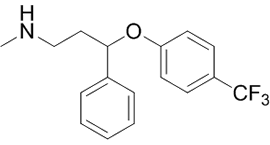
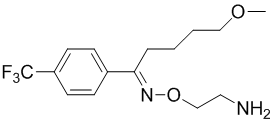
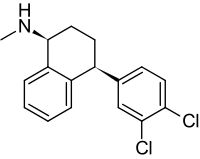
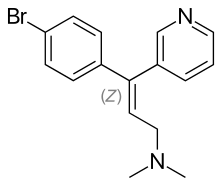
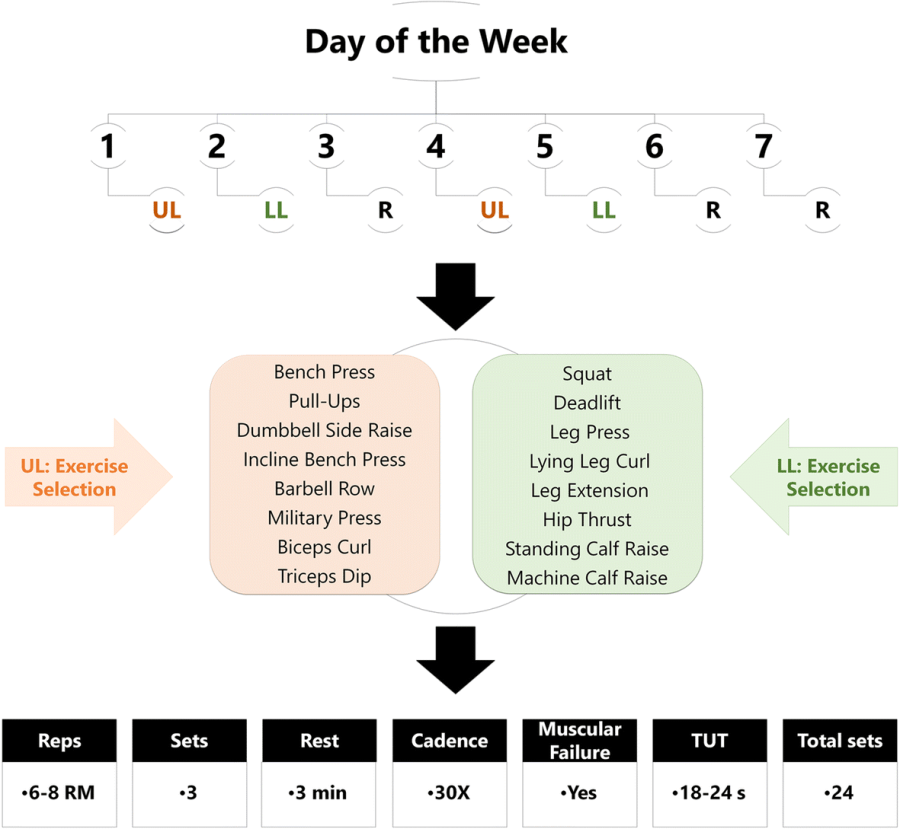
Un maggior utilizzo di AAS da parte di un atleta si traduce in una maggiore probabilità di avere problemi sessuali durante i periodi di non utilizzo. E no, la PCT non sembra rendere esenti da tali possibili problemi. I ricercatori del American Mayo hospital hanno osservato tale effetto in seguito ad uno studio svolto su diverse centinaia di Bodybuilder supplementati farmacologicamente.(1)
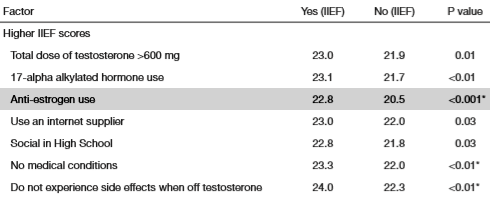
I ricercatori hanno contattato 231 utilizzatori di AAS maschi, attraverso nove forum di bodybuilding, i quali erano pronti a rispondere a domande online sulla loro salute sessuale e sul loro uso di AAS. Per rilevare eventuali disfunzioni erettili, i ricercatori hanno utilizzato una versione abbreviata del questionario IEEF-5. Potete trovarne una copia qui. Gli uomini che totalizzano un punteggio pari o maggiore di 22 non hanno nulla di cui lamentarsi sessualmente parlando, mentre gli uomini che ottengono un punteggio di 17-21 soffrono di un lieve disturbo erettile.
I partecipanti hanno ottenuto un punteggio leggermente superiore sull’IEEF-5 se erano sottoposti ad iniezioni superiori ai 600mg di Testosterone a settimana, se usavano anche un AAS orale o un anti-estrogeno e se erano in buone condizioni di salute. Non molto sorprendente come risultato.
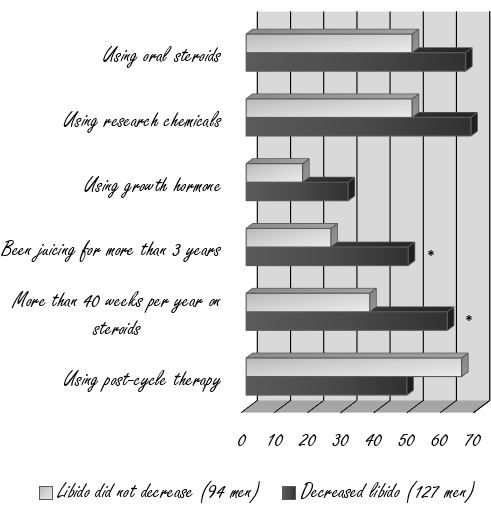
127 utilizzatori hanno affermato che nei loro periodi “off” la libido diminuiva. Questo però non è stato riportato da altri 94 utilizzatori. Quando i ricercatori hanno messo a confronto questi due gruppi, hanno notato che il rischio che si verifichi una riduzione della libido aumentava in modo significativo qualora gli utilizzatori avessero seguito protocolli di AAS per molte settimane all’anno e avessero una lunga storia di utilizzo di questa classe di farmaci.
Lo svolgimento di una PCT era pratica diffusa nel gruppo in cui la libido tra i cicli non diminuiva rispetto al gruppo in cui la libido diminuiva. Tuttavia, questa differenza non era statisticamente significativa.
L’attuale studio rappresenta ad oggi la più grande valutazione sull’impatto dell’utilizzo di AAS ad alto dosaggio e nel lungo periodo sulla funzione sessuale. I risultati dimostrano che l’aumento della durata e della frequenza di utilizzo degli AAS sono associate a più alti tassi di disfunzione erettile de novo e diminuzione della libido dopo l’interruzione d’uso del/i composto/i.
Gli uomini con disfunzione erettile de novo avevano anche maggiori probabilità di riportare altri sintomi legati a bassi livelli di Testosterone, come riduzione della libido, diminuzione dell’energia, depressione, riduzione soggettiva della massa muscolare e aumento soggettivo della massa grassa. Diversamente a ciò, durante l’uso di un dosaggio più alto di Testosterone e l’uso (con tutta probabilità ponderato) di anti-estrogeni si sono osservati punteggi più alti sul questionario IEEF-5.
L’aumento esponenziale di patologie psichiatriche, tra le quali emergono maggiormente ansia e depressione, ha portato ad un consequenziale aumento del consumo di psicofarmaci (una stima riporta che il 20% della popolazione in Italia fa uso di psicofarmaci). Il tema dell’utilizzo di psicofarmaci, come ansiolitici e antidepressivi, è molto attuale e discussa in gran parte d’Europa e negli Stati Uniti. E non sono pochi i medici che si interrogano da tempo sulle cause e i potenziali effetti della loro diffusione. Gli innumerevoli effetti collaterali legati agli psicofarmaci più diffusi (vedi antidepressivi e benzodiazepine) in molti casi, “arginando” (con marcate differenze soggettive) da una parte il malessere primario lamentato dal paziente, tendono a causare diversi problemi più o meno gravi che non solo hanno il potenziale di peggiorare le condizioni di salute del soggetto trattato ma possono anche peggiorarne le condizioni psicologiche. Uno di questi effetti è l’aumento di peso. Di particolare interesse anche per gli atleti, l’aumento di peso e il peggioramento della composizione corporea legato all’uso degli antidepressivi è un tema tutt’altro che chiarito ed è spesso dibattuto senza avere la ben che minima formazione in merito. Scopo di questo articolo è quello di analizzare più da vicino la correlazione tra antidepressivi e aumento del peso corporeo.
Introduzione
I farmaci psicotropi hanno vari effetti collaterali e l’aumento del peso corporeo è uno di questi effetti ed è correlato ad un certo numero di composti facenti parte di questa classe di farmaci. Esiste un’ampia evidenza empirica che mostra la relazione tra la terapia antipsicotica e l’aumento del peso corporeo. Più precisamente gli antipsicotici di seconda generazione (SGA) sono noti per il loro potenziale di causare un significativo aumento del peso corporeo.(1) Tuttavia esiste una carenza di prove per quanto riguarda l’effetto degli antidepressivi sui cambiamenti del peso e della composizione corporea rispetto agli antipsicotici, lasciando aperte ulteriori controversie relative all’effetto di questi farmaci su tale risposta indotta.(1)
La depressione e l’obesità sono ormai due diffusi problemi della salute psicofisica nella società moderna.(2,3) Non ci sono studi che suggeriscono un’associazione positiva tra la depressione e l’obesità. (4-7) Alcuni ricercatori hanno tentato di stabilire i meccanismi alla base per l’associazione positiva tra obesità e depressione. (4,6,8-11) Wild et al. (4), McCarty et al. (8) e Ball et al (9) hanno suggerito che le donne sono più inclini a diventare obese durante gli stati depressivi rispetto agli uomini. Heo et al (10) hanno identificato nel sesso, età e razza dei fattori che contribuiscono all’associazione tra obesità e depressione. Fattori sociali e culturali possono anche contribuire all’aumento di peso e all’obesità che si verificano parallelamente ai disturbi dell’umore.(6) Afari et al (11) hanno svolto uno studio finalizzato a constatare se le influenze genetiche condivise sono responsabili dell’associazione tra queste due condizioni scoprendo un’associazione fenotipica modesta tra la depressione e l’obesità. È stato anche scoperto che i farmaci antidepressivi rappresentano un fattore potenziale per l’induzione dell’aumento di peso nei pazienti depressi. (5,12,13)
Tra antidepressivi TCA (antidepressivi triciclici), IMAO (inibitori delle monoamino ossidasi), e Mirtazapina (antidepressivo di seconda generazione appartenente alla classe farmacologica dei NaSSA) sono noti per dare maggiori problemi legati all’aumento di peso.(14) Alcuni pazienti possono anche aumentare di peso durante l’assunzione di SSRI, in particolare con terapia a base di Paroxetina.(15) Lo scopo di questo articolo è di intraprendere una revisione completa della letteratura riguardante l’effetto degli antidepressivi sui cambiamenti del peso e della composizione corporea, per chiarire se l’aumento di peso si verifica frequentemente con l’uso di antidepressivi e quali antidepressivi sono associati con l’aumento di peso. Gli studi disponibili saranno classificati in base a ciascun antidepressivo al fine di ottenere una prospettiva generale sull’effetto di ciascun farmaco sulle variazioni del peso corporeo. Infine verranno discusse le aree nelle quali la conoscenza è maggiormente carente dei farmaci antidepressivi e gli esiti dell’aumento di peso.
Una ricerca in rete è stata condotta attraverso i database di Medline, Pubmed, Cochrane library e Science Direct ed è stata passata al vaglio la letteratura pubblicata tra il gennaio 1973 e l’agosto 2012 utilizzando come criterio di ricerca due gruppi di parole chiave: antidepressivi, i nomi di ciascuna categoria di antidepressivi, parola chiave del primo gruppo, e, come parola chiave del secondo gruppo, obesità e peso. Gli articoli identificati nella procedura di ricerca sono stati esaminati e gli elenchi di riferimento degli articoli recensiti sono stati cercati manualmente. I riferimenti identificati come rilevanti sono stati recuperati e rivisti. Dopo aver esaminato tutti gli studi, quelli che non avevano alcun contenuto sull’associazione tra antidepressivi e variazioni di peso sono stati esclusi. Gli articoli rimanenti hanno tutti soddisfatto i criteri di ricerca come risultato principale o come risultato secondario. Tra questi articoli, sono stati identificati quarantanove studi empirici tra cui alcuni che prendevano in esame l’applicazione di diversi antidepressivi senza alcuna specificazione e alcuni hanno usato ciascun particolare antidepressivo in gruppi separati. L’ultimo gruppo che contiene risultati più precisi per ciascun antidepressivo specifico è stato enfatizzato nel presente articolo.
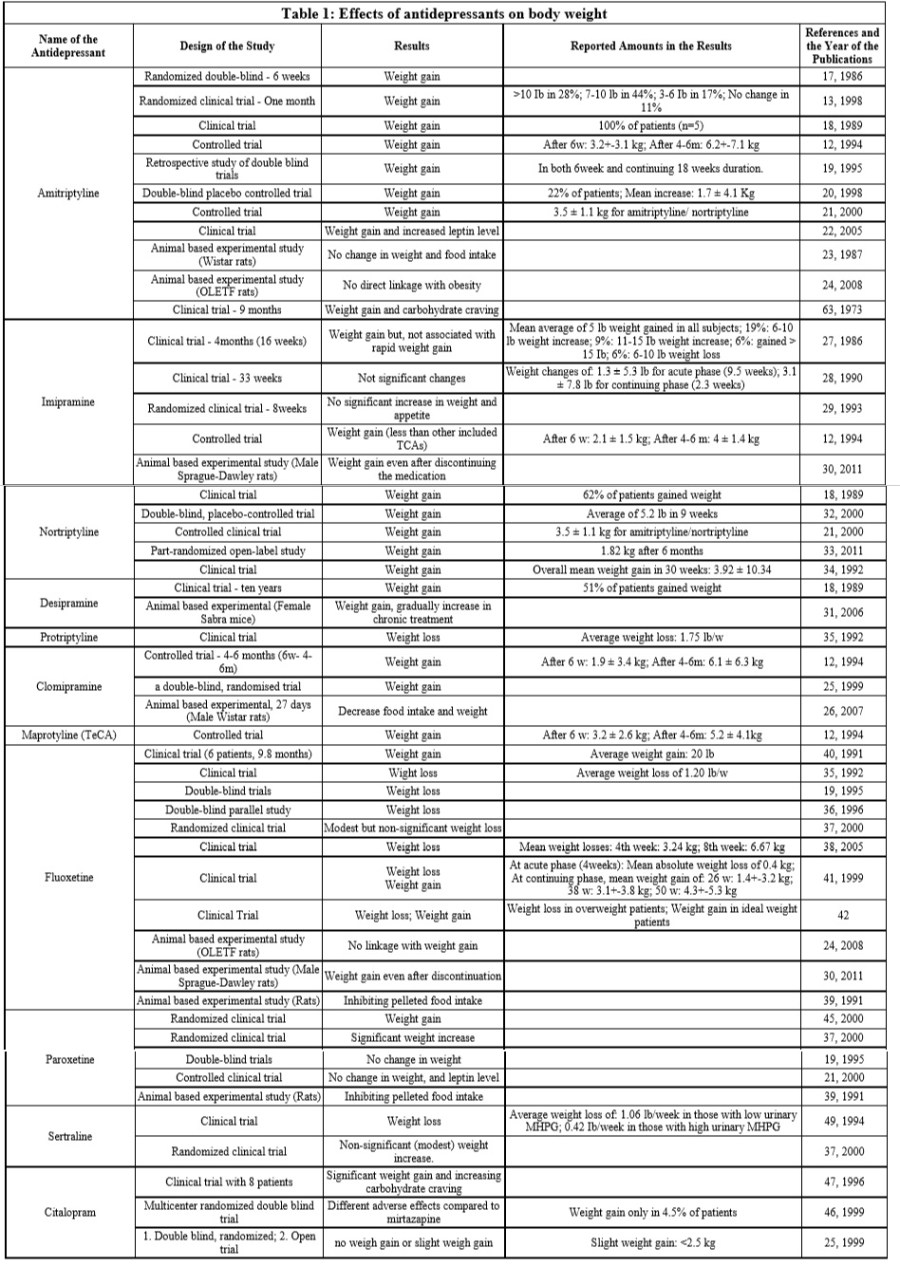
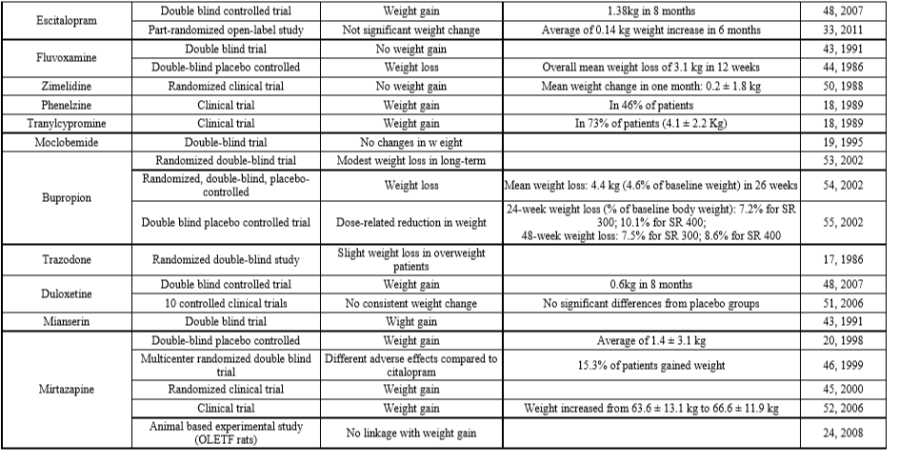
Esistono quarantanove studi empirici in letteratura riguardanti l’effetto degli antidepressivi sui cambiamenti del peso corporeo. Ogni antidepressivo viene discusso separatamente secondo la letteratura pertinente. La tabella 1 illustra il riepilogo dei risultati che sono stati raggiunti attraverso la review* che ha reso possibile la realizzazione di questo articolo.
Esposizione “causa/effetto” dei farmaci antidepressivi presi in esame
Amitriptilina
L’Amitriptilina è un antidepressivo comunemente collegato all’aumento di peso. (12,13,16-22) In uno studio clinico 51 donne depresse trattate con Amitriptilina sono state divise in due gruppi; un gruppo aveva mantenuto l’uso della Amitriptilina per nove mesi mentre un altro gruppo aveva sospeso l’assunzione del farmaco dopo tre mesi. Entrambi i gruppi hanno mostrato un guadagno di peso durante il recupero. Tuttavia, il gruppo trattato con Amitriptilina ha continuato a ingrassare eccessivamente, mentre la cessazione dell’assunzione del farmaco dopo nove mesi di trattamento ha causato una perdita di peso.(16) Il risultato di un altro studio randomizzato in doppio cieco di sei settimane ha mostrato aumenti di peso corporeo significativamente più elevati con l’assunzione di Amitriptilina rispetto al placebo e al Trazodone (quest’ultimo ha causato una leggera perdita di peso).(17) Pande et al (18) hanno scoperto che il 100% dei pazienti trattati con Amitriptilina durante il trattamento ha subito un aumento di peso. L’Amitriptilina induce un notevole aumento di peso nei pazienti, il quale è superiore all’aumento di peso dovuto alla somministrazione di Clomipramina e Imipramina.(12) Un altro studio ha riportato aumenti di peso modesti (1,7 ± 4,1 Kg) nel 22% dei pazienti trattati con Amitriptilina in un doppio-studio clinico controllato con placebo. (20) Berilgen et al (22) hanno riscontrato che l’Amitriptilina causa aumento di peso e aumento dei livelli serici di Leptina suggerendo che l’Amitriptilina può causare resistenza alla Leptina con tutte le conseguenze ad essa correlate. Al contrario, Hinze-Selch et al (21) non hanno riscontrato influenze sui livelli di Leptina con l’uso di TCA compresa l’Amitriptilina, nonostante l’uso di questi composti abbia portato ad un aumento di peso.
Due esperimenti svolti su animali nei quali sono stati utilizzati ratti Wistar (23) e ratti OLETF (24) non furono in grado di riprodurre l’aumento di peso corporeo connesso alla terapia con Amitriptilina e lo studio portò alla conclusione che non vi era alcun legame tra l’aumento di peso e il trattamento con Amitriptilina nei ratti.(23, 24) Ciò mostra che gli studi disponibili nei quali è stato osservato l’effetto della Amitriptilina sugli animali non possono essere usati come modello di paragone per ipotizzare i meccanismi che portano all’aumento di peso con la somministrazione di Amitriptilina osservato in ambiente clinico.
Clomipramina
La Clomipramina induce un aumento di peso nell’uomo come sostenuto da due studi clinici.(12, 25) Al contrario, la Clomipramina ha ridotto l’aumento di peso e l’assunzione di cibo nei ratti Wistar maschi mantenuti su un regime dietetico di auto-selezione con fonti separate di proteine, grassi e carboidrati.(26) Durante i 27 giorni di questo studio, la Clomipramina non ha alterato il consumo alimentare dalle fonti proteiche e lipidiche, ma ha ridotto l’apporto energetico a seguito di una diminuzione nell’assunzione dalla fonte glucidica cosa che si è riflessa positivamente sul peso corporeo.(26)
Imipramina
L’Imipramina non mostra lo stesso impatto nel causare l’aumento del peso corporeo rispetto all’Amitriptilina e alla Clomipramina. Un aumento di peso non significativo è stato il risultato della maggior parte degli studi svolti (27-29). Pesi medi di 2,1 ± 1,5 kg dopo 6 settimane e 4 ± 1,4 kg dopo 4-6 mesi sono stati registrati nei pazienti che assumevano Imipramina in uno studio controllato.(12) In uno studio sperimentale basato sugli animali, l’aumento di peso è stato raggiunto e persino protratto nei ratti dopo l’interruzione della somministrazione della Imipramina.(30)
Desipramina
Il cinquanta per cento dei pazienti trattati con Desipramina, in uno studio clinico durato 10 anni, hanno guadagnato peso.(18) topi Sabra femmina hanno mostrato un graduale aumento del loro peso corporeo durante l’assunzione della Desipramina, rispetto al gruppo dei ratti trattati con placebo.(31)
Nortriptilina
Tutti e cinque gli studi clinici disponibili per la Nortriptilina hanno riportato vari gradi di aumento di peso nei pazienti che assumevano questo farmaco.(18,21,32-34) Un gruppo di ricercatori ha esaminato l’efficacia e la tollerabilità della Nortriptilina in 35 bambini e adolescenti affetti da ADHD (Sindrome da deficit di attenzione e iperattività). Una delle loro scoperte durante lo studio in doppio cieco, controllato con placebo , è stata che la Nortriptilina ha causato un aumento di peso di 5,2 libbre (2,35kg) in media nel corso delle nove settimane dello studio.(32) Hinze-Selch et al (21) hanno riscontrato significativi aumenti di peso con Amitriptilina /Nortriptilina nella loro sperimentazione clinica controllata, mentre non hanno segnalato alcun aumento dei livelli plasmatici di Leptina . Nei pazienti geriatrici, la Nortriptilina non ha causato un marcato aumento di peso poiché un significativo aumento di peso (> 10 lb) si è verificato solo nel 17,2% dei pazienti nel periodo di 30 settimane dello studio.(34) Il 24% dei pazienti dello studio ha mostrato una perdita di peso al di sotto del livello premorboso e il 20,7% non ha mostrato alcun cambiamento di peso.(34)
Protriptilina
Una perdita di peso media di 1,75Ib (793g circa) a settimana è stata raggiunta dai pazienti trattati con Protriptilina in uno studio clinico nel quale è stata osservata l’influenze della Protriptilina sui cambiamenti del peso corporeo nei pazienti con basso livello urinario di 3metossi-4-idrossi-fenilglicole.(35)
Maprotilina (antidepressivo tetraciclico)
Come risultato di uno studio clinico controllato nel quale si sono osservati gli effetti di diversi antidepressivi sulle variazioni del peso corporeo, la Maprotilina e l’Amitriptilina hanno indotto un aumento di peso più marcato mentre gli antidepressivi Imipramina e non triciclici hanno indotto un aumento di peso minore. (12) I pazienti trattati con Maprotilina hanno subito aumenti di peso di 3,2 ± 2,6 kg dopo 6 settimane e 5,2 ± 4,1 kg dopo 4-6 mesi.(12)
Fenelzina e Tranilcipromina
Partendo dall’alto: Fenelzina e Tranilcipromina
Nell’unico studio clinico disponibile attraverso il quale si è osservata l’influenza della Fenelzina sulle variazioni del peso corporeo, ha mostrato che l’aumento di peso è stato raggiunto dal 46% dei pazienti che avevano assunto questo farmaco per dieci anni.(18) Nello stesso studio il Tranilcipromina ha indotto il maggiore aumento di peso ( una media di 4,1 ± 2,2 Kg di aumento di peso nel 73% dei pazienti) rispetto ad altri antidepressivi inclusi Desipramina, Nortriptilina, Amitriptilina e Fenelzina (18).
Moclebomide
L’unico studio disponibile trattante l’effetto del Moclebomide sulle variazioni del peso corporeo ha riportato un aumento di peso non significativo a seguito dell’assunzione di questo farmaco durante il trattamento a breve termine (6 settimane) ed a lungo termine (18 settimane).(19)
Fluoxetina
Esistono risultati contrastanti in letteratura sull’effetto della Fluoxetina sui cambiamenti del peso corporeo. Alcuni ricercatori hanno riportato la perdita di peso in seguito a somministrazione di Fluoxetina (19, 35-38), e alcuni altri hanno osservato un effetto inibitorio sull’assunzione di cibo da parte dei ratti trattati con questo farmaco.(39) Al contrario, altri ricercatori hanno scoperto che la Fluoxetina può sia provocare un aumento di peso (30,40) che non portare alcun cambiamento in questo parametro.(24) Michelson et al (41) hanno anche esaminato l’effetto della Fluoxetina sui cambiamenti di peso corporeo e hanno rilevato sia un aumento e una perdita di peso corporeo, rispettivamente durante le fasi acuta e cronica del trattamento. Durante questo studio clinico durato un anno e nel quale sono stati esaminati 839 pazienti depressi, una modesta perdita di peso si è verificata dopo le 4 settimane iniziali nei pazienti che assumevano Fluoxetina. (41) Nella fase continua e dopo la remissione dei sintomi depressivi tutti i pazienti hanno preso peso e questi aumenti erano simili in entrambi i gruppi, sia in quello trattato con Fluoxetina che nel gruppo Placebo. Pertanto questo può essere dovuto alla remissione e non necessariamente al farmaco in modo diretto.(41) Un altro studio che ha trovato risultati contrastanti per la Fluoxetina è stato effettuato prendendo in considerazione il peso di base dei pazienti depressi.(42) Questo studio ha osservato una perdita di peso nei pazienti in sovrappeso e un aumento del peso corporeo nei pazienti normo peso come risultato del trattamento con Fluoxetina.(42)
Fluvoxamina
In uno studio in doppio cieco, Moon e Jesinger (43) hanno confrontato gli effetti collaterali della Fluvoxamina e della Mianserina in 59 pazienti depressi. Di conseguenza, la Fluvoxamina non ha causato alcun aumento di peso come effetto collaterale mentre la Mianserina ha indotto l’aumento di peso. (43) In un altro studio i cui partecipanti erano 40 donne obese, la Fluvoxamina ha causato una perdita di peso media di 3,1 kg durante 12 settimane. Maggiore, ma non significativamente diverso dal gruppo controllato trattato con placebo.(44) Precisamente, la Fluvoxamina non induce aumenti di peso e può addirittura causare la perdita di peso nelle donne depresse obese.(44)
Paroxetina
La Paroxetina è un altro antidepressivo che ha ottenuto risultati diversi per quanto riguarda la sua influenza sui cambiamenti del peso corporeo. Vi sono due studi clinici randomizzati che riportano aumenti di peso con l’uso di questo farmaco.(37,45) Benkert et al (41) hanno studiato l’efficacia e la tollerabilità della Mirtazapina rispetto alla Paroxetina e hanno scoperto che entrambi i farmaci inducevano aumenti di peso dopo 6 settimane in pazienti depressi e questo effetto era più forte per la Mirtazapina rispetto alla Paroxetina. Cambiamenti nel peso corporeo di 284 pazienti con disturbo depressivo maggiore che sono stati assegnati in modo casuale al trattamento in doppio cieco con Fluoxetina, Sertralina e Paroxetina sono stati osservati in uno studio clinico.(37) Aumenti significativi del peso si sono verificati solo nel gruppo Paroxetina per un totale di 26 e 32 settimane.(37) Al contrario, la Paroxetina non è stata osservata indurre alcun aumento di peso statisticamente significativo nei pazienti depressi durante uno studio retrospettivo comparativo che ha utilizzato registri clinici di studi in doppio cieco.(19) Un altro studio che ha concluso che la Paroxetina non influenza il peso corporeo è quello svolto da Hinze-Selch et al.(21) In questo studio hanno osservato gli effetti di diversi antidepressivi sul peso corporeo, i livelli della Leptina plasmatica, i recettori TNF-a e solubili del TNF. Gli autori hanno riportato che la Paroxetina non ha apportato alcuna modifica nei fattori citati, compreso il peso corporeo, simile agli effetti del trattamento senza farmaci, ma contrario al trattamento con TCA che ha causato aumenti del peso corporeo e dei livelli di Leptina nel plasma. In un altro studio si suggerisce che la Paroxetina inibisca significativamente l’assunzione di cibo nei ratti.(39)
Citalopram
Wade et al hanno condotto due studi di 12 mesi: (i) uno studio randomizzato in doppio cieco nel quale si è usato placebo, Citalopram e Clomipmmina su 279 pazienti con disturbo di panico e (ii) uno studio aperto con Citalopram su 541 pazienti depressi. Nel primo studio i pazienti con disturbo di panico trattati con Citalopram non si sono avvicinati ad un aumento di peso statisticamente significativo e nel trial dove i pazienti depressi hanno assunto il Citalopram esso o non ha causato alcun aumento di peso o a portato ad un leggero aumento di peso (<2,5 kg) nella maggioranza dei pazienti.(25) Gli autori hanno suggerito che i minimi aumenti di peso osservati nei pazienti depressi possono essere il risultato di un aumento dell’appetito legato al miglioramento della loro condizione.(25) Il Citalopram è stato anche causa di un aumento del peso non significativo in un altro studio clinico.(46) Di 18 pazienti che sono stati esaminati per valutare il desiderio di carboidrati causato dal trattamento con Citalopram (SSRI), in una clinica per i disturbi dell’umore, otto soggetti hanno mostrato un aumento significativo della voglia di carboidrati insieme all’aumento di peso subito dopo l’inizio del trattamento.(47)
Escitalopram
Durante uno studio clinico in doppio cieco della durata di otto mesi i pazienti trattati con Escitalopram hanno subito un aumento di peso medio di 1,38 kg, che era superiore all’aumento di peso nei pazienti trattati con Duloxetina.(48) Un aumento di peso non significativo di 0,14 kg in 6 mesi è stato il risultato di un altro studio aperto randomizzato.(33)
Sertralina
La Sertralina può indurre un modesto aumento ponderale nei pazienti depressi.(37) I pazienti obesi possono perdere peso durante l’assunzione di Sertralina in particolare quelli con MHPG urinario basso (3-metossi-4-idrossifenilglicole).(49) Meyerowitz e Jaramillo (1994) in uno studio clinico per valutare l’effetto della Sertralina sul peso corporeo di 23 pazienti depressi e sovrappeso hanno effettuato la misurazione delle loro concentrazioni urinarie di 3-metossi-4-idrossifenilglicole (MHPG). I risultati hanno mostrato che la Sertralina può causare la perdita di peso e con una media di 1,06 libbre/settimana (circa 480g) in pazienti con bassi livelli urinari di MHPG che è risultata significativamente superiore alla perdita media di peso di 0,42 lb/settimana (circa 190g) riscontrata nei pazienti con alti livelli urinari di MHPG.(49)
Zimelidina
L’unico studio disponibile ha riportato che la Zimelidina non ha causato alcun aumento di peso e, in molti casi, ha portato ad una perdita di peso con un cambiamento medio del peso di 0,2 ± 1,8 kg durante un mese di somministrazione di questo antidepressivo in uno studio clinico randomizzato. (50)
Duloxetina
Durante uno studio controllato in doppio cieco, la Duloxetina ha avuto un’incidenza significativamente maggiore nell’aumento anormale del peso durante il trattamento (aumento del 7% del peso rispetto al basale) rispetto al placebo.(48) Durante 10 studi clinici controllati l’effetto della Duloxetina sul peso corporeo dei pazienti con disturbo depressivo maggiore è stato analizzato. (51) I risultati di questi studi non hanno indicato alcun effetto coerente dato dalla Duloxetina sul peso poiché i pazienti trattati con questo farmaco hanno subito una modesta perdita di peso in fase acuta a seguito di un modesto aumento ponderale durante trattamenti più lunghi. (51)
Mianserina
In uno studio in doppio cieco di sei settimane, Moon e Jesinger (43) hanno valutato l’efficacia della Mianserina e della Fluvoxamina in pazienti affetti da episodio depressivo maggiore. La Mianserina ha influenzato la compliance a causa dell’aumento di peso per un periodo più lungo.
Mirtazapina
La Mirtazapina ha mostrato di causare aumenti di peso in diversi studi svolti sull’uomo. (20,45,46,52) Durante uno studio in doppio cieco placebo-controllato svolto su pazienti adulti con disturbo depressivo maggiore, l’efficacia della Mirtazapina è stata confrontata con l’Amitriptilina. Uno dei risultati di questo studio ha evidenziato che l’aumento di peso misurato era più frequente con l’uso di Amitriptilina (22% dei pazienti) rispetto a quanto osservato con l’uso di Mirtazapina (13% dei pazienti).(20) Un altro confronto è stato fatto tra l’efficacia e la tollerabilità della Mirtazapina rispetto alla Paroxetina su 275 pazienti ambulatoriali depressi. I pazienti sono stati assegnati in modo casuale a 6 settimane di trattamento con Mirtazapina o Paroxetina. Il risultato relativo all’aumento di peso ha mostrato un aumento di peso maggiore nel gruppo trattato con Mirtazapina rispetto al gruppo trattato con Paroxetina.(45) Uno studio multicentrico randomizzato in doppio cieco con l’obbiettivo di confrontare l’efficacia e la tollerabilità della Mirtazapina e del Citalopram in pazienti depressi, ha osservato un aumento dell’appetito e del peso corporeo significativo nei pazienti trattati con Mirtazapina (rispettivamente del 8,8% e del 15,3%) rispetto ai pazienti trattati con Citalopram (1,5% e 4,5%).(46) Laimer et al hanno studiato l’influenza del trattamento con Mirtazapina sul peso corporeo, sulla massa grassa, sul metabolismo del glucosio, sul profilo lipoproteico e sulla Leptina mettendo a confronto due gruppi di donne di cui uno era composto da sette donne depresse mentre l’altro era composto da sette donne volontarie mentalmente e fisicamente sane (gruppo di controllo). I risultati hanno confermato che la Mirtazapina causa aumenti significativi del peso corporeo (da una media di 63,6 ± 13,1 kg a un peso corporeo medio di 66,6 ± 11,9 kg), della massa grassa e delle concentrazioni di Leptina nei pazienti trattati.(52) Contrariamente agli studi svolti sull’uomo non ci sono stati collegamenti diretti tra lo sviluppo dell’obesità e la Mirtazapina secondo lo studio controllato sugli animali condotto da Jeon, Joe e Kee (24) i qiali hanno usato i ratti OLETF (Otsuka Long-Evans Tokushima Fatty) per la loro ricerca.
Bupropione
Tre studi clinici randomizzati hanno preso in esame gli effetti del Bupropione sulle variazioni del peso corporeo. In tutti questi studi, i ricercatori hanno concluso che il Bupropione può indurre la perdita di peso nei pazienti trattati. (53-55) In uno studio randomizzato in doppio cieco, la media della perdita di peso è risultata modesta con trattamento a base di Bupropione SR a lungo termine in pazienti con depressione aumentata in risposta all’aumento del peso corporeo al basale.(53) Un altro studio randomizzato, in doppio cieco, controllato con placebo ha valutato l’efficacia del Bupropione SR nella riduzione del peso e dei sintomi depressivi in 422 adulti obesi con sintomi depressivi. Dopo 26 settimane, il gruppo Bupropione SR ha perso in media una maggiore quantità di peso (4,4 kg: 4,6% del peso basale) rispetto al placebo (1,7 kg: 1,8% del peso basale).(54) Nel terzo studio in doppio cieco controllato con placebo della durata di 48 settimane ha mostrato che il Bupropione SR in combinazione con un programma di intervento sullo stile di vita era associato a una riduzione del peso corporeo dose correlata.(55)
Trazodone
In uno studio randomizzato in doppio cieco della durata di 6 settimane è stato osservato che il Trazodone causava una leggera perdita di peso nei pazienti sovrappeso.(17)
Discussione conclusiva
Una significativa associazione positiva è stata riportata tra depressione e obesità in modo più marcato tra le donne.(56) L’aumento di peso corporeo è un noto effetto collaterale legato a numerosi farmaci psicotropi, inclusi gli antidepressivi.(57) Si sono riportati a conoscenza del “grande pubblico” dei vari e, spesso, contrastanti effetti dei diversi antidepressivi sulle variazioni del peso corporeo. I TCA e in particolare l’Amitriptilina, come abbiamo visto, sono noti tra gli antidepressivi per causare l’aumento di peso.(18,58,59) E’ stato anche riportato che l’Amitriptilina è stata ripetutamente segnalata come un induttore dell’aumento di peso negli studi clinici. (12,13,16-22) Tutti i TCA sembrano causare un aumento di peso negli studi clinici eccetto la Protriptilina che ha causato una perdita di peso quando somministrata a pazienti con depressione maggiore.(35) Si pensava che gli SSRI inducessero la perdita di peso piuttosto che l’aumento del peso in contrasto con i TCA.(58) Come osservabile tra i risultati, la maggior parte degli studi riporta una perdita di peso nei pazienti che assumevano la Fluoxetina, tuttavia in alcuni studi è stato riscontrato un aumento di peso con l’uso di questo farmaco. Gli stessi risultati incoerenti si applicano alla Paroxetina.
L’aumento di peso è stato anche osservato negli studi nei quali si sono presi in esame altri SSRI tra cui il Citalopram e l’Ecitalopram. Le ricerche disponibili per gli IMAO hanno riportato un aumento di peso correlato alla Fenelzina e alla Tranilcipromina, ma non è stato osservato alcun cambiamento significativo di peso con il Moclebomide. Il Bupropione è l’unico antidepressivo del quale tutti gli studi confermano l’induzione della perdita di peso in pazienti depressi trattati con questo farmaco.
Alcuni studi hanno suggerito che l’aumento di peso può essere almeno in parte legato alla remissione dalla depressione stessa e non necessariamente causato dai farmaci antidepressivi. (25, 60) A sostegno di questa ipotesi vi è la comune diminuzione dell’appetito durante gli stati depressivi. Tuttavia ci sono prove che gli antidepressivi causano un aumento di peso in pazienti con altri disturbi psichiatrici come ADHD (32) ed emicrania (22).
Anche la durata del trattamento è un fattore importante che può influire sui risultati. Un discreto numero di studi ha riportato differenze tra gli effetti a breve e a lungo termine degli antidepressivi sulle variazioni del peso corporeo. Michelson et al (41) hanno riportato la perdita di peso nei pazienti trattati con Fluoxetina durante la fase acuta (4 settimane) e un aumento di peso negli stessi pazienti durante la fase continua. Un altro studio comprendente 10 trial clinici che hanno esaminato gli effetti della Doluxetina ha riscontrato sia una perdita che un aumento di peso rispettivamente nella fase acuta e cronica, ma in tassi modesti e, quindi, non significativi. La categorizzazione delle fasi acute, croniche, di breve durata, a lungo termine, non è la stessa tra i diversi studi considerando gli effetti dipendenti dal tempo d’assunzione degli antidepressivi e aggiunge maggiori difficoltà nell’interpretazione dei risultati quando si vuole avere un punto di vista generale sulla questione.
L’età è un altro fattore che sembra influenzare la relazione tra farmaci antidepressivi e aumento del peso corporeo. Come osservato nello studio condotto da Corman et al. (34), la Nortriptilina non ha causato un marcato aumento di peso nei pazienti geriatrici e ha causato anche una perdita di peso in alcuni dei soggetti trattati. Contrariamente a ciò, nei pazienti non anziani il trattamento con Nortriptilina ha causato un marcato aumento di peso.(18,21,32)
Il peso basale dei pazienti trattati sembra essere un altro fattore di una certa importanza nel risultato delle variazioni di peso dovute all’uso dei farmaci antidepressivi. Ciò è stato confermato in uno studio clinico condotto da Orzack et al (42) che ha rilevato un aumento di peso tra i pazienti normopeso e una perdita di peso nei pazienti sovrappeso quando trattati con Fluoxetina.
I risultati incoerenti per gli effetti degli antidepressivi sui cambiamenti del peso corporeo che sono stati riportati nella letteratura disponibile possono essere riconducibili al concomitante uso di un farmaco psicotropico da parte dei pazienti psichiatrici co-morbosi. Questo fenomeno può anche essere dovuto a diversi fattori, come le dimensioni del campione e la durata degli studi, o come la sensibilità genetica all’azione del farmaco.
Come ormai risaputo, l’obesità e il sovrappeso possono portare a gravi problemi di salute come lo sviluppo di malattie cardiovascolari. Inoltre, l’aumento di peso causato dai farmaci antidepressivi è una delle principali ragioni per la non conformità dei pazienti con il trattamento e lo scarso esito dello stesso; la lotta all’aumento di peso una volta che si è verificato può essere molto difficile. (57) Quindi comprendere i meccanismi sottostanti che contribuiscono all’effetto degli antidepressivi sui cambiamenti del peso corporeo sono importanti.
I risultati degli studi svolti su animali, come di consueto, non sono sempre coerenti con quelli clinici. Per esempio l’Amitriptilina e la Mirtazapina non hanno avuto alcuna associazione diretta con l’obesità nei ratti e infatti è stato suggerito da Jeon et al (24) che questi due antidepressivi possono regolare i livelli circolanti di Adiponectina e i recettori dell’Adiponectina. L’Amitriptilina è risultata essere anche inefficace nell’aumentare l’assunzione giornaliera di cibo e il peso corporeo dei ratti in una serie di studi sperimentali nonostante l’applicazione di vari dosaggi, vie di somministrazione, composizione della dieta e appetibilità (23). La Clomipramina è stata somministrata cronicamente a ratti wistar maschi esposti a procedura di autoselezione dei macronutrienti portando ad una riduzione del consumo di cibo e dell’aumento del peso corporeo (26) mentre, similmente a quanto osservato con la Amitriptilina e Mirtazapina, la Clomipramina è stata osservata indurre un aumento di peso negli studi clinici. (12,25) Interpretare i risultati degli studi sugli animali per l’induzione dell’ obesità in seguito a somministrazione di farmaci antidepressivi è una sfida anche quando si considerano le situazioni di vita reale per l’uomo. Mastronardi et al (30) hanno tentato di simulare una situazione simile a quella di pazienti con stress/depressione ed esposizione a farmaci antidepressivi a breve termine con un consumo di una dieta ricca di grassi nei ratti. Per ottenere questo tipo di esperimento, i ratti sono stati sottoposti a ripetuti stress (RRS) e al farmaco antidepressivo nel breve termine dopo un lungo periodo di esposizione a una dieta ricca di grassi (30). I risultati hanno mostrato effetti di aumento di peso anche dopo la sospensione degli antidepressivi Imipramina e Fluoxetina nei ratti che hanno tollerato il fenomeno di sensibilizzazione tempo-dipendente. (30) In un altro studio sperimentale su base animale la Desipramina ha provocato da prima la perdita di peso nei ratti e, successivamente, a portato all’aumento di peso nelle fasi continue di trattamento della durata di oltre 3 mesi.(31)
È noto che molte sostanze regolatrici influenzano l’appetito, inclusi neurotrasmettitori come la Noradrenalina, il 5HT, i Neuropeptidi come la Colecistochinina, l’Ormone di Rilascio della Corticotropina, il neuropeptide Y, gli oppioidi e altri peptidi ormone-simili come l’Enterostatina, la Bombesina, l’Amilina e Leptina (59 ). È stato riportato che i TCA sono associati con l’aumento di peso a causa della loro azione antagonizzante sui recettori H1 protratta nel tempo. (61) È stato suggerito che l’induzione dell’aumento di peso osservata con l’uso del Citalopram possa anche essere dovuta alla sua elevata affinità con i recettori H1. Alcuni ricercatori hanno tentato di individuare il ruolo della Leptina nell’associazione tra antidepressivi e obesità.(21, 22) Berilgen et al (22) hanno scoperto che l’Amitriptilina può causare resistenza alla Leptina attraverso meccanismi diversi determinando quindi un aumento dei livelli serici di Leptina e del BMI. Al contrario, in un altro studio l’Amitriptilina non ha causato alcun aumento dei livelli di Leptina mentre induceva un aumento di peso.(21) I meccanismi sottostanti all’aumento di peso dipendente dall’assunzione di farmaci antidepressivi non sono ancora ben compresi e sono necessari ulteriori studi al fine di indagare il ruolo dei neurotrasmettitori e di altri possibili fattori che contribuiscono al induzione dell’aumento di peso causato dall’uso di farmaci antidepressivi. Le aree di controversia, come gli ovvi risultati opposti tra gli studi svolti su animali e gli studi svolti su esseri umani, o i diversi effetti dipendenti dal tempo di esposizione agli antidepressivi sui cambiamenti del peso corporeo, dovranno essere affrontati in studi futuri. Indagini continue e più precise sul fattore tempo-dipendente nell’influenza degli antidepressivi sull’aumento del peso corporeo, come è già stato fatto per gli antipsicotici (61), potrà aiutare a chiarire il ruolo della durata del trattamento con questa classe di farmaci nell’induzione di tale fenomeno. Indagare sugli effetti di altri antidepressivi che non sono stati inclusi negli studi precedenti come la Doxepina, la Trimipramina, la Venlafaxina, il Nefazodone e la Amoxapina è un altro problema che dovrà essere considerato attraverso pertinenti studi futuri riguardanti l’associazione dei farmaci antidepressivi e il cambiamento del peso corporeo.
Gabriel Bellizzi
Riferimenti:
*The Association of Antidepressant Medication and Body Weight Gain. Authors Sara Ranjbar, Faculty of Health and Behavioral Sciences, University of Wollongong, New South Wales, Australia 2522, Nagesh B. Pai, Graduate School of Medicine, and Illawarra Health and Medical Research Institute, University of Wollongong, NSW 2522, Australia, Chao Deng, School of Health Sciences, and Illawarra Health and Medical Research Institute, University of Wollongong, NSW 2522, Australia. Ranjbar S, Pai NB, Deng C. The Association of Antidepressant Medication and Body Weight Gain. Online J Health Allied Scs. 2013;12(1):1. Available at URL: http://www.ojhas.org/issue45/2013-1-1.html
Nihalani N, Schwartz TL, Siddiqui UA, Megna, JL. Obesity and psychotropics, CNS neuroscience & therapeutics. 2012;18;57–63.
Wild B, Herzog W, Lechner S, Niehoff D, Brenner H, Müller H, Rothenbacher D, Stegmaier C, Raum E. Gender specific temporal and cross-sectional associations between BMI-class and symptoms of depression in the elderly. Journal of Psychosomatic Research. 2012;72:376-382.
Kivimäki M, Batty D, Singh-Manoux A, Nabi H, Sabia S, Tabak A, Tasnime N, Akbaraly T, Vahtera J, Marmot M, Jokela M. Association between common mental disorder and obesity over the adult life course. The British journal of psychiatry: the Journal of mental science. 2009;195:149–155.
Simon G, Von Korff M, Saunders K, Miglioretti D, Crane P, Belle G, Kessler R. Association Between Obesity and Psychiatric Disorders in the US Adult Population. Archives of General Psychiatry. 2006;63:824–830.
Roberts RE et al. Prospective association between obesity and depression: evidence from the Alameda County Study. International journal of obesity and related metabolic disorders. Journal of the International Association for the Study of Obesity. 2003;27(4):514.
McCarty CA, Kosterman R, Mason W, McCauley E, Hawkins D, Herrenkohl T, Lengua L. Longitudinal associations among depression, obesity and alcohol use disorders in young adulthood, General Hospital Psychiatry. 2009;31:442–450.
Ball K, Burton N, Brown W. A Prospective Study of Overweight, Physical Activity, and Depressive Symptoms in Young Women. Obesity. 2008;17:66-71.
Heo M, Pietrobelli A, Fontaine KR, Sirey JA, Faith MS. Depressive mood and obesity in US adults: comparison and moderation by sex, age, and race. International Journal of Obesity. 2006;30:513–519.
Afari N, Noonan C, Goldberg J, Roy-Byrne P, Schur E, Golnari G, Buchwald D. Depression and Obesity: Do Shared Genes Explain the Relationship? Depression And Anxiety. 2010;27:799-806.
Kazes M, Danion JM, Grange D, et al. Eating behaviour and depression before and after antidepressant treatment: a prospective, naturalistic study. J Affect disord. 1994;30:193207.
Fernstrom MH, Kupfer DJ. Antidepressant-induced weight gain: a comparison study of four medications. Psychiatry Res. 1998;26:265–271.
Schatzberg A, Cole J, DeBattista C. Manual of Clinical Psychopharmacology [Internet]. 7th Edition, American Psychiatric Publishing, Inc. 1000 Wilson Boulevard, 2012 [cited 2012 Sep 12]. Available from: http://psychiatryonline.org.ezproxy.uow.edu.au/content.a spx?bookid=2§ionid=1359932
Katzung BG, Masters SB, Trevor AJ. Basic & Clinical Pharmacology, McGraw Hill Companies, 12th ed, 2012.
Paykel ES, Mueller PS, De La Vergne PM. Amitriptyline, weight gain and carbohydrate craving: A side effect. Br J Psychiatry. 1973;123:501-507.
Hecht-Orzack M, Cole JO, Friedman L, Bird M, McEachern J. Weight changes in antidepressants: a comparison of amitriptyline and trazodone. Neuropsychobiology. 1986;15:2830.
Joubert AT, Gagiano CA, Joubert G. Antidepressants and weight. Behavioural Pharmacology. 1995;6:30.
Montgomery SA, Reimitz PE, Zivkoz M. Mirtazapine versus amitriptyline in long-term treatment of depression: a doubleblind placebo-controlled study.IntclinPsychopharmacol. 1998;13:63-73.
Hinze-Selch D, Schuld A, Kraus T, Kuhn M, Uhr M, Haack M, Pollmacher T. Effects of antidepressants on weight and on the plasma levels of leptin, TNF-alpha and soluble TNF receptors: a longitudinal study in patients treated with amitriptyline or paroxetine. Neuropsychopharmacology. 2000;23:13–9.
Berilgen MS, Bulut S, Gonen M, Tekatas A, Dag E, Mungen, B. Comparison of the effects of amitriptyline and flunarizine on weight gain and serum leptin, C peptide and insulin levels when used as migraine preventive treatment. Cephalalgia. 2005:25:1048.
Nobrega J, Coscinaa D. Effects of chronic amitriptyline and desipramine on food intake and body weight in rats. Pharmacology Biochemistry and Behavior. 1987;27:105– 112.
Jeon HT, Joe GH, Kee BS. The effects of antidepressants on the weight, blood glucose, leptin, and adiponectin in diabetic (OLETF) and non diabetic (LETO) rats,European Neuropsychopharmacology. 2008;18:S349-S349.
Wade A, Overo KF, Lemming O, et al. Weight monitoring during two long-term trials of citalopram. Presented at the 12th Congress of the European College of Neuropsychopharmacology; London, England, 1999.
Calegari L, Gorenstein C, Gentil V, Planeta CS, Nunes-deSouza R L. Effect of Chronic Treatment with Clomipramine on Food Intake, Macronutrient Selection and Body Weight Gain in Rats, Biological & Pharmaceutical Bulletin. 2007;30:15411546.
Fernstrom MH, Krowinski RL, Kupfer DJ. Chronic imipramine treatment and weight gain, Psychol Res. 1986;17:269–273.
Frank E, Kupfer DJ, Bulik CM, Levenson JA. Imipramine and weight gain during the treatment of recurrent depression. J Affect Disord. 1990;20:165–172.
Balon R, Yeragani V K, Pohl R, et al. Changes in appetite and weight during the pharmacological treatment of patients with panic disorder, Can J Psychiatry. 1993;28:19-22.
Mastronardi C, Paz-Filho GJ, Valdez E, Maestre-Mesa J, Licinio J, Wong M-L. Long-term body weight outcomes of antidepressant-environment interactions, Molecular Psychiatry. 2011;16:265–272.
Gobshtis N, Ben-Shabat S, Fride E. Antidepressant-induced undesirable weight gain: prevention with rimonabant without interference with behavioral effectiveness.European journal of Pharmacology. 2006;554:155–163.
Prince JB, Wilens TE, Biederman JA. A controlled study of nortriptyline in children and adolescents with attention deficit hyperactivity disorder. Journal of Child and Adolescent Psychopharmacology. 2000;10:193–204.
Uher R, Mors O, Hauser J, Rietschel M, Maier W, Kozel D, Henigsberg N, Souery D, Placentino A, Keers R, Gray JM, Dernovsek MZ, Strohmaier J, Larsen ER, et al. Changes in body weight during pharmacological treatment of depression. The International Journal of Neuropsychopharmacology. 2011;14:367-375.
Paradis CF et al. Nortriptyline and weight change in depressed patients over 60. Journal of clinical psychopharmacology. 1992;12(4):246-250.
Meyerowitz W, Jaramillo J. Antidepressant treatment and weight loss, Current Therapeutic Research. 1992;52:169–174.
Daubresse JC, Kolanowski J, Krzentowski G, et al. Usefulness of fluoxetine in obese non-insulin-dependent diabetics: a multicenter study, Obes res. 1996;4:391-396.
Fava M, Judge R, Hoog SL, Nilsson ME, Koke SC. Fluoxetine vs sertraline and paroxetine in major depressive disorder: changes in weight with long-term treatment. J Clin Psychiatry. 2000;61:863–867.
Afkhami-Ardekani M, Sedghi H. Effect of fluoxetine on weight reduction in obese patients. Indian Journal of Clinical Biochemistry. 2005;20:135–138.
Rasmussen J, Johnson A, Stewart B, Palmer K. Paroxetine and fluoxetine on food intake in rats and effect of paroxetine on body weight in depressed patients. European Neuropsychopharmacology. 1991;1:443–444.
Folgelson DL. Weight gain during fluoxetine treatment. Journal of Clinical Psychopharmacology. 1991;11:220.
Michelson D, Amsterdam JD, Quitkin FM, et al. Changes in weight during a 1-year trial of fluoxetine. Am J Psychiatry. 1999;156:1170–1176.
Orzack MH, Friedman LM. Weight changes on fluoxetine as a function of baseline weight in depressed patients. Psychopharmacol Bull. 1990;26:327–330.
Moon CA, Jesinger DK. The effects of psychomotor performance of fluvoxamine versus mianserin in depressed patients in general practice. Brit J Clin Prac. 1991;45:259–262.
Abell CA, Farquhar DL, Galloway SM, Steven F, Philip AE, Munro JF. Placebo controlled double-blind trial of fluvoxamine maleate in the obese. Journal of Psychosomatic Research. 1986;30:143–146.
Leinonen E, Skarstein J, Behnke K. Efficacy and tolerability of mirtazapine versus citalopram: A double blind, randomized study in patients with major depressive disorder, Nordic antidepressant study group. Int Clin Psychopharm. 1999;14:329–337.
Bouwer CD, Harvey BH. Phasic craving for carbohydrate observed with citalopram. Int Clin Psychopharmacol. 1999;11:273–278.
Pigott TA, Prakash A, Arnold LM, Aaronson ST, Mallinckrodt CH, Wohlreich MM. Duloxetine versus escitalopram and placebo: An 8-month, double-blind trial in patients with major depressive disorder. Current Medical Research and Opinion. 2007;23:1303–1318.
Meyerowitz W, Jaramillo J. Sertraline treatment and weight loss. Current Therapeutic Research. 1994;55:1176–1181.
Wise TN et al. Effects of the antidepressant duloxetine on body weight: analyses of 10 clinical studies. Primary care companion to the Journal of clinical psychiatry. 2006;8(5):269278.
Benkert O, Szegedi A, Kohnen R. Mirtazapine compared with paroxetine in major depression. J Clin Psychiatry. 2000;61(9):656-663.
Laimer M, Kramer-Reinstadler K, Rauchenzauner M, LechnerSchoner T, Strauss R, Engl J, Deisenhammer EA, Hinterhuber H, Patsch JR, Ebenbichler CF. Effect of mirtazapine treatment on body composition and metabolism. J Clin Psychiatry. 2006;67:421-424.
Croft H, Houser TL, Jamerson BD, Leadbetter R, BoldenWatson C, Donahue R, Metz A. Effect on body weight of bupropion sustained-release in patients with major depression treated for 52 weeks. Clinical Therapeutics. 2002;24:662-672.
Jain AK, Kaplan RA, Gadde KM et al. Bupropion SR vs. placebo for weight loss in obese patients with depressive symptoms. Obes Res. 2002;10:1049–1056.
Anderson JW, Greenway FL, Fujioka K, Gadde KM, McKenney J, O’Neil PM. Bupropion SR Enhances Weight Loss: A 48-Week Double-Blind, Placebo- Controlled Trial.Obesity Research. 2002;10:633–641.
de Wita L, Luppino F, van Straten A, Penninx B, Zitman F, Cuijpers P. Depression and obesity: A meta-analysis of community-based studies. Psychiatry Research. 2010;178:230– 235.
Rashmi D, Kathleen F. Managing weight gain as a side effect of antidepressant therapy. Cleveland Clinic Journal of Medicine. 2003;70(7):614.
Zimmermann U, Kraus T, Himerich H, et al. Epidemiology, implications and mechanisms underlying drug induced weight gain in psychiatric patients. J Psychiatr Res.2003;37:193-220.
Masand PS. Weight gain associated with psychotropic drugs. Review. Expert Opinion on Pharmacotherapy. 2000;1:7–389.
Benazzi F. Weight gain in depression remitted with antidepressants: pharmacological or recovery effect? PsychotherPsychosom. 1998;67:271–274.
Harvey BH, Bouwer CD. Neuropharmacology of paradoxic weight gain with selective serotonin reuptake inhibitors. Clin Neuropharm. 2000;23:90–97.
Pai N, Deng C, Vella S-L, Castle D, Huange X-F. Are there different neural mechanisms responsible for three stages of weight gain development in anti-psychotic therapy: Temporally based hypothesis. Asian Journal of Psychiatry. 2012; in press.
La Luteolina, un flavonoide contenuto nel rosmarino, timo, prezzemolo e negli agrumi, sembra essere un efficace composto anti-aromatase, almeno secondo uno studio svolto su animali e pubblicato da ricercatori del Centro di Scienze Sanitarie dell’Università di Pechino nel Journal of Pharmacology and Experimental Therapeutics.(1) A differenza dei composti anti-aromatasi sintetici, la Luteolina sembra avere il potenziale di migliorare anche l’equilibrio del Colesterolo.
Già nel 2013, in uno studio in vitro svolto da farmacologi dell’Istituto di biologia di Chengdu, dove si erano prese in esame oltre 100 sostanze al fine di valutarne il potenziale antiestrogenico, la Luteolina risultò la più interessante a tal fine.(2)
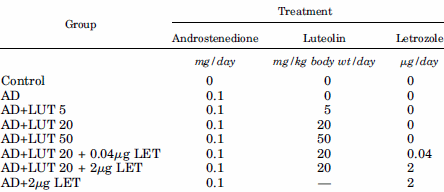
Nello studio del quale ho introdotto brevemente i risultati emersi all’inizio di questo articolo, i ricercatori hanno usato topi di sesso femminile le cui ovaie sono state rimosse chirurgicamente come pratica preliminare per lo svolgimento dell’esperimento. Ad un certo numero di animali sono state somministrate iniezioni giornaliere di Androstenedione [AD]. Come ben sappiamo, l’enzima aromatasi converte l’Androstenedione in Estrone e, attraverso l’azione dell’Estradiolo 17beta-deidrogenasi, in Estradiolo.
Ad alcuni animali è stata anche somministrata per via orale la Luteolina [LUT]. Le dosi utilizzate sono mostrate nella figura riportata di seguito. L’equivalente umano delle dosi utilizzate varia dai 45mg ai 450mg di Luteolina al giorno. Alcuni topi sono stati trattati con iniezioni di Letrozolo [LET], conosciutissimo e potente inibitore dell’aromatasi non steroideo di terza generazione.
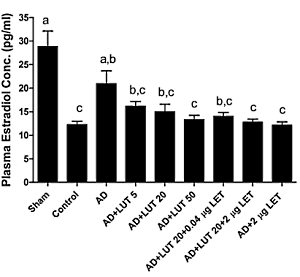
Dopo 12 settimane, i ricercatori hanno misurato le concentrazioni di Estradiolo nel sangue dei topi. La figura seguente mostra che l’effetto anti-aromatasi dato dalla dose più alta di Luteolina era uguale a quello ottenuto con l’uso del Letrozolo.
I ricercatori avevano impiantato nei topi cellule di cancro al seno estradiolo-sensibili. Sia la Luteolina che il Letrozolo hanno inibito la crescita tumorale, ma il Letrozolo ha ottenuto risultati leggermente migliori rispetto alla Luteolina.
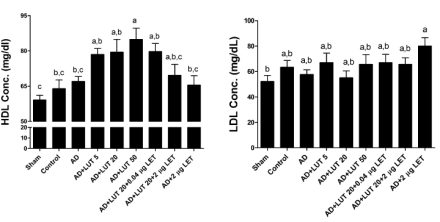
L’uso degli inibitori dell’aromatasi ha tra i suoi effetti collaterali quello di poter causare uno squilibrio delle lipoproteine. Le concentrazioni di LDL aumentano mentre quelle di HDL diminuiscono. La Luteolina, pur agendo attraverso l’inibizione dell’enzima aromatasi, mostra l’effetto opposto sull’equilibrio del Colesterolo. La Luteolina mostra di causare un abbassamento delle concentrazioni di LDL e aumento delle concentrazioni di HDL.
La figura sopra riportata mostra che la Luteolina può anche annullare l’effetto negativo del Letrozolo sui livelli di Colesterolo.
La Luteolina non ha avuto alcun effetto sui livelli di Trigliceridi.
I ricercatori sottolineano il fatto che, sebbene la Luteolina sia ampiamente presenti in diversi alimenti vegetali, i dosaggi utilizzati nello studio qui riportato erano superiori ai livelli normalmente consumati dagli esseri umani. Tuttavia, questo studio potrebbe fornire le basi scientifiche per lo sviluppo nutraceutico o farmacologico di questo flavone.
In fine, i ricercatori concludono che, dato il potenziale di alterazione del rapporto LDL/HDL solitamente associato all’uso a lungo termine degli inibitori dell’aromatasi, la somministrazione di Luteolina può essere un potenziale meccanismo di compensazione senza compromissioni sull’aromatasi.
L’argomento “Cardio a digiuno” è stato da me già trattato in passato, ma in modo condizionato da una certa letteratura di parte. Quindi, è mia intenzione riproporlo in chiave più “neutra” (o, meglio, oggettiva) servendomi della letteratura scientifica oggi disponibile.
I miti nel mondo del BodyBuilding, in particolare, e del Fitness, in generale, sono difficili da debellare. Nonostante le numerose prove a loro discredito continuano a persistere diffondendosi negli spogliatoi delle palestre e nei forum in rete. Ad esempio, nonostante le innumerevoli prove schiaccianti contro l’ipotesi secondo la quale i carboidrati e l’Insulina facciano ingrassare, e nonostante il tasso con il quale tali prove continuano ad accumularsi, molta gente rimane convinta della veridicità di questa credenza.
La pratica del Cardio a digiuno al fine di migliorare la perdita di grasso fa parte di queste credenze che circolano nel mondo del BodyBuilding e del Fitness, nonostante le prove contro tale mito si siano accumulate nel corso del tempo. Sebbene in questo specifico caso le prove contro le affermazioni di presunti vantaggi dati da tale pratica non siano così schiaccianti come per la prima citata ipotesi su carboidrati e Insulina, non esiste alcuna prova che mostri un reale vantaggio nella sua applicazione.
Eppure, molte persone giurano che il cardio a digiuno sia una pratica che apporta reali vantaggi. Il fatto è che non solo non esistono prove che supportano tale affermazione, non ha nemmeno senso applicare tale pratica quando si esamina attentamente il suo reale impatto. Quello che riporterò in seguito mette in chiaro come non vi sia un meccanismo basato sull’evidenza che dimostri che il Cardio a digiuno possa plausibilmente migliorare la perdita di grasso. Nel web sono presenti molti articoli trattanti la questione in modo critico, ma molti di questi non entrano nel dettaglio dei motivi meccanicistici del perché il Cardio a digiuno non porta i vantaggi attribuibili da una certa “credenza da spogliatoio”.
Una semplicistica esposizione del “come perdiamo grasso”
Per illustrare ciò di cui si sta parlando, è necessario come prima cosa parlare di come perdiamo il grasso corporeo. Perdiamo grasso corporeo, semplicisticamente parlando, quando creiamo un deficit energetico, cioè quando la nostra spesa energetica è inferiore rispetto a quella assunta attraverso il cibo. Dal momento che non viene fornita una sufficiente quantità di energia attraverso le fonti alimentari il corpo soddisferà tale mancanza ricavando l’energia necessaria da altre fonti. In questo caso, l’energia verrà ricavata dalle riserve adipose e dagli aminoacidi. Ora, ovviamente, si desidera che tale deficit energetico venga compensato unicamente a carico del grasso di deposito mantenendo intatte le proteine strutturali (vedi massa muscolare), ma, ovviamente, non è così che funzionano le cose, anche se possiamo fare in modo, attraverso un adeguata manipolazione alimentare, che il catabolismo muscolare sia di molto ridotto. Comunque, a fini esemplificativi, supponiamo che il 90% di questa energia provenga dal grasso di deposito mentre il 10% derivi dalla massa magra (quindi anche dai muscoli). Ora, supponiamo di creare un deficit energetico di 500Kcal al giorno (3.500Kcal a settimana). Di quelle 500Kcal, 450 proverranno dal grasso corporeo (50g di grassi) mentre le altre 50 proverranno dalla massa magra (12,5g di proteine). Se si prosegue mantenendo questo deficit calorico su base giornaliera, con tutte le limitazioni adattative e loro gestione, si perderà gradualmente grasso corporeo (e anche un po’ di massa magra).
Il Cardio a digiuno migliorerebbe la perdita di grasso se…
Ora, prendiamo come esempio il contesto che ho appena descritto. Si ha un deficit di 500Kcal al giorno (450 saranno ricavate dal grasso corporeo), e tale deficit si è raggiunto tramite una combinazione di dieta e allenamento Cardio. Supponiamo a questo punto che si decida di inserire il Cardio a digiuno convinti del fatto che ciò aumenterà la perdita di grasso. Bene, il Cardio a digiuno per poter aumentare la perdita di grasso dovrebbe agire almeno attraverso uno di questi tre meccanismi:
Aumentare il dispendio energetico.In questo caso, si aumenta il deficit energetico mantenendo inalterata la quantità di cibo consumata. Si può ipotizzare che il deficit passi da 500Kcal al giorno a 600Kcal al giorno. Come ipotetico risultato, si avrà una spesa di 540Kcal derivanti dai grassi (90%) e 60Kcal provenienti dalla massa magra (10%).
Diminuire l’apporto energetico. In questo caso, si aumenta il deficit energetico attraverso la diminuzione dell’assunzione di cibo, ma mantenendo stabile il dispendio energetico dato dall’attività Cardio. In altre parole, il Cardio a digiuno sopprime in qualche modo l’appetito. Adesso si consumano 100Kcal in meno di quanto si farebbe normalmente, portando il deficit energetico da 500Kcal al giorno a 600Kcal al giorno. Ora, come riportato nel punto 1, si avrà una spesa di 540Kcal derivanti dai grassi e 60Kcal derivanti dalla massa magra.
Migliora la perdita di grasso e preserva la massa magra con lo stesso deficit energetico (un effetto di ripartizione dei tessuti). In questo caso, il deficit energetico giornaliero rimane a 500Kcal. Tuttavia, la percentuale di provenienza dell’energia ricavata dal grasso di deposito aumenta a discapito di quella ricavata dalla massa magra. Supponiamo quindi un 95% dell’energia ricavata dai grassi di deposito (475Kcal) e un 5% dalla massa magra (25Kca).
Questi sono gli unici tre scenari ipotetici in cui il Cardio a digiuno potrebbe aumentare la perdita di grasso rispetto al Cardio non a digiuno. La domanda ora è se una qualsiasi delle 3 ipotesi elencate possa effettivamente accadere con il Cardio a digiuno. Fortunatamente, la scienza applicata ci può fornire la risposta.
Il Cardio a digiuno aumenta la spesa energetica?
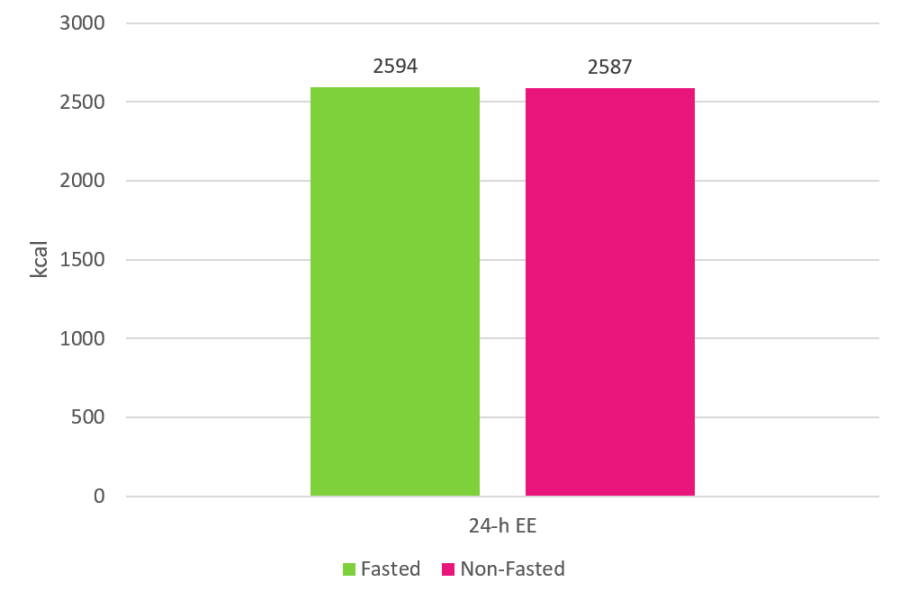
La risposta a questa domanda è no. Quando i ricercatori hanno confrontato le spese energetiche nelle 24 ore di 60 minuti di Cardio a digiuno rispetto a 60 minuti di Cardio non a digiuno, non sono state osservate differenze (se non di un mero scarto di 7Kcal).(1)
Il grafico sopra esposto mostra il dispendio energetico nelle 24 ore per le due condizioni prese in esame nello studio precedentemente citato. Il Cardio a digiuno non ha praticamente aumentato il dispendio energetico, e, quindi, questo non può essere un meccanismo attraverso il quale tale pratica possa migliorare la perdita di grasso. Il punto 1 può essere cancellato dalla lista.
Aumentare il dispendio energetico
Effetto soppressivo sull’appetito
Effetto di ripartizione dei tessuti
Il Cardio a digiuno ha un effetto soppressivo sull’appetito?
La risposta è no. In realtà, esiste uno studio (2) nel quale si è rilevato che la sensazione di appetito era inferiore nei soggetti che praticavano Cardio non a digiuno. Inoltre, l’apporto calorico ad libitum era più basso nelle 24 ore in seguito al Cardio non a digiuno rispetto al Cardio a digiuno.
Anche il punto 2 può essere cancellato dalla lista.
Aumentare il dispendio energetico
Effetto soppressivo sull’appetito
Effetto di ripartizione dei tessuti
Il Cardio a digiuno ha un effetto di ripartizione dei tessuti (aumento della perdita di grasso e risparmio della massa magra per un determinato deficit energetico)?
La ragione principale per cui le persone scelgono di fare Cardio a digiuno è che sono convinte di ossidare più grassi durante l’esercizio rispetto a quello che riuscirebbero a fare con il Cardio non a digiuno.(3) Il problema con questa linea di pensiero è che è al quanto concettualmente limitata. In primo luogo, il grasso che si utilizza come fonte energetica durante l’esercizio fisico non è importante per la perdita di grasso. Altrimenti, non si perderebbe mai grasso corporeo svolgendo attività come l’Interval Training, durante il quale il corpo ha una preferenza nel ricavare energia dalla via metabolica glucidica. Tuttavia, sappiamo perfettamente che si può diminuire la percentuale di grasso corporeo anche attraverso lo svolgimento di pratiche allenanti che usano il metabolismo glucidico come via metabolica preferenziale come, appunto, nel Interval Training, in egual modo a quanto è possibile ottenere con una pratica allenante con preferenza metabolica lipidica come il Cardio LISS, a patto che il deficit energetico sia lo stesso. Non è il tipo di substrato energetico usato durante l’esercizio ad essere importante; piuttosto, è il deficit energetico che viene creato in un periodo di 24 ore (o nel complesso delle 168 ore settimanali).(4)
In secondo luogo, l’aumento dell’ossidazione dei grassi durante il Cardio a digiuno non indica la provenienza di questi ultimi. La metà del grasso ossidato durante una sessione di Cardio LISS proviene dai trigliceridi intramuscolari (“gocce” di grasso immagazzinate all’interno del tessuto muscolare), NON dal grasso viscerale o sottocutaneo.(5) Inoltre, più ci si allena, più il corpo si affida ai Trigliceridi intramuscolari per ricavare energia.(6) Infine, l’esercizio fisico aumenta l’ossidazione dei grassi alimentari consumati dopo che la sessione di allenamento è terminata.(7) Questo è simile al modo in cui una dieta a basso contenuto di carboidrati o chetogenica aumenta l’ossidazione lipidica, ma ciò non aumenta la perdita di grasso se il deficit energetico è uguale a quello creato seguendo una dieta ricca di carboidrati.(8) Quindi, non ci sono prove che dimostrino che svolgere Cardio a digiuno aumenti la perdita di grasso corporeo, anche se c’è un temporaneo aumento dell’ossidazione dei grassi. Se mai, il Cardio a digiuno semplicemente aumenterà l’utilizzo dei Trigliceridi intramuscolari e dei grassi alimentari per ricavare energia.
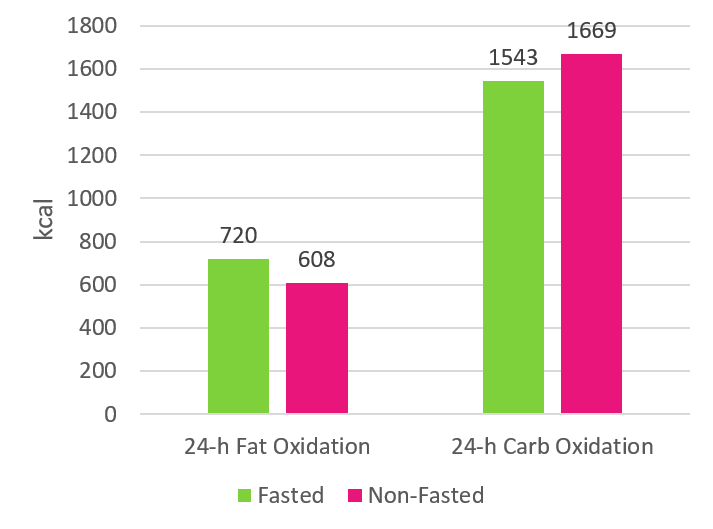
In terzo luogo, mentre il Cardio a digiuno può aumentare l’ossidazione del grasso nelle 24 ore come mostrato nel grafico seguente, lo fa risparmiando i carboidrati, NON le proteine o la massa magra.(9)
Fondamentalmente, ciò che sta accadendo è che, quando si fa Cardio a digiuno, si riduce il glicogeno muscolare di circa il 18% (questa quantità varia a seconda della durata e dell’intensità della seduta allenante).(10) I glucidi consumati successivamente alla seduta di allenamento andranno a reintegrare (tempo variabile dipendente dalle quantità e dalla deplezione di glicogeno creata) quel glicogeno muscolare piuttosto che essere utilizzati come substrato energetico (ecco perché si ossidano meno carboidrati nell’arco di 24 ore come mostrato nel grafico sopra esposto). Poiché il corpo sta immagazzinando carboidrati sotto forma di glicogeno, in tale condizione ci si trova in un “bilancio positivo di carboidrati”. Ma è importante ricordare che i depositi di glicogeno sono limitati. Ciò significa che tale condizione creatasi non può essere protratta per un tempo indeterminato; non è possibile rimanere in uno stato di bilancio positivo dei carboidrati in continuum. Così, il corpo si adatterà nel tempo aumentando la velocità con cui ossida i carboidrati e diminuendo la velocità con cui ossida i grassi.(11) Questo è il motivo per cui occorre pensare a ciò che accade nel lungo periodo; anche ciò che succede nelle 24 ore successive alla sessione non ci dice tutto.
Quando si tratta di perdere grasso corporeo nel tempo, non ci si preoccupa del risparmio glucidico a digiuno; ci si concentra sul risparmio proteico. La ricerca ha dimostrato che il Cardio a digiuno non ha un effetto di risparmio proteico, sia attraverso l’esame dei tassi di ossidazione delle proteine (12) che attraverso l’azoto urinario (13); prodotto di scarto del metabolismo proteico.
Infatti, se si continua a fare Cardio a digiuno, senza bilanciare il consumo di carboidrati e grassi, e senza un consumo sufficiente di calorie e carboidrati al fine di ricostituire il glicogeno muscolare perso durante la sessione allenante, alla fine si raggiungerebbe una marcata deplezione dei depositi di glicogeno muscolare protratta nel tempo. Una volta raggiunto il 50% di riduzione del glicogeno muscolare, il corpo inizia ad ossidare più proteine per ricavare energia.(14) Questa non è di certo una condizione desiderabile dal momento che si vuole perdere grasso conservando il più possibile la massa muscolare.
Pertanto, l’evidenza non supporta un effetto di ripartizione dei tessuti, almeno non quello auspicato. Pertanto, possiamo cancellare anche il punto 3 della lista.
Aumentare il dispendio energetico
Effetto soppressivo sull’appetito
Effetto di ripartizione dei tessuti
Il gioco non vale la candela…
Ora, supponiamo per un momento che esista un effetto di ripartizione dei tessuti dato dal Cardio a digiuno e che l’aumento dell’ossidazione dei grassi provenga in realtà da una maggiore perdita di grasso corporeo. Se si osserva il grafico sopra riportato, si può notare una differenza di 112Kcal ricavate dall’ossidazione lipidica nell’arco delle 24 ore tra Cardio digiunato e Cardio non a digiuno. Supponiamo che il 50% provenga dai Trigliceridi intramuscolari mentre l’altro 50% provenga dal grasso corporeo (viscerale e/o sottocutaneo). Ecco 56Kcal in più provenienti dall’ossidazione del grasso corporeo, che equivale all’incirca a 6g di grasso. Se si è svolto Cardio a digiuno per 5 giorni a settimana, sono circa 30g di grasso corporeo ossidato a settimana. Per perdere un chilo di grasso corporeo in più (454g), sarebbe necessario fare Cardio a digiuno per 60 minuti al giorno, cinque giorni alla settimana per 15 settimane consecutive. Questo non è certo qualcosa che è considerabile come vantaggiosa per la perdita di grasso, soprattutto perché potrebbe facilmente essere annullata da un aumento dell’appetito e da consequenziale aumento dell’apporto calorico.(15) È possibile ottenere risultati migliori semplicemente aumentando il dispendio energetico giornaliero aggiungendo del Cardio durante la giornata e/o riducendo ulteriormente l’apporto calorico.
Risultati nel mondo reale
Naturalmente, tutte queste dimostrazioni teorico-pratiche non hanno un importanza assoluta ed è necessario anche osservare i risultati ottenuti nella vita reale.
La domanda quindi è: cosa succede se si prendono in esame gruppi di persone alle quali viene fatto svolgere un programma di allenamento Cardio a digiuno e non a digiuno e si osserva la loro perdita di grasso? Fortunatamente, i miei colleghi Brad Schoenfeld, Alan Aragon e James Krieger lo hanno fatto, ed hanno pubblicato il loro studio nel 2014.(16) Hanno reclutato 20 giovani soggetti di sesso femmine sottoponendoli a 1 ora di Cardio a digiuno o non digiuno, tre giorni a settimana per 4 settimane. Al termine del test, non hanno riscontrato differenze significative nella perdita di grasso tra i due gruppi.
I ricercatori hanno divulgato anche riprese video dei soggetti presi in esame in questo studio.
Ora, ammettiamo che questo studio sia limitato in dimensioni e durata (solo 4 settimane). Tuttavia, questo non è l’unico studio che non mostra alcuna differenza nella perdita di grasso tra Cardio digiunato e Cardio non a digiuno. Un altro studio di 6 settimane non ha mostrato differenze nella perdita di grasso tra sedute di Interval Training a digiuno e non a digiuno.(17)
Cardio secondo possibilità o preferenze…
Il fatto è che non c’è motivo di svolgere sedute di Cardio a digiuno per cercare di migliorare la perdita di grasso. Per il semplice fatto che non vi sono prove a sostegno di questa tesi, ed i numeri indicano che non si otterrebbe alcun vantaggio significativo sulla perdita di grasso anche se tale vantaggio sussistesse. Gli attuali trial controllati randomizzati, nonostante i loro limiti, non ne supportano l’uso. Il punto è che si dovrebbe semplicemente svolgere la seduta Cardio secondo possibilità o preferenze.
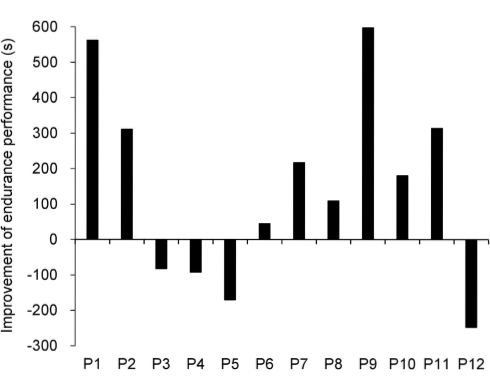
Le radici di ginseng contengono diverse sostanze simili agli steroidi e una di esse è il ginsenoside Rg1. In seguito all’assunzione di 2 capsule contenenti 5 mg di Rg1, sono stati osservati miglioramenti nelle prestazioni in soggetti giovani inattivi. Durante l’esercizio intenso l’effetto è risultato maggiore con una azione protettiva a livello muscolare e un tempo di recupero ridotto.(1)
Gli scienziati dell’Università di Taipei (Taiwan) hanno reclutato studenti di sesso maschile, sani ma non allenati. Li hanno sottoposti ad esercizio fisico, somministrando loro una dose di 5mg di ginsenoside Rg1 la sera prima della sessione allenante, e un’altra dose eguale un’ora prima della sessione. In un’altra occasione gli studenti hanno ricevuto un placebo.
I ricercatori stavano studiando il ginsenoside Rg1 da diverso tempo. La loro ricerca è stata finanziata dal governo di Taiwan e dalla NuLiv, con sede negli Stati Uniti [nulivscience.com]. La NuLiv produce estratti erboristici che successivamente vengono commercializzati sotto forma di integratori alimentari.
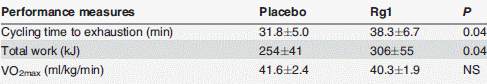
Quando i ricercatori hanno chiesto ai soggetti presi in esame di pedalare il più a lungo possibile ad un’intensità pari all’80% del loro VO2max, hanno osservato che la supplementazione con Rg1 causava un aumento del 20% del tempo di resistenza.
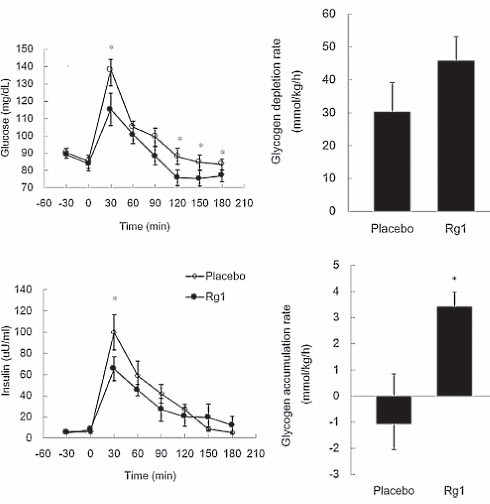
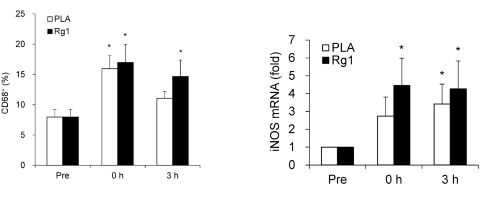
Dopo la sessione allenante, i soggetti dello studio hanno consumato un pasto ricco di carboidrati. Quando i ricercatori hanno analizzato il sangue dei soggetti, hanno notato che lo steroide aveva accelerato l’assorbimento del glucosio [in basso a sinistra]. In basso a destra si può vedere che la supplementazione con ginsenoside Rg1 ha aumentato la quantità di glicogeno nelle cellule muscolari.
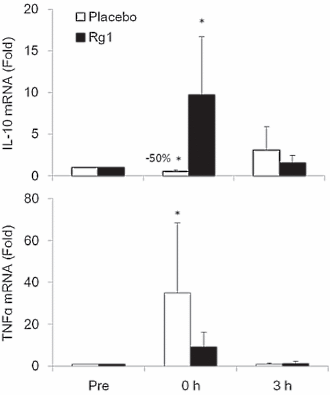
Dopo l’esercizio, la concentrazione cellulare di TNF-alfa dei soggetti trattati con placebo era aumentata. L’Rg1 ha ridotto tale concentrazione. Allo stesso tempo, la sostanza ha indotto le cellule muscolari a produrre più interleuchina-10 (anti-infiammatoria).
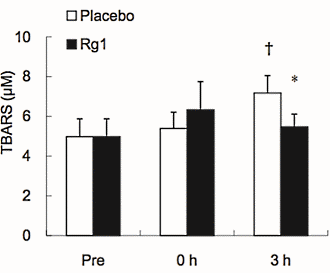
Dopo la sessione allenante, il danno delle cellule muscolari causato dall’attività dei radicali liberi [TBARS] era aumentata nei soggetti trattati con placebo. La supplementazione con ginsenoside Rg1 ha ridotto questo aumento.
I ricercatori affermano che, il risultato principale dello studio è stato quello di dimostrare come la supplementazione con Rg1 pre-workout aumenti significativamente le prestazioni di resistenza durante attività fisica ad alta intensità.
I ricercatori concludono dicendo che, i partecipanti allo studio non allenati sono stati in grado di aumentare la loro capacità di lavoro su un cicloergometro del 20% circa rispetto ai soggetti trattati con placebo. Lo steroide Rg1 può offrire benefici ergogenici per l’uomo.
Un altro studio, svolto dagli stessi ricercatori del precedente, questa volta di recente realizzazione, che ha preso in esame l’azione del Rg1 sugli esseri umani, ha osservato che la combinazione di esercizio fisico intenso e supplementazione con ginseng ha un effetto protettivo a livello muscolare. Lo studio svolto dai biochimici taiwanesi sarà pubblicato sul Journal of Ginseng Research.(2) I ricercatori hanno somministrato ai soggetti dello studio 5mg di ginsenoside Rg1 prima di una intensa sessione su un cicloergometro.
Come per il precedente studio, anche questo è stato in parte finanziato dalla NuLiv Science, [nulivscience.com] e alcuni degli autori sono affiliati alla NuLiv come consulenti scientifici.
Una dei prodotti realizzati dalla NuLiv è l’ActiGin, una combinazione di estratti da Rosa Roxberghii e Panax ginseng. L’ActiGin dovrebbe migliorare le prestazioni sportive, aumentare l’assorbimento di glucosio da parte delle cellule muscolari e accelerare il recupero. Nella pubblicazione della quale stiamo parlando ora si può leggere che questo lavoro è stato finanziato allo scopo di produrre un supplemento ergogenico ActiGin per la Nuliv Science.
Tuttavia, in questo studio i ricercatori non hanno usato l’ActiGin, ma il ginsenoside Rg1. Non mi è stato però possibile constatare il reale contenuto di ginsenoside Rg1 nel ActiGin.
I ricercatori hanno somministrato a 12 studenti maschi non allenati una capsula contenete 5mg di ginsenoside Rg1, un’ora prima che iniziassero la sessione su cicloergometro ad un’intensità pari all’80% del loro VO2max. I soggetti presi in esame dovevano continuare a pedalare fino ad esaurimento. In un’altra occasione, i ricercatori hanno ripetuto l’esperimento, ma somministrando ai soggetti un placebo.
Per la maggior parte dei soggetti presi in esame la supplementazione con Rg1 ha prolungato il loro tempo di resistenza sul cicloergometro. L’evento osservato è paragonabile a quanto riportato per il precedente studio.
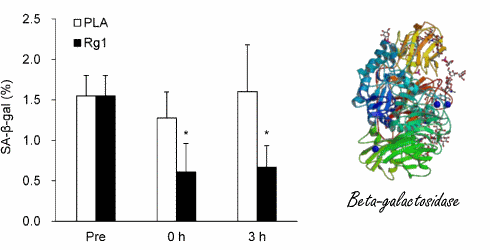
Prima e durante la sessione su cicloergometro, i ricercatori hanno misurato la concentrazione dell’enzima beta-galattosidasi senescente-associato nelle cellule muscolari dei soggetti dello studio. Si tratta di un marker per l’invecchiamento cellulare. Maggiori sono le concentrazioni dell’enzima, più una cellula è senescente.(3)
La combinazione di cicloergometro e supplementazione con ginsenoside Rg1 ha ridotto la concentrazione di beta-galattosidasi.
In basso a sinistra si può notare come il ginsenoside Rg1 può portare ad un “ringiovanimento” muscolare. Se i soggetti dello studio avevano assunto l’Rg1, i ricercatori osservavano una maggiore concentrazione di CD68+ nei loro muscoli dopo lo sforzo. Si tratta di cellule le quali ripuliscono le cellule muscolari danneggiate stimolando le cellule staminali a sostituirle.
I ricercatori concludono affermando che, i risultati dello studio suggeriscono che l’attivazione dei macrofagi dopo supplementazione con Rg1 è associata alla clearance delle cellule senescenti osservate nel muscolo scheletrico umano allenato.
Se non avete ancora letto le precedenti parti componenti questa serie di articoli vi invito a farlo prima di procedere con la lettura di questa quarta ed ultima parte: 1° Parte – 2° Parte – 3° Parte.
Farmacocinetica, Farmacodinamica e Feedback Negativi
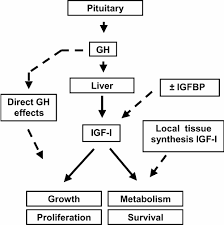
Come già detto, il fegato rappresenta il principale bersaglio del GH, il quale è il principale regolatore della sintesi epatica di IGF-1. Per causare tale effetto, il GH si lega con i GHR localizzati nel dominio extracellulare degli epatociti stimolando successivamente la produzione di IGF-1 endocrino tramite la trascrizione genica, utilizzando la via di segnalazione JAK-STAT. Inoltre, è stato dimostrato che la somministrazione di GH causa una rapida sovraregolazione dell’mRNA del IGF-1 nel fegato.[338]
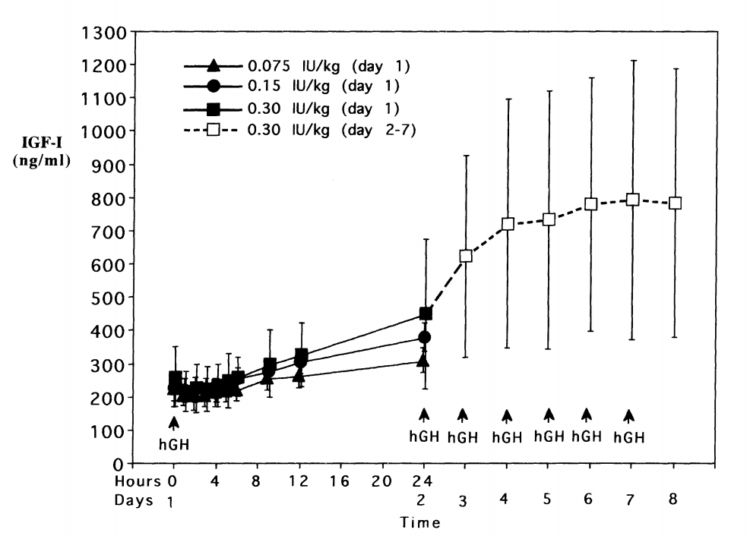
Aumenti dei livelli serici di IGF-1 si verificano molto rapidamente anche in presenza di un grande bolo di rHGH. Incrementi significativi di IGF-1 sono già osservabili dopo 6-12h dall’iniezione.[339] Questi livelli serici di IGF-1 continuano ad aumentare fino a raggiungere il loro punto di saturazione dose-dipendente entro 4-7 giorni, anche quando si utilizzano dosi estremamente elevate che ammontano a 20-30UI al giorno di rHGH.[340] In particolare, il punto di saturazione si è rivelato essere compreso nell’intervallo dei 700-800 ng/mL e sembra suggerire che i livelli endocrini di IGF-1 hanno un tetto massimo negli adulti sani. I meccanismi esatti devono ancora essere chiariti, ma sono probabilmente il risultato dei complessi meccanismi di controllo intrinseci all’Asse GH/IGF-1. Coloro i quali desiderano elevare i livelli endocrini di IGF-1 al fine di ottenerne un vantaggio sull’ipertrofia dovrebbe tenerlo a mente, in quanto vi è un punto in cui l’uso di dosi maggiori di rHGH semplicemente non si traducono in elevati livelli serici di IGF-1. Qui di seguito ho riportato il grafico dello studio di Tanaka il quale mostra la relazione tra l’rhGH e i livelli serici di IGF-:
Ora, vorrei dedicarmi brevemente all’analisi dell’azione del IGF-1autocrino e del perché esso rappresenti un mediatore cruciale del processo ipertrofico, prima di tornare nuovamente a discutere su questioni inerenti alla farmacodinamica e farmacocinetica. La segnalazione recettoriale del IGF-1 è unica nel suo genere, e questo lo si deve al fatto che utilizza due percorsi distinti per stimolare la proliferazione o la differenziazione.[341-343] Questo è un comportamento abbastanza interessante, poiché nessun altro membro della famiglia dei fattori di crescita ha dimostrato di agire in tal modo. Poiché la proliferazione e la differenziazione sono processi opposti, inizialmente era difficile per i ricercatori capire come un singolo fattore di crescita, attraverso un singolo recettore, potesse inviare un segnale che attivasse entrambi.[294] Da quando sono state fatte queste prime scoperte, è stato ulteriormente chiarito che l’IGF-1 non svolge simultaneamente queste azioni. Test su varie linee di coltura cellulare hanno dimostrato che gli effetti proliferativi arrivano prima, durando tra le 24 e le 36 ore. È solo dopo questa fase proliferativa iniziale che si verifica la differenziazione miogenica.[344]
Gli effetti proliferativi mediati dall’IGF-1 sui mioblasti sono noti sin dagli anni ’70, quando vennero osservati per la prima volta nelle cellule epatiche di ratto.[345] Questa stimolazione proliferativa del IGF-1 si traduce in un aumento del numero di cellule, nei livelli di proteine, nella sintesi del DNA, nell’assorbimento di aminoacidico, nell’assorbimento del glucosio e nella soppressione della proteolisi.[346] Nelle colture cellulari umane, l’IGF-1 ha anche dimostrato di aumentare la dimensione dei miotubi indipendentemente dal fatto che i mioblasti proliferino attivamente o che la proliferazione sia cessata. Regola la dimensione dei miotubi attivando la sintesi proteica, inibendo la degradazione proteica e inducendo la fusione delle cellule di riserva.[347-348] La capacità dell’IGF di sopprimere la proteolisi nel muscolo scheletrico, la scomposizione delle proteine in aminoacidi, è stata dimostrata innumerevoli volte nel corso degli anni.[349-352] È stato anche dimostrato che l’IGF-1 induce la proliferazione e la differenziazione delle cellule satelliti in miociti maturi, come determinato da un aumento del numero di miofibre nucleate a livello centrale rispetto a quelle periferiche.[148,353-354]
La capacità dell’IGF-1 autocrino di causare la differenziazione dei mioblasti è stata in realtà una scoperta che potremmo definire quasi “ibrida” dal momento che degli studi svolti negli anni ’60 avevano mostrato che questo effetto si verifica con alti livelli di Insulina.[355] Successivamente è stato dimostrato che gli IGF sono stimolatori molto più potenti nella differenziazione miogenica rispetto all’Insulina e si è concluso che la stessa Insulina agisce realmente come un analogo dell’IGF-1 in questo sistema.[356-357] Gli effetti di differenziazione dati dall’IGF-1 autocrino sono bifasici, con basse concentrazioni che stimolano progressivamente la differenziazione dei mioblasti mentre concentrazioni molto elevate mostrano una cessazione dell’attività di differenziazione. Il limite massimo per la differenziazione sembra attestarsi a circa 100ng/mL per l’IGF-1 e 300ng/mL per IGF-2.[358] Questo effetto non è legato alla proliferazione, poiché non si osservano ulteriori aumenti nel numero complessivo delle cellule.[294] È possibile che le molecole di segnalazione coinvolte nella regolazione negativa del sistema miogenico siano aumentate, ma questa è una affermazione puramente speculativa.[359-360]
La somministrazione di rHGH eleva l’espressione dell’mRNA dell’IGF-1 nel muscolo scheletrico in numerosi modelli cellulari, umani e animali.[127,150,361-364] Ciò avviene abbastanza rapidamente, entro 60 minuti dall’iniezione sottocutanea di rHGH ed i picchi sono segnalati tra le 6 e le 12 ore post iniezione.[363] In questo particolare modello animale citato, il raddoppio della dose di GH non ha portato ad ulteriori aumentati dei livelli di mRNA dell’IGF-1, il che suggerisce che esiste un sistema di regolazione che determina quanto GH sia necessario per stimolare al massimo l’espressione locale dell’IGF-1 nel muscolo scheletrico. In precedenza si è potuto appurare che la differenziazione dei miociti mediata dall’IGF-1 si arresta quando le concentrazioni locali raggiungono circa i 100ng/mL, ma quanto GH è necessario per raggiungere il punto di saturazione dell’espressione dell’mRNA dell’IGF-1?
Gli studi sui miociti umani mostrano che il GH aumenta l’espressione dell’mRNA dell’IGF-1 entro 30-60 minuti con picchi molto più rapidi rispetto a quelli osservati negli studi sugli animali, entro 1-2 ore, usando la via di segnalazione JAK / STAT5b.[365] Questi livelli elevati di mRNA hanno dimostrato di durare fino a 48 ore dopo una singola esposizione al GH. La quantità di GH necessaria per stimolare al massimo l’espressione dell’mRNA dell’IGF-1 è risultata essere una dose compresa tra i 7,5ng/mL e 30ng/ml [366], con una dose media efficace che si attesta a 3ng/ml. Questi numeri sono in linea con gli intervalli di dose fisiologica osservati negli animali, che sono effettivamente compresi tra i 2-100 ng/mL.[367] Inoltre, si collocano esattamente in linea con quanto si osserva endogenamente nell’uomo, con concentrazioni normali di picco comprese tra i 22,4 e 32,4ng/mL.[368-369,436] Ci sono stati casi in cui gli uomini presi in esame hanno mostrato concentrazioni di picco leggermente più alte, ma questi devono essere considerati valori anomali.[370] In ogni caso, ciò che questi dati tendono a suggerire è che il corpo umano è particolarmente adatto a gestire livelli naturali di picco della secrezioni di GH endogeno. Cercare di incidere ulteriormente il sistema elevando i livelli di GH oltre quelli endogeni, unicamente per tentare di potenziare i processi ipertrofici, potrebbe in realtà non tradursi nell’effetto desiderato.
Gli studi che mettono a confronto le infusioni locali con le infusioni sistemiche di GH o IGF-1 sono un po’ più difficili da trovare di quanto si vorrebbe. Le poche sperimentazioni sugli animali che sono riuscito a trovare indicano che l’infusione diretta di GH o IGF-1 nei tessuti bersaglio determina un aumento della massa muscolare. Questo aumento dell’ipertrofia si verifica anche senza che il muscolo bersaglio sia stato sottoposto ad attività motoria.[371-372] Gli studi dimostrano anche che le iniezioni locali di GH portano a livelli sostanzialmente più alti nell’espressione dell’mRNA dell’ IGF-1 locale rispetto alle iniezioni locali di IGF-1, di un fattore di oltre venti.[127] Sono riuscito a trovare uno studio nel quale si confrontavano le risposte dei ratti (attivi e non) all’infusione locale di IGF-1. Il gruppo “IGF-1 plus training” ha mostrato un aumento sia della massa muscolare che della forza locale maggiore rispetto al semplice trattamento in isolamento.[373] Quindi, anche se limitata, la letteratura disponibile è apparentemente in grado di dimostrare che le iniezione locali di GH o IGF-1 hanno effettivamente valore.
Ne ho già parlato diverse volte ma, nel tentativo di imprimere ulteriormente questo concetto, è necessario ricordarsi che i livelli autocrini di IGF-1 sembrano essere molto più importanti dei livelli endocrini di IGF-1 in relazione alla regolazione della massa muscolare. Oltre a questo punto, la sovraespressione dell’IGF-1 autocrino nel muscolo provoca l’ipertrofia delle fibre.[374] La sovraespressione dell’IGF-1 autocrino ha anche mostrato effetti anti-catabolici, con modelli animali tendenti a mostrare una resistenza generale all’atrofia muscolare normalmente osservata con l’invecchiamento.[375] L’IGF-1 localizzato fornisce anche capacità rigenerative indipendenti dall’età nelle cellule muscolari.[376]
Vi sono anche alcune prove convincenti che suggeriscono che l’IGF-1 endocrino agisce direttamente come un regolatore di feedback negativo sulla produzione di IGF-1 autocrino. Questo meccanismo di feedback negativo è dipendente dal pathway PI3K/Akt [377-378]. Inoltre, elevati livelli di IGF-1 endocrino possono anche agire indirettamente per sopprimere la produzione di IGF-1 autocrino. Quindi, in altre parole, non solo l’IGF-1 endocrino ha un impatto diretto minore sulla regolazione della massa muscolare, ma può anche sopprimere l’IGF-1 autocrino che ha impatti maggiori sull’ipertrofia.
Elevati livelli di IGF-1 circolante e, nello specifico, di IGF-1 libero elevati agiscono in modo negativo sul GH determinando un tasso di soppressione della produzione di IGF-1 autocrino a valle.[379] Non è del tutto chiaro, tuttavia, se la regolazione negativa dell’IGF-1 modifichi l’emivita dell’mRNA dell’IGF-1 o influenzi direttamente l’espressione del gene IGF-1. Oltre a questo, è stato anche dimostrato che l’espressione dell’IGF-1 autocrino è sottoregolata nelle cellule muscolari dopo trattamento con IGF-1.[366] È stato anche dimostrato che l’espressione epatica dell’mRNA dell’IGF-1 è sottoregolata dall’esposizione acuta all’IGF-1.[127] Quindi, mantenere livelli endocrini il più possibile soppressi con rispettiva dose di rHGH, elevando contemporaneamente i livelli autocrini, dovrebbe essere un fattore prioritario in un protocollo di GH volto all’ipertrofia.
Il GH è pulsatile per natura sia nell’uomo che nelle specie animali. Quindi, sarebbe logico pensare che molti dei processi intrinseci del corpo saranno tarati in modo tale da rispondere in maniera ottimale all’esposizione al GH in modo simile. In accordo con questa affermazione è stato dimostrato che solo la somministrazione di GH pulsatile, e non l’infusione continua, ha la capacità di stimolare massimamente l’espressione dell’mRNA dell’IGF-1 nel muscolo scheletrico.[366,380-381] È stato anche dimostrato che la somministrazione pulsatile porta ad un aumento del potenziale di crescita postnatale complessivo rispetto all’infusione continua.[89,382] La somministrazione pulsatile può anche portare a livelli endocrini di IGF-1 serici comparabili, o addirittura diminuiti [383], il che è vantaggioso a causa delle potenziali capacità di regolazione negativa che possiede sull’espressione dell’IGF-1 autocrino e che sono state discusse in precedenza. L’evidenza suggerisce anche che il picco stesso, e non necessariamente il numero di picchi, potrebbe essere della massima importanza per i tessuti bersaglio.[384] Per la massima crescita e potenziale ipertrofico, l’evidenza tende a suggerire che creare picchi di GH elevati, e quindi tornare ai livelli basali più volte al giorno, può essere preferibile rispetto a mantenerli elevati per periodi di tempo più lunghi. Questo pratica permette di riprodurre gli schemi secretori in vivo.
I pathways del GH coinvolti nell’anabolismo sono anche suscettibili alla desensibilizzazione, che è parte della fisiologia del GH endogeno.[385] A causa della natura intrinsecamente pulsatile del GH in vivo, l’attività dei recettori e dei pathways sono regolati da un impulso seguito da un periodo di inattività.[386] L’esposizione continua o ripetuta al GH senza un adeguato lasso di tempo refrattario comporterà livelli di attività fortemente soppressi. In effetti, nel corso degli anni numerosi studi hanno dimostrato che tale effetto si verifica. Le cellule ed il tessuto muscolare richiedono un periodo refrattario piuttosto lungo prima che la loro piena risposta al GH venga recuperata. Dopo l’esposizione al GH, le cellule muscolari non sono nemmeno in grado di rispondere alle successive dosi di GH. In realtà, occorrono due ore complete per riprendere parzialmente la reattività nei modelli cellulari, con un totale di 6-8 ore di astinenza dall’uso di GH necessarie per ripristinare la piena sensibilità.[366] Viceversa, quando il GH è micro-dosato in impulsi di dieci minuti, seguiti da intervalli di otto ore, è stato mostrato aumentare progressivamente l’mRNA dell’IGF-1 con ogni impulso successivo.[386]
Questo fenomeno è potenzialmente il risultato di una desensibilizzazione complessiva all’interno della via JAK-STAT5, poiché è stato dimostrato che l’esposizione al GH negli studi sulle cellule epatiche causa resistenza alla successiva attivazione della via STAT5 per 4-8 ore.[387-388] Questo lasso di tempo è sufficiente per sincronizzarsi abbastanza bene con ciò che è stato visto nei modelli di cellule miocitarie citati in precedenza. Nei modelli di cellule epatiche, il GH ha stimolato un significativo aumento dell’espressione del SOCS3, che è un potente inibitore dell’azione del GH.[389]. Poiché il GH non ha avuto alcun effetto sull’espressione del SOCS3 nelle cellule muscolari, questo deve essere un altro meccanismo causante il periodo refrattario. Questo meccanismo può essere dipeso dalla sottoregolazione dei GHR, dall’inibizione mediata da un’altra proteina SOCS, o dall’induzione di una tirosina fosfatasi che semplicemente inattiva la via JAK / STAT.[390] La via JAK-STAT5b, che come ricorderete è intimamente associata al muscolo scheletrico e all’espressione dell’IGF-1, è di natura transitoria – con attivazione massima raggiunta entro 10-30 minuti, seguita da un prolungato periodo di inattivazione.
Una scoperta piuttosto nuova di Xu et al. [391] ha dimostrato che anche distanziare le esposizioni al GH di cinque ore lasciava entrambi i percorsi a valle MEK1/2 e ERK1/2 significativamente soppressi rispetto a tutti i percorsi a monte, a causa di una potenziale disconnessione nella trasduzione del segnale . Ciò è di particolare interesse in quanto questi stessi due percorsi a valle sono stati coinvolti in modo significativo sia nella crescita sia nella proliferazione.[392-393] È stato anche scoperto che l’attivazione indotta da GH di STAT1 e STAT3 è stata desensibilizzata, ma l’esposizione all’Insulina inverte la desensibilizzazione osservata in tutti i percorsi interessati. Anche se non sto per trattare approfonditamente l’Insulina, ci sono un paio di importanti punti da dovere prendere in considerazione. Bisogna comprendere innanzitutto che ci sono molti obiettivi a valle del recettore del GH e molti di questi hanno il potenziale per essere desensibilizzati dopo l’esposizione al GH. Bisogna comprendi anche che l’Insulina possiede l’abilità unica di risensibilizzare molti di questi percorsi. Ciò ha un senso vista la relazione tipo yin-yang tra i due composti. È noto che il GH e l’Insulina possiedono una relazione anabolica sinergica a causa di molti effetti che esercitano l’uno sull’altro. Questo sembra essere soltanto un’anteprima di uno di questi effetti.
Asse GH/IGF-1 – Relazione con altri ormoni
Prima di passare alle note conclusive, vorrei trattare brevemente alcuni altri ormoni connessi a diverso grado con l’Asse GH/IGF-1. Per prima cosa, voglio trattare brevemente l’Asse Tiroideo dal momento che l’inserimento di composti tiroidei insieme al GH è una pratica comune anche durante i protocolli di massa.
Il muscolo scheletrico è il principale bersaglio di segnalazione dell’ormone tiroideo, con trasportatori degli ormoni tiroidei e enzimi di conversione espressi localmente.[394] È ben noto che il GH potenzia la deiodinazione periferica che converte il T4 in T3, riducendo così il T4 e il reverse T3, aumentando contemporaneamente i livelli di T3.[395-398] Ciò che molte persone non riescono a capire è che questo è un effetto transitorio, e studi a lungo termine sembrano indicare che gli effetti mediati dal GH sulla conversione periferica si stabilizzino con il tempo.[399-402]
Via ubiquitina/proteasoma
Invece di proseguire ulteriormente su questo, avendo già trattato la questione nel dettaglio in un mio vecchio articolo, preferirei concentrarmi su alcune pubblicazioni relative alla tiroide che non vengono discusse abbastanza spesso. Gli ormoni tiroidei, per loro natura, sono composti tendenzialmente catabolici in quanto stimolano la disgregazione proteica dell’intero corpo in misura maggiore rispetto alla sintesi proteica.[403] A livello locale, nel muscolo scheletrico stimolano un aumento dell’attività all’interno della via ubiquitina/proteasoma, che è ampiamente coinvolta nella proteolisi.[404-406] Il risultato di questo è un tasso accelerato del turnover proteico e una perdita netta complessiva degli aminoacidi situati all’interno dei muscoli scheletrici.
Inoltre, negli esseri umani, sia gli stati di ipertiroidismo che di ipotiroidismo sono stati associati a livelli di IGF-1 soppressi con una tendenza alla normalizzazione quando viene ristabilita una condizione di eutiroidismo. L’ipertiroidismo è anche associato ad una bassa attività di legame recettoriale del GH, che si ipotizza essere il risultato di una ridotta capacità di elaborazione dei recettori del GH.[407] E’ stato anche ipotizzato che l’ipertiroidismo sia in grado anche di accelerare la clearance del GH urinario.[408] Inoltre, studi su animali hanno dimostrato che gli ormoni tiroidei possono avere importanti effetti soppressivi sulla sintesi di IGF-1 stimolata con il GH.[409] Ovviamente, a causa della complessa relazione che l’Asse Tiroideo ha con l’Asse GH/IGF-1, raggruppando tutte le interazioni che hanno tra loro in pochi paragrafi, trattare l’argomento diventerebbe poco pratico. Tuttavia, quando il corpo della letteratura scientifica viene esaminato nella sua interezza, ci sono molte prove che suggeriscono che la supplementazione con composti tiroidei esogeni potrebbe non essere l’ideale quando l’obiettivo di un individuo è l’ipertrofia, anche se la regolazione del dosaggio dei tiroidei in tale contesto rimane la misura di “sicurezza” più intelligente visto l’impatto negativo sulla funzionalità tiroidea dato dal GH. Comunque, per tutti coloro che sono interessati ad approfondire questo argomento, consiglio di iniziare con la review nella nota seguente.[410]
Miostatina
Mi piacerebbe trattare anche la Miostatin, che rappresenta un argomento molto discusso nei vari forum di BodyBuilding presenti in rete. La sua fama proviene dai risultati ipertrofici espressi dai bovini privi per mutazione del gene della Miostatina, i quali mostrano una massa muscolare significativamente maggiore rispetto ai loro simili non mutati.[411] La Miostatina, un fattore di crescita e differenziazione appartenente alla superfamiglia dei TGF-beta, ha dimostrato di inibire selettivamente la miogenesi, in gran parte tramite il suo effetto soppressivo sulla proliferazione dei mioblasti.[412] È espressa e secreta prevalentemente dal muscolo scheletrico. Come molti sanno, se riesci a sopprimere o inibire la Miostatina, di conseguenza il potenziale ipertrofico aumenta significativamente.
Le mutazioni della Miostatina sono state osservate sia negli animali che nell’uomo. Queste mutazioni del gene della Miostatina portano ad un fenotipo ipertrofico negli animali, come accennato in precedenza.[413-415] L’Asse GH/IGF-1 e la Miostatina sembrano avere una relazione regolativa diretta tra loro, come osservato nei pazienti affetti contemporaneamente da GHD e HIV che mostrano marcati aumenti nell’espressione dell’mRNA della Miostatina.[416] Quindi, è possibile che attraverso una supplementazione di dosi sovrafisiologiche di rHGH si possa indurre una diminuzione dell’mRNA della Miostatina [209,417-419]? Sfortunatamente, nonostante la presenza di alcuni casi studio selezionati, non credo che si abbiano abbastanza dati in questo momento per sapere se ciò possa dare risultati apprezzabili in seguito alla sua applicazione.
Quello che sappiamo è che aumenti dell’espressione dell’mRNA dell’IGF-1 e le concentrazioni circolanti di IGF-1 sono state osservati dopo inibizione della Miostatina.[419-421] Sappiamo anche che l’inibizione della Miostatina tende a causare l’ipertrofia attraverso molte delle stesse modalità osservate con l’IGF-1 autocrino, cioè l’aumento della sintesi proteica e l’attivazione delle cellule satelliti.[422-425] E sappiamo anche che l’ipertrofia indotta dalla sovraespressione dell’IGF-1 o dall’inibizione della Miostatina utilizza la stessa identica via – PI3K/Akt/mTOR.[426-428] Tuttavia, l’IGF-1 non è un requisito per l’ipertrofia indotta dalla Follistatina, tranne nel caso di livelli di Insulina estremamente bassi – come ben sappiamo, la Follistatina è un inibitore della Miostatina [429]. E l’esposizione cronica al GH può in realtà portare ad un’espressione sovrastimolata della Miostatina e del suo recettore.[209]
Quindi quello che possiamo dire, con certezza, è che l’espressione della Miostatina non sarà un fattore diretto o indiretto per quanto riguarda il potenziamento dei processi ipertrofici, né dell’attività contrattile, nei muscoli scheletrici umani.[430] Proprio per questo motivo, non ritengo che sia un fattore sul quale gli atleti debbano eccessivamente concentrarsi, al di fuori di un utile arricchimento delle proprie conoscenze in materia.
Applicazioni pratiche e pensieri conclusivi
Arrivati a questo punto è mia intenzione unire tutto ciò che è stato esposto in questa serie di articoli e esporlo sotto forma di alcuni suggerimenti pratici rivolti a tutti coloro i quali vogliono semplicemente massimizzare la loro capacità ipertrofica.
Ora è chiaro che il GH possiede pochissimi, se non nulli, effetti diretti sull’ipertrofia. Pertanto, qualsiasi protocollo di massa che contempli il suo uso dovrà tenere in considerazione questo punto includendo gli AAS, i quali, per l’appunto, hanno anche una proficua sinergia con il GH. Sia la letteratura scientifica che i dati aneddotici dimostrano chiaramente che l’uso combinato di entrambi i composti ha un massimale ipertrofico significativamente più alto rispetto all’uso singolo. Personalmente, penso che i BodyBuilder dovrebbero sempre optare per l’utilizzo di una base di Testosterone e Boldenone (correttamente rapportati in base a contesto e alle caratteristiche individuali) anche in una fase “Bulk” nella quale viene inserito l’uso del GH. Il Trenbolone può essere considerato come parte “accessoria” di un protocollo di massa, a causa della sua intrinseca difficoltà gestazionale come sostanza anabolizzante. Dovrebbe essere usato con parsimonia e con cautela poiché, insieme ai suoi numerosi punti di forza come composto anabolizzante, presenta alcune limitazioni. Queste limitazioni derivano per lo più, come già accennato, dalla difficile gestione del composto in quanto esso non è facilmente tollerato da una buona parte degli individui. Quindi, se viene usato il Trenbolone, dovrebbe essere inserito calcolando con attenzione il dosaggio e la tolleranza individuale, anche per quanto concerne la tolleranza temporale individuale all’uso di tale composto.
Dopo un periodo d’uso prolungato di dosi sovrafisiologico di AAS (Ciclo+Bridge), è buona cosa procedere con l’interruzione o con una marcata riduzione del numero e degli AAS utilizzati. Detta in parole semplici, questa interruzione può contemplare una completa astinenza dagli AAS, svolgendo un adeguata PCT al fine di ristabilire una omeostasi ormonale fisiologica, o una transizione ad una TRT, metodologia comunemente chiamata “blast and cruise”. La struttura del protocollo di supplementazione farmacologica dovrebbe sempre seguire i principi del dosaggio minimo efficace con aumenti nei dosaggi degli AAS solo nel caso si sia raggiunto il limite di crescita con il precedente dosaggio, assicurandosi che tutte le altre variabili nello stile di vita siano correttamente regolate. L’utilizzo di questo approccio limita il rischio che si sviluppino effetti collaterali indesiderati sul lungo termine.
Quando si decide di usare il GH, la dove ce ne sia la possibilità economica, esso dovrebbe provenire da uno dei prodotti presenti nel mercato farmaceutico. Questi prodotti approvati devono superare anni di studi strettamente controllati per dimostrare la loro sicurezza, purezza ed efficacia su soggetti umani. I progressi tecnologici nel corso degli anni hanno reso molto più facile la produzione di rHGH. Per questo motivo, i produttori ora provengono da tutto il mondo. Spesso questi produttori realizzano ciò che viene definito “GH generico” sui forum, ma tale termine non mi piace molto. Definire qualcosa come “generico” implica che sia una replica perfetta di prodotti legati a specifici marchi farmaceutici approvati dell’agenzia del farmaco che hanno perso il cui brevetto è scaduto, il che non è il caso del GH. Infatti, a causa della natura estremamente complessa del processo di produzione del rHGH, per esempio, la FDA non consente nemmeno l’uso del termine “generico” quando si tratta di rHGH e utilizza invece il termine ” follow-on protein product ” o FOPP.
Spesso questi marchi off-label sono venduti ad un costo molto ridotto, ed è qui che sta il dilemma, in quanto questo può essere molto allettante. Tuttavia, con questo costo ridotto per il consumatore, non ci sarà nemmeno la garanzia del produttore su cosa ci sia realmente nella fiala o persino su come è stato prodotto. Il problema di fondo è che il processo di produzione del rHGH è estremamente complessa, ed è molto facile che nelle fasi di questo processo si commettano errori con conseguenti variazioni nella catena proteica che potenzialmente portano ad effetti indesiderati, o anche a risposte autoimmuni.
Spesso gli atleti si affidano semplicemente ai test serici per misurare i livelli di GH e/o IGF-1 al fine di concludere che un prodotto contenete GH sia “buono”, ma dobbiamo ricordarci che raggiungere livelli ematici ormonali elevati è la parte relativamente facile. Anche le molecole di GH che sono state alterate o danneggiate durante la produzione possono dare questo esito. Tuttavia, queste stesse molecole di GH danneggiate o mutate possono spesso stimolare risposte autoimmuni. Ciò potrebbe indurre il corpo ad avere una risposta recettoriale degradata, che può anche riflettersi sulla secrezione endogena nel tempo. [431-432] Rimane poi il problema del reale contenuto della vial o fiala, la quale può non presentare nessun principio attivo al suo interno.
Il GH dovrebbe essere usato in modo pulsatile, per mimare le condizioni in vivo. Tra queste iniezioni, deve esserci un periodo di refrattarietà o si deve consumare un pasto che abbia un buon stimolo sull’Insulina. Può anche essere utilizzata l’Insulina esogena al fine di bypassare molte delle limitazioni del periodo refrattario, ma questo va oltre lo scopo di questo articolo. Anche se il cumulo delle dosi giornaliere dovrebbe essere sovrafisiologico, le dosi individuali non hanno bisogno di essere ad alto dosaggio, poiché la massima stimolazione dell’IGF-1 autocrino nel tessuto muscolo scheletrico si verifica ben all’interno delle concentrazioni fisiologiche di GH. Aneddoticamente, sembra anche esserci un limite con il quale l’uso di rHGH diventa additivo in presenza di AAS. Potrebbe essere necessario un po’ di ponderata (e supportata da personale qualificato) auto-sperimentazione per scoprire dove si trova questa singola dose di saturazione, ma la maggior parte dei soggetti troverà questo limite tra le 4 e le 8 UI/die. Oltre questo dosaggio, la maggior parte degli utilizzatori tenderà a scoprire che la giustificazione dei costi e il rapporto rischio /beneficio tendono a diminuire rapidamente.
Non bisognerebbe passare troppo tempo a riflettere sul tempo delle iniezioni di GH, dal momento che gli aumenti dei livelli di IGF-1 autocrino avvengono rapidamente e possono rimanere elevati per giorni. Bisognerebbe concentrati invece sul programma di iniezione che si addice meglio al contesto della giornata, tenendo contemporaneamente presenti le linee guida per il periodo di refrattarietà del GH. Si possono anche prendere in considerazione piccole o grandi iniezioni, poiché alcuni potrebbero trovare più pratiche e funzionali iniezioni con un dosaggio inferiore e somministrate più frequenti mentre altri potrebbero preferire iniezioni con un dosaggio maggiore e somministrazioni meno frequenti. Naturalmente, maggiore è il contenuto dell’iniezione, maggiore è la probabilità che si superi la soglia massima di espressione dell’IGF-1 autocrino.
Massimizzare l’espressione dell’IGF-1 autocrino, mentre contemporaneamente si sopprimono i livelli di IGF-1 endocrino, sarà una priorità. Esistono prove a sostegno dell’ipotesi secondo cui un iniezione locale di GH possa aiutare a raggiungere questo obiettivo, con una conseguente minore possibilità di feedback negativo. Sono stati osservati aumenti significativi della massa muscolare in appena due settimane di iniezioni locali di IGF-1.[441]
La dove cause di forza maggiore non lo impediscano, è consigliabile evitare o comunque regolare attentamente l’uso di tutti quei composti che possono avere interazioni negative con l’uso del GH a fini ipertrofici. Inibitori della Aromatasi, Modulatori Selettivi del Recettore degli Estrogeni e T3 hanno tutti dimostrato di avere un potenziale effetto negativo sul processo globale ipertrofico legato al GH e per tale motivo dovrebbero essere usati con parsimonia, se non omessi del tutto dove possibile (vedi in particolare il T3).
Questo non dovrebbe sorprendere nessuno e non dovrebbe nemmeno essere troppo difficile da tenere a mente: allenarsi duramente, allenarsi in modo intelligente e allenarsi in modo coerente. Sebbene non sia stato affrontato direttamente nell’articolo, bisogna capire che l’allenamento contro resistenza ha impatti unici e additivi sull’ipertrofia. In effetti, alcuni di questi meccanismi non sono nemmeno mediati dall’Asse AR e/o GH/IGF-1. [433] Bisogna anche comprendere che non esiste una “magica” routine allenante universalmente applicabile, la chiave di volta sarà la coerenza nel garantire un carico di lavoro adeguato, con elementi di sovraccarico progressivo nel tempo assicurando un adeguato stimolo meccanico gestendo al meglio le variabili allenanti (intensità, volume, densità e intensità percepita). La logica composizione del piano allenante servirà a garantire lo stimolo ipertrofico di base che sarà coadiuvato dall’azione dei composti utilizzati.
Nonostante la mole e l’importanza delle informazioni presentate in questa serie di articoli, è necessario ricordare che i meccanismi d’azione ormonali sono governati da innumerevoli fattori. Anche esaminando l’intero corpo della letteratura scientifica equivarrebbe a poco più che accumulare una serie di utili nozioni garanti di assicurare una solida base conoscitiva sull’argomento la quale rappresenterà un punto di partenza intelligente per l’applicazione pratica, rimanendo sempre soggetti alle variabili di risposta individuale. Seguendo questa linea, i migliori risultati nella pratica spesso provengono da coloro i quali posseggono una buona conoscenza dei principi scientifici e la capacità innata di saperli applicare non solo su se stessi ma, soprattutto, su terzi. Infatti, molto raramente due persone rispondono in modo identico alla supplementazione di ormoni esogeni (e non solo), quindi, non bisogna assolutamente pensare che basti semplicemente applicare su se stessi o su terzi un protocollo che ha portato benefici realmente apprezzabili su un soggetto per ottenere la medesima risposta.
A tal fine, invito atleti e Preparatori a utilizzare queste pubblicazioni come punto di partenza per essere in grado di gestire l’applicazione pratica in modo più consapevole e produttivo. Inoltre, chi ne fosse in grado, può consultare il vasto numero di riferimenti riportati nel corso di queste pubblicazioni è tentare di ragionare sulle mie conclusioni. Ad ogni citazione presente in questa serie di articoli, assicuratevi che il riferimento elencato supporti effettivamente le affermazioni fatte. Mantenere sempre una mente aperta ma con i giusti “filtri”, e cercare di non credere per partito preso ad una singola opinione, specialmente di fronte alle nuove evidenze scientifiche. Infine, è buona cosa verificare sempre la veridicità di quanto è stato affermato.
Punti conclusivi per un corretto utilizzo della chimica e del GH a fini ipertrofici:
Usare il GH in combinazione con gli AAS
Usare GH e AAS di grado farmaceutico la dove ciò è possibile
Assicurarsi una base di Testosterone correttamente rapportata al Boldenone aggiungendo (in base a maturità e tolleranza) il Trenbolone
Iniettare il GH in modo pulsatile, considerare l’opzione delle iniezioni locali nei gruppi carenti
Mantenere un dosaggio complessivo ottimale di GH il quale si attesta tra le 4 e le 8UI/die
Evitare o regolare attentamente l’uso dei composti che possono interagire negativamente con i processi ipertrofici legati al uso di GH (vedi AI, SERM e T3)
Dopo un periodo di tempo (variabile) nel quale si è stati sottoposti a dosaggi ormonali sovrafisiologici optare per una PCT o per una TRT (a seconda delle proprie necessità e priorità)
Essere a conoscenza dei potenziali effetti collaterali legati all’auso/abuso di GH (nausea, vomito, cefalea, ritenzione idrica e sodica, edemi, parestesie, sindrome del tunnel carpale, rigidità articolare, dolori articolari, artrite, dolori muscolari, ipertensione, insulino-resistenza, diabete di tipo II, acromegalia, dilatazione addominale, ipertrofia cardiaca ecc…)
Svolgere regolarmente esami del sangue; sia durante i periodi di picco nell’uso della farmacologia sia nel periodo successivo (vedi PCT/OCT o TRT)
Gestire al meglio le variabili legate agli stressor ambientali, all’allenamento, all’alimentazione e al sonno.
Gabriel Bellizzi
Riferimenti:
338. Mathews LS, Norstedt G, Palmiter RD. Regulation of insulin-like growth factor I gene expression by growth hormone. Proc Natl Acad Sci U S A. 1986 Dec;83(24):9343-7.
339. Keller A, Wu Z, Kratzsch J, Keller E, Blum WF, Kniess A, Preiss R, Teichert J, Strasburger CJ, Bidlingmaier M. Pharmacokinetics and pharmacodynamics of GH: dependence on route and dosage of administration. Eur J Endocrinol. 2007 Jun;156(6):647-53.
340. Tanaka T, Seino Y, Fujieda K, Igarashi Y, Yokoya S, Tachibana K, Ogawa Y. Pharmacokinetics and metabolic effects of high-dose growth hormone administration in healthy adult men. Endocr J. 1999 Aug;46(4):605-12.
341. Quinn LS, Steinmetz B, Maas A, Ong L, Kaleko M. Type-1 insulin-like growth factor receptor overexpression produces dual effects on myoblast proliferation and differentiation. J Cell Physiol. 1994 Jun;159(3):387-98.
342. Coolican SA, Samuel DS, Ewton DZ, McWade FJ, Florini JR. The mitogenic and myogenic actions of insulin-like growth factors utilize distinct signaling pathways. J Biol Chem. 1997 Mar 7;272(10):6653-62.
343. Foulstone EJ, Huser C, Crown AL, Holly JM, Stewart CE. Differential signalling mechanisms predisposing primary human skeletal muscle cells to altered proliferation and differentiation: roles of IGF-I and TNFalpha. Exp Cell Res. 2004 Mar 10;294(1):223-35.
344. Ewton DZ, Roof SL, Magri KA, McWade FJ, Florini JR. IGF-II is more active than IGF-I in stimulating L6A1 myogenesis: greater mitogenic actions of IGF-I delay differentiation. J Cell Physiol. 1994 Nov;161(2):277-84.
345. Florini JR, Nicholson ML, Dulak NC. Effects of peptide anabolic hormones on growth of myoblasts in culture. Endocrinology. 1977 Jul;101(1):32-41.
346. Laviola L, Natalicchio A, Giorgino F. The IGF-I signaling pathway. Curr Pharm Des. 2007;13(7):663-9. Review.
347. Jacquemin V, Furling D, Bigot A, Butler-Browne GS, Mouly V. IGF-1 induces human myotube hypertrophy by increasing cell recruitment. Exp Cell Res. 2004 Sep 10;299(1):148-58.
348. Jacquemin V, Butler-Browne GS, Furling D, Mouly V. IL-13 mediates the recruitment of reserve cells for fusion during IGF-1-induced hypertrophy of human myotubes. J Cell Sci. 2007 Feb 15;120(Pt 4):670-81. Epub 2007 Jan 30.
349. Ballard FJ, Francis GL. Effects of anabolic agents on protein breakdown in L6 myoblasts. Biochem J. 1983 Jan 15;210(1):243-9.
350. Ewton DZ, Falen SL, Florini JR. The type II insulin-like growth factor (IGF) receptor has low affinity for IGF-I analogs: pleiotypic actions of IGFs on myoblasts are apparently mediated by the type I receptor. Endocrinology. 1987 Jan;120(1):115-23.
351. Hembree JR, Hathaway MR, Dayton WR. Isolation and culture of fetal porcine myogenic cells and the effect of insulin, IGF-I, and sera on protein turnover in porcine myotube cultures. J Anim Sci. 1991 Aug;69(8):3241-50.
352. Hong D, Forsberg NE. Effects of serum and insulin-like growth factor I on protein degradation and protease gene expression in rat L8 myotubes. J Anim Sci. 1994 Sep;72(9):2279-88.
353. Florini JR, Ewton DZ, Roof SL. Insulin-like growth factor-I stimulates terminal myogenic differentiation by induction of myogenin gene expression. Mol Endocrinol. 1991 May;5(5):718-24.
354. Musarò A, McCullagh KJ, Naya FJ, Olson EN, Rosenthal N. IGF-1 induces skeletal myocyte hypertrophy through calcineurin in association with GATA-2 and NF-ATc1. Nature. 1999 Aug 5;400(6744):581-5.
355. Haba GDL, Cooper GW, Elting V. HORMONAL REQUIREMENTS FOR MYOGENESIS OF STRIATED MUSCLE IN VITRO: INSULIN AND SOMATOTROPIN. Proceedings of the National Academy of Sciences of the United States of America. 1966;56(6):1719-1723.
356. Florini JR, Ewton DZ. Insulin acts as a somatomedin analog in stimulating myoblast growth in serum-free medium. In Vitro. 1981 Sep;17(9):763-8.
357. Schmid C, Steiner T, Froesch ER. Preferential enhancement of myoblast differentiation by insulin-like growth factors (IGF I and IGF II) in primary cultures of chicken embryonic cells. FEBS Lett. 1983 Sep 5;161(1):117-21.
358. Florini JR, Ewton DZ, Falen SL, Van Wyk JJ. Biphasic concentration dependency of stimulation of myoblast differentiation by somatomedins. Am J Physiol. 1986 May;250(5 Pt 1):C771-8.
359. Quinn LS, Ehsan M, Steinmetz B, Kaleko M. Ligand-dependent inhibition of myoblast differentiation by overexpression of the type-1 insulin-like growth factor receptor. J Cell Physiol. 1993 Sep;156(3):453-61.
360. Olson EN. Signal transduction pathways that regulate skeletal muscle gene expression. Mol Endocrinol. 1993 Nov;7(11):1369-78. Review.
361. Murphy LJ, Bell GI, Friesen HG. Growth hormone stimulates sequential induction of c-myc and insulin-like growth factor I expression in vivo. Endocrinology. 1987 May;120(5):1806-12.
362. Turner JD, Rotwein P, Novakofski J, Bechtel PJ. Induction of mRNA for IGF-I and -II during growth hormone-stimulated muscle hypertrophy. Am J Physiol. 1988 Oct;255(4 Pt 1):E513-7.
363. Isgaard J, Nilsson A, Vikman K, Isaksson OG. Growth hormone regulates the level of insulin-like growth factor-I mRNA in rat skeletal muscle. J Endocrinol. 1989 Jan;120(1):107-12.
364. Bichell DP, Kikuchi K, Rotwein P. Growth hormone rapidly activates insulin-like growth factor I gene transcription in vivo. Mol Endocrinol. 1992 Nov;6(11):1899-908.
365. Sadowski CL, Wheeler TT, Wang LH, Sadowski HB. GH regulation of IGF-I and suppressor of cytokine signaling gene expression in C2C12 skeletal muscle cells. Endocrinology. 2001 Sep;142(9):3890-900.
366. Frost RA, Nystrom GJ, Lang CH. Regulation of IGF-I mRNA and signal transducers and activators of transcription-3 and -5 (Stat-3 and -5) by GH in C2C12 myoblasts. Endocrinology. 2002 Feb;143(2):492-503.
367. MacLeod JN, Pampori NA, Shapiro BH. Sex differences in the ultradian pattern of plasma growth hormone concentrations in mice. J Endocrinol. 1991 Dec;131(3):395-9.
368. Rochiccioli P, Messina A, Tauber MT, Enjaume C. Correlation of the parameters of 24-hour growth hormone secretion with growth velocity in 93 children of varying height. Horm Res. 1989;31(3):115-8.
369. Hansen TK, Gravholt CH, ØRskov H, Rasmussen MH, Christiansen JS, Jørgensen JO. Dose dependency of the pharmacokinetics and acute lipolytic actions of growth hormone. J Clin Endocrinol Metab. 2002 Oct;87(10):4691-8.
370. Baum WF, Klöditz E, Hesse V, Jahreis G, Schneyer U, Giebler H. [Increase in spontaneous growth hormone secretion in asthmatic children–a symptom of atopic disposition?]. Kinderarztl Prax. 1993 Nov;61(9):323-8.
371. Adams GR, McCue SA. Localized infusion of IGF-I results in skeletal muscle hypertrophy in rats. J Appl Physiol (1985). 1998 May;84(5):1716-22.
372. Alzghoul MB, Gerrard D, Watkins BA, Hannon K. Ectopic expression of IGF-I and Shh by skeletal muscle inhibits disuse-mediated skeletal muscle atrophy and bone osteopenia in vivo. FASEB J. 2004 Jan;18(1):221-3. Epub 2003 Nov 3.
373. Lee S, Barton ER, Sweeney HL, Farrar RP. Viral expression of insulin-like growth factor-I enhances muscle hypertrophy in resistance-trained rats. J Appl Physiol (1985). 2004 Mar;96(3):1097-104. Erratum in: J Appl Physiol. 2004 Jun;96(6):2343.
374. Coleman ME, DeMayo F, Yin KC, Lee HM, Geske R, Montgomery C, Schwartz RJ. Myogenic vector expression of insulin-like growth factor I stimulates muscle cell differentiation and myofiber hypertrophy in transgenic mice. J Biol Chem. 1995 May 19;270(20):12109-16.
375. Barton-Davis ER, Shoturma DI, Musaro A, Rosenthal N, Sweeney HL. Viral mediated expression of insulin-like growth factor I blocks the aging-related loss of skeletal muscle function. Proc Natl Acad Sci U S A. 1998 Dec 22;95(26):15603-7.
376. Musarò A, McCullagh K, Paul A, Houghton L, Dobrowolny G, Molinaro M, Barton ER, Sweeney HL, Rosenthal N. Localized Igf-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat Genet. 2001 Feb;27(2):195-200
377. Lewis MI, Bulut Y, Biring MS, Da X, Fournier M. (1999) IGF-I administration prevents corticosteroids-induced diaphragm atrophy in emphysema . Am J Respir Crit Care Med 159:A580
378. Fournier M, Huang ZS, Cercek B, Li H, Bykhovskaya I, Lewis MI. (2000) Administration of insulin-like growth factor-1 (IGF-I) and corticosteroids in emphysematous hamsters: influences on diaphragm IGF-I . Am J Respir Crit Care Med 161:A18
379. Shavlakadze T, Grounds M. Of bears, frogs, meat, mice and men: complexity of factors affecting skeletal muscle mass and fat. Bioessays. 2006 Oct;28(10):994-1009. Review.
380. Maiter D, Underwood LE, Maes M, Davenport ML, Ketelslegers JM. Different effects of intermittent and continuous growth hormone (GH) administration on serum somatomedin-C/insulin-like growth factor I and liver GH receptors in hypophysectomized rats. Endocrinology. 1988 Aug;123(2):1053-9.
381. Isgaard J, Carlsson L, Isaksson OG, Jansson JO. Pulsatile intravenous growth hormone (GH) infusion to hypophysectomized rats increases insulin-like growth factor I messenger ribonucleic acid in skeletal tissues more effectively than continuous GH infusion. Endocrinology. 1988 Dec;123(6):2605-10.
382. Clark RG, Jansson JO, Isaksson O, Robinson IC. Intravenous growth hormone: growth responses to patterned infusions in hypophysectomized rats. J Endocrinol. 1985 Jan;104(1):53-61.
383. Bick T, Hochberg Z, Amit T, Isaksson OG, Jansson JO. Roles of pulsatility and continuity of growth hormone (GH) administration in the regulation of hepatic GH-receptors, and circulating GH-binding protein and insulin-like growth factor-I. Endocrinology. 1992 Jul;131(1):423-9.
384. Weltman A, Weltman JY, Schurrer R, Evans WS, Veldhuis JD, Rogol AD. Endurance training amplifies the pulsatile release of growth hormone: effects of training intensity. J Appl Physiol (1985). 1992 Jun;72(6):2188-96.
385. Flores-Morales A, Greenhalgh CJ, Norstedt G, Rico-Bautista E. Negative regulation of growth hormone receptor signaling. Mol Endocrinol. 2006 Feb;20(2):241-53. Epub 2005 Jul 21. Review.
386. Hartman ML, Veldhuis JD, Thorner MO. Normal control of growth hormone secretion. Horm Res. 1993;40(1-3):37-47. Review.
387. Fernández L, Flores-Morales A, Lahuna O, Sliva D, Norstedt G, Haldosén LA, Mode A, Gustafsson JA. Desensitization of the growth hormone-induced Janus kinase 2 (Jak 2)/signal transducer and activator of transcription 5 (Stat5)-signaling pathway requires protein synthesis and phospholipase C. Endocrinology. 1998 Apr;139(4):1815-24.
388. Gebert CA, Park SH, Waxman DJ. Termination of growth hormone pulse-induced STAT5b signaling. Mol Endocrinol. 1999 Jan;13(1):38-56.
389. Ram PA, Waxman DJ. SOCS/CIS protein inhibition of growth hormone-stimulated STAT5 signaling by multiple mechanisms. J Biol Chem. 1999 Dec 10;274(50):35553-61.
390. Ram PA, Waxman DJ. Role of the cytokine-inducible SH2 protein CIS in desensitization of STAT5b signaling by continuous growth hormone. J Biol Chem.2000 Dec 15;275(50):39487-96.
391. Xu J, Keeton AB, Franklin JL, Li X, Venable DY, Frank SJ, Messina JL. Insulin enhances growth hormone induction of the MEK/ERK signaling pathway. J Biol Chem. 2006 Jan 13;281(2):982-92. Epub 2005 Nov 4.
392. Lewis TS, Shapiro PS, Ahn NG. Signal transduction through MAP kinase cascades. Adv Cancer Res. 1998;74:49-139. Review.
394. Mebis L, Paletta D, Debaveye Y, Ellger B, Langouche L, D’Hoore A, Darras VM, Visser TJ, Van den Berghe G. Expression of thyroid hormone transporters during critical illness. Eur J Endocrinol. 2009 Aug;161(2):243-50.
395. Jørgensen JO, Pedersen SA, Laurberg P, Weeke J, Skakkebaek NE, Christiansen JS. Effects of growth hormone therapy on thyroid function of growth hormone-deficient adults with and without concomitant thyroxine-substituted central hypothyroidism. J Clin Endocrinol Metab. 1989 Dec;69(6):1127-32.
396. Jørgensen JO, Pedersen SB, Børglum J, Møller N, Schmitz O, Christiansen JS, Richelsen B. Fuel metabolism, energy expenditure, and thyroid function in growth hormone-treated obese women: a double-blind placebo-controlled study. Metabolism. 1994 Jul;43(7):872-7.
397. Wolthers T, Grøftne T, Møller N, Christiansen JS, Orskov H, Weeke J, Jørgensen JO. Calorigenic effects of growth hormone: the role of thyroid hormones. J Clin Endocrinol Metab. 1996 Apr;81(4):1416-9.
398. Feldt-Rasmussen U. Interactions between growth hormone and the thyroid gland — with special reference to biochemical diagnosis. Curr Med Chem. 2007;14(26):2783-8. Review.
399. Kalina-Faska B, Kalina M, Koehler B. Effects of recombinant growth hormone therapy on thyroid hormone concentrations. Int J Clin Pharmacol Ther. 2004 Jan;42(1):30-4.
400. Hubina E, Mersebach H, Rasmussen AK, Juul A, Sneppen SB, Góth MI, Feldt-Rasmussen U. Effect of growth hormone replacement therapy on pituitary hormone secretion and hormone replacement therapies in GHD adults. Horm Res. 2004;61(5):211-7. Epub 2004 Jan 30.
401. Seminara S, Stagi S, Candura L, Scrivano M, Lenzi L, Nanni L, Pagliai F, Chiarelli F. Changes of thyroid function during long-term hGH therapy in GHD children. A possible relationship with catch-up growth? Horm Metab Res. 2005 Dec;37(12):751-6.
402. Losa M, Scavini M, Gatti E, Rossini A, Madaschi S, Formenti I, Caumo A, Stidley CA, Lanzi R. Long-term effects of growth hormone replacement therapy on thyroid function in adults with growth hormone deficiency. Thyroid. 2008 Dec;18(12):1249-54.
403. Müller MJ, Seitz HJ. Thyroid hormone action on intermediary metabolism. Part III. Protein metabolism in hyper- and hypothyroidism. Klin Wochenschr. 1984 Feb 1;62(3):97-102.
404. Tawa NE Jr, Odessey R, Goldberg AL. Inhibitors of the proteasome reduce the accelerated proteolysis in atrophying rat skeletal muscles. J Clin Invest. 1997 Jul 1;100(1):197-203. PubMed PMID: 9202072
405. Dace A, Zhao L, Park KS, et al. Hormone binding induces rapid proteasome-mediated degradation of thyroid hormone receptors. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(16):8985-8990.
406. Clément K, Viguerie N, Diehn M, Alizadeh A, Barbe P, Thalamas C, Storey JD, Brown PO, Barsh GS, Langin D. In vivo regulation of human skeletal muscle gene expression by thyroid hormone. Genome Res. 2002 Feb;12(2):281-91.
407. Miell JP, Taylor AM, Zini M, Maheshwari HG, Ross RJ, Valcavi R. Effects of hypothyroidism and hyperthyroidism on insulin-like growth factors (IGFs) and growth hormone- and IGF-binding proteins. J Clin Endocrinol Metab. 1993 Apr;76(4):950-5.
408. Murao K, Takahara J, Sato M, Tamaki M, Niimi M, Ishida T. Relationship between thyroid functions and urinary growth hormone secretion in patients with hyper- and hypothyroidism. Endocr J. 1994 Oct;41(5):517-22.
409. Wolf M, Ingbar SH, Moses AC. Thyroid hormone and growth hormone interact to regulate insulin-like growth factor-I messenger ribonucleic acid and circulating levels in the rat. Endocrinology. 1989 Dec;125(6):2905-14.
410. Laron Z. Interactions between the thyroid hormones and the hormones of the growth hormone axis. Pediatr Endocrinol Rev. 2003 Dec;1 Suppl 2:244-9-discussion 250. Review.
411. Fiems LO. Double Muscling in Cattle: Genes, Husbandry, Carcasses and Meat. Animals : an Open Access Journal from MDPI. 2012;2(3):472-506.
412. Langley B, Thomas M, Bishop A, Sharma M, Gilmour S, Kambadur R. Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J Biol Chem. 2002 Dec 20;277(51):49831-40. Epub 2002 Sep 18.
413. McPherron AC, Lee SJ. Double muscling in cattle due to mutations in the myostatin gene. Proc Natl Acad Sci U S A. 1997 Nov 11;94(23):12457-61.
414. Schuelke M, Wagner KR, Stolz LE, Hübner C, Riebel T, Kömen W, Braun T, Tobin JF, Lee SJ. Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med. 2004 Jun 24;350(26):2682-8.
415. Clop A, Marcq F, Takeda H, Pirottin D, Tordoir X, Bibé B, Bouix J, Caiment F, Elsen JM, Eychenne F, Larzul C, Laville E, Meish F, Milenkovic D, Tobin J, Charlier C, Georges M. A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat Genet. 2006 Jul;38(7):813-8.
416. Gonzalez-Cadavid NF, Taylor WE, Yarasheski K, Sinha-Hikim I, Ma K, Ezzat S, Shen R, Lalani R, Asa S, Mamita M, Nair G, Arver S, Bhasin S. Organization of the human myostatin gene and expression in healthy men and HIV-infected men with muscle wasting. Proc Natl Acad Sci U S A. 1998 Dec 8;95(25):14938-43.
417. Liu W, Thomas SG, Asa SL, Gonzalez-Cadavid N, Bhasin S, Ezzat S. Myostatin is a skeletal muscle target of growth hormone anabolic action. J Clin Endocrinol Metab. 2003 Nov;88(11):5490-6.
418. Oldham JM, Osepchook CC, Jeanplong F, Falconer SJ, Matthews KG, Conaglen JV, Gerrard DF, Smith HK, Wilkins RJ, Bass JJ, McMahon CD. The decrease in mature myostatin protein in male skeletal muscle is developmentally regulated by growth hormone. J Physiol. 2009 Feb 1;587(3):669-77.
419. Williams NG, Interlichia JP, Jackson MF, Hwang D, Cohen P, Rodgers BD. Endocrine actions of myostatin: systemic regulation of the IGF and IGF binding protein axis. Endocrinology. 2011 Jan;152(1):172-80.
420. Winbanks CE, Weeks KL, Thomson RE, Sepulveda PV, Beyer C, Qian H, Chen JL, Allen JM, Lancaster GI, Febbraio MA, Harrison CA, McMullen JR, Chamberlain JS, Gregorevic P. Follistatin-mediated skeletal muscle hypertrophy is regulated by Smad3 and mTOR independently of myostatin. J Cell Biol. 2012 Jun 25;197(7):997-1008.
421. Lach-Trifilieff E, Minetti GC, Sheppard K, Ibebunjo C, Feige JN, Hartmann S, Brachat S, Rivet H, Koelbing C, Morvan F, Hatakeyama S, Glass DJ. An antibody blocking activin type II receptors induces strong skeletal muscle hypertrophy and protects from atrophy. Mol Cell Biol. 2014 Feb;34(4):606-18.
422. Bark TH, McNurlan MA, Lang CH, Garlick PJ. Increased protein synthesis after acute IGF-I or insulin infusion is localized to muscle in mice. Am J Physiol. 1998 Jul;275(1 Pt 1):E118-23.
423. Barton-Davis ER, Shoturma DI, Sweeney HL. Contribution of satellite cells to IGF-I induced hypertrophy of skeletal muscle. Acta Physiol Scand. 1999 Dec;167(4):301-5.
424. Suryawan A, Frank JW, Nguyen HV, Davis TA. Expression of the TGF-beta family of ligands is developmentally regulated in skeletal muscle of neonatal rats. Pediatr Res. 2006 Feb;59(2):175-9.
425. Gilson H, Schakman O, Kalista S, Lause P, Tsuchida K, Thissen JP. Follistatin induces muscle hypertrophy through satellite cell proliferation and inhibition of both myostatin and activin. Am J Physiol Endocrinol Metab. 2009 Jul;297(1):E157-64.
426. Bodine SC, Stitt TN, Gonzalez M, Kline WO, Stover GL, Bauerlein R, Zlotchenko E, Scrimgeour A, Lawrence JC, Glass DJ, Yancopoulos GD. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol. 2001 Nov;3(11):1014-9.
428. Kalista S, Schakman O, Gilson H, Lause P, Demeulder B, Bertrand L, Pende M, Thissen JP. The type 1 insulin-like growth factor receptor (IGF-IR) pathway is mandatory for the follistatin-induced skeletal muscle hypertrophy. Endocrinology. 2012 Jan;153(1):241-53.
429. Barbé C, Kalista S, Loumaye A, Ritvos O, Lause P, Ferracin B, Thissen JP. Role of IGF-I in follistatin-induced skeletal muscle hypertrophy. Am J Physiol Endocrinol Metab. 2015 Sep 15;309(6):E557-67. doi: 10.1152/ajpendo.00098.2015. Epub 2015 Jul 28.
430. Coffey VG, Shield A, Canny BJ, Carey KA, Cameron-Smith D, Hawley JA. Interaction of contractile activity and training history on mRNA abundance in skeletal muscle from trained athletes. Am J Physiol Endocrinol Metab. 2006 May;290(5):E849-55.
431. Moore WV, Leppert P. Role of aggregated human growth hormone (hGH) in development of antibodies to hGH. J Clin Endocrinol Metab. 1980 Oct;51(4):691-7
432. Dannies PS. Protein folding and deficiencies caused by dominant-negative mutants of hormones. Vitam Horm. 2000;58:1-26. Review.
433. DeVol DL, Rotwein P, Sadow JL, Novakofski J, Bechtel PJ. Activation of insulin-like growth factor gene expression during work-induced skeletal muscle growth. Am J Physiol. 1990 Jul;259(1 Pt 1):E89-95.
434. Hermansen K, Bengtsen M, Kjær M, Vestergaard P, Jørgensen JOL. Impact of GH administration on athletic performance in healthy young adults: A systematic review and meta-analysis of placebo-controlled trials. Growth Horm IGF Res. 2017 Jun;34:38-44.
435. de Souza GL, Hallak J. Anabolic steroids and male infertility: a comprehensive review. BJU Int. 2011 Dec;108(11):1860-5.
436. Kraemer WJ, Marchitelli L, Gordon SE, Harman E, Dziados JE, Mello R, Frykman P, McCurry D, Fleck SJ. Hormonal and growth factor responses to heavy resistance exercise protocols. J Appl Physiol (1985). 1990 Oct;69(4):1442-50.
437. Pfeffer LA, Brisson BK, Lei H, Barton ER. The insulin-like growth factor (IGF)-I E-peptides modulate cell entry of the mature IGF-I protein. Mol Biol Cell. 2009 Sep;20(17):3810-7.
438. Mills P, Dominique JC, Lafrenière JF, Bouchentouf M, Tremblay JP. A synthetic mechano growth factor E Peptide enhances myogenic precursor cell transplantation success. Am J Transplant. 2007 Oct;7(10):2247-59.
439. Brisson BK, Barton ER. Insulin-like growth factor-I E-peptide activity is dependent on the IGF-I receptor. PLoS One. 2012;7(9):e45588.
440. Brisson BK, Spinazzola J, Park S, Barton ER. Viral expression of insulin-like growth factor I E-peptides increases skeletal muscle mass but at the expense of strength. Am J Physiol Endocrinol Metab. 2014 Apr 15;306(8):E965-74.
441. Goldspink G, Harridge S. Mechanism for adaptation in skeletal muscle In: Komi P, editor. Strength and power in sport: Olympic encyclopedia of sports medicine. Oxford: Blackwell; 2002. p. 231–51.
442. Janssen JA, Hofland LJ, Strasburger CJ, van den Dungen ES, Thevis M. Potency of Full-Length MGF to Induce Maximal Activation of the IGF-I R Is Similar to Recombinant Human IGF-I at High Equimolar Concentrations. PLoS One. 2016 Mar 18;11(3):e0150453.
Nei primi anni del XXI secolo, negli scaffali dei negozi di integratori (fisici o online) emersero alcuni nuovi supplementi e tra questi emerse l’Acido Tetradeciltioacetico [TTA]. In quel periodo questo particolare composto era uno dei principi attivi, insieme a molte altre sostanze, di alcuni controversi supplementi per la perdita di grasso. La maggior parte di questi prodotti è scomparsa dal mercato, ma l’Acido Tetradeciltiocetico è stato recentemente reinserito nel mercato. Ora, spesso in concentrazioni significativamente più elevate rispetto a un decennio fa, il TTA è spesso l’unico ingrediente attivo presente nei supplementi che lo contengono. E alcuni produttori di integratori vendono tale composto sotto forma di polvere. È quindi interessante approfondire le caratteristiche del composto e di cosa è ipoteticamente in grado di fare.
L’Acido Tetradeciltioacetico è un acido grasso sintetico che non può essere convertito in energia.
Negli anni ’90 gli scienziati scoprirono che l’Acido Tetradeciltioacetico stimolava il metabolismo dei grassi attivando i PPAR-α. I ricercatori hanno scoperto anche che, attraverso lo stesso meccanismo, la sostanza era in grado di inibire l’infiammazione, la crescita delle cellule tumorali e lo sviluppo/peggioramento dell’insulino-resistenza.
Esistono tre tipi di recettori PPAR: α, γ e δ. Il recettore α è espresso a livello epatico. Quando viene attivato, il fegato utilizza più acidi grassi per produrre energia. Il recettore γ è espresso nelle cellule adipose. Se viene stimolato, la cellula adiposa immagazzina acidi grassi. Il recettore δ è espresso a livello muscolare. Se il recettore δ viene stimolato, i muscoli utilizzano maggiormente gli acidi grassi per produrre energia.
Nel 2002, scienziati norvegesi hanno riferito che gli animali presi in esame, e che avevano ricevuto una dose significativa di Acido Tetradeciltioacetico per via orale, non hanno mostrato un aumento della percentuale di grasso corporeo in concomitanza con un regime alimentare ipercalorico.(1) La suddetta pubblicazione ha anche riportato che l’Acido Tetradeciltioacetico non solo stimolava il PPAR- α, ma anche il PPAR- γ e, in misura minore, il PPAR-δ.
Nel 2009 nutrizionisti affiliati all’Università di Oslo, hanno pubblicato un altro studio svolto su animali. In questo studio, i norvegesi hanno fatto ingrassare i ratti dando loro un mangime che conteneva una elevata percentuale di grasso [Lard] per 7 settimane.
La metà dei ratti presi in esame ha ricevuto anche Acido Tetradeciltioacetico [Lard Plus TTA]. Se i topi fossero stati esseri umani adulti, avrebbero consumato giornalmente circa 1,5g di Acido Tetradeciltioacetico.
L’integrazione con TTA ha significativamente inibito l’aumento del peso corporeo degli animali trattati. Tuttavia, però, la supplementazione ha indotto gli animali a consumare più cibo. Tale comportamento è strano, dal momento che la maggior parte degli agonisti PPAR-α diminuiscono l’appetito interferendo con la biosintesi di alcuni neuropeptidi ipotalamici.
Al termine delle 7 settimane del test, i ratti trattati con Acido Tetradeciltioacetico non erano solo più magri dei ratti non trattati. La loro composizione corporea differiva anche dai ratti del gruppo “Lard”: presentavano una miglior massa muscolare e una ridotta percentuale di grasso.
Di seguito viene mostrato come l’Acido Tetradeciltioacetico stimola il metabolismo nei ratti. Nel fegato, l’attività del gene UCP3, che potenzia il metabolismo, ha subito un incremento di non meno di un fattore di duemila.
I ricercatori scrivono che i loro dati sono in accordo nel riconoscere l’attivazione del PPAR- α come mediatore principale degli effetti dell’Acido Tetradeciltiocetico sull’espressione del gene epatico. L’effetto dell’Acido Tetradeciltiocetico sull’assunzione di cibo è risultato opposto a quanto riportato per altri ligandi il PPAR- α e può essere correlato all’attivazione concomitante del PPAR- γ, alla riduzione della segnalazione della Leptina o alla riduzione della sensibilità ipotalamica del malonil-CoA.
Per chiarire ulteriormente gli effetti dell’Acido Tetradeciltioacetico sull’assunzione di cibo e sull’efficienza del cibo, continuano i ricercatori, l’uso di gabbie metaboliche potrebbe chiarire laquestione. Inoltre, dovrebbero essere prese in considerazione le indagini sull’eventualità che l’Acido Tetradeciltioacetico si accumuli nell’ipotalamo e promuova […] i livelli di sazietà regolati dai neuropeptidi.
Quando i primi supplementi per la perdita di grasso contenenti Acido Tetradeciltioacetico sono comparsi nel mercato degli integratori nel primo decennio del XXI secolo, i produttori hanno cercato di prevenire l’aumento dell’appetito combinando il TTA con composti aventi azione anoressizzante l’Oleoietanolamide.
Se quei supplementi funzionassero davvero è un altro discorso. C’è da considerare anche che la quantità di Acido Tetradeciltioacetico in essi contenuta era bassa. Tuttavia, con i nuovi prodotti presenti sul mercato, le dosi di TTA sono molto più alte.
Sebbene in numero limitato, esistono degli studi svolti sull’uomo che hanno osservato l’effetto del TTA… e i risultati ottenuti non sono così promettenti come quelli osservati sugli animali.
Nel 2004, i ricercatori del Rikshospitalet di Oslo hanno somministrato a 10 pazienti affetti da HIV 1g di TTA al giorno per 4 settimane.(2) L’integrazione non ha avuto alcun effetto sulle cellule T, ma ha ridotto la concentrazione di TNF-α, LDL e Trigliceridi nel sangue. Ma la perdita di peso? Non si è verificata.
Nel 2008, i ricercatori norvegesi, affiliati all’Università di Bergen, hanno somministrato a 18 volontari 200mg, 600mg o 1g di TTA al giorno durante uno studio di fase 1 per 7 giorni. Non sono emersi effetti collaterali degni di nota.(3) Per quanto possibile dedurre da un test della ristretta durata di 7 giorni, il TTA è risultato essere sicuro.
Nel 2009, i ricercatori norvegesi dell’Università di Bergen hanno pubblicato un piccolo studio svolto su esseri umani nel quale hanno somministrato a 16 uomini con diabete di tipo II 1g di TTA ogni giorno per 4 settimane. Gli uomini seguivano una dieta apposita o usavano farmaci per il trattamento del diabete. Alcuni di loro usavano anche statine.
Ovviamente, i livelli di colesterolo e trigliceridi degli uomini sono migliorati (effetto da attribuirsi maggiormente ai farmaci utilizzati), ma non hanno mostrato alcuna perdita di peso.
Nella stessa pubblicazione, i ricercatori parlano anche di esperimenti con cellule muscolari umane. In concentrazioni di diverse decine di micromoli, il TTA ha aumentato l’ossidazione degli acidi grassi.
Nelle cellule muscolari la TTA ha aumentato la produzione di Carnitina Palmitoiltransferasi-I, un enzima coinvolto nell’ossidazione degli acidi grassi, e di CD36, una proteina che è coinvolta nell’assorbimento cellulare degli acidi grassi.
Tuttavia, questi effetti apparentemente non sono abbastanza incisivi da causare una perdita di peso. Forse sono annullati da un aumento dell’appetito, che è stato dimostrato negli studi svolti su animali precedentemente esposti.
Come accennato al principio di questo articolo, il TTA è un acido grasso sintetico, che non si trova in natura. Necessita di essere consumato nel ordine dei grammi [una capsula generalmente può contenerne 0,5g]. Il TTA non viene utilizzato come fonte energetica dall’organismo. Inoltre, il TTA è una sostanza con attività farmacologica. È a tutti gli effetti un farmaco sperimentale, la cui sicurezza non è stata ancora studiata adeguatamente. E negli studi sull’uomo, non sembra funzionare.
In seguito a quanto esposto, non considero una supplementazione con TTA una buona idea sia per la probabile inefficacia in termini di perdita di grasso e sia per le incognite di un suo uso sul lungo termine.
Se non avete ancora letto la prima e la seconda parte di questa serie di articoli vi invito a farlo prima di procedere con la lettura di questa terza parte: 1° Parte – 2° Parte.
Effetti diretti di GH e IGF-1 sull’ipertrofia muscolare
Andiamo dritti al punto – di per sé, il GH non causa direttamente ipertrofia muscolare. Questa caratteristica è stata ampiamente osservata per decenni e, finora, nessuno studio attendibile è stato in grado di mostrare un chiaro effetto della somministrazione di rHGH a medio-lungo termine sull’ipertrofia – anche in dosi sovrafisiologiche somministrate ad atleti di alto livello sottoposti ad intensi allenamenti contro resistenza.
Il fatto che questa correlazione non sia stata dimostrata non è certamente dovuto alla mancanza di tentativi in tal senso. Diversi gruppi di ricerca nel corso degli anni hanno tentato di identificare un ipotetica ipertrofia GH-mediata in soggetti adulti sani [48,195-199] o in soggetti anziani [188,198,200-203] senza successo, o in modo inconcludente. Inoltre, è stato dimostrato che gli aumenti della secrezione di GH indotta dall’esercizio in acuto non hanno prodotto cambiamenti nella MPS o nell’ipertrofia [204-205]. È interessante notare che, sebbene la quantità di studi presenti in letteratura siano significativamente meno comuni di quelli in cui è stato somministrato GH, anche la somministrazione sistemica di rhIGF-1 non ha prodotto alcun effetto ipertrofico misurabile sia in soggetti giovani [63,206] che negli anziani [207- 208].
Vi sono prove recenti che suggeriscono che l’esposizione cronica al GH aumenta l’espressione delle vie intramuscolari responsabili dell’atrofia.[209] È ovvio che molte delle caratteristiche anaboliche dimostrate dal GH possono essere compensate da questa maggiore espressione del catabolismo, che potrebbe essere il motivo per cui l’esposizione cronica al GH non porta all’ipertrofia. Potrebbe anche essere responsabile del perché l’esposizione cronica al GH può produrre muscoli meno efficienti e più deboli.[210] Molto semplicemente, questo potrebbe essere ancora un altro fattore nella lunga serie di effetti regolatori negativi intrinseci nell’Asse GH/IGF-1, ma ulteriori studi dovranno essere condotti per chiarire maggiormente questa ipotesi.
Per quelli che difficilmente accettano le informazioni riportate in questa serie di articoli, e vogliono addentrarsi loro stessi nella letteratura scientifica sperando di trovare informazioni che confutino quanto da me riportato, voglio esporre un concetto molto importante da me già espresso nella prima parte di questa serie di articoli. La maggior parte degli studi che hanno preso in esame il GH segnalano un aumento della massa magra nei soggetti dei gruppi trattati con tale ormone. Quindi, ad un lettore inesperto, potrebbero risultare infondate le affermazioni sulla mancanza di un effetto ipertrofico diretto del GH. Bisogna ricordare, però, che il GH causa una non indifferente ritenzione idrica, oltre ad aumentare la massa dei tessuti molli della quale parlerò brevemente. Nello specifico, il GH aumenta la ritenzione di sodio e, di conseguenza, dell’acqua extracellulare, in modo dose-dipendente, attraverso i suoi effetti sul sistema renina-angiotensina.[211] Questi incrementi nella ritenzione idrica e sodica sono stati osservati anche con la somministrazione di IGF-1, poiché l’IGF-1 stesso sembra essere un regolatore chiave del tasso di escrezione renale di sodio.[83,212-213] Quindi il punto della questione è che bisogna stare molto attenti nel trarre conclusioni quando non si ha ben chiara la differenza tra aumento della massa magra e crescita effettiva del tessuto muscolare.
Effetti indiretti di GH e IGF-1 sull’ipertrofia muscolare
Nella sezione precedente si è dimostrato che il GH e l’IGF-1, da soli, non hanno alcun impatto diretto sull’ipertrofia. Tuttavia, questo non significa che questi peptidi non abbiano un ruolo nei processi ipertrofici. L’obiettivo principale di questa sezione è quello di spiegare alcuni dei molti meccanismi che interessano in modo indiretto il GH e l’IGF-1 nei processi ipertrofici, molti dei quali saranno fattori importanti per gli atleti interessati a massimizzare il loro potenziale ipertrofico.
L’Ormone della Crescita è un potente stimolatore della sintesi di collagene sia nei tendini che nei muscoli. Questo effetto è probabilmente mediato dalla capacità dell’IGF-1 autocrino di stimolare i fibroblasti per sintetizzarlo.[214-215] In realtà, questo processo avviene senza che il peptide influenzi la sintesi proteica muscolare, nonostante i livelli di IGF-1 circolante e di quello locale siano significativamente più alti. Questo effetto è anche indotto indipendentemente dall’allenamento contro resistenza ed è stato persino osservato in soggetti immobilizzati hai quali è stato somministrato il GH.[216] La componente connettiva del muscolo scheletrico è vitale per la trasmissione della forza, che è prodotta dalle fibre muscolari, ai tendini e alle ossa affinché si verifichi il movimento. In particolare, il collagene è un importante componente nella trasmissione della forza della matrice extracellulare, che viene continuamente sottoposta a carichi intesi durante i movimenti.
A causa dei potenti effetti del GH sui componenti della matrice extracellulare, si può chiaramente iniziare a capire il perché gli aneddoti nel corso degli anni suggerivano che l’aggiunta del GH in una preparazione farmacologica produceva impatti positivi sulla riduzione del dolore tendineo-articolare. D’altra parte però, questo potrebbe anche essere un fattore che contribuisce in primo luogo al motivo per cui vari effetti collaterali sono segnalati dagli utilizzatori di GH come l’edema dei tessuti molli, il dolore articolare e la sindrome del tunnel carpale.[217-219] Ci sono anche molti che credono che il GH possa accelerare i tempi di recupero dagli infortuni, ma questo è un argomento complesso che verrà discusso in futuro.
Gli impatti del GH sulla sintesi di collagene potrebbero anche essere di grande interesse per gli atleti di forza che non sono necessariamente interessati all’ipertrofia, ma il cui obiettivo principale è la creazione di una condizione favorevole allo spostamento del carico massimale. Stimolare la sintesi del collagene potrebbe aiutare a rafforzare l’intero sistema di supporto dei muscoli scheletrici. Ora, vale la pena aggiungere una piccola nota di chiarimento. Nonostante questo suoni positivo in linea di principio, la supplementazione di GH non ha mai portato direttamente a guadagni di forza in nessuno degli studi svolti su soggetti sani, coprendo vari gruppi.[48,50,184,196-198,200-201,203,220-221] Naturalmente, se il GH fosse usato insieme ad un composto con la capacità di aumentare direttamente la forza, non è difficile supporre che in questo caso l’effetto addizionale potrebbe fare del GH un prezioso componente della preparazione.
Decorina
La Decorina è una proteina strutturale, che risiede principalmente nella matrice extracellulare del muscolo scheletrico e il cui ruolo è correlato alla crescita e alla riparazione dei muscoli.[222-223] Qualche decennio fa è stato dimostrato per la prima volta che la somministrazione di GH potrebbe aumentare direttamente l’espressione del gene della Decorina negli animali [224]. Recentemente, è stato dimostrato che questo effetto si manifesta anche in soggetti umani sottoposti ad allenamenti ricreativi.[225] Nello studio più recente, i livelli di Decorina sono fortemente correlati a quelli di PIIINP, che induce la stimolazione del Decorina GH-mediata che può essere coinvolta nel processo di assemblaggio della matrice di collagene osseo. Questo effetto è più pronunciato negli uomini rispetto alle donne e può essere un sottoprodotto dei livelli più alti di IGF-1 osservati negli uomini, sebbene questa affermazione sia speculativa. L’aumento dell’espressione di Decorina non è stato alterato dall’aggiunta di Testosterone, quindi questo effetto è indipendente dagli androgeni. Dopo aver visto gli effetti che il GH ha sulla sintesi di collagene e Decorina, sta diventando piuttosto chiaro che l’asse GH/IGF-1 è molto più importante per rafforzare la matrice extracellulare di supporto piuttosto che contribuire direttamente alla crescita del tessuto muscolare.
Adenosina Trifosfato (ATP)
La somministrazione acuta di GH in soggetti sani ha anche dimostrato di causare una maggiore produzione di ATP mitocondriale e una maggiore attività della citrato sintasi nel muscolo scheletrico, con una maggiore abbondanza di mRNA muscolari codificanti l’IGF-1.[226] Non solo questo potrebbe essere un fattore che contribuisce al motivo per cui il GH potrebbe avere la capacità di promuovere un aumento dei tassi di spesa energetica giornaliera ma potrebbe anche essere coinvolto nello spostamento verso la preferenza dei lipidi come substrato energetico. Anche se, come abbiamo visto in precedenza, il corpo della letteratura non supporta questa pratica, l’aumento della produzione di ATP mitocondriale potrebbe avere un ruolo nella capacità aerobica.[227]
È stato dimostrato che il GH promuove la fusione dei mioblasti con i miotubi in modelli cellulari [84], un effetto completamente indipendente dalla sovraregolazione locale del IGF-1. Per capire perché questo possa essere importante, bisogna approfondire maggiormente la questione dei fattori cellulari coinvolti nell’ipertrofia del muscolo scheletrico. L’ipertrofia dei muscoli scheletrici negli esseri umani si basa sulle cellule satelliti, che sono cellule dormienti situate all’interno delle miofibre, proprio sotto lo strato di lamina basale nella matrice extracellulare.[148] Una volta attivate queste cellule satellite, spesso con l’esercizio fisico o il danno muscolare, proliferano. Dopo la proliferazione, queste cellule satellite migrano verso siti ove si trova il danno differenziandosi e fondendosi con le miofibre esistenti che forniscono nuovi nuclei per l’ipertrofia e la riparazione. [228] Non è ancora del tutto chiaro se il GH abbia un effetto diretto sulla proliferazione e la differenziazione delle cellule satelliti.[229-230] Vale anche la pena di affermare che ciò che si osserva nelle colture cellulari potrebbe non essere del tutto indicativo di ciò che accade in vivo a causa di vari fattori esterni che non possono essere spiegati in condizioni di laboratorio.
Ci sono due fasi distinte in questa fusione dei mioblasti che si verifica.[85] Il primo sarebbe lo stadio iniziale della differenziazione in cui un sottoinsieme di cellule mononucleate si fondono per formare miotubi nascenti (fusione mioblasto/mioblasto). Questo è seguito dal secondo stadio che coinvolge ulteriori cellule disponibili che si fondono con questi miotubi nascenti e dove avviene la crescita muscolare effettiva (fusione mioblasto/miotubo). È all’interno di quest’ultimo stadio in cui il GH esercita i suoi effetti.
Questa è una scoperta piuttosto interessante, ma ancora una volta, numerosi studi sugli esseri umani non sono riusciti a dimostrare un effetto ipertrofico del GH in condizioni reali. Quindi, possiamo probabilmente dedurre da ciò che gli effetti che il GH ha sulla fusione dei nascenti miotubi non si traducono direttamente nell’ipertrofia. Tuttavia, cosa accadrebbe se si aggiungesse un’altra variabile nell’equazione in grado di creare un ambiente in cui esistessero numeri di celle satelliti potenziati, creando più materiale sul quale l’azione del GH possa manifestarsi?[231]
Introduzione degli AAS
A differenza del GH e del IGF, l’uso degli AAS ha mostrato di avere un impatto pronunciato su ipertrofia e forza. Questo è stato ben noto per decenni anche perché sono stati usati e abusati da atleti fin dalla loro creazione negli anni ’30 (con buona pace dei “puritani” della “old school”).[232] La famiglia degli AAS consiste in un gruppo di potenti composti sintetici che sono simili nella struttura chimica al Testosterone e/o al suo derivato 5α-ridotto (DHT). Vari tipi di singoli composti AAS sono stati creati nel corso degli anni usando come base la molecola naturale di Testosterone manipolandola, quindi, attraverso l’aggiunta di un gruppo etile, metile, idrossile o benzile in uno o più siti lungo la sua struttura.[233-234 ] Alcune delle varianti AAS più facilmente riconoscibili includono i composti metilati in C-17, i quali sono notoriamente dotati di un elevata biodisponibilità orale, e le varianti del 19-nortestosterone nelle quali viene rimosso il gruppo 19-metilico dalla molecola di Testosterone nel tentativo di aumentare la sua attività anabolica diminuendo al contempo la sua androgenicità e tendenza all’aromatizzazione. Conosciuti anche come “19-norsteroidi”, questa famiglia include ben note varianti di AAS come il Nandrolone ed il Trenbolone.
Molte di questi composti sono nati come risultato di un desiderio di aumentare le caratteristiche anaboliche del Testosterone a livello muscolare, abbassando allo stesso tempo gli effetti collaterali androgenici intrinsecamente inerenti alla molecola di Testosterone.[235] In generale, i rischi complessivi degli effetti collaterali derivanti dall’uso cronico di AAS sembrano essere relativamente bassi rispetto a molte sostanze socialmente accettate come l’alcol, il tabacco e vari farmaci da prescrizione.[233,236] Ovviamente, l’uso e l’abuso sono termini che si escludono a vicenda e abusare di qualsiasi sostanza tende a creare un ambiente a più alto rischio di effetti collaterali. A meno che non sia diversamente specificato, da questo punto in poi, parlerò specificamente del Testosterone in quanto è l’ormone sessuale endogeno maschile nativo, nonché l’androgeno più approfonditamente studiato in letteratura. Tanto per avere un riferimento, i maschi adulti sani producono una media di 14-77mg/settimana di Testosterone endogeno. [435]
Testosterone
Il Testosterone è un noto regolatore della massa muscolare, e gli aumenti della massa muscolare mediati dal Testosterone sono associati all’ipertrofia delle fibre, così come ad un aumento delle cellule satelliti e del numero mio nucleare.[231,237-240] Il muscolo scheletrico sembra essere uno dei tessuti più reattivi al Testosterone e si stima che i livelli circolanti di Testosterone rappresentino il fattore causale significativo (incidenza del 40-75%) dei guadagni nella massa muscolare osservati in studi di controllo randomizzati. Se ricordate ciò che è stato affermato e dimostrato nella precedente sezione, il GH possiede un’abilità molto particolare in quanto può aumentare la fusione dei mioblasti durante il processo ipertrofico. La massimizzazione di questa capacità si basa sull’avere un numero adeguato di cellule satellitari disponibili.
I risultati mostrano costantemente che i trattamenti con Testosterone determinano aumenti dose-dipendenti della massa e della forza del muscolo scheletrico, indipendentemente dal fatto che i soggetti presi in esame siano maschi più giovani o più anziani.[241-243] Al contrario, negli studi con soggetti sani giovani in cui il Testosterone endogeno è stato soppresso artificialmente, la forza e la composizione corporea hanno entrambi subito peggioramenti significativi.[244] Per ribadire il nostro precedente punto, gli aumenti della massa muscolare mediati dal Testosterone sono basati sull’ipertrofia e non sono il risultato di una transizione della fibra (cambiando le fibre di tipo I in fibre di tipo II o viceversa) o di una sua scissione.[245] A causa dei loro meccanismi anabolici unici e non sovrapposti, la somministrazione di Testosterone e l’allenamento contro resistenza hanno anche mostrato effetti sinergici e additivi l’uno sull’altro per quanto riguarda lo stimolo dell’aumento della massa muscolare.[246]
Recettore degli Androgeni (AR)
Gli Androgeni mediano principalmente i loro effetti attraverso il gene del recettore degli androgeni (AR) che è espresso in mioblasti, miofibre e cellule satelliti.[247-248] I AR sono stati rilevati anche nelle cellule che supportano i muscoli, come i fibroblasti e le cellule endoteliali. La densità dei AR sembra essere specifica per i gruppi muscolari, con l’allenamento contro resistenza e l’uso di AAS che hanno la capacità di influenzare il numero di AR presenti in questi gruppi muscolari. Oltre ai suoi effetti sulla densità dei AR, l’uso di AAS ha anche dimostrato la capacità di influenzare i livelli di attività dei AR sia in modo acuto che sul lungo termine.[249-250] Questi sono fattori piuttosto importanti da considerare quando ci si imbatte in individui che sostengono che il precedente utilizzo di AAS non offre necessariamente un vantaggio competitivo permanente.
A causa della complessità dell’argomento, ci sono state diverse ipotesi riguardo ai meccanismi con cui l’AAS esercita le sue azioni anaboliche sul muscolo scheletrico.[251] È stato dimostrato che i trattamenti con Testosterone aumentano i tassi di sintesi proteica muscolare (MPS) [252], riducono i tassi di degradazione delle proteine [253] facendo persino in modo che il corpo utilizzi più efficientemente gli amminoacidi prontamente disponibili. Quindi, ancora una volta, questo è un sistema abbastanza complesso che può anche essere semplificato ricordando che gli AAS promuovono l’anabolismo muscolare attraverso la loro capacità di incidere positivamente sull’equilibrio degli aminoacidi.
Via Wnt/B-catenina
È generalmente accettato che gli AAS esercitino i loro effetti anabolizzanti attraverso il legame e attivazione del AR che successivamente attiva cascate di segnalazione a valle che coinvolgono la via Wnt/β-catenina.[254-256]. I Wingless-INT (Wnt) sono una famiglia di glicoproteine secrete che regolano la proliferazione e la differenziazione cellulare.[257-258] I modelli cellulari hanno mostrato che il AR forma un complesso con la beta-catenina che si potenzia in presenza di AAS. [259-260] Una volta attivato questo complesso, migra nei nuclei in cui regola l’espressione dei geni target e la differenziazione delle cellule satelliti.[261-262] Questo è anche il pathway degli AAS in gran parte responsabile della miogenesi, la formazione del tessuto muscolare.[263-265]
Vale la pena notare che l’AAS possiede anche caratteristiche non genomiche che possono influenzare rapidamente numerosi processi ormonali e metabolici al di fuori del classico legame con il recettore. In letteratura sono stati riportati casi di maschi adulti con disturbi di insensibilità agli androgeni, causati da mutazioni del AR, che hanno risposto al Testosterone in modo molto simile ai soggetti sani. Questi casi studio rafforzano l’ipotesi che gli effetti anabolici dell’AAS possano essere mediati indipendentemente dal AR.[266] Le azioni non genomiche degli Androgeni possono in realtà essere un argomento piuttosto affascinante, ma che va oltre lo scopo previsto per questo articolo. Per coloro che vogliono approfondire l’argomento, consiglio di iniziare con le seguenti note di riferimento.[267-268]
AAS e potenziale sinergico con l’Asse GH/IGF-1
Ho riportato molte informazioni utili fino a questo punto, ma è qui che le cose cominciano davvero a farsi interessanti. A questo punto una domanda logica sarebbe: esistono studi svolti su soggetti sani nei quali si sono confrontate le differenze tra trattamenti singoli con GH o Androgeni e trattamenti combinati? Fortunatamente per noi, la risposta è “sì” in quanto vi sono stati alcuni studi, principalmente utilizzando soggetti anziani, sia maschi che femmine. I risultati di ognuno di questi studi dimostrano chiaramente che il GH ha un effetto additivo sui benefici consolidati che la terapia con ormoni sessuali fornisce: l’ipertrofia, la lipolisi, la sintesi del collagene, la funzione fisica, la qualità della vita e altri vari indicatori di prestazione.[187-188,269-270] Dato che è abbastanza chiaro che esiste un effetto additivo, vediamo se è possibile approfondire ulteriormente l’argomento al fine di scoprire alcuni dei meccanismi sottostanti che operano nella sinergia tra Androgeni e GH.
GHRH
Deve essere chiaro che il Testosterone, di per sé, ha un effetto additivo sull’intero Asse GH/IGF-1. Questo è stato osservato sia in modelli umani che animali, con la somministrazione di Testosterone che porta ad un aumento dei livelli circolanti di GH e IGF-1.[241,271-276] Viceversa, il deficit di Testosterone è comunemente associato a livelli significativamente ridotti di IGF-1.[277] L’effetto stimolante del Testosterone sull’Asse GH/IGF-1 sembra essere mediato a livello ipotalamico da una promozione della funzionalità del GHRH.[278]
Inoltre, e questo è bene ricordarlo, gli AAS non aromatizzabili sembrano non possedere lo stesso effetto stimolante sull’Asse GH/IGF-1.[279] Gli inibitori dell’aromatasi (AI), progettati per sopprimere il processo di aromatizzazione degli Androgeni, hanno dimostrato di attenuare direttamente la stimolazione del GH seguente la somministrazione di Testosterone. Questi indizi forniscono prove abbastanza convincenti sul fatto che gli estrogeni locali, derivanti dall’aromatizzazione, svolgono un ruolo fondamentale nella regolazione della secrezione di GH nei maschi.[280-281] Poiché l’aromatasi non è espressa nel fegato, gli AI non influenzano l’azione epatica del GH, mentre influenzano la sua azione a livello del sistema centrale.[282-283] Tuttavia i modulatori selettivi del recettore degli estrogeni (SERM) sono ancora più soppressivi in quanto agiscono ad entrambi i livelli a causa del loro meccanismo d’azione.[284-285]
Anche gli Androgeni che aumentano i livelli serici di Estrogeni, come il Nandrolone (Nor-Estrogeni), mostrano un effetto minimo sui livelli sistemici di GH e IGF-1 rispetto al Testosterone.[286] Ciò è legato ipoteticamente sia al fatto che il Nandrolone è poco soggetto all’aromatizzazione e sia al fatto che la forma estrogenica derivata (Nor-Estrogeno) abbia un ridotto potenziale di attività.[287] Ricordiamoci che l’aromatizzazione sembra essere il passo più cruciale nella stimolazione ipotalamica mediata dagli Androgeni. Ora, è ovvio che un utilizzatore di rHGH deve preoccuparsi parzialmente di questo effetto rispetto ad un non utilizzatore, considerando il fatto che, in tal caso, i livelli ormonali sono controllati quasi esclusivamente da mezzi esogeni. Detto questo, è sempre utile analizzare il quadro generale, soprattutto se l’obiettivo è massimizzare l’ipertrofia.
GHR
Un altro potenziale motivo legato agli aumenti dei livelli di GH e IGF osservati durante i trattamenti con Testosterone, è rappresentato dagli effetti diretti di quest’ultimo sui GHR. Sia gli studi sull’uomo che sugli animali hanno fornito prove del fatto che il Testosterone modifica i GHR sia nel fegato che nei tessuti periferici, migliorandone l’espressione.[288-289] Inoltre, soggetti umani ipopituitari e ipogonadici sottoposti a trattamenti con GH hanno mostrato una aumentata risposta sia all’IGF-1 locale che all’espressione del gene del recettore degli androgeni quando sottoposti anche a trattamento con Testosterone.[187,290-291] Inoltre, i maschi ipopituitari sottoposti a trattamenti con Testosterone hanno mostrato effetti notevoli sull’anabolismo proteico in presenza di GH, con il sito primario di interazione ormonale a livello epatico.[292] Quindi anche quando i livelli ormonali sono carenti, c’è ancora un’interazione molto importante tra il Testosterone e l’Asse GH/IGF-1.
Come accennato in precedenza, il GHR è espresso in quasi tutti i principali tipi di tessuto. Vale la pena sottolineare che il GHR è espresso in quantità molto bassa nel muscolo scheletrico – solo circa il 4-33% dei livelli osservati in altri tessuti. D’altra parte, il recettore del IGF-1 è espresso in maniera molto più elevata nei muscoli scheletrici, proprio come nel tessuto epatico.[293-294] Detto questo, l’aumento della sensibilità del GHR in un ambiente con livelli sovra fisiologici di GH andrà certamente a beneficio del BodyBuilder. I BodyBuilder cercano continuamente di utilizzare quantità elevate di rHGH nel tentativo di massimizzare i processi ipertrofici, e se il GH vede potenziata la sua azione e, quindi, il suo stimolo diretto sulla sintesi e rilascio del IGF-1, il quale opera direttamente sul tessuto muscolare, venendosi a creare quindi una vantaggiosa alterazione dell’Asse GH/IGF-1, l’effetto ipertrofico potenziato sarà indubbio.
IGFBP-4
Gli Androgeni hanno mostrato di avere la capacità di aumentare l’espressione del mRNA del IGF-1 locale nel muscolo scheletrico. Possiamo pertanto ipotizzare che gli Androgeni, in particolare a dosi più elevate, creino un ambiente all’interno del muscolo scheletrico estremamente favorevole alla gestione di livelli più alti di IGF-1 seguenti alla somministrazione di dosi sovrafisioogiche di rHGH. Ci sono persino stati studi sull’uomo che hanno mostrato livelli ridotti di IGFBP-4 locale nei campioni di tessuto muscolare, oltre a maggiori livelli di mRNA del IGF-1. Ciò potrebbe indicare che i cambiamenti hanno avuto luogo in quei muscoli per liberare più IGF-1 locale affinché esso possa legarsi ai suoi recettori.[277,295] Non ho molto approfondito la questione delle proteine leganti l’IGF, ma l’IGFBP-4 inibisce l’azione dell’IGF-1 e, quindi, minore è il livello della proteina legante, maggiori saranno i livelli di IGF-1 libero e attivo.[149,296]
È stato anche dimostrato che il Testosterone promuove l’ipertrofia negli stati deficitari di GH/IGF-1. [297-298] Questo è interessante in quanto dimostra che il Testosterone possiede sia vie metaboliche IGF-indipendenti che IGF-indipendenti nel tessuto muscolare.[299] Inoltre, i modelli cellulari hanno dimostrato che il Testosterone può sovraregolare l’espressione delle varie isoforme di IGF nei muscoli scheletrici, anche in assenza di GH/IGF-1.[298] E, anche se questo è stato dimostrato nei fibroblasti, il Testosterone ha mostrato di aumentare l’espressione del IGFBP-3 – un effetto che è stato ulteriormente migliorato dalla somministrazione di IGF-1.[300] È abbastanza chiaro, comunque, che il Testosterone eserciti effetti sinergici e additivi sull’anabolismo mediato da GH/IGF-1.
Fino ad ora mi sono concentrato sul Testosterone, tuttavia, però, sono stati osservati comportamenti leggermente diversi in relazione alle varianti androgene e al loro impatto sull’espressione sistemica e locale del IGF-1. Volevo analizzare un paio di specifici composti che sono spesso presenti nei protocolli farmacologici: sto parlando del Trenbolone e del Nandrolone. Salvo diverse indicazione, i risultati in seguito esposti provengono da studi svolti su animali.
Nandrolone
La somministrazione di Nandrolone ha costantemente dimostrato di non causare cambiamenti nei livelli di IGF-1 endocrino, nonostante produca simultaneamente un’espressione del IGF-1 muscolare significativamente più alta e un aumento della CSA delle fibre muscolari.[286,301-302] Inoltre, i livelli di IGFBP-3 locali sono stati riportati come significativamente più alti mentre i livelli di IGFBP-4 sono stati significativamente soppressi, il che ci rimanda a quanto esposto in precedenza – cioè ad un fattore che determina un aumento dei livelli di IGF-1 locale libero. In tutti gli studi, la somministrazione di Nandrolone ha portato direttamente ad un aumento dell’ipertrofia, nonostante non abbia avuto alcun impatto sui livelli di IGF-1 sistemici. Ciò rafforza ulteriormente l’ipotesi secondo cui l’IGF-1 endocrino non sia un fattore primario nell’ipertrofia dei muscoli scheletrici e quindi i livelli elevati non sono un prerequisito per l’aumento della massa muscolare.[303-305]
Trenbolone
È stato inoltre universalmente dimostrato che il Trenbolone aumenta i tassi di crescita muscolare in tutte le varie specie nelle quali è stato testato. A differenza del Testosterone e del Nandrolone, non si converte in estrogeno ed è stato suggerito fin dagli anni ’70 che l’aggiunta di Estradiolo al Trenbolone sembra migliorare gli effetti anabolici del composto.[306-307] Ci sono stati anche effetti potenziati sull’ipertrofia quando il Trenbolone è stato somministrato insieme ad un fattore di rilascio dell’Ormone della Crescita (GHRF).[308] Come accennato in precedenza, l’aumento di GH/IGF-1 necessita di livelli adeguati di estrogeni (derivanti principalmente dall’aromatizzazione) affinché questi esercitino uno stimolo dell’asse GH/IGF-1. Poiché la somministrazione di Trenbolone diminuisce intrinsecamente i livelli di Estradiolo, mediante inibizione a feedback negativo del Testosterone attraverso l’Asse Ipotalamo-Ipofisi-Gonadi (HPG) [309], la somministrazione di Estradiolo, tecnicamente, dovrebbe migliorare la funzionalità dell’asse GH/IGF-1. Questo dovrebbe quindi favorire ulteriormente la sinergia anabolica che avrebbe con l’androgeno. Questa ipotesi è in linea con ciò che vari studi hanno dimostrato nel corso degli anni.
Nelle colture cellulari, è stato anche dimostrato che gli Estrogeni alterano direttamente i tassi di MPS e MPB del Trenbolone attraverso meccanismi che coinvolgono sia il recettore estrogenico che il recettore del IGF-1.[310-311] In effetti, in linea di massima, i trattamenti con solo Trenbolone non mostrano aumenti significativi dei livelli endocrini o autocrini del IGF-1. Tuttavia, la co-somministrazione con Estradiolo ha tradizionalmente mostrato aumenti nei livelli di IGF-1 autocrini, similmente a quanto osservato con il Testosterone.[312-314] Questa è solo un’ulteriore prova che suggerisce che l’Estrogeno, sia di origine sistemica che derivata dall’aromatasi, è un componente chiave sia della massima stimolazione dell’Asse GH/IGF-1 che delle capacità anaboliche massimali degli Androgeni.
Proprio come il suo “cugino” 19-nor, il Trenbolone ha anche mostrato una maggiore espressione del fattore di crescita nei tessuti muscolo scheletrici, oltre ad una maggiore reattività dei muscoli a tali fattori di crescita.[315] Il Trenbolone ha anche mostrato di operare un aumento dell’attivazione e della proliferazione delle cellule satelliti in varie specie, in misura simile al Testosterone.[316-317] Ora, essendo a conoscenza delle interazioni legate al GH, si può facilmente giungere alla conclusione che entrambi questi effetti sarebbero vantaggiosi in un protocollo che includesse entrambi i composti.
PIIINP
Tornando al Testosterone, sia il GH che questo Androgeno aumentano i marker di sintesi del collagene come il PIIINP. Inoltre, il Testosterone ha anche dimostrato di potenziare le capacità del GH di aumentare la sintesi di collagene sia nei muscoli che nei tendini.[318] A sostegno di ciò, la somministrazione concomitante di GH e Testosterone in soggetti umani allenati (a livello amatoriale) ha causato aumenti significativi sia dei marker del IGF-1 che del collagene.[319] E’ interessante notare come alcuni di questi stessi marker del collagene dei quali si sta discutendo sono indicatori esatti che vengono esaminati come parte dei test antidoping per la rilevazione del GH.[320-321]
In precedenza ho solo brevemente toccato il percorso JAK-STAT ma, arrivati a questo punto, è necessario trattare l’argomento in maniera leggermente più approfondita. La via JAK-STAT è un componente critico del GH e riguarda sia la trascrizione del gene del IGF-1 che la crescita postnatale. Una delle proteine STAT in particolare, la STAT5, sembra essere intimamente coinvolta anche nella regolazione del muscolo scheletrico.[322] Ci sono due sottoproteine nella famiglia STAT5 e sono indicate come STAT5a e STAT5b. Sebbene siano identiche al 96%, è la variante STAT5b che è abbondante nei muscoli e nel tessuto epatico ed è quindi la proteina specifica su cui mi concentrerò da questo punto in avanti.[323-324]
Una revisione completa del percorso di segnalazione renderebbe questo articolo estremamente prolisso, ma ritengo importante che si comprenda che il percorso JAK/STAT5b è stato ripetutamente dimostrato avere una relazione diretta con l’espressione dell’IGF-1 locale nel tessuto muscolare e sul’ipertrofia.[248,325-332] Per questo motivo, se ci fossero modi per migliorare o ottimizzare questo specifico percorso, sembrerebbe che ciò si possa tradurre non solo in un aumento dell’attivazione del gene del IGF-1 [333-335] ma anche in un maggiore potenziale ipertrofico.
Fortunatamente, alcuni nuovi studi svolti su animali hanno già mostrato come i percorsi AR e JAK-STAT siano intimamente correlati.[248,336] Per essere precisi, il percorso STAT5a/b è a monte e l’AR è un bersaglio diretto a valle tramite la regolazione dell’espressione genica del AR. Studi su esseri umani hanno anche dimostrato che questo si verifica anche nell’uomo, con l’attività della STAT5 correlata positivamente all’espressione AR nelle linee cellulari del cancro alla prostata. [337]
Nella prossima parte di questa serie di articoli, parlerò dei modi in cui è possibile tentare di garantire che la via JAK-STAT5b-AR sia massimamente sensibilizzata, assicurando così che il potenziale ipertrofico sia potenziato quando Androgeni e GH vengono co-somministrati. Inoltre tratterò le applicazioni pratiche dell’uso del GH a fini ipertrofici esponendo infine le mie conclusioni in merito.
Stay tuned!
Gabriel Bellizzi
Riferimenti:
Yarasheski KE, Campbell JA, Smith K, Rennie MJ, Holloszy JO, Bier DM. Effect of growth hormone and resistance exercise on muscle growth in young men. Am J Physiol. 1992 Mar;262(3 Pt 1):E261-7.
Yarasheski KE, Zachwieja JJ, Campbell JA, Bier DM. Effect of growth hormone and resistance exercise on muscle growth and strength in older men. Am J Physiol. 1995 Feb;268(2 Pt 1):E268-76.
Fryburg DA. Insulin-like growth factor I exerts growth hormone- and insulin-like actions on human muscle protein metabolism. Am J Physiol. 1994 Aug;267(2 Pt 1):E331-6.
Ohlsson C, Mohan S, Sjögren K, Tivesten A, Isgaard J, Isaksson O, Jansson JO, Svensson J. The role of liver-derived insulin-like growth factor-I. Endocr Rev. 2009 Aug;30(5):494-535.
Wakelam MJ. The fusion of myoblasts. Biochem J. 1985 May 15;228(1):1-12. Review.
Crist DM, Peake GT, Egan PA, Waters DL. Body composition response to exogenous GH during training in highly conditioned adults. J Appl Physiol (1985). 1988 Aug;65(2):579-84.
Deyssig R, Frisch H, Blum WF, Waldhör T. Effect of growth hormone treatment on hormonal parameters, body composition and strength in athletes. Acta Endocrinol (Copenh). 1993 Apr;128(4):313-8.
Yarasheski KE, Zachweija JJ, Angelopoulos TJ, Bier DM. Short-term growth hormone treatment does not increase muscle protein synthesis in experienced weight lifters. J Appl Physiol (1985). 1993 Jun;74(6):3073-6.
Lange KH, Andersen JL, Beyer N, Isaksson F, Larsson B, Rasmussen MH, Juul A, Bülow J, Kjaer M. GH administration changes myosin heavy chain isoforms in skeletal muscle but does not augment muscle strength or hypertrophy, either alone or combined with resistance exercise training in healthy elderly men. J Clin Endocrinol Metab. 2002 Feb;87(2):513-23.
Ehrnborg C, Ellegård L, Bosaeus I, Bengtsson BA, Rosén T. Supraphysiological growth hormone: less fat, more extracellular fluid but uncertain effects on muscles in healthy, active young adults. Clin Endocrinol (Oxf). 2005 Apr;62(4):449-57.
Rudman D, Feller AG, Nagraj HS, Gergans GA, Lalitha PY, Goldberg AF, Schlenker RA, Cohn L, Rudman IW, Mattson DE. Effects of human growth hormone in men over 60 years old. N Engl J Med. 1990 Jul 5;323(1):1-6.
Taaffe DR, Pruitt L, Reim J, Hintz RL, Butterfield G, Hoffman AR, Marcus R. Effect of recombinant human growth hormone on the muscle strength response to resistance exercise in elderly men. J Clin Endocrinol Metab. 1994 Nov;79(5):1361-6.
Taaffe DR, Jin IH, Vu TH, Hoffman AR, Marcus R. Lack of effect of recombinant human growth hormone (GH) on muscle morphology and GH-insulin-like growth factor expression in resistance-trained elderly men. J Clin Endocrinol Metab. 1996 Jan;81(1):421-5.
Hennessey JV, Chromiak JA, DellaVentura S, Reinert SE, Puhl J, Kiel DP, Rosen CJ, Vandenburgh H, MacLean DB. Growth hormone administration and exercise effects on muscle fiber type and diameter in moderately frail older people. J Am Geriatr Soc. 2001 Jul;49(7):852-8.
West DW, Kujbida GW, Moore DR, Atherton P, Burd NA, Padzik JP, De Lisio M, Tang JE, Parise G, Rennie MJ, Baker SK, Phillips SM. Resistance exercise-induced increases in putative anabolic hormones do not enhance muscle protein synthesis or intracellular signalling in young men. J Physiol. 2009 Nov 1;587(Pt 21):5239-47.
West DW, Burd NA, Tang JE, Moore DR, Staples AW, Holwerda AM, Baker SK, Phillips SM. Elevations in ostensibly anabolic hormones with resistance exercise enhance neither training-induced muscle hypertrophy nor strength of the elbow flexors. J Appl Physiol (1985). 2010 Jan;108(1):60-7.
Fryburg DA, Jahn LA, Hill SA, Oliveras DM, Barrett EJ. Insulin and insulin-like growth factor-I enhance human skeletal muscle protein anabolism during hyperaminoacidemia by different mechanisms. J Clin Invest. 1995 Oct;96(4):1722-9
Butterfield GE, Thompson J, Rennie MJ, Marcus R, Hintz RL, Hoffman AR. Effect of rhGH and rhIGF-I treatment on protein utilization in elderly women. Am J Physiol. 1997 Jan;272(1 Pt 1):E94-9.
Friedlander AL, Butterfield GE, Moynihan S, Grillo J, Pollack M, Holloway L, Friedman L, Yesavage J, Matthias D, Lee S, Marcus R, Hoffman AR. One year of insulin-like growth factor I treatment does not affect bone density, body composition, or psychological measures in postmenopausal women. J Clin Endocrinol Metab. 2001 Apr;86(4):1496-503.
Consitt LA, Saneda A, Saxena G, List EO, Kopchick JJ. Mice overexpressing growth hormone exhibit increased skeletal muscle myostatin and MuRF1 with attenuation of muscle mass. Skelet Muscle. 2017 Sep 4;7(1):17.
Wolf E, Wanke R, Schenck E, Hermanns W, Brem G. Effects of growth hormone overproduction on grip strength of transgenic mice. Eur J Endocrinol. 1995 Dec;133(6):735-40.
Ho KY, Weissberger AJ. The antinatriuretic action of biosynthetic human growth hormone in man involves activation of the renin-angiotensin system. Metabolism. 1990 Feb;39(2):133-7.
Blazer-Yost BL, Cox M. Insulin-like growth factor 1 stimulates renal epithelial Na+ transport. Am J Physiol. 1988 Sep;255(3 Pt 1):C413-7.
Giordano M, DeFronzo RA. Acute effect of human recombinant insulin-like growth factor I on renal function in humans. Nephron. 1995;71(1):10-5.
Ehrnborg C, Lange KH, Dall R, Christiansen JS, Lundberg PA, Baxter RC, Boroujerdi MA, Bengtsson BA, Healey ML, Pentecost C, Longobardi S, Napoli R, Rosén T; GH-2000 Study Group. The growth hormone/insulin-like growth factor-I axis hormones and bone markers in elite athletes in response to a maximum exercise test. J Clin Endocrinol Metab. 2003 Jan;88(1):394-401.
Doessing S, Heinemeier KM, Holm L, Mackey AL, Schjerling P, Rennie M, Smith K, Reitelseder S, Kappelgaard AM, Rasmussen MH, Flyvbjerg A, Kjaer M. Growth hormone stimulates the collagen synthesis in human tendon and skeletal muscle without affecting myofibrillar protein synthesis. J Physiol. 2010 Jan 15;588(Pt 2):341-51.
Boesen AP, Dideriksen K, Couppé C, Magnusson SP, Schjerling P, Boesen M, Kjaer M, Langberg H. Tendon and skeletal muscle matrix gene expression and functional responses to immobilisation and rehabilitation in young males: effect of growth hormone administration. J Physiol. 2013 Dec 1;591(23):6039-52.
Cohn L, Feller AG, Draper MW, Rudman IW, Rudman D. Carpal tunnel syndrome and gynaecomastia during growth hormone treatment of elderly men with low circulating IGF-I concentrations. Clin Endocrinol (Oxf). 1993 Oct;39(4):417-25.
Sullivan DH, Carter WJ, Warr WR, Williams LH. Side effects resulting from the use of growth hormone and insulin-like growth factor-I as combined therapy to frail elderly patients. J Gerontol A Biol Sci Med Sci. 1998 May;53(3):M183-7.
Dickerman RD, Douglas JA, East JW. Bilateral median neuropathy and growth hormone use: a case report. Arch Phys Med Rehabil. 2000 Dec;81(12):1594-5.
Papadakis MA, Grady D, Black D, Tierney MJ, Gooding GA, Schambelan M, Grunfeld C. Growth hormone replacement in healthy older men improves body composition but not functional ability. Ann Intern Med. 1996 Apr 15;124(8):708-16.
Zachwieja JJ, Yarasheski KE. Does growth hormone therapy in conjunction with resistance exercise increase muscle force production and muscle mass in men and women aged 60 years or older? Phys Ther. 1999 Jan;79(1):76-82. Review.
Kishioka Y, Thomas M, Wakamatsu J, Hattori A, Sharma M, Kambadur R, Nishimura T. Decorin enhances the proliferation and differentiation of myogenic cells through suppressing myostatin activity. J Cell Physiol. 2008 Jun;215(3):856-67.
Kanzleiter T, Rath M, Görgens SW, Jensen J, Tangen DS, Kolnes AJ, Kolnes KJ, Lee S, Eckel J, Schürmann A, Eckardt K. The myokine decorin is regulated by contraction and involved in muscle hypertrophy. Biochem Biophys Res Commun. 2014 Jul 25;450(2):1089-94.
Zhang CZ, Li H, Bartold PM, Young WG, Waters MJ. Effect of growth hormone on the distribution of decorin and biglycan during odontogenesis in the rat incisor. J Dent Res. 1995 Oct;74(10):1636-43.
Bahl N, Stone G, McLean M, Ho KKY, Birzniece V. Decorin, a growth hormone regulated protein in humans. Eur J Endocrinol. 2017 Nov 14. pii: EJE-17-0844.
Short KR, Moller N, Bigelow ML, Coenen-Schimke J, Nair KS. Enhancement of muscle mitochondrial function by growth hormone. J Clin Endocrinol Metab. 2008 Feb;93(2):597-604. Epub 2007 Nov 13.
Lange KH, Isaksson F, Juul A, Rasmussen MH, Bülow J, Kjaer M. Growth hormone enhances effects of endurance training on oxidative muscle metabolism in elderly women. Am J Physiol Endocrinol Metab. 2000 Nov;279(5):E989-96.
Chargé SB, Rudnicki MA. Cellular and molecular regulation of muscle regeneration. Physiol Rev. 2004 Jan;84(1):209-38. Review.
Halevy O, Hodik V, Mett A. The effects of growth hormone on avian skeletal muscle satellite cell proliferation and differentiation. Gen Comp Endocrinol. 1996 Jan;101(1):43-52.
Kim H, Barton E, Muja N, Yakar S, Pennisi P, Leroith D. Intact insulin and insulin-like growth factor-I receptor signaling is required for growth hormone effects on skeletal muscle growth and function in vivo. Endocrinology. 2005 Apr;146(4):1772-9. Epub 2004 Dec 23.
Sinha-Hikim I, Roth SM, Lee MI, Bhasin S. Testosterone-induced muscle hypertrophy is associated with an increase in satellite cell number in healthy, young men. Am J Physiol Endocrinol Metab. 2003 Jul;285(1):E197-205. Epub 2003 Apr 1.
Ruzicka, L., Wettstein, A. and Kägi, H. (1935), Sexualhormone VIII. Darstellung von Testosteron unter Anwendung gemischter Ester. HCA, 18: 1478–1482.
Haupt HA, Rovere GD. Anabolic steroids: a review of the literature. Am J Sports Med. 1984 Nov-Dec;12(6):469-84. Review.
Shahidi NT. A review of the chemistry, biological action, and clinical applications of anabolic-androgenic steroids. Clin Ther. 2001 Sep;23(9):1355-90. Review.
Calof OM, Singh AB, Lee ML, Kenny AM, Urban RJ, Tenover JL, Bhasin S. Adverse events associated with testosterone replacement in middle-aged and older men: a meta-analysis of randomized, placebo-controlled trials. J Gerontol A Biol Sci Med Sci. 2005 Nov;60(11):1451-7
Hartgens F, Kuipers H. Effects of androgenic-anabolic steroids in athletes. Sports Med. 2004;34(8):513-54. Review.
Kadi F, Eriksson A, Holmner S, Thornell LE. Effects of anabolic steroids on the muscle cells of strength-trained athletes. Med Sci Sports Exerc. 1999 Nov;31(11):1528-34.
Herbst KL, Bhasin S. Testosterone action on skeletal muscle. Curr Opin Clin Nutr Metab Care. 2004 May;7(3):271-7. Review.
Eriksson A, Kadi F, Malm C, Thornell LE. Skeletal muscle morphology in power-lifters with and without anabolic steroids. Histochem Cell Biol. 2005 Aug;124(2):167-75.
Kadi F. Cellular and molecular mechanisms responsible for the action of testosterone on human skeletal muscle. A basis for illegal performance enhancement. Br J Pharmacol. 2008 Jun;154(3):522-8. Epub 2008 Apr 14. Review.
Bhasin S, Woodhouse L, Casaburi R, Singh AB, Bhasin D, Berman N, Chen X, Yarasheski KE, Magliano L, Dzekov C, Dzekov J, Bross R, Phillips J, Sinha-Hikim I, Shen R, Storer TW. Testosterone dose-response relationships in healthy young men. Am J Physiol Endocrinol Metab. 2001 Dec;281(6):E1172-81.
Woodhouse LJ, Reisz-Porszasz S, Javanbakht M, Storer TW, Lee M, Zerounian H, Bhasin S. Development of models to predict anabolic response to testosterone administration in healthy young men. Am J Physiol Endocrinol Metab. 2003 May;284(5):E1009-17.
Bhasin S, Woodhouse L, Casaburi R, Singh AB, Mac RP, Lee M, Yarasheski KE, Sinha-Hikim I, Dzekov C, Dzekov J, Magliano L, Storer TW. Older men are as responsive as young men to the anabolic effects of graded doses of testosterone on the skeletal muscle. J Clin Endocrinol Metab. 2005 Feb;90(2):678-88. Epub 2004 Nov 23.
Kvorning T, Andersen M, Brixen K, Madsen K. Suppression of endogenous testosterone production attenuates the response to strength training: a randomized, placebo-controlled, and blinded intervention study. Am J Physiol Endocrinol Metab. 2006 Dec;291(6):E1325-32. Epub 2006 Jul 25.
Sinha-Hikim I, Artaza J, Woodhouse L, Gonzalez-Cadavid N, Singh AB, Lee MI, Storer TW, Casaburi R, Shen R, Bhasin S. Testosterone-induced increase in muscle size in healthy young men is associated with muscle fiber hypertrophy. Am J Physiol Endocrinol Metab. 2002 Jul;283(1):E154-64.
Bhasin S, Storer TW, Berman N, Callegari C, Clevenger B, Phillips J, Bunnell TJ, Tricker R, Shirazi A, Casaburi R. The effects of supraphysiologic doses of testosterone on muscle size and strength in normal men. N Engl J Med. 1996 Jul 4;335(1):1-7.
Sinha-Hikim I, Taylor WE, Gonzalez-Cadavid NF, Zheng W, Bhasin S. Androgen receptor in human skeletal muscle and cultured muscle satellite cells: up-regulation by androgen treatment. J Clin Endocrinol Metab. 2004 Oct;89(10):5245-55.
Klover P, Chen W, Zhu BM, Hennighausen L. Skeletal muscle growth and fiber composition in mice are regulated through the transcription factors STAT5a/b: linking growth hormone to the androgen receptor. FASEB J. 2009 Sep;23(9):3140-8.
Sheffield-Moore M, Urban RJ, Wolf SE, Jiang J, Catlin DH, Herndon DN, Wolfe RR, Ferrando AA. Short-term oxandrolone administration stimulates net muscle protein synthesis in young men. J Clin Endocrinol Metab. 1999 Aug;84(8):2705-11.
Kadi F, Bonnerud P, Eriksson A, Thornell LE. The expression of androgen receptors in human neck and limb muscles: effects of training and self-administration of androgenic-anabolic steroids. Histochem Cell Biol. 2000 Jan;113(1):25-9.
de Rooy C, Grossmann M, Zajac JD, Cheung AS. Targeting muscle signaling pathways to minimize adverse effects of androgen deprivation. Endocr Relat Cancer. 2016 Jan;23(1):R15-26.
Brodsky IG, Balagopal P, Nair KS. Effects of testosterone replacement on muscle mass and muscle protein synthesis in hypogonadal men–a clinical research center study. J Clin Endocrinol Metab. 1996 Oct;81(10):3469-75.
Ferrando AA, Sheffield-Moore M, Paddon-Jones D, Wolfe RR, Urban RJ. Differential anabolic effects of testosterone and amino acid feeding in older men. J Clin Endocrinol Metab. 2003 Jan;88(1):358-62.
Bauer ER, Daxenberger A, Petri T, Sauerwein H, Meyer HH. Characterisation of the affinity of different anabolics and synthetic hormones to the human androgen receptor, human sex hormone binding globulin and to the bovine progestin receptor. APMIS. 2000 Dec;108(12):838-46.
Singh R, Artaza JN, Taylor WE, Gonzalez-Cadavid NF, Bhasin S. Androgens stimulate myogenic differentiation and inhibit adipogenesis in C3H 10T1/2 pluripotent cells through an androgen receptor-mediated pathway. Endocrinology. 2003 Nov;144(11):5081-8. Epub 2003 Jul 24.
Yarrow JF, McCoy SC, Borst SE. Tissue selectivity and potential clinical applications of trenbolone (17beta-hydroxyestra-4,9,11-trien-3-one): A potent anabolic steroid with reduced androgenic and estrogenic activity. Steroids. 2010 Jun;75(6):377-89.
Mulholland DJ, Dedhar S, Coetzee GA, Nelson CC. Interaction of nuclear receptors with the Wnt/beta-catenin/Tcf signaling axis: Wnt you like to know? Endocr Rev. 2005 Dec;26(7):898-915. Epub 2005 Aug 26. Review.
Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006 Nov 3;127(3):469-80. Review.
Singh R, Bhasin S, Braga M, Artaza JN, Pervin S, Taylor WE, Krishnan V, Sinha SK, Rajavashisth TB, Jasuja R. Regulation of myogenic differentiation by androgens: cross talk between androgen receptor/ beta-catenin and follistatin/transforming growth factor-beta signaling pathways. Endocrinology. 2009 Mar;150(3):1259-68.
Zhao JX, Hu J, Zhu MJ, Du M. Trenbolone enhances myogenic differentiation by enhancing β-catenin signaling in muscle-derived stem cells of cattle. Domest Anim Endocrinol. 2011 May;40(4):222-9.
Armstrong DD, Esser KA. Wnt/beta-catenin signaling activates growth-control genes during overload-induced skeletal muscle hypertrophy. Am J Physiol Cell Physiol. 2005 Oct;289(4):C853-9. Epub 2005 May 11.
Takada I, Kouzmenko AP, Kato S. Wnt and PPARgamma signaling in osteoblastogenesis and adipogenesis. Nat Rev Rheumatol. 2009 Aug;5(8):442-7.
Cossu G, Borello U. Wnt signaling and the activation of myogenesis in mammals. EMBO J. 1999 Dec 15;18(24):6867-72. Review.
Buckingham M. Skeletal muscle formation in vertebrates. Curr Opin Genet Dev. 2001 Aug;11(4):440-8. Review.
Polesskaya A, Seale P, Rudnicki MA. Wnt signaling induces the myogenic specification of resident CD45+ adult stem cells during muscle regeneration. Cell. 2003 Jun 27;113(7):841-52.
Tincello DG, Saunders PT, Hodgins MB, Simpson NB, Edwards CR, Hargreaves TB, Wu FC. Correlation of clinical, endocrine and molecular abnormalities with in vivo responses to high-dose testosterone in patients with partial androgen insensitivity syndrome. Clin Endocrinol (Oxf). 1997 Apr;46(4):497-506.
Foradori CD, Weiser MJ, Handa RJ. Non-genomic Actions of Androgens. Frontiers in neuroendocrinology. 2008;29(2):169-181.
Lucas-Herald AK, Alves-Lopes R, Montezano AC, Ahmed SF, Touyz RM. Genomic and non-genomic effects of androgens in the cardiovascular system: clinical implications. Clin Sci (Lond). 2017 Jul 1;131(13):1405-1418.
Münzer T, Harman SM, Hees P, Shapiro E, Christmas C, Bellantoni MF, Stevens TE, O’Connor KG, Pabst KM, St Clair C, Sorkin JD, Blackman MR. Effects of GH and/or sex steroid administration on abdominal subcutaneous and visceral fat in healthy aged women and men. J Clin Endocrinol Metab. 2001 Aug;86(8):3604-10.
Sattler FR, Castaneda-Sceppa C, Binder EF, Schroeder ET, Wang Y, Bhasin S, Kawakubo M, Stewart Y, Yarasheski KE, Ulloor J, Colletti P, Roubenoff R, Azen SP. Testosterone and growth hormone improve body composition and muscle performance in older men. J Clin Endocrinol Metab. 2009 Jun;94(6):1991-2001.
Illig R, Prader A. Effect of testosterone on growth hormone secretion in patients with anorchia and delayed puberty. J Clin Endocrinol Metab. 1970 May;30(5):615-8.
Pfeilschifter J, Scheidt-Nave C, Leidig-Bruckner G, Woitge HW, Blum WF, Wüster C, Haack D, Ziegler R. Relationship between circulating insulin-like growth factor components and sex hormones in a population-based sample of 50- to 80-year-old men and women. J Clin Endocrinol Metab. 1996 Jul;81(7):2534-40.
Erfurth EM, Hagmar LE, Sääf M, Hall K. Serum levels of insulin-like growth factor I and insulin-like growth factor-binding protein 1 correlate with serum free testosterone and sex hormone binding globulin levels in healthy young and middle-aged men. Clin Endocrinol (Oxf). 1996 Jun;44(6):659-64.
van Kesteren P, Lips P, Deville W, Popp-Snijders C, Asscheman H, Megens J, Gooren L. The effect of one-year cross-sex hormonal treatment on bone metabolism and serum insulin-like growth factor-1 in transsexuals. J Clin Endocrinol Metab. 1996 Jun;81(6):2227-32.
Veldhuis JD, Keenan DM, Mielke K, Miles JM, Bowers CY. Testosterone supplementation in healthy older men drives GH and IGF-I secretion without potentiating peptidyl secretagogue efficacy. Eur J Endocrinol. 2005 Oct;153(4):577-86.
Lewis MI, Fournier M, Storer TW, Bhasin S, Porszasz J, Ren SG, Da X, Casaburi R. Skeletal muscle adaptations to testosterone and resistance training in men with COPD. J Appl Physiol (1985). 2007 Oct;103(4):1299-310.
Mauras N, Hayes V, Welch S, Rini A, Helgeson K, Dokler M, Veldhuis JD, Urban RJ. Testosterone deficiency in young men: marked alterations in whole body protein kinetics, strength, and adiposity. J Clin Endocrinol Metab. 1998 Jun;83(6):1886-92.
Bondanelli M, Ambrosio MR, Margutti A, Franceschetti P, Zatelli MC, degli Uberti EC. Activation of the somatotropic axis by testosterone in adult men: evidence for a role of hypothalamic growth hormone-releasing hormone. Neuroendocrinology. 2003 Jun;77(6):380-7.
Veldhuis JD, Metzger DL, Martha PM Jr, Mauras N, Kerrigan JR, Keenan B, Rogol AD, Pincus SM. Estrogen and testosterone, but not a nonaromatizable androgen, direct network integration of the hypothalamo-somatotrope (growth hormone)-insulin-like growth factor I axis in the human: evidence from pubertal pathophysiology and sex-steroid hormone replacement. J Clin Endocrinol Metab. 1997 Oct;82(10):3414-20.
Weissberger AJ, Ho KK. Activation of the somatotropic axis by testosterone in adult males: evidence for the role of aromatization. J Clin Endocrinol Metab. 1993 Jun;76(6):1407-12.
Veldhuis JD, Mielke KL, Cosma M, Soares-Welch C, Paulo R, Miles JM, Bowers CY. Aromatase and 5alpha-reductase inhibition during an exogenous testosterone clamp unveils selective sex steroid modulation of somatostatin and growth hormone secretagogue actions in healthy older men. J Clin Endocrinol Metab. 2009 Mar;94(3):973-81.
Yamamoto T, Sakai C, Yamaki J, Takamori K, Yoshiji S, Kitawaki J, Fujii M, Yasuda J, Honjo H, Okada H. Estrogen biosynthesis in human liver–a comparison of aromatase activity for C-19 steroids in fetal liver, adult liver and hepatoma tissues of human subjects. Endocrinol Jpn. 1984 Jun;31(3):277-81.
Hata S, Miki Y, Saito R, Ishida K, Watanabe M, Sasano H. Aromatase in human liver and its diseases. Cancer Med. 2013 Jun;2(3):305-15.
Riggs BL, Hartmann LC. Selective estrogen-receptor modulators — mechanisms of action and application to clinical practice. N Engl J Med. 2003 Feb 13;348(7):618-29. Review. Erratum in: N Engl J Med. 2003 Mar 20;348(12):1192.
Löfgren L, Wallberg B, Wilking N, Fornander T, Rutqvist LE, Carlström K, von Schoultz B, von Schoultz E. Tamoxifen and megestrol acetate for postmenopausal breast cancer: diverging effects on liver proteins, androgens, and glucocorticoids. Med Oncol. 2004;21(4):309-18.
Centrella M, McCarthy TL, Chang WZ, Labaree DC, Hochberg RB. Estren (4-estren-3alpha,17beta-diol) is a prohormone that regulates both androgenic and estrogenic transcriptional effects through the androgen receptor. Mol Endocrinol. 2004 May;18(5):1120-30.
Yu YM, Domené HM, Sztein J, Counts DR, Cassorla F. Developmental changes and differential regulation by testosterone and estradiol of growth hormone receptor expression in the rabbit. Eur J Endocrinol. 1996 Nov;135(5):583-90.
Zung A, Phillip M, Chalew SA, Palese T, Kowarski AA, Zadik Z. Testosterone effect on growth and growth mediators of the GH-IGF-I axis in the liver and epiphyseal growth plate of juvenile rats. J Mol Endocrinol. 1999 Oct;23(2):209-21.
Hayes VY, Urban RJ, Jiang J, Marcell TJ, Helgeson K, Mauras N. Recombinant human growth hormone and recombinant human insulin-like growth factor I diminish the catabolic effects of hypogonadism in man: metabolic and molecular effects. J Clin Endocrinol Metab. 2001 May;86(5):2211-9.
Sculthorpe N, Solomon AM, Sinanan AC, Bouloux PM, Grace F, Lewis MP. Androgens affect myogenesis in vitro and increase local IGF-1 expression. Med Sci Sports Exerc. 2012 Apr;44(4):610-5.
Birzniece V, Meinhardt UJ, Umpleby MA, Handelsman DJ, Ho KK. Interaction between testosterone and growth hormone on whole-body protein anabolism occurs in the liver. J Clin Endocrinol Metab. 2011 Apr;96(4):1060-7.
Mertani HC, Delehaye-Zervas MC, Martini JF, Postel-Vinay MC, Morel G. Localization of growth hormone receptor messenger RNA in human tissues. Endocrine. 1995 Feb;3(2):135-42.
Florini JR, Ewton DZ, Coolican SA. Growth hormone and the insulin-like growth factor system in myogenesis. Endocr Rev. 1996 Oct;17(5):481-517. Review.
Urban RJ, Bodenburg YH, Gilkison C, Foxworth J, Coggan AR, Wolfe RR, Ferrando A. Testosterone administration to elderly men increases skeletal muscle strength and protein synthesis. Am J Physiol. 1995 Nov;269(5 Pt 1):E820-6.
Ewton DZ, Coolican SA, Mohan S, Chernausek SD, Florini JR. Modulation of insulin-like growth factor actions in L6A1 myoblasts by insulin-like growth factor binding protein (IGFBP)-4 and IGFBP-5: a dual role for IGFBP-5. J Cell Physiol. 1998 Oct;177(1):47-57.
Venken K, Movérare-Skrtic S, Kopchick JJ, Coschigano KT, Ohlsson C, Boonen S, Bouillon R, Vanderschueren D. Impact of androgens, growth hormone, and IGF-I on bone and muscle in male mice during puberty. J Bone Miner Res. 2007 Jan;22(1):72-82.
Serra C, Bhasin S, Tangherlini F, Barton ER, Ganno M, Zhang A, Shansky J, Vandenburgh HH, Travison TG, Jasuja R, Morris C. The role of GH and IGF-I in mediating anabolic effects of testosterone on androgen-responsive muscle. Endocrinology. 2011 Jan;152(1):193-206.
Spangenburg EE, Le Roith D, Ward CW, Bodine SC. A functional insulin-like growth factor receptor is not necessary for load-induced skeletal muscle hypertrophy. J Physiol. 2008 Jan 1;586(1):283-91.
Yoshizawa A, Clemmons DR. Testosterone and insulin-like growth factor (IGF) I interact in controlling IGF-binding protein production in androgen-responsive foreskin fibroblasts. J Clin Endocrinol Metab. 2000 Apr;85(4):1627-33.
Gayan-Ramirez G, Rollier H, Vanderhoydonc F, Verhoeven G, Gosselink R, Decramer M. Nandrolone decanoate does not enhance training effects but increases IGF-I mRNA in rat diaphragm. J Appl Physiol (1985). 2000 Jan;88(1):26-34.
Lewis MI, Horvitz GD, Clemmons DR, Fournier M. Role of IGF-I and IGF-binding proteins within diaphragm muscle in modulating the effects of nandrolone. Am J Physiol Endocrinol Metab. 2002 Feb;282(2):E483-90
Salmons S. Myotrophic effects of an anabolic steroid in rabbit limb muscles. Muscle Nerve. 1992 Jul;15(7):806-12.
Bisschop A, Gayan-Ramirez G, Rollier H, Dekhuijzen PN, Dom R, de Bock V, Decramer M. Effects of nandrolone decanoate on respiratory and peripheral muscles in male and female rats. J Appl Physiol (1985). 1997 Apr;82(4):1112-8
Lewis MI, Fournier M, Yeh AY, Micevych PE, Sieck GC. Alterations in diaphragm contractility after nandrolone administration: an analysis of potential mechanisms. J Appl Physiol (1985). 1999 Mar;86(3):985-92.
Heitzman RJ. The effectiveness of anabolic agents in increasing rate of growth in farm animals; report on experiments in cattle. Environ Qual Saf Suppl. 1976;(5):89-98. Review.
Buttery, P., Vernon, B., & Pearson, J. (1978). Anabolic agents—some thoughts on their mode of action. Proceedings of the Nutrition Society, 37(3), 311-315.
Hongerholt DD, Crooker BA, Wheaton JE, Carlson KM, Jorgenson DM. Effects of a growth hormone-releasing factor analogue and an estradiol-trenbolone acetate implant on somatotropin, insulin-like growth factor I, and metabolite profiles in growing Hereford steers. J Anim Sci. 1992 May;70(5):1439-48.
Tan RS, Scally MC. Anabolic steroid-induced hypogonadism–towards a unified hypothesis of anabolic steroid action. Med Hypotheses. 2009 Jun;72(6):723-8.
Kamanga-Sollo E, White ME, Hathaway MR, Weber WJ, Dayton WR. Effect of Estradiol-17beta on protein synthesis and degradation rates in fused bovine satellite cell cultures. Domest Anim Endocrinol. 2010 Jul;39(1):54-62.
Kamanga-Sollo E, Thornton KJ, White ME, Dayton WR. Role of G protein-coupled estrogen receptor-1, matrix metalloproteinases 2 and 9, and heparin binding epidermal growth factor-like growth factor in estradiol-17β-stimulated bovine satellite cell proliferation. Domest Anim Endocrinol. 2014 Oct;49:20-6.
Dunn JD, Johnson BJ, Kayser JP, Waylan AT, Sissom EK, Drouillard JS. Effects of flax supplementation and a combined trenbolone acetate and estradiol implant on circulating insulin-like growth factor-I and muscle insulin-like growth factor-I messenger RNA levels in beef cattle. J Anim Sci. 2003 Dec;81(12):3028-34.
Pampusch MS, Johnson BJ, White ME, Hathaway MR, Dunn JD, Waylan AT, Dayton WR. Time course of changes in growth factor mRNA levels in muscle of steroid-implanted and nonimplanted steers. J Anim Sci. 2003 Nov;81(11):2733-40.
Pampusch MS, White ME, Hathaway MR, Baxa TJ, Chung KY, Parr SL, Johnson BJ, Weber WJ, Dayton WR. Effects of implants of trenbolone acetate, estradiol, or both, on muscle insulin-like growth factor-I, insulin-like growth factor-I receptor, estrogen receptor-{alpha}, and androgen receptor messenger ribonucleic acid levels in feedlot steers. J Anim Sci. 2008 Dec;86(12):3418-23.
Thompson SH, Boxhorn LK, Kong WY, Allen RE. Trenbolone alters the responsiveness of skeletal muscle satellite cells to fibroblast growth factor and insulin-like growth factor I. Endocrinology. 1989 May;124(5):2110-7.
Johnson BJ, Halstead N, White ME, Hathaway MR, DiCostanzo A, Dayton WR. Activation state of muscle satellite cells isolated from steers implanted with a combined trenbolone acetate and estradiol implant. J Anim Sci. 1998 Nov;76(11):2779-86.
Dalbo VJ, Roberts MD, Mobley CB, Ballmann C, Kephart WC, Fox CD, Santucci VA, Conover CF, Beggs LA, Balaez A, Hoerr FJ, Yarrow JF, Borst SE, Beck DT. Testosterone and trenbolone enanthate increase mature myostatin protein expression despite increasing skeletal muscle hypertrophy and satellite cell number in rodent muscle. Andrologia. 2017 Apr;49(3).
Bhasin S, He EJ, Kawakubo M, Schroeder ET, Yarasheski K, Opiteck GJ, Reicin A, Chen F, Lam R, Tsou JA, Castaneda-Sceppa C, Binder EF, Azen SP, Sattler FR. N-terminal propeptide of type III procollagen as a biomarker of anabolic response to recombinant human GH and testosterone. J Clin Endocrinol Metab. 2009 Nov;94(11):4224-33.
Nelson AE, Meinhardt U, Hansen JL, Walker IH, Stone G, Howe CJ, Leung KC, Seibel MJ, Baxter RC, Handelsman DJ, Kazlauskas R, Ho KK. Pharmacodynamics of growth hormone abuse biomarkers and the influence of gender and testosterone: a randomized double-blind placebo-controlled study in young recreational athletes. J Clin Endocrinol Metab. 2008 Jun;93(6):2213-22.
Holt RI. Detecting growth hormone misuse in athletes. Indian J Endocrinol Metab. 2013 Oct;17(Suppl 1):S18-22.
Tan SH, Lee A, Pascovici D, Care N, Birzniece V, Ho K, Molloy MP, Khan A. Plasma biomarker proteins for detection of human growth hormone administration in athletes. Sci Rep. 2017 Aug 30;7(1):10039.
Jørgensen JO, Jessen N, Pedersen SB, Vestergaard E, Gormsen L, Lund SA, Billestrup N. GH receptor signaling in skeletal muscle and adipose tissue in human subjects following exposure to an intravenous GH bolus. Am J Physiol Endocrinol Metab. 2006 Nov;291(5):E899-905.
Liu X, Robinson GW, Gouilleux F, Groner B, Hennighausen L. Cloning and expression of Stat5 and an additional homologue (Stat5b) involved in prolactin signal transduction in mouse mammary tissue. Proc Natl Acad Sci U S A. 1995 Sep 12;92(19):8831-5.
Hennighausen L, Robinson GW. Interpretation of cytokine signaling through the transcription factors STAT5A and STAT5B. Genes Dev. 2008 Mar 15;22(6):711-21.
Eshet R, Laron Z, Pertzelan A, Arnon R, Dintzman M. Defect of human growth hormone receptors in the liver of two patients with Laron-type dwarfism. Isr J Med Sci. 1984 Jan;20(1):8-11.
Teglund S, McKay C, Schuetz E, van Deursen JM, Stravopodis D, Wang D, Brown M, Bodner S, Grosveld G, Ihle JN. Stat5a and Stat5b proteins have essential and nonessential, or redundant, roles in cytokine responses. Cell. 1998 May 29;93(5):841-50.
Hwa V, Little B, Adiyaman P, Kofoed EM, Pratt KL, Ocal G, Berberoglu M, Rosenfeld RG. Severe growth hormone insensitivity resulting from total absence of signal transducer and activator of transcription 5b. J Clin Endocrinol Metab. 2005 Jul;90(7):4260-6. Epub 2005 Apr 12.
Rowland JE, Lichanska AM, Kerr LM, White M, d’Aniello EM, Maher SL, Brown R, Teasdale RD, Noakes PG, Waters MJ. In vivo analysis of growth hormone receptor signaling domains and their associated transcripts. Mol Cell Biol. 2005 Jan;25(1):66-77. Erratum in: Mol Cell Biol. 2005 Mar;25(5):2072.
Klover P, Hennighausen L. Postnatal body growth is dependent on the transcription factors signal transducers and activators of transcription 5a/b in muscle: a role for autocrine/paracrine insulin-like growth factor I. Endocrinology. 2007 Apr;148(4):1489-97.
Barclay JL, Kerr LM, Arthur L, Rowland JE, Nelson CN, Ishikawa M, d’Aniello EM, White M, Noakes PG, Waters MJ. In vivo targeting of the growth hormone receptor (GHR) Box1 sequence demonstrates that the GHR does not signal exclusively through JAK2. Mol Endocrinol. 2010 Jan;24(1):204-17.
Hwa V, Nadeau K, Wit JM, Rosenfeld RG. STAT5b deficiency: lessons from STAT5b gene mutations. Best Pract Res Clin Endocrinol Metab. 2011 Feb;25(1):61-75.
Varco-Merth B, Feigerlová E, Shinde U, Rosenfeld RG, Hwa V, Rotwein P. Severe growth deficiency is associated with STAT5b mutations that disrupt protein folding and activity. Mol Endocrinol. 2013 Jan;27(1):150-61.
Davey HW, Xie T, McLachlan MJ, Wilkins RJ, Waxman DJ, Grattan DR. STAT5b is required for GH-induced liver IGF-I gene expression. Endocrinology. 2001 Sep;142(9):3836-41.
Woelfle J, Chia DJ, Rotwein P. Mechanisms of growth hormone (GH) action. Identification of conserved Stat5 binding sites that mediate GH-induced insulin-like growth factor-I gene activation. J Biol Chem. 2003 Dec 19;278(51):51261-6. Epub 2003 Oct 7.
Woelfle J, Billiard J, Rotwein P. Acute control of insulin-like growth factor-I gene transcription by growth hormone through Stat5b. J Biol Chem. 2003 Jun 20;278(25):22696-702. Epub 2003 Apr 7.
MacLean HE, Chiu WS, Notini AJ, Axell AM, Davey RA, McManus JF, Ma C, Plant DR, Lynch GS, Zajac JD. Impaired skeletal muscle development and function in male, but not female, genomic androgen receptor knockout mice. FASEB J. 2008 Aug;22(8):2676-89.
Tan SH, Dagvadorj A, Shen F, Gu L, Liao Z, Abdulghani J, Zhang Y, Gelmann EP, Zellweger T, Culig Z, Visakorpi T, Bubendorf L, Kirken RA, Karras J, Nevalainen MT. Transcription factor Stat5 synergizes with androgen receptor in prostate cancer cells. Cancer Res. 2008 Jan 1;68(1):236-48.
de Souza GL, Hallak J. Anabolic steroids and male infertility: a comprehensive review. BJU Int. 2011 Dec;108(11):1860-5.
Sono ormai alcuni decenni che i fautori de “le calorie non contano” e quelli, conservatori, de “le calorie contano” si scontrano cercando di argomentare (più o meno) scientificamente le loro affermazioni. Tralasciando la questione non poco importante della trattazione superficiale che spesso caratterizza entrambe le fazioni, la schiera dei “negazionisti delle calorie” tocca livelli di scientismo e di contraddizione abbastanza evidenti (mai letto un loro libro?). Essi cercano spiegazioni (all’apparenza) plausibili per avvalorare la loro tesi. Una di esse è perfettamente rappresentata da un recente studio svolto da scienziati dello sport dell’Università di Malaga i quali hanno osservato l’effetto di una dieta chetogenica ipercalorica su atleti di forza.(1)
I ricercatori hanno reclutato 24 uomini in buona salute, che si erano allenati con pesi per almeno 2 anni, e gli hanno sottoposti ad un identico allenamento finalizzato all’ipertrofia per 8 settimane: il piano allenante era suddiviso in 2 giorni di Upper Body e 2 giorni Lower Body, con 72 ore di riposo tra le sessioni per favorire il recupero. I ricercatori hanno diviso i partecipanti allo studio in 3 gruppi e hanno dato ad ogni gruppo una dieta diversa.
Panoramica del protocollo di allenamento. WK: allenamento (microciclo); UL: Upper-Limb; LL: Lower-Limb; R: Riposo; 30 X: 3s nell’eccentrica e movimento esplosivo durante la fase concentrica.
I soggetti del primo gruppo (gruppo di controllo), ovviamente, hanno continuato a mangiare come era loro abitudine [CG].
I soggetti del secondo gruppo hanno ricevuto un piano alimentare il quale presentava un introito proteico pari a 2g di proteine per chilogrammo di peso corporeo al giorno, e un carico glucidico di massimo 42g di carboidrati al giorno [KD]. L’apporto calorico era alto. Ogni giorno i soggetti presi in esame dovevano ingerire più di 3000Kcal.
In fine, i soggetti del terzo gruppo hanno ricevuto un piano alimentare più o meno “normale” e con un buon apporto proteico [NKD]. Anche i soggetti di questo gruppo dovevano ingerire più di 3000Kcal al giorno.
Al termine del periodo di test, è stato osservato che i soggetti sottoposti ad una dieta a basso contenuto di carboidrati avevano ridotto la loro massa grassa generale, compreso il grasso nella cavità addominale. Quest’ultimo tipo di grasso è noto per essere un fattore predittivo per lo sviluppo di diverse patologie. La massa magra di questi soggetti è rimasta costante.
I ricercatori hanno concluso dicendo che, secondo i loro risultati, i soggetti sottoposti a un allenamento contro-resistenza durante una dieta chetogenica hanno sperimentato una maggiore riduzione della massa grassa e del tessuto adiposo viscerale, rispetto al gruppo che ha seguito un regime alimentare non chetogenico. La maggiore riduzione del tessuto adiposo viscerale può avere una certa rilevanza clinica a causa della sua associazione inversa al rischio cardio-metabolico.
I ricercatori continuano dicendo che, sono necessari ulteriori studi per valutare i vantaggi di questa combinazione (allenamento contro-resistenza e dieta chetogenica) in soggetti con un eccesso di massa grassa, con particolare attenzione alla significativa riduzione riportata a carico del tessuto adiposo viscerale, che potrebbe essere di grande beneficio per questa fascia di popolazione dato anche il fatto che la massa corporea magra è mantenuta. E aggiungono che, in effetti, questa ricerca non ha mostrato cambiamenti significativi sulla massa magra, nonostante la condizione iperenergetica e l’elevato apporto proteico (2,0g/kg/die) assunto dai soggetti sottoposti ad allenamenti contro-resistenza del gruppo “dieta chetogenica”. Quindi, essi concludono, non poco banalmente, che gli approcci dietetici a basso contenuto di carboidrati non sarebbero una strategia ottimale per la costruzione di massa muscolare in uomini allenati nelle condizioni poste da questo studio (protocollo di allenamento contro-resistenza focalizzato sulla tensione meccanica della durata di 8 settimane).
Immagino già la “standing ovation” dei detrattori della termodinamica… mantenete la calma, lo studio qui brevemente esposto ha diverse limitazioni….
Le limitazioni del presente studio vanno dalle limitate misurazioni dei risultati, dal numero ridotto di partecipanti e dal tempo del test (8 settimane); tutti fattori che di per se riducono il valore dello studio. E, cosa assai rilevante, durante lo studio non sono state eseguite valutazioni sulla corretta assunzione calorica dei soggetti presi in esame e non è stata presa in considerazione la soppressione dell’appetito derivante da una dieta ricca di proteine e grassi. Quindi, è possibile, anzi è molto probabile, che si siano verificate variazioni nell’assunzione calorica anche se i partecipanti sono stati istruiti a seguire specifiche raccomandazioni dietetiche. In parole povere, i soggetti sottoposti a regime chetogenico hanno, con tutta probabilità, seguito un regime leggermente ipocalorico in modo indiretto come risposta alle variabili indotte dalla specifica strategia alimentare. Ed ecco spiegato, in modo assai più scientifico, il perché del calo della massa grassa e il mantenimento della massa muscolare. Quest’ultimo elemento trova la sua logica spiegazione nell’introito proteico giornaliero (2g/Kg/die) il quale ha garantito un effetto di “protein sparing”. Il numero limitato delle settimane del test (8 settimane) è stato senza dubbio incisivo anche in questo. E non dimentichiamoci del fatto che, guarda caso, non si è verificato alcun aumento della massa muscolare ne della massa corporea in generale. Evento questo decisamente ai “confini della realtà” se rapportato a soggetti in fisiologia sottoposti ad un regime ipercalorico (di qualsiasi natura esso sia).
Bomba Calorimetrica
Sebbene l’uomo non sia una macchina calorimetrica, che un acido polinsaturo non equivale ad uno saturo, ecc… bisognerebbe tuttavia ricordarsi che facciamo parte di questo universo e come tutta la materia rispondiamo alle Leggi delle Termodinamica e come tutti gli essere viventi a quelle della Bioenergia:
“Queste leggi descrivono come il calore (o energia termica) viene convertito da e per diversi tipi di energia, e l’effetto che questo può avere sulle varie forme di materia.”
Detto in termini semplici, quando si mangia si introduce dell’energia, questa energia non può sparire nel nulla.
Anche se un soggetto possa avere problemi d’assorbimento intestinali, disfunzioni delle proteine mitocondriali UCP2-3, una grave insulino-resistenza, o soffra di ipotiroidismo in ogni caso l’energia che introduce si trasforma sempre seguendo le leggi universali.
Ogni soggetto ha un proprio equilibrio energetico, quanto sia elevato questo equilibrio dipende dallo stato metabolico. Questi due fattori si influenzano a vicenda e sono imprescindibili l’uno dall’alto.
Semplice schema rappresentante le varie componenti di un corretto piano alimentare con il loro grado di importanza.
Per quanto vi possa irritare, alla fine della giostra, una caloria rimane pur sempre una caloria…