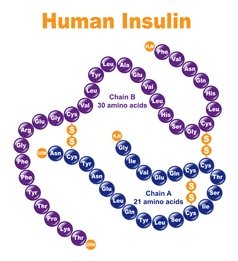
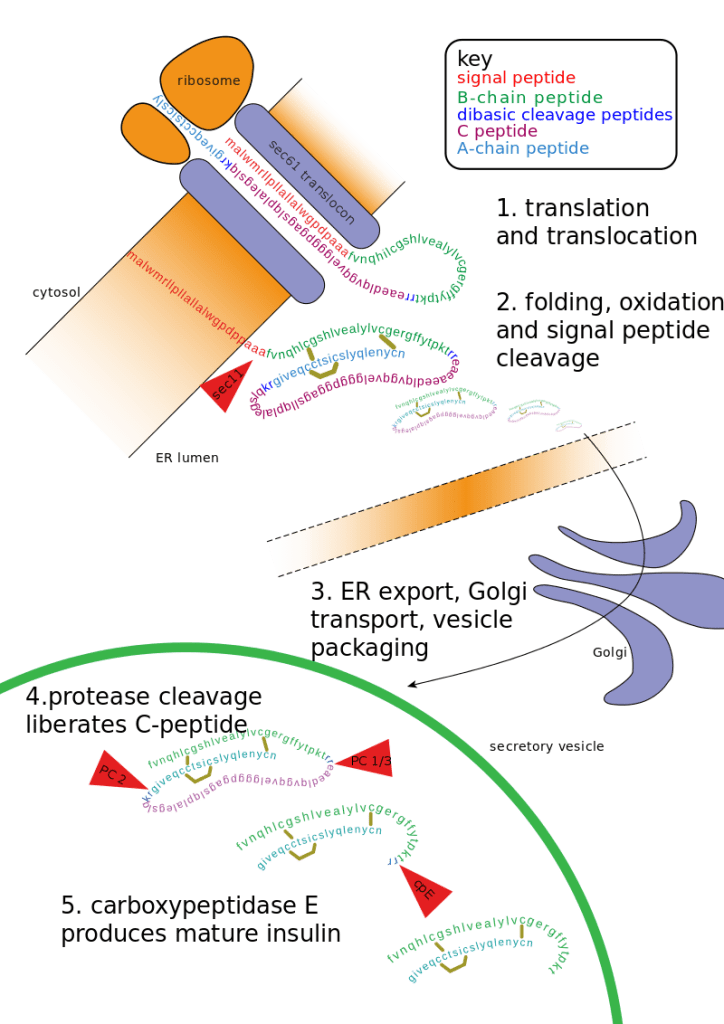
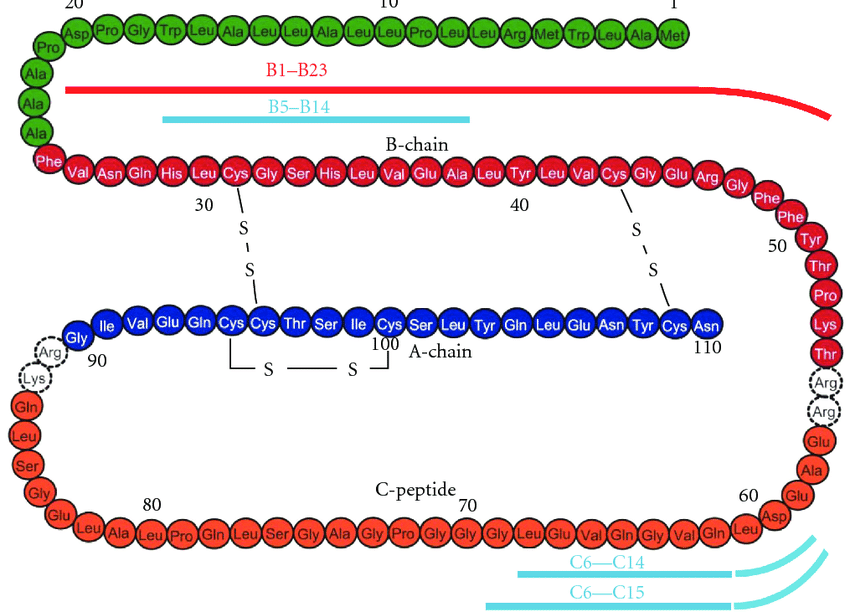
Introduzione:
Il BodyBuilding si differenzia dagli sport di prestazione perché il giorno della gara gli atleti vengono giudicati in base all’aspetto piuttosto che alle capacità atletiche. I bodybuilder posano sul palco dove vengono giudicati per la muscolatura, la definizione e la simmetria. Nel corso di una stagione, i bodybuilder attraversano tre fasi diverse: la fase di crescita muscolare (Off-Season), la dieta per la competizione (preparazione alla gara) e la gara stessa. La maggior parte della letteratura riguarda la fase di dieta pre-gara e la peak week.[1]
Tuttavia, la letteratura scientifica sulle raccomandazioni alimentari per i bodybuilder durante la Off-Season è carente. Si tratta di una lacuna importante, poiché la maggior parte della carriera di un bodybuilder si svolge in questa fase, in cui l’obiettivo è aumentare la massa muscolare riducendo al minimo l’aumento eccessivo della massa grassa. I bodybuilder sono noti per avere atteggiamenti rigidi nei confronti della selezione degli alimenti, della frequenza dei pasti, dei tempi di alimentazione e dell’integrazione [2]. Storicamente, le informazioni sull’alimentazione e l’integrazione sono state trasmesse dalle riviste di bodybuilding e dai concorrenti di successo, ma recentemente sono emerse più informazioni attraverso Internet e i forum [3,4]. Di conseguenza, molte delle strategie alimentari utilizzate dai bodybuilder non hanno un solido supporto scientifico e la letteratura scientifica dimostra che alcune di queste strategie, tra cui l’uso massiccio di farmaci, ma anche di integratori più in generale, possono essere ovviamente dannosi per la salute [5,6,7].
Poiché i bodybuilder trascorrono la maggior parte del loro tempo in Off-Season, è evidente la necessità di raccomandazioni nutrizionali e di supplementazione, sia OTC che PEDs, il più possibile “sicure” e basate sull’evidenza per questa popolazione. È stato inoltre dimostrato che alcuni bodybuilder, e non soltanto i concorrenti di alto livello nel bodybuilding “Natural”, potrebbero essere interessati a informazioni basate sull’evidenza [8]. Con il supporto della review realizzata e pubblicata da Juma Iraki et al. che tratta del Off-Season a livello alimentare e integrativo, lo scopo di questo articolo sarà quello di riportare quanto evidenziato dalla letteratura scientifica sugli argomenti relativi all’alimentazione e all’integrazione alimentare e supplementazione PEDs rilevanti per i bodybuilder nella Off-Season e di fornire raccomandazioni pratiche sull’assunzione di energia, macronutrienti, frequenza dei pasti, tempistica dei nutrienti, integratori alimentari e PEDs .
Transizione dalla dieta pre-gara/peak week alla dieta in Off-Season – Reverse Diet Vs. Recovery Diet:
Il primo step che il bodybuilder si trova davanti è la gestione del passaggio da una dieta ipocalorica ad una ipercalorica. Ed è in questo frangente che emergono due strategie simili all’apparenza ma in realtà diverse: la “Recovery Diet” e la “Reverse Diet”.

Ora, molto semplicemente, la “Recovery Diet” consiste in un graduale aumento calorico ma di consistenza tale che l’atleta esca dalla condizione di ipocalorica nel giro di due settimane circa. Con la “Reverse Diet”, invece, abbiamo sempre un graduale aumento calorico ma caratterizzato da una ridotta consistenza dello stesso (si parla di circa 100Kcal/die a settimana). In questo caso specifico, il bodybuilder rimarrebbe in ipocalorica per diverse settimane con possibile emersione di problemi psicofisici legati al protrarsi dello stato stressorio.
Quindi, con il termine “Recovery Diet” ci riferiamo ad uno schema alimentare avente l’obiettivo generale di RECUPERARE da un periodo di dieta cronica sperimentato durante la preparazione alla gara. La “Recovery Diet” incoraggia i bodybuilder a guadagnare il 5-10% del loro peso di gara nelle prime 4-8 settimane successive all’evento. Questo con l’intento di accelerare l’aumento di grasso corporeo e far rientrare il soggetto in un range di grasso corporeo “sano”, fisiologico, il prima possibile. In seguito, si consiglia agli atleti di rallentare il ritmo di aumento del peso e di mantenere un surplus controllato, con un aumento medio dello 0,5-1% del peso corporeo al mese passando pienamente nella Off-Season. Questo fino a quando non raggiungono un punto in cui un ulteriore aumento di peso è considerato improduttivo. Con il termine “Reverse Diet” ci si riferisce ad una strategia la quale può ancora essere attuata con discreti vantaggi per aiutare un agonista a recuperare dopo il contest. Tuttavia, se rispettata e seguita correttamente, piccoli aumenti di cibo di ~100 Kcal/die a settimana potrebbero comunque protrarre il deficit calorico del soggetto, prolungando così il periodo di dieta ipocalorica. Sebbene questa possa essere una strategia utile in alcune circostanze, ad esempio durante l’avvicinamento alla competizione, le modalità di applicazione non permettono un recupero di una bf salubre in tempi ottimali. È risaputo che un bodybuilder in condizioni di picco non è necessariamente al massimo della salute, e questo è in gran parte correlato al livello di grasso corporeo. Accettare un certo aumento di grasso avrà effetti positivi su tutti gli aspetti della Off-Season come le prestazioni in allenamento, i marcatori ormonali, la disponibilità di energia, la qualità del sonno e, inoltre, sarà vantaggioso sulla longevità complessiva dello sport praticato.
In definitiva, se si parte da body fat estremamente basse, tipiche da gara, allora la “Recovery Diet” è la scelta migliore per shiftare dal regime ipocalorico che ha caratterizzato il periodo di preparazione alla gara a quello ipercalorico del Off-Season. Discorso diverso se ci troviamo di fronte ad un soggetto amatoriale, con una body fat del 8-10% arrivato al termine del percorso di “Cut”. In questo caso la “Reverse Diet” è la scelta più funzionale permettendo un controllo migliore degli incrementi calorici evitando che la massa grassa sfori eccessivamente e che il lavoro precedentemente svolto in “Cut” venga facilmente e totalmente compromesso. Anche “ibridazioni” con aumenti settimanali di 45-50g di CHO die possono essere applicati con buoni risultati.
Energia:
Durante la Off-Season, l’obiettivo principale di un bodybuilder è quello di aumentare la massa muscolare riducendo al minimo l’aumento della massa grassa attraverso l’uso di allenamenti contro-resistenza e il mantenimento di un bilancio energetico positivo. Per valutare con precisione il fabbisogno energetico dei bodybuilder durante la bassa stagione, è necessario considerare il volume, la frequenza e l’intensità dell’allenamento. Durante la fase off-season, è stato riportato che i bodybuilder si allenano alla resistenza 5-6 volte a settimana, esercitando ogni gruppo muscolare 1-2 volte a settimana [9]. È stato inoltre riferito che seguono una routine di allenamento ad alto volume con 4-5 esercizi per gruppo muscolare, eseguendo 3-6 serie per esercizio, 7-12 ripetizioni massime (RM) per ogni serie con 1-2 minuti di riposo tra le serie. La durata della sessione di allenamento è stata indicata in ~40-90 minuti. Tuttavia, i piani di allenamento possono variare notevolmente da atleta ad atleta. È necessario valutare anche l’apporto calorico medio dei bodybuilder. Nella fase off-season, l’apporto energetico è di solito sostanzialmente più elevato rispetto alla fase di dieta: tra i bodybuilder maschi è stato riportato un apporto medio di ~3800 kcal/giorno durante la fase off-season e di ~2400 kcal/giorno durante la fase di dieta [2].
- Bilancio energetico positivo:
È stato dimostrato che un bilancio energetico positivo ha un importante effetto anabolico, anche in assenza di allenamento contro-resistenza [10]. Tuttavia, la combinazione di un bilancio energetico positivo con l’allenamento contro-resistenza rappresenta il metodo più efficace per garantire che gli effetti anabolici siano diretti all’aumento della massa muscolo-scheletrica [11,12]. L’entità del surplus energetico ideale per guadagnare massa muscolare limitando l’accumulo di tessuto adiposo può variare in base allo stato di allenamento. Nei soggetti non allenati, è stato dimostrato che un surplus energetico sostanziale di circa 2.000 kcal, combinato con l’allenamento contro-resistenza, fornisce un robusto aumento di peso, in cui il contributo della massa magra (LBM) può raggiungere il 100% [12]. Tuttavia, nei soggetti allenati, un surplus energetico sostanziale potrebbe non essere necessario o vantaggioso. Uno studio condotto su atleti d’élite ha esaminato l’effetto delle indicazioni dietetiche sui cambiamenti della composizione corporea tra gli atleti d’élite quando l’allenamento contro-resistenza è stato combinato con diverse entità di surplus energetico. Un gruppo con un peso corporeo medio di 75kg ha consumato energia ad libitum (2964 kcal) per raggiungere un surplus molto ridotto, mentre un secondo gruppo con un peso corporeo medio di 71kg ha ricevuto una consulenza dietetica e ha consumato ~600 kcal in più rispetto al gruppo ad libitum [13].
Entrambi i gruppi hanno seguito lo stesso programma di allenamento contro-resistenza di 4 giorni alla settimana per un periodo di 8-12 settimane. I ricercatori hanno ipotizzato che il gruppo ipercalorico avrebbe avuto un aumento maggiore del peso corporeo e della LBM. Sebbene il gruppo ipercalorico abbia ottenuto un aumento maggiore della LBM rispetto a quelli che mangiavano ad libitum, questo non ha raggiunto la significatività statistica (1,7kg contro 1,2kg, rispettivamente). Inoltre, rispetto al gruppo che mangiava a sazietà, hanno registrato un aumento significativamente maggiore della massa grassa (1,1kg contro 0,2kg, rispettivamente). I ricercatori hanno concluso che un surplus di 200-300 kcal al giorno negli atleti altamente allenati potrebbe essere più appropriato di 500 kcal per minimizzare il rischio di inutili aumenti di grasso corporeo. I soggetti non allenati, più lontani dal loro tetto genetico di massa muscolare, possono essere in grado di aumentare i muscoli a un ritmo più veloce rispetto agli individui allenati.
Il tasso di crescita muscolare può rallentare con l’avanzare dell’età [14]. Pertanto, un maggiore surplus energetico può essere più vantaggioso per i bodybuilder alle prime armi, mentre i bodybuilder avanzati potrebbero trarre maggiore beneficio da diete ipercaloriche conservative per limitare inutili aumenti di grasso corporeo. Studi precedenti hanno raccomandato ai bodybuilder di consumare una dieta leggermente ipercalorica, con un aumento dell’apporto energetico di circa il 15% rispetto al mantenimento nella Off-Season [15]. Tuttavia, ciò non tiene conto della storia di allenamento e del livello di esperienza del singolo bodybuilder. Poiché la capacità di aumentare la massa muscolare è limitata, un surplus aggressivo può portare a un inutile aumento del grasso corporeo, che aumenterebbe la durata o la gravità dei successivi periodi di preparazione alle gare, aumentando di conseguenza la durata o la gravità della scarsa disponibilità energetica. Pertanto, il numero di calorie che un bodybuilder consuma al di sopra del livello di mantenimento può essere stabilito in base al livello di esperienza e poi regolato in base al tasso di aumento di peso e ai cambiamenti nella composizione corporea. Dato che i bodybuilder spesso aumentano rapidamente di peso dopo una gara, potrebbe essere utile avere un obiettivo di aumento di peso per settimana e regolarsi di conseguenza [16,17].
Tuttavia, come detto precedentemente, inizialmente, dopo la gara, potrebbe essere utile un aumento di peso più rapido per aiutare a riportare il concorrente a uno stato di salute sia psicologico che fisiologico, prima che il tasso di aumento di peso venga rallentato per limitare l’accumulo eccessivo di tessuto adiposo. Nella letteratura scientifica si raccomanda di puntare a un aumento di peso di circa 0,25-0,5 kg a settimana per cercare di aumentare la LBM e ridurre al minimo l’aumento della massa grassa [14,18]. Per un bodybuilder avanzato, un potenziale aumento di 2kg di peso corporeo su base mensile potrebbe essere eccessivo e comportare un’inutile accumulazione di grasso corporeo; pertanto, questo tasso dovrebbe essere considerato con cautela. Sulla base delle prove attuali, potrebbe essere opportuno raccomandare ai bodybuilder di consumare una dieta leggermente ipercalorica (~10-20% sopra le calorie di mantenimento) nella Off-Season e raccomandare ai bodybuilder avanzati di puntare all’estremità inferiore di questa raccomandazione, o addirittura di essere più conservativi se si verificano aumenti sostanziali della massa grassa. Dato che i bodybuilder consumano in media 45 kcal/kg durante la bassa stagione, il surplus raccomandato equivale a circa 42-48 kcal/kg [2]. Potrebbe essere utile puntare a un aumento di peso di circa 0,25-0,5% del peso corporeo a settimana, regolando al contempo l’apporto energetico in base alle variazioni della composizione corporea. Inoltre, potrebbe essere più appropriato considerare le variazioni di peso medie settimanali basate su pesate giornaliere (o più volte alla settimana) per limitare gli errori delle fluttuazioni giornaliere del peso che possono verificarsi durante la settimana. Una volta determinato il surplus calorico, il passo successivo sarà quello di distribuire le calorie tra proteine, grassi e carboidrati.
Proteine:

Il turnover proteico del muscolo scheletrico è il rapporto tra la sintesi proteica muscolare (MPS) e la degradazione proteica muscolare (MPB). L’ipertrofia del muscolo scheletrico richiede un equilibrio netto in cui la MPS supera la MPB. L’esercizio contro-resistenza fornisce lo stimolo di tensione iniziale che induce l’ipertrofia risultante dall’aumento cumulativo della MPS dopo l’esercizio cronico [19]; tuttavia, l’aumento della massa grassa (FFM) può essere limitato se l’apporto proteico giornaliero è insufficiente [20]. Oltre alla quantità totale consumata al giorno, i ricercatori hanno ipotizzato che la qualità delle proteine possa aumentare il guadagno muscolare indotto dall’allenamento contro-resistenza [21]. Pertanto, entrambi questi argomenti saranno discussi nelle sezioni seguenti.
- Introito proteico giornaliero:
Mentre l’attuale RDA per le proteine negli individui sani sedentari è di 0,8 g/kg, in una meta-analisi del 2018 di Morton e colleghi [22] è stato osservato che il doppio di questa quantità massimizza l’ipertrofia indotta dall’allenamento contro-resistenza. Inoltre, gli autori hanno osservato che “potrebbe essere prudente raccomandare ~2,2g di proteine/kg/die per coloro che cercano di massimizzare i guadagni di FFM indotti dall’allenamento contro-resistenza”, poiché 2,2g/kg era l’estremità superiore del limite di confidenza [22] e le differenze individuali impongono che alcuni atleti abbiano un fabbisogno proteico più elevato di altri [23]. Inoltre, la raccomandazione “meglio prevenire che curare” è probabilmente sicura, vista l’assenza di danni apparenti in studi di 1-2 anni tra i sollevatori che consumavano apporti proteici di almeno 2,2 g/kg [24,25]. Infine, la media e il limite superiore di confidenza del 95% per il fabbisogno proteico utilizzando la tecnica di ossidazione degli aminoacidi con indicatore tra i bodybuilder maschi nei giorni di non allenamento sono stati riportati rispettivamente come 1,7 e 2,2g/kg [26], che è simile al fabbisogno tra le donne quando è normalizzato alla FFM [27].
Tuttavia, è stato riportato che i bodybuilder consumano fino a 4,3g/kg di proteine al giorno tra i soggetti di sesso maschile e 2,8g/kg tra quelli di sesso femminile, superando di gran lunga queste raccomandazioni [2]. Le linee guida precedentemente fornite per i bodybuilder nella Off-Season erano di consumare il 25-30% del loro apporto energetico dalle proteine [15]. Potrebbe essere ragionevole opporsi all’indicazione di raccomandazioni basate su percentuali dell’apporto energetico totale, poiché un individuo con un peso non particolarmente elevato ma con un alto fabbisogno energetico potrebbe finire per consumare proteine che superano di gran lunga quelle necessarie e quindi richieste. Inoltre, questo può portare a un’assunzione insufficiente di carboidrati e grassi se l’atleta mira a un apporto calorico specifico. Pertanto, potrebbe essere più appropriato raccomandare un fabbisogno proteico basato sul peso corporeo. Pertanto, i bodybuilder dovrebbero consumare un minimo di 1,6g/kg di proteine nella Off-Season, anche se un obiettivo più vicino a 2,2 g/kg potrebbe garantire una risposta ottimizzata in modo più coerente in una maggiore percentuale di atleti.
E per i “Doped”? Dovremo ormai sapere che la fisiologia di base è la medesima per ogni individuo con le consuete variabili. Detto ciò, l’uso di PEDs va si ad alterare la fisiologia ma in questo specifico ambito, ossia introito proteico per massimizzare lo stimolo ipertrofico, hanno una azione di perfezionamento dell'”economia proteica cellulare”: in parole più semplici, sembra che l’uso di AAS porti ad una migliore resa nell’utilizzo degli amminoacidi scissi e assorbiti dalle proteine alimentari. Di conseguenza, a parità di apporto proteico, la veicolazione degli amminoacidi a scopo plastico è maggiore come minore è l’attività catabolica. Ciò significa che abusare delle proteine, in special modo durante una fase ipercalorica, perchè si è sotto AAS potrebbe risultare più inutile di quanto non lo sia in contesto “Natural”.
Infine, ed è necessario sottolinearlo, tra i bodybuilder che lottano con la fame in Off-Season e che di conseguenza assumono quantità caloriche che portano a un aumento di peso più rapido e all’accumulo di grasso in eccesso, un apporto proteico più elevato può essere utile (se non controindicato per motivi clinici). In uno studio condotto da Antonio e colleghi, i partecipanti ad allenamenti contro-resistenza che consumavano più proteine (4,4g/kg al giorno) e più calorie hanno guadagnato una quantità simile di FFM, ma non hanno guadagnato ulteriore grasso corporeo rispetto al gruppo che consumava meno proteine e meno calorie [28]. Allo stesso modo, in uno studio di follow-up, un gruppo che consumava 3,4g/kg di proteine al giorno ha guadagnato una quantità simile di FFM, ma ha perso una percentuale maggiore di grasso corporeo rispetto a un gruppo a basso contenuto proteico, ancora una volta, nonostante un apporto energetico più elevato [29]. Gli autori di questi studi sulla “vita libera” hanno ipotizzato che i loro risultati fossero dovuti a un aumento della termogenesi indotta dalla dieta attraverso protocolli alimentari ad alto contenuto proteico. Tuttavia, ciò è in contrasto con uno studio di Bray e colleghi del 2012 sul reparto metabolico, più strettamente controllato, in cui il contenuto proteico della dieta influenzava la percentuale di massa corporea acquisita, mentre la massa corporea totale era dettata dal solo contenuto energetico della dieta [30].
Pertanto, mentre la termogenesi indotta dalla dieta potrebbe essere significativamente più elevata con assunzioni di proteine nell’intervallo di 3 g/kg o superiore, la perdita di grasso o la mancanza di aumento di peso osservata da Antonio e colleghi, nonostante un apporto energetico più elevato, potrebbe con più probabilità riflettere l’effetto saziante di assunzioni proteiche molto elevate che diminuiscono l’assunzione calorica effettiva, piuttosto che un aumento della sola termogenesi.
- Qualità delle Proteine:
Gli aminoacidi essenziali (EAA) sono gli unici aminoacidi necessari per stimolare il processo di MPS [31]. Sebbene tutti gli aminoacidi forniscano i “mattoni” necessari per la sintesi di nuovi tessuti, l’aminoacido Leucina in particolare sembra essere particolarmente importante come “innesco metabolico” della MPS [32]. È stato suggerito che una concentrazione sufficiente di Leucina è necessaria per raggiungere una “soglia di Leucina” che è richiesta per stimolare al massimo la MPS [33]. In breve, dal punto di vista della costruzione muscolare, le fonti proteiche che innescano una consistente risposta della MPS (quantità sufficiente di Leucina) e forniscono i mattoni essenziali per la costruzione di nuovo tessuto muscolare (contengono l’intero spettro di aminoacidi essenziali in abbondanza) possono essere considerate di “qualità superiore”.
Sebbene l’effetto meccanicistico della Leucina sulle MPS esuli dallo scopo di questo articolo, si invitano i lettori a leggere una rassegna che tratta questo argomento in dettaglio [34]. In generale, su una base di grammo per grammo, le fonti proteiche di origine animale contengono in genere più Leucina ed EAA, anche se ci sono eccezioni degne di nota. Le proteine della soia, uno dei più comuni integratori proteici di origine vegetale, contengono tutti gli EAA, ma in una quantità inferiore per grammo rispetto alle proteine del latte e quindi, in uno studio, hanno prodotto un aumento minore delle MPS rispetto al siero di latte dopo un’ingestione acuta [35]. È interessante notare che in questo stesso studio la soia ha prodotto un aumento maggiore delle MPS rispetto alla caseina, anch’essa una proteina casearia di “alta qualità”, presumibilmente a causa della più lenta velocità di digestione della caseina [35]. Rammentate sempre la differenza tra risposta “acuta” e “cronica”. Per l’appunto, ciò significa che, sebbene il contenuto di Leucina e di EAA di una fonte proteica debba essere preso in considerazione, la risposta acuta alla MPS non è l’unica variabile legata all’ipertrofia a lungo termine. Infatti, una proteina di alta qualità ma “lenta” come la caseina produce inizialmente una risposta MPS di minore ampiezza. Tuttavia, la caseina (e altre proteine a lenta digestione) può produrre un’area MPS sotto la curva simile o maggiore se osservata longitudinalmente rispetto a una fonte proteica “veloce” come il siero di latte, che determina un aumento iniziale maggiore e poi una brusca riduzione [36].
Inoltre, la risposta acuta della MPS a un determinato tipo di proteina non deve essere vista in una prospettiva riduzionista. Nel mondo reale si consumano quotidianamente più porzioni di varie fonti proteiche, rendendo probabilmente superflue alcune di queste distinzioni nel profilo aminoacidico e nella cinetica di digestione. Infatti, in una meta-analisi che ha confrontato i cambiamenti longitudinali della composizione corporea con diversi tipi di integratori proteici, non sono state riscontrate differenze significative tra i partecipanti che consumavano soia rispetto al siero di latte, ad altre proteine del latte o alle proteine isolate del manzo [37].
Come dimostrato in uno studio che ha messo a confronto gruppi che consumavano proteine dopo l’allenamento (in aggiunta a una dieta già composta dal 25% di proteine), sia che venissero forniti 48g di proteine del siero del latte (contenenti 5,5g di Leucina), sia che venissero forniti 48g di proteine del riso (contenenti 3,8g di Leucina), non è stato osservato alcun impatto sui cambiamenti della composizione corporea tra i gruppi dopo otto settimane [38]. Pertanto, se consumate in quantità sufficienti (soprattutto se si considera l’apporto proteico totale giornaliero), la qualità delle proteine di un singolo pasto è meno preoccupante. Tuttavia, se si volesse consumare una dieta dominata da fonti proteiche di origine vegetale, esistono alternative alla soia e al riso. Ad esempio, le proteine isolate del pisello sono ricche di EAA e di Leucina. In uno studio di 12 settimane, un gruppo che consumava 50g di proteine isolate di pisello al giorno ha registrato un aumento maggiore dello spessore muscolare indotto dall’allenamento di resistenza rispetto al placebo, non significativamente diverso da un gruppo che consumava 50g di siero di latte [39].
Pertanto, nel contesto delle indicazioni di questo articolo, la qualità delle proteine può essere un problema solo se si utilizza la fascia bassa delle linee guida sulle proteine (1,6g/kg) o se si consuma una dieta a base prevalentemente vegetale. In entrambi i casi, potrebbe essere utile integrare con fonti proteiche ricche di Leucina e di EAA, a seconda delle preferenze alimentari (ad esempio, proteine del latte o del pisello se si è vegani), per garantire la risposta attesa della MPS all’assunzione di proteine.
Grassi:

Il grasso è un nutriente fondamentale per molte funzioni dell’organismo. Tuttavia, non si sa molto dell’effetto dei grassi alimentari sull’ipertrofia del muscolo scheletrico. È stato riportato che l’assunzione di grassi alimentari tra i bodybuilder varia dall’8 al 33% delle calorie totali [2]. Sebbene i trigliceridi intramuscolari possano fungere da substrato energetico durante l’allenamento di resistenza, non sono un fattore limitante poiché i substrati derivano principalmente da processi anaerobici [40]. Di interesse per il bodybuilder, è dimostrato che negli atleti allenati contro-resistenza [41] e nei giocatori di hockey [42] le diete a basso contenuto di carboidrati (30-45% dell’energia o meno) possono influire sul rapporto Testosterone libero/Cortisolo (fTC), il che potrebbe avere un impatto negativo sul recupero. D’altra parte, la riduzione dei grassi alimentari nelle diete isocaloriche da ~30-40% a ~15-25% ha portato a riduzioni significative ma modeste dei livelli di Testosterone [43,44,45,46].
Tuttavia, non è chiaro se le variazioni di Testosterone all’interno di intervalli normali influenzino in modo significativo l’aumento della massa muscolare [47]. Nonostante la possibilità che i livelli di testosterone possano essere più elevati quando si consuma una percentuale maggiore di energia proveniente dai grassi alimentari, i cambiamenti effettivi nella massa muscolare durante gli studi longitudinali di individui allenati alla resistenza che seguono diete “chetogeniche” ad alto contenuto di grassi sono stati costantemente inferiori rispetto ad approcci moderati o a basso contenuto di grassi con ampi carboidrati [48,49,50,51]. Non è ancora stato chiarito se ciò sia dovuto a cambiamenti nella capacità di esercizio, ad alterazioni del rapporto fTC o a qualche altro meccanismo legato alla componente ad alto contenuto di grassi o a basso contenuto di carboidrati della dieta.
Tuttavia, ciò indica che forse si dovrebbe consumare una proporzione più moderata di grassi nella dieta, piuttosto che un apporto basso o alto. In letteratura sono state proposte raccomandazioni del 15-20% e del 20-30% delle calorie provenienti dai grassi alimentari [15,52]. Tuttavia, sono necessarie ulteriori ricerche per stabilire l’effetto e la quantità ottimale di grassi alimentari per favorire l’ipertrofia muscolare.
Sulla base delle evidenze attuali, può essere prudente raccomandare che i grassi alimentari rappresentino il 20-35% delle calorie, in linea con le raccomandazioni dell’American College of Sports Medicine per gli atleti [53], che nella maggior parte dei casi corrispondono a circa 0,5-1,5 g/kg/giorno. Inoltre, va notato che un apporto sufficiente di proteine e carboidrati non deve essere compromesso da un’elevata assunzione di grassi nella dieta.
Anche la qualità dei grassi, come gli essenziali omega 3 e gli omega 6, potrebbe essere importante per i bodybuilder. Se l’apporto di questi acidi grassi è sufficiente, non è necessario integrarli con una dieta di alta qualità contenente buone fonti di acidi grassi. Tuttavia, per alcuni potrebbe essere difficile assumere le quantità ottimali. Per questo motivo, l’argomento verrà trattato in modo più approfondito nella sezione dedicata agli integratori alimentari.
Carboidrati:

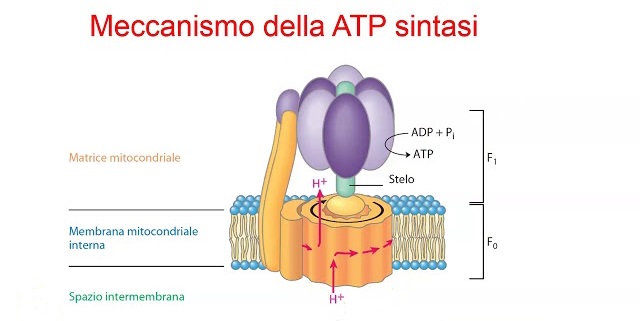
A differenza delle proteine e dei grassi, i carboidrati sono considerati non essenziali per la dieta umana perché l’organismo è in grado di produrre il glucosio necessario ai tessuti attraverso la gluconeogenesi [54]. Tuttavia, l’assunzione di carboidrati ha un ruolo importante nella dieta del bodybuilder come regolatore degli ormoni tiroidei e come contributo al fabbisogno di micronutrienti [55,56]. Inoltre, una dieta a basso contenuto di carboidrati potrebbe limitare la rigenerazione dell’adenosina trifosfato (ATP) e limitare la capacità dei muscoli di contrarsi con una forza elevata [57,58]. Durante l’esercizio ad alta intensità, il glicogeno muscolare è il principale contributore di substrato energetico ed è stato dimostrato che la glicolisi fornisce circa l’80% del fabbisogno di ATP di una serie di flessioni del gomito se portata al cedimento muscolare [59]. Nonostante ciò, parte del glicogeno utilizzato durante questo tipo di esercizio può essere risintetizzato dal lattato, il che potrebbe ridurre il fabbisogno di carboidrati. È stato inoltre dimostrato che l’allenamento contro-resistenza riduce il glicogeno muscolare del 24-40% in una singola sessione [59,60].
La quantità esaurita può variare in base alla durata, all’intensità e al lavoro svolto, ma l’allenamento tipico del bodybuilding con ripetizioni più elevate e carichi moderati sembra causare la maggiore riduzione delle scorte di glicogeno muscolare [61]. Inoltre, è stato suggerito che quando le scorte di glicogeno sono troppo basse (~70 mmol/kg), ciò può inibire il rilascio di calcio e accelerare l’insorgenza della fatica muscolare [62]. Un basso livello di glicogeno muscolare riduce significativamente il numero di ripetizioni eseguite quando si eseguono tre serie di Squat all’80% di 1RM [57].
Tuttavia, è stato dimostrato che il consumo di una dieta contenente 7,7 g/kg/die di carboidrati per 48 ore prima di una sessione di allenamento non ha un effetto maggiore sulle prestazioni rispetto a 0,37g/kg/die quando si eseguono 15 serie a 15RM di esercizi per la parte inferiore del corpo [63]. Analogamente, un altro studio ha rilevato che una dieta con il 70% di carboidrati rispetto a una dieta con il 50% di carboidrati non ha un effetto maggiore sulle prestazioni durante l’esercizio sopramassimale; tuttavia, una dieta composta dal 25% di carboidrati ha ridotto significativamente le prestazioni [64].
Inoltre, visti gli effetti negativi a lungo termine sulla massa muscolare osservati di recente in studi su popolazioni allenate alla resistenza che seguono diete chetogeniche [49,51], potrebbe essere prudente per i bodybuilder assicurarsi semplicemente un apporto sufficiente di carboidrati, visti questi risultati disparati. Pertanto, mentre le diete a moderato e alto contenuto di carboidrati sono probabilmente appropriate per il bodybuilding, le diete a bassissimo contenuto di carboidrati possono essere dannose per l’allenamento.
Nei bodybuilder maschi, sono stati riportati apporti medi di carboidrati pari a 5,3g/kg/giorno durante la Off-Season [2]. Tuttavia, non sono state stabilite le quantità ottimali di carboidrati per i bodybuilder. In letteratura sono state proposte raccomandazioni per gli sport di forza, tra cui il bodybuilding, con assunzioni di 4-7g/kg/giorno e 5-6g/kg [15,65]. I carboidrati sembrano essere importanti per il bodybuilder, ma per ottenere benefici possono essere necessarie solo quantità moderate. Pertanto, dopo aver destinato le calorie alle proteine (1,6-2,2g/kg/die) e ai grassi (0,5-1,5g/kg/die), le restanti calorie dovrebbero essere destinate ai carboidrati. Tuttavia, sulla base delle prove attuali, potrebbe essere ragionevole consumare quantità sufficienti di carboidrati nell’intervallo ≥3-5g/kg/giorno, se possibile.
Sono necessarie ulteriori ricerche tra i bodybuilder per stabilire se l’assunzione abituale di carboidrati, superiore o inferiore a quella osservata, possa produrre ulteriori benefici. La Tabella sottostante riassume le raccomandazioni per le calorie e i macronutrienti.

Distribuzione e timing dei nutrienti:

Si dice che i bodybuilder consumino in media sei pasti al giorno [66]; tuttavia, non esistono studi che esaminino specificamente quale possa essere la frequenza ottimale dei pasti per questa popolazione [65]. Questa elevata frequenza dei pasti si basa sulla convinzione di un maggiore stato di anabolismo e persino di un migliore utilizzo dei nutrienti durante il giorno, che potrebbe tradursi in un miglioramento della composizione corporea.
Il concetto di temporizzazione dell’assunzione di proteine per massimizzare l’ipertrofia comprende diverse strategie di dosaggio. La prima a comparire in letteratura è stata il consumo di proteine in prossimità dell’allenamento contro-resistenza. I picchi di MPS sono più elevati in questo periodo quando si consumano proteine; pertanto, questa strategia è stata proposta per migliorare l’efficienza della riparazione e del rimodellamento del muscolo scheletrico [31]. Inoltre, a causa dell'”effetto muscolo pieno”, per cui un ulteriore apporto di proteine non aumenta la MPS finché non è trascorso un tempo sufficiente, distribuire uniformemente l’assunzione di proteine tra più pasti è un’altra strategia studiata per massimizzare la MPS totale giornaliera [67]. Infine, il consumo prima di andare a letto di proteine a lenta digestione (come la caseina) per evitare periodi catabolici prolungati durante il sonno è la strategia proposta più di recente per migliorare il bilancio proteico netto giornaliero [68], sebbene si sia dimostrata inutile nel perseguire il fine o, per lo meno, non molto diversa dalla risultante di una assunzione di isolate in un contesto alimentare con parità nel totale proteico giornaliero. Ciascuna di queste tre strategie sarà discussa in seguito.
- Dosaggio proteico:
Il periodo post-allenamento consente un picco della MPS più elevato quando si consumano proteine [31] e per raggiungere il picco di MPS può essere necessaria un’adeguata dose di Leucina “soglia” [32]. Diversi studi hanno esaminato il dosaggio proteico necessario per massimizzare la MPS dopo l’allenamento [69,70,71]. In uno studio sono stati consumati 0, 5, 10, 20 o 40g di proteine d’uovo intere dopo l’esercizio contro-resistenza della parte inferiore del corpo, con 20g che stimolavano al massimo la MPS [69]. Risultati simili sono stati riscontrati anche in un altro studio, in cui 20 g di siero di latte sono stati sufficienti a stimolare al massimo i tassi post-assorbitivi di MPS sia a riposo che dopo un lavoro unilaterale delle gambe all’80% del 1RM [70]. Inoltre, 40g di siero di latte non hanno prodotto ulteriori aumenti di MPS in questo studio e hanno portato all’ossidazione amminoacidica e alla produzione di urea.
Tuttavia, uno studio recente ha rilevato che, durante l’esecuzione di esercizi contro-resistenza per tutto il corpo al 75% del 1RM, 40g di siero di latte hanno prodotto una risposta MPS significativamente più elevata rispetto a 20g [71]. Esiste quindi una relazione tra il volume di tessuto muscolare danneggiato e stimolato e l’assunzione adeguata di proteine. È interessante notare che gli autori di una meta-analisi del 2013 hanno osservato che, nonostante gli studi con traccianti a breve termine mostrassero risposte nella MPS maggiori quando le proteine venivano consumate nella “finestra anabolica” post-allenamento, negli studi longitudinali sull’allenamento non è stato riscontrato alcun effetto significativo sull’ipertrofia quando si controllava l’apporto proteico totale giornaliero, indipendentemente dal fatto che le proteine fossero consumate all’interno della “finestra anabolica” o al di fuori di essa [72].
- Nutrient Timing:
Analogamente, i ricercatori di uno studio tracciante a breve termine che ha esaminato il dosaggio delle proteine nel corso di 12 ore hanno riportato una maggiore area sotto la curva della MPS quando sono state consumate quattro dosi di proteine del siero di latte da 20g ogni tre ore rispetto a due dosi da 40g a distanza di sei ore e otto dosi da 10g ogni ora e mezza [73]. In teoria, data la soglia oltre la quale le proteine supplementari consumate in una singola seduta non contribuiscono ulteriormente alla MPS [69] e a causa del “periodo refrattario” postprandiale durante il quale la MPS non può essere nuovamente stimolata al massimo [67], si potrebbe concludere che un bodybuilder dovrebbe raggiungere, ma non superare, questa dose soglia ogni poche ore per massimizzare l’ipertrofia a lungo termine. Tuttavia, gli autori di una review sistematica del 2018 sugli integratori proteici, comprendente 34 studi randomizzati e controllati, hanno riportato guadagni di massa magra simili tra i gruppi che utilizzavano un programma di dosaggio con i pasti (che comportava un minor numero di dosi di proteine di entità elevata) e tra i pasti (che comportava un maggior numero di dosi di proteine di entità moderata) [74].
È interessante notare che i dati che esaminano l’alimentazione proteica notturna mostrano uno distacco simile tra gli studi meccanicistici a breve termine e gli interventi di allenamento a lungo termine. Nel 2012 è stata condotta la prima ricerca che esaminava la risposta acuta all’alimentazione notturna con caseina [68]. Gli autori hanno riportato che 40g di caseina consumati prima di andare a letto sono stati digeriti, assorbiti e hanno stimolato la MPS e migliorato l’equilibrio proteico dell’intero corpo durante il periodo notturno in misura maggiore rispetto al placebo. Negli anni successivi sono stati pubblicati altri studi in acuto che hanno confermato [75] e riconfermato questi risultati in una popolazione più anziana [76]. Nel 2015, gli autori del primo studio longitudinale hanno riportato un aumento della forza e dell’ipertrofia in un gruppo a cui era stato somministrato un supplemento proteico notturno rispetto a un gruppo placebo [77].
Tuttavia, la quantità totale di proteine giornaliere non è stata equiparata, in quanto il gruppo con proteine notturne ha consumato 1,9g/kg/giorno, mentre il gruppo placebo ha consumato solo 1,3g/kg. È importante notare che in entrambi gli unici studi longitudinali con corrispondenza proteica che hanno confrontato l’integrazione notturna di caseina con i gruppi che hanno assunto l’integrazione prima, non sono state riportate differenze significative nell’aumento della FFM tra i gruppi [78,79]. Pertanto, la domanda è la stessa per ogni strategia di distribuzione: perché ci sono ripetuti distacchi tra gli studi meccanicistici a breve termine sulle MPS e le ricerche a lungo termine che esaminano l’effettiva ipertrofia? La risposta potrebbe risiedere nei metodi utilizzati negli studi sulla MPS, in quanto i partecipanti sono a digiuno, ricevono solo proteine in polvere in isolamento, spesso viene loro somministrato del siero di latte (che viene digerito molto rapidamente) e vengono osservati per brevi periodi. Questi contesti di laboratorio determinano tempi di digestione e cinetiche degli aminoacidi diversi da quelli che si verificano nel “mondo reale”. In particolare, in queste condizioni di laboratorio i livelli di base degli aminoacidi nel corpo sono più bassi del normale e la digestione e il successivo apporto di aminoacidi al muscolo sono più rapidi.
In condizioni di vita libera, le proteine vengono consumate principalmente da fonti alimentari intere, più volte al giorno e insieme ad altri alimenti, il che ritarda lo svuotamento gastrico. Per questi motivi, gli aminoacidi vengono titolati nel flusso sanguigno in modo più lento e costante; pertanto, in condizioni normali, le scorte sono quasi sempre prontamente disponibili [80]. Pertanto, l’efficacia della “finestra anabolica” e persino delle strategie di distribuzione delle proteine potrebbe non tradursi nella pratica. Inoltre, le limitazioni specifiche del laboratorio si estendono anche agli studi sull’alimentazione notturna. Si consideri, ad esempio, che 26g di proteine provenienti da una bistecca magra determinano un aumento sostenuto della MPS che dura almeno sei ore (l’intero periodo di tempo studiato) [81].
Inoltre, 26g sono solo il ~37% della dose di proteine contenuta in media in una cena americana [82], che richiederebbe più tempo per essere digerita a causa della maggiore porzione di proteine e dell’aggiunta di fibre, lipidi e altri nutrienti che ritarderebbero ulteriormente la digestione [80]. Pertanto, il tipico pasto finale potrebbe già soddisfare lo scopo di un frullato di caseina. Detto questo, nonostante queste discrepanze tra MPS e risultati della composizione corporea, non c’è nulla di male nel tentare queste strategie, soprattutto se attuate in modo pragmatico e senza introdurre ulteriori oneri logistici nel proprio programma quotidiano.
Pertanto, potrebbe essere prudente consigliare ai bodybuilder di suddividere l’assunzione giornaliera di 1,6-2,2 g/kg di proteine in più pasti contenenti ciascuno ~0,40-0,55g/kg [80] e di fare in modo che uno di questi pasti avvenga entro 1-2 ore prima o dopo l’allenamento, mentre un’alimentazione costituita da una fonte proteica e non proteica venga consumata 1-2 ore prima di dormire. Ad esempio, un bodybuilder di 90 kg potrebbe consumare 40-50g di proteine alle 8-9 del mattino per la colazione, allenarsi alle 11, consumare 40-50g di proteine alle 12-13 per il pranzo/post-allenamento, 40-50g di proteine a cena tra le 17-18, e poi un pasto finale di 40-50g di proteine non contenenti fonti proteiche grasse alle 21-10 prima di andare a letto entro le 23.
I carboidrati consumati prima dell’allenamento sono spesso una strategia utilizzata dagli atleti per migliorare le prestazioni negli esercizi ad alta intensità. La completa risintesi del glicogeno può essere raggiunta entro 24 ore da un allenamento che depaupera il glicogeno se si consumano quantità sufficienti di carboidrati [83]. Tuttavia, solo il 24-40% del glicogeno muscolare viene esaurito dopo un allenamento contro-resistenza [59,60]. Pertanto, una quantità di ≥3-5g/kg di carboidrati al giorno sarebbe probabilmente sufficiente per la risintesi del glicogeno. Questo elevato apporto giornaliero di carboidrati probabilmente riduce anche l’impatto della tempistica dei carboidrati pre-allenamento sulle prestazioni dell’esercizio.
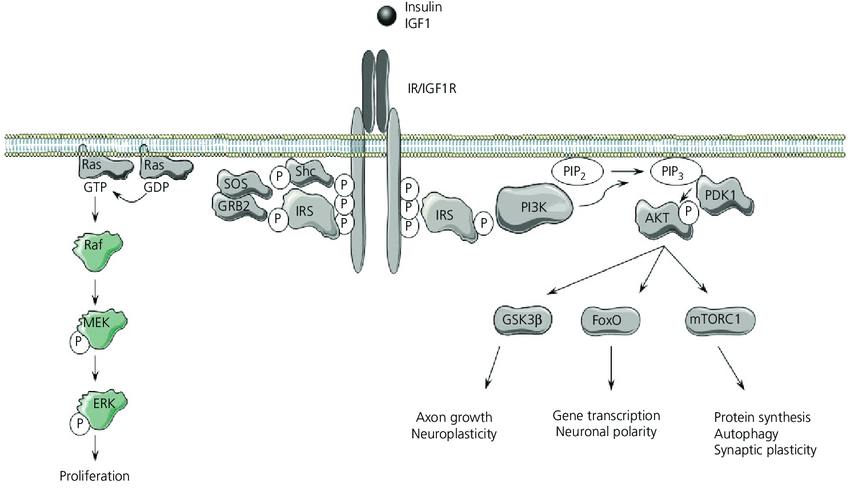
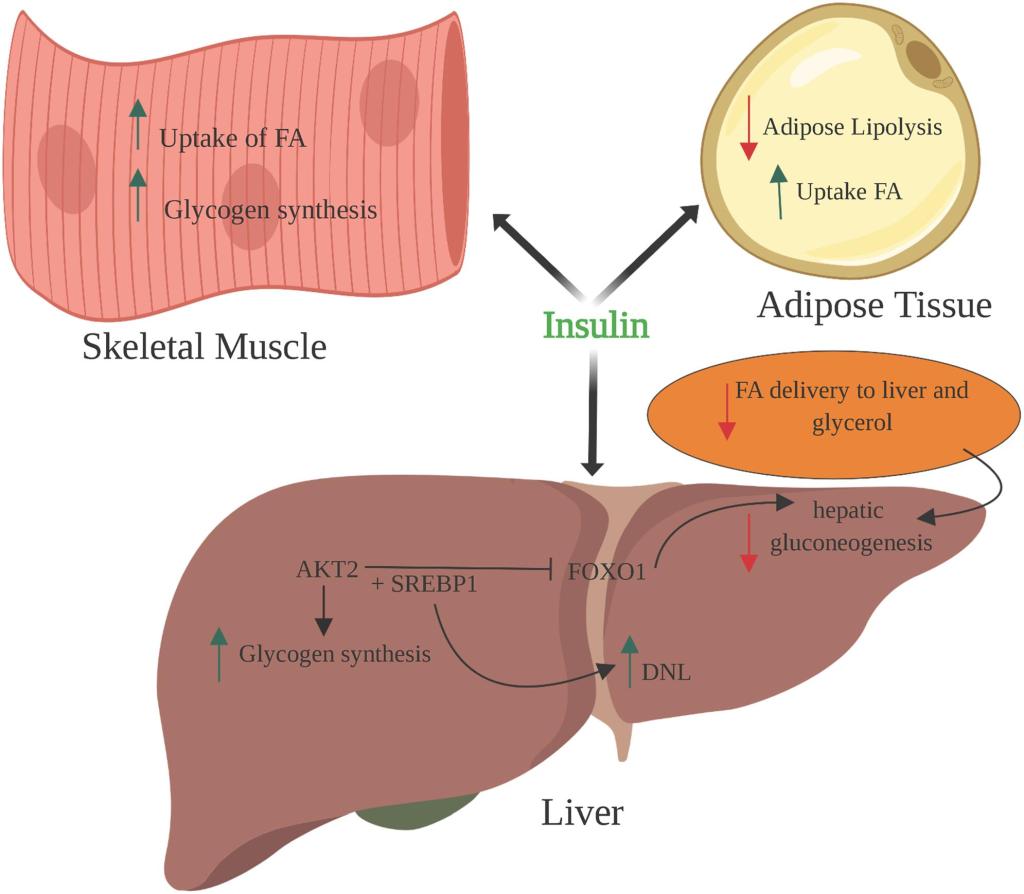
Spesso si sostiene che il consumo di carboidrati con le proteine dopo l’allenamento abbia un effetto anabolico dovuto alla secrezione di Insulina. Sebbene sia stato dimostrato che l’Insulina ha effetti anabolici [84], a livelli fisiologici il suo rilascio ha uno scarso impatto sull’anabolismo post-esercizio [85]. Inoltre, diversi studi non hanno evidenziato ulteriori effetti sulla sintesi proteica muscolare post-esercizio quando i carboidrati sono combinati con gli aminoacidi [86,87].
Inoltre, per i bodybuilder che non hanno bisogno di enfatizzare il rifornimento di glicogeno, le proteine aumentano la MPS post-allenamento a livelli massimi anche senza l’aggiunta di carboidrati [86,87]. Anche se il consumo di carboidrati nel post-allenamento non è certo dannoso, è improbabile che questo favorisca l’ipertrofia a lungo termine, come discusso in precedenti review [1,88]. Pertanto, è meglio concentrarsi sul consumo di un’adeguata quantità di carboidrati giornalieri e basare la distribuzione dei carboidrati intorno all’allenamento sulle preferenze personali.
Supplementazione OTC:

In un recente sondaggio condotto tra i bodybuilder, è stato riportato che tutti i partecipanti assumevano integratori alimentari [9]. Gli integratori alimentari più comuni erano: integratori di proteine (86%), creatina (68%), aminoacidi a catena ramificata (67%), glutammina (42%), vitamine (40%), olio di pesce (37%) e prodotti contenenti caffeina/efedrina (24%).
Sebbene gli integratori proteici siano molto popolari tra i bodybuilder, vengono utilizzati prevalentemente come gli alimenti interi per raggiungere gli obiettivi proteici. Pertanto, non verranno discussi in dettaglio. I lettori sono invitati a leggere la posizione dell’ISSN su questo argomento [89]. Inoltre, la trattazione di tutti gli integratori comunemente utilizzati dai bodybuilder esula dallo scopo di questo articolo. L’attenzione si concentrerà piuttosto sugli integratori alimentari che potrebbero potenzialmente produrre un effetto ergogenico e sugli integratori che possono garantire un apporto sufficiente di micronutrienti e acidi grassi essenziali.
- Creatina Monoidrato:

La Creatin-fosfato si trova in alte concentrazioni nel muscolo scheletrico e cardiaco, dove agisce come fonte di energia [90]. La Creatina può essere ottenuta anche attraverso la dieta nei soggetti che consumano carne; tuttavia, le concentrazioni di Creatina nella carne si riducono con la cottura [91].
Numerosi studi hanno osservato un aumento della massa e della forza muscolare in seguito a fasi di carico di Creatina, in genere di 20g al giorno per circa una settimana, spesso seguite da fasi di mantenimento di 2-3g di Creatina al giorno [92]. Tuttavia, la fase di carico potrebbe non essere necessaria. È stato dimostrato che la saturazione della Creatina muscolare dopo un’integrazione di 3g di Creatina Monoidrato per 28 giorni è simile al consumo di Creatina Monoidrato dopo la tipica fase di carico [93].
La maggior parte degli individui non raggiunge i 3g giornalieri con la dieta e può essere necessaria un’integrazione. Esistono numerose forme di Creatina negli integratori in commercio, tra le quali la Creatina Monoidrato è la più studiata. Le versioni più recenti di Creatina, come la kre-alkalyn [94] e la Creatina etil-estere [95], non si sono dimostrate superiori alla Creatina Monoidrato, nonostante abbiano in genere un prezzo più elevato. Pertanto, si raccomanda il consumo di 3-5g di Creatina Monoidrato al giorno. La tempistica di assunzione della Creatina non sembra avere importanza, poiché la saturazione delle riserve di Creatin-fosfato richiede circa 28 giorni per raggiungere le concentrazioni massime quando si consumano 3g al giorno e non ha un effetto in acuto [93].
- Caffeina:

Uno degli integratori alimentari più utilizzati dai bodybuilder sono gli stimolanti, in particolare la Caffeina [9]. Oltre ad aumentare l’eccitazione [96], la Caffeina può ridurre il dolore e lo sforzo percepito durante l’esercizio [97] e migliora la gestione del Calcio, aumentando la potenza [98]. Studi sull’allenamento contro-resistenza hanno rilevato che la Caffeina riduce la fatica e aumenta la forza [99,100]. Tuttavia, non tutti gli studi hanno dimostrato un effetto ergogenico sull’allenamento contro-resistenza [101]. Gli studi che hanno dimostrato un effetto ergogenico hanno utilizzato dosaggi elevati di caffeina (5-6 mg/kg), che sono al limite superiore di quello che è considerato un dosaggio sicuro [99,100]. Tuttavia, può essere consigliabile consumare il dosaggio minimo efficace per individuo, poiché l’assunzione regolare può generare tolleranza [102]. A causa dell’effetto acuto della Caffeina, è consigliabile assumerla circa 1 ora prima dell’esercizio fisico [99]. Tuttavia, l’emivita della Caffeina è di circa 3-9 ore; pertanto, può essere consigliabile consumare la Caffeina all’inizio della giornata per favorire un sonno sano se l’esercizio fisico viene svolto più tardi nel corso della giornata [103]. Sono necessarie ulteriori ricerche per trovare un consenso sull’uso della Caffeina nell’allenamento contro-resistenza, ma sulla base delle prove attuali un dosaggio di 5-6 mg/kg consumato prima dell’esercizio potrebbe produrre un effetto ergogenico sulle prestazioni nell’allenamento contro-resistenza.
- Beta-Alanina:

È stato dimostrato che l’ingestione di 4-6 g di beta-alanina aumenta i livelli di carnosina muscolare [104]. La carnosina agisce come tampone del pH nel muscolo scheletrico e può ritardare l’inizio dell’affaticamento muscolare durante l’esercizio ad alta intensità [105]. Una meta-analisi ha concluso che la beta-alanina potrebbe produrre effetti ergogenici durante l’esercizio ad alta intensità della durata di 60-240 secondi [104]. Inoltre, non sono stati riscontrati effetti benefici negli esercizi di durata inferiore a 60 secondi. La maggior parte degli studi inclusi nella meta-analisi riguardava l’esercizio di resistenza.
Tuttavia, è dimostrato che l’integrazione di beta-alanina può migliorare la resistenza muscolare negli atleti allenati alla resistenza [105] e può migliorare la composizione corporea [106]. Sono necessari ulteriori studi per esaminare l’effetto ergogenico della beta-alanina sulla composizione corporea e sulle prestazioni. Tuttavia, dato che i bodybuilder si allenano spesso con più di 10 ripetizioni per serie e spesso includono tecniche di intensità come drop set, pause di riposo, myo reps e altre, la beta-alanina potrebbe apportare un beneficio alla resistenza di queste serie [9].
Pertanto, potrebbe essere ragionevole per un bodybuilder consumare 3-5 g di beta alanina al giorno durante le fasi di allenamento ad alte ripetizioni o nelle fasi di allenamento in cui si incorporano diverse tecniche di intensità che prolungano la durata di un set. Come la creatina monoidrato, la beta-alanina non ha un effetto acuto, in quanto le concentrazioni di carnosina muscolare richiedono circa 4 settimane per raggiungere concentrazioni tali da produrre un effetto ergogenico, a condizione che se ne consumi una quantità sufficiente al giorno [104].
- Citrullina Malato:

Recentemente, la Citrullina Malato ha guadagnato popolarità tra i bodybuilder. Il potenziale effetto ergogenico è dovuto all’aumento del flusso ematico al muscolo, alla produzione di ATP e alla potenziale capacità della Citrullina Malato di agire come agente tampone [107]. È stato dimostrato che il consumo di 8g di Citrullina Malato aumenta le ripetizioni fino al cedimento del 50% [107,108,109,110], riduce l’indolenzimento muscolare del 40% [107] e migliora la forza massimale e la potenza anaerobica [111].
Tuttavia, non tutti gli studi hanno osservato effetti ergogenici del consumo di Citrullina Malato. Due studi recenti non hanno mostrato un miglioramento delle prestazioni, un aumento della risposta del gonfiore muscolare dovuto all’allenamento, un’attenuazione della fatica o un aumento dell’attenzione e dell’energia in seguito all’integrazione di Citrullina Malato in uomini allenati contro-resistenza a livello amatoriale [112,113].
Una recente meta-analisi di Trexler et al. ha analizzato 12 studi sullla CM per le prestazioni di forza e potenza [114]. Sebbene abbiano riscontrato solo una piccola dimensione dell’effetto (0,20), hanno concluso che questo potrebbe essere rilevante per gli atleti di alto livello in cui i risultati delle competizioni si decidono su margini ridotti, come i culturisti agonisti di alto livello. Si consiglia di assumere la Citrullina Malato circa 60 minuti prima dell’esercizio fisico per consentire un assorbimento sufficiente.
Sono necessarie ulteriori ricerche per determinare l’efficacia della Citrullina Malato nell’esercizio contro-resistenza. Allo stato attuale, i dati indicano un effetto benefico o neutro sulle prestazioni. Pertanto, sulla base delle prove attuali, 8g al giorno di Citrullina Malato consumati prima dell’esercizio potrebbero avere dei benefici interessanti per i bodybuilder.
- Alfa-GPC:

L’Alfa-GPC (alfa-glicerofosfocolina o colina alfoscerato) è un fosfolipide contenente colina. Quando viene ingerita, l’Alfa-GPC viene metabolizzata in colina e glicerolo-1-fosfato. La colina è un precursore dell’acetilcolina, un neurotrasmettitore coinvolto nella memoria, nell’attenzione e nella contrazione dei muscoli scheletrici. Il glicerolo-1-fosfato serve a sostenere le membrane cellulari.[https://pubmed.ncbi.nlm.]
L’Alfa-GPC sembra attraversare facilmente la barriera emato-encefalica e viene assorbito rapidamente. Attualmente è il miglior colinergico per aumentare i livelli plasmatici e cerebrali di colina.[https://pubmed.ncbi.nlm.]
L’integrazione orale di Alfa-GPC è interessante soprattutto per scopi nootropici o di potenziamento cognitivo. Esistono numerosi studi sui roditori che supportano questo effetto, ma non è ancora stato dimostrato negli esseri umani altrimenti sani. Negli anziani affetti da demenza lieve o moderata – che comporta un’alterazione della neurotrasmissione colinergica – l’Alfa-GPC migliora i sintomi cognitivi (ad esempio, disturbi della memoria e dell’attenzione).[https://pubmed.ncbi.nlm] L’Alfa-GPC può anche migliorare l’efficacia degli inibitori dell’acetilcolinesterasi (cioè i farmaci che aumentano la disponibilità di acetilcolina rallentandone la degradazione), utilizzati per il trattamento della malattia di Alzheimer.[https://pubmed.ncbi.nlm.]
Gli atleti sono un’altra popolazione che può trarre beneficio dall’integrazione di Alfa-GPC. Prove preliminari suggeriscono che l’alfa-GPC aumenta la potenza del salto verticale.[https://jissn.biomedcentral.com][https://pubmed.ncbi.nlm.] Inoltre, uno studio pilota ha riportato che l’Alfa-GPC ha aumentato il picco di forza nella panca, ma non la potenza di picco o il tasso di sviluppo della forza.[Ziegenfuss T, Landis J, Hofheins JJ Int Soc Sports Nutr.] Attualmente non è chiaro se l’Alfa-GPC aumenti la forza isometrica, ma i dati empirici e aneddotici sono incoraggianti [https://pubmed.ncbi.nlm.]
L’integrazione di un dosaggio pari a 600mg di Alpha-GPC prima di un test di potenza (spinte su panca) ha riportato un miglioramento della potenza del 14% rispetto al placebo quando assunta 45 minuti prima dell’attività; si trattava di uno studio pilota.[http://www.jissn.com] In media si è notato che il dosaggio di Alfa-GPC efficacie per trarre miglioramenti nella forza è nel range dei 300-600mg 45-30 minuti prima della seduta allenante.
- Multi Vitaminico-Multi Minerale:

Storicamente, i bodybuilder hanno utilizzato diete restrittive che eliminano alimenti o interi gruppi di alimenti. Di conseguenza, sono comuni numerose carenze di vitamine e minerali. Nei bodybuilder a dieta sono state osservate carenze di Calcio, vitamina D, Zinco, Ferro e altre ancora [115,116,117]. Tuttavia, la maggior parte della letteratura sulle pratiche alimentari dei bodybuilder risale agli anni ’80 e ’90; pertanto, sono necessari dati più recenti [2].
Più di recente, le pratiche alimentari dei bodybuilder che seguono una dieta tradizionale restrittiva sono state confrontate con quelle degli agonisti che utilizzano un approccio dietetico basato sui macronutrienti, in cui nessun alimento o gruppo alimentare è off limits [118]. Non sorprende che i concorrenti che utilizzano un approccio dietetico più flessibile presentino meno carenze di micronutrienti. In particolare, la vitamina E, la vitamina K e le proteine sono risultate significativamente inferiori nelle donne che utilizzavano approcci dietetici rigidi rispetto a quelle che utilizzavano approcci più flessibili. Nel presente articolo, specie se si parla di Off-Season, si raccomanda di utilizzare un approccio dietetico flessibile, in cui nessun alimento o gruppo viene eliminato dalla dieta.
In questo modo, è meno probabile che si verifichino carenze di micronutrienti, soprattutto se si considera che le atlete in Off-Season hanno a disposizione una maggiore quantità di calorie rispetto a quelle a dieta per un contest, il che dovrebbe consentire loro di incorporare una maggiore varietà di alimenti.
Ciononostante, può essere consigliabile raccomandare un integratore multivitaminico/minerale a basso dosaggio (≤100% RDA) come misura di sicurezza per prevenire eventuali carenze di micronutrienti, sottolineando al contempo il consumo di una buona varietà di alimenti al giorno per soddisfare il fabbisogno di micronutrienti.
- Omega 3 (EPA-DHA):

Gli acidi grassi polinsaturi con un doppio legame a tre atomi di distanza dal gruppo metilico terminale sono noti come ω-3 o acidi grassi omega-3 (O3). Un basso apporto di O3 nelle diete occidentali rispetto ad altre fonti di grassi alimentari (come gli acidi grassi omega-6) è associato a un peggioramento della salute multispettrale negli studi epidemiologici [119]. Pertanto, è interessante concentrarsi specificamente sulle modifiche della dieta per fornire acidi eicosapentaenoici e docosaesaenoici (EPA e DHA) – la carenza alimentare più comune nel mondo occidentale; ma vale la pena notare che la misurazione, l’interazione e l’effetto di O3 e acidi grassi omega-6 in relazione alla salute non sono chiari e vanno oltre lo scopo di questo articolo. Per una rassegna si rimanda ad altra pubblicazione [120].
Oltre alla salute, c’è interesse per i potenziali effetti anabolici degli integratori di EPA e DHA [121], che di solito vengono forniti attraverso l’olio di pesce o, in alcuni casi, l’olio di alghe. Tuttavia, ci sono dati contrastanti sulla capacità dell’olio di pesce di aumentare la risposta della sintesi proteica muscolare all’ingestione di proteine. Mentre un articolo di revisione del 2014 ha evidenziato una serie di studi secondo cui l’olio di pesce può aumentare la risposta [122], uno studio recente non ha rilevato alcun effetto sulla risposta della MPS a una sessione di allenamento contro-resistenza e all’ingestione di proteine dopo l’allenamento [123]. Inoltre, i dati sull’ipertrofia longitudinale sono pochi [124] e gli studi sulle prestazioni dell’allenamento contro-resistenza sono contrastanti [125] e in gran parte non applicabili o difficili da valutare a causa dell’uso di partecipanti non allenati o di allenamenti non standardizzati ed ecologicamente non realistici rispetto al bodybuilding.
In una recente review che affronta specificamente la questione se gli integratori di O3 possano o meno aumentare l’ipertrofia [126], gli autori hanno concluso che attualmente non ci sono prove sufficienti per fare tale affermazione. Sebbene siano necessarie ulteriori ricerche prima di poter raccomandare l’integrazione di O3 (o di alterazioni della dieta) a fini di costruzione muscolare, i benefici per la salute dell’integrazione di O3 sono degni di nota. Ad esempio, recenti meta-analisi hanno riportato che l’integrazione di olio di pesce riduce i sintomi della depressione [127], diminuisce il rischio di morte cardiaca [128], riduce la pressione sanguigna [129] e diminuisce la circonferenza vita [130]. Pertanto, gli atleti estetici possono prendere in considerazione l’integrazione giornaliera di olio di pesce (o di alghe) (1.5-2.5g di EPA/DHA) per la salute generale e multi spettro, ma sono necessari studi futuri per formulare raccomandazioni relative alle prestazioni nel bodybuilding.
- Acido Arachidonico (AA):

L’Acido Arachidonico (AA) è l’acido grasso omega-6 più rilevante dal punto di vista biologico e, nella membrana lipidica di una cellula, è l’acido grasso che viene confrontato con i due acidi grassi dell’olio di pesce (EPA e DHA) nella costituzione di un rapporto omega-3:6. Dati recenti suggeriscono un’assunzione giornaliera di 50-250mg di Acido Arachidonico[https://www.ncbi.nlm.][https://www.ncbi.nlm.][https://www.ncbi.nlm.][https://www.ncbi.nlm.] con alcune fonti che stimano livelli fino a 500mg al giorno;[https://www.ncbi.nlm.] l’assunzione di Acido Arachidonico sembra essere inferiore nei vegetariani[https://www.ncbi.nlm.].
Si ritiene che l’Acido Arachidonico sia importante per il metabolismo del muscolo scheletrico, poiché si pensa che i fosfolipidi della membrana del sarcoplasma riflettano la dieta,[https://www.ncbi.nlm.][https://www.ncbi.nlm.] l’allenamento stesso sembra alterare il contenuto di fosfolipidi del muscolo (indipendentemente dalla composizione delle fibre muscolari[https://www.ncbi.nlm.] e associato a un rapporto omega 6:3 più basso[https://www.ncbi.nlm.][https://www.ncbi.nlm.]) e gli eicosanoidi dell’Acido Arachidonico interagiscono con la sintesi proteica muscolare attraverso i loro recettori.
L’Acido Arachidonico segnala la sintesi proteica muscolare attraverso una via dipendente dalla COX-2 (che suggerisce il coinvolgimento delle prostaglandine)[https://www.ncbi.nlm.] che è associata ad aumenti sia della prostaglandina E2 (PGE2) che del PGF(2α),[https://www.ncbi.nlm.][https://www.ncbi.nlm.][https://www.ncbi.nlm.][https://www.ncbi.nlm.] anche se l’incubazione con PGE2 o PGF(2α) isolati non sembra replicare pienamente gli effetti ipertrofici dell’Acido Arachidonico. [https://www.ncbi.nlm.] PGE2 e PGF(2α) sono indotti anche dall’esercizio fisico (nello specifico, dallo stiramento delle cellule muscolari in vitro[https://www.ncbi.nlm.]) ed è stato osservato sia nel siero[https://pubmed.ncbi.nlm.][https://www.ncbi.nlm.] che a livello intramuscolare (quadruplicato, da 0,95+/-0,26ng/mL a 3,97+/-0. La capacità del riflesso da stiramento di aumentare le concentrazioni di PGE2 e PGF(2α)[https://www.ncbi.nlm.] potrebbe essere dovuta semplicemente al fatto che lo stiramento aumenta l’attività delle COX2.[https://www.ncbi.nlm.][https://www.ncbi.nlm.]
Va notato che l’integrazione di 1.500mg di Acido Arachidonico (rispetto a una dieta di controllo contenente 200mg dello stesso) per 49 giorni ha aumentato la secrezione di PGE2 da parte di cellule immunitarie stimolate (del 50-100%) in giovani uomini altrimenti sani,[https://www.ncbi.nlm.] ma la rilevanza di questo studio per il muscolo scheletrico non è nota. Questo studio ha anche osservato che, senza stimolazione, non c’erano differenze significative tra i gruppi.[https://www.ncbi.nlm.] Altrove, è stata osservata una tendenza all’aumento delle concentrazioni sieriche di PGE2 a riposo in uomini allenati a cui sono stati somministrati 1.000mg di Acido Arachidonico per 50 giorni.[https://www.ncbi.nlm.]
L’Acido Arachidonico, attraverso gli eicosanoidi noti come PGF(2α) e PGE2, stimola la sintesi proteica muscolare. Sono prodotti a partire dall’Acido Arachidonico, ma normalmente non formano i rispettivi eicosanoidi per la costruzione del muscolo finché la cellula non viene stimolata da un fattore di stress (come il riflesso di stiramento di una cellula muscolare) che ne induce la produzione.
Il recettore per il PGF(2α) (recettore FP) sembra essere sovraregolato dagli inibitori della COX1 (l’acetaminofene utilizzato in questo studio)[https://www.ncbi.nlm.] e si ritiene che una maggiore segnalazione del PGF(2α) sia alla base del miglioramento della sintesi proteica muscolare osservato nei soggetti anziani con farmaci antinfiammatori. La supplementazione di Acido Arachidonico non sembra influenzare la quantità di recettori FP nei giovani;[https://www.ncbi.nlm.] mentre l’esercizio fisico stesso può aumentare il contenuto di recettori EP3, né gli inibitori della COX1[https://www.ncbi.nlm.] né l’Acido Arachidonico[https://www.ncbi.nlm.] sembrano influenzarlo ulteriormente.
Tuttavia, è stato riscontrato che l’uso di inibitori della COX2 (nei giovani) sopprime l’aumento di PGF(2α) indotto dall’esercizio fisico (Ibuprofene e Acetaminofene)[https://www.ncbi.nlm.][https://www.ncbi.nlm.] e di PGE2,[https://www.ncbi.nlm.] il che si pensa sia dovuto al fatto che la conversione da PGH2 in questi metaboliti dipende dall’attività della COX2.
Poiché la produzione di questi eicosanoidi dipende dall’enzima COX2, si ritiene che l’inibizione di questo enzima riduca gli effetti anabolizzanti dell’esercizio fisico se assunto prima dello stesso.
L’acido arachidonico (così come l’EPA dall’olio di pesce) non ha compromesso l’assorbimento del glucosio nelle cellule muscolari isolate e 10μM di acido grasso sono in grado di attenuare la resistenza all’Insulina indotta dai grassi saturi; [https://pubmed.ncbi.nlm.] un fenomeno osservato con i grassi saturi a 18 o più catene di carbonio[https://www.ncbi.nlm.] che non sembra applicarsi agli acidi grassi polinsaturi di uguale lunghezza di catena[https://www.ncbi.nlm.][https://www.ncbi.nlm.] ed è probabilmente legato all’aumento delle ceramidi intracellulari[https://www.ncbi.nlm.] che compromettono la segnalazione di Akt[https://www.ncbi.nlm.][https://www.ncbi.nlm.] e riducono l’assorbimento di glucosio mediato da GLUT4 con l’Insulina.[https://www.ncbi.nlm.]
L’Acido Arachidonico e i grassi polinsaturi omega-3 sono entrambi associati a una migliore sensibilità all’Insulina delle cellule muscolari, che potrebbe essere secondaria alla riduzione dei livelli di grassi saturi nella membrana lipidica e quindi alla riduzione delle concentrazioni intracellulari di ceramidi. È possibile che ciò non sia correlato agli eicosanoidi o al rapporto omega-3:6.
In 31 uomini allenati, sottoposti a un programma di sollevamento pesi e a una dieta standardizzata (500kcal in eccesso con 2g/kg di proteine) con 1g di Acido Arachidonico al giorno o placebo, l’integrazione per 50 giorni è sembrata aumentare la potenza di picco (7,1%) e la potenza media (3,6%) al test di Wingate, ma non è riuscita a influenzare positivamente la massa muscolare o le misure di potenza del sollevamento pesi (bench press e leg press).[https://www.ncbi.nlm.]
Attualmente non ci sono prove sufficienti per raccomandare una dose ideale di integrazione di Acido Arachidonico, ma aneddoticamente si usa un dosaggio di circa 1.500 mg da assumere 45 minuti prima dell’allenamento per un periodo medio di 8 settimane. Non è certo che si tratti di una dose ottimale o che sia necessaria la tempistica.
Va inoltre notato che per le persone affette da patologie infiammatorie croniche, come l’artrite reumatoide o le malattie infiammatorie intestinali, la dose ideale di Acido Arachidonico può essere in realtà una sua restrizione dietetica. Nei casi di malattie infiammatorie, l’integrazione di Acido Arachidonico è probabilmente controindicata.
Raccomandazioni per gli integratori alimentari e il dosaggio per i bodybuilder in Off-Season:
- Creatina Monoidrato= 3-5g/die;
- Beta-Alanina= 3-5g/die;
- Citrullina Malato= 8g/pre-workout;
- Alfa-GPC= 300-600mg/pre-workout;
- Caffeina= 5-6mg/Kg/pre-workout (media standard tra 200 e 600mg/die);
- Multi Vitaminico – Multi Minerale= ≤100% RDA/die;
- Omega 3 (EPA-DHA)= 1.5-2.5g/die;
- Acido Arachidonico= 1.5g/pre-workout.
Supplementazione PEDs:

Una cosa occorre premettere prima di procedere con la descrizione delle molecole più utilizzate nel contesto della Off-Season: non esistono PEDs esclusivamente confinabili in uno dei contesti della programmazione di un bodybuilder. Esiste il grado di versatilità il quale sta ad indicare quanto una molecola possa essere gestita con facilità in situazioni preparatorie differenti. Esistono molecole che per caratteristiche possono dare vantaggi maggiori in Off-Season/Bulk per via di alcune loro caratteristiche che in altro contesto, per esempio il pre-contest, risulterebbero più complesse da gestire. Ma questo non significa che tali molecole siano generalemnte da considerarsi “off-limitz” in un altra fase della preparazione annuale.
Premesso ciò, l’attenzione in questo paragrafo si concentrerà sui principali PEDs usati in Off-Season.
- Testosterone (in sospensione ed esterificato):

Tra tutti gli AAS, il Testosterone è quello che non ha bisogno di particolari presentazioni. Si tratta dell’ormone sessuale maschile per antonomasia. Nell’uomo, il Testosterone svolge un ruolo fondamentale nello sviluppo dei tessuti riproduttivi maschili, come i testicoli e la prostata, oltre a promuovere le caratteristiche sessuali secondarie, come l’aumento della massa muscolare e ossea e la crescita dei peli. Inoltre, in entrambi i sessi, il Testosterone è coinvolto nella salute e nel benessere, compresi gli stati d’animo, il comportamento e la prevenzione dell’osteoporosi in cooperazione con l’Estradiolo. Livelli insufficienti di Testosterone negli uomini possono portare ad anomalie, tra cui la fragilità e la perdita ossea.
In generale, il Testosterone promuove la sintesi proteica e quindi la crescita dei tessuti dotati di recettori per gli androgeni. Il Testosterone può essere descritto come avente effetti virilizzanti e anabolizzanti (anche se queste descrizioni categoriali sono in qualche modo arbitrarie, poiché vi è una grande sovrapposizione reciproca tra di essi).
- Gli effetti anabolizzanti comprendono la crescita della massa e della forza muscolare, l’aumento della densità e della resistenza ossea e la stimolazione della crescita lineare e della maturazione ossea.
- Gli effetti androgeni comprendono la maturazione degli organi sessuali, in particolare del pene, e la formazione dello scroto nel feto, e dopo la nascita (di solito nella pubertà) l’approfondimento della voce, la crescita dei peli del viso (come la barba) e dei peli ascellari. Molti di questi effetti rientrano nella categoria dei caratteri sessuali secondari maschili.
Al principio degli anni 30 del novecento avvenne la sintesi chimica del Testosterone, quando Butenandt e G. Hanisch pubblicarono un articolo che descriveva “Un metodo per preparare il Testosterone dal colesterolo”. Solo una settimana dopo, il terzo gruppo, Ruzicka e A. Wettstein, annunciò una domanda di brevetto in un documento “Sulla preparazione artificiale dell’ormone testicolare Testosterone (Androsten-3-one-17-ol).” Ruzicka e Butenandt ricevettero il premio Nobel per la chimica nel 1939 per il loro lavoro.
Gli studi clinici sull’uomo, che prevedevano dosi PO (per via orale) di Methyltestosterone o iniezioni di Testosterone Propionato, iniziarono già nel 1937. Il Testosterone Propionato è menzionato in una lettera all’editore della rivista Strength and Health nel 1938; questo è il primo riferimento noto a un AAS in una rivista statunitense di sollevamento pesi o Bodybuilding.
Lo sviluppo delle proprietà di costruzione muscolare del Testosterone proseguì negli anni ’40, in Unione Sovietica e nei paesi del blocco orientale come la Germania dell’Est, dove sono stati utilizzati programmi di AAS per migliorare le prestazioni dei sollevatori di pesi olimpici e di altri dilettanti già prima degli anni ’50. In risposta al successo dei sollevatori di pesi russi, il medico della squadra olimpica statunitense John Ziegler lavorò con un equipe di chimici per sviluppare un AAS con effetti androgeni ridotti. Ma questa è un altra storia.
L’uso del Testosterone nello sport si diffuse tra gli anni ’50 e gli anni ’60. Le forme utilizzate nei primi tempi erano il Testosterone in sospensione e il Testosterone Propionato, che rappresentano con il Methyltestosterone (Testosterone metilato in C-17) le forme più datate dell’ormone in questione (1935).
In ambito culturistico, il Testosterone rappresenta un AAS sufficientemente versatile in maniera dose-dipendente e sensibilità-dipendente dal momento che il dosaggio dovrebbe essere tarato in base alle risposte metaboliche soggettive alle quali è soggetto l’ormone (vedi, ad esempio, aromatizzazione in estrogeni). Questo ultimo punto è di estrema importanza al fine di evitare l’uso/abuso di AI (Inibitori dell’Aromatasi) e/o SERM (Modulatori Selettivi del Recettore degli Estrogeni). Oltre a peggiorare potenzialmente il quadro lipidico, sommandosi all’azione degli AAS utilizzati, essi riducono l’espressione epatica di IGF-1 cosa che può ridurre la risposta anabolizzante del protocollo PEDs. Nei soggetti caratterizzati da una elevata sensibilità all’attività estrogenica, le procedure applicate vedono: 1) l’uso di Raloxifene o Tamoxifene (SERM) a dosi sufficienti a impedire la comparsa o il peggioramento di una ginecomastia in stadio iniziale già presente e non ancora asportata chirurgicamente 2) l’uso di dosi fisiologiche di Testosterone come base onde evitare la comparsa di stati letargici, affaticabilità, disfunzioni sessuali ecc 3) l’uso di un “mix” composto da Testosterone e Boldenone (vedi in seguito) tale da poter usufruire della bassa e diversa sensibilità all’azione dell’Enzima Aromatasi su quest’ultimo riuscendo ad avere un controllo estrogenico teoricamente migliore (Testosterone e Boldenone mostrano qualità anabolizzanti intrinseche simili).
In un contesto Off-Season, quindi, vista l’importanza della presenza di un buon livello di Estradiolo sia sul complesso degli effetti anabolizzanti ricercati sia per la sua attività sessuale e cerebrale, il Testosterone andrebbe inizialmente calibrato sul soggetto e nel caso affiancato da dosi altrettanto ben tarate di SERM la dove ne risultasse un reale bisogno.
L’uso di un estere che garantisca un rilascio graduale della molecola (vedi Enantato o Cypionato) risulta la scelta migliore al fine di creare una soglia ematica stabile e esente da picchi e cali che possono risultare controproducenti a livello psicofisico. Tenere sempre in considerazione l’emivita di una molecola è uno dei punti fondamentali per sfruttarla al meglio. Nel caso degli esteri sopra citati, una divisione del dosaggio settimanale in due somministrazioni uguali distanziate da quattro-cinque giorni l’una dall’altra risulta una pratica ottimale allo scopo di creare una soglia ematica stabile.
I dosaggi comunemente utilizzati, parlando di molecole esterificate, vanno da 200mg ad 1g a settimana. Per quanto riguarda il Testosterone in sospensione, le dosi comunemente utilizzate vanno dai 175mg ai 700mg a settimana.

Il Boldenone [1,4-androstadiene-3-one,17b-ol], commercializzato con il nome di Equipoise, Ganabol, Equigan, Ultragan, e Boldane, è uno steroide anabolizzante-androgeno spesso legato all’estere Undecylenato. Strutturalmente molto simile al Testosterone, il Boldenone differisce da questo per il doppio legame tra C1 e C2.
La Ciba brevettò il Boldenone nel 1949. Successivamente, negli anni ’50 e ’60, sviluppò diversi esteri sperimentali del farmaco. Uno di questi era il Boldenone Undecilenato, che fu introdotto per uso clinico con il marchio Parenabol e fu utilizzato alla fine degli anni ’60 e all’inizio degli anni ’70. Tuttavia, fu sospeso prima della fine degli anni ’70. Ad oggi l’uso del Boldenone è legale in alcuni paesi in campo veterinario.
Essendo una molecola che ha mostrato una bassa tendenza alla conversione in Estradiolo, come accennato nella sezione dedicata al Testosterone, viene spesso utilizzata come agente “mix” da abbinare come base al Testosterone al fine di avere un maggiore controllo sui livelli estrogenici.
Se qualcuno volesse usare 500mg di Testosterone, ma non potrebbe usare un tale dosaggio dal momento che presenta particolare difficoltà nella gestione estrogenica in specie senza l’uso di AI come Exemestane o Anastrozolo, una conclusione a cui molti superficialmente sono giunti è che si potrebbe semplicemente usare il Boldenone al dosaggio sopra citato per ridurre della metà l’attività estrogenica, ma comunque supportare un’adeguata produzione di Estradiolo. Ma quando si approfondisci l’ipotesi e la si testa sul campo, è davvero così che stanno le cose? In realtà no, o, comunque, la media delle variabili di risposta spinge a confermare una maggiore validità nel “mixare” Testosterone e Boldenone coprendo la dose base calcolata in precedenza, e con variazione di percentuale T:B ratio da 1:1 a 2:1.
Comunque, oltre a rappresentare genericamente una discreta molecola sia in in preparazione alla gara che in Off-Season, I dosaggi utilizzati si settano nel range tra i 200mg ed i 500mg a settimana, spesso abbinato ad una dose variabile (vedi sopra) di Testosterone.

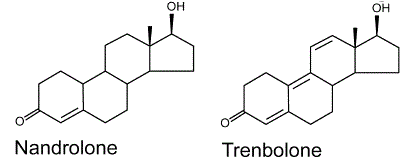
Il Nandrolone, noto anche come 19-nortestosterone, è uno Steroide Androgeno Anabolizzante (AAS) utilizzato sotto forma di molecola legata a esteri come quello Decanoato (nome commerciale Deca-Durabolin) e il Fenilpropionato (nome commerciale Durabolin). Gli esteri del Nandrolone sono utilizzati nel trattamento di anemie, cachessia (sindrome da deperimento), osteoporosi, cancro al seno e per altre indicazioni mediche.
Il Nandrolone è stato sintetizzato per la prima volta nel 1950. È stato introdotto per la prima volta nel mercato farmaceutico, come Nandrolone Fenilpropionato, nel 1959, e poi come Nandrolone Decanoato nel 1962, seguito da ulteriori esteri.
Il Nandrolone ha una bassa affinità di interazione con l’Enzima Aromatasi convertendo in Estrone, un estrogeno molto meno potente dell’Estradiolo, circa 10 volte meno attivo, e, come tale, è un estrogeno relativamente debole. In una condizione di somministrazione del Nandrolone senza una base di Testosterone, i livelli di Estradiolo calerebbero marcatamente a favore di un aumento del Estrone il quale non potrebbe però sostituire nelle diverse attività tissutali il prima citato E2. Le conseguenze negative si verificherebbero dall’attività sessuale all’attività neurosteroidea.
Infatti, un effetto da non sottovalutare con l’uso di Nandrolone è il suo impatto sul SNC. L’impatto del Nandrolone sul Sistema Nervoso Centrale è stato osservato scientificamente. Nello studio intitolato “The Impact of Nandrolone Decanoate on the Central Nervous System” vengono descritti chiaramente i numerosi effetti psicologici di questa molecola. Essi comprendono e influenzano:
1- Aggressività
2- Ansia, paura e stress
3- Ricompensa e dipendenza
4- Apprendimento, memoria e capacità di lavoro
5- Locomozione e attività fisica
6- Effetti sulla HPAA (Asse Ipotalamo-Pituitaria-Surrene)
7- Effetto sui neurotrasmettitori: Recettore Acido γ-Aminobutirrico Tipo A (GABAA); Recettori 5-idrossitriptamina (5-HT) e 5-HT; Recettori della Dopamina e Recettori Oppioidi.
Questo effetto, unito alla modesta potenzialità anabolizzante se confrontata con altre molecole anche della stessa famiglia, fa pendere l’ago della bilancia verso gli svantaggi d’uso piuttosto che i vantaggi. Sebbene vi sia un rapporto tra Testosterone e Nandrolone finalizzato a ridurre la comparsa di questi effetti avversi (ratio T:N = 2:1) su un buon numero di soggetti risulta dare comunque problemi rilevanti.
Il suo uso principale in Off-Season comprende dosaggi medi tra i 200mg ed i 400mg a settimana, con un adeguato rapporto con il Testosterone. Se utilizzato a fini di recupero articolare viene usato a dosaggi di 100mg a settimana, e con tali dosaggi difficilmente emergono i problemi sopra elencati a patto che ci sia una base di Testosterone.

Il Drostanolone, noto anche come 2α-metil-5α-diidrotestosterone (2α-metil-DHT) o come 2α-metil-5α-androstan-17β-ol-3-one, è uno steroide androstano sintetico e un derivato del DHT. Si tratta nello specifico di DHT con un gruppo metile in posizione C2α. La forma esterificata Drostanolone Propionato è stata usata in passato nel trattamento del cancro al seno nelle donne per via della sua attività antiestrogenica. Questa azione il Drostanolone la esplica sia agendo come antagonista del recettore degli estrogeni e sia come inibitore dell’Enzima Aromatasi. Ed è proprio per questo motivo che una molecola generalmente relegata all’uso in “Cut” o pre-gara trova un suo uso funzionale in Off-Season. La sua attività AI è comunque moderata ma sufficiente in un buon numero di soggetti per evitare l’aggiunta di SERM e/o AI di altro genere. L’attività AI moderata sembra non incidere negativamente in modo sensibile sull’Asse GH/IGF1.
L’effetto miotrofico risulta simile a quello osservato con il Methenolone, in generale moderatamente inferiore al Testosterone. I dosaggi utilizzati in Off-Season per il controllo estrogenico sono nel range dei 200-400mg a settimana (diviso in due iniezioni distanziate da 4-5 giorni) per l’estere Enantato, mentre per il Propionato 150-350mg a settimana (dosi a giorni alterni o giornaliere).

Il Trenbolone, noto anche come 19-nor-δ9,11-testosterone o come estra-4,9,11-trien-17β-ol-3-one, è uno steroide sintetico e un derivato del Nandrolone (19-nortestosterone) sintetizzato per la prima volta nel 1963. Si tratta nello specifico di Nandrolone con due doppi legami aggiuntivi nel nucleo steroideo. Gli esteri del Trenbolone, che hanno un estere in posizione C17β, includono il Trenbolone Acetato, il Trenbolone Enantato, Il Trenbolone Hexahydrobenzylcarbonato e il Trenbolone Undecanoato. Il Trenbolone Acetato (marchi Finajet, Finaplix, e altri) e il Trenbolone Hexahydrobenzylcarbonato (marchi Parabolan, Hexabolan), sono o sono stati commercializzati per uso veterinario e clinico nell’uomo. Il Trenbolone Acetato è utilizzato in medicina veterinaria nel bestiame per aumentare la crescita muscolare e l’appetito degli animali, mentre il Trenbolone Hexahydrobenzylcarbonato è stato utilizzato in passato a livello clinico nell’uomo, ma ora non è più commercializzato.
Si tratta di uno degli AAS più versatili in assoluto, con un ottima resa tanto in preparazione alla gara quanto in Off-Season. L’enorme potenziale anabolizzante del Trenbolone, così come dei suoi analoghi, è stato riportato fin dagli anni ’60. La sua diffusione nel Bodybuilding è iniziata circa negli anni ’80 del secolo scorso. La sua elevata potenzialità miotrofica, lipolitica e di spinta mentale lo resero in poco tempo estremamente popolare tra i culturisti.
In Off-Season viene utilizzato nelle sue forme eseterificate Enantato e Hexahydrobenzylcarbonato a dosaggi nell’ordine dei 100-400mg a settimana (divisa in due somministrazioni distanziate l’una dall’altra da 4-5 giorni), sebbene il trend d’oltre oceano è arrivato a dosaggi decisamente eccessivi e nell’ordine del grammo. Per l’esetere Acetato i dosaggi medi vanno da 150mg a 350mg a settimana con dosaggi a giorni alterni o giornalieri.
E’ necessario ricordare ai lettori che gli effetti collaterali a livello del SNC possono verificarsi in alcuni punti come nel caso del Nandrolone sebbene i vantaggi rendano il Trenbolone più bilanciato tra sides e vantaggi.

Il Trestolone, noto anche come 7α-metil-19-nortestosterone (MENT) o come 7α-metilestr-4-en-17β-ol-3-one, è uno steroide sintetico e un derivato del Nandrolone (19-nortestosterone). È una forma modificata del Nandrolone con un gruppo metile in posizione C7α. Tra gli AAS strettamente correlati vi sono il 7α-metil-19-norandrostenedione (MENT dione, trestione), un pro-ormone androgeno del Trestolone, e il Dimetandrolone (7α, 11β-dimetil-19-nortestosterone), il derivato metilato C11β del Trestolone, nonché il Mibolerone (7α,17α-dimetil-19-nortestosterone) e il Dimetiltrienolone (7α,17α-dimetil-δ9,11-19-nortestosterone). Anche il progestinico Tibolone (7α-metil-17α-etinil-δ5(10)-19-nortestosterone) è strettamente correlato al Trestolone.
Il Trestolone è stato descritto per la prima volta nel 1963. Tuttavia, non è stato successivamente studiato fino al 1990. Lo sviluppo del Trestolone per un potenziale uso nella contraccezione ormonale maschile e nella terapia sostitutiva degli androgeni è stato avviato nel 1993 ed è proseguito in seguito. Non sembra che siano stati condotti ulteriori sviluppi dal 2013. Il Trestolone è stato sviluppato dal Population Council, un’organizzazione non governativa senza scopo di lucro dedicata alla salute riproduttiva.
Come AAS, il Trestolone è un agonista del recettore degli androgeni (AR), analogamente agli androgeni come il Testosterone e il Diidrotestosterone (DHT). Questo AAS presenta spiccate proprietà anticortisolemiche sia attraverso l’inibizione enzimatica sia per attività antagonista recettoriale. Il Trestolone non è un substrato per la 5α-reduttasi e quindi non è potenziato o inattivato nei cosiddetti tessuti “androgeni” come la pelle, i follicoli piliferi e la ghiandola prostatica. Come tale, ha un elevato rapporto tra attività anabolica e androgena, analogamente ad altri derivati del Nandrolone. Il Trestolone è un substrato per l’Aromatasi e quindi produce come metabolita l’estrogeno 7α-metilestradiolo. Tuttavia, il Trestolone ha solo una debole attività estrogenica e una quantità che sembrerebbe essere insufficiente per scopi terapici sostitutivi, come evidenziato dalla diminuzione della densità minerale ossea negli uomini trattati con esso per l’ipogonadismo.
Il potenziale anabolizzante del Trestolone ha mostrato un grado di superiorità miotrofica rispetto al Trenbolone. Le sue caratteristiche ne fanno prediligere l’uso in Off-Season/Bulk. I dosaggi utilizzati con la forma Acetato sono nell’ordine dei 150-350mg a settimana con una cadenza nelle somministrazioni a giorni alterni. Sebbene sia più rara da reperire, la forma Enantato è utilizzato nel range dei 200-400mg a settimana divisi in somministrazioni ogni 4-5 giorni.

L’Oxymetholone, noto anche come 2-idrossimetilene-17α-metil-4,5α-diidrotestosterone (2-idrossimetilene-17α-metil-DHT) o come 2-idrossimetilene-17α-metil-5α-androstan-17β-olo-3-one, è uno steroide androstanico sintetico e un derivato 17α-alchilato del DHT.
L’Oxymetholone è stato descritto per la prima volta in un articolo del 1959 da scienziati della Syntex. È stato introdotto per uso medico dalla Syntex e dalla Imperial Chemical Industries nel Regno Unito con il marchio Anapolon nel 1961. L’Oxymetholone è stato introdotto anche con i marchi Adroyd (Parke-Davis) nel 1961 e Anadrol (Syntex) nel 1962. Il farmaco è stato commercializzato negli Stati Uniti nei primi anni ’60.
Come altri AAS, l’Oxymetholone è un agonista del recettore degli androgeni (AR). Non è un substrato per la 5α-reduttasi (dal momento che è già 5α-ridotto) ed è uno substrato scarso per il 3α-idrossisteroide deidrogenasi (3α-HSD), e quindi mostra un alto rapporto di attività anabolizzante rispetto all’effetto androgenico.
Data la sua derivanza dal DHT, l’Oxymetholone non è un substrato per l’Enzima Aromatasi e quindi non può essere aromatizzato in metaboliti estrogenici. Tuttavia, caratteristica unica tra i derivati del DHT, l’Oxymetholone è comunque associato a un’estrogenicità relativamente elevata ed è noto per avere il potenziale di produrre effetti collaterali estrogenici come ginecomastia (anche se non comune) e ritenzione idrica. È stato suggerito che questo può essere una conseguenza del legame diretto a l’attivazione del recettore degli estrogeni da parte dell’Oxymetholone (estrogenicità intrinseca). L’Oxymetholone non possiede alcuna attività progestinica significativa. Per via della caratteristica attività estrogenica intrinseca, con l’uso di Oxymetholone è spesso necessario l’uso di un SERM onde avere un controllo sulla aumentata attività estrogenica.
A causa della sua struttura 17α-alchilata, l’Oxymetholone è epatotossico. L’uso a lungo termine del farmaco può causare una varietà di disturbi gravi, tra cui l’epatite, il cancro al fegato e la cirrosi; pertanto si raccomandano test periodici di funzionalità epatica per coloro che assumono l’Oxymetholone a fini terapeutici. Questa molecola ha ottenuto, infatti, la nomea di essere uno tra gli AAS più epatotossici. Ciò deriva da i dosaggi comunemente, ed erroneamente, utilizzati in contesto culturistico. Si parla di dosaggi che facilmente sforano i 150mg/die.
Osservazioni e esaminazione di diversi referti di esami ematici hanno evidenziato una soglia di “vantaggio/svantaggio” a favore del primo con un dosaggio calcolato con la formula 1mg/Kg. Genericamente, però, il dosaggio standard e conservativo si attesta nel range dei 50-100mg/die per non più di 28 giorni consecutivi, al fine di ridurre l’impatto negativo sul fegato e lipidemia.

Il Methandrostenolone, noto anche come 17α-metil-δ1-testosterone o come 17α-metilandrost-1,4-dien-17β-ol-3-one, è uno steroide androstanico sintetico e un derivato 17α-alchilato del Testosterone. È una modifica del Testosterone con un gruppo metile in posizione C17α e un doppio legame aggiuntivo tra le posizioni C1 e C2. Il farmaco è anche il derivato 17α-metilato del Boldenone (δ1-testosterone) e l’analogo δ1 del Methyltestosterone (17α-metiltestosterone).
Il Methandrostenolone è stato descritto per la prima volta nel 1955. È stato sintetizzato dai ricercatori dei laboratori CIBA di Basilea, in Svizzera. La CIBA depositò un brevetto statunitense nel 1957 e iniziò a commercializzare il farmaco sotto il nome di Dianabol nel 1958 negli Stati Uniti. Inizialmente veniva prescritto alle vittime di ustioni e agli anziani. Tra i primi utilizzatori vi furono i giocatori dell’Oklahoma University e l’allenatore dei San Diego Chargers Sid Gillman, che somministrò il Dianabol alla sua squadra a partire dal 1963.
Anche se il primo a somministrare il Methandrostenolone agli atleti fu il Dr. John Ziegler, personaggio che ebbe non poca importanza nella storia dell’uso degli AAS negli Stati Uniti. Ziegler contribuì a facilitare l’adozione degli AAS in generale, e del Dianabol in particolare, da parte degli atleti americani. Ziegler fu la prima persona a somministrare il Dianabol agli atleti competitivi poco dopo la sua introduzione da parte della CIBA nel 1958. Ebbe accesso al laboratorio CIBA a Summit (New Jersey) nel corso degli anni 50’ e somministrava già ai pesisti il Testosterone Propionato per “scopi di ricerca”. Da li il passo fu breve per la diffusione a macchia d’olio di questo AAS tra i culturisti.
Data la sua principale modifica strutturale, ossia la metilazione in C-17, il Methandrostenolone mostra un aumentata stabilità del legame recettoriale aumentando così l’affinità sia al AR sia, successivamente all’aromatizzazione nel suo metabolita 17-Methylestradiolo, per i recettori estrogenici rendendo il composto molto più estrogenico del Testosterone. Tale caratteristiche migliora però il potenziale proliferativo dei AR e l’influenza positiva sulla sintesi di IGF-1. Da non dimenticare è il suo significativo impatto anticortisolemico.
Trattandosi di una molecola con una discreta tendenza all’aromatizzazione, il suo uso tipico la vede inserita nelle fasi Off-Season. Il calcolo del dosaggio, per via dati aneddotici e osservativi raccolti, lo si ottiene con la formula 5mg/12Kg di peso corporeo. Trattandosi di un composto orale metilato in C-17 se ne scoraggia l’utilizzo oltre i 28 giorni consecutivi onde ridurre l’impatto negativo su fegato e lipidemia. Data la sua emivita di circa 4h, il dosaggio giornaliero dovrebbe essere diviso in più assunzioni distribuite durante l’arco della giornata.
- hGH:

L’Ormone della Crescita (GH) o Somatotropina, noto anche come Ormone della Crescita Umano (hGH o HGH), è un ormone peptidico che stimola la crescita, la riproduzione e la rigenerazione cellulare nell’uomo e in altri animali. È quindi importante per lo sviluppo umano. Il GH stimola anche la produzione di IGF-1 e aumenta la concentrazione di glucosio e acidi grassi liberi nel sangue. È un tipo di mitogeno specifico solo per i recettori di alcuni tipi di cellule. Il GH è un polipeptide a catena singola di 191 aminoacidi che viene sintetizzato, immagazzinato e secreto dalle cellule somatotrope nelle ali laterali dell’ipofisi anteriore.
Una forma ricombinante di hGH, chiamata Somatropina, viene utilizzata come farmaco da prescrizione per il trattamento dei disturbi della crescita nei bambini e della carenza di Ormone della Crescita negli adulti. Molte delle funzioni dell’hGH rimangono sconosciute.
Nel suo ruolo di agente anabolizzante, l’hGH è stato utilizzato dagli sportivi agonisti almeno dal 1982, quando la sola forma disponibile era quella derivata dall’Ipofisi dei cadaveri, ed è stato vietato dal CIO e dall’NCAA. L’analisi tradizionale delle urine non è in grado di rilevare il doping con HGH, pertanto il divieto è stato applicato solo all’inizio degli anni 2000, quando sono stati sviluppati test del sangue in grado di distinguere tra hGH naturale e artificiale.
In ambiente bodybuilding, l’hGH viene utilizzato in Off-Season (dai soggetti meglio informati) a dosaggi nel range delle 4-8UI al giorno o 8-16UI a giorni alterni. La somministrazione in concomitanza con l’uso di Insulina ha mostrato effetti sinergici molto evidenti che trovano la loro origine nel miglioramento della sintesi di IGF-1 e della sua frazione libera quindi attiva. Ricordo inoltre che l’uso di hGH può causare una sottoregolazione della funzionalità tiroidea per via del feedback negativo causato da un aumento della conversione del T4 in T3 per azione del GH. L’uso di T4, nel periodo d’uso in Off-Season, è in alcuni casi una necessità.

Il Fattore di Crescita Insulino-Simile 1 (IGF-1), chiamato anche Somatomedina C, è un ormone dalla struttura molecolare simile a quella dell’insulina che svolge un ruolo importante nella crescita infantile e ha effetti anabolici negli adulti. L’IGF-1 è costituito da 70 aminoacidi in una singola catena con tre ponti disolfuro intramolecolari.
L’IGF-1 è prodotto principalmente dal fegato. La produzione è stimolata dall’Ormone della Crescita (GH). La maggior parte dell’IGF-1 è legata a una delle 6 proteine di legame (IGF-BP). L’IGFBP-1 è regolato dall’Insulina. L’IGF-1 viene prodotto durante tutta la vita; i tassi più alti di produzione di IGF-1 si verificano durante la crescita puberale. I livelli più bassi si verificano nell’infanzia e nella vecchiaia.
L’IGF-1 lega e attiva il proprio recettore, l’IGF-1R, attraverso l’espressione sulla superficie cellulare delle tirosin-chinasi recettoriali (RTK) e segnala ulteriormente attraverso molteplici cascate di trasduzione intracellulare. L’IGF-1R è l’induttore che svolge un ruolo critico nella modulazione degli effetti metabolici dell’IGF-1 per la senescenza e la sopravvivenza cellulare. L’IGF-1 è responsabile di stimolare la crescita di tutti i tipi di cellule e di provocare effetti metabolici significativi. Un importante effetto metabolico dell’IGF-1 è la sua capacità di segnalare alle cellule che sono disponibili nutrienti sufficienti per l’ipertrofia e la divisione cellulare. Questi segnali consentono inoltre all’IGF-1 di inibire l’apoptosi cellulare e di aumentare la produzione di proteine cellulari. I recettori dell’IGF-1 sono ubiquitari, il che consente che i cambiamenti metabolici causati dall’IGF-1 si verifichino in tutti i tipi di cellule. Gli effetti metabolici dell’IGF-1 sono di vasta portata e possono coordinare il metabolismo delle proteine, dei carboidrati e dei grassi in una varietà di tipi di cellule diverse. La regolazione degli effetti metabolici dell’IGF-1 sui tessuti bersaglio è coordinata anche con altri ormoni, come l’Ormone della Crescita e l’Insulina.
L’IGF-1 da DNA ricombinante è disponibile principalmente in due diversi formati/varianti, lr3 e DES. È importante ricordare che, a prescindere dalla variante, tutti funzionano a livello sistemico nell’organismo e che, nonostante la somministrazione dell’ormone per via intramuscolare direttamente in un muscolo specifico, non genererà una crescita localizzata misurabile.
Ovviamente tralascerò di descrivere l’IGF-1 bioidentico commercializzato come Mecasermina dal momento che la sua farmacocinetica è identica a quella del IGF-1 endogeno. Dirò soltanto che mediamente viene utilizzato in dosi giornaliere nel range tra 60-1.000mcg post-workout. L’emivita di questa forma di IGF-1 è di circa 5.8h.
IGF-1 LR3: Questa forma è la variante di IGF-1 più comune e molto popolare sul mercato e utilizzata da bodybuilder e atleti di altre discipline. Contiene IGF-1 bioidentico costituito dalla catena originale di 70 aminoacidi, ma con 13 aminoacidi in più all’estremità N, per un totale di 83 aminoacidi. Possiede anche una seconda modifica, in cui un’Arginina si trova in 3a posizione invece dell’Acido Glutammico originale. Il risultato di queste modifiche è che l’IGF-1 continua a svolgere la sua attività originaria sul recettore dell’IGF-1 nei tessuti corporei e ha un’affinità di legame molto bassa per le proteine leganti l’IGF menzionate in precedenza. Inoltre, presenta una vita attiva significativamente più lunga, di circa 20-30 ore, rispetto a quella dell’IGF-1 di 12-15 ore. L’insieme di questi fattori ha dimostrato che l’LR3 ha un’efficacia circa tre volte superiore a quella dell’IGF-1.
I dosaggi medi utilizzati per questa forma sono nel range dei 40-80mcg/die. A causa della sua lunga vita attiva nell’organismo, la variante LR3 non dovrebbe essere somministrata più di una volta al giorno per il semplice fatto che non risulta necessario. Nei giorni di allenamento, il dosaggio di IGF-1 è solitamente somministrato subito dopo l’allenamento. La scelta è a discrezione dell’utilizzatore, in quanto può essere benissimo somministrato sia prima che dopo (solo prima dell’allenamento o solo dopo l’allenamento). E’ possibile comunque dividere il dosaggio giornaliero in due somministrazioni nell’arco della giornata, il dosaggio giornaliero completo può essere diviso quindi a metà tra i due (ad esempio, 20mcg prima dell’allenamento e 20mcg dopo l’allenamento, per un totale di 40mcg al giorno). Nei giorni di non allenamento, può essere somministrato in qualsiasi momento della giornata.
IGF-1 DES: Conosciuto anche come DES(1-3)IGF-1, questa è la forma di IGF-1 comunemente conosciuta come ad azione molto rapida e di solito è la meno preferita tra le due. Le sue modifiche rispetto alla molecola originale di IGF-1 sono tali da farle mancare i primi 3 aminoacidi all’N terminale, il che conferisce all’IGF-1 DES un totale di 67 aminoacidi nella sua catena rispetto ai 70 originali. Questa modifica garantisce all’IGF-1 DES una ridotta affinità di legame per le proteine leganti l’IGF menzionate in precedenza, oltre a una maggiore forza di legame e potenziale miotrofico, circa dieci volte superiore a quella dell’IGF-1 originale e cinque volte superiore a quella dell’IGF-1 LR3. A differenza dell’IGF-1 LR3, l’IGF-1 DES ha un’emivita molto più breve, di circa 20-30 minuti. Grazie alla sua attività più rapida e alla maggiore forza/potenza, la variante DES dell’IGF-1 è comunemente ritenuta in grado di ottenere una crescita muscolare localizzata nel sito in cui viene iniettata. Sebbene ciò sia in parte vero, gli studi hanno dimostrato che, come l’IGF-1 in generale, agisce a livello sistemico una volta raggiunti i capillari e il flusso sanguigno. Quindi l’effetto localizzato è minimo e non significativamente differente dall’effetto sistemico.
Il dosaggio della variante DES è leggermente più variabile rispetto a quello del LR3. Per l’IGF-1 DES, il dosaggio varia da 50 a 150 mcg al giorno. A causa della sua emivita molto più breve rispetto alla variante LR3, è possibile utilizzare dosaggi più elevati con una ipotetica riduzione del rischio di effetti a lungo termine sull’organismo, anche se è necessario usare comunque cautela. Può essere utilizzato nello stesso modo dell’IGF-1 LR3 post-workout, ed è infatti comunemente usato in questo modo a causa della sua breve emivita.
Entrambe le forme di IGF-1 possono essere somministrate per via intramuscolare o sottocutanea. L’uso di una delle due forme non deve superare la durata di 30 giorni prima di una pausa di almeno 2 settimane, anche se fare pause più lunghe di 2 settimane tra un ciclo di IGF-1 e l’altro è l’opzione migliore. Questo non solo per ridurre il rischio di effetti sulla salute a lungo termine, ma anche per garantire che i recettori dell’IGF-1 tornino ad un grado di sensibilità ottimale e, quindi, a “rispondere” correttamente dopo un ciclo.
- Insulina (Humalog e Lantus):

L’insulina è un ormone peptidico prodotto dalle cellule beta delle isole pancreatiche. Regola il metabolismo dei carboidrati, dei grassi e delle proteine promuovendo l’assorbimento del glucosio dal sangue nelle cellule del fegato, dei grassi e dei muscoli scheletrici. In questi tessuti il glucosio assorbito viene convertito in glicogeno attraverso la glicogenesi o in grassi (trigliceridi) attraverso la lipogenesi o, nel caso del fegato, in entrambi. La produzione e la secrezione di glucosio da parte del fegato sono fortemente inibite da alte concentrazioni di Insulina nel sangue. L’Insulina circolante influisce anche sulla sintesi di proteine in un’ampia varietà di tessuti. È quindi un ormone anabolico e anticatabolico, che promuove la conversione di piccole molecole nel sangue in grandi molecole all’interno delle cellule. Bassi livelli di Insulina nel sangue hanno l’effetto opposto, favorendo un diffuso catabolismo, soprattutto del grasso corporeo di riserva.
La maggior parte dei bodybuilder utilizza una sola forma di Insulina (ad azione rapida o ultra-rapida), anche se alcuni utilizzano anche un’Insulina a lunga durata d’azione o in monoterapia insulinica o in conbinazione con le forme ad azione rapida o ultra-rapida.
L’Humalog® (Insulina Lispro) è senza dubbio la forma di Insulina più diffusa tra i bodybuilder insieme all’Humulin-R. L’Humalog è un analogo a breve durata d’azione dell’Insulina umana, in particolare l’analogo Lys(B28) Pro(B29) dell’Insulina che si crea quando gli aminoacidi in posizione 28 e 29 sono invertiti. È considerata equipotente all’Insulina solubile normale su base unitaria, ma con un’attività più rapida. L’inizio dell’azione del farmaco in seguito alla somministrazione sottocutanea è di circa 10-15 minuti e il suo picco d’effetto viene raggiunto in 30-90 minuti.
La durata d’azione totale è compresa tra 3-5 ore. L’Insulina lispro viene solitamente utilizzata come supplemento a un prodotto a base di Insulina a più lunga durata d’azione, fornendo un farmaco ad azione rapida che può essere assunto prima o subito dopo i pasti per imitare la secrezione insulinica naturale dell’organismo. Molti atleti ritengono che la sua breve finestra d’effetto la renda un farmaco insulinico ideale per
scopi dopanti, in quanto la maggior parte dell’azione può essere concentrata nel periodo successivo all’allenamento sfruttando l’assimilazione dei nutrienti durante la così detta “finestra anabolica”. Proprio al fine di potenziare la “finestra anabolica”, l’Humalog viene usata in concomitanza del GH il quale viene somministrato in una tempistica tale che i due picchi di rilascio (curva ematica massima) si “incrocino” andando a creare un affetto additivo di potenziamento della sintesi epatica di IGF-1 e della sua attività per via della riduzione dei trasportatori IGFBP.
Tuttavia, l’uso di una base insulinica composta da Insuline Glargine (Lantus), con una vita attiva di 24-26.5h, la quale sembra avere effetti di maggiore affinità di legame per il recettore del IGF-1 rispetto all’Insulina umana regolare o uno dei qualsiasi altri analoghi, viene da alcuni inserita nei protocolli Off-Season.
I dosaggi di Insulina non andrebbero calcolati in modo distaccato dal piano alimentare e dal suo contenuto glucidico. Se il margine di “sicurezza” indica un assunzione di 10-15g di Carboidrati per UI di Insulina, questi non dovrebbero essere addizionati al piano alimentare già tarata in surplus calorico. Il calcolo delle unità dovrebbe essere tarato sul quantitativo glucidico della dieta e sul rapporto con il peso corporeo dell’atleta. Facciamo un semplice esempio: Soggetto di 90Kg = formula 1UI ogni 10Kg di peso = 9UI massime somministrabili per pasto e in base alla vita attiva della forma utilizzata = assicurarsi che il pasto appena successivo alla somministrazione dell’Insulina a questo dosaggio sia pari o superiore ai 90g di Carboidrati.
Il monitoraggio della glicemia attraverso un glucometro è ovviamente d’obbligo in un protocollo di Insulina.
Nota: tali informazioni esposte non rappresentano in nessun modo un parere medico ne tanto meno una prescrizione e/o incentivo all’uso di sostanze dopanti e illegali. Le descrizioni presentate per i PEDs solitamente più utilizzati in Off-Season sono sintetiche sia per motivi di “Off Topic” sia per ragioni legate alla loro descrizione approfondita in altri articoli presenti nel database di questo sito. In queste pubblicazioni potrete trovare informazioni inerenti anche agli affetti collaterali connessi ad un uso/abuso “off-label” dei diversi PEDs.
Conclusioni:
Per concludere e fare una sintesi delle nozioni esposte in questo articolo, dobbiamo ricordarci che i bodybuilder in Off-Season dovrebbero concentrarsi sul consumo di una dieta leggermente ipercalorica (~10-20% sopra le calorie di mantenimento) con l’obiettivo di guadagnare ~0,25-0,5% del peso corporeo a settimana per un “Natural”, mentre nel caso di un “Doped” la soglia può spostarsi tra l’1-2% con variabili connesse a risposte genetiche differenziali e anzianità nella carriera culturistica (principiante, intermedio e avanzato). In ogni caso, in una fetta maggioritaria di praticanti, ai bodybuilder avanzati si consiglia di essere più prudenti con il surplus calorico e il tasso di aumento di peso settimanale. L’assunzione di proteine nella dieta è raccomandata a 1,6-2,2 g/kg/giorno, con particolare attenzione a una quantità sufficiente di proteine a ogni pasto (0,40-0,55 g/kg/pasto) e a una distribuzione uniforme nell’arco della giornata (3-6 pasti). Per i “Doped”, in alcuni casi, l’introito proteico può essere portato, con minimi vantaggi in contesto ipercalorico, a 2,5g/Kg con le medesime linee guida di suddivisione per numero di pasti. I grassi alimentari devono essere consumati a livelli moderati, né troppo bassi né troppo alti (0,5-1,5 g/kg/die), per evitare un rapporto fTC sfavorevole e per prevenire riduzioni dei livelli di testosterone. Nei “Doped” l’obbiettivo con i lipidi è principalmente quello di assumerne una dose necessaria, e altamente qualitativa, al fine di assimilare vitamine liposolubili, per substrato strutturale, per sintesi di eicosanoidi (vedi assunzione EPA, DHA e AA), protezioni epidermide e capelli; di conseguenza attenersi ad un dosaggio medio pari a 35-50g/die. Dopo che le calorie sono state distribuite tra Proteine e Grassi, le restanti calorie dovrebbero provenire dai Carboidrati, assicurandosi di consumarne una quantità sufficiente (≥3-5 g/kg/giorno). Si possono ottenere benefici maggiori consumando proteine (0,40-0,55 g/kg/pasto) in prossimità delle sessioni di allenamento (1-2 ore prima dell’esercizio ed entro 1-2 ore dopo l’esercizio). È opportuno prendere in considerazione la Creatina Monoidrato (3-5 g/giorno) e la Caffeina (5-6 mg/kg), in quanto possono produrre effetti ergogenici per i bodybuilder. Inoltre, Beta-Alanina (3-5 g/die) e Citrullina Malato (8 g/die) sono integratori alimentari che possono essere presi in considerazione in quanto potenzialmente utili per i bodybuilder, a seconda dei regimi di allenamento individuali. I bodybuilder che non sono in grado di assumere un apporto sufficiente di micronutrienti e acidi grassi essenziali nella loro dieta dovrebbero prendere in considerazione l’integrazione di questi nutrienti per evitare carenze. Il limite principale di questo articolo è la mancanza di studi su larga scala e a lungo termine sui bodybuilder durante la Off-Season. Sono necessarie ulteriori ricerche su questa popolazione per ottimizzare la nutrizione e le raccomandazioni sugli integratori alimentari.
Gabriel Bellizzi
Riferimenti:
1. Helms E.R., Aragon A.A., Fitschen P.J. Evidence-based recommendations for natural bodybuilding contest preparation: Nutrition and supplementation. J. Int. Soc. Sports Nutr. 2014;11:20. doi: 10.1186/1550-2783-11-20. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
2. Spendlove J., Mitchell L., Gifford J., Hackett D., Slater G., Cobley S., O’Connor H. Dietary Intake of Competitive Bodybuilders. Sports Med. 2015;45:1041–1063. doi: 10.1007/s40279-015-0329-4. [PubMed] [CrossRef] [Google Scholar]
3. Cho S., Lee H., Kim K. Physical Characteristics and Dietary Patterns of Strength Athletes; Bodybuilders, Weight Lifters. [(accessed on 25 March 2019)];Korean J. Community Nutr. 2007 12:864–872. Available online: https://www.komci.org/GSResult.php?RID=0106KJCN%2F2007.12.6.864&DT=6 [Google Scholar]
4. Philen R.M., Ortiz D.I., Auerbach S.B., Falk H. Survey of Advertising for Nutritional Supplements in Health and Bodybuilding Magazines. JAMA. 1992;268:1008. doi: 10.1001/jama.1992.03490080082029. [PubMed] [CrossRef] [Google Scholar]
5. Giampreti A., Lonati D., Locatelli C., Rocchi L., Campailla M.T. Acute neurotoxicity after yohimbine ingestion by a bodybuilder. [(accessed on 25 March 2019)];Clin. Toxicol. 2009 47:827–829. doi: 10.1080/15563650903081601. Available online: https://www.ncbi.nlm.nih.gov/pubmed/19640235 [PubMed] [CrossRef] [Google Scholar]
6. Grunewald K.K., Bailey R.S. Commercially Marketed Supplements for Bodybuilding Athletes. Sports Med. 1993;15:90–103. doi: 10.2165/00007256-199315020-00003. [PubMed] [CrossRef] [Google Scholar]
7. Della Guardia L., Cavallaro M., Cena H. The risks of self-made diets: The case of an amateur bodybuilder. J. Int. Soc. Sports Nutr. 2015;12:5. doi: 10.1186/s12970-015-0077-8. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
8. Mitchell L., Hackett D., Gifford J., Estermann F., O’Connor H. Do Bodybuilders Use Evidence-Based Nutrition Strategies to Manipulate Physique? [(accessed on 25 March 2019)];Sports. 2017 5:76. doi: 10.3390/sports5040076. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5969027/ [PMC free article] [PubMed] [CrossRef] [Google Scholar]
9. Hackett D.A., Johnson N.A., Chow C.-M. Training Practices and Ergogenic Aids Used by Male Bodybuilders. J. Strength Cond. Res. 2013;27:1609–1617. doi: 10.1519/JSC.0b013e318271272a. [PubMed] [CrossRef] [Google Scholar]
10. Forbes G.B., Brown M.R., Welle S.L., Lipinski B.A. Deliberate overfeeding in women and men: Energy cost and composition of the weight gain. Br. J. Nutr. 1986;56:1–9. doi: 10.1079/BJN19860080. [PubMed] [CrossRef] [Google Scholar]
11. Kreider R.B., Klesges R., Harmon K., Ramsey L., Bullen D., Wood L., Almada A., Grindstaff P., Li Y. Effects of Ingesting Supplements Designed to Promote Lean Tissue Accretion on Body Composition during Resistance Training. Int. J. Sport Nutr. 1996;6:234–246. doi: 10.1123/ijsn.6.3.234. [PubMed] [CrossRef] [Google Scholar]
12. Rozenek R., Ward P., Long S., Garhammer J. Effects of high-calorie supplements on body composition and muscular strength following resistance training. J. Sports Med. Phys. Fit. 2002;42:340–347. [PubMed] [Google Scholar]
13. Garthe I., Raastad T., Refsnes P.E., Sundgot-Borgen J. Effect of nutritional intervention on body composition and performance in elite athletes. Eur. J. Sport Sci. 2013;13:295–303. doi: 10.1080/17461391.2011.643923. [PubMed] [CrossRef] [Google Scholar]
14. American College og Sports Medicine American College of Sports Medicine position stand. Progression models in resistance training for healthy adults. [(accessed on 25 March 2019)];Med. Sci. Sport. Exerc. 2009 41:687–708. doi: 10.1249/MSS.0b013e3181915670. Available online: https://www.ncbi.nlm.nih.gov/pubmed/19204579 [PubMed] [CrossRef] [Google Scholar]
15. Lambert C.P., Frank L.L., Evans W.J., Lambert D.C.P. Macronutrient Considerations for the Sport of Bodybuilding. Sports Med. 2004;34:317–327. doi: 10.2165/00007256-200434050-00004. [PubMed] [CrossRef] [Google Scholar]
16. Walberg-Rankin J., Edmonds C.E., Gwazdauskas F.C. Diet and Weight Changes of Female Bodybuilders Before and After Competition. Int. J. Sport Nutr. 1993;3:87–102. doi: 10.1123/ijsn.3.1.87. [PubMed] [CrossRef] [Google Scholar]
17. Lamar-Hildebrand N., Saldanha L., Endres J. Dietary and exercise practices of college-aged female bodybuilders. J. Am. Diet. Assoc. 1989;89:1308–1310. [PubMed] [Google Scholar]
18. Houston M.E. Gaining Weight: The Scientific Basis of Increasing Skeletal Muscle Mass. Can. J. Appl. Physiol. 1999;24:305–316. doi: 10.1139/h99-024. [PubMed] [CrossRef] [Google Scholar]
19. Phillips S.M. A Brief Review of Critical Processes in Exercise-Induced Muscular Hypertrophy. Sports Med. 2014;44:71–77. doi: 10.1007/s40279-014-0152-3. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
20. Campbell B.I., Aguilar D., Conlin L., Vargas A., Schoenfeld B.J., Corson A., Gai C., Best S., Galvan E., Couvillion K. Effects of High Versus Low Protein Intake on Body Composition and Maximal Strength in Aspiring Female Physique Athletes Engaging in an 8-Week Resistance Training Program. Int. J. Sport Nutr. Exerc. Metab. 2018;28:580–585. doi: 10.1123/ijsnem.2017-0389. [PubMed] [CrossRef] [Google Scholar]
21. Morton R.W., McGlory C., Phillips S.M. Nutritional interventions to augment resistance training-induced skeletal muscle hypertrophy. Front. Physiol. 2015;6:1–9. doi: 10.3389/fphys.2015.00245. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
22. Morton R.W., Murphy K.T., McKellar S.E., Schoenfeld B.J., Henselmans M., Helms E., Aragon A.A., Devries M.C., Banfield L., Krieger J.W., et al. A systematic review, meta-analysis and meta-regression of the effect of protein supplementation on resistance training-induced gains in muscle mass and strength in healthy adults. [(accessed on 25 March 2019)];Br. J. Sports Med. 2018 52:376–384. doi: 10.1136/bjsports-2017-097608. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28698222 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
23. Houltham S.D., Rowlands D.S. A snapshot of nitrogen balance in endurance-trained women. Appl. Physiol. Nutr. Metab. 2014;39:219–225. doi: 10.1139/apnm-2013-0182. [PubMed] [CrossRef] [Google Scholar]
24. Antonio J., Ellerbroek A. Case Reports on Well-Trained Bodybuilders: Two Years on a High Protein Diet. [(accessed on 25 March 2019)];JEPonline. 2018 21:14–24. Available online: https://www.asep.org/asep/asep/JEPonlineFEBRUARY2018_Antonio.pdf [Google Scholar]
25. Antonio J., Ellerbroek A., Silver T., Vargas L., Peacock C. The effects of a high protein diet on indices of health and body composition—A crossover trial in resistance-trained men. J. Int. Soc. Sports Nutr. 2016;13:8. doi: 10.1186/s12970-016-0114-2. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
26. Bandegan A., Courtney-Martin G., Rafii M., Pencharz P.B., Lemon P.W. Indicator Amino Acid–Derived Estimate of Dietary Protein Requirement for Male Bodybuilders on a Nontraining Day Is Several-Fold Greater than the Current Recommended Dietary Allowance. J. Nutr. 2017;147:850–857. doi: 10.3945/jn.116.236331. [PubMed] [CrossRef] [Google Scholar]
27. Malowany J.M., West D.W.D., Williamson E., Volterman K.A., Sawan S.A., Mazzulla M., Moore D.R. Protein to Maximize Whole-Body Anabolism in Resistance-trained Females after Exercise. Med. Sci. Sports Exerc. 2019;51:798–804. doi: 10.1249/MSS.0000000000001832. [PubMed] [CrossRef] [Google Scholar]
28. Antonio J., Peacock C.A., Ellerbroek A., Fromhoff B., Silver T. The effects of consuming a high protein diet (4.4 g/kg/d) on body composition in resistance-trained individuals. J. Int. Soc. Sports Nutr. 2014;11:19. doi: 10.1186/1550-2783-11-19. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
29. Antonio J., Ellerbroek A., Silver T., Orris S., Scheiner M., Gonzalez A., Peacock C.A. A high protein diet (3.4 g/kg/d) combined with a heavy resistance training program improves body composition in healthy trained men and women—A follow-up investigation. J. Int. Soc. Sports Nutr. 2015;12:39. doi: 10.1186/s12970-015-0100-0. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
30. Bray G.A., Smith S.R., de Jonge L., Xie H., Rood J., Martin C.K., Most M., Brock C., Mancuso S., Redman L.M. Effect of dietary protein content on weight gain, energy expenditure, and body composition during overeating: A randomized controlled trial. [(accessed on 25 March 2019)];JAMA. 2012 307:47–55. doi: 10.1001/jama.2011.1918. Available online: https://www.ncbi.nlm.nih.gov/pubmed/22215165 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
31. Tipton K.D., Ferrando A.A., Phillips S.M., Doyle D., Wolfe R.R. Postexercise net protein synthesis in human muscle from orally administered amino acids. Am. J. Physiol. Metab. 1999;276:628–634. doi: 10.1152/ajpendo.1999.276.4.E628. [PubMed] [CrossRef] [Google Scholar]
32. Rieu I., Balage M., Sornet C., Giraudet C., Pujos E., Grizard J., Mosoni L., Dardevet D. Leucine supplementation improves muscle protein synthesis in elderly men independently of hyperaminoacidaemia. J. Physiol. 2006;575:305–315. doi: 10.1113/jphysiol.2006.110742. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
33. Burd N.A., Tang J.E., Moore D.R., Phillips S.M. Exercise training and protein metabolism: Influences of contraction, protein intake, and sex-based differences. [(accessed on 25 March 2019)];J. Appl. Physiol. 2008 106:1692–1701. doi: 10.1152/japplphysiol.91351.2008. Available online: https://www.ncbi.nlm.nih.gov/pubmed/19036897 [PubMed] [CrossRef] [Google Scholar]
34. Drummond M.J., Dreyer H.C., Fry C.S., Glynn E.L., Rasmussen B.B. Nutritional and contractile regulation of human skeletal muscle protein synthesis and mTORC1 signaling. J. Appl. Physiol. 2009;106:1374–1384. doi: 10.1152/japplphysiol.91397.2008. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
35. Tang J.E., Moore D.R., Kujbida G.W., Tarnopolsky M.A., Phillips S.M. Ingestion of whey hydrolysate, casein, or soy protein isolate: Effects on mixed muscle protein synthesis at rest and following resistance exercise in young men. J. Appl. Physiol. 2009;107:987–992. doi: 10.1152/japplphysiol.00076.2009. [PubMed] [CrossRef] [Google Scholar]
36. Kanda A., Nakayama K., Sanbongi C., Nagata M., Ikegami S., Itoh H. Effects of Whey, Caseinate, or Milk Protein Ingestion on Muscle Protein Synthesis after Exercise. Nutrients. 2016;8:339. doi: 10.3390/nu8060339. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
37. Messina M., Lynch H., Dickinson J.M., Reed K.E. No Difference Between the Effects of Supplementing With Soy Protein Versus Animal Protein on Gains in Muscle Mass and Strength in Response to Resistance Exercise. Int. J. Sport Nutr. Exerc. Metab. 2018;28:674–685. doi: 10.1123/ijsnem.2018-0071. [PubMed] [CrossRef] [Google Scholar]
38. Joy J.M., Lowery R.P., Wilson J.M., Purpura M., De Souza E.O., Mc Wilson S., Kalman D.S., Dudeck J.E., Jäger R. The effects of 8 weeks of whey or rice protein supplementation on body composition and exercise performance. Nutr. J. 2013;12:86. doi: 10.1186/1475-2891-12-86. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
39. Babault N., Paizis C., Deley G., Guérin-Deremaux L., Saniez M.-H., Lefranc-Millot C., Allaert F.A. Pea proteins oral supplementation promotes muscle thickness gains during resistance training: A double-blind, randomized, Placebo-controlled clinical trial vs. Whey protein. J. Int. Soc. Sports Nutr. 2015;12:1692. doi: 10.1186/s12970-014-0064-5. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
40. Tesch P.A. Glycogen and triglyceride utilization in relation to muscle metabolic characteristics in men performing heavy-resistance exercise. Graefe’s Arch. Clin. Exp. Ophthalmol. 1990;61:5–10. [PubMed] [Google Scholar]
41. Lane A.R., Duke J.W., Hackney A.C. Influence of dietary carbohydrate intake on the free testosterone: Cortisol ratio responses to short-term intensive exercise training. [(accessed on 25 March 2019)];Eur. J. Appl. Physiol. 2010 108:1125–1131. doi: 10.1007/s00421-009-1220-5. Available online: https://www.ncbi.nlm.nih.gov/pubmed/20091182 [PubMed] [CrossRef] [Google Scholar]
42. Tegelman R., Aberg T., Pousette A., Carlström K. Effects of a diet regimen on pituitary and steroid hormones in male ice hockey players. [(accessed on 25 March 2019)];Int. J. Sports Med. 1992 13:420–430. doi: 10.1055/s-2007-1021292. Available online: https://www.ncbi.nlm.nih.gov/pubmed/1387870 [PubMed] [CrossRef] [Google Scholar]
43. Dorgan J.F., Judd J.T., Longcope C., Brown C., Schatzkin A., Clevidence B.A., Campbell W.S., Nair P.P., Franz C., Kahle L., et al. Effects of dietary fat and fiber on plasma and urine androgens and estrogens in men: A controlled feeding study. Am. J. Clin. Nutr. 1996;64:850–855. doi: 10.1093/ajcn/64.6.850. [PubMed] [CrossRef] [Google Scholar]
44. Hämäläinen E., Adlercreutz H., Puska P., Pietinen P. Decrease of serum total and free testosterone during a low-fat high-fibre diet. J. Steroid Biochem. 1983;18:369–370. doi: 10.1016/0022-4731(83)90117-6. [PubMed] [CrossRef] [Google Scholar]
45. Hämäläinen E., Adlercreutz H., Puska P., Pietinen P. Diet and serum sex hormones in healthy men. J. Steroid Biochem. 1984;20:459–464. doi: 10.1016/0022-4731(84)90254-1. [PubMed] [CrossRef] [Google Scholar]
46. Wang C., Catlin D.H., Starcevic B., Heber D., Ambler C., Berman N., Lucas G., Leung A., Schramm K., Lee P.W.N., et al. Low-Fat High-Fiber Diet Decreased Serum and Urine Androgens in Men. J. Clin. Endocrinol. Metab. 2005;90:3550–3559. doi: 10.1210/jc.2004-1530. [PubMed] [CrossRef] [Google Scholar]
47. Morton R.W., Sato K., Gallaugher M.P.B., Oikawa S.Y., McNicholas P.D., Fujita S., Phillips S.M. Muscle Androgen Receptor Content but Not Systemic Hormones Is Associated With Resistance Training-Induced Skeletal Muscle Hypertrophy in Healthy, Young Men. Front. Physiol. 2018;9:9. doi: 10.3389/fphys.2018.01373. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
48. Tinsley G.M., Willoughby D.S. Fat-Free Mass Changes During Ketogenic Diets and the Potential Role of Resistance Training. Int. J. Sport Nutr. Exerc. Metab. 2016;26:78–92. doi: 10.1123/ijsnem.2015-0070. [PubMed] [CrossRef] [Google Scholar]
49. Vargas S., Romance R., Petro J.L., Bonilla D.A., Galancho I., Espinar S., Kreider R.B., Benítez-Porres J. Efficacy of ketogenic diet on body composition during resistance training in trained men: A randomized controlled trial. J. Int. Soc. Sports Nutr. 2018;15:31. doi: 10.1186/s12970-018-0236-9. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
50. Kephart W.C., Pledge C.D., Roberson P.A., Mumford P.W., Romero M.A., Mobley C.B., Martin J.S., Young K.C., Lowery R.P., Wilson J.M., et al. The Three-Month Effects of a Ketogenic Diet on Body Composition, Blood Parameters, and Performance Metrics in CrossFit Trainees: A Pilot Study. Sports. 2018;6:1. doi: 10.3390/sports6010001. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
51. Greene D.A., Varley B.J., Hartwig T.B., Chapman P., Rigney M. A Low-Carbohydrate Ketogenic Diet Reduces Body Mass Without Compromising Performance in Powerlifting and Olympic Weightlifting Athletes. [(accessed on 26 March 2019)];J. Strength Cond. Res. 2018 32:3373–3382. Available online: https://www.ncbi.nlm.nih.gov/pubmed/30335720 [PubMed] [Google Scholar]
52. Bird S. Strength Nutrition: Maximizing Your Anabolic Potential. Strength Cond. J. 2010;32:80–86. doi: 10.1519/SSC.0b013e3181d5284e. [CrossRef] [Google Scholar]
53. American Dietetic Association. Dietitians of Canada. American College of Sports Medicine. Rodriguez N.R., Di Marco N.M., Langley S. American College of Sports Medicine position stand. Nutrition and athletic performance. [(accessed on 26 March 2019)];Med. Sci. Sports Exerc. 2009 41:709–731. Available online: https://www.ncbi.nlm.nih.gov/pubmed/19225360 [PubMed] [Google Scholar]
54. Chung S.T., Chacko S.K., Sunehag A.L., Haymond M.W. Measurements of Gluconeogenesis and Glycogenolysis: A Methodological Review. Diabetes. 2015;64:3996–4010. doi: 10.2337/db15-0640. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
55. Azizi F. Effect of dietary composition on fasting-induced changes in serum thyroid hormones and thyrotropin. Metabolism. 1978;27:935–942. doi: 10.1016/0026-0495(78)90137-3. [PubMed] [CrossRef] [Google Scholar]
56. Mathieson R.A., Walberg J.L., Gwazdauskas F.C., Hinkle D.E., Gregg J.M. The effect of varying carbohydrate content of a very-low-caloric diet on resting metabolic rate and thyroid hormones. Metabolism. 1986;35:394–398. doi: 10.1016/0026-0495(86)90126-5. [PubMed] [CrossRef] [Google Scholar]
57. Leveritt M., Abernethy P.J. Effects of Carbohydrate Restriction on Strength Performance. J. Strength Cond. Res. 1999;13:52–57. [Google Scholar]
58. Jacobs I., Kaiser P., Tesch P. Muscle strength and fatigue after selective glycogen depletion in human skeletal muscle fibers. Graefe’s Arch. Clin. Exp. Ophthalmol. 1981;46:47–53. doi: 10.1007/BF00422176. [PubMed] [CrossRef] [Google Scholar]
59. Ray S., Sale D.G., Lee P., Garner S., MacDougall J.D., McCartney N. Muscle Substrate Utilization and Lactate Production During Weightlifting. Can. J. Appl. Physiol. 1999;24:209–215. [PubMed] [Google Scholar]
60. Tesch P.A., Colliander E.B., Kaiser P. Muscle metabolism during intense, heavy-resistance exercise. Graefe’s Arch. Clin. Exp. Ophthalmol. 1986;55:362–366. doi: 10.1007/BF00422734. [PubMed] [CrossRef] [Google Scholar]
61. Pascoe D.D., Costill D.L., Fink W.J., Robergs R.A., Zachwieja J.J. Glycogen resynthesis in skeletal muscle following resistive exercise. Med. Sci. Sports Exerc. 1993;25:349. doi: 10.1249/00005768-199303000-00009. [PubMed] [CrossRef] [Google Scholar]
62. Ørtenblad N., Westerblad H., Nielsen J. Muscle glycogen stores and fatigue. [(accessed on 26 March 2019)];J. Physiol. 2013 591:4405–4413. doi: 10.1113/jphysiol.2013.251629. Available online: https://www.ncbi.nlm.nih.gov/pubmed/23652590 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
63. Mitchell J.B., DiLauro P.C., Pizza F.X., Cavender D.L. The Effect of Preexercise Carbohydrate Status on Resistance Exercise Performance. Int. J. Sport Nutr. 1997;7:185–196. doi: 10.1123/ijsn.7.3.185. [PubMed] [CrossRef] [Google Scholar]
64. Lima-Silva A.E., Silva-Cavalcante M.D., Oliveira R.S., Kiss M.A., Pires F.O., Bertuzzi R., Bishop D. Effects of a low- or a high-carbohydrate diet on performance, energy system contribution, and metabolic responses during supramaximal exercise. Appl. Physiol. Nutr. Metab. 2013;38:928–934. doi: 10.1139/apnm-2012-0467. [PubMed] [CrossRef] [Google Scholar]
65. Vega F., Jackson R. Dietary habits of bodybuilders and other regular exercisers. Nutr. Res. 1996;16:3–10. doi: 10.1016/0271-5317(95)02054-3. [CrossRef] [Google Scholar]
66. Chappell A.J., Simper T., Barker M.E. Nutritional strategies of high level natural bodybuilders during competition preparation. J. Int. Soc. Sports Nutr. 2018;15:4. doi: 10.1186/s12970-018-0209-z. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
67. Atherton P.J., Etheridge T., Watt P.W., Wilkinson D., Selby A., Rankin D., Smith K., Rennie M.J. Muscle full effect after oral protein: Time-dependent concordance and discordance between human muscle protein synthesis and mTORC1 signaling. Am. J. Clin. Nutr. 2010;92:1080–1088. doi: 10.3945/ajcn.2010.29819. [PubMed] [CrossRef] [Google Scholar]
68. Res P.T., Groen B., Pennings B., Beelen M., Wallis G.A., Gijsen A.P., Senden J.M., Van Loon L.J. Protein ingestion before sleep improves postexercise overnight recovery. [(accessed on 25 March 2019)];Med. Sci. Sports Exerc. 2012 44:1560–1569. doi: 10.1249/MSS.0b013e31824cc363. Available online: https://www.ncbi.nlm.nih.gov/pubmed/22330017 [PubMed] [CrossRef] [Google Scholar]
69. Moore D.R., Robinson M.J., Fry J.L., Tang J.E., Glover E.I., Wilkinson S.B., Prior T., Tarnopolsky M.A., Phillips S.M. Ingested protein dose response of muscle and albumin protein synthesis after resistance exercise in young men. [(accessed on 25 March 2019)];Am. J. Clin. Nutr. 2009 89:161–168. doi: 10.3945/ajcn.2008.26401. Available online: https://www.ncbi.nlm.nih.gov/pubmed/19056590 [PubMed] [CrossRef] [Google Scholar]
70. Witard O.C., Jackman S.R., Breen L., Smith K., Selby A., Tipton K.D. Muscle protein synthesis rates subsequent to a meal in response to increasing doses of whey protein at rest and after. [(accessed on 25 March 2019)];Am. J. Clin. Nutr. 2014 99:86–95. doi: 10.3945/ajcn.112.055517. Available online: https://www.ncbi.nlm.nih.gov/pubmed/24257722 [PubMed] [CrossRef] [Google Scholar]
71. Macnaughton L.S., Wardle S.L., Witard O.C., McGlory C., Hamilton D.L., Jeromson S., Lawrence C.E., Wallis G.A., Tipton K.D. The response of muscle protein synthesis following whole-body resistance exercise is greater following 40 g than 20 g of ingested whey protein. Physiol. Rep. 2016;4:e12893. doi: 10.14814/phy2.12893. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
72. Schoenfeld B.J., Aragon A.A., Krieger J.W. The effect of protein timing on muscle strength and hypertrophy: A meta-analysis. J. Int. Soc. Sports Nutr. 2013;10:53. doi: 10.1186/1550-2783-10-53. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
73. Areta J.L., Burke L.M., Ross M.L., Camera D.M., West D.W.D., Broad E.M., Jeacocke N.A., Moore D.R., Stellingwerff T., Phillips S.M., et al. Timing and distribution of protein ingestion during prolonged recovery from resistance exercise alters myofibrillar protein synthesis. J. Physiol. 2013;591:2319–2331. doi: 10.1113/jphysiol.2012.244897. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
74. Hudson J.L., Bergia R.E., Campbell W.W. Effects of protein supplements consumed with meals, versus between meals, on resistance training–induced body composition changes in adults: A systematic review. Nutr. Rev. 2018;76:461–468. doi: 10.1093/nutrit/nuy012. [PubMed] [CrossRef] [Google Scholar]
75. Trommelen J., Kouw I.W.K., Holwerda A.M., Snijders T., Halson S.L., Rollo I., Verdijk L.B., Van Loon L.J.C. Pre-sleep dietary protein-derived amino acids are incorporated in myofibrillar protein during post-exercise overnight recovery. [(accessed on 25 March 2019)];Am. J. Physiol. Metab. 2018 1:457–467. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28536184 [Google Scholar]
76. Kouw I.W., Holwerda A.M., Trommelen J., Kramer I.F., Bastiaanse J., Halson S.L., Wodzig W.K., Verdijk L.B., Van Loon L.J. Protein Ingestion before Sleep Increases Overnight Muscle Protein Synthesis Rates in Healthy Older Men: A Randomized Controlled Trial. [(accessed on 25 March 2019)];J. Nutr. 2017 147:2252–2261. doi: 10.3945/jn.117.254532. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28855419 [PubMed] [CrossRef] [Google Scholar]
77. Snijders T., Res P.T., Smeets J.S., Van Vliet S., Van Kranenburg J., Maase K., Kies A.K., Verdijk L.B., Van Loon L.J. Protein ingestion before sleep increases muscle mass and strength gains during prolonged resistance-type exercise training in healthy young men. [(accessed on 25 March 2019)];J. Nutr. 2015 145:1178–1184. doi: 10.3945/jn.114.208371. Available online: https://www.ncbi.nlm.nih.gov/pubmed/25926415 [PubMed] [CrossRef] [Google Scholar]
78. Joy J.M., Vogel R.M., Broughton K.S., Kudla U., Kerr N.Y., Davison J.M., Wildman R.E.C., DiMarco N.M. Daytime and nighttime casein supplements similarly increase muscle size and strength in response to resistance training earlier in the day: A preliminary investigation. J. Int. Soc. Sports Nutr. 2018;15:24. doi: 10.1186/s12970-018-0228-9. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
79. Antonio J., Ellerbroek A., Peacock C., Silver T. Casein Protein Supplementation in Trained Men and Women: Morning versus Evening. Int. J. Exerc. Sci. 2017;10:479–486. [PMC free article] [PubMed] [Google Scholar]
80. Schoenfeld B.J., Aragon A.A. How much protein can the body use in a single meal for muscle-building? Implications for daily protein distribution. J. Int. Soc. Sports Nutr. 2018;15:10. doi: 10.1186/s12970-018-0215-1. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
81. Pennings B., Groen B.B., Van Dijk J.-W., De Lange A., Kiskini A., Kuklinski M., Senden J.M., Van Loon L.J. Minced beef is more rapidly digested and absorbed than beef steak, resulting in greater postprandial protein retention in older men. Am. J. Clin. Nutr. 2013;98:121–128. doi: 10.3945/ajcn.112.051201. [PubMed] [CrossRef] [Google Scholar]
82. Kim I.Y., Schutzler S., Schrader A., Spencer H.J., Azhar G., Ferrando A.A., Wolfe R.R. The anabolic response to a meal containing different amounts of protein is not limited by the maximal stimulation of protein synthesis in healthy young adults. [(accessed on 25 March 2019)];Am. J. Physiol. Metab. 2016 310:73–80. doi: 10.1152/ajpendo.00365.2015. Available online: https://www.ncbi.nlm.nih.gov/pubmed/26530155 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
83. Jentjens R., Jeukendrup A.E. Determinants of Post-Exercise Glycogen Synthesis During Short-Term Recovery. Sports Med. 2003;33:117–144. doi: 10.2165/00007256-200333020-00004. [PubMed] [CrossRef] [Google Scholar]
84. Biolo G., Williams B.D., Fleming R.Y., Wolfe R.R. Insulin action on muscle protein kinetics and amino acid transport during recovery after resistance exercise. Diabetes. 1999;48:949–957. doi: 10.2337/diabetes.48.5.949. [PubMed] [CrossRef] [Google Scholar]
85. Greenhaff P.L., Karagounis L.G., Peirce N., Simpson E.J., Hazell M., Layfield R., Wackerhage H., Smith K., Atherton P., Selby A., et al. Disassociation between the effects of amino acids and insulin on signaling, ubiquitin ligases, and protein turnover in human muscle. Am. J. Physiol. Metab. 2008;295:E595–E604. doi: 10.1152/ajpendo.90411.2008. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
86. Glynn E.L., Fry C.S., Timmerman K.L., Drummond M.J., Volpi E., Rasmussen B.B., Leroy J.L., Gadsden P., De Cossío T.G., Gertler P. Addition of Carbohydrate or Alanine to an Essential Amino Acid Mixture Does Not Enhance Human Skeletal Muscle Protein Anabolism123. J. Nutr. 2013;143:307–314. doi: 10.3945/jn.112.168203. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
87. Koopman R., Beelen M., Stellingwerff T., Pennings B., Saris W.H.M., Kies A.K., Kuipers H., Van Loon L.J.C. Coingestion of carbohydrate with protein does not further augment postexercise muscle protein synthesis. Am. J. Physiol. Metab. 2007;293:E833–E842. doi: 10.1152/ajpendo.00135.2007. [PubMed] [CrossRef] [Google Scholar]
88. Aragon A.A., Schoenfeld B.J. Nutrient timing revisited: Is there a post-exercise anabolic window? J. Int. Soc. Sports Nutr. 2013;10:5. doi: 10.1186/1550-2783-10-5. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
89. Jäger R., Kerksick C.M., Campbell B.I., Cribb P.J., Wells S.D., Skwiat T.M., Purpura M., Ziegenfuss T.N., Ferrando A.A., Arent S.M., et al. International Society of Sports Nutrition position stand: Protein and exercise. [(accessed on 25 March 2019)];J. Int. Soc. Sport. Nutr. 2017 4:20. doi: 10.1186/s12970-017-0177-8. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28642676 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
90. Darrabie M.D., Arciniegas A.J.L., Mishra R., Bowles D.E., Jacobs D.O., Santacruz L. AMPK and substrate availability regulate creatine transport in cultured cardiomyocytes. Am. J. Physiol. Metab. 2011;300:870–876. doi: 10.1152/ajpendo.00554.2010. [PubMed] [CrossRef] [Google Scholar]
91. Purchas R., Busboom J., Wilkinson B. Changes in the forms of iron and in concentrations of taurine, carnosine, coenzyme Q10, and creatine in beef longissimus muscle with cooking and simulated stomach and duodenal digestion. Meat Sci. 2006;74:443–449. doi: 10.1016/j.meatsci.2006.03.015. [PubMed] [CrossRef] [Google Scholar]
92. Branch J.D. Effect of Creatine Supplementation on Body Composition and Performance: A Meta-analysis. Int. J. Sport Nutr. Exerc. Metab. 2003;13:198–226. doi: 10.1123/ijsnem.13.2.198. [PubMed] [CrossRef] [Google Scholar]
93. Hultman E., Söderlund K., Timmons J.A., Cederblad G., Greenhaff P.L. Muscle creatine loading in men. [(accessed on 25 March 2019)];J. Appl. Physiol. Soc. 1996 81:232–237. doi: 10.1152/jappl.1996.81.1.232. Available online: https://www.ncbi.nlm.nih.gov/pubmed/8828669 [PubMed] [CrossRef] [Google Scholar]
94. Jagim A.R., Oliver J.M., Sanchez A., Galvan E., Fluckey J., Riechman S., Greenwood M., Kelly K., Meininger C., Rasmussen C., et al. A buffered form of creatine does not promote greater changes in muscle creatine content, body composition, or training adaptations than creatine monohydrate. J. Int. Soc. Sports Nutr. 2012;9:43. doi: 10.1186/1550-2783-9-43. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
95. Spillane M., Schoch R., Cooke M., Harvey T., Greenwood M., Kreider R., Willoughby D.S., Cooke M. The effects of creatine ethyl ester supplementation combined with heavy resistance training on body composition, muscle performance, and serum and muscle creatine levels. J. Int. Soc. Sports Nutr. 2009;6:6. doi: 10.1186/1550-2783-6-6. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
96. Childs E., De Wit H., Wit H. Subjective, behavioral, and physiological effects of acute caffeine in light, nondependent caffeine users. Psychopharmacology. 2006;185:514–523. doi: 10.1007/s00213-006-0341-3. [PubMed] [CrossRef] [Google Scholar]
97. Bellar D., Kamimori G.H., Glickman E.L. The Effects of Low-Dose Caffeine on Perceived Pain During a Grip to Exhaustion Task. J. Strength Cond. Res. 2011;25:1225–1228. doi: 10.1519/JSC.0b013e3181d9901f. [PubMed] [CrossRef] [Google Scholar]
98. Davis J.K., Green J.M. Caffeine and anaerobic performance: Ergogenic value and mechanisms of action. [(accessed on 25 March 2019)];Sport. Med. 2009 39:813–832. doi: 10.2165/11317770-000000000-00000. Available online: https://www.ncbi.nlm.nih.gov/pubmed/19757860 [PubMed] [CrossRef] [Google Scholar]
99. Wickwire P.J., McLester J.R., Gendle S., Hudson G., Pritchett R.C., Laurent C.M., Green J.M. Effects of Caffeine on Repetitions to Failure and Ratings of Perceived Exertion during Resistance Training. Int. J. Sports Physiol. Perform. 2007;2:250–259. [PubMed] [Google Scholar]
100. Duncan M.J., Oxford S.W. The effect of caffeine ingestion on mood state and bench press performance to failure. [(accessed on 25 March 2019)];J. Strength Cond. Res. 2001 25:178–185. doi: 10.1519/JSC.0b013e318201bddb. Available online: https://www.ncbi.nlm.nih.gov/pubmed/22124354 [PubMed] [CrossRef] [Google Scholar]
101. Williams A.D., Cribb P.J., Cooke M.B., Hayes A. The Effect of Ephedra and Caffeine on Maximal Strength and Power in Resistance-Trained Athletes. J. Strength Cond. Res. 2008;22:464–470. doi: 10.1519/JSC.0b013e3181660320. [PubMed] [CrossRef] [Google Scholar]
102. Tarnopolsky M.A., Atkinson S.A., MacDougall J.D., Sale D.G., Sutton J.R. Physiological responses to caffeine during endurance running in habitual caffeine users. Med. Sci. Sports Exerc. 1989;21:418–424. doi: 10.1249/00005768-198908000-00013. [PubMed] [CrossRef] [Google Scholar]
103. Blanchard J., Sawers S.J.A. The absolute bioavailability of caffeine in man. Eur. J. Clin. Pharmacol. 1983;24:93–98. doi: 10.1007/BF00613933. [PubMed] [CrossRef] [Google Scholar]
104. Hobson R.M., Saunders B., Ball G., Harris R.C., Sale C. Effects of β-alanine supplementation on exercise performance: A meta-analysis. Amino Acids. 2012;43:25–37. doi: 10.1007/s00726-011-1200-z. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
105. Hoffman J., Ratamess N.A., Ross R., Kang J., Magrelli J., Neese K., Faigenbaum A.D., Wise J.A. Beta-alanine and the hormonal response to exercise. [(accessed on 25 March 2019)];Int. J. Sports Med. 2008 29:952–958. doi: 10.1055/s-2008-1038678. Available online: https://www.ncbi.nlm.nih.gov/pubmed/18548362 [PubMed] [CrossRef] [Google Scholar]
106. Hoffman J., Ratamess N., Kang J., Mangine G., Faigenbaum A., Stout J. Effect of creatine and β-alanine supplementation on performance and endocrine responses in strength/power athletes. [(accessed on 25 March 2019)];Int. J. Sport Nutr. Exerc. Metab. 2006 16:430–446. doi: 10.1123/ijsnem.16.4.430. Available online: https://www.ncbi.nlm.nih.gov/pubmed/17136944 [PubMed] [CrossRef] [Google Scholar]
107. Pérez-Guisado J., Jakeman P.M. Citrulline Malate Enhances Athletic Anaerobic Performance and Relieves Muscle Soreness. J. Strength Cond. Res. 2010;24:1215–1222. doi: 10.1519/JSC.0b013e3181cb28e0. [PubMed] [CrossRef] [Google Scholar]
108. Wax B., Kavazis A.N., Weldon K., Sperlak J. Effects of Supplemental Citrulline Malate Ingestion During Repeated Bouts of Lower-Body Exercise in Advanced Weightlifters. J. Strength Cond. Res. 2015;29:786–792. doi: 10.1519/JSC.0000000000000670. [PubMed] [CrossRef] [Google Scholar]
109. Wax B., Kavazis A.N., Luckett W. Effects of Supplemental Citrulline-Malate Ingestion on Blood Lactate, Cardiovascular Dynamics and Resistance Exercise Performance in Trained Males. [(accessed on 25 March 2019)];J. Diet. 2016 13:269–282. doi: 10.3109/19390211.2015.1008615. Available online: https://www.ncbi.nlm.nih.gov/pubmed/25674699 [PubMed] [CrossRef] [Google Scholar]
110. Glenn J.M., Gray M., Wethington L.N., Stone M.S., Stewart R.W., Jr., Moyen N.E. Acute citrulline malate supplementation improves upper- and lower-body submaximal weightlifting exercise performance in resistance-trained females. [(accessed on 25 March 2019)];Eur. J. Nutr. 2017 56:775–784. doi: 10.1007/s00394-015-1124-6. Available online: https://www.ncbi.nlm.nih.gov/pubmed/26658899 [PubMed] [CrossRef] [Google Scholar]
111. Glenn J.M., Gray M., Jensen A., Stone M.S., Vincenzo J.L. Acute citrulline-malate supplementation improves maximal strength and anaerobic power in female, masters athletes tennis players. Eur. J. Sport Sci. 2016;16:1–9. doi: 10.1080/17461391.2016.1158321. [PubMed] [CrossRef] [Google Scholar]
112. Gonzalez A.M., Spitz R.W., Ghigiarelli J.J., Sell K.M., Mangine G.T. Acute Effect of Citrulline Malate Supplementation on Upper-Body Resistance Exercise Performance in Recreationally Resistance-Trained Men. J. Strength Cond. Res. 2018;32:3088–3094. doi: 10.1519/JSC.0000000000002373. [PubMed] [CrossRef] [Google Scholar]
113. Farney T.M., Bliss M.V., Hearon C.M., Salazar D.A. The Effect of Citrulline Malate Supplementation On Muscle Fatigue Among Healthy Participants. J. Strength Cond. Res. 2017:1. doi: 10.1519/JSC.0000000000002356. [PubMed] [CrossRef] [Google Scholar]
114. Trexler E.T., Persky A.M., Ryan E.D., Schwartz T.A., Stoner L., Smith-Ryan A.E. Acute Effects of Citrulline Supplementation on High-Intensity Strength and Power Performance: A Systematic Review and Meta-Analysis. Sports Med. 2019;49:707–718. doi: 10.1007/s40279-019-01091-z. [PubMed] [CrossRef] [Google Scholar]
115. Kleiner S.M., Bazzarre T.L., Litchford M.D. Metabolic profiles, diet, and health practices of championship male and female bodybuilders. J. Am. Diet. Assoc. 1990;90:962–967. [PubMed] [Google Scholar]
116. Kleiner S.M., Bazzarre T.L., Ainsworth B.E. Nutritional Status of Nationally Ranked Elite Bodybuilders. Int. J. Sport Nutr. 1994;4:54–69. doi: 10.1123/ijsn.4.1.54. [PubMed] [CrossRef] [Google Scholar]
117. Sandoval W.M., Heyward V.H. Food Selection Patterns of Bodybuilders. Int. J. Sport Nutr. 1991;1:61–68. doi: 10.1123/ijsn.1.1.61. [PubMed] [CrossRef] [Google Scholar]
118. Ismaeel A., Weems S., Willoughby D.S. A Comparison of the Nutrient Intakes of Macronutrient-Based Dieting and Strict Dieting Bodybuilders. Int. J. Sport Nutr. Exerc. Metab. 2018;28:502–508. doi: 10.1123/ijsnem.2017-0323. [PubMed] [CrossRef] [Google Scholar]
119. Nelson J.R., Raskin S. The eicosapentaenoic acid:arachidonic acid ratio and its clinical utility in cardiovascular disease. Postgrad. Med. 2019;131:268–277. doi: 10.1080/00325481.2019.1607414. [PubMed] [CrossRef] [Google Scholar]
120. Harris W.S. The Omega-6: Omega-3 ratio: A critical appraisal and possible successor. [(accessed on 15 June 2019)];Prostaglandins Leukot Essent Fatty Acids. 2018 132:34–40. doi: 10.1016/j.plefa.2018.03.003. Available online: https://www.ncbi.nlm.nih.gov/m/pubmed/29599053/ [PubMed] [CrossRef] [Google Scholar]
121. Tachtsis B., Camera D., Lacham-Kaplan O. Potential Roles of n-3 PUFAs during Skeletal Muscle Growth and Regeneration. Nutrients. 2018;10:309. doi: 10.3390/nu10030309. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
122. Di Girolamo F.G., Situlin R., Mazzucco S., Valentini R., Toigo G., Biolo G. Omega-3 fatty acids and protein metabolism: Enhancement of anabolic interventions for sarcopenia. [(accessed on 15 June 2019)];Curr. Opin. Clin. Nutr. Metab Care. 2014 17:145–150. doi: 10.1097/MCO.0000000000000032. Available online: https://www.ncbi.nlm.nih.gov/pubmed/24500439 [PubMed] [CrossRef] [Google Scholar]
123. McGlory C., Wardle S.L., Macnaughton L.S., Witard O.C., Scott F., Dick J., Bell J.G., Phillips S.M., Galloway S.D.R., Hamilton D.L., et al. Fish oil supplementation suppresses resistance exercise and feeding-induced increases in anabolic signaling without affecting myofibrillar protein synthesis in young men. Physiol. Rep. 2016;4:e12715. doi: 10.14814/phy2.12715. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
124. Crestani D.M., Bonin E.F.R., Barbieri R.A., Zagatto A.M., Higino W.P., Milion F. Chronic supplementation of omega-3 can improve body composition and maximal strength, but does not change the resistance to neuromuscular fatigue. [(accessed on 15 June 2019)];Sport Sci. Health. 2017 13:259–265. doi: 10.1007/s11332-016-0322-9. Available online: https://link.springer.com/article/10.1007/s11332-016-0322-9 [CrossRef] [Google Scholar]
125. Lewis E.J.H., Radonic P.W., Wolever T.M.S., Wells G.D. 21 days of mammalian omega-3 fatty acid supplementation improves aspects of neuromuscular function and performance in male athletes compared to olive oil placebo. J. Int. Soc. Sports Nutr. 2015;12:28. doi: 10.1186/s12970-015-0089-4. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
126. Rossato L.T., Schoenfeld B.J., De Oliveira E.P. Is there sufficient evidence to supplement omega-3 fatty acids to increase muscle mass and strength in young and older adults? Clin. Nutr. 2019 doi: 10.1016/j.clnu.2019.01.001. [PubMed] [CrossRef] [Google Scholar]
127. Mocking R.J.T., Harmsen I., Assies J., Koeter M.W.J., Ruhé H.G., Schene A.H. Meta-analysis and meta-regression of omega-3 polyunsaturated fatty acid supplementation for major depressive disorder. Transl. Psychiatry. 2016;6:e756. doi: 10.1038/tp.2016.29. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
128. Maki K.C., Palacios O.M., Bell M., Toth P.P. Use of supplemental long-chain omega-3 fatty acids and risk for cardiac death: An updated meta-analysis and review of research gaps. J. Clin. Lipidol. 2017;11:1152–1160.e2. doi: 10.1016/j.jacl.2017.07.010. [PubMed] [CrossRef] [Google Scholar]
129. Miller P.E., Van Elswyk M., Alexander D.D. Long-Chain Omega-3 Fatty Acids Eicosapentaenoic Acid and Docosahexaenoic Acid and Blood Pressure: A Meta-Analysis of Randomized Controlled Trials. Am. J. Hypertens. 2014;27:885–896. doi: 10.1093/ajh/hpu024. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
130. Du S., Jin J., Fang W., Su Q. Does Fish Oil Have an Anti-Obesity Effect in Overweight/Obese Adults? A Meta-Analysis of Randomized Controlled Trials. PLoS ONE. 2015;10:e0142652. doi: 10.1371/journal.pone.0142652. [PMC free article] [PubMed] [CrossRef] [Google Scholar]