DISCLAIMER: Il presente articolo è a solo scopo educativo, di intrattenimento e informativo. Non rappresenta in alcun modo una forma di incitamento all’uso/abuso di sostanze dopanti. L’autore ed il sito, per tanto, è esentato da qualsiasi responsabilità dipendente dalla libera scelta individuale.
Introduzione ad un dilemma…
Chiunque frequenti l’ambiente del Bodybuilding e del Fitness avrà letto o sentito almeno una volta nella vita espressioni del genere “se mi dopassi sarei anche io così [indicando Flex Wheeler]” o “ho provato di tutto e senza farmaci non riuscirò ad ottenere risultati”. Andando poi ad approfondire la storia di ognuno di questi soggetti si scopre in percentuale quasi assoluta che si tratta di individui nella norma (o al di sotto) frustrati e/o con personalità deboli, speranzosi omini che attendono placidamente che accada una svolta miracolosa nella loro banale e piatta esistenza e, cosa molto importante, con il minimo dello sforzo (meglio se nessuno).
Nella mia esperienza come ricercatore e operatore nel campo della cultura fisica in qualità di Preparatore Atletico, ho assistito a innumerevoli casi in cui un soggetto aspirava al miglioramento della propria composizione corporea trascurando, consciamente o inconsciamente, le basi fondamentali rappresentate da Nutrizione e Allenamento baipassandole in vista della possibile prescrizione di una pillola miracolosa capace di renderlo/a possessore della forma fisica ambita.
Tralasciando l’ovvio ragionamento che spinge ogni essere umano dotato di un minimo d’intelletto verso la comprensione che la genetica è il blocco d’argilla sul quale si va ad operare, ma le sue qualità e difetti sono presenti in modo eterogeneo nella popolazione mondiale, e ciò non è modificabile nemmeno con la farmacologia più oculata, quando ci si trova davanti al bivio tra “pillola rossa” (PEDs) e “pillola blu” (drug free) bisogna essere pienamente consapevoli non solo del fattore illegalità ma del fattore conoscitivo. Purtroppo, la politica del terrore ha operato in modo fallimentare nel goffo intento di allontanare dalla scelta “rossa”, e ciò si è tradotto in un numero sensibile di soggetti abusatori con tutte le conseguenze cliniche derivanti.
Se un individuo non ha raggiunto un livello di maturità sportiva tale da conferirgli una gestione corretta della nutrizione e della periodizzazione allenante (gestione delle variabili volume, intensità, densità ecc…), è molto meglio per lui/lei rivedere i suoi programmi e scegliere ancora la “pillola blu”. Capita, a volte, di incontrare persone decise ad intraprendere la via del “lato oscuro” che, dopo una approfondita chiacchierata sulla gestione dei suddetti fattori, rivede le proprie posizioni.
Per tutti coloro i quali sono immersi nel dilemma della scelta, vi espongo alcuni punti per rendere l’eventuale decisione meno rischiosa anche se pur sempre illegale nel “bel paese”…
“Pillola blu o pillola rossa?” I punti da tenere in considerazione per una scelta consapevole:
#1 Raggiungere una adeguata maturità sportiva
Per “maturità sportiva”, in particolare riferimento al BodyBuilding, si intende la capacità del atleta di sapersi alimentare e allenare correttamente con piena gestione delle proprie potenzialità fisiologiche/genetiche. Questa è la base, se viene a mancare ciò non solo la vostra esperienza finirà per deludervi e rendervi ancora di più dei frustrati, ma potrebbe rovinosamente portarvi ad un abuso cronico a senso inesorabilmente negativo…
#2 I PEDs non faranno miracoli
Una cosa da tenere bene a mente, e questo non dovrebbe interessare solo gli aspiranti “doped”, è che l’uso di PEDs non renderà diversi da ciò che rientra nelle potenzialità espressive del proprio patrimonio genetico. Certamente le caratteristiche genetiche verranno “iperespresse”, nel bene e nel male, dall’uso di PEDs ma non vi sarà nessun miracolo! Migliorerete ma non sarete ne più ne meno di ciò che potete essere!
Un esempio per capire come la base genetica faccia la differenza anche con protocolli che, ad oggi, spesso non raggiungono nemmeno i livelli del “bridge” più soft..
#3 Ridurre la percentuale di grasso corporeo
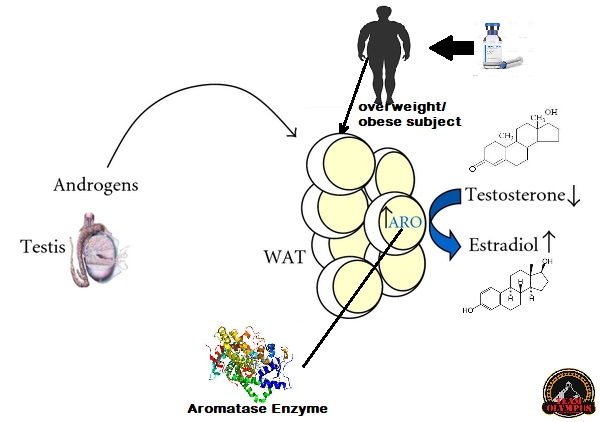
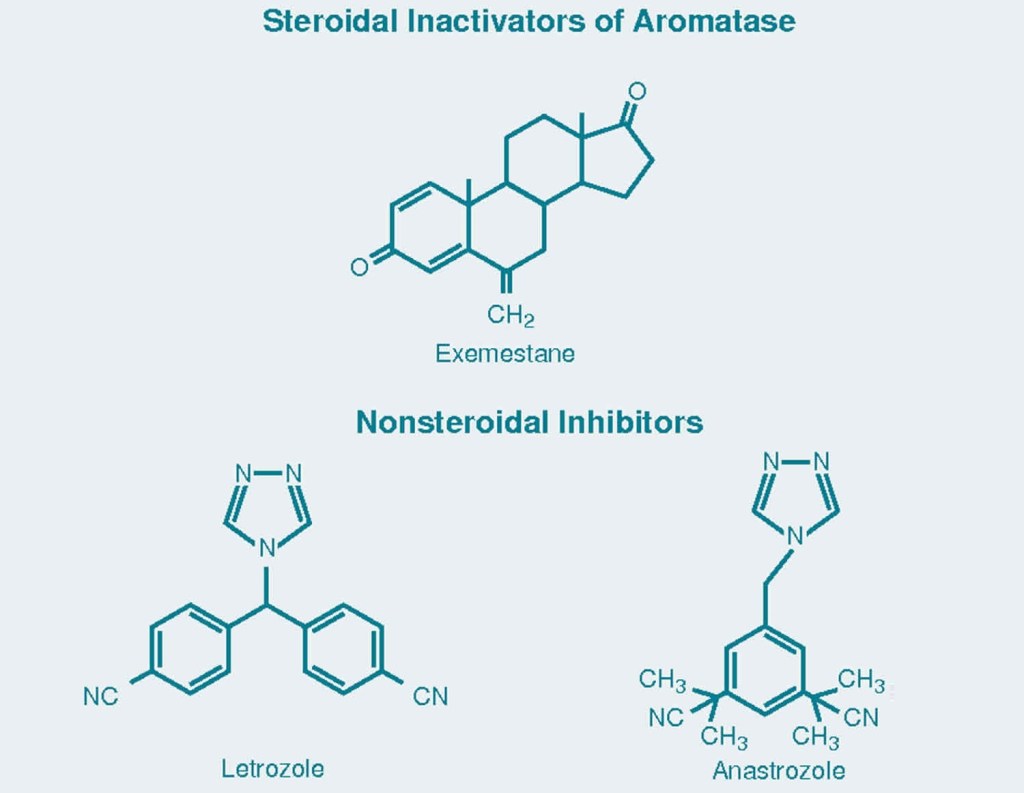
Il tessuto adiposo rappresenta uno dei siti dove il Testosterone, ed altri AAS soggetti all’aromatizzazione, viene convertito in Estradiolo. Soggetti con percentuali di grasso corporeo elevate vedrebbero una alterazione marcata della Testosterone:Estradiolo ratio a favore della componente estrogenica, con conseguenze quali alterazione del comportamento sessuale (impotenza, difficoltà nel raggiungere e/o mantenere l’erezione), ritenzione idrica, accumulo di grasso con modello femminile e ginecomastia. E no, l’uso di DHT derivati o di SARM non steroidei senza una base di Testosterone non risolverebbe il problema o, per lo meno, porterebbe ad altre conseguenze negative, che pur non comprendendo, per esempio, ritenzione idrica e ginecomastia, interesserebbero l’attività sessuale e la condizione psichica del soggetto trattato. [1]
Schema esemplificato del processo di aromatizzazione degli androgeni aumentati in un soggetto con percentuale di grasso corporeo alta.
Allo stesso tempo, i rischi cardiovascolari della somministrazione di AAS- come il possibile aumento esponenziale del Ematocrito, l’aumento del LDL e Trigliceridi a discapito di una riduzione del HDL, e l’aumento della pressione sanguigna – sarebbero già presenti in certa misura quando la body fat è già alta e sarebbero quindi soggetti ad un repentino aggravamento.
Se la percentuale di grasso è relativamente alta, si dovrebbe prima di tutto considerare di migliorare la composizione corporea con una adeguata routine alimentare e allenante (senza farmaci) prima di iniziare solo a pensare all’uso di AAS. Sicuramente ciò renderà la scelta più efficace e meno rischiosa.
Nel caso fosse necessario sottolinearlo, no, non è saggio nemmeno utilizzare agenti PEDs a fini lipolitici e/o antiadipogenici e/o termogenici (compresi gli Ormoni Tiroidei). A meno che non siate affetti da ipotiroidismo, e in questo caso la terapia vi dovrebbe essere stilata dal vostro medico, per ridurre in modo sensibile la body fat non sono necessari i farmaci!
#4 Controllare se si ha una storia familiare di trombosi (o qualsiasi altra malattia cardiovascolare)
Molte malattie cardiovascolari hanno una componente di base genetica. Uno stile di vita sano può ridurne sensibilmente la loro insorgenza, ma l’uso di AAS può causare l’attivazione di specifici geni implicati nella comparsa di malattie cardio-circolatorie. Caratteristico dell’interazione tra AAS e geni specifici è un caso studio ben documentato che ricercatori americani hanno pubblicato sul “Blood Coagulation & Fibrinolysis”.[2]
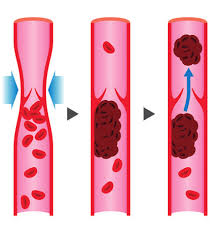
Trombosi venosa
Oltre all’attivazione genica diretta dagli AAS, e nociva per il sistema cardio-circolatorio, vi sono altre condizioni negative innescate dall’uso/abuso di Steroidi Anabolizzanti, e di altri PEDs, come, per esempio, l’aumento del tasso di coagulazione, l’incremento eccessivo dell’Ematocrito con aumento pressorio, rigidità dell’endotelio vascolare con perdita di efficienza strutturale e aumento della pressione ematica con incremento delle possibilità di danno strutturale dei componenti del sistema interessato.
#5 Inserire delle sedute di allenamento cardio prima, durante e dopo l’uso di PEDs
Un moderato allenamento cardiovascolare è sicuramente una delle migliori strategie preventive contro la comparsa di malattie cardio-circolatorie. Tale tipologia di allenamento può portare un miglioramento e/o riduzione delle alterazioni lipidiche ematiche del praticante, fornendo un, seppur minimo, tampone all’azione negativa degli AAS e SARM non steroidei sui livelli di LDL (aumento), Trigliceridi (aumento) e HDL (diminuzione). Secondo quanto riportato da una interessante review del 2013, l’abbinamento di sedute cardio e in sala pesi possono avere una azione additiva benefica sui livelli di LDL, Trigliceridi e HDL.[3]
Risulta interessante anche quanto emerso da alcuni studi su animali a seguito dei quali si è osservato un significativo grado di protezione dato dall’allenamento cardio negli esemplari trattati con AAS.[4]
#6 Assicurarsi di rimanere ben idratati
Oltre ad agevolare il mantenimento di un Ematocrito migliore, una buona idratazione risulta positiva sulla pressione di lavoro renale nel filtraggio del sangue. Diversi AAS come il Trenbolone e i metilati in C-17 presentano una particolare resistenza metabolica che, oltre a causare un aumentato stress epatico, può portare ad una sofferenza renale sfociabile nel patologico. Si è osservato come una combinazione di AAS, dieta iperproteica e supplementazione di Creatina possa aumentare l’incidenza di problemi renali.[5] In un soggetto in fisiologia, la sola dieta ad altro contenuto proteico e la supplementazione di Creatina non hanno mostrato nessun grado di pericolosità, soprattutto sul breve/medio termine.
#7 Non usare “droghe ricreative”
A livello globale, il numero di decessi tra gli abusatori di AAS è in aumento. Alcuni, troppo superficialmente, dicono che questo sia dovuto al fatto che sempre più uomini e donne usano AAS, ma questa è solo una spiegazione dozzinale e limitata. Il sospetto ricade soprattutto sulle modalità di approccio dei consumatori di AAS: i dosaggi sono drammaticamente aumentati e un numero crescente di individui combina PEDs con “droghe ricreative”. Ed è su questi due ultimi punti che risiede la spiegazione principale dell’aumento statistico prima menzionato. Soprattutto la combinazione di PEDs e le così dette “droghe ricreative” risulta essere probabilmente un fattore significativo, come evidenziato alcuni anni fa da ricercatori australiani. Nel loro studio sono state analizzate tutte le morti documentate tra i consumatori di AAS a Sydney tra il 1997 e il 2012, scoprendo che le droghe ricreative come la cocaina avevano avuto un ruolo nella schiacciante molteplicità dei casi. Dagli studi sugli animali ora sappiamo della possibilità che la co-assunzione di un AAS come il Nandrolone con la cocaina vede moltiplicati gli effetti cardiotossici rispetto ai singoli composti.[6] E secondo studi in vitro la combinazione di Testosterone e cocaina aumenterebbe la possibilità di formazione di coaguli nel flusso ematico. [7]
#8 Corretta modalità di iniezione e herpes labiale
Gli utilizzatori di AAS a volte sviluppano ascessi, ma non sempre dovuti alla bassa qualità dei prodotti utilizzati.
Alcuni medici ritengono che gli utilizzatori di AAS dovrebbero effettivamente ricevere una formazione sulle tecniche di iniezione corrette, onde evitare embolie oleose o ascessi per cattiva gestione igienica della procedura. [8]
Molti utilizzatori ancora non sanno che disturbi come l’herpes labiale rendono le iniezioni ancora più rischiose. Perchè? Il virus che causa l’herpes labiale, come altri patogeni, riduce l’efficienza del sistema immunitario, fornendo così terreno fertile per infezioni batteriche i cui microorganismi scatenanti vengono inoculati nel corpo del soggetto attraverso l’iniezione in modo diretto o indiretto.
#9 Non fare affidamento sugli integratori
Secondo un buon numero di studi svolti su animali, alcuni integratori proteggono dagli effetti collaterali degli AAS. Secondo alcune ricerche, la Taurina, la Vitamina C ed E proteggono i testicoli durante un ciclo e la vitamina C e il cacao proteggono la prostata.
L’utilità dei risultati provenienti da questi studi è limitata per tre motivi:
A. gli animali da laboratorio non sono esseri umani, e
B. le dosi utilizzate e rapportate ad un essere umano sono quasi sempre molto inferiori rispetto a quelle utilizzate dai “doped”, e
C. la ricerca in campo psicologico mostra che l’uso di integratori stimola comportamenti rischiosi e malsani. I supplementi fanno pensare agli utilizzatori di essere invulnerabili e di non dover comportarsi in modo sano ed attento.[9]
Gli integratori possono aiutare a creare una mentalità che non si dovrebbe avere da utilizzatore consapevole di AAS.
Ovviamente, alcuni supplementi “protettivi” utilizzati dai soggetti meglio informati hanno un potenziale di “tamponare” in modo discreto alcune alterazioni legate all’uso di AAS e SARM come, ma non limitato a, Riso Rosso fermantato (controllo lipidico) [10], Silimarina (epatoprotezione), NAC (epatoprotezione) [11], Niacina (controllo lipidico) ecc…
#10 Ridurre al minimo (se non eliminare) il consumo di alcolici
Potrebbe sembrare un indicazione superflua ma non lo è.
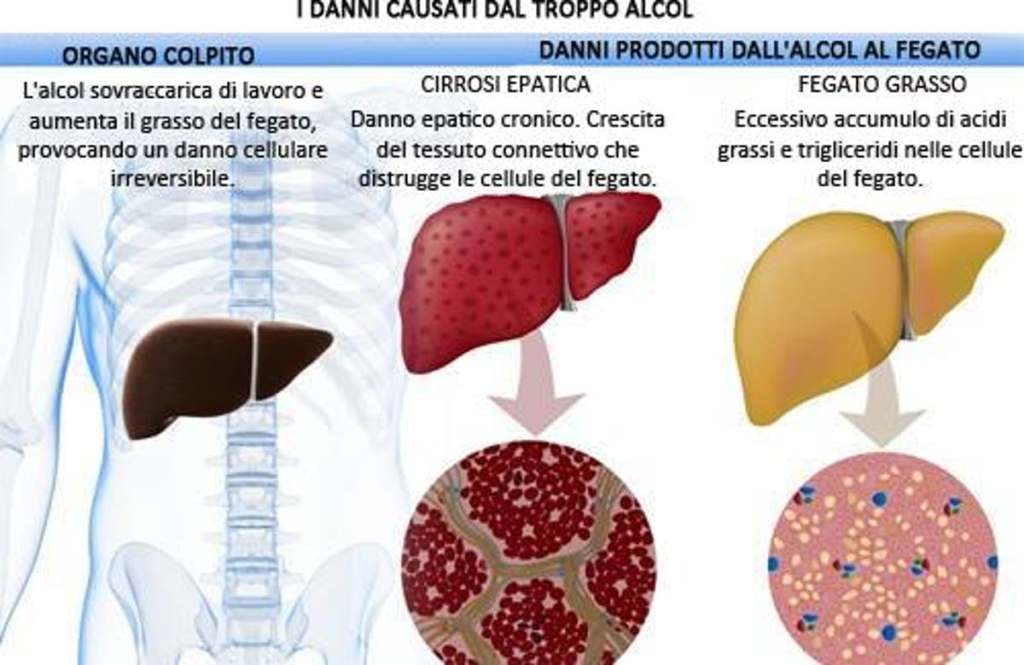
L’abuso di alcol è indubbiamente uno dei problemi sociali più diffusi. Uno dei problemi correlati all’abuso di alcol e l’epatopatia alcolica. Questo stato patologico è derivante da un processo infiammatorio progressivo ai danni del fegato legato al consumo eccessivo di alcolici. È una malattia a più stadi. La steatosi provoca un ingrossamento del fegato causato da un accumulo di trigliceridi, spesso senza sintomi per molto tempo. I rischi correlati sono la steatosi (fegato grasso), l’epatite alcolica e la cirrosi epatica. Il rapporto con l’alcolismo è complesso. Non tutti i bevitori, infatti, hanno danni al fegato, anche se sono altamente probabili. La causa è da rinvenire in una trasformazione dell’alcol (etanolo) in sostanze tossiche che danneggiano il fegato in maniera irreversibile e cronica, con un rischio elevato di insufficienza epatica e di cancro, fino alla necessità di un trapianto di fegato.
In acuto, invece, l’alcol può essere una causa di alterazione delle transaminasi ma non si può sapere se e con quale modalità si potrebbero innalzare: dipende molto dalla risposta individuale dell’organismo. In caso di stress preesistente, di causa iatrogena e/o alimentare, si può presentare una alterazione significativa. [12]
Il primo caso è una consequenziale possibile se eventi stressori concomitanti si presentano in cronico. Ed è semplice giungere alla conclusione che l’uso di AAS, specie se metilati, possa comportare un aumentato stress epatico che potrebbe degenerare in peliosi epatica, cirrosi ecc…
Che siate “doped” o “natural”, per ragioni legate e non, dovreste evitare di consumare più di 25g per gli uomini, o 12,5g per le donne, di Etanolo al giorno.
#11 Sottoporsi a regolari controlli medici pre, intra e post utilizzo
Il monitoraggio della salute dovrebbe essere la base fondante del comportamento del utilizzatore consapevole e minimamente attento ai potenziali rischi nei quali potrebbe imbattersi.
Gli esami di controllo sono i seguenti:
Esami ematici e delle urine (comprendenti il quadro ormonale secondo necessità);
Elettrocardiogramma ogni 6 mesi circa;
Elettrocardiogramma sotto sforzo (prima di iniziare);
Ecocardiogramma ogni 6 mesi circa;
Coronarografia ogni 6 mesi circa;
Monitoraggio della pressione ematica;
TAC addome completa ogni 6 mesi circa.
Ovviamente, ogni accertamento , al di la degli esami ematici, deve essere gestito in base alle esigenze soggettive, caratteristiche e tipo di PEDs utilizzati.
#12 Essere seguiti da personale qualificato
Fin troppa gente è stata salutisticamente deturpata da gorilla di spogliatoio a mala pena consapevoli dell’esistenza dei macronutrienti e che, nonostante ciò, si sono improvvisati farmacisti. Donne divenuti uomini e uomini divenuti simili a cagne in calore per via di orrende ginecomastie. Evitate il fai da te e l’affidarsi a semianalfabeti … la somaticità sopra la norma è cosa diversa dall’intelligenza e alla competenza in biologia, biochimica e farmacologia… senza offesa per tutti quelli che “io mi facevo e ho vinto! Senzia scienzia!” …
#13 Pensare seriamente al post ciclo prima del ciclo
Molti aspiranti “doped” non considerano il fattore post ciclo. La maggior parte di loro è convinta che la PCT sarà una facile soluzione alla sottoregolazione dell’Asse HPT, ma in realtà non è proprio così. Esistono diversi casi studio che mostrano come gli ex utilizzatori abbiano spesso livelli di Testosterone inferiori rispetto al pre-utilizzo anche a distanza di anni dal cessato uso di AAS. Sembra che i fattori che aumentano le possibilità e il grado di tale effetto sul lungo termine siano:
Tempo di somministrazione;
Età
Molecole utilizzate (con maggiore impatto negativo dato dai19-norsteroidi come il Nandrolone per via della lunga permanenza dei metaboliti nel sistema).
Tutto ciò è indipendente dalla qualità della PCT, anche se essa può avere dei riscontri positivi specie nel primo periodo di stacco dagli AAS. Le alterazioni ormonali legate ad una alterazione dell’Asse HPT comprendono depressione, ansia, bassa libido, difficoltà nel raggiungere e mantenere l’erezione, stanchezza cronica ecc…
Per questa ragione molti scelgono di entrare in TRT (Terapia Sostitutiva del Testosterone) dopo il primo ciclo.
Quale conclusione?…
Se mai non dovesse bastare il disclaimer, questo articolo non rappresenta in alcun modo un consiglio e, ne tanto meno, un incitamento all’uso di sostanze dopanti! E’ semplicemente a fine divulgativo con l’obbiettivo di far comprendere a più persone possibili che la scelta di intraprendere coscientemente certe pratiche (illegali) necessita di una sufficiente (e veritiera) conoscenza del argomento.
Quindi? Leggete e comprendete correttamente ciò che ho riportato in sintesi fruibile ad un largo pubblico… Pensate prima di tutto ad alimentarvi e allenarvi in modo ottimale!
La conoscenza della Verità rende liberi dalla cattiva informazione, dagli strumenti commerciali e dal relativismo… Negarla è semplice e pericolosa manifestazione di profonda ignoranza… di VERO NEGAZIONISMO!
Se avete una buona conoscenza della lingua inglese e volete approfondire l’argomento PEDs e Sport, potete leggere il libro ANABOLICS 11th Edition di William Llewellyn
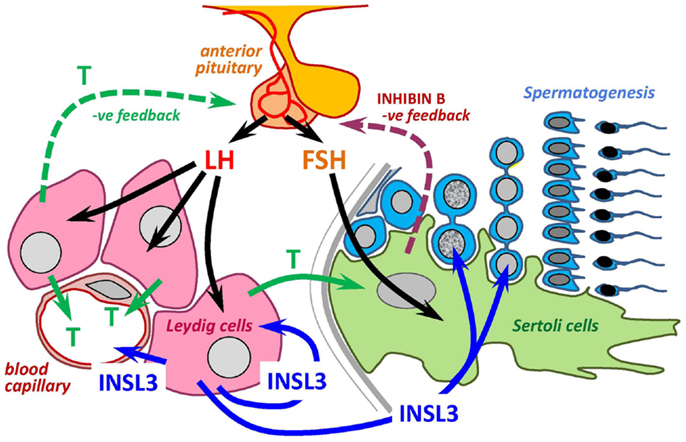
Anni dopo l’interruzione dell’uso di AAS da parte di soggetti di sesso maschile questi ultimi potrebbero riscontrare ancora una condizione di ipo-funzionamento testicolare. La concentrazione ematica del fattor isulino simile 3 (INSL3), un peptide del quale si sa ancora poco, rende chiaro quanto possa durare l’impatto endocrinologico dato dall’uso di AAS. Ciò risulta particolarmente evidente dai risultati di uno studio danese, al quale hanno partecipato 132 bodybuilder.[1]
Fattore Insulino Simile-3 (INSL3)
Dettagli dello studio
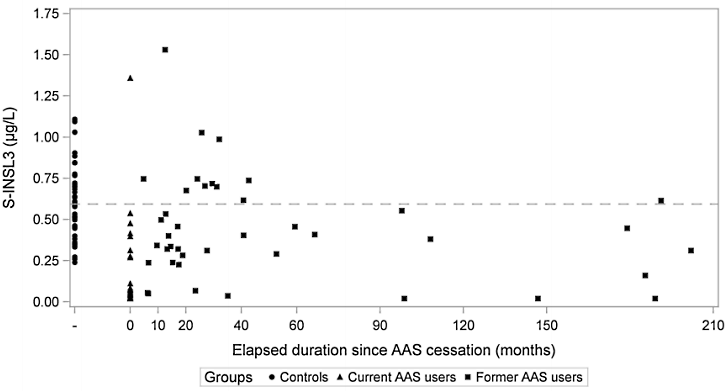
I ricercatori, un gruppo di endocrinologi dell’Università di Copenaghen, hanno osservato 132 uomini di età compresa tra 18 e 50 anni che si allenavano con i pesi. Questi uomini sono stati divisi in 3 gruppi formati all’incirca dallo stesso numero di individui:
Un gruppo non aveva mai usato AAS;
un secondo gruppo era sotto ciclo di AAS durante lo studio;
infine, il terzo gruppo aveva usato AAS in passato.
Il partecipante medio allo studio in quest’ultimo gruppo aveva smesso di usare AAS 32 mesi prima. I ricercatori hanno misurato la concentrazione di INSL3 nel sangue dei soggetti partecipanti allo studio. Quell’ormone, che di per sé ha anche proprietà anabolizzanti, è prodotto dai testicoli. Gli endocrinologi sospettano che i testicoli siano più sani in quanto producono più INSL3 e pensano persino che questo peptide sia un marker di riferimento maggiormente importante per la valutazione della vitalità dei testicoli rispetto al Testosterone.
Risultati dello studio
Dei tre gruppi sotto osservazione, i soggetti che utilizzavano AAS nel periodo dello studio avevano la più bassa concentrazione ematica di INSL3. Tuttavia, i soggetti che avevano usato AAS in passato avevano anch’essi meno INSL3 sierico rispetto a coloro che non avevano mai utilizzato AAS:
I non utilizzatori avevano una media di 0,59mcg di INSL3 per litro;
gli ex utenti 0,39mcg per litro.
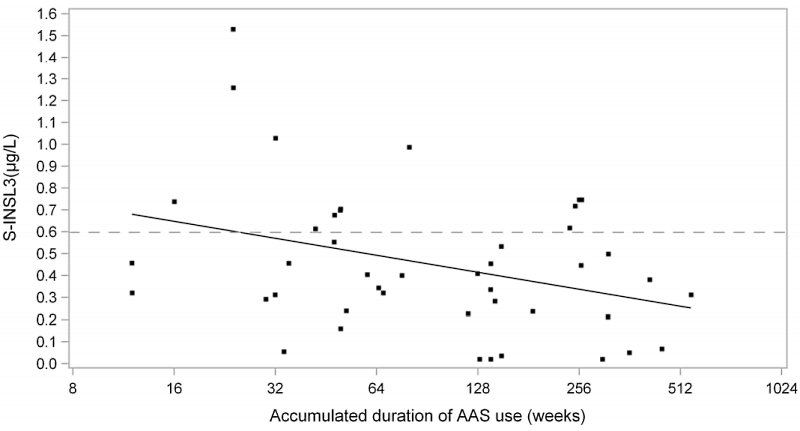
Più a lungo gli ex utilizzatori erano stati sotto AAS in passato, minore era il INSL3 rilevato attraverso esame ematico.
I dati riportati nella figura sopra sono di notevole interesse. Si guardi all’associazione tra il passare del tempo dopo l’ultima somministrazione di AAS da una parte e la concentrazione sierica di INSL3, dall’altro. Bene, si nota una consequenziale en significativa risposta…
Contrariamente, è ovvio che se non ci fosse stato alcun legame significativo il INSL3 non avrebbe alcuna valenza come marker per la valutazione dello stato di salute dei testicoli.
Conclusioni
Il principale ricercatore dello studio, Jon Jarlov Rasmussen, in un comunicato stampa ha affermato che è ancora dibattuto se l’uso off-label di AAS provochi una carenza di Testosterone sul lungo termine. I loro risultati suggeriscono, però, la presenza di una capacità gonadica compromessa sul lungo termine nei precedenti utilizzatori di AAS. I risultati sollevano la questione se alcuni precedenti utilizzatori di AAS debbano ricevere una terapia di stimolazione medica per aumentare la capacità funzionale delle cellule di Leydig nei testicoli.[2]
Rasmussen ipotizza come base di trattamento inibitori dell’Aromatasi e SERM.
Tempo fa, usando come fonte un brevetto del 1955 dell’americana Upjohn, ho trattato brevemente un particolare gruppo di AAS derivati del Nandrolone con un gruppo 2-α-metile.[1] Inizialmente si poteva ipotizzare che dopo la pubblicazione di quel brevetto nessuno avesse preso in considerazione gli analoghi del 2-α-methyl-Nandrolone. Ma, invece, esiste uno studio giapponese dei primi anni ’60 che analizza le caratteristiche di tale modifica nello scheletro carbossilico del Nandrolone.[2]
Dettagli dello studio:
Il prima citato studio è stato pubblicato nel 1964 in un articolo è frutto del lavoro di ricercatori della società farmaceutica giapponese Dainippon Pharmaceutical . Questo articolo descrive, tra le altre cose, una serie di esperimenti sugli animali con l’utilizzo di diversi AAS che sono descritti anche nel brevetto della Upjohn del 1955. I ricercatori nipponici hanno testato i loro AAS su ratti da laboratorio maschi sterilizzati sottoponendoli ad iniezioni per 7 giorni. Successivamente, i ricercatori hanno misurato il peso del muscolo levator ani per stimare (con un grosso margine d’errore) l’effetto anabolico degli ormoni sintetici inoculati. Per avere un’idea degli effetti collaterali androgeni, hanno anche misurato il peso della prostata degli animali (anche qui la stima non gode della migliore attendibilità).
Risultati:
L’effetto anabolico degli analoghi del 2-α-methyl-Nandrolone era inferiore a quello del Testosterone, ma il loro effetto androgeno era molto inferiore (per l’inattendibilità dei dati riporto ai link sopra riportati). Il rapporto tra effetto anabolico-androgeno [Androgeno:Anabolico ratio = valore indice terapeutico] era quindi un fattore 3-5 più favorevole di quello del Testosterone.
Conclusioni e potenziali effetti collaterali:
La struttura di molte delle sostanze di cui sopra ricorda quella del designer steroid Methyldrostanolone (commercializzato per la prima volta nel 2005 con il nome di Superdrol). Ma a differenza del 2-α-methyl-Nandrolone e derivati, Il Methyldrostanolone, la cui formula strutturale si può vedere di seguito, è un AAS con due metilazioni: una in C-2 (come i sopra citati analoghi) e una in C-17. Tale caratteristica lo rende un AAS epatotossico che, se abusato, può causare danni al fegato oltre che, potenzialmente, ai reni. Poi, ovviamente, il Methyldrostanolone, non è un derivato del Nandrolone, ma del DHT.
Nonostante questa significativa differenza, la epatotossicità degli AAS riportati nell’elenco sopra sia di grado lieve/moderato ma comunque presente. Sulla base di ciò che sappiamo sui derivati del Nandrolone metilati in C-17, questi potrebbero essere anche più epatotossici del Methyldrostanolone. Ma, lo ripeto, questo non è potenzialmente correlabile all’effetto a livello epatico del 5827 o 2-α-methyl-Nandrolone. Questo AAS probabilmente non è solo lieve sul fegato, ma può anche avere effetti collaterali androgeni più moderati rispetto al Nandrolone.
2-α-methyl-Nandrolone
Altri effetti collaterali? I ricercatori hanno anche determinato l’effetto estrogenico e progestinico dei loro AAS. Non era rilevante. Ciò è conforme al contenuto del brevetto della Upjohn.
Ma è disponibile sul mercato “nero” o “grigio”? A quanto ne so no, e anche se lo fosse non è un AAS dal grande potenziale capace di essere concorrenziale con altre molecole con una miotroficità molto più elevata e già presenti sul mercato da decenni.
“Per la prima volta, le persone possono ottenere attraverso i farmaci ciò che era possibile solo attraverso un intervento chirurgico per dimagrire “, è l’affermazione sensazionalistica estratta da un comunicato stampa della endocrinologa e professoressa britannica Rachel Batterham.[1] Il farmaco di cui parla la Batterham è il Semaglutide. In uno studio pubblicato sul New England Journal of Medicine, è stato riportato che i soggetti obesi hanno perso in media 15 libbre (circa 6,80Kg) quando trattati con Semaglutide.[2]
Semaglutide? Una spiegazione d’obbligo…
Semaglutide
Il Semaglutide, venduto con i marchi Ozempic e Rybelsus, è un farmaco antidiabetico utilizzato nello specifico per il trattamento del diabete di tipo 2. [3][4]
Il Semaglutide agisce come il peptide Glucagone simile-1 umano (GLP-1) in modo che aumenti la secrezione di Insulina, migliorando di conseguenza il metabolismo glucidico cellulare. È distribuito sotto forma di soluzione iniettabile da praticarsi sottocute all’interno di una penna predosata. Uno dei suoi vantaggi rispetto ad altri farmaci antidiabetici è che ha una lunga durata d’azione, quindi è sufficiente solo un’iniezione una volta alla settimana. [5]
Una versione iniettabile (Ozempic) è stata approvata per uso medico negli Stati Uniti nel dicembre 2017,[6] e nell’Unione Europea,[7] Canada,[8] e Giappone nel 2018. Una versione che viene assunta oralmente (Rybelsus) è stata approvata per uso medico negli Stati Uniti nel settembre 2019,[9] e nell’Unione Europea nell’aprile 2020. [10] E’ il primo trattamento a base di una proteina con affinità per il recettore del peptide glucagone simile 1 approvato per l’uso negli Stati Uniti che non ha bisogno di essere iniettato, ed è stato prodotto dalla Novo Nordisk.
Il Semaglutide è chimicamente simile al GLP-1 umano, con una somiglianza del 94%. Le uniche differenze sono due sostituzioni di amminoacidi nelle posizioni 8 e 34, dove l’Alanina e la Lisina sono sostituiti rispettivamente dall’acido 2-amminoisobutirrico e dall’Arginina. [11] La sostituzione degli amminoacidi nella posizione 8 previene la rottura chimica da parte di un enzima dipeptidile peptidasi-4. Inoltre, la Lisina in posizione 26 è nella sua forma derivata (acilata con diacid stearico). L’acilazione con un distanziale e una catena di diacidi grassi C-18 aumenta il legame del farmaco con la proteina del sangue albumina, il che consente una vita attiva più lunga della molecola. La sua emivita è di circa 7 giorni (165-184 ore), quindi è sufficiente un’iniezione una volta alla settimana. [5][12]
Come precedentemente accennato, il Semaglutide è un agonista del recettore del peptide Glucagone simile-1. Aumenta la sintesi di Insulina, che ovviamente abbassa il livello della glicemia ematica. [13] Sembra anche aumentare la crescita delle cellule β nel pancreas, che sono i siti di sintesi del Insulina. [12] Dall’altra parte inibisce il Glucagone, ormone con effetto iperglicemizzante. Inoltre riduce l’assunzione di cibo abbassando l’appetito e rallentando la digestione gastrica. [14] In questo modo può agire come agente per la riduzione del grasso corporeo. [15]
In sintesi, nel corpo, il GLP-1 viene rilasciato quando il cibo raggiunge la fine dell’intestino tenue e i livelli di glucosio aumentano. Nel cervello il GLP-1 inibisce l’appetito, e nel pancreas stimola il rilascio di Insulina. Il GLP-1 somministrato per via esogena viene rapidamente eliminato dal corpo, ma il Semaglutide presentando le prima citate modifiche strutturali, rimane in circolo per un periodo di tempo prolungato. Gli enzimi che scompongono il GLP-1 in singoli amminoacidi hanno difficoltà nella lisi del Semaglutide.
Studio:
Tornando a parlare nello specifico dello studio citato al principio di questo articolo, la Batterham ei suoi colleghi hanno reclutato 1.961 persone di età superiore ai 18 anni con un BMI ≥30 sottoponendole al loro studio per 68 settimane. Una parte dei soggetti è stata trattata con un placebo, mentre un’altra parte di essi ha ricevuto una iniezione settimanale di Semaglutide.
La dose iniziale per iniezione era di 0,25mg di Semaglutide, ma la dose è aumentata gradualmente fino a 2,4mg nelle prime 16 settimane dello studio. Alcuni soggetti non sono stati in grado di tollerare questa dose di 2,4mg e hanno continuato a usarne una quantità inferiore.
I ricercatori hanno utilizzato un prodotto Novo-Nordisk. Come sappiamo ormai, la Novo-Nordisk produce Semaglutide sia in forma orale che iniettabile. Diabetologi ed endocrinologi somministrano la molecola in questione a una dose inferiore alle persone con diabete di tipo 2.
Nota: lo studio potrebbe godere di credibilità limitata visto che è statpo sponsorizzato dalla Novo-Nordisk.
Risultati dello studio:
Il soggetto medio del gruppo sperimentale ha perso poco più di 15 libbre (circa 6.80Kg). Ciò è principalmente dovuto al fatto che il Semiglutide riduce l’appetito.
La somministrazione di Semaglutide ha anche comportato una certa perdita di massa corporea magra (questa legata alla mancanza di una adeguata routine allenante contro-resistenza). Tuttavia, poiché i soggetti hanno perso significativamente più massa grassa rispetto alla massa corporea magra, il Semaglutide ha migliorato la composizione corporea anche grazie al miglioramento della ripartizione calorica (vedi anche miglioramento del insulino-sensibilità che ne è alla base). I soggetti nel gruppo sperimentale hanno sperimentato una significativa riduzione del girovita. Inoltre, il loro equilibrio del colesterolo è ovviamente migliorato. Infine, i soggetti hanno anche segnalato un aumento della loro qualità di vita.
Effetti collaterali riscontrati con il Semaglutide:
Una piccola percentuale dei soggetti nel gruppo sperimentale si è ritirata a causa di effetti collaterali come nausea, costipazione, diarrea e vomito. Questo non è sorprendente, perché gli analoghi del GLP-1 rallentano la funzione intestinale. Secondo lo studio del NEJM, il Semaglutide è relativamente sicuro.
Bisognerebbe anche tenere presente che il Semaglutide è un nuovo farmaco, i cui effetti collaterali non sono stati ancora del tutto identificati. Sappiamo di altri agonisti del GLP-1 più vecchi, che potrebbero avere effetti collaterali che gli utilizzatori dovrebbero prendere seriamente in considerazione. Ci sono alcune indicazioni che i vecchi analoghi del GLP-1 possono danneggiare i reni in alcuni soggetti. [16] Inoltre, la FDA statunitense è a conoscenza di alcuni casi in cui gli utilizzatori di analoghi del GLP-1 hanno sviluppato un’infiammazione del pancreas. Alcuni di questi casi sono stati fatali.[17]
Breve conclusione sul Semaglutide nel BodyBuilding:
Molti di voi si staranno chiedendo come questo farmaco possa (e in quale circostanza) essere utilizzato per migliorare la composizione corporea. A riguardo, tenendo in considerazione quanto pocanzi esposto, si può essere spinti a pensare che tale peptide possa trovare uso funzionale in fasi ipercaloriche permettendo il mantenimento di un miglior partizionamento calorico anche se, a dire il vero, la soppressione dell’appetito e il rallentamento dello svuotamento gastrico non sono le migliori condizioni da avere in un momento come il “Bulk”. Potrebbe quindi essere funzionale in una leggera ipocalorica o isocalorica seguente una fase “Bulk” con finalità di miglioramento marcato della sensibilità insulinica, della ripartizione calorica ed un effetto sulla soppressione dell’appetito che può tornare utile in tali contesti.
In conclusione? Lasciatelo (almeno per il momento) ai diabetici e obesi…
In rete sono consultabili articoli a tema DNP, scritti da improvvisati biochimici o da qualche “guru” del settore Fitness e BodyBuilding, nei quali si espongono “consigli” sulla riduzione degli effetti collaterali legati all’uso di questo disaccoppiante della fosforilazione ossidativa. Alcuni di questi sono incentrati sulla caratteristica del DNP di aumentare massivamente i radicali liberi. Per ovviare a ciò, si riportano mix integrativi caratterizzati da antiossidanti come la Vitamina C, il NAC o direttamente il Glutatione. Ma tale pratica svolge un reale effetto riduttivo sui potenziali danni di una concentrazione elevata di radicali liberi DNP dipendente? Se si legge lo studio svolto su animali che i biologi finlandesi hanno pubblicato su “Comparative Biochemistry and Physiology Part C”, la risposta a questa specifica domanda potrebbe essere negativa o, comunque, l’effetto potrebbe essere poco rilevante nel contesto del tentativo di ridurre gli effetti collaterali del nitrocomposto in questione.[1] Lo so, stiamo parlando di uno studio su animali, ma ci offre comunque una visione d’insieme abbastanza concreta per porre ipotesi sull’efficacia di questa supplementazione sull’impatto negativo correlato all’uso di DNP.
2,4-dinitrofenolo
Dettagli dello studio:
Antoine Stier dell’Università di Turku in Finlandia ha svolto il suo esperimento utilizzando i fringuelli zebra. Gli animali sono stati divisi in due gruppi: al primo era stata somministrata acqua potabile per 4 anni mentre all’altro gruppo ha ricevuto acqua potabile contenente DNP. Ai fringuelli sono stati somministrati 4mg di DNP per chilo di peso corporeo al giorno. Se il dosaggio usato nei fringuelli fosse stato trasposto agli esseri umani, essi avrebbero ricevuto solo una frazione di quella dose. Il metabolismo dei fringuelli zebra è molto più elevato di quello dell’uomo.
Fringuelli zebra
Risultati dello studio:
I fringuelli trattati con il DNP, ovviamente, risposero alla somministrazione con un calo del peso e della massa grassa. Allo stesso tempo, i fringuelli del gruppo DNP vivevano il 21% in meno rispetto ai fringuelli del gruppo di controllo [solo acqua potabile].
Di per sé, la durata della vita più breve dei fringuelli zebra trattati con DNP non sorprende. Il DNP è un dissipatore di ATP sotto forma di calore, il che causa una produzione massiva di radicali liberi nei mitocondri. Da qui il consiglio di assumere quantità significative di antiossidanti con il DNP . Ma, stranamente, la concentrazione del perossido di idrogeno (uno dei due radicali liberi più conosciuti a contenuto d’ossigeno [ROS da Reacting Oxygen Species]) non è aumentata nei fringuelli zebra del gruppo DNP.
Conclusione:
Lo studio evidenzia che, anche a una dose moderata […], un trattamento cronico con DNP può abbreviare la durata della vita. Il DNP promuove il flusso di protoni non solo attraverso la membrana mitocondriale, ma anche attraverso la membrana plasmatica. Questo potrebbe essere un elemento chiave che spiega l’impatto negativo del DNP sulla durata della vita, ma potrebbe essere potenzialmente risolto utilizzando disaccoppiatori di prossima generazione (ad esempio BAM15) specifici per la membrana mitocondriale. Ulteriori studi che indagano i percorsi molecolari e fisiologici attraverso i quali il DNP riduce la durata della vita nei fringuelli zebra sarebbero utili per consentire indagini mirate di effetti deleteri subletali in altri modelli animali e potenzialmente nell’uomo. Il presente studio dovrebbe essere un potenziale segnale di avvertimento per gli attuali utilizzatori di DNP e sollevare domande per gli scienziati che indagano sull’uso del DNP come medicinale.
Quindi è inutile una supplementazione a base di antiossidanti per attenuare gli effetti collaterali cellulari legati alla somministrazione di DNP? Beh, dai risultati sembrerebbe che la questione radicali liberi sia addirittura molto secondaria nella lista degli effetti collaterali DNP-dipendenti ma, ed è da tenere in considerazione, parliamo di uno studio effettuato utilizzando una specie con caratteristiche nettamente differenti da quella umana e, oltretutto, lo studio è stato svolto con dosaggi di DNP somministrati in cronico (4 anni). Ovviamente, gli utilizzatori di DNP usano tale composto solo per brevi periodi di tempo; parliamo di una media di 2 settimane consecutive. Detto ciò, l’assunzione di antiossidanti non è da considerarsi (al momento) del tutto inutile visto il tasso di stressor aggiuntivi ai quali il soggetto in trattamento con DNP viene sottoposto.
Nota:l’effetto collaterale predominante e incisivo (non che letale) del DNP rimane lo shock termico da dose letale la quale è molto vicina alla dose comunemente utilizzata a scopi termogenici.
Come intuibile dal titolo, questa volta il mio intento non sarà quello di esporre una semplice descrizione “commerciale” del Methenolone, ma ben si una sua dettagliata descrizione molecolare e bio-attiva a livello muscolare.
Per molti professionisti e profani, il Methenolone è un AAS “debole” con una capacità miotrofica poco inferiore a quella del Testosterone. Questo effetto è generalmente attribuito al metabolismo di questa molecola. In particolare, la cosiddetta 3α-riduzione che è nota avvenire nel muscolo scheletrico, e che rende AAS come il Mesterolone, soggetto a questa riduzione, con un valore miotrofico dipendente dal legame AR molto scarso. Ma, dal momento che si sta comunque parlando di una 3α-riduzione, vorrei anche sottolineare che questo è probabilmente il motivo per cui l’AAS Methenolone ha proprietà di stimolo ipertrofico della massa muscolare così relativamente deboli. Si lega all’AR tanto quanto il Testosterone e in uno studio dove veniva misurata l’attività androgena, la sua potenza relativa è persino superiore a quella del Testosterone.[1] Allora come si può considerare questo AAS come una molecola debole? Bene, questo studio è stato svolto utilizzando cellule del osteosarcoma, e non cellule muscolo-scheletriche. Le cellule muscolo-scheletriche esprimono l’enzima 3α-HSD e infatti i metaboliti primari del Methenolone sono 3α-ridotti. Questi metaboliti 3α ridotti sono il risultato di una singola reazione chimica, catalizzata dal 3α-HSD. Questo praticamente non avviene con il Testosterone, il quale è un cattivo substrato per questo enzima. Questi risultati suggeriscono che il Methenolone subisce modifiche strutturali in misura apprezzabile nel muscolo scheletrico, rendendolo meno potente in questo tessuto. Quindi, la sua attività nel muscolo scheletrico (e in altri tessuti che esprimono il 3α-HSD) subisce una riduzione, mentre ciò non si verifica nei tessuti che ne sono privi, rendendolo quindi una cattiva scelta per chi cerca AAS con un effetto miotrofico rilevante.
No, l’articolo non finisce qui… siamo solo all’introduzione…
In questo articolo cercherò di presentare le prove disponibili in merito alle affermazioni sopra esposte in modo equilibrato. Esaminerò ciò che si sa sul metabolismo del Methenolone, le insidie che possono esserci nei test degli androgeni e alcuni degli studi clinici che hanno utilizzato il Methenolone.
Methenolone – caratteristiche molecolari e metabolismo – :
Prima di entrare nel dettaglio del metabolismo del Methenolone, è utile esaminare brevemente la sua formula strutturale. Dopo tutto, il suo metabolismo è il risultato della sua struttura chimica.
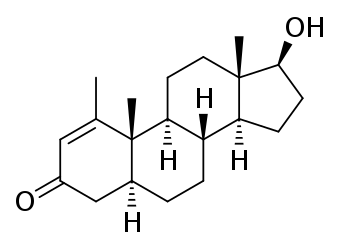
Il Methenolone [17beta-Hydroxy-1-methyl-5alpha-androst-1-en-3-one] è stato descritto per la prima volta nel 1960.(2) Squibb introdusse il farmaco (nella forma orale e iniettabile) negli Stati Uniti nel 1962.(3) Si trattava e, ovviamente, si tratta di un AAS derivato dal DHT con valore androgeno/anabolizzante pari a 44-57:88 . Si tratta, inoltre, di una molecola presente in natura, sebbene essa si trovi all’interno delle ghiandole surrenali dei felini domestici in gravidanza.(4)
Methenolone
Nota: Il Methenolone, essendo una molecola 5-alfa ridotta (in quanto derivato del DHT) non subisce alcuna aromatizzazione (5) e non presenta alcuna attività estrogenica misurabile.
Le caratteristiche chimiche che sono importanti sono il gruppo cheto (= O) al carbonio 3, l’atomo di idrogeno α-orientato (-H) al carbonio 5, il gruppo metile (-CH3) attaccato al carbonio 1 e il doppio legame (C = C) tra gli atomi di carbonio 1 e 2.
Il gruppo cheto al carbonio 3 e l’atomo di idrogeno α-orientato al carbonio 5 sono presenti anche nel Diidrotestosterone (DHT). Gli steroidi con queste due caratteristiche chimiche generalmente formano buoni substrati per enzimi che riducono il gruppo cheto al carbonio 3. Questi enzimi sono indicati come 3α / 3β-idrossisteroide deidrogenasi. Il risultato è un gruppo idrossile (-OH). Il gruppo 3-cheto è molto importante in termini di affinità di legame per il recettore degli androgeni. Quasi tutti gli steroidi anabolizzanti comuni hanno un gruppo 3-cheto: Testosterone, Nandrolone, Trenbolone, Boldenone, Drostanolone, Oxandrolone, Fluoxymesterone, Oxymetholone, Methandienone, 4-Chlorodehydromethyltestosterone, ecc. La ragione di ciò è che l’atomo di ossigeno può funzionare come un accettore di legami idrogeno per gli amminoacidi carichi del recettore degli androgeni che si trovano nelle immediate vicinanze dell’anello A quando si legano [6]. Se riduci questo atomo di ossigeno, non ci sono coppie di elettroni che funzionino come accettori di legami idrogeno, e quindi l’affinità di legame diminuisce enormemente. Lo Stanozololo potrebbe essere un’eccezione strutturale, poiché non ha un gruppo 3-cheto (né un gruppo idrossile li), ma accade la stessa cosa. Il secondo atomo di azoto dell’anello pirazolico dello Stanozololo può funzionare come accettore di legami idrogeno. Il messaggio da portare a casa qui è che, una volta che uno steroide vede ridotto il suo gruppo 3-cheto, è effettivamente reso inutile.
3α-riduzione nell’anello A dello scheletro carbossilico
Gli esseri umani esprimono 4 diversi cosiddetti isoenzimi che possono catalizzare la riduzione 3α [7]. Questi isoenzimi sono denominati da AKR1C1 a AKR1C4. Tutti sono in grado di catalizzare la riduzione 3α, ma differiscono per la distribuzione tissutale e la specificità del substrato. Questo è in netto contrasto nel ratto, che esprime solo un singolo enzima in grado di ridurre il 3α. A causa della presenza di più enzimi in grado di catalizzare la riduzione del 3α, nonché del fatto che il singolo nei ratti non è identico a nessuno dei 4 isoenzimi nell’uomo, è necessario prestare molta attenzione quando si estrapola un dato dai ratti e lo si trasferisce all’uomo.
Gli isoenzimi AKR1C2 e AKR1C4 sono quelli che sembrano dimostrare la maggiore attività di 3α-riduzione. AKR1C4 è espresso in gran parte nel fegato, ma solo in misura minore nel polmone, nella prostata, nella ghiandola mammaria, nel cervello, nell’intestino tenue e nei testicoli.[7] In quanto tale, AKR1C4 sembra essere una forma specifica del fegato. AKR1C2 d’altra parte, oltre ad essere espresso nel fegato, è anche espresso in larga misura nei polmoni e nella prostata. Alcune espressioni più piccole, ma significative, si verificano anche in altri tessuti.
“E i muscoli-scheletrici?”. Sfortunatamente, lo studio che ho menzionato sopra non ha verificato questo punto. Un altro studio ha verificato la presenza nel muscolo scheletrico, ma lo ha fatto solo per AKR1C1 e AKR1C4.[8] Entrambi non sono stati rilevati nel muscolo scheletrico. Ancora un altro studio ha scoperto che AKR1C3 è espresso nei muscoli e che la sua espressione attraverso l’mRNA è sovraregolata con la terapia con Testosterone (250 mg di Sustanon ogni due settimane). [9] Non sono riuscito a trovare nessuno studio che esamini l’espressione del AKR1C2 nel muscolo scheletrico.
Finora abbiamo stabilito che almeno uno, e forse due, degli isoenzimi che possono ridurre gli steroidi 3α sono espressi nel muscolo scheletrico. Esistono quindi anche studi che dimostrano un’attività significativa di questi enzimi nel muscolo scheletrico? Un vecchio lavoro tedesco lo ha dimostrato elegantemente iniettando per via endovenosa DHT tracciato a tre partecipanti al fine di monitorare il suo destino metabolico [10]. I campioni di tessuto muscolare sono stati raccolti da 20 a 60 minuti dopo l’iniezione. Il 29,4-51,0% dello steroide marcato è stato identificato come 3α-androstandiolo (il metabolita 3α-ridotto del DHT) in questi campioni. Sono stati identificati anche un paio di altri metaboliti e solo dal 18,1 al 26,2% di DHT era rimasto integro. Ciò dimostra infatti una significativa modifica del DHT che si verifica nel tessuto muscolo scheletrico come risultato della riduzione del suo gruppo 3-cheto. Come nota a margine, hanno fatto la stessa cosa con il Testosterone marcato, di cui il 69,5-79,8% dello steroide marcato era ancora identificato come Testosterone nei campioni di tessuto muscolare.
E il Methenolone? Come accennato all’inizio di questa sezione dell’articolo, esso presenta un gruppo cheto al carbonio 3 e un atomo di idrogeno α-orientato al carbonio 5. Il motivo per cui non segue necessariamente lo stesso destino del DHT, è a causa delle altre 2 alterazioni : il gruppo metile attaccato al carbonio 1 e il doppio legame tra il carbonio 1 e 2. Cosa si sa del metabolismo del Methenolone? La tabella seguente proviene da uno studio condotto dal produttore del Methenolone (Schering, oggi Bayer). [11]
Le parole “stark gehemmt” sono tedesche e stanno per “fortemente inibito”. Come tale, questa tabella suggerisce che la riduzione 3α del Methenolone è fortemente inibita. Ciò è quanto seguirebbe dalla tabella 5 dello stesso documento. La Tabella 5 elenca 2 potenziali metaboliti 3α-ridotti del Methenolone che hanno controllato. Infatti, uno di loro, 1- (α) -Methyl-androstan-3α-ol-17-on (Methylandrosterone), è stato elencato come non rilevabile. L’altro, androstan-3α-ol-17-on (androsterone), è stato elencato come (+), il che significa che ne hanno trovato una traccia. Lo stesso Methenolone è stato recuperato nella misura massima, elencato come +++ nella tabella. (Indubbiamente questo è stato coniugato, poiché la ricerca successiva non è stata in grado di recuperare il Methenolone immodificato dall’urina dopo l’iniezione [12].)
Il Methylandrosterone è una forma 3α ridotta di Methenolone che manca del doppio legame tra il carbonio 1 e 2. L’altro metabolita, Androsterone, manca inoltre del gruppo 1α-metile. Ma questi non sono gli unici potenziali metaboliti 3α-ridotti del Methenolone. In effetti, ricerche successive hanno dimostrato che dopo la somministrazione orale, il metabolita principale del Methenolone era il 3α-idrossi-1-metilen-5a-andorstan-17-one [13]. In effetti, è stato escreto in misura maggiore del 50% circa rispetto allo stesso Methenolone (coniugato). In quanto tale, la nota che la riduzione 3α è “fortemente inibita” nella tabella sopra si basa su uno screening incompleto dei metaboliti e dovrebbe essere scartata come falsa data la ricerca che è emersa in seguito.
Tuttavia, si nota che hanno estratto l’1,62-1,65% del Methenolone ingerito come immodificato nelle urine e il 2,4% della dose di Methenolone escreta come metabolita 3α-ridotto (3α-idrossi-1-metilen-5α-andorstan-17-one ) entro 7 giorni dall’ingestione. Non menzionano la % di altri metaboliti, ma si può dedurre che cumulativamente costituissero non più dell’1% della dose di Methenolone. Tutto sommato, circa il 95% della dose ingerita rimane dispersa. Ciò può essere spiegato da due fattori:
Mancano metaboliti aggiuntivi
Escrezione attraverso altre vie (feci attraverso il riciclo entroepatico).
Ricerche svolte successivamente. e che utilizzano una tecnica all’avanguardia (LiQuiD ChRoMaToGrApHy QuAdRuPoLe TiMe-Of-FliGhT mAsS sPeCtRoMeTry aka LC-QTOFMS), hanno effettivamente trovato diversi metaboliti aggiuntivi dopo l’iniezione di Methenolone [14]. Tuttavia, non hanno eseguito un’analisi quantitativa, quindi non può essere derivata la parte % mancante della dose di Methenolone. È mia opinione che la maggior parte finisca nelle feci piuttosto che i metaboliti non rilevati costituiscano la maggior parte della dose non quantificabile. Inoltre, lo studio [12] non ha trovato Methenolone letteralmente invariato nelle urine (hanno trovato Methenolone coniugato senza altre alterazioni chimiche). Ciò significa che il Methenolone immodificato riportato in altri studi deve essere stato un miscuglio con un metabolita diverso, oppure i gruppi sulfo o acido glucuronico sono stati rimossi dopo la raccolta del campione di urina. In ogni caso, il messaggio da portare a casa è che il Methenolone si 3α-riduce significativamente, ma in misura minore rispetto al DHT. Le domande senza risposta sono in che misura ciò avvenga nel muscolo scheletrico (dati i diversi isoenzimi) e con quale velocità. Queste domande sono senza risposta con i dati attuali.
Approfondimenti da non trascurare:
Dato che il Methenolone si 3α-riduce e il muscolo scheletrico esprime enzimi che possono catalizzare questa reazione, si può sostenere che l’effetto del Methenolone nel muscolo scheletrico diminuisce. Non è chiaro in che misura ciò accada. Inoltre, altri tessuti sensibili agli androgeni che esprimono enzimi in grado di farlo diminuiranno anche il suo effetto in questi. Si può quindi sostenere che in alcuni tessuti sensibili agli androgeni anche il suo effetto sarà ridotto. Quello che sto cercando di dire qui è che da un lato può essere dannoso per la suo cosiddetta Anabolizzante/Androgeno ratio, mentre dall’altro potrebbe essere utile, a seconda del tessuto.
Quantificare la anabolico-androgeno ratio per un determinato AAS è qualcosa che alcuni test hanno cercato di fare. L’esempio principale di tale analisi è il dosaggio Hershberger. Questo test è stato descritto per la prima volta da Hershberger e dai suoi colleghi nel 1953 [15]. Per lo svolgimento si prendono dei ratti e li si castra per sbarazzarsi del Testosterone endogeno prodotto che potrebbe interferire con i risultati. Quindi si inietta loro l’AAS sotto ricerca e si aspetta un certo periodo di tempo (generalmente 8 giorni) prima di uccidere i ratti e sezionarli. Si sezionano per pesare il muscolo levator ani, la prostata ventrale e le vescicole seminali. Si suppone che l’aumento di peso del muscolo levator ani rifletta l’attività anabolica del composto e si suppone invece che l’aumento di peso della prostata ventrale e delle vescicole seminali rifletta la sua attività androgena.
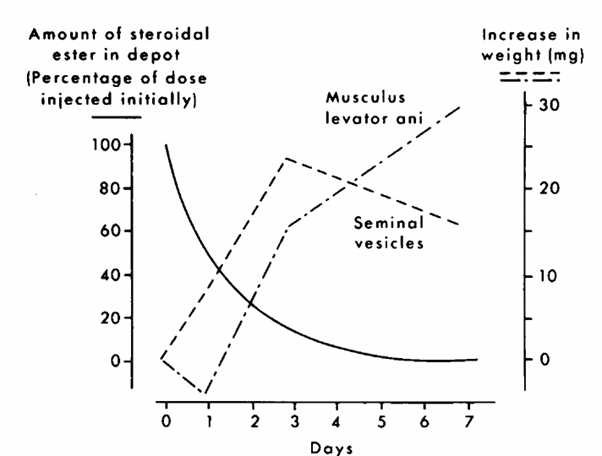
I problemi con questo test sono numerosi. Innanzitutto, scegliere il muscolo levator ani come sostituto del muscolo scheletrico è un non lieve difetto di forma per l’esecuzione di uno studio con variabili accettabili, non per nulla ha ricevuto alcune critiche dalla comunità scientifica. Questo muscolo, a quanto pare, non è un normale muscolo scheletrico. È un muscolo fortemente dipendente dagli androgeni che fa parte del sistema riproduttivo del ratto (in realtà è il muscolo bulbocavernoso). Un articolo di Keith Hayes copre ampiamente questo aspetto [16]. In un certo senso, si potrebbe quindi sostenere che un aumento di peso del muscolo bulbocavernoso è rappresentativo dell’attività androgenica piuttosto che dell’attività anabolica. Un altro problema è che il muscolo bulbocavernoso e le vescicole seminali rispondono in modo diverso alle diverse concentrazioni di un androgeno. Come tale, il rapporto misurato sarà diverso a seconda della dose utilizzata e del momento in cui sono state effettuate le misurazioni. Ciò è ben illustrato nella figura seguente presa da van der Vies dopo l’iniezione di Nandrolone [17].
Uno dei motivi per cui il test Hershberger è fallace.
Durante i primi 3 giorni, la concentrazione è sufficientemente elevata da stimolare la crescita sia delle vescicole seminali che del muscolo bulbocavernoso. Tuttavia, dopo tre giorni la concentrazione non è abbastanza alta da mantenere questa crescita per le vescicole seminali e le loro dimensioni diminuiscono nuovamente. Tuttavia, il muscolo bulbocavernoso è ancora sufficientemente stimolato per continuare a crescere di dimensioni. In quanto tale, la androgeno/anabolico ratio misurata il giorno 3 sarà molto diversa da quella che si misurerà il giorno 7, nonostante si stia utilizzando lo stesso composto. Ciò evidenzia anche che organi diversi rispondono semplicemente in modo diverso a seconda della concentrazione del composto. Anche se ci fosse un modo accurato per determinare una androgeno/anabolico ratio, estrapolarlo da concentrazioni fisiologiche a concentrazioni sovrafisiologiche sarebbe fortemente soggetto ad errori interpretativi, se non completamente sbagliato!
Continuando con questo ragionamento logico, è un presupposto errato che la crescita della prostata ventrale o delle vescicole seminali sia rappresentativa di tutti gli altri effetti androgeni. Non ci sono prove a sostegno di questa ipotesi (anzi, le prove esistenti sono contrarie).
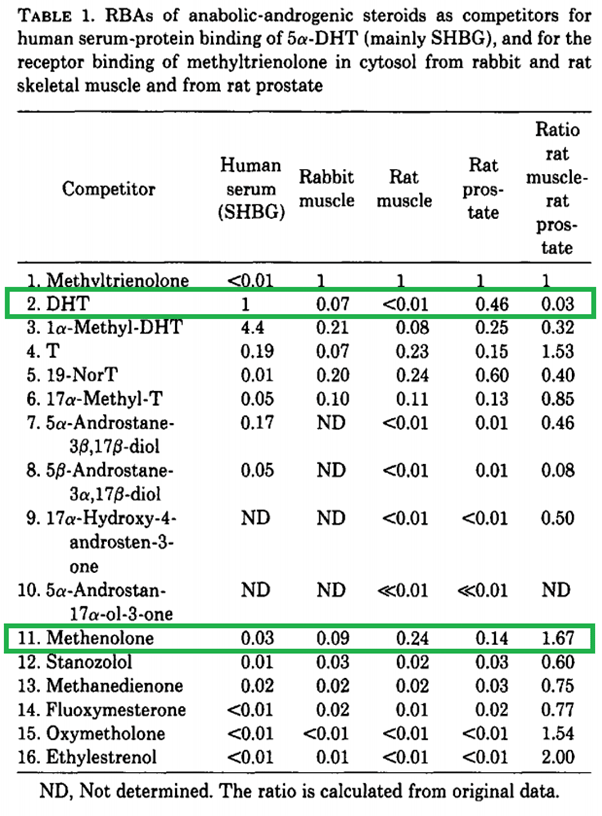
Infine, ovviamente, i ratti non sono esseri umani. È un altro enorme presupposto che i tessuti omologhi negli esseri umani risponderebbero allo stesso modo ad un AAS come osservato nel ratto. Nessun valore deve essere attribuito ai rapporti provenienti dal dosaggio Hershberger. Un altro metodo consiste nel misurare l’affinità di legame relativa di uno steroide in vari tessuti. Saartok et al. ha utilizzato questo metodo determinando l’affinità di legame relativa del Methenolone nel muscolo del coniglio, nel muscolo del ratto e nella prostata del ratto, rispetto al composto di riferimento (Methyltrienolone) [18]. Gli autori erano consapevoli dei problemi con il muscolo bulbocavernoso e, di conseguenza, sembra che abbiano utilizzato i muscoli soleo e gastrocnemio. I risultati sono stati i seguenti:
Affinità di legame relativa di diversi steroidi dallo studio di Saartok et al.
Diamo prima un’occhiata al DHT e confrontiamolo con Testosterone. Nel muscolo di ratto, l’affinità di legame è molto inferiore a quella del Testosterone. Ciò è molto probabilmente dovuto alla rapida metabolizzazione attraverso la 3α-riduzione nel muscolo scheletrico (come ipotizzato anche dagli autori). Tuttavia, l’affinità di legame è simile a quella del Testosterone nel muscolo del coniglio. Ciò evidenzia una notevole differenza tra le due specie animali. Apparentemente si verifica una 3α-riduzione del DHT significativamente inferiore nel muscolo scheletrico del coniglio. Ora, se guardiamo al Methenolone, le affinità di legame sono nello stesso campo di applicazione di quelle del Testosterone. Questo significa che non si 3α-riduce negli esseri umani? Assolutamente no. Come accennato in precedenza in questo articolo, ci sono diversi enzimi che agiscono in tal senso e che differiscono nella loro specificità del substrato rispetto ai ratti, e questo studio da solo mostra già una notevole differenza tra due specie animali quando si guarda al DHT.
L’inutilità di questo test per avere un’idea della androgeno/anabolico ratio (o della sola attività anabolica) negli esseri umani è ulteriormente evidenziata dalle affinità di legame ridicolmente basse di alcuni degli altri AAS elencati.
Un test finale di cui vorrei discutere è il dosaggio CALUX del recettore degli androgeni (AR) [19]. Il test è stato sviluppato per rilevare nuove sostanze dopanti negli atleti piuttosto che essere utilizzato per derivare questi rapporti o l’attività anabolica di un composto. Allo scopo di rilevare nuove sostanze dopanti, è perfetto. Fondamentalmente hanno modificato una cellula in modo tale che emani una bioluminescenza quando viene attivato il recettore degli androgeni. Lo hanno fatto co-trasfettando la cellula con un gene della luciferasi che è sotto il controllo trascrizionale del recettore degli androgeni. A questo scopo, hanno utilizzato cellule di osteosarcoma. Tratterò questo a breve, perché è uno dei problemi principali riguardanti il Methenolone dal momento che queste cellule chiaramente non hanno alcuna significativa attività nella 3α-riduzione. Ciò è dimostrato dal fatto che il DHT si presenta come un AAS estremamente potente in questo test. Quindi, si può anche leggere quello che hanno misurato con il Methenolone, ma ciò non dirà nulla su quello che realmente accade nel muscolo scheletrico con questo AAS.
Studi clinici:
In fine, quello che si vorrebbe sapere è come il Methenolone si sovrappone a un altro AAS in termini di effetti collaterali e crescita muscolare in uno studio clinico. Esistono studi del genere? Sì, ma effettuati in pazienti con cancro al seno e in assenza di misurazioni della crescita muscolare in un contesto clinico, praticamente irrilevante per i bodybuilder. Un’altra cosa che è possibile fare per dare risposta al quesito iniziale, è quella di confrontare ciò che è stato riportato negli studi clinici senza un confronto diretto tra AAS e mettere in rapporto ad altri studi che hanno utilizzato altri composti in funzione di confronto potenziale. Certo, questo è già un tentativo fallito in quanto il confronto tra i dati, specialmente con campioni di dimensioni inferiori, è molto problematico.
C’è uno studio che avrebbe potuto essere fantastico. Si tratta di una ricerca svolta reclutando 47 uomini i quali sono stati randomizzati a ricevere un placebo, Methenolone Acetato, placebo+ Methenolone Acetato e Methenolone Acetato + Allenamento [20]. Lo studio è durato ben 16 settimane. Hanno eseguito misurazioni della forza e del peso dei soggetti. Allora perché non c’è da entusiasmarsi? Perché hanno dato il Methenolone (20mg al giorno) per via orale invece che tramite iniezioni. Sebbene abbia una certa biodisponibilità orale, non è sicuramente all’altezza di quella di un AAS 17a metilato (la biodisponibilità del Methenolone Acetato in forma orale è molto simile a quella del Mesterolone assunto per la medesima via [3% circa]). In quanto tale, viene metabolizzato troppo rapidamente per esercitare un effetto apprezzabile a quel dosaggio. In effetti, gli autori non hanno riscontrato differenze tra i gruppi in nessuna delle misurazioni.
Un altro studio ha esaminato l’effetto di 100mg di Methenolone Enantato mediante iniezione intramuscolare una volta alla settimana per 6 settimane [19]. Tuttavia, è stato fatto insieme alla terapia riabilitativa in pazienti con emiplegia 1-8 mesi dopo l’ictus. Cioè, pazienti con paralisi completa di metà del corpo (ovviamente la terapia riabilitativa mirata alla metà del corpo non paralizzata.). Lo studio non era in cieco e non randomizzato ed i partecipanti ai gruppi placebo e Methenolone erano anziani (in media dai 62 ai 67 anni di età). Le misurazioni della forza erano aumentate, così come l’area della sezione trasversale muscolare delle cosce. È difficile, se non impossibile, dire come si comporta rispetto ad altri AAS. La “colpevolezza” del Methenolone non è tanto nel “non funzionare”: perchè, in effetti, funziona. Il colpevole è che la sua attività nel muscolo scheletrico potrebbe diminuire, rendendo non ottimale per acutizzare l’ipertrofia muscolare (anche se, in una certa misura, lo fa).
Esiste uno studio nel quale pazienti con cancro al seno (donne) hanno ricevuto Methenolone Enantato tramite iniezione intramuscolare [21]. Le donne hanno ricevuto prima 3x 400mg a settimana, dopodiché il dosaggio è stato ridotto a 3x 200mg o 3x 300mg a settimana. La terapia su queste donne è durata, in media, 7,7 mesi. C’è poco che può essere derivato da questo studio, a parte il fatto che il Methenolone causa un aumento del colesterolo totale in modo abbastanza significativo. Questo è qualcosa di condiviso con altri AAS, specie se DHT derivati, non soggetti ad aromatizzazione e orali metilati in C-17. E forse questo effetto potrebbe essere più pronunciato nelle donne, ma non è chiaro. Ad ogni modo, ho trovato uno studio in cui hanno fatto un confronto tra AAS in pazienti con carcinoma mammario avanzato [22]. Un gruppo ha ricevuto un dosaggio molto elevato di Methenolone (3x 400mg a settimana) e l’altro ha ricevuto un alto dosaggio di Testosterone Propionato (3x 100mg a settimana). Il 48% dei pazienti nel gruppo Methenolone ha avuto un miglioramento oggettivo del cancro al seno (e chiaramente un tempo di sopravvivenza più lungo), mentre nessuno ha mostrato miglioramenti nel gruppo Testosterone. È tutto molto interessante, certo, ma questo non ci porta oltre per quanto riguarda il confronto con altri composti in termini di ipertrofia muscolare. Se qualcuno è a conoscenza di qualche vecchio e oscuro studio clinico di confronto tra AAS, con presenza del Methenolone rispetto ad un altro AAS in persone sane, in cui è stata misurata la composizione corporea o la forza, per favore mi contattati.
Conclusioni:
Finora abbiamo stabilito alcune cose sul Methenolone.
Il Methenolone viene modificato in misura apprezzabile mediante la 3α-riduzione, come evidenziato dal suo metabolita primario 3α-idrossi-1-metilen-5a-andorstan-17-one. Una volta 3α-ridotto, la sua attività è praticamente nulla. Ci sono 4 isoenzimi nell’uomo che sono distinti dal singolo enzima nei ratti che catalizza questa reazione e variano l’uno dall’altro in termini di specificità del substrato e distribuzione tissutale. Ciò rende l’estrapolazione dei dati dai ratti (o da qualsiasi altro animale per questa materia) non attendibile. Di questi 4 isoenzimi, almeno 1 è espresso nel muscolo scheletrico, e possibilmente 2. Mentre il Methenolone si 3α-riduce, lo fa in misura minore rispetto al DHT. Non è noto in che misura ciò avvenga nel muscolo scheletrico e con quale velocità. I dati attuali non sono sufficienti per rispondere a queste importanti domande. I dosaggi degli androgeni sono essenzialmente buoni, ma hanno tutti difetti significativi, rendendoli notoriamente inaffidabili per farci affidamento e per dire se un determinato composto è più anabolico o androgeno di un altro nella pratica. Pertanto, da questi studi non è possibile ricavare informazioni affidabili riguardo il Methenolone.
Ci sono alcuni studi clinici svolti con utilizzo del Methenolone, e uno in particolare sarebbe stato estremamente interessante; se non avessero somministrato il Methenolone per via orale invece di iniettarlo. La mancanza di prove di confronto tra AAS con partecipanti sani in cui viene misurata la composizione corporea o la forza muscolare rende impossibile rispondere a qualsiasi domanda su come il Methenolone si relazioni ad altri AAS.
Comunque sia, la maggior parte delle persone mi ha descritto il Methenolone come un AAS relativamente mite. Ciò implica che “devono” abbinarlo con altri AAS, o dosarlo massivamente, per un effetto apprezzabile sull’ipertrofia muscolare. Questa è stata la ragione principale per cui ho ricollegato il possibile “effetto mite” alla 3α-riduzione: dal momento che i feedback riportatimi descrivevano il Methenolone come un AAS lieve, e dato che la molecola è soggetta alla 3α-riduzione nel muscolo scheletrico, la spiegazione più probabile si trova in questa caratteristica. Tuttavia, sono sicuro che altre persone non saranno d’accordo con questa descrizione sull’effetto del Methenolone e / o con quello che ne ho ricavato. Infine, che vi piaccia oppure no, stanno crescendo le prove che qualunque AAS sia denunciato sull’etichetta della fiala o multidose acquistato nel mercato nero, con una discreta probabilmente non la conterrà (a meno che tu non l’abbia preso direttamente da una farmacia, o da un seller con collegamenti esteri “sicuri”, ma questa non è la regola nel torbido mondo delle UGL).
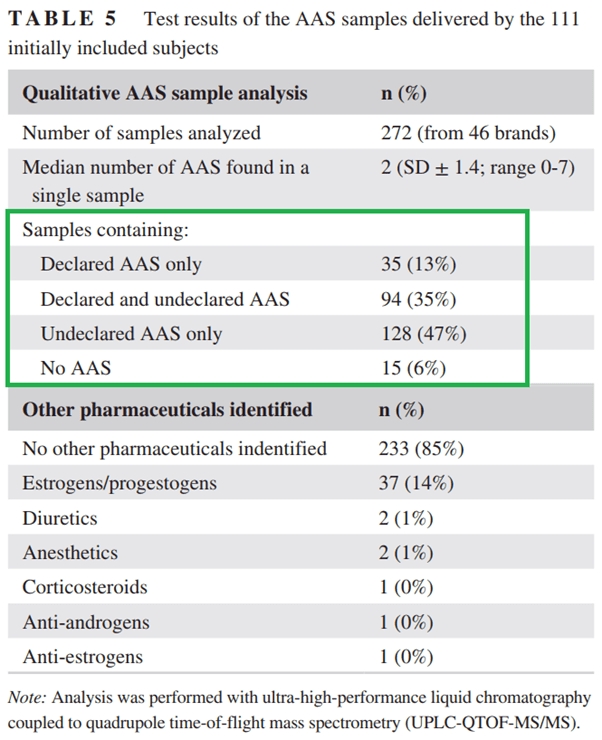
Un recente studio condotto dalla clinica ambulatoriale per utilizzatori di steroidi anabolizzanti nei Paesi Bassi ha esaminato 272 campioni di AAS di 46 diversi marchi forniti loro da i soggetti che si sono arruolati nella loro sperimentazione clinica [23]. I risultati possono essere trovati nella tabella tratta dal documento qui sotto:
Qualità degli AAS esaminati nello studio HAARLEM
Sorprendentemente, solo il 13% degli AAS forniti conteneva solo la molecola dichiarata. Attenzione però, questa era un’analisi qualitativa. Quindi questo non significa nemmeno che questi AAS siano stati dosati come dichiarato sull’etichetta! Vi sento dire: “Sì, ma quelli sono fake, il mio è reale, ho un fornitore affidabile”. Guarda caso, tutti i soggetti della ricerca in questione erano estremamente convinti che il loro AAS fosse buono come descritto sull’etichetta. Per loro era impensabile che i prodotti in loro possesso non fossero quelli riportati. Avevano il miglior fornitore sulla piazza. Tuttavia, si sbagliavano. Gli autori dell’articolo menzionano anche questo: “Il tentativo di aggirare prodotti AAS inaffidabili acquistando da un marchio o rivenditore apparentemente” affidabile “- un argomento sempre interessante per gli utilizzatori di AAS – sembra insignificante poiché nessun marchio ha mostrato un’affidabilità costante”. Risultati simili sono stati pubblicati nel 2005 dall’autorità antidoping olandese dopo aver esaminato 336 prodotti [24] e in Svezia, quando hanno analizzato oltre 1.000 campioni di prodotti dopanti sequestrati al confine [25]. L’ombra del mercato nero degli AAS (che continuerà ad esistere fin tanto che la conoscenza verrà oscurata dalle ideologie di sistema) si aggiunge all’inaffidabilità dei resoconti personali sull’utilizzo di AAS, al di là degli ovvi inconvenienti legati ai dati aneddotici. La maggior parte degli utilizzatori di AAS semplicemente non assume ciò che pensa di assumere.
Nota riflessiva: Durante un intervista dove si trattava l’argomento su quali AAS i bodybuilder della “Golden Era” assumevano, Steve Davis ha affermato:
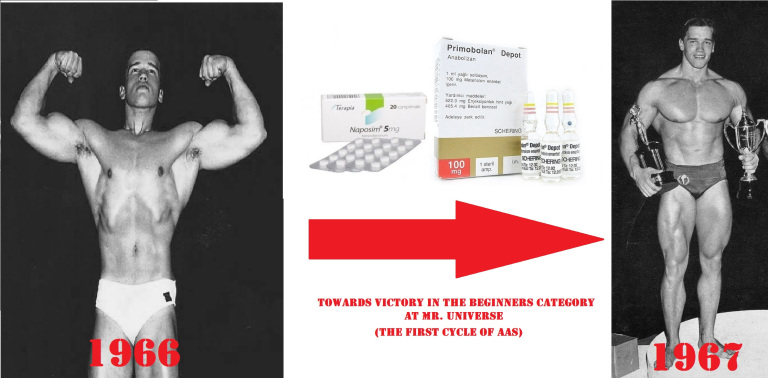
Steve Davis: Sai, nella mia epoca, era consuetudine assumere 3 Dianabol al giorno e una iniezione a settimana.
Ric Drasin: È esattamente così.
Steve Davis: E abbiamo sentito che alcuni austriaci (vedi Arnold Schwarzenegger) che assumevano 4 Dianabol al giorno e una iniezione alla settimana (Methenolone NdR).
Allora, visto che il Methenolone Enantato è stato commercializzato fin dal suo ingresso nel mercato farmaceutico in fiale da 100mg/ml, e che il Methandrostenolone (Dianabol) era comunemente venduto in compresse da 5mg, un tipico ciclo di Schwarzenegger avrebbe potuto essere composto da 100mg/week di Methenolone Enantato e 20mg/die di Methandrostenolone…
Lo stesso Arnold, nella sua autobiografia “Total Recall – My Unbelievably True Life Story”, scrive che la sua scoperta degli AAS risale a prima della competizione Mr.Universe del 1967 mentre faceva ricerche sui metodi di allenamento dei tedeschi dell’est e dei sovietici. Correva voce che stessero usando farmaci per migliorare le prestazioni e per ottenere risultati superiori dai loro sollevatori di pesi, lanciatori e nuotatori e presto scoprì che gli AAS erano i farmaci che stavano usando.
Arnold continua raccontando di quando si recò dal medico per chiedere una prescrizione di AAS. Ha semplicemente chiesto:
Puoi farmi provare?
Il dottore acconsentì…. e gli fu poi prescritta un’iniezione ogni due settimane e delle pillole da prendere giornalmente.
Tenete presente che questo è stato il primo ciclo di AAS di Arnold in assoluto.
I suoi dosaggi potrebbero essere aumentati nei cicli successivi e probabilmente ha sperimentato diversi anabolizzanti ad un certo punto almeno una o due volte.
Sebbene Arnold non abbia precisato esattamente ciò che gli era stato prescritto, è possibile ipotizzarlo con un certo margine di attendibilità utilizzando alcune informazioni utili sulla pratica di somministrazione di ciascun composto (orale e iniettabile) e quale tipo di farmaci rientrerebbero con molta probabilità in queste due categorie in quel preciso momento storico, senza tralasciare tutte le altre informazioni provenienti da interviste di altri campioni della “Golden Era”. A questo punto, sappiamo che stava usando un composto iniettabile e un composto orale.
In un’altra intervista Arnold più o meno sottintende di aver usato 3 Dianabol (15mg ca.) al giorno. Ed è noto nella comunità del bodybuilding che Arnold era un utilizzatore di Dianabol nel corso della giornata.
Arnold Schwarzenegger in posa sul palco del Mr. Olympia 1970 che determino la sua prima vittoria (delle 7) nel contest di maggiore livello nel BodyBuilding.
Dedicato ai buon temponi sognatori che “mi inietto un pò di genetica”…
3- Methenolone acetate, Summary of information for clinical investigators, New Brunswick, NJ.The Squibb Institute for Medical Research, May 30, 1962. Methenolone enanthate, Summary of information for clinical investigators, New Brunswick, NJ. The Squibb Institute for Medical Research, April 15, 1962.
6- Pereira de Jésus-Tran, Karine, et al. “Comparison of crystal structures of human androgen receptor ligand‐binding domain complexed with various agonists reveals molecular determinants responsible for binding affinity.” Protein Science 15.5 (2006): 987-999.
7- Penning, Trevor M., et al. “Human 3α-hydroxysteroid dehydrogenase isoforms (AKR1C1–AKR1C4) of the aldo-keto reductase superfamily: functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones.” Biochemical journal 351.1 (2000): 67-77.
8- O’CONNOR, Tania, et al. “Major differences exist in the function and tissue-specific expression of human aflatoxin B1 aldehyde reductase and the principal human aldo-keto reductase AKR1 family members.” Biochemical journal 343.2 (1999): 487-504.
9- Gharahdaghi, Nima, et al. “Testosterone therapy induces molecular programming augmenting physiological adaptations to resistance exercise in older men.” Journal of Cachexia, Sarcopenia and Muscle 10.6 (2019): 1276-1294.
10-Becker, H., et al. “In vivo uptake and metabolism of 3H-testosterone and 3H-5α-dihydrotestosterone by human benign prostatic hypertrophy.” European Journal of Endocrinology 71.3 (1972): 589-599.
11-Langecker, H. “BEZIEHUNGEN ZWISCHEN SUBSTITUTION IM RING A UND ABBAU IM STOFFWECHSEL BEI VERWANDTEN DES TESTOSTERONS.” European Journal of Endocrinology 41.4 (1962): 494-506.
12-Goudreault, Danielle, and Robert Massé. “Studies on anabolic steroids—4. Identification of new urinary metabolites of methenolone acetate (primobolan®) in human by gas chromatography/mass spectrometry.” The Journal of steroid biochemistry and molecular biology 37.1 (1990): 137-154.
13-He, Genye, et al. “New long term metabolite in human urine for metenolone misuse by liquid chromatography quadrupole time-of-flight mass spectrometry.” Steroids 105 (2016): 1-11.
14-Hershberger, L. G., Elva G. Shipley, and Roland K. Meyer. “Myotrophic activity of 19-nortestosterone and other steroids determined by modified levator ani muscle method.” Proceedings of the Society for Experimental Biology and Medicine 83.1 (1953): 175-180.
15-Hayes, Keith J. “The so-called ‘levator ani’ of the rat.” European Journal of Endocrinology 48.3 (1965): 337-347.
16-Van der Vies, J. “Implications of basic pharmacology in the therapy with esters of nandrolone.” European Journal of Endocrinology 110.3_Suppla (1985): S38-S44.
17-Saartok, Tönu, Erik Dahlberg, and Jan-Åke Gustafsson. “Relative binding affinity of anabolic-androgenic steroids: comparison of the binding to the androgen receptors in skeletal muscle and in prostate, as well as to sex hormone-binding globulin.” Endocrinology 114.6 (1984): 2100-2106.
18-Houtman, Corine J., et al. “Detection of anabolic androgenic steroid abuse in doping control using mammalian reporter gene bioassays.” Analytica chimica acta 637.1-2 (2009): 247-258.
19-Fowler JR, William M., Gerald W. Gardner, and Glen H. Egstrom. “Effect of an anabolic steroid on physical performance of young men.” Journal of Applied Physiology 20.5 (1965): 1038-1040.
20-Shimodozono, Megumi, et al. “Addition of an anabolic steroid to strength training promotes muscle strength in the nonparetic lower limb of poststroke hemiplegia patients.” International Journal of Neuroscience 120.9 (2010): 617-624.
21-Garbrecht, M., et al. “Hyperlipoproteinämie bei additiver Behandlung des metastasierenden Mammakarzinoms mit Metenolonönanthat.” DMW-Deutsche Medizinische Wochenschrift 106.13 (1981): 400-403.
22-Kennedy, B. J., and John W. Yarbro. “Effect of methenolone enanthate (NSC‐64967) in advanced cancer of the breast.” Cancer 21.2 (1968): 197-201.
23-Smit, Diederik L., et al. “Baseline characteristics of the HAARLEM study: 100 male amateur athletes using anabolic androgenic steroids.” Scandinavian Journal of Medicine & Science in Sports 30.3 (2020): 531-539.
25-Weber, Christina, et al. “Qualitative and semiquantitative analysis of doping products seized at the Swiss border.” Substance use & misuse 52.6 (2017): 742-753.
In questo articolo ho intenzione di trattare due “designer-steroid”(DS), il Methylepitiostanolo e Cyanostano. Per la pima molecola (Methylepitiostanolo) mi soffermerò sull’aspetto dei potenziali effetti collaterali ad essa legati e manifestatisi in numerosi casi [1] mentre, nel secondo caso (Cyanostano) sottolineerò la scarsità di letteratura di riferimento e quanto la mancanza di essa possa essere fonte di problemi per l’utilizzatore. [2]
Methylepitiostanolo e i potenziali effetti del suo abuso:
Come già riportato in un mio vecchio articolo dedicato a questa molecola, il Methylepitiostanolo (commercialmente noto con i nomi di Epistane, Hemapolin, Havoc, Epi Plex, ecc…), è anche conosciuto come 17α-Methylepitiostanolo o 2α, 3α-epithio-17α-metil-5α-androstan-17β-olo, è uno steroide steroide androgeno-anabolizzante sintetico (AAS) derivato 17α-metilato del Epitiostanolo, un AAS con azione antiestrogenica utilizzato nel trattamento del cancro al seno in Giappone; e in modo simile al Mepitiostano (estere Epitiostanolo 17-methyloxycyclopentyl), è una versione attiva per via orale del Epitiostanolo. Il Methylepitiostanolo è una forma modificata del Diidrotestosterone (DHT), differendo dalla molecola di partenza per l’aggiunta di un gruppo metilico in posizione C-17, che contribuisce alla protezione dell’AAS durante somministrazione orale e successivo transito epatico e ne aumenta la refrattarietà di legame con le SHBG, e la sostituzione del 3-cheto gruppo rende la molecola maggiormente potente nel legame recettoriale aumentando di conseguenza il suo potenziale Anabolizzante e Androgeno nel tessuto muscolo-scheletrico.
Nota: la Androgeno:Anabolico ratio del Methylepitiostanolo è pari a 91:1100 (riferimento al Methyltestosterone orale 94-130:115-150)
Methylepitiostanolo
Il Methylepitiostanolo è stato descritto per la prima volta nel 1966, durante una serie di ricerche sulla modifica dell’anello-A dei derivati del Androstane. Nello stesso anno è stata analizzata la sua potenza androgena e anabolizzante attraverso studi su ratti normali, dimostrando spiccate proprietà anabolizzanti con un potere androgeno tutto sommato contenuto. I risultati del test sono stati probabilmente più simili a quelli del Desoxymethyltestosterone (Madol), anche se il Methylepitiostanolo mostra la metà del valore androgenico. Sebbene i risultati dei test sugli animali erano molto favorevoli, questo composto non arrivò fino al punto di essere testato su soggetti umani. Come successo per molti altri AAS, il Methylepitiostanolo è stato esaminato ma non è stato mai immesso sul mercato dei farmaci da prescrizione. Per quarant’anni, del composto se ne persero quasi totalmente le tracce, resistendo solo come elemento di interesse per la ricerca.
Il Methylepitiostanolo riemerse dall’oscurità della ricerca alla fine del 2006, quando una nuova società denominata Recomp Performance Nutrition lo introdusse sul mercato statunitense dei prodotti OTC con il nome commerciale di Havoc. Viene venduto come prodotto OTC a causa del fatto che i brevetti del mercato degli integratori alimentari non sono strettamente regolamentati, e il composto in questione non è mai stato classificato (in particolare secondo la legge), come uno steroide anabolizzante. Mentre i regolamenti che impedirebbero la vendita di un nuovo farmaco non approvato come integratore alimentare esistono, non hanno lo stesso peso come le leggi sul controllo degli steroidi anabolizzanti, e sono da sempre non applicate in modo aggressivo.
Nel 2015, i ricercatori dell’Università di Malaga hanno pubblicato 25 casi studio i quali riportavano i danni epatici osservati negli utilizzatori di AAS spagnoli e ispanoamericani.[3] I casi risalgono al periodo 2001-2013.
Nello studio, l’AAS che ha mostrato di causare più frequentemente danni al fegato è stato lo Stanozololo. Non sorprende, dal momento che lo Stanozololo è uno degli AAS più abusati in ambiente sportivo. Più sorprendente (per posizione) è stato il numero 2 nella Top 5 degli AAS tossici per il fegato compilata dai ricercatori. Si trattava, appunto, del Methylepitiostanolo, uno DS che sappiamo benissimo essere ancora presente nel mercato.
Nel 2020, altri ricercatori affiliati all’Instituto de Investigacion Sanitaria La Fe hanno pubblicato altri quattro casi di danno epatico da Methylepitiostanolo.[4]
Generalmente, quando gli AAS orali causano danni epatici, essi coinvolgono la colestasi. Ciò significa che il fegato non può più trasportare correttamente la bile nell’intestino perché i dotti biliari sono bloccati. Il corpo produce la bile dal colesterolo e il corpo si sbarazza del colesterolo attraverso la bile. Se l’espulsione della bile è compromessa, si deteriora anche l’equilibrio del colesterolo.
Allo stesso tempo, anche lo smaltimento della bilirubina è compromessa. La bilirubina viene rilasciata durante la degradazione dei globuli rossi. La bilirubina viene espulsa dal corpo attraverso la bile. Nello studio del 2020, la quantità totale di acidi biliari nel corpo degli utilizzatori di Methylepitiostanolo è aumentata di un fattore da 14 a 61. La figura seguente mostra che la concentrazione di bilirubina in tre degli utilizzatori ha superato i 20mg per decilitro.
A quella concentrazione, la bilirubina può danneggiare i reni. Questo è accaduto ad un altro bodybuilder di 40 anni che utilizzava il Methylepitiostanolo, sul cui caso la nefrologa Monica Milla Castellanos ha pubblicato un caso di studio nel 2018.[5]
L’uomo presentava i classici sintomi da danno epatico, come ittero e prurito. La concentrazione di bilirubina nel sangue era alta. Inizialmente, la sua concentrazione di creatinina era normale. Un livello normale di creatinina oscilla tra 0,84 e 1,21. Dopo pochi giorni, tuttavia, il livello di creatinina era salito a livelli preoccupanti. La ricercatrice ha rilevato la bilirubina nelle urine dell’uomo.
Apparentemente l’alta concentrazione di bilirubina ha danneggiato i reni dell’uomo, quindi i medici hanno deciso di trattare il paziente con farmaci come i corticosteroidi e, quando ciò si è rivelato non essere di sufficiente impatto terapeutico, il trattamento è passato alla terapia dialitica [MARS, CVVHD].
I danni ai reni causati dall’abuso di AAS non sono rari. Nello studio dell’Università di Malaga sopra menzionato, il 31% degli utilizzatori di AAS con danni al fegato ha sviluppato anche danni ai reni.
Adesso passiamo al Cyanostano…
Cyanostano caratteristiche e “zone d’ombra”:
Il Cyanostano è strutturalmente simile al Methyldrostanolone (Superdrol), differendo da esso per la presenza di una sostituzione del legame 2α-metilico con un legame cianidrico (caratteristica per l’appunto mancante nel Methyldrostanolone e che lo rende stabile nel 3-Chetogruppo e, di conseguenza, più anabolizzante).
Cyanostano
Le aziende che vendono il Cyanostano affermano che la molecola sia uno steroide anabolizzante molto efficace. L’affermazione principale è che la Androgeno:Anabolico ratio del Cyanostano sia sorprendentemente alto. Alcuni speculatori, senza citare nemmeno le fonti, hanno riportato una A:A ratio pari a 45:800.
Chiunque voglia scoprire su quale ricerca scientifica si basano queste affermazioni rimarrà sorpreso. Se si cercano informazioni sul Cyanostano nel lavoro di Julius Vida “Androgens and Anabolic Agents”, ci si imbatte in uno steroide anabolizzante che assomiglia al Cyanostano e ha le proprietà che l’industria degli integratori attribuisce allo stesso.
Tuttavia, questo promettente AAS non è il Cyanostano. Il Cyanostano può essere descritto come 2-ciano-metil-DHT, mentre l’agente anabolizzante descritto da Vida è il 2-ciano-madolo. Vida trovò le informazioni riportate in un articolo che i ricercatori della società farmaceutica americana Syntex avevano pubblicato sul Journal of Organic Chemistry nel 1964.[6]
E in quell’articolo ci si imbatte nel Cyanostano. Già nella prima pagina. È lo steroide 1a. Modificando questo steroide, precedentemente descritto in un articolo del 1960 su “Chemistry & Industry” (di cui non sono riuscito a procurarmi una copia), il dipartimento di ricerca della Syntex aveva realizzato una serie di analoghi delta-2 che erano piuttosto attivi. Almeno secondo i loro studi sugli animali.
I nuovi analoghi delta-2 sono “un po ‘più androgeni” del Testosterone e del Methyltestosterone e “hanno un’attività miotrofica che è molte volte quella degli standard di riferimento”. Questi standard di riferimento sono ovviamente il Testosterone e il Methyltestosterone. Il Cyanostano in sé non è molto promettente, e lo si può capire leggendo le prime righe dell’articolo.
Eh no, non penso che i ricercatori si siano sbagliati confondendo le molecole tra loro…
Conclusioni
Qualcuno si domanda ancora perchè la liberalizzazione dei farmaci per il miglioramento delle prestazioni sia una cattiva idea quanto si è dimostrata esserlo quella di vietarli e dare spazio alla criminalità organizzata di lucrare sul loro mercato nero. Si tratta di molecole aventi impatto marcato e differenziato sull’intero organismo, non si tratta di prodigiosi elisir che trasformano roiti umani in atleti dall’estetica sopra la media, e certamente non lo fanno in condizioni di forte abuso. In conclusione, abbandonate la filosofia del “il fine giustifica i mezzi” e, piuttosto, abbracciate quella “di necessità virtù”… chi vuole capire capisca …
Sfogliando superficialmente la letteratura medica si può giungere all’erronea conclusione secondo cui gli effetti collaterali consequenziali all’uso/abuso di AAS si limitino principalmente alla sfera fisica e, quindi, organica e sistemica. Ma andando ad approfondire la ricerca in merito ci si imbatte in non pochi casi studio i quali non riportano solamente i classici effetti collaterali correlati agli AAS: aumentato rischio di malattie cardiovascolari, disturbi endocrinologici e, se si parla soprattutto di AAS orali metilati in C-17, anomalie epatiche. Un altro effetto collaterale dovrebbe essere preso in considerazione, anche perchè potrebbe peggiorare, e non di poco, la qualità della vita dell’utilizzatore; sto parlando degli effetti collaterali di natura psicologica. Nel 2019, i ricercatori dell’Università di Oslo hanno pubblicato i risultati di una loro ricerca nella quale si evidenziava che gli effetti collaterali più comuni degli AAS non sono fisici, ma di natura psicologica.[1]
Prima di trattare lo studio, però, è necessario comprendere la correlazione tra cervello e attività steroidea.
I Neurosteroidi
I neurosteroidi, noti anche come steroidi neuroattivi, sono steroidi endogeni o esogeni che alterano rapidamente l’eccitabilità neuronale attraverso l’interazione con canali ionici ligando-dipendenti e altri recettori della superficie cellulare. [2] [3] Il termine neurosteroide è stato coniato dal fisiologo francese Étienne-Émile Baulieu e si riferisce agli steroidi sintetizzati nel cervello. [4] [5] Per essere più precisi, il termine steroide neuroattivo si riferisce agli steroidi che possono essere sintetizzati nel cervello, o sono sintetizzati da una ghiandola endocrina, partendo dalla quale raggiungono il cervello attraverso il flusso sanguigno e hanno effetti sulla funzione cerebrale. [6] Il termine steroidi neuroattivi è stato coniato per la prima volta nel 1992 da Steven Paul e Robert Purdy. Oltre alle loro azioni sui recettori della membrana neuronale, alcuni di questi steroidi possono anche esercitare effetti sull’espressione genica attraverso i recettori degli ormoni steroidei nucleari. I neurosteroidi hanno una vasta gamma di potenziali applicazioni cliniche dalla sedazione al trattamento dell’epilessia [7] e delle lesioni cerebrali traumatiche. [8] [9] Il Ganaxolone, un analogo sintetico del neurosteroide endogeno Allopregnanolone, è in fase di studio per il trattamento dell’epilessia. [10]
Dr. Étienne-Émile Baulieu
I neurosteroidi sono sintetizzati ovviamente dal colesterolo, che viene convertito in Pregnenolone e poi in tutti gli altri steroidi endogeni. I neurosteroidi sono prodotti nel cervello dopo la sintesi locale o per conversione di steroidi surrenali di derivazione periferica o steroidi gonadici. Si accumulano soprattutto nelle cellule gliali mieliniche, dal colesterolo o dai precursori steroidei importati da fonti periferiche. [11] [12] La 5α-reduttasi di tipo I e la 3α-idrossisteroide deidrogenasi sono coinvolte nella biosintesi dei neurosteroidi inibitori, mentre la 3β-idrossisteroide deidrogenasi e l’idrossisteroide sulfotransferasi sono coinvolte nella produzione di neurosteroidi eccitatori. [4]
3α-Idrossisteroide deidrogenasi (3α-HSDoaldo-keto reductasi family 1 member C4)
Sulla base delle differenze di attività e struttura, i neurosteroidi possono essere ampiamente classificati in diversi gruppi principali. [4]
Neurosteroidi inibitori
Questi neurosteroidi esercitano azioni inibitorie sulla neurotrasmissione. Agiscono come modulatori allosterici positivi del recettore GABAA (in particolare isoforme contenenti subunità δ) e possiedono, in nessun ordine particolare, attività antidepressiva, ansiolitica, riducente lo stress, gratificante, [13] prosociale, [14] antiaggressiva, [15] prosessuale, [13] sedativa, pro-sonno, [16] cognitiva e con disturbi della memoria, analgesica, [17] effetti anestetici, anticonvulsivanti, neuroprotettivi e neurogeni. [4] I principali esempi includono il Tetraidrodeossicorticosterone (THDOC), l’Androstano 3α-androstandiolo, il Colestano colesterolo e il Pregnanolone (Eltanolone), Allopregnanolone (3α, 5α-THP). [18] [19]
Tetrahydrodeoxycorticosterone
Neurosteroidi eccitatori
Questi neurosteroidi hanno effetti eccitatori sulla neurotrasmissione. Agiscono come potenti modulatori allosterici negativi del recettore GABAA, modulatori allosterici deboli positivi del recettore NMDA e / o agonisti del recettore σ1 e per lo più hanno effetti antidepressivi, ansiogeni, cognitivi e di potenziamento della memoria, convulsivi, neuroprotettivi e neurogenici . [4] I principali esempi includono il Pregnenolon Solfato (PS), l’Epipregnanolone e l’Isopregnanolone (Sepranolone), gli androstani Deidroepiandrosterone (DHEA; Prasterone) e il Deidroepiandrosterone Solfato (DHEA-S; Prasterone Solfato) e il Colestano 24S-Hydroxycholesterol (Sidrossicol) (Selettivo per il recettore NMDA; molto potente). [20]
Sintesi del 24S-hydroxycholesterol dal colesterolo, catalizzato dall’enzima CYP46A1.
Feromoni
I feromoni sono neurosteroidi che influenzano l’attività cerebrale, in particolare la funzione ipotalamica, tramite l’attivazione delle cellule del recettore vomeronasale. [21] [22] [23] Includono androstani Androstadienol, Androstadienone, Androstenol e Androstenone e Estrane Estratetraenolo.
Androstenone (5α-androst-16-en-3-one)
Altri neurosteroidi
Alcuni altri steroidi endogeni, come il Pregnenolone, [24] Progesterone, [25] [26] Estradiolo, [6] e il Corticosterone sono anch’essi neurosteroidi. Tuttavia, a differenza di quelli sopra elencati, questi neurosteroidi non modulano i recettori GABAA o NMDA e invece influenzano vari altri recettori della superficie cellulare e bersagli non genomici. Inoltre, molti steroidi endogeni, tra cui Pregnenolone, Progesterone, Corticosterone, Desossicorticosterone, DHEA e Testosterone, vengono metabolizzati in (altri) neurosteroidi, funzionando efficacemente come cosiddetti proneurosteroidi.
Progesterone
AAS derivati dal 19-nortestosterone/progestinici
In questa categoria vengono inseriti tutti gli AAS con struttura simile al Progesterone e che interagiscono a diverso grado con i recettori dello stesso, anche a livello neuronale. Non solo il Nandrolone è stato osservato avere effetti significativi a livello cerebrale e sul SNC ma anche altri composti della stessa famiglia (derivati del 19-nortestosterone), come il Trenbolone, hanno mostrato effetti di tipo misto eccitatorio-depressivo. Tali effetti, che si manifestano con maggior enfasi se la molecola in questione va a creare uno squilibrio endocrino maggiormente in contrapposizione con quello mediamente mantenuto in fisiologia (vedi anche rapporto Testosterone:Progestinici), possono essere i seguenti [27]:
1- Aggressività 2- Ansia, paura e stress 3- Ricompensa e dipendenza 4- Alterazione dell’apprendimento, memoria e capacità di lavoro 5- Alterazione della locomozione e attività fisica 6- Effetti sulla HPAA (Asse Ipotalamo-Pituitaria-Surrene) 7- Effetto sui neurotrasmettitori: Recettore Acido γ-Aminobutirrico Tipo A (GABAA); Recettori 5-idrossitriptamina (5-HT) e 5-HT; Recettori della Dopamina e Recettori Oppioidi.
Trenbolone
Alcune delle principali funzioni biologiche note dei neurosteroidi includono la modulazione della plasticità neurale, [28] processi di apprendimento e memoria, [29] comportamento, [30] [31] e suscettibilità alle crisi [32], nonché risposte a stress, ansia e depressione . [33] I neurosteroidi sembrano anche svolgere un ruolo importante in vari comportamenti sessualmente dimorfici e risposte emotive. [34]
Lo stress acuto aumenta i livelli di neurosteroidi inibitori come l’Allopregnanolone e questi neurosteroidi sono noti per contrastare molti degli effetti dello stress. [35] Questo è simile al caso delle endorfine, che vengono rilasciate in risposta allo stress e al dolore fisico e contrastano gli effetti soggettivi negativi di tali stati. Pertanto, è stato suggerito che una delle funzioni biologiche di questi neuromodulatori potrebbe essere quella di aiutare a mantenere l’omeostasi emotiva. [30] [36] Lo stress cronico è stato associato a livelli ridotti di Allopregnanolone e reattività allo stress di Allopregnanolone alterata, disturbi psichiatrici e disregolazione dell’asse ipotalamo-ipofisi-surrene. [33] [35]
Si ritiene che le fluttuazioni dei livelli di neurosteroidi inibitori durante il ciclo mestruale e la gravidanza svolgano un ruolo importante in una varietà di condizioni femminili, tra cui sindrome premestruale (PMS), disturbo disforico premestruale (PMDD), depressione postpartum (PPD), psicosi postpartum e l’epilessia catameniale. [37] [38] [39] Inoltre, si pensa che i cambiamenti nei livelli di neurosteroidi possano essere coinvolti nei cambiamenti di umore, ansia e desiderio sessuale che si verificano durante la pubertà in entrambi i sessi e durante la menopausa nelle donne. [4] [40] [41]
Livelli elevati di neurosteroidi inibitori, vale a dire Allopregnanolone, possono produrre effetti paradossali, come umore negativo, ansia, irritabilità e aggressività. [42] [43] [44] [45] Ciò sembra essere dovuto al fatto che questi neurosteroidi, come altri modulatori allosterici positivi del recettore GABAA come le benzodiazepine, i barbiturici e l’etanolo, [36] [44] possiedono azioni bifasiche a forma di U – livelli moderati (nell’intervallo di 1,5– Alloprogesterone totale 2 nM / L, che sono approssimativamente equivalenti ai livelli della fase luteale) inibiscono l’attività del recettore GABAA, mentre concentrazioni più basse e più alte facilitano l’attività del recettore. [42] [43]
E’ interessante notare come alcuni farmaci antidepressivi come la Fluoxetina e la Fluvoxamina, che si ritiene generalmente influenzino la depressione agendo come inibitori selettivi della ricaptazione della serotonina (SSRI), sono stati osservati agire come normalizzatori dei livelli di alcuni neurosteroidi (che sono frequentemente carenti nei pazienti depressi) a dosi nelle quali non presentano attività nell’influenzare la ricaptazione della Serotonina. Ciò suggerisce che anche altre azioni che coinvolgono i neurosteroidi possono essere in gioco nell’efficacia di questi farmaci contro la depressione. [45] [46]
Fluoxetina
Inoltre, le benzodiazepine possono influenzare il metabolismo dei neurosteroidi in virtù della loro azione sulla proteina traslocatrice (TSPO; “recettore periferico delle benzodiazepine”). [47] Le azioni farmacologiche delle benzodiazepine a livello del recettore GABAA sono simili a quelle dei neurosteroidi. I fattori che influenzano la capacità delle singole benzodiazepine di alterare i livelli di neurosteroidi possono dipendere dal fatto che il singolo farmaco benzodiazepinico interagisca con TSPO. Alcune benzodiazepine possono anche inibire gli enzimi neurosteroidogenici riducendo la sintesi dei neurosteroidi. [48]
Adesso che avete una panoramica di cosa sia un Neurosteroide e come esso possa agire in base al tipo di molecola, passiamo allo studio prima citato…
Dettagli dello studio
I ricercatori hanno utilizzato i dati di 232 utilizzatori di AAS e 60 loro amici e familiari. Queste persone, hanno contattato il progetto Steroid del governo norvegese nel 2015-2019. Il progetto Steroid mira a indirizzare gli utilizzatore di AAS con problemi medici verso specialisti al fine di cercare di consentire loro di cessarne l’uso.
In questo studio, apparso su Substance Abuse Treatment, Prevention, and Policy nel 2019, i ricercatori hanno fatto il punto della situazione sui problemi degli utilizzatori che li hanno contattati.
Risultati dello studio
Il 77,2% degli utilizzatori ha contattato lo Steroid Project e ha deciso di smettere di usarli. Quando i ricercatori hanno confrontato i soggetti che avevano cessato l’utilizzo con gli utilizzatori che volevano continuare a percorrere la strada della supplementazione farmacologica, hanno scoperto che i primi erano più grandi, avevano maggiori probabilità di avere figli e hanno anche segnalato problemi medici più spesso. Bene, questo ha senso.
La figura seguente mostra i problemi segnalati più frequentemente dagli utilizzatori. Erano di natura psicologica piuttosto che fisica. Depressione, cambiamento di comportamento e ansia sono stati i tre effetti collaterali più frequentemente riportati.
Gli amici e i familiari hanno visto principalmente effetti collaterali come cambiamento comportamentale, aggressività e depressione tra gli utenti.
Conclusioni
I ricercatori concludono scrivendo che i problemi contro cui gli utilizzatori di AAS stanno lottando sono piuttosto complessi. I programmi di riduzione del danno, o programmi che vogliono incoraggiare gli utilizzatori a smettere, devono tenerne conto.
I problemi della sfera psicologica si possono presentare sia durante il ciclo (a diverso grado) che nel post ciclo, sebbene una corretta PCT possa smorzarne il grado almeno fin tano che essa persiste (il vero recupero avviene nei mesi successivi alla PCT, ed è qui che gli effetti di una omeostasi ancora alterata si fanno percepire; vedi fase OCT).
Quindi, anche voi lettori, che siate utilizzatori o intenzionati a farlo, dovreste mettere sul piatto della bilancia anche l’impatto psicologico AAS-dipendente/AAS-correlato…
Nota: anche l’uso di SARM, attraverso il feedback negativo AR-dipendente a livello ipotalamico, possono causare i medesimi squilibri nella sintesi ed attività dei neurosteroidi.
12-^ Mellon SH, Griffin LD (2002). “Neurosteroids: biochemistry and clinical significance”. Trends in Endocrinology and Metabolism. 13(1): 35–43.
13- Rougé-Pont F, Mayo W, Marinelli M, Gingras M, Le Moal M, Piazza PV (July 2002). “The neurosteroid allopregnanolone increases dopamine release and dopaminergic response to morphine in the rat nucleus accumbens”. The European Journal of Neuroscience. 16 (1): 169–73. doi:10.1046/j.1460-9568.2002.02084.x. PMID12153544. S2CID9953445.
16- ^ Terán-Pérez G, Arana-Lechuga Y, Esqueda-León E, Santana-Miranda R, Rojas-Zamorano JÁ, Velázquez Moctezuma J (October 2012). “Steroid hormones and sleep regulation”. Mini Reviews in Medicinal Chemistry. 12 (11): 1040–8. doi:10.2174/138955712802762167. PMID23092405.
17- ^ Patte-Mensah C, Meyer L, Taleb O, Mensah-Nyagan AG (February 2014). “Potential role of allopregnanolone for a safe and effective therapy of neuropathic pain”. Progress in Neurobiology. 113: 70–8. doi:10.1016/j.pneurobio.2013.07.004. PMID23948490. S2CID207407077.
23- ^ Baulieu E, Schumacher M (2000). “Progesterone as a neuroactive neurosteroid, with special reference to the effect of progesterone on myelination”. Steroids. 65 (10–11): 605–12. doi:10.1016/s0039-128x(00)00173-2. PMID11108866. S2CID14952168.
33- ^ Jump up to:ab Bäckström T, Andersson A, Andreé L, Birzniece V, Bixo M, Björn I, Haage D, Isaksson M, Johansson IM, Lindblad C, Lundgren P, Nyberg S, Odmark IS, Strömberg J, Sundström-Poromaa I, Turkmen S, Wahlström G, Wang M, Wihlbäck AC, Zhu D, Zingmark E (December 2003). “Pathogenesis in menstrual cycle-linked CNS disorders”. Annals of the New York Academy of Sciences. 1007 (1): 42–53. Bibcode:2003NYASA1007…42B. doi:10.1196/annals.1286.005. PMID14993039. S2CID20995334.
38- ^ Andréen L, Sundström-Poromaa I, Bixo M, Nyberg S, Bäckström T (August 2006). “Allopregnanolone concentration and mood–a bimodal association in postmenopausal women treated with oral progesterone”. Psychopharmacology. 187 (2): 209–21.
39- Bäckström T, Haage D, Löfgren M, Johansson IM, Strömberg J, Nyberg S, Andréen L, Ossewaarde L, van Wingen GA, Turkmen S, Bengtsson SK (September 2011). “Paradoxical effects of GABA-A modulators may explain sex steroid induced negative mood symptoms in some persons”. Neuroscience. 191: 46–54. doi:10.1016/j.neuroscience.2011.03.061. PMID21600269. S2CID38928854.
40- ^ Jump up to:ab Andréen L, Nyberg S, Turkmen S, van Wingen G, Fernández G, Bäckström T (September 2009). “Sex steroid induced negative mood may be explained by the paradoxical effect mediated by GABAA modulators”. Psychoneuroendocrinology. 34 (8): 1121–32. doi:10.1016/j.psyneuen.2009.02.003. PMID19272715. S2CID22259026.
41- ^ Jump up to:ab Bäckström T, Bixo M, Johansson M, Nyberg S, Ossewaarde L, Ragagnin G, Savic I, Strömberg J, Timby E, van Broekhoven F, van Wingen G (February 2014). “Allopregnanolone and mood disorders”. Progress in Neurobiology. 113: 88–94.
44-^ Pinna G, Costa E, Guidotti A (June 2006). “Fluoxetine and norfluoxetine stereospecifically and selectively increase brain neurosteroid content at doses that are inactive on 5-HT reuptake”. Psychopharmacology. 186 (3): 362–72. doi:10.1007/s00213-005-0213-2. PMID16432684. S2CID7799814.
Iniziamo questo 2021 parlando di due SARM non steroidei relativamente nuovi; il GLPG0492 e LGD-2226. Il primo SARM in questione, GLPG0492, è stato oggetto di ricerca da parte della società belga-olandese Galalagos la quale ha condotto studi durante il decennio appena trascorso su questo composto in forma orale. La Galalagos ha persino condotto studi clinici di fase 1 sul GLPG0492, ma senza che questi passassero alle fasi successive.[1] Per quanto riguarda LGD-2226, invece, ad oggi sono disponibili pochi dati e nessuno studio svolto su esseri umani.[2]
Studi su animali con il GLPG0492
Non esistono ad oggi studi in letteratura in cui il GLPG0492 è stato somministrato ad esseri umani, ma esistono alcuni studi sugli animali.[3]
Il primo di questi studi sugli animali è apparso nel 2012. In questo documento la Galalagos ha descritto il GLPG0492 come un SARM disponibile per via orale che approssimava l’effetto di potenziamento muscolare del Testosterone legato all’estere Propionato somministrato tramite iniezione, con solo una frazione dell’effetto androgeno di quest’ultimo (androgeno:anabolico ratio del Testosterone 100:100).[4]
Studi su esseri umani?
La prima pubblicazione del 2012 afferma già che il “GLPG0492 è ora in fase di sperimentazione clinica di fase 1”. Secondo un comunicato stampa diffuso da Galalagos il 12/2/2010, i primi esperimenti di farmacocinetica, in cui i soggetti hanno ricevuto singole dosi da 0,5 a 120mg, hanno avuto esito positivo. La Galalagos ha sperimentato formulazioni orali solide e liquide. Su clinicaltrials.gov [5] si può vedere che nel 2010-2012 la Galalagos aveva già completato 3 studi clinici di fase 1 con somministrazioni orali di GLPG0492. I risultati non sono stati pubblicati in articoli sottoposti a review paritaria, per quanto ne sappia.
Test
Si potrebbe sospettare che i risultati di questi studi siano stati deludenti. Eppure i ricercatori al di fuori delle Galalagos stanno prendendo sul serio il GLPG0492. L’Unione Europea ha recentemente incaricato alcuni chimici della Queen’s University di Belfast al fine di sviluppare un test per rilevare l’uso di GLPG0492 nei bovini e nei cavalli da corsa. [6] Apparentemente Bruxelles non esclude che il mercato illegale del doping non inizierà a commercializzare il GLPG0492 in futuro. Due anni prima che la UE si interessasse a questo SARM, nel 2018, biochimici tedeschi avevano pubblicato un test antidoping commissionato dalla WADA. [7] Lo sviluppo di test richiede tempo e denaro. Succede solo quando gli scienziati hanno indicazioni che un farmaco ha un potenziale. Quindi è possibile che il GLPG0492 potrebbe comparire come supplemento del “mercato grigio” …
E LGD-2226?
Nonostante LGD-2226 circoli dal 2019, esiste pochissima letteratura scientifica su di esso. Una volta letta la ricerca di Jeffrey Miner et al – tutti affiliati alla US Ligand Pharmaceuticals – pubblicata su Endocrinology nel 2007, si conosce praticamente tutto SARM fino a questo momento.
Attività androgena
Nelle piastre di Petri, LGD-2226 difficilmente stimolava la produzione di PSA nelle cellule della prostata. Ciò suggerisce che LGD-2226 ha uno scarso effetto androgeno.
I ricercatori hanno somministrato LGD-2226 a dosi diverse a ratti castrati per due settimane e hanno confrontato l’effetto della SARM con quello del Testosterone.
La linea 100 nelle figure sottostanti indica il livello che ti aspetteresti in esemplari intatti.
A una dose giornaliera di oltre 4-5mg per chilogrammo di peso corporeo, la massa muscolare dei ratti inizia ad aumentare al di sopra del livello “100”.
Se rapportiamo questo dosaggio ad un essere umano adulto, otteniamo un dosaggio di circa 50mg/die.
Tuttavia, la pubblicazione del 2007 contiene anche una cifra basata sulla somministrazione nell’arco di sei settimane. In questo studio sugli animali, una dose orale giornaliera di 1-3mg per chilogrammo di peso corporeo è già sufficiente per stimolare la sviluppo della massa muscolare al di sopra del livello “100” riscontrato negli animali da laboratorio intatti. L’equivalente umano di questa dose è di circa 25mg di LGD-2226 al giorno.
Ed è il dosaggio comune con il quale questo SARM viene commercializato negli store online.
Conclusione
Margine di sicurezza per questi due SARM? Ignoto per il GLPG0492, sebbene si ipotizzi che LGD-2226 possa avere un impatto molto simile al più conosciuto e diffuso LGD-4033.
Come ben sappiamo, gli Inibitori dell’Aromatasi (AI) sono farmaci che possono abbassare il livello degli estrogeni nel flusso ematico riducendone di conseguenza l’attività tissutale-metabolica. Ovviamente, la loro azione inibitoria è esplicata attraverso l’inattivazione di uno specifico enzima. Si tratta infatti dell’Enzima Aromatasi. Detto in modo semplicistico e riduttivo, l’Enzima Aromatasi converte gli Androgeni in Estrogeni. Ad esempio, converte il Testosterone in Estradiolo.
Ci sono attualmente in circolazione tre AI divenuti estremamente popolari in ambito sportivo, e soprattutto nel BodyBuilding. Questi sono: Exemestane (Aromasin), Anastrozolo (Arimidex) e Letrozolo (Femera). Sono comunemente usati nel trattamento del cancro al seno. Sono state quindi svolte molte ricerche sulle donne. Nelle donne, gli AI hanno mostrato un grado di soppressione estrogenica significativamente marcato. Ad esempio, l’Exemestane sopprime i livelli di Estradiolo del 92% nei pazienti con carcinoma mammario in postmenopausa.[1] Allo stesso modo, il Letrozolo e l’Anastrozolo riducono i livelli di Estradiolo di quasi il 90%, anche in questo caso nei pazienti con carcinoma mammario in postmenopausa.[2]
Per questo motivo, nel presente articolo non mi accingerò ad esporre una semplice disamina degli effetti generali o specifici degli inibitori del Aromatasi (cosa già fatta, e nemmeno molto semplicisticamente, qualche anno fa). E’ mia intenzione, invece, trattare con il supporto della letteratura scientifica ad oggi disponibile il reale impatto che questi farmaci hanno sugli individui di sesso maschile, sia in fisiologia che durante l’uso di AAS esogeni soggetti all’Enzima Aromatasi.
Effetto degli AI negli individui di sesso maschile in fisiologia:
Pochi studi hanno osservato l’efficacia degli Inibitori dell’Aromatasi per ridurre i livelli di estrogeni negli uomini. I loro risultati differiscono sensibilmente da quelli riscontrati negli studi sulle donne.
Uno studio condotto su giovani uomini ha rilevato che 25mg di Exemestane sopprimevano i livelli di Estradiolo del 62%, 12 ore dopo l’assunzione.[3] Dopo una somministrazione regolare protratta per dieci giorni, è stata riscontrata una soppressione di solo il 38% nelle 24 ore seguenti l’ultima dose somministrata. In particolare, raddoppiare il dosaggio a 50mg/die non ha portato a una maggiore diminuzione dei livelli di Estradiolo.
Risultati simili si osservano in giovani uomini trattati con Anastrozolo per 10 giorni.[4] La somministrazione giornaliera di Anastrozolo a 0,5 e 1 mg porta ad una diminuzione dei livelli di Estradiolo di circa il 50%. Sebbene di dubbia rilevanza, gli autori non hanno menzionato quante ore dopo l’ultima dose sono state effettuate le misurazioni.
Infine, il Letrozolo ha mostrato la stessa efficacia dell’Exemestane e dell’Anastrozolo. Dopo 28 giorni di trattamento con Letrozolo alla dose di 2,5mg/die, i livelli di Estradiolo hanno subito una riduzione del 46% negli uomini giovani e del 62% negli uomini anziani.[5] Non è chiaro se la differenza tra giovani uomini e uomini anziani sia una differenza reale. Gli autori non hanno eseguito alcun test statistico per valutare matematicamente ciò. In linea con questi risultati, un altro studio ha rilevato che i livelli di Estradiolo erano stati ridotti del 56% negli uomini trattati con 2,5mg di Letrozolo al giorno per 4 settimane.[6]
In sintesi, si potrebbe affermare che i tre popolari AI sono dotati di pari efficaci nel diminuire gli estrogeni negli individui di sesso maschile in fisiologia.
Effetto degli AI nei soggetti di sesso maschile sottoposti a somministrazioni sovrafisiologiche di AAS:
Gli studi di cui sopra sono stati condotti su uomini in fisiologia con i loro livelli endogeni di Testosterone nel range di normalità. Ma la situazione sembra essere diversa quando il Testosterone endogeno viene sostituito con il Testosterone esogeno. Finkelstein et al. ha esaminato gli effetti di quantità crescenti di Testosterone con e senza Inibitori dell’Aromatasi su diversi fattori.[7] Un totale di 198 uomini sani sono stati assegnati in modo casuale a ricevere un placebo, 1,25g, 2,5g, 5g o 10g/die di Testosterone in gel per 16 settimane, e altri 202 hanno ricevuto l’Anastrozolo in combinazione con la dose di Testosterone. La produzione endogena di Testosterone era stata soppressa da iniezioni di Goserelina Acetato.
Meccanismo d’azione schematizzato della Goserelina Acetato
L’immagine seguente mostra l’effetto sui livelli di Testosterone ed Estradiolo in riferimento alle diverse somministrazioni. Si noti che le barre blu si riferiscono agli uomini che ricevevano solo Testosterone mentre le barre rosse quelli che lo ricevevano in combinazione con Anastrozolo.
Tratto da Finkelstein et al. [7]
Come si può vedere, i livelli di Estradiolo sono maggiormente diminuiti rispetto ai numeri discussi in precedenza. Ciò suggerisce che la soppressione dell’Estradiolo da parte degli Inibitori dell’Aromatasi è più marcata negli uomini trattati con Testosterone esogeno in modo “dose dipendente”, e non solamente attribuibile ad una ratio derivante da livelli sovrafisiologici di AAS soggetti all’aromatizzazione.
Qual’è il nesso causale che determina la differenza di risposta tra soggetti in fisiologia e soggetti trattati con Testosterone esogeno?
Sfortunatamente, non esiste una ricerca diretta che risponda in modo chiaro e inequivocabile a questa domanda. Ma se si dovesse ipotizzare, per i dati emersi, sembrerebbe che, banalmente, l’efficacia degli AI negli uomini sia direttamente proporzionata al livello di AAS circolanti soggetti all’Aromatasi il quale causerebbe un incremento dell’espressione enzimatica a livello testicolare e adiposo. Una parte sostanziale dell’Estradiolo è prodotta dall’attività dell’Aromatasi nei testicoli. Nei testicoli, le concentrazioni di Testosterone arrivano a livelli circa 100 volte superiori a quelli presenti nel circolo ematico. Poiché gli AI devono inibire in modo competitivo l’Aromatasi, i dosaggi potrebbero dover essere molto più alti per portare a una significativa inibizione enzimatica nei testicoli. Ma, ovviamente, questa è un’ipotesi al momento non scientificamente dimostrata, sebbene rimanga una delle più plausibile.
Riferimenti:
1- J. Geisler, N. King, G. Anker, G. Ornati, E. Di Salle, P. Lønning, and M. Dowsett. In vivo inhibition of aromatization by exemestane, a novel irreversible aromatase inhibitor, in postmenopausal breast cancer patients. Clinical Cancer Research, 4(9):2089–2093, 1998. 2- J. Geisler, B. Haynes, G. Anker, M. Dowsett, and P. Lonning. Influence of letrozole and anastrozole on total body aromatization and plasma estrogen levels in postmenopausal breast cancer patients evaluated in a randomized, cross-over study. Journal of Clinical Oncology, 20(3):751–757, 2002. 3- N. Mauras, J. Lima, D. Patel, A. Rini, E. di Salle, A. Kwok, and B. Lippe. Pharmacokinetics and dose finding of a potent aromatase inhibitor, aromasin (exemestane), in young males. The Journal of Clinical Endocrinology & Metabolism, 88(12):5951–5956, 2003. 4- N. Mauras, K. O. O’Brien, K. O. Klein, and V. Hayes. Estrogen suppression in males: metabolic effects. The Journal of Clinical Endocrinology & Metabolism, 85(7):2370–2377, 2000. 5- G. G. T’sjoen, V. A. Giagulli, H. Delva, P. Crabbe, D. De Bacquer, and J.-M. Kaufman. Comparative assessment in young and elderly men of the gonadotropin response to aromatase inhibition. The Journal of Clinical Endocrinology & Metabolism, 90(10):5717–5722, 2005. 6- G. Raven, F. H. de Jong, J.-M. Kaufman, and W. de Ronde. In men, peripheral estradiol levels directly reflect the action of estrogens at the hypothalamo-pituitary level to inhibit gonadotropin secretion. The Journal of Clinical Endocrinology & Metabolism, 91(9):3324–3328, 2006. 7- J. S. Finkelstein, H. Lee, S.-A. M. Burnett-Bowie, J. C. Pallais, E. W. Yu, L. F. Borges, B. F. Jones, C. V. Barry, K. E. Wulczyn, B. J. Thomas, et al. Gonadal steroids and body composition, strength, and sexual function in men. New England Journal of Medicine, 369(11):1011–1022, 2013.