
Se non avete ancora letto le precedenti parti componenti questa serie di articoli vi invito a farlo prima di procedere con la lettura di questa quarta ed ultima parte: 1° Parte – 2° Parte – 3° Parte.
Farmacocinetica, Farmacodinamica e Feedback Negativi

Come già detto, il fegato rappresenta il principale bersaglio del GH, il quale è il principale regolatore della sintesi epatica di IGF-1. Per causare tale effetto, il GH si lega con i GHR localizzati nel dominio extracellulare degli epatociti stimolando successivamente la produzione di IGF-1 endocrino tramite la trascrizione genica, utilizzando la via di segnalazione JAK-STAT. Inoltre, è stato dimostrato che la somministrazione di GH causa una rapida sovraregolazione dell’mRNA del IGF-1 nel fegato.[338]
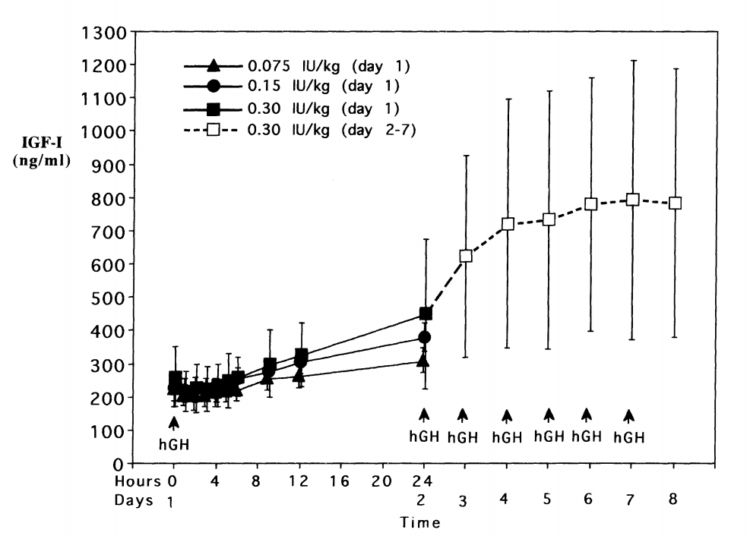
Aumenti dei livelli serici di IGF-1 si verificano molto rapidamente anche in presenza di un grande bolo di rHGH. Incrementi significativi di IGF-1 sono già osservabili dopo 6-12h dall’iniezione.[339] Questi livelli serici di IGF-1 continuano ad aumentare fino a raggiungere il loro punto di saturazione dose-dipendente entro 4-7 giorni, anche quando si utilizzano dosi estremamente elevate che ammontano a 20-30UI al giorno di rHGH.[340] In particolare, il punto di saturazione si è rivelato essere compreso nell’intervallo dei 700-800 ng/mL e sembra suggerire che i livelli endocrini di IGF-1 hanno un tetto massimo negli adulti sani. I meccanismi esatti devono ancora essere chiariti, ma sono probabilmente il risultato dei complessi meccanismi di controllo intrinseci all’Asse GH/IGF-1. Coloro i quali desiderano elevare i livelli endocrini di IGF-1 al fine di ottenerne un vantaggio sull’ipertrofia dovrebbe tenerlo a mente, in quanto vi è un punto in cui l’uso di dosi maggiori di rHGH semplicemente non si traducono in elevati livelli serici di IGF-1. Qui di seguito ho riportato il grafico dello studio di Tanaka il quale mostra la relazione tra l’rhGH e i livelli serici di IGF-:
Ora, vorrei dedicarmi brevemente all’analisi dell’azione del IGF-1autocrino e del perché esso rappresenti un mediatore cruciale del processo ipertrofico, prima di tornare nuovamente a discutere su questioni inerenti alla farmacodinamica e farmacocinetica. La segnalazione recettoriale del IGF-1 è unica nel suo genere, e questo lo si deve al fatto che utilizza due percorsi distinti per stimolare la proliferazione o la differenziazione.[341-343] Questo è un comportamento abbastanza interessante, poiché nessun altro membro della famiglia dei fattori di crescita ha dimostrato di agire in tal modo. Poiché la proliferazione e la differenziazione sono processi opposti, inizialmente era difficile per i ricercatori capire come un singolo fattore di crescita, attraverso un singolo recettore, potesse inviare un segnale che attivasse entrambi.[294] Da quando sono state fatte queste prime scoperte, è stato ulteriormente chiarito che l’IGF-1 non svolge simultaneamente queste azioni. Test su varie linee di coltura cellulare hanno dimostrato che gli effetti proliferativi arrivano prima, durando tra le 24 e le 36 ore. È solo dopo questa fase proliferativa iniziale che si verifica la differenziazione miogenica.[344]
Gli effetti proliferativi mediati dall’IGF-1 sui mioblasti sono noti sin dagli anni ’70, quando vennero osservati per la prima volta nelle cellule epatiche di ratto.[345] Questa stimolazione proliferativa del IGF-1 si traduce in un aumento del numero di cellule, nei livelli di proteine, nella sintesi del DNA, nell’assorbimento di aminoacidico, nell’assorbimento del glucosio e nella soppressione della proteolisi.[346] Nelle colture cellulari umane, l’IGF-1 ha anche dimostrato di aumentare la dimensione dei miotubi indipendentemente dal fatto che i mioblasti proliferino attivamente o che la proliferazione sia cessata. Regola la dimensione dei miotubi attivando la sintesi proteica, inibendo la degradazione proteica e inducendo la fusione delle cellule di riserva.[347-348] La capacità dell’IGF di sopprimere la proteolisi nel muscolo scheletrico, la scomposizione delle proteine in aminoacidi, è stata dimostrata innumerevoli volte nel corso degli anni.[349-352] È stato anche dimostrato che l’IGF-1 induce la proliferazione e la differenziazione delle cellule satelliti in miociti maturi, come determinato da un aumento del numero di miofibre nucleate a livello centrale rispetto a quelle periferiche.[148,353-354]

La capacità dell’IGF-1 autocrino di causare la differenziazione dei mioblasti è stata in realtà una scoperta che potremmo definire quasi “ibrida” dal momento che degli studi svolti negli anni ’60 avevano mostrato che questo effetto si verifica con alti livelli di Insulina.[355] Successivamente è stato dimostrato che gli IGF sono stimolatori molto più potenti nella differenziazione miogenica rispetto all’Insulina e si è concluso che la stessa Insulina agisce realmente come un analogo dell’IGF-1 in questo sistema.[356-357] Gli effetti di differenziazione dati dall’IGF-1 autocrino sono bifasici, con basse concentrazioni che stimolano progressivamente la differenziazione dei mioblasti mentre concentrazioni molto elevate mostrano una cessazione dell’attività di differenziazione. Il limite massimo per la differenziazione sembra attestarsi a circa 100ng/mL per l’IGF-1 e 300ng/mL per IGF-2.[358] Questo effetto non è legato alla proliferazione, poiché non si osservano ulteriori aumenti nel numero complessivo delle cellule.[294] È possibile che le molecole di segnalazione coinvolte nella regolazione negativa del sistema miogenico siano aumentate, ma questa è una affermazione puramente speculativa.[359-360]
La somministrazione di rHGH eleva l’espressione dell’mRNA dell’IGF-1 nel muscolo scheletrico in numerosi modelli cellulari, umani e animali.[127,150,361-364] Ciò avviene abbastanza rapidamente, entro 60 minuti dall’iniezione sottocutanea di rHGH ed i picchi sono segnalati tra le 6 e le 12 ore post iniezione.[363] In questo particolare modello animale citato, il raddoppio della dose di GH non ha portato ad ulteriori aumentati dei livelli di mRNA dell’IGF-1, il che suggerisce che esiste un sistema di regolazione che determina quanto GH sia necessario per stimolare al massimo l’espressione locale dell’IGF-1 nel muscolo scheletrico. In precedenza si è potuto appurare che la differenziazione dei miociti mediata dall’IGF-1 si arresta quando le concentrazioni locali raggiungono circa i 100ng/mL, ma quanto GH è necessario per raggiungere il punto di saturazione dell’espressione dell’mRNA dell’IGF-1?
Gli studi sui miociti umani mostrano che il GH aumenta l’espressione dell’mRNA dell’IGF-1 entro 30-60 minuti con picchi molto più rapidi rispetto a quelli osservati negli studi sugli animali, entro 1-2 ore, usando la via di segnalazione JAK / STAT5b.[365] Questi livelli elevati di mRNA hanno dimostrato di durare fino a 48 ore dopo una singola esposizione al GH. La quantità di GH necessaria per stimolare al massimo l’espressione dell’mRNA dell’IGF-1 è risultata essere una dose compresa tra i 7,5ng/mL e 30ng/ml [366], con una dose media efficace che si attesta a 3ng/ml. Questi numeri sono in linea con gli intervalli di dose fisiologica osservati negli animali, che sono effettivamente compresi tra i 2-100 ng/mL.[367] Inoltre, si collocano esattamente in linea con quanto si osserva endogenamente nell’uomo, con concentrazioni normali di picco comprese tra i 22,4 e 32,4ng/mL.[368-369,436] Ci sono stati casi in cui gli uomini presi in esame hanno mostrato concentrazioni di picco leggermente più alte, ma questi devono essere considerati valori anomali.[370] In ogni caso, ciò che questi dati tendono a suggerire è che il corpo umano è particolarmente adatto a gestire livelli naturali di picco della secrezioni di GH endogeno. Cercare di incidere ulteriormente il sistema elevando i livelli di GH oltre quelli endogeni, unicamente per tentare di potenziare i processi ipertrofici, potrebbe in realtà non tradursi nell’effetto desiderato.
Gli studi che mettono a confronto le infusioni locali con le infusioni sistemiche di GH o IGF-1 sono un po’ più difficili da trovare di quanto si vorrebbe. Le poche sperimentazioni sugli animali che sono riuscito a trovare indicano che l’infusione diretta di GH o IGF-1 nei tessuti bersaglio determina un aumento della massa muscolare. Questo aumento dell’ipertrofia si verifica anche senza che il muscolo bersaglio sia stato sottoposto ad attività motoria.[371-372] Gli studi dimostrano anche che le iniezioni locali di GH portano a livelli sostanzialmente più alti nell’espressione dell’mRNA dell’ IGF-1 locale rispetto alle iniezioni locali di IGF-1, di un fattore di oltre venti.[127] Sono riuscito a trovare uno studio nel quale si confrontavano le risposte dei ratti (attivi e non) all’infusione locale di IGF-1. Il gruppo “IGF-1 plus training” ha mostrato un aumento sia della massa muscolare che della forza locale maggiore rispetto al semplice trattamento in isolamento.[373] Quindi, anche se limitata, la letteratura disponibile è apparentemente in grado di dimostrare che le iniezione locali di GH o IGF-1 hanno effettivamente valore.
Ne ho già parlato diverse volte ma, nel tentativo di imprimere ulteriormente questo concetto, è necessario ricordarsi che i livelli autocrini di IGF-1 sembrano essere molto più importanti dei livelli endocrini di IGF-1 in relazione alla regolazione della massa muscolare. Oltre a questo punto, la sovraespressione dell’IGF-1 autocrino nel muscolo provoca l’ipertrofia delle fibre.[374] La sovraespressione dell’IGF-1 autocrino ha anche mostrato effetti anti-catabolici, con modelli animali tendenti a mostrare una resistenza generale all’atrofia muscolare normalmente osservata con l’invecchiamento.[375] L’IGF-1 localizzato fornisce anche capacità rigenerative indipendenti dall’età nelle cellule muscolari.[376]
Vi sono anche alcune prove convincenti che suggeriscono che l’IGF-1 endocrino agisce direttamente come un regolatore di feedback negativo sulla produzione di IGF-1 autocrino. Questo meccanismo di feedback negativo è dipendente dal pathway PI3K/Akt [377-378]. Inoltre, elevati livelli di IGF-1 endocrino possono anche agire indirettamente per sopprimere la produzione di IGF-1 autocrino. Quindi, in altre parole, non solo l’IGF-1 endocrino ha un impatto diretto minore sulla regolazione della massa muscolare, ma può anche sopprimere l’IGF-1 autocrino che ha impatti maggiori sull’ipertrofia.
Elevati livelli di IGF-1 circolante e, nello specifico, di IGF-1 libero elevati agiscono in modo negativo sul GH determinando un tasso di soppressione della produzione di IGF-1 autocrino a valle.[379] Non è del tutto chiaro, tuttavia, se la regolazione negativa dell’IGF-1 modifichi l’emivita dell’mRNA dell’IGF-1 o influenzi direttamente l’espressione del gene IGF-1. Oltre a questo, è stato anche dimostrato che l’espressione dell’IGF-1 autocrino è sottoregolata nelle cellule muscolari dopo trattamento con IGF-1.[366] È stato anche dimostrato che l’espressione epatica dell’mRNA dell’IGF-1 è sottoregolata dall’esposizione acuta all’IGF-1.[127] Quindi, mantenere livelli endocrini il più possibile soppressi con rispettiva dose di rHGH, elevando contemporaneamente i livelli autocrini, dovrebbe essere un fattore prioritario in un protocollo di GH volto all’ipertrofia.

Il GH è pulsatile per natura sia nell’uomo che nelle specie animali. Quindi, sarebbe logico pensare che molti dei processi intrinseci del corpo saranno tarati in modo tale da rispondere in maniera ottimale all’esposizione al GH in modo simile. In accordo con questa affermazione è stato dimostrato che solo la somministrazione di GH pulsatile, e non l’infusione continua, ha la capacità di stimolare massimamente l’espressione dell’mRNA dell’IGF-1 nel muscolo scheletrico.[366,380-381] È stato anche dimostrato che la somministrazione pulsatile porta ad un aumento del potenziale di crescita postnatale complessivo rispetto all’infusione continua.[89,382] La somministrazione pulsatile può anche portare a livelli endocrini di IGF-1 serici comparabili, o addirittura diminuiti [383], il che è vantaggioso a causa delle potenziali capacità di regolazione negativa che possiede sull’espressione dell’IGF-1 autocrino e che sono state discusse in precedenza. L’evidenza suggerisce anche che il picco stesso, e non necessariamente il numero di picchi, potrebbe essere della massima importanza per i tessuti bersaglio.[384] Per la massima crescita e potenziale ipertrofico, l’evidenza tende a suggerire che creare picchi di GH elevati, e quindi tornare ai livelli basali più volte al giorno, può essere preferibile rispetto a mantenerli elevati per periodi di tempo più lunghi. Questo pratica permette di riprodurre gli schemi secretori in vivo.
I pathways del GH coinvolti nell’anabolismo sono anche suscettibili alla desensibilizzazione, che è parte della fisiologia del GH endogeno.[385] A causa della natura intrinsecamente pulsatile del GH in vivo, l’attività dei recettori e dei pathways sono regolati da un impulso seguito da un periodo di inattività.[386] L’esposizione continua o ripetuta al GH senza un adeguato lasso di tempo refrattario comporterà livelli di attività fortemente soppressi. In effetti, nel corso degli anni numerosi studi hanno dimostrato che tale effetto si verifica. Le cellule ed il tessuto muscolare richiedono un periodo refrattario piuttosto lungo prima che la loro piena risposta al GH venga recuperata. Dopo l’esposizione al GH, le cellule muscolari non sono nemmeno in grado di rispondere alle successive dosi di GH. In realtà, occorrono due ore complete per riprendere parzialmente la reattività nei modelli cellulari, con un totale di 6-8 ore di astinenza dall’uso di GH necessarie per ripristinare la piena sensibilità.[366] Viceversa, quando il GH è micro-dosato in impulsi di dieci minuti, seguiti da intervalli di otto ore, è stato mostrato aumentare progressivamente l’mRNA dell’IGF-1 con ogni impulso successivo.[386]

Questo fenomeno è potenzialmente il risultato di una desensibilizzazione complessiva all’interno della via JAK-STAT5, poiché è stato dimostrato che l’esposizione al GH negli studi sulle cellule epatiche causa resistenza alla successiva attivazione della via STAT5 per 4-8 ore.[387-388] Questo lasso di tempo è sufficiente per sincronizzarsi abbastanza bene con ciò che è stato visto nei modelli di cellule miocitarie citati in precedenza. Nei modelli di cellule epatiche, il GH ha stimolato un significativo aumento dell’espressione del SOCS3, che è un potente inibitore dell’azione del GH.[389]. Poiché il GH non ha avuto alcun effetto sull’espressione del SOCS3 nelle cellule muscolari, questo deve essere un altro meccanismo causante il periodo refrattario. Questo meccanismo può essere dipeso dalla sottoregolazione dei GHR, dall’inibizione mediata da un’altra proteina SOCS, o dall’induzione di una tirosina fosfatasi che semplicemente inattiva la via JAK / STAT.[390] La via JAK-STAT5b, che come ricorderete è intimamente associata al muscolo scheletrico e all’espressione dell’IGF-1, è di natura transitoria – con attivazione massima raggiunta entro 10-30 minuti, seguita da un prolungato periodo di inattivazione.
Una scoperta piuttosto nuova di Xu et al. [391] ha dimostrato che anche distanziare le esposizioni al GH di cinque ore lasciava entrambi i percorsi a valle MEK1/2 e ERK1/2 significativamente soppressi rispetto a tutti i percorsi a monte, a causa di una potenziale disconnessione nella trasduzione del segnale . Ciò è di particolare interesse in quanto questi stessi due percorsi a valle sono stati coinvolti in modo significativo sia nella crescita sia nella proliferazione.[392-393] È stato anche scoperto che l’attivazione indotta da GH di STAT1 e STAT3 è stata desensibilizzata, ma l’esposizione all’Insulina inverte la desensibilizzazione osservata in tutti i percorsi interessati. Anche se non sto per trattare approfonditamente l’Insulina, ci sono un paio di importanti punti da dovere prendere in considerazione. Bisogna comprendere innanzitutto che ci sono molti obiettivi a valle del recettore del GH e molti di questi hanno il potenziale per essere desensibilizzati dopo l’esposizione al GH. Bisogna comprendi anche che l’Insulina possiede l’abilità unica di risensibilizzare molti di questi percorsi. Ciò ha un senso vista la relazione tipo yin-yang tra i due composti. È noto che il GH e l’Insulina possiedono una relazione anabolica sinergica a causa di molti effetti che esercitano l’uno sull’altro. Questo sembra essere soltanto un’anteprima di uno di questi effetti.
Asse GH/IGF-1 – Relazione con altri ormoni

Prima di passare alle note conclusive, vorrei trattare brevemente alcuni altri ormoni connessi a diverso grado con l’Asse GH/IGF-1. Per prima cosa, voglio trattare brevemente l’Asse Tiroideo dal momento che l’inserimento di composti tiroidei insieme al GH è una pratica comune anche durante i protocolli di massa.
Il muscolo scheletrico è il principale bersaglio di segnalazione dell’ormone tiroideo, con trasportatori degli ormoni tiroidei e enzimi di conversione espressi localmente.[394] È ben noto che il GH potenzia la deiodinazione periferica che converte il T4 in T3, riducendo così il T4 e il reverse T3, aumentando contemporaneamente i livelli di T3.[395-398] Ciò che molte persone non riescono a capire è che questo è un effetto transitorio, e studi a lungo termine sembrano indicare che gli effetti mediati dal GH sulla conversione periferica si stabilizzino con il tempo.[399-402]

Invece di proseguire ulteriormente su questo, avendo già trattato la questione nel dettaglio in un mio vecchio articolo, preferirei concentrarmi su alcune pubblicazioni relative alla tiroide che non vengono discusse abbastanza spesso. Gli ormoni tiroidei, per loro natura, sono composti tendenzialmente catabolici in quanto stimolano la disgregazione proteica dell’intero corpo in misura maggiore rispetto alla sintesi proteica.[403] A livello locale, nel muscolo scheletrico stimolano un aumento dell’attività all’interno della via ubiquitina/proteasoma, che è ampiamente coinvolta nella proteolisi.[404-406] Il risultato di questo è un tasso accelerato del turnover proteico e una perdita netta complessiva degli aminoacidi situati all’interno dei muscoli scheletrici.
Inoltre, negli esseri umani, sia gli stati di ipertiroidismo che di ipotiroidismo sono stati associati a livelli di IGF-1 soppressi con una tendenza alla normalizzazione quando viene ristabilita una condizione di eutiroidismo. L’ipertiroidismo è anche associato ad una bassa attività di legame recettoriale del GH, che si ipotizza essere il risultato di una ridotta capacità di elaborazione dei recettori del GH.[407] E’ stato anche ipotizzato che l’ipertiroidismo sia in grado anche di accelerare la clearance del GH urinario.[408] Inoltre, studi su animali hanno dimostrato che gli ormoni tiroidei possono avere importanti effetti soppressivi sulla sintesi di IGF-1 stimolata con il GH.[409] Ovviamente, a causa della complessa relazione che l’Asse Tiroideo ha con l’Asse GH/IGF-1, raggruppando tutte le interazioni che hanno tra loro in pochi paragrafi, trattare l’argomento diventerebbe poco pratico. Tuttavia, quando il corpo della letteratura scientifica viene esaminato nella sua interezza, ci sono molte prove che suggeriscono che la supplementazione con composti tiroidei esogeni potrebbe non essere l’ideale quando l’obiettivo di un individuo è l’ipertrofia, anche se la regolazione del dosaggio dei tiroidei in tale contesto rimane la misura di “sicurezza” più intelligente visto l’impatto negativo sulla funzionalità tiroidea dato dal GH. Comunque, per tutti coloro che sono interessati ad approfondire questo argomento, consiglio di iniziare con la review nella nota seguente.[410]

Mi piacerebbe trattare anche la Miostatin, che rappresenta un argomento molto discusso nei vari forum di BodyBuilding presenti in rete. La sua fama proviene dai risultati ipertrofici espressi dai bovini privi per mutazione del gene della Miostatina, i quali mostrano una massa muscolare significativamente maggiore rispetto ai loro simili non mutati.[411] La Miostatina, un fattore di crescita e differenziazione appartenente alla superfamiglia dei TGF-beta, ha dimostrato di inibire selettivamente la miogenesi, in gran parte tramite il suo effetto soppressivo sulla proliferazione dei mioblasti.[412] È espressa e secreta prevalentemente dal muscolo scheletrico. Come molti sanno, se riesci a sopprimere o inibire la Miostatina, di conseguenza il potenziale ipertrofico aumenta significativamente.
Le mutazioni della Miostatina sono state osservate sia negli animali che nell’uomo. Queste mutazioni del gene della Miostatina portano ad un fenotipo ipertrofico negli animali, come accennato in precedenza.[413-415] L’Asse GH/IGF-1 e la Miostatina sembrano avere una relazione regolativa diretta tra loro, come osservato nei pazienti affetti contemporaneamente da GHD e HIV che mostrano marcati aumenti nell’espressione dell’mRNA della Miostatina.[416] Quindi, è possibile che attraverso una supplementazione di dosi sovrafisiologiche di rHGH si possa indurre una diminuzione dell’mRNA della Miostatina [209,417-419]? Sfortunatamente, nonostante la presenza di alcuni casi studio selezionati, non credo che si abbiano abbastanza dati in questo momento per sapere se ciò possa dare risultati apprezzabili in seguito alla sua applicazione.

Quello che sappiamo è che aumenti dell’espressione dell’mRNA dell’IGF-1 e le concentrazioni circolanti di IGF-1 sono state osservati dopo inibizione della Miostatina.[419-421] Sappiamo anche che l’inibizione della Miostatina tende a causare l’ipertrofia attraverso molte delle stesse modalità osservate con l’IGF-1 autocrino, cioè l’aumento della sintesi proteica e l’attivazione delle cellule satelliti.[422-425] E sappiamo anche che l’ipertrofia indotta dalla sovraespressione dell’IGF-1 o dall’inibizione della Miostatina utilizza la stessa identica via – PI3K/Akt/mTOR.[426-428] Tuttavia, l’IGF-1 non è un requisito per l’ipertrofia indotta dalla Follistatina, tranne nel caso di livelli di Insulina estremamente bassi – come ben sappiamo, la Follistatina è un inibitore della Miostatina [429]. E l’esposizione cronica al GH può in realtà portare ad un’espressione sovrastimolata della Miostatina e del suo recettore.[209]
Quindi quello che possiamo dire, con certezza, è che l’espressione della Miostatina non sarà un fattore diretto o indiretto per quanto riguarda il potenziamento dei processi ipertrofici, né dell’attività contrattile, nei muscoli scheletrici umani.[430] Proprio per questo motivo, non ritengo che sia un fattore sul quale gli atleti debbano eccessivamente concentrarsi, al di fuori di un utile arricchimento delle proprie conoscenze in materia.
Applicazioni pratiche e pensieri conclusivi
Arrivati a questo punto è mia intenzione unire tutto ciò che è stato esposto in questa serie di articoli e esporlo sotto forma di alcuni suggerimenti pratici rivolti a tutti coloro i quali vogliono semplicemente massimizzare la loro capacità ipertrofica.
Ora è chiaro che il GH possiede pochissimi, se non nulli, effetti diretti sull’ipertrofia. Pertanto, qualsiasi protocollo di massa che contempli il suo uso dovrà tenere in considerazione questo punto includendo gli AAS, i quali, per l’appunto, hanno anche una proficua sinergia con il GH. Sia la letteratura scientifica che i dati aneddotici dimostrano chiaramente che l’uso combinato di entrambi i composti ha un massimale ipertrofico significativamente più alto rispetto all’uso singolo. Personalmente, penso che i BodyBuilder dovrebbero sempre optare per l’utilizzo di una base di Testosterone e Boldenone (correttamente rapportati in base a contesto e alle caratteristiche individuali) anche in una fase “Bulk” nella quale viene inserito l’uso del GH. Il Trenbolone può essere considerato come parte “accessoria” di un protocollo di massa, a causa della sua intrinseca difficoltà gestazionale come sostanza anabolizzante. Dovrebbe essere usato con parsimonia e con cautela poiché, insieme ai suoi numerosi punti di forza come composto anabolizzante, presenta alcune limitazioni. Queste limitazioni derivano per lo più, come già accennato, dalla difficile gestione del composto in quanto esso non è facilmente tollerato da una buona parte degli individui. Quindi, se viene usato il Trenbolone, dovrebbe essere inserito calcolando con attenzione il dosaggio e la tolleranza individuale, anche per quanto concerne la tolleranza temporale individuale all’uso di tale composto.
Dopo un periodo d’uso prolungato di dosi sovrafisiologico di AAS (Ciclo+Bridge), è buona cosa procedere con l’interruzione o con una marcata riduzione del numero e degli AAS utilizzati. Detta in parole semplici, questa interruzione può contemplare una completa astinenza dagli AAS, svolgendo un adeguata PCT al fine di ristabilire una omeostasi ormonale fisiologica, o una transizione ad una TRT, metodologia comunemente chiamata “blast and cruise”. La struttura del protocollo di supplementazione farmacologica dovrebbe sempre seguire i principi del dosaggio minimo efficace con aumenti nei dosaggi degli AAS solo nel caso si sia raggiunto il limite di crescita con il precedente dosaggio, assicurandosi che tutte le altre variabili nello stile di vita siano correttamente regolate. L’utilizzo di questo approccio limita il rischio che si sviluppino effetti collaterali indesiderati sul lungo termine.
Quando si decide di usare il GH, la dove ce ne sia la possibilità economica, esso dovrebbe provenire da uno dei prodotti presenti nel mercato farmaceutico. Questi prodotti approvati devono superare anni di studi strettamente controllati per dimostrare la loro sicurezza, purezza ed efficacia su soggetti umani. I progressi tecnologici nel corso degli anni hanno reso molto più facile la produzione di rHGH. Per questo motivo, i produttori ora provengono da tutto il mondo. Spesso questi produttori realizzano ciò che viene definito “GH generico” sui forum, ma tale termine non mi piace molto. Definire qualcosa come “generico” implica che sia una replica perfetta di prodotti legati a specifici marchi farmaceutici approvati dell’agenzia del farmaco che hanno perso il cui brevetto è scaduto, il che non è il caso del GH. Infatti, a causa della natura estremamente complessa del processo di produzione del rHGH, per esempio, la FDA non consente nemmeno l’uso del termine “generico” quando si tratta di rHGH e utilizza invece il termine ” follow-on protein product ” o FOPP.
Spesso questi marchi off-label sono venduti ad un costo molto ridotto, ed è qui che sta il dilemma, in quanto questo può essere molto allettante. Tuttavia, con questo costo ridotto per il consumatore, non ci sarà nemmeno la garanzia del produttore su cosa ci sia realmente nella fiala o persino su come è stato prodotto. Il problema di fondo è che il processo di produzione del rHGH è estremamente complessa, ed è molto facile che nelle fasi di questo processo si commettano errori con conseguenti variazioni nella catena proteica che potenzialmente portano ad effetti indesiderati, o anche a risposte autoimmuni.
Spesso gli atleti si affidano semplicemente ai test serici per misurare i livelli di GH e/o IGF-1 al fine di concludere che un prodotto contenete GH sia “buono”, ma dobbiamo ricordarci che raggiungere livelli ematici ormonali elevati è la parte relativamente facile. Anche le molecole di GH che sono state alterate o danneggiate durante la produzione possono dare questo esito. Tuttavia, queste stesse molecole di GH danneggiate o mutate possono spesso stimolare risposte autoimmuni. Ciò potrebbe indurre il corpo ad avere una risposta recettoriale degradata, che può anche riflettersi sulla secrezione endogena nel tempo. [431-432] Rimane poi il problema del reale contenuto della vial o fiala, la quale può non presentare nessun principio attivo al suo interno.
Il GH dovrebbe essere usato in modo pulsatile, per mimare le condizioni in vivo. Tra queste iniezioni, deve esserci un periodo di refrattarietà o si deve consumare un pasto che abbia un buon stimolo sull’Insulina. Può anche essere utilizzata l’Insulina esogena al fine di bypassare molte delle limitazioni del periodo refrattario, ma questo va oltre lo scopo di questo articolo. Anche se il cumulo delle dosi giornaliere dovrebbe essere sovrafisiologico, le dosi individuali non hanno bisogno di essere ad alto dosaggio, poiché la massima stimolazione dell’IGF-1 autocrino nel tessuto muscolo scheletrico si verifica ben all’interno delle concentrazioni fisiologiche di GH. Aneddoticamente, sembra anche esserci un limite con il quale l’uso di rHGH diventa additivo in presenza di AAS. Potrebbe essere necessario un po’ di ponderata (e supportata da personale qualificato) auto-sperimentazione per scoprire dove si trova questa singola dose di saturazione, ma la maggior parte dei soggetti troverà questo limite tra le 4 e le 8 UI/die. Oltre questo dosaggio, la maggior parte degli utilizzatori tenderà a scoprire che la giustificazione dei costi e il rapporto rischio /beneficio tendono a diminuire rapidamente.
Non bisognerebbe passare troppo tempo a riflettere sul tempo delle iniezioni di GH, dal momento che gli aumenti dei livelli di IGF-1 autocrino avvengono rapidamente e possono rimanere elevati per giorni. Bisognerebbe concentrati invece sul programma di iniezione che si addice meglio al contesto della giornata, tenendo contemporaneamente presenti le linee guida per il periodo di refrattarietà del GH. Si possono anche prendere in considerazione piccole o grandi iniezioni, poiché alcuni potrebbero trovare più pratiche e funzionali iniezioni con un dosaggio inferiore e somministrate più frequenti mentre altri potrebbero preferire iniezioni con un dosaggio maggiore e somministrazioni meno frequenti. Naturalmente, maggiore è il contenuto dell’iniezione, maggiore è la probabilità che si superi la soglia massima di espressione dell’IGF-1 autocrino.
Massimizzare l’espressione dell’IGF-1 autocrino, mentre contemporaneamente si sopprimono i livelli di IGF-1 endocrino, sarà una priorità. Esistono prove a sostegno dell’ipotesi secondo cui un iniezione locale di GH possa aiutare a raggiungere questo obiettivo, con una conseguente minore possibilità di feedback negativo. Sono stati osservati aumenti significativi della massa muscolare in appena due settimane di iniezioni locali di IGF-1.[441]
La dove cause di forza maggiore non lo impediscano, è consigliabile evitare o comunque regolare attentamente l’uso di tutti quei composti che possono avere interazioni negative con l’uso del GH a fini ipertrofici. Inibitori della Aromatasi, Modulatori Selettivi del Recettore degli Estrogeni e T3 hanno tutti dimostrato di avere un potenziale effetto negativo sul processo globale ipertrofico legato al GH e per tale motivo dovrebbero essere usati con parsimonia, se non omessi del tutto dove possibile (vedi in particolare il T3).
Questo non dovrebbe sorprendere nessuno e non dovrebbe nemmeno essere troppo difficile da tenere a mente: allenarsi duramente, allenarsi in modo intelligente e allenarsi in modo coerente. Sebbene non sia stato affrontato direttamente nell’articolo, bisogna capire che l’allenamento contro resistenza ha impatti unici e additivi sull’ipertrofia. In effetti, alcuni di questi meccanismi non sono nemmeno mediati dall’Asse AR e/o GH/IGF-1. [433] Bisogna anche comprendere che non esiste una “magica” routine allenante universalmente applicabile, la chiave di volta sarà la coerenza nel garantire un carico di lavoro adeguato, con elementi di sovraccarico progressivo nel tempo assicurando un adeguato stimolo meccanico gestendo al meglio le variabili allenanti (intensità, volume, densità e intensità percepita). La logica composizione del piano allenante servirà a garantire lo stimolo ipertrofico di base che sarà coadiuvato dall’azione dei composti utilizzati.
Nonostante la mole e l’importanza delle informazioni presentate in questa serie di articoli, è necessario ricordare che i meccanismi d’azione ormonali sono governati da innumerevoli fattori. Anche esaminando l’intero corpo della letteratura scientifica equivarrebbe a poco più che accumulare una serie di utili nozioni garanti di assicurare una solida base conoscitiva sull’argomento la quale rappresenterà un punto di partenza intelligente per l’applicazione pratica, rimanendo sempre soggetti alle variabili di risposta individuale. Seguendo questa linea, i migliori risultati nella pratica spesso provengono da coloro i quali posseggono una buona conoscenza dei principi scientifici e la capacità innata di saperli applicare non solo su se stessi ma, soprattutto, su terzi. Infatti, molto raramente due persone rispondono in modo identico alla supplementazione di ormoni esogeni (e non solo), quindi, non bisogna assolutamente pensare che basti semplicemente applicare su se stessi o su terzi un protocollo che ha portato benefici realmente apprezzabili su un soggetto per ottenere la medesima risposta.
A tal fine, invito atleti e Preparatori a utilizzare queste pubblicazioni come punto di partenza per essere in grado di gestire l’applicazione pratica in modo più consapevole e produttivo. Inoltre, chi ne fosse in grado, può consultare il vasto numero di riferimenti riportati nel corso di queste pubblicazioni è tentare di ragionare sulle mie conclusioni. Ad ogni citazione presente in questa serie di articoli, assicuratevi che il riferimento elencato supporti effettivamente le affermazioni fatte. Mantenere sempre una mente aperta ma con i giusti “filtri”, e cercare di non credere per partito preso ad una singola opinione, specialmente di fronte alle nuove evidenze scientifiche. Infine, è buona cosa verificare sempre la veridicità di quanto è stato affermato.
Punti conclusivi per un corretto utilizzo della chimica e del GH a fini ipertrofici:
- Usare il GH in combinazione con gli AAS
- Usare GH e AAS di grado farmaceutico la dove ciò è possibile
- Assicurarsi una base di Testosterone correttamente rapportata al Boldenone aggiungendo (in base a maturità e tolleranza) il Trenbolone
- Iniettare il GH in modo pulsatile, considerare l’opzione delle iniezioni locali nei gruppi carenti
- Mantenere un dosaggio complessivo ottimale di GH il quale si attesta tra le 4 e le 8UI/die
- Evitare o regolare attentamente l’uso dei composti che possono interagire negativamente con i processi ipertrofici legati al uso di GH (vedi AI, SERM e T3)
- Dopo un periodo di tempo (variabile) nel quale si è stati sottoposti a dosaggi ormonali sovrafisiologici optare per una PCT o per una TRT (a seconda delle proprie necessità e priorità)
- Essere a conoscenza dei potenziali effetti collaterali legati all’auso/abuso di GH (nausea, vomito, cefalea, ritenzione idrica e sodica, edemi, parestesie, sindrome del tunnel carpale, rigidità articolare, dolori articolari, artrite, dolori muscolari, ipertensione, insulino-resistenza, diabete di tipo II, acromegalia, dilatazione addominale, ipertrofia cardiaca ecc…)
- Svolgere regolarmente esami del sangue; sia durante i periodi di picco nell’uso della farmacologia sia nel periodo successivo (vedi PCT/OCT o TRT)
- Gestire al meglio le variabili legate agli stressor ambientali, all’allenamento, all’alimentazione e al sonno.
Gabriel Bellizzi
Riferimenti:
338. Mathews LS, Norstedt G, Palmiter RD. Regulation of insulin-like growth factor I gene expression by growth hormone. Proc Natl Acad Sci U S A. 1986 Dec;83(24):9343-7.
339. Keller A, Wu Z, Kratzsch J, Keller E, Blum WF, Kniess A, Preiss R, Teichert J, Strasburger CJ, Bidlingmaier M. Pharmacokinetics and pharmacodynamics of GH: dependence on route and dosage of administration. Eur J Endocrinol. 2007 Jun;156(6):647-53.
340. Tanaka T, Seino Y, Fujieda K, Igarashi Y, Yokoya S, Tachibana K, Ogawa Y. Pharmacokinetics and metabolic effects of high-dose growth hormone administration in healthy adult men. Endocr J. 1999 Aug;46(4):605-12.
341. Quinn LS, Steinmetz B, Maas A, Ong L, Kaleko M. Type-1 insulin-like growth factor receptor overexpression produces dual effects on myoblast proliferation and differentiation. J Cell Physiol. 1994 Jun;159(3):387-98.
342. Coolican SA, Samuel DS, Ewton DZ, McWade FJ, Florini JR. The mitogenic and myogenic actions of insulin-like growth factors utilize distinct signaling pathways. J Biol Chem. 1997 Mar 7;272(10):6653-62.
343. Foulstone EJ, Huser C, Crown AL, Holly JM, Stewart CE. Differential signalling mechanisms predisposing primary human skeletal muscle cells to altered proliferation and differentiation: roles of IGF-I and TNFalpha. Exp Cell Res. 2004 Mar 10;294(1):223-35.
344. Ewton DZ, Roof SL, Magri KA, McWade FJ, Florini JR. IGF-II is more active than IGF-I in stimulating L6A1 myogenesis: greater mitogenic actions of IGF-I delay differentiation. J Cell Physiol. 1994 Nov;161(2):277-84.
345. Florini JR, Nicholson ML, Dulak NC. Effects of peptide anabolic hormones on growth of myoblasts in culture. Endocrinology. 1977 Jul;101(1):32-41.
346. Laviola L, Natalicchio A, Giorgino F. The IGF-I signaling pathway. Curr Pharm Des. 2007;13(7):663-9. Review.
347. Jacquemin V, Furling D, Bigot A, Butler-Browne GS, Mouly V. IGF-1 induces human myotube hypertrophy by increasing cell recruitment. Exp Cell Res. 2004 Sep 10;299(1):148-58.
348. Jacquemin V, Butler-Browne GS, Furling D, Mouly V. IL-13 mediates the recruitment of reserve cells for fusion during IGF-1-induced hypertrophy of human myotubes. J Cell Sci. 2007 Feb 15;120(Pt 4):670-81. Epub 2007 Jan 30.
349. Ballard FJ, Francis GL. Effects of anabolic agents on protein breakdown in L6 myoblasts. Biochem J. 1983 Jan 15;210(1):243-9.
350. Ewton DZ, Falen SL, Florini JR. The type II insulin-like growth factor (IGF) receptor has low affinity for IGF-I analogs: pleiotypic actions of IGFs on myoblasts are apparently mediated by the type I receptor. Endocrinology. 1987 Jan;120(1):115-23.
351. Hembree JR, Hathaway MR, Dayton WR. Isolation and culture of fetal porcine myogenic cells and the effect of insulin, IGF-I, and sera on protein turnover in porcine myotube cultures. J Anim Sci. 1991 Aug;69(8):3241-50.
352. Hong D, Forsberg NE. Effects of serum and insulin-like growth factor I on protein degradation and protease gene expression in rat L8 myotubes. J Anim Sci. 1994 Sep;72(9):2279-88.
353. Florini JR, Ewton DZ, Roof SL. Insulin-like growth factor-I stimulates terminal myogenic differentiation by induction of myogenin gene expression. Mol Endocrinol. 1991 May;5(5):718-24.
354. Musarò A, McCullagh KJ, Naya FJ, Olson EN, Rosenthal N. IGF-1 induces skeletal myocyte hypertrophy through calcineurin in association with GATA-2 and NF-ATc1. Nature. 1999 Aug 5;400(6744):581-5.
355. Haba GDL, Cooper GW, Elting V. HORMONAL REQUIREMENTS FOR MYOGENESIS OF STRIATED MUSCLE IN VITRO: INSULIN AND SOMATOTROPIN. Proceedings of the National Academy of Sciences of the United States of America. 1966;56(6):1719-1723.
356. Florini JR, Ewton DZ. Insulin acts as a somatomedin analog in stimulating myoblast growth in serum-free medium. In Vitro. 1981 Sep;17(9):763-8.
357. Schmid C, Steiner T, Froesch ER. Preferential enhancement of myoblast differentiation by insulin-like growth factors (IGF I and IGF II) in primary cultures of chicken embryonic cells. FEBS Lett. 1983 Sep 5;161(1):117-21.
358. Florini JR, Ewton DZ, Falen SL, Van Wyk JJ. Biphasic concentration dependency of stimulation of myoblast differentiation by somatomedins. Am J Physiol. 1986 May;250(5 Pt 1):C771-8.
359. Quinn LS, Ehsan M, Steinmetz B, Kaleko M. Ligand-dependent inhibition of myoblast differentiation by overexpression of the type-1 insulin-like growth factor receptor. J Cell Physiol. 1993 Sep;156(3):453-61.
360. Olson EN. Signal transduction pathways that regulate skeletal muscle gene expression. Mol Endocrinol. 1993 Nov;7(11):1369-78. Review.
361. Murphy LJ, Bell GI, Friesen HG. Growth hormone stimulates sequential induction of c-myc and insulin-like growth factor I expression in vivo. Endocrinology. 1987 May;120(5):1806-12.
362. Turner JD, Rotwein P, Novakofski J, Bechtel PJ. Induction of mRNA for IGF-I and -II during growth hormone-stimulated muscle hypertrophy. Am J Physiol. 1988 Oct;255(4 Pt 1):E513-7.
363. Isgaard J, Nilsson A, Vikman K, Isaksson OG. Growth hormone regulates the level of insulin-like growth factor-I mRNA in rat skeletal muscle. J Endocrinol. 1989 Jan;120(1):107-12.
364. Bichell DP, Kikuchi K, Rotwein P. Growth hormone rapidly activates insulin-like growth factor I gene transcription in vivo. Mol Endocrinol. 1992 Nov;6(11):1899-908.
365. Sadowski CL, Wheeler TT, Wang LH, Sadowski HB. GH regulation of IGF-I and suppressor of cytokine signaling gene expression in C2C12 skeletal muscle cells. Endocrinology. 2001 Sep;142(9):3890-900.
366. Frost RA, Nystrom GJ, Lang CH. Regulation of IGF-I mRNA and signal transducers and activators of transcription-3 and -5 (Stat-3 and -5) by GH in C2C12 myoblasts. Endocrinology. 2002 Feb;143(2):492-503.
367. MacLeod JN, Pampori NA, Shapiro BH. Sex differences in the ultradian pattern of plasma growth hormone concentrations in mice. J Endocrinol. 1991 Dec;131(3):395-9.
368. Rochiccioli P, Messina A, Tauber MT, Enjaume C. Correlation of the parameters of 24-hour growth hormone secretion with growth velocity in 93 children of varying height. Horm Res. 1989;31(3):115-8.
369. Hansen TK, Gravholt CH, ØRskov H, Rasmussen MH, Christiansen JS, Jørgensen JO. Dose dependency of the pharmacokinetics and acute lipolytic actions of growth hormone. J Clin Endocrinol Metab. 2002 Oct;87(10):4691-8.
370. Baum WF, Klöditz E, Hesse V, Jahreis G, Schneyer U, Giebler H. [Increase in spontaneous growth hormone secretion in asthmatic children–a symptom of atopic disposition?]. Kinderarztl Prax. 1993 Nov;61(9):323-8.
371. Adams GR, McCue SA. Localized infusion of IGF-I results in skeletal muscle hypertrophy in rats. J Appl Physiol (1985). 1998 May;84(5):1716-22.
372. Alzghoul MB, Gerrard D, Watkins BA, Hannon K. Ectopic expression of IGF-I and Shh by skeletal muscle inhibits disuse-mediated skeletal muscle atrophy and bone osteopenia in vivo. FASEB J. 2004 Jan;18(1):221-3. Epub 2003 Nov 3.
373. Lee S, Barton ER, Sweeney HL, Farrar RP. Viral expression of insulin-like growth factor-I enhances muscle hypertrophy in resistance-trained rats. J Appl Physiol (1985). 2004 Mar;96(3):1097-104. Erratum in: J Appl Physiol. 2004 Jun;96(6):2343.
374. Coleman ME, DeMayo F, Yin KC, Lee HM, Geske R, Montgomery C, Schwartz RJ. Myogenic vector expression of insulin-like growth factor I stimulates muscle cell differentiation and myofiber hypertrophy in transgenic mice. J Biol Chem. 1995 May 19;270(20):12109-16.
375. Barton-Davis ER, Shoturma DI, Musaro A, Rosenthal N, Sweeney HL. Viral mediated expression of insulin-like growth factor I blocks the aging-related loss of skeletal muscle function. Proc Natl Acad Sci U S A. 1998 Dec 22;95(26):15603-7.
376. Musarò A, McCullagh K, Paul A, Houghton L, Dobrowolny G, Molinaro M, Barton ER, Sweeney HL, Rosenthal N. Localized Igf-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat Genet. 2001 Feb;27(2):195-200
377. Lewis MI, Bulut Y, Biring MS, Da X, Fournier M. (1999) IGF-I administration prevents corticosteroids-induced diaphragm atrophy in emphysema . Am J Respir Crit Care Med 159:A580
378. Fournier M, Huang ZS, Cercek B, Li H, Bykhovskaya I, Lewis MI. (2000) Administration of insulin-like growth factor-1 (IGF-I) and corticosteroids in emphysematous hamsters: influences on diaphragm IGF-I . Am J Respir Crit Care Med 161:A18
379. Shavlakadze T, Grounds M. Of bears, frogs, meat, mice and men: complexity of factors affecting skeletal muscle mass and fat. Bioessays. 2006 Oct;28(10):994-1009. Review.
380. Maiter D, Underwood LE, Maes M, Davenport ML, Ketelslegers JM. Different effects of intermittent and continuous growth hormone (GH) administration on serum somatomedin-C/insulin-like growth factor I and liver GH receptors in hypophysectomized rats. Endocrinology. 1988 Aug;123(2):1053-9.
381. Isgaard J, Carlsson L, Isaksson OG, Jansson JO. Pulsatile intravenous growth hormone (GH) infusion to hypophysectomized rats increases insulin-like growth factor I messenger ribonucleic acid in skeletal tissues more effectively than continuous GH infusion. Endocrinology. 1988 Dec;123(6):2605-10.
382. Clark RG, Jansson JO, Isaksson O, Robinson IC. Intravenous growth hormone: growth responses to patterned infusions in hypophysectomized rats. J Endocrinol. 1985 Jan;104(1):53-61.
383. Bick T, Hochberg Z, Amit T, Isaksson OG, Jansson JO. Roles of pulsatility and continuity of growth hormone (GH) administration in the regulation of hepatic GH-receptors, and circulating GH-binding protein and insulin-like growth factor-I. Endocrinology. 1992 Jul;131(1):423-9.
384. Weltman A, Weltman JY, Schurrer R, Evans WS, Veldhuis JD, Rogol AD. Endurance training amplifies the pulsatile release of growth hormone: effects of training intensity. J Appl Physiol (1985). 1992 Jun;72(6):2188-96.
385. Flores-Morales A, Greenhalgh CJ, Norstedt G, Rico-Bautista E. Negative regulation of growth hormone receptor signaling. Mol Endocrinol. 2006 Feb;20(2):241-53. Epub 2005 Jul 21. Review.
386. Hartman ML, Veldhuis JD, Thorner MO. Normal control of growth hormone secretion. Horm Res. 1993;40(1-3):37-47. Review.
387. Fernández L, Flores-Morales A, Lahuna O, Sliva D, Norstedt G, Haldosén LA, Mode A, Gustafsson JA. Desensitization of the growth hormone-induced Janus kinase 2 (Jak 2)/signal transducer and activator of transcription 5 (Stat5)-signaling pathway requires protein synthesis and phospholipase C. Endocrinology. 1998 Apr;139(4):1815-24.
388. Gebert CA, Park SH, Waxman DJ. Termination of growth hormone pulse-induced STAT5b signaling. Mol Endocrinol. 1999 Jan;13(1):38-56.
389. Ram PA, Waxman DJ. SOCS/CIS protein inhibition of growth hormone-stimulated STAT5 signaling by multiple mechanisms. J Biol Chem. 1999 Dec 10;274(50):35553-61.
390. Ram PA, Waxman DJ. Role of the cytokine-inducible SH2 protein CIS in desensitization of STAT5b signaling by continuous growth hormone. J Biol Chem.2000 Dec 15;275(50):39487-96.
391. Xu J, Keeton AB, Franklin JL, Li X, Venable DY, Frank SJ, Messina JL. Insulin enhances growth hormone induction of the MEK/ERK signaling pathway. J Biol Chem. 2006 Jan 13;281(2):982-92. Epub 2005 Nov 4.
392. Lewis TS, Shapiro PS, Ahn NG. Signal transduction through MAP kinase cascades. Adv Cancer Res. 1998;74:49-139. Review.
393. Cobb MH. MAP kinase pathways. Prog Biophys Mol Biol. 1999;71(3-4):479-500. Review.
394. Mebis L, Paletta D, Debaveye Y, Ellger B, Langouche L, D’Hoore A, Darras VM, Visser TJ, Van den Berghe G. Expression of thyroid hormone transporters during critical illness. Eur J Endocrinol. 2009 Aug;161(2):243-50.
395. Jørgensen JO, Pedersen SA, Laurberg P, Weeke J, Skakkebaek NE, Christiansen JS. Effects of growth hormone therapy on thyroid function of growth hormone-deficient adults with and without concomitant thyroxine-substituted central hypothyroidism. J Clin Endocrinol Metab. 1989 Dec;69(6):1127-32.
396. Jørgensen JO, Pedersen SB, Børglum J, Møller N, Schmitz O, Christiansen JS, Richelsen B. Fuel metabolism, energy expenditure, and thyroid function in growth hormone-treated obese women: a double-blind placebo-controlled study. Metabolism. 1994 Jul;43(7):872-7.
397. Wolthers T, Grøftne T, Møller N, Christiansen JS, Orskov H, Weeke J, Jørgensen JO. Calorigenic effects of growth hormone: the role of thyroid hormones. J Clin Endocrinol Metab. 1996 Apr;81(4):1416-9.
398. Feldt-Rasmussen U. Interactions between growth hormone and the thyroid gland — with special reference to biochemical diagnosis. Curr Med Chem. 2007;14(26):2783-8. Review.
399. Kalina-Faska B, Kalina M, Koehler B. Effects of recombinant growth hormone therapy on thyroid hormone concentrations. Int J Clin Pharmacol Ther. 2004 Jan;42(1):30-4.
400. Hubina E, Mersebach H, Rasmussen AK, Juul A, Sneppen SB, Góth MI, Feldt-Rasmussen U. Effect of growth hormone replacement therapy on pituitary hormone secretion and hormone replacement therapies in GHD adults. Horm Res. 2004;61(5):211-7. Epub 2004 Jan 30.
401. Seminara S, Stagi S, Candura L, Scrivano M, Lenzi L, Nanni L, Pagliai F, Chiarelli F. Changes of thyroid function during long-term hGH therapy in GHD children. A possible relationship with catch-up growth? Horm Metab Res. 2005 Dec;37(12):751-6.
402. Losa M, Scavini M, Gatti E, Rossini A, Madaschi S, Formenti I, Caumo A, Stidley CA, Lanzi R. Long-term effects of growth hormone replacement therapy on thyroid function in adults with growth hormone deficiency. Thyroid. 2008 Dec;18(12):1249-54.
403. Müller MJ, Seitz HJ. Thyroid hormone action on intermediary metabolism. Part III. Protein metabolism in hyper- and hypothyroidism. Klin Wochenschr. 1984 Feb 1;62(3):97-102.
404. Tawa NE Jr, Odessey R, Goldberg AL. Inhibitors of the proteasome reduce the accelerated proteolysis in atrophying rat skeletal muscles. J Clin Invest. 1997 Jul 1;100(1):197-203. PubMed PMID: 9202072
405. Dace A, Zhao L, Park KS, et al. Hormone binding induces rapid proteasome-mediated degradation of thyroid hormone receptors. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(16):8985-8990.
406. Clément K, Viguerie N, Diehn M, Alizadeh A, Barbe P, Thalamas C, Storey JD, Brown PO, Barsh GS, Langin D. In vivo regulation of human skeletal muscle gene expression by thyroid hormone. Genome Res. 2002 Feb;12(2):281-91.
407. Miell JP, Taylor AM, Zini M, Maheshwari HG, Ross RJ, Valcavi R. Effects of hypothyroidism and hyperthyroidism on insulin-like growth factors (IGFs) and growth hormone- and IGF-binding proteins. J Clin Endocrinol Metab. 1993 Apr;76(4):950-5.
408. Murao K, Takahara J, Sato M, Tamaki M, Niimi M, Ishida T. Relationship between thyroid functions and urinary growth hormone secretion in patients with hyper- and hypothyroidism. Endocr J. 1994 Oct;41(5):517-22.
409. Wolf M, Ingbar SH, Moses AC. Thyroid hormone and growth hormone interact to regulate insulin-like growth factor-I messenger ribonucleic acid and circulating levels in the rat. Endocrinology. 1989 Dec;125(6):2905-14.
410. Laron Z. Interactions between the thyroid hormones and the hormones of the growth hormone axis. Pediatr Endocrinol Rev. 2003 Dec;1 Suppl 2:244-9-discussion 250. Review.
411. Fiems LO. Double Muscling in Cattle: Genes, Husbandry, Carcasses and Meat. Animals : an Open Access Journal from MDPI. 2012;2(3):472-506.
412. Langley B, Thomas M, Bishop A, Sharma M, Gilmour S, Kambadur R. Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J Biol Chem. 2002 Dec 20;277(51):49831-40. Epub 2002 Sep 18.
413. McPherron AC, Lee SJ. Double muscling in cattle due to mutations in the myostatin gene. Proc Natl Acad Sci U S A. 1997 Nov 11;94(23):12457-61.
414. Schuelke M, Wagner KR, Stolz LE, Hübner C, Riebel T, Kömen W, Braun T, Tobin JF, Lee SJ. Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med. 2004 Jun 24;350(26):2682-8.
415. Clop A, Marcq F, Takeda H, Pirottin D, Tordoir X, Bibé B, Bouix J, Caiment F, Elsen JM, Eychenne F, Larzul C, Laville E, Meish F, Milenkovic D, Tobin J, Charlier C, Georges M. A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat Genet. 2006 Jul;38(7):813-8.
416. Gonzalez-Cadavid NF, Taylor WE, Yarasheski K, Sinha-Hikim I, Ma K, Ezzat S, Shen R, Lalani R, Asa S, Mamita M, Nair G, Arver S, Bhasin S. Organization of the human myostatin gene and expression in healthy men and HIV-infected men with muscle wasting. Proc Natl Acad Sci U S A. 1998 Dec 8;95(25):14938-43.
417. Liu W, Thomas SG, Asa SL, Gonzalez-Cadavid N, Bhasin S, Ezzat S. Myostatin is a skeletal muscle target of growth hormone anabolic action. J Clin Endocrinol Metab. 2003 Nov;88(11):5490-6.
418. Oldham JM, Osepchook CC, Jeanplong F, Falconer SJ, Matthews KG, Conaglen JV, Gerrard DF, Smith HK, Wilkins RJ, Bass JJ, McMahon CD. The decrease in mature myostatin protein in male skeletal muscle is developmentally regulated by growth hormone. J Physiol. 2009 Feb 1;587(3):669-77.
419. Williams NG, Interlichia JP, Jackson MF, Hwang D, Cohen P, Rodgers BD. Endocrine actions of myostatin: systemic regulation of the IGF and IGF binding protein axis. Endocrinology. 2011 Jan;152(1):172-80.
420. Winbanks CE, Weeks KL, Thomson RE, Sepulveda PV, Beyer C, Qian H, Chen JL, Allen JM, Lancaster GI, Febbraio MA, Harrison CA, McMullen JR, Chamberlain JS, Gregorevic P. Follistatin-mediated skeletal muscle hypertrophy is regulated by Smad3 and mTOR independently of myostatin. J Cell Biol. 2012 Jun 25;197(7):997-1008.
421. Lach-Trifilieff E, Minetti GC, Sheppard K, Ibebunjo C, Feige JN, Hartmann S, Brachat S, Rivet H, Koelbing C, Morvan F, Hatakeyama S, Glass DJ. An antibody blocking activin type II receptors induces strong skeletal muscle hypertrophy and protects from atrophy. Mol Cell Biol. 2014 Feb;34(4):606-18.
422. Bark TH, McNurlan MA, Lang CH, Garlick PJ. Increased protein synthesis after acute IGF-I or insulin infusion is localized to muscle in mice. Am J Physiol. 1998 Jul;275(1 Pt 1):E118-23.
423. Barton-Davis ER, Shoturma DI, Sweeney HL. Contribution of satellite cells to IGF-I induced hypertrophy of skeletal muscle. Acta Physiol Scand. 1999 Dec;167(4):301-5.
424. Suryawan A, Frank JW, Nguyen HV, Davis TA. Expression of the TGF-beta family of ligands is developmentally regulated in skeletal muscle of neonatal rats. Pediatr Res. 2006 Feb;59(2):175-9.
425. Gilson H, Schakman O, Kalista S, Lause P, Tsuchida K, Thissen JP. Follistatin induces muscle hypertrophy through satellite cell proliferation and inhibition of both myostatin and activin. Am J Physiol Endocrinol Metab. 2009 Jul;297(1):E157-64.
426. Bodine SC, Stitt TN, Gonzalez M, Kline WO, Stover GL, Bauerlein R, Zlotchenko E, Scrimgeour A, Lawrence JC, Glass DJ, Yancopoulos GD. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol. 2001 Nov;3(11):1014-9.
427. Rommel C, Bodine SC, Clarke BA, Rossman R, Nunez L, Stitt TN, Yancopoulos GD, Glass DJ. Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat Cell Biol. 2001 Nov;3(11):1009-13.
428. Kalista S, Schakman O, Gilson H, Lause P, Demeulder B, Bertrand L, Pende M, Thissen JP. The type 1 insulin-like growth factor receptor (IGF-IR) pathway is mandatory for the follistatin-induced skeletal muscle hypertrophy. Endocrinology. 2012 Jan;153(1):241-53.
429. Barbé C, Kalista S, Loumaye A, Ritvos O, Lause P, Ferracin B, Thissen JP. Role of IGF-I in follistatin-induced skeletal muscle hypertrophy. Am J Physiol Endocrinol Metab. 2015 Sep 15;309(6):E557-67. doi: 10.1152/ajpendo.00098.2015. Epub 2015 Jul 28.
430. Coffey VG, Shield A, Canny BJ, Carey KA, Cameron-Smith D, Hawley JA. Interaction of contractile activity and training history on mRNA abundance in skeletal muscle from trained athletes. Am J Physiol Endocrinol Metab. 2006 May;290(5):E849-55.
431. Moore WV, Leppert P. Role of aggregated human growth hormone (hGH) in development of antibodies to hGH. J Clin Endocrinol Metab. 1980 Oct;51(4):691-7
432. Dannies PS. Protein folding and deficiencies caused by dominant-negative mutants of hormones. Vitam Horm. 2000;58:1-26. Review.
433. DeVol DL, Rotwein P, Sadow JL, Novakofski J, Bechtel PJ. Activation of insulin-like growth factor gene expression during work-induced skeletal muscle growth. Am J Physiol. 1990 Jul;259(1 Pt 1):E89-95.
434. Hermansen K, Bengtsen M, Kjær M, Vestergaard P, Jørgensen JOL. Impact of GH administration on athletic performance in healthy young adults: A systematic review and meta-analysis of placebo-controlled trials. Growth Horm IGF Res. 2017 Jun;34:38-44.
435. de Souza GL, Hallak J. Anabolic steroids and male infertility: a comprehensive review. BJU Int. 2011 Dec;108(11):1860-5.
436. Kraemer WJ, Marchitelli L, Gordon SE, Harman E, Dziados JE, Mello R, Frykman P, McCurry D, Fleck SJ. Hormonal and growth factor responses to heavy resistance exercise protocols. J Appl Physiol (1985). 1990 Oct;69(4):1442-50.
437. Pfeffer LA, Brisson BK, Lei H, Barton ER. The insulin-like growth factor (IGF)-I E-peptides modulate cell entry of the mature IGF-I protein. Mol Biol Cell. 2009 Sep;20(17):3810-7.
438. Mills P, Dominique JC, Lafrenière JF, Bouchentouf M, Tremblay JP. A synthetic mechano growth factor E Peptide enhances myogenic precursor cell transplantation success. Am J Transplant. 2007 Oct;7(10):2247-59.
439. Brisson BK, Barton ER. Insulin-like growth factor-I E-peptide activity is dependent on the IGF-I receptor. PLoS One. 2012;7(9):e45588.
440. Brisson BK, Spinazzola J, Park S, Barton ER. Viral expression of insulin-like growth factor I E-peptides increases skeletal muscle mass but at the expense of strength. Am J Physiol Endocrinol Metab. 2014 Apr 15;306(8):E965-74.
441. Goldspink G, Harridge S. Mechanism for adaptation in skeletal muscle In: Komi P, editor. Strength and power in sport: Olympic encyclopedia of sports medicine. Oxford: Blackwell; 2002. p. 231–51.
442. Janssen JA, Hofland LJ, Strasburger CJ, van den Dungen ES, Thevis M. Potency of Full-Length MGF to Induce Maximal Activation of the IGF-I R Is Similar to Recombinant Human IGF-I at High Equimolar Concentrations. PLoS One. 2016 Mar 18;11(3):e0150453.
