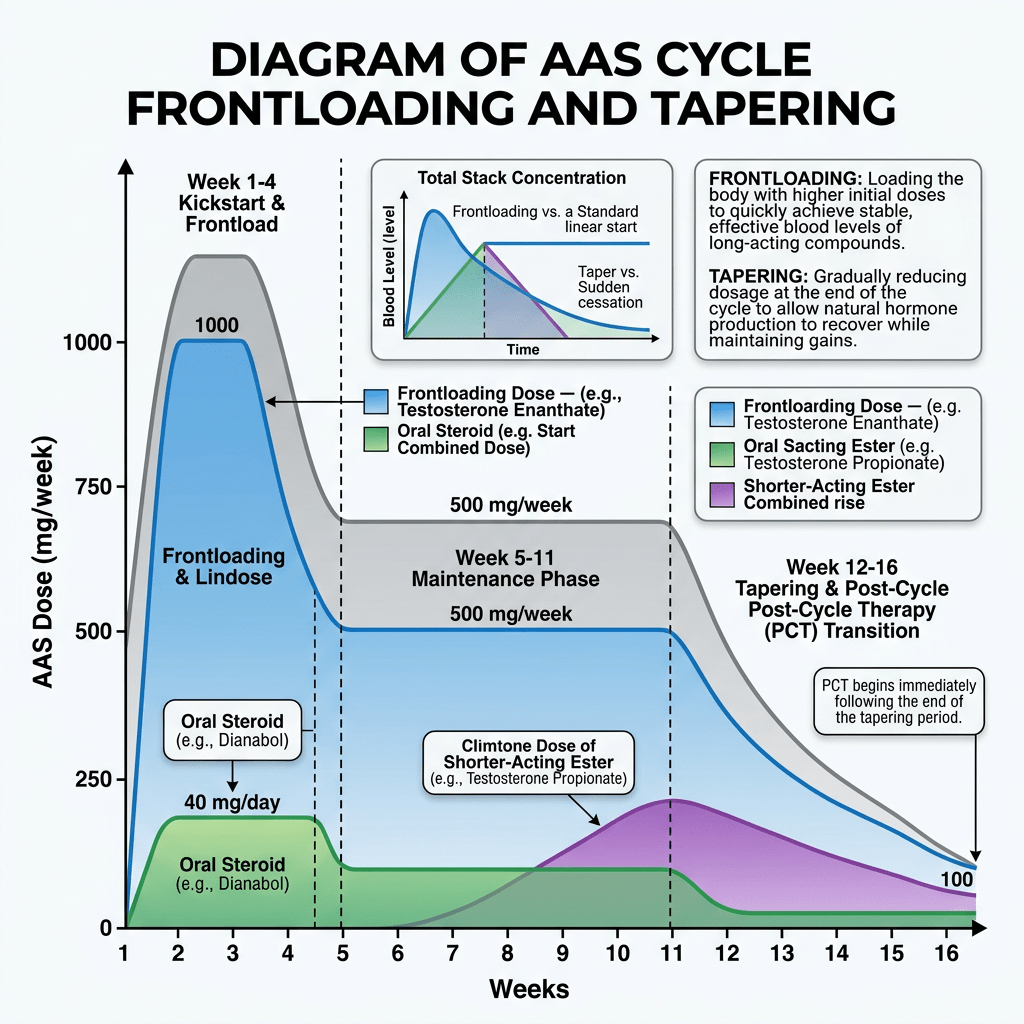
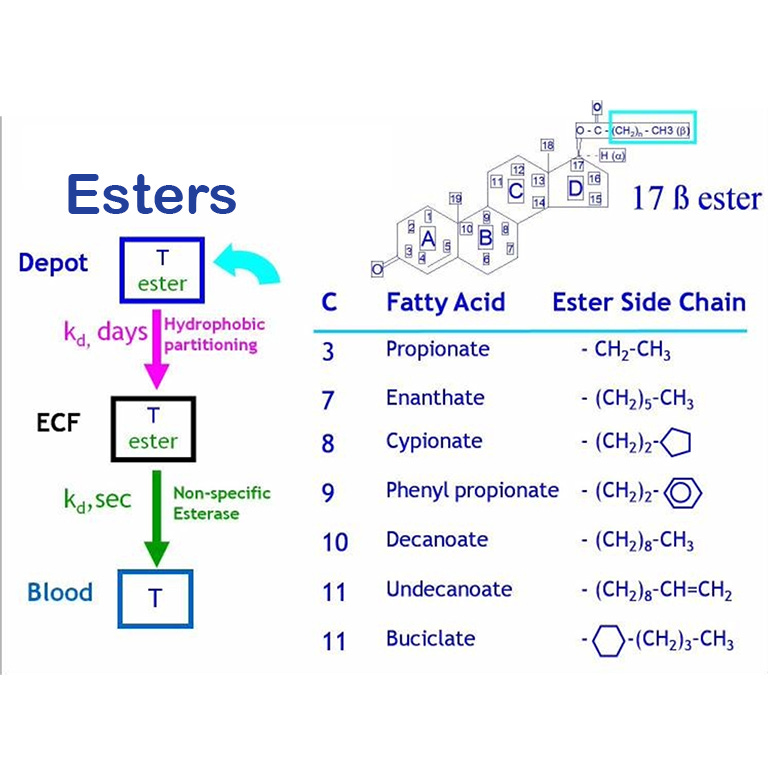
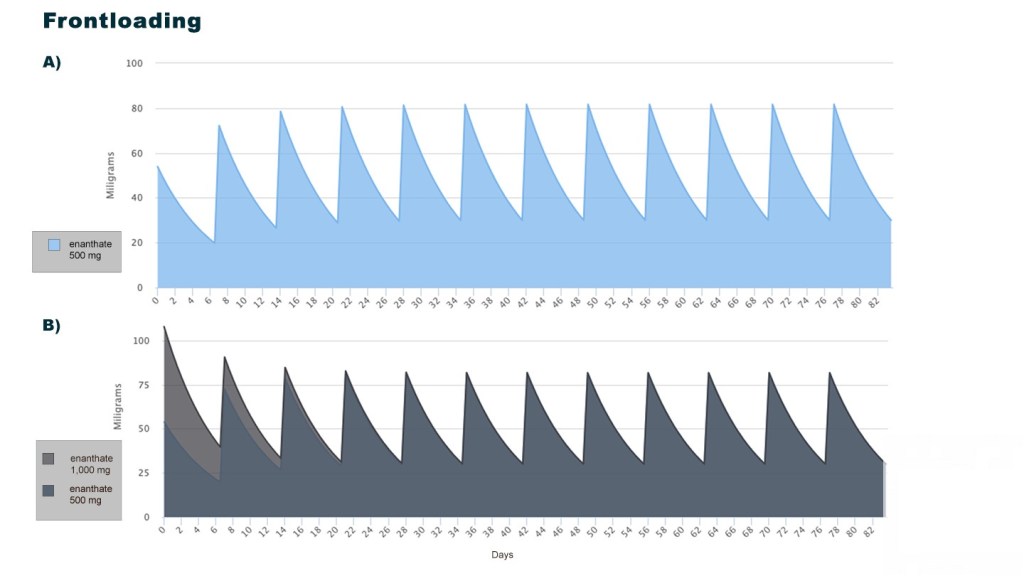
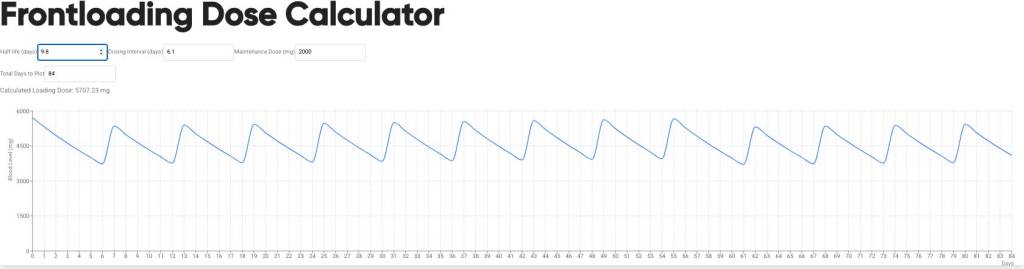
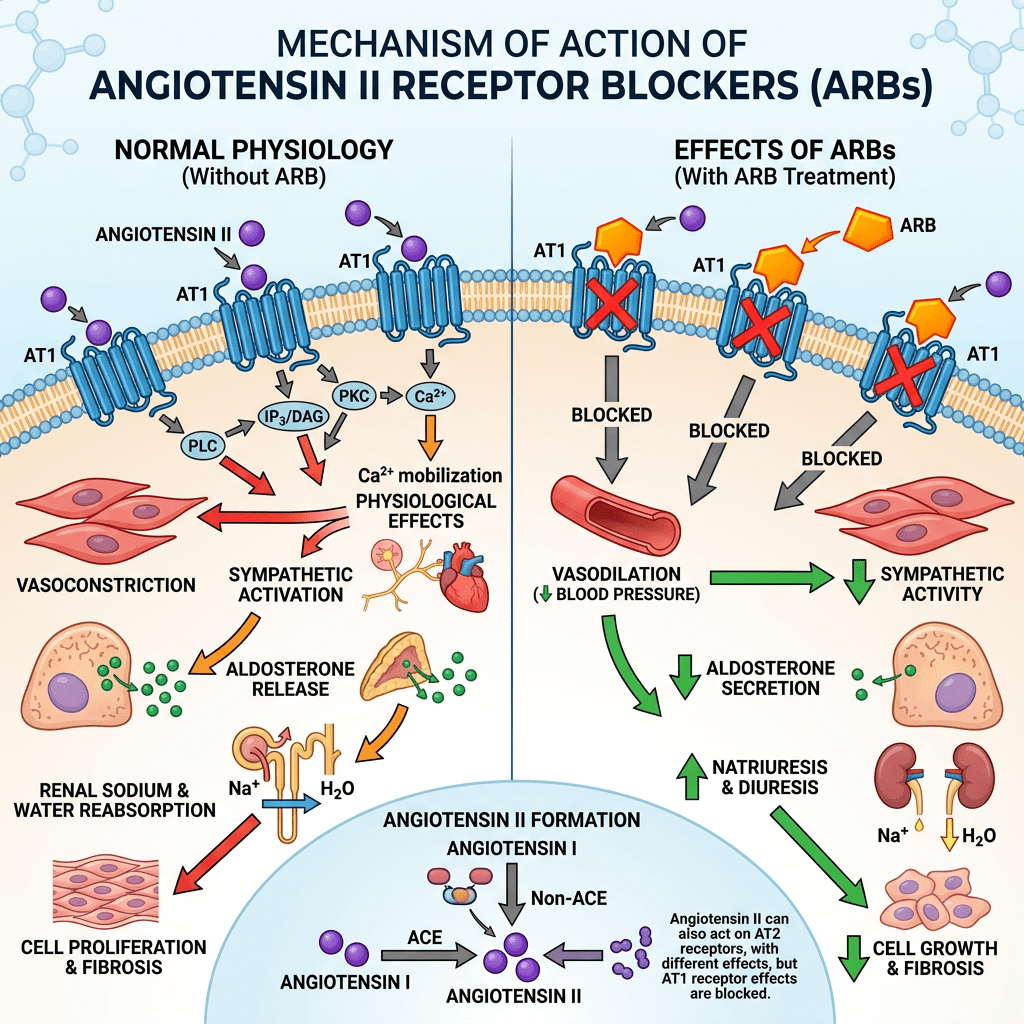
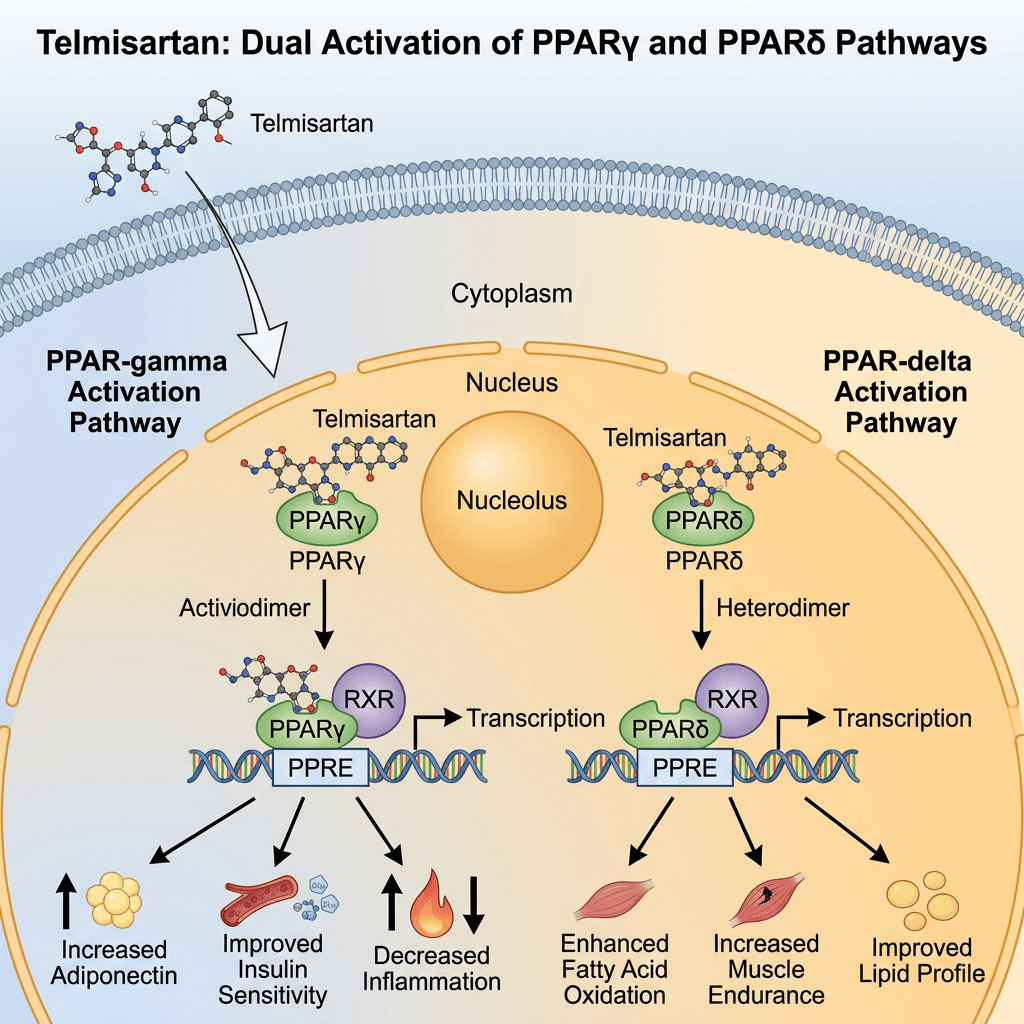
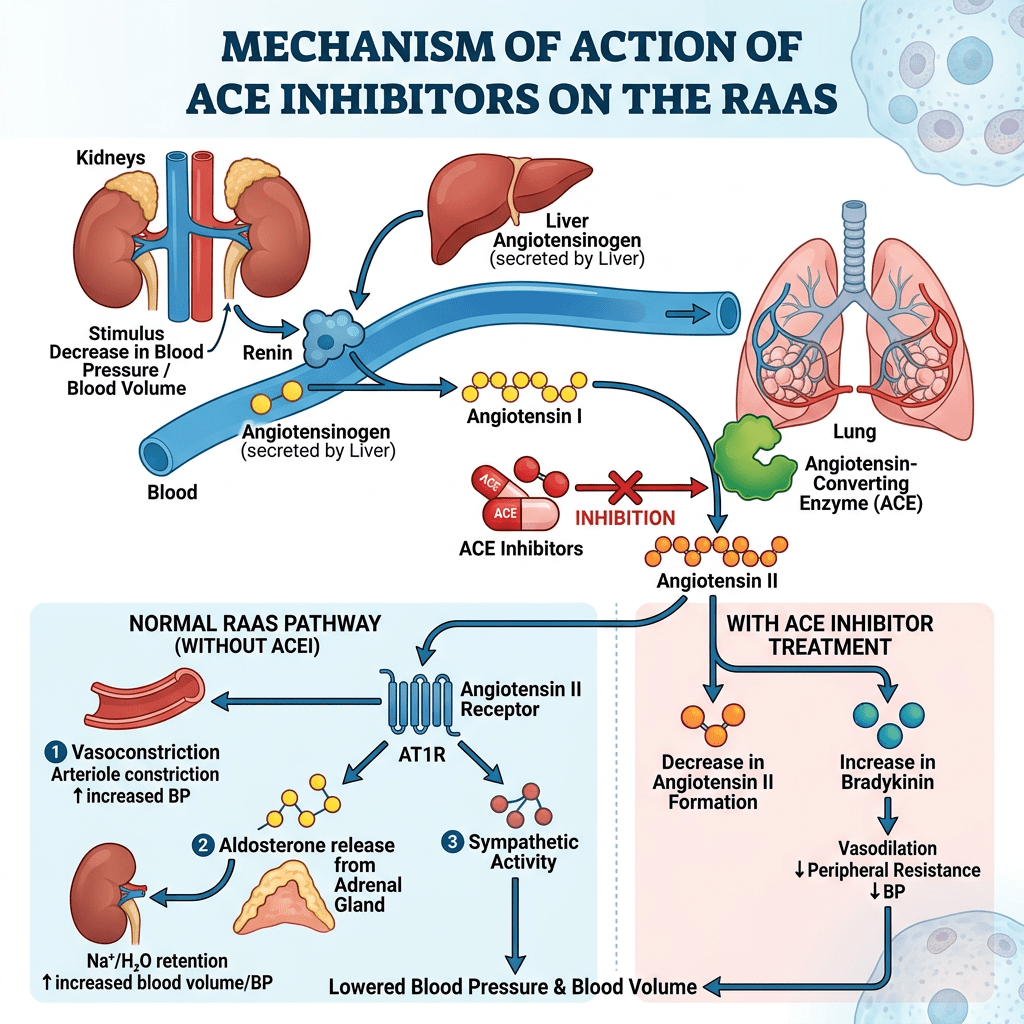
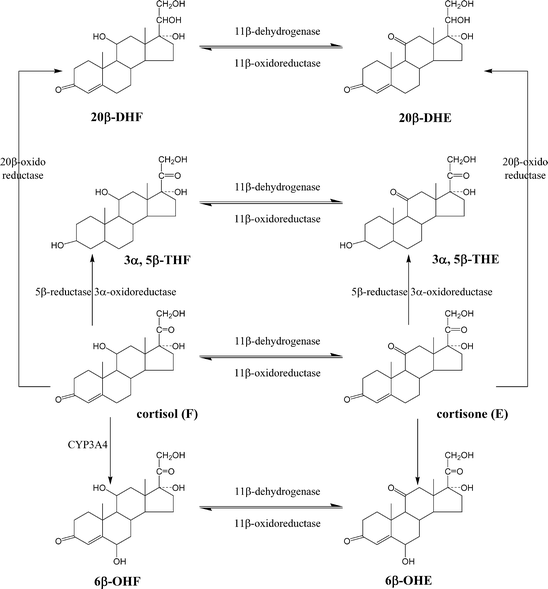
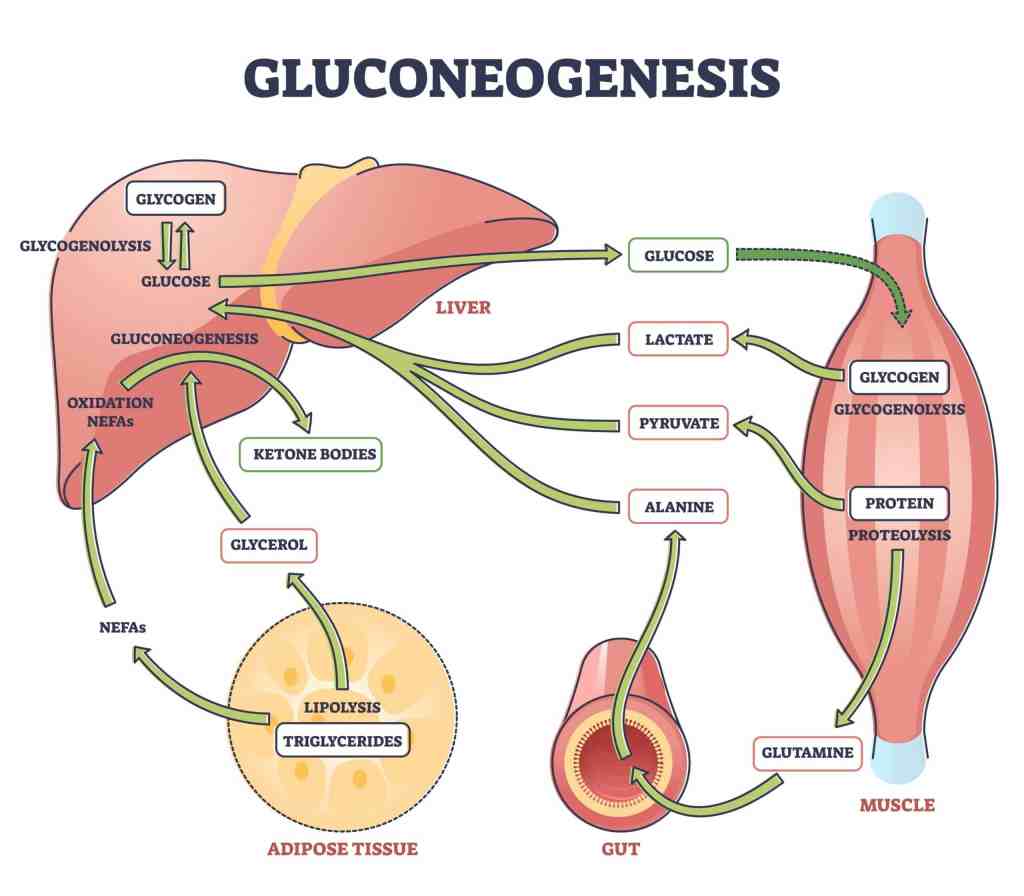
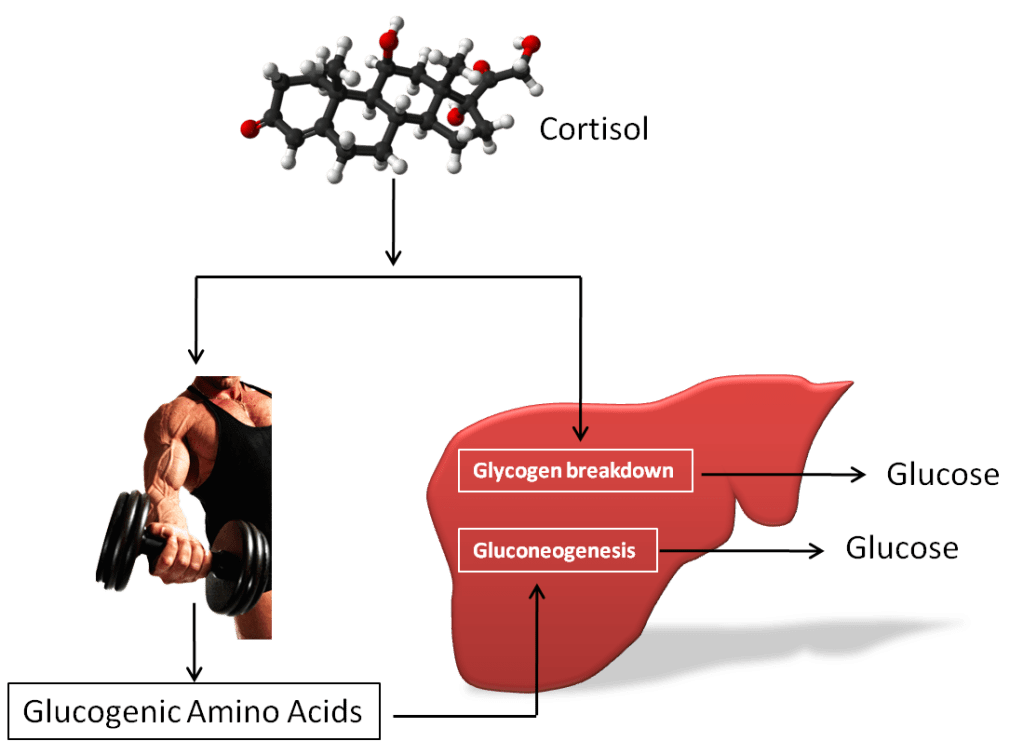
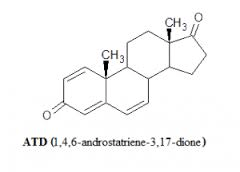
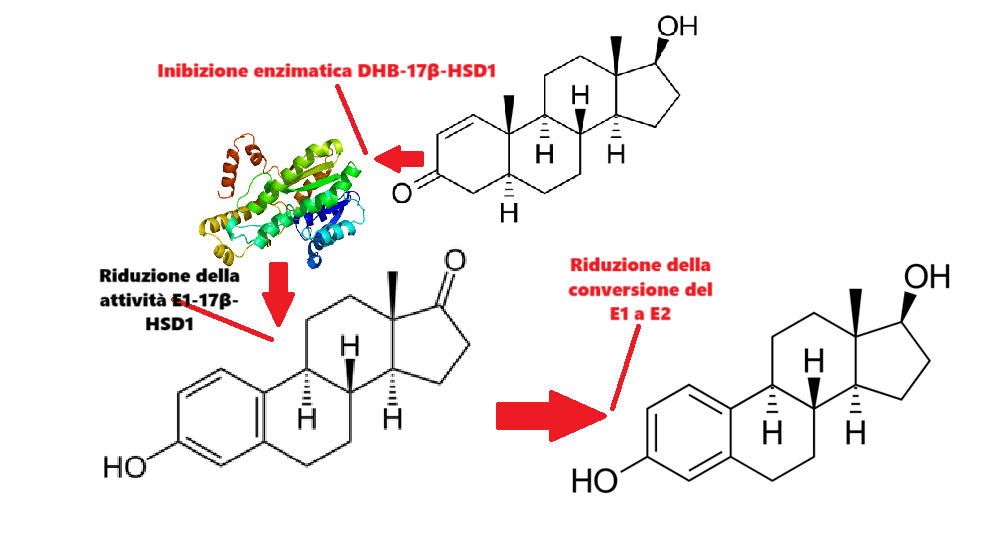
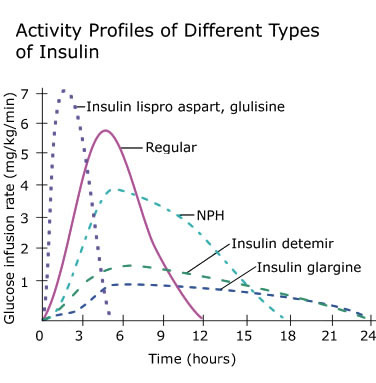
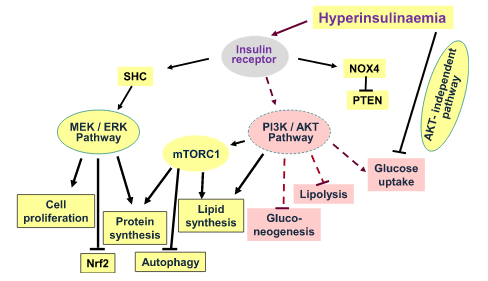
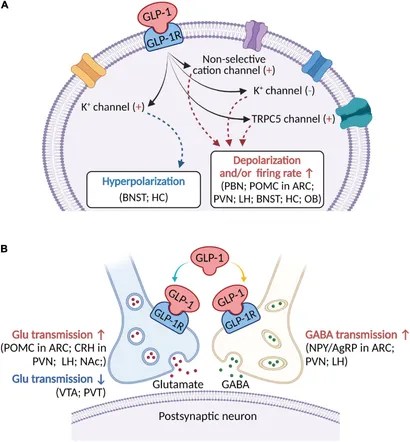
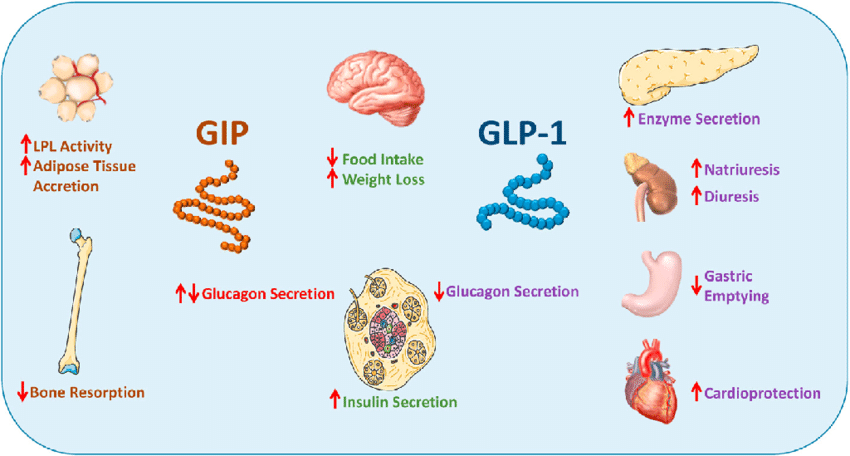
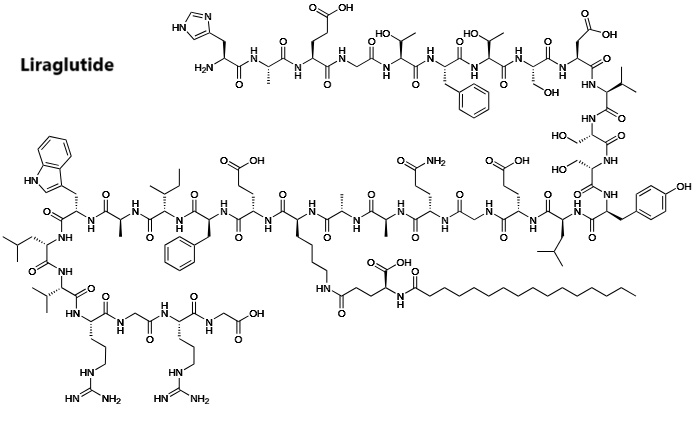
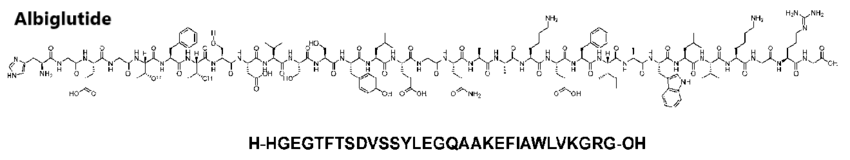
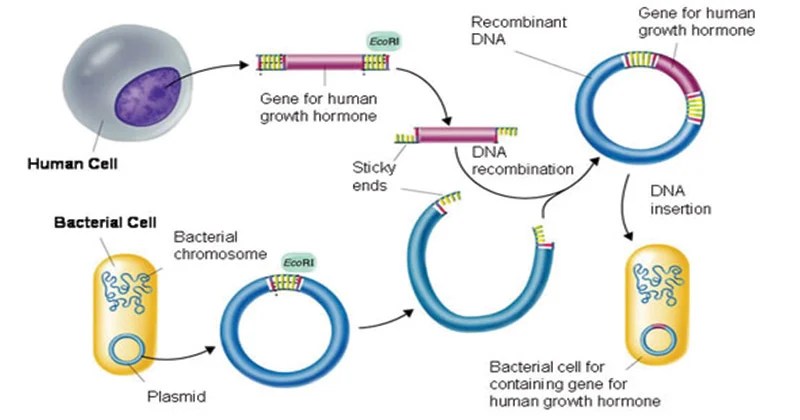
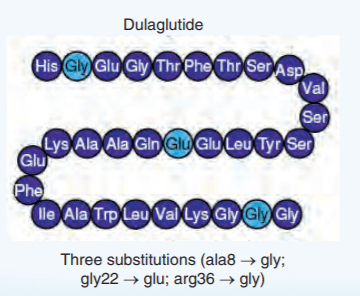
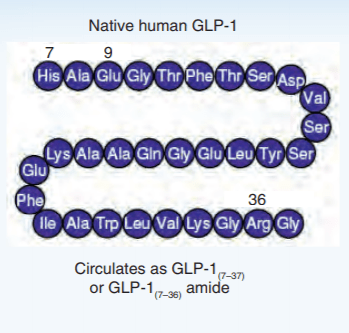
Introduzione:
Chi mi conosce da più tempo sa bene che l’argomento enhanced in contesto femminile è stato spesso trattato in modo approfondito in diversi miei lavori; da articoli a video fino ai seminari. Trattandosi di un argomento complesso e proseguendo nella ricerca teorico-pratica negli anni, ho ritenuto utile ritornare sull’argomento anche per continuare a sfatare quei miti da “pitecus palestriculus” che continuano a rendere il bodybuilding femminile più una sorta di “Casablkanca” che uno sport estetico, così come è nato e dovrebbe essere.
In questo articolo si tratterà quindi di ottimizzazione farmacologica nel Bodybuilding femminile per le prestazioni e l’estetica basata su solide basi scientifiche. Tutto ciò richiederà una ricca introduzione sulla fisiologia ormonale nelle donne, cosa che permetterà meglio di comprendere: metabolismo degli estrogeni, degli androgeni, sensibilità recettoriale, periodizzazione nel OMC [ovulatory-menstrual cycle] , protocolli per la PCOS, strategie per la menopausa e selezione di composti con dati farmacocinetici ecc… . Niente bias.
In questa prima parte inizieremo dalle basi della steroidogenesi per poi proseguire con la diversità d’impatto dei vari sistemi ormonali steroidei fino alle differenze dimorfiche dell’Asse hGH/IGF1 e della distribuzione e mobilitazione del tessuto adiposo… Impegnativo ma di estrema utilità se si vuole diventare lucidi conoscitori dell’argomento trattato…
Le basi della steroidogenesi nella donna
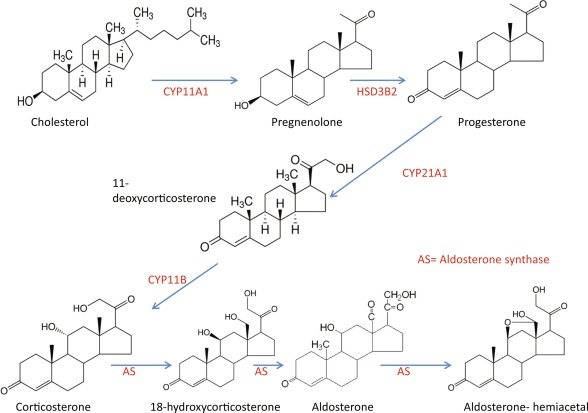
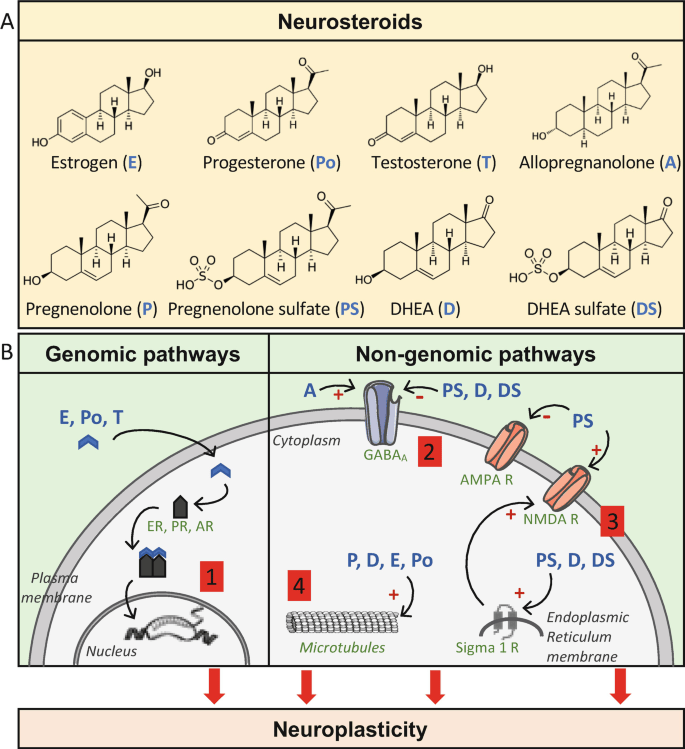
La biosintesi degli steroidi [vedi steroidogenesi] nelle donne è il processo metabolico articolato in più fasi che [similmente a quanto avviene nell’uomo] converte il Colesterolo in ormoni steroidei — tra cui progestinici, androgeni, estrogeni, glucocorticoidi e mineralcorticoidi — principalmente all’interno delle ovaie, delle ghiandole surrenali e dei tessuti bersaglio periferici.

- Substrato primario fondamentale e prima fase
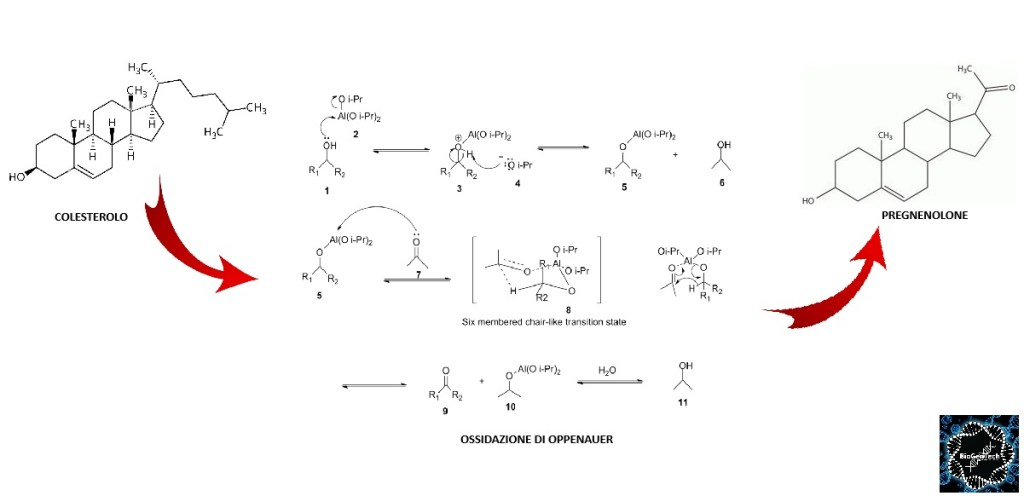
- Colesterolo: il precursore universale di tutti gli ormoni steroidei.
- Sintesi del Pregnenolone: l’enzima mitocondriale CYP11A1 scinde la catena laterale del Colesterolo, formando il Pregnenolone, che funge da molecola “di genesi” per tutte le vie metaboliche successive.
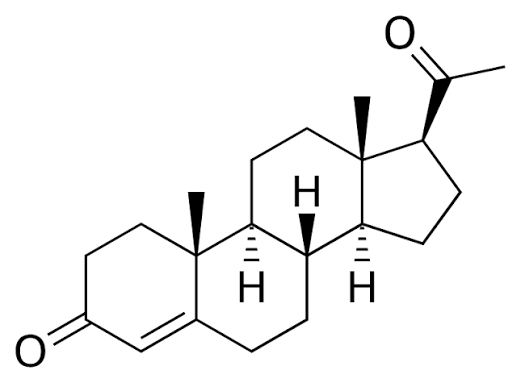
- Pregnenolone – lo steroide progenitore –

Il Pregnenolone fu sintetizzato per la prima volta da Adolf Butenandt e dai suoi collaboratori nel 1934.
Il Pregnenolone è noto chimicamente anche come pregn-5-en-3β-ol-20-one. Come altri steroidi, è costituito da quattro idrocarburi ciclici interconnessi. Il composto contiene gruppi funzionali chetonici e idrossilici, due ramificazioni metiliche e un doppio legame in posizione C5, nell’anello idrocarburico B. Come molti ormoni steroidei, è idrofobico. Il derivato solfato, il Pregnenolone Solfato, è idrosolubile.
Il 3β-diidroprogesterone (pregn-4-en-3β-ol-20-one) è un isomero del Pregnenolone in cui il doppio legame in C5 è stato sostituito da un doppio legame in C4.
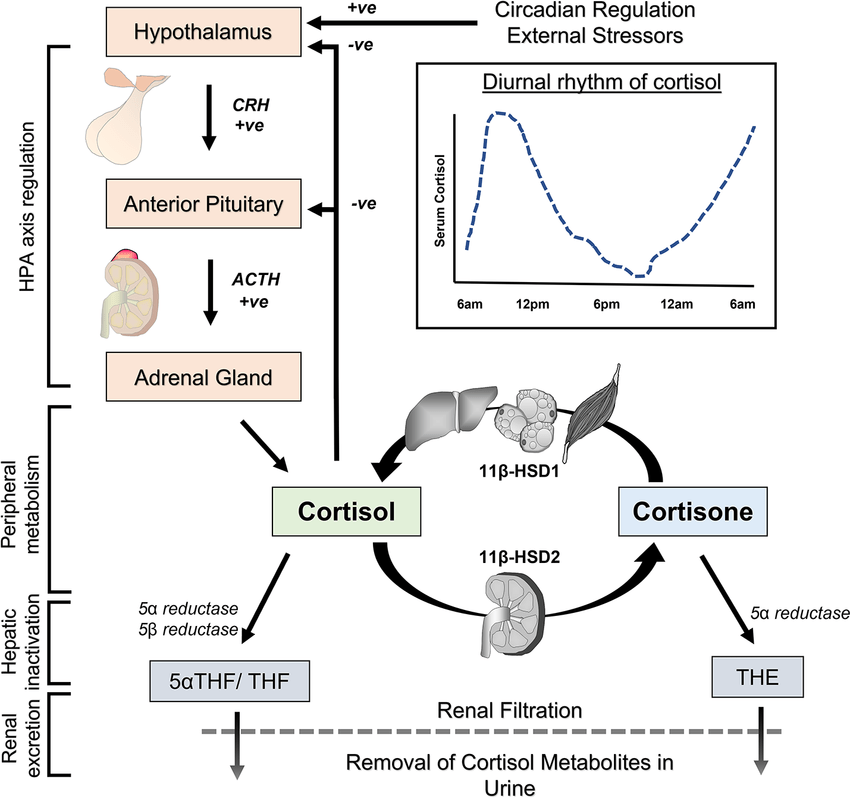
Come abbiamo visto nel precedente paragrafo, il Pregnenolone viene sintetizzato dal Colesterolo. Questa conversione comporta l’idrossilazione della catena laterale nelle posizioni C20 e C22, con scissione della catena laterale. L’enzima che svolge questa funzione è il citocromo P450scc, situato nei mitocondri e regolato dagli ormoni trofici dell’ipofisi anteriore, quali l’Ormone Adrenocorticotropo [ACTH], l’Ormone Follicolo-Stimolante [FSH] e l’Ormone Luteinizzante [LH], nelle ghiandole surrenali e nelle gonadi. Nella trasformazione del Colesterolo in Pregnenolone sono presenti due intermedi, il 22R-idrossicolesterolo e il 20α,22R-diidrossicolesterolo, e tutte e tre le fasi della trasformazione sono catalizzate dal P450scc. Il Pregnenolone viene prodotto principalmente nelle ghiandole surrenali, nelle gonadi e nel cervello. Sebbene il Pregnenolone venga prodotto anche nelle gonadi e nel cervello, la maggior parte del Pregnenolone circolante deriva dalla corteccia surrenale.
Il Pregnenolone è lipofilo e attraversa facilmente la barriera emato-encefalica. Ciò è in contrasto con il Pregnenolone Solfato, che non attraversa la barriera emato-encefalica.
Dopo la sua sintesi il Pregnenolone subisce un ulteriore metabolismo steroideo secondo una delle seguenti vie:
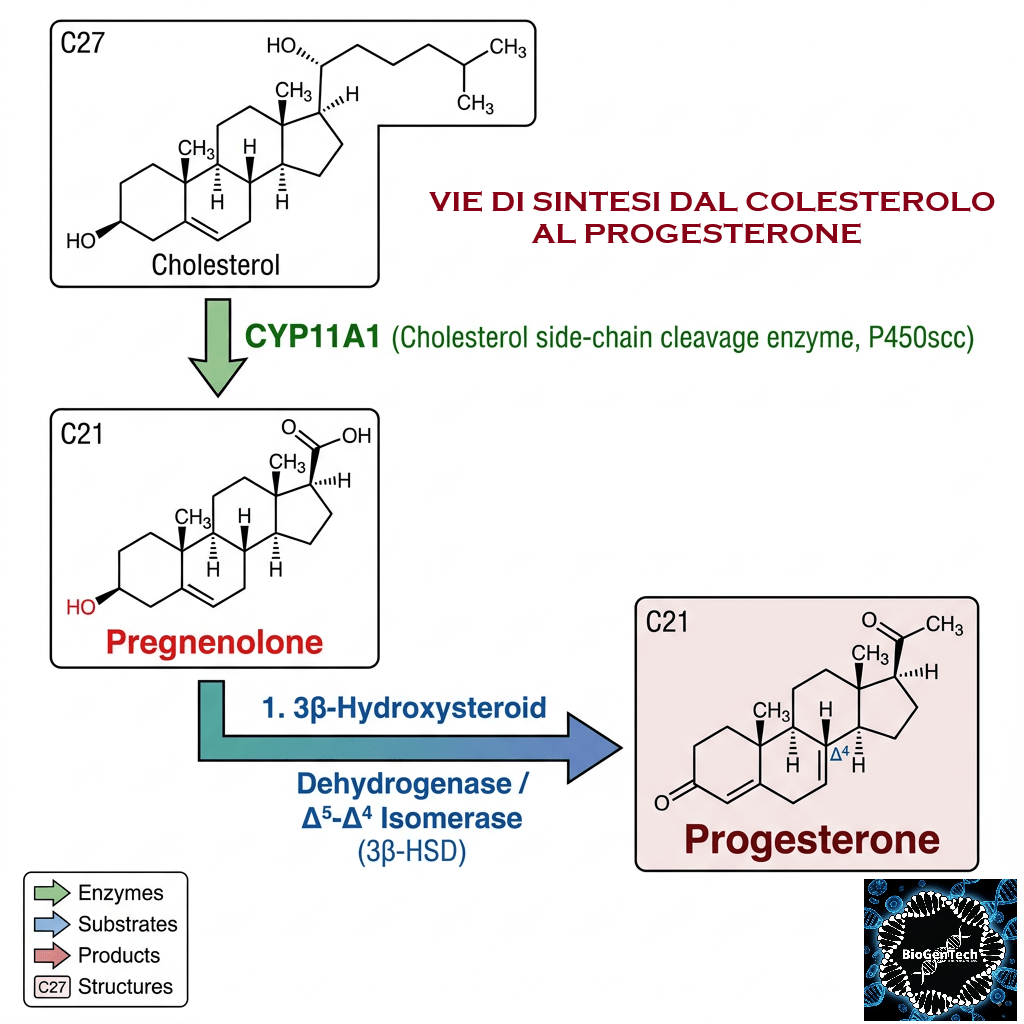
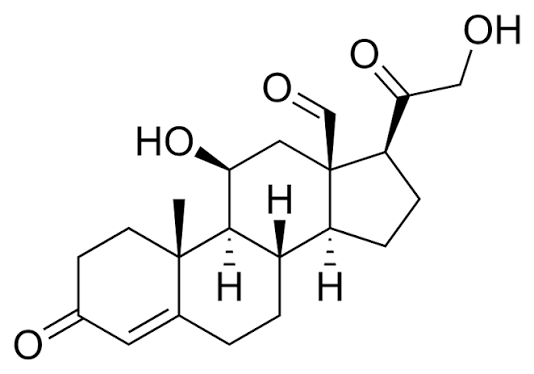
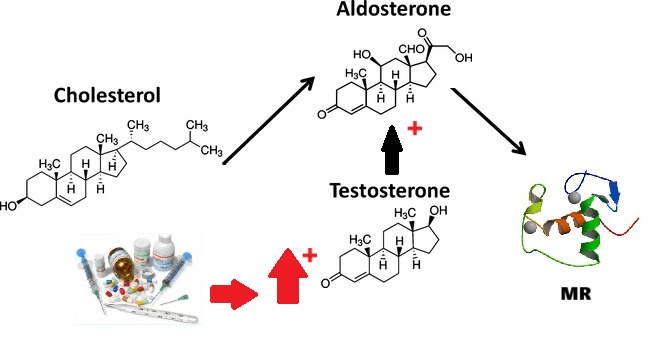
- Il Pregnenolone può essere convertito in Progesterone. La fase enzimatica cruciale si articola in due fasi, che coinvolgono una 3β-idrossisteroide deidrogenasi e una Δ5-4 isomerasi. Quest’ultima trasferisce il doppio legame dalla posizione C5 alla posizione C4 sull’anello A. Il Progesterone costituisce il punto di ingresso nella via Δ4, che porta alla produzione di 17α-idrossiprogesterone e Androstenedione, precursori del Testosterone e dell’Estrone. Anche l’Aldosterone e i corticosteroidi derivano dal Progesterone o dai suoi derivati.
- Il Pregnenolone può essere convertito in 17α-idrossipregnenolone dall’enzima 17α-idrossilasi (CYP17A1). Seguendo questa via, denominata via Δ5, il passo successivo è la conversione in Deidroepiandrosterone (DHEA) tramite la 17,20-liasi (CYP17A1). Il DHEA è il precursore dell’Androstenedione.
- Il Pregnenolone può essere convertito in Androstadienolo dalla 16-ene sintasi (CYP17A1).
- Il Pregnenolone può essere convertito in solfato di Pregnenolone dalla steroide solfotransferasi, e questa conversione può essere invertita dalla steroide solfatasi.
I livelli ematici circolanti ritenuti normali per il Pregnenolone sono i seguenti:
- Uomini: da 10 a 200ng/dL
- Donne: da 10 a 230ng/dL
- Bambini: da 10 a 48ng/dL
- Ragazzi adolescenti: da 10 a 50ng/dL
- Ragazze adolescenti: da 15 a 84ng/dL
È stato riscontrato che i livelli medi di Pregnenolone non differiscono in modo significativo nelle donne in postmenopausa e negli uomini anziani (rispettivamente 40 e 39ng/dL).
Il Pregnenolone è un endocannabinoide allosterico, in quanto agisce come modulatore allosterico negativo del recettore CB1. Il Pregnenolone è coinvolto in un ciclo naturale di feedback negativo contro l’attivazione del recettore CB1 negli animali. Impedisce agli agonisti del recettore CB1, come il tetraidrocannabinolo (THC), il principale principio attivo della cannabis, di attivare completamente il recettore CB1. Un composto correlato, l’AEF0117, è stato derivato dal Pregnenolone ed è più specifico per questo tipo di attività.
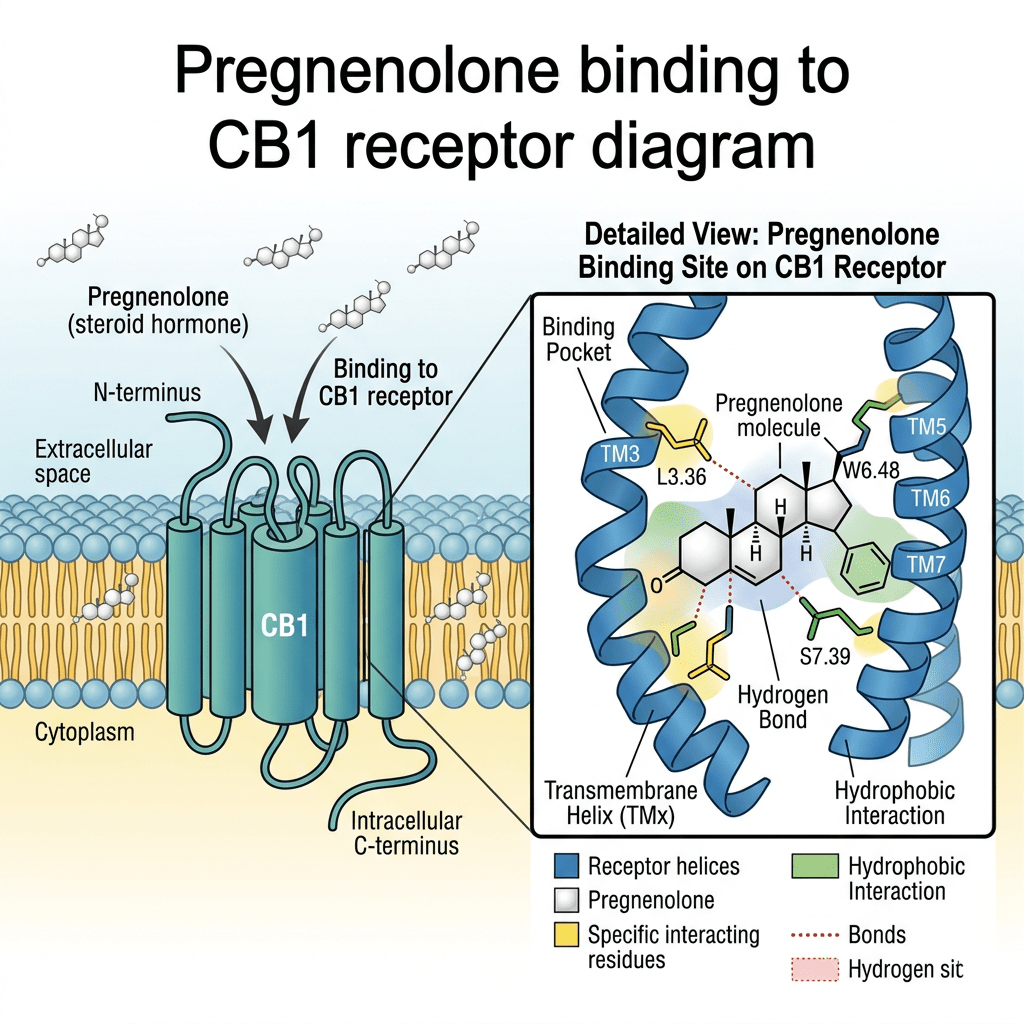
È stato riscontrato che il pregnenolone si lega con elevata affinità, dell’ordine dei nanomolari, alla proteina 2 associata ai microtubuli (MAP2) nel cervello. A differenza del pregnenolone, il solfato di pregnenolone non si è legato ai microtubuli. Il progesterone, invece, si è legato con un’affinità simile a quella del pregnenolone, sebbene, a differenza di quest’ultimo, non abbia aumentato il legame della MAP2 alla tubulina. È stato osservato che il pregnenolone induce la polimerizzazione dei microtubuli nelle colture neuronali e aumenta la crescita dei neuriti nelle cellule PC12 trattate con il fattore di crescita nervoso. Pertanto, il pregnenolone potrebbe controllare la formazione e la stabilizzazione dei microtubuli nei neuroni e potrebbe influenzare sia lo sviluppo neurale durante la fase prenatale sia la plasticità neurale durante l’invecchiamento.
Sebbene il pregnenolone di per sé non possieda queste attività, il suo metabolita, il solfato di pregnenolone, è un modulatore allosterico negativo del recettore GABAA nonché un modulatore allosterico positivo del recettore NMDA. Inoltre, è stato dimostrato che il solfato di pregnenolone attiva il canale ionico TRPM3 (Transient Receptor Potential M3) negli epatociti e nelle isole pancreatiche, provocando l’ingresso di calcio e il conseguente rilascio di insulina.
È stato dimostrato che il Pregnenolone agisce come agonista del recettore X dei pregnani. Il Pregnenolone non presenta attività progestinica, corticosteroidea, estrogenica, androgenica o antiandrogenica.
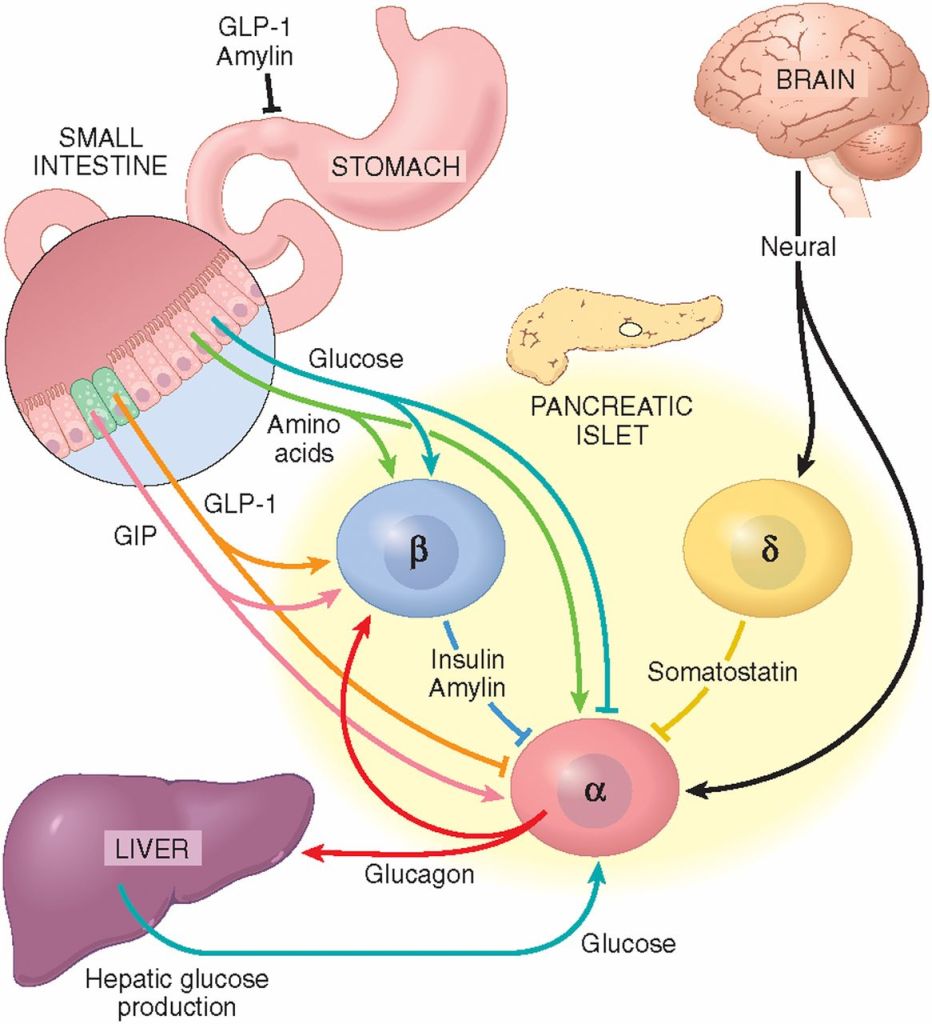
- Principali sedi di produzione steroidea negli individui di sesso femminile
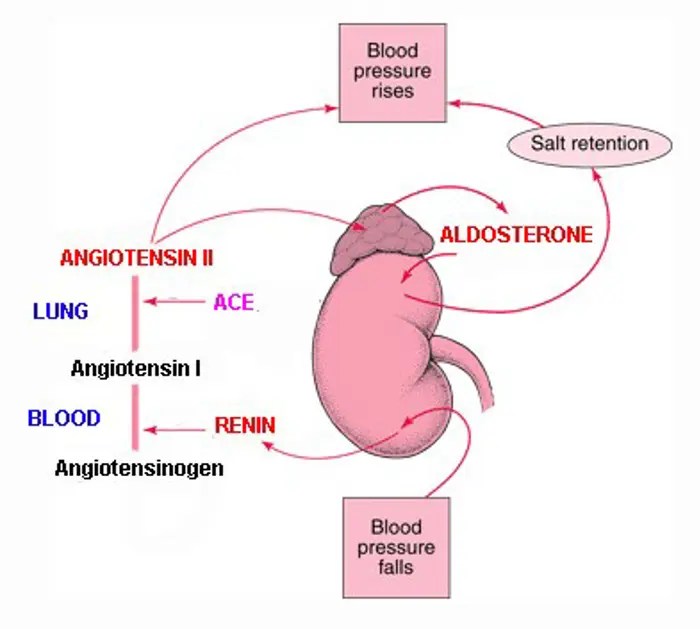
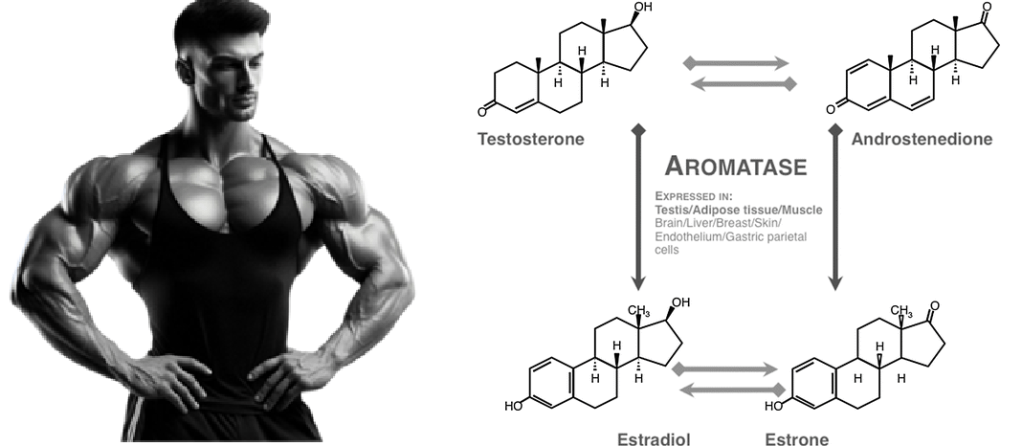
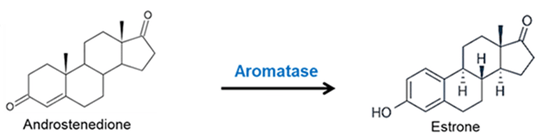
- Ovaie: utilizzano un sistema a due cellule e due gonadotropine (LH e FSH). L’LH stimola le cellule della teca a produrre androgeni (Androstenedione), mentre l’FSH induce le cellule della granulosa a utilizzare l’enzima Aromatasi per convertire tali androgeni in estrogeni.
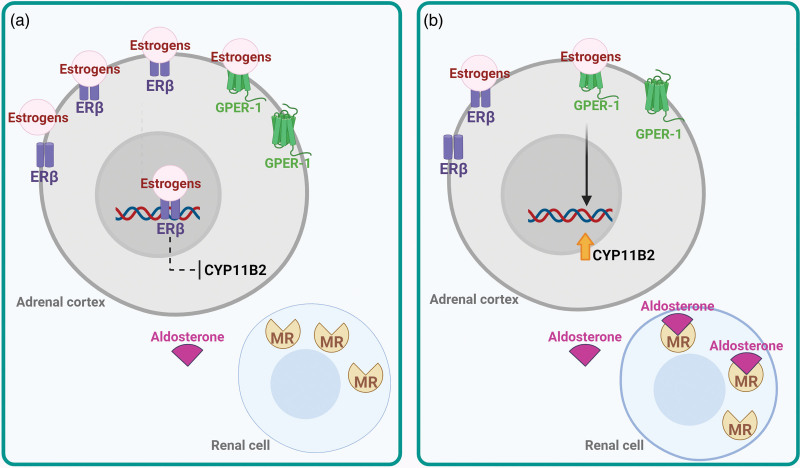
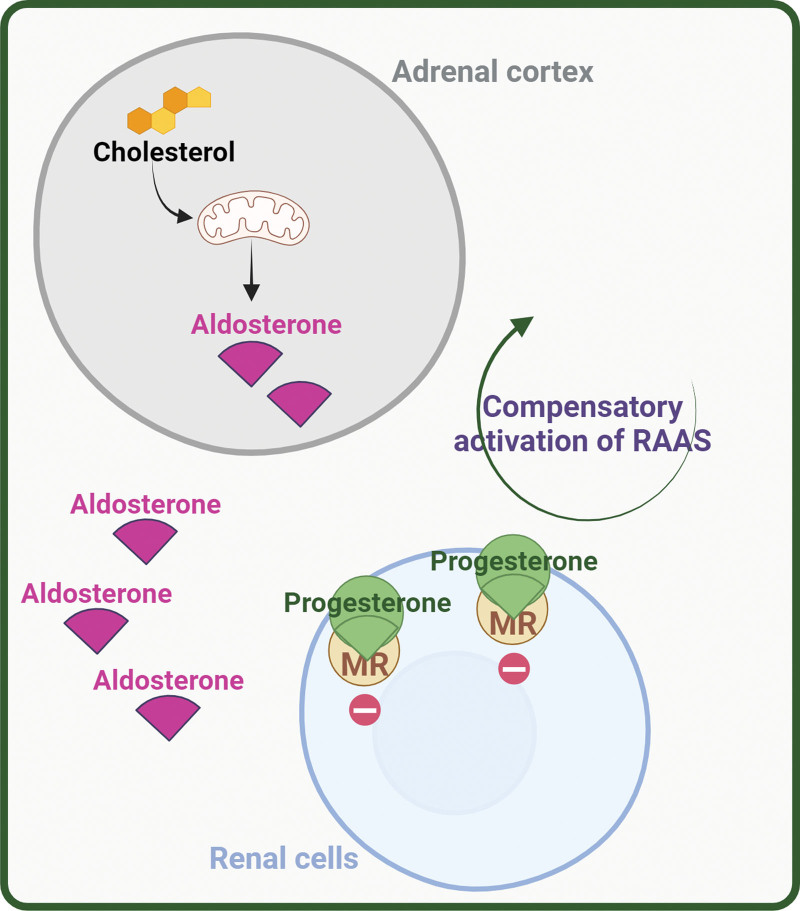
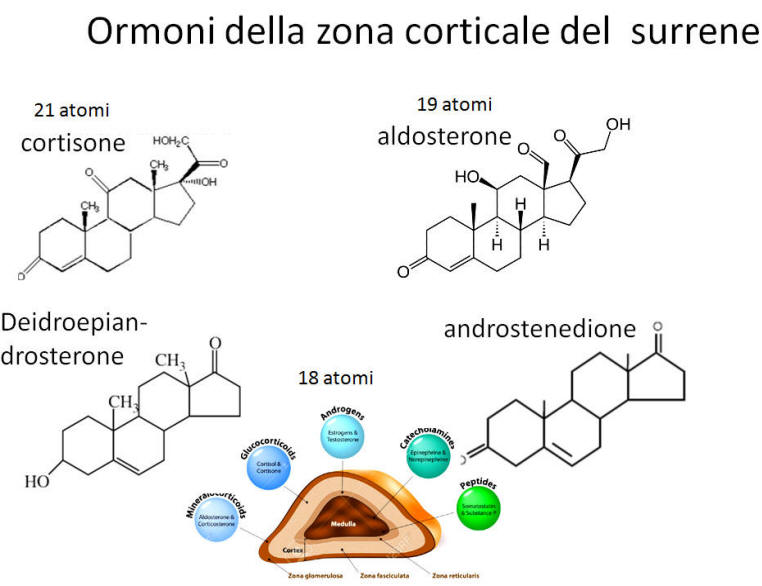
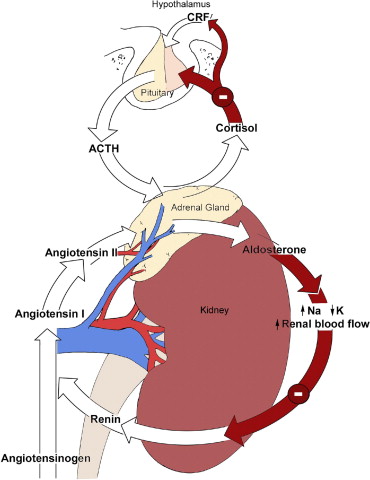
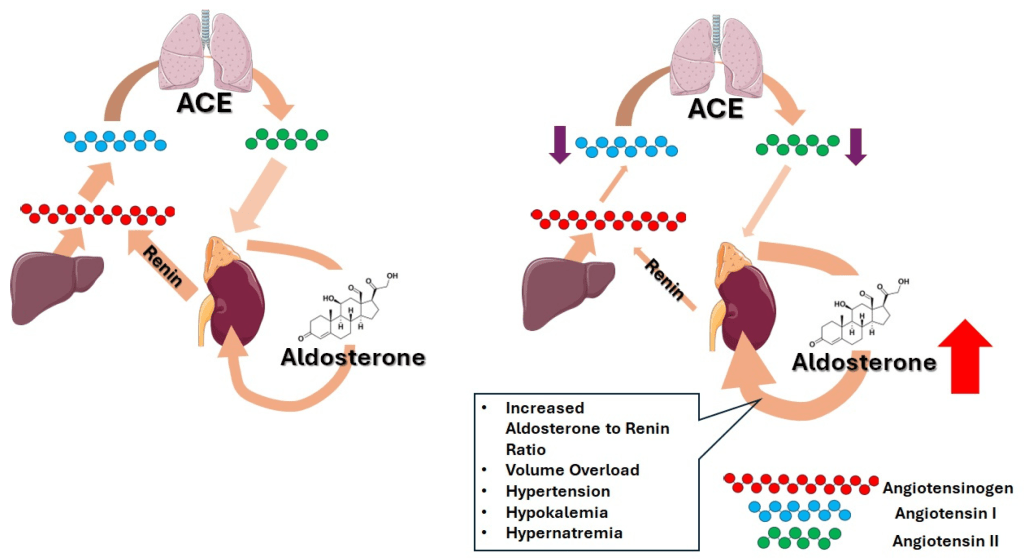
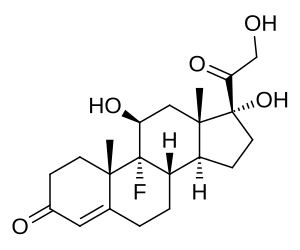
- Corteccia surrenale: produce mineralcorticoidi (Aldosterone), glucocorticoidi (Cortisolo) e androgeni surrenali (DHEA e Androstenedione).
- Tessuti periferici (intracrinologia): il tessuto adiposo, la pelle e il fegato convertono localmente i precursori surrenali inattivi (come il DHEA) in steroidi sessuali attivi. Questa sintesi locale diventa la fonte dominante di estrogeni dopo la menopausa.
- Placenta: funge da fonte endocrina temporanea durante la gravidanza per sintetizzare grandi quantità di Progesterone ed estrogeni.
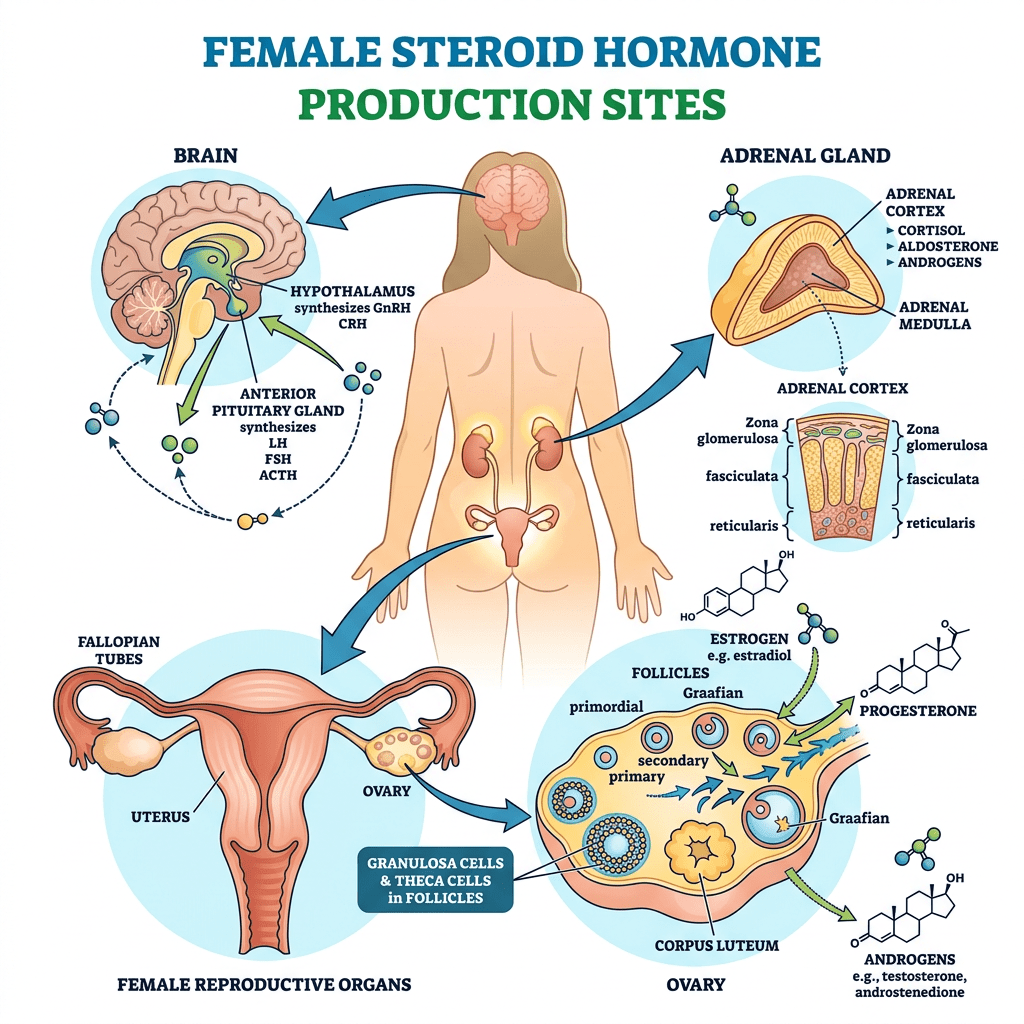
- Principali classi di ormoni prodotti
- Progestinici: Progesterone (regola il rivestimento uterino e il ciclo mestruale).
- Androgeni: DHEA, Androstenedione e Testosterone (precursori degli estrogeni e fattori determinanti per la salute delle ossa e dei muscoli e per la libido).
- Estrogeni: Estradiolo, Estrone ed Estriolo (ormoni sessuali femminili primari che regolano la salute riproduttiva e metabolica).
- Corticosteroidi: Cortisolo (stress e metabolismo) e Aldosterone (pressione sanguigna ed equilibrio elettrolitico).
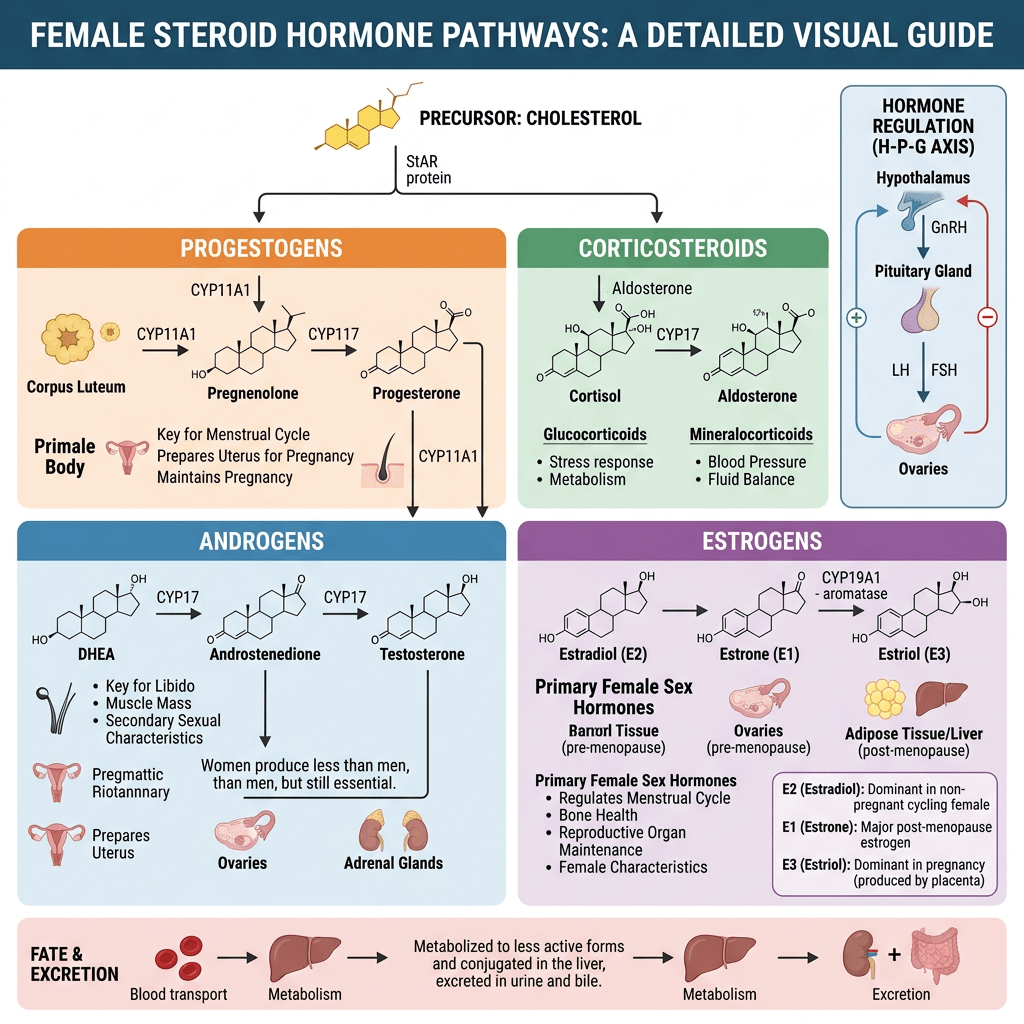
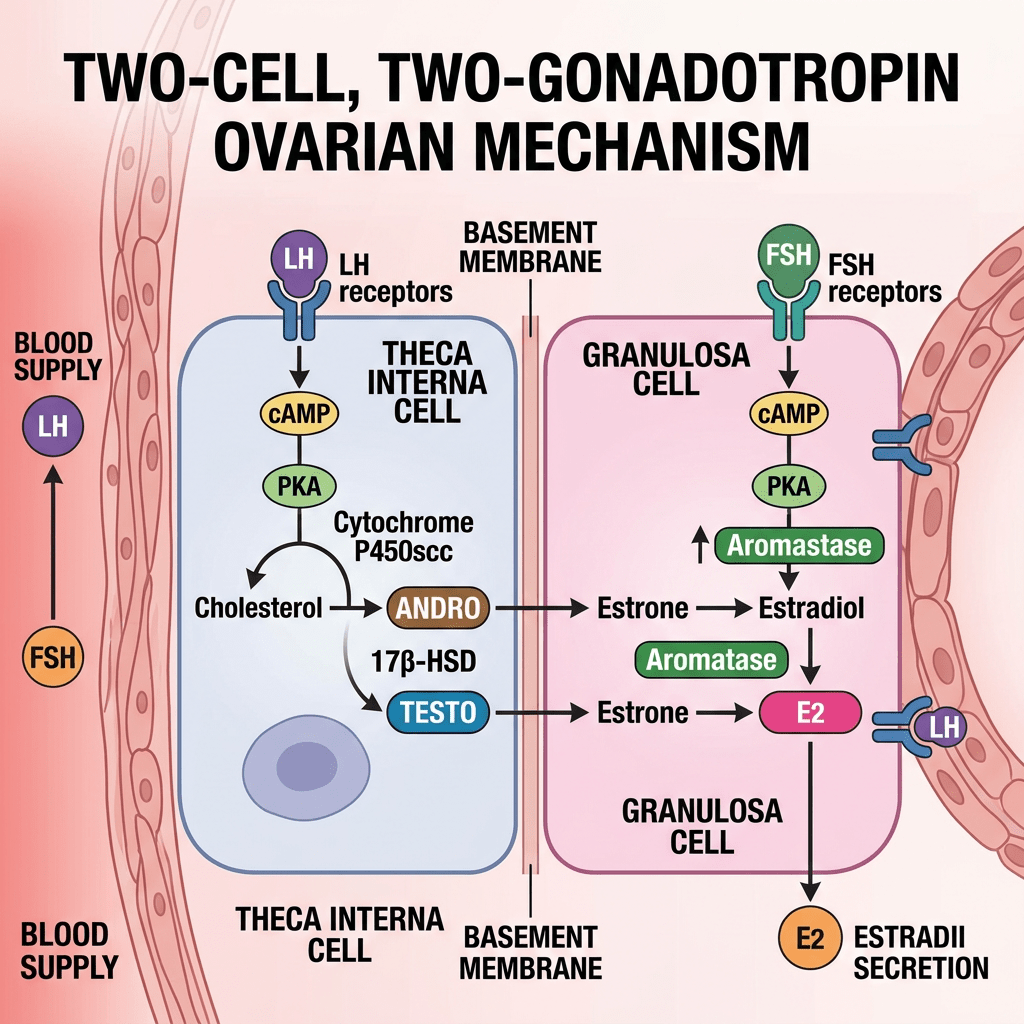
- Two-cell, two-gonadotropin theory?
La Two-cell, two-gonadotropin theory [Teoria delle due cellule e delle due gonadotropine] spiega come le ovaie collaborino utilizzando due tipi distinti di cellule e due ormoni ipofisari per sintetizzare gli estrogeni.
- Meccanismo fondamentale
Questo sistema supera una limitazione specifica: nessuno dei due tipi di cellule possiede tutti gli enzimi necessari per convertire autonomamente il colesterolo in estrogeni.
- Cellule della teca (strato esterno): stimolate dall’LH, assorbono il Colesterolo e lo convertono in androgeni (principalmente Androstenedione e una piccola quantità di Testosterone). Le cellule della teca sono prive dell’Enzima Aromatasi, pertanto non possono convertire questi androgeni in estrogeni. Gli androgeni, invece, si diffondono attraverso la membrana basale nelle vicine cellule della granulosa.
- Cellule della granulosa (strato interno): stimolate dall’FSH. Queste cellule sono prive dell’enzima chiave (CYP17A1) necessario per convertire i progestinici in androgeni, il che significa che non possono produrre autonomamente i propri precursori degli androgeni. Tuttavia, sono ricche di Aromatasi. Assorbono gli androgeni ricevuti dalle cellule della teca e li aromatizzano (convertono) in estrogeni (principalmente Estradiolo).
- Significato clinico
- Sviluppo follicolare: questa produzione ormonale coordinata stimola la crescita del follicolo ovarico e prepara il rivestimento uterino a una potenziale gravidanza.
- Ovulazione: l’aumento dei livelli di Estradiolo derivante da questo processo finisce per innescare il picco di LH necessario per l’ovulazione.
- Sindrome dell’ovaio policistico (PCOS): uno squilibrio in questo sistema — spesso caratterizzato da un’eccessiva stimolazione delle cellule della teca da parte dell’LH — porta a una sovrapproduzione di androgeni, causando i sintomi classici della PCOS (iperandrogenismo e anovulazione).
L’insulino-resistenza stimola direttamente e indirettamente le cellule della teca ovarica a produrre una quantità eccessiva di androgeni, rappresentando il meccanismo chiave alla base dell’iperandrogenismo nella PCOS.
Mentre i tessuti come i muscoli e il fegato diventano resistenti all’Insulina, le cellule della teca mantengono un’alta sensibilità a questo ormone, subendo gli effetti negativi dei livelli elevati di Insulina nel sangue.
L’iperinsulinemia compensatoria agisce direttamente sulle cellule della teca attraverso i recettori dell’Insulina e i recettori del IGF-1.
- Aumento di CYP11A1: L’Insulina accelera l’attività dell’enzima di clivaggio della catena laterale del Colesterolo, aumentando la conversione del Colesterolo in Pregnenolone.
- Aumento di CYP17A1: L’Insulina stimola fortemente l’attività della 17α-idrossilasi/17,20-liasi. Questo è l’enzima chiave che converte i progestinici in androgeni (Androstenedione e Testosterone).
L’Insulina non agisce da sola, ma potenzia la risposta della teca ai segnali ipofisari.
- Amplificazione del segnale: L’Insulina aumenta il numero di recettori per l’LH sulle cellule della teca.
- Iper-reattività: Di conseguenza, anche normali o lievi picchi di LH provocano una produzione massiccia e sproporzionata di androgeni rispetto a quanto avverrebbe in condizioni di normale sensibilità insulinica.
L’insulina altera la biodisponibilità degli ormoni a livello sistemico agendo sul fegato.
- Inibizione della SHBG: Alti livelli di Insulina sopprimono la produzione epatica della globulina legante gli ormoni sessuali (SHBG).
- Più testosterone libero: Con meno SHBG disponibile nel sangue per legare e “disattivare” gli androgeni, la quota di Testosterone libero (la frazione biologicamente attiva che causa irsutismo, acne e blocca l’ovulazione) aumenta drasticamente.
L’iperandrogenismo e l’anovulazione cronica sono strettamente legati in un circolo vizioso: l’eccesso di androgeni altera sia la maturazione fisica dei follicoli nell’ovaio, sia i segnali ormonali che partono dal cervello.
- Reclutamento eccessivo: Gli androgeni stimolano inizialmente la crescita di troppi piccoli follicoli precoci.
- Mancanza di selezione: L’eccesso di androgeni locali impedisce ai follicoli di rispondere correttamente all’FSH (ormone follicolo-stimolante). Senza il giusto segnale dell’FSH, nessun follicolo riesce a completare la maturazione e a diventare dominante.
- Aspetto policistico: I follicoli si bloccano a uno stadio intermedio (circa 2-9 mm) e si accumulano nella corteccia ovarica, dando all’ovaio il classico aspetto “policistico” all’ecografia.
- Accelerazione del generatore di impulsi: Gli androgeni (spesso convertiti localmente in estrogeni) riducono la sensibilità dell’ipotalamo al progesterone.
- Iperproduzione di LH: Questo causa un aumento della frequenza dei battiti del GnRH (ormone rilasciante le gonadotropine), che stimola l’ipofisi a produrre troppo LH rispetto all’FSH.
- Mancanza del picco ovulatorio: Poiché i livelli di LH sono costantemente alti e l’FSH è basso, viene a mancare il picco acuto di LH necessario a far scoppiare il follicolo per liberare l’ovocita.
- Iperplasia della teca: Lo strato di cellule della teca si ispessisce, producendo ancora più androgeni.
- Ispessimento della tonaca albuginea: La capsula esterna dell’ovaio diventa più densa e fibrosa, creando una vera e propria barriera meccanica che ostacola la rottura del follicolo e la successiva ovulazione.
Il ruolo degli androgeni nella salute e nel benessere delle donne
Sia negli uomini che nelle donne, gli androgeni endogeni — tra cui il Testosterone, il DHT, l’Androstenedione (A), il DHEA e il DHEA-S) — vengono sintetizzati in vari tessuti, tra cui:
- le ghiandole surrenali,
- le ovaie,
- i testicoli,
- la placenta,
- il cervello e
- la pelle.
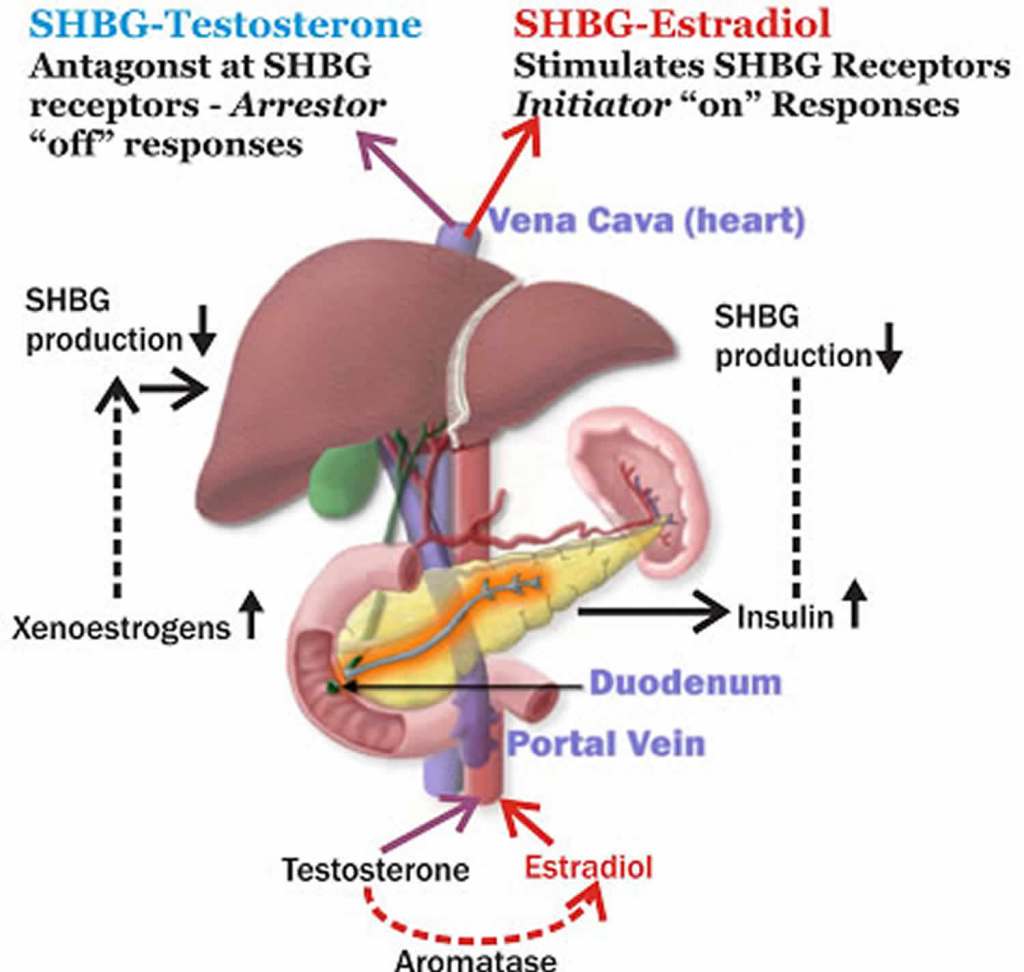
Nelle donne, il Testosterone circolante deriva in parte dalla secrezione ovarica e surrenale; in confronto, quantità simili derivano dalla conversione enzimatica dell’A e del DHEA-S. Nell’ovaio, la produzione di Testosterone aumenta durante le fasi follicolari e raggiunge i livelli massimi al momento dell’ovulazione e nella fase Luteale; le ghiandole surrenali, al contrario, producono solo piccole quantità di Testosterone. La maggior parte del Testosterone circolante è legata in modo reversibile alle proteine plasmatiche, tra cui:
- la globulina legante gli ormoni sessuali (SHBG) (50–60%) e
- l’albumina (40–50%).
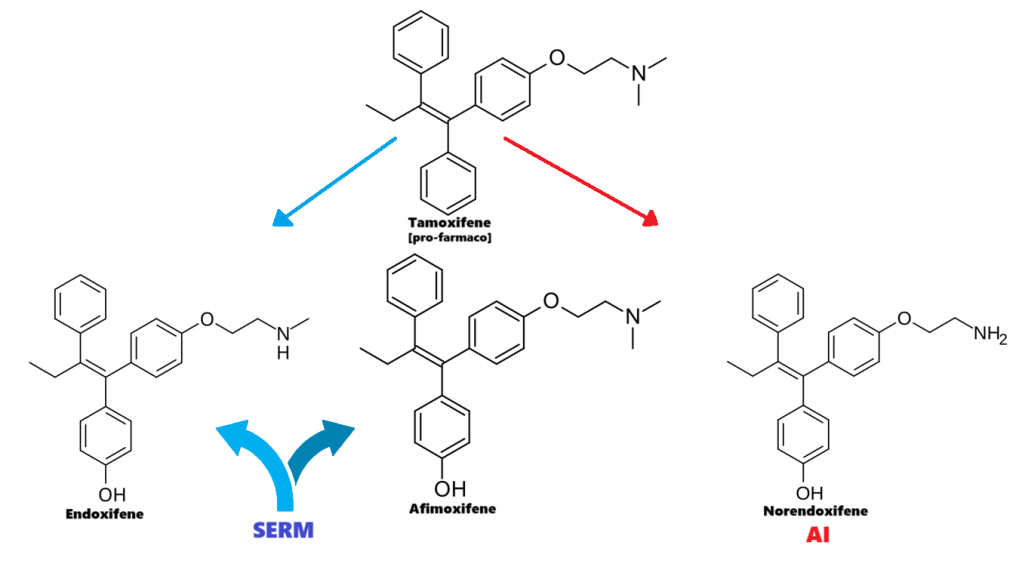
Solo l’1–2% del Testosterone plasmatico è libero e biodisponibile; inoltre, questa frazione libera può essere ulteriormente ridotta dall’aumento dei livelli di SHBG indotto dalla terapia sostitutiva con estrogeni o l’uso di SERM.
La metà dei livelli circolanti di Testosterone e DHT deriva dalla conversione enzimatica dei precursori androgeni surrenali circolanti (DHEA-S, DHEA e A). La conversione del Testosterone in DHT è mediata dalla 5α-reduttasi (isoforme di tipo I e II). Tra questi androgeni, il DHT presenta la maggiore affinità per il recettore degli androgeni (AR) ed è il più potente dal punto di vista biologico tra gli androgeni endogeni. Il DHT non può essere ulteriormente aromatizzato in estrogeni, il che ne aumenta l’emivita.
- Il DHEA plasmatico deriva da:
- secrezione delle cellule della zona reticolare della corteccia surrenale (50%),
- secrezione ovarica (20%) e
- metabolismo periferico del DHEA-S (30%).
Il DHEA stesso è instabile e può essere convertito in Androstenedione (A) dalla 3β-idrossisteroide deidrogenasi (3βHSD); successivamente, l’Androstenedione può essere convertito in Testosterone dalla 17β-idrossisteroide deidrogenasi (17βHSD), nonché in DHT dalle 5α-reduttasi presenti nel tessuto endometriale.
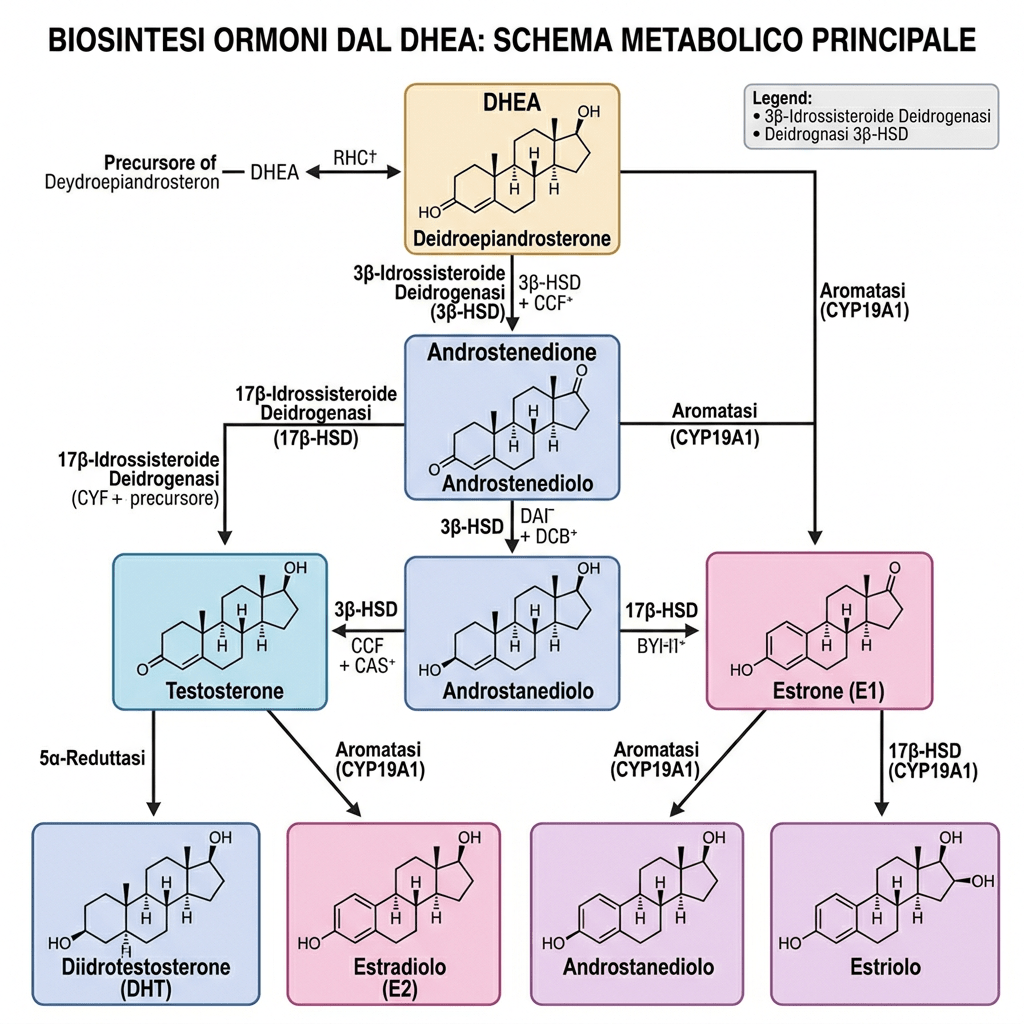
Nelle donne, circa la metà del DHEA circolante deriva dai pre-androgeni; in particolare, la zona reticolare rappresenta l’80% del DHEA-S presente nella circolazione femminile, mentre la parte restante proviene dalle ovaie. Il principale pre-androgeno è il DHEA-S, che viene convertito in DHEA, DHT ed Estrogeni attraverso specifici passaggi enzimatici. Il DHEA-S plasmatico, presente in concentrazioni significative, funge da ampio serbatoio di substrato per la conversione in DHEA, androgeni e/o estrogeni nei tessuti periferici. Le concentrazioni plasmatiche significative di DHEA-S sono, in parte, una conseguenza del suo forte legame con l’albumina, che ne prolunga l’emivita; di conseguenza, le concentrazioni plasmatiche di DHEA-S riflettono la produzione di androgeni da parte delle ghiandole surrenali. La secrezione di DHEA-S è principalmente sotto il controllo dell’asse ipotalamo-ipofisario ed è stimolata dall’ACTH; tuttavia, la sua secrezione è modulata da altri ormoni quali l’Estradiolo, la Prolattina e l’IGF-1.
In larga misura, il Testosterone deriva dal metabolismo dell’Androstene tramite la 17βHSD (tipo 5).
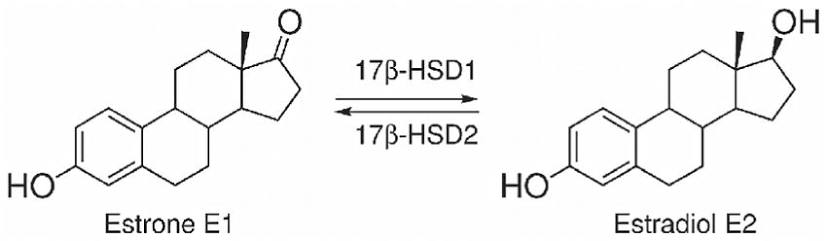
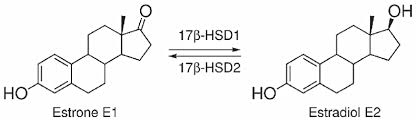
Le 17β-idrossisteroide deidrogenasi (17β-HSD, HSD17B) (EC 1.1.1.51), note anche come 17-chetosteroidi reduttasi (17-KSR), sono un gruppo di ossidoreduttasi alcoliche che catalizzano la riduzione dei 17-chetosteroidi e la deidrogenazione dei 17β-idrossisteroidi nella steroidogenesi e nel metabolismo degli steroidi. Ciò comprende l’interconversione tra DHEA e Androstenediolo, tra Androstenedione e Testosterone, e tra Estrone ed Estradiolo.
Le principali reazioni catalizzate dalla 17β-HSD (come la conversione dell’Androstenedione in Testosterone) sono reazioni di idrogenazione (riduzione), piuttosto che reazioni di deidrogenazione (ossidazione).
La 17β-idrossisteroide deidrogenasi di tipo 5 (17βHSD5), nota anche come membro C3 della famiglia 1 delle aldo-cheto reduttasi (AKR1C3), è un enzima chiave che converte gli androgeni surrenali inattivi in Testosterone e DHT attivi. Essa stimola la produzione locale di ormoni e la crescita delle cellule tumorali nel carcinoma prostatico resistente alla castrazione.
Funzione, ruolo e importanza clinica:
- Conversione steroidea: converte l’Androstenedione e il DHEA in androgeni attivi.
- Localizzazione tissutale: presente nella prostata, nel fegato, nel seno e nei tessuti riproduttivi.
- Collegamento con le patologie: favorisce la crescita tumorale e la resistenza al trattamento nei tumori della prostata e della mammella.
- Bersaglio farmacologico: i ricercatori prendono di mira questo enzima per bloccare la produzione di Testosterone all’interno dei tumori quando la terapia ormonale standard fallisce.
- Inibitori: farmaci sperimentali come l’ASP9521 sono oggetto di studio per bloccare l’attività di questo specifico enzima.
Come già riportato, nelle donne, livelli plasmatici elevati di Testosterone sono correlati all’incidenza della sindrome dell’ovaio policistico (PCOS), osservata in circa il 20% delle giovani donne. La PCOS è associata a oligomenorrea, disfunzione ovulatoria e infertilità. L’iperandrogenismo nella PCOS è un fattore critico che predispone le donne all’obesità, all’insulino-resistenza e alla sindrome metabolica.
Nelle donne adulte, i livelli plasmatici di androgeni e di SHBG diminuiscono progressivamente con l’avanzare dell’età, con riduzioni marcate e significative correlate alla menopausa; a titolo esemplificativo, i livelli di Testosterone totale nelle donne di età compresa tra i 65 e i 74 anni sono pari a circa un terzo di quelli osservati nelle ventenni. In confronto, i livelli di Testosterone libero diminuiscono con l’età del 90%, mentre i livelli di DHEA-S e A diminuiscono entrambi di circa un terzo. Nelle donne in premenopausa, le ovaie sono la fonte principale di Estradiolo, che funge da ormone estrogenico predominante circolante agendo sui tessuti bersaglio distali. Nelle donne in postmenopausa, a causa della cessazione della funzione ovarica, gli estrogeni vengono prodotti in una serie di siti extragonadici, tra cui il tessuto adiposo, le ossa, il cervello, l’endotelio vascolare e le cellule muscolari lisce dell’aorta, dove agiscono localmente in modo paracrino o intracrino. La loro concentrazione locale nei tessuti dipende da una fonte esterna di precursori androgenici, poiché i tessuti extragonadici non sono in grado di convertire il Colesterolo in steroidi.
Il fatto che l’ovaio postmenopausale abbia un’attività endocrina significativa nella produzione di androgeni è in qualche modo controverso. Alcuni studi hanno riportato che i livelli circolanti di androgeni (ovvero T, A, DHEA e DHEA-S) sono estremamente rilevanti nel -fornire substrato per la biosintesi degli estrogeni periferici, attraverso l’aromatizzazione periferica di A e T. Poiché l’attività dell’Aromatasi rimane inalterata nelle donne sottoposte a ovariectomia, è stato ipotizzato che l’ovaio in postmenopausa non sia una fonte significativa di produzione di androgeni. Al contrario, altri studi hanno osservato che l’ovariectomia bilaterale ha determinato una riduzione marcata e prolungata dei livelli sia di T totale che di T biodisponibile, suggerendo che l’ovaio in postmenopausa rappresenti una fonte fondamentale di androgeni per tutto l’arco della vita delle donne anziane.
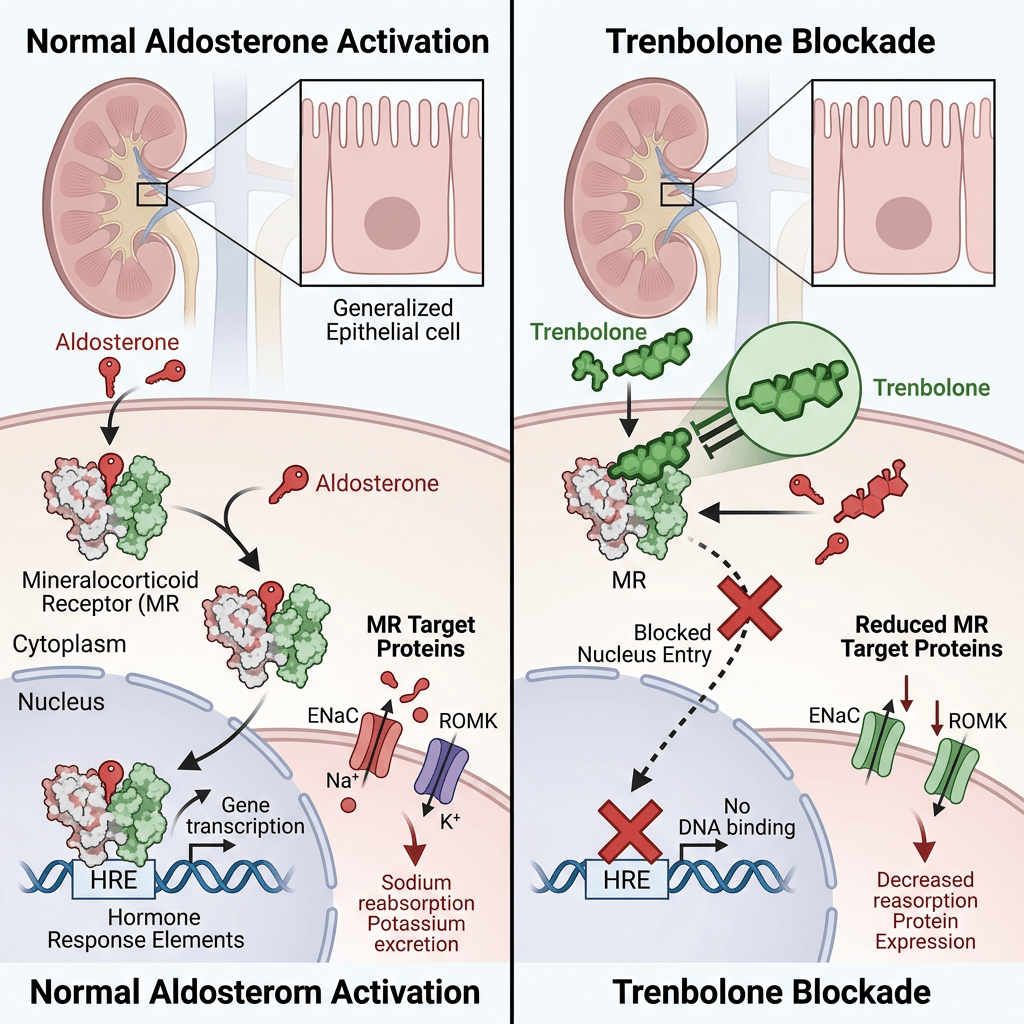
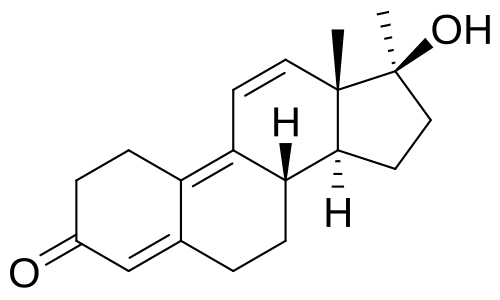
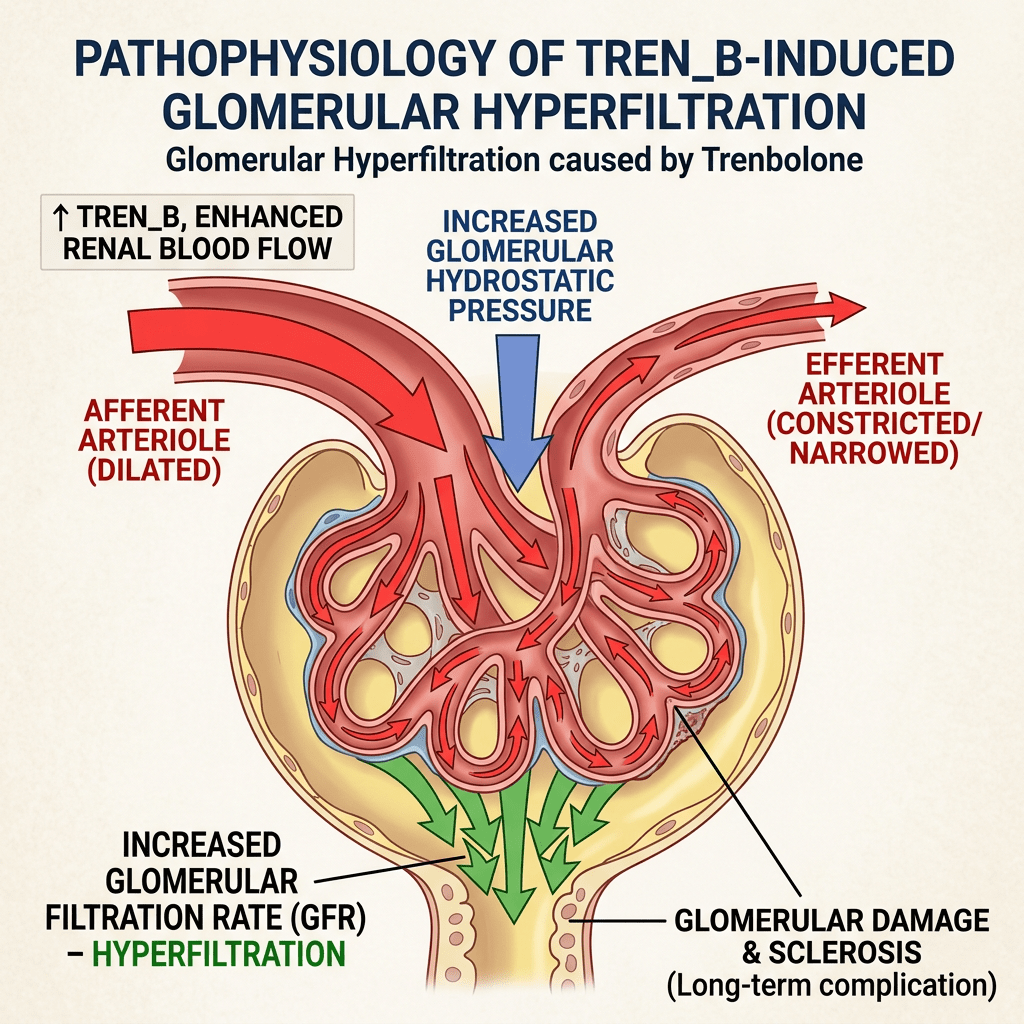
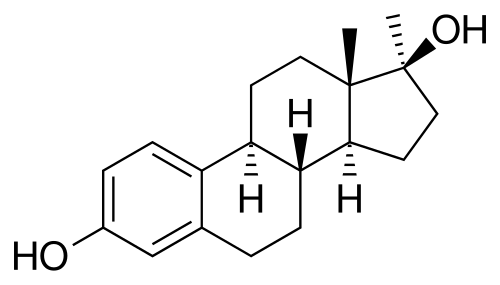
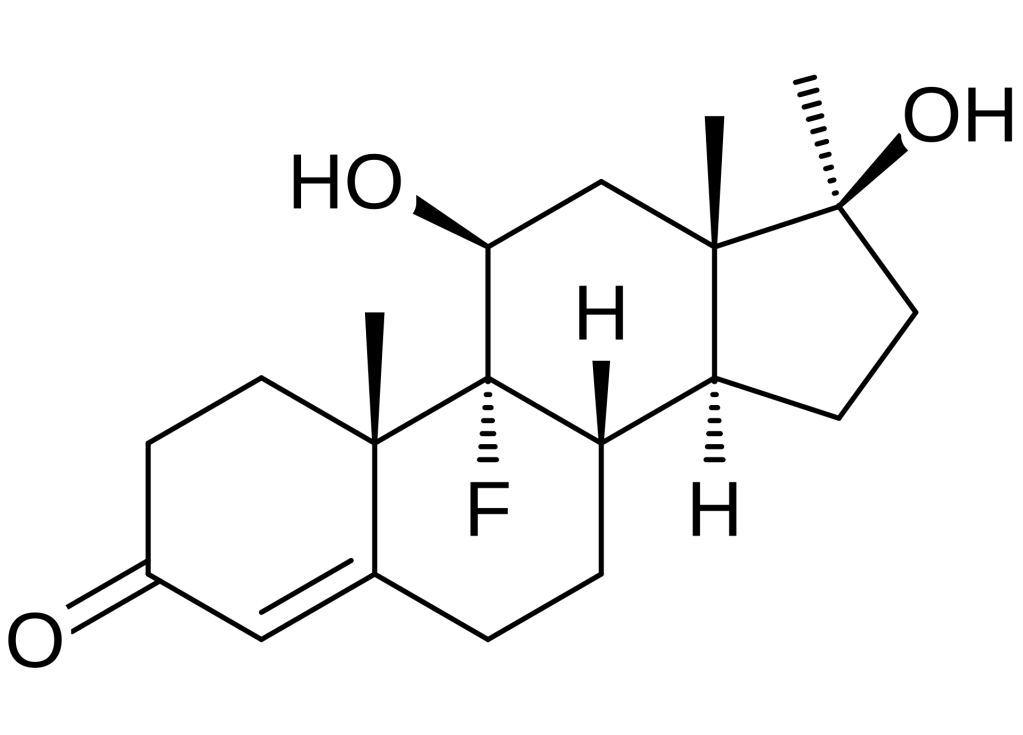
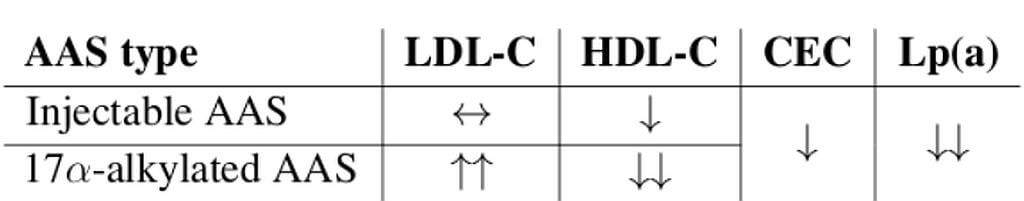
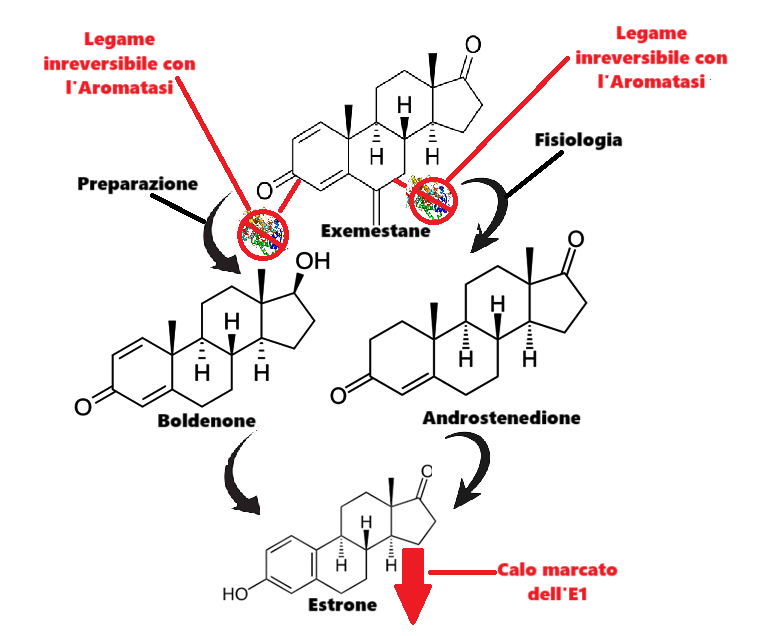
Da un punto di vista farmacologico, gli AAS, che possono derivare dal Testosterone, dal DHT o dal 19-nortestosterone, includono l’Oxandrolone, lo Stanozololo, il Nandrolone, il Trenbolone e altre molecole applicate attualmente ma soprattutto in passato anche in pazienti di sesso femminile.
Gli AAS sono classificati principalmente in due classi farmacologiche:
- androgeni aromatizzabili, come il Testosterone, che viene metabolizzato in Estradiolo dall’Aromatasi, il quale interagisce poi sia con l’ERα e l’ERβ, e
- androgeni non aromatizzabili, come il DHT, che si lega esclusivamente al AR. Gli androgeni sintetici, come l’Oxandrolone e lo Stanozololo, non sono soggetti all’aromatizzazione, il che ne priva il potenziale estrogenico.
Una delle difficoltà più complesse negli studi clinici che coinvolgono gli ormoni gonadici è la misurazione accurata e affidabile dei livelli plasmatici di ormoni. Inoltre, poiché le concentrazioni plasmatiche di androgeni nelle donne sono basse, i test immunologici per il testosterone disponibili in commercio forniscono solitamente risultati contraddittori. Di conseguenza, la misurazione accurata dei livelli plasmatici di testosterone, DHT ed estradiolo nelle donne richiede un metodo alternativo convalidato, come la cromatografia liquida accoppiata alla spettrometria di massa in tandem (LC-MS/MS), che è tecnicamente molto più complessa e costosa rispetto a un test immunologico commerciale.
In un sistema omeostatico fisiologico femminile, gli androgeni (come il Testosterone) sono ormoni fondamentali per il benessere psicofisico dell’individuo, in tutte le fasi della vita e con le corrette modifiche terapeutiche. Favoriscono il desiderio sessuale, contribuiscono alla formazione di ossa e muscoli forti, aumentano l’energia e proteggono la salute del cuore e del cervello in ottimale rapporto con l’attività estrogenica [vedi soprattutto Estradiolo]. Livelli equilibrati migliorano l’umore generale, la vitalità e le funzioni fisiche.
- Principali benefici per la salute
- Salute sessuale: migliora la libido, l’afflusso di sangue ai genitali, l’eccitazione e la soddisfazione orgasmica.
- Resistenza ossea: aiuta a mantenere la massa ossea e favorisce un sano rimodellamento osseo.
- Muscoli e metabolismo: favorisce il trofismo muscolare, la forza fisica e l’equilibrio metabolico.
- Cervello e umore: contribuisce alle funzioni cognitive, all’energia e al benessere psicologico generale.
- Usi medici e contesto
- Supporto alla menopausa: il Testosterone a basso dosaggio [3.5mg/week] viene talvolta utilizzato per trattare il disturbo da desiderio sessuale ipoattivo (HSDD) nelle donne in postmenopausa, quando altri trattamenti non risultano sufficienti.
- Rimedio alla carenza: può aiutare a ripristinare l’energia e la funzione sessuale nelle donne con insufficienza androgenica accertata (ad esempio in seguito alla rimozione chirurgica delle ovaie).
- Precauzioni: dosi elevate possono causare effetti collaterali quali acne, crescita di peli sul viso, abbassamento del tono della voce, ipertrofia clitoridea ecc… . Sono in corso ricerche sulla sicurezza a lungo termine.
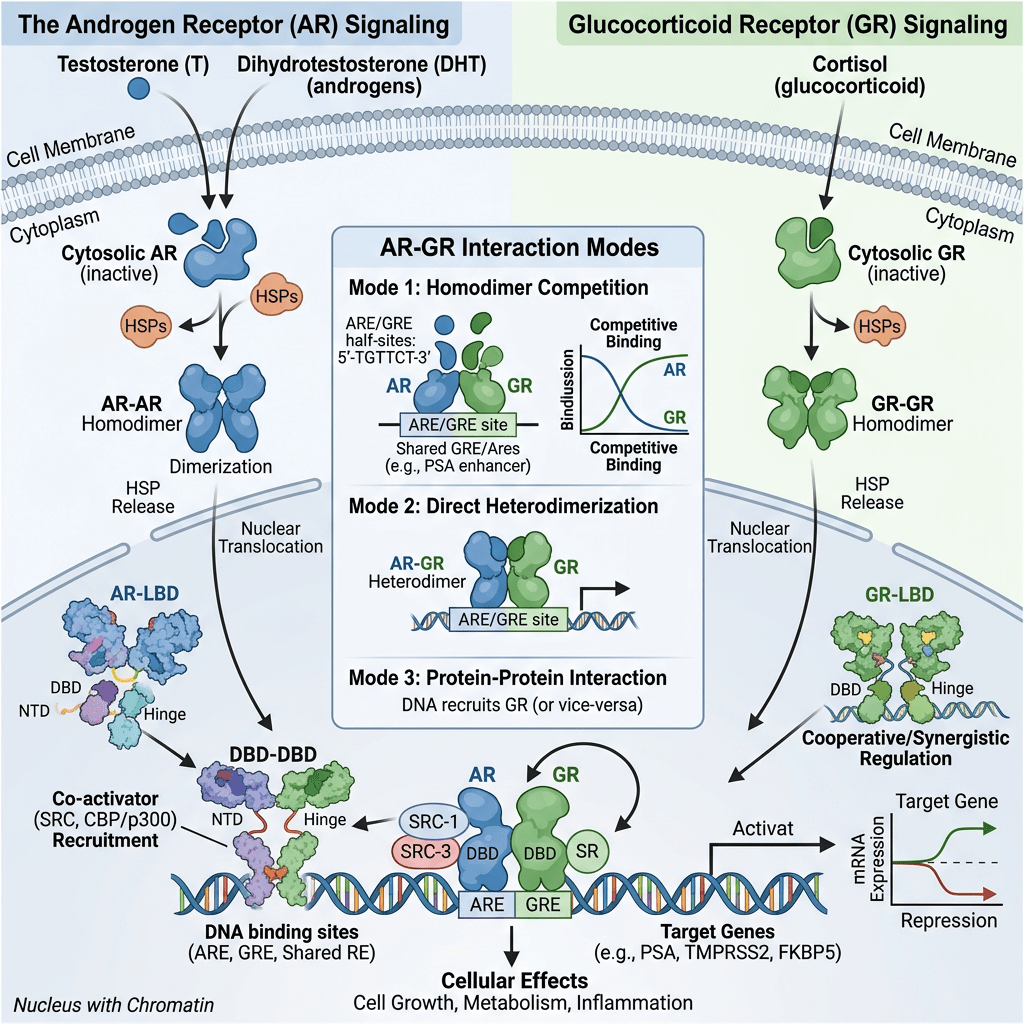
AR e sensibilità agli androgeni nelle donne
Sebbene le donne presentino naturalmente livelli di Testosterone circolante molto più bassi rispetto agli uomini, si osserva una maggiore sensibilità a livello degli organi-bersaglio o dei tessuti agli androgeni. Questa maggiore reattività fa sì che anche livelli ematici di poco alterati di questi ormoni possano comunque provocare segni fisici evidenti, sia a livello di acne, diradamento dei capelli in soggetti predisposti o una crescita eccessiva di peli, che a livello muscolo-scheletrico.
L’AR umano è un regolatore della trascrizione appartenente alla famiglia dei recettori nucleari. La struttura del gene dell’AR, l’mRNA maturo sottoposto a splicing e i domini proteici presentano somiglianze con altri membri della famiglia: i recettori degli estrogeni e i recettori del progesterone. L’AR comprende 8 esoni diversi che formano la proteina AR, costituita da 4 domini, ciascuno con una funzione specifica:
- il dominio N-terminale (NTD);
- il dominio di legame al DNA (DBD);
- una regione cerniera (HR); e
- il dominio C-terminale di legame al ligando (LBD).
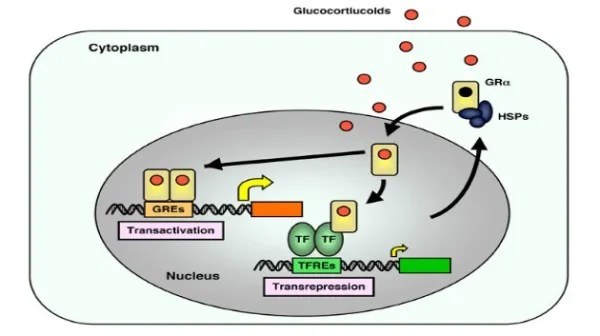
Quando l’AR è presente nel citosol, rimane nella sua forma trascrizionalmente inattiva, stabilizzata dalla proteina chaperone molecolare HSP90 (proteina da shock termico 90). Una volta che un ligando (come il Testosterone o il DHT) si lega all’LBD, l’AR si dissocia dall’HSP90. Successivamente, si dimerizza e si traslocca nel nucleo, dove si lega all’elemento di risposta agli androgeni nella regione regolatoria dei suoi geni bersaglio, modulandone così l’espressione.
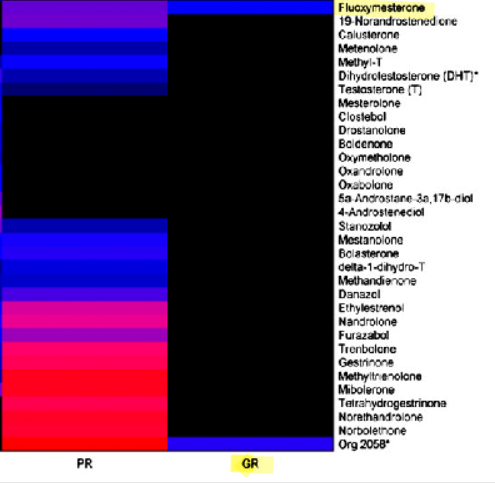
Alla luce dell’elevata presenza di AR nelle donne (vedi tabella sopra), è opportuno analizzare in modo approfondito il loro ruolo fondamentale nella patogenesi dell’iperandrogenismo. È quindi importante individuare le manifestazioni cliniche più comuni. Una di queste manifestazioni è l’irsutismo, che colpisce fino al 15% di tutte le donne e il 70-80% delle donne affette da iperandrogenismo. I metodi di valutazione visiva del tipo di crescita dei peli, come il punteggio di Ferriman-Gallwey, sono efficaci nel determinare la gravità dell’irsutismo e nel distinguerlo dall’ipertricosi (crescita eccessiva di peli indipendenti dagli androgeni). La virilizzazione rappresenta un processo rapido in cui si manifestano gravi caratteristiche cliniche di un marcato eccesso di androgeni endogeni o esogeni. Nell’ambito di uno spettro più ampio, può includere: irsutismo, acne, alopecia androgenetica, ingrossamento del clitoride, abbassamento del tono della voce, aumento della massa muscolare, riduzione delle dimensioni del seno e, frequentemente, amenorrea. Inoltre, l’iperandrogenismo è correlato a numerose alterazioni metaboliche, ad esempio: iperinsulinemia, insulino-resistenza, iperglicemia, dislipidemia e aumento del rischio di aterosclerosi. Queste caratteristiche costituiscono le componenti fondamentali della sindrome metabolica. È importante sottolineare che le variazioni dei livelli di AR nell’iperandrogenismo devono essere mediate da vie regolate dai AR.
Un esempio dell’importanza dell’AR è il rischio confermato di cancro al seno nelle donne in premenopausa con concentrazioni sieriche elevate di Testosterone, Androstenedione e DHEA-S. Pertanto, le nuove terapie contro il cancro al seno prendono di mira l’AR per la sua espressione osservata nei tre principali sottotipi di cancro al seno. È importante sottolineare che l’AR si è già dimostrato un bersaglio promettente per le terapie antiandrogeniche nel cancro al seno triplo-negativo, il più aggressivo.
I AR possono presentare variabili espressive maggiori nel sistema muscolo-scheletrico delle donne, sebbene la vera peculiarità più volte osservata riguarda l’attivazione/sensibilità recettoriale. Questo ci da come risultante una maggiore risposta dose/tempo agli androgeni/AAS rispetto ai soggetti di sesso maschile. Sembrerebbe trattarsi di una caratteristica regolativa/compensativa in risposta a concentrazioni androgene dimorfiche inferiori.
Consideriamo, infatti, che negli uomini il Testosterone totale è tipicamente tra 300 e 1000ng/dL [range standard 3ng/mL-11.5ng/mL], mentre nelle donne varia tra 15 e 76ng/dL [range standard 0,15 e 0,76ng/ml]. Gli uomini presentano quindi livelli circa 10-20 volte superiori rispetto alle donne.
Discorso inverso sembra poter essere fatto con gli estrogeni per gli uomini. Nelle donne in età fertile l’Estradiolo [E2] varia tra 15 e 350pg/mL (con picchi più alti), mentre negli uomini i valori normali sono compresi tra 10 e 40pg/mL con un range soglia massimo di 60pg/mL. Le donne presentano quindi concentrazioni medie da 5 a 10 volte superiori di E2 rispetto agli uomini. Si noti che piccole variazioni nei livelli di E2 nell’uomo possono generare effetti clinici evidenti (ciò avviene in variazione soggettiva alla sensibilità individuale insieme a quella intrinseca di genere). Si è affermato che ciò sia dovuto più che altro alla stretta finestra di eufunzione rispetto ad una maggiore sensibilità biologica intrinseca rispetto alla donna. Nonostante ciò, molti casi studio hanno evidenziato che:
- Presenza di sensibilità intrinseca: nonostante la comparsa di problematiche legate ad eccessi o carenze di E2 mostrano variabili soggettive legate a fattori a monte [vedi, per esempio, tasso d’espressione dell’Aromatasi e risposta epigenetica ad alterazioni iatrogene comprendenti l’espressione dei gene che regola il ER], una costante sembra essere una spiccata sensibilità a variazioni minime su tessuti/sistemi diversi.
- Similitudini di risposta alle alterazioni: come nelle donne variazioni anche minime nelle concentrazioni di AAS danno risposte evidenti [seppur variabili] anche nell’uomo le fluttuazioni di E2 [in alcuni casi la sensibilità interessa anche il meno attivo E1] danno risposte evidenti sebbene differenziate da grado e area tissutale/sistemica.
Nel complesso, stiamo vivendo un’era entusiasmante caratterizzata da nuove scoperte nel campo dei AR e dei loro molteplici ruoli nelle donne. Quanto esposto in questo paragrafo evidenzia che la conoscenza della segnalazione degli AR e delle alterazioni della struttura dei recettori è fondamentale per comprendere la risposta androgenica, tanto in terapia quanto in campo del miglioramento delle prestazioni. Pertanto, gli sforzi scientifici futuri saranno in parte volti a chiarire ulteriormente l’importanza degli AR nella salute, nelle prestazioni [ed estetica] e nelle patologie delle donne rivestiranno un’importanza fondamentale.
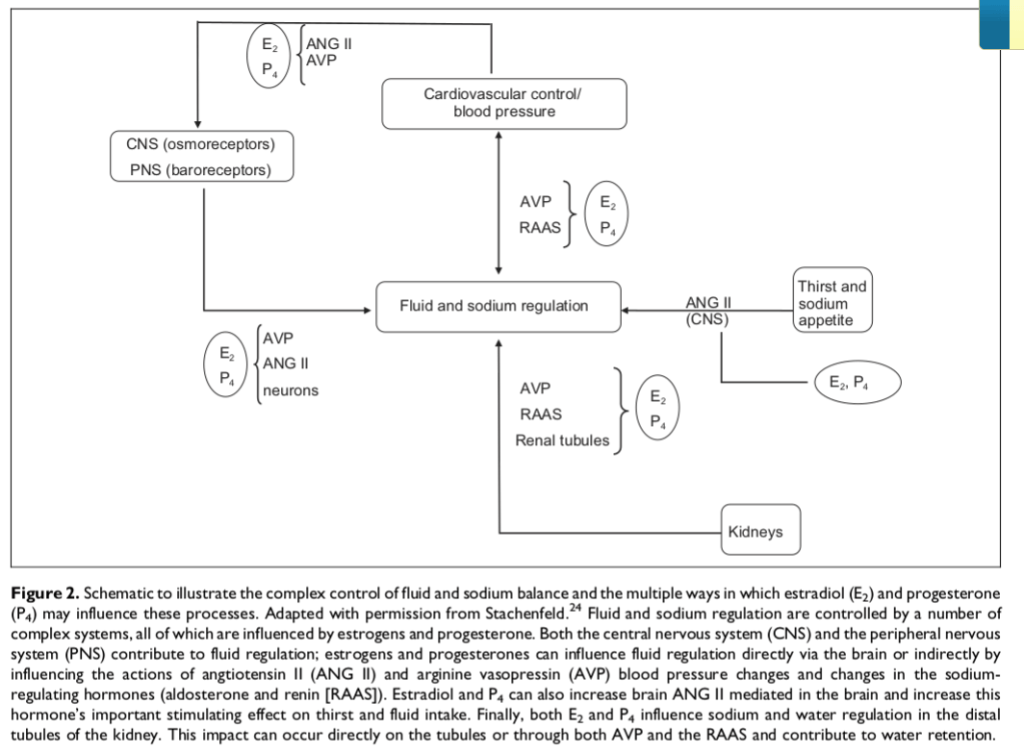
Asse hGH/IGF-1, E2 e dimorfismi sessuali
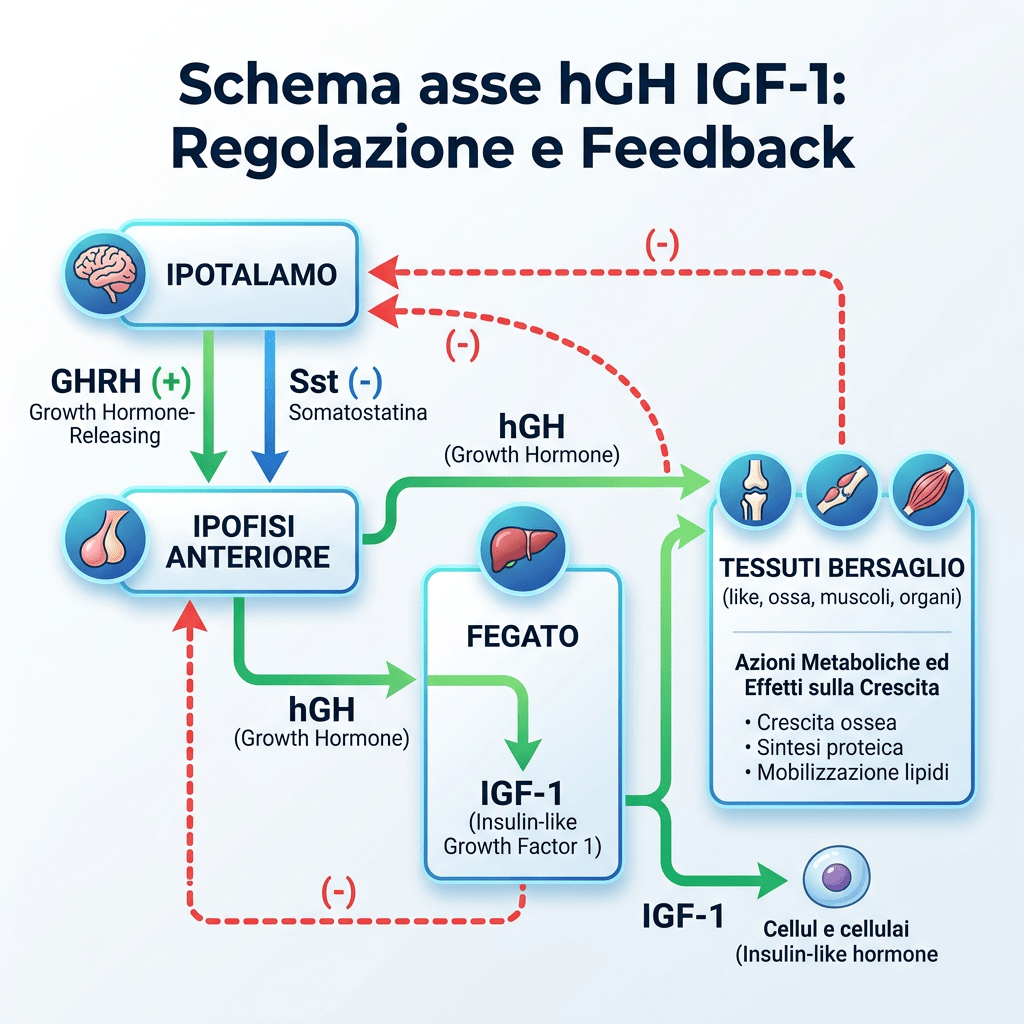
L’Asse Ormone della Crescita/Fattore di Crescita Insulino-Simile-1 [Asse hGH/IGF-1] svolge un ruolo fondamentale nella promozione della crescita lineare nei bambini, mentre negli adulti il suo ruolo è principalmente di rilevanza metabolica. L’hGH viene secreto dalla ghiandola pituitaria e stimola la sintesi e la secrezione dell’IGF-I, che a sua volta inibisce la secrezione del hGH attraverso un meccanismo di feedback negativo. Il fegato è il principale responsabile del pool circolante di IGF-I, delle sei proteine leganti l’IGF (da IGFBP-1 a -6) e della subunità labile in ambiente acido (ALS). In circolo, circa il 99% del pool di IGF-I è legato con elevata affinità alle IGFBP, che circolano in eccesso molare rispetto all’IGF-I; ciò spiega perché meno dell’1% circoli come IGF-I libero e non legato. Fungendo da vettori dell’IGF-I circolante, le IGFBP e l’ALS prolungano l’emivita dell’IGF-I e ne modulano l’accesso ai tessuti, controllandone così l’azione.
- Interazione dell’Insulina
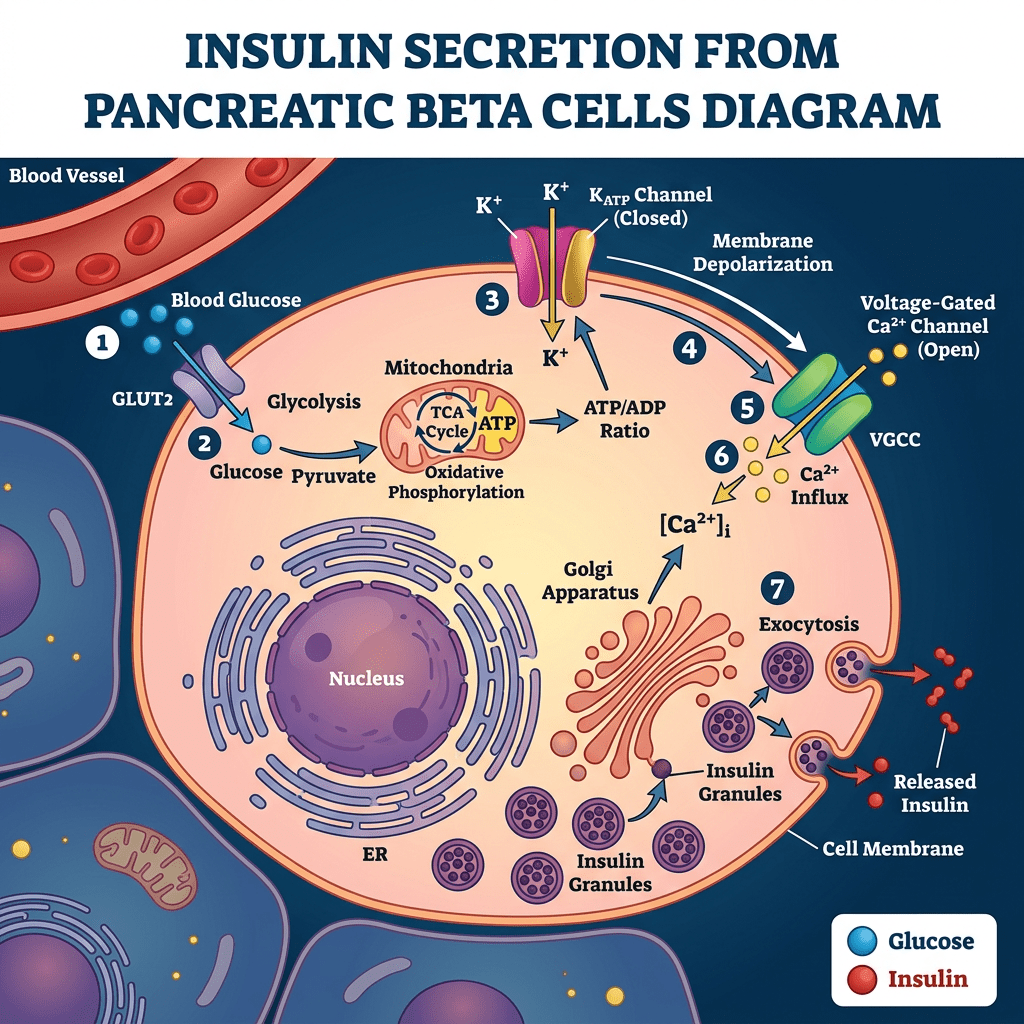
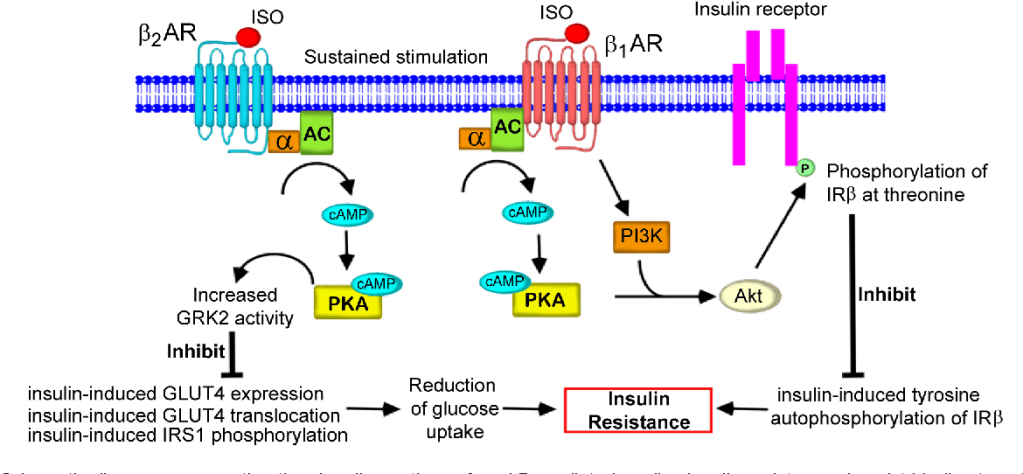
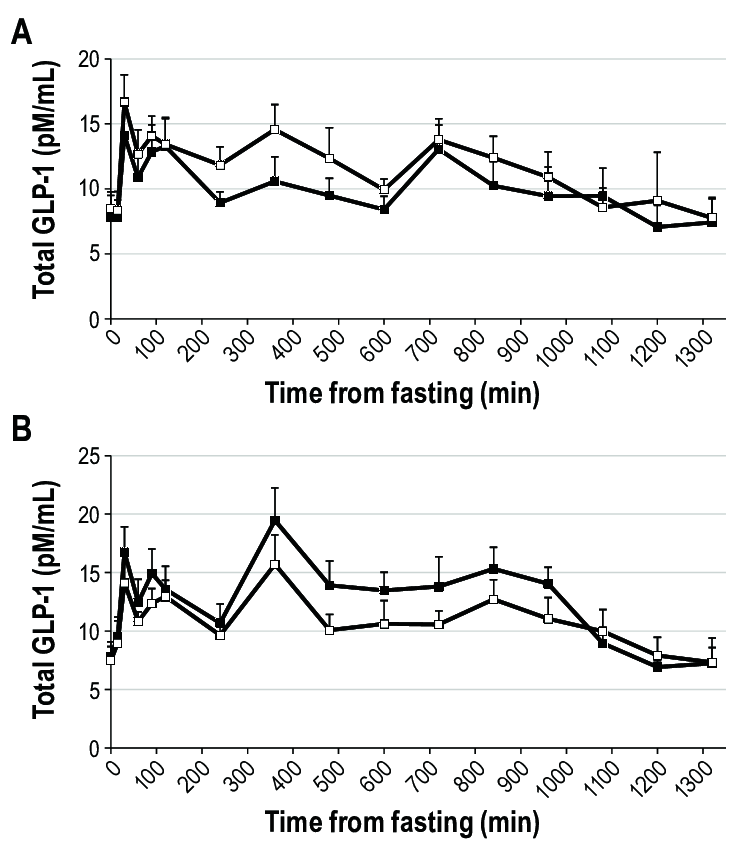
L’Insulina, una volta secreta dalle cellule β del pancreas, viene trasportata dalla vena porta direttamente al fegato, determinando un’elevata esposizione degli epatociti. In particolare, il fegato è uno dei principali organi bersaglio degli effetti metabolici dell’Insulina ed esercita un’estrazione di primo passaggio che può raggiungere l’85% dell’Insulina convogliata dalla vena porta. Inoltre, grazie alla sua capacità di regolare la sensibilità epatica al hGH, il trasporto intra-portale dell’Insulina si è rivelato un fattore coadiuvante essenziale per un’efficace sintesi epatica di IGF-I indotta dal hGH. Infatti, si può considerare la capacità dell’Insulina di controllare la sensibilità epatica al hGH come una normale risposta fisiologica ai cambiamenti nutrizionali. Durante il digiuno, con livelli ridotti di Insulina intraportale, la sensibilità epatica al hGH diminuisce, determinando una riduzione dei livelli sierici di IGF-I nonostante un aumento compensatorio della secrezione di hGH. Al contrario, l’eccesso alimentare aumenta i livelli di Insulina intraportale, potenziando la sensibilità epatica al hGH in misura tale che una secrezione relativamente bassa di hGH sia sufficiente a mantenere la sintesi e la secrezione epatiche di IGF-I a livelli normali, nonché livelli circolanti normali di IGF-I. Questa risposta del hGH ai cambiamenti nutrizionali è fisiologicamente appropriata in quanto porta ad un aumento dell’insulino-resistenza mediata dal hGH che previene l’ipoglicemia durante il digiuno e ad una diminuzione dell’insulino-resistenza mediata dal hGH durante i periodi di sovralimentazione. Tuttavia, anche alcune patologie (ad esempio, l’obesità, la sindrome di Cushing, il diabete di tipo 1 [T1D] e l’anoressia nervosa) e alcuni farmaci di uso comune [ad esempio, i glucocorticoidi e gli agonisti del GLP-1RA)] che influenzano la secrezione di Insulina da parte delle cellule β pancreatiche possono alterare l’asse hGH/IGF-I. Poiché il hGH, l’IGF-I e l’Insulina modulano continuamente la secrezione e l’azione reciproca sia in condizioni di salute che di malattia, una buona comprensione dei meccanismi alla base di queste interazioni riveste importanza sia clinica che al fine del miglioramento delle prestazioni/estetica.
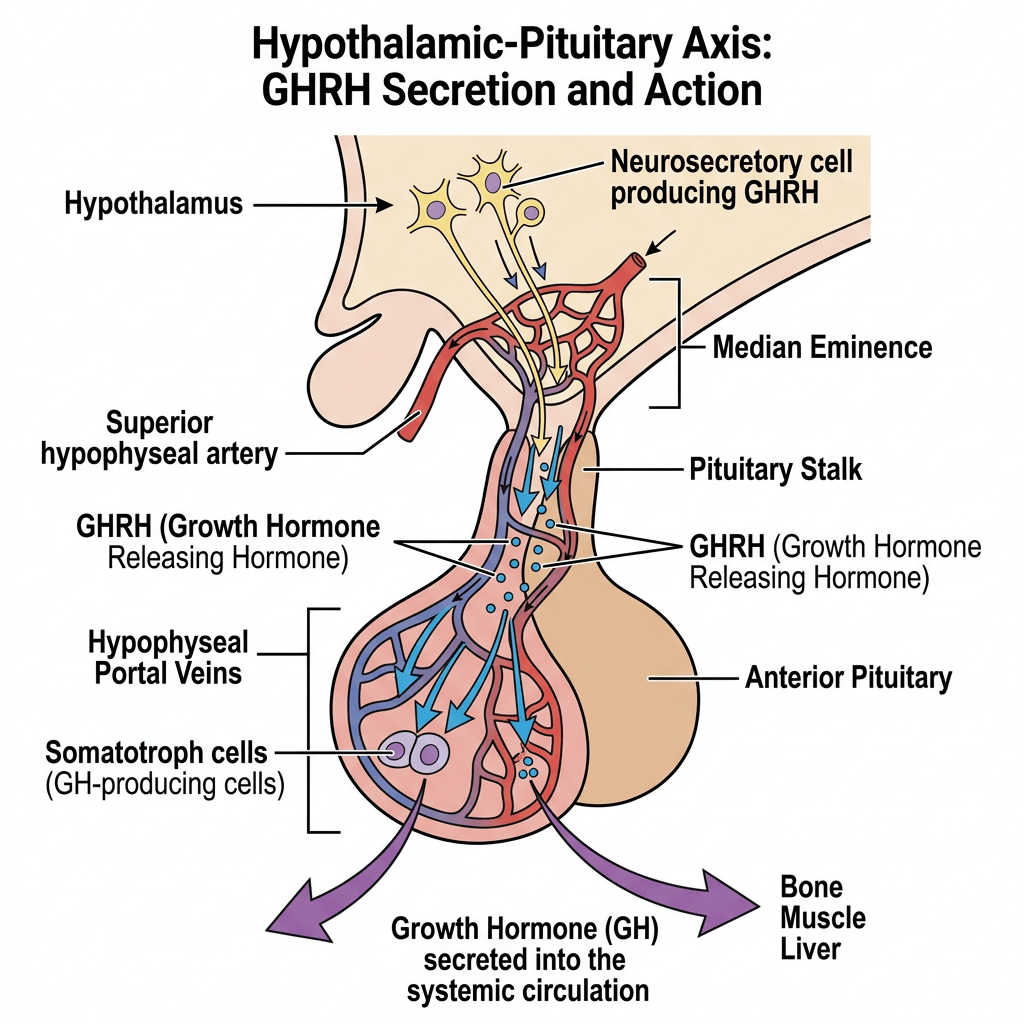
La secrezione ipofisaria dell’hGH è regolata dalla stimolazione del GHRH e dall’inibizione della Somatostatina, entrambi secreti dall’ipotalamo. La secrezione dell’hGH è episodica e pulsatile, con livelli che variano tra picchi e valli e livelli molto bassi tra un impulso e l’altro. L’hGH stimola la sintesi e la secrezione dell’IGF-I, che agisce a sua volta sull’ipofisi inibendo la secrezione di hGH. Il IGF-I influenza la regolazione della secrezione di hGH a livello dell’ipotalamo inibendo l’espressione genica del GHRH e stimolando la secrezione di Somatostatina, mentre a livello dell’ipofisi l’IGF-I inibisce la secrezione spontanea e quella stimolata dal GHRH del hGH. A differenza della natura episodica e pulsatile della secrezione di hGH, l’IGF-I viene secreto in modo continuo, ha un’emivita più lunga [per via del legame con le proteine di trasporto IGFP] e presenta concentrazioni più stabili nel sangue. Pertanto, l’IGF-I viene utilizzato come biomarcatore dello stato secretorio del hGH, poiché i suoi livelli riflettono la secrezione integrata di hGH nell’arco delle 24 ore.
Oltre all’Insulina, esistono altri ormoni in grado di influenzare la secrezione ipofisaria di hGH, quali la Grelina, gli estrogeni e gli androgeni. La Grelina, un peptide gastrico con potenti proprietà secretagoghe del hGH, amplifica la secrezione ipotalamica di GHRH e ne potenzia gli effetti di stimolazione del hGH ipofisario. Gli estrogeni stimolano la secrezione ipofisaria di hGH, ma inibiscono l’azione del hGH sul fegato sopprimendo la segnalazione del GHR. Inoltre, gli estrogeni possono potenziare l’azione della Grelina, mentre gli androgeni potenziano le azioni periferiche del hGH. Infine, la secrezione ipofisaria di hGH è inversamente correlata all’adiposità viscerale intra-addominale attraverso meccanismi che dipendono dai flussi di acidi grassi liberi (FFA).
L’Insulina inibisce la secrezione di hGH, ma i meccanismi alla base di questo fenomeno non sono ancora ben chiari e si ipotizza che agiscano a diversi livelli. A livello dell’ipofisi, si è visto che l’Insulina sopprime direttamente la secrezione ipofisaria di hGH indipendentemente dal recettore dell’IGF-I (IGF-IR). A livello epatico, sembrerebbe che l’Insulina stimola la sintesi della proteina GHR e il legame del hGH al GHR in modo dose-dipendente. Analogamente, è stata osservata una ridotta espressione epatica del GHR e un ridotto legame del hGH in seguito all’induzione del diabete, e che il trattamento con Insulina facilita il legame del hGH. Queste possibilità evidenziano il ruolo chiave che l’Insulina svolge nella modulazione della risposta epatica al hGH e sostengono l’idea che l’Insulina abbia un ruolo importante nella regolazione della sensibilità epatica al hGH attraverso la sua capacità di influenzare direttamente l’espressione del GHR e il legame del hGH. Tuttavia, per quanto a nostra conoscenza, questa ipotesi deve ancora essere inconfutabilmente dimostrata nell’uomo.
Inoltre, oltre a regolare i livelli circolanti di IGF-I attraverso il suo effetto sulla sensibilità epatica al GH, l’insulina inibisce anche la sintesi e la secrezione epatica della proteina legante l’IGF-1 (IGFBP-1), un inibitore dell’azione dell’IGF-I (7). Pertanto, la relazione tra insulina e IGFBP-1 è inversa (34), per cui l’IGFBP-1 è bassa in condizioni di iperinsulinemia ma elevata in condizioni di ipoinsulinemia (35). Poiché i livelli di IGFBP-1 fluttuano nel corso della giornata (36), ciò ha portato all’ipotesi che l’IGFBP-1 colleghi l’azione dell’IGF-I all’apporto nutrizionale. Tuttavia, i dati attuali indicano che l’IGFBP-1 funge più da “freno” all’azione dell’IGF-I, inibendo la segnalazione anabolica durante la deplezione nutrizionale, piuttosto che da “acceleratore” dell’azione dell’IGF-I durante l’eccesso alimentare. La logica è che, in circostanze fisiologiche normali, il calo dell’IGFBP-1 dal suo livello massimo stimolato durante il digiuno notturno (34) al livello osservato dopo l’assunzione di cibo, su base molare, è relativamente modesto e porta solo a lievi aumenti dell’IGF-I libero sierico (7, 37, 38). Un’altra spiegazione è che le concentrazioni molari delle altre cinque IGFBP superano di gran lunga la concentrazione dell’IGFBP-1 (39) e, non essendo completamente sature, le altre cinque IGFBP sono pienamente in grado di sequestrare l’aumento dell’IGF-I libero secondario alle riduzioni dell’IGFBP-1 circolante. Tuttavia, quando i livelli di IGFBP-1 sono cronicamente ridotti in presenza di iperinsulinemia intraportale prolungata (ad esempio, obesità, sindrome di Cushing e terapia con glucocorticoidi), entra in gioco l’effetto “acceleratore” dell’IGFBP-1, che determina un aumento dell’IGF-I libero sierico. Al contrario, durante un’ipoinsulinemia intraportale prolungata (ad esempio, malnutrizione, anoressia nervosa e diabete di tipo 1), i livelli di IGFBP-1 aumentano di diverse volte, portando a una maggiore formazione di complessi tra IGF-I e IGFBP-1 e a una netta riduzione dell’IGF-I libero (effetto “freno”).
- Interazione dell’E2
Il 17β-estradiolo [E2] ha azioni fisiologiche che non si limitano agli organi riproduttivi sia nelle femmine che nei maschi (Simpson et al., 2005; Barros e Gustafsson, 2011). Il fegato è un bersaglio diretto degli estrogeni poiché esprime il ERα, che è collegato, tra l’altro, all’omeostasi dei lipidi e del glucosio (Foryst-Ludwig e Kintscher, 2010; Barros e Gustafsson, 2011; Faulds et al., 2012) e alla crescita corporea (Vidal et al., 2000). Gli estrogeni possono modulare l’azione del hGH nel fegato agendo a livello centrale, regolando la secrezione ipofisaria di hGH, e, a livello periferico, modulando la segnalazione del hGH. La maggior parte degli studi precedenti si è concentrata sull’influenza degli estrogeni sulla secrezione ipofisaria di hGH (Kerrigan e Rogol, 1992). È stato dimostrato che il rilascio ipofisario di hGH, specifico per sesso, ha un forte impatto sulla regolazione trascrizionale epatica (Mode e Gustafsson, 2006). Tuttavia, esistono anche prove evidenti che gli estrogeni modulano l’azione del hGH a livello dell’espressione e della segnalazione del GHR. In particolare, è stato dimostrato che l’E2 induce il soppressore della segnalazione delle citochine (SOCS)-2, il quale a sua volta regola negativamente la via di segnalazione GHR-Janus chinasi (JAK)-2-trasduttore di segnale e attivatore della trascrizione (STAT)-5 (Leung et al., 2004; Santana-Farre et al., 2008). Questo fenomeno è clinicamente rilevante poiché la via di segnalazione GHR-JAK2-STAT-5 riveste particolare importanza nella regolazione delle azioni endocrine, metaboliche e legate alla differenziazione sessuale del hGH nel fegato. È importante sottolineare che l’alterazione della via di segnalazione GHR-JAK2-STAT5 è associata a cambiamenti metabolici epatici che includono steatosi epatica, fibrosi e carcinoma epatocellulare (Baik et al., 2011). Questa interazione è rilevante anche in considerazione della diffusa esposizione degli esseri umani agli estrogeni o a composti correlati agli estrogeni (Wolthers et al., 2001). In questo lavoro riassumeremo le molteplici conseguenze biologiche che possono manifestarsi a seguito dell’interazione tra E2 e hGH nel fegato.
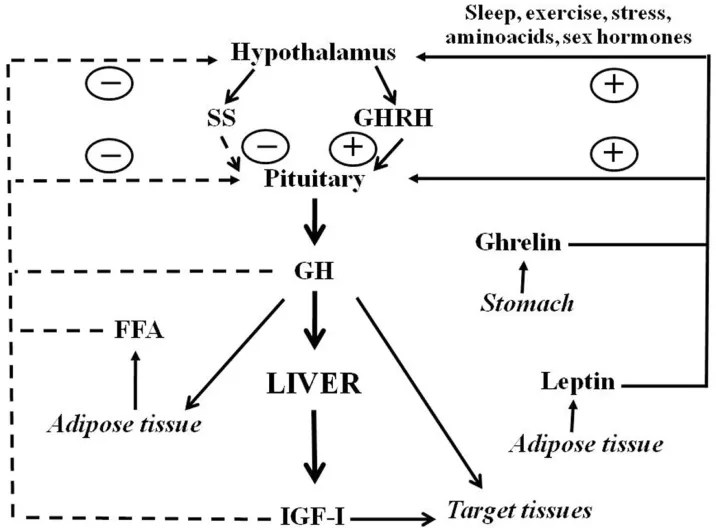
- Dimorfismo sessuale
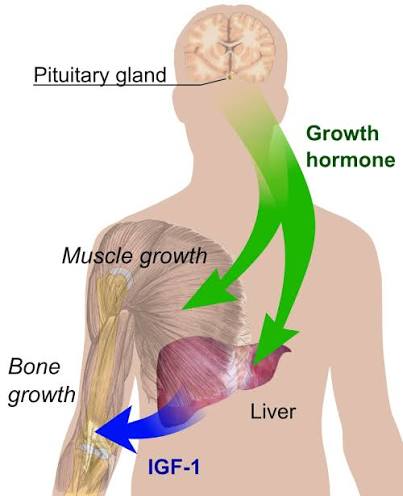
Gli ormoni sessuali determinano un modello di secrezione dell’hGH ipofisario dipendente dal sesso, che svolge un ruolo fondamentale nella definizione e nel mantenimento del dimorfismo sessuale della trascrizione genica epatica (Mode e Gustafsson, 2006). Le analisi genomiche e bioinformatiche hanno contribuito a chiarire i meccanismi molecolari coinvolti nella trascrizione genica epatica regolata dal hGH (Flores-Morales et al., 2001; Tollet-Egnell et al., 2004; Ståhlberg et al., 2005; Waxman e O’Connor, 2006; Wauthier et al., 2010). L’espressione dipendente dal sesso e la regolazione da parte del hGH caratterizzano diverse famiglie di geni epatici coinvolti nel metabolismo degli endo- e xenobiotici, nonché in importanti funzioni metaboliche (ad esempio, il metabolismo dei lipidi); il 20–30% di tutti i geni epatici presenta un modello di espressione specifico per sesso. La maggior parte di queste differenze epatiche legate al sesso è spiegata dal modello di secrezione del hGH specifico delle femmine, attraverso l’induzione di trascritti predominanti nelle femmine e la soppressione di quelli predominanti nei maschi. È stato dimostrato che la somministrazione continua di hGH aumenta l’espressione epatica del fattore di trascrizione SREBP-1c e dei suoi geni bersaglio a valle (Tollet-Egnell et al., 2001), nonché la sintesi epatica di trigliceridi (TG) e la secrezione di VLDL (Sjoberg et al., 1996). Come menzionato in precedenza, le azioni del hGH nel fegato determinano un aumento della lipogenesi (cioè l’induzione di SREBP-1c) e una diminuzione dell’ossidazione dei lipidi (cioè l’inibizione del PPARα), oltre a favorire la crescita anabolica nei tessuti periferici (cioè muscoli, ossa) (Flores-Morales et al., 2001; Tollet-Egnell et al., 2004; Ståhlberg et al., 2005). Per quanto riguarda la presente revisione, gli estrogeni provocano effetti opposti sul metabolismo dei lipidi e del glucosio, il che rappresenta un punto rilevante delle interazioni regolatorie tra estrogeni e hGH.
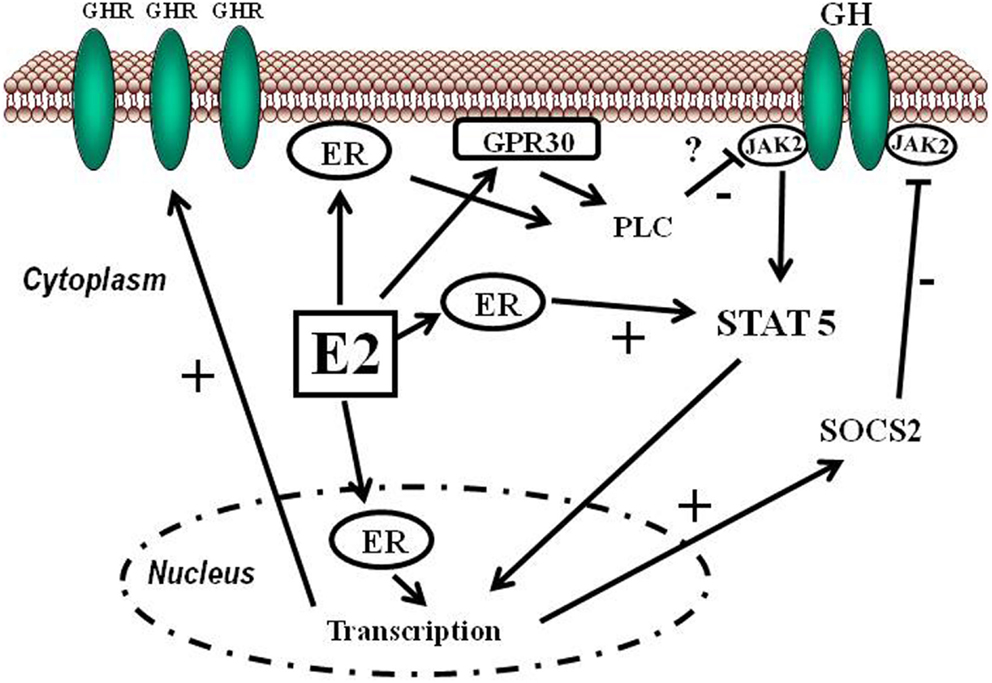
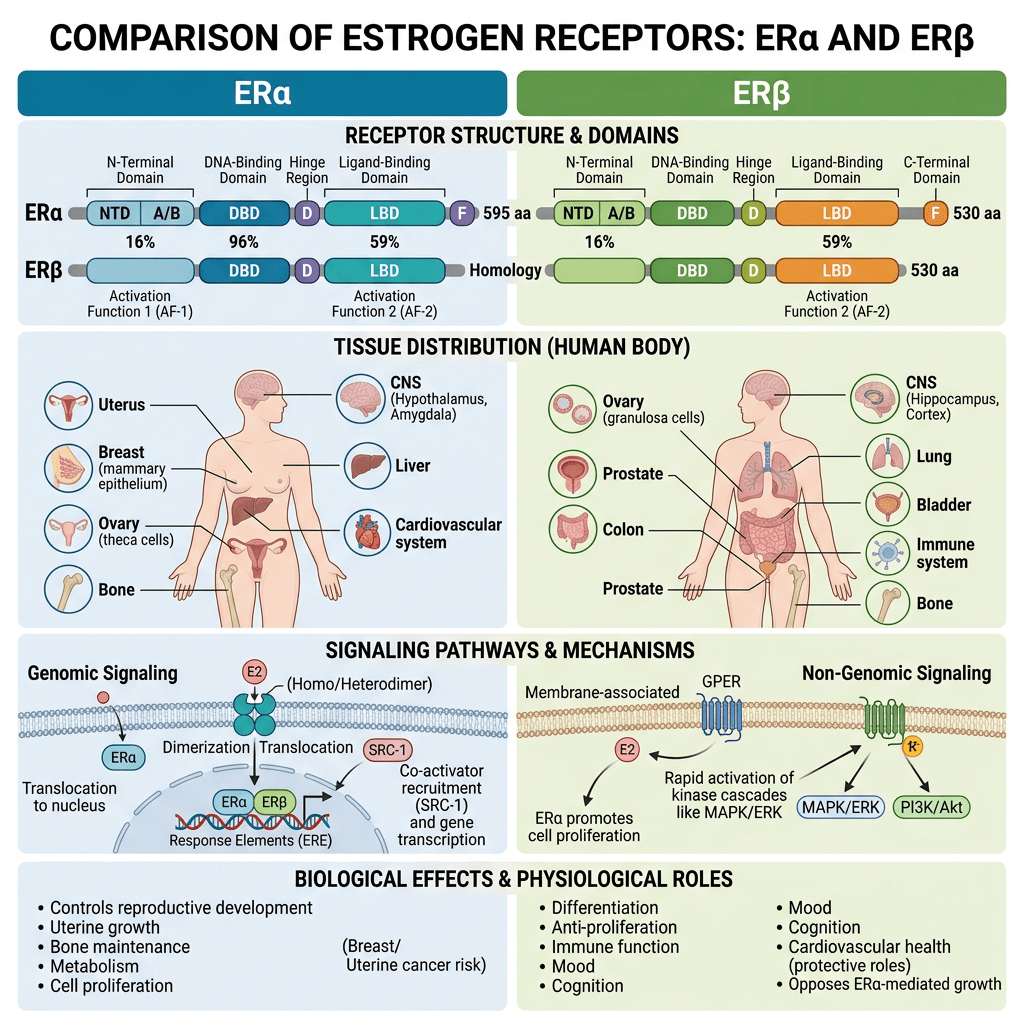
- Il fegato, un bersaglio fisiologico degli estrogeni
La segnalazione degli estrogeni può essere mediata da diversi recettori (Heldring et al., 2007). La maggior parte degli effetti estrogenici noti è mediata dall’interazione diretta degli estrogeni con i fattori di trascrizione che si legano al DNA, ERα ed ERβ. La segnalazione classica degli estrogeni avviene attraverso il legame diretto dei dimeri di ER agli elementi sensibili agli estrogeni (ES) nelle regioni regolatorie dei geni bersaglio degli estrogeni, seguito dall’attivazione del meccanismo trascrizionale nel sito di inizio della trascrizione. Inoltre, gli estrogeni possono modulare l’espressione genica attraverso un secondo meccanismo in cui gli ER interagiscono con altri fattori di trascrizione, come STAT5, tramite un processo denominato «crosstalk dei fattori di trascrizione». Gli estrogeni possono anche esercitare effetti attraverso meccanismi non genomici, che comportano l’attivazione di vie di segnalazione a valle delle chinasi, quali PKA, PKC e MAPK, tramite ER localizzati sulla membrana. Un recettore orfano accoppiato alla proteina G (GPR)-30 presente nella membrana cellulare media la segnalazione non genomica e rapida degli estrogeni. Infine, l’E2 presenta un’affinità simile per l’ERα e l’ERβ e questi recettori vengono attivati da un’ampia gamma di ligandi, tra cui i modulatori selettivi dei recettori degli estrogeni (SERM) (ad es. il raloxifene) e molti altri composti. L’ERβ è espresso nell’ovaio, nella prostata, nel polmone, nel tratto gastrointestinale, nella vescica, nel sistema ematopoietico e nel sistema nervoso centrale, mentre l’ERα è espresso principalmente nei tessuti riproduttivi, nei reni, nelle ossa, nel tessuto adiposo bianco e nel fegato. Il fegato esprime ERα ma livelli quasi impercettibili di ERβ, il che indica che le azioni specifiche degli estrogeni nel fegato possono essere riprodotte utilizzando agonisti selettivi di ERα come il propil-pirazolo-triolo (PPT) (Lundholm et al., 2008). Nel complesso, i dati sopra menzionati indicano che i meccanismi coinvolti nella segnalazione degli ER sono influenzati dal fenotipo cellulare, dal gene bersaglio e dall’attività o dall’interazione con altre reti di segnalazione. Il fegato rappresenta un sito in cui possono svilupparsi interazioni fisiologicamente e terapeuticamente rilevanti tra estrogeni e GH. Particolarmente rilevante è l’interazione degli estrogeni con la via di segnalazione GHR-JAK2-STAT5 nella regolazione della crescita somatica, del metabolismo dei lipidi e del glucosio e della “sessualità epatica”.
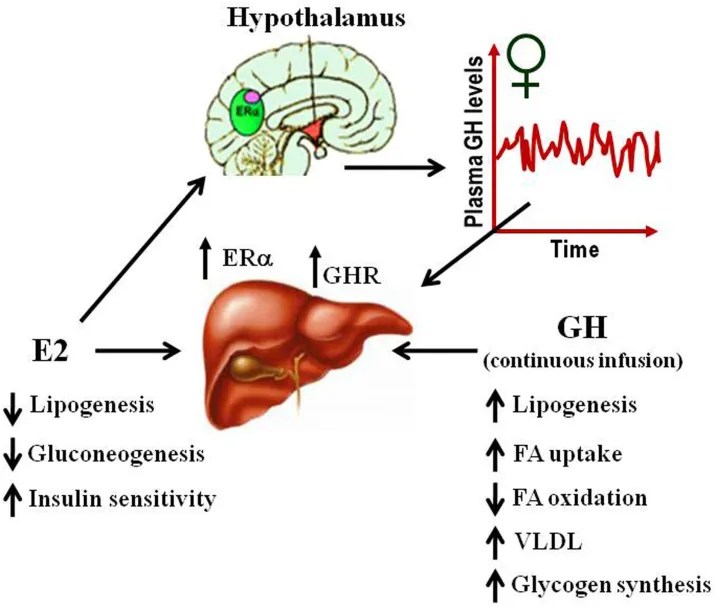
Effetti fisiologici dell’E2 e del hGH sul metabolismo dei lipidi e del glucosio nel fegato.
- Crescita somatica e composizione corporea
È ben noto che gli steroidi sessuali e l’ormone della crescita (GH) interagiscono strettamente per regolare la crescita puberale (Kerrigan e Rogol, 1992). È interessante notare che la perdita di ERα (ERαKO), ma non di ERβ, media importanti effetti degli estrogeni sullo scheletro dei topi maschi durante la crescita e la maturazione (Vidal et al., 2000). Un fenotipo simile a quello dei topi ERαKO si riscontra nei topi o negli esseri umani con deficit di aromatasi, che presentano una carenza di estrogeni (Riggs et al., 2002). Inoltre, le differenze di composizione corporea legate al genere sono in parte mediate dagli steroidi sessuali che modulano l’asse GH-IGF-I (LeRoith, 2009; Rogol, 2010; Birzniece et al., 2011). Ciò è supportato dall’osservazione che le differenze di genere nella composizione corporea emergono durante la crescita puberale. Inoltre, l’efficacia dell’attività del GH è modulata dagli estrogeni anche in età adulta. Ciò è esemplificato dal fatto che le donne rispondono in misura minore rispetto agli uomini al trattamento con GH (Burman et al., 1997); il trattamento con GH induce un maggiore aumento della massa magra e una maggiore riduzione della massa grassa, oppure un maggiore aumento degli indici di ricambio osseo e della massa ossea, nei pazienti maschi affetti da GHD rispetto alle pazienti di sesso femminile. Rilevante per la fisiologia del GH è l’alterazione della biodisponibilità dell’IGF-I mediante somministrazione orale di dosi farmacologiche di estrogeni [rivisto da Leung et al., 2004]. La disponibilità e l’attività dell’IGF-I nei tessuti sono regolate dalle proteine leganti l’IGF (IGFBP) (Kaplan e Cohen, 2007; LeRoith e Yakar, 2007; Ohlsson et al., 2009). L’IGF-I circola quasi interamente sotto forma di complesso ternario legato all’IGFBP-3 e all’ALS, entrambi fortemente regolati dal GH a livello epatico. Questo complesso ternario regola la biodisponibilità dell’IGF-I. Anche l’IGFBP-1 è una proteina di origine epatica che si lega alla piccola frazione di IGF-I libero e ne attenua l’effetto ipoglicemico (Lewitt et al., 1991). Contrariamente al suo effetto soppressivo sull’ALS e sull’IGF-I, la somministrazione orale di estrogeni aumenta l’IGFBP-1 circolante. È prevedibile che l’aumento dell’IGFBP-1 riduca ulteriormente la frazione libera di IGF-I, il che dovrebbe ridurne l’attività. È interessante notare che l’attivazione della via di segnalazione GH-STAT5b induce l’espressione di ALS e IGF-I ma inibisce l’IGFBP-1 (Ono et al., 2007). Pertanto, l’inibizione della via di segnalazione GHR-JAK2-STAT5 nel fegato (vedi sotto) contribuisce molto probabilmente agli effetti degli estrogeni su IGF-I, ALS e IGFBP-1. Di conseguenza, gli estrogeni esercitano effetti profondi sulle IGFBP di origine epatica quando somministrati per via orale, il che molto probabilmente modifica le azioni biologiche dell’IGF-I. Inoltre, la somministrazione orale di dosi farmacologiche di estrogeni può inibire gli effetti metabolici regolati dal GH (ad esempio, l’ossidazione dei lipidi, la sintesi proteica) (Huang e O’Sullivan, 2009). Questi effetti sul metabolismo e sulla composizione corporea sono attenuati dalla somministrazione transdermica, il che suggerisce che il fegato sia la sede principale del controllo regolatorio da parte degli estrogeni.
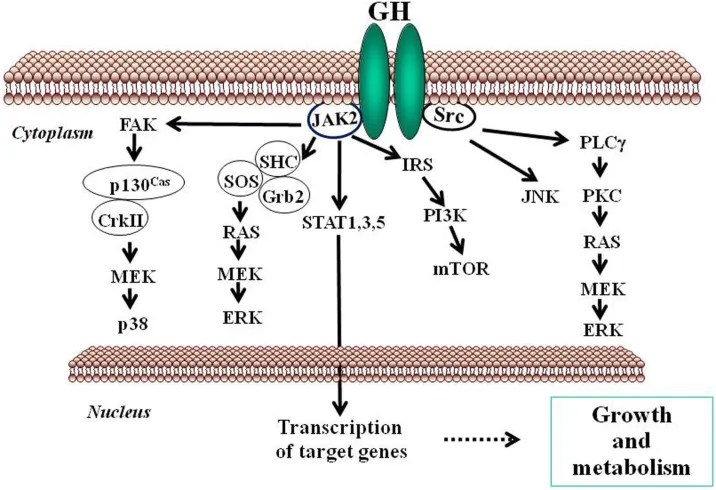
Gli estrogeni possono modulare l’azione del GH sul fegato influenzandone la risposta, il che comporta variazioni nell’espressione epatica del recettore GHR e un’interazione con la via di segnalazione JAK2-STAT5 attivata dal GH (Leung et al., 2004) (Figura 3). In particolare, l’E2 può indurre l’espressione di SOCS2 e SOCS3, che a loro volta regolano negativamente la via di segnalazione GHR-JAK2-STAT5, portando a una riduzione dell’attività trascrizionale nel fegato. Pertanto, oltre alla regolazione da parte dell’E2 del modello dimorfico sessuale della secrezione ipofisaria di GH, l’induzione dell’espressione delle SOCS e l’inibizione della via di segnalazione JAK2-STAT5 costituiscono un meccanismo molto rilevante che, in parte, potrebbe spiegare come gli estrogeni inibiscano direttamente gli effetti del GH in diverse azioni regolate da STAT5 (ad esempio, crescita somatica, composizione corporea, metabolismo e funzioni epatiche legate al genere). Abbiamo osservato che la somministrazione a lungo termine di dosi fisiologiche di E2 a ratti maschi affetti da GHD (ipotiroidei) regola diversi membri della famiglia SOCS attraverso una complessa interazione con il GH e gli ormoni tiroidei (Santana-Farre et al., 2008). Ipoteticamente, anche altri membri dei regolatori negativi della famiglia STAT potrebbero contribuire all’interazione degli estrogeni con la segnalazione del GH nel fegato. Ciò si spiega con la stimolazione da parte di ERα dell’espressione di PIAS3, che si lega a STAT3 e ne blocca l’attività di legame al DNA. È interessante notare che l’attivazione di ER da parte di E2, seguita dall’interazione diretta di ER con STAT5, possa anche inibire l’attività trascrizionale dipendente da STAT5 (Faulds et al., 2001; Wang et al., 2004). D’altra parte, è stato dimostrato che l’attivazione di ERα o ERβ da parte dell’E2, attraverso meccanismi non genomici, induce un programma trascrizionale dipendente da STAT5 (e STAT3) nelle cellule endoteliali (Bjornstrom e Sjoberg, 2005). Nel complesso, questi studi hanno evidenziato un’interazione diretta tra la segnalazione dell’ER e quella di STAT5 e dimostrano inoltre che le conseguenze funzionali di questa interazione dipendono dalle precise condizioni dell’ambiente intracellulare.
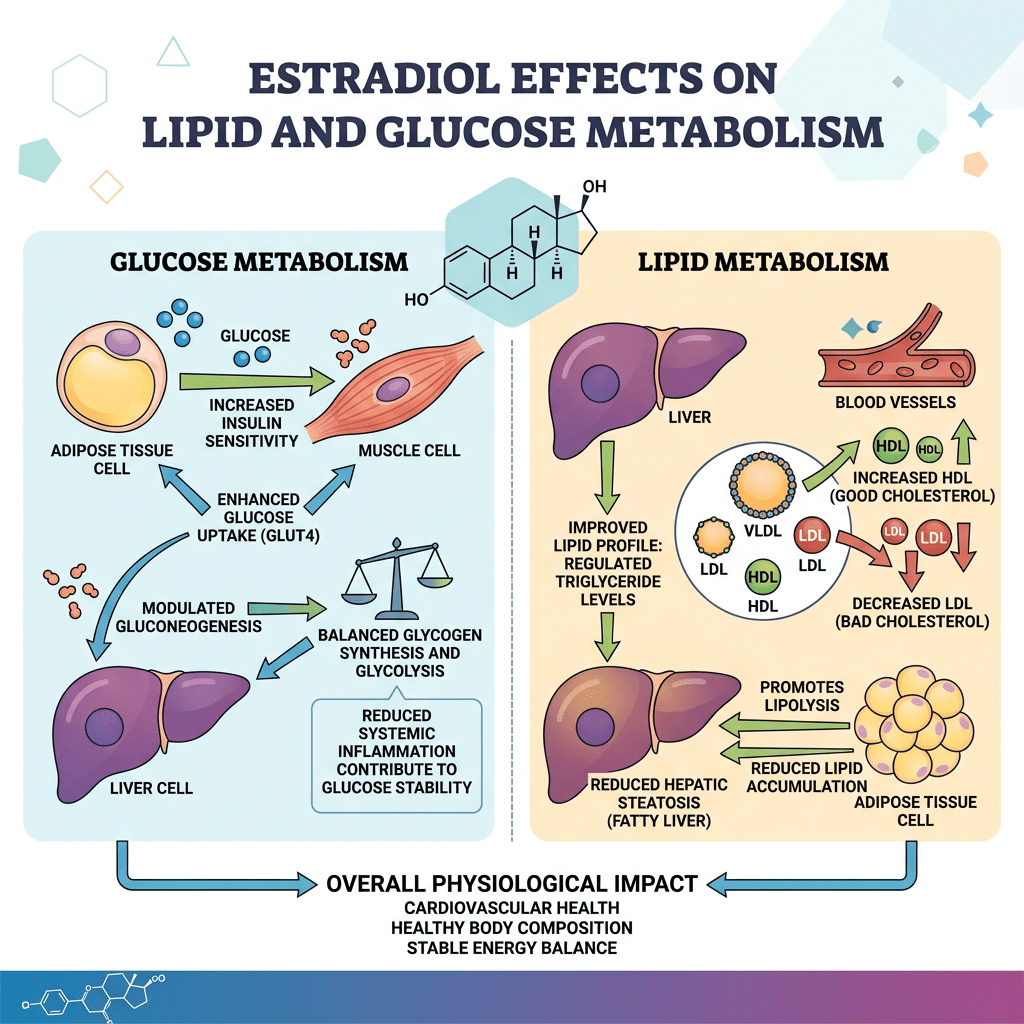
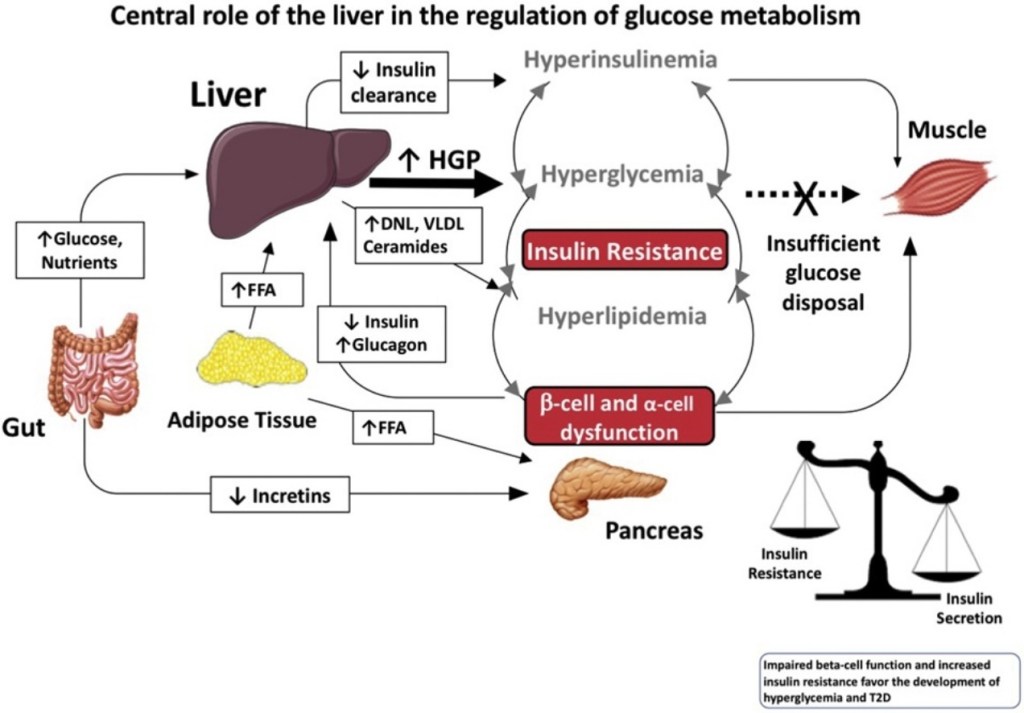
Diversi studi hanno suggerito che la segnalazione mediata dall’E2 possa svolgere un ruolo importante nel controllo del metabolismo dei lipidi e del glucosio (Barros e Gustafsson, 2011; Faulds et al., 2012). Studi condotti sia sull’uomo che sui roditori indicano che livelli alterati di estrogeni o dei loro recettori possono portare a un fenotipo simile alla sindrome metabolica (ovvero, insulino-resistenza, adiposità, dislipidemia). Ad esempio, le donne in postmenopausa hanno una probabilità tre volte maggiore di sviluppare anomalie associate alla sindrome metabolica rispetto alle donne in premenopausa (Eshtiaghi et al., 2010). Inoltre, è stato dimostrato che la terapia ormonale sostitutiva a base di estrogeni e progestinici nelle donne in postmenopausa riduce il tessuto adiposo viscerale, la glicemia a digiuno e i livelli di insulina (Munoz et al., 2002). Osservazioni cliniche su maschi con deficit di ERα o con livelli ridotti di aromatasi hanno evidenziato lo sviluppo di aumento del peso corporeo, insulino-resistenza e iperinsulinemia (Smith et al., 1994; Jones et al., 2007). Analogamente, i topi ERαKO e quelli con deficit di aromatasi sviluppano insulino-resistenza, adiposità intra-addominale, steatosi e compromissione dell’ossidazione dei lipidi nel fegato, condizioni che possono essere invertite dal trattamento con E2 (Heine et al., 2000; Simpson et al., 2005; Jones et al., 2007). L’influenza benefica degli estrogeni sulla normalizzazione dell’omeostasi lipidica e glicemica è evidenziata anche nei topi ob/ob e in quelli alimentati con una dieta ricca di grassi, modelli di obesità e diabete di tipo 2. In entrambi i modelli, il trattamento con E2 migliora la tolleranza al glucosio e la sensibilità all’insulina e riduce il peso nei topi alimentati con una dieta ricca di grassi (Gao et al., 2006; Bryzgalova et al., 2008). Studi condotti su topi ERαKO hanno dimostrato che l’ERα media principalmente gli effetti metabolici benefici degli estrogeni, quali l’inibizione della lipogenesi, il miglioramento della sensibilità all’insulina e della tolleranza al glucosio e la riduzione dell’adiposità (Barros e Gustafsson, 2011; Faulds et al., 2012). Al contrario, i topi ERβKO non mostrano alterazioni della sensibilità all’insulina e/o del peso corporeo. Tuttavia, esistono alcune evidenze secondo cui l’ERβ potrebbe essere dannoso per il mantenimento di una regolare omeostasi del glucosio e dei lipidi. Oltre alle osservazioni relative all’ERαKO, è stato riportato che l’ablazione selettiva dell’ERα nella regione ipotalamica del cervello o nelle cellule ematopoietiche/mieloidi determina un aumento del peso corporeo e una ridotta tolleranza al glucosio (Ribas et al., 2011; Xu et al., 2011). L’insulino-resistenza nei topi ERαKO è in gran parte localizzata nel fegato e comporta un aumento del contenuto lipidico e della produzione epatica di glucosio. Sorprendentemente, l’ablazione selettiva di ERα a livello epatico (LERKO) non ha riprodotto il fenotipo osservato nei topi ERαKO (Matic et al., 2013). I topi LERKO non hanno aumentato il peso corporeo né hanno sviluppato intolleranza al glucosio o insulino-resistenza, nemmeno in seguito a una dieta ricca di grassi. Gli autori ipotizzano la presenza di uno o più meccanismi compensatori non identificati oppure che la resistenza all’insulina epatica si verifichi come effetto secondario dell’ablazione della segnalazione dell’E2 in altri tipi cellulari. Inoltre, il trattamento dei topi ob/ob con l’agonista selettivo dell’ERα PPT può migliorare la tolleranza al glucosio e la sensibilità all’insulina, il che conferma il ruolo fondamentale della segnalazione dell’ERα nel controllo dell’omeostasi del glucosio. Anche la segnalazione estrogenica tramite GPR-30 è stata implicata nella produzione di insulina e nell’omeostasi del glucosio (Mårtensson et al., 2009). Come menzionato in precedenza, l’assenza di E2 o della segnalazione GHR-JAK2-STAT5 causa adiposità e steatosi epatica, che possono essere attenuate rispettivamente dalla somministrazione di E2 (Heine et al., 2000; Simpson et al., 2005; Jones et al., 2007) o con la terapia sostitutiva con GH (LeRoith e Yakar, 2007), rispettivamente. Ciò suggerisce che la segnalazione dell’E2 e del GH regoli reti cellulari sovrapposte correlate al controllo fisiologico del metabolismo dei lipidi e del glucosio.
- Punti chiave sulla relazione “riduttiva” dell’attività del hGH e dell’IGF-1 E2 correlata
E’ importante, al fine della comprensione del presente lavoro, che l’E2 riduce l’azione periferica dell’hGH nel fegato, in particolare se vi si creano concentrazioni elevate in loco d’organo. Sebbene gli estrogeni aumentino la secrezione di hGH da parte dell’ipofisi, inibiscono contemporaneamente la segnalazione del hGH nei tessuti epatici, riducendo la produzione di mediatori a valle come l’IGF-1.
- I meccanismi di down-regulation del hGH da parte dell’E2 sono:
- Inibizione della via JAK/STAT: l’estradiolo sopprime la segnalazione indotta dal GH attraverso la via JAK2-STAT5.
- Induzione delle SOCS: gli estrogeni stimolano l’espressione delle proteine soppressive della segnalazione delle citochine (SOCS), come la SOCS2, che bloccano attivamente le cascate di segnalazione del GHR.
- Dipendenza nelle concentrazioni di E2 epatico iatrogeno dipendente: l’instaurarsi di alte concentrazioni di E2 o composti correlati a livello epatico esercita un forte effetto di primo passaggio epatico che attenua efficacemente la produzione sistemica di IGF-1, mentre la condizione fisiologica evita in gran parte, ma non del tutto, questa profonda down-regulation epatica.
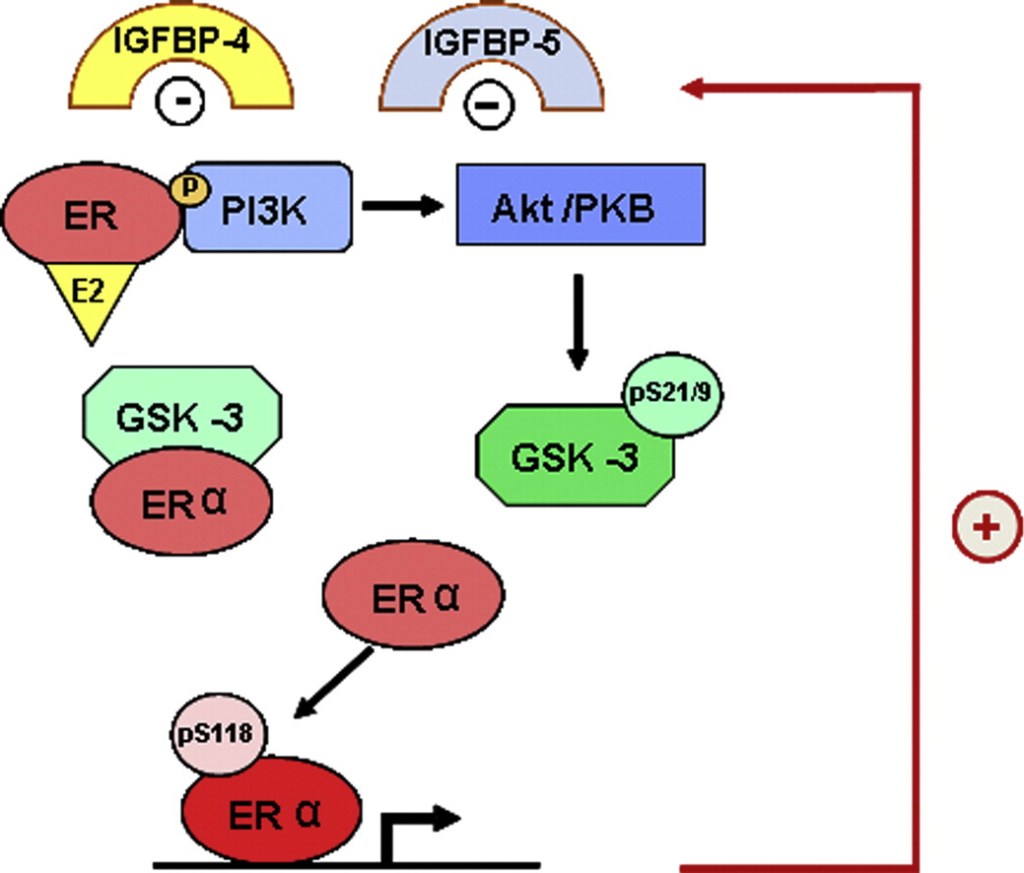
Inoltre, l’E2 può aumentare i livelli di specifiche proteine leganti il fattore di crescita insulino-simile (IGFBP), ovvero l’IGFBP-1 e l’IGFBP-3. Queste proteine leganti contribuiscono a regolare la quantità di IGF-1 disponibile/attiva per le cellule.
- Effetti dell’E2 su specifiche IGFBP
- IGFBP-1: Gli estrogeni aumentano generalmente i livelli sierici di IGFBP-1, il che può ridurre la quantità di IGF-1 libero e attivo in circolazione. L’estradiolo per via orale ha un effetto stimolante particolarmente forte sulla produzione epatica di IGFBP-1 a causa del passaggio diretto attraverso la circolazione portale di primo passaggio.
- IGFBP-3: Gli studi dimostrano che l’estradiolo può aumentare la sintesi e i livelli sierici di IGFBP-3, agendo in sinergia con le vie dell’ormone della crescita in vari tessuti.
- Altre IGFBP: L’estradiolo può anche aumentare i livelli locali o sistemici di IGFBP-2 e IGFBP-4 in specifici tessuti bersaglio come il cervello, l’ipofisi o la cartilagine ossea.
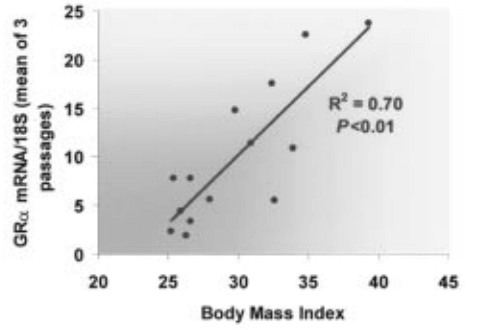
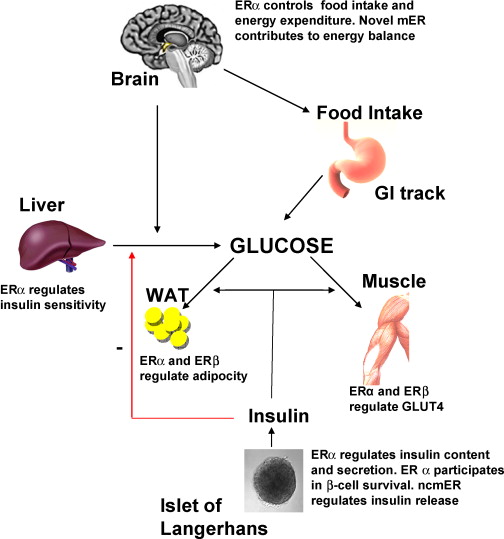
Differenze di genere nella biologia del tessuto adiposo
In ambito clinico, le differenze di genere nell’adiposità e nel metabolismo variano a seconda delle fasi della vita. Prima della pubertà, vi è poca differenza nella distribuzione del tessuto adiposo, ma durante la pubertà precoce, le differenze nella traiettoria del peso e nella composizione corporea diventano più evidenti. Ad esempio, gli uomini tendono a sviluppare una maggiore distribuzione centrale del grasso durante la transizione dall’adolescenza alla giovane età adulta. Le donne in premenopausa hanno livelli più elevati di estrogeni che hanno un effetto protettivo contro l’aumento di peso, aumentando il dispendio energetico e distribuendo il tessuto adiposo nelle regioni sottocutanee. Parte di questo aumento del dispendio energetico deriva dall’effetto estrogenico del “browning” o attività metabolica del tessuto adiposo. Il BAT è metabolicamente più attivo a causa dell’aumento del numero di mitocondri. Ciò è stato dimostrato in campioni bioptici e nell’aumento dell’espressione genica per la funzione mitocondriale. Le donne in postmenopausa aumentano di peso in parte a causa della naturale diminuzione dell’E2 endogeno durante la menopausa. Questa riduzione del dispendio energetico può essere prevenuta dalla terapia sostitutiva con estrogeni; Tuttavia, il rischio di malattie metaboliche non sembra migliorare. Questa risposta agli estrogeni può essere sessualmente dimorfica poiché gli estrogeni in vitro aumentano l’espressione sia di ERα che di ERβ negli adipociti sottocutanei delle donne, ma aumentano solo ERα negli adipociti sottocutanei e viscerali degli uomini.
Le donne con livelli di SHBG notevolmente inferiori e alti livelli di Testosterone biodisponibile presentavano un aumento del tessuto adiposo addominale e viscerale. Negli uomini, l’aumento del testosterone biodisponibile diminuiva l’obesità addominale e il profilo di rischio metabolico. Lo studio SWAN ha dimostrato che le donne nere avevano livelli di SHBG più elevati rispetto alle donne bianche, mentre sia le donne nere che quelle ispaniche avevano un indice di androgeni liberi (FAI) inferiore rispetto alle donne bianche, il che è coerente con la maggiore prevalenza di obesità riscontrata nelle donne nere e ispaniche. Le donne cinesi avevano livelli di SHBG significativamente inferiori e un FAI più elevato rispetto alle donne bianche, il che è coerente con la minore prevalenza di obesità riscontrata nelle donne cinesi. Essere consapevoli dell’influenza del gruppo etnico e del genere sull’adiposità può essere utile per identificare il rischio individuale e la scelta della gestione migliore del soggetto.
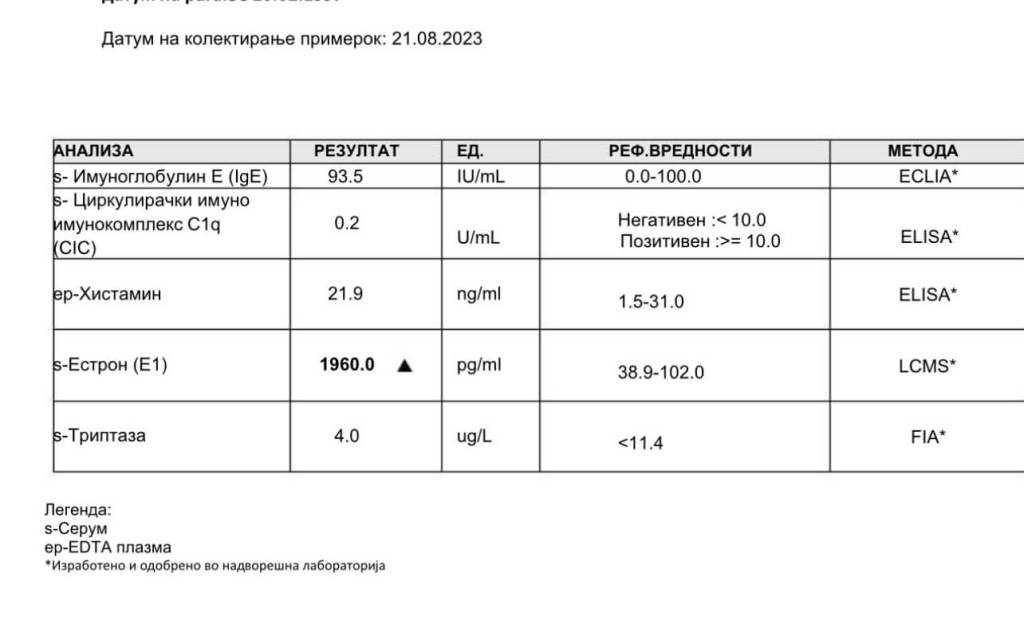
Nelle donne in postmenopausa, il tessuto adiposo bianco (WAT) diventa il principale loco di sintesi di estrogeni per l’organismo, la cui produzione dipende da una significativa espressione e attività dell’Aromatasi. In particolare, nel WAT, l’Aromatasi converte l’Estrone [E1], un prodotto aromatico derivato dall’Androstenedione, quest’ultimo secreto dalla ghiandola surrenale, in E2 tramite la classe di enzimi 17-beta idrossisteroide deidrogenasi (17β-HSD). Tuttavia, i livelli di estrogeni prodotti da questa via metabolica non sono in grado di compensare la perdita della produzione ovarica di estrogeni; di conseguenza, la terapia ormonale sostitutiva (TOS) può essere necessaria per le donne in postmenopausa. Ciononostante, il 17β-estradiolo (17β-E2) è la principale forma di estrogeno circolante e biologicamente attiva. È anche la forma più studiata nella regolazione del tessuto adiposo e, pertanto, utilizzeremo il termine generico estrogeno per riferirci al 17β-estradiolo in senso lato.
Oltre all’attivazione tradizionale degli ER, gli estrogeni possono anche associarsi e attivare un recettore degli estrogeni legato alla membrana e accoppiato a una proteina G chiamato GPER o GPER30. Il GPER sembra essere fondamentale per guidare i percorsi indipendenti dagli ER e media gli effetti non genomici degli estrogeni. In seguito all’attivazione del GPER da parte del 17β-estradiolo, vengono attivate le vie MAPK (Erk1/2) e dell’adenilato ciclasi per promuovere varie attività cellulari. In particolare, è stato suggerito che il GPER promuova l’inibizione dell’apoptosi indotta dallo stress ossidativo mediata dagli estrogeni, l’aumento della crescita cellulare attraverso la stimolazione dell’espressione della ciclina D e la sovraregolazione della produzione del fattore di crescita nervoso nei macrofagi. Questo repertorio biologico unico e diversificato ha forti implicazioni sulla biologia e sulla progressione del cancro al seno. Questa gamma di attività esiste anche per il tessuto adiposo, e tutti gli ER e il GPER3 sono espressi all’interno di vari depositi di WAT. Tuttavia, l’ERα è stato il più ampiamente studiato e sembra guidare la maggior parte delle funzioni del WAT correlate agli estrogeni. Al contrario, i ruoli di ERβ e GPER non sono ben caratterizzati; tuttavia, entrambi i recettori degli estrogeni sembrano regolare il metabolismo.
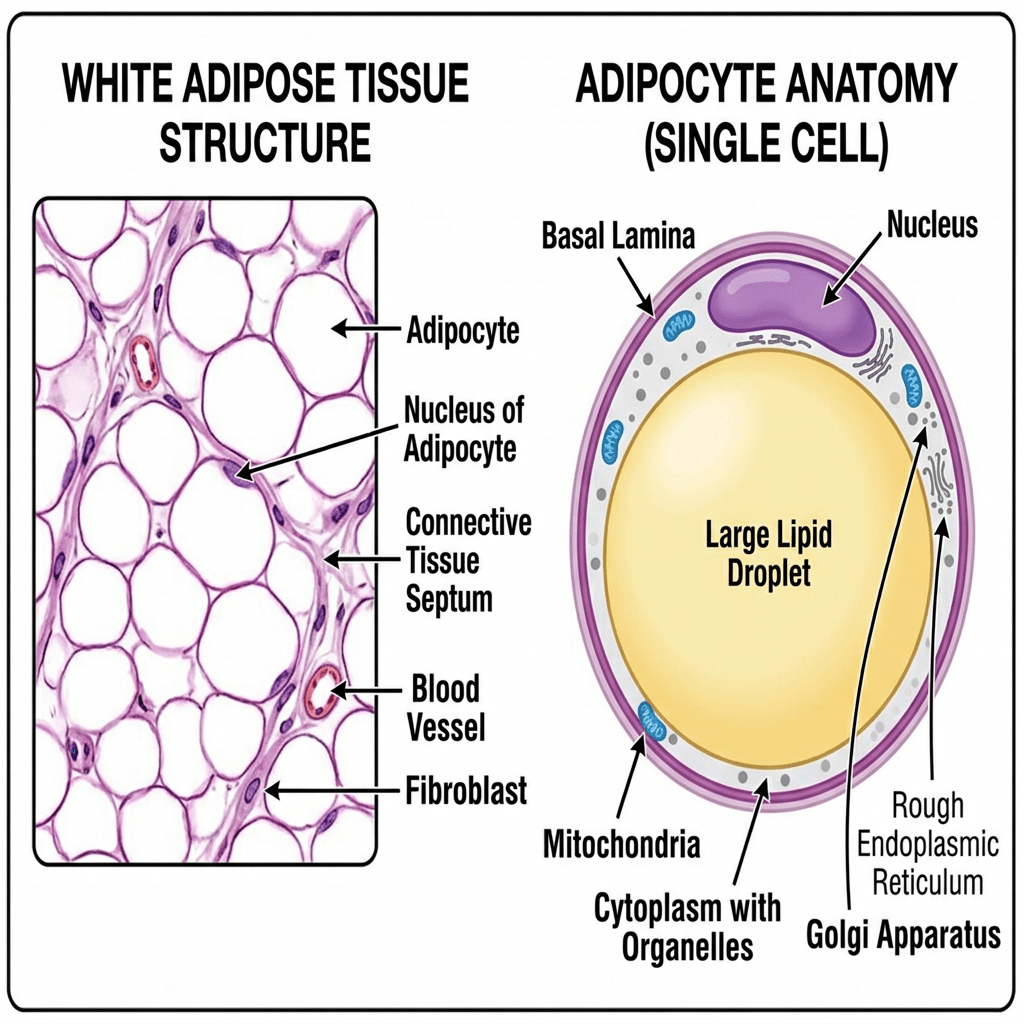
Il tessuto adiposo bianco (WAT) è disperso e non contiguo in tutto il corpo, rappresentando la potenziale eterogeneità di questo organo. Ad esempio, il WAT può essere suddiviso in due ampie localizzazioni anatomiche, sottocutaneo e viscerale, che possono essere ulteriormente separate in compartimenti distinti chiamati depositi. Il grasso sottocutaneo si trova sotto il derma, mentre il grasso viscerale circonda gli organi interni all’interno della cavità corporea . Questa macroscopica distribuzione anatomica sembra avere anche significative implicazioni metaboliche. Il grasso sottocutaneo sembra essere metabolicamente protettivo, mentre il grasso viscerale contribuisce alla disregolazione metabolica . Questa distribuzione anatomica protettiva del WAT è particolarmente rilevante tra uomini e donne. Ad esempio, le donne tendono ad avere il 10-20% in più di grasso corporeo rispetto agli uomini con lo stesso indice di massa corporea (BMI) . Tuttavia, le donne in premenopausa accumulano preferenzialmente grasso sottocutaneo nella parte inferiore del corpo, nei fianchi e nelle cosce e presentano una ridotta adiposità viscerale . Inoltre, le donne in premenopausa sono più protette dallo sviluppo di malattie metaboliche, probabilmente a causa dell’aumento del rapporto tra grasso sottocutaneo e grasso viscerale. Al contrario, gli uomini spesso accumulano grasso viscerale in eccesso, che porta a disturbi metabolici e malattie cardiovascolari . Tuttavia, le donne in postmenopausa spesso accumulano grasso viscerale riducendo al contempo i depositi di tessuto adiposo bianco sottocutaneo. Questo effetto è dovuto principalmente alla mancanza di estrogeni, che predispone le donne alle malattie metaboliche . Infatti, studi che utilizzano i dati del National Health and Nutrition Examination Survey (NHANES), i dati di valutazione nutrizionale più estesi disponibili su interviste ed esami fisici, hanno dimostrato che un aumento dell’adiposità viscerale o centralizzata è associato al rischio più significativo di mortalità nelle donne . Uno studio britannico ha ulteriormente supportato questa nozione dimostrando un rischio simile di mortalità e adiposità viscerale nelle donne . In generale, un elevato deposito di grasso viscerale è associato ai più alti rischi di malattie metaboliche e morte prematura, indipendentemente dal sesso. Pertanto, si ritiene che i cambiamenti complessivi nei siti di accumulo del tessuto adiposo e le risposte sessualmente dimorfiche che controllano la disposizione del grasso corporeo spieghino perché gli uomini sviluppano malattie cardiometaboliche prima delle donne.
Cosa potrebbe spiegare queste differenze metaboliche e l’adiposità tra uomini e donne? Sembra che dipenda da dove avviene la crescita del tessuto adiposo e dalla biodisponibilità degli estrogeni. Prove crescenti da studi su esseri umani e roditori hanno dimostrato che livelli più elevati di estrogeni aumentano l’espansione del tessuto adiposo bianco sottocutaneo e smorzano la crescita del tessuto adiposo bianco viscerale. Oltre alla distribuzione del grasso corporeo, i livelli di estrogeni ovarici possono ulteriormente proteggere dai segnali obesogeni e dalle malattie metaboliche . La ricerca ha dimostrato che la riduzione dei livelli circolanti di estrogeni dovuta alla menopausa o all’ovariectomia aumenta il rischio di sviluppare obesità, diabete di tipo 2 e malattie cardiovascolari. Nei roditori e negli esseri umani, la terapia sostitutiva con estradiolo o HRT inverte l’obesità riducendo la massa di grasso viscerale, migliorando così la forma fisica metabolica. Negli esseri umani, l’HRT ha dimostrato di avere numerosi effetti metabolici benefici nelle donne in post-menopausa. Ad esempio, in uno studio di 3 anni, la terapia ormonale sostitutiva (TOS) ha ridotto statisticamente i livelli di glucosio a digiuno e ha abbassato significativamente l’incidenza del diabete . Inoltre, la TOS ha aumentato l’HDL e ridotto l’LDL, determinando profili lipidici più sani. Inoltre, le donne in postmenopausa che assumevano estrogeni hanno mostrato una diminuzione dell’adiposità viscerale, che ha ridotto il loro rischio di malattie cardiovascolari. In accordo con questi studi, i roditori ovariectomizzati sottoposti a terapia sostitutiva con estradiolo hanno mostrato una riduzione dell’assunzione di cibo e un aumento del dispendio energetico, proteggendoli dall’accumulo di massa grassa. È interessante notare che queste osservazioni non si limitano alle donne; la ricerca suggerisce che anche gli uomini e i topi maschi traggono beneficio dall’attività e dalla segnalazione degli estrogeni. Ad esempio, studi hanno dimostrato che la perdita della segnalazione degli estrogeni nei maschi promuove l’obesità e compromette il metabolismo del glucosio. Inoltre, la terapia ormonale di transizione di genere nelle donne trans che assumono estrogeni mostra una distribuzione del grasso corporeo più femminile e un rapporto vita-fianchi inferiore. Pertanto, gli estrogeni circolanti contribuiscono alla distribuzione, al rimodellamento e al mantenimento del grasso corporeo, ma come vengono mantenuti i livelli di estrogeni circolanti e l’attività dei recettori degli estrogeni?
La modulazione degli estrogeni ha un impatto sull’adiposità nelle donne in pre e postmenopausa, ma questo effetto è guidato dai recettori degli estrogeni (ER)? Nelle donne in premenopausa in sovrappeso o obese, si possono osservare differenze nell’espressione dei recettori degli estrogeni nei tessuti adiposi addominali e glutei. Questi dati suggeriscono che la disponibilità dei recettori potrebbe essere fondamentale per contrastare i depositi di grasso regionali in risposta a un bilancio energetico positivo. Uno studio sull’uomo che confrontava le differenze regionali nel tessuto sottocutaneo addominale e femorale in donne in pre e postmenopausa ha rilevato che le differenze metaboliche dipendevano dall’espressione dell’isotipo dei recettori degli estrogeni. Ovvero, queste differenze metaboliche (cioè la sensibilità all’insulina) derivavano dalle alterazioni nel rapporto tra ERα e ERβ tra i due depositi di grasso. Un rapporto ERα/ERβ più elevato è stato osservato nelle donne in premenopausa rispetto alle donne in postmenopausa. Inoltre, il trattamento di soggetti sia pre che postmenopausali con 17β-estradiolo ha aumentato il rapporto ERα/ERβ all’interno del WAT, suggerendo che i cambiamenti mediati dagli estrogeni nel rapporto ER sono fondamentali per indurre la sensibilità all’insulina del WAT. Sebbene interessanti e fisiologicamente rilevanti, i meccanismi che governano questa risposta devono ancora essere chiariti. Un potenziale meccanismo regolatorio che spiega questi cambiamenti nel rapporto ERα:ERβ potrebbe essere dovuto a un silenziamento potenziato del promotore di ERα tramite metilazione del DNA. Questo sembra verificarsi nell’aorta del ratto, ma mancano studi che esaminino la metilazione del promotore di ERα nel grasso e non può essere collegato a malattie metaboliche. Ciò che è chiaro è il costante aumento dell’adiposità, in particolare del WAT viscerale, e l’alterato metabolismo energetico dei topi maschi e femmine privi di ERα. Questo obesogeno e l’alterato equilibrio energetico nei topi privi di ERα sono ulteriormente accentuati quando alimentati con una dieta ricca di grassi.
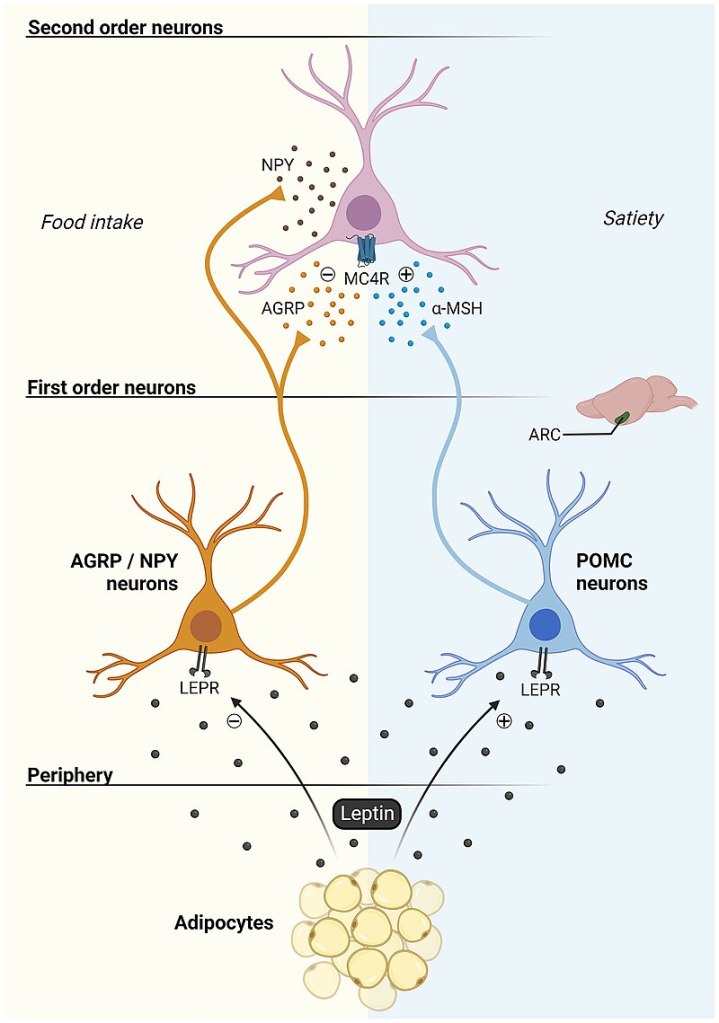
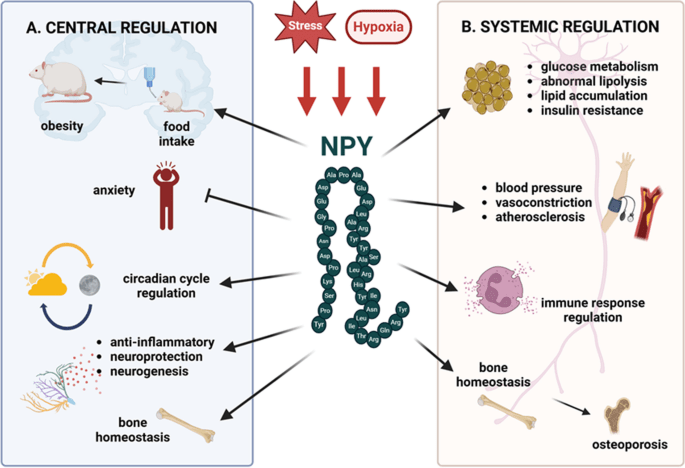
Tuttavia, l’aumento dell’adiposità nei topi ERα nulli potrebbe essere indipendente dalla biologia del tessuto adiposo bianco e potrebbe essere collegato alla regolazione ipotalamica degli estrogeni e dei recettori degli estrogeni (ER). Ad esempio, la riduzione dell’espressione di ERα nel nucleo ventromediale (VMN) ha causato un aumento di peso attraverso un’alterazione del dispendio energetico indipendentemente dall’assunzione di cibo. Inoltre, la riduzione dell’espressione di ERα nel VMN provoca disfunzioni metaboliche nei ratti. Tuttavia, gli effetti indotti dagli estrogeni sull’assunzione di cibo possono essere osservati in risposta a microiniezioni a basso dosaggio di 17β-estradiolo nel cervello. In accordo con ciò, alcuni studi hanno suggerito che gli estrogeni diminuiscono i peptidi oressigeni per ridurre l’assunzione di cibo. Ad esempio, una riduzione del neuropeptide Y (NPY), un potente stimolatore dell’appetito, è stata associata ad un aumento dei livelli circolanti di estrogeni e all’attività dei recettori degli estrogeni mediata dagli estrogeni . Nello specifico, la trascrizione di NPY corrisponde al rapporto ERα:ERβ presente nell’ipotalamo. Quando il rapporto è alto, viene trascritto meno NPY; al contrario, quando il rapporto è basso, viene prodotto NPY . Inoltre, le variazioni del livello dell’ormone della fame grelina sono state associate a diverse fasi del ciclo ovarico . In particolare, l’infusione di grelina durante il diestro uno e il diestro due stimola l’assunzione di cibo, ma non durante il proestro o l’estro, in coincidenza con il picco dei livelli di estrogeni. A sostegno di questa nozione, i topi knockout per il recettore della grelina ovariectomizzati non sviluppano iperfagia o aumento di peso corporeo. Ciò suggerisce che la riduzione dei livelli circolanti di estrogeni aumenta l’assunzione di cibo liberando la grelina da un effetto inibitorio tonico degli estrogeni. Pertanto, gli estrogeni normalmente sopprimono la funzione della grelina per bloccare l’assunzione di cibo, ma in assenza di estrogeni, la secrezione di grelina è incontrollata, mediando la risposta iperfagica. Non è ancora chiaro in che modo gli estrogeni influenzino l’alimentazione mediata dalla grelina e se questi effetti possano derivare dalla segnalazione, dalla secrezione o dall’espressione dei recettori della grelina.
Mentre l’ablazione del gene ERα porta a una risposta metabolica disfunzionale distinta e robusta, la delezione del gene ERβ appare meno evidente. È interessante notare che la delezione di ERβ in tutto il corpo non ha praticamente alcun effetto sull’adiposità o sul bilancio energetico . Questa osservazione ha portato all’ipotesi che ERβ promuovesse l’obesità e i disturbi metabolici. Tale osservazione è stata ulteriormente supportata da un aumento di 10 volte della concentrazione di 17β-estradiolo nei topi ERα nulli, il che suggerisce un aumento del flusso di segnalazione degli estrogeni attraverso ERβ. A sostegno di questa visione, i topi ERβ nulli ovariectomizzati erano protetti dall’obesità . Tuttavia, studi successivi hanno dimostrato che i topi ERβ nulli erano in realtà più suscettibili all’obesità ma protetti contro l’insulino-resistenza. Questo effetto obesogeno sui topi ERβ nulli è stato ulteriormente accentuato dall’ovariectomia. In linea con questi studi sui roditori, uno studio genetico umano ha rivelato cinque polimorfismi a singolo nucleotide (SNP) nell’ERβ associati all’obesità sia nei maschi che nelle femmine. Tuttavia, i meccanismi molecolari e le caratteristiche regolatorie dell’ERβ nell’obesità indotta dalla dieta devono ancora essere chiariti. Ma nuovi studi stanno iniziando a suggerire che l’ERβ potrebbe avere un ruolo metabolicamente protettivo regolando l’attività dei mitocondri del tessuto adiposo bianco (WAT). Ad esempio, nei roditori, i ligandi specifici dell’ERβ sembrano aumentare il dispendio energetico e l’attività dei mitocondri del WAT, anche in assenza di estrogeni circolanti . Inoltre, i cambiamenti nei geni di accumulo di grasso e nelle adipochine sono stati correlati all’attivazione dell’ERβ rispetto all’ERα o al rapporto tra i due recettori . Nel complesso, questi studi suggerirebbero che sia l’ERα che l’ERβ sono necessari per un corretto metabolismo energetico al fine di mediare la flessibilità metabolica. Nel loro insieme, l’attivazione degli isotipi del recettore degli estrogeni (ER), il profilo di espressione dell’ER, l’interazione tra il recettore dell’ER e il DNA e le attività non genomiche degli estrogeni rappresentano aree di ricerca cruciali che contribuiranno a risolvere i quesiti ancora irrisolti.
Oltre alle risposte mediate dal genoma ligando-recettore, gli estrogeni possono anche avere risposte non genomiche mediate da GPER (recettore degli estrogeni legato alla membrana e accoppiato a proteine G). GPER è espresso nelle membrane intracellulari e, come altri GPCR, accoppia la segnalazione degli estrogeni a cambiamenti nell’attività dell’adenilato ciclasi, della chinasi e dei canali ionici. In particolare, GPER può anche regolare indirettamente l’espressione dei geni bersaglio. GPER è espresso in una moltitudine di tessuti; tuttavia, GPER non sembra essere coinvolto nella riproduzione mediata dagli estrogeni . Questo perché i topi privi di GPER sono fertili, mentre i topi privi di ERα sono infertili . All’interno del tessuto adiposo, i ruoli metabolici di GPER rimangono elusivi, ma negli ultimi anni sono state raccolte maggiori informazioni, che sono state recentemente esaminate in . In breve, alcuni studi mostrano che la carenza di GPER porta a una riduzione del peso corporeo e della crescita ossea nelle femmine, e altri studi hanno riportato un aumento significativo della massa grassa quando il GPER viene eliminato . Al contrario, diversi studi hanno mostrato un effetto del GPER o della sua attivazione sul peso corporeo anche in risposta a una dieta ricca di grassi . Nel complesso, la capacità degli estrogeni di regolare la massa grassa e la distribuzione sessualmente dimorfica dipende dall’espressione del recettore, dalla biodisponibilità e sintesi degli estrogeni e dall’età.
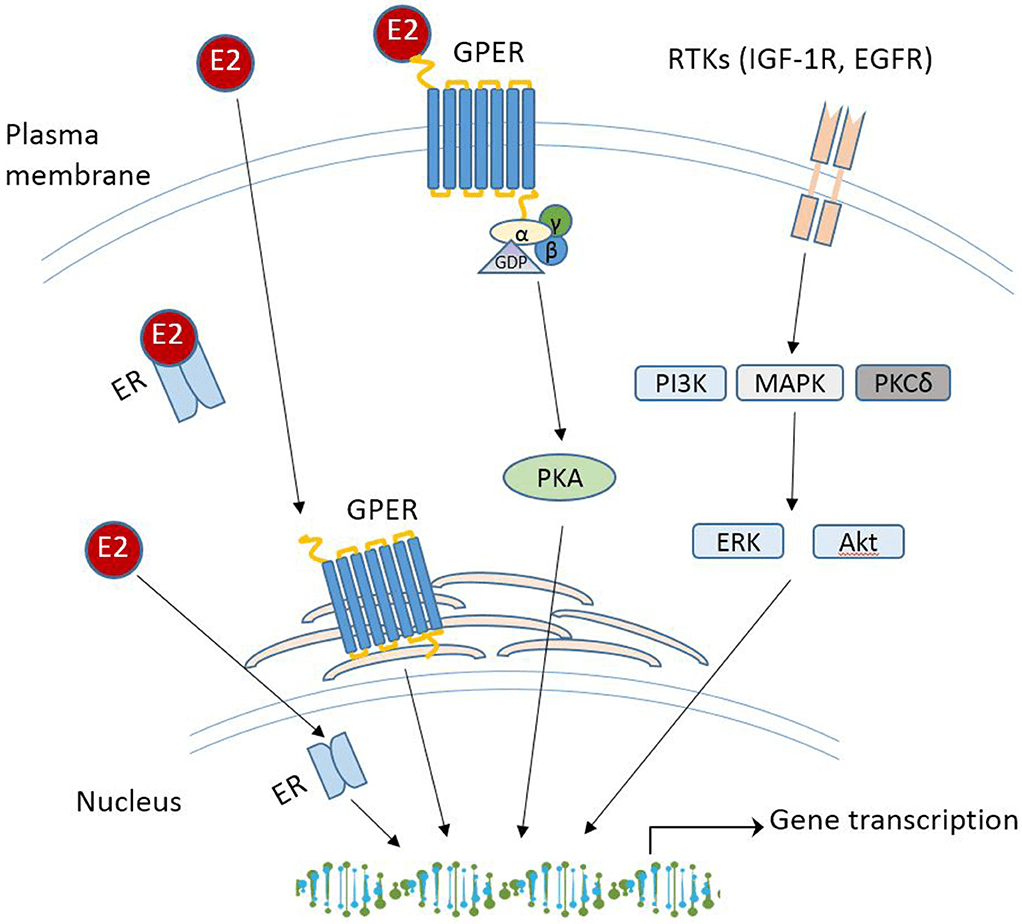
Tuttavia, gli estrogeni sembrano avere un ruolo diretto nella soppressione dei geni di accumulo lipidico e nell’attivazione preferenziale delle vie lipolitiche. Ad esempio, negli adipociti e nelle cellule tumorali, è stato dimostrato che il recettore ER interagisce direttamente con il recettore gamma attivato dai proliferatori dei perossisomi (Pparγ), un fattore chiave nell’accumulo di lipidi e nell’adipogenesi, bloccandone l’attività trascrizionale. Il blocco dell’attività di Pparγ ha diverse implicazioni sia nel compartimento delle cellule progenitrici adipose che in quello degli adipociti. Ad esempio, il blocco dell’attività di Pparγ mediato dagli estrogeni nelle cellule progenitrici adipose impedirebbe l’adipogenesi e ostacolerebbe il potenziale beneficio metabolico dell’iperplasia. Al contrario, la soppressione dell’attività trascrizionale di Pparγ negli adipociti maturi da parte degli estrogeni reprimerebbe la biogenesi lipidica e i geni di sensibilizzazione all’insulina. Come notato in precedenza, questi effetti degli estrogeni su Pparγ potrebbero essere specifici per il deposito e potrebbero avere ulteriori passaggi di regolazione trascrizionale che controllano i geni bersaglio di Pparg. Oltre a Pparγ, è stato dimostrato che gli estrogeni e i loro livelli reprimono direttamente la trascrizione del gene della lipoproteina lipasi (LPL) e diminuiscono l’attività della LPL attraverso modifiche post-trascrizionali . Ad esempio, le analisi cliniche dei livelli sierici di trigliceridi mostrano un aumento nelle donne in postmenopausa, ma vengono normalizzati o addirittura ridotti in risposta alla terapia ormonale sostitutiva (HRT) . Analogamente, nelle donne obese, una minore attività della LPL a digiuno è stata associata a livelli più elevati di estrogeni circolanti. È fondamentale sottolineare che l’attività della LPL è responsabile della conversione dei trigliceridi in acidi grassi liberi, consentendo l’assorbimento di acidi grassi liberi nei tessuti non epatici. Pertanto, la riduzione dell’espressione della LPL o la diminuzione della sua attività attraverso modifiche post-trascrizionali attenuerà l’assorbimento di acidi grassi liberi. Tuttavia, non è chiaro se gli estrogeni regolino l’attività e l’espressione genica della lipoproteina lipasi (LPL) in modo specifico per il tessuto adiposo bianco (WAT). In generale, sembrano mancare studi di trascrizione genetica su larga scala che esaminino il ruolo della segnalazione estrogenica nella regolazione dell’espressione genica degli adipociti sottocutanei e viscerali. Studi incentrati sulla comprensione dell’interazione tra steroidi sessuali e geni potrebbero contribuire a chiarire queste associazioni, così come studi osservazionali per descrivere le differenze nella distribuzione e nell’attività del tessuto adiposo bianco maschile e femminile.
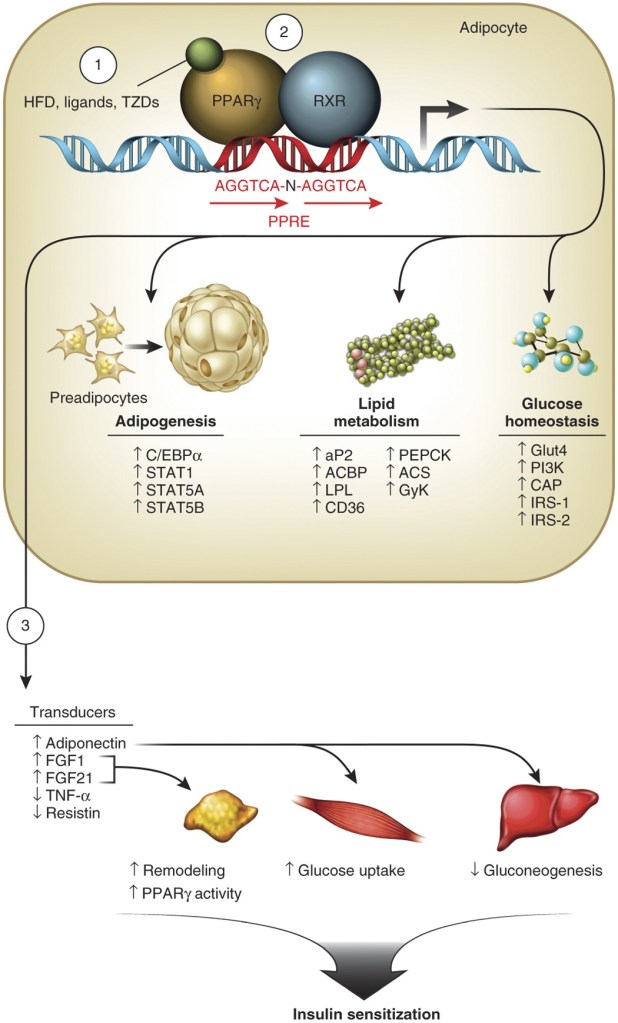
Gli estrogeni potrebbero anche facilitare i tassi di lipolisi degli adipociti. L’attivazione dell’ERα da parte degli estrogeni sembra aumentare l’espressione dei recettori adrenergici alfa2A antilipolitici solo nel tessuto adiposo bianco sottocutaneo, ma non in quello viscerale . Inoltre, l’esame della distribuzione del grasso in donne affette da sindrome dell’ovaio policistico (PCOS), una patologia in cui le ovaie producono una quantità eccessiva di androgeni, ha mostrato un’espansione preferenziale del tessuto adiposo bianco viscerale con una riduzione del tessuto adiposo bianco sottocutaneo. Si ritiene che questi cambiamenti nell’immagazzinamento e nel rimodellamento del tessuto adiposo nelle pazienti con PCOS siano dovuti ai livelli plasmatici di androgeni, che bloccano la lipolisi e stimolano la lipogenesi nei compartimenti viscerali. Inoltre, i topi ovariectomizzati trattati con estradiolo hanno mostrato risposte lipolitiche potenziate, favorendo l’ossidazione degli acidi grassi liberi e non il loro accumulo. Questo effetto lipolitico potrebbe essere attribuito ai cambiamenti nell’espressione genica indotti dagli estrogeni nel recettore nucleare dell’ormone dell’ossidazione degli acidi grassi, Pparδ, e nelle sue vie di ossidazione degli acidi grassi . Inoltre, l’ossidazione lipidica potrebbe richiedere l’attività non genomica dell’estradiolo per attivare rapidamente la proteina chinasi attivata da AMP. Tuttavia, sono necessarie ulteriori informazioni per comprendere l’intero spettro degli effetti degli estrogeni sulla lipolisi, sui tassi di lipolisi e sull’espressione genica dei geni lipolitici e lipogenici.
Poiché gli estrogeni possono regolare la crescita sottocutanea e bloccare l’espansione del tessuto adiposo viscerale, i ricercatori hanno esaminato se gli estrogeni modulano il potenziale angiogenico di vari depositi di tessuto adiposo. Infatti, le donne in postmenopausa tendono ad avere una riduzione del flusso sanguigno nel tessuto adiposo, aumentando la loro suscettibilità alle malattie metaboliche. I dati suggeriscono anche che l’attivazione dell’ER regola positivamente l’espressione genica del VEGFA. In accordo con ciò, il blocco dell’attività dell’ER ha mostrato una diminuzione dell’espressione genica del VEGFA, con conseguente ipertrofia degli adipociti, infiammazione e insulino-resistenza. È stato anche dimostrato che la segnalazione degli estrogeni regola l’angiotensinogeno nel fegato. È stato dimostrato che l’angiotensinogeno regola il flusso e la pressione sanguigna ed è spesso elevato nei pazienti obesi, aumentando l’ipertensione indotta dall’obesità. Coerentemente con questa nozione, l’angiotensinogeno è espresso e secreto dagli adipociti e aumenta all’interno degli adipociti ipertrofici. Inoltre, nei modelli murini, sembra che gli adipociti siano la principale fonte di angiotensinogeno e possano spiegare le variazioni della pressione sanguigna sistolica in risposta a una dieta ricca di grassi. Tuttavia, non è chiaro se gli estrogeni siano un mediatore dell’espressione e dell’attività dell’angiotensinogeno. È ben documentato che gli estrogeni possono regolare la biologia dei vasi sanguigni nel contesto dell’aterosclerosi e dell’ipertensione e che le donne in postmenopausa perdono la protezione da queste condizioni. Ad esempio, il tessuto adiposo perivascolare regola il tono vascolare, ma nelle donne in postmenopausa il tessuto adiposo bianco perivascolare si espande, aumentando la dissezione arteriosa. Sebbene questi indizi evidenzino che gli estrogeni regolano la biologia e il tono vascolare nel tessuto adiposo bianco e in altri sistemi di organi, sembra che i meccanismi molecolari e trascrizionali debbano ancora essere completamente determinati.
- La variabile dell’E2 sulla α2-AR:β2-AR ratio
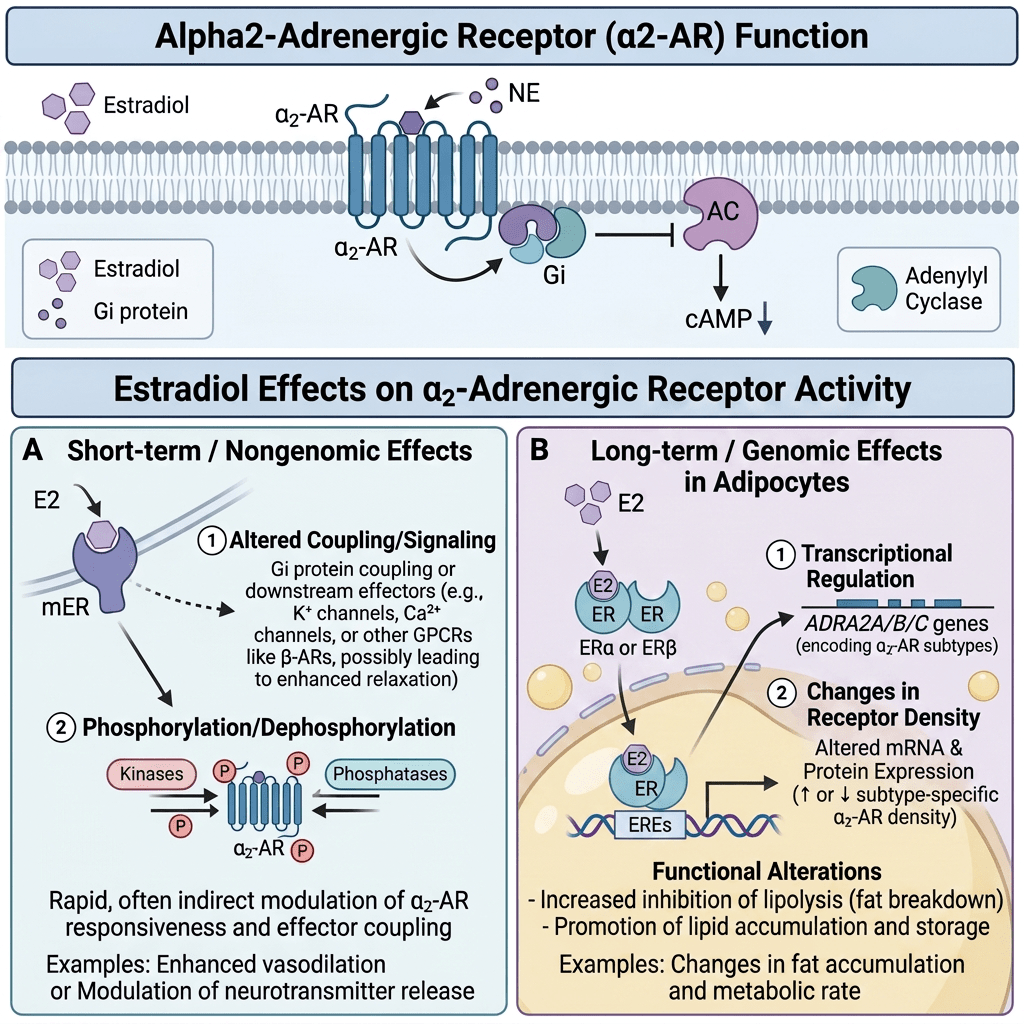
È stato dimostrato che il progesterone aumenta la sintesi dei recettori β2-AR durante la gravidanza e il numero di proteine G attivate, motivo per cui può essere combinato con gli agonisti dei recettori β2-AR nella minaccia di parto pretermine. L’espressione dei recettori α1-AR nel miometrio è influenzata dagli ormoni steroidei sessuali femminili, principalmente dagli estrogeni. Il 17β-estradiolo diminuisce l’espressione dei recettori α1A-AR, ma non influenza l’espressione dei recettori α1D-AR. Gli estrogeni svolgono un ruolo importante nell’influenza diretta sui sottotipi di recettori α2-AR, che sono anche coinvolti nel meccanismo della lipolisi.
I recettori α2-AR sono stati suddivisi in sottotipi α2A, α2B e α2C. Tutti e tre i sottotipi recettoriali sono accoppiati alla subunità α della proteina Gi sensibile alla tossina della pertosse e diminuiscono l’attività dell’adenilato ciclasi (AC) e le correnti di Ca2+ voltaggio-dipendenti, attivando allo stesso tempo le correnti di K+ dipendenti dal recettore. La stimolazione di questi recettori porta all’inibizione del feedback presinaptico del rilascio di (-)-noradrenalina sui neuroni adrenergici e media una varietà di funzioni cellulari, come la vasocostrizione, l’aumento della pressione sanguigna e la nocicezione. In determinate circostanze, i recettori α2-AR possono accoppiarsi non solo alle proteine Gi ma anche alle proteine Gs, con conseguente attivazione dell’AC.
Nel tessuto adiposo sottocutaneo (sc), gli estrogeni possono aumentare il numero di recettori α2A-AR, che sono coinvolti nell’inibizione della lipolisi. Ciò può contribuire al tipico modello di distribuzione del grasso femminile, in cui il grasso viene immagazzinato maggiormente a livello sottocutaneo piuttosto che viscerale. Inoltre, una condizione estrogenica non ben manipolata può rendere, dato questo effetto, più difficile raggiungere definizioni estremizzate.
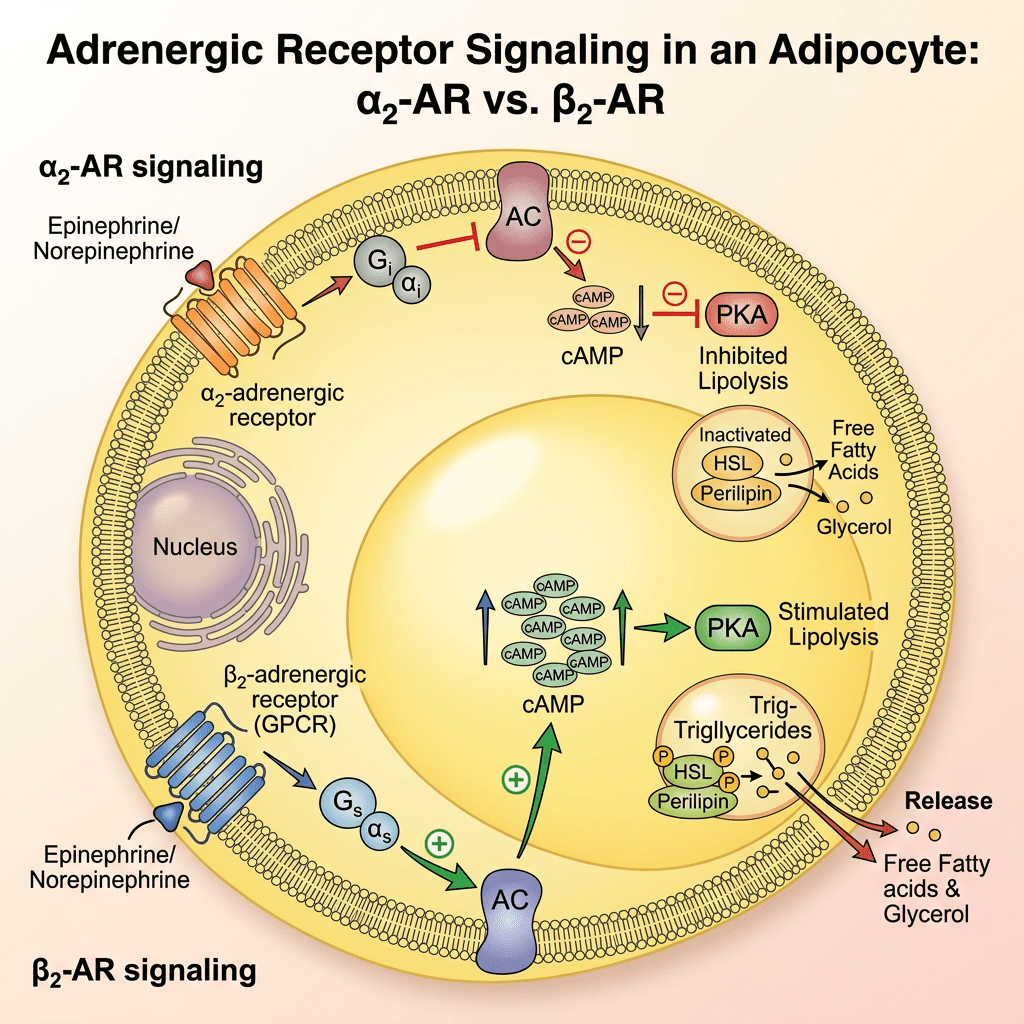
E’ stato infatti osservato che l’aumento del numero di recettori α2A-AR causa una risposta lipolitica attenuata dell’Epinefrina negli adipociti sc; al contrario, non è stato osservato alcun effetto degli estrogeni sull’espressione dell’mRNA dei recettori α2A-AR negli adipociti del deposito di grasso intra-addominale. Dai risultati ci viene mostrato che gli estrogeni abbassano la risposta lipolitica nel deposito di grasso sc aumentando il numero di recettori α2A-AR, mentre gli estrogeni non sembrano influenzare la lipolisi negli adipociti del deposito di grasso intra-addominale. Questi risultati dimostrano che gli estrogeni attenuano la risposta lipolitica attraverso la sovraregolazione del numero di recettori α2A-AR antilipolitici solo nel sc e non nei depositi di grasso viscerale. Pertanto, questi risultati offrono una spiegazione del modo in cui gli estrogeni, se non ben modulati, rendono molto più ostica la mobilitazione del grasso sc in specie nella donna.[https://pubmed.ncbi.nlm.nih.gov/15070958/]
Quindi, gli estrogeni regolano positivamente i α2A-AR direttamente nel tessuto adiposo sottocutaneo umano tramite il ERα. Questo meccanismo aumenta l’attività antilipolitica, riduce la degradazione del grasso in risposta alle catecolamine come l’Epinefrina e contribuisce a mantenere la tipica distribuzione del grasso nella parte inferiore del corpo femminile e nel tessuto adiposo sottocutaneo.
- Meccanismo d’azione
- Specificità recettoriale: Gli estrogeni agiscono attraverso il ERα per modulare il numero di recettori adrenergici [vedi ratio tra α2-AR (antilipolitico) e β2-AR (lipolitico) a favore del α2-AR].
- Specificità del deposito: L’aumento dell’espressione si verifica esclusivamente nei depositi di grasso sottocutaneo (sc) piuttosto che in quelli viscerali o intra-addominali.
- Sottoregolazione della lipolisi: Un numero maggiore di recettori α2A-AR (che inibiscono l’adenilato ciclasi) attenua l’effetto mobilizzante del grasso dell’Epinefrina negli adipociti sottocutanei.
- Implicazioni fisiologiche
- Distribuzione del grasso: Inibendo la mobilizzazione del grasso specificamente nelle aree sottocutanee (regioni gluteo-femorali), gli estrogeni favoriscono l’accumulo di grasso nei depositi della parte inferiore del corpo rispetto all’accumulo viscerale.
- Dualismo tra protezione metabolica e limitazione estetica: Questa regolazione dell’accumulo sito-specifica impedisce al grasso di accumularsi negli spazi intra-addominali, proteggendo così dai rischi metabolici associati all’obesità viscerale. Ciò nonostante, in un contesto di miglioramento della composizione corporea in specie nel BodyBuilding, questa caratteristica risulta limitante richiedendo specifiche modifiche nella preparazione.
Equilibrio dei recettori adrenergici
Le catecolamine (Adrenalina e Noradrenalina) si legano sia ai recettori β stimolatori che ai recettori α2A inibitori presenti sugli adipociti:
- Gli adipociti sottocutanei esprimono un’elevata densità di recettori α2A adrenergici, in particolare nelle donne a causa della segnalazione estrogenica. Questo agisce come un “freno” molecolare che smorza la degradazione del grasso.
- Gli adipociti viscerali esprimono un’elevata densità di recettori β₁ e β₂ e livelli inferiori di recettori α2A, rendendoli altamente sensibili alla stimolazione che induce la scissione del grasso.
Una nota sui Glucocorticoidi e Mineralocorticoidi nella donna
Le donne presentano generalmente concentrazioni basali totali più elevate di glucocorticoidi (Cortisolo) a causa del legame con le proteine, mentre le loro concentrazioni di mineralcorticoidi (Aldosterone) fluttuano ciclicamente e sono altamente sensibili alle interazioni tra ormoni sessuali. Al contrario, gli uomini mostrano concentrazioni basali più stabili, ma presentano picchi molto più netti e più elevati nei livelli di glucocorticoidi quando sottoposti a stress acuto.
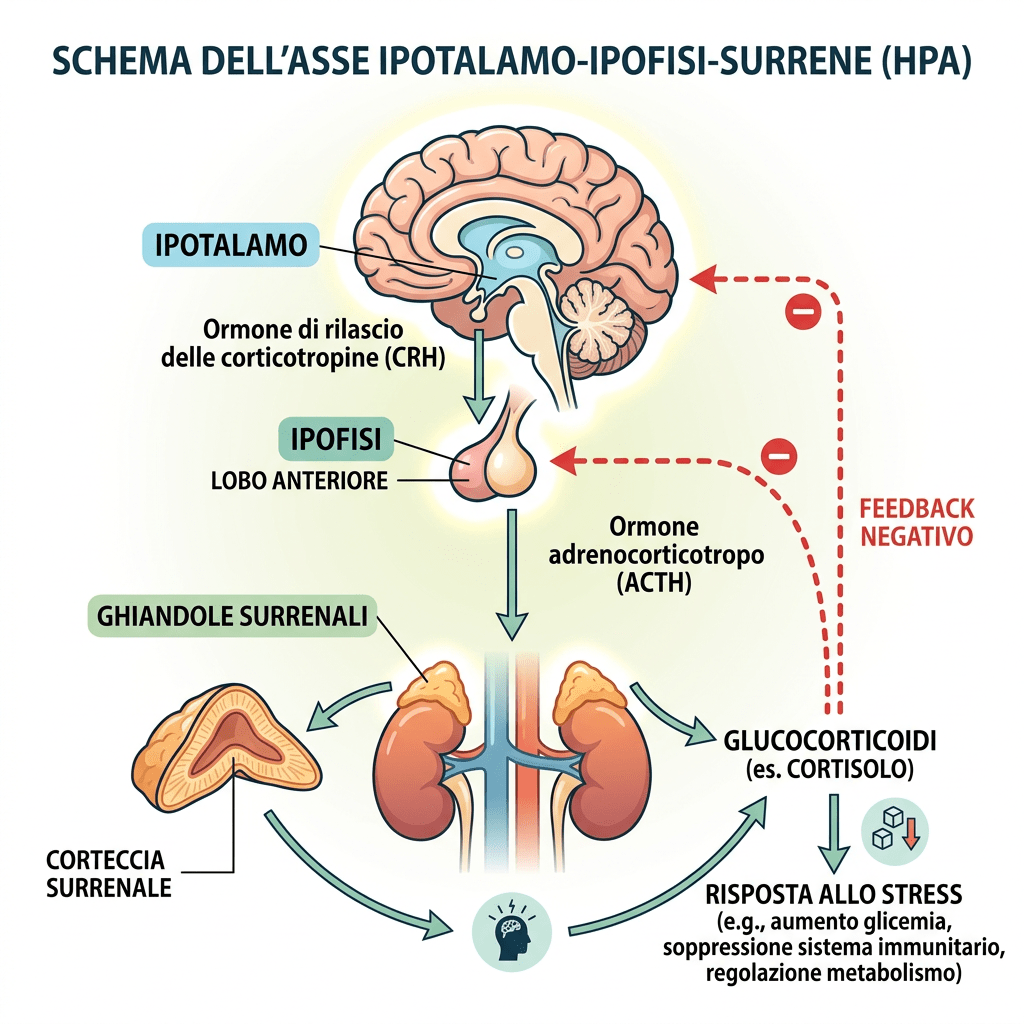
- Concentrazioni di glucocorticoidi (Cortisolo)
Sebbene entrambi i sessi mantengano il cortisolo entro un intervallo fisiologico ampio e simile (da 5 a 25 mcg/dL al mattino), le dinamiche di concentrazione e i meccanismi di regolazione sottostanti differiscono:
- Cortisolo totale vs. libero: le donne presentano livelli di cortisolo circolante totale più elevati principalmente perché gli estrogeni stimolano la produzione di globulina legante il cortisolo (CBG) nel fegato. Tuttavia, la concentrazione di cortisolo “libero” (biologicamente attivo) rimane altamente comparabile tra uomini e donne sani.
- Reattività allo stress acuto: quando esposti a stress psicologico o fisico acuto, gli uomini mostrano un picco di concentrazione di glucocorticoidi significativamente più forte e più rapido. Le donne mostrano una risposta acuta del cortisolo più attenuata, tranne durante la fase luteale del ciclo mestruale, quando la loro risposta allo stress rispecchia da vicino quella degli uomini.
- Ritmo circadiano: entrambi i sessi seguono un ritmo circadiano preciso, ma le donne mostrano frequentemente una risposta del cortisolo al risveglio (CAR) leggermente più pronunciata, ovvero un picco mattutino più ripido subito dopo il risveglio.
- Concentrazioni di mineralcorticoidi (Aldosterone)
Le concentrazioni basali di mineralcorticoidi sono fortemente dimorfiche a causa dell’impatto diretto delle fluttuazioni ormonali sul sistema renina-angiotensina-aldosterone (RAAS):
- Variabilità ciclica vs. stabilità: Gli uomini mantengono concentrazioni basali di mineralcorticoidi molto stabili giorno per giorno. Le donne, invece, sperimentano enormi fluttuazioni a seconda del ciclo mestruale. Durante la fase luteale, le concentrazioni di aldosterone raddoppiano o triplicano perché il progesterone blocca i recettori dei mineralcorticoidi, costringendo l’organismo a produrre più aldosterone per mantenere l’equilibrio del sodio.
- Selettività e sensibilità tissutale: Gli uomini mostrano naturalmente una maggiore sensibilità alle variazioni della pressione sanguigna indotte dal sale. Le donne beneficiano di meccanismi estrogenici protettivi che limitano l’iperattivazione dei recettori dei mineralcorticoidi nel sistema cardiovascolare, il che significa che una concentrazione simile di aldosterone causa meno danni vascolari nelle donne rispetto agli uomini.
- Differenze a livello di recettori e meccanismi di feedback
Il modo in cui queste concentrazioni influenzano l’organismo è determinato dall’espressione dei recettori nel cervello e negli organi. Gli uomini presentano una concentrazione basale più elevata di recettori dei glucocorticoidi (GR) e recettori dei mineralcorticoidi (MR) in aree come l’ippocampo. Di conseguenza, gli uomini possiedono un meccanismo di feedback negativo altamente efficiente che interrompe rapidamente la risposta allo stress. Le donne presentano un sistema di feedback più graduale, il che significa che, sebbene le loro concentrazioni di ormoni dello stress non raggiungano picchi così elevati, il ritorno alla normalità richiede più tempo.
- Appetito e chimica cerebrale: fattori scatenanti comportamentali
Lo stress cronico altera le vie neurali che regolano la fame in modo diverso nei due sessi:
- Donne (Iperfagia edonica): L’aumento del Cortisolo interagisce con gli estrogeni alterando profondamente i centri cerebrali che regolano l’appetito. Le donne stressate producono livelli più elevati dell’ormone della fame Grelina e mostrano un declino più rapido dell’attività della corteccia prefrontale (il centro di controllo degli impulsi del cervello). I dati epidemiologici di uno studio mostrano che le donne sperimentano aumenti significativamente maggiori dell’indice di massa corporea (BMI) in seguito a importanti eventi stressanti rispetto agli uomini. Sperimentano un’intensa voglia di cibi iper-appetitosi e ad alta densità calorica (zucchero e grassi).
- Uomini (Strategie di coping basate su sostanze): Gli uomini stressati sono biologicamente meno inclini all’alimentazione compulsiva indotta dallo stress. Al contrario, come evidenziato dalla ricerca sui meccanismi di coping, gli uomini sono più propensi a ricorrere all’alcol o al fumo per far fronte alla pressione cronica. Questo introduce calorie liquide “vuote” che aggirano i normali segnali di sazietà e accelerano ulteriormente l’accumulo di grasso viscerale nel fegato e nell’addome.
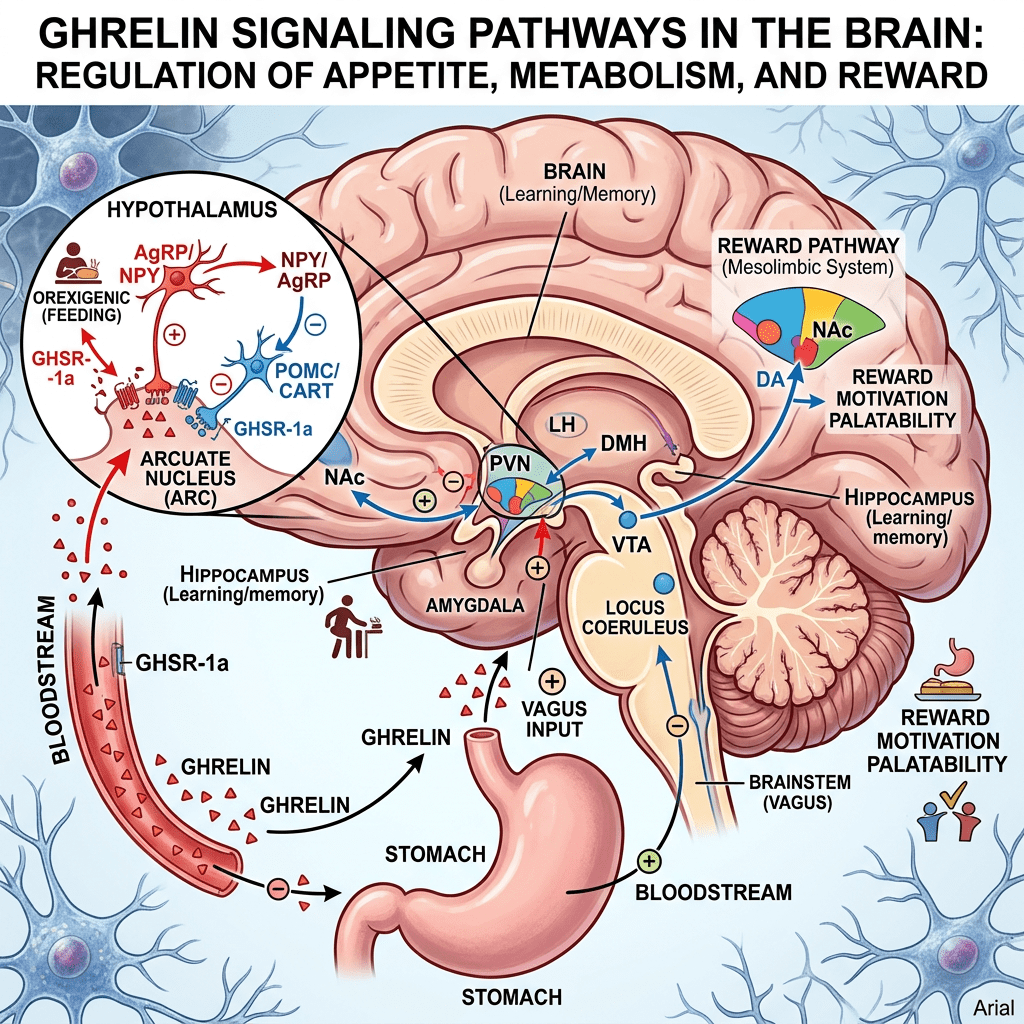
- Il paradosso del RAAS nella donna in età fertile
Nelle donne in età fertile si verifica un fenomeno unico: l’attività del RAAS è naturalmente più alta durante la fase luteale del ciclo mestruale, ma senza causare ipertensione.
- Il ruolo del progesterone: Durante la fase luteale, i livelli di progesterone si impennano. Il progesterone è un potente antagonista competitivo dei recettori dei mineralocorticoidi (MR); in parole semplici, blocca i recettori dell’aldosterone.
- La risposta compensatoria: Per evitare una massiva perdita di sodio e una conseguente disidratazione, il corpo della donna risponde attivando fortemente il RAAS. La produzione di renina e aldosterone raddoppia o triplica per superare il blocco del progesterone.
- La protezione degli estrogeni: Nonostante l’elevata concentrazione di componenti del RAAS in questa fase, gli estrogeni stimolano la produzione di ossido nitrico e promuovono la vasodilatazione, proteggendo i vasi sanguigni e impedendo l’aumento della pressione arteriosa.
Il vero rischio clinico legato a un aumento dell’attività del RAAS nella donna si manifesta dopo la menopausa.
- Perdita del freno estrogenico: Nonostante l’E2 aumenti l’espressione del Angiotensiogeno epatico, con il calo degli estrogeni in menopausa, svanisce l’effetto vasodilatatore protettivo. L’angiotensina II inizia a stringere i vasi in modo incontrollato.
- Spostamento verso il recettore AT1: Il deficit di estrogeni sposta l’equilibrio del RAAS verso l’attivazione dei recettori AT1 (che promuovono infiammazione, vasocostrizione e fibrosi) a discapito dei recettori AT2 (protettivi).
- Conseguenze cliniche: Questo cambiamento molecolare spiega perché, dopo la menopausa, le donne mostrano una suscettibilità accelerata all’ipertensione sensibile al sale, all’ipertrofia cardiaca e alla rigidità arteriosa rispetto agli uomini della stessa età.
Sintesi sul dimorfismo sessuale ormonale-metabolico
Le differenze ormonali e metaboliche tra uomo e donna costituiscono il fondamento fisiologico della dimorfia sessuale, influenzando la composizione corporea, l’utilizzo dei substrati energetici, la risposta all’esercizio e la suscettibilità a diverse condizioni patologiche e pratiche cliniche o PEDs.
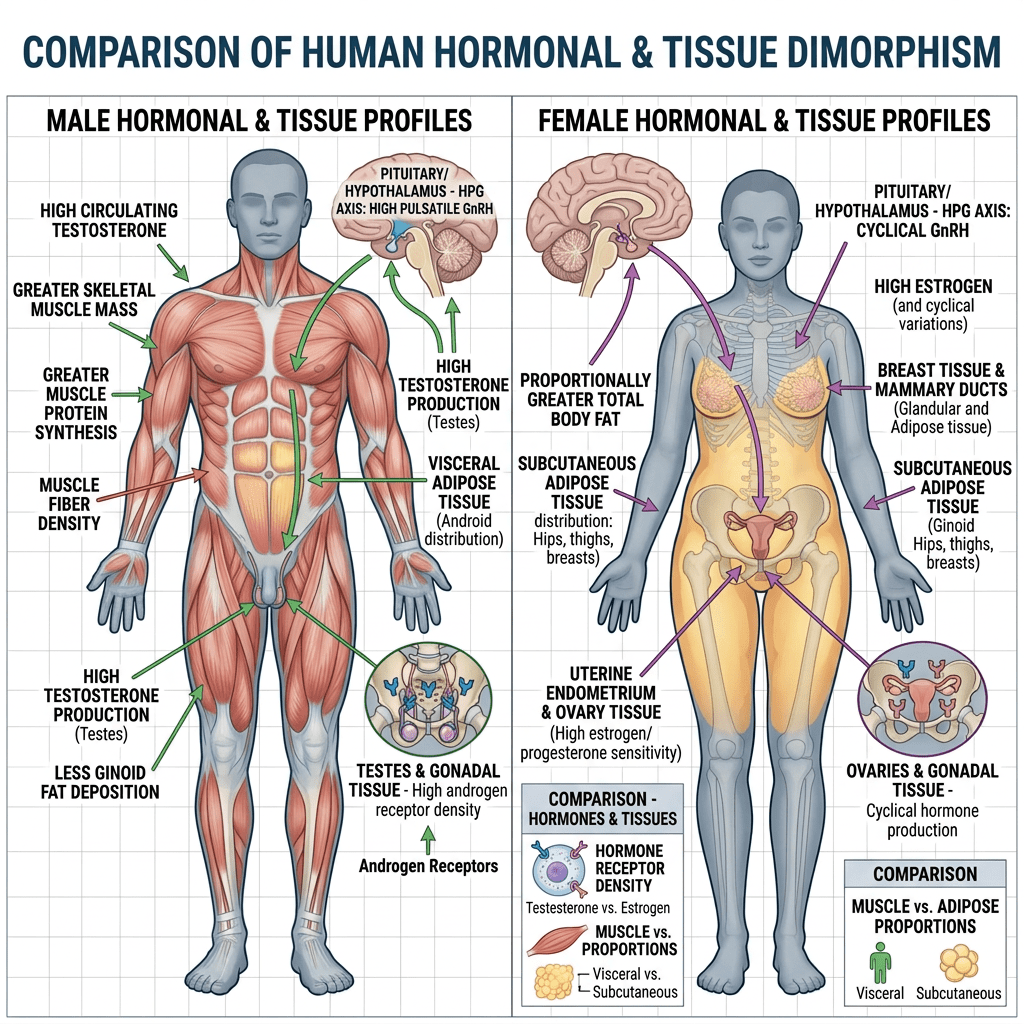
- Profilo Ormonale
Le principali differenze endocrine risiedono nei livelli basali degli steroidi sessuali e nel loro pattern di secrezione.
- Androgeni vs. Estrogeni e Progesterone Testosterone (Uomo): Prodotto principalmente dalle cellule di Leydig nei testicoli (95%) e dalle ghiandole surrenali. I livelli plasmatici fisiologici nell’uomo adulto variano indicativamente tra 300 e 1000 ng/dL. Il testosterone stimola la sintesi proteica muscolare via recettore AR, la densità ossea, la lipolisi e la produzione di eritropoietina (EPO).
- Estrogeni (E2 / Estradiolo) e Progesterone (Donna): Nelle donne in età fertile, i livelli di estradiolo oscillano ciclicamente tra 30 e 400 pg/mL a seconda della fase del ciclo mestruale (follicolare vs. luteale/ovulatoria). Gli estrogeni hanno un ruolo protettivo sul sistema cardiovascolare, modulano la sensibilità insulinica, migliorano la salute ossea e favoriscono l’accumulo di grasso periferico/ginoide.
- Pattern di Secrezione: Nell’uomo la secrezione dell’asse HHG (ipotalamo-ipofisi-gonadi) segue un ritmo circadiano (con picco nelle prime ore del mattino). Nella donna in età fertile segue un ritmo infradiano (ciclo mestruale di ~28 giorni) con marcate fluttuazioni nella sensibilità ai nutrienti e nell’idratazione corporea.
2. Distribuzione del Grasso:
- Uomo (Androide): Predominanza di accumulo viscerale e addominale. Il grasso viscerale è più lipoliticamente attivo e pro-infiammatorio.
- Donna (Ginoide): Predominanza di accumulo sottocutaneo su fianchi e glutei/cosce, guidato dagli estrogeni e dall’attività della lipoproteina lipasi (LPL) gluteo-femorale.
3. Ossidazione dei Nutrienti durante l’Esercizio
- Ossidazione dei Lipidi (Donna): Durante l’esercizio aerobico a pari intensità relativa (%VO2max), le donne ossidano una quota maggiore di grassi rispetto ai carboidrati. L’estradiolo attiva vie di segnalazione (es. AMPK) che promuovono l’ingresso degli acidi grassi nei mitocondri.
- Risparmio del Glicogeno (Donna): Questo maggior ricorso ai lipidi permette alla donna di risparmiare il glicogeno muscolare ed epatico, conferendo una maggiore resistenza alla fatica nei lavori di lunga durata/ultra-endurance.
- Ossidazione dei Carboidrati (Uomo): Gli uomini dipendono in misura maggiore dai carboidrati (glicogenolisi e glicolisi) durante il lavoro submassimale e massimale. Sensibilità Insulinica e Metabolismo del Glucosio
- Donne: Presentano una sensibilità insulinica muscolare mediamente superiore nell’arco della vita (fino alla menopause), in parte grazie all’azione protettiva degli estrogeni e alla maggiore densità capillare per unità di massa muscolare.
- Uomini: Tendono a sviluppare più facilmente insulino-resistenza sistemica in presenza di surplus calorico, a causa del rapido accumulo di tessuto adiposo viscerale e intraepatico.
La transizione verso la menopausa e la sua fase conclamata rappresentano un reset neuroendocrino e metabolico profondo. Con la cessazione della funzione ovarica follicolare, la brusca caduta degli estrogeni e del progesterone — unita a una complessa riorganizzazione della sensibilità recettoriale — altera radicalmente l’omeostasi glucidica, lipidica e la composizione corporea.
4. Il Quadro Endocrino e il Declino degli Steroidi Sessuali
Con la menopausa, l’asse ipotalamo-ipofisi-gonadi (HPG) subisce una drastica de-soppressione per assenza del feedback negativo indotto dagli steroidi ovarici:
- Estradiolo (E2): Crolla da livelli ciclici (fino a 300-400pg/mL) a valori costantemente bassi (<10-20pg/mL). L’Estrone (E1), derivante dalla conversione periferica dell’Androstenedione da parte dell’aromatasi nel tessuto adiposo, diventa l’estrogeno circolante dominante, pur essendo un agonista recettoriale nettamente meno potente di E2.
- Progesterone: Scompare quasi completamente a causa dell’assenza del corpo luteo e dell’ovulazione.
5. Androgeni (Testosterone e DHEA):
- Il Testosterone varia in modo meno brusco rispetto agli estrogeni: l’ovaio in menopausa continua a produrne piccole quote guidate dalle alte concentrazioni di LH, unitamente alla produzione surrenalica. Ciò genera un iperandrogenismo relativo (aumento del rapporto Testosterone/Estradiolo libero).
- SHBG (Sex Hormone-Binding Globulin): La riduzione della sintesi epatica di SHBG — indotta dal deficit estrogenico — aumenta la frazione libera e biologicamente attiva sia degli estrogeni residui sia degli androgeni.
6. Sensibilità Recettoriale e Rimodellamento Metabolico
A. L’impatto della carenza di steroidi sessuali si esplica attraverso la perdita della segnalazione dei recettori estrogenici ERα e ERβ nei tessuti metabolicamente attivi (muscolo scheletrico, fegato, tessuto adiposo e sistema vascolare).
B. Shift della Distribuzione Adipocitaria: Da Ginoide ad Androide Inattivazione della LPL Gluteo-Femorale: Gli estrogeni mantengono attiva la lipoproteina lipasi (LPL) nel distretto sottocutaneo inferiore. Senza E2, la LPL sottocutanea riduce la propria attività, mentre aumenta il deposito lipidico nel distretto viscerale (addominale). Infiammazione di Basso Grado: Il tessuto adiposo viscerale espanso recluta macrofagi M1 pro-infiammatori e secerne citochine TNF-alpha, IL-6, favorendo uno stato flogistico cronico che peggiora ulteriormente l’insulino-resistenza sistemica.
C. Profilo Lipidico e Salute Cardiovascolare Down-regulation dei Recettori LDL Epatici: La mancanza di estrogeni riduce l’espressione dei recettori per le LDL a livello epatico, portando a un incremento significativo del colesterolo LDL e dei trigliceridi ematici, unito a una contrazione o alterazione funzionale delle HDL. Tono Vascolare: Viene meno la stimolazione della sintesi di ossido nitrico endoteliale (eNOS) mediate da ERα, aumentando la rigidità arteriosa e la pressione arteriosa sistemica.
7. Risposta agli Steroidi Sessuali Esogeni
La sensibilità del tessuto bersaglio alla somministrazione esogena di steroidi cambia significativamente in relazione alla tempistica di avvio del trattamento e al contesto recettoriale.
- Terapia Ormonale Sostitutiva (TOS / HRT) La “Finestra d’Opportunità” (Timing Hypothesis): Se la TOS (a base di E2 bioidentico, affiancato da progesterone/progestinico a protezione endometriale nelle donne non isterectomizzate) viene avviata precocemente nella perimenopausa o nei primi anni di menopausa (<60 anni o <10 anni dalla menopausa), ripristina la sensibilità insulinica, previene l’accumulo di grasso viscerale, preserva la densità minerale ossea (BMD) e protegge la funzione endoteliale.
- Somministrazione Tardiva: L’avvio della TOS a distanza di molti anni dall’avvenuta menopausa trova vasi e recettori in uno stato indurito/pro-infiammatorio, riducendo i benefici metabolici e aumentando il rischio tromboembolico e cardiovascolare.
- Via di Somministrazione: La via transdermica per l’Estradiolo evita il primo passaggio epatico, non altera i fattori della coagulazione e la SHBG, e garantisce una risposta metabolica e un profilo di sicurezza sovrapponibili alla fisiologia.
8. Sensibilità agli Androgeni Esogeni
- In caso di deficit sintomatico di libido e affaticamento marcato, micro-dosi di Testosterone (sotto stretta supervisione medica) mostrano un’elevata sensibilità recettoriale nel tessuto osseo, muscolare e cerebrale. Tuttavia, la risposta della donna in menopausa agli androgeni è estremamente sensibile alle dosi: nonostante non sembri manifestarsi come nel periodo pre-menopausa, sottili sovradossaggi portano rapidamente a segni di virilizzazione, riduzione delle HDL e ritenzione idrica, data la differente densità/sensibilità recettoriale presumibilmente correlata al volume di distribuzione ridotto rispetto all’uomo.
Fine Prima Parte…
Gabriel Bellizzi [CEO BioGenTech]
Riferimenti:
- https://pmc.ncbi.nlm.nih.gov/articles/PMC9635380/
- Butenandt, Adolf; Westphal, Ulrich; Cobler, Heinz (12 September 1934). “Über einen Abbau des Stigmasterins zu corpus-luteum-wirksamen Stoffen; ein Beitrag zur Konstitution des Corpusluteum-Hormons (Vorläuf. Mitteil.)”. Berichte der Deutschen Chemischen Gesellschaft (A and B Series). 67 (9): 1611–1616.
- Henderson E, Weinberg M, Wright WA (April 1950). “Pregnenolone”. J. Clin. Endocrinol. Metab. 10 (4): 455–74.
- “The Quest Diagnostics Manual. Endocrinology. Test Selection and Interpretation. Fourth Edition”. docplayer.net. p. 152. Retrieved 15 September 2023.