Ovviamente, l’aumento della produzione intracellulare di MGF non è l’unica, ma certamente una nuova e importantissima via attraverso la quale gli AAS promuovono “attivamente” la crescita muscolare. Secondo uno studio del 2013 del Dipartimento di Riabilitazione e Medicina Fisica della Graduate School of Medical and Dental Sciences dell’Università di Kagoshima in Giappone (Ikeda. 2013) [1], gli agenti anabolizzanti come il Methenolone, che, come sappiamo, è un AAS presente in natura e classificato dalla WADA, con moderate proprietà androgene, presentano questa caratteristica.
Crescita muscolare indotta dalla tensione meccanica:
Per i roditori dello studio in questione, gli scienziati hanno settato il dosaggio somministrato a circa 10mg/kg per esemplare. Successivamente, i muscoli gastrocnemio destro sono stati allungati (sotto tensione) ripetutamente mediante dorsiflessione manuale della caviglia 15 volte al minuto per 15 minuti. I muscoli controlaterali non sono stati allungati come controllo. Nei ratti di controllo (n=6), il gastrocnemio è stato allungato come nel gruppo di trattamento, ma non è stato somministrato Methenolone. Ventiquattro ore dopo la procedura, i ratti sono stati soppressi mediante iniezione di una dose letale di Pentobarbital di sodio e i loro muscoli gastrocnemici mediali sono stati rimossi da entrambi i lati. In realtà, per lo scopo dell’analisi non sarebbe stato necessario sopprimere gli animali, poiché l’estrazione della ” variante autocrina specifica dello splicing IGF-I del fattore di crescita meccanica” è qualcosa che si può misurare da una biopsia muscolare. Quindi, l’unico argomento contro uno studio sull’uomo è probabilmente il dosaggio (alto) e la somministrazione generale di AAS a soggetti umani.
Effetti del trattamento su MGF, MyoD, Miogenina (a.u.) in ratti con/senza iniezione di Methenolone (Ikeda. 2013)
Con gli effetti altamente significativi sul MGF e quelli non significativi sulla MyoD e sulla Miogenina, entrambe coinvolte nel reclutamento di nuovi nuclei muscolari dal pool di cellule staminali (cellule satelliti) nella muscolatura, il risultato dello studio è ancora di natura generica e quasi certamente si applicherà anche all’uomo.
Fare stretching per la crescita muscolare?
Non è l’atto dello stretching, ma piuttosto l’usura delle cellule che viene interpretata come un lavoro intenso a indurre la crescita muscolare e soprattutto l’adattamento strutturale: in altre parole, uno “allungamento durante un sollevamento” (vedi Tensione Meccanica). Gli effetti a valle di queste reazioni intracrine (=confinate all’interno della cellula stessa) vanno ben oltre il semplice pompaggio di più proteine nella struttura muscolare esistente. In uno studio precedente, Ikeda et al. hanno già dimostrato che lo stretching continuo o ripetitivo di breve durata dei muscoli per 1 settimana aumenta i livelli di espressione dell’mRNA di MyoD, Miogenina e MyHC embrionale rispetto a quelli dei muscoli non sottoposti a tele procedura. (Ikeda. 2003)[2]
Sezione di una fibra muscolare scheletrica di mammifero – mionucleo (turchese), mitocondri (blu), rettilo sarcoplasmatico (marrone), tubuli (arancione), miofibrille (rosato) Artista: Lesley Skeates. Originariamente da Gray’s Anatomy 29a ed. Elsevier. 2008
Queste ultime sono i marcatori della spesso osannata attivazione, reclutamento e rifornimento delle “cellule staminali” o “satelliti” del muscolo che sono ciò che permette di crescere oltre il limite naturale, un limite che rende il muscolo degli animali Miostatina-deficienti enorme, ma disfunzionale – un risultato diretto dei cambiamenti strutturali che non sono in grado di tenere il passo con il costante afflusso di proteine. Poiché gli effetti dell’MGF sono correlati agli importanti effetti di facilitazione della forza dell’esercizio fisico e la causa di fondo dei cambiamenti è una semplice tensione della muscolatura, i risultati pongono un’ulteriore enfasi sulla necessità di “stressare” adeguatamente il tessuto muscolare per realizzare gli adattamenti epigenetici indotti dall’esercizio fisico.
Qual è allora il risultato esatto dello studio?
Questo studio non fa altro che confermare (a buon grado di riscontro umano) il complesso ruolo degli AAS nella crescita del muscolo scheletrico. Una dimostrazione in più di come l’assetto ormonale (anabolizzante) sia coadiuvante e cooperativo con IGF-1, MGF, hGH, Miostatina e AAS come attori di spicco con i loro ruoli specifici nell’ipetrofia del muscolo scheletrico.
Fonte immagine:
La Figura qui sopra, offre un’anticipazione di ciò di cui sto parlando.
Nonostante lo studio sia stato ricavato da una ricerca sui roditori con un agente anabolizzante “leggero”, non c’è dubbio che i risultati dello studio in questione siano rilevanti anche per gli atleti enhanced e ci sono buone prove che ciò si si possa applicare anche agli atleti di sesso femminile.
Gabriel Bellizzi [CEO BioGenTech]
Riferimenti:
Ikeda S et al. The Effect of Anabolic Steroid Administration on Passive Stretching-Induced Expression of Mechano-Growth Factor in Skeletal Muscle. The Scientific World Journal. 2013: Article ID 313605.
Ikeda S, Yoshida A, Matayoshi S, Tanaka N. Repetitive stretch induces c-fos and myogenin mRNA within several hours in skeletal muscle removed from rats. Arch Phys Med Rehabil. 2003 Mar;84(3):419-23.
Nella prima parte abbiamo discusso del impatto sull’ipertrofia muscolare ormone-correlata dato dal numero, densità e sensibilità dei Recettori degli Androgeni [AR] espressi in modo variabile secondo caratteristiche genetiche individuali. In questa seconda ed ultima parte tratteremo della mutazione del gene della Miostatina e del suo impatto nella suddivisione tra “High” e “Low” gainers/responders.
Introduzione alla mutazione del gene della Miostatina:
Un altro fattore da considerare sarebbe quello della Miostatina e sulla mutazione del suo gene regolatore.
Il gene della Miostatina (MSTN) è un gene che fornisce le istruzioni per la produzione della proteina Miostatina.
La Miostatina regola la crescita del muscolo scheletrico limitandola quando necessario. A sua volta, impedisce all’organismo di aumentare troppo la massa muscolare anche attraverso la regolazione del catabolismo muscolare.
La ricerca attuale che circonda la Miostatina si basa sul suo trattamento di controllo per le malattie degenerative del sistema muscolo-scheletrico.
Per coincidenza, gli animali che presentano mutazioni nel gene codificante MSTN mostrano una maggiore massa muscolare, forza e, in alcune circostanze, anche una riduzione del grasso corporeo.
Esempi di carenze di miostatina si trovano in modelli di roditori da esperimenti e nell’industria zootecnica con bovini carenti di Miostatina, come già accennato nel precedente articolo.
I topi privi del gene per la sintesi della Miostatina hanno una massa muscolare circa doppia rispetto ai topi normali [1].
Confronto tra topi wild-type e F66/Mstn-/- [mutazione del gene della Miostatina].
Gli inibitori della Miostatina sono stati proposti da molti come la più promettente nuova area scientifica nel contesto del bodybuilding, nonché come un trattamento alternativo potenzialmente migliore per le malattie da deterioramento muscolare.
Gli esemplari di Belgian Blu presentano una mutazione del gene della Miostatina, che impedisce il corretto funzionamento del ciclo di feedback di inibizione della crescita muscolare.
Questa mutazione interferisce con il deposito di grasso e può portare a un’accelerazione della crescita muscolare magra.
L’accelerazione della crescita muscolare nei Belgian Blues è dovuta principalmente ai cambiamenti fisiologici delle cellule muscolari (fibre) dell’animale, che passano da una modalità di crescita ipertrofica a una iperplasica.
Questa crescita avviene nel feto e fa sì che un vitello nasca con un numero di fibre muscolari due volte superiore a quello di un vitello senza mutazione del gene della Miostatina [2].
(A) Analisi di sequenziamento dei tipi di mutazione biallelica MSTN nei vitelli clonati. I tipi di mutazione biallelica MSTN consistevano in una delezione di 6 bp in un allele (gli ultimi 4 bp dell’esone 1 e i primi 2 bp dell’introne 1) e in una delezione di 117 bp (posizioni nucleotidiche 8-124 nell’introne 1) e un’inserzione di 9 bp (gli ultimi 2 bp dell’esone 1 e i primi 7 bp dell’introne 1, AG GCACGGG) nell’altro allele, che erano coerenti con la colonia 6. Le lettere rosse rappresentano l’esone 1 di MSTN e le lettere blu rappresentano l’introne 1 di MSTN. (B) I vitelli con mutazioni bialleliche di MSTN mostravano il fenotipo doppio muscoloso e non presentavano effetti negativi. Nei cerchi rossi, la massa muscolare del vitello mutante MSTN (a sinistra) era maggiore di quella del vitello wild-type (a destra). (C) Sezioni trasversali del muscolo quadricipite colorate con ematossilina ed eosina. Le fibre muscolari dei vitelli con mutazioni bialleliche di MSTN (a sinistra) erano ipertrofiche, rispetto a quelle dei vitelli wild-type (a destra). Tutti gli animali avevano un mese di età alla data del prelievo dei campioni di tessuto.
Il paradosso dell’aumento della Miostatina in risposta agli Androgeni: Anche se probabilmente esistono altri meccanismi di controregolazione nell’organismo che inibiscono la crescita muscolare eccessiva, il fattore principale sembra essere l’aumento della Miostatina. La Miostatina aumenta per impedire l’aumento di massa muscolare non salutare.
In uno studio sono stati valutati gli effetti del Testosterone e del Trenbolone esogeni sui livelli di Miostatina [3]. Questo studio ha dimostrato che dopo 29 giorni di somministrazione di Testosterone o Trenbolone, i livelli di proteina Miostatina erano più alti del 197% nel gruppo castrato e Testosterone e del 209% nel gruppo castrato e Trenbolone rispetto al placebo.
C’è un motivo per cui questo meccanismo è presente nell’organismo umano e non è possibile crescere in modo lineare. I meccanismi omeostatici del corpo cercheranno sempre di ristabilire l’equilibrio, la dove in grado. Quindi, come già detto, la Miostatina è un inibitore della crescita che aumenta in presenza di androgeni in misura dose-dipendente.
In base alle ricerche attuali, sembra che quanto più alta è la dose di anabolizzanti esogeni, tanto maggiore è il potenziale di crescita muscolare e, di conseguenza, tanto più alta sarà la Miostatina per inibire tassi spropositati di crescita muscolare.
In uno studio che ha valutato l’effetto di dosi graduate di Testosterone sui livelli di Miostatina in uomini giovani e anziani, i livelli di Miostatina erano significativamente più alti al giorno 56 rispetto al basale in entrambi i gruppi [4].
E’ singolare constatare che l’aumento di Miostatina si manifesti a grado significativo dopo 29 giorni di somministrazione cronica di AAS. In effetti, inizialmente la risposta è inversa, cioè inibitoria.
L’ipotesi della Miostatina non è scientificamente teorizzabile al momento. Essa presenta alcune lacune nei dati che contraddicono i suoi effetti di inibizione della crescita muscolare.
Tuttavia, sulla base di ciò che sappiamo finora, la ricerca suggerisce che è più che probabile che sia il principale meccanismo di regolazione coinvolto nella risposta alla crescita muscolare rispetto all’attivazione del Recettore degli Androgeni. È infatti noto che la Miostatina regola negativamente la massa muscolare nei topi, nei bovini, nei cani e nell’uomo [5].
Mutazioni del gene della Miostatina e influenza sui progressi nel bodybuilding:
E’ stato condotto un piccolo studio per scoprire se le mutazioni dello SNP rs1805086 hanno un impatto sulla popolazione maschile che pratica il bodybuilding dal punto di vista dell’ipertrofia muscolare e delle prestazioni muscolari [6].
L’obiettivo secondario era quello di ipotizzare se le mutazioni rare siano più diffuse in coloro che decidono di scegliere uno sport come il bodybuilding, dal momento che la ricerca indica che le mutazioni del MSTN possono indurre un maggiore aumento della massa muscolare e una riduzione del grasso corporeo.
Il polimorfismo Lys(K)153Arg(R) nell’esone 2 (rs1805086, sostituzione 2379 A>G) del gene della Miostatina (MSTN) è candidato a influenzare i fenotipi del muscolo scheletrico ed è elencato su SNPedia come il genotipo a maggior rischio di causare l’ipertrofia muscolare legata alla Miostatina [7, 8].
Il 17% del gruppo di soggetti aveva una mutazione (AG), l’83% aveva l’esito comune (AA) e lo 0% (0) aveva due mutazioni (GG).
I soggetti con genotipo AG avevano una circonferenza media del braccio di 46,37 cm rispetto agli AA che avevano una media di 42,02 cm.
I soggetti con il genotipo AG avevano un punteggio medio di pull-up max di 21, rispetto agli AA che avevano una media di 12.
I soggetti con genotipo AG avevano una media di flessioni massime pari a 61 rispetto agli AA che avevano una media di 40.
Lo studio mostra chiaramente che i soggetti con una mutazione sono rari, tuttavia la mutazione sembra dare al soggetto un vantaggio in termini di prestazioni e di dimensioni rispetto a quelli con il risultato comune.
Un altro studio ha ottenuto risultati simili valutando i polimorfismi A55T e K153R [9].
I ricercatori di questo ultimo studio hanno affermato che i loro risultati indicano che gli individui con genotipo AT + TT del polimorfismo A55T hanno mostrato un aumento significativo dello spessore dei bicipiti (0,292 ± 0,210 cm, P = 0,03), ma non dei quadricipiti (0,254 ± 0,198 cm, P = 0,07), rispetto ai portatori del genotipo AA.
Per il polimorfismo K153R, gli aumenti degli spessori sia del bicipite (0,300 ± 0,131 cm) che del quadricipite (0,421 ± 0,281 cm) erano significativamente più elevati tra gli individui con genotipo KR rispetto a quelli con genotipo KK (P < 0,01 per entrambi i muscoli).
I risultati ottenuti suggeriscono quindi una possibile associazione tra i due polimorfismi e l’ipertrofia muscolare indotta dall’allenamento di forza tra gli uomini di etnia cinese Han.
Il polimorfismo K153R è lo stesso polimorfismo Lys(K)153Arg(R) nell’esone 2 (rs1805086, sostituzione 2379 A>G) del gene della Miostatina (MSTN) valutato nel primo studio citato.
Fotografie di un bambino con mutazione del gene della Miostatina all’età di sei giorni e sette mesi (pannello A), ecografie (pannello B) e analisi morfometriche (pannello C) dei muscoli del paziente e di un neonato di controllo e pedigree del paziente (pannello D). Le punte di freccia nel pannello A indicano i muscoli sporgenti della coscia e del polpaccio del paziente. Nel pannello B, una sezione trasversale ultrasonografica (trasduttore lineare, 10 MHz) attraverso la parte centrale della coscia rivela le differenze tra il paziente e un neonato di controllo della stessa età, sesso e peso. VL indica il vasto laterale, VI il vasto intermedio, VM il vasto mediale, RF il retto femorale e F il femore. Nel pannello C, i ritracciamenti dei contorni dei muscoli e i risultati dell’analisi morfometrica dei piani delle sezioni muscolari dei due neonati rivelano differenze marcate. Il pannello D mostra il pedigree del paziente. I simboli solidi indicano i membri della famiglia che sono eccezionalmente forti, secondo le informazioni della loro storia clinica. I simboli quadrati indicano i membri della famiglia di sesso maschile e i cerchi quelli di sesso femminile.
Gli SNP influenzano l’ipertrofia muscolare correlata alla Miostatina:
Secondo SNPedia, questi 3 SNP sono sicuramente correlati all’ipertrofia muscolare legata alla Miostatina:
L’SNP rs1805086, in particolare, è quello più comunemente esaminato in relazione ai risultati del bodybuilding.
Viene spesso citato nelle discussioni sul “gene del bodybuilder”.
Il genotipo AA dello SNP rs1805086 è considerato quello comunemente presente, mentre gli alleli di rischio sono il genotipo GG dello SNP rs1805086.
La malattia letteralmente elencata come esito potenziale del possesso di questo genotipo di rischio è l’ipertrofia muscolare legata alla Miostatina.
Avere un solo allele G è raro, ed essere omozigoti per esso è molto raro.
E’ stato ipotizzato che Flex Wheeler avesse probabilmente il genotipo GG più raro per l’SNP rs1805086.
Victor Conte, Flex Wheeler e la “sua mutazione”:
Si presume che Flex Wheeler abbia partecipato a uno studio condotto in collaborazione con il dipartimento di genetica umana dell’Università di Pittsburgh, che ha coinvolto 62 uomini.
Durante questo studio, Flex avrebbe scoperto di avere una mutazione molto rara della Miostatina nella posizione dell’esone 2 del gene.
Flex Wheeler
In teoria, questa presunta mutazione genetica impediva al suo organismo di produrre quantità normali di Miostatina, determinando di conseguenza un numero di fibre muscolari molto più elevato rispetto agli uomini nella media.
Gli animali e gli esseri umani con livelli di Miostatina inibiti hanno costantemente dimostrato di avere livelli di muscolatura molto più elevati rispetto alle loro controparti non inibite, e sulla base di ciò non è assurdo supporre che i mostri di genetica nel bodybuilding abbiano sviluppato il loro fisico come risultato anche di una mutazione genetica simile.
In teoria, chi ha bassi livelli di Miostatina potrebbe continuare a progredire a ritmi che sarebbero impossibili per chi ha livelli normali del peptide.
Il risultato finale di livelli cronicamente bassi di Miostatina potrebbe essere un aumento muscolare sostanzialmente maggiore a parità di variabili.
Victor Conte è una delle persone associate allo studio sulla mutazione della Miostatina condotto su Flex Wheeler e su una serie di altri bodybuilder professionisti IFBB.
Il 99% di coloro che nella comunità del bodybuilding discutono della carenza di Miostatina di Flex fanno riferimento a una lettera scritta nell’ottobre 1998 da Victor Conte.
Non è chiaro se questa lettera sia legittima e inalterata, ma per quanto possa valere, la considereremo legittima in quanto è quella che è circolata nella comunità del bodybuilding per anni.
1 ottobre 1998
Oggetto: Flex Wheeler
A chi può interessare:
Scrivo questa lettera su richiesta di Flex Wheeler.
Vorrei innanzitutto fornirvi alcune informazioni di base sui Laboratori BALCO. BALCO lavora con atleti olimpici e professionisti d’élite da oltre quindici anni. BALCO ha fornito test e consulenze a oltre 250 giocatori della NFL, tra cui l’intera squadra dei Denver Broncos, campione del Super Bowl 1998, e l’intera squadra dei Miami Dolphins. BALCO lavora con atleti professionisti in molti sport, tra cui tennis (Michael Chang, Jim Courier, ecc.), hockey, bodybuilding (10 dei 16 concorrenti di Mr. Olympia 1998), atletica leggera, calcio e basket (Seattle SuperSonics).
Nell’ultimo anno i Laboratori BALCO hanno effettuato test e monitoraggi di routine su Flex. Sono stati eseguiti esami come quelli ematochimici (SMAC), emocromo completo (CBC), PSA, livelli di ormoni anabolizzanti, genotipizzazione e analisi complete degli elementi nutrizionali. I risultati dei test di Flex sono stati confrontati con quelli di altri ventiquattro bodybuilder professionisti e nel complesso il suo profilo è tra i più sani. In sostanza, Flex gode di ottima salute e ha dimostrato la disciplina necessaria per mantenere un livello di preparazione ottimale.
Flex ha partecipato a uno studio condotto di recente in collaborazione con il Dipartimento di Genetica Umana dell’Università di Pittsburgh, che ha coinvolto 62 uomini che hanno ottenuto aumenti di massa muscolare insolitamente elevati in risposta all’allenamento della forza (extreme responders). Flex era uno dei soli nove rispondenti estremi che presentavano la rarissima “mutazione della Miostatina”. Il gene della Miostatina regola il peptide che “limita la crescita muscolare”. In particolare, Flex presentava la forma più rara di mutazione della Miostatina nella posizione “esone 2” del gene. Ciò significa semplicemente che Flex ha un numero molto maggiore di fibre muscolari rispetto agli altri soggetti o alla popolazione normale. Riteniamo che questi siano i primi risultati di una mutazione della Miostatina nell’uomo e i risultati di questo studio di riferimento sono già stati presentati per la pubblicazione. In Flex è stato anche riscontrato un tipo di gene IGF-1 molto insolito. Infatti, Flex è stato l’unico partecipante allo studio a non avere una “corrispondenza”. Tutti gli altri rispondenti estremi avevano almeno altri tre soggetti con un gene IGF-1 corrispondente. Sulla base del profilo genetico unico di Flex, abbiamo intenzione di pubblicare rapidamente un documento scientifico che riveli il suo genotipo completo in modo dettagliato. La pubblicazione dei suoi straordinari dati genetici dovrebbe generare un’enorme esposizione mediatica.
Spero che queste informazioni siano utili e vi prego di chiamarmi se posso esservi d’aiuto.
Cordiali saluti,
/Victor Conte
Victor Conte
Presidente
BALCO Laboratories, Inc.
Da sinistra: Flex Wheeler, Victor Conte e Gunter Schlierkamp
Lo studio sulla mutazione della Miostatina condotto su Flex Wheeler e altri professionisti IFBB:
Questo studio è comunemente citato, ma devo ancora venire a conoscenza di qualcuno che lo abbia effettivamente trovato e che abbia incrociato i dati in esso contenuti con le affermazioni fatte nella lettera di Victor Conte.
Ma, facendo qualche ricerca, l’ho trovato.
Lo studio si chiama “frequent sequence variation in the human myostatin (GDF8) gene as a marker for analysis of muscle-related phenotypes” [10].
In base a quanto dichiarato da Victor nella sua lettera, c’erano nove rispondenti estremi con una mutazione molto rara della Miostatina.
Si suppone che Flex Wheeler avesse la mutazione più rara di tutte nella posizione dell’esone 2 del gene, che lo rendeva unico rispetto a tutti gli altri individui dello studio.
Soggetti dello studio:
Il sequenziamento di regioni selezionate del gene della Miostatina e la genotipizzazione di varianti comuni sono stati eseguiti in un campione di confronto di 96 soggetti caucasici e 96 afroamericani selezionati a caso dalla popolazione generale.
Altri 72 individui sono stati sottoposti a screening per la presenza di una variante comune dell’esone 2.
Centocinquantatré soggetti, tra cui 127 uomini (32 afroamericani, 91 caucasici e 4 asiatici) e 26 donne (9 afroamericani, 16 caucasici e 1 asiatico), sono stati classificati in base all’entità dell’aumento della massa muscolare registrato con l’allenamento della forza.
I soggetti erano costituiti da:
18 culturisti di livello mondiale (classificati tra i primi 100 al mondo)
25 culturisti agonisti non classificati tra i primi 100
7 sollevatori di potenza d’élite
9 giocatori di calcio universitari
55 soggetti non allenati in precedenza, ai quali è stato misurato il volume del muscolo quadricipite mediante risonanza magnetica prima e dopo 9 settimane di allenamento di resistenza pesante degli estensori del ginocchio
61 non atleti, che sono stati interrogati sulla loro capacità di aumentare la massa muscolare in risposta a un allenamento di forza intenso e prolungato.
5 dei 18 bodybuilder di livello mondiale erano concorrenti di Mr. Olympia, classificati tra i primi 10 al mondo.
Il punteggio di 5 è stato assegnato a coloro che erano bodybuilder di livello mondiale e a coloro che avevano aumentato la massa muscolare dei quadricipiti di oltre 400 cm³ dopo solo 9 settimane di allenamento della forza, mentre il punteggio di 0 è stato assegnato a coloro che non avevano registrato un aumento notevole della massa muscolare dopo un allenamento della forza vigoroso per almeno 6 mesi.
Diciotto soggetti hanno ricevuto un punteggio di 5, mentre 13 hanno ricevuto un punteggio di 0. I restanti soggetti hanno avuto una valutazione intermedia.
Le valutazioni dei restanti soggetti si collocano tra questi due estremi.
62 soggetti con valutazione 4 o 5 sono stati classificati come responder estremi e sono stati confrontati con 48 soggetti con valutazione 0 o 1, classificati come non responder.
I soggetti sono stati anche raggruppati e confrontati per etnia.
Le informazioni sulle variazioni della massa muscolare con l’allenamento della forza nei restanti soggetti sono state ottenute attraverso le stime della massa priva di grasso valutate con l’assorbimetria a raggi X a doppia energia o l’idrodensitometria oppure, nel caso di bodybuilder agonisti, sollevatori di potenza, giocatori di calcio e non atleti, attraverso i dati del questionario sui precedenti successi nelle competizioni di bodybuilding e/o sulle variazioni della massa muscolare con l’allenamento della forza.
Risultati dello studio:
Senza annoiarvi con i dettagli meno rilevanti dello studio, la parte più rilevante è la conclusione.
La mancanza di una relazione significativa tra i genotipi della Miostatina e la risposta complessiva della massa muscolare all’allenamento della forza suggerisce che la risposta non è influenzata in modo significativo dalla variazione del locus della Miostatina.
Tuttavia, è interessante notare che tre dei non responder afroamericani erano omozigoti per l’allele meno comune (Arg) nel sito K153R dell’esone 2, mentre nessuno dei responder era omozigote per questo allele.
Tre delle cinque mutazioni che causano il fenotipo del muscolo doppio nei bovini si verificano nell’esone 2 e sono recessive, ma due sono mutazioni di terminazione della catena e una è una delezione, che dovrebbe produrre una proteina della Miostatina non funzionale.
Per stabilire se le variazioni nel gene della Miostatina influenzino fenotipi muscolari diversi dall’aumento della massa muscolare in risposta all’allenamento per la forza, sono necessari ulteriori approfondimenti.
L’allele Arg, meno comune, a cui si fa riferimento nelle conclusioni dello studio, è la mutazione che ci si aspetterebbe da Flex Wheeler.
Ma non sembra che ce l’abbia.
A metà dello studio si parla di ciò che potrebbe evidenziare la vera radice della superiorità genetica di Flex.
Tra i sei cambiamenti nucleotidici, due, P198A e l’introne 2 A/G, sono stati osservati in un singolo individuo e due, I225T e E164K, sono stati osservati in due individui, sempre eterozigoti con l’allele wildtype.
Gli altri due erano presenti nella popolazione generale come polimorfismi comuni.
Le varianti (A55T) e (K153R) sono comuni in entrambi i gruppi etnici, con l’allele meno frequente che ha una frequenza da tre a quattro volte superiore negli afroamericani.
Questi siti variabili sono potenzialmente in grado di alterare la funzione del prodotto genico della Miostatina e potrebbero alterare la ripartizione dei nutrienti negli individui eterozigoti o omozigoti per l’allele della variante.
Possiamo presumere che Flex Wheeler abbia due cambiamenti nucleotidici, P198A e l’introne 2 A/G.
Questa è l’unica nota dell’intera pubblicazione che distingue un individuo dello studio dagli altri.
Quelle che possiamo presumere essere le variazioni nucleotidiche di Flex Wheeler non sono nemmeno menzionate nell’elenco di SNPedia dei genotipi a rischio correlati.
L’unico vago riferimento che abbiamo è in uno studio che ha esaminato l’associazione tra le varianti esoniche MSTN e la potenza “esplosiva” delle gambe in 214 studenti universitari maschi [11].
E in quello studio l’unica cosa menzionata è che nessun soggetto dello studio presentava la variante esonica P198A di MSTN.
Sembra che, nonostante l’allele non comune (Arg) nel sito K153R dell’esone 2 sia il fulcro della maggior parte dei lavori sulla miostatina e sia stato considerato la radice del “gene del bodybuilder”, alla fine dei conti non sembra avere un impatto così significativo sulla risposta della crescita muscolare all’allenamento come molti pensavano.
La mancanza di una relazione significativa tra i genotipi della miostatina e la massa muscolare complessiva è molto significativa, dato che questo studio includeva 5 bodybuilder del calibro di Mr. Olympia e diversi altri professionisti IFBB di alto livello.
La cosa più interessante da notare è che tre dei non rispondenti afroamericani erano omozigoti per l’allele meno comune (Arg) nel sito K153R dell’esone 2, mentre nessuno dei rispondenti era omozigote per questo allele.
Tra le variazioni GDF8 identificate nell’uomo, il polimorfismo Lys(K)153Arg(R) nell’esone 2 (rs1805086, sostituzione 2379 A>G) del gene della Miostatina (MSTN) è candidato a influenzare i fenotipi del muscolo scheletrico [12].
Tuttavia, nessuno dei bodybuilder extreme responder era omozigote per questo allele.
Nel primo video ho detto che il genotipo AG in generale è raro.
Nello studio che ho descritto all’inizio dell’articolo, è stato riscontrato un impatto significativo sulle dimensioni e sulla forza muscolare.
A rigor di logica, si potrebbe ipotizzare che il genotipo GG (ancora più raro) comporti una mancanza di miostatina e un livello di crescita muscolare pazzesco.
In base a questo studio, però, non sembra essere così.
3 dei soggetti che hanno avuto una scarsa risposta all’allenamento e una crescita muscolare inferiore (non rispondenti) erano quelli che avevano questo genotipo raro.
Solo tre individui presentavano cambiamenti nucleotidici estremamente rari.
Tra questi c’è colui che presumo sia Flex, che presenta due alterazioni nucleotidiche, P198A e l’introne 2 A/G, e altri due individui con alterazioni nucleotidiche I225T e E164K, tutti eterozigoti con l’allele wildtype.
Ciò lascia due culturisti di alto livello del calibro di Mr. Olympia, diversi altri culturisti professionisti IFBB di livello mondiale e molti altri atleti d’élite con genotipi MSTN che hanno dimostrato di avere un impatto minimo sulla risposta della crescita muscolare all’allenamento in questo studio.
Le altre due variazioni nucleotidiche che causano il doppio fenotipo muscolare nei bovini sono le varianti A55T e K153R e sono presenti nella popolazione generale come polimorfismi comuni.
Questi siti variabili hanno dimostrato di poter alterare la funzione del prodotto genico della miostatina e potrebbero alterare la ripartizione dei nutrienti in individui eterozigoti o omozigoti per l’allele della variante.
Tuttavia, i dati di questo studio dimostrano che non esiste una relazione significativa tra i genotipi della miostatina e la risposta complessiva della massa muscolare all’allenamento della forza.
Inconsistenza dei dati tecnici nella lettera di Victor Conte:
Non si sa da dove provengano le affermazioni contenute nella lettera scritta da Victor.
Egli sostiene che Flex Wheeler aveva la forma più rara di mutazione della Miostatina nell’esone 2 del gene.
Ma se guardiamo lo studio stesso, si legge che 3 dei non responders erano omozigoti.
Nessuno dei responders era omozigote.
Flex Wheeler sarebbe stato senza dubbio classificato come un responder estremo, eppure non era uno degli individui con la variazione GDF8 nell’uomo che ci aspetteremmo di vedere in un individuo carente di Miostatina.
In base a ciò, possiamo presumere che si tratti dell’individuo menzionato nello studio con due variazioni nucleotidiche, P198A e l’introne 2 A/G.
Victor ha anche menzionato come “nove soggetti con risposta estrema presentavano la rarissima mutazione della Miostatina”.
Dai dati si evince che solo tre individui presentavano mutazioni nucleotidiche non comuni, non nove, mentre il resto dei soggetti presentava polimorfismi comuni presenti nella popolazione generale.
Inoltre, tra le mutazioni citate, anche se un numero maggiore di bodybuilder di alto livello presentasse mutazioni degne di nota, la conclusione dello studio afferma comunque che non esiste una relazione significativa tra i genotipi della Miostatina e la risposta complessiva della massa muscolare all’allenamento della forza.
Nella sua lettera, Victor ha anche affermato che Flex è uno dei bodybuilder professionisti più sani tra quelli che ha monitorato e che gode di ottima salute.
Abbiamo eseguito esami che comprendono la chimica del sangue (SMAC), l’emocromo completo (CBC), il PSA, i livelli di ormoni anabolizzanti, la genotipizzazione e un’analisi completa degli elementi nutrizionali.
I risultati dei test di Flex sono stati confrontati con quelli di altri ventiquattro bodybuilder professionisti e nel complesso il suo profilo è tra i più sani.
In sostanza, Flex gode di ottima salute e ha dimostrato la disciplina necessaria per mantenere un livello di preparazione ottimale.
Questo articolo è stato scritto il 1° ottobre 1998.
Se conoscete la storia di Flex Wheeler, saprete che ha dovuto smettere di gareggiare dopo aver scoperto, nel 1999, di essere affetto da glomerulosclerosi focale segmentaria (una forma di malattia renale) e si è ritirato poco dopo.
Non so come una cosa così grave possa essere trascurata a tal punto.
Mi fa dubitare della legittimità di questa lettera.
Se Flex era davvero sull’orlo di un’insufficienza renale, non capisco come sia stato possibile stabilire che era uno dei bodybuilder più sani seguiti da Victor, e come questi test approfonditi non l’abbiano rilevato.
La prima cosa che mi viene in mente è la curiosità di sapere se c’era o meno una qualche forma di guadagno associata a questa vicenda.
Negli anni ’90, l’industria degli integratori era impazzita.
Gli steroidi erano venduti legalmente al banco, e si potevano fare affermazioni ridicole e false su praticamente tutto ciò che si voleva e poi vendere prodotti basati su questo.
Le affermazioni false esistono ancora oggi, ma oggi abbiamo a disposizione le risorse necessarie per capire la spazzatura che ci viene propinata, mentre negli anni ’90 nessuno ne sapeva di più e un integratore che inibisce la miostatina e che può farvi diventare grossi come Flex Wheeler avrebbe probabilmente fatto il botto.
Forse questa ipotesi è molto lontana da quelle che erano le reali intenzioni, ma non capisco quale possa essere stata la motivazione di questa lettera, o quale sia il suo scopo.
È del tutto possibile che stessero pensando di collaborare per creare una sorta di integratore inibitore della Miostatina basato sul genotipo unico di Flex.
Conclusioni:
Non so se l’ulteriore pubblicazione di cui parla Victor nella lettera sia mai stata realizzata.
Sulla base del profilo genetico unico di Flex, abbiamo intenzione di pubblicare rapidamente un articolo scientifico che riveli il suo genotipo completo in modo dettagliato.
La pubblicazione dei suoi notevoli dati genetici dovrebbe generare un’enorme esposizione mediatica.
Presumo che questo progetto sia stato probabilmente accantonato dopo i problemi di salute di Flex verificatisi nel 1999.
Non so quale fosse l’obiettivo di questa lettera e ci sono diverse incongruenze tra la lettera e lo studio vero e proprio che necessitano di ulteriori chiarimenti per poter fare affermazioni conclusive.
A chi era indirizzata questa lettera e perché Flex Wheeler ha chiesto di scriverla?
A parte il mistero di questa lettera, che mi interessa relativamente, sembra che possiamo almeno concludere, sulla base dei risultati dello studio, che la maggior parte delle mutazioni del gene della Miostatina non sembra essere il fattore di differenziazione tra i migliori atleti responder estremi del calibro di Mr. Olympia e persone comuni, o almeno non il solo.
La complessità della biochimica e delle risposte genetiche non interessano quasi mai un solo fattore ma più fattori correlati aventi tra loro influenza diretta e/o indiretta.
Con molta probabilità, sia il fattore di mutazione del gene della Miostatina che il numero, la densità e sensibilità dei AR nel muscolo scheletrico rappresentino due delle maggiori determinanti di separazione tra lo spettro di soggetti che vanno dai rarissimi “No Responders” agli altrettanto rari “Freak”.
Prima che qualcuno di voi cambi sport perchè scoraggiato dalle evidenze, ho da darvi una buona e scontata notizia. Quale? Che tra voi, con molta probabilità, vi siano alcuni convinti di essere dei low responders ma in realtà rientrano nella media. È molto probabile che non stiate migliorando come vorreste, o a causa di aspettative irrealistiche, o perché state facendo alcune cose decisamente controproducenti al miglioramento della condizione ipertrofica muscolare.
Assicuratevi di…
Consumare un surplus calorico adeguato e ben tarato. Quanto meno siete geneticamente portati per la costruzione di muscoli, tanto maggiore sarà la cura della percentuale di macronutrienti (in particolare proteine, ma anche di carboidrati) del surplus necessario.
Cercate di assumere da 1.5 a 2,5 grammi di proteine per chilo di peso corporeo. Questo vi garantirà la quantità di proteine necessaria per avviare i processi ipertrofici al vostro ritmo ottimale. Un consumo eccessivo di proteine, superando queste linee guida, non accelererà la crescita muscolare, a meno che non siate “resistenti all’anabolismo”: in questo caso la quota proteica può aumentare fino a 3g/Kg. Quando si raggiungono i 2,5 g/Kg peso, il puntare sull’aggiunta di carboidrati è più vantaggioso.
Cercate di dormire otto ore di qualità a notte. Potreste arrivare a sette ogni tanto e va bene, ma una media di otto è ottimale. L’assunzione di più carboidrati a fine giornata può aiutare a dormire meglio aumentando il trasporto del Triptofano a livello cerebrale e con esso migliorare la sintesi di Serotonina e Melatonina.
Nella maggior parte dei casi, l’obiettivo principale dell’allenamento dovrebbe essere quello di fare meglio dell’ultima volta. Questo può significare usare più peso almeno in alcuni esercizi [carico progressivo]. Ma può anche significare fare più ripetizioni con lo stesso peso [aumento del volume], fare lo stesso carico e le stesse ripetizioni con meno riposo tra le serie [aumentare la densità] e assicurarsi di eseguire meglio gli esercizi.
Non esagerate con il volume. In caso di dubbio, fate circa 24 serie settimanali per i distretti come petto e schiena e 12 serie per i distretti come bicipiti, tricipiti e spalle.
Non fate più di quanto vi permette il vostro adattamento. Se oltre le quattro sedute settimanali vedete che i recuperi non sono ottimali e iniziano ad emergere problemi di stanchezza cronica e calo della prestazione, concentratevi sul volume di lavoro adatto a voi. In questo modo riuscirete a rendere al massimo delle vostre capacità ad ogni allenamento con miglioramenti tra i mesocicli.
Non pensate che l’uso di PEDs vi risolva i problemi. Dopo la lettura di questi due articoli dovreste aver capito che il farmaco esalta determinati caratteri genetici ma non li cambia. Inoltre, prima di prendere in considerazione un eventuale (ed illegale) uso di PEDs assicuratevi o di essere seguiti da anni da un professionista degno di tale appellativo oppure di essere in possesso delle conoscenze necessarie per gestire nel migliore dei modi i pilastri fondanti del bodybuilding, la dieta e l’allenamento.
E no… L’utilizzo di inibitori della Miostatina non vi renderà immuni dalle limitazioni date da una deficienza del gene MSTN. Al massimo, e torniamo sempre al solito discorso che giova sempre sottolineare, ridurranno l’attività della Miostatina, che è cosa molto variabile e ben diversa dall’avere una mutazione del gene in questione…
Per concludere, non state troppo a cruciarvi sulle vostre limitatezze genetiche, il lamentarsi e il negare lo stato delle cose non cambierà nulla. Piuttosto, sarebbe molto più produttivo agire iniziando ad essere consapevoli di ciò che si è con lo scopo di fare il meglio nei limiti delle proprie possibilità, qualunque esse siano. Certamente, le informazioni che ho esposto in questi due articoli, per coloro in grado di comprenderle, non sono semplicemente finalizzate ad una compressione dei limiti individuali, ma sono poste anche in modo tale da permettere di agire seguendo le scelte logiche migliori per raggiungere gli stessi.
L’ACE-031 può rientrare a pieno titolo nel “club” delle molecole PEDs semisconosciute. Un peptide praticamente unico nel panorama “doped”, sicuramente promettente, specie nel BodyBuilding, ma del quale se ne parla poco.
Nel 2013 sembrava che la ricerca sul ACE-031 fosse stata definitivamente interrotta, nonostante funzionasse piuttosto bene.
Le aziende farmaceutiche Acceleron Pharma e Shire misero in pausa la ricerca sull’inibitore della Miostatina ACE-031 [Acceleronpharma.com 2 maggio 2013]. E questo evento risultò piuttosto strano. In un comunicato stampa congiunto rilasciato qualche tempo dopo il sopra citato annuncio, Muscle & Nerve aveva pubblicato uno studio che dimostrava che l’ACE-031 è un composto che un culturista supplementato farmacologicamente aggiungerebbe volentieri al suo “arsenale”.
L’ACE-031 iniettabile è un recettore sintetico dell’Attivina di Tipo IIB. Anche le cellule muscolari hanno questo recettore. È destinato a proteine come la Miostatina, il GDF11 e l’Attivina A e B. Se la Miostatina si lega al recettore dell’Attivina di Tipo IIB, la crescita delle fibre muscolari si riduce. Nelle circostanze “giuste” la Miostatina arriva addirittura a degradare il muscolo-scheletrico.
Se si somministra l’ACE-031, questo non accade o, comunque, l’effetto viene marcatamente ridotto. Il recettore sintetico dell’Attivina di Tipo IIB si lega con il tristemente noto peptide Miostatina impedendo a quest’ultimo di legarsi al sito recettore della cellula e compiere la sua attività di riduzione ipertrofica e degradazione del tessuto muscolo-scheletrico.
ACE-031 e “recettori esca”:
Come accennato pocanzi, l’ACE-031 non è altro che un “recettore esca”. Un recettore esca è un recettore in grado di riconoscere e legare in modo efficiente specifici fattori di crescita o citochine, ma non è strutturalmente in grado di segnalare o attivare il complesso recettoriale previsto. Agisce come un inibitore, legando un ligando e impedendogli di legarsi al suo recettore abituale. I recettori esca partecipano a un metodo comune di inibizione del segnale e sono anche abbondanti nei tessuti maligni, costituendo un argomento significativo nella ricerca sul cancro.[1]
“Recettori esca”: si legano ai ligandi e inibiscono la segnalazione attraverso i recettori veri e propri.
IL1R2 è stato uno dei primi recettori esca identificati.[2] [3] Lega IL1A e IL1B e inibisce il loro legame con IL1R1, impedendo la risposta infiammatoria che è generalmente promossa dal legame delle interleuchine di tipo 1 con il recettore 1 dell’interleuchina di tipo I.[4]
Un altro membro di questa categoria è il recettore DcR3, conosciuto anche come TNFRSF6, che si trova principalmente nei tessuti maligni umani.[5] Agisce come recettore esca per i membri delle citochine TNF: FasL, LIGHT e TL1A, inibendo la capacità delle citochine di segnalare la morte cellulare o l’apoptosi.
TNFRSF6
Il VEGFR-1 è una tirosin-chinasi recettoriale che modula negativamente l’angiogenesi agendo come recettore esca.[6] La caratteristica di “esca” del VEGFR-1 è necessaria per lo sviluppo e l’angiogenesi normali. Il VEGFR-1 inibisce l’attività del VEGFR-2 sequestrando il VEGF, impedendo così al VEGFR-2 di legarsi al VEGF.
Quindi eccoci di nuovo con ACE-031. Esso è stato studiato in quanto è un recettore esca ingegnerizzato con attività inibitoria della Miostatina potenzialmente utile nel tentativo di trattare i bambini affetti da distrofia muscolare di Duchenne (DMD). Il recettore ACE-031 circola al di fuori della membrana della fibra muscolare. Poiché questo recettore si lega alla Miostatina, riduce la quantità di questo peptide che può legarsi al recettore nativo nella membrana (ActRIIB), impedendo alla Miostatina di fornire il segnale che limita la crescita muscolare e ne promuove il catabolismo.[7]
I principali studi su ACE-031:
Nel 2007 Acceleron Pharma aveva grandi aspettative su ACE-031. All’epoca l’azienda aveva condotto solo studi sugli animali. Tuttavia, nel marzo 2013 AP ha pubblicato uno studio sull’uomo in cui 48 donne sane di età compresa tra 45 e 75 anni hanno ricevuto una singola iniezione con 0.02, 0.05, 0.1, 0.3, 1 o 3 mg di ACE-031 per kg di peso corporeo. Il composto ha circolato per alcune settimane nell’organismo dei soggetti trattati. L’emivita è stata stimata essere di 10-15 giorni. Tuttavia, questa singola iniezione ha prodotto una crescita muscolare. La dose di 3mg/kg ha mostrato un aumento del volume muscolare del 5%. La massa magra è aumentata del 3% [poco più di un chilo] e sembra anche diminuire la massa grassa.
L’iniezione ha ridotto la Leptina e aumentato la concentrazione di Adiponectina. Ciò suggerisce che l’ACE-031 riduce la massa grassa.
Inoltre, è aumentato l’inibitore della Miostatina, i livelli di fosfatasi alcalina specifica per le ossa [BSAP] nel sangue e si è ridotto quello del telopeptide C-terminale del collagene di tipo 1 [CTX]. Ciò suggerisce che l’ACE-031 rende le ossa più forti. Negli studi sugli animali con RAP-031, la versione per topi di ACE-031, Acceleron è riuscita a dimostrare questi effetti. [Endocrinology. 2010 Sep; 151 (9) :4289-300].
Se si legge lo studio su Muscle & Nerve, ci si chiede perché mai la Acceleron abbia interrotto lo sviluppo di ACE-031. E perché non agisce legalmente contro tutti gli store online che si puliscono le terga con i brevetti di Acceleron e vendono l’ACE-031 a un prezzo al quale una normale azienda farmaceutica non può trarre alcun profitto.[Muscle Nerve. 2013 Mar; 47 (3) :416-23.]
La risposta si trova in un messaggio sul sito web dell’Associazione per la Distrofia Muscolare. [Quest.mda.org 2 maggio 2013] In esso si legge che nel 2011, durante uno studio [NCT01099761] in cui i ricercatori somministravano l’ACE 031 a bambini affetti da malattie muscolari, sono emersi effetti collaterali che hanno costretto i ricercatori a interrompere lo studio.
“Gli eventi avversi che i partecipanti alla sperimentazione hanno subito – piccoli sanguinamenti del naso e delle gengive e dilatazione dei vasi sanguigni della pelle – non sono stati considerati di per sé pericolosi. Tuttavia, le aziende e le agenzie regolatorie coinvolte affermano di aver bisogno di comprendere appieno questi eventi prima di continuare gli studi clinici sull’ACE-031”. “
Un altro strano effetto collaterale è stato rivelato nello studio pubblicato su Muscle & Nerve. È emerso che la somministrazione di ACE-031 abbia ridotto fortemente la concentrazione di FSH nelle donne partecipanti. I ricercatori non ne conoscono la causa e le possibili conseguenze.
Sembrava che ACE-031 fosse stato definitivamente accantonato dalla ricerca fino alla pubblicazione nel 2017 di uno studio sul recettore esca , sempre su Muscle Nerve [Myostatin inhibitor ACE-031 treatment of ambulatory boys with Duchenne muscular dystrophy: Results of a randomized, placebo-controlled clinical trial]. L’ACE-031 è stato somministrato per via sottocutanea ogni 2-4 settimane a ragazzi affetti da DMD [distrofia muscolare di Duchenne] in uno studio randomizzato, in doppio cieco, controllato con placebo, a dose crescente. L’obiettivo primario era la valutazione della sicurezza. Gli obiettivi secondari comprendevano la caratterizzazione della farmacocinetica e della farmacodinamica.
L’ACE-031, durante lo studio, non è stato associato a eventi avversi gravi o molto gravi. Lo studio è stato interrotto dopo il secondo regime di dosaggio a causa di potenziali problemi di sicurezza legati a epistassi e teleangectasie. È stata rilevata una tendenza al mantenimento della distanza del test del cammino di 6 minuti (6MWT) nei gruppi ACE-031 rispetto al calo osservato nel gruppo placebo (non statisticamente significativo), nonché una tendenza all’aumento della massa magra e della densità minerale ossea (BMD) e alla riduzione della massa grassa.
Anche in questo studio, l’uso dell’ACE-031 ha dimostrato tendenze per gli effetti farmacodinamici sulla massa magra, sulla massa grassa, sulla BMD e sul 6MWT (6-minute walk test). Ma, come successo in precedenza, gli eventi avversi non correlati ai muscoli hanno contribuito alla decisione di interrompere lo studio. Nonostante l’inibizione della Miostatina è un approccio terapeutico promettente per la DMD.
Neanche lo studio su MYO-029, il miostatinblokker della Wyeth, ha avuto successo. Nel 2008 uno studio deludente ha dimostrato che gli adulti con distrofia muscolare, dopo la somministrazione di MYO-029, non sono diventati più forti. [Ann Neurol. 2008 May, 63 (5) :561-71] e la Wyeth ha interrotto lo sviluppo del MYO-029.
Uso nel BodyBuilding e conclusioni:
Ora sappiamo che questo “recettore esca” può favorire lo sviluppo del muscolo-scheletrico legandosi alla Miostatina ed impedendo a questa di esercitare la sua azione di controllo e catabolismo muscolare. Sappiamo inoltre che gli studi effettuati su esseri umani sono stati promettenti ma non sufficientemente sicuri da permetterne uno sviluppo completo. I casi di epistassi e teleangectasie hanno spinto i ricercatori ad interrompere la ricerca. Ma come spesso accade, ogni qualvolta nel panorama scientifico si affaccia una molecola potenzialmente vantaggiosa per lo sportivo, e per il BodyBuilder in particolare, anche se la ricerca si interrompe non si può dire lo stesso per quella svolta illegalmente da improvvisate cavie umane. E questo evento si è verificato anche per l’ACE-031.
Partendo dalle prove emerse durante gli studi, sappiamo che una dose di 3mg/Kg ha comportato un aumento del volume muscolare del 5%, un aumento della massa muscolare del 3% e sembra portare anche a una riduzione della massa grassa. La molecola sembra ridurre la concentrazione di Leptina, condizione che potrebbe portare ad uno scompenso nella regolazione fame/sazietà, ed un aumento dell’Adiponectina, la quale è correlata ad un miglioramento della sensibilità all’Insulina.
Prove sul campo raccolte negli ultimi anni, hanno permesso di quantificare i dosaggi mediamente efficaci per un Bodybuilder e i tempi di somministrazione: 1-3mg per chilogrammo di peso corporeo ogni 15 giorni è risultato essere il range standard per ottenere i migliori risultati possibili. Per quanto concerne la lunghezza del trattamento, si presume che l’uso debba essere circoscritto in un arco temporale di circa 5-6 settimane, limite di conservazione che non dovrebbe essere superato.
Ricordo che il principale effetto collaterale di ACE-031 è la dilatazione dei vasi sanguigni. Tuttavia, questo effetto collaterale, se contenuto, non sembra avere svantaggi. Inoltre, l’uso di ACE-031 può causare epistassi e gengive sanguinanti. Non sono noti altri effetti collaterali. I soggetti emofiliaci sono a forte rischio emorragico potenziale con l’uso di ACE-031.
Anche se dovrebbe essere scontato, ribadisco il fatto che nessuno sta invitando all’uso sperimentale ed illegale di una molecola della quale, oltretutto, si sa poco. Le informazioni ivi presenti sono a puro scopo divulgativo e non rappresentano in alcun modo prescrizioni mediche e affini.
Gabriel Bellizzi
Riferimenti:
Decoy Receptor”. Encyclopedia of Cancer. Springer Berlin Heidelberg. 2012. p. 1070.
La capacità di riacquisire la condizione della massa muscolare precedente a un periodo di deallenamento o inattività fisica è noto come “memoria muscolare”. Quindi, se un soggetto ha avuto una condizione muscolare ottimale (vedi muscoli più ipertrofici) in passato, ciò lo aiuterà a riportarli nuovamente nelle precedenti condizioni una volta ripreso un regolare stimolo allenante. Il concetto di memoria muscolare si basa in buona parte su qualcosa chiamato permanenza mio-nucleare. Il ‘mio’ in ‘mionucleare’ si riferisce al ‘muscolo’ e il ‘nucleare’ si riferisce alla parola ‘nucleo’: un organello della cellula. Prima di esplorare ulteriormente il concetto di memoria muscolare, e come gli AAS si leghino a questo, cerchiamo prima di rispolverare un po’ di concetti utili sui nuclei muscolari o mionuclei.
Informazioni di base sui nuclei muscolari/mionuclei:
Le cellule muscolo-scheletriche sono le singole cellule contrattili all’interno di un muscolo e sono spesso definite fibre muscolari.[1] Un singolo muscolo come il bicipite in un giovane individuo di sesso maschile adulto contiene circa 253.000 fibre muscolari.[2]
Sezione 3D di una fibra del muscolo-scheletrico
Le fibre muscolo-scheletriche sono le uniche cellule muscolari multinucleate con i nuclei spesso indicati come mionuclei . Ciò si verifica durante la miogenesi con la fusione di mioblasti, ciascuno dei quali contribuisce a un nucleo.[3] La fusione dipende da proteine muscolo-specifiche note come fusogeni chiamate myomaker e myomerger .[4]
Molti nuclei sono necessari alla cellula muscolo-scheletrica per le grandi quantità di proteine ed enzimi necessari per essere prodotti per il normale funzionamento della cellula. Una singola fibra muscolare può contenere da centinaia a migliaia di nuclei.[5] Una fibra muscolare ad esempio nel bicipite umano con una lunghezza di 10cm può avere fino a 3000 nuclei.[5] A differenza di una cellula non muscolare in cui il nucleo è posizionato centralmente, il mionucleo è allungato e si trova vicino al sarcolemma . I mionuclei sono disposti in modo abbastanza uniforme lungo la fibra con ciascun nucleo che ha il proprio dominio mionucleare dove è responsabile del supporto del volume del citoplasma in quella particolare sezione della miofibra.[4,5]
Un gruppo di cellule staminali muscolari conosciute come cellule miosatelliti, anche cellule satelliti che si trovano tra la membrana basale e il sarcolemma delle fibre muscolari, sono normalmente quiescenti ma possono essere attivate dall’esercizio o anche condizioni patologiche per fornire mionuclei aggiuntivi per la crescita o la riparazione muscolare.[6]
Detto più semplicemente, i muscoli sono costituiti da un insieme di fibre muscolari. Ogni fibra muscolare, o cellula muscolare, contiene più nuclei, l’organello di una cellula che contiene il DNA ed è il luogo dove avviene il processo di trascrizione dei geni. La maggior parte degli altri tipi di cellule umane contiene solo un nucleo, o in alcuni casi addirittura nessun nucleo (globuli rossi/Eritrociti). Per dare un’idea di quanti nuclei si stia parlando: le fibre muscolari di ratto contengono da 44 a 116 nuclei per millimetro di lunghezza della fibra, con le fibre muscolari di tipo 1 che contengono più nuclei per millimetro delle fibre muscolari di tipo 2.[7] Il numero sembra più basso negli esseri umani, come riportato da un ricercatore il quale segnala la presenza di circa 30 nuclei per millimetro di lunghezza della fibra nel muscolo del bicipite brachiale.[8] Come tali, le fibre muscolari possono contenere migliaia di mionuclei, dato che possono estendersi per diversi centimetri di lunghezza.
Poiché i nuclei cellulari delle fibre muscolari non sono in grado di dividersi (cioè sono differenziati terminalmente), le fibre muscolari dipendono dalle cellule satelliti circostanti per l’aggiunta di nuovi nuclei. Essenzialmente, le cellule satelliti sono cellule staminali delle fibre muscolari che si trovano schiacciate tra il sarcolemma (la membrana cellulare di una fibra muscolare) e la lamina basale (uno strato di matrice extracellulare che è avvolto intorno al sarcolemma). Sono stati scoperti e descritti per la prima volta da Alexander Mauro nella letteratura scientifica nel 1961.[9] Usando un microscopio elettronico, egli vide delle cellule “incastrate” tra il sarcolemma delle fibre muscolari di rana e la lamina basale. Le descrisse aventi una scarsità di citoplasma, con il nucleo che costituisce quasi l’intero volume della cellula satellite. Ha continuato a speculare sull’origine e sul ruolo delle cellule satelliti, toccando brevemente l’idea che potrebbero essere coinvolte nella risposta al trauma inflitto a una fibra muscolare. Cosa che, in effetti, sono.[10]
L’ipotesi del dominio mionucleare e la permanenza mionucleare
La scoperta delle cellule satelliti e il loro ruolo nella rigenerazione muscolare fanno sorgere la domanda sulla misura in cui le cellule satelliti sono coinvolte nell’ipertrofia. Un’ipotesi chiamata “ipotesi del dominio mionucleare” si è agganciata a questo quesito. Essa postula che un mionucleo controlla una quantità limitata di citoplasma, e quindi, affinché la crescita muscolare abbia luogo, i mionuclei devono essere aggiunti alla fibra muscolare per sostenerla. Tre osservazioni chiave hanno sostenuto questa ipotesi, vale a dire:
L’esposizione alle radiazioni γ rende le cellule satellite incapaci di dividersi e inibisce fortemente l’ipertrofia da sovraccarico nei modelli animali, mantenendo intatto il metabolismo cellulare o la sintesi proteica [11].
I prodotti (organelli, membrane e proteine strutturali) derivati da un nucleo rimangono localizzati nelle sue vicinanze [12].
Il rapporto citoplasma/mionucleo rimane abbastanza costante [13].
Questo implicherebbe un aumento del numero di mionuclei con la crescita di una fibra muscolare (ipertrofia), mentre diminuirebbe con una perdita di dimensioni della stessa (atrofia). Tuttavia, vari studi su animali suggeriscono che i mionuclei non si perdono durante l’atrofia.[14] Così è nato il paradigma della permanenza mionucleare: una volta che i mionuclei sono guadagnati con l’ipertrofia, non vengono persi di nuovo con il deallenamento. Questo potrebbe potenzialmente permettere alle fibre muscolari di ricrescere in modo più efficiente durante il successivo riallenamento e quindi servire come un meccanismo di “memoria muscolare”.
Il concetto di memoria muscolare basato sulla permanenza mionucleare illustrato da Bruusgaard et al.
AAS e permanenza mionucleare:
E gli AAS? Ciò che è chiaro è che l’uso di AAS aumenta il numero di mionuclei. Dosaggi crescenti di Testosterone Enantato portano ad un aumento del numero di mionuclei per mm di fibra muscolare.[15] Questo effetto non è poi così sorprendente: si osserva semplicemente questo effetto con praticamente tutte le modalità di induzione ipertrofica.
Ma che dire della loro permanenza? Questi mionuclei permangono una volta che la massa muscolare diminuisce di nuovo? In un esperimento su animali, da me già riportato anni fa, topi femmina sono stati trattati con Testosterone Propionato per 2 settimane, che ha portato a un aumento del 66% del numero di mionuclei e un aumento del 77% della fibra muscolare CSA [16]. La massa muscolare è tornata alla normalità dopo la successiva interruzione della somministrazione di Testosterone, ma il numero di mionuclei è rimasto elevato per almeno 3 mesi. 3 mesi potrebbe non sembrare molto, ma sulla scala temporale di un topo lo sono: i topi che hanno usato per lo studio vivono per circa 2 anni. Comunque, dopo questi 3 mesi, quando i topi sono stati sottoposti a sovraccarico per induzione ipertrofica, la CSA delle fibre muscolari è aumentata del 30% dopo 6 giorni, mentre quella dei topi di controllo non è aumentata significativamente. Dopo questo, la massa muscolare è aumentata in parallelo tra entrambi i gruppi, ma la CSA era ancora più alta del 20% nel gruppo che era stato precedentemente trattato con Testosterone dopo 14 giorni. Anche se questo non prova un nesso causale tra il numero più alto di mionuclei e l’ipertrofia, è comunque un’osservazione interessante.
Si noti come il gruppo che è stato trattato con Testosterone per 2 settimane, circa 3 mesi prima ha mostrato un forte aumento della massa muscolare rapidamente ottenuto in risposta al sovraccarico.
E negli esseri umani? Due studi hanno valutato questo e sono stati portati all’attenzione da Alexander Kolliari-Turner, uno studente con dottorato di ricerca presso la School of Sport and Health Sciences of the University of Brighton nel Regno Unito. Una è una tesi di master e l’altra è una tesi di dottorato.
Nella tesi di dottorato di Anders Eriksson [17], sono stati reclutati quattro gruppi di soggetti. Un gruppo di soggetti sedentari che fungeva da controllo (gruppo C), un gruppo di PowerLifter natural (gruppo P), un gruppo di powerlifter che usano AAS (gruppo PAS), e un gruppo di PowerLifter che hanno precedentemente usato AAS (gruppo PREV). I mionuclei per fibra muscolare sono stati determinati nei muscoli vasto laterale e trapezio. Il gruppo PREV aveva interrotto l’uso di AAS da almeno un anno (con una media di 8 anni). Infatti, l’area delle fibre muscolari misurata nel gruppo PREV era paragonabile a quella del gruppo P, e notevolmente più piccola di quella del gruppo PAS.
La distribuzione del dominio nucleare (nr. di nuclei per fibra diviso per l’area della fibra) per gruppo si trova nell’immagine qui sotto. Se ci fosse una permanenza dei mioonuclei, ci si aspetterebbe un dominio nucleare più piccolo, cioè più nuclei rispetto all’area delle fibre, nel gruppo PREV rispetto agli altri gruppi.
Chiaramente questo non è il caso del vasto laterale, ma è il caso del trapezio. È difficile dire cosa causa questa apparente discrepanza tra i due muscoli. O qualche proprietà che differisce tra i due muscoli, o il suo modo di utilizzo dopo la cessazione dell’uso di AAS, forse ha portato a apparente permanenza mionucleare nel muscolo trapezio.
Va notato, tuttavia, che questo era uno studio trasversale con un piccolo numero di soggetti (32 in totale). L’ideale sarebbe avere uno studio prospettico che valuti questo, anche se ciò è estremamente difficile su lunghi periodi di tempo, in quanto potrebbe richiedere almeno un anno o più prima che i cambiamenti diventino evidenti. In alternativa, anche uno studio trasversale con un gruppo di soggetti più grande sarebbe piuttosto interessante. Indipendentemente da ciò, questo presta una certa credibilità alla permanenza dei mionuclei negli esseri umani come risultato dell’uso di steroidi anabolizzanti in muscoli selezionati.
In una tesi di laurea di Lindholm et al. sono stati reclutati tre gruppi di soggetti: attuali consumatori di AAS (gruppo CAS), ex consumatori di AAS (gruppo FAS) e controllo allenati alla resistenza (gruppo CON) [18]. Gli ex consumatori di AAS avevano smesso di usarli per una media di 6,5 anni. In questo studio, sono state prese solo biopsie del muscolo vasto laterale. In particolare, non c’erano differenze significative nella CSA delle fibre muscolari tra i tre gruppi. Questo è senza dubbio il risultato delle dimensioni relativamente piccole del gruppo (34 soggetti in totale; un errore di tipo 2).
Una piccola, ma significativa, differenza nel dominio mio-nucleare è stata trovata tra le fibre muscolari di tipo 2 del gruppo FAS rispetto al gruppo CON, come si può vedere nella figura sottostante:
Questo suggerisce una permanenza mionucleare? Forse. La differenza era piccola e può essere facilmente spiegata anche dalla natura trasversale dello studio (e non c’era alcuna differenza rispetto agli attuali utilizzatori di AAS).
Le prove finora sono scarse. In ogni caso, quando si guarda alla permanenza mionucleare in generale, l’evidenza generale indica che questa regge a breve termine, ma mancano prove per il lungo termine [19]. Inoltre, non è chiaro se la permanenza mionucleare possa aiutare o meno il ritorno alla condizione muscolo-scheletrica precedente. E visti i dati di cui sopra, il dibattito sul fatto che l’uso di AAS porti o meno alla manifestazione della memoria muscolare come risultato della permanenza mionucleare, è tutt’altro che risolto.
Conclusione:
Come osservazione conclusiva: c’è anche un concetto di memoria muscolare basato su qualcosa di diverso dalla permanenza mionucleare, vale a dire, la memoria epigenetica.[20] In breve, questa si riferisce a modifiche apportate al DNA senza influenzare la sua sequenza nucleotidica, quindi senza cambiare il codice genetico. Ciò comporta l’aggiunta (o la rimozione) di gruppi metilici ai nucleotidi di Citosina e Adenina o modifiche degli istoni (ad esempio, metilazione o acetilazione di residui di aminoacidi delle proteine istoniche). Il risultato di ciò è che influisce sull’espressione genica. Questo potrebbe forse essere trattato in un futuro articolo, dato che più ricerche vengono gradualmente pubblicate su questa nuova ed interessante strada ipotetica.
A proposito di “memoria epigenetica”: questa figura illustra un modello di sviluppo della persistenza batterica basato sulla presenza di un potenziale effetto di “memoria” epigenetica che include l’eredità stabile di certi modelli di metilazione del DNA. Lo stato di metilazione del DNA cellulare potrebbe portare alla conservazione di alcuni profili di espressione genica che favoriscono la dormienza, conservati in alcune cellule dopo il risveglio dalla dormienza. Cinetica di uccisione bifasica adattata da. (A) Popolazione originale di cellule metabolicamente attive che potrebbero contenere un’intrinseca eterogeneità fenotipica. (B) Quando incontra lo stress, la maggior parte delle cellule metabolicamente attive muore, mentre una piccola frazione di cellule entra nello stato di persistenza. La popolazione di persister può essere in qualche modo eterogenea, cioè formata da diversi percorsi (stocastico contro specifico). (C) Dopo gli stimoli nutrizionali/la rimozione dello stress, alcuni persister si risvegliano. Qui, la maggior parte dei persister inizia rapidamente la crescita e si divide in cellule regolari e metabolicamente attive. Tuttavia, alcune cellule potrebbero sperimentare un effetto di “memoria” epigenetica. Qui, lo stato di metilazione del DNA di alcuni siti che si trovano a monte di regioni codificanti regolate per esprimere tratti che favoriscono la dormienza potrebbe essere mantenuto dopo la replicazione del DNA. (D) A livello di popolazione totale, la popolazione finale dopo il risveglio potrebbe essere ugualmente suscettibile allo stress come la popolazione originale in (A). Tuttavia, a livello di singola cellula, alcune cellule potrebbero contenere un effetto di “memoria” legato alla dormienza, basato sull’eredità di alcuni tratti epigenetici dipendenti dalla metilazione del DNA. (E) L’esistenza di un effetto di “memoria” epigenetica potrebbe potenzialmente aumentare la frequenza dei persister nel tempo durante ripetuti cicli di stress.
Klein, CS; Marsh, GD; Petrella, RJ; Rice, CL (July 2003). “Muscle fiber number in the biceps brachii muscle of young and old men”. Muscle & Nerve. 28 (1): 62–8.
Tseng, Brian S., Christine E. Kasper, and V. Reggie Edgerton. “Cytoplasm-to-myonucleus ratios and succinate dehydrogenase activities in adult rat slow and fast muscle fibers.” Cell and tissue research 275.1 (1994): 39-49.
Schmalbruch H. Skeletal Muscle. Berlin: Springer-Verlag; 1985.
Mauro, Alexander. “Satellite cell of skeletal muscle fibers.” The Journal of Cell Biology 9.2 (1961): 493-495.
Forcina, Laura, et al. “An overview about the biology of skeletal muscle satellite cells.” Current genomics 20.1 (2019): 24-37.
Rosenblatt, J. David, David Yong, and David J. Parry. “Satellite cell activity is required for hypertrophy of overloaded adult rat muscle.” Muscle & nerve 17.6 (1994): 608-613.
Pavlath, Grace K., et al. “Localization of muscle gene products in nuclear domains.” Nature 337.6207 (1989): 570-573.
Allen, David L., Roland R. Roy, and V. Reggie Edgerton. “Myonuclear domains in muscle adaptation and disease.” Muscle & nerve 22.10 (1999): 1350-1360.
Gundersen, Kristian, and Jo C. Bruusgaard. “Nuclear domains during muscle atrophy: nuclei lost or paradigm lost?.” The Journal of physiology 586.11 (2008): 2675-2681.
Sinha-Hikim, Indrani, et al. “Testosterone-induced muscle hypertrophy is associated with an increase in satellite cell number in healthy, young men.” American Journal of Physiology-Endocrinology and Metabolism 285.1 (2003): E197-E205.
Egner, Ingrid M., et al. “A cellular memory mechanism aids overload hypertrophy in muscle long after an episodic exposure to anabolic steroids.” The Journal of physiology 591.24 (2013): 6221-6230.
Eriksson, Anders. Strength training and anabolic steroids: a comparative study of the trapezius, a shoulder muscle and the vastus lateralis, a thigh muscle, of strength trained athletes. PhD Diss. 2006.
Lindholm, Jesper Bøgh, et al. Effects of Long-Term Supplementation of Androgen Anabolic Steroids on Human Skeletal Muscle – Evidence for Muscle Memory? Master’s Thesis, 2019.
Snijders, Tim, et al. “The concept of skeletal muscle memory: Evidence from animal and human studies.” Acta Physiologica 229.3 (2020): e13465.
Seaborne, Robert A., et al. “Human skeletal muscle possesses an epigenetic memory of hypertrophy.” Scientific reports 8.1 (2018): 1-17.
Verso la fine del primo decennio del presente secolo, una “presunta” nuova classe di farmaci con attività anabolizzante ha iniziato a diffondersi in diverse discipline sportive , dal ciclismo a, ovviamente, il Bodybuilding. Sto parlando ovviamente dei SARMs, acronimo di Selective Androgen Receptor Modulators (in italiano, Modulatori Selettivi del Recettore degli Androgeni, SARM).
Essendo molecole sperimentali e non ancora commercializzate come farmaci da prescrizione per uso umano, i SARM si sono diffusi rapidamente in tutto il mondo grazie anche alla vendita da parte degli store online UK e USA (dove la vendita di supplementi contenenti tali molecole è legale).
Non ci volle molto tempo prima che un “alone leggendario” avvolgesse i SARM ed i loro presunti o reali effetti. I SARM vennero in breve pubblicizzati come il “doping ideale” con tutti gli effetti positivi degli steroidi anabolizzanti, pur non avendo alcun svantaggio o effetto collaterale legato a questi ultimi.
In generale, gli effetti positivi principali degli AAS sono considerati essere l’effetto anabolizzante sulla massa muscolare e l’effetto stimolante sul miglioramento della densità minerale ossea. Tutti gli altri effetti cosi detti androgeni sono generalmente considerati indesiderati. Anche se, ovviamente, ciò dipende in gran parte dal grado con il quale essi si verificano (ma anche dal sesso e dalla disciplina praticata dall’utilizzatore).
Ad esempio, gli AAS inducono l’Eritropoiesi, il processo di biosintesi degli Eritrociti (globuli rossi). Questo porta ad un aumento dell’Ematocrito che, quando diventa troppo alto, ossia oltre la soglia del 53-54%, vede arrestati i suoi effetti benefici sulla resistenza vedendo aumentato sensibilmente il rischio di trombosi venosa. Tuttavia, se si eliminasse completamente qualsiasi effetto stimolante sull’eritropoiesi, l’ematocrito potrebbe diventare troppo basso, in specie se viene a mancare un fattore compensativo alla riduzione indotta. Di conseguenza, si finirebbe per essere anemici. Quindi anche alcuni di quegli effetti indesiderati degli AAS sono “voluti” in una certa misura. Ma i paradossi della selettività non terminano con questo, ovviamente. Per semplicità, tuttavia, tratterò il discorso più avanti nel presente articolo.
Il punto della questione è: i SARM danno veramente un vantaggio in quanto a rapporto tra effetti positivi e collaterali rispetto agli AAS? La risposta richiede una spiegazione dettagliata della storia, delle caratteristiche e degli effetti, constatati sia in ambito clinico che “off-label”, legati ai SARM.
Nozioni iniziali sui SARM.
Come la maggior parte di voi saprà, SARM sono una classe di ligandi selettivi del recettore degli androgeni (AR).[1]
Nonostante un certo numero di persone sia convinta che i SARM siano stati sintetizzati circa venti anni fa, e che non abbiano nulla a che vedere nel loro sviluppo con gli AAS, la realtà è che il termine si riferisce ad un macrogruppo di molecole affini al AR con un valore terapeutico (vedi potenziale androgeno e anabolizzante) superiore a 1, cioè al Testosterone. Per questa ragione esistono due gruppi di SARM: i SARM steroidei ed i SARM non-steroidei. Di conseguenza, tutti i derivati del Testosterone, del DHT, compresi i 19-Norsteroidi, che sono stati modificati strutturalmente al fine di accentuarne le caratteristiche anabolizzanti e ridurne quelle androgene sono considerabili quali SARM steroidei.
Due esempi tipici di SARM steroideo e non-steroideo
Gli sforzi iniziali per sviluppare SARM steroidei, basati su modifiche della molecola di Testosterone, risalgono agli anni ’40. L’era moderna dei SARM non steroidei è stata scatenata da un lavoro indipendente presso la Ligand Pharmaceuticals (2, 3) e l’Università del Tennessee.(4, 5) Gli scienziati della Ligand Pharmaceuticals sono stati i primi a sviluppare una serie di Chinolinoni ciclici con attività anabolica sul muscolo scheletrico e un certo grado di selettività tissutale.(2, 6, 7, 8) La scoperta di Dalton e Miller che le Aril Propionammidi con somiglianze strutturali con il Bicalutamide e l’Idrossiflutammide potrebbero innescare l’attività trascrizionale AR-dipendente ha fornito la prima guida per lo sviluppo della classe di SARM diaril propionammidi.(4, 5) Il decennio successivo a questi primi sforzi ha visto l’emergere di un gran numero di SARM non steroidei praticamente da tutte le principali aziende farmaceutiche.(9)
Fondamenti logici nella ricerca dei SARM non-steroidei
Il Testosterone, il principale ligando per il Recettore degli Androgeni, svolge una varietà di funzioni fisiologiche nell’uomo (10): è essenziale, anche per via della sua conversione in DHT, al fine di mantenere una corretta funzione sessuale, lo sviluppo delle cellule germinali e gli organi sessuali accessori. Il Testosterone interagisce ovviamente anche con il muscolo scheletrico, grasso, ossa, emopoiesi, coagulazione, metabolismo dei lipidi, proteine e carboidrati e comportamenti psicosessuali e cognitivi. Sebbene la carenza di androgeni negli uomini adulti sia il disturbo più diffuso della alterazione nella segnalazione AR (11), il principale impulso per lo sviluppo dei SARM è legato allo sfruttamento dei potenziali effetti anabolici di questi composti sul muscolo scheletrico e sull’osso.
Come ben sappiamo, man mano che uomini e donne invecchiano, perdono massa muscolare scheletrica, forza, potenza (12, 13), principalmente a causa della perdita preferenziale delle fibre muscolari di tipo 2 (14), e la densità ossea. La perdita di massa muscolare e forza associata all’età aumenta il rischio di cadute, fratture, limitazione della mobilità, disabilità fisica e scarsa qualità della vita (15, 16). Il declino funzionale e la dipendenza negli anziani gravano pesantemente sui servizi e sui costi sanitari. Nonostante l’elevata prevalenza di limitazioni funzionali e disabilità tra gli individui più anziani, i geriatri praticanti hanno poche scelte terapeutiche per il trattamento degli individui più anziani con limitazioni funzionali e disabilità fisica. Allo stesso modo, il decorso di molte malattie croniche, come la malattia polmonare ostruttiva cronica, la malattia renale allo stadio terminale, l’insufficienza cardiaca congestizia e alcuni tipi di cancro, è punteggiato da perdita di massa muscolare e limitazioni funzionali fisiche, che contribuiscono indipendentemente a sintomi, limitazione della mobilità e disabilità. Pertanto, c’è un enorme bisogno insoddisfatto di funzioni che promuovano terapie anabolizzanti che possano migliorare la funzione fisica e ridurre il peso della disabilità.
Tra le varie terapie anabolizzanti candidate ad applicazione in fase di sviluppo, quella con SARM non steroidei è la più recente in corso di sviluppo. La somministrazione di Testosterone aumenta la massa muscolare scheletrica e la massima forza volontaria in uomini sani, con carenza di androgeni (17-18) ed eugonadici (19, 20) e anziani (21), e negli uomini con molti disturbi cronici (22, 23). Gli effetti anabolizzanti del Testosterone sulla massa e sulla forza dei muscoli scheletrici sono correlati alla dose di Testosterone e alle sue concentrazioni ematiche (20, 21, 24, 25). Pertanto, il potenziale per ottenere il rimodellamento del muscolo scheletrico e l’aumento della massa e della forza del muscolo scheletrico con la somministrazione di androgeni è notevole. Tuttavia, la somministrazione di dosi sovrafisiologiche di androgeni è associata ad un’elevata frequenza di effetti avversi dose-dipendenti, come eritrocitosi, edema delle gambe ed eventi prostatici (21, 26). Pertanto, agenti terapeutici come i SARM non steroidei con la cui somministrazione possono far ottenere effetti anabolizzanti sul muscolo scheletrico e sull’osso senza gli effetti avversi limitanti riscontrati con dosaggi di Testosterone aventi il medesimo effetto terapeutico sarebbero attraenti come terapie anabolizzanti d’elezione (27, 28, 29). Il riconoscimento di queste potenziali opportunità per lo sviluppo di nuove terapie per le limitazioni funzionali e disabilità associate a disturbi cronici, invecchiamento e osteoporosi ha guidato gli sforzi farmaceutici per sviluppare SARM non steroidei.
Il raggiungimento della selettività dei tessuti
Storicamente sono stati utilizzati due approcci generali per ottenere la selettività tissutale dell’azione degli Androgeni. Il primo approccio consiste nello sviluppare un SARM con un profilo di attività desiderato e la selettività tissutale. Il secondo approccio è quello di chiarire i meccanismi di azione degli androgeni sul muscolo scheletrico e sulla Prostata e di identificare le molecole di segnalazione che sono a valle del recettore degli androgeni e che attivano le vie coinvolte nell’ipertrofia del muscolo scheletrico, ma non della Prostata.
SARM steroidei: relazioni struttura-attività
Come accennato in precedenza, strutturalmente, i SARM possono essere classificati in SARM steroidei e non steroidei. I SARM steroidei si formano modificando la struttura chimica della molecola di Testosterone (vedi figura seguente).
È stato riconosciuto negli anni ’40 che la sostituzione di un metile in posizione C-17 ritarda il metabolismo presistemico del Testosterone, estendendone l’emivita e rendendolo attivo per via orale. Pertanto, un certo numero di androgeni orali, come il Methylterstosterone, hanno una metilazione in C-17. Tuttavia, gli androgeni 17-alfa alchilati somministrati per via orale, sono potenzialmente epatotossici e abbassano notevolmente il colesterolo HDL plasmatico.
La rimozione del gruppo 19-metile aumenta l’attività anabolizzante del Testosterone (Figura sopra). Pertanto, il 19-nortestosterone ha costituito la base della serie di molecole derivate del Nandrolone. Il Nandrolone è ridotto dalla 5-α reduttasi nei tessuti bersaglio a un androgeno meno potente, il Diidronandrolone (DHN), ma è meno suscettibile all’aromatizzazione in estrogeni convertendo primariamente nel poco attivo Estrone.
Le sostituzioni alchiliche 7-alfa rendono il Testosterone meno suscettibile alla 5-α riduzione e ne aumentano la selettività tissutale rispetto alla Prostata. Pertanto, il 7-alfa metil, 19-nortestosterone ha attività anabolica teoricamente superiore all’attività androgena, sebbene i test fatti sono stati svolti su topi attraverso il ben poco affidabile se rapportato all’uomo “test di Hershberger” (per approfondimenti clicca qui). Comunque, altre molecole di questa serie con gruppi alchilici variabili sono state studiate per la loro attività anabolica.
Il Testosterone viene eliminato rapidamente dalla circolazione e ha una breve emivita. L’esterificazione del gruppo ossidrile 17-β rende la molecola più idrofoba; più lunga è la catena laterale dell’estere, maggiore è l’idrofobicità. Quando gli esteri idrossilici 17-β del Testosterone vengono somministrati attraverso un iniezione intramuscolare in una sospensione oleosa, vengono rilasciati lentamente dal deposito oleoso nella circolazione. Il lento rilascio di esteri idrossilici 17-β dal deposito oleoso estende la loro durata d’azione. Tuttavia, la de-esterificazione degli esteri di Testosterone non limita la velocità della metabolizzazione molecolare; in breve, l’emivita del Testosterone Enantato nel plasma non è significativamente diversa da quella del Testosterone non esterificato una volta scissa l’esterificazione. Allo stesso modo, l’esterificazione del Nandrolone per formare il Nandrolone Decanoato aumenta la sua emivita.
Molecola di Testosterone legata ad un estere Enantato.
L’Oxandrolone è un AAS orale derivato dal DHT che ha un sostituente metilico 17-alfa. La sostituzione del secondo carbonio con l’ossigeno aumenta la stabilità del 3-cheto gruppo e ne aumenta l’attività anabolizzante. Non aromatizza in estrogeno e ha mostrato una bassa attività androgena. Indi, esso è un altro esempio di SARM steroideo.
Struttura molecolare del Oxandrolone
SARM non-steroidei
Gli sforzi pionieristici degli scienziati della Ligand Pharmaceuticals e dell’Università del Tennessee hanno fornito le prime basi della scoperta dei SARM non-steroidei. Da allora, sono state esplorate una serie di categorie strutturali di SARM farmacofori: aril-propionamide (GTX, Inc.), idantoina biciclica (BMS), chinolinoni (Ligand Pharmaceuticals), analoghi della tetraidrochinolina (Kaken Pharmaceuticals, Inc.), benizimidazolo, imidazolopirazolo. , indolo e derivati pirazolina (Johnson e Johnson), derivati azasteroidali (Merck) e derivati anilina, diaril anilina e bezoxazepinoni (GSK) (vedi figura seguente). Poiché è stata pubblicata solo una parte della ricerca sulla scoperta, è probabile che esistano categorie strutturali aggiuntive. Una recente review di Narayanan et al fornisce un eccellente trattato delle strutture dei SARM (28).
Le modifiche strutturali degli analoghi dell’aril propionammide bicalutamide e idrossiflutamide hanno portato alla scoperta della prima generazione di SARM. I composti S1 e S4 in questa serie si legano al AR con elevata affinità e dimostrano selettività tissutale nel impreciso test di Hershberger che utilizza un modello di ratto castrato (30, 31). In questo modello di ratto castrato, sia S1 che S4 hanno prevenuto l’atrofia indotta dalla castrazione del muscolo levat ani e hanno agito come deboli agonisti nella Prostata (30, 31, 32). Alla dose di 3 mg/kg/die, S4 ha parzialmente ripristinato il peso della prostata a < 20% di quello intatto, ma ha ripristinato completamente il peso del levator ani, la forza dei muscoli scheletrici, la densità minerale ossea, la forza ossea e la massa corporea magra e ha soppresso LH e FSH (33, 34). S4 ha anche prevenuto la perdita ossea indotta dall’ovariectomia nel modello di osteoporosi femminile di ratto (35). La capacità dei SARM di promuovere sia la forza muscolare che la forza meccanica ossea costituisce un vantaggio unico rispetto ad altre terapie per l’osteoporosi che aumentano solo la densità ossea.
S1 e S4 sono agonisti parziali; quindi, in ratti maschi intatti (31), S1 e S4 competono con gli androgeni endogeni (o esogeni) e agiscono come antagonisti nella Prostata, tali SARM con attività antagonista o bassa attività intrinseca nella Prostata potrebbero essere utili nel trattamento dell’IPB o del cancro alla Prostata. Gli effetti soppressivi di questa classe di SARM sulla secrezione di gonadotropine nei ratti suggeriscono una potenziale applicazione per la contraccezione maschile.(31)
SARM non-steroideo S4 (Andarina)
Il legame etereo e la sostituzione della posizione-para dell’anello B sono fondamentali per l’attività agonista dei SARM aril propionammidi (30). Sulla base delle strutture cristalline, i composti con legame etereo sembrano adattare una conformazione più compatta rispetto alla bicalutamide a causa della formazione di un legame H intramolecolare, consentendo all’anello B di evitare il conflitto sterico con la catena laterale di W741 nel AR e potenzialmente spiegando l’attività agonista.(36)
I derivati dell’idantoina, sviluppati dal gruppo BMS (37), hanno una struttura ad anello A simile a quella della bicalutamide. Il gruppo ciano o nitro di queste molecole interagisce con Q711 e R752 (38, 39). L’anello benzenico o gruppo naftile, insieme all’anello idantoico, si sovrappone al piano steroideo, mentre l’azoto dell’anello idantoinico forma un legame H con N705. BMS-564929 lega al AR con alta affinità e alta specificità. BMS-564929 ha dimostrato attività anabolizzante nel muscolo levator ani e un alto grado di selettività tissutale, come indicato da una ED50 sostanzialmente più elevata per la Prostata. I derivati dell’idantoina sono potenti soppressori dell’LH. BMS-564929 è disponibile per via orale nell’uomo, con un’emivita di 8-14 ore. L’emivita prolungata di questi ligandi nei ratti può spiegare la dose più bassa necessaria per ottenere effetti farmacologici; differenze nelle attività in vivo di SARM che condividono affinità di legame e attività in vitro simili possono essere correlate alle differenze nella farmacocinetica e nell’esposizione al farmaco.(40)
Hanada et al (41) della Kaken Pharmaceutical Co. hanno riportato una serie di derivati della tetraidrochinolina come agonisti dell’AR nell’osso. Sebbene questi composti mostrino un’elevata affinità per l’AR e una forte attività agonista nella Prostata e nel levator ani, hanno dimostrato una scarsa selettività tra i tessuti androgeni e anabolici (41). Una significativa attività farmacologica in vivo è stata osservata solo ad alte dosi sottocutanee.(28, 41)
I composti ligandi LGD2226 e LGD 2941 che sono derivati biciclici del 6-anilino chinolinone hanno mostrato attività anabolica sul muscolo levator ani, nonché sulla massa ossea e sulla forza, pur avendo scarso effetto sulla dimensione della Prostata in un modello preclinico di roditori (42, 43, 44). È stato anche dimostrato che LGD2226 mantiene il comportamento riproduttivo maschile nel modello di roditore castrato (42). Gli scienziati della Johnson e Johnson hanno sostituito il legante propionammidico con elementi ciclici come pirazoli, benzimidazoli, indoli e mimetici propionanilidi ciclici (45). Gli scienziati della Merck hanno sviluppato una serie di derivati 4-azasteroidali e butanammidi (28). Ulteriori composti sono stati sviluppati da altre aziende farmaceutiche, ma una discussione dettagliata di ciascun composto esula dallo scopo di questo articolo.
Meccanismi di selettività tissutale dei SARM
Narayanan et al hanno confrontato le vie attivate da un aril propionamide SARM, S-22, con quelle attivate dal DHT (46) e hanno scoperto che S-22 e DHT attivavano diverse vie di segnalazione distinte. S-22 e DHT differivano significativamente nel reclutamento del AR e dei suoi co-regolatori come potenziatore del PSA. L’S-22 differiva anche dal DHT nell’induzione della rapida fosforilazione di diverse chinasi (46). Tuttavia, i meccanismi che contribuiscono all’attivazione trascrizionale tessuto-specifica e alla selettività degli effetti biologici dei SARM rimangono poco compresi. Sono state proposte tre ipotesi generali, anche se queste ipotesi non si escludono a vicenda. L’ipotesi del co-attivatore presuppone che il repertorio di proteine co-regolatrici che si associa al AR legato al SARM differisce da quello associato al AR legato al Testosterone che porta all’attivazione trascrizionale di un insieme di geni regolati in modo differenziale.
Antigene Prostatico Specifico (Prostate Specific Antigen, PSA)
L’ipotesi conformazionale afferma che le differenze funzionali nelle classi di ligandi (agonisti, antagonisti e SARM) si riflettono in stati conformazionalmente distinti con partizionamento termodinamico distinto. Il legame con il ligando induce specifici cambiamenti conformazionali nel dominio di legame del ligando, che potrebbe modulare la topologia di superficie e le successive interazioni proteina-proteina tra AR e altri co-regolatori coinvolti nell’attivazione trascrizionale genomica o proteine citosoliche coinvolte nella segnalazione non genomica. Le differenze nella conformazione del recettore ligando-specifico e le interazioni proteina-proteina potrebbero portare a una regolazione genica tessuto-specifica, a causa di potenziali cambiamenti nelle interazioni con ARE, co-regolatori o fattori di trascrizione. Le interazioni proteina-proteina indotte dal ligando contribuiscono alle interazioni tra le estremità amminiche e carbossiliche del AR (cioè l’interazione N/C) e il reclutamento di co-attivatori (47). Entrambe le interazioni sono mediate dall’interazione tra la regione AF2 del AR ed i motivi di legame FXXLF o LXXLL (48). Il solco idrofobo presente nella regione AF2 del AR LBD sembra essere più favorevole per il legame della fenilalanina, il che suggerisce che l’interazione N/C è preferita. Sebbene la conformazione AR-LBD legata al SARM non steroideo non sia stata ben caratterizzata, Sathya et al (49) hanno riportato che alcuni SARM steroidei che hanno attività agonista in vitro inducono un cambiamento conformazionale attivante senza facilitare le interazioni N/C. Questi dati suggeriscono che il cambiamento conformazionale specifico del ligando è ottenibile con ligandi sintetici.
(A) Il gene AR consiste di 8 esoni che codificano per il recettore degli androgeni con un prodotto genico della dimensione tipica di 919 amminoacidi. Il AR è composto da un dominio N-terminale (NTD), un dominio di legame al DNA centrale (DBD), una regione a cerniera corta e un LBD C-terminale. (B) LBD comprende una struttura elicoidale 12 che racchiude una tasca centrale di legame dell’ormone (HBP), un secondo dominio della funzione di attivazione (AF2) che si trova all’estremità carbossi-terminale dell’LBD e un sito di legame scoperto di recente, funzione di legame 3 (BF3). La conformazione adottata dell’H12 è inequivocabilmente associata al meccanismo d’azione molecolare dei ligandi legati all’HBP. (C) Come mostrato nella struttura complessa di Diidrotestosterone (DHT) e AR-LBD, l’AR HBP è composto principalmente da residui idrofobici (palla verde) che possono formare forti interazioni non polari con il DHT. L’ancoraggio proteina-ligando può essere ulteriormente stabilizzato da una rete di legami idrogeno (linea tratteggiata blu) che coinvolge i residui polari R752, Q711, N705 e T877.[fonte immagine https://www.researchgate.net/%5D
Bohl et al (36) hanno riportato che la bicalutamide adotta una conformazione molto piegata nel AR. Sebbene l’anello A e il legame ammidico della molecola di bicalutamide si sovrappongano al piano steroideo, l’anello B della bilcautammide si piega lontano dal piano, puntando verso la parte superiore della tasca di legame del ligando (LBP), che costituisce una caratteristica strutturale unica di questo classe di leganti (36). Il gruppo ciano dell’anello A forma legami H con Q711 e R752, simile al 3-cheto gruppo nel 5α-DHT (36). Il gruppo idrossile chirale forma legami H con L704 e N705, imitando l’anello C e il gruppo 17β-OH nel 5α-DHT (36). Queste interazioni di legame H sono fondamentali per un’elevata affinità di legame. Lievi modifiche strutturali possono cambiare il ligando da antagonista AR ad agonista. Il legame idrogeno favorevole tra il ligando e la catena laterale T877, le caratteristiche strutturali che imitano il 3-cheto gruppo del Testosterone e le interazioni idrofobiche sono fondamentali affinché il ligando si leghi con alta affinità e stimoli l’azione del AR. La struttura cristallina a raggi X del AR legato a S-1 ha rivelato che la catena laterale W741 è spostata dall’anello B per espandere la tasca di legame in modo che il composto si orienti verso la regione AF2 (50). Il ripiegamento proteico del AR legato al SARM è lo stesso che si tratti di un SARM steroideo e non steroideo (50). Non è chiaro come l’interazione ligando-recettore determini l’attività agonista o antagonista del ligando.
La selettività tissutale dei SARM potrebbe anche essere correlata a differenze nella loro distribuzione tissutale, potenziali interazioni con la 5α-reduttasi o l’aromatasi CYP19, o l’espressione tessuto-specifica di co-regolatori (51). Tuttavia, studi di autoradiografia con derivati di bicalutamide e idantoina (52) hanno mostrato che non si accumulano preferenzialmente nei tessuti “anabolizzanti”. L’azione del Testosterone in alcuni tessuti androgeni è amplificata dalla sua conversione in 5α-DHT (53); i SARM non steroidei non fungono da substrati per la 5α-reduttasi. La selettività tissutale dei SARM potrebbe essere correlata all’espressione tessuto-specifica delle proteine co-regolatorie. Allo stesso modo, alcune differenze delle azioni dei SARM rispetto al Testosterone potrebbero essere correlate all’incapacità dei SARM non steroidei di subire l’aromatizzazione.
Esperienza di studi preclinici e clinici con i SARM di prima generazione
Un gran numero di SARM candidati sono stati sottoposti a studi preclinici di verifica teorica e tossicologici e sono entrati in studi clinici di fase I e II (27, 28). Gli studi preclinici hanno rivelato una promettente selettività dei tessuti; tuttavia, poiché molti di questi dati generati dalle aziende farmaceutiche sono rimasti inediti, i confronti della potenza relativa e della selettività dei tessuti tra i diversi SARM sono difficili da convalidare.
Un certo numero di SARM di prima generazione sono stati testati in prove di fase I. Questi composti sono stati posizionati per studi di efficacia precoci per il trattamento dell’osteoporosi, la fragilità ossea, la cachessia del cancro e le limitazioni funzionali associate all’invecchiamento. Inoltre, i SARM che inibiscono potentemente le gonadotropine, ma risparmiano l’attività a livello della Prostata, hanno suscitato una certa attrattiva come candidati per la contraccezione maschile. È stato proposto l’uso di SARM per il trattamento delle sindromi da carenza di androgeni negli uomini; i vantaggi relativi ai SARM rispetto al Testosterone per questa indicazione non sono immediatamente evidenti e risultano limitati. Molte funzioni biologiche del Testosterone, in particolare i suoi effetti sulla libido e sul comportamento, sulle ossa e sui lipidi plasmatici, richiedono la sua aromatizzazione in estrogeni; poiché i SARM attualmente disponibili non sono né aromatizzabili né 5-alfa riducibili, questi composti risultano fortemente limitati come base terapica di sostituzione androgena in andropausa e dovrebbero affrontare una barra normativa in salita per l’approvazione in quanto sarebbero tenuti a dimostrare efficacia e sicurezza in molti più domini di azione degli androgeni rispetto a quanto richiesto dalle formulazioni di Testosterone la quale si conosce per effetti diretti ed indiretti in condizione terapeutica sostitutiva degli androgeni endogeni.
Alle dosi che sono state testate, i SARM di prima generazione inducono modesti guadagni di massa corporea magra in volontari sani, che non sono affatto vicini ai guadagni molto maggiori nella massa muscolare scheletrica riportati con dosi sovrafisiologiche di Testosterone. I modesti guadagni da 1,0 a 1,5 kg di massa magra con i SARM di prima generazione in 4-6 settimane dovrebbero essere confrontati con i guadagni di 5-7 kg di massa magra con dosi da 300 e 600mg di Testosterone Enantato (pari approssimativamente a 216mg e 432mg di Testosterone effettivo rispettivamente). Tuttavia, è possibile che la prossima generazione di molecole SARM avrà maggiore potenza e selettività rispetto ai SARM di prima generazione, ma ad oggi non sussiste ancora dimostrazione a riguardo.
Raggiungimento della selettività e spiegazione dei meccanismi d’azione
Un altro approccio per ottenere la selettività d’azione è chiarire i meccanismi dell’azione del Testosterone sulla Prostata e identificare le molecole a valle associate all’attivazione della segnalazione AR nel muscolo scheletrico, ma non nella Prostata. Attraverso la comprensione di questi meccanismi, potrebbe essere possibile identificare molecole candidate che prendono di mira aspetti specifici della cascata di segnalazione AR.
Le analisi delle biopsie muscolari di uomini trattati con dosi graduate di testosterone hanno rivelato che la somministrazione di testosterone induce ipertrofia delle fibre muscolari sia di tipo I che di tipo II (54, 55); I cambiamenti nelle aree trasversali di entrambe le fibre di tipo I e II sono correlati alla dose di Testosterone e alle concentrazioni di Testosterone totale e libero (54). Tuttavia, né il numero assoluto né la proporzione relativa delle fibre di tipo I e II cambiano durante la somministrazione di Testosterone.
Poiché le cellule satellite muscolari sono state implicate nell’ipertrofia del muscolo scheletrico e nell’aumento del numero mionucleare (56), sono state quantificate le cellule satellite e il numero mionucleare mediante microscopia elettronica, utilizzando metodi di conteggio diretto e orientamento spaziale nelle biopsie del vasto laterale ottenute al basale e dopo 20- settimane di trattamento con un agonista del GnRH e dosi graduate di Testosterone Enantato. Il numero assoluto e percentuale di cellule satellite a 20 settimane era significativamente maggiore del basale negli uomini che ricevevano dosi sovrafisiologiche di Testosterone (57). La variazione del numero di cellule satellite era correlata alle variazioni dei livelli di Testosterone totale e libero (57). Quindi, l’ipertrofia delle fibre muscolari indotta dal Testosterone è associata ad un aumento delle cellule satellite e del numero di mionuclei.
Il Testosterone e il DHT promuovono la differenziazione delle cellule staminali mesenchimali multipotenti in linea miogenica e inibiscono la loro differenziazione in linea adipogenica (58, 59). Il Testosterone inibisce anche la differenziazione dei pre-adipociti in adipociti (59, 60). Altri hanno suggerito che l’ipertrofia indotta dal Testosterone sia causata dalla stimolazione della sintesi proteica e dall’inibizione della degradazione proteica (61, 62). Testosterone e DHT promuovono l’associazione del ligando AR con il suo co-attivatore, β-catenina; questa interazione stabilizza la β-catenina, promuove la sua traslocazione nel nucleo e l’associazione con TCF-4, e l’attivazione trascrizionale di un certo numero di geni bersaglio Wnt (63). La β-catenina svolge un ruolo essenziale nel mediare gli effetti del Testosterone sulla differenziazione miogenica. Il Testosterone sovra-regola l’espressione della Follistatina in vivo e in vitro (63); l’infusione della proteina Follistatina ricombinante aumenta la massa muscolare e diminuisce la massa grassa nei topi castrati. Il Testosterone sovra-regola l’SMAD 7 e sotto-regola la segnalazione del SMAD mediata dal TGFβ e i geni bersaglio del TGFβ (63). La Follistatina inibisce l’azione di diversi membri della famiglia del TGFβ. Questi studi supportano l’ipotesi che gli effetti del Testosterone siano trasmessi in modo incrociato dalla via Wnt alla via TGFβ-SMAD attraverso la Follistatina. Pertanto, è possibile che molecole candidate come la Follistatina che sono a valle del AR e β-catenina e che mediano gli effetti del Testosterone sul muscolo possano fornire la selettività desiderata degli effetti anabolici. La via di segnalazione mediata dal AR a valle della β-catenina può essere un interessante serbatoio di bersagli candidati per lo sviluppo di farmaci anabolizzanti selettivi.
Molecola di Follistatina
Ostacoli normativi allo sviluppo dei SARM
Negli studi di fase I e II, i SARM di prima generazione hanno mostrato riduzioni significative delle concentrazioni di colesterolo HDL e SHBG e lievi aumenti transitori di AST e ALT. Non è chiaro se gli aumenti delle transaminasi riflettano la tossicità epatica di primo passaggio tipica degli androgeni somministrati per via orale o un effetto di classe sulla trascrizione del gene AST. Allo stesso modo, la soppressione del colesterolo HDL potrebbe riflettere gli effetti combinati della via di somministrazione orale e la mancanza di aromatizzazione. È possibile che una via di somministrazione sistemica – transdermica o intramuscolare – possa attenuare il potenziale di aumento delle transaminasi e riduzioni di HDL-C.
Globulina Legante gli Ormoni Sessuali (in inglese sex hormone-binding globulin o SHBG)
Mentre il percorso normativo per l’approvazione dei farmaci per l’osteoporosi è stato ben delineato a causa della precedenza stabilita dai farmaci precedentemente approvati, il percorso per l’approvazione delle terapie anabolizzanti che promuovono la suddetta funzione non è stato chiaramente stabilito. Sono in corso sforzi considerevoli per generare un consenso su indicazioni, risultati di efficacia negli studi cardine e differenze clinicamente importanti minime nei risultati di efficacia chiave; questi sforzi dovrebbero facilitare le prove di efficacia delle molecole candidate. Ma il risultato, ad oggi, non è molto promettente.
Allora i SARM non-steroidei sono tessuto-selettivi?
Ammetto che quanto esposto fino ad ora non è propriamente “masticabile” da tutti, ed è per questo che vi renderò la comprensione più facile.
Allora, un modo per ottenere la selettività tissutale è tramite un fapping molecolare che implica l’attivazione del recettore degli androgeni (AR) specificamente nel tessuto muscolare. Mentre l’AR è lo stesso in tutti i tessuti, il contesto cellulare è diverso: puoi immaginare che il contenuto di una cellula muscolare sia abbastanza diverso da quello di una cellula della ghiandola sebacea. Quando l’AR viene attivato per indurre la trascrizione genica, che alla fine porterà ai guadagni muscolari, entrano in gioco molte altre proteine. Queste proteine coinvolte nella trascrizione sono i cosiddetti coregolatori trascrizionali. Chiamiamoli cofattori in breve. Questi possono aiutare nella trascrizione (coattivatori) o reprimerla (corepressori). Quei cofattori, e le loro proporzioni, che vengono reclutati da un AR attivato, possono variare da un tessuto all’altro. Questo dipende, in parte, da quale molecola è legata all’AR. In quanto tale, un SARM potrebbe essere in grado di reclutare un gruppo di cofattori che porteranno a una trascrizione genica minima o nulla nel tessuto A (Prostata), mentre portano alla trascrizione genica completa nel tessuto B (Muscolo).
Quanto detto sopra sembra comunque piuttosto complesso, e lo è, ma non mi è possibile comunicare a gesti per spiegarvi una cosa che è di base complessa. Comunque sia, come si fa a sapere quale tipo di ligando per l’AR interagisce con quali cofattori e in che misura? Non lo fa, si dovrebbero eseguire test quasi infiniti sul composto in questione per determinarlo effettivamente. E questo processo sembra richiedere molto tempo. Tuttavia, questo è attualmente pubblicizzato come uno dei motivi per cui i SARM – in sostanza avendolo scoperto per “caso” – esercitano i loro effetti specifici sui tessuti. Ad esempio, è stato dimostrato che l’antiandrogeno steroideo TSAA-291 esercita un’attività tessuto-specifica che coincide con profili di reclutamento di coregolatori differenziali rispetto al Diidrotestosterone (DHT) [64]. Tuttavia, poiché non hanno confrontato altri AAS, potrebbe anche essere che avrebbero visto diversi profili di reclutamento di coregolatori con altri AAS. Pertanto, è difficile vedere quanto sia effettivamente rilevante per le proprietà specifiche dei SARM. Dopotutto, la correlazione non implica la causalità.
Oxendolone (TSAA-291)
Andando avanti con la semplificazione pratica del concetto di selettività specifica, un altro modo in cui un SARM potrebbe esercitare tale specificità tissutale è attraverso il la sua via di metabolizzazione. Una molecola viene metabolizzata dall’azione degli enzimi. E la presenza di tali enzimi metabolizzanti può differire da un tessuto all’altro. Ad esempio, questo è molto evidente con la metabolizzazione del Testosterone. Il Testosterone è suscettibile di metabolizzazione per riduzione sul suo quinto atomo di carbonio. Questa riduzione è catalizzata dall’enzima 5α-reduttasi. Il risultato di questa riduzione è il più potente androgeno Diidrotestosterone (DHT). Pertanto, l’effetto del testosterone viene amplificato nei tessuti che esprimono questo enzima. Sfortunatamente, il muscolo scheletrico non è uno di quei tessuti. E, in effetti, il DHT viene degradato nel molto debole androgeno 3α-Androstanediolo dall’enzima 3α-HSD nel muscolo [65], diminuendo così il suo effetto in loco.
3α-idrossisteroide deidrogenasi ( 3α-HSD o aldo-cheto reduttasi famiglia 1 membro C4)
Tuttavia, questo aspetto è leggermente diverso per i SARM. Gli enzimi steroidogeni, come la 5α-reduttasi e la 3α-HSD, non hanno effetto sui SARM non steroidei. Gli enzimi che metabolizzano i SARM variano da una classe di SARM all’altra. Come tale, deve essere studiato per ogni SARM, analizzandone il modo in cui viene metabolizzato e con quale velocità ciò si verifica nei vari tessuti. Questo risulta essere più banale per la maggior parte degli AAS sui quali possiamo ampiamente prevederlo. Contrariamente, risulta difficile per lo sviluppo dei SARM non steroidei.
3α-Androstanediolo
Infine, è noto che gli AAS possono esercitare anche effetti non genomici [66]. Come suggerisce il nome, questi sono effetti che non sono mediati dalla trascrizione genica. Pertanto, questi effetti si verificano molto rapidamente (entro secondi/minuti dopo l’esposizione di una cellula ad essa). Alcune ricerche indicano che il recettore degli androgeni localizzato nella membrana plasmatica, così come altri recettori legati alla membrana, mediano questi effetti. Ipoteticamente è possibile che AAS – e per estensione SARM – siano in grado di influenzare le vie di segnalazione a seconda del contesto cellulare, cioè gli effetti potrebbero differire da una cellula all’altra: specificità del tessuto.
Più di 20 anni di ricerca sui SARM ma nessuna approvazione clinica
Sapere queste cose è interessante e utile per comprendere l’attività di tali molecole, ma tali attività ci mostrano di essere ben lungi (ancora) dal possedere la chiave di volta nello sviluppo di SARM terapeuticamente e pienamente efficaci. Ma almeno abbiamo una base attraverso la quale i SARM potrebbero effettivamente funzionare. Tuttavia, dopo oltre 2 decenni di ricerca sui SARM [67], nessuno è stato approvato dalla Food and Drug Administration (FDA). E no, non c’entra “bIg PhaRma”, complottaro da tastiera.
Parte del motivo per cui ciò avviene può essere ricondotto al modo in cui i ricercatori hanno esaminato i potenziali SARM. Come ho riportato in un mio precedente articolo, la anabolico:androgeno ratio, come valutato dal test di Hershberger, è pressoché inutile. Eppure questo test è stato utilizzato dalle aziende farmaceutiche per decidere se perseguire o meno la ricerca su determinati SARM di particolare interesse, queste aziende includono la GTx, Inc. con lo sviluppo del Enobosarm (GTx-024) [68], la GlaxoSmithKline con lo sviluppo del GSK2881078 [69 ], la Takeda Pharmaceutical Company con lo sviluppo del SARM-2f [70], la Aska Pharmaceuticals con lo sviluppo del S42 [71], e la Merck & Co, Inc con lo sviluppo del MK-4541 [72], ecc.
Non si sono forse già visti risultati ridicolmente buoni con AAS convenzionali in passato utilizzando questi test? Si, e non per una molecola. Ad esempio, si dice che lo Stanozololo abbia un rapporto anabolico/androgeno circa 10 volte superiore a quello del Testosterone, mentre il Methyldrostanolone ha circa un rapporto anabolico/androgeno 20 volte superiore [73]. Tuttavia, come sappiamo, queste molecole non sono considerate SARM sito-specifici e non sono scevre da eventuali effetti androgenizzanti. Perché? Perchè uno studio con molteplici variabili svolto su roditori non può essere rapportato correttamente all’uomo, come ho spiegato nell’articolo dedicato alla anabolico:androgeno ratio.
Un ulteriore problema con la ricerca sui SARM emerge quando si esaminano gli studi clinici. Poiché i SARM vengono sviluppati per superare gli AAS convenzionali, non ci si aspetterebbe forse che essi vengano confrontati con gli AAS convenzionali negli studi clinici? Per qualche ragione, in tutti gli studi clinici con i SARM, questi vengono confrontati con un placebo. Se si vuole valutare l’efficacia reale di una molecola rispetto ad un altra, non lo si fa confrontandola solo ad un placebo, o forse solo inizialmente lo si farebbe, come in una sperimentazione pilota per risparmiare sui costi, e per valutare se ne vale la pena o meno. Questi studi mostrano comunemente guadagni marginali (nell’ordine di 1kg) di LBM in un periodo di diverse settimane/mesi con una corrispondente buona tollerabilità. Anche gli AAS convenzionali sono generalmente ben tollerati e aumentano marginalmente l’LBM quando vengono somministrati a basso dosaggio, niente di sconvolgente in questo. La Ligand Pharmaceuticals ha persino trovato la necessità di menzionare quanto segue nella conclusione del loro abstract di studio che copre gli effetti del loro SARM LGD-4033: “LGD-4033 era sicuro, aveva un profilo farmacocinetico favorevole e un aumento della massa corporea magra anche durante questo breve periodo senza cambiamento nell’antigene prostatico specifico”. Cosa si aspettavano in poche settimane di trattamento con il loro SARM? Anche 600mg di Testosterone Enantato a settimana per 20 settimane non aumentano l’antigene prostatico specifico (PSA) negli uomini giovani [74, 75] o negli uomini più anziani [76].
LGD-4033
Se l’unico requisito ricercato è che un SARM non steroideo sia più efficace di un placebo pur essendo ben tollerato, ce l’hanno fatta. Ma praticamente tutti gli AAS convenzionali sono anche più efficaci di un placebo pur essendo ben tollerati. Superare il placebo non è mai stato l’obiettivo dello sviluppo dei SARM, quindi perché gli studi testa a testa sono ancora gravemente carenti? Forse perchè non vi è superiorità ne negli effetti benefici e nel rapporto tra benefici e rischi sistemici? …
Conclusioni:
I SARM si basano sulla selettività dei tessuti per esercitare i loro effetti anabolici (costruzione muscolare), mantenendo gli effetti collaterali al minimo assoluto. Dopotutto, gli effetti collaterali si riducono in gran parte, ma non totalmente, all’azione androgena nei tessuti diversi dai muscoli. I SARM possono esercitare questi effetti tessuto-specifici attraverso circa tre diversi meccanismi. Uno sfrutta le differenze nelle molecole tra i diversi tipi di cellule che “aiutano” un SARM ad avviare la trascrizione genica. Un altro si basa su enzimi di espressione tessuto-specifici che metabolizzano il SARM. Un terzo si basa sugli effetti non genomici che potrebbero essere mediati da un SARM che, ancora una volta, potrebbe variare da un tipo di cellula all’altro.
Poiché questi processi biochimici sono estremamente difficile da prevedere in anticipo, le aziende farmaceutiche devono esaminare molte molecole per vedere quale potrebbe essere la soluzione migliore. Nessun SARM è stato ancora approvato e credo che ciò sia in parte dovuto a questo processo di screening che si basa su metodi obsoleti e imperfetti come il test di Hershberger e all’incapacità di sopperire all’attività fisiologica del DHT e dell’Estradiolo, i quali subiscono una marcata soppressione consequenziale al abbassamento dei livelli di Testosterone endogeno. Questo punto deve essere sicuramente migliorato. Ed è quindi questa la strada che dovrebbe intraprendere la ricerca sui SARM.
Negli sport, ed in particolare nel Bodybuilding, l’uso dei SARM non steroidei, dopo l’iniziale eccitazione per le promesse commerciali affiancate al loro uso da parte dei rivenditori e brand, sono caduti in un uso più che altro amatoriale, da parte di persone poco informate in materia e dalla mente facilmente manipolabile dalla pubblicità e informazioni incomplete se non del tutto errate.
L’unico ambito in cui i SARM non steroidei hanno visto un certo potenziale è nel culturismo femminile. In questa circostanza, le molecole più testate, prima su tutte l’Ostarina, ha mostrato un certo vantaggio se l’obbiettivo era quello di aumenti contenuti del tessuto muscolare e la mancanza di possibili effetti mascolinizzanti alle dosi comprese tra 5 e 10mg/die.
Nell’uso maschile i SARM hanno lasciato una serie di delusioni e promesse non mantenute. In monoterapia il loro uso ha portato ad atleti con problemi non indifferenti nella sfera sessuale, con difficoltà di raggiungimento e mantenimento dell’erezione, letargia, stanchezza cronica, affaticabilità, depressione e stati ansiosi. Tutti sintomi legati ad un calo significativo del DHT e del Estradiolo, con conseguente riduzione o mancanza della loro, per esempio, attività a livello cerebrale (neurosteroideo).
Di conseguenza, utilizzare uno o più SARM senza una base esogena di Testosterone (o, per lo meno, di hCG) è una totale pazzia! E, comunque, l’uso dei SARM come aggiunte ad un ciclo di classici AAS iniettabili non risulta quasi mai all’altezza delle aspettative di risposta ipertrofica rispetto all’uso, per esempio, di AAS orali come starter e/o finisher. Ovviamente la valutazione si basa anche e soprattutto sul rapporto effetti collaterali:benefici in contesto preparatorio correttamente impostato.
Inoltre, gli effetti collaterali a livello epatico e della lipidemia ematica non sono estranei all’uso di SARM non steroidei, sebbene essi si mostrino a diverso grado di entità molecola-dipendente e dose-dipendente. La stessa Ostarina aveva mostrato lievi alterazioni di ALT e AST con riduzione del HDL al dosaggio di 3mg in studi clinici; la molecola in ambito “physique” viene assunta ad un dosaggio nel range di 10-20mg/die, e l’impatto sulle transaminasi, colesterolo totale, LDL e HDL osservato attraverso esami ematici mostrano variazioni significative e variabili in misura soggettiva.
Il SARM non steroideo con il più alto carico di effetti collaterali è risultato essere LGD4033, il quale, in diversi casi studio, ha mostrato di poter causare forte stress epatico oltre che alterare sensibilmente la lipidemia ematica. Nel caso di questa molecola, si è osservato anche una perdita della selettività con possibile comparsa di effetti androgenicizzanti. Complice di questi riscontri è soprattutto l’abuso che se ne fa della molecola, sforando i dosaggi efficaci e contenitivi (2-8mg/die) a favore di somministrazioni elevate (≥10mg/die).
Anche il RAD140 sembra non essere privo di effetti collaterali significativi a livello epatico, nonostante il suo potenziale effetto protettivo sulla Prostata che, a dosaggi minimi (5mg/die) potrebbe avere un riscontro terapeutico preventivo per l’ipertrofia prostatica.
SARM non-steroideo RAD140
Lascerei perdere discorsi ipotetici su altri SARM comunemente utilizzati dagli atleti (specialmente amatori) ma che alle spalle sono privi di studi clinici (vedi, per esempio, l’S23) e, quindi, di dati oggettivi sulle possibili attività nell’uomo. L’unica eccezione tra questi la fa, forse, il SARM steroideo YK11, il quale sembra essere gestibile a dosi di 5-10mg/die con un buon rapporto tra benefici ed alterazioni dei marker ematici.
SARM steroideo YK11
Per concludere, mi sembra di avervi dato sufficienti informazioni per valutare correttamente i SARM e deporli con cognizione logica dall'”altarino” di innocuità sul quale brand e venditori li hanno posti e dove una parte di voi continua a tenerli.
Gabriel Bellizzi
Riferimenti:
Mohler ML, Bohl CE, Jones A, Coss CC, Narayanan R, He Y, Hwang DJ, Dalton JT, Miller DD (June 2009). “Nonsteroidal selective androgen receptor modulators (SARMs): dissociating the anabolic and androgenic activities of the androgen receptor for therapeutic benefit”. Journal of Medicinal Chemistry. 52(12): 3597–617.
Yin D, Gao W, Kearbey JD, Xu H, Chung K, He Y, Marhefka CA, Veverka KA, Miller DD, Dalton JT (March 2003). “Pharmacodynamics of selective androgen receptor modulators”. The Journal of Pharmacology and Experimental Therapeutics. 304 (3): 1334–40.
Manfredi MC, Bi Y, Nirschl AA, Sutton JC, Seethala R, Golla R, Beehler BC, Sleph PG, Grover GJ, Ostrowski J, Hamann LG (August 2007). “Synthesis and SAR of tetrahydropyrrolo[1,2-b][1,2,5]thiadiazol-2(3H)-one 1,1-dioxide analogues as highly potent selective androgen receptor modulators”. Bioorganic & Medicinal Chemistry Letters. 17 (16): 4487–90.
Zhang X, Li X, Allan GF, Sbriscia T, Linton O, Lundeen SG, Sui Z (August 2007). “Design, synthesis, and in vivo SAR of a novel series of pyrazolines as potent selective androgen receptor modulators”. Journal of Medicinal Chemistry. 50 (16): 3857–69.
Long YO, Higuchi RI, Caferro TR, Lau TL, Wu M, Cummings ML, Martinborough EA, Marschke KB, Chang WY, López FJ, Karanewsky DS, Zhi L (May 2008). “Selective androgen receptor modulators based on a series of 7H-[1,4]oxazino[3,2-g]quinolin-7-ones with improved in vivo activity”. Bioorganic & Medicinal Chemistry Letters. 18 (9): 2967–71.
Bhasin S, Cunningham GR, Hayes FJ, Matsumoto AM, Snyder PJ, Swerdloff RS, Montori VM. Testosterone therapy in adult men with androgen deficiency syndromes: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2006;91:1995–2010.
Baumgartner RN. Body composition in healthy aging. Annals of the New York Academy of Sciences. 2000;904:437–448.
Bassey EJ, Fiatarone MA, O’Neill EF, Kelly M, Evans WJ, Lipsitz LA. Leg extensor power and functional performance in very old men and women. Clin Sci (Lond) 1992;82:321–327.
. Lexell J, Downham D, Sjostrom M. Distribution of different fibre types in human skeletal muscles. A statistical and computational study of the fibre type arrangement in m. vastus lateralis of young, healthy males. Journal of the neurological sciences. 1984;65:353–365.
Orwoll E, Lambert LC, Marshall LM, Blank J, Barrett-Connor E, Cauley J, Ensrud K, Cummings SR. Endogenous testosterone levels, physical performance, and fall risk in older men. Arch Intern Med. 2006;166:2124–2131.
Bhasin S, Storer TW, Berman N, Yarasheski KE, Clevenger B, Phillips J, Lee WP, Bunnell TJ, Casaburi R. Testosterone replacement increases fat-free mass and muscle size in hypogonadal men. J Clin Endocrinol Metab. 1997;82:407–413.
Snyder PJ, Peachey H, Berlin JA, Hannoush P, Haddad G, Dlewati A, Santanna J, Loh L, Lenrow DA, Holmes JH, Kapoor SC, Atkinson LE, Strom BL. Effects of testosterone replacement in hypogonadal men. J Clin Endocrinol Metab. 2000;85:2670–2677.
. Bhasin S, Storer TW, Berman N, Callegari C, Clevenger B, Phillips J, Bunnell TJ, Tricker R, Shirazi A, Casaburi R. The effects of supraphysiologic doses of testosterone on muscle size and strength in normal men. N Engl J Med. 1996;335:1–7.
. Bhasin S, Woodhouse L, Casaburi R, Singh AB, Bhasin D, Berman N, Chen X, Yarasheski KE, Magliano L, Dzekov C, Dzekov J, Bross R, Phillips J, Sinha-Hikim I, Shen R, Storer TW. Testosterone dose-response relationships in healthy young men. Am J Physiol Endocrinol Metab. 2001;281:E1172–1181.
Bhasin S, Woodhouse L, Casaburi R, Singh AB, Mac RP, Lee M, Yarasheski KE, Sinha-Hikim I, Dzekov C, Dzekov J, Magliano L, Storer TW. Older men are as responsive as young men to the anabolic effects of graded doses of testosterone on the skeletal muscle. J Clin Endocrinol Metab. 2005;90:678–688.
Casaburi R, Bhasin S, Cosentino L, Porszasz J, Somfay A, Lewis MI, Fournier M, Storer TW. Effects of testosterone and resistance training in men with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2004;170:870–878.
Johansen KL, Mulligan K, Schambelan M. Anabolic effects of nandrolone decanoate in patients receiving dialysis: a randomized controlled trial. Jama. 1999;281:1275–1281.
Woodhouse LJ, Reisz-Porszasz S, Javanbakht M, Storer TW, Lee M, Zerounian H, Bhasin S. Development of models to predict anabolic response to testosterone administration in healthy young men. Am J Physiol Endocrinol Metab. 2003;284:E1009–1017.
Storer TW, Magliano L, Woodhouse L, Lee ML, Dzekov C, Dzekov J, Casaburi R, Bhasin S. Testosterone dose-dependently increases maximal voluntary strength and leg power, but does not affect fatigability or specific tension. J Clin Endocrinol Metab. 2003;88:1478–1485.
Calof O, Singh AB, Lee ML, Urban RJ, Kenny AM, Tenover JL, Bhasin S. Adverse events associated with testosterone supplementation of odler men. J Greontol Med Sci. 2005 in press.
Bhasin S, Calof OM, Storer TW, Lee ML, Mazer NA, Jasuja R, Montori VM, Gao W, Dalton JT. Drug insight: Testosterone and selective androgen receptor modulators as anabolic therapies for chronic illness and aging. Nature Clinical Practice Endocrinology & Metabolism. 2006;2:146–159.
Narayanan R, Mohler ML, Bohl CE, Miller DD, Dalton JT. Selective androgen receptor modulators in preclinical and clinical development. Nuclear receptor signaling. 2008;6:e010. An excellent treatise of SARM chemistry and structure-activity relationships.
Negro-Vilar A. Selective androgen receptor modulators (SARMs): a novel approach to androgen therapy for the new millennium. J Clin Endocrinol Metab. 1999;84:3459–3462.
Yin D, He Y, Perera MA, Hong SS, Marhefka C, Stourman N, Kirkovsky L, Miller DD, Dalton JT. Key structural features of nonsteroidal ligands for binding and activation of the androgen receptor. Mol Pharmacol. 2003;63:211–223.
Gao W, Kearbey JD, Nair VA, Chung K, Parlow AF, Miller DD, Dalton JT. Comparison of the pharmacological effects of a novel selective androgen receptor modulator, the 5alpha-reductase inhibitor finasteride, and the antiandrogen hydroxyflutamide in intact rats: new approach for benign prostate hyperplasia. Endocrinology. 2004;145:5420–5428.
Gao W, Reiser PJ, Coss CC, Phelps MA, Kearbey JD, Miller DD, Dalton JT. Selective Androgen Receptor Modulator (SARM) Treatment Improves Muscle Strength and Body Composition, and Prevents Bone Loss in Orchidectomized Rats. Endocrinology 2005
Gao W, Reiser PJ, Coss CC, Phelps MA, Kearbey JD, Miller DD, Dalton JT. Selective androgen receptor modulator treatment improves muscle strength and body composition and prevents bone loss in orchidectomized rats. Endocrinology. 2005;146:4887–4897.
Gao W, Reiser PJ, Kearbey JD, Phelps MA, Coss CC, Miller DD, Dalton JT. Effects of Novel Selective Androgen Receptor Modulator (SARM) on Skeletal Muscle Mass and Strength in Castrated Male Rats. The Endocrine Society; New Orleans: 2004.
Kearbey JD, Gao W, Narayanan R, Fisher SJ, Wu D, Miller DD, Dalton JT. Selective Androgen Receptor Modulator (SARM) treatment prevents bone loss and reduces body fat in ovariectomized rats. Pharmaceutical research. 2007;24:328–335.
Bohl CE, Gao W, Miller DD, Bell CE, Dalton JT. Structural basis for antagonism and resistance of bicalutamide in prostate cancer. Proc Natl Acad Sci U S A. 2005;102:6201–6206. An important paper that describes the structural basis of antagonism of bicalutamide based on the crystal structure.
Hamann LG, Manfredi MC, Sun C, Krystek SR, Jr, Huang Y, Bi Y, Augeri DJ, Wang T, Zou Y, Betebenner DA, Fura A, Seethala R, Golla R, Kuhns JE, Lupisella JA, Darienzo CJ, Custer LL, Price JL, Johnson JM, Biller SA, Zahler R, Ostrowski J. Tandem optimization of target activity and elimination of mutagenic potential in a potent series of N-aryl bicyclic hydantoin-based selective androgen receptor modulators. Bioorganic & medicinal chemistry letters. 2007;17:1860–1864.
Manfredi MC, Bi Y, Nirschl AA, Sutton JC, Seethala R, Golla R, Beehler BC, Sleph PG, Grover GJ, Ostrowski J, Hamann LG. Synthesis and SAR of tetrahydropyrrolo[1,2-b][1,2,5]thiadiazol-2(3H)-one 1,1-dioxide analogues as highly potent selective androgen receptor modulators. Bioorganic & medicinal chemistry letters. 2007;17:4487–4490.
Ostrowski J, Kuhns JE, Lupisella JA, Manfredi MC, Beehler BC, Krystek SR, Jr, Bi Y, Sun C, Seethala R, Golla R, Sleph PG, Fura A, An Y, Kish KF, Sack JS, Mookhtiar KA, Grover GJ, Hamann LG. Pharmacological and x-ray structural characterization of a novel selective androgen receptor modulator: potent hyperanabolic stimulation of skeletal muscle with hypostimulation of prostate in rats. Endocrinology. 2007;148:4–12.
Kim J, Wu D, Hwang DJ, Miller DD, Dalton JT. The para substituent of S-3-(phenoxy)-2-hydroxy-2-methyl-N-(4-nitro-3-trifluoromethyl-phenyl)-prop ionamides is a major structural determinant of in vivo disposition and activity of selective androgen receptor modulators. The Journal of pharmacology and experimental therapeutics. 2005;315:230–239.
Hanada K, Furuya K, Yamamoto N, Nejishima H, Ichikawa K, Nakamura T, Miyakawa M, Amano S, Sumita Y, Oguro N. Bone anabolic effects of S-40503, a novel nonsteroidal selective androgen receptor modulator (SARM), in rat models of osteoporosis. Biol Pharm Bull. 2003;26:1563–1569.
Miner JN, Chang W, Chapman MS, Finn PD, Hong MH, Lopez FJ, Marschke KB, Rosen J, Schrader W, Turner R, van Oeveren A, Viveros H, Zhi L, Negro-Vilar A. An orally active selective androgen receptor modulator is efficacious on bone, muscle, and sex function with reduced impact on prostate. Endocrinology. 2007;148:363–373.
van Oeveren A, Motamedi M, Mani NS, Marschke KB, Lopez FJ, Schrader WT, Negro-Vilar A, Zhi L. Discovery of 6-N,N-bis(2,2,2-trifluoroethyl)amino-4-trifluoromethylquinolin-2(1H)-one as a novel selective androgen receptor modulator. Journal of medicinal chemistry. 2006;49:6143–6146.
van Oeveren A, Motamedi M, Martinborough E, Zhao S, Shen Y, West S, Chang W, Kallel A, Marschke KB, Lopez FJ, Negro-Vilar A, Zhi L. Novel selective androgen receptor modulators: SAR studies on 6-bisalkylamino-2-quinolinones. Bioorganic & medicinal chemistry letters. 2007;17:1527–1531.
Ng RA, Lanter JC, Alford VC, Allan GF, Sbriscia T, Lundeen SG, Sui Z. Synthesis of potent and tissue-selective androgen receptor modulators (SARMs): 2-(2,2,2)-Trifluoroethyl-benzimidazole scaffold. Bioorganic & medicinal chemistry letters. 2007;17:1784–1787.
Narayanan R, Coss CC, Yepuru M, Kearbey JD, Miller DD, Dalton JT. Steroidal androgens and nonsteroidal, tissue-selective androgen receptor modulator, S-22, regulate androgen receptor function through distinct genomic and nongenomic signaling pathways. Mol Endocrinol. 2008;22:2448–2465. This paper showed that DHT and SARMs activate distinct signaling pathways.
Masiello D, Chen SY, Xu Y, Verhoeven MC, Choi E, Hollenberg AN, Balk SP. Recruitment of beta-catenin by wild-type or mutant androgen receptors correlates with ligand-stimulated growth of prostate cancer cells. Mol Endocrinol. 2004;18:2388–2401.
Song LN, Herrell R, Byers S, Shah S, Wilson EM, Gelmann EP. Beta-catenin binds to the activation function 2 region of the androgen receptor and modulates the effects of the N-terminal domain and TIF2 on ligand-dependent transcription. Mol Cell Biol. 2003;23:1674–1687.
Sathya G, Chang CY, Kazmin D, Cook CE, McDonnell DP. Pharmacological uncoupling of androgen receptor-mediated prostate cancer cell proliferation and prostate-specific antigen secretion. Cancer Res. 2003;63:8029–8036.
Sathya G, Chang CY, Kazmin D, Cook CE, McDonnell DP. Pharmacological uncoupling of androgen receptor-mediated prostate cancer cell proliferation and prostate-specific antigen secretion. Cancer Res. 2003;63:8029–8036.
Bohl CE, Wu Z, Miller DD, Bell CE, Dalton JT. Crystal structure of the T877A human androgen receptor ligand-binding domain complexed to cyproterone acetate provides insight for ligand-induced conformational changes and structure-based drug design. J Biol Chem. 2007;282:13648–13655.
Heinlein CA, Chang C. Androgen receptor (AR) coregulators: an overview. Endocr Rev. 2002;23:175–200.
Hamann LG. Discovery and preclinical profile of a highly potent and muscle selective androgen receptor modulator (SARM). 227th National Meeting of the American Chemical Society Medicinal Chemistry Division.2004.
Sinha-Hikim I, Artaza J, Woodhouse L, Gonzalez-Cadavid N, Singh AB, Lee MI, Storer TW, Casaburi R, Shen R, Bhasin S. Testosterone-induced increase in muscle size in healthy young men is associated with muscle fiber hypertrophy. Am J Physiol Endocrinol Metab. 2002;283:E154–164.
Kadi F, Eriksson A, Holmner S, Thornell LE. Effects of anabolic steroids on the muscle cells of strength-trained athletes. Medicine and science in sports and exercise. 1999;31:1528–1534.
Sinha-Hikim I, Roth SM, Lee MI, Bhasin S. Testosterone-induced muscle hypertrophy is associated with an increase in satellite cell number in healthy, young men. Am J Physiol Endocrinol Metab. 2003;285:E197–205.
Singh R, Artaza JN, Taylor WE, Gonzalez-Cadavid NF, Bhasin S. Androgens stimulate myogenic differentiation and inhibit adipogenesis in C3H 10T1/2 pluripotent cells through an androgen receptor-mediated pathway. Endocrinology. 2003;144:5081–5088. This paper was the first to report that androgens regulate myogenic differentiation of mesenchymal multipotent cells.
Gupta V, Bhasin S, Guo W, Singh R, Miki R, Chauhan P, Choong K, Tchkonia T, Lebrasseur NK, Flanagan JN, Hamilton JA, Viereck JC, Narula NS, Kirkland JL, Jasuja R. Effects of dihydrotestosterone on differentiation and proliferation of human mesenchymal stem cells and preadipocytes. Molecular and cellular endocrinology. 2008;296:32–40.
Singh R, Artaza JN, Taylor WE, Braga M, Yuan X, Gonzalez-Cadavid NF, Bhasin S. Testosterone inhibits adipogenic differentiation in 3T3-L1 cells: nuclear translocation of androgen receptor complex with beta-catenin and T-cell factor 4 may bypass canonical Wnt signaling to down-regulate adipogenic transcription factors. Endocrinology. 2006;147:141–154.
Brodsky IG, Balagopal P, Nair KS. Effects of testosterone replacement on muscle mass and muscle protein synthesis in hypogonadal men–a clinical research center study. J Clin Endocrinol Metab. 1996;81:3469–3475.
Ferrando AA, Sheffield-Moore M, Yeckel CW, Gilkison C, Jiang J, Achacosa A, Lieberman SA, Tipton K, Wolfe RR, Urban RJ. Testosterone administration to older men improves muscle function: molecular and physiological mechanisms. Am J Physiol Endocrinol Metab. 2002;282:E601–607.
Singh R, Bhasin S, Braga M, Artaza JN, Pervin S, Taylor WE, Krishnan V, Sinha SK, Rajavashisth TB, Jasuja R. Regulation of Myogenic Differentiation by Androgens: Cross-Talk between Androgen Receptor/{beta}-Catenin and Follistatin/TGF-{beta} Signaling Pathways. Endocrinology. 2008 This paper describes the important role of beta-catenin/Wnt pathway in mediating the effects of testosterone on myogenic differentiation and the role of follistatin in cross-communicating the signal from Wnt to TGFbeta/SMAD pathway.
Hikichi, Yukiko, et al. “Selective androgen receptor modulator activity of a steroidal antiandrogen TSAA-291 and its cofactor recruitment profile.” European journal of pharmacology 765 (2015): 322-331.
Becker, H., et al. “In vivo uptake and metabolism of 3H-testosterone and 3H-5α-dihydrotestosterone by human benign prostatic hypertrophy.” European Journal of Endocrinology 71.3 (1972): 589-599.
Foradori, C. D., M. J. Weiser, and R. J. Handa. “Non-genomic actions of androgens.” Frontiers in neuroendocrinology 29.2 (2008): 169-181.
Negro-Vilar, Andres. “Selective androgen receptor modulators (SARMs): a novel approach to androgen therapy for the new millennium.” The Journal of Clinical Endocrinology & Metabolism 84.10 (1999): 3459-3462.
Kim, Juhyun, et al. “The para substituent of S-3-(phenoxy)-2-hydroxy-2-methyl-N-(4-nitro-3-trifluoromethyl-phenyl)-propionamides is a major structural determinant of in vivo disposition and activity of selective androgen receptor modulators.” Journal of Pharmacology and Experimental Therapeutics 315.1 (2005): 230-239.
Neil, David, et al. “GSK2881078, a SARM, produces dose-dependent increases in lean mass in healthy older men and women.” The Journal of Clinical Endocrinology & Metabolism 103.9 (2018): 3215-3224.
Aikawa, Katsuji, et al. “Synthesis and biological evaluation of novel selective androgen receptor modulators (SARMs) Part III: Discovery of 4-(5-oxopyrrolidine-1-yl) benzonitrile derivative 2f as a clinical candidate.” Bioorganic & medicinal chemistry 25.13 (2017): 3330-3349.
Min, Liu, et al. “A novel synthetic androgen receptor ligand, S42, works as a selective androgen receptor modulator and possesses metabolic effects with little impact on the prostate.” Endocrinology 150.12 (2009): 5606-5616.
Schmidt, Azriel, et al. “Identification of an anabolic selective androgen receptor modulator that actively induces death of androgen-independent prostate cancer cells.” The Journal of steroid biochemistry and molecular biology 143 (2014): 29-39.
Basaria, Shehzad, et al. “The safety, pharmacokinetics, and effects of LGD-4033, a novel nonsteroidal oral, selective androgen receptor modulator, in healthy young men.” Journals of Gerontology Series A: Biomedical Sciences and Medical Sciences 68.1 (2013): 87-95.
Bhasin, Shalender, et al. “Testosterone dose-response relationships in healthy young men.” American Journal of Physiology-Endocrinology And Metabolism (2001).
Bhasin, Shalender, et al. “Effect of testosterone supplementation with and without a dual 5α-reductase inhibitor on fat-free mass in men with suppressed testosterone production: a randomized controlled trial.” Jama 307.9 (2012): 931-939.
Bhasin, Shalender, et al. “Older men are as responsive as young men to the anabolic effects of graded doses of testosterone on the skeletal muscle.” The Journal of Clinical Endocrinology & Metabolism 90.2 (2005): 678-688.
Siamo pienamente a conoscenza dell’impatto che una condizione di stress cronicamente protratto, sia fisico che psichico, ha sulla attivazione dell’Asse Ipotalamo-Ipofisi-Surrene (nota anche nella denominazione inglese di Hypothalamic–Pituitary–Adrenal Axis; abbreviato in HPA Axis) e la sintesi di Cortisolo con conseguenze negative anche a livello fisico con un accentuato stato catabolico del tessuto proteico del muscolo-scheletrico. Ma lo stress cronico potrebbe compromettere maggiormente la capacità di sviluppo della massa contrattile rispetto a quanto normalmente potremmo essere indotti a pensare.
Asse Ipotalamo-Ipofisi-Surrene (HPA). Sperimentare un fattore di stress ambientale, come percepito dal cervello, provoca l’attivazione dell’Asse HPA. L’Ipotalamo secernerà quindi l’Ormone di Rilascio della Corticotropina (CRH). Nel lobo anteriore dell’Ipofisi, il CRH stimola la secrezione dell’Ormone Adrenocorticotropo (ACTH). La corteccia delle ghiandole surrenali produrrà quindi glucocorticoidi (Cortisolo nell’uomo) in risposta all’ACTH. Il Cortisolo genererà quindi una risposta allo stress.
In un piccolo studio svolto su topi presso la University of Colorado e pubblicato nel settembre 2010 sul American Journal of Physiology, è stato riportato che lo stress causa una iper-espressione del gene della Miostatina.[1]
Miostatina
Sappiamo benissimo che la Miostatina, espressa nell’uomo dal gene MSTN, è un peptide che nel muscolo maturo inibisce l’Akt, una chinasi sufficiente a causare l’ipertrofia muscolare, in parte attraverso l’attivazione della sintesi proteica mentre stimola la produzione di ubiquitina ligasi, proteine che regolano la disgregazione proteica muscolare. Tuttavia, l’Akt non è responsabile di tutti gli effetti ipertrofici muscolari osservati che sono mediati dall’inibizione della miostatina[2] Pertanto la Miostatina agisce in due modi: inibendo la sintesi proteica indotta dal Akt e stimolando la degradazione proteica regolata dall’ubiquitina.
Caratteristiche dello studio
I ricercatori hanno fatto la loro scoperta quando hanno posto dei topi in una gabbia diversa ogni giorno per una settimana [CS], o li hanno messi per un breve periodo di tempo ogni giorno in una piccola “camera restrittiva” [RS]. Quest’ultimo trattamento risulta essere particolarmente doloroso per questi animali.
Dopo 7 giorni la massa muscolare di entrambi i gruppi era diminuita, ma in modo maggiore dei topi RS.
La sigla “TA” sta per muscolo del polpaccio tibiale anteriore, “SOL” per soleo. BC = massa muscolare all’inizio dell’esperimento, HC = massa muscolare in un gruppo di controllo di topi che sono stati pesati ogni giorno, ma non hanno ricevuto stimoli stressanti.
I ricercatori notarono che i muscoli dei topi sottoposti a stress iniziavano a produrre più Miostatina il giorno 1, in particolare i topi RS.
Poiché volevano sapere se la Miostatina svolgesse davvero un ruolo così importante nello stress psicologico, i ricercatori hanno ripetuto il loro esperimento con topi geneticamente modificati che non producevano Miostatina [MSTN KO]. Questi sono i topi massivamente ipertrofici le cui foto sono reperibili in rete. Essi provengono dal laboratorio dell’esperto di Miostatina Se-Jin Lee.
I topi non modificati sono topi WT.
Conclusioni
I ricercatori hanno concluso che lo stress psicologico può portare all’obesità e all’indebolimento muscolare.
Le diminuzioni della massa muscolare possono contribuire ovviamente a un cambiamento nella composizione corporea che può favorire l’obesità. Una perdita di massa muscolare magra riduce la quantità di tessuto metabolicamente attivo disponibile per la degradazione ossidativa dei substrati energetici.
Inoltre, una diminuzione della massa muscolo-scheletrica in risposta allo stress psicologico può anche predisporre il muscolo scheletrico a una maggiore probabilità o gravità di lesioni.
Ma nell’uomo le conseguenze dello stress psicofisico possono essere le medesime sui livelli aumentati di Miostatina osservati nei topi? Non possiamo ancora sapere con certezza quanto e come ciò possa influire sull’espressione genica MSTN nell’uomo, ma possiamo ipotizzare che ciò sia potenzialmente verificabile. Inoltre ciò non dovrebbe nemmeno stupire. Un organismo sottoposto a stress cronico tende a ricercare una omeostasi anche attraverso la riduzione del dispendio energetico tramite una riduzione del tessuto metabolicamente attivo. Gli esempi non mancano in letteratura. Pensate per esempio ai soggetti denutriti o stanziati in ambienti inospitali.
2- Sartori R, Gregorevic P, Sandri M (September 2014). “TGFβ and BMP signaling in skeletal muscle: potential significance for muscle-related disease”. Trends in Endocrinology and Metabolism.
DISCLAIMER: Il presente articolo è a solo scopo educativo, di intrattenimento e informativo. Non rappresenta in alcun modo una forma di incitamento all’uso/abuso di sostanze dopanti. L’autore ed il sito, per tanto, è esentato da qualsiasi responsabilità dipendente dalla libera scelta individuale.
Introduzione ad un dilemma…
Chiunque frequenti l’ambiente del Bodybuilding e del Fitness avrà letto o sentito almeno una volta nella vita espressioni del genere “se mi dopassi sarei anche io così [indicando Flex Wheeler]” o “ho provato di tutto e senza farmaci non riuscirò ad ottenere risultati”. Andando poi ad approfondire la storia di ognuno di questi soggetti si scopre in percentuale quasi assoluta che si tratta di individui nella norma (o al di sotto) frustrati e/o con personalità deboli, speranzosi omini che attendono placidamente che accada una svolta miracolosa nella loro banale e piatta esistenza e, cosa molto importante, con il minimo dello sforzo (meglio se nessuno).
Nella mia esperienza come ricercatore e operatore nel campo della cultura fisica in qualità di Preparatore Atletico, ho assistito a innumerevoli casi in cui un soggetto aspirava al miglioramento della propria composizione corporea trascurando, consciamente o inconsciamente, le basi fondamentali rappresentate da Nutrizione e Allenamento baipassandole in vista della possibile prescrizione di una pillola miracolosa capace di renderlo/a possessore della forma fisica ambita.
Tralasciando l’ovvio ragionamento che spinge ogni essere umano dotato di un minimo d’intelletto verso la comprensione che la genetica è il blocco d’argilla sul quale si va ad operare, ma le sue qualità e difetti sono presenti in modo eterogeneo nella popolazione mondiale, e ciò non è modificabile nemmeno con la farmacologia più oculata, quando ci si trova davanti al bivio tra “pillola rossa” (PEDs) e “pillola blu” (drug free) bisogna essere pienamente consapevoli non solo del fattore illegalità ma del fattore conoscitivo. Purtroppo, la politica del terrore ha operato in modo fallimentare nel goffo intento di allontanare dalla scelta “rossa”, e ciò si è tradotto in un numero sensibile di soggetti abusatori con tutte le conseguenze cliniche derivanti.
Se un individuo non ha raggiunto un livello di maturità sportiva tale da conferirgli una gestione corretta della nutrizione e della periodizzazione allenante (gestione delle variabili volume, intensità, densità ecc…), è molto meglio per lui/lei rivedere i suoi programmi e scegliere ancora la “pillola blu”. Capita, a volte, di incontrare persone decise ad intraprendere la via del “lato oscuro” che, dopo una approfondita chiacchierata sulla gestione dei suddetti fattori, rivede le proprie posizioni.
Per tutti coloro i quali sono immersi nel dilemma della scelta, vi espongo alcuni punti per rendere l’eventuale decisione meno rischiosa anche se pur sempre illegale nel “bel paese”…
“Pillola blu o pillola rossa?” I punti da tenere in considerazione per una scelta consapevole:
#1 Raggiungere una adeguata maturità sportiva
Per “maturità sportiva”, in particolare riferimento al BodyBuilding, si intende la capacità del atleta di sapersi alimentare e allenare correttamente con piena gestione delle proprie potenzialità fisiologiche/genetiche. Questa è la base, se viene a mancare ciò non solo la vostra esperienza finirà per deludervi e rendervi ancora di più dei frustrati, ma potrebbe rovinosamente portarvi ad un abuso cronico a senso inesorabilmente negativo…
#2 I PEDs non faranno miracoli
Una cosa da tenere bene a mente, e questo non dovrebbe interessare solo gli aspiranti “doped”, è che l’uso di PEDs non renderà diversi da ciò che rientra nelle potenzialità espressive del proprio patrimonio genetico. Certamente le caratteristiche genetiche verranno “iperespresse”, nel bene e nel male, dall’uso di PEDs ma non vi sarà nessun miracolo! Migliorerete ma non sarete ne più ne meno di ciò che potete essere!
Un esempio per capire come la base genetica faccia la differenza anche con protocolli che, ad oggi, spesso non raggiungono nemmeno i livelli del “bridge” più soft..
#3 Ridurre la percentuale di grasso corporeo
Il tessuto adiposo rappresenta uno dei siti dove il Testosterone, ed altri AAS soggetti all’aromatizzazione, viene convertito in Estradiolo. Soggetti con percentuali di grasso corporeo elevate vedrebbero una alterazione marcata della Testosterone:Estradiolo ratio a favore della componente estrogenica, con conseguenze quali alterazione del comportamento sessuale (impotenza, difficoltà nel raggiungere e/o mantenere l’erezione), ritenzione idrica, accumulo di grasso con modello femminile e ginecomastia. E no, l’uso di DHT derivati o di SARM non steroidei senza una base di Testosterone non risolverebbe il problema o, per lo meno, porterebbe ad altre conseguenze negative, che pur non comprendendo, per esempio, ritenzione idrica e ginecomastia, interesserebbero l’attività sessuale e la condizione psichica del soggetto trattato. [1]
Schema esemplificato del processo di aromatizzazione degli androgeni aumentati in un soggetto con percentuale di grasso corporeo alta.
Allo stesso tempo, i rischi cardiovascolari della somministrazione di AAS- come il possibile aumento esponenziale del Ematocrito, l’aumento del LDL e Trigliceridi a discapito di una riduzione del HDL, e l’aumento della pressione sanguigna – sarebbero già presenti in certa misura quando la body fat è già alta e sarebbero quindi soggetti ad un repentino aggravamento.
Se la percentuale di grasso è relativamente alta, si dovrebbe prima di tutto considerare di migliorare la composizione corporea con una adeguata routine alimentare e allenante (senza farmaci) prima di iniziare solo a pensare all’uso di AAS. Sicuramente ciò renderà la scelta più efficace e meno rischiosa.
Nel caso fosse necessario sottolinearlo, no, non è saggio nemmeno utilizzare agenti PEDs a fini lipolitici e/o antiadipogenici e/o termogenici (compresi gli Ormoni Tiroidei). A meno che non siate affetti da ipotiroidismo, e in questo caso la terapia vi dovrebbe essere stilata dal vostro medico, per ridurre in modo sensibile la body fat non sono necessari i farmaci!
#4 Controllare se si ha una storia familiare di trombosi (o qualsiasi altra malattia cardiovascolare)
Molte malattie cardiovascolari hanno una componente di base genetica. Uno stile di vita sano può ridurne sensibilmente la loro insorgenza, ma l’uso di AAS può causare l’attivazione di specifici geni implicati nella comparsa di malattie cardio-circolatorie. Caratteristico dell’interazione tra AAS e geni specifici è un caso studio ben documentato che ricercatori americani hanno pubblicato sul “Blood Coagulation & Fibrinolysis”.[2]
Trombosi venosa
Oltre all’attivazione genica diretta dagli AAS, e nociva per il sistema cardio-circolatorio, vi sono altre condizioni negative innescate dall’uso/abuso di Steroidi Anabolizzanti, e di altri PEDs, come, per esempio, l’aumento del tasso di coagulazione, l’incremento eccessivo dell’Ematocrito con aumento pressorio, rigidità dell’endotelio vascolare con perdita di efficienza strutturale e aumento della pressione ematica con incremento delle possibilità di danno strutturale dei componenti del sistema interessato.
#5 Inserire delle sedute di allenamento cardio prima, durante e dopo l’uso di PEDs
Un moderato allenamento cardiovascolare è sicuramente una delle migliori strategie preventive contro la comparsa di malattie cardio-circolatorie. Tale tipologia di allenamento può portare un miglioramento e/o riduzione delle alterazioni lipidiche ematiche del praticante, fornendo un, seppur minimo, tampone all’azione negativa degli AAS e SARM non steroidei sui livelli di LDL (aumento), Trigliceridi (aumento) e HDL (diminuzione). Secondo quanto riportato da una interessante review del 2013, l’abbinamento di sedute cardio e in sala pesi possono avere una azione additiva benefica sui livelli di LDL, Trigliceridi e HDL.[3]
Risulta interessante anche quanto emerso da alcuni studi su animali a seguito dei quali si è osservato un significativo grado di protezione dato dall’allenamento cardio negli esemplari trattati con AAS.[4]
#6 Assicurarsi di rimanere ben idratati
Oltre ad agevolare il mantenimento di un Ematocrito migliore, una buona idratazione risulta positiva sulla pressione di lavoro renale nel filtraggio del sangue. Diversi AAS come il Trenbolone e i metilati in C-17 presentano una particolare resistenza metabolica che, oltre a causare un aumentato stress epatico, può portare ad una sofferenza renale sfociabile nel patologico. Si è osservato come una combinazione di AAS, dieta iperproteica e supplementazione di Creatina possa aumentare l’incidenza di problemi renali.[5] In un soggetto in fisiologia, la sola dieta ad altro contenuto proteico e la supplementazione di Creatina non hanno mostrato nessun grado di pericolosità, soprattutto sul breve/medio termine.
#7 Non usare “droghe ricreative”
A livello globale, il numero di decessi tra gli abusatori di AAS è in aumento. Alcuni, troppo superficialmente, dicono che questo sia dovuto al fatto che sempre più uomini e donne usano AAS, ma questa è solo una spiegazione dozzinale e limitata. Il sospetto ricade soprattutto sulle modalità di approccio dei consumatori di AAS: i dosaggi sono drammaticamente aumentati e un numero crescente di individui combina PEDs con “droghe ricreative”. Ed è su questi due ultimi punti che risiede la spiegazione principale dell’aumento statistico prima menzionato. Soprattutto la combinazione di PEDs e le così dette “droghe ricreative” risulta essere probabilmente un fattore significativo, come evidenziato alcuni anni fa da ricercatori australiani. Nel loro studio sono state analizzate tutte le morti documentate tra i consumatori di AAS a Sydney tra il 1997 e il 2012, scoprendo che le droghe ricreative come la cocaina avevano avuto un ruolo nella schiacciante molteplicità dei casi. Dagli studi sugli animali ora sappiamo della possibilità che la co-assunzione di un AAS come il Nandrolone con la cocaina vede moltiplicati gli effetti cardiotossici rispetto ai singoli composti.[6] E secondo studi in vitro la combinazione di Testosterone e cocaina aumenterebbe la possibilità di formazione di coaguli nel flusso ematico. [7]
#8 Corretta modalità di iniezione e herpes labiale
Gli utilizzatori di AAS a volte sviluppano ascessi, ma non sempre dovuti alla bassa qualità dei prodotti utilizzati.
Alcuni medici ritengono che gli utilizzatori di AAS dovrebbero effettivamente ricevere una formazione sulle tecniche di iniezione corrette, onde evitare embolie oleose o ascessi per cattiva gestione igienica della procedura. [8]
Molti utilizzatori ancora non sanno che disturbi come l’herpes labiale rendono le iniezioni ancora più rischiose. Perchè? Il virus che causa l’herpes labiale, come altri patogeni, riduce l’efficienza del sistema immunitario, fornendo così terreno fertile per infezioni batteriche i cui microorganismi scatenanti vengono inoculati nel corpo del soggetto attraverso l’iniezione in modo diretto o indiretto.
#9 Non fare affidamento sugli integratori
Secondo un buon numero di studi svolti su animali, alcuni integratori proteggono dagli effetti collaterali degli AAS. Secondo alcune ricerche, la Taurina, la Vitamina C ed E proteggono i testicoli durante un ciclo e la vitamina C e il cacao proteggono la prostata.
L’utilità dei risultati provenienti da questi studi è limitata per tre motivi:
A. gli animali da laboratorio non sono esseri umani, e
B. le dosi utilizzate e rapportate ad un essere umano sono quasi sempre molto inferiori rispetto a quelle utilizzate dai “doped”, e
C. la ricerca in campo psicologico mostra che l’uso di integratori stimola comportamenti rischiosi e malsani. I supplementi fanno pensare agli utilizzatori di essere invulnerabili e di non dover comportarsi in modo sano ed attento.[9]
Gli integratori possono aiutare a creare una mentalità che non si dovrebbe avere da utilizzatore consapevole di AAS.
Ovviamente, alcuni supplementi “protettivi” utilizzati dai soggetti meglio informati hanno un potenziale di “tamponare” in modo discreto alcune alterazioni legate all’uso di AAS e SARM come, ma non limitato a, Riso Rosso fermantato (controllo lipidico) [10], Silimarina (epatoprotezione), NAC (epatoprotezione) [11], Niacina (controllo lipidico) ecc…
#10 Ridurre al minimo (se non eliminare) il consumo di alcolici
Potrebbe sembrare un indicazione superflua ma non lo è.
L’abuso di alcol è indubbiamente uno dei problemi sociali più diffusi. Uno dei problemi correlati all’abuso di alcol e l’epatopatia alcolica. Questo stato patologico è derivante da un processo infiammatorio progressivo ai danni del fegato legato al consumo eccessivo di alcolici. È una malattia a più stadi. La steatosi provoca un ingrossamento del fegato causato da un accumulo di trigliceridi, spesso senza sintomi per molto tempo. I rischi correlati sono la steatosi (fegato grasso), l’epatite alcolica e la cirrosi epatica. Il rapporto con l’alcolismo è complesso. Non tutti i bevitori, infatti, hanno danni al fegato, anche se sono altamente probabili. La causa è da rinvenire in una trasformazione dell’alcol (etanolo) in sostanze tossiche che danneggiano il fegato in maniera irreversibile e cronica, con un rischio elevato di insufficienza epatica e di cancro, fino alla necessità di un trapianto di fegato.
In acuto, invece, l’alcol può essere una causa di alterazione delle transaminasi ma non si può sapere se e con quale modalità si potrebbero innalzare: dipende molto dalla risposta individuale dell’organismo. In caso di stress preesistente, di causa iatrogena e/o alimentare, si può presentare una alterazione significativa. [12]
Il primo caso è una consequenziale possibile se eventi stressori concomitanti si presentano in cronico. Ed è semplice giungere alla conclusione che l’uso di AAS, specie se metilati, possa comportare un aumentato stress epatico che potrebbe degenerare in peliosi epatica, cirrosi ecc…
Che siate “doped” o “natural”, per ragioni legate e non, dovreste evitare di consumare più di 25g per gli uomini, o 12,5g per le donne, di Etanolo al giorno.
#11 Sottoporsi a regolari controlli medici pre, intra e post utilizzo
Il monitoraggio della salute dovrebbe essere la base fondante del comportamento del utilizzatore consapevole e minimamente attento ai potenziali rischi nei quali potrebbe imbattersi.
Gli esami di controllo sono i seguenti:
Esami ematici e delle urine (comprendenti il quadro ormonale secondo necessità);
Elettrocardiogramma ogni 6 mesi circa;
Elettrocardiogramma sotto sforzo (prima di iniziare);
Ecocardiogramma ogni 6 mesi circa;
Coronarografia ogni 6 mesi circa;
Monitoraggio della pressione ematica;
TAC addome completa ogni 6 mesi circa.
Ovviamente, ogni accertamento , al di la degli esami ematici, deve essere gestito in base alle esigenze soggettive, caratteristiche e tipo di PEDs utilizzati.
#12 Essere seguiti da personale qualificato
Fin troppa gente è stata salutisticamente deturpata da gorilla di spogliatoio a mala pena consapevoli dell’esistenza dei macronutrienti e che, nonostante ciò, si sono improvvisati farmacisti. Donne divenuti uomini e uomini divenuti simili a cagne in calore per via di orrende ginecomastie. Evitate il fai da te e l’affidarsi a semianalfabeti … la somaticità sopra la norma è cosa diversa dall’intelligenza e alla competenza in biologia, biochimica e farmacologia… senza offesa per tutti quelli che “io mi facevo e ho vinto! Senzia scienzia!” …
#13 Pensare seriamente al post ciclo prima del ciclo
Molti aspiranti “doped” non considerano il fattore post ciclo. La maggior parte di loro è convinta che la PCT sarà una facile soluzione alla sottoregolazione dell’Asse HPT, ma in realtà non è proprio così. Esistono diversi casi studio che mostrano come gli ex utilizzatori abbiano spesso livelli di Testosterone inferiori rispetto al pre-utilizzo anche a distanza di anni dal cessato uso di AAS. Sembra che i fattori che aumentano le possibilità e il grado di tale effetto sul lungo termine siano:
Tempo di somministrazione;
Età
Molecole utilizzate (con maggiore impatto negativo dato dai19-norsteroidi come il Nandrolone per via della lunga permanenza dei metaboliti nel sistema).
Tutto ciò è indipendente dalla qualità della PCT, anche se essa può avere dei riscontri positivi specie nel primo periodo di stacco dagli AAS. Le alterazioni ormonali legate ad una alterazione dell’Asse HPT comprendono depressione, ansia, bassa libido, difficoltà nel raggiungere e mantenere l’erezione, stanchezza cronica ecc…
Per questa ragione molti scelgono di entrare in TRT (Terapia Sostitutiva del Testosterone) dopo il primo ciclo.
Quale conclusione?…
Se mai non dovesse bastare il disclaimer, questo articolo non rappresenta in alcun modo un consiglio e, ne tanto meno, un incitamento all’uso di sostanze dopanti! E’ semplicemente a fine divulgativo con l’obbiettivo di far comprendere a più persone possibili che la scelta di intraprendere coscientemente certe pratiche (illegali) necessita di una sufficiente (e veritiera) conoscenza del argomento.
Quindi? Leggete e comprendete correttamente ciò che ho riportato in sintesi fruibile ad un largo pubblico… Pensate prima di tutto ad alimentarvi e allenarvi in modo ottimale!
La conoscenza della Verità rende liberi dalla cattiva informazione, dagli strumenti commerciali e dal relativismo… Negarla è semplice e pericolosa manifestazione di profonda ignoranza… di VERO NEGAZIONISMO!
Se avete una buona conoscenza della lingua inglese e volete approfondire l’argomento PEDs e Sport, potete leggere il libro ANABOLICS 11th Edition di William Llewellyn
Ricercatori danesi hanno sviluppato un analogo della Follistatina che ha mostrato, nei topi, di causare un aumento della massa muscolare specifica del 19% in una settimana.(1) Questa forma di Follistatina sembrerebbe priva dell’effetto collaterale che ha frenato la ricerca con questa classe di composti.
La ricerca sugli inibitori della Miostatina è tutt’ora in corso, ma, come già accennato, è stata rallentata dalla scoperta che almeno un tipo di inibitori della Miostatina ha l’effetto collaterale di aumentare l’eritropoiesi, con conseguente aumento dell’ematocrito e sue complicanze. Stiamo parlando quindi di composti sintetici con attività di legame con i recettori IIA e IIB. Queste sostanze hanno azione antagonista della Miostatina. Il meccanismo attraverso il quale questi composti causano un incremento dell’eritropoiesi non è al momento noto.
Per questa ragione, i ricercatori danesi, presso l’ospedale universitario di Aarhus, hanno iniziato la ricerca per individuare un altro inibitore della Miostatina che fosse privo dell’effetto di aumento della eritropoiesi. Così, hanno tentato la via della modifica strutturale della molecola di Follistatina. Come ormai risaputo, la Follistatina è una glicoproteina sintetizzata dalle cellule muscolari, e che controlla l’effetto della Miostatina impedendo a quest’ultima di potersi legare al proprio recettore.
Al principio della ricerca, non venne modificata la forma regolare ma dall’analogo FST-dHBS.(2) A questo analogo è stata aggiunta poi una porzione di catena amminoacidica: la regione Fc della molecola IgG.
In vitro, la molecola di Follistatina modificata ha neutralizzato l’Activina A e B, la Miostatina e il GDF11 – ugualmente alla Follistatina non modificata.
La somministrazione della Follistatina regolare non è molto efficiente. La sostanza [FST315] in breve tempo non è più rilevabile nel flusso ematico. Tuttavia, dopo la somministrazione della forma modificata [FST-dHBS-hFc] i livelli ematici del composto hanno raggiunto concentrazioni elevate. Inoltre, una settimana dopo l’inoculazione nei topi di una dose elevata, la molecola modificata [FST-dHBS-mFc] era ancora rilevabile nel sangue.
Quando i ricercatori hanno trattato i topi con la forma modificata di Follistatina per una settimana, il peso corporeo degli animali ha subito un incremento dell’11%; il peso del gastrocnemio era aumentato del 19%. La nuova molecola ha avuto un effetto anabolizzante maggiore rispetto al ActRIIA-mFC [ActRIIA = activin type IIA receptor].
Durante la settimana dell’esperimento, i ricercatori hanno sottoposto i topi a tre iniezioni nell’intestino tenue di Follistatina modificata. Questo potrebbe suggerire che questa molecola potrebbe persino essere attiva per via orale. La dose somministrata era di 10mg per chilogrammo di peso corporeo. Qui potete leggere come calcolare l’equivalente umano della dose utilizzata.
A differenza del ActRIIA-mFC, il nuovo analogo della Follistatina non ha mostrato di avere effetti sul livello dei globuli rossi e, di conseguenza, sull’ematocrito.
In sintesi, la FST-DHBS-mFc rappresenta una molecola di segnalazione del recettore dell’Activina che causa un aumento della massa muscolo-scheletrica senza però influenzare negativamente globuli rossi, emoglobina ed ematocrito.
I ricercatori concludono dicendo che questo apre la possibilità di un uso terapeutico di composti con attività di segnale del recettore dell’Activina in pazienti con ematocrito normale o alto e pone l’FST-DHBS-mFc in una posizione unica tra gli altri farmaci candidati che interferiscono con le vie dei 3 ligandi cardine: l’Activina A, la Miostatina e il GDF11.
Se non avete ancora letto le precedenti parti componenti questa serie di articoli vi invito a farlo prima di procedere con la lettura di questa quarta ed ultima parte: 1° Parte – 2° Parte – 3° Parte.
Farmacocinetica, Farmacodinamica e Feedback Negativi
Come già detto, il fegato rappresenta il principale bersaglio del GH, il quale è il principale regolatore della sintesi epatica di IGF-1. Per causare tale effetto, il GH si lega con i GHR localizzati nel dominio extracellulare degli epatociti stimolando successivamente la produzione di IGF-1 endocrino tramite la trascrizione genica, utilizzando la via di segnalazione JAK-STAT. Inoltre, è stato dimostrato che la somministrazione di GH causa una rapida sovraregolazione dell’mRNA del IGF-1 nel fegato.[338]
Aumenti dei livelli serici di IGF-1 si verificano molto rapidamente anche in presenza di un grande bolo di rHGH. Incrementi significativi di IGF-1 sono già osservabili dopo 6-12h dall’iniezione.[339] Questi livelli serici di IGF-1 continuano ad aumentare fino a raggiungere il loro punto di saturazione dose-dipendente entro 4-7 giorni, anche quando si utilizzano dosi estremamente elevate che ammontano a 20-30UI al giorno di rHGH.[340] In particolare, il punto di saturazione si è rivelato essere compreso nell’intervallo dei 700-800 ng/mL e sembra suggerire che i livelli endocrini di IGF-1 hanno un tetto massimo negli adulti sani. I meccanismi esatti devono ancora essere chiariti, ma sono probabilmente il risultato dei complessi meccanismi di controllo intrinseci all’Asse GH/IGF-1. Coloro i quali desiderano elevare i livelli endocrini di IGF-1 al fine di ottenerne un vantaggio sull’ipertrofia dovrebbe tenerlo a mente, in quanto vi è un punto in cui l’uso di dosi maggiori di rHGH semplicemente non si traducono in elevati livelli serici di IGF-1. Qui di seguito ho riportato il grafico dello studio di Tanaka il quale mostra la relazione tra l’rhGH e i livelli serici di IGF-:
Ora, vorrei dedicarmi brevemente all’analisi dell’azione del IGF-1autocrino e del perché esso rappresenti un mediatore cruciale del processo ipertrofico, prima di tornare nuovamente a discutere su questioni inerenti alla farmacodinamica e farmacocinetica. La segnalazione recettoriale del IGF-1 è unica nel suo genere, e questo lo si deve al fatto che utilizza due percorsi distinti per stimolare la proliferazione o la differenziazione.[341-343] Questo è un comportamento abbastanza interessante, poiché nessun altro membro della famiglia dei fattori di crescita ha dimostrato di agire in tal modo. Poiché la proliferazione e la differenziazione sono processi opposti, inizialmente era difficile per i ricercatori capire come un singolo fattore di crescita, attraverso un singolo recettore, potesse inviare un segnale che attivasse entrambi.[294] Da quando sono state fatte queste prime scoperte, è stato ulteriormente chiarito che l’IGF-1 non svolge simultaneamente queste azioni. Test su varie linee di coltura cellulare hanno dimostrato che gli effetti proliferativi arrivano prima, durando tra le 24 e le 36 ore. È solo dopo questa fase proliferativa iniziale che si verifica la differenziazione miogenica.[344]
Gli effetti proliferativi mediati dall’IGF-1 sui mioblasti sono noti sin dagli anni ’70, quando vennero osservati per la prima volta nelle cellule epatiche di ratto.[345] Questa stimolazione proliferativa del IGF-1 si traduce in un aumento del numero di cellule, nei livelli di proteine, nella sintesi del DNA, nell’assorbimento di aminoacidico, nell’assorbimento del glucosio e nella soppressione della proteolisi.[346] Nelle colture cellulari umane, l’IGF-1 ha anche dimostrato di aumentare la dimensione dei miotubi indipendentemente dal fatto che i mioblasti proliferino attivamente o che la proliferazione sia cessata. Regola la dimensione dei miotubi attivando la sintesi proteica, inibendo la degradazione proteica e inducendo la fusione delle cellule di riserva.[347-348] La capacità dell’IGF di sopprimere la proteolisi nel muscolo scheletrico, la scomposizione delle proteine in aminoacidi, è stata dimostrata innumerevoli volte nel corso degli anni.[349-352] È stato anche dimostrato che l’IGF-1 induce la proliferazione e la differenziazione delle cellule satelliti in miociti maturi, come determinato da un aumento del numero di miofibre nucleate a livello centrale rispetto a quelle periferiche.[148,353-354]
La capacità dell’IGF-1 autocrino di causare la differenziazione dei mioblasti è stata in realtà una scoperta che potremmo definire quasi “ibrida” dal momento che degli studi svolti negli anni ’60 avevano mostrato che questo effetto si verifica con alti livelli di Insulina.[355] Successivamente è stato dimostrato che gli IGF sono stimolatori molto più potenti nella differenziazione miogenica rispetto all’Insulina e si è concluso che la stessa Insulina agisce realmente come un analogo dell’IGF-1 in questo sistema.[356-357] Gli effetti di differenziazione dati dall’IGF-1 autocrino sono bifasici, con basse concentrazioni che stimolano progressivamente la differenziazione dei mioblasti mentre concentrazioni molto elevate mostrano una cessazione dell’attività di differenziazione. Il limite massimo per la differenziazione sembra attestarsi a circa 100ng/mL per l’IGF-1 e 300ng/mL per IGF-2.[358] Questo effetto non è legato alla proliferazione, poiché non si osservano ulteriori aumenti nel numero complessivo delle cellule.[294] È possibile che le molecole di segnalazione coinvolte nella regolazione negativa del sistema miogenico siano aumentate, ma questa è una affermazione puramente speculativa.[359-360]
La somministrazione di rHGH eleva l’espressione dell’mRNA dell’IGF-1 nel muscolo scheletrico in numerosi modelli cellulari, umani e animali.[127,150,361-364] Ciò avviene abbastanza rapidamente, entro 60 minuti dall’iniezione sottocutanea di rHGH ed i picchi sono segnalati tra le 6 e le 12 ore post iniezione.[363] In questo particolare modello animale citato, il raddoppio della dose di GH non ha portato ad ulteriori aumentati dei livelli di mRNA dell’IGF-1, il che suggerisce che esiste un sistema di regolazione che determina quanto GH sia necessario per stimolare al massimo l’espressione locale dell’IGF-1 nel muscolo scheletrico. In precedenza si è potuto appurare che la differenziazione dei miociti mediata dall’IGF-1 si arresta quando le concentrazioni locali raggiungono circa i 100ng/mL, ma quanto GH è necessario per raggiungere il punto di saturazione dell’espressione dell’mRNA dell’IGF-1?
Gli studi sui miociti umani mostrano che il GH aumenta l’espressione dell’mRNA dell’IGF-1 entro 30-60 minuti con picchi molto più rapidi rispetto a quelli osservati negli studi sugli animali, entro 1-2 ore, usando la via di segnalazione JAK / STAT5b.[365] Questi livelli elevati di mRNA hanno dimostrato di durare fino a 48 ore dopo una singola esposizione al GH. La quantità di GH necessaria per stimolare al massimo l’espressione dell’mRNA dell’IGF-1 è risultata essere una dose compresa tra i 7,5ng/mL e 30ng/ml [366], con una dose media efficace che si attesta a 3ng/ml. Questi numeri sono in linea con gli intervalli di dose fisiologica osservati negli animali, che sono effettivamente compresi tra i 2-100 ng/mL.[367] Inoltre, si collocano esattamente in linea con quanto si osserva endogenamente nell’uomo, con concentrazioni normali di picco comprese tra i 22,4 e 32,4ng/mL.[368-369,436] Ci sono stati casi in cui gli uomini presi in esame hanno mostrato concentrazioni di picco leggermente più alte, ma questi devono essere considerati valori anomali.[370] In ogni caso, ciò che questi dati tendono a suggerire è che il corpo umano è particolarmente adatto a gestire livelli naturali di picco della secrezioni di GH endogeno. Cercare di incidere ulteriormente il sistema elevando i livelli di GH oltre quelli endogeni, unicamente per tentare di potenziare i processi ipertrofici, potrebbe in realtà non tradursi nell’effetto desiderato.
Gli studi che mettono a confronto le infusioni locali con le infusioni sistemiche di GH o IGF-1 sono un po’ più difficili da trovare di quanto si vorrebbe. Le poche sperimentazioni sugli animali che sono riuscito a trovare indicano che l’infusione diretta di GH o IGF-1 nei tessuti bersaglio determina un aumento della massa muscolare. Questo aumento dell’ipertrofia si verifica anche senza che il muscolo bersaglio sia stato sottoposto ad attività motoria.[371-372] Gli studi dimostrano anche che le iniezioni locali di GH portano a livelli sostanzialmente più alti nell’espressione dell’mRNA dell’ IGF-1 locale rispetto alle iniezioni locali di IGF-1, di un fattore di oltre venti.[127] Sono riuscito a trovare uno studio nel quale si confrontavano le risposte dei ratti (attivi e non) all’infusione locale di IGF-1. Il gruppo “IGF-1 plus training” ha mostrato un aumento sia della massa muscolare che della forza locale maggiore rispetto al semplice trattamento in isolamento.[373] Quindi, anche se limitata, la letteratura disponibile è apparentemente in grado di dimostrare che le iniezione locali di GH o IGF-1 hanno effettivamente valore.
Ne ho già parlato diverse volte ma, nel tentativo di imprimere ulteriormente questo concetto, è necessario ricordarsi che i livelli autocrini di IGF-1 sembrano essere molto più importanti dei livelli endocrini di IGF-1 in relazione alla regolazione della massa muscolare. Oltre a questo punto, la sovraespressione dell’IGF-1 autocrino nel muscolo provoca l’ipertrofia delle fibre.[374] La sovraespressione dell’IGF-1 autocrino ha anche mostrato effetti anti-catabolici, con modelli animali tendenti a mostrare una resistenza generale all’atrofia muscolare normalmente osservata con l’invecchiamento.[375] L’IGF-1 localizzato fornisce anche capacità rigenerative indipendenti dall’età nelle cellule muscolari.[376]
Vi sono anche alcune prove convincenti che suggeriscono che l’IGF-1 endocrino agisce direttamente come un regolatore di feedback negativo sulla produzione di IGF-1 autocrino. Questo meccanismo di feedback negativo è dipendente dal pathway PI3K/Akt [377-378]. Inoltre, elevati livelli di IGF-1 endocrino possono anche agire indirettamente per sopprimere la produzione di IGF-1 autocrino. Quindi, in altre parole, non solo l’IGF-1 endocrino ha un impatto diretto minore sulla regolazione della massa muscolare, ma può anche sopprimere l’IGF-1 autocrino che ha impatti maggiori sull’ipertrofia.
Elevati livelli di IGF-1 circolante e, nello specifico, di IGF-1 libero elevati agiscono in modo negativo sul GH determinando un tasso di soppressione della produzione di IGF-1 autocrino a valle.[379] Non è del tutto chiaro, tuttavia, se la regolazione negativa dell’IGF-1 modifichi l’emivita dell’mRNA dell’IGF-1 o influenzi direttamente l’espressione del gene IGF-1. Oltre a questo, è stato anche dimostrato che l’espressione dell’IGF-1 autocrino è sottoregolata nelle cellule muscolari dopo trattamento con IGF-1.[366] È stato anche dimostrato che l’espressione epatica dell’mRNA dell’IGF-1 è sottoregolata dall’esposizione acuta all’IGF-1.[127] Quindi, mantenere livelli endocrini il più possibile soppressi con rispettiva dose di rHGH, elevando contemporaneamente i livelli autocrini, dovrebbe essere un fattore prioritario in un protocollo di GH volto all’ipertrofia.
Il GH è pulsatile per natura sia nell’uomo che nelle specie animali. Quindi, sarebbe logico pensare che molti dei processi intrinseci del corpo saranno tarati in modo tale da rispondere in maniera ottimale all’esposizione al GH in modo simile. In accordo con questa affermazione è stato dimostrato che solo la somministrazione di GH pulsatile, e non l’infusione continua, ha la capacità di stimolare massimamente l’espressione dell’mRNA dell’IGF-1 nel muscolo scheletrico.[366,380-381] È stato anche dimostrato che la somministrazione pulsatile porta ad un aumento del potenziale di crescita postnatale complessivo rispetto all’infusione continua.[89,382] La somministrazione pulsatile può anche portare a livelli endocrini di IGF-1 serici comparabili, o addirittura diminuiti [383], il che è vantaggioso a causa delle potenziali capacità di regolazione negativa che possiede sull’espressione dell’IGF-1 autocrino e che sono state discusse in precedenza. L’evidenza suggerisce anche che il picco stesso, e non necessariamente il numero di picchi, potrebbe essere della massima importanza per i tessuti bersaglio.[384] Per la massima crescita e potenziale ipertrofico, l’evidenza tende a suggerire che creare picchi di GH elevati, e quindi tornare ai livelli basali più volte al giorno, può essere preferibile rispetto a mantenerli elevati per periodi di tempo più lunghi. Questo pratica permette di riprodurre gli schemi secretori in vivo.
I pathways del GH coinvolti nell’anabolismo sono anche suscettibili alla desensibilizzazione, che è parte della fisiologia del GH endogeno.[385] A causa della natura intrinsecamente pulsatile del GH in vivo, l’attività dei recettori e dei pathways sono regolati da un impulso seguito da un periodo di inattività.[386] L’esposizione continua o ripetuta al GH senza un adeguato lasso di tempo refrattario comporterà livelli di attività fortemente soppressi. In effetti, nel corso degli anni numerosi studi hanno dimostrato che tale effetto si verifica. Le cellule ed il tessuto muscolare richiedono un periodo refrattario piuttosto lungo prima che la loro piena risposta al GH venga recuperata. Dopo l’esposizione al GH, le cellule muscolari non sono nemmeno in grado di rispondere alle successive dosi di GH. In realtà, occorrono due ore complete per riprendere parzialmente la reattività nei modelli cellulari, con un totale di 6-8 ore di astinenza dall’uso di GH necessarie per ripristinare la piena sensibilità.[366] Viceversa, quando il GH è micro-dosato in impulsi di dieci minuti, seguiti da intervalli di otto ore, è stato mostrato aumentare progressivamente l’mRNA dell’IGF-1 con ogni impulso successivo.[386]
Questo fenomeno è potenzialmente il risultato di una desensibilizzazione complessiva all’interno della via JAK-STAT5, poiché è stato dimostrato che l’esposizione al GH negli studi sulle cellule epatiche causa resistenza alla successiva attivazione della via STAT5 per 4-8 ore.[387-388] Questo lasso di tempo è sufficiente per sincronizzarsi abbastanza bene con ciò che è stato visto nei modelli di cellule miocitarie citati in precedenza. Nei modelli di cellule epatiche, il GH ha stimolato un significativo aumento dell’espressione del SOCS3, che è un potente inibitore dell’azione del GH.[389]. Poiché il GH non ha avuto alcun effetto sull’espressione del SOCS3 nelle cellule muscolari, questo deve essere un altro meccanismo causante il periodo refrattario. Questo meccanismo può essere dipeso dalla sottoregolazione dei GHR, dall’inibizione mediata da un’altra proteina SOCS, o dall’induzione di una tirosina fosfatasi che semplicemente inattiva la via JAK / STAT.[390] La via JAK-STAT5b, che come ricorderete è intimamente associata al muscolo scheletrico e all’espressione dell’IGF-1, è di natura transitoria – con attivazione massima raggiunta entro 10-30 minuti, seguita da un prolungato periodo di inattivazione.
Una scoperta piuttosto nuova di Xu et al. [391] ha dimostrato che anche distanziare le esposizioni al GH di cinque ore lasciava entrambi i percorsi a valle MEK1/2 e ERK1/2 significativamente soppressi rispetto a tutti i percorsi a monte, a causa di una potenziale disconnessione nella trasduzione del segnale . Ciò è di particolare interesse in quanto questi stessi due percorsi a valle sono stati coinvolti in modo significativo sia nella crescita sia nella proliferazione.[392-393] È stato anche scoperto che l’attivazione indotta da GH di STAT1 e STAT3 è stata desensibilizzata, ma l’esposizione all’Insulina inverte la desensibilizzazione osservata in tutti i percorsi interessati. Anche se non sto per trattare approfonditamente l’Insulina, ci sono un paio di importanti punti da dovere prendere in considerazione. Bisogna comprendere innanzitutto che ci sono molti obiettivi a valle del recettore del GH e molti di questi hanno il potenziale per essere desensibilizzati dopo l’esposizione al GH. Bisogna comprendi anche che l’Insulina possiede l’abilità unica di risensibilizzare molti di questi percorsi. Ciò ha un senso vista la relazione tipo yin-yang tra i due composti. È noto che il GH e l’Insulina possiedono una relazione anabolica sinergica a causa di molti effetti che esercitano l’uno sull’altro. Questo sembra essere soltanto un’anteprima di uno di questi effetti.
Asse GH/IGF-1 – Relazione con altri ormoni
Prima di passare alle note conclusive, vorrei trattare brevemente alcuni altri ormoni connessi a diverso grado con l’Asse GH/IGF-1. Per prima cosa, voglio trattare brevemente l’Asse Tiroideo dal momento che l’inserimento di composti tiroidei insieme al GH è una pratica comune anche durante i protocolli di massa.
Il muscolo scheletrico è il principale bersaglio di segnalazione dell’ormone tiroideo, con trasportatori degli ormoni tiroidei e enzimi di conversione espressi localmente.[394] È ben noto che il GH potenzia la deiodinazione periferica che converte il T4 in T3, riducendo così il T4 e il reverse T3, aumentando contemporaneamente i livelli di T3.[395-398] Ciò che molte persone non riescono a capire è che questo è un effetto transitorio, e studi a lungo termine sembrano indicare che gli effetti mediati dal GH sulla conversione periferica si stabilizzino con il tempo.[399-402]
Via ubiquitina/proteasoma
Invece di proseguire ulteriormente su questo, avendo già trattato la questione nel dettaglio in un mio vecchio articolo, preferirei concentrarmi su alcune pubblicazioni relative alla tiroide che non vengono discusse abbastanza spesso. Gli ormoni tiroidei, per loro natura, sono composti tendenzialmente catabolici in quanto stimolano la disgregazione proteica dell’intero corpo in misura maggiore rispetto alla sintesi proteica.[403] A livello locale, nel muscolo scheletrico stimolano un aumento dell’attività all’interno della via ubiquitina/proteasoma, che è ampiamente coinvolta nella proteolisi.[404-406] Il risultato di questo è un tasso accelerato del turnover proteico e una perdita netta complessiva degli aminoacidi situati all’interno dei muscoli scheletrici.
Inoltre, negli esseri umani, sia gli stati di ipertiroidismo che di ipotiroidismo sono stati associati a livelli di IGF-1 soppressi con una tendenza alla normalizzazione quando viene ristabilita una condizione di eutiroidismo. L’ipertiroidismo è anche associato ad una bassa attività di legame recettoriale del GH, che si ipotizza essere il risultato di una ridotta capacità di elaborazione dei recettori del GH.[407] E’ stato anche ipotizzato che l’ipertiroidismo sia in grado anche di accelerare la clearance del GH urinario.[408] Inoltre, studi su animali hanno dimostrato che gli ormoni tiroidei possono avere importanti effetti soppressivi sulla sintesi di IGF-1 stimolata con il GH.[409] Ovviamente, a causa della complessa relazione che l’Asse Tiroideo ha con l’Asse GH/IGF-1, raggruppando tutte le interazioni che hanno tra loro in pochi paragrafi, trattare l’argomento diventerebbe poco pratico. Tuttavia, quando il corpo della letteratura scientifica viene esaminato nella sua interezza, ci sono molte prove che suggeriscono che la supplementazione con composti tiroidei esogeni potrebbe non essere l’ideale quando l’obiettivo di un individuo è l’ipertrofia, anche se la regolazione del dosaggio dei tiroidei in tale contesto rimane la misura di “sicurezza” più intelligente visto l’impatto negativo sulla funzionalità tiroidea dato dal GH. Comunque, per tutti coloro che sono interessati ad approfondire questo argomento, consiglio di iniziare con la review nella nota seguente.[410]
Miostatina
Mi piacerebbe trattare anche la Miostatin, che rappresenta un argomento molto discusso nei vari forum di BodyBuilding presenti in rete. La sua fama proviene dai risultati ipertrofici espressi dai bovini privi per mutazione del gene della Miostatina, i quali mostrano una massa muscolare significativamente maggiore rispetto ai loro simili non mutati.[411] La Miostatina, un fattore di crescita e differenziazione appartenente alla superfamiglia dei TGF-beta, ha dimostrato di inibire selettivamente la miogenesi, in gran parte tramite il suo effetto soppressivo sulla proliferazione dei mioblasti.[412] È espressa e secreta prevalentemente dal muscolo scheletrico. Come molti sanno, se riesci a sopprimere o inibire la Miostatina, di conseguenza il potenziale ipertrofico aumenta significativamente.
Le mutazioni della Miostatina sono state osservate sia negli animali che nell’uomo. Queste mutazioni del gene della Miostatina portano ad un fenotipo ipertrofico negli animali, come accennato in precedenza.[413-415] L’Asse GH/IGF-1 e la Miostatina sembrano avere una relazione regolativa diretta tra loro, come osservato nei pazienti affetti contemporaneamente da GHD e HIV che mostrano marcati aumenti nell’espressione dell’mRNA della Miostatina.[416] Quindi, è possibile che attraverso una supplementazione di dosi sovrafisiologiche di rHGH si possa indurre una diminuzione dell’mRNA della Miostatina [209,417-419]? Sfortunatamente, nonostante la presenza di alcuni casi studio selezionati, non credo che si abbiano abbastanza dati in questo momento per sapere se ciò possa dare risultati apprezzabili in seguito alla sua applicazione.
Quello che sappiamo è che aumenti dell’espressione dell’mRNA dell’IGF-1 e le concentrazioni circolanti di IGF-1 sono state osservati dopo inibizione della Miostatina.[419-421] Sappiamo anche che l’inibizione della Miostatina tende a causare l’ipertrofia attraverso molte delle stesse modalità osservate con l’IGF-1 autocrino, cioè l’aumento della sintesi proteica e l’attivazione delle cellule satelliti.[422-425] E sappiamo anche che l’ipertrofia indotta dalla sovraespressione dell’IGF-1 o dall’inibizione della Miostatina utilizza la stessa identica via – PI3K/Akt/mTOR.[426-428] Tuttavia, l’IGF-1 non è un requisito per l’ipertrofia indotta dalla Follistatina, tranne nel caso di livelli di Insulina estremamente bassi – come ben sappiamo, la Follistatina è un inibitore della Miostatina [429]. E l’esposizione cronica al GH può in realtà portare ad un’espressione sovrastimolata della Miostatina e del suo recettore.[209]
Quindi quello che possiamo dire, con certezza, è che l’espressione della Miostatina non sarà un fattore diretto o indiretto per quanto riguarda il potenziamento dei processi ipertrofici, né dell’attività contrattile, nei muscoli scheletrici umani.[430] Proprio per questo motivo, non ritengo che sia un fattore sul quale gli atleti debbano eccessivamente concentrarsi, al di fuori di un utile arricchimento delle proprie conoscenze in materia.
Applicazioni pratiche e pensieri conclusivi
Arrivati a questo punto è mia intenzione unire tutto ciò che è stato esposto in questa serie di articoli e esporlo sotto forma di alcuni suggerimenti pratici rivolti a tutti coloro i quali vogliono semplicemente massimizzare la loro capacità ipertrofica.
Ora è chiaro che il GH possiede pochissimi, se non nulli, effetti diretti sull’ipertrofia. Pertanto, qualsiasi protocollo di massa che contempli il suo uso dovrà tenere in considerazione questo punto includendo gli AAS, i quali, per l’appunto, hanno anche una proficua sinergia con il GH. Sia la letteratura scientifica che i dati aneddotici dimostrano chiaramente che l’uso combinato di entrambi i composti ha un massimale ipertrofico significativamente più alto rispetto all’uso singolo. Personalmente, penso che i BodyBuilder dovrebbero sempre optare per l’utilizzo di una base di Testosterone e Boldenone (correttamente rapportati in base a contesto e alle caratteristiche individuali) anche in una fase “Bulk” nella quale viene inserito l’uso del GH. Il Trenbolone può essere considerato come parte “accessoria” di un protocollo di massa, a causa della sua intrinseca difficoltà gestazionale come sostanza anabolizzante. Dovrebbe essere usato con parsimonia e con cautela poiché, insieme ai suoi numerosi punti di forza come composto anabolizzante, presenta alcune limitazioni. Queste limitazioni derivano per lo più, come già accennato, dalla difficile gestione del composto in quanto esso non è facilmente tollerato da una buona parte degli individui. Quindi, se viene usato il Trenbolone, dovrebbe essere inserito calcolando con attenzione il dosaggio e la tolleranza individuale, anche per quanto concerne la tolleranza temporale individuale all’uso di tale composto.
Dopo un periodo d’uso prolungato di dosi sovrafisiologico di AAS (Ciclo+Bridge), è buona cosa procedere con l’interruzione o con una marcata riduzione del numero e degli AAS utilizzati. Detta in parole semplici, questa interruzione può contemplare una completa astinenza dagli AAS, svolgendo un adeguata PCT al fine di ristabilire una omeostasi ormonale fisiologica, o una transizione ad una TRT, metodologia comunemente chiamata “blast and cruise”. La struttura del protocollo di supplementazione farmacologica dovrebbe sempre seguire i principi del dosaggio minimo efficace con aumenti nei dosaggi degli AAS solo nel caso si sia raggiunto il limite di crescita con il precedente dosaggio, assicurandosi che tutte le altre variabili nello stile di vita siano correttamente regolate. L’utilizzo di questo approccio limita il rischio che si sviluppino effetti collaterali indesiderati sul lungo termine.
Quando si decide di usare il GH, la dove ce ne sia la possibilità economica, esso dovrebbe provenire da uno dei prodotti presenti nel mercato farmaceutico. Questi prodotti approvati devono superare anni di studi strettamente controllati per dimostrare la loro sicurezza, purezza ed efficacia su soggetti umani. I progressi tecnologici nel corso degli anni hanno reso molto più facile la produzione di rHGH. Per questo motivo, i produttori ora provengono da tutto il mondo. Spesso questi produttori realizzano ciò che viene definito “GH generico” sui forum, ma tale termine non mi piace molto. Definire qualcosa come “generico” implica che sia una replica perfetta di prodotti legati a specifici marchi farmaceutici approvati dell’agenzia del farmaco che hanno perso il cui brevetto è scaduto, il che non è il caso del GH. Infatti, a causa della natura estremamente complessa del processo di produzione del rHGH, per esempio, la FDA non consente nemmeno l’uso del termine “generico” quando si tratta di rHGH e utilizza invece il termine ” follow-on protein product ” o FOPP.
Spesso questi marchi off-label sono venduti ad un costo molto ridotto, ed è qui che sta il dilemma, in quanto questo può essere molto allettante. Tuttavia, con questo costo ridotto per il consumatore, non ci sarà nemmeno la garanzia del produttore su cosa ci sia realmente nella fiala o persino su come è stato prodotto. Il problema di fondo è che il processo di produzione del rHGH è estremamente complessa, ed è molto facile che nelle fasi di questo processo si commettano errori con conseguenti variazioni nella catena proteica che potenzialmente portano ad effetti indesiderati, o anche a risposte autoimmuni.
Spesso gli atleti si affidano semplicemente ai test serici per misurare i livelli di GH e/o IGF-1 al fine di concludere che un prodotto contenete GH sia “buono”, ma dobbiamo ricordarci che raggiungere livelli ematici ormonali elevati è la parte relativamente facile. Anche le molecole di GH che sono state alterate o danneggiate durante la produzione possono dare questo esito. Tuttavia, queste stesse molecole di GH danneggiate o mutate possono spesso stimolare risposte autoimmuni. Ciò potrebbe indurre il corpo ad avere una risposta recettoriale degradata, che può anche riflettersi sulla secrezione endogena nel tempo. [431-432] Rimane poi il problema del reale contenuto della vial o fiala, la quale può non presentare nessun principio attivo al suo interno.
Il GH dovrebbe essere usato in modo pulsatile, per mimare le condizioni in vivo. Tra queste iniezioni, deve esserci un periodo di refrattarietà o si deve consumare un pasto che abbia un buon stimolo sull’Insulina. Può anche essere utilizzata l’Insulina esogena al fine di bypassare molte delle limitazioni del periodo refrattario, ma questo va oltre lo scopo di questo articolo. Anche se il cumulo delle dosi giornaliere dovrebbe essere sovrafisiologico, le dosi individuali non hanno bisogno di essere ad alto dosaggio, poiché la massima stimolazione dell’IGF-1 autocrino nel tessuto muscolo scheletrico si verifica ben all’interno delle concentrazioni fisiologiche di GH. Aneddoticamente, sembra anche esserci un limite con il quale l’uso di rHGH diventa additivo in presenza di AAS. Potrebbe essere necessario un po’ di ponderata (e supportata da personale qualificato) auto-sperimentazione per scoprire dove si trova questa singola dose di saturazione, ma la maggior parte dei soggetti troverà questo limite tra le 4 e le 8 UI/die. Oltre questo dosaggio, la maggior parte degli utilizzatori tenderà a scoprire che la giustificazione dei costi e il rapporto rischio /beneficio tendono a diminuire rapidamente.
Non bisognerebbe passare troppo tempo a riflettere sul tempo delle iniezioni di GH, dal momento che gli aumenti dei livelli di IGF-1 autocrino avvengono rapidamente e possono rimanere elevati per giorni. Bisognerebbe concentrati invece sul programma di iniezione che si addice meglio al contesto della giornata, tenendo contemporaneamente presenti le linee guida per il periodo di refrattarietà del GH. Si possono anche prendere in considerazione piccole o grandi iniezioni, poiché alcuni potrebbero trovare più pratiche e funzionali iniezioni con un dosaggio inferiore e somministrate più frequenti mentre altri potrebbero preferire iniezioni con un dosaggio maggiore e somministrazioni meno frequenti. Naturalmente, maggiore è il contenuto dell’iniezione, maggiore è la probabilità che si superi la soglia massima di espressione dell’IGF-1 autocrino.
Massimizzare l’espressione dell’IGF-1 autocrino, mentre contemporaneamente si sopprimono i livelli di IGF-1 endocrino, sarà una priorità. Esistono prove a sostegno dell’ipotesi secondo cui un iniezione locale di GH possa aiutare a raggiungere questo obiettivo, con una conseguente minore possibilità di feedback negativo. Sono stati osservati aumenti significativi della massa muscolare in appena due settimane di iniezioni locali di IGF-1.[441]
La dove cause di forza maggiore non lo impediscano, è consigliabile evitare o comunque regolare attentamente l’uso di tutti quei composti che possono avere interazioni negative con l’uso del GH a fini ipertrofici. Inibitori della Aromatasi, Modulatori Selettivi del Recettore degli Estrogeni e T3 hanno tutti dimostrato di avere un potenziale effetto negativo sul processo globale ipertrofico legato al GH e per tale motivo dovrebbero essere usati con parsimonia, se non omessi del tutto dove possibile (vedi in particolare il T3).
Questo non dovrebbe sorprendere nessuno e non dovrebbe nemmeno essere troppo difficile da tenere a mente: allenarsi duramente, allenarsi in modo intelligente e allenarsi in modo coerente. Sebbene non sia stato affrontato direttamente nell’articolo, bisogna capire che l’allenamento contro resistenza ha impatti unici e additivi sull’ipertrofia. In effetti, alcuni di questi meccanismi non sono nemmeno mediati dall’Asse AR e/o GH/IGF-1. [433] Bisogna anche comprendere che non esiste una “magica” routine allenante universalmente applicabile, la chiave di volta sarà la coerenza nel garantire un carico di lavoro adeguato, con elementi di sovraccarico progressivo nel tempo assicurando un adeguato stimolo meccanico gestendo al meglio le variabili allenanti (intensità, volume, densità e intensità percepita). La logica composizione del piano allenante servirà a garantire lo stimolo ipertrofico di base che sarà coadiuvato dall’azione dei composti utilizzati.
Nonostante la mole e l’importanza delle informazioni presentate in questa serie di articoli, è necessario ricordare che i meccanismi d’azione ormonali sono governati da innumerevoli fattori. Anche esaminando l’intero corpo della letteratura scientifica equivarrebbe a poco più che accumulare una serie di utili nozioni garanti di assicurare una solida base conoscitiva sull’argomento la quale rappresenterà un punto di partenza intelligente per l’applicazione pratica, rimanendo sempre soggetti alle variabili di risposta individuale. Seguendo questa linea, i migliori risultati nella pratica spesso provengono da coloro i quali posseggono una buona conoscenza dei principi scientifici e la capacità innata di saperli applicare non solo su se stessi ma, soprattutto, su terzi. Infatti, molto raramente due persone rispondono in modo identico alla supplementazione di ormoni esogeni (e non solo), quindi, non bisogna assolutamente pensare che basti semplicemente applicare su se stessi o su terzi un protocollo che ha portato benefici realmente apprezzabili su un soggetto per ottenere la medesima risposta.
A tal fine, invito atleti e Preparatori a utilizzare queste pubblicazioni come punto di partenza per essere in grado di gestire l’applicazione pratica in modo più consapevole e produttivo. Inoltre, chi ne fosse in grado, può consultare il vasto numero di riferimenti riportati nel corso di queste pubblicazioni è tentare di ragionare sulle mie conclusioni. Ad ogni citazione presente in questa serie di articoli, assicuratevi che il riferimento elencato supporti effettivamente le affermazioni fatte. Mantenere sempre una mente aperta ma con i giusti “filtri”, e cercare di non credere per partito preso ad una singola opinione, specialmente di fronte alle nuove evidenze scientifiche. Infine, è buona cosa verificare sempre la veridicità di quanto è stato affermato.
Punti conclusivi per un corretto utilizzo della chimica e del GH a fini ipertrofici:
Usare il GH in combinazione con gli AAS
Usare GH e AAS di grado farmaceutico la dove ciò è possibile
Assicurarsi una base di Testosterone correttamente rapportata al Boldenone aggiungendo (in base a maturità e tolleranza) il Trenbolone
Iniettare il GH in modo pulsatile, considerare l’opzione delle iniezioni locali nei gruppi carenti
Mantenere un dosaggio complessivo ottimale di GH il quale si attesta tra le 4 e le 8UI/die
Evitare o regolare attentamente l’uso dei composti che possono interagire negativamente con i processi ipertrofici legati al uso di GH (vedi AI, SERM e T3)
Dopo un periodo di tempo (variabile) nel quale si è stati sottoposti a dosaggi ormonali sovrafisiologici optare per una PCT o per una TRT (a seconda delle proprie necessità e priorità)
Essere a conoscenza dei potenziali effetti collaterali legati all’auso/abuso di GH (nausea, vomito, cefalea, ritenzione idrica e sodica, edemi, parestesie, sindrome del tunnel carpale, rigidità articolare, dolori articolari, artrite, dolori muscolari, ipertensione, insulino-resistenza, diabete di tipo II, acromegalia, dilatazione addominale, ipertrofia cardiaca ecc…)
Svolgere regolarmente esami del sangue; sia durante i periodi di picco nell’uso della farmacologia sia nel periodo successivo (vedi PCT/OCT o TRT)
Gestire al meglio le variabili legate agli stressor ambientali, all’allenamento, all’alimentazione e al sonno.
Gabriel Bellizzi
Riferimenti:
338. Mathews LS, Norstedt G, Palmiter RD. Regulation of insulin-like growth factor I gene expression by growth hormone. Proc Natl Acad Sci U S A. 1986 Dec;83(24):9343-7.
339. Keller A, Wu Z, Kratzsch J, Keller E, Blum WF, Kniess A, Preiss R, Teichert J, Strasburger CJ, Bidlingmaier M. Pharmacokinetics and pharmacodynamics of GH: dependence on route and dosage of administration. Eur J Endocrinol. 2007 Jun;156(6):647-53.
340. Tanaka T, Seino Y, Fujieda K, Igarashi Y, Yokoya S, Tachibana K, Ogawa Y. Pharmacokinetics and metabolic effects of high-dose growth hormone administration in healthy adult men. Endocr J. 1999 Aug;46(4):605-12.
341. Quinn LS, Steinmetz B, Maas A, Ong L, Kaleko M. Type-1 insulin-like growth factor receptor overexpression produces dual effects on myoblast proliferation and differentiation. J Cell Physiol. 1994 Jun;159(3):387-98.
342. Coolican SA, Samuel DS, Ewton DZ, McWade FJ, Florini JR. The mitogenic and myogenic actions of insulin-like growth factors utilize distinct signaling pathways. J Biol Chem. 1997 Mar 7;272(10):6653-62.
343. Foulstone EJ, Huser C, Crown AL, Holly JM, Stewart CE. Differential signalling mechanisms predisposing primary human skeletal muscle cells to altered proliferation and differentiation: roles of IGF-I and TNFalpha. Exp Cell Res. 2004 Mar 10;294(1):223-35.
344. Ewton DZ, Roof SL, Magri KA, McWade FJ, Florini JR. IGF-II is more active than IGF-I in stimulating L6A1 myogenesis: greater mitogenic actions of IGF-I delay differentiation. J Cell Physiol. 1994 Nov;161(2):277-84.
345. Florini JR, Nicholson ML, Dulak NC. Effects of peptide anabolic hormones on growth of myoblasts in culture. Endocrinology. 1977 Jul;101(1):32-41.
346. Laviola L, Natalicchio A, Giorgino F. The IGF-I signaling pathway. Curr Pharm Des. 2007;13(7):663-9. Review.
347. Jacquemin V, Furling D, Bigot A, Butler-Browne GS, Mouly V. IGF-1 induces human myotube hypertrophy by increasing cell recruitment. Exp Cell Res. 2004 Sep 10;299(1):148-58.
348. Jacquemin V, Butler-Browne GS, Furling D, Mouly V. IL-13 mediates the recruitment of reserve cells for fusion during IGF-1-induced hypertrophy of human myotubes. J Cell Sci. 2007 Feb 15;120(Pt 4):670-81. Epub 2007 Jan 30.
349. Ballard FJ, Francis GL. Effects of anabolic agents on protein breakdown in L6 myoblasts. Biochem J. 1983 Jan 15;210(1):243-9.
350. Ewton DZ, Falen SL, Florini JR. The type II insulin-like growth factor (IGF) receptor has low affinity for IGF-I analogs: pleiotypic actions of IGFs on myoblasts are apparently mediated by the type I receptor. Endocrinology. 1987 Jan;120(1):115-23.
351. Hembree JR, Hathaway MR, Dayton WR. Isolation and culture of fetal porcine myogenic cells and the effect of insulin, IGF-I, and sera on protein turnover in porcine myotube cultures. J Anim Sci. 1991 Aug;69(8):3241-50.
352. Hong D, Forsberg NE. Effects of serum and insulin-like growth factor I on protein degradation and protease gene expression in rat L8 myotubes. J Anim Sci. 1994 Sep;72(9):2279-88.
353. Florini JR, Ewton DZ, Roof SL. Insulin-like growth factor-I stimulates terminal myogenic differentiation by induction of myogenin gene expression. Mol Endocrinol. 1991 May;5(5):718-24.
354. Musarò A, McCullagh KJ, Naya FJ, Olson EN, Rosenthal N. IGF-1 induces skeletal myocyte hypertrophy through calcineurin in association with GATA-2 and NF-ATc1. Nature. 1999 Aug 5;400(6744):581-5.
355. Haba GDL, Cooper GW, Elting V. HORMONAL REQUIREMENTS FOR MYOGENESIS OF STRIATED MUSCLE IN VITRO: INSULIN AND SOMATOTROPIN. Proceedings of the National Academy of Sciences of the United States of America. 1966;56(6):1719-1723.
356. Florini JR, Ewton DZ. Insulin acts as a somatomedin analog in stimulating myoblast growth in serum-free medium. In Vitro. 1981 Sep;17(9):763-8.
357. Schmid C, Steiner T, Froesch ER. Preferential enhancement of myoblast differentiation by insulin-like growth factors (IGF I and IGF II) in primary cultures of chicken embryonic cells. FEBS Lett. 1983 Sep 5;161(1):117-21.
358. Florini JR, Ewton DZ, Falen SL, Van Wyk JJ. Biphasic concentration dependency of stimulation of myoblast differentiation by somatomedins. Am J Physiol. 1986 May;250(5 Pt 1):C771-8.
359. Quinn LS, Ehsan M, Steinmetz B, Kaleko M. Ligand-dependent inhibition of myoblast differentiation by overexpression of the type-1 insulin-like growth factor receptor. J Cell Physiol. 1993 Sep;156(3):453-61.
360. Olson EN. Signal transduction pathways that regulate skeletal muscle gene expression. Mol Endocrinol. 1993 Nov;7(11):1369-78. Review.
361. Murphy LJ, Bell GI, Friesen HG. Growth hormone stimulates sequential induction of c-myc and insulin-like growth factor I expression in vivo. Endocrinology. 1987 May;120(5):1806-12.
362. Turner JD, Rotwein P, Novakofski J, Bechtel PJ. Induction of mRNA for IGF-I and -II during growth hormone-stimulated muscle hypertrophy. Am J Physiol. 1988 Oct;255(4 Pt 1):E513-7.
363. Isgaard J, Nilsson A, Vikman K, Isaksson OG. Growth hormone regulates the level of insulin-like growth factor-I mRNA in rat skeletal muscle. J Endocrinol. 1989 Jan;120(1):107-12.
364. Bichell DP, Kikuchi K, Rotwein P. Growth hormone rapidly activates insulin-like growth factor I gene transcription in vivo. Mol Endocrinol. 1992 Nov;6(11):1899-908.
365. Sadowski CL, Wheeler TT, Wang LH, Sadowski HB. GH regulation of IGF-I and suppressor of cytokine signaling gene expression in C2C12 skeletal muscle cells. Endocrinology. 2001 Sep;142(9):3890-900.
366. Frost RA, Nystrom GJ, Lang CH. Regulation of IGF-I mRNA and signal transducers and activators of transcription-3 and -5 (Stat-3 and -5) by GH in C2C12 myoblasts. Endocrinology. 2002 Feb;143(2):492-503.
367. MacLeod JN, Pampori NA, Shapiro BH. Sex differences in the ultradian pattern of plasma growth hormone concentrations in mice. J Endocrinol. 1991 Dec;131(3):395-9.
368. Rochiccioli P, Messina A, Tauber MT, Enjaume C. Correlation of the parameters of 24-hour growth hormone secretion with growth velocity in 93 children of varying height. Horm Res. 1989;31(3):115-8.
369. Hansen TK, Gravholt CH, ØRskov H, Rasmussen MH, Christiansen JS, Jørgensen JO. Dose dependency of the pharmacokinetics and acute lipolytic actions of growth hormone. J Clin Endocrinol Metab. 2002 Oct;87(10):4691-8.
370. Baum WF, Klöditz E, Hesse V, Jahreis G, Schneyer U, Giebler H. [Increase in spontaneous growth hormone secretion in asthmatic children–a symptom of atopic disposition?]. Kinderarztl Prax. 1993 Nov;61(9):323-8.
371. Adams GR, McCue SA. Localized infusion of IGF-I results in skeletal muscle hypertrophy in rats. J Appl Physiol (1985). 1998 May;84(5):1716-22.
372. Alzghoul MB, Gerrard D, Watkins BA, Hannon K. Ectopic expression of IGF-I and Shh by skeletal muscle inhibits disuse-mediated skeletal muscle atrophy and bone osteopenia in vivo. FASEB J. 2004 Jan;18(1):221-3. Epub 2003 Nov 3.
373. Lee S, Barton ER, Sweeney HL, Farrar RP. Viral expression of insulin-like growth factor-I enhances muscle hypertrophy in resistance-trained rats. J Appl Physiol (1985). 2004 Mar;96(3):1097-104. Erratum in: J Appl Physiol. 2004 Jun;96(6):2343.
374. Coleman ME, DeMayo F, Yin KC, Lee HM, Geske R, Montgomery C, Schwartz RJ. Myogenic vector expression of insulin-like growth factor I stimulates muscle cell differentiation and myofiber hypertrophy in transgenic mice. J Biol Chem. 1995 May 19;270(20):12109-16.
375. Barton-Davis ER, Shoturma DI, Musaro A, Rosenthal N, Sweeney HL. Viral mediated expression of insulin-like growth factor I blocks the aging-related loss of skeletal muscle function. Proc Natl Acad Sci U S A. 1998 Dec 22;95(26):15603-7.
376. Musarò A, McCullagh K, Paul A, Houghton L, Dobrowolny G, Molinaro M, Barton ER, Sweeney HL, Rosenthal N. Localized Igf-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat Genet. 2001 Feb;27(2):195-200
377. Lewis MI, Bulut Y, Biring MS, Da X, Fournier M. (1999) IGF-I administration prevents corticosteroids-induced diaphragm atrophy in emphysema . Am J Respir Crit Care Med 159:A580
378. Fournier M, Huang ZS, Cercek B, Li H, Bykhovskaya I, Lewis MI. (2000) Administration of insulin-like growth factor-1 (IGF-I) and corticosteroids in emphysematous hamsters: influences on diaphragm IGF-I . Am J Respir Crit Care Med 161:A18
379. Shavlakadze T, Grounds M. Of bears, frogs, meat, mice and men: complexity of factors affecting skeletal muscle mass and fat. Bioessays. 2006 Oct;28(10):994-1009. Review.
380. Maiter D, Underwood LE, Maes M, Davenport ML, Ketelslegers JM. Different effects of intermittent and continuous growth hormone (GH) administration on serum somatomedin-C/insulin-like growth factor I and liver GH receptors in hypophysectomized rats. Endocrinology. 1988 Aug;123(2):1053-9.
381. Isgaard J, Carlsson L, Isaksson OG, Jansson JO. Pulsatile intravenous growth hormone (GH) infusion to hypophysectomized rats increases insulin-like growth factor I messenger ribonucleic acid in skeletal tissues more effectively than continuous GH infusion. Endocrinology. 1988 Dec;123(6):2605-10.
382. Clark RG, Jansson JO, Isaksson O, Robinson IC. Intravenous growth hormone: growth responses to patterned infusions in hypophysectomized rats. J Endocrinol. 1985 Jan;104(1):53-61.
383. Bick T, Hochberg Z, Amit T, Isaksson OG, Jansson JO. Roles of pulsatility and continuity of growth hormone (GH) administration in the regulation of hepatic GH-receptors, and circulating GH-binding protein and insulin-like growth factor-I. Endocrinology. 1992 Jul;131(1):423-9.
384. Weltman A, Weltman JY, Schurrer R, Evans WS, Veldhuis JD, Rogol AD. Endurance training amplifies the pulsatile release of growth hormone: effects of training intensity. J Appl Physiol (1985). 1992 Jun;72(6):2188-96.
385. Flores-Morales A, Greenhalgh CJ, Norstedt G, Rico-Bautista E. Negative regulation of growth hormone receptor signaling. Mol Endocrinol. 2006 Feb;20(2):241-53. Epub 2005 Jul 21. Review.
386. Hartman ML, Veldhuis JD, Thorner MO. Normal control of growth hormone secretion. Horm Res. 1993;40(1-3):37-47. Review.
387. Fernández L, Flores-Morales A, Lahuna O, Sliva D, Norstedt G, Haldosén LA, Mode A, Gustafsson JA. Desensitization of the growth hormone-induced Janus kinase 2 (Jak 2)/signal transducer and activator of transcription 5 (Stat5)-signaling pathway requires protein synthesis and phospholipase C. Endocrinology. 1998 Apr;139(4):1815-24.
388. Gebert CA, Park SH, Waxman DJ. Termination of growth hormone pulse-induced STAT5b signaling. Mol Endocrinol. 1999 Jan;13(1):38-56.
389. Ram PA, Waxman DJ. SOCS/CIS protein inhibition of growth hormone-stimulated STAT5 signaling by multiple mechanisms. J Biol Chem. 1999 Dec 10;274(50):35553-61.
390. Ram PA, Waxman DJ. Role of the cytokine-inducible SH2 protein CIS in desensitization of STAT5b signaling by continuous growth hormone. J Biol Chem.2000 Dec 15;275(50):39487-96.
391. Xu J, Keeton AB, Franklin JL, Li X, Venable DY, Frank SJ, Messina JL. Insulin enhances growth hormone induction of the MEK/ERK signaling pathway. J Biol Chem. 2006 Jan 13;281(2):982-92. Epub 2005 Nov 4.
392. Lewis TS, Shapiro PS, Ahn NG. Signal transduction through MAP kinase cascades. Adv Cancer Res. 1998;74:49-139. Review.
394. Mebis L, Paletta D, Debaveye Y, Ellger B, Langouche L, D’Hoore A, Darras VM, Visser TJ, Van den Berghe G. Expression of thyroid hormone transporters during critical illness. Eur J Endocrinol. 2009 Aug;161(2):243-50.
395. Jørgensen JO, Pedersen SA, Laurberg P, Weeke J, Skakkebaek NE, Christiansen JS. Effects of growth hormone therapy on thyroid function of growth hormone-deficient adults with and without concomitant thyroxine-substituted central hypothyroidism. J Clin Endocrinol Metab. 1989 Dec;69(6):1127-32.
396. Jørgensen JO, Pedersen SB, Børglum J, Møller N, Schmitz O, Christiansen JS, Richelsen B. Fuel metabolism, energy expenditure, and thyroid function in growth hormone-treated obese women: a double-blind placebo-controlled study. Metabolism. 1994 Jul;43(7):872-7.
397. Wolthers T, Grøftne T, Møller N, Christiansen JS, Orskov H, Weeke J, Jørgensen JO. Calorigenic effects of growth hormone: the role of thyroid hormones. J Clin Endocrinol Metab. 1996 Apr;81(4):1416-9.
398. Feldt-Rasmussen U. Interactions between growth hormone and the thyroid gland — with special reference to biochemical diagnosis. Curr Med Chem. 2007;14(26):2783-8. Review.
399. Kalina-Faska B, Kalina M, Koehler B. Effects of recombinant growth hormone therapy on thyroid hormone concentrations. Int J Clin Pharmacol Ther. 2004 Jan;42(1):30-4.
400. Hubina E, Mersebach H, Rasmussen AK, Juul A, Sneppen SB, Góth MI, Feldt-Rasmussen U. Effect of growth hormone replacement therapy on pituitary hormone secretion and hormone replacement therapies in GHD adults. Horm Res. 2004;61(5):211-7. Epub 2004 Jan 30.
401. Seminara S, Stagi S, Candura L, Scrivano M, Lenzi L, Nanni L, Pagliai F, Chiarelli F. Changes of thyroid function during long-term hGH therapy in GHD children. A possible relationship with catch-up growth? Horm Metab Res. 2005 Dec;37(12):751-6.
402. Losa M, Scavini M, Gatti E, Rossini A, Madaschi S, Formenti I, Caumo A, Stidley CA, Lanzi R. Long-term effects of growth hormone replacement therapy on thyroid function in adults with growth hormone deficiency. Thyroid. 2008 Dec;18(12):1249-54.
403. Müller MJ, Seitz HJ. Thyroid hormone action on intermediary metabolism. Part III. Protein metabolism in hyper- and hypothyroidism. Klin Wochenschr. 1984 Feb 1;62(3):97-102.
404. Tawa NE Jr, Odessey R, Goldberg AL. Inhibitors of the proteasome reduce the accelerated proteolysis in atrophying rat skeletal muscles. J Clin Invest. 1997 Jul 1;100(1):197-203. PubMed PMID: 9202072
405. Dace A, Zhao L, Park KS, et al. Hormone binding induces rapid proteasome-mediated degradation of thyroid hormone receptors. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(16):8985-8990.
406. Clément K, Viguerie N, Diehn M, Alizadeh A, Barbe P, Thalamas C, Storey JD, Brown PO, Barsh GS, Langin D. In vivo regulation of human skeletal muscle gene expression by thyroid hormone. Genome Res. 2002 Feb;12(2):281-91.
407. Miell JP, Taylor AM, Zini M, Maheshwari HG, Ross RJ, Valcavi R. Effects of hypothyroidism and hyperthyroidism on insulin-like growth factors (IGFs) and growth hormone- and IGF-binding proteins. J Clin Endocrinol Metab. 1993 Apr;76(4):950-5.
408. Murao K, Takahara J, Sato M, Tamaki M, Niimi M, Ishida T. Relationship between thyroid functions and urinary growth hormone secretion in patients with hyper- and hypothyroidism. Endocr J. 1994 Oct;41(5):517-22.
409. Wolf M, Ingbar SH, Moses AC. Thyroid hormone and growth hormone interact to regulate insulin-like growth factor-I messenger ribonucleic acid and circulating levels in the rat. Endocrinology. 1989 Dec;125(6):2905-14.
410. Laron Z. Interactions between the thyroid hormones and the hormones of the growth hormone axis. Pediatr Endocrinol Rev. 2003 Dec;1 Suppl 2:244-9-discussion 250. Review.
411. Fiems LO. Double Muscling in Cattle: Genes, Husbandry, Carcasses and Meat. Animals : an Open Access Journal from MDPI. 2012;2(3):472-506.
412. Langley B, Thomas M, Bishop A, Sharma M, Gilmour S, Kambadur R. Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J Biol Chem. 2002 Dec 20;277(51):49831-40. Epub 2002 Sep 18.
413. McPherron AC, Lee SJ. Double muscling in cattle due to mutations in the myostatin gene. Proc Natl Acad Sci U S A. 1997 Nov 11;94(23):12457-61.
414. Schuelke M, Wagner KR, Stolz LE, Hübner C, Riebel T, Kömen W, Braun T, Tobin JF, Lee SJ. Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med. 2004 Jun 24;350(26):2682-8.
415. Clop A, Marcq F, Takeda H, Pirottin D, Tordoir X, Bibé B, Bouix J, Caiment F, Elsen JM, Eychenne F, Larzul C, Laville E, Meish F, Milenkovic D, Tobin J, Charlier C, Georges M. A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat Genet. 2006 Jul;38(7):813-8.
416. Gonzalez-Cadavid NF, Taylor WE, Yarasheski K, Sinha-Hikim I, Ma K, Ezzat S, Shen R, Lalani R, Asa S, Mamita M, Nair G, Arver S, Bhasin S. Organization of the human myostatin gene and expression in healthy men and HIV-infected men with muscle wasting. Proc Natl Acad Sci U S A. 1998 Dec 8;95(25):14938-43.
417. Liu W, Thomas SG, Asa SL, Gonzalez-Cadavid N, Bhasin S, Ezzat S. Myostatin is a skeletal muscle target of growth hormone anabolic action. J Clin Endocrinol Metab. 2003 Nov;88(11):5490-6.
418. Oldham JM, Osepchook CC, Jeanplong F, Falconer SJ, Matthews KG, Conaglen JV, Gerrard DF, Smith HK, Wilkins RJ, Bass JJ, McMahon CD. The decrease in mature myostatin protein in male skeletal muscle is developmentally regulated by growth hormone. J Physiol. 2009 Feb 1;587(3):669-77.
419. Williams NG, Interlichia JP, Jackson MF, Hwang D, Cohen P, Rodgers BD. Endocrine actions of myostatin: systemic regulation of the IGF and IGF binding protein axis. Endocrinology. 2011 Jan;152(1):172-80.
420. Winbanks CE, Weeks KL, Thomson RE, Sepulveda PV, Beyer C, Qian H, Chen JL, Allen JM, Lancaster GI, Febbraio MA, Harrison CA, McMullen JR, Chamberlain JS, Gregorevic P. Follistatin-mediated skeletal muscle hypertrophy is regulated by Smad3 and mTOR independently of myostatin. J Cell Biol. 2012 Jun 25;197(7):997-1008.
421. Lach-Trifilieff E, Minetti GC, Sheppard K, Ibebunjo C, Feige JN, Hartmann S, Brachat S, Rivet H, Koelbing C, Morvan F, Hatakeyama S, Glass DJ. An antibody blocking activin type II receptors induces strong skeletal muscle hypertrophy and protects from atrophy. Mol Cell Biol. 2014 Feb;34(4):606-18.
422. Bark TH, McNurlan MA, Lang CH, Garlick PJ. Increased protein synthesis after acute IGF-I or insulin infusion is localized to muscle in mice. Am J Physiol. 1998 Jul;275(1 Pt 1):E118-23.
423. Barton-Davis ER, Shoturma DI, Sweeney HL. Contribution of satellite cells to IGF-I induced hypertrophy of skeletal muscle. Acta Physiol Scand. 1999 Dec;167(4):301-5.
424. Suryawan A, Frank JW, Nguyen HV, Davis TA. Expression of the TGF-beta family of ligands is developmentally regulated in skeletal muscle of neonatal rats. Pediatr Res. 2006 Feb;59(2):175-9.
425. Gilson H, Schakman O, Kalista S, Lause P, Tsuchida K, Thissen JP. Follistatin induces muscle hypertrophy through satellite cell proliferation and inhibition of both myostatin and activin. Am J Physiol Endocrinol Metab. 2009 Jul;297(1):E157-64.
426. Bodine SC, Stitt TN, Gonzalez M, Kline WO, Stover GL, Bauerlein R, Zlotchenko E, Scrimgeour A, Lawrence JC, Glass DJ, Yancopoulos GD. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol. 2001 Nov;3(11):1014-9.
428. Kalista S, Schakman O, Gilson H, Lause P, Demeulder B, Bertrand L, Pende M, Thissen JP. The type 1 insulin-like growth factor receptor (IGF-IR) pathway is mandatory for the follistatin-induced skeletal muscle hypertrophy. Endocrinology. 2012 Jan;153(1):241-53.
429. Barbé C, Kalista S, Loumaye A, Ritvos O, Lause P, Ferracin B, Thissen JP. Role of IGF-I in follistatin-induced skeletal muscle hypertrophy. Am J Physiol Endocrinol Metab. 2015 Sep 15;309(6):E557-67. doi: 10.1152/ajpendo.00098.2015. Epub 2015 Jul 28.
430. Coffey VG, Shield A, Canny BJ, Carey KA, Cameron-Smith D, Hawley JA. Interaction of contractile activity and training history on mRNA abundance in skeletal muscle from trained athletes. Am J Physiol Endocrinol Metab. 2006 May;290(5):E849-55.
431. Moore WV, Leppert P. Role of aggregated human growth hormone (hGH) in development of antibodies to hGH. J Clin Endocrinol Metab. 1980 Oct;51(4):691-7
432. Dannies PS. Protein folding and deficiencies caused by dominant-negative mutants of hormones. Vitam Horm. 2000;58:1-26. Review.
433. DeVol DL, Rotwein P, Sadow JL, Novakofski J, Bechtel PJ. Activation of insulin-like growth factor gene expression during work-induced skeletal muscle growth. Am J Physiol. 1990 Jul;259(1 Pt 1):E89-95.
434. Hermansen K, Bengtsen M, Kjær M, Vestergaard P, Jørgensen JOL. Impact of GH administration on athletic performance in healthy young adults: A systematic review and meta-analysis of placebo-controlled trials. Growth Horm IGF Res. 2017 Jun;34:38-44.
435. de Souza GL, Hallak J. Anabolic steroids and male infertility: a comprehensive review. BJU Int. 2011 Dec;108(11):1860-5.
436. Kraemer WJ, Marchitelli L, Gordon SE, Harman E, Dziados JE, Mello R, Frykman P, McCurry D, Fleck SJ. Hormonal and growth factor responses to heavy resistance exercise protocols. J Appl Physiol (1985). 1990 Oct;69(4):1442-50.
437. Pfeffer LA, Brisson BK, Lei H, Barton ER. The insulin-like growth factor (IGF)-I E-peptides modulate cell entry of the mature IGF-I protein. Mol Biol Cell. 2009 Sep;20(17):3810-7.
438. Mills P, Dominique JC, Lafrenière JF, Bouchentouf M, Tremblay JP. A synthetic mechano growth factor E Peptide enhances myogenic precursor cell transplantation success. Am J Transplant. 2007 Oct;7(10):2247-59.
439. Brisson BK, Barton ER. Insulin-like growth factor-I E-peptide activity is dependent on the IGF-I receptor. PLoS One. 2012;7(9):e45588.
440. Brisson BK, Spinazzola J, Park S, Barton ER. Viral expression of insulin-like growth factor I E-peptides increases skeletal muscle mass but at the expense of strength. Am J Physiol Endocrinol Metab. 2014 Apr 15;306(8):E965-74.
441. Goldspink G, Harridge S. Mechanism for adaptation in skeletal muscle In: Komi P, editor. Strength and power in sport: Olympic encyclopedia of sports medicine. Oxford: Blackwell; 2002. p. 231–51.
442. Janssen JA, Hofland LJ, Strasburger CJ, van den Dungen ES, Thevis M. Potency of Full-Length MGF to Induce Maximal Activation of the IGF-I R Is Similar to Recombinant Human IGF-I at High Equimolar Concentrations. PLoS One. 2016 Mar 18;11(3):e0150453.
Se si assume una dose di Terbutalina poco più alta della normale dose terapeutica utilizzata dai soggetti asmatici, è potenzialmente possibile riscontrare un aumento della massa muscolare pari ad 1KG in un mese. Questo, ovviamente, può accadere se ci si allena contro resistenza in modo serio, ma anche quando si svolge sufficiente attività fisica. Gli scienziati dello sport dell’Università di Copenaghen sono arrivati a questa conclusione in seguito ad uno studio svolto su esseri uomani, pubblicato sullo Scandinavian Journal of Medicine & Science in Sports. (1)
La Terbutalina, come il Clenbuterolo e il Salbutamolo, è un beta-2 agonista. I beta-2 agonisti, come suggerisce il nome, si legano selettivamente al recettore beta-2 adrenergico. Quando questo recettore viene attivato, la muscolatura liscia si rilassa e le vie respiratorie si dilatano.
Molti beta-2 agonisti presentano anche un effetto anti-catabolico, il quale (teoricamente) permette agli utilizzatori di aumentare o conservare la massa muscolare (effetto solitamente limitato dalla dose necessaria richiesta e da un aumento del Cortisolo). Come ben sappiamo, gli atleti supplementari farmacologicamente spesso usano i beta-2 agonisti per agevolare la riduzione del grasso corporeo.
Se gli atleti hanno l’asma, è consentito loro usare la Terbutalina. Da quanto si sa, le dosi terapeutiche di Terbutalina non hanno alcun effetto sulla massa muscolare. Ma i ricercatori danesi si sono chiesti cosa succederebbe se gli atleti utilizzassero una dose più elevata di Terbutalina di quanto sia normalmente necessario per il trattamento dell’asma.
I ricercatori, che tra l’altro erano pagati dal governo danese e dalla WADA, hanno reclutato per l’esperimento 66 uomini sani e attivi tra i 18 ed i 36 anni. I soggetti si allenavano per 2-5 ore a settimana. Alcuni giocavano a calcio, altri praticavano jogging o andavano in bici tutti i giorni da e verso il lavoro. I ricercatori hanno diviso i soggetti in 3 gruppi.
Ai soggetti del primo gruppo non sono state applicate modifiche alla loro routine di attività fisica [Habitual].
I soggetti del secondo gruppo sono stati sottoposti ad allenamenti di resistenza. Per 4 settimane, 3 volte a settimana, i soggetti eseguivano un allenamento a intervalli, che consisteva in 3 sessioni da 10 minuti di Cyclette con un’intensità dell’85% del VO2max. Ogni sessione si concludeva con uno sprint finale di 30 secondi. Dopo l’allenamento, i soggetti consumavano uno shake contenente 30g di Whey.
I soggetti del terzo gruppo sono stati sottoposti ad allenamenti contro resistenza 3 volte a settimana. Ogni seduta allenante consisteva il una “Full Body”, in cui i soggetti allenavano i loro principali gruppi muscolari con esercizi di base come leg-press, bench-press, extensions, military-press, lunges, lat-pulldowns, leg-curl e low-row. I soggetti hanno eseguito serie da 12 ripetizioni riposandosi per 2 minuti tra le serie. Dopo l’allenamento, i soggetti consumavano uno shake contenente 30g di Whey.
In ogni gruppo, metà dei soggetti presi in esame assumeva la Terbutalina. Il farmaco è stato somministrato per via inalatoria. I soggetti, per l’esattezza, hanno usato il Bricanyl Turbohaler della AstraZeneca. Ogni giorno i soggetti trattati inalavano 8 erogazioni da 0,5mg di Terbutalina (4mg totali al giorno).
Nei soggetti del primo gruppo e nei soggetti che si allenavano contro resistenza, la somministrazione di Terbutalina a causato un aumento della massa corporea magra di poco più di un chilo. Questo non si è verificato nei soggetti del gruppo sottoposto ad allenamenti di resistenza.
La Terbutalina, hai dosaggi utilizzati nello studio, non ha avuto effetti sulla massa grassa.
I ricercatori scrivono che, il presente studio mostra come l’inalazione quotidiana di un beta2-agonista comunemente somministrato aumenta la massa magra in individui che non sono di per sé allenanti, ma che piuttosto mantengono un basso livello di attività fisica, e che sperimentano un aumentano additivo della massa magra se combinata con esercizi contro resistenza.
I ricercatori proseguono scrivendo che, si tratta di una problematica per l’antidoping e stabilire una soglia delle concentrazioni di Terbutalina nelle urine dovrebbe essere una priorità per la WADA, al fine di evitare un eccessivo uso improprio al fine di un aumento dell’ipertrofia muscolare da parte degli atleti che hanno accesso alla Terbutalina.
Concludendo, i ricercatori affermano che, i dati del presente studio dovrebbero suggerire cautela ai medici che trattano gli atleti d’élite come inalazioni di Terbutalina in dosi di 4 mg/die le quali, come riportato sopra, possono potenziare gli adattamenti dell’allenamento contro resistenza.
Nel 2015, gli stessi ricercatori danesi hanno pubblicato uno studio nel quale i volontari hanno assunto un dosaggio consistente di Terbutalina in compresse. Gli effetti di ricomposizione corporea sono stati decisamente migliori rispetto a quelli legati al precedente studio qui esposto. (2)
I ricercatori hanno reclutato per lo studio 18 maschi attivi di vent’anni. I soggetti praticavano ciclismo, corsa o fitness e si allenavano per 4-8 ore a settimana. Durante l’esperimento i soggetti reclutati hanno continuato a svolgere le loro attività motorie abituali.
I ricercatori hanno diviso i soggetti in 2 gruppi. Per 4 settimane un gruppo ha ricevuto un placebo e l’altro la Terbutalina.
La dose terapeutica massima di Terbutalina per gli adulti è di 15mg/die, suddivisa in tre assunzioni. I ricercatori hanno somministrato ai soggetti del test una dose pari a 2-3 volte il dosaggio terapeutico massimo: per ogni 30 kg di peso corporeo i soggetti assumevano 5mg di Terbutalina Solfato ogni giorno, due volte al giorno. I ricercatori hanno utilizzato il Bricanyl Retard della AstraZeneca.
La forza isometrica [Massima contrazione volontaria] che i soggetti potevano esprimere durante l’esecuzione alla leg extension è rimasta costante nel gruppo placebo, mentre ha subito un aumento di 97 Newton nel gruppo Terbutalina.
Durante uno sprint su Cyclette di 30 secondi, i soggetti del gruppo Terbutalina [TER] hanno potuto sviluppare una potenza maggiore dopo 4 settimane. Ciò significa che sono diventati più veloci. Questo non è accaduto nel gruppo placebo [PLA].
La massa corporea magra nel gruppo trattato con Terbutalina era aumentata di 2Kg rispetto al gruppo placebo. Allo stesso tempo, la massa grassa nel gruppo trattato con Terbutalina era diminuita di 1kg rispetto al gruppo placebo.
Nelle cellule muscolari, prelevate dalle gambe dei soggetti dello studio, i ricercatori hanno scoperto un aumento delle proteine muscolari della catena pesante della miosina I e della catena pesante della miosina II nel gruppo trattato con Terbutalina.
Nelle cellule muscolari, la Terbutalina non ha avuto alcun effetto sul espressione della Miostatina, famoso peptide deputato alla regolazione dell’ipertrofia muscolare. Tuttavia, il farmaco ha aumentato la produzione di Follistatina. E, come ormai ben sappiamo, la Follistatina esercita un azione inibitoria nei confronti della Miostatina.
Ovviamente, la dose di Terbutalina utilizzata non è priva di effetti collaterali. Nella prima settimana dello studio, alcuni soggetti hanno sperimentato tremori, palpitazioni e irrequietezza. Dopo la prima settimana, gli effetti collaterali sono scomparsi.
I ricercatori si sono premurati di concludere dicendo che, dato l’impatto sul miglioramento delle prestazioni in seguito ad uso acuto e cronico di beta-2-adrenergici sulla massa muscolare, la forza e la potenza durante l’esercizio massimale, sembra logico che l’uso sistemico di beta-2-agonisti debba rimanere nell’elenco delle sostanze proibite negli sport competitivi.


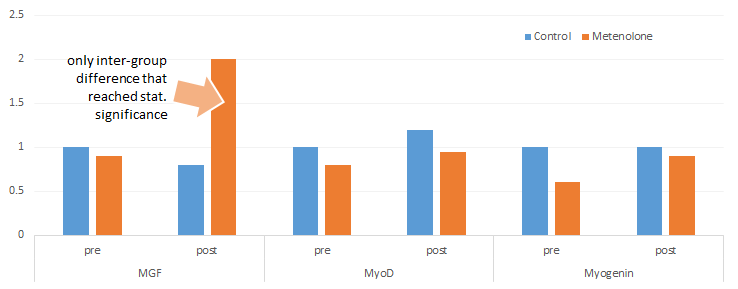














































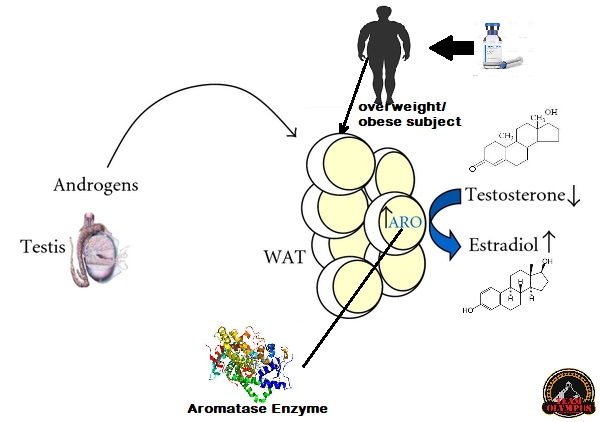



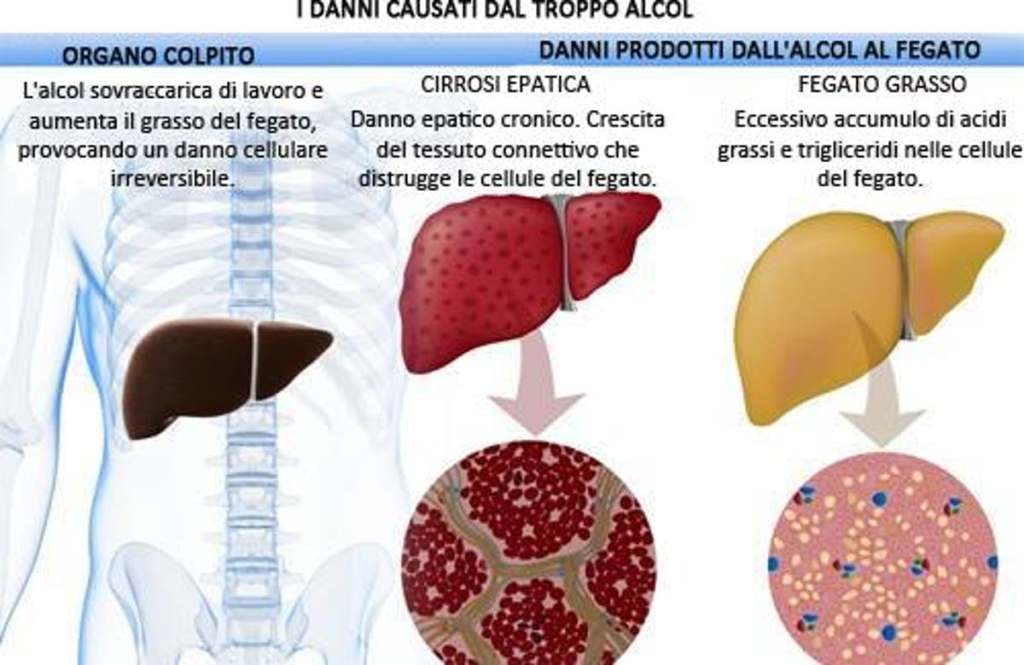





























{kind=link}