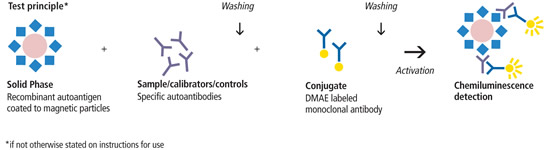
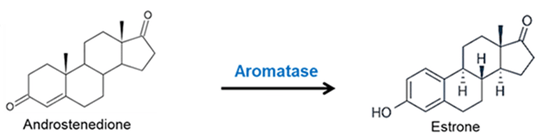
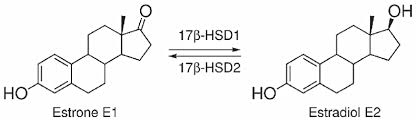
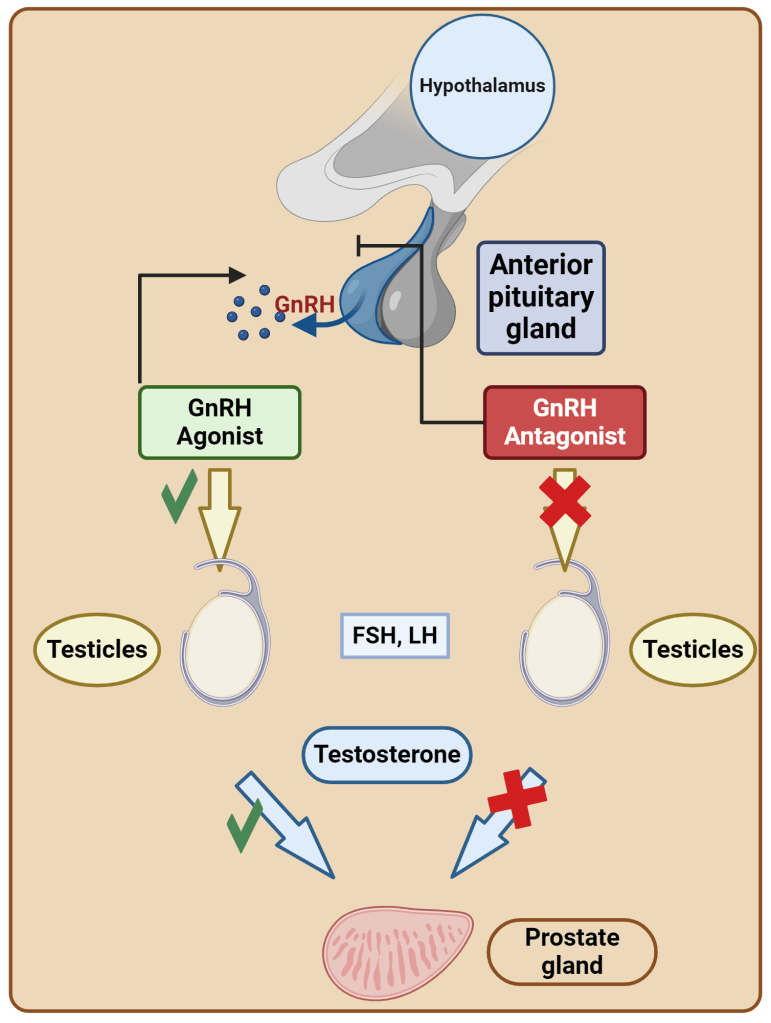
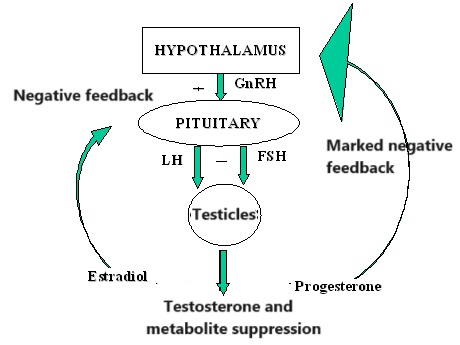
Introduzione agli aspetti pleiotropici degli estrogeni:
L’importanza della componente estrogenica, e nella fattispecie dell’estrogeno maggiormente attivo Estradiolo [E2], nel maschio è ormai nota e viene approfondita anche da studi di nuova pubblicazione.
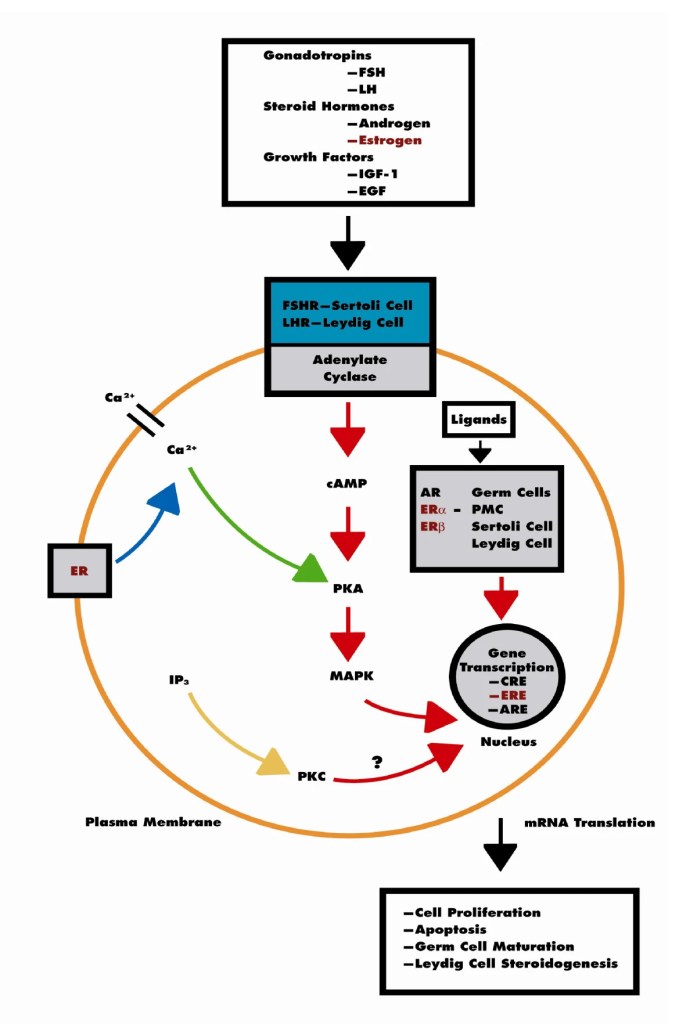
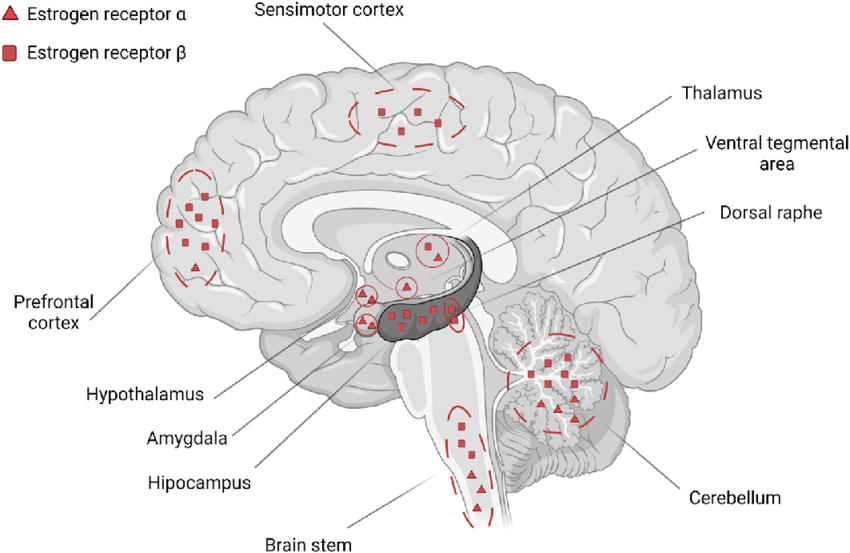
Nei maschi, gli estrogeni esercitano effetti pleiotropici agendo su diversi tessuti e organi, tra cui il sistema riproduttivo. Nell’uomo, gli estrogeni sono in grado di esercitare la loro azione a diversi livelli attraverso il tratto riproduttivo e su diverse cellule riproduttive. Tuttavia, la regolazione della riproduzione maschile umana è complessa e il ruolo degli estrogeni è meno chiaro rispetto ai topi. Durante la vita fetale e perinatale, gli estrogeni agiscono sul sistema nervoso centrale modulando lo sviluppo di alcune aree cerebrali deputate al controllo del comportamento sessuale maschile in termini di definizione dell’identità di genere, sviluppo dell’orientamento sessuale ed evoluzione del normale comportamento sessuale maschile adulto. Questo effetto organizzativo e centrale degli estrogeni è particolarmente significativo in altre specie (soprattutto roditori e montoni), ma probabilmente meno importante negli uomini, dove i fattori psicosociali diventano più determinanti.[https://pmc.ncbi.nlm.nih.gov/]
L‘Estradiolo negli uomini è quindi essenziale per modulare la libido, la funzione erettile e la spermatogenesi. I recettori degli Estrogeni, così come l’Aromatasi, sono abbondanti nel cervello, nel pene e nei testicoli, organi importanti per la funzione sessuale. Nel cervello, la sintesi dell’Estradiolo è elevata nelle aree correlate all’attività sessuale. Inoltre, nel pene, i recettori degli Estrogeni si trovano in tutto il corpus cavernosum con elevata concentrazione intorno ai fasci neurovascolari. Un livello basso di Testosterone e elevato di Estrogeni aumenta l’incidenza della disfunzione erettile indipendentemente l’uno dall’altro. Nei testicoli, la spermatogenesi è modulata a tutti i livelli dagli Estrogeni, a partire dall’asse ipotalamo-ipofisi-gonadi, seguita dalle cellule di Leydig, Sertoli e germinali e terminando con l’epitelio ductale, l’epididimo e lo sperma maturo. La regolazione delle cellule testicolari mediante l’Estradiolo mostra sia un’influenza inibitoria che una stimolatoria, indicando un intricata sinfonia di modulazione dose-dipendente e temporalmente sensibile.[https://www.researchgate.net]
In genere si ritiene che gli estrogeni e il Testosterone siano i principali steroidi sessuali che regolano il metabolismo osseo rispettivamente nelle donne e negli uomini. Si sono infatti osservati uomini portatori di mutazioni omozigoti nel gene ER-alfa e uomini con mutazioni omozigoti nel gene dell’Aromatasi presentare osteopenia, epifisi non fuse e indici elevati di turnover osseo. Sebbene questi risultati indichino che gli estrogeni svolgono un ruolo nella regolazione dello scheletro maschile, hanno lasciato irrisolto il problema se gli estrogeni agiscano sullo scheletro maschile principalmente per migliorare l’acquisizione di massa ossea durante la crescita e la maturazione, o se agiscano anche per ritardare la perdita di massa ossea negli individui che invecchiano. Per risolvere questo problema, diversi studi osservazionali trasversali hanno messo in relazione la densità minerale ossea (BMD) con gli steroidi sessuali negli uomini anziani, scoprendo che gli estrogeni si correlavano meglio del Testosterone con la BMD. Inoltre, recenti studi longitudinali indicano che gli estrogeni biodisponibili si correlano meglio del Testosterone sia con l’aumento della BMD negli uomini giovani sia con la perdita di BMD negli uomini anziani. Tuttavia, questi studi osservazionali non dimostrano la causalità, che richiede studi interventistici diretti.[https://pubmed.ncbi.nlm.nih.gov/]
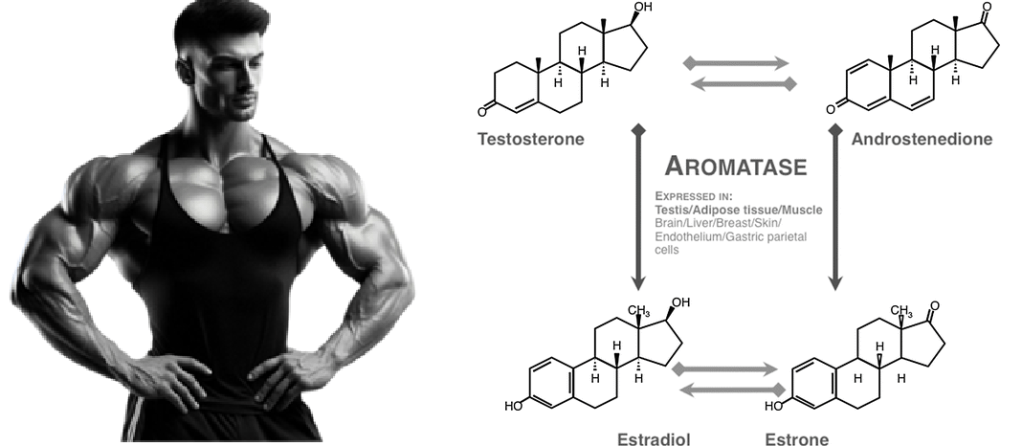
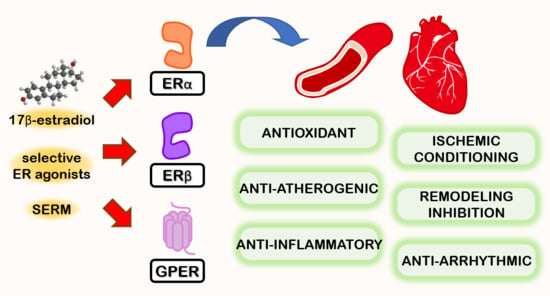
Sono stati osservati effetti favorevoli, oltre che a livello sessuale e sulla fisiologia ossea, sulla salute cerebrale e cardiovascolare correlata ad un adeguato livello di E2, mentre è stato seriamente sospettato un potenziale ruolo nella patologia prostatica dell’uomo che invecchia in seguito a dis-regolazione della T:E ratio. Gli estrogeni nell’uomo sono prevalentemente i prodotti dell’aromatizzazione periferica o in sede ghiandolare degli androgeni testicolari e surrenali. Gli estrogeni esercitano effetti sul cervello: sulla funzione cognitiva, sulla coordinazione dei movimenti, sul dolore e sullo stato affettivo, e sono forse protettivi nei confronti della malattia di Alzheimer. Gli effetti degli estrogeni sul sistema cardiovascolare includono quelli sui profili lipidici, sulla distribuzione del grasso, sui fattori endocrini/paracrini prodotti dalla parete vascolare (come endoteline, ossido nitrico), sulle piastrine, sui fattori infiammatori e sulla coagulazione.[https://pubmed.ncbi.nlm.nih.gov/]
Gli Estrogeni possono giocare un ruolo significativamente importante nella promozione di uno stato anabolico influenzando l’utilizzo del Glucosio nel tessuto muscolare. Ciò avviene attraverso un’alterazione del livello di glucosio 6-fosfato deidrogenasi disponibile, un enzima direttamente legato all’uso del glucosio per la crescita e il recupero del tessuto muscolare. Più specificamente, il G6PD è l’enzima che catalizza la prima reazione della via dei pentoso fosfati (definita anche Shunt dell’Esosomonofosfato [HMP shunt] o PPP da Pentose phosphate pathway), un processo metabolico citoplasmatico, parallelo alla glicolisi, in grado di generare NADPH e zuccheri pentosi (a 5 atomi di carbonio). Durante il periodo di rigenerazione tissutale seguente il danno muscolare, i livelli di G6PD aumentano considerevolmente, il che è ritenuto essere un meccanismo che il corpo attua per migliorare il recupero quando necessario. Sorprendentemente, si osserva che l’Estrogeno è direttamente legato al livello di G6PD che deve essere messo a disposizione delle cellule in questa fase di recupero. In sintesi, gli Estrogeni svolgono anche una azione metabolica accelerando la sintesi degli acidi nucleici, delle proteine e del glicogeno. Anche in questo caso, però, un livello eccessivo di E2 sembrerebbe poter causare alterazioni negative della sensibilità all’Insulina per via di una cronicità di stimolo e una sottoregolazione consequenziale.
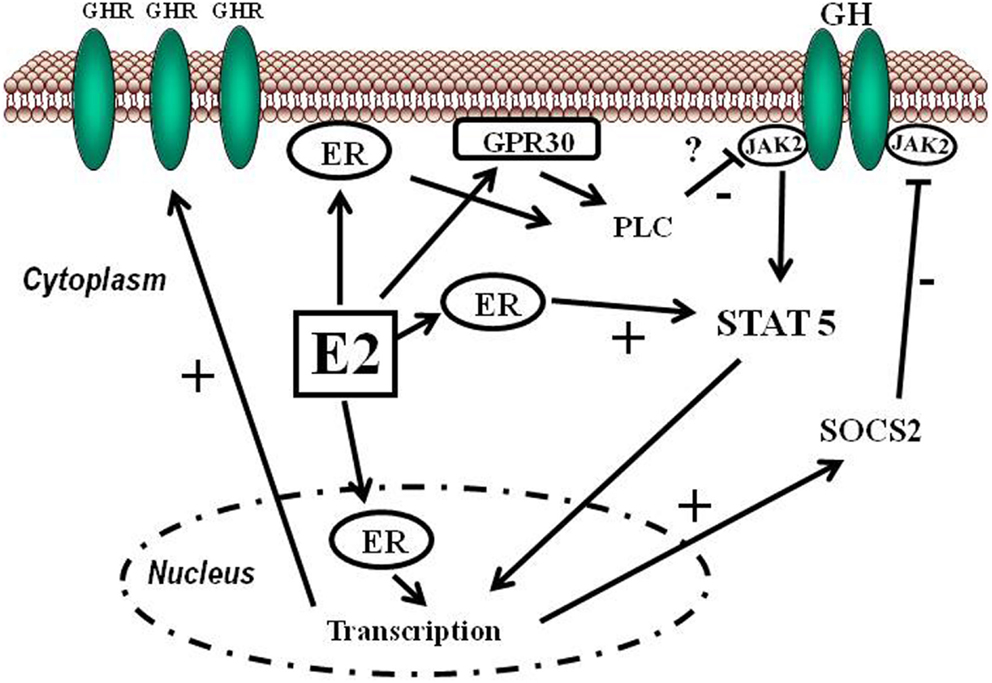
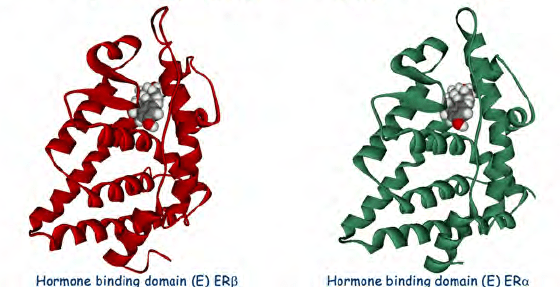
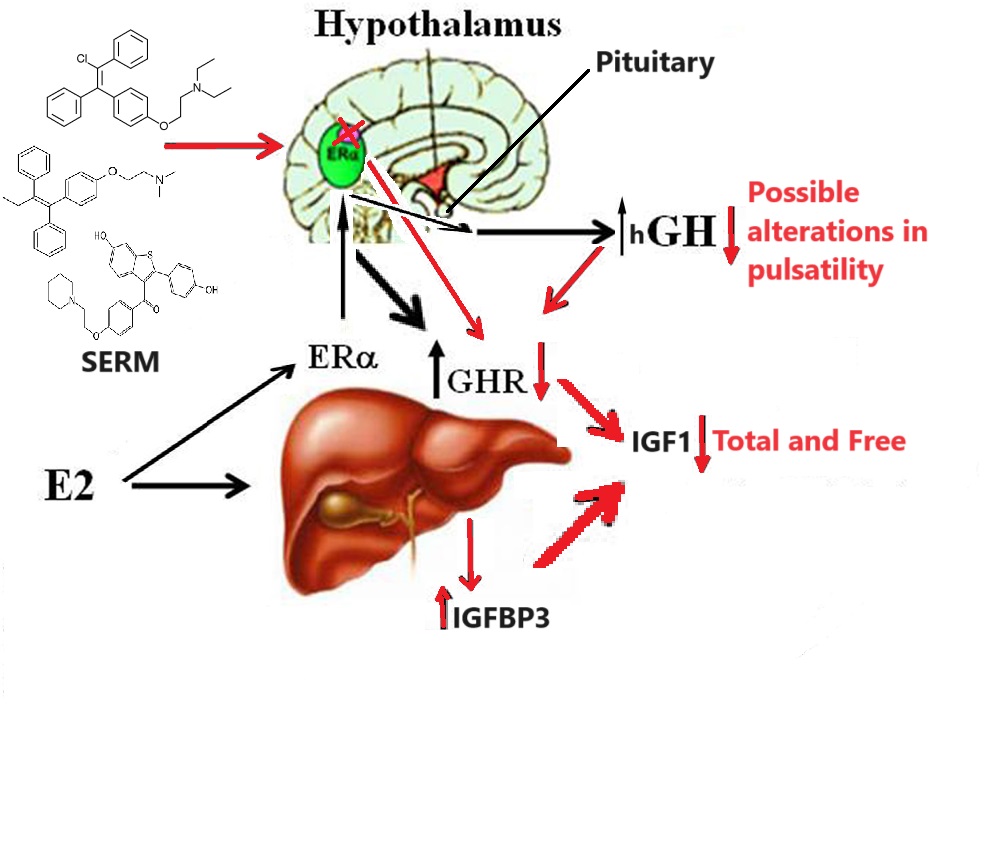
Sappiamo che gli estrogeni possono anche svolgere un ruolo importante nell’influenzare l’attività dell’Asse hGH/IGF-1. L’E2 ha un’affinità simile per l’ERα e l’ERβ e questi recettori sono attivati da un’ampia gamma di ligandi, tra cui i SERM (ad esempio, il Raloxifene e il Tamoxifene) e molti altri composti. L’ERβ è espresso nell’ovaio, nella prostata, nel polmone, nel tratto gastrointestinale, nella vescica, nel sistema ematopoietico e nel sistema nervoso centrale, mentre l’ERα è espresso principalmente nei tessuti riproduttivi, nel rene, nell’osso, nel tessuto adiposo bianco e nel fegato. Il fegato esprime ERα ma livelli quasi irrilevanti di ERβ, il che indica che le azioni specifiche degli estrogeni nel fegato possono essere imitate utilizzando agonisti selettivi di ERα come il Propilpirazolo-triolo (PPT) (Lundholm et al., 2008). Nel complesso, i dati sopra citati indicano che i meccanismi coinvolti nella segnalazione ER sono influenzati dal fenotipo cellulare, dal gene bersaglio e dall’attività o dal crosstalk con altre reti di segnalazione. Il fegato rappresenta un sito in cui si possono sviluppare interazioni fisiologicamente e terapeuticamente rilevanti tra estrogeni e hGH. Particolarmente rilevante è l’interazione degli estrogeni con la via di segnalazione GHR-JAK2-STAT5 nella regolazione della crescita somatica, del metabolismo dei lipidi e del glucosio e della “sessualità epatica”.
L’E2 può regolare le azioni del hGH nel fegato modulando la reattività del hGH, che comprende cambiamenti nell’espressione del GHR epatico e il crosstalk con la via di segnalazione JAK2-STAT5 attivata dal hGH. Gli estrogeni possono indurre l’espressione di SOCS2, che a sua volta inibisce negativamente la via di segnalazione GHR-JAK2-STAT5.
Inoltre, le differenze di composizione corporea legate al genere sono in parte mediate dagli steroidi sessuali che modulano l’Asse hGH/IGF-I (LeRoith, 2009; Rogol, 2010; Birzniece et al., 2011). Ciò è supportato dall’osservazione che le differenze di genere nella composizione corporea emergono al momento della crescita puberale. Inoltre, l’efficienza dell’attività del hGH è modulata anche dagli estrogeni in età adulta. Questo è esemplificato dal fatto che le donne sono meno reattive degli uomini al trattamento con hGH (Burman et al., 1997); il trattamento con hGH induce un maggiore aumento della massa magra e una diminuzione della massa grassa, o un maggiore aumento degli indici di turnover osseo e della massa ossea, nei pazienti GHD maschi rispetto alle femmine. È rilevante per la fisiologia del hGH l’alterazione della biodisponibilità dell’IGF-I dovuta alla somministrazione orale di dosi farmacologiche di estrogeni [rivisto da Leung et al., 2004]. La disponibilità e l’attività tissutale dell’IGF-I sono regolate dalle proteine leganti l’IGF (IGFBPs) (Kaplan e Cohen, 2007; LeRoith e Yakar, 2007; Ohlsson et al., 2009).
L’IGF-I circola quasi interamente come complesso ternario legato all’IGFBP-3 e all’ALS, entrambi fortemente regolati dal hGH nel fegato. Questo complesso ternario regola la biodisponibilità dell’IGF-I. Anche l’IGFBP-1 è una proteina di derivazione epatica che lega la piccola frazione di IGF-I libero e attenua l’effetto ipoglicemizzante del fattore di crescita (Lewitt et al., 1991). In contrasto con il suo effetto soppressivo sulla SLA e sull’IGF-I, la somministrazione orale di estrogeni aumenta l’IGFBP-1 circolante. Si può prevedere che l’effetto dell’aumento di IGFBP-1 riduca ulteriormente la frazione libera di IGF-I, con conseguente riduzione della sua attività. È interessante notare che l’attivazione della segnalazione GH-STAT5b induce l’espressione di ALS e IGF-I, ma inibisce IGFBP-1 (Ono et al., 2007). Pertanto, l’inibizione della via di segnalazione GHR-JAK2-STAT5 nel fegato (vedi sotto), molto probabilmente contribuisce agli effetti degli estrogeni su IGF-I, ALS e IGFBP-1. Pertanto, gli estrogeni esercitano effetti profondi sulle IGFPB derivate dal fegato quando vengono somministrati per via orale, che molto probabilmente modificano le azioni biologiche dell’IGF-I. Inoltre, la somministrazione orale di dosi farmacologiche di estrogeni può inibire gli effetti metabolici regolati dal GH (ad esempio, ossidazione lipidica, sintesi proteica) (Huang e O’Sullivan, 2009). Questi effetti sul metabolismo e sulla composizione corporea sono attenuati dalla somministrazione transdermica, suggerendo che il fegato è il principale sito di controllo regolatorio da parte degli estrogeni. Si rammenta, però, che in una condizione di iperestrogenemia [>45-60pg/dL] porta automaticamente ad un aumento delle concentrazioni di E2 a livello epatico con interferenze negative sull’Asse hGH/IGF1.
Gli estrogeni possono modulare le azioni del hGH sul fegato attraverso la modulazione della responsività al hGH, che include cambiamenti nell’espressione del GHR epatico e il crosstalk con la via di segnalazione JAK2-STAT5 attivata dal GH (Leung et al., 2004). In particolare, l’E2 può indurre l’espressione di SOCS2 e SOCS3, che a sua volta regola negativamente la via di segnalazione GHR-JAK2-STAT5, con conseguente riduzione dell’attività trascrizionale nel fegato. Pertanto, oltre alla regolazione da parte dell’E2 del modello di dimorfismo sessuale della secrezione ipofisaria di hGH, l’induzione dell’espressione di SOCS e l’inibizione della segnalazione JAK2-STAT5 è un meccanismo molto rilevante che, in parte, potrebbe spiegare come gli estrogeni inibiscano direttamente gli effetti del hGH in diverse azioni regolate da STAT5 (ad esempio, crescita somatica, composizione corporea, metabolismo e funzioni epatiche legate al sesso). Ipoteticamente, anche altri membri dei regolatori negativi della famiglia STAT possono contribuire all’interazione degli estrogeni con la segnalazione del hGH nel fegato. Ciò si spiega con la stimolazione da parte di ERα dell’espressione di PIAS3, che si lega e blocca l’attività di legame al DNA di STAT3. È interessante notare che l’attivazione di ER da parte di E2, seguita dall’interazione diretta di ER con STAT5, può anche inibire l’attività trascrizionale STAT5-dipendente (Faulds et al., 2001; Wang et al., 2004). D’altra parte, è stato dimostrato che l’attivazione di ERα o ERβ da parte di E2, attraverso meccanismi non genomici, induce un programma trascrizionale STAT5 (e STAT3) dipendente nelle cellule endoteliali (Bjornstrom e Sjoberg, 2005). Nel complesso, questi studi hanno dimostrato l’esistenza di un’interazione diretta tra la segnalazione di ER e STAT5, dimostrando inoltre che le conseguenze funzionali di questo crosstalk dipendono dal preciso contesto dell’ambiente intracellulare.
Se vi sentite confusi per ciò che è stato detto, dal momento che siete stati convinti da numerosi “top coach” che l’E2 deve rimanere alto per garantire una migliore risposta anabolica complessiva, e che i SERM causano una sottoregolazione dell’Asse hGH/IGF1 in quanto antagonizzano i ER, beh… le cose sono più complesse di così. Ad esempio, il Tamoxifene ha effetti prevalentemente antiestrogenici nelle mammelle, ma prevalentemente estrogenici nell’utero e nel fegato. Egualmente, il Raloxifene ha un’attività estrogenica in alcuni tessuti, come le ossa e il fegato, e un’attività antiestrogenica in altri tessuti, come il seno e l’utero. Ed è quindi l’aumento dell’attività recettoriale indotta da E2 o SERM ha causare alterazioni dell’Asse hGH/IGF1. In conclusione, la regolazione del E2 è essenziale per garantire ottimali risposte dell’Asse hGH/IGF1, evitando eccessi o cali eccessivamente prolungati e/o fuori contesto.
Un altro beneficio correlato a ottimali livelli di E2 riguarda la possibilità di indurre un aumento della concentrazione dei AR in alcuni tessuti. Ciò è stato dimostrato in studi svolti su ratti che hanno esaminato gli effetti degli Estrogeni sui AR cellulari in animali sottoposti ad orchiectomia (rimozione dei testicoli, spesso effettuata per diminuire la produzione endogena di Androgeni). Secondo lo studio, la somministrazione di Estrogeni ha determinato un aumento del legame recettoriale nel muscolo levator ani del Metribolone pari al 480%. E qui c’è il primo “inghippo” dal momento che il levator ani è parte dell’apparato sessuale dell’animale e non è paragonabile al muscolo-scheletrico umano. Infatti, lo studio ha esaminato l’effetto sui AR dato dagli Estrogeni nei tessuti musco scheletrici veloci (tibialis anterior e extensor digitorum longus), ma senza notare lo stesso aumento visto nel levator ani.[Modulation of the cytosolic androgen receptor in striated muscle by sex steroids. Endocrinology. 1984 Sep;115(3):862-6.] Se vi sia riscontro nell’uomo questo è comunque limitato alle possibilità di espressione genica del soggetto e, probabilmente, l’incremento degli AR non risulterebbe paragonabile a quello sperimentabile con dosi sovrafisiologiche di AAS.
Recettore degli Androgeni.
Sicuramente il mancato controllo del fattore estrogenico è tanto deleterio quanto può esserlo una sua soppressione marcata. Un’altra importante funzione degli estrogeni in ambo i sessi è la sua capacità di promuovere uno stato mentale di vigilanza. L’abuso di AI, con la marcata soppressione del E2 consequenziale, si manifestano stati di stanchezza. In tali condizioni, l’atleta, anche se sta seguendo un ciclo correttamente formulato, potrebbe non essere in grado di massimizzare i propri risultati di miglioramento della condizione fisica a causa di un’incapacità di allenarsi con pieno vigore. Questo effetto è talvolta anche soprannominato “letargia steroidea” o “Steroid Fatigue”. La ragione principale per cui ciò accade è legata all’importante azione di supporto all’attività della Serotonina data dall’E2. La Serotonina è uno dei principali neurotrasmettitori del corpo, di essenziale importanza per un adeguata lucidità mentale e un regolare ciclo sonno / veglia.[ Effect of estrogen-serotonin interactions on mood and cognition. Zenab Amin et al. Behav Cogn Neurosci Reviews 4(1) 2005:43-58] L’alterazione di questo neurotrasmettitore è associata anche alla sindrome da affaticamento cronico, e ciò ci fa comprendere quanto possa essere incisiva in particolare per la stanchezza. L’abbassamento dei livelli estrogenici nella menopausa è stata associata anche alla stanchezza, così come l’uso clinico di inibitori dell’aromatasi più recenti (e più potenti) come l’Anastrozolo, il Letrozolo, l’Exemestane, e il Fadrozolo in alcuni pazienti. L’uso di AAS non aromatizzabili e/o SARM non steroidei possono causare questo effetto, il quale, in questa circostanza, è dovuto alla soppressione/sottoregolazione della produzione endogena di Testosterone (riduzione del substrato principale nell’uomo per la sintesi di E2 per via della attività dell’enzima Aromatasi).
Gli estrogeni possono influire sui livelli di colesterolo, influenzando potenzialmente la salute cardiovascolare. In particolare, gli estrogeni possono influire sul colesterolo LDL (“cattivo”) e sul colesterolo HDL (“buono”); alcuni studi suggeriscono un legame tra livelli più elevati di estrogeni e una diminuzione del LDL e un aumento del HDL negli uomini. Il termine “elevati” non dovrebbe essere fuorviante dal momento che alterazioni metaboliche come quelle descritte in precedenza e correlate ad un eccessivo incremento del E2, potrebbero vedere sensibilmente ridotto questo effetto positivo.
In fine, sappiamo che se non si dispone di una quantità sufficiente di estrogeni rispetto ai livelli di androgeni nell’organismo, i livelli di cardiotossicità e neurotossicità saranno significativamente più alti di quelli che si avrebbero se si mantenessero livelli ottimali di estrogeni. Dal punto di vista del Bodybuilding, gli estrogeni a livello ottimale (tarato sul soggetto) sono necessari per ottimizzare la crescita muscolare, l’insulino-sensibilità e la sintesi di IGF-1 e fattori di crescita/segnalazione cellulare. Ricordiamoci, però, che un dosaggio fisiologico di Testosterone, e sua successiva aromatizzazione, risulta essere neuroprotettivo. Il Testosterone amplificava la neurotossicità solo a dosaggi sovrafisiologici anche senza AI. Sebbene l’aromatizzazione del Testosterone in E2 prevenga, seppur non marcatamente, una quantità significativa di danno neuronale, si può osservare chiaramente che le concentrazioni sovrafisiologiche di Testosterone esacerbano la neurotossicità in ogni caso e che i livelli sovrafisiologici di estrogeni non forniscono un aumento dose-dipendente della neuroprotezione.
Ci sono sempre dei limiti… sempre un fio da pagare. Sicuramente, come si può ben capire, un incremento spropositato di E2 in condizione di sovrafisiologia d’uso di AAS non garantirebbe altro che il sommarsi di altri effetti avversi.
Dopo questa “carrelata” di effetti positivi e limiti annessi al E2, e agli estrogeni in generale, possiamo entrare nel vivo della questione “gestione degli estrogeni” partendo dalla clinica, ossia dalla TRT.
Gestione estrogenica in TRT
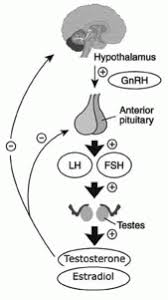
Condizioni di bassi livelli di Testosterone si verificano nel 6-25% degli uomini. Secondo le linee guida dell’American Urological Association, il trattamento del Testosterone basso [vedi TRT] è indicato se i livelli sono inferiori alla soglia di 300ng/dL [3ng/mL] con segni o sintomi associati.2 Uno degli effetti collaterali più comuni riscontrabili nei pazienti in TRT, oltre alle variazioni dell’Ematocrito, è l’aumento dell’E2 attraverso l’attività dell’enzima Aromatasi (aromatizzazione), che converte il Testosterone in E2.
Sebbene la manifestazione di questa problematica può essere esacerbata o scatenata dalla cosomministrazione di hCG, le prime misure adottate per la sua gestione comprendo 1) modifiche nel dosaggio e/o nella cadenza di somministrazione e 2) modificando, se vi sono le circostanze a richiederlo, lo stile di vita del paziente portandolo ad una riduzione della massa grassa e, di conseguenza, dell’espressione dell’Aromatasi. Tuttavia, ciò potrebbe non essere sufficiente.
Si è notato, infatti, in diversi casi, che il tasso di aromatizzazione subiva un aumento nel corso del primo anno di trattamento clinico. Questo aumento non sembra essere correlato ad un aumento sensibile della massa grassa. Si è, di conseguenza, ipotizzato che tale risposta facesse parte di un adattamento epigenetico (o un tentativo in tal senso) in risposta a concentrazioni stabili di Testosterone in cronico. In definitiva, in questi casi, anche l’uso di ancillari steroidei [vedi Mesterolone e Drostanolone] non mostra miglioramenti assoluti nei livelli di E2, sebbene tissutalmente il Drostanolone a 100mg/settimana abbia mostrato una egregia attività controllo sull’azione estrogenica.
Molecola di Anastrozolo
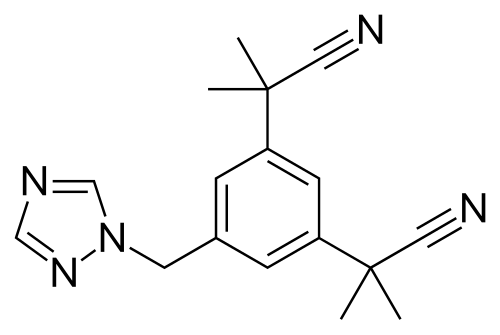
In questi casi la soluzione per il paziente è l’aggiunta di un inibitore dell’Aromatasi come l’Anastrozolo (AZ). L’Anastrozolo è un triazolo benzilico non steroideo. È noto anche come α,α,α’,α’-tetrametil-5-(1H-1,2,4-triazolo-1-ilmetil)-m-benziacetonitrile. L’Anastrozolo è strutturalmente correlato al Letrozolo, al Fadrozolo e al Vorozolo, tutti classificati come azoli.[Environmental Health Perspectives: Supplements.]
Struttura molecolare dell’Enzima Aromatasi
L’Anastrozolo agisce legandosi reversibilmente all’enzima Aromatasi e, attraverso un’inibizione competitiva, blocca la conversione degli androgeni in estrogeni nei tessuti periferici (extragonadici). È stato riscontrato che il farmaco raggiunge un’inibizione dell’Aromatasi compresa tra il 96,7% e il 97,3% al dosaggio di 1 mg/die e il 98,1% al dosaggio di 10mg/die nell’uomo. [Pertanto, 1 mg/die è considerato il dosaggio minimo necessario per ottenere la soppressione massima dell’Aromatasi con l’Anastrozolo. Questa diminuzione dell’attività dell’Aromatasi si traduce in una riduzione di almeno l’85% dei livelli di E2 nelle donne in postmenopausa. I livelli di corticosteroidi e altri steroidi surrenali non sono influenzati dall’Anastrozolo.
Bisogna specificare, però, che soggetti i quali mantengono una attività gonadale con l’uso di hCG durante la TRT risultano meno responsivi al tasso di inibizione del farmaco. Questo accade, con buona probabilità, perché una parte sostanziale dell’Estradiolo è prodotta dall’attività dell’Aromatasi nei testicoli. Nei testicoli, le concentrazioni di Testosterone [stimolate in questo caso dal hCG] arrivano a livelli circa 100 volte superiori a quelli presenti nel circolo ematico. Poiché gli AI devono inibire in modo competitivo l’Aromatasi, i dosaggi potrebbero dover essere più alti per portare a una significativa inibizione enzimatica nei testicoli. Infatti, in questi casi, la somministrazione giornaliera di Anastrozolo a 0,5 e 1mg porta ad una diminuzione dei livelli di Estradiolo di circa il 50% .
Molecola di Letrozolo
Interessante, a tal proposito, è il risultato osservato dopo 28 giorni di trattamento con Letrozolo alla dose di 2,5mg/die, dove i livelli di Estradiolo hanno subito una riduzione del 46% negli uomini giovani e del 62% negli uomini anziani. Uomini anziani con una ridotta attività gonadale.[https://pmc.ncbi.nlm.nih.gov/]
In generale, comunque, l’AZ è indicato per ritardare la maturazione epifisaria nei ragazzi adolescenti, ma è stato utilizzato nei maschi infertili con alterazioni dei livelli di Testosterone e livelli elevati di E2 per preservare la spermatogenesi. Il profilo degli effetti collaterali dell’AZ è stato ampiamente studiato nelle donne ed è un farmaco ben tollerato.4 L’elevato livello di E2 è più comunemente associato alla ginecomastia, ma non è l’unico meccanismo responsabile sebbene risulti il primario.5 L’AZ è stato utilizzato per trattare gli uomini con E2 elevato, ma attualmente esistono prove e/o linee guida limitate per la gestione ottimale degli elevati livelli di E2 negli uomini in TRT.
Nell’uomo vi sono due tipi principali di estrogeni: il potente Estradiolo e il meno potente, in senso di attività biologica, Estrone [E1]. Le quantità sono misurate in picogrammi per millilitro (pg/ml). Le medie tipiche di ciascuno di essi sono:
In uno studio pubblicato su Sex Med nel 2021, è stato riportato che su 1708 uomini in TRT, 51 (3%) sono stati trattati con AZ (AZ+). Di questi, 7 (14%) sono stati esclusi (3 precedentemente in trattamento con AZ e 4 con storia di cancro al seno). Un totale di 44 (2,6%) ha mostrato livelli elevati di estradiolo (range 40-165 pg/mL) ed è stato incluso. Il tempo mediano di somministrazione della TRT prima dell’inizio dell’AZ è stato di 11,96 mesi (IQR 4,63-31,44). I restanti 1657 uomini (97,0%) non hanno ricevuto il trattamento con AZ (AZ-).
Di conseguenza, in tale studio, solo il 2,6% degli uomini presentava un E2 tale da giustificare il trattamento con AZ. L’AZ è stato utilizzato negli uomini con una deficienza del Testosterone nel tentativo di ridurre la conversione del T in E2, pur mantenendo la fertilità.6 E’ stato utilizzato un cut-off per l’uso dell’AZ, ma la letteratura non supporta un cut-off chiaro o un’indicazione per l’inizio della terapia di abbassamento dell’E2. Similmente, in un campione nazionale, il 3,5% (1.200/34.016) degli uomini è stato trattato con un AI.
Sebbene gli uomini dello studio che ricevevano AZ, e quindi con un E2 più alto, avevano tassi maggiori, anche se non statisticamente significativi, di OSA e che tale condizione è correlato all’obesità e che gli uomini con obesità hanno livelli fisiologici maggiori di Aromatasi, aumentando così la conversione di T in E2, in corso di TRT anche in soggetti normopeso e con body fat contenute (se non basse) possono presentare condizioni di iperestrogenemia in specie dopo 12 mesi di trattamento. Probabilmente, questa risposta adattativa è la conseguenza di un tentativo di adattamento regolatorio della omeostasi ormonale in una condizione dove i livelli di T sono stabili e non caratterizzati [come nei soggetti in fisiologia funzionale] da fluttuazioni nelle 24h e nei mesi dell’anno [vedi variabili stagionali].
Un dato importante che andrebbe preso in considerazione, è che nello studio i soggetti non trattati con AZ avevano una terapia a base di Testosterone in soluzione ad uso topico, mentre gli uomini che utilizzavano AZ erano sottoposti ad una terapia a base di Testosterone somministrato per via intramuscolare. Ciò riflette alcuni dati che suggeriscono che aumenti statisticamente significativi di E2 sono stati osservati negli uomini in terapia iniettiva 3 mesi dopo l’inizio del TRT.
Quasi tutti gli uomini trattati con AZ hanno avuto un recupero dell’E2 entro i livelli normali (<40 pg/mL) con il contemporaneo mantenimento di un T sierico. Ciò evidenzia l’importanza di una regolazione appropriata, dati gli importanti processi fisiologici regolati dall’E2.5 Mentre l’AZ è comunemente usato come terapia aggiuntiva per aumentare i livelli di T negli uomini in cui è importante la conservazione della spermatogenesi e quindi evitare il Testosterone esogeno è fondamentale in diversi casi, nello studio discusso gli AI non hanno avuto un impatto sui livelli di Testosterone. Tuttavia, altri studi suggeriscono che la co-somministrazione di un’IA con Testosterone esogeno aumenta il T sierico. Tutti questi pazienti, però, hanno ricevuto un impianto di T, il che suggerisce una risposta differenziale in base al tipo di TRT.
I soggetti trattati con AZ hanno ricevuto una dose del AI pari a 0,5mg tre volte alla settimana (off-label). La regressione logistica è stata utilizzata per determinare i fattori predittivi di una maggiore probabilità di risposta al trattamento con Anastrozolo, definita come punteggio composito (riduzione dell’Estradiolo a meno di 60pg/mL e diminuzione di 20pg/mL dei livelli di Estradiolo).
Punti chiave per la gestione delE2 in TRT:
La modifica primaria è la riduzione della body fat;
Controllo più accurato del dosaggio terapeutico di Testosterone;
Controllo e/o modifica della dose e somministrazione di hCG [nonché valutarne i pro e i contro in funzione anche dell’età del paziente];
Aggiunta di un ancillare steroideo a dosi minime efficaci con attività di controllo del E2 [vedi, ad esempio, il Drostanolone o il Mesterolone];
L’ultimo intervento consiste nell’inserimento di un AI come l’Anastrozolo al dosaggio di 0,5mg per 3 volte a settimana.
Gestione estrogenica nell'”Enhanced”:
Trattato l’aspetto clinico della gestione estrogenica in contesto TRT/HRT, passiamo ora al contesto PEDs o, meglio, al contesto Enhanced BodyBuilding.
Il mondo della cultura fisica tout court non è mai stata immune alle mode generate da convinzioni rese “dogmatiche” perché affermate dal “guru” del settore. Questa tendenza ha e colpisce anche la questione della gestione estrogenica. Si passa dalla fobia e tendenza a mantenere i livelli di E2 cronicamente bassi all’esatto opposto con iperestrogenemia mantenuta con la convinzione che l’atleta ne gioverà. E’ superfluo dire che ambo le posizioni sono errate.
Come abbiamo potuto appurare all’inizio del presente articolo, gli estrogeni, ed in particolare l’E2, hanno una attività pleiotropica. Quindi? Quindi, ciò significa che può avere effetti multipli e, a volte, apparentemente non correlati, sullo sviluppo di determinate risposte organiche. Anche in questo caso, la frase mal tradotta e diffusa attribuita a Paracelso “E’ la dose che fa il veleno” può benissimo essere applicata anche al fattore estrogenico. E dal momento che ogni Preparatore dovrebbe avere una formazione di Biologia, Biochimica, Farmacologia, Andrologia e Endocrinologia, esso deve indirizzare tali conoscenze al fine di comprendere il soggetto interessato e operare al fine di trovare un settaggio ideale di E2. In poche parole, l’atleta dovrebbe essere un nuovo libro con informazioni aggiuntive e a se stanti.
Ricordiamoci, quindi, che negli uomini in fisiologia, i testicoli producono circa il 20% degli estrogeni circolanti. Il resto proviene dalla produzione locale da parte dei tessuti adiposi, cerebrali, cutanei e ossei, che esprimono l’Aromatasi (T ⇒ E₂).. Le concentrazioni di T nel sangue periferico degli uomini, pari a ~20nM, sono di almeno due ordini di grandezza superiori alle concentrazioni di E₂ (30-200pM). [Cooke PS, Nanjappa MK, Ko C, Prins GS, Hess RA. Estrogens in Male Physiology. Physiol Rev. 2017 Jul].
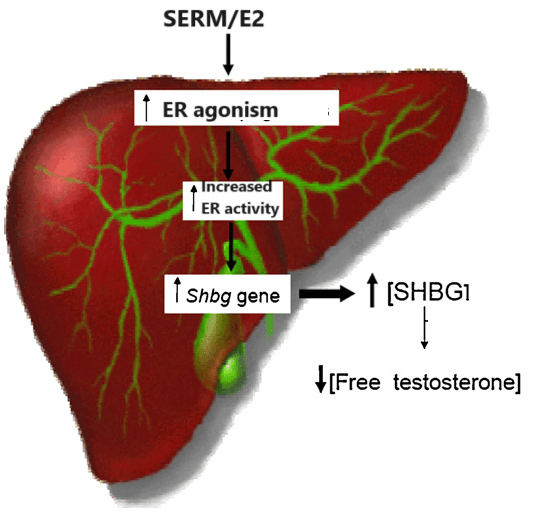
Mentre gli estrogeni esogeni e un loro eccesso endogeno causano patologie riproduttive maschili, gli estrogeni endogeni nel giusto assetto sono fondamentali per il funzionamento sessuale maschile. In parte, ciò è dovuto a un drastico aumento della SHBG (che riduce la biodisponibilità del T). [Damewood MD, Bellantoni JJ, Bachorik PS, Kimball AW Jr, Rock JA. Exogenous estrogen effect on lipid/lipoprotein cholesterol in transsexual males. J Endocrinol Invest. 1989 Jul-Aug;12(7):449-54.].
Un punto a sfavore della scelta deleteria di ridurre marcatamente l’E2 in cronico risiede senza dubbio nell’effetto positivo degli estrogeni nella lipidemia ematica.
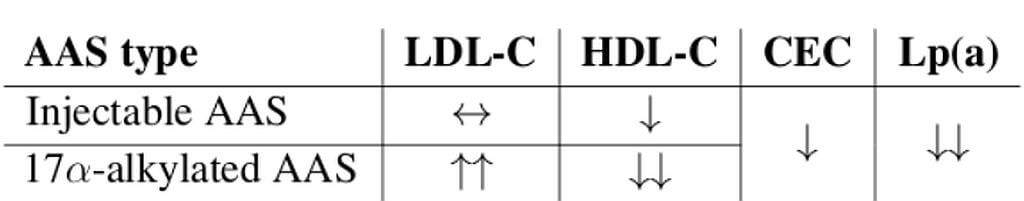
Sappiamo che gli effetti degli AAS sui lipidi sono modulati attraverso:
(A) ↑ l’attività della trigliceride lipasi epatica (HTGLA), riducendo così le lipoproteine ad alta densità (↓HDL-C), e
(B) ↑Apo B, quindi ↑LDL-C.
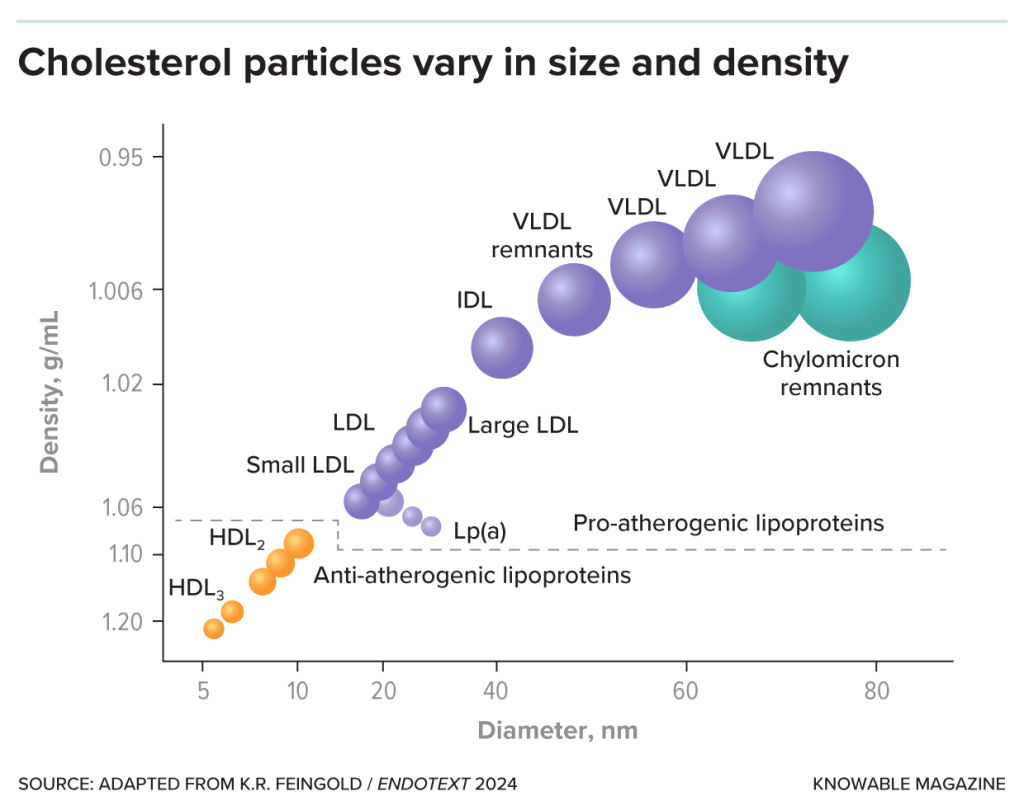
In primo luogo, per quanto riguarda (A): la lipasi epatica (HL) è un enzima secreto dal fegato che libera gli acidi grassi dal triacilglicerolo e dai fosfolipidi che fanno parte delle lipoproteine, comprese le lipoproteine ad alta densità (HDL). Gli AAS, aumentando la sua attività (HTGLA), inducono un passaggio da HDL₂ più grandi a HDL₃ più piccole, suscettibili di ulteriore degradazione, e quindi riducono le HDL. [Thompson, P. D. (1989).].
In secondo luogo, per quanto riguarda (B): l’Apo B è fortemente associata alle lipoproteine a bassissima densità (VLDL; una classe di particelle maggiormente aterosclerotica) e fornisce indicazioni sulle LDL effettivamente presenti nella circolazione sanguigna, forse perché gli AAS aumentano la secrezione epatica di queste lipoproteine. [Hartgens, F. (2004).].
Sezione della tabella presa da “Bond P, Smit DL, de Ronde W. Anabolic-androgenic steroids: How do they work and what are the risks? Front Endocrinol (Lausanne)”
La dislipidemia è caratterizzata da ↑LDL-C e ↓HDL-C.
Struttura molecolare della HTGLA
Gli AAS 17α-alchilati non aromatizzabili (metilati in C17), poiché non sono aromatizzabili o sono resistenti all’aromatizzazione e quindi non apportano benefici estrogenici ai lipidi (estrogeni ↓HTGLA), e poiché sono metabolizzati principalmente nel fegato, avendo maggiori effetti sulle proteine epatiche (ad es., HL), sono più dislipidemici e aterosclerotici degli androgeni parenterali (soprattutto Testosterone). [Friedl, K. E., Hannan, C. J., Jones, R. E., and Plymate, S. R. (1990).].
In particolare, i metilati in C-17 possono ↑Apo B, [Hartgens, F. (2004).] – che è associata a VLDL e LDL effettivamente in circolo – forse attraverso la ↑ secrezione epatica di queste lipoproteine.
Gli estrogeni prodotti dagli androgeni aromatizzanti, in particolare il T, favoriscono i lipidi riducendo l’HTGLA, provocando un flusso netto verso particelle HDL₂ più grandi, migliorando così la dislipidemia e l’aterosclerosi. Ovviamente vi è un “collo di bottiglia” determinato dal dosaggio totale di AAS utilizzati, dal protrarsi di una condizione metabolicamente alterata la quale si verifica in ipercalorica e in condizione di iperestrogenemia protratta.
Per fare un altro esempio sulla necessità di mantenere una adeguata attività estrogenica sul lungo termine possiamo osservare l’effetto dell’E2 sul tessuto adiposo e il metabolismo energetico. I ricercatori utilizzano topi maschi knock-out per il recettore degli estrogeni (ERKO) per studiare gli effetti dell’E₂, senza alcuna influenza confondente da parte del Testosterone. I topi maschi ERKO possiedono depositi di tessuto adiposo (AT; tessuto grasso) che sono aumentati del 100% entro 9-12 mesi (approssimativamente la mezza età negli esseri umani). Questo aumento del tessuto adiposo riflette sia l’iperplasia che l’ipertrofia degli adipociti ed è accompagnato da intolleranza al glucosio e insulino-resistenza (IR). I topi maschi ERαKO, quelli con l’ER-α eliminato ma con l’ER-β intatto, hanno mostrato infiammazione ↑AT, dimensioni degli adipociti e alterata tolleranza al glucosio rispetto ai topi maschi normali. Parliamo, ovviamente, di effetti possibili in contesto di deficienze.
E’ interessante notare come il metabolismo del glucosio per kg di muscolo è del 45% più elevato nelle donne e questo è probabilmente mediato da ER-α. [Physiol Rev. 2017 Jul]. Negli uomini, gli effetti metabolici benefici del T sono mediati più dal suo prodotto aromatico (E₂) che dagli androgeni (E₂ > T ↓ di deposito nel AT). Circa il 20% degli estrogeni circolanti nell’uomo deriva dalla sintesi e dalla secrezione testicolare (nelle cellule di Leydig) e il resto dall’attività dell’Aromatasi periferica. [Adv Exp Med Biol. 2017].
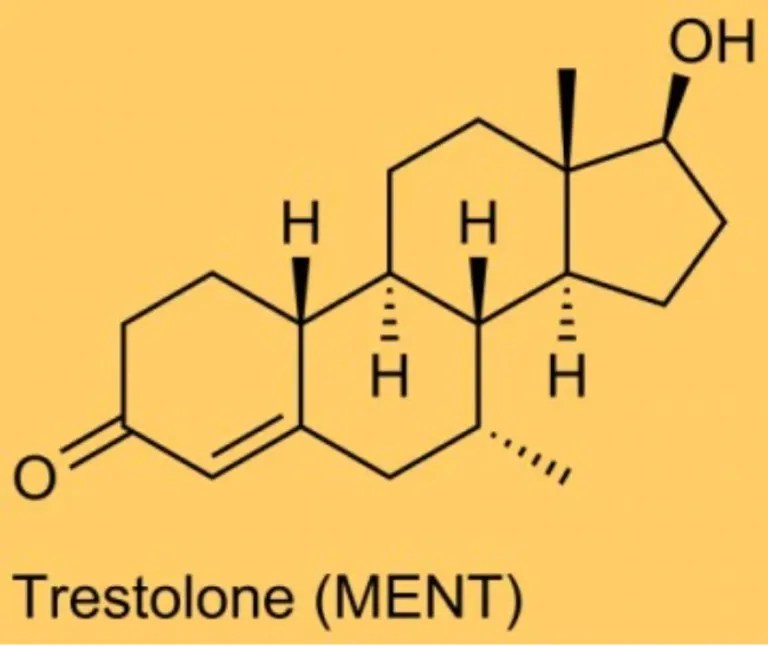
In pratica, per gli uomini che utilizzano dosi sovrafisiologiche di androgeni aromatizzabili (ad esempio, T, Nandrolone [Deca, NPP], Boldenone [EQ], Trestolone [MENT], Metandienone [Dianabol]), ciò significa che i siti extra-testicolari dell’Aromatasi, in particolare le cellule adipose, sono prevalentemente responsabili degli effetti associati agli estrogeni (estrogenicità). I bodybuilder che associano gli estrogeni a modifiche sfavorevoli della massa grassa non hanno di per sé torto. Infatti, se da un lato gli estrogeni regolano l’aumento della massa grassa, dall’altro la ridistribuiscono in modo più femminile, in particolare sui glutei e sui fianchi, piuttosto che nella zona addominale, un modello di distribuzione del grasso più maschile… Nuovamente, si tratta di quantità e tempo…
Inoltre, l’ER-α e -β sono espressi nel muscolo scheletrico umano in modo ubiquitario, nelle miofibre, nelle cellule endoteliali e nelle cellule satelliti. L’allenamento di contro-resistenza aumenta ER-α e -β nel muscolo scheletrico umano, suggerendo che la loro espressione è alterata dalle richieste funzionali del muscolo.
Mentre ER-α regola principalmente i lipidi del sangue e il rimodellamento osseo, si ritiene che ER-β medi il rimodellamento nel tessuto muscolare.
L’estradiolo (E₂) è un ligando “potente” per ER-β. Il suo tasso di dissociazione (κd) per questo recettore è di 2,08 nM nell’uomo. [Perkins, M. S., Louw-du Toit, R., and Africander, D. (2017). ]. Il suo tasso di dissociazione (κd) per ER-α, in confronto, è di soli 0,24 nM nell’uomo. [Perkins, M. S., Louw-du Toit, R., and Africander, D. (2017).]. Questo può spiegare gran parte del suo effetto anabolico nel muscolo.
Gli estrogeni sono componenti combinati di regimi steroidei somministrati a bovini maschi castrati per stimolare la crescita muscolare e migliorare la qualità della carne, il che indica che gli estrogeni hanno effetti anabolici sulla massa muscolare maschile. [Veldhuis, J. D., and Bowers, C. Y. (2003).]. Tuttavia, è importante notare le principali differenze tra i bovidi e l’uomo. Nei bovidi, l’E₂ esogeno aumenta in modo dose-dipendente l’IGF-I, ma nell’uomo l’effetto dell’E₂ sull’IGF-I è parabolico, soggetto a una forma a U inversa quando si traccia la dose (cioè la concentrazione ematica) sull’ascissa (asse delle ascisse) rispetto alla risposta (IGF-I) sull’ordinata (asse delle ordinate). Ciò è dovuto al fatto che gli estrogeni (ad esempio, l’E₂) diminuiscono la biodisponibilità dell’IGF-I aumentando la IGFBP-1. [Veldhuis, J. D., and Bowers, C. Y. (2003).]. Un altra volta emerge la necessità di un certo controllo estrogenico.
Nota: La terapia sostitutiva degli estrogeni (ERT; HRT) sembra facilitare la crescita del muscolo scheletrico nelle donne in postmenopausa, probabilmente attraverso effetti sulle cellule satelliti del muscolo. Dopo la menopausa, le donne subiscono un calo cronico dei livelli di estrogeni. Questo fenomeno è associato a una riduzione della sensibilità agli stimoli anabolici (ad esempio, l’allenamento di resistenza) che è reversibile con la ERT. Sebbene la ERT possa effettivamente diminuire la sintesi proteica muscolare al basale, aumenta la crescita dopo l’allenamento contro-resistenza, cioè la risposta anabolica. [Published 2019 Jan 15. ].
Gli estrogeni hanno anche un effetto drammatico sulla funzione muscolo-scheletrica, dato il loro ruolo nello sviluppo, nella maturazione e nell’invecchiamento di ossa, tendini e legamenti.
Gli estrogeni migliorano la massa e la forza muscolare e aumentano il contenuto di collagene nei tessuti connettivi. Tuttavia, a differenza di quanto accade nelle ossa e nei muscoli, dove gli estrogeni migliorano la funzione, nei tendini e nei legamenti gli estrogeni ↓rigidità, quindi ↓ prestazione e potenza e ↑ rischio di lesioni ai legamenti. [Published 2019 Jan 15.]
Mentre questo articolo si concentra sugli estrogeni, un articolo di prossima pubblicazione scritto da questo autore relativo agli AAS e al collagene, alle articolazioni e alle ossa (per-composto) approfondirà gli effetti di particolari androgeni aromatizzabili (ad esempio, Testosterone, Nandrolone, Metandienone) e non aromatizzabili (ad esempio, Stanozololo, Oxandrolone), del rhGH e di altri agenti anabolizzanti sui tessuti connettivi, compresi tendini, legamenti e ossa. Questi effetti sono diversi per classe e per composto. Mentre alcuni AAS e altri agenti anabolizzanti apportano benefici ai tessuti connettivi, altri li danneggiano.
Ricordiamoci che, sebbene gli androgeni abbiano effetti significativi sulle ossa maschili, gli estrogeni sono più importanti per la crescita e il mantenimento delle ossa. [Cauley, J. A. (2015).]. L’E₂ è essenziale per la mineralizzazione, la massa e il ricambio osseo normali negli uomini. [Cauley, J. A. (2015).].
L’aromatizzazione del T ⇒ E₂ è essenziale per gli effetti del T (T + E₂ > T) sull’osso (perdita + BMD). L’E₂ è chiaramente necessaria per la normale crescita e il mantenimento dell’osso negli uomini e l’E₂ media alcuni effetti del T sull’omeostasi ossea . L’azione finale degli estrogeni sullo scheletro è quella di diminuire il rimodellamento e il riassorbimento osseo, mantenendo la formazione ossea. [Cauley, J. A. (2015).].
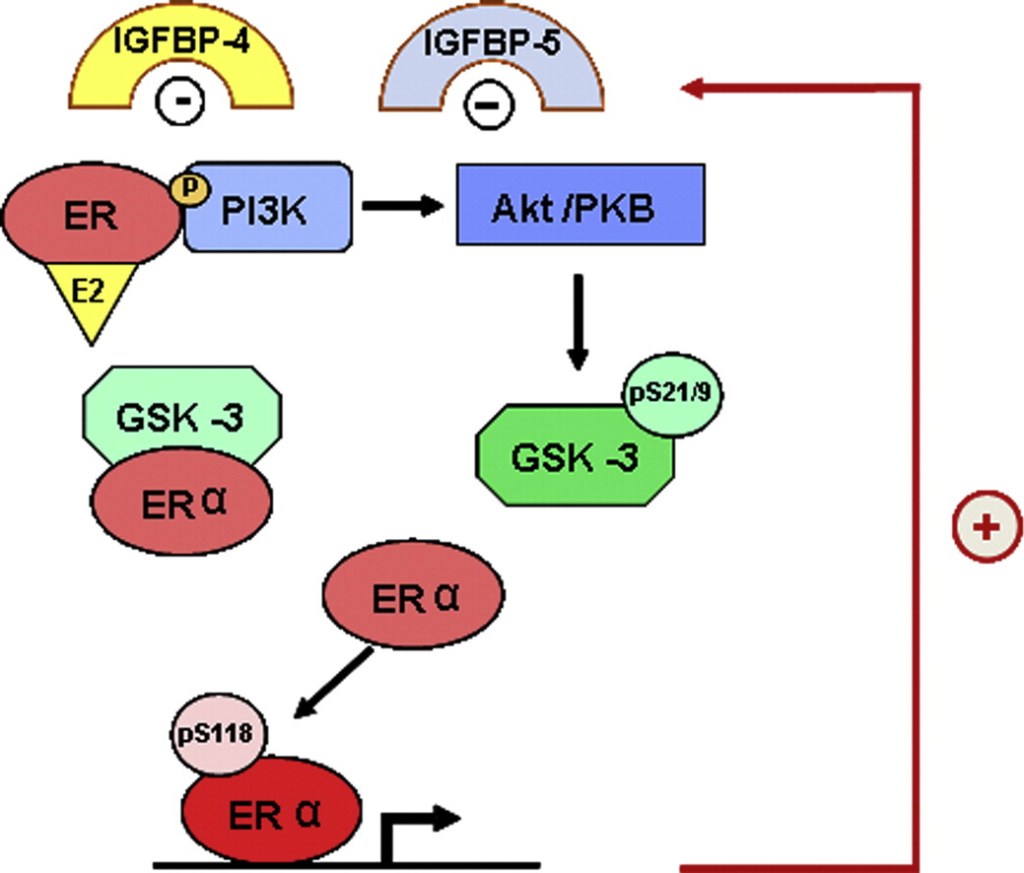
Gli estrogeni aumentano anche in parte il contenuto di collagene del tendine attraverso un effetto indiretto sull’IGF-I. Gli estrogeni modulano direttamente sia l’IGF-I che le IGFBP (Hansen, et al., 2009b) e quindi possono, tramite l’IGF-I, influenzare il contenuto di collagene attraverso un aumento della sintesi di proteine del collagene tramite la produzione della LARP6 (La-related protein 6) (Blackstone, et al., 2014). LARP6 è una proteina legante aumentata dall’IGF-I, che si lega direttamente all’mRNA del collagene di tipo I e aumenta specificamente la traduzione del collagene di tipo I.
Sia nel tendine che nel legamento, la composizione in peso secco è costituita principalmente da collagene – tra il 60 e l’85% nel tendine e circa il 75% nel legamento. Di questo collagene, la maggior parte è di tipo I: 60% nel tendine e fino all’85% nel legamento.
La rigidità dei legamenti è una buona cosa (✓), in quanto è utile per mantenere la stabilità dell’articolazione e il rischio di lesioni in questi tendini che collegano le ossa alle altre ossa. Al contrario, poiché il tendine collega l’osso rigido al muscolo conforme, un tendine più rigido non è sempre vantaggioso.
La rigidità del tendine è mista: i tendini più rigidi favoriscono le prestazioni, ma aumentano anche il rischio di lesioni. In termini di prestazioni (ad esempio, forza, potenza, sprint), l’aumento della rigidità del tendine trasmette più velocemente le forze muscolari all’osso, aumentando così le prestazioni; tuttavia, questo interesse deve essere bilanciato dal potenziale di concentrazione delle deformazioni all’interno del muscolo, con conseguente rottura. Quando un muscolo collegato a un tendine lasso si contrae, il tendine si allunga in modo flessibile mentre il muscolo si accorcia. Un tendine rigido, invece, non si allunga, ma è costretto ad allungarsi durante la contrazione, causando forze eccentriche. Ciò significa che in un muscolo collegato a un tendine rigido, un carico eccentrico maggiore per un determinato movimento aumenta il rischio di lesioni.
Gli estrogeni diminuiscono la rigidità dei tendini e dei legamenti, riducendo le prestazioni e aumentando il rischio di rottura dei legamenti. Un altra questione di “dose”.
Nel contesto sessuale/riproduttivo, la segnalazione ER-α nell’uomo è di supporto:
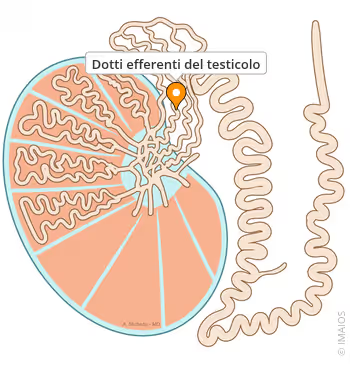
✓ Dotti efferenti e funzioni epididimali.
✓ Trasporto di ioni e riassorbimento di H₂O, necessari per sostenere il normale funzionamento dello sperma (riproduzione maschile).
✖ Concentrazioni di FSH e LH. [Bond, P. Article: Regulation of Testosterone Production. Aug 2021.].
Come l’E₂ nei confronti dell’IGF-I, l’effetto degli estrogeni sulla funzione sessuale è parabolico e soggetto a una forma a U inversa quando si traccia la dose (cioè la concentrazione nel sangue) sull’ascissa (asse delle ascisse) rispetto alla funzione sessuale (per esempio, libido, qualità erettile, fertilità) sull’asse delle ordinate (asse delle ordinate). Questo perché le concentrazioni alle quali l’E₂ sostiene i dotti efferenti, il funzionamento epididimale e la qualità e motilità degli spermatozoi sono basse, mentre quando gli estrogeni sono elevati (come nel caso di dosi sovrafisiologiche di T), le concentrazioni di FSH e LH e l’intera cascata del funzionamento ipotalamo-ipofisario gonadico iniziano a essere regolate negativamente. L’estradiolo è un ormone più fortemente soppressivo del T. [Veldhuis, J. D., and Bowers, C. Y. (2003).].
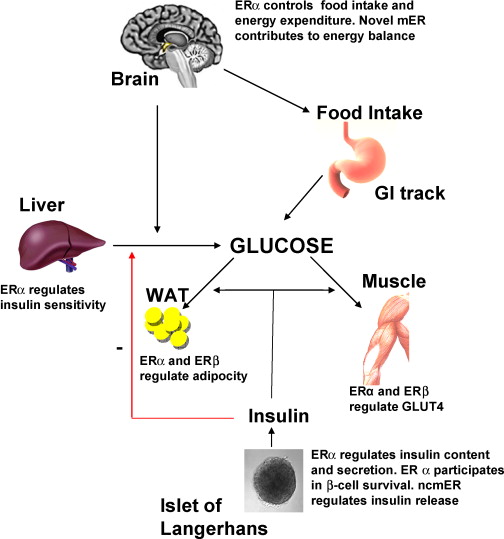
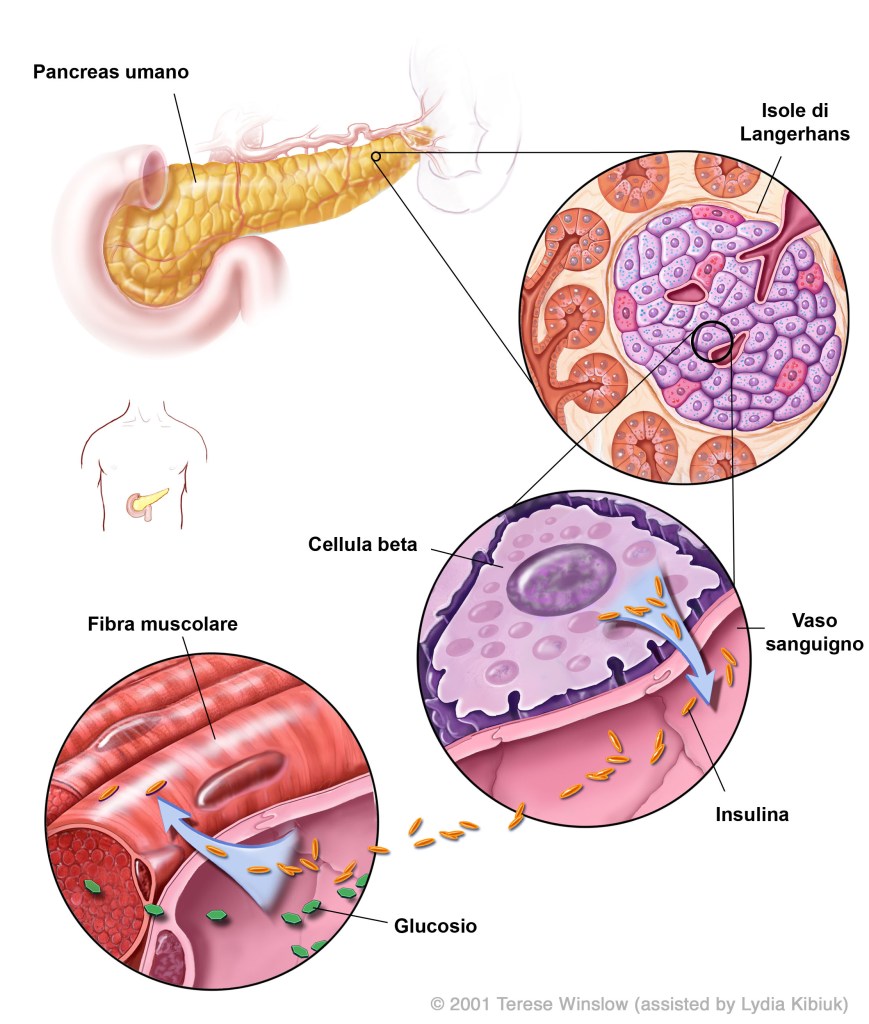
Gli estrogeni hanno effetti positivi sulla funzione delle β-cellule pancreatiche e sull’incidenza del diabete mellito, mediati in parte dagli effetti degli estrogeni sull’apoptosi delle β-cellule, sul contenuto di Insulina delle β-cellule, sull’espressione del gene dell’insulina e sul rilascio di insulina.
Non è assolutamente chiaro se gli estrogeni abbiano un effetto positivo o negativo sul sistema cardiovascolare. Mentre alcuni effetti protettivi del T sono mediati indirettamente attraverso l’E₂, la diminuzione del T sierico è più fortemente associata a rischi più elevati di morte per malattie cardiovascolari negli uomini rispetto alle variazioni dell’E₂ sierico. Al contrario, livelli sierici di E₂ più elevati sono stati riportati in uomini con malattia coronarica e arresto cardiaco improvviso.
A differenza delle femmine, l’E₂ inibisce la guarigione delle ferite nei maschi attraverso l’ER-α.
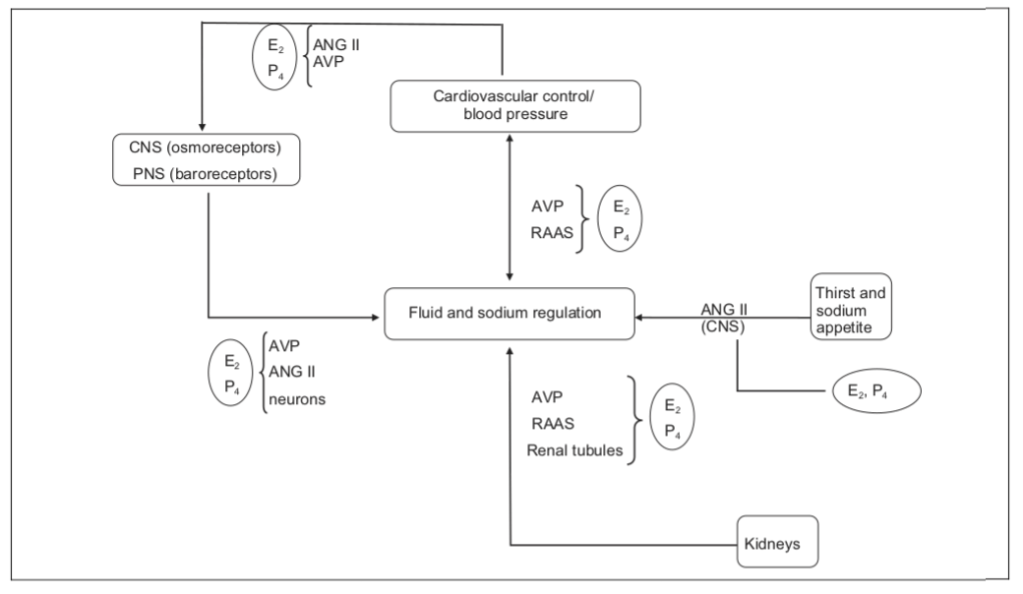
Risaputo è il fatto che l’E₂ influenza la ritenzione di liquidi agendo sul SNC (regolando la sete e l’assunzione di liquidi e sodio) e sul RAAS (agendo principalmente sull’aldosterone). [Curtis, K. S. (2015).].
Adesso la questione dovrebbe essere più chiara:
L’E2 deve essere mantenuto entro range di concentrazioni e attività/sensibilità ottimali per il soggetto al fine di sfruttare i benefici additivi e diretti di adeguati livelli di E2;
Ogni eccesso di E2, più si allontana dal punto 0 (ottimale) pende verso la comparsa di effetti collaterali maggiori degli eventuali benefici residui;
Il punto chiave primario nella modulazione funzionale dell’E2 per il miglioramento della composizione corporea risulta essere la manipolazione in difetto (abbassamento significativo) per previ periodi e livelli mantenuti regolari secondo sensibilità individuale (mantenimento livelli ottimali) nel lungo termine.
Gestione estrogenica in Bulk/Off-Season:
Abbiamo passato in rassegna i lati positivi e potenzialmente additivi al miglioramento della composizione corporea mantenendo livelli adeguati di E2. Abbiamo altresì visto, che vi è sempre un “collo di bottiglia”, un limite oltre il quale un aumento di E2 causa esclusivamente, o maggiormente, effetti collaterali controproducenti al miglioramento della condizione dell’atleta.
E’ corretto quindi partire esaminando le migliori possibilità di gestione estrogenica in una fase della preparazione annuale dove, solitamente, i bodybuilder, o i loro preparatori, trascurano il fattore estrogenico convinti di poter ottenere vantaggi soprattutto sulla ipertrofia ottenibile.
In questo caso, ovviamente, vanno tenute in considerazione le seguenti variabili soggettive:
Body Fat [variabile genetica-alimentare];
Tasso di aromatizzazione [variabile genetica];
Utilizzo di hCG [variabile iatrogena];
Uso di AAS con attività estrogenica intrinseca (quindi non secondaria alla aromatizzazione) come l’Oxymetholone [variabile iatrogena];
Presenza di un “accenno” di ginecomastia o sua precedente comparsa e successiva regressione [variabile genetica];
Nel caso del primo punto, la questione si risolve generalmente con un break diet/mini Cut della lunghezza dipendente dalla condizione della body fat. Per quanto riguarda, invece, il tasso di aromatizzazione, il quale è connesso all’espressione genica dell’enzima Aromatasi, essa può subire anche adattamenti epigenetici sul lungo termine anche, come accennato nella sezione dedicata alla TRT, con dosi di Testosterone entro il limite massimo fisiologico. L’atleta che presenta questa caratteristica predominante tra i punti sopradetti, e manifesta gli effetti tipici di una iperestrogenemia [ritenzione idrica con massivo aumento pressorio, sviluppo ginecomastico, aumento della massa grassa soprattutto con distribuzione a modello femminile ecc…] dovrà avere molta più accortezza nel dosaggio degli AAS aromatizzabili. Se il soggetto, non presenta un accenno di ginecomastia o non ha presentato tale accenno in passato [fatto regredire farmacologicamente] potrà intervenire con una minima dose efficace di AI [es. 0.5mg di Anastrozolo da 3 a 4 volte a settimana] in misura dipendente dalla dose di AAS aromatizzabili utilizzati e al monitoraggio dei livelli di E2. Se invece il soggetto presenta un accenno di ginecomastia [ovviamente non operabile] o ha presentato tale accenno in passato [fatto regredire farmacologicamente] potrà iniziare con l’uso di un SERM calibrato secondo dosaggio degli AAS aromatizzabili [generalmente da 10 a 30mg di Tamoxifene/die e 60mg/die di Raloxifene]. Ovviamente, anche la presenza di hCG può esacerbare tale situazione. In questo caso le scelte possono essere 1) interruzione d’uso dell’hCG per il tempo di durata della preparazione 2) riduzione del dosaggio e/o dilatazione della tempistica di somministrazione 3) aumentare la dose di AI in funzione dell’alterazione della risposta data. E’ inutile dire che la miglior scelta ricade spesso nel punto 2. Nel caso in cui sia presente un AAS con attività estrogenica intrinseca [quindi indipendente dall’attività della Aromatasi, come l’Oxymetholone] il soggetto, specie se presenta una certa sensibilità recettoriale, dovrebbe preventivamente assumere un SERM al dosaggio minimo efficace [es. 10-20mg di Tamoxifene] al fine di evitare eventuali comparse di ginecomastia legata alla suddetta molecola.
E per quanto riguarda le donne in Off-Season/Bulk?
Ormai sappiamo che la manipolazione della attività estrogenica mostra maggiore impatto sul miglioramento della composizione corporea nelle donne. L’Estradiolo, come estrogeno dominante, attraverso il legame con le subunità recettoriali influenza altre vie ormonali come l’Asse GH/IGF1. Infatti, nonostante la produzione giornaliera di hGH è circa 2 volte superiore nelle giovani donne rispetto agli uomini e varia di 20 volte in base allo sviluppo sessuale e all’età, la sua azione (compresa quella del IGF1) è strettamente influenzata dall’attività estrogenica (in senso inversamente proporzionale per concentrazione epatica di E2). Gli estrogeni inibiscono l’attivazione della via JAK/STAT da parte del hGH. L’inibizione è dose-dipendente e deriva dalla soppressione della fosforilazione di JAK2 indotta dal hGH, con conseguente riduzione dell’attività trascrizionale di STAT3 e STAT5. Gli estrogeni stimolano l’espressione di SOCS-2, che a sua volta inibisce l’azione di JAK2. L’espressione di SOCS-2 è sovra-regolata dagli estrogeni in modo ERα-dipendente. Dato il ruolo centrale di JAK2 nell’attivazione di molteplici cascate di segnalazione del GHR, gli estrogeni possono influenzare anche altre vie a valle per esercitare un impatto più ampio sull’azione del hGH e sulla riduzione del IGF-1 libero per via anche di un aumento delle IGFBP.
Gestione estrogenica in Cut/Pre-Contest:
Quando si parla di Cut e/o Pre-Contest, la riduzione marcata del fattore estrogenico per un breve periodo di tempo può risultare favorevole al miglioramento marcato della condizione.
“Spessore della pelle” e ritenzione idrica:
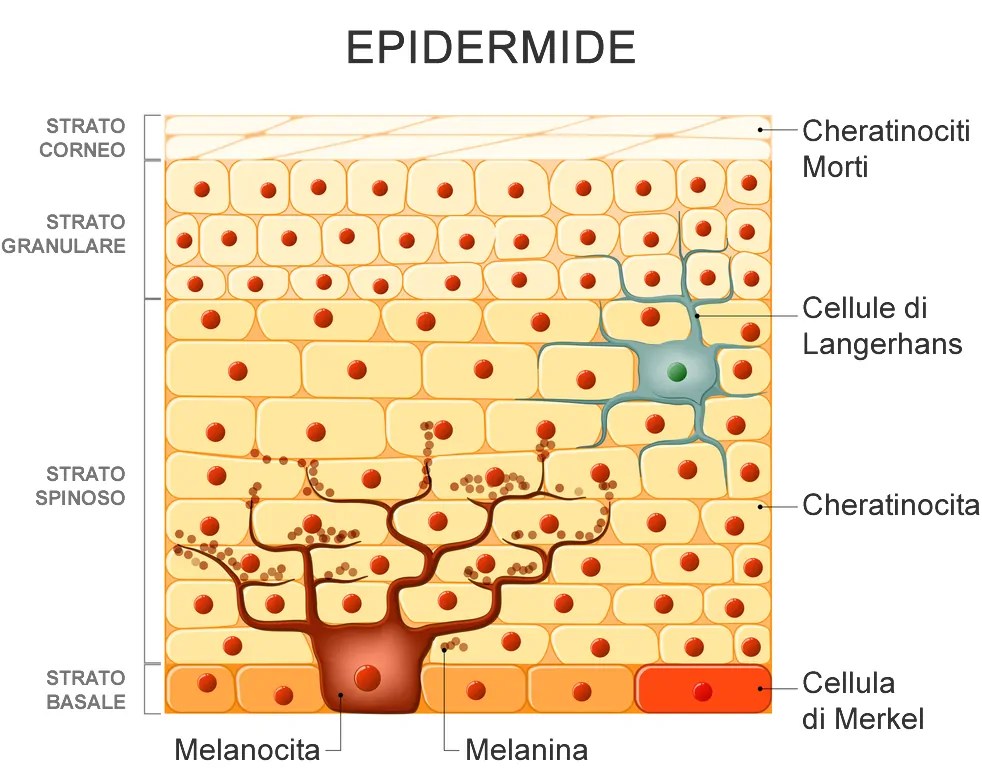
Quando si tratta di raggiungere la condizione da gara, l’eliminazione del grasso e dell’acqua sottocutanei sono i due obiettivi più importanti, in quanto hanno il più grande effetto complessivo sul aspetto. Questi non sono gli unici fattori, però. Variabili come la pienezza muscolare, la durezza e la densità muscolare, e le striature giocano un ruolo nel determinare il giudizio degli osservatori. Un altro fattore, anche se molto meno frequentemente esposto rispetto ai precedenti, è lo spessore della pelle.
La pelle è composta da tre strati; l’epidermide (strato più esterno), il derma (lo strato centrale), e il tessuto sottocutaneo (lo strato più interno). Anche se lo strato sottocutaneo è tecnicamente considerato parte della pelle, è li dove è conservato il grasso sottocutaneo ed è quindi molto variabile in termini di contenuto totale ed è distinto dagli altri due strati, che sono ciò che noi di solito pensiamo quando sentiamo la parola “pelle”. Il derma è facilmente il più spesso, dal momento che costituisce circa il 90% dello spessore totale della pelle e di conseguenza, è molto più adatto a sfocare la definizione muscolare che l’epidermide. Tuttavia, entrambi gli strati contribuiscono a questo effetto, rendendo necessaria la loro riduzione al minimo essenziale per la visualizzazione massima dei dettagli muscolari. Purtroppo, sia l’rhGH che gli estrogeni possono avere un effetto profondo sullo spessore della pelle, rendendo la loro cattiva gestione potenzialmente controproducente per raggiungere le condizioni ottimali per la gara.
Tralasciando l’effetto del rhGH, che al momento ci interessa “poco”, gli estrogeni sulla sintesi di collagene non sono di certo migliori, dal momento che interessano sia il derma e l’epidermide attraverso percorsi multipli.[https://pubmed.ncbi.nlm.nih.gov/] Nel valutare i suoi effetti sul derma, troviamo che gli estrogeni operano attraverso uno degli stessi meccanismi del hGH sul aumento della sintesi del collagene, ma il modo in cui si compie questo processo è un po’ diverso. In questo caso, gli estrogeni stimolano i fibroblasti dermici (cellule all’interno dello strato del derma che generano il tessuto connettivo), una funzione primaria è produrre collagene. In uno studio, gli estrogeni hanno dimostrato di aumentare la produzione di collagene di tipo I del 76%. Anche se non è così drammatica come con il rhGH, questo è ancora un aumento di tutto rispetto, soprattutto alla luce della capacità degli estrogeni di promuovere la sintesi di acido ialuronico. Direttamente coinvolto nella idratazione cutanea, un aumento dei livelli di acido ialuronico si traduce in un aumento del contenuto di acqua dermica e una successiva espansione del volume della pelle. In uno studio, la somministrazione di estrogeni ha mostrato un aumento della sintesi di acido ialuronico a un pieno 70% nel giro di due settimane.
Gli Estrogeni hanno anche pronunciati effetti sull’epidermide, aumentando lo spessore della pelle attraverso tre meccanismi distinti. Il primo è la stimolazione dell’attività mitotica nei cheratinociti; il tipo cellulare principale trovato nell’epidermide (i cheratinociti costituiscono circa il 90% di tutte le cellule epidermiche). In parole povere, questo significa che l’estrogeno induce la proliferazione dei cheratinociti tramite scissione cellulare, portando a un aumento complessivo del numero di cheratinociti presenti nella pelle. Il secondo è inibendo direttamente l’apoptosi (morte cellulare) dei cheratinociti e l’ottundimento della produzione di chemochine; molecole infiammatorie che possono potenzialmente contribuire alla distruzione della cellula. Infine, gli estrogeni svolgono un ruolo nella idratazione epidermica, volumizzando questo strato di pelle.
Eliminare gli effetti negativi degli estrogeni sullo spessore della pelle è fortunatamente un compito tutto sommato facilmente gestibile, in quanto richiede il mantenimento dei propri livelli di estrogeni entro l’intervallo massimo (come indicato sopra). E’ importante notare che gli effetti negativi degli estrogeni sullo spessore della pelle possono richiedere diversi mesi affinché vangano eliminati completamente, quindi il mantenimento di un elevato livello di estrogeni durante i precedenti mesi di preparazione alla gara per poi farli calare fino al livello minimo solo un paio di settimane prima dell’esibizione non è l’ideale. Per tutti coloro che usano grandi dosi di AAS aromatizzabili, o per i soggetti particolarmente sensibili, per la maggior parte della preparazione, è buona cosa tenere questo concetto in mente.
Basandomi sui dati raccolti sul campo da diversi atleti, e con la conferma anche di esperti del settore come Mike Arnold, la cosa migliore sarebbe optare per il mantenimento di base un livello di estrogeni normale (<60ng/dL con range ideale 30-40ng/dL) per poi calare i livelli a 10-20ng nell’ultimo paio di settimane prima della gara. Oltre a ciò, facendo calare i livelli verso il basso a circa 10ng nelle ultime settimane, si elimina quasi completamente la ritenzione idrica persistente che potrebbe ancora essere un problema, lasciando un livello di estrogeni sufficiente a garantire un ottimale accumulo di glicogeno in concerto additivo con altri PEDs cosomministrati [i dettagli su questo ultimo punto saranno trattati più avanti].
Effetto sui recettori α2-AR e “grasso testardo”:
Nel tessuto adiposo sottocutaneo (sc), gli estrogeni possono aumentare il numero di recettori α2A-AR, che sono coinvolti nell’inibizione della lipolisi. Ciò può contribuire al tipico modello di distribuzione del grasso femminile, in cui il grasso viene immagazzinato maggiormente a livello sottocutaneo piuttosto che viscerale. Inoltre, una condizione estrogenica non ben manipolata può rendere , dato questo effetto, più difficile raggiungere definizioni estremizzate.
E’ stato infatti osservato che l’aumento del numero di recettori α2A-AR causa una risposta lipolitica attenuata dell’epinefrina negli adipociti sc; al contrario, non è stato osservato alcun effetto degli estrogeni sull’espressione dell’mRNA dei recettori α2A-AR negli adipociti del deposito di grasso intra-addominale. Dai risultati ci viene mostrato che gli estrogeni abbassano la risposta lipolitica nel deposito di grasso sc aumentando il numero di recettori α2A-AR, mentre gli estrogeni non sembrano influenzare la lipolisi negli adipociti del deposito di grasso intra-addominale. Questi risultati dimostrano che gli estrogeni attenuano la risposta lipolitica attraverso la sovraregolazione del numero di recettori α2A-AR antilipolitici solo nel sc e non nei depositi di grasso viscerale. Pertanto, questi risultati offrono una spiegazione del modo in cui gli estrogeni, se non ben modulati, rendono molto più ostica la mobilitazione del grasso sc.[https://pubmed.ncbi.nlm.nih.gov/15070958/]
Quindi, per avere un ottimale controllo sulla manipolazione del numero e densità dei α2A-AR al fine di facilitare la mobilitazione e l’uso dei depositi adiposi sc nelle aree particolarmente ostiche, oltre all’uso concomitante di Yohimbina e ACE II inibitori, il mantenimento per circa 8-12 settimane di un range del E2 pari a 20-30pg/dL risulta particolarmente funzionale.
Composizione strategica di AAS/SERM/AI per il maggior controllo estrogenico nel Pre-Contest/Cut:
Un ulteriore dettaglio di design della preparazione alla gara o Cut risiede nel corpo dei PEDs componenti la stessa. Vi sono, infatti, AAS che sono particolarmente funzionali al massimo controllo della attività estrogenica e che trovano il loro migliore inserimento proprio in quei momenti della preparazione dove la riduzione marcata della componente estrogenica è essenziale per il “tocco finale” per il palco, lo shooting fotografico o per un semplice obbiettivo personale da amatore.
Boldenone Undecylenato:
Molecola di Boldenone
Il Boldenone [1,4-androstadiene-3-one,17b-ol] è uno steroide anabolizzante-androgeno spesso legato all’estere Undecylenato. Strutturalmente molto simile al Testosterone, il Boldenone differisce da questo per il doppio legame tra C1 e C2. Tale modifica rende la molecola un substrato molto meno affine all’enzima 5-α reduttasi rispetto al Testosterone, ed era teoria comune che riducesse anche il tasso di aromatizzazione della molecola.
Ed è per questo ultimo punto che molti ritengono che il Boldenone possa essere utilizzato come base “Mix” con il Testosterone o in sostituzione ad esso per coloro che sono inclini agli effetti collaterali estrogenici. E, in teoria, utilizzando il Boldenone, si potrebbe ridurre il rischio di sviluppare effetti collaterali correlati ad un livello elevato di estrogeni poiché dovrebbe aromatizzare circa la metà del Testosterone. Vista la riduzione marcata del E2 si era ipotizzato che uno (o più) dei suoi metaboliti agisca come un Inibitore dell’Aromatasi (AI). Secondo questa ipotesi, i metaboliti del Boldenone sono in realtà la causa del ridotto impatto dell’Enzima Aromatasi su questa molecola e su altri substrati soggetti come il Testosterone. Ma, i limiti di verifica hanno reso questa ipotesi piuttosto traballante; i livelli sierici di Estradiolo sono stati spesso determinati utilizzando kit di test immunologico per elettrochemiluminescenza (ECLIA). Parliamo di uno dei peggiori metodi di test ematico dal momento che è soggetto a influenze ormonali non ricercate o a limitazione di precisione della conta ormonale.
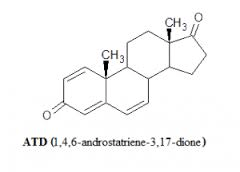
In letteratura, passando al vaglio i vari metaboliti del Boldenone, il famoso “AI” del mercato grigio ATD non era elencato nello studio sui metaboliti umani condotto sul Boldenone [https://pubmed.ncbi.nlm.nih.gov/]. Proprio come con l’ADD, l’ATD ha dimostrato di essere sia un metabolita del Boldenone che di metabolizzare in Boldenone [https://pubmed.ncbi.nlm.nih.gov/]. In uno studio in vitro è stato dimostrato che l’ATD riduce significativamente la biosintesi degli estrogeni.[https://pubmed.ncbi.nlm.nih.gov/] Il problema, però, è che l’ATD non risulta essere un metabolita significativamente espresso nell’uomo e, nel pratico, la sua potenzialità AI è spesso stata deludente o ad un tasso blando. La risultante della ricerca, quindi, non solo non chiarisce se il Boldenone agisca come un AI ma anche se esso aromatizza in Estradiolo ad un dato tasso.
Rappresentazione dei passaggi componenti il test immunologico per elettrochemiluminescenza (ECLIA).
Le cose diventano più chiare quando i campioni ematici vengono analizzati con test LC/MS-MS ultra sensibile e non l’ECLIA. Analizzando i campioni di un utilizzatore sottoposto ad unciclo comprendente circa 850mg di Boldenone Undecylenato e 250mg di Testosterone Enantato a settimana, i risultati mostravano un significativo aumento dei livelli di Estrone, con un risultato di 662pg/mL, con il limite massimo dell’intervallo di riferimento pari a 65pg/mL. Il livello di Estradiolo non era rilevabile con meno di 2,5pg/mL. Sappiamo, inoltre, che il Testosterone utilizzato dal soggetto in questione era realmente Testosterone (1431 ng/dL), poiché, per l’appunto, i campioni ematici sono stati sottoposti ad un test specifico LC/MS-MS, che è il gold standard per verificare il totale esatto del Testosterone evitando il rilevamento incrociato di altri anabolizzanti. La quantità di Testosterone rilevata, normalmente, porterebbe ad un livello di Estradiolo medio-alto, non di certo così basso come è risultato. Fortunatamente, con il risultato del esame del sangue comprendente l’Estrone [E1], possiamo finalmente risolvere l’enigma su ciò che con molta probabilità accede realmente.
E’ stato osservato come anche con uno schema di dosaggio quasi identico tra Testosterone e Boldenone (vedi esempio 300mg di Boldenone e 400mg di Testosterone) i livelli di Estradiolo risultano generalmente bassi.
Quindi, a questo punto, si potrebbe ipotizzare che il Boldenone non aromatizza effettivamente in Estradiolo, né inibisca l’enzima Aromatasi ma, piuttosto, esso potrebbe competere con il Testosterone nell’interazione con l’Aromatasi dando come prodotto aromatico l’E1.
Il Boldenone sembra avere una maggiore tendenza alla conversione in Estrone e una forza di legame all’enzima Aromatasi dose-dipendente
L’E1 è un estrogeno molto meno potente dell’estradiolo e, in quanto tale, è un estrogeno relativamente debole.[Kuhl H (August 2005).] Secondo uno studio, le affinità di legame relative dell’estrone per l’ERα e l’ERβ umani erano rispettivamente il 4,0% e il 3,5% di quelle dell’Estradiolo. Secondo uno studio, l’affinità di legame relativa dell’E1 per l’ERα e l’ERβ umani è pari al 4,0% e al 3,5% di quella dell’E2, rispettivamente, e la capacità transattiva relativa dell’E1 per l’ERα e l’ERβ è pari al 2,6% e al 4,3% di quella dell’E2, rispettivamente.[Escande A, Pillon A, Servant N, Cravedi JP, Larrea F, Muhn P, et al. (May 2006).] In accordo, l’attività estrogenica dell’estrone è stata riportata a circa il 4% di quella dell’Estradiolo.
La via principale attraverso la quale l’Estrone viene biosintetizzato coinvolge l’Androstenedione come intermedio, con quest’ultimo che viene convertito in Estrone dall’enzima Aromatasi. Questo è il punto chiave da ricordare nel contesto di questa anamnesi di design dei componenti PEDs della Preparazione alla gara o Cut.
In definitiva, il Boldenone, con tutta probabilità, ha una funzione di “ormone esca” per l’enzima Aromatasi. Sappiamo però che, probabilmente, la sua conversione in estrogeno lo vede convertirsi prevalentemente in Estrone [E1] e non in Estradiolo [E2]. Sappiamo che l’Estrone può convertirsi in Estradiolo (e viceversa) ma che il tasso in cui ciò avviene è molto basso. Siamo a conoscenza del fatto che l’E1 è un estrogeno molto meno potente dell’E2 e, come tale, è un estrogeno relativamente debole.[Kuhl H (August 2005), Escande A et al. (May 2006), Ruggiero RJ, Likis FE (2002)]. Ciò, darà come risultante, una attività estrogenica tissutale [genomica e non genomica] nettamente ridotta anche con un dosaggio di Boldenone non particolarmente elevato [es. 300-500mg/week].
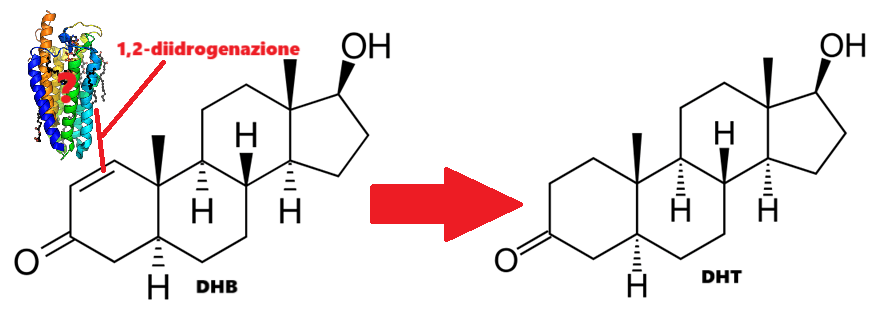
DHB:
Molecola di DHB
L’1-Testosterone (maggiormente noto come Dihydroboldenone, abbreviato in DHB), è uno steroide anabolizzante-androgeno sintetico (AAS) e un derivato 5α-ridotto del Boldenone (Δ1-testosterone). Si differenzia dal prodotto 5α-ridotto del Testosterone, il DHT, per la presenza di un doppio legame C1-C2 nell’anello A dello scheletro carbossilico.[William Llewellyn (2009).] Il DHB possiede una potente affinità di legame con il AR ma non presenta particolare selettività tissutale e ha un’elevata capacità di stimolare la transattivazione AR-dipendente. In vivo, a differenza del Testosterone Propionato, il DHB aumenta anche il peso del fegato con possibile disfunzionalità d’organo [dose e tempo dipendente].[Friedel A et al. (August 2006).]
Il DHB è strutturalmente simile al Methenolone (Primobolan/Rimobolan) e allo Stenbolone. In quanto 1-ene, il DHB è un AAS le cui caratteristiche anabolizzanti predominano su quelle androgene (mascolinizzanti, calvizie, ecc.).
Differenze e similitudini molecolari tra Methenolone e DHB
È interessante notare che il DHB metabolizza principalmente a DHT (5α-diidrotestosterone; DHT), pur non essendo soggetto a nuove interazione con la 5α-reduttasi, ed è particolarmente epatotossico – cosa insolita per un AAS iniettabile – sia direttamente, con prove dell’aumento delle dimensioni del fegato (visto prevalentemente negli animali), sia indirettamente, dato che i produttori e i chimici usano tipicamente solventi industriali come il guaiacolo, una sostanza chimica pericolosa e probabilmente cancerogena, per mantenere il DHB in soluzione.
Struttura Proteine C-reattiva
La particolare intollerabilità e i controversi effetti anabolizzanti del DHB, considerato da una fazione di fedelissimi come un “Tren lite” e da un’altra fazione di detrattori che lo considerano fondamentalmente un Methenolone meno selettivo [della stessa classe di 1-eni] ma con dolore post-iniezione (PIP) e innalzamento della Proteina C-reattiva (CRP), lo classificano tra gli AAS meno utili per i bodybuilder. Eppure, c’è una caratteristica che, a “piccole dosi” e “strategicamente” può rendere il DHB una molecola opzionale da inserire nella preparazione per il controllo estrogenico.
Per introdurre questa potenzialità del DHB, vorrei sottolineare quanto accennato in precedenza sul principale metabolita di questo AAS: il DHT. Il DHB metabolizza principalmente in DHT, pur non essendo suscettibile alla attività della 5α-reduttasi [Fragkaki et al. C. (2009).]. Come ben sappiamo, specialmente in passato, il DHT veniva utilizzato per trattare le pazienti con cancro al seno estrogeno dipendente. L’aumento di questa molecola in risposta ai processi metabolici ai quali è soggetto il DHB causa un controllo estrogenico a livello dell’attività tissutale.
Esistono solo spiegazioni imperfette per la sintesi di DHT come metabolita del DHB. Peter Bond ha comunicato via e-mail di essere rimasto sorpreso dall’identificazione da parte di Fragkaki del DHT come metabolita primario dell’1-testosterone, poiché suggerisce che qualche enzima finora non identificato è responsabile della 1,2-diidrogenazione di questo androst-1-ene-3-one. [Bond, Peter 2 April 2022].
Processo ipotetico dato dall’enzima “X” che interagendo con il DHB avvia la 1,2-diidrogenazione dando come metabolita principale il DHT.
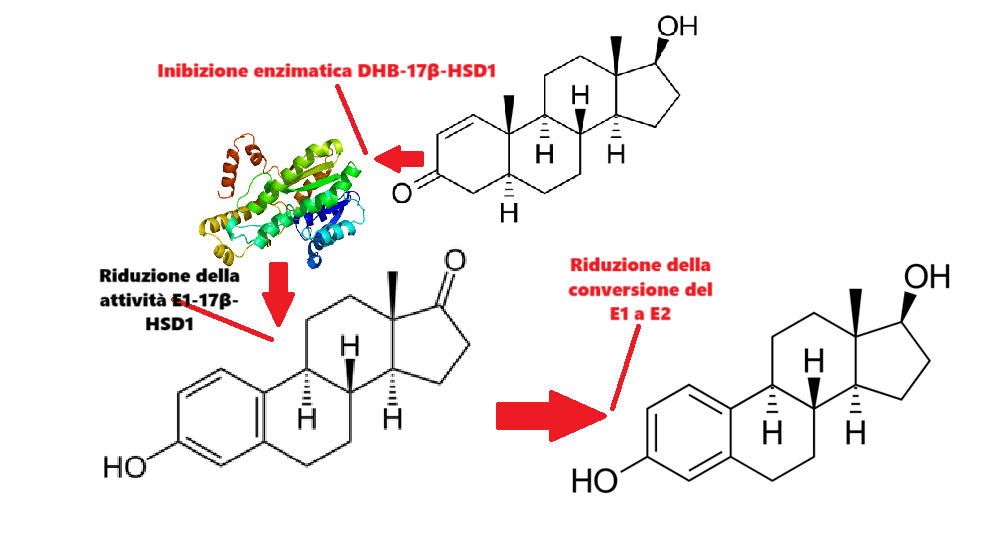
Ma l’attività propria del DHB sulla componente estrogenica dipende da una capacità di aumentare l’E1.
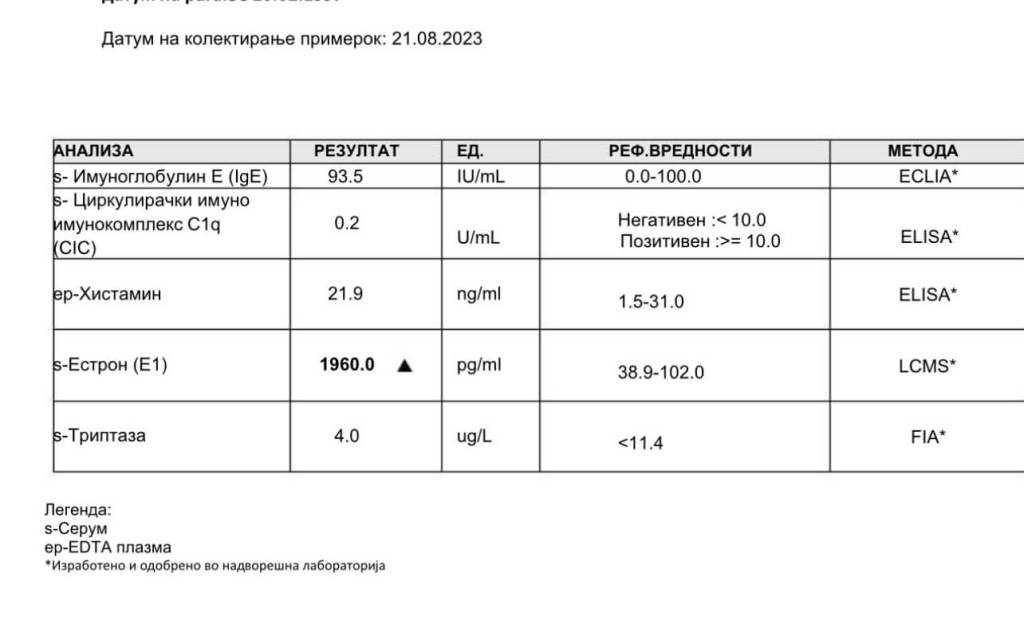
Come si può vedere dalla sopraesposta griglia, la LCMS sensibile per l’Estrone (E1) mostra un valore notevole di 1.960,0 pg/mL per l’E1 in un individuo che utilizza dosi fino a 700mg di DHB i.m. q.w. con una costante di 100mg di Testosterone i.m. q.w. Tali alterazioni sono state viste anche con dosi di DHB inferiori [es. 200-400mg/week] anche se l’impatto è ovviamente a grado dose-dipendente.
L’ipotesi è che il DHB possa agire come inibitore della 17β-HSD1, aumentando così il DHT e l’E1 e diminuendo l’E2. L’E2 stimola, mentre il DHT inibisce la crescita del cancro al seno e lo sviluppo della ginecomastia (modulando l’attività tissutale e sistemica del E2).
Struttura della 17β-HSD1
La 17β-idrossisteroide deidrogenasi 1 (17β-HSD1) è un enzima che nell’uomo è codificato dal gene HSD17B1.[Luu-The V et al. Feb 1990] Questo enzima ossida o riduce il gruppo C17 idrossi/cheto di androgeni ed estrogeni ed è quindi in grado di regolare la potenza di questi steroidi sessuali.
Questo enzima è responsabile dell’interconversione di Estrone (E1) ad Estradiolo (E2) e dell’interconversione di Androstenedione a Testosterone:
L’isozima 17β-HSD1 umano è altamente specifico per gli estrogeni rispetto agli androgeni, mentre l’isozima dei roditori è meno specifico.[Saloniemi T et al. 2012]
Il DHT, strutturalmente simile al DHB, è un noto, anche se debole, inibitore della 17β-HSD1. Affinché gli androgeni DHT o Testosterone (T) diventino un buon ligando per questo enzima, devono imitare più da vicino la forma planare dell’anello A dell’E2 e fornire un ampio nucleo idrofobico. [He, W., Gauri, M., Li, T., Wang, R., & Lin, S.-X. (2016).]. Il DHB soddisfa entrambi i requisiti, differenziandosi dal DHT per un doppio legame C-1- C-2, che gli conferisce un anello A più simile a quello degli estrogeni, e dal Testosterone per la sua ampia spina dorsale idrofobica di 5α-androstene. Questi 5α-androsteni hanno un’affinità per l’AR 173 volte maggiore rispetto alla loro controparte orientata verso il β; ad esempio, il DHT ha una maggiore planarità ed è meno ingombrante rispetto al T per inserirsi nella cavità idrofobica del sito di legame sull’AR. [Liao, S., Hung, S. C., Tymoczko, J. L., & Liang, T. (1976)].
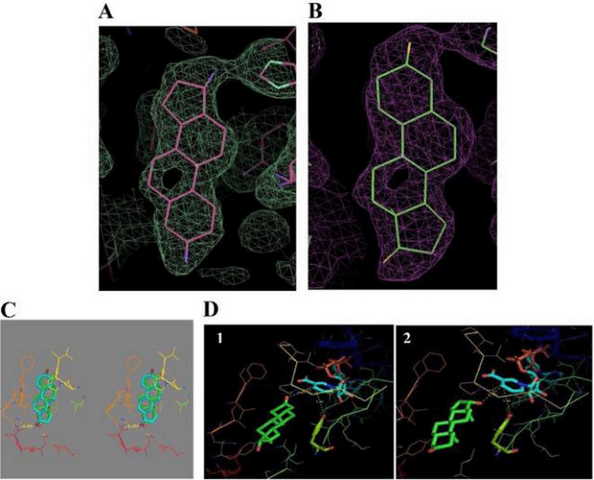
Legame 17β-HSD1 da parte del DHT: (A) – (E) Cristallizzazione 3D del complesso DHT/17β-HSD1 e dati di docking.
L’enzima 17β-HSD1 è altamente espresso nel tessuto adiposo e nella prostata degli uomini. La sua funzione è quella di catalizzare la riduzione dell’estrogeno debole E1 all’estrogeno forte E2 e la sua espressione è correlata positivamente all’attivazione dell’E1 e ai livelli di E2. L’inibizione della 17β-HSD1 riduce l’E2, anche se l’abolizione (a 0,00pg/mL) richiederebbe l’inibizione anche dell’Aromatasi e della Solfatasi [Thomas MP et al. J Steroid Biochem Mol Biol. 2013].
La 17β-HSD1 catalizza l’ultima fase della biosintesi degli estrogeni attivi (E2 e 5-androstene-3β,17β-diolo), mentre l’E1 è il suo substrato primario, l’enzima ha una certa attività per substrati non estrogenici. Il DHT è un substrato non riconosciuto dalla 17β-HSD1 e si lega in modo simile all’E2, con un’orientazione simile, ma con alcune differenze per accogliere un’ulteriore massa (per esempio, il gruppo metilico C-19). Il complesso (17β-HSD1-DHT) ha siti di legame con gli steroidi ben definiti e il valore Km noto per il DHT è 17 volte superiore a quello dell’E2 (quindi ci vuole più DHT per saturare l’enzima) e l’attività specifica 26 volte inferiore. [Lin, S. X., et al. 1999].
Azione schematizzata del DHB come inibitore della 17β-HSD1
In conclusione, il DHB, se inibisce la 17β-HSD1, lo fa in modo contenuto – certamente non con una specificità sufficiente per essere commercializzato per uso clinico nel trattamento del cancro al seno. Sebbene la letteratura citata suggerisca che gli androgeni C19 non sono inibitori della 17β-HSD1 altamente potenti e selettivi – in modo da consentire un buon legame e un’elevata velocità di reazione – gli androgeni sono ligandi attivi per l’enzima. Per massimizzare il legame, un inibitore progettato per essere specifico per la 17β-HSD1 deve imitare la forma planare dell’anello A dell’E2 e mantenere un ampio nucleo idrofobico che possa interagire con il sito di legame steroideo dell’enzima. [Lin, S. X., et al. 1999]. L’anello A del DHB, più simile a quello degli estrogeni, e l’ampio backbone idrofobico del 5α-androstene soddisfano questi requisiti, rendendolo un androgeno C19 teoricamente più potente del DHT nell’inibire la 17β-HSD1.
Anastrozolo e Exemestane:
Dell’Anastrozolo abbiamo già parlato nel paragrafo in riferimento alla gestione estrogenica durante una TRT. Abbiamo ribadito che in condizione di attività testicolare mantenuta per via d’uso dell’hCG, l’Anastrozolo darà una soppressione del E2 al dosaggi tra 0,5 e 1mg/die di circa il 50% . Data questa variabile, generalmente si interrompe la somministrazione di hCG 21-14 giorni prima del contest in concomitanza con la sospensione d’uso a 14 giorni dal contest del Testosterone. L’ambiente creato, in calo di substrati aromatizzabili a E2 e stimolanti l’espressione del Aromatasi [vedi hCG], il potenziale dell’Anastrozolo non solo aumenta in % di rapporto con i substrati con cui competere ma anche in termini di efficacia per concentrazione.
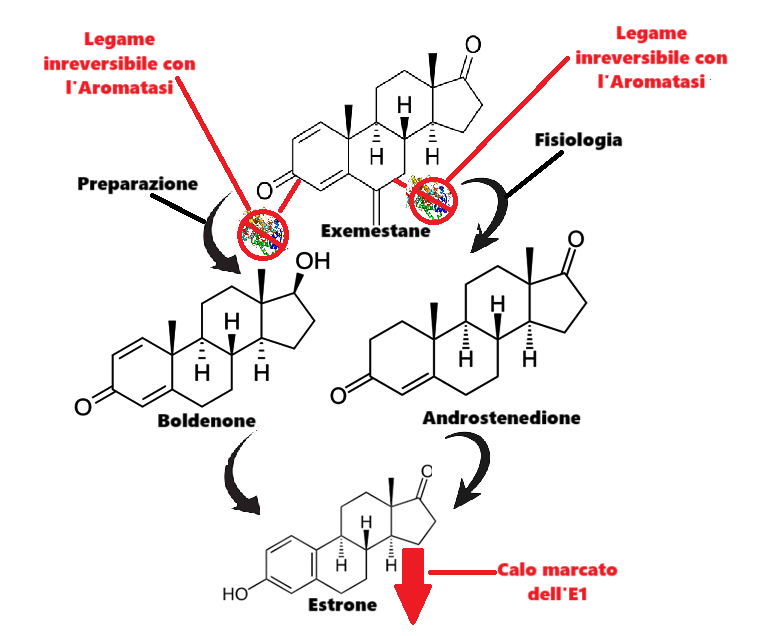
Come abbiamo visto in precedenza, il Boldenone sembra essere particolarmente soggetto alla conversione nel debole E1. Sebbene questo estrogeno sia di debole potenza biologica, con dosi di 500mg> di Boldenone potrebbe giocare un ruolo di “fino” la sua riduzione. Sicuramente starete pensando che basta l’Anastrozolo anche per questo scopo, e non è del tutto sbagliato come ragionamento. Però, e c’è un però, esiste un AI che potrebbe lavorare in sinergia con l’Anastrozolo nelle ultime settimane pre-contest e “occuparsi” maggiormente della componente E1. Sto parlando del Exemestane, comunemente conosciuto come Aromasin.
Molecola di Exemestane
L’Exemestane è un inibitore orale steroideo dell’Aromatasi. L’Exemestane è un inattivatore irreversibile steroideo di tipo I dell’Aromatasi, strutturalmente correlato al substrato naturale 4-androstenedione. Agisce come falso substrato per l’enzima Aromatasi e viene trasformato in un intermedio che si lega irreversibilmente al sito attivo dell’enzima causandone l’inattivazione, un effetto noto anche come “inibizione suicida”. Essendo strutturalmente simile ai bersagli enzimatici, l’Exemestane si lega in modo permanente agli enzimi, impedendo loro di convertire gli androgeni in estrogeni.[Jasek, W, ed. (2007)]
Uno studio condotto su giovani maschi adulti ha rilevato che il tasso di soppressione degli estrogeni per l’Exemestane variava dal 35% per l’E2 al 70% per l’E1.[Mauras N et al. December 2003] Ed è proprio questa la caratteristica che rende sensato l’uso del Exemestane in combinazione, e in questa fase, con l’Anastrozolo.
Ma perchè questa caratteristica?
L’Exemestane è noto chimicamente come 6-metilideneandrosta-1,4-diene-3,17-dione. Come gli inibitori dell’Aromatasi Formestano e Atamestano, l’Exemestane è uno steroide strutturalmente simile al 4-androstenedione, il substrato naturale dell’Aromatasi. Si distingue dalla molecola A4 solo per il gruppo metilidenico in posizione 6 e per un ulteriore doppio legame in posizione C1-2. Si è quindi ipotizzato che la sua similitudine al A4 sia una possibile risposta alla maggiore soppressione del E1 rispetto al E2.
Molecola di Androstenedione
L’Androstenedione, o 4-androstenedione (abbreviato in A4 o Δ4-dione), noto anche come androst-4-ene-3,17-dione, è un ormone steroide endogeno debole e intermedio nella biosintesi dell’Estrone e del Testosterone a partire dal DHEA. È strettamente correlato all’Androstenediolo (androst-5-ene-3β,17β-diolo). Esso è un substrato diretto per l’E1 che, a sua volta, può essere convertito in E2 per via della attività della 17β-HSD1 [vedi sezione dedicata al DHB]; tuttavia il tasso di conversione è generalmente basso.
Questa similitudine nella struttura steroidea sembra indicare una ipotetica competizione maggiore e migliore con il 4A piuttosto che con il Testosterone, con la risultante della maggiore soppressione del E1 rispetto all’E2.
Oltre alla similitudine conformazionale con il 4A, l’Exemestane condivide una similitudine tipica degli “1-ene” e del Boldenone: il doppio legame in C-1-C-2.
Unendo i due “punti di similitudine molecolare” possiamo ipotizzare una maggior competizione di legame specifica con il Boldenone e una ulteriore e addizionale diminuzione del livello estrogenico assoluto anche a carico del E1. I test fatti per verificare la teoria sono stati soddisfacenti, e il “tocco dry” raggiunto dai soggetti sperimentali era generalmente maggiore rispetto a coloro che non hanno applicato questa strategia.
Competizione schematizzata del Exemestane per il legame con l’enzima Aromatasi nei confronti dei due substrati “similari”, Boldenone e Androstenedione.
Un possibile approccio con questo “AI mix” prevede l’uso alternato di 0,5mg di Anastrozolo e 12,5-25mg di Exemestane per le ultime due settimane di preparazione alla gara.
Un altra ipotesi che si unisce a quella sopra esposta, vede una attività inibitoria maggiore sulla Solfatasi steroidea da parte del Exemestane e una conseguente riduzione dell’Estrone attivo.
La proteina codificata da questo gene catalizza la conversione dei precursori steroidei solfatati in steroidi liberi. Ciò include il DHEA solfato, l’Estrone solfato, il Pregnenolone solfato e il Colesterolo solfato, tutti nelle loro forme non coniugate (DHEA, Estrone, pregnenolone e colesterolo, rispettivamente).[4][5] La proteina codificata si trova nel reticolo endoplasmatico, dove è presente come omodimero.
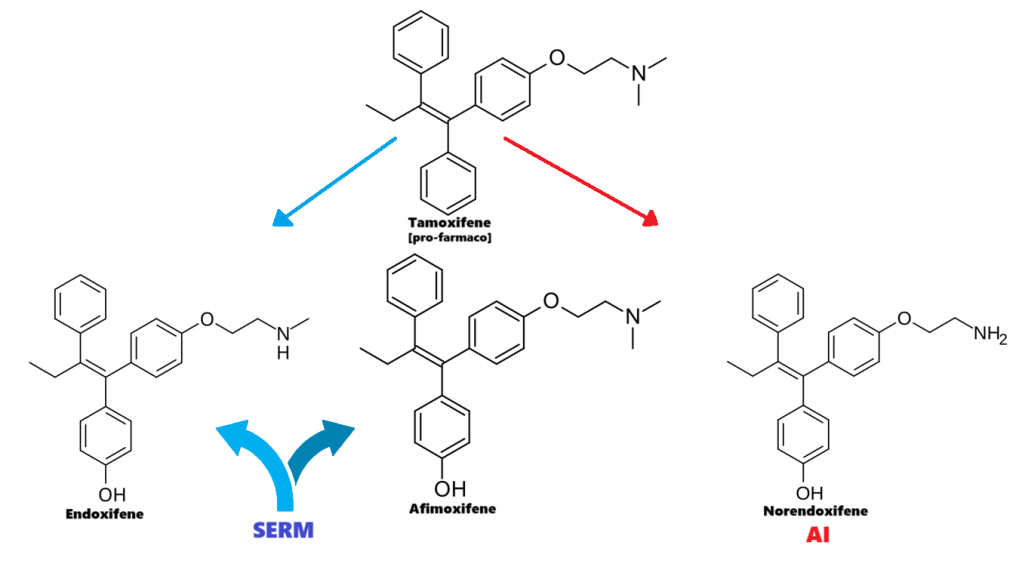
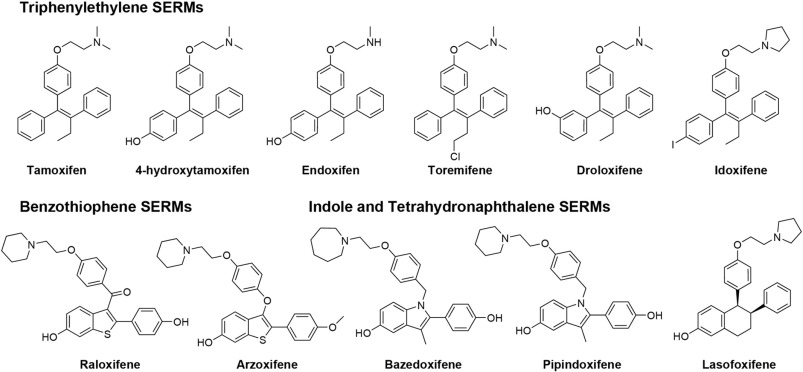
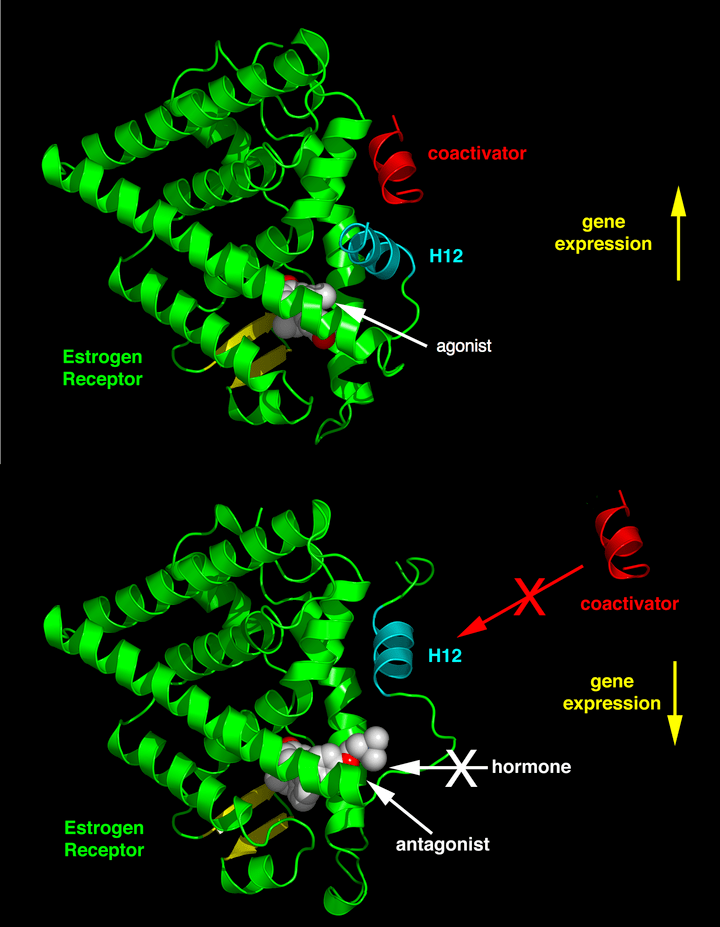
Tamoxifene:
Molecola di Tamoxifene
Il Tamoxifene è un pro-farmaco i cui metaboliti agiscono come modulatori selettivi dei recettori degli estrogeni (SERM), ovvero come agonista parziale dei recettori degli estrogeni (ER), e come Inibitori della Aromatasi (AI). Ha un’attività mista estrogenica e antiestrogenica, con un profilo di effetti diverso a seconda dei tessuti. Ad esempio, il prodotto metabolico del Tamoxifene ha effetti prevalentemente antiestrogenici nel seno, ma prevalentemente estrogenici nell’utero e nel fegato. Nel tessuto mammario, agisce come antagonista ER, inibendo la trascrizione dei geni che rispondono agli estrogeni.[Wang DYet al. (February 2004)] Un effetto collaterale benefico del Tamoxifene è che previene la perdita di massa ossea agendo come agonista ER (cioè imitando gli effetti degli estrogeni) in questo tipo di cellule. Pertanto, inibendo gli osteoclasti, previene l’osteoporosi.
Come accennato, il Tamoxifene agisce come pro-farmaco di metaboliti attivi come l’Endoxifene (4-idrossi-N-desmetiltamoxifene) e l’Afimoxifene (4-idrossitammoxifene; 4-OHT).[Klein DJ et al. (November 2013).] Questi metaboliti hanno un’affinità per gli ER da 30 a 100 volte superiore a quella del Tamoxifene stesso.[Ahmad A et al. (December 2010).] L’Afimoxifene ha mostrato di avere il 178% e il 338% dell’affinità dell’estradiolo per l’ERα e l’ERβ, rispettivamente. [Kuhl H (August 2005).] Le potenze antiestrogeniche dell’Endoxifene e dell’Afimoxifene sono molto simili. Tuttavia, l’Endoxifene è presente in concentrazioni molto più elevate rispetto all’Afimoxifene ed è ora ritenuto la principale forma attiva del Tamoxifene nell’organismo. Il Norendoxifene (4-idrossi-N,N-didesmetiltamoxifene), un altro metabolita attivo del Tamoxifene, è risultato agire come un potente inibitore competitivo dell’Aromatasi (IC50 = 90 nM) ed è con tutta probabilità coinvolto nell’attività antiestrogenica complessiva del Tamoxifene.
Metaboliti epatici principali del Tamoxifene
Il suo inserimento è “preliminare”, ovvero avviene fin dal principio della preparazione ad un dosaggio contenuto e comunemente assestato sui 10-20mg/die. La sua azione è in funzione “preparatoria” al tocco finale delle fasi conclusive della preparazione al contest. Come già detto, inserendolo con tempistiche adeguate il Tamoxifene contribuisce alla riduzione dei α2A-AR adipocitari facilitando la mobilitazione e l’uso (dipendente da dieta e fattori termogenici) del “grasso testardo” in combinazione potenziale con Yohimbina e ACE II inibitori.
Un altra cosa particolarmente interessante riguardante il Tamoxifene, o, meglio i suoi metaboliti attivi, è che può influenzare positivamente il metabolismo del glucosio, in particolare nel muscolo scheletrico. Alcuni studi suggeriscono che il Tamoxifene può avere un impatto sulla tolleranza al glucosio e sulla sensibilità all’insulina, con alcune evidenze che indicano un aumento della resistenza all’insulina in determinati contesti, soprattutto in presenza di obesità. Tuttavia, altre ricerche indicano che il Tamoxifene può migliorare la tolleranza al glucosio, evidenziando la complessità dei suoi effetti sul metabolismo del glucosio. In pratica, l’Afimoxifene agisce come un agonista del ER nel muscolo-scheletrico sopperendo al drastico calo del E2 nella fase Cut/Pre-Contest mantenendo i vantaggi dell’estrogeno la dove maggiormente servono all’atleta. Inoltre, alcuni studi su animali hanno dimostrato che il Tamoxifene può proteggere dai danni muscolari causati dalle contrazioni migliorando il recupero post esercizio, migliorare la forza muscolare e persino migliorare la struttura muscolare.
Ricordiamoci che la maggior parte degli effetti dati dai metaboliti del Tamoxifene deriva dal loro legame ad alta affinità con ERα ed Erβ, che sono espressi nei tessuti estrogeno-responsivi di maschi e femmine, compreso il muscolo scheletrico. Esistono diverse isoforme di ERβ. L’ERβ1 è considerata l’isoforma fisiologicamente attiva, mentre l’ERβ2, un’isoforma più lunga con un’affinità molto ridotta per gli estrogeni, agisce in modo dominante negativo per gli altri ER. L’espressione di ERβ è stata riscontrata nelle fibre muscolari e nei capillari dell’uomo. I principali effetti fisiologici degli estrogeni nei muscoli scheletrici sono, come abbiamo già detto, coadiuvanti l’aumento della forza e il recupero muscolare.
La presenza dei recettori ERβ nelle fibre muscolari e nei capillari è cruciale nella risposta fisiologica all’attività trascrizionale ER-mediata e ai conseguenti effetti biologici. Si ritiene che un meccanismo d’azione sia costituito, nel muscolo, da un potenziamento della miogenesi e dell’angiogenesi mediata dal fattore di crescita endoteliale vascolare (VEGF). Questi effetti ER-mediati possono favorire la riparazione e l’adattamento del tessuto muscolare dopo l’allenamento (Wiik et al., 2005). I recettori degli estrogeni regolano la trascrizione genica attraverso i classici elementi di risposta agli estrogeni (ERE) e i geni AP-1 responsivi. Il Tamoxifene agisce come antagonista ER sugli ERE ma come agonista ERβ/agonista parziale sui geni responsivi della proteina attivatrice 1 (AP-1), responsabili di diverse funzioni nella crescita e nella proliferazione cellulare (Jain e Koh 2010). In condizione di ipoestrogenemia, la componente di parzialità agonista diventa predominante. Infatti, clinicamente, gli agonisti parziali possono essere utilizzati per attivare i recettori in modo da dare una risposta submassimale desiderata quando sono presenti quantità inadeguate del ligando endogeno, oppure possono ridurre la sovrastimolazione dei recettori quando sono presenti quantità eccessive del ligando endogeno.[Zhu BT (April 2005).]
Relazione tra attività percentuale e concentrazione di agonisti completi e parziali
* In caso nella preparazione siano presenti AAS sintetizzati e usati in clinica per il trattamento del cancro al seno estrogeno dipendente, quali Drostanolone e/o Methenolone, l’uso di SERM e AI andrebbe con ulteriore attenzione calibrato per ovvie attività riduttive sulla funzione estrogenica connesse a queste due molecole. Nel caso in cui venga utilizzato il Methylepitiostanolo, il quale è la forma metilata di un AAS utilizzato in medicina per il trattamento del cancro al seno (Epitiostanolo), e che possiede una marcata attività AI, l’uso di Anasttrozolo risulta superfluo mentre (per i motivi sopra citati) l’uso di Exemestane rimane funzionale.
Punti importanti:
Se un soggetto non presenta particolare sensibilità all’attività dell’E2, l’uso del Boldenone mixato al Testosterone [rapporto da tarare sul soggetto] e l’aggiunta terminale di solo Anastrozolo EOD sono di gran lunga sufficienti;
Eliminare i substrati altamente aromatizzabili nelle ultime due settimane pre-contest, nel caso sopra detto, possono limitarsi alla sola omissione d’uso del hCG;
E’ consigliabile eseguire monitoraggi con esami ematici per E2 e E1 durante il percorso preparatorio onde poter valutare aggiustamenti dello stesso;
Gli effetti collaterali connessi ad un marcato abbassamento estrogenico comprendono peggioramento del profilo lipidico, affaticamento, umore basso, sbalzi di umore, letargia, dolori articolari e diminuzione della funzione sessuale;
Questo tipo di pratiche NON SONO PER PRINCIPIANTI O INTERMEDI MA PER ATLETI ESPERTI E DI ALTO LIVELLO!
Nulla di ciò che è stato scritto rappresenta una indicazione o un incitamento all’uso di farmaci dopanti e illegali!
E per quanto riguarda le donne in Cut/Pre-contest?
Di quanto sopra elencato, per un efficace controllo estrogenico in una atleta è sufficiente l’uso combinato di un SERM e un AI. Il Tamoxifene può essere sostituito con il Raloxifene anche se di minore potenza d’impatto. Il Letrozolo risulta invece una potenziale sostituzione al Anastrozolo, diversamente dagli uomini. In caso l’atleta abbia tra gli AAS in uso Drostanolone e/o Methenolone, il discorso fatto pocanzi continua ad essere valido e andranno calibrate con maggiore accuratezza, mgXmg, il SERM e l’AI inseriti. Anche in questo caso occorre dare particolarmente attenzione ai tempi di soppressione e allo scalo graduale delle molecole regolatrici al termine della preparatzione.
Conclusioni:
Abbiamo visto come gestire un eventuale iperestrogenemia sia per variabili di condizione della composizione-corporea e sia adattativa in TRT e come poter gestire al meglio in termini di vantaggi sulla composizione corporea il fattore estrogenico in Bulk/Off-Season e in Cut/Pre-Contest.
Quindi, i bodybuilder in Off-Season/Bulk beneficiano di un aumento controllato dell’E₂ grazie a una migliore risposta agli stimoli anabolici e alla riparazione (effetti sul danno muscolare). Un adeguato controllo del E2 anche in questa fase rende più facile la riduzione/gestione dei sides legati ad un aumento della attività estrogenica come la ritenzione di liquidi e una distribuzione del grasso più femminile, sui fianchi e sui glutei.
L’Aromatizzazione dei substrati soggetti porta ad un aumento dell’IGF-I e quindi delle possibilità di crescita muscolare, ma i livelli di E₂ nel sangue devono essere ottimizzati piuttosto che massimizzati a causa della forma parabolica, a U inversa, della curva E₂/IGF-I. Occorre tuttavia tenere conto del carico di allenamento, a causa degli effetti deleteri degli estrogeni sulla meccanica di tendini e legamenti (riduzione della rigidità). L’uso eccessivo di farmaci e non strategico di inibitori dell’aromatasi (AI) può avere conseguenze indesiderate attraverso la riduzione degli estrogeni, in particolare sul metabolismo osseo, sulla gestione dei lipidi e sulle potenzialità ipertrofiche muscolari, in particolare sulla risposta anabolica all’allenamento. Ecco perchè l’unico momento funzionale ad una maggiore soppressione della attività estrogenica, come abbiamo visto, risulta essere limitata al Cut/Pre-Contest. E’ consigliabile, quindi, cercare di mantenere un range ottimale del E2 per la maggior parte dell’anno. Tale range dovrebbe attestarsi tra i 40-60pg/dL ± 30pg [ossia con una punta massima di tolleranza pari a 90pg/ml] dipendente dalla sensibilità soggettiva.
Gabriel Bellizzi [CEO BioGenTech]
Approfondimenti bibliografici:
Cooke PS, Nanjappa MK, Ko C, Prins GS, Hess RA. Estrogens in Male Physiology. Physiol Rev. 2017 Jul 1;97(3):995-1043. doi:10.1152/physrev.00018.2016.
Chidi-Ogbolu N, Baar K. Effect of Estrogen on Musculoskeletal Performance and Injury Risk. Front Physiol. 2019;9:1834. Published 2019 Jan 15. doi:10.3389/fphys.2018.01834
Zhang Y, Stefanovic B. mTORC1 phosphorylates LARP6 to stimulate type I collagen expression. Sci Rep. 2017 Jan 23;7:41173. doi:10.1038/srep41173.
Kim, J. H., Cho, H. T., and Kim, Y. J. (2014). The role of estrogen in adipose tissue metabolism: insights into glucose homeostasis regulation [Review]. Endocrine Journal, 61(11), 1055–1067. doi:10.1507/endocrj.ej14-0262
Eyster, K. M. (Ed.). (2016). Estrogen Receptors. Methods in Molecular Biology. doi:10.1007/978-1-4939-3127-9
Cauley, J. A. (2015). Estrogen and bone health in men and women. Steroids, 99, 11–15. doi:10.1 016/j.steroids.2014.12.010
Veldhuis, J. D., and Bowers, C. Y. (2003). Human GH pulsatility: An ensemble property regulated by age and gender. Journal of Endocrinological Investigation, 26(9), 799–813. doi:10.1007/bf03345229
Damewood MD, Bellantoni JJ, Bachorik PS, Kimball AW Jr, Rock JA. Exogenous estrogen effect on lipid/lipoprotein cholesterol in transsexual males. J Endocrinol Invest. 1989 Jul-Aug;12(7):449-54. doi:10.1007/BF03350728.
Rubinow KB. Estrogens and Body Weight Regulation in Men. Adv Exp Med Biol. 2017;1043:285-313. doi:10.1007/978-3-319-70178-3_14
Stachenfeld, N. S. (2014). Hormonal Changes During Menopause and the Impact on Fluid Regulation. Reproductive Sciences, 21(5), 555–561. doi:10.1177/1933719113518992
Curtis, K. S. (2015). Estradiol and osmolality: Behavioral responses and central pathways. Physiology and Behavior, 152, 422–430. doi:10.1016/j.physbeh.2015.06.017
Warner M, Fan X, Strom A, Wu W, Gustafsson JÅ. 25 years of ERβ: a personal journey. J Mol Endocrinol. 2021 Oct 15;68(1):R1-R9. doi: 10.1530/JME-21-0121
Kley, H. K., Deselaers, T., Peerenboom, H., and Kruskemer, H. L. (1980). Enhanced Conversion of Androstenedione to Estrogens in Obese Males*. The Journal of Clinical Endocrinology and Metabolism, 51(5), 1128–1132. doi:10.1210/jcem-51-5-1128
Thompson, P. D. (1989). Contrasting Effects of Testosterone and Stanozolol on Serum Lipoprotein Levels. JAMA: The Journal of the American Medical Association, 261(8), 1165. doi:10.1001/jama.1989.03420080085036
Friedl, K. E., Hannan, C. J., Jones, R. E., and Plymate, S. R. (1990). High-density lipoprotein cholesterol is not decreased if an aromatizable androgen is administered. Metabolism, 39(1), 69–74. doi:10.1016/0026-0495(90)90150-b
Hartgens, F. (2004). Effects of androgenic-anabolic steroids on apolipoproteins and lipoprotein (a). British Journal of Sports Medicine, 38(3), 253–259. doi:10.1136/bjsm.2003.000199
Perkins, M. S., Louw-du Toit, R., and Africander, D. (2017). A comparative characterization of estrogens used in hormone therapy via estrogen receptor (ER)-α and -β. The Journal of Steroid Biochemistry and Molecular Biology, 174, 27–39. doi:10.1016/j.jsbmb.2017.07.022
Bond P, Smit DL, de Ronde W. Anabolic-androgenic steroids: How do they work and what are the risks? Front Endocrinol (Lausanne). 2022 Dec 19;13:1059473. doi: 10.3389/fendo.2022.1059473. PMID: 36644692; PMCID: PMC9837614.
Lin, S. X., Han, Q., Azzi, A., Zhu, D.-W., Gongloff, A., & Campbell, R. L. (1999). 3D-structure of human estrogenic 17β-HSD1: binding with various steroids. The Journal of Steroid Biochemistry and Molecular Biology, 69(1-6), 425–429. doi:10.1016/s0960-0760(99)00062-x
Del Trestolone [MENT; Methylnortestosterone; 7α-methyl-19-nortestosterone; 7α-CH₃-NorT) ho già parlato sia in un articolo che in un video della rubrica “The Lord Of The PEDs”. In entrambi i lavori sono stati esposti in generale pregi e difetti della molecola in questione. In questo articolo, invece, vorrei soffermarmi sulle caratteristiche uniche del Trestolone, le quali, e lo si vedrà, non gli conferiscono particolari e reali vantaggi d’uso nel Bodybuilding, a differenza, per esempio, del Trenbolone, dell’Oxandrolone o del Fluoxymesterone. Piuttosto, le sue peculiarità si combinano in modo sfavorevole per qualsiasi uso pratico nel bodybuilding, attraverso l’influenza sullo sviluppo della ginecomastia, dell’equilibrio dei liquidi e della pressione sanguigna.
Le caratteristiche uniche del Trestolone:
Effetti estrogenici per aromatizzazione al prodotto aromatico più potente del 17β-estradiolo: il 7α-methylestradiolo (7α-ME);
Effetti gestageni dovuti alla forte attivazione del recettore del Progesterone (PR):
Che si combinano sinergicamente per produrre:
1. Effetti ginecomastici (crescita del tessuto mammario negli uomini)
2. Effetti edematosi (ritenzione di liquidi; “gonfiore”) e
3. Effetti ipertensivi (pressione sanguigna elevata, in particolare sistolica, cioè pressione da contrazione cardiaca).
Prima di discutere gli effetti edematosi del Trestolone (ritenzione di liquidi; “gonfiore”), dobbiamo innanzitutto esaminare i fattori che sono alla base della ritenzione di liquidi (effetti estrogenici e gestagenici/progestinici), prima di discutere il modo in cui questi stessi fattori sono alla base dei suoi particolari effetti ipertensivi (aumento della pressione arteriosa, in particolare della pressione sistolica; cioè, aumento della pressione da contrazione cardiaca).
Effetti estrogenici:
Struttura molecolare del 7α-Methylestradiolo
Il lettore nella media, ormai, dovrebbe essere al corrente sul fatto che il Trestolone aromatizza in 7α-methylestradiolo (7α-ME). [1].
Dalla Teoria delle Potenze Estrogeniche Dipendenti dai Composti (Per-AAS) e Individuali (Per-Utilizzatore) (Modello di Type-IIx), l’estrogenicità si riferisce agli effetti associati all’attivazione di ERα e β (quest’ultima generalmente opposta alla prima), e dipende da Fattori Dipendenti dai Composti (Per-AAS) e Individuali (Per-Utilizzatori) che determinano sia (A) i livelli ematici effettivi che (B) gli effetti a livello tissutale dei prodotti aromatici di un AAS.
Tabella 1: Il più potente di tutti, il 7α-ME produce ½ della risposta di crescita massima (EC₅₀) nel tessuto mammario umano (T47Dco) a una sola attivazione: una concentrazione di 4,4 picomoli (pM). Come il Trenbolone, ma in modo peggiore, esso fa di più con minor quantità.[1]
Tuttavia, in letteratura, viene riportato che il Trestolone possiede solo una debole attività estrogenica data da un tasso di aromatizzazione che sembrerebbe essere insufficiente per scopi sostitutivi, come dimostrato dalla diminuzione della densità minerale ossea negli uomini trattati con questo farmaco per l’ipogonadismo. Dobbiamo, però, anche considerare il tasso di degradazione del 7α-methylestradiolo prodotto, nonché la potenza estrogenica dei metaboliti risultanti che vanno a sommarsi alla potenziale attività estrogenica di altri AAS aromatizzabili co-somministrati [vedi Oxymetholone e la sua attività estrogenica intrinseca e il Methandrostenolone con il suo metabolita estrogenico 17α-methylestradiolo] e l’attività gestatinica propria del Trestolone.
Il prodotto aromatico del Trestolone, il 7α-ME, ha una potenza più che quadrupla (“efficace”, una cosa negativa in questo caso) rispetto al 17β-estradiolo (E2) nelle cellule con presenza di ER.[1] L’efficacia si determina misurando l’effetto, ad esempio la crescita (in questo caso, nelle cellule di cancro al seno). La EC₅₀ (EC50) è determinata dalle concentrazioni alle quali il ligando innesca la crescita e può essere confermata da misurazioni della progressione del ciclo cellulare (cioè l’ingresso nella fase S durante il ciclo cellulare).
L’affinità di legame (IC₅₀) del prodotto aromatico del Trestolone, 7α-ME, è pari al 102% di quella dell’E2, che in letteratura viene tipicamente utilizzato come composto di riferimento per il legame con l’ER, data la sua notevole efficacia, potenza e affinità per il recettore ER-α. [1].
Confrontando il tasso di aromatizzazione tra Trestolone e Nandrolone, Attardi et al. hanno scoperto che, “[a] 180 min, circa il 23% del Trestolone è stato convertito in 7α-ME e circa il 13% del [Nandrolone] in E2”. Poiché il Nandrolone ha un tasso di aromatizzazione del 20% rispetto al Testosterone (T), e che presenta una maggior tendenza alla conversione in Estrone (E1), possiamo dedurre che il Trestolone aromatizza in 7α-ME circa il 35% rispetto al T [che aromatizza in E2], con una potenza quattro volte superiore a quella dell’E2, cioè per provocare la crescita delle cellule del cancro al seno. La semplice moltiplicazione del tasso di aromatizzazione (35%) × EC50(7α-methylestradiolo) × RBA(7α-methylestradiolo) ≈ Il potenziale di crescita del Trestolone nelle cellule con presenza di ER è superiore del 40% rispetto al T. [2]
Differenze strutturali tra una molecola di Estrone (E1) e di Estradiolo (E2)
La deduzione, quindi, supporta le segnalazioni degli utilizzatori secondo cui il Trestolone è potentemente estrogenico. Matematicamente, possiamo affermare che 50mg al giorno di Trestolone Enantato ≈ estrogenico quanto 500mg di Testosterone Enantato alla settimana.
Inoltre, come descritto nella sezione seguente, gli effetti gestageni del Trestolone potenziano notevolmente i suoi effetti ipertensivi ed edematosi (tendenza a trattenere liquidi).
Il Trestolone è un androgeno potentemente progestinico (“ gestagenico”) che possiede il 27,5% della potenza di Androcur™ [Ciproterone Acetato] – un farmaco antiandrogeno e progestinico usato per trattare le patologie androgeno dipendenti, tra cui l’acne, l’irsutismo e il cancro alla prostata – per attivare il recettore del progesterone (PR). [11].
I progestinici contribuiscono alla soppressione dell’asse ipotalamo-ipofisi-gonadi (HPG) disregolando le gonadotropine ipotalamiche attraverso la segnalazione del dendro KNDy, interrompendo la pulsatilità del GnRH e inibendo la secrezione di LH ipofisario [e FSH], inibendo così la sintesi e la secrezione di Testosterone (endogeno). [12][13] I progestinici sintetici utilizzati in ambito contraccettivo traggono la loro efficacia da questa caratteristica. Bebb et al. hanno randomizzato uomini sani a ricevere Testosterone Enantato (100mg settimanali) o lo stesso dosaggio di Testosterone Enantato in combinazione con il progestinico Levonorgestrel, la cui aggiunta ha praticamente soppresso la secrezione di LH e FSH. [14] La diminuzione di LH e FSH può causare ipogonadismo secondario, con conseguente diminuzione del rapporto androgeni/estrogeni (A:E), causando ginecomastia. [15]
Gli effetti dei progestinici sono legati alle loro interazioni con i recettori: recettori degli androgeni (AR) (ad esempio, acne, effetti lipidici); recettori dei glucocorticoidi (GR) (ad esempio, ritenzione di sodio e acqua, gonfiore); o recettori dei mineralocorticoidi (MR) (ad esempio, diminuzione della ritenzione idrica e del peso). I progestinici antiandrogeni possono agire in diversi modi. Possono esercitare un’inibizione competitiva dell’AR, oppure legarsi all’enzima 5-α reduttasi e quindi interagire con la conversione del Testosterone in DHT (il suo metabolita fortemente androgeno)[16].
Il Progesterone e i suoi derivati e i “progestinici-mimici” (ad esempio, il Trestolone) si legano moderatamente all’AR in modo competitivo (cioè antagonista). [17]. I derivati del Progesterone alterano le risposte tissutali mediate da AR e PR, ma non da ER. [17]
Gli estrogeni regolano la sintesi di PR. [18] Inoltre, l’attivazione del PR è stata collegata a una ridotta espressione dell’AR, ostacolando così l’inibizione della crescita del tessuto mammario mediata dagli androgeni osservata in condizioni di normale omeostasi ormonale. [19].
Il Progesterone e i suoi derivati possono ulteriormente ma indirettamente causare ginecomastia potenziando l’effetto dell’E2 sui tessuti mammari. [20].
In sintesi, le caratteristiche discusse – effetti estrogenici e gestagenici del Trestolone – sono alla base dei suoi potenti effetti edematosi e ipertensivi.
Effetti edematosi:
L’edema, o ritenzione di liquidi, è il metro di paragone dell’eccessiva estrogenicità nei bodybuilder che fanno uso di AAS.
Figura 2: Schema che illustra il controllo complesso dell’equilibrio dei liquidi e del sodio e i molteplici modi in cui l’Estradiolo (E2) e il Progesterone (P4) possono influenzare questi processi. Adattato con il permesso di Stachenfeld. La regolazione dei fluidi e del sodio è controllata da una serie di sistemi complessi, tutti influenzati da estrogeni e Progesterone. Sia il sistema nervoso centrale (SNC) che il sistema nervoso periferico (PNS) contribuiscono alla regolazione dei fluidi; gli estrogeni e i progestinici possono influenzare la regolazione dei fluidi direttamente attraverso il cervello o indirettamente influenzando le azioni dell’Angiotensina II (ANG II) e dell’Arginina Vasopressina (AVP) e dei cambiamenti negli ormoni che regolano il sodio (Aldosterone e Renina [RAAS]). Estradiolo e Progesterone/Progestinici, possono anche aumentare l’Angiotesina II cerebrale mediato nel cervello e aumentare l’importante effetto stimolante di questo ormone sulla sete e sull’assunzione di liquidi. Infine, sia E2 che P4, influenzano la regolazione del sodio e dell’acqua nei tubuli distali del rene. Questo impatto può verificarsi direttamente sui tubuli o sia attraverso l’AVP che il RAAS e contribuire alla ritenzione idrica.
Il Trestolone si sostituisce a E₂ e P₄ per promuovere l’equilibrio dei fluidi attraverso molteplici meccanismi. [10]
Il diagramma qui sopra illustra come il Trestolone agisca in modo molteplice per promuovere la ritenzione di liquidi (edema) e l’ipertensione (aumento della pressione arteriosa sistolica; aumento della pressione da contrazione cardiaca). Il Trestolone è analogo all’E₂ (Estradiolo; E2) e al P₄ (Progesterone) nei confronti del 7α-ME e nella sua potenza del 29,2% nell’attivare la PR come il P₄. [11]
Il Trestolone, quindi, promuove la ritenzione di liquidi agendo su:
1. Il cervello e il sistema nervoso centrale (SNC) per aumentare la sete e il bisogno di assumere sodio attraverso la segnalazione dell’Angiotensina II, e 2. I reni agendo su: 1. gli ormoni antidiuretici, ad esempio l’Arginina Vasopressina (AVP), riducendo la minzione 2. il sistema renina-angiotensina-aldosterone (RAAS), aumentando il bilancio del sodio, e/o 3. i tubuli renali, favorendo la ritenzione di sodio e acqua. [10]
Effetti ipertensivi:
Nella Figura 1 è illustrato anche, in parte, come il Trestolone, al posto dell’E₂ (Estradiolo; E2) e del P₄ (Progesterone), favorisca l’aumento della pressione arteriosa. Lo fa aumentando lo squilibrio dei liquidi (cioè il “gonfiore”), come i suoi effetti edematosi, e agendo sugli osmorecettori del sistema nervoso centrale (SNC) e sui barorecettori del sistema nervoso periferico (SNP), nonché agendo sull’Angiotensina II, sull’AVP e sui neuroni di vari sistemi. [10]
– Aumento della pressione sistolica
Il Trestolone aumenta significativamente la pressione arteriosa sistolica (cioè la pressione da contrazione cardiaca) a una dose settimanale inferiore a 2mg. [21]
La pressione del polso (Pp) è la differenza (mmHg) tra pressione sistolica e diastolica. Una pressione sistolica normale è quindi di 40 mmHg (120 mmHg – 80 mmHg = 40 mmHg).
La pressione sistolica , o del polso (Pp), rappresenta la forza pressoria che il cuore genera ogni volta che si contrae, o la compliance arteriosa (C). Se la pressione del polso è normale a 40 mmHg, una Pp < 25% della pressione sistolica è bassa o ristretta, mentre una Pp > 100 mmHg è alta o estesa.
Una variazione della pressione sistolica (ΔPp) è proporzionale alla variazione del volume (V) (ΔV) ma inversamente proporzionale alla compliance arteriosa (C):
ΔPp = Δ V/C
Poiché la variazione di volume è dovuta al volume della gittata (SV) del sangue espulso dal ventricolo sinistro, possiamo approssimare la pressione del polso come:
Pp = SV/C
Un giovane adulto normale a riposo ha un volume di gittata (SV) di circa 80 mL. La compliance arteriosa (C) è di circa 2mL/mmHg, il che conferma che la pressione normale del polso è di circa 40 mmHg.
Il Trestolone, quindi, induce un aumento della pressione sistolica, aumentando il volume della gittata.
Effetti ematologici:
– Aumento del Ematocrito [HCT]
Il Trestolone, aumentando la ritenzione di sodio e di liquidi, e aumenta il volume plasmatico. Inoltre, aumenta rapidamente l’Emoglobina.
L’Emoglobina (Hb) è una proteina che si lega agli eritrociti (RBC) all’O₂ (Hb 13,5 – 17g/dL [uomini], 12 – 15,5g/dL [donne]).
L’Ematocrito (HCT) rappresenta la % del volume sanguigno occupato dagli eritrociti (RBC) [uomini 41-51%, donne 36-47%].
L’Ematocrito (HCT) è correlato all’Emoglobina (Hb) mediante la formula di base:
Hb (g/dL) × 3 ≈ HCT (%)
Esempio: Hb di 15g/dL ≈ HCT del 45%.
I livelli di emoglobina sono stati significativamente aumentati (149 ± 2,9 g/L → 154 ± 3,3; dimensione dell’effetto: 1,724; %Δ: +3,35%; intervallo di confidenza del 95%) con il Trestolone (~ 2 mg q.w.) a 12 settimane, e un andamento simile (da 0,44 a 0,46; dimensione dell’effetto: 2; %Δ: +4,5%; non significativo) è stato osservato nell’Ematocrito, che però non ha raggiunto la significatività statistica. [21] Nel gruppo Testosterone (~ 120mg q.w.), invece, è stato osservato un aumento progressivo più lento della concentrazione di Emoglobina, che è diventato significativo solo a 48 settimane. Nel gruppo Testosterone si è registrato anche un aumento complessivo significativo dell’Ematocrito, sebbene nessuno dei singoli punti di trattamento fosse significativamente diverso dal pre-trattamento (0,45 ± 0,01). [21]
Conclusioni:
Le caratteristiche di base del Trestolone – la sua potente estro- e gesta- genicità – pongono le basi per i suoi forti effetti edematosi e ipertensivi, in modo tale che i suoi effetti ginecomastici, gli effetti ginecomastici derivanti dall’estrogenicità e gli effetti ginecomastici derivanti dall’antiandrogenicità, non possono essere ignorati. Attraverso il suo metabolita aromatico 7α-ME, sopprime potentemente le gonadotropine (LH, FSH), diminuendo la A:E ratio, sinergizzando con le caratteristiche gestageniche con una potenza pari a quella degli antiandrogeni farmaceutici (ad esempio, Androcur™ [Ciproterone Acetato]), stimolando la crescita direttamente del tessuto mammario attraverso l’ER e indirettamente attraverso l’azione gestagenica e antiandrogena nella modalità del Progesterone (con una potenza di quasi ⅓ per mg). Il Trestolone favorisce in particolare il “gonfiore da ritenzione” attraverso la sua azione sui reni (influenzando negativamente la regolazione della ritenzione di liquidi e sodio) e sul cervello (aumentando la sete e l’ingestione di sodio) e favorisce in particolare l’ipertensione, soprattutto l’aumento della pressione sistolica, aumentando il volume di gittata attraverso l’aumento del volume plasmatico e dell’Ematocrito, cioè la viscosità o lo densità del sangue.
Questo articolo potrebbe prevedibilmente servire da deterrente all’uso di questo agente praticamente inutile (se paragonato alle altre molecole AAS) da parte dei bodybuilder dal momento che presenta alcuni aspetti unici e non accettabili nell’insieme di una corretta valutazione della molecola.
Se lo confrontiamo con il Trenbolone, non abbiamo alcun vantaggio nel suo inserimento sostitutivo: oltre al potenziale neurotossico e cardiotossico, come abbiamo visto, si aggiungono problematiche peculiari date dalla molecola che ne rendono un ipotetico uso nettamente difficile da gestire.
Gabriel Bellizzi [CEO BioGenTech]
Riferimenti:
Articles by Type-IIx
[1] Attardi BJ, Pham TC, Radler LC, Burgenson J, Hild SA, Reel JR. Dimethandrolone (7alpha,11beta-dimethyl-19-nortestosterone) and 11beta-methyl-19-nortestosterone are not converted to aromatic A-ring products in the presence of recombinant human aromatase. J Steroid Biochem Mol Biol. 2008;110(3-5):214-222. doi:10.1016/j.jsbmb.2007.11.009
[2] Ryan, Kenneth J. “Biological aromatization of steroids.” Journal of Biological Chemistry 234.2. (1959): 268-272.
[3] Navarro, V. M., Gottsch, M. L., Chavkin, C., Okamura, H., Clifton, D. K., and Steiner, R. A. (2009). Regulation of Gonadotropin-Releasing Hormone Secretion by Kisspeptin/Dynorphin/Neurokinin B Neurons in the Arcuate Nucleus of the Mouse. Journal of Neuroscience, 29(38), 11859–11866. doi:10.1523/jneurosci.1569-09.2009
[4] Girmus, R. L., and Wise, M. E. (1992). Progesterone Directly Inhibits Pituitary Luteinizing Hormone Secretion in an Estradiol-dependent Manner1. Biology of Reproduction, 46(4), 710–714. doi:10.1095/biolreprod46.4.710
[5] Bebb, R. A., Anawalt, B. D., Christensen, R. B., Paulsen, C. A., Bremner, W. J., and Matsumoto, A. M. (1996). Combined administration of levonorgestrel and testosterone induces more rapid and effective suppression of spermatogenesis than testosterone alone: a promising male contraceptive approach. The Journal of Clinical Endocrinology and Metabolism, 81(2), 757–762. doi:10.1210/jcem.81.2.8636300
[6] Sitruk-Ware, R. (2004). Pharmacological profile of progestins. Maturitas, 47(4), 277–283. doi:1
0.1016/j.maturitas.2004.01.001
[7] Fang, H., Tong, W., Branham, W. S., Moland, C. L., Dial, S. L., Hong, H., … Sheehan, D. M. (2003). Study of 202 Natural, Synthetic, and Environmental Chemicals for Binding to the Androgen Receptor. Chemical Research in Toxicology, 16(10), 1338–1358. doi:10.1021/tx030011g
[8] Eyster, K. M. (Ed.). (2016). Estrogen Receptors. Methods in Molecular Biology. doi:10.1007/978-1-4939-3127-9
[9] Sansone, A., Romanelli, F., Sansone, M., Lenzi, A., and Di Luigi, L. (2016). Gynecomastia and hormones. Endocrine, 55(1), 37–44. doi:10.1007/s12020-016-0975-9
[10] Zhou, J., Ng, S., Adesanya-Famuiya, O., Anderson, K., and Bondy, C. A. (2000). Testosterone inhibits estrogen-induced mammary epithelial proliferation and suppresses estrogen receptor expression. The FASEB Journal, 14(12), 1725–1730. doi:10.1096/fj.99-0863com
[11] Houtman, C. J., Sterk, S. S., van de Heijning, M. P. M., Brouwer, A., Stephany, R. W., van der Burg, B., and Sonneveld, E. (2009). Detection of anabolic androgenic steroid abuse in doping control using mammalian reporter gene bioassays. Analytica Chimica Acta, 637(1-2), 247–258. doi:10.1016/j.aca.2008.09.037
[12] Navarro, V. M., Gottsch, M. L., Chavkin, C., Okamura, H., Clifton, D. K., and Steiner, R. A. (2009). Regulation of Gonadotropin-Releasing Hormone Secretion by Kisspeptin/Dynorphin/Neurokinin B Neurons in the Arcuate Nucleus of the Mouse. Journal of Neuroscience, 29(38), 11859–11866. doi:10.1523/jneurosci.1569-09.2009
[13] Girmus, R. L., and Wise, M. E. (1992). Progesterone Directly Inhibits Pituitary Luteinizing Hormone Secretion in an Estradiol-dependent Manner1. Biology of Reproduction, 46(4), 710–714. doi:10.1095/biolreprod46.4.710
[14] Bebb, R. A., Anawalt, B. D., Christensen, R. B., Paulsen, C. A., Bremner, W. J., and Matsumoto, A. M. (1996). Combined administration of levonorgestrel and testosterone induces more rapid and effective suppression of spermatogenesis than testosterone alone: a promising male contraceptive approach. The Journal of Clinical Endocrinology and Metabolism, 81(2), 757–762. doi:10.1210/jcem.81.2.8636300
[16] Sitruk-Ware, R. (2004). Pharmacological profile of progestins. Maturitas, 47(4), 277–283. doi:10.1016/j.maturitas.2004.01.001
[17] Fang, H., Tong, W., Branham, W. S., Moland, C. L., Dial, S. L., Hong, H., … Sheehan, D. M. (2003). Study of 202 Natural, Synthetic, and Environmental Chemicals for Binding to the Androgen Receptor. Chemical Research in Toxicology, 16(10), 1338–1358. doi:10.1021/tx030011g
[18] Eyster, K. M. (Ed.). (2016). Estrogen Receptors. Methods in Molecular Biology. Doi:10.1007/978-1-4939-3127-9
[19] Sansone, A., Romanelli, F., Sansone, M., Lenzi, A., and Di Luigi, L. (2016). Gynecomastia and hormones. Endocrine, 55(1), 37–44. doi:10.1007/s12020-016-0975-9
[20] Zhou, J., Ng, S., Adesanya-Famuiya, O., Anderson, K., and Bondy, C. A. (2000). Testosterone inhibits estrogen-induced mammary epithelial proliferation and suppresses estrogen receptor expression. The FASEB Journal, 14(12), 1725–1730. doi:10.1096/fj.99-0863com
[21] Walton, M. J., Kumar, N., Baird, D. T., Ludlow, H., & Anderson, R. A. (2007). 7 -Methyl-19-Nortestosterone (MENT) vs Testosterone in Combination With Etonogestrel Implants for Spermatogenic Suppression in Healthy Men. Journal of Andrology, 28(5), 679–688. doi:10.2164/jandrol.107.002683
La maggior parte degli “addetti ai lavori” e degli atleti, è perfettamente a conoscenza del fatto che una “base” di Testosterone sia necessaria all’interno di un ciclo di AAS/SARM al fine di avere un adeguato livello di metaboliti connessi [vedi E2 e DHT] evitando o riducendo quei problemi legati ad un loro marcato calo: alterazioni dell’umore, letargia, sonnolenza, spossatezza, ridotta libido, difficoltà a raggiungere e mantenere l’erezione ecc… .
Esistono altresì soggetti che decidono di non avvalersi dell’uso di una base di Testosterone optando, per esempio, per una somministrazione “rivista” di hCG. Ma vi sono altri, i così detti “agofobici” [si, esistono…si dopano e hanno paura dell’ago] che cercano di ripiegare con l’uso spesso fallimentare di DHEA [il quale, attraverso la conversione in Androstenediolo e Androstenedione converte maggiormente in E1 che a sua volta possiede una scarsa tendenza alla conversione nel più utile E2. Altri decidono di usare il Clomifene Citrato (Clomid®) o l’Enclomifene Citrato (Androxal®) per cercare di mantenere una attività dell’Asse HPT tale da garantire loro adeguati livelli di E2.
Sappiamo benissimo che i SERM agiscono a livello dei ER ipotalamici stimolando il rilascio di GnRH e, successivamente, a livello ipofisario, di LH e FSH. E’ infatti pratica comune nella PCT utilizzare tali farmaci per avere una risposta di “recupero” iniziale della produzione endogena di Testosterone dopo l’uso di AAS e loro azione soppressiva del sistema endocrino in questione.
A questo punto la domanda è: è possibile che l’uso di SERM come il Clomifene Citrato o il suo enantiomero attivo Enclomifene possa avere una risposta terapeutica anche durante l’uso di AAS?
Facciamo un pò di ripasso e cerchiamo di arrivare ad una conclusione logica e, per lo meno, accademica …
SERM e loro caratteristiche:
Siti di legame [ERα e ERβ]
I SERM sono agonisti parziali competitivi dell’ER.[1] I diversi tessuti hanno gradi diversi di sensibilità all’attività degli estrogeni endogeni, quindi i SERM producono effetti estrogenici o antiestrogenici a seconda del tessuto specifico in questione e della percentuale di attività intrinseca (IA) del SERM. [2] Un esempio di SERM con un’elevata IA e quindi con effetti prevalentemente estrogenici è il clorotrianisene, mentre un esempio di SERM con una bassa IA e quindi con effetti prevalentemente antiestrogenici è l’etamoxitripetolo. SERM come il clomifene e il tamoxifene sono relativamente più a metà strada per quanto riguarda l’IA e l’equilibrio tra attività estrogenica e antiestrogenica. Il raloxifene è un SERM più antiestrogenico del tamoxifene; entrambi sono estrogenici nelle ossa, ma il raloxifene è antiestrogenico nell’utero mentre il tamoxifene è estrogenico in questa parte del corpo.[2]
Da sinistra a destra: ERβ e ERα .
I SERM agiscono sul recettore degli estrogeni (ER), che è un attivatore trascrizionale intracellulare ligando-dipendente e appartiene alla famiglia dei recettori nucleari.[4] Sono stati identificati due diversi sottotipi di ER, ERα e ERβ. ERα è considerato il principale mezzo in cui i segnali estrogenici vengono trasdotti a livello trascrizionale ed è l’ER predominante nel tratto riproduttivo femminile e nelle ghiandole mammarie, mentre ERβ si trova principalmente nelle cellule endoteliali vascolari, nell’osso e nel tessuto prostatico maschile.[5] È noto che la concentrazione di ERα ed ERβ è diversa nei tessuti durante lo sviluppo, l’invecchiamento o lo stato patologico.[6] Molte caratteristiche sono simili tra questi due tipi, come le dimensioni (~600 e 530 aminoacidi) e la struttura. ERα ed ERβ condividono circa il 97% dell’identità di sequenza aminoacidica nel dominio che lega il DNA e circa il 56% nel dominio che lega il ligando.[4][6] La differenza principale dei domini che legano il ligando è determinata da Leu-384 e Met-421 in ERα, che sono sostituiti da Met-336 e Ile-373, rispettivamente, in ERβ.[7] La variazione è maggiore sull’N-terminus tra ERα ed ERβ.[8]
Strutture chimiche di diverse classi di SERM (Trifeniletilene, Benzotiofene, Indolo e Tetraidronaftalene).
Il dominio di legame al DNA è costituito da due sottodomini. Uno ha un box prossimale che è coinvolto nel riconoscimento del DNA, mentre l’altro contiene un box distale responsabile della dimerizzazione DNA-dipendente del dominio DNA-binding. La sequenza del box prossimale è identica tra ERα ed ERβ, il che indica una specificità e un’affinità simili tra i due sottogruppi. Le proteine globulari del dominio DNA-binding contengono otto cisteine e consentono una coordinazione tetraedrica di due ioni zinco. Questa coordinazione rende possibile il legame di ER con gli elementi di risposta agli estrogeni.[5] Il dominio legante il ligando è una struttura globulare a tre strati composta da 11 eliche e contiene una tasca per il ligando naturale o sintetico.[5][4] I fattori che influenzano l’affinità di legame sono principalmente la presenza di una frazione fenolica, la dimensione e la forma molecolare, i doppi legami e l’idrofobicità.[9]
Il posizionamento differenziale dell’elica 12 della funzione attivante 2 (AF-2) nel dominio di legame del ligando da parte del ligando legato determina se il ligando ha un effetto agonista o antagonista. Nei recettori legati all’agonista, l’elica 12 è posizionata adiacentemente alle eliche 3 e 5. Le eliche 3, 5 e 12 insieme formano una superficie di legame per un motivo NR box contenuto nei coattivatori con la sequenza canonica LXXLL (dove L rappresenta la leucina o l’isoleucina e X è un amminoacido qualsiasi).
I recettori non bloccati (apo) o i recettori legati a ligandi antagonisti allontanano l’elica 12 dalla superficie di legame LXXLL, il che porta al legame preferenziale di un motivo più lungo ricco di leucina, LXXXIXXX(I/L), presente sui corepressori NCoR1 o SMRT. Inoltre, alcuni cofattori si legano all’ER attraverso i terminali, il sito di legame del DNA o altri siti di legame. Pertanto, un composto può essere un agonista ER in un tessuto ricco di coattivatori ma un antagonista ER in tessuti ricchi di corepressori.[4]
Meccanismo d’azione
I composti estrogenici coprono uno spettro di attività che va da:
Agonisti completi (agonisti in tutti i tessuti) come l’ormone endogeno naturale Estradiolo
Agonisti misti/antagonisti (agonisti in alcuni tessuti e antagonisti in altri) come il Tamoxifene (SERM).
Antagonisti puri (antagonisti in tutti i tessuti), come il Fulvestrant.
I SERM sono noti per stimolare l’azione estrogenica in tessuti come il fegato, le ossa e il sistema cardiovascolare, ma anche per bloccare l’azione degli estrogeni laddove la stimolazione non è auspicabile, come nel seno e nell’utero. [10] Questa attività agonistica o antagonistica provoca vari cambiamenti strutturali dei recettori, con conseguente attivazione o repressione dei geni bersaglio degli estrogeni.[10][11] I SERM interagiscono con i recettori diffondendosi nelle cellule e legandosi alle subunità ERα o ERβ, con conseguente dimerizzazione e cambiamenti strutturali dei recettori. Ciò facilita l’interazione dei SERM con gli elementi di risposta agli estrogeni, che portano all’attivazione di geni inducibili dagli estrogeni e mediano gli effetti di questi ultimi.[10]
Impatto dei SERM sul omeostasi del Colesterolo.
La caratteristica unica dei SERM è la loro attività selettiva per tessuti e cellule. Ci sono sempre più prove a sostegno del fatto che l’attività dei SERM è determinata principalmente dal reclutamento selettivo di corepressori e coattivatori ai geni bersaglio dell’ER in specifici tipi di tessuti e cellule.[11][12] I SERM possono avere un impatto sulla stabilità delle proteine dei coattivatori e possono anche regolarne l’attività attraverso modifiche post-traslazionali come la fosforilazione. Molteplici vie di segnalazione della crescita, come HER2, PKC, PI3K e altre, sono downregolate in risposta al trattamento anti-estrogeno. Il coattivatore 3 dei recettori steroidei (SRC-3) viene fosforilato da chinasi attivate che ne potenziano l’attività di coattivatore, influenzano la crescita cellulare e contribuiscono alla resistenza ai farmaci.[12]
Il rapporto tra ERα ed ERβ in un sito bersaglio può essere un altro modo per determinare l’attività dei SERM. Alti livelli di proliferazione cellulare sono ben correlati con un alto rapporto ERα:ERβ, ma la repressione della proliferazione cellulare è correlata alla dominanza di ERβ su ERα. Il rapporto tra ER nel tessuto mammario neoplastico e normale potrebbe essere importante quando si considera la chemioprofilassi con i SERM.[10][11]
Per quanto riguarda le differenze tra ERα ed ERβ, sono importanti la Funzione di Attivazione 1 (AF-1) e la Funzione di Attivazione 2 (AF-2). Insieme svolgono un ruolo importante nell’interazione con altre proteine co-regolatrici che controllano la trascrizione genica.[10] AF-1 si trova nella terminazione amminica dell’ER ed è omologa solo al 20% in ERα ed ERβ. D’altra parte, AF-2 è molto simile in ERα e ERβ, e solo un aminoacido è diverso. Gli studi hanno dimostrato che scambiando le regioni di AF-1 in ERα e ERβ, si ottengono differenze specifiche nell’attività di trascrizione. In generale, i SERM possono attivare parzialmente geni ingegnerizzati attraverso ERα da un elemento del recettore degli estrogeni, ma non attraverso ERβ.[10][11] Tuttavia, il raloxifene e la forma attiva del tamoxifene possono stimolare geni reporter regolati da AF-1 sia in ERα che in ERβ.
La scoperta dell’esistenza di due sottotipi di ER ha portato alla sintesi di una serie di ligandi specifici per il recettore in grado di attivare o disattivare un particolare recettore. Tuttavia, la forma esterna del complesso risultante è ciò che diventa il catalizzatore per modificare la risposta di un tessuto bersaglio a un SERM.[10][11]
La cristallografia a raggi X di estrogeni o antiestrogeni ha mostrato come i ligandi programmino il complesso recettoriale per interagire con altre proteine. Il dominio legante dell’ER dimostra come i ligandi promuovano e impediscano il legame del coattivatore in base alla forma del complesso estrogeno o antiestrogeno. L’ampia gamma di ligandi che si legano all’ER può creare uno spettro di complessi ER completamente estrogenici o antiestrogenici in uno specifico sito bersaglio.[11] Il risultato principale del legame di un ligando all’ER è un riarrangiamento strutturale della tasca di legame del ligando, principalmente nell’AF-2 della regione C-terminale. Il legame dei ligandi all’ER porta alla formazione di una tasca idrofobica che regola i cofattori e la farmacologia del recettore. Il corretto ripiegamento del dominio di legame con i ligandi è necessario per l’attivazione della trascrizione e per l’interazione di ER con una serie di coattivatori.
Basi strutturali del meccanismo d’azione degli agonisti e degli antagonisti dei recettori degli estrogeni. Le strutture qui mostrate sono del dominio di legame del ligando (LBD) del recettore degli estrogeni (diagramma a fumetti verde) complessato con l’agonista Dietilstilbestrolo (in alto, PDB: 3ERD) o con l’antagonista 4-idrossitamossifene (in basso, 3ERT). I ligandi sono rappresentati come sfere che riempiono lo spazio (bianco = carbonio, rosso = ossigeno). Quando un agonista è legato a un recettore nucleare, l’alfa elica C-terminale della LBD (H12; azzurro) è posizionata in modo tale che una proteina coattivatrice (rosso) possa legarsi alla superficie della LBD. Qui è mostrata solo una piccola parte della proteina coattivatrice, la cosiddetta scatola NR contenente il motivo di sequenza aminoacidica LXXLL. Gli antagonisti occupano la stessa cavità di legame del ligando del recettore nucleare. Tuttavia, i ligandi antagonisti hanno un’estensione della catena laterale che sposta stericamente H12 per occupare all’incirca la stessa posizione nello spazio in cui si legano i coattivatori. Di conseguenza, il legame del coattivatore alla LBD viene bloccato.
I coattivatori non sono solo partner proteici che collegano tra loro i siti di un complesso. I coattivatori svolgono un ruolo attivo nel modificare l’attività di un complesso. La modificazione post-traduzionale dei coattivatori può dar luogo a un modello dinamico di azione degli ormoni steroidei attraverso molteplici vie chinasiche avviate dai recettori dei fattori di crescita della superficie cellulare. Sotto la guida di una moltitudine di rimodellatori proteici per formare un complesso multiproteico di coattivatori in grado di interagire con l’ER fosforilato in uno specifico sito promotore genico, il core coactivator deve prima reclutare una serie specifica di coattivatori. Le proteine che il core coactivator assembla come complesso di coattivatori hanno attività enzimatiche individuali per metilare o acetilare le proteine adiacenti. I substrati ER o il coenzima A possono essere poliubiquitinati da più cicli della reazione oppure, a seconda delle proteine di legame, possono essere ulteriormente attivati o degradati dal proteasoma 26S.[10]
Di conseguenza, per avere una trascrizione genica efficace, programmata e mirata dalla struttura e dallo stato di fosforilazione dell’ER e dei coattivatori, è necessario un processo dinamico e ciclico di capacità di rimodellamento per l’assemblaggio trascrizionale, dopo il quale il complesso di trascrizione viene poi istantaneamente distrutto dal proteasoma.[10]
Effetti sull’Asse HPT
Gli estrogeni sono un importante regolatore dell’Asse HPT. L’ipofisi si trova al di fuori della barriera ematoencefalica e accumula alti livelli di SERM. Inoltre, i SERM possono bloccare l’aumento di peso dell’ipofisi indotto dagli estrogeni [12], suggerendo un’azione anti-estrogenica. Antagonizzando i recettori estrogenici e bloccando l’attivazione di questi da parte del E2, i SERM stimolano il rilascio da parte dell’Ipotalamo di GnRH che a sua volta induce la sintesi ed il rilascio di Ormone Luteinizzante [LH] e Ormone Follicolo Stimolante [FSH]. Ciò, di conseguenza, aumenta la sintesi testicolare di Testosterone e la spermatogenesi.
Ciclo di feedback negativo dell’Asse HPT E2 dipendente.
L’affinità del Clomifene per l’ER rispetto all’estradiolo varia dallo 0,1 al 12% in diversi studi, un valore simile a quello del tamoxifene (0,06-16%).[13][14][15] Il 4-idrossiclomifene, uno dei principali metaboliti attivi del Clomifene/Enclomifene, e l’Afimoxifene (4-idrossitamoxifene), uno dei principali metaboliti attivi del Tamoxifene, mostrano rispettivamente l’89-251% e il 41-246% dell’affinità dell’Estradiolo per l’ER nelle cellule di cancro al seno MCF-7 umano. [16] L’affinità per l’ER degli isomeri del 4-idrossiclomifene era del 285% per l'(E)-4-idrossiclomifene e del 16% per lo (Z)-4-idrossiclomifene rispetto all’Estradiolo. [16] Il 4-idrossi-N-desmetilclomifene ha un’affinità simile a quella del 4-idrossi-clomifene per l’ER.[17] In uno studio, l’affinità del Clomifene e dei suoi metaboliti per l’ERα era di ~100 nM per il Clomifene, ~2,4 nM per il 4-idrossi-clomifene, ~125 nM per l’N-desmetilclomifene e ~1,4 nM per il 4-idrossi-N-desmetilclomifene.[17]
Anche se il Clomifene ha un certo effetto estrogenico, dato dalla componente di Zuclomifene, si ritiene che la proprietà antiestrogenica sia la fonte principale della stimolazione dell’ovulazione, data dal Enclomifene. Il Clomifene sembra agire soprattutto nell’ipotalamo, dove esaurisce gli ER ipotalamici e blocca l’effetto di feedback negativo dell’Estradiolo endogeno circolante, che a sua volta determina un aumento della frequenza degli impulsi ipotalamici dell’ormone di rilascio delle gonadotropine (GnRH) e delle concentrazioni circolanti di ormone follicolo-stimolante (FSH) e ormone luteinizzante (LH).
Negli uomini normali, è stato riscontrato che 50mg/die di Clomifene per 8 mesi aumentano i livelli di Testosterone di circa 870ng/dL negli uomini più giovani e di circa 490ng/dL negli uomini più anziani.[18] I livelli di Estradiolo aumentano di 62pg/mL negli uomini più giovani e di 40pg/mL negli uomini più anziani.[18] Questi risultati suggeriscono che gli effetti progonadotropi del Clomifene sono più forti negli uomini più giovani che in quelli più anziani. Negli uomini con ipogonadismo, il Clomifene è risultato in grado di aumentare i livelli di Testosterone da 293 a 362ng/dL e i livelli di Estradiolo da 5,5 a 13pg/mL.[18] In un ampio studio clinico su uomini con bassi livelli di Testosterone (<400ng/dL), 25mg/die di Clomifene [circa 15.5mg di Enclomifene] hanno aumentato i livelli di Testosterone da 309ng/dL a 642ng/dL dopo 3 mesi di terapia. Non sono stati osservati cambiamenti significativi nei livelli di colesterolo HDL, trigliceridi, glucosio a digiuno o Prolattina, sebbene i livelli di colesterolo totale siano diminuiti significativamente.[18][19]
E’ di interesse sottolineare che la miscela racemica del Clomifene è composta per il 38% da Zuclomifene e per il 62% da Enclomifene. Lo Zuclomifene è lo stereoisomero (Z) del Clomifene, mentre l’Enclomifene è lo stereoisomero (E). Lo Zuclomifene è leggermente estrogenico, e a differenza dell’Enclomifene, esso ha azione antigonadotropa a causa dell’attivazione del recettore degli estrogeni con successiva riduzione dei livelli di Testosterone negli uomini. È inoltre circa cinque volte più potente dell’Enclomifene nell’indurre l’ovulazione nelle donne.
Il primo studio pubblicato sul Enclomifene comprendeva solo 12 uomini e non era in cieco [20]. In altre parole, sia i partecipanti che i ricercatori sapevano quale trattamento stavano ricevendo gli uomini. I partecipanti erano uomini con ipogonadismo secondario trattati in precedenza con Testosterone topico. Sono stati randomizzati a ricevere nuovamente Testosterone topico o Enclomifene (25mg al giorno).
Dopo sei mesi di trattamento, i livelli di Testosterone erano praticamente gli stessi tra i gruppi: 545ng/dL (18,9nmol/L) nel gruppo che riceveva il gel e 525ng/dL (18,2nmol/L) nel gruppo che riceveva l’Enclomifene. Anche i livelli di Testosterone libero sono aumentati e sono rimasti praticamente invariati tra i gruppi. Inoltre, e naturalmente, il numero di spermatozoi è stato ridotto negli uomini che ricevevano Testosterone, con numeri intorno ai 20milioni/mL. Inoltre, come previsto, il numero di spermatozoi è aumentato negli uomini che hanno ricevuto l’Enclomifene, con una media di circa 150milioni/mL.
Due interessanti studi [21][22]sull’Enclomifene hanno utilizzato lo stesso protocollo e l’aspetto forse più interessante è stata la dimensione del campione: 256 soggetti in totale. L’intervento è durato 16 settimane e i soggetti del gruppo Enclomifene hanno ricevuto 12,5mg al giorno e sono stati trattati fino a 25mg al giorno se i livelli di Testosterone non erano aumentati ad almeno 450ng/dL (15,6nmol/L) alla quarta settimana. La dose è stata aumentata per la metà dei soggetti che ricevevano l’Enclomifene. A questo punto le cose iniziano a farsi interessanti: sebbene metà dei soggetti sia stata modificata nel dosaggio alla quarta settimana, non è successo assolutamente nulla con la concentrazione media di Testosterone:
E, in effetti, alla fine dell’intervento, la media del gruppo era appena al di sotto del valore limite di 450ng/dL (15,6nmol/L) per l’up-titration. Infine, 29 degli 85 uomini del gruppo Enclomifene non hanno visto il loro Testosterone aumentare al di sopra del valore limite di ipogonadismo di 300ng/dL (10,4nmol/L) dopo 16 settimane di trattamento. Inoltre, i ricercatori hanno fatto un lavoro non propriamente apprezzabile nel trattare correttamente il gruppo che utilizzava il gel di Testosterone, come si può vedere dalla concentrazione media di Testosterone di quel gruppo.
E’ interessante notare che il Clomifene mostra in realtà risultati molto simili, anche mg per mg, a quelli dell’Enclomifene.
Uso dei SERM nella terapia per la fertilità in pazienti sottoposti a TRT
Uno studio ha assegnato i pazienti oligozoospermici a due gruppi di trattamento: (1) 20mg/die di Tamoxifene Citrato e 120mg/die di Testosterone Undecanoato [forma orale; pari a 75.9mg di Testosterone effettivo con una biodisponibilità del 8% = 6.072mg circa di principio attivo in circolo nelle 24h] (n = 106) e (2) trattamento con placebo (n = 106) per 6 mesi. Nel gruppo Tamoxifene/T, il numero totale di spermatozoi è aumentato da una mediana [25°, 75° percentile] di 27,1 × 106 cellule/mL [9,4, 54,0 × 106 cellule/mL] a 61,5 × 106 cellule/mL [28,2, 119,6 × 106 cellule/mL], la motilità progressiva è aumentata dal 29,7% ± 12,0% al 41,6% ± 13,1% e la morfologia normale è aumentata dal 41,2% ± 14,0% al 56,6% ± 11,5% dopo 6 mesi. Il tasso di gravidanza spontanea è stato del 33,9% nel gruppo Tamoxifene/T e del 10,3% nel gruppo placebo. Questo metodo di somministrazione concomitante di Testosterone e SERM potrebbe essere efficace nel mantenere la fertilità in una certa fetta di pazienti sottoposti a TRT. L’uso concomitante di hCG o Clomifene [o altro SERM] durante la TRT potrebbe non essere ottimale negli uomini in cerca di fertilità.[https://www.mdpi.com/1648-9144/60/2/275]
E’ interessante anche un piccolo studio del 1979 che ha preso in esame l’effetto delle somministrazione cronica di Clomifene in concomitanza con diversi androgeni…
Nelle osservazioni dello studio, l’infusione di Testosterone (T; 7,5mg/die per 4 giorni) ha prodotto un calo del 40% delle concentrazioni sieriche di LH e FSH. L’infusione di estradiolo (E2) in dosi equivalenti a quelle derivate dal T infuso (45μg/die) ha provocato un calo dell’LH sierico pari al 60% di quello osservato con il T, indicando che la maggior parte della soppressione dell’LH mediata dal T può essere attribuita alla sua aromatizzazione a E. Anche l’infusione di diidrotestosterone ha provocato una diminuzione del 35% dell’LH sierico medio e una diminuzione del numero di impulsi spontanei di LH simile a quella osservata con il T, a sostegno di un ruolo della componente androgenica pura nella soppressione dell’LH mediata dal T. Durante la terapia cronica con Clomifene, né il T né l’E2, se somministrati in dosi pari al doppio del loro tasso di produzione medio negli uomini normali, né gli androgeni non aromatizzabili, il Diidrotestosterone e il Fluoxymesterone, in dosi equipotenti al T infuso, sono stati in grado di sopprimere i livelli sierici di LH e FSH o di alterare le risposte di LH e FSH alla somministrazione di GnRH. La resistenza della gonadotropina alla soppressione da parte degli androgeni durante il blocco del Clomifene rimane ma con probabili variabili dose-temporali.[https://www.researchgate.net/]
Punti chiave
Abbiamo ripassato la funzionalità documentata del Clomifene e dell’Enclomifene di causare un aumento del GnRH con conseguente incremento di LH, FSH, Tetstosterone (e metaboliti annessi) e spermatogenesi in soggetti sani e ipogonadici [ipogonadismo secondario e AAS-indotto]. Ma durante l’uso di AAS/SARM è possibile avere una risposta terapeutica?
Oltre ai dati riportati in contesto TRT e SERM, se leggiamo con attenzione i dati sopra riportati, con una risposta di legame con effetto antagonista del ER ipotalamico dei mataboliti del Clomifene/Enclomifene del 285%, possiamo ipotizzare che la sua efficacia in presenza di molecole aromatizzabili sia proporzionale ai livelli di E2 o di suoi più potenti analoghi metilati in C7α o in C17α in circolo. In assenza di queste e in cosomministrazione con molecole non aromatizzabili, il suo potenziale di legame risulterebbe analogo al contesto di non utilizzo di AAS.
Possiamo chiuderla qui con un “si, ha una azione terapeutica anche in cosomministrazione con AAS/SARM, specie se non aromatizzabili!”? Purtroppo no, perchè il controllo dell’attività dell’Asse HPT non è regolato solo ed esclusivamente dal feedback negativo del E2.
I fattori che sopprimo l’Asse HPT
Come detto pocanzi, la sottoregolazione/soppressione dell’Asse HPT non è solo dipendente dal feedback negativo dato da un aumento del E2 circolante. Infatti, i fattori che influenzano la sottoregolazione/soppressione dell’Asse HPT sono:
L’origine del AAS, e di conseguenza…
Il tasso di conversione del AAS ad estrogeno, attraverso l’enzima aromatasi in alcuni tessuti (adiposo, mammario)
L’attività estrogenica intrinseca della molecola
L’attività progestinica dell’AAS
Dose e tempo d’uso/abuso del AAS
Attività androgena del AAS
Come possiamo vedere, oltre al fattore estrogenico vi sono quello diretto dall’AAS, la sua attività progestinica e la sua affinità con l’AR.
Sebbene l’utilizzatore del “tampone SERM” per cercare di garantirsi livelli di E2 e DHT nella norma (indi minimamente funzionali) raramente utilizza progestinici, la cosa non è impossibile vista la presenza di PH/AAS orali con attività progestinica [vedi 19-Nor-5-androstenediolo, MENTDIONE, MENT, Trenbolone Acetato, Metribolone ecc…].
Struttura molecolare del 19-nor-5-androstenediolo, noto anche come estr-5-ene-3β,17β-diolo, il proormone del Nandrolone e di altri 19-norandrostani.
Il Progesterone svolge inoltre un ruolo cruciale nell’Asse HPT. Durante la fase luteale, l’ipotalamo rilascia l’ormone di rilascio delle gonadotropine (GnRH), che agisce su una ghiandola chiamata ipofisi anteriore. Una quantità eccessiva di Progesterone o la presenza di Progestinici provoca un’inibizione a feedback negativo a livello ipotalamico/ipofisario, con conseguente cessazione marcata del rilascio di ormoni; maggiore di quella riscontrata con il ciclo di feedback del E2. Questo processo, nella maggior parte dei casi (se non in una estrema maggioranza con uno scarto di possibilità limitato) non è compensabile con l’uso di SERM.
Un altro fattore che interviene a livello del feedback negativo dell’Asse HPT risiede della attività AR della molecola. Di conseguenza, dovrebbe essere chiaro che anche farmaci puramente androgeni o essenzialmente anabolizzanti e con forte potenziale di legame con il AR [vedi SARM non steroidei] possono causare una sotto-regolazione della funzionalità dell’Asse HPT, quindi con meccanismi indipendenti dalla aromatizzazione della molecola.
Infatti, gli AAS [ed i SARM non steroidei] attraversano la barriera ematoencefalica e si legano ai recettori Ipotalamici. Ciò comporterà una marcata soppressione dell’HPTA per via di intermediari quali i peptidi oppioidi endogeni.
Quindi, bisogna sapere che l’attività di soppressione/sottoregolazione dell’Asse HPT androgeno-dipendente ha come intermediari i peptidi oppioidi endogeni, con attività principale da parte della Beta-Endorfina, delle Encefaline e Dinorfine attraverso il legame con i recettori oppioidi μ.
Recettori μ-opioidi attivi e inattivi
Tale effetto ridurrà comunque l’efficacia terapeutica dei SERM utilizzati anche se questi limiteranno il feedback negativo del E2. In breve, lo stimolo del GnRH e, di conseguenza, di LH e FSH saranno potenzialmente ridotti in rapporto AAS-dipendente e dose-dipendente. Ciò significa che non sarà possibile garantire livelli adeguati di E2 secondari alla aromatizzazione del Testosterone stimolato dalla attività del LH legata alla somministrazione di Clomifene o Enclomifene.
Struttura molecolare del Fluoxymesterone
Con l’uso del Fluoxymesterone le cose si complicherebbero ulteriormente. La sua capacità inibitiva sull’Asse HPT è più marcata di quella esercitata dal Methyltestosterone, nonostante non sia aromatizzabile, e si manifesta maggiormente a livello testicolare. Nel range dei 20mg/die non sembra mostrare un significativo impatto su FSH e LH ma già sul Testosterone circolante. Il Fluoxymesterone possiede una biodisponibilità del 100%, dovuta alla metilazione in posizione 17α la quale inibisce il metabolismo epatico per ossidazione enzimatica del 17β-idrossile, consentendo l’assorbimento nel flusso sanguigno della molecola. Come molti altri steroidi metilati in C-17, il Fluoxymesterone presenta una scarsa affinità con i recettori AR, ciononostante le sue azioni sono mediate dal recettore degli androgeni, molto probabilmente a causa della sua prolungata emivita plasmatica che è di circa 9,2 ore.(Seth Roberts “Anabolic Pharmacology”. 2009)
Effetto dei SERM sull’Asse hGH/IGF1
Esistono poche differenze tra i vari SERM nell’influenzare negativamente l’Asse hGH/IGF1, in quanto è stato riportato che il Raloxifene ha indotto una minore diminuzione dei livelli di IGF1 rispetto al Tamoxifene, considerando che entrambi i farmaci sono stati somministrati a un dosaggio massimo di 120mg/die e 20mg/die, rispettivamente [94].
Cozzi et al. [95] hanno provato per la prima volta a utilizzare il tamoxifene come possibile trattamento dell’acromegalia; nel 1997 hanno trattato 19 soggetti acromegalici (6 maschi, 13 femmine) per due mesi con un dosaggio crescente, fino a raggiungere i 40 mg/die. L’IGF1 medio è diminuito del 29,5%, con un range compreso tra il 18% e il 60%, in 13 dei 19 pazienti, raggiungendo un controllo ormonale completo in quattro di essi (21%). I livelli di GH sono leggermente aumentati rispetto al basale, mentre dopo la sospensione del tamoxifene l’IGF1 sierico è prontamente aumentato. Molti anni dopo, Balili et al. [31] hanno riportato che 17 pazienti (15 maschi e 2 femmine) con acromegalia resistente sono stati trattati con tamoxifene (dose massima 40mg/die) per un periodo mediano di quattro mesi. È stata evidenziata una riduzione significativa dell’IGF1 nell’82% dei pazienti, raggiungendo il controllo della malattia nel 47% dei casi. I livelli sierici di IGF1 si sono ridotti del 17,5%, mentre i livelli di GH non hanno subito variazioni significative.
Schema semplificato dell’azione di E2 e SERM sull’Asse hGH/IGF1
Duarte et al. [35] nel 2016 hanno studiato 16 maschi con acromegalia non controllata, dimostrando l’efficacia del Clomifene Citrato (CC) come terapia aggiuntiva a SRL o Cabergolina. I pazienti sono stati trattati per tre mesi con CC 50mg/die, mostrando una riduzione media dei livelli di IGF1 del 41% (con valori compresi tra il 16,8% e il 68,3%), che ha portato il 44% dei pazienti a raggiungere il controllo ormonale. Gli estrogeni e i SERM hanno ampiamente dimostrato una significativa attività di riduzione dell’IGF1.
Le concentrazioni plasmatiche seriali di hGH sono state misurate ogni 20 minuti per 24 ore prima e dopo la somministrazione di Clomifene Citrato (100mg/die per 7 giorni) a quattro soggetti sani maschi giovani adulti. Il numero di episodi secretori di hGH e l’entità del picco delle concentrazioni plasmatiche durante la veglia e il sonno sono diminuiti dopo i periodi di trattamento con Clomifene Citrato.[https://www.sciencedirect.com/science/article/abs]
In uno studio sono stati inclusi sette bracci, comprendenti donne in postmenopausa con diabete mellito di tipo 2, donne in postmenopausa con cancro al seno, donne sane in postmenopausa e uomini anziani sani. La terapia con Raloxifene ha ridotto significativamente i livelli di IGF-1 (WMD: -2,92 nmol/L, 95% CI: -3,49, -2,35, p < 0,001) rispetto al placebo. Il dosaggio di raloxifene ˃60mg/die (WMD: -3,29 ng/mL, 95% CI: -3,50-3,08, I2 = 0,0%) ha ridotto i livelli di IGF-1 più di 60 mg/die (WMD: -2,29 ng/mL, 95% CI: -2,90 -1,69, I2 = 16%). Inoltre, la durata dell’intervento ˃26 settimane (WMD: -3,48 ng/mL, 95% CI: -5,26 a -1,69, I2 = 0,0%) ha ridotto i livelli di IGF-1 più di ˂26 settimane (WMD: -2,55 ng/mL, 95% CI: -3,31 a -1,79, I2 = 92%). Al contrario, i risultati complessivi del modello a effetti casuali non hanno suggerito un cambiamento significativo nei livelli di IGFBP-3 con la terapia con raloxifene. La terapia con Raloxifene ha ridotto significativamente i livelli sierici di IGF-1, ma senza variazioni nei livelli di IGFPB-3.[https://www.sciencedirect.com/science/article/abs/pii/S1096637421000447]
Il Tamoxifene è in grado di ridurre l’IGF-1 biodisponibile (calcolato come rapporto tra IGF-1 e IGF-BP3) per almeno 18 mesi. Sebbene le concentrazioni di IGF-1 non si siano ridotte in modo significativo, le concentrazioni della sua principale proteina legante IGF-BP3 sono aumentate in modo significativo, riducendo così la quantità di IGF-1 disponibile. Tuttavia, il rapporto tra IGF-1 e IGF-BP3 non era significativamente ridotto rispetto al basale a 27 mesi, per cui l’effetto di un trattamento più lungo resta da chiarire. Anche il Tamoxifene ha aumentato significativamente le concentrazioni di IGF-BP1 rispetto al basale dopo 18 mesi di trattamento. Questo aumento è stato osservato anche in altri studi.
In alcuni studi sul Tamoxifene è stata notata paradossalmente un’assenza di effetti sulle concentrazioni di IGF-1 a differenza di altri studi che hanno dimostrato una riduzione dell’IGF-1 da parte del Tamoxifene. Questo potrebbe essere il risultato del numero ridotto di pazienti degli studi in questione o della selezione della popolazione. Tuttavia, uno studio non ha mostrato un effetto sull’IGF-1 a un follow-up mediano di 29 mesi. Questi ricercatori avevano osservato una diminuzione significativa dei valori di IGF-1 dopo sei mesi di trattamento con Tamoxifene e i loro dati indicano un effetto limitato dopo un trattamento a lungo termine. Anche altri dati da campioni più piccoli indicano una riduzione iniziale (sebbene non significativa) dell’IGF-1, che si perde con l’aumentare del tempo di follow-up. Ciò indica un effetto potenzialmente importante della durata del trattamento sull’esito e sottolinea la necessità di ulteriori studi longitudinali con periodi di follow-up rigorosamente tempificati.
Uno studio a lungo termine controllato con placebo ha mostrato una riduzione significativa dell’IGF-1 dopo un follow-up medio di 27 mesi (follow-up minimo di tre mesi), ma non sono stati prelevati campioni longitudinali. È possibile che i campioni provenienti dagli studi di prevenzione con Tamoxifene in corso (come l’IBIS) vengano utilizzati per ulteriori ricerche sugli effetti del Tamoxifene sul sistema IGF. In alcuni studi i campioni utilizzati non erano a digiuno e questo può essere importante perché i valori possono fluttuare in base all’assunzione di nutrienti.
Il meccanismo con cui il Tamoxifene altera lo stato dell’IGF non è stato completamente chiarito. Tuttavia, si ritiene che il Tamoxifene alteri i valori di IGF-1 riducendo la produzione di hGH da parte dell’ipofisi, abbassando così la quantità di IGF-1 prodotta dal fegato [endocrina] e rilasciata in circolo. Sappiamo che il Tamoxifene ha anche un’azione diretta come antagonista dell’E2 in diversi tessuti del corpo oltre che sulle cellule del cancro al seno, e sembrerebbe alterare la quantità di IGF-1 e di proteine leganti rilasciate dalle cellule stesse.
Il Tamoxifene, quindi, può aumentare l’IGF-BP1, l’IGF-BP3 e ridurre il rapporto tra IGF-1 e IGF-BP3. Gli effetti a lungo termine dell’uso del Tamoxifene sullo stato dell’IGF devono ancora essere stabiliti. Non è ancora del tutto chiaro quando e per quanto tempo il Tamoxifene può ridurre l’IGF-1 circolante.[https://www.ncbi.nlm.nih.gov/]
Aumento delle SHBG
L’effetto del Clomifene Citrato (CC) sulle SHBG è stato studiato in 10 pazienti oligozoospermici con varicocele e 6 uomini normospermici. Le SHBG plasmatiche, Testosterone (T), Estradiolo (E2), FSH, LH. Prolattina (Prl), Tiroxina (T4) e 17-OH-progesterone (17-OH-P) sono stati determinati prima e durante la terapia. La concentrazione di SHBG è aumentata da 38,1 ± 18,3 a 54,3 ± 16,0 nmol/l (P < 0,01), mentre il T e l’E2 hanno mostrato aumenti significativi da 31,2 ± 10,8 nmol/***l e 24,6 ± 5,4 pg/ml a 52,0 ± 3,6 e 43,3 ± 14,9, rispettivamente nei pazienti oligozoospermici, con aumenti simili osservati negli uomini normospermici. L’FSH, l’LH e il 17-OH-P sono risultati marcatamente elevati durante la somministrazione di CC, mentre Prl e T4 sono rimasti invariati. I risultati di questo studio indicano che la CC provoca un aumento della concentrazione di SHBG, probabilmente correlato anche all’aumento della concentrazione di E2. Questa variazione della SHBG, combinata con l’attività estrogenica intrinseca del CC, potrebbe essere uno dei fattori responsabili, attraverso una diminuzione del T libero e uno squilibrio tra T ed E2, della mancanza di un effetto significativo sui parametri della qualità seminale nei pazienti così trattati. [https://onlinelibrary.wiley.com/doi/abs/]
Schema semplificato dell’azione dei SERM e E2 sull’espressione del gene SHBG e sintesi delle SHBG.
In uno studio, tredici pazienti sono stati sottoposti a trattamento con Tamoxifene dopo la classificazione secondo Nydick (gruppo 1). Il gruppo 2 era composto da otto pazienti seguiti senza trattamento. La ginecomastia era presente bilateralmente in 15 pazienti. In entrambi i gruppi si è verificata una riduzione statisticamente significativa delle dimensioni del seno. Si è verificata una diminuzione significativa della SHBG sierica solo nel gruppo 2. Questi risultati suggeriscono che la SHBG sierica è aumentata dal trattamento con Tamoxifene negli adolescenti maschi trattati. I livelli di SHBG sono diminuiti per tutta la durata del follow-up nei pazienti che sono guariti con o senza trattamento. Tuttavia, questa diminuzione era statisticamente significativa nel gruppo non trattato, ma non in quello trattato con Tamoxifene. In conclusione, è stato suggerito che il calo puberale dei livelli di SHBG sia attenuato dal trattamento con tamoxifene somministrato per la ginecomastia puberale, poiché il Tamoxifene aumenta i livelli di SHBG negli adolescenti maschi.[https://pubmed.ncbi.nlm.nih.gov/15379424/]
Ma gli Inibitori della Aromatasi?
Gli IA possono essere in alcuni casi un modo efficace per controllare i livelli di E2 durante la TRT. Tuttavia, il dosaggio necessario per mantenere i livelli di E2 nell’intervallo ottimale dipende da ciascun individuo e richiede un attento monitoraggio da parte di un professionista sanitario. Ma in un contesto di alterazione del ciclo di feedbeack negativo del E2, specie se cosomministrati con AAS non aromatizzabili, possono portare a peggioramento delle condizioni più che ad una risposta positiva nel mantenimento di una certa attività dell’Asse HPT.
Conclusioni:
Nonostante la ricerca abbia mostrato in studi su animali sottoposti a somministrazione di AAS (Oxymetholone) abbinata al Clomifene Citrato una qualche conservazione del Testosterone endogeno [Growth-hormone-secretagogue-GHRP-6-and-clomiphene?redirectedFrom=fulltext], e che nelle terapie per la fertilità in soggetti in TRT, o in soggetti trattati per brevi periodi con AAS e.v., la somministrazione di Clomifene Citrato ha mostrato un effetto misurabile [ma qui parliamo comunque di condizioni più che altro “mimiche-fisiologiche”], sul campo la misurazione dell’efficacia della somministrazione di SERM (soprattutto Clomifene e Enclomifene) per mantenere una certa sintesi endogena di Testosterone e consequenzialmente dei suoi metaboliti E2 e DHT, non è lineare e chiara, sia per la difficile identificazione della qualità dei PEDs utilizzati e sia per la difficolta di svolgere esami ematici che non siano basati sul fallace (ormonalmente) metodo ECLIA/ELISA. La rara possibilità (almeno in Italia) di poter accedere a laboratori dove sono svolti test LC/MS-MS ultra sensibile [vedi spettrometria di massa accoppiata] limita le valutazioni precise necessarie dal momento che con i metodi sopra citati ormoni diversi possono essere letti come il medesimo ormone. Nonostante ciò, siamo stati in grado di notare degli effetti terapeutici sufficienti con cicli a medio/basso dosaggio di AAS come Oxandrolone e Stanozololo [media 30mg/die]. In altre circostanze, e in una buona fetta di popolazione, l’andamento dell’efficacia variava all’interno dello stesso arco temporale del ciclo al quale i soggetti si sottoponevano.
Basandoci sulla ricerca diretta, possiamo teoricamente elencare gli AAS/SARM/PH e DS con l’effetto ipoteticamente raggiungibile in combinazione con SERM:
Effetto buono
Oxandrolone [=30mg di media]
Stanozololo [=20mg di media]
Methyldrostanolone [=30mg di media]
4-clorodeidrometiltestosterone [=40mg di media]
Ostarina [=20mg di media]
RAD140 [=20mg di media]
Effetto discreto/moderato
Testosterone Undecanoato [<120mg/die di media]
Methandrostenolone [<20mg di media]
Oxymetholone [<50mg di media]
LGD4033 [<10mg di media]
Effetto non sufficiente
Fluoxymesterone [≥10mg di media]
MENTDIONE [≥50mg di media]
MENT [≥25mg di media]
Metribolone [≥250mcg di media]
Norethandrolone [≥20mg di media]
Trenbolone Acetato (orale) [≥25mg di media]
19-Nor-5-androstenediolo [≥50mg di media]
Chi sceglie di prendere la “via del Enhanced” e la sua paura principale è basata sulle iniezioni beh, forse è meglio che abbandoni tale possibile scelta… no?…
Paradossalmente, è di gran lunga più funzionale l’inserimento di piccole dosi di Methandrostenolone [15mg/die circa] come base “sostitutiva” del Testosterone compensando il DHT con la versione metilata in C1 di questo, il Mesterolone.
Kremoser C, Albers M, Burris TP, Deuschle U, Koegl M (Oct 2007). “Panning for SNuRMs: using cofactor profiling for the rational discovery of selective nuclear receptor modulators”. Drug Discovery Today. 12 (19–20): 860–9. doi:10.1016/j.drudis.2007.07.025. PMID17933688.
Rosano C, Stec-Martyna E, Lappano R, Maggiolini M (2011). “Structure-based approach for the discovery of novel selective estrogen receptor modulators”. Current Medicinal Chemistry. 18 (8): 1188–94. doi:10.2174/092986711795029645. PMID21291367.
Xu X, Yang W, Li Y, Wang Y (Jan 2010). “Discovery of estrogen receptor modulators: a review of virtual screening and SAR efforts”. Expert Opinion on Drug Discovery. 5 (1): 21–31. doi:10.1517/17460440903490395. PMID22823969. S2CID207492889.
Musa MA, Khan MO, Cooperwood JS (2007). “Medicinal chemistry and emerging strategies applied to the development of selective estrogen receptor modulators (SERMs)”. Current Medicinal Chemistry. 14 (11): 1249–61. doi:10.2174/092986707780598023. PMID17504144.
Lewis JS, Jordan VC (Dec 2005). “Selective estrogen receptor modulators (SERMs): mechanisms of anticarcinogenesis and drug resistance”. Mutation Research. 591 (1–2): 247–63. doi:10.1016/j.mrfmmm.2005.02.028. PMID16083919.
Fang H, Tong W, Shi LM, Blair R, Perkins R, Branham W, et al. (March 2001). “Structure-activity relationships for a large diverse set of natural, synthetic, and environmental estrogens”. Chemical Research in Toxicology. 14 (3): 280–94. doi:10.1021/tx000208y. PMID11258977.
Baumann RJ, Bush TL, Cross-Doersen DE, Cashman EA, Wright PS, Zwolshen JH, et al. (March 1998). “Clomiphene analogs with activity in vitro and in vivo against human breast cancer cells”. Biochemical Pharmacology. 55 (6): 841–51. doi:10.1016/s0006-2952(97)00574-1. PMID9586957.
Sutherland RL, Watts CK, Ruenitz PC (October 1986). “Definition of two distinct mechanisms of action of antiestrogens on human breast cancer cell proliferation using hydroxytriphenylethylenes with high affinity for the estrogen receptor”. Biochemical and Biophysical Research Communications. 140 (2): 523–9. doi:10.1016/0006-291x(86)90763-1. PMID3778464.
Obach RS (April 2013). “Pharmacologically active drug metabolites: impact on drug discovery and pharmacotherapy”. Pharmacological Reviews. 65 (2): 578–640. doi:10.1124/pr.111.005439. PMID23406671. S2CID720243.
Kaminetsky, Jed, et al. “Oral enclomiphene citrate stimulates the endogenous production of testosterone and sperm counts in men with low testosterone: comparison with testosterone gel.” The journal of sexual medicine 10.6 (2013): 1628-1635.
Kim, Edward D., Andrew McCullough, and Jed Kaminetsky. “Oral enclomiphene citrate raises testosterone and preserves sperm counts in obese hypogonadal men, unlike topical testosterone: restoration instead of replacement.” BJU international 117.4 (2016): 677-685.
Earl, Joshua A., and Edward D. Kim. “Enclomiphene citrate: A treatment that maintains fertility in men with secondary hypogonadism.” Expert review of endocrinology & metabolism 14.3 (2019): 157-165.
*Nota per il lettore: la tesi di seguito esposta si affianca a quanto già ipotizzato dal “web writer”, nonché coach, autore e ricercatore, Type-IIx di MesoRx .
Introduzione:
Abbiamo imparato che il Boldenone, con tutta probabilità, ha una funzione di “ormone esca” per l’enzima Aromatasi. Sappiamo però che, probabilmente, la sua conversione in estrogeno lo vede convertirsi prevalentemente in Estrone [E1] e non in Estradiolo [E2]. Sappiamo che l’Estrone può convertirsi in Estradiolo (e viceversa) ma che il tasso in cui ciò avviene è molto basso. Siamo a conoscenza del fatto che l’E1 è un estrogeno molto meno potente dell’E2 e, come tale, è un estrogeno relativamente debole.[Kuhl H (August 2005), Escande A et al. (May 2006), Ruggiero RJ, Likis FE (2002)] Secondo uno studio, le affinità di legame relative dell’E1 per l’ERα e l’ERβ umani erano rispettivamente il 4,0% e il 3,5% di quelle dell’E2, e le capacità transazionali relative dell’E1 all’ERα e all’ERβ erano rispettivamente il 2,6% e il 4,3% di quelle dell’E2. [ Escande A et al. (May 2006)] In accordo, l’attività estrogenica dell’Estrone è stata riportata a circa il 4% di quella dell’Estradiolo.[Kuhl H (August 2005)] Non sicuramente una caratteristica favorevole per l’uso di una molecola senza la presenza di una base di Testosterone e/o hCG.
Farmacocinetica schematizzata del Boldenone UndecylenatoConversione del Boldenone in Estrone attraverso l’interazione con l’enzima Aromatasi.
Conosciamo molto bene anche il Methenolone che, come derivato del DHT, non è soggetto ad aromatizzazione e quindi non ha la propensione a produrre effetti collaterali estrogenici come la ginecomastia.[William Llewellyn (2011). Anabolics] Come AAS, il Methenolone è antigonadotropo e esercita una soppressione dell’Asse HPT causando ipogonadismo reversibile e infertilità.[van Breda E et al. (Apr 2003)] Essendo un derivato del DHT conserva alcune caratteristiche antiestrogeniche, sebbene esse siano inferiori a quelle osservate con altre molecole simili come il Drostanolone. Queste proprietà, in un ambiente già predisposto a carenza di E2 [vedi mancanza di una base di Testosterone, mancato utilizzo di hCG e/o dosi sufficienti di questa, presenza di una molecola con marcati tassi di conversione in E1] non fanno altro che portare ad effetti avversi tipici dell’ipoestrogenemia [vedi, ad esempio, letargia, debolezza, dolori articolari, bassa libido, difficoltà a raggiungere e mantenere l’erezione ecc…].
Da sinistra: struttura molecolare del Methenolone privo di legame con l’estere e struttura molecolare dell’AAS legata all’estere Enantato.
Magari avete esperienza nell’uso di Boldenone Undecilenato, di Methenolone Enantato, o forse anche delle due molecole in combinazione ( magari con altri AAS). Forse potrete aver visto riportati i feedback degli utilizzatori in qualche forum in rete, o potreste anche essere a conoscenza di qualcuno che ha avuto effetti completamente diversi dai vostri con l’uso degli stessi farmaci. Nel primo caso (testimonianze su internet), avete, forse, ritenuto che questi utilizzatori si siano probabilmente somministrati prodotti non contenenti le suddette molecole (sperando di non essere voi gli interessati da ciò!). Nel secondo caso, in cui qualcuno che conoscete bene e capite che non ha alcuna motivazione per cui mentire e che sta usando AAS indubbiamente autentici (ad esempio, autenticati da HP/LC) vi riferisce allo stesso modo effetti completamente diversi da quelli da voi riscontrati.
Ma come stanno le cose? – come possono persone diverse sperimentare effetti così marcatamente diversi, persino opposti, dalla stessa molecola (o dalle stesse molecole) a dosi simili?
Non resta che:
Affrontare questa domanda, in modo rigoroso, per rivelarci ciò che non era immediatamente evidente e, auspicabilmente, imparare alcuni fatti preziosi come risultato.
Fornire soluzioni a coloro che sperimentano sintomi intollerabili di bassa estrogenicità come conseguenza dell’uso non medico di AAS.
Tesi Teoria delle potenze estrogeniche dipendenti dalla molecola (per-AAS) e individualizzate (per utilizzatore):
Gli effetti di ogni AAS sull’estrogenicità (effetti associati all’attivazione di ER- α e β) dipendono da fattori dipendenti dalla molecola (per-AAS) e individualizzati (per-utilizzatore) che determinano sia
A. i livelli ematici effettivi che
B. gli effetti a livello tissutale dei prodotti aromatici di ogni AAS.
I prodotti aromatici consequenziali ai processi biochimici degli AAS vanno da quelli nulli (cioè non aromatizzabili), all’E1 (Estrone), un estrogeno debole, all’E2 (Estradiolo), un estrogeno potente (il più potente tra quelli endogeni) di cui tutti i lettori conoscono almeno l’esistenza e che è associato ai classici effetti estrogenici (sia che l’E2 sia “crashato” o meno), fino agli estrogeni non endogeni e altamente potenti come il 7α-metilestradiolo (il prodotto aromatico notevolmente potente del MENT, o anche noto come Trestolone).
Gli effetti di ciascun AAS (alla sua dose e durata) e dei suoi prodotti aromatici (alle loro concentrazioni e durate) determinano l’Androgeno/Estrogeno ratio (A/E), un indicatore degli effetti sistemici generali degli AAS (diretti e collaterali); ad esempio, ginecomastia. Il “braccio” androgeno del rapporto A/E è il prodotto della potenza dell’AAS di attivare l’AR alla sua area sotto la curva (AUC), come nmol×h/L. Il “braccio” estrogenico del rapporto A/E ha due aspetti: effetti estrogenici e antiestrogenici. Per quanto riguarda gli effetti estrogenici, questi sono il prodotto della concentrazione e della durata (AUC come nmol×h/L) dei prodotti aromatici (cioè gli estrogeni) e delle loro capacità di attivare ER- α e β. Reciprocamente, gli effetti antiestrogenici, che sono effetti intrinseci della classe degli AAS ben consolidati nell’uomo e negli animali, derivano dagli effetti ipofisari (cioè antigonadotropi) e tissutali locali (ad esempio, impediscono l’assorbimento degli estrogeni) degli AAS, che si ricollegano al “braccio” degli androgeni.
Gli effetti individualizzati (per utilizzatore) degli AAS sull’estrogenicità dipendono in gran parte da tre (3) fattori ereditabili discreti (cioè, il risultato del proprio fenotipo genetico) che sono soggetti a un’ampia variazione interindividuale (differenze tra utilizzatori): il profilo ormonale legante¹, l’espressione dell’isozima 17β-HSD e l’espressione dell’Aromatasi³. In primo luogo, il profilo ormonale legante dell’utilizzatore (cioè le attività di SHBG, albumina, α₁ glicoproteina acida, globulina legante i corticosteroidi) determina le attività di E1/E2 liberi (estrogeni liberi) e il rapporto E1/E2 liberi:androgeni. In secondo luogo, questo profilo ormonale vincolante¹ interagisce con la velocità di aromatizzazione dell’AAS (Vmax) e la lunghezza della catena di esteri (cioè logP e idrofobicità) quando le concentrazioni del farmaco raggiungono lo stato stazionario, influenzando il gradiente di concentrazione degli estrogeni attivi (E1 ed E2 liberi) poiché l’esterasi libera l’ormone progenitore dal profarmaco mediante idrolisi attiva nel sangue intero [4]. In terzo luogo, l’espressione dell’isoenzima 17β-HSD dell’utilizzatore determina il flusso netto di E1 ( estrogeno debole) rispetto all’E2 (estrogeno potente). Infine, l’espressione dell’Aromatasi dell’utilizzatore – in parte modificabile dall’autoregolazione della massa grassa – determina le concentrazioni assolute di estrogeni (E1 ed E2).
Nota: non lasciatevi dissuadere da questa presentazione così massiccia dei fattori che influenzano le concentrazioni di estrogeni nel sangue e le attività estrogeniche a livello tissutale, poiché non li abbiamo ancora analizzati. Continuate a leggere: questi fattori verranno illustrati man mano che procederemo.
Divergenza negli effetti estrogenici del Boldenone e del Methenolone; e i limiti dei livelli circolanti come indice della regolazione estrogenica tessuto-specifica:
Da referti di casi reali raccolti in rete, i cui soggetti proprietari hanno riferito l’uso di Boldenone e/o Methenolone.
Quattro (4) casi distinti in cui non è stata utilizzata alcuna molecola AI:
1- Innalzamento dell’E2 e dell’E1 sierici con 800mg di Boldenone Undecylenato, 600mg di Trenbolone e 300mg di Testosterone:
Boldenone Undecylenato (800mg) + Trenbolone Enantato (600mg) + Testosterone Enantato (300mg). Analisi del sangue: Estrone (E1): 1.352 pmol/L (Intervallo di riferimento: < 250 pmol/L), cioè 365,6 pg/mL (Molto alto).
2-Elevazioni dell’E2 sierica da 300 mg di Primo, 300 mg di Test:
*Methenolone Enantato + Testosterone Enantato analisi del sangue con E2 basso-moderato
3-Riduzione dell’E2 sotto la norma con 750mg di Testosterone Enantato, 500mg di Boldenone Undecylenato, 400mg di Methenolone Enantato:
Che trarre qualsiasi deduzione (per non parlare delle conclusioni) da questi risultati divergenti è un azzardo. Essi ci indicano una sola cosa: semplicemente che il Boldenone Undecylenato e/o il Methenolone (Enantato) sembrano abbassare l’estrogenicità riflessa dagli esami del sangue in alcuni casi e che per caratteristiche molecolari i meccanismi sono di natura sicuramente diversa.
I risultati di queste analisi del sangue illustrano i rischi di trarre inferenze o conclusioni dalle analisi del sangue di laboratorio postate in rete da diversi utilizzatori.
Dopo che il lettore avrà compreso i limiti dei livelli circolanti come indice della regolazione degli estrogeni specifica per i tessuti, verrà spiegato – nel modo più parsimonioso possibile rispetto alle prove e alla domanda – i fattori che influenzano le concentrazioni di estrogeni nel sangue e le attività estrogeniche a livello tissutale, al fine di “dare un’occhiata sotto il velo” a ciò che potrebbe guidare questa divergenza negli effetti estrogenici del Boldenone e del Methenolone.
Limiti dei livelli circolanti di estrogeni come indice della regolazione estrogenica tessuto-specifica:
[10]
AD: Androstenedione
Struttura molecolare del Androstenedione.
La regolazione della produzione e del metabolismo degli estrogeni nei tessuti periferici è consentita dall’espressione locale dell’Aromatasi (CYP19A1), che converte gli androgeni in estrogeni (T ⇒ E2 e AD ⇒ E1 [l’E2 è l’estrogeno più prevalente nell’uomo; ciò può spiegare la maggiore tollerabilità del Boldenone nelle donne]). Gli estrogeni possono inoltre essere convertiti in solfati di estrogeni e in esteri acilici grassi di estrogeni tramite estrogeno solfotransferasi (EST) e acil-transferasi, rispettivamente. Infine, questi derivati degli estrogeni possono essere riconvertiti in estrogeni progenitori attraverso l’attività della solfatasi steroidea (sulfatasi) e della lipasi [10].
Il tessuto adiposo (AT) è particolarmente ricco di esteri acilici grassi degli estrogeni e, di conseguenza, possiede un ampio sistema di tamponamento che consente la regolazione locale della produzione e del metabolismo degli estrogeni… In particolare, in uno studio condotto su uomini obesi, le concentrazioni di esteri acilici grassi dell’E2 sono risultate correlate nel siero e nel grasso (Wang, et al., 2013) [10], indicando probabilmente che i livelli di estrogeni nel siero influenzano il contenuto di estrogeni immagazzinati nell’AT, ma la conversione in forme bioattive è regolata localmente [10].
Diversi studi clinici hanno dimostrato una dissociazione tra i livelli di estrogeni circolanti e quelli intra-adiposi, anche negli uomini (Blankenstein, et al., 1992; Belanger, et al., 2006; Deslypere, et al., 1985; Wang, et al., 2013) [10].
Fattori confondenti nei dati dell’estrogenicità di Boldenone e/o Methenolone:
In questo articolo si ragionerà sui fattori che determinano un fenomeno di apparenti contraddizioni multiple – per comprendere una realtà (cioè la nostra) in cui praticamente tutti dicono la “verità”, affermando di aver assunto quelli che ritengono essere gli stessi farmaci a dosi comparabili, eppure, sorprendentemente, l’estrogenicità (un fattore coinvolto nella tollerabilità) differisce tra gli individui. I fattori in gioco sono i seguenti:
Le analisi ematiche di laboratorio possono non riflettere l’estrogenicità perché sono coinvolti meccanismi a livello tissutale (ad esempio, blocco dell’assorbimento degli estrogeni, attività intra- ed endocrina).
Variazione interindividuale del profilo ormonale legante¹, dell’espressione dell’isoenzima 17β-HSD² e dell’espressione dell’Aromatasi³, per non parlare di fattori come l’espressione del ER (cioè la densità o il numero), ad esempio nel tessuto mammario (fattori che sono coinvolti nella tollerabilità).
Incompletezza degli esami ematici di laboratorio in cui viene utilizzato il Boldenone (ad esempio, le misure di E2 nel siero sono insufficienti senza le misure di E1).
Contraffazione o presenza di altra molecola nel prodotto (ad es. Methenolone viene sostituito da Testosterone o Drostanolone).
Differenze nella lunghezza dell’estere (ad esempio, Boldenone Cypionato vs. Undecylenato) che riflettono il logP: coefficiente di ripartizione e la lipofilia: polarità; profondità di iniezione (ad esempio, nello spazio sottocutaneo vs. intramuscolare profondo) e sito di somministrazione che differiscono nel flusso sanguigno e quindi nell’attività dell’esterasi, influenzando indirettamente il tasso di reazioni dell’Aromatasi.
Le presunte autodichiarazioni dei professionisti del fitness che traggono un reddito dalla generazione di notizie sui media possono essere motivate da travisamenti e/o frodi al fine di aumentare gli introiti pubblicitari come minimo, se non per integrare le loro scoperte scintillanti e nuove nel loro portafoglio utilizzandole come insegna o segno distintivo, su cui il loro lavoro (ad esempio, video su YouTube, scritti) sarà identificato e distinto.
Fattori che influenzano le concentrazioni di estrogeni nel sangue e le attività estrogeniche a livello tissutale:
Fattori dipendenti dalle molecole (Per-AAS)
Prodotti aromatici e loro capacità di attivare ER- α e β.
a) Boldenone =[Aromatasi]=> E1 (Estrone, un estrogeno debole, 2% di potenza ER-α rispetto all’E2) ed E2 (Estradiolo, il 17β-OH lo rende 50 volte più potente dell’E2) {aromatizza in E1 ed E2}.
b) Methenolone =X[Aromatasi] {non aromatizza}, quindi non supera:
Effetti antiestrogenici che sono effetti di classe degli AAS, specie nei DHT derivati:
a) inibizione delle gonadotropine secrete dall’ipofisi (che riducono indirettamente gli estrogeni) e
b) blocco diretto dell’attività degli estrogeni a livello degli organi bersaglio, impedendo l’assorbimento degli estrogeni, ad esempio, nelle cellule sinoviali, causando sintomi di “articolazione secca e dolorante”. È questo l’effetto che rende il Methenolone [1], [2] – e prima che venisse sospeso – Drostanolone [3], così efficace per il cancro al seno metastatico resistente al trattamento.
Boldenone Undecylenato: a) velocità di aromatizzazione (Vmax) ridotta rispetto al Boldenone libero.
Km: pari alla concentrazione del substrato (ascissa; valori dell’asse delle ascisse) quando la velocità è la metà della velocità massima (1/2Vmax; ordinata; valori dell’asse delle ordinate).
T: Testosterone
L’aromatizzazione è ostacolata (rispetto al T) per gli androsta-1,4-diene-3-oni (come il Boldenone; Undecylenato.), per cui procede lentamente [17].
T =[Aromatasi]=> E2, Κm = 1,83nM, secondo la cinetica di Michaelis-Menten [18].
Non conosciamo il Km per l’attività dell’Aromatasi in vivo rispetto al Boldenone Undecylenato. Sappiamo però che l’enzima Aromatasi è saturabile, per cui al di sopra di una certa dose, che dipende dall’espressione³ o dal numero di proteine dell’Aromatasi (e dal profilo ormonale di legame¹), tale dose non causerà ulteriori aumenti degli estrogeni attivi (E2 ed E1 liberi). Poiché il Boldenone Undecylenato è soggetto a un’aromatizzazione ostacolata, la sua velocità di reazione (Vmax) deve essere relativamente rallentata. Di conseguenza, la sua Km in vivo deve essere spostata verso destra (rispetto a quella di T/E2) e richiede concentrazioni maggiori di T per la saturazione dell’Aromatasi. Questo ci dice che, rispetto al T, sono necessarie dosi più elevate di Boldenone prima che l’Aromatasi si saturi (non è soggetto ad alcun aumento di E2 a dosi superiori al punto di saturazione).
Inoltre sappiamo anche che il 40% in più di Vmax dell’Aromatasi in rapporto al T negli uomini anziani rispetto a quelli giovani è stato praticamente interamente spiegato dalla massa grassa e dalle SHBG (cioè il profilo ormonale legato¹).[18] Poiché l’Aromatasi è espressa anche negli adipociti (cellule grasse), il cui numero è soggetto ad aumentare a causa della lipogenesi di nuove cellule grasse (adipociti), il mantenimento di una bassa percentuale di grasso corporeo per tutta la vita è un fattore importante che può essere controllato dal soggetto. È importante capire che le cellule adipose non vengono distrutte dalla restrizione calorica: l’aspetto visivo di una bassa percentuale di grasso corporeo dopo una dieta ipocalorica non riflette la perdita di numero di adipociti, ma solo la riduzione delle riserve di lipidi all’interno di tali cellule. Solo la lisazione o il congelamento (ad esempio, lisazione chimica come Kybella, CoolSculpting, mesoterapia ecc.) per la successiva rimozione attraverso le feci o la liposuzione (rimozione fisica) delle cellule di grasso distruggono effettivamente queste cellule, in modo tale che si verifichi una riduzione dell’aromatizzazione.
Interconversione di E2 ed E1 da parte della 17β-HSD dopo somministrazione i.m. di Boldenone Undecylenato.
Fattori individuali (per utilizzatore):
A seconda del profilo ormonale legato di un individuo¹, il rilascio più lento dal deposito per il Boldenone Undecylenato prima di raggiungere lo stato stazionario determinerà quasi certamente una riduzione dell’attività dell’Aromatasi.
A seconda dell’espressione dell’isozima 17β-HSD di un individuo², il flusso netto di estrogeni potrebbe produrre E1 > E2 dopo la somministrazione di Boldenone Undecylenato, con il risultato che gli estrogeni prevalenti nella circolazione sanguigna sono molto più deboli rispetto all’E2.
A seconda dell’espressione dell’Aromatasi³ di un individuo, la tollerabilità dell’estrogenicità da parte di androgeni aromatizzabili (ad esempio, il Boldenone) dipende in parte dal numero di Aromatasi.
Figura: Previsione del target molecolare del Methenolone (Primobolan/Rimobolan):
Nota: sebbene vi siano prove (Figura, sopra) che il Methenolone Enantato abbia un’alta probabilità di legare l’Aromatasi (citocromo P450 19A1) (probabilità dell’88%) – la cui inibizione competitiva ridurrebbe l’E2 sierica – e una bassa probabilità di legare la 17β-HSD1, la 17β-HSD2 e la 17β-HSD3 – non farò supposizioni su questi potenziali meccanismi per gli effetti sull’estrogenicità, perché il modello semplicemente non ne ha bisogno. Inoltre, non sappiamo quale modalità di legame utilizzerebbe né la sua rilevanza biologica. È dominio esclusivo della “bro-science” impegnarsi in queste speculazioni sconsiderate.
Fattori individuali per utilizzatore
Fattori individuali (definizioni):
¹: profilo ormonale legato: Le attività di SHBG, albumina, α₁ glicoproteina acida e globulina legante i corticosteroidi influenzano le porzioni inattive legate rispetto a quelle attive libere di androgeni ed estrogeni. ²: Espressione dell’isoenzima 17β-HSD: Il numero relativo di isozimi 17β-HSD di tipo 1 e di tipo 2 determina le proporzioni relative e i livelli assoluti di E2 ed E1 circolanti, rispettivamente. ³: Espressione dell’Aromatasi: Il numero assoluto di proteine Aromatasi determina i livelli di prodotti aromatici (cioè estrogeni).
17β-HSD
Struttura del 17β-HSD
La 17β-HSD è un gruppo di enzimi che interconvertono gli steroidi (estrogeni, androgeni) con un gruppo cheto in posizione 17 (ad esempio, E1, AD) e quelli con un gruppo idrossi nella stessa posizione (ad esempio, E2, T).
Tutti gli enzimi 17β-HSD catalizzano l’ossidazione o la riduzione del carbonio in posizione 17 nel substrato steroideo:
preferenze diverse per il substrato (ad esempio, E1, E2, T, 3β-diolo, DHT) funzioni fisiologiche distinte (Jansson, 2009) [15]. Nell’uomo sono state identificate dodici (12) 17β-HSD… alcune catalizzano reazioni di substrati non steroidei… se il substrato è steroideo, la reazione è di ossidazione o riduzione, a seconda del cofattore e della localizzazione cellulare [16].
Per evitare di sovraccaricare il lettore con informazioni troppo complesse, questo lavoro si concentrerà sulle prime due (2) isoforme principali della 17β-HSD (tipo 1 e tipo 2).
La 17β-HSD1 (tipo 1), sotto il controllo del gene A1-Q327, catalizza la riduzione degli steroidi (estrogeni, androgeni) con un 17-cheto a uno che ha un gruppo idrossi nella stessa posizione. Quindi, da E1 (Estrone) =[17β-HSD1]=> E2 (Estradiolo), e da AD =[17β-HSD1]=> T.
L’espressione della 17β-HSD1 è correlata positivamente all’attivazione dell’E1 e ai livelli di E2 [15] e la sua inibizione li riduce. Inibizione della 17β-HSD1 => ↓E2 [16].
La 17β-HSD2 (tipo 2) inverte le reazioni della 17β-HSD1 (cioè, E2 =[17β-HSD2]=> E1 e E3 =[17β-HSD2]=> 16α-idrossiestrone) e converte il T =[17β-HSD2]=> AD (Androstenedione), ossidando il 17-idrossile per sostituire il C-17 con un gruppo 17-cheto.
La sovraespressione relativa della 17β-HSD2 e la sottoespressione della 17β-HSD1 producono l’effetto netto di un aumento dell’Estrone (E1), soggetto a variazioni interindividuali nel metabolismo.
Aromatasi
Struttura enzima Aromatasi
L’enzima Aromatasi, sotto il controllo del gene CYP19A1, è presente in vari tessuti dell’uomo… tra cui gonadi, cervello e tessuto adiposo (4) [20].
L’aromatasi è l’unico enzima umano in grado di aromatizzare l’anello A degli steroidi, convertendo così gli androgeni in estrogeni [21].
Questo enzima scinde il 19-metile dall’AAS e riconfigura l’anello A dello steroide in modo da formare tre doppi legami alternati. Questa configurazione dell’anello A è descritta come aromatica (pertanto, questo processo è definito aromatizzazione).
Negli uomini, esiste una variazione della popolazione nell’altezza e nell’espressione del gene dell’Aromatasi [22]. Questo ha senso perché gli estrogeni prodotti dall’aromatizzazione del T endogeno in E2 sono fondamentali per la crescita e il mantenimento delle ossa negli uomini.
Sintomi di bassa estrogenicità
Articolazioni “secche” e doloranti (artralgia) – Gli estrogeni hanno naturalmente proprietà antinocicettive che potrebbero essere, da una prospettiva teleologica, una caratteristica di design per conferire alle donne la tolleranza al dolore durante il parto, quando i livelli di estrogeni sono naturalmente aumentati [8]. Si ritiene che ciò sia mediato da neuroni del midollo spinale contenenti oppioidi che esprimono ER (24) [8]. I dati sugli animali dimostrano che i topi ovariectomizzati presentano un turnover accelerato della cartilagine (25) che può contribuire alla riduzione dell’ammortizzazione articolare [8]. Gli estrogeni sopprimono la produzione di citochine infiammatorie, mentre una riduzione degli estrogeni aumenta i livelli di citochine infiammatorie come IL-1 e TNF-α (26)… Le cellule sinoviali esprimono l’Aromatasi e, quando questa catalizza la conversione dall’Androstenedione (AD) all’Estrone (E1) e all’Estradiolo (E2), l’espressione di IL-6 si riduce nell’articolazione (28) [8]. Pertanto, un basso livello di estrogeni, e di conseguenza di IA, può provocare un aumento relativo della produzione di IL-6, che notoriamente agisce come citochina pro- e anti-infiammatoria. È anche nota per essere uno dei mediatori chiave dell’aumento della perdita ossea nelle donne in post-menopausa (29) [8].
Perdita ossea – Gli estrogeni svolgono un ruolo fondamentale nel prevenire la perdita di contenuto/densità minerale ossea. Sebbene gli androgeni abbiano effetti significativi sull’osso maschile, gli estrogeni sono più importanti per la crescita e il mantenimento dell’osso… L’E2 è essenziale per la normale mineralizzazione, massa e turnover dell’osso, ma non per la crescita lineare dell’osso negli uomini (648, 649) [9].
Resistenza all’Insulina – Il metabolismo del glucosio per kg di muscolo è più alto del 45% nelle donne (756) (probabilmente mediato da ER-α) [9]. Negli uomini, gli effetti metabolici benefici del Testosterone sono mediati più dal suo prodotto aromatico (E2) che dagli androgeni (E2 > T nell’accumulo di ↓AT)… ~15% degli estrogeni circolanti deriva dalla sintesi e dalla secrezione testicolare (cellule di Leydig) e il resto dall’attività dell’Aromatasi periferica… [9].
Aumento del grasso corporeo (↑AT; AT: tessuto adiposo) – Negli uomini, l’E2 regola le riserve di grasso corporeo > T. I topi maschi ERKO: Estrogen Receptor Knockout (ER null) hanno mostrato depositi di AT superiori del 100% a 9-12 mesi di età (invecchiati)… riflette sia l’iperplasia che l’ipertrofia degli adipociti (281) e si accompagna a intolleranza al glucosio e resistenza all’Insulina (IR) [9]. I topi maschi ERαKO presentano infiammazione del ↑AT, dimensioni degli adipociti e alterata tolleranza al glucosio [9].
Disfunzioni sessuali – La segnalazione ER-α nell’uomo supporta: i dotti efferenti e le funzioni epididimali; il trasporto di ioni e il riassorbimento di H₂O necessari per sostenere il normale funzionamento degli spermatozoi (riproduzione maschile); il cervello, l’adipe, il muscolo scheletrico, le ossa, i tessuti cardiovascolari e immunitari [9].
Nota: mentre gli estrogeni esogeni causano patologie riproduttive maschili [9], gli estrogeni endogeni (a livelli normali di T) sono fondamentali per il funzionamento sessuale maschile.
Ridotta reattività del muscolo scheletrico agli stimoli anabolici – Questa affermazione non è attualmente supportata dalle prove relative ai sintomi di bassa estrogenicità indotti dagli AAS. Nonostante sia un luogo comune tra i bodybuilder che l’uso di AI/SERM, attraverso l’azione antiestrogenica nel muscolo scheletrico, riduca l’anabolismo muscolare; o che l’E2 molto alto promuova l’anabolismo muscolare – queste affermazioni non sono supportate da alcuna prova reale (vale a dire, sottoposte a un design di studio rigoroso e a metodi probabilistici e statistici per distinguere causa, effetto e casualità). Ciò che è dimostrato è che la terapia estrogenica sostitutiva (HRT, in letteratura; diversa dalla TRT) aumenta la sintesi proteica muscolare (MPS) indotta dall’allenamento contro-resistenza (RT), ma a scapito della MPS basale (ad es, La sostituzione degli estrogeni nelle donne in post-menopausa riduce la MPS nelle 24 ore) [10]… Mentre le prove nei ruminanti (cioè nei bovini) supportano l’E2 esogeno + androgeni (ad esempio, impianti di Trenbolone Acetato), questo è, come la HRT (sostituzione degli estrogeni) nelle donne in post-menopausa, non analogo agli AAS negli uomini sani.
Poiché le donne in post-menopausa sono invecchiate e in genere non ricorrono alla terapia ormonale sostitutiva (estrogeni) per periodi di anni dopo la cessazione delle mestruazioni, la semplice associazione tra bassi estrogeni e attenuata reattività agli stimoli anabolici è più probabilmente legata ad altri fattori legati all’età che non alla riduzione degli estrogeni (ad esempio, la diminuzione della capacità rigenerativa delle cellule satelliti e la diminuzione dell’espressione dell’mRNA di IGF-IEc nel muscolo scheletrico).
Poiché i ruminanti non sperimentano un aumento dell’IGFBP-1 in risposta all’E2 esogeno come gli esseri umani [11], che riduce la disponibilità di IGF-I libero e scatena (endogenamente) la secrezione di GH tramite il ritiro del feedback, qualsiasi connessione estrogeno-anabolismo nel muscolo scheletrico umano è, nella migliore delle ipotesi, tenue e probabilmente un mero fattore terziario, legato invece al T endogeno e al processo di aromatizzazione (che aumenta l’IGF-I) piuttosto che al suo prodotto aromatico. Gli estrogeni (ad esempio, l’E2) aumentano in modo dose-dipendente l’IGFBP-1, motivo per cui le donne hanno livelli di GH endogeno molto più elevati ma livelli di IGF-I proporzionalmente più bassi rispetto agli uomini in base alla superficie corporea (una risposta ridotta al GH) [13], e per cui le donne che assumono contraccettivi ormonali (cioè estrogeni) devono titolare le dosi di rhGH per vedere i benefici sulla crescita e sul metabolismo, ad esempio nella carenza di Ormone della Crescita nell’adulto [14]. Nelle donne in premenopausa, l’Etinilestradiolo orale riduce i livelli di IGF-I fino a una media del 30% (24-27) [13].
I casi di Boldenone Undecylenato e Methenolone Enantato
L’uso di Methenolone Enantato e/o Boldenone Undecylenato può provocare sintomi di bassa estrogenicità, che possono (o meno) essere riflessi da concentrazioni di E2 inferiori alla norma.
Adattamento di Methenolone Enantato e/o Boldenone Undecylenato alla tesi qui esposta
Vedere Teoria delle potenze estrogeniche (modello teorico):
Ogni AAS influisce sul flusso netto di estrogenicità attraverso i suoi particolari effetti sulle concentrazioni di estrogeni nel sangue e sulle attività estrogeniche a livello tissutale nei seguenti modi:
Methenolone:
Struttura molecolare del Methenolone
Il Methenolone, in quanto AAS non aromatizzabile, non converte in estrogeni. Di conseguenza, a dosi moderate/elevate, i suoi effetti sul flusso netto di estrogeni rispetto agli aspetti degli effetti dipendenti dal composto (per-AAS) saranno marcatamente anti-estrogenici – l’inibizione delle gonadotropine secrete dall’ipofisi (che riducono indirettamente gli estrogeni nell’uomo attraverso la soppressione della sintesi e della secrezione di T endogeno [steroidogenesi] da cui dipende la biosintesi dell’Estradiolo [E2] nell’uomo), e il blocco diretto dell’attività degli estrogeni a livello degli organi bersaglio, impedendo l’assorbimento degli estrogeni nelle cellule (ad es. g., cellule sinoviali, causando sintomi di “articolazione secca e dolorante”).
Boldenone:
Struttura molecolare del Boldenone.
Il Boldenone, rispetto al Testosterone, aromatizza maggiormente in Estrone (E1) e scarsamente in Estradiolo (E2) [5]. L’E1 è un estrogeno debole perché manca del gruppo 17β-OH dell’E2 e possiede appena il 2% della potenza dell’E2 nel transattivare l’ER-α [6]. Poiché l’espressione dell’isoenzima 17β-HSD² dell’individuo determina il flusso netto dell’equilibrio E1/E2, è particolarmente determinante nel caso degli effetti del Boldenone sul flusso netto di estrogenicità.
Il Boldenone è soggetto a una grande variazione interindividuale rispetto a tutti e tre i fattori enumerati (profilo ormonale legato¹, espressione dell’isozima 17β-HSD² ed espressione dell’Aromatasi³). La sua Vmax relativamente lenta (velocità di reazione dell’Aromatasi), l’aromatizzazione maggiore in E1 (un estrogeno debole) e minore in E2, le sue porzioni libere o legate e il numero assoluto di Aromatasi sono fattori che determinano un’ampia divergenza degli effetti del Boldenone sull’estrogenicità.
Gestione dell’estrogenicità
Per visualizzare il modo in cui l’utente dovrebbe approcciarsi alla gestione dell’estrogenicità si può ricorrere a un semplice modello: la curva a U inversa:
Figura: Un modello semplificato – la curva a U.
L’asse x è correlato all’attivazione ER a livello tissutale, che potrebbe non essere riflessa dalle concentrazioni di estrogeni nel sangue. L’asse y riflette la tollerabilità. L’area sotto la curva agli estremi (troppo bassa o troppo alta) è caratteristicamente intollerabile. La gestione dell’estrogenicità è quindi un “problema Goldilocks”. L’estrogenicità non può essere troppo bassa o troppo alta, ma deve essere “giusta” rispetto alla tollerabilità individuale.
La sezione che segue è di carattere pratico: un diagramma di flusso decisionale a cui l’utilizzatore può fare riferimento in caso di sospetta bassa estrogenicità (“crash E2”).
Pratica – Un diagramma di flusso del processo decisionale per affrontare la bassa estrogenicità derivante dall’uso di Boldenone e/o Methenolone:
Diagramma di flusso indicativo/esemplificativo del processo decisionale di fronte a sintomi di bassa estrogenicità.
Conclusioni:
L’estrogenicità (sintomi associati all’attivazione dell’ER) degli AAS è soggetta a effetti per-AAS e per utilizzatore. Il Methenolone, in quanto AAS non aromatizzabile e DHT derivato, agisce come antiestrogeno e androgeno. Il Boldenone è un composto interessante proprio per il fatto che è soggetto a effetti divergenti tra gli utilizzatori, che dipendono da fattori quali il profilo ormonale di legame¹, l’espressione dell’isoenzima 17β-HSD² e l’espressione dell’Aromatasi³. Le analisi del sangue di laboratorio spesso non sono sufficientemente precise per gli utilizzatori di AAS che cercano di capire l’estrogenicità a causa di fattori che includono gli effetti locali sui tessuti e le dissociazioni tra intra- ed endocrinologia. È per questo motivo che l’auto interpretazione delle analisi del sangue e il loro utilizzo per dettare il dosaggio e le pratiche dei farmaci ancillari (vedi SERM e/o AI) – che sono più spesso cattiva “bro-science” che medicina – piuttosto che rimanere semplicemente in sintonia con la tollerabilità di questi agenti e lavorare attraverso il diagramma di flusso presentato come necessario, porta il più delle volte a un frustrante gioco di “whack-a-mole” per gli utilizzatori di AAS.
Gabriel Bellizzi [CEO BioGenTech]
Riferimenti e fonti:
Primo and/or EQ Symptoms of Low vs. High Estrogens, Explained, and a Practical Flowchart of Decisionmaking for Symptoms of Low Estrogens as a Result of Primo and/or EQ Use. By Type-IIx
[1] Suchowsky, GK, Junkmann, K. [Anabolic steroids and their side-effects]. Acta Endocrinol (Copenh). 1962 Jan;39:68-78. German. PMID: 13918121.
[2] Junkmann, K, Suchowsky, G. [Research on anabolic-active steroids]. Arzneimittelforschung. 1962 Mar;12:214-8. German. PMID: 14452833.
[3] Trams, G. (1977). Effect of drostanolone propionate on the binding of oestradiol and dihydrotestosterone by normal and malignant target tissues. European Journal of Cancer (1965), 13(2), 149–153. doi:10.1016/0014-2964(77)90193-1
[4] Kalicharan, R. W., Bout, M. R., Oussoren, C., and Vromans, H. (2016). Where does hydrolysis of nandrolone decanoate occur in the human body after release from an oil depot? International Journal of Pharmaceutics, 515(1-2), 721–728. doi:10.1016/j.ijpharm.2016.10.068
[5] Gual, C., Morato, T., Hayano, M., Gut, M., and Dorfman, R. I. (1962). Biosynthesis of Estrogens. Endocrinology, 71(6), 920–925. doi:10.1210/endo-71-6-920
[6] Houtman, C. J., Sterk, S. S., van de Heijning, M. P. M., Brouwer, A., Stephany, R. W., van der Burg, B., and Sonneveld, E. (2009). Detection of anabolic androgenic steroid abuse in doping control using mammalian reporter gene bioassays. Analytica Chimica Acta, 637(1-2), 247–258. doi:10.1016/j.aca.2008.09.037
[7] Thomas MP, Potter BV. The structural biology of oestrogen metabolism. J Steroid Biochem Mol Biol. 2013;137:27-49. doi:10.1016/j.jsbmb.2012.12.014 [8] P. Niravath, Aromatase inhibitor-induced arthralgia: a review, Annals of Oncology, Volume 24, Issue 6, 2013, Pages 1443-1449, ISSN 0923-7534, doi.org/10.1093/annonc/mdt037.
[9] Cooke PS, Nanjappa MK, Ko C, Prins GS, Hess RA. Estrogens in Male Physiology. Physiol Rev. 2017 Jul 1;97(3):995-1043. doi:10.1152/physrev.00018.2016.
[10] Rubinow KB. Estrogens and Body Weight Regulation in Men. Adv Exp Med Biol. 2017;1043:285-313. doi:10.1007/978-3-319-70178-3_14
[11] Chidi-Ogbolu N, Baar K. Effect of Estrogen on Musculoskeletal Performance and Injury Risk. Front Physiol. 2019;9:1834. Published 2019 Jan 15. doi:10.3389/fphys.2018.01834
[12] Veldhuis, J. D., and Bowers, C. Y. (2003). Human GH pulsatility: An ensemble property regulated by age and gender. Journal of Endocrinological Investigation, 26(9), 799–813. doi:10.1007/bf03345229
[13] Chanson, P., Arnoux, A., Mavromati, M., Brailly-Tabard, S., Massart, C., … Young, J. (2016). Reference Values for IGF-I Serum Concentrations: Comparison of Six Immunoassays. The Journal of Clinical Endocrinology and Metabolism, 101(9), 3450–3458. doi:10.1210/jc.2016-1257
[14] Cook, D. M., Ludlam, W. H., and Cook, M. B. (1999). Route of Estrogen Administration Helps to Determine Growth Hormone (GH) Replacement Dose in GH-Deficient Adults1. The Journal of Clinical Endocrinology and Metabolism, 84(11), 3956–3960. doi:10.1210/jcem.84.11.6113
[15] He, W., Gauri, M., Li, T., Wang, R., and Lin, S.-X. (2016). Current knowledge of the multifunctional 17β-hydroxysteroid dehydrogenase type 1 (HSD17B1). Gene, 588(1), 54–61. doi:10.1016/j.gene.2016.04.031
[16] Thomas MP, Potter BV. The structural biology of oestrogen metabolism. J Steroid Biochem Mol Biol. 2013;137:27-49. doi:10.1016/j.jsbmb.2012.12.014
[17] Gual, C., Morato, T., Hayano, M., Gut, M., and Dorfman, R. I. (1962). Biosynthesis of Estrogens. Endocrinology, 71(6), 920–925. doi:10.1210/endo-71-6-920
[18] Lakshman, K. M., Kaplan, B., Travison, T. G., Basaria, S., Knapp, P. E., Singh, A. B., … Bhasin, S. (2010). The Effects of Injected Testosterone Dose and Age on the Conversion of Testosterone to Estradiol and Dihydrotestosterone in Young and Older Men. The Journal of Clinical Endocrinology and Metabolism, 95(8), 3955–3964. doi:10.1210/jc.2010-0102
[19] Fouad Mansour M, Pelletier M, Boulet MM, Mayrand D, Brochu G, Lebel S, Poirier D, Fradette J, Cianflone K, Luu-The V, Tchernof A. Oxidative activity of 17β-hydroxysteroid dehydrogenase on testosterone in male abdominal adipose tissues and cellular localization of 17β-HSD type 2. Mol Cell Endocrinol. 2015 Oct 15;414:168-76. doi: 10.1016/j.mce.2015.06.016. Epub 2015 Jun 26.
[20] Attardi BJ, Pham TC, Radler LC, Burgenson J, Hild SA, Reel JR. Dimethandrolone (7alpha,11beta-dimethyl-19-nortestosterone) and 11beta-methyl-19-nortestosterone are not converted to aromatic A-ring products in the presence of recombinant human aromatase. J Steroid Biochem Mol Biol. 2008;110(3-5):214-222. doi:10.1016/j.jsbmb.2007.11.009 [21] Thomas MP, Potter BV. The structural biology of oestrogen metabolism. J Steroid Biochem Mol Biol. 2013;137:27-49. doi:10.1016/j.jsbmb.2012.12.014
[22] Ellis, J. A., Stebbing, M., and Harrap, S. B. (2001). Significant Population Variation in Adult Male Height Associated with the Y Chromosome and the Aromatase Gene. The Journal of Clinical Endocrinology and Metabolism, 86(9), 4147–4150. doi:10.1210/jcem.86.9.7875
Nel 1986 i ricercatori hanno svolto studi approfonditi sull’ossido nitrico (NO), un potente vasodilatatore che può migliorare la circolazione e la salute del cuore. I ricercatori della Pfizer iniziarono a sperimentare farmaci chiamati inibitori della PDE-5 che potenziano e perpetuano gli effetti di dilatazione dei vasi sanguigni dell’NO.
Il loro obiettivo, all’epoca, era quello di trovare un trattamento per l’angina. Il primo farmaco fu il Sildenafil citrato, ma le sperimentazioni dimostrarono che la sua efficacia nel trattamento della patologia era modesta. Tuttavia, i ricercatori hanno iniziato a esaminare le note che descrivevano gli effetti collaterali del farmaco. Ed ecco che molti soggetti hanno riferito di aver sperimentato erezioni durature. Pfizer cambiò rapidamente marcia e avviò studi pilota sugli effetti del Sildenafil citrato sulla disfunzione erettile. Il Viagra, nome commerciale del Sildenafil, fu presto approvato dalla FDA.
Non sono gli anziani hanno beneficiato di questo effetto. Infatti, uomini più giovani si sono affezionati al farmaco, come hanno fatto con i suoi cugini Cialis [Tadalafil] e Levitra [Vardenafil], perché i farmaci in questione aiutavano a gestire l’ansia da prestazione e riducevano i tempi morti tra un episodio sessuale e l’altro.
Confronto tra le strutture di cGMP, Sildenafil e altri inibitori della PDE5. a | Il substrato nativo, cGMP. b | Sildenafil. c | Vardenafil e Tadalafil. cGMP, guanosina monofosfato ciclico; PDE-5, fosfo-diesterasi di tipo 5.
Ma ci sono altri motivi per cui gli uomini potrebbero usare questa classe di farmaci. Non sono solo legati alla salute sessuale, ma anche al Bodybuilding. Infatti, ci sono prove sufficienti per sostenere l’idea di assumere questi farmaci ogni giorno, come qualsiasi altro integratore ritenuto “base” nella preparazione di un bodybuilder.
Caratteristiche dei PDE-5 inibitori:
Un inibitore della fosfodiesterasi di tipo 5 (inibitore della PDE-5) è un farmaco vasodilatatore che agisce bloccando l’azione degradativa della fosfodiesterasi di tipo 5 (PDE-5) specifica per il cGMP sul GMP ciclico nelle cellule muscolari lisce che rivestono i vasi sanguigni che riforniscono vari tessuti. Questi farmaci dilatano i corpi cavernosi del pene, facilitando l’erezione con la stimolazione sessuale, e sono utilizzati nel trattamento della disfunzione erettile (DE). Il Sildenafil è stato il primo trattamento orale efficace disponibile per la DE. Poiché la PDE-5 è presente anche nella muscolatura liscia delle pareti delle arteriole polmonari, due inibitori della PDE-5, il Sildenafil e il Tadalafil, sono approvati dalla FDA per il trattamento dell’ipertensione polmonare. Dal 2019 si stanno apprezzando i più ampi benefici cardiovascolari degli inibitori della PDE-5.[https://www.ncbi.nlm.nih.gov/]
Schema della via dell’Ossido Nitrico (NO)/guanosina monofosfato ciclico (cGMP)/ nucleotide ciclico fosfodiesterasi 5 (PDE-5) e del sito d’azione degli inibitori della PDE-5.
Parte del processo fisiologico di vasodilatazione prevede il rilascio di ossido nitrico (NO) da parte delle cellule endoteliali vascolari, che poi si diffonde alle vicine cellule muscolari lisce vascolari. Lì, l’NO attiva la guanilato ciclasi solubile che converte la guanosina trifosfato (GTP) in guanosina monofosfato ciclico (cGMP), il principale effettore del sistema. Ad esempio, nel pene, il rilascio di NO ad alti livelli dalle cellule endoteliali e dai nervi penieni durante la stimolazione sessuale porta al rilassamento della vascolarizzazione liscia dei corpi cavernosi, causando una vasocongestione e un’erezione prolungata.[https://www.ncbi.nlm.nih.gov/]
Gli inibitori della PDE-5 prolungano l’azione del cGMP inibendo la sua degradazione da parte dell’enzima PDE-5, presente in tutto il corpo. Nel pene, gli inibitori della PDE-5 potenziano gli effetti del cGMP per prolungare l’erezione e aumentare la soddisfazione sessuale, mentre nel muscolo scheletrico aumentano l’iperemia del tessuto per via della vasodilatazione.[https://www.nejm.org/] Tuttavia, gli inibitori della PDE-5 non provocano erezioni senza stimolazione sessuale.
Oltre agli effetti emodinamici, gli inibitori della PDE-5 hanno dimostrato in diversi esperimenti proprietà antinfiammatorie, antiossidanti, antiproliferative e metaboliche.[https://www.ncbi.nlm.nih.gov/] Ma sono ovviamente necessari studi più ampi e a lungo termine per stabilirne l’efficacia e la sicurezza rispetto ad altri farmaci in altre patologie.
Quindi l’uso di questa classe di farmaci nel Bodybuilding si limita al classico trattamento per la disfunzione erettile e il pompaggio muscolare? Non esattamente.
Sicuramente, il potenziale additivo dei PDE-5 inibitori per lo stimolo massimo del “pump”, in specie in combinazione con Citrullina, nel pre-palco può incidere positivamente sugli ultimi ritocchi del “look” dell’atleta. Ricordo inoltre che un maggiore afflusso di sangue al tessuto muscolare significa un migliore pompaggio dato dall’esercizio contro-resistenza e un maggiore afflusso di sostanze nutritive ai muscoli, il che è positivo per la performance, il recupero e la crescita muscolare.
Ma i potenziali non si fermano qui:
Tadalafil
Uno studio del 2005 ha rilevato che dosi di Tadalafil da 10 e 20mg, assunte in media 10 volte al mese, riducevano significativamente i livelli di Estradiolo, ma solo negli uomini che non avevano troppo grasso corporeo – quelli con un IMC inferiore a 27 (1). Gli uomini con più grasso corporeo hanno livelli di Aromatasi più elevati e convertono maggiormente il Testosterone in Estradiolo, indipendentemente dal Tadalafil assunto.
Uno studio sugli effetti del Sildenafil su 140 uomini con un basso livello di Testosterone, di età compresa tra i 40 e i 70 anni, ha rilevato che il farmaco ha aumentato i livelli di Testosterone di circa 100 punti (2). Sebbene una parte di questo aumento dell’ormone maschile possa essere dovuta alla mancata conversione in Estradiolo di una parte del Testosterone, una percentuale di questo aumento sembra derivare anche da una maggiore produzione di Testosterone da parte dei testicoli.
Il Sildenafil riduce lo stress ossidativo indotto dal diabete e migliora la sensibilità all’Insulina. (3) Questo esperimento, a differenza degli altri, è stato condotto sui ratti, ma è probabile che funzioni in modo simile anche nell’uomo.
L’ipotesi che i farmaci che influenzano il flusso sanguigno possano essere utili per la crescita muscolare negli adulti più anziani, ha spinto il Dipartimento di Medicina Interna dell’Università del Texas Medical Branch ha condurre uno studio.
Time-line dello studio
Secondo i ricercatori, le riduzioni della funzione muscolare scheletrica si verificano nel corso di un invecchiamento sano, ma anche con la sedentarietà o con diverse malattie come il cancro, la distrofia muscolare e l’insufficienza cardiaca. Tuttavia, non esistono terapie farmacologiche accettate per migliorare la funzione muscolare scheletrica compromessa.
L’ossido nitrico può influenzare la funzione del muscolo scheletrico attraverso effetti sull’accoppiamento eccitazione-contrazione, sulla funzione miofibrillare, sulla perfusione e sul metabolismo.
I soggetti dello studio erano di mezza età, non allenati e per lo più in sovrappeso, e dovevano assumere un’integrazione giornaliera di Sildenafil per otto giorni, mentre si è analizzato l’effetto sulla sintesi proteica muscolare (il processo che guida la crescita muscolare) e sulla funzione muscolare rispetto a un placebo.
Lo studio ha dimostrato che l’aumento della segnalazione dell’ossido nitrico-guanosina monofosfato ciclico mediante la somministrazione giornaliera a breve termine dell’inibitore della fosfodiesterasi 5, il Sildenafil, aumenta la sintesi proteica, altera l’espressione proteica e la nitrosilazione e riduce la fatica nel muscolo scheletrico umano.
Questi risultati suggeriscono che gli inibitori della fosfodiesterasi 5 rappresentano un valido intervento farmacologico per migliorare la funzione muscolare. Ciò che è stato rilevato, infatti, è che Il Sildenafil aumenta la sintesi proteica muscolare e riduce l’affaticamento muscolare.
Effetti del trattamento con Sildenafil sulla funzione muscolare scheletrica. (A) Forza isometrica degli estensori del ginocchio (percentuale media del giorno di riferimento ± errore standard (SE)) dopo 8 giorni di trattamento, determinata con la dinamometria. (B) Forza isocinetica (120° al secondo) degli estensori del ginocchio (percentuale media del giorno di riferimento ± SE) dopo 8 giorni di trattamento, determinata con la dinamometria. (C) Ripetizioni riuscite (percentuale media del giorno di riferimento ± SE) durante contrazioni isocinetiche affaticanti (120° al secondo) dopo 8 giorni di trattamento. *p = 0,016 rispetto al placebo, t-test non accoppiato, N = 6 placebo, 5 sildenafil. Il numero individuale di ripetizioni riuscite prima (pre) e dopo (post) il trattamento per i soggetti che hanno ricevuto il placebo (pannello superiore) e il Sildenafil (pannello inferiore) è mostrato a destra.Effetti del trattamento con Sildenafil sul proteoma del muscolo scheletrico. (A) Sintesi proteica del muscolo scheletrico (media ± SE) dopo 8 giorni di trattamento, determinata utilizzando l’approccio precursore-prodotto per determinare il tasso di sintesi frazionale. *p = 0,004 rispetto al placebo, t-test non accoppiato, N = 6 placebo, 5 Sildenafil. Percorsi canonici (B) e funzionali (C) influenzati in modo differenziato da sildenafil e placebo, determinati utilizzando l’Ingenuity Pathways Analysis (IPA) dell’espressione proteica in campioni di biopsia del muscolo scheletrico (sono mostrati i 6 percorsi principali). Percorsi canonici (D) e funzionali (E) influenzati in modo differenziato dal Sildenafil e dal placebo, determinati utilizzando l’IPA della S-nitrosilazione delle proteine nei campioni di biopsia del muscolo scheletrico (sono indicati i sei percorsi principali).
L’affaticamento del muscolo scheletrico nel primo giorno di trattamento non era statisticamente diverso dal basale o diverso tra i gruppi di trattamento. Tuttavia, gli scienziati hanno ammesso che dopo otto giorni di trattamento, i soggetti del gruppo Sildenafil hanno completato un numero significativamente maggiore di ripetizioni di successo rispetto al basale in rapporto a quelli che hanno ricevuto il placebo durante contrazioni isocinetiche massimali ripetute.
Essendo un farmaco già approvato e con un eccellente record di sicurezza, i risultati di questo studio suggeriscono che il Sildenafil, e possibilmente altri inibitori della fosfodiesterasi 5, rappresenta una potenziale strategia farmacologica per migliorare la funzione del muscolo scheletrico.
Jeff Nippard
Jeff Nippard, un famoso blogger di fitness su YouTube, ha cercato di mettere le cose in chiaro. Per prima cosa ha contattato Jorn Tromellen, ricercatore sul metabolismo muscolare. Tromellen ha rivelato che in realtà i risultati sono meno impressionanti di quanto sembri. Il Sildenafil non è paragonabile agli AAS, ovviamente, in quanto quando si assume il Sildenafil la sintesi proteica aumenta nel giro di un’ora o due. Mentre gli AAS la stimolano in modo significativo (vedi attività genomica) per tutta la vita attiva del farmaco.
Tuttavia, questo non era sufficiente per Nippard, così si è rivolto a colui che ha effettivamente usato il Sildenafil: il compianto John Meadows.
John Meadows
Meadows ha fatto uso di Sildenafil per tutta la sua carriera agonistica. La prima cosa che ha ammesso è che il livello “di lavoro” del Sildenafil dipendono dalla quantità di cibo presente nello stomaco.
Lui ammette che, gli atleti lo usano prevalentemente per avere più “pump” prima di salire sul palco.
Poi Meadows ha confrontato le sensazioni generali dopo l’assunzione di Testosterone e Sildenafil e, ovviamente, non sono neanche lontanamente paragonabili. Considero questa deduzione al pari dell’affermazione secondo cui l’uomo non possa respirare sottacqua senza attrezzatura apposita… banalità…
La verità è che questo studio ha lasciato ancora più domande rispetto a prima. I risultati sembrano molto promettenti, ma rimangono ancora alcune perplessità.
Lo studio è stato condotto su persone non allenate. Quindi la prima domanda è: Funzionerebbe anche su soggetti più giovani e/o allenati?.
E ancora: sappiamo che avviene un aumento della sintesi proteica, ma non è chiaro se ciò sia a carico delle fibre miofibrillari o di altri tessuti.
Possibili effetti avversi dall’uso di inibitori del PDE-5:
Tutti gli inibitori della PDE-5 sono generalmente ben tollerati.[1] La comparsa di effetti collaterali, o reazioni avverse al farmaco (ADR), con gli inibitori della PDE-5 dipende dalla dose e dal tipo di agente.[https://www.ncbi.nlm.nih.gov/] La cefalea è una ADR molto comune, che si verifica nel >10% dei pazienti. Altre ADR comuni sono: vertigini, vampate di calore, dispepsia, congestione nasale o rinite.[6] Anche il mal di schiena e i dolori muscolari sono più comuni nei pazienti che assumono Tadalafil.[https://www.ncbi.nlm.nih.gov/]
Nel 2007, la Food and Drug Administration (FDA) statunitense ha annunciato l’aggiunta di un’avvertenza sulla possibile perdita improvvisa dell’udito alle etichette dei farmaci inibitori della PDE-5.[https://www.fda.gov/]
Dal 2007 sono emerse prove che suggeriscono che gli inibitori della PDE-5 possono causare una neuropatia ottica anteriore,[https://doi.org/1] anche se l’aumento del rischio assoluto è piccolo.[https://www.ncbi.nlm.nih.gov/]
Infine, si teme che gli inibitori della PDE-5 possano aumentare il rischio di mortalità neonatale nelle donne in gravidanza, e sono stati sospesi gli studi sull’uso dei farmaci per la restrizione della crescita fetale.[https://www.ncbi.nlm.nih.gov/]
Gli inibitori della PDE5 sono metabolizzati principalmente dal sistema enzimatico del citocromo P450, in particolare dal CYP3A4. Esiste la possibilità di interazioni avverse con altri farmaci che inibiscono o inducono il CYP3A4, tra cui gli inibitori della proteasi dell’HIV, il Ketoconazolo e l’Itraconazolo,[Australian Medicines Handbook 2006.] anche se la co-somministrazione non è stata collegata a cambiamenti nella sicurezza o nell’efficacia di entrambi gli agenti. [La combinazione con nitrovasodilatatori come la nitroglicerina e il PETN è controindicata perché può verificarsi ipotensione potenzialmente pericolosa per la vita.[Haberfeld H, ed. (2009). Austria-Codex (in German) (2009/2010 ed.). Vienna: Österreichischer Apothekerverlag.] Gli inibitori della PDE5 non interagiscono sinergicamente con altri farmaci antipertensivi.[https://www.ncbi.nlm.nih.gov/]
Conclusioni:
L’uso sporadico di questi farmaci (meno di 8-10 volte al mese) potrebbe non conferire effetti duraturi sulla salute. Tuttavia, il Tadalafil è approvato per l’uso una volta al giorno ed è ragionevole pensare che i pazienti, giovani o anziani, che hanno una tale prescrizione e lo usano ogni giorno, stiano raccogliendo alcuni, se non tutti, i benefici di cui sopra.
Tuttavia, se ulteriori ricerche confermeranno o aggiungeranno ulteriori elementi positivi all’elenco, potremmo arrivare al punto in cui i medici raccomanderanno quasi universalmente l’uso pressoché quotidiano di questi farmaci.
Per il momento, l’applicazione del Sildenafil nel Bodybuilding si è dimostrata più redditizia come “NO booster” potenziato e coadiuvato dalla Citrullina sia come mezzo per aumentare marcatamente il “pump” sul palco e sia per aumentare l’afflusso ematico nei muscoli (vedi ossigeno e nutrienti) durante il workout.
Diversamente, piccole dosi di Tadalafil possono garantire un contenuto controllo estrogenico in soggetti con body fat contenute, senza il rischio di incorrere il alterazioni lipidiche ematiche.
Gabriel Bellizzi [CEO BioGenTech]
Riferimenti:
Greco EA et al. Testosterone:Estradiol ratio changes associated with long-term tadalafil administration: a pilot study. J Sex Med. 2006 Jul;3(4):716-722. PubMed.
Spitzer M et al. Sildenafil increases serum testosterone levels by a direct action on the testes. Andrology. 2013 Nov;1(6):913-8. PubMed.
Milani E et al. Reduction of diabetes-induced oxidative stress by phos- phodiesterase inhibitors in rats. Comp Biochem Physiol C Toxicol Pharmacol. 2005 Feb;140(2):251-5. PubMed.
Come deducibile dal titolo, questo articolo si concentra sugli effetti degli androgeni sulla libido nelle donne.
Nell’articolo precedente, abbiamo discusso di come il Testosterone (T) e il suo prodotto della 5α-reduttasi, il DHT, nonché l’Estradiolo, esercitino un chiaro effetto organizzatore e attivatore sul comportamento sessuale, compreso il desiderio sessuale (libido). In questa sede ci concentriamo sugli effetti degli androgeni sulla libido nelle donne, dove gli effetti possono essere meno consistenti e robusti, nonostante sia interessante notare una maggiore reattività comportamentale agli androgeni nelle donne. [1]. [2].
Le donne e gli uomini differiscono ampiamente per quanto riguarda la libido. Pochi sosterrebbero, nonostante alcune variazioni e l’esistenza di valori anomali relativi, che gli uomini non siano caratteristicamente più virili delle donne. Eppure, nonostante un ampio consenso tra i ricercatori biologi e biomedici, così come tra i non addetti ai lavori, sul fatto che il Testosterone sia implicato nelle differenze di sesso nella libido e che anzi la aumenti, in letteratura si trovano prove sorprendentemente contrastanti su questo argomento, in particolare per quanto riguarda le differenze nelle e tra le donne.
Effetto soglia
Mentre negli uomini (nonostante una forte evidenza del contrario), la visione medica prevalente è che essi sono soggetti a un effetto soglia del Testosterone sul comportamento (compresa la libido); questo modello non si applica alle donne. Le donne, quindi, non sono soggette ad alcun effetto soglia o soglia minima, al di sopra del quale le concentrazioni di T non contribuiscono all’aumento della libido, o al di sotto del quale si manifestano i sintomi di una carenza di androgeni, con conseguente patologia, rispettivamente. Ciò è dovuto in parte alla mancanza di intervalli di riferimento stabiliti per le concentrazioni di T normali o sane e alla mancanza di un’adeguata sensibilità del test per rilevare le basse concentrazioni che potrebbero costituire la soglia minima per le concentrazioni di Testosterone (poiché sarebbero molto basse).
Ancora più interessante, però, è la mancanza di un limite superiore teorico, o tetto, per gli effetti comportamentali degli androgeni nelle donne.
Punti di apparente contraddizione negli effetti del Testosterone sulla libido nelle donne
Esistono diverse linee di risposta apparentemente contraddittorie della libido al Testosterone nelle donne rispetto alle loro controparti maschili.
Le donne sono fortemente influenzate dai cosiddetti affetti (umore, benessere ed energia) sulla libido. Dati i potenti effetti dell’umore sulla libido nelle donne, la depressione, l’ansia e lo stress aumentano la produzione surrenale di precursori del Testosterone e di altri ormoni surrenali nelle donne; mentre negli uomini questi stati d’animo negativi diminuiscono la produzione testicolare di Testosterone (che la produzione dei precursori surrenali non può superare). [1]. Eppure, una minoranza significativa di uomini dimostra un aumento paradossale della libido in stati di ansia e persino di depressione, mentre le donne non dimostrano apparentemente questo aumento paradossale della libido.
Un ostacolo fondamentale che presenta difficoltà nello studio degli effetti degli androgeni sulla libido è che la risposta genitale femminile (secrezione vaginale) è fondamentalmente dissociata dalla cognizione o dalla percezione dell’eccitazione sessuale. Si osserva spesso che la risposta genitale non è percepita come un aumento dell’eccitazione sessuale nelle donne, mentre negli uomini la risposta erettile aumenta in modo inequivocabile la libido, contribuendo a modulare ulteriormente il desiderio e la funzione (contribuendo al mantenimento dell’erezione). [1]. Eppure, il trattamento delle disfunzioni sessuali nelle donne attraverso il miglioramento dei sintomi della secchezza vaginale può essere secondo solo al miglioramento degli affetti (umore, benessere ed energia) nell’efficacia terapeutica.
Il normale ciclo mestruale ovulatorio dell’adulto. [3].
Tuttavia, l’effetto degli ormoni sulla libido femminile è innegabilmente potente. Durante il ciclo ovulatorio-mestruale (OMC), la fase follicolare tardiva, appena prima dell’ovulazione, è caratterizzata da LH e T elevati, Progesterone basso, E2 in aumento (da 5pg/mL nella fase follicolare precoce a un picco di 200-500pg/mL appena prima dell’ovulazione) che diminuisce bruscamente, e FSH in concomitante aumento. [3]. Le donne riferiscono assiduamente che la libido aumenta costantemente nella settimana precedente l’ovulazione e raggiunge un picco intorno al momento dell’ovulazione, per poi essere seguita da un calo precipitoso nella settimana successiva. [6]. Durante l’OMC dell’adulto, la produzione ovarica di T segue un andamento ciclico in cui i livelli di T aumentano durante la fase follicolare e raggiungono un picco approssimativamente per il terzo medio dell’OMC, diminuendo durante l’ultimo terzo (fase luteale) per raggiungere il nadir nei primi giorni della fase follicolare successiva. All’interno del terzo medio, i livelli di T possono essere relativamente stabili o comparire in picchi peri-ovulatori. [1]. In questo caso, il T può avere un’influenza di controllo; oppure, E2, LH e FSH possono essere i fattori principali dell’aumento della libido.
Effetti dei contraccettivi orali sul ciclo mestruale ovulatorio. [3].
I contraccettivi orali dovrebbero teoricamente ridurre la libido riducendo il T libero (gli estrogeni esogeni, spesso associati a un progestinico) e aumentando le SHBG (che lega il T e l’E2). I progestinici esercitano generalmente effetti antiandrogeni, sono associati a una diminuzione dell’espressione dell’AR e la combinazione con gli estrogeni aumenta l’espressione della PR. [4]. [5]. Sebbene i contraccettivi orali siano associati a una diminuzione della libido con una certa rilevanza, l’effetto non è robusto e si osservano eccezioni. Queste sono solitamente attribuite a differenze qualitative nella popolazione di donne che utilizzano contraccettivi orali, che possono avere un’ansia di tratto più bassa e quindi affetti più positivi (umore, benessere ed energia), una ridotta prevalenza di problemi sessuali e comportamenti e atteggiamenti sessuali generalmente più permissivi (forse anche un Testosterone endogeno di base più alto).
Effetti della menopausa sul ciclo ovulatorio mestruale [3].
Gli effetti più evidenti del potenziamento della libido nelle donne da parte del Testosterone derivano dalla somministrazione della TOS (terapia ormonale sostitutiva) a donne senza problemi sessuali. Tuttavia, ciò comporta ancora una certa confusione. La popolazione di donne a cui viene prescritta la TOS è generalmente in perimenopausa (in fase di transizione verso la riduzione, la diminuzione o la totale abolizione pratica della secrezione di E2 e Progesterone che caratterizza la menopausa). Le femmine di ratto ovariectomizzate a cui viene somministrato il solo E2 (Estradiolo Benzoato) sono moderatamente ricettive dal punto di vista sessuale, mentre il trattamento con E2 + Progesterone le rende pienamente ricettive e proattive dal punto di vista sessuale. [6]. La terapia ormonale sostitutiva per le donne somministrata come estrogeno + Testosterone (e in genere Testosterone sovrafisiologico) aumenta costantemente la libido nelle donne senza problemi sessuali. Mentre gli estrogeni esogeni riducono il Testosterone libero e gli estrogeni biodisponibili, aumentando di fatto le SHBG, il Testosterone esogeno aumenta il Testosterone libero e gli estrogeni biodisponibili riducendo le SHBG e l’aromatizzazione del T a E2. Sorge spontanea la domanda: qual è il contributo – positivo, negativo o neutro – dell’aumento degli estrogeni biodisponibili sulla libido? E nelle donne rispetto agli uomini?
Inoltre, la terapia ormonale sostitutiva di solito migliora l’umore, il benessere e l’energia: data l’influenza dominante degli affetti sulla libido nelle donne, gli effetti dell’E2+T sono solo indiretti, in quanto migliorano questi fattori?
Inoltre, con la somministrazione a lungo termine della TOS alle donne in menopausa e in postmenopausa, si verifica alla fine (dopo almeno diversi mesi) una diminuzione del miglioramento della libido. Questo fenomeno è stato variamente attribuito a tre fattori potenzialmente confondenti:
Che sia l’aumento in sé o il grado di variazione delle concentrazioni di Testosterone, piuttosto che la quantità assoluta di Testosterone, a determinare l’aumento della libido; una volta che le variazioni delle concentrazioni di T si riducono, sembrerebbe che la libido si riduca o si riduca in modo analogo.
Che esiste una diminuzione della sensibilità all’AR legata all’età (nelle donne come negli uomini) e che, con l’avanzare dell’età, la loro sensibilità diminuisce.
Che vi sia una desensibilizzazione agli effetti comportamentali (cioè alla libido) degli androgeni. Bancroft e colleghi hanno proposto l’ipotesi della desensibilizzazione come quadro ipotetico per spiegare le apparenti contraddizioni degli effetti Testosterone-libido nelle donne. [1]. [2].
Ipotesi di desensibilizzazione
Un tentativo teorico di spiegare le differenze di sesso negli effetti degli androgeni sulla libido. [1]. [2].:
1-La maggiore variabilità della sensibilità agli androgeni nelle donne potrebbe derivare da una maggiore variabilità genetica nelle donne, sulla base del fatto che in esse la risposta comportamentale agli steroidi gonadici è meno determinante rispetto agli uomini.
2-Una delle conseguenze dei livelli di T molto più elevati negli uomini è che essi mostrano effetti mascolinizzanti, come l’aumento della crescita e della massa muscolare, che dipendono dagli effetti anabolizzanti periferici del T. È stato ipotizzato che se i maschi fossero sensibili agli effetti del T sul sistema nervoso centrale come le femmine, gli effetti comportamentali di questi livelli mascolinizzanti sarebbero disadattivi (si veda al precedente articolo sugli effetti degli steroidi anabolizzanti-androgeni sulla libido negli uomini: Funzione del recettore degli androgeni nel SNC (maschi)). Pertanto, nel maschio è necessario ridurre la reattività agli effetti degli androgeni nel cervello.
3-L’esposizione a livelli di T sostanzialmente più elevati durante lo sviluppo fetale e anche durante le prime settimane postnatali [il picco perinatale] potrebbe essere responsabile della desensibilizzazione del SNC agli effetti del T nel maschio. Tale desensibilizzazione agirebbe presumibilmente a livello genomico piuttosto che nella fase recettoriale dell’azione ormonale… e a breve termine, sia l’esposizione al T che al DHT determina una sovraregolazione dell’AR. Una conseguenza di tale desensibilizzazione nell’uomo sarebbe che le variazioni geneticamente determinate nella risposta dei recettori del SNC al T verrebbero “appiattite”, consentendo livelli molto più elevati di T dalla pubertà in poi senza iperstimolazione dei meccanismi del SNC.
4-Senza questa desensibilizzazione nelle femmine, la variabilità genetica di base sarebbe più evidente, a livelli molto più bassi di T, e si manifesterebbe come una maggiore variabilità nella reattività comportamentale, dimostrata a partire dal primo sviluppo adolescenziale.
5-L’evidenza di studi su donne con iperplasia surrenalica congenita (CAH), in particolare la varietà a perdita di sale associata a livelli più elevati di T durante lo sviluppo fetale, mostra non solo un certo grado di mascolinizzazione del comportamento, ma anche bassi livelli di interesse sessuale. Sebbene in questi casi vi sia una serie di fattori che potrebbero compromettere il normale sviluppo sessuale, questa evidenza è coerente con l’esistenza di un certo grado di desensibilizzazione agli elevati livelli fetali di T, che diminuiscono e rimangono bassi dopo la nascita quando la CAH viene trattata.
6-Una domanda interessante è se questo ipotetico meccanismo di desensibilizzazione sia un “effetto organizzativo” dell’alto T che è operativo solo durante lo sviluppo precoce, o se tale soppressione sia possibile se l’esposizione ad alti livelli avviene più tardi nello sviluppo. In molti degli studi sulla TOS esaminati in precedenza sono state riportate prove di “tolleranza” agli [effetti comportamentali sulla libido del] T sovrafisiologico. Ciò suggerisce che tale desensibilizzazione potrebbe verificarsi anche più tardi nella vita, almeno in una certa misura… Tuttavia, è possibile che nelle donne, con l’avanzare dell’età, si verifichi un declino della sensibilità all’AR paragonabile a quello riscontrato negli uomini.
Conclusione
Mentre gli androgeni endogeni (T e DHT) esercitano un chiaro effetto sulla libido negli uomini, nelle donne gli effetti del T sono meno chiari, nonostante una maggiore reattività comportamentale, dovuta a una maggiore sensibilità agli effetti dell’umore, dell’energia e del benessere, nonché ai capricci di una complessa interazione tra ciclo ovulatorio-mestruale e comportamento.
Gli androgeni sovrafisiologici aumentano generalmente la libido negli uomini (anche in quelli normali e sani) e nelle donne, ma le modifiche chimiche degli androgeni possono influire sul fatto che determinati androgeni esercitino un effetto di potenziamento o addirittura di soppressione (ad esempio, il Nandrolone) sulla libido sia negli uomini che nelle donne.
È indispensabile che il lettore comprenda l’importante ruolo della Dopamina e del sistema eccitatorio e la modulazione della libido da parte degli ormoni steroidei attraverso i circuiti della dopamina. Per approfondire questo aspetto della libido negli esseri umani (sia negli uomini che nelle donne), consultare la sezione Dopamina e libido (dal precedente articolo sugli effetti degli steroidi anabolizzanti-androgeni sulla libido maschile). Sebbene sia difficile distinguere gli effetti del Testosterone sul comportamento sessuale nelle donne da quelli degli estrogeni, di cui il Testosterone esogeno aumenta la biodisponibilità, esistono prove inconfutabili che il Testosterone a dosi sovrafisiologiche aumenta la libido nelle donne senza problemi sessuali.
L’ipotesi della desensibilizzazione è un quadro teorico per spiegare le apparenti contraddizioni tra i sessi (e tra le donne) negli effetti degli androgeni sulla libido.
Gli uomini sono semplici, il Testosterone governa chiaramente la funzione sessuale e la libido (con una certa influenza dell’aromatizzazione in Estradiolo, in particolare nel SNC e nel cervello). Le donne sono più sfaccettate nelle dinamiche del loro ambiente ormonale e per trarre qualsiasi inferenza sugli effetti ormonali sul comportamento femminile è necessario un modello teorico ricco di sfumature, un osservatore informato e una lente granulare, anche solo per tentare una descrizione razionale.
Gabriel Bellizzi [CEO BioGenTech]
Riferimenti:
Fonte studi articolo Type-IIx
[1] Bancroft, J. Androgens and sexual function in men and women. In: Bremner, W., Bagattel, C., eds. Androgens in health and disease. Totowa: Humana Press.
[2] Bancroft, J. (2002). Sexual effects of androgens in women: some theoretical considerations. Fertility and Sterility, 77, 55–59. doi:10.1016/s0015-0282(02)02961-8
[3] Chidi-Ogbolu N, Baar K. (2019). Effect of Estrogen on Musculoskeletal Performance and Injury Risk. Front Physiol.;9:1834. doi:10.3389/fphys.2018.01834
[4] Eyster, K. M. (Ed.). (2016). Estrogen Receptors. Methods in Molecular Biology. doi:10.1007/978-1-4939-3127-9
[5] Sansone, A., Romanelli, F., Sansone, M., Lenzi, A., & Di Luigi, L. (2016). Gynecomastia and hormones. Endocrine, 55(1), 37–44. doi:10.1007/s12020-016-0975-9
[6] Pfaus, J. G. (2009). Pathways of Sexual Desire. The Journal of Sexual Medicine, 6(6), 1506–1533. doi:10.1111/j.1743-6109.2009.01309.x
In questo articolo mi concentrerò sugli effetti degli androgeni sulla libido negli uomini (nel prossimo articolo analizzerò gli effetti degli androgeni sulla libido nelle donne). Verrà inoltre presentato il Nandrolone come controesempio al generale aumento della libido da parte degli androgeni e verranno enumerati e discussi i meccanismi putativi che ostacolano la libido e la funzione sessuale. Infine, verrà discusso il neurotrasmettitore dopamina e la sua influenza sul sistema eccitatorio, in particolare la sua relazione con la soppressione della libido da parte del Nandrolone. Si ipotizza un nuovo meccanismo in base al quale il Nandrolone contribuisce probabilmente a sopprimere la libido attraverso un aumento del metabolismo della dopamina.
Negli uomini, gli androgeni endogeni Testosterone (T) e il suo prodotto della 5α-reduttasi, il DHT, nonché l’Estradiolo, esercitano un chiaro effetto organizzatore e attivatore sul comportamento sessuale, compreso il desiderio sessuale (libido) che è riconducibile allo spermarca (la comparsa della prima eiaculazione) e alla maturazione (la pubertà nel maschio è associata a un aumento di 18 volte delle concentrazioni di Testosterone endogeno). [1].
Gli uomini si differenziano ampiamente dalle donne per quanto riguarda la libido. Pochi sosterrebbero, nonostante alcune variazioni e l’esistenza di relativi outlier, che gli uomini non siano caratteristicamente più virili delle donne. Eppure, nonostante un ampio consenso tra i ricercatori biologi e biomedici, così come tra i non addetti ai lavori, sul fatto che il Testosterone sia implicato nelle differenze di sesso nella libido e che anzi migliori la libido, in letteratura si trovano prove sorprendentemente contrastanti su questo argomento, in particolare per quanto riguarda le differenze nelle e tra le donne. Gli effetti particolari degli androgeni sulla libido nelle donne sono discussi nella seconda parte di questa serie.
Definizioni
Libido: desiderio o interesse sessuale che nasce dall’eccitazione e dalla risposta centrale, che si manifesta con pensieri sul sesso accompagnati da una risposta genitale. [1].
Funzione sessuale: concetto più ampio che comprende le misure della libido (ad esempio, il Male Sexual Health Questionnaire: desiderio sessuale [comprehensive]), come un aspetto o una componente.
Gli steroidi anabolizzanti-androgeni (AAS) o gli androgeni migliorano, con differenze tra molecole, la funzione sessuale (agendo sul SNC e sui tessuti genitali). [2].
Effetto soglia
È chiaro che la TRT (terapia sostitutiva del Testosterone) somministrata terapeuticamente a un uomo ipogonadico allevierà quasi certamente i sintomi della scarsa libido, in assenza di una patologia organica della funzione sessuale.
Concettualmente, la visione medica prevalente è che le concentrazioni di Testosterone negli uomini sono soggette a un effetto soglia, con una linea di base stabilita di concentrazioni normali di Testosterone totale (TT) e Testosterone libero (fT) (normale TT 450 – 1.000 ng/dL e fT 1 – 2% di TT), al di sotto della quale prevalgono gli effetti negativi sulla libido e sulla funzione sessuale; e al di sopra della quale si manifestano pochi cambiamenti comportamentali.
La TRT per alleviare i sintomi dell’ipogonadismo (compresa la scarsa libido) mostra effetti chiari e consistenti, aumentando generalmente la frequenza dei rapporti sessuali e della masturbazione. Tuttavia, è necessario fare importanti distinzioni tra i pazienti che utilizzano la TRT. In primo luogo, la TRT viene spesso prescritta a uomini anziani, altrimenti sani e con relazioni stabili, il che aumenta naturalmente le opportunità di rapporti sessuali. La TRT viene prescritta più frequentemente in culture che hanno una visione liberale della masturbazione e ne riportano onestamente la frequenza. In effetti, gli uomini differiscono da una cultura all’altra per quanto riguarda la manifestazione della libido come comportamento masturbatorio [3]; e la popolazione dei consumatori di AAS o androgeni (ad esempio, per obiettivi di miglioramento del fisico o delle prestazioni) può essere qualitativamente diversa da quella della TRT. Pertanto, le misure dei cambiamenti della libido causati dagli androgeni devono necessariamente essere misurate in modo da non essere influenzate da questi fattori culturali o socio-relazionali.
Esiste davvero un “effetto tetto” ai livelli endogeni, al di sopra del quale gli androgeni non hanno alcun effetto sulla libido?
È interessante notare che le prove suggeriscono fortemente l’assenza di un limite superiore teorico, o tetto massimo, per gli effetti comportamentali degli androgeni. I dati relativi agli uomini normali e sani (la popolazione a cui si rivolge la visione medica prevalente) suggeriscono che non esiste un limite superiore teorico, o almeno che, se tale limite esiste, è di gran lunga superiore anche ai livelli endogeni normali di androgeni circolanti:
Anderson e colleghi hanno dimostrato che una dose settimanale di 200mg di Testosterone Enantato aumenta l’interesse sessuale in uomini normali e sani. In particolare, i risultati hanno mostrato un aumento dei punteggi della Sottoscala 2 della Sexual Experience Scale, che misura l’entità in cui un individuo cerca o permette (piuttosto che evita o rifiuta) stimoli sessuali di tipo audiovisivo o immaginario; si tratta, quindi, di un indice di interesse sessuale indipendente dall’interazione con un partner (maggiore validità rispetto alla frequenza del coito, poiché la disponibilità di un partner romantico influenza la frequenza delle attività sessuali; la frequenza della masturbazione è influenzata culturalmente; ecc.) [4].
Su e colleghi hanno dimostrato che una dose giornaliera di 240mg di Metiltestosterone ha aumentato l’eccitazione sessuale su scala analogica visiva (VAS) in uomini normali e sani. [5].
Moss e colleghi hanno dimostrato che gli atleti maschi che fanno uso di androgeni si impegnano in una maggiore frequenza di rapporti sessuali e raggiungono un numero più elevato di eiaculazioni settimanali (tutti i soggetti avevano la disponibilità di un partner sessuale) rispetto alle loro controparti che non fanno uso di androgeni. [6].
Funzione del recettore degli androgeni nel SNC (maschi)
I modelli di knockout del recettore degli androgeni (ARKO) sono uno strumento utile per studiare la funzione dei T/androgeni. Nei tessuti del SNC, l’eliminazione del recettore AR (espressione nulla) provoca nei roditori maschi un comportamento privo di attività sessuale e aggressività. [7].
Ciò ha implicazioni teleologiche: non è solo un costrutto sociale che ci si aspetta che gli uomini, entro i limiti della società, siano l’inseguitore romantico (chiedere un appuntamento, piuttosto che lo faccia la donna); e forse anche che mostrino aggressività in camera da letto (essere un gentiluomo per le strade, ma una bestia tra le lenzuola). È probabile che le pressioni ambientali e l’interesse della specie umana per la riproduzione si basino sul fatto che gli uomini affermino il loro ruolo sessuale nella competizione per le compagne e che questa competizione sia necessariamente legata alla prestanza fisica. Come spesso accade, la società interagisce con la biologia, definendo i confini dell’aggressività maschile e della ricerca sessuale.
L’ipotesi della desensibilizzazione (che verrà approfondita nel prossimo articolo) descrive un modello teorico che trae inferenze su alcuni probabili processi biologici ed epigenetici che determinano la desensibilizzazione comportamentale agli androgeni nei maschi a causa dell’aumento perinatale del Testosterone (e, di conseguenza, la manifestazione di comportamenti potenzialmente disadattivi se tale desensibilizzazione viene abbandonata, in età adulta, quando i livelli di T aumentano di 18 volte). In particolare, Bancroft e colleghi sostengono che “l’esposizione a livelli di T sostanzialmente più elevati durante lo sviluppo fetale e anche durante le prime settimane postnatali [il picco perinatale] potrebbe essere responsabile della desensibilizzazione del SNC agli effetti del T nel maschio. Tale desensibilizzazione agirebbe presumibilmente a livello genomico piuttosto che nella fase recettoriale dell’azione ormonale… e a breve termine, sia l’esposizione al T che al DHT determina una sovraregolazione dell’AR. Una conseguenza di tale desensibilizzazione nel maschio consisterebbe nelle variazioni geneticamente determinate nella reattività dei recettori del SNC al T che verrebbero “appiattite”, consentendo livelli molto più elevati di T dalla pubertà in poi senza iperstimolazione dei meccanismi del SNC.” [1].
La diminuzione della libido Nandrolone-correlata:
Un farmaco in particolare, utilizzato clinicamente e terapeuticamente negli uomini e nelle donne, che rappresenta un controesempio all’apparente aumento della libido da parte degli androgeni è il Nandrolone. Infatti, è spesso associato a una riduzione della libido. [8]. Una logica non dichiarata – forse poco rassicurante – del suo uso negli uomini con HIV, piuttosto che il Testosterone, è quella di ridurre la libido (e quindi di ridurre le interazioni sessuali tra uomini gay per ridurre la diffusione della malattia).
Hulsbæk e colleghi hanno somministrato il Nandrolone in dosi diverse a tre gruppi: (1) uomini il cui Testosterone totale (TT) era ≥11 nmol/L (100 mg di Nandrolone Decanoato ogni tre settimane), (2) uomini il cui TT era <11 nmol/L (200mg di Nandrolone Decanoato ogni tre settimane) e (3) donne (50mg ogni tre settimane), per 12 settimane. I risultati non hanno mostrato alcuna incidenza di aumento della libido da parte del Nandrolone Decanoato (0/9 donne e 0/3 uomini; 1 incidenza di aumento della libido è stata riportata nel gruppo placebo, a cui è stato somministrato un veicolo inattivo a base di olio senza androgeni). [9].
Prolattina
È stato affermato che è improbabile che il Nandrolone diminuisca la libido aumentando la Prolattina. Mentre alti livelli di Prolattina negli uomini, come nel caso degli adenomi che secernono Prolattina, sono associati a sintomi di ipogonadismo (cioè bassa libido) e persino galattorrea (lattazione), è improbabile che il Nandrolone aumenti la Prolattina (almeno alle dosi utilizzate nella pratica comune). In generale, gli androgeni aromatizzabili (ad esempio, Testosterone [10], MENT [11]) mostrano una tendenza (una tendenza, piuttosto che un effetto significativo) ad aumentare la Prolattina sierica come conseguenza dei loro prodotti aromatici (cioè gli estrogeni) che agiscono come fattori stimolanti la secrezione di Prolattina dall’ipofisi anteriore [10]. Il Nandrolone a dosi inferiori non sembra avere effetti significativi sulla Prolattina sierica (probabilmente a causa di livelli di E2 inferiori alla norma) [12].
Al contrario, gli androgeni non aromatizzabili (ad esempio, Trenbolone, Oxandrolone, ecc.) probabilmente riducono la Prolattina sierica. Questa è un’osservazione empirica basata sui risultati di analisi del sangue umano, nonché la base di un’ipotesi prevalente, a conoscenza di questo autore, avanzata per la prima volta da De Las Heras e colleghi nel 1979. [13].:
Poiché è stato riportato che la secrezione di Prolattina nel ratto maschio è pulsatile (17), l’analisi delle differenze tra i livelli basali di Prolattina basata su una singola determinazione può essere fuorviante. Una possibilità alternativa è che alcuni androgeni siano in realtà inibitori della secrezione di Prolattina. Nei nostri studi, i valori più bassi tra tutti i gruppi sono stati ottenuti negli animali trattati con Diidrotestosterone o Androstanediolo, anche se le differenze non hanno mai raggiunto la significatività. Nolin et al. (11) hanno riportato che il Diidrotestosterone ha soppresso in modo significativo i livelli di Prolattina in ratti femmina intatti.
Tra i fattori che probabilmente influenzano l’incapacità del Nandrolone di aumentare – e persino di diminuire – la libido vi sono [8]:
Estrogeni*: Il nandrolone tende a determinare livelli di estrogeni sub-normali negli uomini a dosi terapeutiche fino a 200mg settimanali. Dopo 6 settimane, l’Estradiolo sierico (E2) si è ridotto a 11 ± 9pg/mL con una dose settimanale di 100mg di Nandrolone Decanoato e a 14 ± 4pg/mL con una dose settimanale di 200mg di Nandrolone Decanoato in uomini normali [14]. L’influenza degli estrogeni e dell’Estradiolo sulla libido non è stata stabilita, né negli uomini né nelle donne; tuttavia, alcune indicazioni suggeriscono una curva a forma di U inversa rispetto alle concentrazioni di Estradiolo e alla libido (con concentrazioni troppo basse e troppo alte che causano riduzioni della libido).
5α-riduzione a DHN, un androgeno indebolito: Il Nandrolone, a differenza del Testosterone che viene convertito in DHT, più potente, nel SNC e nei tessuti sessuali dalla 5α-reduttasi, diminuisce la sua potenza androgena in questi organi bersaglio producendo DHN (5α-diidronandrolone). È stato dimostrato che gli inibitori della 5α-reduttasi (ad esempio, Dutasteride, Finasteride) che riducono la 5α-riduzione del T in DHT sono associati a una riduzione della libido negli uomini sani. [15].
Feedback negativo (inibizione) delle gonadotropine: Il Nandrolone, privo del gruppo metile C-19 del Testosterone, presenta un’omologia più ampia per la superfamiglia dei recettori nucleari, compreso il Recettore del Progesterone (PR), dati gli effetti di questa modifica sulle sue proprietà stereochimiche e sulla sua forma conformazionale. Pertanto, contribuisce a disregolare la secrezione di gonadotropine regolata dal GnRH ipotalamico, che comprende l’interazione tra la secrezione dell’Ormone Luteinizzante (LH) e dell’Ormone Follicolo-Stimolante (FSH) dall’ipotalamo [regolatori positivi], della globulina legante gli ormoni sessuali (SHBG) e dell’inibina dalle cellule del Sertoli [regolatori negativi] e del T dalle cellule di Leydig [regolatore negativo], in modo più pluripotente rispetto al Testosterone. Il Nandrolone serve a disregolare questo sistema di regolazione agendo per: –esercitare un feedback negativo sull’ipofisi (secrezione di LH) attraverso la sua aromatizzazione, anche se ridotta, in Estradiolo e maggiormente in Estrone -rallentare la frequenza degli impulsi dell’ormone di rilascio delle gonadotropine (GnRH) ipotalamico mediante un’azione androgena ed estrogenica -disregolare la regolazione ipotalamica del T e delle gonadotropine attraverso la segnalazione/pulsatilità del dendro KNDy come analogo del progestinico o del Progesterone -aumentano l’espressione della Prolattina ad alte dosi (attraverso il suo prodotto aromatico, l’Estrone e, in misura minore, l’Estradiolo) – l’elevata Prolattina sierica ha un ruolo terziario nella riduzione della libido (principalmente agendo sull’ipotalamo). [16].
Aumento del metabolismo della Dopamina (cioè della sua degradazione netta): A conoscenza di questo autore, si tratta di un nuovo meccanismo putativo, mai proposto prima, per il modo in cui il Nandrolone può contribuire alla riduzione della libido: il Nandrolone aumenta l’acido omovanillico (HVA) nel siero dell’uomo [14], riflettendo il suo metabolismo (cioè la sua degradazione), ed è probabilmente correlato alla riduzione del numero di recettori della Dopamina, L’acido omovanillico (HVA) sierico è cambiato significativamente con il Nandrolone Decanoato a 100mg settimanali (+17,6 ± 7,7 pmol/L) e con il Nandrolone Decanoato a 300mg settimanali (+11,0 ± 3,3 pmol/L), ma non nei gruppi di Testosterone Enantato a 100mg o di Testosterone Enantato a 300mg settimanali [14]. Quindi, anche a dosaggi clinici terapeutici, esiste un effetto del Nandrolone sul metabolismo della Dopamina. Gli stimoli erotici di natura audiovisiva riducono l’HVA [17], riflettendo l’aumento dell’attività dopaminergica associata all’eccitazione sessuale. Ne consegue quindi che, se l’attività dopaminergica è parte integrante della libido e delle manifestazioni della risposta genitale e dei pensieri sul sesso derivanti dall’eccitazione centrale (riflessa dalla diminuzione dell’HVA), gli effetti del Nandrolone sul metabolismo della Dopamina (riflessi dall’aumento dell’HVA) potrebbero determinare una riduzione dell’eccitazione sessuale.
*Il ruolo esatto dell’Estradiolo in ogni area della funzione sessuale maschile, compresa la libido, la funzione erettile e la spermatogenesi, è difficile da determinare con esattezza. Un complesso equilibrio di Testosterone, Estradiolo, Aromatasi ed ER nei testicoli, nel pene e nel cervello conferma un’interazione ormonale indispensabile e altamente regolata degli estrogeni nell’uomo. Gli ER e l’Aromatasi condividono le posizioni topografiche con i feromoni nel cervello, rendendo chiaro che gli estrogeni contribuiscono allo sviluppo sessuale precoce e al comportamento sessuale in età adulta. Gli estrogeni possono sostenere la libido e influenzare la quantità di recettori della serotonina nel cervello, modulando l’umore, lo stato mentale, la cognizione e le emozioni. La funzione erettile è influenzata negativamente dall’esposizione agli estrogeni nelle prime fasi dello sviluppo del pene e l’esposizione all’Estradiolo nel pene maturo porta a un aumento della permeabilità vascolare con conseguente aumento della disfunzione erettile. La disfunzione erettile dovuta a una maggiore esposizione all’Estradiolo è indipendente dal livello di Testosterone. Inoltre, la spermatogenesi dipende in qualche misura dall’Estradiolo, poiché tutte le cellule coinvolte nel processo di produzione dello sperma contengono Aromatasi ed esprimono ER. Infine, i livelli di Estradiolo devono essere presi in considerazione quando si trattano uomini con TRT, poiché i livelli di estradiolo inferiori a 5ng/dl sono correlati a una diminuzione della libido.
Inoltre, l’Estrone, che rappresenta il maggior metabolita della aromatizzazione del Nandrolone, essendo meno potente del Estradiolo (circa il 4% dell’attività estrogenica del E2) nelle attività tissutali, aggrava la condizione di riduzione della libido e della funzionalità erettile negli utilizzatori di questo progestinico .
Dopamina e libido
Il sistema eccitatorio stimola la libido, mentre il sistema inibitorio stimola la ricompensa sessuale, la sedazione e la sazietà. Il nucleo del sistema eccitatorio si trova nei sistemi cerebrali della Dopamina (DA) (incertoipotalamico e mesolimbico) che collegano l’ipotalamo e i sistemi limbici e comprende le Melanocortine (MC), l’Ossitocina (OT) e la Noradrenalina (NE). Il sistema inibitorio contiene i sistemi cerebrali degli oppioidi, degli Endocannabinoidi (ECB) e della Serotonina (5-HT), che si attivano durante i periodi di inibizione sessuale e bloccano il sistema eccitatorio. [18].
I farmaci che stimolano l’attivazione della DA ipotalamica o che bloccano il rilascio di ECB o 5-HT e/o il legame postsinaptico possono stimolare la libido. [18].
Gli ormoni steroidei attivano i meccanismi di eccitazione sessuale dirigendo la sintesi di enzimi e recettori per i sistemi neurochimici interattivi di DA, NE, MC e OT, che agiscono nelle regioni cerebrali ipotalamiche e limbiche per stimolare l’eccitazione sessuale, l’attenzione e i comportamenti. L’attivazione di questi sistemi neurochimici eccitatori smorza l’influenza dei meccanismi inibitori, quali:
gli oppioidi endogeni rilasciati nella corteccia, nel sistema limbico, nell’ipotalamo e nel mesencefalo durante un orgasmo o una ricompensa sessuale (che inducono un periodo refrattario e una diminuzione dell’espressione dell’AR nelle regioni ipotalamiche e limbiche)
ECB che mediano la sedazione e
5-HT, che viene elevata in queste regioni per indurre refrattarietà e sazietà sessuale. [18].
L’Estradiolo (E2) facilita il rilascio di DA e il Testosterone (T) potenzia la sintesi di Ossido Nitrico che controlla il rilascio di DA nei ratti (86-88). [18]. Pertanto, gli ormoni steroidei endogeni sembrano porre le basi – [un effetto priming] – per un aumento della sintesi e del rilascio di DA durante i periodi in cui la risposta sessuale potrebbe essere potenziata. [18].
Il lavoro comportamentale (ad esempio, il corteggiamento negli esseri umani o l’attraversamento di griglie elettrificate per raggiungere una compagna nei ratti maschi) per acquisire la ricompensa sessuale, considerato analogo alla libido nella ricerca osservazionale sugli animali, è ridotto dalla castrazione, indicando che l’azione degli steroidi gonadici nel cervello è necessaria per lo sviluppo e/o il mantenimento di questo comportamento strumentale. Il lavoro comportamentale per acquisire la ricompensa sessuale (cioè la libido) è analogamente ridotto da lesioni all’amigdala basolaterale, una regione cerebrale che concentra gli steroidi, e dalla somministrazione di un antagonista della Dopamina al nucleo accumbens all’interno del sistema limbico. Questo comportamento strumentale viene ripristinato nei ratti maschi con lesioni all’amigdala basale dall’infusione di anfetamina nel nucleo accumbens, indicando che il rilascio mesolimbico di DA è parte integrante della libido. [18].
Esiste un nesso preciso tra gli ormoni steroidei (ad esempio, androgeni ed estrogeni), l’attività della Dopamina e la libido. Gli steroidi sessuali endogeni innescano la sintesi e il rilascio di Dopamina nei centri cerebrali chiave per sostenere le funzioni sessuali e la Dopamina stimola il sistema eccitatorio centrale per governare la libido e il conseguente comportamento sessuale.
Conclusione
Gli androgeni endogeni (Testosterone e DHT) esercitano un chiaro effetto sulla libido negli uomini; effetto nel quale è implicato anche l’Estradiolo. Gli androgeni sovrafisiologici generalmente aumentano la libido negli uomini (anche in quelli normali e sani), ma le modifiche chimiche degli androgeni possono influire sul fatto che determinati androgeni esercitino un effetto di aumento o addirittura di soppressione della libido. In generale, l’eccezione conferma la regola per quanto riguarda l’affermazione che gli androgeni tendono ad aumentare la libido negli uomini.
Il Testosterone, in quanto ormone sessuale maschile primario, ha la funzione biologica di controllare l’espressione del comportamento sessuale e aggressivo maschile, che deve necessariamente essere collegato alla prestanza fisica per quanto riguarda i vantaggi adattativi e competitivi per promuovere la sopravvivenza della specie umana.
I meccanismi dell’influenza degli androgeni sulla libido coinvolgono il neurotrasmettitore Dopamina e la sua attivazione del sistema eccitatorio, nonché gli effetti indiretti degli estrogeni, l’amplificazione periferica e la diminuzione, le gonadotropine dell’Asse Ipotalamo-Ipofisi-Gonadi, gli effetti sul metabolismo della Dopamina e persino gli effetti terziari sulla libido della Prolattina.
Per gli uomini è semplice: il Testosterone governa chiaramente la funzione sessuale e la libido (con una certa influenza dell’aromatizzazione in Estradiolo, in particolare nel SNC e nel cervello).
Nella prossimo articolo, analizzeremo come le donne siano più sfaccettate nelle dinamiche del loro ambiente ormonale e le ramificazioni dei cambiamenti ormonali del ciclo ovulatorio-mestruale; l’influenza predominante degli affetti (umore, benessere ed energia) sulla libido femminile; i problemi che derivano dalla difficoltà intrinseca di disgiungere gli effetti degli androgeni da quelli degli estrogeni nelle donne; l’ipotesi della desensibilizzazione e il suo potere esplicativo nel descrivere le differenze negli effetti degli androgeni sulla libido nelle donne rispetto agli uomini e tra le donne, nonché altri fattori che influenzano la libido e che sono correlati alle difficoltà nell’assegnare la causalità agli effetti degli androgeni esogeni sulla libido femminile.
Gabriel Bellizzi [CEO BioGenTech]
Riferimenti:
Fonte studi articolo Type-IIx
[1] Bancroft, J. Androgens and sexual function in men and women. In: Bremner, W., Bagattel, C., eds. Androgens in health and disease. Totowa: Humana Press.
[2] Isidori, A. M., Buvat, J., Corona, G., Goldstein, I., Jannini, E. A., Lenzi, A., … Maggi, M. (2014). A Critical Analysis of the Role of Testosterone in Erectile Function: From Pathophysiology to Treatment—A Systematic Review. European Urology, 65(1), 99–112. doi:10.1016/j.eururo.2013.08.048
[3] Anderson, R. A., Martin, C. W., Kung, A. W. C., Everington, D., Pun, T. C., Tan, K. C. B., … Baird, D. T. (1999). 7α-Methyl-19-Nortestosterone Maintains Sexual Behavior and Mood in Hypogonadal Men1. The Journal of Clinical Endocrinology & Metabolism, 84(10), 3556–3562. doi:10.1210/jcem.84.10.6028
[4] Anderson, R. A., Bancroft, J., & Wu, F. C. (1992). The effects of exogenous testosterone on sexuality and mood of normal men. The Journal of Clinical Endocrinology & Metabolism, 75(6), 1503–1507. doi:10.1210/jcem.75.6.1464655
[5] Su, T.-P. (1993). Neuropsychiatric Effects of Anabolic Steroids in Male Normal Volunteers. JAMA: The Journal of the American Medical Association, 269(21), 2760. doi:10.1001/jama.1993.03500210060032
[6] Moss, H. B., Panzak, G. L., & Tarter, R. E. (1993). Sexual functioning of male anabolic steroid abusers. Archives of Sexual Behavior, 22(1), 1–12. doi:10.1007/bf01552908
[7] Sato, T., Matsumoto, T., Kawano, H., Watanabe, T., Uematsu, Y., Sekine, K., … Kato, S. (2004). Brain masculinization requires androgen receptor function. Proceedings of the National Academy of Sciences, 101(6), 1673–1678. doi:10.1073/pnas.0305303101
[8] Pan, M. M., & Kovac, J. R. (2016). Beyond testosterone cypionate: evidence behind the use of nandrolone in male health and wellness. Translational Andrology and Urology, 5(2), 213–219. doi:10.21037/tau.2016.03.03
[9] Hulsbæk, S., Bandholm, T., Ban, I., Foss, N. B., Jensen, J.-E. B., Kehlet, H., & Kristensen, M. T. (2021). Feasibility and preliminary effect of anabolic steroids in addition to strength training and nutritional supplement in rehabilitation of patients with hip fracture: a randomized controlled pilot trial (HIP-SAP1 trial). BMC Geriatrics, 21(1). doi:10.1186/s12877-021-02273-z
[10] Sodi, R., Fikri, R., Diver, M., Ranganath, L., & Vora, J. (2005). Testosterone replacement-induced hyperprolactinaemia: case report and review of the literature. Annals of Clinical Biochemistry, 42(2), 153–159. doi:10.1258/0004563053492784
[11] Segaloff, A., Weeth, J. B., Cuningham, M., & Meyer, K. K. (1964). Hormonal therapy in cancer of the breast.XXIII. Effect of 7α-methyl-19-nortestosterone acetate and testosterone propionate on clinical course and hormonal excretion. Cancer, 17(10), 1248–1253. doi:10.1002/1097-0142(196410)17:10<1248::aid-cncr2820171005>3.0.co;2-a
[12] Maeda Y, Nakanishi T, Ozawa K, Kijima Y, Nakayama I, Shoji T, Sasaoka T. Anabolic steroid-associated hypogonadism in male hemodialysis patients. Clin Nephrol. 1989 Oct;32(4):198-201. PMID: 2805460.
[13] De Las Heras, F., & Negro-vilar, A. (1979). Effect of Aromatizable Androgens and Estradiol on Prolactin Secretion in Prepuberal Male Rats. Archives of Andrology, 2(2), 135–139. doi:10.3109/01485017908987305
[14] Hannan, C.J., Friedl, K., Zold, A., Kettler, T., & Plymate, S. (1991). Psychological and serum homovanillic acid changes in men administered androgenic steroids. Psychoneuroendocrinology, 16(4), 335–343. doi:10.1016/0306-4530(91)90019-p
[15] Lee S, Lee YB, Choe SJ, Lee WS. Adverse Sexual Effects of Treatment with Finasteride or Dutasteride for Male Androgenetic Alopecia: A Systematic Review and Meta-analysis. Acta Derm Venereol. 2019 Jan 1;99(1):12-17. doi:10.2340/00015555-3035
[17] Meston, C. M., & McCall, K. M. (2005). Dopamine and Norepinephrine Responses to Film-Induced Sexual Arousal in Sexually Functional and Sexually Dysfunctional Women. Journal of Sex & Marital Therapy, 31(4), 303–317. doi:10.1080/00926230590950217
[18] Pfaus, J. G. (2009). Pathways of Sexual Desire. The Journal of Sexual Medicine, 6(6), 1506–1533. doi:10.1111/j.1743-6109.2009.01309.x
E’ solo di recente che si è iniziato a trattare in maniera più dettagliata dei potenziali effetti collaterali neuronali degli AAS. Infatti, ci si è sempre e solo concentrati sull’impatto che queste molecole possono avere, per esempio, sulla lipidemia ematica, sulla funzionalità epatica e renale , tralasciando tutte quelle alterazioni che possono emergere a livello comportamentale e plastico-cerebrale, che sono certamente da non sottovalutare. Trattai già dell’attività neurosteroidea, vi rimando quindi alla lettura di quell’articolo per ulteriori approfondimenti.
Vista la scarsa diffusioni di valide informazioni sulla attività neuronale degli AAS, un mito comune che circola nella comunità del bodybuilding e dei pazienti in TRT è che il Testosterone sia intrinsecamente neuroprotettivo e che sia in questo unico rispetto a tutti gli altri steroidi androgeni anabolizzanti.
Ma il motivo per cui il Testosterone è neuroprotettivo è semplicemente perché esso aromatizza ad un tasso tale da fornire una quantità di Estradiolo sufficiente a bilanciare l’Androgenicità esercitata dallo stesso Testosterone e dal DHT.
I dati ottenuti in modelli di roditori che utilizzano cellule corticali lo suggeriscono in modo molto convincente, mostrando come l’inibitore dell’aromatasi Anastrozolo (Arimidex) elimini completamente gli effetti neuroprotettivi del Testosterone [1].
AAS sintetici e Testosterone a confronto:
Sono stati pubblicati molti studi che suggeriscono quanto gli AAS sintetici siano peggiori rispetto al Testosterone per quanto riguarda le malattie cardiovascolari, la neurotossicità e una miriade di altri esiti organici deleteri.
Ritengo che una parte significativa di questi dati sia in parte estremizzata, data la mancanza di osservazioni di risposta seguenti alla somministrazione di estrogeni esogeni. In definitiva, il Testosterone è l’Androgeno più sicuro a dosaggi fisiologici. Il DHEA segue il grado favorevole di sicurezza, ma questo è un altro discorso.
Tuttavia, si ipotizza che molti AAS non siano così pericolosi come siamo portati a credere. Attenzione però, con questo non si intende assolutamente dire che essi abbiano un ottimale margine di sicurezza.
L’ipotesi è che alcune molecole non sono intrinsecamente più pericolose del Testosterone, ma è la loro mancanza di aromatizzazione, di 5-α riduzione o di diversa affinità per i recettori fuori bersaglio a renderli più pericolosi.
L’effetto sui Recettori degli Estrogeni (ER) e l’affinità dell’AAS per l’attività dell’Enzima Aromatasi sono i fattori principali che incidono sulla validità di un ormone in un contesto di monoterapia.
I derivati del DHT non possono essere convertiti dall’Enzima Aromatasi in un estrogeno come l’Estradiolo.
Il Nandrolone (19-nortestosterone) e i suoi derivati (19-nor) hanno ciascuno la propria affinità (o mancanza di affinità) per i ER e l’interazione con l’Aromatasi, che spesso si traduce in livelli di estrogeni inferiori alla media (esistono eccezioni come il Trestolone/MENT il quale si converte nel potente 7α-methyl-Estradiolo).
In sostanza, credo che alcuni AAS possano risultare significativamente più cardiotossici e neurotossici nei dati perché vengono sempre utilizzati da soli con una quantità insufficiente di estrogeni per bilanciare l’attività androgenica.
L’importanza di mantenere livelli ottimali di Estradiolo:
Livelli correttamente bilanciati di estrogeni sono necessari per la libido, la qualità dell’erezione, la vasodilatazione, la salute cardiovascolare, la salute del cervello, la salute delle ossa e molte altre funzioni sistemiche.
Nelle donne il rischio di malattie cardiovascolari aumenta notevolmente dopo la menopausa.
Non è una coincidenza che la maggior parte delle donne che sviluppano malattie cardiache lo facciano dopo che la produzione di estrogeni è scesa a livelli inferiori a quelli maschili.
Se non si dispone di una quantità sufficiente di estrogeni rispetto ai livelli di androgeni nell’organismo, i livelli di cardiotossicità e neurotossicità saranno significativamente più alti di quelli che si avrebbero se si mantenessero livelli ottimali di estrogeni.
Dal punto di vista del Bodybuilding, gli estrogeni sono necessari per ottimizzare la crescita muscolare, l’insulino-sensibilità e la sintesi di IGF-1 e fattori di crescita/segnalazione cellulare.
Per questo motivo gli AAS altamente aromatizzabili possono indirettamente determinare un maggiore potenziale di crescita e sono spesso classificati come composti “Bulking”.
Aneddoticamente, molti bodybuilder riferiscono di essere cresciuti al massimo durante l’Off-Season, quando i loro livelli di estrogeni erano sufficientemente alti.
La logica di inserimento di una “base” di Testosterone:
Il Testosterone non ha una selettività tissutale e, in realtà, è un modesto “costruttore muscolare” milligrammo per milligrammo rispetto ad altri AAS sintetici sviluppati negli anni successivi alla sua scoperta.
Per quanto riguarda la ritenzione di azoto, sulla carta non è superiore a molti AAS.
Tuttavia, il Testosterone aromatizza in Estradiolo a un ritmo strettamente regolato, è bioidentico e il corpo sa esattamente cosa fare con esso.
Inoltre, il corpo sa quanto Testosterone legare con le SHBG, quanto liberarne e mettere a disposizione dei tessuti, nonché quanto 5α ridurne a DHT per antagonizzare l’attivazione dei Recettori degli Estrogeni nel caso in cui questa vada fuori controllo.
Dal punto di vista del bodybuilding, il Testosterone è sottovalutato sotto molti aspetti.
Tuttavia, in un contesto di salute generale, longevità e bodybuilding, il Testosterone non può essere battuto a dosaggi terapeutici.
Utilizzando una base di Testosterone o una fonte di estrogeni sufficiente, le carenze di altri agenti anabolizzanti possono essere attenuate in una certa misura, motivo per cui il Testosterone è la base della maggior parte dei cicli di SARM steroidei e non steroidei.
Bilancio tra Testosterone, DHT ed Estradiolo:
La steroidogenesi nell’organismo si esplica come un’enorme orchestra volta alla regolazione di innumerevoli funzioni. Essa è molto più complessa della semplice sintesi di Testosterone, estrogeni e DHT.
La steroidogenesi umana, con le principali classi di ormoni steroidei, i singoli steroidi e le vie enzimatiche.[Häggström M, Richfield D (2014). “Diagram of the pathways of human steroidogenesis”. WikiJournal of Medicine.] I cambiamenti nella struttura molecolare da un precursore sono evidenziati in bianco.
Anche a livello acuto, l’equilibrio tra androgeni ed estrogeni nell’organismo è strettamente regolato e viene attuato per garantire una ottimizzazione della salute.
Questo equilibrio diventa sempre più disfunzionale con l’età, uno stile di vita scorretto, un’alimentazione scorretta, una cattiva igiene del sonno e numerosi altri fattori.
Tuttavia, come detto precedentemente, l’organismo sa esattamente cosa fare con il Testosterone, come creare una quantità ottimale di estrogeni e quanto Testosterone ridurre in DHT per contrastare l’eccesso di estrogeni e sostenere le caratteristiche sessuali secondarie maschili.
Quando si confrontano i dati clinici su un AAS sintetico con il Testosterone in un contesto di monoterapia, bisogna considerare che questi studi utilizzano l’AAS sintetico da solo, non con estrogeni esogeni o qualsiasi ormone supplementare che potrebbe essere necessario per bilanciare la sua androgenicità, la mancanza di attività estrogenica e/o l’interazione con l’Aromatasi.
Ovviamente, se si prende un composto che non aromatizza a sufficienza in estrogeni e lo si confronta con l’androgeno bioidentico che il nostro corpo sa aromatizzare e 5α ridurre in modo perfettamente bilanciato, si può solo immaginare quale sarà la scelta migliore data dall’osservazione comparativa in termini di cardiotossicità e neurotossicità.
La neurotossicità di Testosterone, Nandrolone e Stanozololo:
Confrontando l’effetto del Testosterone con quello del 19-nortestosterone (Nandrolone) e dello Stanozololo (Winstrol) sulla neurotossicità, si vede chiaramente che sono gli estrogeni a proteggere i neuroni del cervello, non il Testosterone in se.
Strutture molecolari di Testosterone, Nandrolone e Stanozololo
Nello studio del 2007 di Rosamaria Orlando et al., un dosaggio fisiologico di Testosterone risultava essere neuroprotettivo [1]. Il Testosterone amplificava la neurotossicità solo a dosaggi sovrafisiologici.
L’effetto neuroprotettivo di un dosaggio fisiologico di Testosterone è stato completamente eliminato quando è stato co-somministrato l’inibitore dell’Aromatasi Anastrozolo (Arimidex), suggerendo che la tossicità intrinseca del Testosterone come androgeno è controbilanciata solo dalla sua aromatizzazione in 17β-estradiolo.
Struttura molecolare di Estradiolo
A differenza del Testosterone, il Nandrolone non presenta un tasso di aromatizzazione sufficiente in termini assoluti (quantità) e specifici (tipo di estrogeno) mentre lo Stanozololo non subisce a nessun grado l’aromatizzazione.
Come prevedibile, il Nandrolone e lo Stanozololo sono risultati entrambi neurotossici a ogni singola dose valutata, indipendentemente dalla co-somministrazione o meno di Anastrozolo.
L’antiandrogeno Flutamide è stato in grado di attenuare la neurotossicità di tutti e tre gli androgeni, rafforzando così ulteriormente il fatto che dosaggi fisiologici di androgeni senza una quantità sufficiente di estrogeni a controbilanciarne gli effetti, o dosaggi sovrafisiologici di androgeni possono facilitare la morte neuronale.
Nessuno degli steroidi androgeni anabolizzanti di questo studio è risultato tossico in assenza di NMDA (recettore N-metil-D-aspartato), suggerendo quindi che il meccanismo attraverso il quale gli androgeni non controbilanciati facilitano la morte neuronale è una maggiore vulnerabilità agli insulti eccitotossici.
Rappresentazione grafica semplificata della struttura del recettore NMDA
Effetto neuroprotettivo del Testosterone a dosaggi fisiologici senza Anastrozolo:
A dosaggi fisiologici senza la presenza di un inibitore dell’Aromatasi, il Testosterone ha dimostrato di avere un effetto neuroprotettivo.
Spesso si ritiene che sia l’androgeno stesso (Testosterone) a proteggere il cervello. Tuttavia, l’inibitore dell’Aromatasi Anastrozolo ha eliminato completamente tutti gli effetti neuroprotettivi del Testosterone allo stesso dosaggio fisiologico.
L’Anastrozolo ha esacerbato la neurotossicità a ogni singolo dosaggio di Testosterone quando è stato co-somministrato.
Ciò suggerisce che il Testosterone non è un androgeno unico e con attività che neuroprotettiva maggiore rispetto a tutti gli altri AAS, ma che è la sua aromatizzazione in estrogeni a essere neuroprotettiva.
Neurotossicità del Testosterone a dosaggi soprafisiologici con e senza Anastrozolo:
A dosaggi sovrafisiologici il Testosterone ha dimostrato di esacerbare la neurotossicità. Sebbene la sua aromatizzazione in estrogeni prevenga comunque una quantità significativa di morte neuronale, possiamo vedere chiaramente che le concentrazioni sovrafisiologiche di Testosterone esacerbano la neurotossicità in ogni caso e che i livelli sovrafisiologici di estrogeni non forniscono un aumento dose-dipendente della neuroprotezione.
Quindi, i dati suggeriscono che le concentrazioni fisiologiche di Testosterone facilitano la neuroprotezione cerebrale attraverso l’aromatizzazione in estrogeni, ma c’è una soglia per questa neuroprotezione e le concentrazioni sovrafisiologiche non sono comunque neurologicamente salutari.
La neurotossicità del Nandrolone è indipendente dall’uso di Anastrozolo:
Il Nandrolone ha esacerbato la neurotossicità a tutti i dosaggi, indipendentemente dal fatto che sia stata valutata una concentrazione bassa o alta.
Inoltre, la co-somministrazione di Anastrozolo non ha avuto alcun impatto sulla neurotossicità del Nandrolone in questo modello, a qualsiasi dosaggio.
Ciò suggerisce che il Nandrolone non aromatizza in estrogeni ad un tasso sufficiente, né attiva i recettori degli estrogeni da solo ad un grado soddisfacente per fornire gli effetti neuroprotettivi di livelli sani di estrogeni.
Sarebbe probabilmente necessaria una fonte di estrogeni da co-somministrare con il Nandrolone per poterlo considerare una valida alternativa alla monoterapia in un contesto di HRT o di ciclo con un certo margine di “sicurezza”.
La neurotossicità dello Stanozololo è indipendente dall’uso di Anastrozolo:
Lo Stanozololo ha esacerbato la neurotossicità a tutti i dosaggi, indipendentemente dal fatto che si valutasse una concentrazione bassa o alta.
Inoltre, la co-somministrazione di Anastrozolo non ha avuto alcun impatto sulla neurotossicità dello Stanozololo in questo modello, a qualsiasi dosaggio.
Sappiamo già che lo Stanozololo non è soggetto ad aromatizzazione.
Anche in questo caso, i dati suggeriscono che sarebbe necessaria una fonte di estrogeni da co-somministrare con lo Stanozololo per poterlo considerare una valida alternativa alla monoterapia in un contesto di HRT o di ciclo con un certo margine di “sicurezza”.
Attenuazione della neurotossicità con co-somministrazione di antiandrogeni:
Gli androgeni non controbilanciati da una quantità sufficiente di estrogeni sono cardiotossici e neurotossici.
Per questo motivo la Flutamide (un anti-androgeno) è stata in grado di eliminare la neurotossicità del Nandrolone e dello Stanozololo.
Gli antiandrogeni hanno una risposta dose-dipendente proprio come gli AAS, quindi si verifica una competizione tra gli antiandrogeni e gli androgeni per il legame e l’attivazione del Recettore degli Androgeni (AR).
Gli antiandrogeni agiscono come antagonisti competitivi dell’AR o come steroidi sintetici di fortuna, anche se con un’androgenicità significativamente ridotta o pressoché assente.
In pratica, a seconda dell’antiandrogeno utilizzato, o enzimatico (vedi inibitori della 5α-reduttasi come la Finasteride) o competitivo (vedi Bicalutamide, Flutamide ecc…), essi agiranno riducendo l’attività degli androgeni a livello sistemico (orali) o topico (soluzione da applicare).
L’efficacia dell’antiandrogeno nell’inibire il legame degli androgeni con l’AR si basa sull’affinità di legame, sulla costante di legame, sull’emivita, sul dosaggio utilizzato e su una miriade di altri fattori.
La Flutamide è un antagonista selettivo del recettore degli androgeni non-steroideo, competitivo e “silenzioso” dell’AR. Si tratta di un antiandrogeno primitivo e di livello inferiore rispetto agli sviluppi più recenti della medicina, tuttavia è ancora efficace nell’impedire agli androgeni di legarsi ai recettori bersaglio specifici.
Per questo motivo la Flutamide è stata in grado di eliminare completamente la neurotossicità del Nandrolone e dello Stanozololo a tutti i dosaggi. Impedendo al Nandrolone e allo Stanozololo di legarsi ai recettori degli androgeni, essi non sono più in grado di innescare la trascrizione e, quindi, manifestare i loro effetti nei tessuti.
I dati relativi alla Flutamide e all’Anastrozolo rafforzano il fatto che il Nandrolone non converte in estrogeni ad un tasso sufficiente da fornire la neuroprotezione necessaria per evitare la morte neuronale. Con o senza inibitore dell’Aromatasi, il Nandrolone aggrava la neurotossicità allo stesso modo.
Senza un inibitore dell’Aromatasi ma con un antiandrogeno, la Neurotossicità del Nandrolone viene eliminata completamente. Ed è probabile che lo stesso valga anche per la cardiotossicità intrinseca del Nandrolone.
Quanto detto vale, prevedibilmente, anche per lo Stanozololo, che non è soggetto ad aromatizzazione.
È qui che tutti gli studi che dimostrano quanto il Nandrolone abbia un influenza negativa per il cuore e il cervello vengono messi in discussione, poiché gli esiti negativi riscontrati nei dati potrebbero non essere stati così drastici se fosse stata co-somministrata una fonte di estrogeni.
Lo stesso dosaggio di Flutamide non è stato in grado di compensare completamente la neurotossicità del Testosterone a dosaggi sovrafisiologici senza la presenza di Anastrozolo.
Quando i dosaggi di Testosterone superano le concentrazioni fisiologiche, la vulnerabilità alla neurotossicità e alla cardiotossicità sale vertiginosamente.
Una quantità eccessiva di qualsiasi cosa nell’organismo è dannosa, e il Testosterone non è esente da questo problema solo perché è l’ormone bioidentico che produciamo naturalmente e che è soggetto ad aromatizzazione in estrogeni.
L’aromatizzazione degli androgeni in estrogeni regola la neurotossicità:
Riflettendo sui dati con e senza inibitore dell’Aromatasi, possiamo vedere chiaramente che sono gli estrogeni ad esercitare la neuroprotezione, non il Testosterone.
Molti pensano erroneamente che il Testosterone sia un androgeno unico che si lega all’AR in qualche modo speciale per proteggere il cervello e che gli altri AAS lo danneggino. Ma i dati ci mostrano che le cose siano proprio così nette.
Infatti, i dati mostrino chiaramente che con la co-somministrazione di Anastrozolo l’effetto neuroprotettivo viene annullato, mentre senza Anastrozolo si ha un effetto neuroprotettivo.
Se si dispone di una quantità sufficiente di estrogeni per bilanciare gli androgeni nel corpo, si ottiene un livello stabile e ottimale di neuroprotezione, che si riflette nel modo in cui il nostro corpo regola l’aromatizzazione endogena degli androgeni.
Ma se si ha un livello sovrafisiologico di androgeni o si inibisce l’Aromatasi così da impedire la sintesi di una quantità di estrogeni necessari per svolgere le funzioni organiche, la neurotossicità aumenta indipendentemente dal fatto che si stia valutando il Testosterone e non un AAS sintetico non bioidentico. E tutto ciò si ricollega alla logica dell’utilizzo di una base di Testosterone durante il ciclo o di una fonte sufficiente di estrogeni esogeni in caso di carenza.
Questo rafforza anche il fatto che gli inibitori dell’aromatasi sono più che altro deleteri se usati senza una reale necessità.
Sarebbe opportuno fare tutto il possibile per evitare l’uso di inibitori dell’Aromatasi.
Se si ha bisogno di un inibitore dell’Aromatasi, è spesso probabile che ciò sia legato, per esempio, ad un dosaggio troppo alto di Testosterone (o altri AAS soggetti ad aromatizzazione), o ad una percentuale di grasso troppo alta (più grasso = più Aromatasi). Altre volte ciò può dipendere da un polimorfismo genetico che determina un metabolismo degli ormoni sessuali alterato o da una sovraespressione epigenetico-dipendente alla modificata omeostasi ormonale.
In definitiva, la probabilità che vi sia il bisogno di un AI per gestire una dose terapeutica di Testosterone, se i soprariportati punti sono ottimizzati, non è così probabile.
In un contesto diBodybuilding con dosaggi sovrafisiologici, ritengo inoltre che nella maggior parte dei casi (quindi non in tutti) l’uso di un AI solo per poter utilizzare una dose “troppo alta” di Testosterone sia una strategia sbagliata.
È opportuno assumere estrogeni esogeni per prevenire la neurotossicità e la cardiotossicità?
Il fatto che siano gli estrogeni a fornire protezione neurologica, e non il Testosterone, non significa assolutamente che si debba iniziare ad assumere pillole anticoncezionali come fossero caramelle. Gli estrogeni non controbilancianti nell’organismo sono a loro volta cancerogeni. C’è un motivo per cui i primi trattamenti per il cancro al seno sono stati SERM e gli AI. Inoltre, gli estrogeni non forniscono neuroprotezione in modo dipendente dalla dose.
C’è un punto di equilibrio per ogni cosa nell’organismo, e una quantità eccessiva di qualsiasi cosa può essere dannosa. Un pretrattamento di 4 giorni con basse concentrazioni 0,01 μM (10 nM) di 17β-estradiolo è stato sostanzialmente neuroprotettivo contro la tossicità NMDA.
Tuttavia, è possibile notare chiaramente dal grafico che non si è verificata una diminuzione dose-dipendente della neurotossicità.
La neuroprotezione è stata significativamente inferiore con 1μM di 17β-estradiolo rispetto al dosaggio molto più basso di 0,01 μM di 17β-estradiolo.
L’organismo ha un sistema strettamente regolato in cui è necessaria una certa quantità di estrogeni per le funzioni fisiologiche. Una quantità eccessiva di estrogeni senza una quantità sufficiente di androgeni può provocare lo sviluppo di tumori, ginecomastia e diversi altri problemi.
Troppo pochi estrogeni ed eccessivi androgeni possono causare malattie cardiovascolari, morte neuronale e altrettanti problemi.
Quindi, cosa concludere?
Quindi, se si utilizza un androgeno senza una quantità sufficiente di estrogeni opposti per bilanciarlo, non solo si inibisce la crescita muscolare e la perdita di grasso, ma si mette il corpo in uno stato di salute che si deteriora ancora più rapidamente di quello in cui si sarebbe già trovato semplicemente a causa dei livelli di androgeni sovrafisiologici.
Se si utilizza una dose di Testosterone da TRT, sarebbe meglio non inibire inutilmente l’Aromatasi.
Inoltre, se si utilizza un AAS che o non è un potente substrato per l’aromatasi o non è soggetto ad essa (e questo interessa tutti i SARM steroidei e non steroidei), sarebbe meglio aggiungere al ciclo una base di Testosterone o una fonte di estrogeni adeguata. La co-somministrazione di DHEA non è garante di una risultante estrogenica ematica adeguata per via di variabili enzimatiche legate alla conversione dell’androgeno surrenalico a Androstenedione e Estradiolo.
La cosa interessante da valutare sarebbe se tutti gli AAS precedentemente descritti dalla letteratura riportata come deleteri per il cuore e il cervello sarebbero ancora descritti come tali se un dosaggio adeguato di estrogeni esogeni venisse usato insieme ad essi negli studi corrispondenti.
Questo apre sicuramente nuove prospettive per potenziali alternative alla HRT.
Nel 1957 sono stati pubblicati i primi risultati del Framingham Heart Study [1]. Si trattava (o dovrei dire si tratta, visto che è ancora in corso) di uno studio epidemiologico che cercava di individuare i fattori di rischio per le malattie cardiovascolari. Lo studio prende il nome dalla città di Framingham, Massachusetts, negli Stati Uniti. Sono stati reclutati 5.209 residenti della città di età compresa tra i 30 e i 62 anni. Diversi dati di questo gruppo di persone (coorte) sono stati raccolti nel tempo per scoprire questi fattori di rischio. Nella loro importante pubblicazione, hanno identificato tre fattori di rischio per le malattie cardiovascolari: ipertensione, obesità e ipercolesterolemia (alti livelli di colesterolo).
Ai fini di questo articolo, mi concentrerò sul Colesterolo. Prima di parlare dell’effetto degli Steroidi Anabolizzanti Androgeni sul colesterolo HDL, fornirò alcune informazioni di base.
Relazione tra malattie cardiovascolari e colesterolo LDL e HDL:
Dopo i risultati iniziali del Framingham Heart Study, il ruolo del colesterolo nello sviluppo del rischio di malattie cardiovascolari è stato ulteriormente chiarito. Un primo passo avanti in quest’area di ricerca è stata la suddivisione del colesterolo in colesterolo a bassa densità (LDL) e colesterolo ad alta densità (HDL) e il loro rispettivo contributo al rischio di malattie cardiovascolari. Queste due frazioni di colesterolo sono note al grande pubblico rispettivamente come colesterolo “cattivo” e “buono”.
Il colesterolo LDL elevato è stato associato a un aumento del rischio di malattie cardiovascolari. Dopo decenni di ricerche, una pletora di prove ha stabilito con certezza che questa associazione è causale [2]. In effetti, la terapia per abbassare le LDL, ad esempio con l’uso di statine, è una pietra miliare del trattamento delle dislipidemie. L’associazione tra colesterolo HDL e rischio di malattie cardiovascolari è opposta a quella del colesterolo LDL: un colesterolo HDL elevato è risultato associato a una diminuzione del rischio di malattie cardiovascolari. Gli studi epidemiologici rilevano una riduzione del rischio cardiovascolare di circa il 2-3% per ogni aumento di 1 mg/dL di colesterolo HDL [3].
A differenza del colesterolo LDL, tuttavia, non sembra esistere un legame causale diretto tra i livelli di colesterolo HDL e il rischio di malattie cardiovascolari [4]. Studi genetici sull’uomo, in cui sono state analizzate alcune mutazioni genetiche che portano a livelli più o meno elevati di colesterolo HDL, non hanno dimostrato chiaramente un’associazione con il rischio di malattie cardiovascolari. Questo sarebbe stato prevedibile se ci fosse stato un legame causale diretto. Lo scollamento tra i livelli di colesterolo HDL e il rischio di malattie cardiovascolari è diventato forse più dolorosamente evidente negli studi clinici sui farmaci. Sono stati sviluppati (o esistevano già) alcuni farmaci che aumentano i livelli di colesterolo HDL in modo significativo, ma non riescono a ridurre la mortalità o l’incidenza di eventi cardiovascolari, come ictus o infarto del miocardio [5]. Ciò include anche l’uso di integratori da banco, come la Niacina [5, 6].
Nota: quando si parla di “colesterolo LDL”, “colesterolo HDL” o “colesterolo VLDL” ci si riferisce alle lipoproteine trasportatrici. La sigla VLDL sta per “very low density lipoproteins”, LDL per “low density lipoproteins” e HDL per “high density lipoproteins”. La densità a cui si fa riferimento è legata al loro contenuto lipidico. In particolare la densità è tanto più bassa quanto maggiori sono i trigliceridi racchiusi all’interno della particella. Ne deriva che le VLDL sono lipoproteine ad alto contenuto in trigliceridi, le LDL sono lipoproteine a basso contenuto in trigliceridi e le HDL sono lipoproteine estremamente povere di trigliceridi. In compenso LDL e HDL sono caratterizzate da un alto contenuto in colesterolo. Ognuna di queste lipoproteine ricopre ruoli diversi. Le VLDL hanno il compito di trasferire trigliceridi dal fegato ai tessuti; in particolare, dopo essere state sintetizzate nel fegato, vengono riversate nel circolo ematico e cedute soprattutto al tessuto muscolare e a quello adiposo. LDL ed HDL trasportano il colesterolo nel circolo sanguigno. Mentre le LDL hanno lo scopo di cederlo ai tessuti, le HDL sono deputate alla rimozione del colesterolo presente in eccesso nel plasma. Le VLDL vengono sintetizzate soprattutto nelle cellule epatiche (epatociti) e trasportano principalmente Trigliceridi di origine endogena.
Le IDL (lipoproteine a densità intermedia) sono il prodotto del catabolismo parziale delle VLDL. Sono più piccole (da 25 a 30 nm), hanno più Colesterolo e meno Trigliceridi. Hanno una densità compresa tra 1,006 e 1,019 g/ml e migrano elettroforeticamente con le ß-globuline. Per ogni molecola tipica di VLDL che viene degradata, viene prodotta una IDL.
Per la stima del VLDL-C è necessario dividere il valore dei Trigliceridi misurato per 5, nel caso in cui siano espressi in mg/dL, o 2.2, nel caso siano espressi in mmol/L. Nella maggior parte dei casi, la formula consente di effettuare una stima accurata del reale valore del VLDL-C.
AAS e riduzione del HDL:
Ora siete un po’ più informati sulla relazione tra malattie cardiovascolari e colesterolo LDL e HDL. Diversi studi interventistici hanno esaminato l’effetto dell’uso di AAS sul colesterolo. Peter Bond ha fatto un piccolo riassunto di questi studi nel suo libro “Book on Steroids” il quale riporto nella tabella sottostante. Sebbene non tutti gli studi abbiano riscontrato una diminuzione statisticamente significativa del colesterolo HDL (↔️), molti lo fanno e nel complesso mostrano inequivocabilmente una diminuzione. Ciò è particolarmente vero per gli AAS orali, che sembrano avere l’effetto maggiore sul colesterolo HDL.
In uno studio, condotto dal gruppo di Bhasin [11], sono stati somministrati dosaggi graduali di Testosterone (25, 50 125, 300 e 600mg di Testosterone Enantato alla settimana). Gli autori hanno quindi potuto valutare se esisteva una relazione dose-risposta tra il dosaggio di Testosterone e il colesterolo HDL, e così è stato. Hanno riscontrato una moderata relazione inversa (r = -0,40) tra i livelli di Testosterone e il colesterolo HDL. Quindi, almeno fino a una dose compresa tra 300 e 600mg settimanali, più alto è il dosaggio, maggiore è la diminuzione del colesterolo HDL.
In un recente studio, 100 consumatori di AAS sono stati seguiti nel tempo durante l’autosomministrazione di questa classe di farmaci. Il dosaggio medio, basato sulle informazioni riportate sull’etichetta, era di 898mg a settimana, rendendo così il loro ciclo di AAS abbastanza rappresentativo dell’uso comune da parte dei bodybuilder. Le misurazioni sono state effettuate prima, durante, 3 mesi dopo la fine del ciclo e 1 anno dopo l’inizio del ciclo. Il colesterolo HDL è diminuito di 0,4 mmol/L (da 1,2 a 0,8) durante l’uso. Si tratta di una diminuzione sostanziale. I valori erano tornati ai valori di base 3 mesi dopo la cessazione dell’uso di AAS.
Meccanismo attraverso il quale gli AAS abbassano l’HDL:
Si ritiene che gli steroidi anabolizzanti riducano il colesterolo HDL aumentando l’attività di un enzima chiamato lipasi epatica [7, 8, 9, 10]. Si tratta di un enzima prodotto principalmente dal fegato. Essendo una lipasi, catalizza le reazioni di idrolisi dei lipidi. In particolare, scinde gli acidi grassi dal triacilglicerolo (Trigliceride) e i fosfolipidi dalle particelle lipoproteiche, come il colesterolo HDL. Idrolizzando il triacilglicerolo e i fosfolipidi dal colesterolo HDL, riduce le dimensioni di queste particelle. Queste particelle più piccole vengono catabolizzate a un ritmo più elevato [12].
Thompson et al. hanno esaminato queste sottofrazioni di colesterolo HDL che differiscono per dimensioni [8]. Hanno misurato i livelli di colesterolo HDL2 e HDL3: le particelle di colesterolo HDL2 sono più grandi e di densità inferiore rispetto a quelle HDL3. Gli uomini partecipanti hanno ricevuto 200mg di Testosterone Enantato alla settimana o 6mg di Stanozololo orale (Winstrol) al giorno per 6 settimane in un design crossover. I risultati sono stati i seguenti:
*Differenza significativa (P < 0,05) rispetto al valore basale.
Come si può notare, la maggiore diminuzione relativa è stata osservata nella frazione HDL2 più grande a seguito del trattamento con Stanozololo. Al contrario, il Testosterone non ha mostrato una diminuzione statisticamente significativa nella frazione HDL2, ma ha fatto altrettanto nella frazione HDL3 più piccola. Non è del tutto chiaro cosa provochi la diminuzione di questa frazione.
Quando dei bodybuilder sono stati randomizzati a ricevere 200mg di Nandrolone Decanoato alla settimana o un placebo [13]. Non sono stati riscontrati cambiamenti statisticamente significativi nel colesterolo totale, nel colesterolo LDL e nel colesterolo HDL. Analogamente, non sono stati riscontrati cambiamenti significativi nelle sottofrazioni di colesterolo HDL2 e HDL3. In particolare, nella stessa pubblicazione, gli autori riferiscono anche di uno studio in cui hanno seguito un gruppo di atleti di forza che si autosomministravano steroidi anabolizzanti. Sono stati utilizzati diversi composti in vari dosaggi, ma vale la pena sottolineare che la maggior parte di essi comprendeva anche uno steroide anabolizzante orale (soprattutto Stanozololo). In questo caso, il colesterolo HDL è sceso in picchiata: da 1,08 mmol/L a 0,43 mmol/L dopo 8 settimane. La sottofrazione di colesterolo HDL2 è scesa da 0,21 a 0,05 e la sottofrazione di colesterolo HDL3 è scesa da 0,87 a 0,40 mmol/L.
Effetto degli AAS sulla funzione del colesterolo HDL:
Dato il legame tra l’effetto di un farmaco sui livelli di colesterolo HDL e il rischio di malattie cardiovascolari, la ricerca ha iniziato a concentrarsi sulla funzione del colesterolo HDL. Il colesterolo HDL è il protagonista di un processo chiamato trasporto inverso del colesterolo. Nell’aterosclerosi, il colesterolo si accumula nelle cellule del sistema immunitario (macrofagi) e nelle cellule muscolari lisce che circondano i vasi sanguigni [14]. Queste cellule, a loro volta, diventano le cosiddette cellule schiumose, che segnano il punto di partenza dell’aterosclerosi. Le particelle di colesterolo HDL sono in grado di raccogliere il colesterolo da queste cellule – efflusso di colesterolo. L’efflusso del colesterolo dalle cellule schiumose nelle particelle di colesterolo HDL è uno dei modi in cui si ritiene che il colesterolo HDL eserciti i suoi effetti protettivi sulle arterie. Il colesterolo HDL raccolto può poi essere riportato al fegato, che lo incorpora nella bile e può quindi essere secreto nelle feci. Allo stesso modo, le particelle di colesterolo HDL possono trasferire parte del loro contenuto alle particelle LDL, che possono finire nuovamente nelle cellule schiumose o essere assorbite dal fegato.
Esistono metodi per misurare la capacità di efflusso del colesterolo HDL e l’idea attuale è che la sua modulazione possa influire sul rischio di malattie cardiovascolari, contrariamente ai livelli di colesterolo HDL in sé. Esistono diversi modi in cui il colesterolo HDL può assorbire il colesterolo dalle cellule schiumose. Uno di questi coinvolge un trasportatore chiamato ATP-binding casette transporter A1 (ABCA1), che si ritiene sia il più importante [15, 16]. Contribuiscono anche altri trasportatori, come ABCG1 e il recettore scavenger B1, oltre alla diffusione semplice.
Diamo uno sguardo agli studi che hanno valutato l’impatto dell’uso di steroidi anabolizzanti sulla capacità di efflusso del colesterolo HDL. In uno studio (non controllato), uomini anziani ipogonadici sono stati randomizzati alla TRT con o senza Dutasteride (un inibitore della 5a-reduttasi) [17]. Dopo 3 mesi, la TRT era riuscita a riportare i livelli di Testosterone di questi uomini all’interno del range di normalità. Il colesterolo HDL e la capacità di efflusso del colesterolo HDL sono rimasti inalterati.
Un altro studio, randomizzato e controllato, ha applicato un approccio leggermente diverso [18]. Uomini sani, di età compresa tra i 19 e i 55 anni, sono stati castrati medicalmente per sopprimere completamente la loro produzione endogena. In seguito, hanno ricevuto un placebo, una TRT a basso dosaggio, una TRT sostitutiva completa o una TRT sostitutiva completa con Letrozolo, un inibitore dell’Aromatasi che inibisce la conversione del Testosterone in Estradiolo. Il colesterolo HDL è aumentato leggermente nel gruppo placebo e in quello a basso dosaggio, mentre è rimasto inalterato nei due gruppi che hanno ricevuto una dose sostitutiva completa. Inoltre, mentre è stata riscontrata una piccola diminuzione della capacità di efflusso di ABCA1 nel gruppo che ha ricevuto anche il Letrozolo, non sono stati osservati cambiamenti negli altri tre gruppi. A causa delle dimensioni ridotte dei gruppi, è possibile che un piccolo effetto non sia stato notato.
E i dosaggi elevati? Sfortunatamente, esiste un solo studio che ha esaminato questo aspetto, ed era di natura trasversale (si tratta di misurazioni effettuate in un solo momento, il che rende impossibile/difficile trarre conclusioni)[19]. I ricercatori hanno confrontato le misurazioni di un gruppo di utilizzatori di AAS con quelle di non utilizzatori e controlli sedentari, che avevano un’età corrispondente. I consumatori di AAS erano forti utilizzatori, avendo fatto uso di AAS in media per circa 8 anni con un dosaggio medio di (apparentemente) 2,5g settimanali. La capacità delle HDL di effluire il colesterolo dai macrofagi è risultata inferiore del 13% nei consumatori di AAS rispetto ai non consumatori con allenamento della forza. Anche in questo caso, a causa della natura trasversale dello studio, è difficile dire se questo sia causale.
Conclusioni:
Gli AAS riducono il colesterolo HDL, in modo dose-dipendente, e questo sembra verificarsi in modo particolarmente marcato con gli AAS orali17α-alchilati. Non è certo come questo si traduca in un rischio di malattia cardiovascolare, in quanto esiste una discrepanza tra la capacità di un farmaco di alterare il colesterolo HDL e il suo effetto su di esso. La correlazione tra i livelli di colesterolo HDL misurati e il rischio di malattie cardiovascolari non è causale. I ricercatori ritengono che la capacità di efflusso del colesterolo HDL possa avere una migliore capacità predittiva, oltre a essere causalmente correlata. Pertanto, i farmaci che influiscono sulla capacità di efflusso potrebbero influenzare il rischio di malattie cardiovascolari. L’effetto degli AAS su questo aspetto non è ancora così chiaro a causa della scarsità di dati, soprattutto per quanto riguarda i dosaggi sovrafisiologici. Alcuni dati suggeriscono che potrebbero avere un impatto negativo sulla capacità di efflusso del colesterolo. Uno studio di coorte longitudinale probabilmente risponderà a questa domanda con maggiore certezza in futuro. Se la capacità di efflusso del colesterolo HDL diminuisce effettivamente in seguito all’uso di AAS, questa diminuzione indotta dagli AAS potrebbe essere dannosa per la salute cardiovascolare.
Certo, vi sono farmaci, non che integratori erboristici da banco, con azione di “tampone” della dislipidemia ematica. Ma ciò non elimina il problema lo rallenta nella sua potenziale comparsa soprattutto agendo sui rapporti tra i marcatori del profilo lipidico ematico. Ciò significa che potenzialmente, e il condizionale è d’obbligo vista la sensibile differenza soggettiva riscontrabile, l’uso di Monacolina-K, Niacina e EPA, nei corretti dosaggi, potrà causare una riduzione del HDL leggermente/moderatamente inferiore rispetto all’utilizzatore meno accorto, con conseguente alterazione delle ratio HDL:LDL, HDL:Trigliceridi e HDL:Colesterolo totale “rallentata” e meno marcata. Di per se questa pratica supplementativa potrebbe portare anche ad una riduzione anche della capacità di efflusso del colesterolo HDL, ma non vi sono, ad oggi, conferme inoppugnabili che ciò avvenga.
Ah, quasi dimenticavo di ricordare ai meno informati che anche i SARM non steroidei (vedi Ostarina, LGD-4033, ecc…) alterano il profilo lipidico ematico a diverso grado. Anche i SERM (Tamoxifene, Clomifene, Raloxifene ecc…) e AI (Letrozolo, Anastrozolo, Exemestane ecc…) hanno un potenziale di alterazione della lipidemia ematica.
Gabriel Bellizzi
Riferimenti:
T. R. Dawber, F. E. Moore, and G. V. Mann. Measuring the risk of coronary heart disease in adult population groups: Ii. coronary heart disease in the framingham study. American Journal of Public Health and the Nations Health, 47(4 Pt 2):4, 1957
Ference, Brian A., et al. “Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel.” European heart journal 38.32 (2017): 2459-2472.
Gordon, David J., et al. “High-density lipoprotein cholesterol and cardiovascular disease. Four prospective American studies.” Circulation 79.1 (1989): 8-15.
Rader, Daniel J., and G. Kees Hovingh. “HDL and cardiovascular disease.” The Lancet 384.9943 (2014): 618-625.
Keene, Daniel, et al. “Effect on cardiovascular risk of high density lipoprotein targeted drug treatments niacin, fibrates, and CETP inhibitors: meta-analysis of randomised controlled trials including 117 411 patients.” Bmj 349 (2014).
Schandelmaier, Stefan, et al. “Niacin for primary and secondary prevention of cardiovascular events.” Cochrane Database of Systematic Reviews 6 (2017).
Friedl, Karl E., et al. “High-density lipoprotein cholesterol is not decreased if an aromatizable androgen is administered.” Metabolism 39.1 (1990): 69-74.
Thompson, Paul D., et al. “Contrasting effects of testosterone and stanozolol on serum lipoprotein levels.” Jama 261.8 (1989): 1165-1168.
Zmuda, Joseph M., et al. “The effect of testosterone aromatization on high-density lipoprotein cholesterol level and postheparin lipolytic activity.” Metabolism 42.4 (1993): 446-450.
Herbst, Karen L., et al. “Testosterone administration to men increases hepatic lipase activity and decreases HDL and LDL size in 3 wk.” American Journal of Physiology-Endocrinology and Metabolism 284.6 (2003): E1112-E1118.
Singh, Atam B., et al. “The effects of varying doses of T on insulin sensitivity, plasma lipids, apolipoproteins, and C-reactive protein in healthy young men.” The Journal of Clinical Endocrinology & Metabolism 87.1 (2002): 136-143.
Jin, Weijun, Dawn Marchadier, and Daniel J. Rader. “Lipases and HDL metabolism.” Trends in Endocrinology & Metabolism 13.4 (2002): 174-178.
Hartgens, F., et al. “Effects of androgenic-anabolic steroids on apolipoproteins and lipoprotein (a).” British journal of sports medicine 38.3 (2004): 253-259.
Ouimet, Mireille, Tessa J. Barrett, and Edward A. Fisher. “HDL and reverse cholesterol transport: Basic mechanisms and their roles in vascular health and disease.” Circulation research 124.10 (2019): 1505-1518.
Du, Xian-Ming, et al. “HDL particle size is a critical determinant of ABCA1-mediated macrophage cellular cholesterol export.” Circulation research 116.7 (2015): 1133-1142.
Adorni, Maria Pia, et al. “The roles of different pathways in the release of cholesterol from macrophages.” Journal of lipid research 48.11 (2007): 2453-2462.
Rubinow, Katya B., et al. “Testosterone replacement in hypogonadal men alters the HDL proteome but not HDL cholesterol efflux capacity.” Journal of lipid research 53.7 (2012): 1376-1383.
Rubinow, Katya B., et al. “Sex steroids mediate discrete effects on HDL cholesterol efflux capacity and particle concentration in healthy men.” Journal of clinical lipidology 12.4 (2018): 1072-1082.
de Souza, Francis Ribeiro, et al. “Diminished cholesterol efflux mediated by HDL and coronary artery disease in young male anabolic androgenic steroid users.” Atherosclerosis 283 (2019): 100-105.
Attualmente gli unici trattamenti approvati per l’ipogonadismo o la carenza di Testosterone sono la Terapia Sostitutiva con Testosterone (TRT) e la terapia con gonadotropina corionica umana (hCG). Tra le due, la TRT è sicuramente quella più comunemente prescritta. Uno dei motivi è rappresentato dal fatto che l’hCG è inefficace nell’ipogonadismo primario, un tipo di ipogonadismo in cui la causa è l’insufficienza testicolare. Questo esclude circa il 15% dei casi di carenza di Testosterone [1]. Altri motivi possono essere il fatto che l’hCG richiede iniezioni frequenti (di solito tre volte alla settimana) ed è più costoso di alcune alternative alla TRT.
Un problema centrale della TRT non affiancata dall’uso regolare di hCG (o abbinamento dell’hCG con l’hMG) è che sopprime la spermatogenesi e quindi porta all’infertilità in un numero considerevole di uomini. Inoltre, le dimensioni dei testicoli diminuiscono. Per gli uomini che desiderano preservare la fertilità e le dimensioni dei testicoli, la TRT in modalità priva di hCG è ovviamente un candidato non ideale. Sebbene questo aspetto sia meno importante per gli uomini più anziani che possono beneficiare della TRT, in quanto è meno probabile che abbiano in programma di avere figli, è un problema importante per gli uomini giovani che desiderano trattare l’ipogonadismo.
Come discusso nel mio precedente articolo sulla fertilità durante l’uso di AAS o in TRT, il Testosterone sopprime la secrezione di LH e FSH, con conseguente inibizione della spermatogenesi. Parte di questa soppressione è mediata dalla conversione del Testosterone in Estradiolo. Si potrebbero quindi aumentare i livelli di Testosterone annullando l’effetto soppressivo dell’Estradiolo sull’ipotalamo e, conseguentemente, sull’ipofisi. In effetti, l’uso di modulatori selettivi dei recettori degli estrogeni (SERM) – che esercitano un’azione antagonista sui recettori degli estrogeni nell’ipotalamo e nell’ipofisi – porta a un forte aumento di LH, FSH e Testosterone negli uomini con ipogonadismo secondario [2]. Allo stesso modo, l’uso di inibitori dell’Aromatasi – che impediscono al Testosterone di essere convertito in Estradiolo dall’azione dell’Enzima Aromatasi – porta a un aumento di LH, FSH e Testosterone negli uomini trattati [3]. Una conseguenza di ciò è che la spermatogenesi può essere preservata sebbene l’uso di AI sia maggiormente deleterio per il profilo lipidico ematico.
Enclomifene nell’Ipogonadismo Secondario:
Per inserire tra le opzioni terapeutiche per il trattamento dell’ipogonadismo secondario l’Enclomifene, un’azienda farmaceutica, la Repros Therapeutics Inc. ha tentato di farlo approvare dalla FDA. Prima di continuare a parlare di come si è svolto il processo, vorrei fornire alcune informazioni sul SERM in questione: l’Enclomifene Citrato (nome commerciale Androxal, successivamente ribattezzato EnCyzix).
Negli anni ’60 è stato scoperto che un farmaco chiamato clomifene citrato induce l’ovulazione. In quanto tale, poteva essere utilizzato come modalità di trattamento per promuovere la fertilità in caso di anovulazione o oligovulazione. Già all’epoca si sapeva che il clomifene agisce aumentando il rilascio di gonadotropine (LH e FSH) [4]. Per questo motivo, i ricercatori hanno iniziato a valutarne l’effetto anche negli uomini sulla spermatogenesi e sul testosterone. Nei decenni successivi, numerosi studi hanno dimostrato la sua efficacia nello stimolare la produzione di testosterone negli uomini ipogonadici. Tuttavia, il clomifene non è stato approvato dalla FDA per il trattamento dell’ipogonadismo. Tuttavia, viene prescritto off-label per questa indicazione e la linea guida 2018 per la valutazione e la gestione della carenza di testosterone dell’American Urological Association ne sostiene condizionatamente l’uso come alternativa alla TRT [5].
Un problema legato al trattamento con Clomifene è che, nonostante il significativo aumento dei livelli di Testosterone, i dati sul suo effetto sulla riduzione dei sintomi dell’ipogonadismo sono contrastanti [6]. Studi su larga scala e di buona qualità potrebbero chiarire questi aspetti e forse fare luce su quali pazienti potrebbero trarre i maggiori benefici dal suo utilizzo. Poiché il brevetto del farmaco è scaduto da tempo e vengono prodotti farmaci generici, le aziende farmaceutiche non sono molto attratte dagli investimenti. Pertanto, questi studi potrebbero non venir mai realizzati.
Come accade per molti altri farmaci, anche il Clomifene è una miscela racemica. Ciò significa che è costituito da una molecola di tipo “levogiro” e una di tipo “destrogiro”. In genere solo uno di questi stereoisomeri, come vengono chiamati, è il composto attivo. E ciò dà come risultato che si adatta meglio al recettore su cui agisce. Come un guanto si adatta solo a una mano e non all’altra, il tipo “levogiro” è più efficace nel legarsi a un recettore “levogiro” rispetto allo stereoisomero “destrogiro”. Il Clomifene è costituito dagli stereoisomeri Zuclomifene (nell’immagine sotto a sinistra) e, come alcuni di voi già sapranno, l’Enclomifene (nell’immagine sotto a destra):
Da sinistra: Zuclomifene e Enclomifene.
In generale, lo Zuclomifene è considerato un agonista del recettore degli estrogeni, mentre l’Enclomifene è considerato un potente antagonista degli estrogeni [7]. L’Enclomifene può quindi essere considerato lo stereoisomero attivo del Clomifene. L’idea dell’Enclomifene privo dello stereoisomero Zuclomifene, quindi, è quella di avere qualcosa di più efficace e sicuro del Clomifene. Tuttavia, l’aspetto più importante è che Repros Therapeutics Inc. potrebbe brevettarne l’uso terapeutico per il trattamento dell’ipogonadismo maschile.
Per richiedere l’approvazione della FDA, l’azienda farmaceutica ha dovuto condurre alcuni studi clinici. Il primo studio pubblicato comprendeva solo 12 uomini e non era in cieco [8]. In altre parole, sia i partecipanti che i ricercatori sapevano quale trattamento stavano ricevendo gli uomini. I partecipanti erano uomini con ipogonadismo secondario trattati in precedenza con Testosterone topico. Sono stati randomizzati a ricevere nuovamente Testosterone topico o Enclomifene (25mg al giorno).
Dopo sei mesi di trattamento, i livelli di Testosterone erano praticamente gli stessi tra i gruppi: 545ng/dL (18,9nmol/L) nel gruppo che riceveva il gel e 525ng/dL (18,2nmol/L) nel gruppo che riceveva l’Enclomifene. Anche i livelli di Testosterone libero sono aumentati e sono rimasti praticamente invariati tra i gruppi. Inoltre, e naturalmente, il numero di spermatozoi è stato ridotto negli uomini che ricevevano Testosterone, con numeri intorno ai 20milioni/mL. Inoltre, come previsto, il numero di spermatozoi è aumentato negli uomini che hanno ricevuto l’Enclomifene, con una media di circa 150milioni/mL.
Sono stati condotti un paio di studi clinici successivi. Forse il più interessante è stato quello pubblicato nel 2016, rivolto a uomini ipogonadici obesi [9]. Il documento comprende due studi paralleli randomizzati, in doppio cieco, a doppio braccio e controllati con placebo. Si tratta di un’affermazione che lascia a bocca aperta e credo che il termine “doppio cieco” richieda qualche spiegazione. Nel precedente studio di cui mi sono occupato, ho detto che era di natura non cieca. Quindi i partecipanti e i ricercatori sapevano quale trattamento stava ricevendo ciascun soggetto. Di solito, quando si confrontano due farmaci diversi, si possono semplicemente mettere in cieco i soggetti (e i ricercatori) dando ai gruppi capsule, o pastiglie, o altro identici. Tuttavia, il gel di Testosterone è un gel, mentre l’Enclomifene è una compressa da inghiottire. Quindi non è possibile farlo. Per poter effettuare uno studio come questo in cieco, è necessario somministrare a entrambi i gruppi sia le pastiglie che il gel. Quindi un gruppo riceve un gel placebo e l’Enclomifene, mentre l’altro gruppo riceve un gel di Testosterone e una compressa placebo. Ovvero, doppio braccio. (E poiché lo studio era controllato con placebo, un gruppo ha ricevuto un gel e una compressa placebo).
I due studi descritti in questo articolo hanno utilizzato lo stesso protocollo e l’aspetto forse più interessante è stata la dimensione del campione: 256 soggetti in totale! Finalmente si è capito qualcosa. L’intervento è durato 16 settimane e i soggetti del gruppo Enclomifene hanno ricevuto 12,5mg al giorno e sono stati trattati fino a 25mg al giorno se i livelli di Testosterone non erano aumentati ad almeno 450ng/dL (15,6nmol/L) alla quarta settimana. La dose è stata aumentata per la metà dei soggetti che ricevevano l’Enclomifene. A questo punto le cose iniziano a farsi interessanti: sebbene metà dei soggetti sia stata modificata nel dosaggio alla quarta settimana, non è successo assolutamente nulla con la concentrazione media di Testosterone:
E, in effetti, alla fine dell’intervento, la media del gruppo era appena al di sotto del valore limite di 450ng/dL (15,6nmol/L) per l’up-titration. Infine, 29 degli 85 uomini del gruppo Enclomifene non hanno visto il loro Testosterone aumentare al di sopra del valore limite di ipogonadismo di 300ng/dL (10,4nmol/L) dopo 16 settimane di trattamento. Inoltre, i ricercatori hanno fatto un LAVORO ORRIBILE nel trattare correttamente il gruppo che utilizzava il gel di Testosterone, come si può vedere dalla concentrazione media di Testosterone di quel gruppo. Quasi come se l’avessero fatto apposta per far sì che il gruppo Enclomifene facesse meglio in alcune misurazioni… (anche se si tratta di uno studio a doppio braccio, è comunque possibile istruire i pazienti in modo scorretto con l’applicazione del gel).
È importante notare che gli unici endpoint erano i livelli di Testosterone, LH e FSH e la concentrazione di sperma. Non sono stati analizzati endpoint clinicamente rilevanti, come il desiderio sessuale, la funzione erettile, la stanchezza/vitalità, ecc. A quanto pare, nemmeno negli altri studi (pubblicati). O, forse, sono stati analizzati, ma semplicemente non sono stati riportati nei risultati dello studio perché erano deludenti. E penso che potrebbe essere stata la seconda ipotesi, visto che la FDA non ha approvato il farmaco per il trattamento dell’ipogonadismo secondario, a causa della mancanza di un miglioramento sintomatico misurabile [10]. Anche l’equivalente della FDA nell’UE, l’EMA, ha rifiutato l’autorizzazione all’immissione in commercio dell’Enclomifene qualche tempo dopo, con preoccupazioni simili:
“Il CHMP [Comitato per i Medicinali per Uso Umano] ha osservato che, sebbene gli studi abbiano mostrato un aumento dei livelli di Testosterone con EnCyzix [Enclomifene], non hanno esaminato se EnCyzix migliorasse sintomi quali la densità e resistenza ossea, l’aumento di peso, l’impotenza e la libido. Inoltre, il farmaco comporta un rischio di tromboembolismo venoso (problemi dovuti alla formazione di coaguli di sangue nelle vene)”.
E il Clomifene mostra in realtà risultati molto simili, anche mg per mg, a quelli dell’Enclomifene. Non riassumerò qui l’intera letteratura sul Clomifene, ma prendiamo ad esempio uno studio di Katz et al. in cui 86 giovani uomini ipogonadici hanno ricevuto il Clomifene Citrato a 25mg o 50mg a giorni alterni per una media di 19 mesi e hanno visto aumentare il Testosterone totale del 152% (da 192 ng/dL a 485 ng/dL) [11]. In particolare, il Testosterone libero è aumentato di ben il 332%. Se consideriamo un altro studio condotto su uomini obesi, il Testosterone è aumentato del 98% (da 303ng/dL a 599ng/dL) con 25mg al giorno [12]. In termini di aumento del Testosterone, l’Enclomifene non sembra avere un vantaggio rispetto al Clomifene (non sono riuscito a trovare uno studio di confronto testa a testa).
Conclusioni:
Quindi, per concludere, purtroppo non esiste ancora un’alternativa approvata dalla FDA oltre all’hCG o alla TRT per il trattamento dell’ipogonadismo. E con ciò, gli uomini ipogonadici che cercano un trattamento saranno vincolati alle iniezioni di hCG ( e spesso anche hMG) se desiderano preservare la fertilità durante la TRT. Forse i SERM (attuali) sono solo un vicolo cieco, poiché il loro antagonismo con gli estrogeni contrasta anche gli effetti positivi. Infatti, come hanno dimostrato elegantemente Finkelstein et al., l’aggiunta di un inibitore dell’Aromatasi a un gel di Testosterone ha un impatto negativo sul grasso corporeo e sulla funzione sessuale [13]. Avrebbero dovuto inserire anche l’aumento della neurotossicità e cardiotossicità da carenza di Estradiolo, oltre a stati depressivi e condizioni annesse.
Gabriel Bellizzi
Riferimenti:
Tajar, Abdelouahid, et al. “Characteristics of secondary, primary, and compensated hypogonadism in aging men: evidence from the European Male Ageing Study.” The Journal of Clinical Endocrinology & Metabolism 95.4 (2010): 1810-1818.
Wheeler, Karen M., et al. “Clomiphene citrate for the treatment of hypogonadism.” Sexual medicine reviews 7.2 (2019): 272-276.
De Ronde, Willem, and Frank H. de Jong. “Aromatase inhibitors in men: effects and therapeutic options.” Reproductive Biology and Endocrinology 9.1 (2011): 1-7.
Jungck, Edwin C., et al. “Effect of clomiphene citrate on spermatogenesis in the human: a preliminary report.” Obstetrical & Gynecological Survey 19.3 (1964): 520.
Mulhall, John P., et al. “Evaluation and management of testosterone deficiency: AUA guideline.” The Journal of urology 200.2 (2018): 423-432.
Scovell, Jason M., and Mohit Khera. “Testosterone replacement therapy versus clomiphene citrate in the young hypogonadal male.” European urology focus 4.3 (2018): 321-323.
Fontenot, Gregory K., Ronald D. Wiehle, and Joseph S. Podolski. “Differential effects of isomers of clomiphene citrate on reproductive tissues in male mice.” BJU Int 117.2 (2016): 344-50.
Kaminetsky, Jed, et al. “Oral enclomiphene citrate stimulates the endogenous production of testosterone and sperm counts in men with low testosterone: comparison with testosterone gel.” The journal of sexual medicine 10.6 (2013): 1628-1635.
Kim, Edward D., Andrew McCullough, and Jed Kaminetsky. “Oral enclomiphene citrate raises testosterone and preserves sperm counts in obese hypogonadal men, unlike topical testosterone: restoration instead of replacement.” BJU international 117.4 (2016): 677-685.
Earl, Joshua A., and Edward D. Kim. “Enclomiphene citrate: A treatment that maintains fertility in men with secondary hypogonadism.” Expert review of endocrinology & metabolism 14.3 (2019): 157-165.
Katz, Darren J., et al. “Outcomes of clomiphene citrate treatment in young hypogonadal men.” BJU International-British Journal of Urology 110.4 (2012): 573.
Pelusi, Carla, et al. “Clomiphene citrate effect in obese men with low serum testosterone treated with metformin due to dysmetabolic disorders: a randomized, double-blind, placebo-controlled study.” PLoS One 12.9 (2017): e0183369.
Finkelstein, Joel S., et al. “Gonadal steroids and body composition, strength, and sexual function in men.” New England Journal of Medicine 369.11 (2013): 1011-1022.