizzati ed ecologicamente non realistici rispetto al bodybuilding.
In una recente review che affronta specificamente la questione se gli integratori di O3 possano o meno aumentare l’ipertrofia [126], gli autori hanno concluso che attualmente non ci sono prove sufficienti per fare tale affermazione. Sebbene siano necessarie ulteriori ricerche prima di poter raccomandare l’integrazione di O3 (o di alterazioni della dieta) a fini di costruzione muscolare, i benefici per la salute dell’integrazione di O3 sono degni di nota. Ad esempio, recenti meta-analisi hanno riportato che l’integrazione di olio di pesce riduce i sintomi della depressione [127], diminuisce il rischio di morte cardiaca [128], riduce la pressione sanguigna [129] e diminuisce la circonferenza vita [130]. Pertanto, gli atleti estetici possono prendere in considerazione l’integrazione giornaliera di olio di pesce (o di alghe) (1.5-2.5g di EPA/DHA) per la salute generale e multi spettro, ma sono necessari studi futuri per formulare raccomandazioni relative alle prestazioni nel bodybuilding.
Acido Arachidonico (AA):
L’Acido Arachidonico (AA) è l’acido grasso omega-6 più rilevante dal punto di vista biologico e, nella membrana lipidica di una cellula, è l’acido grasso che viene confrontato con i due acidi grassi dell’olio di pesce (EPA e DHA) nella costituzione di un rapporto omega-3:6. Dati recenti suggeriscono un’assunzione giornaliera di 50-250mg di Acido Arachidonico[https://www.ncbi.nlm.][https://www.ncbi.nlm.][https://www.ncbi.nlm.][https://www.ncbi.nlm.] con alcune fonti che stimano livelli fino a 500mg al giorno;[https://www.ncbi.nlm.] l’assunzione di Acido Arachidonico sembra essere inferiore nei vegetariani[https://www.ncbi.nlm.].
Si ritiene che l’Acido Arachidonico sia importante per il metabolismo del muscolo scheletrico, poiché si pensa che i fosfolipidi della membrana del sarcoplasma riflettano la dieta,[https://www.ncbi.nlm.][https://www.ncbi.nlm.] l’allenamento stesso sembra alterare il contenuto di fosfolipidi del muscolo (indipendentemente dalla composizione delle fibre muscolari[https://www.ncbi.nlm.] e associato a un rapporto omega 6:3 più basso[https://www.ncbi.nlm.][https://www.ncbi.nlm.]) e gli eicosanoidi dell’Acido Arachidonico interagiscono con la sintesi proteica muscolare attraverso i loro recettori.
L’Acido Arachidonico segnala la sintesi proteica muscolare attraverso una via dipendente dalla COX-2 (che suggerisce il coinvolgimento delle prostaglandine)[https://www.ncbi.nlm.] che è associata ad aumenti sia della prostaglandina E2 (PGE2) che del PGF(2α),[https://www.ncbi.nlm.][https://www.ncbi.nlm.][https://www.ncbi.nlm.][https://www.ncbi.nlm.] anche se l’incubazione con PGE2 o PGF(2α) isolati non sembra replicare pienamente gli effetti ipertrofici dell’Acido Arachidonico. [https://www.ncbi.nlm.] PGE2 e PGF(2α) sono indotti anche dall’esercizio fisico (nello specifico, dallo stiramento delle cellule muscolari in vitro[https://www.ncbi.nlm.]) ed è stato osservato sia nel siero[https://pubmed.ncbi.nlm.][https://www.ncbi.nlm.] che a livello intramuscolare (quadruplicato, da 0,95+/-0,26ng/mL a 3,97+/-0. La capacità del riflesso da stiramento di aumentare le concentrazioni di PGE2 e PGF(2α)[https://www.ncbi.nlm.] potrebbe essere dovuta semplicemente al fatto che lo stiramento aumenta l’attività delle COX2.[https://www.ncbi.nlm.][https://www.ncbi.nlm.]
Va notato che l’integrazione di 1.500mg di Acido Arachidonico (rispetto a una dieta di controllo contenente 200mg dello stesso) per 49 giorni ha aumentato la secrezione di PGE2 da parte di cellule immunitarie stimolate (del 50-100%) in giovani uomini altrimenti sani,[https://www.ncbi.nlm.] ma la rilevanza di questo studio per il muscolo scheletrico non è nota. Questo studio ha anche osservato che, senza stimolazione, non c’erano differenze significative tra i gruppi.[https://www.ncbi.nlm.] Altrove, è stata osservata una tendenza all’aumento delle concentrazioni sieriche di PGE2 a riposo in uomini allenati a cui sono stati somministrati 1.000mg di Acido Arachidonico per 50 giorni.[https://www.ncbi.nlm.]
L’Acido Arachidonico, attraverso gli eicosanoidi noti come PGF(2α) e PGE2, stimola la sintesi proteica muscolare. Sono prodotti a partire dall’Acido Arachidonico, ma normalmente non formano i rispettivi eicosanoidi per la costruzione del muscolo finché la cellula non viene stimolata da un fattore di stress (come il riflesso di stiramento di una cellula muscolare) che ne induce la produzione.
Il recettore per il PGF(2α) (recettore FP) sembra essere sovraregolato dagli inibitori della COX1 (l’acetaminofene utilizzato in questo studio)[https://www.ncbi.nlm.] e si ritiene che una maggiore segnalazione del PGF(2α) sia alla base del miglioramento della sintesi proteica muscolare osservato nei soggetti anziani con farmaci antinfiammatori. La supplementazione di Acido Arachidonico non sembra influenzare la quantità di recettori FP nei giovani;[https://www.ncbi.nlm.] mentre l’esercizio fisico stesso può aumentare il contenuto di recettori EP3, né gli inibitori della COX1[https://www.ncbi.nlm.] né l’Acido Arachidonico[https://www.ncbi.nlm.] sembrano influenzarlo ulteriormente.
Tuttavia, è stato riscontrato che l’uso di inibitori della COX2 (nei giovani) sopprime l’aumento di PGF(2α) indotto dall’esercizio fisico (Ibuprofene e Acetaminofene)[https://www.ncbi.nlm.][https://www.ncbi.nlm.] e di PGE2,[https://www.ncbi.nlm.] il che si pensa sia dovuto al fatto che la conversione da PGH2 in questi metaboliti dipende dall’attività della COX2.
Poiché la produzione di questi eicosanoidi dipende dall’enzima COX2, si ritiene che l’inibizione di questo enzima riduca gli effetti anabolizzanti dell’esercizio fisico se assunto prima dello stesso.
L’acido arachidonico (così come l’EPA dall’olio di pesce) non ha compromesso l’assorbimento del glucosio nelle cellule muscolari isolate e 10μM di acido grasso sono in grado di attenuare la resistenza all’Insulina indotta dai grassi saturi; [https://pubmed.ncbi.nlm.] un fenomeno osservato con i grassi saturi a 18 o più catene di carbonio[https://www.ncbi.nlm.] che non sembra applicarsi agli acidi grassi polinsaturi di uguale lunghezza di catena[https://www.ncbi.nlm.][https://www.ncbi.nlm.] ed è probabilmente legato all’aumento delle ceramidi intracellulari[https://www.ncbi.nlm.] che compromettono la segnalazione di Akt[https://www.ncbi.nlm.][https://www.ncbi.nlm.] e riducono l’assorbimento di glucosio mediato da GLUT4 con l’Insulina.[https://www.ncbi.nlm.]
L’Acido Arachidonico e i grassi polinsaturi omega-3 sono entrambi associati a una migliore sensibilità all’Insulina delle cellule muscolari, che potrebbe essere secondaria alla riduzione dei livelli di grassi saturi nella membrana lipidica e quindi alla riduzione delle concentrazioni intracellulari di ceramidi. È possibile che ciò non sia correlato agli eicosanoidi o al rapporto omega-3:6.
In 31 uomini allenati, sottoposti a un programma di sollevamento pesi e a una dieta standardizzata (500kcal in eccesso con 2g/kg di proteine) con 1g di Acido Arachidonico al giorno o placebo, l’integrazione per 50 giorni è sembrata aumentare la potenza di picco (7,1%) e la potenza media (3,6%) al test di Wingate, ma non è riuscita a influenzare positivamente la massa muscolare o le misure di potenza del sollevamento pesi (bench press e leg press).[https://www.ncbi.nlm.]
Attualmente non ci sono prove sufficienti per raccomandare una dose ideale di integrazione di Acido Arachidonico, ma aneddoticamente si usa un dosaggio di circa 1.500 mg da assumere 45 minuti prima dell’allenamento per un periodo medio di 8 settimane. Non è certo che si tratti di una dose ottimale o che sia necessaria la tempistica.
Va inoltre notato che per le persone affette da patologie infiammatorie croniche, come l’artrite reumatoide o le malattie infiammatorie intestinali, la dose ideale di Acido Arachidonico può essere in realtà una sua restrizione dietetica. Nei casi di malattie infiammatorie, l’integrazione di Acido Arachidonico è probabilmente controindicata.
Raccomandazioni per gli integratori alimentari e il dosaggio per i bodybuilder in Off-Season:
Creatina Monoidrato= 3-5g/die;
Beta-Alanina= 3-5g/die;
Citrullina Malato= 8g/pre-workout;
Alfa-GPC= 300-600mg/pre-workout;
Caffeina= 5-6mg/Kg/pre-workout (media standard tra 200 e 600mg/die);
Multi Vitaminico – Multi Minerale= ≤100% RDA/die;
Omega 3 (EPA-DHA)= 1.5-2.5g/die;
Acido Arachidonico= 1.5g/pre-workout.
Supplementazione PEDs:
Una cosa occorre premettere prima di procedere con la descrizione delle molecole più utilizzate nel contesto della Off-Season: non esistono PEDs esclusivamente confinabili in uno dei contesti della programmazione di un bodybuilder. Esiste il grado di versatilità il quale sta ad indicare quanto una molecola possa essere gestita con facilità in situazioni preparatorie differenti. Esistono molecole che per caratteristiche possono dare vantaggi maggiori in Off-Season/Bulk per via di alcune loro caratteristiche che in altro contesto, per esempio il pre-contest, risulterebbero più complesse da gestire. Ma questo non significa che tali molecole siano generalemnte da considerarsi “off-limitz” in un altra fase della preparazione annuale.
Premesso ciò, l’attenzione in questo paragrafo si concentrerà sui principali PEDs usati in Off-Season.
Tra tutti gli AAS, il Testosterone è quello che non ha bisogno di particolari presentazioni. Si tratta dell’ormone sessuale maschile per antonomasia. Nell’uomo, il Testosterone svolge un ruolo fondamentale nello sviluppo dei tessuti riproduttivi maschili, come i testicoli e la prostata, oltre a promuovere le caratteristiche sessuali secondarie, come l’aumento della massa muscolare e ossea e la crescita dei peli. Inoltre, in entrambi i sessi, il Testosterone è coinvolto nella salute e nel benessere, compresi gli stati d’animo, il comportamento e la prevenzione dell’osteoporosi in cooperazione con l’Estradiolo. Livelli insufficienti di Testosterone negli uomini possono portare ad anomalie, tra cui la fragilità e la perdita ossea.
In generale, il Testosterone promuove la sintesi proteica e quindi la crescita dei tessuti dotati di recettori per gli androgeni. Il Testosterone può essere descritto come avente effetti virilizzanti e anabolizzanti (anche se queste descrizioni categoriali sono in qualche modo arbitrarie, poiché vi è una grande sovrapposizione reciproca tra di essi).
Gli effetti anabolizzanti comprendono la crescita della massa e della forza muscolare, l’aumento della densità e della resistenza ossea e la stimolazione della crescita lineare e della maturazione ossea.
Gli effetti androgeni comprendono la maturazione degli organi sessuali, in particolare del pene, e la formazione dello scroto nel feto, e dopo la nascita (di solito nella pubertà) l’approfondimento della voce, la crescita dei peli del viso (come la barba) e dei peli ascellari. Molti di questi effetti rientrano nella categoria dei caratteri sessuali secondari maschili.
Al principio degli anni 30 del novecento avvenne la sintesi chimica del Testosterone, quando Butenandt e G. Hanisch pubblicarono un articolo che descriveva “Un metodo per preparare il Testosterone dal colesterolo”. Solo una settimana dopo, il terzo gruppo, Ruzicka e A. Wettstein, annunciò una domanda di brevetto in un documento “Sulla preparazione artificiale dell’ormone testicolare Testosterone (Androsten-3-one-17-ol).” Ruzicka e Butenandt ricevettero il premio Nobel per la chimica nel 1939 per il loro lavoro.
Gli studi clinici sull’uomo, che prevedevano dosi PO (per via orale) di Methyltestosterone o iniezioni di Testosterone Propionato, iniziarono già nel 1937. Il Testosterone Propionato è menzionato in una lettera all’editore della rivista Strength and Health nel 1938; questo è il primo riferimento noto a un AAS in una rivista statunitense di sollevamento pesi o Bodybuilding.
Lo sviluppo delle proprietà di costruzione muscolare del Testosterone proseguì negli anni ’40, in Unione Sovietica e nei paesi del blocco orientale come la Germania dell’Est, dove sono stati utilizzati programmi di AAS per migliorare le prestazioni dei sollevatori di pesi olimpici e di altri dilettanti già prima degli anni ’50. In risposta al successo dei sollevatori di pesi russi, il medico della squadra olimpica statunitense John Ziegler lavorò con un equipe di chimici per sviluppare un AAS con effetti androgeni ridotti. Ma questa è un altra storia.
L’uso del Testosterone nello sport si diffuse tra gli anni ’50 e gli anni ’60. Le forme utilizzate nei primi tempi erano il Testosterone in sospensione e il Testosterone Propionato, che rappresentano con il Methyltestosterone (Testosterone metilato in C-17) le forme più datate dell’ormone in questione (1935).
In ambito culturistico, il Testosterone rappresenta un AAS sufficientemente versatile in maniera dose-dipendente e sensibilità-dipendente dal momento che il dosaggio dovrebbe essere tarato in base alle risposte metaboliche soggettive alle quali è soggetto l’ormone (vedi, ad esempio, aromatizzazione in estrogeni). Questo ultimo punto è di estrema importanza al fine di evitare l’uso/abuso di AI (Inibitori dell’Aromatasi) e/o SERM (Modulatori Selettivi del Recettore degli Estrogeni). Oltre a peggiorare potenzialmente il quadro lipidico, sommandosi all’azione degli AAS utilizzati, essi riducono l’espressione epatica di IGF-1 cosa che può ridurre la risposta anabolizzante del protocollo PEDs. Nei soggetti caratterizzati da una elevata sensibilità all’attività estrogenica, le procedure applicate vedono: 1) l’uso di Raloxifene o Tamoxifene (SERM) a dosi sufficienti a impedire la comparsa o il peggioramento di una ginecomastia in stadio iniziale già presente e non ancora asportata chirurgicamente 2) l’uso di dosi fisiologiche di Testosterone come base onde evitare la comparsa di stati letargici, affaticabilità, disfunzioni sessuali ecc 3) l’uso di un “mix” composto da Testosterone e Boldenone (vedi in seguito) tale da poter usufruire della bassa e diversa sensibilità all’azione dell’Enzima Aromatasi su quest’ultimo riuscendo ad avere un controllo estrogenico teoricamente migliore (Testosterone e Boldenone mostrano qualità anabolizzanti intrinseche simili).
In un contesto Off-Season, quindi, vista l’importanza della presenza di un buon livello di Estradiolo sia sul complesso degli effetti anabolizzanti ricercati sia per la sua attività sessuale e cerebrale, il Testosterone andrebbe inizialmente calibrato sul soggetto e nel caso affiancato da dosi altrettanto ben tarate di SERM la dove ne risultasse un reale bisogno.
L’uso di un estere che garantisca un rilascio graduale della molecola (vedi Enantato o Cypionato) risulta la scelta migliore al fine di creare una soglia ematica stabile e esente da picchi e cali che possono risultare controproducenti a livello psicofisico. Tenere sempre in considerazione l’emivita di una molecola è uno dei punti fondamentali per sfruttarla al meglio. Nel caso degli esteri sopra citati, una divisione del dosaggio settimanale in due somministrazioni uguali distanziate da quattro-cinque giorni l’una dall’altra risulta una pratica ottimale allo scopo di creare una soglia ematica stabile.
I dosaggi comunemente utilizzati, parlando di molecole esterificate, vanno da 200mg ad 1g a settimana. Per quanto riguarda il Testosterone in sospensione, le dosi comunemente utilizzate vanno dai 175mg ai 700mg a settimana.
Il Boldenone [1,4-androstadiene-3-one,17b-ol], commercializzato con il nome di Equipoise, Ganabol, Equigan, Ultragan, e Boldane, è uno steroide anabolizzante-androgeno spesso legato all’estere Undecylenato. Strutturalmente molto simile al Testosterone, il Boldenone differisce da questo per il doppio legame tra C1 e C2.
La Ciba brevettò il Boldenone nel 1949. Successivamente, negli anni ’50 e ’60, sviluppò diversi esteri sperimentali del farmaco. Uno di questi era il Boldenone Undecilenato, che fu introdotto per uso clinico con il marchio Parenabol e fu utilizzato alla fine degli anni ’60 e all’inizio degli anni ’70. Tuttavia, fu sospeso prima della fine degli anni ’70. Ad oggi l’uso del Boldenone è legale in alcuni paesi in campo veterinario.
Se qualcuno volesse usare 500mg di Testosterone, ma non potrebbe usare un tale dosaggio dal momento che presenta particolare difficoltà nella gestione estrogenica in specie senza l’uso di AI come Exemestane o Anastrozolo, una conclusione a cui molti superficialmente sono giunti è che si potrebbe semplicemente usare il Boldenone al dosaggio sopra citato per ridurre della metà l’attività estrogenica, ma comunque supportare un’adeguata produzione di Estradiolo. Ma quando si approfondisci l’ipotesi e la si testa sul campo, è davvero così che stanno le cose? In realtà no, o, comunque, la media delle variabili di risposta spinge a confermare una maggiore validità nel “mixare” Testosterone e Boldenone coprendo la dose base calcolata in precedenza, e con variazione di percentuale T:B ratio da 1:1 a 2:1.
Comunque, oltre a rappresentare genericamente una discreta molecola sia in in preparazione alla gara che in Off-Season, I dosaggi utilizzati si settano nel range tra i 200mg ed i 500mg a settimana, spesso abbinato ad una dose variabile (vedi sopra) di Testosterone.
Il Nandrolone, noto anche come 19-nortestosterone, è uno Steroide Androgeno Anabolizzante (AAS) utilizzato sotto forma di molecola legata a esteri come quello Decanoato (nome commerciale Deca-Durabolin) e il Fenilpropionato (nome commerciale Durabolin). Gli esteri del Nandrolone sono utilizzati nel trattamento di anemie, cachessia (sindrome da deperimento), osteoporosi, cancro al seno e per altre indicazioni mediche.
Il Nandrolone è stato sintetizzato per la prima volta nel 1950. È stato introdotto per la prima volta nel mercato farmaceutico, come Nandrolone Fenilpropionato, nel 1959, e poi come Nandrolone Decanoato nel 1962, seguito da ulteriori esteri.
Il Nandrolone ha una bassa affinità di interazione con l’Enzima Aromatasi convertendo in Estrone, un estrogeno molto meno potente dell’Estradiolo, circa 10 volte meno attivo, e, come tale, è un estrogeno relativamente debole. In una condizione di somministrazione del Nandrolone senza una base di Testosterone, i livelli di Estradiolo calerebbero marcatamente a favore di un aumento del Estrone il quale non potrebbe però sostituire nelle diverse attività tissutali il prima citato E2. Le conseguenze negative si verificherebbero dall’attività sessuale all’attività neurosteroidea.
Infatti, un effetto da non sottovalutare con l’uso di Nandrolone è il suo impatto sul SNC. L’impatto del Nandrolone sul Sistema Nervoso Centrale è stato osservato scientificamente. Nello studio intitolato “The Impact of Nandrolone Decanoate on the Central Nervous System” vengono descritti chiaramente i numerosi effetti psicologici di questa molecola. Essi comprendono e influenzano:
1- Aggressività 2- Ansia, paura e stress 3- Ricompensa e dipendenza 4- Apprendimento, memoria e capacità di lavoro 5- Locomozione e attività fisica 6- Effetti sulla HPAA (Asse Ipotalamo-Pituitaria-Surrene) 7- Effetto sui neurotrasmettitori: Recettore Acido γ-Aminobutirrico Tipo A (GABAA); Recettori 5-idrossitriptamina (5-HT) e 5-HT; Recettori della Dopamina e Recettori Oppioidi.
Questo effetto, unito alla modesta potenzialità anabolizzante se confrontata con altre molecole anche della stessa famiglia, fa pendere l’ago della bilancia verso gli svantaggi d’uso piuttosto che i vantaggi. Sebbene vi sia un rapporto tra Testosterone e Nandrolone finalizzato a ridurre la comparsa di questi effetti avversi (ratio T:N = 2:1) su un buon numero di soggetti risulta dare comunque problemi rilevanti.
Il suo uso principale in Off-Season comprende dosaggi medi tra i 200mg ed i 400mg a settimana, con un adeguato rapporto con il Testosterone. Se utilizzato a fini di recupero articolare viene usato a dosaggi di 100mg a settimana, e con tali dosaggi difficilmente emergono i problemi sopra elencati a patto che ci sia una base di Testosterone.
Il Drostanolone, noto anche come 2α-metil-5α-diidrotestosterone (2α-metil-DHT) o come 2α-metil-5α-androstan-17β-ol-3-one, è uno steroide androstano sintetico e un derivato del DHT. Si tratta nello specifico di DHT con un gruppo metile in posizione C2α. La forma esterificata Drostanolone Propionato è stata usata in passato nel trattamento del cancro al seno nelle donne per via della sua attività antiestrogenica. Questa azione il Drostanolone la esplica sia agendo come antagonista del recettore degli estrogeni e sia come inibitore dell’Enzima Aromatasi. Ed è proprio per questo motivo che una molecola generalmente relegata all’uso in “Cut” o pre-gara trova un suo uso funzionale in Off-Season. La sua attività AI è comunque moderata ma sufficiente in un buon numero di soggetti per evitare l’aggiunta di SERM e/o AI di altro genere. L’attività AI moderata sembra non incidere negativamente in modo sensibile sull’Asse GH/IGF1.
L’effetto miotrofico risulta simile a quello osservato con il Methenolone, in generale moderatamente inferiore al Testosterone. I dosaggi utilizzati in Off-Season per il controllo estrogenico sono nel range dei 200-400mg a settimana (diviso in due iniezioni distanziate da 4-5 giorni) per l’estere Enantato, mentre per il Propionato 150-350mg a settimana (dosi a giorni alterni o giornaliere).
Il Trenbolone, noto anche come 19-nor-δ9,11-testosterone o come estra-4,9,11-trien-17β-ol-3-one, è uno steroide sintetico e un derivato del Nandrolone (19-nortestosterone) sintetizzato per la prima volta nel 1963. Si tratta nello specifico di Nandrolone con due doppi legami aggiuntivi nel nucleo steroideo. Gli esteri del Trenbolone, che hanno un estere in posizione C17β, includono il Trenbolone Acetato, il Trenbolone Enantato, Il Trenbolone Hexahydrobenzylcarbonato e il Trenbolone Undecanoato. Il Trenbolone Acetato (marchi Finajet, Finaplix, e altri) e il Trenbolone Hexahydrobenzylcarbonato (marchi Parabolan, Hexabolan), sono o sono stati commercializzati per uso veterinario e clinico nell’uomo. Il Trenbolone Acetato è utilizzato in medicina veterinaria nel bestiame per aumentare la crescita muscolare e l’appetito degli animali, mentre il Trenbolone Hexahydrobenzylcarbonato è stato utilizzato in passato a livello clinico nell’uomo, ma ora non è più commercializzato.
Si tratta di uno degli AAS più versatili in assoluto, con un ottima resa tanto in preparazione alla gara quanto in Off-Season. L’enorme potenziale anabolizzante del Trenbolone, così come dei suoi analoghi, è stato riportato fin dagli anni ’60. La sua diffusione nel Bodybuilding è iniziata circa negli anni ’80 del secolo scorso. La sua elevata potenzialità miotrofica, lipolitica e di spinta mentale lo resero in poco tempo estremamente popolare tra i culturisti.
In Off-Season viene utilizzato nelle sue forme eseterificate Enantato e Hexahydrobenzylcarbonato a dosaggi nell’ordine dei 100-400mg a settimana (divisa in due somministrazioni distanziate l’una dall’altra da 4-5 giorni), sebbene il trend d’oltre oceano è arrivato a dosaggi decisamente eccessivi e nell’ordine del grammo. Per l’esetere Acetato i dosaggi medi vanno da 150mg a 350mg a settimana con dosaggi a giorni alterni o giornalieri.
E’ necessario ricordare ai lettori che gli effetti collaterali a livello del SNC possono verificarsi in alcuni punti come nel caso del Nandrolone sebbene i vantaggi rendano il Trenbolone più bilanciato tra sides e vantaggi.
Il Trestolone, noto anche come 7α-metil-19-nortestosterone (MENT) o come 7α-metilestr-4-en-17β-ol-3-one, è uno steroide sintetico e un derivato del Nandrolone (19-nortestosterone). È una forma modificata del Nandrolone con un gruppo metile in posizione C7α. Tra gli AAS strettamente correlati vi sono il 7α-metil-19-norandrostenedione (MENT dione, trestione), un pro-ormone androgeno del Trestolone, e il Dimetandrolone (7α, 11β-dimetil-19-nortestosterone), il derivato metilato C11β del Trestolone, nonché il Mibolerone (7α,17α-dimetil-19-nortestosterone) e il Dimetiltrienolone (7α,17α-dimetil-δ9,11-19-nortestosterone). Anche il progestinico Tibolone (7α-metil-17α-etinil-δ5(10)-19-nortestosterone) è strettamente correlato al Trestolone.
Il Trestolone è stato descritto per la prima volta nel 1963. Tuttavia, non è stato successivamente studiato fino al 1990. Lo sviluppo del Trestolone per un potenziale uso nella contraccezione ormonale maschile e nella terapia sostitutiva degli androgeni è stato avviato nel 1993 ed è proseguito in seguito. Non sembra che siano stati condotti ulteriori sviluppi dal 2013. Il Trestolone è stato sviluppato dal Population Council, un’organizzazione non governativa senza scopo di lucro dedicata alla salute riproduttiva.
Come AAS, il Trestolone è un agonista del recettore degli androgeni (AR), analogamente agli androgeni come il Testosterone e il Diidrotestosterone (DHT). Questo AAS presenta spiccate proprietà anticortisolemiche sia attraverso l’inibizione enzimatica sia per attività antagonista recettoriale. Il Trestolone non è un substrato per la 5α-reduttasi e quindi non è potenziato o inattivato nei cosiddetti tessuti “androgeni” come la pelle, i follicoli piliferi e la ghiandola prostatica. Come tale, ha un elevato rapporto tra attività anabolica e androgena, analogamente ad altri derivati del Nandrolone. Il Trestolone è un substrato per l’Aromatasi e quindi produce come metabolita l’estrogeno 7α-metilestradiolo. Tuttavia, il Trestolone ha solo una debole attività estrogenica e una quantità che sembrerebbe essere insufficiente per scopi terapici sostitutivi, come evidenziato dalla diminuzione della densità minerale ossea negli uomini trattati con esso per l’ipogonadismo.
Il potenziale anabolizzante del Trestolone ha mostrato un grado di superiorità miotrofica rispetto al Trenbolone. Le sue caratteristiche ne fanno prediligere l’uso in Off-Season/Bulk. I dosaggi utilizzati con la forma Acetato sono nell’ordine dei 150-350mg a settimana con una cadenza nelle somministrazioni a giorni alterni. Sebbene sia più rara da reperire, la forma Enantato è utilizzato nel range dei 200-400mg a settimana divisi in somministrazioni ogni 4-5 giorni.
L’Oxymetholone, noto anche come 2-idrossimetilene-17α-metil-4,5α-diidrotestosterone (2-idrossimetilene-17α-metil-DHT) o come 2-idrossimetilene-17α-metil-5α-androstan-17β-olo-3-one, è uno steroide androstanico sintetico e un derivato 17α-alchilato del DHT. L’Oxymetholone è stato descritto per la prima volta in un articolo del 1959 da scienziati della Syntex. È stato introdotto per uso medico dalla Syntex e dalla Imperial Chemical Industries nel Regno Unito con il marchio Anapolon nel 1961. L’Oxymetholone è stato introdotto anche con i marchi Adroyd (Parke-Davis) nel 1961 e Anadrol (Syntex) nel 1962. Il farmaco è stato commercializzato negli Stati Uniti nei primi anni ’60.
Come altri AAS, l’Oxymetholone è un agonista del recettore degli androgeni (AR). Non è un substrato per la 5α-reduttasi (dal momento che è già 5α-ridotto) ed è uno substrato scarso per il 3α-idrossisteroide deidrogenasi (3α-HSD), e quindi mostra un alto rapporto di attività anabolizzante rispetto all’effetto androgenico.
Data la sua derivanza dal DHT, l’Oxymetholone non è un substrato per l’Enzima Aromatasi e quindi non può essere aromatizzato in metaboliti estrogenici. Tuttavia, caratteristica unica tra i derivati del DHT, l’Oxymetholone è comunque associato a un’estrogenicità relativamente elevata ed è noto per avere il potenziale di produrre effetti collaterali estrogenici come ginecomastia (anche se non comune) e ritenzione idrica. È stato suggerito che questo può essere una conseguenza del legame diretto a l’attivazione del recettore degli estrogeni da parte dell’Oxymetholone (estrogenicità intrinseca). L’Oxymetholone non possiede alcuna attività progestinica significativa. Per via della caratteristica attività estrogenica intrinseca, con l’uso di Oxymetholone è spesso necessario l’uso di un SERM onde avere un controllo sulla aumentata attività estrogenica.
A causa della sua struttura 17α-alchilata, l’Oxymetholone è epatotossico. L’uso a lungo termine del farmaco può causare una varietà di disturbi gravi, tra cui l’epatite, il cancro al fegato e la cirrosi; pertanto si raccomandano test periodici di funzionalità epatica per coloro che assumono l’Oxymetholone a fini terapeutici. Questa molecola ha ottenuto, infatti, la nomea di essere uno tra gli AAS più epatotossici. Ciò deriva da i dosaggi comunemente, ed erroneamente, utilizzati in contesto culturistico. Si parla di dosaggi che facilmente sforano i 150mg/die.
Osservazioni e esaminazione di diversi referti di esami ematici hanno evidenziato una soglia di “vantaggio/svantaggio” a favore del primo con un dosaggio calcolato con la formula 1mg/Kg. Genericamente, però, il dosaggio standard e conservativo si attesta nel range dei 50-100mg/die per non più di 28 giorni consecutivi, al fine di ridurre l’impatto negativo sul fegato e lipidemia.
Il Methandrostenolone, noto anche come 17α-metil-δ1-testosterone o come 17α-metilandrost-1,4-dien-17β-ol-3-one, è uno steroide androstanico sintetico e un derivato 17α-alchilato del Testosterone. È una modifica del Testosterone con un gruppo metile in posizione C17α e un doppio legame aggiuntivo tra le posizioni C1 e C2. Il farmaco è anche il derivato 17α-metilato del Boldenone (δ1-testosterone) e l’analogo δ1 del Methyltestosterone (17α-metiltestosterone).
Il Methandrostenolone è stato descritto per la prima volta nel 1955. È stato sintetizzato dai ricercatori dei laboratori CIBA di Basilea, in Svizzera. La CIBA depositò un brevetto statunitense nel 1957 e iniziò a commercializzare il farmaco sotto il nome di Dianabol nel 1958 negli Stati Uniti. Inizialmente veniva prescritto alle vittime di ustioni e agli anziani. Tra i primi utilizzatori vi furono i giocatori dell’Oklahoma University e l’allenatore dei San Diego Chargers Sid Gillman, che somministrò il Dianabol alla sua squadra a partire dal 1963.
Anche se il primo a somministrare il Methandrostenolone agli atleti fu il Dr. John Ziegler, personaggio che ebbe non poca importanza nella storia dell’uso degli AAS negli Stati Uniti. Ziegler contribuì a facilitare l’adozione degli AAS in generale, e del Dianabol in particolare, da parte degli atleti americani. Ziegler fu la prima persona a somministrare il Dianabol agli atleti competitivi poco dopo la sua introduzione da parte della CIBA nel 1958. Ebbe accesso al laboratorio CIBA a Summit (New Jersey) nel corso degli anni 50’ e somministrava già ai pesisti il Testosterone Propionato per “scopi di ricerca”. Da li il passo fu breve per la diffusione a macchia d’olio di questo AAS tra i culturisti.
Data la sua principale modifica strutturale, ossia la metilazione in C-17, il Methandrostenolone mostra un aumentata stabilità del legame recettoriale aumentando così l’affinità sia al AR sia, successivamente all’aromatizzazione nel suo metabolita 17-Methylestradiolo, per i recettori estrogenici rendendo il composto molto più estrogenico del Testosterone. Tale caratteristiche migliora però il potenziale proliferativo dei AR e l’influenza positiva sulla sintesi di IGF-1. Da non dimenticare è il suo significativo impatto anticortisolemico.
Trattandosi di una molecola con una discreta tendenza all’aromatizzazione, il suo uso tipico la vede inserita nelle fasi Off-Season. Il calcolo del dosaggio, per via dati aneddotici e osservativi raccolti, lo si ottiene con la formula 5mg/12Kg di peso corporeo. Trattandosi di un composto orale metilato in C-17 se ne scoraggia l’utilizzo oltre i 28 giorni consecutivi onde ridurre l’impatto negativo su fegato e lipidemia. Data la sua emivita di circa 4h, il dosaggio giornaliero dovrebbe essere diviso in più assunzioni distribuite durante l’arco della giornata.
L’Ormone della Crescita (GH) o Somatotropina, noto anche come Ormone della Crescita Umano (hGH o HGH), è un ormone peptidico che stimola la crescita, la riproduzione e la rigenerazione cellulare nell’uomo e in altri animali. È quindi importante per lo sviluppo umano. Il GH stimola anche la produzione di IGF-1 e aumenta la concentrazione di glucosio e acidi grassi liberi nel sangue. È un tipo di mitogeno specifico solo per i recettori di alcuni tipi di cellule. Il GH è un polipeptide a catena singola di 191 aminoacidi che viene sintetizzato, immagazzinato e secreto dalle cellule somatotrope nelle ali laterali dell’ipofisi anteriore.
Una forma ricombinante di hGH, chiamata Somatropina, viene utilizzata come farmaco da prescrizione per il trattamento dei disturbi della crescita nei bambini e della carenza di Ormone della Crescita negli adulti. Molte delle funzioni dell’hGH rimangono sconosciute.
Nel suo ruolo di agente anabolizzante, l’hGH è stato utilizzato dagli sportivi agonisti almeno dal 1982, quando la sola forma disponibile era quella derivata dall’Ipofisi dei cadaveri, ed è stato vietato dal CIO e dall’NCAA. L’analisi tradizionale delle urine non è in grado di rilevare il doping con HGH, pertanto il divieto è stato applicato solo all’inizio degli anni 2000, quando sono stati sviluppati test del sangue in grado di distinguere tra hGH naturale e artificiale.
In ambiente bodybuilding, l’hGH viene utilizzato in Off-Season (dai soggetti meglio informati) a dosaggi nel range delle 4-8UI al giorno o 8-16UI a giorni alterni. La somministrazione in concomitanza con l’uso di Insulina ha mostrato effetti sinergici molto evidenti che trovano la loro origine nel miglioramento della sintesi di IGF-1 e della sua frazione libera quindi attiva. Ricordo inoltre che l’uso di hGH può causare una sottoregolazione della funzionalità tiroidea per via del feedback negativo causato da un aumento della conversione del T4 in T3 per azione del GH. L’uso di T4, nel periodo d’uso in Off-Season, è in alcuni casi una necessità.
Il Fattore di Crescita Insulino-Simile 1 (IGF-1), chiamato anche Somatomedina C, è un ormone dalla struttura molecolare simile a quella dell’insulina che svolge un ruolo importante nella crescita infantile e ha effetti anabolici negli adulti. L’IGF-1 è costituito da 70 aminoacidi in una singola catena con tre ponti disolfuro intramolecolari.
L’IGF-1 è prodotto principalmente dal fegato. La produzione è stimolata dall’Ormone della Crescita (GH). La maggior parte dell’IGF-1 è legata a una delle 6 proteine di legame (IGF-BP). L’IGFBP-1 è regolato dall’Insulina. L’IGF-1 viene prodotto durante tutta la vita; i tassi più alti di produzione di IGF-1 si verificano durante la crescita puberale. I livelli più bassi si verificano nell’infanzia e nella vecchiaia.
L’IGF-1 lega e attiva il proprio recettore, l’IGF-1R, attraverso l’espressione sulla superficie cellulare delle tirosin-chinasi recettoriali (RTK) e segnala ulteriormente attraverso molteplici cascate di trasduzione intracellulare. L’IGF-1R è l’induttore che svolge un ruolo critico nella modulazione degli effetti metabolici dell’IGF-1 per la senescenza e la sopravvivenza cellulare. L’IGF-1 è responsabile di stimolare la crescita di tutti i tipi di cellule e di provocare effetti metabolici significativi. Un importante effetto metabolico dell’IGF-1 è la sua capacità di segnalare alle cellule che sono disponibili nutrienti sufficienti per l’ipertrofia e la divisione cellulare. Questi segnali consentono inoltre all’IGF-1 di inibire l’apoptosi cellulare e di aumentare la produzione di proteine cellulari. I recettori dell’IGF-1 sono ubiquitari, il che consente che i cambiamenti metabolici causati dall’IGF-1 si verifichino in tutti i tipi di cellule. Gli effetti metabolici dell’IGF-1 sono di vasta portata e possono coordinare il metabolismo delle proteine, dei carboidrati e dei grassi in una varietà di tipi di cellule diverse. La regolazione degli effetti metabolici dell’IGF-1 sui tessuti bersaglio è coordinata anche con altri ormoni, come l’Ormone della Crescita e l’Insulina.
L’IGF-1 da DNA ricombinante è disponibile principalmente in due diversi formati/varianti, lr3 e DES. È importante ricordare che, a prescindere dalla variante, tutti funzionano a livello sistemico nell’organismo e che, nonostante la somministrazione dell’ormone per via intramuscolare direttamente in un muscolo specifico, non genererà una crescita localizzata misurabile.
Ovviamente tralascerò di descrivere l’IGF-1 bioidentico commercializzato come Mecasermina dal momento che la sua farmacocinetica è identica a quella del IGF-1 endogeno. Dirò soltanto che mediamente viene utilizzato in dosi giornaliere nel range tra 60-1.000mcg post-workout. L’emivita di questa forma di IGF-1 è di circa 5.8h.
IGF-1 LR3: Questa forma è la variante di IGF-1 più comune e molto popolare sul mercato e utilizzata da bodybuilder e atleti di altre discipline. Contiene IGF-1 bioidentico costituito dalla catena originale di 70 aminoacidi, ma con 13 aminoacidi in più all’estremità N, per un totale di 83 aminoacidi. Possiede anche una seconda modifica, in cui un’Arginina si trova in 3a posizione invece dell’Acido Glutammico originale. Il risultato di queste modifiche è che l’IGF-1 continua a svolgere la sua attività originaria sul recettore dell’IGF-1 nei tessuti corporei e ha un’affinità di legame molto bassa per le proteine leganti l’IGF menzionate in precedenza. Inoltre, presenta una vita attiva significativamente più lunga, di circa 20-30 ore, rispetto a quella dell’IGF-1 di 12-15 ore. L’insieme di questi fattori ha dimostrato che l’LR3 ha un’efficacia circa tre volte superiore a quella dell’IGF-1.
I dosaggi medi utilizzati per questa forma sono nel range dei 40-80mcg/die. A causa della sua lunga vita attiva nell’organismo, la variante LR3 non dovrebbe essere somministrata più di una volta al giorno per il semplice fatto che non risulta necessario. Nei giorni di allenamento, il dosaggio di IGF-1 è solitamente somministrato subito dopo l’allenamento. La scelta è a discrezione dell’utilizzatore, in quanto può essere benissimo somministrato sia prima che dopo (solo prima dell’allenamento o solo dopo l’allenamento). E’ possibile comunque dividere il dosaggio giornaliero in due somministrazioni nell’arco della giornata, il dosaggio giornaliero completo può essere diviso quindi a metà tra i due (ad esempio, 20mcg prima dell’allenamento e 20mcg dopo l’allenamento, per un totale di 40mcg al giorno). Nei giorni di non allenamento, può essere somministrato in qualsiasi momento della giornata.
IGF-1 DES: Conosciuto anche come DES(1-3)IGF-1, questa è la forma di IGF-1 comunemente conosciuta come ad azione molto rapida e di solito è la meno preferita tra le due. Le sue modifiche rispetto alla molecola originale di IGF-1 sono tali da farle mancare i primi 3 aminoacidi all’N terminale, il che conferisce all’IGF-1 DES un totale di 67 aminoacidi nella sua catena rispetto ai 70 originali. Questa modifica garantisce all’IGF-1 DES una ridotta affinità di legame per le proteine leganti l’IGF menzionate in precedenza, oltre a una maggiore forza di legame e potenziale miotrofico, circa dieci volte superiore a quella dell’IGF-1 originale e cinque volte superiore a quella dell’IGF-1 LR3. A differenza dell’IGF-1 LR3, l’IGF-1 DES ha un’emivita molto più breve, di circa 20-30 minuti. Grazie alla sua attività più rapida e alla maggiore forza/potenza, la variante DES dell’IGF-1 è comunemente ritenuta in grado di ottenere una crescita muscolare localizzata nel sito in cui viene iniettata. Sebbene ciò sia in parte vero, gli studi hanno dimostrato che, come l’IGF-1 in generale, agisce a livello sistemico una volta raggiunti i capillari e il flusso sanguigno. Quindi l’effetto localizzato è minimo e non significativamente differente dall’effetto sistemico.
Il dosaggio della variante DES è leggermente più variabile rispetto a quello del LR3. Per l’IGF-1 DES, il dosaggio varia da 50 a 150 mcg al giorno. A causa della sua emivita molto più breve rispetto alla variante LR3, è possibile utilizzare dosaggi più elevati con una ipotetica riduzione del rischio di effetti a lungo termine sull’organismo, anche se è necessario usare comunque cautela. Può essere utilizzato nello stesso modo dell’IGF-1 LR3 post-workout, ed è infatti comunemente usato in questo modo a causa della sua breve emivita.
Entrambe le forme di IGF-1 possono essere somministrate per via intramuscolare o sottocutanea. L’uso di una delle due forme non deve superare la durata di 30 giorni prima di una pausa di almeno 2 settimane, anche se fare pause più lunghe di 2 settimane tra un ciclo di IGF-1 e l’altro è l’opzione migliore. Questo non solo per ridurre il rischio di effetti sulla salute a lungo termine, ma anche per garantire che i recettori dell’IGF-1 tornino ad un grado di sensibilità ottimale e, quindi, a “rispondere” correttamente dopo un ciclo.
L’insulina è un ormone peptidico prodotto dalle cellule beta delle isole pancreatiche. Regola il metabolismo dei carboidrati, dei grassi e delle proteine promuovendo l’assorbimento del glucosio dal sangue nelle cellule del fegato, dei grassi e dei muscoli scheletrici. In questi tessuti il glucosio assorbito viene convertito in glicogeno attraverso la glicogenesi o in grassi (trigliceridi) attraverso la lipogenesi o, nel caso del fegato, in entrambi. La produzione e la secrezione di glucosio da parte del fegato sono fortemente inibite da alte concentrazioni di Insulina nel sangue. L’Insulina circolante influisce anche sulla sintesi di proteine in un’ampia varietà di tessuti. È quindi un ormone anabolico e anticatabolico, che promuove la conversione di piccole molecole nel sangue in grandi molecole all’interno delle cellule. Bassi livelli di Insulina nel sangue hanno l’effetto opposto, favorendo un diffuso catabolismo, soprattutto del grasso corporeo di riserva.
La maggior parte dei bodybuilder utilizza una sola forma di Insulina (ad azione rapida o ultra-rapida), anche se alcuni utilizzano anche un’Insulina a lunga durata d’azione o in monoterapia insulinica o in conbinazione con le forme ad azione rapida o ultra-rapida.
L’Humalog® (Insulina Lispro) è senza dubbio la forma di Insulina più diffusa tra i bodybuilder insieme all’Humulin-R. L’Humalog è un analogo a breve durata d’azione dell’Insulina umana, in particolare l’analogo Lys(B28) Pro(B29) dell’Insulina che si crea quando gli aminoacidi in posizione 28 e 29 sono invertiti. È considerata equipotente all’Insulina solubile normale su base unitaria, ma con un’attività più rapida. L’inizio dell’azione del farmaco in seguito alla somministrazione sottocutanea è di circa 10-15 minuti e il suo picco d’effetto viene raggiunto in 30-90 minuti. La durata d’azione totale è compresa tra 3-5 ore. L’Insulina lispro viene solitamente utilizzata come supplemento a un prodotto a base di Insulina a più lunga durata d’azione, fornendo un farmaco ad azione rapida che può essere assunto prima o subito dopo i pasti per imitare la secrezione insulinica naturale dell’organismo. Molti atleti ritengono che la sua breve finestra d’effetto la renda un farmaco insulinico ideale per scopi dopanti, in quanto la maggior parte dell’azione può essere concentrata nel periodo successivo all’allenamento sfruttando l’assimilazione dei nutrienti durante la così detta “finestra anabolica”. Proprio al fine di potenziare la “finestra anabolica”, l’Humalog viene usata in concomitanza del GH il quale viene somministrato in una tempistica tale che i due picchi di rilascio (curva ematica massima) si “incrocino” andando a creare un affetto additivo di potenziamento della sintesi epatica di IGF-1 e della sua attività per via della riduzione dei trasportatori IGFBP.
Tuttavia, l’uso di una base insulinica composta da Insuline Glargine (Lantus), con una vita attiva di 24-26.5h, la quale sembra avere effetti di maggiore affinità di legame per il recettore del IGF-1 rispetto all’Insulina umana regolare o uno dei qualsiasi altri analoghi, viene da alcuni inserita nei protocolli Off-Season.
I dosaggi di Insulina non andrebbero calcolati in modo distaccato dal piano alimentare e dal suo contenuto glucidico. Se il margine di “sicurezza” indica un assunzione di 10-15g di Carboidrati per UI di Insulina, questi non dovrebbero essere addizionati al piano alimentare già tarata in surplus calorico. Il calcolo delle unità dovrebbe essere tarato sul quantitativo glucidico della dieta e sul rapporto con il peso corporeo dell’atleta. Facciamo un semplice esempio: Soggetto di 90Kg = formula 1UI ogni 10Kg di peso = 9UI massime somministrabili per pasto e in base alla vita attiva della forma utilizzata = assicurarsi che il pasto appena successivo alla somministrazione dell’Insulina a questo dosaggio sia pari o superiore ai 90g di Carboidrati. Il monitoraggio della glicemia attraverso un glucometro è ovviamente d’obbligo in un protocollo di Insulina.
Nota: tali informazioni esposte non rappresentano in nessun modo un parere medico ne tanto meno una prescrizione e/o incentivo all’uso di sostanze dopanti e illegali. Le descrizioni presentate per i PEDs solitamente più utilizzati in Off-Season sono sintetiche sia per motivi di “Off Topic” sia per ragioni legate alla loro descrizione approfondita in altri articoli presenti nel database di questo sito. In queste pubblicazioni potrete trovare informazioni inerenti anche agli affetti collaterali connessi ad un uso/abuso “off-label” dei diversi PEDs.
Conclusioni:
Per concludere e fare una sintesi delle nozioni esposte in questo articolo, dobbiamo ricordarci che i bodybuilder in Off-Season dovrebbero concentrarsi sul consumo di una dieta leggermente ipercalorica (~10-20% sopra le calorie di mantenimento) con l’obiettivo di guadagnare ~0,25-0,5% del peso corporeo a settimana per un “Natural”, mentre nel caso di un “Doped” la soglia può spostarsi tra l’1-2% con variabili connesse a risposte genetiche differenziali e anzianità nella carriera culturistica (principiante, intermedio e avanzato). In ogni caso, in una fetta maggioritaria di praticanti, ai bodybuilder avanzati si consiglia di essere più prudenti con il surplus calorico e il tasso di aumento di peso settimanale. L’assunzione di proteine nella dieta è raccomandata a 1,6-2,2 g/kg/giorno, con particolare attenzione a una quantità sufficiente di proteine a ogni pasto (0,40-0,55 g/kg/pasto) e a una distribuzione uniforme nell’arco della giornata (3-6 pasti). Per i “Doped”, in alcuni casi, l’introito proteico può essere portato, con minimi vantaggi in contesto ipercalorico, a 2,5g/Kg con le medesime linee guida di suddivisione per numero di pasti. I grassi alimentari devono essere consumati a livelli moderati, né troppo bassi né troppo alti (0,5-1,5 g/kg/die), per evitare un rapporto fTC sfavorevole e per prevenire riduzioni dei livelli di testosterone. Nei “Doped” l’obbiettivo con i lipidi è principalmente quello di assumerne una dose necessaria, e altamente qualitativa, al fine di assimilare vitamine liposolubili, per substrato strutturale, per sintesi di eicosanoidi (vedi assunzione EPA, DHA e AA), protezioni epidermide e capelli; di conseguenza attenersi ad un dosaggio medio pari a 35-50g/die. Dopo che le calorie sono state distribuite tra Proteine e Grassi, le restanti calorie dovrebbero provenire dai Carboidrati, assicurandosi di consumarne una quantità sufficiente (≥3-5 g/kg/giorno). Si possono ottenere benefici maggiori consumando proteine (0,40-0,55 g/kg/pasto) in prossimità delle sessioni di allenamento (1-2 ore prima dell’esercizio ed entro 1-2 ore dopo l’esercizio). È opportuno prendere in considerazione la Creatina Monoidrato (3-5 g/giorno) e la Caffeina (5-6 mg/kg), in quanto possono produrre effetti ergogenici per i bodybuilder. Inoltre, Beta-Alanina (3-5 g/die) e Citrullina Malato (8 g/die) sono integratori alimentari che possono essere presi in considerazione in quanto potenzialmente utili per i bodybuilder, a seconda dei regimi di allenamento individuali. I bodybuilder che non sono in grado di assumere un apporto sufficiente di micronutrienti e acidi grassi essenziali nella loro dieta dovrebbero prendere in considerazione l’integrazione di questi nutrienti per evitare carenze. Il limite principale di questo articolo è la mancanza di studi su larga scala e a lungo termine sui bodybuilder durante la Off-Season. Sono necessarie ulteriori ricerche su questa popolazione per ottimizzare la nutrizione e le raccomandazioni sugli integratori alimentari.
4. Philen R.M., Ortiz D.I., Auerbach S.B., Falk H. Survey of Advertising for Nutritional Supplements in Health and Bodybuilding Magazines. JAMA. 1992;268:1008. doi: 10.1001/jama.1992.03490080082029. [PubMed] [CrossRef] [Google Scholar]
5. Giampreti A., Lonati D., Locatelli C., Rocchi L., Campailla M.T. Acute neurotoxicity after yohimbine ingestion by a bodybuilder. [(accessed on 25 March 2019)];Clin. Toxicol. 2009 47:827–829. doi: 10.1080/15563650903081601. Available online: https://www.ncbi.nlm.nih.gov/pubmed/19640235 [PubMed] [CrossRef] [Google Scholar]
7. Della Guardia L., Cavallaro M., Cena H. The risks of self-made diets: The case of an amateur bodybuilder. J. Int. Soc. Sports Nutr. 2015;12:5. doi: 10.1186/s12970-015-0077-8. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
9. Hackett D.A., Johnson N.A., Chow C.-M. Training Practices and Ergogenic Aids Used by Male Bodybuilders. J. Strength Cond. Res. 2013;27:1609–1617. doi: 10.1519/JSC.0b013e318271272a. [PubMed] [CrossRef] [Google Scholar]
10. Forbes G.B., Brown M.R., Welle S.L., Lipinski B.A. Deliberate overfeeding in women and men: Energy cost and composition of the weight gain. Br. J. Nutr. 1986;56:1–9. doi: 10.1079/BJN19860080. [PubMed] [CrossRef] [Google Scholar]
11. Kreider R.B., Klesges R., Harmon K., Ramsey L., Bullen D., Wood L., Almada A., Grindstaff P., Li Y. Effects of Ingesting Supplements Designed to Promote Lean Tissue Accretion on Body Composition during Resistance Training. Int. J. Sport Nutr. 1996;6:234–246. doi: 10.1123/ijsn.6.3.234. [PubMed] [CrossRef] [Google Scholar]
12. Rozenek R., Ward P., Long S., Garhammer J. Effects of high-calorie supplements on body composition and muscular strength following resistance training. J. Sports Med. Phys. Fit. 2002;42:340–347. [PubMed] [Google Scholar]
13. Garthe I., Raastad T., Refsnes P.E., Sundgot-Borgen J. Effect of nutritional intervention on body composition and performance in elite athletes. Eur. J. Sport Sci. 2013;13:295–303. doi: 10.1080/17461391.2011.643923. [PubMed] [CrossRef] [Google Scholar]
14. American College og Sports Medicine American College of Sports Medicine position stand. Progression models in resistance training for healthy adults. [(accessed on 25 March 2019)];Med. Sci. Sport. Exerc. 2009 41:687–708. doi: 10.1249/MSS.0b013e3181915670. Available online: https://www.ncbi.nlm.nih.gov/pubmed/19204579 [PubMed] [CrossRef] [Google Scholar]
15. Lambert C.P., Frank L.L., Evans W.J., Lambert D.C.P. Macronutrient Considerations for the Sport of Bodybuilding. Sports Med. 2004;34:317–327. doi: 10.2165/00007256-200434050-00004. [PubMed] [CrossRef] [Google Scholar]
16. Walberg-Rankin J., Edmonds C.E., Gwazdauskas F.C. Diet and Weight Changes of Female Bodybuilders Before and After Competition. Int. J. Sport Nutr. 1993;3:87–102. doi: 10.1123/ijsn.3.1.87. [PubMed] [CrossRef] [Google Scholar]
17. Lamar-Hildebrand N., Saldanha L., Endres J. Dietary and exercise practices of college-aged female bodybuilders. J. Am. Diet. Assoc. 1989;89:1308–1310. [PubMed] [Google Scholar]
18. Houston M.E. Gaining Weight: The Scientific Basis of Increasing Skeletal Muscle Mass. Can. J. Appl. Physiol. 1999;24:305–316. doi: 10.1139/h99-024. [PubMed] [CrossRef] [Google Scholar]
19. Phillips S.M. A Brief Review of Critical Processes in Exercise-Induced Muscular Hypertrophy. Sports Med. 2014;44:71–77. doi: 10.1007/s40279-014-0152-3. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
20. Campbell B.I., Aguilar D., Conlin L., Vargas A., Schoenfeld B.J., Corson A., Gai C., Best S., Galvan E., Couvillion K. Effects of High Versus Low Protein Intake on Body Composition and Maximal Strength in Aspiring Female Physique Athletes Engaging in an 8-Week Resistance Training Program. Int. J. Sport Nutr. Exerc. Metab. 2018;28:580–585. doi: 10.1123/ijsnem.2017-0389. [PubMed] [CrossRef] [Google Scholar]
22. Morton R.W., Murphy K.T., McKellar S.E., Schoenfeld B.J., Henselmans M., Helms E., Aragon A.A., Devries M.C., Banfield L., Krieger J.W., et al. A systematic review, meta-analysis and meta-regression of the effect of protein supplementation on resistance training-induced gains in muscle mass and strength in healthy adults. [(accessed on 25 March 2019)];Br. J. Sports Med. 2018 52:376–384. doi: 10.1136/bjsports-2017-097608. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28698222 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
23. Houltham S.D., Rowlands D.S. A snapshot of nitrogen balance in endurance-trained women. Appl. Physiol. Nutr. Metab. 2014;39:219–225. doi: 10.1139/apnm-2013-0182. [PubMed] [CrossRef] [Google Scholar]
25. Antonio J., Ellerbroek A., Silver T., Vargas L., Peacock C. The effects of a high protein diet on indices of health and body composition—A crossover trial in resistance-trained men. J. Int. Soc. Sports Nutr. 2016;13:8. doi: 10.1186/s12970-016-0114-2. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
26. Bandegan A., Courtney-Martin G., Rafii M., Pencharz P.B., Lemon P.W. Indicator Amino Acid–Derived Estimate of Dietary Protein Requirement for Male Bodybuilders on a Nontraining Day Is Several-Fold Greater than the Current Recommended Dietary Allowance. J. Nutr. 2017;147:850–857. doi: 10.3945/jn.116.236331. [PubMed] [CrossRef] [Google Scholar]
27. Malowany J.M., West D.W.D., Williamson E., Volterman K.A., Sawan S.A., Mazzulla M., Moore D.R. Protein to Maximize Whole-Body Anabolism in Resistance-trained Females after Exercise. Med. Sci. Sports Exerc. 2019;51:798–804. doi: 10.1249/MSS.0000000000001832. [PubMed] [CrossRef] [Google Scholar]
28. Antonio J., Peacock C.A., Ellerbroek A., Fromhoff B., Silver T. The effects of consuming a high protein diet (4.4 g/kg/d) on body composition in resistance-trained individuals. J. Int. Soc. Sports Nutr. 2014;11:19. doi: 10.1186/1550-2783-11-19. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
29. Antonio J., Ellerbroek A., Silver T., Orris S., Scheiner M., Gonzalez A., Peacock C.A. A high protein diet (3.4 g/kg/d) combined with a heavy resistance training program improves body composition in healthy trained men and women—A follow-up investigation. J. Int. Soc. Sports Nutr. 2015;12:39. doi: 10.1186/s12970-015-0100-0. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
30. Bray G.A., Smith S.R., de Jonge L., Xie H., Rood J., Martin C.K., Most M., Brock C., Mancuso S., Redman L.M. Effect of dietary protein content on weight gain, energy expenditure, and body composition during overeating: A randomized controlled trial. [(accessed on 25 March 2019)];JAMA. 2012 307:47–55. doi: 10.1001/jama.2011.1918. Available online: https://www.ncbi.nlm.nih.gov/pubmed/22215165 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
31. Tipton K.D., Ferrando A.A., Phillips S.M., Doyle D., Wolfe R.R. Postexercise net protein synthesis in human muscle from orally administered amino acids. Am. J. Physiol. Metab. 1999;276:628–634. doi: 10.1152/ajpendo.1999.276.4.E628. [PubMed] [CrossRef] [Google Scholar]
32. Rieu I., Balage M., Sornet C., Giraudet C., Pujos E., Grizard J., Mosoni L., Dardevet D. Leucine supplementation improves muscle protein synthesis in elderly men independently of hyperaminoacidaemia. J. Physiol. 2006;575:305–315. doi: 10.1113/jphysiol.2006.110742. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
33. Burd N.A., Tang J.E., Moore D.R., Phillips S.M. Exercise training and protein metabolism: Influences of contraction, protein intake, and sex-based differences. [(accessed on 25 March 2019)];J. Appl. Physiol. 2008 106:1692–1701. doi: 10.1152/japplphysiol.91351.2008. Available online: https://www.ncbi.nlm.nih.gov/pubmed/19036897 [PubMed] [CrossRef] [Google Scholar]
34. Drummond M.J., Dreyer H.C., Fry C.S., Glynn E.L., Rasmussen B.B. Nutritional and contractile regulation of human skeletal muscle protein synthesis and mTORC1 signaling. J. Appl. Physiol. 2009;106:1374–1384. doi: 10.1152/japplphysiol.91397.2008. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
35. Tang J.E., Moore D.R., Kujbida G.W., Tarnopolsky M.A., Phillips S.M. Ingestion of whey hydrolysate, casein, or soy protein isolate: Effects on mixed muscle protein synthesis at rest and following resistance exercise in young men. J. Appl. Physiol. 2009;107:987–992. doi: 10.1152/japplphysiol.00076.2009. [PubMed] [CrossRef] [Google Scholar]
36. Kanda A., Nakayama K., Sanbongi C., Nagata M., Ikegami S., Itoh H. Effects of Whey, Caseinate, or Milk Protein Ingestion on Muscle Protein Synthesis after Exercise. Nutrients. 2016;8:339. doi: 10.3390/nu8060339. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
37. Messina M., Lynch H., Dickinson J.M., Reed K.E. No Difference Between the Effects of Supplementing With Soy Protein Versus Animal Protein on Gains in Muscle Mass and Strength in Response to Resistance Exercise. Int. J. Sport Nutr. Exerc. Metab. 2018;28:674–685. doi: 10.1123/ijsnem.2018-0071. [PubMed] [CrossRef] [Google Scholar]
38. Joy J.M., Lowery R.P., Wilson J.M., Purpura M., De Souza E.O., Mc Wilson S., Kalman D.S., Dudeck J.E., Jäger R. The effects of 8 weeks of whey or rice protein supplementation on body composition and exercise performance. Nutr. J. 2013;12:86. doi: 10.1186/1475-2891-12-86. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
39. Babault N., Paizis C., Deley G., Guérin-Deremaux L., Saniez M.-H., Lefranc-Millot C., Allaert F.A. Pea proteins oral supplementation promotes muscle thickness gains during resistance training: A double-blind, randomized, Placebo-controlled clinical trial vs. Whey protein. J. Int. Soc. Sports Nutr. 2015;12:1692. doi: 10.1186/s12970-014-0064-5. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
40. Tesch P.A. Glycogen and triglyceride utilization in relation to muscle metabolic characteristics in men performing heavy-resistance exercise. Graefe’s Arch. Clin. Exp. Ophthalmol. 1990;61:5–10. [PubMed] [Google Scholar]
41. Lane A.R., Duke J.W., Hackney A.C. Influence of dietary carbohydrate intake on the free testosterone: Cortisol ratio responses to short-term intensive exercise training. [(accessed on 25 March 2019)];Eur. J. Appl. Physiol. 2010 108:1125–1131. doi: 10.1007/s00421-009-1220-5. Available online: https://www.ncbi.nlm.nih.gov/pubmed/20091182 [PubMed] [CrossRef] [Google Scholar]
42. Tegelman R., Aberg T., Pousette A., Carlström K. Effects of a diet regimen on pituitary and steroid hormones in male ice hockey players. [(accessed on 25 March 2019)];Int. J. Sports Med. 1992 13:420–430. doi: 10.1055/s-2007-1021292. Available online: https://www.ncbi.nlm.nih.gov/pubmed/1387870 [PubMed] [CrossRef] [Google Scholar]
43. Dorgan J.F., Judd J.T., Longcope C., Brown C., Schatzkin A., Clevidence B.A., Campbell W.S., Nair P.P., Franz C., Kahle L., et al. Effects of dietary fat and fiber on plasma and urine androgens and estrogens in men: A controlled feeding study. Am. J. Clin. Nutr. 1996;64:850–855. doi: 10.1093/ajcn/64.6.850. [PubMed] [CrossRef] [Google Scholar]
44. Hämäläinen E., Adlercreutz H., Puska P., Pietinen P. Decrease of serum total and free testosterone during a low-fat high-fibre diet. J. Steroid Biochem. 1983;18:369–370. doi: 10.1016/0022-4731(83)90117-6. [PubMed] [CrossRef] [Google Scholar]
45. Hämäläinen E., Adlercreutz H., Puska P., Pietinen P. Diet and serum sex hormones in healthy men. J. Steroid Biochem. 1984;20:459–464. doi: 10.1016/0022-4731(84)90254-1. [PubMed] [CrossRef] [Google Scholar]
46. Wang C., Catlin D.H., Starcevic B., Heber D., Ambler C., Berman N., Lucas G., Leung A., Schramm K., Lee P.W.N., et al. Low-Fat High-Fiber Diet Decreased Serum and Urine Androgens in Men. J. Clin. Endocrinol. Metab. 2005;90:3550–3559. doi: 10.1210/jc.2004-1530. [PubMed] [CrossRef] [Google Scholar]
47. Morton R.W., Sato K., Gallaugher M.P.B., Oikawa S.Y., McNicholas P.D., Fujita S., Phillips S.M. Muscle Androgen Receptor Content but Not Systemic Hormones Is Associated With Resistance Training-Induced Skeletal Muscle Hypertrophy in Healthy, Young Men. Front. Physiol. 2018;9:9. doi: 10.3389/fphys.2018.01373. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
48. Tinsley G.M., Willoughby D.S. Fat-Free Mass Changes During Ketogenic Diets and the Potential Role of Resistance Training. Int. J. Sport Nutr. Exerc. Metab. 2016;26:78–92. doi: 10.1123/ijsnem.2015-0070. [PubMed] [CrossRef] [Google Scholar]
49. Vargas S., Romance R., Petro J.L., Bonilla D.A., Galancho I., Espinar S., Kreider R.B., Benítez-Porres J. Efficacy of ketogenic diet on body composition during resistance training in trained men: A randomized controlled trial. J. Int. Soc. Sports Nutr. 2018;15:31. doi: 10.1186/s12970-018-0236-9. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
50. Kephart W.C., Pledge C.D., Roberson P.A., Mumford P.W., Romero M.A., Mobley C.B., Martin J.S., Young K.C., Lowery R.P., Wilson J.M., et al. The Three-Month Effects of a Ketogenic Diet on Body Composition, Blood Parameters, and Performance Metrics in CrossFit Trainees: A Pilot Study. Sports. 2018;6:1. doi: 10.3390/sports6010001. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
51. Greene D.A., Varley B.J., Hartwig T.B., Chapman P., Rigney M. A Low-Carbohydrate Ketogenic Diet Reduces Body Mass Without Compromising Performance in Powerlifting and Olympic Weightlifting Athletes. [(accessed on 26 March 2019)];J. Strength Cond. Res. 2018 32:3373–3382. Available online: https://www.ncbi.nlm.nih.gov/pubmed/30335720 [PubMed] [Google Scholar]
52. Bird S. Strength Nutrition: Maximizing Your Anabolic Potential. Strength Cond. J. 2010;32:80–86. doi: 10.1519/SSC.0b013e3181d5284e. [CrossRef] [Google Scholar]
53. American Dietetic Association. Dietitians of Canada. American College of Sports Medicine. Rodriguez N.R., Di Marco N.M., Langley S. American College of Sports Medicine position stand. Nutrition and athletic performance. [(accessed on 26 March 2019)];Med. Sci. Sports Exerc. 2009 41:709–731. Available online: https://www.ncbi.nlm.nih.gov/pubmed/19225360 [PubMed] [Google Scholar]
54. Chung S.T., Chacko S.K., Sunehag A.L., Haymond M.W. Measurements of Gluconeogenesis and Glycogenolysis: A Methodological Review. Diabetes. 2015;64:3996–4010. doi: 10.2337/db15-0640. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
55. Azizi F. Effect of dietary composition on fasting-induced changes in serum thyroid hormones and thyrotropin. Metabolism. 1978;27:935–942. doi: 10.1016/0026-0495(78)90137-3. [PubMed] [CrossRef] [Google Scholar]
56. Mathieson R.A., Walberg J.L., Gwazdauskas F.C., Hinkle D.E., Gregg J.M. The effect of varying carbohydrate content of a very-low-caloric diet on resting metabolic rate and thyroid hormones. Metabolism. 1986;35:394–398. doi: 10.1016/0026-0495(86)90126-5. [PubMed] [CrossRef] [Google Scholar]
57. Leveritt M., Abernethy P.J. Effects of Carbohydrate Restriction on Strength Performance. J. Strength Cond. Res. 1999;13:52–57. [Google Scholar]
58. Jacobs I., Kaiser P., Tesch P. Muscle strength and fatigue after selective glycogen depletion in human skeletal muscle fibers. Graefe’s Arch. Clin. Exp. Ophthalmol. 1981;46:47–53. doi: 10.1007/BF00422176. [PubMed] [CrossRef] [Google Scholar]
59. Ray S., Sale D.G., Lee P., Garner S., MacDougall J.D., McCartney N. Muscle Substrate Utilization and Lactate Production During Weightlifting. Can. J. Appl. Physiol. 1999;24:209–215. [PubMed] [Google Scholar]
60. Tesch P.A., Colliander E.B., Kaiser P. Muscle metabolism during intense, heavy-resistance exercise. Graefe’s Arch. Clin. Exp. Ophthalmol. 1986;55:362–366. doi: 10.1007/BF00422734. [PubMed] [CrossRef] [Google Scholar]
63. Mitchell J.B., DiLauro P.C., Pizza F.X., Cavender D.L. The Effect of Preexercise Carbohydrate Status on Resistance Exercise Performance. Int. J. Sport Nutr. 1997;7:185–196. doi: 10.1123/ijsn.7.3.185. [PubMed] [CrossRef] [Google Scholar]
64. Lima-Silva A.E., Silva-Cavalcante M.D., Oliveira R.S., Kiss M.A., Pires F.O., Bertuzzi R., Bishop D. Effects of a low- or a high-carbohydrate diet on performance, energy system contribution, and metabolic responses during supramaximal exercise. Appl. Physiol. Nutr. Metab. 2013;38:928–934. doi: 10.1139/apnm-2012-0467. [PubMed] [CrossRef] [Google Scholar]
65. Vega F., Jackson R. Dietary habits of bodybuilders and other regular exercisers. Nutr. Res. 1996;16:3–10. doi: 10.1016/0271-5317(95)02054-3. [CrossRef] [Google Scholar]
66. Chappell A.J., Simper T., Barker M.E. Nutritional strategies of high level natural bodybuilders during competition preparation. J. Int. Soc. Sports Nutr. 2018;15:4. doi: 10.1186/s12970-018-0209-z. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
67. Atherton P.J., Etheridge T., Watt P.W., Wilkinson D., Selby A., Rankin D., Smith K., Rennie M.J. Muscle full effect after oral protein: Time-dependent concordance and discordance between human muscle protein synthesis and mTORC1 signaling. Am. J. Clin. Nutr. 2010;92:1080–1088. doi: 10.3945/ajcn.2010.29819. [PubMed] [CrossRef] [Google Scholar]
68. Res P.T., Groen B., Pennings B., Beelen M., Wallis G.A., Gijsen A.P., Senden J.M., Van Loon L.J. Protein ingestion before sleep improves postexercise overnight recovery. [(accessed on 25 March 2019)];Med. Sci. Sports Exerc. 2012 44:1560–1569. doi: 10.1249/MSS.0b013e31824cc363. Available online: https://www.ncbi.nlm.nih.gov/pubmed/22330017 [PubMed] [CrossRef] [Google Scholar]
69. Moore D.R., Robinson M.J., Fry J.L., Tang J.E., Glover E.I., Wilkinson S.B., Prior T., Tarnopolsky M.A., Phillips S.M. Ingested protein dose response of muscle and albumin protein synthesis after resistance exercise in young men. [(accessed on 25 March 2019)];Am. J. Clin. Nutr. 2009 89:161–168. doi: 10.3945/ajcn.2008.26401. Available online: https://www.ncbi.nlm.nih.gov/pubmed/19056590 [PubMed] [CrossRef] [Google Scholar]
70. Witard O.C., Jackman S.R., Breen L., Smith K., Selby A., Tipton K.D. Muscle protein synthesis rates subsequent to a meal in response to increasing doses of whey protein at rest and after. [(accessed on 25 March 2019)];Am. J. Clin. Nutr. 2014 99:86–95. doi: 10.3945/ajcn.112.055517. Available online: https://www.ncbi.nlm.nih.gov/pubmed/24257722 [PubMed] [CrossRef] [Google Scholar]
71. Macnaughton L.S., Wardle S.L., Witard O.C., McGlory C., Hamilton D.L., Jeromson S., Lawrence C.E., Wallis G.A., Tipton K.D. The response of muscle protein synthesis following whole-body resistance exercise is greater following 40 g than 20 g of ingested whey protein. Physiol. Rep. 2016;4:e12893. doi: 10.14814/phy2.12893. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
72. Schoenfeld B.J., Aragon A.A., Krieger J.W. The effect of protein timing on muscle strength and hypertrophy: A meta-analysis. J. Int. Soc. Sports Nutr. 2013;10:53. doi: 10.1186/1550-2783-10-53. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
73. Areta J.L., Burke L.M., Ross M.L., Camera D.M., West D.W.D., Broad E.M., Jeacocke N.A., Moore D.R., Stellingwerff T., Phillips S.M., et al. Timing and distribution of protein ingestion during prolonged recovery from resistance exercise alters myofibrillar protein synthesis. J. Physiol. 2013;591:2319–2331. doi: 10.1113/jphysiol.2012.244897. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
74. Hudson J.L., Bergia R.E., Campbell W.W. Effects of protein supplements consumed with meals, versus between meals, on resistance training–induced body composition changes in adults: A systematic review. Nutr. Rev. 2018;76:461–468. doi: 10.1093/nutrit/nuy012. [PubMed] [CrossRef] [Google Scholar]
75. Trommelen J., Kouw I.W.K., Holwerda A.M., Snijders T., Halson S.L., Rollo I., Verdijk L.B., Van Loon L.J.C. Pre-sleep dietary protein-derived amino acids are incorporated in myofibrillar protein during post-exercise overnight recovery. [(accessed on 25 March 2019)];Am. J. Physiol. Metab. 2018 1:457–467. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28536184 [Google Scholar]
76. Kouw I.W., Holwerda A.M., Trommelen J., Kramer I.F., Bastiaanse J., Halson S.L., Wodzig W.K., Verdijk L.B., Van Loon L.J. Protein Ingestion before Sleep Increases Overnight Muscle Protein Synthesis Rates in Healthy Older Men: A Randomized Controlled Trial. [(accessed on 25 March 2019)];J. Nutr. 2017 147:2252–2261. doi: 10.3945/jn.117.254532. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28855419 [PubMed] [CrossRef] [Google Scholar]
77. Snijders T., Res P.T., Smeets J.S., Van Vliet S., Van Kranenburg J., Maase K., Kies A.K., Verdijk L.B., Van Loon L.J. Protein ingestion before sleep increases muscle mass and strength gains during prolonged resistance-type exercise training in healthy young men. [(accessed on 25 March 2019)];J. Nutr. 2015 145:1178–1184. doi: 10.3945/jn.114.208371. Available online: https://www.ncbi.nlm.nih.gov/pubmed/25926415 [PubMed] [CrossRef] [Google Scholar]
78. Joy J.M., Vogel R.M., Broughton K.S., Kudla U., Kerr N.Y., Davison J.M., Wildman R.E.C., DiMarco N.M. Daytime and nighttime casein supplements similarly increase muscle size and strength in response to resistance training earlier in the day: A preliminary investigation. J. Int. Soc. Sports Nutr. 2018;15:24. doi: 10.1186/s12970-018-0228-9. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
79. Antonio J., Ellerbroek A., Peacock C., Silver T. Casein Protein Supplementation in Trained Men and Women: Morning versus Evening. Int. J. Exerc. Sci. 2017;10:479–486. [PMC free article] [PubMed] [Google Scholar]
80. Schoenfeld B.J., Aragon A.A. How much protein can the body use in a single meal for muscle-building? Implications for daily protein distribution. J. Int. Soc. Sports Nutr. 2018;15:10. doi: 10.1186/s12970-018-0215-1. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
81. Pennings B., Groen B.B., Van Dijk J.-W., De Lange A., Kiskini A., Kuklinski M., Senden J.M., Van Loon L.J. Minced beef is more rapidly digested and absorbed than beef steak, resulting in greater postprandial protein retention in older men. Am. J. Clin. Nutr. 2013;98:121–128. doi: 10.3945/ajcn.112.051201. [PubMed] [CrossRef] [Google Scholar]
82. Kim I.Y., Schutzler S., Schrader A., Spencer H.J., Azhar G., Ferrando A.A., Wolfe R.R. The anabolic response to a meal containing different amounts of protein is not limited by the maximal stimulation of protein synthesis in healthy young adults. [(accessed on 25 March 2019)];Am. J. Physiol. Metab. 2016 310:73–80. doi: 10.1152/ajpendo.00365.2015. Available online: https://www.ncbi.nlm.nih.gov/pubmed/26530155 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
83. Jentjens R., Jeukendrup A.E. Determinants of Post-Exercise Glycogen Synthesis During Short-Term Recovery. Sports Med. 2003;33:117–144. doi: 10.2165/00007256-200333020-00004. [PubMed] [CrossRef] [Google Scholar]
84. Biolo G., Williams B.D., Fleming R.Y., Wolfe R.R. Insulin action on muscle protein kinetics and amino acid transport during recovery after resistance exercise. Diabetes. 1999;48:949–957. doi: 10.2337/diabetes.48.5.949. [PubMed] [CrossRef] [Google Scholar]
85. Greenhaff P.L., Karagounis L.G., Peirce N., Simpson E.J., Hazell M., Layfield R., Wackerhage H., Smith K., Atherton P., Selby A., et al. Disassociation between the effects of amino acids and insulin on signaling, ubiquitin ligases, and protein turnover in human muscle. Am. J. Physiol. Metab. 2008;295:E595–E604. doi: 10.1152/ajpendo.90411.2008. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
86. Glynn E.L., Fry C.S., Timmerman K.L., Drummond M.J., Volpi E., Rasmussen B.B., Leroy J.L., Gadsden P., De Cossío T.G., Gertler P. Addition of Carbohydrate or Alanine to an Essential Amino Acid Mixture Does Not Enhance Human Skeletal Muscle Protein Anabolism123. J. Nutr. 2013;143:307–314. doi: 10.3945/jn.112.168203. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
87. Koopman R., Beelen M., Stellingwerff T., Pennings B., Saris W.H.M., Kies A.K., Kuipers H., Van Loon L.J.C. Coingestion of carbohydrate with protein does not further augment postexercise muscle protein synthesis. Am. J. Physiol. Metab. 2007;293:E833–E842. doi: 10.1152/ajpendo.00135.2007. [PubMed] [CrossRef] [Google Scholar]
88. Aragon A.A., Schoenfeld B.J. Nutrient timing revisited: Is there a post-exercise anabolic window? J. Int. Soc. Sports Nutr. 2013;10:5. doi: 10.1186/1550-2783-10-5. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
89. Jäger R., Kerksick C.M., Campbell B.I., Cribb P.J., Wells S.D., Skwiat T.M., Purpura M., Ziegenfuss T.N., Ferrando A.A., Arent S.M., et al. International Society of Sports Nutrition position stand: Protein and exercise. [(accessed on 25 March 2019)];J. Int. Soc. Sport. Nutr. 2017 4:20. doi: 10.1186/s12970-017-0177-8. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28642676 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
90. Darrabie M.D., Arciniegas A.J.L., Mishra R., Bowles D.E., Jacobs D.O., Santacruz L. AMPK and substrate availability regulate creatine transport in cultured cardiomyocytes. Am. J. Physiol. Metab. 2011;300:870–876. doi: 10.1152/ajpendo.00554.2010. [PubMed] [CrossRef] [Google Scholar]
91. Purchas R., Busboom J., Wilkinson B. Changes in the forms of iron and in concentrations of taurine, carnosine, coenzyme Q10, and creatine in beef longissimus muscle with cooking and simulated stomach and duodenal digestion. Meat Sci. 2006;74:443–449. doi: 10.1016/j.meatsci.2006.03.015. [PubMed] [CrossRef] [Google Scholar]
92. Branch J.D. Effect of Creatine Supplementation on Body Composition and Performance: A Meta-analysis. Int. J. Sport Nutr. Exerc. Metab. 2003;13:198–226. doi: 10.1123/ijsnem.13.2.198. [PubMed] [CrossRef] [Google Scholar]
93. Hultman E., Söderlund K., Timmons J.A., Cederblad G., Greenhaff P.L. Muscle creatine loading in men. [(accessed on 25 March 2019)];J. Appl. Physiol. Soc. 1996 81:232–237. doi: 10.1152/jappl.1996.81.1.232. Available online: https://www.ncbi.nlm.nih.gov/pubmed/8828669 [PubMed] [CrossRef] [Google Scholar]
94. Jagim A.R., Oliver J.M., Sanchez A., Galvan E., Fluckey J., Riechman S., Greenwood M., Kelly K., Meininger C., Rasmussen C., et al. A buffered form of creatine does not promote greater changes in muscle creatine content, body composition, or training adaptations than creatine monohydrate. J. Int. Soc. Sports Nutr. 2012;9:43. doi: 10.1186/1550-2783-9-43. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
95. Spillane M., Schoch R., Cooke M., Harvey T., Greenwood M., Kreider R., Willoughby D.S., Cooke M. The effects of creatine ethyl ester supplementation combined with heavy resistance training on body composition, muscle performance, and serum and muscle creatine levels. J. Int. Soc. Sports Nutr. 2009;6:6. doi: 10.1186/1550-2783-6-6. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
96. Childs E., De Wit H., Wit H. Subjective, behavioral, and physiological effects of acute caffeine in light, nondependent caffeine users. Psychopharmacology. 2006;185:514–523. doi: 10.1007/s00213-006-0341-3. [PubMed] [CrossRef] [Google Scholar]
97. Bellar D., Kamimori G.H., Glickman E.L. The Effects of Low-Dose Caffeine on Perceived Pain During a Grip to Exhaustion Task. J. Strength Cond. Res. 2011;25:1225–1228. doi: 10.1519/JSC.0b013e3181d9901f. [PubMed] [CrossRef] [Google Scholar]
98. Davis J.K., Green J.M. Caffeine and anaerobic performance: Ergogenic value and mechanisms of action. [(accessed on 25 March 2019)];Sport. Med. 2009 39:813–832. doi: 10.2165/11317770-000000000-00000. Available online: https://www.ncbi.nlm.nih.gov/pubmed/19757860 [PubMed] [CrossRef] [Google Scholar]
99. Wickwire P.J., McLester J.R., Gendle S., Hudson G., Pritchett R.C., Laurent C.M., Green J.M. Effects of Caffeine on Repetitions to Failure and Ratings of Perceived Exertion during Resistance Training. Int. J. Sports Physiol. Perform. 2007;2:250–259. [PubMed] [Google Scholar]
100. Duncan M.J., Oxford S.W. The effect of caffeine ingestion on mood state and bench press performance to failure. [(accessed on 25 March 2019)];J. Strength Cond. Res. 2001 25:178–185. doi: 10.1519/JSC.0b013e318201bddb. Available online: https://www.ncbi.nlm.nih.gov/pubmed/22124354 [PubMed] [CrossRef] [Google Scholar]
101. Williams A.D., Cribb P.J., Cooke M.B., Hayes A. The Effect of Ephedra and Caffeine on Maximal Strength and Power in Resistance-Trained Athletes. J. Strength Cond. Res. 2008;22:464–470. doi: 10.1519/JSC.0b013e3181660320. [PubMed] [CrossRef] [Google Scholar]
102. Tarnopolsky M.A., Atkinson S.A., MacDougall J.D., Sale D.G., Sutton J.R. Physiological responses to caffeine during endurance running in habitual caffeine users. Med. Sci. Sports Exerc. 1989;21:418–424. doi: 10.1249/00005768-198908000-00013. [PubMed] [CrossRef] [Google Scholar]
103. Blanchard J., Sawers S.J.A. The absolute bioavailability of caffeine in man. Eur. J. Clin. Pharmacol. 1983;24:93–98. doi: 10.1007/BF00613933. [PubMed] [CrossRef] [Google Scholar]
104. Hobson R.M., Saunders B., Ball G., Harris R.C., Sale C. Effects of β-alanine supplementation on exercise performance: A meta-analysis. Amino Acids. 2012;43:25–37. doi: 10.1007/s00726-011-1200-z. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
105. Hoffman J., Ratamess N.A., Ross R., Kang J., Magrelli J., Neese K., Faigenbaum A.D., Wise J.A. Beta-alanine and the hormonal response to exercise. [(accessed on 25 March 2019)];Int. J. Sports Med. 2008 29:952–958. doi: 10.1055/s-2008-1038678. Available online: https://www.ncbi.nlm.nih.gov/pubmed/18548362 [PubMed] [CrossRef] [Google Scholar]
106. Hoffman J., Ratamess N., Kang J., Mangine G., Faigenbaum A., Stout J. Effect of creatine and β-alanine supplementation on performance and endocrine responses in strength/power athletes. [(accessed on 25 March 2019)];Int. J. Sport Nutr. Exerc. Metab. 2006 16:430–446. doi: 10.1123/ijsnem.16.4.430. Available online: https://www.ncbi.nlm.nih.gov/pubmed/17136944 [PubMed] [CrossRef] [Google Scholar]
108. Wax B., Kavazis A.N., Weldon K., Sperlak J. Effects of Supplemental Citrulline Malate Ingestion During Repeated Bouts of Lower-Body Exercise in Advanced Weightlifters. J. Strength Cond. Res. 2015;29:786–792. doi: 10.1519/JSC.0000000000000670. [PubMed] [CrossRef] [Google Scholar]
109. Wax B., Kavazis A.N., Luckett W. Effects of Supplemental Citrulline-Malate Ingestion on Blood Lactate, Cardiovascular Dynamics and Resistance Exercise Performance in Trained Males. [(accessed on 25 March 2019)];J. Diet. 2016 13:269–282. doi: 10.3109/19390211.2015.1008615. Available online: https://www.ncbi.nlm.nih.gov/pubmed/25674699 [PubMed] [CrossRef] [Google Scholar]
110. Glenn J.M., Gray M., Wethington L.N., Stone M.S., Stewart R.W., Jr., Moyen N.E. Acute citrulline malate supplementation improves upper- and lower-body submaximal weightlifting exercise performance in resistance-trained females. [(accessed on 25 March 2019)];Eur. J. Nutr. 2017 56:775–784. doi: 10.1007/s00394-015-1124-6. Available online: https://www.ncbi.nlm.nih.gov/pubmed/26658899 [PubMed] [CrossRef] [Google Scholar]
111. Glenn J.M., Gray M., Jensen A., Stone M.S., Vincenzo J.L. Acute citrulline-malate supplementation improves maximal strength and anaerobic power in female, masters athletes tennis players. Eur. J. Sport Sci. 2016;16:1–9. doi: 10.1080/17461391.2016.1158321. [PubMed] [CrossRef] [Google Scholar]
112. Gonzalez A.M., Spitz R.W., Ghigiarelli J.J., Sell K.M., Mangine G.T. Acute Effect of Citrulline Malate Supplementation on Upper-Body Resistance Exercise Performance in Recreationally Resistance-Trained Men. J. Strength Cond. Res. 2018;32:3088–3094. doi: 10.1519/JSC.0000000000002373. [PubMed] [CrossRef] [Google Scholar]
113. Farney T.M., Bliss M.V., Hearon C.M., Salazar D.A. The Effect of Citrulline Malate Supplementation On Muscle Fatigue Among Healthy Participants. J. Strength Cond. Res. 2017:1. doi: 10.1519/JSC.0000000000002356. [PubMed] [CrossRef] [Google Scholar]
114. Trexler E.T., Persky A.M., Ryan E.D., Schwartz T.A., Stoner L., Smith-Ryan A.E. Acute Effects of Citrulline Supplementation on High-Intensity Strength and Power Performance: A Systematic Review and Meta-Analysis. Sports Med. 2019;49:707–718. doi: 10.1007/s40279-019-01091-z. [PubMed] [CrossRef] [Google Scholar]
115. Kleiner S.M., Bazzarre T.L., Litchford M.D. Metabolic profiles, diet, and health practices of championship male and female bodybuilders. J. Am. Diet. Assoc. 1990;90:962–967. [PubMed] [Google Scholar]
116. Kleiner S.M., Bazzarre T.L., Ainsworth B.E. Nutritional Status of Nationally Ranked Elite Bodybuilders. Int. J. Sport Nutr. 1994;4:54–69. doi: 10.1123/ijsn.4.1.54. [PubMed] [CrossRef] [Google Scholar]
117. Sandoval W.M., Heyward V.H. Food Selection Patterns of Bodybuilders. Int. J. Sport Nutr. 1991;1:61–68. doi: 10.1123/ijsn.1.1.61. [PubMed] [CrossRef] [Google Scholar]
118. Ismaeel A., Weems S., Willoughby D.S. A Comparison of the Nutrient Intakes of Macronutrient-Based Dieting and Strict Dieting Bodybuilders. Int. J. Sport Nutr. Exerc. Metab. 2018;28:502–508. doi: 10.1123/ijsnem.2017-0323. [PubMed] [CrossRef] [Google Scholar]
119. Nelson J.R., Raskin S. The eicosapentaenoic acid:arachidonic acid ratio and its clinical utility in cardiovascular disease. Postgrad. Med. 2019;131:268–277. doi: 10.1080/00325481.2019.1607414. [PubMed] [CrossRef] [Google Scholar]
120. Harris W.S. The Omega-6: Omega-3 ratio: A critical appraisal and possible successor. [(accessed on 15 June 2019)];Prostaglandins Leukot Essent Fatty Acids. 2018 132:34–40. doi: 10.1016/j.plefa.2018.03.003. Available online: https://www.ncbi.nlm.nih.gov/m/pubmed/29599053/ [PubMed] [CrossRef] [Google Scholar]
121. Tachtsis B., Camera D., Lacham-Kaplan O. Potential Roles of n-3 PUFAs during Skeletal Muscle Growth and Regeneration. Nutrients. 2018;10:309. doi: 10.3390/nu10030309. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
122. Di Girolamo F.G., Situlin R., Mazzucco S., Valentini R., Toigo G., Biolo G. Omega-3 fatty acids and protein metabolism: Enhancement of anabolic interventions for sarcopenia. [(accessed on 15 June 2019)];Curr. Opin. Clin. Nutr. Metab Care. 2014 17:145–150. doi: 10.1097/MCO.0000000000000032. Available online: https://www.ncbi.nlm.nih.gov/pubmed/24500439 [PubMed] [CrossRef] [Google Scholar]
123. McGlory C., Wardle S.L., Macnaughton L.S., Witard O.C., Scott F., Dick J., Bell J.G., Phillips S.M., Galloway S.D.R., Hamilton D.L., et al. Fish oil supplementation suppresses resistance exercise and feeding-induced increases in anabolic signaling without affecting myofibrillar protein synthesis in young men. Physiol. Rep. 2016;4:e12715. doi: 10.14814/phy2.12715. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
124. Crestani D.M., Bonin E.F.R., Barbieri R.A., Zagatto A.M., Higino W.P., Milion F. Chronic supplementation of omega-3 can improve body composition and maximal strength, but does not change the resistance to neuromuscular fatigue. [(accessed on 15 June 2019)];Sport Sci. Health. 2017 13:259–265. doi: 10.1007/s11332-016-0322-9. Available online: https://link.springer.com/article/10.1007/s11332-016-0322-9 [CrossRef] [Google Scholar]
125. Lewis E.J.H., Radonic P.W., Wolever T.M.S., Wells G.D. 21 days of mammalian omega-3 fatty acid supplementation improves aspects of neuromuscular function and performance in male athletes compared to olive oil placebo. J. Int. Soc. Sports Nutr. 2015;12:28. doi: 10.1186/s12970-015-0089-4. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
126. Rossato L.T., Schoenfeld B.J., De Oliveira E.P. Is there sufficient evidence to supplement omega-3 fatty acids to increase muscle mass and strength in young and older adults? Clin. Nutr. 2019 doi: 10.1016/j.clnu.2019.01.001. [PubMed] [CrossRef] [Google Scholar]
127. Mocking R.J.T., Harmsen I., Assies J., Koeter M.W.J., Ruhé H.G., Schene A.H. Meta-analysis and meta-regression of omega-3 polyunsaturated fatty acid supplementation for major depressive disorder. Transl. Psychiatry. 2016;6:e756. doi: 10.1038/tp.2016.29. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
128. Maki K.C., Palacios O.M., Bell M., Toth P.P. Use of supplemental long-chain omega-3 fatty acids and risk for cardiac death: An updated meta-analysis and review of research gaps. J. Clin. Lipidol. 2017;11:1152–1160.e2. doi: 10.1016/j.jacl.2017.07.010. [PubMed] [CrossRef] [Google Scholar]
129. Miller P.E., Van Elswyk M., Alexander D.D. Long-Chain Omega-3 Fatty Acids Eicosapentaenoic Acid and Docosahexaenoic Acid and Blood Pressure: A Meta-Analysis of Randomized Controlled Trials. Am. J. Hypertens. 2014;27:885–896. doi: 10.1093/ajh/hpu024. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
130. Du S., Jin J., Fang W., Su Q. Does Fish Oil Have an Anti-Obesity Effect in Overweight/Obese Adults? A Meta-Analysis of Randomized Controlled Trials. PLoS ONE. 2015;10:e0142652. doi: 10.1371/journal.pone.0142652. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
La capacità di riacquisire la condizione della massa muscolare precedente a un periodo di deallenamento o inattività fisica è noto come “memoria muscolare”. Quindi, se un soggetto ha avuto una condizione muscolare ottimale (vedi muscoli più ipertrofici) in passato, ciò lo aiuterà a riportarli nuovamente nelle precedenti condizioni una volta ripreso un regolare stimolo allenante. Il concetto di memoria muscolare si basa in buona parte su qualcosa chiamato permanenza mio-nucleare. Il ‘mio’ in ‘mionucleare’ si riferisce al ‘muscolo’ e il ‘nucleare’ si riferisce alla parola ‘nucleo’: un organello della cellula. Prima di esplorare ulteriormente il concetto di memoria muscolare, e come gli AAS si leghino a questo, cerchiamo prima di rispolverare un po’ di concetti utili sui nuclei muscolari o mionuclei.
Informazioni di base sui nuclei muscolari/mionuclei:
Le cellule muscolo-scheletriche sono le singole cellule contrattili all’interno di un muscolo e sono spesso definite fibre muscolari.[1] Un singolo muscolo come il bicipite in un giovane individuo di sesso maschile adulto contiene circa 253.000 fibre muscolari.[2]
Sezione 3D di una fibra del muscolo-scheletrico
Le fibre muscolo-scheletriche sono le uniche cellule muscolari multinucleate con i nuclei spesso indicati come mionuclei . Ciò si verifica durante la miogenesi con la fusione di mioblasti, ciascuno dei quali contribuisce a un nucleo.[3] La fusione dipende da proteine muscolo-specifiche note come fusogeni chiamate myomaker e myomerger .[4]
Molti nuclei sono necessari alla cellula muscolo-scheletrica per le grandi quantità di proteine ed enzimi necessari per essere prodotti per il normale funzionamento della cellula. Una singola fibra muscolare può contenere da centinaia a migliaia di nuclei.[5] Una fibra muscolare ad esempio nel bicipite umano con una lunghezza di 10cm può avere fino a 3000 nuclei.[5] A differenza di una cellula non muscolare in cui il nucleo è posizionato centralmente, il mionucleo è allungato e si trova vicino al sarcolemma . I mionuclei sono disposti in modo abbastanza uniforme lungo la fibra con ciascun nucleo che ha il proprio dominio mionucleare dove è responsabile del supporto del volume del citoplasma in quella particolare sezione della miofibra.[4,5]
Un gruppo di cellule staminali muscolari conosciute come cellule miosatelliti, anche cellule satelliti che si trovano tra la membrana basale e il sarcolemma delle fibre muscolari, sono normalmente quiescenti ma possono essere attivate dall’esercizio o anche condizioni patologiche per fornire mionuclei aggiuntivi per la crescita o la riparazione muscolare.[6]
Detto più semplicemente, i muscoli sono costituiti da un insieme di fibre muscolari. Ogni fibra muscolare, o cellula muscolare, contiene più nuclei, l’organello di una cellula che contiene il DNA ed è il luogo dove avviene il processo di trascrizione dei geni. La maggior parte degli altri tipi di cellule umane contiene solo un nucleo, o in alcuni casi addirittura nessun nucleo (globuli rossi/Eritrociti). Per dare un’idea di quanti nuclei si stia parlando: le fibre muscolari di ratto contengono da 44 a 116 nuclei per millimetro di lunghezza della fibra, con le fibre muscolari di tipo 1 che contengono più nuclei per millimetro delle fibre muscolari di tipo 2.[7] Il numero sembra più basso negli esseri umani, come riportato da un ricercatore il quale segnala la presenza di circa 30 nuclei per millimetro di lunghezza della fibra nel muscolo del bicipite brachiale.[8] Come tali, le fibre muscolari possono contenere migliaia di mionuclei, dato che possono estendersi per diversi centimetri di lunghezza.
Poiché i nuclei cellulari delle fibre muscolari non sono in grado di dividersi (cioè sono differenziati terminalmente), le fibre muscolari dipendono dalle cellule satelliti circostanti per l’aggiunta di nuovi nuclei. Essenzialmente, le cellule satelliti sono cellule staminali delle fibre muscolari che si trovano schiacciate tra il sarcolemma (la membrana cellulare di una fibra muscolare) e la lamina basale (uno strato di matrice extracellulare che è avvolto intorno al sarcolemma). Sono stati scoperti e descritti per la prima volta da Alexander Mauro nella letteratura scientifica nel 1961.[9] Usando un microscopio elettronico, egli vide delle cellule “incastrate” tra il sarcolemma delle fibre muscolari di rana e la lamina basale. Le descrisse aventi una scarsità di citoplasma, con il nucleo che costituisce quasi l’intero volume della cellula satellite. Ha continuato a speculare sull’origine e sul ruolo delle cellule satelliti, toccando brevemente l’idea che potrebbero essere coinvolte nella risposta al trauma inflitto a una fibra muscolare. Cosa che, in effetti, sono.[10]
L’ipotesi del dominio mionucleare e la permanenza mionucleare
La scoperta delle cellule satelliti e il loro ruolo nella rigenerazione muscolare fanno sorgere la domanda sulla misura in cui le cellule satelliti sono coinvolte nell’ipertrofia. Un’ipotesi chiamata “ipotesi del dominio mionucleare” si è agganciata a questo quesito. Essa postula che un mionucleo controlla una quantità limitata di citoplasma, e quindi, affinché la crescita muscolare abbia luogo, i mionuclei devono essere aggiunti alla fibra muscolare per sostenerla. Tre osservazioni chiave hanno sostenuto questa ipotesi, vale a dire:
L’esposizione alle radiazioni γ rende le cellule satellite incapaci di dividersi e inibisce fortemente l’ipertrofia da sovraccarico nei modelli animali, mantenendo intatto il metabolismo cellulare o la sintesi proteica [11].
I prodotti (organelli, membrane e proteine strutturali) derivati da un nucleo rimangono localizzati nelle sue vicinanze [12].
Il rapporto citoplasma/mionucleo rimane abbastanza costante [13].
Questo implicherebbe un aumento del numero di mionuclei con la crescita di una fibra muscolare (ipertrofia), mentre diminuirebbe con una perdita di dimensioni della stessa (atrofia). Tuttavia, vari studi su animali suggeriscono che i mionuclei non si perdono durante l’atrofia.[14] Così è nato il paradigma della permanenza mionucleare: una volta che i mionuclei sono guadagnati con l’ipertrofia, non vengono persi di nuovo con il deallenamento. Questo potrebbe potenzialmente permettere alle fibre muscolari di ricrescere in modo più efficiente durante il successivo riallenamento e quindi servire come un meccanismo di “memoria muscolare”.
Il concetto di memoria muscolare basato sulla permanenza mionucleare illustrato da Bruusgaard et al.
AAS e permanenza mionucleare:
E gli AAS? Ciò che è chiaro è che l’uso di AAS aumenta il numero di mionuclei. Dosaggi crescenti di Testosterone Enantato portano ad un aumento del numero di mionuclei per mm di fibra muscolare.[15] Questo effetto non è poi così sorprendente: si osserva semplicemente questo effetto con praticamente tutte le modalità di induzione ipertrofica.
Ma che dire della loro permanenza? Questi mionuclei permangono una volta che la massa muscolare diminuisce di nuovo? In un esperimento su animali, da me già riportato anni fa, topi femmina sono stati trattati con Testosterone Propionato per 2 settimane, che ha portato a un aumento del 66% del numero di mionuclei e un aumento del 77% della fibra muscolare CSA [16]. La massa muscolare è tornata alla normalità dopo la successiva interruzione della somministrazione di Testosterone, ma il numero di mionuclei è rimasto elevato per almeno 3 mesi. 3 mesi potrebbe non sembrare molto, ma sulla scala temporale di un topo lo sono: i topi che hanno usato per lo studio vivono per circa 2 anni. Comunque, dopo questi 3 mesi, quando i topi sono stati sottoposti a sovraccarico per induzione ipertrofica, la CSA delle fibre muscolari è aumentata del 30% dopo 6 giorni, mentre quella dei topi di controllo non è aumentata significativamente. Dopo questo, la massa muscolare è aumentata in parallelo tra entrambi i gruppi, ma la CSA era ancora più alta del 20% nel gruppo che era stato precedentemente trattato con Testosterone dopo 14 giorni. Anche se questo non prova un nesso causale tra il numero più alto di mionuclei e l’ipertrofia, è comunque un’osservazione interessante.
Si noti come il gruppo che è stato trattato con Testosterone per 2 settimane, circa 3 mesi prima ha mostrato un forte aumento della massa muscolare rapidamente ottenuto in risposta al sovraccarico.
E negli esseri umani? Due studi hanno valutato questo e sono stati portati all’attenzione da Alexander Kolliari-Turner, uno studente con dottorato di ricerca presso la School of Sport and Health Sciences of the University of Brighton nel Regno Unito. Una è una tesi di master e l’altra è una tesi di dottorato.
Nella tesi di dottorato di Anders Eriksson [17], sono stati reclutati quattro gruppi di soggetti. Un gruppo di soggetti sedentari che fungeva da controllo (gruppo C), un gruppo di PowerLifter natural (gruppo P), un gruppo di powerlifter che usano AAS (gruppo PAS), e un gruppo di PowerLifter che hanno precedentemente usato AAS (gruppo PREV). I mionuclei per fibra muscolare sono stati determinati nei muscoli vasto laterale e trapezio. Il gruppo PREV aveva interrotto l’uso di AAS da almeno un anno (con una media di 8 anni). Infatti, l’area delle fibre muscolari misurata nel gruppo PREV era paragonabile a quella del gruppo P, e notevolmente più piccola di quella del gruppo PAS.
La distribuzione del dominio nucleare (nr. di nuclei per fibra diviso per l’area della fibra) per gruppo si trova nell’immagine qui sotto. Se ci fosse una permanenza dei mioonuclei, ci si aspetterebbe un dominio nucleare più piccolo, cioè più nuclei rispetto all’area delle fibre, nel gruppo PREV rispetto agli altri gruppi.
Chiaramente questo non è il caso del vasto laterale, ma è il caso del trapezio. È difficile dire cosa causa questa apparente discrepanza tra i due muscoli. O qualche proprietà che differisce tra i due muscoli, o il suo modo di utilizzo dopo la cessazione dell’uso di AAS, forse ha portato a apparente permanenza mionucleare nel muscolo trapezio.
Va notato, tuttavia, che questo era uno studio trasversale con un piccolo numero di soggetti (32 in totale). L’ideale sarebbe avere uno studio prospettico che valuti questo, anche se ciò è estremamente difficile su lunghi periodi di tempo, in quanto potrebbe richiedere almeno un anno o più prima che i cambiamenti diventino evidenti. In alternativa, anche uno studio trasversale con un gruppo di soggetti più grande sarebbe piuttosto interessante. Indipendentemente da ciò, questo presta una certa credibilità alla permanenza dei mionuclei negli esseri umani come risultato dell’uso di steroidi anabolizzanti in muscoli selezionati.
In una tesi di laurea di Lindholm et al. sono stati reclutati tre gruppi di soggetti: attuali consumatori di AAS (gruppo CAS), ex consumatori di AAS (gruppo FAS) e controllo allenati alla resistenza (gruppo CON) [18]. Gli ex consumatori di AAS avevano smesso di usarli per una media di 6,5 anni. In questo studio, sono state prese solo biopsie del muscolo vasto laterale. In particolare, non c’erano differenze significative nella CSA delle fibre muscolari tra i tre gruppi. Questo è senza dubbio il risultato delle dimensioni relativamente piccole del gruppo (34 soggetti in totale; un errore di tipo 2).
Una piccola, ma significativa, differenza nel dominio mio-nucleare è stata trovata tra le fibre muscolari di tipo 2 del gruppo FAS rispetto al gruppo CON, come si può vedere nella figura sottostante:
Questo suggerisce una permanenza mionucleare? Forse. La differenza era piccola e può essere facilmente spiegata anche dalla natura trasversale dello studio (e non c’era alcuna differenza rispetto agli attuali utilizzatori di AAS).
Le prove finora sono scarse. In ogni caso, quando si guarda alla permanenza mionucleare in generale, l’evidenza generale indica che questa regge a breve termine, ma mancano prove per il lungo termine [19]. Inoltre, non è chiaro se la permanenza mionucleare possa aiutare o meno il ritorno alla condizione muscolo-scheletrica precedente. E visti i dati di cui sopra, il dibattito sul fatto che l’uso di AAS porti o meno alla manifestazione della memoria muscolare come risultato della permanenza mionucleare, è tutt’altro che risolto.
Conclusione:
Come osservazione conclusiva: c’è anche un concetto di memoria muscolare basato su qualcosa di diverso dalla permanenza mionucleare, vale a dire, la memoria epigenetica.[20] In breve, questa si riferisce a modifiche apportate al DNA senza influenzare la sua sequenza nucleotidica, quindi senza cambiare il codice genetico. Ciò comporta l’aggiunta (o la rimozione) di gruppi metilici ai nucleotidi di Citosina e Adenina o modifiche degli istoni (ad esempio, metilazione o acetilazione di residui di aminoacidi delle proteine istoniche). Il risultato di ciò è che influisce sull’espressione genica. Questo potrebbe forse essere trattato in un futuro articolo, dato che più ricerche vengono gradualmente pubblicate su questa nuova ed interessante strada ipotetica.
A proposito di “memoria epigenetica”: questa figura illustra un modello di sviluppo della persistenza batterica basato sulla presenza di un potenziale effetto di “memoria” epigenetica che include l’eredità stabile di certi modelli di metilazione del DNA. Lo stato di metilazione del DNA cellulare potrebbe portare alla conservazione di alcuni profili di espressione genica che favoriscono la dormienza, conservati in alcune cellule dopo il risveglio dalla dormienza. Cinetica di uccisione bifasica adattata da. (A) Popolazione originale di cellule metabolicamente attive che potrebbero contenere un’intrinseca eterogeneità fenotipica. (B) Quando incontra lo stress, la maggior parte delle cellule metabolicamente attive muore, mentre una piccola frazione di cellule entra nello stato di persistenza. La popolazione di persister può essere in qualche modo eterogenea, cioè formata da diversi percorsi (stocastico contro specifico). (C) Dopo gli stimoli nutrizionali/la rimozione dello stress, alcuni persister si risvegliano. Qui, la maggior parte dei persister inizia rapidamente la crescita e si divide in cellule regolari e metabolicamente attive. Tuttavia, alcune cellule potrebbero sperimentare un effetto di “memoria” epigenetica. Qui, lo stato di metilazione del DNA di alcuni siti che si trovano a monte di regioni codificanti regolate per esprimere tratti che favoriscono la dormienza potrebbe essere mantenuto dopo la replicazione del DNA. (D) A livello di popolazione totale, la popolazione finale dopo il risveglio potrebbe essere ugualmente suscettibile allo stress come la popolazione originale in (A). Tuttavia, a livello di singola cellula, alcune cellule potrebbero contenere un effetto di “memoria” legato alla dormienza, basato sull’eredità di alcuni tratti epigenetici dipendenti dalla metilazione del DNA. (E) L’esistenza di un effetto di “memoria” epigenetica potrebbe potenzialmente aumentare la frequenza dei persister nel tempo durante ripetuti cicli di stress.
Klein, CS; Marsh, GD; Petrella, RJ; Rice, CL (July 2003). “Muscle fiber number in the biceps brachii muscle of young and old men”. Muscle & Nerve. 28 (1): 62–8.
Tseng, Brian S., Christine E. Kasper, and V. Reggie Edgerton. “Cytoplasm-to-myonucleus ratios and succinate dehydrogenase activities in adult rat slow and fast muscle fibers.” Cell and tissue research 275.1 (1994): 39-49.
Schmalbruch H. Skeletal Muscle. Berlin: Springer-Verlag; 1985.
Mauro, Alexander. “Satellite cell of skeletal muscle fibers.” The Journal of Cell Biology 9.2 (1961): 493-495.
Forcina, Laura, et al. “An overview about the biology of skeletal muscle satellite cells.” Current genomics 20.1 (2019): 24-37.
Rosenblatt, J. David, David Yong, and David J. Parry. “Satellite cell activity is required for hypertrophy of overloaded adult rat muscle.” Muscle & nerve 17.6 (1994): 608-613.
Pavlath, Grace K., et al. “Localization of muscle gene products in nuclear domains.” Nature 337.6207 (1989): 570-573.
Allen, David L., Roland R. Roy, and V. Reggie Edgerton. “Myonuclear domains in muscle adaptation and disease.” Muscle & nerve 22.10 (1999): 1350-1360.
Gundersen, Kristian, and Jo C. Bruusgaard. “Nuclear domains during muscle atrophy: nuclei lost or paradigm lost?.” The Journal of physiology 586.11 (2008): 2675-2681.
Sinha-Hikim, Indrani, et al. “Testosterone-induced muscle hypertrophy is associated with an increase in satellite cell number in healthy, young men.” American Journal of Physiology-Endocrinology and Metabolism 285.1 (2003): E197-E205.
Egner, Ingrid M., et al. “A cellular memory mechanism aids overload hypertrophy in muscle long after an episodic exposure to anabolic steroids.” The Journal of physiology 591.24 (2013): 6221-6230.
Eriksson, Anders. Strength training and anabolic steroids: a comparative study of the trapezius, a shoulder muscle and the vastus lateralis, a thigh muscle, of strength trained athletes. PhD Diss. 2006.
Lindholm, Jesper Bøgh, et al. Effects of Long-Term Supplementation of Androgen Anabolic Steroids on Human Skeletal Muscle – Evidence for Muscle Memory? Master’s Thesis, 2019.
Snijders, Tim, et al. “The concept of skeletal muscle memory: Evidence from animal and human studies.” Acta Physiologica 229.3 (2020): e13465.
Seaborne, Robert A., et al. “Human skeletal muscle possesses an epigenetic memory of hypertrophy.” Scientific reports 8.1 (2018): 1-17.
Se si domandasse al bodybuilder nella media quale sia l’oggetto di maggiore interesse nella sua programmazione per il “Bulk” quasi sicuramente, dopo aver detto “raggiungere la massima ipertrofia”, direbbe “ridurre al minimo l’aumento della massa grassa”. Ed è più che logico visto che l’atleta con un minimo di senso logico sa che un peggioramento marcato della “bf” in una fase ipercalorica si tradurrà mediamente in un tempo generalmente più lungo in ipocalorica con restrizioni caloriche (o output calorici) più elevate ed un rischio relativamente aumentato di veder persa più massa contrattile o, peggio ancora, il dover calcare con i PEDs per rientrare nei tempi della preparazione al contest (o a qualsiasi evento dove il soggetto vuole essere nella sua “Top Condition”).
Tralasciando la pratica della “Break Diet”, altamente funzionale se gestita correttamente, esiste una strategia alimentare piuttosto datata ma, complici anche “neo-trafilettari” impomatati, riemersa recentemente, ovvero seguire un regime alimentare “High Fat” nel periodo di “Bulk”.
Siamo soliti collegare le diete “Low Carbs” e “High Fat” nel periodo di restrizione calorica, sapendo benissimo che il vantaggio assoluto esiste solo e soltanto con pratiche supplementative mirate (PEDs), parlando sempre di atleti e non di soggetti in sovrappeso o obesi che inizialmente giovano nel seguire una dieta Chetogenica o iperproteica in quanto a compliance insulino-resistenza correlata. Ma in ipercalorica? Possiamo veramente aspettarci una qualità migliore in quanto aumento della massa muscolare ed il peso guadagnato rispetto ad una dieta con prevalenza glucidica?
Con questo articolo cercherò di dare una risposta la più oggettiva possibile avvalendomi della conoscenza scientifica in nostro possesso che riguarda il metabolismo lipidico, l’impatto dei lipidi sulla massa muscolo-scheletrica, sulle prestazioni e i raffronti con il metabolismo glucidico partendo però dalla storia pratica della metodica alimentare qui trattata…
Back to the Future: Mauro Di Pasquale e la sua “Soluzione Anabolica”
Chi è che non ricorda Mauro Di Pasquale e il suo (a detta dell’autore) “Santo Graal” delle diete, “La Dieta Metabolica”? E la sua versione per i Bodybuilder ” La Soluzione Anabolica”? Immagino che molti di voi conosceranno entrambi i libri e i concetti viziati da bias in essi contenuti.
Visto l’argomento partirò proprio da qui…
Per chi non lo sapesse, Mauro G. Di Pasquale è un Powerlifter campione del mondo, autore di articoli sul bodybuilding e opinionista. Di Pasquale è stato assistente professore all’Università di Toronto dal 1988 al 1998. Ha tenuto conferenze e fatto ricerche sulle prestazioni atletiche, sugli integratori alimentari e sull’uso di farmaci nello sport. Ha conseguito una laurea con lode in scienze biologiche, specializzandosi in biochimica molecolare (1968) e ha una laurea in medicina (1971) – entrambe presso l’Università di Toronto. Di Pasquale è certificato come Medical Review Officer (MRO) dal Medical Review Officer Certification Council (MROCC). Era il MRO per la National Association for Stock Car Auto Racing (NASCAR). Dal 1997 al 1999 Di Pasquale si è occupato di redazione, ricerca e sviluppo di prodotti per le Scienze Sperimentali e Applicate (EAS). Come autore di bodybuilding, Di Pasquale ha scritto migliaia di articoli per molte grandi riviste di bodybuilding e fitness come Muscle & Fitness e Iron Man;[1-4] I suoi articoli e libri sono stati tradotti in lingua italiana e pubblicati in Italia da Sandro Ciccarelli per la rivista Olympian’s News.
Di Pasquale divenne anche un oppositore all’uso del doping pubblicizzando i suoi regimi alimentari e integrativi come “sostitutivi salutari” dei farmaci per il miglioramento delle prestazioni.
Da destra: Mauro di Pasquale insieme al bodybuilder professionista Eddie Robinson.
Nonostante la titolatura sopra esposta, Di Pasquale divulgò alcune teorie decisamente opinabili abbracciando la filosofia delle diete “Low Carb” come soluzione universale per perdere grasso e aumentare la massa muscolare. Egli rappresenta una sorta di “paladino” per la fazione dei sostenitori della così detta “ipotesi dell’Insulina”, tra l’altro smentita scientificamente, la quale ipotizza che non sia l’eccesso energetico (calorie) a causare l’aumento di peso/grasso ma i Carboidrati ed il loro stimolo sull’Insulina. Peccato però che la termodinamica ci dimostri il contrario, ovvero che è l’eccesso energetico ha causare una conservazione dello stesso e non il semplice stimolo ormonale (sul quale, tra l’altro, ci sarebbe molto da dire). Oltretutto, alcune fonti proteiche (vedi prodotti lattiero caseari) hanno un impatto insulinico maggiore del pane bianco, sebbene questo venga regolato da una concomitante risposta del Glucagone. Comunque sia, di quest’ultimo punto, il Di Pasquale sembra non curarsene più di tanto, similmente a quanto fa della risposta insulinica data dalla coingestione di grassi e proteine preoccupandosi del carico glucidico da ciclicizzare (cosa, quest’ultima, condivisibile ma da contestualizzare).
Ma arriviamo al dunque sul suo approccio “High Fat” in Bulk…
Nel 1995 pubblica il libro “The Anabolic Diet“, pubblicato poi in Italia con il nome di “La Soluzione Anabolica”.
Il libro in questione espone le vedute dell’autore, accuratamente servite per vendere il prodotto e convincere il lettore, dividendo in sei fasi la preparazione:
Fase di Inizio;
Fase di Massa;
Fase di Forza;
Fase di Definizione;
Fase Pregara;
Fase di Recupero.
Tralasciando le fasi 1, 3,4,5 e 6 che, per argomento trattato nel presente articolo, non ci interessano, concentriamoci sulla “Fase di Massa“.
Dopo aver trovato la quota calorica di “mantenimento”, procedendo con i primi 12 giorni sostituendo le kcal tipicamente consumate dai Carboidrati con Grassi e Proteine riducendo significativamente i primi (es. 1g/CHO-Pro = 4Kcal; 1g Fat = 9Kcal; 400g di CHO/120g di Pro/50g di Fat = circa 30g di CHO, 220g/die di Pro e 134g/die di Grassi), si dovrebbe passare alla prima “ricarica di Carboidrati” seguita a sua volta da 5-6 giorni “High Fat” e 1 o 2 giorni “High Carbs” per 3-4 settimane.
A questo punto l’autore consiglia di permettere al peso un aumento del 15% oltre il peso ideale (es. Peso ideale = 98Kg, peso da raggiungere = 113Kg [+15%]) premurandosi di avvisare il lettore che non deve commettere l’errore di mangiare eccessivamente e vanificare il potenziale della sua strategia di farvi aumentare di massa magra con poco incremento della massa grassa…
Quindi, il bodybuilder dovrebbe seguire lo schema “High Fat” nei giorni feriali ed effettuare le “ricariche” glucidiche nel fine settimana, con un monte calorico indicativo pari a 55Kcal per Kg di peso corporeo desiderato (es. 90Kg = 4950Kcal/die). E le differenze di risposta delle vie metaboliche? Capacità di gestione delle Kcal connesse alle richieste prestative e al livello dell’atleta?… Punti non pervenuti… Per lo meno, il Di Pasquale dice che se il soggetto presenta difficoltà nell’ingestione di grossi quantitativi calorici giornalieri può far diventare il totale calorico da raggiungere un obbiettivo settimanale, abbandonando saggiamente il concetto forviante di “evento in acuto”.
Nelle indicazioni viene riportata anche la soglia del grasso corporeo da non superare. La quota riportata è del 10%… Ora, fatta eccezione per soggetti con un set point adipocitario sensibilmente basso, arrivare a pesare il 15% in più del peso ideale mantenendo una “bf” del 10% è cosa non proprio semplice, leggermente di più se parliamo di atleti “Natural”. Molto più logico sarebbe stato indicare una soglia tra il 12 ed il 15%. Ma l’autore scrive che la Fase di Massa deve essere interrotta quando si raggiunge il peso desiderato o il 10% di “bf”… Se siete agonisti dovete interromperla se arrivate al limite delle 12 settimane dalla gara.
Per quanto concerne l’obbiettivo di incremento settimanale di peso, viene indicato come “ideale” 1Kg a settimana.
Ma quanti glucidi dovrebbero essere consumati durante il/i giorno/i di “ricarica”? Di Pasquale afferma che si possono raggiungere “quantità enormi” di CHO e Kcal nel week and, anche fino a 12.000Kcal tanto il sabato quanto la domenica. Inoltre sia la quota di CHO giornaliera che il numero di giorni con rialzo glucidico possono essere aumentati in base alle risposte. E già questo punto denota una mancanza di funzionalità di base del suo piano alimentare “High Fat” per una “massa pulita” che però va oltre il concetto di “pulito” arrivando al “nodo metabolico”: il vantaggio indiscutibile del substrato glucidico per sostenere l’attività fisica in uno sport contro-resistenza che sfrutta prevalentemente un metabolismo anaerobico glicolitico.
Percentuali di ripartizione dei macronutrienti durante i giorni “High Fat” e le “ricariche di Carboidrati”. (fonte immagine “THE ANABOLIC DIET”, pag. 30 Capitolo 3).
Lascerò da parte la sfilza di integratori marca “MetabolicDiet”, che per lo più altro non sono che Creatina e Proteine in polvere, e le riduttive, ridicole e confutate chiacchiere sulla manipolazione ormonale “simil-doped”, riflettendo direttamente su quanto detto fino a questo punto.
Ora, abbiamo una dieta per il “Bulk” a tutti gli effetti “High Fat” per 3-4/5 della settimana con delle “ricariche glucidiche” di base di 1 o 2 giorni ma incrementabili a bisogno. Ricordo che questa dieta ha la bellezza di 26 anni, ed il sottoscritto, ai tempi in cui ero fortemente tentato a prestar orecchio più alla “carne ed al sangue” piuttosto che alla logica e al buon senso, la conosce e osserva i suoi effetti sugli atleti che l’hanno voluta sperimentare da circa 15 anni. Beh, posso assicurarvi che i risultati con questo tipo di alimentazione in “Bulk” erano apprezzabili (questo non vuol dire che erano spettacolari o che riflettevano le promesse dell’autore) principalmente in due contesti:
Alteti “doped” con un set point adipocitario basso e una media di 3 copiose “ricariche” a settimana;
Atleti “natural” con un set point adipocitario basso e una media di 3 copiose “ricariche” a settimana e un minimo di CHO giornalieri pari a 2-3g/Kg.;
Atleti di sesso femminile con variabili sul numero di “ricariche” e tempi di svolgimento delle medesime.
Ed i casi osservati con risultanti apprezzabili, comunque, sono pochi rispetto ad una maggioranza di individui non significativamente avvantaggiati dal protocollo.
Come potete vedere, il concetto di base della dieta crolla inesorabilmente sotto il peso di dati empirici raccolti direttamente ed indirettamente nel corso degli anni.
“Mah Gabriel! Esistono altre metodologie “High Fat” per una “massa pulita” e funzionano!” Alt, non ti agitare piccolo “troll”, perchè con il termine “High Fat” mi stai dicendo “tutto e niente”, un pò come succede quando si parla di “dieta iperproteica”.
Si possono considerare “High Fat” tutte le diete che in ipercalorica superano il 20-25% delle calorie giornaliere dai grassi! E’ ovvio che possono esistere degli abissi programmatici tra uno schema alla Di Pasquale e un altro che presenta linee di percentuali macro-caloriche nei range sopra indicati.
Prima di proseguire con esempi di schemi alimentari “High Fat” ben più logici rispetto a quanto presentato ne “La Soluzione Anabolica”, e considerare le argomentazioni a favore di questo tipo di dieta ipercalorica, vediamo come i diversi acidi grassi influenzano la massa muscolare. Passiamo a un pò di teoria per tornare successivamente alla pratica con maggiori conoscenze…
La modulazione nella massa e funzione del muscolo-scheletrico dei lipidi
È importante iniziare il discorso sottolineando che vi sono prove crescenti che supportano un ruolo degli acidi grassi e dei loro intermedi lipidici derivati nella regolazione della massa e della funzione del muscolo scheletrico. E’ mia intenzione quindi discutere le prove relative a quei percorsi che sono coinvolti nella riduzione, aumento e/o conservazione della massa muscolare scheletrica da parte dei lipidi.
L’evidenza di diversi studi suggerisce che gli acidi grassi saturi e insaturi possono agire per regolare in modo differenziale la massa e la funzione del muscolo scheletrico. Ad esempio, è stato dimostrato che l’esposizione dei miotubi C2C12 al palmitato (C16:0), l’acido grasso saturo circolante più abbondante, riduce il diametro del miotubo e sopprime la segnalazione dell’Insulina. [2] In accordo con questo, è stato riportato che la fornitura di palmitato nelle cellule muscolari induce l’espressione di geni pro-atrofici come atrogin-1/MAFbx, in concomitanza con una maggiore localizzazione nucleare del suo regolatore trascrizionale FoxO3. [3] Al contrario, l’applicazione di acido docosaesaenoico (DHA), un acido grasso polinsaturo omega-3 (PUFA), non ha alterato la morfologia dei miotubi quando applicato da solo ed è stato dimostrato che contromodula l’atrofia indotta dal palmitato nei miotubi C2C12. [2] Coerentemente con ciò, uno studio separato ha riportato il miglioramento della degradazione proteica indotta dal palmitato nei miotubi C2C12 dopo il co-trattamento con DHA. [3] In particolare, ciò ha coinciso con la capacità del DHA di mitigare la localizzazione nucleare potenziata di FoxO3 e l’espressione genica di atrogin-1/MAFbx in risposta alla fornitura di palmitato. [3]
In accordo con questi risultati nelle cellule muscolari di coltura, diversi studi in vivo hanno anche riportato la capacità degli acidi grassi insaturi di trasmettere risposte benefiche che agiscono per prevenire l’atrofia muscolare. Ad esempio, l’alimentazione di topi portatori dell’adenocarcinoma del colon-26, un modello animale di cachessia tumorale, con una dieta integrata con acido linoleico coniugato, ha dimostrato di preservare la massa muscolare del gastrocnemio. [4] In particolare, questo effetto protettivo ha coinciso con una riduzione dell’espressione del recettore TNF-α del muscolo scheletrico, suggerendo che i PUFA possono agire per prevenire l’atrofia muscolare, almeno in parte, riducendo le azioni cataboliche della citochina TNF-α. [4 , 5] In uno studio separato, l’integrazione alimentare con acido eicosapentaenoico (EPA; C20:5 (n-3)) ha attenuato la degradazione proteica nel muscolo gastrocnemio di topi portatori del tumore MAC16 che induce la cachessia. [6] Il trattamento con EPA è stato riportato anche per evitare riduzioni dell’artrite indotta e aumento del peso del muscolo gastrocnemio nei ratti dopo somministrazione di adiuvante di Freund, concomitante con la normalizzazione dell’espressione genica di atrogin-1 / MAFbx e MuRF1. [7] Inoltre, i criceti distrofici alimentati con una dieta arricchita in acido α-linolenico PUFA (ALA) (C18:3(n-6)) hanno mostrato miglioramenti nella morfologia e nella funzione muscolare, compreso l’ingrossamento delle miofibre . [8] In accordo con questi risultati, è stato anche dimostrato che i PUFA omega-3 e omega-6 aumentano la fosforilazione di p70S6K1 a Thr389, indicativo della sua maggiore attività, durante la differenziazione miogenica dei miociti L6. [9] Insieme, questi studi supportano l’idea che gli acidi grassi insaturi possono fornire protezione contro l’atrofia muscolare in risposta a varie condizioni patologiche e potenzialmente migliorare le condizione trofiche in soggetti sani. Inoltre, questi risultati evidenziano le risposte distinte che gli acidi grassi saturi e insaturi inducono rispettivamente per promuovere o contrastare l’atrofia muscolare e la degradazione proteica.
Un certo numero di differenti vie di segnalazione e/o intermedi sono stati implicati come potenziali mediatori della atrofia muscolare, che a loro volta possono essere regolati in risposta alla assunzione di acidi grassi (vedi Figura seguente). Ad esempio, è noto che il palmitato agisce come un potente repressore della segnalazione diretta di PKB/Akt nel muscolo scheletrico, almeno in parte attraverso la sua capacità di indurre l’accumulo di intermedi lipidici tossici come la Ceramide. [10 , 11] Infatti, tali sfingolipidi possono agire stimolando le isoforme della proteina fosfatasi 2A (PP2A) o della proteina chinasi C (PKC) atipica (PKCζ) per inibire PKB/Akt. [12] In accordo con ciò, è stato riportato che l’atrofia del miotubo C2C12 indotta da TNF-α coincide con livelli elevati di Ceramide intracellulare, [13] mentre è stato dimostrato che il blocco della sintesi di Ceramide attenua l’atrofia muscolare indotta dal TNF-α nei miotubi L6, oltre a proteggere i topi contro l’atrofia del muscolo scheletrico tumore indotto ( via impianto di carcinoma C26) in vivo. [13] In particolare, queste risposte benefiche hanno contribuito a una maggiore sintesi proteica e a una diminuzione della proteolisi, in concomitanza con una ridotta espressione del gene atrogin-1/MAFbx tramite la funzione Foxo3 soppressa, nonché una maggiore abbondanza di mediatori chiave della sintesi proteica tra cui S6K1 e PKB/Akt. [13] Inoltre, è stato riportato che la fornitura esogena di Ceramide nelle cellule muscolari L6 riduce i livelli proteici del fattore di trascrizione miogenico miogenina attraverso l’ inibizione della fosfolipasi D, mentre l’inibizione della sintesi di Ceramide migliora l’espressione della miogenina e accelera la formazione di miotubi. [14]Uno studio di Turpin e colleghi ha anche dimostrato un aumento del contenuto di Ceramide muscolare dopo l’infusione acuta (5 h) di intralipid®, che ha coinciso con l’attivazione della segnalazione pro-apoptotica, come dimostrato dall’aumento dell’attività della caspasi-3 nel muscolo gastrocnemio. [15] Tuttavia, il ruolo della Ceramide nel promuovere questo aumento dell’apoptosi muscolare guidato dai lipidi non è stato studiato, ad esempio mediante la co-somministrazione di inibitori della sintesi della Ceramide. In alternativa, livelli elevati di Ceramide associati all’iperlipidemia possono anche agire per sopprimere la sintesi proteica inducendo l’espressione e/o l’attività di repressori chiave della segnalazione mTORC1-S6K come Regulated in Development e DNA Damage 1 (REDD1). [16 , 17]In particolare, va anche evidenziato che il ganglioside GM3 (trisialotetrahexosylganglioside), un glicosfingolipide contenente acido sialico derivato dalla Ceramide, è stato anche implicato come regolatore negativo della crescita e/o differenziazione del muscolo scheletrico, in concomitanza con la sua capacità segnalata di modificare l’azione dell’insulina alterando la funzione del recettore specifico (Recettore dell’Insulina). [18 , 19 , 20 , 21] Inoltre, è stato dimostrato che un altro lipide derivato dalla Ceramide, ceramide-1-fosfato, stimola la proliferazione dei mioblasti C2C12 attraverso un meccanismo che coinvolge l’attivazione di Akt, mTOR e ERK1/2. [22] In effetti, un ulteriore lavoro che utilizza topi carenti di GM3 sintasi, l’enzima responsabile della sintesi di GM3, potrebbe far luce sul ruolo di questo ganglioside nel controllo della massa muscolare scheletrica, ad esempio in risposta all’obesità e/o all’invecchiamento.
Riassunto delle vie che mediano l’atrofia muscolare da acidi grassi saturi. L’esposizione delle cellule muscolari ad acidi grassi saturi come il palmitato (C16:0) provoca l’accumulo intracellulare di intermedi lipidici tossici come Ceramide e Diacilglicerolo. (A) L’aumento dei livelli di Ceramide può portare all’inibizione della proteina chinasi B/Akt attraverso l’attivazione di isoforme atipiche della proteina chinasi C (ξ/λ) e/o della proteina fosfatasi 2A. Inoltre, la Ceramide agisce come precursore per la sintesi del glicosfingolipide GM3 che ha dimostrato di compromettere la funzione del recettore dell’Insulina. Inoltre, la Ceramide può anche agire per modulare l’assorbimento dei nutrienti, ad esempio reprimendo l’espressione del trasportatore amminoacidico neutro SNAT2, riducendo così l’apporto cellulare di amminoacidi. (B) È stato dimostrato che la stimolazione indotta da diacilglicerolo della protein chinasi Cθ promuove la fosforilazione della serina dell’IRS-1, con conseguente sua funzione compromessa. La risultante inibizione della protein chinasi B/Akt a sua volta può portare alla repressione della sintesi proteica attraverso la soppressione del segnale meccanicistico bersaglio della rapamicina (mTOR)/p70-S6 chinasi 1-dipendente (C), l’attivazione del forkhead box O (FoxO ) fattori di trascrizione e induzione dei loro geni atrofici bersaglio (D) e/o l’attivazione della proteolisi dipendente dalla caspasi (E). Inoltre, la stimolazione della segnalazione proinfiammatoria da parte degli acidi grassi saturi a catena lunga può portare alla sovraregolazione dei geni atrofici dipendente dal fattore nucleare kappa B (F).
Oltre agli sfingolipidi, i diacilgliceroli (DAG) sono una classe alternativa di lipidi che possono essere generati in risposta alla ingestione di acidi grassi. In particolare, l’aumento dei livelli di DAG è stato associato allo sviluppo dell’insulino-resistenza, fattore di per se limitante sul corretto ripartizionamento calorico a favore del miocita e, quindi, sulla sintesi proteica . [23] sono stati rilevati Inoltre, un aumento dei livelli di DAG muscolare dopo infusione di lipidi nei topi, con concomitante aumento della attività nel muscolo gastrocnemio della caspasi-3 .[15] Sebbene si sappia poco sul ruolo dei DAG nella regolazione della massa muscolare scheletrica, è stato riportato che l’attivazione meccanica ex-vivo della DAG chinasiζ (DGKζ), un enzima che catalizza la conversione di DAG in acido fosfatidico (PA), favorisce un aumento Segnalazione dipendente da mTOR e ipertrofia associata nel muscolo estensore lungo delle dita (EDL) di topo isolato, in concomitanza con la capacità riportata di PA di legare e attivare direttamente mTOR. [24 , 25] In accordo con ciò, è stato anche dimostrato che la sovraespressione cardiaca specifica di DGKζ migliora l’atrofia miocardica nei topi diabetici indotti da streptozotocina. [26] Pertanto, questi risultati suggeriscono che l’attivazione e/o la sovraespressione di DGKζ può fornire un mezzo per stimolare i tassi di sintesi proteica e le risposte ipertrofiche, e quindi migliorare le perdite di massa muscolare, sia riducendo i livelli cellulari di DAG e/o aumentando l’attivazione della segnalazione mTOR indotta dal PA. È importante sottolineare che il lavoro futuro potrebbe comportare lo studio dei potenziali effetti benefici della sovraespressione di DGKζ nel muscolo come mezzo per contrastare l’atrofia muscolare indotta dall’età e/o dalla dieta. Inoltre, modelli animali che mostrano livelli elevati di DAG nel muscolo scheletrico, compresi i topi che sono carenti di lipasi ormone-sensibile (HSL), [27] possono anche essere utili per chiarire il ruolo di DAG nell’atrofia del muscolo scheletrico.
Un’altra considerazione importante riguarda la possibilità che specie di DAG distinte possano avere un impatto diverso sulle vie coinvolte nella regolazione della massa muscolare, ad esempio come determinato dalla composizione dei gruppi acilici grassi che si esterificano a livello sn-1,2, sn‐ 1,3, o le posizioni sn-2,3 della base di glicerolo del DAG. [28 , 29] Infatti, è stato dimostrato che il trattamento dei miotubi L6 di ratto con palmitato porta ad aumenti significativi dei livelli cellulari di alcune specie di DAG, nonché del contenuto totale di DAG cellulare. [30] Inoltre, è stato dimostrato che il co-trattamento con l’acido grasso monoinsaturo (MUFA) palmitoleato (C16:1) sopprime selettivamente gli aumenti indotti dal palmitato nei livelli di specie DAG contenenti porzioni di acidi grassi saturi C18:0 e C20:0, in coincidenza con l’azione antinfiammatoria del MUFA. [30] Sebbene non determinati in questo studio, stereoisomeri distinti di DAG possono anche regolare in modo differenziale la segnalazione anabolica/catabolica muscolare. Per supportare questa nozione, è stato riportato che gli stereoisomeri sn-1,2 DAG (rispetto agli isomeri sn-1,3) sono più potenti nell’attivare le vie di segnalazione legate all’insulino-resistenza, inclusa l’attivazione della PKC. [31] Insieme, questi studi forniscono prove emergenti che alcune molecole/isomeri DAG possono svolgere un ruolo più importante nello sviluppo dell’atrofia muscolare, ad esempio promuovendo la resistenza all’insulina e/o aumentando la spinta proinfiammatoria. Tuttavia, sarà necessario un ulteriore lavoro per determinare quali di queste molecole di DAG, se presenti, sono responsabili delle azioni di atrofia muscolare. Nel tentativo di affrontare questo problema, studi futuri potrebbero comportare il trattamento di cellule muscolari in coltura con diverse molecole/stereoisomeri DAG al fine di determinare i loro effetti sulla miogenesi e/o sull’atrofia muscolare. In alternativa, un ulteriore lavoro può anche incorporare un’analisi lipidomica dettagliata di varie specie di DAG intramuscolari in tessuti isolati da modelli animali di atrofia muscolare, oltre a monitorare potenziali cambiamenti nella loro abbondanza a seguito di interventi noti per aumentare la massa muscolare (ad es. apporto dietetico di PUFA o aumento dell’attività fisica). Infatti, se tali studi dovessero rivelare un ruolo chiave per l’accumulo di DAG nello sviluppo dell’atrofia del muscolo scheletrico, il lavoro successivo potrebbe implicare la determinazione dell’origine di tali specie di DAG, ad esempio inibendo l’attività degli enzimi implicati nella formazione del triacilglicerolo dal DAG ( sintesi di TAG) (es. glicerolo fosfato transferasi (GPAT), acilglicerolfosfato aciltransferasi (AGPAT) e lipina), o alterando l’attività di enzimi implicati nell’idrolisi di TAG e/o DAG (es. lipasi dei trigliceridi adiposa (ATGL) o HSL). A questo fine, i lavori di Badin e collaboratori hanno riportato un’elevata abbondanza di proteine ATGL nel muscolo scheletrico di individui diabetici di tipo 2 rispetto a soggetti di controllo magri, nonché una ridotta espressione di HSL muscolare in individui obesi.[32] Inoltre, gli autori dello stesso studio hanno ulteriormente dimostrato che la sovraespressione di ATGL o l’inibizione dell’attività dell’HSL nei miotubi primari umani determinava l’accumulo di DAG cellulare e una compromissione associata nella segnalazione dell’Insulina. Tuttavia, in questo studio non è stato determinato se questi cambiamenti nei livelli di DAG siano collegati all’atrofia muscolare.
Oltre a modulare la segnalazione diretta di PKB/Akt e/o mTORC1, gli acidi grassi e/oi loro lipidi derivati possono ulteriormente contribuire alla perdita muscolare modulando il trasporto dei nutrienti (aminoacidi) e/o la segnalazione associata. Ad esempio, lavori svolti da diversi gruppi di ricerca hanno dimostrato la capacità della Ceramide di sottoregolare l’espressione e/o l’attività dei principali trasportatori di nutrienti, incluso il trasportatore di amminoacidi neutri SNAT2 (SLC38A2). [33 , 34] In tal modo, gli acidi grassi che agiscono attraverso tali intermedi lipidici possono agire per compromettere l’assorbimento degli aminoacidi, contribuendo così a una perdita di massa muscolare. È interessante notare che in uno studio separato, è stato dimostrato che l’incubazione di miotubi L6 di ratto con acido linoleico (C18:2) limita la sovraregolazione adattativa dell’espressione e dell’attività di SNAT2 in risposta alla carenza di aminoacidi. [35] In particolare, questa riduzione indotta da acidi grassi nell’attività di trasporto del Sistema A è stata mediata da una maggiore ubiquitinazione e degradazione proteasomica della proteina SNAT2. [35] Al contrario, in uno studio separato di Li e collaboratori, è stato riportato che l’espressione dell’mRNA dei transcettori di aminoacidi LAT1 (un trasportatore di aminoacidi di tipo L) e SNAT2 è sovra-regolata nel longissimus dorsi di suini alimentati con diete n. -6 e n-3 PUFA.[36] Quindi, è possibile che gli acidi grassi e/o i loro lipidi derivati possano funzionare per modulare le strategie adattative che vengono utilizzate da tessuti come il muscolo scheletrico, al fine di massimizzare o minimizzare l’assorbimento di nutrienti durante condizioni di digiuno o privazione di nutrienti e, presumibilmente, tale meccanismo può subire alterazioni durante stati di sovra-alimentazione.
È importante sottolineare che la letteratura attuale descrive prove che suggeriscono che gli acidi grassi insaturi possono agire per contrastare i mediatori pro-atrofici, compresi quelli attivati in seguito all’esposizione agli acidi grassi saturi. Ad esempio, è stato riportato che MUFA e PUFA prevengono le riduzioni indotte dal palmitato della sensibilità all’Insulina e veicolano effetti antinfiammatori nelle cellule del muscolo scheletrico. [30 , 37, 38] In effetti, la regolazione trascrizionale dipendente da NF-kB è stata implicata nel promuovere l’atrofia muscolare da disuso nel muscolo soleo di ratto aumentando l’attivazione mediata da FoxO del promotore MuRF1. [39] Inoltre, uno studio recente ha dimostrato che la diminuzione del segnale anabolico nel muscolo scheletrico di topi anziani coincideva con l’accumulo di Ceramide intramuscolare e DAG, nonché con una maggiore abbondanza di mRNA di TNF-α. [40] È interessante notare che la somministrazione di olio di pesce ai suinetti in fase di svezzamento, che ha portato all’arricchimento di EPA, DHA e contenuto totale di PUFA omega-3 all’interno del muscolo gastrocnemio, ha coinciso con una riduzione dei livelli di TNF-α muscolare e una ridotta espressione del recettore Toll-like 4 (TLR4), un recettore bersaglio per gli acidi grassi saturi che stimola la segnalazione proinfiammatoria in risposta alla sua attivazione. [41] In particolare, è stato riportato che la stimolazione del TLR4 da parte del suo ligando lipopolisaccaride induce il catabolismo muscolare nei miotubi C2C12 attraverso l’attivazione delle vie ubiquitina-proteasoma e autofagia-lisosoma. [42] Inoltre, è stato dimostrato che il trattamento con DHA in cellule muscolari umane co-coltivate con macrofagi attenua il contenuto proteico indotto dai macrofagi di Fn14, un modulatore positivo dell’espressione di MuRF-1. [43 , 44] Pertanto, sulla base di questi risultati, è concepibile che le azioni antinfiammatorie riportate degli acidi grassi insaturi nelle cellule muscolari scheletriche possano contribuire, almeno in parte, alla loro capacità di preservare la massa e/o la funzione muscolare.
In particolare, queste azioni protettive possono essere collegate a miglioramenti della funzione mitocondriale, la cui compromissione è stata suggerita dal tipo di dieta e/o all’atrofia muscolare indotta dall’età. [45 , 46] Ad esempio, un recente studio di Roseno e colleghi ha riportato che una dieta ricca di grassi a breve termine (3 settimane) ha aumentato l’atrofia muscolare da denervazione nei topi inducendo la degradazione proteica nel soleo ricco di mitocondri, ma non nel muscolo EDL glicolitico. In particolare, la denervazione di 14 giorni ha indotto una perdita del contenuto proteico mitocondriale nel soleo ma non nell’EDL, indipendentemente dalla dieta. Pertanto, questi risultati suggeriscono che la perdita di mitocondri indotta dalla denervazione e la compromissione della funzione mitocondriale indotta da una dieta ricca di grassi possono combinarsi per promuovere l’atrofia del muscolo scheletrico. Al contrario, uno studio indipendente di Tardif e collaboratori ha dimostrato che i ratti anziani alimentati con una dieta arricchita di oleati mostrano notevoli miglioramenti nella sensibilità all’insulina e un aumento della sintesi proteica muscolare, in concomitanza con una maggiore espressione di geni implicati nella stimolazione della ossidazione mitocondriale, compreso il recettore attivato dal proliferatore dei perossisomi PPARα e PPARβ, nonché CPT-1β. [47 , 48] Inoltre, è stato dimostrato che i miotubi C2C12 trattati con PUFA acido linolenico e ALA mostrano una maggiore attivazione del AMPK, un altro regolatore positivo chiave della -ossidazione mitocondriale. [49] Inoltre, è stato riportato che il DHA inibisce la degradazione proteica nei miotubi C2C12 attraverso una via PPARγ-dipendente. [50] Infatti, la capacità ossidativa mitocondriale potenziata e/o preservata, come precedentemente riportato in risposta alla sola o co-fornitura di acidi grassi insaturi, può anche aiutare a prevenire l’accumulo intramuscolare di intermedi lipotossici come la Ceramide che sono stati implicati nella promozione dell’atrofia muscolare. [51 , 52] Inoltre, è possibile che l’integrazione di PUFA possa agire per alterare le proprietà contrattili e metaboliche muscolari, ad esempio promuovendo un passaggio da fibre glicolitiche veloci a fibre lente (ossidative), cosa non propriamente positiva per un atleta di potenza. A sostegno di questa idea, lavori precedenti hanno dimostrato che l’alimentazione di ratti Wistar con una dieta arricchita in PUFA n-3 porta alla sovraregolazione delle proteine implicate nell’attivazione del metabolismo ossidativo (ad es. nel muscolo EDL, un tessuto muscolare dominante di tipo veloce). [53] È interessante notare che questo cambiamento metabolico mediato da PUFA ha coinciso anche con livelli ridotti di proteine dell’isoforma di tipo veloce MyHC-2b (catena pesante della miosina 2b) nel muscolo EDL. Pertanto, è concepibile che uno spostamento mediato dai PUFA verso un tipo di fibra muscolare a lenta ossidazione possa contribuire, almeno in parte, a guadagni benefici nella massa muscolare e/o nella funzione metabolica.
In alternativa, la regolazione della massa muscolare da parte dei lipidi può anche comportare la modulazione dell’autofagia, un meccanismo omeostatico che facilita la degradazione e il riciclaggio di proteine e organelli attraverso il macchinario lisosomiale. [54] In particolare, è stato riportato che un aumento della degradazione autofagica coincide con l’atrofia muscolare in varie condizioni e/o patologie tra cui cancro, [55] denervazione, [56] e invecchiamento. [55 , 57] Inoltre, è stato dimostrato che un’alimentazione ricca di grassi a breve termine (3 settimane) aumenta l’abbondanza di marcatori autofagosomiali nel soleo denervato dei topi. In accordo con ciò, Yuzefovych e collaboratori hanno dimostrato un aumento dell’autofagia nei miotubi L6 a seguito della somministrazione di palmitato. Pertanto, sebbene non sia ancora stato stabilito un collegamento diretto in vivo , è ipotizzabile che il turnover proteico alterato tramite l’ autofagia possa, almeno in parte, mediare le alterazioni indotte dai lipidi nella massa muscolare.
Va inoltre evidenziato che alcuni acidi grassi insaturi possono alterare il tasso di proliferazione delle cellule satelliti che funzionano come cellule progenitrici miogeniche necessarie per la crescita e la rigenerazione muscolare. Ad esempio, è stato dimostrato che DHA ed EPA inibiscono la proliferazione dei mioblasti C2C12 e delle cellule satelliti isolate dal muscolo di tacchino. [58 , 59] In particolare, queste azioni di soppressione della crescita sono state collegate a livelli ridotti di ciclina E e CDK2, proteine che svolgono un ruolo critico nella progressione del ciclo cellulare, nonché all’attivazione soppressa di ERK1/2, una proteina chinasi attivata da mitogeni implicata nel promuovere la crescita e la divisione cellulare. [59 , 60] Al contrario, è stato dimostrato che l’alimentazione di criceti distrofici carenti di δ-sarcoglicano con una dieta arricchita in ALA (un PUFA omega-3) aumenta la proliferazione e la differenziazione delle cellule satellite nel muscolo EDL, in concomitanza con un miglioramento dell’istologia muscolare. In particolare, queste risposte benefiche hanno coinciso con la capacità dell’ALA di aumentare la proporzione di miofibre α-MHC positive nel muscolo scheletrico dei criceti distrofici, insieme a una riduzione dell’espressione di β-MHC, contribuendo così alla conservazione di un α/β più fisiologico. Rapporto MHC. Inoltre, nello stesso studio, è stato anche dimostrato che l’integrazione di ALA alimentare previene l’accumulo aberrante citoplasmatico di proteine di membrana chiave nei muscoli adduttori dei criceti distrofici, inclusa la caveolina-3, una proteina coinvolta nella regolazione dell’adesione cellulare e della riparazione della membrana, nonché essendo implicato nel controllo della differenziazione muscolare e della segnalazione indotta dall’insulina. [61 , 62] Infatti, dato che le aberrazioni nella funzione e/o localizzazione della caveolina-3 sono state associate a vari fenotipi di malattie del muscolo scheletrico, [63 , 64 , 65 , 66 , 67] è plausibile che gli acidi grassi e/o i loro derivati lipidici possano influenzare la proliferazione delle cellule satellite e/o la differenziazione muscolare, almeno in parte, alterando la funzione e/o la localizzazione subcellulare delle isoforme della caveolina, nonché di altri componenti chiave della membrana strutturale.
Gli effetti degli acidi grassi sulla massa muscolare e sulla differenziazione possono essere mediati anche da una serie di metaboliti lipidici derivati. Ad esempio, è stato dimostrato che la capacità dell’acido arachidonico PUFA (C20; 4n-6) di aumentare le dimensioni, il contenuto mionucleare e il contenuto proteico dei miotubi C2C12 è mediata dall’attività della cicloossigenasi-2 (COX-2), implicando dipendenza da sintesi delle prostaglandine a valle. [68] In accordo con ciò, è stato riportato che la crescita indotta dall’acido arachidonico dei miociti C2C12 coincide con l’aumento della secrezione degli eicosanoidi PGF2α e PGE. [68]È interessante notare che diversi studi hanno anche documentato il ruolo positivo che le prostaglandine svolgono nel promuovere eventi precoci sulla superficie cellulare, inclusa l’adesione cellula-cellula, che successivamente mediano la fusione dei mioblasti nei miotubi. [69 , 70] Infatti, importanti studi di follow-up possono comportare la determinazione dell’esatta identità dei bersagli molecolari attraverso i quali le prostaglandine mediano le loro azioni, ad esempio agendo sui recettori prostanoidi legati alla proteina G (es. EP1). [69 , 71] Al contrario, è stato recentemente riportato che un altro metabolita lipidico derivato dall’acido arachidonico noto come 2-arachidonoilglicerolo (2-AG), un ligando lipidico endogeno chiave del sistema endocannabinoide, inibisce la differenziazione delle cellule satellite primarie umane e dei mioblasti murini C2C12 prendendo di mira il recettore-G ‐ 1 dei cannabinoidi accoppiati a proteine e sua successiva inibizione dei canali Kv7.4. [72] Pertanto, non si può escludere il possibile coinvolgimento di tali intermedi lipidici nella regolazione del catabolismo muscolare in risposta a determinate condizioni patologiche o alimentari.
L’aumento dell’adiposità osservato nell’invecchiamento umano è stato collegato a risposte sintetiche proteiche muscolari alterate negli individui anziani. [73] Pertanto, è ipotizzabile che i cambiamenti nei livelli e/o nella composizione dei lipidi possano contribuire all’atrofia muscolare in queste condizioni ma non limitatamente ad esse. A sostegno di questa idea, ci sono prove che suggeriscono che la modifica della composizione alimentare può avere un impatto sulla massa muscolare e/o sulla sua funzione negli esseri umani. Ad esempio, uno studio di McGlory e collaboratori ha riportato che il consumo di olio di pesce ha aumentato il contenuto di PUFA omega-3 del muscolo ( Vastus lateralis) in individui maschi sani, che ha coinciso con un’elevata espressione di proteine di segnalazione anabolizzanti incluso mTOR. [74] Inoltre, è stato riportato che l’inclusione di MUFA e PUFA nella dieta riduce l’espressione di geni lipogenici nel muscolo scheletrico di soggetti insulino-resistenti, in concomitanza con ridotti tassi di sintesi frazionaria di DAG intramuscolare e triacilgliceroli. [75] In particolare, queste risposte benefiche possono essere collegate a miglioramenti nella sensibilità all’insulina veicolati dall’integrazione alimentare di PUFA omega-3 negli esseri umani. [76 , 77 , 78]
E’ stato riportato che l’integrazione alimentare con acidi grassi insaturi può anche agire per migliorare le prestazioni fisiche e/o aumentare gli effetti metabolici benefici associati all’esercizio, in particolare in individui sedentari o non allenati . [79] A sostegno di questa idea, è stato dimostrato che un basso apporto alimentare di olio di tonno promuove la resistenza all’affaticamento muscolare nei ratti, in concomitanza con un aumento selettivo del contenuto di fosfolipidi della membrana e DHA all’interno del muscolo gastrocnemio. [80 , 81] Pertanto, un approccio più integrato che coinvolga modifiche al consumo di grassi nella dieta e un aumento dell’esercizio fisico può fornire una strategia più efficace per alleviare gli effetti deleteri associati all’atrofia muscolare. Ma stiamo comunque parlando di effetti apprezzabili su soggetti sedentari e non allenati, non su culturisti intermedi o avanzati.
Tirando le somme di quanto fino qui esposto, c’è un certo interesse giustificato sul ruolo importante che gli acidi grassi e/oi loro derivati lipidici possono svolgere nella modulazione della massa muscolare e della sua funzione. Nel complesso, le prove presentate indicano che gli acidi grassi saturi agiscono per trasmettere effetti dannosi sulla funzione muscolare, ad esempio alterando o riducendo la sintesi proteica e accentuando il catabolismo. Al contrario, è stato dimostrato che diversi acidi grassi insaturi contrastano molte delle azioni pro-cataboliche associate alla assunzione di acidi grassi saturi. Tuttavia, sarà necessari ulteriori studi per delineare le vie e i processi alla base dell’atrofia muscolare indotta da acidi grassi, nonché quelli che mediano miglioramenti nella funzione muscolare in risposta alla fornitura di acidi grassi insaturi (cioè aumento della sintesi proteica, ridotta atrofia, miglioramento della funzione metabolica). A tal fine, strategie volte ad alterare il contenuto e/o la composizione lipidica intramuscolare in quelle condizioni che possono favorire l’atrofia muscolare (es. aumento dell’obesità, invecchiamento, inattività fisica), ad esempio sopprimendo l’accumulo di mediatori lipidici come le Ceramidi (es. de novosintesi di ceramide) o prostaglandine (ad es. inibendo l’attività della COX-2), possono fornire informazioni utili sul ruolo che diverse classi di lipidi svolgono nella modulazione della massa e della funzione muscolare. È importante sottolineare che tale lavoro potrebbe implicare l’uso di modelli animali pertinenti o soggetti umani che richiederebbero di prendere in considerazione fattori come il background genetico, la composizione della dieta e l’apporto calorico. Inoltre, questi studi possono anche comportare la determinazione di potenziali alterazioni indotte dai lipidi dell’architettura muscolare e della composizione del tipo di fibra che possono influenzare la forza muscolare, nonché il monitoraggio dei cambiamenti nella segnalazione intramuscolare e nei metaboliti all’interno di specifici tipi di fibre muscolari. Insieme a questo, ulteriori lavori che esplorano il ruolo degli acidi grassi e degli intermedi lipidici nella regolazione della proliferazione, sarebbe necessario eseguire la differenziazione e/o la funzione delle cellule satellite derivate da muscoli umani e dei miotubi primari al fine di effettuare confronti appropriati con i dati ottenuti in altri modelli sperimentali (ad es. miotubi C2C12), che hanno dimostrato di mostrare differenze funzionali (ad es. grado di maturazione).[82] Nel complesso , i dati ottenuti da tali studi possono portare allo sviluppo di nuove strategie terapeutiche per contrastare l’atrofia muscolare e/o migliorare la capacità rigenerativa a seguito di lesioni o malattie.
Potenziali meccanismi attraverso i quali gli acidi grassi insaturi possono contrastare l’obesità e/o l’atrofia del muscolo scheletrico indotta da acidi grassi saturi. Livelli elevati di acidi grassi saturi e/o citochine proinfiammatorie, ad esempio durante l’obesità e ipoteticamente anche nel sovrappeso marcato, possono favorire lo sviluppo dell’atrofia del muscolo scheletrico attraverso varie vie e processi come indicato (A). È importante sottolineare che gli acidi grassi monoinsaturi e polinsaturi hanno dimostrato di contrastare queste azioni pro-atrofiche, che possono essere mediate dalla loro capacità di aumentare la sensibilità all’insulina e la produzione di eicosanoidi protettivi, oltre a migliorare la capacità ossidativa mitocondriale e la sintesi proteica, mentre riducono contemporaneamente la spinta proinfiammatoria (B).
Torniamo alla pratica: tesi, antitesi e sintesi del “Bulk High Fat”.
Quindi, cosa concludere da tutto ciò che ho esposto proveniente dalla letteratura scientifica se parliamo di Bodybuilding e alimentazione “High Fat” in ipercalorica? Quanto segue:
La cosa certa, e risaputa in tutti i contesti dietetici, è quella di preoccuparsi di ridurre l’assunzione di acidi grassi saturi a circa e non oltre il 10% del totale calorico giornaliero (soprattutto l’AcidoPalmitico, Miristico e Laurico);
Concentrare l’assunzione di acidi grassi utilizzando fonti principalmente di Omega-3 (con particolare attenzione ad assumere EPA e DHA in un range tra 1 e 5g/die)
Assumere una buona dose di GLA (Omega-6) la quale può portare ad una migliore risposta nella biosintesi di prostaglandine anti-infiammatorie;
Possibilità di assumere Acido Arachidonico (Omega-6) in un range di dosaggio tra i 100mg ed 1.5g/die (quest’ultimo dosaggio va calibrato con un apporto di EPA/DHA pari ad un minimo di 2,5g/die) che può portare ad un miglioramento della biosintesi di prostaglandine anti-infiammatorie e con attività anabolizzante (vedi, per esempio, la PGF2-α responsabile con la PGE2 della regolazione della proteolisi e della sintesi proteica; così come della proliferazione, differenziazione e fusione delle celle satellite);
Uno schema di ripartizionamento dei macronutrienti potrebbe essere il seguente: 30-40% di Grassi, 30-20% di Carboidrati e 40-30% di Proteine.
Metabolismo e vie di segnalazione dell’Acido Arachidonico. Sono riassunte 3 delle 4 principali vie metaboliche dell’Acido Arachidonico: il ciclo di Terra, la conversione in prostaglandine che richiede l’attività della cicloossigenasi e la conversione in leucotrieni e lipossine tramite l’attività della 5-lipossigenasi. Non è mostrato il metabolismo del citocromo P-450 dell’Acido Arachidonico. I processi enzimatici sono mostrati in rosso, i substrati ei prodotti sono mostrati in nero e i recettori di superficie della proteina G delle cellule sono mostrati in verde. Gli effetti noti dell’attivazione del recettore sull’adenosina monofosfato ciclico (cAMP) a valle o sui livelli di calcio intracellulare sono mostrati in basso. COX-1, cicloossigenasi-1; COX-2, cicloossigenasi-2; 5-LO, 5-lipossigenasi; FLAP, proteina attivante la 5-lipossigenasi; PGH2, PGD2, PGE2, PGF2a e PGI2, sono prostaglandine H2, D2, E2, F2a e I2, rispettivamente; TXA2: trombossano A2; acido 5-HpETE, 5S-idroperossi-6E,8Z,11Z,14Z-eicosatetraenoico; acido 5-HETE, 5S-idrossi6E,8Z,11Z,14Z-eicosatetraenoico; LTA4, LTB4, LTC4, LTD4 e LTE4 sono i leucotrieni A4, B4, C4, D4 ed E4, rispettivamente; LXA4 e LXB4, lipossine A4 e B4; DP1, EP1-EP4, FP e IP, recettori delle prostaglandine; TP, recettore del trombossano A2; BLT1 e BLT2, recettore dei leucotrieni B4; CysLT1 e CysLT2: recettori dei cisteinil leucotrieni; ALXR: recettore delle lipossine.
Partendo da questi punti si hanno delle “linee guida” finalizzate alla qualità degli acidi grassi e alla quantità media percentuale.
Ora, mettiamo il caso che un soggetto per caratteristiche genetiche non riesca a gestire grosse quantità di glucidi e abbia un buon metabolismo lipidico sul quale poter puntare per eventuali surplus calorici.
Il soggetto in questione si trova bene con un quantitativo di CHO pari a 3g/Kg, e pesando 80Kg la suo quota glucidica giornaliera ammonta a 240g. L’assunzione proteica giornaliera è pari a 2,5g/Kg, per un totale di 200g.
Il suo TDEE di mantenimento è di circa 2.500Kcal. Punta ad un surplus calorico giornaliero di 500Kcal (obbiettivo 3000Kcal/die). Dal momento che sa già quanti CHO e proteine dovrà assumere e a quante calorie ammontano (1760Kcal totali da CHO e Pro) il restante lo andrà ad assumere dai Grassi (1240Kcal = circa 138g di Fat).
Totale macronutrienti del soggetto:
Carboidrati: 240g
Proteine: 200g
Grassi: 138g (di cui il 10% di Saturi ed il restante 90% di insaturi, polinsaturi e monoinsaturi; presenza di 5g/die di EPA e DHA, 300mg/die di GLA e 1.5g/die di Acido Arachidonico per 8-12 settimane).
Il soggetto in questione seguirà questo schema per circa 4 settimana senza effettuare alcuna giornata con un aumento dei CHO e una riduzione della componente proteica e lipidica. In base alle risposte ottenute rimodulerà a bisogno il suo schema alimentare.
Ecco, questo è un banalissimo esempio di come una dieta “Bulk High Fat” possa essere per lo meno impostata in modo logico, anche per quanto riguarda il tipo ipotetico di soggetto menzionato. Nonostante ciò, e vi parlo da persona che ha analizzato i risultati di svariati schemi similari a questo, un vantaggio vero e proprio sulla qualità dell’aumento di peso non è praticamente mai emerso, o per lo meno mai in modo sufficientemente apprezzabile.
A questo punto ci sarebbe anche da riportare quanto affermato in uno studio dal Dr. Jose Antonio, PhD, ricercatore della ISSN, che ha affermato come non vi siano aumenti maggiori di grasso corporeo tra una dieta ipercalorica “High Fat” e una “High Carbs” dopo circa 14 giorni.[https://italia-podcast.it/] Questa affermazione va comunque presa come una ipotesi nata da osservazione di alcuni studi. Sebbene tra la teoria e la pratica spesso c’è differenza, quando si parla di surplus calorico e aumento del grasso corporeo sappiamo tutti quanto gli acidi grassi alimentari vengano facilmente stoccati negli adipociti sotto forma di trigliceridi di deposito quando in surplus calorico e ingeriti in quantità significative con carboidrati. Questi ultimi, o meglio il glucosio, in eccesso, in un soggetto in salute vengono in gran parte dissipati in calore e solo un 5-10% è coinvolto nei processi di De Novo Lipogenesi (DNL).
Alcuni sostenitori delle diete “High Fat” in “Bulk” affermano che tali strategie alimentari portino ad un miglioramento della “durezza muscolare”, anche grazie ad un basso stimolo insulinico e consequenziale riduzione della ritenzione idrica, e della pienezza dei ventri data dai Trigliceridi intra-muscolari. Peccato che, Insulina o meno, gli acidi grassi in eccesso vengono captati e depositati negli adipociti per intervento, tra l’altro, della Proteina Stimolante l’Acilazione (ASP), la quale ha azione insulino-indipendente. I Trigliceridi intra-muscolari (IMTG) danno si un effetto di pienezza muscolare dal momento che legano in piccola parte acqua, ma parliamo di un effetto additivo al ben maggiore volume dato dal glicogeno muscolare e apprezzabile a “bf” basse, da pre-contest per intenderci.
Come molti di voi già sapranno, la sintesi proteica aumenta principalmente:
In seguito ad adattamenti in risposta a sedute di allenamento contro-resistenza correttamente svolte generando tensione meccanica, stress metabolico e danno muscolare;
Mangiando sufficienti calorie;
Assumendo un quantitativo adeguato di proteine (media 1,5-2,5g/Kg).
Raggiungere il primo punto, però, diventa molto più difficile se si consuma una quota glucidica bassa. Le diete ricche di grassi possono fornire molte calorie e proteine, ma risultano per la stragrande maggioranza dei soggetti disfunzionali alle prestazioni in sala pesi. Ricordo, per l’ennesima volta, che un bodybuilder è un atleta che sfrutta primariamente il metabolismo glucidico al fine della massima prestazione essendo praticante di uno sport anaerobico alattacido e lattacido.
Durante l’esercizio intenso, i muscoli si basano principalmente sul metabolismo glucidico. E come certamente saprete, il glicogeno è un polimero di glucosio immagazzinato nel muscolo-scheletrico e nel fegato, ed è scomposto nelle sue unità di glucosio e utilizzato come substrato energetico durante l’esercizio nel distretto muscolare di deposito oppure venendo rilasciato nel flusso ematico dal deposito epatico.
Le richieste di carboidrati nella dieta variano da soggetto a soggetto, ma un culturista che si allena seriamente richiede in media una consistente quantità di glucidi per mantenere le sue riserve di glicogeno e sostenere una buona prestazione, motivo per cui le diete ricche di grassi e povere di carboidrati riducono significativamente i livelli di questo polimero. Ed i problemi annessi emergono soprattutto quando ci si allena con pesi consistenti.
Fare solo 6-9 serie può ridurre i livelli di glicogeno muscolare di circa il 40% [83] e, se ci si allena con livelli già bassi, è ragionevole presumere che si farà fatica a sollevare più peso.
Ma cosa mostrano gli studi sugli atleti? La maggior parte degli studi dimostrano che le diete ricche di grassi e povere di carboidrati riducono le prestazioni atletiche [84, 85, 86] mentre altri affermano che non vi è alcuna differenza [87]e persino alcuni che mostrano un aumento delle prestazioni.[88] Tuttavia, ci sono altri motivi per pensare che le diete ad alto contenuto di carboidrati potrebbero essere superiori per l’aumento della massa muscolare.
La ricerca mostra che la disponibilità di glicogeno influenza direttamente la sintesi proteica ei tassi di degradazione.[89] In poche parole: le diete ricche di grassi e povere di carboidrati comportano livelli inferiori di sintesi proteica rispetto a quelle ad alto contenuto di carboidrati.
Un altro modo in cui i carboidrati influenzano favorevolmente l’equilibrio proteico muscolare ha a che fare con la produzione di Insulina.
L’insulina è un ormone peptidico rilasciato dal pancreas che agevola l’assorbimento dei nutrienti dal sangue alle cellule. Possiede, in fisiologia, proprietà prettamente anticataboliche, il che significa che quando i livelli di Insulina sono elevati, viene soppressa la lisi delle proteine muscolari.[90]
Ora, poiché la produzione di Insulina è stimolata ingerendo cibo, e mangiando carboidrati in particolare, ma anche proteine con stimoli differenti dati dalla composizione amminoacidica, non sorprende che le persone che seguono una dieta ricca di carboidrati abbiano generalmente livelli ottimali di Insulina rispetto alle persone che seguono una dieta povera di carboidrati e ricca di grassi.
La formazione del complesso Insulina (di colore rosso) – Recettore dell’Insulina (di colore blu) determina una cascata di segnali per l’esplicazione della funzione dell’ormone. Ispirato da discussioni e cifre in Siddle (2011). L’immagine del complesso Recettore Insulina-Insulina è adattata da Goodsell (2015).
I carboidrati sono principalmente energetici e se non si è molto attivi, non si necessita di loro quantità elevate. Tuttavia, livelli ottimali di insulina (e di sensibilità insulinica) sono altamente desiderabili se si sta cercando di costruire massa muscolare, semplicemente perché si viene a creare un ambiente più anabolico in cui i muscoli possono vedere agevolata la loro crescita. E questa non è solo teoria.
La ricerca condotta dagli scienziati della Ball State University ha scoperto che bassi livelli di glicogeno muscolare (che è inevitabile con una dieta povera di carboidrati e ricca di grassi) compromettono la segnalazione cellulare post-allenamento relativa alla crescita muscolare.[91]
Un altro studio condotto da ricercatori dell’Università della Carolina del Nord ha scoperto che, se combinata con l’esercizio quotidiano, una dieta povera di carboidrati e ricca di grassi aumenta i livelli di Cortisolo a riposo e diminuisce i livelli di Testosterone libero.[92] Sicuramente una situazione compromettente soprattutto per un “Natural”. Quando si tratta di costruire massa muscolare, ciò che si vuole sono bassi livelli di Cortisolo a riposo e alti livelli di Testosterone libero, l’esatto opposto di ciò che porta una dieta a basso contenuto di carboidrati.
Quanto detto aiuta a spiegare i risultati di altri studi sulla questione dei carboidrati e della composizione corporea e delle prestazioni.
Ad esempio, uno studio condotto da ricercatori dell’Università del Rhode Island ha esaminato come l’assunzione di carboidrati a basso e ad alto contenuto di carboidrati influenzasse il danno muscolare indotto dall’esercizio, il recupero della forza e il metabolismo proteico di tutto il corpo dopo un intenso allenamento. Quello che hanno scoperto è che i soggetti con una dieta a basso contenuto di carboidrati hanno perso più forza, si sono ripresi più lentamente e hanno mostrato livelli più bassi di sintesi proteica. Vale anche la pena notare che il gruppo “a basso contenuto di carboidrati” (e quindi ad alto contenuto di grassi) non era poi così “Low Carbs”. Stavano ingerendo circa 220g di carboidrati al giorno contro i 350g assunti dal gruppo con dieta ad alto contenuto di carboidrati.[93] E questi effetti diventano ancora più pronunciati man mano che l’assunzione di carboidrati diminuisce.
Ancora un altro studio degno di nota è stato condotto dai ricercatori della McMaster University, che ha confrontato la dieta ad alto e basso contenuto di carboidrati con soggetti che eseguono allenamenti quotidiani per le gambe. I soggetti che seguono una dieta a basso contenuto di carboidrati (30% delle calorie giornaliere) hanno mostrato tassi più elevati di degradazione proteica e tassi più bassi di sintesi proteica rispetto ai soggetti a dieta ricca di carboidrati (60% delle calorie giornaliere), con conseguente minore crescita muscolare complessiva.[94]
Conclusionie ultime indicazioni:
La linea di fondo è questa: Alcuni studi dimostrano che le diete ricche di grassi possono funzionare per la costruzione muscolare, ma un approccio più ideale e redditizio sembra rimanere quello di seguire una dieta ricca di carboidrati secondo capacità individuali.
I dati empirici ci suggeriscono mediamente la stessa medesima cosa, senza però cadere nell’errore di valutare le innumerevoli variabili soggettive in un unico macro-gruppo di risposta positiva ai regimi prevalentemente glucidici.
Questo significa, in poche parole, che la soggettività genetica non va mai sottovalutata o peggio rinnegata per via di futili credenze pseudoscientifiche o convinzioni personali.
Ciò non toglie, però, che i regimi ipercalorici “High Fat” non portano a sostanziali benefici se non ad un ristretto numero di individui caratterizzati da ipotetiche alterazioni metaboliche rispetto alla media della popolazione. Ma anche in questi casi il “tetto favorevole” del “High Fat” si attesta mediamente al 40% delle calorie totali giornaliere con un totale glucidico del 30-25%.
Ciò che dobbiamo ricordare è che i test alimentari soggettivi con modulazioni macro-caloriche sono utili al fine di attestare una quota singolarmente funzionale per ogni macronutriente. Solo in questo modo, e con una valutazione oggettiva, possiamo capire fin dove possiamo spingerci in quanto a quantità di macronutrienti al fine di ottenerne il massimo dei vantaggi prestativi ed estetici. Questo può significare differenze non da poco in un piano ipercalorico tra Grassi e Carboidrati.
Altri tre esempi di variazione nella percentuale dei macronutrienti in funzione delle risposte soggettive.
Personalmente, ritengo che il range della quota lipidica dovrebbe coprire un arco di grammatura da 0,6 a 2g/Kg. Sempre su base soggettiva e rapporto funzionale tra carboidrati e grassi.
In regimi ipercalorici, con l’obbiettivo di non perdere la flessibilità metabolica, si possono utilizzare, oltre alla “C:G ratio” di Ludovico Lemme, uno schema d’esempio di rapporto come segue:
CHO 4-5g/Kg= Fat 0,6g/Kg
CHO 5-6g/Kg= Fat 0,8g/Kg
CHO 6-7g/Kg= Fat 1g/Kg
CHO 7-8g/Kg= Fat 1,5/Kg
CHO 8-10g/Kg= Fat 2g/Kg
E se volessi tarare il quantitativo lipidico per un regime “Bulk High Fat”? Le linee indicative sarebbero:
CHO 3-4g/Kg= Fat 1-1,2g/Kg
CHO 2.5-3g/Kg= Fat 1,5-1,6g/Kg
CHO 2-2.5g/Kg= Fat 1,8-2g/Kg
CHO 0,5-2g/Kg= Fat 2-2,5g/Kg
Queste sono semplici indicazioni generali estrapolate da dati raccolti empiricamente, nulla di scolpito nella roccia o inconfutabilmente dimostrato.
Prima di chiudere con il presente articolo, è logico che vi dica di fare attenzioni alle fonti lipidiche che assumete o che andrete ad assumere (cosa già detta in precedenza). Nessuno vi sta dicendo di evitare la carne rossa (tagli magri) ma di concentrarvi soprattutto su fonti lipidiche quali tuorlo d’uovo, Olio Extravergine di Oliva, EPA, DHA e MCT (controllandone la composizione ed evitando l’olio di Cocco ricco di Acido Laurico). Fonti proteiche quali salmone selvatico, sgombro, pesce spada e carni da allevamenti “Grass Feed” sono pienamente consigliate, se alla propria portata economica.
Gabriel Bellizzi
Riferimenti:
Dr. Mauro Di Pasquale, bodybuilding.com, accessed February 21, 2007. His articles and books have been translated into Italian language and published in Italy by Sandro Ciccarelli Olympian’s News magazine.
Woodworth‐Hobbs ME, Hudson MB, Rahnert JA, Zheng B, Franch HA, Price SR. Docosahexaenoic acid prevents palmitate‐induced activation of proteolytic systems in C2C12 myotubes. J Nutr Biochem 2014;25:868–74.
Graves E, Hitt A, Pariza MW, Cook ME, McCarthy DO. Conjugated linoleic acid preserves gastrocnemius muscle mass in mice bearing the colon‐26 adenocarcinoma. Res Nurs Health 2005;28:48–55.
Langen RC, Schols AM, Kelders MC, van der Velden JL, Wouters EF, Janssen‐Heininger YM. Muscle wasting and impaired muscle regeneration in a murine model of chronic pulmonary inflammation. Am J Respir Cell Mol Biol 2006;35:689–96.
Whitehouse AS, Smith HJ, Drake JL, Tisdale MJ. Mechanism of attenuation of skeletal muscle protein catabolism in cancer cachexia by eicosapentaenoic acid. Cancer Res 2001;61:3604–9.
Castillero E, Martin AI, Lopez‐Menduina M, Villanua MA, Lopez‐Calderon A. Eicosapentaenoic acid attenuates arthritis‐induced muscle wasting acting on atrogin‐1 and on myogenic regulatory factors. Am J Physiol Regul Integr Comp Physiol 2009;297:R1322–31.
Fiaccavento R, Carotenuto F, Vecchini A, Binaglia L, Forte G, Capucci E, et al. An omega‐3 fatty acid‐enriched diet prevents skeletal muscle lesions in a hamster model of dystrophy. Am J Pathol 2010;177:2176–84.
Briolay A, Jaafar R, Nemoz G, Bessueille L. Myogenic differentiation and lipid‐raft composition of L6 skeletal muscle cells are modulated by PUFAs. Biochim Biophys Acta 2013;1828:602–13.
Watson ML, Coghlan M, Hundal HS. Modulating serine palmitoyl transferase (SPT) expression and activity unveils a crucial role in lipid‐induced insulin resistance in rat skeletal muscle cells. Biochem J 2009;417:791–801.
. Yuzefovych L, Wilson G, Rachek L. Different effects of oleate vs. palmitate on mitochondrial function, apoptosis, and insulin signaling in L6 skeletal muscle cells: role of oxidative stress. Am J Physiol Endocrinol Metab 2010;299:E1096–105.
Mahfouz R, Khoury R, Blachnio‐Zabielska A, Turban S, Loiseau N, Lipina C, et al. Characterising the inhibitory actions of ceramide upon insulin signaling in different skeletal muscle cell models: a mechanistic insight. PLoS One 2014;9:e101865.
De Larichaudy J, Zufferli A, Serra F, Isidori AM, Naro F, Dessalle K, et al. TNF‐alpha‐ and tumor‐induced skeletal muscle atrophy involves sphingolipid metabolism. Skelet Muscle 2012;2:2.
Mebarek S, Komati H, Naro F, Zeiller C, Alvisi M, Lagarde M, et al. Inhibition of de novo ceramide synthesis upregulates phospholipase D and enhances myogenic differentiation. J Cell Sci 2007;120:407–16.
Turpin SM, Ryall JG, Southgate R, Darby I, Hevener AL, Febbraio MA, et al. Examination of ‘lipotoxicity’ in skeletal muscle of high‐fat fed and ob/ob mice. J Physiol 2009;587:1593–605.
Williamson DL, Dungan CM, Jadhav KS. High lipid concentrations regulate skeletal muscle REDD1. FASEB J 2013;27:Supplement 942.1.
Gordon BS, Williamson DL, Lang CH, Jefferson LS, Kimball SR. Nutrient‐induced stimulation of protein synthesis in mouse skeletal muscle is limited by the mTORC1 repressor REDD1. J Nutr 2015;145:708–13.
Leskawa KC, Erwin RE, Buse PE, Hogan EL. Glycosphingolipid biosynthesis during myogenesis of rat L6 cells in vitro. Mol Cell Biochem 1988;83:47–54.
Anastasia L, Papini N, Colazzo F, Palazzolo G, Tringali C, Dileo L, et al. NEU3 sialidase strictly modulates GM3 levels in skeletal myoblasts C2C12 thus favoring their differentiation and protecting them from apoptosis. J Biol Chem 2008;283:36265–71.
Papini N, Anastasia L, Tringali C, Dileo L, Carubelli I, Sampaolesi M, et al. MmNEU3 sialidase over‐expression in C2C12 myoblasts delays differentiation and induces hypertrophic myotube formation. J Cell Biochem 2012;113:2967–78.
Lipina C, Hundal HS. Ganglioside GM3 as a gatekeeper of obesity‐associated insulin resistance: evidence and mechanisms. FEBS Lett 2015;589:3221–7.
Gangoiti P, Bernacchioni C, Donati C, Cencetti F, Ouro A, Gomez‐Munoz A, et al. Ceramide 1‐phosphate stimulates proliferation of C2C12 myoblasts. Biochimie 2012;94:597–607.
Szendroedi J, Yoshimura T, Phielix E, Koliaki C, Marcucci M, Zhang D, et al. Role of diacylglycerol activation of PKCtheta in lipid‐induced muscle insulin resistance in humans. Proc Natl Acad Sci U S A 2014;111:9597–602.
You JS, Lincoln HC, Kim CR, Frey JW, Goodman CA, Zhong XP, et al. The role of diacylglycerol kinase zeta and phosphatidic acid in the mechanical activation of mammalian target of rapamycin (mTOR) signaling and skeletal muscle hypertrophy. J Biol Chem 2014;289:1551–63.
Shad BJ, Smeuninx B, Atherton PJ, Breen L. The mechanistic and ergogenic effects of phosphatidic acid in skeletal muscle. Appl Physiol Nutr Metab 2015;40:1233–41.
Bilim O, Takeishi Y, Kitahara T, Arimoto T, Niizeki T, Sasaki T, et al. Diacylglycerol kinase zeta inhibits myocardial atrophy and restores cardiac dysfunction in streptozotocin‐induced diabetes mellitus. Cardiovasc Diabetol 2008;7:2.
Haemmerle G, Zimmermann R, Hayn M, Theussl C, Waeg G, Wagner E, et al. Hormone‐sensitive lipase deficiency in mice causes diglyceride accumulation in adipose tissue, muscle, and testis. J Biol Chem 2002;277:4806–15.
Marignani PA, Epand RM, Sebaldt RJ. Acyl chain dependence of diacylglycerol activation of protein kinase C activity in vitro. Biochem Biophys Res Commun 1996;225:469–73.
Eichmann TO, Kumari M, Haas JT, Farese RV Jr, Zimmermann R, Lass A, et al. Studies on the substrate and stereo/regioselectivity of adipose triglyceride lipase, hormone‐sensitive lipase, and diacylglycerol‐O‐acyltransferases. J Biol Chem 2012;287:41446–57.
Macrae K, Stretton C, Lipina C, Blachnio‐Zabielska A, Baranowski M, Gorski J, et al. Defining the role of DAG, mitochondrial function, and lipid deposition in palmitate‐induced proinflammatory signaling and its counter‐modulation by palmitoleate. J Lipid Res 2013;54:2366–78.
Rando RR, Young N. The stereospecific activation of protein kinase C. Biochem Biophys Res Commun 1984;122:818–23.
Badin PM, Louche K, Mairal A, Liebisch G, Schmitz G, Rustan AC, et al. Altered skeletal muscle lipase expression and activity contribute to insulin resistance in humans. Diabetes 2011;60:1734–42.
Hyde R, Hajduch E, Powell DJ, Taylor PM, Hundal HS. Ceramide down‐regulates System A amino acid transport and protein synthesis in rat skeletal muscle cells. FASEB J 2005;19:461–3.
Guenther GG, Peralta ER, Rosales KR, Wong SY, Siskind LJ, Edinger AL. Ceramide starves cells to death by downregulating nutrient transporter proteins. Proc Natl Acad Sci U S A 2008;105:17402–7.
Nardi F, Hoffmann TM, Stretton C, Cwiklinski E, Taylor PM, Hundal HS. Proteasomal modulation of cellular SNAT2 (SLC38A2) abundance and function by unsaturated fatty acid availability. J Biol Chem 2015;290:8173–84.
Li F, Duan Y, Li Y, Tang Y, Geng M, Oladele OA, et al. Effects of dietary n‐6:n‐3 PUFA ratio on fatty acid composition, free amino acid profile and gene expression of transporters in finishing pigs. Br J Nutr 2015;113:739–48.
Coll T, Eyre E, Rodriguez‐Calvo R, Palomer X, Sanchez RM, Merlos M, et al. Oleate reverses palmitate‐induced insulin resistance and inflammation in skeletal muscle cells. J Biol Chem 2008;283:11107–16.
Salvado L, Coll T, Gomez‐Foix AM, Salmeron E, Barroso E, Palomer X, et al. Oleate prevents saturated‐fatty‐acid‐induced ER stress, inflammation and insulin resistance in skeletal muscle cells through an AMPK‐dependent mechanism. Diabetologia 2013;56:1372–82.
Wu CL, Cornwell EW, Jackman RW, Kandarian SC. NF‐kappaB but not FoxO sites in the MuRF1 promoter are required for transcriptional activation in disuse muscle atrophy. Am J Physiol Cell Physiol 2014;306:C762–7.
Rivas DA, McDonald DJ, Rice NP, Haran PH, Dolnikowski GG, Fielding RA. Diminished anabolic signaling response to insulin induced by intramuscular lipid accumulation is associated with inflammation in aging but not obesity. Am J Physiol Regul Integr Comp Physiol 2016;310:R561–9.
Liu Y, Chen F, Odle J, Lin X, Zhu H, Shi H, et al. Fish oil increases muscle protein mass and modulates Akt/FOXO, TLR4, and NOD signaling in weanling piglets after lipopolysaccharide challenge. J Nutr 2013;143:1331–9.
Doyle A, Zhang G, Abdel Fattah EA, Eissa NT, Li YP. Toll‐like receptor 4 mediates lipopolysaccharide‐induced muscle catabolism via coordinate activation of ubiquitin–proteasome and autophagy–lysosome pathways. FASEB J 2011;25:99–110.
Mittal A, Bhatnagar S, Kumar A, Lach‐Trifilieff E, Wauters S, Li H, et al. The TWEAK‐Fn14 system is a critical regulator of denervation‐induced skeletal muscle atrophy in mice. J Cell Biol 2010;188:833–49.
Finlin BS, Varma V, Nolen GT, Dube J, Starnes CP, Rasouli N, et al. DHA reduces the atrophy‐associated Fn14 protein in differentiated myotubes during coculture with macrophages. J Nutr Biochem 2012;23:885–91.
Jang YC, Lustgarten MS, Liu Y, Muller FL, Bhattacharya A, Liang H, et al. Increased superoxide in vivo accelerates age‐associated muscle atrophy through mitochondrial dysfunction and neuromuscular junction degeneration. FASEB J 2010;24:1376–90.
Gouspillou G, Sgarioto N, Kapchinsky S, Purves‐Smith F, Norris B, Pion CH, et al. Increased sensitivity to mitochondrial permeability transition and myonuclear translocation of endonuclease G in atrophied muscle of physically active older humans. FASEB J 2014;28:1621–33.
Muoio DM, Way JM, Tanner CJ, Winegar DA, Kliewer SA, Houmard JA, et al. Peroxisome proliferator‐activated receptor‐alpha regulates fatty acid utilization in primary human skeletal muscle cells. Diabetes 2002;51:901–9.
Tardif N, Salles J, Landrier JF, Mothe‐Satney I, Guillet C, Boue‐Vaysse C, et al. Oleate‐enriched diet improves insulin sensitivity and restores muscle protein synthesis in old rats. Clin Nutr 2011;30:799–806.
Park SY, Kim MH, Ahn JH, Lee SJ, Lee JH, Eum WS, et al. The stimulatory effect of essential fatty acids on glucose uptake involves both Akt and AMPK activation in C2C12 skeletal muscle cells. Korean J Physiol Pharmacol: Off J Korean Phys Soc Korean Soc Pharm 2014;18:255–61.
Wang Y, Lin QW, Zheng PP, Zhang JS, Huang FR. DHA inhibits protein degradation more efficiently than EPA by regulating the PPARgamma/NFkappaB pathway in C2C12 myotubes. BioMed Res Int 2013;2013:318981.
Lanza IR, Blachnio‐Zabielska A, Johnson ML, Schimke JM, Jakaitis DR, Lebrasseur NK, et al. Influence of fish oil on skeletal muscle mitochondrial energetics and lipid metabolites during high‐fat diet. Am J Physiol Endocrinol Metab 2013;304:E1391–403.
Lim JH, Gerhart‐Hines Z, Dominy JE, Lee Y, Kim S, Tabata M, et al. Oleic acid stimulates complete oxidation of fatty acids through protein kinase A‐dependent activation of SIRT1‐PGC1alpha complex. Nutr Metab 2013;288:7117–26.
Mizunoya W, Iwamoto Y, Shirouchi B, Sato M, Komiya Y, Razin FR, et al. Dietary fat influences the expression of contractile and metabolic genes in rat skeletal muscle. PLoS One 2013;8:e80152.
Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011;147:728–41.
Penna F, Costamagna D, Pin F, Camperi A, Fanzani A, Chiarpotto EM, et al. Autophagic degradation contributes to muscle wasting in cancer cachexia. Am J Pathol 2013;182:1367–78.
O’Leary MF, Vainshtein A, Carter HN, Zhang Y, Hood DA. Denervation‐induced mitochondrial dysfunction and autophagy in skeletal muscle of apoptosis‐deficient animals. Am J Physiol Cell Physiol 2012;303:C447–54.
Wenz T, Rossi SG, Rotundo RL, Spiegelman BM, Moraes CT. Increased muscle PGC‐1alpha expression protects from sarcopenia and metabolic disease during aging. Proc Natl Acad Sci U S A 2009;106:20405–10.
McFarland DC, Velleman SG, Pesall JE, Coy CS. Effect of lipids on avian satellite cell proliferation, differentiation and heparan sulfate proteoglycan expression. Comp Biochem Physiol A Mol Integr Physiol 2011;159:188–95.
Peng Y, Zheng Y, Zhang Y, Zhao J, Chang F, Lu T, et al. Different effects of omega‐3 fatty acids on the cell cycle in C2C12 myoblast proliferation. Mol Cell Biochem 2012;367:165–73.
Schevzov G, Kee AJ, Wang B, Sequeira VB, Hook J, Coombes JD, et al. Regulation of cell proliferation by ERK and signal‐dependent nuclear translocation of ERK is dependent on Tm5NM1‐containing actin filaments. Mol Biol Cell 2015;26:2475–90, doi:10.1091/mbc.E14-10-1453.
Parton RG, Way M, Zorzi N, Stang E. Caveolin‐3 associates with developing T‐tubules during muscle differentiation. J Cell Biol 1997;136:137–54.
Oshikawa J, Otsu K, Toya Y, Tsunematsu T, Hankins R, Kawabe J, et al. Insulin resistance in skeletal muscles of caveolin‐3‐null mice. Proc Natl Acad Sci U S A 2004;101:12670–5.
Galbiati F, Volonte D, Minetti C, Chu JB, Lisanti MP. Phenotypic behavior of caveolin‐3 mutations that cause autosomal dominant limb girdle muscular dystrophy (LGMD‐1C). Retention of LGMD‐1C caveolin‐3 mutants within the golgi complex. J Biol Chem 1999;274:25632–41.
Minetti C, Bado M, Broda P, Sotgia F, Bruno C, Galbiati F, et al. Impairment of caveolae formation and T‐system disorganization in human muscular dystrophy with caveolin‐3 deficiency. Am J Pathol 2002;160:265–70.
Park DS, Woodman SE, Schubert W, Cohen AW, Frank PG, Chandra M, et al. Caveolin‐1/3 double‐knockout mice are viable, but lack both muscle and non‐muscle caveolae, and develop a severe cardiomyopathic phenotype. Am J Pathol 2002;160:2207–17.
Capozza F, Combs TP, Cohen AW, Cho YR, Park SY, Schubert W, et al. Caveolin‐3 knockout mice show increased adiposity and whole body insulin resistance, with ligand‐induced insulin receptor instability in skeletal muscle. Am J Physiol Cell Physiol 2005;288:C1317–31.
Gazzerro E, Sotgia F, Bruno C, Lisanti MP, Minetti C. Caveolinopathies: from the biology of caveolin‐3 to human diseases. Eur J Hum Genet: EJHG 2010;18:137–45.
Markworth JF, Cameron‐Smith D. Arachidonic acid supplementation enhances in vitro skeletal muscle cell growth via a COX‐2‐dependent pathway. Am J Physiol Cell Physiol 2013;304:C56–67.
Hausman RE, Velleman SG. Prostaglandin E1 receptors on chick embryo myoblasts. Biochem Biophys Res Commun 1981;103:213–8.
Santini MT, Indovina PL, Hausman RE. Prostaglandin dependence of membrane order changes during myogenesis in vitro. Biochim Biophys Acta 1988;938:489–92.
Hausman RE, elGendy H, Craft F. Requirement for G protein activity at a specific time during embryonic chick myogenesis. Cell Differ Dev: The Official J Int Soc Dev Biol 1990;29:13–20.
Iannotti FA, Silvestri C, Mazzarella E, Martella A, Calvigioni D, Piscitelli F, et al. The endocannabinoid 2‐AG controls skeletal muscle cell differentiation via CB1 receptor‐dependent inhibition of Kv7 channels. Proc Natl Acad Sci U S A 2014;111:E2472–81.
Murton AJ, Marimuthu K, Mallinson JE, Selby AL, Smith K, Rennie MJ, et al. Obesity appears to be associated with altered muscle protein synthetic and breakdown responses to increased nutrient delivery in older men, but not reduced muscle mass or contractile function. Diabetes 2015;64:3160–71.
McGlory C, Galloway SD, Hamilton DL, McClintock C, Breen L, Dick JR, et al. Temporal changes in human skeletal muscle and blood lipid composition with fish oil supplementation. Prostaglandins Leukot Essent Fatty Acids 2014;90:199–206.
Jans A, van Hees AM, Gjelstad IM, Sparks LM, Tierney AC, Riserus U, et al. Impact of dietary fat quantity and quality on skeletal muscle fatty acid metabolism in subjects with the metabolic syndrome. Metabolism 2012;61:1554–65.
Rasic‐Milutinovic Z, Perunicic G, Pljesa S, Gluvic Z, Sobajic S, Djuric I, et al. Effects of N‐3 PUFAs supplementation on insulin resistance and inflammatory biomarkers in hemodialysis patients. Ren Fail 2007;29:321–9.
Dangardt F, Chen Y, Gronowitz E, Dahlgren J, Friberg P, Strandvik B. High physiological omega‐3 fatty acid supplementation affects muscle fatty acid composition and glucose and insulin homeostasis in obese adolescents. J Nutr Metab 2012;2012:395757.
Jimenez‐Gomez Y, Cruz‐Teno C, Rangel‐Zuniga OA, Peinado JR, Perez‐Martinez P, Delgado‐Lista J, et al. Effect of dietary fat modification on subcutaneous white adipose tissue insulin sensitivity in patients with metabolic syndrome. Mol Nutr Food Res 2014;58:2177–88.
Kawabata F, Neya M, Hamazaki K, Watanabe Y, Kobayashi S, Tsuji T. Supplementation with eicosapentaenoic acid‐rich fish oil improves exercise economy and reduces perceived exertion during submaximal steady‐state exercise in normal healthy untrained men. Biosci Biotechnol Biochem 2014;78:2081–8.
Henry R, Peoples GE, McLennan PL. Muscle fatigue resistance in the rat hindlimb in vivo from low dietary intakes of tuna fish oil that selectively increase phospholipid n‐3 docosahexaenoic acid according to muscle fibre type. Br J Nutr 2015;114:873–84.
Peoples GE, McLennan PL. Long‐chain n‐3 DHA reduces the extent of skeletal muscle fatigue in the rat in vivo hindlimb model. Br J Nutr 2014;111:996–1003.
Langelaan ML, Boonen KJ, Rosaria‐Chak KY, van der Schaft DW, Post MJ, Baaijens FP. Advanced maturation by electrical stimulation: differences in response between C2C12 and primary muscle progenitor cells. J Tissue Eng Regen Med 2011;5:529–39.
DISCLAIMER: Il presente articolo è a solo scopo educativo, di intrattenimento e informativo. Non rappresenta in alcun modo una forma di incitamento all’uso/abuso di sostanze dopanti. L’autore ed il sito, per tanto, è esentato da qualsiasi responsabilità dipendente dalla libera scelta individuale.
Introduzione ad un dilemma…
Chiunque frequenti l’ambiente del Bodybuilding e del Fitness avrà letto o sentito almeno una volta nella vita espressioni del genere “se mi dopassi sarei anche io così [indicando Flex Wheeler]” o “ho provato di tutto e senza farmaci non riuscirò ad ottenere risultati”. Andando poi ad approfondire la storia di ognuno di questi soggetti si scopre in percentuale quasi assoluta che si tratta di individui nella norma (o al di sotto) frustrati e/o con personalità deboli, speranzosi omini che attendono placidamente che accada una svolta miracolosa nella loro banale e piatta esistenza e, cosa molto importante, con il minimo dello sforzo (meglio se nessuno).
Nella mia esperienza come ricercatore e operatore nel campo della cultura fisica in qualità di Preparatore Atletico, ho assistito a innumerevoli casi in cui un soggetto aspirava al miglioramento della propria composizione corporea trascurando, consciamente o inconsciamente, le basi fondamentali rappresentate da Nutrizione e Allenamento baipassandole in vista della possibile prescrizione di una pillola miracolosa capace di renderlo/a possessore della forma fisica ambita.
Tralasciando l’ovvio ragionamento che spinge ogni essere umano dotato di un minimo d’intelletto verso la comprensione che la genetica è il blocco d’argilla sul quale si va ad operare, ma le sue qualità e difetti sono presenti in modo eterogeneo nella popolazione mondiale, e ciò non è modificabile nemmeno con la farmacologia più oculata, quando ci si trova davanti al bivio tra “pillola rossa” (PEDs) e “pillola blu” (drug free) bisogna essere pienamente consapevoli non solo del fattore illegalità ma del fattore conoscitivo. Purtroppo, la politica del terrore ha operato in modo fallimentare nel goffo intento di allontanare dalla scelta “rossa”, e ciò si è tradotto in un numero sensibile di soggetti abusatori con tutte le conseguenze cliniche derivanti.
Se un individuo non ha raggiunto un livello di maturità sportiva tale da conferirgli una gestione corretta della nutrizione e della periodizzazione allenante (gestione delle variabili volume, intensità, densità ecc…), è molto meglio per lui/lei rivedere i suoi programmi e scegliere ancora la “pillola blu”. Capita, a volte, di incontrare persone decise ad intraprendere la via del “lato oscuro” che, dopo una approfondita chiacchierata sulla gestione dei suddetti fattori, rivede le proprie posizioni.
Per tutti coloro i quali sono immersi nel dilemma della scelta, vi espongo alcuni punti per rendere l’eventuale decisione meno rischiosa anche se pur sempre illegale nel “bel paese”…
“Pillola blu o pillola rossa?” I punti da tenere in considerazione per una scelta consapevole:
#1 Raggiungere una adeguata maturità sportiva
Per “maturità sportiva”, in particolare riferimento al BodyBuilding, si intende la capacità del atleta di sapersi alimentare e allenare correttamente con piena gestione delle proprie potenzialità fisiologiche/genetiche. Questa è la base, se viene a mancare ciò non solo la vostra esperienza finirà per deludervi e rendervi ancora di più dei frustrati, ma potrebbe rovinosamente portarvi ad un abuso cronico a senso inesorabilmente negativo…
#2 I PEDs non faranno miracoli
Una cosa da tenere bene a mente, e questo non dovrebbe interessare solo gli aspiranti “doped”, è che l’uso di PEDs non renderà diversi da ciò che rientra nelle potenzialità espressive del proprio patrimonio genetico. Certamente le caratteristiche genetiche verranno “iperespresse”, nel bene e nel male, dall’uso di PEDs ma non vi sarà nessun miracolo! Migliorerete ma non sarete ne più ne meno di ciò che potete essere!
Un esempio per capire come la base genetica faccia la differenza anche con protocolli che, ad oggi, spesso non raggiungono nemmeno i livelli del “bridge” più soft..
#3 Ridurre la percentuale di grasso corporeo
Il tessuto adiposo rappresenta uno dei siti dove il Testosterone, ed altri AAS soggetti all’aromatizzazione, viene convertito in Estradiolo. Soggetti con percentuali di grasso corporeo elevate vedrebbero una alterazione marcata della Testosterone:Estradiolo ratio a favore della componente estrogenica, con conseguenze quali alterazione del comportamento sessuale (impotenza, difficoltà nel raggiungere e/o mantenere l’erezione), ritenzione idrica, accumulo di grasso con modello femminile e ginecomastia. E no, l’uso di DHT derivati o di SARM non steroidei senza una base di Testosterone non risolverebbe il problema o, per lo meno, porterebbe ad altre conseguenze negative, che pur non comprendendo, per esempio, ritenzione idrica e ginecomastia, interesserebbero l’attività sessuale e la condizione psichica del soggetto trattato. [1]
Schema esemplificato del processo di aromatizzazione degli androgeni aumentati in un soggetto con percentuale di grasso corporeo alta.
Allo stesso tempo, i rischi cardiovascolari della somministrazione di AAS- come il possibile aumento esponenziale del Ematocrito, l’aumento del LDL e Trigliceridi a discapito di una riduzione del HDL, e l’aumento della pressione sanguigna – sarebbero già presenti in certa misura quando la body fat è già alta e sarebbero quindi soggetti ad un repentino aggravamento.
Se la percentuale di grasso è relativamente alta, si dovrebbe prima di tutto considerare di migliorare la composizione corporea con una adeguata routine alimentare e allenante (senza farmaci) prima di iniziare solo a pensare all’uso di AAS. Sicuramente ciò renderà la scelta più efficace e meno rischiosa.
Nel caso fosse necessario sottolinearlo, no, non è saggio nemmeno utilizzare agenti PEDs a fini lipolitici e/o antiadipogenici e/o termogenici (compresi gli Ormoni Tiroidei). A meno che non siate affetti da ipotiroidismo, e in questo caso la terapia vi dovrebbe essere stilata dal vostro medico, per ridurre in modo sensibile la body fat non sono necessari i farmaci!
#4 Controllare se si ha una storia familiare di trombosi (o qualsiasi altra malattia cardiovascolare)
Molte malattie cardiovascolari hanno una componente di base genetica. Uno stile di vita sano può ridurne sensibilmente la loro insorgenza, ma l’uso di AAS può causare l’attivazione di specifici geni implicati nella comparsa di malattie cardio-circolatorie. Caratteristico dell’interazione tra AAS e geni specifici è un caso studio ben documentato che ricercatori americani hanno pubblicato sul “Blood Coagulation & Fibrinolysis”.[2]
Trombosi venosa
Oltre all’attivazione genica diretta dagli AAS, e nociva per il sistema cardio-circolatorio, vi sono altre condizioni negative innescate dall’uso/abuso di Steroidi Anabolizzanti, e di altri PEDs, come, per esempio, l’aumento del tasso di coagulazione, l’incremento eccessivo dell’Ematocrito con aumento pressorio, rigidità dell’endotelio vascolare con perdita di efficienza strutturale e aumento della pressione ematica con incremento delle possibilità di danno strutturale dei componenti del sistema interessato.
#5 Inserire delle sedute di allenamento cardio prima, durante e dopo l’uso di PEDs
Un moderato allenamento cardiovascolare è sicuramente una delle migliori strategie preventive contro la comparsa di malattie cardio-circolatorie. Tale tipologia di allenamento può portare un miglioramento e/o riduzione delle alterazioni lipidiche ematiche del praticante, fornendo un, seppur minimo, tampone all’azione negativa degli AAS e SARM non steroidei sui livelli di LDL (aumento), Trigliceridi (aumento) e HDL (diminuzione). Secondo quanto riportato da una interessante review del 2013, l’abbinamento di sedute cardio e in sala pesi possono avere una azione additiva benefica sui livelli di LDL, Trigliceridi e HDL.[3]
Risulta interessante anche quanto emerso da alcuni studi su animali a seguito dei quali si è osservato un significativo grado di protezione dato dall’allenamento cardio negli esemplari trattati con AAS.[4]
#6 Assicurarsi di rimanere ben idratati
Oltre ad agevolare il mantenimento di un Ematocrito migliore, una buona idratazione risulta positiva sulla pressione di lavoro renale nel filtraggio del sangue. Diversi AAS come il Trenbolone e i metilati in C-17 presentano una particolare resistenza metabolica che, oltre a causare un aumentato stress epatico, può portare ad una sofferenza renale sfociabile nel patologico. Si è osservato come una combinazione di AAS, dieta iperproteica e supplementazione di Creatina possa aumentare l’incidenza di problemi renali.[5] In un soggetto in fisiologia, la sola dieta ad altro contenuto proteico e la supplementazione di Creatina non hanno mostrato nessun grado di pericolosità, soprattutto sul breve/medio termine.
#7 Non usare “droghe ricreative”
A livello globale, il numero di decessi tra gli abusatori di AAS è in aumento. Alcuni, troppo superficialmente, dicono che questo sia dovuto al fatto che sempre più uomini e donne usano AAS, ma questa è solo una spiegazione dozzinale e limitata. Il sospetto ricade soprattutto sulle modalità di approccio dei consumatori di AAS: i dosaggi sono drammaticamente aumentati e un numero crescente di individui combina PEDs con “droghe ricreative”. Ed è su questi due ultimi punti che risiede la spiegazione principale dell’aumento statistico prima menzionato. Soprattutto la combinazione di PEDs e le così dette “droghe ricreative” risulta essere probabilmente un fattore significativo, come evidenziato alcuni anni fa da ricercatori australiani. Nel loro studio sono state analizzate tutte le morti documentate tra i consumatori di AAS a Sydney tra il 1997 e il 2012, scoprendo che le droghe ricreative come la cocaina avevano avuto un ruolo nella schiacciante molteplicità dei casi. Dagli studi sugli animali ora sappiamo della possibilità che la co-assunzione di un AAS come il Nandrolone con la cocaina vede moltiplicati gli effetti cardiotossici rispetto ai singoli composti.[6] E secondo studi in vitro la combinazione di Testosterone e cocaina aumenterebbe la possibilità di formazione di coaguli nel flusso ematico. [7]
#8 Corretta modalità di iniezione e herpes labiale
Gli utilizzatori di AAS a volte sviluppano ascessi, ma non sempre dovuti alla bassa qualità dei prodotti utilizzati.
Alcuni medici ritengono che gli utilizzatori di AAS dovrebbero effettivamente ricevere una formazione sulle tecniche di iniezione corrette, onde evitare embolie oleose o ascessi per cattiva gestione igienica della procedura. [8]
Molti utilizzatori ancora non sanno che disturbi come l’herpes labiale rendono le iniezioni ancora più rischiose. Perchè? Il virus che causa l’herpes labiale, come altri patogeni, riduce l’efficienza del sistema immunitario, fornendo così terreno fertile per infezioni batteriche i cui microorganismi scatenanti vengono inoculati nel corpo del soggetto attraverso l’iniezione in modo diretto o indiretto.
#9 Non fare affidamento sugli integratori
Secondo un buon numero di studi svolti su animali, alcuni integratori proteggono dagli effetti collaterali degli AAS. Secondo alcune ricerche, la Taurina, la Vitamina C ed E proteggono i testicoli durante un ciclo e la vitamina C e il cacao proteggono la prostata.
L’utilità dei risultati provenienti da questi studi è limitata per tre motivi:
A. gli animali da laboratorio non sono esseri umani, e
B. le dosi utilizzate e rapportate ad un essere umano sono quasi sempre molto inferiori rispetto a quelle utilizzate dai “doped”, e
C. la ricerca in campo psicologico mostra che l’uso di integratori stimola comportamenti rischiosi e malsani. I supplementi fanno pensare agli utilizzatori di essere invulnerabili e di non dover comportarsi in modo sano ed attento.[9]
Gli integratori possono aiutare a creare una mentalità che non si dovrebbe avere da utilizzatore consapevole di AAS.
Ovviamente, alcuni supplementi “protettivi” utilizzati dai soggetti meglio informati hanno un potenziale di “tamponare” in modo discreto alcune alterazioni legate all’uso di AAS e SARM come, ma non limitato a, Riso Rosso fermantato (controllo lipidico) [10], Silimarina (epatoprotezione), NAC (epatoprotezione) [11], Niacina (controllo lipidico) ecc…
#10 Ridurre al minimo (se non eliminare) il consumo di alcolici
Potrebbe sembrare un indicazione superflua ma non lo è.
L’abuso di alcol è indubbiamente uno dei problemi sociali più diffusi. Uno dei problemi correlati all’abuso di alcol e l’epatopatia alcolica. Questo stato patologico è derivante da un processo infiammatorio progressivo ai danni del fegato legato al consumo eccessivo di alcolici. È una malattia a più stadi. La steatosi provoca un ingrossamento del fegato causato da un accumulo di trigliceridi, spesso senza sintomi per molto tempo. I rischi correlati sono la steatosi (fegato grasso), l’epatite alcolica e la cirrosi epatica. Il rapporto con l’alcolismo è complesso. Non tutti i bevitori, infatti, hanno danni al fegato, anche se sono altamente probabili. La causa è da rinvenire in una trasformazione dell’alcol (etanolo) in sostanze tossiche che danneggiano il fegato in maniera irreversibile e cronica, con un rischio elevato di insufficienza epatica e di cancro, fino alla necessità di un trapianto di fegato.
In acuto, invece, l’alcol può essere una causa di alterazione delle transaminasi ma non si può sapere se e con quale modalità si potrebbero innalzare: dipende molto dalla risposta individuale dell’organismo. In caso di stress preesistente, di causa iatrogena e/o alimentare, si può presentare una alterazione significativa. [12]
Il primo caso è una consequenziale possibile se eventi stressori concomitanti si presentano in cronico. Ed è semplice giungere alla conclusione che l’uso di AAS, specie se metilati, possa comportare un aumentato stress epatico che potrebbe degenerare in peliosi epatica, cirrosi ecc…
Che siate “doped” o “natural”, per ragioni legate e non, dovreste evitare di consumare più di 25g per gli uomini, o 12,5g per le donne, di Etanolo al giorno.
#11 Sottoporsi a regolari controlli medici pre, intra e post utilizzo
Il monitoraggio della salute dovrebbe essere la base fondante del comportamento del utilizzatore consapevole e minimamente attento ai potenziali rischi nei quali potrebbe imbattersi.
Gli esami di controllo sono i seguenti:
Esami ematici e delle urine (comprendenti il quadro ormonale secondo necessità);
Elettrocardiogramma ogni 6 mesi circa;
Elettrocardiogramma sotto sforzo (prima di iniziare);
Ecocardiogramma ogni 6 mesi circa;
Coronarografia ogni 6 mesi circa;
Monitoraggio della pressione ematica;
TAC addome completa ogni 6 mesi circa.
Ovviamente, ogni accertamento , al di la degli esami ematici, deve essere gestito in base alle esigenze soggettive, caratteristiche e tipo di PEDs utilizzati.
#12 Essere seguiti da personale qualificato
Fin troppa gente è stata salutisticamente deturpata da gorilla di spogliatoio a mala pena consapevoli dell’esistenza dei macronutrienti e che, nonostante ciò, si sono improvvisati farmacisti. Donne divenuti uomini e uomini divenuti simili a cagne in calore per via di orrende ginecomastie. Evitate il fai da te e l’affidarsi a semianalfabeti … la somaticità sopra la norma è cosa diversa dall’intelligenza e alla competenza in biologia, biochimica e farmacologia… senza offesa per tutti quelli che “io mi facevo e ho vinto! Senzia scienzia!” …
#13 Pensare seriamente al post ciclo prima del ciclo
Molti aspiranti “doped” non considerano il fattore post ciclo. La maggior parte di loro è convinta che la PCT sarà una facile soluzione alla sottoregolazione dell’Asse HPT, ma in realtà non è proprio così. Esistono diversi casi studio che mostrano come gli ex utilizzatori abbiano spesso livelli di Testosterone inferiori rispetto al pre-utilizzo anche a distanza di anni dal cessato uso di AAS. Sembra che i fattori che aumentano le possibilità e il grado di tale effetto sul lungo termine siano:
Tempo di somministrazione;
Età
Molecole utilizzate (con maggiore impatto negativo dato dai19-norsteroidi come il Nandrolone per via della lunga permanenza dei metaboliti nel sistema).
Tutto ciò è indipendente dalla qualità della PCT, anche se essa può avere dei riscontri positivi specie nel primo periodo di stacco dagli AAS. Le alterazioni ormonali legate ad una alterazione dell’Asse HPT comprendono depressione, ansia, bassa libido, difficoltà nel raggiungere e mantenere l’erezione, stanchezza cronica ecc…
Per questa ragione molti scelgono di entrare in TRT (Terapia Sostitutiva del Testosterone) dopo il primo ciclo.
Quale conclusione?…
Se mai non dovesse bastare il disclaimer, questo articolo non rappresenta in alcun modo un consiglio e, ne tanto meno, un incitamento all’uso di sostanze dopanti! E’ semplicemente a fine divulgativo con l’obbiettivo di far comprendere a più persone possibili che la scelta di intraprendere coscientemente certe pratiche (illegali) necessita di una sufficiente (e veritiera) conoscenza del argomento.
Quindi? Leggete e comprendete correttamente ciò che ho riportato in sintesi fruibile ad un largo pubblico… Pensate prima di tutto ad alimentarvi e allenarvi in modo ottimale!
La conoscenza della Verità rende liberi dalla cattiva informazione, dagli strumenti commerciali e dal relativismo… Negarla è semplice e pericolosa manifestazione di profonda ignoranza… di VERO NEGAZIONISMO!
Se avete una buona conoscenza della lingua inglese e volete approfondire l’argomento PEDs e Sport, potete leggere il libro ANABOLICS 11th Edition di William Llewellyn
La Niacina è largamente utilizzata dagli atleti supplementati chimicamente, in special modo da coloro i quali usano molecole con un potenziale negativo marcato sui lipidi ematici. Ma come spesso capita, gli utilizzatori non conoscono a sufficienza le caratteristiche di ciò che assumono, e questa essenziale vitamina del gruppo B (B3) non è da meno. Per la maggior parte degli individui tanto basta sapere che una sua integrazione si traduce in livelli migliorati di Colesterolo e Trigliceridi. Purtroppo, però, si trascurano caratteristiche importanti la cui conoscenza può fare la differenza tra un uso più o meno funzionale per la salute sistemica. Infatti, un effetto collaterale dell’integrazione di Niacina è un peggioramento della resistenza all’insulina, cosa che limita i benefici di tale supplementazione sulla salute cardiovascolare se non vengono prese adeguate precauzioni.
Prima di correre a defenestrare in preda al panico la vostra Niacina, leggete con attenzione (e comprendete) le informazioni che seguono…
Introduzione alla Niacina (vitamina B3)
Niacina
La Niacina, nota anche come Acido Nicotinico, è un composto organico e una forma di vitamina B3, un micronutriente essenziale per l’essere umano. [1] La Niacina ha formula bruta C6H5NO2 e appartiene al gruppo dell’acido piridinecarbossilico.[1] Come precursore di NAD e NADP, la Niacina è coinvolta nella riparazione del DNA.[2] La Niacina viene assunta attraverso la dieta da una varietà di alimenti interi e trasformati, con il più alto contenuto in alimenti confezionati fortificati, carne, pollame, pesce rosso come tonno e salmone, con minori quantità nelle noci, legumi e semi. [1] [3] La Niacina come integratore alimentare viene anche utilizzata per trattare la pellagra, una malattia causata da una sua carenza. Segni e sintomi includono lesioni della pelle e della bocca, anemia, mal di testa e stanchezza.[4] Molti paesi richiedono la sua aggiunta alla farina di grano o ad altri cereali, riducendo così il rischio di pellagra.[1][5] Come vitamina, le raccomandazioni di dosaggio giornaliero indicate in diversi paesi sono 14-18mg/die per gli adulti, quota sufficiente per soddisfare le esigenze delle persone sane. [6] [7] [8]
Sebbene la Niacina e la Nicotinamide (Niacinamide) siano identiche nella loro attività vitaminica, la Nicotinamide non ha gli stessi effetti farmacologici, modificanti i lipidi o gli effetti collaterali della Niacina, cioè quando la Niacina assume il gruppo -amide, non riduce il Colesterolo né causa vampate di calore.[9][10] La Nicotinamide è raccomandata come trattamento per la carenza di Niacina poiché può essere somministrata in quantità correttive senza causare l’effetto negativo del rossore.[11]
La Niacina è anche un farmaco di prescrizione. Quantità molto superiori all’assunzione dietetica raccomandata per le funzioni vitaminiche ridurranno i Trigliceridi nel sangue e le lipoproteine a bassa densità (LDL-C) e aumenteranno le lipoproteine ad alta densità (HDL-C). Ne esistono due forme: Niacina a rilascio immediato e a rilascio prolungato. Le quantità iniziali di prescrizione sono di 500mg/die, con possibilità di essere aumentate nel tempo fino a raggiungere l’effetto terapeutico ricercato. Le dosi a rilascio immediato possono arrivare fino a 3g/die; quelle a rilascio prolungato fino a 2g/die. [12] Nonostante i comprovati cambiamenti lipidici, la Niacina non è stata trovata utile per ridurre il rischio di malattie cardiovascolari nei soggetti già in trattamento con statine. [13] Una review del 2010 aveva concluso che l’efficacia della Niacina si osservava in mono-terapia, [14] ma una review del 2017 che incorporava il doppio del numero degli studi ha concluso che la Niacina su prescrizione, pur influenzando i livelli lipidici, non riduceva la mortalità per tutte le cause, la mortalità cardiovascolare, gli infarti del miocardio, né ictus fatali o non fatali. [15] È stato dimostrato che la Niacina da prescrizione provoca epatotossicità [16] e aumenta il rischio di diabete di tipo 2. [17] [18] Le prescrizioni di Niacina negli Stati Uniti avevano raggiunto il picco nel 2009, a 9,4 milioni, in calo a 1,3 milioni entro il 2017.[19]
Niacina, flusso ematico, pressione e vasodilatazione
Uno studio sulla supplementazione di Niacina che ha valutato il flusso sanguigno dell’avambraccio non è riuscito a trovare un effetto significativo fino a 1g al giorno somministrati nel corso di due settimane in soggetti altrimenti sani, [20] e 1.5g di Niacina a rilascio prolungato negli uomini con sindrome metabolica non sono riusciti a influenzare la dilatazione flusso- mediata (FMD). [21] Un altro studio non è riuscito a trovare un effetto significativo in un intero gruppo di pazienti affetti da afta epizootica, mentre in un gruppo di pazienti con malattia coronarica ha riscontrato un miglioramento in un sottogruppo con bassi livelli HDL-C. [22]
In soggetti con bassi livelli di HDL-C, è stato osservato che 1g di Niacina a rilascio prolungato per una settimana aumenta il flusso sanguigno (via FMD) del 4,5%; questo meccanismo non era correlato alle Prostaglandini, poiché il Laropiprant (un inibitore della Prostaglandine D2) non ha influenzato l’effetto. [23] Questo effetto ha anche coinciso con un aumento della bilirubina indiretta (ma non totale) del 62%. [23] Poiché la bilirubina del acido biliare è un antiossidante endoteliale, [24] e poiché i benefici della niacina sulla funzione endoteliale in questo studio sono stati ritenuti dipendenti dall’ossido nitrico, [23] è stato ipotizzato che un effetto conservativo della bilirubina sulla biodisponibilità dell’ossido nitrico sia alla base della beneficio osservato. Sia l’aumento della bilirubina che il miglioramento del flusso sanguigno si sono dissipati una settimana dopo l’interruzione della Niacina.[23]
Laropiprant
I soggetti che in precedenza avevano subito infarto del miocardio, a seguito del trattamento con Niacina (con Laropiprant) hanno riscontrato un aumento del flusso sanguigno dipendente dall’ossido nitrico (FMD) dopo dodici settimane di terapia insieme a un miglioramento della vasodilatazione indotta da nitroglicerina, entrambe non correlate con alterazioni dei trigliceridi. [25] Miglioramenti simili nel flusso sanguigno sono stati osservati in pazienti con infezione da HIV e con bassi livelli di HDL-C trattati con la sola Niacina. [26]
Prostaglandine D2 (PGD2)
È noto che la Niacina influenza il diametro dei vasi sanguigni, in particolare per via della sua reazione vasodilatativa cutanea (allargamento dei vasi nella pelle), che ha portato a ipotizzare che potrebbe influenzare la pressione sanguigna aumentando il diametro delle arterie e vene. Tuttavia, una review [27] ha notato che un possibile effetto di riduzione della pressione arteriosa della Niacina è indipendente dalla Prostaglandine che media il rossore, nota come PGD2.
È stato osservato che le infusioni di Niacina riducono acutamente la pressione sanguigna negli ipertesi senza alcun effetto nei soggetti con pressione sanguigna normale ed è stata associata ad un aumento della gittata cardiaca e della frequenza cardiaca che era simile in entrambi i gruppi. [28] Un altro studio ha confermato questo risultato, scoprendo che la pressione arteriosa ambulatoriale di 24 ore non sembra essere influenzata da un supplemento di Niacina fino a 1g nell’arco di due settimane in soggetti altrimenti sani. [20]
In termini di effetti della Niacina in cronico sulla pressione sanguigna, una review [27] che ha valutato gli studi che hanno misurato la pressione sanguigna negli ipertesi [29] [30] [31] [32] non ha notato alcun effetto statisticamente significativo nella riduzione della pressione sanguigna associata alla supplementazione di Niacina, sebbene questi studi in quanto a metodologie di misurazione sulle variazioni della pressione sanguigna non fossero ideali secondo gli autori della review. Tuttavia, la review ha osservato che in un ampio studio (il Coronary Drug Project), che inizialmente non è riuscito a trovare alcuna influenza della terapia con Niacina sulla pressione arteriosa, [32] ha osservato variazioni sensibili soltanto sui soggetti con sindrome metabolica. Questi presentavano un lieve riduzione di 2,2mmHg della pressione arteriosa sistolica con una moderata riduzione di 2,9mmHg della pressione diastolica. [33] Un’analisi post-hoc di un altro studio clinico [34] ha rilevato che la pressione arteriosa sistolica è stata abbassata di 2,2mmHg e la pressione sistolica di 2,7 rispetto al placebo nei pazienti dislipidemici trattati per 24 settimane. [35]
Niacina, Trigliceridi, Colesterolo e Aterosclerosi
Apolipoproteina B
La Niacina sembra abbassare i trigliceridi nel sangue inibendo sia la sintesi degli acidi grassi sia la loro esterificazione epatica per formare i trigliceridi, il che aumenta il tasso di degradazione dell’apolipoproteina B riducendo la sua secrezione dalle cellule epatiche. [36] Un meccanismo con cui la Niacina fa questo è attraverso l’inibizione diretta e non competitiva della diacilglicerolo aciltransferasi 2 (DGAT2), l’enzima finale nella sintesi dei trigliceridi nelle cellule epatiche, senza inibizione della DGAT1. [37]
Si è visto che gli effetti della Niacina sulla sintesi dei trigliceridi influenzano i livelli sierici di lipoproteine a densità molto bassa (vLDL-C), dove la terapia con Niacina per 16 settimane in soggetti con malattia del fegato grasso non alcolica (NAFLD) sembra ridurre le vLDL-C nel siero così come i complessi con trigliceridi (vLDL-TG) e apolipoproteina B (vLDL-ApoB) rispetto al placebo e con una potenza paragonabile al fenofibrato. [38] La Niacina lo fa riducendo la secrezione epatica di vLDL-C, sebbene ciò non aumenti la quantità di trigliceridi nel fegato anche nello stato di NAFLD. [38]
Oltre ai suoi effetti sul fegato, la Niacina può anche sopprimere il rilascio di acidi grassi liberi dal tessuto adiposo [39] che normalmente verrebbero reesterificati come trigliceridi nel fegato e quindi secreti via vLDL. [40] Tuttavia, questo meccanismo specifico, che è mediato dal recettore HM74A, [39] non sembra essere rilevante per le proprietà riducenti dei trigliceridi della Niacina. [41]
I benefici sui livelli di trigliceridi possono verificarsi entro una settimana dall’inizio della supplementazione con Niacina a rilascio prolungato (1g), sebbene in misura minore di circa il 4%. [23]
L’integrazione di 1.5-2g di Niacina a rilascio prolungato per due anni con follow-up di un anno nelle persone in terapia con statine caratterizzate da bassi livelli di HDL-C ha mostrato una riduzione dei trigliceridi del 28,6% (statina da sola dell’8,1%). [42]
Esiste un fenomeno noto come “rimbalzo degli acidi grassi” associato alla supplementazione di Niacina, in quanto l’azione iniziale del composto sul suo recettore (HM74A) nel tessuto adiposo può determinare una minore lipolisi e una minore secrezione di acidi grassi non esterificati (NEFA) nel sangue [43] e una migliore conservazione adiposa; [44] si tratta di fenomeni prontamente reversibili in quanto in un giorno di esposizione continua vi è un aumento netto del NEFA piuttosto che la sua soppressione [45] [46] [47] e alterazioni nel NEFA possono non riflette alterazioni dei trigliceridi.
Il primo meccanismo pensato per spiegare il miglioramento del profilo sierico di colesterolo in seguito alla supplementazione di Niacina è stato attraverso la riduzione del rilascio di acidi grassi non esterificati (NEFA) dai tessuti, che non è più considerato un probabile meccanismo in quanto l’integrazione di niacina in cronico è associata ad un aumento, piuttosto che alla soppressione, di NEFA mentre il recettore HM74A appare superfluo in termini di effetti della Niacina nei topi con altri ligandi del HM74A (Acipimox [48] e MK-0354 [49]) che si sono mostrati rispettivamente meno efficaci o inefficaci sul colesterolo. Attualmente si ritiene che l’influenza della Niacina sui NEFA nel siero non sia un fattore determinante nel modo in cui influenza i livelli di colesterolo nel corpo, con le teorie attuali che ipotizzano che il fattore sia determinato dalla sua sintesi e dal suo tasso di catabolismo.
Il primo potenziale meccanismo prevede la sintesi di HDL-C nel fegato attraverso l’aumento della trascrizione del gene ABCA1 (che dipende dal legame LXRα alla regione del promotore DR4 di questo gene). [50] L’attività di ABCA1 promuove la “lipidazione” della principale proteina dell’HDL nota come apolipoproteina AI (ApoAI) aumentando il tasso che associa ai fosfolipidi e al colesterolo, [51] [52] un passaggio obbligatorio nella sintesi dell’HDL-C che è aumentato di 500-1000µM con Niacina in vitro. [50] Questo meccanismo non è stato confermato, poiché mentre l’ApoAI può essere aumentato parallelamente all’aumento dell’HDL-C in soggetti trattati con Niacina e con livelli di HDL-C bassi di base, [53] LXRα sembra richiedere un coattivatore (PPARγ) per esercitare questi effetti, [54] che è attivato dal recettore della Niacina. [55] Tuttavia, l’attività del recettore della Niacina non è stata richiesta per i suoi effetti sui livelli di colesterolo, suggerendo che altri meccanismi potrebbero essere rilevanti.
PPARγ
L’altra teoria relativa alla sintesi di HDL dalla Niacina afferma che ciò dipenda dalla proteina di trasferimento dell’estere del colesterolo (CETP) nonostante la riduzione del colesterolo totale e dei trigliceridi non richieda per entrambe questa proteina. [56] [57] CETP è una proteina che facilita il trasferimento di lipidi tra diverse lipoproteine (generalmente donando un trigliceride da vLDL a HDL e prendendo un estere di colesterolo in un processo noto come trasporto inverso di colesterolo. [58]) La Niacina riduce l’espressione di CETP nel fegato e la sua attività nel sangue dei topi; [56] una riduzione del CETP aumenta la quantità di HDL-C nel sangue poiché i tassi di catabolismo dell’HDL / LDL riflettono l’attività del trasporto inverso del colesterolo e raggiungono rapidamente l’equilibrio, [59] e se il CETP è ridotto allora sarebbe necessario più HDL per normalizzare i tassi di trasporto inverso del colesterolo. Questo meccanismo può anche essere correlato a LXRα, poiché mentre un eteromero di LXRα con il recettore nucleare di vitamina A (RXR) attiva l’elemento DR4 aumenta la CETP [60] la Niacina agevola l’eterodimerizzazione di LXRα e PPARγ che attiva ancora DR4, ma in un modo che promuove l’efflusso di colesterolo. [61-44] Questa eterodimerizzazione competitiva [62] non è stata ancora dimostrata sperimentalmente, e lo studio che ha utilizzato dosi di Niacina da 2g nell’uomo non è riuscito a trovare un’influenza sull’attività del CETP nel siero nonostante un aumento dell’HDL. [63]
L’ultimo potenziale meccanismo per l’aumento dell’HDL non consiste nel suo incremento di sintesi ma piuttosto nel preservare il colesterolo HDL già sintetizzato arricchito con apoAI, riducendo il tasso in cui la lipoproteina viene assunta nelle cellule epatiche nonostante la donazione di colesterolo dall’HDL a queste cellule sia inalterata a causa della riduzione dell’espressione del recettore (catena beta sintasi ATP) che normalmente trasporta l’HDL nella cellula. [64] Questa ipotesi funziona meglio con le osservazioni che suggeriscono che il ridotto catabolismo dell’HDL è il principale fattore determinante dei suoi livelli più elevati, [65] e influenza anche l’apoA1 poiché la sua clearance dal sangue e l’assorbimento da parte dei reni sono ridotti. [66]
Una supplementazione di Niacina a rilascio prolungato (1g) della durata di una settimana in soggetti con bassi livelli di HDL-C non sembra essere sufficiente da aumentare sensibilmente i livelli totali di HDL-C, sebbene sia stata notata una riduzione della dimensione media delle particelle; [23] le variazioni di HDL -C possono mediare un miglioramento della vasodilatazione dipendente dall’ossido nitrico, sebbene sia stato anche osservato un aumento della bilirubina indiretta. [23]
L’integrazione prolungata di Niacina nei diabetici è associata ad un aumento della quantità e delle dimensioni particellari dell’HDL-C (32,7%) mentre le particelle di dimensioni più piccole sono diminuite (8,2%). [67]
È stato osservato che la Niacina conferisce un effetto protettivo sulla mortalità cardiovascolare poiché una metanalisi [68] ha osservato che negli studi su soggetti con malattia coronarica la terapia con Niacina era associata a un minor rischio di rivascolarizzazione dell’arteria coronarica (RR di 0,31; IC al 95% di 0,15-0,63), infarto miocardico non fatale (RR di 0,72; IC al 95% di 0,60-0,86) e attacco ischemico transitorio (RR di 0,76; IC al 95% di 0,61-0,94) mentre la riduzione della mortalità complessiva non è riuscita a raggiungere significatività statistica (RR 0,883; IC 95% di 0,773-1,008). I sette studi inclusi in questa meta-analisi [32] [29] [31] [30] (e un follow-up [69]) hanno totalizzato 5137 pazienti che utilizzavano anche vari prodotti farmaceutici della classe di statine e fibrati .
In uno studio i cui partecipanti erano in terapia con statine e avevano bassi livelli di colesterolo HDL è stato rilevato che 1.5-2g di Niacina a rilascio prolungato sono stati in grado di fornire benefici additivi nel miglioramento dell’HDL-C (20%) e nella riduzione dell’LDL-C (17%) rispetto al placebo, sebbene per quanto riguarda l’endpoint clinico predeterminato (morte o ricovero in ospedale) sia la Niacina che il placebo avevano una uguale quantità di responder. [70] Questo studio ha rilevato un’alta percentuale di pazienti con sindrome metabolica (80%) e commenti [71] hanno suggerito che a causa di una possibile capacità della Niacina a rilascio prolungato di deteriorare l’insulino-resistenza [72] che i suoi benefici potrebbero essere compensati da questo effetto avverso, mentre lo studio stesso ha suggerito che i benefici delle statine hanno sostituito i benefici della Niacina.
Mentre uno studio precedente che utilizzava alte dosi di Niacina a rilascio immediato (3g) ha riscontrato una riduzione della morte del 14% rispetto al placebo insieme a una riduzioni del colesterolo totale, [32] ed è stato osservato che questa riduzione è simile per grandezza agli studi che combinano statine con placebo.
Studi in vitro suggeriscono che la Niacina potrebbe in teoria prevenire la formazione di placche aterosclerotiche riducendo l’infiammazione e il danno alla parete endoteliale attraverso diversi meccanismi. Limitate ricerche su animali hanno mostrato che la Niacina nella dieta, a concentrazioni paragonabili a quelle utilizzate per ridurre il colesterolo, riduce la deposizione della placca sulla parete dell’arteria e ritarda l’aterosclerosi.[73][74][75][76][77][78][79][80]
Niacina e sue interazioni con il metabolismo del glucosio
L’assunzione prolungata di Niacina è stata osservata causare una riduzione della sensibilità all’insulina, causando un aumento compensativo della produzione di insulina da parte delle cellule β del pancreas per mantenere i livelli di glucosio nel sangue. [81] La Niacina non sembra avere effetti diretti sulle cellule β pancreatiche, tuttavia, poiché la perfusione negli isolotti pancreatici (isole di Langerhans) di ratto isolati con Niacina in vitro non ha influenzato la secrezione di insulina. [82] Ciò indica che la Niacina aumenta la produzione di insulina mediante un meccanismo indiretto, secondario a causare insulino-resistenza periferica. È stato osservato che la supplementazione induce resistenza all’insulina a dosi comprese tra 500mg e 1g, che rientrano nell’intervallo di dosaggio che conferisce effetti di riduzione del colesterolo. [83]
In particolare, sembra che sia necessaria una supplementazione cronica di Niacina per aumentare la produzione di Insulina, poiché in uno studio è stato dimostrato che la supplementazione acuta riduce i livelli di questo peptide in soggetti altrimenti sani prima di un picco dopo un giorno, [84] mentre altri studi in acuto hanno notato un effetto minimo o nullo sui livelli di Insulina. [85] [86] [87] [88]
Gli effetti dell’integrazione cronica di Niacina sui livelli di Insulina possono anche dipendere dalla popolazione. È stato osservato che la Niacina provoca iperinsulinemia in soggetti che invecchiano altrimenti sani [83] (1g / die) ed è stato dimostrato che quasi raddoppiano i livelli di Insulina nei soggetti con NAFLD (2g / giorno [89] [90]). Nei pazienti con sindrome metabolica, l’integrazione di Niacina a 6 settimane di somministrazione alla dose di 1.5g / die ha aumentato i livelli di Insulina del 30%. [91]
Nei soggetti obesi con malattia del fegato grasso non alcolico (NAFLD), l’integrazione giornaliera di Niacina a rilascio prolungato (titolata fino a 2g) per 16 settimane sembrava aumentare lo stato di resistenza all’insulina nel fegato, nei muscoli e nel tessuto adiposo [89] con un effetto inibitorio sulle azioni dell’Insulina nel fegato notate negli uomini non diabetici con dislipidemia. [92]
Adiponectina
Negli uomini adulti con sindrome metabolica, è stato osservato che la Niacina a rilascio prolungato alla dose di 1.5g ostacola in modo significativo la sensibilità all’Insulina, valutata dall’HOMA-IR (42%), che è stata associata ad un aumento dell’Insulina sierica nonostante un aumento dell’Adiponectina sierica. [91] Questo è stato notato anche in un altro studio (aumento del 22% dell’HOMA-IR), in cui l’Aspirina assunta insieme alla Niacina non ha impedito la comparsa di una ridotta sensibilità all’Insulina. [93]
Questo effetto può persistere in soggetti altrimenti sani, poiché i soggetti trattati con 1g di Niacina per due settimane a cui veniva somministrato un clamp iperinsulinaemico-euglicemico richiedono meno glucosio per mantenere l’omeostasi, il che è indicativo di una riduzione dell’assorbimento del glucosio (attraverso un aumento dell’Insulino-resistenza). [94]
La resistenza all’Insulina indotta dalla Niacina è stata inizialmente attribuita a un effetto di rebound nel tessuto adiposo in cui un aumento del rilascio di acidi grassi non esterificati (NEFA) da parte della Niacina compromette gli effetti della segnalazione dell’Insulina. [95] [96] Ciò è plausibile, poiché la resistenza all’Insulina può essere indotta con infusione di NEFA in 24 ore nei roditori. [97] Altre fonti suggeriscono che la resistenza all’Insulina non è associata al rebound del NEFA, poiché i soggetti con NAFLD che sperimentano resistenza all’Insulina dalla terapia con Niacina non hanno necessariamente un aumento del NEFA nel siero. [89].
Modello ipotetico per i ruoli intracellulari del DGAT1 e DGAT2.
Un’altra possibile opzione è che la Niacina può inibire in modo non competitivo l’enzima noto come diacilglicerolo aciltransferasi 2 (DGAT2) con un IC50 di 100 µM (potenza simile a circa 300 µM). [98] L’inibizione di questo enzima non causa di per sé resistenza all’insulina con la somministrazione di Niacina, [92] ma poiché il DGAT catalizza il primo stadio della sintesi dei trigliceridi, la sua inibizione può favorire l’accumulo di diacilglicerolo (DAG) che è la molecola che si ritiene spieghi parzialmente la resistenza all’insulina data dalla Niacina. [92] Poiché l’aumento del DAG nelle cellule del fegato sopprime la segnalazione dell’Insulina, [99-162] l’inibizione mediata dalla Niacina del DGAT2 provoca insulino-resistenza, [98] [89] ostacolando così la capacità dell’Insulina di sopprimere la sintesi di glucosio e promuovendo indirettamente uno stato di iperglicemia.
Sebbene l’integrazione cronica di alte dosi di Niacina riduca la sensibilità all’Insulina, ciò non è associato a variazioni dei livelli di glucosio a digiuno. [90] Ciò può essere spiegato da un aumento compensativo della sintesi di Insulina che contrasta la resistenza alla stessa, lasciando sostanzialmente invariati i livelli di glucosio nel sangue. [81]
L’attivazione del recettore della Niacina (HM74A) da parte di alcuni altri agonisti sembra ridurre rapidamente il glucosio sierico nei diabetici migliorando la sensibilità all’Insulina [100] o comunque migliorando i tassi di smaltimento del glucosio. [101] Ciò indica che lo stesso recettore della Niacina può avere effetti benefici sul metabolismo del glucosio e che la resistenza all’Insulina indotta dalla Niacina non si verifica tramite l’attivazione del HM74A.
Quando si osserva il muscolo scheletrico, è stato dimostrato che la terapia con Niacina induce resistenza all’Insulina in questo tessuto in soggetti obesi con NAFLD (2g al giorno nel corso di 16 settimane). Uno studio svolto su ratti a digiuno (il digiuno aumenta la concentrazione plasmatica di acidi grassi non esterificati (NEFA), similmente alla somministrazione di Niacina [102-135] e diminuisce il glicogeno del muscolo scheletrico [103]) in cui sono stati accuratamente somministrati 20mg/kg di Niacina ha mostrato che il glicogeno nel soleo era ridotto mentre il gastrocnemius e il fegato non sono stati influenzati. [103]
Metilgliossale
Quando il processo di glicazione è testato in vitro, la Niacina ha avuto solo effetti inibitori minori sulla glicazione dell’albumina sierica bovina da un noto agente glicante (Metilgliossale [104]) nonostante altri antiossidanti testati come lo Zinco (10-25 µg / mL) avessero più potenti benefici. [105]
È importante sottolineare che qualsiasi effetto della Niacina sulla glicazione in vitro deve essere interpretato con l’avvertenza che la Niacina riduce la sensibilità all’Insulina. Mentre la resistenza all’Insulina indotta dalla Niacina è ben compensata in soggetti sani giovani, lasciando sostanzialmente invariati i livelli di glucosio nel sangue, [81] la compensazione delle cellule β del pancreas negli individui più anziani o in quelli con ridotta tolleranza al glucosio era incompleta in uno studio, [83] causando aumenti nei livelli ematici di glucosio. Pertanto, la misura in cui la Niacina possa influenzare la glicazione in vivo non è chiara e probabilmente dipendente dalla popolazione.
Obesità e massa grassa
L’Adiponectina, un’adipochina nota per migliorare la sensibilità all’Insulina, per essere cardioprotettiva e ritenuta anche antiobesogena, [106] è aumentata in risposta all’attivazione mediata dalla Niacina del recettore HM74A nei topi. [107] La produzione di Adiponectina indotta dalla Niacina è stata rapida in questo studio, aumentando i livelli di questa adipochina del 37% entro 10 minuti da una dose di 30mg / kg per iniezione. I livelli sierici hanno raggiunto il picco dopo 60 minuti e sono rimasti elevati al di sopra del basale fino a 24 ore dopo la somministrazione. [107]
Leptina
È noto anche che la Leptina è aumentata in seguito alla somministrazione di Niacina nell’uomo [91], il che si ritiene si verifichi tramite un meccanismo simile poiché l’agonista farmaceutico HM74A Acipimox induce anch’esso la secrezione di Leptina dal tessuto adiposo in vitro [108] e in vivo. [109]
È stato osservato che la supplementazione di Niacina nel corso di sei settimane negli uomini obesi aumenta l’Adiponectina sierica del 43-56%, con circa metà dell’aumento rappresentato dalla forma ad alto peso molecolare [93] [91] insieme a un aumento del 26,8% della Leptina [91 ] senza cambiamenti osservabili nella Resistina. [91] L’Adiponectina è stata osservata aumentare di circa il 30% in soggetti obesi con NAFLD in risposta alla terapia con Niacina (fino a 2g al giorno), che era correlata con un aumento dell’Insulino-resistenza, [90] portando all’ipotesi che i due meccanismi siano intrecciati, forse come risposta adattativa. [90]
Resistina
Lo “spillover” degli acidi grassi risultante da una conservazione inefficiente del grasso dopo un pasto aumenta i lipidi sierici non esterificati (NEFA), [110] che influenzano negativamente la sensibilità all’Insulina epatica, aumentando la produzione di VLDL e potenzialmente svolgono un ruolo causale nella steatosi epatica. [111] [112] La somministrazione in acuto di Niacina (285 mg per via endovenosa) nell’uomo durante l’alimentazione ha dimostrato di ridurre lo spillover degli acidi grassi, promuovendo l’assorbimento del grasso alimentare nel tessuto adiposo e riducendo i Trigliceridi sierici e i NEFA. [113]
Al contrario, è stato osservato che un trattamento prolungato con Niacina, noto per favorire la resistenza all’Insulina nell’uomo, induce la resistenza all’Insulina adipocitaria, [114] che favorirebbe lo spillover degli acidi grassi, aumentando i livelli sierici di NEFA.[115]
Glucosio-6-fosfato deidrogenasi (G6PD)
È stato osservato che la Nicotinamide sopprime la differenziazione degli adipociti 3T3-L1 in modo dipendente dalla concentrazione con un range superiore a 10mM (il valore ED50), raggiungendo la soppressione completa a 20mM dopo nove giorni. [116] Si ritiene che ciò sia correlato a un effetto inibitorio sulla poli (ADP-ribosio) sintetasi, [116] che la Nicotinamide inibisce a 50µM mentre la Niacina non lo fa. [117] Quando aggiunta dopo differenziazione e in condizioni di glucosio elevato, la Nicotinamide sembra inibire il glucosio-6-fosfato deidrogenasi (G6PD) e prevenire il normale stress ossidativo. [118]
Il recettore dell’Acido Nicotinico è espresso negli dipociti in cui la sua attivazione sopprime l’adenilato ciclasi. [119] Questo effetto sembra essere circa il 30% più efficace negli adipociti rispetto ad altre linee cellulari (milza). [120] Poiché l’attivazione di questo recettore inibisce l’adenilato ciclasi, [119] e i fenolici che agiscono su di esso riducono anch’essi i tassi di lipolisi, [35] l’effetto complessivo dell’Acido Nicotinico sarebbe quello di ridurre la lipolisi negli adipociti, almeno a breve termine.
A lungo termine, tuttavia, il recettore dell’Acido Nicotinico può essere desensibilizzato con esposizione cronica a un agonista [121] e uno studio sui topi ha evidenziato che gli adipociti che sono diventati insulino-resistenti dopo la terapia con Niacina hanno mostrato una maggiore reattività dei recettori adrenergici (β1 e β2) all’aumentare dei livelli di cAMP nella cellula adiposa, [114] (il cAMP viene normalmente soppresso dalla Niacina che agisce sul recettore GRP109A [119]). Ciò potrebbe essere stato correlato alla sottoregolazione dei geni mediata dalla Niacina nella via di segnalazione dell’Insulina incluso il PDE3B, che normalmente degrada il cAMP, [114] una potenziale risposta adattativa nelle cellule adipose che è stata osservata avere la funzione di normalizzare i tassi di lipolisi (nei ratti sotto l’infusione di Niacina) . [97]
Un piccolo studio su sette partecipanti altrimenti sani che assumevano Niacina a 500mg/die, e aumentando la dose a 2g nel corso di due settimane ha mostrato una riduzione dei tassi di ossidazione dei grassi. [94] Tuttavia, a causa di un aumento dei tassi di ossidazione dei carboidrati, non vi era alcuna differenza netta nel tasso metabolico tra Niacina e placebo [94].
I topi privi di PARP-1 sembrano avere tassi metabolici più alti e una minore massa grassa; in assenza di PARP, aumentano le concentrazioni di NAD +, attivando le SIRT1 che quindi lavorano per deacetilare varie proteine (PGC-1α e FOXO1) per promuovere il dispendio energetico attraverso un metabolismo ossidativo aumentato e un incremento dei mitocondri.[122]
La SIRT2 e la SIRT3 non sono influenzate dalla bassa attività del PARP-1, [122] e l’inibizione della ribosilazione dell’ADP con altri mezzi come il knockdown NMNAT-1 sembra conferire anche effetti antiobesità nei roditori. [182] L’alimentazione aumenta acutamente l’attività del PARP-1 nei topi e ostacola transitoriamente l’attività della SIRT1, [122] che si pensa sia correlata al PARP-1 che ha la priorità per l’uso dei donatori di NAD +.
La supplementazione orale di Nicotinamide Riboside a 400mg/kg nel topo sembra aumentare il contenuto di NAD + nel muscolo scheletrico similmente a quanto avviene alla stessa dose di Niacina (Nicotinamide Mononucleotide inefficace in questo organo) [123] e sembra aumentare il dispendio energetico nei topi nutriti con un alto contenuto di grassi insieme all’aumento dell’attività dei geni bersaglio di FOXO1, suggerendo che l’integrazione orale è efficace. [123]
Esistono prove limitate nell’uomo che valutano gli effetti della Niacina sul tasso metabolico, sebbene l’estremità inferiore del dosaggio farmacologico della niacina (1g) in soggetti altrimenti sani non sia riuscito ad aumentare il tasso metabolico rispetto al placebo. [94]
Niacina, muscolo scheletrico e prestazioni fisiche
La somministrazione di Niacina nell’uomo ha dimostrato di aumentare l’espressione dei fattori di trascrizione PPARδ e PPARγ coactivator-1α (PGC-1α) nel muscolo scheletrico. [124] Poiché questi fattori di trascrizione sono importanti regolatori del metabolismo ossidativo e della biogenesi mitocondriale, [125] [126] questo suggerisce che l’integrazione con Niacina può svolgere un ruolo nella resistenza dei muscoli scheletrici.
Gli studi sugli animali hanno supportato questa idea, in cui è stato dimostrato che l’integrazione di Niacina provoca una transizione di fibre muscolari dal tipo II (contrazione rapida) al tipo I (contrazione lenta), aumentando anche il numero complessivo di fibre di tipo I nei muscoli scheletrici nello Zucker (ratto) obeso [127] e suini in crescita [128] (750mg di Niacina/kg di dieta) e pecore (1g di Niacina al giorno). [129] Questo effetto può essere limitato a determinati modelli animali, tuttavia, poiché studi su ratti sani hanno dimostrato che la Niacina ha un effetto trascurabile sulla distribuzione del tipo di fibra muscolare o sul fenotipo metabolico. [130] Inoltre, nonostante la Niacina aumenti l’espressione dei fattori di trascrizione pro-ossidativa nell’uomo, [124] fino ad oggi nessuno studio ha dimostrato che migliora le prestazioni o la capacità di resistenza del muscolo scheletrico.
Tuttavia, come substrato per la sintesi di NAD +, un’adeguata presenza di Niacina può supportare indirettamente il metabolismo ossidativo e la resistenza muscolare. In soggetti altrimenti sani, un lieve esercizio fisico sembra essere associato ad un aumento delle concentrazioni di NAD + nel sangue rispetto a uno stato di riposo (indipendente da qualsiasi integrazione [131]) mentre, quando testato in un esercizio lieve nei roditori, portava anche ad un aumento del NAD + nel sangue prima che diminuisse durante un esercizio ad esaurimento, [131] che è stato notato anche nel muscolo scheletrico. [132] A questo livello di esaurimento c’è un concomitante aumento del contenuto di NADH nel muscolo scheletrico [133] [134] che è stato proposto [135] indicativo di una riduzione del trasferimento di elettroni dal NADH alla sintesi di ATP.
È stato inoltre proposto [135] che da quando l’esercizio aumenta l’ossidazione nei tessuti stimolati e i fattori di stress ossidativo sono noti per alterare l’attività del ciclo di Kreb (TCA) [136] e la catena di trasporto degli elettroni (compresa la NADH deidrogenasi [137]) che forniscono antiossidanti aumenterebbe la resistenza secondaria alla conservazione della cinetica intramuscolare di NAD + / NADH. Quando si forniscono 36mg di picnogenolo [135] come antiossidante durante l’esercizio fisico fino all’esaurimento, sembra che la diminuzione del NAD + nel sangue sia stata invertita in un aumento con gli effetti (sia la diminuzione che l’aumento in attesa di integrazione) più marcati negli atleti allenati. [135]
Impatto organico della Niacina e principali effetti collaterali
Indometacina
In uno studio svolto sui ratti, la Nicotinamide ad un dosaggio di 20mg/kg somministrata un’ora prima di una dose di Indometacina che induceva ulcerazioni allo stomaco ha impedito l’ulcerazione a un livello paragonabile sia al controllo (nessuna induzione di ulcere) sia al farmaco di riferimento di 400mg/kg di sucralfato, che agisce localmente per forma una superficie protettiva per lo stomaco. [138] Questo effetto si è verificato insieme alla conservazione dell’attività del glutatione, alla riduzione della perossidazione lipidica e al miglioramento del muco gastrico. [138] Simili effetti protettivi contro l’ulcerazione indotta da etanolo e stress sono stati notati altrove, con il metabolita primario della Nicotinamide (1-metilnicotinamide; MNA). [139] Questo effetto gastroprotettivo è stato associato con un aumento dell’attività delle prostaglandine, in particolare la PGI2, [139] e nicotinamide, nonché il suo metabolita MNA sono stati implicati nell’aumento del flusso sanguigno gastrico [139] e nella riduzione della permeabilità microvascolare [138] dopo l’ulcerazione.
Interleuchina 10 (IL-10)
Nel colon dei topi, il recettore della Niacina (GPR109A) è necessario per la proliferazione ottimale delle cellule T CD4 + e la produzione di IL-10, che si traduce in un effetto antiinfiammatorio. [140] Questo effetto antinfiammatorio guidato dal GPR109A è mediato dal butirrato, l’acido grasso a catena corta del colon [140], che è un agonista del GPR109A ed è prodotto attraverso la fermentazione della fibra alimentare da parte dei batteri nel colon. [141] [142]
L’effetto riducente dei Trigliceridi dato dalla Niacina sembra da doversi ricondurre al fegato, dove la secrezione di lipoproteine a bassissima densità (vLDL) è ridotta; poiché le vLDL normalmente trasportano i Trigliceridi dal fegato ad altri tessuti, riducendo la secrezione di vLDL ciò si traduce in un livello sierico di Trigliceridi inferiori. [89] La diminuzione della secrezione di vLDL può essere secondaria all’inibizione della lipolisi nel tessuto adiposo, poiché l’aumento cronico di acidi grassi liberi nel siero può regolare negativamente la secrezione di vLDL. [143]
Sembra che l’integrazione di Niacina in acuto (che riduce gli acidi grassi liberi nel siero) sopprime anche la produzione di vLDL e la sua complessazione con i trigliceridi. [144] Ciò suggerisce un altro possibile meccanismo, che può verificarsi attraverso la soppressione acuta del PGC-1β, [145] una proteina nota per promuovere la secrezione di Trigliceridi dal fegato in risposta all’ingestione di grassi nella dieta. [146] In accordo con quest’ultimo meccanismo, la somministrazione di Niacina con un pasto ad alto contenuto di grassi sembra ridurre il picco normale dei trigliceridi postprandiali. [147]
Regioni regolatorie, fattori di trascrizione e molecole di segnalazione (citochine, fattori di crescita, metaboliti, farmaci) che modulano l’espressione del gene ABCA1 nei macrofagi e in altri tessuti. Le frecce e le linee di blocco indicano rispettivamente l’attivazione e la repressione.
Non è confermato come la Niacina riduca le vLDL-C, ma la sua capacità di stimolare l’attività del gene ABCA1 e aumentare il suo contenuto proteico nelle cellule del fegato è alla base dell’aumento dell’HDL-C, [148] che è noto anche per sopprimere la secrezione di vLDL-C. [57] La Niacina (2g per 16 settimane), nonostante riduca le vLDL-C e il complesso con Trigliceridi, non sembra aumentare significativamente il contenuto di trigliceridi intraepatici in soggetti con malattia del fegato grasso non alcolica (NAFLD). [149]
La Niacina sembra anche agire sulle cellule del fegato per promuovere l’accumulo di diacilglicerolo (DAG), che è associato all’insulino-resistenza localizzata. [92] La resistenza all’insulina nelle cellule del fegato riduce l’effetto soppressivo dell’insulina sulla sintesi del glucosio, con conseguente aumento dell’efflusso di glucosio dal fegato nel sangue. [150] Poiché gli stadi iniziali dell’insulino-resistenza indotta dalla Niacina (prima degli aumenti dell’insulina basale e del glucosio) sono stati associati a un fabbisogno ridotto di glucosio per bilanciare un morsetto euglicemico iperinsulinaemico, [109] questo suggerisce che l’inizio dell’insulino-resistenza avviene a livello del fegato. Il ruolo preciso del DAG in questo processo è tuttavia incerto. Mentre il DAG promuove la resistenza all’insulina attraverso l’attivazione di PKCε, [151] l’attivazione di questa proteina non è stata osservata nelle cellule del fegato che sono diventate insulino-resistenti con la Niacina. [92]
TRANSAMINASI. Enzimi intracellulari prodotti principalmente dagli epatociti. normalmente presenti in circolo a bassi livelli nel sangue. Aumentano in caso di danno cellulare. Indici molto sensibili ma moderatamente specifici di danno epatico. ALT è un marker più specifico di danno epatocellulare. (localizzazione citoplasmatica e più lunga emivita)
Nota: La Niacina in dosi terapeutiche può causare aumenti modesti delle transaminasi sieriche e della bilirubina non coniugata, entrambi biomarcatori del danno epatico. Le modifiche vengono invertite se il trattamento farmacologico viene interrotto e di solito si risolvono anche quando si continua l’assunzione. [152] [153] [154] Tuttavia, meno comunemente, la forma di rilascio prolungato del farmaco può portare a gravi epatotossicità, con insorgenza in giorni o settimane. I primi sintomi di gravi danni al fegato includono nausea, vomito e dolore addominale, seguiti da ittero e prurito. Si ritiene che il meccanismo sia una tossicità diretta della Niacina sierica elevata. Abbassare la dose o passare alla forma a rilascio immediato può risolvere i sintomi. In rari casi la lesione è grave e progredisce fino a insufficienza epatica. [152]
È noto che la Niacina rende le cellule β pancreatiche (un tipo di cellula specializzata che secerne insulina in risposta al glucosio) meno sensibile al glucosio sierico. [81] Inoltre, la normale riduzione della sensibilità al glucosio delle cellule β del pancreas associata all’invecchiamento può essere ulteriormente esacerbata dalla supplementazione di Niacina (500mg-1g due volte al giorno). [83] Anche se sembra esserci un aumento compensativo della secrezione di insulina nella risposta alla Niacina [83], in un modello di primati con diabete di tipo I, [155] questo non è stato sufficiente a ridurre la glicemia a livelli normali, con conseguente lieve iperglicemia e iperinsulinemia dopo due settimane di integrazione.[83]
Va notato che una linea cellulare coinvolta nel rossore cutaneo tipico della Niacina, nota come Langerhans, [156] [157] è diversa dall’area del pancreas nota come “Isole di Langerhans”.
Nota: il rossore dato dalla Niacina – una dilatazione a breve termine delle arteriole della pelle, che causa il colore della pelle rossastra – di solito dura circa 15-30 minuti, anche se a volte può persistere per settimane con uso cronico e di forme a lento rilascio. In genere, il viso è maggiormente interessato, ma la reazione può estendersi al collo e alla parte superiore del torace. La causa è la dilatazione dei vasi sanguigni [158] [93] dovuta all’aumento della prostaglandina GD2 (PGD2) e serotonina. [159] [160] [161] [162] Si pensava spesso che il rossore riguardasse l’istamina, ma è stato dimostrato che l’istamina non è coinvolta nella reazione. [159] Il rossore a volte è accompagnato da una sensazione di prurito, in particolare, nelle aree coperte da indumenti. [93]
Acido Acetilsalicilico (Aspirina)
La prevenzione del rossore richiede l’alterazione o il blocco della via mediata dalle prostaglandine. [93] [163] L’Aspirina [165-325mg] assunta mezz’ora prima della Niacina riduce fortemente il rossore, così come l’Ibuprofene [200mg] (una riduzione della frequenza e intensità del rossore fino al 50%). L’assunzione di Niacina ai pasti aiuta anche a ridurre questo effetto collaterale. [93] La tolleranza acquisita aiuterà a ridurre l’effetto; dopo diverse settimane a dosaggio costante, la maggior parte delle persone non ha più esperienza di vampate di calore. [93] Sono state sviluppate forme di Niacina a rilascio lento o “prolungato” per ridurre questi effetti collaterali. [164] [165]
Conclusioni sulla supplementazione di Niacina
Le informazioni che abbiamo a disposizione sulla Niacina e la sua supplementazione, ci presentano un composto senz’altro utile per il controllo o riassesto del quadro lipidico ma allo stesso tempo questa molecola risulta di una complessità d’azione biochimica non trascurabile. Il suo peggiorare l’insulino-resistenza in cronico ma con un maggior picco in acuto, picco che sembra venire controbilanciato da altri fattori come l’aumento della Adiponectina. Lo stesso effetto sulla riduzione della lipolisi può destare preoccupazione nell’atleta, specie se questo si trova in una fase ipocalorica con il principale intento di ridurre la massa grassa. Anche in questo caso sembrerebbe che l’effetto si manifesti in acuto per poi rientrare in condizioni pre-utilizzo. Ciò che è quasi certo, è che le osservazioni sul campo non hanno fatto emergere grosse differenze nell’alterazione della composizione corporea, sia con l’uso della Niacina in regimi ipercalorici che ipocalorici. L’utilizzo di GDA (in specie Berberina) anche alle dosi base efficaci potrebbe essere un “tampone” sufficienti a compensare almeno in parte i possibili peggioramenti dei parametri dell’insulino resistenza. I controlli della glicemia basale e della insulinemia a digiuno sono indicatori da tenere sotto controllo durante l’uso di Niacina. Non è da trascurare la possibilità che la Niacina possa influenzare lo “shift” dalle fibre muscolari di tipo II a quelle di tipo I, cosa non auspicabile per un Bodybuilder o altro atleta di forza.
In definitiva, considerando i dosaggi efficaci e la migliore azione in combinazione con statine (vedi Monacolina K), l’assunzione di Niacina può essere mantenuta con un certo margine di sicurezza tra i 500mg ed 1g/die, ovviamente tarando il dosaggio in risposta agli esami ematici di controllo.
Gabriel Bellizzi
Riferimenti:
“Niacin”. Micronutrient Information Center, Linus Pauling Institute, Oregon State University, Corvallis, OR. 8 October 2018. Retrieved 16 September 2019.
Hegyi J, Schwartz RA, Hegyi V (January 2004). “Pellagra: dermatitis, dementia, and diarrhea”. International Journal of Dermatology. 43 (1): 1–5.
“Why fortify?”. Food Fortification Initiative. 2017. Retrieved 4 April 2017.
Institute of Medicine (1998). “Niacin”. Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline. Washington, DC: The National Academies Press. pp. 123–149.
Bruckert E, Labreuche J, Amarenco P (June 2010). “Meta-analysis of the effect of nicotinic acid alone or in combination on cardiovascular events and atherosclerosis”. Atherosclerosis. 210 (2): 353–61.
“Niacin”. IN: LiverTox: Clinical and Research Information on Drug-Induced Liver Injury (Internet). Bethesda, MD: National Institute of Diabetes and Digestive and Kidney Diseases. February 2014.
D: Per quanto tempo è necessario utilizzare le PGF-2, e come ciclicizzarne l’uso?
R: I Bodybuilder sono così abituati a ciclicizzare gli AAS che è legittimo interrogarsi sulla necessità di ciclicizzare le PGF-2. Dopo più di 50 anni di utilizzo continuo di steroidi anabolizzanti e di ricerca scientifica su questi, nessuno è stato in grado di elaborare un modello di ciclicizzazion…e degli AAS universale che potesse soddisfare tutti; anche se il range dei 28-42 giorni per ciclo risulta lo schema più accreditato nella somma rischio/beneficio. Quindi, non c’è da aspettarsi che dopo una breve esperienza (rispetto agli AAS) dell’uso di PGF2 esista un modello/schema ciclico universalmente ritenuto ottimale. Quindi non resta altro che analizzare l’uso delle PGF-2 in modo razionale. Per esempio, se si è in periodo “off” da AAS con intervalli di due mesi alla volta, il ciclo di PGF-2 potrebbe essere pianificato durare quei due mesi, al fine di trasformare questo periodo generalmente di “stallo” in un’opportunità per mettere massa muscolare addizionale. Un altro modo d’uso ciclico delle PGF-2 è quello di utilizzarle per circa 21-28giorni, con iniezioni localizzate a rotazione o mirate ad un gruppo muscolare carente. L’utilizzo di PGF2 non è di certo dei più confortevoli per via della necessità di effettuare più iniezioni al giorno. La maggior parte dei Culturisti non sono abbastanza motivati a sottoporsi a iniezioni più volte al giorno per più di 60-90 giorni alla volta. Così, anche in base a questo va pianificata la lunghezza del ciclo. Risulterebbe controproducente per un Bodybuilder usare un farmaco con riluttanza. Per quanto riguarda la necessità di interrompere l’utilizzo di PGF-2 è necessario, come sempre, contestualizzare. Per quanto mi riguarda, considero necessario e molto producente ciclicizzare qualsiasi tipo di farmaco o integratore da banco (fatte le dovute eccezioni). E’ ovvio che se stiamo parlando di un Bodybuilder agonista, la necessità di utilizzo del farmaco risulterà più lunga rispetto ad un Bodybuilder amatoriale che basa il suo protocollo sulla necessità di apparire al meglio per la spiaggia. Quindi, in media, cicli di 21-28 giorni, da tre iniezioni giornaliere, risultano essere i più tollerabili e gestibili dalla maggior parte degli utilizzatori.
D: Quanto tempo ci vuole per vedere i primi risultati?
R:Esteticamente parlando, meno di una settimana. La maggior parte dei primi risultati sono dovuti alle proprietà diuretiche delle prostaglandine. Ci si accorge dell’estremo pompaggio muscolare, rendendo il movimento dei muscoli interessati difficile. Il “Pump” muscolare ottenuto con PGF-2 è molto più marcato rispetto a quello ottenuto con gli AAS. Si potrà rapidamente notare che la forma complessiva dei muscoli interessati diventerà molto più gradevole. Per capire di cosa sto parlando, scattare una foto dei muscoli dove viene iniettata la PGF-2 prima e dopo, poi confrontatele . Si noterà che i muscoli saranno più pieni e rotondi. In una o due settimane di PGF-2, è possibile ottenere un ottima qualità/rotondità muscolare dei gruppi interessati. Con PGF-2 si possono ottenere guadagni di peso corporeo di circa 2,5Kg. Questo aumento di peso non è certamente molto, ma ricordiamoci che la PGF-2 non è un farmaco per l’accumulo di peso . Comunque, a questi 2,5Kg bisogna sottrarre 1-1,5Kg di acqua e di grasso la cui perdita è dovuta all’azione di PGF-2. In termini di guadagno di peso corporeo puro, non è comunque molto impressionante. D’altra parte, un aumento della massa magre e una riduzione concomitante del grasso/acqua è il modo migliore e più veloce per raggiungere una forma fisica di alto livello. Per quanto riguarda la perdita di grasso, dipende naturalmente della dieta e dagli altri farmaci co-somministrati (ad esempio insulina). È difficile fare una stima poiché è impossibile distinguere esattamente la perdita d’acqua e quella di grasso. Diciamo che in media ci si può sbarazzare di 2,5/5kg tra acqua e grasso nel giro di 8 – 10 settimane con un ciclo moderato.
D: Quale deve essere il dosaggio di PGF-2?
R:I dosaggi medi utilizzati da chi si cimenta per la prima volta nell’uso di PGF-2 dovrebbero mantenersi entro 0,5-1mg per iniezione divisi tra i gruppi interessati per tre volte al giorno. Le dosi possono salire a 2mg per pancia muscolare. La dose complessiva si aggira tra i 3 e i 10mg/die. E’ sempre meglio cominciare con i dosaggi minimi efficaci per poi aumentare gradualmente ad ogni protocollo.
D: PGF-2 viene rilevato nei test anti-doping?
R:Finora, PGF-2 non viene rilevato. Dubito che sarà mai rilevato. Si tratta di un puro costruttore muscolare a brevissima azione, e non un miglioratore della prestazione. In realtà, tende a ridurre la resistenza e le prestazioni. Inoltre, può essere molto difficile da rilevare vista la brevissima vita attiva. D’altra parte, PGF-2 altera gli ormoni steroidei in molti modi. Ad esempio, potrebbe alterare il rapporto Testosterone/Epitestosterone.
D:Come evitare gli effetti collaterali intestinali di PGF-2 ? R: E’ consigliabile evitare le iniezioni in prossimità dell’intestino, così da evitare l’effetto del PGF-2 a livello intestinale con conseguenti gravi problemi di evacuazione.
Da oltre un decennio si sono diffuse, partendo dalla cerchia ristretta del BodyBuilding di vertice per poi arrivare nel cassetto del “culturista medio”, delle nuove strategie di supplementazione chimica capaci di incrementare ancora di più il già esasperato limite della massa e della potenza muscolare raggiungibile riducendo allo stesso tempo il grasso corporeo. Dopo i già trattati GH, IGF-1, Insulina: effetti e applicazioni vorrei parlarvi di altre molecole che sono state incluse nel novero dei grandi anabolizzanti non steroidei, le Prostaglandine. E’ dalla mia prima lettura dei lavori del dott. Berry Sears (2005) che conosco questa classe di sostanze, le loro differenze ed effetti fisiologici. Ed è proprio di queste sostanze, delle loro caratteristiche e delle loro applicazione, che tratterò in questo articolo.
Storia e caratteristiche delle Prostaglandine
Si tratta di una classe di sostanze che il nostro organismo produce spontaneamente a livello locale, direttamente nei tessuti.
Le Prostaglandine insieme ai Trombosani, alle Prostacicline e ai Leucotrieni fanno parte della famiglia degli Eicosanoidi che si formano nei nostri tessuti a partire dagli acidi grassi in seguito alla loro ossigenazione da parte del l’enzima ciclo -ossigenasi. Gli Eicosanoidi sono presenti praticamente in ogni tessuto del l’organismo dove esercitano sia un’azione autocrina (agiscono sulle stesse cellule che li hanno prodotti ), che paracrina (agiscono sulle cellule circostanti ). Non sono veri e propri ormoni e neppure neurotrasmettitori, ma sono in ogni caso considerati corollario del sistema endocrino. La scoperta delle Prostaglandine risale al 1930 quando due ginecologi di New York osservarono come il fluido seminale maschile fosse in grado di stimolare “in vitro” la contrazione dei tessuti muscolari dell’utero. Un paio d’anni più tardi in Svezia, Ulf Von Euler ne confermò l’effetto sulla muscolatura viscerale liscia e osservò la riduzione della pressione sanguigna a seguito della loro infusione. Le misteriose sostanze presero così il nome di Prostaglandine dalla prostata, l’organo che produce il fluido seminale. Il composto di sintesi originariamente elaborato in laboratorio con metodi molto onerosi fu sostituito a partire dal 1969 con Prostaglandine di origine naturale di cui sono ricche le gorgonie. Ad oggi la produzione di sintesi è così efficace e poco costosa da non richiedere il ricorso ad altre fonti.
ALCUNE DELLE FUNZIONI PIU’ CONOSCIUTE DELLE PROSTAGLANDINE
Infiammazione e dolore. Le Prostaglandine promuovono molti aspetti della risposta infiammatoria. Sono inoltre associate all’insorgere della febbre.
Sistema riproduttivo.Influenzano l’ovulazione, le funzioni del corpo luteo nelle ovaie e le contrazioni uterine. Il loro eccesso è responsabile del travaglio prematuro, dell’endometriosi, della dismenorrea e di altre affezioni ginecologiche. Iniezioni di prostaglandine nei corpi cavernosi del pene, curano la disfunzione erettile maschile.
Intestino. Le Prostaglandine inibiscono la secrezione gastrica e influiscono sull’ assorbimento dei fluidi.
Sistema respiratorio. A seconda del tipo prodotto possono produrre vasocostrizione o vasodilatazione degli alveoli polmonari. Influiscono sull’asma.
Vasi sanguigni. Alcune Prostaglandine sono vasodilatatrici, altre vasocostrittrici, l’effetto è determinato dal predominare delle une o delle altre.
Trombi. I trombosani stimolano una eccessiva produzione di piastrine, predisponendo i vasi ad una eccessiva aggregazione e vasocostrizione. Le prostacicline prodotte dall’endotelio vascolare inibiscono il processo.
Reni. Prodotte dalla parte midollare del surrene aumentano il flusso del sangue, l’escrezione di acqua e di sali.
Sintesi proteica.Regolano la sintesi del muscolo scheletrico. Le PGF2 provocano catabolismo, mentre le PGF2-alfa stimolano l’anabolismo.
Adipogenesi.La formazione degli adipociti è inibita dalle PGF2-alfa.
Le Prostaglandine sono messaggeri intercellulari naturali. Senza di esse, molte delle azioni delle sostanze anabolizzanti non riescono a favorire la sintesi proteica. Il rapporto fra l’aumento di alcuni livelli di Prostaglandine e la riduzione del catabolismo è un fatto clinico. Possono quindi essere in parte il ripetitore fra i siti recettori e la traduzione per risposte specifiche, ovvero i messaggeri secondari. La ricerca ha mostrato che le Prostaglandine esercitano nelle cellule un’azione anabolizzante diretta piuttosto forte. Introducendo le Prostaglandine come il PGF2-alfa e il PGE-2 nelle cellule muscolari, la sintesi proteica si verifica a un ritmo incredibile. Se insieme al PGF2-alfa e il PGE-2 è introdotta anche l’Insulina, si verifica una potente risposta sinergica. Ciò pone alcune Prostaglandine fra gli attivatori anabolizzanti più potenti. Ciò indica anche che le potenti azioni anabolizzanti di IGF-1, Insulina e amminoacidi (per citarne alcuni) sono tutte mediate in un modo o in un altro dalle Prostaglandine.
A questo punto è utile spiegare il processo di produzione delle Prostaglandine. Alcune Prostaglandine come le PGE-2 sono sintetizzate partendo da un acido grasso presente nelle membrane plasmatiche delle cellule, l’Acido Aranchidonico. In condizioni normali, di non allenamento, la maggior parte dell’Acido Aranchidonico è presente in forma esterificata (legata) all’interno dei fosfolipidi della membrana. Dal momento che solo l’Acido Aranchidonico libero (non legato) può produrre Prostaglandine della serie PGE-2, la loro sintesi di base è molto lenta. Un po’ come è considerato normale l’equilibrio tra sintesi e degradazione proteica (omeostasi). In seguito all’azione dell’enzima fosfolipasi A2 , l’Acido Aranchidonico esterificato (legato) si libera. L’Acido Aranchidonico libero è convertito in PGE-2 da un altro gruppo di enzimi detti ciclossigenasi (COX). Il COX-1 e il COX-2 si trovano entrambi all’interno della maggior parte delle cellule e sono responsabili per il rilascio normale di Prostaglandine. Comunque, solitamente il COX-2 non è presente nelle cellule in condizioni normali. Durante un allenamento (condizione di stress), il COX-2 è sintetizzato rapidamente e ciò a sua volta stimola fortemente il rilascio di Prostaglandine. Alcune Prostaglandine sono mediatori dell’infiammazione ma in questo articolo ci concentreremo sul PGF2-alfa, un potente stimolante anabolizzante (gli inibitori specifici del COX-2 come il Celebrex bloccano parzialmente questa attività). (Le Prostaglandine non sono depositate come succede per alcuni ormoni. Sono sintetizzate velocemente e poi distrutte velocemente. Non ci sono riserve di Prostaglandine).
PGF2-alfa e crescita muscolare
La stimolazione meccanica intermittente dello stretching muscolare, provoca la produzione a livello locale di PGF2 e PGF2-alfa. Le prime aumentano la degradazione muscolare, le seconde la sintesi di nuovo tessuto. L’ ipertrofia infatti è prodotta usualmente da un moderato catabolismo e dal successivo incremento della sintesi proteica. Le prostaglandine in questione creano appunto queste condizioni. Il meccanismo coinvolge le proteine G che, incassate nella membrana cellulare, incrementano la produzione di ciclo-ossigenasi, l’enzima che forma le prostaglandine a partire dall’acido arachidonico. Va ricordato come la modulazione della sintesi muscolare avvenga sostanzialmente a due livelli, la cosiddetta fase breve e la fase lunga. Nella fase breve l’alterazione del ritmo di sintesi avviene grazie all’aumento dei ribosomi e dei fattori eucariotici di iniziazione (eIFs), già dopo pochi minuti dallo stimolo scatenante. La fase lunga si attua con la proliferazione dei nuclei cellulari e coinvolge ormoni e fattori di crescita (GH, IGF-1) che attivano le cellule staminali. Naturalmente (senza chimica esogena) occorrono fino a quindici giorni perché questa fase si concluda. Riguardo alla fase breve, il ruolo delle PGF2-alfa è ipotizzato in base alla loro influenza sui flussi di calcio, sull ‘attività del canale ionico della membrana plasmatica e sui nucleotidi. Sono tutti fattori fondamentali per lo sviluppo di nuovo tessuto muscolare. Nella fase lunga le PGF2-alfa sono potenti induttrici della proliferazione cellulare e dei nuclei che le cellule contengono. Si tratta di un’azione fondamentale perché per svilupparsi rapidamente le fibre devono produrre molto mRNA, un processo a carico del nucleo cellulare. Quindi più nuclei ci sono, più mRNA viene prodotto.
Comunque, oltre allo stretching un altro metodo naturale efficace per indurre la sintesi di Prostaglandine è il bruciore muscolare intenso. Questo perché l’accumulo di acido lattico stimola il rilascio di Prostaglandine.
Da più di un decennio circolano nel mercato nero le prostaglandine sintetizzate. Gli atleti che si sono sottoposti a protocolli contenenti PGF2-alfa iniettato in siti specifici 3-5 volte al giorno hanno sperimentato trasformazioni incredibili. Le iniezioni sono state fatte direttamente nel gruppo muscolare allenato quel giorno (iniezione post-workout, solitamente con Insulina ad azione rapida come l’Humulin-R o l’Humalog). A causa dell’indolenzimento estremo, la parte corporea sottoposta all’iniezione non è stata allenabile per alcuni giorni. Dato che le Prostaglandine sono sito specifico, la crescita dell’addome non sembra verificarsi come con l’IGF-1.
La PGF2-alfa è la Prostaglandine più attiva. Alcuni atleti di élite hanno sperimentato una marcata ipertrofia muscolare (crescita) con una dose di 1-2mg per 3-5 volte al girono (3-10mg in totale al giorno). L’effetto collaterale è stato un forte indolenzimento in tutto il muscolo dove era stata iniettata la Prostaglandine (come un grosso crampo, molto peggiore del Testosterone in sospensione o del Winstrol Depot). E’ prassi consueta evitare di iniettare il PGF2-alfa in prossimità dell’intestino (le Prostaglandine della serie 2 fanno si che i muscoli lisci come l’intestino e lo stomaco si contraggano fortemente, qualcuno ha sperimentato gravi problemi di evacuazione). Se iniettata, una piccola quantità di PGF2-alfa raggiunge il sangue, ciò spiega la crescita complessiva sperimentata. Comunque, il motivo per il quale sono necessarie iniezioni così frequenti è l’emivita molto breve delle Prostaglandine.
Alcune note interessanti:
Il Cortisolo (sia naturale che sintetico) inibisce la produzione di Prostaglandine bloccando la fosfolipasi A2 (l’enzima che mette a disposizione l’acido arachidonico) e la ciclo-ossigenasi (converte l’acido arachidonico in prostaglandine).
L’Insulina stimola la sintesi proteica e la fosfolipasi. Questo determina un incremento delle PGF2- alfa ma al tempo stesso sopprime la produzione di GLUT4 il principale transporter del glucosio nella cellula muscolare.
Gli Acidi grassi omega-3 (olio di pesce, olio di lino) diminuiscono la produzione di prostaglandine infiammatorie. Gli acidi grassi omega-6 (olii di granoturco e di oliva) aumentano viceversa la produzione di Prostaglandine pro e anti-infiammatorie.
I farmaci anti infiammatori non steroidei (FANS) come aspirina, ibuprofene, naproxene, inibiscono l ‘attività della ciclo-ossigenasi e quindi la produzione di Prostaglandine. Si è osservato tuttavia che l’uso di questi farmaci in concomitanza con l’infusione esogena di PGF2-alfa ne incrementa l’effetto anabolico.
L’uso degli AAS con le Prostaglandine si è rivelato una “parziale” cattiva idea. Spesso gli utilizzatori hanno affermato che i loro muscoli avevano difficoltà a funzionare a causa dei forti pompaggi. Il vantaggio è stato che, dopo i cicli di PGF2-alfa, i cicli di AAS si sono fatti maggiormente efficaci. Secondo parte della letteratura disponibile, l’uso del PGF2-alfa sembra aumentare il numero dei recettori a un ritmo molto accelerato.
Il PGF2-alfa ha impedito gli effetti di aumento del grasso dei protocolli di Insulina attraverso la differenziazione durante l’aumento del TGF (Fattore di Crescita Trasformante). Ciò si è rivelato un vantaggio durante i periodi pre-gara o per i soggetti insulino-resistenti. Nelle donne, il PGF2-alfa ha indotto forti crampi mestruali.
L’acido arachidonico si trova nella carne rossa in quantità piuttosto alte. Sfortunatamente, l’attività di un enzima detto 5-lipossigenasi (5-LO) sull’acido arachidonico in presenza degli estrogeni è stata legata al cancro alla prostata. Un fatto interessante è che l’inibizione dell’enzima 5-LO stimola l’apoptosi massiccia (morte cellulare programmata) nelle cellule cancerose della prostata umana. Il ginger sembra inibire questo enzima e gli acidi grassi omega-3 hanno un effetto protettivo contro il cancro alla prostata.
Nel muscolo non allenato iniezioni locali di 0,9 -1,9 mcg/Kg/die di IGF-1 equivalgono allo stimolo dell’allenamento intenso. Aumenta l’area trasversa della fibra muscolare, il deposito proteico, il contenuto di DNA. Si tratta di risultati ottenibili con dosaggi minimi, ma localizzati. Aggiungere le PGF2-alfa accelera il processo coadiuvando come si è visto l’azione dell’IGF-1 con risultati maggiori.
Come detto in precedenza, l’allenamento intenso e lo stretching innescano il rilascio di Prostaglandine attraverso l’azione sugli enzimi A2 responsabili per la loro formazione: la fosfolipasia A2 aumenta e ciò si traduce in una maggiore formazione di acido aranchidonico libero (non legato) all’interno delle cellule muscolari. Poi le contrazioni muscolari aumentano il COX-2 che trasforma l’acido aranchidonico libero in Prostaglandine. Il processo può continuare per giorni dopo un allenamento intenso. Inizialmente, le Prostaglandina che aumenta di più è il PGF-2alfa, anche la sintesi di FGF-2 (Fattore di crescita dei fibroblasti-2) aumenta ma in misura minore, almeno inizialmente. Con la guarigione degli infortuni indotti dall’allenamento, la produzione di PGF-2alfa aumenta progressivamente (e ciò è responsabile per la gran parte della reazione anabolica all’allenamento).
Un modo legale e molto meno efficace per stimolare le Prostaglandine è aumentare l’assunzione di acidi grassi essenziali (EFA). In particolare, l’Acido Linoleico, un acido grasso Omega-6, e l’Acido Alfa- Linolenico, un acido grasso Omega-3. L’Acido Gamma Linoleico (GLA) è un altro acido grasso essenziale per la sintesi di PGF-2. Gli EFA non sono solamente legati alla salute e alla prestazione. Questi EFA sono estremamente essenziali, tanto da essere vitali. Purtroppo, la dieta media del BodyBuilder è molto povera di EFA. Il rapporto migliore sembra essere 3:1, ovvero 6gr di Omega-3 e 2gr di Omega-6 al giorno per la maggior parte dei BodyBuilders che si allenano duramente. La fonte migliore di entrambi è l’olio di semi di canapa perché contiene naturalmente il rapporto 3:1. Anche l’olio di semi di lino e l’olio di enagra sono delle ottime fonti, il secondo è un ottima fonte di GLA. Assumendo molto olio di canapa è possibile risultare positivi per la marijuana. Comunque, l’assunzione di 1 cucchiaio di olio di lino, 1 cucchiaio di olio di oliva extravergine, 2gr di olio di pesce e 1gr di olio di borraggine per tre volte al giorno risultano efficaci per lo stimolo naturale e benefico delle Prostaglandine.
Conclusioni
La PGF2-alfa non è attualmente approvato dalla FDA per l’uso umano. Prodotti contenenti PGF2-alfa dovrebbero essere considerati pericolosi per le donne e devono essere maneggiati con estrema cura. La PGF2-alfa è facilmente assorbito attraverso la pelle e può causare difetti alla nascita e/o l’aborto istantanea. Le prostaglandine d’uso oggi negli esseri umani sono della classe “E” e vengono somministrate alle donne per l’aborto o per indurre il travaglio. Le prostaglandine sono utilizzate anche per l’impotenza negli uomini. In tal caso la PGE-1 viene iniettata direttamente nel pene per provocare l’erezione. La PGF2-alfa è stata testata su diverse specie animali, dalle scimmie ai cavalli. Nella maggior parte dei casi gli effetti collaterali hanno compreso: aumento della temperatura corporea, vomito e diarrea, costrizione bronchiale, confusione, perdita di coordinazione, tachicardia, e pressione sanguigna bassa, solo per citarne alcuni. La PGF2-alfa non è tossica, e come già detto ha un emivita nel siero di pochi minuti. In Italia è commercializzato come farmaco veterinario sotto il nome di DINOLYTIC (10ml, 5mg/ml), ma si può trovare da farmacia estera sotto il nome di Lutalyse. Ovviamente, l’uso delle PGF2-alfa dovrebbe essere limitato ai culturisti avanzati, e non di certo all’atleta principiante/intermedio o all’amatoriale che non ha scopi agonistici. Prima di arrivare a contemplare l’uso di PGF2-alfa, come di Insulina e IGF-1, bisogna aver raggiunto il proprio potenziale massimo con le tecniche meno avanzate.
Comunque, la ricerca da me esposta evidenzia come la crescita muscolare sia un meccanismo molto complesso. Chiarisce definitivamente come le tensioni muscolari intense fino alla degradazione del tessuto siano stimolo indispensabile all’ipertrofia e suggerisce come opportuni interventi nutrizionali, aumento dei cibi ricchi di acidi grassi omega-6 (olio d’oliva, olio di granoturco), e degli omega-3 (olio di pesce), aiutino ad incrementare il processo.
Gabriel Bellizzi
Riferimenti
Chemical Muscle Enhancement. A.L.Rea.
Costruire la bestia perfetta (Chemical Muscle Enhancement II). A.L.Rea.
WILLIAM LLEWELLYN’S ANABOLICS – 10th Edition
PROSTAGLANDINE PGF2-alfa. L’anabolico locale di nuova generazione. BIG, 2001. Giovanni Cianti