Se non avete letto ancora la prima e la seconda parte di questa serie di articoli vi invito a farlo: 1° Parte – 2° Parte.
- Cromo: caratteristiche e possibili applicazioni.

Il Cromo è un minerale essenziale nella dieta umana ed è comunemente utilizzato come integratore alimentare (es. Picolinato o Polinicotinato) per migliorare la sensibilità all’insulina nei soggetti sani o nei soggetti diabetici. (1)
Il Cromo può essere trovato nel:
- Colostro bovino (sotto forma di un oligopeptide di cromodulina ricco di zinco, con un atomo di Cromo per quattro amminoacidi (2) (3)) che fornisce 220mcg di Cromo per 1.035g di proteine (193ng/g di proteine) (2)
• Latte scremato, ad una concentrazione di 252mcg di Cromo per 1.172g di proteine (215ng/g di proteine) (2)
Il Cromo è sia un minerale dietetico che un elemento (Cr) con più valenze. La forma completamente ossidata di Cromo (Cr (VI)), che è esavalente (+6 stato di ossidazione), è altamente tossica e impiegata in una varietà di applicazioni industriali.(4) Dato l’alto grado di tossicità, il Cromo esavalente non viene mai usato come integratore. Le forme supplementari di Cromo comprendono il bivalente (Cr (II)) o il trivalente (Cr (III)), quest’ultima è la forma più stabile.(1)
Il quantitativo di Cromo assunto con la dieta dovrebbe essere almeno di 0,005-0,2mg(5-20mcg) al giorno al fine di prevenirne il deficit, e l’assunzione giornaliera raccomandata è di 21-25mcg per le donne e di 25-35mcg per gli uomini con la fascia di età tra i 18 ed i 45 anni che richiede quantità verso il punto più alto dell’intervallo riportato.(5) Le donne di tutte le età che stanno allattando richiedono un’assunzione giornaliera di Cromo pari a 45mcg.(5) La dose raccomandata per i bambini da 1 a 3 anni è 11mcg/die mentre dai 4 agli 8 anni il dosaggio sale a 15mcg/die.(5)
Le concentrazioni standard di Cromo circolante in uno stato non carente sono state misurate nell’intervallo di 2,8-45mcg/L nel sangue intero e 0,12-2,1mcg/L nel siero.(6)
Una carenza di Cromo può essere indotta con una nutrizione parenterale totale a lungo termine (TPN) priva del minerale, e può essere invertita con una supplementazione di 150mcg di Cromo al giorno aggiunti al TPN come riscontrato attraverso un caso studio (7) e 250mcg al giorno per 2 settimane seguite da una dose di mantenimento pari a 20μg al giorno per 18 mesi in un altro.(8) I principali sintomi da carenza di Cromo in questi particolari casi si manifestavano attraverso un compromessa tolleranza al glucosio e una riduzione dell’insulino-sensibilità associata alla perdita di peso, così come la neuropatia e l’encefalopatia che erano reversibili con il reintegro del minerale. (8)(7)
Quindi, una grave carenza di Cromo è associata a sintomi simili a quelli riscontrati nel diabete di tipo I (alterata tolleranza al glucosio e perdita di peso) e nella neuropatia, e può essere invertita con la somministrazione del minerale.
Le carenze subcliniche di Cromo sono associate all’insulino resistenza, poiché le concentrazioni di questo minerale sono risultate inferiori nei diabetici rispetto ai soggetti di controllo (9) (tuttavia, l’evidenza è eterogenea per il diabete gestazionale (10)(11)). Le diete con un assunzione cronica di zuccheri (35% delle calorie giornaliere) sono state associate ad una accelerata perdita di Cromo attraverso le urine (Cromo urinario) (12) sebbene le diete composte da cibi ad alto indice glicemico non abbiano influenzato in modo significativo l’eliminazione del Cromo attraverso le urine in soggetti sani, pur mostrando una tendenza nell’arco di sei giorni.(13)

Si ritiene che questa perdita accelerata di Cromo attraverso le urine si verifichi per via del rilascio di Cromodulina (LMWCr; Low-molecular-weight chromium-binding substance) nel flusso ematico da parte delle cellule insulino-sensibili, con conseguente eliminazione urinaria.(14) La Cromodulina è un peptide che esiste all’interno delle cellule. Quando combinato con il Cromo immesso nelle cellule dal flusso sanguigno, amplifica la segnalazione dell’insulina legandosi ai recettori insulinici stimolati dall’ormone.(14) La Cromodulina lega lo ione cromo ad altissima affinità, formando un complesso che può essere separato solo in condizioni non fisiologiche. Una volta che i livelli di insulina scendono, tuttavia, i recettori dell’insulina non hanno più bisogno di essere sensibilizzati, quindi l’intero complesso deve essere eliminato nel suo insieme.(14) (15)Questa ipotesi è supportata dal rilevamento della Cromodulina nelle urine (16) e dalla sua stretta correlazione con i tassi di secrezione dell’Insulina e l’esposizione in condizioni non complementari.(16) (17) (18)
Le concentrazioni urinarie di Cromo risultano elevate in seguito ad allenamenti di resistenza (con un aumento di cinque volte dopo due ore di corsa, ma con solo un aumento di due volte nel corso delle ventiquattro ore) in un modo che non è correlato ad un aumento dell’insulina serica o ad un aumento di qualsiasi altro ione urinario.(18) Questa condizione, nonostante l’assenza di significativi livelli di Insulina, è nota per richiedere un maggiore assorbimento di glucosio nel tessuto muscolare sostenuto da un maggiore rilascio di glucosio da parte del fegato.(19)
Il Cromo trivalente (che si trova negli integratori) sembra avere effetti tossici a concentrazioni superiori a 20mcg/mL nel siero o nelle cellule; questa tossicità è associata al danno ossidativo al DNA.(20) Questo è lo stesso meccanismo mediante il quale il cromo esavalente esprime la sua tossicità, con l’unica differenza che quest’ultimo è tossico a concentrazioni molto più basse (21), in particolare dopo inalazione durante un impiego che comporta la sua manipolazione. (22) (4)
Con il termine Cromo Picolinato ci si riferisce al Cromo nello stato trivalente (Cr (III)) il quale è legato a tre molecole di acido picolinico, un analogo strutturale della Niacina. Questa forma di Cromo è altamente stabile (23), a parte una possibile degradazione indotta dall’acido, che rimuove una molecola di picolinato e porta a due ioni di cromo che si legano insieme. (24) I ligandi picolinati sono in una posizione tale che il Cr (III) può essere ridotto in Cr (II) nella coltura cellulare senza perdere il picolinato (25), una proprietà che sembra essere unica per il picolinato rispetto ad altre forme supplementari (Cloruro e Nicotinato) e si pensa che sia alla base delle possibili proprietà cancerogene indotte da alte concentrazioni.(26)
Si ritiene che il Cromo Picolinato sia fisiologicamente inattivo fino ad avvenuta liberazione della molecola di Cromo (26), suggerendo che esso funga da “pro farmaco” al Cromo.
Il Cromo è noto per essere presente nel lievito, dove svolge un ruolo fisiologico importante.(27) (28) All’interno delle cellule del lievito si trova il “Fattore di Tolleranza al Glucosio” (GTF) (29), che è stato inizialmente derivato dal lievito di birra.(30) Il GTF può essere purificato dai lieviti dopo l’estrazione metanolica e la successiva filtrazione, ottenendo un insieme di molecole di dimensioni variabili da 1.000 a 3.500 Da. (31)(32) I principali componenti attivi in questo set di molecole sono considerati l’acido trivalente al cromo nicotinico insieme ad alcuni aminoacidi (Glicina, L-cisteina e Acido Glutammico).(33) L’apporto alimentare del lievito sembra conferire alcuni dei benefici dati dall’integrazione di Cromo, probabilmente a causa dell’ingestione di GTF e Cromo.(29)
Si ritiene che il Cromo presente nel lievito sia acido cromo-nicotinico, sebbene possano esistere altre forme di Cromo nel lievito che non sono state ancora rilevate.

L’Acido Nicotinico di Cromo (noto anche come Cromo Polinicotinato), forma altamente assimilabile di Cromo, è composto da Cromo legato all’Acido Nicotinico (Niacina o Vitamina B3) e si dice che abbia effetti sulla riduzione del Colesterolo.(34)(35) Negli studi in cui il Colesterolo è stato ridotto in seguito all’assunzione di Cromo Polinicotinato, non è stato trovano necessariamente un nesso benefico dato dal miglioramento del metabolismo glucidico (34)(35), suggerendo che è la Niacina a causare questi effetti.
Il Cromo Dinicocisteinato (CDNC) è un complesso dello ione Cromo con l’aminoacido L-cisteina. Uno studio che ha confrontato l’effetto di 400mcg di CDNC con 400mcg di Cromo Picolinato ha rilevato miglioramenti nei livelli di Insulina e della sensibilità a questa solo con il CDNC. (36)
Uno dei principali meccanismi che si ritiene correlato all’integrazione con Cromo comporta la modulazione della via di segnalazione dell’Insulina.(37) [38] Questo è stato scoperto per la prima volta quando è stato identificato un oligopeptide legante il Cromo a basso peso molecolare che ha aumentato gli effetti dell’Insulina e l’ossidazione del glucosio. (38)[39] Chiamato anche LMCr o Cromodulina (39), questo oligopeptide viene sintetizzato nel fegato dei ratti dopo iniezioni di Cromo (40) e ha una massa di circa 1500 kDa. (39)(41)

È stato rilevato che la Cromodulina ha aumentato la segnalazione di Insulina in presenza di un livello di quest’ultima pari a 5-8 volte superiore rispetto all’attività basale, senza influenzarne la segnalazione in assenza di Insulina.(42) La deplezione di Cromo da parte della Cromodulina ne blocca l’attività (42) che si correla positivamente con il contenuto del minerale nel peptide. Inoltre, altri minerali non sono riusciti a replicarne gli effetti. (43)

In definitiva, la Cromodulina aumenta l’autofosforilazione del Recettore dell’Insulina. La segnalazione del Recettore dell’Insulina richiede che l’Insulina o un mimetico (qualcosa che si comporti come l’Insulina) si leghi alla subunità α extracellulare del recettore (44) che consente alla subunità β intracellulare di essere autofosforilata. (44) La Cromodulina sembra agire intracellularmente nella subunità β del Recettore dell’Insulina.(14)
Le funzioni cromo-dipendenti della Cromodulina sono probabilmente la ragione biologica per la quale il Cromo è un minerale essenziale (45), sebbene la natura essenziale del Cromo sia stata recentemente contestata.(46)
L’Adenosina Monofosfato Chinasi (AMPK) è un sensore chiave dello stato energetico cellulare, il quale monitora costantemente i livelli di ATP al fine di mantenere l’omeostasi metabolica. L’AMPK si attiva durante gli stati di carenza energetica (caratterizzato da un aumento della AMP:ATP ratio) dove coordina il metabolismo degli acidi grassi e del glucosio in modo anti-obesità e anti-diabetico.(47) Quando attivato, l’AMPK sopprime le vie anaboliche come la sintesi proteica, di trigliceridi e di acidi grassi attivando contemporaneamente percorsi catabolici come la glicolisi e l’ossidazione degli acidi grassi per aumentare la produzione di ATP. (48)
È stato notato che il Cromo (trivalente con D-fenilalanina) attiva l’AMPK nel suo sito catalitico (Thr172) nei cardiomiociti e nelle cellule muscolo-scheletriche a 25μM, suggerendo che i complessi organici del Cromo possono essere nuovi attivatori della via dell’AMPK.(49)
L’assorbimento del Cromo alimentare è inversamente correlato all’assunzione, variando dallo 0,4% al 2,0%, con l’assorbimento più efficiente (2%) a un apporto dietetico inferiore di circa 10mcg negli uomini adulti.(50) Questo diminuisce a circa lo 0,5% quando l’assunzione con il cibo raggiunge i 40mcg che sembra essere il limite, dato che l’assunzione di Cromo nel range di 40-240mcg ha un assorbimento di circa lo 0,4%. (50) (51)

L’assorbimento del Cromo è influenzato da una serie di fattori dietetici. Nei ratti, l’assorbimento del Cromo sembra essere ostacolato dalla coingestione dei fitati, che impedisce il trasporto e l’assorbimento attraverso l’intestino. (52) È stato dimostrato che il deficit di Zinco aumenta l’assorbimento del Cromo, che è aumentato nei ratti carenti di Zinco e ridotto dallo Zinco supplementare (53), suggerendo che questi due minerali possono competere per l’assorbimento. L’assorbimento del Cromo nei ratti è anche potenziato dall’ossalato, un acido organico presente in molte verdure e cereali. (52) Sebbene sia informativo, occorre prestare attenzione quando si estrapolano i risultati dagli studi sui ratti rapportandoli all’uomo, poiché studi recenti hanno rilevato che l’assorbimento di Cromo alimentare nell’uomo è significativamente maggiore rispetto a quanto avviene nei ratti per numerosi complessi di cromo testati.(54)[55]

Gli amminoacidi sembrano migliorare l’assorbimento del Cromo alimentare poiché formano complessi che migliorano l’assorbimento riducendo la tendenza del Cromo a precipitare nel liquido intestinale alcalino.(1) L’assorbimento del Cromo negli esseri umani è anche significativamente aumentato in presenza di Acido Ascorbico e Acido Nicotinico.(1)
Nei diabetici di tipo II, un integrazione giornaliera di Cromo (come cromo Picolinato) pari a 1.000mcg è risultata sufficiente a portare i livelli del minerale a digiuno nel siero da 2,40 ± 0,19 vs 0,16 ± 0,05ng/dL al basale dopo 12 settimane e 2,62 ± 0,09ng/ dL vs 0,17 +/- 0,04ng /dL al basale dopo l’integrazione di 24 settimane.(55)

La transferrina è una proteina di trasporto del siero nota per legarsi ai minerali (in particolare il Ferro). È stato notato che presenta affinità per il Cromo trivalente.(56) Per ogni molecola di transferrina si legano due ioni di cromo. (57)(58) Si pensa che la transferrina doni il Cromo all’oligopeptide Cromodulina.(59) Anche se studi precedenti hanno suggerito che la Cromodulina dona il Cromo alla transferrina. Questo lavoro però è stato condotto a temperature più elevate, che potrebbero aver causato la degradazione della Cromodulina.(60) Tuttavia, studi più recenti hanno dimostrato che la Cromodulina non rilascia il Cromo alla transferrina. (59) Poiché la transferrina rilascia ioni all’interno di una cellula dopo l’endocitosi (61), sembra che la Cromodulina accetti e trattenga questi ioni dalla transferrina.
La supplementazione con Cromo determina un aumento dell’eliminazione urinaria del minerale.(55)

Uno studio svolto su ratti ha osservato che i livelli tossici di Cromo (100mcg/kg assunto con il cibo) sembrano bioaccumularsi di più con il Cromo Cloruro rispetto al Cromo Picolinato, in parte dipendente da un più alto tasso di escrezione osservato con il Picolinato.(62) Ciò è stato ipotizzato essere dovuto all’Acido Picolinico, che è stato osservato aumentare l’eliminazione di minerali come lo Zinco.(63)
Diversi studi hanno suggerito che la supplementazione con Cromo può promuovere una riduzione dell’appetito, con conseguente diminuzione del consumo di cibo, sia negli animali che negli esseri umani. Una recente meta-analisi di 10 studi randomizzati, in doppio cieco, controllati con placebo ha concluso che il Cromo Picolinato ha un effetto sulla riduzione del peso relativamente modesto, ma significativo rispetto al placebo (64), suggerendo un possibile effetto sulla soppressione dell’appetito. I meccanismi associati all’effetto anoressizzante dato dall’uso del Cromo sono attualmente sconosciuti, sebbene sia stato ipotizzato che si verifichi attraverso l’azione di specifici neurotrasmettitori nel cervello deputati al controllano l’appetito e il comportamento alimentare. (65)(66)
Ciò è stato confermato in un recente studio condotto su donne in sovrappeso adulte che hanno riportato voglie di carboidrati intense (almeno due volte a settimana). La supplementazione giornaliera con 1.000mcg di Cromo (come Picolinato) nel corso di otto settimane ha comportato una maggiore riduzione dell’assunzione di cibo (25%) rispetto al placebo (8%).(67) La riduzione dell’assunzione di cibo era associata ad una diminuzione della fame e dell’appetito, tuttavia la composizione dei macronutrienti non era influenzata e questi cambiamenti erano indipendenti da qualsiasi effetto sulla sensibilità all’insulina.(67) In uno studio parallelo condotto dallo stesso gruppo di ricerca, è stato riscontrato che la somministrazione periferica di Cromo nei ratti (tramite iniezione IP) ha comportato solo una modesta diminuzione dell’assunzione di cibo, rispetto a una significativa riduzione dose-dipendente dell’assunzione di cibo quando somministrato a livello centrale (direttamente nel cervello). (67) Nel suo insieme, questo lavoro suggerisce che, come detto pocanzi, la supplementazione con Cromo può promuovere una riduzione dell’apporto di cibo attraverso l’azione di neurotrasmettitori nel cervello che controllano l’appetito e il comportamento alimentare.
Nei pazienti con depressione atipica (che è un particolare sottogruppo di depressione associato a maggiore assunzione di cibo, sonnolenza e reattività dell’umore (68)), 600mcg di Cromo Picolinato per otto settimane non hanno influenzato significativamente la maggior parte dei sintomi depressivi. Tuttavia, ci sono stati significativi miglioramenti nella voglia di carboidrati e nell’assunzione di cibo con un effetto maggiore in coloro i quali il desiderio di carboidrati era maggiore al basale. (66) Nelle persone con disturbo da alimentazione incontrollata, il tasso di declino della frequenza di binging era maggiore con 1.000mcg di Cromo rispetto al placebo e 600mcg, sebbene la riduzione complessiva non abbia raggiunto la significatività statistica.(69)
La supplementazione con 1.000mcg di Cromo (come Picolinato) in due dosi suddivise per 24 settimane nei diabetici di tipo II non ha influenzato significativamente la gluconeogenesi epatica rispetto al placebo. (55) (La gluconeogenesi epatica è spesso patologicamente elevata nei diabetici (70)).
Quando i diabetici consumavano 200mcg di Cromo (come Cloruro) al giorno aggiunto a un prodotto di latte in polvere per 16 settimane, i livelli di glucosio e di insulina nel sangue erano significativamente ridotti mentre la sensibilità all’insulina migliorava. (71) I risultati di questo studio erano tuttavia specifici per genere, in quanto miglioramenti significativi nei suddetti marker dell’omeostasi del glucosio si sono verificati solo in soggetti di sesso maschile. (71)
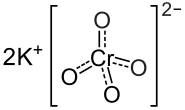
Come detto in precedenza, la Cromodulina è un oligopeptide endogeno (41) contenente Cromo che media positivamente la segnalazione del recettore dell’insulina in presenza di Insulina.(40) L’iniezione di Cromo (come Cromato di Potassio) nei ratti aumenta le concentrazioni urinarie e fecali di questo oligopeptide. (72) Tuttavia, la Cromodulina urinaria non sembra essere saturata in condizioni basali, il che implica che più Cromo potrebbe essere legato all’oligopeptide.(72) Poiché la potenza della Cromodulina nel potenziare la segnalazione dell’Insulina è correlata con la quantità di Cromo legata ad esso (43), e le iniezioni di Cromato di Potassio nei ratti determinano una rapida associazione con la Cromodulina (60)(73), è possibile che l’aumento di Cromo alimentare possa aumentare l’attività di questo oligopeptide.
Operando partendo dal presupposto che le assunzioni tipiche di Cromo nella dieta sono insufficienti per saturare la Cromodulina, la supplementazione con Cromo potrebbe teoricamente migliorare la segnalazione dell’Insulina tramite l’aumento del legame cromo-cromodulina.
Negli studi nei quali è stato utilizzato il Cromo trivalente, sembra esserci un aumento dell’attività della chinasi del recettore insulinico (in presenza di Insulina) quando il cromo 1-10μM viene aggiunto alla coltura di cellule di mammifero.(74) Questo aumento è indipendente da qualsiasi influenza diretta sulla fosforilazione o autofosforilazione (74) e distinta da quella della Cromodulina, che influenza l’autofosforilazione. (42)

Alcuni complessi con Cromo trivalente hanno interazioni minori con il Recettore dell’Insulina, con complessi legati a piccole molecole endogene come Istidinato, Lattato, Acetato o Propionato che mostrano effetti inibitori minori a concentrazioni intorno a 100μM. Di questi complessi, il Cromo Propionato sembra essere il più potente, mostrando effetti inibitori a concentrazioni fino a 1μM. (75)
Lo stesso Cromo è stato implicato nel potenziare la segnalazione dell’Insulina, sebbene il meccanismo con gli ioni Cromo sembra differire da quello osservato con la Cromodulina e richiede una concentrazione significativamente più alta. Lo stesso Cromo non sembra influenzare direttamente il Recettore dell’Insulina come la lattina di Cromodulina.
La Fosfo-tirosin–fosfatasi 1B (PTP1B) è un regolatore negativo del segnale del Recettore dell’Insulina (76) che può essere soppresso dal Cromo endogeno. Anche se la Cromodulina è stata osservata promuovere l’attività della PTP della membrana in uno studio precedente (77), ci sono molti enzimi PTP endogeni e il PTP1B non è stato specificamente esaminato in questo studio. È stato dimostrato che il Cromo trivalente inibisce il PTP1B del 21-33% nelle cellule di epatoma umano e di ratto (78), suggerendo che il Cromo può potenziare la segnalazione dell’Insulina sopprimendo la defosforilazione mediata dal PTB1B nel Recettore dell’Insulina. Al contrario, uno studio più recente ha osservato che il Cromo non è riuscito a inibire l’attività della fosfatasi PTP1B umana ricombinante in un sistema in vitro puro, suggerendo che il Cromo può potenziare la segnalazione dell’Insulina da meccanismi distinti da qualsiasi effetto sul PTP1B. (74)
In uno studio in vivo, ratti obesi diabetici trattati con 80mcg/kg di Cromo (come Picolinato) hanno subito una diminuzione complessiva dell’attività della PTP1B e dell’espressione proteica correlata ad un aumento della segnalazione dell’Insulina nel muscolo scheletrico.(79) Questa diminuzione non è stata osservata nei ratti magri ai quali è stato somministrato il Cromo alla stessa dose.(79)

La fosforilazione dell’IRS-1, un importante trasduttore della segnalazione dell’Insulina che è inibito dalla fosforilazione a Serine307 (80), non è influenzato dal Cromo a 10μM in varie forme trivalenti.(75) Inoltre, l’espressione della proteina IRS è rimasta inalterata con una supplementazione di Cromo fino a 80mcg /kg nei ratti.(79) In assenza di Insulina, tuttavia, la segnalazione basale del IRS-1 è leggermente aumentata a 10 μM di Cromo, che si pensa sia dovuta alla diminuzione della fosforilazione del Serine307 (75) dal Jun NH (2) -terminal kinase (JNK). (37) Il JNK regola negativamente la segnalazione del IRS tramite fosforilazione a Serine307 (80) (81), che è aumentata nei topi obesi, (82) (83) causando insulino-resistenza. In particolare, l’attenuazione mediata dal JNK della segnalazione dell’Insulina nei ratti obesi è soppressa dal Cromo. (82)(83)
L’attivazione del JNK sopra riportata potrebbe essere ricondotta teoricamente allo stress del reticolo endoplasmatico (ER) (84), e gli agenti che riducono lo stress del ER attenuano anche i sintomi diabetici. (85)(86) È noto che lo stress del ER aumenta nelle cellule degli animali obesi e diabetici ed è curabile con il Cromo.(82)
Per riassumere i concetti esposti, sappiamo che l’interazioni del Cromo con il PTP1B, un regolatore negativo dell’attività del Recettore dell’Insulina, non sono ben compresi. Alcuni studi suggeriscono che il Cromo potrebbe non avere effetti apprezzabili sulla segnalazione del PTP1B. È possibile, tuttavia, che il Cromo sopprima l’attenuazione JNK-mediata della segnalazione dell’Insulina nel contesto di uno stato di insulino-resistenza preesistente.
Il Cromo non sembra aumentare l’espressione del Recettore dell’Insulina in presenza o assenza di Insulina, suggerendo che i suoi effetti sulla segnalazione dell’Insulina avvengono indipendentemente da eventuali cambiamenti nei livelli dei recettori insulinici. (87) (75) Inoltre, quando incubato con Insulina, il Cromo non influenza l’interazione dell’Insulina con il suo recettore.(74) Ciò suggerisce che il Cromo non influisce sulla sensibilità all’Insulina aumentando l’affinità del Recettore dell’Insulina.
Uno studio preliminare condotto nel 1992 ha rivelato che il Cromo aumenta l’internalizzazione dell’Insulina a 1μM (418ng/ml), effetto associato ad una maggiore fluidità della membrana e non replicato con altre chelazioni di Cromo o Zinco Picolinato. (88) La scoperta che l’Insulina è internalizzata nella cellula è stata successivamente rivelata come un importante meccanismo di feedback negativo per la segnalazione del Recettore dell’Insulina. Dopo che l’Insulina si lega con il suo recettore, il complesso del recettore insulinico viene internalizzato dall’endocitosi (89), innescando la degradazione dell’Insulina (90) e riducendo efficacemente il numero di recettori dell’insulina presenti sulla superficie cellulare come meccanismo per attenuare la risposta insulinica.(91)
In breve, dopo avvenuto legame con il suo recettore sulla superficie della cellula, l’Insulina innesca il movimento del complesso del Recettore dell’Insulina all’interno della cellula. Questo riduce il numero di recettori insulinici presenti sulla superficie cellulare e funziona come un meccanismo di feedback negativo per limitare la risposta della segnalazione insulinica.
In risposta a un test orale di tolleranza al glucosio, una supplementazione di 200mcg di Cromo per otto settimane non ha aumentato la risposta all’Insulina in soggetti diabetici di tipo II quando misurata dopo 10 minuti (71) mentre ad un dosaggio di 1.000mcg (come Picolinato) in soggetti non diabetici con sindrome metabolica per oltre 16 settimane ha aumentato la risposta all’Insulina nonostante non sia stato rilevato altro cambiamento nei biomarcatori del diabete. (92)
Uno studio ha osservato che, nonostante l’incapacità di trovare miglioramenti statisticamente significativi nella sensibilità all’Insulina per l’intero gruppo di soggetti presi in esame, il 46% degli individui che avevano un grado di insulino resistenza più elevato presentavano un miglioramento della sensibilità all’Insulina del 10%. (55) In particolare, non vi era alcuna differenza nell’assorbimento o cinetica del Cromo tra responder e non responder (55), suggerendo che la supplementazione con Cromo può aumentare la sensibilità all’Insulina in soggetti con insulino resistenza.
Inoltre, è stato osservato che una supplementazione di 1.000mcg di Cromo (come Picolinato) per 24 settimane in soggetti con diabete di tipo II riduce leggermente le concentrazioni di lipidi intramuscolari rispetto al placebo. (55) Poiché l’accumulo cronico di lipidi nel tessuto muscolare è una delle numerose cause patologiche dell’insulino-resistenza (93), anche questo lavoro suggerisce che la supplementazione con Cromo può aumentare la sensibilità all’Insulina in coloro che sono già insulino-resistenti.

Una meta-analisi di studi condotti su diabetici di tipo II trattati con >250mcg di Cromo per un periodo superiore ai tre mesi non ha rilevato alcuna influenza sul HbA1c rispetto al trattamento con placebo. (94) Ciò è in contrasto con precedenti revisioni che valutato solamente studi condotti su diabetici con un HbA1c basale superiore al 7%, in cui la supplementazione con Cromo ha determinato una riduzione dell’HbA1c dello 0,34% rispetto al placebo. (95) Altre revisioni hanno rilevato riduzioni dello 0,6% (96), e fino allo 0,9% quando sono state incluse tutte le forme di diabete e gradi di insulino resistenza.(97) Va notato, tuttavia, che alcune di queste analisi comprendevano prove della durata inferiore a tre mesi (96), che potrebbero non essere sufficienti per misurare i cambiamenti nel HbA1c. (94)
A seconda della popolazione studiata e del tipo e della qualità degli studi osservati, è dimostrato che il Cromo influisce in modo eterogeneo sui livelli di emoglobina A1C.
La supplementazione con 400 o 800mcg di Cromo (come Picolinato) insieme a un pasto di prova in adulti sani ha ridotto l’area del glucosio sotto la curva (AUC) del 30-36% nei responder, con la dose bassa più efficace.(97) In particolare, i responder sono stati classificati come soggetti aventi un consumo di carne e latte relativamente più basso (97), suggerendo che il Cromo può influenzare il metabolismo del glucosio postprandiale negli individui con livelli di Cromo basali inferiori. La riduzione del glucosio non è stata associata ad alcun cambiamento nell’Insulina, escludendo un effetto insulinogeno, e si è verificata in persone senza un metabolismo del glucosio alterato.(97)
Nella meta-analisi dove sono stati vagliati gli studi che valutavano la supplementazione di cromo oltre ai 250mcg nei diabetici di tipo II per un periodo di tre mesi (o più lungo)(94), i sette studi inclusi nella meta-analisi (55)(98)(99)(100)(101)(102)(103) non hanno mostrato una riduzione dei livelli di HbA1c nel siero nonostante una lieve riduzione della glicemia (RR di -0,95 e un IC 95% da -1,4 a -0,5).(94)
Un’analisi dei dati osservazionali del National Health and Nutrition Examination Survey (NHANES) ha rilevato che le persone che hanno consumato un integratore alimentare contenente Cromo avevano una probabilità inferiore di sviluppare il diabete (OR = 0,73), definito avendo un livello di HbA1c superiore a 6,5. L’uso di integratori in generale non ha avuto un effetto statisticamente significativo sulle probabilità di sviluppare diabete in questo studio.(104)
L’aggiunta di 400mcg di Cromo (come Picolinato) a una bevanda contenente carboidrati prima di un shuttle run test in uomini sani e attivi non ha modificato i benefici della bevanda contenente carboidrati rispetto al controllo, suggerendo che non vi è alcun beneficio aggiuntivo. (105)
Uno studio nel quale è stata somministrata una dose di 600mcg di Cromo (come Picolinato) ogni giorno per un mese prima di un esercizio di deplezione del glicogeno ha rilevato che immediatamente dopo l’esercizio e nell’ora successiva il gruppo trattato aveva livelli di lattato significativamente più alti rispetto al placebo. (106) In un altro studio che utilizzava un modello shuttle-run exercise, questo aumento di lattato non si è verificato con oltre 75 minuti di test a seguito del consumo di 400mcg di Cromo Picolinato o carboidrati o acqua (gruppo di controllo). (105) Inoltre, la concentrazione di lattato e il grado di fatica era simile in questo studio tra i due gruppi e il controllo. (105)

Il Glicogeno Sintasi è l’enzima responsabile della conversione del glucosio in glicogeno, la forma di deposito dei carboidrati nel corpo. Allo stesso modo, la fosforilasi di glicogeno è coinvolta nello scomporre queste riserve di carboidrati in glucosio per produrre energia. A causa dei suoi effetti sul metabolismo del glucosio, il Cromo è stato studiato per il suo impatto sulle riserve di glicogeno. Prove preliminari hanno rivelato che i ratti supplementati con Cromo avevano una minore dispersione del glicogeno epatico rispetto al gruppo di controllo durante il digiuno. (107) Successivamente, si è notato che il Cromo aumentava l’attività dell’enzima glicogeno sintasi nel muscolo e nel fegato dei ratti addestrati rispetto al gruppo di controllo non supplementato, ma la glicogeno fosforilasi non era influenzata.(108)
Negli adulti sovrappeso e leggermente allenati o sedentari la somministrazione di 600mcg di Cromo (come Picolinato) per un mese insieme ad una dieta standardizzata con gli ultimi due giorni progettati per esaurire il glicogeno, la supplementazione non ha modificato i livelli di glicogeno o il tasso di risintesi (da un carboidrato contenuto in una bevande) rispetto al placebo.(106)
Secondo la meta-analisi sul peso nei diabetici di tipo II supplementati con Cromo (oltre 250mcg) per oltre tre mesi, non vi è stata alcuna alterazione significativa del peso rispetto al placebo, nonostante una modesta riduzione del glucosio nel sangue.(94) Al contrario, un’altra meta-analisi ha rilevato che gli adulti sovrappeso e obesi che hanno integrato con il cromo picolinato hanno ridotto il peso corporeo nell’intervallo di dosaggio di 200-1.000 μg, indipendentemente dallo stato diabetico. La perdita di peso è stata tuttavia molto modesta, per un totale di soli 1,1 kg (IC del 95% nell’intervallo 0,4-1,7 kg).(109) Da notare, quest’ultima meta-analisi ha ritenuto la qualità delle prove non ottimale, mettendo in discussione gli effetti mediati dal cromo sulla perdita di peso.(109)
Uno studio ha rilevato che l’aumento di peso associato alla terapia con Sulfonilurea (0,9 kg su 10 mesi) nei diabetici è stato mitigato dalla cosomministrazione di 1.000mcg di Cromo. (102) È importante sottolineare che questi risultati possono essere limitati a coloro che sono sottoposti a terapia Sulfonilurea. Quando i soggetti diabetici che non erano stati trattati con il medicinale ricevevano istruzioni per seguire una dieta di mantenimento del peso, la supplementazione di 1.000mcg di Cromo Picolinato non modificavano l’assunzione di cibo, l’appetito o il peso corporeo. (55)
Il Cromo è stato anche usato nel tentativo di mitigare l’aumento di peso associato alla cessazione del fumo, poiché le persone che smettono di fumare spesso tendono ad aumentare di peso.(110) Questo studio ha utilizzato l’Hypericum perforatum (900 mg) come primo aiuto anti-fumo e poi ha diviso i soggetti dello studio in gruppi trattati con Cromo o placebo. Sfortunatamente, la tendenza del Cromo ad attenuare l’aumento di peso non ha potuto essere testata con sufficiente potenza, a causa dei bassi tassi di successo con l’erba di cui sopra. Tuttavia, gli effetti del Cromo erano promettenti, con una probabilità di attenuare l’aumento di peso da 5,76 kg a 2,7kg dopo sei mesi.(111)
Il Cromo può anche ridurre la perossidazione lipidica in alcune popolazioni. Sono necessari però ulteriori studi per determinarne la dose appropriata e chi potrebbe trarne reale beneficio.
Come accennato in precedenza, Il Cromo Picolinato, più di altre forme trivalenti di Cromo, ha la capacità di formare proossidanti che possono potenzialmente causare danni al DNA. La rilevanza per la supplementazione orale standard non è nota, poiché la concentrazione richiesta per danneggiare il DNA (livello alto di 50μM) è significativamente più alta di quella osservata nel sangue dopo l’ingestione orale di integratori. Inoltre, gli studi su soggetti umani non hanno notato danni al DNA con dosi supplementari standard (200-400mcg/die).
Il Cromo si accumula nei testicoli dei ratti quando iniettato, anche se i possibili benefici o danni nei testicoli con integrazione orale di Cromo non sono stati studiati. Il Cromo esavalente, la forma tossica non presente negli integratori, è noto per essere tossico per i testicoli.
Esistono diverse altre azioni potenziali legate all’uso del Cromo che sono state scientificamente documentate. Per ovvie ragioni, legate soprattutto all’argomento principale trattato in questa serie di articoli, ho omesso diversi studi di un certo interesse. Per chiunque volesse approfondire cliccare qui.
In seguito alle numerose informazioni riportate nel presente articolo, possiamo con una certa sicurezza concludere che una supplementazione di Cromo risulta maggiormente incisiva in caso di carenza del minerale a causa di una insufficiente assunzione con gli alimenti o in condizioni di insulino resistenza (sia “pre-diabetica” che nella condizione diabetica). Il potenziale anoressizzante del Cromo rappresenta sicuramente un elemento di vantaggio durante una dieta ipocalorico (specie se ipoglucidica). Il Cromo sembrerebbe avere anche una certa azione sul miglioramento dello stoccaggio del glicogeno e sulla sua preservazione, anche se la cosa, in realtà, non è mai stata riscontrata nell’uomo. Se tale azione fosse possibile o significativa, si potrebbe riflettere positivamente a livello prestativo ma che in un contesto di “scarico del glicogeno” potrebbe risultare limitante.
A questo punto la domanda che si ripresenta è “come si possono utilizzare queste informazioni per pianificare l’uso del Cromo”?
- Vista la sua efficacia in soggetti patologici e/o in condizioni di insulino resistenza non patologica, l’uso temporalmente ridotto di 1mg (stand alone) o protratto di 400-600mcg (in combinazione con altri GDA; vedi possibile azione additiva con la Berberina e ALA) di Cromo Picolinato al giorno può portare a dei vantaggi in quei soggetti con una insulino-resistenza di base genetica, cioè individui con una tolleranza glucidica limitata rispetto alla media, o durante regimi ipercalorici (vedi peggioramento dell’insulino-resistenza durante regimi ipercalorici).
- Dosi contenute di Cromo Picolinato (200-400mcg/die) assunte durante periodi ipocalorici possono aiutare il soggetto trattato a tollerare la riduzione calorica per via dell’effetto anoressizzante dato dall’uso di questo composto.
- In combinazione con altri GDA durante e nel periodo successivo (periodo “protocollare” di 4 settimane) all’uso di Insulina esogena.
Alcuni effetti collaterali comuni riscontrati con l’uso del Cromo Picolinato (dose correlato) possono includere insonnia, cambiamenti di umore, irritabilità e mal di testa.
Altri effetti collaterali possibilmente riscontrabili con l’uso di alte dosi di Cromo Picolinato includono problemi di coordinamento o di equilibrio, problemi di concentrazione o difficoltà di pensiero, e sintomi legati a problemi epatici (che comprendono: nausea; mal di stomaco nella zona superiore; prurito; stanchezza; perdita di appetito; urina di colore scuro; ingiallimento della pelle o degli occhi (ittero)).
Chiedere prontamente assistenza medica di emergenza se si verificano segni di anafilassi, una reazione allergica grave che può includere orticaria, difficoltà di respirazione o gonfiore del viso, delle labbra, della lingua o della gola.
Esiste un caso studio di una donna che in seguito all’ingestione di 1200-2400mcg di Cromo (come Picolinato) per 4-5 mesi mostrava sintomi di danno renale. (112) In un altro caso studio, un Bodybuilder aveva sviluppato rabdomiolisi associata all’assunzione di 1.200mcg di Cromo Picolinato per due giorni.(113)
La possibile comparsa di questi effetti avversi può essere evitata con una attenta calibrazione della dose giornaliera di Cromo. Se ne sconsiglia quindi un assunzione superiore a 1mg/die (dose quest’ultima comunque relegabile a periodi d’uso brevi). Una supplementazione giornaliera di 200-400mcg di Cromo Picolinato è generalmente ben tollerata con una punta massima di dosaggio di 600mcg/die.
Per ottenere una migliore biodisponibilità del composto, il Cromo andrebbe assunto lontano dalla somministrazione di integratori di Zinco o con pasti contenenti fonti ricche di fitati (vedi cereali integrali e legumi; il cui contenuto di fitati può comunque essere ridotto con, ad esempio, l’ammollo e la adeguata cottura).
Come detto la volta scorsa per l’Acido Alfa Lipoico, anche con l’uso di Cromo Picolinato (o altra forma) è essenziale la cura del dosaggio e la ponderatezza nell’utilizzo.
Fine 3° Parte…
Gabriel Bellizzi
Riferimenti:
- Lukaski HC. Chromium as a supplement. Annu Rev Nutr. (1999)
- Yamamoto A, Wada O, Suzuki H. Purification and properties of biologically active chromium complex from bovine colostrum. J Nutr. (1988)
- Yamamoto A, Wada O, Suzuki H. Separation of biologically active chromium complex from cow colostrum. Tohoku J Exp Med. (1987)
- Hexavalent Chromium.
- Trumbo P, et al. Dietary reference intakes: vitamin A, vitamin K, arsenic, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. J Am Diet Assoc. (2001)
- Iyengar V, Woittiez J. Trace elements in human clinical specimens: evaluation of literature data to identify reference values. Clin Chem. (1988)
- Freund H, Atamian S, Fischer JE. Chromium deficiency during total parenteral nutrition. JAMA. (1979)
- Jeejeebhoy KN, et al. Chromium deficiency, glucose intolerance, and neuropathy reversed by chromium supplementation, in a patient receiving long-term total parenteral nutrition. Am J Clin Nutr. (1977)
- Davies S, et al. Age-related decreases in chromium levels in 51,665 hair, sweat, and serum samples from 40,872 patients–implications for the prevention of cardiovascular disease and type II diabetes mellitus. Metabolism. (1997)
- Sundararaman PG, et al. Serum chromium levels in gestational diabetes mellitus. Indian J Endocrinol Metab. (2012)
- Woods SE, et al. Serum chromium and gestational diabetes. J Am Board Fam Med. (2008)
- Kozlovsky AS, et al. Effects of diets high in simple sugars on urinary chromium losses. Metabolism. (1986)
- Hajifaraji M, Leeds AR. The effect of high and low glycemic index diets on urinary chromium in healthy individuals: a cross-over study. Arch Iran Med. (2008)
- Vincent JB. The biochemistry of chromium. J Nutr. (2000)
- Davis CM, Vincent JB. Isolation and characterization of a biologically active chromium oligopeptide from bovine liver. Arch Biochem Biophys. (1997)
- Clodfelder BJ, et al. The trail of chromium(III) in vivo from the blood to the urine: the roles of transferrin and chromodulin. J Biol Inorg Chem. (2001)
- Anderson RA, et al. Urinary chromium excretion of human subjects: effects of chromium supplementation and glucose loading. Am J Clin Nutr. (1982)
- Anderson RA, et al. Effect of Exercise (Running) on Serum Glucose, Insulin, Glucagon, and Chromium Excretion. Diabetes. (1982)
- Wahren J, et al. Glucose metabolism during leg exercise in man. J Clin Invest. (1971)
- Bagchi D, et al. Comparative induction of oxidative stress in cultured J774A.1 macrophage cells by chromium picolinate and chromium nicotinate. Res Commun Mol Pathol Pharmacol. (1997)
- Wise SS, Holmes AL, Wise JP Sr. Hexavalent chromium-induced DNA damage and repair mechanisms. Rev Environ Health. (2008)
- Zhang XH, et al. Chronic occupational exposure to hexavalent chromium causes DNA damage in electroplating workers. BMC Public Health. (2011)
- Kingry KF, Royer AC, Vincent JB. Nuclear magnetic resonance studies of chromium(III) pyridinecarboxylate complexes. J Inorg Biochem. (1998)
- Stearns DM, Armstrong WH. Mononuclear and binuclear chromium(III) picolinate complexes. Inorg Chem. (1992)
- Yuen G, Heaster H, Hoggard PE. Amine spectrochemical properties in tris(aminocarboxylate) complexes of chromium(III). Inorg Chim Acta. (1983)
- Speetjens JK, et al. The nutritional supplement chromium(III) tris(picolinate) cleaves DNA. Chem Res Toxicol. (1999)
- Raspor P, et al. The influence of chromium compounds on yeast physiology (a review). Acta Microbiol Immunol Hung. (2000)
- Pas M, et al. Uptake of chromium(III) and chromium(VI) compounds in the yeast cell structure. Biometals. (2004)
- Grant AP, McMullen JK. The effect of brewers yeast containing glucose tolerance factor on the response to treatment in Type 2 diabetics. A short controlled study. Ulster Med J. (1982)
- Schwarz K, Mertz W. A glucose tolerance factor and its differentiation from factor 3. Arch Biochem Biophys. (1957)
- Mirsky N, Weiss A, Dori Z. Chromium in biological systems, I. Some observations on glucose tolerance factor in yeast. J Inorg Biochem. (1980)
- Weksler-Zangen S, et al. Glucose tolerance factor extracted from yeast: oral insulin-mimetic and insulin-potentiating agent: in vivo and in vitro studies. Br J Nutr. (2012)
- Toepfer EW, et al. Preparation of chromium-containing material of glucose tolerance factor activity from brewer’s yeast extracts and by synthesis. J Agric Food Chem. (1976)
- Preuss HG, et al. Effects of niacin-bound chromium and grape seed proanthocyanidin extract on the lipid profile of hypercholesterolemic subjects: a pilot study. J Med. (2000)
- Thomas VL, Gropper SS. Effect of chromium nicotinic acid supplementation on selected cardiovascular disease risk factors. Biol Trace Elem Res. (1996)
- Jain SK, et al. Effect of chromium dinicocysteinate supplementation on circulating levels of insulin, TNF-α, oxidative stress, and insulin resistance in type 2 diabetic subjects: randomized, double-blind, placebo-controlled study. Mol Nutr Food Res. (2012)
- Hua Y, et al. Molecular mechanisms of chromium in alleviating insulin resistance. J Nutr Biochem. (2012)
- Yamamoto A, Wada O, Ono T. Isolation of a biologically active low-molecular-mass chromium compound from rabbit liver. Eur J Biochem. (1987)
- Vincent JB. Quest for the molecular mechanism of chromium action and its relationship to diabetes. Nutr Rev. (2000)
- Yamamoto A, Wada O, Ono T. A low-molecular-weight, chromium-binding substance in mammals. Toxicol Appl Pharmacol. (1981)
- Chen Y, et al. Characterization of the organic component of low-molecular-weight chromium-binding substance and its binding of chromium. J Nutr. (2011)
- Davis CM, Vincent JB. Chromium oligopeptide activates insulin receptor tyrosine kinase activity. Biochemistry. (1997)
- Yamamoto A, Wada O, Manabe S. Evidence that chromium is an essential factor for biological activity of low-molecular-weight, chromium-binding substance. Biochem Biophys Res Commun. (1989)
- Myers MG Jr, White MF. The new elements of insulin signaling. Insulin receptor substrate-1 and proteins with SH2 domains. Diabetes. (1993)
- Vincent JB. Recent advances in the nutritional biochemistry of trivalent chromium. Proc Nutr Soc. (2004)
- Vincent JB. Chromium: celebrating 50 years as an essential element. Dalton Trans. (2010)
- Rutter GA, Da Silva Xavier G, Leclerc I. Roles of 5′-AMP-activated protein kinase (AMPK) in mammalian glucose homoeostasis. Biochem J. (2003)
- Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev. (2011)
- Zhao P, et al. A newly synthetic chromium complex-chromium (D-phenylalanine)3 activates AMP-activated protein kinase and stimulates glucose transport. Biochem Pharmacol. (2009)
- Anderson RA, Kozlovsky AS. Chromium intake, absorption and excretion of subjects consuming self-selected diets. Am J Clin Nutr. (1985)
- Bunker VW, et al. The uptake and excretion of chromium by the elderly. Am J Clin Nutr. (1984)
- Chen NS, Tsai A, Dyer IA. Effect of chelating agents on chromium absorption in rats. J Nutr. (1973)
- Hahn CJ, Evans GW. Absorption of trace metals in the zinc-deficient rat. Am J Physiol. (1975)
- Laschinsky N, et al. Bioavailability of chromium(III)-supplements in rats and humans. Biometals. (2012)
- Cefalu WT, et al. Characterization of the metabolic and physiologic response to chromium supplementation in subjects with type 2 diabetes mellitus. Metabolism. (2010)
- HOPKINS LL Jr, SCHWARZ K. CHROMIUM (3) BINDING TO SERUM PROTEINS, SPECIFICALLY SIDEROPHILIN. Biochim Biophys Acta. (1964)
- Ainscough EW, et al. Studies on human lactoferrin by electron paramagnetic resonance, fluorescence, and resonance Raman spectroscopy. Biochemistry. (1980)
- Aisen P, Aasa R, Redfield AG. The chromium, manganese, and cobalt complexes of transferrin. J Biol Chem. (1969)
- Sun Y, et al. The binding of trivalent chromium to low-molecular-weight chromium-binding substance (LMWCr) and the transfer of chromium from transferrin and chromium picolinate to LMWCr. J Biol Inorg Chem. (2000)
- Yamamoto A, Wada O, Ono T. Distribution and chromium-binding capacity of a low-molecular-weight, chromium-binding substance in mice. J Inorg Biochem. (1984)
- Harding C, Heuser J, Stahl P. Receptor-mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. J Cell Biol. (1983)
- Yoshida M, et al. Tissue accumulation and urinary excretion of chromium in rats fed diets containing graded levels of chromium chloride or chromium picolinate. J Toxicol Sci. (2010)
- Seal CJ, Heaton FW. Effect of dietary picolinic acid on the metabolism of exogenous and endogenous zinc in the rat. J Nutr. (1985)
- Pittler MH, Stevinson C, Ernst E. Chromium picolinate for reducing body weight: meta-analysis of randomized trials. Int J Obes Relat Metab Disord. (2003)
- Attenburrow MJ, et al. Chromium treatment decreases the sensitivity of 5-HT2A receptors. Psychopharmacology (Berl). (2002)
- Docherty JP, et al. A double-blind, placebo-controlled, exploratory trial of chromium picolinate in atypical depression: effect on carbohydrate craving. J Psychiatr Pract. (2005)
- Anton SD, et al. Effects of chromium picolinate on food intake and satiety. Diabetes Technol Ther. (2008)
- Singh T, Williams K. Atypical depression. Psychiatry (Edgmont). (2006)
- Brownley KA, et al. A double-blind, randomized pilot trial of chromium picolinate for binge eating disorder: results of the Binge Eating and Chromium (BEACh) study. J Psychosom Res. (2013)
- Gastaldelli A, et al. Influence of obesity and type 2 diabetes on gluconeogenesis and glucose output in humans: a quantitative study. Diabetes. (2000)
- Pei D, et al. The influence of chromium chloride-containing milk to glycemic control of patients with type 2 diabetes mellitus: a randomized, double-blind, placebo-controlled trial. Metabolism. (2006)
- Wu GY, Wada O. Studies on a specific chromium binding substance (a low-molecular-weight chromium binding substance) in urine (author’s transl). Sangyo Igaku. (1981)
- Wada O, et al. Low-molecular-weight, chromium-binding substance in rat lungs and its possible role in chromium movement. Ind Health. (1983)
- Wang H, Kruszewski A, Brautigan DL. Cellular chromium enhances activation of insulin receptor kinase. Biochemistry. (2005)
- Mackowiak P, et al. Evaluation of insulin binding and signaling activity of newly synthesized chromium(III) complexes in vitro. Mol Med Rep. (2010)
- Ukkola O, Santaniemi M. Protein tyrosine phosphatase 1B: a new target for the treatment of obesity and associated co-morbidities. J Intern Med. (2002)
- Davis CM, Sumrall KH, Vincent JB. A biologically active form of chromium may activate a membrane phosphotyrosine phosphatase (PTP). Biochemistry. (1996)
- Goldstein BJ, et al. Enhancement of post-receptor insulin signaling by trivalent chromium in hepatoma cells is associated with differential inhibition of specific protein-tyrosine phosphatases. J Trace Elem Exp Med. (2001)
- Wang ZQ, et al. Chromium picolinate enhances skeletal muscle cellular insulin signaling in vivo in obese, insulin-resistant JCR:LA-cp rats. J Nutr. (2006)
- Aguirre V, et al. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307). J Biol Chem. (2000)
- Solinas G, et al. Saturated fatty acids inhibit induction of insulin gene transcription by JNK-mediated phosphorylation of insulin-receptor substrates. Proc Natl Acad Sci U S A. (2006)
- Sreejayan N, et al. Chromium alleviates glucose intolerance, insulin resistance, and hepatic ER stress in obese mice. Obesity (Silver Spring). (2008)
- Chen WY, et al. Chromium supplementation enhances insulin signalling in skeletal muscle of obese KK/HlJ diabetic mice. Diabetes Obes Metab. (2009)
- Ozcan U, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. (2004)
- Engin F, Hotamisligil GS. Restoring endoplasmic reticulum function by chemical chaperones: an emerging therapeutic approach for metabolic diseases. Diabetes Obes Metab. (2010)
- Ozcan U, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. (2006)
- Yang X, et al. Insulin-sensitizing and cholesterol-lowering effects of chromium (D-Phenylalanine)3. J Inorg Biochem. (2006)
- Evans GW, Bowman TD. Chromium picolinate increases membrane fluidity and rate of insulin internalization. J Inorg Biochem. (1992)
- Gorden P, et al. Intracellular translocation of iodine-125-labeled insulin: direct demonstration in isolated hepatocytes. Science. (1978)
- McClain DA. Mechanism and role of insulin receptor endocytosis. Am J Med Sci. (1992)
- Geiger D, et al. Down-regulation of insulin receptors is related to insulin internalization. Exp Cell Res. (1989)
- Iqbal N, et al. Chromium picolinate does not improve key features of metabolic syndrome in obese nondiabetic adults. Metab Syndr Relat Disord. (2009)
- Morino K, Petersen KF, Shulman GI. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes. (2006)
- Abdollahi M, et al. Effect of chromium on glucose and lipid profiles in patients with type 2 diabetes; a meta-analysis review of randomized trials. J Pharm Pharm Sci. (2013)
- Patal PC, Cardino MT, Jimeno CA. A meta-analysis on the effect of chromium picolinate on glucose and lipid profiles among patients with type 2 diabetes mellitus. Philipp J Intern Med. (2010)
- Balk EM, et al. Effect of chromium supplementation on glucose metabolism and lipids: a systematic review of randomized controlled trials. Diabetes Care. (2007)
- Broadhurst CL, Domenico P. Clinical studies on chromium picolinate supplementation in diabetes mellitus–a review. Diabetes Technol Ther. (2006)
- Frauchiger MT, Wenk C, Colombani PC. Effects of acute chromium supplementation on postprandial metabolism in healthy young men. J Am Coll Nutr. (2004)
- Ghosh D, et al. Role of chromium supplementation in Indians with type 2 diabetes mellitus. J Nutr Biochem. (2002)
- Kleefstra N, et al. Chromium treatment has no effect in patients with poorly controlled, insulin-treated type 2 diabetes in an obese Western population: a randomized, double-blind, placebo-controlled trial. Diabetes Care. (2006)
- Lai MH. Antioxidant effects and insulin resistance improvement of chromium combined with vitamin C and e supplementation for type 2 diabetes mellitus. J Clin Biochem Nutr. (2008)
- Martin J, et al. Chromium picolinate supplementation attenuates body weight gain and increases insulin sensitivity in subjects with type 2 diabetes. Diabetes Care. (2006)
- Racek J, et al. Influence of chromium-enriched yeast on blood glucose and insulin variables, blood lipids, and markers of oxidative stress in subjects with type 2 diabetes mellitus. Biol Trace Elem Res. (2006)
- McIver DJ, et al. Risk of Type 2 Diabetes Is Lower in US Adults Taking Chromium-Containing Supplements. J Nutr. (2015)
- Volek JS, et al. Effects of chromium supplementation on glycogen synthesis after high-intensity exercise. Med Sci Sports Exerc. (2006)
- Davis JM, Welsh RS, Alerson NA. Effects of carbohydrate and chromium ingestion during intermittent high-intensity exercise to fatigue. Int J Sport Nutr Exerc Metab. (2000)
- Roginski EE, Mertz W. Effects of Chromium (III) Supplementation on Glucose and Amino Acid Metabolism in Rats Fed a Low Protein Diet. J Nutr. ()
- Campbell WW, et al. Exercise training and dietary chromium effects on glycogen, glycogen synthase, phosphorylase and total protein in rats. J Nutr. (1989)
- Tian H, et al. Chromium picolinate supplementation for overweight or obese adults. Cochrane Database Syst Rev. (2013)
- Meyers AW, et al. Are weight concerns predictive of smoking cessation? A prospective analysis. J Consult Clin Psychol. (1997)
- Parsons A, et al. A proof of concept randomised placebo controlled factorial trial to examine the efficacy of St John’s wort for smoking cessation and chromium to prevent weight gain on smoking cessation. Drug Alcohol Depend. (2009)
- Cerulli J, et al. Chromium picolinate toxicity. Ann Pharmacother. (1998)
- Martin WR, Fuller RE. Suspected chromium picolinate-induced rhabdomyolysis. Pharmacotherapy. (1998)