Introduzione:
Nel 1957 sono stati pubblicati i primi risultati del Framingham Heart Study [1]. Si trattava (o dovrei dire si tratta, visto che è ancora in corso) di uno studio epidemiologico che cercava di individuare i fattori di rischio per le malattie cardiovascolari. Lo studio prende il nome dalla città di Framingham, Massachusetts, negli Stati Uniti. Sono stati reclutati 5.209 residenti della città di età compresa tra i 30 e i 62 anni. Diversi dati di questo gruppo di persone (coorte) sono stati raccolti nel tempo per scoprire questi fattori di rischio. Nella loro importante pubblicazione, hanno identificato tre fattori di rischio per le malattie cardiovascolari: ipertensione, obesità e ipercolesterolemia (alti livelli di colesterolo).

Ai fini di questo articolo, mi concentrerò sul Colesterolo. Prima di parlare dell’effetto degli Steroidi Anabolizzanti Androgeni sul colesterolo HDL, fornirò alcune informazioni di base.
Relazione tra malattie cardiovascolari e colesterolo LDL e HDL:
Dopo i risultati iniziali del Framingham Heart Study, il ruolo del colesterolo nello sviluppo del rischio di malattie cardiovascolari è stato ulteriormente chiarito. Un primo passo avanti in quest’area di ricerca è stata la suddivisione del colesterolo in colesterolo a bassa densità (LDL) e colesterolo ad alta densità (HDL) e il loro rispettivo contributo al rischio di malattie cardiovascolari. Queste due frazioni di colesterolo sono note al grande pubblico rispettivamente come colesterolo “cattivo” e “buono”.
Il colesterolo LDL elevato è stato associato a un aumento del rischio di malattie cardiovascolari. Dopo decenni di ricerche, una pletora di prove ha stabilito con certezza che questa associazione è causale [2]. In effetti, la terapia per abbassare le LDL, ad esempio con l’uso di statine, è una pietra miliare del trattamento delle dislipidemie. L’associazione tra colesterolo HDL e rischio di malattie cardiovascolari è opposta a quella del colesterolo LDL: un colesterolo HDL elevato è risultato associato a una diminuzione del rischio di malattie cardiovascolari. Gli studi epidemiologici rilevano una riduzione del rischio cardiovascolare di circa il 2-3% per ogni aumento di 1 mg/dL di colesterolo HDL [3].
A differenza del colesterolo LDL, tuttavia, non sembra esistere un legame causale diretto tra i livelli di colesterolo HDL e il rischio di malattie cardiovascolari [4]. Studi genetici sull’uomo, in cui sono state analizzate alcune mutazioni genetiche che portano a livelli più o meno elevati di colesterolo HDL, non hanno dimostrato chiaramente un’associazione con il rischio di malattie cardiovascolari. Questo sarebbe stato prevedibile se ci fosse stato un legame causale diretto. Lo scollamento tra i livelli di colesterolo HDL e il rischio di malattie cardiovascolari è diventato forse più dolorosamente evidente negli studi clinici sui farmaci. Sono stati sviluppati (o esistevano già) alcuni farmaci che aumentano i livelli di colesterolo HDL in modo significativo, ma non riescono a ridurre la mortalità o l’incidenza di eventi cardiovascolari, come ictus o infarto del miocardio [5]. Ciò include anche l’uso di integratori da banco, come la Niacina [5, 6].
Nota: quando si parla di “colesterolo LDL”, “colesterolo HDL” o “colesterolo VLDL” ci si riferisce alle lipoproteine trasportatrici. La sigla VLDL sta per “very low density lipoproteins”, LDL per “low density lipoproteins” e HDL per “high density lipoproteins”. La densità a cui si fa riferimento è legata al loro contenuto lipidico. In particolare la densità è tanto più bassa quanto maggiori sono i trigliceridi racchiusi all’interno della particella. Ne deriva che le VLDL sono lipoproteine ad alto contenuto in trigliceridi, le LDL sono lipoproteine a basso contenuto in trigliceridi e le HDL sono lipoproteine estremamente povere di trigliceridi. In compenso LDL e HDL sono caratterizzate da un alto contenuto in colesterolo. Ognuna di queste lipoproteine ricopre ruoli diversi. Le VLDL hanno il compito di trasferire trigliceridi dal fegato ai tessuti; in particolare, dopo essere state sintetizzate nel fegato, vengono riversate nel circolo ematico e cedute soprattutto al tessuto muscolare e a quello adiposo. LDL ed HDL trasportano il colesterolo nel circolo sanguigno. Mentre le LDL hanno lo scopo di cederlo ai tessuti, le HDL sono deputate alla rimozione del colesterolo presente in eccesso nel plasma. Le VLDL vengono sintetizzate soprattutto nelle cellule epatiche (epatociti) e trasportano principalmente Trigliceridi di origine endogena.

Hanno una densità compresa tra 1,006 e 1,019 g/ml e migrano elettroforeticamente con le ß-globuline. Per ogni molecola tipica di VLDL che viene degradata, viene prodotta una IDL.
Per la stima del VLDL-C è necessario dividere il valore dei Trigliceridi misurato per 5, nel caso in cui siano espressi in mg/dL, o 2.2, nel caso siano espressi in mmol/L. Nella maggior parte dei casi, la formula consente di effettuare una stima accurata del reale valore del VLDL-C.
AAS e riduzione del HDL:
Ora siete un po’ più informati sulla relazione tra malattie cardiovascolari e colesterolo LDL e HDL. Diversi studi interventistici hanno esaminato l’effetto dell’uso di AAS sul colesterolo. Peter Bond ha fatto un piccolo riassunto di questi studi nel suo libro “Book on Steroids” il quale riporto nella tabella sottostante. Sebbene non tutti gli studi abbiano riscontrato una diminuzione statisticamente significativa del colesterolo HDL (↔️), molti lo fanno e nel complesso mostrano inequivocabilmente una diminuzione. Ciò è particolarmente vero per gli AAS orali, che sembrano avere l’effetto maggiore sul colesterolo HDL.

In uno studio, condotto dal gruppo di Bhasin [11], sono stati somministrati dosaggi graduali di Testosterone (25, 50 125, 300 e 600mg di Testosterone Enantato alla settimana). Gli autori hanno quindi potuto valutare se esisteva una relazione dose-risposta tra il dosaggio di Testosterone e il colesterolo HDL, e così è stato. Hanno riscontrato una moderata relazione inversa (r = -0,40) tra i livelli di Testosterone e il colesterolo HDL. Quindi, almeno fino a una dose compresa tra 300 e 600mg settimanali, più alto è il dosaggio, maggiore è la diminuzione del colesterolo HDL.
In un recente studio, 100 consumatori di AAS sono stati seguiti nel tempo durante l’autosomministrazione di questa classe di farmaci. Il dosaggio medio, basato sulle informazioni riportate sull’etichetta, era di 898mg a settimana, rendendo così il loro ciclo di AAS abbastanza rappresentativo dell’uso comune da parte dei bodybuilder. Le misurazioni sono state effettuate prima, durante, 3 mesi dopo la fine del ciclo e 1 anno dopo l’inizio del ciclo. Il colesterolo HDL è diminuito di 0,4 mmol/L (da 1,2 a 0,8) durante l’uso. Si tratta di una diminuzione sostanziale. I valori erano tornati ai valori di base 3 mesi dopo la cessazione dell’uso di AAS.
Meccanismo attraverso il quale gli AAS abbassano l’HDL:
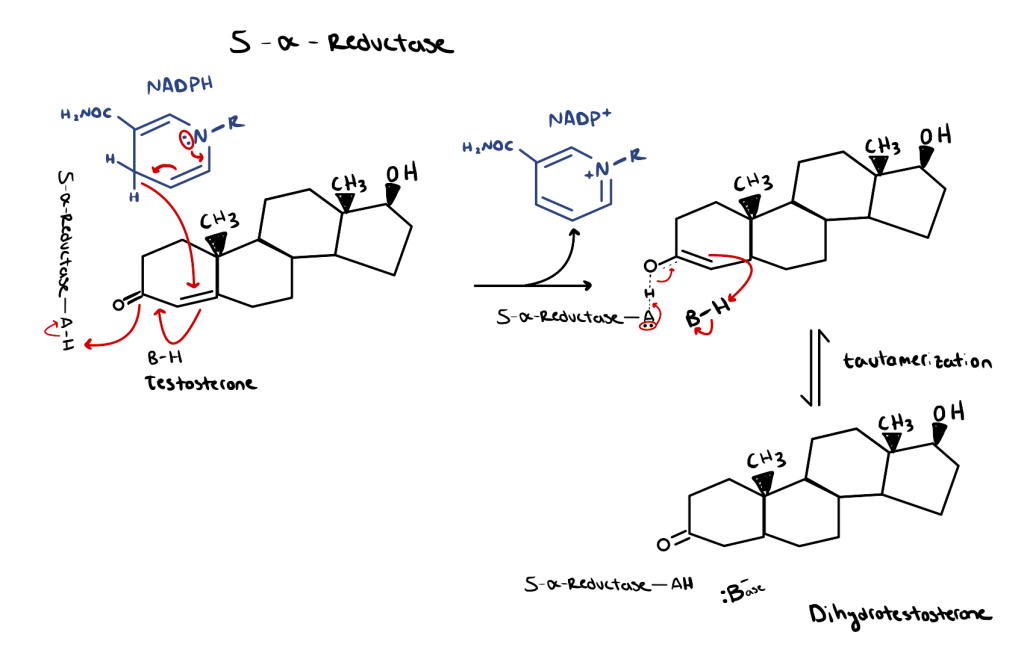
Si ritiene che gli steroidi anabolizzanti riducano il colesterolo HDL aumentando l’attività di un enzima chiamato lipasi epatica [7, 8, 9, 10]. Si tratta di un enzima prodotto principalmente dal fegato. Essendo una lipasi, catalizza le reazioni di idrolisi dei lipidi. In particolare, scinde gli acidi grassi dal triacilglicerolo (Trigliceride) e i fosfolipidi dalle particelle lipoproteiche, come il colesterolo HDL. Idrolizzando il triacilglicerolo e i fosfolipidi dal colesterolo HDL, riduce le dimensioni di queste particelle. Queste particelle più piccole vengono catabolizzate a un ritmo più elevato [12].
Thompson et al. hanno esaminato queste sottofrazioni di colesterolo HDL che differiscono per dimensioni [8]. Hanno misurato i livelli di colesterolo HDL2 e HDL3: le particelle di colesterolo HDL2 sono più grandi e di densità inferiore rispetto a quelle HDL3. Gli uomini partecipanti hanno ricevuto 200mg di Testosterone Enantato alla settimana o 6mg di Stanozololo orale (Winstrol) al giorno per 6 settimane in un design crossover. I risultati sono stati i seguenti:

Come si può notare, la maggiore diminuzione relativa è stata osservata nella frazione HDL2 più grande a seguito del trattamento con Stanozololo. Al contrario, il Testosterone non ha mostrato una diminuzione statisticamente significativa nella frazione HDL2, ma ha fatto altrettanto nella frazione HDL3 più piccola. Non è del tutto chiaro cosa provochi la diminuzione di questa frazione.
Quando dei bodybuilder sono stati randomizzati a ricevere 200mg di Nandrolone Decanoato alla settimana o un placebo [13]. Non sono stati riscontrati cambiamenti statisticamente significativi nel colesterolo totale, nel colesterolo LDL e nel colesterolo HDL. Analogamente, non sono stati riscontrati cambiamenti significativi nelle sottofrazioni di colesterolo HDL2 e HDL3. In particolare, nella stessa pubblicazione, gli autori riferiscono anche di uno studio in cui hanno seguito un gruppo di atleti di forza che si autosomministravano steroidi anabolizzanti. Sono stati utilizzati diversi composti in vari dosaggi, ma vale la pena sottolineare che la maggior parte di essi comprendeva anche uno steroide anabolizzante orale (soprattutto Stanozololo). In questo caso, il colesterolo HDL è sceso in picchiata: da 1,08 mmol/L a 0,43 mmol/L dopo 8 settimane. La sottofrazione di colesterolo HDL2 è scesa da 0,21 a 0,05 e la sottofrazione di colesterolo HDL3 è scesa da 0,87 a 0,40 mmol/L.
Effetto degli AAS sulla funzione del colesterolo HDL:
Dato il legame tra l’effetto di un farmaco sui livelli di colesterolo HDL e il rischio di malattie cardiovascolari, la ricerca ha iniziato a concentrarsi sulla funzione del colesterolo HDL. Il colesterolo HDL è il protagonista di un processo chiamato trasporto inverso del colesterolo. Nell’aterosclerosi, il colesterolo si accumula nelle cellule del sistema immunitario (macrofagi) e nelle cellule muscolari lisce che circondano i vasi sanguigni [14]. Queste cellule, a loro volta, diventano le cosiddette cellule schiumose, che segnano il punto di partenza dell’aterosclerosi. Le particelle di colesterolo HDL sono in grado di raccogliere il colesterolo da queste cellule – efflusso di colesterolo. L’efflusso del colesterolo dalle cellule schiumose nelle particelle di colesterolo HDL è uno dei modi in cui si ritiene che il colesterolo HDL eserciti i suoi effetti protettivi sulle arterie. Il colesterolo HDL raccolto può poi essere riportato al fegato, che lo incorpora nella bile e può quindi essere secreto nelle feci. Allo stesso modo, le particelle di colesterolo HDL possono trasferire parte del loro contenuto alle particelle LDL, che possono finire nuovamente nelle cellule schiumose o essere assorbite dal fegato.

Esistono metodi per misurare la capacità di efflusso del colesterolo HDL e l’idea attuale è che la sua modulazione possa influire sul rischio di malattie cardiovascolari, contrariamente ai livelli di colesterolo HDL in sé. Esistono diversi modi in cui il colesterolo HDL può assorbire il colesterolo dalle cellule schiumose. Uno di questi coinvolge un trasportatore chiamato ATP-binding casette transporter A1 (ABCA1), che si ritiene sia il più importante [15, 16]. Contribuiscono anche altri trasportatori, come ABCG1 e il recettore scavenger B1, oltre alla diffusione semplice.
Diamo uno sguardo agli studi che hanno valutato l’impatto dell’uso di steroidi anabolizzanti sulla capacità di efflusso del colesterolo HDL. In uno studio (non controllato), uomini anziani ipogonadici sono stati randomizzati alla TRT con o senza Dutasteride (un inibitore della 5a-reduttasi) [17]. Dopo 3 mesi, la TRT era riuscita a riportare i livelli di Testosterone di questi uomini all’interno del range di normalità. Il colesterolo HDL e la capacità di efflusso del colesterolo HDL sono rimasti inalterati.
Un altro studio, randomizzato e controllato, ha applicato un approccio leggermente diverso [18]. Uomini sani, di età compresa tra i 19 e i 55 anni, sono stati castrati medicalmente per sopprimere completamente la loro produzione endogena. In seguito, hanno ricevuto un placebo, una TRT a basso dosaggio, una TRT sostitutiva completa o una TRT sostitutiva completa con Letrozolo, un inibitore dell’Aromatasi che inibisce la conversione del Testosterone in Estradiolo. Il colesterolo HDL è aumentato leggermente nel gruppo placebo e in quello a basso dosaggio, mentre è rimasto inalterato nei due gruppi che hanno ricevuto una dose sostitutiva completa. Inoltre, mentre è stata riscontrata una piccola diminuzione della capacità di efflusso di ABCA1 nel gruppo che ha ricevuto anche il Letrozolo, non sono stati osservati cambiamenti negli altri tre gruppi. A causa delle dimensioni ridotte dei gruppi, è possibile che un piccolo effetto non sia stato notato.
E i dosaggi elevati? Sfortunatamente, esiste un solo studio che ha esaminato questo aspetto, ed era di natura trasversale (si tratta di misurazioni effettuate in un solo momento, il che rende impossibile/difficile trarre conclusioni)[19]. I ricercatori hanno confrontato le misurazioni di un gruppo di utilizzatori di AAS con quelle di non utilizzatori e controlli sedentari, che avevano un’età corrispondente. I consumatori di AAS erano forti utilizzatori, avendo fatto uso di AAS in media per circa 8 anni con un dosaggio medio di (apparentemente) 2,5g settimanali. La capacità delle HDL di effluire il colesterolo dai macrofagi è risultata inferiore del 13% nei consumatori di AAS rispetto ai non consumatori con allenamento della forza. Anche in questo caso, a causa della natura trasversale dello studio, è difficile dire se questo sia causale.
Conclusioni:
Gli AAS riducono il colesterolo HDL, in modo dose-dipendente, e questo sembra verificarsi in modo particolarmente marcato con gli AAS orali17α-alchilati. Non è certo come questo si traduca in un rischio di malattia cardiovascolare, in quanto esiste una discrepanza tra la capacità di un farmaco di alterare il colesterolo HDL e il suo effetto su di esso. La correlazione tra i livelli di colesterolo HDL misurati e il rischio di malattie cardiovascolari non è causale. I ricercatori ritengono che la capacità di efflusso del colesterolo HDL possa avere una migliore capacità predittiva, oltre a essere causalmente correlata. Pertanto, i farmaci che influiscono sulla capacità di efflusso potrebbero influenzare il rischio di malattie cardiovascolari. L’effetto degli AAS su questo aspetto non è ancora così chiaro a causa della scarsità di dati, soprattutto per quanto riguarda i dosaggi sovrafisiologici. Alcuni dati suggeriscono che potrebbero avere un impatto negativo sulla capacità di efflusso del colesterolo. Uno studio di coorte longitudinale probabilmente risponderà a questa domanda con maggiore certezza in futuro. Se la capacità di efflusso del colesterolo HDL diminuisce effettivamente in seguito all’uso di AAS, questa diminuzione indotta dagli AAS potrebbe essere dannosa per la salute cardiovascolare.
Certo, vi sono farmaci, non che integratori erboristici da banco, con azione di “tampone” della dislipidemia ematica. Ma ciò non elimina il problema lo rallenta nella sua potenziale comparsa soprattutto agendo sui rapporti tra i marcatori del profilo lipidico ematico. Ciò significa che potenzialmente, e il condizionale è d’obbligo vista la sensibile differenza soggettiva riscontrabile, l’uso di Monacolina-K, Niacina e EPA, nei corretti dosaggi, potrà causare una riduzione del HDL leggermente/moderatamente inferiore rispetto all’utilizzatore meno accorto, con conseguente alterazione delle ratio HDL:LDL, HDL:Trigliceridi e HDL:Colesterolo totale “rallentata” e meno marcata. Di per se questa pratica supplementativa potrebbe portare anche ad una riduzione anche della capacità di efflusso del colesterolo HDL, ma non vi sono, ad oggi, conferme inoppugnabili che ciò avvenga.
Ah, quasi dimenticavo di ricordare ai meno informati che anche i SARM non steroidei (vedi Ostarina, LGD-4033, ecc…) alterano il profilo lipidico ematico a diverso grado. Anche i SERM (Tamoxifene, Clomifene, Raloxifene ecc…) e AI (Letrozolo, Anastrozolo, Exemestane ecc…) hanno un potenziale di alterazione della lipidemia ematica.
Gabriel Bellizzi
Riferimenti:
- T. R. Dawber, F. E. Moore, and G. V. Mann. Measuring the risk of coronary heart disease in adult population groups: Ii. coronary heart disease in the framingham study. American Journal of Public Health and the Nations Health, 47(4 Pt 2):4, 1957
- Ference, Brian A., et al. “Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel.” European heart journal 38.32 (2017): 2459-2472.
- Gordon, David J., et al. “High-density lipoprotein cholesterol and cardiovascular disease. Four prospective American studies.” Circulation 79.1 (1989): 8-15.
- Rader, Daniel J., and G. Kees Hovingh. “HDL and cardiovascular disease.” The Lancet 384.9943 (2014): 618-625.
- Keene, Daniel, et al. “Effect on cardiovascular risk of high density lipoprotein targeted drug treatments niacin, fibrates, and CETP inhibitors: meta-analysis of randomised controlled trials including 117 411 patients.” Bmj 349 (2014).
- Schandelmaier, Stefan, et al. “Niacin for primary and secondary prevention of cardiovascular events.” Cochrane Database of Systematic Reviews 6 (2017).
- Friedl, Karl E., et al. “High-density lipoprotein cholesterol is not decreased if an aromatizable androgen is administered.” Metabolism 39.1 (1990): 69-74.
- Thompson, Paul D., et al. “Contrasting effects of testosterone and stanozolol on serum lipoprotein levels.” Jama 261.8 (1989): 1165-1168.
- Zmuda, Joseph M., et al. “The effect of testosterone aromatization on high-density lipoprotein cholesterol level and postheparin lipolytic activity.” Metabolism 42.4 (1993): 446-450.
- Herbst, Karen L., et al. “Testosterone administration to men increases hepatic lipase activity and decreases HDL and LDL size in 3 wk.” American Journal of Physiology-Endocrinology and Metabolism 284.6 (2003): E1112-E1118.
- Singh, Atam B., et al. “The effects of varying doses of T on insulin sensitivity, plasma lipids, apolipoproteins, and C-reactive protein in healthy young men.” The Journal of Clinical Endocrinology & Metabolism 87.1 (2002): 136-143.
- Jin, Weijun, Dawn Marchadier, and Daniel J. Rader. “Lipases and HDL metabolism.” Trends in Endocrinology & Metabolism 13.4 (2002): 174-178.
- Hartgens, F., et al. “Effects of androgenic-anabolic steroids on apolipoproteins and lipoprotein (a).” British journal of sports medicine 38.3 (2004): 253-259.
- Ouimet, Mireille, Tessa J. Barrett, and Edward A. Fisher. “HDL and reverse cholesterol transport: Basic mechanisms and their roles in vascular health and disease.” Circulation research 124.10 (2019): 1505-1518.
- Du, Xian-Ming, et al. “HDL particle size is a critical determinant of ABCA1-mediated macrophage cellular cholesterol export.” Circulation research 116.7 (2015): 1133-1142.
- Adorni, Maria Pia, et al. “The roles of different pathways in the release of cholesterol from macrophages.” Journal of lipid research 48.11 (2007): 2453-2462.
- Rubinow, Katya B., et al. “Testosterone replacement in hypogonadal men alters the HDL proteome but not HDL cholesterol efflux capacity.” Journal of lipid research 53.7 (2012): 1376-1383.
- Rubinow, Katya B., et al. “Sex steroids mediate discrete effects on HDL cholesterol efflux capacity and particle concentration in healthy men.” Journal of clinical lipidology 12.4 (2018): 1072-1082.
- de Souza, Francis Ribeiro, et al. “Diminished cholesterol efflux mediated by HDL and coronary artery disease in young male anabolic androgenic steroid users.” Atherosclerosis 283 (2019): 100-105.