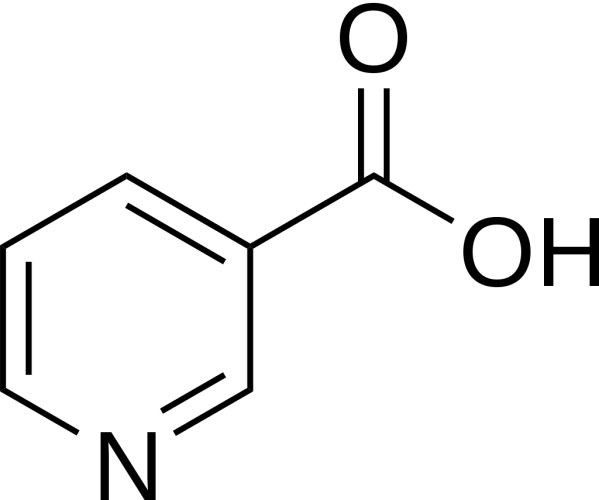
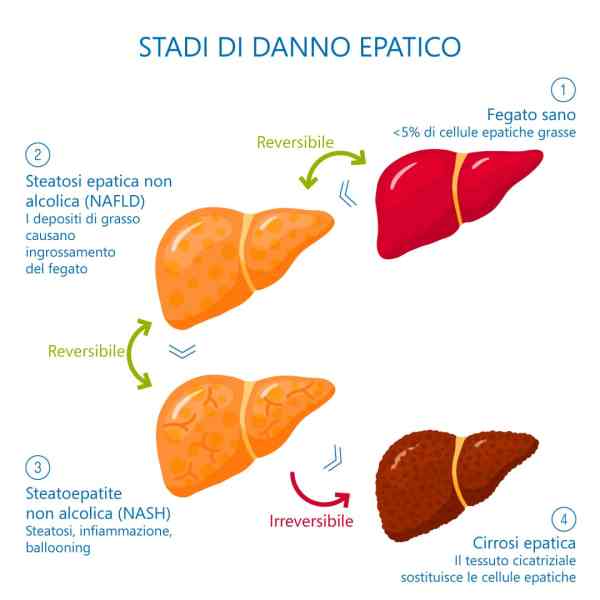
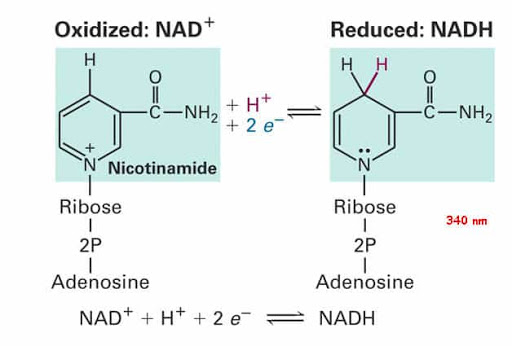
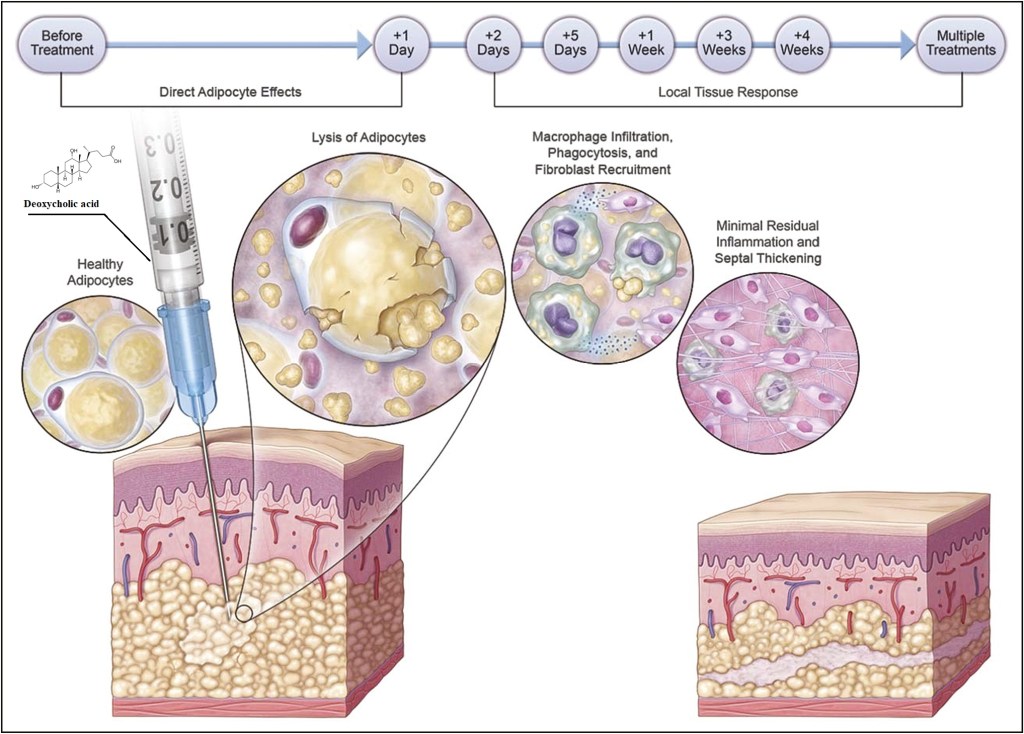
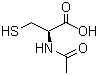
Il fegato è un organo importante ed è vitale per la sopravvivenza del soggetto. È responsabile di diverse e importanti funzioni nel corpo umano. Produce acidi biliari e proteine plasmatiche, immagazzina glicogeno e produce glucosio attraverso la gluconeogenesi, gioca un ruolo nel sistema immunitario, metabolizza un numero elevato di molecole, ecc. Quindi, si, avete capito bene: è importante. Quando qualcosa risulta dannosa per il fegato, essa si indica come epatotossico (dal greco hêpar-atos, fegato). Un chiaro esempio è l’alcol. Gli alcolisti tendono a sviluppare una malattia del fegato a un certo punto della loro vita. Tuttavia, molti farmaci da prescrizione, o anche over-the-counter, possono essere epatotossici, come l’Acetaminofene. E, come è ben dimostrato, anche gli AAS possono essere epatotossici, anche se specifici. Come sembra, solo quelli con una specifica alterazione chimica sembrano essere maggiormente epatotossici – in particolare, quelli che presentano una metilazione in pozione C-17α.
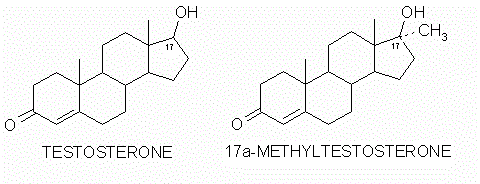
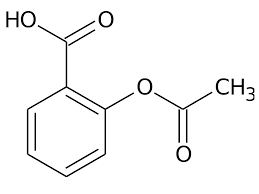
Modifica della struttura carbossilica del Testosterone (sinistra) in posizione C-17α (destra).
In questo articolo tratterò principalmente ciò che sembra causare questa epatotossicità indotta da AAS. L’effetto epatotossico può essere riscontrato attraverso l’osservazione dei cambiamenti nei marcatori ematici del danno epatico, come Alanina Transaminasi (ALAT), Aspartato Transaminasi (ASAT), γ-glutamiltransferasi (GGT) e la Fosfatasi Alcalina (ALP). Una nota di cautela deve essere presa in considerazione quando si interpretano gli aumenti di ALAT e ASAT, poiché entrambi aumenteranno anche a causa del intyenso lavoro muscolare [1]. È bene sapere che in questi casi, ASAT sarà di solito più alto del ALAT, mantenendo un rapporto ASAT/ALAT superiore a 1. Quindi, quando questi aumentano con un rapporto inferiore a 1, si può essere più sicuri che il danno muscolare non è il colpevole dell’alterazione. Idealmente, nessun esercizio (contro-resistenza) viene svolto 1-2 settimane prima dell’esame del sangue per escludere il danno muscolare muscolare come causa dell’innalzamento, sebbene ciò dipenda anche dall’intensità del allenamento. In rari casi, il danno al fegato potrebbe avanzare clinicamente fino allo sviluppo di ittero colestatico [2]. In questo caso, un prodotto della degradazione dei globuli rossi (bilirubina) si accumula nel corpo. L’ittero può essere osservato visivamente (tono giallo della pelle e della sclera degli occhi), e si possono sviluppare sintomi come nausea, vomito, dolore allo stomaco e prurito. Inoltre, alcuni rari casi di peliosis hepatis (Peliosi Epatica) sono stati segnalati verificarsi come risultato dell’uso di AAS orali ad alte dosi [3]. Questa è una condizione nella quale si vengono a formare cisti piene di sangue nel fegato. La sospensione dell’AAS in questione è solitamente sufficiente e porterà alla scomparsa di queste caratteristiche cliniche entro pochi mesi. In casi più gravi, tuttavia, potrebbero richiedere un intervento chirurgico. Infine, alcuni casi in letteratura hanno riportato un’associazione tra uso di AAS e carcinoma epatico [4] e adenoma [5].
Ho già trattato in passato tale problematica legata all’uso di AAS, ma questa volta voglio trattare la questione più nello specifico, analizzando le due ipotesi che ruotano intorno all’epatotossicità AAS-dipendente: “ipotesi dello stress ossidativo” e “ipotesi di coniugazione dell’anello D”.
L’ipotesi dello stress ossidativo:
L’ipotesi dello stress ossidativo che tratterò qui si basa su un documento che William Llewellyn, Peter Van Mol e Peter Bond hanno pubblicato [6]. Lo stress ossidativo è qualcosa che si pensa possa risultare nell’epatotossicità osservata con l’uso di AAS, e se l’ipotesi è vera, dà qualche opportunità per contrastarla in modo migliore. Quindi, cominciamo con spiegare quello che è lo stress ossidativo. Lo stress ossidativo è descritto da Helmut Sies come un disturbo nell’equilibrio pro-ossidante-antiossidante a favore del primo [7], che si riduce a molecole contenenti ossigeno, che sono altamente reattive (specie reattive dell’ossigeno [ROS]), sopraffacendo il sistema antiossidante. Poiché le ROS sono così altamente reattive, possono reagire con molecole come lipidi, proteine, carboidrati e acidi nucleici (elementi costitutivi del DNA). Quando si dice “reagire con queste molecole”, si intende che danneggia queste molecole (estremamente semplificato, ma è sufficiente per far comprendere il processo). Questi ROS provengono da varie reazioni catalizzate da enzimi come la respirazione cellulare (l’ossidazione dei macronutrienti per fornire energia), altri processi metabolici e radiazioni. La fonte primaria di ROS all’interno di una cellula sono i mitocondri, il che non è sorprendente dato che i mitocondri sono le “centrali energetiche” della cellula. È il posto nella cellula dove i carboidrati alimentari, gli acidi grassi e le proteine (o, meglio, gli amminoacidi che le compongono) finiscono per essere ossidate per produrre energia in un processo chiamato fosforilazione ossidativa. Come suggerisce il nome, la fosforilazione ossidativa ossida e richiede ossigeno per farlo. Questo processo, tuttavia, non è perfetto. Per non complicare troppo le cose al lettore, non mi addentrerò nelle complessità delle reazioni chimiche, ma fondamentalmente, questo processo può produrre ROS come sottoprodotto (superossido in particolare). Le cellule del corpo sono dotate di meccanismi per tenere a bada questi ROS generati (la parte antiossidante dell’equazione). In circostanze normali questo porta ad un sottile equilibrio tra i due. Avere qualche ROS qua e là nelle cellule è normale. Essi giocano un ruolo essenziale nel normale funzionamento di vari processi vitali [8]. Tuttavia, il problema nasce quando questo equilibrio si altera a favore della parte proossidante dell’equazione: lo stress ossidativo. Questo è il momento in cui i ROS prendono il sopravvento, per così dire, e possono iniziare a creare il caos nella cellula. Quanto sopra è un quadro un po’ troppo semplificato. Ci sono diversi tipi di ROS (radicali liberi e non radicali). Ciò che conta è dove si trovano questi ROS nella cellula e come evolvono nel tempo. Inoltre, questo interagisce con il sistema antiossidativo delle cellule, il che complica ulteriormente il quadro. Ma credo che quanto sopra sia sufficiente per dare una buona comprensione di tutto questo. Ciò che conta è che l’epatotossicità indotta da AAS è stata ripetutamente dimostrata essere associata allo stress ossidativo nelle cellule epatiche (fegato) di modelli animali [9]. Questo fa sorgere la domanda: è solo un’associazione, o c’è una relazione causale con l’epatotossicità indotta da AAS? Dopo aver scavato nella letteratura, sono emersi alcuni studi che sembrano sostenere una relazione causale. Uno studio svolto su un carcinoma prostatico umano epiteliale (22Rv1) ha collegato l’attivazione del recettore degli androgeni (AR) a un aumento dei ROS basali [10]. Più tardi, lo stesso gruppo ha pubblicato una ricerca applicando un disegno di studio simile. Questo studio ha confermato i precedenti risultati e ha anche dimostrato che l’aumento dei ROS è dovuto a un aumento indotto dall’AAS nella β-ossidazione mitocondriale degli acidi grassi [11]. Quindi, l’attivazione di l’AR porta a una maggiore ossidazione degli acidi grassi nei mitocondri, con conseguente maggiore produzione di ROS come sottoprodotto. Da notare che questo studio ha anche trovato un aumento dell’mRNA della carnitina palmitoiltransferasi (CPT1). Tutto quello che dovete sapere è che la CPT1 è considerata essere l’enzima che regola la velocità nel processo di ossidazione mitocondriale degli acidi grassi. Quindi, se si aumenta la CPT1, si aumenta l’ossidazione mitocondriale degli acidi grassi. Ora, le cellule del cancro alla prostata non sono cellule del fegato, ovviamente. Ma ciò che è interessante è che l’AAS 17α-alchilato Fluoxymesterone e Metilandrostanolone hanno dimostrato di aumentare l’attività del CPT1 nel fegato di ratto [12]. Inoltre, se si guardano agli epatociti di ratto (cellule epatiche) trattati con AAS 17α-alchilati, si vedrà il gonfiore dei mitocondri e solo cristae leggermente definite [13]. (Le criste sono quelle pieghe caratteristiche della membrana interna dei mitocondri). Infatti, la produzione di ROS è una causa nota di gonfiore mitocondriale, e il gonfiore è un fattore importante che porta alla successiva morte cellulare [14]. Quindi, apparentemente, suggerisce un potenziale ruolo dello stress ossidativo. Questo non vuol dire che qualsiasi aumento nella produzione di energia di una cellula sia negativo. Usando i muscoli aumenta anche la produzione di energia nelle cellule muscolari. Di conseguenza, più ROS vengono prodotti anche in queste cellule. In contrasto con l’aumento di ROS indotto dall’AAS nelle cellule del fegato, questi aumenti sono transitori invece che continui. Inoltre, le cellule muscolari differiscono nei loro meccanismi antiossidanti per gestire questa condizione. Quindi, normalmente, questo non è assolutamente un problema. Tuttavia, l’esercizio intenso e prolungato può anche provocare danni ossidativi alle molecole delle cellule muscolari [15].
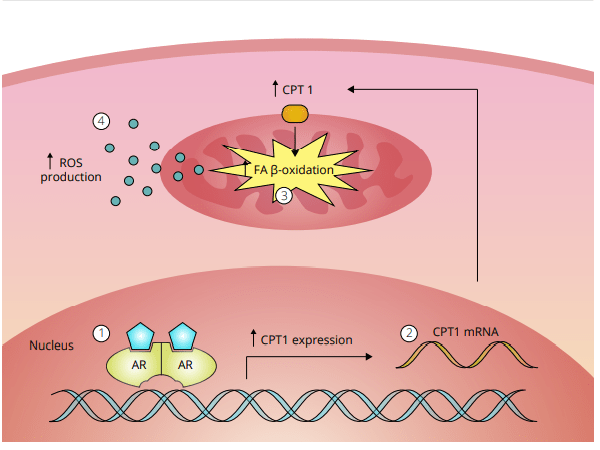
L’ipotesi dello stress ossidativo nella epatotossicità indotta da AAS come descritto da Bond et al. [49]. 1 Un androgeno si lega a, e attiva, il recettore degli androgeni (AR) nelle cellule epatiche. Questo porta a 2 la sovra-regolazione della Carnitina Palmitoiltransferasi 1 (CPT1), l’enzima che regola il tasso di β-ossidazione degli acidi grassi (FA). Si pensa che questo porti a 3 un aumento della β-ossidazione degli acidi grassi nei mitocondri. Di conseguenza, 4 la produzione di specie reattive dell’ossigeno (ROS) è aumentata. L’aumento dei ROS poi danneggia i mitocondri, il che sembra essere alla base dell’epatotossicità indotta dall’AAS.
Ora, se si integrassero gli antiossidanti (mitocondriali), si allevierebbe questo danno? Può darsi. Mentre non c’è un trial di buona qualità che valuti questo, uno studio osservazionale su 320 atleti dimostra qualcosa del genere [16]. In breve, gli utilizzatori di AAS che hanno preso un supplemento contenente alcuni composti antiossidanti non ha mostrato alcun aumento dei marcatori di danno epatico dopo il ciclo rispetto a quelli che non hanno assunto quel supplemento. Ancora una volta, questo sarebbe in linea con lo stress ossidativo che gioca un ruolo causale nell’epatotossicità indotta da AAS. Infine, sembra che l’epatotossicità indotta da AAS potrebbe essere legata all’attivazione del AR nelle cellule epatiche. In un vecchio studio del 1964, Marquardt et al. non sono riusciti a dimostrare che l’AAS non 17α-alchilato produce test di funzionalità epatica anormali [17]. Infatti, gli AAS 17α-alchilati mostrano segni di epatotossicità in diversi studi, mentre non si vede questo con AAS non-17αalchilati, nemmeno con un alto dosaggio di 600 mg di Testosterone Enantato settimanale [18]. La 17α-alchilazione sembra quasi necessaria per rendere epatotossico un AAS, probabilmente perché è l’unica alterazione che lo rende sufficientemente biodisponibile per via orale. E, di conseguenza, porta ad alte concentrazioni del composto nel fegato. Ma possiamo individuare le differenze tra i vari AAS 17α-alchilati che riguardano la loro capacità di attivare l’AR? Certamente sembra così. In generale, sembra che sia vero quanto segue:
Epatotossicità = resistenza alla decomposizione epatica×potenza di attivazione del AR
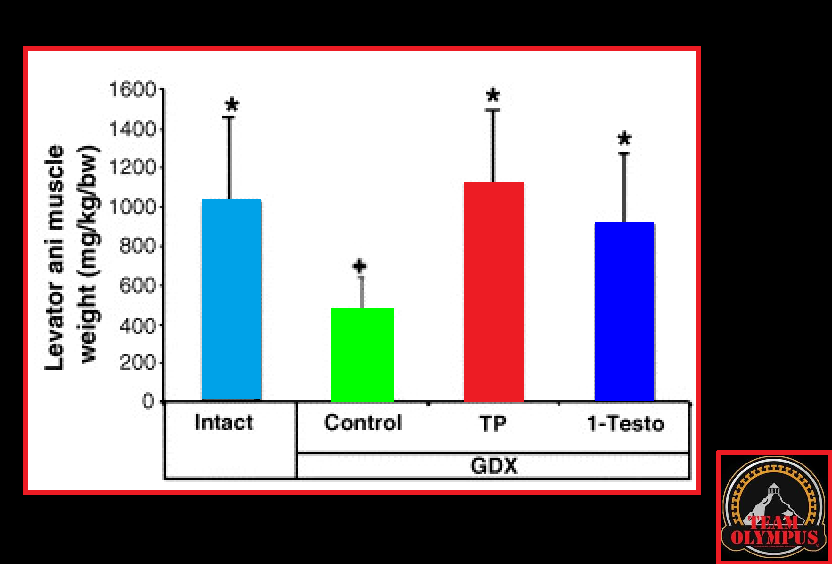
Quindi, facciamo un esempio. Il Methyltrienolone (R1881) ha un’affinità molto alta per l’AR, ha un’alta potenza per la transattivazione dell’AR [19], ed è fortemente resistente al metabolismo epatico. Come tale, è un composto ideale per un saggio dei siti di legame agli androgeni [20]. Infatti, un studio clinico che impiega un basso dosaggio dello steroide (≤1 mg al giorno) ha dimostrato un significativo aumento dei marcatori di danno epatico entro due settimane [21]. Gli autori lo hanno definito “(…) attualmente lo steroide più epatotossico”. Lo steroide 17α-alchilato meno epatotossico è solitamente considerato l’Oxandrolone. Anche con alti dosaggi fino a 80mg al giorno, mostra solo deboli segni di epatotossicità [22]. Mentre lo steroide è abbastanza resistente al metabolismo epatico [23], ha una bassa affinità per il AR [23]. La sua potenza relativa in termini di transattivazione AR è anche quasi 100 volte inferiore a quella del Methyltrienolone [19]. Allo stesso modo, anche l’Oxymetholone ha una bassa affinità per l’AR [23] e la sua potenza in termini di transattivazione AR è molto simile a quella dell’Oxandrolone [19]. Non sorprende che mostri segni di epatotossicità solo in una minoranza di pazienti, nonostante gli alti dosaggi (100-150 mg al giorno) [24].
L’ipotesi di coniugazione dell’anello D:
Avete mai sfogliato il libro Doping in Sports di Thieme e Hemmersbach? [25] In questo libro gli autori notano che non c’è correlazione tra la tossicità epatica e gli effetti farmacologici primari (cioè gli effetti anabolizzanti) – il che è sufficientemente ovvio perché gli AAS non 17α-alchilati sono rapidamente metabolizzati nel fegato, quindi la loro concentrazione in loco non sarebbe come quella dei 17α-alchilati. Naturalmente, non si troverà una correlazione se si guarda solo a questo fattore. Bisogna anche prendere in considerazione la sua resistenza al metabolismo epatico come è stato fatto con l’ipotesi dello stress ossidativo descritta sopra.
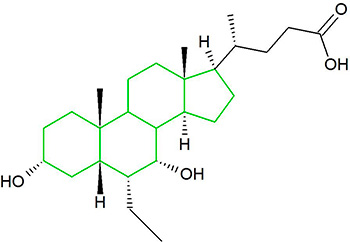
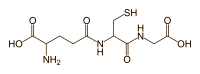
In ogni caso, questo ha portato gli autori a formulare un’alternativa ipotesi di ciò che causa l’epatotossicità indotta da AAS. E sembrava essere l’unica. Essi suggeriscono che l’epatotossicità è probabilmente dovuta alla coniugazione dell’anello D con l’acido glucuronico. Questo processo è chiamato glucuronidazione ed è una cosiddetta comune reazione di fase 2 nel metabolismo del farmaco. Rende la molecola madre più solubile in acqua, facilitando così la sua escrezione nelle urine.
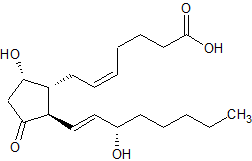
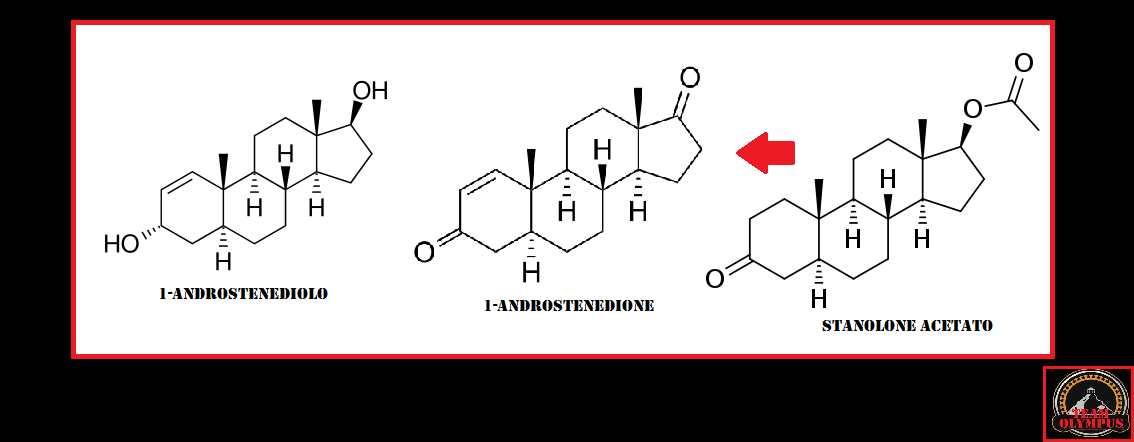
Il gruppo 17β-glucuronide (in blu) attaccato al anello D di uno steroide 17α-metilato (gruppo 17α-metilico in rosso).
È semplicemente l’attaccamento (coniugazione) dell’acido glucuronico alla molecola madre (vedi figura sopra). Quando il Testosterone con un gruppo 17β-glucuronide (così come diversi estrogeni con questa modifica) viene iniettato nel ratto, il flusso biliare è inibito [521]. Presumibilmente, perché questi composti condividono somiglianze strutturali con gli acidi biliari, questi composti competono con gli acidi biliari per legarsi a certi recettori. Tuttavia, a parte questo, non c’è molta sostanza per sostenere questa ipotesi come la ragione per l’epatotossicità indotta da AAS, soprattutto perché molti degli AAS non 17α-alchilati, compreso il Testosterone, subiscono la glucuronizzazione del loro gruppo 17β-idrossi. Eppure questi non sono sensibilmente epatotossici. Infatti, la 17βglucuronidazione è stata identificata solo per alcuni AAS 17α-alchilati, e sembra che essi subiscono questo processo solo in piccola misura [26]. Così, ironicamente, se questa ipotesi fosse vera, o significativa, ci si aspetterebbe l’epatotossicità con il Testosterone ma non con gli AAS 17α-alchilati.
Conclusioni sulle ipotesi esposte:
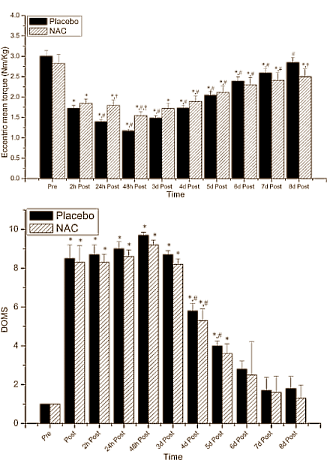
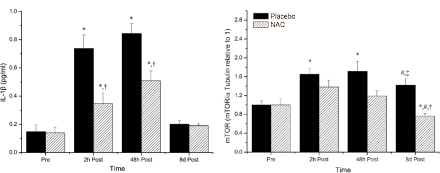
Non è sicuramente una novità per l’utilizzatore medio, ma anche per il semplice soggetto interessato all’argomento PEDs, che gli AAS metilati in C-17 (17α-alchilati) abbiano un effetto epatotossico con lievi variabili tra molecole aventi la stessa modifica strutturale. E non è nemmeno una rivelazione che la supplementazione con antiossidanti (vedi NAC e Silimarina) possa ridurre tale effetto. Di conseguenza, l’ipotesi dello stress ossidativo sembra essere la principale causa del epatotossicità AAS-indotta. Ma non l’unico fattore.
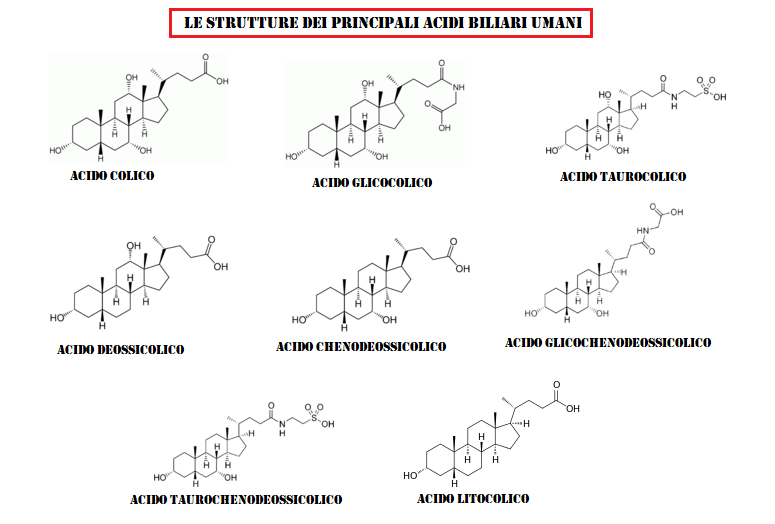
Nell’ultimo decennio si è aggiunto ai classici composti antiossidanti l’uso di acidi biliari come l’Acido Ursodesossicolico e l’Acido Tauroursodesossicolico assunti oralmente.
L’Acido Ursodesossicolico è un acido biliare secondario che deriva dal metabolismo dell’acido colico da parte del microbiota umano intestinale. Il suo nome deriva dal fatto che è il principale acido biliare negli orsi (dal latino ursus). In biologia e biochimica lo si etichetta con l’acronimo UDCA. Il nome completo del UDCA è Acido 3α,7β-diidrossi-5β-colanoico.[27]
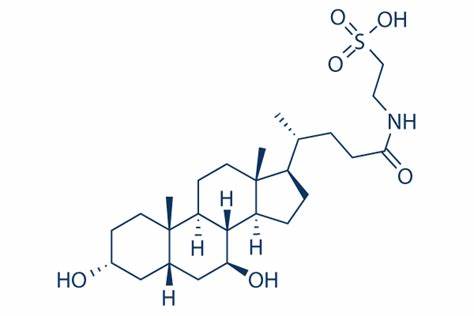
Acido Ursodesossicolico (UDCA)
L’Acido Tauroursodesossicolico (TUDCA) è un acido biliare ambifilico. È la forma coniugata di Taurina ed il precedentemente citato Acido Ursodeossicolico (UDCA). Il nome completo del TUDCA è 2-{(4R)-4-[(1R,3aS,3bR,4S,5aS,7R,9aS,9bS,11aR)-4,7-Dihydroxy-9a,11a-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-1-yl]pentanamido} acido etan-1-sulfonico.[28]
Acido Tauroursodesossicolico (TUDCA)
l’UDCA è approvato per il trattamento della cirrosi biliare primaria.[1][2] Di conseguenza, l’Acido Ursodesossicolico (UDCA) ha mostrato effetti epatoprotettivi. Tuttavia, i suoi meccanismi molecolari sottostanti rimangono poco chiari. Per tale motivazione, sono stati condotti alcuni studi come quello di Da Jung Kim et al. nel quale è stato osservato l’effetto epatoprotettivo dell’UDCA e della vitamina E utilizzando la metabolomica e l’analisi metagenomica. In questo studio, sono stati analizzati campioni di sangue e urine di pazienti con obesità e disfunzione epatica. Nove pazienti sono stati assegnati in modo casuale a ricevere UDCA (300 mg due volte al giorno), e 10 soggetti hanno ricevuto la vitamina E (400 UI due volte al giorno) per 8 settimane. L’UDCA ha migliorato significativamente i punteggi della funzionalità epatica dopo 4 settimane di trattamento e ha ridotto efficacemente i livelli epatici di acido Desossicolico e di microRNA-122 nel siero. Per comprendere meglio il suo meccanismo protettivo, è stato condotto uno studio di metabolomica globale ed è stato scoperto che l’UDCA ha regolato le tossine uremiche (acido ippurico, solfato di p-cresolo e metaboliti derivati dall’indolo), gli antiossidanti (solfato di ascorbato e N-acetil-L-cisteina) e il percorso fenilalanina/tirosina. Inoltre, il coinvolgimento del microbioma, in particolare di Lactobacillus e Bifidobacterium, è stato dimostrato attraverso l’analisi metagenomica delle vescicole extracellulari derivate dai batteri. Nel frattempo, il trattamento con vitamina E non ha portato a tali alterazioni, tranne che ha ridotto le tossine uremiche e la disfunzione epatica. I nostri risultati hanno suggerito che entrambi i trattamenti erano efficaci nel migliorare la funzione epatica, anche se attraverso meccanismi diversi.
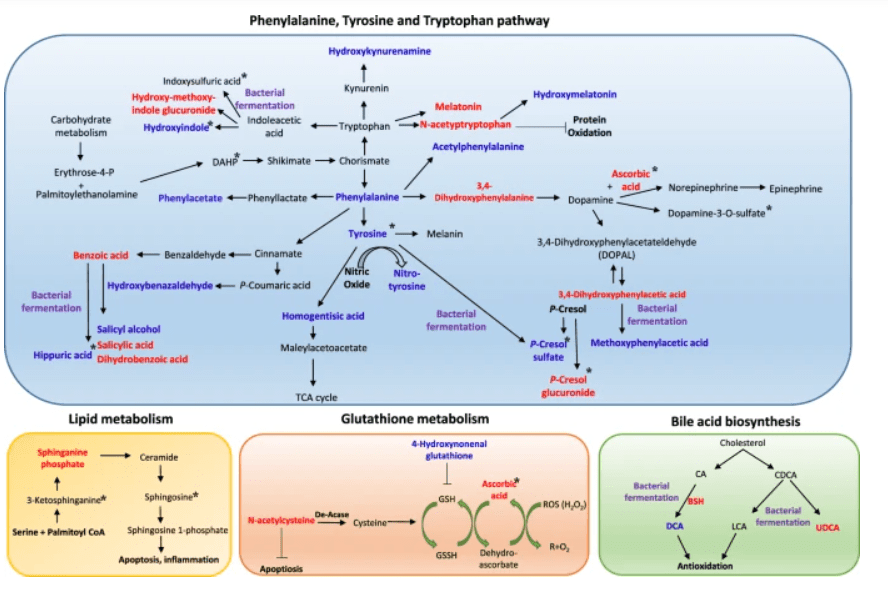
Schema dei potenziali meccanismi terapeutici del trattamento con UDCA. L’analisi metabolomica ha rivelato che l’UDCA riduce i principali composti nei percorsi fenilalanina/tirosina e triptofano, tra cui fenilalanina, fenilacetato, acetilfenilalanina, aldeide 3,4-idrossifenilacetato, dopamina-3-O-solfato, idrossibenzaldeide, p-cresolo solfato, idrossicynurenamina, idrossindolo e acido ippurico, nel plasma e nelle urine. I metaboliti intermedi degli aminoacidi aromatici come l’idrossimelatonina, l’acido benzoico e l’acido salicilico sono stati aumentati. I forti antiossidanti come l’ascorbato, l’acetiltriptofano e la N-acetil-L-cisteina erano elevati. Inoltre, la disintossicazione delle tossine uremiche tramite glucuronidazione (idrossimetossiindolo glucuronide e p-cresolo glucuronide) è stata osservata dopo il trattamento UDCA. Tuttavia, la vitamina E ha ridotto l’acido indolo-propionico, il solfato di indoxile, la 3-ketosphinganina e la sfingosina, che non sono stati regolati dall’UDCA. Il colore blu indica una diminuzione del livello del metabolita, e il colore rosso indica un aumento del livello del metabolita dopo il trattamento UDCA. I metaboliti che sono cambiati dopo il trattamento con vitamina E sono contrassegnati da un asterisco (*). I metaboliti che sono stati possibilmente regolati da modifiche batteriche sono contrassegnati da un colore viola.
Inoltre, si sa che l’UDCA a livello epatico stimola la secrezione di ATP da parte degli epatociti[29]; sebbene il significato di quest’azione non è ancora noto. Si sa però che interagisce col sistema dei citocromi P450 e che riduce la Glicuronazione degli estrogeni sintetici e non solo.[30] Vi ricorda qualcosa? Esatto! L’ipotesi di coniugazione dell’anello D e la sua potenzialità di essere parte dell’effetto epatotossico AAS-indotto! Se a ciò aggiungiamo che l’UDCA possiede la capacità di attivare direttamente il recettore per i glucocorticoidi, che contribuirebbe ad allargare i meccanismi della sua azione anticolestatica ed antinfiammatoria sul parenchima epatico [31], e che stimola la sintesi del glutatione (GSH), potente antiossidante endogeno, attraverso l’intervento delle chinasi dipendenti dai fosfoinositidi (PI-3K e PKB) [32], ciò fa si che l’UDCA risulti la chiave di volta nella protezione epatica durante l’uso di AAS con marcata resistenza al metabolismo epatico in abbinamento ai largamente utilizzati NAC (precursone ad alta biodisponibilità del Glutatione) e Silimarina.
Quanto detto non rappresenta ne un consiglio medico ne una scusa per abusare di AAS di qualsiasi tipo! Si tratta semplicemente della divulgazione di informazioni che la seria ricerca scientifica ha permesso di estrapolare, per il momento…
Gabriel Bellizzi
Riferimenti:
W. J. Meyer, A. Webb, C. A. Stuart, J. W. Finkelstein, B. Lawrence, and P. A. Walker. Physical and hormonal evaluation of transsexual patients: a longitudinal study. Archives of sexual behavior, 15(2):121–138, 1986.
A. M. Elsharkawy, S. McPherson, S. Masson, A. D. Burt, R. T. Dawson, and M. Hudson. Cholestasis secondary to anabolic steroid use in young men. Bmj, 344, 2012.
J. Nadell and J. Kosek. Peliosis hepatis. twelve cases associated with oral androgen therapy. Archives of pathology & laboratory medicine, 101(8):405–410, 1977.
F. L. Johnson, K. Lerner, M. Siegel, J. Feagler, P. Majerus, J. Hartmann, and E. D. Thomas. Association of androgenic-anabolic steroid therapy with development of hepatocellular carcinoma. The Lancet, 300(7790):1273–1276, 1972.
L. Hernandez-Nieto, M. Bruguera, J. A. Bombi, L. Camacho, and C. Rozman. Benign liver-cell adenom associated with long-term administration of an androgenic-anabolic steroid (methandienone). Cancer,40(4):1761–1764, 1977.
P. Bond, W. Llewellyn, and P. Van Mol. Anabolic androgenic steroid-induced hepatotoxicity. Medical Hypotheses, 93:150–153, 2016.
H. Sies et al. Oxidative stress: introductory remarks. Oxidative stress, 501:1–8, 1985.
K. Brieger, S. Schiavone, F. J. Miller Jr, and K.-H. Krause. Reactive oxygen species: from health to disease. Swiss medical weekly, 142:w13659, 2012.
S. P. Frankenfeld, L. P. Oliveira, V. H. Ortenzi, I. C. Rego-Monteiro, E. A. Chaves, A. C. Ferreira, A. C. Leitáo, D. P. Carvalho, and R. S. Fortunato. The anabolic androgenic steroid nandrolone decanoate disrupts redox homeostasis in liver, heart and kidney of male wistar rats. PloS one, 9(9):e102699, 2014.
J. H. Pinthus, I. Bryskin, J. Trachtenberg, J.-P. Luz, G. Singh, E. Fridman, and B. C. Wilson. Androgen induces adaptation to oxidative stress in prostate cancer: implications for treatment with radiation therapy. Neoplasia, 9(1):68–80, 2007.
H. Lin, J.-P. Lu, P. Laflamme, S. Qiao, B. Shayegan, I. Bryskin, L. Monardo, B. C. Wilson, G. Singh, and J. H. Pinthus. Inter-related in vitro effects of androgens, fatty acids and oxidative stress in prostate cancer: a mechanistic model supporting prevention strategies. International journal of oncology, 37(4):761–766, 2010.
M. Guzmán, A. Saborido, J. Castro, F. Molano, and A. Megias. Treatment with anabolic steroids increases the activity of the mitochondrial outer carnitine palmitoyltransferase in rat liver and fast-twitch muscle. Biochemical pharmacology, 41(5):833–835, 1991.
R. Gragera, A. Saborido, F. Molano, L. Jimenez, E. Muñiz, and A. Megias. Ultrastructural changes induced by anabolic steroids in liver of trained rats. Histology and histopathology, 1993.
X. Chapa-Dubocq, V. Makarov, and S. Javadov. Simple kinetic model of mitochondrial swelling in cardiac cells. Journal of cellular physiology, 233(7):5310–5321, 2018.
S. K. Powers, L. L. Ji, A. N. Kavazis, and M. J. Jackson. Reactive oxygen species: impact on skeletal muscle. Comprehensive Physiology, 1(2):941–969, 2011.
T. A. Pagonis, G. N. Koukoulis, C. S. Hadjichristodoulou, P. N. Toli, and N. V. Angelopoulos. Multivitamins and phospholipids complex protects the hepatic cells from androgenic-anabolic-steroids-induced toxicity. Clinical Toxicology, 46(1):57–66, 2008.
G. H. Marquardt, C. E. Logan, W. G. Tomhave, and R. M. Dowben. Failure of non-17-alkylated anabolic steroids to produce abnormal liver function tests. The Journal of Clinical Endocrinology & Metabolism, 24(12):1334–1336, 1964.
S. Bhasin, L. Woodhouse, R. Casaburi, A. B. Singh, D. Bhasin, N. Berman, X. Chen, K. E. Yarasheski, L. Magliano, C. Dzekov, et al. Testosterone dose-response relationships in healthy young men. American Journal of Physiology-Endocrinology And Metabolism, 281(6):E1172–E1181, 2001.
C. J. Houtman, S. S. Sterk, M. P. Van de Heijning, A. Brouwer, R. W. Stephany, B. Van der Burg, and E. Sonneveld. Detection of anabolic androgenic steroid abuse in doping control using mammalian reporter gene bioassays. Analytica chimica acta, 637(1-2):247–258, 2009.
C. Bonne and J.-P. Raynaud. Assay of androgen binding sites by exchange with methyltrienolone (r 1881). Steroids, 27(4):497–507, 1976.
H. L. Krüskemper and G. Noell. Liver toxicity of a new anabolic agent: methyltrienolone (17α-methyl-4, 9, 11-estratriene-17β-ol-3-one). Steroids, 8(1):13–24, 1966.
C. Grunfeld, D. P. Kotler, A. Dobs, M. Glesby, S. Bhasin, O. S. Group, et al. Oxandrolone in the treatment of hiv-associated weight loss in men: a randomized, double-blind, placebo-controlled study. JAIDS Journal of Acquired Immune Deficiency Syndromes, 41(3):304–314, 2006.
J. A. Kemppainen, E. Langley, C.-i. Wong, K. Bobseine, W. R. Kelce, and E. M. Wilson. Distinguishing androgen receptor agonists and antagonists: distinct mechanisms of activation by medroxyprogesterone acetate and dihydrotestosterone. Molecular Endocrinology, 13(3):440–454, 1999.
U. R. Hengge, K. Stocks, S. Faulkner, H. Wiehler, C. Lorenz, W. Jentzen, D. Hengge, and G. Ringham. Oxymetholone for the treatment of hiv-wasting: a double-blind, randomized, placebo-controlled phase iii trial in eugonadal men and women. HIV clinical trials, 4:150–163, 2003.
A. Sansone, F. Romanelli, M. Sansone, A. Lenzi, and L. Di Luigi. Gynecomastia and hormones. Endocrine, 55(1):37–44, 2017.
W. Schänzer. Metabolism of anabolic androgenic steroids. Clinical chemistry, 42(7):1001–1020, 1996.
Boatright, Jeffrey H.; Nickerson, John M.; Moring, Anisha G.; Pardue, Machelle T. (2009). “Bile acids in treatment of ocular disease”. Journal of Ocular Biology, Diseases, and Informatics. 2 (3): 149–159.
Nathanson MH et al. Stimulation of ATP secretion in the liver by therapeutic bile acids. Biochem J. 2001; 358(Pt 1):1-5.
Weitzel C et al. Ursodeoxycholic acid induced activation of the glucocorticoid receptor in primary rat hepatocytes. Eur J Gastroenterol Hepatol. 2005 Feb; 17(2):169-77.
Sanchez Pozzi EJ et al. Ursodeoxycholate reduces ethinylestradiol glucuronidation in the rat: role in prevention of estrogen-induced cholestasis. J Pharmacol Exp Ther. 2003 Jul; 306(1):279-86.
Arisawa S et al. Ursodeoxycholic acid induces glutathione synthesis through activation of PI3K/Akt pathway in HepG2 cells. Biochem Pharmacol. 2009 Mar 1;77(5):858-66.
DISCLAIMER: Il presente articolo è a solo scopo educativo, di intrattenimento e informativo. Non rappresenta in alcun modo una forma di incitamento all’uso/abuso di sostanze dopanti. L’autore ed il sito, per tanto, è esentato da qualsiasi responsabilità dipendente dalla libera scelta individuale.
Introduzione ad un dilemma…
Chiunque frequenti l’ambiente del Bodybuilding e del Fitness avrà letto o sentito almeno una volta nella vita espressioni del genere “se mi dopassi sarei anche io così [indicando Flex Wheeler]” o “ho provato di tutto e senza farmaci non riuscirò ad ottenere risultati”. Andando poi ad approfondire la storia di ognuno di questi soggetti si scopre in percentuale quasi assoluta che si tratta di individui nella norma (o al di sotto) frustrati e/o con personalità deboli, speranzosi omini che attendono placidamente che accada una svolta miracolosa nella loro banale e piatta esistenza e, cosa molto importante, con il minimo dello sforzo (meglio se nessuno).
Nella mia esperienza come ricercatore e operatore nel campo della cultura fisica in qualità di Preparatore Atletico, ho assistito a innumerevoli casi in cui un soggetto aspirava al miglioramento della propria composizione corporea trascurando, consciamente o inconsciamente, le basi fondamentali rappresentate da Nutrizione e Allenamento baipassandole in vista della possibile prescrizione di una pillola miracolosa capace di renderlo/a possessore della forma fisica ambita.
Tralasciando l’ovvio ragionamento che spinge ogni essere umano dotato di un minimo d’intelletto verso la comprensione che la genetica è il blocco d’argilla sul quale si va ad operare, ma le sue qualità e difetti sono presenti in modo eterogeneo nella popolazione mondiale, e ciò non è modificabile nemmeno con la farmacologia più oculata, quando ci si trova davanti al bivio tra “pillola rossa” (PEDs) e “pillola blu” (drug free) bisogna essere pienamente consapevoli non solo del fattore illegalità ma del fattore conoscitivo. Purtroppo, la politica del terrore ha operato in modo fallimentare nel goffo intento di allontanare dalla scelta “rossa”, e ciò si è tradotto in un numero sensibile di soggetti abusatori con tutte le conseguenze cliniche derivanti.
Se un individuo non ha raggiunto un livello di maturità sportiva tale da conferirgli una gestione corretta della nutrizione e della periodizzazione allenante (gestione delle variabili volume, intensità, densità ecc…), è molto meglio per lui/lei rivedere i suoi programmi e scegliere ancora la “pillola blu”. Capita, a volte, di incontrare persone decise ad intraprendere la via del “lato oscuro” che, dopo una approfondita chiacchierata sulla gestione dei suddetti fattori, rivede le proprie posizioni.
Per tutti coloro i quali sono immersi nel dilemma della scelta, vi espongo alcuni punti per rendere l’eventuale decisione meno rischiosa anche se pur sempre illegale nel “bel paese”…
“Pillola blu o pillola rossa?” I punti da tenere in considerazione per una scelta consapevole:
#1 Raggiungere una adeguata maturità sportiva
Per “maturità sportiva”, in particolare riferimento al BodyBuilding, si intende la capacità del atleta di sapersi alimentare e allenare correttamente con piena gestione delle proprie potenzialità fisiologiche/genetiche. Questa è la base, se viene a mancare ciò non solo la vostra esperienza finirà per deludervi e rendervi ancora di più dei frustrati, ma potrebbe rovinosamente portarvi ad un abuso cronico a senso inesorabilmente negativo…
#2 I PEDs non faranno miracoli
Una cosa da tenere bene a mente, e questo non dovrebbe interessare solo gli aspiranti “doped”, è che l’uso di PEDs non renderà diversi da ciò che rientra nelle potenzialità espressive del proprio patrimonio genetico. Certamente le caratteristiche genetiche verranno “iperespresse”, nel bene e nel male, dall’uso di PEDs ma non vi sarà nessun miracolo! Migliorerete ma non sarete ne più ne meno di ciò che potete essere!
Un esempio per capire come la base genetica faccia la differenza anche con protocolli che, ad oggi, spesso non raggiungono nemmeno i livelli del “bridge” più soft..
#3 Ridurre la percentuale di grasso corporeo
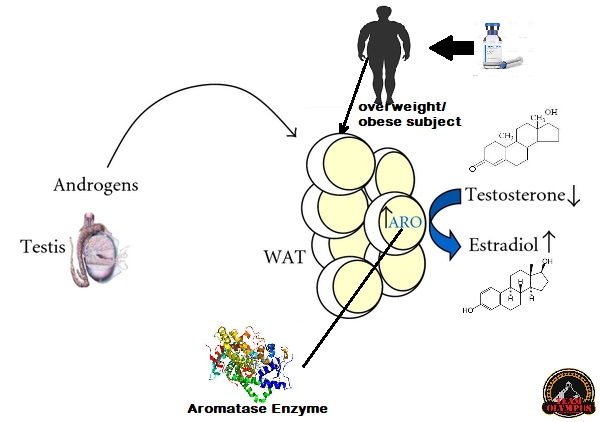
Il tessuto adiposo rappresenta uno dei siti dove il Testosterone, ed altri AAS soggetti all’aromatizzazione, viene convertito in Estradiolo. Soggetti con percentuali di grasso corporeo elevate vedrebbero una alterazione marcata della Testosterone:Estradiolo ratio a favore della componente estrogenica, con conseguenze quali alterazione del comportamento sessuale (impotenza, difficoltà nel raggiungere e/o mantenere l’erezione), ritenzione idrica, accumulo di grasso con modello femminile e ginecomastia. E no, l’uso di DHT derivati o di SARM non steroidei senza una base di Testosterone non risolverebbe il problema o, per lo meno, porterebbe ad altre conseguenze negative, che pur non comprendendo, per esempio, ritenzione idrica e ginecomastia, interesserebbero l’attività sessuale e la condizione psichica del soggetto trattato. [1]
Schema esemplificato del processo di aromatizzazione degli androgeni aumentati in un soggetto con percentuale di grasso corporeo alta.
Allo stesso tempo, i rischi cardiovascolari della somministrazione di AAS- come il possibile aumento esponenziale del Ematocrito, l’aumento del LDL e Trigliceridi a discapito di una riduzione del HDL, e l’aumento della pressione sanguigna – sarebbero già presenti in certa misura quando la body fat è già alta e sarebbero quindi soggetti ad un repentino aggravamento.
Se la percentuale di grasso è relativamente alta, si dovrebbe prima di tutto considerare di migliorare la composizione corporea con una adeguata routine alimentare e allenante (senza farmaci) prima di iniziare solo a pensare all’uso di AAS. Sicuramente ciò renderà la scelta più efficace e meno rischiosa.
Nel caso fosse necessario sottolinearlo, no, non è saggio nemmeno utilizzare agenti PEDs a fini lipolitici e/o antiadipogenici e/o termogenici (compresi gli Ormoni Tiroidei). A meno che non siate affetti da ipotiroidismo, e in questo caso la terapia vi dovrebbe essere stilata dal vostro medico, per ridurre in modo sensibile la body fat non sono necessari i farmaci!
#4 Controllare se si ha una storia familiare di trombosi (o qualsiasi altra malattia cardiovascolare)
Molte malattie cardiovascolari hanno una componente di base genetica. Uno stile di vita sano può ridurne sensibilmente la loro insorgenza, ma l’uso di AAS può causare l’attivazione di specifici geni implicati nella comparsa di malattie cardio-circolatorie. Caratteristico dell’interazione tra AAS e geni specifici è un caso studio ben documentato che ricercatori americani hanno pubblicato sul “Blood Coagulation & Fibrinolysis”.[2]
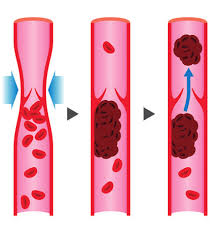
Trombosi venosa
Oltre all’attivazione genica diretta dagli AAS, e nociva per il sistema cardio-circolatorio, vi sono altre condizioni negative innescate dall’uso/abuso di Steroidi Anabolizzanti, e di altri PEDs, come, per esempio, l’aumento del tasso di coagulazione, l’incremento eccessivo dell’Ematocrito con aumento pressorio, rigidità dell’endotelio vascolare con perdita di efficienza strutturale e aumento della pressione ematica con incremento delle possibilità di danno strutturale dei componenti del sistema interessato.
#5 Inserire delle sedute di allenamento cardio prima, durante e dopo l’uso di PEDs
Un moderato allenamento cardiovascolare è sicuramente una delle migliori strategie preventive contro la comparsa di malattie cardio-circolatorie. Tale tipologia di allenamento può portare un miglioramento e/o riduzione delle alterazioni lipidiche ematiche del praticante, fornendo un, seppur minimo, tampone all’azione negativa degli AAS e SARM non steroidei sui livelli di LDL (aumento), Trigliceridi (aumento) e HDL (diminuzione). Secondo quanto riportato da una interessante review del 2013, l’abbinamento di sedute cardio e in sala pesi possono avere una azione additiva benefica sui livelli di LDL, Trigliceridi e HDL.[3]
Risulta interessante anche quanto emerso da alcuni studi su animali a seguito dei quali si è osservato un significativo grado di protezione dato dall’allenamento cardio negli esemplari trattati con AAS.[4]
#6 Assicurarsi di rimanere ben idratati
Oltre ad agevolare il mantenimento di un Ematocrito migliore, una buona idratazione risulta positiva sulla pressione di lavoro renale nel filtraggio del sangue. Diversi AAS come il Trenbolone e i metilati in C-17 presentano una particolare resistenza metabolica che, oltre a causare un aumentato stress epatico, può portare ad una sofferenza renale sfociabile nel patologico. Si è osservato come una combinazione di AAS, dieta iperproteica e supplementazione di Creatina possa aumentare l’incidenza di problemi renali.[5] In un soggetto in fisiologia, la sola dieta ad altro contenuto proteico e la supplementazione di Creatina non hanno mostrato nessun grado di pericolosità, soprattutto sul breve/medio termine.
#7 Non usare “droghe ricreative”
A livello globale, il numero di decessi tra gli abusatori di AAS è in aumento. Alcuni, troppo superficialmente, dicono che questo sia dovuto al fatto che sempre più uomini e donne usano AAS, ma questa è solo una spiegazione dozzinale e limitata. Il sospetto ricade soprattutto sulle modalità di approccio dei consumatori di AAS: i dosaggi sono drammaticamente aumentati e un numero crescente di individui combina PEDs con “droghe ricreative”. Ed è su questi due ultimi punti che risiede la spiegazione principale dell’aumento statistico prima menzionato. Soprattutto la combinazione di PEDs e le così dette “droghe ricreative” risulta essere probabilmente un fattore significativo, come evidenziato alcuni anni fa da ricercatori australiani. Nel loro studio sono state analizzate tutte le morti documentate tra i consumatori di AAS a Sydney tra il 1997 e il 2012, scoprendo che le droghe ricreative come la cocaina avevano avuto un ruolo nella schiacciante molteplicità dei casi. Dagli studi sugli animali ora sappiamo della possibilità che la co-assunzione di un AAS come il Nandrolone con la cocaina vede moltiplicati gli effetti cardiotossici rispetto ai singoli composti.[6] E secondo studi in vitro la combinazione di Testosterone e cocaina aumenterebbe la possibilità di formazione di coaguli nel flusso ematico. [7]
#8 Corretta modalità di iniezione e herpes labiale
Gli utilizzatori di AAS a volte sviluppano ascessi, ma non sempre dovuti alla bassa qualità dei prodotti utilizzati.
Alcuni medici ritengono che gli utilizzatori di AAS dovrebbero effettivamente ricevere una formazione sulle tecniche di iniezione corrette, onde evitare embolie oleose o ascessi per cattiva gestione igienica della procedura. [8]
Molti utilizzatori ancora non sanno che disturbi come l’herpes labiale rendono le iniezioni ancora più rischiose. Perchè? Il virus che causa l’herpes labiale, come altri patogeni, riduce l’efficienza del sistema immunitario, fornendo così terreno fertile per infezioni batteriche i cui microorganismi scatenanti vengono inoculati nel corpo del soggetto attraverso l’iniezione in modo diretto o indiretto.
#9 Non fare affidamento sugli integratori
Secondo un buon numero di studi svolti su animali, alcuni integratori proteggono dagli effetti collaterali degli AAS. Secondo alcune ricerche, la Taurina, la Vitamina C ed E proteggono i testicoli durante un ciclo e la vitamina C e il cacao proteggono la prostata.
L’utilità dei risultati provenienti da questi studi è limitata per tre motivi:
A. gli animali da laboratorio non sono esseri umani, e
B. le dosi utilizzate e rapportate ad un essere umano sono quasi sempre molto inferiori rispetto a quelle utilizzate dai “doped”, e
C. la ricerca in campo psicologico mostra che l’uso di integratori stimola comportamenti rischiosi e malsani. I supplementi fanno pensare agli utilizzatori di essere invulnerabili e di non dover comportarsi in modo sano ed attento.[9]
Gli integratori possono aiutare a creare una mentalità che non si dovrebbe avere da utilizzatore consapevole di AAS.
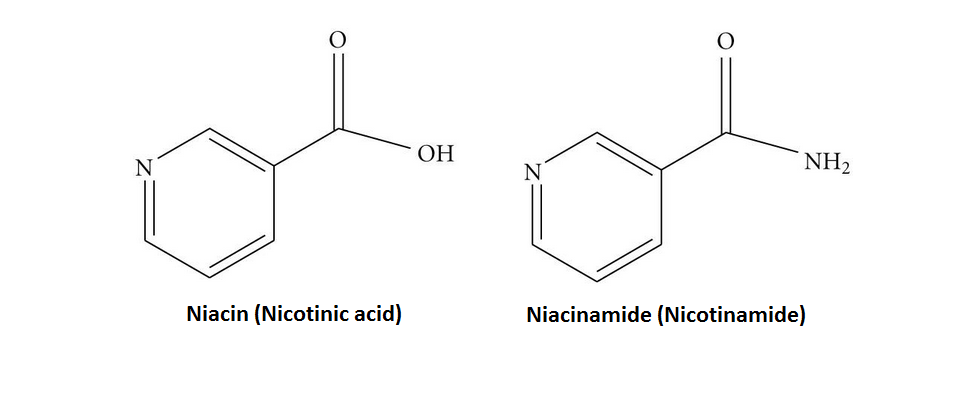
Ovviamente, alcuni supplementi “protettivi” utilizzati dai soggetti meglio informati hanno un potenziale di “tamponare” in modo discreto alcune alterazioni legate all’uso di AAS e SARM come, ma non limitato a, Riso Rosso fermantato (controllo lipidico) [10], Silimarina (epatoprotezione), NAC (epatoprotezione) [11], Niacina (controllo lipidico) ecc…
#10 Ridurre al minimo (se non eliminare) il consumo di alcolici
Potrebbe sembrare un indicazione superflua ma non lo è.
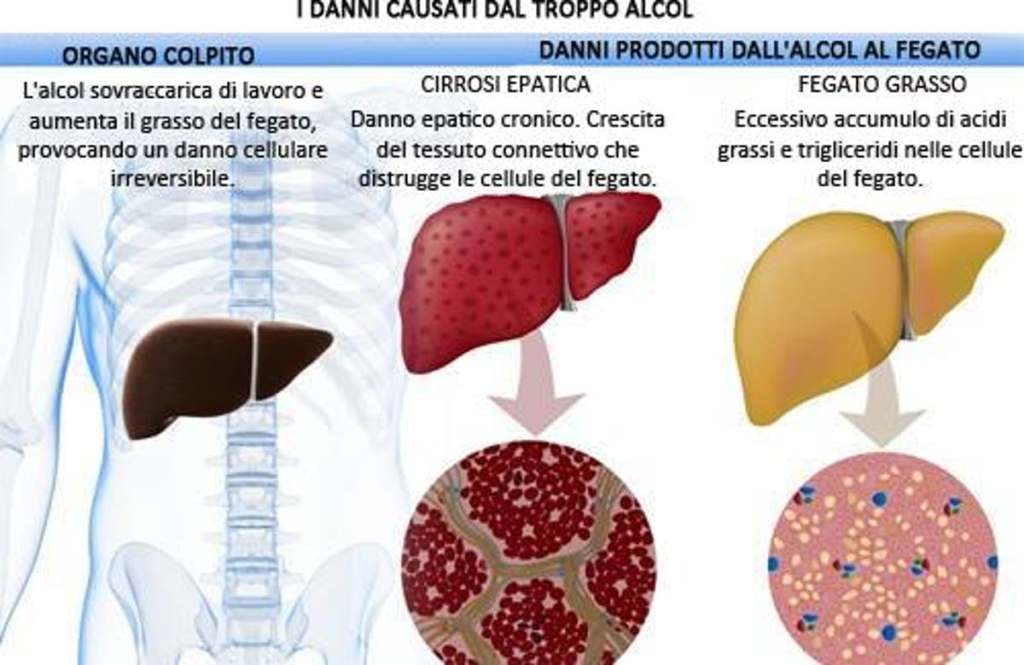
L’abuso di alcol è indubbiamente uno dei problemi sociali più diffusi. Uno dei problemi correlati all’abuso di alcol e l’epatopatia alcolica. Questo stato patologico è derivante da un processo infiammatorio progressivo ai danni del fegato legato al consumo eccessivo di alcolici. È una malattia a più stadi. La steatosi provoca un ingrossamento del fegato causato da un accumulo di trigliceridi, spesso senza sintomi per molto tempo. I rischi correlati sono la steatosi (fegato grasso), l’epatite alcolica e la cirrosi epatica. Il rapporto con l’alcolismo è complesso. Non tutti i bevitori, infatti, hanno danni al fegato, anche se sono altamente probabili. La causa è da rinvenire in una trasformazione dell’alcol (etanolo) in sostanze tossiche che danneggiano il fegato in maniera irreversibile e cronica, con un rischio elevato di insufficienza epatica e di cancro, fino alla necessità di un trapianto di fegato.
In acuto, invece, l’alcol può essere una causa di alterazione delle transaminasi ma non si può sapere se e con quale modalità si potrebbero innalzare: dipende molto dalla risposta individuale dell’organismo. In caso di stress preesistente, di causa iatrogena e/o alimentare, si può presentare una alterazione significativa. [12]
Il primo caso è una consequenziale possibile se eventi stressori concomitanti si presentano in cronico. Ed è semplice giungere alla conclusione che l’uso di AAS, specie se metilati, possa comportare un aumentato stress epatico che potrebbe degenerare in peliosi epatica, cirrosi ecc…
Che siate “doped” o “natural”, per ragioni legate e non, dovreste evitare di consumare più di 25g per gli uomini, o 12,5g per le donne, di Etanolo al giorno.
#11 Sottoporsi a regolari controlli medici pre, intra e post utilizzo
Il monitoraggio della salute dovrebbe essere la base fondante del comportamento del utilizzatore consapevole e minimamente attento ai potenziali rischi nei quali potrebbe imbattersi.
Gli esami di controllo sono i seguenti:
Esami ematici e delle urine (comprendenti il quadro ormonale secondo necessità);
Elettrocardiogramma ogni 6 mesi circa;
Elettrocardiogramma sotto sforzo (prima di iniziare);
Ecocardiogramma ogni 6 mesi circa;
Coronarografia ogni 6 mesi circa;
Monitoraggio della pressione ematica;
TAC addome completa ogni 6 mesi circa.
Ovviamente, ogni accertamento , al di la degli esami ematici, deve essere gestito in base alle esigenze soggettive, caratteristiche e tipo di PEDs utilizzati.
#12 Essere seguiti da personale qualificato
Fin troppa gente è stata salutisticamente deturpata da gorilla di spogliatoio a mala pena consapevoli dell’esistenza dei macronutrienti e che, nonostante ciò, si sono improvvisati farmacisti. Donne divenuti uomini e uomini divenuti simili a cagne in calore per via di orrende ginecomastie. Evitate il fai da te e l’affidarsi a semianalfabeti … la somaticità sopra la norma è cosa diversa dall’intelligenza e alla competenza in biologia, biochimica e farmacologia… senza offesa per tutti quelli che “io mi facevo e ho vinto! Senzia scienzia!” …
#13 Pensare seriamente al post ciclo prima del ciclo
Molti aspiranti “doped” non considerano il fattore post ciclo. La maggior parte di loro è convinta che la PCT sarà una facile soluzione alla sottoregolazione dell’Asse HPT, ma in realtà non è proprio così. Esistono diversi casi studio che mostrano come gli ex utilizzatori abbiano spesso livelli di Testosterone inferiori rispetto al pre-utilizzo anche a distanza di anni dal cessato uso di AAS. Sembra che i fattori che aumentano le possibilità e il grado di tale effetto sul lungo termine siano:
Tempo di somministrazione;
Età
Molecole utilizzate (con maggiore impatto negativo dato dai19-norsteroidi come il Nandrolone per via della lunga permanenza dei metaboliti nel sistema).
Tutto ciò è indipendente dalla qualità della PCT, anche se essa può avere dei riscontri positivi specie nel primo periodo di stacco dagli AAS. Le alterazioni ormonali legate ad una alterazione dell’Asse HPT comprendono depressione, ansia, bassa libido, difficoltà nel raggiungere e mantenere l’erezione, stanchezza cronica ecc…
Per questa ragione molti scelgono di entrare in TRT (Terapia Sostitutiva del Testosterone) dopo il primo ciclo.
Quale conclusione?…
Se mai non dovesse bastare il disclaimer, questo articolo non rappresenta in alcun modo un consiglio e, ne tanto meno, un incitamento all’uso di sostanze dopanti! E’ semplicemente a fine divulgativo con l’obbiettivo di far comprendere a più persone possibili che la scelta di intraprendere coscientemente certe pratiche (illegali) necessita di una sufficiente (e veritiera) conoscenza del argomento.
Quindi? Leggete e comprendete correttamente ciò che ho riportato in sintesi fruibile ad un largo pubblico… Pensate prima di tutto ad alimentarvi e allenarvi in modo ottimale!
La conoscenza della Verità rende liberi dalla cattiva informazione, dagli strumenti commerciali e dal relativismo… Negarla è semplice e pericolosa manifestazione di profonda ignoranza… di VERO NEGAZIONISMO!
Se avete una buona conoscenza della lingua inglese e volete approfondire l’argomento PEDs e Sport, potete leggere il libro ANABOLICS 11th Edition di William Llewellyn
Al principio del mese di giugno di quest’anno ho riportato alcuni casi studio i quali facevano emergere il potenziale effetto epatotossico dato dall’uso dei SARM RAD-140 e LGD-4033. Il caso studio riguardante LGD-4033 non era di per sé convincente, poiché il Bodybuilder in questione era solito consumare discrete quantità di alcol. Di recente, i medici del Baylor College of Medicine negli Stati Uniti hanno segnalato un altro caso di danno epatico legato all’uso di LGD-4033.[1] E in questo nuovo caso studio, non ci sono altri fattori esplicativi del problema.
Il soggetto protagonista del caso studio è un Bodybuilder di 32 anni che ha riferito ai medici di aver usato 10mg/die di LGD-4033 in forma liquida per 15 giorni. Dopo di che aveva cominciato a lamentare malessere e interruppe il suo ciclo con il suddetto SARM. L’uomo aveva dolori di stomaco e nausea, oltre a prurito e ittero. Le sue feci erano grigie, e aveva perso l’appetito. Quando si è rivolto ai medici, l’uomo aveva già perso 18Kg.
Questi sono classici sintomi da danno epatico. Infatti, quando i medici hanno scansionato la cavità addominale del Bodybuilder, hanno notato che il fegato dell’uomo era più grande del normale. Una biopsia ha mostrato che il fegato del aveva cicatrici in alcuni punti. I dotti biliari, che trasportano i sali biliari all’intestino, erano ostruiti.
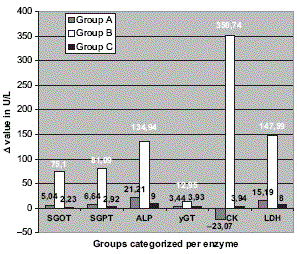
Nelle settimane successive, i medici hanno monitorato quattro marker del danno epatico nel sangue del Bodybuilder. La figura seguente mostra che le condizioni del fegato dell’uomo sono lentamente migliorate.
Come avevo già riportato nell’articolo di giugno, secondo uno studio del 2012 condotto dai produttori del LGD-4033, questo SARM non è significativamente dannoso per il fegato. Ma in questo studio, i soggetti non hanno ricevuto più di 1mg/die di LGD-4033.[2]
Le aziende che vendono SARM online e alcuni guru del bodybuilding raccomandano dosi significativamente più elevate di 1mg/die. Ad esempio, il paziente del quale si è parlato in questo articolo ha assunto dieci volte la dose più alta testata nello studio del 2012. Con tutta probabilità, un dosaggio di LGD-4033 di tale entità o superiore rappresenta uno stress epatico eccessivo, in specie se l’utilizzatore presenta una marcata sensibilità e manca di una efficace epatoprotezione (comunque non garante di immunità da effetti collaterali a livello epatico).
Alcuni utilizzatori di LGD-4033 hanno pubblicato il loro esame ematico sui forum presenti in rete. Sembra che non mostrino segni di danno epatico, ma l’affidabilità di certi dati è assai scarsa.
Forse il Bodybuilder in questione stava usando un prodotto contaminato o fake. Non tutti i SARM negli store online sono prodotti con le giuste misure di controllo, come riportato da una recente ricerca inglese e americana.[3]
E’ anche possibile che il Bodybuilder del caso studio stava usando altre sostanze oltre al solo LGD-4033 e non ne ha fatto menzione ai medici che lo hanno preso in cura. Le possibilità sono diverse ma ciò che è sufficientemente certo è che l’uso di LGD-4033 ad alte dosi, per vie metaboliche intrinseche, è un potenziale fattore causale per stress e danno epatico.
La Niacina è largamente utilizzata dagli atleti supplementati chimicamente, in special modo da coloro i quali usano molecole con un potenziale negativo marcato sui lipidi ematici. Ma come spesso capita, gli utilizzatori non conoscono a sufficienza le caratteristiche di ciò che assumono, e questa essenziale vitamina del gruppo B (B3) non è da meno. Per la maggior parte degli individui tanto basta sapere che una sua integrazione si traduce in livelli migliorati di Colesterolo e Trigliceridi. Purtroppo, però, si trascurano caratteristiche importanti la cui conoscenza può fare la differenza tra un uso più o meno funzionale per la salute sistemica. Infatti, un effetto collaterale dell’integrazione di Niacina è un peggioramento della resistenza all’insulina, cosa che limita i benefici di tale supplementazione sulla salute cardiovascolare se non vengono prese adeguate precauzioni.
Prima di correre a defenestrare in preda al panico la vostra Niacina, leggete con attenzione (e comprendete) le informazioni che seguono…
Introduzione alla Niacina (vitamina B3)
Niacina
La Niacina, nota anche come Acido Nicotinico, è un composto organico e una forma di vitamina B3, un micronutriente essenziale per l’essere umano. [1] La Niacina ha formula bruta C6H5NO2 e appartiene al gruppo dell’acido piridinecarbossilico.[1] Come precursore di NAD e NADP, la Niacina è coinvolta nella riparazione del DNA.[2] La Niacina viene assunta attraverso la dieta da una varietà di alimenti interi e trasformati, con il più alto contenuto in alimenti confezionati fortificati, carne, pollame, pesce rosso come tonno e salmone, con minori quantità nelle noci, legumi e semi. [1] [3] La Niacina come integratore alimentare viene anche utilizzata per trattare la pellagra, una malattia causata da una sua carenza. Segni e sintomi includono lesioni della pelle e della bocca, anemia, mal di testa e stanchezza.[4] Molti paesi richiedono la sua aggiunta alla farina di grano o ad altri cereali, riducendo così il rischio di pellagra.[1][5] Come vitamina, le raccomandazioni di dosaggio giornaliero indicate in diversi paesi sono 14-18mg/die per gli adulti, quota sufficiente per soddisfare le esigenze delle persone sane. [6] [7] [8]
Sebbene la Niacina e la Nicotinamide (Niacinamide) siano identiche nella loro attività vitaminica, la Nicotinamide non ha gli stessi effetti farmacologici, modificanti i lipidi o gli effetti collaterali della Niacina, cioè quando la Niacina assume il gruppo -amide, non riduce il Colesterolo né causa vampate di calore.[9][10] La Nicotinamide è raccomandata come trattamento per la carenza di Niacina poiché può essere somministrata in quantità correttive senza causare l’effetto negativo del rossore.[11]
La Niacina è anche un farmaco di prescrizione. Quantità molto superiori all’assunzione dietetica raccomandata per le funzioni vitaminiche ridurranno i Trigliceridi nel sangue e le lipoproteine a bassa densità (LDL-C) e aumenteranno le lipoproteine ad alta densità (HDL-C). Ne esistono due forme: Niacina a rilascio immediato e a rilascio prolungato. Le quantità iniziali di prescrizione sono di 500mg/die, con possibilità di essere aumentate nel tempo fino a raggiungere l’effetto terapeutico ricercato. Le dosi a rilascio immediato possono arrivare fino a 3g/die; quelle a rilascio prolungato fino a 2g/die. [12] Nonostante i comprovati cambiamenti lipidici, la Niacina non è stata trovata utile per ridurre il rischio di malattie cardiovascolari nei soggetti già in trattamento con statine. [13] Una review del 2010 aveva concluso che l’efficacia della Niacina si osservava in mono-terapia, [14] ma una review del 2017 che incorporava il doppio del numero degli studi ha concluso che la Niacina su prescrizione, pur influenzando i livelli lipidici, non riduceva la mortalità per tutte le cause, la mortalità cardiovascolare, gli infarti del miocardio, né ictus fatali o non fatali. [15] È stato dimostrato che la Niacina da prescrizione provoca epatotossicità [16] e aumenta il rischio di diabete di tipo 2. [17] [18] Le prescrizioni di Niacina negli Stati Uniti avevano raggiunto il picco nel 2009, a 9,4 milioni, in calo a 1,3 milioni entro il 2017.[19]
Niacina, flusso ematico, pressione e vasodilatazione
Uno studio sulla supplementazione di Niacina che ha valutato il flusso sanguigno dell’avambraccio non è riuscito a trovare un effetto significativo fino a 1g al giorno somministrati nel corso di due settimane in soggetti altrimenti sani, [20] e 1.5g di Niacina a rilascio prolungato negli uomini con sindrome metabolica non sono riusciti a influenzare la dilatazione flusso- mediata (FMD). [21] Un altro studio non è riuscito a trovare un effetto significativo in un intero gruppo di pazienti affetti da afta epizootica, mentre in un gruppo di pazienti con malattia coronarica ha riscontrato un miglioramento in un sottogruppo con bassi livelli HDL-C. [22]
In soggetti con bassi livelli di HDL-C, è stato osservato che 1g di Niacina a rilascio prolungato per una settimana aumenta il flusso sanguigno (via FMD) del 4,5%; questo meccanismo non era correlato alle Prostaglandini, poiché il Laropiprant (un inibitore della Prostaglandine D2) non ha influenzato l’effetto. [23] Questo effetto ha anche coinciso con un aumento della bilirubina indiretta (ma non totale) del 62%. [23] Poiché la bilirubina del acido biliare è un antiossidante endoteliale, [24] e poiché i benefici della niacina sulla funzione endoteliale in questo studio sono stati ritenuti dipendenti dall’ossido nitrico, [23] è stato ipotizzato che un effetto conservativo della bilirubina sulla biodisponibilità dell’ossido nitrico sia alla base della beneficio osservato. Sia l’aumento della bilirubina che il miglioramento del flusso sanguigno si sono dissipati una settimana dopo l’interruzione della Niacina.[23]
Laropiprant
I soggetti che in precedenza avevano subito infarto del miocardio, a seguito del trattamento con Niacina (con Laropiprant) hanno riscontrato un aumento del flusso sanguigno dipendente dall’ossido nitrico (FMD) dopo dodici settimane di terapia insieme a un miglioramento della vasodilatazione indotta da nitroglicerina, entrambe non correlate con alterazioni dei trigliceridi. [25] Miglioramenti simili nel flusso sanguigno sono stati osservati in pazienti con infezione da HIV e con bassi livelli di HDL-C trattati con la sola Niacina. [26]
Prostaglandine D2 (PGD2)
È noto che la Niacina influenza il diametro dei vasi sanguigni, in particolare per via della sua reazione vasodilatativa cutanea (allargamento dei vasi nella pelle), che ha portato a ipotizzare che potrebbe influenzare la pressione sanguigna aumentando il diametro delle arterie e vene. Tuttavia, una review [27] ha notato che un possibile effetto di riduzione della pressione arteriosa della Niacina è indipendente dalla Prostaglandine che media il rossore, nota come PGD2.
È stato osservato che le infusioni di Niacina riducono acutamente la pressione sanguigna negli ipertesi senza alcun effetto nei soggetti con pressione sanguigna normale ed è stata associata ad un aumento della gittata cardiaca e della frequenza cardiaca che era simile in entrambi i gruppi. [28] Un altro studio ha confermato questo risultato, scoprendo che la pressione arteriosa ambulatoriale di 24 ore non sembra essere influenzata da un supplemento di Niacina fino a 1g nell’arco di due settimane in soggetti altrimenti sani. [20]
In termini di effetti della Niacina in cronico sulla pressione sanguigna, una review [27] che ha valutato gli studi che hanno misurato la pressione sanguigna negli ipertesi [29] [30] [31] [32] non ha notato alcun effetto statisticamente significativo nella riduzione della pressione sanguigna associata alla supplementazione di Niacina, sebbene questi studi in quanto a metodologie di misurazione sulle variazioni della pressione sanguigna non fossero ideali secondo gli autori della review. Tuttavia, la review ha osservato che in un ampio studio (il Coronary Drug Project), che inizialmente non è riuscito a trovare alcuna influenza della terapia con Niacina sulla pressione arteriosa, [32] ha osservato variazioni sensibili soltanto sui soggetti con sindrome metabolica. Questi presentavano un lieve riduzione di 2,2mmHg della pressione arteriosa sistolica con una moderata riduzione di 2,9mmHg della pressione diastolica. [33] Un’analisi post-hoc di un altro studio clinico [34] ha rilevato che la pressione arteriosa sistolica è stata abbassata di 2,2mmHg e la pressione sistolica di 2,7 rispetto al placebo nei pazienti dislipidemici trattati per 24 settimane. [35]
Niacina, Trigliceridi, Colesterolo e Aterosclerosi
Apolipoproteina B
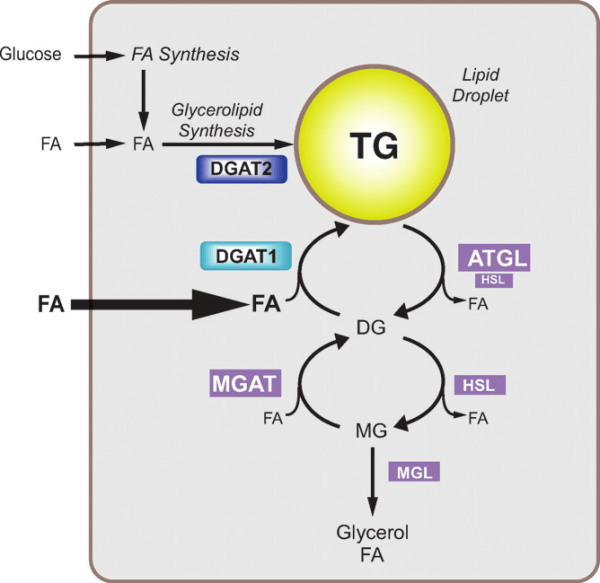
La Niacina sembra abbassare i trigliceridi nel sangue inibendo sia la sintesi degli acidi grassi sia la loro esterificazione epatica per formare i trigliceridi, il che aumenta il tasso di degradazione dell’apolipoproteina B riducendo la sua secrezione dalle cellule epatiche. [36] Un meccanismo con cui la Niacina fa questo è attraverso l’inibizione diretta e non competitiva della diacilglicerolo aciltransferasi 2 (DGAT2), l’enzima finale nella sintesi dei trigliceridi nelle cellule epatiche, senza inibizione della DGAT1. [37]
Si è visto che gli effetti della Niacina sulla sintesi dei trigliceridi influenzano i livelli sierici di lipoproteine a densità molto bassa (vLDL-C), dove la terapia con Niacina per 16 settimane in soggetti con malattia del fegato grasso non alcolica (NAFLD) sembra ridurre le vLDL-C nel siero così come i complessi con trigliceridi (vLDL-TG) e apolipoproteina B (vLDL-ApoB) rispetto al placebo e con una potenza paragonabile al fenofibrato. [38] La Niacina lo fa riducendo la secrezione epatica di vLDL-C, sebbene ciò non aumenti la quantità di trigliceridi nel fegato anche nello stato di NAFLD. [38]
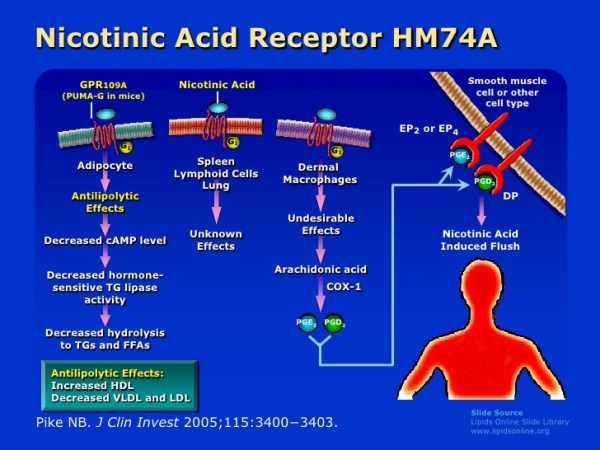
Oltre ai suoi effetti sul fegato, la Niacina può anche sopprimere il rilascio di acidi grassi liberi dal tessuto adiposo [39] che normalmente verrebbero reesterificati come trigliceridi nel fegato e quindi secreti via vLDL. [40] Tuttavia, questo meccanismo specifico, che è mediato dal recettore HM74A, [39] non sembra essere rilevante per le proprietà riducenti dei trigliceridi della Niacina. [41]
I benefici sui livelli di trigliceridi possono verificarsi entro una settimana dall’inizio della supplementazione con Niacina a rilascio prolungato (1g), sebbene in misura minore di circa il 4%. [23]
L’integrazione di 1.5-2g di Niacina a rilascio prolungato per due anni con follow-up di un anno nelle persone in terapia con statine caratterizzate da bassi livelli di HDL-C ha mostrato una riduzione dei trigliceridi del 28,6% (statina da sola dell’8,1%). [42]
Esiste un fenomeno noto come “rimbalzo degli acidi grassi” associato alla supplementazione di Niacina, in quanto l’azione iniziale del composto sul suo recettore (HM74A) nel tessuto adiposo può determinare una minore lipolisi e una minore secrezione di acidi grassi non esterificati (NEFA) nel sangue [43] e una migliore conservazione adiposa; [44] si tratta di fenomeni prontamente reversibili in quanto in un giorno di esposizione continua vi è un aumento netto del NEFA piuttosto che la sua soppressione [45] [46] [47] e alterazioni nel NEFA possono non riflette alterazioni dei trigliceridi.
Il primo meccanismo pensato per spiegare il miglioramento del profilo sierico di colesterolo in seguito alla supplementazione di Niacina è stato attraverso la riduzione del rilascio di acidi grassi non esterificati (NEFA) dai tessuti, che non è più considerato un probabile meccanismo in quanto l’integrazione di niacina in cronico è associata ad un aumento, piuttosto che alla soppressione, di NEFA mentre il recettore HM74A appare superfluo in termini di effetti della Niacina nei topi con altri ligandi del HM74A (Acipimox [48] e MK-0354 [49]) che si sono mostrati rispettivamente meno efficaci o inefficaci sul colesterolo. Attualmente si ritiene che l’influenza della Niacina sui NEFA nel siero non sia un fattore determinante nel modo in cui influenza i livelli di colesterolo nel corpo, con le teorie attuali che ipotizzano che il fattore sia determinato dalla sua sintesi e dal suo tasso di catabolismo.
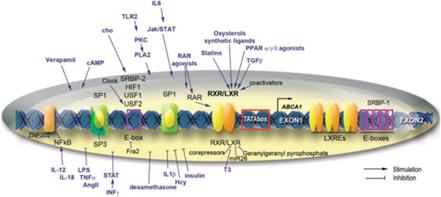
Il primo potenziale meccanismo prevede la sintesi di HDL-C nel fegato attraverso l’aumento della trascrizione del gene ABCA1 (che dipende dal legame LXRα alla regione del promotore DR4 di questo gene). [50] L’attività di ABCA1 promuove la “lipidazione” della principale proteina dell’HDL nota come apolipoproteina AI (ApoAI) aumentando il tasso che associa ai fosfolipidi e al colesterolo, [51] [52] un passaggio obbligatorio nella sintesi dell’HDL-C che è aumentato di 500-1000µM con Niacina in vitro. [50] Questo meccanismo non è stato confermato, poiché mentre l’ApoAI può essere aumentato parallelamente all’aumento dell’HDL-C in soggetti trattati con Niacina e con livelli di HDL-C bassi di base, [53] LXRα sembra richiedere un coattivatore (PPARγ) per esercitare questi effetti, [54] che è attivato dal recettore della Niacina. [55] Tuttavia, l’attività del recettore della Niacina non è stata richiesta per i suoi effetti sui livelli di colesterolo, suggerendo che altri meccanismi potrebbero essere rilevanti.
PPARγ
L’altra teoria relativa alla sintesi di HDL dalla Niacina afferma che ciò dipenda dalla proteina di trasferimento dell’estere del colesterolo (CETP) nonostante la riduzione del colesterolo totale e dei trigliceridi non richieda per entrambe questa proteina. [56] [57] CETP è una proteina che facilita il trasferimento di lipidi tra diverse lipoproteine (generalmente donando un trigliceride da vLDL a HDL e prendendo un estere di colesterolo in un processo noto come trasporto inverso di colesterolo. [58]) La Niacina riduce l’espressione di CETP nel fegato e la sua attività nel sangue dei topi; [56] una riduzione del CETP aumenta la quantità di HDL-C nel sangue poiché i tassi di catabolismo dell’HDL / LDL riflettono l’attività del trasporto inverso del colesterolo e raggiungono rapidamente l’equilibrio, [59] e se il CETP è ridotto allora sarebbe necessario più HDL per normalizzare i tassi di trasporto inverso del colesterolo. Questo meccanismo può anche essere correlato a LXRα, poiché mentre un eteromero di LXRα con il recettore nucleare di vitamina A (RXR) attiva l’elemento DR4 aumenta la CETP [60] la Niacina agevola l’eterodimerizzazione di LXRα e PPARγ che attiva ancora DR4, ma in un modo che promuove l’efflusso di colesterolo. [61-44] Questa eterodimerizzazione competitiva [62] non è stata ancora dimostrata sperimentalmente, e lo studio che ha utilizzato dosi di Niacina da 2g nell’uomo non è riuscito a trovare un’influenza sull’attività del CETP nel siero nonostante un aumento dell’HDL. [63]
L’ultimo potenziale meccanismo per l’aumento dell’HDL non consiste nel suo incremento di sintesi ma piuttosto nel preservare il colesterolo HDL già sintetizzato arricchito con apoAI, riducendo il tasso in cui la lipoproteina viene assunta nelle cellule epatiche nonostante la donazione di colesterolo dall’HDL a queste cellule sia inalterata a causa della riduzione dell’espressione del recettore (catena beta sintasi ATP) che normalmente trasporta l’HDL nella cellula. [64] Questa ipotesi funziona meglio con le osservazioni che suggeriscono che il ridotto catabolismo dell’HDL è il principale fattore determinante dei suoi livelli più elevati, [65] e influenza anche l’apoA1 poiché la sua clearance dal sangue e l’assorbimento da parte dei reni sono ridotti. [66]
Una supplementazione di Niacina a rilascio prolungato (1g) della durata di una settimana in soggetti con bassi livelli di HDL-C non sembra essere sufficiente da aumentare sensibilmente i livelli totali di HDL-C, sebbene sia stata notata una riduzione della dimensione media delle particelle; [23] le variazioni di HDL -C possono mediare un miglioramento della vasodilatazione dipendente dall’ossido nitrico, sebbene sia stato anche osservato un aumento della bilirubina indiretta. [23]
L’integrazione prolungata di Niacina nei diabetici è associata ad un aumento della quantità e delle dimensioni particellari dell’HDL-C (32,7%) mentre le particelle di dimensioni più piccole sono diminuite (8,2%). [67]
È stato osservato che la Niacina conferisce un effetto protettivo sulla mortalità cardiovascolare poiché una metanalisi [68] ha osservato che negli studi su soggetti con malattia coronarica la terapia con Niacina era associata a un minor rischio di rivascolarizzazione dell’arteria coronarica (RR di 0,31; IC al 95% di 0,15-0,63), infarto miocardico non fatale (RR di 0,72; IC al 95% di 0,60-0,86) e attacco ischemico transitorio (RR di 0,76; IC al 95% di 0,61-0,94) mentre la riduzione della mortalità complessiva non è riuscita a raggiungere significatività statistica (RR 0,883; IC 95% di 0,773-1,008). I sette studi inclusi in questa meta-analisi [32] [29] [31] [30] (e un follow-up [69]) hanno totalizzato 5137 pazienti che utilizzavano anche vari prodotti farmaceutici della classe di statine e fibrati .
In uno studio i cui partecipanti erano in terapia con statine e avevano bassi livelli di colesterolo HDL è stato rilevato che 1.5-2g di Niacina a rilascio prolungato sono stati in grado di fornire benefici additivi nel miglioramento dell’HDL-C (20%) e nella riduzione dell’LDL-C (17%) rispetto al placebo, sebbene per quanto riguarda l’endpoint clinico predeterminato (morte o ricovero in ospedale) sia la Niacina che il placebo avevano una uguale quantità di responder. [70] Questo studio ha rilevato un’alta percentuale di pazienti con sindrome metabolica (80%) e commenti [71] hanno suggerito che a causa di una possibile capacità della Niacina a rilascio prolungato di deteriorare l’insulino-resistenza [72] che i suoi benefici potrebbero essere compensati da questo effetto avverso, mentre lo studio stesso ha suggerito che i benefici delle statine hanno sostituito i benefici della Niacina.
Mentre uno studio precedente che utilizzava alte dosi di Niacina a rilascio immediato (3g) ha riscontrato una riduzione della morte del 14% rispetto al placebo insieme a una riduzioni del colesterolo totale, [32] ed è stato osservato che questa riduzione è simile per grandezza agli studi che combinano statine con placebo.
Studi in vitro suggeriscono che la Niacina potrebbe in teoria prevenire la formazione di placche aterosclerotiche riducendo l’infiammazione e il danno alla parete endoteliale attraverso diversi meccanismi. Limitate ricerche su animali hanno mostrato che la Niacina nella dieta, a concentrazioni paragonabili a quelle utilizzate per ridurre il colesterolo, riduce la deposizione della placca sulla parete dell’arteria e ritarda l’aterosclerosi.[73][74][75][76][77][78][79][80]
Niacina e sue interazioni con il metabolismo del glucosio
L’assunzione prolungata di Niacina è stata osservata causare una riduzione della sensibilità all’insulina, causando un aumento compensativo della produzione di insulina da parte delle cellule β del pancreas per mantenere i livelli di glucosio nel sangue. [81] La Niacina non sembra avere effetti diretti sulle cellule β pancreatiche, tuttavia, poiché la perfusione negli isolotti pancreatici (isole di Langerhans) di ratto isolati con Niacina in vitro non ha influenzato la secrezione di insulina. [82] Ciò indica che la Niacina aumenta la produzione di insulina mediante un meccanismo indiretto, secondario a causare insulino-resistenza periferica. È stato osservato che la supplementazione induce resistenza all’insulina a dosi comprese tra 500mg e 1g, che rientrano nell’intervallo di dosaggio che conferisce effetti di riduzione del colesterolo. [83]
In particolare, sembra che sia necessaria una supplementazione cronica di Niacina per aumentare la produzione di Insulina, poiché in uno studio è stato dimostrato che la supplementazione acuta riduce i livelli di questo peptide in soggetti altrimenti sani prima di un picco dopo un giorno, [84] mentre altri studi in acuto hanno notato un effetto minimo o nullo sui livelli di Insulina. [85] [86] [87] [88]
Gli effetti dell’integrazione cronica di Niacina sui livelli di Insulina possono anche dipendere dalla popolazione. È stato osservato che la Niacina provoca iperinsulinemia in soggetti che invecchiano altrimenti sani [83] (1g / die) ed è stato dimostrato che quasi raddoppiano i livelli di Insulina nei soggetti con NAFLD (2g / giorno [89] [90]). Nei pazienti con sindrome metabolica, l’integrazione di Niacina a 6 settimane di somministrazione alla dose di 1.5g / die ha aumentato i livelli di Insulina del 30%. [91]
Nei soggetti obesi con malattia del fegato grasso non alcolico (NAFLD), l’integrazione giornaliera di Niacina a rilascio prolungato (titolata fino a 2g) per 16 settimane sembrava aumentare lo stato di resistenza all’insulina nel fegato, nei muscoli e nel tessuto adiposo [89] con un effetto inibitorio sulle azioni dell’Insulina nel fegato notate negli uomini non diabetici con dislipidemia. [92]
Adiponectina
Negli uomini adulti con sindrome metabolica, è stato osservato che la Niacina a rilascio prolungato alla dose di 1.5g ostacola in modo significativo la sensibilità all’Insulina, valutata dall’HOMA-IR (42%), che è stata associata ad un aumento dell’Insulina sierica nonostante un aumento dell’Adiponectina sierica. [91] Questo è stato notato anche in un altro studio (aumento del 22% dell’HOMA-IR), in cui l’Aspirina assunta insieme alla Niacina non ha impedito la comparsa di una ridotta sensibilità all’Insulina. [93]
Questo effetto può persistere in soggetti altrimenti sani, poiché i soggetti trattati con 1g di Niacina per due settimane a cui veniva somministrato un clamp iperinsulinaemico-euglicemico richiedono meno glucosio per mantenere l’omeostasi, il che è indicativo di una riduzione dell’assorbimento del glucosio (attraverso un aumento dell’Insulino-resistenza). [94]
La resistenza all’Insulina indotta dalla Niacina è stata inizialmente attribuita a un effetto di rebound nel tessuto adiposo in cui un aumento del rilascio di acidi grassi non esterificati (NEFA) da parte della Niacina compromette gli effetti della segnalazione dell’Insulina. [95] [96] Ciò è plausibile, poiché la resistenza all’Insulina può essere indotta con infusione di NEFA in 24 ore nei roditori. [97] Altre fonti suggeriscono che la resistenza all’Insulina non è associata al rebound del NEFA, poiché i soggetti con NAFLD che sperimentano resistenza all’Insulina dalla terapia con Niacina non hanno necessariamente un aumento del NEFA nel siero. [89].
Modello ipotetico per i ruoli intracellulari del DGAT1 e DGAT2.
Un’altra possibile opzione è che la Niacina può inibire in modo non competitivo l’enzima noto come diacilglicerolo aciltransferasi 2 (DGAT2) con un IC50 di 100 µM (potenza simile a circa 300 µM). [98] L’inibizione di questo enzima non causa di per sé resistenza all’insulina con la somministrazione di Niacina, [92] ma poiché il DGAT catalizza il primo stadio della sintesi dei trigliceridi, la sua inibizione può favorire l’accumulo di diacilglicerolo (DAG) che è la molecola che si ritiene spieghi parzialmente la resistenza all’insulina data dalla Niacina. [92] Poiché l’aumento del DAG nelle cellule del fegato sopprime la segnalazione dell’Insulina, [99-162] l’inibizione mediata dalla Niacina del DGAT2 provoca insulino-resistenza, [98] [89] ostacolando così la capacità dell’Insulina di sopprimere la sintesi di glucosio e promuovendo indirettamente uno stato di iperglicemia.
Sebbene l’integrazione cronica di alte dosi di Niacina riduca la sensibilità all’Insulina, ciò non è associato a variazioni dei livelli di glucosio a digiuno. [90] Ciò può essere spiegato da un aumento compensativo della sintesi di Insulina che contrasta la resistenza alla stessa, lasciando sostanzialmente invariati i livelli di glucosio nel sangue. [81]
L’attivazione del recettore della Niacina (HM74A) da parte di alcuni altri agonisti sembra ridurre rapidamente il glucosio sierico nei diabetici migliorando la sensibilità all’Insulina [100] o comunque migliorando i tassi di smaltimento del glucosio. [101] Ciò indica che lo stesso recettore della Niacina può avere effetti benefici sul metabolismo del glucosio e che la resistenza all’Insulina indotta dalla Niacina non si verifica tramite l’attivazione del HM74A.
Quando si osserva il muscolo scheletrico, è stato dimostrato che la terapia con Niacina induce resistenza all’Insulina in questo tessuto in soggetti obesi con NAFLD (2g al giorno nel corso di 16 settimane). Uno studio svolto su ratti a digiuno (il digiuno aumenta la concentrazione plasmatica di acidi grassi non esterificati (NEFA), similmente alla somministrazione di Niacina [102-135] e diminuisce il glicogeno del muscolo scheletrico [103]) in cui sono stati accuratamente somministrati 20mg/kg di Niacina ha mostrato che il glicogeno nel soleo era ridotto mentre il gastrocnemius e il fegato non sono stati influenzati. [103]
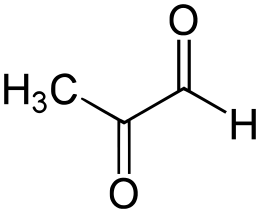
Metilgliossale
Quando il processo di glicazione è testato in vitro, la Niacina ha avuto solo effetti inibitori minori sulla glicazione dell’albumina sierica bovina da un noto agente glicante (Metilgliossale [104]) nonostante altri antiossidanti testati come lo Zinco (10-25 µg / mL) avessero più potenti benefici. [105]
È importante sottolineare che qualsiasi effetto della Niacina sulla glicazione in vitro deve essere interpretato con l’avvertenza che la Niacina riduce la sensibilità all’Insulina. Mentre la resistenza all’Insulina indotta dalla Niacina è ben compensata in soggetti sani giovani, lasciando sostanzialmente invariati i livelli di glucosio nel sangue, [81] la compensazione delle cellule β del pancreas negli individui più anziani o in quelli con ridotta tolleranza al glucosio era incompleta in uno studio, [83] causando aumenti nei livelli ematici di glucosio. Pertanto, la misura in cui la Niacina possa influenzare la glicazione in vivo non è chiara e probabilmente dipendente dalla popolazione.
Obesità e massa grassa
L’Adiponectina, un’adipochina nota per migliorare la sensibilità all’Insulina, per essere cardioprotettiva e ritenuta anche antiobesogena, [106] è aumentata in risposta all’attivazione mediata dalla Niacina del recettore HM74A nei topi. [107] La produzione di Adiponectina indotta dalla Niacina è stata rapida in questo studio, aumentando i livelli di questa adipochina del 37% entro 10 minuti da una dose di 30mg / kg per iniezione. I livelli sierici hanno raggiunto il picco dopo 60 minuti e sono rimasti elevati al di sopra del basale fino a 24 ore dopo la somministrazione. [107]
Leptina
È noto anche che la Leptina è aumentata in seguito alla somministrazione di Niacina nell’uomo [91], il che si ritiene si verifichi tramite un meccanismo simile poiché l’agonista farmaceutico HM74A Acipimox induce anch’esso la secrezione di Leptina dal tessuto adiposo in vitro [108] e in vivo. [109]
È stato osservato che la supplementazione di Niacina nel corso di sei settimane negli uomini obesi aumenta l’Adiponectina sierica del 43-56%, con circa metà dell’aumento rappresentato dalla forma ad alto peso molecolare [93] [91] insieme a un aumento del 26,8% della Leptina [91 ] senza cambiamenti osservabili nella Resistina. [91] L’Adiponectina è stata osservata aumentare di circa il 30% in soggetti obesi con NAFLD in risposta alla terapia con Niacina (fino a 2g al giorno), che era correlata con un aumento dell’Insulino-resistenza, [90] portando all’ipotesi che i due meccanismi siano intrecciati, forse come risposta adattativa. [90]
Resistina
Lo “spillover” degli acidi grassi risultante da una conservazione inefficiente del grasso dopo un pasto aumenta i lipidi sierici non esterificati (NEFA), [110] che influenzano negativamente la sensibilità all’Insulina epatica, aumentando la produzione di VLDL e potenzialmente svolgono un ruolo causale nella steatosi epatica. [111] [112] La somministrazione in acuto di Niacina (285 mg per via endovenosa) nell’uomo durante l’alimentazione ha dimostrato di ridurre lo spillover degli acidi grassi, promuovendo l’assorbimento del grasso alimentare nel tessuto adiposo e riducendo i Trigliceridi sierici e i NEFA. [113]
Al contrario, è stato osservato che un trattamento prolungato con Niacina, noto per favorire la resistenza all’Insulina nell’uomo, induce la resistenza all’Insulina adipocitaria, [114] che favorirebbe lo spillover degli acidi grassi, aumentando i livelli sierici di NEFA.[115]
Glucosio-6-fosfato deidrogenasi (G6PD)
È stato osservato che la Nicotinamide sopprime la differenziazione degli adipociti 3T3-L1 in modo dipendente dalla concentrazione con un range superiore a 10mM (il valore ED50), raggiungendo la soppressione completa a 20mM dopo nove giorni. [116] Si ritiene che ciò sia correlato a un effetto inibitorio sulla poli (ADP-ribosio) sintetasi, [116] che la Nicotinamide inibisce a 50µM mentre la Niacina non lo fa. [117] Quando aggiunta dopo differenziazione e in condizioni di glucosio elevato, la Nicotinamide sembra inibire il glucosio-6-fosfato deidrogenasi (G6PD) e prevenire il normale stress ossidativo. [118]
Il recettore dell’Acido Nicotinico è espresso negli dipociti in cui la sua attivazione sopprime l’adenilato ciclasi. [119] Questo effetto sembra essere circa il 30% più efficace negli adipociti rispetto ad altre linee cellulari (milza). [120] Poiché l’attivazione di questo recettore inibisce l’adenilato ciclasi, [119] e i fenolici che agiscono su di esso riducono anch’essi i tassi di lipolisi, [35] l’effetto complessivo dell’Acido Nicotinico sarebbe quello di ridurre la lipolisi negli adipociti, almeno a breve termine.
A lungo termine, tuttavia, il recettore dell’Acido Nicotinico può essere desensibilizzato con esposizione cronica a un agonista [121] e uno studio sui topi ha evidenziato che gli adipociti che sono diventati insulino-resistenti dopo la terapia con Niacina hanno mostrato una maggiore reattività dei recettori adrenergici (β1 e β2) all’aumentare dei livelli di cAMP nella cellula adiposa, [114] (il cAMP viene normalmente soppresso dalla Niacina che agisce sul recettore GRP109A [119]). Ciò potrebbe essere stato correlato alla sottoregolazione dei geni mediata dalla Niacina nella via di segnalazione dell’Insulina incluso il PDE3B, che normalmente degrada il cAMP, [114] una potenziale risposta adattativa nelle cellule adipose che è stata osservata avere la funzione di normalizzare i tassi di lipolisi (nei ratti sotto l’infusione di Niacina) . [97]
Un piccolo studio su sette partecipanti altrimenti sani che assumevano Niacina a 500mg/die, e aumentando la dose a 2g nel corso di due settimane ha mostrato una riduzione dei tassi di ossidazione dei grassi. [94] Tuttavia, a causa di un aumento dei tassi di ossidazione dei carboidrati, non vi era alcuna differenza netta nel tasso metabolico tra Niacina e placebo [94].
I topi privi di PARP-1 sembrano avere tassi metabolici più alti e una minore massa grassa; in assenza di PARP, aumentano le concentrazioni di NAD +, attivando le SIRT1 che quindi lavorano per deacetilare varie proteine (PGC-1α e FOXO1) per promuovere il dispendio energetico attraverso un metabolismo ossidativo aumentato e un incremento dei mitocondri.[122]
La SIRT2 e la SIRT3 non sono influenzate dalla bassa attività del PARP-1, [122] e l’inibizione della ribosilazione dell’ADP con altri mezzi come il knockdown NMNAT-1 sembra conferire anche effetti antiobesità nei roditori. [182] L’alimentazione aumenta acutamente l’attività del PARP-1 nei topi e ostacola transitoriamente l’attività della SIRT1, [122] che si pensa sia correlata al PARP-1 che ha la priorità per l’uso dei donatori di NAD +.
La supplementazione orale di Nicotinamide Riboside a 400mg/kg nel topo sembra aumentare il contenuto di NAD + nel muscolo scheletrico similmente a quanto avviene alla stessa dose di Niacina (Nicotinamide Mononucleotide inefficace in questo organo) [123] e sembra aumentare il dispendio energetico nei topi nutriti con un alto contenuto di grassi insieme all’aumento dell’attività dei geni bersaglio di FOXO1, suggerendo che l’integrazione orale è efficace. [123]
Esistono prove limitate nell’uomo che valutano gli effetti della Niacina sul tasso metabolico, sebbene l’estremità inferiore del dosaggio farmacologico della niacina (1g) in soggetti altrimenti sani non sia riuscito ad aumentare il tasso metabolico rispetto al placebo. [94]
Niacina, muscolo scheletrico e prestazioni fisiche
La somministrazione di Niacina nell’uomo ha dimostrato di aumentare l’espressione dei fattori di trascrizione PPARδ e PPARγ coactivator-1α (PGC-1α) nel muscolo scheletrico. [124] Poiché questi fattori di trascrizione sono importanti regolatori del metabolismo ossidativo e della biogenesi mitocondriale, [125] [126] questo suggerisce che l’integrazione con Niacina può svolgere un ruolo nella resistenza dei muscoli scheletrici.
Gli studi sugli animali hanno supportato questa idea, in cui è stato dimostrato che l’integrazione di Niacina provoca una transizione di fibre muscolari dal tipo II (contrazione rapida) al tipo I (contrazione lenta), aumentando anche il numero complessivo di fibre di tipo I nei muscoli scheletrici nello Zucker (ratto) obeso [127] e suini in crescita [128] (750mg di Niacina/kg di dieta) e pecore (1g di Niacina al giorno). [129] Questo effetto può essere limitato a determinati modelli animali, tuttavia, poiché studi su ratti sani hanno dimostrato che la Niacina ha un effetto trascurabile sulla distribuzione del tipo di fibra muscolare o sul fenotipo metabolico. [130] Inoltre, nonostante la Niacina aumenti l’espressione dei fattori di trascrizione pro-ossidativa nell’uomo, [124] fino ad oggi nessuno studio ha dimostrato che migliora le prestazioni o la capacità di resistenza del muscolo scheletrico.
Tuttavia, come substrato per la sintesi di NAD +, un’adeguata presenza di Niacina può supportare indirettamente il metabolismo ossidativo e la resistenza muscolare. In soggetti altrimenti sani, un lieve esercizio fisico sembra essere associato ad un aumento delle concentrazioni di NAD + nel sangue rispetto a uno stato di riposo (indipendente da qualsiasi integrazione [131]) mentre, quando testato in un esercizio lieve nei roditori, portava anche ad un aumento del NAD + nel sangue prima che diminuisse durante un esercizio ad esaurimento, [131] che è stato notato anche nel muscolo scheletrico. [132] A questo livello di esaurimento c’è un concomitante aumento del contenuto di NADH nel muscolo scheletrico [133] [134] che è stato proposto [135] indicativo di una riduzione del trasferimento di elettroni dal NADH alla sintesi di ATP.
È stato inoltre proposto [135] che da quando l’esercizio aumenta l’ossidazione nei tessuti stimolati e i fattori di stress ossidativo sono noti per alterare l’attività del ciclo di Kreb (TCA) [136] e la catena di trasporto degli elettroni (compresa la NADH deidrogenasi [137]) che forniscono antiossidanti aumenterebbe la resistenza secondaria alla conservazione della cinetica intramuscolare di NAD + / NADH. Quando si forniscono 36mg di picnogenolo [135] come antiossidante durante l’esercizio fisico fino all’esaurimento, sembra che la diminuzione del NAD + nel sangue sia stata invertita in un aumento con gli effetti (sia la diminuzione che l’aumento in attesa di integrazione) più marcati negli atleti allenati. [135]
Impatto organico della Niacina e principali effetti collaterali
Indometacina
In uno studio svolto sui ratti, la Nicotinamide ad un dosaggio di 20mg/kg somministrata un’ora prima di una dose di Indometacina che induceva ulcerazioni allo stomaco ha impedito l’ulcerazione a un livello paragonabile sia al controllo (nessuna induzione di ulcere) sia al farmaco di riferimento di 400mg/kg di sucralfato, che agisce localmente per forma una superficie protettiva per lo stomaco. [138] Questo effetto si è verificato insieme alla conservazione dell’attività del glutatione, alla riduzione della perossidazione lipidica e al miglioramento del muco gastrico. [138] Simili effetti protettivi contro l’ulcerazione indotta da etanolo e stress sono stati notati altrove, con il metabolita primario della Nicotinamide (1-metilnicotinamide; MNA). [139] Questo effetto gastroprotettivo è stato associato con un aumento dell’attività delle prostaglandine, in particolare la PGI2, [139] e nicotinamide, nonché il suo metabolita MNA sono stati implicati nell’aumento del flusso sanguigno gastrico [139] e nella riduzione della permeabilità microvascolare [138] dopo l’ulcerazione.
Interleuchina 10 (IL-10)
Nel colon dei topi, il recettore della Niacina (GPR109A) è necessario per la proliferazione ottimale delle cellule T CD4 + e la produzione di IL-10, che si traduce in un effetto antiinfiammatorio. [140] Questo effetto antinfiammatorio guidato dal GPR109A è mediato dal butirrato, l’acido grasso a catena corta del colon [140], che è un agonista del GPR109A ed è prodotto attraverso la fermentazione della fibra alimentare da parte dei batteri nel colon. [141] [142]
L’effetto riducente dei Trigliceridi dato dalla Niacina sembra da doversi ricondurre al fegato, dove la secrezione di lipoproteine a bassissima densità (vLDL) è ridotta; poiché le vLDL normalmente trasportano i Trigliceridi dal fegato ad altri tessuti, riducendo la secrezione di vLDL ciò si traduce in un livello sierico di Trigliceridi inferiori. [89] La diminuzione della secrezione di vLDL può essere secondaria all’inibizione della lipolisi nel tessuto adiposo, poiché l’aumento cronico di acidi grassi liberi nel siero può regolare negativamente la secrezione di vLDL. [143]
Sembra che l’integrazione di Niacina in acuto (che riduce gli acidi grassi liberi nel siero) sopprime anche la produzione di vLDL e la sua complessazione con i trigliceridi. [144] Ciò suggerisce un altro possibile meccanismo, che può verificarsi attraverso la soppressione acuta del PGC-1β, [145] una proteina nota per promuovere la secrezione di Trigliceridi dal fegato in risposta all’ingestione di grassi nella dieta. [146] In accordo con quest’ultimo meccanismo, la somministrazione di Niacina con un pasto ad alto contenuto di grassi sembra ridurre il picco normale dei trigliceridi postprandiali. [147]
Regioni regolatorie, fattori di trascrizione e molecole di segnalazione (citochine, fattori di crescita, metaboliti, farmaci) che modulano l’espressione del gene ABCA1 nei macrofagi e in altri tessuti. Le frecce e le linee di blocco indicano rispettivamente l’attivazione e la repressione.
Non è confermato come la Niacina riduca le vLDL-C, ma la sua capacità di stimolare l’attività del gene ABCA1 e aumentare il suo contenuto proteico nelle cellule del fegato è alla base dell’aumento dell’HDL-C, [148] che è noto anche per sopprimere la secrezione di vLDL-C. [57] La Niacina (2g per 16 settimane), nonostante riduca le vLDL-C e il complesso con Trigliceridi, non sembra aumentare significativamente il contenuto di trigliceridi intraepatici in soggetti con malattia del fegato grasso non alcolica (NAFLD). [149]
La Niacina sembra anche agire sulle cellule del fegato per promuovere l’accumulo di diacilglicerolo (DAG), che è associato all’insulino-resistenza localizzata. [92] La resistenza all’insulina nelle cellule del fegato riduce l’effetto soppressivo dell’insulina sulla sintesi del glucosio, con conseguente aumento dell’efflusso di glucosio dal fegato nel sangue. [150] Poiché gli stadi iniziali dell’insulino-resistenza indotta dalla Niacina (prima degli aumenti dell’insulina basale e del glucosio) sono stati associati a un fabbisogno ridotto di glucosio per bilanciare un morsetto euglicemico iperinsulinaemico, [109] questo suggerisce che l’inizio dell’insulino-resistenza avviene a livello del fegato. Il ruolo preciso del DAG in questo processo è tuttavia incerto. Mentre il DAG promuove la resistenza all’insulina attraverso l’attivazione di PKCε, [151] l’attivazione di questa proteina non è stata osservata nelle cellule del fegato che sono diventate insulino-resistenti con la Niacina. [92]
TRANSAMINASI. Enzimi intracellulari prodotti principalmente dagli epatociti. normalmente presenti in circolo a bassi livelli nel sangue. Aumentano in caso di danno cellulare. Indici molto sensibili ma moderatamente specifici di danno epatico. ALT è un marker più specifico di danno epatocellulare. (localizzazione citoplasmatica e più lunga emivita)
Nota: La Niacina in dosi terapeutiche può causare aumenti modesti delle transaminasi sieriche e della bilirubina non coniugata, entrambi biomarcatori del danno epatico. Le modifiche vengono invertite se il trattamento farmacologico viene interrotto e di solito si risolvono anche quando si continua l’assunzione. [152] [153] [154] Tuttavia, meno comunemente, la forma di rilascio prolungato del farmaco può portare a gravi epatotossicità, con insorgenza in giorni o settimane. I primi sintomi di gravi danni al fegato includono nausea, vomito e dolore addominale, seguiti da ittero e prurito. Si ritiene che il meccanismo sia una tossicità diretta della Niacina sierica elevata. Abbassare la dose o passare alla forma a rilascio immediato può risolvere i sintomi. In rari casi la lesione è grave e progredisce fino a insufficienza epatica. [152]
È noto che la Niacina rende le cellule β pancreatiche (un tipo di cellula specializzata che secerne insulina in risposta al glucosio) meno sensibile al glucosio sierico. [81] Inoltre, la normale riduzione della sensibilità al glucosio delle cellule β del pancreas associata all’invecchiamento può essere ulteriormente esacerbata dalla supplementazione di Niacina (500mg-1g due volte al giorno). [83] Anche se sembra esserci un aumento compensativo della secrezione di insulina nella risposta alla Niacina [83], in un modello di primati con diabete di tipo I, [155] questo non è stato sufficiente a ridurre la glicemia a livelli normali, con conseguente lieve iperglicemia e iperinsulinemia dopo due settimane di integrazione.[83]
Va notato che una linea cellulare coinvolta nel rossore cutaneo tipico della Niacina, nota come Langerhans, [156] [157] è diversa dall’area del pancreas nota come “Isole di Langerhans”.
Nota: il rossore dato dalla Niacina – una dilatazione a breve termine delle arteriole della pelle, che causa il colore della pelle rossastra – di solito dura circa 15-30 minuti, anche se a volte può persistere per settimane con uso cronico e di forme a lento rilascio. In genere, il viso è maggiormente interessato, ma la reazione può estendersi al collo e alla parte superiore del torace. La causa è la dilatazione dei vasi sanguigni [158] [93] dovuta all’aumento della prostaglandina GD2 (PGD2) e serotonina. [159] [160] [161] [162] Si pensava spesso che il rossore riguardasse l’istamina, ma è stato dimostrato che l’istamina non è coinvolta nella reazione. [159] Il rossore a volte è accompagnato da una sensazione di prurito, in particolare, nelle aree coperte da indumenti. [93]
Acido Acetilsalicilico (Aspirina)
La prevenzione del rossore richiede l’alterazione o il blocco della via mediata dalle prostaglandine. [93] [163] L’Aspirina [165-325mg] assunta mezz’ora prima della Niacina riduce fortemente il rossore, così come l’Ibuprofene [200mg] (una riduzione della frequenza e intensità del rossore fino al 50%). L’assunzione di Niacina ai pasti aiuta anche a ridurre questo effetto collaterale. [93] La tolleranza acquisita aiuterà a ridurre l’effetto; dopo diverse settimane a dosaggio costante, la maggior parte delle persone non ha più esperienza di vampate di calore. [93] Sono state sviluppate forme di Niacina a rilascio lento o “prolungato” per ridurre questi effetti collaterali. [164] [165]
Conclusioni sulla supplementazione di Niacina
Le informazioni che abbiamo a disposizione sulla Niacina e la sua supplementazione, ci presentano un composto senz’altro utile per il controllo o riassesto del quadro lipidico ma allo stesso tempo questa molecola risulta di una complessità d’azione biochimica non trascurabile. Il suo peggiorare l’insulino-resistenza in cronico ma con un maggior picco in acuto, picco che sembra venire controbilanciato da altri fattori come l’aumento della Adiponectina. Lo stesso effetto sulla riduzione della lipolisi può destare preoccupazione nell’atleta, specie se questo si trova in una fase ipocalorica con il principale intento di ridurre la massa grassa. Anche in questo caso sembrerebbe che l’effetto si manifesti in acuto per poi rientrare in condizioni pre-utilizzo. Ciò che è quasi certo, è che le osservazioni sul campo non hanno fatto emergere grosse differenze nell’alterazione della composizione corporea, sia con l’uso della Niacina in regimi ipercalorici che ipocalorici. L’utilizzo di GDA (in specie Berberina) anche alle dosi base efficaci potrebbe essere un “tampone” sufficienti a compensare almeno in parte i possibili peggioramenti dei parametri dell’insulino resistenza. I controlli della glicemia basale e della insulinemia a digiuno sono indicatori da tenere sotto controllo durante l’uso di Niacina. Non è da trascurare la possibilità che la Niacina possa influenzare lo “shift” dalle fibre muscolari di tipo II a quelle di tipo I, cosa non auspicabile per un Bodybuilder o altro atleta di forza.
In definitiva, considerando i dosaggi efficaci e la migliore azione in combinazione con statine (vedi Monacolina K), l’assunzione di Niacina può essere mantenuta con un certo margine di sicurezza tra i 500mg ed 1g/die, ovviamente tarando il dosaggio in risposta agli esami ematici di controllo.
Gabriel Bellizzi
Riferimenti:
“Niacin”. Micronutrient Information Center, Linus Pauling Institute, Oregon State University, Corvallis, OR. 8 October 2018. Retrieved 16 September 2019.
Hegyi J, Schwartz RA, Hegyi V (January 2004). “Pellagra: dermatitis, dementia, and diarrhea”. International Journal of Dermatology. 43 (1): 1–5.
“Why fortify?”. Food Fortification Initiative. 2017. Retrieved 4 April 2017.
Institute of Medicine (1998). “Niacin”. Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline. Washington, DC: The National Academies Press. pp. 123–149.
Bruckert E, Labreuche J, Amarenco P (June 2010). “Meta-analysis of the effect of nicotinic acid alone or in combination on cardiovascular events and atherosclerosis”. Atherosclerosis. 210 (2): 353–61.
“Niacin”. IN: LiverTox: Clinical and Research Information on Drug-Induced Liver Injury (Internet). Bethesda, MD: National Institute of Diabetes and Digestive and Kidney Diseases. February 2014.
L’1-Testosterone ( noto anche come Dihydroboldenone o, più semplicemente, come DHB) ha suscitato molto interesse negli ultimi anni* tra gli appartenenti alla comunità del Bodybuilding. La continua ricerca di nuove molecole dai maggiori potenziali spinge molti atleti facenti parte di questa “sottocultura” a sperimentare nuovi farmaci e/o diversi protocolli di applicazione di questi. Come ovvio che sia, per mancanza di preparazione degli interessati, queste sperimentazioni, e ciò accade molto spesso, si traducono in diffusione di dati non contestualizzati e privi di prove concretamente riconducibili al test su una specifica molecola. Fortunatamente, esiste una piccolissima percentuale di ricercatori indipendenti all’interno di questa comunità ed essi si preoccupano di valutare tutte le variabili in gioco, dalla certezza della molecola testata alla sua contestualizzazione d’uso (es. se essa è usata in mono-terapia o in co-somministrazione ecc…).
*In vero, il DHB è stato venduto legalmente online negli Stati Uniti, in forma prevalentemente orale, forma dalla scarsa biodisponibilità, dal 2002, anno in cui il brevetto del farmaco venne rilasciato per la vendita nel mercato degli integratori alimentari. Anche se tecnicamente la sua legalità era discutibile, la popolarità del nuovo composto è stata rapida e innumerevoli prodotti OTC come “Xtreme” ebbero largo seguito. Ciò durò fino al 2005, quando venne riclassificato come farmaco e inserito all’interno del Controlled Substances Act.
Tornando al punto originario del discorso, il quale ha la finalità di introdurre il lettore all’argomento trattato in questo articolo, il DHB, se attentamente analizzato, risulta decisamente ridimensionato nelle sue potenzialità e valutato sotto un altra luce in riferimento ai suoi possibili effetti avversi rispetto a quanto riportato dai comuni articolisti di settore. Motivo per cui, ora, torno nuovamente sull’argomento DHB, molecola da me già trattata tempo fa su questo sito, in modo maggiormente analitico su alcuni ed essenziali punti.
Ma iniziamo con ordine…
Caratteristiche base del 1-Testosterone
L’1-Testosterone (abbreviato in 1-Testo, 1-T), chiamato anche (nome IUPAC) 4,5α-dihydro-δ1-testosterone (Δ1-DHT) o 5α-androst-1-en-17β-ol-3-one, è uno steroide androgeno-anabolizzante (AAS), uno steroide androstano sintetico, che si differenzia dal Testosterone per l’aggiunta di un doppio legame in posizione C1-C2 e la mancanza del doppio legame in posizione C4-C5 nell’anello A. Il DHB è descrivibile più semplicemente, anche se pur sempre in modo incompleto, come il metabolita 5α-ridotto del Boldenone. [1] Venne sintetizzato negli anni 60 del XX Secolo e descritto per la prima volta nella letteratura medica occidentale nell’anno 1962.[2]
Il doppio legame in C1-C2 presente nello scheletro carbossilico del DHB stabilizza il ketogruppo in C3, essenziale per il legame androgeno.
Due proormoni del 1-Testosterone sono l’1-Androstenediolo e l’1-Androstenedione, l’ultimo dei quali può essere sintetizzato dallo Stanolone Acetato.[3]
Il Mesabolone è un chetale a base di 1-Testosterone.
L’1-Testosterone è anche noto per essere usato per sintetizzare il Metanolone e il Metenolone (comunemente noto come Primoboloan/Rimobolan).
L’aggiunta di un metile in posizione C17 da come risultato la molecola di Methyl-1-Testosterone.
Per via delle sue caratteristiche strutturali, l’1-Testosterone non converte in estrogeno, anche se non ci sono prove che all’interno del corpo (quindi in vivo) possa essere inserito un doppio legame in C4. [4,5] Ciò che sappiamo proviene dai dati aneddotici i quali suggeriscono che l’1-Testosterone sia un AAS con una attività estrogenica irrilevante. Effetti collaterali estrogeno-dipendenti, come ginecomastia, accumuli di grasso con modello femminile e maggiore ritenzione idrica non sono generalmente osservati quando il DHB viene somministrato in mono-terapia.
Essendo un AAS 5α-ridotto, esso non è soggetto all’enzima 5α-reduttasi e, di conseguenza, la sua attività androgena non può essere manipolata dall’uso di inibitori enzimatici con bersaglio l’enzima precedentemente indicato. Quindi, l’androgenicità relativa del 1-Testostosterone non è influenzata dall’uso di Finasteride o Dutasteride).
La Anabolico/Androgeno ratio dell’1-Testosterone riporta un valore pari a 200/100 in riferimento al Testosterone Propionato 100/100. Tale rapporto risulta però dubbio anche per il semplice fatto che il metodo attraverso il quale è stato estrapolato non è chiaro. Ma su questo ritorneremo più avanti.
I più sapranno sicuramente che il Boldenone è un derivato del Testosterone il quale è stato sintetizzato modificando la struttura del substrato di derivazione aggiungendo un doppio legame tra gli atomi di carbonio 1 e 2 dell’anello A dello scheletro carbossilico.
Questo legame rallenta drasticamente l’aromatizzazione della molecola rendendola e un substrato scarsamente affina anche per la 5α-reduttasi.
Sebbene il Boldenone rimanga un substrato affine alla 5α-reduttasi potendo essere quindi convertito in DHB, anche con la somministrazione di elevati dosaggi la quantità del metabolita derivante non è sostanziale.
La malsana pratica di utilizzare mega-dosaggi di Boldenone nel tentativo di ottenere concentrazioni sieriche moderate di DHB è stata applicata da molti culturisti in passato.
Poiché l’ormone progenitore (Boldenone) è un substrato così scarsamente affine per la 5α-reduttasi, un numero crescente di culturisti ha iniziato a optare semplicemente per l’uso diretto del DHB.
La scarsa e poco incoraggiante letteratura scientifica sul DHB
Esistono pochissimi dati clinici che valutano l’efficacia o il profilo di sicurezza del DHB.
C’è solo uno studio [6] a cui possiamo fare riferimento il quale mostra in realtà come il DHB sia paragonabile al Testosterone in un ambiente clinico controllato.
Questo studio è stato condotto sul 1-Testosterone (DHB) dopo che esso venne classificato come sostanza sottoposta a controllo e non più commercializzabile negli Stati Uniti.
L’obiettivo dello studio era stabilire che il DHB non è un “proormone”, come era stato affermato da molte società di integratori prima del suo ritiro dal mercato della supplementazione OTC.
Valutando l’effetto del DHB sui tessuti sensibili agli androgeni nel corpo attraverso il Recettore degli Androgeni (AR), lo studio ha stabilito che il DHB è a tutti gli effetti uno Steroide Androgeno Anabolizzante.
Mentre la premessa dello studio aveva lo scopo di esplorare semplicemente la transattivazione dipendente dall’AR mediata dal DHB, l’effetto misurato attraverso l’azione della molecola su vari tessuti del corpo rispetto al Testosterone ha fatto luce su quanto sia selettivo ed efficace il DHB.
Dopo essere stati orchiectomizzati (rimozione chirurgica dei testicoli), i ratti castrati , prima di essere utilizzati per l’esperimento, sono stati lasciati nelle loro gabbie per 7 giorni al fine di consentire un sufficiente declino ormonale.
Questo processo consente ai ricercatori di ridurre le variabili influenti la valutazione della ricerca e di somministrare androgeni esogeni, quindi di pesare diversi organi del corpo e misurare il grado di impatto di un farmaco specifico sul tessuto muscolare, sui tessuti sensibili agli androgeni come la prostata e sugli organi vitali.
Ai ratti dello studio è stato somministrato giornalmente per via sottocutanea un placebo, 1mg/kg/bw/giorno di Testosterone Propionato (TP) o 1mg/kg/bw/giorno di DHB (1-Testo).
Milligrammo per milligrammo, il DHB ha dimostrato di indurre una crescita muscolare leggermente inferiore rispetto al Testosterone, stimolando altrettanto la prostata e la vescicola seminale.
Effetto del Testosterone Propionato (TP) e del 1-Testosterone (1-Testo) sul muscolo Levator Ani dei ratti orchiectomizzati.
Ciò significa che per replicare lo stesso livello di crescita muscolare, il DHB dovrebbe essere somministrato a dosi più elevate del Testosterone.
Tornando alla precedentemente introdotta questione della presunta Anabolico/Androgeno ratio, l’ipotetico valore di 200/100 non si sa bene da dove provenga e come sia stato ricavato. Ciò che risulta piuttosto chiaro è che gli unici dati clinici che ho potuto trovare, e attraverso i quali mi è stato possibile valutare la selettività dei tessuti da parte del DHB, certamente non riflettono tale potenziale.
In effetti sembra essere meno selettivo a livello dei tessuti rispetto al Testosterone e complessivamente un po’ meno anabolico.
Osservando la struttura molecolare del DHB si evince una bassa resistenza alla disattivazione epatica di primo passaggio dovuta alla mancanza di modifiche atte a alterare il metabolismo dell’AAS. Sappiamo che la tossicità epatica si presenta a livelli significativi con l’uso di AAS metilati in C17, anche se molecole come il Trenbolone, aventi per modifiche strutturali non correlate a metilazione, presentano una certa resistenza alla disattivazione epatica causando un certo grado di tossicità potenziale.
I segni chiave della epatotossicità comprendono l’elevazione degli enzimi epatici e, in alcuni casi, persino l’ingrossamento del fegato.
Nello studio qui discusso, il gruppo di topi trattati con Testosterone Propionato non ha mostrato alcun segno di tossicità epatica. Tuttavia, il gruppo trattato con DHB ha avuto un aumento significativo del peso del fegato.
Effetto del Testosterone Propionato (TP) e del 1-Testosterone (1-Testo) sull’aumento del peso del fegato nei ratti orchiectomizzati.
Anche se quanto riscontrato a livello epatico nei topi trattati con DHB non sia una prova assoluta della potenziale tossicità del DHB, l’ipertrofia epatica potrebbe compromettere il funzionamento dell’organo se cronicizzata. Come accennaio tempo a dietro nella scheda tecnica del 1-Testosterone, qualcuno ipotizza (senza documentazione a convalidarlo) che l’ipertrofia epatica DHB-dipendente non è dovuta a fenomeni steatosici (“fegato grasso”) o tumorali, ma semplicemente alla capacità dell’1-Testosterone di legarsi ai recettori androgeni epatici; caratteristica comune ad uno dei prima citati anabolizzanti usatio come substrato di sintesi per il DHB, il Metenolone il quale in passato era ampiamente usato nella cura delle lesioni causate dalla cirrosi epatica.
Valutazione della molecola
Come valutare il DHB alla luce della (scarsa) letteratura disponibile e dei dati aneddotici provenienti dagli atleti (per lo meno quelli affidabili)?
Una cosa che si nota particolarmente nella scelta degli AAS da parte dei bodybuilder è che questi ultimi tendono a optare per molecole non aromatizzabili, terrorizzati dall’ignoranza che aleggia sugli estrogeni e sulla loro corretta gestione. Sebbene un AAS non aromatizzabile si dimostri discretamente più gestibile sotto alcuni aspetti, la mancanza di attività estrogenica indiretta può causare una serie di problemi che vanno dalla sfera psicologica alla disfunzione erettile. Tali effetti sono semplicemente evitabili, insieme a quelli legati ad una condizione di iperestrogenemia, gestendo i fattori estrogenici attraverso dosaggi intelligentemente settati e, nel caso, l’inserimento di anti aromatase e/o SERM. La scelta di una molecola dovrebbe basarsi maggiormente sui potenziali che potrebbero offrire sotto l’aspetto della sicurezza e potenza in rapporto alle altre molecole disponibili. In breve, AAS che sono più selettivi nei tessuti bersaglio e anabolizzanti del Testosterone.
Sebbene il DHB non sia un substrato soggetto all’enzima aromatasi, ciò non è, per esempio, particolarmente vantaggioso per un suo ipotetico uso in monoterapia, poiché richiederebbe la somministrazione esogena di Estradiolo (o di un substrato soggetto alla conversione in esso) al fine di evitare la comparsa di effetti collaterali psicofisici dovuti ad un basso livello estrogenico.
In conclusione, non vi sono dati certi sull’effetto che il DHB può avere sul corpo umano fatta eccezione per le testimonianze raccolta nei forum e, questa volta con qualche valenza in più, dai dati registrati dai ricercatori indipendenti che alternano i libri alla ghisa. Gli unici dati “certi” che abbiamo provengono, come visto, dal modello preclinico di roditori, e non sono nemmeno così promettenti.
Sebbene io non sia contrario alla sperimentazione indipendente, con questa molecola tale pratica può permettersela soltanto chi è in possesso di approfondite conoscenze in campo medico con particolarità nell’Endocrinologia, Andrologia e Farmacologia. I meno esperti, e meno portati, dovrebbero limitarsi alla scelta di molecole con effetti terapeutici riportati basati sul profilo preclinico registrato e anche in sperimentazione umana.
Il DHB nella pratica si è dimostrato una molecola discreta in “Cut” con una capacità miotropica stimabile con quella del Metenolone e del Boldenone.
Ma le sue limitazioni ad oggi persistono e sono:
Nessuno studio svolto su esseri umani e sul/i quale/i poter fare riferimento;
Leggermente meno anabolico e selettivo a livello tissutale rispetto al Testosterone, almeno in base agli studi sui topi;
Potenzialmente deleterio per il fegato sul lungo termine anche in forma iniettabile.
Occorrerebbe svolgere studi più approfonditi, ma dubito che qualcuno con mezzi adegiuati e possibilità maggiori rispetto ad un piccolo ricercatore indipendente si possa muovere in tal senso, anche perché non vi sono necessità che spingerebbero a ciò.
Gabriel Bellizzi
Riferimenti:
1- William Llewellyn (2009). Anabolics (9 ed.). Molecular Nutrition. p. 22,135.
2- Friedel A, Geyer H, Kamber M, Laudenbach-Leschowsky U, Schänzer W, Thevis M, Vollmer G, Zierau O, Diel P (August 2006). “17beta-hydroxy-5alpha-androst-1-en-3-one (1-testosterone) is a potent androgen with anabolic properties”. Toxicol. Lett. 165 (2): 149–55.
3- Zhang H, Qiu Z (December 2006). “An efficient synthesis of 5α-androst-1-ene-3,17-dione”. Steroids. 71 (13–14): 1088–90.
4- 358. K. J. Ryan. Acta Endocrinol. 35, Suppl. 51,697 (1960).
5- C. Gual, T. Morato, M. Gut, R.1. Dorfman, Endocrinology 71,920 (1962). 17beta-hydroxy-5alpha-androst-1-en-3-one (l-testosterone) is a potent androgen with anabolic properties. Friedel A, Geyer H, Kamber M.
Gli acidi biliari sono acidi steroidei che si trovano principalmente nella bile dei mammiferi e di altri vertebrati. Diverse forme molecolari di acidi biliari possono essere sintetizzate nel fegato da diverse specie.[1] Gli acidi biliari sono coniugati con Taurina o Glicina nel fegato e i sali di Sodio e di Potassio di questi acidi biliari coniugati sono chiamati sali biliari.[2][3][4]
Gli acidi biliari primari sono quelli sintetizzati dal fegato. Gli acidi biliari secondari derivano da azioni batteriche nel colon. Nell’uomo, l’Acido Taurocholico e l’Acido Glicocolico (derivati dell’Acido Colico) e l’Acido Taurochenodesossicolico e l’Acido Glicocchenodesossicolico (derivati dell’Acido Chenodesossicolico) sono i principali sali biliari nella bile e hanno approssimativamente la stessa concentrazione in essa.[5] Si trovano anche i sali coniugati dei loro derivati 7-alfa-deidrossilati, Acido Desossicolico e Acido Litocolico, con derivati degli acidi Colico, Chenodesossicolico e Desossicolico che rappresentano oltre il 90% degli acidi biliari umani.[5]
Enzimi di sintesi dell’acido biliare. Nell’immagine sono mostrati i 16 enzimi e le 17 reazioni che catalizzano la conversione del Colesterolo in un sale biliare coniugato. Sono mostrate la struttura del Colesterolo e un generico sale biliare coniugato. R = OH o H; X = Glicina o Taurina.
Gli acidi biliari rappresentano circa l’80% dei composti organici presenti nella bile (altri sono Fosfolipidi e Colesterolo).[5] Una maggiore secrezione di acidi biliari produce un aumento del flusso biliare. La funzione principale degli acidi biliari è quella di consentire la digestione di grassi e oli alimentari agendo come un tensioattivo che li emulsiona in micelle, [6] consentendo loro di essere sospesi colloidalmente nel chimo prima dell’ulteriore processazione. Hanno anche azioni ormonali sistemiche (in tutto il corpo), in particolare attraverso il Recettore X farnesoide e GPBAR1 (noto anche come TGR5).[7]
La sintesi degli acidi biliari si verifica nelle cellule epatiche che sintetizzano gli acidi biliari primari (Acido Colico e Acido Chenodesossicolico nell’uomo) mediante ossidazione del Colesterolo citocromo P450-mediata in un processo suddiviso in più fasi. Circa 600 mg di sali biliari vengono sintetizzati quotidianamente per sostituire gli acidi biliari persi nelle feci, anche se, come verrà descritto di seguito, ne vengono secreti quantitativi molto maggiori, riassorbiti nell’intestino e riciclati. La fase di limitazione della velocità in sintesi è costituita dall’aggiunta di un gruppo idrossilico in 7a posizione del nucleo steroideo da parte dell’enzima colesterolo 7 alfa-idrossilasi. Questo enzima è sotto-regolato dall’Acido Colico, sovra-regolato dal Colesterolo ed è inibito dall’azione dell’ormone Ileale FGF15/19.[2][3]
Circolazione enteropatica degli acidi biliari
Prima di secernere uno qualsiasi degli acidi biliari (primario o secondario), le cellule epatiche li coniugano con uno dei due aminoacidi, Glicina o Taurina, per formare un totale di 8 possibili acidi biliari coniugati. Questi acidi biliari coniugati sono spesso indicati come sali biliari a causa delle loro proprietà acido-base fisiologicamente importanti. Il pKa degli acidi biliari non coniugati è compreso tra 5 e 6,5, [4] e il pH del duodeno varia tra 3 e 5, quindi quando gli acidi biliari non coniugati si trovano nel duodeno, sono quasi sempre protonati (forma HA), il che li rende relativamente insolubili in acqua. Gli acidi biliari coniugati con gli aminoacidi riducono la pKa del coniugato acido biliare/aminoacido tra 1 e 4. Pertanto gli acidi biliari coniugati sono quasi sempre nella loro forma deprotonata (A-) nel duodeno, il che li rende molto più idro-solubile e molto più in grado di adempiere alla loro funzione fisiologica di grassi emulsionanti.[8][9]
Quando questi sali biliari vengono secreti nel lume intestinale, la disidrossilazione parziale batterica e la rimozione dei gruppi Glicina e Taurina formano gli acidi biliari secondari, l’Acido Desossicolico e l’Acido Litocolico. L’Acido Colico viene convertito in Acido Desossicolico e l’Acido Chenodesossicolico in Acido Litocolico. Tutti e quattro questi acidi biliari possono essere riportati nel flusso sanguigno, tornare nel fegato ed essere nuovamente secreti in un processo noto come circolazione enteroepatica.[2][3]
Come molecole anfipatiche con regioni idrofobiche e idrofile, i sali biliari coniugati si trovano in una condizione lipidica/acquosa e, alla giusta concentrazione, formano micelle.[9] La solubilità aggiunta dei sali biliari coniugati aiuta nella loro funzione prevenendo il riassorbimento passivo nell’intestino tenue. Di conseguenza, la concentrazione di acidi/sali biliari nell’intestino tenue è abbastanza elevata da formare micelle e solubilizzare i lipidi. Con il termine “concentrazione micellare critica” ci si riferisce sia a una proprietà intrinseca dell’acido biliare stesso sia alla quantità di acido biliare necessaria per adempiere alla funzionalità nella formazione spontanea e dinamica delle micelle.[9] Le micelle contenenti l’acido biliare aiutano le lipasi a digerire i lipidi e li avvicinano alla membrana dell’orletto a spazzola nell’intestino, con conseguente assorbimento dei grassi.[6]
La sintesi degli acidi biliari è una delle principali vie del metabolismo del Colesterolo nella maggior parte delle specie diverse dall’uomo. Il corpo produce circa 800mg di Colesterolo al giorno (con variabili determinate anche dal tipo di alimentazione seguita dall’individuo) e circa la metà di questi viene utilizzata per la sintesi di 400-600mg di acidi biliari al giorno. Gli esseri umani adulti secernono nell’intestino tra i 12 ed i 18g di acidi biliari ogni giorno, principalmente dopo i pasti. La dimensione del pool di acidi biliari è compresa tra i 4 ed i 6g, il che significa che gli acidi biliari vengono riciclati più volte al giorno. Circa il 95% degli acidi biliari viene riassorbito dal trasporto attivo nell’Ileo e riciclato nel fegato per un’ulteriore secrezione nel sistema biliare e nella cistifellea. Questa circolazione enteroepatica degli acidi biliari consente un basso tasso di sintesi ma con grandi quantità che vengono secrete nell’intestino.[5]
Gli acidi biliari hanno altre funzioni, tra cui l’eliminazione del Colesterolo dal corpo, nel flusso biliare per eliminare alcuni cataboliti (compresa la bilirubina), come emulsionante per le vitamine liposolubili e consentirne il loro assorbimento, favorire la motilità e la riduzione della flora batterica presente nell’intestino tenue e il tratto biliare.[5]
Gli acidi biliari hanno azioni metaboliche nel corpo simili a quelle degli ormoni, agendo attraverso due recettori specifici, i prima citati Recettore X farnesoide e il Recettore degli Acidi Biliari accoppiato con Proteine G/TGR5.[7][10] Si legano in modo meno specifico ad alcuni altri recettori ed è stata osservata una loro azione nella regolazione dell’attività di alcuni enzimi [11], canali ionici [12] e la sintesi di diverse sostanze tra cui Etanolamidi di acidi grassi endogeni.
I sali biliari costituiscono una grande famiglia di molecole, ed esse sono formate da una struttura steroidea con quattro anelli, una catena laterale a cinque o otto atomi di Carbonio che termina in un Acido Carbossilico e diversi gruppi idrossilici, il cui numero e orientamento sono diversi tra gli specifici sali biliari.[1] I quattro anelli sono classificati con le lettere A, B, C e D, dal più lontano al più vicino alla catena laterale con il gruppo carbossilico. L’anello D è più piccolo di un carbonio rispetto agli altri tre. La struttura è comunemente rappresentata con A a sinistra e D a destra. I gruppi idrossilici possono essere in una delle due configurazioni: in alto (o fuori), definita beta (β; spesso disegnata per convenzione come linea continua) o in basso, definita alfa (α; visualizzata come linea tratteggiata). Tutti gli acidi biliari hanno un gruppo 3-idrossile, derivato dalla molecola madre, il Colesterolo, in cui il 3-idrossile è beta. [1]
Da sinistra: struttura steroidea del Colesterolo rappresentata con i quattro anelli (A-B-C-D) e la sua struttura con la numerazione degli atomi secondo le indicazione della IUPAC.
Il primo passo nella via classica della sintesi epatica degli acidi biliari è l’aggiunta enzimatica di un gruppo idrossilico in posizione 7α da parte del Colesterolo 7α-idrossilasi (CYP7A1) che forma 7α-idrossicolesterolo. Questo viene quindi metabolizzato in 7α-idrossi-4-colesten-3-one. Ci sono più passaggi nella sintesi di un acido biliare che richiedono in tutto 14 enzimi.[3] Ciò comporta l’alterazione del legame tra i primi due anelli steroidei (A e B), che fa piegare la molecola; in questo processo, il 3-idrossile viene convertito nell’orientamento α. L’acido biliare a 24 atomi di Carbonio più semplice ha due gruppi idrossilici nelle posizioni 3α e 7α. Questo è l’Acido 3α, 7α-diidrossi-5β-colan-24-oico o, come più comunemente noto, Acido Chenodesossicolico. Questo acido biliare fu inizialmente isolato dall’oca domestica, dal cui nome deriva la porzione “cheno” del nome della molecola. Il 5β nel nome indica l’orientamento del legame tra gli anelli A e B del nucleo steroideo (in questo caso, sono piegati). Il termine “colan” indica una particolare struttura steroidea di 24 atomi di Carbonio e “acido 24-oico” indica che l’Acido Carbossilico si trova in posizione 24, alla fine della catena laterale. L’Acido Chenodesossicolico è sintetizzato da molte specie ed è il prototipo dell’acido biliare funzionale.[2][3]
Una via alternativa (acida) della sintesi dell’acido biliare è iniziata dalla sterolo mitocondriale 27-idrossilasi (CYP27A1), espresso nel fegato, nei macrofagi e in altri tessuti. Il CYP27A1 contribuisce in modo significativo alla sintesi totale dell’acido biliare catalizzando l’ossidazione della catena laterale dello sterolo, dopo di che la scissione di un’unità a tre atomi di Carbonio nei perossisomi porta alla formazione di un acido biliare C24. Anche le vie minori iniziate dalla 25-idrossilasi nel fegato e dalla 24-idrossilasi nel cervello possono contribuire alla sintesi dell’acido biliare. La 7α-idrossilasi (CYP7B1) genera ossisteroli, che possono essere ulteriormente convertiti nel fegato in CDCA.[2][3]
L’Acido Colico, 3α, 7α, 12α-triidrossi-5β-colan-24-oico, l’acido biliare più abbondante nell’uomo e in molte altre specie, è stato scoperto prima dell’Acido Chenodesossicolico. È un acido tri-idrossile-biliare con 3 gruppi idrossilici (3α, 7α e 12α). Nella sua sintesi epatica, l’idrossilazione 12α viene eseguita dall’azione aggiuntiva del CYP8B1. Come già descritto, la scoperta dell’Acido Chenodesossicolico (con 2 gruppi ossidrilici) ha reso la denominazione di questo nuovo acido biliare “Acido Desossicolico” in quanto aveva un gruppo idrossilico in meno rispetto all’Acido Colico.[2][3]
L’Acido Desossicolico è formato dall’Acido Colico mediante 7-deidrossilazione, risultando in 2 gruppi idrossilici (3α e 12α). Questo processo con Acido Chenodesossicolico da come risultato un acido biliare con solo un gruppo idrossilico 3α, chiamato Acido Litocolico (lito = pietra) che è stato isolato e classificato per la prima volta attraverso l’analisi di calcoli prelevati da un polpaccio. È scarsamente solubile in acqua e piuttosto tossico per le cellule.[2][3]
Diverse famiglie di vertebrati sono adatte ad utilizzare le modifiche della maggior parte delle posizioni sul nucleo steroideo e sulla catena laterale della struttura dell’acido biliare. Per evitare i problemi associati alla produzione di Acido Litocolico, la maggior parte delle specie è in grado di aggiunge un terzo gruppo ossidrilico all’Acido Chenodesossicolico. La successiva rimozione del gruppo idrossilico in posizione 7α da parte dei batteri intestinali si tradurrà quindi in un acido biliare diidrossile meno tossico ma ancora funzionale. Si ipotizza che nel corso del tempo, le locazioni per il posizionamento del terzo gruppo ossidrilico nei vertebrati si sia modificato. Sembrerebbe che, inizialmente, la posizione 16α fosse la principale, in particolare negli uccelli. Successivamente, questa posizione avrebbe lasciato il posto in un gran numero di specie alla posizione 12α. I primati (e gli esseri umani) utilizzano il 12α per la loro terza posizione del gruppo ossidrilico, producendo Acido Colico. Nei topi e in altri roditori, l’idrossilazione 6β forma acidi muricolici (α o β a seconda della posizione dell’idrossile 7). I suini hanno una idrossilazione 6α nell’Acido Ecolico (acido 3α, 6α, 7α-triidrossi-5β-cholanoico) e altre specie hanno un gruppo ossidrilico nella posizione 23 della catena laterale.
L’Acido Ursodesossicolico è stato inizialmente isolato dalla bile di orso, la quale è stata usata in medicina per secoli. La sua struttura ricorda l’Acido Chenodesossicolico ma con il gruppo 7-idrossile in posizione β.[1]
L’Acido Obeticolico, l’acido 6α-etil-chenodesossicolico, è un acido biliare semisintetico con maggiore attività come agonista del FXR che è oggetto di ricerche come agente farmacologico.
Acido Obeticolico (OCA). Questo acido biliare semisintetico ha dimostrato di migliorare i livelli ematici di ALT e ALP nei soggetti non responsivi al trattamento con Acido Ursodesossicolico (UDCA), ed è l’unica terapia di seconda linea autorizzata. Negli studi clinici l’OCA ha migliorato i risultati degli esami epatici nell’87% dei soggetti trattati ed è stato di aiuto quasi nel 50% dei pazienti.
Gli acidi biliari fungono anche da ormoni steroidei, secreti dal fegato, assorbiti dall’intestino e con varie azioni metaboliche dirette nel corpo attraverso il recettore nucleare FXR, noto anche con il nome genico NR1H4. [14][15][16] Un altro recettore degli acidi biliari è il recettore della membrana cellulare noto come recettore 1 o TGR5 degli acidi biliari accoppiato con proteine G. Molte delle loro funzioni come molecole di segnalazione nel fegato e nell’intestino sono mediate attivando il FXR, mentre l’interazione con il TGR5 può avviare coinvolgimenti nelle funzioni metaboliche, endocrine e neurologiche.[7]
In quanto tensioattivi o detergenti, gli acidi biliari sono potenzialmente tossici per le cellule e quindi le loro concentrazioni sono strettamente regolate. L’attivazione del FXR nel fegato inibisce la sintesi di acidi biliari ed è un meccanismo di controllo a feedback quando i livelli di acidi biliari sono troppo alti. In secondo luogo, l’attivazione del FXR da parte degli acidi biliari durante l’assorbimento nell’intestino aumenta la trascrizione e la sintesi di FGF19, che quindi inibisce la sintesi epatica di acidi biliari.[17]
Regolazione FXR e TGR5-mediata della sintesi degli acidi biliari e del metabolismo dei lipidi e del glucosio nel fegato e nell’intestino.
Prove emergenti associano l’attivazione del FXR con modifiche del metabolismo dei trigliceridi, del metabolismo del glucosio e della crescita del fegato.[7][18]
Gli acidi biliari si legano ad alcune altre proteine oltre ai loro prima citati recettori ormonali (FXR e TGR5) e ai loro trasportatori. Tra questi target proteici, l’enzima N-acil fosfatidiletanolammina specifica fosfolipasi D (NAPE-PLD) genera ammidi lipidici bioattivi (ad esempio l’Anandamide Cannabinoide Endogeno) che svolgono ruoli importanti in diversi percorsi fisiologici tra cui stress e risposte al dolore, appetito e durata della vita. La NAPE-PLD regola un dialogo di segnali incrociati diretti tra ammide lipidica e fisiologia degli acidi biliari.[13]
Poiché gli acidi biliari sono costituiti da Colesterolo endogeno, l’interruzione della circolazione enteroepatica degli acidi biliari abbasserà il Colesterolo. I sequestranti degli acidi biliari si legano a questi ultimi nell’intestino, prevenendone il riassorbimento. Così facendo, il Colesterolo endogeno viene deviato nella produzione di nuovi acidi biliari, riducendo così i livelli di Colesterolo. Gli acidi biliari sequestrati vengono quindi escreti nelle feci.[19]
I test per gli acidi biliari sono utili sia nella medicina umana che in quella veterinaria, poiché aiutano nella diagnosi di una serie di condizioni patologiche, tra cui vi sono i tipi di colestasi come la colestasi intraepatica della gravidanza, shunt portosistemico e displasia microvascolare epatica nei cani.[20] Anomalie strutturali o funzionali del sistema biliare provocano un aumento della bilirubina (ittero) e degli acidi biliari nel sangue. Gli acidi biliari sono correlati alla comparsa di prurito che è comune in condizioni colestatiche come nella Cirrosi Biliare Primaria (PBC), Colangite Sclerosante Primaria o Colestasi Intraepatica della Gravidanza.[21] Il trattamento con Acido Ursodesossicolico è stato usato per molti anni nel trattamento di questi disturbi colestatici.[22][23]
La relazione tra acidi biliari e saturazione del Colesterolo nelle precipitazioni di bile e Colesterolo nella formazione di calcoli biliari è stata ampiamente studiata. I calcoli biliari possono derivare da una maggiore saturazione di Colesterolo o bilirubina o da stasi biliare. Concentrazioni più basse di acidi biliari o fosfolipidi nella bile riducono la solubilità del Colesterolo e portano alla formazione di microcristalli. La terapia orale con acido Chenodesossicolico e/o Acido Ursodesossicolico è stata utilizzata per dissolvere i calcoli biliari di Colesterolo.[24][25][26] I calcoli possono ripresentarsi quando il trattamento viene interrotto. La terapia con un acido biliare può essere utile per prevenire i calcoli in determinate circostanze, come in seguito alla chirurgia bariatrica.[27]
Concentrazioni eccessive di acidi biliari nel colon sono una causa di diarrea cronica. Si manifesta comunemente quando l’Ileo è anormale o è stato rimosso chirurgicamente, come nella malattia di Crohn, o causa una condizione che ricorda la sindrome dell’intestino irritabile predominante nella diarrea (IBS-D). Questa condizione di diarrea da acido biliare/malassorbimento di acido biliare può essere diagnosticata dal test SeHCAT e trattata con sequestranti di acido biliare.[28]
Gli acidi biliari possono avere una certa importanza nello sviluppo del cancro del colon-retto.[29] L’Acido Desossicolico (DCA) risulta aumentato nelle sue concentrazioni nel colon dell’uomo in risposta a una dieta ricca di grassi.[30] Nelle popolazioni con un’alta incidenza di carcinoma del colon-retto, le concentrazioni fecali di acidi biliari sono più elevate, [31] [32] e questa associazione suggerisce che una maggiore esposizione del colon agli acidi biliari potrebbe svolgere un ruolo nello sviluppo del cancro. In un confronto particolare, le concentrazioni fecali di DCA nei nativi africani del Sudafrica (che seguono una dieta povera di grassi) rispetto agli afroamericani (che seguono una dieta ricca di grassi) erano 7,30 contro 37,51 nmol/g di feci umide.[33] I nativi africani del Sud Africa hanno un basso tasso di incidenza di cancro al colon inferiore a 1: 100.000, [34] rispetto all’alto tasso di incidenza per i maschi afroamericani che è di 72: 100.000.[35]
Attraverso studi sperimentali sono stati ipotizzati meccanismi che potrebbero regolare le alte concentrazioni di acidi biliari al carcinoma del colon. L’esposizione delle cellule del colon ad alte concentrazioni di DCA aumenta la formazione di specie reattive dell’ossigeno, causando stress ossidativo e aumentando anche il danno nel DNA.[36] I topi nutriti con una dieta con aggiunta di DCA che mimava i livelli di DCA del colon negli esseri umani che seguono una dieta ricca di grassi hanno sviluppato neoplasie del colon, tra cui adenomi e adenocarcinomi (tumori), a differenza dei topi alimentati con una dieta di controllo che causa un decimo delle concentrazioni di DCA nel colon e non hanno sviluppato neoplasie del colon.[37][38]
Gli effetti dell’Acido Ursodesossicolico (UDCA) nella modifica del rischio di sviluppo del carcinoma del colon-retto sono stati esaminati in diversi studi, in particolare nella colangite sclerosante primitiva e nella malattia infiammatoria intestinale, con risultati variabili in parte correlati al dosaggio.[39][40] La variazione genetica dell’enzima chiave di sintesi dell’acido biliare, CYP7A1, ha influenzato l’efficacia dell’UDCA nella prevenzione dell’adenoma colorettale in un ampio studio.[41]
Gli acidi biliari possono essere utilizzati in soluzione iniettabile da somministrare sottocute per il trattamento delle adiposità localizzate (vedi Mesoterapia). L’Acido Desossicolico in soluzione iniettabile ha ricevuto l’approvazione della FDA per il trattamento del grasso submentale.[42] Gli studi di fase III hanno mostrato risposte significative sebbene molti soggetti abbiano avuto lievi reazioni avverse come lividi, gonfiore, dolore, intorpidimento, eritema e rigidità intorno all’area trattata.[43][44] Della correlazione tra acidi biliari e tessuto adiposo, punto di particolare interesse in questo articolo, ne parlerò a breve.
Acido Ursodesossicolico
Gli atleti che utilizzano PED conoscono ormai da tempo il potenziale “epatoprotettivo” dato dall’uso degli acidi biliari. L’Acido Tauroursodesossicolico (TUDCA) e l’Acido Ursodesossicolico (UDCA) sono generalmente utilizzati come parte integrante dei “mix” epatoprotettivi, in specie durante l’utilizzo di farmaci orali con una forte resistenza al metabolismo epatico (vedi, per esempio, AAS metilati in C17). Infatti, l’UDCA ha mostrato di:
stimolare la secrezione di ATP da parte degli epatociti e di interagire quindi col sistema dei citocromi P450 riducendo la glicuronazione degli estrogeni sintetici (cosa che spiega in parte i suoi effetti benefici sulla colestasi epatica). Gli utilizzatori di AAS potrebbero non essere consapevoli del rischio aumentato di sviluppare calcoli biliari durante l’uso di molecole aromatizzabili e dell’aiuto nell’arginare il fenomeno correlato ad un aumento dei livelli estrogenici sul fattore in questione dato dal UDCA [45];
attivare direttamente il recettore per i glucocorticoidi, il che contribuirebbe ad allargare i meccanismi della sua azione anticolestatica ed antinfiammatoria sul parenchima epatico;
di fungere (come già accennato in precedenza) da agonista parziale del recettore FXRalpha coinvolto nell’espressione di proteine ed enzimi protettivi e/o regolatori del metabolismo intermedio riducendo la colestasi e l’intossicazione epatica stimolando dei meccanismi endogeni di difesa;
stimolare la sintesi del Glutatione (GSH), potente antiossidante endogeno, attraverso l’intervento delle chinasi dipendenti dai Fosfoinositidi (PI-3K e PKB). Questo meccanismo potrebbe giustificarne l’impiego corrente nelle epatiti croniche;
attivare un fattore di trascrizione particolare chiamato Nrf-2, presente allo stato di riposo nel citoplasma di tutte le cellule. Esso si attiva solo quando vi sono variazioni dello stato ossidoriduzione cellulare, ovvero la genesi di eccessive quantità di radicali liberi dell’ossigeno, o delle tossine esterne chiamate (xenobiotici). In risposta a questi stimoli, l’Nrf-2 entra nel nucleo cellulare ed induce una batteria di geni coinvolti nella protezione cellulare dallo stress ossidativo. Si pensa quindi che l’UDCA possa agire da ligando endogeno dell’Nrf-2, anche se non esistono ancora prove dirette o conclusive. Anche questo meccanismo ha azione protettiva sul tessuto epato-biliare.[46]
Come detto pocanzi, un altro uso degli acidi biliari conosciuto dai più in ambito sportivo e, soprattutto, culturistico, è quello, già accennato in precedenza, del trattamento mesoterapico. In particolare, Il Desossicolato di Sodio usato da solo o miscelato con Fosfatidilcolina, viene utilizzato nel trattamento delle adiposità localizzate (soprattutto nella zona submentale e periombellicale) in alternativa all’escissione chirurgica.[47]
Lo sviluppo della ricerca scientifica sugli acidi biliari e metabolismo/composizione corporea
Pratica di utilizzo degli acidi biliari meno conosciuta, ed ancora “embrionale” nella sua applicazione, è quella che vede la loro applicazione per il miglioramento della composizione corporea attraverso assunzione orale.
Già nel 2006 uno studio pubblicato sulla rivista Nature aveva rivelato che gli acidi biliari potevano aumentare il tasso metabolico e la perdita di peso nei topi, lasciando intravedere la possibilità che ciò si potesse verificare anche nell’uomo.[48] Mentre gli acidi biliari (AB) erano da tempo noti per essere essenziali nell’assorbimento dei lipidi nella dieta e nel catabolismo del Colesterolo, come abbiamo precedentemente visto, negli anni precedenti a questo studio era emerso un ruolo importante degli acidi biliari come molecole di segnalazione. Gli AB, come già detto, attivano le vie della protein chinasi attivate dal mitogeno, sono ligandi per il recettore TGR5 (GPCR) accoppiato alle proteine G e attivano i recettori dell’ormone nucleare come il recettore alfa X farnesoide (FXR-alfa; NR1H4). Il FXR-alfa regola il riciclo enteroepatico e la biosintesi degli AB controllando l’espressione di geni come il partner eterodimero breve (SHP; NR0B2) che inibisce l’attività di altri recettori nucleari. L’induzione SHP mediata da FXR-alfa è anche alla base della sottoregolazione della biosintesi di acidi grassi e dei Trigliceridi a livello epatico e della produzione di lipoproteine a bassissima densità mediata dalla proteina 1c legante gli elementi regolatori degli steroli. Ciò indicava che l’assunzione di AB avrebbe potuto essere in grado di funzionare al di là del semplice controllo dell’omeostasi degli stessi nelle vie metaboliche generali. Nello studio viene mostrato che la somministrazione di AB nei topi aumenta il dispendio energetico nel tessuto adiposo bruno, prevenendo l’obesità e il peggioramento della resistenza all’insulina. Questo nuovo effetto metabolico degli AB dipende in modo critico dall’induzione della sintesi dell’ormone tiroideo tri-iodotironina (T3) AMP-ciclico-dipendente attraverso l’attivazione l’enzima iodotironina deiodinasi di tipo 2 (D2). Il trattamento degli adipociti bruni e dei miociti muscolo-scheletrici umani con AB mostrò un aumento dell’attività del D2 e del consumo di ossigeno. Questi effetti sono indipendenti dall’FXR-alfa e sono invece mediati dall’aumentata produzione di cAMP che deriva dal legame degli AB con il recettore TGR5 accoppiato con proteine G. Sia nei roditori che nell’uomo, i tessuti più termogenicamente importanti sono specificamente presi di mira da questo meccanismo poiché coesprimono sia l’enzima D2 che il TGR5. Dallo studio emerse quindi che la via di segnalazione AB-TGR5-cAMP-D2 è un meccanismo cruciale per l’ottimizzazione dell’omeostasi energetica che può essere mirata a migliorare il controllo metabolico.
Gli acidi biliari stimolano l’espressione del D2 negli adipociti bruni. Rappresentazione schematica della via dell’acido biliare-TGR5 – D2 negli adipociti bruni Gli acidi biliari nella circolazione generale derivati dalla circolazione enteroepatica possono potenzialmente stimolare l’aumento del TAMP del cAMP, portando così ad un aumento dell’espressione del D2 nei tessuti in cui entrambe le proteine sono coespresse, ad esempio nel BAT e nel muscolo scheletrico. Questo percorso ha dimostrato di aumentare il dispendio energetico e proteggere dall’obesità indotta dalla dieta nei topi. Riprodotto in parte con il permesso di Baxter e Webb: Nature 439: 402–403, 2006 (446).
Dal 2006 ad oggi la ricerca sugli AB ed i loro effetti a livello metabolico-energetico sono proseguiti con ulteriori conferme sulla loro applicabilità per il trattamento dell’obesità negli esseri umani.
Uno studio del 2018 [49] ha evidenziato come il TGR5 sia un mediatore della conversione del tessuto adiposo bianco sottocutaneo (scWAT) in beige (metabolicamente simile al tessuto adiposo marrone) attraverso molteplici stimoli ambientali, tra cui l’esposizione al freddo e una alimentazione ricca di grassi. Inoltre, la somministrazione di mimetici degli acidi biliari TGR5-selettivi nei topi tenuti in ambiente termoneutrale porta alla comparsa di marker adipocitari beige e aumenta il contenuto mitocondriale nel tessuto scWAT dei topi Tgr5+/+ ma non nei loro simili Tgr5 -/- che si trovavano con loro e, quindi, esposti alle medesime condizioni ambientali. Questo fenotipo viene ricapitolato in vitro in adipociti differenziati, in cui l’attivazione del TGR5 aumenta la disponibilità di acidi grassi liberi attraverso la lipolisi, aumentando quindi la β-ossidazione e l’attività termogenica. La segnalazione del TGR5 induce anche la fissione mitocondriale attraverso il percorso ERK/DRP1, migliorando ulteriormente la respirazione mitocondriale. Nel loro insieme, questi dati identificano il TGR5 come un bersaglio farmacologico per promuovere la conversione del tessuto adiposo bianco in beige con potenziali applicazioni nella gestione dei disturbi metabolici.
Un altro studio, sempre del 2018, ha approfondito le dinamiche legate allo spiccato effetto metabolico e saziante, molto simile a quello ottenuto tramite bypass gastrico Roux-en-Y (RYGB), correlato alla diversione biliare verso l’Ileo (GB-IL) in modelli animali (roditori) obesi.[50] I ricercatori volevano appurare se i benefici metabolici di queste procedure (bypass gastrico) fossero mediate dall’aumento degli acidi biliari, dal momento che variazioni parallele del peso corporeo e altre variabili confondenti limitavano una possibile interpretazione oggettiva. Per fare ciò hanno utilizzato topi TGR5 -/- o FXRalfa -/- sottoposti ad una alimentazione ricca di grassi, confrontandoli con topi “selvaggi” con un antagonista del recettore per il polipeptide glucagone-simile 1 (Glp-1r) sottoposti ad una dieta chow (ricca in cereali e fibre). Il GB-IL ha indotto la perdita di peso e ha migliorato la tolleranza al glucosio assunto oralmente nei topi Tgr5 – / -, ma non nei topi FxrΔ -/- alimentati con una dieta ricca di grassi, suggerendo un ruolo dell’FXR intestinale. Il GB-IL in topi di tipo “selvaggio” alimentati con cibo convenzionale ha indotto miglioramenti della tolleranza al glucosio e al controllo della glicemia ematica indipendenti dal peso e secondari alla risposta aumentata dell’insulina. I miglioramenti erano concomitanti con un aumento dei livelli di GLP-1 linfatico nello stato a digiuno e un aumento dei livelli del batterio intestinale Akkermansia muciniphila. I miglioramenti nella glicemia a digiuno dopo il GB-IL sono stati mitigati con exendin-9, un antagonista del recettore del GLP-1 o Colestiramina, un sequestrante degli acidi biliari. Gli effetti glucoregolatori del GB-IL sono stati persi nei topi Glp-1r -/- a livello sistemico.
Confronto tra le caratteristiche anatomiche e il flusso di cibo e bile prima (Controllo) e dopo RYGB e GB-IL. (A) In risposta a un pasto, la bile della cistifellea e i succhi pancreatici vengono rilasciati nel duodeno (arancione) dove aiutano la scomposizione e l’assorbimento dei grassi alimentari mentre attraversano l’intestino tenue (digiuno e ileo). Gli acidi biliari vengono riassorbiti nell’ileo terminale (blu) in una circolazione enteroepatica elaborata. (B) Dopo RYGB, il cibo ingerito (punteggiato di viola) e la bile (frecce verdi interrotte) formano un mix (frecce rotte nere) ritardando l’assorbimento dei lipidi al digiuno prossimale / medio. (C) Nella diversione biliare verso l’ileo la miscelazione di nutrienti e bile viene ritardata fino all’ileo terminale.
Per avere una panoramica dettagliata ed estremamente chiara sui potenziali effetti metabolici e, con sequenzialmente, sulla composizione corporea dati dall’uso di acidi biliari ci viene in aiuto una interessantissima review che vaglia tutti gli studi svolti fino al 2018 sulle possibili applicazioni farmacologiche degli acidi biliari e dei loro derivati nel trattamento della sindrome metabolica, pubblicata sulla rivista “Frontiers in Pharmacology” del 3 Dicembre 2018.[51] Anche in questo caso, glia autori sottolineano come, oltre alle classiche funzioni degli acidi biliari nella digestione e nella solubilizzazione dei nutrienti e dei farmaci lipofili nell’intestino tenue (Mikov e Fawcett, 2006; Mircioiu et al., 2012), ci sono prove emergenti che indicano il ruolo degli acidi biliari e il loro derivati come segnalatori, molecole endocrine che esercitano una varietà di effetti metabolici attraverso percorsi complessi e intrecciati, diventando così una classe di moecole molto interessanti per la ricerca volta al vaglio di nuovi trattamenti per la sindrome metabolica (Taoka et al., 2016; Chávez-Talavera et al., 2017; de Boer et al., 2018; Molinaro et al., 2018; Shapiro et al., 2018). Questa review fornisce una panoramica delle attuali conoscenze relative alle implicazioni degli acidi biliari nel metabolismo del glucosio, dei lipidi e delle vie energetiche, nonché la potenziale applicazione degli acidi biliari nel trattamento della sindrome metabolica con raccomandazioni per ulteriori studi.
Nella review è nuovamente riportato come il ricircolo enteroepatico degli acidi biliari amplifica il flusso transepatico degli acidi biliari che attivano il recettore X farnesoide (FXR), che ormai sappiamo essere il fattore di trascrizione più significativo coinvolto nella regolazione della biosintesi e del trasporto degli acidi biliari. Inoltre, il flusso transintestinale di acidi biliari riassorbiti attiva la FXR intestinale, regolando il ricircolo enteroepatico di queste molecole anfifiliche. Questi eventi epatici e intestinali prevengono il sovraccarico e l’accumulo di acidi biliari negli epatociti evitando così la possibilità che si creino concentrazioni tossiche prevenendo così potenziali lesioni epatocellulari, diminuendo la loro stessa sintesi e assorbimento, mantenendo allo stesso tempo quantità sufficienti di acidi biliari nell’albero biliare e nel lume intestinale per l’emulsificazione dei lipidi alimentari . Inoltre, l’assorbimento epatocellulare degli acidi biliari è incompleto e la gamma micromolare delle concentrazioni si riversa dal portale alla circolazione sistemica attraverso anastomosi venose. Queste concentrazioni sistemiche sono sufficienti per interagire con diversi recettori nucleari attualmente identificati; FXR, recettore X della gravidanza (PXR), recettore della vitamina D (VDR) e recettore costitutivo del androstano (CAR), nonché il già più volte citato TGR5 (o GPBAR) che esercita effetti di segnalazione sistemici oltre i tessuti enteroepatici (Gioiello et al., 2014; Comeglio et al., 2017).
E’ interessante notare come la transattivazione ligando-dipendente di geni bersaglio da parte degli acidi biliari è indotta dal legame dell’acido biliare come ligandi endogeni più potenti, con il CDCA come il più potente agonista, mentre l’UDCA, idrofilo, non attiva l’FXR (Kemper, 2011). La potenza degli acidi biliari naturali per il FXR è riassunta nella tabella seguente (secondo Gioiello et al., 2014). Nell’intestino, il FXR attiva la trascrizione del enterokine, fattore di crescita dei fibroblasti-19 (FGF-19 o ortologo FGF-15 roditore) attraverso SHP, governando gli acidi biliari post-prandiale e il metabolismo dei nutrienti (Mertens et al., 2017).
SRC-1
Inoltre, come endobiotici, gli acidi biliari possono attivare la PXR (NR1I2, cromosoma 3q13.33), che possiede la capacità di interagire con una vasta gamma di composti idrofobici strutturalmente diversi tra cui farmaci, integratori alimentari e inquinanti ambientali. La PXR-non attivata più comunemente crea complessi inibitori con il mediatore silenziante per il recettore dell’acido retinoico e il recettore dell’ormone tiroideo (SMRT), SHP e deacetylases dell’istone (Pavlovic et al., 2017). Dopo il legame con il ligando, il recettore subisce cambiamenti conformazionali e il rilascio di fattori corepressori induce l’acetilazione dell’istone o il reclutamento di proteine co-attivanti come il SRC–1 (steroid receptor coactivator–1) (Pavek, 2016; Buchman et al., 2018).
Acidi biliari e metabolismo glucidico
Nell’ultimo decennio, un numero crescente di prove ha fortemente indicato che gli acidi biliari regolano il metabolismo glucidico postprandiale mostrando anche capacità di segnalazione antidiabetica esplicata attraverso i recettori attivati dall’acido biliare, oltre che a migliorare il ripiegamento e la funzione delle proteine, e agli effetti antiapoptotico (Hylemon et al., 2009).
Diagramma schematico del recettore (I) FXR nel fegato, (II) recettore FXR nell’intestino, (IIIA) recettore TGR5 nell’intestino, (IIIB) recettore TGR5 nella cistifellea e (IIIC) recettore TGR5 negli adipociti e nei muscoli.
Vi sono, tuttavia, alcune discrepanze irrisolte in letteratura tra i risultati ottenuti relativi all’implicazione degli acidi biliari nel metabolismo glucidico. Gli studi iniziali che hanno identificato le implicazioni del FXR nel metabolismo del glucosio hanno dimostrato che gli acidi biliari o gli agonisti sintetici specifici del FXR hanno indotto l’espressione del tasso di controllo dell’enzima della gluconeogenesi, fosfoenolpiruvato carbossinasi (PEPCK), aumentando anche la produzione totale di glucosio nell’epatocita umano e di ratto come nei topi in vivo (Stayrook et al., 2005). Al contrario, Yamagata et al. (2004) hanno dimostrato che gli acidi biliari sopprimono l’espressione dei geni della gluconeogenesi PEPCK, glucosio-6-fosfatasi (G6Pase) e fruttosio 1,6-bis-fosfatasi (FBP1) attraverso l’interazione tra SHP con il fattore nucleare degli epatociti 4 (HNF-4) o fattore di trascrizione della fronte Foxo1, sia in vivo che in vitro. La riduzione dell’espressione del PEPCK sembra essere una possibilità interessante per il trattamento del diabete di tipo 2, dato che questa patologia è caratterizzato da un aumento della produzione di glucosio epatico e da iperglicemia (Cariou et al., 2005). Inoltre, l’attivazione epatica del FXR porta ad un aumento dell’attività della glicogeno sintasi mediante fosforilazione e inattivazione della glicogeno sintasi chinasi 3 (GSK3b) (Mencarelli e Fiorucci, 2010).
GW4064
Nello studio del 2006 citato in precedenza (di Zhang et al.) è stato mostrato che la carenza del FXR nei topi è associata a intolleranza al glucosio e resistenza all’insulina manifestate da iperglicemia, ridotta tolleranza al glucosio e grave riduzione della sensibilità insulinica nel fegato, nei muscoli e nel tessuto adiposo. Di conseguenza, l’attivazione del FXR da parte dell’agonista sintetico non steroideo GW4064 nei topi (30mg/kg due volte al giorno) ha significativamente ridotto la produzione di glucosio epatico, abbassato i livelli di glucosio nel sangue, aumentato la glicogenesi, migliorato la sintesi e la secrezione di insulina, migliorato la sensibilità all’Insulina a livello centrale (epatico) e periferico negli animali (Zhang et al., 2006).
L’attivazione indotta del FXR nella trascrizione e nella secrezione di Insulina nelle cellule β del pancreas sono regolate da diversi meccanismi che coinvolgono effetti sia genomici che non genomici. Gli effetti genomici dell’attivazione del FXR si basano sull’induzione del KLF11, che si è dimostrato essere un fattore essenziale per la trascrizione del gene dell’Insulina. Gli effetti non genomici dell’attivazione del FXR nelle cellule βTC6 trasmettono sull’aumento della fosforilazione di Akt e nella traslocazione del trasportatore di glucosio di tipo 2 (GLUT2), un membro delle proteine di membrana che facilita il trasporto del glucosio lungo un gradiente di concentrazione alla membrana plasmatica, aumentando l’assorbimento di glucosio da parte delle cellule β del pancreas (Renga et al., 2010). Inoltre, l’attivazione del FXR induce l’espressione del trasportatore di glucosio GLUT4 nel fegato, la cui espressione risulta ridotta, sia nei soggetti diabetici di tipo 1 che 2 (Garvey et al., 1991, 1992). L’attivazione del FXR è stata in grado di sovraregolare l’espressione dei GLUT4 attraverso il FXRE nel promotore del gene GLUT4. Shen et al. (2008) hanno riferito che l’attivazione del FXR da parte del CDCA in concentrazione di 10 μM potrebbe indurre la trascrizione del GLUT4 nelle linee cellulari 3T3-L1 e HepG2 e aumentare l’espressione della proteina GLUT 4 nei topi C57BL / 6J trattati con CDCA (20 mg / kg / giorno ). Complessivamente, questi cambiamenti hanno comportato una riduzione del livello di glucosio plasmatico, una riduzione della gluconeogenesi epatica e un aumento della sintesi epatica di glicogeno (Mencarelli e Fiorucci, 2010).
Vie di segnalazione intrecciate mediate dagli acidi biliari nel metabolismo del glucosio. L’attivazione da parte degli acidi biliari delle vie di segnalazione FXR e TGR-5 inibisce la gluconeogenesi e promuove la sintesi del glicogeno nel fegato, promuove il rilascio di insulina stimolato dal glucosio nel pancreas, aumenta il dispendio energetico soprattutto nei muscoli scheletrici e nel tessuto adiposo bruno. Nel cervello, la segnalazione degli acidi biliari-TGR5 media la sazietà.
Gli acidi biliari possono anche influenzare l’omeostasi del glucosio in modo indipendente dal FXR (Stanimirov et al., 2012). Oltre al ruolo nel dispendio energetico, è stato dimostrato che l’attivazione del recettore di membrana TGR5 da parte degli acidi biliari aumenta la secrezione intestinale di GLP-1 dalle cellule L endero-endocrine, sia in vitro che in vivo, stimolando la rilascio di Insulina dalle cellule β pancreatiche senza rilascio di Glucagone dalle cellule α e la riduzione della glicemia postprandiale (Katsuma et al., 2005; Kumar et al., 2012; Duboc et al., 2014). Inoltre, Maruyama et al. (2006) hanno mostrato che i topi TGR5-null hanno una riduzione del 25% nelle dimensioni del pool di acido biliare, mentre i topi TGR5-null femmine hanno mostrato un significativo accumulo di grasso con aumento di peso corporeo rispetto a quello dei topi wild-type quando nutriti con una dieta ricca di grassi. La figura seguente mostra chiaramente i percorsi intrecciati delle implicazioni degli acidi biliari nel metabolismo del glucosio mediato dalla segnalazione FXR e TGR5.
L’evidenza genetica in vitro e in vivo indica un forte legame causale tra la capacità funzionale del reticolo endoplasmatico e gli effetti dell’Insulina. Pertanto, la modulazione della funzione del reticolo endoplasmatico potrebbe fornire un nuovo approccio per il trattamento del diabete (Ozcan et al., 2006). Ozcan et al. (2006) hanno mostrato che il tauro-UDCA migliora la resistenza all’Insulina attenuando lo stress del reticolo endoplasmatico negli animali affetti da diabete di tipo 2 con conseguente normalizzazione dell’iperglicemia, miglioramento della sensibilità sistemica all’insulina e dell’azione dell’insulina in vari tessuti.
L’efficacia degli agonisti sintetici del FXR come potenziale terapia per il diabete mellito di tipo 2 è stata dimostrata in uno studio clinico di fase II già menzionato condotto da Mudaliar et al. (2013) nel quale è stato dimostrano che la somministrazione di OCA a pazienti con diabete mellito di tipo 2 e NAFLD per 6 settimane è stata ben tollerata, migliorando la sensibilità all’insulina e riducendo i marker del infiammazione e la fibrosi epatica.
Come già visto, recentemente, la ricerca dei meccanismi alla base dei rapidi miglioramenti glicemici a seguito di procedure chirurgiche bariatriche [bypass gastrico Roux-en-Y (RYGB) e gastrectomia a manica verticale] in pazienti con obesità patologica e diabete di tipo 2 ha confermato i significativi effetti fisiologici degli acidi biliari nella omeostasi del glucosio (Bradley et al., 2012; Pournaras et al., 2012). Queste procedure hanno causato un aumento degli acidi biliari liberi sia nel siero che nell’intestino inferiore (Madsbad et al., 2014; Raghow, 2015). L’aumentata concentrazione di acidi biliari liberi nel lume intestinale ha avuto un effetto diretto sulle Incretine attraverso l’asse TGR5 – GLP-1, migliorando la secrezione di Insulina, nonché attivando il FXR intestinale e il suo target diretto a valle FGF-15/19 – un ormone peptidico postprandiale che migliora la tolleranza al glucosio, una proteina la cui espressione è, altrimenti, ridotta nei pazienti diabetici (Jansen et al., 2011; Schaap, 2012; Madsbad and Holst, 2014). Gli effetti benefici mediati dai recettori degli acidi biliari FXR e TGR5 in seguito a chirurgia bariatrica sono stati ulteriormente confermati poiché in assenza di segnalazione FXR in topi null-FXR o segnalazione GLP-1 antagonizzata dal exendin- (9-39), gli effetti benefici della chirurgia bariatrica sul peso corporeo e il metabolismo del glucosio sono stati annullati (Salehi et al., 2011; Ryan et al., 2014). L’aumento delle concentrazioni di acido biliare libero nelle parti inferiori del tratto intestinale dopo RYGB crea un ambiente adatto per la crescita di batteri tolleranti la bile come il gruppo tassonomico Proteobacter phylum (Osto et al., 2013). La crescita eccessiva dei Proteobacter provoca una diminuzione delle specie secondarie, ceppi batterici che generano attraverso i loro processi metabolici acidi biliari più tossici, con conseguente aumento dei livelli di acido biliare primario (Acido Chenodeossicolico) nel plasma e un pronunciato effetto sulle Incretine (Vrieze et al., 2014).
Acidi biliari e microbiota intestinale
Il tratto intestinale umano è colonizzato da un insieme diversificata di microorganismi, con batteri come membri più numerosi. La composizione del microbiota intestinale è specifica per l’individuo ma rimane relativamente costante nel tempo (Stojančević et al., 2014). Alterazioni del microbioma intestinale umano possono avere un ruolo nello sviluppo di alcune patologie (DeGruttola et al., 2016). Il microbiota intestinale regola il metabolismo dell’ospite producendo numerosi metaboliti che segnalano attraverso i loro recettori affini, influenzando così il peso corporeo, il metabolismo degli acidi biliari, l’attività pro infiammatoria, la resistenza all’Insulina e la modulazione degli ormoni intestinali (Han e Lin, 2014; Wahlstrom et al., 2016). Un’importante classe di metaboliti prodotti dal microbiota sono appunto gli acidi biliari. L’idrolasi batterica dei Sali biliari (BSH), presente in tutte le principali specie batteriche nell’intestino umano, effettua la deconugazione del sale biliare aumentando così la resistenza alla tossicità biliare, mentre la 7α-deidrossilasi converte gli acidi biliari primari CA e CDCA nella bile negli acidi secondaria DCA e LCA, rispettivamente (Wahlstrom et al., 2016). Altre conversioni dei sali biliari eseguite dal microbiota intestinale sono l’ossidazione e l’epimerizzazione dei gruppi idrossilici, 7-disidrossilazione, esterificazione e desolfatazione, contribuendo così alla grande diversità chimica e al pool di acidi biliari più idrofobi (Ridlon et al., 2016). Inoltre, gli acidi biliari possono modulare la composizione del microbiota intestinale sia direttamente, distruggendo le strutture delle membrane batteriche, sia indirettamente, tramite l’attivazione del FXR promuovendo la trascrizione di agenti antimicrobici come iNOS e IL-18 nell’intestino tenue e inibendo così la crescita batterica (Inagaki et al., 2006; Wahlstrom et al., 2016).
Considerando la complessa relazione tra acidi biliari e microbiota intestinale, non sorprende che oltre ai cambiamenti nella composizione degli acidi biliari, i pazienti diabetici abbiano anche una diversa composizione e attività del microbiota intestinale rispetto agli individui sani (Quercia et al., 2014 ). Sia il diabete mellito di tipo 1 che quello di tipo 2 sono associati a una riduzione della diversità microbica complessiva, caratterizzata da riduzione di Firmicutes e batteri produttori di butirrato, nonché da una funzione alterata della barriera intestinale e da una maggiore permeabilità intestinale (DeGruttola et al., 2016; Knip e Siljander, 2016).
La supplementazione con batteri probiotici ha portato a una modulazione benefica del metabolismo dell’ospite in termini di secrezione di GLP-1 stimolata da specifici metaboliti batterici come gli acidi grassi a catena corta (SCFA) attraverso il meccanismo GPR41/43-dipendente (Everard e Cani, 2014). Inoltre, la somministrazione di batteri probiotici ha ridotto l’assunzione di cibo e protetto dall’aumento di peso corporeo e dal peggioramento dell’insulino resistenza nei modelli animali obesi e diabetici (Yadav et al., 2013). Gli studi condotti hanno dimostrato che un approccio multi-terapeutico che utilizza una combinazione di probiotici e acidi biliari come terapia aggiuntiva in un modello di ratto con diabete mellito di tipo 2 ha esercitato una regolazione glicemica ancora migliore e ha portato ad alleviare le complicanze rispetto a ciascun singolo trattamento (Al- Salami et al., 2008, 2012). Gli effetti sinergici degli acidi biliari, probiotici e terapia antidiabetica attuale sono esaminati in dettaglio da Mikov et al. (2017) indicando la potenziale applicazione di questa combinazione nei disturbi metabolici con particolare enfasi sul diabete mellito.
Pertanto, la manipolazione terapeutica del microbiota intestinale mediante integrazione probiotica con effetti secondari sulla composizione del pool di acidi biliari rappresenta una strategia promettente per il trattamento di tali condizioni. Sebbene ulteriori studi siano altamente raccomandati per svelare gli esatti meccanismi responsabili degli effetti benefici della co-somministrazione di acidi biliari-probiotici, l’attivazione di complesse vie di segnalazione FXR e TGR5 è proposta come una delle possibili spiegazioni.
A questo punto è utile precisare che nella composizione corporea il microbiota è un primo “ostacolo” alle calorie assunte. Più esso risulta in equilibrio e in salute e maggiore sembra essere la capacità batterica di esercitare un “blocco” nei confronti dell’eccesso calorico. Il microbiota in condizioni di salute agisce anche sul senso di sazietà, attraverso una segnalazione che parte dagli enterociti (le cellule dell’intestino) per giungere ai centri ipotalamici della fame. Questo avviene perché il microbiota converte le fibre alimentari in acidi grassi a corta catena, i quali sono il nutriente principale degli enterociti. In questo modo le cellule intestinali inviano al cervello il segnale di sazietà.
Acidi biliari e metabolismo lipidico
Come prodotto principale del catabolismo del Colesterolo, gli acidi biliari esercitano effetti profondi, non solo sul metabolismo del Colesterolo, ma anche sul metabolismo dei triacilgliceroli, regolando quindi il metabolismo di varie specie di lipoproteine. L’aumentata sintesi di acidi biliari aumenta l’utilizzo del colesterolo come substrato. Attivando il FXR gli acidi biliari inibiscono il CYP7A1, l’enzima che limita la velocità della sintesi degli acidi biliari e del catabolismo del Colesterolo negli epatociti. Di conseguenza, l’integrazione a lungo termine di 750mg o 375mg/die di CDCA nei pazienti con malattia dei calcoli biliari provoca un modesto aumento del livello di colesterolo lipoproteico a bassa densità (LDL) (Schoenfield e Lachin, 1981). L’aumento del LDL si è verificato nell’85,2% dei pazienti che ricevevano 750mg/die e, nell’82,8% dei pazienti che ricevevano 375mg/die, tuttavia, l’incremento del 67,0% è stato registrato in un gruppo di pazienti che assumevano placebo, probabilmente come conseguenza principale legata alla malattia. D’altra parte, studi in vitro dimostrano che l’agonista del FXR CDCA (250 μM) stabilizza l’mRNA del recettore LDL e aumenta l’attività del recettore LDL nella linea cellulare epatica umana in coltura aumentando così l’assorbimento e la clearance delle particelle di LDL (Nakahara et al., 2002). I cambiamenti nel livello di Colesterolo circolante attraverso l’attivazione del FXR in vivo sono distinti tra modelli animali (roditori) e umani. Vale a dire che, l’espressione del CYP7a1 nei roditori è opposta agli umani nei quali è regolata da due recettori nucleari, il recettore X del fegato-α (LXR-α) e il FXR, entrambi espressi abbondantemente nel fegato. Il LXR-α può essere attivato da derivati del Colesterolo tra cui il 24 (S), 25-epossicolesterolo e il 24 (S) -idrossicolcololo, e in seguito alla sua attivazione LXR interagisce con un elemento di risposta all’interno del promotore del CYP7a1, stimolando in tal modo l’espressione genica. La traduzione degli effetti osservati nei roditori è stata sconcertante poiché in queste specie il Colesterolo circolante è prevalentemente “impacchettato” sotto forma di lipoproteine HDL, contrariamente alle specie LDL dominanti nell’uomo. Nei topi chimerici i cui fegati contengono principalmente epatociti umani e un profilo lipoproteico “umanizzato”, il trattamento con un potente agonista FXR specifico, il derivato dell’acido biliare semisintetico OCA (10mg/kg/giorno), porta all’aumento della riduzione circolante di LDL e HDL , analogamente all’attivazione del FXR nell’uomo. L’aumento del LDL è correlato alla ridotta attività regolante della proteina 2 legante gli elementi sterolici (SREBP-2) e alla sua espressione genica bersaglio, inclusa una significativa sottoregolazione nell’espressione della proteina del recettore LDL (Papazyan et al., 2018). La somministrazione di OCA, 25 o 50mg/die per 2 settimane, durante gli studi clinici ha prodotto effetti simili (Mudaliar et al., 2013; Walters et al., 2015).
La riduzione del livello di HDL mediante attivazione del FXR [utilizzando chow integrato con Acido Taurocolico allo 0,5% p/p (TLCA) per un periodo di 6 giorni] può essere spiegata dalla repressione del gene dell’apolipoproteina A-I, nonché dall’etero-scambio di esteri del colesterolo e triacilgliceroli tra le HDL plasmatiche e le lipoproteine contenenti ApoB inducendo l’espressione della proteina di trasferimento dell’estere del colesterolo (colesteril estere)(Lambert et al., 2003; Gautier et al., 2013). D’altra parte, il targeting FXR può anche rafforzare il trasporto inverso del Colesterolo, un processo nel trasporto del Colesterolo dai tessuti e cellule periferici agli epatociti e al sistema biliare, al fine di eliminare il Colesterolo attraverso la via intestinale. L’analisi Northern blot su campioni di fegato di topi maschi C57BL/6, alimentati con una dieta integrata all’1% per un mese, ha rivelato che gli effetti sopra menzionati sono mediati tramite la proteina di trasferimento dei fosfolipidi, una proteina che media il rilascio di fosfolipidi e Colesterolo dalle lipoproteine LDL a HDL. Gli effetti osservati sono anche una conseguenza dei cambiamenti nell’espressione del recettore scavenger-B1 (SR-B1), che è coinvolto nel riconoscimento delle particelle di HDL e nel loro assorbimento da parte degli epatociti (Urizar et al., 2000). Inoltre, è stato dimostrato che l’attivazione diretta del target FXR, l’enterokina FGF15/19, stimola la consistente secrezione di Colesterolo nel lume intestinale attraverso l’eterodimero di sterolo adenosina trifosfato (ATP)-legante i membri della sottofamiglia G 5/8 (ABCG5 / G8) nei topi (de Boer et al., 2017). Questa scoperta ha potenziali implicazioni nello sviluppo di strategie volte alla riduzione del riassorbimento intestinale del Colesterolo. La somministrazione di 10 o 25mg di OCA al giorno in volontari sani ha indotto un aumento sostenuto della concentrazione sierica di LDL e una riduzione dell’HDL, con un leggero aumento del livello di Colesterolo totale indipendentemente dalla dose (Pencek et al., 2016). Cambiamenti simili sono stati osservati in pazienti con diabete mellito di tipo 2 e in pazienti con steatoepatite non alcolica confermata per biopsia (Mudaliar et al., 2013; Neuschwander-Tetri et al., 2015). Tuttavia, nei pazienti con colangite biliare primitiva trattati con 5-10mg di OCA al giorno per 1 anno, l’aumento di LDL e la riduzione del livello di HDL era minore in ampiezza e transitorio (Nevens et al., 2016). Gli effetti dell’attivazione del FXR mediante somministrazione ripetuta di 5, 10 o 25mg di OCA al giorno per 2 settimane o 20 giorni sono stati stimati nella sperimentazione clinica che recluta volontari sani (Pencek et al., 2016). I risultati osservati in questo studio hanno confermato i risultati di precedenti studi clinici in termini di lievi disturbi dello stato lipidico; vale a dire, il trattamento con OCA ha indotto una riduzione dose-indipendente di HDL, come risultato della riduzione della concentrazione di particelle HDL piccole e medie e un aumento del livello di colesterolo LDL (Pencek et al., 2016). Anche se l’attivazione del FXR da parte degli acidi biliari e dei derivati degli acidi biliari può indurre un fenotipo potenzialmente aterogenico in termini di spostamento delle frazioni di lipoproteine, sono altamente necessari ampi studi clinici multicentrici randomizzati per chiarire l’impatto clinico di tali effetti dislipidemici trattati con acidi biliari o derivati dell’acido biliare.
D’altra parte, è stato dimostrato che l’integrazione di acidi biliari con CA allo 0,5% (w/w) per 8 settimane modula il metabolismo dei triacilgliceroli attraverso diversi meccanismi distinti, principalmente attraverso l’attivazione del FXR. Attivando gli acidi biliari il FXR vanno a reprimere la sintesi di de novo triacilglicerolo attraverso il target downstream FXR, l’inibizione mediata dal SHP della trascrizione dell’elemento regolatore sterolo che lega la proteina-1c (SREBP-1c), un fattore chiave che controlla la trascrizione di diversi geni che regolano la sintesi di acidi grassi e triacilgliceroli, incluso un principale enzima lipogenico nel fegato, acido grasso sintasi (Watanabe et al., 2004). È stato anche dimostrato che l’attivazione del FXR induce l’espressione del recettore-α del perossisoma attivato dal proliferatore (PPAR-α) e dell’isoenzima-4 piruvato deidrogenasi chinasi che promuove l’ossidazione degli acidi grassi, mentre mediante l’inibizione SHP-mediata si verifica l’inibizione dell’espressione proteica microsomiale di trasferimento del triacilglicerolo per ridurre la sintesi di VLDL (Pan et al., 2010). Inoltre, il FXR promuove l’attività dell’enzima ancorato alla superficie luminale delle cellule endoteliali vascolari, una lipoproteina lipasi (LPL) che catalizza quindi l’idrolisi dei triacilgliceroli inducendo l’espressione di apoC-II attivanti LPL in topi C57BL/6J alimentati per 3 settimane con una dieta contenente lo 0,5% di Colato di Sodio. Inoltre, l’attivazione del FXR ha indotto l’espressione del recettore VLDL, facilitando la clearance delle particelle VLDL nelle cellule HepG2 incubate con 50μM di CDCA per 24 ore (Jiao et al., 2015). Oltre a ridurre i sintomi della diarrea da acido biliare nei pazienti, l’integrazione orale di 25mg di OCA al giorno per 2 settimane ha comportato un aumento del livello di FGF-19 e una diminuzione di triacilgliceroli, il che implica il ruolo potenziale dell’FGF-19 sul metabolismo dei triacilgliceroli (Walters et al., 2015).
L’ulteriore complessità della rete di segnalazione tra gli acidi biliari e la rete del triacilglicerolo nasce dalle interazioni con il microbiota intestinale, che è comunemente cambiato nelle persone alimentate con una dieta di tipo occidentale e nell’obesità. In effetti, anche la somministrazione a breve termine di antibiotici non assorbibili orali ha provocato disbiosi intestinale seguita dalla diminuzione delle quantità di acidi biliari secondari DCA e LCA le concentrazioni epatiche e ridotto livello sierico di triacilglicerolo, che non si è ripreso nemmeno dopo l’integrazione con acido biliare (Kuno et al., 2018). I risultati dello studio suggeriscono che gli acidi biliari secondari prodotti dalle specie batteriche intestinali esercitano un ruolo regolatorio significativo nel mantenimento dei livelli sierici di triacilgliceroli e del metabolismo nell’ospite.
Acidi grassi e fegato grasso non alcolico dipendente
Gli effetti positivi dati dall’attivazione del FXR e del TGR5 mediata dagli acidi biliari in una vasta gamma di processi metabolici, tra cui il metabolismo del glucosio e la segnalazione dell’Insulina, il metabolismo dei trigliceridi e del colesterolo, come abbiamo appena visto, nonché sull’infiammazione, focalizzano l’interesse della ricerca sugli acidi biliari per lo sviluppo di un’efficace terapia strategica per trattare la patologia del fegato grasso non alcolica.
Grazie alla sua caratterizzazione e valutazione preclinica, l’OCA è diventato uno dei ligandi più comunemente utilizzati per la decodifica della rete di segnalazione FXR sia in vivo che in vitro. Durante gli studi preclinici la somministrazione di OCA ha avuto effetti benefici significativi in numerosi disturbi del sistema enteroepatico, incluso il miglioramento della colestasi indotta dagli estrogeni, la fibrosi epatica, la NASH, la resistenza all’insulina, l’ipertensione portale, la riduzione dell’infiammazione intestinale e il miglioramento della funzione della barriera ileale durante la colestasi, il miglioramento della diarrea cronica indotta dell’acido biliare (Fiorucci et al., 2004, 2005; Adorini et al., 2012; Verbeke et al., 2014; Walters et al., 2015). È stato dimostrato che la somministrazione per via orale di 25mg di OCA per 72 settimane migliora le caratteristiche istologiche del fegato di pazienti con NASH durante la seconda fase della sperimentazione clinica FLINT (Neuschwander-Tetri et al., 2015). Gli effetti benefici dell’attivazione del FXR nei pazienti con NAFLD comprendono una migliore sensibilità insulinica epatica e di conseguenza un aumento della sintesi di glicogeno, una diminuzione della de novo lipogenesi nel fegato, una migliore sensibilità all’insulina adiposa (riduzione degli effetti lipotossici) e una migliore funzione del recettore attivato dal proliferatore del perossisoma -gamma (PPAR-γ) e PPAR-α, che regolano il metabolismo degli acidi grassi e del glucosio sia nel tessuto adiposo che nel fegato (Fiorucci et al., 2007). L’integrazione dietetica con OCA (10mg/kg/giorno) per il modello animale NASH sottoposto a regime alimentare dieta fast food, che imita la sindrome metabolica, presenta micro RNA-21 ablato (miR21) e PPAR-α attivato che ha portato a una significativa riduzione della steatosi, infiammazione e lipo -apoptosi, svelando il riassetto dell’asse miR21/PPAR-α nel fegato e nei tessuti muscolari mediante attivazione del FXR dato dal OCA (Rodrigues et al., 2017). L’attivazione da parte del OCA (10mg/kg/giorno, somministrato per via orale per 7 settimane) del FXR e del TGR5 mediante un intervento dietetico di 8 settimane contenente 30mg/kg/giorno di INT-777, ha portato ad un miglioramento della steatosi epatica e dell’insulina resistenza nel modello animale (roditori) con NAFLD e obeso (Thomas et al., 2009; Cipriani et al., 2010). La somministrazione di OCA (25, 50mg/die per os per 6 settimane) in pazienti con diabete mellito di tipo 2 e NAFLD ha ridotto significativamente il peso corporeo, ha migliorato la sensibilità all’insulina e ridotto il livello sierico di gamma-glutamiltransferasi, mentre l’aumento sierico dell’FGF-19 enterokine intestinale ha confermato l’attivazione del FXR nei pazienti (Mudaliar et al., 2013). Inoltre, l’OCA (10mg/kg) ha prevenuto l’infiammazione epatica prevenendo l’immunomodulazione e l’infiammazione mediate dal fattore nucleare dannoso κB. Inoltre, attraverso l’inibizione dell’attivazione delle cellule stellate epatiche, l’incubazione con 10μM di OCA ha impedito la progressione verso la fibrosi epatica e lo sviluppo della cirrosi (Goto et al., 2018). Il trattamento con questo agonista del FXR ha comportato un miglioramento delle caratteristiche biochimiche e istologiche del fegato nei pazienti con NASH. Queste funzioni indicano che il FXR è un bersaglio terapeutico interessante per le malattie del fegato (Massafra e van Mil, 2018). Ulteriori studi clinici randomizzati di grandi dimensioni sono altamente desiderabili per confermare gli effetti del “bussare alla porta del FXR” come un potenziale approccio terapeutico in cui il cambiamento nella dimensione e nella composizione del pool di acidi biliari può essere sfruttato per il trattamento dei disturbi metabolici.
Acidi biliari , obesità e patologie cardiometaboliche
È stato riscontrato che un aumento del livello di acidi biliari circolante negli individui obesi è correlato positivamente con l’indice di massa corporea (Prinz et al., 2015). Inoltre, l’insulino-resistenza è associata alla sottoregolazione mediata dal Foxo-1 del CYP8B1 con conseguente deplezione del pool di acido biliare 12α-idrossilato, che può essere spiegato dalla degradazione del Foxo indotta dall’iperglicemia e quindi dalla mancanza di attivazione del CYP8B1, mentre nel diabete di tipo 2 la concentrazione nei pazienti trattati con DCA è risultata elevata (Brufau et al., 2010; Haeusler et al., 2013). Pertanto, gli interventi che manipolano la composizione del pool di acidi biliari potrebbero rappresentare nuove strategie terapeutiche nel trattamento della resistenza all’insulina. I cambiamenti nella dimensione e nella composizione del pool di acidi biliari dopo la chirurgia bariatrica in soggetti obesi si riflettono nel miglioramento dell’omeostasi metabolica (Penney et al., 2015). È quindi ragionevole presumere che l’integrazione con acidi biliari o derivati dell’acido biliare, modificando la segnalazione dell’acido biliare, possa essere considerata come un potenziale intervento cardioprotettivo che migliora il metabolismo e diminuisce il livello infiammatorio. Infatti, l’attivazione del TGR5 nei macrofagi e nelle cellule endoteliali da parte di livelli micromolari di acidi biliari circolanti sia durante il digiuno che nello stato postprandiale quando gli acidi biliari circolanti bilaterali raggiungono il picco di concentrazione, esercitano effetti anti-aterogenici e inibiscono l’aterosclerosi e la malattia coronarica (Steiner et al ., 2011). Il recettore TGR5, espresso negli adipociti, regola il dispendio energetico e il peso corporeo (van Nierop et al., 2017). Il GLP1 indotto dagli acidi biliari esercita anche effetti benefici sulla funzione endoteliale, sulla pressione sanguigna, sul metabolismo miocardico, sulla frazione di eiezione ventricolare sinistra, sull’aterosclerosi e sulla risposta al danno ossidativo indotto dalla riperfusione ischemica (Kang e Jung, 2016). La procedura RYGBP nei pazienti obesi comporta un aumento del livello di GLP1 con effetti cardioprotettivi. Inoltre, un aumento del flusso intestinale di acidi biliari a seguito di RYGBP porta all’attivazione del FXR intestinale e del suo target a valle, Enterokine FGF15/19, che ha dimostrato di reprimere l’espressione genica apo(a) mediante la cascata mediata dalla fosforilazione ERK1/2, e la riduzione del livello circolante di lipoproteine altamente aterogeniche (a) (Chennamsetty et al., 2012). La somministrazione e sovraespressione di FGF19 o FGF19 ricombinante in topi diabetici con deficit di Leptina ha dimostrato di ridurre l’aumento di peso, invertire la condizione diabetica, aumentare il tasso di ossidazione dei lipidi diminuendo anche il contenuto epatico di trigliceridi (Fu et al., 2004). Le procedure di chirurgia bariatrica comportano anche un aumento dei livelli circolanti delle specie di acido biliare, sia in stato di digiuno che postprandiale, nonché cambiamenti qualitativi nella composizione del pool di acidi biliari, a causa di una maggiore sintesi e riassorbimento epatico e riduzione dell’eliminazione intestinale o da cambiamenti nella composizione del microbiota (De Giorgi et al., 2015; Dutia et al., 2016). Di conseguenza, l’aumento della concentrazione di acidi biliari sia nel lume intestinale che nella circolazione sistemica, come potenti ligandi per FXR e TGR5, media il miglioramento del tasso metabolico, del glucosio e del metabolismo lipidico, aumenta la termogenesi con conseguente riduzione del peso corporeo, migliora l’infiammazione sistemica e promuove persino la conversione del tessuto adiposo bianco nel metabolicamente più attivo tessuto adiposo beige, simile al tessuto adiposo marrone (Fang et al., 2015). Questi miglioramenti metabolici implicano che il cambiamento nella composizione e nelle dimensioni del pool degli acidi biliari mediante approccio farmacologico o chirurgia metabolica influisce sul metabolismo sistemico con esito favorevole, suggerendo un nuovo approccio terapeutico nel trattamento dell’obesità e dei componenti della sindrome metabolica. Tuttavia, dato che gli acidi biliari attivano recettori nucleari multipli e possibilmente più di un GPCR, un’accurata dissezione e una valutazione approfondita delle vie di segnalazione mediata dagli acidi biliari su modalità specifiche dei tessuti dovrebbero fornire informazioni utili nel futuro sviluppo di nuovi derivati specifici e selettivi degli acidi biliari come nuovi agenti terapeutici nel trattamento della sindrome metabolica.
Regolazione dipendente dal recettore e indipendente dal recettore delle vie metaboliche e di segnalazione da parte degli acidi biliari nella sindrome metabolica. Gli effetti dei recettori FXR e TGR5 attivati dagli acidi biliari sul glucosio, sui lipidi e sul metabolismo energetico, nonché sugli eventi vascolari associati all’aterosclerosi. FXR e TGR5 hanno un numero significativo di effetti sovrapposti attualmente identificati. D’altro canto, gli effetti dell’UDCA non sono associati all’attivazione di questi recettori degli acidi biliari, mentre l’UDCA esercita vari effetti fisiologici / farmacologici associati alle sue proprietà strutturali specifiche.
Giunti a questo punto abbiamo appurato che le evidenze emergenti dell’ultimo decennio hanno indicato il ruolo degli acidi biliari come molecole di segnalazione endocrine che regolano il glucosio, i lipidi e il metabolismo energetico attraverso percorsi complessi e correlati che includono principalmente la cascata di segnalazione FXR e TGR5. Pertanto, la modulazione di questi percorsi di segnalazione utilizzando gli acidi biliari e i loro derivati e la co-somministrazione con batteri probiotici con effetti secondari sulla composizione del pool di acido biliare è diventata un’area di particolare interesse nella ricerca moderna che offre un nuovo approccio per il trattamento della sindrome metabolica. Tuttavia, molti dei risultati ottenuti derivano da studi condotti su modelli animali che dovrebbero essere presi in considerazione con una attenta interpretazione dei risultati a causa delle principali differenze nel metabolismo degli acidi biliari e nella composizione del microbiota intestinale tra animali e umani. Inoltre, le differenze interindividuali nella composizione del microbiota intestinale, vale a dire l’impronta batterica specifica in alcuni individui, contribuiscono anche a profili di acidi biliari altamente specifici nel singolo soggetto, che influenzano in modo differenziato la patogenesi della malattia e, presumibilmente, la risposta a interventi preventivi e terapeutici correlati agli acidi biliari, che richiedono ulteriori studi e chiarimenti. Da un punto di vista terapeutico, è altamente auspicabile un approfondimento degli effetti metabolici di ciascuna delle specie di acido biliare naturale e sintetico in vivo. I probiotici, come potenziale strategia per modulare la composizione del microbiota intestinale, dovrebbero essere ulteriormente studiati per aiutare il ripristino del metabolismo degli acidi biliari e potenzialmente aiutare nel trattamento dei disturbi metabolici. La ricerca futura dovrebbe basarsi su approcci metabolomici, proteomici e lipidomici in popolazioni sia sane che malate al fine di identificare biomarcatori correlati all’acido biliare che possano essere utili per la previsione della terapia correlata all’acido biliare di diversi disturbi metabolici.
Conclusioni pratiche “sperimentali” per l’uso degli acidi biliari nel miglioramento della composizione corporea
Ma all’atto pratico, l’atleta interessato al miglioramento della composizione corporea come può trarre qualcosa di applicabile in concreto da tutto quello che è stato fino ad ora riportato?
Ovviamente, come spesso è capitato e capita nell’ambiente del BodyBuilding, la “sperimentazione” di nuovi PED non è un comportamento inusuale. E l’applicazione d’uso “sperimentale” degli acidi biliari assunti oralmente al fine di migliorare la composizione corporea ha interessato diversi atleti fin dalle prime pubblicazioni scientifiche.
Alcune pratiche speculari vedono l’uso concomitante della Caffeina durante il periodo di assunzione degli acidi biliari al fine di aumentare l’attività del cAMP (Adenosina Monofosfato Ciclico) stimolato dall’attivazione del TGR5 indotta dall’acido bilare. Pertanto, l’efficacia degli acidi biliari potrebbe essere potenziata assumendo contemporaneamente una quantità ragionevole di Caffeina o altre Xantine. In tessuti come il grasso marrone, nei muscoli scheletrici e nel fegato, l’attivazione del TGR5 aumenta le concentrazioni dell’enzima Deiodinasi 2 (D2) nella cellula.[52] Come ben sappiamo, la D2 converte la forma dell’ormone tiroideo poco attiva T4, che ha per l’appunto un modesto effetto sul metabolismo, nella forma più attiva T3. Questo effetto si tradurrebbe in un vantaggio sia per l’atleta “Natural” in ipocalorica sia nell’atleta che utilizza T4 sintetico nella medesima circostanza. Pertanto, l’utilizzo di acidi biliari in concomitanza con l’uso di T4 porterebbe ad una maggiore conversione di questo in T3. In una fase preparatoria dove l’atleta usa il T4 insieme al GH, il quale ne aumenta già la conversione in T3, l’effetto potrebbe essere additivo. L’effetto in questione è stato osservato con l’uso di Acido Colico e Acido Chenodesossicolico, anche se è potenzialmente riproducibile con l’uso di Acido Ursodesossicolico o Tauroursodesossicolico.
cAMP
Per chi fosse totalmente digiuno di biochimica, il cAMP è il mediatore intracellulare degli effetti dell’ormone. Mentre l’ormone è considerato il primo messaggero che agisce sulle cellule dotate di recettori per l’ormone stesso il cAMP è il secondo messaggero, uguale per tutte le cellule e per tutti gli ormoni che porta il messaggio all’interno della cellula.
Dai risultati di cui sopra, ci sono suggerimenti che indicano i fattori che possono migliorare o compromettere gli effetti sulla composizione corporea legati agli acidi biliari. Come accennato in precedenza, la Caffeina può aumentare l’effetto del segnale del TGR5 agendo sul cAMP nei tessuti. [53] Di conseguenza, per esempio, l’uso di beta-2 agonisti (es. Clenbuterolo) può accentuare ulteriormente l’effetto degli acidi biliari. [53][54] (15-17) Gli Estrogeni e i contraccettivi orali alterano la produzione di acidi biliari, diminuendo in particolare la produzione di Acido Chenodesossicolico; le Statine che abbassano il Colesterolo riducono la produzione totale di acidi biliari fino al 30%.[55][56] Il consumo di alcol riduce la produzione di acidi biliari e gli alcolisti cronici hanno una marcata riduzione della produzione di acidi biliari a causa in parte di un danno epatico avanzato. [57] Gli acidi biliari non raggiungono concentrazioni significative nei soggetti che seguono una dieta povera di grassi. Una dieta ricca di grassi e ricca di proteine induce un aumento del 50% nella produzione di acidi biliari e gli effetti metabolici potrebbero essere ancora maggiori se la fonte proteica è ricca di Taurina e Glicina. [58][59]
Coloro che seguono regimi alimentari ipocalorici “Low-Fat” con uno schema di digiuno intermittente potrebbero notare ulteriori benefici dalla supplementazione con acidi biliari. Questa ipotesi nasce dal fatto che, a digiuno, l’attività della D2 è bassa e le concentrazioni sieriche di acidi biliari sono al minimo, in particolare tra gli obesi. Sebbene non sia stato analizzato in una camera metabolica, è ragionevole ipotizzare che l’integrazione con un acido biliare, insieme a Caffeina o altra molecola con caratteristiche lipolitiche e termogeniche, possa aumentare la beta ossidazione e la termogenesi (dissipazione dell’energia sotto forma di calore), rispetto a un integrazione priva della componente AB durante il digiuno. Ma ciò potrebbe essere vero anche con regimi differenti al digiuno intermittente “Low-Fat”.
Gli acidi biliari hanno una migliore assimilazione se assunti durante un pasto composto da un moderato quantitativo di grassi ed enzimi che ne favoriscono la digestione. In questo modo si verificherà un aumento significativo post-prandiale degli acidi biliari nel flusso ematico. Gli acidi biliari possono anche essere assunti a digiuno e possono aumentare l’effetto beta ossidativo indotto dall’esercizio fisico svolto in tale contesto, sebbene non molto significativo di per se. Le dosi dovrebbero essere contenute , poiché anche i livelli prodotti endogenamente possono provocare ripercussioni negative a livello del colon, dove i batteri convertono gli acidi biliari in metaboliti più tossici. Alcuni acidi biliari sono usati in medicina alla dose di 10 mg/kg/die e sono considerati sicuri – 1g per una persona di 100Kg; gli effetti collaterali legati ad un sovradosaggio includono diarrea ed in cronico portano ad alterazioni degli enzimi epatici. Di conseguenza, la dose di 1g/die è considerabile come il “tetto massimo” di sicurezza. Inoltre, c’è da ricordarsi che l’eccesso delle concentrazioni di acidi biliari dato dalla supplementazione ridurrà marcatamente la loro sintesi endogena, limitandone il beneficio complessivo. In medio stat virtus!
E, ovviamente, evitate tassativamente di darvi al “fai da te” o di affidarvi alla guida di qualche “pitecus gimnicus”…
Jump up to: abcd Fiorucci S, Mencarelli A, Palladino G, Cipriani S (November 2009). “Bile-acid-activated receptors: targeting TGR5 and farnesoid-X-receptor in lipid and glucose disorders”. Trends Pharmacol. Sci. 30 (11): 570–80. doi:1016/j.tips.2009.08.001. PMID19758712.
‘Essentials of Medical Biochemistry, Lieberman, Marks and Smith, eds, p432, 2007’
Wiemuth D, Sahin H, Falkenburger BH, Lefevre CM, Wasmuth HE, Grunder S (2012). “BASIC–a bile acid-sensitive ion channel highly expressed in bile ducts”. FASEB J. 26 (10): 4122–30. doi:1096/fj.12-207043. PMID22735174.
Makishima M, Okamoto AY, Repa JJ, et al. (May 1999). “Identification of a nuclear receptor for bile acids”. Science. 284 (5418): 1362–5. doi:1126/science.284.5418.1362. PMID10334992.
Parks DJ, Blanchard SG, Bledsoe RK, et al. (May 1999). “Bile acids: natural ligands for an orphan nuclear receptor”. Science. 284 (5418): 1365–8. doi:1126/science.284.5418.1365. PMID10334993.
Wang H, Chen J, Hollister K, Sowers LC, Forman BM (May 1999). “Endogenous bile acids are ligands for the nuclear receptor FXR/BAR”. Mol. Cell. 3 (5): 543–53. doi:1016/s1097-2765(00)80348-2. PMID10360171.
Kim, I; Ahn, SH; Inagaki, T; Choi, M; Ito, S; Guo, GL; Kliewer, SA; Gonzalez, FJ (2007). “Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine”. Journal of Lipid Research. 48 (12): 2664–72. doi:1194/jlr.M700330-JLR200. PMID17720959.
Davidson MH (2011). “A systematic review of bile acid sequestrant therapy in children with familial hypercholesterolemia”. J Clin Lipidol. 5 (2): 76–81. doi:1016/j.jacl.2011.01.005. PMID21392720.
Allen L, Stobie D, Mauldin GN, Baer KE (January 1999). “Clinicopathologic features of dogs with hepatic microvascular dysplasia with and without portosystemic shunts: 42 cases (1991-1996)”. J. Am. Vet. Med. Assoc. 214 (2): 218–20. PMID9926012.
Poupon RE, Balkau B, Eschwège E, Poupon R (May 1991). “A multicenter, controlled trial of ursodiol for the treatment of primary biliary cirrhosis. UDCA-PBC Study Group”. N. Engl. J. Med. 324 (22): 1548–54. doi:1056/NEJM199105303242204. PMID1674105.
Glantz A, Marschall HU, Lammert F, Mattsson LA (December 2005). “Intrahepatic cholestasis of pregnancy: a randomized controlled trial comparing dexamethasone and ursodeoxycholic acid”. Hepatology. 42 (6): 1399–405. doi:10.1002/hep.20952. PMID16317669.
Danzinger RG, Hofmann AF, Schoenfield LJ, Thistle JL (January 1972). “Dissolution of cholesterol gallstones by chenodeoxycholic acid”. N. Engl. J. Med. 286 (1): 1–8. doi:10.1056/NEJM197201062860101. PMID5006919.
Thistle JL, Hofmann AF (September 1973). “Efficacy and specificity of chenodeoxycholic acid therapy for dissolving gallstones”. N. Engl. J. Med. 289 (13): 655–9. doi:10.1056/NEJM197309272891303. PMID4580472.
Petroni ML, Jazrawi RP, Pazzi P, et al. (January 2001). “Ursodeoxycholic acid alone or with chenodeoxycholic acid for dissolution of cholesterol gallstones: a randomized multicentre trial. The British-Italian Gallstone Study group”. Aliment. Pharmacol. Ther. 15 (1): 123–8. doi:10.1046/j.1365-2036.2001.00853.x. PMID11136285.
Uy MC, Talingdan-Te MC, Espinosa WZ, Daez ML, Ong JP (December 2008). “Ursodeoxycholic acid in the prevention of gallstone formation after bariatric surgery: a meta-analysis”. Obes Surg. 18 (12): 1532–8. doi:10.1007/s11695-008-9587-7. PMID18574646.
Pattni, S; Walters, JR (2009). “Recent advances in the understanding of bile acid malabsorption”. British Medical Bulletin. 92: 79–93. doi:10.1093/bmb/ldp032. PMID19900947.
Degirolamo C, Modica S, Palasciano G, Moschetta A (2011). “Bile acids and colon cancer: Solving the puzzle with nuclear receptors”. Trends Mol Med. 17 (10): 564–72. doi:10.1016/j.molmed.2011.05.010. PMID21724466.
Reddy BS, Hanson D, Mangat S, et al. (September 1980). “Effect of high-fat, high-beef diet and of mode of cooking of beef in the diet on fecal bacterial enzymes and fecal bile acids and neutral sterols”. J. Nutr. 110 (9): 1880–7. doi:10.1093/jn/110.9.1880. PMID7411244.
Ou J, DeLany JP, Zhang M, Sharma S, O’Keefe SJ (2012). “Association between low colonic short-chain fatty acids and high bile acids in high colon cancer risk populations”. Nutr Cancer. 64 (1): 34–40. doi:10.1080/01635581.2012.630164. PMID22136517.
O’Keefe SJ, Kidd M, Espitalier-Noel G, Owira P (May 1999). “Rarity of colon cancer in Africans is associated with low animal product consumption, not fiber”. Am. J. Gastroenterol. 94 (5): 1373–80. doi:10.1111/j.1572-0241.1999.01089.x. PMID10235221.
Prasad AR, Prasad S, Nguyen H, Facista A, Lewis C, Zaitlin B, Bernstein H, Bernstein C. Novel diet-related mouse model of colon cancer parallels human colon cancer. World J Gastrointest Oncol. 2014 Jul 15;6(7):225-43. doi: 10.4251/wjgo.v6.i7.225. PMID25024814
Singh S, Khanna S, Pardi DS, Loftus EV, Talwalkar JA (2013). “Effect of ursodeoxycholic acid use on the risk of colorectal neoplasia in patients with primary sclerosing cholangitis and inflammatory bowel disease: a systematic review and meta-analysis”. Inflamm. Bowel Dis. 19 (8): 1631–8. doi:10.1097/MIB.0b013e318286fa61. PMID23665966.
Henriksson P, Einarsson K, et al. Estrogen-induced gallstone formation in males. Relation to changes in serum and biliary lipids during hormonal treatment of prostatic carcinoma. J Clin Invest 1989;84:811-6.
Sigma Aldrich; rev. del 12.07.2012
Duncan D, Rotunda AM (2011). “Injectable therapies for localized fat loss: state of the art”. Clin Plast Surg. 38 (3): 489–501, vii. doi:10.1016/j.cps.2011.02.005.
Watanabe M, Houten SM, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 2006;439:484-9.
Teodoro JS, Zouhar P, et al. Enhancement of brown fat thermogenesis using chenodeoxycholic acid in mice. Int J Obes 2013 Dec 6. [E-pub, ahead of print]
Dulloo AG, Seydoux J, et al. Potentiation of the thermogenic antiobesity effects of ephedrine by dietary methylxanthines: adenosine antagonism or phosphodiesterase inhibition? Metabolism 1992;41:1233-41.
van der Lans AA, Hoeks J, et al. Cold acclimation recruits human brown fat and increases nonshivering thermogenesis. J Clin Invest 2013;123:3395-403.
Everson GT, Fennessey P, et al. Contraceptive steroids alter the steady-state kinetics of bile acids. J Lipid Res 1988;29:68-76.
Bertolotti M, Zambianchi L, et al. Influence of newly synthesized cholesterol on bile acid synthesis during chronic inhibition of bile acid absorption. Hepatology 2003;38:939-46.
Monroe P, Vlahcevic ZR, et al. Effects of acute and chronic ethanol intake on bile acid metabolism. Alcohol Clin Exp Res 1981;5:92-100.
Bortolotti M, Kreis R, et al. High protein intake reduces intrahepatocellular lipid deposition in humans. Am J Clin Nutr 2009;90:1002-10.
Liaset B, Hao Q, et al. Nutritional regulation of bile acid metabolism is associated with improved pathological characteristics of the metabolic syndrome. J Biol
Come risaputo, il fattore primario che rende un AAS epatotossico è la presenza di una metilazione in C-17. L’esposizione prolungata nel tempo o ad alte dosi a molecole con tale metilazione può causare danni al fegato.
Per analizzare e trovare una soluzione al problema, bisogna necessariamente partire dalle basi.
Il parenchima epatico è la parte del fegato che filtra le tossine presenti nel sangue. In sostanza, è un laboratorio biochimico, in cui avvengono diversi processi:
1.Produzione della bile, responsabile per la digestione degli acidi grassi 2. Biosintesi dei fattori di coagulazione (vitamina K), responsabile dei meccanismi di coagulazione 3.Sintesi della maggior parte delle proteine che circolano nel plasma, tra cui l’albumina e la maggior parte delle globuline. 4.Deposito di glicogeno come approvvigionamento energetico (glicogenesi), scomposizione del glicogeno in glucosio (glicogenolisi) e sintesi di glucosio da fonti non glucidiche (gluconeogenesi) 5.Sintesi delle lipoproteine (HLD & LDL), responsabile per l’eterogenesi -amartomatosi infiammatoria nel endotelio arterioso. 6. Produzione ormonale ( IGF1 – Somatomedina C) 7.Deposito di Vitamine (Cianocobalammina-B12) 8.Pulizia e disintossicazione da sostanze chimiche (paracetamolo, farmaci antinfiammatori non steroidei-NSAID, glucocorticoidi, alcool-etanolo, AAS).
Aspetto tridimensionale del fegato normale di MIT OCW.
Ogni volta che una sostanza chimica entra nella circolazione passando in seguito per la vena porta del fegato, l’organo deve rilevare se questa è una sostanza tossica o qualcosa di utile per il sistema. Il fegato metabolizza molti farmaci che non causano problemi epatici. Tuttavia, alcuni AAS possono essere responsabile di una vasta gamma di tipi di problemi epatici e lesioni all’organo. I marcatori biochimici come gli enzimi epatici, noti come transaminasi (alanina transaminasi -ALT / SGOT e aspartato transaminasi-AST / SGPT), fosfatasi alcalina (ALP), γ-glutamil transpeptidasi (GGT), lattato deidrogenasi (LDH) e bilirubina ) sono spesso usati per indicare il danno epatico. Il danno epatico può essere definito da un aumento del:
• (a) livello della ALT superiore di tre volte il limite superiore al normale,
• (b) livello della ALP più del doppio del limite superiore del normale, o
• (c) livello totale di bilirubina superiore al doppio del limite superiore al normale, quando è associato ad un aumento ALT o ALP. Il danno epatico è ulteriormente caratterizzato nel tipo epatocellulari (elevazione ALT) e nel tipo colestatico (aumento ALP).
Gli AAS secondo la loro tossicità epatica, primariamente, sono classificati in due categorie principali:
• (a) Gli AAS metilati in C-17 (la maggior parte dei composti orali),
• (b) Gli AAS non metilati in C-17 (la maggior parte dei composti iniettabili).
Uno dei rari AAS iniettabili metilati in C-17 è lo Stanozololo (Winstrol Depot), mentre i maggiori composti orali non metilati in C-17 sono:
Mesterolone (Proviron)
Methenolone (Primobolan Acetato)
Testosterone Undecaonate (Andriol)
Tra gli AAS metilati in C-17 più forti troviamo:
Methyltrienolone (M3)
Fluoxymesterone (Halotestin)
Oxymetholone (Anadrol 50)
Methandrostenolone-Methandienone (Dianabol)
Methyltestosterone (Teston)
Stanozolol (Winstrol)
Methyldrostanolone (Superdrol)
Methylstenbolone (Ultradrol)
Methy-1-testosterone (M1T)
Methylhydroxynandrolone (MOHN)
Methylepitiostanolo (Epistane)
Chlorodehydromethyltestosterone (Oral Turinabol)
Mibolerone (Cheque Drops)
Tetrahydrogestrinone (THG)
Un caso a parte nei composti metilati in C-17 lo fa l’Oxandrolone, per via del suo assorbimento non a totale carico del fegato.
Tutti gli AAS sono responsabili della distorsione dell’indice ateromatico e dell’alterazione del rapporto delle lipoproteine (HDL / LDL). Questo eventualmente può portare allo sviluppo di malattie cardiovascolari (CVD). La metilazione della molecola steroidea assicura ad essa la capacità di resistere a qualsiasi ambiente ostile, come i succhi gastrici (acido). Quando la sostanza entra nel parenchima epatico, attraverso la circolazione, il fegato deve metabolizzarla. La metilazione è qualcosa di difficile da scomporre, pertanto questo mette sotto stress gli epatociti. Ciò può comportare un aumento dei livelli sierici di AST e ALT (> 100).
Gli AAS iniettabile che presentano una metilazione in C-17 (come lo Stanozololo iniettabile) mostrano una tossicità epatica leggermente inferiore. Una delle ragioni per cui ciò avviene è dovuta al fatto che la molecola così somministrata bypassa il passaggio attraverso la vena porta del fegato (e la conseguente disattivazione di primo passaggio) entrando nel flusso ematico dalla circolazione intramuscolare (via parenterale). Ciò influenza indirettamente il fegato, quindi lo stress è ridotto. Oltre allo Stanozololo, vi sono altri AAS iniettabili metilati in C-17 e venduti nel mercato nero (vedi, ad esempio, l’Oxymetholone e il Methandrostenolone iniettabile). Un metodo complicato affinché un AAS orale metilato risulti meno tossico per il fegato implica che il farmaco venga assunto per via sublinguale, cioè sotto la lingua. Questa specifica zona nella cavità orale è piena di piccoli vasi sanguigni. Si verifica di conseguenza uno spiccato assorbimento del farmaco; quindi lo steroide entra direttamente nel flusso sanguigno, in modo simile a quanto avviene con la somministrazione intramuscolare. In questo modo, la sostanza non raggiunge concentrazioni elevate a livello epatico come dopo somministrazione orale causando un minore stress epatico (anche se ancora significativo).
C’è da aggiungere che, molecole prive di metilazione in C-17 possono, a diverso grado, causare stress epatico: per esempio, il Trenbolone, che non presenta alcuna metilazione, possiede comunque un forte livello di resistenza alla disattivazione epatica, e una significativa tossicità epatica è stata osservato nei Bodybuilder che abusano del Trenbolone. Anche il Boldenone Undecilenato ha mostrato, in studi su animali, di causare danno epatico transitorio, nonostante anche questa molecola sia priva di una metilazione in C-17.
Gli AAS sono stati implicati in quattro distinte forme di lesione epatica:
1. Epatite farmacologica; In questo caso, le transaminasi epatiche sono elevate (> 100). Queste elevazioni sono attribuite all’assunzione di AAS orali, e sono di solito asintomatiche, transitorie e i livelli ritornano alle concentrazioni di base entro alcune settimane dalla cessazione del farmaco. Tali elevazioni sono state maggiormente legate ad AAS quali Fluoxymesterone e Oxymetholone.
Occasionalmente questo aumento è erroneamente diagnosticato come danno muscolare / rabdomiolisi (CPK> 500), piuttosto che come disfunzione epatica, in quanto sia gli epatociti che le cellule muscolari possiedono recettori per gli enzimi SGOT e SGPT. Recentemente, gli studi hanno dimostrato che il GGT è l’enzima più distintivo per la rilevazione della disfunzione epatica.
2.Colestasi; una condizione in cui si verifica ittero. In questo caso non vi è una corretta eliminazione della bile, attraverso il canale biliare e i tubuli biliari intracellulari del parenchima epatico. L’insorgenza è di solito accompagnata da sviluppo di nausea, stanchezza e prurito seguite da urine scure-marroni (urobilinogeno elevato) e ittero (bilirubina elevata). L’ittero può protrarsi anche dopo l’interruzione dell’assunzione di AAS. Tipicamente, sono presenti elevazioni seriche di marcatori colestatici come ALP, γGT, e la bilirubina diretta / indiretta. La biopsia del fegato mostra tipicamente la colestasi con infiammazione, necrosi epatocellulare e iperplasia degli epatociti. Questo fenotipo clinico di colestasi è tipico dell’uso/abuso di AAS.
3.Peliosi epatica; è una rara sindrome in cui i lobi del fegato sono coperti da noduli che contengono cisti piene di sangue. Il fegato può essere ingrossato, e di colore rosso intenso. I livelli degli enzima nel siero sono solitamente normali o lievemente elevati. I pazienti possono lamentare dolore al lato superiore destro. Questa è una condizione critica e talvolta fatale. Tuttavia, la peliosi epatica associata ad abuso di AAS ,in genere, subisce un inversione quando il paziente interrompe l’uso/abuso di AAS.
4.Carcinoma epatocellulare; la complicazione più grave dell’utilizzo di AAS è lo sviluppo di tumori epatici, adenoma (HCA) o carcinoma epatocellulare (HCC). E’ generalmente diagnosticato tramite esame obiettivo, esame del sangue, TC (tomografia computerizzata), ecografia, risonanza magnetica, angiografia epatica e biopsia. Anche se nelle fasi iniziali non dà alcun segno di sé, via via che la malattia si diffonde, però, iniziano a comparire i sintomi specifici, tra i quali il dolore alla parte superiore dell’addome, che si può irradiare anche alla schiena e alle spalle, l’ingrossamento del ventre, la perdita di peso e di appetito, la nausea, il vomito, la sensazione di sazietà, la stanchezza, l’ittero (ovvero il colore giallo della pelle), la colorazione scura delle urine e la febbre. Mentre il Fluoxymesterone è associato alla formazione di HCA, altre sostanze come l’Oxymetholone e il Methyltestosterone possono portare allo sviluppo di HCC. Ci sono diverse strategie terapeutiche per il HCC senza metastasi. In generale il trapianto di fegato è la terapia di scelta per i pazienti selezionati con HCC senza la possibilità di metastasi extraepatica. L’abuso di AAS per un lungo periodo di tempo è un fattore di rischio per lo sviluppo di HCC e quindi gli utilizzatori devono essere ben monitorati. L’ecografia epatica periodica sembra essere una procedura di screening adeguata per rilevare lo sviluppo di lesioni epatiche.
La steatosi epatica è una condizione dovuta principalmente a motivi metabolici. L’obesità e la sindrome metabolica di solito portano all’accumulo di grasso nel parenchima epatico. In alcuni casi, il fegato grasso può essere accompagnato da infiammazione epatica e morte delle cellule epatiche (steatoepatite). Questo caso è reversibile, con un’adeguata alimentazione e l’integrazione di fattori lipotropici (colina, inositolo). La supplementazione fornisce una prevenzione medica e assicura che gli enzimi epatici non siano molto elevati. L’acido Ursodeossicolico (UDCA) sembra essere estremamente utile nei casi di colestasi, in cui avviene l’ittero. Questi sali biliari hanno la capacità di ridurre la concentrazione di bilirubina, diminuire la tossicità del pool biliare e i valori colestatici inversi (ALP, GGT).
Altri potenti antiossidanti, come il Glutatione (iniettabili) o la N-Acetil-Cisteina (NAC), sono inoltre utili. La NAC è un precursore del Glutatione e serve come agente disintossicante del fegato. La NAC serve ad aumentare le riserve di Glutatione nel corpo, insieme con il Glutatione stesso; entrambi si legano direttamente ai metaboliti tossici. Prodotti erboristici come la Silimarina (Cardo Mariano) e il carciofo sono noti come potenti antiossidanti in particolare a livello epatico. Anche l’Acido Alfa Lipoico è un potente agente contro i radicali liberi e lo stress ossidativo. Il Tarassaco è un’erba diuretica che ripulisce dalle tossine attraverso l’urina. Ci sono diversi prodotti contenenti formule per la protezione epatica sul mercato. Personalmente, preferisco evitare i preparati commerciali a causa della loro inadeguatezza nei dosaggi dei vari componenti (cosa spesso riscontrata). Prediligo la creazione di formule per l’epatoprotezione basate sulle esigenze del caso (dal tipo di ciclo e dalle molecole utilizzate). Un’ottima formulazione che ha dimostrato un enorme potenziale prevede l’uso di Silimarina (400-600mg), NAC (3g) e Deursil (300-450mg) a dosaggi adeguati. Tuttavia, queste formulazioni devono essere utilizzate con una dieta bassa in grassi saturi, senza il consumo di alcool né un consumo eccessivo di acetaminofene / paracetamolo. Almeno mentre il soggetto in questione è sotto ciclo di AAS.
L’N-acetilcisteina (NAC) è un forte antiossidante e diversi atleti lo assumono anche per proteggere i loro muscoli dagli effetti negativi dello sforzo estremo, come dolore e infiammazione. Ma sembra che inibisca anche il recupero muscolare, come riportato dagli scienziati dello sport della Democritus University of Thrace in Grecia. (1)
Glutatione
Il peptide glutatione aiuta gli enzimi nelle cellule muscolari a svolgere le loro funzioni di protezione. Se si assume la N-acetilcisteina, le cellule utilizzano questo aminoacido come un precursore per la sintesi di glutatione.
Più le cellule muscolari contengono glutatione, peggiore sarà la funzione del fattore di trascrizione NF-kB. Di conseguenza, una supplementazione post-allenamento con N-acetilcisteina può indebolire le reazioni infiammatorie avviate da parte delle cellule del sistema immunitario che ripuliscono le cellule muscolari danneggiate.
Questo potenzia la prestazione o no? Questa è la domanda che i ricercatori si sono posti. Per rispondere a essa hanno eseguito un esperimento con 10 sportivi amatoriali, che hanno eseguito due sedute di allenamento estremo per i quadricipite: hanno svolto 20 serie da 15 ripetizioni su una leg-extension.
In un’occasione i soggetti dello studio hanno assunto 20 mg di N-acetilcisteina per kg di peso corporeo ogni giorno per otto giorni dopo il loro allenamento. Il dosaggio dei soggetti è stato suddiviso in tre dosi giornaliere. In un’altra occasione i soggetti hanno assunto un placebo.
Durante gli otto giorni in cui è stato assunto il supplemento dopo la sessione di allenamento si sono notati aumenti delle concentrazioni di glutatione nei muscoli e una riduzione del TBARS – un indicatore dell’attività dei radicali liberi.
La cosa suona bene, ma nei giorni dopo l’allenamento, il recupero [resistenza] era meno buono quando i soggetti avevano assunto N-acetilcisteina. D’altra parte, la N-acetilcisteina ha ridotto il dolore muscolare [DOMS].
La N-acetilcisteina ha ridotto la secrezione delle proteine infiammatorie come il CRP, l’interleuchina 1-beta e l’interleuchina-6 [In basso a sinistra]. Inoltre, la N-acetilcisteina ha anche ridotto l’attività di segnalazione delle molecole anabolizzanti come l’Akt, l’mTOR e il MyoD nelle cellule muscolari [In basso a destra].
I ricercatori hanno scritto che i loro risultati confermano quanto suggerito da studi precedenti e cioè che l’uso pesante di antiossidanti può avere un effetto negativo sulle prestazioni muscolare ed il recupero, probabilmente alterando vie di segnalazione che mediano l’infiammazione muscolare ed il recupero e la biogenesi mitocondriale e successivamente il metabolismo energetico.
Quindi, si potrebbe dire, che non solo “è la dose che fa il veleno” ma che ciò dipende anche da quando la “dose” la si prende…
Visionando alcuni studi tra i quali il “Milk Thistle Constituents Inhibit Raloxifene Intestinal Glucuronidation: A Potential Clinically Relevant Natural Product–Drug Interaction”, emerge palesemente che la Silimarina, integratore largamente usato per la sua azione a livello epatico, presenta azione estrogenica. Infatti, la Silimarina ha effetti estrogenici a causa della sua interazione su i recettori ERb. Tale effetto è dato in particolare da alcuni fitocomposti (nel dettaglio quercitina e taxifolina) i quali legandosi a tali recettori (ERb) si comportano come deboli agonisti, da due a quattro volte meno potenti dell’Estradiolo, al pari dello Zearalenone che invece è una micotossina che esplica i medesimi effetti e la si trova nei cereali e nel mais infetti (facente parte della vasta gamma degli Xenoestrogeni). Per via dei flavolignani silibinina A e B contenuti nell’estratto, la Silimarina inibisce l’enzima UGT (UDP-glucuronosiltransferasi) nelle varie isoforme tra cui anche la 1A1 che è quella responsabile della glucuronazione della bilirubina (e del suo smaltimento) e come ampiamente comprovato, un deficit di questo enzima porta all’accumulo di bilirubina che è ciò che succede nel morbo di Gilbert. Dunque la Silimarina inibisce tale enzima, riducendo la capacità epatica di detossificazione dalla bilirubina ma anche da altre sostanze che usano tale enzima come ad esempio è il caso del Raloxifene*, la cui concentrazione aumenta fino a 5 volte rispetto al normale.
Ci sono anche altri estratti che esercitano un azione di inibizione simile sull’UGT, tra i quali vi sono:
Di conseguenza sono tutti estratti che i soggetti con Gilbert non dovrebbero assumere, o quantomeno soprattutto se in concomitanza usano farmaci o altre sostanze che sono substrato dell’enzima UGT.
* Il Raloxifene è un farmaco che rientra nella categoria dei modulatori selettivi del recettore degli estrogeni (come il più famoso Tamoxifene), uno dei principi attivi creati per contrastare gli effetti negativi degli estrogeni nella cura dell’osteoporosi nella fase successiva alla menopausa.
La protezione epatica è una delle precauzioni primarie che l’atleta supplementato chimicamente prende in considerazione quando è in procinto di cominciare un ciclo con AAS (specie se metilati in C-17). Oltre ai classici Silimarina e NAC esistono dei preparati che contengono diversi principi attivi i quali dovrebbero proteggere il fegato e che gli atleti supplementati chimicamente utilizzano sempre più spesso. Ricercatori greci dell’Università della Tessaglia hanno eseguito uno studio volto ad esaminare se il così detto “Composto N” (prodotto da Natterman), che risulta essere Essentiale Forte, abbia un reale potenziale nella protezione epatica durante l’uso di AAS. (1)
Una confezione del suddetto prodotto contiene una ventina di capsule, ciascuna delle quali contenente 300 mg di Fosfatidilcolina poliene. Questa probabilmente derivata dalla Fosfatidilcolina della soia.
Inoltre, ogni capsula contiene 6 mg di vitamina B1, 6 mg di vitamina B2, 6 mg di vitamina B6, 6 mcg di vitamina B12, 30 mg di nicotinammide e 6 mg di vitamina E.
I ricercatori greci, che tra l’altro avevano svolto una ricerca sugli effetti psicologici degli steroidi anabolizzanti, hanno coinvolto nell’esperimento trecentoventi atleti. La metà di loro, centosessanta atleti, usava AAS. A questi atleti supplementati chimicamente, i ricercatori somministrarono Essentiale Forte: due capsule al giorno assunte con il cibo.
Tutti gli atleti supplementari chimicamente hanno assunto AAS monitorati dai ricercatori per otto settimane. L’elenco che riporto nella figura seguente vi darà un’idea di ciò che stavano usando i culturisti greci.
Durante il periodo di otto settimane i ricercatori hanno misurato le concentrazioni dei seguenti enzimi nel sangue degli atleti: aspartato aminotransferasi (AST / SGOT), alanina aminotransferasi (ALT / SGPT), lattato deidrogenasi (LDH), fosfatasi alcalina (ALP), gamma-glutamiltranspeptidasi ( gamma-GT) e creatin-chinasi (CK).
Ovviamente, più le concentrazioni di enzimi nel sangue erano elevate, più lo stress epatico era elevato.
La figura seguente mostra cosa è successo alle concentrazioni degli enzimi epatici:
Gruppo A = atleti supplementari chimicamente e che hanno assunto Essentiale Forte.
Gruppo B = atleti supplementari chimicamente che non hanno assunto un supplemento per la protezione epatica.
Gruppo C = atleti “natural”.
I ricercatori non sono sicuri di come la Fosfatidilcolina e le vitamine presenti nel prodotto proteggano il fegato degli utilizzatori di AAS. Si sospetta che il mix rinforzi le membrane delle cellule epatiche. Il fegato degli utilizzatori di AAS deve lavorare più duramente del normale per neutralizzare tutte le sostanze alle quali sono sottoposti. E come risultato le cellule del fegato diminuiscono la loro capacità di metabolizzare i grassi. Il fegato diventa grasso perché le cellule del fegato non bruciano efficientemente il grasso [beta-ossidazione.] Una protezione delle membrane cellulari costituite da catene di acidi grassi viene meno. Il supplemento dello studio aiuta le cellule del fegato a svolgere queste funzioni.
Il risultato dello studio è sicuramente interessante, ma ad alcuni viene difficile credere che una semplice assunzione di pillole di vitamina B e Fisfatidilcolina siano in grado di proteggere il fegato degli utilizzatori di AAS. Gli stessi ricercatori greci ebbero difficoltà nel far pubblicare questo studio.
Ecco perché usano frasi prolisse come: “. I risultati della nostra coorte di individui che esercitano in modo simile suggeriscono che i fosfolipidi polinsaturi in combinazione con vitamine del complesso B proteggono le cellule epatiche dai danni AAS-indotti”. Secondo i ricercatori greci il “Composto N” non è un integratore, ma “un agente farmaceutico controllato”.
Per concludere, con i feedbak raccolti da diversi utilizzatori di AAS, so per esperienza che l’Essentiale Forte ha un discreto potenziale nella protezione epatica durante l’uso di AAS.
In seguito a ricerche svolte, anche se in piccolo, ho potuto constatare l’ottima efficacia in termini di protezione epatica della combinazione Silimarina, NAC e Acido Tauroursodesossicolico Biidrato anche in concomitanza con cicli di AAS metilati della durata di 8 settimane (non provateci, nonostante che i valori degli enzimi epatici sono risultati contenuti non lo erano quelli del colesterolo, e marcato fu anche l’abbassamento dell’HDL).