Ricercatori australiani presso la University of New South Wales stanno testando un nuovo farmaco per la perdita di peso che agisce similmente al DNP (2,4-dinitrofenolo), ma senza i numerosi effetti collaterali connessi a quest’ultimo. Il loro lavoro è stato recentemente pubblicato su Nature Communications.[1]
BAM15?
Finché l’epidemia di obesità continuerà a crescere, la ricerca di farmaci per la perdita di peso continuerà. Parte di questa ricerca si svolge tenendo come riferimento il pericoloso ma estremamente efficace DNP. Sarebbe possibile sintetizzare una molecola che sia efficace come DNP, ma non così pericolosa?
Il DNP è un disaccoppiatore della fosforilizzazione ossidativa: interferisce con la resintesi di ATP nel mitocondrio causando una significativa dispersione di energia sotto forma di calore.
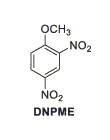
Ricercatori americani presso la Yale University stanno studiando un analogo metilato del DNP, il DNPME.[2]
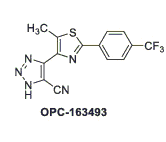
La giapponese Otsuka Pharmaceutical sta conducendo esperimenti con OPC-163493.[3]
I ricercatori australiani che hanno pubblicato la ricerca che qui si sta trattando, stanno svolgendo esperimenti sul N5, N6-bis (2-fluorofenil) [1,2,5] oxadiazolo [3,4-b] pirazina-5,6-diammina. In breve: BAM15 .
Effetti del BAM15 nei topi
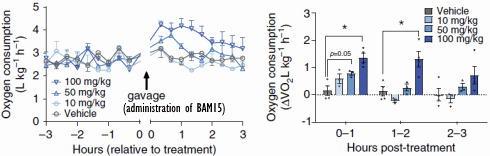
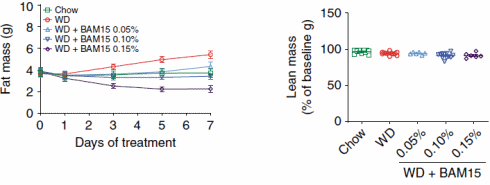
I ricercatori hanno somministrato ai topi dosi orali di BAM15 e hanno osservato che nelle ore successive alla somministrazione, il consumo di ossigeno degli animali – e quindi il loro consumo calorico – era aumentato di alcune decine di punti percentuale. L’effetto è stato temporaneo. Questo perché l’emivita del BAM15 nei topi è di sole 1,7 ore.
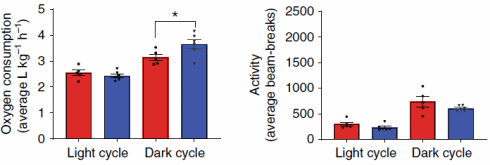
In altri esperimenti, in cui i topi sono stati alimentati con cibo contenente BAM15, i ricercatori hanno scoperto che la molecola aumentava il dispendio calorico solo di notte. Ciò non sorprende, perché i topi sono animali notturni e quindi preferiscono mangiare quando è buio. Il BAM15 non ha aumentato la quantità di attività fisica svolta.
Il dosaggio utilizzato nei topi trasposto in un essere umano adulto corrisponderebbe a circa 1g/die di BAM15.
In un altro esperimento, i ricercatori hanno nutrito i topi con zucchero e grasso addizionali [WD], rendendo gli animali più grassi. Se i topi venivano trattati con il BAM15, la loro massa grassa aumentava di meno, o per nulla, o addirittura diminuiva. L’equivalente umano delle dosi alle quali si sono verificati questi effetti è stato rispettivamente di 500mg, 1g e 1,5g di BAM15 al giorno.
Il BAM15 non ha aumentato la temperatura corporea dei topi, non ha ridotto la massa magra o aumentato l’attività dei radicali liberi. Per quanto i ricercatori hanno potuto scoprire, il BAM15 aumenta anche la sensibilità all’Insulina.
Conclusioni
Concludendo, i ricercatori riportano che il BAM15 rappresenta un raro disaccoppiatore mitocondriale che previene e inverte l’obesità senza influire sull’assunzione di cibo o sulla massa magra. Una limitazione del BAM15 è la bassa solubilità acquosa, ma questa proprietà non ha influito sulla biodisponibilità orale e infatti la bassa solubilità acquosa è un parametro importante che consente al BAM15 di penetrare nelle membrane cellulari ed entrare nei mitocondri. Un’altra limitazione del BAM15 è l’emivita di 1,7 ore e le direzioni future esamineranno le strategie di formulazione per migliorare l’esposizione. Collettivamente, i dati qui presentati supportano l’ulteriore sviluppo del BAM15 come potenziale terapeutico per l’obesità e le malattie metaboliche.
Non sono un amante delle diete “prefabbricate”, impostate su “dogmi” traballanti e letture parziali di studi mal interpretati. C’è stato un periodo della mia vita nel quale mi “innamorai” di un idea alimentare che mi portò dalla Dieta a Zona di Barry Sears alla Paleo Diet di Loren Cordain. Ma, fortunatamente, gli errori di gioventù insegnano ed è più di un decennio, da quando studio scienze della nutrizione, che mi sono totalmente discostato da una certa visione scientista dell’alimentazione.
Allora perchè dedicare un articolo ad una dieta “prefabbricata”? Beh, perchè l’onestà intellettuale non mi manca e quando c’è da riconoscere un merito, anche se può essere contornato da bislacche affermazioni, sono il primo a riconoscerlo.
Sicuramente, non scrivo questo articolo perchè penso che la “Vertical Diet” sia il must delle diete, e nemmeno perchè è opera di Stan Efferding, un Bodybuilder e PowerLifter d’élite, oppure perchè Hafthor Bjornsson, l’uomo più forte del mondo 2018, e Brian Shaw, l’uomo più forte del mondo per quattro volte, la seguono.
Ho già accennato il mio punto di vista su questa metodica alimentare in un post pubblicato qualche tempo fa su Instagram. Questa volta, però, merito del numero di caratteri qui esprimibili, avrò la possibilità di poter approfondire l’argomento in modo esaustivo.
Cos’è la “Vertical Diet”?
Stan “The White Rhino” Efferding (nato il 6 novembre 1967 a Portland, Oregon) è un Bodybuilder professionista americano IFBB ed un PowerLifter competitivo nella Southern Powerlifting Federation (SPF). Ha detenuto il record assoluto di powerlifting del mondo nella classe 275 libbre senza fasciatura del ginocchio (2.226,6 libbre) e nello Squat senza fasciatura del ginocchio (854 libbre). Grazie alla sua enorme forza fisica, partecipa regolarmente a gare di PowerLifting professionale e durante la sua carriera nel Bodybuilding competitivo professionistico, Efferding veniva spesso definito il “bodybuilder più forte del mondo”.
La “Vertical Diet” è stata sviluppata da Stan Efferding, un powerlifter d’élite, per migliorare le prestazioni di Bodybuilder, PowerLifter e atleti di alto livello.
Nel programma alimentare viene affermato anche che la presente strategia risulta ideale per i comuni frequentatori di palestra che stanno cercando di aumentare la massa muscolare o perdere peso.
A differenza delle tradizionali diete “orizzontali” che enfatizzano la varietà dietetica in numerosi gruppi alimentari, la “Vertical Diet” si concentra su un numero limitato di alimenti ricchi di nutrienti di alta qualità.
Secondo Efferding, limitare la varietà rende il corpo più efficiente nel digerire e assorbire i nutrienti, il che dovrebbe migliorare la crescita muscolare, il recupero, la salute dell’intestino e il metabolismo.
Detto questo, le sue affermazioni non sono supportate da valide prove scientifiche .
Alimenti nella “Vertical Diet”
Se c’è una cosa che rende questa metodica alimentare degna di nota è l’enfasi straordinariamente grande che l’autore da sull’ottimizzazione della digestione.
Si concentra su carne rossa, riso bianco e alimenti a basso contenuto di FODMAP.
In breve, la “Vertical Diet” riguarda il consumo di alimenti ricchi di nutrienti che sono facilmente digeribili per aiutare l’atleta perdere, guadagnare o mantenere il peso, massimizzare gli allenamenti e ottenere un migliore assorbimento dei nutrienti. L’obiettivo è quello di mantenere l’attenzione principale su micronutrienti come vitamine, minerali e antiossidanti.
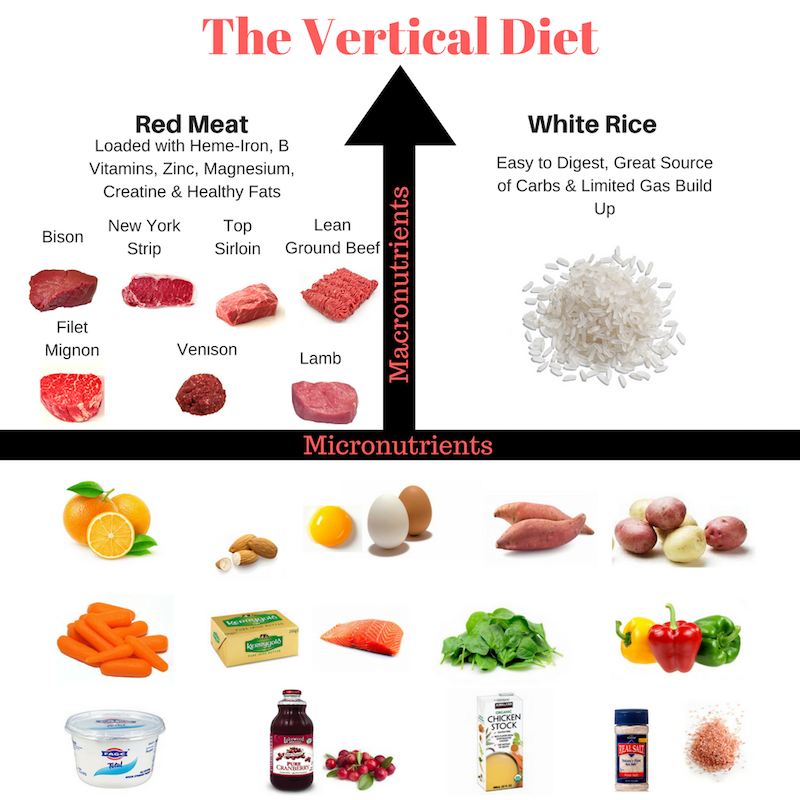
Si chiama “Vertical Diet” perché quando è disposta graficamente, sembra una “T” rovesciata: alimenti base e alimenti “ancillari”. Come si può vedere nell’immagine riportata di seguito gli alimenti principali, posti in verticale, sono carne rossa e riso bianco.
Perché?
Ci sono pochi dubbi sul fatto che il riso bianco venga digerito molto facilmente dalla maggior parte delle persone, praticamente senza fibre e grassi è un “carboidrato pulito”: l’autore giustifica la sua scelta anche in base a vecchie credenze sulla “nocività” della lectina o acido fitico – chiamati “antinutrienti”, di cui parleremo di seguito – contenuti in alcuni alimenti di origine vegetale.
La carne rossa è stata scelta da Efferding perchè considerata il tipo di carne più nutriente, ricca di ferro eme, vitamine del gruppo B, Zinco, Magnesio, Creatina e Grassi sani (se grass fed).
Si può rimanere un pò sorpresi di vedere il Magnesio nell’elenco dato che le verdure a foglia verde e i legumi (sconsigliati da Efferding) sono generalmente considerati le migliori fonti. Duecento grammi ne contengono 50mg di questo importante minerale, anche se questo è solo il 12% del RDA – quindi, assumendo molti degli altri alimenti raccomandati, probabilmente è possibile raggiungere l’assunzione di 400mg al giorno.
Sotto i pilastri della dieta rappresentati da carne rossa e riso bianco, si trova la “base della “T”: frutta, patate, spinaci, peperoni rossi, carote, succo e alcuni altri prodotti animali come burro, brodo e pesce grasso.
Quindi l’enfasi è sulla carne rossa, riso e vegetali. Ma c’è un lungo elenco di cibi da “non mangiare”. E la compliance si riduce ulteriormente.
Ricapitolando, gli alimenti su cui ruota la “Vertical Diet” sono:
Riso: solo bianco
Carni rosse: manzo, agnello, bisonte e carne di cervo (grass fed)
Frutta: principalmente arance, succo d’arancia 100%, mirtilli e succo di mirtillo 100% – ma tutti i frutti sono ammessi
Patate: patate bianche e dolci
Verdure a basso contenuto di FODMAP: carote, sedano, zucchine, cetrioli, peperoni, melanzane, spinaci, zucca, ecc.
Oli e grassi: olio extra vergine di oliva, olio di cocco, olio di avocado, burro, noci
Pesce grasso: il consumo di salmone selvaggio dell’Alaska è fortemente incoraggiato
Uova: uova intere
Latticini: yogurt intero, latte intero, formaggio
Sodio: brodo di ossa, brodo di pollo, sale da tavola iodato
Pollame: pollo, tacchino
Possibili scelte solo in determinate modalità
Avena: solo se messa in ammollo e/o fermentata
Legumi: fagioli e altri legumi, solo se messi in ammollo e/o fermentati
Come si può notare, la dieta incoraggia a consumare cibi di alta qualità, come carni provenienti da animali allevati al pascolo, uova da galline ruspanti e frutta e verdura biologiche.
Cosa non mangiare nella “Vertical Diet”
Grani
L’autore, sempre condizionato da conoscenze parziali, vieta il consumo di fonti di acido fitico e glutine (che non causa alcun tipo di problema in soggetti sani), di conseguenza la dieta non include il consumo di pasta, cereali o pane, a meno che non sia pane fermentato a lievitazione naturale.
Avena
A meno che non sia messa a mollo e fermentata, sempre per la questione della riduzione degli “antinutrienti”.
Legumi
Considerata una fonte di lectine, e proteine dalla ridotta biodisponibilità per via del ricco contenuto di fibre. Possono essere consumati se messi in ammollo e fermentati.
Oli vegetali trasformati
Una scelta piuttosto non controversa: queste sono fonti ricche di acidi grassi Omega-6, già abbondantemente consumati nella dieta occidentale e che sono stati fortemente collegati a infiammazione, malattie cardiache e obesità. (1) (2) (3)
Caffè
Efferding afferma che il caffè provoca disidratazione e compromette la digestione.
Dice di non mangiare cibi che piacciono, ma di mangiare cibi che mi piacciono. Avere una ottimale salute intestinale e una digestione efficiente potrebbe far sentire meglio della soddisfazione provata consumando cibi appaganti il palato… almeno secondo l’autore.
Polialcoli (Polioli)
Dolcificanti naturali come l’Eritritolo e lo Xilitolo, piuttosto popolari nelle barrette proteiche e nei gelati dietetici. Come per i legumi, sono noti per poter causare indigestione in alcune persone, anche se non è un fenomeno così diffuso.
Verdure ad alto contenuto di Raffinosio
Forse questa è l’esclusione più controversa, che include verdure crocifere come broccoli, cavolfiori e asparagi. Il colpevole è il Raffinosio, un certo zucchero presente in alcune verdure, cereali e legumi che è resistente alla digestione e talvolta può causare gas.
Aglio e cipolla
Sono alimenti ricchi di FODMAP (sigla che sta per “Fermentable Oligosaccharides, Disaccharides, Monosaccharides and Polyols” ovvero Oligosaccaridi, Disaccaridi, Monosaccaridi Fermentabili e Polioli) .
Acqua alcalinizzata
Secondo l’autore, questa bufala commerciale, può alterare la digestione.
Bisogna tenere presente che la dieta consente il consumo di piccole quantità di alcuni di questi alimenti purché il soggetto possa digerirli senza alcun sintomo digestivo, come gas o gonfiore.
Tuttavia, gli oli vegetali trasformati non sono mai ammessi.
Quindi cos’è un alimento FODMAP?
I FODMAP sono carboidrati fermentabili il cui consumo è limitato tra le persone con disturbi digestivi
La “Vertical Diet” è fondamentalmente una dieta a basso contenuto di FODMAP.
Come già accennato, FODMAP è l’acronimo di “Fermentable Oligosaccharides, Disaccharides, Monosaccharides and Polyols” ovvero Oligosaccaridi, Disaccaridi, Monosaccaridi Fermentabili e Polioli, che si riferisce ai carboidrati a catena corta che sono spesso limitati tra le persone con disturbi digestivi come la sindrome dell’intestino irritabile. (4) (5) Sono tipi di fibre e attraversano il sistema non digerite; i batteri intestinali li usano come substrato energetico, producendo idrogeno gassoso nel processo. (6) In alcune persone, ciò può attirare liquidi nell’intestino e causare diarrea e per queste persone le diete povere o a basso contenuto di FODMAP possono essere una manna dal cielo.
Poiché la “Vertical Diet” prevede macronutrienti facilmente digeribili, i cibi ricchi di FODMAP sono limitati o preparati in modo da ridurre il disagio digestivo. Le verdure come spinaci, cetrioli, peperoni e patate sono migliori delle verdure crocifere per aumentare i risultati positivi della dieta secondo quanto affermato da Efferding.
Non ci sono molti dati su popolazioni sane che usano una dieta a basso contenuto di FODMAP, ma alcune ricerche hanno mostrato una riduzione dei gas fermentativi e miglioramenti nella salute dell’intestino, e uno studio ha persino scoperto che una dieta low-FODMAP migliora i sintomi gastrointestinali legati all’esercizio fisico. (7) (8) Questa è solo una congettura, ma questo potrebbe essere una alimentazione utile per le persone che hanno bisogno di digerire una quantità straordinariamente alta di calorie.
Lista degli alimenti ricchi in FODMAP
Nota: si ricordi che anche nella dieta FODMAP, la quale non è una classica dieta ad eliminazione di determinati alimenti ma è un regime alimentare strutturato su schemi piuttosto rigidi, prevede in seguito il reintegro, dopo un dato lasso di tempo, degli alimenti eliminati (FODMAP). Infatti non è consigliabile eliminare definitivamente tutti i cibi che contengono FODMAP, principalmente per la riduzione nella varietà della dieta e, come avvalorato da diversi studi, si registrano dei potenziali effetti benefici, soprattutto a medio-lungo termine, dei FODMAP e degli alimenti che li contengono (vedi, per esempio, i legumi).
Lectine, acido fitico e “antinutrienti”
Questi composti possono ridurre l’assorbimento di alcuni minerali
Questo può essere risolto consumandoli con vitamina C e / o tenendoli in ammollo e/o facendoli fermentare.
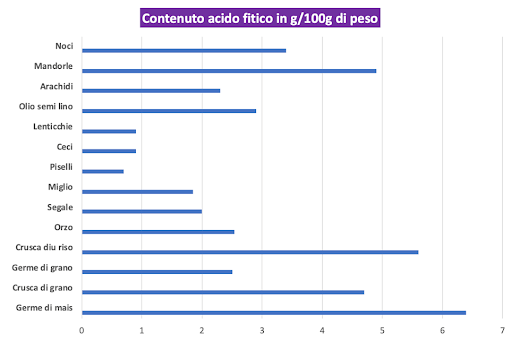
Lectine e acido fitico sono spesso denominati “antinutrienti” perché, a concentrazioni rilevanti, si legano e compromettono la digestione di minerali come magnesio, ferro e zinco. (9)(10)
Questo effetto viene quasi completamente eliminato se si consuma della Vitamina C nello stesso pasto, inoltre la lectina ha alcuni collegamenti per ridurre i rischi di cancro e l’acido fitico è un antiossidante che è stato associato a minori rischi di malattie cardiovascolari e calcoli renali. (11) (12) (13) (14) (15) (16) Ecco il motivo per il quale alcune persone consumano integratori di acido fitico.
E’ di facile conclusione il fatto che lectine e acido fitico non sono il “male assoluto” come propagandato, per esempio, dai fanatici della “Paleo Diet”. L’idea che questi composti siano assolutamente dannosi per tutti non è basata sui dati reali. Comunque sia, il consumo di legumi e grani nella “Vertical Diet” è consentita solo attraverso precise modalità di preparazione, come la messa in ammollo o la fermentazione, pratiche che aiutano a ridurre la presenza di “antinutrienti”. (17) (18)
Settaggio calorico nella “Vertical Diet”
Fortunatamente, questa volta non ci troviamo di fronte ad una dieta di classica foggia commerciale nella quale troneggia lo slogan “le calorie non contano”. In questo l’autore mantiene un filo logico.
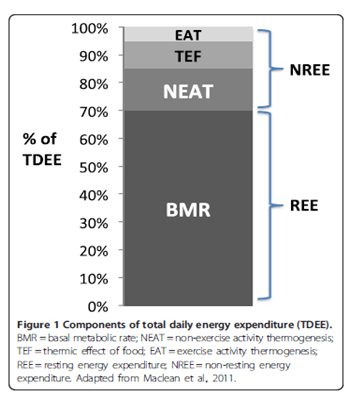
Infatti, inizialmente è consigliato il calcolo della spesa calorica basale (BMR). Successivamente si addiziona al risultato del calcolo del BMR il dispendio calorico derivante dall’attività sportiva e extra-sportiva (NEAT). Quindi, una volta ottenuto il proprio TDEE, a seconda dell’obbiettivo prefissato, si aggiungono o tolgono calorie.
Viene consigliato anche di tarare la dieta in un periodo di prova nel quale il corpo si adatterà alle nuove variabili alimentari. Se in questo periodo si inizia a sentire fame tra i pasti, il soggetto dovrebbe “andare in verticale” aggiungendo più calorie. Questo processo ha lo scopo di supportare maggiori guadagni muscolari, un recupero più rapido e sessioni di allenamento più intense e/o frequenti.
Il numero esatto di calorie aggiuntive si basa sulle esigenze di allenamento e comporta l’aumento delle porzioni di riso e carne o un pasto aggiuntivo durante il giorno.
Una volta che si inizia di nuovo ad avere fame tra i pasti, va ripetuto questo processo fino a raggiungere il peso o la massa muscolare prefissata come obiettivo.
Macronutrienti nella “Vertical Diet”
di base, la “Vertical Diet” è relativamente povera di grassi e ricca di carboidrati
Le calorie totali dipendono dal metabolismo basale e dal carico di lavoro soggettivo, ma i macronutrienti di base in questa strategia alimentare sono così gestiti:
Proteine: 2g per chilo di peso corporeo
Grassi: 0,6g per chilo di peso corporeo
Carboidrati: il resto delle calorie.
Supponiamo che un soggetto pesi 80Kg e abbia una spesa calorica giornaliera di 3000Kcal. La quantità dei macronutrienti sarebbe la seguente:
E’ ovvio che il calcolo del proprio monte calorico necessario ai fini dell’obbiettivo che ci si è prefissati è un componente importante di ogni piano nutrizionale, e questa dieta non fa eccezione.
Si noti che questa è di base una dieta relativamente povera di grassi, così progettata per gli atleti, che in genere hanno bisogno di fare più spazio nei loro macronutrienti per l’inserimento di una buona quota di carboidrati rispetto alle persone sedentarie.
La “Vertical Diet” non è statica nella ripartizione calorica, tanto che risulta versatile non solo per la sua applicabilità sia nei periodi “Bulk” che in quelli +”Cut”, ma anche per quanto riguarda le percentuali di macronutrienti e la loro gestione. Infatti può essere adattata a regimi come le Low Carb, IF o Paleo.
Benefici potenziali della “Vertical Diet”
Ovviamente un surplus calorico è essenziale per aumentare la massa muscolare (5).
Concentrandosi su alimenti facilmente digeribili, la “Vertical Diet” rende più facile mangiare pasti frequenti e ipercalorici senza sperimentare effetti collaterali digestivi.
Inoltre, la dieta enfatizza l’aumento dell’assunzione di carboidrati, fattore che contribuisce ad aumentare la massa muscolare (19, 20, 21).
Gli studi dimostrano che un’adeguata assunzione di carboidrati prima dell’allenamento può migliorare le prestazioni atletiche. I carboidrati possono anche aumentare la sintesi proteica e ridurre il catabolismo muscolare (20, 21).
Le diete povere di FODMAP – alimenti limitati nella “Vertical Diet” – hanno dimostrato di ridurre significativamente i sintomi legati a problemi digestivi, come gonfiore, crampi allo stomaco, costipazione e diarrea, specie nelle persone con sindrome dell’intestino irritabile (IBS). (22, 23)
I Bodybuilder e altri atleti che hanno bisogno di consumare pasti frequenti e ipercalorici possono anche trarne beneficio, in quanto i cibi a basso contenuto di FODMAP riducono il rischio di gonfiore addominale. In caso contrario, il gonfiore può limitare il consumo di cibo compromettendo il raggiungimento della quota calorica prefissata per l’aumento della massa muscolare.
Tuttavia, alcuni alimenti ricchi di FODMAP sono ammessi nella “Vertical Diet”, tra cui latte, yogurt, mele, ciliegie, fichi e altri frutti.
Pertanto, i soggetti con IBS dovrebbero evitare anche questi alimenti.
Potenziali aspetti negativi della “Vertical Diet”
È importante notare che la “Vertical Diet” ha numerosi aspetti negativi, tra cui:
Basso contenuto di fibre. Un’adeguata assunzione di fibre aiuta a raggiungere il senso di sazietà (quando ricercato), indirettamente la salute cardiovascolare e direttamente la salute dell’apparato digerente. Può anche ridurre il rischio di comparsa di malattie croniche, come il diabete di tipo 2 e alcune forme di cancro (24, 25, 26).
Basso contenuto di prebiotici. Nonostante si affermi che questa dieta migliori la salute dell’intestino, essa esclude molte importanti fonti di prebiotici – fibra alimentare che alimenta i batteri benefici nell’intestino – tra cui, e non solo, aglio, cipolle e orzo.(24, 27)
Limitata nella varietà degli alimenti. La “Vertical Diet” è restrittiva e ripetitiva, rendendo difficile l’aderenza sul lungo termine. Può anche portare a carenze nutrizionali se non pianificata correttamente.(28, 29)
Inappropriato per vegetariani o vegani. Poiché la “Vertical Diet” enfatizza l’assunzione di carne rossa limitando l’assunzione di verdure, cereali e legumi, non è adatta per persone vegetariane o vegane.
Costosa da seguire. Mentre il riso bianco è generalmente economico, gli altri componenti della “Vertical Diet” possono essere costosi, soprattutto considerando la raccomandazione di acquistare solo alimenti di alta qualità, come carne di manzo “grass fed” e prodotti biologici.
Esempio di un piano alimentare “Vertical Diet”
Ecco un menu “Vertical Diet” di esempio di 3 giorni . Non sono specificate le quantità per alimento tranne che gli ml di succo o brodo, e il numero di pasti può variare in base al regime di allenamento e al fabbisogno calorico.
Giorno 1
Pasto 1: uova intere strapazzate con formaggio, peperoni rossi, spinaci e sale, servite con carotine crude, mandorle crude e 120ml di succo di mirtillo
Pasto 2: filetto di manzo macinato e riso bianco cotto nel brodo di pollo, più 120ml di succo d’arancia
Pasto 3: petto di pollo e patata dolce servito con 120 ml di succo d’arancia
Pasto 4: bistecca grass-fed con riso bianco cotto nel brodo di pollo e 120ml di succo di mirtillo
Spuntino: yogurt greco e carotine.
Giorno 2
Pasto 1: uova intere strapazzate con formaggio, spinaci, peperoni rossi e brodo di ossa, servite con patate lesse e 120ml di succo di mirtillo
Pasto 2: bisonte macinato con riso bianco, patata dolce e brodo di ossa, insieme a 120ml di succo d’arancia
Pasto 3: petto di pollo con riso bianco, patata dolce, brodo di ossa e un’arancia
Pasto 4: bistecca grass-fed con riso bianco, patate, zucchine e brodo di ossa, servito con 120ml di succo di mirtillo
Spuntino: latte intero e carotine.
Giorno 3
Pasto 1: uova intere strapazzate con formaggio, spinaci, peperoni rossi e sale, insieme all’avena messa in ammollo durante la notte con base di yogurt, latte e miele e noci opzionali
Pasto 2: bistecca di controfiletto macinata con riso bianco, peperoni e brodo di pollo, servito con 120ml di succo di mirtillo
Pasto 3: salmone dell’Atlantico selvaggio con riso bianco, spinaci, peperoni e brodo di pollo, oltre a carotine e 120ml di succo d’arancia
Pasto 4: bistecca grass-fed con riso bianco, patate dolci e brodo di pollo, oltre a 120ml di succo di mirtillo
Spuntino: yogurt greco e frutti di bosco.
Tiriamo le somme sulla “Vertical Diet”
Come abbiamo visto, la “Vertical Diet” ha lo scopo di aiutare i Bodybuilder ed altri atleti ad aumentare la massa muscolare e migliorare le prestazioni o a perdere peso.
Include alimenti facilmente digeribili per aiutare il corpo ad assorbire i nutrienti in modo più efficiente e prevenire effetti collaterali digestivi, come il gonfiore. Per aumentare l’assunzione di proteine e carboidrati, l’autore sottolinea di consumare porzioni sempre più grandi di carne rossa e riso bianco.
La “Vertical Diet” è severamente restrittiva, costosa da seguire e povera di fibre complessive e prebiotiche, oltre che a presentarsi come un generico piano alimentare basato su alcuni punti non adeguatamente contestualizzati. La compliance non è facilitata ed è una dieta estremamente difficili da mantenere sul lungo termine.
Ma quando si guarda a una dieta così focalizzata sull’aiutare a digerire il cibo nel modo più efficiente possibile, non è difficile capire perché possa avere delle grandi potenzialità applicative in regimi alimentari ipercalorici. Con una dieta ad elevato contenuto calorico, i cibi ricchi di FODMAP, i cibi ricchi di fibre e cibi estremamente grassi – tutti esclusi o severamente limitati nella “Vertical Diet” – potrebbero potenzialmente rendere più difficile consumare e assimilare la quantità di cibo necessaria per ottenere i risultati agognati. La maggior parte delle persone che hanno provato ad aumentare di peso con piani alimentari contenenti legumi affermeranno che ad un certo punto, la fibra smette di essere un fattore benefico a livello digestivo.
Non si può comunque negare che la maggior parte degli alimenti limitati in questa dieta hanno più benefici che svantaggi. Detto questo, la “Vertical Diet” consente una varietà abbastanza ampia di frutta, verdura, verdure a foglia verde, carne e fonti di acidi grassi Omega-3, sufficienti per rendere improbabili le carenze nutrizionali con un regime ben pianificato. E non esiste alcuna regola nel piano dietetico contro gli integratori per arginare eventuali carenze.
Personalmente considero la “Vertical Diet” un ottima opzione nutrizionale per tutti coloro i quali presentano facilmente difficoltà digestive. La sua applicazione migliore risulta essere in “Bulk” per via della considerevole quantità di cibo richiesta. In fasi “Cut” non la ritengo una dieta particolarmente vantaggiosa a meno che non si parli di soggetti con IBS o con problemi digestivi.
In conclusione, Stan Efferding ha messo insieme nozioni logicamente intuitive (vedi garantire una migliore digestione e assorbimento dei nutrienti) con una serie di limitazioni non sempre veritiere e/o applicabili vantaggiosamente (vedi, per esempio, eliminazione dei cibi contenenti glutine a prescindere dalla presenza di celiachia). Nulla di nuovo sotto il sole… nonostante una piccola ma meritata nota positiva sull’importanza della corretta assimilazione di ciò di cui ci si nutre…
Se volete acquistare l’Ebook della “Vertica Diet” cliccate qui.
Gabriel Bellizzi
Riferimenti:
Simopoulos AP, et al. An Increase in the Omega-6/Omega-3 Fatty Acid Ratio Increases the Risk for Obesity. Nutrients. 2016 Mar 2;8(3):128.
Patterson E, et al. Health implications of high dietary omega-6 polyunsaturated Fatty acids. J Nutr Metab. 2012;2012:539426.
Okuyama H, et al. Omega3 fatty acids effectively prevent coronary heart disease and other late-onset diseases–the excessive linoleic acid syndrome. World Rev Nutr Diet. 2007;96:83-103.
Barrett JS, et al. Fermentable oligosaccharides, disaccharides, monosaccharides and polyols (FODMAPs) and nonallergic food intolerance: FODMAPs or food chemicals? Therap Adv Gastroenterol. 2012 Jul;5(4):261-8.
Turco R, et al. Does a low FODMAPs diet reduce symptoms of functional abdominal pain disorders? A systematic review in adult and paediatric population, on behalf of Italian Society of Pediatrics. Ital J Pediatr. 2018 May 15;44(1):53.
Ong DK, et al. Manipulation of dietary short chain carbohydrates alters the pattern of gas production and genesis of symptoms in irritable bowel syndrome. J Gastroenterol Hepatol. 2010 Aug;25(8):1366-73.
Sloan TJ, et al. A low FODMAP diet is associated with changes in the microbiota and reduction in breath hydrogen but not colonic volume in healthy subjects. PLoS One. 2018 Jul 26;13(7):e0201410.
Wiffin M, et al. Effect of a short-term low fermentable oligiosaccharide, disaccharide, monosaccharide and polyol (FODMAP) diet on exercise-related gastrointestinal symptoms. J Int Soc Sports Nutr. 2019 Jan 15;16(1):1.
Schuchardt JP, et al. Intestinal Absorption and Factors Influencing Bioavailability of Magnesium-An Update. Curr Nutr Food Sci. 2017 Nov;13(4):260-278.
Jiang QL, et al. Plant lectins, from ancient sugar-binding proteins to emerging anti-cancer drugs in apoptosis and autophagy. Cell Prolif. 2015 Feb;48(1):17-28.
Davidsson L. Approaches to improve iron bioavailability from complementary foods. J Nutr. 2003 May;133(5 Suppl 1):1560S-2S.
Siegenberg D, et al. Ascorbic acid prevents the dose-dependent inhibitory effects of polyphenols and phytates on nonheme-iron absorption. Am J Clin Nutr. 1991 Feb;53(2):537-41.
Schlemmer U, et al. Phytate in foods and significance for humans: food sources, intake, processing, bioavailability, protective role and analysis. Mol Nutr Food Res. 2009 Sep;53 Suppl 2:S330-75.
Omoruyi FO, et al. The potential benefits and adverse effects of phytic Acid supplement in streptozotocin-induced diabetic rats. Adv Pharmacol Sci. 2013;2013:172494.
Grases F, et al. Phytate (IP6) is a powerful agent for preventing calcifications in biological fluids: usefulness in renal lithiasis treatment. Anticancer Res. 1999 Sep-Oct;19(5A):3717-22.
Gupta RK, et al. Reduction of phytic acid and enhancement of bioavailable micronutrients in food grains. J Food Sci Technol. 2015 Feb;52(2):676-84.
Liang J, et al. Effects of soaking, germination and fermentation on phytic acid, total and in vitro soluble zinc in brown rice. Food Chem. 2008 Oct 15;110(4):821-8.
Chi si interessa in modo approfondito di supplementazione farmacologica nello sport, penserà di avere una conoscenza discretamente completa su una delle pratiche più conosciute nell’ambiente, vale a dire la PCT (Post Cycle Therapy). Questo tentativo di recupero della propria funzionalità dell’Asse HPT ha subito perfezionamenti nel corso degli ultimi decenni. Si è passati da una illogica accozzaglia di farmaci tra i quali spiccavano il Mesterolone e l’Oxandrolone insieme ai classici SERM e hCG, ad una logica sequenza di composti strutturata sugli andamenti della curva ematica delle molecole utilizzate durante il ciclo e all’azione sinergica e ordinata di hCG seguito da Tamoxifene Citrato e Clomifene Citrato, con la recente aggiunta di Inibitori dell’Aromatasi (AI). Vedi PCT Scally.
Da qualche tempo, però, circola la voce secondo la quale il piano di recupero ormonale dell’HPTA può essere migliorato con l’inserimento di un altra classe di farmaci. Questa classe di farmaci è quella degli Antiandrogeni.
Prima di svelarvi il nesso che ha spinto qualche mente speculatrice a partorire tale idea, è corretto darvi una base di cultura generale sugli Antiandrogeni per concludere con la spiegazione del perchè un loro possibile inserimento in una PCT possa essere favorevole…forse…
Una panoramica sugli Antiandrogeni
Gli Antiandrogeni, noti anche come antagonisti degli androgeni o bloccanti del Testosterone, sono una classe di farmaci che impediscono agli androgeni come il Testosterone e il Dihydrotestosterone (DHT) di mediare i loro effetti biologici nel corpo. Agiscono bloccando il Recettore degli Androgeni (AR) e/o inibendo o sopprimendo la produzione di androgeni.[1][2] Possono essere pensati come gli opposti funzionali degli agonisti AR, come ad esempio gli Steroidi Anabolizzanti Androgeni (AAS) e i Modulatori Selettivi del Recettore degli Androgeni (SARM). Gli antiandrogeni sono uno dei tre tipi di antagonisti degli ormoni sessuali, gli altri sono antiestrogeni e antiprogestinici.[3]
Gli Antiandrogeni sono usati per trattare una serie di condizioni androgeno-dipendenti. [4] Nei maschi, gli Antiandrogeni sono usati nel trattamento del cancro alla prostata, ipertrofia prostatica, perdita di capelli, desiderio sessuale eccessivamente elevato, impulsi sessuali insoliti e problematici e pubertà precoce.[4][5] Nelle donne, gli antiandrogeni sono usati per trattare l’acne, la seborrea, l’eccessiva crescita dei peli, la perdita dei capelli e gli alti livelli di androgeni, come quelli che si verificano nella sindrome dell’ovaio policistico (PCOS).[4] Gli antiandrogeni sono anche usati come componente della terapia ormonale femminizzante per i transgender e come bloccanti della pubertà nelle ragazze transgender.[4]
Ciproterone Acetato
Le Antigonadotropine come gli Estrogeni e i progestinici furono entrambe introdotte per la prima volta negli anni ’30. [6] Gli effetti benefici della deprivazione di androgeni attraverso la castrazione chirurgica o la terapia con estrogeni ad alte dosi sul cancro alla prostata furono scoperti nel 1941. [7][8] antagonisti del AR furono scoperti per la prima volta nei primi anni ’60.[9] Il Ciproterone Acetato è un antiandrogeno steroideo scoperto nel 1961 e introdotto nel 1973 ed è spesso descritto come il primo antiandrogeno commercializzato. [10] [11] Tuttavia, lo Spironolattone fu introdotto nel 1959, [12] [13], sebbene i suoi effetti antiandrogeni non fossero stati riconosciuti o sfruttati fin da subito e fossero originariamente considerati un’azione indesiderata fuori bersaglio del farmaco.[14] Oltre allo Spironolattone, il Clormadinone Acetato e il Megestrolo Acetato sono antiandrogeni steroidei che sono più deboli del Ciproterone Acetato ma sono stati introdotti precedentemente, negli anni ’60. [15] [16] [17] Altri primi antiandrogeni steroidei che sono stati sviluppati in questo periodo ma che non sono mai stati commercializzati includono il Benorterone (SKF-7690; 17α-metil-B-Nortestosterone), BOMT (Ro 7-2340), il Ciproterone (SH-80881) e il Trimetiltrienolone (R- 2956).[18][19]
Flutamide
La Flutamide è un antiandrogena non steroideo descritto per la prima volta nel 1967. [20] Fu introdotto sul mercato nel 1983 ed è stato il primo antiandrogeno non steroideo commercializzato. [21] [22] Un altro antiandrogeno precoce non steroideo, [23] DIMP (Ro 7-8117), che è strutturalmente correlato alla Talidomide [24] ed è un antiandrogeno relativamente debole, [25] [26] fu descritto per la prima volta nel 1973 e non fu mai commercializzato. [27] La Flutamide è stata seguita dalla Nilutamide nel 1989 e dalla Bicalutamide nel 1995. [28] Oltre a questi tre farmaci, che sono stati considerati antiandrogeni non steroidei di prima generazione, gli antiandrogeni non steroidei di seconda generazione Enzalutamide e Apalutamide sono stati introdotti rispettivamente nel 2012 e nel 2018. [29] [30] [31] Differiscono dai precedenti antiandrogeni non steroidei, in particolar modo per il fatto che sono molto più efficaci.[30]
Aminoglutetimide
Gli inibitori della sintesi androgena Aminoglutetimide e Ketoconazolo furono commercializzati per la prima volta rispettivamente nel 1960 e nel 1977 [32] [33] e il più recente farmaco Abiraterone Acetato è stato introdotto nel sul mercato nel 2011. [34] I modulatori del GnRH furono introdotti per la prima volta negli anni ’80. [35] Gli inibitori della 5α-reduttasi Finasteride e Dutasteride sono stati introdotti sul mercato rispettivamente nel 1992 e nel 2002.[36] [37] L’Elagolix, il primo modulatore GnRH attivo per via orale ad essere commercializzato, è stato introdotto sul mercato nel 2018. [38]
Abiraterone Acetato
Quindi, gli antiandrogeni possono essere suddivisi in diversi tipi in base alla struttura chimica, inclusi antiandrogeni steroidei, antiandrogeni non steroidei e peptidi. Gli antiandrogeni steroidei comprendono composti come il Ciproterone Acetato, lo Spironolattone, l’Estradiolo, l’Abiraterone Acetato e la Finasteride; antiandrogeni non steroidei includono composti come il Bicalutamide, l’Elagolix, il Dietilstilbestrolo, l’Aminoglutetimide e Ketoconazolo; e i peptidi includono analoghi del GnRH come Leuprorelina e il Cetrorelix.
Gli Antiandrogeni si dividono in cinque gruppi principali: [39]
Antagonisti del recettore degli androgeni: farmaci che si legano direttamente al AR bloccando il legame con l’ormone bersaglio.[40][41] Questi farmaci comprendono gli antiandrogeni steroidei Ciproterone Acetato, Megestrolo Acetato, Clormadinone Acetato, Spironolattone, Oxendolone e Osaterone Acetato (veterinario) e gli antiandrogeni non steroidei Flutamide, Bicalutamide, Nilutamide, Topilutamide, Enzalutamide e Apalutamide. [41][40] ] [42] A parte il Ciproterone Acetato e il Clormadinone Acetato, alcuni altri progestinici usati nei contraccettivi orali e / o nella TOS in menopausa tra cui Dienogest, Drospirenone, Medrogestone, Nomegestrolo Acetato, Promegestone e Trimegestone hanno anche vari gradi di attività AR-antagonista. [43] [44] [45]
Inibitori della sintesi degli Androgeni: farmaci che inibiscono direttamente la biosintesi enzimatica di androgeni come Testosterone e/o DHT. [46] [47] Gli esempi includono gli inibitori del CYP17A1 Ketoconazolo, Abiraterone Acetato e Seviteronel, [46] l’inibitore del CYP11A1 (P450scc) Aminoglutetimidico , [46] e gli inibitori della 5α-reduttasi Finasteride, Dutasteride, Epristeride, Alfatradiolo e il blando Saw Palmetto (Palmetto Seghettato).[88] Numerosi altri antiandrogeni, tra cui Ciproterone Acetato, Spironolattone, Medrogestone, Flutamide, Nilutamide e Bifluranolo, sono anche noti per inibire debolmente la sintesi degli Androgeni.
Antigonadotropici: farmaci che sopprimono il rilascio di gonadotropine indotto dall’ormone di rilascio delle gonadotropine (GnRH) e conseguente attivazione della produzione di androgeni gonadici. [2] [48] Gli esempi includono modulatori del GnRH come Leuprorelina (un agonista del GnRH) e Cetrorelix (un antagonista del GnRH), [90] progestinici come Allilestrenolo, Clormadinone Acetato, Ciproterone Acetato, Gestonorone Caproato, Idrossiprogesterone Caproato, Medroxyprogesterone Acetato, Megestrol Acetato, Osaterone Acetato (veterinario), e Oxendolone, [49] [50] ed estrogeni come Estradiolo, esteri dell’Estradiolo, Etinilestradiolo, Estrogeni coniugati e Dietilstilbestrolo. [2] [49]
Miscellanei: farmaci che si oppongono agli effetti degli androgeni con mezzi diversi da quelli sopra indicati. Esempi includono Estrogeni, in particolare sintetici orali (ad esempio Etinilestradiolo, Dietilstilbestrolo), che stimolano la produzione di globulina legante gli ormoni sessuali (SHBG) nel fegato e quindi diminuiscono i livelli liberi e quindi bioattivi di Testosterone e DHT; anticorticotropine come i glucocorticoidi, che sopprimono la produzione indotta dall’ormone adrenocorticotropo (ACTH) di androgeni surrenali; e immunogeni e vaccini contro l’Androstenedione come l’albumina Ovandrotone e l’albumina Androstenedione, che riducono i livelli di androgeni attraverso la generazione di anticorpi contro il precursore androgeno androstenedione (usato solo in medicina veterinaria).
Come si è potuto vedere, alcuni antiandrogeni combinano molti dei meccanismi di cui sopra. [39] [51] Un esempio è l’antiandrogeno steroideo Ciproterone Acetato, che è un potente antagonista AR, un potente progestinico e quindi antigonadotropico, un glucocorticoide debole e quindi anticorticotropo e un inibitore debole della sintesi degli androgeni. [39] [51] [52] [53]
Per ovvie ragioni di sintesi, la lista sopra include antagonisti AR, inibitori della sintesi degli androgeni e progestinici commercializzati per l’uso o ampiamente usati come antiandrogeni, ma non include specificatamente agonisti del GnRH, antagonisti del GnRH, inibitori della 5α-reduttasi o Estrogeni.
La classe degli Antagonisti del Recettore degli Androgeni è di nostro particolare interesse…
Gli antagonisti del AR agiscono legandosi direttamente e sostituendo in modo competitivo gli androgeni come il Testosterone e il DHT dal AR, impedendo così loro di attivare il recettore e mediare i loro effetti biologici. [40] [41] Gli antagonisti del AR, come abbiamo già visto, sono classificati in due tipi, in base alla struttura chimica: steroidei e non steroidei. [54] [42] [40] [41] [55] Gli antagonisti di AR steroide sono strutturalmente correlati agli ormoni steroidei come Testosterone e Progesterone, mentre gli antagonisti del AR non steroidei non sono steroidi e sono strutturalmente distinti. Gli antagonisti del AR steroidei tendono ad avere azioni ormonali fuori bersaglio a causa della loro somiglianza strutturale con altri ormoni steroidei. [55] Al contrario, gli antagonisti del AR non steroidei sono selettivi per l’AR e non hanno attività ormonale fuori bersaglio. [55] Per questo motivo, a volte sono descritti come antiandrogeni “puri”. [55]
Spironolattone
Sebbene siano descritti come antiandrogeni e in effetti mostrano solo tali effetti in generale, la maggior parte o tutti gli antagonisti AR steroidei non sono in realtà antagonisti inattivi del AR ma piuttosto sono agonisti parziali deboli e sono in grado di attivare il recettore in assenza di agonisti AR più potenti come Testosterone e DHT. [40] [47] [55] [56] Ciò può avere implicazioni cliniche nel contesto specifico del trattamento del cancro alla prostata. [40] [55] Ad esempio, gli antagonisti del AR steroidei sono in grado di aumentare il peso della prostata e accelerare la crescita delle cellule tumorali della prostata in assenza di più potenti agonisti dell’AR, [40] [55] e lo Spironolattone ha dimostrato di accelerare la progressione del cancro alla prostata nei casi clinici [57] [58] Inoltre, mentre il Ciproterone Acetato produce genitali ambigui attraverso la femminilizzazione nei feti maschi quando somministrato ad animali in gravidanza, [59] è stato osservato che causa la mascolinizzazione dei genitali dei feti femminili di animali in gravidanza. [40] A differenza degli antagonisti AR steroidei, gli antagonisti AR non steroidei sono antagonisti inattivi del AR e, quindi, non attivano il recettore. [60] [47] [61] [55] Questo potrebbe essere il motivo per cui hanno una maggiore efficacia rispetto agli antagonisti del AR steroidei nel trattamento del cancro alla prostata ed è un motivo importante per cui li hanno ampiamente sostituiti per questa indicazione in medicina. [60] [47] [61] [55]
Bicalutamide
Gli antiandrogeni non steroidei hanno un’affinità relativamente bassa per il AR rispetto ai ligandi AR steroidei. [47] [61] [62] Ad esempio, la Bicalutamide ha circa il 2% dell’affinità di DHT per il AR e circa il 20% dell’affinità del CPA per il AR. [62] Nonostante la loro bassa affinità con il AR, tuttavia, la mancanza di un’attività agonista parziale debole degli NSAA sembra migliorare la loro potenza rispetto agli antiandrogeni steroidei. [62] [63] Ad esempio, sebbene la Flutamide abbia un’affinità circa 10 volte inferiore per il AR rispetto al CPA, mostra una potenza pari o leggermente maggiore al CPA come antiandrogeno nei biotest. [62] [63] Inoltre, le concentrazioni terapeutiche circolanti di antiandrogeni non steroidei sono molto elevate, nell’ordine di migliaia di volte superiori a quelle di Testosterone e DHT, e ciò consente loro di competere efficacemente e bloccare la segnalazione del AR. [64]
Gli antagonisti del AR non possono legarsi o bloccare i recettori degli androgeni di membrana (mARs), che sono distinti dal AR nucleare classico. [65] [66] [67] Tuttavia, le mARs non sembrano essere coinvolte nella mascolinizzazione. Ciò è evidenziato dal fenotipo perfettamente femminile di donne con sindrome da insensibilità agli androgeni completa. [68] [69] Queste donne hanno un cariotipo 46, XY (cioè geneticamente “maschio”) e alti livelli di androgeni ma possiedono un AR difettoso e per questo motivo non mascolinizzano mai. [68] [69] Sono descritti come altamente femminili, sia fisicamente che mentalmente e comportamentalmente. [70] [71] [72]
Perchè questo interesse per gli Antagonisti del Recettore degli Androgeni?
Asse Ipotalamo-Ipofisi-Testicoli
Piccolo ripasso sul controllo omeostatico ormonale riferito all’Asse HPT.
Con Asse Ipotalamo-Ipofisi-Testicoli (HPTA) ci si riferisce alla connessione tra ipotalamo, ghiandola pituitaria e testicoli come se queste singole ghiandole endocrine fossero una singola entità. Poiché queste ghiandole spesso agiscono in concerto, i fisiologi e gli endocrinologi ritengono conveniente e descrittivo parlare di esse come di un unico sistema.
L’asse HPTA svolge una parte critica nello sviluppo e nella regolazione di un certo numero di sistemi del corpo, come i sistemi riproduttivi e immunitari. Le fluttuazioni di questo asse causano variazioni negli ormoni prodotti da ciascuna ghiandola e hanno diversi effetti locali e sistemici nel corpo.
In breve, l’asse HPTA rappresenta un sistema di stimolazione/inibizione degli ormoni prodotti dalle rispettiva strutture:
Ipotalamo: GnRH (ormone di rilascio delle gonadotropine; in inglese Gonadotropin-releasing hormone).
Ipofisi (o ghiandola Pituitaria): dalle cellule beta e gamma rispettivamente l’ormone follicolo-stimolante (FSH) e l’ormone luteinizzante (LH).
Come ben sappiamo, diversi AAS sono derivati sintetici del Testosterone, il principale androgeno nei maschi. Il Testosterone sopprime marcatamente l’HPTA, mentre altri derivati lo fanno in misura maggiore o minore.
I fattori che contribuiscono alla soppressione dell’HPTA sono:
L’origine del AAS
Il tasso di conversione del AAS ad estrogeno, attraverso l’Enzima Aromatasi in alcuni tessuti (adiposo, mammario)
Dose e tempo d’uso/abuso del AAS
Attività androgena del AAS
Bingo! Ci siete arrivati adesso? In ogni caso andiamo avanti…
Conosciamo tutti il feedback negativo indotto dagli estrogeni a livello ipotalamico.
Estradiolo
Gli estrogeni (principalmente E2-beta Estradiolo) causano un feedback negativo sull’ipotalamo per la produzione di GnRH, che a sua volta stimola LH che stimola la sintesi di Testosterone nelle cellule Leydig nei testicoli. Pertanto, gli AAS fortemente soggetti all’aromatizzazione o che posseggono una attività estrogenica intrinseca (Oxymetholone, Methyltestosterone, Testosterone, Methandienone ecc…) influenzano marcatamente la funzione dell’HPTA.
Ed ecco perchè l’uso di SERM causa un incremento del GnRH, e consequenzialmente del LH e FSH, bloccando il legame recettoriale estrogenico ipotalamico inducendo un feedback positivo.
Gli AAS con alta affinità con il AR si legano fortemente ad esso. Gli AAS attraversano la barriera ematoencefalica e si legano ai recettori sull’ipotalamo. Ciò comporterà una marcata soppressione dell’HPTA. L’attività androgena si traduce nelle caratteristiche sessuali secondarie (crescita dei peli e della barba, allargamento delle spalle e il rafforzarsi dei muscoli, l’ingrandimento del pene, dei testicoli e della prostata.)
E qui entrano in gioco gli Antagonisti del Recettore Androgeno che, agendo similmente ai SERM, causano un incremento della secrezione di LH. Tale incremento è stato osservato in diversi studi tra i quali uno svolto su animali nel 1989, nel quale si era utilizzata la Flutamide.[73] L’effetto indotto è quindi progonadotropico.[74]
Ed è da ciò che è nata l’idea di inserire piccole quantità per un breve lasso di tempo di Antiandrogeni (nello specifico Antagonisti del Recettore degli Androgeni non steroidei) nel protocollo PCT al fine di potenziarne gli affetti.
Nota: Gli effetti collaterali degli antiandrogeni variano a seconda del tipo di antiandrogeno – ovvero se si tratta di un antagonista AR selettivo o un inibitore della biosintesi androgena – nonché dalla presenza di attività fuori bersaglio terapico dell’antiandrogeno in questione. [74][75] Ad esempio, mentre gli antiandrogeni antigonadotropici come i modulatori del GnRH e il Ciproterone Acetato sono associati a disfunzione sessuale pronunciata e osteoporosi negli uomini, gli antagonisti selettivi del AR come la Bicalutamide non sono associati all’osteoporosi e sono stati correlati solo a una disfunzione sessuale minima. [74] [76] [77] Queste differenze sono ritenute una conseguenza del fatto che le antigonadotropine sopprimono i livelli di androgeni e, per estensione, dei livelli dei metaboliti bioattivi degli androgeni come estrogeni e neurosteroidi, mentre gli antagonisti selettivi del AR neutralizzano gli effetti degli androgeni ma lasciano intatti i livelli degli stessi (e di fatto i loro metaboliti) potendo persino aumentarli a causa dei loro effetti progonadotropici.[74] Come altro esempio, gli antiandrogeni steroidei Ciproterone Acetato e Spironolattone possiedono azioni off-target tra cui attività progestinica, antimineralocorticoide e / o glucocorticoide in aggiunta alla loro attività antiandrogena, e queste attività off-target possono provocare ulteriori effetti collaterali.[75]
Nei maschi, i principali effetti collaterali degli antiandrogeni sono la demasculinizzazione e la femminilizzazione.[78] Questi effetti collaterali includono dolore al seno / lipomastia e ginecomastia (sviluppo del seno / ingrossamento), riduzione della crescita / densità dei peli corporei, riduzione della massa e della forza muscolare, cambiamenti femminili nella massa e nella distribuzione del grasso e riduzione della lunghezza del pene e delle dimensioni dei testicoli. [78] I tassi di ginecomastia negli uomini con monoterapia antagonista selettiva del AR sono stati stimati tra il 30 e l’85%. [79] Inoltre, gli antiandrogeni possono causare infertilità, osteoporosi, vampate di calore, disfunzione sessuale (inclusa perdita di libido e disfunzione erettile), depressione, affaticamento, anemia e riduzione del volume spermatico / eiaculato nei maschi.[78] Al contrario, gli effetti collaterali degli antagonisti selettivi del AR nelle donne sono minimi. [80] [81] Tuttavia, gli antiandrogeni antigonadotropici come il Ciproterone Acetato possono produrre ipoestrogenismo, amenorrea e osteoporosi nelle donne in premenopausa, tra gli altri effetti collaterali. [82] [83] [84]
Numerosi antiandrogeni sono stati associati a epatotossicità. [85] Questi includono, in varia misura, Ciproterone Acetato, Flutamide, Nilutamide, Bicalutamide, Aminoglutetimide e Ketoconazolo. [85] Al contrario, Spironolattone, Enzalutamide, [86] e altri antiandrogeni non sono associati a epatotossicità. Tuttavia, sebbene non presentino un rischio di epatotossicità, lo Spironolattone ha un rischio di causare iperkaliemia e l’Enzalutamide ha un rischio di causare convulsioni.
Conclusioni
E’ ovvio che si sta parlando di pura teoria, lungi dall’essere dimostrata come terapeuticamente valida. Ma, per amor di conoscenza, ho ritenuto utile trattare l’argomento in modo tale che meno persone si facessero strane e confuse idee a riguardo, magari dopo essersi imbattuti nel “bongo” da spogliatoio che, con atteggiamento del primate dominante, dispensa consigli applicativi di un qualcosa per lui difficilmente comprensibile.
Gabriel Bellizzi
Riferimenti:
Mowszowicz I (1989). “Antiandrogens. Mechanisms and paradoxical effects”. Ann. Endocrinol. Paris. 50 (3): 50(3):189–99.
Brueggemeier, Robert W. (2006). “Sex Hormones (Male): Analogs and Antagonists”. Encyclopedia of Molecular Cell Biology and Molecular Medicine.
Student S, Hejmo T, Poterała-Hejmo A, Leśniak A, Bułdak R (January 2020). “Anti-androgen hormonal therapy for cancer and other diseases”. Eur. J. Pharmacol. 866: 172783.
Gillatt D (2006). “Antiandrogen treatments in locally advanced prostate cancer: are they all the same?”. J Cancer Res Clin Oncol. 1: S17-26.
William Figg; Cindy H. Chau; Eric J. Small (14 September 2010). Drug Management of Prostate Cancer. Springer Science & Business Media. pp. 71–72, 75, 91–96.
Benno Clemens Runnebaum; Thomas Rabe; Ludwig Kiesel (6 December 2012). Female Contraception: Update and Trends. Springer Science & Business Media. pp. 136–.
Liu, Bo; Su, Lei; Geng, Jingkun; Liu, Junjie; Zhao, Guisen (2010). “Developments in Nonsteroidal Antiandrogens Targeting the Androgen Receptor”. ChemMedChem. 5 (10): 1651–1661.
Heyns, W.; G., Verhoeven; De Moor, P. (1976). “Androgen binding in rat uterus cytosol. Study of the specificity”. Journal of Steroid Biochemistry. 7 (5): 335–343.
Boris, A.; Scott, J. W.; DeMartino, L.; Cox, D. C. (1973). “Endocrine Profile of a Nonsteroidal Antiandrogen N-(3,5-Dimethyl-4-Isoxazolylmethyl)Phthalimide (Dimp)”. European Journal of Endocrinology. 72 (3): 604–614.
Menon MP, Higano CS (2013). “Enzalutamide, a second generation androgen receptor antagonist: development and clinical applications in prostate cancer”. Curr Oncol Rep. 15 (2): 69–75.
Kolvenbag, Geert J. C. M.; Furr, Barrington J. A. (2009). “Nonsteroidal Antiandrogens”. In V. Craig Jordan; Barrington J. A. Furr (eds.). Hormone Therapy in Breast and Prostate Cancer. Humana Press. pp. 347–368.
Shen, Howard C.; Taplin, Mary-Ellen; Balk, Steven P. (2010). “Androgen Receptor Antagonists”. Drug Management of Prostate Cancer: 71–81.
Kolvenbag, Geert J. C. M.; Furr, Barrington J. A. (2009). “Nonsteroidal Antiandrogens”. In V. Craig Jordan; Barrington J. A. Furr (eds.). Hormone Therapy in Breast and Prostate Cancer. Humana Press. pp. 347–368.
William Figg; Cindy H. Chau; Eric J. Small (14 September 2010). Drug Management of Prostate Cancer. Springer Science & Business Media. pp. 71–72, 75, 91–96.
Peter B. Farmer; John M. Walker (6 December 2012). The Molecular Basis of Cancer. Springer Science & Business Media. pp. 232–.
de Lignières B, Silberstein S (April 2000). “Pharmacodynamics of oestrogens and progestogens”. Cephalalgia: An International Journal of Headache. 20 (3): 200–7.
^William Ledger; William D. Schlaff; Thierry G. Vancaillie (11 December 2014). Chronic Pelvic Pain. Cambridge University Press. pp. 55–.
Louise Hanna; Tom Crosby; Fergus Macbeth (19 November 2015). Practical Clinical Oncology. Cambridge University Press. pp. 37–.
Mahler C, Verhelst J, Denis L (May 1998). “Clinical pharmacokinetics of the antiandrogens and their efficacy in prostate cancer”. Clin Pharmacokinet. 34 (5): 405–17.
Schröder, Fritz H.; Radlmaier, Albert (2009). “Steroidal Antiandrogens”. In V. Craig Jordan; Barrington J. A. Furr (eds.). Hormone Therapy in Breast and Prostate Cancer. Humana Press. pp. 325–346.
Poyet P, Labrie F (October 1985). “Comparison of the antiandrogenic/androgenic activities of flutamide, cyproterone acetate and megestrol acetate”. Molecular and Cellular Endocrinology. 42 (3): 283–8.
Luthy IA, Begin DJ, Labrie F (1988). “Androgenic activity of synthetic progestins and spironolactone in androgen-sensitive mouse mammary carcinoma (Shionogi) cells in culture”. Journal of Steroid Biochemistry. 31 (5): 845–52.
Flynn T, Guancial EA, Kilari M, Kilari D (2016). “Case Report: Spironolactone Withdrawal Associated With a Dramatic Response in a Patient With Metastatic Castrate-Resistant Prostate Cancer”. Clin Genitourin Cancer. 15 (1): e95–e97.
Caubet JF, Tosteson TD, Dong EW, Naylon EM, Whiting GW, Ernstoff MS, Ross SD (1997). “Maximum androgen blockade in advanced prostate cancer: a meta-analysis of published randomized controlled trials using nonsteroidal antiandrogens”. Urology. 49 (1): 71–8. Because steroidal antiandrogens such as cyproterone acetate have intrinsic androgenic activity and lower antiandrogenic activity than the NSAAs such as flutamide and nilutamide,39–43 it is not surprising that the two classes of antiandrogens may have different efficacies.
Singh SM, Gauthier S, Labrie F (February 2000). “Androgen receptor antagonists (antiandrogens): structure-activity relationships”. Current Medicinal Chemistry. 7 (2): 211–47.
Ayub M, Levell MJ (August 1989). “The effect of ketoconazole related imidazole drugs and antiandrogens on [3H] R 1881 binding to the prostatic androgen receptor and [3H]5 alpha-dihydrotestosterone and [3H]cortisol binding to plasma proteins”. J. Steroid Biochem. 33 (2): 251–5.
Yamasaki K, Sawaki M, Noda S, Muroi T, Takakura S, Mitoma H, Sakamoto S, Nakai M, Yakabe Y (2004). “Comparison of the Hershberger assay and androgen receptor binding assay of twelve chemicals”. Toxicology. 195 (2–3): 177–86.
William B. Pratt (1994). The Anticancer Drugs. Oxford University Press. pp. 220–. In patients receiving flutamide at the usual dosage of 250 mg every 8 hours, the minimal plasma concentration of hydroxyflutamide is about 5 uM, which is 5,000 times the plasma concentration of testosterone (1 nM) in patients treated with an LHRH agonist.127 As hydroxyflutamide is only one percent as potent as testosterone in competing for binding to the androgen receptor,126 a plasma level of 5 uM hydroxyflutamide is required to ensure effective competition.127 […] Both cyproterone acetate and flutamide have been demonstrated to be effective therapy (roughly equivalent to an estrogen) when used alone in the treatment of carcinoma of the prostate.123
Bennett NC, Gardiner RA, Hooper JD, Johnson DW, Gobe GC (2010). “Molecular cell biology of androgen receptor signalling”. Int. J. Biochem. Cell Biol. 42 (6): 813–27.
Iversen P, Melezinek I, Schmidt A (2001). “Nonsteroidal antiandrogens: a therapeutic option for patients with advanced prostate cancer who wish to retain sexual interest and function”. BJU Int. 87 (1): 47–56.
Higano CS (2003). “Side effects of androgen deprivation therapy: monitoring and minimizing toxicity”. Urology. 61 (2 Suppl 1): 32–8.
Di Lorenzo G, Autorino R, Perdonà S, De Placido S (December 2005). “Management of gynaecomastia in patients with prostate cancer: a systematic review”. Lancet Oncol. 6 (12): 972–9.
Erem C (2013). “Update on idiopathic hirsutism: diagnosis and treatment”. Acta Clin Belg. 68 (4): 268–74.
W. Futterweit (6 December 2012). Polycystic Ovarian Disease. Springer Science & Business Media. pp. 282–.
Katsambas AD, Dessinioti C (2010). “Hormonal therapy for acne: why not as first line therapy? facts and controversies”. Clin. Dermatol. 28 (1): 17–23.
Thole Z, Manso G, Salgueiro E, Revuelta P, Hidalgo A (2004). “Hepatotoxicity induced by antiandrogens: a review of the literature”. Urol. Int. 73 (4): 289–95.
Keating GM (March 2015). “Enzalutamide: a review of its use in chemotherapy-naïve metastatic castration-resistant prostate cancer”. Drugs & Aging. 32 (3): 243–9.
Nel comune pensare dell’uomo (e dell’atleta) medio, il Dihydrotestosterone (DHT) è, al pari degli Estrogeni, visto come un ormone tendenzialmente negativo, da ridurre il più possibile. Ovviamente questa visione è a dir poco ristretta dal momento che valuta l’attività del suddetto metabolita del Testosterone solamente in quelle circostanze dove un suo consistente livello può causare, specie nei soggetti predisposti o in determinate circostanze multifattoriali, acne, perdita accelerata dei capelli e ipertrofia prostatica (ovviamente parliamo di soggetti di sesso maschile). Inoltre, il DHT è considerato un metabolita pressoché insignificante nel miglioramento delle prestazioni, soprattutto per quanto concerne l’ipertrofia muscolare. Ma è veramente così limitato il suo impatto per un atleta? ..
Per rispondere a questo quesito nel presente articolo, in modo simile a quanto già feci nell’articolo dedicato agli Estrogeni, esporrò una panoramica dettagliata di tutto ciò che concerne il Dihydrotestosterone e le sue caratteristiche anche alla luce di recenti ed interessanti studi.
Cos’è il DHT?
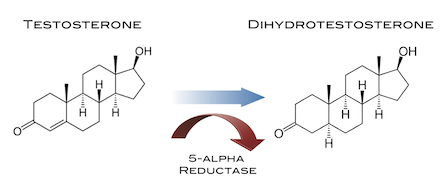
Il Dihydrotestosterone (DHT, 5α-dihydrotestosterone, 5α-DHT, Androstanolone o Stanolone) è uno steroide con caratteristiche fortemente androgene, principalmente ottenuto dalla 5α-riduzione del Testosterone. Infatti, l’enzima 5α-reduttasi catalizza la formazione di DHT dal Testosterone in alcuni tessuti tra cui la ghiandola prostatica, le vescicole seminali, le epididimidi, la pelle, i follicoli piliferi, il fegato e il cervello. Questo enzima media la riduzione del doppio legame C4-5 del Testosterone. Rispetto al Testosterone, il DHT è considerevolmente più potente come agonista del recettore degli androgeni (AR), seppure limitato da percorsi enzimatici.
Oltre al suo ruolo di ormone naturale, il DHT è stato usato come farmaco, ad esempio nel trattamento di bassi livelli di Androgeni negli uomini (vedi Androstanolone).
Il DHT nella Storia
Adolf Friedrich Johann Butenandt (24 marzo 1903-18 gennaio 1995) fu un biochimico tedesco. Nel 1939 gli fu assegnato il premio Nobel per la chimica per il suo “lavoro sugli ormoni sessuali”. Inizialmente respinse il premio a causa della politica nazional-socialista, accettandolo solo nel 1949 dopo la seconda guerra mondiale.
Il DHT fu sintetizzato per la prima volta da Adolf Butenandt e dai suoi colleghi nel 1935. [1][2] Venne ottenuto mediante idrogenazione del Testosterone [3], che era stato scoperto all’inizio di quell’anno.[4] Il DHT è stato introdotto per uso medico come AAS nel 1953 ed è stato inizialmente notato per essere più potente del Testosterone ma con maggiore androgenicità.[5][6][7] Ma il suo potenziale androgeno non fu chiaro fino al 1956, quando venne dimostrato che veniva sintetizzato dal Testosterone negli omogenati di fegato di ratto.[2][8] Inoltre, l’importanza biologica del DHT non è stata realizzata fino agli inizi degli anni ’60, quando si è scoperto che era prodotto dalla 5α-riduzione del Testosterone circolante nei tessuti bersaglio come la ghiandola prostatica e le vescicole seminali risultando più potente del Testosterone in test biologici.[9][10][11][12] Le funzioni biologiche del DHT nell’uomo sono state definite in modo molto più chiaro alla scoperta e alla caratterizzazione del deficit di 5α-reduttasi di tipo II nel 1974.[13] Il DHT è stato l’ultimo importante ormone sessuale, gli altri sono Testosterone, Estradiolo e Progesterone, ad essere scoperto, ed è unico in quanto risulta essere il solo ormone sessuale principale che agisce fondamentalmente come ormone intracrino e paracrino piuttosto che come ormone endocrino.[12]
Biosintesi e distribuzione
Il DHT, noto anche come 5α-androstan-17β-ol-3-one, è uno steroide androstano presente in natura con un gruppo chetonico nella posizione C3 e un gruppo idrossile nella posizione C17β. È il derivato del Testosterone in cui il doppio legame tra le posizioni C4 e C5 è stato ridotto o idrogenato.
Differenze strutturali tra Testosterone e DHT
Il DHT è sintetizzato irreversibilmente dal Testosterone dall’enzima 5α-reduttasi. [14] [15] Ciò si verifica in vari tessuti tra cui i genitali (pene, scroto, clitoride, grandi labbra), [16] prostata, pelle, follicoli piliferi, fegato e cervello. [14] Circa il 5-7% del Testosterone subisce una 5α-riduzione in DHT [17] [18], e circa 200-300μg di DHT vengono sintetizzati giornalmente nel corpo. La maggior parte del DHT è prodotta nei tessuti periferici come la pelle e il fegato, mentre la maggior parte del DHT circolante proviene specificamente dal fegato. I testicoli e la ghiandola prostatica contribuiscono relativamente poco alle concentrazioni di DHT nel circolo ematico.[14]
Ghiandola sebacea
Esistono due isoforme principali di 5α-reduttasi, la SRD5A1 (tipo I) e la SRD5A2 (tipo II), quest’ultimo isoenzima ha una maggiore importanza biologica.[14] Esiste anche una terza forma di 5α-reduttasi: SRD5A3. [19] L’SRD5A2 è maggiormente espressa nei genitali, nella ghiandola prostatica, nelle epididimidi, nelle vescicole seminali, nella pelle genitale, nei follicoli piliferi del viso, del torace [20][21] e nel fegato, mentre si osserva un’espressione più bassa in alcune aree del cervello, pelle non genitale / follicoli piliferi, testicoli e reni. L’SRD5A1 è maggiormente espressa nei follicoli non genitali della pelle / dei capelli, nel fegato e in alcune aree del cervello, mentre sono presenti livelli più bassi nella prostata, nelle epididimidi, nelle vescicole seminali, nella pelle genitale, nei testicoli, nelle ghiandole surrenali e nei reni.[14] Nella pelle, la 5α-reduttasi è espressa in ghiandole sebacee, ghiandole sudoripare, cellule epidermiche e follicoli piliferi.[20][21] Entrambi gli isoenzimi sono espressi nei follicoli piliferi del cuoio capelluto [22], sebbene l’SRD5A2 predomina in queste cellule.[21] Il sottotipo SRD5A2 è l’isoforma quasi esclusivamente espressa nella ghiandola prostatica.[23][24]
Globuline leganti gli ormoni sessuali (SHBG)
Il legame del DHT con le proteine plasmatiche è superiore al 99%. Negli uomini, circa lo 0,88% del DHT non è legato e quindi libero, mentre nelle donne in premenopausa, circa lo 0,47-0,48% non è legato. Negli uomini, il DHT è legato per il 49,7% alla globulina legante gli ormoni sessuali (SHBG), il 39,2% per l’albumina e lo 0,22% per la globulina legante i corticosteroidi (CBG), mentre nelle donne in premenopausa il DHT è legato per il 78,1-78,4% alle SHBG, 21,0-21,3% all’albumina e lo 0,12% al CBG. Nella tarda gravidanza, solo lo 0,07% del DHT non è legato nelle donne; Il 97,8% è legato alle SHBG mentre il 2,15% è legato all’albumina e lo 0,04% è legato al CBG. [25][26] Il DHT ha un’affinità maggiore per le SHBG rispetto al Testosterone, all’Estradiolo o qualsiasi altro ormone steroideo.[27][26]
Funzioni e attività biologiche del DHT
Il DHT è biologicamente importante per la differenziazione sessuale dei genitali maschili durante l’embriogenesi, la maturazione del pene e dello scroto durante la pubertà, la crescita dei peli nel viso, nel corpo e dei peli pubici e lo sviluppo e il mantenimento della ghiandola prostatica e delle vescicole seminali. Come già accennato, è principalmente sintetizzato per via della 5α-riduzione del Testosterone in alcuni tessuti ed è il principale androgeno nei genitali, nella ghiandola prostatica, nelle vescicole seminali, nella pelle e nei follicoli piliferi. [28]
3α-Hydroxysteroide dehydrogenasi (3α-HSD)
Il DHT esplica una segnalazione principalmente in maniera intracrina e paracrina nei tessuti in cui viene sintetizzato, svolgendo un ruolo secondario, sebbene non trascurabile, come ormone endocrino circolante.[29][30][31] I livelli circolanti di DHT sono 1/10 e 1/20 di quelli del Testosterone in termini di concentrazioni totali e libere, rispettivamente [32], mentre i livelli locali di DHT possono essere fino a 10 volte quelli del Testosterone nei tessuti con alta espressione del 5α-reduttasi come la prostata.[33] Inoltre, a differenza del Testosterone, il DHT viene inattivato dalla 3α-idrossisteroide deidrogenasi (3α-HSD) nell’androgeno 3α-androstanediolo molto debole in vari tessuti come quello muscolare, adiposo e epatico, tra gli altri [31][34][35], e in relazione a questo, è generalmente stato riportato che il DHT è un agente anabolico molto scarso quando somministrato esogenamente come farmaco. [36] Ma su questo ci torneremo più avanti.
Progressione della alopecia androgenetica
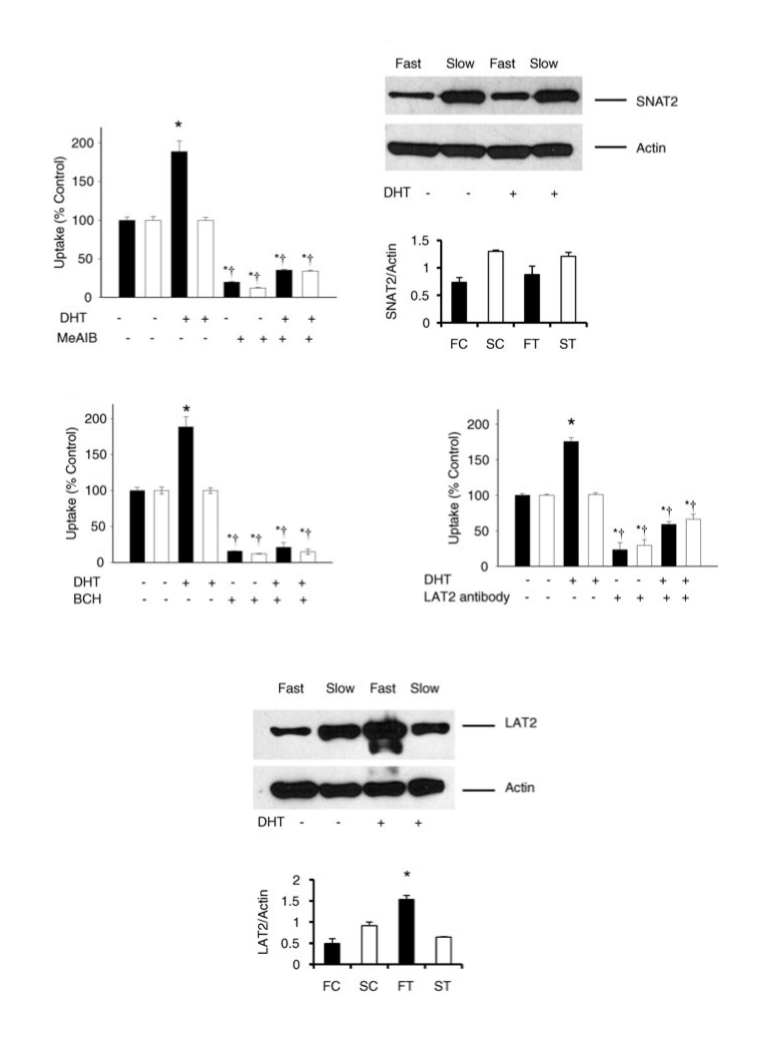
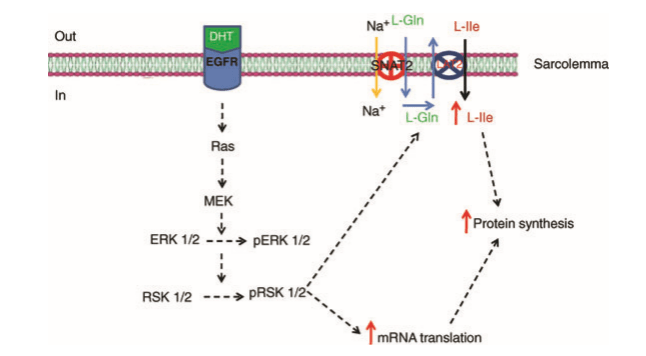
Oltre alle funzioni biologiche di base, il DHT svolge anche un importante ruolo causale in una serie di condizioni dipendenti dagli androgeni, tra cui le condizioni inerenti alla crescita della peluria come l’irsutismo (eccessiva crescita dei peli sul viso / corpo) e anche la perdita di capelli (alopecia androgenetica o calvizie) e malattie della prostata come l’iperplasia prostatica benigna (IPB) e il carcinoma prostatico.[28] Gli inibitori della 5α-reduttasi, che impediscono la sintesi di DHT, sono efficaci nella prevenzione e nel trattamento di queste condizioni, sebbene siano accompagnati da pesanti effetti collaterali.[37][38][39][40] Inoltre, il DHT può svolgere una funzione nel reclutamento e nella funzione del trasportatore di aminoacidi nel muscolo scheletrico.[41] Ed anche su questo punto torneremo tra poco.
Recettore Estrogeno beta (ERβ)
È stato scoperto che i metaboliti del DHT agiscono come neurosteroidi con la propria attività biologica indipendente dall’AR.[42] Il 3α-Androstanediol è un potente modulatore allosterico positivo del recettore GABAA, mentre il 3β-androstanediol è un potente e selettivo agonista del sottotipo ERβ del Recettore degli Estrogeni (ER).[42] Questi metaboliti possono svolgere un ruolo importante negli effetti centrali del DHT e per estensione del Testosterone, inclusi i loro effetti antidepressivi, ansiolitici, gratificanti / edonici, antistress e pro-cognitivi.[42][43] Ed è soprattutto grazie all’azione neurosteroidea dei metaboliti del DHT a conferire a questa molecola i suoi benefici sull’aumento della forza muscolare e del focus mentale, entrambe caratteristiche ricercate negli sport di potenza e propedeutiche ad un migliore stimolo ipertrofico indotto dall’allenamento contro-resistenza.
Il DHT è un potente agonista dell’AR ed è in effetti il ligando endogeno più potente conosciuto per questo recettore. Ha un’affinità (Kd) compresa tra 0,25 e 0,5 nM per la RA umana, che è circa 2-3 volte superiore a quella del Testosterone (Kd = 0,4 a 1,0 nM) [44] e 15-30 volte superiore a quella degli androgeni surrenali.[45] Inoltre, il tasso di dissociazione del DHT dall’AR è 5 volte più lento di quello del Testosterone.[46 L’EC50 del DHT per l’attivazione dell’AR è 0,13 nM, che è circa 5 volte più forte di quello del Testosterone (EC50 = 0,66 nM).[47] Nei biotest, il DHT è risultato essere da 2,5 a 10 volte più potente del Testosterone.[44]
L’emivita di eliminazione del DHT nel corpo (53 minuti) è più lunga di quella del Testosterone (34 minuti), e ciò potrebbe spiegare alcune delle differenze nella loro potenza.[48] Uno studio sul trattamento transdermico con DHT e Testosterone ha riportato emivite terminali rispettivamente di 2,83 ore e 1,29 ore.[49]
A differenza di altri androgeni come il Testosterone, il DHT non può essere convertito dall’enzima aromatasi in estrogeno come l’Estradiolo. Pertanto, viene spesso utilizzato in contesti di ricerca per distinguere tra gli effetti del testosterone causati dal legame con l’AR e quelli causati dalla conversione del Testosterone in Estradiolo e il successivo legame e attivazione del ER.[50] Sebbene il DHT non possa essere aromatizzato, viene comunque trasformato in metaboliti con significativa affinità e attività ER. Questi sono 3α-androstanediolo e 3β-androstanediolo, che sono agonisti predominanti dell’ERβ.[51] Determinano l’effetto anti-estrogenico attribuito al DHT.
I livelli sierici di DHT sono circa il 10% di quelli del Testosterone, ma i livelli nella ghiandola prostatica sono da 5 a 10 volte superiori a quelli del Testosterone a causa di una conversione di oltre il 90% di quest’ultimo in DHT da parte della 5α-reduttasi espressa localmente.[33] Per questo motivo, e oltre al fatto che il DHT è molto più potente come agonista dell’AR rispetto al Testosterone [44], il DHT è considerato il principale androgeno della ghiandola prostatica.[33]
3α-androstanediol
Il DHT è inattivato nel fegato e nei tessuti extraepatici come la pelle in 3α-androstanediol dall’enzima 3α-idrossistoidea deidrogenasi, e in 3β-androstanediol dall’enzimi 3β-idrossisteroidide deidrogenasi.[34][52]Questi metaboliti vengono a loro volta convertiti, rispettivamente, in Androsterone ed Epiandrosterone, quindi coniugati (tramite glucuronidazione e/o solfatazione), rilasciati in circolazione ed escreti nelle urine.[34]
Come già detto, a differenza del Testosterone, il DHT non può essere aromatizzato in estrogeno come l’Estradiolo e, per questo motivo, non ha propensione ad esercitare effetti estrogenici.[53] Quindi, il DHT viene escreto nelle urine sotto forma di metaboliti, come i coniugati di 3α-androstanediol e Androsterone.[54][34]
Uso del DHT in medicina
Il DHT è disponibile in formulazioni farmaceutiche per uso medico come steroide anabolizzante androgeno (AAS) con finalità prettamente androgene.[55] È usato come ancillare principalmente nel trattamento dell’ipogonadismo maschile.[56] Quando usato come farmaco, il DHT viene chiamato Androstanolone (INN) o Stanolone (BAN) [55] [57] [58], e viene venduto sotto nomi commerciali differenti come Andractim. [55] [57] [58] [56] [59] La disponibilità di DHT farmaceutica è limitata; non è disponibile negli Stati Uniti o in Canada, [60] [61] ma è disponibile in alcuni paesi europei. [58] [56] Le formulazioni disponibili di DHT includono compresse orali o sublinguali, gel topici e, come esteri in olio, iniettabili come Androstanolone propionato e Androstanolone Valerato.[55] [56] [59]
L’Androstanolone è disponibile in formulazioni farmaceutiche per uso medico come androgeno.[4] È usato principalmente come forma ancillare nella terapia sostitutiva degli androgeni nel trattamento dell’ipogonadismo maschile ed è specificamente approvato per questa indicazione in alcuni paesi.[62] [13] [63] [64] [65] [66] [67] Non è più raccomandato come solo farmaco nelle terapie sostitutive degli androgeni a causa delle differenze biologiche con il Testosterone come la mancanza di effetti Estrogenici e effetti androgeni parziali.[68] L’Androstanolone topico è utile nel trattamento della ginecomastia.[69] Allo stesso modo, l’Androstanolone Enantato tramite iniezione intramuscolare è risultato efficace nel trattamento della ginecomastia puberale persistente.[70] Il farmaco è stato anche usato come gel topico per il trattamento del pene piccolo nei ragazzi pre e peripubertali con sindrome da insensibilità agli androgeni lieve o parziale.[71] [72] [73]
Drostanolone Propionato
L’Androstanolone è risultato efficace nel trattamento del carcinoma mammario in fase avanzata nelle donne negli anni ’50, sebbene fosse utilizzato in dosi molto elevate e causasse una grave virilizzazione.[74] [75] [76] È stato usato in sospensione acquosa microcristallina mediante iniezione intramuscolare.[77] [78] [79] Poco dopo, il Drostanolone Propionato (2α-Methylandrostanolone Propionato) fu sviluppato per questo uso al fine di sostituire l’Androstanolone a causa della sua superiore farmacodinamica e fu introdotto per questa indicazione negli Stati Uniti e in Europa nei primi anni ’60.[80] [81] [82] [83]
L’Androstanolone è stato usato alla dose di 25mg per via sublinguale da due a tre volte al giorno nella terapia sostitutiva con androgeni per gli uomini.[84] Questo è anche il dosaggio di Androstanolone comunemente utilizzato nel trattamento di individui si sesso maschile.[84]
La questione DHT e ipertrofia muscolare
La sarcopenia, caratterizzata da una perdita di massa muscolare, ossea, forza e resistenza, si verifica con l’invecchiamento e disturbi medici cronici come l’infezione da virus dell’immunodeficienza umana (HIV) e la terapia a lungo termine con glucocorticoidi sistemici (Gcc). D’altra parte, la somministrazione di Testosterone negli uomini più anziani e negli uomini con infezione da HIV con perdita di peso che hanno basse concentrazioni di Testosterone (Bhasin et al. 2001), nonché gli uomini che richiedono un trattamento sistemico a lungo termine di Gcc (Truhan e Ahmed 1989) aumentano il grasso corporeo- massa magra e forza muscolare.
Recettore degli Androgeni
Il muscolo scheletrico è uno dei tessuti bersaglio per l’azione anabolica degli androgeni. Recettori degli Androgeni (AR), localizzati nelle cellule muscolari e adipose, cellule nervose e pluripotenti mesenchimali che risiedono nel tessuto muscolare, probabilmente mediano gli effetti degli androgeni aumentando la massa muscolare, la sintesi proteica, il contenuto ribosomiale, le aree mitocondriali, il numero mioonucleare, il numero di cellule satellite e la miogenesi delle cellule mesenchimali pluripotenti riducendo la degradazione delle proteine e l’adipogenesi delle cellule mesenchimali pluripotenti (Herbst & Bhasin 2004). Sul ligando che si lega all’AR intracellulare, il complesso androgeno-AR viene traslocato nel nucleo e si lega a sequenze specifiche di DNA, elementi di risposta agli androgeni, con conseguente trascrizione di geni specifici (Michel & Baulieu 1980, Simental et al. 1991). Gli androgeni hanno anche azioni rapide non genomiche nel muscolo (Estrada et al. 2000, 2003), tra cui il recettore di membrana accoppiato alla proteina G, il recettore dell’inositolo 1,4,5-trisfosfato (IP3), lo ione calcio (Ca2 +) e la cascata della fosforilazione della proteina chinasi mitogeno-attivata (MAPK) / proteina chinasi regolata da segnali extracellulari (ERK). Oltre al suo ruolo nella contrazione muscolare, si ritiene che Ca2+ intracellulare regola l’espressione genica nel muscolo scheletrico (Estrada et al. 2001, Araya et al. 2003). Pertanto, le azioni genomiche e non genomiche degli androgeni sono responsabili della trascrizione dei geni sensibili agli androgeni (ARG).
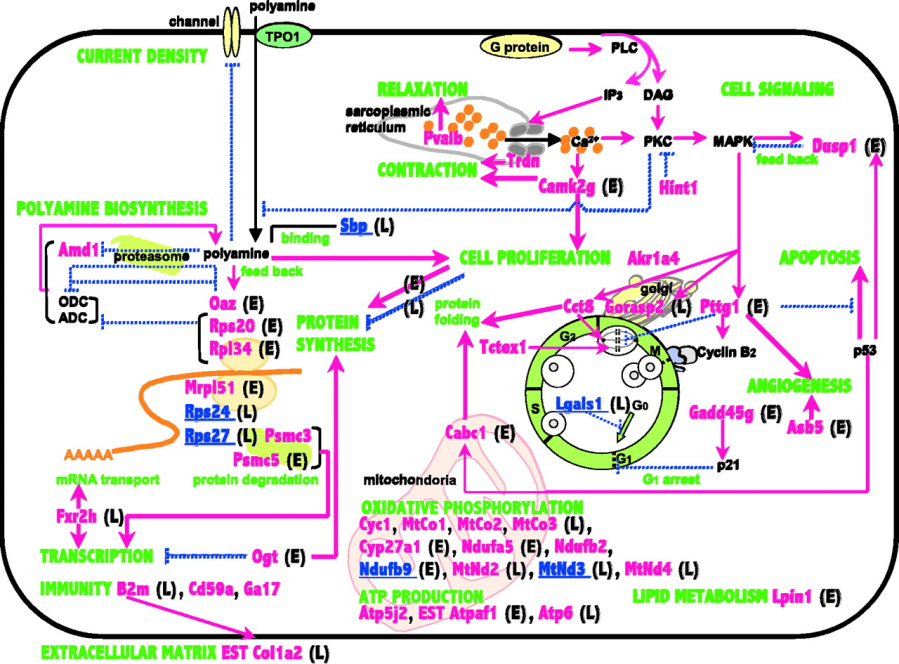
Tuttavia, i meccanismi molecolari dell’effetto anabolico degli androgeni nel muscolo scheletrico sono mal compresi. Con l’avvento dell’analisi seriale dell’espressione genica (SAGE) (Velculescu et al. 1995), sono sorte nuove possibilità per l’analisi del trascrittoma su larga scala. Usando questo metodo, si sono precedentemente studiati i meccanismi molecolari responsabili dell’atrofia muscolare causata dall’immobilizzazione nei ratti (St-Amand et al. 2001), nonché il profilo di espressione genica degli uomini allenati per la resistenza (Yoshioka et al. 2003). In un interessante studio del 2006 [85], si sono studiati gli effetti della castrazione (GDX) e del DHT sull’espressione genica globale nel muscolo scheletrico dei topi maschi usando la strategia SAGE. Le trascrizioni modulate DHT sono coinvolte nel rilascio di Ca2 +, nella segnalazione cellulare, nella proliferazione cellulare, nella sintesi di mRNA e proteine e nel metabolismo energetico. Questi risultati costituiscono un primo passo verso una comprensione precisa dei meccanismi molecolari coinvolti negli effetti fisiologici degli androgeni nel muscolo scheletrico.
Topo C57BL6
Nello studio, è stato asportato il muscolo gastrocnemio destro dai topi C57BL6 di età compresa tra 12 e 14 settimane. Gli animali sono stati tenuti con luci accese da 0715 a 1915h, e hanno avuto accesso all’acqua ad libitum. Nessun trattamento è stato eseguito su 26 topi intatti. Il GDX è stato eseguito 7 giorni prima della raccolta di organi in ciascuno dei 14 topi dai gruppi GDX e DHT. I topi del gruppo GDX hanno ricevuto un i.p. della soluzione del veicolo (0,4% (p / v) Methocel A15 LV Premium / 5% etanolo; Dow Chemicals Co, Laval, Quebec, Canada) 24 ore prima della morte, mentre una dose fisiologica di DHT (0,1 mg / topo) è stato iniettato 1, 3, 6 e 24 ore prima della loro uccisione (gruppi DHT 1 h, DHT 3 h, DHT 6 he DHT 24 h). Il muscolo gastrocnemio destro è stato campionato da ciascun topo e messo insieme per l’analisi dello stesso gruppo per eliminare le variazioni inter-individuali ed estrarre quantità sufficienti di mRNA. I tessuti sono stati conservati a -80 ° C fino all’estrazione dell’RNA.
Poliammina Spermina
Gli ormoni anabolizzanti stimolano la crescita muscolare principalmente aumentando la sintesi proteica (Rooyackers & Nair 1997). In questo studio, la condizione GDX ha represso l’espressione del membro della famiglia delle proteine da shock termico 7 (Hspb7), mentre l’iniezione di DHT ha regolato verso l’alto cinque geni che codificano proteine ribosomiali e chaperoni (Mrpl51, Rpl34, Rps20, Cct8 e Cabc1) entro 3 ore e modulando altre tre trascrizioni (Fxr2h, Rps24 e Rps27) a 24 h. Oltre alla sintesi proteica, Rpl34 e Rps20 sono implicati nella biosintesi delle poliammine (Panagiotidis et al. 1995). La proteina Cct8 la cui espressione è fortemente dipendente dalla crescita cellulare (Yokota et al. 1999) piega le proteine appena sintetizzate, compresi i componenti cellulari necessari per la crescita cellulare (Thulasiraman et al. 1999), oltre a comportarsi come proteina associata ai microtubuli (Roobol et al. 1999). Mrpl51 è codificato dal DNA mitocondriale e Cabc1 codifica una proteina mitocondriale essenziale per la corretta conformazione e funzionamento dei complessi proteici nella catena respiratoria (Iiizumi et al. 2002). In effetti, le espressioni di 18 trascrizioni relative alla produzione di OxPhos e ATP sono state sovra-regolate dal DHT in questo studio. Questi dati suggeriscono che il DHT aumenta la sintesi proteica e la stabilizzazione in parallelo con la crescita cellulare entro 3 ore nei topi in vivo.
Le trascrizioni modulate 24h dopo il trattamento con DHT nel presente studio suggeriscono quanto segue:
l’aumento del trasporto di mRNA tra citoplasma e nucleolo da parte delle proteine fragili correlate all’X (FMRP) (Tamanini et al. 1999);
la soppressione della degradazione dell’mRNA da parte della proteina ribosomiale S27 (Revenkova et al. 1999);
la diminuzione della sintesi proteica poiché la proteina ribosomiale S24 è strettamente coinvolta sia nei processi di iniziazione che di allungamento durante la sintesi proteica (Bommer et al. 1988).
Il fatto che il tasso di sintesi proteica diminuisca drasticamente durante la mitosi nelle cellule di mammifero potrebbe spiegare i dati.
La degradazione delle proteine da parte del proteasoma 26S è essenziale per la progressione del ciclo cellulare, il metabolismo delle poliammine e la presentazione della catena pesante di classe I del maggiore complesso di istocompatibilità (MHC) sulla superficie cellulare. Il DHT ha indotto Psmc3 e Psmc5 le cui proteine sono i componenti integrali della subunità normativa 19S del proteasoma 26S, che potrebbe riflettere l’induzione della proliferazione cellulare e la modulazione dell’immunità da DHT.
La regolazione trascrizionale è un punto di controllo essenziale per diverse funzioni cellulari come la proliferazione cellulare, la differenziazione, la trasformazione e l’apoptosi. Il DHT ha sovra-regolato cinque fattori trascrizionali (Ogt, Pttg1, Psmc3, Psmc5 e Smyd2) entro 3 ore dopo il trattamento e il Fxr2h a 24h. L’attivazione della cascata MAPK provoca la traslocazione della proteina citoplasmatica Pttg1 nel nucleo (Pei 2000) dove la proteina Pttg1 transattiva i geni bersaglio che promuovono la proliferazione cellulare (Pei 2001). Le proteine di Psmc3 e Psmc5 suggeriscono ruoli nella transattivazione del recettore dell’ormone tiroideo (Ishizuka et al. 2001). La condizione GDX ha anche ridotto il livello di espressione del Ttr, il cui prodotto è una proteina plasmatica omotetramericana che trasporta tiroxina e retinolo. Sebbene gli ormoni tiroidei siano essenziali durante la crescita, sia un eccesso che una carenza causano un degrado muscolare da meccanismi sconosciuti (Rooyackers & Nair 1997). La proteina Fxr2h mostra una forte attivazione della trascrizione (Hillman & Gecz 2001). La glicosilazione delle proteine nucleari e citoplasmatiche è una modifica post-traduzionale diffusa e reversibile nelle cellule eucariotiche. La glicosilazione intracellulare dei residui di serina e treonina è catalizzata dalla proteina di Ogt, che regola un numero di funzioni cellulari tra cui l’attivazione trascrizionale (geni bersaglio p53) / repressione (RNA polimerasi II) e l’attivazione traslazionale (Wells et al. 2003). La presenza nel gene Smyd2 di domini SET e MYND sarebbe in accordo con gli effetti rispettivamente sulla deacetilazione e metilazione dell’istone (Sims et al. 2002). Pertanto, i risultati suggeriscono che almeno alcune delle azioni del DHT si verificano attraverso l’attivazione o la repressione dei regolatori trascrizionali.
Via di segnalazione MAPK
Nel muscolo scheletrico, il Ca2+ svolge un ruolo chiave nella contrazione e nel rilassamento. Il presente studio ha dimostrato che il trattamento con DHT ha aumentato l’espressione della Pvalb, una proteina di legame Ca2+ ad alta affinità che agisce come fattore di rilassamento muscolare dopo la contrazione, e Trdn che forma un complesso quaternario con il recettore della ryanodina, junctina e calsequestrina nel lume del reticolo sarcoplasmatico ( SR) per il buffering passivo di Ca2+ luminale SR, nonché un rilascio Ca2+ attivo dal processo SR durante l’accoppiamento eccitazione-contrazione. Topi transgenici che sovraesprimono Trdn1 nel cuore mostrano ipertrofia cardiaca con rilassamento alterato e contrattilità attenuata (Kirchhefer et al. 2001). Pertanto, l’induzione di entrambi i fattori di rilassamento muscolare e di contrazione potrebbe contribuire a una generazione di energia generalmente osservata negli atleti che assumono steroidi anabolizzanti.
Fosfolipasi C
La depolarizzazione delle cellule muscolari provoca anche un rilascio transitorio lento di Ca2 +, che è mediato dalla fosfolipasi C (PLC) e IP3 tramite i recettori IP3 (Estrada et al. 2001, Powell et al. 2001) e porta alla fosforilazione di ERK1 / 2 (Powell et al. 2001). Negli osteoblasti, il DHT attiva la proteina Gβ4 accoppiata al PLC-β2 che aumenta la formazione di IP3 e diacilglicerolo (DAG) e innesca il rilascio di Ca2 + intracellulare dal reticolo endoplasmatico (Zagar et al. 2004). Gli aumenti dei livelli di DAG e Ca2 + regolano l’attività della proteina chinasi C (PKC) che stimola ERK1 / 2 attraverso l’attivazione di MAPK chinasi 1/2 (Zagar et al. 2004). I risultati hanno mostrato le induzioni di Camk2g, Dusp1 e Hint1 entro 3 ore dall’iniezione di DHT. Ca2 + multifunzionale / proteinodinasi dipendente dalla calmodulina (CaMKII) media le risposte cellulari alla Ca2 + intracellulare ed è implicato nel controllo di funzioni essenziali quali trasmissione sinaptica, canali ionici, trascrizione genica e progressione del ciclo cellulare (Santella 1998, Anderson 2005). Le cellule proliferanti (Tombes & Krystal 1997) e il cuore ipertrofico con maggiore contrattilità (Colomer et al. 2003) esprimono l’isoforma CaMKIIγ codificata da Camk2g. Dusp1 (chiamato anche CL100 o MAPK fosfatasi 1) è stato originariamente identificato come un gene precoce immediato indotto da mitogeni (Charles et al. 1992, Keyse & Emslie 1992), e il suo livello di trascrizione riflette l’attivazione di ERK1 / 2 (Camps et al. 2000).
Proteina Hint1
La Hint1, una proteina che interagisce con la PKC che originariamente si pensava inibisse quest’ultima, può svolgere un ruolo di soppressore del tumore (Su et al. 2003). Abbiamo osservato la sovra-regolazione di Mpp6, Oaz, Psmc3 e Psmc5 entro 3 ore dal trattamento con DHT. La funzione di Mpp6, un membro della sottofamiglia guanilato chinasi (MAGUK) associata alla membrana p55, non è ancora nota. Tuttavia, il MAGUK interagisce con i recettori del glutammato e vari canali ionici (Godreau et al. 2004). Le poliammine (spermine, spermidina e putrescina) interagiscono anche con alcuni canali ionici e controllano i livelli intracellulari di Ca2 + (Williams 1997). La biosintesi della poliammina nelle cellule di mammifero inizia con una produzione di putrescina da parte dell’ornitina decarbossilasi (ODC). Quando i livelli di poliammina intracellulare aumentano, l’antizima ODC, codificato da Oaz, si lega all’OCD e facilita il suo rapido degrado da parte del proteosoma 26S (Thomas & Thomas 2003). Pertanto, le induzioni di Oaz così come Psmc3 e Psmc5, che sono componenti integranti del proteasoma 26S, possono riflettere il livello aumentato di poliammine intracellulari. Inoltre, le modulazioni di queste trascrizioni e di Mpp6 entro 3 ore, lo stesso decorso di Camk2g, suggeriscono la loro partecipazione al controllo di Ca2 + intracellulare. Inoltre, il percorso Ras / MAPK controlla la trascrizione di Cct8 (Yamazaki et al. 2003) che è stata indotta a 1 ora dopo il trattamento con DHT nel presente studio. Nel loro insieme, il secondo messaggero, ovvero Ca2 + intracellulare, e le sue cascate a valle tra cui PKC e MAPK, che sono essenziali per la regolazione della crescita cellulare, sembrano essere modulati dal DHT.
Le cellule satellite / mioblasti all’interno del tessuto muscolo-scheletrico proliferano in seguito all’esposizione a fattori di crescita e a seguito di lesioni muscolari, ma smettono di dividersi quando si fondono con fibre muscolari preesistenti. La fusione è generalmente accoppiata con l’inizio della proliferazione cellulare. Nel presente studio, la trascrizioni sovra-regolate da parte del DHT e correlate all’entrata in fase S (Pttg1) (Nasmyth et al. 2000), assemblaggio di microtubuli (Cct8) (Roobol et al. 1999), formazione del fuso bipolare (Tctex1) (Vaisberg et al 1993), uguale segregazione cromosomica (Pttg1) (Nasmyth et al. 2000), accatastamento di Golgi cisternae (Gorasp2) (Shorter et al. 1999) e disintossicazione dei metaboliti reattivi prodotti durante la proliferazione cellulare (Akr1a4) (Barski et al. 2004 ), suggeriscono un’induzione della proliferazione cellulare da parte del DHT. D’altra parte, il DHT ha sotto-regola un fattore cistostatico, Lgals1, che mantiene G0 e controlla la traversata G2 (Wells & Mallucci 1991). Il fuso mitotico richiede il montaggio / smontaggio di icrotubuli e l’azione di complessi motori come il dynein (Vaisberg et al. 1993). La proteina Tctex1 è una catena leggera del complesso motorio dynein (Tai et al. 1998). Il Cct8 aumenta durante la transizione G1 / S attraverso la prima fase S (Yokota et al. 1999). Pttg1, protezione umana, si accumula all’inizio della fase S con picchi nelle fasi G2 – M, e previene l’attivazione prematura delle separine durante la mitosi (Nasmyth et al. 2000).
Il DHT sovra-regola Psmc3, Dusp1, Gadd45g e Pttg1 nelle presenti condizioni. La sovraespressione di Psmc3 aumenta le proteine p53 e p21 (Pollice et al. 2004). In risposta al danno al DNA e ad altri stress, il soppressore del tumore p53 induce l’arresto del ciclo cellulare o l’apoptosi a seconda dei contesti cellulari specifici (Yu & Zhang 2005). In risposta al danno al DNA, p53 promuove la riparazione del DNA influenzando il percorso di riparazione dell’escissione del DNA e arrestando le cellule in G1 attraverso l’induzione di p21 che contribuiscono a fornire più tempo per la riparazione (Smith & Seo 2002), mentre anche l’arresto G1 mediato da p53 si verifica per induzione di Dusp1 in assenza di danni al DNA (Li et al. 2003). Le proteine codificate da Gadd45g interagiscono con p21 e sopprimono la crescita cellulare senza alcuna evidenza di apoptosi (Nakayama et al. 1999). L’arresto della crescita mediato dagli inibitori del ciclo cellulare p21 e Gadd45 inibisce la risposta apoptotica indotta da bersagli apoptotici di p53 (Yu & Zhang 2005). Inoltre, la protezione codificata da Pttg1 inibisce la capacità di p53 di indurre la morte cellulare (Bernal et al. 2002). Nel loro insieme, il DHT potrebbe promuovere l’arresto di G1 senza indurre l’apoptosi, almeno secondo ciò che è emerso dal presente studio.
S-adenosilmetionina decarbossilasi (SAMDC)
Il presente studio riporta l’induzione di Amd1 che codifica per la S-adenosilmetionina decarbossilasi (SAMDC), Oaz, Rps20 e Rpl34, nonché Psmc3 e Psmc5 dopo l’iniezione di DHT. D’altra parte, il DHT sotto-regolato Sbp il cui prodotto è correlato all’accumulo di spermatozoi (Moruzzi et al. 1982). I composti policristici sintetizzati dagli enzimi che limitano la velocità, ODC e SAMDC, sono cruciali per la crescita e la proliferazione delle cellule di mammifero. L’antizima ODC codificato da Oaz, così come le proteine ribosomiali L34 e S20, inibiscono le decarbossilasi di arginina e ODC (Panagiotidis et al. 1995). ODC e SAMDC sono degradati dal proteasoma 26S (Yerlikaya e Stanley 2004) che è codificato da Psmc3 e Psmc5. L’antizima ODC inibisce anche l’assorbimento della poliammina e stimola l’escrezione (Sakata et al. 2000). Inoltre, l’assorbimento della poliammina è inibito dal PKC ed è stimolato dalla sua inibizione (Dot et al. 2000). Per coincidenza, Hint1, che inibisce la PKC, aveva un modello di modulazione simile a quello di Oaz con il trattamento a base di DHT. Nel loro insieme, le modulazioni di Pttg1, Cct8, Tctex1, Gorasp2, Akr1a4, Lgals1, Amd1, Oaz, Rpl34, Rps20, Psmc3, Psmc5 e Sbp mediante dal DHT nel presente studio potrebbero riflettere la proliferazione di cellule satelliti / mioblasti nel muscolo scheletrico .
Inoltre, la proteina Pttg1 induce angiogenesi sia in vitro che in vivo (Ishikawa et al. 2001). Nel presente studio, il DHT ha sovra-regolato Pttg1 e Asb5, che è una nuova proteina implicata nell’inizio dell’arteriogenesi (Boengler et al. 2003). La vascolarizzazione è un importante fattore determinante dell’approvvigionamento energetico e della rimozione dei rifiuti durante la contrazione muscolare e la sua stimolazione da parte del DHT in questo studio è quindi in accordo con altri dati presentati.
Lpin1
Nel metabolismo lipidico, la condizione GDX ha spento la Apina2 e il DHT a sovra-regolato la Lpin1. L’apolipoproteina A-II codificata da Apoa2, la seconda proteina più abbondante delle particelle di lipoproteine ad alta densità (HDL), esercita un marcato effetto sul legame HDL e sull’assorbimento selettivo dei lipidi da parte dei recettori scavenger di classe B. Nel topo, l’espressione migliorata di Lpin1 nel muscolo scheletrico promuove l’obesità diminuendo il dispendio energetico dell’intero corpo e l’utilizzo dei grassi, nonché inducendo resistenza all’insulina (Phan & Reue 2005). Contrariamente a quanto accade nel muscolo, la sovraespressione di Lpin1 nel tessuto adiposo provoca obesità senza insulino-resistenza (Phan & Reue 2005). Ho precedentemente riportato che il livello di espressione di Lpin1 nel tessuto adiposo rimane inalterato con il trattamento a base di DHT (Bolduc et al. 2004). L’induzione di Lpin1 solo nel muscolo potrebbe suggerire che il carboidrato fosse usato per aumentare la produzione di OxPhos e ATP. Ulteriori studi sono necessari per chiarire questo intrigante meccanismo.
Le prime risposte all’iniezione di DHT (DHT 1, 3 e 6 h) sono l’induzione sia dei fattori di rilassamento muscolare (Pvalb) che di contrazione (Trdn) che modulano i livelli intracellulari di Ca2+ . Il DHT ha anche indotto le trascrizioni relative alla segnalazione cellulare come Ca2 + (Camk2g), PKC (Hint1) e percorsi MAPK (Dusp1) nonché la biosintesi della poliammina (Amd1, Oaz, Psmc3, Rps20 e Rpl34), proliferazione cellulare (Akr1a4, Cct8 Pttg1 e Tctex1), arresto del ciclo cellulare (Gadd45g), p53 (Cabc1, Dusp1 e Pttg1) e angiogenesi (Asb5 e Pttg1). L’induzione di mRNA correlati alla trascrizione (Ogt, Psmc3 e Psmc5), sintesi proteica (Mrpl51, Ogt, Rps20 e Rpl34), modifica (Cabc1, Cct8 e Ogt) e degradazione (Psmc3 e Psmc5), fosforilazione ossidativa (Cyc1, MtCo1, in questi punti sono stati osservati anche MtCo2, Cyp27a1, Ndufa5 e Ndufb2), produzione di ATP (Atp5j2 e EST Atpaf1), metabolismo lipidico (Lpin1) e immunità (Cd59a e Ga17). Tuttavia, l’induzione di trascrizioni relative alla segnalazione di Ca2+, MAPK, arresto del ciclo cellulare, p53, sintesi proteica e angiogenesi non è più significativa dopo 24 ore dall’iniezione di DHT mentre le trascrizioni relative alla progressione del ciclo cellulare sono ancora sovraregolate. Inoltre, le trascrizioni relative alla sintesi proteica (Rps24 e Rps27) erano sotto-regolate. Questi risultati indicano che l’iniezione di DHT induce la generazione di energia, la sintesi proteica, la funzione mitocondriale e la proliferazione di cellule satellite / mioblasti a livello trascrizionale in vivo, supportando precedenti risultati di un’azione anabolica del composto in questione.
Panoramica degli ARG nel muscolo scheletrico. Le trascrizioni sovra e sotto regolate dal trattamento con DHT sono mostrate rispettivamente in rosa e in blu. Le trascrizioni che mostrano solo una risposta precoce (DHT 1 ora, 3 ore e 6 ore) o tardiva (DHT 24 ore) sono indicate rispettivamente come (E) o (L). Le linee continue e tratteggiate rappresentano rispettivamente attivazione / induzione e inibizione. Akr1a4, aldo-keto reductasi famiglia 1 membro A4; Amd1, S-adenosilmetionina decarbossilasi 1; Asb5, ripetizione di ankyrin e proteina 5 contenente scatola SOCs; Atp5j2, ATP sintasi subunità mitocondriale F0 complessa f isoforma 2; Atp6, ATP sintasi 6; Atpaf1, fattore di assemblaggio complesso mitocondriale F1 sintasi ATP 1; B2m, microglobulina β-2; Cabc1, attività di accompagnatore ABC1 del complesso bc1 simile; Camk2g, protein chinasi IIγ calcio / calmodulina-dipendente; Cct8, subunità chaperonin 8; Cd59a, antigene CD59a; Col1a2, procollagene tipo I α2; Cyc1, citocromo c-1; Cyp27a1, citocromo P450 famiglia 27 sottofamiglia un polipeptide 1; DAG, diacilglicerolo; Dusp1, fosfatasi 1 a doppia specificità; EST, tag di sequenza espressi; Fxr2h, fragile 2 ritardo mentale X gene 2; Ga17, proteina cellulare dendritica GA17; Gadd45g, arresto della crescita e DNA inducibile 45γ; Gorasp2, Golgi riassemblando la proteina 2; Suggerimento 1, proteina 1 di legame alla triade di nucleotidi di istidina; IP3, inositolo 1,4,5-trisfosfato; Lgals1, lectina legante il galattosio solubile 1; Lpin1, lipina 1; MAPK, protein chinasi attivata dal mitogeno; Mrpl51, proteina ribosomiale L51; MtCo1, citocromo c ossidasi 1; MtCo2, citocromo c ossidasi 2; MtCo3, citocromo c ossidasi 3; MtNd2, NADH deidrogenasi 2; MtNd3, NADH deidrogenasi 3; MtNd4, NADH deidrogenasi 4; Ndufa5, NADH deidrogenasi 1α sottocomplex 5; Ndufb2, NADH deidrogenasi 1β sottocomplex 2; Ndufb9, NADH deidrogenasi 1β sottocomplex 9; Oaz, ornitina decarbossilasi antizima; Ogt, N-acetilglucosamina transferasi legata all’O; PKC, proteina chinasi C; PLC, fosfolipasi C; Psmc, subunità ATPase 26S proteasoma; Pvalb, parvalbumina; Rp, proteina ribosomiale; Pttg1, trasformazione del tumore pituitario 1; Sbp, proteina legante lo sperminozoo; Tctex1, testicolo complesso t espresso 1; TPO1, trasportatore di poliammina; Trdn, triadin.
Un altro studio interessante del 2011 e pubblicato sul “The Journal of Physiology” [86] ci offre ulteriori indizzi sul potenziale del DHT nell’ipertrofia muscolare.
Il gruppo di ricerca che ha svolto questo studio ne aveva in precedenza condotto un altro attraverso il quale avevano dimostrato che il Dihydrotestosterone (DHT), ma non il Testosterone, aumenta la produzione di forza nei muscoli a contrazione rapida e la diminuisce in quelli a contrazione lenta. Questi risultati hanno loro suggerito che il DHT poteva essere un androgeno con capacità ben superiori a quelle comunemente attribuiteli. Nel presente studio, i ricercatori hanno esaminato gli effetti di questi ormoni sul trasporto degli aminoacidi nei fasci muscolari dell’apparato muscolo-scheletrico a rapida contrazione del topo. I risultati mostrano che il DHT aumenta la sintesi proteica e l’aumento delle proteine che trasportano aminoacidi essenziali in fasci muscolari a contrazione rapida. Questo non è stato osservato con il Testosterone.
E’ stato osservato che il DHT esercita azioni acute/non genomiche nel muscolo-scheletrico di mammiferi adulti le cui funzioni fisiologiche sono state capite di recente, pertanto l’obiettivo primario di questo studio era di osservare gli effetti acuti / non genomici del DHT sul uptacke di aminoacidi e sugli eventi di trasduzione del segnale cellulare alla base di queste azioni in fasci muscolari scheletrici a contrazione rapida e lenta di topo. Gli aminoacidi marcati con 14C sono stati usati per studiare gli effetti del DHT e del Testosterone (T) sull’assorbimento degli aminoacidi e sono stati usati interventi farmacologici per determinare gli eventi di trasduzione del segnale cellulare che mediano queste azioni. Mentre il T non ha avuto alcun effetto sull’assorbimento di isoleucina (Ile) e acido α-metilamminoisobutirrico (MeAIB) in entrambi i tipi di fibre, il DHT ha aumentato il loro assorbimento nei fasci di fibre a contrazione rapida. Questo effetto è stato invertito dagli inibitori della traslazione proteica, del recettore del prodotto per la crescita epidemica (EGFR), del sistema A, del sistema L, del mTOR e del MEK. Tuttavia, è stato relativamente correlato agli inibitori della trascrizione, dei recettori degli androgeni e dell’IP3K / Akt. In aggiunta, il trattamento con DHT ha aumentato l’espressione della LAT2 e l’insufflazione fosforilata del GRFR, mentre il miscuglio rapido-twitch si mescola e in entrambi i tipi di ERK1 / 2, RSK1 / 2 e ATF2. Inoltre, ha diminuito la fosforilazione di eEF2 e ha aumentato l’incorporazione di proteine di ferro in entrambi i tipi di fibre. La maggior parte di questi effetti è stata superata dagli inibitori di EGFR e MEK. Da questi risultati si ipotizza che un’altra funzione fisiologica delle azioni acute/non genomiche del DHT nelle fibre muscolari isolate dei mammiferi è quella di stimolare l’assorbimento di aminoacidi. Questo effetto è mediato dall’EGFR e comporta l’attivazione della via MAPK e un aumento dell’espressione LAT2.
IGF-1
Un risultato chiave nel presente studio è stato l’osservazione che il trattamento di piccoli fasci di fibre muscolari scheletriche isolati dall’EDL e dal soleo di topi femmine adulti con concentrazioni fisiologiche (630pgml − 1) di DHT, per 1 ora, significativamente (P = 0,001) ha aumentato l’assorbimento di Ile e MeAiB nei fasci di fibre isolati dall’EDL ma non in quelli isolati dal soleo. Sebbene la somministrazione acuta di ormoni come l’insulina (Biolo et al. 1995), il fattore di crescita insulino-simile 1 (IGF-1) (Fryburg et al. 1995) e l’ormone della crescita (Fryburg et al. 1995) hanno dimostrato di aumentare la sintesi proteica e per promuovere l’assorbimento di aminoacidi nel muscolo scheletrico umano, è la prima volta che è stato dimostrato un aumento dell’assorbimento di aminoacidi in risposta alla somministrazione acuta di uno steroide anabolizzante-androgeno nel muscolo scheletrico dei mammiferi adulti. Solo altri due studi hanno precedentemente osservato gli effetti dell’assorbimento di aminoacidi. Entrambi gli studi hanno utilizzato soggetti umani e non sono stati in grado di dimostrare alcun cambiamento nell’assorbimento degli aminoacidi in gruppi muscolari interi (Bhasin et al. 1997; Ferrando et al. 1998). Sebbene nel presente studio siano state utilizzate piccole fibre muscolari e il T sia stata applicato direttamente ai fasci di fibre, è importante notare che questo ormone non ha avuto alcun effetto sull’assorbimento degli aminoacidi in entrambi i tipi di fibre. Al contrario, il trattamento dei fasci di fibre con DHT ha comportato un marcato aumento dell’assorbimento di Ile e MeAIB solo nei fasci di fibre a contrazione rapida (Fig. 1). In precedenza, abbiamo anche dimostrato che il T non ha effetti acuti sulla produzione di forza in fasci muscolari di topo isolati intatti, mentre il DHT ha aumentato la produzione di forza nei fasci di fibre a contrazione rapida, ma l’ha diminuita in quelli a contrazione lenta (Hamdi & Mutungi, 2010). Nel loro insieme, questi risultati suggeriscono che il T potrebbe non avere effetti acuti / non genomici nelle fasce muscolari scheletriche dei mammiferi adulti.
Come già detto, nella maggior parte dei tessuti il T viene convertito in DHT dall’enzima 5 α-reduttasi. Pertanto, è probabile che tutti gli effetti acuti del T osservati in precedenza nei miociti in coltura (Estrada et al. 2003) potrebbero essere stati esercitati dal DHT. In effetti, è stato precedentemente suggerito che gli effetti anabolici del T nei muscoli scheletrici umani possano essere indiretti o secondari al rilascio di un altro ormone come IGF-1 (Ferrando et al. 1998). Con la presente si precisa che oltre all’IGF-1, gli effetti del T nel muscolo scheletrico dei mammiferi possono anche essere esercitati attraverso il rilascio di DHT. I ricercatori suggeriscono anche che il DHT è il principale steroide anabolizzante-androgeno nei muscoli scheletrici dei mammiferi adulti.
Nonostante la maggior parte degli studi è stata effettuata su fasci muscolari di topi femmina, in precedenza era già stato dimostrato che il DHT ha effetti simili sulla produzione di forza nei fasci muscolari scheletrici di topi maschi e femmine (Hamdi & Mutungi, 2010).
I risultati riportati in questo studio (vedi figura seguente) mostrano che sebbene il DHT aumenti l’attività del SNAT2, ciò non influisce sulla sua espressione. Al contrario, aumenta l’attività e l’espressione del LAT2 (vedi Fig. 3). Inoltre, il trattamemto del fascio di fibre muscolari con il classico sistema di inibitori A e L-typeaminoacid ha portato a una marcata riduzione dell’assorbimento basale di L- [U-14C] Ile in entrambi i tipi di fibre e ha completamente soppresso l’aumento indotto da DHT nell’assorbimento di Ile.
Gli effetti del DHT sull’assorbimento di aminoacidi nei fasci di fibre muscolari di topo sono parzialmente mediati attraverso un trasportatore di aminoacidi del sistema A e sono mediati attraverso LAT2.
Effetti simili sono stati osservati anche quando i fasci di fibre sono stati trattati con la soluzione di Ringer contenente anticorpi contro il LAT2, suggerendo che gli effetti del DHT sull’Ile sono mediati attraverso questo trasportatore. Da queste osservazioni si ipotizza che i due trasportatori (LAT2, SNAT2) siano in qualche modo collegati. Pertanto, modulando l’espressione e l’attività di LAT2, anche il DHT sembra regolare indirettamente l’attività di SNAT2.
Recettore del Fattore di Crescita dell’Epidermide (EGFR)
In precedenza, era stato dimostrato che le azioni acute/ non genomiche del DHT sulla produzione di forza nei fasci muscolo-scheletrici di topi adulti sono mediate attraverso il Recettore del Fattore di Crescita dell’Epidermide (EGFR) e comportano l’attivazione di ERK1 / 2 (Hamdi & Mutungi, 2010). È interessante notare che i risultati qui riportati suggeriscono che le azioni acute/non genomiche del DHT sull’assorbimento di aminoacidi nelle fibre muscolari dei mammiferi adulti sono mediate attraverso lo stesso recettore e lo stesso percorso. Inoltre, il pretrattamento dei fasci di fibre con Ciproterone o Flutamide in modo significativo (P <0,05) ha attenuato gli effetti del DHT sull’assorbimento dell’Ile senza sopprimerlo completamente. In precedenza, è stato suggerito che il Testosterone può attivare la via MAPK tramite un recettore degli androgeni accoppiato con proteina G (Estrada et al. 2003). Tuttavia, se questo meccanismo, comunemente indicato come transattivazione, è quello che media gli effetti acuti/non genomici del DHT nelle fibre muscolari dei mammiferi adulti è incerto e sono necessarie ulteriori ricerche per chiarirlo.
Sistema regolatorio di segnalazione ERK1/2.
Dopo l’attivazione, ERK1 / 2 può rimanere nel citosol o traslare nel nucleo dove svolge un ruolo critico nella regolazione dell’espressione genica e della replicazione del DNA (Brunet et al. 1999). Nel nucleo, ERK1 / 2 fosforila una serie di target, inclusi molti fattori di trascrizione e una famiglia di chinasi correlate a RSK, le chinasi proteiche attivate dallo stress e dal mitogeno (MSK) (Deaketal.1998). Anche se nel presente studio è stato esaminato un gran numero di target a valle di ERK1 / 2, il trattamento con DHT ha aumentato soltanto la fosforilazione di RSK1 / 2 e ATF2 . Inoltre, il DHT non ha avuto alcun effetto sulla fosforilazione degli altri modulatori MAPK. Per i ricercatori, questi risultati suggeriscono che le azioni acute / non genomiche del DHT nei muscoli scheletrici dei mammiferi adulti sono esercitate principalmente attraverso l’EGFR e comportano l’attivazione della modulazione ERK1 / 2 del percorso MAPK. L’ipotesi è che il DHT, direttamente o indirettamente, attivi l’EGFR che a sua volta attiva la via MAPK che porta ad un aumento del trasporto di aminoacidi nelle fibre aumentando anche la sintesi proteica in esse. Inoltre, il DHT sembra avere un attività principale sulle fibre a contrazione rapida.
Negli animali adulti, un aumento della massa muscolo scheletrica (ipertrofia) si verifica principalmente a causa di un aumento delle dimensioni piuttosto che del numero di fibre muscolari ed è generalmente ritenuto che sia regolata dal percorso Akt / mTOR (Glass, 2003). Il percorso Akt / mTOR è attivato da molti stimoli tra cui fattori di crescita, altri ormoni e sostanze nutritive. Inoltre, la sua attivazione culmina nel blocco del apoptosi, induzione della sintesi proteica, trascrizione genica e proliferazione cellulare (Dann etal.2007). Pertanto, è stato stimato che il DHT e il T potessero attivare questo percorso. L’assorbimento di amminoacidi indotto dal DHT nelle fibre a contrazione rapida senza la sua completa soppressione, suggerisce che le azioni acute / non genomiche del DHT non sono mediate attraverso Akt. Invece, i risultati che vengono presentati nel presente studio suggeriscono che il DHT aumenta la sintesi proteica regolando la traduzione dell’mRNA già presente nelle cellule. In effetti, il trattamento dei fasci di fibre muscolari con DHT ha portato a una marcata riduzione della fosforilazione di eEF2 e ad un moderato aumento (∼50%) della sintesi proteica nei fasci di fibre a contrazione rapida.
È anche degno di nota il fatto che il pretrattamento dei fasci di fibre con l’inibitore specifico del mTOR non solo ha ridotto l’assorbimento basale di Ile, ma ha anche soppresso completamente l’aumento indotto dal DHT dell’assorbimento di Ile nei gruppi di fibre a contrazione rapida. Per i ricercatori questi risultati suggeriscono che alcuni degli effetti acuti / non genomici del DHT sono probabilmente mediati attraverso mTOR. In effetti, numerosi studi hanno suggerito che mTOR è il legame tra la disponibilità di aminoacidi e una maggiore sintesi proteica (Beugnet et al. 2003; Avruch et al. 2009). Tuttavia, la rapamicina può anche indurre l’autofagia (Ravikumar et al. 2004). Pertanto, un’altra possibilità è che i suoi effetti siano dovuti all’accumulo di aminoacidi nelle fibre muscolari.
Via di segnalazione cellulare che media gli effetti del DHT sul trasporto di aminoacidi nelle fibre muscolo-scheletriche dei mammiferi. Un diagramma schematico che mostra la via di segnalazione cellulare ipotizzata che media gli effetti acuti del DHT sull’amminoacido nei fasci muscolari dei mammiferi. L’ipotesi è che il DHT, attraverso un meccanismo sconosciuto, attiva l’EGFR e questo porta all’attivazione di RSK1 / 2 da parte di ERK1 / 2. L’RSK1 / 2 attivato aumenta quindi l’attività e l’espressione di LAT2 (cerchio blu con una croce), che a sua volta aumenta il trasporto di Ile nei fasci muscolari. Inoltre, RSK1 / 2 migliora la traduzione dell’mRNA in proteine. Si noti che il trasporto di Ile è accoppiato a quello di piccoli aminoacidi neutri come la glutammina (Gln) che vengono trasportati nella cellula dai trasportatori di aminoacidi del sistema A come SNAT2 (cerchio rosso con una croce). Pertanto, un aumento dell’attività di LAT2 aumenta indirettamente l’attività di SNAT2. Gli eventuali effetti di tutti questi processi sono di aumentare la sintesi proteica e quindi la massa muscolare, specialmente nelle fibre muscolari a contrazione rapida.
In conclusione, ci sono state prove che dimostrano che un’altra funzione fisiologica delle azioni acute / non genomiche del DHT nelle fibre muscolari dei mammiferi adulti è quella di aumentare l’assorbimento di aminoacidi essenziali. Sebbene non si possa escludere completamente il coinvolgimento del recettore degli androgeni, i principali risultati suggeriscono che l’aumento dell’assorbimento di aminoacidi e la sintesi proteica sono mediati attraverso l’EGFR e comportano l’attivazione del modulo ERK1 / 2 del pathway MAPK che a sua volta attiva RSK1 / 2 portando ad un aumento dell’espressione del trasportatore di aminoacidi di tipo L LAT2.
Piccola parentesi sul Mesterolone
Arrivati a questo punto, prima di trattare le conclusioni sul DHT, vorrei riportarvi due interessanti studi nei quali si è osservata l’azione di una forma di DHT metilata in C-1, il noto Mesterolone, sulla massa muscolare.
Senza dilungarsi troppo in preamboli, come ben sappiamo, il Mesterolone (nome commerciale Proviron) [1 alpha-methyl-17 beta- hydroxy-5 alpha-androstan-3-one] è un AAS derivato dal Dihydrotestosterone (DHT). A differenza del DHT il Mesterolone presenta l’aggiunta di un gruppo metilico in C1 (similmente al Metenolone) cosa che ne aumenta discretamente la biodisponibilità orale, senza però apportare eccessivo stress epatico. Questa alterazione però non aumenta la stabilità del C3 chetogruppo, essenziale per l’attività anabolizzante a livello muscolare mediata dal legame recettoriale, come il suo precursore.
Ora, sappiamo anche che ci sono frequenti segnalazioni di abuso di Mesterolone negli sport umani ed equini per il miglioramento della prestazione. Tuttavia, sono disponibili informazioni limitate su come questo farmaco esercita i suoi effetti sul muscolo scheletrico.
Le cellule satellite (SC) sono cellule staminali miogeniche mononucleari che contribuiscono alla crescita e alla riparazione dei muscoli postnatali. Poiché l’attivazione delle SC e la successiva differenziazione nei nuovi myonuclei sono un evento importante durante l’ipertrofia muscolare. In uno studio pubblicato nel maggio del 2012 [87], si è osservata l’influenza del Mesterolone sulla distribuzione delle SC all’interno del muscolo pettorale dei polli (Gallus gallus ). Nello specifico, questo studio ha testato le ipotesi secondo cui che il Mesterolone induca l’ipertrofia del muscolo scheletrico aviario e che aumenti anche il numero di SC nel muscolo scheletrico aviario. Sono state utilizzate solide tecniche immunocitochimiche e analisi morfometriche per calcolare il numero di SC e myonuclei. Inoltre, sono stati misurati i livelli di concentrazione di DNA e proteine Pax7 per confermare i risultati immunocitochimici. Il Mesterolone ha aumentato significativamente la massa pettorale e le dimensioni delle fibre. Tutti gli indici delle SC e il numero di myonuclei sono aumentati significativamente con la somministrazione di Mesterolone. Inoltre, negli uccelli trattati con Mesterolone sono state riscontrate una maggiore concentrazione di DNA ed espressione della proteina Pax7. Questo studio indica che il Mesterolone può indurre ipertrofia del muscolo scheletrico aviario e che questo è correlato con un aumento del numero di SC. I ricercatori suggeriscono che le SC siano intermediari cellulari chiave per l’ipertrofia muscolare indotta da Mesterolone. L’ipotesi, che necessità di ulteriori ricerche, è che tale effetto possa riscontrarsi anche in altre specie compreso l’uomo.
Identificazione immunofluorescente di SC nelle sezioni trasversali dei muscoli pettorali ottenuti da polli di controllo e trattati con Mesterolone. (A) e (C) rappresentano una sezione ottenuta dal muscolo pettorale di un esemplare di controllo. (B) e (D) rappresentano una sezione ottenuta dal muscolo pettorale di un esemplare trattato con Mesterolone. (A) e (B) mostrano tutti i nuclei in blu (colorazione DAPI) e le lamine basali delle fibre muscolari in rosso (colorazione anti-laminina). (C) e (D) mostrano SC in verde (etichettatura anti-Pax7) e le lamine basali in rosso. Barre di scala = 50 μm.
In un altro studio pubblicato nel 2010 [88], la microscopia ottica ed elettronica e la morfometria quantitativa sono state utilizzate per determinare gli effetti dell’esercizio e del Mesterolone sul muscolo soleo dei topi. Sia l’esercizio fisico che il Mesterolone hanno causato una significativa ipertrofia delle fibre muscolari extrafusali. L’ipertrofia delle fibre di tipo I era maggiore di quella delle fibre di tipo II. Non c’era iperplasia. I mitocondri erano più numerosi e più grandi rispetto a quanto osservato nei muscoli degli animali sedentari. La capillarità è aumentata e sono comparse piccole fibre muscolari nucleate centralmente, di solito in piccoli gruppi e molto spesso nei muscoli degli animali esposti al Mesterolone. Una piccola percentuale di cellule satelliti mostrava segni di attivazione ma nei muscoli degli animali trattati con Mesterolone c’era una massa muscolare maggiore dopo l’esercizio. I muscoli di animali che erano stati entrambi fatti esercitati e trattati con Mesterolone presentavano i maggiori cambiamenti: la massa muscolare e l’ipertrofia delle fibre muscolari erano maggiori rispetto a tutti gli altri gruppi di animali, la capillarità era maggiore e> il 30% di tutte le cellule satellitari riconosciute mostrava segni di attivazione. Gruppi di piccole fibre muscolari nucleate centralmente sono state comunemente osservate in questi muscoli. Sembravano essere il risultato di lacerazioni rigenerate in fibre muscolari esistenti. Sia con l’esercizio fisico sia con il Mesterolone, da solo o in combinazione, si è verificato un aumento della percentuale di fibre muscolari di tipo I e una diminuzione della proporzione di tipo II.
Entrambi gli studi esposti, sebbene non siano di grande impatto, fanno sicuramente nascere il dubbio in coloro i quali hanno sempre valutato le possibili azioni di una molecola in modo ristretto, sia per mancanza di una conoscenza sufficienza in materia sia per limitatezza nel formulare ipotesi estrapolate dall’osservazione.
Ora, non sto sicuramente dicendo che un culturista trattato con solo Mesterolone possa ambire a grandi cambiamenti nella composizione corporea. Sto semplicemente ipotizzando, che le nozioni in merito alle attività degli AAS sono datate e parziali, anche perchè si basano per lo più, tanto quanto gli studi esposti, sulla capacità di legame degli AAS testati osservata nella prostata e nel levator ani dei topi. Inoltre, il Mesterolone ha visto una riscoperta come farmaco ancillare nella TRT o come anti-depressivo funzionale in alcune tipologie di depressione.
In fondo, il peggior difetto del Mesterolone è dipeso dalla sua consuetudinaria via di somministrazione…
Nota: la biodisponibilità orale del Mesterolone è stata stimata al 3%.
Tiriamo le somme…con possibili critiche…
Già sento i soliti limitati in intelletto e pazienza, sempre pronti a parlare senza cognizione di causa, urlare al “esiste lo studio X che afferma che l’aggiunta di Dutasteride con dosi sovrafisiologiche di Testosterone non cambia il risultato!” … Lo conosco anche io capre!… A proposito, vediamo di cosa si tratta…
Testosterone Enantato
Il principale studio è quello pubblicato su JAMA nel 2012.[89] Si tratta di uno studio in doppio cieco, randomizzato, controllato con placebo. Lo studio è stato diretto dal dott. Bhasin e colleghi, i quali hanno reclutato 139 uomini sani, di età compresa tra 18 e 50 anni, con livelli normali di Testosterone.
Ai pazienti è stata assegnata 1 su 4 dosi di Testosterone Enantato (50, 125, 300 o 600 mg / settimana) più 2,5mg/die di Dutasteride o sono stati assegnati a 1 su 4 gruppi a cui è stato somministrato 1 di 4 dosi di Testosterone Enantato (50, 125, 300 o 600 mg / settimana) più placebo per 20 settimane.
Dutasteride
Il cambiamento nella massa magra rispetto al basale, misurata dall’assorbtiometria a raggi X a doppia energia, è stato il risultato primario. I ricercatori hanno anche misurato i livelli di Testosterone, la forza muscolare, la funzione sessuale, la produzione di sebo e l’acne.
La forza nel Leg Press e del petto sono state misurate con il metodo 1 ripetizione massimale con l’uso di macchine Keizer.
La funzione sessuale è stata valutata con l’indice internazionale della funzione erettile e il questionario sulla salute sessuale maschile per una valutazione più completa del desiderio sessuale.
Il volume della prostata è stato misurato con la risonanza magnetica 1.5-Tesla.
La produzione di sebo è stata misurata con l’uso di nastri di sebo. La scala Palatzi è stata utilizzata per valutare l’acne.
139 uomini furono assegnati in modo casuale; 102 (54 nei gruppi placebo e 48 nei gruppi dutasteride) hanno completato l’intervento di 20 settimane.
Gli uomini assegnati alla Dutasteride erano simili al basale degli uomini assegnati al placebo. Inoltre, i livelli di Testosterone totale e libero non differivano tra i due gruppi.
L’aumento della massa magra ottenuta dai gruppi Dutasteride+Testosterone era di 0,6 kg (IC 95%, da -0,1 a 1,2 kg) quando ricevevano 50 mg/settimana di Testosterone Enantato, 2,6 kg (IC 95%, 0,9 – 4,3 kg) per 125mg/settimana, 5,8 kg (IC 95%, 4,8 – 6,9 kg) per 300mg/settimana e 7,1kg (IC 95%, 6,0 – 8,2 kg) per 600 mg/settimana.
La massa magra ottenuta dai gruppi placebo+Testosterone era di 0,8 kg (IC 95%, da -0,1 a 1,7 kg) quando ricevevano 50mg/settimana di Testosterone Enantato, 3,5 kg (IC 95%, 2,1-4,8 kg) per 125 mg/settimana, 5,7 kg (IC 95%, 4,8 – 6,5 kg) per 300mg/settimana e 8,1 kg (IC 95%, 6,7 – 9,5 kg) per 600mg/settimana.
Le differenze dose-dipendente tra i gruppi Dutasteride e placebo per la massa magra non erano significative (P = .18).
I cambiamenti nella massa grassa, nella forza muscolare, nella funzione sessuale, nel volume della prostata, nella produzione di sebo e nei livelli di ematocrito e lipidi non differivano tra i gruppi.
La frequenza complessiva di eventi avversi, inclusa la frequenza di disfunzione sessuale, acne, vampate di calore e dolorabilità mammaria era simile nei gruppi placebo e dutasteride.
Misure in riferimento alla composizione corporea e alla forza muscolare raccolte nello studio trattato. Nei grafici a sinistra, i marker indicati hanno un margine di errore con intervalli di confidenza al 95%. Nei grafici a destra (variazione dei livelli di Testosterone), le scale dell’asse orizzontale sono state spostate (offset) di 1000ng/dL e quindi è stata applicata una trasformazione della radice quadrata. Il livellamento semiparametrico è stato ottenuto utilizzando modelli di additivi generalizzati. Le aree ombreggiate indicano intervalli di confidenza al 95%.
Bene…possiamo cestinare tutto quanto ipotizzato in precedenza? Visto anche il tipo di studio esposto? Non esattamente …
Ora, da notare la tipologia di soggetti arruolati e il loro grado di “maturità sportiva”, più specificatamente negli allenamenti contro resistenza. Essa non è chiaramente specificata e la misurazione dell’espressione della forza in soggetti poco o per nulla allenati non è un grande parametro sebbene lo scopo dello studio fosse quello di constatare l’impatto nella forza e ipertrofia dato da un protocollo a dosaggio differenziato di Testosterone Enantato con o senza un inibitore della 5α-reduttasi (Dutasteride).
Secondo punto da notare, la mancanza di misurazione specifica della massa muscolare. Infatti, la composizione corporea dei partecipanti è stata valutata per i parametri di massa grassa e massa magra. Come risaputo, quando si parla di massa magra si prende anche in considerazione l’acqua. Sebbene la differenza sia bassa, probabilmente essa è da ricollegare anche al fatto che le dosi di Testosterone utilizzate erano differenti e con limite a 600mg di Testosterone Enantato a settimana (pari a 432mg di Testosterone), e che tale dosaggio non sempre crea iperestrogenemia marcata (considerando anche un lasso di tempo contenuto) tale da causare accumuli sensibili di acqua extracellulare. Considerando poi che uno degli effetti collaterali collegati all’uso di inibitore della 5α-reduttasi sia proprio un aumento degli estrogeni circolanti (per aumento del substrato di sintesi) con ripercussioni a carico di (e non solo limitate a) ritenzione idrica, accumulo di grasso con modello ginoide e ginecomastia, i dubbi sulle metodiche di analisi dei partecipanti allo studio diventano senz’altro dubbie.
Da notare che il Dutasteride è stato osservato ridurre il DHT sierico del >90% e quello intraprostatico del 94% alla dose di 0.5mg/die.[90] Nello studio presentato, i partecipanti assegnati ai gruppi Dutasteride e Dutasteride+Testosterone erano trattati con una dose di Dutasteride pari a 2,5mg/die.
I ricercatori volevano condurre uno studio meticolosamente progettato ed eseguito per rispondere a una domanda vecchia di quasi 5 decenni: il Testosterone, il DHT o entrambi a livelli fisiologici e soprafisiologici sono importanti per il guadagno della massa magra? Purtroppo sembra che non siano stati sufficientemente meticolosi…
Non è di certo mia intenzione far valere di più uno studio svolto in vitro o su animali rispetto ad uno svolto su esseri umani. E’ invece mia intenzione far riflettere il lettore sul design di uno studio, ed in base al livello di questo poter valutare quanto in esso riportato.
Un altra critica riduttiva che potrebbe essere mossa contro le nuove ipotesi di attività del DHT potrebbe essere riferita all’enzima 3α-Hydroxysteroide dehydrogenasi (3α-HSD). Si dimentica però che tale enzima è un “limitatore” dell’attività del DHT a livello di tessuti come quello muscolare e adiposo. Infatti, la sua azione non esclude assolutamente la potenziale attività in acuto e non genomica del DHT nei tessuti dove l’enzima 3α-HSD viene espresso. E’ possibile che la cooperatività del DHT con il Testosterone sia di tipo sequenziale: attività prettamente recettoriale e in cronico (sebbene sotto regolazione organica) data dal Testosterone preceduta e seguita da una attività acuta e non genomica data dal DHT.
Inoltre, ed è una cosa molto importante da tenere in considerazione, la ricerca è tutt’ora in essere. Esistono ipotesi diverse con il loro bagaglio di ricerca. Ciò che il lettore preparato può fare è semplicemente soppesare le informazioni raccolte al fine di farsi un idea il più concreta possibile della questione.
Sebbene la ricerca scientifica abbia fatto enormi passi avanti nell’ultimo secolo, siamo ancora lungi dal conoscere tutto… anche quando si parla di ormoni e la loro attività diretta e indiretta…
Nota conclusiva: è ovvio che non si mette in dubbio l’utilità del controllo terapeutico del DHT in condizioni patologiche (vedi iperplasia prostatica maligna) o preventive in particolari soggetti. Negli individui in fisiologia e senza particolari predisposizioni, l’uso di inibitori della 5α-reduttasi potrebbe risultare una scelta controproducente sotto molteplici aspetti.
Gabriel Bellizzi
Riferimenti:
Schnitzer R (1 January 1967). Experimental Chemotherapy. Elsevier Science. pp. 156–. ISBN 978-0-323-14611-1.
Krüskemper H (22 October 2013). Anabolic Steroids. Elsevier. pp. 12–. ISBN 978-1-4832-6504-9.
Taylor WN (16 January 2002). Anabolic Steroids and the Athlete, 2d ed. McFarland. pp. 178–. ISBN 978-0-7864-1128-3.
William Andrew Publishing (2007). Pharmaceutical Manufacturing Encyclopedia. William Andrew Pub. ISBN 978-0-8155-1526-5.
Newsweek. Newsweek. 1953.
New and Nonofficial Drugs. Lippincott. 1958.
Rubin BL, Dorfman RI (1956). “In vitro conversion of testosterone to 17beta-hydroxyandrostan-3-one”. Proc. Soc. Exp. Biol. Med. 91 (4): 585–6. doi:10.3181/00379727-91-22337. PMID 13323010.
Agmo A (18 April 2011). Functional and Dysfunctional Sexual Behavior: A Synthesis of Neuroscience and Comparative Psychology. Academic Press. pp. 196–. ISBN 978-0-08-054938-5.
Oreopoulos DG, Michelis M, Herschorn S (6 December 2012). Nephrology and Urology in the Aged Patient. Springer Science & Business Media. pp. 495–. ISBN 978-94-011-1822-4.
Webster GF, Rawlings AV (17 May 2007). Acne and Its Therapy. CRC Press. pp. 168–. ISBN 978-1-4200-1841-7.
Smith LB, Mitchell RT, McEwan IJ (1 October 2013). Testosterone: From Basic Research to Clinical Applications. Springer Science & Business Media. pp. 5–. ISBN 978-1-4614-8978-8.
Azzouni F, Mohler J (2012). “Role of 5α-reductase inhibitors in benign prostatic diseases”. Prostate Cancer Prostatic Dis. 15 (3): 222–30.
Melmed S (2016). Williams Textbook of Endocrinology. Elsevier Health Sciences. pp. 621, 711.
Blume-Peytavi U, Whiting DA, Trüeb RM (26 June 2008). Hair Growth and Disorders. Springer Science & Business Media. pp. 161–162.
Rhoades RA, Bell DR (18 January 2012). Medical Phisiology: Principles for Clinical Medicine. Lippincott Williams & Wilkins. pp. 690–.
Rakel D (12 April 2012). Integrative Medicine E-Book. Elsevier Health Sciences. pp. 321–.
Morrison MF (4 May 2000). Hormones, Gender and the Aging Brain: The Endocrine Basis of Geriatric Psychiatry. Cambridge University Press. pp. 17–.
Azzouni F, Godoy A, Li Y, Mohler J (2012). “The 5 alpha-reductase isozyme family: a review of basic biology and their role in human diseases”. Advances in Urology. 2012: 1–18.
Zouboulis CC, Chen WC, Thornton MJ, Qin K, Rosenfield R (2007). “Sexual hormones in human skin”. Horm. Metab. Res. 39 (2): 85–95.
Bolognia JL, Jorizzo JL, Schaffer JV (8 June 2012). Dermatology E-Book. Elsevier Health Sciences. pp. 1094–. ISBN 978-0-7020-5182-1.
Murphy MJ (24 March 2011). Molecular Diagnostics in Dermatology and Dermatopathology. Springer Science & Business Media. pp. 373–.
eam SJ, Scott LJ (2008). “Dutasteride: a review of its use in the management of prostate disorders”. Drugs. 68 (4): 463–85.
Heesakkers J, Chapple C, Ridder DD, Farag F (24 February 2016). Practical Functional Urology. Springer. pp. 280–.
Nieschlag E, Behre HM, Nieschlag S (26 July 2012). Testosterone: Action, Deficiency, Substitution. Cambridge University Press. pp. 61–.
Dunn JF, Nisula BC, Rodbard D (July 1981). “Transport of steroid hormones: binding of 21 endogenous steroids to both testosterone-binding globulin and corticosteroid-binding globulin in human plasma”. J. Clin. Endocrinol. Metab. 53 (1): 58–68.
Williams DA, Foye WO, Lemke TL (2002). Foye’s Principles of Medicinal Chemistry. Lippincott Williams & Wilkins. pp. 707–.
Coutts, S. B.; Kicman, A. T.; Hurst, D. T.; Cowan, D. A. (1997-11-01). “Intramuscular administration of 5α-dihydrotestosterone heptanoate: changes in urinary hormone profile”. Clinical Chemistry. 43 (11): 2091–2098.
Marks LS (2004). “5α-reductase: history and clinical importance”. Rev Urol. 6 Suppl 9: S11–21.
Horton R (1992). “Dihydrotestosterone is a peripheral paracrine hormone”. J. Androl. 13 (1): 23–7. doi:10.1002/j.1939-4640.1992.tb01621.x (inactive 2020-03-09).
Wilson JD (1996). “Role of dihydrotestosterone in androgen action”. Prostate Suppl. 6: 88–92.
Swerdloff RS, Dudley RE, Page ST, Wang C, Salameh WA (2017). “Dihydrotestosterone: Biochemistry, Physiology, and Clinical Implications of Elevated Blood Levels”. Endocr. Rev. 38 (3): 220–254.
Bhasin S (13 February 1996). Pharmacology, Biology, and Clinical Applications of Androgens: Current Status and Future Prospects. John Wiley & Sons. pp. 72–.
Hay ID, Wass JA (26 January 2009). Clinical Endocrine Oncology. John Wiley & Sons. pp. 37–.
Melmed S (2016). Williams Textbook of Endocrinology. Elsevier Health Sciences. pp. 621, 711.
Jin Y, Penning TM (2001). “Steroid 5alpha-reductases and 3alpha-hydroxysteroid dehydrogenases: key enzymes in androgen metabolism”. Best Pract. Res. Clin. Endocrinol. Metab. 15 (1): 79–94.
Llewellyn W (2009). Anabolics. Molecular Nutrition Llc. pp. 19, 163.
Blume-Peytavi U, Whiting DA, Trüeb RM (26 June 2008). Hair Growth and Disorders. Springer Science & Business Media. pp. 161–162.
Azzouni F, Mohler J (2012). “Role of 5α-reductase inhibitors in benign prostatic diseases”. Prostate Cancer Prostatic Dis. 15 (3): 222–30.
Azzouni F, Mohler J (2012). “Role of 5α-reductase inhibitors in prostate cancer prevention and treatment”. Urology. 79 (6): 1197–205.
Lotti F, Maggi M (28 April 2015). “Hormonal Treatment for Skin Androgen-Related Disorders”. In Katsambas A, Lotti T, Dessinioti C, D’Erme AM (eds.). European Handbook of Dermatological Treatments. Springer. pp. 1451–1464.
Wendowski, Oskar; Redshaw, Zoe; Mutungi, Gabriel (February 2017). “Dihydrotestosterone treatment rescues the decline in protein synthesis as a result of sarcopenia in isolated mouse skeletal muscle fibres”. Journal of Cachexia, Sarcopenia and Muscle. 8 (1): 48–56.
Kohtz AS, Frye CA (2012). Dissociating behavioral, autonomic, and neuroendocrine effects of androgen steroids in animal models. Methods Mol. Biol. Methods in Molecular Biology. 829. pp. 397–431.
Mozayani A, Raymon L (18 September 2011). Handbook of Drug Interactions: A Clinical and Forensic Guide. Springer Science & Business Media. pp. 656–.
Hemat RA (2004). Principles Of Orthomolecularism. Urotext. p. 426.
Grino PB, Griffin JE, Wilson JD (February 1990). “Testosterone at high concentrations interacts with the human androgen receptor similarly to dihydrotestosterone”. Endocrinology. 126 (2): 1165–72.
Wilderer PA (1 September 2010). “Bioassays for Estrogenic and Androgenic Effects of Water Constituents”. Treatise on Water Science, Four-Volume Set. Newnes. pp. 1805–.
Diamanti-Kandarakis E (1999). “Current aspects of antiandrogen therapy in women”. Current Pharmaceutical Design. 5 (9): 707–23. PMID 10495361.
von Deutsch DA, Abukhalaf IK, Lapu-Bula R (15 October 2003). “Anabolic Doping Agents”. In Mozayani A, Raymon L (eds.). Handbook of Drug Interactions: A Clinical and Forensic Guide. Springer Science & Business Media. pp. 510–.
Swerdloff RS, Wang C (October 1998). “Dihydrotestosterone: a rationale for its use as a non-aromatizable androgen replacement therapeutic agent”. Baillière’s Clinical Endocrinology and Metabolism. 12 (3): 501–6.
Kohtz AS, Frye CA (2012). Dissociating behavioral, autonomic, and neuroendocrine effects of androgen steroids in animal models. Methods Mol. Biol. Methods in Molecular Biology. 829. pp. 397–431.
Rizner TL, Lin HK, Peehl DM, Steckelbroeck S, Bauman DR, Penning TM (July 2003). “Human type 3 3alpha-hydroxysteroid dehydrogenase (aldo-keto reductase 1C2) and androgen metabolism in prostate cells”. Endocrinology. 144 (7): 2922–32.
Weiner IB, Gallagher M (2003). Handbook of Psychology, Biological Psychology. John Wiley & Sons. pp. 333–.
Schill W, Comhaire FH, Hargreave TB (26 August 2006). Andrology for the Clinician. Springer Science & Business Media. pp. 243–.
Hyde TE, Gengenbach MS (2007). Conservative Management of Sports Injuries. Jones & Bartlett Learning. pp. 1100–.
“Androstanolone Drug Profile”. Adis Insight. 4 December 2006.
Elks J (14 November 2014). The Dictionary of Drugs: Chemical Data: Chemical Data, Structures and Bibliographies. Springer. pp. 640–.
Index Nominum 2000: International Drug Directory. Taylor & Francis. January 2000. pp. 63–.
List PH, Hörhammer L (12 March 2013). Chemikalien und Drogen: Teil B: R, S. Springer-Verlag. pp. 523–.
“Drugs@FDA: FDA Approved Drug Products”. United States Food and Drug Administration. Retrieved 16 November 2016.
“Drug Product Database – Health Canada”. Health Canada. Retrieved 13 November 2016.
Blume-Peytavi U, Whiting DA, Trüeb RM (26 June 2008). Hair Growth and Disorders. Springer Science & Business Media. pp. 161–162.
Azzouni F, Mohler J (2012). “Role of 5α-reductase inhibitors in prostate cancer prevention and treatment”. Urology. 79 (6): 1197–205.
Lotti F, Maggi M (28 April 2015). “Hormonal Treatment for Skin Androgen-Related Disorders”. In Katsambas A, Lotti T, Dessinioti C, D’Erme AM (eds.). European Handbook of Dermatological Treatments. Springer.
Wendowski, Oskar; Redshaw, Zoe; Mutungi, Gabriel (February 2017). “Dihydrotestosterone treatment rescues the decline in protein synthesis as a result of sarcopenia in isolated mouse skeletal muscle fibres”. Journal of Cachexia, Sarcopenia and Muscle. 8 (1): 48–56.
Kohtz AS, Frye CA (2012). Dissociating behavioral, autonomic, and neuroendocrine effects of androgen steroids in animal models. Methods Mol. Biol. Methods in Molecular Biology. 829. pp. 397–431.
Llewellyn W (2009). Anabolics. Molecular Nutrition Llc. pp. 19, 163.
Okeigwe I, Kuohung W (2014). “5-Alpha reductase deficiency: a 40-year retrospective review”. Curr Opin Endocrinol Diabetes Obes. 21 (6): 483–7.
Heesakkers J, Chapple C, Ridder DD, Farag F (24 February 2016). Practical Functional Urology. Springer. pp. 280–.
Imperato-McGinley J, Zhu YS (2002). “Androgens and male physiology the syndrome of 5alpha-reductase-2 deficiency”. Mol. Cell. Endocrinol. 198 (1–2): 51–9.
Coutts, S. B.; Kicman, A. T.; Hurst, D. T.; Cowan, D. A. (1997-11-01). “Intramuscular administration of 5α-dihydrotestosterone heptanoate: changes in urinary hormone profile”. Clinical Chemistry. 43 (11): 2091–2098.
Imperato-McGinley J, Peterson RE, Gautier T, Sturla E (1979). “Androgens and the evolution of male-gender identity among male pseudohermaphrodites with 5alpha-reductase deficiency”. N. Engl. J. Med. 300 (22): 1233–7.
Kang HJ, Imperato-McGinley J, Zhu YS, Rosenwaks Z (2014). “The effect of 5α-reductase-2 deficiency on human fertility”. Fertil. Steril. 101 (2): 310–6.
Katz MD, Cai LQ, Zhu YS, Herrera C, DeFillo-Ricart M, Shackleton CH, Imperato-McGinley J (1995). “The biochemical and phenotypic characterization of females homozygous for 5 alpha-reductase-2 deficiency”. J. Clin. Endocrinol. Metab. 80 (11): 3160–7.
Cilotti A, Danza G, Serio M (2001). “Clinical application of 5alpha-reductase inhibitors”. J. Endocrinol. Invest. 24 (3): 199–203.
Bradbury R (30 January 2007). Cancer. Springer Science & Business Media. pp. 49–.Burchum J, Rosenthal L (2 December 2014). Lehne’s Pharmacology for Nursing Care. Elsevier Health Sciences. pp. 803–.
Bostwick DG, Cheng L (24 January 2014). Urologic Surgical Pathology. Elsevier Health Sciences. pp. 492–.
Sun J, Xiang H, Yang LL, Chen JB (2011). “A review on steroidal 5α-reductase inhibitors for treatment of benign prostatic hyperplasia”. Curr. Med. Chem. 18 (23): 3576–89.
Torres F (2015). Androgenetic, diffuse and senescent alopecia in men: practical evaluation and management. Curr. Probl. Dermatol. Current Problems in Dermatology. 47. pp. 33–44.
Check JH, Cohen R (2015). “An update on the treatment of female alopecia and the introduction of a potential novel therapy”. Clin Exp Obstet Gynecol. 42 (4): 411–5.
Blume-Peytavi U, Whiting DA, Trüeb RM (26 June 2008). Hair Growth and Disorders. Springer Science & Business Media. pp. 182, 369.
Check JH, Cohen R (2015). “An update on the treatment of female alopecia and the introduction of a potential novel therapy”. Clin Exp Obstet Gynecol. 42 (4): 411–5.
Blume-Peytavi U, Whiting DA, Trüeb RM (26 June 2008). Hair Growth and Disorders. Springer Science & Business Media. pp. 182, 369.