Pip: Benvenuti a BioGenTech Sport and Health, dove oggi ci immergiamo nella fisiologia femminile con la stessa leggerezza con cui si affronta un manuale di biochimica avanzata — cioè, nessuna.
Mara: Gabriel Bellizzi, CEO di BioGenTech, ha pubblicato la prima parte di un lavoro approfondito sull'ottimizzazione farmacologica nel bodybuilding femminile: steroidogenesi, dimorfismi ormonali, asse hGH/IGF-1 e biologia del tessuto adiposo. Iniziamo proprio da lì.
Fisiologia e dimorfismi nel bodybuilding femminile
Pip: Il punto di partenza del lavoro è chiaro: per capire come agiscono i farmaci nelle donne, bisogna prima capire come funziona la fisiologia ormonale femminile, perché applicare schemi maschili a un sistema diverso non è ottimizzazione — è improvvisazione.
Mara: L'articolo lo dice esplicitamente fin dall'introduzione: l'obiettivo è trattare di "ottimizzazione farmacologica nel Bodybuilding femminile per le prestazioni e l'estetica basata su solide basi scientifiche", coprendo metabolismo degli estrogeni, degli androgeni, sensibilità recettoriale, periodizzazione nel ciclo ovulatorio-mestruale, protocolli per la PCOS e strategie per la menopausa.
Pip: Quindi il fondamento è la steroidogenesi: tutto parte dal colesterolo, convertito in pregnenolone dall'enzima mitocondriale CYP11A1, e da lì si diramano tutte le vie verso progestinici, androgeni ed estrogeni.
Mara: Esatto. E la produzione ovarica segue la cosiddetta "two-cell, two-gonadotropin theory": le cellule della teca, stimolate dall'LH, producono androgeni che non possono aromatizzare autonomamente; le cellule della granulosa, stimolate dall'FSH, ricevono quegli androgeni e li convertono in estradiolo grazie all'aromatasi. Nessuna delle due cellule può farlo da sola.
Pip: La PCOS è l'esempio patologico di questo sistema in cortocircuito — l'insulino-resistenza iperstimola le cellule della teca, i livelli di LH salgono, e si produce un eccesso di androgeni che blocca l'ovulazione e crea quel quadro classico di follicoli bloccati a stadio intermedio.
Mara: Il testo descrive il meccanismo con precisione: l'iperinsulinemia aumenta l'attività di CYP11A1 e CYP17A1, amplifica la risposta della teca all'LH e sopprime la SHBG epatica, aumentando il testosterone libero circolante. Il risultato è un circolo vizioso tra iperandrogenismo e anovulazione cronica.
Pip: E qui emerge una delle osservazioni più pratiche del lavoro: nelle donne i recettori degli androgeni mostrano una sensibilità maggiore rispetto agli uomini, probabilmente come meccanismo compensativo alle concentrazioni androgene strutturalmente più basse.
Mara: I numeri lo confermano: il testosterone totale negli uomini varia tra 300 e 1000 ng/dL, nelle donne tra 15 e 76 ng/dL — un rapporto di circa 10-20 volte. Nonostante questo, anche piccole variazioni nei livelli di androgeni esogeni producono risposte cliniche evidenti nelle donne, sia estetiche che metaboliche.
Pip: Il che spiega perché dosi che per un uomo sarebbero quasi omeopatiche in contesto femminile richiedono una gestione completamente diversa.
Mara: L'articolo poi affronta l'asse hGH/IGF-1 e il ruolo dell'estradiolo su di esso. L'E2 aumenta la secrezione ipofisaria di hGH ma inibisce contemporaneamente la segnalazione epatica attraverso la via JAK2-STAT5, inducendo le proteine SOCS. In pratica, concentrazioni elevate di estradiolo — specialmente per via orale con effetto di primo passaggio epatico — riducono la produzione sistemica di IGF-1.
Mara: Sul tessuto adiposo, il dimorfismo è altrettanto marcato: le donne in premenopausa accumulano preferenzialmente grasso sottocutaneo gluteo-femorale grazie agli estrogeni, che sovraregolano i recettori α2A-AR antilipolitici specificamente nel grasso sottocutaneo. Questo protegge dal rischio metabolico viscerale ma rende più difficile la mobilizzazione del grasso periferico in contesto agonistico.
Pip: Un sistema ottimizzato per la sopravvivenza metabolica che diventa un ostacolo quando l'obiettivo è la definizione estrema — e che richiede strategie specifiche, non la semplice trasposizione di protocolli maschili.
Mara: La menopausa rappresenta infine un reset neuroendocrino profondo: il crollo di E2 sposta la distribuzione adiposa da ginoide ad androide, peggiora il profilo lipidico, riduce la sensibilità insulinica e altera la risposta agli steroidi esogeni. Il lavoro indica che la finestra terapeutica ottimale per la TOS si colloca nei primi anni dalla menopausa, prima che vasi e recettori entrino in uno stato pro-infiammatorio irreversibile.
Pip: La seconda parte del lavoro — dove si entra nei protocolli pratici — sarà il banco di prova di tutto questo impianto teorico.
Mara: Quello che emerge da questa prima parte è che la fisiologia femminile non è una variante semplificata di quella maschile — è un sistema distinto, ciclico e altamente sensibile alle concentrazioni ormonali.
Pip: E capirlo è il prerequisito per qualsiasi discorso serio sulla farmacologia applicata. La prossima parte promette di mettere tutto questo in pratica.
Nel dibattito scientifico l’origine e sviluppo della vita sulla terra è tutt’altro che arrivato ad un punto chiaro e incontestabile. Oggi, la maggior parte della comunità scientifica ha abbracciato la filosofia materialista e casualista, comunemente chiamata evoluzionismo. Ciò nonostante, con l’avanzare delle evidenze scientifiche in campo paleontologico, biochimico e genetico la crisi della fazione materialista/casualista si è aggravata specie con la nascita del movimento scientifico dell’Intelligence Design il quale, al contrario dei preponderanti e disperati evoluzionisti, applica rigorosamente il metodo scientifico e l’osservazione e comprensione oggettiva e lucida dei dati emergenti dalla ricerca.
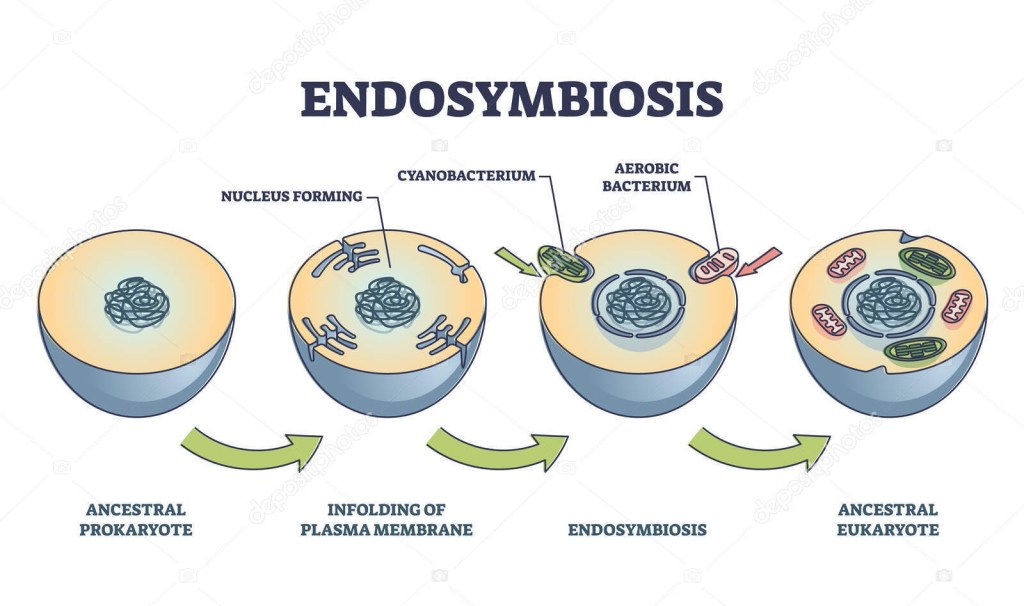
Tra le varie teorie (non dimostrate) del panorama evoluzionista ve ne è una che prende il nome di “endosimbiosi”. In breve, L’endosimbiosi (dal greco: ἔνδον = dentro; συν = insieme; βιος = vita) sarebbe una particolare forma di interazione biologica, una simbiosi mutualistica nella quale un organismo (di solito unicellulare) vive all’interno di un altro organismo.
Endosymbiosis process stages with symbiotic living organisms outline diagram. Labeled educational biological evolution theory steps with ancestral prokaryote evolving and eukaryote vector illustration
L’endosimbiosi è un’interazione di carattere mutualistico: entrambe le specie che interagiscono traggono vantaggio dall’interazione; questo mutuo beneficio distingue la simbiosi mutualistica dal parassitismo e dal commensalismo. Stiamo praticamente parlando di una trasmissione di cooperatività mutualistica generativa o generazionale.
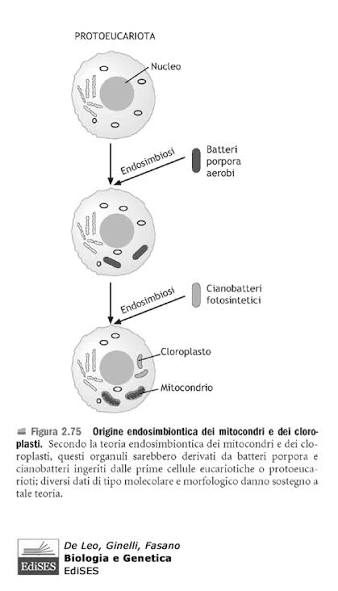
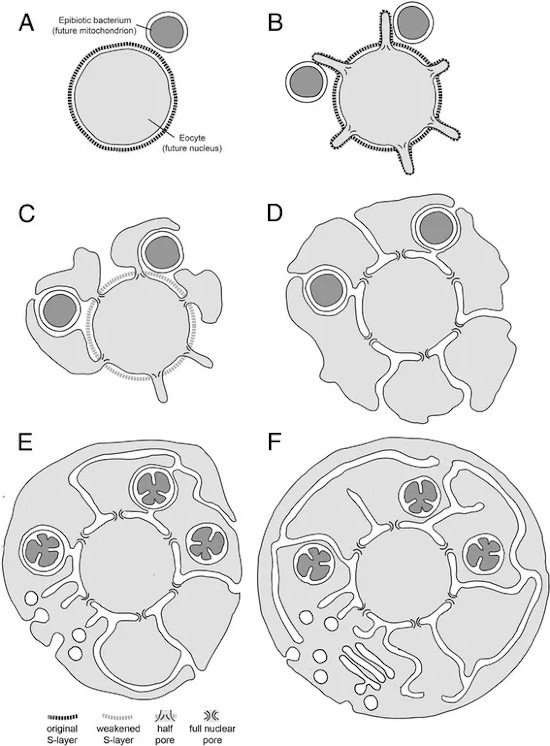
Alcuni scienziati hanno supposto che un’antica endosimbiosi abbia originato alcune caratteristiche permanenti degli organismi attuali, formulando la teoria endosimbiotica (chiamata anche teoria endosimbiontica o teoria dell’endosimbionte) riguardo alle origini di alcuni organismi.
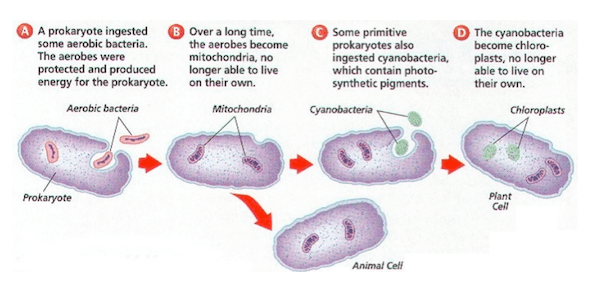
L’ipotesi è che alcuni organismi biologici siano stati ingeriti da altri organismi e poiché ne trassero un vantaggio evoluzionistico di sopravvivenza reciproco, svilupparono una relazione simbiotica permanente che nelle generazioni è divenuta indissolubile e imprescindibile; come esempio viene postulato che, nel passato remoto del teorico Precambriano, un batterio aerobico (che richiede ossigeno) sia stato ingerito da un batterio anaerobio (per il quale forse l’ossigeno era tossico) acquisendo un vantaggio reciproco e che continuando la loro relazione mutualistica abbiano superato evoluzionisticamente gli altri organismi in quell’ambiente; nel tempo il batterio interno ha perso o spostato materiale genetico nel nucleo dell’ospitante, per la codifica di tutto ciò che non era più necessario o superfluo.
Ma stiamo parlando di una teoria stabilizzata su estese sperimentazioni, osservazioni scientifiche ed analisi genetiche?
Breve approfondimento sulla endosimbiosi
Lynn Margulis.
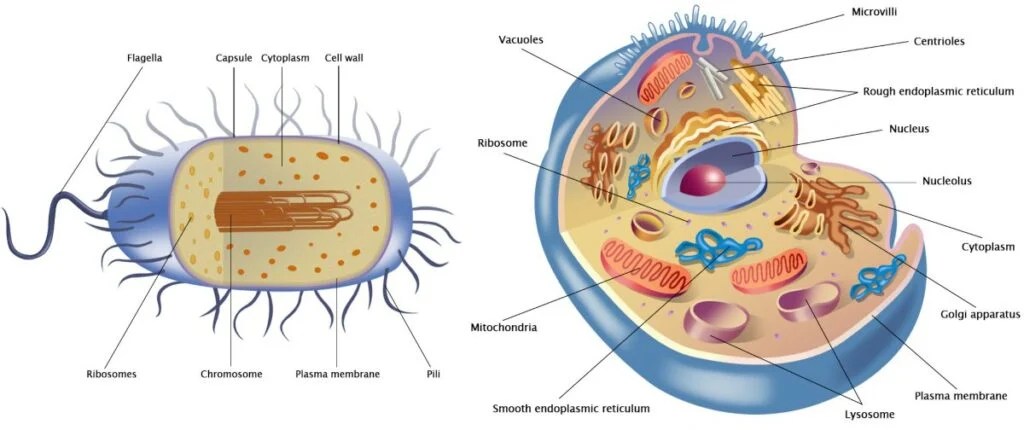
Come sappiamo, le cellule si dividono in due gruppi principali: procarioti (più piccola e semplice, ha il DNA sparso nel citoplasma (in una zona detta nucleoide) e non possiede organuli) ed eucarioti (hanno un nucleo ben definito che racchiude il DNA ed è ricca di organuli interni). La teoria dell’evoluzione postula che i procarioti si siano evoluti in eucarioti. Esiste un enorme divario tra questi due tipi di cellule che non avrebbe potuto essere colmato da forme di transizione inquanto avrebbero dovuto essere frutto di tentativi mutageni enormi e impossibilità o bassissima sopravvivenza. Il tentativo più diffuso di spiegare questo divario è la teoria dell’endosimbiosi di Lynn Margulis. I suoi sostenitori ipotizzano che alcune cellule proto-eucariotiche abbiano inglobato dei procarioti e che, successivamente, i proteobatteri inglobati si siano evoluti in organelli negli eucarioti primitivi. In questo articolo vengono esaminati i numerosi problemi principali di questa teoria, giungendo alla conclusione che essa è ampiamente accettata solo perché rappresenta l’ipotesi evolutiva più plausibile e non per via di prove empiriche. Infatti, come documentato in questo articolo, esistono numerose prove contrarie alla teoria dell’endosimbiosi.
Le differenze tra la struttura delle cellule procariote ed eucariote
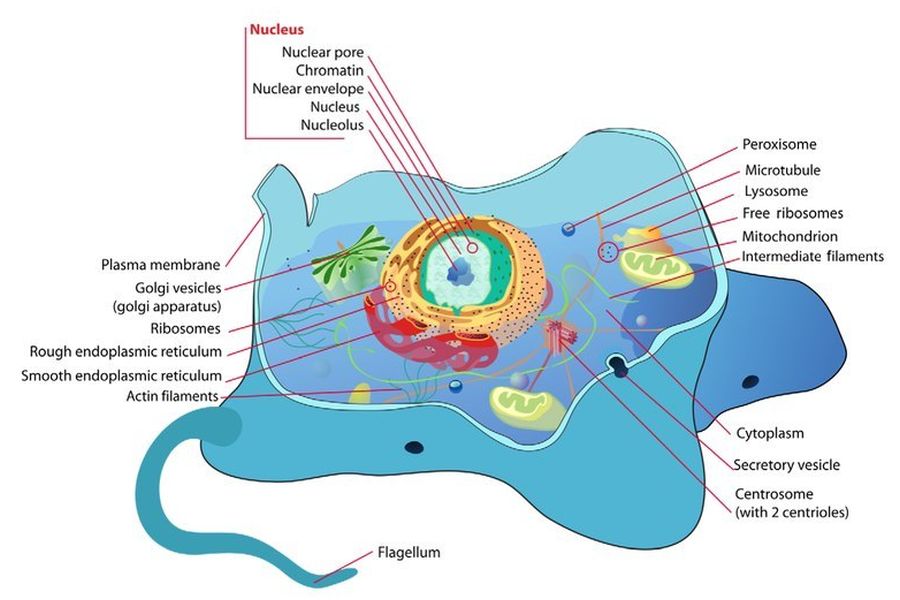
Comprendere l’evoluzione della complessità cellulare eucariotica è una delle grandi sfide della biologia moderna. A differenza dei procarioti, le cellule eucariotiche sono altamente compartimentalizzate e contengono molti organelli delimitati da membrana, assenti nei batteri o negli archei (procarioti unicellulari non batterici). Oltre a numerose differenze genetiche e molecolari, nei procarioti non si trovano organelli compartimentalizzati complessi. Gli evoluzionisti generalmente tentano di spiegare come ciò sia avvenuto attraverso l’endosimbiosi, sostenendo che un antico archeo abbia inglobato un proteobatterio che alla fine ha dato origine al primo organello, il Mitocondrio.
L’endosimbiosi è un fenomeno in cui un organismo vive all’interno di un altro in una relazione di mutuo beneficio, come si osserva nel caso di alcuni batteri che vivono all’interno delle termiti, ad esempio. In questo articolo, il termine “endosimbiosi” non si riferirà a tali fenomeni osservati, bensì all’ipotesi ampiamente accettata dal compianto Professor Lynn Margulis per spiegare l’origine dei Mitocondri, che è l’oggetto principale di questo lavoro (a volte indicata come “endosimbiosi primaria”). Essa viene invocata nel tentativo di colmare il divario esistente tra le cellule prive di organelli compartimentalizzati (procarioti) e quelle che li possiedono (eucarioti). Naturalmente, le differenze vanno ben oltre i Mitocondri. Alcuni degli altri organelli che un eucariote possiede, a differenza dei procarioti, sono il nucleo, il nucleolo, il reticolo endoplasmatico rugoso e liscio, l’apparato di Golgi, i centrioli, i perossisomi e i lisosomi.
Origine dei plastidi e teoria seriale dell’endosimbiosi
Secondo questa teoria dell’endosimbiosi, in un passato remoto un batterio procariote avrebbe inglobato un ipotetico proteobatterio, che sarebbe rimasto all’interno dell’ospite instaurando una relazione simbiotica. Ciò significa che centinaia di geni sarebbero stati in qualche modo modificati, assumendo funzioni completamente nuove. Inoltre, la teoria suggerisce che migliaia di geni del proteobatterio sarebbero stati trasferiti nel nucleo della cellula protoeucariote, mentre molti altri sarebbero stati scartati.
Il Mitocondrio come esempio di endosimbiosi(?)
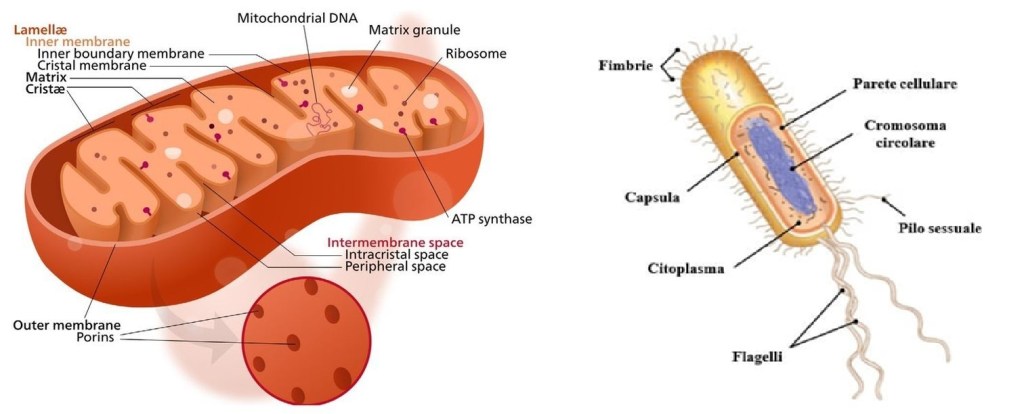
Il Mitocondrio (pl. mitocondri) è un organello presente nelle cellule della maggior parte degli eucarioti, come animali, piante e funghi. I Mitocondri hanno una struttura a doppia membrana e utilizzano la respirazione aerobica per generare adenosina trifosfato (ATP), che viene utilizzato in tutta la cellula come fonte di energia chimica. Furono scoperti da Albert von Kölliker nel 1857 nei muscoli volontari degli insetti. Il termine Mitocondrio, che significa “granulo filamentoso”, fu coniato da Carl Benda nel 1898. Il Mitocondrio è comunemente soprannominato “centrale energetica della cellula”, un’espressione resa popolare da Philip Siekevitz in un articolo del 1957 su Scientific American con lo stesso titolo.
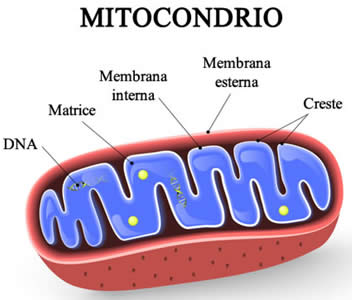
I Mitocondri hanno generalmente una sezione trasversale compresa tra 0,75 e 3 μm², ma variano considerevolmente in dimensioni e struttura. A meno che non vengano colorati specificamente, non sono visibili. Il Mitocondrio è composto da compartimenti che svolgono funzioni specializzate. Questi compartimenti o regioni includono la membrana esterna, lo spazio intermembrana, la membrana interna, le creste e la matrice.
Oltre a fornire energia cellulare, i Mitocondri sono coinvolti in altre funzioni, come la segnalazione, la differenziazione cellulare e la morte cellulare, nonché nel mantenimento del controllo del ciclo cellulare e della crescita cellulare. La biogenesi mitocondriale è a sua volta coordinata temporalmente con questi processi cellulari.
I Mitocondri sono implicati in disturbi e condizioni umane come malattie mitocondriali,, disfunzioni cardiache, insufficienza cardiaca e autismo.
Il numero di Mitocondri in una cellula varia notevolmente a seconda dell’organismo, del tessuto e del tipo di cellula. Un globulo rosso maturo non ha mitocondri, mentre una cellula epatica può averne più di 2000.
Sebbene la maggior parte del DNA di una cellula eucariotica sia contenuta nel nucleo cellulare, il Mitocondrio ha un proprio genoma (“mitogenoma”) simile ai genomi batterici [sottolineiamo SIMILE]. Questa scoperta ha portato all’accettazione di una parte della comunità scientifica della simbiogenesi (teoria endosimbiotica) – secondo la quale gli antenati procarioti a vita libera dei Mitocondri moderni si sono fusi permanentemente con le cellule eucariotiche in un lontano passato, evolvendosi in modo tale che gli animali, le piante, i funghi e gli altri eucarioti moderni respirino per generare energia cellulare.
Seguendo la “logica” endosimbiontica, i Mitocondri, organuli delle cellule eucariotiche, si sarebbero quindi originati come organismi procarioti esterni, introdottisi nella cellula come endosimbionti, circa 1,5 miliardi di anni fa. I mitocondri si sarebbero sviluppati da proteobacteria (in particolare, Rickettsiales o affini e forse da un batterio molto vicino a Rickettsia prowazekii.) Alcune ipotesi del fatto che i mitocondri si originarono da antiche endosimbiosi di batteri sono ad esempio:
I mitocondri contengono DNA diverso da quello del nucleo cellulare e simile a quello dei bacteria; si nota la presenza di un DNA circolare a doppia elica e la presenza di ribosomi propri e di una doppia membrana. Come i batteri, i mitocondri non hanno istoni ed i loro ribosomi sono sensibili ad alcuni antibiotici (come il cloramfenicolo). In più i mitocondri sono organelli semiautonomi in quanto replicano, per scissione binaria, autonomamente rispetto alla cellula.
Sono circondati da due o più membrane, la più interna delle quali mostra una composizione differente da quella delle altre membrane della cellula; si nota la presenza di molecole di cardiolipina ed assenza di colesterolo; la sua composizione è simile a quella di una membrana cellulare procariotica.
Nuovi mitocondri si formano solamente attraverso un processo simile alla scissione binaria. In alcuni casi i mitocondri possono essere distrutti da alcuni agenti ambientali o disfunzioni patogenetiche senza tuttavia danneggiare la cellula che li ospita, e comunque non si rigenerano.
La maggior parte della struttura interna e la biochimica dei mitocondri è molto simile a quella dei batteri. L’idea filogenetica, basata sui genomi di batteri, mitocondri ed eucarioti, suggerisce che i mitocondri siano strettamente derivati da bacteria.
L’analisi della sequenza del DNA e la teoria filogenetica suggeriscono che il DNA nucleare probabilmente contiene geni che vennero dai batteri/mitocondri originali inglobati.
Alcune proteine codificate nei nuclei sono trasportate agli organelli e i mitocondri hanno genomi piccoli se paragonati a quelli dei batteri. Ciò concorda con l’idea di un incremento della dipendenza sull’ospite eucariote dopo la formazione di un’endosimbiosi. La maggior parte dei geni dei batteri/organelli inglobati è andata perduti se inutile o si è spostata nel nucleo. La maggior parte dei geni necessari per le funzioni mitocondriali si trova nel nucleo. Molti hanno avuto origine dalla endosimbiosi batterica.
Se i mitocondri si fossero originati ex novo, dovrebbero averlo fatto molteplici volte, nel qual caso la loro rispettiva somiglianza è difficilmente spiegabile. Molti protisti contengono batteri ospitati secondari che sono stati acquisiti da altri eucarioti contenenti mitocondri, e non direttamente.
Tra gli eucarioti più antichi, i mitocondri sono maggiormente somiglianti ai batteri primigeni.
I ribosomi dei mitocondri sono come quelli trovati nei batteri (70S).
Le proteine originate dai mitocondri usano, come quelle dei batteri, N-formilmetionina come amminoacido iniziale.
Per colmare, o meglio, giustificare il fatto che non possono sopravvivere in ossigeno o fuori dalla cellula, si gioca la carta della perdita “funzionale” di molti geni necessari per la sopravvivenza, accaduto nel corso di un lungo intervallo di tempo in cui i mitocondri hanno co-abitato con i loro ospiti; i geni e i sistemi che non erano più necessari sarebbero stati semplicemente eliminati, o in molti casi trasferiti nel genoma ospite (infatti questi trasferimenti costituiscono un importante mezzo per la cellula ospite di regolare l’attività mitocondriale). Secondo recenti osservazioni si è ipotizzato che gli endosimbionti mitocondriali possono sopravvivere almeno per un po’ a vita libera in fluidi corporei stabili, come il sangue, e che siano in grado di trasferirsi da cellula a cellula in caso di necessità, quindi sempre passando per uno stadio a vita libera, anche se breve.
Ma le cose non sono “liquidabili” in questo modo…
L’abisso del DNA procariotico-eucariotico
I procarioti contengono una singola molecola di DNA circolare che occupa la regione del nucleoide e spesso anche piccoli anelli di DNA extracromosomico a doppio filamento chiamati plasmidi. Il DNA dei procarioti è “profondamente diverso da quello degli eucarioti” che contengono “da due a quattro genomi di DNA nucleare, cloroplastico, microtubulare e mitocondriale separati e trasmessi indipendentemente”. Il contrasto procariotico-eucariotico è così grande da costituire “la più grande singola discontinuità evolutiva” della vita conosciuta, e “l’origine degli eucarioti è rimasta una delle questioni più enigmatiche, controverse e impegnative dell’evoluzione”. Rimangono solo due teorie naturalistiche sull’evoluzione degli organelli; tutte le altre sono state effettivamente confutate.
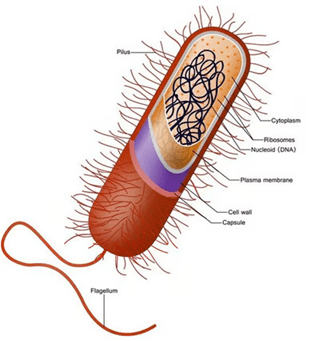
Rappresentazione semplificata di una cellula batterica e del suo interno.
La prima teoria è l’ipotesi autogena, ovvero quella dell'”autogenerazione”, che postula che gli organelli si siano evoluti gradualmente da un organello precursore attraverso la selezione naturale delle mutazioni. Questa visione non trova riscontro in tutti gli organelli esistenti e, di conseguenza, è stata ampiamente respinta e sostituita da una qualche forma di endosimbiosi.
La seconda teoria è quella dell’endosimbiosi, detta anche teoria dell’endosimbiosi seriale (SET), simbiogenesi o ipotesi xenogena. I batteri inglobati si sono successivamente evoluti all’interno dei loro ospiti per acquisire funzioni specializzate, alcuni diventando mitocondri che alla fine hanno assunto il ruolo di fornire energia “caricando” l’ADP in ATP. Questo avviene tramite l’enzima ATP sintasi, una “macchina” molecolare presente sia negli eucarioti che nei procarioti. I procarioti sono organismi microscopici unicellulari che hanno esigenze energetiche molto inferiori rispetto agli eucarioti.
L’apparato di sintesi dell’ATP in un procariote è incorporato nella sua membrana cellulare, come sarebbe accaduto anche per il batterio inglobatore, che in questa fase della teoria è ancora un procariote. Al contrario, gli eucarioti utilizzano i mitocondri per caricare l’ADP, quindi quando l’antico archeo avrebbe presumibilmente inglobato un proteobatterio che sarebbe poi diventato un mitocondrio, doveva comunque soddisfare il proprio fabbisogno energetico durante l’evoluzione da procariote a eucariote. Inoltre, solo una “piccola frazione delle proteine necessarie per la propagazione e la funzione dei mitocondri è codificata dai loro genomi, mentre i geni nucleari codificano la stragrande maggioranza”. Il fulcro della spiegazione dell’endosimbiosi per l’abisso tra procarioti ed eucarioti è il DNA mitocondriale.
Dall’endosimbiosi agli organismi multicellulari
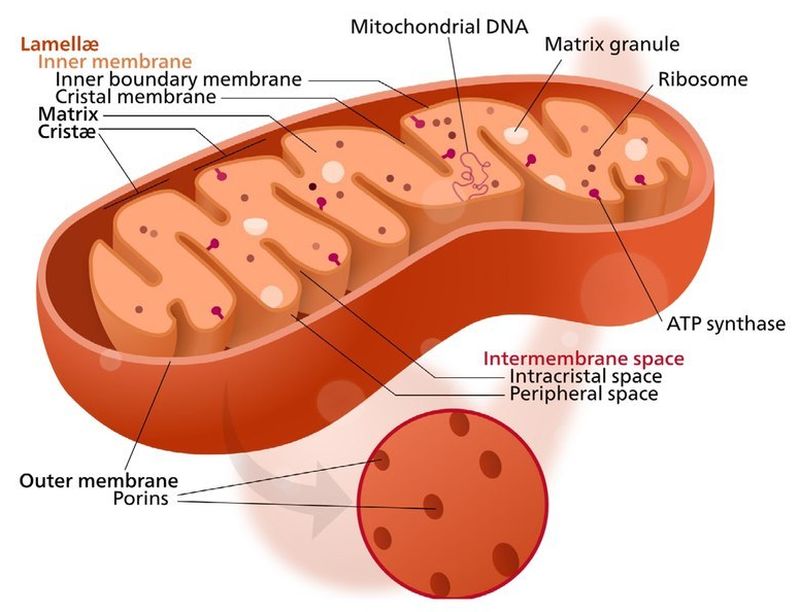
Schema semplificato di un mitocondrio. Si noti la notevole differenza rispetto alla cellula batterica sopra riportata.
È noto da tempo che “il principio dell’endosimbiosi fu proposto più di un secolo fa, ma fu generalmente considerato una ‘fantasia divertente'”. Questa visione rimase sostanzialmente invariata fino al lavoro di Lynn Margulis, che sviluppò l’idea dell’endosimbiosi in modo molto dettagliato. Grazie al suo lavoro e alla sua influenza, l’endosimbiosi è passata da un’idea oscura e scarsamente accettata alla teoria più popolare sull’origine degli organelli oggi.
Lo scenario comune dell’endosimbiosi postula che le cellule eucariotiche a vita libera si siano infine unite in comunità oggi chiamate organismi multicellulari.11 I problemi emersero poco dopo la proposta della teoria dell’endosimbiosi. Ad esempio, ulteriori ricerche hanno scoperto che i mitocondri di funghi, piante e animali erano così diversi che l’endosimbiosi deve essersi verificata indipendentemente molte volte, il che non fa che moltiplicare le probabilità, già di per sé molto improbabili, che lo scenario endosimbiotico si verifichi anche solo una volta.
Margulis predisse che il DNA mitocondriale sarebbe stato diverso dal DNA nucleare, ma che sarebbe invece consistito in una miscela di sequenze provenienti da geni eubatterici e archeali. Nel 1963, si scoprì che i mitocondri possedevano DNA indipendente dal DNA nucleare. Un altro fattore importante a sostegno dell’endosimbiosi fu la scoperta, negli anni ’60, del DNA plastidiale (cpDNA) nei cloroplasti delle piante, chiamato plastoma, che secondo i sostenitori rendeva più plausibile il meccanismo dell’endosimbiosi per l’origine degli organelli.15
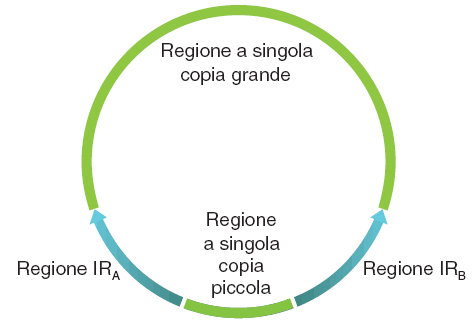
Rappresentazione schematica del DNA plastidiale.
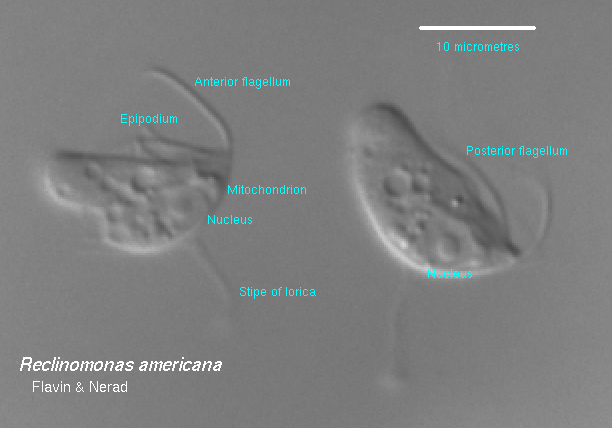
Si ritiene comunemente che gli organelli chiamati mitocondri siano comparsi una sola volta nella storia evolutiva, ma nonostante la loro comune origine, i DNA mitocondriali “L’architettura del genoma e il contenuto genico variano ampiamente tra gli eucarioti.” I ricercatori hanno scoperto che il mtDNA più grande si trova in un protozoo d’acqua dolce, Reclinomonas americana, che ha 69.034 nucleotidi e 97 geni che codificano 67 proteine, tra cui almeno 18 proteine non precedentemente note per essere codificate nei mitocondri. L’endosimbiosi deve quindi postulare che i batteri inglobati abbiano perso il 96-99% delle loro proteine, da circa 1.600 a meno di 67, a seconda dello specifico protobatterio inglobato. Per gli esseri umani, solo 37 geni sono essenziali per la respirazione cellulare.
Inoltre, i confronti tra proteine eucariotiche presenti negli organelli eucariotici hanno rivelato che non si tratta semplicemente di una miscela di sequenze provenienti da archei ed eubatteri, come previsto dall’endosimbiosi, ma che sono spesso uniche, in contrasto con l’idea che i genomi eucariotici siano una combinazione di geni eubatterici e archeali.
Anche tutti gli altri esempi esaminati mostrano notevoli differenze tra i genomi mitocondriali e batterici, come previsto date le differenze proteiche descritte da Kirkland in un paragrafo precedente. Persino per l’esempio più noto (mtDNA di R. americana), esistono enormi differenze tra i suoi 69 kbp (97 geni) e sia il genoma di 580 kbp (470 geni) di Mycoplasma genitalium, sia il genoma di 1.830 kbp (1.743 geni) di Haemophilus influenzae. Inoltre,
“Il confronto tra i genomi di Mycoplasma e Haemophilus ha suggerito che il loro diverso contenuto genico riflette ‘profonde differenze… tra questi due organismi’… In questo contesto, il genoma mitocondriale di Reclinomonas può essere considerato un esempio estremo di riduzione del genoma eubatterico, in cui gli unici geni rimanenti sono correlati all’espressione genica mitocondriale (trascrizione, elaborazione dell’RNA e traduzione) e alla biogenesi dei complessi proteici necessari per il trasporto di elettroni e la fosforilazione ossidativa accoppiata (inclusi i componenti implicati nel trasporto e nella biosintesi delle proteine mitocondriali).”¹⁹
In breve, questi pochi esempi illustrano l’abisso esistente tra il DNA batterico e il mtDNA, che è solo uno dei molti problemi dell’endosimbiosi.
Somiglianze e differenze tra mitocondri e batteri
La teoria dell’endosimbiosi si basa in gran parte sull’omologia tra organelli e batteri. Ad esempio, ogni mitocondrio possiede un genoma circolare come quello dei batteri, ma molto più piccolo e privo di proteine istoniche. Il DNA mitocondriale (mtDNA) si trova solitamente nella matrice mitocondriale, sebbene a volte sia attaccato alla membrana mitocondriale interna.
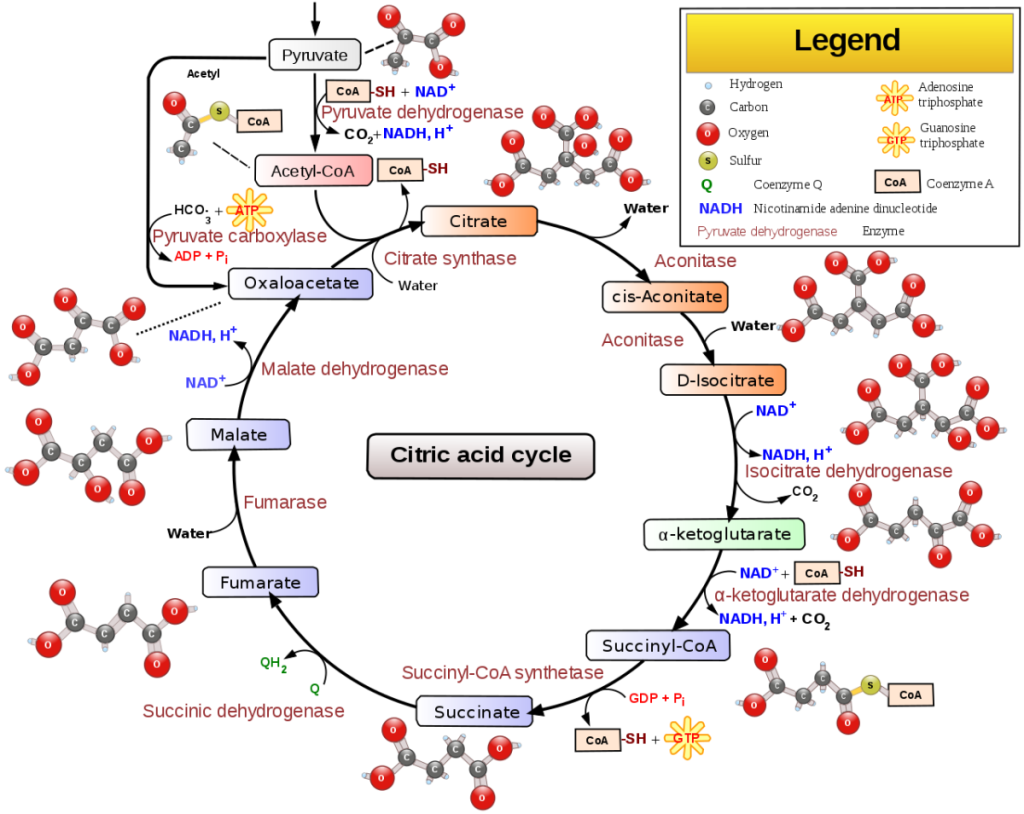
I mitocondri assomigliano molto ai batteri aerobici viola per dimensioni e forma. Entrambi utilizzano l’ossigeno per la produzione di ATP tramite il ciclo di Krebs. Alcuni antibiotici che uccidono i batteri inibiscono anche le funzioni mitocondriali. Queste somiglianze generali da sole non dimostrano l’endosimbiosi perché, come documentato di seguito, esistono molte differenze significative, spesso importanti e critiche.
Rappresentazione schematica del Ciclo di Krebs
Un fattore a favore dell’endosimbiosi è la composizione della membrana. La membrana esterna sia dei cloroplasti che dei mitocondri presenta somiglianze strutturali e chimiche con la membrana cellulare procariotica. Ricerche successive, tuttavia, hanno determinato che le membrane mitocondriali sono solo superficialmente simili alle membrane cellulari procariotiche. Una differenza risiede nel fatto che il proteobatterio che si presume sia penetrato nel protoeucariote avrebbe avuto una singola membrana, mentre i mitocondri moderni ne possiedono una doppia (interna ed esterna). La doppia membrana non è facoltativa, ma fondamentale per la sua funzione di caricamento dell’ADP. La membrana interna contiene numerose pieghe a forma di piastra chiamate creste, che presentano sacche membranose contenenti enzimi. Le creste possono essere esclusivamente lamellari o esclusivamente tubulari, ma alcuni mitocondri contengono entrambi i tipi. Un’altra differenza è che la membrana mitocondriale interna ha una composizione chimica diversa da quella dei procarioti, ma è identica a quella degli eucarioti, contrariamente a quanto previsto dalla teoria dell’endosimbiosi.
Christian de Duve (1917–2013)
Margulis propose “che gli eucarioti si fossero formati a seguito di una graduale unione multi-endosimbiotica con i procarioti. Al contrario, altri, come de Duve e Stanier, proposero che la fagotrofia, che richiede un citoscheletro dinamico, un sistema endomembranoso e la perdita della parete cellulare rigida procariotica, si fosse evoluta prima dell’endosimbiosi”. In contrasto con questa proposta, la filogenetica basata sul mtDNA concluse in seguito che i mitocondri avrebbero potuto evolversi una sola volta da un α-proteobatterio. La visione dell’endosimbiosi come evoluzione degli organelli è ampiamente accettata non per via di prove empiriche, ma perché nessun’altra teoria è neanche lontanamente plausibile. Per questo motivo, Battley descrive l’endosimbiosi come “nella migliore delle ipotesi provvisoria”.
Poiché non esistono prove fisiche per la maggior parte delle fasi della transizione dalla cellula procariotica alla cellula eucariotica, si ricorre a ragionamenti astratti (ad esempio, i mitocondri e i cloroplasti possiedono un piccolo plasmide di DNA che superficialmente assomiglia al DNA procariotico) come supporto. In realtà, il DNA degli organelli è più simile ai geni nucleari eucariotici. Un esempio ben noto di alcuni geni degli organelli che assomigliano ai geni nucleari eucariotici è la presenza di introni, che sono raramente presenti nei geni procariotici.
L’endosimbiosi non risolve il problema dell’origine degli organelli.
Sebbene l’origine endosimbiotica dei mitocondri e dei cloroplasti sia ormai un dogma consolidato nei manuali, le proposte secondo cui la maggior parte degli “altri compartimenti cellulari siano il risultato della simbiosi… non sono altrettanto ampiamente accettate”. Per la maggior parte degli altri organelli, l’endosimbiosi è considerata da molti ricercatori una spiegazione implausibile per la loro origine. (È anche una spiegazione inadeguata per il mitocondrio e il cloroplasto, sebbene la teoria dell’evoluzione del cloroplasto sarà trattata in un articolo separato).
L’endosimbiosi non è una spiegazione adeguata per i mitocondri
Un grave problema scientifico dell’ipotesi dell’endosimbiosi è che, fin dalla sua prima formulazione, la teoria si è rivelata (e rimane tuttora) non verificabile.26 Il problema è che l’endosimbiosi “non propone un meccanismo reale e la maggior parte dei libri di testo presenta l’immagine semplicistica di una cellula che ingloba un’altra cellula che diventa un mitocondrio”. In realtà, oggi è molto meno plausibile di quando fu proposta per la prima volta, poiché oggi si sa molto di più sugli organelli (ad esempio i mitocondri) e sui batteri.
Tra gli altri problemi fondamentali della teoria vi sono: cosa ha impedito alla cellula ospite di digerire l’organismo invasore? e: da dove provengono le numerose altre strutture necessarie alla sopravvivenza di una cellula eucariotica? Ad esempio, i microtubuli non sono spiegati dalla teoria, sebbene siano necessari per la divisione cellulare e la motilità nelle cellule eucariotiche. De Duve osserva che non si sa nulla sull’evoluzione del sistema del citoscheletro cellulare, che richiede molte nuove innovazioni per funzionare. Simili prove citate a sostegno della teoria secondo cui i batteri spirocheti avrebbero dato origine ai flagelli sono problematiche. La tubulina, il componente principale dei microtubuli nelle cellule eucariotiche, non è stata trovata in nessun procariote. Per questi motivi, la maggior parte dei biologi evoluzionisti rifiuta l’idea che flagelli, tubulina e la maggior parte delle altre strutture cellulari abbiano avuto origine per endosimbiosi. Nella migliore delle ipotesi, l’endosimbiosi spiega l’origine di uno o due organelli. Ma affinché una cellula eucariotica funzioni, è necessario un insieme completamente nuovo di strutture, tutte le quali devono evolversi simultaneamente per garantire l’integrità funzionale.
L’endosimbiosi tenta di spiegare l’origine evolutiva di un solo organello nelle cellule animali. Ad oggi, dopo 150 anni di tentativi, l’origine evolutiva delle 17 strutture di base mostrate nel diagramma sopra, incluso il mitocondrio, non ha una spiegazione universalmente accettata. Per la maggior parte delle 17 strutture, esistono solo ipotesi provvisorie. Tutte queste strutture sono necessarie, in qualche forma, all’esistenza di una cellula eucariotica.
Principali differenze tra i ribosomi e il mitoribosoma
Un argomento fondamentale a sostegno della teoria endosimbiotica sull’origine degli organelli era che la struttura dei ribosomi mitocondriali è “nettamente diversa” da quella dei ribosomi eucariotici, e che i ribosomi mitocondriali assomigliano a quelli dei procarioti. Contrariamente alla teoria endosimbiotica, tuttavia, i ribosomi mitocondriali dei mammiferi e le sequenze di amminoacidi che li compongono sono completamente diversi dalle corrispondenti caratteristiche nei procarioti. I ribosomi mitocondriali sono infatti così diversi dai ribosomi batterici da essere denominati mitoribosomi.
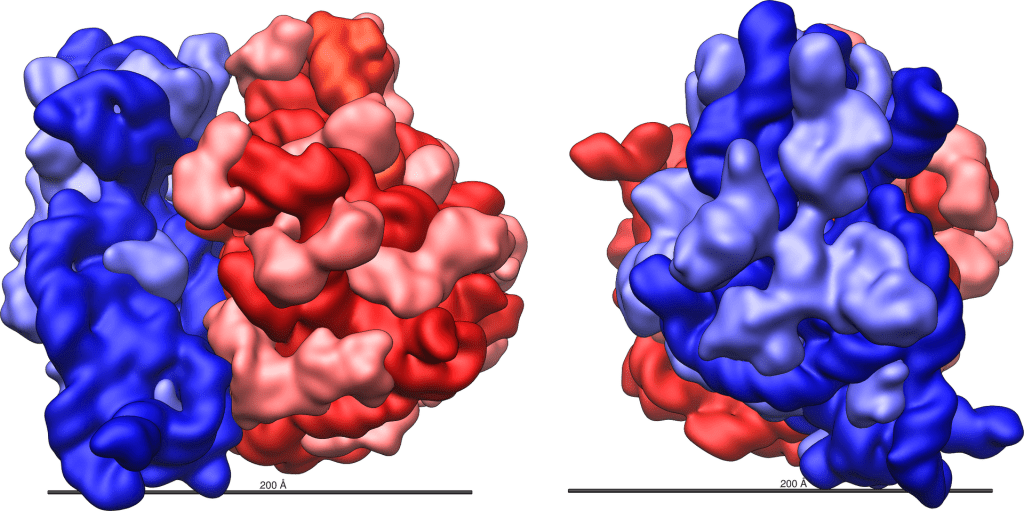
Ribosoma 70S di Escherichia coli. In rosso la subunità grande e in blu quella piccola. La scala è 200 Ångström (20nm). I colori più chiari (azzurro e rosa) indicano le proteine.
Quando l’endosimbiosi fu proposta per la prima volta, si presumeva che i ribosomi esistessero solo in due forme: una varietà più piccola, 70S, utilizzata nei procarioti, e un ribosoma più grande, 80S, utilizzato negli eucarioti. La S in 70S si riferisce all’unità di misura del coefficiente di sedimentazione. Il coefficiente di sedimentazione misura le differenze morfologiche di base, una quantità correlata alla dimensione della particella che è pari alla velocità terminale di avanzamento della particella quando centrifugata in un mezzo fluido standard, divisa per la forza centrifuga che agisce su di essa. Ci si aspettava che i ribosomi utilizzati nei mitocondri dei mammiferi assomigliassero ai ribosomi 70S dei procarioti, data la loro simile dimensione. Invece, i ricercatori hanno scoperto che “i ribosomi mitocondriali dei mammiferi (55S) differiscono inaspettatamente dai ribosomi batterici (70S) e citoplasmatici (80S), così come da altri tipi di ribosomi mitocondriali”.
La struttura completa del ribosoma mitocondriale 55S dei mammiferi
I mitocondri utilizzano un sistema necessario per la produzione di proteine che è anch’esso molto diverso da quello dei batteri. I ribosomi mitocondriali, chiamati mitoribosomi, sono descritti come mini-ribosomi “in miniatura”, contenenti una mini-RNA polimerasi e persino un mini-DNA. I mitoribosomi si differenziano dai ribosomi procariotici per il contenuto di RNA e proteine, nonché per la posizione e la funzione delle diverse componenti ribosomiali, e presentano differenze significative nella sequenza del DNA (in particolare nelle regioni che non entrano in contatto con il tRNA o con la catena polipeptidica in crescita). Tra le caratteristiche peculiari del ribosoma mitocondriale si annoverano nuovi mRNA che processano l’mRNA mitocondriale e un nuovo sito di legame per il guanosina trifosfato (GTP) utilizzato durante l’allungamento della catena polipeptidica.
Altre differenze tra i ribosomi batterici e i mitoribosomi riguardano la loro struttura e i loro processi di assemblaggio. Le proteine costituiscono una porzione maggiore dei mitoribosomi rispetto ai ribosomi procariotici. Alcune di queste proteine si trovano in posizioni inedite e svolgono funzioni diverse rispetto a quelle del ribosoma procariotico. Uno dei tanti esempi è rappresentato dal mitoribosoma 55S, tenuto insieme da 15 ponti intersubunitari, di cui solo sei simili a quelli utilizzati nei procarioti.33 Inoltre, 33 delle 81 proteine identificate finora nei mitoribosomi umani non hanno omologhi nei ribosomi procariotici.
Questi esempi delle numerose differenze tra il ribosoma procariotico e quello mitocondriale illustrano ulteriormente l’abisso che separa i due tipi di ribosoma. Si potrebbero documentare altri esempi e senza dubbio ne verranno scoperti altri con ulteriori ricerche.
Il problema della fagocitosi
Un altro problema è che la fagocitosi avrebbe presumibilmente introdotto un proteobatterio nei procarioti attraverso la “fagocitosi” di batteri, ma “la natura precisa della cellula ospite che si è associata a questo endosimbionte è, tuttavia, una questione ancora aperta”.¹ Il problema è che gli archei – in realtà tutti i procarioti – “non sono in grado di eseguire la fagocitosi e non c’è motivo di credere che abbiano mai avuto tali capacità”. Di conseguenza, “il modo in cui l’endosimbionte è entrato nel suo ospite è un enigma”. Il motivo per cui i procarioti non possiedono la capacità di fagocitosi è che si tratta di un processo complesso che richiede
Rappresentazione schematizzata della Fagocitosi
“… una parete cellulare flessibile, un citoscheletro interno dinamico con proteine motrici che interagiscono con un complesso sistema endomembranoso, lisosomi che gemmano dal complesso di Golgi e sono diretti verso i vacuoli alimentari e particelle racchiuse in una coppa fagocitica basata sulla polimerizzazione spazialmente controllata dell’actina. “I caratteri sono assenti nei procarioti.”
Una soluzione teorica a questo problema suggerisce che l’organismo invasore “fosse un piccolo α-proteobatterio (facoltativo) aerobico, che penetrava e si replicava nel periplasma dell’ospite, diventando in seguito i mitocondri cellulari.” Questa storia, per ora, non trova riscontro empirico.
Altre differenze tra i mitocondri e i batteri
I mitocondri sono organelli importanti perché contengono i meccanismi e gli enzimi necessari per convertire il cibo in una molecola energetica chiamata adenosina trifosfato (ATP). Il processo enzimatico mediante il quale i mitocondri convertono il cibo in adenosina trifosfato (ATP) è chiamato fosforilazione ossidativa. Il processo finale prevede la conversione dell’adenosina difosfato (ADP) nella molecola ad alta energia ATP. I mitocondri producono il 90% dell’energia cellulare e la loro disfunzione causa diverse malattie che colpiscono il sistema nervoso centrale e, in seguito, altri sistemi.38 I mitocondri producono questa energia a partire da grassi, zuccheri e proteine; sono presenti in tutte le cellule umane, ad eccezione dei globuli rossi anucleati.
Differenze tra Mitocondrio e un batterio
Definiti “le centrali energetiche della cellula”, sappiamo ora che le cellule più attive, come quelle muscolari, epatiche e dei tubuli renali, contengono un gran numero di mitocondri. Al contrario, le cellule meno attive, come quelle che secernono muco, contengono un numero relativamente basso di mitocondri. Altre funzioni dei mitocondri sono di tipo regolatorio, tra cui il controllo del livello di calcio citoplasmatico.39 Sono inoltre coinvolti in specifici processi di sintesi lipidica.
Le membrane interne contengono un ampio insieme di enzimi che convertono il cibo in ADP carico attraverso una serie di reazioni chiamate ciclo di Krebs o ciclo dell’acido citrico, che producono fosforilazione ossidativa. La regione interna del mitocondrio, chiamata matrice, è riempita di un gel contenente numerosi enzimi di diverso tipo. Le membrane interna ed esterna differiscono sia per attività enzimatica che per composizione lipidica. Alcuni enzimi, come l’ATPasi, sono permanentemente attaccati alla membrana mitocondriale. In breve, i mitocondri sono molto diversi dai batteri.
Geni che controllano i mitocondri
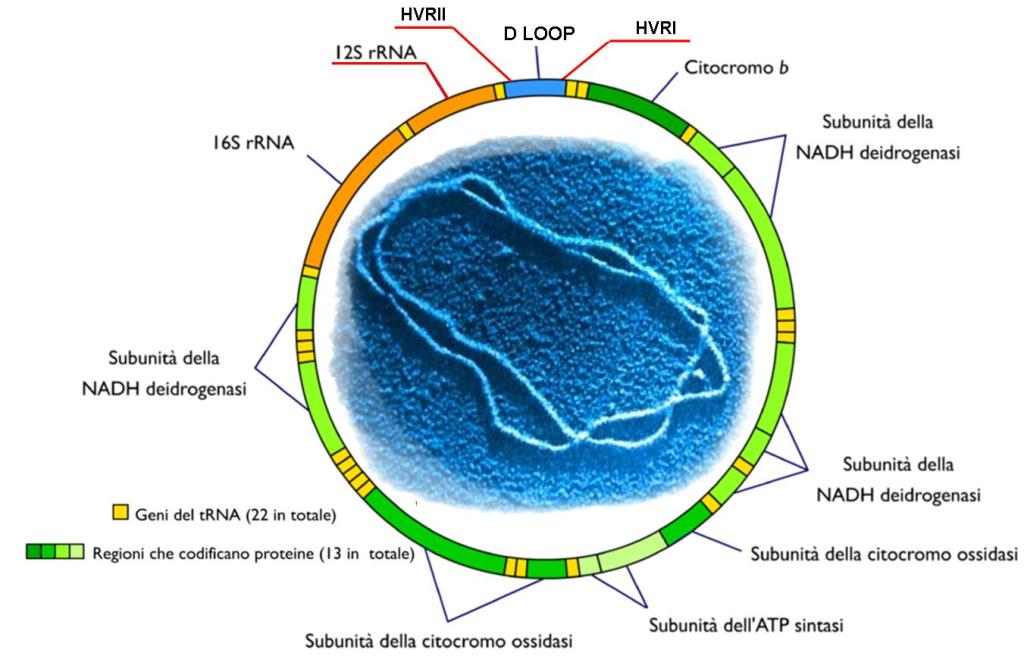
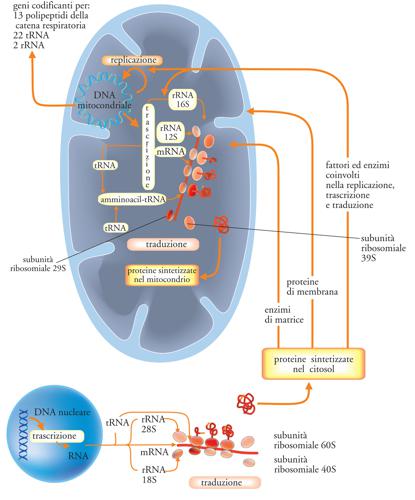
Come già accennato, il mitocondrio è un organello unico perché contiene il proprio DNA (mtDNA) sotto forma di plasmidi. Il DNA mitocondriale è utilizzato esclusivamente per le funzioni dell’organello stesso, in particolare per controllare, seppur non completamente, la propria replicazione.
Il mtDNA umano è stato completamente sequenziato. I suoi 16.569 paia di basi codificano per 37 geni, tra cui solo 13 geni che codificano per proteine, 22 geni per tRNA e due geni per rRNA, tutti essenziali. I sistemi respiratori non possono funzionare senza la presenza di tutte queste proteine e tRNA. Questi sono solo alcuni dei geni necessari ai mitocondri umani. Circa il 90% delle proteine importate dal citoplasma sono codificate nel nucleo, il che indica un elevato livello di integrazione che rende l’endosimbiosi insostenibile.
DNA mitocondriale
Poiché la maggior parte dei geni che controllano i mitocondri non si trova negli organelli stessi, ma nel nucleo cellulare, i sostenitori dell’endosimbiosi devono postulare un trasferimento di geni dagli organelli al nucleo della cellula ospite. Questo problema non è di poco conto: “la migrazione dei geni dagli endosimbionti al nucleo è notevole perché sembra aver sollevato più difficoltà di quante ne abbia risolte”. Un altro problema è:
“… in quale forma i geni trasferiti compiono fisicamente questo viaggio intracellulare: come RNA, come cDNA, come frammenti di DNA degli organelli o come interi cromosomi degli organelli? Le attuali teorie si concentrano sul cDNA come veicolo, basandosi su alcuni esempi tratti dalle piante. Ma altri meccanismi, che implicano il trasferimento diretto del DNA dai cromosomi degli organelli, potrebbero anche spiegare i dati disponibili”.
L’analogia non è dissimile dall’ipotizzare il trasferimento di una piccola casa in una casa più grande per spiegare le stanze di quest’ultima, quando queste possono essere spiegate più facilmente, anche da un punto di vista evolutivo, ipotizzando la loro costruzione individuale e separata. Questa preoccupazione è significativa in quanto i geni nei mitocondri rappresentavano una delle principali prove originali a sostegno della teoria dell’endosimbiosi. Da un punto di vista darwiniano, l’ipotesi che l’endosimbiosi ha sostituito, ovvero il processo di infiltrazione delle membrane all’interno delle cellule ospiti formando tutti gli organelli, incluso il nucleo, appare più plausibile e quindi ha ancora dei sostenitori. Man mano che i problemi con l’endosimbiosi si accumulano, l’ipotesi dell’infiltrazione delle membrane potrebbe tornare in auge.
In breve, la genetica suggerisce che questo trasferimento genico deve essersi verificato se l’endosimbiosi ha avuto luogo. I veri problemi sono: come sono sopravvissute le cellule pre-eucariotiche fino al trasferimento dei geni, come sono sopravvissute prima di questo trasferimento e perché e come sono stati trasferiti. La replicazione mitocondriale sembra richiedere il sistema di controllo nucleare perché, per quanto ne sappiamo, è universale. Tuttavia, il motivo per cui molti geni importanti per la funzione mitocondriale si trovino nel nucleo e come vi siano arrivati è oggetto di molte speculazioni evoluzionistiche.
Una delle scoperte più inaspettate è stata la scarsità di geni che potrebbero supportare l’endosimbiosi. Uno studio ha scoperto, contrariamente alle aspettative della teoria, che i confronti con l’organismo, chiamato endosimbionte α-proteobatterico, ampiamente ritenuto essere l’endosimbionte batterico
“… hanno identificato un nucleo conservato di proteine derivate dall’endosimbionte α-proteobatterico che ha dato origine al mitocondrio ed è stata la fonte del genoma mitocondriale negli eucarioti contemporanei. Un risultato sorprendente delle analisi filogenetiche è la proporzione relativamente piccola (10-20%) del proteoma mitocondriale che mostra una chiara ascendenza α-proteobatterica. Una grande frazione di proteine mitocondriali ha tipicamente omologhi rilevabili [similarità di sequenza] solo in altri eucarioti e si presume rappresenti proteine emerse specificamente all’interno degli eucarioti.”
I ricercatori hanno concluso:
“Comprendere l’origine e l’evoluzione del mitocondrio rimane una sfida, nonostante le Un’ondata di dati e intuizioni biochimiche, cellulari, di biologia molecolare e filogenetica, accumulate nei quasi cinque decenni trascorsi dalla moderna rinascita della consolidata ipotesi dell’endosimbiosi; l’idea che questo organello sia un batterio endosimbionte addomesticato e altamente rielaborato. L’abbondanza di informazioni relative all’evoluzione delle cellule eucariotiche (in particolare e più recentemente i dati di sequenza) e le divergenze su come i dati vengono analizzati e interpretati hanno generato una pletora di idee, spesso contrastanti, su quando e come, in un contesto endosimbiontico, si sia originato il mitocondrio.
Mitocondri dipendenti dal genoma nucleare
Un altro problema importante dell’endosimbiosi è che il genoma del mtDNA non è indipendente, ma è funzionalmente integrato con il genoma nucleare. Sono necessarie oltre 90 proteine per produrre i ribosomi mitocondriali, e quasi tutte vengono fornite all’organello dal nucleo dell’ospite. (Tra le fonti consultate, nessuna è stata in grado di fornire un numero specifico che, al momento della stesura di questo testo, rimane sconosciuto). Questi geni sono codificati dal DNA nucleare; le proteine risultanti vengono sintetizzate nel citosol cellulare e poi trasportate singolarmente all’interno dell’organello.
I mitocondri umani “devono importare il 99% delle loro proteine dal citoplasma”.51 Sarebbe molto più semplice (e quindi, secondo la “legge della parsimonia”, una spiegazione migliore) far evolvere i mitocondri da zero piuttosto che incorporare un organismo indipendente che richiederebbe: 1) la perdita della maggior parte dei suoi geni; 2) l’evoluzione di molti nuovi geni, il che implica: 3) Il fatto che la maggior parte dei geni necessari al funzionamento debba essersi evoluta originariamente nel nucleo e che, senza questi geni, il mitocondrio non potrebbe funzionare. Inoltre, l’utilizzo del sistema a due geni, nucleare e mitocondriale, richiede l’evoluzione di un macchinario di importazione estremamente complesso che coinvolge recettori di superficie complessi, sistemi di legame e un sistema di segnalazione mirato.
Un altro problema complesso è che alcuni mitocondri utilizzano codoni genetici diversi da tutti i codici batterici ed eucariotici. Ad esempio, il codone CUA normalmente codifica per la leucina, ma nei mitocondri del lievito codifica per la treonina. In realtà, esistono molteplici codici per codificare la treonina, e sono presenti modelli di riconoscimento dei codoni più complessi per altri amminoacidi. Per questi motivi, alcuni ipotizzano che il codice mitocondriale si sia evoluto in alcuni mitocondri di lievito, poiché, per quanto ne sappiamo, tutti i mitocondri di lievito presentano le stesse differenze di codice rispetto ai batteri. Man mano che verranno completati ulteriori sequenziamenti genetici su diversi mitocondri di altri organismi, è probabile che vengano scoperte altre differenze di codifica.
Un codice mitocondriale identico a quello utilizzato nei batteri, ma diverso da quello utilizzato negli eucarioti, se così fosse, potrebbe avvalorare l’ipotesi dell’endosimbiosi. Tuttavia, la differenza effettivamente esistente ha spinto alcuni evoluzionisti a ipotizzare che il codice mitocondriale sia più “primitivo” e quello batterico più evoluto. Contrariamente a questa visione, la differenza è meglio spiegata dall’ipotesi che il codice mitocondriale sia progettato per le esigenze specifiche dei mitocondri, in particolare per impedire lo scambio di geni del mtDNA con geni nucleari.
Spostare il problema altrove
Un altro problema importante della teoria dell’endosimbiosi è che non risolve il problema dell’evoluzione degli organelli. Al contrario, evita il problema perché parte dall’esistenza di un sistema complesso e funzionante che non è in grado di spiegare. A fini argomentativi,
«… supponiamo che la simbiosi immaginata da Margulis sia stata in realtà un evento comune nel corso della storia della vita. La domanda importante per noi biochimici è: la simbiosi può spiegare l’origine di sistemi biochimici complessi?
«Chiaramente no. L’essenza della simbiosi è l’unione di due cellule separate, o due sistemi separati, entrambi già funzionanti. Nel caso del mitocondrio, una cellula vitale preesistente è entrata in una relazione simbiotica con un’altra cellula simile. Né Margulis né nessun altro ha offerto una spiegazione dettagliata di come si siano originate le cellule preesistenti.»
Inoltre, i sostenitori della teoria simbiotica devono «presupporre che le cellule invasive fossero già in grado di produrre energia da sostanze nutritive; Essi [presuppongono] esplicitamente che la cellula ospite fosse già in grado di mantenere un ambiente interno stabile che avrebbe avvantaggiato il simbionte.”
Margulis e Sagan proposero che le prime cellule eucariotiche fossero i protottisti: le amebe, le diatomee, le alghe giganti e le alghe rosse. Queste creature eucariotiche, tuttavia, sono per molti aspetti più simili agli eucarioti di livello “superiore” che ai procarioti. Anche se la teoria dell’endosimbiosi non si adatta ai fatti esaminati in questo articolo, essa viene comunque periodicamente riproposta quando le teorie alternative si rivelano errate.
Carl Richard Woese (1928 – 2012)
Un’altra indicazione dei problemi dell’endosimbiosi è l’ampio disaccordo tra i ricercatori sui meccanismi alla base del concetto. Ad esempio, Margulis e Sagan notano che alcuni batteri sono stati rinominati “archei” da Carl Woese, una terminologia ora ampiamente accettata.
“Questa classificazione rifiuta l’endosimbiosi ed è una “negazione della loro natura batterica” perché Ciò comporta l’elevazione del gruppo degli “archei” a uno status paritario rispetto agli altri batteri e a tutti gli eucarioti. Il risultato è la formazione di tre gruppi fondamentali chiamati domini, o superregni, il che contraddice la teoria dell’endosimbiosi.58
Un’altra preoccupazione è che l’endosimbiosi implichi un’evoluzione relativamente rapida, in contrasto con l’evoluzione graduale darwiniana. Dato che non esiste una spiegazione gradualista plausibile per l’evoluzione degli eucarioti, gli evoluzionisti sono stati spinti a combinare posizioni gradualiste e non gradualiste darwiniane. O’Malley scrive che, come sosteneva Maynard Smith, l’endosimbiosi è spiegata dalle mutazioni standard e dalle macromutazioni.59 Altri ricercatori sostengono che si debbano tenere in considerazione altri fattori, rendendo l’evoluzione degli eucarioti ancora più complessa (e di conseguenza meno probabile). Ad esempio, Edgar aggiunge che “l’evoluzione concomitante del sistema redox dell’acido L-ascorbico dovrebbe essere considerata un fattore chiave che ha portato all’evoluzione degli eucarioti multicellulari e rimane coinvolta nel mantenimento della multicellularità e di molte altre caratteristiche eucariotiche”.
Perché l’evoluzione degli organelli è [praticamente] impossibile
Behe sostiene che il divario esistente tra cellule eucariotiche e procariotiche non potrà mai essere colmato a causa della complessità irriducibile. La complessità anche di una macchina semplice può essere ridotta solo fino a un certo punto: al di sotto di tale limite, la macchina non può funzionare. L’esempio classico è una trappola per topi standard, che deve avere almeno cinque componenti principali per funzionare: una piattaforma, una barra di supporto, un martello, un fermo e una molla.
Una trappola per topi non funzionerà finché non saranno presenti tutte le sue parti necessarie, ognuna delle quali deve essere progettata correttamente per articolarsi con le altre. Un’ipotesi contraria è quella di proporre che alcune di queste parti possano essere eliminate con vari metodi, come ad esempio inchiodando la trappola al pavimento. Questo approccio non elimina una parte, ma la sostituisce con un’altra; il pavimento viene utilizzato come base. Allo stesso modo, gli organelli non funzioneranno se non esiste ogni parte necessaria, correttamente progettata e prodotta, e tutte queste parti devono essere assemblate correttamente per formare un sistema operativo.
Gli organelli sono strutture molto complesse, costituite da migliaia di parti complesse più piccole, e il problema della complessità irriducibile è molto probabilmente valido anche per ogni singola parte di ciascun organello. Una cellula non può sopravvivere senza i ribosomi, ognuno dei quali contiene migliaia o addirittura decine di migliaia di molecole, ognuna delle quali deve essere assemblata secondo specifiche precise. Pertanto, la vita cellulare è impossibile finché tutte le sue parti necessarie non vengono prodotte e assemblate correttamente. Sebbene il DNA sia descritto come rappresentante di “un’intelligenza massiccia… [esso] non ha di per sé né futuro né presente. Il DNA senza una cellula che lo sostenga ed esprima non ha alcun significato fisiologico”.
Pochi scienziati si sono persino cimentati nello speculare sui dettagli delle forme di transizione tra gli ipotetici pre-organelli, per non parlare della presentazione di prove delle migliaia di forme di transizione necessarie a creare uno scenario plausibile che possa collegare le cellule a vita libera con le cellule dotate di organelli utilizzate negli organismi multicellulari.
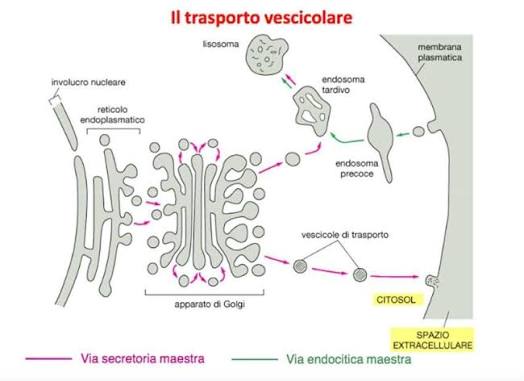
Il sistema di trasporto cellulare è un altro esempio che illustra perché il concetto di complessità irriducibile renda impossibile l’evoluzione degli organelli. Dopo essere state prodotte, le proteine non fluttuano liberamente all’interno della cellula, ma devono essere trasportate da un meccanismo appropriato ovunque siano necessarie. Due meccanismi comuni per il trasporto delle proteine sono il trasporto controllato e il trasporto vescicolare.
Il trasporto controllato richiede la costruzione di una porta tra il citoplasma e la membrana nucleare e un sensore chimico (una proteina dotata del tag di identificazione corretto). Quando il pacchetto proteico si avvicina al sensore, questo apre la porta, permettendo alla proteina di passare. Questo meccanismo di controllo richiede che la proteina abbia il tag di identificazione appropriato e una porta programmata per aprirsi in risposta. La porta stessa contiene a sua volta molte parti, introducendo così un ulteriore livello di complessità irriducibile. Ciascuno di questi componenti del trasporto controllato è complesso e consiste di migliaia di parti a livello molecolare, tutte necessarie affinché il sistema di trasporto controllato funzioni.
Anche il sistema di trasporto vescicolare utilizza una serie di sensori appositamente progettati. Invece di una porta, il tag di identificazione appropriato fa sì che la membrana del compartimento si prolunghi verso l’esterno, distaccandosi e formando una vescicola che circonda completamente la proteina. La vescicola di trasporto si sposta quindi verso una destinazione predeterminata dal suo tag di identificazione. Se il tag della vescicola e il sensore di identificazione corrispondono, un altro sensore riconosce la vescicola, che si fonde con il compartimento.
Successivamente, il processo di distacco viene invertito per consentire alle proteine di essere trasportate all’interno del nuovo compartimento. La complessità quasi certamente irriducibile del sistema deve includere due complessi sistemi di sensori, due tag di identificazione e il recipiente stesso. A un livello superiore, ogni tag di identificazione del sensore e i recipienti di trasporto sono, a livello molecolare, anch’essi costituiti da migliaia di componenti, ognuna delle quali è probabilmente un esempio di complessità irriducibile. La vescicola deve contenere tutte le strutture che le consentono di gemmare dal compartimento originale e poi di unirsi a un altro compartimento.
Altri problemi dell’endosimbiosi
La teoria standard dell’endosimbiosi è stata recentemente oggetto di critiche da più fronti e alcuni ricercatori stanno ora proponendo una nuova teoria per spiegare l’evoluzione degli organelli. Alcuni di questi scienziati ritengono che una nuova teoria “potrebbe risolvere alcuni problemi persistenti della teoria prevalente” dell’endosimbiosi. I dettagli di questa nuova teoria sono ancora vaghi. Ammettono che, anche se una nuova teoria fosse argomentata in modo elegante, probabilmente ci saranno molte difficoltà che una nuova ipotesi non prenderà in considerazione. Gli evoluzionisti concludono anche che potremmo dover ammettere che
“… il mitocondrio è stato un fortunato incidente. In primo luogo, la cellula ancestrale – probabilmente un archeobatterio, come suggeriscono recenti analisi genetiche – acquisì la capacità di inglobare e digerire molecole complesse. Iniziò a predare i suoi compagni microbici. A un certo punto, tuttavia, questa cellula predatrice non digeriva completamente la sua preda, e ne risultò una cellula ancora più efficiente quando un pasto destinato a diventare preda si stabilì permanentemente e divenne il mitocondrio.”
Inoltre, per decenni gli scienziati hanno creduto che
“… avessero esempi dei diretti discendenti di quegli eucarioti primitivi: alcuni protisti privi di mitocondri. Ma recenti analisi dei geni in questi organismi suggeriscono che anche loro un tempo possedevano mitocondri, ma li persero in seguito (Science, 12 settembre 1997, p. 1604). Questi risultati suggeriscono che gli eucarioti potrebbero in qualche modo aver acquisito i loro mitocondri prima di aver sviluppato la capacità di inglobare e digerire altre cellule.”
Un altro problema dell’endosimbiosi è l’ampio disaccordo tra i ricercatori sul meccanismo. Una sintesi ha concluso:
“… che i modelli ‘mitocondriali precoci’, che postulano l’acquisizione del protomitocondrio da parte di un ospite archeale, sono più plausibili dei modelli ‘mitocondriali tardivi’. Tuttavia, poiché i procarioti non sono in grado di effettuare la fagocitosi, tali modelli non sono riusciti a suggerire un meccanismo ragionevole attraverso il quale l’endosimbionte abbia avuto accesso al suo ospite.”
Una soluzione a tutti questi problemi è dare più tempo all’evoluzione. Come spiega il professor Edgar, ci sono voluti 0,5 miliardi di anni perché i procarioti (batteri e archei) sviluppassero “una biochimica piuttosto complessa e alcune caratteristiche eucariotiche”, ma
“… la transizione dai procarioti unicellulari agli eucarioti multicellulari e aerobici ha richiesto altri 2,5 miliardi di anni per iniziare. Il fattore o i fattori chiave che alla fine hanno causato questa transizione a lungo ritardata sono una questione che è stata al centro di numerose ricerche e un argomento di discussione per molti anni.”
Conclusioni:
Esistono due gruppi principali di organismi: procarioti ed eucarioti. Non sono mai stati rinvenuti organismi intermedi tra di loro, con organelli parzialmente sviluppati. Salvo possibili eccezioni minori, ciò che si trova è o l’assenza di organelli, oppure organelli completamente funzionali e sviluppati. “Non esistono anelli mancanti tra eucarioti e batteri, né nei fossili né negli organismi viventi.” Inoltre, è persino difficile ipotizzare, attraverso la costruzione di storie fantasiose, come possano esistere i legami tra procarioti ed eucarioti.
L’endosimbiosi postula che i mitocondri fossero un tempo batteri a vita libera e che “nelle prime fasi dell’evoluzione le cellule eucariotiche ancestrali si nutrissero semplicemente dei loro futuri partner.” Sia la teoria della conversione graduale che quella dell’endosimbiosi richiedono migliaia di forme di transizione, ognuna delle quali conferisce alla cellula un vantaggio competitivo rispetto alle cellule non modificate.
L’idea dell’endosimbiosi è popolare non per via delle prove empiriche, ma perché nessun’altra ipotesi è minimamente plausibile. La completa assenza di prove fossili e di altro tipo rappresenta un ulteriore problema. Pertanto, il professor Battley descrive la nozione di endosimbiosi come “provvisoria nella migliore delle ipotesi”. Un problema principale dell’endosimbiosi è che è sempre stata impossibile da verificare. Ulteriori ricerche e conoscenze hanno spinto un ricercatore all’avanguardia in questo campo a concludere nel 1998 che gli studi
“… pubblicati negli ultimi due o tre anni, molti dei quali derivanti da progetti di sequenziamento del genoma, suggeriscono che sia giunto il momento di una nuova teoria. In particolare, sta emergendo che i genomi nucleari eucariotici contengono molti geni di origine batterica (a volte α-proteobatterica) che non hanno nulla a che fare con le funzioni mitocondriali. Inoltre, gli eucarioti privi di mitocondri, che eravamo giunti a considerare diretti discendenti degli antichi proto-eucarioti, presentano geni mitocondriali nei loro genomi nucleari.”
La teoria dell’endosimbiosi è stata attaccata da molte altre parti, e senza dubbio questi attacchi continueranno. Il direttore del Laboratorio di Genetica dell’Università di Clemson ha definito l’endosimbiosi una teoria in crisi che, nella migliore delle ipotesi, spiega ben poco dell’evoluzione degli eucarioti dai procarioti. L’origine dell’intero nuovo sistema cellulare, gli eucarioti, rimane ancora da spiegare. Ha concluso:
“… le sequenze di molti genomi eucariotici mostrano ora chiaramente che i repertori genici necessari al funzionamento dei mitocondri non derivano dai batteri, ma sono straordinariamente unici per il tipo di organismo in cui si trovano. Sebbene esistano alcune somiglianze genetiche, queste corrispondenze sono plausibilmente spiegate dal concetto ingegneristico standard di riutilizzo del codice: codice comune per risolvere problemi simili. La grande quantità di dati genomici sta ora distruggendo completamente l’idea dell’evoluzione su tutti i fronti, persino nell’ambito dell’endosimbiosi, una delle teorie preferite dai laicisti.”
La teoria dell’endosimbiosi presenta seri problemi. I mitocondri differiscono dai batteri: geneticamente, strutturalmente, funzionalmente e operativamente. Inoltre, il tempo non si è rivelato utile per spiegare queste differenze all’interno dei parametri della teoria, ma ha solo accentuato il contrasto tra le due.
Questa analisi può solo delineare alcuni dei principali problemi relativi al tentativo di colmare il divario tra procarioti ed eucarioti attraverso il concetto di endosimbiosi. Per criticare efficacemente questa idea problematica, sarebbe necessario un intero articolo per ciascuna delle aree problematiche in questione.
Gabriel Bellizzi [CEO BioGenTech]
Riferimenti:
Archibald, J., Endosymbiosis and eukaryotic cell evolution, Current Biology 25(19):R911–R921, 5 October 2015; R911.
Helder, M., Endosymbiotic theory, Creation Dialogue 24(4):6, December 1997.
Spuhler, J.N., Evolution of mitochondrial DNA in monkeys, apes, and humans, Yearbook of Physical Anthropology 31:15–48, 1988; p. 16.
Davidov, Y. and Jurkevitch, E., Predation between prokaryotes and the origin of eukaryotes, Bioessays 31(7):748–757, July 31, 2009; p. 748.
Spuhler, ref. 3, p. 18.
Palmer, J. et al., It takes teamwork: how endosymbiosis changed life on earth, evolution.berkeley.edu/evolibrary/article/endosymbiosis_01, 2007.
Berg, O. and Kirkland, C.G., Why mitochondrial genes are most often found in nuclei, Molecular Biology and Evolution 17(6):951–961, June 2000; p. 95, academic.oup.com/mbe/article/17/6/951/1037844.
Davidov and Jurkevitch, ref. 4, pp. 748–749.
Cavalier-Smith, T., Our symbiotic origins? The Sciences 37(4):46–47, 1997; p. 46.
Margulis, L., Origin of eukaryotic cells: evidence and research implications for a theory of the origin and evolution of microbial, plant, and animal cells on the Precambrian earth, Yale University Press, New Haven, CT, 1970.
Hoagland, M. et al., Exploring the Way Life Works: The science of biology, Random House, New York, p. 72, 1995.
Spuhler, ref. 3, p. 19.
Gray, M.W., Mitochondrial evolution, Cold Spring Harbor Perspectives in Biology 4(9):a011403, 2012.
Spuhler, ref. 3, p. 15.
Davidov and Jurkevitch, ref. 4, p. 748–749.
Burger, G. and Lang, B.F., Parallels in genome evolution in mitochondria and bacterial symbionts, IUBMB Life 55(4–5):205–212, April–May 2003.
Lang, B. et al., An ancestral mitochondrial DNA resembling a eubacterial genome in miniature, Nature 387:493–497, May 1997; p. 493.
Kurland, C.G. et al., Genomics and the irreducible nature of eukaryote cells, Science, 312:1011–1014, 2006.
Lang et al., ref. 17, p. 496.
Davidov and Jurkevitch, ref. 4, p. 748.
Lewin, B., Genes VI, Oxford University Press, Oxford, UK, 1997.
Battley, E.H., Book review of Dexter et al., Tracing the History of Eukaryotic Cells: The enigmatic smile, Columbia University Press, New York, in The Quarterly Review of Biology 71(2):275–276, June 1994, p. 276.
Vallès, Y. et al., Group II introns break new boundaries: presence in a bilaterian’s genome, PloS ONE 3(1):e1488, February 2008 ǀ doi:10.1371/journal.pone.0001488.
See for example Bergstrom, C.T. and Dugatkin, L.A., Evolution, Norton, New York, pp. 442–445, 2016.
Behe, M., Darwin’s Black Box: The biochemical challenge to evolution, Basic Books, New York, p. 189, 1996.
Margulis, L., The origin of plant and animal cells, American Scientist 59(2):230–235, March–April 1971, p. 230.
de Roos, A.A., Critique on the endosymbiotic theory for the origin of mitochondria, Telic Thoughts, 17 October 2007; researchgate.net/publication/322228552_A_critique_on_the_endosymbiotic_theory_for_the_origin_of_mitochondria.
Mears, J.A., et al., A structural model for the large subunit of the mammalian mitochondrial ribosome, J. Molecular Biology 358(1):193–212, April 2006.
O’Brien, T.W., Evolution of a protein-rich mitochondrial ribosome: implications for human genetic disease, Gene 286(1):73–79, 6 March 2002.
O’Brien, T., Properties of human mitochondrial ribosomes, IUBMB Life 55(9):505–513. 2003.
Mears et al., ref. 28, p. 193.
Frank-Kamenetskii, M.D., Unraveling DNA: The most important molecule of life, Perseus Books, Reading, MA, p. 68, 1997.
Sharma, M.R., et al., Structure of the mammalian mitochondrial ribosome reveals an expanded functional role for its component proteins, Cell 115(1):97–108, 2003.
Smits, P., et al., Reconstructing the evolution of the mitochondrial ribosomal proteome, Nucleic Acids Research 35(14):4686–4703, July 2007.
Davidov and Jurevitch, ref. 4, pp. 748, 750.
Davidov and Jurevitch, ref. 4, p. 750.
Davidov and Jurevitch, ref. 4, pp. 748–749.
Wallace, D.C., Mitochondrial DNA in aging and disease, Scientific American, New York, 1997.
Rizzuto, R., Mitochondria as sensors and regulators of calcium signaling, Nature Reviews Molecular Cell Biology 13:566–578, 2012.
Hoffmeister, M. et al., Mitochondrial trans-2-Enoyl-CoA reductase of wax ester fermentation from Euglena gracilis defines a new family of enzymes involved in lipid synthesis, J. Biological Chemistry 280:4329–4338, 2004.
Frank-Kamenetskii, ref. 32, p. 68–69.
Bogenhagen, D.F. et al., Kinetics and mechanism of mammalian mitochondrial ribosome assembly, Cell Replication 22(7):1935–1944, February 13, 2018.
Bogenhagen, ref. 42, p. 1935.
Helder, ref. 2, pp. 6–7.
de Duve, C., The birth of complex cells, Scientific American 274(4):50–57, April 1996, p. 57.
Henze, K. and Martin, W., How do mitochondrial genes get into the nucleus? TRENDS in Genetics 17(7):383–387, July 2001, p. 383.
Helder, ref. 2, p. 7.
Henze and Martin, ref. 46, pp. 286–387.
Gray, M.W., Mosaic nature of the mitochondrial proteome: implications for the origin and evolution of mitochondria, PNAS 112(33):10133–10138, August 2015, p. 10133.
Gray, ref. 49, p. 10133–10134.
Schatz, G., Just follow the acid chain, Nature 388(6638):121–122, July 1997, p. 121.
Dietmeier, K. et al., Tom5 functionally links mitochondrial preprotein receptors to the general import pore, Nature 388:195–200, July 1997.
Su, D. et al., An unusual tRNAThr derived from tRNAHis reassigns in yeast mitochondria the CUN codons to threonine, Nucleic Acid Research 39(11):4866–4874, 2011: ǀ doi:10.1093/nar/gkr073; and Gray, M. and Doolittle, W.F., Has the endosymbiont hypothesis been proven? Microbiological Reviews 46(1):1–42, 1982.
Miranda, I. et al., Evolution of the genetic code in yeasts, Yeast 23(3):203–213, 2006.
Behe, ref. 25, p. 189.
Margulis, L. and Sagan, D., What is Life? Simon & Schuster, New York, p. 154, 1995.
Margulis and Sagan, ref. 56, p. 91.
Margulis, L. and Sagan, D., Acquiring Genomes: A theory of the origins of species, Basic Books, p. 154, 2002.
O’Malley, M.A., Endosymbiosis and its implications for evolutionary theory, PNAS 112(33):10270–10277, 2015; p. 10270.
Edgar, J.A., L-ascorbic acid and the evolution of multicellular eukaryotes, J. Theoretical Biology 476:62–73, September 2019.
Huang, L., Massa, L., and Karle, J., Drug target interaction energies by the kernel energy method in aminoglycoside drugs and ribosomal A site RNA targets, PNAS 104(11):4261–4266. 2006.
Kornberg, A., For the Love of Enzymes: The odyssey of a biochemist, Harvard University Press, Cambridge, MA, p. 316, 1989.
Vogel, G., Did the first complex cell eat hydrogen? Science 279(5357):1633–1634, March 1998, p. 1633.
Davidov and Jurkevitch, ref. 4, p. 755.
Brown, C., Are there organelles in bacteria? CRSQ 42(2):129–130, September 2005.
Margulis and Sagan, ref. 58, p. 139.
Tuszynski J.A., Molecular and Cellular Biophysics, CRC Press, Boca Raton, FL, p. 207, 2007.
Margulis, ref. 26, p. 230.
Doolittle, W.F., A paradigm gets shifty, Nature 392:15–16, March 1998; p. 15.
Mathieu, L.G. and Sonea, S., A powerful bacterial world. ScienceDirect (Endeavour) 19(3):112–117, 1995; sciencedirect.com/science/article/abs/pii/016093279597496U.
Tomkins, J.P., Endosymbiosis: a theory in crisis, Acts & Facts 44(11):13, November 30, 2015.
Nel 1986 i ricercatori hanno svolto studi approfonditi sull’ossido nitrico (NO), un potente vasodilatatore che può migliorare la circolazione e la salute del cuore. I ricercatori della Pfizer iniziarono a sperimentare farmaci chiamati inibitori della PDE-5 che potenziano e perpetuano gli effetti di dilatazione dei vasi sanguigni dell’NO.
Il loro obiettivo, all’epoca, era quello di trovare un trattamento per l’angina. Il primo farmaco fu il Sildenafil citrato, ma le sperimentazioni dimostrarono che la sua efficacia nel trattamento della patologia era modesta. Tuttavia, i ricercatori hanno iniziato a esaminare le note che descrivevano gli effetti collaterali del farmaco. Ed ecco che molti soggetti hanno riferito di aver sperimentato erezioni durature. Pfizer cambiò rapidamente marcia e avviò studi pilota sugli effetti del Sildenafil citrato sulla disfunzione erettile. Il Viagra, nome commerciale del Sildenafil, fu presto approvato dalla FDA.
Non sono gli anziani hanno beneficiato di questo effetto. Infatti, uomini più giovani si sono affezionati al farmaco, come hanno fatto con i suoi cugini Cialis [Tadalafil] e Levitra [Vardenafil], perché i farmaci in questione aiutavano a gestire l’ansia da prestazione e riducevano i tempi morti tra un episodio sessuale e l’altro.
Confronto tra le strutture di cGMP, Sildenafil e altri inibitori della PDE5. a | Il substrato nativo, cGMP. b | Sildenafil. c | Vardenafil e Tadalafil. cGMP, guanosina monofosfato ciclico; PDE-5, fosfo-diesterasi di tipo 5.
Ma ci sono altri motivi per cui gli uomini potrebbero usare questa classe di farmaci. Non sono solo legati alla salute sessuale, ma anche al Bodybuilding. Infatti, ci sono prove sufficienti per sostenere l’idea di assumere questi farmaci ogni giorno, come qualsiasi altro integratore ritenuto “base” nella preparazione di un bodybuilder.
Caratteristiche dei PDE-5 inibitori:
Un inibitore della fosfodiesterasi di tipo 5 (inibitore della PDE-5) è un farmaco vasodilatatore che agisce bloccando l’azione degradativa della fosfodiesterasi di tipo 5 (PDE-5) specifica per il cGMP sul GMP ciclico nelle cellule muscolari lisce che rivestono i vasi sanguigni che riforniscono vari tessuti. Questi farmaci dilatano i corpi cavernosi del pene, facilitando l’erezione con la stimolazione sessuale, e sono utilizzati nel trattamento della disfunzione erettile (DE). Il Sildenafil è stato il primo trattamento orale efficace disponibile per la DE. Poiché la PDE-5 è presente anche nella muscolatura liscia delle pareti delle arteriole polmonari, due inibitori della PDE-5, il Sildenafil e il Tadalafil, sono approvati dalla FDA per il trattamento dell’ipertensione polmonare. Dal 2019 si stanno apprezzando i più ampi benefici cardiovascolari degli inibitori della PDE-5.[https://www.ncbi.nlm.nih.gov/]
Schema della via dell’Ossido Nitrico (NO)/guanosina monofosfato ciclico (cGMP)/ nucleotide ciclico fosfodiesterasi 5 (PDE-5) e del sito d’azione degli inibitori della PDE-5.
Parte del processo fisiologico di vasodilatazione prevede il rilascio di ossido nitrico (NO) da parte delle cellule endoteliali vascolari, che poi si diffonde alle vicine cellule muscolari lisce vascolari. Lì, l’NO attiva la guanilato ciclasi solubile che converte la guanosina trifosfato (GTP) in guanosina monofosfato ciclico (cGMP), il principale effettore del sistema. Ad esempio, nel pene, il rilascio di NO ad alti livelli dalle cellule endoteliali e dai nervi penieni durante la stimolazione sessuale porta al rilassamento della vascolarizzazione liscia dei corpi cavernosi, causando una vasocongestione e un’erezione prolungata.[https://www.ncbi.nlm.nih.gov/]
Gli inibitori della PDE-5 prolungano l’azione del cGMP inibendo la sua degradazione da parte dell’enzima PDE-5, presente in tutto il corpo. Nel pene, gli inibitori della PDE-5 potenziano gli effetti del cGMP per prolungare l’erezione e aumentare la soddisfazione sessuale, mentre nel muscolo scheletrico aumentano l’iperemia del tessuto per via della vasodilatazione.[https://www.nejm.org/] Tuttavia, gli inibitori della PDE-5 non provocano erezioni senza stimolazione sessuale.
Oltre agli effetti emodinamici, gli inibitori della PDE-5 hanno dimostrato in diversi esperimenti proprietà antinfiammatorie, antiossidanti, antiproliferative e metaboliche.[https://www.ncbi.nlm.nih.gov/] Ma sono ovviamente necessari studi più ampi e a lungo termine per stabilirne l’efficacia e la sicurezza rispetto ad altri farmaci in altre patologie.
Quindi l’uso di questa classe di farmaci nel Bodybuilding si limita al classico trattamento per la disfunzione erettile e il pompaggio muscolare? Non esattamente.
Sicuramente, il potenziale additivo dei PDE-5 inibitori per lo stimolo massimo del “pump”, in specie in combinazione con Citrullina, nel pre-palco può incidere positivamente sugli ultimi ritocchi del “look” dell’atleta. Ricordo inoltre che un maggiore afflusso di sangue al tessuto muscolare significa un migliore pompaggio dato dall’esercizio contro-resistenza e un maggiore afflusso di sostanze nutritive ai muscoli, il che è positivo per la performance, il recupero e la crescita muscolare.
Ma i potenziali non si fermano qui:
Tadalafil
Uno studio del 2005 ha rilevato che dosi di Tadalafil da 10 e 20mg, assunte in media 10 volte al mese, riducevano significativamente i livelli di Estradiolo, ma solo negli uomini che non avevano troppo grasso corporeo – quelli con un IMC inferiore a 27 (1). Gli uomini con più grasso corporeo hanno livelli di Aromatasi più elevati e convertono maggiormente il Testosterone in Estradiolo, indipendentemente dal Tadalafil assunto.
Uno studio sugli effetti del Sildenafil su 140 uomini con un basso livello di Testosterone, di età compresa tra i 40 e i 70 anni, ha rilevato che il farmaco ha aumentato i livelli di Testosterone di circa 100 punti (2). Sebbene una parte di questo aumento dell’ormone maschile possa essere dovuta alla mancata conversione in Estradiolo di una parte del Testosterone, una percentuale di questo aumento sembra derivare anche da una maggiore produzione di Testosterone da parte dei testicoli.
Il Sildenafil riduce lo stress ossidativo indotto dal diabete e migliora la sensibilità all’Insulina. (3) Questo esperimento, a differenza degli altri, è stato condotto sui ratti, ma è probabile che funzioni in modo simile anche nell’uomo.
L’ipotesi che i farmaci che influenzano il flusso sanguigno possano essere utili per la crescita muscolare negli adulti più anziani, ha spinto il Dipartimento di Medicina Interna dell’Università del Texas Medical Branch ha condurre uno studio.
Time-line dello studio
Secondo i ricercatori, le riduzioni della funzione muscolare scheletrica si verificano nel corso di un invecchiamento sano, ma anche con la sedentarietà o con diverse malattie come il cancro, la distrofia muscolare e l’insufficienza cardiaca. Tuttavia, non esistono terapie farmacologiche accettate per migliorare la funzione muscolare scheletrica compromessa.
L’ossido nitrico può influenzare la funzione del muscolo scheletrico attraverso effetti sull’accoppiamento eccitazione-contrazione, sulla funzione miofibrillare, sulla perfusione e sul metabolismo.
I soggetti dello studio erano di mezza età, non allenati e per lo più in sovrappeso, e dovevano assumere un’integrazione giornaliera di Sildenafil per otto giorni, mentre si è analizzato l’effetto sulla sintesi proteica muscolare (il processo che guida la crescita muscolare) e sulla funzione muscolare rispetto a un placebo.
Lo studio ha dimostrato che l’aumento della segnalazione dell’ossido nitrico-guanosina monofosfato ciclico mediante la somministrazione giornaliera a breve termine dell’inibitore della fosfodiesterasi 5, il Sildenafil, aumenta la sintesi proteica, altera l’espressione proteica e la nitrosilazione e riduce la fatica nel muscolo scheletrico umano.
Questi risultati suggeriscono che gli inibitori della fosfodiesterasi 5 rappresentano un valido intervento farmacologico per migliorare la funzione muscolare. Ciò che è stato rilevato, infatti, è che Il Sildenafil aumenta la sintesi proteica muscolare e riduce l’affaticamento muscolare.
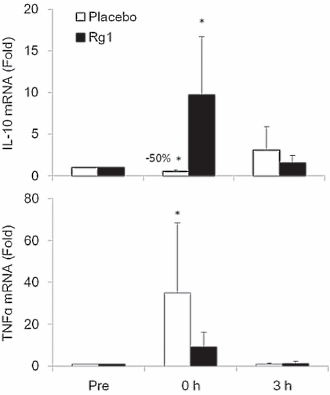
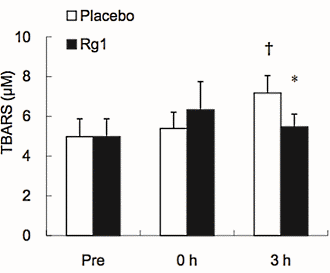
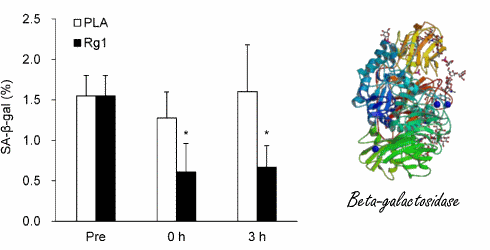
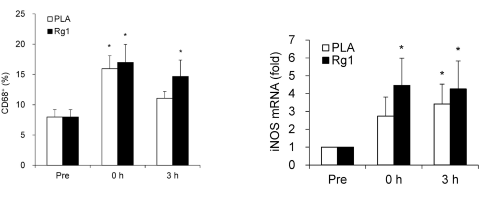
Effetti del trattamento con Sildenafil sulla funzione muscolare scheletrica. (A) Forza isometrica degli estensori del ginocchio (percentuale media del giorno di riferimento ± errore standard (SE)) dopo 8 giorni di trattamento, determinata con la dinamometria. (B) Forza isocinetica (120° al secondo) degli estensori del ginocchio (percentuale media del giorno di riferimento ± SE) dopo 8 giorni di trattamento, determinata con la dinamometria. (C) Ripetizioni riuscite (percentuale media del giorno di riferimento ± SE) durante contrazioni isocinetiche affaticanti (120° al secondo) dopo 8 giorni di trattamento. *p = 0,016 rispetto al placebo, t-test non accoppiato, N = 6 placebo, 5 sildenafil. Il numero individuale di ripetizioni riuscite prima (pre) e dopo (post) il trattamento per i soggetti che hanno ricevuto il placebo (pannello superiore) e il Sildenafil (pannello inferiore) è mostrato a destra.Effetti del trattamento con Sildenafil sul proteoma del muscolo scheletrico. (A) Sintesi proteica del muscolo scheletrico (media ± SE) dopo 8 giorni di trattamento, determinata utilizzando l’approccio precursore-prodotto per determinare il tasso di sintesi frazionale. *p = 0,004 rispetto al placebo, t-test non accoppiato, N = 6 placebo, 5 Sildenafil. Percorsi canonici (B) e funzionali (C) influenzati in modo differenziato da sildenafil e placebo, determinati utilizzando l’Ingenuity Pathways Analysis (IPA) dell’espressione proteica in campioni di biopsia del muscolo scheletrico (sono mostrati i 6 percorsi principali). Percorsi canonici (D) e funzionali (E) influenzati in modo differenziato dal Sildenafil e dal placebo, determinati utilizzando l’IPA della S-nitrosilazione delle proteine nei campioni di biopsia del muscolo scheletrico (sono indicati i sei percorsi principali).
L’affaticamento del muscolo scheletrico nel primo giorno di trattamento non era statisticamente diverso dal basale o diverso tra i gruppi di trattamento. Tuttavia, gli scienziati hanno ammesso che dopo otto giorni di trattamento, i soggetti del gruppo Sildenafil hanno completato un numero significativamente maggiore di ripetizioni di successo rispetto al basale in rapporto a quelli che hanno ricevuto il placebo durante contrazioni isocinetiche massimali ripetute.
Essendo un farmaco già approvato e con un eccellente record di sicurezza, i risultati di questo studio suggeriscono che il Sildenafil, e possibilmente altri inibitori della fosfodiesterasi 5, rappresenta una potenziale strategia farmacologica per migliorare la funzione del muscolo scheletrico.
Jeff Nippard
Jeff Nippard, un famoso blogger di fitness su YouTube, ha cercato di mettere le cose in chiaro. Per prima cosa ha contattato Jorn Tromellen, ricercatore sul metabolismo muscolare. Tromellen ha rivelato che in realtà i risultati sono meno impressionanti di quanto sembri. Il Sildenafil non è paragonabile agli AAS, ovviamente, in quanto quando si assume il Sildenafil la sintesi proteica aumenta nel giro di un’ora o due. Mentre gli AAS la stimolano in modo significativo (vedi attività genomica) per tutta la vita attiva del farmaco.
Tuttavia, questo non era sufficiente per Nippard, così si è rivolto a colui che ha effettivamente usato il Sildenafil: il compianto John Meadows.
John Meadows
Meadows ha fatto uso di Sildenafil per tutta la sua carriera agonistica. La prima cosa che ha ammesso è che il livello “di lavoro” del Sildenafil dipendono dalla quantità di cibo presente nello stomaco.
Lui ammette che, gli atleti lo usano prevalentemente per avere più “pump” prima di salire sul palco.
Poi Meadows ha confrontato le sensazioni generali dopo l’assunzione di Testosterone e Sildenafil e, ovviamente, non sono neanche lontanamente paragonabili. Considero questa deduzione al pari dell’affermazione secondo cui l’uomo non possa respirare sottacqua senza attrezzatura apposita… banalità…
La verità è che questo studio ha lasciato ancora più domande rispetto a prima. I risultati sembrano molto promettenti, ma rimangono ancora alcune perplessità.
Lo studio è stato condotto su persone non allenate. Quindi la prima domanda è: Funzionerebbe anche su soggetti più giovani e/o allenati?.
E ancora: sappiamo che avviene un aumento della sintesi proteica, ma non è chiaro se ciò sia a carico delle fibre miofibrillari o di altri tessuti.
Possibili effetti avversi dall’uso di inibitori del PDE-5:
Tutti gli inibitori della PDE-5 sono generalmente ben tollerati.[1] La comparsa di effetti collaterali, o reazioni avverse al farmaco (ADR), con gli inibitori della PDE-5 dipende dalla dose e dal tipo di agente.[https://www.ncbi.nlm.nih.gov/] La cefalea è una ADR molto comune, che si verifica nel >10% dei pazienti. Altre ADR comuni sono: vertigini, vampate di calore, dispepsia, congestione nasale o rinite.[6] Anche il mal di schiena e i dolori muscolari sono più comuni nei pazienti che assumono Tadalafil.[https://www.ncbi.nlm.nih.gov/]
Nel 2007, la Food and Drug Administration (FDA) statunitense ha annunciato l’aggiunta di un’avvertenza sulla possibile perdita improvvisa dell’udito alle etichette dei farmaci inibitori della PDE-5.[https://www.fda.gov/]
Dal 2007 sono emerse prove che suggeriscono che gli inibitori della PDE-5 possono causare una neuropatia ottica anteriore,[https://doi.org/1] anche se l’aumento del rischio assoluto è piccolo.[https://www.ncbi.nlm.nih.gov/]
Infine, si teme che gli inibitori della PDE-5 possano aumentare il rischio di mortalità neonatale nelle donne in gravidanza, e sono stati sospesi gli studi sull’uso dei farmaci per la restrizione della crescita fetale.[https://www.ncbi.nlm.nih.gov/]
Gli inibitori della PDE5 sono metabolizzati principalmente dal sistema enzimatico del citocromo P450, in particolare dal CYP3A4. Esiste la possibilità di interazioni avverse con altri farmaci che inibiscono o inducono il CYP3A4, tra cui gli inibitori della proteasi dell’HIV, il Ketoconazolo e l’Itraconazolo,[Australian Medicines Handbook 2006.] anche se la co-somministrazione non è stata collegata a cambiamenti nella sicurezza o nell’efficacia di entrambi gli agenti. [La combinazione con nitrovasodilatatori come la nitroglicerina e il PETN è controindicata perché può verificarsi ipotensione potenzialmente pericolosa per la vita.[Haberfeld H, ed. (2009). Austria-Codex (in German) (2009/2010 ed.). Vienna: Österreichischer Apothekerverlag.] Gli inibitori della PDE5 non interagiscono sinergicamente con altri farmaci antipertensivi.[https://www.ncbi.nlm.nih.gov/]
Conclusioni:
L’uso sporadico di questi farmaci (meno di 8-10 volte al mese) potrebbe non conferire effetti duraturi sulla salute. Tuttavia, il Tadalafil è approvato per l’uso una volta al giorno ed è ragionevole pensare che i pazienti, giovani o anziani, che hanno una tale prescrizione e lo usano ogni giorno, stiano raccogliendo alcuni, se non tutti, i benefici di cui sopra.
Tuttavia, se ulteriori ricerche confermeranno o aggiungeranno ulteriori elementi positivi all’elenco, potremmo arrivare al punto in cui i medici raccomanderanno quasi universalmente l’uso pressoché quotidiano di questi farmaci.
Per il momento, l’applicazione del Sildenafil nel Bodybuilding si è dimostrata più redditizia come “NO booster” potenziato e coadiuvato dalla Citrullina sia come mezzo per aumentare marcatamente il “pump” sul palco e sia per aumentare l’afflusso ematico nei muscoli (vedi ossigeno e nutrienti) durante il workout.
Diversamente, piccole dosi di Tadalafil possono garantire un contenuto controllo estrogenico in soggetti con body fat contenute, senza il rischio di incorrere il alterazioni lipidiche ematiche.
Gabriel Bellizzi [CEO BioGenTech]
Riferimenti:
Greco EA et al. Testosterone:Estradiol ratio changes associated with long-term tadalafil administration: a pilot study. J Sex Med. 2006 Jul;3(4):716-722. PubMed.
Spitzer M et al. Sildenafil increases serum testosterone levels by a direct action on the testes. Andrology. 2013 Nov;1(6):913-8. PubMed.
Milani E et al. Reduction of diabetes-induced oxidative stress by phos- phodiesterase inhibitors in rats. Comp Biochem Physiol C Toxicol Pharmacol. 2005 Feb;140(2):251-5. PubMed.
È noto ai più che la Creatina porta a ritenzione idrica intracellulare. Ma vi sono alcuni che si pongono una domanda interessante e più completa: come è realmente distribuita quest’acqua? C’è un totale indirizzamento e accumulo intracellulare o vi sono percentuali di accumulo differenti tra l’interno (intracellulare) e l’esterno (extracellulare) del miocita (cellula muscolare)?
La Creatina è prevalentemente immagazzinata nelle cellule muscolari ed è una sostanza osmoticamente attiva. Quindi aumenta l’osmolalità delle cellule. Di conseguenza, le cellule attireranno acqua per osmosi. Logicamente, sembra molto ragionevole supporre che l’acqua sia immagazzinata insieme alla Creatina (depositi di Fosfocreatina e Creatina libera) all’interno delle cellule muscolari.
Alcune ricerche hanno verificato la logicità di tale ragionamento. Dopo tutto, è necessario testare le proprie ipotesi per avvalorarle. Ci sono diverse tecniche che possono essere utilizzate per mettere alla prova questo ragionamento logico. In letteratura troverete le seguenti quattro tecniche utilizzate:
Analisi di bioimpedenza a multifrequenza (MBIA)
Risonanza magnetica (MR)
Analisi di diluizione isotopica (IDA)
Assorbimetria a raggi X a doppia energia (DEXA)
Quindi è utile indagare attraverso la letteratura scientifica e vedere cosa hanno da dire queste tecniche di analisi sulla questione della compartimentalizzazione dell’acqua legata all’assunzione di Creatina.
Analisi di bioimpedenza a multifrequenza (MBIA): la Creatina causa ritenzione idrica intracellulare.
Come ben sappiamo, il corpo umano è composto da vari tipi di tessuti, come il tessuto adiposo, il tessuto osseo, il tessuto muscolare, ecc. Questi tessuti differiscono notevolmente nella quantità di fluido che contengono e nella loro conducibilità elettrica. La MBIA si basa su quest’ultimo principio, le differenze di conduttività.
In breve, piccole scosse elettriche con frequenze variabili sono erogate attraverso il corpo. Si suppone che si possa stimare il volume del fluido extracellulare con la bassa frequenza e il volume totale del fluido con l’alta frequenza. Sottraendo il volume extracellulare dal volume totale si ottiene il volume del fluido intracellulare. Ci sono però molti inconvenienti con questo metodo, ma non li tratterò qui al fine di evitare di discostarmi eccessivamente dai concetti essenziali che questo articolo si pone di esporre.
Il MBIA è stato utilizzato da Ziegenfuss et al. [1]. Ai soggetti sono stati somministrati 0,07g di Creatina per kg di massa grassa per tre giorni. Per un individuo di 70kg con il 15% di grasso corporeo questo ammonterebbe a 21g al giorno. Quindi, in pratica, i ricercatori hanno somministrato ai soggetti dello studio un dosaggio di carico di Creatina consuetudinario. E’ stato quindi osservato un aumento non statisticamente significativo (P=0,07) del fluido corporeo totale del 2%. Il volume del fluido intracellulare è aumentato significativamente del 3% e il volume del fluido extracellulare è rimasto invariato. Questi risultati suggeriscono che la ritenzione idrica legata all’assunzione di Creatina è effettivamente limitata al compartimento del fluido intracellulare.
Risonanza magnetica (MR): la Creatina trattiene l’acqua all’interno delle cellule.
Le MBIA stimano il contenuto di acqua di tutto il corpo. Le tecniche di risonanza magnetica permettono al ricercatore di controllare una particolare area del corpo. La MR sfrutta il fatto che i protoni hanno un momento di dipolo magnetico. Questi momenti di dipolo dei protoni si allineano con un campo magnetico quando questo è presente. Tale allineamento può avvenire nella direzione del campo magnetico (spin up) o contro di esso (spin down). Quando si rilascia un impulso di radiofrequenza su questi protoni, questi momenti di dipolo cambieranno direzione. Quando l’impulso di radiofrequenza non c’è più, ricadono nella loro vecchia direzione (quella del campo magnetico). Questa “ricaduta” richiede un certo tempo e questo varia a seconda del tessuto. Questi tempi sono anche chiamati tempi di rilassamento. Si presume che questi tempi di rilassamento differiscano tra il compartimento extracellulare e intracellulare. Di conseguenza, le misure di questi tempi di rilassamento possono fornire informazioni sui volumi dei compartimenti extracellulare e intracellulare.
Questa tecnica è stata utilizzata da Saab et al. [2]. I ricercatori hanno somministrato ai soggetti 20g di creatina al giorno per 5 giorni e hanno esaminato il muscolo flexor digitorum profundus (un muscolo dell’avambraccio). Gli autori hanno trovato un aumento della concentrazione di protoni che corrisponde al tempo di rilassamento che si adatta al compartimento intracellulare, ma non a quello extracellulare. Come tale, gli autori concludono che l’integrazione di Creatina trattiene l’acqua all’interno delle cellule.
Analisi di diluizione isotopica (IDA): la Creatina causa ritenzione idrica intra ed extracellulare.
Gli isotopi di un particolare elemento hanno lo stesso numero di protoni ma differiscono nel loro numero di neutroni. Di conseguenza, si comportano fisiologicamente allo stesso modo, ma è possibile distinguerli nelle misurazioni a causa del diverso numero di neutroni. Che cosa ha a che fare questo con le misurazioni dei fluidi corporei? Per esempio, prendiamo un isotopo dell’idrogeno: il deuterio. Quando due molecole di deuterio si combinano con l’ossigeno, si ottiene acqua pesante (ossido di deuterio). Se bevi un bicchiere di acqua pesante, sarai in grado di distinguere queste molecole di acqua pesante dalle molecole di acqua normale. Si lascia bere e si aspetta da 2 a 6 ore che si diffonda uniformemente in tutto il corpo. Poi si preleva del sangue e se ne misura la concentrazione. Poi si determina la concentrazione di acqua pesante con un contatore a scintillazione, e voilà. Con un po’ di calcoli si conosce la quantità totale di acqua nel corpo!
Si può fare la stessa cosa con un isotopo diverso che non può entrare nelle cellule, quindi si può calcolare il volume di acqua extracellulare. Uno di questi isotopi è il bromuro di sodio. E poi, sottraendo il volume d’acqua extracellulare dal volume d’acqua totale, conoscerete il volume d’acqua intracellulare.
Questo è un metodo molto affidabile ed è stato applicato da Powers et al. per studiare la ritenzione idrica della creatina [3]. I risultati sono stati in qualche modo inaspettati. Hanno misurato un aumento del fluido corporeo totale 7 e 28 giorni dopo l’assunzione di creatina. Non è una sorpresa. Tuttavia, non hanno misurato alcuna alterazione nella distribuzione dei fluidi. Il che significa che entrambi i compartimenti del fluido extracellulare e intracellulare sono aumentati in proporzione l’uno all’altro.
Questo sembra in contrasto con i due studi precedenti che ho citato sopra. Una notevole differenza con lo studio MR è che quello ha guardato localmente solo un singolo muscolo. IDA misura la distribuzione dei fluidi di tutto il corpo, proprio come MBIA. MBIA d’altra parte è sicuramente meno affidabile della IDA.
Assorbimetria a raggi X a doppia energia (DEXA) e Bioimpedenza bioelettrica spettrale: la Creatina non causa alterazioni della ritenzione idrica extracellulare.
L’assorbimetria a raggi X a doppia energia (DXA, o DEXA) è un mezzo per misurare la densità minerale ossea (BMD) utilizzando l’imaging spettrale. Due fasci di raggi X, con diversi livelli di energia, sono puntati sulle ossa del paziente. Quando l’assorbimento dei tessuti molli viene sottratto, la densità minerale ossea (BMD) può essere determinata dall’assorbimento di ogni raggio da parte dell’osso. L’assorbimetria a raggi X a doppia energia è la tecnologia di misurazione della densità ossea più utilizzata e più studiata.
Le scansioni DEXA possono anche essere utilizzate per misurare la composizione corporea totale e il contenuto di grasso con un alto grado di precisione paragonabile alla pesata idrostatica, con alcune importanti avvertenze.[4] Dalle scansioni DEXA, si può anche generare un’immagine a bassa risoluzione “fat shadow”, che dà un’impressione generale della distribuzione del grasso in tutto il corpo.[5] È stato suggerito che, pur misurando molto accuratamente i minerali e i tessuti molli magri (LST), la DEXA può fornire risultati distorti a causa del suo metodo di calcolo indiretto della massa grassa sottraendola dalla LST e/o dalla massa cellulare corporea (BCM) che la DEXA effettivamente misura.[6]
Tuttavia, le scansioni DEXA sono state suggerite come strumenti utili per diagnosticare condizioni con una distribuzione anomala del grasso, come la lipodistrofia parziale familiare.[7][8][5] Sono anche utilizzate per valutare l’adiposità nei bambini, soprattutto per condurre ricerche cliniche.[9] In definitiva, sufficientemente affidabile per chiarire la questione qui trattata.
Nello studio di Alex S Ribeiro et al.[10]gli autori hanno voluto confrontare gli effetti dell’integrazione di Creatina (Cr) combinata con l’allenamento contro-resistenza sulla massa muscolo-scheletrica (SMM), l’acqua corporea totale, l’acqua intracellulare (ICW) e l’acqua extracellulare (ECW) in uomini allenati contro-resistenza, nonché determinare se il rapporto SMM/ICW cambia in risposta all’uso di questo supplemento ergogenico. Ventisette uomini allenati contro-resistenza hanno ricevuto Cr (n = 14) o placebo (n = 13) per 8 settimane. Durante lo stesso periodo, i soggetti hanno eseguito due routine di allenamento contro-resistenza divise in quattro sedute a settimana. L’SMM è stato stimato dal tessuto molle magro appendicolare valutato mediante assorbimetria a raggi X a doppia energia (DEXA). L’acqua corporea totale, ICW e ECW sono stati determinati dall’impedenza bioelettrica spettrale. Entrambi i gruppi hanno mostrato miglioramenti (p < .05) nella SMM, acqua corporea totale e ICW, con valori maggiori osservati per il gruppo Cr rispetto al placebo. L’ECW è aumentata in modo simile in entrambi i gruppi (p < .05). Il rapporto SMM/ICW non è cambiato in nessuno dei due gruppi (p > .05), mentre il rapporto SMM/ECW è diminuito solo nel gruppo Cr (p < .05). È stata osservata una correlazione positiva (p < .05) tra i cambiamenti SMM e ICW (r = .71). I risultati degli autori suggeriscono che l’aumento della massa muscolare indotto dalla Cr combinato con l’allenamento contro-resistenza avviene senza alterazione del rapporto tra ICW e SMM negli uomini allenati con la resistenza.
Conclusioni:
Che l’assunzione di Creatina porta alla ritenzione di liquidi è stato suggerito dall’intero corpo documentale riportato. La logica impone che il fluido sia contenuto all’interno delle cellule (muscolari) e non all’esterno delle cellule (extracellulare). Le misurazioni MBIA, MR e DEXA supportano questo punto di vista. Tuttavia, l’IDA, che potrebbe essere considerata una misura standard per la distribuzione dei fluidi, suggerisce che la ritenzione di fluidi avviene proporzionalmente sia nel compartimento extracellulare che in quello intracellulare. Non sembra esserci una buona spiegazione per questo, tranne quanto onestamente riportato dagli stessi ricercatori.
Infatti, essi affermano che l’assunzione di calorie e fluidi dei soggetti in esame non è stata registrata , e le differenze in uno di questi fattori potrebbero aver influenzato i cambiamenti nella massa corporea e nel bilancio dei fluidi. Inoltre, ogni soggetto era coinvolto in un programma di allenamento contro-resistenza individualizzato. Questi protocolli di allenamento non sono stati controllati; tuttavia, il volume di ogni sessione di allenamento è stato registrato. A causa della grande variabilità tra i soggetti, non è stato possibile stabilire una relazione tra il volume di allenamento e i cambiamenti della massa corporea e dell’equilibrio dei fluidi. Inoltre, i protocolli di allenamento prima dell’integrazione non sono stati registrati, quindi non è stato possibile fare confronti. Pertanto, è possibile che le differenze nel volume di allenamento abbiano anche influenzato i cambiamenti nella massa corporea e nell’equilibrio dei fluidi.
Ora, anche alla luce dei dati raccolti negli anni e provenienti da preparazioni alla gara di atleti di diverso livello ma sempre nell’ambito culturistico, sono propenso a seguire quanto riportato dai risultati delle misurazioni MBIA, MR e DEXA. Ulteriori ricerche potrebbero sicuramente chiarire questa discrepanza. Ma, ritornando sul discorso empirico/aneddotico, non mi sono mai imbattuto in condizioni di alterata ritenzione extracellulare in atleti supplementati in cronico con dosi pari a 3-5g/die di Creatina quando tutte le variabili di incidenza maggioritaria (vedi, ad esempio, regolare assunzione di liquidi, elettroliti e corretto bilancio acqua:sodio) erano sotto controllo, soprattutto in condizioni di ipocalorica. Alcuni ipotizzano che dosi elevate di Creatina possano causare un certo grado di ritenzione idrica extracellulare dovuta alla saturazione cellulare. Si tratta però di pure supposizioni basate, oltretutto, su possibilità remote e dettate da soli due scenari: 1) Fase di “carico” della Creatina e 2) ignoranza del soggetto (o di chi per lui) sul range di dose efficace del supplemento; in questo caso potrebbero con tutta probabilità aggiungersi altre variabili che influenzano la ritenzione idrica e che sono indipendenti dalla supplementazione di Creatina.
Gabriel Bellizzi
Riferimenti:
Ziegenfuss, Tim N., Lonnie M. Lowery, and Peter WR Lemon. “Acute fluid volume changes in men during three days of creatine supplementation.” J Exerc Physiol 1.3 (1998): 1-9. APA
Saab, George, et al. “Changes in Human Muscle Transverse Relaxation Following Short‐Term Creatine Supplementation.” Experimental physiology 87.3 (2002): 383-389.
Powers, Michael E., et al. “Creatine supplementation increases total body water without altering fluid distribution.” Journal of athletic training 38.1 (2003): 44.
Per i più informati, è ormai appurato il fatto che la soppressione dell’attività dell’Asse HPT, per via dell’uso di AAS, è determinata da:
L’origine del AAS
Il tasso di conversione del AAS ad estrogeno, attraverso l’enzima Aromatasi in alcuni tessuti (adiposo, mammario)
Dose e tempo d’uso/abuso del AAS
Attività androgena del AAS.
Di conseguenza, dovrebbe essere chiaro che anche farmaci puramente androgeni o essenzialmente anabolizzanti e con forte potenziale di legame con il AR [vedi SARM non steroidei] possono causare una sotto-regolazione della funzionalità dell’Asse HPT, quindi con meccanismi indipendenti dalla aromatizzazione della molecola.
Infatti, gli AAS [ed i SARM non steroidei] attraversano la barriera ematoencefalica e si legano ai recettori Ipotalamici. Ciò comporterà una marcata soppressione dell’HPTA per via di intermediari quali i peptidi oppioidi endogeni.
Da qualche tempo, e per i risultati ottenuti nel trattamento di soggetti dipendenti da droghe e alcool, è emerso il potenziale degli antagonisti dei recettori oppiacei, come il Naltrexone, di bloccare l’attività dei peptidi oppioidi endogeni sull’Ipotalamo e, consequenzialmente, impedire una riduzione del rilascio di GnRH e, di conseguenza, di LH e FSH.
Questo indubbio potenziale ha attirato l’interesse degli utilizzatori di AAS, sempre in cerca, e a ragione, di un modo per ridurre gli effetti collaterali in largo spetro dato dall’uso di PEDs.
In questo articolo descriverò come i recettori oppioidi situati nel Ipotalamo agiscano causando una riduzione gonadotropica e, di rimando, androgena. Tratterò in dettaglio il farmaco Naltrexone e dove questo potrebbe realmente trovare applicazione.
Ma prima un breve ripasso sul HPTA…
Dettaglia sul HPTA:
Con Asse-Ipotalamo-Ipofisi-Testicoli (HPTA) ci si riferisce alla connessione tra ipotalamo, ghiandola pituitaria e testicoli come se queste singole ghiandole endocrine fossero una singola entità. Poiché queste ghiandole spesso agiscono in concerto, i fisiologi e gli endocrinologi ritengono conveniente e descrittivo parlare di esse come di un unico sistema.
L’asse HPTA svolge una parte critica nello sviluppo e nella regolazione di un certo numero di sistemi del corpo, come i sistemi riproduttivi e immunitari. Le fluttuazioni di questo asse causano variazioni negli ormoni prodotti da ciascuna ghiandola e hanno diversi effetti locali e sistemici nel corpo.
In breve, l’asse HPT rappresenta un sistema di stimolazione/inibizione degli ormoni prodotti dalle rispettiva strutture:
Ipotalamo: GnRH (ormone di rilascio delle gonadotropine; in inglese Gonadotropin-releasing hormone).
Ipofisi (o ghiandola Pituitaria): dalle cellule beta e gamma rispettivamente l’ormone follicolo-stimolante (FSH) e l’ormone luteinizzante (LH).
Come ben sappiamo, diversi AAS sono derivati sintetici del Testosterone, il principale androgeno nei maschi. Il Testosterone sopprime marcatamente l’HPTA, mentre altri derivati lo fanno in misura maggiore o minore.
In questo specifico caso ci concentreremo sulla soppressione/sottoregolazione del HPTA Androgeno-dipendente.
Come accennato pocanzi, l’effetto sotto-regolatore dato dagli androgeni a livello Ipotalamico è mediato dai peptidi oppioidi endogeni. Ma come si verifica la disfunzione endocrina derivata dalla attivazione dei recettori oppioidi?
Oppioidi e disfunzione endocrina:
Gli oppioidi sono stati usati per secoli come metodo principale per alleviare il dolore intenso, in particolare il dolore acuto e il dolore da cancro avanzato. Gli oppioidi somministrati regolarmente 24 ore su 24 continuano a essere il cardine nella gestione del dolore associato alle condizioni di fine vita e sono sanciti nella scala analgesica dell’Organizzazione Mondiale della Sanità. Cinquemila anni fa il Papaver somniferumfu veniva coltivato dai Sumeri. Da allora si è intrecciato con l’esperienza umana di molte culture successive, fungendo da benedizione ma anche da maledizione, come analgesico e ansiolitico, stupefacente e musa. Su di esso sono state combattute guerre; qualcuno potrebbe dire che questo continua ad essere perpetrato. Nel 1804 Friedrich Sertürner isolò la morfina e nel 1843 Alexander Wood utilizzò la prima forma iniettabile di questa.
Le proprietà analgesiche degli oppioidi hanno alleviato la sofferenza di innumerevoli persone e il loro ruolo nella gestione del dolore acuto e del dolore oncologico è ben consolidato. Tuttavia, circa il 50% dei riceventi con dolore non oncologico soffre di effetti avversi e nel 20% di questi ciò porta all’interruzione della terapia. (1) Gli effetti avversi, ben noti sia agli operatori sanitari che ai non addetti, comprendono sonnolenza, costipazione, nausea, prurito e depressione respiratoria. Gli effetti avversi meno noti includono instabilità cardiovascolare, mal di testa e spasmi muscolari sia della muscolatura liscia che striata, che portano a ritenzione urinaria, rallentamento della motilità intestinale e scatti mioclonici. (2) Meno ancora saranno a conoscenza della potenziale compromissione della funzione immunitaria, nota per essere specifica per gli oppioidi.(3) I medici del dolore probabilmente conosceranno l’iperalgesia indotta da oppioidi, anche se raramente viene diagnosticata.
Gli effetti a lungo termine meno conosciuti sono la soppressione della funzione endocrina e l’effetto sulla funzione cognitiva.(3) Con l’invecchiamento della popolazione, l’aumento dei tassi di sopravvivenza al cancro e l’uso crescente di oppioidi per il dolore persistente non oncologico, il numero di prescrizioni di oppioidi nella comunità è aumentato da 6 milioni a 15 milioni nel Regno Unito tra il 1999 e il 2008 (NHS Information Center). Gli Stati Uniti hanno il 5% della popolazione mondiale ma consumano l’80% dell’offerta globale di oppioidi. Il numero di overdose fatali accidentali correlate a oppioidi da prescrizione negli Stati Uniti è ora superiore a quello per l’uso ricreativo di eroina e cocaina combinati. (4)
È per ciò fondamentale che tutti coloro che gestiscono e prescrivono oppioidi per il dolore persistente siano consapevoli degli effetti a lungo termine degli oppioidi sulla funzione endocrina e siano in grado di diagnosticare carenze, monitorare i livelli ormonali, comprendere e spiegare al paziente le possibili conseguenze di queste carenze, sforzarsi di ridurre e sospendere gli oppioidi quando appropriato e collaborare con altri medici per sostituire le carenze ormonali.
Effetti endocrini degli oppioidi:
Nel 1895 il reverendo RH Graves (5) notò come “l’oppio mangiava la virilità dell’individuo” e nel 1925 il chirurgo generale HS Cumming (6) affermò che “l’oppio rende effeminato un uomo”. Katz (7) ha già citato Charles Bruce, che, nel 1839, definì i tossicodipendenti da oppio in Assam “più effeminati delle donne”. (8)
È stato riportato in molte occasioni che gli oppioidi, somministrati per qualsiasi via, sopprimono l’asse ipotalamo-ipofisi-gonadi e hanno un impatto misurabile sulla funzione gonadica. (7)
L’ipotalamo, come sappiamo, è fondamentale per la regolazione degli ormoni sessuali. Esercita il suo controllo attraverso la secrezione dell’ormone di rilascio delle gonadotropine (GnRH) dall’area preottica nella circolazione portale ipofisaria all’eminenza mediana. Il GnRH stimola il rilascio dell’ormone follicolo-stimolante (FSH) e dell’ormone luteinizzante (LH) dall’ipofisi anteriore attivando il proprio recettore del GnRH, che, attraverso l’aumento dei livelli di calcio e proteina chinasi C, porta alla formazione e secrezione di FSH e LH (Figura seguente).
Rappresentazione del normale funzionamento dell’asse ipotalamo-ipofisi-gonadi femminile e maschile (9) . (Riprodotto con il permesso di NIAAA).
Il GnRH viene normalmente rilasciato a impulsi in tutti i vertebrati studiati. Questi impulsi producono picchi circadiani essenziali per il corretto funzionamento del sistema riproduttivo determinando quando ciascun ormone viene rilasciato. La secrezione non pulsatile di GnRH provoca una sottoregolazione dell’ipofisi e porta a una secrezione di LH deficitaria. 7
L’FSH è responsabile della crescita precoce dei follicoli ovarici nelle femmine e del mantenimento dell’epitelio spermatogeno nei maschi. L’LH è responsabile della maturazione finale dei follicoli e della loro secrezione di estrogeni, nonché dell’ovulazione e della formazione iniziale del corpo luteo nella femmina. Nel maschio stimola le cellule di Leydig a secernere testosterone. 10 Questi ormoni sono necessari anche per lo sviluppo appropriato dell’essere umano – fisiologicamente, fisicamente e socialmente.
L’asse ipotalamo-ipofisario è costantemente sotto l’effetto di molteplici sostanze tra cui neurotrasmettitori, ormoni steroidei e oppioidi endogeni. Gli oppioidi esogeni esercitano un effetto sugli stessi recettori degli oppioidi endogeni e hanno dimostrato di interferire con il rilascio (compresa la sua natura pulsatile) di GnRH. 7 , 10 , 11 Il naloxone ha mostrato un aumento dei livelli di GnRH, e quindi un aumento della concentrazione di LH e della frequenza del polso, che ha suggerito un livello basale di inibizione della secrezione di LH a base di oppioidi. È stato suggerito che la morfina inibisca la biosintesi del GnRH. 11 Gli oppioidi riducono anche il feedback negativo degli steroidi sessuali sull’ipofisi anteriore, così come la sua risposta al GnRH. 7Al contrario, la secrezione di FSH non è, o solo in minima parte, influenzata.
Con la riduzione dei livelli di LH, il testosterone e l’estradiolo si abbassano proporzionalmente. Li Shizhen scrisse dell’oppio nel suo Compendium of Materia Medica (1578) che “le persone lo usano per l’arte del sesso, in particolare per “arrestare l’emissione seminale”” 11 . Gli studi sugli animali hanno aggiunto credito all’osservazione di Shizhen, dimostrando una ricettività sessuale inibita in entrambi i sessi di ratti con oppioidi e il contrario con naloxone. 12 Ciò sembra essere dovuto alla diminuzione dell’eccitazione piuttosto che all’impotenza. In modo allarmante, studi correlati hanno mostrato che l’esposizione prepuberale agli oppioidi inibiva la maturazione sessuale. 13 Nell’uomo, in seguito al trattamento con oppioidi per via orale e intratecale, si sono verificate irregolarità mestruali, inclusa l’amenorrea. Daniele14 ha notato che c’era anche una diminuzione degli androgeni surrenali, e quindi spiegando la diminuzione della libido e delle prestazioni sessuali così spesso incontrate in coloro che sono esposti a oppioidi a lungo termine. Sembra che la diminuzione del comportamento sessuale possa anche essere dovuta all’azione diretta degli oppioidi sui recettori µ e ∂ nell’ipotalamo. 15 , 16
Il termine “ormoni sessuali” comprende gli steroidi sessuali (testosterone ed estradiolo) e gli ormoni non steroidei (GnRH, FSH e LH).
Il testosterone è il principale ormone dei testicoli, sintetizzato dal colesterolo nelle cellule di Leydig e dall’androstenedione nella corteccia surrenale. La sua secrezione è controllata da LH a 4–9 mg/die. Piccole quantità sono secrete dalle ovaie e forse dalla corteccia surrenale nelle donne. È legato per il 98% alle proteine (globulina e albumina leganti gli steroidi gonadici) nel plasma. Solo il testosterone libero e debolmente legato all’albumina è disponibile per agire sui recettori degli androgeni. Alcune cellule bersaglio convertono il testosterone in diidrotestosterone, che forma complessi ormone-recettore più stabili. Nei maschi il testosterone ha un ritmo circadiano, con i livelli più alti al mattino. La variazione massima può essere dell’ordine del 35%.
Il testosterone impartisce un feedback negativo sul rilascio di LH dall’ipofisi. Sviluppa e mantiene le caratteristiche sessuali secondarie maschili e incoraggia i comportamenti sessuali maschili. È anabolico, aumenta il tasso di crescita e, insieme all’FSH, promuove la spermatogenesi. Fa tutto questo legandosi ai recettori intracellulari e formando complessi che si legano al DNA, facilitando così una certa espressione genica.
Gli estrogeni (17ß-estradiolo, estrene ed estriolo) sono i principali steroidi sessuali femminili e sono biosintetizzati dal testosterone e dall’androstenedione. Sono secreti dalle cellule della granulosa nei follicoli ovarici. Il novantotto per cento degli estrogeni è legato alle proteine. Sono metabolizzati dal fegato ed escreti nelle urine. Gli estrogeni facilitano lo sviluppo dei follicoli ovarici, aiutano nella regolazione del ciclo mestruale e nei necessari cambiamenti nell’anatomia interna femminile e hanno effetti anabolici sull’utero e sulle tube di Falloppio. Diminuiscono i livelli di FSH e alterano i livelli di LH. Sono responsabili dei comportamenti sessuali femminili e della libido e sono in gran parte responsabili dello sviluppo del seno. Questi effetti, combinati con l’assenza di androgeni, portano a caratteristiche sessuali secondarie femminili.
In sintesi, gli oppioidi portano a una diminuzione della secrezione di GnRH, che a sua volta porta a livelli ridotti di LH. Ciò si traduce in una diminuzione della secrezione di testosterone ed estradiolo, che porta ai segni e sintomi elencati nell’immagine sottostante. È importante notare che questi cambiamenti si sviluppano nel corso di settimane o anni.
Diagnosi di ipogonadismo nel maschio:
La diagnosi di ipogonadismo dipende dall’anamnesi e dall’esame insieme ai test di laboratorio. Nel maschio postpuberale le potenziali cause di ipogonadismo primario menzionate nella tabella sottostante possono causare difficoltà diagnostiche, soprattutto per quanto riguarda la loro graduale insorgenza. Se si sospetta l’ipogonadismo, è quindi importante differenziare l’insufficienza gonadica primaria da quella dell’asse ipotalamo-ipofisario.
Per la seconda consultazione internazionale dell’Organizzazione mondiale della sanità sulla disfunzione erettile, che ha considerato il ritmo circadiano e la natura pulsante della secrezione di Testosterone, dovrebbero essere prelevati due campioni di sangue tra le 8:00 e le 11:00 (quando si presume che i livelli di Testosterone siano al massimo, sebbene il ritmo circadiano può diminuire con l’aumentare dell’età e con variazioni tra sportivi e sedentari). I campioni devono essere inviati per la misurazione del testosterone sierico, della globulina legante gli ormoni sessuali (SHBG), della prolattina, dei livelli di FSH e LH.
Livelli di FSH/LH da normali ad alti potrebbero indicare un ipogonadismo primario (vedi tabella sopra). È importante misurare il livello di FSH poiché ha un’emivita più lunga e dimostra una minore variabilità rispetto all’LH. Ipogonadismo secondario (Tabella seguente) è indicato da bassi livelli di testosterone e livelli di FSH/LH da normali a bassi ( Riquadro seguante).
Con l’invecchiamento si riduce la fluttuazione diurna del testosterone sierico (le variabili vi sono nella popolazione sportivamente attiva). Il livello scende notevolmente durante il giorno, rafforzando l’importanza del campionamento mattutino. Nell’ipogonadismo in questa fascia di età i livelli di testosterone totale possono essere normali se i livelli di globulina legante gli ormoni sessuali (SHBG) sono aumentati. I livelli di SHBG aumentano con l’età e quindi riducono la biodisponibilità del testosterone.
I dati recenti del Massachusetts Male Aging Study (MMAS) forniscono prospettive sui normali intervalli di androgeni, come mostrato nella tabella seguente.
Diagnosi di ipogonadismo nella femmina:
Allo stesso modo, l’ipogonadismo nelle femmine può essere dovuto a un asse ipotalamo-ipofisario o a un difetto gonadico primario. I segni ei sintomi sono strettamente legati al ciclo mestruale negli anni postmenarca e premenopausale. I segni comuni includono oligomenorrea, amenorrea e mancato concepimento. Possono verificarsi sintomi più sottili come vampate di calore e ansia, raramente con cambiamenti nella distribuzione dei peli pubici e nella dimensione del seno 21 . Sebbene gli effetti clinici possano essere gravi per le donne come per gli uomini, i cambiamenti esteriori o visibili non sono così evidenti e non sono stati studiati in dettaglio, specialmente nelle donne in postmenopausa.
Sebbene sia stato dimostrato che la carenza di androgeni è sintomatica nelle donne che utilizzano la terapia con oppioidi intratecali e in quelle in trattamento con metadone di mantenimento, i livelli di testosterone non sono stati misurati di routine. Si ritiene che il diidroepiandrosterone (DHEA) sia abbassato ed è noto che i livelli di LH sono notevolmente ridotti. Il DHEA è un marker della produzione di androgeni surrenali e circa il 50% degli androgeni prodotti nella femmina sono di origine surrenale. Vi è una scarsità di dati per quanto riguarda i livelli ormonali nelle donne e sono necessari ulteriori studi per quantificare il significato clinico di questo aspetto potenzialmente molto interessante della carenza di androgeni indotta da oppioidi (OPIAD).
Effetto degli oppioidi sugli ormoni surrenali:
Il dolore acuto e cronico, come risposte fisiologiche, determina un aumento della secrezione dell’ormone adrenocorticotropo (ACTH) e del cortisolo. Tuttavia, in diversi studi è stato riscontrato che l’uso cronico di oppioidi esogeni riduce i livelli di ACTH e cortisolo e le risposte del cortisolo alle sfide dell’adrenocorticotropina. 19 Gli oppioidi influenzano anche i ritmi circadiani della secrezione di cortisolo, determinando un aumento persistente dei livelli di ACTH e cortisolo e alla fine attenuando la risposta allo stress. 20 Anche i livelli di deidroepiandrosterone solfato (DHEAS), un precursore degli androgeni surrenali, sono stati notevolmente ridotti nei consumatori cronici di oppioidi sia maschi che femmine. 14 , 18Occasionalmente, la somministrazione di oppioidi può causare insufficienza surrenalica franca, ma i fattori di rischio per questo sono attualmente sconosciuti. 15
Prolattina:
La somministrazione acuta di oppioidi stimola il rilascio di prolattina dall’ipofisi anteriore attraverso un effetto a livello dell’ipotalamo. 15 Questo effetto può essere bloccato dalla metoclopramide, suggerendo che sia mediato da meccanismi dopaminergici. L’effetto della somministrazione a lungo termine di oppioidi sulla prolattina è meno chiaro. C’è un aumento occasionale del rilascio di prolattina, probabilmente dipendente dal tipo di oppioide. Il significato clinico di questo è sconosciuto, ma può causare galattorrea.
Ormoni tiroidei:
In generale, gli oppioidi non sembrano alterare in modo significativo la funzione tiroidea, 19 sebbene possano stimolare l’ormone stimolante la tiroide (TSH) attraverso l’ipotalamo. 15 Questo non è importante negli individui con tiroxina libera normale, ma gli individui con ipotiroidismo possono avere risposte prolungate ed esagerate agli oppioidi. 22
Ormone della crescita:
La somministrazione acuta di oppioidi porta ad un aumento della secrezione dell’ormone della crescita (GH), attraverso meccanismi che coinvolgono i recettori degli oppioidi, i livelli di feedback e la trascrizione genica. La dose minima richiesta è di circa 15 mg di morfina. 15 Abs et al. 23 hanno riscontrato una carenza di GH in circa il 15% dei pazienti che ricevevano oppioidi intratecali a lungo termine, ma non in tutti i pazienti. È stato dimostrato che il naloxone inibisce il GH nei soggetti sani, ma lo aumenta nelle donne obese. L’effetto della somministrazione cronica di oppioidi sul GH è complesso e attualmente poco conosciuto, ma sembra essere correlato agli ormoni sessuali, alla composizione corporea e al grado di insulino-resistenza. 15
Vasopressina:
È stato scoperto che il tramadolo causa iponatriemia attraverso il rilascio di vasopressina indotto dalla serotonina. 24 L’effetto degli oppioidi sull’ipofisi posteriore non è chiaro: sono stati riscontrati livelli di vasopressina sia aumentati che diminuiti, a seconda dello stato di idratazione. 22
Ossitocina:
Studi su donne in gravidanza hanno dimostrato che la morfina inibisce la produzione di ossitocina nelle prime fasi del travaglio e durante l’allattamento al seno dopo il parto. 25
Obesità e diabete:
Un numero crescente di dati suggerisce che gli oppioidi svolgono un ruolo nella regolazione dell’assunzione di cibo e della scelta del cibo, e forse la ricompensa associata ai cibi di buon gusto, attraverso meccanismi centrali. 15 L’uso cronico di oppioidi è associato ad aumento di peso, iperglicemia e peggioramento del diabete. Questa può essere un’azione centrale attraverso il sistema nervoso simpatico e una ridotta secrezione di insulina. 26 L’ipogonadismo è associato ad un aumento della resistenza all’insulina e al rischio di diabete mellito, 15 un rischio che è migliorato dalla sostituzione del testosterone. 27
Metabolismo delle catecolamine:
La terapia con oppioidi aumenta la secrezione di catecolamine attraverso l’ipotalamo e il tronco cerebrale. I pazienti che assumono oppioidi a lungo termine devono essere sottoposti a screening per l’ipertensione. 22
Metabolismo osseo:
Esistono molti fattori di rischio per la diminuzione della densità minerale ossea e l’osteoporosi nei pazienti trattati con oppioidi, tra cui possibile scarso stato nutrizionale, ipogonadismo, inibizione degli osteoblasti, ridotta sintesi di osteocalcina, calcio anormale e ormone paratiroideo e aumento del riassorbimento osseo, mediato dall’interleuchina 1. rappresenta un aumento del rischio di frattura ossea nei pazienti che assumono oppioidi. 3
Via di somministrazione:
Gli oppioidi per uso cronico vengono somministrati per via orale o intratecale. Occasionalmente possono essere utilizzate altre vie, ad esempio l’iniezione ripetuta, sebbene questa pratica non sia raccomandata. 28
Cambiamenti ormonali che si verificano dopo la somministrazione intratecale sono stati riportati da diversi autori. 23 , 29 , 30
Si stima che il 90% dei pazienti che assumono oppioidi intratecali svilupperà ipogonadismo. Gli oppioidi orali hanno lo stesso effetto, sebbene l’inizio dell’azione possa essere più lento.
Tipo e dose di oppioide:
Le classi di oppioidi differiscono nel loro effetto sulla soppressione delle gonadi. Tramadolo e buprenorfina non alterano significativamente i livelli di testosterone negli animali e nell’uomo e la buprenorfina non sopprime il cortisolo sierico.
L’incidenza di ipogonadismo era maggiore nei sopravvissuti al cancro che ricevevano una dose equivalente o superiore a 200 mg di morfina al giorno per almeno 1 anno rispetto ai sopravvissuti di pari età non in terapia con oppioidi, suggerendo che gli effetti sono correlati alla dose. 31 Anche la durata della terapia con oppioidi sembra aumentare la possibilità di soppressione ormonale, sebbene ciò debba essere studiato ulteriormente.
Esiste una possibile differenza di genere negli effetti endocrini, tendente a una maggiore sintomatologia nelle donne, ma sono necessari ulteriori studi. 22
Quando e cosa si misura prima di iniziare una terapia con oppioidi:
Prima di iniziare la terapia cronica con oppioidi si raccomanda di misurare quanto segue:
pressione sanguigna;
elettroliti (soprattutto se si utilizza tramadolo);
livelli di glucosio a digiuno;
funzione tiroidea (per escludere l’ipotiroidismo);
livelli di testosterone, globulina legante l’ormone sessuale, LH/FSH ed estradiolo; e
densità ossea (in un gruppo “a rischio”).
Monitoraggio:
Non ci sono standard accettati, ma sembra ragionevole ripetere i test di cui sopra ogni 6 mesi.
Considera i livelli di cortisolo nel sangue, DHEA, ACTH e GH a digiuno mattutino. (Ricorda che un livello di cortisolo nel sangue a digiuno anormalmente alto può rappresentare la perdita della variazione diurna e dovresti chiedere consiglio.)
Ripetere la densità ossea ogni anno nel gruppo “a rischio”.
Misurare i livelli di prolattina se c’è galattorrea.
Terapia sostitutiva:
Non esistono standard accettati per la gestione della disfunzione endocrina indotta da oppioidi.
L’opzione migliore è ridurre gradualmente e ritirare gli oppioidi e monitorare la risposta per un periodo di alcuni mesi, se appropriato. Non è noto se il cambio di oppioidi sia di qualche beneficio. La buprenorfina sembra avere un effetto minore sugli ormoni surrenali ma ha un effetto maggiore sul TSH rispetto alla morfina. La risposta a diversi oppioidi è in gran parte sconosciuta al momento della scrittura.
Se non è possibile ottenere l’astinenza da oppiacei e il paziente presenta sintomi definiti di disfunzione endocrina, si raccomanda la sostituzione ormonale, con il monitoraggio da parte di un endocrinologo.
Il testosterone può essere sostituito, sia negli uomini che nelle donne, come cerotto o gel transdermico o per iniezione. È necessario un attento monitoraggio poiché gli effetti collaterali includono reazioni in sede, policitemia e aumento del rischio di cancro alla prostata negli uomini e irregolarità mestruali e irsutismo nelle donne.
La terapia sostitutiva con estrogeni è meglio monitorata da un ginecologo.
Cosa estrapolare di utile per un utilizzatore di PEDs da quanto detto?
Innanzi tutto bisogna sapere che l’attività di soppressione/sottoregolazione dell’Asse HPT androgeno-dipendente ha come intermediari i peptidi oppioidi endogeni, con attività principale da parte della Beta-Endorfina, delle Encefaline e Dinorfine attraverso il legame con i recettori oppioidi μ.
Quindi, di conseguenza, comprendere i meccanismi e gli effetti dell’uso di oppioidi sulla omeostasi ormonale, soprattutto riguardante l’HPTA, ci permette di capire meglio il legame tra attività AR di una molecola e suoi effetti mediati a livello ipotalamico che causano una sottoregolazione/soppressione della funzione del Asse HPT, anche se la molecola non è aromatizzabile e non possiede attività estrogenica e/o progestinica.
Adesso possiamo approfondire il discorso passando all’analisi del antagonisti degli oppioidi Naltrexone.
Caratteristiche del Naltrexone:
Il Naltrexone, venduto con tra gli altri con i nomi commerciali di ReVia e Vivitrol, è un farmaco utilizzato principalmente per gestire l’abuso di alcol o il disturbo da uso di oppioidi, riducendo le voglie e le sensazioni di euforia associate al disturbo da uso di sostanze “compensative”.[32] È stato anche osservato essere efficace nel trattamento di altre dipendenze e può essere utilizzato per loro in modalità off-label. [33] Una persona dipendente da oppioidi non dovrebbe ricevere il Naltrexone prima della disintossicazione.[4] Viene assunto per bocca o tramite iniezione intra-muscolo.[32] Gli effetti iniziano entro 30 minuti dalla somministrazione.[32] Una diminuzione del desiderio di oppioidi può richiedere alcune settimane per verificarsi.[32]
Il Naltrexone è un antagonista degli oppioidi e funziona bloccando gli effetti degli oppioidi, sia quelli endogeni che esogeni.[32]
Il Naltrexone è stato prodotto per la prima volta nel 1965 ed è stato approvato per uso medico negli Stati Uniti nel 1984.[32][34] Il Naltrexone, come Naltrexone/bupropione (nome commerciale Contrave), è anche usato per trattare l’obesità.[35]
Il Naltrexone, noto anche come N-ciclopropil-metilnorossimorfone, è un derivato dell’Ossimorfone (14-idrossi-diidromorfone). È specificamente il derivato dell’Ossimorfone in cui il sostituto metilico dell’ammina terziaria è sostituito con il Metilciclopropano. Il farmaco strettamente correlato, il Metilnaltrexone (N-metilnaltrexone), è usato per trattare la costipazione indotta dagli oppioidi, ma non tratta la dipendenza in quanto non attraversa la barriera emato-encefalica e, quindi, non è di nostro interesse per l’ipotetico utilizzo in ambito di PEDs. Il Nalmefene (6-desossi-6-metilenaltrexone) è simile al Naltrexone ed è usato per gli stessi scopi. Il Naltrexone non deve essere confuso con il Naloxone (N-allylnoroxymorphone), che è usato in casi di emergenza di overdose da oppioidi. Altri antagonisti degli oppioidi correlati al Naltrexone includono 6β-naltrexol (6β-idrossinaltrexone), Samidorphan (3-carbossamido-4-idrossinaltrexone), β-funaltrexamine (Naltrexone Fumarato Metil Estere), Nalodeina (N-allylnorcodeine), Nalorphine (N-allylnormorphine), e Nalbuphine (N-cyclobutylmethyl-14-hydroxydihydronormorphine).
Farmacocinetica del Naltrexone:
L’assorbimento del Naltrexone con la somministrazione orale è rapido e quasi completo (96%).[36] La biodisponibilità del Naltrexone con la somministrazione orale è dal 5 al 60% a causa di un esteso metabolismo di primo passaggio.[37][38] Le concentrazioni di picco del naltrexone sono da 19 a 44 μg/L dopo una singola dose orale di 100 mg e il tempo per le concentrazioni di picco del naltrexone e del 6β-naltrexol (metabolita) è entro 1 ora. [37][38][36] Aumenti lineari nelle concentrazioni circolanti di naltrexone e 6β-naltrexolo in un intervallo di dosi orali da 50 a 200 mg.[37] Il naltrexone non sembra essere accumulato con la somministrazione orale ripetuta una volta al giorno e non vi è alcun cambiamento nel tempo di picco delle concentrazioni con la somministrazione ripetuta.[37]
6β-naltrexolo
Il legame alle proteine plasmatiche del naltrexone è di circa il 20% in un intervallo di concentrazione di naltrexone da 0,1 a 500 μg/L.[37][36] Il suo volume apparente di distribuzione a 100 mg per via orale è di 16,1 L/kg dopo una singola dose e 14,2 L/kg con dosi ripetute.[37]
Il naltrexone viene metabolizzato nel fegato principalmente dalle diidrodiol deidrogenasi in 6β-naltrexolo (6β-idrossinaltrexone).[37][38] I livelli di 6β-naltrexolo sono da 10 a 30 volte superiori a quelli del naltrexone con la somministrazione orale a causa del vasto metabolismo di primo passaggio. [39] Al contrario, l’esposizione al 6β-naltrexolo è solo circa 2 volte superiore a quella del naltrexone con l’iniezione intramuscolare di naltrexone in microsfere (nome commerciale Vivitrol).[40] Il 6β-Naltrexolo è un antagonista dei recettori degli oppioidi in modo simile al naltrexone e mostra un profilo di legame comparabile ai recettori degli oppioidi. [41] Tuttavia, il 6β-naltrexolo è selettivo a livello periferico e attraversa il cervello molto meno facilmente del naltrexone.[41] In ogni caso, il 6β-naltrexolo mostra ancora una certa attività centrale e può contribuire significativamente alle azioni centrali del naltrexone orale. [41][37] Altri metaboliti del naltrexone includono il 2-idrossi-3-metossi-6β-naltrexolo e il 2-idrossi-3-metossinaltrexone.[37] Dopo la loro formazione, i metaboliti del naltrexone sono ulteriormente metabolizzati per coniugazione con l’acido glucuronico per formare glucuronidi.[37] Il Naltrexone non è metabolizzato dal sistema del citocromo P450 e ha un basso potenziale di interazioni farmacologiche.[42]
L’eliminazione del naltrexone è biesponenziale e rapida nelle prime 24 ore seguita da un terzo declino estremamente lento dopo 24 ore.[37] Le emivite di eliminazione rapida del naltrexone e del suo metabolita 6β-naltrexolo sono rispettivamente di circa 4 ore e 13 ore.[43] Nelle compresse orali Contrave, che contengono anche bupropione e sono descritte come a rilascio prolungato, l’emivita del naltrexone è di 5 ore. [44] L’emivita di eliminazione lenta in fase terminale del naltrexone è di circa 96 ore.[45] Come microsfere di Naltrexone per iniezione intramuscolare (Vivitrol), le emivite di eliminazione del naltrexone e del 6β-naltrexolo sono entrambe di 5-10 giorni.[46] Mentre il naltrexone orale viene somministrato quotidianamente, il naltrexone in microsfere per iniezione intramuscolare è adatto alla somministrazione una volta ogni 4 settimane o una volta al mese.[46]
Livelli ematici di Naltrexone dopo una dose orale di 50mg allo stato stazionario durante il trattamento con 50mg/giorno di Naltrexone.Livelli ematici di Naltrexone dopo una dose di 380mg in microsfere (Vivitrol) per iniezione intramuscolare allo stato stazionario durante il trattamento mensile con 380mg di Naltrexone in microsfere.
Il Naltrexone e i suoi metaboliti sono escreti nelle urine.[36]
Farmacodinamica del Naltrexone:
Il Naltrexone e il suo metabolita attivo 6β-naltrexolo sono antagonisti competitivi dei recettori oppioidi.[47][48] Il naltrexone è specificamente un antagonista preferenzialmente del recettore μ-opioide (MOR), in misura minore del recettore κ-opioide (KOR), e in misura molto minore del recettore δ-opioide (DOR). [47] Tuttavia, il naltrexone non è in realtà un antagonista silenzioso di questi recettori, ma agisce invece come un debole agonista parziale, con valori di Emax dal 14 al 29% al MOR, dal 16 al 39% al KOR, e dal 14 al 25% al DOR in diversi studi.[48][49][50] In accordo con il suo agonismo parziale, sebbene il naltrexone sia descritto come un puro antagonista dei recettori oppioidi, ha mostrato alcune prove di deboli effetti oppioidi in studi clinici e preclinici.[37]
Da solo, il naltrexone agisce come un antagonista o un debole agonista parziale dei recettori oppioidi.[48] In combinazione con agonisti del MOR come la morfina, tuttavia, il naltrexone sembra diventare un agonista inverso del MOR.[48] Al contrario, il naltrexone rimane un antagonista neutro (o un debole agonista parziale) del KOR e DOR. [In contrasto con il naltrexone, il 6β-naltrexolo è puramente un antagonista neutro dei recettori oppioidi.[51] L’agonismo inverso MOR del naltrexone quando è co-presente con agonisti MOR può in parte spiegare la sua capacità di far precipitare l’astinenza in individui dipendenti da oppioidi.[51][48] Ciò può essere dovuto alla soppressione della segnalazione MOR basale attraverso l’agonismo inverso.[51][48]
L’occupazione dei recettori degli oppioidi nel cervello da parte del Naltrexone è stata studiata utilizzando la tomografia a emissione di positroni (PET).[52][53] Il Naltrexone alla dose di 50 mg/giorno è risultato occupare circa il 90-95% dei MOR del cervello e il 20-35% dei DOR del cervello. [52] Il Naltrexone alla dose di 100 mg/giorno è stato osservato raggiungere l’87% e il 92% di occupazione cerebrale del KOR in diversi studi.[54][53][55] Per simulazione, una dose più bassa di Naltrexone di 25 mg/giorno potrebbe essere prevista per raggiungere circa il 60% di occupazione cerebrale del KOR ma ancora vicino al 90% di occupazione del MOR. [53] In uno studio sulla durata del blocco del MOR con il Naltrexone, il farmaco con una singola dose da 50mg ha mostrato un blocco del 91% del legame cerebrale al [11C]carfentanil (un ligando selettivo del MOR) a 48 ore (2 giorni), un blocco dell’80% a 72 ore (3 giorni), un blocco del 46% a 120 ore (5 giorni) e un blocco del 30% a 168 ore (7 giorni). [8][9] Il tempo di dimezzamento del blocco MOR del cervello da parte del Naltrexone in questo studio era da 72 a 108 ore (3. 0 a 4,5 giorni).[56][45] Sulla base di questi risultati, dosi di Naltrexone anche inferiori a 50mg/giorno dovrebbero raggiungere un’occupazione dei MOR cerebrali praticamente completa.[8][9] Il blocco dei MOR cerebrali con Naltrexone è molto più duraturo rispetto ad altri antagonisti oppioidi come il Naloxone (tempo di dimezzamento di ~ 1,7 ore per via intranasale) o il Nalmefene (tempo di dimezzamento di ~ 29 ore).[56][57][58]
Nalmefene
L’emivita di occupazione dei MOR cerebrali e la durata dell’effetto clinico del Naltrexone sono molto più lunghe di quanto suggerito dalla sua emivita di eliminazione plasmatica.[56][59][45] Una singola dose orale di 50mg di Naltrexone ha mostrato di bloccare i MOR cerebrali e gli effetti oppioidi per almeno 48-72 ore. [59][45][60] L’emivita del blocco dei MOR cerebrali da parte del Naltrexone (72-108 ore) è molto più lunga della componente di clearance plasmatica rapida del Naltrexone e del 6β-naltrexolo (~4-12 ore), ma è stato riportato che corrisponde bene alla fase terminale più lunga della clearance plasmatica del Naltrexone (96 ore). [56][45][61] Come possibilità alternativa, la prolungata occupazione cerebrale dei MOR da parte degli antagonisti oppioidi come il Naltrexone e il Nalmefene può essere dovuta alla lenta dissociazione dai MOR conseguente alla loro affinità MOR molto alta (<1,0 nM).[58][62]
Il Naltrexone blocca gli effetti degli agonisti MOR come la Morfina, l’Eroina e l’Idromorfone nell’uomo attraverso il suo antagonismo MOR.[37][42] Dopo una singola dose di 100mg di Naltrexone, gli effetti soggettivi e oggettivi dell’Eroina sono stati bloccati del 90% a 24 ore, con un blocco che è diminuito fino a 72 ore.[1] Allo stesso modo, da 20 a 200mg di Naltrexone hanno antagonizzato in modo dose-dipendente gli effetti dell’Eroina fino a 72 ore. [37] Il Naltrexone blocca anche gli effetti degli agonisti KOR come la Salvinorina A, la Pentazocina e il Butorfanolo negli esseri umani attraverso il suo antagonismo KOR.[63][64][65][66] Oltre agli oppioidi, il Naltrexone è stato osservato bloccare o ridurre gli effetti gratificanti e altri effetti di altre droghe euforizzanti tra cui l’alcol,[67] la Nicotina,[68] e le Anfetamine.[69]
Come visto in precedenza, i recettori degli oppioidi sono coinvolti nella regolazione neuroendocrina.[37] Gli agonisti MOR producono aumenti dei livelli di Prolattina e diminuzioni dei livelli di Ormone Luteinizzante (LH) e Testosterone. [37] Dosi di Naltrexone da 25 a 150mg/giorno sono state osservate produrre aumenti significativi nei livelli di β-endorfina, Cortisolo e LH, cambiamenti equivoci nei livelli di Prolattina e Testosterone, e nessun cambiamento significativo nei livelli di ormone Adrenocorticotropico (ACTH) o Ormone Follicolo-Stimolante (FSH). [37] Il Naltrexone influenza l’Asse Ipotalamo-Ipofisi-Surrene (Asse HPT) probabilmente attraverso l’interferenza con la segnalazione del recettore degli oppioidi da parte delle endorfine.
Si pensa che il blocco dei MOR sia il meccanismo d’azione del Naltrexone nella gestione della dipendenza da oppioidi – blocca o attenua reversibilmente gli effetti degli oppioidi. Si pensa anche che sia coinvolto nell’efficacia del Naltrexone nella dipendenza da alcol riducendo gli effetti euforici di quest’ultimo. Il ruolo della modulazione del KOR da parte del Naltrexone nella sua efficacia per la dipendenza da alcol non è chiaro, ma questa azione potrebbe anche essere coinvolta in base alla teoria e agli studi sugli animali.[70][71]
Oltre ai recettori degli oppioidi, il Naltrexone si lega e agisce come antagonista del recettore del fattore di crescita degli oppioidi (OGFR) e del recettore toll-like 4 (TLR4) e interagisce con siti di legame ad alta e bassa affinità nella filamina-A (FLNA).[72][73][74][75] Si dice che dosi molto basse di Naltrexone (<0. 001-1 mg/die) interagiscono con FLNA, basse dosi (da 1 a 5 mg/die) producono antagonismo di TLR4, e dosi cliniche standard (da 50 a 100 mg/die) esercitano antagonismo del recettore degli oppioidi e OGFR.[72][74] Le interazioni del Naltrexone con FLNA e TLR4 sono ritenute coinvolte negli effetti terapeutici del Naltrexone a basse dosi.[72]
Farmacogenetica del Naltrexone: Prove provvisorie suggeriscono che la storia familiare e la presenza del polimorfismo Asn40Asp predice l’efficacia del Naltrexone.[76][77]
Effetti collaterali del Naltrexone:
Gli effetti collaterali più comuni riportati con il Naltrexone sono disturbi gastrointestinali come diarrea e crampi addominali.[36] Questi effetti avversi sono analoghi ai sintomi dell’astinenza da oppioidi, poiché il blocco del recettore μ-opioide aumenterà la motilità gastrointestinale.
Gli effetti collaterali del naltrexone per incidenza sono i seguenti:[36]
Più del 10%: difficoltà a dormire, ansia, nervosismo, dolori addominali/crampi, nausea e/o vomito, poca energia, dolori articolari/muscolari e mal di testa.[36]
Meno del 10%: perdita di appetito, diarrea, costipazione, sete, aumento di energia, sensazione di depressione, irritabilità, vertigini, eruzione cutanea, eiaculazione ritardata, disfunzione erettile e brividi.[36]
Una varietà di altri eventi avversi sono stati riportati anche con un’incidenza inferiore all’1%.[36] È stato segnalato che il Naltrexone può causare danni al fegato se somministrato a dosi più elevate di quelle raccomandate.[52] È oggetto di un avvertimento della FDA per questo raro effetto collaterale.
Ipotesi d’uso del Naltrexone in contesto PEDs:
Ora, abbiamo tutte le informazioni necessarie per poter comprendere come un antagonista dei recettori oppioidi possa portare a dei possibili vantaggi durante l’uso di PEDs o, meglio, di alcuni farmaci rientranti in tale categoria.
Riassumendo in breve:
Sappiamo che la sottoregolazione/soppressione del Asse HPT avviene anche in modo androgeno-dipendente: quindi, anche nel caso di molecole non aromatizzabili e non aventi attività estrogeniche intrinseche o progestiniche;
Sappiamo che i peptidi oppioidi endogeni agiscono da intermediari dell’attività androgena ipotalamica: attività che vede una maggiore interazione da parte delle Encefaline, Beta-Endorfine e Dinorfine con i recettori oppioidi mu (μ) (MOR OP3(I));
Sappiamo che il Naltrexone può bloccare l’attività dei delle Encefaline, Beta-Endorfine e Dinorfine con i recettori oppioidi mu (μ) (MOR OP3(I));
Sappiamo che tale blocco recettoriale può tradursi in una mancata (o per lo meno marcata e significativa) sottoregolazione/soppressione androgeno-dipendente del Asse HPT.
Se a ciò aggiungiamo che tale potenziale sarebbe al quanto insignificante in un contesto d’uso di AAS aromatizzabili e/o aventi attività estrogenica intrinseca e/o progestinica, anche volendoci abbinare un SERM il gioco non varrebbe la candela, l’unica possibilità d’uso con ritorno “positivo” si verificherebbe senza dubbio con l’uso di soli SARM non steroidei o, tutt’al più, con l’uso di DHT derivati senza la sopramenzionata caratteristica di attività estrogenica intrinseca (quindi niente Oxymetholone).
Per quanto rimanga un idiozia per un atleta di sesso maschile intraprendere protocolli di soli SARM non steroidei o AAS non-aromatizzabili, o progestinici, senza una base per lo meno fisiologica di Testosterone o l’uso di hCG onde garantire almeno un sufficiente “equilibrio” etsrogenico e di DHT, rappresenti il culmine dell’ignoranza e dell’incompetenza (se parliamo di “preparatori”), per i “testofobici” e gli “agofobici” l’uso del Naltrexone potrebbe (e, sottolineo, il condizionale “potrebbe”) garantire una conservazione sufficiente dell’attività del Asse HPT.
Nonostante vi siano scarsi lavori che hanno osservato l’attività cerebrale dei SARM non steroidei, e che uno in particolare abbia affermato che l’Ostarina non passi in modo significativo la barriera ematoencefalica, questa categoria di PEDs è per lo più strutturata su modifiche della molecola antiandrogena Bicalutamide la quale oltrepassa la barriera ematoencefalica.
Molecole di Ostarina (sinistra) e Bicalutamide (destra) a confronto.
Sebbene le modifiche strutturali determinino, per ovvie ragioni, una diversa farmacodinamica, il fatto che la sottoregolazione del HPTA, misurata anche a dosi di 3mg/die di Ostarina, sia con tutta probabilità androgeno-dipendente a livello ipotalamico [androgeno dipendente in senso di legame con i AR non nel senso di effetto androgeno genico] mi spinge ad ipotizzare una potenziale utilità del Naltrexone. Senza considerare che SARM non steroidei come LGD 4033, nonostante abbiano caratteristiche strutturali diverse dall’Ostarina o dalla Andarina, causino sottoregolazioni molto più marcate dei SARM non steroidei con struttura base del Bicalutamide.
LGD4033
Conclusioni:
Questo articolo è un esposizione accurata di una ipotesi e nulla più! Per quanto sensata possa essa essere, non abbiamo attualmente documentazione scientifica che dimostri inconfutabilmente che tale pratica sia funzionale e vantaggiosa o tantomeno sicura.
Il Naltrexone è un farmaco con una serie di potenziali effetti avversi (vedi sopra). Alcuni di essi potrebbero, in alcuni soggetti, vanificare i vantaggi sulla sfera sessuale ottenuti da un ipotetica conservazione del attività del HPTA. Da considerare anche la cura della dose somministrata per possibile danno epatico da sovradosaggio.
Gabriel Bellizzi
Riferimenti:
Moore AR, McQuay HJ. Prevalenza di eventi avversi da oppioidi nel dolore cronico non maligno: revisione sistematica di studi randomizzati di oppioidi orali . Artrite Res Ther 2005; 7 : 1046–1051. [ Articolo gratuito PMC ] [ PubMed ] [ Google Scholar ]2.
Furlan AD, Sandoval JA, Mailis-Gagnon A, et al. Oppioidi per il dolore cronico non oncologico: una meta-analisi di efficacia ed effetti collaterali . Can Med Assoc J 2006; 174 : 1589–1594. [ Articolo gratuito PMC ] [ PubMed ] [ Google Scholar ]3.
Raghavan S, Harvey AD, Humble SR. Nuovi effetti collaterali e implicazioni per gli oppioidi per la terapia a lungo termine . Tendenze Anaesth Crit Care 2011; 1 : 18–21. [ Google Scholar ]4.
Okie S. Un’ondata di oppioidi, una marea crescente di morti . N inglese J Med 2010; 363 ( 21 ): 1981–1983. [ PubMed ] [ Google Scholar ]5.
Graves RH. Quarant’anni in Cina, Baltimora: Woodward , 1895. [ Google Scholar ]6.
Cumming HS, Control of DrugAddiction Mainly a Police Problem, The American City Magazine (novembre 1925, fascicolo 126, riquadro 3, sottoserie 1, serie 3.7.
Katz N, Mazer NA. L’impatto degli oppioidi sul sistema endocrino . Clin J Dolore 2009; 25 ( 2 ): 170–175. [ PubMed ] [ Google Scholar ]8.
Bruce CA. Relazione sulla produzione del tè e sull’estensione e la produzione delle piantagioni di tè nell’Assam . Calcutta: : Bishop’s College Press, 1839. [ Google Scholar ]9.
Hiller-Sturmhofel S, Bartke A. Il sistema endocrino: una panoramica . Alcol Health Res World 1998; 22 ( 3 ): 153–164. [ Articolo gratuito PMC ] [ PubMed ] [ Google Scholar ]10.
Ganong WF. Rassegna di fisiologia medica . USA: Lange/McGraw-Hill, 2005. [ Google Scholar ]11.
Luo Xiwen tr. Bencao Gangmu: Compendio di Materia Medica . Foreign Languages Press, 2003. Pubblicato per la prima volta nel 157812.
Van Vugt DA, Baby N, Stewart M, Reid RL. L’effetto stimolante paradossale della morfina sulla secrezione di LH è dose-dipendente e reversibile al naloxone . Neuroendocrinologia 1989; 50 : 109–116. [ PubMed ] [ Google Scholar ]13.
De la Rosa RE, Hennessey JV. Ipogonadismo e metadone: ipogonadismo ipotalamico dopo l’uso a lungo termine di metadone ad alte dosi . Pratica Endocr 1996; 2 :4–7. [ PubMed ] [ Google Scholar ]14.
Daniel HW. Deficit di DHEAS durante il consumo di oppioidi prescritti ad azione prolungata: evidenza dell’inibizione indotta da oppioidi della produzione di androgeni surrenali . J Dolore 2006; 7 ( 12 ): 901–907. [ PubMed ] [ Google Scholar ]15.
Vuong C, Van Uum SHM, O’Dell L, et al. Gli effetti degli oppioidi e degli analoghi degli oppioidi sui sistemi endocrini animali e umani . Endocr Rev 2010; 31 ( 1 ): 98–132. [ Articolo gratuito PMC ] [ PubMed ] [ Google Scholar ]16.
Van Furth WR, van Emst MG, van Ree JM. Oppioidi e comportamento sessuale dei ratti maschi: coinvolgimento dell’area preottica mediale . Comportamento Neurosci 1995; 109 ; 123–134. [ PubMed ] [ Google Scholar ]17.
Daniell Harry W. Le linee guida cliniche della Endocrine Society sulla terapia con testosterone negli uomini adulti con sindromi da carenza di androgeni . J Clin Metab endocrinolo 2010; 95 ( 6 ): 2536–2559. [ PubMed ] [ Google Scholar ]18.
O’Donne AB, Araujo AB, McKinlay JB. La salute degli uomini che invecchiano normalmente: The Massachusetts Male Aging Study (1987–2004) . Scad. Gerontolo 2004; ( 7 ): 975–984. [ PubMed ] [ Google Scholar ]19.
Katz N. L’impatto degli oppioidi sul sistema endocrino . Round di gestione del dolore 2005; 1 ( 9 ). www.painmanagementrounds.org (visitato il 5 febbraio 2012) [ Google Scholar ]20.
Faccinetti F, Grasso A, Petraglia F, et al. Ridotta ritmicità circadiana di β-lipotropina, β-endorfina e ACTH nei tossicodipendenti . Acta endocrinolo 1984; 105 ( 2 ): 149–155. [ PubMed ] [ Google Scholar ]21.
Daniel HW. Endocrinopatia da oppioidi nelle donne che consumano oppioidi ad azione prolungata prescritti per il controllo del dolore non maligno . J Dolore 2008; 9 ( 1 ): 28–36. [ PubMed ] [ Google Scholar ]22.
Thosani S, Jiminez C. Alterazioni biochimiche dell’asse neuroendocrino indotte da oppioidi . Esperto Rev Endocrinol Metab 2011; 6 ( 5 ): 705–713. [ Google Scholar ]23.
Abs R, Verhelst J, Maeyaert J, et al. Conseguenze endocrine della somministrazione intratecale a lungo termine di oppioidi . J Clin Endo Metab 2000; 85 ( 6 ): 2215–2222. [ PubMed ] [ Google Scholar ]24.
Sarret D, Le Berre JP, Zemraoui N. Iponatriemia indotta da Tramadol . Am J Kidney Dis 2008; 52 ( 5 ): 1026. [ PubMed ] [ Google Scholar ]25.
Lindow SW, Hendricks MS, Nugent FA, et al. La morfina sopprime la risposta all’ossitocina nelle donne che allattano . Gynecol Obstet Invest 1999; 48 ( 1 ): 33–37. [ PubMed ] [ Google Scholar ]26.
Giugliano D. Morfina, peptidi oppioidi e funzione delle isole pancreatiche . Cura del diabete 1984; 7 : 92–98. [ PubMed ] [ Google Scholar ]27.
Kapoor D, Goodwin E, Channer KS, Jones TH. La terapia sostitutiva del testosterone migliora la resistenza all’insulina, il controllo glicemico, l’adiposità viscerale e l’ipercolesterolemia negli uomini ipogonadici con diabete di tipo 2 . Eur J Endocrinolo 2006; 154 : 899–906. [ PubMed ] [ Google Scholar ]28. Oppiacei per il dolore persistente; buona pratica . 2010.
The British Pain Society, Facoltà di Medicina del Dolore del Royal College of Anesthetists, Royal College of General Practitioners e Facoltà di Medicina delle Dipendenze del Royal College of Psychiatrists . Disponibile su: http://www.britishpainsociety.org/book_opioid_main.pdf (visitato il 5 febbraio 2012)
29. Roberts LJ, Finch PM, Pullan PT, et al. Soppressione dell’ormone sessuale da parte degli oppioidi intratecali: uno studio prospettico . Clin J Dolore 2002; 18 : 144–148. [ PubMed ] [ Google Scholar ]
30.Thimineur MA, Kravitz E, Vodapally MS. Trattamento intratecale con oppioidi per il dolore cronico non maligno: uno studio prospettico di 3 anni . Dolore 2004; 109 : 242–249. [ PubMed ] [ Google Scholar ]
31.Rajagopal A, Vasilopoulou-Sellin R, Palmer JL, et al. Ipogonadismo sintomatico nei sopravvissuti maschi al cancro con esposizione cronica agli oppioidi . Cancro 2004; 100 ( 4 ): 851–858. [ PubMed ] [ Google Scholar ]
37. Gonzalez JP, Brogden RN (March 1988). “Naltrexone. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic efficacy in the management of opioid dependence”. Drugs. 35 (3): 192–213.
38. Lee MW, Fujioka K (August 2009). “Naltrexone for the treatment of obesity: review and update”. Expert Opin Pharmacother. 10 (11): 1841–5.
39. Davis MP, Glare PA, Hardy J (28 May 2009). Opioids in Cancer Pain. Oxford University Press. pp. 41–.
40. Dunbar JL, Turncliff RZ, Dong Q, Silverman BL, Ehrich EW, Lasseter KC (March 2006). “Single- and multiple-dose pharmacokinetics of long-acting injectable naltrexone”. Alcohol Clin Exp Res. 30 (3): 480–90.
41.Hipkin, R. William; Dolle, Roland E. (2010). “Opioid Receptor Antagonists for Gastrointestinal Dysfunction”. Annual Reports in Medicinal Chemistry. Vol. 45. Elsevier. pp. 142–155.
42.Sevarino, Kevin A.; Kosten, Thomas R. (2009). “Naltrexone for Initiation and Maintenance of Opiate Abstinence”. Opiate Receptors and Antagonists. Humana Press. pp. 227–245.
48.Wang D, Sun X, Sadee W (May 2007). “Different effects of opioid antagonists on mu-, delta-, and kappa-opioid receptors with and without agonist pretreatment”. J Pharmacol Exp Ther. 321 (2): 544–52.
51.Sadée W, Wang D, Bilsky EJ (February 2005). “Basal opioid receptor activity, neutral antagonists, and therapeutic opportunities”. Life Sci. 76 (13): 1427–37.
52.Ray LA, Chin PF, Miotto K (March 2010). “Naltrexone for the treatment of alcoholism: clinical findings, mechanisms of action, and pharmacogenetics”. CNS Neurol Disord Drug Targets. 9 (1): 13–22.
53.de Laat B, Nabulsi N, Huang Y, O’Malley SS, Froehlich JC, Morris ED, Krishnan-Sarin S (June 2020). “Occupancy of the kappa opioid receptor by naltrexone predicts reduction in drinking and craving”. Mol Psychiatry. 26 (9): 5053–5060.
54.Placzek MS (August 2021). “Imaging Kappa Opioid Receptors in the Living Brain with Positron Emission Tomography”. Handb Exp Pharmacol. Handbook of Experimental Pharmacology. 271: 547–577.
56.Colasanti, Alessandro; Lingford-Hughes, Anne; Nutt, David (2013). “Opioids Neuroimaging”. In Miller, Peter M. (ed.). Biological Research on Addiction. Comprehensive Addictive Behaviors and Disorders. Vol. 2. Elsevier. pp. 675–687.
57.van Waarde, Aren; Absalom, Anthony R.; Visser, Anniek K. D.; Dierckx, Rudi A. J. O. (30 September 2020). “Positron Emission Tomography (PET) Imaging of Opioid Receptors”. PET and SPECT of Neurobiological Systems. Springer International Publishing. pp. 749–807.
58.Soyka M, Rösner S (November 2010). “Nalmefene for treatment of alcohol dependence”. Expert Opin Investig Drugs. 19 (11): 1451–9.
61.Miotto K, McCann M, Basch J, Rawson R, Ling W (2002). “Naltrexone and dysphoria: fact or myth?”. Am J Addict. 11 (2): 151–60.
62.Ingman K, Hagelberg N, Aalto S, Någren K, Juhakoski A, Karhuvaara S, Kallio A, Oikonen V, Hietala J, Scheinin H (December 2005). “Prolonged central mu-opioid receptor occupancy after single and repeated nalmefene dosing”. Neuropsychopharmacology. 30 (12): 2245–53.
65.Preston KL, Bigelow GE (February 1993). “Differential naltrexone antagonism of hydromorphone and pentazocine effects in human volunteers”. J Pharmacol Exp Ther. 264 (2): 813–23.
66.Clark SD, Abi-Dargham A (October 2019). “The Role of Dynorphin and the Kappa Opioid Receptor in the Symptomatology of Schizophrenia: A Review of the Evidence”. Biol Psychiatry. 86 (7): 502–511.
67.Unterwald EM (September 2008). “Naltrexone in the treatment of alcohol dependence”. J Addict Med. 2 (3): 121–7.
68.Modesto-Lowe V, Van Kirk J (August 2002). “Clinical uses of naltrexone: a review of the evidence”. Exp Clin Psychopharmacol. 10 (3): 213–27.
69.Lam L, Anand S, Li X, Tse ML, Zhao JX, Chan EW (April 2019). “Efficacy and safety of naltrexone for amfetamine and methamfetamine use disorder: a systematic review of randomized controlled trials”. Clin Toxicol (Phila). 57 (4): 225–233.
70.Soyka M, Friede M, Schnitker J (March 2016). “Comparing Nalmefene and Naltrexone in Alcohol Dependence: Are there any Differences? Results from an Indirect Meta-Analysis”. Pharmacopsychiatry. 49 (2): 66–75.
74. Lee B, Elston DM (June 2019). “The uses of naltrexone in dermatologic conditions”. J Am Acad Dermatol. 80 (6): 1746–1752.
75.Zagon IS, Verderame MF, McLaughlin PJ (February 2002). “The biology of the opioid growth factor receptor (OGFr)”. Brain Res Brain Res Rev. 38 (3): 351–76.
77.Garbutt JC, Greenblatt AM, West SL, Morgan LC, Kampov-Polevoy A, Jordan HS, Bobashev GV (August 2014). “Clinical and biological moderators of response to naltrexone in alcohol dependence: a systematic review of the evidence”. Addiction. 109 (8): 1274–84.
La società farmaceutica Novartis sta conducendo studi con l’antagonista della Miostatina Bimagrumab, del quale ho già parlato in un articolo del 2017. Gli studi in cui i soggetti patologici, affetti da miosite da corpi inclusi (IBM), sono stati trattati con Bimagrumab sono risultati non terapeuticamente responsivi [1] ma un recente studio suggerisce che questo nuovo farmaco possa avere più applicazioni oltre ad un uso per aumentare l’anabolismo muscolare. Si è osservato, infatti, che il Bimagrumab riduce la massa grassa.[2]
Cos’è il Bimagrumab?:
Bimagrumab (BYM338) è un anticorpo monoclonale umano sviluppato da Novartis per il trattamento della perdita e la debolezza muscolare patologica. Il 20 agosto 2013, è stato annunciato che Bimagrumab aveva ricevuto una designazione di terapia rivoluzionaria per la miosite sporadica da corpi inclusi (sIBM) dalla Food and Drug Administration degli Stati Uniti. [3] Nel 2014, Bimagrumab è entrato nello sviluppo di Fase II, con alcune ricerche che indicano effetti clinici. [4] Novartis ha presentato domanda nel 2016 per l’approvazione della FDA per il trattamento dei pazienti con sIBM con Bimagrumab. [5] Come accennato precedentemente, nell’aprile 2016, Novartis ha annunciato che Bimagrumab aveva fallito uno studio di Fase IIb / III per la miosite sporadica da corpi inclusi. Nel gennaio 2021, lo studio che andremo a trattare in questo articolo, l’uso di Bimagrumab ha confermato la sua sicurezza ed efficace per il trattamento dell’adiposità in eccesso e dei disturbi metabolici in pazienti adulti con obesità e diabete di tipo II.
Modalità di legame del bimagrumab con ActRIIA e ActRIIB. Strutture cristalline del complesso bimagrumab Fv (nastro blu chiaro / scuro) con (A) ActRIIA umana LBD (nastro dorato) o (B) ActRIIB LBD umano (nastro rosso), mostrate nello stesso orientamento. (C) Struttura cristallina del complesso ActRIIB LBD di topo con activin-β umana (file PDB 1S4Y) (35), mostrato con lo stesso orientamento di A, B e D. (D) Sovrapposizione del complesso ActRIIA Fv (oro nastro) e complesso ActRIIB Fv (nastro rosso). (E) Impronta di activin (superficie grigia, calcolata dall’archivio PDB 1S4Y) e (F) Bimagrumab su ActRIIB LBD. Notare l’ampia sovrapposizione tra le due superfici di rilegatura.
Caratteristiche dello studio:
I ricercatori hanno diviso un gruppo di circa 50 obesi, alcuni dei quali con diabete di tipo II, in 2 gruppi. Per 48 settimane, hanno somministrato ai soggetti di un gruppo il Bimagrumab con una flebo ogni 4 settimane, alla dose di 10mg per chilogrammo di peso corporeo. La dose massima che i soggetti hanno ricevuto è stata di 1.2g per somministrazione. I soggetti in un gruppo di controllo hanno ricevuto un’infusione senza una sostanza attiva (placebo). Entrambi i gruppi hanno ricevuto informazioni da seguire concernenti alimentazione sana e lo svolgimento di esercizio fisico.
Risultati:
Alla fine delle 48 settimane, i soggetti nel gruppo sperimentale avevano perso 3,4Kg di grasso corporeo. Ciò ammontava al 21% della loro massa grassa. I soggetti del gruppo placebo non hanno praticamente perso grasso corporeo in modo significativo.
Allo stesso tempo, i soggetti che hanno ricevuto il Bimagrumab hanno guadagnato quasi 4Kg di massa corporea magra [sebbene non sia specificata quanta massa muscolare effettiva], mentre i soggetti dell’altro gruppo hanno perso quasi 0.5Kg di massa corporea magra.
Effetti collaterali riscontrati:
L’inibitore della Miostatina non è risultato privo di effetti collaterali. Ogni 10 partecipanti allo studio che avevano ricevuto il Bimagrumab, 4 manifestarono diarrea. Un numero uguale di partecipanti allo studio ha riportato spasmi muscolari.
Conclusioni:
I ricercatori riportano che, 48 settimane di esposizione al Bimagrumab, un inibitore di anticorpi di ActRII, sono state sicure ed efficaci per il trattamento dell’eccesso di adiposità e disturbi metabolici in pazienti adulti con obesità e diabete di tipo 2. Mentre il blocco o knockout degli anticorpi di ActRII nei modelli animali è accompagnato da un marcato aumento della massa muscolo-scheletrica, questo studio conferma che l’inibizione di questo recettore nei partecipanti umani porta non solo ad un aumento della massa magra ma anche a una profonda diminuzione del grasso corporeo, insieme a miglioramenti nel controllo glicemico.
L’inibizione di ActRII può fornire un nuovo percorso per la gestione farmacologica dell’eccesso di adiposità e dei disturbi metabolici associati.
Chiunque abbia assistito ad una competizione di BodyBuilding, indipendentemente dal livello, e in special modo negli ultimi decenni, si sarà accorto di “anomalie” decisamente antiestetiche in un buon numero di partecipanti. Non parlo di asimmetrie, ritenzione idrica o percentuali di grasso discutibili in un contest, ma della Ginecomastia. Tralasciando le gare di campagna stile “camporella tra paesani”, e i “naturnazzi” che ogni tanto possono destare qualche sospetto, la diffusione di casi di Ginecomastia è diventata una “piaga” che, oltretutto, sembra non destare disagio ai “bonghi”, ed ai loro pseudo preparatori, i quali postano tranquillamente foto mostranti palesi ipertrofie mammarie. Ora, ovviamente ciò è dovuto all’ignoranza in materia farmacologica e applicativa dei PED da parte dei prima citati “preparatori” (anche noti) e/o degli atleti. La mancata conoscenza delle basi necessarie per gestire una supplementazione farmacologica rende praticamente impossibile prevenire la comparsa di Ginecomastia iatrogeno-dipendente. Per tale motivo, ho ritenuto utile scrivere un articolo finalizzato alla conoscenza dettagliata di questo inestetismo e di come possa essere evitato o, in altri casi, trattato quando il bisturi non è ancora diventato l’ultima risorsa.
“La bellezza è Verità, la Verità è bellezza: questo è tutto ciò che voi sapete in terra e tutto ciò che vi occorre sapere.”
John Keats.
Cos’è la Ginecomastia e quali sono le sue caratteristiche?
Ginecomastia adolescenziale in un soggetto con una massa grassa ridotta (1951); nessuna presenza di lipomastia.
La Ginecomastia (detta anche Ginaecomastia) è una malattia del sistema endocrino che si manifesta con un aumento non canceroso delle dimensioni del tessuto mammario maschile.[1][2] A causa della sua comparsa, può verificarsi disagio psicologico o disforia da parte del soggetto interessato.
Lo sviluppo della Ginecomastia è solitamente associato a cambiamenti puberali benigni. Tuttavia, il 75% dei casi di Ginecomastia puberale si risolve entro due anni dall’esordio senza trattamento.[2] In rari casi, è noto che la Ginecomastia si manifesta in associazione a determinati stati patologici.[3] Le cause patologiche della ginecomastia sono diverse e possono includere la sindrome di Klinefelter, alcuni tipi di cancro, disturbi endocrini, disfunzione metabolica, vari farmaci o possono verificarsi a causa di un calo naturale o indotta della produzione di Testosterone.[4][5] Si ritiene che i disturbi del sistema endocrino che portano ad un aumento del rapporto estrogeni / androgeni siano responsabili dello sviluppo della Ginecomastia.[3] Ciò può verificarsi anche se i livelli di estrogeni e androgeni sono entrambi appropriati, ma il rapporto è alterato.[3] La diagnosi si basa su segni e sintomi.
Un caso eccezionale di ginecomastia estrema in un uomo di 63 anni, trattato con l’antiandrogeno non steroideo Flutamide per il cancro alla prostata. (a) con Flutammide; (b) dopo l’interruzione della Flutamide. Più del 90% dei casi di ginecomastia da uso di antiandrogeni non steroidei inclusa la Futamide sono da lievi a moderati. Notare la presenza di lipomastia data da percentuali elevate di grasso corporeo.
La condizione comunemente si risolve da sola e la gestione conservativa della Ginecomastia è spesso tutto ciò che risulta necessario. Il trattamento medico della Ginecomastia che persistente per un periodo di oltre due anni è spesso considerato inefficace. Farmaci come gli inibitori dell’Aromatasi si sono dimostrati efficaci in rari casi di Ginecomastia derivante da disturbi come la sindrome da eccesso di Aromatasi o la sindrome di Peutz-Jeghers, [5] ma di solito è necessaria la rimozione chirurgica del tessuto in eccesso.[6]
La Ginecomastia fisiologica si sviluppa nei ragazzi adolescenti fino al 70%.[4] [3] I neonati hanno spesso Ginecomastia temporanea a causa dell’influenza degli ormoni materni. I maschi adolescenti hanno spesso ginecomastia temporanea a causa di cambiamenti ormonali durante la pubertà.
La caratteristica classica della Ginecomastia è l’ingrandimento del seno maschile con tessuto toracico sottocutaneo morbido, comprimibile e mobile palpato sotto l’areola del capezzolo in contrasto con il tessuto adiposo più morbido, non sempre accompagnato da lipomastia. [7] Questo allargamento può verificarsi su un lato o su entrambi. [8] Le fossette della pelle e la retrazione del capezzolo non sono caratteristiche tipiche della Ginecomastia. [9] Anche la secrezione lattiginoso dal capezzolo non è un riscontro tipico, ma può essere visto in un individuo ginecomastico con un tumore che secerne Prolattina o in condizioni iperprolattiniche derivanti da squilibrio ormonale iatrogeno-indotto o patologico.[3] Un aumento del diametro dell’areola e l’asimmetria del tessuto toracico sono altri possibili segni di Ginecomastia.[10]
I maschi con Ginecomastia, come accennato in precedenza, possono apparire ansiosi o stressati a causa delle preoccupazioni circa la possibilità di avere un cancro al seno.[11][12]
Quali possono essere le cause della comparsa della Ginecomastia?
Si ritiene che la Ginecomastia sia causata da un rapporto alterato tra estrogeni e androgeni mediato da un aumento della produzione di estrogeni, una diminuzione della produzione di androgeni o una combinazione di questi due fattori.[6] L’estrogeno agisce come un ormone della crescita sul tessuto mammario maschile causandone un aumento delle dimensioni.[13] La causa della Ginecomastia è sconosciuta nel 25% circa dei casi. [14] Si stima che i farmaci causino il 10-25% dei casi di Ginecomastia. [15]
Alcuni problemi di salute negli uomini come malattie epatiche, insufficienza renale o bassi livelli di Testosterone possono causare la crescita del seno negli individui interessati. I farmaci e le malattie epatiche sono la causa più comune di Ginecomastia negli adulti.[16] Altri farmaci noti per causare Ginecomastia includono il Metadone; antagonisti dell’Aldosterone (Spironolattone ed Eplerenone); Farmaci contro l’HIV; chemioterapia contro il cancro; trattamento ormonale per il cancro alla prostata; farmaci per il trattamento del bruciore di stomaco e ulcera; bloccanti dei canali del calcio; farmaci antifungini come il Ketoconazolo; antibiotici come il Metronidazolo; antidepressivi triciclici come l’Amitriptilina; ed erbe come lavanda, olio di melaleuca e dong quai. [17] L’insetticida Fenotrina possiede attività antiandrogena ed è stato associata alla comparsa di Ginecomastia.[18][19]
Le cause della comparsa di Ginecomastia sono quattro:
Fisiologica:
Molti neonati di entrambi i sessi mostrano sviluppo del seno alla nascita o nelle prime settimane di vita. [20] Durante la gravidanza, la placenta converte l’ormone androgeno Deidroepiandrosterone (DHEA) e la sua forma coniugata allo Zolfo Deidroepiandrosterone Solfato (DHEA-S) rispettivamente negli ormoni estrogenici Estrone ed Estradiolo; dopo che questi estrogeni sono stati prodotti dalla placenta, vengono trasferiti nella circolazione ematica del bambino, portando così alla ginecomastia temporanea nel soggetto. [13] [21] In alcuni neonati il latte neonatale (noto anche come “latte di strega”) può essere secreto.[11] La Ginecomastia temporanea osservata nei neonati di solito si risolve dopo due o tre settimane.[13]
DHEA-Solfato
La Ginecomastia negli adolescenti di solito inizia tra i 10 ed i 12 anni e comunemente scompare dopo 18 mesi.[13]
La diminuzione dei livelli di Testosterone e un aumento del livello di tessuto adiposo sottocutaneo, con maggiore espressione dell’Enzima Aromatasi consequenziale, visto come parte del normale processo di invecchiamento può portare alla Ginecomastia negli uomini più anziani. Questa è anche nota come Ginecomastia senile.[13] L’aumento del tessuto adiposo, e la maggiore espressione dell’Enzima Aromatasi, in questi uomini porta ad una aumentata conversione degli ormoni androgeni come il Testosterone in estrogeni. [13]
Enzima Aromatasi
Quando il corpo umano viene privato di un’alimentazione adeguata, i livelli di Testosterone diminuiscono, mentre le ghiandole surrenali per sintesi di conversione biochimica continuano a produrre estrogeni, provocando così uno squilibrio ormonale. [13] La ginecomastia può verificarsi anche una volta ripresa la normale alimentazione (questo è noto come Ginecomastia da rialimentazione). [13]
Una piccola percentuale di casi di Ginecomastia maschile può essere osservata con rari disturbi ereditari come l’atrofia muscolare spinale e bulbare e la rarissima sindrome da eccesso di Aromatasi. [22] [23]
Non fisiologica:
Si stima che circa il 10-25% dei casi di Ginecomastia derivi dall’uso di farmaci, ed essi sono noti come Ginecomastia non fisiologica. [9] [14] I farmaci noti per causare ginecomastia includono Gimetidina, Ketoconazolo, analoghi dell’Ormone di rilascio delle Gonadotropine, Ormone della Crescita Umano, Gonadotropina Corionica Umana, inibitori della 5α-reduttasi come Finasteride e Dutasteride, alcuni estrogeni usati per il cancro alla Prostata, antiandrogeni come Bicalutamide, Flutamide e Spironolattone e AAS aromatizzabili, aventi azione estrogenica intrinseca e progestinici. [24][25][26][27] [28] [29]
I farmaci che sono probabilmente associati alla Ginecomastia includono i bloccanti dei canali del Calcio come il Verapamil, Amlodipina e Nifedipina; Risperidone, Olanzapina [9] [29], Alcol, Oppioidi, Efavirenz, agenti alchilanti e Omeprazolo. [9] [30] Alcuni componenti dei prodotti per la cura della pelle personale come l’Olio essenziale di lavanda [31] o l’Olio dell’Albero del Tè e alcuni integratori alimentari come il Dong Quai e il Tribulus Terrestris sono stati ipoteticamente associati alla ginecomastia.[14] Questi ultimi agenti scatenanti sono quasi del tutto puramente ipotetici.
Bodybuilder con ginecomastia di IV grado. Liposcultura del torace con escissione completa della ghiandola. A destra, 3 mesi dopo l’intervento con retrazione cutanea spontanea.
Malattie croniche:
Le persone con insufficienza renale sono spesso malnutrite, il che può contribuire allo sviluppo della Ginecomastia. La dialisi può attenuare la malnutrizione dovuta a insufficienza renale. Inoltre, molti pazienti con insufficienza renale sperimentano uno squilibrio ormonale dovuto alla soppressione della produzione di Testosterone e danni ai testicoli causati da alti livelli di urea, noto anche come ipogonadismo associato all’uremia. [14] [32]
Negli individui con insufficienza epatica o cirrosi, la capacità del fegato di metabolizzare correttamente gli ormoni come gli estrogeni può essere compromessa. Inoltre, quelli con epatopatia alcolica sono ulteriormente messi a rischio di sviluppo di Ginecomastia; L’Etanolo può interrompere direttamente la sintesi del Testosterone e la presenza di Fitoestrogeni nelle bevande alcoliche può anche contribuire a un rapporto estrogeno / testosterone più elevato. [14] Anche condizioni che possono causare malassorbimento come la fibrosi cistica o la colite ulcerosa possono portare alla comparsa di Ginecomastia. [14]
Tumori:
I tumori testicolari come i tumori alle cellule di Leydig o i tumori a cellule del Sertoli [33] (come nella sindrome di Peutz-Jeghers) [4] o il coriocarcinoma secernente hCG [30] possono provocare Ginecomastia. Altri tumori come i tumori surrenali, i tumori della ghiandola pituitaria (come un prolattinoma) o il cancro del polmone, possono produrre ormoni che alterano l’equilibrio ormonale maschile-femminile portando alla comparsa di Ginecomastia.
Gli individui con cancro alla Prostata trattati con terapia di deprivazione androgenica possono sperimentare Ginecomastia. [34]
Uno sguardo alla fisiopatologia
La prominenza del seno può derivare dall’allargamento del tessuto ghiandolare dello stesso, del tessuto adiposo toracico (grasso) e della pelle o da una combinazione di questi tratti. [30] Come accade negli individui di sesso femminile, gli estrogeni stimolano la crescita del tessuto mammario nei soggetti di sesso maschile.[6] Oltre a stimolare direttamente la crescita del tessuto mammario maschile, gli estrogeni riducono indirettamente la secrezione di Testosterone sopprimendo la secrezione dell’Ormone Luteinizzante, con conseguente diminuzione della secrezione testicolare di Testosterone. [6] Inoltre, gli estrogeni possono aumentare i livelli ematici della Globulina Legante l’Ormone Sessuale (SHBG), che lega il Testosterone libero (la principale forma attiva) determinando una diminuzione dell’azione del Testosterone nel tessuto mammario maschile. [6]
Globulina Legante l’Ormone Sessuale (SHBG)
L’ipogonadismo primario (che indica un problema intrinseco con i testicoli) porta ad una diminuzione della sintesi del Testosterone e ad una maggiore conversione del Testosterone in Estradiolo, portando potenzialmente ad un aspetto ginecomastico. [13] La sindrome di Klinefelter è un notevole esempio di un disturbo che causa ipogonadismo e Ginecomastia ed ha un rischio più elevato di correlazione al cancro al seno nei maschi (20-50 volte superiore rispetto ai maschi senza il disturbo).[35] L’ipogonadismo centrale (che indica un problema ipotalamico-ipofisario) porta a una diminuzione della produzione e del rilascio dell’Ormone Luteinizzante (LH, un segnale stimolatorio per la sintesi del Testosterone) che porta a una diminuzione della produzione di Testosterone ed Estradiolo nei testicoli. [13]
Gli individui con cirrosi o altra malattia epatica cronica possono sviluppare Ginecomastia per diversi motivi. I cirrotici tendono ad avere una maggiore secrezione dell’ormone androgeno Androstenedione da parte delle ghiandole surrenali, una maggiore conversione di questo ormone in vari tipi di estrogeni, [6] e un aumento dei livelli di SHBG, che porta a una diminuzione dei livelli ematici di Testosterone libero e bioattivo. [13] Circa il 10–40% delle persone con malattia di Graves (una forma comune di ipertiroidismo) soffre di Ginecomastia. [13] Una maggiore conversione del Testosterone in estrogeni mediante una maggiore attività dell’Aromatasi, [6] livelli aumentati di SHBG e una maggiore produzione di Testosterone ed Estradiolo da parte dei testicoli a causa di livelli elevati di LH causano Ginecomastia. Un corretto trattamento dell’ipertiroidismo può portare alla risoluzione della Ginecomastia derivante da questa condizione. [13]
È noto che i farmaci causano Ginecomastia attraverso diversi meccanismi. Questi meccanismi includono un aumento dei livelli di Estrogeni, azione simil-estrogenica, diminuzione dei livelli di Testosterone o di altri androgeni, blocco dei recettori degli androgeni, aumento dei livelli di Prolattina o attraverso vie non identificate.[13] Alti livelli di Prolattina nel sangue (che possono verificarsi a causa di alcuni tumori o come effetto collaterale di alcuni farmaci) sono stati associati alla Ginecomastia. [13] Un alto livello di Prolattina nel sangue può inibire il rilascio dell’Ormone di Rilascio delle Gonadotropine e quindi causare ipogonadismo centrale. [6] [13] I Recettori per la Prolattina e altri ormoni inclusi il Fattore di Crescita Insulino-Simile 1, il Fattore di Crescita Insulino-Simile 2, l’Ormone Luteinizzante, il Progesterone e la Gonadotropina Corionica Umana sono stati identificati nel tessuto mammario maschile, ma l’impatto di questi vari ormoni sullo sviluppo della Ginecomastia non lo è ben compreso. [6]
Fattore di Crescita Insulino-Simile (IGF-1)
Per diagnosticare la ginecomastia, un medico richiede un’anamnesi completa e un esame fisico. Aspetti importanti dell’esame obiettivo includono la valutazione del tessuto mammario maschile con la palpazione per valutare il cancro al seno e la pseudoginecomastia (ingrossamento del tessuto mammario maschile dovuto esclusivamente all’eccesso di tessuto adiposo), la valutazione delle dimensioni e dello sviluppo del pene, la valutazione dello sviluppo testicolare e una valutazione per masse che sollevano il sospetto di cancro ai testicoli e il corretto sviluppo delle caratteristiche sessuali secondarie come la quantità e la distribuzione dei peli pubici e ascellari. [6] La Ginecomastia di solito si presenta con un coinvolgimento bilaterale del tessuto mammario, ma può manifestarsi anche unilateralmente. [14]
Una revisione dei farmaci o delle sostanze per il miglioramento delle prestazioni che un individuo assume può rivelare la causa della Ginecomastia. [14] Le indagini di laboratorio consigliate per trovare la causa alla base della Ginecomastia includono test per aspartato transaminasi e alanina transaminasi per escludere malattie epatiche, creatinina sierica per determinare se è presente danno renale e livelli di TSH per valutare l’ipertiroidismo. Se questi test di laboratorio iniziali non riescono a scoprire la causa della Ginecomastia, devono essere controllati ulteriori test per valutare un equilibrio ormonale sottostante dovuto a ipogonadismo o un tumore testicolare, compresi i livelli totali e liberi di Testosterone, Ormone Luteinizzante, Ormone Follicolo Stimolante, Estradiolo, Gonadotropina Corionica Umana-β (β-hCG) e Prolattina. [14]
Alti livelli di Prolattina sono rari nelle persone con ginecomastia. [14] Se i livelli di β-hCG sono anormalmente alti, è necessario eseguire un’ecografia dei testicoli per verificare la presenza di segni di un tumore testicolare secernente ormone. [14] Marcatori ormonali di tumori testicolari, surrenali o altri come il 17-chetosteroide urinario o il deidroepiandrosterone sierico possono essere controllati anche se vi è evidenza di squilibrio ormonale all’esame obiettivo. Se questa valutazione non rivela la causa della Ginecomastia, allora è considerata Ginecomastia idiopatica (di causa non chiara).[14]
Gonadotropina Corionica Umana-β (β-hCG)
Altre cause di ingrossamento del seno maschile come mastite, [14] [36] cancro al seno, pseudoginecomastia, lipoma, cisti sebacea, cisti dermoide, ematoma, metastasi, ectasia duttale, necrosi adiposa o amartoma sono tipicamente escluse prima di procedere con la diagnosi. [14] Un’altra condizione che può essere confusa con la Ginecomastia è l’allargamento dei muscoli pettorali.
La mammografia è il metodo di scelta per l’esame radiologico del tessuto mammario maschile nella diagnosi di Ginecomastia quando si sospetta un cancro al seno all’esame obiettivo. [6] [10] Tuttavia, poiché il cancro al seno è una causa rara di ingrossamento del tessuto mammario negli uomini, la mammografia è raramente necessaria. [6] Se viene eseguita la mammografia e questa non rivela reperti suggerenti la presenza di cancro al seno, in genere non sono necessarie ulteriori indagini in tal senso [10]. Se si ritiene che un tumore delle ghiandole surrenali o dei testicoli sia responsabile della Ginecomastia, può essere eseguito un esame ecografico di queste strutture organiche.[6]
Le prime caratteristiche istologiche che ci si aspetta di vedere all’esame del tessuto ginecomastico ottenuto mediante biopsia con aspirazione con ago sottile includono: proliferazione e allungamento dei dotti, aumento del tessuto connettivo, aumento dell’infiammazione e del gonfiore che circonda i dotti e un aumento dei fibroblasti nel tessuto connettivo. [13] La Ginecomastia cronica può mostrare diverse caratteristiche istologiche quali aumento della fibrosi del tessuto connettivo, aumento del numero di dotti, minore infiammazione rispetto allo stadio acuto della Ginecomastia, aumento del grasso subareolare e ialinizzazione dello stroma. [12] [13] Quando viene eseguito un intervento chirurgico, la ghiandola viene regolarmente inviata al laboratorio per confermare la presenza di Ginecomastia e per verificare la presenza di tumori al microscopio. L’utilità dell’esame patologico del tessuto mammario rimosso da pazienti adolescenti maschi affetti da Ginecomastia è stata recentemente messa in dubbio a causa della rarità del cancro al seno in questa popolazione. [37]
Lo spettro di gravità della Ginecomastia viene classificato come segue:[38]
Grado I: ingrandimento minore, nessun eccesso di pelle; Grado II: moderato ingrandimento, nessun eccesso di pelle; Grado III: moderato ingrossamento, eccesso di pelle; Grado IV: marcato ingrossamento, eccesso di pelle.
Una analisi ravvicinata delle cause della Ginecomastia non fisiologica farmaco indotta
Androgeni ed Estrogeni
Sembra esserci un consenso generale in letteratura sul fatto che la causa primaria della Ginecomastia sia uno squilibrio tra l’azione androgenica ed estrogenica sul tessuto mammario.[39] Detto semplicemente: gli estrogeni ne causano un aumento mentre gli androgeni lo inibiscono. Come tale, la Ginecomastia può svilupparsi quando c’è un eccesso relativo o assoluto dell’azione estrogenica o una diminuzione relativa o assoluta dell’azione androgenica a livello del tessuto mammario.
Questo è il motivo principale a causa del quale diversi utilizzatori di AAS potrebbero sviluppare Ginecomastia. Se ci si somministra Testosterone a dosi sovrafisiologiche, non si sta soltanto aumentando la parte androgenica dell’equazione. Parte di quel Testosterone verrà convertito in estrogeni dall’azione dell’Enzima Aromatasi. Non è quindi raro osservare livelli sovrafisiologici di Estradiolo nel sangue di soggetti trattati con dosi sovrafisiologiche di Testosterone. In realtà, è abbastanza normale trovare livelli di Estradiolo aumentati in tali circostanze. Tuttavia, va notato che ciò non riduce il rapporto androgeni / estrogeni. Anzi, lo aumenta fortemente, come si può vedere nella figura sottostante (si noti che mostra il rapporto tra Estradiolo e Testoserone).
Rapporto sierico E2 / T totale e rapporto E2 / T libero in risposta alla somministrazione di dosi graduali di Testosterone Enantato in uomini giovani (barre nere) e anziani (barre bianche). ∗ indica una differenza significativa tra uomini giovani e anziani. Abbreviazioni: T, Testosterone; TE, Testosterone Enantato; E2, Estradiolo. Figura ridisegnata da Lakshman et al. [40]
Sembra quindi improbabile che una diminuzione del rapporto androgeni / estrogeni sia la causa della Ginecomastia durante l’utilizzo di AAS. Sembra ragionevole presumere che in tali situazioni il colpevole sia un eccesso assoluto di estrogeni.
Dopo un ciclo, tuttavia, ci sarà uno stato transitorio di ipogonadismo iatrogeno (carenza di Testosterone). Ciò significa che il soggetto si ritroverà con una carenza di androgeni assoluta e probabilmente anche un eccesso relativo di estrogeni. Durante questo periodo di tempo si ha un rischio maggiore di sviluppare Ginecomastia. L’applicazione di una PCT correttamente impostata può ridurre sensibilmente questa condizione, anche se al termine di questa fase l’Asse HPT non sarà completamente efficiente e si potranno verificare alterazioni della Testosterone/Estrogeni ratio con conseguente Ginecomastia.
Prolattina
La Prolattina è un ormone che sembra in gran parte irrilevante nella fisiologia maschile. Tuttavia, può svolgere un ruolo importante nella fisiopatologia. La Prolattina è un ormone peptidico che viene rilasciato in grandi quantità dall’Ipofisi durante gli ultimi mesi di gravidanza e dopo il parto. È un ormone lattogeno, nel senso che stimola la produzione di latte del seno. Inoltre, ha un forte effetto soppressivo sulla secrezione di LH e FSH.[41] Questo è il motivo principale per cui può essere coinvolto nello sviluppo della Ginecomastia negli uomini. Vale a dire, a causa dell’effetto soppressivo che ha su LH e FSH, può causare ipogonadismo secondario. Di conseguenza, il rapporto androgeni / estrogeni diminuisce, il che può portare alla comparsa di Ginecomastia.
Prolattina
Il ruolo diretto della Prolattina nella Ginecomastia, se esiste, è come minimo non chiaro. Se gioca un ruolo, per esempio, sembra essere effettivamente irrilevante nella maggior parte dei casi, semplicemente perché il recettore della Prolattina sembra essere espresso nel tessuto mammario solo in una minoranza di casi di Ginecomastia.[42] E’ quindi difficile aspettarsi una interazione minimamente significativa nello sviluppo della Ginecomastia da parte della Prolattina dal momento che il suo recettore non è generalmente espresso nel tessuto mammario. Oltre a questo, sembrano esserci solo alcune speculazioni basate sugli effetti della regolazione incrociata con l’Ormone della Crescita e il Progesterone nelle linee cellulari di cancro al seno.[43] Inoltre, uno studio ha scoperto che l’incubazione di due linee cellulari di cancro al seno con Prolattina ha ridotto l’mRNA del recettore degli androgeni e l’attività di legame [44]. Tuttavia, se ho imparato una cosa dalla biologia delle cellule cancerose, è che si comportano in modo terribilmente diverso ai segnali esterni rispetto alle cellule normali per quanto riguarda la crescita.
Inoltre: le alterazioni della Prolattina sono facilmente inducibili in un ciclo di AAS, dal momento che la secrezione di questo peptide sembra avere correlazioni con gli squilibri estrogenici e progestinici. Ovviamente, se la Prolattina non ha effetti significativi sulla Ginecomastia li ha sulla possibile comparsa di lattazione quando i suoi livelli vengono mantenuti cronicamente alti.
Un’altra cosa da precisare è che il comportamento compulsivo di alcuni utilizzatori di AAS fobici della Ginecomastia, cioè quello di controllare costantemente i loro capezzoli, spremendoli e talvolta pizzicandoli, non è un idea così intelligente. Tale comportamento ha anche il potenziale per aumentare la Prolattina in una certa misura e può infiammare il tessuto sottostante (che sembrerà più consistente). Stranamente, mi sono imbattuto in alcuni casi in letteratura in cui l ‘”auto-manipolazione del seno” apparentemente aveva causato Galattorrea (secrezione mammaria).[45]
Progesterone e Progestinici
Il Progesterone è un ormone steroideo e, come la Prolattina, sembra giocare in gran parte un ruolo irrilevante nella fisiologia maschile. Ancora una volta, non fraintendete questa mia affermazione, non sto dicendo che non ha nessun effetto. Infatti, il Progesterone ha effetti nel corpo maschile. In particolare a livello cerebrale.[46]
Progesterone
Comunque, c’è stata qualche preoccupazione riguardo al Progesterone è alla possibilità che esso possa essere coinvolto nello sviluppo della Ginecomastia. Ovviamente, gli AAS non aumentano realmente i livelli di Progesterone. Esistono però alcuni AAS i quali hanno una certa affinità per il recettore del progesterone, dando loro in tal modo la possibilità di avere un’azione progestinica. È stato scoperto che alcuni AAS attivano il recettore del progesterone in uno studio genetico svolto su mammiferi.[47] Stiamo parlando della classe di AAS derivati dal 19-Nortestosterone. Esempi noti di queste molecole sono il famosissimo Nandrolone, il Mibolerone (meglio conosciuto con il nome commerciale di Check Drops), il Trenbolone e il Metiltrienolone, che sembrano abbastanza potente anche a basse concentrazioni, intorno a 1 nmol / L. Altri AAS, come il Testosterone, lo Stanozololo, l’Oxandrolone, il Methenolone e molti altri, non attivano il recettore del Progesterone (PR). Un dato interessante riguarda il fatto che lo Stanozololo sembra avere una azione antiprogestinica data dalla sua possibilità di legarsi al PR come una molecola antagonista.
Vorrei ricordare, inoltre, che l’effetto progestinico osservato negli AAS sopra elencati diventa significativo a dosaggi elevati.
Quindi, è un po ‘difficile dire se questi AAS citati effettivamente attivino in modo significativo il PR anche negli esseri umani, ma almeno il test biologico conferisce una certa credibilità a tali affermazioni. Ma c’è credibilità nell’affermazione secondo la quale questi AAS, tramite un’azione progestinica, possano concorrere a causare Ginecomastia? Il Progesterone sembra controllare la proliferazione e la morfogenesi dell’epitelio luminale della mammella.[48] O, almeno, sembra che lo faccia nelle donne. L’epitelio luminale è formato da cellule che compongono i dotti lattiferi. Tuttavia, non sembra avere un ruolo nello sviluppo del tessuto lobulare / ghiandolare (che è responsabile della massa che si vede e/o sente con la Ginecomastia). In quanto tale, sembra improbabile che giochi un ruolo diretto nello sviluppo della Ginecomastia. Ciò che rimane è un ruolo indiretto ed è stato ipotizzato che il Progesterone aumenti l’effetto dell’Estradiolo sul tessuto mammario. Anche se non sono stato in grado di trovare dati provenienti da studi su esseri umani, sembra che tale effetto non si manifesti nei primati. [49] Tutto sommato, non sembra esserci alcuna prova certa a sostegno del ruolo dell’attivazione del recettore del progesterone nello sviluppo della Ginecomastia negli utilizzatori di AAS. In effetti, non è stata osservata Ginecomastia in uno studio sui contraccettivi maschili in cui gli uomini presi in esame sono stati trattati con 100mg di Testosterone Enantato a settimana in combinazione con un alto dosaggio del progestinico Levonogestrel (0,5mg al giorno) per 6 mesi.[50] Se l’assunzione di Levonogestrel non ha causato la comparsa di Ginecomastia, viene difficile pensare che ciò possa verificarsi con l’uso un AAS progestinico. Fino a che non avremo dati certi, rimarrà comunque un ipotesi, sebbene di ipotetica difficile dimostrabilità.
Trattamento della Ginecomastia
In sostanza ci sono due modalità di trattamento per la Ginecomastia. Uno è finalizzato al trattamento dell’eccesso relativo o assoluto degli Estrogeni (trattamento endocrino). Il farmaco di elezione per questo approccio risulta essere il Modulatore Selettivo del Recettore degli Estrogeni (SERM) Tamoxifene (Nolvadex), dal momento che in letteratura vi sono abbondanti documentazioni sulla sua applicazione per tale scopo.
Un problema con il trattamento endocrino, tuttavia, è che non è sempre così efficace. Uno dei motivi è che la ginecomastia cambia nel tempo. Il tessuto alla fine subirà fibrosi (irreversibile) e ialinizzazione.[51] Ciò renderà il tessuto effettivamente insensibile al trattamento endocrino. È difficile dire quanto tempo ci vorrà prima che la Ginecomastia smetta di rispondere a questo tipo di trattamento, ma in letteratura viene menzionato un periodi da 1 anno [52] a 2 anni.[53] Tuttavia, alcuni autori hanno riportato il successo del trattamento endocrino in ginecomastie presenti da più di 2 anni.[54]
L’altra opzione è la chirurgia. Sebbene indubbiamente efficace, può avere alcuni aspetti negativi. Uno di questi è economico, poiché è piuttosto costosa e tende a non essere coperta dalla maggior parte delle assicurazioni sanitarie o dal SSN.
In tale sede mi concentrerò sul trattamento endocrino della Ginecomastia. Come ho descritto in precedenza, il principale colpevole risiede nell’azione estrogenica. Dato ciò, mi concentrerò sui SERM e sugli inibitori dell’Aromatasi (AI).
Modulatori Selettivi del Recettore degli Estrogeni (SERM)
Sono tre i SERM maggiormente utilizzati negli studi clinici per il trattamento della Ginecomastia: Tamoxifene (Nolvadex), Clomifene (Clomid) e Raloxifene (Evista). La maggior parte di questi studi ha visto l’utilizzo dei SERM nel trattamento della Ginecomastia puberale o della Ginecomastia indotta dal trattamento anti-androgeno nei soggetti con cancro alla prostata. Tuttavia, questo non ha importanza per quanto riguarda l’opzione di trattamento da scegliere. A costo di sembrare un po troppo semplicistico, dal punto di vista del trattamento la ginecomastia è ginecomastia.
Tamoxifene Citrato
Una review riportata che 10 – 20mg di Tamoxifene al giorno per 3-9 mesi hanno avuto un’efficacia fino al 90% nella risoluzione della Ginecomastia [53]. Un’altra review indica che il Tamoxifene provoca una regressione parziale in circa l’80% e una regressione completa in circa il 60% dei casi con Ginecomastia [55]. È interessante notare che, in un recente studio di coorte che includeva 81 pazienti con Ginecomastia idiopatica, il 90,1% ha avuto una risoluzione completa della stessa in risposta al trattamento con Tamoxifene (10 mg al giorno) [56]. La cosa interessante di questo studio è che la durata media del ipertrofia ghiandolare prima del trattamento (vale a dire il tempo di presenza della Ginecomastia), era in realtà più lunga in coloro che avevano una risoluzione completa (22 mesi) rispetto a coloro i quali soffrivano del problema da meno tempo (17 mesi ). Tuttavia, sembrava esserci una tendenza verso una dimensione maggiore della Ginecomastia (misurata con ultrasuoni) in coloro che non avevano raggiunto una risoluzione completa. Sfortunatamente, la dimensione della coorte era troppo piccola per dimostrare una differenza statistica significativa nella dimensione della Ginecomastia, nonostante la differenza fosse più che doppia. Tuttavia, suggerisce che la dimensione, piuttosto che da quanto tempo è stata presente, è un indicatore migliore del successo della terapia endocrina nella Ginecomastia idiopatica. Infine, l’alta percentuale di successo in questo studio potrebbe essere stata correlata alla durata del trattamento. La maggior parte degli studi dura solo uno o un paio di mesi. Gli autori di questo studio quindi scrivono che hanno regolato la durata del trattamento in base alla risposta dei pazienti piuttosto che utilizzando un protocollo di trattamento rigido. La durata media del trattamento nei soggetti in cui la Ginecomastia è stata risolta è risultato essere di quasi 7 mesi, con alcune variazioni (deviazione standard di 4,8 mesi).
Tamoxifene Citrato
Quello che dovrebbe essere tenuto bene a mente è che la situazione è diversa quando la ginecomastia si sviluppa durante un ciclo di AAS. Se ci si basa sulle testimonianze presenti in rete, sembra che quasi tutti abbiano manifestato la ginecomastia durante il loro ciclo di AAS. Ma cosa dice effettivamente la letteratura? Sfortunatamente, molto poco. Gli studi condotti da Bhasin et al. con dosaggi fino a 600mg/week di Testosterone Enantato (432mg di Testosterone effettivo) fino a 20 settimane non ha nemmeno elencato la ginecomastia come effetto collaterale. Tuttavia, un ampio studio menziona esplicitamente la presenza di ginecomastia. [57] Un totale di 271 soggetti hanno ricevuto 200mg/week di Testosterone Enantato per un minimo di 6 mesi. Solo 9 (circa il 3%) di questi soggetti ha sviluppato ginecomastia.
La possibilità di comparsa di una ginecomastia effettiva potrebbe anche essere maggiore a causa di prodotti contaminati. Un recente documento aveva testato qualitativamente 272 campioni di AAS per il loro contenuto come parte di uno studio prospettico sugli utilizzatori di AAS.[58-8] Il documento rileva che il 14% dei campioni conteneva estrogeni / progestinici. (Sfortunatamente non viene fatta alcuna distinzione tra i due. L’Estradiolo Valerato è stato trovato in campioni contenenti Boldenone [comunicazione personale]). Ovviamente, si è soggetti ad un rischio maggiore di sviluppare ginecomastia se si somministrano estrogeni.
Nel caso in cui si sviluppi ginecomastia durante un ciclo, potrebbe essere necessario un dosaggio più elevato di Tamoxifene (Nolvadex) rispetto a quanto descritto sopra. Il Tamoxifene, detto molto semplicemente, agisce prevalentemente come un antagonista competitivo per il sito recettore dell’Estradiolo. Quindi, se ci sono più estrogeni in circolo, si necessiterà anche di più Tamoxifene per raggiungere concentrazioni terapeutiche tali da poter efficacemente competere con l’E2 (Estradiolo) e ottenere una rioduzione dell’attività tissutale estrogenica. 10mg/die sembrano un dosaggio appropriato in condizioni fisiologiche o poco sopra il livello di soglia in molti soggetti. Tuttavia, se la ginecomastia si manifesta durante un ciclo con Testosterone a dosaggi superiori o uguali a 200mg/week, i livelli di Estradiolo potrebbero rivelarsi molto più alti in specie per i soggetti più tendenti all’aromatizzazione. Sembra che i livelli di Estradiolo aumentino a malapena quando i livelli di Testosterone sono aumentati oltre i 100nmol/L.[59] Che è più o meno la concentrazione che si raggiunge con 500mg/week di Testosterone Enantato . Dato che un dosaggio da 75 a 100mg a settimana è approssimativamente in linea con le concentrazioni fisiologiche, si potrebbe sostenere che sia necessario aggiungere 10mg/die di Tamoxifene per ogni 100mg/week di Testosterone Enantato (fino a un massimo di 500 mg), sebbene siano i controlli dei marker ematici di riferimento a rappresentare l’indicatore per la logicità dell’inserimento di un farmaco per il controllo estrogenico. Ma, andando per ipotesi, frutto della raccolta di letteralmente centinaia di dati empirici, poiché l’aumento dell’Estradiolo con l’incremento delle dosi di Testosterone non è lineare, si potrebbe dire che con un dosaggio tra i 30 ed i 40mg di Tamoxifene al giorno si rimanga in una sufficiente risposta terapeutica per il trattamento della ginecomastia con alti dosaggi di Testosterone (≥ 500mg a settimana) durante un ciclo. Tuttavia, è anche immaginabile che alcuni potrebbero richiedere dosaggi maggiori, a causa delle differenze nel metabolismo del Tamoxifene e ai tassi di aromatizzazione soggettivi.
Clomifene Citrato (Clomid)
Clomifene Citrato
La documentazione sul trattamento della ginecomastia con Clomifene Citrato e la sua efficacia è estremamente scarsa. Citerò qui solo l’abstract dello studio più recente in riferimento alla questione qui discussa (1983!).[60] Ovviamente, il motivo per cui ci sono così pochi dati sul trattamento della ginecomastia con il Clomifene è perché esso non ha mostrato un sufficiente grado di efficacia essendo più affine ai recettori ipotalamici piuttosto che ha quelli del tessuto mammario.
Dodici ragazzi, di età compresa tra 12 e 19 anni, con ginecomastia persistente sono stati trattati con l’antiestrogeno, il Clomifene Citrato, alla dose di 50mg/die per via orale per 1-3 mesi. La dimensione media del seno è diminuita dallo 0% al 36%, con solo cinque ragazzi che hanno registrato una riduzione superiore al 20%. Cinque ragazzi hanno successivamente richiesto una mastoplastica riduttiva. I livelli di gonadotropine urinarie, testosterone sierico ed estradiolo sono aumentati significativamente durante la terapia. Poiché il rapporto tra testosterone ed estradiolo è rimasto invariato durante il trattamento, gli effetti antiestrogeni sono stati raggiunti principalmente a livello del tessuto mammario. Il Clomifene Citrato (Clomid) ad una dose di 50mg/die ha determinato solo piccole riduzioni della ginecomastia puberale persistente e non è stata una terapia medica soddisfacente per la condizione.
Raloxifene
Il Raloxifene (Evista) è un SERM relativamente nuovo, appartenente a una classe diversa di SERM rispetto al Tamoxifene (Nolvadex) e al Clomifene (Clomid): benzotiofeni invece del derivato trifeniletilene / cloroetilene. Sono stato in grado di trovare solo uno studio clinico in cui il Raloxifene è stato utilizzato nel trattamento della ginecomastia [61]. Sfortunatamente, non è stato uno studio randomizzato controllato (RCT), ma una review retrospettiva, che lascia aperti a tutti i tipi di pregiudizi e dubbi che rendono impossibile trarre conclusioni definitive e certe.
Raloxifene
Inibitori dell’aromatasi
Dato il ruolo centrale degli estrogeni nello sviluppo della ginecomastia, gli inibitori dell’aromatasi (AI) sembrano molecole altamente indicate nel trattamento di questa condizione. Dopo tutto, inibiscono l’enzima (aromatasi) che converte gli androgeni in estrogeni. Ad esempio, converte il Testosterone nell’estrogeno predominante, l’Estradiolo. Sfortunatamente, solo pochi studi hanno esaminato l’uso degli AI nel trattamento della ginecomastia.
In un RCT in doppio cieco, 1mg/die del AI Anastrozolo (Arimidex) è stato confrontato con il placebo per il trattamento della ginecomastia puberale [62]. Il periodo di trattamento totale è stato di 6 mesi. Alla fine del periodo di trattamento, ad avere una risposta è stato il 38,5% delle pazienti che hanno ricevuto Anastrozolo, ed il 31,4% di quelle che hanno ricevuto placebo (definita come una riduzione> 50% del volume mammario totale dopo 6 mesi). Questa differenza tra i gruppi non era statisticamente significativa. L’Anastrozolo ha determinato un forte aumento del rapporto tra Testosterone ed Estradiolo. Tuttavia, circa la metà dei soggetti ha avuto ginecomastia per almeno due anni, il che li rende meno sospettabili di rispondere al trattamento endocrino. Ciò potrebbe aver in parte spiegato i risultati, supponendo che l’Anastrozolo abbia effettivamente funzionato come, d’altra parte, sembra avere fatto con un numero sostanziale di atleti trattati.
Anastrozolo
Nonostante ciò, anche altri studi sembrano suggerire che l’Anastrozolo non funzioni bene per questo scopo. Un’altra situazione in cui si verifica spesso la ginecomastia è durante il trattamento del cancro alla prostata. Nello specifico, il trattamento con la Bicalutamide (anti-androgeno). In un RCT in doppio cieco, i pazienti con carcinoma prostatico hanno ricevuto Bicalutamide con un placebo o in combinazione con Tamoxifene (20 mg al giorno) o Anastrozolo (1 mg al giorno) per 48 settimane [63]. La ginecomastia si è sviluppata nel 73% dei pazienti che ricevevano placebo, nel 10% dei pazienti che ricevevano Tamoxifene e nel 51% dei pazienti che ricevevano Anastrozolo. In quanto tale, l’Anastrozolo sembra inadatto per la prevenzione dello sviluppo della ginecomastia, mentre il Tamoxifene sembra molto efficace. Va notato che una carenza assoluta dell’azione degli androgeni è il segno distintivo nel trattamento della Bicalutamide piuttosto che l’eccesso di estrogeni (come nel caso dell’uso di AAS). Tuttavia, il Tamoxifene ha dimostrato una grande efficacia.
In accordo con questi risultati, Saltzstein et al. ha osservato che il Tamoxifene è molto efficace sia nella prevenzione che nel trattamento della ginecomastia indotta da Bicalutamide, mentre l’Anastrozolo ha mostrato solo un’efficacia minima [64]. Non sono stato in grado di trovare studi clinici che valutino l’efficacia degli altri due AI comuni, Exemestane e il Letrozolo, per il trattamento della ginecomastia. Data la potenza simile di questi due inibitori dell’aromatasi rispetto all’Anastrozolo nel ridurre gli estrogeni sierici (vedi il mio articolo L’efficacia degli inibitori dell’aromatasi negli uomini), sembra improbabile che siano da soli più benefici dell’Anastrozolo a questo proposito.
Conclusioni
In conclusione, il Tamoxifene (Nolvadex) è il farmaco di punta nel trattamento endocrino per la ginecomastia. Gli inibitori dell’aromatasi (da soli) mancano di ampie prove e le scarse prove che ci sono suggeriscono che sono solo minimamente efficaci. Considerando la ragionevole percentuale di campioni AAS che potrebbero contenere anche estrogeni (e quindi non richiedere alcuna conversione dell’aromatasi), gli AI sembrano ancora meno efficaci in monoterapia.
Parlando di dati puramente empirici raccolti in un decennio di ricerche indipendenti, la combinazione di Tamoxifene e Anastrozolo (sebbene esiste una competitività di riassorbimento che determina una riduzione d’efficacia del 30% del Anastrozolo; cosa non presente con AI steroidei) ha mostrato un alta percentuale di successo nella regressione della ginecomastia. Anche l’uso del Raloxifene è stato usato spesso con successo per la prevenzione e il trattamento della ginecomastia (spesso in coppia con AI).
Il giorno in cui non si vedranno più ginecomastie in un contest di Bodybuilding, sarà il giorno in cui l’intelligenza della “teoria applicata alla pratica”, indispensabile per un professionista veramente competente e competitivo, avrà trionfato su una categoria, sfortunatamente ancora dominante, composta da gorilla brandizzati, con nessuna concezione sulla nutrizione e la supplementazione (e forse anche su allenamenti logici), ed i mantenuti aventi l’unico talento risiedente nel loro pubblicizzarsi e piazzare specchietti per le allodole…
^ Jump up to:ab Cordova A, Moschella F (2008). “Algorithm for clinical evaluation and surgical treatment of gynaecomastia”. J Plast Reconstr Aesthet Surg. 61 (1): 41–9.
Barros, Alfredo Carlos Simões Dornellas de; Sampaio, Marcelo de Castro Moura (2012). “Gynecomastia: physiopathology, evaluation and treatment”. São Paulo Medical Journal. 130 (3): 187–197. Reinforcing the evidence suggesting that there is a relationship between chemicals and GM, it is worthwhile mentioning the epidemic onset observed among Haitian refugees in 1981 about four months after arrival in United States detention centers.22 After analyzing all identifiable environmental exposures, it was then found that phenothrin, a multi-insecticide contained in sprays that they had used was the causative agent.23 It is now widely known that phenothrin has antiandrogenic activity.
^ Steven A. Brody MD; D. Lynn Loriaux MD (2003). “Epidemic Of Gynecomastia Among Haitian Refugees: Exposure To An Environmental Antiandrogen”. Endocrine Practice. 9 (5): 370–375.
^ Gillatt, David (2006). “Antiandrogen treatments in locally advanced prostate cancer: are they all the same?”. Journal of Cancer Research and Clinical Oncology. 132 (S1): 17–26. doi:10.1007/s00432-006-0133-5. ISSN0171-5216. PMID16845534. S2CID23888640. Unlike CPA, nonsteroidal antiandrogens appear to be better tolerated than castration, allowing patients to maintain sexual activity, physical ability, and bone mineral density, but these agents have a higher incidence of gynecomastia and breast pain (mild to moderate in > 90% of cases).
^ Goldspiel BR, Kohler DR (1990). “Flutamide: An Antiandrogen for Advanced Prostate Cancer”. Ann Pharmacother. 24 (6): 616–623. doi:10.1177/106002809002400612. PMID2193461. S2CID26125621. […] They [in patients treated with flutamide] observed mild gynecomastia in 30 patients (57%), moderate gynecomastia in 19 (36%), and massive gynecomastia in four patients (8%). Complaints of nipple and areolar tenderness were noted in 50/53 patients (94%%).” Airhart et al. reported that 42% of patients receiving flutamide 750 mg/d or 1500 mg/d developed gynecomastia within 12 weeks of starting treatment with an apparent direct correlation between the dose of flutamide administered and the severity of gynecomastia. In another study, two of five evaluable patients developed moderate gynecomastia with mild tenderness at four and eight weeks after starting flutamide 750 mg/d. Patients with pre-existing gynecomastia as a result of previous endocrine therapy with estrogens sustained no worsening of their gynecomastia and may have improved symptomatically.” Keating et al.
^ Diaz A, Luque L, Badar Z, Kornic S, Danon M (2016). “Prepubertal gynecomastia and chronic lavender exposure: report of three cases”. J. Pediatr. Endocrinol. Metab. 29 (1): 103–107.
^ Fukami, M; Miyado, M; Nagasaki, K; Shozu, M; Ogata, T (March 2014). “Aromatase excess syndrome: a rare autosomal dominant disorder leading to pre- or peri-pubertal onset gynecomastia”. Pediatric Endocrinology Reviews. 11 (3): 298–305. PMID24716396.
^ Mayo Clinic Staff (2010). “Tests and diagnosis”. Mayo Clinic. Retrieved 3 February 2013.
H. S. Narula and H. E. Carlson. Gynaecomastia—pathophysiology, diagnosis and treatment. Nature Reviews Endocrinology, 10(11):684, 2014
K. M. Lakshman, B. Kaplan, T. G. Travison, S. Basaria, P. E. Knapp, A. B. Singh, M. P. LaValley, N. A. Mazer, and S. Bhasin. The effects of injected testosterone dose and age on the conversion of testosterone to estradiol and dihydrotestosterone in young and older men. The Journal of Clinical Endocrinology & Metabolism, 95(8):3955–3964, 2010.
M. De Rosa, S. Zarrilli, A. Di Sarno, N. Milano, M. Gaccione, B. Boggia, G. Lombardi, and A. Colao. Hyperprolactinemia in men. Endocrine, 20(1-2):75–82, 2003.
M. Ferreira, M. Mesquita, M. Quaresma, and S. Andre. Prolactin receptor expression in gynaecomastia and male breast carcinoma. Histopathology, 53(1):56–61, 2008.
A. Sansone, F. Romanelli, M. Sansone, A. Lenzi, and L. Di Luigi. Gynecomastia and hormones. Endocrine, 55(1):37–44, 2017.
C. J. Ormandy, R. E. Hall, D. L. Manning, J. F. Robertson, R. W. Blamey, P. A. Kelly, R. I. Nicholson, and R. L. Sutherland. Coexpression and cross-regulation of the prolactin receptor and sex steroid hormone receptors in breast cancer. The Journal of Clinical Endocrinology & Metabolism, 82(11):3692–3699, 1997.
R. D. Rohn. Benign galactorrhea/breast discharge in adolescent males probably due to breast selfmanipulation. Journal of Adolescent Health Care, 5(3):210–212, 1984
M. Schumacher, C. Mattern, A. Ghoumari, J. Oudinet, P. Liere, F. Labombarda, R. Sitruk-Ware, A. F. De Nicola, and R. Guennoun. Revisiting the roles of progesterone and allopregnanolone in the nervous system: resurgence of the progesterone receptors. Progress in neurobiology, 113:6–39, 2014.
C. J. Houtman, S. S. Sterk, M. P. Van de Heijning, A. Brouwer, R. W. Stephany, B. Van der Burg, and E. Sonneveld. Detection of anabolic androgenic steroid abuse in doping control using mammalian reporter gene bioassays. Analytica chimica acta, 637(1-2):247–258, 2009
Obr, Alison E., and Dean P. Edwards. “The biology of progesterone receptor in the normal mammary gland and in breast cancer.” Molecular and cellular endocrinology 357.1-2 (2012): 4-17.
J. Zhou, S. Ng, O. Adesanya-Famuiya, K. Anderson, and C. A. Bondy. Testosterone inhibits estrogen induced mammary epithelial proliferation and suppresses estrogen receptor expression. The FASEB Journal, 14(12):1725–1730, 2000.
R. A. Bebb, B. D. Anawalt, R. B. Christensen, C. A. Paulsen, W. J. Bremner, and A. M. Matsumoto. Combined administration of levonorgestrel and testosterone induces more rapid and effective suppression of spermatogenesis than testosterone alone: a promising male contraceptive approach. The Journal of Clinical Endocrinology & Metabolism, 81(2):757–762, 1996.
G. L. Nicolis, R. S. Modlinger, and J. L. Gabrilove. A study of the histopathology of human gynecomastia. The Journal of Clinical Endocrinology & Metabolism, 32(2):173–178, 1971
R. Mathur and G. D. Braunstein. Gynecomastia: pathomechanisms and treatment strategies. Hormone Research in Paediatrics, 48(3):95–102, 1997
H. S. Narula and H. E. Carlson. Gynaecomastia—pathophysiology, diagnosis and treatment. Nature Reviews Endocrinology, 10(11):684, 2014.
E. Devoto, M. Madariaga, X. Lioi, and N. Mardones. Influence of size and duration of gynecomastia on its response to treatment with tamoxifen. Revista medica de Chile, 135(12):1558–1565, 2007.
G. D. Braunstein. Gynecomastia. New England Journal of Medicine, 357(12):1229–1237, 2007.
G. S. Mannu, M. Sudul, J. H. Bettencourt-Silva, S. M. Tsoti, G. Cunnick, and S. F. Ahmed. Role of tamoxifen in idiopathic gynecomastia: A 10-year prospective cohort study. The breast journal, 24(6):1043–1045, 2018.
F. C. Wu, T. M. Farley, A. Peregoudov, G. M. Waites, et al. Effects of testosterone enanthate in normal men: experience from a multicenter contraceptive efficacy study. Fertility and sterility, 65(3):626–636, 1996.
Smit, Diederik L., et al. “Baseline characteristics of the HAARLEM study: 100 male amateur athletes using anabolic androgenic steroids.” Scandinavian journal of medicine & science in sports (2019).
K. M. Lakshman, B. Kaplan, T. G. Travison, S. Basaria, P. E. Knapp, A. B. Singh, M. P. LaValley, N. A. Mazer, and S. Bhasin. The effects of injected testosterone dose and age on the conversion of testosterone to estradiol and dihydrotestosterone in young and older men. The Journal of Clinical Endocrinology & Metabolism, 95(8):3955–3964, 2010.
P. V. Plourde, H. E. Kulin, and S. J. Santner. Clomiphene in the treatment of adolescent gynecomastia: clinical and endocrine studies. American Journal of Diseases of Children, 137(11):1080–1082, 1983.
S. E. Lawrence, K. A. Faught, J. Vethamuthu, and M. L. Lawson. Beneficial effects of raloxifene and tamoxifen in the treatment of pubertal gynecomastia. The Journal of pediatrics, 145(1):71–76, 2004.
P. V. Plourde, E. O. Reiter, H.-C. Jou, P. E. Desrochers, S. D. Rubin, B. B. Bercu, F. B. Diamond Jr, P. F. Backeljauw, and A. G. Study. Safety and efficacy of anastrozole for the treatment of pubertal gynecomastia: a randomized, double-blind, placebo-controlled trial. The Journal of Clinical Endocrinology & Metabolism, 89(9):4428–4433, 2004.
F. Boccardo, A. Rubagotti, M. Battaglia, P. Di Tonno, F. Selvaggi, G. Conti, G. Comeri, A. Bertaccini, G. Martorana, P. Galassi, et al. Evaluation of tamoxifen and anastrozole in the prevention of gynecomastia and breast pain induced by bicalutamide monotherapy of prostate cancer. Journal of clinical oncology, 23(4):808–815, 2005.
D. Saltzstein, P. Sieber, T. Morris, and J. Gallo. Prevention and management of bicalutamide-induced gynecomastia and breast pain: randomized endocrinologic and clinical studies with tamoxifen and anastrozole. Prostate cancer and prostatic diseases, 8(1):75, 2005.
I prodotti contenenti Pau d’arco – un estratto ricavato dalla corteccia dell’albero sudamericano Tabebuia avellandae – sono venduti come supplementi per rafforzare il sistema immunitario, ma secondo i ricercatori dello Strang Cancer Prevention Center di New York, questo estratto erboristico possiede anche un effetto anti-estrogenico.(1)
Lapachol
Le popolazioni native del sudamerica usano il Pau d’arco, spesso come tè, da centinaia di anni contro le malattie infettive. L’estratto di questa pianta contiene Naftachinoni come il Lapachol e gli Antrochinoni, che possono avere un effetto tossico. Se si somministrano dosi elevate di Lapachol agli animali da laboratorio durante la gravidanza, i loro embrioni muoiono.(2)
Gli oncologi molecolari hanno esaminato il potenziale del Pau d’arco come composto contro il cancro. I ricercatori dello Strang Cancer Prevention Center hanno esposto le cellule del cancro al seno MCF-7, che crescono velocemente in rapporto alle concentrazioni di Estradiolo con le quali entrano in contatto, ad un estratto di Pau d’arco a base acquosa. Più concentrato era l’estratto, più risultava micidiale per le cellule tumorali.
Quando i ricercatori hanno esaminato l’attività di importanti geni nelle cellule, hanno osservato come il Pau d’arco aveva inibito le cellule tumorali: aveva aumentato l’attività del gene per l’enzima CYP1A1 di un fattore di 19,8 [la figura sotto è logaritmica, non lineare ]. L’attività del gene per il CYP1B1 è aumentata di un fattore di 7,9.
Il CYP1A1 è, per quanto riguarda l’Estradiolo, un enzima ad azione “positiva”. Converte l’Estradiolo nell’innocuo 2-idrossi-estradiolo. Il CYP1B1 è invece un enzima con azione negativa in quanto converte l’Estradiolo in 4-idrossi-estradiolo. Il 4-idrossi-estradiolo è anch’esso meno attivo dell’Estradiolo, ma rappresenta una preoccupazione oncologica, poiché può legarsi al DNA, causando mutazioni che portano la conversione di una cellula sana in una cellula cancerosa.
Un altro dato interessante riguardante l’effetto d’uso del Pau d’arco è legato al fumo: i fumatori con una maggiore espressione genica del CYP1A1 tendono a sviluppare il cancro più frequentemente rispetto agli altri fumatori.
Il Pau d’arco sembra avere il potenziale per essere un utile supplemento finalizzato al controllo dell’attività estrpogenica, con la possibilità di essere abbinato all’uso di farmaci specifici (vedi SERM e AI), così da poterne – in teoria – gestire diversamente le dosi, durante l’uso di AAS o nelle fasi successive (vedi PCT). Ma essendo i dati inerenti all’effetto indiretto del Pau d’arco sul metabolismo del Estradiolo molto scarsi e limitati nell’applicazione, queste ipotesi rimangono semplici supposizioni che potrebbero facilmente non trovare riscontri significativi nell’uomo.
Le radici di ginseng contengono diverse sostanze simili agli steroidi e una di esse è il ginsenoside Rg1. In seguito all’assunzione di 2 capsule contenenti 5 mg di Rg1, sono stati osservati miglioramenti nelle prestazioni in soggetti giovani inattivi. Durante l’esercizio intenso l’effetto è risultato maggiore con una azione protettiva a livello muscolare e un tempo di recupero ridotto.(1)
Gli scienziati dell’Università di Taipei (Taiwan) hanno reclutato studenti di sesso maschile, sani ma non allenati. Li hanno sottoposti ad esercizio fisico, somministrando loro una dose di 5mg di ginsenoside Rg1 la sera prima della sessione allenante, e un’altra dose eguale un’ora prima della sessione. In un’altra occasione gli studenti hanno ricevuto un placebo.
I ricercatori stavano studiando il ginsenoside Rg1 da diverso tempo. La loro ricerca è stata finanziata dal governo di Taiwan e dalla NuLiv, con sede negli Stati Uniti [nulivscience.com]. La NuLiv produce estratti erboristici che successivamente vengono commercializzati sotto forma di integratori alimentari.
Quando i ricercatori hanno chiesto ai soggetti presi in esame di pedalare il più a lungo possibile ad un’intensità pari all’80% del loro VO2max, hanno osservato che la supplementazione con Rg1 causava un aumento del 20% del tempo di resistenza.
Dopo la sessione allenante, i soggetti dello studio hanno consumato un pasto ricco di carboidrati. Quando i ricercatori hanno analizzato il sangue dei soggetti, hanno notato che lo steroide aveva accelerato l’assorbimento del glucosio [in basso a sinistra]. In basso a destra si può vedere che la supplementazione con ginsenoside Rg1 ha aumentato la quantità di glicogeno nelle cellule muscolari.
Dopo l’esercizio, la concentrazione cellulare di TNF-alfa dei soggetti trattati con placebo era aumentata. L’Rg1 ha ridotto tale concentrazione. Allo stesso tempo, la sostanza ha indotto le cellule muscolari a produrre più interleuchina-10 (anti-infiammatoria).
Dopo la sessione allenante, il danno delle cellule muscolari causato dall’attività dei radicali liberi [TBARS] era aumentata nei soggetti trattati con placebo. La supplementazione con ginsenoside Rg1 ha ridotto questo aumento.
I ricercatori affermano che, il risultato principale dello studio è stato quello di dimostrare come la supplementazione con Rg1 pre-workout aumenti significativamente le prestazioni di resistenza durante attività fisica ad alta intensità.
I ricercatori concludono dicendo che, i partecipanti allo studio non allenati sono stati in grado di aumentare la loro capacità di lavoro su un cicloergometro del 20% circa rispetto ai soggetti trattati con placebo. Lo steroide Rg1 può offrire benefici ergogenici per l’uomo.
Un altro studio, svolto dagli stessi ricercatori del precedente, questa volta di recente realizzazione, che ha preso in esame l’azione del Rg1 sugli esseri umani, ha osservato che la combinazione di esercizio fisico intenso e supplementazione con ginseng ha un effetto protettivo a livello muscolare. Lo studio svolto dai biochimici taiwanesi sarà pubblicato sul Journal of Ginseng Research.(2) I ricercatori hanno somministrato ai soggetti dello studio 5mg di ginsenoside Rg1 prima di una intensa sessione su un cicloergometro.
Come per il precedente studio, anche questo è stato in parte finanziato dalla NuLiv Science, [nulivscience.com] e alcuni degli autori sono affiliati alla NuLiv come consulenti scientifici.
Una dei prodotti realizzati dalla NuLiv è l’ActiGin, una combinazione di estratti da Rosa Roxberghii e Panax ginseng. L’ActiGin dovrebbe migliorare le prestazioni sportive, aumentare l’assorbimento di glucosio da parte delle cellule muscolari e accelerare il recupero. Nella pubblicazione della quale stiamo parlando ora si può leggere che questo lavoro è stato finanziato allo scopo di produrre un supplemento ergogenico ActiGin per la Nuliv Science.
Tuttavia, in questo studio i ricercatori non hanno usato l’ActiGin, ma il ginsenoside Rg1. Non mi è stato però possibile constatare il reale contenuto di ginsenoside Rg1 nel ActiGin.
I ricercatori hanno somministrato a 12 studenti maschi non allenati una capsula contenete 5mg di ginsenoside Rg1, un’ora prima che iniziassero la sessione su cicloergometro ad un’intensità pari all’80% del loro VO2max. I soggetti presi in esame dovevano continuare a pedalare fino ad esaurimento. In un’altra occasione, i ricercatori hanno ripetuto l’esperimento, ma somministrando ai soggetti un placebo.
Per la maggior parte dei soggetti presi in esame la supplementazione con Rg1 ha prolungato il loro tempo di resistenza sul cicloergometro. L’evento osservato è paragonabile a quanto riportato per il precedente studio.
Prima e durante la sessione su cicloergometro, i ricercatori hanno misurato la concentrazione dell’enzima beta-galattosidasi senescente-associato nelle cellule muscolari dei soggetti dello studio. Si tratta di un marker per l’invecchiamento cellulare. Maggiori sono le concentrazioni dell’enzima, più una cellula è senescente.(3)
La combinazione di cicloergometro e supplementazione con ginsenoside Rg1 ha ridotto la concentrazione di beta-galattosidasi.
In basso a sinistra si può notare come il ginsenoside Rg1 può portare ad un “ringiovanimento” muscolare. Se i soggetti dello studio avevano assunto l’Rg1, i ricercatori osservavano una maggiore concentrazione di CD68+ nei loro muscoli dopo lo sforzo. Si tratta di cellule le quali ripuliscono le cellule muscolari danneggiate stimolando le cellule staminali a sostituirle.
I ricercatori concludono affermando che, i risultati dello studio suggeriscono che l’attivazione dei macrofagi dopo supplementazione con Rg1 è associata alla clearance delle cellule senescenti osservate nel muscolo scheletrico umano allenato.
Se non avete ancora letto le precedenti parti componenti questa serie di articoli vi invito a farlo prima di procedere con la lettura di questa quarta ed ultima parte: 1° Parte – 2° Parte – 3° Parte.
Farmacocinetica, Farmacodinamica e Feedback Negativi
Come già detto, il fegato rappresenta il principale bersaglio del GH, il quale è il principale regolatore della sintesi epatica di IGF-1. Per causare tale effetto, il GH si lega con i GHR localizzati nel dominio extracellulare degli epatociti stimolando successivamente la produzione di IGF-1 endocrino tramite la trascrizione genica, utilizzando la via di segnalazione JAK-STAT. Inoltre, è stato dimostrato che la somministrazione di GH causa una rapida sovraregolazione dell’mRNA del IGF-1 nel fegato.[338]
Aumenti dei livelli serici di IGF-1 si verificano molto rapidamente anche in presenza di un grande bolo di rHGH. Incrementi significativi di IGF-1 sono già osservabili dopo 6-12h dall’iniezione.[339] Questi livelli serici di IGF-1 continuano ad aumentare fino a raggiungere il loro punto di saturazione dose-dipendente entro 4-7 giorni, anche quando si utilizzano dosi estremamente elevate che ammontano a 20-30UI al giorno di rHGH.[340] In particolare, il punto di saturazione si è rivelato essere compreso nell’intervallo dei 700-800 ng/mL e sembra suggerire che i livelli endocrini di IGF-1 hanno un tetto massimo negli adulti sani. I meccanismi esatti devono ancora essere chiariti, ma sono probabilmente il risultato dei complessi meccanismi di controllo intrinseci all’Asse GH/IGF-1. Coloro i quali desiderano elevare i livelli endocrini di IGF-1 al fine di ottenerne un vantaggio sull’ipertrofia dovrebbe tenerlo a mente, in quanto vi è un punto in cui l’uso di dosi maggiori di rHGH semplicemente non si traducono in elevati livelli serici di IGF-1. Qui di seguito ho riportato il grafico dello studio di Tanaka il quale mostra la relazione tra l’rhGH e i livelli serici di IGF-:
Ora, vorrei dedicarmi brevemente all’analisi dell’azione del IGF-1autocrino e del perché esso rappresenti un mediatore cruciale del processo ipertrofico, prima di tornare nuovamente a discutere su questioni inerenti alla farmacodinamica e farmacocinetica. La segnalazione recettoriale del IGF-1 è unica nel suo genere, e questo lo si deve al fatto che utilizza due percorsi distinti per stimolare la proliferazione o la differenziazione.[341-343] Questo è un comportamento abbastanza interessante, poiché nessun altro membro della famiglia dei fattori di crescita ha dimostrato di agire in tal modo. Poiché la proliferazione e la differenziazione sono processi opposti, inizialmente era difficile per i ricercatori capire come un singolo fattore di crescita, attraverso un singolo recettore, potesse inviare un segnale che attivasse entrambi.[294] Da quando sono state fatte queste prime scoperte, è stato ulteriormente chiarito che l’IGF-1 non svolge simultaneamente queste azioni. Test su varie linee di coltura cellulare hanno dimostrato che gli effetti proliferativi arrivano prima, durando tra le 24 e le 36 ore. È solo dopo questa fase proliferativa iniziale che si verifica la differenziazione miogenica.[344]
Gli effetti proliferativi mediati dall’IGF-1 sui mioblasti sono noti sin dagli anni ’70, quando vennero osservati per la prima volta nelle cellule epatiche di ratto.[345] Questa stimolazione proliferativa del IGF-1 si traduce in un aumento del numero di cellule, nei livelli di proteine, nella sintesi del DNA, nell’assorbimento di aminoacidico, nell’assorbimento del glucosio e nella soppressione della proteolisi.[346] Nelle colture cellulari umane, l’IGF-1 ha anche dimostrato di aumentare la dimensione dei miotubi indipendentemente dal fatto che i mioblasti proliferino attivamente o che la proliferazione sia cessata. Regola la dimensione dei miotubi attivando la sintesi proteica, inibendo la degradazione proteica e inducendo la fusione delle cellule di riserva.[347-348] La capacità dell’IGF di sopprimere la proteolisi nel muscolo scheletrico, la scomposizione delle proteine in aminoacidi, è stata dimostrata innumerevoli volte nel corso degli anni.[349-352] È stato anche dimostrato che l’IGF-1 induce la proliferazione e la differenziazione delle cellule satelliti in miociti maturi, come determinato da un aumento del numero di miofibre nucleate a livello centrale rispetto a quelle periferiche.[148,353-354]
La capacità dell’IGF-1 autocrino di causare la differenziazione dei mioblasti è stata in realtà una scoperta che potremmo definire quasi “ibrida” dal momento che degli studi svolti negli anni ’60 avevano mostrato che questo effetto si verifica con alti livelli di Insulina.[355] Successivamente è stato dimostrato che gli IGF sono stimolatori molto più potenti nella differenziazione miogenica rispetto all’Insulina e si è concluso che la stessa Insulina agisce realmente come un analogo dell’IGF-1 in questo sistema.[356-357] Gli effetti di differenziazione dati dall’IGF-1 autocrino sono bifasici, con basse concentrazioni che stimolano progressivamente la differenziazione dei mioblasti mentre concentrazioni molto elevate mostrano una cessazione dell’attività di differenziazione. Il limite massimo per la differenziazione sembra attestarsi a circa 100ng/mL per l’IGF-1 e 300ng/mL per IGF-2.[358] Questo effetto non è legato alla proliferazione, poiché non si osservano ulteriori aumenti nel numero complessivo delle cellule.[294] È possibile che le molecole di segnalazione coinvolte nella regolazione negativa del sistema miogenico siano aumentate, ma questa è una affermazione puramente speculativa.[359-360]
La somministrazione di rHGH eleva l’espressione dell’mRNA dell’IGF-1 nel muscolo scheletrico in numerosi modelli cellulari, umani e animali.[127,150,361-364] Ciò avviene abbastanza rapidamente, entro 60 minuti dall’iniezione sottocutanea di rHGH ed i picchi sono segnalati tra le 6 e le 12 ore post iniezione.[363] In questo particolare modello animale citato, il raddoppio della dose di GH non ha portato ad ulteriori aumentati dei livelli di mRNA dell’IGF-1, il che suggerisce che esiste un sistema di regolazione che determina quanto GH sia necessario per stimolare al massimo l’espressione locale dell’IGF-1 nel muscolo scheletrico. In precedenza si è potuto appurare che la differenziazione dei miociti mediata dall’IGF-1 si arresta quando le concentrazioni locali raggiungono circa i 100ng/mL, ma quanto GH è necessario per raggiungere il punto di saturazione dell’espressione dell’mRNA dell’IGF-1?
Gli studi sui miociti umani mostrano che il GH aumenta l’espressione dell’mRNA dell’IGF-1 entro 30-60 minuti con picchi molto più rapidi rispetto a quelli osservati negli studi sugli animali, entro 1-2 ore, usando la via di segnalazione JAK / STAT5b.[365] Questi livelli elevati di mRNA hanno dimostrato di durare fino a 48 ore dopo una singola esposizione al GH. La quantità di GH necessaria per stimolare al massimo l’espressione dell’mRNA dell’IGF-1 è risultata essere una dose compresa tra i 7,5ng/mL e 30ng/ml [366], con una dose media efficace che si attesta a 3ng/ml. Questi numeri sono in linea con gli intervalli di dose fisiologica osservati negli animali, che sono effettivamente compresi tra i 2-100 ng/mL.[367] Inoltre, si collocano esattamente in linea con quanto si osserva endogenamente nell’uomo, con concentrazioni normali di picco comprese tra i 22,4 e 32,4ng/mL.[368-369,436] Ci sono stati casi in cui gli uomini presi in esame hanno mostrato concentrazioni di picco leggermente più alte, ma questi devono essere considerati valori anomali.[370] In ogni caso, ciò che questi dati tendono a suggerire è che il corpo umano è particolarmente adatto a gestire livelli naturali di picco della secrezioni di GH endogeno. Cercare di incidere ulteriormente il sistema elevando i livelli di GH oltre quelli endogeni, unicamente per tentare di potenziare i processi ipertrofici, potrebbe in realtà non tradursi nell’effetto desiderato.
Gli studi che mettono a confronto le infusioni locali con le infusioni sistemiche di GH o IGF-1 sono un po’ più difficili da trovare di quanto si vorrebbe. Le poche sperimentazioni sugli animali che sono riuscito a trovare indicano che l’infusione diretta di GH o IGF-1 nei tessuti bersaglio determina un aumento della massa muscolare. Questo aumento dell’ipertrofia si verifica anche senza che il muscolo bersaglio sia stato sottoposto ad attività motoria.[371-372] Gli studi dimostrano anche che le iniezioni locali di GH portano a livelli sostanzialmente più alti nell’espressione dell’mRNA dell’ IGF-1 locale rispetto alle iniezioni locali di IGF-1, di un fattore di oltre venti.[127] Sono riuscito a trovare uno studio nel quale si confrontavano le risposte dei ratti (attivi e non) all’infusione locale di IGF-1. Il gruppo “IGF-1 plus training” ha mostrato un aumento sia della massa muscolare che della forza locale maggiore rispetto al semplice trattamento in isolamento.[373] Quindi, anche se limitata, la letteratura disponibile è apparentemente in grado di dimostrare che le iniezione locali di GH o IGF-1 hanno effettivamente valore.
Ne ho già parlato diverse volte ma, nel tentativo di imprimere ulteriormente questo concetto, è necessario ricordarsi che i livelli autocrini di IGF-1 sembrano essere molto più importanti dei livelli endocrini di IGF-1 in relazione alla regolazione della massa muscolare. Oltre a questo punto, la sovraespressione dell’IGF-1 autocrino nel muscolo provoca l’ipertrofia delle fibre.[374] La sovraespressione dell’IGF-1 autocrino ha anche mostrato effetti anti-catabolici, con modelli animali tendenti a mostrare una resistenza generale all’atrofia muscolare normalmente osservata con l’invecchiamento.[375] L’IGF-1 localizzato fornisce anche capacità rigenerative indipendenti dall’età nelle cellule muscolari.[376]
Vi sono anche alcune prove convincenti che suggeriscono che l’IGF-1 endocrino agisce direttamente come un regolatore di feedback negativo sulla produzione di IGF-1 autocrino. Questo meccanismo di feedback negativo è dipendente dal pathway PI3K/Akt [377-378]. Inoltre, elevati livelli di IGF-1 endocrino possono anche agire indirettamente per sopprimere la produzione di IGF-1 autocrino. Quindi, in altre parole, non solo l’IGF-1 endocrino ha un impatto diretto minore sulla regolazione della massa muscolare, ma può anche sopprimere l’IGF-1 autocrino che ha impatti maggiori sull’ipertrofia.
Elevati livelli di IGF-1 circolante e, nello specifico, di IGF-1 libero elevati agiscono in modo negativo sul GH determinando un tasso di soppressione della produzione di IGF-1 autocrino a valle.[379] Non è del tutto chiaro, tuttavia, se la regolazione negativa dell’IGF-1 modifichi l’emivita dell’mRNA dell’IGF-1 o influenzi direttamente l’espressione del gene IGF-1. Oltre a questo, è stato anche dimostrato che l’espressione dell’IGF-1 autocrino è sottoregolata nelle cellule muscolari dopo trattamento con IGF-1.[366] È stato anche dimostrato che l’espressione epatica dell’mRNA dell’IGF-1 è sottoregolata dall’esposizione acuta all’IGF-1.[127] Quindi, mantenere livelli endocrini il più possibile soppressi con rispettiva dose di rHGH, elevando contemporaneamente i livelli autocrini, dovrebbe essere un fattore prioritario in un protocollo di GH volto all’ipertrofia.
Il GH è pulsatile per natura sia nell’uomo che nelle specie animali. Quindi, sarebbe logico pensare che molti dei processi intrinseci del corpo saranno tarati in modo tale da rispondere in maniera ottimale all’esposizione al GH in modo simile. In accordo con questa affermazione è stato dimostrato che solo la somministrazione di GH pulsatile, e non l’infusione continua, ha la capacità di stimolare massimamente l’espressione dell’mRNA dell’IGF-1 nel muscolo scheletrico.[366,380-381] È stato anche dimostrato che la somministrazione pulsatile porta ad un aumento del potenziale di crescita postnatale complessivo rispetto all’infusione continua.[89,382] La somministrazione pulsatile può anche portare a livelli endocrini di IGF-1 serici comparabili, o addirittura diminuiti [383], il che è vantaggioso a causa delle potenziali capacità di regolazione negativa che possiede sull’espressione dell’IGF-1 autocrino e che sono state discusse in precedenza. L’evidenza suggerisce anche che il picco stesso, e non necessariamente il numero di picchi, potrebbe essere della massima importanza per i tessuti bersaglio.[384] Per la massima crescita e potenziale ipertrofico, l’evidenza tende a suggerire che creare picchi di GH elevati, e quindi tornare ai livelli basali più volte al giorno, può essere preferibile rispetto a mantenerli elevati per periodi di tempo più lunghi. Questo pratica permette di riprodurre gli schemi secretori in vivo.
I pathways del GH coinvolti nell’anabolismo sono anche suscettibili alla desensibilizzazione, che è parte della fisiologia del GH endogeno.[385] A causa della natura intrinsecamente pulsatile del GH in vivo, l’attività dei recettori e dei pathways sono regolati da un impulso seguito da un periodo di inattività.[386] L’esposizione continua o ripetuta al GH senza un adeguato lasso di tempo refrattario comporterà livelli di attività fortemente soppressi. In effetti, nel corso degli anni numerosi studi hanno dimostrato che tale effetto si verifica. Le cellule ed il tessuto muscolare richiedono un periodo refrattario piuttosto lungo prima che la loro piena risposta al GH venga recuperata. Dopo l’esposizione al GH, le cellule muscolari non sono nemmeno in grado di rispondere alle successive dosi di GH. In realtà, occorrono due ore complete per riprendere parzialmente la reattività nei modelli cellulari, con un totale di 6-8 ore di astinenza dall’uso di GH necessarie per ripristinare la piena sensibilità.[366] Viceversa, quando il GH è micro-dosato in impulsi di dieci minuti, seguiti da intervalli di otto ore, è stato mostrato aumentare progressivamente l’mRNA dell’IGF-1 con ogni impulso successivo.[386]
Questo fenomeno è potenzialmente il risultato di una desensibilizzazione complessiva all’interno della via JAK-STAT5, poiché è stato dimostrato che l’esposizione al GH negli studi sulle cellule epatiche causa resistenza alla successiva attivazione della via STAT5 per 4-8 ore.[387-388] Questo lasso di tempo è sufficiente per sincronizzarsi abbastanza bene con ciò che è stato visto nei modelli di cellule miocitarie citati in precedenza. Nei modelli di cellule epatiche, il GH ha stimolato un significativo aumento dell’espressione del SOCS3, che è un potente inibitore dell’azione del GH.[389]. Poiché il GH non ha avuto alcun effetto sull’espressione del SOCS3 nelle cellule muscolari, questo deve essere un altro meccanismo causante il periodo refrattario. Questo meccanismo può essere dipeso dalla sottoregolazione dei GHR, dall’inibizione mediata da un’altra proteina SOCS, o dall’induzione di una tirosina fosfatasi che semplicemente inattiva la via JAK / STAT.[390] La via JAK-STAT5b, che come ricorderete è intimamente associata al muscolo scheletrico e all’espressione dell’IGF-1, è di natura transitoria – con attivazione massima raggiunta entro 10-30 minuti, seguita da un prolungato periodo di inattivazione.
Una scoperta piuttosto nuova di Xu et al. [391] ha dimostrato che anche distanziare le esposizioni al GH di cinque ore lasciava entrambi i percorsi a valle MEK1/2 e ERK1/2 significativamente soppressi rispetto a tutti i percorsi a monte, a causa di una potenziale disconnessione nella trasduzione del segnale . Ciò è di particolare interesse in quanto questi stessi due percorsi a valle sono stati coinvolti in modo significativo sia nella crescita sia nella proliferazione.[392-393] È stato anche scoperto che l’attivazione indotta da GH di STAT1 e STAT3 è stata desensibilizzata, ma l’esposizione all’Insulina inverte la desensibilizzazione osservata in tutti i percorsi interessati. Anche se non sto per trattare approfonditamente l’Insulina, ci sono un paio di importanti punti da dovere prendere in considerazione. Bisogna comprendere innanzitutto che ci sono molti obiettivi a valle del recettore del GH e molti di questi hanno il potenziale per essere desensibilizzati dopo l’esposizione al GH. Bisogna comprendi anche che l’Insulina possiede l’abilità unica di risensibilizzare molti di questi percorsi. Ciò ha un senso vista la relazione tipo yin-yang tra i due composti. È noto che il GH e l’Insulina possiedono una relazione anabolica sinergica a causa di molti effetti che esercitano l’uno sull’altro. Questo sembra essere soltanto un’anteprima di uno di questi effetti.
Asse GH/IGF-1 – Relazione con altri ormoni
Prima di passare alle note conclusive, vorrei trattare brevemente alcuni altri ormoni connessi a diverso grado con l’Asse GH/IGF-1. Per prima cosa, voglio trattare brevemente l’Asse Tiroideo dal momento che l’inserimento di composti tiroidei insieme al GH è una pratica comune anche durante i protocolli di massa.
Il muscolo scheletrico è il principale bersaglio di segnalazione dell’ormone tiroideo, con trasportatori degli ormoni tiroidei e enzimi di conversione espressi localmente.[394] È ben noto che il GH potenzia la deiodinazione periferica che converte il T4 in T3, riducendo così il T4 e il reverse T3, aumentando contemporaneamente i livelli di T3.[395-398] Ciò che molte persone non riescono a capire è che questo è un effetto transitorio, e studi a lungo termine sembrano indicare che gli effetti mediati dal GH sulla conversione periferica si stabilizzino con il tempo.[399-402]
Via ubiquitina/proteasoma
Invece di proseguire ulteriormente su questo, avendo già trattato la questione nel dettaglio in un mio vecchio articolo, preferirei concentrarmi su alcune pubblicazioni relative alla tiroide che non vengono discusse abbastanza spesso. Gli ormoni tiroidei, per loro natura, sono composti tendenzialmente catabolici in quanto stimolano la disgregazione proteica dell’intero corpo in misura maggiore rispetto alla sintesi proteica.[403] A livello locale, nel muscolo scheletrico stimolano un aumento dell’attività all’interno della via ubiquitina/proteasoma, che è ampiamente coinvolta nella proteolisi.[404-406] Il risultato di questo è un tasso accelerato del turnover proteico e una perdita netta complessiva degli aminoacidi situati all’interno dei muscoli scheletrici.
Inoltre, negli esseri umani, sia gli stati di ipertiroidismo che di ipotiroidismo sono stati associati a livelli di IGF-1 soppressi con una tendenza alla normalizzazione quando viene ristabilita una condizione di eutiroidismo. L’ipertiroidismo è anche associato ad una bassa attività di legame recettoriale del GH, che si ipotizza essere il risultato di una ridotta capacità di elaborazione dei recettori del GH.[407] E’ stato anche ipotizzato che l’ipertiroidismo sia in grado anche di accelerare la clearance del GH urinario.[408] Inoltre, studi su animali hanno dimostrato che gli ormoni tiroidei possono avere importanti effetti soppressivi sulla sintesi di IGF-1 stimolata con il GH.[409] Ovviamente, a causa della complessa relazione che l’Asse Tiroideo ha con l’Asse GH/IGF-1, raggruppando tutte le interazioni che hanno tra loro in pochi paragrafi, trattare l’argomento diventerebbe poco pratico. Tuttavia, quando il corpo della letteratura scientifica viene esaminato nella sua interezza, ci sono molte prove che suggeriscono che la supplementazione con composti tiroidei esogeni potrebbe non essere l’ideale quando l’obiettivo di un individuo è l’ipertrofia, anche se la regolazione del dosaggio dei tiroidei in tale contesto rimane la misura di “sicurezza” più intelligente visto l’impatto negativo sulla funzionalità tiroidea dato dal GH. Comunque, per tutti coloro che sono interessati ad approfondire questo argomento, consiglio di iniziare con la review nella nota seguente.[410]
Miostatina
Mi piacerebbe trattare anche la Miostatin, che rappresenta un argomento molto discusso nei vari forum di BodyBuilding presenti in rete. La sua fama proviene dai risultati ipertrofici espressi dai bovini privi per mutazione del gene della Miostatina, i quali mostrano una massa muscolare significativamente maggiore rispetto ai loro simili non mutati.[411] La Miostatina, un fattore di crescita e differenziazione appartenente alla superfamiglia dei TGF-beta, ha dimostrato di inibire selettivamente la miogenesi, in gran parte tramite il suo effetto soppressivo sulla proliferazione dei mioblasti.[412] È espressa e secreta prevalentemente dal muscolo scheletrico. Come molti sanno, se riesci a sopprimere o inibire la Miostatina, di conseguenza il potenziale ipertrofico aumenta significativamente.
Le mutazioni della Miostatina sono state osservate sia negli animali che nell’uomo. Queste mutazioni del gene della Miostatina portano ad un fenotipo ipertrofico negli animali, come accennato in precedenza.[413-415] L’Asse GH/IGF-1 e la Miostatina sembrano avere una relazione regolativa diretta tra loro, come osservato nei pazienti affetti contemporaneamente da GHD e HIV che mostrano marcati aumenti nell’espressione dell’mRNA della Miostatina.[416] Quindi, è possibile che attraverso una supplementazione di dosi sovrafisiologiche di rHGH si possa indurre una diminuzione dell’mRNA della Miostatina [209,417-419]? Sfortunatamente, nonostante la presenza di alcuni casi studio selezionati, non credo che si abbiano abbastanza dati in questo momento per sapere se ciò possa dare risultati apprezzabili in seguito alla sua applicazione.
Quello che sappiamo è che aumenti dell’espressione dell’mRNA dell’IGF-1 e le concentrazioni circolanti di IGF-1 sono state osservati dopo inibizione della Miostatina.[419-421] Sappiamo anche che l’inibizione della Miostatina tende a causare l’ipertrofia attraverso molte delle stesse modalità osservate con l’IGF-1 autocrino, cioè l’aumento della sintesi proteica e l’attivazione delle cellule satelliti.[422-425] E sappiamo anche che l’ipertrofia indotta dalla sovraespressione dell’IGF-1 o dall’inibizione della Miostatina utilizza la stessa identica via – PI3K/Akt/mTOR.[426-428] Tuttavia, l’IGF-1 non è un requisito per l’ipertrofia indotta dalla Follistatina, tranne nel caso di livelli di Insulina estremamente bassi – come ben sappiamo, la Follistatina è un inibitore della Miostatina [429]. E l’esposizione cronica al GH può in realtà portare ad un’espressione sovrastimolata della Miostatina e del suo recettore.[209]
Quindi quello che possiamo dire, con certezza, è che l’espressione della Miostatina non sarà un fattore diretto o indiretto per quanto riguarda il potenziamento dei processi ipertrofici, né dell’attività contrattile, nei muscoli scheletrici umani.[430] Proprio per questo motivo, non ritengo che sia un fattore sul quale gli atleti debbano eccessivamente concentrarsi, al di fuori di un utile arricchimento delle proprie conoscenze in materia.
Applicazioni pratiche e pensieri conclusivi
Arrivati a questo punto è mia intenzione unire tutto ciò che è stato esposto in questa serie di articoli e esporlo sotto forma di alcuni suggerimenti pratici rivolti a tutti coloro i quali vogliono semplicemente massimizzare la loro capacità ipertrofica.
Ora è chiaro che il GH possiede pochissimi, se non nulli, effetti diretti sull’ipertrofia. Pertanto, qualsiasi protocollo di massa che contempli il suo uso dovrà tenere in considerazione questo punto includendo gli AAS, i quali, per l’appunto, hanno anche una proficua sinergia con il GH. Sia la letteratura scientifica che i dati aneddotici dimostrano chiaramente che l’uso combinato di entrambi i composti ha un massimale ipertrofico significativamente più alto rispetto all’uso singolo. Personalmente, penso che i BodyBuilder dovrebbero sempre optare per l’utilizzo di una base di Testosterone e Boldenone (correttamente rapportati in base a contesto e alle caratteristiche individuali) anche in una fase “Bulk” nella quale viene inserito l’uso del GH. Il Trenbolone può essere considerato come parte “accessoria” di un protocollo di massa, a causa della sua intrinseca difficoltà gestazionale come sostanza anabolizzante. Dovrebbe essere usato con parsimonia e con cautela poiché, insieme ai suoi numerosi punti di forza come composto anabolizzante, presenta alcune limitazioni. Queste limitazioni derivano per lo più, come già accennato, dalla difficile gestione del composto in quanto esso non è facilmente tollerato da una buona parte degli individui. Quindi, se viene usato il Trenbolone, dovrebbe essere inserito calcolando con attenzione il dosaggio e la tolleranza individuale, anche per quanto concerne la tolleranza temporale individuale all’uso di tale composto.
Dopo un periodo d’uso prolungato di dosi sovrafisiologico di AAS (Ciclo+Bridge), è buona cosa procedere con l’interruzione o con una marcata riduzione del numero e degli AAS utilizzati. Detta in parole semplici, questa interruzione può contemplare una completa astinenza dagli AAS, svolgendo un adeguata PCT al fine di ristabilire una omeostasi ormonale fisiologica, o una transizione ad una TRT, metodologia comunemente chiamata “blast and cruise”. La struttura del protocollo di supplementazione farmacologica dovrebbe sempre seguire i principi del dosaggio minimo efficace con aumenti nei dosaggi degli AAS solo nel caso si sia raggiunto il limite di crescita con il precedente dosaggio, assicurandosi che tutte le altre variabili nello stile di vita siano correttamente regolate. L’utilizzo di questo approccio limita il rischio che si sviluppino effetti collaterali indesiderati sul lungo termine.
Quando si decide di usare il GH, la dove ce ne sia la possibilità economica, esso dovrebbe provenire da uno dei prodotti presenti nel mercato farmaceutico. Questi prodotti approvati devono superare anni di studi strettamente controllati per dimostrare la loro sicurezza, purezza ed efficacia su soggetti umani. I progressi tecnologici nel corso degli anni hanno reso molto più facile la produzione di rHGH. Per questo motivo, i produttori ora provengono da tutto il mondo. Spesso questi produttori realizzano ciò che viene definito “GH generico” sui forum, ma tale termine non mi piace molto. Definire qualcosa come “generico” implica che sia una replica perfetta di prodotti legati a specifici marchi farmaceutici approvati dell’agenzia del farmaco che hanno perso il cui brevetto è scaduto, il che non è il caso del GH. Infatti, a causa della natura estremamente complessa del processo di produzione del rHGH, per esempio, la FDA non consente nemmeno l’uso del termine “generico” quando si tratta di rHGH e utilizza invece il termine ” follow-on protein product ” o FOPP.
Spesso questi marchi off-label sono venduti ad un costo molto ridotto, ed è qui che sta il dilemma, in quanto questo può essere molto allettante. Tuttavia, con questo costo ridotto per il consumatore, non ci sarà nemmeno la garanzia del produttore su cosa ci sia realmente nella fiala o persino su come è stato prodotto. Il problema di fondo è che il processo di produzione del rHGH è estremamente complessa, ed è molto facile che nelle fasi di questo processo si commettano errori con conseguenti variazioni nella catena proteica che potenzialmente portano ad effetti indesiderati, o anche a risposte autoimmuni.
Spesso gli atleti si affidano semplicemente ai test serici per misurare i livelli di GH e/o IGF-1 al fine di concludere che un prodotto contenete GH sia “buono”, ma dobbiamo ricordarci che raggiungere livelli ematici ormonali elevati è la parte relativamente facile. Anche le molecole di GH che sono state alterate o danneggiate durante la produzione possono dare questo esito. Tuttavia, queste stesse molecole di GH danneggiate o mutate possono spesso stimolare risposte autoimmuni. Ciò potrebbe indurre il corpo ad avere una risposta recettoriale degradata, che può anche riflettersi sulla secrezione endogena nel tempo. [431-432] Rimane poi il problema del reale contenuto della vial o fiala, la quale può non presentare nessun principio attivo al suo interno.
Il GH dovrebbe essere usato in modo pulsatile, per mimare le condizioni in vivo. Tra queste iniezioni, deve esserci un periodo di refrattarietà o si deve consumare un pasto che abbia un buon stimolo sull’Insulina. Può anche essere utilizzata l’Insulina esogena al fine di bypassare molte delle limitazioni del periodo refrattario, ma questo va oltre lo scopo di questo articolo. Anche se il cumulo delle dosi giornaliere dovrebbe essere sovrafisiologico, le dosi individuali non hanno bisogno di essere ad alto dosaggio, poiché la massima stimolazione dell’IGF-1 autocrino nel tessuto muscolo scheletrico si verifica ben all’interno delle concentrazioni fisiologiche di GH. Aneddoticamente, sembra anche esserci un limite con il quale l’uso di rHGH diventa additivo in presenza di AAS. Potrebbe essere necessario un po’ di ponderata (e supportata da personale qualificato) auto-sperimentazione per scoprire dove si trova questa singola dose di saturazione, ma la maggior parte dei soggetti troverà questo limite tra le 4 e le 8 UI/die. Oltre questo dosaggio, la maggior parte degli utilizzatori tenderà a scoprire che la giustificazione dei costi e il rapporto rischio /beneficio tendono a diminuire rapidamente.
Non bisognerebbe passare troppo tempo a riflettere sul tempo delle iniezioni di GH, dal momento che gli aumenti dei livelli di IGF-1 autocrino avvengono rapidamente e possono rimanere elevati per giorni. Bisognerebbe concentrati invece sul programma di iniezione che si addice meglio al contesto della giornata, tenendo contemporaneamente presenti le linee guida per il periodo di refrattarietà del GH. Si possono anche prendere in considerazione piccole o grandi iniezioni, poiché alcuni potrebbero trovare più pratiche e funzionali iniezioni con un dosaggio inferiore e somministrate più frequenti mentre altri potrebbero preferire iniezioni con un dosaggio maggiore e somministrazioni meno frequenti. Naturalmente, maggiore è il contenuto dell’iniezione, maggiore è la probabilità che si superi la soglia massima di espressione dell’IGF-1 autocrino.
Massimizzare l’espressione dell’IGF-1 autocrino, mentre contemporaneamente si sopprimono i livelli di IGF-1 endocrino, sarà una priorità. Esistono prove a sostegno dell’ipotesi secondo cui un iniezione locale di GH possa aiutare a raggiungere questo obiettivo, con una conseguente minore possibilità di feedback negativo. Sono stati osservati aumenti significativi della massa muscolare in appena due settimane di iniezioni locali di IGF-1.[441]
La dove cause di forza maggiore non lo impediscano, è consigliabile evitare o comunque regolare attentamente l’uso di tutti quei composti che possono avere interazioni negative con l’uso del GH a fini ipertrofici. Inibitori della Aromatasi, Modulatori Selettivi del Recettore degli Estrogeni e T3 hanno tutti dimostrato di avere un potenziale effetto negativo sul processo globale ipertrofico legato al GH e per tale motivo dovrebbero essere usati con parsimonia, se non omessi del tutto dove possibile (vedi in particolare il T3).
Questo non dovrebbe sorprendere nessuno e non dovrebbe nemmeno essere troppo difficile da tenere a mente: allenarsi duramente, allenarsi in modo intelligente e allenarsi in modo coerente. Sebbene non sia stato affrontato direttamente nell’articolo, bisogna capire che l’allenamento contro resistenza ha impatti unici e additivi sull’ipertrofia. In effetti, alcuni di questi meccanismi non sono nemmeno mediati dall’Asse AR e/o GH/IGF-1. [433] Bisogna anche comprendere che non esiste una “magica” routine allenante universalmente applicabile, la chiave di volta sarà la coerenza nel garantire un carico di lavoro adeguato, con elementi di sovraccarico progressivo nel tempo assicurando un adeguato stimolo meccanico gestendo al meglio le variabili allenanti (intensità, volume, densità e intensità percepita). La logica composizione del piano allenante servirà a garantire lo stimolo ipertrofico di base che sarà coadiuvato dall’azione dei composti utilizzati.
Nonostante la mole e l’importanza delle informazioni presentate in questa serie di articoli, è necessario ricordare che i meccanismi d’azione ormonali sono governati da innumerevoli fattori. Anche esaminando l’intero corpo della letteratura scientifica equivarrebbe a poco più che accumulare una serie di utili nozioni garanti di assicurare una solida base conoscitiva sull’argomento la quale rappresenterà un punto di partenza intelligente per l’applicazione pratica, rimanendo sempre soggetti alle variabili di risposta individuale. Seguendo questa linea, i migliori risultati nella pratica spesso provengono da coloro i quali posseggono una buona conoscenza dei principi scientifici e la capacità innata di saperli applicare non solo su se stessi ma, soprattutto, su terzi. Infatti, molto raramente due persone rispondono in modo identico alla supplementazione di ormoni esogeni (e non solo), quindi, non bisogna assolutamente pensare che basti semplicemente applicare su se stessi o su terzi un protocollo che ha portato benefici realmente apprezzabili su un soggetto per ottenere la medesima risposta.
A tal fine, invito atleti e Preparatori a utilizzare queste pubblicazioni come punto di partenza per essere in grado di gestire l’applicazione pratica in modo più consapevole e produttivo. Inoltre, chi ne fosse in grado, può consultare il vasto numero di riferimenti riportati nel corso di queste pubblicazioni è tentare di ragionare sulle mie conclusioni. Ad ogni citazione presente in questa serie di articoli, assicuratevi che il riferimento elencato supporti effettivamente le affermazioni fatte. Mantenere sempre una mente aperta ma con i giusti “filtri”, e cercare di non credere per partito preso ad una singola opinione, specialmente di fronte alle nuove evidenze scientifiche. Infine, è buona cosa verificare sempre la veridicità di quanto è stato affermato.
Punti conclusivi per un corretto utilizzo della chimica e del GH a fini ipertrofici:
Usare il GH in combinazione con gli AAS
Usare GH e AAS di grado farmaceutico la dove ciò è possibile
Assicurarsi una base di Testosterone correttamente rapportata al Boldenone aggiungendo (in base a maturità e tolleranza) il Trenbolone
Iniettare il GH in modo pulsatile, considerare l’opzione delle iniezioni locali nei gruppi carenti
Mantenere un dosaggio complessivo ottimale di GH il quale si attesta tra le 4 e le 8UI/die
Evitare o regolare attentamente l’uso dei composti che possono interagire negativamente con i processi ipertrofici legati al uso di GH (vedi AI, SERM e T3)
Dopo un periodo di tempo (variabile) nel quale si è stati sottoposti a dosaggi ormonali sovrafisiologici optare per una PCT o per una TRT (a seconda delle proprie necessità e priorità)
Essere a conoscenza dei potenziali effetti collaterali legati all’auso/abuso di GH (nausea, vomito, cefalea, ritenzione idrica e sodica, edemi, parestesie, sindrome del tunnel carpale, rigidità articolare, dolori articolari, artrite, dolori muscolari, ipertensione, insulino-resistenza, diabete di tipo II, acromegalia, dilatazione addominale, ipertrofia cardiaca ecc…)
Svolgere regolarmente esami del sangue; sia durante i periodi di picco nell’uso della farmacologia sia nel periodo successivo (vedi PCT/OCT o TRT)
Gestire al meglio le variabili legate agli stressor ambientali, all’allenamento, all’alimentazione e al sonno.
Gabriel Bellizzi
Riferimenti:
338. Mathews LS, Norstedt G, Palmiter RD. Regulation of insulin-like growth factor I gene expression by growth hormone. Proc Natl Acad Sci U S A. 1986 Dec;83(24):9343-7.
339. Keller A, Wu Z, Kratzsch J, Keller E, Blum WF, Kniess A, Preiss R, Teichert J, Strasburger CJ, Bidlingmaier M. Pharmacokinetics and pharmacodynamics of GH: dependence on route and dosage of administration. Eur J Endocrinol. 2007 Jun;156(6):647-53.
340. Tanaka T, Seino Y, Fujieda K, Igarashi Y, Yokoya S, Tachibana K, Ogawa Y. Pharmacokinetics and metabolic effects of high-dose growth hormone administration in healthy adult men. Endocr J. 1999 Aug;46(4):605-12.
341. Quinn LS, Steinmetz B, Maas A, Ong L, Kaleko M. Type-1 insulin-like growth factor receptor overexpression produces dual effects on myoblast proliferation and differentiation. J Cell Physiol. 1994 Jun;159(3):387-98.
342. Coolican SA, Samuel DS, Ewton DZ, McWade FJ, Florini JR. The mitogenic and myogenic actions of insulin-like growth factors utilize distinct signaling pathways. J Biol Chem. 1997 Mar 7;272(10):6653-62.
343. Foulstone EJ, Huser C, Crown AL, Holly JM, Stewart CE. Differential signalling mechanisms predisposing primary human skeletal muscle cells to altered proliferation and differentiation: roles of IGF-I and TNFalpha. Exp Cell Res. 2004 Mar 10;294(1):223-35.
344. Ewton DZ, Roof SL, Magri KA, McWade FJ, Florini JR. IGF-II is more active than IGF-I in stimulating L6A1 myogenesis: greater mitogenic actions of IGF-I delay differentiation. J Cell Physiol. 1994 Nov;161(2):277-84.
345. Florini JR, Nicholson ML, Dulak NC. Effects of peptide anabolic hormones on growth of myoblasts in culture. Endocrinology. 1977 Jul;101(1):32-41.
346. Laviola L, Natalicchio A, Giorgino F. The IGF-I signaling pathway. Curr Pharm Des. 2007;13(7):663-9. Review.
347. Jacquemin V, Furling D, Bigot A, Butler-Browne GS, Mouly V. IGF-1 induces human myotube hypertrophy by increasing cell recruitment. Exp Cell Res. 2004 Sep 10;299(1):148-58.
348. Jacquemin V, Butler-Browne GS, Furling D, Mouly V. IL-13 mediates the recruitment of reserve cells for fusion during IGF-1-induced hypertrophy of human myotubes. J Cell Sci. 2007 Feb 15;120(Pt 4):670-81. Epub 2007 Jan 30.
349. Ballard FJ, Francis GL. Effects of anabolic agents on protein breakdown in L6 myoblasts. Biochem J. 1983 Jan 15;210(1):243-9.
350. Ewton DZ, Falen SL, Florini JR. The type II insulin-like growth factor (IGF) receptor has low affinity for IGF-I analogs: pleiotypic actions of IGFs on myoblasts are apparently mediated by the type I receptor. Endocrinology. 1987 Jan;120(1):115-23.
351. Hembree JR, Hathaway MR, Dayton WR. Isolation and culture of fetal porcine myogenic cells and the effect of insulin, IGF-I, and sera on protein turnover in porcine myotube cultures. J Anim Sci. 1991 Aug;69(8):3241-50.
352. Hong D, Forsberg NE. Effects of serum and insulin-like growth factor I on protein degradation and protease gene expression in rat L8 myotubes. J Anim Sci. 1994 Sep;72(9):2279-88.
353. Florini JR, Ewton DZ, Roof SL. Insulin-like growth factor-I stimulates terminal myogenic differentiation by induction of myogenin gene expression. Mol Endocrinol. 1991 May;5(5):718-24.
354. Musarò A, McCullagh KJ, Naya FJ, Olson EN, Rosenthal N. IGF-1 induces skeletal myocyte hypertrophy through calcineurin in association with GATA-2 and NF-ATc1. Nature. 1999 Aug 5;400(6744):581-5.
355. Haba GDL, Cooper GW, Elting V. HORMONAL REQUIREMENTS FOR MYOGENESIS OF STRIATED MUSCLE IN VITRO: INSULIN AND SOMATOTROPIN. Proceedings of the National Academy of Sciences of the United States of America. 1966;56(6):1719-1723.
356. Florini JR, Ewton DZ. Insulin acts as a somatomedin analog in stimulating myoblast growth in serum-free medium. In Vitro. 1981 Sep;17(9):763-8.
357. Schmid C, Steiner T, Froesch ER. Preferential enhancement of myoblast differentiation by insulin-like growth factors (IGF I and IGF II) in primary cultures of chicken embryonic cells. FEBS Lett. 1983 Sep 5;161(1):117-21.
358. Florini JR, Ewton DZ, Falen SL, Van Wyk JJ. Biphasic concentration dependency of stimulation of myoblast differentiation by somatomedins. Am J Physiol. 1986 May;250(5 Pt 1):C771-8.
359. Quinn LS, Ehsan M, Steinmetz B, Kaleko M. Ligand-dependent inhibition of myoblast differentiation by overexpression of the type-1 insulin-like growth factor receptor. J Cell Physiol. 1993 Sep;156(3):453-61.
360. Olson EN. Signal transduction pathways that regulate skeletal muscle gene expression. Mol Endocrinol. 1993 Nov;7(11):1369-78. Review.
361. Murphy LJ, Bell GI, Friesen HG. Growth hormone stimulates sequential induction of c-myc and insulin-like growth factor I expression in vivo. Endocrinology. 1987 May;120(5):1806-12.
362. Turner JD, Rotwein P, Novakofski J, Bechtel PJ. Induction of mRNA for IGF-I and -II during growth hormone-stimulated muscle hypertrophy. Am J Physiol. 1988 Oct;255(4 Pt 1):E513-7.
363. Isgaard J, Nilsson A, Vikman K, Isaksson OG. Growth hormone regulates the level of insulin-like growth factor-I mRNA in rat skeletal muscle. J Endocrinol. 1989 Jan;120(1):107-12.
364. Bichell DP, Kikuchi K, Rotwein P. Growth hormone rapidly activates insulin-like growth factor I gene transcription in vivo. Mol Endocrinol. 1992 Nov;6(11):1899-908.
365. Sadowski CL, Wheeler TT, Wang LH, Sadowski HB. GH regulation of IGF-I and suppressor of cytokine signaling gene expression in C2C12 skeletal muscle cells. Endocrinology. 2001 Sep;142(9):3890-900.
366. Frost RA, Nystrom GJ, Lang CH. Regulation of IGF-I mRNA and signal transducers and activators of transcription-3 and -5 (Stat-3 and -5) by GH in C2C12 myoblasts. Endocrinology. 2002 Feb;143(2):492-503.
367. MacLeod JN, Pampori NA, Shapiro BH. Sex differences in the ultradian pattern of plasma growth hormone concentrations in mice. J Endocrinol. 1991 Dec;131(3):395-9.
368. Rochiccioli P, Messina A, Tauber MT, Enjaume C. Correlation of the parameters of 24-hour growth hormone secretion with growth velocity in 93 children of varying height. Horm Res. 1989;31(3):115-8.
369. Hansen TK, Gravholt CH, ØRskov H, Rasmussen MH, Christiansen JS, Jørgensen JO. Dose dependency of the pharmacokinetics and acute lipolytic actions of growth hormone. J Clin Endocrinol Metab. 2002 Oct;87(10):4691-8.
370. Baum WF, Klöditz E, Hesse V, Jahreis G, Schneyer U, Giebler H. [Increase in spontaneous growth hormone secretion in asthmatic children–a symptom of atopic disposition?]. Kinderarztl Prax. 1993 Nov;61(9):323-8.
371. Adams GR, McCue SA. Localized infusion of IGF-I results in skeletal muscle hypertrophy in rats. J Appl Physiol (1985). 1998 May;84(5):1716-22.
372. Alzghoul MB, Gerrard D, Watkins BA, Hannon K. Ectopic expression of IGF-I and Shh by skeletal muscle inhibits disuse-mediated skeletal muscle atrophy and bone osteopenia in vivo. FASEB J. 2004 Jan;18(1):221-3. Epub 2003 Nov 3.
373. Lee S, Barton ER, Sweeney HL, Farrar RP. Viral expression of insulin-like growth factor-I enhances muscle hypertrophy in resistance-trained rats. J Appl Physiol (1985). 2004 Mar;96(3):1097-104. Erratum in: J Appl Physiol. 2004 Jun;96(6):2343.
374. Coleman ME, DeMayo F, Yin KC, Lee HM, Geske R, Montgomery C, Schwartz RJ. Myogenic vector expression of insulin-like growth factor I stimulates muscle cell differentiation and myofiber hypertrophy in transgenic mice. J Biol Chem. 1995 May 19;270(20):12109-16.
375. Barton-Davis ER, Shoturma DI, Musaro A, Rosenthal N, Sweeney HL. Viral mediated expression of insulin-like growth factor I blocks the aging-related loss of skeletal muscle function. Proc Natl Acad Sci U S A. 1998 Dec 22;95(26):15603-7.
376. Musarò A, McCullagh K, Paul A, Houghton L, Dobrowolny G, Molinaro M, Barton ER, Sweeney HL, Rosenthal N. Localized Igf-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat Genet. 2001 Feb;27(2):195-200
377. Lewis MI, Bulut Y, Biring MS, Da X, Fournier M. (1999) IGF-I administration prevents corticosteroids-induced diaphragm atrophy in emphysema . Am J Respir Crit Care Med 159:A580
378. Fournier M, Huang ZS, Cercek B, Li H, Bykhovskaya I, Lewis MI. (2000) Administration of insulin-like growth factor-1 (IGF-I) and corticosteroids in emphysematous hamsters: influences on diaphragm IGF-I . Am J Respir Crit Care Med 161:A18
379. Shavlakadze T, Grounds M. Of bears, frogs, meat, mice and men: complexity of factors affecting skeletal muscle mass and fat. Bioessays. 2006 Oct;28(10):994-1009. Review.
380. Maiter D, Underwood LE, Maes M, Davenport ML, Ketelslegers JM. Different effects of intermittent and continuous growth hormone (GH) administration on serum somatomedin-C/insulin-like growth factor I and liver GH receptors in hypophysectomized rats. Endocrinology. 1988 Aug;123(2):1053-9.
381. Isgaard J, Carlsson L, Isaksson OG, Jansson JO. Pulsatile intravenous growth hormone (GH) infusion to hypophysectomized rats increases insulin-like growth factor I messenger ribonucleic acid in skeletal tissues more effectively than continuous GH infusion. Endocrinology. 1988 Dec;123(6):2605-10.
382. Clark RG, Jansson JO, Isaksson O, Robinson IC. Intravenous growth hormone: growth responses to patterned infusions in hypophysectomized rats. J Endocrinol. 1985 Jan;104(1):53-61.
383. Bick T, Hochberg Z, Amit T, Isaksson OG, Jansson JO. Roles of pulsatility and continuity of growth hormone (GH) administration in the regulation of hepatic GH-receptors, and circulating GH-binding protein and insulin-like growth factor-I. Endocrinology. 1992 Jul;131(1):423-9.
384. Weltman A, Weltman JY, Schurrer R, Evans WS, Veldhuis JD, Rogol AD. Endurance training amplifies the pulsatile release of growth hormone: effects of training intensity. J Appl Physiol (1985). 1992 Jun;72(6):2188-96.
385. Flores-Morales A, Greenhalgh CJ, Norstedt G, Rico-Bautista E. Negative regulation of growth hormone receptor signaling. Mol Endocrinol. 2006 Feb;20(2):241-53. Epub 2005 Jul 21. Review.
386. Hartman ML, Veldhuis JD, Thorner MO. Normal control of growth hormone secretion. Horm Res. 1993;40(1-3):37-47. Review.
387. Fernández L, Flores-Morales A, Lahuna O, Sliva D, Norstedt G, Haldosén LA, Mode A, Gustafsson JA. Desensitization of the growth hormone-induced Janus kinase 2 (Jak 2)/signal transducer and activator of transcription 5 (Stat5)-signaling pathway requires protein synthesis and phospholipase C. Endocrinology. 1998 Apr;139(4):1815-24.
388. Gebert CA, Park SH, Waxman DJ. Termination of growth hormone pulse-induced STAT5b signaling. Mol Endocrinol. 1999 Jan;13(1):38-56.
389. Ram PA, Waxman DJ. SOCS/CIS protein inhibition of growth hormone-stimulated STAT5 signaling by multiple mechanisms. J Biol Chem. 1999 Dec 10;274(50):35553-61.
390. Ram PA, Waxman DJ. Role of the cytokine-inducible SH2 protein CIS in desensitization of STAT5b signaling by continuous growth hormone. J Biol Chem.2000 Dec 15;275(50):39487-96.
391. Xu J, Keeton AB, Franklin JL, Li X, Venable DY, Frank SJ, Messina JL. Insulin enhances growth hormone induction of the MEK/ERK signaling pathway. J Biol Chem. 2006 Jan 13;281(2):982-92. Epub 2005 Nov 4.
392. Lewis TS, Shapiro PS, Ahn NG. Signal transduction through MAP kinase cascades. Adv Cancer Res. 1998;74:49-139. Review.
394. Mebis L, Paletta D, Debaveye Y, Ellger B, Langouche L, D’Hoore A, Darras VM, Visser TJ, Van den Berghe G. Expression of thyroid hormone transporters during critical illness. Eur J Endocrinol. 2009 Aug;161(2):243-50.
395. Jørgensen JO, Pedersen SA, Laurberg P, Weeke J, Skakkebaek NE, Christiansen JS. Effects of growth hormone therapy on thyroid function of growth hormone-deficient adults with and without concomitant thyroxine-substituted central hypothyroidism. J Clin Endocrinol Metab. 1989 Dec;69(6):1127-32.
396. Jørgensen JO, Pedersen SB, Børglum J, Møller N, Schmitz O, Christiansen JS, Richelsen B. Fuel metabolism, energy expenditure, and thyroid function in growth hormone-treated obese women: a double-blind placebo-controlled study. Metabolism. 1994 Jul;43(7):872-7.
397. Wolthers T, Grøftne T, Møller N, Christiansen JS, Orskov H, Weeke J, Jørgensen JO. Calorigenic effects of growth hormone: the role of thyroid hormones. J Clin Endocrinol Metab. 1996 Apr;81(4):1416-9.
398. Feldt-Rasmussen U. Interactions between growth hormone and the thyroid gland — with special reference to biochemical diagnosis. Curr Med Chem. 2007;14(26):2783-8. Review.
399. Kalina-Faska B, Kalina M, Koehler B. Effects of recombinant growth hormone therapy on thyroid hormone concentrations. Int J Clin Pharmacol Ther. 2004 Jan;42(1):30-4.
400. Hubina E, Mersebach H, Rasmussen AK, Juul A, Sneppen SB, Góth MI, Feldt-Rasmussen U. Effect of growth hormone replacement therapy on pituitary hormone secretion and hormone replacement therapies in GHD adults. Horm Res. 2004;61(5):211-7. Epub 2004 Jan 30.
401. Seminara S, Stagi S, Candura L, Scrivano M, Lenzi L, Nanni L, Pagliai F, Chiarelli F. Changes of thyroid function during long-term hGH therapy in GHD children. A possible relationship with catch-up growth? Horm Metab Res. 2005 Dec;37(12):751-6.
402. Losa M, Scavini M, Gatti E, Rossini A, Madaschi S, Formenti I, Caumo A, Stidley CA, Lanzi R. Long-term effects of growth hormone replacement therapy on thyroid function in adults with growth hormone deficiency. Thyroid. 2008 Dec;18(12):1249-54.
403. Müller MJ, Seitz HJ. Thyroid hormone action on intermediary metabolism. Part III. Protein metabolism in hyper- and hypothyroidism. Klin Wochenschr. 1984 Feb 1;62(3):97-102.
404. Tawa NE Jr, Odessey R, Goldberg AL. Inhibitors of the proteasome reduce the accelerated proteolysis in atrophying rat skeletal muscles. J Clin Invest. 1997 Jul 1;100(1):197-203. PubMed PMID: 9202072
405. Dace A, Zhao L, Park KS, et al. Hormone binding induces rapid proteasome-mediated degradation of thyroid hormone receptors. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(16):8985-8990.
406. Clément K, Viguerie N, Diehn M, Alizadeh A, Barbe P, Thalamas C, Storey JD, Brown PO, Barsh GS, Langin D. In vivo regulation of human skeletal muscle gene expression by thyroid hormone. Genome Res. 2002 Feb;12(2):281-91.
407. Miell JP, Taylor AM, Zini M, Maheshwari HG, Ross RJ, Valcavi R. Effects of hypothyroidism and hyperthyroidism on insulin-like growth factors (IGFs) and growth hormone- and IGF-binding proteins. J Clin Endocrinol Metab. 1993 Apr;76(4):950-5.
408. Murao K, Takahara J, Sato M, Tamaki M, Niimi M, Ishida T. Relationship between thyroid functions and urinary growth hormone secretion in patients with hyper- and hypothyroidism. Endocr J. 1994 Oct;41(5):517-22.
409. Wolf M, Ingbar SH, Moses AC. Thyroid hormone and growth hormone interact to regulate insulin-like growth factor-I messenger ribonucleic acid and circulating levels in the rat. Endocrinology. 1989 Dec;125(6):2905-14.
410. Laron Z. Interactions between the thyroid hormones and the hormones of the growth hormone axis. Pediatr Endocrinol Rev. 2003 Dec;1 Suppl 2:244-9-discussion 250. Review.
411. Fiems LO. Double Muscling in Cattle: Genes, Husbandry, Carcasses and Meat. Animals : an Open Access Journal from MDPI. 2012;2(3):472-506.
412. Langley B, Thomas M, Bishop A, Sharma M, Gilmour S, Kambadur R. Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J Biol Chem. 2002 Dec 20;277(51):49831-40. Epub 2002 Sep 18.
413. McPherron AC, Lee SJ. Double muscling in cattle due to mutations in the myostatin gene. Proc Natl Acad Sci U S A. 1997 Nov 11;94(23):12457-61.
414. Schuelke M, Wagner KR, Stolz LE, Hübner C, Riebel T, Kömen W, Braun T, Tobin JF, Lee SJ. Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med. 2004 Jun 24;350(26):2682-8.
415. Clop A, Marcq F, Takeda H, Pirottin D, Tordoir X, Bibé B, Bouix J, Caiment F, Elsen JM, Eychenne F, Larzul C, Laville E, Meish F, Milenkovic D, Tobin J, Charlier C, Georges M. A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat Genet. 2006 Jul;38(7):813-8.
416. Gonzalez-Cadavid NF, Taylor WE, Yarasheski K, Sinha-Hikim I, Ma K, Ezzat S, Shen R, Lalani R, Asa S, Mamita M, Nair G, Arver S, Bhasin S. Organization of the human myostatin gene and expression in healthy men and HIV-infected men with muscle wasting. Proc Natl Acad Sci U S A. 1998 Dec 8;95(25):14938-43.
417. Liu W, Thomas SG, Asa SL, Gonzalez-Cadavid N, Bhasin S, Ezzat S. Myostatin is a skeletal muscle target of growth hormone anabolic action. J Clin Endocrinol Metab. 2003 Nov;88(11):5490-6.
418. Oldham JM, Osepchook CC, Jeanplong F, Falconer SJ, Matthews KG, Conaglen JV, Gerrard DF, Smith HK, Wilkins RJ, Bass JJ, McMahon CD. The decrease in mature myostatin protein in male skeletal muscle is developmentally regulated by growth hormone. J Physiol. 2009 Feb 1;587(3):669-77.
419. Williams NG, Interlichia JP, Jackson MF, Hwang D, Cohen P, Rodgers BD. Endocrine actions of myostatin: systemic regulation of the IGF and IGF binding protein axis. Endocrinology. 2011 Jan;152(1):172-80.
420. Winbanks CE, Weeks KL, Thomson RE, Sepulveda PV, Beyer C, Qian H, Chen JL, Allen JM, Lancaster GI, Febbraio MA, Harrison CA, McMullen JR, Chamberlain JS, Gregorevic P. Follistatin-mediated skeletal muscle hypertrophy is regulated by Smad3 and mTOR independently of myostatin. J Cell Biol. 2012 Jun 25;197(7):997-1008.
421. Lach-Trifilieff E, Minetti GC, Sheppard K, Ibebunjo C, Feige JN, Hartmann S, Brachat S, Rivet H, Koelbing C, Morvan F, Hatakeyama S, Glass DJ. An antibody blocking activin type II receptors induces strong skeletal muscle hypertrophy and protects from atrophy. Mol Cell Biol. 2014 Feb;34(4):606-18.
422. Bark TH, McNurlan MA, Lang CH, Garlick PJ. Increased protein synthesis after acute IGF-I or insulin infusion is localized to muscle in mice. Am J Physiol. 1998 Jul;275(1 Pt 1):E118-23.
423. Barton-Davis ER, Shoturma DI, Sweeney HL. Contribution of satellite cells to IGF-I induced hypertrophy of skeletal muscle. Acta Physiol Scand. 1999 Dec;167(4):301-5.
424. Suryawan A, Frank JW, Nguyen HV, Davis TA. Expression of the TGF-beta family of ligands is developmentally regulated in skeletal muscle of neonatal rats. Pediatr Res. 2006 Feb;59(2):175-9.
425. Gilson H, Schakman O, Kalista S, Lause P, Tsuchida K, Thissen JP. Follistatin induces muscle hypertrophy through satellite cell proliferation and inhibition of both myostatin and activin. Am J Physiol Endocrinol Metab. 2009 Jul;297(1):E157-64.
426. Bodine SC, Stitt TN, Gonzalez M, Kline WO, Stover GL, Bauerlein R, Zlotchenko E, Scrimgeour A, Lawrence JC, Glass DJ, Yancopoulos GD. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol. 2001 Nov;3(11):1014-9.
428. Kalista S, Schakman O, Gilson H, Lause P, Demeulder B, Bertrand L, Pende M, Thissen JP. The type 1 insulin-like growth factor receptor (IGF-IR) pathway is mandatory for the follistatin-induced skeletal muscle hypertrophy. Endocrinology. 2012 Jan;153(1):241-53.
429. Barbé C, Kalista S, Loumaye A, Ritvos O, Lause P, Ferracin B, Thissen JP. Role of IGF-I in follistatin-induced skeletal muscle hypertrophy. Am J Physiol Endocrinol Metab. 2015 Sep 15;309(6):E557-67. doi: 10.1152/ajpendo.00098.2015. Epub 2015 Jul 28.
430. Coffey VG, Shield A, Canny BJ, Carey KA, Cameron-Smith D, Hawley JA. Interaction of contractile activity and training history on mRNA abundance in skeletal muscle from trained athletes. Am J Physiol Endocrinol Metab. 2006 May;290(5):E849-55.
431. Moore WV, Leppert P. Role of aggregated human growth hormone (hGH) in development of antibodies to hGH. J Clin Endocrinol Metab. 1980 Oct;51(4):691-7
432. Dannies PS. Protein folding and deficiencies caused by dominant-negative mutants of hormones. Vitam Horm. 2000;58:1-26. Review.
433. DeVol DL, Rotwein P, Sadow JL, Novakofski J, Bechtel PJ. Activation of insulin-like growth factor gene expression during work-induced skeletal muscle growth. Am J Physiol. 1990 Jul;259(1 Pt 1):E89-95.
434. Hermansen K, Bengtsen M, Kjær M, Vestergaard P, Jørgensen JOL. Impact of GH administration on athletic performance in healthy young adults: A systematic review and meta-analysis of placebo-controlled trials. Growth Horm IGF Res. 2017 Jun;34:38-44.
435. de Souza GL, Hallak J. Anabolic steroids and male infertility: a comprehensive review. BJU Int. 2011 Dec;108(11):1860-5.
436. Kraemer WJ, Marchitelli L, Gordon SE, Harman E, Dziados JE, Mello R, Frykman P, McCurry D, Fleck SJ. Hormonal and growth factor responses to heavy resistance exercise protocols. J Appl Physiol (1985). 1990 Oct;69(4):1442-50.
437. Pfeffer LA, Brisson BK, Lei H, Barton ER. The insulin-like growth factor (IGF)-I E-peptides modulate cell entry of the mature IGF-I protein. Mol Biol Cell. 2009 Sep;20(17):3810-7.
438. Mills P, Dominique JC, Lafrenière JF, Bouchentouf M, Tremblay JP. A synthetic mechano growth factor E Peptide enhances myogenic precursor cell transplantation success. Am J Transplant. 2007 Oct;7(10):2247-59.
439. Brisson BK, Barton ER. Insulin-like growth factor-I E-peptide activity is dependent on the IGF-I receptor. PLoS One. 2012;7(9):e45588.
440. Brisson BK, Spinazzola J, Park S, Barton ER. Viral expression of insulin-like growth factor I E-peptides increases skeletal muscle mass but at the expense of strength. Am J Physiol Endocrinol Metab. 2014 Apr 15;306(8):E965-74.
441. Goldspink G, Harridge S. Mechanism for adaptation in skeletal muscle In: Komi P, editor. Strength and power in sport: Olympic encyclopedia of sports medicine. Oxford: Blackwell; 2002. p. 231–51.
442. Janssen JA, Hofland LJ, Strasburger CJ, van den Dungen ES, Thevis M. Potency of Full-Length MGF to Induce Maximal Activation of the IGF-I R Is Similar to Recombinant Human IGF-I at High Equimolar Concentrations. PLoS One. 2016 Mar 18;11(3):e0150453.