
Introduzione
Se si domandasse al bodybuilder nella media quale sia l’oggetto di maggiore interesse nella sua programmazione per il “Bulk” quasi sicuramente, dopo aver detto “raggiungere la massima ipertrofia”, direbbe “ridurre al minimo l’aumento della massa grassa”. Ed è più che logico visto che l’atleta con un minimo di senso logico sa che un peggioramento marcato della “bf” in una fase ipercalorica si tradurrà mediamente in un tempo generalmente più lungo in ipocalorica con restrizioni caloriche (o output calorici) più elevate ed un rischio relativamente aumentato di veder persa più massa contrattile o, peggio ancora, il dover calcare con i PEDs per rientrare nei tempi della preparazione al contest (o a qualsiasi evento dove il soggetto vuole essere nella sua “Top Condition”).
Tralasciando la pratica della “Break Diet”, altamente funzionale se gestita correttamente, esiste una strategia alimentare piuttosto datata ma, complici anche “neo-trafilettari” impomatati, riemersa recentemente, ovvero seguire un regime alimentare “High Fat” nel periodo di “Bulk”.
Siamo soliti collegare le diete “Low Carbs” e “High Fat” nel periodo di restrizione calorica, sapendo benissimo che il vantaggio assoluto esiste solo e soltanto con pratiche supplementative mirate (PEDs), parlando sempre di atleti e non di soggetti in sovrappeso o obesi che inizialmente giovano nel seguire una dieta Chetogenica o iperproteica in quanto a compliance insulino-resistenza correlata. Ma in ipercalorica? Possiamo veramente aspettarci una qualità migliore in quanto aumento della massa muscolare ed il peso guadagnato rispetto ad una dieta con prevalenza glucidica?
Con questo articolo cercherò di dare una risposta la più oggettiva possibile avvalendomi della conoscenza scientifica in nostro possesso che riguarda il metabolismo lipidico, l’impatto dei lipidi sulla massa muscolo-scheletrica, sulle prestazioni e i raffronti con il metabolismo glucidico partendo però dalla storia pratica della metodica alimentare qui trattata…
Back to the Future: Mauro Di Pasquale e la sua “Soluzione Anabolica”
Chi è che non ricorda Mauro Di Pasquale e il suo (a detta dell’autore) “Santo Graal” delle diete, “La Dieta Metabolica”? E la sua versione per i Bodybuilder ” La Soluzione Anabolica”? Immagino che molti di voi conosceranno entrambi i libri e i concetti viziati da bias in essi contenuti.
Visto l’argomento partirò proprio da qui…
Per chi non lo sapesse, Mauro G. Di Pasquale è un Powerlifter campione del mondo, autore di articoli sul bodybuilding e opinionista. Di Pasquale è stato assistente professore all’Università di Toronto dal 1988 al 1998. Ha tenuto conferenze e fatto ricerche sulle prestazioni atletiche, sugli integratori alimentari e sull’uso di farmaci nello sport. Ha conseguito una laurea con lode in scienze biologiche, specializzandosi in biochimica molecolare (1968) e ha una laurea in medicina (1971) – entrambe presso l’Università di Toronto. Di Pasquale è certificato come Medical Review Officer (MRO) dal Medical Review Officer Certification Council (MROCC). Era il MRO per la National Association for Stock Car Auto Racing (NASCAR). Dal 1997 al 1999 Di Pasquale si è occupato di redazione, ricerca e sviluppo di prodotti per le Scienze Sperimentali e Applicate (EAS). Come autore di bodybuilding, Di Pasquale ha scritto migliaia di articoli per molte grandi riviste di bodybuilding e fitness come Muscle & Fitness e Iron Man;[1-4] I suoi articoli e libri sono stati tradotti in lingua italiana e pubblicati in Italia da Sandro Ciccarelli per la rivista Olympian’s News.
Di Pasquale divenne anche un oppositore all’uso del doping pubblicizzando i suoi regimi alimentari e integrativi come “sostitutivi salutari” dei farmaci per il miglioramento delle prestazioni.

Nonostante la titolatura sopra esposta, Di Pasquale divulgò alcune teorie decisamente opinabili abbracciando la filosofia delle diete “Low Carb” come soluzione universale per perdere grasso e aumentare la massa muscolare. Egli rappresenta una sorta di “paladino” per la fazione dei sostenitori della così detta “ipotesi dell’Insulina”, tra l’altro smentita scientificamente, la quale ipotizza che non sia l’eccesso energetico (calorie) a causare l’aumento di peso/grasso ma i Carboidrati ed il loro stimolo sull’Insulina. Peccato però che la termodinamica ci dimostri il contrario, ovvero che è l’eccesso energetico ha causare una conservazione dello stesso e non il semplice stimolo ormonale (sul quale, tra l’altro, ci sarebbe molto da dire). Oltretutto, alcune fonti proteiche (vedi prodotti lattiero caseari) hanno un impatto insulinico maggiore del pane bianco, sebbene questo venga regolato da una concomitante risposta del Glucagone. Comunque sia, di quest’ultimo punto, il Di Pasquale sembra non curarsene più di tanto, similmente a quanto fa della risposta insulinica data dalla coingestione di grassi e proteine preoccupandosi del carico glucidico da ciclicizzare (cosa, quest’ultima, condivisibile ma da contestualizzare).
Ma arriviamo al dunque sul suo approccio “High Fat” in Bulk…
Nel 1995 pubblica il libro “The Anabolic Diet“, pubblicato poi in Italia con il nome di “La Soluzione Anabolica”.

Il libro in questione espone le vedute dell’autore, accuratamente servite per vendere il prodotto e convincere il lettore, dividendo in sei fasi la preparazione:
- Fase di Inizio;
- Fase di Massa;
- Fase di Forza;
- Fase di Definizione;
- Fase Pregara;
- Fase di Recupero.
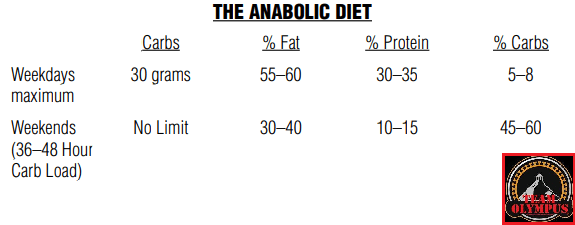
Tralasciando le fasi 1, 3,4,5 e 6 che, per argomento trattato nel presente articolo, non ci interessano, concentriamoci sulla “Fase di Massa“.
Dopo aver trovato la quota calorica di “mantenimento”, procedendo con i primi 12 giorni sostituendo le kcal tipicamente consumate dai Carboidrati con Grassi e Proteine riducendo significativamente i primi (es. 1g/CHO-Pro = 4Kcal; 1g Fat = 9Kcal; 400g di CHO/120g di Pro/50g di Fat = circa 30g di CHO, 220g/die di Pro e 134g/die di Grassi), si dovrebbe passare alla prima “ricarica di Carboidrati” seguita a sua volta da 5-6 giorni “High Fat” e 1 o 2 giorni “High Carbs” per 3-4 settimane.
A questo punto l’autore consiglia di permettere al peso un aumento del 15% oltre il peso ideale (es. Peso ideale = 98Kg, peso da raggiungere = 113Kg [+15%]) premurandosi di avvisare il lettore che non deve commettere l’errore di mangiare eccessivamente e vanificare il potenziale della sua strategia di farvi aumentare di massa magra con poco incremento della massa grassa…
Quindi, il bodybuilder dovrebbe seguire lo schema “High Fat” nei giorni feriali ed effettuare le “ricariche” glucidiche nel fine settimana, con un monte calorico indicativo pari a 55Kcal per Kg di peso corporeo desiderato (es. 90Kg = 4950Kcal/die). E le differenze di risposta delle vie metaboliche? Capacità di gestione delle Kcal connesse alle richieste prestative e al livello dell’atleta?… Punti non pervenuti… Per lo meno, il Di Pasquale dice che se il soggetto presenta difficoltà nell’ingestione di grossi quantitativi calorici giornalieri può far diventare il totale calorico da raggiungere un obbiettivo settimanale, abbandonando saggiamente il concetto forviante di “evento in acuto”.
Nelle indicazioni viene riportata anche la soglia del grasso corporeo da non superare. La quota riportata è del 10%… Ora, fatta eccezione per soggetti con un set point adipocitario sensibilmente basso, arrivare a pesare il 15% in più del peso ideale mantenendo una “bf” del 10% è cosa non proprio semplice, leggermente di più se parliamo di atleti “Natural”. Molto più logico sarebbe stato indicare una soglia tra il 12 ed il 15%. Ma l’autore scrive che la Fase di Massa deve essere interrotta quando si raggiunge il peso desiderato o il 10% di “bf”… Se siete agonisti dovete interromperla se arrivate al limite delle 12 settimane dalla gara.
Per quanto concerne l’obbiettivo di incremento settimanale di peso, viene indicato come “ideale” 1Kg a settimana.
Ma quanti glucidi dovrebbero essere consumati durante il/i giorno/i di “ricarica”? Di Pasquale afferma che si possono raggiungere “quantità enormi” di CHO e Kcal nel week and, anche fino a 12.000Kcal tanto il sabato quanto la domenica. Inoltre sia la quota di CHO giornaliera che il numero di giorni con rialzo glucidico possono essere aumentati in base alle risposte. E già questo punto denota una mancanza di funzionalità di base del suo piano alimentare “High Fat” per una “massa pulita” che però va oltre il concetto di “pulito” arrivando al “nodo metabolico”: il vantaggio indiscutibile del substrato glucidico per sostenere l’attività fisica in uno sport contro-resistenza che sfrutta prevalentemente un metabolismo anaerobico glicolitico.
Lascerò da parte la sfilza di integratori marca “MetabolicDiet”, che per lo più altro non sono che Creatina e Proteine in polvere, e le riduttive, ridicole e confutate chiacchiere sulla manipolazione ormonale “simil-doped”, riflettendo direttamente su quanto detto fino a questo punto.
Ora, abbiamo una dieta per il “Bulk” a tutti gli effetti “High Fat” per 3-4/5 della settimana con delle “ricariche glucidiche” di base di 1 o 2 giorni ma incrementabili a bisogno. Ricordo che questa dieta ha la bellezza di 26 anni, ed il sottoscritto, ai tempi in cui ero fortemente tentato a prestar orecchio più alla “carne ed al sangue” piuttosto che alla logica e al buon senso, la conosce e osserva i suoi effetti sugli atleti che l’hanno voluta sperimentare da circa 15 anni. Beh, posso assicurarvi che i risultati con questo tipo di alimentazione in “Bulk” erano apprezzabili (questo non vuol dire che erano spettacolari o che riflettevano le promesse dell’autore) principalmente in due contesti:
- Alteti “doped” con un set point adipocitario basso e una media di 3 copiose “ricariche” a settimana;
- Atleti “natural” con un set point adipocitario basso e una media di 3 copiose “ricariche” a settimana e un minimo di CHO giornalieri pari a 2-3g/Kg.;
- Atleti di sesso femminile con variabili sul numero di “ricariche” e tempi di svolgimento delle medesime.
Ed i casi osservati con risultanti apprezzabili, comunque, sono pochi rispetto ad una maggioranza di individui non significativamente avvantaggiati dal protocollo.
Come potete vedere, il concetto di base della dieta crolla inesorabilmente sotto il peso di dati empirici raccolti direttamente ed indirettamente nel corso degli anni.
“Mah Gabriel! Esistono altre metodologie “High Fat” per una “massa pulita” e funzionano!” Alt, non ti agitare piccolo “troll”, perchè con il termine “High Fat” mi stai dicendo “tutto e niente”, un pò come succede quando si parla di “dieta iperproteica”.
Si possono considerare “High Fat” tutte le diete che in ipercalorica superano il 20-25% delle calorie giornaliere dai grassi! E’ ovvio che possono esistere degli abissi programmatici tra uno schema alla Di Pasquale e un altro che presenta linee di percentuali macro-caloriche nei range sopra indicati.
Prima di proseguire con esempi di schemi alimentari “High Fat” ben più logici rispetto a quanto presentato ne “La Soluzione Anabolica”, e considerare le argomentazioni a favore di questo tipo di dieta ipercalorica, vediamo come i diversi acidi grassi influenzano la massa muscolare. Passiamo a un pò di teoria per tornare successivamente alla pratica con maggiori conoscenze…
La modulazione nella massa e funzione del muscolo-scheletrico dei lipidi
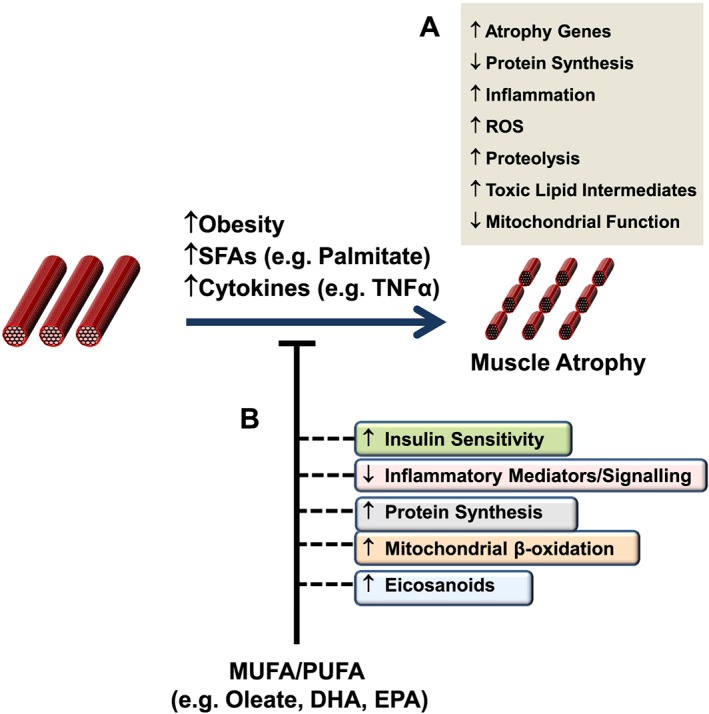
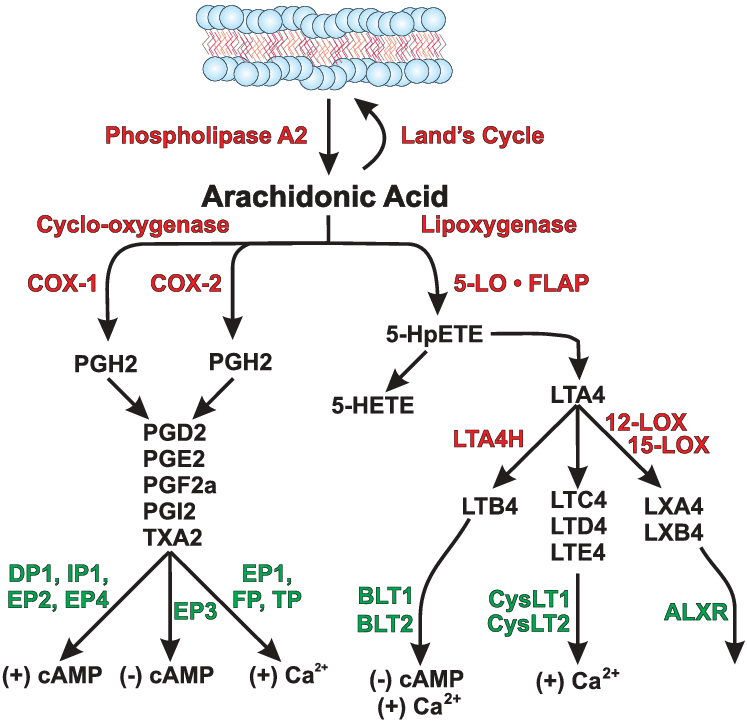
È importante iniziare il discorso sottolineando che vi sono prove crescenti che supportano un ruolo degli acidi grassi e dei loro intermedi lipidici derivati nella regolazione della massa e della funzione del muscolo scheletrico. E’ mia intenzione quindi discutere le prove relative a quei percorsi che sono coinvolti nella riduzione, aumento e/o conservazione della massa muscolare scheletrica da parte dei lipidi.
L’evidenza di diversi studi suggerisce che gli acidi grassi saturi e insaturi possono agire per regolare in modo differenziale la massa e la funzione del muscolo scheletrico. Ad esempio, è stato dimostrato che l’esposizione dei miotubi C2C12 al palmitato (C16:0), l’acido grasso saturo circolante più abbondante, riduce il diametro del miotubo e sopprime la segnalazione dell’Insulina. [2] In accordo con questo, è stato riportato che la fornitura di palmitato nelle cellule muscolari induce l’espressione di geni pro-atrofici come atrogin-1/MAFbx, in concomitanza con una maggiore localizzazione nucleare del suo regolatore trascrizionale FoxO3. [3] Al contrario, l’applicazione di acido docosaesaenoico (DHA), un acido grasso polinsaturo omega-3 (PUFA), non ha alterato la morfologia dei miotubi quando applicato da solo ed è stato dimostrato che contromodula l’atrofia indotta dal palmitato nei miotubi C2C12. [2] Coerentemente con ciò, uno studio separato ha riportato il miglioramento della degradazione proteica indotta dal palmitato nei miotubi C2C12 dopo il co-trattamento con DHA. [3] In particolare, ciò ha coinciso con la capacità del DHA di mitigare la localizzazione nucleare potenziata di FoxO3 e l’espressione genica di atrogin-1/MAFbx in risposta alla fornitura di palmitato. [3]

In accordo con questi risultati nelle cellule muscolari di coltura, diversi studi in vivo hanno anche riportato la capacità degli acidi grassi insaturi di trasmettere risposte benefiche che agiscono per prevenire l’atrofia muscolare. Ad esempio, l’alimentazione di topi portatori dell’adenocarcinoma del colon-26, un modello animale di cachessia tumorale, con una dieta integrata con acido linoleico coniugato, ha dimostrato di preservare la massa muscolare del gastrocnemio. [4] In particolare, questo effetto protettivo ha coinciso con una riduzione dell’espressione del recettore TNF-α del muscolo scheletrico, suggerendo che i PUFA possono agire per prevenire l’atrofia muscolare, almeno in parte, riducendo le azioni cataboliche della citochina TNF-α. [4 , 5] In uno studio separato, l’integrazione alimentare con acido eicosapentaenoico (EPA; C20:5 (n-3)) ha attenuato la degradazione proteica nel muscolo gastrocnemio di topi portatori del tumore MAC16 che induce la cachessia. [6] Il trattamento con EPA è stato riportato anche per evitare riduzioni dell’artrite indotta e aumento del peso del muscolo gastrocnemio nei ratti dopo somministrazione di adiuvante di Freund, concomitante con la normalizzazione dell’espressione genica di atrogin-1 / MAFbx e MuRF1. [7] Inoltre, i criceti distrofici alimentati con una dieta arricchita in acido α-linolenico PUFA (ALA) (C18:3(n-6)) hanno mostrato miglioramenti nella morfologia e nella funzione muscolare, compreso l’ingrossamento delle miofibre . [8] In accordo con questi risultati, è stato anche dimostrato che i PUFA omega-3 e omega-6 aumentano la fosforilazione di p70S6K1 a Thr389, indicativo della sua maggiore attività, durante la differenziazione miogenica dei miociti L6. [9] Insieme, questi studi supportano l’idea che gli acidi grassi insaturi possono fornire protezione contro l’atrofia muscolare in risposta a varie condizioni patologiche e potenzialmente migliorare le condizione trofiche in soggetti sani. Inoltre, questi risultati evidenziano le risposte distinte che gli acidi grassi saturi e insaturi inducono rispettivamente per promuovere o contrastare l’atrofia muscolare e la degradazione proteica.
Un certo numero di differenti vie di segnalazione e/o intermedi sono stati implicati come potenziali mediatori della atrofia muscolare, che a loro volta possono essere regolati in risposta alla assunzione di acidi grassi (vedi Figura seguente). Ad esempio, è noto che il palmitato agisce come un potente repressore della segnalazione diretta di PKB/Akt nel muscolo scheletrico, almeno in parte attraverso la sua capacità di indurre l’accumulo di intermedi lipidici tossici come la Ceramide. [10 , 11] Infatti, tali sfingolipidi possono agire stimolando le isoforme della proteina fosfatasi 2A (PP2A) o della proteina chinasi C (PKC) atipica (PKCζ) per inibire PKB/Akt. [12] In accordo con ciò, è stato riportato che l’atrofia del miotubo C2C12 indotta da TNF-α coincide con livelli elevati di Ceramide intracellulare, [13] mentre è stato dimostrato che il blocco della sintesi di Ceramide attenua l’atrofia muscolare indotta dal TNF-α nei miotubi L6, oltre a proteggere i topi contro l’atrofia del muscolo scheletrico tumore indotto ( via impianto di carcinoma C26) in vivo. [13] In particolare, queste risposte benefiche hanno contribuito a una maggiore sintesi proteica e a una diminuzione della proteolisi, in concomitanza con una ridotta espressione del gene atrogin-1/MAFbx tramite la funzione Foxo3 soppressa, nonché una maggiore abbondanza di mediatori chiave della sintesi proteica tra cui S6K1 e PKB/Akt. [13] Inoltre, è stato riportato che la fornitura esogena di Ceramide nelle cellule muscolari L6 riduce i livelli proteici del fattore di trascrizione miogenico miogenina attraverso l’ inibizione della fosfolipasi D, mentre l’inibizione della sintesi di Ceramide migliora l’espressione della miogenina e accelera la formazione di miotubi. [14]Uno studio di Turpin e colleghi ha anche dimostrato un aumento del contenuto di Ceramide muscolare dopo l’infusione acuta (5 h) di intralipid®, che ha coinciso con l’attivazione della segnalazione pro-apoptotica, come dimostrato dall’aumento dell’attività della caspasi-3 nel muscolo gastrocnemio. [15] Tuttavia, il ruolo della Ceramide nel promuovere questo aumento dell’apoptosi muscolare guidato dai lipidi non è stato studiato, ad esempio mediante la co-somministrazione di inibitori della sintesi della Ceramide. In alternativa, livelli elevati di Ceramide associati all’iperlipidemia possono anche agire per sopprimere la sintesi proteica inducendo l’espressione e/o l’attività di repressori chiave della segnalazione mTORC1-S6K come Regulated in Development e DNA Damage 1 (REDD1). [16 , 17]In particolare, va anche evidenziato che il ganglioside GM3 (trisialotetrahexosylganglioside), un glicosfingolipide contenente acido sialico derivato dalla Ceramide, è stato anche implicato come regolatore negativo della crescita e/o differenziazione del muscolo scheletrico, in concomitanza con la sua capacità segnalata di modificare l’azione dell’insulina alterando la funzione del recettore specifico (Recettore dell’Insulina). [18 , 19 , 20 , 21] Inoltre, è stato dimostrato che un altro lipide derivato dalla Ceramide, ceramide-1-fosfato, stimola la proliferazione dei mioblasti C2C12 attraverso un meccanismo che coinvolge l’attivazione di Akt, mTOR e ERK1/2. [22] In effetti, un ulteriore lavoro che utilizza topi carenti di GM3 sintasi, l’enzima responsabile della sintesi di GM3, potrebbe far luce sul ruolo di questo ganglioside nel controllo della massa muscolare scheletrica, ad esempio in risposta all’obesità e/o all’invecchiamento.

Oltre agli sfingolipidi, i diacilgliceroli (DAG) sono una classe alternativa di lipidi che possono essere generati in risposta alla ingestione di acidi grassi. In particolare, l’aumento dei livelli di DAG è stato associato allo sviluppo dell’insulino-resistenza, fattore di per se limitante sul corretto ripartizionamento calorico a favore del miocita e, quindi, sulla sintesi proteica . [23] sono stati rilevati Inoltre, un aumento dei livelli di DAG muscolare dopo infusione di lipidi nei topi, con concomitante aumento della attività nel muscolo gastrocnemio della caspasi-3 .[15] Sebbene si sappia poco sul ruolo dei DAG nella regolazione della massa muscolare scheletrica, è stato riportato che l’attivazione meccanica ex-vivo della DAG chinasiζ (DGKζ), un enzima che catalizza la conversione di DAG in acido fosfatidico (PA), favorisce un aumento Segnalazione dipendente da mTOR e ipertrofia associata nel muscolo estensore lungo delle dita (EDL) di topo isolato, in concomitanza con la capacità riportata di PA di legare e attivare direttamente mTOR. [24 , 25] In accordo con ciò, è stato anche dimostrato che la sovraespressione cardiaca specifica di DGKζ migliora l’atrofia miocardica nei topi diabetici indotti da streptozotocina. [26] Pertanto, questi risultati suggeriscono che l’attivazione e/o la sovraespressione di DGKζ può fornire un mezzo per stimolare i tassi di sintesi proteica e le risposte ipertrofiche, e quindi migliorare le perdite di massa muscolare, sia riducendo i livelli cellulari di DAG e/o aumentando l’attivazione della segnalazione mTOR indotta dal PA. È importante sottolineare che il lavoro futuro potrebbe comportare lo studio dei potenziali effetti benefici della sovraespressione di DGKζ nel muscolo come mezzo per contrastare l’atrofia muscolare indotta dall’età e/o dalla dieta. Inoltre, modelli animali che mostrano livelli elevati di DAG nel muscolo scheletrico, compresi i topi che sono carenti di lipasi ormone-sensibile (HSL), [27] possono anche essere utili per chiarire il ruolo di DAG nell’atrofia del muscolo scheletrico.
Un’altra considerazione importante riguarda la possibilità che specie di DAG distinte possano avere un impatto diverso sulle vie coinvolte nella regolazione della massa muscolare, ad esempio come determinato dalla composizione dei gruppi acilici grassi che si esterificano a livello sn-1,2, sn‐ 1,3, o le posizioni sn-2,3 della base di glicerolo del DAG. [28 , 29] Infatti, è stato dimostrato che il trattamento dei miotubi L6 di ratto con palmitato porta ad aumenti significativi dei livelli cellulari di alcune specie di DAG, nonché del contenuto totale di DAG cellulare. [30] Inoltre, è stato dimostrato che il co-trattamento con l’acido grasso monoinsaturo (MUFA) palmitoleato (C16:1) sopprime selettivamente gli aumenti indotti dal palmitato nei livelli di specie DAG contenenti porzioni di acidi grassi saturi C18:0 e C20:0, in coincidenza con l’azione antinfiammatoria del MUFA. [30] Sebbene non determinati in questo studio, stereoisomeri distinti di DAG possono anche regolare in modo differenziale la segnalazione anabolica/catabolica muscolare. Per supportare questa nozione, è stato riportato che gli stereoisomeri sn-1,2 DAG (rispetto agli isomeri sn-1,3) sono più potenti nell’attivare le vie di segnalazione legate all’insulino-resistenza, inclusa l’attivazione della PKC. [31] Insieme, questi studi forniscono prove emergenti che alcune molecole/isomeri DAG possono svolgere un ruolo più importante nello sviluppo dell’atrofia muscolare, ad esempio promuovendo la resistenza all’insulina e/o aumentando la spinta proinfiammatoria. Tuttavia, sarà necessario un ulteriore lavoro per determinare quali di queste molecole di DAG, se presenti, sono responsabili delle azioni di atrofia muscolare. Nel tentativo di affrontare questo problema, studi futuri potrebbero comportare il trattamento di cellule muscolari in coltura con diverse molecole/stereoisomeri DAG al fine di determinare i loro effetti sulla miogenesi e/o sull’atrofia muscolare. In alternativa, un ulteriore lavoro può anche incorporare un’analisi lipidomica dettagliata di varie specie di DAG intramuscolari in tessuti isolati da modelli animali di atrofia muscolare, oltre a monitorare potenziali cambiamenti nella loro abbondanza a seguito di interventi noti per aumentare la massa muscolare (ad es. apporto dietetico di PUFA o aumento dell’attività fisica). Infatti, se tali studi dovessero rivelare un ruolo chiave per l’accumulo di DAG nello sviluppo dell’atrofia del muscolo scheletrico, il lavoro successivo potrebbe implicare la determinazione dell’origine di tali specie di DAG, ad esempio inibendo l’attività degli enzimi implicati nella formazione del triacilglicerolo dal DAG ( sintesi di TAG) (es. glicerolo fosfato transferasi (GPAT), acilglicerolfosfato aciltransferasi (AGPAT) e lipina), o alterando l’attività di enzimi implicati nell’idrolisi di TAG e/o DAG (es. lipasi dei trigliceridi adiposa (ATGL) o HSL). A questo fine, i lavori di Badin e collaboratori hanno riportato un’elevata abbondanza di proteine ATGL nel muscolo scheletrico di individui diabetici di tipo 2 rispetto a soggetti di controllo magri, nonché una ridotta espressione di HSL muscolare in individui obesi.[32] Inoltre, gli autori dello stesso studio hanno ulteriormente dimostrato che la sovraespressione di ATGL o l’inibizione dell’attività dell’HSL nei miotubi primari umani determinava l’accumulo di DAG cellulare e una compromissione associata nella segnalazione dell’Insulina. Tuttavia, in questo studio non è stato determinato se questi cambiamenti nei livelli di DAG siano collegati all’atrofia muscolare.
Oltre a modulare la segnalazione diretta di PKB/Akt e/o mTORC1, gli acidi grassi e/oi loro lipidi derivati possono ulteriormente contribuire alla perdita muscolare modulando il trasporto dei nutrienti (aminoacidi) e/o la segnalazione associata. Ad esempio, lavori svolti da diversi gruppi di ricerca hanno dimostrato la capacità della Ceramide di sottoregolare l’espressione e/o l’attività dei principali trasportatori di nutrienti, incluso il trasportatore di amminoacidi neutri SNAT2 (SLC38A2). [33 , 34] In tal modo, gli acidi grassi che agiscono attraverso tali intermedi lipidici possono agire per compromettere l’assorbimento degli aminoacidi, contribuendo così a una perdita di massa muscolare. È interessante notare che in uno studio separato, è stato dimostrato che l’incubazione di miotubi L6 di ratto con acido linoleico (C18:2) limita la sovraregolazione adattativa dell’espressione e dell’attività di SNAT2 in risposta alla carenza di aminoacidi. [35] In particolare, questa riduzione indotta da acidi grassi nell’attività di trasporto del Sistema A è stata mediata da una maggiore ubiquitinazione e degradazione proteasomica della proteina SNAT2. [35] Al contrario, in uno studio separato di Li e collaboratori, è stato riportato che l’espressione dell’mRNA dei transcettori di aminoacidi LAT1 (un trasportatore di aminoacidi di tipo L) e SNAT2 è sovra-regolata nel longissimus dorsi di suini alimentati con diete n. -6 e n-3 PUFA.[36] Quindi, è possibile che gli acidi grassi e/o i loro lipidi derivati possano funzionare per modulare le strategie adattative che vengono utilizzate da tessuti come il muscolo scheletrico, al fine di massimizzare o minimizzare l’assorbimento di nutrienti durante condizioni di digiuno o privazione di nutrienti e, presumibilmente, tale meccanismo può subire alterazioni durante stati di sovra-alimentazione.
È importante sottolineare che la letteratura attuale descrive prove che suggeriscono che gli acidi grassi insaturi possono agire per contrastare i mediatori pro-atrofici, compresi quelli attivati in seguito all’esposizione agli acidi grassi saturi. Ad esempio, è stato riportato che MUFA e PUFA prevengono le riduzioni indotte dal palmitato della sensibilità all’Insulina e veicolano effetti antinfiammatori nelle cellule del muscolo scheletrico. [30 , 37, 38] In effetti, la regolazione trascrizionale dipendente da NF-kB è stata implicata nel promuovere l’atrofia muscolare da disuso nel muscolo soleo di ratto aumentando l’attivazione mediata da FoxO del promotore MuRF1. [39] Inoltre, uno studio recente ha dimostrato che la diminuzione del segnale anabolico nel muscolo scheletrico di topi anziani coincideva con l’accumulo di Ceramide intramuscolare e DAG, nonché con una maggiore abbondanza di mRNA di TNF-α. [40] È interessante notare che la somministrazione di olio di pesce ai suinetti in fase di svezzamento, che ha portato all’arricchimento di EPA, DHA e contenuto totale di PUFA omega-3 all’interno del muscolo gastrocnemio, ha coinciso con una riduzione dei livelli di TNF-α muscolare e una ridotta espressione del recettore Toll-like 4 (TLR4), un recettore bersaglio per gli acidi grassi saturi che stimola la segnalazione proinfiammatoria in risposta alla sua attivazione. [41] In particolare, è stato riportato che la stimolazione del TLR4 da parte del suo ligando lipopolisaccaride induce il catabolismo muscolare nei miotubi C2C12 attraverso l’attivazione delle vie ubiquitina-proteasoma e autofagia-lisosoma. [42] Inoltre, è stato dimostrato che il trattamento con DHA in cellule muscolari umane co-coltivate con macrofagi attenua il contenuto proteico indotto dai macrofagi di Fn14, un modulatore positivo dell’espressione di MuRF-1. [43 , 44] Pertanto, sulla base di questi risultati, è concepibile che le azioni antinfiammatorie riportate degli acidi grassi insaturi nelle cellule muscolari scheletriche possano contribuire, almeno in parte, alla loro capacità di preservare la massa e/o la funzione muscolare.
In particolare, queste azioni protettive possono essere collegate a miglioramenti della funzione mitocondriale, la cui compromissione è stata suggerita dal tipo di dieta e/o all’atrofia muscolare indotta dall’età. [45 , 46] Ad esempio, un recente studio di Roseno e colleghi ha riportato che una dieta ricca di grassi a breve termine (3 settimane) ha aumentato l’atrofia muscolare da denervazione nei topi inducendo la degradazione proteica nel soleo ricco di mitocondri, ma non nel muscolo EDL glicolitico. In particolare, la denervazione di 14 giorni ha indotto una perdita del contenuto proteico mitocondriale nel soleo ma non nell’EDL, indipendentemente dalla dieta. Pertanto, questi risultati suggeriscono che la perdita di mitocondri indotta dalla denervazione e la compromissione della funzione mitocondriale indotta da una dieta ricca di grassi possono combinarsi per promuovere l’atrofia del muscolo scheletrico. Al contrario, uno studio indipendente di Tardif e collaboratori ha dimostrato che i ratti anziani alimentati con una dieta arricchita di oleati mostrano notevoli miglioramenti nella sensibilità all’insulina e un aumento della sintesi proteica muscolare, in concomitanza con una maggiore espressione di geni implicati nella stimolazione della ossidazione mitocondriale, compreso il recettore attivato dal proliferatore dei perossisomi PPARα e PPARβ, nonché CPT-1β. [47 , 48] Inoltre, è stato dimostrato che i miotubi C2C12 trattati con PUFA acido linolenico e ALA mostrano una maggiore attivazione del AMPK, un altro regolatore positivo chiave della -ossidazione mitocondriale. [49] Inoltre, è stato riportato che il DHA inibisce la degradazione proteica nei miotubi C2C12 attraverso una via PPARγ-dipendente. [50] Infatti, la capacità ossidativa mitocondriale potenziata e/o preservata, come precedentemente riportato in risposta alla sola o co-fornitura di acidi grassi insaturi, può anche aiutare a prevenire l’accumulo intramuscolare di intermedi lipotossici come la Ceramide che sono stati implicati nella promozione dell’atrofia muscolare. [51 , 52] Inoltre, è possibile che l’integrazione di PUFA possa agire per alterare le proprietà contrattili e metaboliche muscolari, ad esempio promuovendo un passaggio da fibre glicolitiche veloci a fibre lente (ossidative), cosa non propriamente positiva per un atleta di potenza. A sostegno di questa idea, lavori precedenti hanno dimostrato che l’alimentazione di ratti Wistar con una dieta arricchita in PUFA n-3 porta alla sovraregolazione delle proteine implicate nell’attivazione del metabolismo ossidativo (ad es. nel muscolo EDL, un tessuto muscolare dominante di tipo veloce). [53] È interessante notare che questo cambiamento metabolico mediato da PUFA ha coinciso anche con livelli ridotti di proteine dell’isoforma di tipo veloce MyHC-2b (catena pesante della miosina 2b) nel muscolo EDL. Pertanto, è concepibile che uno spostamento mediato dai PUFA verso un tipo di fibra muscolare a lenta ossidazione possa contribuire, almeno in parte, a guadagni benefici nella massa muscolare e/o nella funzione metabolica.
In alternativa, la regolazione della massa muscolare da parte dei lipidi può anche comportare la modulazione dell’autofagia, un meccanismo omeostatico che facilita la degradazione e il riciclaggio di proteine e organelli attraverso il macchinario lisosomiale. [54] In particolare, è stato riportato che un aumento della degradazione autofagica coincide con l’atrofia muscolare in varie condizioni e/o patologie tra cui cancro, [55] denervazione, [56] e invecchiamento. [55 , 57] Inoltre, è stato dimostrato che un’alimentazione ricca di grassi a breve termine (3 settimane) aumenta l’abbondanza di marcatori autofagosomiali nel soleo denervato dei topi. In accordo con ciò, Yuzefovych e collaboratori hanno dimostrato un aumento dell’autofagia nei miotubi L6 a seguito della somministrazione di palmitato. Pertanto, sebbene non sia ancora stato stabilito un collegamento diretto in vivo , è ipotizzabile che il turnover proteico alterato tramite l’ autofagia possa, almeno in parte, mediare le alterazioni indotte dai lipidi nella massa muscolare.
Va inoltre evidenziato che alcuni acidi grassi insaturi possono alterare il tasso di proliferazione delle cellule satelliti che funzionano come cellule progenitrici miogeniche necessarie per la crescita e la rigenerazione muscolare. Ad esempio, è stato dimostrato che DHA ed EPA inibiscono la proliferazione dei mioblasti C2C12 e delle cellule satelliti isolate dal muscolo di tacchino. [58 , 59] In particolare, queste azioni di soppressione della crescita sono state collegate a livelli ridotti di ciclina E e CDK2, proteine che svolgono un ruolo critico nella progressione del ciclo cellulare, nonché all’attivazione soppressa di ERK1/2, una proteina chinasi attivata da mitogeni implicata nel promuovere la crescita e la divisione cellulare. [59 , 60] Al contrario, è stato dimostrato che l’alimentazione di criceti distrofici carenti di δ-sarcoglicano con una dieta arricchita in ALA (un PUFA omega-3) aumenta la proliferazione e la differenziazione delle cellule satellite nel muscolo EDL, in concomitanza con un miglioramento dell’istologia muscolare. In particolare, queste risposte benefiche hanno coinciso con la capacità dell’ALA di aumentare la proporzione di miofibre α-MHC positive nel muscolo scheletrico dei criceti distrofici, insieme a una riduzione dell’espressione di β-MHC, contribuendo così alla conservazione di un α/β più fisiologico. Rapporto MHC. Inoltre, nello stesso studio, è stato anche dimostrato che l’integrazione di ALA alimentare previene l’accumulo aberrante citoplasmatico di proteine di membrana chiave nei muscoli adduttori dei criceti distrofici, inclusa la caveolina-3, una proteina coinvolta nella regolazione dell’adesione cellulare e della riparazione della membrana, nonché essendo implicato nel controllo della differenziazione muscolare e della segnalazione indotta dall’insulina. [61 , 62] Infatti, dato che le aberrazioni nella funzione e/o localizzazione della caveolina-3 sono state associate a vari fenotipi di malattie del muscolo scheletrico, [63 , 64 , 65 , 66 , 67] è plausibile che gli acidi grassi e/o i loro derivati lipidici possano influenzare la proliferazione delle cellule satellite e/o la differenziazione muscolare, almeno in parte, alterando la funzione e/o la localizzazione subcellulare delle isoforme della caveolina, nonché di altri componenti chiave della membrana strutturale.
Gli effetti degli acidi grassi sulla massa muscolare e sulla differenziazione possono essere mediati anche da una serie di metaboliti lipidici derivati. Ad esempio, è stato dimostrato che la capacità dell’acido arachidonico PUFA (C20; 4n-6) di aumentare le dimensioni, il contenuto mionucleare e il contenuto proteico dei miotubi C2C12 è mediata dall’attività della cicloossigenasi-2 (COX-2), implicando dipendenza da sintesi delle prostaglandine a valle. [68] In accordo con ciò, è stato riportato che la crescita indotta dall’acido arachidonico dei miociti C2C12 coincide con l’aumento della secrezione degli eicosanoidi PGF2α e PGE. [68]È interessante notare che diversi studi hanno anche documentato il ruolo positivo che le prostaglandine svolgono nel promuovere eventi precoci sulla superficie cellulare, inclusa l’adesione cellula-cellula, che successivamente mediano la fusione dei mioblasti nei miotubi. [69 , 70] Infatti, importanti studi di follow-up possono comportare la determinazione dell’esatta identità dei bersagli molecolari attraverso i quali le prostaglandine mediano le loro azioni, ad esempio agendo sui recettori prostanoidi legati alla proteina G (es. EP1). [69 , 71] Al contrario, è stato recentemente riportato che un altro metabolita lipidico derivato dall’acido arachidonico noto come 2-arachidonoilglicerolo (2-AG), un ligando lipidico endogeno chiave del sistema endocannabinoide, inibisce la differenziazione delle cellule satellite primarie umane e dei mioblasti murini C2C12 prendendo di mira il recettore-G ‐ 1 dei cannabinoidi accoppiati a proteine e sua successiva inibizione dei canali Kv7.4. [72] Pertanto, non si può escludere il possibile coinvolgimento di tali intermedi lipidici nella regolazione del catabolismo muscolare in risposta a determinate condizioni patologiche o alimentari.
L’aumento dell’adiposità osservato nell’invecchiamento umano è stato collegato a risposte sintetiche proteiche muscolari alterate negli individui anziani. [73] Pertanto, è ipotizzabile che i cambiamenti nei livelli e/o nella composizione dei lipidi possano contribuire all’atrofia muscolare in queste condizioni ma non limitatamente ad esse. A sostegno di questa idea, ci sono prove che suggeriscono che la modifica della composizione alimentare può avere un impatto sulla massa muscolare e/o sulla sua funzione negli esseri umani. Ad esempio, uno studio di McGlory e collaboratori ha riportato che il consumo di olio di pesce ha aumentato il contenuto di PUFA omega-3 del muscolo ( Vastus lateralis) in individui maschi sani, che ha coinciso con un’elevata espressione di proteine di segnalazione anabolizzanti incluso mTOR. [74] Inoltre, è stato riportato che l’inclusione di MUFA e PUFA nella dieta riduce l’espressione di geni lipogenici nel muscolo scheletrico di soggetti insulino-resistenti, in concomitanza con ridotti tassi di sintesi frazionaria di DAG intramuscolare e triacilgliceroli. [75] In particolare, queste risposte benefiche possono essere collegate a miglioramenti nella sensibilità all’insulina veicolati dall’integrazione alimentare di PUFA omega-3 negli esseri umani. [76 , 77 , 78]
E’ stato riportato che l’integrazione alimentare con acidi grassi insaturi può anche agire per migliorare le prestazioni fisiche e/o aumentare gli effetti metabolici benefici associati all’esercizio, in particolare in individui sedentari o non allenati . [79] A sostegno di questa idea, è stato dimostrato che un basso apporto alimentare di olio di tonno promuove la resistenza all’affaticamento muscolare nei ratti, in concomitanza con un aumento selettivo del contenuto di fosfolipidi della membrana e DHA all’interno del muscolo gastrocnemio. [80 , 81] Pertanto, un approccio più integrato che coinvolga modifiche al consumo di grassi nella dieta e un aumento dell’esercizio fisico può fornire una strategia più efficace per alleviare gli effetti deleteri associati all’atrofia muscolare. Ma stiamo comunque parlando di effetti apprezzabili su soggetti sedentari e non allenati, non su culturisti intermedi o avanzati.
Tirando le somme di quanto fino qui esposto, c’è un certo interesse giustificato sul ruolo importante che gli acidi grassi e/oi loro derivati lipidici possono svolgere nella modulazione della massa muscolare e della sua funzione. Nel complesso, le prove presentate indicano che gli acidi grassi saturi agiscono per trasmettere effetti dannosi sulla funzione muscolare, ad esempio alterando o riducendo la sintesi proteica e accentuando il catabolismo. Al contrario, è stato dimostrato che diversi acidi grassi insaturi contrastano molte delle azioni pro-cataboliche associate alla assunzione di acidi grassi saturi. Tuttavia, sarà necessari ulteriori studi per delineare le vie e i processi alla base dell’atrofia muscolare indotta da acidi grassi, nonché quelli che mediano miglioramenti nella funzione muscolare in risposta alla fornitura di acidi grassi insaturi (cioè aumento della sintesi proteica, ridotta atrofia, miglioramento della funzione metabolica). A tal fine, strategie volte ad alterare il contenuto e/o la composizione lipidica intramuscolare in quelle condizioni che possono favorire l’atrofia muscolare (es. aumento dell’obesità, invecchiamento, inattività fisica), ad esempio sopprimendo l’accumulo di mediatori lipidici come le Ceramidi (es. de novosintesi di ceramide) o prostaglandine (ad es. inibendo l’attività della COX-2), possono fornire informazioni utili sul ruolo che diverse classi di lipidi svolgono nella modulazione della massa e della funzione muscolare. È importante sottolineare che tale lavoro potrebbe implicare l’uso di modelli animali pertinenti o soggetti umani che richiederebbero di prendere in considerazione fattori come il background genetico, la composizione della dieta e l’apporto calorico. Inoltre, questi studi possono anche comportare la determinazione di potenziali alterazioni indotte dai lipidi dell’architettura muscolare e della composizione del tipo di fibra che possono influenzare la forza muscolare, nonché il monitoraggio dei cambiamenti nella segnalazione intramuscolare e nei metaboliti all’interno di specifici tipi di fibre muscolari. Insieme a questo, ulteriori lavori che esplorano il ruolo degli acidi grassi e degli intermedi lipidici nella regolazione della proliferazione, sarebbe necessario eseguire la differenziazione e/o la funzione delle cellule satellite derivate da muscoli umani e dei miotubi primari al fine di effettuare confronti appropriati con i dati ottenuti in altri modelli sperimentali (ad es. miotubi C2C12), che hanno dimostrato di mostrare differenze funzionali (ad es. grado di maturazione).[82] Nel complesso , i dati ottenuti da tali studi possono portare allo sviluppo di nuove strategie terapeutiche per contrastare l’atrofia muscolare e/o migliorare la capacità rigenerativa a seguito di lesioni o malattie.
Torniamo alla pratica: tesi, antitesi e sintesi del “Bulk High Fat”.
Quindi, cosa concludere da tutto ciò che ho esposto proveniente dalla letteratura scientifica se parliamo di Bodybuilding e alimentazione “High Fat” in ipercalorica? Quanto segue:
- La cosa certa, e risaputa in tutti i contesti dietetici, è quella di preoccuparsi di ridurre l’assunzione di acidi grassi saturi a circa e non oltre il 10% del totale calorico giornaliero (soprattutto l’Acido Palmitico, Miristico e Laurico);
- Concentrare l’assunzione di acidi grassi utilizzando fonti principalmente di Omega-3 (con particolare attenzione ad assumere EPA e DHA in un range tra 1 e 5g/die)
- Assumere una buona dose di GLA (Omega-6) la quale può portare ad una migliore risposta nella biosintesi di prostaglandine anti-infiammatorie;
- Possibilità di assumere Acido Arachidonico (Omega-6) in un range di dosaggio tra i 100mg ed 1.5g/die (quest’ultimo dosaggio va calibrato con un apporto di EPA/DHA pari ad un minimo di 2,5g/die) che può portare ad un miglioramento della biosintesi di prostaglandine anti-infiammatorie e con attività anabolizzante (vedi, per esempio, la PGF2-α responsabile con la PGE2 della regolazione della proteolisi e della sintesi proteica; così come della proliferazione, differenziazione e fusione delle celle satellite);
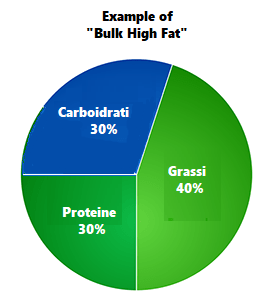
- Uno schema di ripartizionamento dei macronutrienti potrebbe essere il seguente: 30-40% di Grassi, 30-20% di Carboidrati e 40-30% di Proteine.
Partendo da questi punti si hanno delle “linee guida” finalizzate alla qualità degli acidi grassi e alla quantità media percentuale.
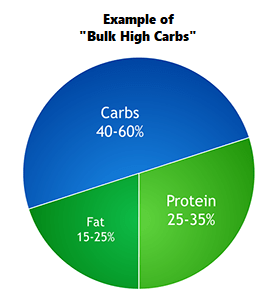
Ora, mettiamo il caso che un soggetto per caratteristiche genetiche non riesca a gestire grosse quantità di glucidi e abbia un buon metabolismo lipidico sul quale poter puntare per eventuali surplus calorici.
Il soggetto in questione si trova bene con un quantitativo di CHO pari a 3g/Kg, e pesando 80Kg la suo quota glucidica giornaliera ammonta a 240g. L’assunzione proteica giornaliera è pari a 2,5g/Kg, per un totale di 200g.
Il suo TDEE di mantenimento è di circa 2.500Kcal. Punta ad un surplus calorico giornaliero di 500Kcal (obbiettivo 3000Kcal/die). Dal momento che sa già quanti CHO e proteine dovrà assumere e a quante calorie ammontano (1760Kcal totali da CHO e Pro) il restante lo andrà ad assumere dai Grassi (1240Kcal = circa 138g di Fat).
Totale macronutrienti del soggetto:
- Carboidrati: 240g
- Proteine: 200g
- Grassi: 138g (di cui il 10% di Saturi ed il restante 90% di insaturi, polinsaturi e monoinsaturi; presenza di 5g/die di EPA e DHA, 300mg/die di GLA e 1.5g/die di Acido Arachidonico per 8-12 settimane).
Il soggetto in questione seguirà questo schema per circa 4 settimana senza effettuare alcuna giornata con un aumento dei CHO e una riduzione della componente proteica e lipidica. In base alle risposte ottenute rimodulerà a bisogno il suo schema alimentare.
Ecco, questo è un banalissimo esempio di come una dieta “Bulk High Fat” possa essere per lo meno impostata in modo logico, anche per quanto riguarda il tipo ipotetico di soggetto menzionato. Nonostante ciò, e vi parlo da persona che ha analizzato i risultati di svariati schemi similari a questo, un vantaggio vero e proprio sulla qualità dell’aumento di peso non è praticamente mai emerso, o per lo meno mai in modo sufficientemente apprezzabile.
A questo punto ci sarebbe anche da riportare quanto affermato in uno studio dal Dr. Jose Antonio, PhD, ricercatore della ISSN, che ha affermato come non vi siano aumenti maggiori di grasso corporeo tra una dieta ipercalorica “High Fat” e una “High Carbs” dopo circa 14 giorni.[https://italia-podcast.it/] Questa affermazione va comunque presa come una ipotesi nata da osservazione di alcuni studi. Sebbene tra la teoria e la pratica spesso c’è differenza, quando si parla di surplus calorico e aumento del grasso corporeo sappiamo tutti quanto gli acidi grassi alimentari vengano facilmente stoccati negli adipociti sotto forma di trigliceridi di deposito quando in surplus calorico e ingeriti in quantità significative con carboidrati. Questi ultimi, o meglio il glucosio, in eccesso, in un soggetto in salute vengono in gran parte dissipati in calore e solo un 5-10% è coinvolto nei processi di De Novo Lipogenesi (DNL).
Alcuni sostenitori delle diete “High Fat” in “Bulk” affermano che tali strategie alimentari portino ad un miglioramento della “durezza muscolare”, anche grazie ad un basso stimolo insulinico e consequenziale riduzione della ritenzione idrica, e della pienezza dei ventri data dai Trigliceridi intra-muscolari. Peccato che, Insulina o meno, gli acidi grassi in eccesso vengono captati e depositati negli adipociti per intervento, tra l’altro, della Proteina Stimolante l’Acilazione (ASP), la quale ha azione insulino-indipendente. I Trigliceridi intra-muscolari (IMTG) danno si un effetto di pienezza muscolare dal momento che legano in piccola parte acqua, ma parliamo di un effetto additivo al ben maggiore volume dato dal glicogeno muscolare e apprezzabile a “bf” basse, da pre-contest per intenderci.
Come molti di voi già sapranno, la sintesi proteica aumenta principalmente:
- In seguito ad adattamenti in risposta a sedute di allenamento contro-resistenza correttamente svolte generando tensione meccanica, stress metabolico e danno muscolare;
- Mangiando sufficienti calorie;
- Assumendo un quantitativo adeguato di proteine (media 1,5-2,5g/Kg).
Raggiungere il primo punto, però, diventa molto più difficile se si consuma una quota glucidica bassa. Le diete ricche di grassi possono fornire molte calorie e proteine, ma risultano per la stragrande maggioranza dei soggetti disfunzionali alle prestazioni in sala pesi. Ricordo, per l’ennesima volta, che un bodybuilder è un atleta che sfrutta primariamente il metabolismo glucidico al fine della massima prestazione essendo praticante di uno sport anaerobico alattacido e lattacido.
Durante l’esercizio intenso, i muscoli si basano principalmente sul metabolismo glucidico. E come certamente saprete, il glicogeno è un polimero di glucosio immagazzinato nel muscolo-scheletrico e nel fegato, ed è scomposto nelle sue unità di glucosio e utilizzato come substrato energetico durante l’esercizio nel distretto muscolare di deposito oppure venendo rilasciato nel flusso ematico dal deposito epatico.

Le richieste di carboidrati nella dieta variano da soggetto a soggetto, ma un culturista che si allena seriamente richiede in media una consistente quantità di glucidi per mantenere le sue riserve di glicogeno e sostenere una buona prestazione, motivo per cui le diete ricche di grassi e povere di carboidrati riducono significativamente i livelli di questo polimero. Ed i problemi annessi emergono soprattutto quando ci si allena con pesi consistenti.
Fare solo 6-9 serie può ridurre i livelli di glicogeno muscolare di circa il 40% [83] e, se ci si allena con livelli già bassi, è ragionevole presumere che si farà fatica a sollevare più peso.
Ma cosa mostrano gli studi sugli atleti? La maggior parte degli studi dimostrano che le diete ricche di grassi e povere di carboidrati riducono le prestazioni atletiche [84, 85, 86] mentre altri affermano che non vi è alcuna differenza [87]e persino alcuni che mostrano un aumento delle prestazioni.[88] Tuttavia, ci sono altri motivi per pensare che le diete ad alto contenuto di carboidrati potrebbero essere superiori per l’aumento della massa muscolare.
La ricerca mostra che la disponibilità di glicogeno influenza direttamente la sintesi proteica ei tassi di degradazione.[89] In poche parole: le diete ricche di grassi e povere di carboidrati comportano livelli inferiori di sintesi proteica rispetto a quelle ad alto contenuto di carboidrati.
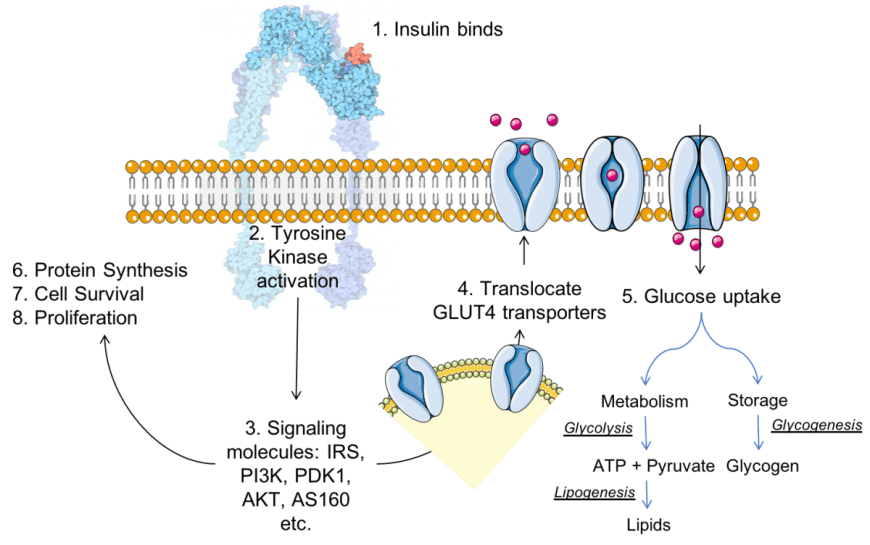
Un altro modo in cui i carboidrati influenzano favorevolmente l’equilibrio proteico muscolare ha a che fare con la produzione di Insulina.
L’insulina è un ormone peptidico rilasciato dal pancreas che agevola l’assorbimento dei nutrienti dal sangue alle cellule. Possiede, in fisiologia, proprietà prettamente anticataboliche, il che significa che quando i livelli di Insulina sono elevati, viene soppressa la lisi delle proteine muscolari.[90]
Ora, poiché la produzione di Insulina è stimolata ingerendo cibo, e mangiando carboidrati in particolare, ma anche proteine con stimoli differenti dati dalla composizione amminoacidica, non sorprende che le persone che seguono una dieta ricca di carboidrati abbiano generalmente livelli ottimali di Insulina rispetto alle persone che seguono una dieta povera di carboidrati e ricca di grassi.
I carboidrati sono principalmente energetici e se non si è molto attivi, non si necessita di loro quantità elevate. Tuttavia, livelli ottimali di insulina (e di sensibilità insulinica) sono altamente desiderabili se si sta cercando di costruire massa muscolare, semplicemente perché si viene a creare un ambiente più anabolico in cui i muscoli possono vedere agevolata la loro crescita. E questa non è solo teoria.
La ricerca condotta dagli scienziati della Ball State University ha scoperto che bassi livelli di glicogeno muscolare (che è inevitabile con una dieta povera di carboidrati e ricca di grassi) compromettono la segnalazione cellulare post-allenamento relativa alla crescita muscolare.[91]
Un altro studio condotto da ricercatori dell’Università della Carolina del Nord ha scoperto che, se combinata con l’esercizio quotidiano, una dieta povera di carboidrati e ricca di grassi aumenta i livelli di Cortisolo a riposo e diminuisce i livelli di Testosterone libero.[92] Sicuramente una situazione compromettente soprattutto per un “Natural”. Quando si tratta di costruire massa muscolare, ciò che si vuole sono bassi livelli di Cortisolo a riposo e alti livelli di Testosterone libero, l’esatto opposto di ciò che porta una dieta a basso contenuto di carboidrati.
Quanto detto aiuta a spiegare i risultati di altri studi sulla questione dei carboidrati e della composizione corporea e delle prestazioni.
Ad esempio, uno studio condotto da ricercatori dell’Università del Rhode Island ha esaminato come l’assunzione di carboidrati a basso e ad alto contenuto di carboidrati influenzasse il danno muscolare indotto dall’esercizio, il recupero della forza e il metabolismo proteico di tutto il corpo dopo un intenso allenamento. Quello che hanno scoperto è che i soggetti con una dieta a basso contenuto di carboidrati hanno perso più forza, si sono ripresi più lentamente e hanno mostrato livelli più bassi di sintesi proteica. Vale anche la pena notare che il gruppo “a basso contenuto di carboidrati” (e quindi ad alto contenuto di grassi) non era poi così “Low Carbs”. Stavano ingerendo circa 220g di carboidrati al giorno contro i 350g assunti dal gruppo con dieta ad alto contenuto di carboidrati.[93] E questi effetti diventano ancora più pronunciati man mano che l’assunzione di carboidrati diminuisce.
Ancora un altro studio degno di nota è stato condotto dai ricercatori della McMaster University, che ha confrontato la dieta ad alto e basso contenuto di carboidrati con soggetti che eseguono allenamenti quotidiani per le gambe. I soggetti che seguono una dieta a basso contenuto di carboidrati (30% delle calorie giornaliere) hanno mostrato tassi più elevati di degradazione proteica e tassi più bassi di sintesi proteica rispetto ai soggetti a dieta ricca di carboidrati (60% delle calorie giornaliere), con conseguente minore crescita muscolare complessiva.[94]
Conclusioni e ultime indicazioni:
La linea di fondo è questa: Alcuni studi dimostrano che le diete ricche di grassi possono funzionare per la costruzione muscolare, ma un approccio più ideale e redditizio sembra rimanere quello di seguire una dieta ricca di carboidrati secondo capacità individuali.
I dati empirici ci suggeriscono mediamente la stessa medesima cosa, senza però cadere nell’errore di valutare le innumerevoli variabili soggettive in un unico macro-gruppo di risposta positiva ai regimi prevalentemente glucidici.
Questo significa, in poche parole, che la soggettività genetica non va mai sottovalutata o peggio rinnegata per via di futili credenze pseudoscientifiche o convinzioni personali.
Ciò non toglie, però, che i regimi ipercalorici “High Fat” non portano a sostanziali benefici se non ad un ristretto numero di individui caratterizzati da ipotetiche alterazioni metaboliche rispetto alla media della popolazione. Ma anche in questi casi il “tetto favorevole” del “High Fat” si attesta mediamente al 40% delle calorie totali giornaliere con un totale glucidico del 30-25%.
Ciò che dobbiamo ricordare è che i test alimentari soggettivi con modulazioni macro-caloriche sono utili al fine di attestare una quota singolarmente funzionale per ogni macronutriente. Solo in questo modo, e con una valutazione oggettiva, possiamo capire fin dove possiamo spingerci in quanto a quantità di macronutrienti al fine di ottenerne il massimo dei vantaggi prestativi ed estetici. Questo può significare differenze non da poco in un piano ipercalorico tra Grassi e Carboidrati.

Personalmente, ritengo che il range della quota lipidica dovrebbe coprire un arco di grammatura da 0,6 a 2g/Kg. Sempre su base soggettiva e rapporto funzionale tra carboidrati e grassi.
In regimi ipercalorici, con l’obbiettivo di non perdere la flessibilità metabolica, si possono utilizzare, oltre alla “C:G ratio” di Ludovico Lemme, uno schema d’esempio di rapporto come segue:
- CHO 4-5g/Kg= Fat 0,6g/Kg
- CHO 5-6g/Kg= Fat 0,8g/Kg
- CHO 6-7g/Kg= Fat 1g/Kg
- CHO 7-8g/Kg= Fat 1,5/Kg
- CHO 8-10g/Kg= Fat 2g/Kg
E se volessi tarare il quantitativo lipidico per un regime “Bulk High Fat”? Le linee indicative sarebbero:
- CHO 3-4g/Kg= Fat 1-1,2g/Kg
- CHO 2.5-3g/Kg= Fat 1,5-1,6g/Kg
- CHO 2-2.5g/Kg= Fat 1,8-2g/Kg
- CHO 0,5-2g/Kg= Fat 2-2,5g/Kg
Queste sono semplici indicazioni generali estrapolate da dati raccolti empiricamente, nulla di scolpito nella roccia o inconfutabilmente dimostrato.
Prima di chiudere con il presente articolo, è logico che vi dica di fare attenzioni alle fonti lipidiche che assumete o che andrete ad assumere (cosa già detta in precedenza). Nessuno vi sta dicendo di evitare la carne rossa (tagli magri) ma di concentrarvi soprattutto su fonti lipidiche quali tuorlo d’uovo, Olio Extravergine di Oliva, EPA, DHA e MCT (controllandone la composizione ed evitando l’olio di Cocco ricco di Acido Laurico). Fonti proteiche quali salmone selvatico, sgombro, pesce spada e carni da allevamenti “Grass Feed” sono pienamente consigliate, se alla propria portata economica.
Gabriel Bellizzi
Riferimenti:
- Dr. Mauro Di Pasquale, bodybuilding.com, accessed February 21, 2007. His articles and books have been translated into Italian language and published in Italy by Sandro Ciccarelli Olympian’s News magazine.
- Bryner RW, Woodworth‐Hobbs ME, Williamson DL, Alway SE. Docosahexaenoic Acid protects muscle cells from palmitate‐induced atrophy. ISRN Obes 2012;2012:647348.
- Woodworth‐Hobbs ME, Hudson MB, Rahnert JA, Zheng B, Franch HA, Price SR. Docosahexaenoic acid prevents palmitate‐induced activation of proteolytic systems in C2C12 myotubes. J Nutr Biochem 2014;25:868–74.
- Graves E, Hitt A, Pariza MW, Cook ME, McCarthy DO. Conjugated linoleic acid preserves gastrocnemius muscle mass in mice bearing the colon‐26 adenocarcinoma. Res Nurs Health 2005;28:48–55.
- Langen RC, Schols AM, Kelders MC, van der Velden JL, Wouters EF, Janssen‐Heininger YM. Muscle wasting and impaired muscle regeneration in a murine model of chronic pulmonary inflammation. Am J Respir Cell Mol Biol 2006;35:689–96.
- Whitehouse AS, Smith HJ, Drake JL, Tisdale MJ. Mechanism of attenuation of skeletal muscle protein catabolism in cancer cachexia by eicosapentaenoic acid. Cancer Res 2001;61:3604–9.
- Castillero E, Martin AI, Lopez‐Menduina M, Villanua MA, Lopez‐Calderon A. Eicosapentaenoic acid attenuates arthritis‐induced muscle wasting acting on atrogin‐1 and on myogenic regulatory factors. Am J Physiol Regul Integr Comp Physiol 2009;297:R1322–31.
- Fiaccavento R, Carotenuto F, Vecchini A, Binaglia L, Forte G, Capucci E, et al. An omega‐3 fatty acid‐enriched diet prevents skeletal muscle lesions in a hamster model of dystrophy. Am J Pathol 2010;177:2176–84.
- Briolay A, Jaafar R, Nemoz G, Bessueille L. Myogenic differentiation and lipid‐raft composition of L6 skeletal muscle cells are modulated by PUFAs. Biochim Biophys Acta 2013;1828:602–13.
- Watson ML, Coghlan M, Hundal HS. Modulating serine palmitoyl transferase (SPT) expression and activity unveils a crucial role in lipid‐induced insulin resistance in rat skeletal muscle cells. Biochem J 2009;417:791–801.
- . Yuzefovych L, Wilson G, Rachek L. Different effects of oleate vs. palmitate on mitochondrial function, apoptosis, and insulin signaling in L6 skeletal muscle cells: role of oxidative stress. Am J Physiol Endocrinol Metab 2010;299:E1096–105.
- Mahfouz R, Khoury R, Blachnio‐Zabielska A, Turban S, Loiseau N, Lipina C, et al. Characterising the inhibitory actions of ceramide upon insulin signaling in different skeletal muscle cell models: a mechanistic insight. PLoS One 2014;9:e101865.
- De Larichaudy J, Zufferli A, Serra F, Isidori AM, Naro F, Dessalle K, et al. TNF‐alpha‐ and tumor‐induced skeletal muscle atrophy involves sphingolipid metabolism. Skelet Muscle 2012;2:2.
- Mebarek S, Komati H, Naro F, Zeiller C, Alvisi M, Lagarde M, et al. Inhibition of de novo ceramide synthesis upregulates phospholipase D and enhances myogenic differentiation. J Cell Sci 2007;120:407–16.
- Turpin SM, Ryall JG, Southgate R, Darby I, Hevener AL, Febbraio MA, et al. Examination of ‘lipotoxicity’ in skeletal muscle of high‐fat fed and ob/ob mice. J Physiol 2009;587:1593–605.
- Williamson DL, Dungan CM, Jadhav KS. High lipid concentrations regulate skeletal muscle REDD1. FASEB J 2013;27:Supplement 942.1.
- Gordon BS, Williamson DL, Lang CH, Jefferson LS, Kimball SR. Nutrient‐induced stimulation of protein synthesis in mouse skeletal muscle is limited by the mTORC1 repressor REDD1. J Nutr 2015;145:708–13.
- Leskawa KC, Erwin RE, Buse PE, Hogan EL. Glycosphingolipid biosynthesis during myogenesis of rat L6 cells in vitro. Mol Cell Biochem 1988;83:47–54.
- Anastasia L, Papini N, Colazzo F, Palazzolo G, Tringali C, Dileo L, et al. NEU3 sialidase strictly modulates GM3 levels in skeletal myoblasts C2C12 thus favoring their differentiation and protecting them from apoptosis. J Biol Chem 2008;283:36265–71.
- Papini N, Anastasia L, Tringali C, Dileo L, Carubelli I, Sampaolesi M, et al. MmNEU3 sialidase over‐expression in C2C12 myoblasts delays differentiation and induces hypertrophic myotube formation. J Cell Biochem 2012;113:2967–78.
- Lipina C, Hundal HS. Ganglioside GM3 as a gatekeeper of obesity‐associated insulin resistance: evidence and mechanisms. FEBS Lett 2015;589:3221–7.
- Gangoiti P, Bernacchioni C, Donati C, Cencetti F, Ouro A, Gomez‐Munoz A, et al. Ceramide 1‐phosphate stimulates proliferation of C2C12 myoblasts. Biochimie 2012;94:597–607.
- Szendroedi J, Yoshimura T, Phielix E, Koliaki C, Marcucci M, Zhang D, et al. Role of diacylglycerol activation of PKCtheta in lipid‐induced muscle insulin resistance in humans. Proc Natl Acad Sci U S A 2014;111:9597–602.
- You JS, Lincoln HC, Kim CR, Frey JW, Goodman CA, Zhong XP, et al. The role of diacylglycerol kinase zeta and phosphatidic acid in the mechanical activation of mammalian target of rapamycin (mTOR) signaling and skeletal muscle hypertrophy. J Biol Chem 2014;289:1551–63.
- Shad BJ, Smeuninx B, Atherton PJ, Breen L. The mechanistic and ergogenic effects of phosphatidic acid in skeletal muscle. Appl Physiol Nutr Metab 2015;40:1233–41.
- Bilim O, Takeishi Y, Kitahara T, Arimoto T, Niizeki T, Sasaki T, et al. Diacylglycerol kinase zeta inhibits myocardial atrophy and restores cardiac dysfunction in streptozotocin‐induced diabetes mellitus. Cardiovasc Diabetol 2008;7:2.
- Haemmerle G, Zimmermann R, Hayn M, Theussl C, Waeg G, Wagner E, et al. Hormone‐sensitive lipase deficiency in mice causes diglyceride accumulation in adipose tissue, muscle, and testis. J Biol Chem 2002;277:4806–15.
- Marignani PA, Epand RM, Sebaldt RJ. Acyl chain dependence of diacylglycerol activation of protein kinase C activity in vitro. Biochem Biophys Res Commun 1996;225:469–73.
- Eichmann TO, Kumari M, Haas JT, Farese RV Jr, Zimmermann R, Lass A, et al. Studies on the substrate and stereo/regioselectivity of adipose triglyceride lipase, hormone‐sensitive lipase, and diacylglycerol‐O‐acyltransferases. J Biol Chem 2012;287:41446–57.
- Macrae K, Stretton C, Lipina C, Blachnio‐Zabielska A, Baranowski M, Gorski J, et al. Defining the role of DAG, mitochondrial function, and lipid deposition in palmitate‐induced proinflammatory signaling and its counter‐modulation by palmitoleate. J Lipid Res 2013;54:2366–78.
- Rando RR, Young N. The stereospecific activation of protein kinase C. Biochem Biophys Res Commun 1984;122:818–23.
- Badin PM, Louche K, Mairal A, Liebisch G, Schmitz G, Rustan AC, et al. Altered skeletal muscle lipase expression and activity contribute to insulin resistance in humans. Diabetes 2011;60:1734–42.
- Hyde R, Hajduch E, Powell DJ, Taylor PM, Hundal HS. Ceramide down‐regulates System A amino acid transport and protein synthesis in rat skeletal muscle cells. FASEB J 2005;19:461–3.
- Guenther GG, Peralta ER, Rosales KR, Wong SY, Siskind LJ, Edinger AL. Ceramide starves cells to death by downregulating nutrient transporter proteins. Proc Natl Acad Sci U S A 2008;105:17402–7.
- Nardi F, Hoffmann TM, Stretton C, Cwiklinski E, Taylor PM, Hundal HS. Proteasomal modulation of cellular SNAT2 (SLC38A2) abundance and function by unsaturated fatty acid availability. J Biol Chem 2015;290:8173–84.
- Li F, Duan Y, Li Y, Tang Y, Geng M, Oladele OA, et al. Effects of dietary n‐6:n‐3 PUFA ratio on fatty acid composition, free amino acid profile and gene expression of transporters in finishing pigs. Br J Nutr 2015;113:739–48.
- Coll T, Eyre E, Rodriguez‐Calvo R, Palomer X, Sanchez RM, Merlos M, et al. Oleate reverses palmitate‐induced insulin resistance and inflammation in skeletal muscle cells. J Biol Chem 2008;283:11107–16.
- Salvado L, Coll T, Gomez‐Foix AM, Salmeron E, Barroso E, Palomer X, et al. Oleate prevents saturated‐fatty‐acid‐induced ER stress, inflammation and insulin resistance in skeletal muscle cells through an AMPK‐dependent mechanism. Diabetologia 2013;56:1372–82.
- Wu CL, Cornwell EW, Jackman RW, Kandarian SC. NF‐kappaB but not FoxO sites in the MuRF1 promoter are required for transcriptional activation in disuse muscle atrophy. Am J Physiol Cell Physiol 2014;306:C762–7.
- Rivas DA, McDonald DJ, Rice NP, Haran PH, Dolnikowski GG, Fielding RA. Diminished anabolic signaling response to insulin induced by intramuscular lipid accumulation is associated with inflammation in aging but not obesity. Am J Physiol Regul Integr Comp Physiol 2016;310:R561–9.
- Liu Y, Chen F, Odle J, Lin X, Zhu H, Shi H, et al. Fish oil increases muscle protein mass and modulates Akt/FOXO, TLR4, and NOD signaling in weanling piglets after lipopolysaccharide challenge. J Nutr 2013;143:1331–9.
- Doyle A, Zhang G, Abdel Fattah EA, Eissa NT, Li YP. Toll‐like receptor 4 mediates lipopolysaccharide‐induced muscle catabolism via coordinate activation of ubiquitin–proteasome and autophagy–lysosome pathways. FASEB J 2011;25:99–110.
- Mittal A, Bhatnagar S, Kumar A, Lach‐Trifilieff E, Wauters S, Li H, et al. The TWEAK‐Fn14 system is a critical regulator of denervation‐induced skeletal muscle atrophy in mice. J Cell Biol 2010;188:833–49.
- Finlin BS, Varma V, Nolen GT, Dube J, Starnes CP, Rasouli N, et al. DHA reduces the atrophy‐associated Fn14 protein in differentiated myotubes during coculture with macrophages. J Nutr Biochem 2012;23:885–91.
- Jang YC, Lustgarten MS, Liu Y, Muller FL, Bhattacharya A, Liang H, et al. Increased superoxide in vivo accelerates age‐associated muscle atrophy through mitochondrial dysfunction and neuromuscular junction degeneration. FASEB J 2010;24:1376–90.
- Gouspillou G, Sgarioto N, Kapchinsky S, Purves‐Smith F, Norris B, Pion CH, et al. Increased sensitivity to mitochondrial permeability transition and myonuclear translocation of endonuclease G in atrophied muscle of physically active older humans. FASEB J 2014;28:1621–33.
- Muoio DM, Way JM, Tanner CJ, Winegar DA, Kliewer SA, Houmard JA, et al. Peroxisome proliferator‐activated receptor‐alpha regulates fatty acid utilization in primary human skeletal muscle cells. Diabetes 2002;51:901–9.
- Tardif N, Salles J, Landrier JF, Mothe‐Satney I, Guillet C, Boue‐Vaysse C, et al. Oleate‐enriched diet improves insulin sensitivity and restores muscle protein synthesis in old rats. Clin Nutr 2011;30:799–806.
- Park SY, Kim MH, Ahn JH, Lee SJ, Lee JH, Eum WS, et al. The stimulatory effect of essential fatty acids on glucose uptake involves both Akt and AMPK activation in C2C12 skeletal muscle cells. Korean J Physiol Pharmacol: Off J Korean Phys Soc Korean Soc Pharm 2014;18:255–61.
- Wang Y, Lin QW, Zheng PP, Zhang JS, Huang FR. DHA inhibits protein degradation more efficiently than EPA by regulating the PPARgamma/NFkappaB pathway in C2C12 myotubes. BioMed Res Int 2013;2013:318981.
- Lanza IR, Blachnio‐Zabielska A, Johnson ML, Schimke JM, Jakaitis DR, Lebrasseur NK, et al. Influence of fish oil on skeletal muscle mitochondrial energetics and lipid metabolites during high‐fat diet. Am J Physiol Endocrinol Metab 2013;304:E1391–403.
- Lim JH, Gerhart‐Hines Z, Dominy JE, Lee Y, Kim S, Tabata M, et al. Oleic acid stimulates complete oxidation of fatty acids through protein kinase A‐dependent activation of SIRT1‐PGC1alpha complex. Nutr Metab 2013;288:7117–26.
- Mizunoya W, Iwamoto Y, Shirouchi B, Sato M, Komiya Y, Razin FR, et al. Dietary fat influences the expression of contractile and metabolic genes in rat skeletal muscle. PLoS One 2013;8:e80152.
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011;147:728–41.
- Penna F, Costamagna D, Pin F, Camperi A, Fanzani A, Chiarpotto EM, et al. Autophagic degradation contributes to muscle wasting in cancer cachexia. Am J Pathol 2013;182:1367–78.
- O’Leary MF, Vainshtein A, Carter HN, Zhang Y, Hood DA. Denervation‐induced mitochondrial dysfunction and autophagy in skeletal muscle of apoptosis‐deficient animals. Am J Physiol Cell Physiol 2012;303:C447–54.
- Wenz T, Rossi SG, Rotundo RL, Spiegelman BM, Moraes CT. Increased muscle PGC‐1alpha expression protects from sarcopenia and metabolic disease during aging. Proc Natl Acad Sci U S A 2009;106:20405–10.
- McFarland DC, Velleman SG, Pesall JE, Coy CS. Effect of lipids on avian satellite cell proliferation, differentiation and heparan sulfate proteoglycan expression. Comp Biochem Physiol A Mol Integr Physiol 2011;159:188–95.
- Peng Y, Zheng Y, Zhang Y, Zhao J, Chang F, Lu T, et al. Different effects of omega‐3 fatty acids on the cell cycle in C2C12 myoblast proliferation. Mol Cell Biochem 2012;367:165–73.
- Schevzov G, Kee AJ, Wang B, Sequeira VB, Hook J, Coombes JD, et al. Regulation of cell proliferation by ERK and signal‐dependent nuclear translocation of ERK is dependent on Tm5NM1‐containing actin filaments. Mol Biol Cell 2015;26:2475–90, doi:10.1091/mbc.E14-10-1453.
- Parton RG, Way M, Zorzi N, Stang E. Caveolin‐3 associates with developing T‐tubules during muscle differentiation. J Cell Biol 1997;136:137–54.
- Oshikawa J, Otsu K, Toya Y, Tsunematsu T, Hankins R, Kawabe J, et al. Insulin resistance in skeletal muscles of caveolin‐3‐null mice. Proc Natl Acad Sci U S A 2004;101:12670–5.
- Galbiati F, Volonte D, Minetti C, Chu JB, Lisanti MP. Phenotypic behavior of caveolin‐3 mutations that cause autosomal dominant limb girdle muscular dystrophy (LGMD‐1C). Retention of LGMD‐1C caveolin‐3 mutants within the golgi complex. J Biol Chem 1999;274:25632–41.
- Minetti C, Bado M, Broda P, Sotgia F, Bruno C, Galbiati F, et al. Impairment of caveolae formation and T‐system disorganization in human muscular dystrophy with caveolin‐3 deficiency. Am J Pathol 2002;160:265–70.
- Park DS, Woodman SE, Schubert W, Cohen AW, Frank PG, Chandra M, et al. Caveolin‐1/3 double‐knockout mice are viable, but lack both muscle and non‐muscle caveolae, and develop a severe cardiomyopathic phenotype. Am J Pathol 2002;160:2207–17.
- Capozza F, Combs TP, Cohen AW, Cho YR, Park SY, Schubert W, et al. Caveolin‐3 knockout mice show increased adiposity and whole body insulin resistance, with ligand‐induced insulin receptor instability in skeletal muscle. Am J Physiol Cell Physiol 2005;288:C1317–31.
- Gazzerro E, Sotgia F, Bruno C, Lisanti MP, Minetti C. Caveolinopathies: from the biology of caveolin‐3 to human diseases. Eur J Hum Genet: EJHG 2010;18:137–45.
- Markworth JF, Cameron‐Smith D. Arachidonic acid supplementation enhances in vitro skeletal muscle cell growth via a COX‐2‐dependent pathway. Am J Physiol Cell Physiol 2013;304:C56–67.
- Hausman RE, Velleman SG. Prostaglandin E1 receptors on chick embryo myoblasts. Biochem Biophys Res Commun 1981;103:213–8.
- Santini MT, Indovina PL, Hausman RE. Prostaglandin dependence of membrane order changes during myogenesis in vitro. Biochim Biophys Acta 1988;938:489–92.
- Hausman RE, elGendy H, Craft F. Requirement for G protein activity at a specific time during embryonic chick myogenesis. Cell Differ Dev: The Official J Int Soc Dev Biol 1990;29:13–20.
- Iannotti FA, Silvestri C, Mazzarella E, Martella A, Calvigioni D, Piscitelli F, et al. The endocannabinoid 2‐AG controls skeletal muscle cell differentiation via CB1 receptor‐dependent inhibition of Kv7 channels. Proc Natl Acad Sci U S A 2014;111:E2472–81.
- Murton AJ, Marimuthu K, Mallinson JE, Selby AL, Smith K, Rennie MJ, et al. Obesity appears to be associated with altered muscle protein synthetic and breakdown responses to increased nutrient delivery in older men, but not reduced muscle mass or contractile function. Diabetes 2015;64:3160–71.
- McGlory C, Galloway SD, Hamilton DL, McClintock C, Breen L, Dick JR, et al. Temporal changes in human skeletal muscle and blood lipid composition with fish oil supplementation. Prostaglandins Leukot Essent Fatty Acids 2014;90:199–206.
- Jans A, van Hees AM, Gjelstad IM, Sparks LM, Tierney AC, Riserus U, et al. Impact of dietary fat quantity and quality on skeletal muscle fatty acid metabolism in subjects with the metabolic syndrome. Metabolism 2012;61:1554–65.
- Rasic‐Milutinovic Z, Perunicic G, Pljesa S, Gluvic Z, Sobajic S, Djuric I, et al. Effects of N‐3 PUFAs supplementation on insulin resistance and inflammatory biomarkers in hemodialysis patients. Ren Fail 2007;29:321–9.
- Dangardt F, Chen Y, Gronowitz E, Dahlgren J, Friberg P, Strandvik B. High physiological omega‐3 fatty acid supplementation affects muscle fatty acid composition and glucose and insulin homeostasis in obese adolescents. J Nutr Metab 2012;2012:395757.
- Jimenez‐Gomez Y, Cruz‐Teno C, Rangel‐Zuniga OA, Peinado JR, Perez‐Martinez P, Delgado‐Lista J, et al. Effect of dietary fat modification on subcutaneous white adipose tissue insulin sensitivity in patients with metabolic syndrome. Mol Nutr Food Res 2014;58:2177–88.
- Kawabata F, Neya M, Hamazaki K, Watanabe Y, Kobayashi S, Tsuji T. Supplementation with eicosapentaenoic acid‐rich fish oil improves exercise economy and reduces perceived exertion during submaximal steady‐state exercise in normal healthy untrained men. Biosci Biotechnol Biochem 2014;78:2081–8.
- Henry R, Peoples GE, McLennan PL. Muscle fatigue resistance in the rat hindlimb in vivo from low dietary intakes of tuna fish oil that selectively increase phospholipid n‐3 docosahexaenoic acid according to muscle fibre type. Br J Nutr 2015;114:873–84.
- Peoples GE, McLennan PL. Long‐chain n‐3 DHA reduces the extent of skeletal muscle fatigue in the rat in vivo hindlimb model. Br J Nutr 2014;111:996–1003.
- Langelaan ML, Boonen KJ, Rosaria‐Chak KY, van der Schaft DW, Post MJ, Baaijens FP. Advanced maturation by electrical stimulation: differences in response between C2C12 and primary muscle progenitor cells. J Tissue Eng Regen Med 2011;5:529–39.
- https://pubmed.ncbi.nlm.nih.gov/2055849/
- https://pubmed.ncbi.nlm.nih.gov/16141377/
- https://onlinelibrary.wiley.com/doi/abs/10.1111/j.1748-1716.1979.tb06438.x
- https://pubmed.ncbi.nlm.nih.gov/16357078/
- https://pubmed.ncbi.nlm.nih.gov/11782652/
- https://pubmed.ncbi.nlm.nih.gov/11402254/
- https://pubmed.ncbi.nlm.nih.gov/20489032/
- https://pubmed.ncbi.nlm.nih.gov/3298320/
- https://pubmed.ncbi.nlm.nih.gov/15879168/
- https://pubmed.ncbi.nlm.nih.gov/20091182/
- http://www.asep.org/asep/asep/JEPonlineDec2009Lamont.doc
- https://pubmed.ncbi.nlm.nih.gov/20091182/
