Introduzione:

La maggior parte degli “addetti ai lavori” e degli atleti, è perfettamente a conoscenza del fatto che una “base” di Testosterone sia necessaria all’interno di un ciclo di AAS/SARM al fine di avere un adeguato livello di metaboliti connessi [vedi E2 e DHT] evitando o riducendo quei problemi legati ad un loro marcato calo: alterazioni dell’umore, letargia, sonnolenza, spossatezza, ridotta libido, difficoltà a raggiungere e mantenere l’erezione ecc… .
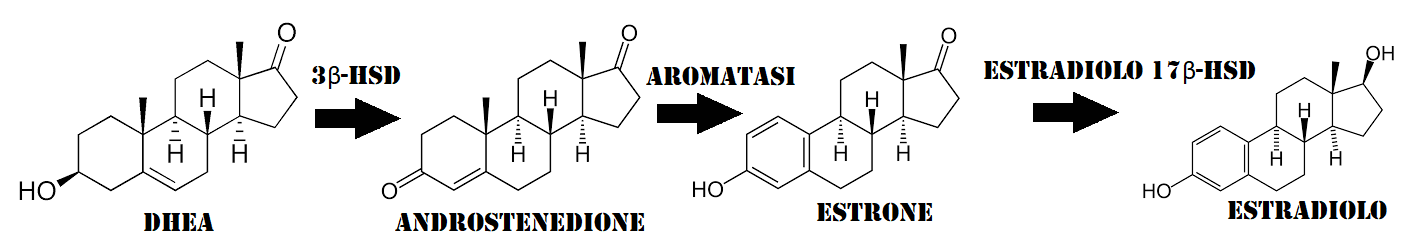
Esistono altresì soggetti che decidono di non avvalersi dell’uso di una base di Testosterone optando, per esempio, per una somministrazione “rivista” di hCG. Ma vi sono altri, i così detti “agofobici” [si, esistono…si dopano e hanno paura dell’ago] che cercano di ripiegare con l’uso spesso fallimentare di DHEA [il quale, attraverso la conversione in Androstenediolo e Androstenedione converte maggiormente in E1 che a sua volta possiede una scarsa tendenza alla conversione nel più utile E2. Altri decidono di usare il Clomifene Citrato (Clomid®) o l’Enclomifene Citrato (Androxal®) per cercare di mantenere una attività dell’Asse HPT tale da garantire loro adeguati livelli di E2.
Sappiamo benissimo che i SERM agiscono a livello dei ER ipotalamici stimolando il rilascio di GnRH e, successivamente, a livello ipofisario, di LH e FSH. E’ infatti pratica comune nella PCT utilizzare tali farmaci per avere una risposta di “recupero” iniziale della produzione endogena di Testosterone dopo l’uso di AAS e loro azione soppressiva del sistema endocrino in questione.
A questo punto la domanda è: è possibile che l’uso di SERM come il Clomifene Citrato o il suo enantiomero attivo Enclomifene possa avere una risposta terapeutica anche durante l’uso di AAS?
Facciamo un pò di ripasso e cerchiamo di arrivare ad una conclusione logica e, per lo meno, accademica …
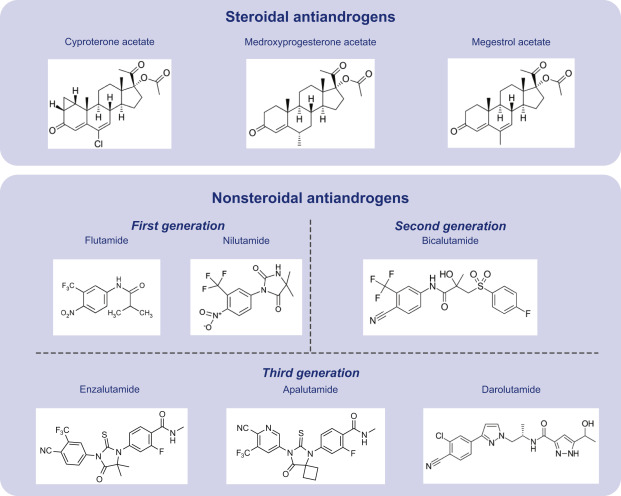
SERM e loro caratteristiche:
- Siti di legame [ERα e ERβ]
I SERM sono agonisti parziali competitivi dell’ER.[1] I diversi tessuti hanno gradi diversi di sensibilità all’attività degli estrogeni endogeni, quindi i SERM producono effetti estrogenici o antiestrogenici a seconda del tessuto specifico in questione e della percentuale di attività intrinseca (IA) del SERM. [2] Un esempio di SERM con un’elevata IA e quindi con effetti prevalentemente estrogenici è il clorotrianisene, mentre un esempio di SERM con una bassa IA e quindi con effetti prevalentemente antiestrogenici è l’etamoxitripetolo. SERM come il clomifene e il tamoxifene sono relativamente più a metà strada per quanto riguarda l’IA e l’equilibrio tra attività estrogenica e antiestrogenica. Il raloxifene è un SERM più antiestrogenico del tamoxifene; entrambi sono estrogenici nelle ossa, ma il raloxifene è antiestrogenico nell’utero mentre il tamoxifene è estrogenico in questa parte del corpo.[2]
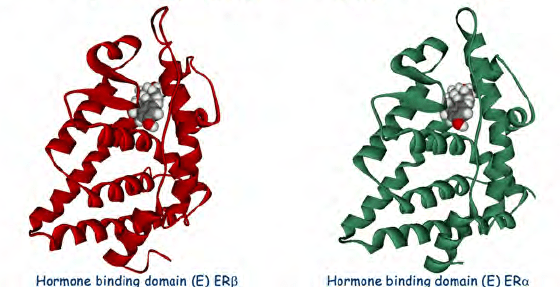
I SERM agiscono sul recettore degli estrogeni (ER), che è un attivatore trascrizionale intracellulare ligando-dipendente e appartiene alla famiglia dei recettori nucleari.[4] Sono stati identificati due diversi sottotipi di ER, ERα e ERβ. ERα è considerato il principale mezzo in cui i segnali estrogenici vengono trasdotti a livello trascrizionale ed è l’ER predominante nel tratto riproduttivo femminile e nelle ghiandole mammarie, mentre ERβ si trova principalmente nelle cellule endoteliali vascolari, nell’osso e nel tessuto prostatico maschile.[5] È noto che la concentrazione di ERα ed ERβ è diversa nei tessuti durante lo sviluppo, l’invecchiamento o lo stato patologico.[6] Molte caratteristiche sono simili tra questi due tipi, come le dimensioni (~600 e 530 aminoacidi) e la struttura. ERα ed ERβ condividono circa il 97% dell’identità di sequenza aminoacidica nel dominio che lega il DNA e circa il 56% nel dominio che lega il ligando.[4][6] La differenza principale dei domini che legano il ligando è determinata da Leu-384 e Met-421 in ERα, che sono sostituiti da Met-336 e Ile-373, rispettivamente, in ERβ.[7] La variazione è maggiore sull’N-terminus tra ERα ed ERβ.[8]
Il dominio di legame al DNA è costituito da due sottodomini. Uno ha un box prossimale che è coinvolto nel riconoscimento del DNA, mentre l’altro contiene un box distale responsabile della dimerizzazione DNA-dipendente del dominio DNA-binding. La sequenza del box prossimale è identica tra ERα ed ERβ, il che indica una specificità e un’affinità simili tra i due sottogruppi. Le proteine globulari del dominio DNA-binding contengono otto cisteine e consentono una coordinazione tetraedrica di due ioni zinco. Questa coordinazione rende possibile il legame di ER con gli elementi di risposta agli estrogeni.[5] Il dominio legante il ligando è una struttura globulare a tre strati composta da 11 eliche e contiene una tasca per il ligando naturale o sintetico.[5][4] I fattori che influenzano l’affinità di legame sono principalmente la presenza di una frazione fenolica, la dimensione e la forma molecolare, i doppi legami e l’idrofobicità.[9]
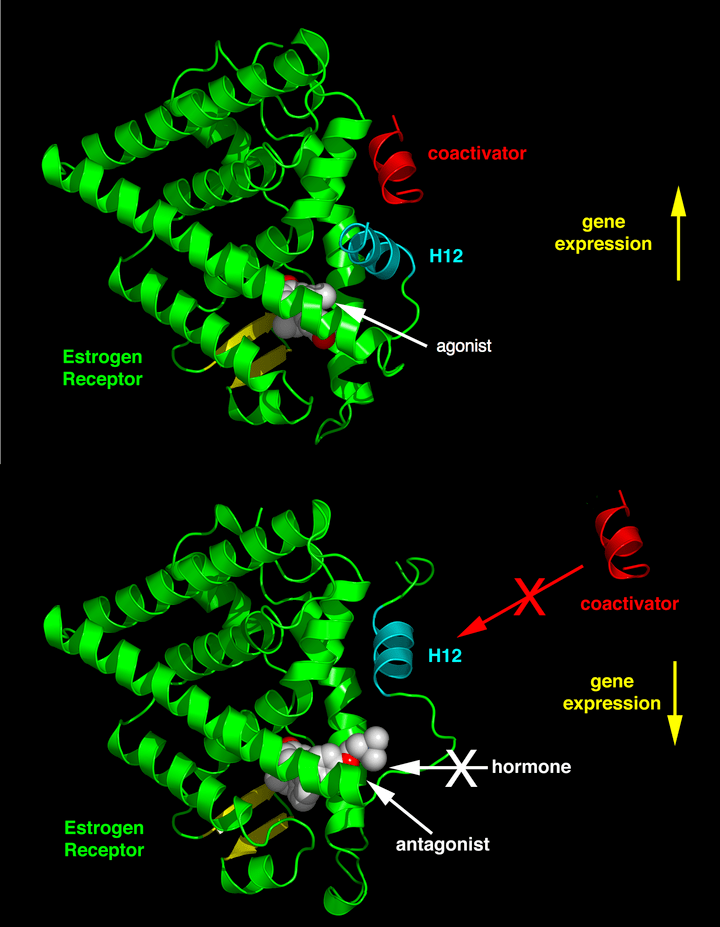
Il posizionamento differenziale dell’elica 12 della funzione attivante 2 (AF-2) nel dominio di legame del ligando da parte del ligando legato determina se il ligando ha un effetto agonista o antagonista. Nei recettori legati all’agonista, l’elica 12 è posizionata adiacentemente alle eliche 3 e 5. Le eliche 3, 5 e 12 insieme formano una superficie di legame per un motivo NR box contenuto nei coattivatori con la sequenza canonica LXXLL (dove L rappresenta la leucina o l’isoleucina e X è un amminoacido qualsiasi).
I recettori non bloccati (apo) o i recettori legati a ligandi antagonisti allontanano l’elica 12 dalla superficie di legame LXXLL, il che porta al legame preferenziale di un motivo più lungo ricco di leucina, LXXXIXXX(I/L), presente sui corepressori NCoR1 o SMRT. Inoltre, alcuni cofattori si legano all’ER attraverso i terminali, il sito di legame del DNA o altri siti di legame. Pertanto, un composto può essere un agonista ER in un tessuto ricco di coattivatori ma un antagonista ER in tessuti ricchi di corepressori.[4]
- Meccanismo d’azione
I composti estrogenici coprono uno spettro di attività che va da:
- Agonisti completi (agonisti in tutti i tessuti) come l’ormone endogeno naturale Estradiolo
- Agonisti misti/antagonisti (agonisti in alcuni tessuti e antagonisti in altri) come il Tamoxifene (SERM).
- Antagonisti puri (antagonisti in tutti i tessuti), come il Fulvestrant.
I SERM sono noti per stimolare l’azione estrogenica in tessuti come il fegato, le ossa e il sistema cardiovascolare, ma anche per bloccare l’azione degli estrogeni laddove la stimolazione non è auspicabile, come nel seno e nell’utero. [10] Questa attività agonistica o antagonistica provoca vari cambiamenti strutturali dei recettori, con conseguente attivazione o repressione dei geni bersaglio degli estrogeni.[10][11] I SERM interagiscono con i recettori diffondendosi nelle cellule e legandosi alle subunità ERα o ERβ, con conseguente dimerizzazione e cambiamenti strutturali dei recettori. Ciò facilita l’interazione dei SERM con gli elementi di risposta agli estrogeni, che portano all’attivazione di geni inducibili dagli estrogeni e mediano gli effetti di questi ultimi.[10]
La caratteristica unica dei SERM è la loro attività selettiva per tessuti e cellule. Ci sono sempre più prove a sostegno del fatto che l’attività dei SERM è determinata principalmente dal reclutamento selettivo di corepressori e coattivatori ai geni bersaglio dell’ER in specifici tipi di tessuti e cellule.[11][12] I SERM possono avere un impatto sulla stabilità delle proteine dei coattivatori e possono anche regolarne l’attività attraverso modifiche post-traslazionali come la fosforilazione. Molteplici vie di segnalazione della crescita, come HER2, PKC, PI3K e altre, sono downregolate in risposta al trattamento anti-estrogeno. Il coattivatore 3 dei recettori steroidei (SRC-3) viene fosforilato da chinasi attivate che ne potenziano l’attività di coattivatore, influenzano la crescita cellulare e contribuiscono alla resistenza ai farmaci.[12]
Il rapporto tra ERα ed ERβ in un sito bersaglio può essere un altro modo per determinare l’attività dei SERM. Alti livelli di proliferazione cellulare sono ben correlati con un alto rapporto ERα:ERβ, ma la repressione della proliferazione cellulare è correlata alla dominanza di ERβ su ERα. Il rapporto tra ER nel tessuto mammario neoplastico e normale potrebbe essere importante quando si considera la chemioprofilassi con i SERM.[10][11]
Per quanto riguarda le differenze tra ERα ed ERβ, sono importanti la Funzione di Attivazione 1 (AF-1) e la Funzione di Attivazione 2 (AF-2). Insieme svolgono un ruolo importante nell’interazione con altre proteine co-regolatrici che controllano la trascrizione genica.[10] AF-1 si trova nella terminazione amminica dell’ER ed è omologa solo al 20% in ERα ed ERβ. D’altra parte, AF-2 è molto simile in ERα e ERβ, e solo un aminoacido è diverso. Gli studi hanno dimostrato che scambiando le regioni di AF-1 in ERα e ERβ, si ottengono differenze specifiche nell’attività di trascrizione. In generale, i SERM possono attivare parzialmente geni ingegnerizzati attraverso ERα da un elemento del recettore degli estrogeni, ma non attraverso ERβ.[10][11] Tuttavia, il raloxifene e la forma attiva del tamoxifene possono stimolare geni reporter regolati da AF-1 sia in ERα che in ERβ.
La scoperta dell’esistenza di due sottotipi di ER ha portato alla sintesi di una serie di ligandi specifici per il recettore in grado di attivare o disattivare un particolare recettore. Tuttavia, la forma esterna del complesso risultante è ciò che diventa il catalizzatore per modificare la risposta di un tessuto bersaglio a un SERM.[10][11]
La cristallografia a raggi X di estrogeni o antiestrogeni ha mostrato come i ligandi programmino il complesso recettoriale per interagire con altre proteine. Il dominio legante dell’ER dimostra come i ligandi promuovano e impediscano il legame del coattivatore in base alla forma del complesso estrogeno o antiestrogeno. L’ampia gamma di ligandi che si legano all’ER può creare uno spettro di complessi ER completamente estrogenici o antiestrogenici in uno specifico sito bersaglio.[11] Il risultato principale del legame di un ligando all’ER è un riarrangiamento strutturale della tasca di legame del ligando, principalmente nell’AF-2 della regione C-terminale. Il legame dei ligandi all’ER porta alla formazione di una tasca idrofobica che regola i cofattori e la farmacologia del recettore. Il corretto ripiegamento del dominio di legame con i ligandi è necessario per l’attivazione della trascrizione e per l’interazione di ER con una serie di coattivatori.
I coattivatori non sono solo partner proteici che collegano tra loro i siti di un complesso. I coattivatori svolgono un ruolo attivo nel modificare l’attività di un complesso. La modificazione post-traduzionale dei coattivatori può dar luogo a un modello dinamico di azione degli ormoni steroidei attraverso molteplici vie chinasiche avviate dai recettori dei fattori di crescita della superficie cellulare. Sotto la guida di una moltitudine di rimodellatori proteici per formare un complesso multiproteico di coattivatori in grado di interagire con l’ER fosforilato in uno specifico sito promotore genico, il core coactivator deve prima reclutare una serie specifica di coattivatori. Le proteine che il core coactivator assembla come complesso di coattivatori hanno attività enzimatiche individuali per metilare o acetilare le proteine adiacenti. I substrati ER o il coenzima A possono essere poliubiquitinati da più cicli della reazione oppure, a seconda delle proteine di legame, possono essere ulteriormente attivati o degradati dal proteasoma 26S.[10]
Di conseguenza, per avere una trascrizione genica efficace, programmata e mirata dalla struttura e dallo stato di fosforilazione dell’ER e dei coattivatori, è necessario un processo dinamico e ciclico di capacità di rimodellamento per l’assemblaggio trascrizionale, dopo il quale il complesso di trascrizione viene poi istantaneamente distrutto dal proteasoma.[10]
- Effetti sull’Asse HPT
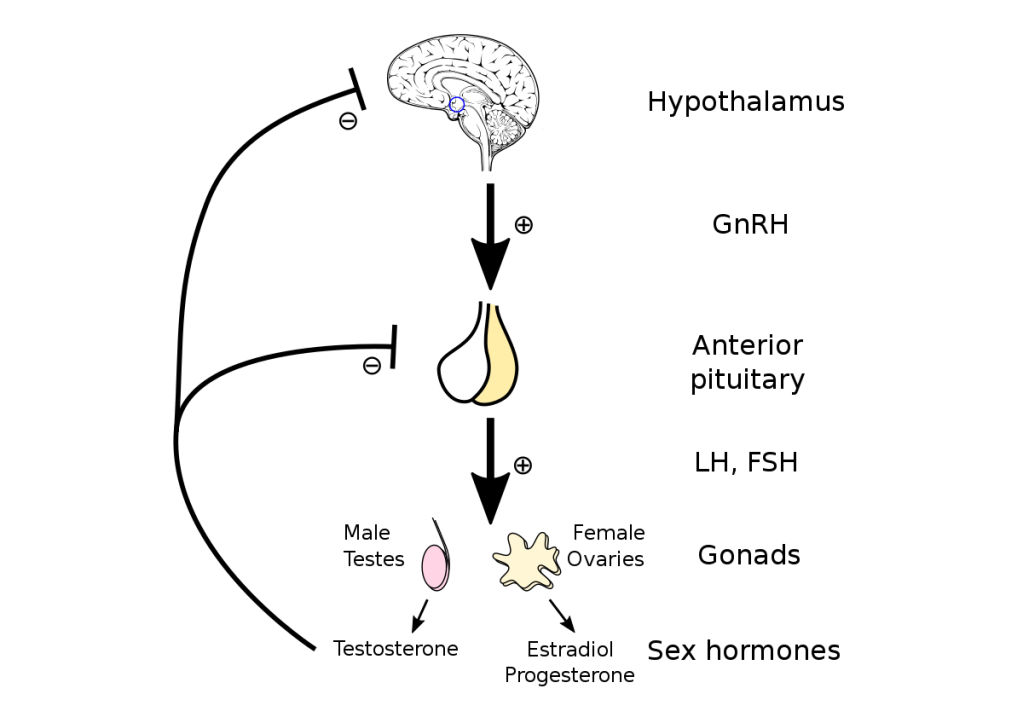
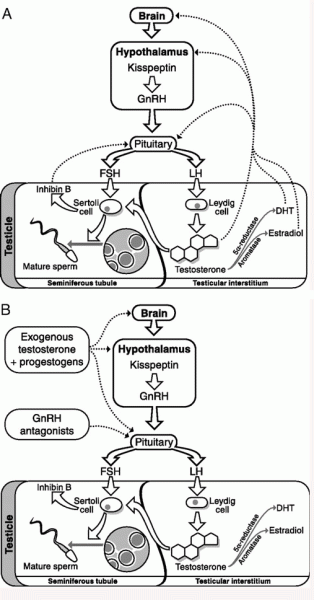
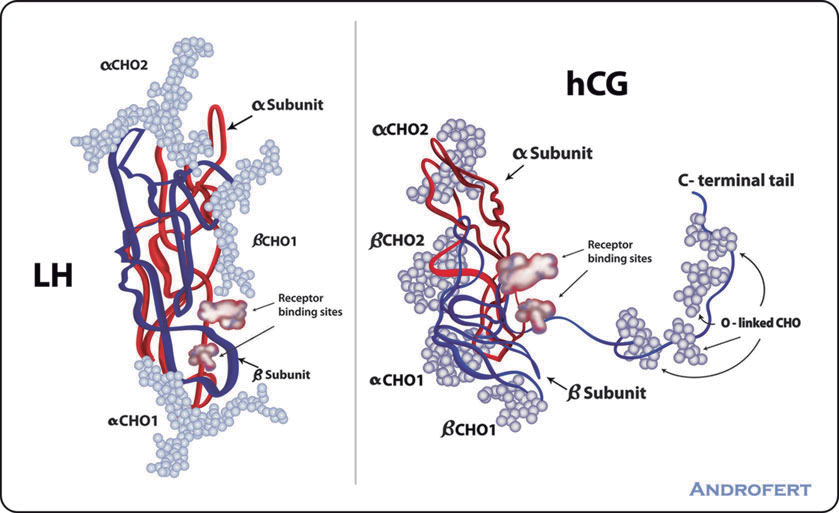
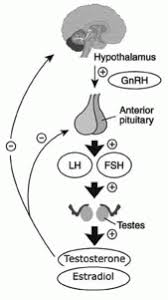
Gli estrogeni sono un importante regolatore dell’Asse HPT. L’ipofisi si trova al di fuori della barriera ematoencefalica e accumula alti livelli di SERM. Inoltre, i SERM possono bloccare l’aumento di peso dell’ipofisi indotto dagli estrogeni [12], suggerendo un’azione anti-estrogenica. Antagonizzando i recettori estrogenici e bloccando l’attivazione di questi da parte del E2, i SERM stimolano il rilascio da parte dell’Ipotalamo di GnRH che a sua volta induce la sintesi ed il rilascio di Ormone Luteinizzante [LH] e Ormone Follicolo Stimolante [FSH]. Ciò, di conseguenza, aumenta la sintesi testicolare di Testosterone e la spermatogenesi.
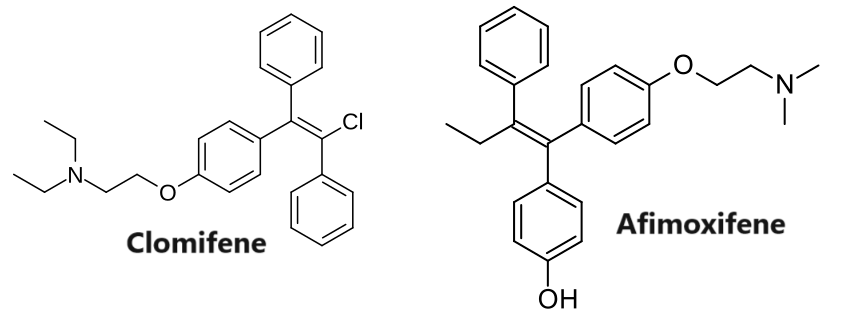
L’affinità del Clomifene per l’ER rispetto all’estradiolo varia dallo 0,1 al 12% in diversi studi, un valore simile a quello del tamoxifene (0,06-16%).[13][14][15] Il 4-idrossiclomifene, uno dei principali metaboliti attivi del Clomifene/Enclomifene, e l’Afimoxifene (4-idrossitamoxifene), uno dei principali metaboliti attivi del Tamoxifene, mostrano rispettivamente l’89-251% e il 41-246% dell’affinità dell’Estradiolo per l’ER nelle cellule di cancro al seno MCF-7 umano. [16] L’affinità per l’ER degli isomeri del 4-idrossiclomifene era del 285% per l'(E)-4-idrossiclomifene e del 16% per lo (Z)-4-idrossiclomifene rispetto all’Estradiolo. [16] Il 4-idrossi-N-desmetilclomifene ha un’affinità simile a quella del 4-idrossi-clomifene per l’ER.[17] In uno studio, l’affinità del Clomifene e dei suoi metaboliti per l’ERα era di ~100 nM per il Clomifene, ~2,4 nM per il 4-idrossi-clomifene, ~125 nM per l’N-desmetilclomifene e ~1,4 nM per il 4-idrossi-N-desmetilclomifene.[17]
Anche se il Clomifene ha un certo effetto estrogenico, dato dalla componente di Zuclomifene, si ritiene che la proprietà antiestrogenica sia la fonte principale della stimolazione dell’ovulazione, data dal Enclomifene. Il Clomifene sembra agire soprattutto nell’ipotalamo, dove esaurisce gli ER ipotalamici e blocca l’effetto di feedback negativo dell’Estradiolo endogeno circolante, che a sua volta determina un aumento della frequenza degli impulsi ipotalamici dell’ormone di rilascio delle gonadotropine (GnRH) e delle concentrazioni circolanti di ormone follicolo-stimolante (FSH) e ormone luteinizzante (LH).

Negli uomini normali, è stato riscontrato che 50mg/die di Clomifene per 8 mesi aumentano i livelli di Testosterone di circa 870ng/dL negli uomini più giovani e di circa 490ng/dL negli uomini più anziani.[18] I livelli di Estradiolo aumentano di 62pg/mL negli uomini più giovani e di 40pg/mL negli uomini più anziani.[18] Questi risultati suggeriscono che gli effetti progonadotropi del Clomifene sono più forti negli uomini più giovani che in quelli più anziani. Negli uomini con ipogonadismo, il Clomifene è risultato in grado di aumentare i livelli di Testosterone da 293 a 362ng/dL e i livelli di Estradiolo da 5,5 a 13pg/mL.[18] In un ampio studio clinico su uomini con bassi livelli di Testosterone (<400ng/dL), 25mg/die di Clomifene [circa 15.5mg di Enclomifene] hanno aumentato i livelli di Testosterone da 309ng/dL a 642ng/dL dopo 3 mesi di terapia. Non sono stati osservati cambiamenti significativi nei livelli di colesterolo HDL, trigliceridi, glucosio a digiuno o Prolattina, sebbene i livelli di colesterolo totale siano diminuiti significativamente.[18][19]
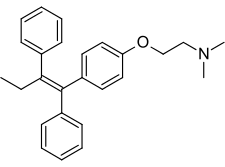
E’ di interesse sottolineare che la miscela racemica del Clomifene è composta per il 38% da Zuclomifene e per il 62% da Enclomifene. Lo Zuclomifene è lo stereoisomero (Z) del Clomifene, mentre l’Enclomifene è lo stereoisomero (E). Lo Zuclomifene è leggermente estrogenico, e a differenza dell’Enclomifene, esso ha azione antigonadotropa a causa dell’attivazione del recettore degli estrogeni con successiva riduzione dei livelli di Testosterone negli uomini. È inoltre circa cinque volte più potente dell’Enclomifene nell’indurre l’ovulazione nelle donne.
Il primo studio pubblicato sul Enclomifene comprendeva solo 12 uomini e non era in cieco [20]. In altre parole, sia i partecipanti che i ricercatori sapevano quale trattamento stavano ricevendo gli uomini. I partecipanti erano uomini con ipogonadismo secondario trattati in precedenza con Testosterone topico. Sono stati randomizzati a ricevere nuovamente Testosterone topico o Enclomifene (25mg al giorno).
Dopo sei mesi di trattamento, i livelli di Testosterone erano praticamente gli stessi tra i gruppi: 545ng/dL (18,9nmol/L) nel gruppo che riceveva il gel e 525ng/dL (18,2nmol/L) nel gruppo che riceveva l’Enclomifene. Anche i livelli di Testosterone libero sono aumentati e sono rimasti praticamente invariati tra i gruppi. Inoltre, e naturalmente, il numero di spermatozoi è stato ridotto negli uomini che ricevevano Testosterone, con numeri intorno ai 20milioni/mL. Inoltre, come previsto, il numero di spermatozoi è aumentato negli uomini che hanno ricevuto l’Enclomifene, con una media di circa 150milioni/mL.
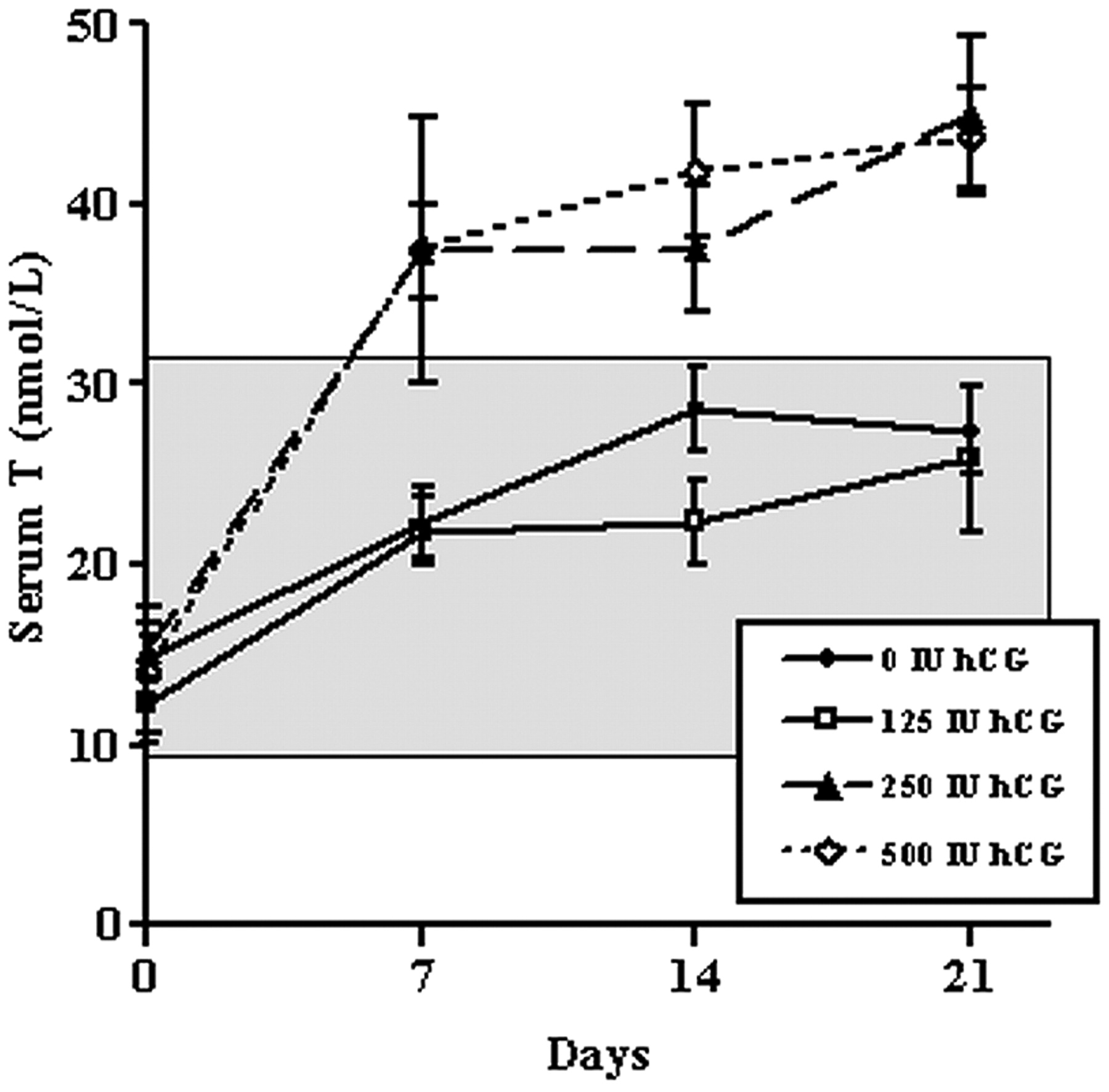
Due interessanti studi [21][22]sull’Enclomifene hanno utilizzato lo stesso protocollo e l’aspetto forse più interessante è stata la dimensione del campione: 256 soggetti in totale. L’intervento è durato 16 settimane e i soggetti del gruppo Enclomifene hanno ricevuto 12,5mg al giorno e sono stati trattati fino a 25mg al giorno se i livelli di Testosterone non erano aumentati ad almeno 450ng/dL (15,6nmol/L) alla quarta settimana. La dose è stata aumentata per la metà dei soggetti che ricevevano l’Enclomifene. A questo punto le cose iniziano a farsi interessanti: sebbene metà dei soggetti sia stata modificata nel dosaggio alla quarta settimana, non è successo assolutamente nulla con la concentrazione media di Testosterone:
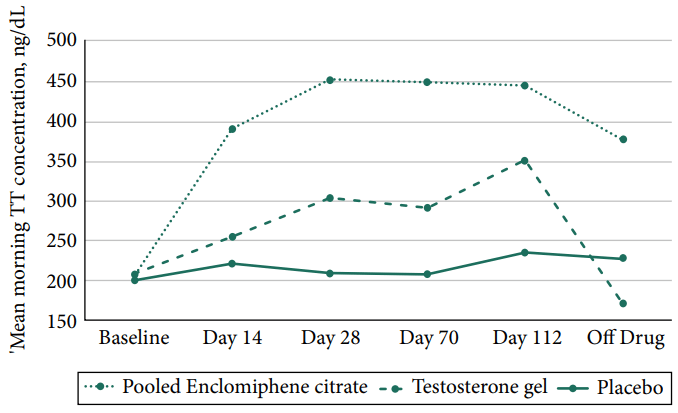
E, in effetti, alla fine dell’intervento, la media del gruppo era appena al di sotto del valore limite di 450ng/dL (15,6nmol/L) per l’up-titration. Infine, 29 degli 85 uomini del gruppo Enclomifene non hanno visto il loro Testosterone aumentare al di sopra del valore limite di ipogonadismo di 300ng/dL (10,4nmol/L) dopo 16 settimane di trattamento. Inoltre, i ricercatori hanno fatto un lavoro non propriamente apprezzabile nel trattare correttamente il gruppo che utilizzava il gel di Testosterone, come si può vedere dalla concentrazione media di Testosterone di quel gruppo.
E’ interessante notare che il Clomifene mostra in realtà risultati molto simili, anche mg per mg, a quelli dell’Enclomifene.
Uso dei SERM nella terapia per la fertilità in pazienti sottoposti a TRT
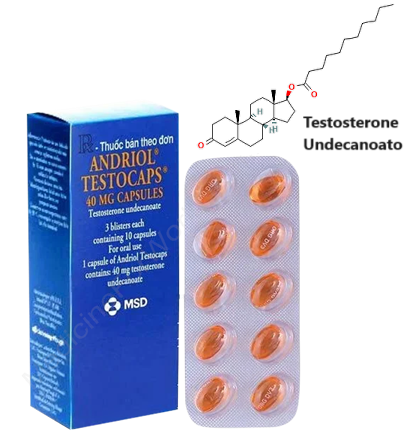
Uno studio ha assegnato i pazienti oligozoospermici a due gruppi di trattamento: (1) 20mg/die di Tamoxifene Citrato e 120mg/die di Testosterone Undecanoato [forma orale; pari a 75.9mg di Testosterone effettivo con una biodisponibilità del 8% = 6.072mg circa di principio attivo in circolo nelle 24h] (n = 106) e (2) trattamento con placebo (n = 106) per 6 mesi. Nel gruppo Tamoxifene/T, il numero totale di spermatozoi è aumentato da una mediana [25°, 75° percentile] di 27,1 × 106 cellule/mL [9,4, 54,0 × 106 cellule/mL] a 61,5 × 106 cellule/mL [28,2, 119,6 × 106 cellule/mL], la motilità progressiva è aumentata dal 29,7% ± 12,0% al 41,6% ± 13,1% e la morfologia normale è aumentata dal 41,2% ± 14,0% al 56,6% ± 11,5% dopo 6 mesi. Il tasso di gravidanza spontanea è stato del 33,9% nel gruppo Tamoxifene/T e del 10,3% nel gruppo placebo. Questo metodo di somministrazione concomitante di Testosterone e SERM potrebbe essere efficace nel mantenere la fertilità in una certa fetta di pazienti sottoposti a TRT. L’uso concomitante di hCG o Clomifene [o altro SERM] durante la TRT potrebbe non essere ottimale negli uomini in cerca di fertilità.[https://www.mdpi.com/1648-9144/60/2/275]
E’ interessante anche un piccolo studio del 1979 che ha preso in esame l’effetto delle somministrazione cronica di Clomifene in concomitanza con diversi androgeni…
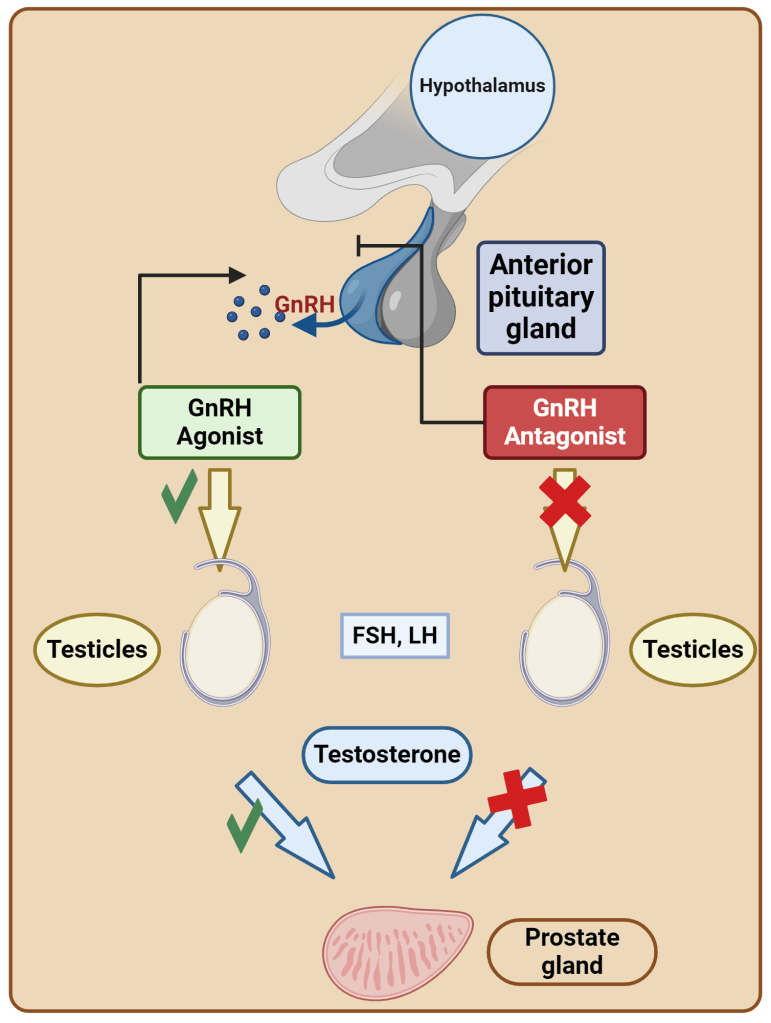
Nelle osservazioni dello studio, l’infusione di Testosterone (T; 7,5mg/die per 4 giorni) ha prodotto un calo del 40% delle concentrazioni sieriche di LH e FSH. L’infusione di estradiolo (E2) in dosi equivalenti a quelle derivate dal T infuso (45μg/die) ha provocato un calo dell’LH sierico pari al 60% di quello osservato con il T, indicando che la maggior parte della soppressione dell’LH mediata dal T può essere attribuita alla sua aromatizzazione a E. Anche l’infusione di diidrotestosterone ha provocato una diminuzione del 35% dell’LH sierico medio e una diminuzione del numero di impulsi spontanei di LH simile a quella osservata con il T, a sostegno di un ruolo della componente androgenica pura nella soppressione dell’LH mediata dal T. Durante la terapia cronica con Clomifene, né il T né l’E2, se somministrati in dosi pari al doppio del loro tasso di produzione medio negli uomini normali, né gli androgeni non aromatizzabili, il Diidrotestosterone e il Fluoxymesterone, in dosi equipotenti al T infuso, sono stati in grado di sopprimere i livelli sierici di LH e FSH o di alterare le risposte di LH e FSH alla somministrazione di GnRH. La resistenza della gonadotropina alla soppressione da parte degli androgeni durante il blocco del Clomifene rimane ma con probabili variabili dose-temporali.[https://www.researchgate.net/]
- Punti chiave
Abbiamo ripassato la funzionalità documentata del Clomifene e dell’Enclomifene di causare un aumento del GnRH con conseguente incremento di LH, FSH, Tetstosterone (e metaboliti annessi) e spermatogenesi in soggetti sani e ipogonadici [ipogonadismo secondario e AAS-indotto]. Ma durante l’uso di AAS/SARM è possibile avere una risposta terapeutica?
Oltre ai dati riportati in contesto TRT e SERM, se leggiamo con attenzione i dati sopra riportati, con una risposta di legame con effetto antagonista del ER ipotalamico dei mataboliti del Clomifene/Enclomifene del 285%, possiamo ipotizzare che la sua efficacia in presenza di molecole aromatizzabili sia proporzionale ai livelli di E2 o di suoi più potenti analoghi metilati in C7α o in C17α in circolo. In assenza di queste e in cosomministrazione con molecole non aromatizzabili, il suo potenziale di legame risulterebbe analogo al contesto di non utilizzo di AAS.
Possiamo chiuderla qui con un “si, ha una azione terapeutica anche in cosomministrazione con AAS/SARM, specie se non aromatizzabili!”? Purtroppo no, perchè il controllo dell’attività dell’Asse HPT non è regolato solo ed esclusivamente dal feedback negativo del E2.
I fattori che sopprimo l’Asse HPT
Come detto pocanzi, la sottoregolazione/soppressione dell’Asse HPT non è solo dipendente dal feedback negativo dato da un aumento del E2 circolante. Infatti, i fattori che influenzano la sottoregolazione/soppressione dell’Asse HPT sono:
- L’origine del AAS, e di conseguenza…
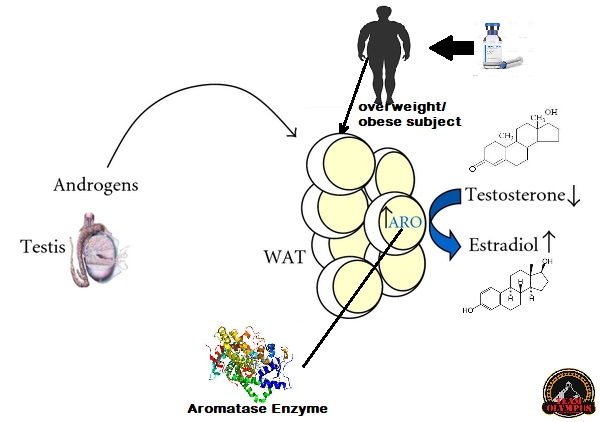
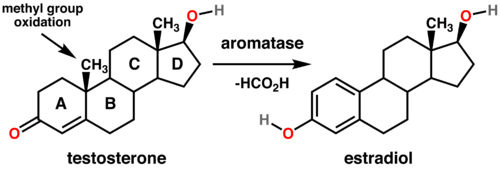
- Il tasso di conversione del AAS ad estrogeno, attraverso l’enzima aromatasi in alcuni tessuti (adiposo, mammario)
- L’attività estrogenica intrinseca della molecola
- L’attività progestinica dell’AAS
- Dose e tempo d’uso/abuso del AAS
- Attività androgena del AAS
Come possiamo vedere, oltre al fattore estrogenico vi sono quello diretto dall’AAS, la sua attività progestinica e la sua affinità con l’AR.
Sebbene l’utilizzatore del “tampone SERM” per cercare di garantirsi livelli di E2 e DHT nella norma (indi minimamente funzionali) raramente utilizza progestinici, la cosa non è impossibile vista la presenza di PH/AAS orali con attività progestinica [vedi 19-Nor-5-androstenediolo, MENTDIONE, MENT, Trenbolone Acetato, Metribolone ecc…].
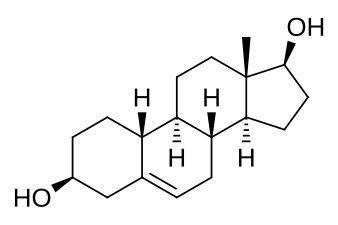
Il Progesterone svolge inoltre un ruolo cruciale nell’Asse HPT. Durante la fase luteale, l’ipotalamo rilascia l’ormone di rilascio delle gonadotropine (GnRH), che agisce su una ghiandola chiamata ipofisi anteriore. Una quantità eccessiva di Progesterone o la presenza di Progestinici provoca un’inibizione a feedback negativo a livello ipotalamico/ipofisario, con conseguente cessazione marcata del rilascio di ormoni; maggiore di quella riscontrata con il ciclo di feedback del E2. Questo processo, nella maggior parte dei casi (se non in una estrema maggioranza con uno scarto di possibilità limitato) non è compensabile con l’uso di SERM.
Un altro fattore che interviene a livello del feedback negativo dell’Asse HPT risiede della attività AR della molecola. Di conseguenza, dovrebbe essere chiaro che anche farmaci puramente androgeni o essenzialmente anabolizzanti e con forte potenziale di legame con il AR [vedi SARM non steroidei] possono causare una sotto-regolazione della funzionalità dell’Asse HPT, quindi con meccanismi indipendenti dalla aromatizzazione della molecola.
Infatti, gli AAS [ed i SARM non steroidei] attraversano la barriera ematoencefalica e si legano ai recettori Ipotalamici. Ciò comporterà una marcata soppressione dell’HPTA per via di intermediari quali i peptidi oppioidi endogeni.
Quindi, bisogna sapere che l’attività di soppressione/sottoregolazione dell’Asse HPT androgeno-dipendente ha come intermediari i peptidi oppioidi endogeni, con attività principale da parte della Beta-Endorfina, delle Encefaline e Dinorfine attraverso il legame con i recettori oppioidi μ.
Tale effetto ridurrà comunque l’efficacia terapeutica dei SERM utilizzati anche se questi limiteranno il feedback negativo del E2. In breve, lo stimolo del GnRH e, di conseguenza, di LH e FSH saranno potenzialmente ridotti in rapporto AAS-dipendente e dose-dipendente. Ciò significa che non sarà possibile garantire livelli adeguati di E2 secondari alla aromatizzazione del Testosterone stimolato dalla attività del LH legata alla somministrazione di Clomifene o Enclomifene.
Con l’uso del Fluoxymesterone le cose si complicherebbero ulteriormente. La sua capacità inibitiva sull’Asse HPT è più marcata di quella esercitata dal Methyltestosterone, nonostante non sia aromatizzabile, e si manifesta maggiormente a livello testicolare. Nel range dei 20mg/die non sembra mostrare un significativo impatto su FSH e LH ma già sul Testosterone circolante. Il Fluoxymesterone possiede una biodisponibilità del 100%, dovuta alla metilazione in posizione 17α la quale inibisce il metabolismo epatico per ossidazione enzimatica del 17β-idrossile, consentendo l’assorbimento nel flusso sanguigno della molecola. Come molti altri steroidi metilati in C-17, il Fluoxymesterone presenta una scarsa affinità con i recettori AR, ciononostante le sue azioni sono mediate dal recettore degli androgeni, molto probabilmente a causa della sua prolungata emivita plasmatica che è di circa 9,2 ore.(Seth Roberts “Anabolic Pharmacology”. 2009)
Effetto dei SERM sull’Asse hGH/IGF1
Esistono poche differenze tra i vari SERM nell’influenzare negativamente l’Asse hGH/IGF1, in quanto è stato riportato che il Raloxifene ha indotto una minore diminuzione dei livelli di IGF1 rispetto al Tamoxifene, considerando che entrambi i farmaci sono stati somministrati a un dosaggio massimo di 120mg/die e 20mg/die, rispettivamente [94].
Cozzi et al. [95] hanno provato per la prima volta a utilizzare il tamoxifene come possibile trattamento dell’acromegalia; nel 1997 hanno trattato 19 soggetti acromegalici (6 maschi, 13 femmine) per due mesi con un dosaggio crescente, fino a raggiungere i 40 mg/die. L’IGF1 medio è diminuito del 29,5%, con un range compreso tra il 18% e il 60%, in 13 dei 19 pazienti, raggiungendo un controllo ormonale completo in quattro di essi (21%). I livelli di GH sono leggermente aumentati rispetto al basale, mentre dopo la sospensione del tamoxifene l’IGF1 sierico è prontamente aumentato.
Molti anni dopo, Balili et al. [31] hanno riportato che 17 pazienti (15 maschi e 2 femmine) con acromegalia resistente sono stati trattati con tamoxifene (dose massima 40mg/die) per un periodo mediano di quattro mesi. È stata evidenziata una riduzione significativa dell’IGF1 nell’82% dei pazienti, raggiungendo il controllo della malattia nel 47% dei casi. I livelli sierici di IGF1 si sono ridotti del 17,5%, mentre i livelli di GH non hanno subito variazioni significative.
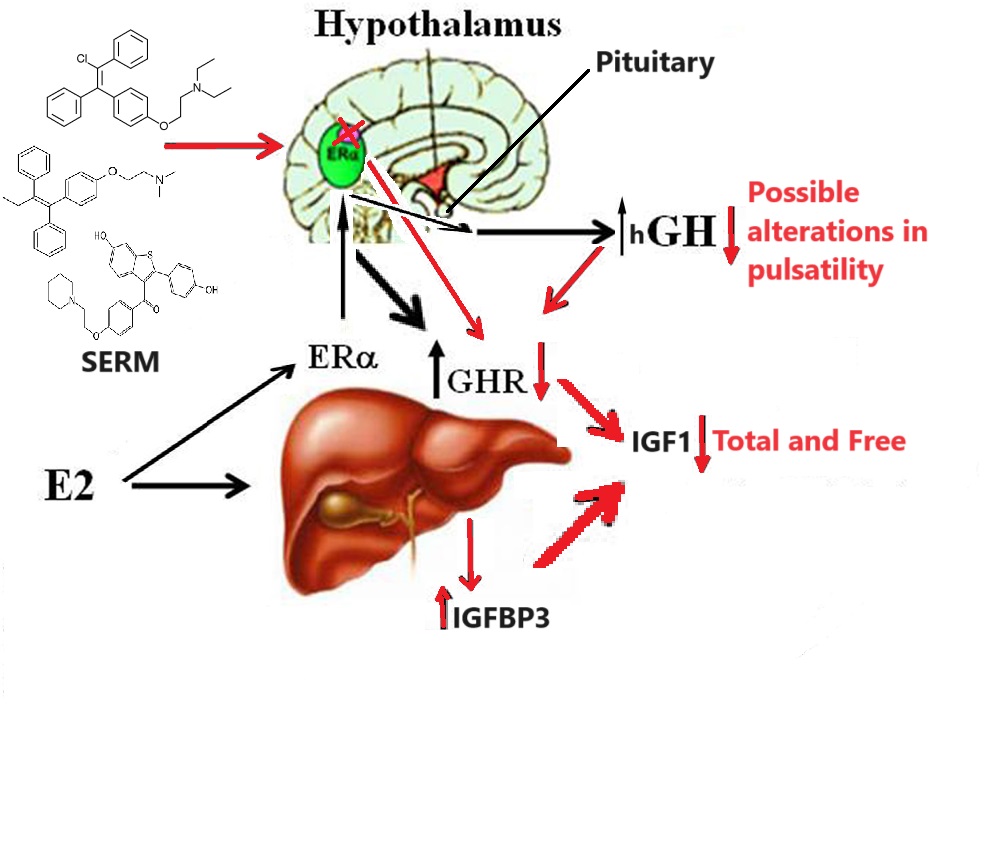
Duarte et al. [35] nel 2016 hanno studiato 16 maschi con acromegalia non controllata, dimostrando l’efficacia del Clomifene Citrato (CC) come terapia aggiuntiva a SRL o Cabergolina. I pazienti sono stati trattati per tre mesi con CC 50mg/die, mostrando una riduzione media dei livelli di IGF1 del 41% (con valori compresi tra il 16,8% e il 68,3%), che ha portato il 44% dei pazienti a raggiungere il controllo ormonale.
Gli estrogeni e i SERM hanno ampiamente dimostrato una significativa attività di riduzione dell’IGF1.
Le concentrazioni plasmatiche seriali di hGH sono state misurate ogni 20 minuti per 24 ore prima e dopo la somministrazione di Clomifene Citrato (100mg/die per 7 giorni) a quattro soggetti sani maschi giovani adulti. Il numero di episodi secretori di hGH e l’entità del picco delle concentrazioni plasmatiche durante la veglia e il sonno sono diminuiti dopo i periodi di trattamento con Clomifene Citrato.[https://www.sciencedirect.com/science/article/abs]
In uno studio sono stati inclusi sette bracci, comprendenti donne in postmenopausa con diabete mellito di tipo 2, donne in postmenopausa con cancro al seno, donne sane in postmenopausa e uomini anziani sani. La terapia con Raloxifene ha ridotto significativamente i livelli di IGF-1 (WMD: -2,92 nmol/L, 95% CI: -3,49, -2,35, p < 0,001) rispetto al placebo. Il dosaggio di raloxifene ˃60mg/die (WMD: -3,29 ng/mL, 95% CI: -3,50-3,08, I2 = 0,0%) ha ridotto i livelli di IGF-1 più di 60 mg/die (WMD: -2,29 ng/mL, 95% CI: -2,90 -1,69, I2 = 16%). Inoltre, la durata dell’intervento ˃26 settimane (WMD: -3,48 ng/mL, 95% CI: -5,26 a -1,69, I2 = 0,0%) ha ridotto i livelli di IGF-1 più di ˂26 settimane (WMD: -2,55 ng/mL, 95% CI: -3,31 a -1,79, I2 = 92%). Al contrario, i risultati complessivi del modello a effetti casuali non hanno suggerito un cambiamento significativo nei livelli di IGFBP-3 con la terapia con raloxifene. La terapia con Raloxifene ha ridotto significativamente i livelli sierici di IGF-1, ma senza variazioni nei livelli di IGFPB-3.[https://www.sciencedirect.com/science/article/abs/pii/S1096637421000447]
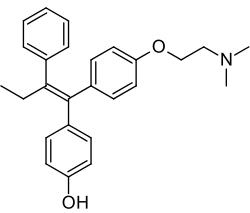
Il Tamoxifene è in grado di ridurre l’IGF-1 biodisponibile (calcolato come rapporto tra IGF-1 e IGF-BP3) per almeno 18 mesi. Sebbene le concentrazioni di IGF-1 non si siano ridotte in modo significativo, le concentrazioni della sua principale proteina legante IGF-BP3 sono aumentate in modo significativo, riducendo così la quantità di IGF-1 disponibile. Tuttavia, il rapporto tra IGF-1 e IGF-BP3 non era significativamente ridotto rispetto al basale a 27 mesi, per cui l’effetto di un trattamento più lungo resta da chiarire. Anche il Tamoxifene ha aumentato significativamente le concentrazioni di IGF-BP1 rispetto al basale dopo 18 mesi di trattamento. Questo aumento è stato osservato anche in altri studi.
In alcuni studi sul Tamoxifene è stata notata paradossalmente un’assenza di effetti sulle concentrazioni di IGF-1 a differenza di altri studi che hanno dimostrato una riduzione dell’IGF-1 da parte del Tamoxifene. Questo potrebbe essere il risultato del numero ridotto di pazienti degli studi in questione o della selezione della popolazione. Tuttavia, uno studio non ha mostrato un effetto sull’IGF-1 a un follow-up mediano di 29 mesi. Questi ricercatori avevano osservato una diminuzione significativa dei valori di IGF-1 dopo sei mesi di trattamento con Tamoxifene e i loro dati indicano un effetto limitato dopo un trattamento a lungo termine. Anche altri dati da campioni più piccoli indicano una riduzione iniziale (sebbene non significativa) dell’IGF-1, che si perde con l’aumentare del tempo di follow-up. Ciò indica un effetto potenzialmente importante della durata del trattamento sull’esito e sottolinea la necessità di ulteriori studi longitudinali con periodi di follow-up rigorosamente tempificati.
Uno studio a lungo termine controllato con placebo ha mostrato una riduzione significativa dell’IGF-1 dopo un follow-up medio di 27 mesi (follow-up minimo di tre mesi), ma non sono stati prelevati campioni longitudinali. È possibile che i campioni provenienti dagli studi di prevenzione con Tamoxifene in corso (come l’IBIS) vengano utilizzati per ulteriori ricerche sugli effetti del Tamoxifene sul sistema IGF. In alcuni studi i campioni utilizzati non erano a digiuno e questo può essere importante perché i valori possono fluttuare in base all’assunzione di nutrienti.
Il meccanismo con cui il Tamoxifene altera lo stato dell’IGF non è stato completamente chiarito. Tuttavia, si ritiene che il Tamoxifene alteri i valori di IGF-1 riducendo la produzione di hGH da parte dell’ipofisi, abbassando così la quantità di IGF-1 prodotta dal fegato [endocrina] e rilasciata in circolo. Sappiamo che il Tamoxifene ha anche un’azione diretta come antagonista dell’E2 in diversi tessuti del corpo oltre che sulle cellule del cancro al seno, e sembrerebbe alterare la quantità di IGF-1 e di proteine leganti rilasciate dalle cellule stesse.
Il Tamoxifene, quindi, può aumentare l’IGF-BP1, l’IGF-BP3 e ridurre il rapporto tra IGF-1 e IGF-BP3. Gli effetti a lungo termine dell’uso del Tamoxifene sullo stato dell’IGF devono ancora essere stabiliti. Non è ancora del tutto chiaro quando e per quanto tempo il Tamoxifene può ridurre l’IGF-1 circolante.[https://www.ncbi.nlm.nih.gov/]
- Aumento delle SHBG
L’effetto del Clomifene Citrato (CC) sulle SHBG è stato studiato in 10 pazienti oligozoospermici con varicocele e 6 uomini normospermici. Le SHBG plasmatiche, Testosterone (T), Estradiolo (E2), FSH, LH. Prolattina (Prl), Tiroxina (T4) e 17-OH-progesterone (17-OH-P) sono stati determinati prima e durante la terapia. La concentrazione di SHBG è aumentata da 38,1 ± 18,3 a 54,3 ± 16,0 nmol/l (P < 0,01), mentre il T e l’E2 hanno mostrato aumenti significativi da 31,2 ± 10,8 nmol/***l e 24,6 ± 5,4 pg/ml a 52,0 ± 3,6 e 43,3 ± 14,9, rispettivamente nei pazienti oligozoospermici, con aumenti simili osservati negli uomini normospermici. L’FSH, l’LH e il 17-OH-P sono risultati marcatamente elevati durante la somministrazione di CC, mentre Prl e T4 sono rimasti invariati. I risultati di questo studio indicano che la CC provoca un aumento della concentrazione di SHBG, probabilmente correlato anche all’aumento della concentrazione di E2. Questa variazione della SHBG, combinata con l’attività estrogenica intrinseca del CC, potrebbe essere uno dei fattori responsabili, attraverso una diminuzione del T libero e uno squilibrio tra T ed E2, della mancanza di un effetto significativo sui parametri della qualità seminale nei pazienti così trattati. [https://onlinelibrary.wiley.com/doi/abs/]
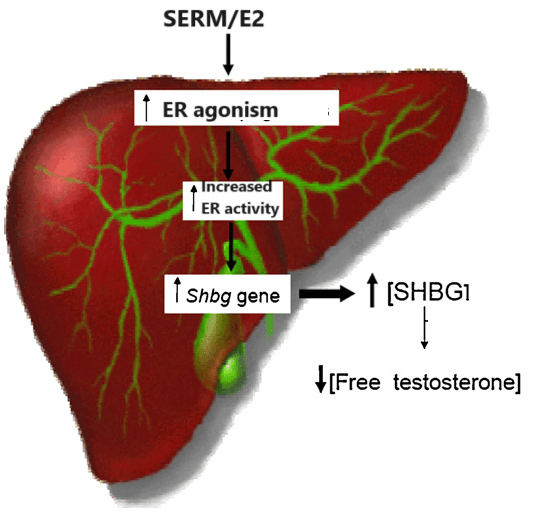
In uno studio, tredici pazienti sono stati sottoposti a trattamento con Tamoxifene dopo la classificazione secondo Nydick (gruppo 1). Il gruppo 2 era composto da otto pazienti seguiti senza trattamento. La ginecomastia era presente bilateralmente in 15 pazienti. In entrambi i gruppi si è verificata una riduzione statisticamente significativa delle dimensioni del seno. Si è verificata una diminuzione significativa della SHBG sierica solo nel gruppo 2. Questi risultati suggeriscono che la SHBG sierica è aumentata dal trattamento con Tamoxifene negli adolescenti maschi trattati. I livelli di SHBG sono diminuiti per tutta la durata del follow-up nei pazienti che sono guariti con o senza trattamento. Tuttavia, questa diminuzione era statisticamente significativa nel gruppo non trattato, ma non in quello trattato con Tamoxifene. In conclusione, è stato suggerito che il calo puberale dei livelli di SHBG sia attenuato dal trattamento con tamoxifene somministrato per la ginecomastia puberale, poiché il Tamoxifene aumenta i livelli di SHBG negli adolescenti maschi.[https://pubmed.ncbi.nlm.nih.gov/15379424/]
Ma gli Inibitori della Aromatasi?
Gli IA possono essere in alcuni casi un modo efficace per controllare i livelli di E2 durante la TRT. Tuttavia, il dosaggio necessario per mantenere i livelli di E2 nell’intervallo ottimale dipende da ciascun individuo e richiede un attento monitoraggio da parte di un professionista sanitario. Ma in un contesto di alterazione del ciclo di feedbeack negativo del E2, specie se cosomministrati con AAS non aromatizzabili, possono portare a peggioramento delle condizioni più che ad una risposta positiva nel mantenimento di una certa attività dell’Asse HPT.
Conclusioni:

Nonostante la ricerca abbia mostrato in studi su animali sottoposti a somministrazione di AAS (Oxymetholone) abbinata al Clomifene Citrato una qualche conservazione del Testosterone endogeno [Growth-hormone-secretagogue-GHRP-6-and-clomiphene?redirectedFrom=fulltext], e che nelle terapie per la fertilità in soggetti in TRT, o in soggetti trattati per brevi periodi con AAS e.v., la somministrazione di Clomifene Citrato ha mostrato un effetto misurabile [ma qui parliamo comunque di condizioni più che altro “mimiche-fisiologiche”], sul campo la misurazione dell’efficacia della somministrazione di SERM (soprattutto Clomifene e Enclomifene) per mantenere una certa sintesi endogena di Testosterone e consequenzialmente dei suoi metaboliti E2 e DHT, non è lineare e chiara, sia per la difficile identificazione della qualità dei PEDs utilizzati e sia per la difficolta di svolgere esami ematici che non siano basati sul fallace (ormonalmente) metodo ECLIA/ELISA. La rara possibilità (almeno in Italia) di poter accedere a laboratori dove sono svolti test LC/MS-MS ultra sensibile [vedi spettrometria di massa accoppiata] limita le valutazioni precise necessarie dal momento che con i metodi sopra citati ormoni diversi possono essere letti come il medesimo ormone. Nonostante ciò, siamo stati in grado di notare degli effetti terapeutici sufficienti con cicli a medio/basso dosaggio di AAS come Oxandrolone e Stanozololo [media 30mg/die]. In altre circostanze, e in una buona fetta di popolazione, l’andamento dell’efficacia variava all’interno dello stesso arco temporale del ciclo al quale i soggetti si sottoponevano.
Basandoci sulla ricerca diretta, possiamo teoricamente elencare gli AAS/SARM/PH e DS con l’effetto ipoteticamente raggiungibile in combinazione con SERM:
- Effetto buono
- Oxandrolone [=30mg di media]
- Stanozololo [=20mg di media]
- Methyldrostanolone [=30mg di media]
- 4-clorodeidrometiltestosterone [=40mg di media]
- Ostarina [=20mg di media]
- RAD140 [=20mg di media]
- Effetto discreto/moderato
- Testosterone Undecanoato [<120mg/die di media]
- Methandrostenolone [<20mg di media]
- Oxymetholone [<50mg di media]
- LGD4033 [<10mg di media]
- Effetto non sufficiente
- Fluoxymesterone [≥10mg di media]
- MENTDIONE [≥50mg di media]
- MENT [≥25mg di media]
- Metribolone [≥250mcg di media]
- Norethandrolone [≥20mg di media]
- Trenbolone Acetato (orale) [≥25mg di media]
- 19-Nor-5-androstenediolo [≥50mg di media]
Chi sceglie di prendere la “via del Enhanced” e la sua paura principale è basata sulle iniezioni beh, forse è meglio che abbandoni tale possibile scelta… no?…
Paradossalmente, è di gran lunga più funzionale l’inserimento di piccole dosi di Methandrostenolone [15mg/die circa] come base “sostitutiva” del Testosterone compensando il DHT con la versione metilata in C1 di questo, il Mesterolone.
Amedeo Bellizzi [CEO BioGenTech]
Riferimenti:
- Cameron JL, Cameron AM (20 November 2013). Current Surgical Therapy. Elsevier Health Sciences. pp. 582–. ISBN 978-0-323-22511-3.
- Huang X, Aslanian RG (19 April 2012). Case Studies in Modern Drug Discovery and Development. John Wiley & Sons. pp. 392–394. ISBN 978-1-118-21967-6.
- Kremoser C, Albers M, Burris TP, Deuschle U, Koegl M (Oct 2007). “Panning for SNuRMs: using cofactor profiling for the rational discovery of selective nuclear receptor modulators”. Drug Discovery Today. 12 (19–20): 860–9. doi:10.1016/j.drudis.2007.07.025. PMID 17933688.
- Rosano C, Stec-Martyna E, Lappano R, Maggiolini M (2011). “Structure-based approach for the discovery of novel selective estrogen receptor modulators”. Current Medicinal Chemistry. 18 (8): 1188–94. doi:10.2174/092986711795029645. PMID 21291367.
- Nilsson S, Koehler KF, Gustafsson JÅ (Oct 2011). “Development of subtype-selective oestrogen receptor-based therapeutics”. Nature Reviews. Drug Discovery. 10 (10): 778–92. doi:10.1038/nrd3551. PMID 21921919. S2CID 23043739.
- Koehler KF, Helguero LA, Haldosén LA, Warner M, Gustafsson JA (May 2005). “Reflections on the discovery and significance of estrogen receptor beta”. Endocrine Reviews. 26 (3): 465–78. doi:10.1210/er.2004-0027. PMID 15857973.
- Dutertre M, Smith CL (Nov 2000). “Molecular mechanisms of selective estrogen receptor modulator (SERM) action”. The Journal of Pharmacology and Experimental Therapeutics. 295 (2): 431–7. PMID 11046073.
- Xu X, Yang W, Li Y, Wang Y (Jan 2010). “Discovery of estrogen receptor modulators: a review of virtual screening and SAR efforts”. Expert Opinion on Drug Discovery. 5 (1): 21–31. doi:10.1517/17460440903490395. PMID 22823969. S2CID 207492889.
- Musa MA, Khan MO, Cooperwood JS (2007). “Medicinal chemistry and emerging strategies applied to the development of selective estrogen receptor modulators (SERMs)”. Current Medicinal Chemistry. 14 (11): 1249–61. doi:10.2174/092986707780598023. PMID 17504144.
- Lewis JS, Jordan VC (Dec 2005). “Selective estrogen receptor modulators (SERMs): mechanisms of anticarcinogenesis and drug resistance”. Mutation Research. 591 (1–2): 247–63. doi:10.1016/j.mrfmmm.2005.02.028. PMID 16083919.
- Feng Q, O’Malley BW (Nov 2014). “Nuclear receptor modulation–role of coregulators in selective estrogen receptor modulator (SERM) actions”. Steroids. 90: 39–43. doi:10.1016/j.steroids.2014.06.008. PMC 4192004. PMID 24945111.
- Wittliff JL, Kerr II DA, Andres SA (2005). “Estrogens IV: Estrogen-Like Pharmaceuticals”. In Wexler P (ed.). Encyclopedia of Toxicology, 2nd Edition. Vol. Dib–L. Elsevier. pp. 254–258. ISBN 9780080548005.
- Blair RM, Fang H, Branham WS, Hass BS, Dial SL, Moland CL, et al. (March 2000). “The estrogen receptor relative binding affinities of 188 natural and xenochemicals: structural diversity of ligands”. Toxicological Sciences. 54 (1): 138–53. doi:10.1093/toxsci/54.1.138. PMID 10746941.
- Fang H, Tong W, Shi LM, Blair R, Perkins R, Branham W, et al. (March 2001). “Structure-activity relationships for a large diverse set of natural, synthetic, and environmental estrogens”. Chemical Research in Toxicology. 14 (3): 280–94. doi:10.1021/tx000208y. PMID 11258977.
- Baumann RJ, Bush TL, Cross-Doersen DE, Cashman EA, Wright PS, Zwolshen JH, et al. (March 1998). “Clomiphene analogs with activity in vitro and in vivo against human breast cancer cells”. Biochemical Pharmacology. 55 (6): 841–51. doi:10.1016/s0006-2952(97)00574-1. PMID 9586957.
- Sutherland RL, Watts CK, Ruenitz PC (October 1986). “Definition of two distinct mechanisms of action of antiestrogens on human breast cancer cell proliferation using hydroxytriphenylethylenes with high affinity for the estrogen receptor”. Biochemical and Biophysical Research Communications. 140 (2): 523–9. doi:10.1016/0006-291x(86)90763-1. PMID 3778464.
- Obach RS (April 2013). “Pharmacologically active drug metabolites: impact on drug discovery and pharmacotherapy”. Pharmacological Reviews. 65 (2): 578–640. doi:10.1124/pr.111.005439. PMID 23406671. S2CID 720243.
- Trost LW, Khera M (July 2014). “Alternative treatment modalities for the hypogonadal patient”. Current Urology Reports. 15 (7): 417. doi:10.1007/s11934-014-0417-2. PMID 24817260. S2CID 20304701.
- Rambhatla A, Mills JN, Rajfer J (2016). “The Role of Estrogen Modulators in Male Hypogonadism and Infertility”. Reviews in Urology. 18 (2): 66–72. doi:10.3909/riu0711 (inactive 31 January 2024). PMC 5010627. PMID 27601965.
- Kaminetsky, Jed, et al. “Oral enclomiphene citrate stimulates the endogenous production of testosterone and sperm counts in men with low testosterone: comparison with testosterone gel.” The journal of sexual medicine 10.6 (2013): 1628-1635.
- Kim, Edward D., Andrew McCullough, and Jed Kaminetsky. “Oral enclomiphene citrate raises testosterone and preserves sperm counts in obese hypogonadal men, unlike topical testosterone: restoration instead of replacement.” BJU international 117.4 (2016): 677-685.
- Earl, Joshua A., and Edward D. Kim. “Enclomiphene citrate: A treatment that maintains fertility in men with secondary hypogonadism.” Expert review of endocrinology & metabolism 14.3 (2019): 157-165.