Nonostante decenni di “lotta al doping” esso rimane assai diffuso, e non solo nelle competizioni di alto livello. L’errore alla base di questa campagna mediatico-salutistica è stata la generalizzazione; ossia fornire informazioni imprecise, accentuando i possibili sides senza però premurarsi di una vera e propria informativa preventiva chiara, veritiera ed efficace. In poche parole, quello che non si è fatto è dire: “l’uso di PEDs ha una serie di possibili effetti collaterali di gravità dipendente dal tipo di molecola, dal tempo e dalle modalità di assunzione”. Tutto ciò accompagnato da un manuale scientificamente corretto e di facile comprensione, contenente informazioni utili riguardanti la materia PEDs tale da permettere una migliore comprensione della questione che, a sua volta, renda possibile una più consapevole scelta individuale. Ma ciò non è stato fatto. Con l’unica eccezione di alcuni esperti indipendenti che nel corso degli anni hanno pubblicato libri e scritto articoli di una certa utilità.
Lo scopo di questa serie di articoli sarà quello di arginare il fenomeno dell’abuso dei PEDs, cosa che sta degenerando e che sta mostrando i suoi peggiori effetti su atleti di ambo i sessi.
Per la prima pubblicazione di questa nuova serie iniziamo con l’Oxymetholone…
Una (sempre utile) introduzione alla molecola di Oxymetholone:
L’Oxymetholone, noto anche come 2-idrossimetilene-17α-metil-4,5α-diidrotestosterone (2-idrossimetilene-17α-metil-DHT) o come 2-idrossimetilene-17α-metil-5α-androstan-17β-ol-3-one, è uno steroide androstano sintetico e un derivato 17α-alchilato del DHT.[1][2][3]
Le informazioni disponibili sulla farmacocinetica di questo AAS sono limitate.[4] Sembra essere ben assorbito con la somministrazione orale.[4] L’Oxymetholone ha affinità molto bassa per le globuline leganti gli ormoni sessuali nel siero umano (SHBG), meno del 5% di quella del Testosterone e meno dell’1% di quella del DHT. [5] Il farmaco viene metabolizzato nel fegato tramite ossidazione in posizione C2, riduzione in posizione C3, idrossilazione in posizione C17 e coniugazione. [4][6] Il gruppo C2 idrossimetilene del Oxymetholone può essere scisso per formare il Mestanolone (17α-metil-DHT), che può contribuire agli effetti della molecola precursore.[3] L’emivita del Oxymetholone è sconosciuta sebbene vi siano alcune ipotesi a riguardo.[6] L’Oxymetholone e suoi metaboliti vengono eliminati attraverso le urine.[5][6]
Come altri AAS, l’Oxymetholone è un agonista del recettore degli androgeni (AR).[3] Non è un substrato per la 5α-reduttasi (dal momento che è già 5α-ridotto) ed è uno substrato scarso per il 3α-idrossisteroide deidrogenasi (3α-HSD), e quindi mostra un alto rapporto di attività anabolizzante rispetto all’effetto androgenico.[3]
Data la sua derivanza dal DHT, l’Oxymetholone non è un substrato per l’enzima Aromatasi e quindi non può essere aromatizzato in metaboliti estrogenici.[3] Tuttavia, caratteristica unica tra i derivati del DHT, l’Oxymetholone è comunque associato a un’estrogenicità relativamente elevata ed è noto per avere il potenziale di produrre effetti collaterali estrogenici come ginecomastia (raramente) e ritenzione idrica. [3][7][8][9] È stato suggerito che questo può essere una conseguenza del legame diretto a l’attivazione del recettore degli estrogeni da parte dell’Oxymetholone (estrogenicità intrinseca).[3] L’Oxymetholone non possiede alcuna attività progestinica significativa.[3]
A causa della sua struttura 17α-alchilata, l’Oxymetholone è epatotossico.[3] L’uso a lungo termine del farmaco può causare una varietà di disturbi gravi, tra cui l’epatite, il cancro al fegato e la cirrosi; pertanto si raccomandano test periodici di funzionalità epatica per coloro che assumono l’Oxymetholone a fini terapeutici.[10] Questa molecola ha ottenuto, infatti, la nomea di essere uno tra gli AAS più epatotossici. Ciò deriva da i dosaggi comunemente, ed erroneamente, utilizzati in contesto culturistico. Si parla di dosaggi che facilmente sforano i 100-150mg/die. Ma tali dosaggi sono realmente vantaggiosi in termini di guadagni ipertrofici specie se messi in rapporto con gli effetti collaterali possibilmente verificabili? Questa domanda può ottenere una risposta sufficientemente esaustiva attraverso i risultati di uno studio che ha messo a confronto gli effetti di una dose di Oxymetholone da 50mg/die e una da 100mg/die.[11]
Oxymetholone – 50mg Vs. 100mg:
In questo studio, possiamo vedere i cambiamenti nel peso corporeo, nella massa magra, e la perdita di grasso in risposta a un dosaggio moderato e alto di Oxymetholone (50 mg vs 100 mg).
I cambiamenti nella composizione corporea sono mostrati per i gruppi placebo (barre nere), 50mg di Oxymetholone al giorno (barre bianche) e 100mg al giorno (barre grigie). I numeri sopra le barre rappresentano i cambiamenti assoluti medi e le barre di errore sono ± 1 SE. Per la massa corporea magra totale (LBM) e il grasso totale, le differenze tra i 3 gruppi erano significative (P <0,0001, ANOVA a una via). * Differenze significative rispetto al placebo, P ≤ 0,001.
Come ci si aspetterebbe, il gruppo placebo non ha guadagnato massa magra, né ha perso grasso corporeo.
Il gruppo trattato con 50mg di Oxymetholone ha guadagnato 3,3Kg di massa magra e ha perso 2,6kg di grasso.
Il gruppo trattato con 100mg di Oxymetholone ha guadagnato 4,2Kg di massa magra e ha perso 2,5kg di grasso.
I cambiamenti nella composizione regionale (n = 16) sono mostrati per i gruppi placebo, 50mg/die e 100mg/die. A: i numeri sopra le barre rappresentano i cambiamenti assoluti medi per il grasso del tronco mediante assorbimetria a raggi X a doppia energia (DEXA). B: le barre rappresentano i cambiamenti assoluti medi (kg) per la LBM dell’arto superiore (braccio destro più braccio sinistro) mediante DEXA. C: area della sezione trasversale del muscolo totale prossimale (barre grigie) e posteriore (barre nere) dei muscoli della coscia tramite risonanza magnetica. Le barre di errore sono ± 1 SE. * Differenza significativa rispetto al placebo, P ≤ 0,005. .
Guardando la massa corporea magra, è possibile vedere che quando si confrontano i due gruppi di dosaggio, il gruppo da 100mg ha guadagnato solo 0,9kg di massa corporea magra in più rispetto al gruppo da 50mg.
Questo dopo tre mesi di esposizione al doppio della quantità di farmaco.
Se si confrontano i biomarcatori tra i due gruppi, è possibile vedere che l’effetto di 100mg di Oxymetholone ha avuto sui livelli di ALT e AST era molto più deleterio rispetto al gruppo di 50 mg.
Caratteristiche di base della popolazione dello studio
Come molti di voi già sapranno, l’alanina aminotransferasi (ALT) e l’aspartato aminotransferasi (AST) sono biomarcatori comunemente usati per valutare i danni al fegato.
La somministrazione di un dosaggio di Oxymetholone doppio rispetto al basale di 50mg ha prodotto un ulteriore 27% di crescita muscolare relativa (la massa magra non è composta solo dal muscolo scheletrico!), ma ha provocato un picco 3.4x più alto di ALT e un picco 2.7x più alto nei livelli di AST.
Il calo del HDL è stato simile in entrambi i gruppi 50mg/die e 100mg/die.
Quelli sono solo biomarcatori con valore diagnostico per un eventuale danno epatico ma non sono indicativi di ciò che comporta la variabile del dosaggio sull’ipertrofia ventricolare, o altri fattori comunemente trascurati che dovrebbero essere utilizzati per valutare la salute cardiovascolare.
Anche se è possibile che gli aumenti di massa magra misurati dalla DEXA fossero legati in buona parte alla ritenzione idrica causata dalla terapia con Oxymetholone, i notevoli aumenti di forza muscolare misurati con il metodo 1-RM nei gruppi da 50 e 100mg/die (8,2-18,4%) suggeriscono che gli aumenti di massa magra erano probabilmente dovuti all’accrescimento di proteine miofibrillari oltre che alla semplice massa magra totale, poiché la forza è in una certa misura legata alle dimensioni dei muscoli. Inoltre, i membri del gruppo di ricerca hanno riferito che i cambiamenti nella massa magra appendicolare tramite DEXA sono quantitativamente correlati ai cambiamenti nella forza muscolare scheletrica in risposta a stimoli anabolici. In effetti, nel presente studio, sono stati in grado di corroborare questa relazione dimostrando che gli aumenti significativi del tessuto magro della parte superiore del corpo mediante scansione DEXA appendicolare erano altamente correlati con i cambiamenti nella forza della parte superiore del corpo come valutato da esercizi di Chest Press e Lat Pull-Down. Inoltre, i cambiamenti nella forza muscolare massima volontaria per gli esercizi della parte superiore del corpo hanno mostrato una risposta legata alla dose.
I cambiamenti relativi (%) nella forza sono mostrati per i gruppi placebo (barre nere), 50mg/giorno Oxymetholone (barre bianche) e 100mg/giorno Oxymetholone (barre grigie). I numeri sopra le barre rappresentano il cambiamento relativo (%) dal basale alla settimana 12 per le prove di forza massima a 1 ripetizione. Le barre di errore rappresentano ± 1 SE dalla media. * Differenza significativa rispetto al placebo, P < 0,05; † differenza significativa rispetto al placebo con il test di Wilcoxon, P < 0,02.
Al contrario, c’erano guadagni non significativi tra i tre gruppi di trattamento per la forza degli arti inferiori (3,9-12,0%), coerentemente con la mancanza di un aumento significativo della massa magra degli arti inferiori mediante scansione DEXA. Tuttavia, c’era una differenza quasi significativa (P = 0,052) tra i gruppi per il cambiamento del area della sezione trasversale del muscolo (CSA) dei muscoli della coscia tramite la risonanza magnetica, suggerendo che la terapia dello studio può aver influenzato positivamente i muscoli degli arti inferiori. È possibile che i test di forza di gruppi muscolari multipli e di grandi dimensioni, come quelli utilizzati con l’esercizio Leg Press, siano meno sensibili ai modesti cambiamenti nella massa muscolare, e lo studio potrebbe non aver avuto sufficiente potenza per rilevare piccoli ma significativi guadagni nelle estremità inferiori. Si ipotizza che ciò sia dovuto al fatto che i grandi muscoli delle gambe sono abitualmente utilizzati più frequentemente per sostenere il carico (ad esempio, camminare, alzarsi da una sedia) rispetto ai muscoli dell’estremità superiore negli adulti più anziani. Piccoli ma significativi guadagni nella forza e nella massa muscolare della parte inferiore del corpo possono essere meno dimostrabili che per i muscoli della parte superiore del corpo, che possono essere utilizzati meno per il lavoro ad alto volume e più inclini alla sarcopenia nelle persone anziane. Inoltre, i muscoli degli arti superiori, rispetto ai muscoli degli arti inferiori, hanno proporzioni maggiori di fibre a contrazione rapida di tipo II, che possono essere perse preferibilmente con l’invecchiamento. Inoltre, uno studio longitudinale in uomini anziani ha mostrato che le fibre di tipo I sono state perse principalmente nel vasto laterale della gamba, portando all’ipotesi che ci potrebbe essere una maggiore perdita di fibre di tipo II nelle braccia con l’invecchiamento. Così la risposta agli stimoli anabolici può essere più facilmente dimostrabile nelle estremità superiori di questa popolazione.
C’erano anche significative ma simili diminuzioni del grasso corporeo totale di 2,6 ± 1,2 e 2,5 ± 1,6 kg nei gruppi di 50 e 100mg al giorno, rispettivamente. Una parte importante del miglioramento dell’adiposità riguardava la diminuzione del grasso del tronco (1,7 ± 1,0 e 2,2 ± 0,9 kg nei due rispettivi gruppi di trattamento attivo). Una riduzione significativa del grasso del tronco potrebbe influenzare favorevolmente i fattori di rischio per le malattie cardiovascolari. Anche se ci aspetteremmo che la riduzione del grasso addominale si rifletta in una migliore sensibilità all’insulina, le misure indirette (HOMA-IR e QUICKI) potrebbero non essere state abbastanza sensibili. È anche possibile che ci fossero troppo pochi soggetti in ogni gruppo per rilevare cambiamenti piccoli ma significativi.
Ci sono ragioni teoriche per temere che l’eccesso di androgeni possa provocare o essere associato all’insulino-resistenza, anche se questa relazione è stata dimostrata solo in donne con sindrome dell’ovaio policistico. Non è stata misurata direttamente la sensibilità all’insulina né con il clamp euglicemico iperinsulinemico né con test di tolleranza al glucosio endovena a campionamento frequente. Tuttavia, le misure indirette della sensibilità insulinica (insulina a digiuno, HOMA-IR, QUICKI) non hanno mostrato prove di resistenza insulinica.
Cosa estrapolare?
Questo studio però presenta alcune limitazioni che possono averne influenzato i risultati. In primo luogo, la piccola dimensione del campione di meno di una dozzina di soggetti per gruppo può aver limitato la capacità di rilevare piccoli ma importanti cambiamenti in variabili come la massa magra (LBM) delle estremità inferiori e il CSA della muscolatura della coscia. Allo stesso modo, è possibile che le differenze osservate per i cambiamenti nella LBM totale e nella forza avrebbero potuto essere significative tra i gruppi di trattamento con dimensioni del campione maggiori. Quest’ultimo avrebbe fornito ulteriore supporto alla nostra supposizione di una risposta dose-dipendente con l’Oxymetholone. In secondo luogo, la popolazione rappresentava uomini adulti più anziani, che sono stati caratterizzati come a rischio di sarcopenia legata all’età sulla base dei rapporti che mostrano la perdita di massa e forza muscolare con l’invecchiamento. Tuttavia, i soggetti non sono stati reclutati per la perdita di peso, la fragilità o l’ipogonadismo palese di per sé, dal momento che è stato dimostrato che gli uomini più giovani con concentrazioni di Testosterone normali possono ottenere aumenti apprezzabili della massa muscolare e della forza dopo l’integrazione di androgeni. Inoltre, ci sono prove che la sintesi proteica miofibrillare nelle persone anziane può essere significativamente aumentata a livelli paragonabili a quelli raggiunti nelle persone più giovani in risposta a un potente stimolo anabolico. Infine, poiché l’Oxymetholone è un AAS 17-metilato che provoca un elevato effetto di primo passaggio nel fegato, e che nel presente studio non sono state prese misure di contenimento per l’epatotossicità potenziale, i risultati di AST e ALT ottenuti rappresentano solamente modelli privi di ancillari volti ad una epatoprotezione.
Conclusioni sul dosaggio “ottimale” di Oxymetholone:
Evidenziati i limiti dello studio, pur prendendo i dati ivi riportati universalmente rapportabili al basale d’uso della molecola (es. vedi epatotossicità), possiamo giungere, grazie all’ausilio di dati empirici raccolti negli anni attraverso indagini svolte sulle preparazioni di svariati atleti di medio e alto livello, ad identificare un dosaggio con una ratio “efficacia:rischio (E:R)” favorevole per l’atleta.
Un dato è emerso preponderante nel corso delle indagini svolte: quale fosse il peso dell’atleta e il suo condizionamento atletico, nonché l’utilizzo di una adeguata epatoprotezione e controllo della dislipidemia, il margine della ratio E:R diveniva evidentemente sfavorevole oltre i 150mg/die. Indi per cui, i dosaggi elevati raggiunti da certi atleti, arrivando a picchi di 200-300mg/die, sono risultati inutili al miglioramento delle risposte anabolizzanti complessive e inficianti per il corretto svolgimento della stessa preparazione (vedi, ad esempio, marcata inappetenza e nausea).
Dosaggi standard per un atleta di sesso maschile non dovrebbero discostarsi dal range 50-100mg/die, considerando che la taratura del “dosaggio ideale” si è ottenuta calcolando la dose individuale con la formula 1mg/Kg di peso corporeo. Ovviamente, l’assicurarsi una adeguata protezione epatica e lipidica è il punto parallelo da raggiungere.
Nelle atlete, invece, vista la loro maggiore sensibilità agli aumenti degli androgeni circolanti, la “dose ideale” si è attestata a 25mg/die con punte massime (anche se non necessarie) di 50mg/die. A tal proposito, vorrei ricordare che l’Oxymetholone è risultato essere una molecola più vantaggiosa nel controllo degli effetti collaterali androgenizzanti rispetto a composti quali Methenolone e Boldenone.
La linea tra abuso e uso è spesso molto sottile, ma nel caso del Oxymetholone essa si mostra sufficientemente marcata…
Pavlatos AM, Fultz O, Monberg MJ, Vootkur A (June 2001). “Review of oxymetholone: a 17alpha-alkylated anabolic-androgenic steroid”. Clinical Therapeutics. 23 (6): 789–801, discussion 771.
Saartok T, Dahlberg E, Gustafsson JA (June 1984). “Relative binding affinity of anabolic-androgenic steroids: comparison of the binding to the androgen receptors in skeletal muscle and in prostate, as well as to sex hormone-binding globulin”. Endocrinology. 114 (6): 2100–6.
Hengge UR, Stocks K, Wiehler H, Faulkner S, Esser S, Lorenz C, et al. (March 2003). “Double-blind, randomized, placebo-controlled phase III trial of oxymetholone for the treatment of HIV wasting”. AIDS. 17 (5): 699–710.
Cortesgallegos V, Castaneda G, Alonso R, Perezpasten E, Reyeslugo V, Barron C, Mondragon L, Villalpando S (January 1982). “Spontaneous and Oxymetholone-Induced Gynecomastia”. Journal of Andrology. C/O Allen Press, Inc Po Box 368, Lawrence, Ks 66044: Amer Soc Andrology, Inc. 3 (1): 33.
Villalpando S, Mondragon L, Barron C, Reyeslugo U, Perezpasten E, Alonso R, Castaneda G, Gallegos V (January 1982). “5-Alpha Reductase Blockade May Be Responsible for Spontaneous and Oxymetholone-Induced Gynecomastia”. Archivos de Investigacion Medica. Social Apdo Postal 73-032, Mexico Df 03020, Mexico: Inst Mexicano Seguro. 13 (2): s13.
Il fegato è un organo importante ed è vitale per la sopravvivenza del soggetto. È responsabile di diverse e importanti funzioni nel corpo umano. Produce acidi biliari e proteine plasmatiche, immagazzina glicogeno e produce glucosio attraverso la gluconeogenesi, gioca un ruolo nel sistema immunitario, metabolizza un numero elevato di molecole, ecc. Quindi, si, avete capito bene: è importante. Quando qualcosa risulta dannosa per il fegato, essa si indica come epatotossico (dal greco hêpar-atos, fegato). Un chiaro esempio è l’alcol. Gli alcolisti tendono a sviluppare una malattia del fegato a un certo punto della loro vita. Tuttavia, molti farmaci da prescrizione, o anche over-the-counter, possono essere epatotossici, come l’Acetaminofene. E, come è ben dimostrato, anche gli AAS possono essere epatotossici, anche se specifici. Come sembra, solo quelli con una specifica alterazione chimica sembrano essere maggiormente epatotossici – in particolare, quelli che presentano una metilazione in pozione C-17α.
Modifica della struttura carbossilica del Testosterone (sinistra) in posizione C-17α (destra).
In questo articolo tratterò principalmente ciò che sembra causare questa epatotossicità indotta da AAS. L’effetto epatotossico può essere riscontrato attraverso l’osservazione dei cambiamenti nei marcatori ematici del danno epatico, come Alanina Transaminasi (ALAT), Aspartato Transaminasi (ASAT), γ-glutamiltransferasi (GGT) e la Fosfatasi Alcalina (ALP). Una nota di cautela deve essere presa in considerazione quando si interpretano gli aumenti di ALAT e ASAT, poiché entrambi aumenteranno anche a causa del intyenso lavoro muscolare [1]. È bene sapere che in questi casi, ASAT sarà di solito più alto del ALAT, mantenendo un rapporto ASAT/ALAT superiore a 1. Quindi, quando questi aumentano con un rapporto inferiore a 1, si può essere più sicuri che il danno muscolare non è il colpevole dell’alterazione. Idealmente, nessun esercizio (contro-resistenza) viene svolto 1-2 settimane prima dell’esame del sangue per escludere il danno muscolare muscolare come causa dell’innalzamento, sebbene ciò dipenda anche dall’intensità del allenamento. In rari casi, il danno al fegato potrebbe avanzare clinicamente fino allo sviluppo di ittero colestatico [2]. In questo caso, un prodotto della degradazione dei globuli rossi (bilirubina) si accumula nel corpo. L’ittero può essere osservato visivamente (tono giallo della pelle e della sclera degli occhi), e si possono sviluppare sintomi come nausea, vomito, dolore allo stomaco e prurito. Inoltre, alcuni rari casi di peliosis hepatis (Peliosi Epatica) sono stati segnalati verificarsi come risultato dell’uso di AAS orali ad alte dosi [3]. Questa è una condizione nella quale si vengono a formare cisti piene di sangue nel fegato. La sospensione dell’AAS in questione è solitamente sufficiente e porterà alla scomparsa di queste caratteristiche cliniche entro pochi mesi. In casi più gravi, tuttavia, potrebbero richiedere un intervento chirurgico. Infine, alcuni casi in letteratura hanno riportato un’associazione tra uso di AAS e carcinoma epatico [4] e adenoma [5].
Ho già trattato in passato tale problematica legata all’uso di AAS, ma questa volta voglio trattare la questione più nello specifico, analizzando le due ipotesi che ruotano intorno all’epatotossicità AAS-dipendente: “ipotesi dello stress ossidativo” e “ipotesi di coniugazione dell’anello D”.
L’ipotesi dello stress ossidativo:
L’ipotesi dello stress ossidativo che tratterò qui si basa su un documento che William Llewellyn, Peter Van Mol e Peter Bond hanno pubblicato [6]. Lo stress ossidativo è qualcosa che si pensa possa risultare nell’epatotossicità osservata con l’uso di AAS, e se l’ipotesi è vera, dà qualche opportunità per contrastarla in modo migliore. Quindi, cominciamo con spiegare quello che è lo stress ossidativo. Lo stress ossidativo è descritto da Helmut Sies come un disturbo nell’equilibrio pro-ossidante-antiossidante a favore del primo [7], che si riduce a molecole contenenti ossigeno, che sono altamente reattive (specie reattive dell’ossigeno [ROS]), sopraffacendo il sistema antiossidante. Poiché le ROS sono così altamente reattive, possono reagire con molecole come lipidi, proteine, carboidrati e acidi nucleici (elementi costitutivi del DNA). Quando si dice “reagire con queste molecole”, si intende che danneggia queste molecole (estremamente semplificato, ma è sufficiente per far comprendere il processo). Questi ROS provengono da varie reazioni catalizzate da enzimi come la respirazione cellulare (l’ossidazione dei macronutrienti per fornire energia), altri processi metabolici e radiazioni. La fonte primaria di ROS all’interno di una cellula sono i mitocondri, il che non è sorprendente dato che i mitocondri sono le “centrali energetiche” della cellula. È il posto nella cellula dove i carboidrati alimentari, gli acidi grassi e le proteine (o, meglio, gli amminoacidi che le compongono) finiscono per essere ossidate per produrre energia in un processo chiamato fosforilazione ossidativa. Come suggerisce il nome, la fosforilazione ossidativa ossida e richiede ossigeno per farlo. Questo processo, tuttavia, non è perfetto. Per non complicare troppo le cose al lettore, non mi addentrerò nelle complessità delle reazioni chimiche, ma fondamentalmente, questo processo può produrre ROS come sottoprodotto (superossido in particolare). Le cellule del corpo sono dotate di meccanismi per tenere a bada questi ROS generati (la parte antiossidante dell’equazione). In circostanze normali questo porta ad un sottile equilibrio tra i due. Avere qualche ROS qua e là nelle cellule è normale. Essi giocano un ruolo essenziale nel normale funzionamento di vari processi vitali [8]. Tuttavia, il problema nasce quando questo equilibrio si altera a favore della parte proossidante dell’equazione: lo stress ossidativo. Questo è il momento in cui i ROS prendono il sopravvento, per così dire, e possono iniziare a creare il caos nella cellula. Quanto sopra è un quadro un po’ troppo semplificato. Ci sono diversi tipi di ROS (radicali liberi e non radicali). Ciò che conta è dove si trovano questi ROS nella cellula e come evolvono nel tempo. Inoltre, questo interagisce con il sistema antiossidativo delle cellule, il che complica ulteriormente il quadro. Ma credo che quanto sopra sia sufficiente per dare una buona comprensione di tutto questo. Ciò che conta è che l’epatotossicità indotta da AAS è stata ripetutamente dimostrata essere associata allo stress ossidativo nelle cellule epatiche (fegato) di modelli animali [9]. Questo fa sorgere la domanda: è solo un’associazione, o c’è una relazione causale con l’epatotossicità indotta da AAS? Dopo aver scavato nella letteratura, sono emersi alcuni studi che sembrano sostenere una relazione causale. Uno studio svolto su un carcinoma prostatico umano epiteliale (22Rv1) ha collegato l’attivazione del recettore degli androgeni (AR) a un aumento dei ROS basali [10]. Più tardi, lo stesso gruppo ha pubblicato una ricerca applicando un disegno di studio simile. Questo studio ha confermato i precedenti risultati e ha anche dimostrato che l’aumento dei ROS è dovuto a un aumento indotto dall’AAS nella β-ossidazione mitocondriale degli acidi grassi [11]. Quindi, l’attivazione di l’AR porta a una maggiore ossidazione degli acidi grassi nei mitocondri, con conseguente maggiore produzione di ROS come sottoprodotto. Da notare che questo studio ha anche trovato un aumento dell’mRNA della carnitina palmitoiltransferasi (CPT1). Tutto quello che dovete sapere è che la CPT1 è considerata essere l’enzima che regola la velocità nel processo di ossidazione mitocondriale degli acidi grassi. Quindi, se si aumenta la CPT1, si aumenta l’ossidazione mitocondriale degli acidi grassi. Ora, le cellule del cancro alla prostata non sono cellule del fegato, ovviamente. Ma ciò che è interessante è che l’AAS 17α-alchilato Fluoxymesterone e Metilandrostanolone hanno dimostrato di aumentare l’attività del CPT1 nel fegato di ratto [12]. Inoltre, se si guardano agli epatociti di ratto (cellule epatiche) trattati con AAS 17α-alchilati, si vedrà il gonfiore dei mitocondri e solo cristae leggermente definite [13]. (Le criste sono quelle pieghe caratteristiche della membrana interna dei mitocondri). Infatti, la produzione di ROS è una causa nota di gonfiore mitocondriale, e il gonfiore è un fattore importante che porta alla successiva morte cellulare [14]. Quindi, apparentemente, suggerisce un potenziale ruolo dello stress ossidativo. Questo non vuol dire che qualsiasi aumento nella produzione di energia di una cellula sia negativo. Usando i muscoli aumenta anche la produzione di energia nelle cellule muscolari. Di conseguenza, più ROS vengono prodotti anche in queste cellule. In contrasto con l’aumento di ROS indotto dall’AAS nelle cellule del fegato, questi aumenti sono transitori invece che continui. Inoltre, le cellule muscolari differiscono nei loro meccanismi antiossidanti per gestire questa condizione. Quindi, normalmente, questo non è assolutamente un problema. Tuttavia, l’esercizio intenso e prolungato può anche provocare danni ossidativi alle molecole delle cellule muscolari [15].
L’ipotesi dello stress ossidativo nella epatotossicità indotta da AAS come descritto da Bond et al. [49]. 1 Un androgeno si lega a, e attiva, il recettore degli androgeni (AR) nelle cellule epatiche. Questo porta a 2 la sovra-regolazione della Carnitina Palmitoiltransferasi 1 (CPT1), l’enzima che regola il tasso di β-ossidazione degli acidi grassi (FA). Si pensa che questo porti a 3 un aumento della β-ossidazione degli acidi grassi nei mitocondri. Di conseguenza, 4 la produzione di specie reattive dell’ossigeno (ROS) è aumentata. L’aumento dei ROS poi danneggia i mitocondri, il che sembra essere alla base dell’epatotossicità indotta dall’AAS.
Ora, se si integrassero gli antiossidanti (mitocondriali), si allevierebbe questo danno? Può darsi. Mentre non c’è un trial di buona qualità che valuti questo, uno studio osservazionale su 320 atleti dimostra qualcosa del genere [16]. In breve, gli utilizzatori di AAS che hanno preso un supplemento contenente alcuni composti antiossidanti non ha mostrato alcun aumento dei marcatori di danno epatico dopo il ciclo rispetto a quelli che non hanno assunto quel supplemento. Ancora una volta, questo sarebbe in linea con lo stress ossidativo che gioca un ruolo causale nell’epatotossicità indotta da AAS. Infine, sembra che l’epatotossicità indotta da AAS potrebbe essere legata all’attivazione del AR nelle cellule epatiche. In un vecchio studio del 1964, Marquardt et al. non sono riusciti a dimostrare che l’AAS non 17α-alchilato produce test di funzionalità epatica anormali [17]. Infatti, gli AAS 17α-alchilati mostrano segni di epatotossicità in diversi studi, mentre non si vede questo con AAS non-17αalchilati, nemmeno con un alto dosaggio di 600 mg di Testosterone Enantato settimanale [18]. La 17α-alchilazione sembra quasi necessaria per rendere epatotossico un AAS, probabilmente perché è l’unica alterazione che lo rende sufficientemente biodisponibile per via orale. E, di conseguenza, porta ad alte concentrazioni del composto nel fegato. Ma possiamo individuare le differenze tra i vari AAS 17α-alchilati che riguardano la loro capacità di attivare l’AR? Certamente sembra così. In generale, sembra che sia vero quanto segue:
Epatotossicità = resistenza alla decomposizione epatica×potenza di attivazione del AR
Quindi, facciamo un esempio. Il Methyltrienolone (R1881) ha un’affinità molto alta per l’AR, ha un’alta potenza per la transattivazione dell’AR [19], ed è fortemente resistente al metabolismo epatico. Come tale, è un composto ideale per un saggio dei siti di legame agli androgeni [20]. Infatti, un studio clinico che impiega un basso dosaggio dello steroide (≤1 mg al giorno) ha dimostrato un significativo aumento dei marcatori di danno epatico entro due settimane [21]. Gli autori lo hanno definito “(…) attualmente lo steroide più epatotossico”. Lo steroide 17α-alchilato meno epatotossico è solitamente considerato l’Oxandrolone. Anche con alti dosaggi fino a 80mg al giorno, mostra solo deboli segni di epatotossicità [22]. Mentre lo steroide è abbastanza resistente al metabolismo epatico [23], ha una bassa affinità per il AR [23]. La sua potenza relativa in termini di transattivazione AR è anche quasi 100 volte inferiore a quella del Methyltrienolone [19]. Allo stesso modo, anche l’Oxymetholone ha una bassa affinità per l’AR [23] e la sua potenza in termini di transattivazione AR è molto simile a quella dell’Oxandrolone [19]. Non sorprende che mostri segni di epatotossicità solo in una minoranza di pazienti, nonostante gli alti dosaggi (100-150 mg al giorno) [24].
L’ipotesi di coniugazione dell’anello D:
Avete mai sfogliato il libro Doping in Sports di Thieme e Hemmersbach? [25] In questo libro gli autori notano che non c’è correlazione tra la tossicità epatica e gli effetti farmacologici primari (cioè gli effetti anabolizzanti) – il che è sufficientemente ovvio perché gli AAS non 17α-alchilati sono rapidamente metabolizzati nel fegato, quindi la loro concentrazione in loco non sarebbe come quella dei 17α-alchilati. Naturalmente, non si troverà una correlazione se si guarda solo a questo fattore. Bisogna anche prendere in considerazione la sua resistenza al metabolismo epatico come è stato fatto con l’ipotesi dello stress ossidativo descritta sopra.
In ogni caso, questo ha portato gli autori a formulare un’alternativa ipotesi di ciò che causa l’epatotossicità indotta da AAS. E sembrava essere l’unica. Essi suggeriscono che l’epatotossicità è probabilmente dovuta alla coniugazione dell’anello D con l’acido glucuronico. Questo processo è chiamato glucuronidazione ed è una cosiddetta comune reazione di fase 2 nel metabolismo del farmaco. Rende la molecola madre più solubile in acqua, facilitando così la sua escrezione nelle urine.
Il gruppo 17β-glucuronide (in blu) attaccato al anello D di uno steroide 17α-metilato (gruppo 17α-metilico in rosso).
È semplicemente l’attaccamento (coniugazione) dell’acido glucuronico alla molecola madre (vedi figura sopra). Quando il Testosterone con un gruppo 17β-glucuronide (così come diversi estrogeni con questa modifica) viene iniettato nel ratto, il flusso biliare è inibito [521]. Presumibilmente, perché questi composti condividono somiglianze strutturali con gli acidi biliari, questi composti competono con gli acidi biliari per legarsi a certi recettori. Tuttavia, a parte questo, non c’è molta sostanza per sostenere questa ipotesi come la ragione per l’epatotossicità indotta da AAS, soprattutto perché molti degli AAS non 17α-alchilati, compreso il Testosterone, subiscono la glucuronizzazione del loro gruppo 17β-idrossi. Eppure questi non sono sensibilmente epatotossici. Infatti, la 17βglucuronidazione è stata identificata solo per alcuni AAS 17α-alchilati, e sembra che essi subiscono questo processo solo in piccola misura [26]. Così, ironicamente, se questa ipotesi fosse vera, o significativa, ci si aspetterebbe l’epatotossicità con il Testosterone ma non con gli AAS 17α-alchilati.
Conclusioni sulle ipotesi esposte:
Non è sicuramente una novità per l’utilizzatore medio, ma anche per il semplice soggetto interessato all’argomento PEDs, che gli AAS metilati in C-17 (17α-alchilati) abbiano un effetto epatotossico con lievi variabili tra molecole aventi la stessa modifica strutturale. E non è nemmeno una rivelazione che la supplementazione con antiossidanti (vedi NAC e Silimarina) possa ridurre tale effetto. Di conseguenza, l’ipotesi dello stress ossidativo sembra essere la principale causa del epatotossicità AAS-indotta. Ma non l’unico fattore.
Nell’ultimo decennio si è aggiunto ai classici composti antiossidanti l’uso di acidi biliari come l’Acido Ursodesossicolico e l’Acido Tauroursodesossicolico assunti oralmente.
L’Acido Ursodesossicolico è un acido biliare secondario che deriva dal metabolismo dell’acido colico da parte del microbiota umano intestinale. Il suo nome deriva dal fatto che è il principale acido biliare negli orsi (dal latino ursus). In biologia e biochimica lo si etichetta con l’acronimo UDCA. Il nome completo del UDCA è Acido 3α,7β-diidrossi-5β-colanoico.[27]
Acido Ursodesossicolico (UDCA)
L’Acido Tauroursodesossicolico (TUDCA) è un acido biliare ambifilico. È la forma coniugata di Taurina ed il precedentemente citato Acido Ursodeossicolico (UDCA). Il nome completo del TUDCA è 2-{(4R)-4-[(1R,3aS,3bR,4S,5aS,7R,9aS,9bS,11aR)-4,7-Dihydroxy-9a,11a-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-1-yl]pentanamido} acido etan-1-sulfonico.[28]
Acido Tauroursodesossicolico (TUDCA)
l’UDCA è approvato per il trattamento della cirrosi biliare primaria.[1][2] Di conseguenza, l’Acido Ursodesossicolico (UDCA) ha mostrato effetti epatoprotettivi. Tuttavia, i suoi meccanismi molecolari sottostanti rimangono poco chiari. Per tale motivazione, sono stati condotti alcuni studi come quello di Da Jung Kim et al. nel quale è stato osservato l’effetto epatoprotettivo dell’UDCA e della vitamina E utilizzando la metabolomica e l’analisi metagenomica. In questo studio, sono stati analizzati campioni di sangue e urine di pazienti con obesità e disfunzione epatica. Nove pazienti sono stati assegnati in modo casuale a ricevere UDCA (300 mg due volte al giorno), e 10 soggetti hanno ricevuto la vitamina E (400 UI due volte al giorno) per 8 settimane. L’UDCA ha migliorato significativamente i punteggi della funzionalità epatica dopo 4 settimane di trattamento e ha ridotto efficacemente i livelli epatici di acido Desossicolico e di microRNA-122 nel siero. Per comprendere meglio il suo meccanismo protettivo, è stato condotto uno studio di metabolomica globale ed è stato scoperto che l’UDCA ha regolato le tossine uremiche (acido ippurico, solfato di p-cresolo e metaboliti derivati dall’indolo), gli antiossidanti (solfato di ascorbato e N-acetil-L-cisteina) e il percorso fenilalanina/tirosina. Inoltre, il coinvolgimento del microbioma, in particolare di Lactobacillus e Bifidobacterium, è stato dimostrato attraverso l’analisi metagenomica delle vescicole extracellulari derivate dai batteri. Nel frattempo, il trattamento con vitamina E non ha portato a tali alterazioni, tranne che ha ridotto le tossine uremiche e la disfunzione epatica. I nostri risultati hanno suggerito che entrambi i trattamenti erano efficaci nel migliorare la funzione epatica, anche se attraverso meccanismi diversi.
Schema dei potenziali meccanismi terapeutici del trattamento con UDCA. L’analisi metabolomica ha rivelato che l’UDCA riduce i principali composti nei percorsi fenilalanina/tirosina e triptofano, tra cui fenilalanina, fenilacetato, acetilfenilalanina, aldeide 3,4-idrossifenilacetato, dopamina-3-O-solfato, idrossibenzaldeide, p-cresolo solfato, idrossicynurenamina, idrossindolo e acido ippurico, nel plasma e nelle urine. I metaboliti intermedi degli aminoacidi aromatici come l’idrossimelatonina, l’acido benzoico e l’acido salicilico sono stati aumentati. I forti antiossidanti come l’ascorbato, l’acetiltriptofano e la N-acetil-L-cisteina erano elevati. Inoltre, la disintossicazione delle tossine uremiche tramite glucuronidazione (idrossimetossiindolo glucuronide e p-cresolo glucuronide) è stata osservata dopo il trattamento UDCA. Tuttavia, la vitamina E ha ridotto l’acido indolo-propionico, il solfato di indoxile, la 3-ketosphinganina e la sfingosina, che non sono stati regolati dall’UDCA. Il colore blu indica una diminuzione del livello del metabolita, e il colore rosso indica un aumento del livello del metabolita dopo il trattamento UDCA. I metaboliti che sono cambiati dopo il trattamento con vitamina E sono contrassegnati da un asterisco (*). I metaboliti che sono stati possibilmente regolati da modifiche batteriche sono contrassegnati da un colore viola.
Inoltre, si sa che l’UDCA a livello epatico stimola la secrezione di ATP da parte degli epatociti[29]; sebbene il significato di quest’azione non è ancora noto. Si sa però che interagisce col sistema dei citocromi P450 e che riduce la Glicuronazione degli estrogeni sintetici e non solo.[30] Vi ricorda qualcosa? Esatto! L’ipotesi di coniugazione dell’anello D e la sua potenzialità di essere parte dell’effetto epatotossico AAS-indotto! Se a ciò aggiungiamo che l’UDCA possiede la capacità di attivare direttamente il recettore per i glucocorticoidi, che contribuirebbe ad allargare i meccanismi della sua azione anticolestatica ed antinfiammatoria sul parenchima epatico [31], e che stimola la sintesi del glutatione (GSH), potente antiossidante endogeno, attraverso l’intervento delle chinasi dipendenti dai fosfoinositidi (PI-3K e PKB) [32], ciò fa si che l’UDCA risulti la chiave di volta nella protezione epatica durante l’uso di AAS con marcata resistenza al metabolismo epatico in abbinamento ai largamente utilizzati NAC (precursone ad alta biodisponibilità del Glutatione) e Silimarina.
Quanto detto non rappresenta ne un consiglio medico ne una scusa per abusare di AAS di qualsiasi tipo! Si tratta semplicemente della divulgazione di informazioni che la seria ricerca scientifica ha permesso di estrapolare, per il momento…
Gabriel Bellizzi
Riferimenti:
W. J. Meyer, A. Webb, C. A. Stuart, J. W. Finkelstein, B. Lawrence, and P. A. Walker. Physical and hormonal evaluation of transsexual patients: a longitudinal study. Archives of sexual behavior, 15(2):121–138, 1986.
A. M. Elsharkawy, S. McPherson, S. Masson, A. D. Burt, R. T. Dawson, and M. Hudson. Cholestasis secondary to anabolic steroid use in young men. Bmj, 344, 2012.
J. Nadell and J. Kosek. Peliosis hepatis. twelve cases associated with oral androgen therapy. Archives of pathology & laboratory medicine, 101(8):405–410, 1977.
F. L. Johnson, K. Lerner, M. Siegel, J. Feagler, P. Majerus, J. Hartmann, and E. D. Thomas. Association of androgenic-anabolic steroid therapy with development of hepatocellular carcinoma. The Lancet, 300(7790):1273–1276, 1972.
L. Hernandez-Nieto, M. Bruguera, J. A. Bombi, L. Camacho, and C. Rozman. Benign liver-cell adenom associated with long-term administration of an androgenic-anabolic steroid (methandienone). Cancer,40(4):1761–1764, 1977.
P. Bond, W. Llewellyn, and P. Van Mol. Anabolic androgenic steroid-induced hepatotoxicity. Medical Hypotheses, 93:150–153, 2016.
H. Sies et al. Oxidative stress: introductory remarks. Oxidative stress, 501:1–8, 1985.
K. Brieger, S. Schiavone, F. J. Miller Jr, and K.-H. Krause. Reactive oxygen species: from health to disease. Swiss medical weekly, 142:w13659, 2012.
S. P. Frankenfeld, L. P. Oliveira, V. H. Ortenzi, I. C. Rego-Monteiro, E. A. Chaves, A. C. Ferreira, A. C. Leitáo, D. P. Carvalho, and R. S. Fortunato. The anabolic androgenic steroid nandrolone decanoate disrupts redox homeostasis in liver, heart and kidney of male wistar rats. PloS one, 9(9):e102699, 2014.
J. H. Pinthus, I. Bryskin, J. Trachtenberg, J.-P. Luz, G. Singh, E. Fridman, and B. C. Wilson. Androgen induces adaptation to oxidative stress in prostate cancer: implications for treatment with radiation therapy. Neoplasia, 9(1):68–80, 2007.
H. Lin, J.-P. Lu, P. Laflamme, S. Qiao, B. Shayegan, I. Bryskin, L. Monardo, B. C. Wilson, G. Singh, and J. H. Pinthus. Inter-related in vitro effects of androgens, fatty acids and oxidative stress in prostate cancer: a mechanistic model supporting prevention strategies. International journal of oncology, 37(4):761–766, 2010.
M. Guzmán, A. Saborido, J. Castro, F. Molano, and A. Megias. Treatment with anabolic steroids increases the activity of the mitochondrial outer carnitine palmitoyltransferase in rat liver and fast-twitch muscle. Biochemical pharmacology, 41(5):833–835, 1991.
R. Gragera, A. Saborido, F. Molano, L. Jimenez, E. Muñiz, and A. Megias. Ultrastructural changes induced by anabolic steroids in liver of trained rats. Histology and histopathology, 1993.
X. Chapa-Dubocq, V. Makarov, and S. Javadov. Simple kinetic model of mitochondrial swelling in cardiac cells. Journal of cellular physiology, 233(7):5310–5321, 2018.
S. K. Powers, L. L. Ji, A. N. Kavazis, and M. J. Jackson. Reactive oxygen species: impact on skeletal muscle. Comprehensive Physiology, 1(2):941–969, 2011.
T. A. Pagonis, G. N. Koukoulis, C. S. Hadjichristodoulou, P. N. Toli, and N. V. Angelopoulos. Multivitamins and phospholipids complex protects the hepatic cells from androgenic-anabolic-steroids-induced toxicity. Clinical Toxicology, 46(1):57–66, 2008.
G. H. Marquardt, C. E. Logan, W. G. Tomhave, and R. M. Dowben. Failure of non-17-alkylated anabolic steroids to produce abnormal liver function tests. The Journal of Clinical Endocrinology & Metabolism, 24(12):1334–1336, 1964.
S. Bhasin, L. Woodhouse, R. Casaburi, A. B. Singh, D. Bhasin, N. Berman, X. Chen, K. E. Yarasheski, L. Magliano, C. Dzekov, et al. Testosterone dose-response relationships in healthy young men. American Journal of Physiology-Endocrinology And Metabolism, 281(6):E1172–E1181, 2001.
C. J. Houtman, S. S. Sterk, M. P. Van de Heijning, A. Brouwer, R. W. Stephany, B. Van der Burg, and E. Sonneveld. Detection of anabolic androgenic steroid abuse in doping control using mammalian reporter gene bioassays. Analytica chimica acta, 637(1-2):247–258, 2009.
C. Bonne and J.-P. Raynaud. Assay of androgen binding sites by exchange with methyltrienolone (r 1881). Steroids, 27(4):497–507, 1976.
H. L. Krüskemper and G. Noell. Liver toxicity of a new anabolic agent: methyltrienolone (17α-methyl-4, 9, 11-estratriene-17β-ol-3-one). Steroids, 8(1):13–24, 1966.
C. Grunfeld, D. P. Kotler, A. Dobs, M. Glesby, S. Bhasin, O. S. Group, et al. Oxandrolone in the treatment of hiv-associated weight loss in men: a randomized, double-blind, placebo-controlled study. JAIDS Journal of Acquired Immune Deficiency Syndromes, 41(3):304–314, 2006.
J. A. Kemppainen, E. Langley, C.-i. Wong, K. Bobseine, W. R. Kelce, and E. M. Wilson. Distinguishing androgen receptor agonists and antagonists: distinct mechanisms of activation by medroxyprogesterone acetate and dihydrotestosterone. Molecular Endocrinology, 13(3):440–454, 1999.
U. R. Hengge, K. Stocks, S. Faulkner, H. Wiehler, C. Lorenz, W. Jentzen, D. Hengge, and G. Ringham. Oxymetholone for the treatment of hiv-wasting: a double-blind, randomized, placebo-controlled phase iii trial in eugonadal men and women. HIV clinical trials, 4:150–163, 2003.
A. Sansone, F. Romanelli, M. Sansone, A. Lenzi, and L. Di Luigi. Gynecomastia and hormones. Endocrine, 55(1):37–44, 2017.
W. Schänzer. Metabolism of anabolic androgenic steroids. Clinical chemistry, 42(7):1001–1020, 1996.
Boatright, Jeffrey H.; Nickerson, John M.; Moring, Anisha G.; Pardue, Machelle T. (2009). “Bile acids in treatment of ocular disease”. Journal of Ocular Biology, Diseases, and Informatics. 2 (3): 149–159.
Nathanson MH et al. Stimulation of ATP secretion in the liver by therapeutic bile acids. Biochem J. 2001; 358(Pt 1):1-5.
Weitzel C et al. Ursodeoxycholic acid induced activation of the glucocorticoid receptor in primary rat hepatocytes. Eur J Gastroenterol Hepatol. 2005 Feb; 17(2):169-77.
Sanchez Pozzi EJ et al. Ursodeoxycholate reduces ethinylestradiol glucuronidation in the rat: role in prevention of estrogen-induced cholestasis. J Pharmacol Exp Ther. 2003 Jul; 306(1):279-86.
Arisawa S et al. Ursodeoxycholic acid induces glutathione synthesis through activation of PI3K/Akt pathway in HepG2 cells. Biochem Pharmacol. 2009 Mar 1;77(5):858-66.
Di AAS/SARM e saturazione recettoriale se ne parla spesso negli ambienti del culturismo “Enhancement“, nei social e nelle community online. Il problema è sempre il medesimo però, il quale colpisce altre argomentazioni le quali richiedono un certo livello culturale per essere trattate: se ne parla in modo confuso e male. Fortunatamente, però, su “Reddit” si tengono discussioni valide, e con letteratura al seguito, riguardo questo argomento, con persone “addette ai lavori”.
Quindi, l’obiettivo di questo breve articolo è principalmente quello di riportare i chiarimenti scientificamente supportati per ciò che concerne l’uso di AAS/SARM e la saturazione dei Recettori degli Androgeni.
Prima di proseguire, è giusto ricordare che ho una vasta conoscenza di biochimica e genetica e faccio ricerca e divulgazione scientifica da anni. Di conseguenza, le mie affermazioni non sono in alcun modo un “punto di vista” dal momento che, ed i miei lavori lo testimoniano già a sufficienza, ho una comprensione alquanto decente di ciò che viene riportato nelle pubblicazioni scientifiche.[1]
Vi espongo di seguito i “punti chiave” necessari per comprendere la questione AAS/SARM e saturazione AR:
I Recettori degli Androgeni nella maggior parte dei tessuti sono saturi all’estremità inferiore del normale intervallo fisiologico di Testosterone.
Nonostante questa saturazione, la crescita muscolare e la diminuzione della massa grassa è ancora legata al Testosterone in modo dipendente dalla dose, anche a livelli sovrafisiologici.
L’aumento della sintesi proteica non è l’unico (e forse non il principale) meccanismo attraverso il quale il Testosterone causa la crescita del muscolo-scheletrico.
Gli Androgeni sembrano causare un aumento delle cellule satelliti e dei mioonuclei nei muscoli. L’aggiunta di mionuclei alle fibre muscolari è uno dei meccanismi principali con cui essi crescono in dimensione. Questo aumento delle cellule satelliti e dei mionuclei avviene attraverso un percorso dipendente dal Recettore degli Androgeni.
In molti tessuti, l’aumento della concentrazione di Androgeni porta a un aumento della densità dei Recettori degli Androgeni. Questo può aiutare a dare una spiegazione alla possibilità di crescita potenziale maggiore “off cycle” attraverso il precedente uso di anabolizzanti. A tal proposito ricordiamoci anche della così detta “Memoria Muscolare”.[2]
L’aumento delle cellule satellite deriva dalla differenziazione delle cellule staminali mesodermiche pluripotenti. Queste sono le stesse cellule che si differenziano in adipociti (cellule del tessuto adiposo, quindi grasso). L’aumento della differenziazione di queste cellule in cellule satellite (che generano mionuclei) spiega il perché dosi più elevate di Androgeni portano a una diminuzione della massa grassa.
L’aumento delle cellule satelliti e dei mionuclei nella fibra muscolare è più che raddoppiato quando si confronta la somministrazione di 300mg vs. 600mg di Testosterone Enantato. Queste, ovviamente, sono già dosi sovrafisiologiche e questo dimostra l’opposto dei rendimenti decrescenti; tuttavia c’è ancora probabilmente un “collo di bottiglia” sconosciuto a questa differenziazione.
Notare le frecce nella figura C, che denotano fibre muscolari divise in un PowerLifter che aveva usato AAS nei precedenti 10 anni. Le fibre più piccole contenevano una isoforma in via di sviluppo della miosina (cioè miosina fetale), suggerendo che erano in realtà fibre di nuova formazione da iperplasia. La teoria qui esposta è che le fibre hanno una certa soglia di crescita, e che una volta raggiunta questa soglia, alla fine si dividono per formare nuove fibre. Con le tradizionali pratiche di allenamento “Natty”, non sembra che i PL raggiungano questa soglia; ma con l’uso di AAS, la crescita può diventare così accentuata che si verifica l’iperplasia (si noti la differenza di dimensioni delle fibre tra il PL “juiced” in Figura A e il sollevatore”Natty” in Figura B). Anche se mancano prove oggettive e inconfutabili, è logico supporre che le fibre aggiunte (e AR sovraespressi) vengano mantenute, anche se il sollevatore interrompe l’uso di AAS. La questione della possibile ipotrofia di queste nuove fibre una volta cessato l’uso di AAS è un altra possibilità.
Conclusioni:
Dosi più elevate di AAS/SARM o abbinamento di questi porteranno a risultati migliori? Ancora non lo sappiamo con certezza, sebbene i dati empirici ci portino ad una parziale conclusione favorevole al quesito posto. Per esempio, sappiamo che il Ki (con tale sigla ci si riferisce al potenziale di legame/saturazione del AR dose-dipendente) del RAD-140 è di 7nM (rispetto a 29nM del Testosterone e i 10nM del DHT).[3] Questo però non ci dà l’efficacia del ligando, ne il tasso di dissociazione (il testosterone si dissocia dal recettore degli androgeni a un tasso 5x rispetto al DHT nonostante abbia un Ki 2,9x maggiore [4]), ma se dovessimo usarlo come unico parametro di misurazione dell’efficacia, sembrerebbe così. Prendendo il tasso di biodisponibilità proposto del 65-75% (vedi riferimento Ki) del RAD nelle scimmie come punto di riferimento per gli esseri umani, un ciclo proposto di 10mg/die (concentrazione stabile intorno ai 25mg), sembrerebbe poter dare ancora dei benefici (e dei danni in termini di effetti collaterali) da dosi più elevate.
Un altro aspetto che non conosciamo è legato agli effetti AR-indipendenti del testosterone. Ci sono state proposte che collegano alcuni degli effetti del testosterone al suo antagonismo degli effetti dei glucocorticoidi attraverso il legame a bassa affinità con il recettore dei glucocorticoidi. Per quanto ne so, non abbiamo alcun indizio circa l’affinità di cui qualsiasi SARMs legano questo recettore.
Non sappiamo in termini assoluti se abbinare AAS/SARM apporti vantaggi superiori alla monoterapia, sebbene, e lo ripeto, i risultati empirici ci portano verso una risposta almeno parzialmente positiva. Ciò che bisogna evitare di fare, è smettere di usare affermazioni semplicistiche e riduttive come “la saturazione dei AR è il fattore principale che determina il tasso soggettivo di ipertrofia muscolare ottenibile”.
Esiste una interessantissima pubblicazione la quale suggerisce che sono le concentrazioni di Recettori degli Androgeni e non i livelli ormonali il fattore limitante della crescita muscolare a livelli fisiologici. Per l’appunto, LIVELLI FISIOLOGICI! Ancora una volta, vi ricordo di tenere a mente che gli androgeni sovraregolano i Recettori degli Androgeni in modo dose dipendente.[5]
OPK-88004 è un nuovo SARM non steroideo sviluppato dalla Transition Therapeutics, e che è stato acquistato dalla OKPO nel 2016.
Struttura molecolare del OPK-88004
Uno studio di recente pubblicazione svolto su questo SARM sembra aver mostrato che causa un aumento dose-dipendente della massa muscolare, che diminuisce la massa grassa e aumenta anche la quantità di Testosterone libero. Prima di eccitarvi troppo sull’ultimo punto, vediamo nel dettaglio lo studio. Caratteristiche dello studio:
In questo studio controllato con placebo, randomizzato, in doppio cieco, 114 uomini, di età ≥19 anni, che avevano subito una prostatectomia radicale per un cancro alla prostata di basso grado e localizzato all’organo, PSA non rilevabile (<0,1 ng/mL) per ≥2 anni dopo la prostatectomia radicale e carenza di Testosterone sono stati randomizzati per gradi a placebo [0mg] o 1, 5, o 15mg/die di OPK-88004 per 12 settimane. I risultati includevano la recidiva del PSA, l’attività sessuale, il desiderio sessuale, la funzione erettile, la composizione corporea, la forza muscolare e le misure della funzione fisica, l’umore, la fatica e i marker ossei.
Risultati dello studio:
I partecipanti avevano un’età media di 67,5 anni e una grave disfunzione sessuale (punteggi medi della funzione erettile e del dominio del desiderio sessuale 7,3 e 14,6, rispettivamente). Nessun partecipante ha avuto recidive di PSA o eritrocitosi. OPK-88004 è stato associato a un aumento correlato alla dose della massa magra [non specificatamente muscolare] (P <0,001) e appendicolare (P <0,001) e a una diminuzione significativamente maggiore della percentuale di grasso corporeo (P <0,001) e della fosfatasi alcalina nel siero (P <0,001) rispetto al placebo. I cambiamenti nell’attività sessuale, il desiderio sessuale, la funzione erettile, l’umore, l’affaticamento, le prestazioni fisiche e i marker ossei non differiscono tra i gruppi (P = 0,73).
Risultati dei test per valutare il miglioramento delle prestazioni fisiche.Variabili ormonali riscontrate durante lo studio.
Conclusioni sul OPK-88004:
La somministrazione di OPK-88004 è stata sicura e non è stata associata alla recidiva del PSA in uomini con deficit di androgeni che erano stati sottoposti a prostatectomia radicale per cancro alla prostata confinato all’organo. OPK-88004 ha aumentato la massa corporea magra e diminuito la massa grassa, ma non ha migliorato i sintomi sessuali o le prestazioni fisiche.
In conseguenza dei dati estrapolati dallo studio ivi esposto, e nonostante i dati di sicurezza a breve termine siano rassicuranti, questo SARM non steroideo mostra i difetti dei suoi predecessori:
Nonostante vi sia un aumento del Testosterone libero che potrebbe interessare maggiormente il pubblico rispetto al Testosterone totale, non bisogna dimenticarsi del fatto che gli Androgeni possono interagire con le attività cellulari anche attraverso interazioni non genomiche (non mediate direttamente dal recettore androgeno), le quali avvengono anche con l’ormone legato all’albumina (trasportatore ematico che lega l’ormone sessuale; pari circa al 55-35% del Testosterone). Inoltre, anche SARM non steroidei “datati” come l’Ostarina hanno mostrato le medesime caratteristiche sui livelli di Testosterone.
La diminuzione dell’Estradiolo (E2) può causare, in misura dipendente dall’entità del calo e dalla sensibilità individuale, molteplici problemi come depressione, letargia, affaticabilità, ansia, o disfuzione erettile (o difficolta a raggiungere l’erezione e/o a mantenerla) e riduzione della libido.
Problemi legati ai precedenti sono riscontrabili dalla riduzione del DHT, in maniera sempre dipendente dall’entità del calo e dalla sensibilità individuale. A tal proposito si veda la Testosterone:Estradiolo ratio o la più approfondita DHT:Estradiolo ratio.
Non è un caso che il trattamento con il suddetto SARM non abbia portato ad un aumento della funzione sessuale.
La mancanza di miglioramento nelle prestazioni fisiche e dei marker ossei, non lo rende molto allettante.
L’unico punto interessante rimane la riduzione della massa grassa, essendo l’aumento della massa magra estremamente generico e non inducibile a specifiche miotrofiche. Nonostante ciò, i possibili svantaggi superano di netto il suddetto vantaggio che, oltretutto, è riscontrabile in maniera accentuata in un “vecchio” SARM non steroideo, L’Andarina (S4).
Ora, come si può vedere, analizzando con logica tutti i dati in nostro possesso, possiamo valutare concretamente questo nuovo SARM non steroideo e rilegarlo in una posizione di basso interesse sia per uso terapeutico che “off-label”. Ma la ricerca effettuata non è letteralmente da buttare. I dati raccolti devono spingere la ricerca a migliorare queste molecole, sviluppando nuovi SARM che migliorino non solo il trofismo muscolo-scheletrico ma che portino ad una ottimale funzione sessuale e delle prestazioni psicofisiche.
Verso la fine del primo decennio del presente secolo, una “presunta” nuova classe di farmaci con attività anabolizzante ha iniziato a diffondersi in diverse discipline sportive , dal ciclismo a, ovviamente, il Bodybuilding. Sto parlando ovviamente dei SARMs, acronimo di Selective Androgen Receptor Modulators (in italiano, Modulatori Selettivi del Recettore degli Androgeni, SARM).
Essendo molecole sperimentali e non ancora commercializzate come farmaci da prescrizione per uso umano, i SARM si sono diffusi rapidamente in tutto il mondo grazie anche alla vendita da parte degli store online UK e USA (dove la vendita di supplementi contenenti tali molecole è legale).
Non ci volle molto tempo prima che un “alone leggendario” avvolgesse i SARM ed i loro presunti o reali effetti. I SARM vennero in breve pubblicizzati come il “doping ideale” con tutti gli effetti positivi degli steroidi anabolizzanti, pur non avendo alcun svantaggio o effetto collaterale legato a questi ultimi.
In generale, gli effetti positivi principali degli AAS sono considerati essere l’effetto anabolizzante sulla massa muscolare e l’effetto stimolante sul miglioramento della densità minerale ossea. Tutti gli altri effetti cosi detti androgeni sono generalmente considerati indesiderati. Anche se, ovviamente, ciò dipende in gran parte dal grado con il quale essi si verificano (ma anche dal sesso e dalla disciplina praticata dall’utilizzatore).
Ad esempio, gli AAS inducono l’Eritropoiesi, il processo di biosintesi degli Eritrociti (globuli rossi). Questo porta ad un aumento dell’Ematocrito che, quando diventa troppo alto, ossia oltre la soglia del 53-54%, vede arrestati i suoi effetti benefici sulla resistenza vedendo aumentato sensibilmente il rischio di trombosi venosa. Tuttavia, se si eliminasse completamente qualsiasi effetto stimolante sull’eritropoiesi, l’ematocrito potrebbe diventare troppo basso, in specie se viene a mancare un fattore compensativo alla riduzione indotta. Di conseguenza, si finirebbe per essere anemici. Quindi anche alcuni di quegli effetti indesiderati degli AAS sono “voluti” in una certa misura. Ma i paradossi della selettività non terminano con questo, ovviamente. Per semplicità, tuttavia, tratterò il discorso più avanti nel presente articolo.
Il punto della questione è: i SARM danno veramente un vantaggio in quanto a rapporto tra effetti positivi e collaterali rispetto agli AAS? La risposta richiede una spiegazione dettagliata della storia, delle caratteristiche e degli effetti, constatati sia in ambito clinico che “off-label”, legati ai SARM.
Nozioni iniziali sui SARM.
Come la maggior parte di voi saprà, SARM sono una classe di ligandi selettivi del recettore degli androgeni (AR).[1]
Nonostante un certo numero di persone sia convinta che i SARM siano stati sintetizzati circa venti anni fa, e che non abbiano nulla a che vedere nel loro sviluppo con gli AAS, la realtà è che il termine si riferisce ad un macrogruppo di molecole affini al AR con un valore terapeutico (vedi potenziale androgeno e anabolizzante) superiore a 1, cioè al Testosterone. Per questa ragione esistono due gruppi di SARM: i SARM steroidei ed i SARM non-steroidei. Di conseguenza, tutti i derivati del Testosterone, del DHT, compresi i 19-Norsteroidi, che sono stati modificati strutturalmente al fine di accentuarne le caratteristiche anabolizzanti e ridurne quelle androgene sono considerabili quali SARM steroidei.
Due esempi tipici di SARM steroideo e non-steroideo
Gli sforzi iniziali per sviluppare SARM steroidei, basati su modifiche della molecola di Testosterone, risalgono agli anni ’40. L’era moderna dei SARM non steroidei è stata scatenata da un lavoro indipendente presso la Ligand Pharmaceuticals (2, 3) e l’Università del Tennessee.(4, 5) Gli scienziati della Ligand Pharmaceuticals sono stati i primi a sviluppare una serie di Chinolinoni ciclici con attività anabolica sul muscolo scheletrico e un certo grado di selettività tissutale.(2, 6, 7, 8) La scoperta di Dalton e Miller che le Aril Propionammidi con somiglianze strutturali con il Bicalutamide e l’Idrossiflutammide potrebbero innescare l’attività trascrizionale AR-dipendente ha fornito la prima guida per lo sviluppo della classe di SARM diaril propionammidi.(4, 5) Il decennio successivo a questi primi sforzi ha visto l’emergere di un gran numero di SARM non steroidei praticamente da tutte le principali aziende farmaceutiche.(9)
Fondamenti logici nella ricerca dei SARM non-steroidei
Il Testosterone, il principale ligando per il Recettore degli Androgeni, svolge una varietà di funzioni fisiologiche nell’uomo (10): è essenziale, anche per via della sua conversione in DHT, al fine di mantenere una corretta funzione sessuale, lo sviluppo delle cellule germinali e gli organi sessuali accessori. Il Testosterone interagisce ovviamente anche con il muscolo scheletrico, grasso, ossa, emopoiesi, coagulazione, metabolismo dei lipidi, proteine e carboidrati e comportamenti psicosessuali e cognitivi. Sebbene la carenza di androgeni negli uomini adulti sia il disturbo più diffuso della alterazione nella segnalazione AR (11), il principale impulso per lo sviluppo dei SARM è legato allo sfruttamento dei potenziali effetti anabolici di questi composti sul muscolo scheletrico e sull’osso.
Come ben sappiamo, man mano che uomini e donne invecchiano, perdono massa muscolare scheletrica, forza, potenza (12, 13), principalmente a causa della perdita preferenziale delle fibre muscolari di tipo 2 (14), e la densità ossea. La perdita di massa muscolare e forza associata all’età aumenta il rischio di cadute, fratture, limitazione della mobilità, disabilità fisica e scarsa qualità della vita (15, 16). Il declino funzionale e la dipendenza negli anziani gravano pesantemente sui servizi e sui costi sanitari. Nonostante l’elevata prevalenza di limitazioni funzionali e disabilità tra gli individui più anziani, i geriatri praticanti hanno poche scelte terapeutiche per il trattamento degli individui più anziani con limitazioni funzionali e disabilità fisica. Allo stesso modo, il decorso di molte malattie croniche, come la malattia polmonare ostruttiva cronica, la malattia renale allo stadio terminale, l’insufficienza cardiaca congestizia e alcuni tipi di cancro, è punteggiato da perdita di massa muscolare e limitazioni funzionali fisiche, che contribuiscono indipendentemente a sintomi, limitazione della mobilità e disabilità. Pertanto, c’è un enorme bisogno insoddisfatto di funzioni che promuovano terapie anabolizzanti che possano migliorare la funzione fisica e ridurre il peso della disabilità.
Tra le varie terapie anabolizzanti candidate ad applicazione in fase di sviluppo, quella con SARM non steroidei è la più recente in corso di sviluppo. La somministrazione di Testosterone aumenta la massa muscolare scheletrica e la massima forza volontaria in uomini sani, con carenza di androgeni (17-18) ed eugonadici (19, 20) e anziani (21), e negli uomini con molti disturbi cronici (22, 23). Gli effetti anabolizzanti del Testosterone sulla massa e sulla forza dei muscoli scheletrici sono correlati alla dose di Testosterone e alle sue concentrazioni ematiche (20, 21, 24, 25). Pertanto, il potenziale per ottenere il rimodellamento del muscolo scheletrico e l’aumento della massa e della forza del muscolo scheletrico con la somministrazione di androgeni è notevole. Tuttavia, la somministrazione di dosi sovrafisiologiche di androgeni è associata ad un’elevata frequenza di effetti avversi dose-dipendenti, come eritrocitosi, edema delle gambe ed eventi prostatici (21, 26). Pertanto, agenti terapeutici come i SARM non steroidei con la cui somministrazione possono far ottenere effetti anabolizzanti sul muscolo scheletrico e sull’osso senza gli effetti avversi limitanti riscontrati con dosaggi di Testosterone aventi il medesimo effetto terapeutico sarebbero attraenti come terapie anabolizzanti d’elezione (27, 28, 29). Il riconoscimento di queste potenziali opportunità per lo sviluppo di nuove terapie per le limitazioni funzionali e disabilità associate a disturbi cronici, invecchiamento e osteoporosi ha guidato gli sforzi farmaceutici per sviluppare SARM non steroidei.
Il raggiungimento della selettività dei tessuti
Storicamente sono stati utilizzati due approcci generali per ottenere la selettività tissutale dell’azione degli Androgeni. Il primo approccio consiste nello sviluppare un SARM con un profilo di attività desiderato e la selettività tissutale. Il secondo approccio è quello di chiarire i meccanismi di azione degli androgeni sul muscolo scheletrico e sulla Prostata e di identificare le molecole di segnalazione che sono a valle del recettore degli androgeni e che attivano le vie coinvolte nell’ipertrofia del muscolo scheletrico, ma non della Prostata.
SARM steroidei: relazioni struttura-attività
Come accennato in precedenza, strutturalmente, i SARM possono essere classificati in SARM steroidei e non steroidei. I SARM steroidei si formano modificando la struttura chimica della molecola di Testosterone (vedi figura seguente).
È stato riconosciuto negli anni ’40 che la sostituzione di un metile in posizione C-17 ritarda il metabolismo presistemico del Testosterone, estendendone l’emivita e rendendolo attivo per via orale. Pertanto, un certo numero di androgeni orali, come il Methylterstosterone, hanno una metilazione in C-17. Tuttavia, gli androgeni 17-alfa alchilati somministrati per via orale, sono potenzialmente epatotossici e abbassano notevolmente il colesterolo HDL plasmatico.
La rimozione del gruppo 19-metile aumenta l’attività anabolizzante del Testosterone (Figura sopra). Pertanto, il 19-nortestosterone ha costituito la base della serie di molecole derivate del Nandrolone. Il Nandrolone è ridotto dalla 5-α reduttasi nei tessuti bersaglio a un androgeno meno potente, il Diidronandrolone (DHN), ma è meno suscettibile all’aromatizzazione in estrogeni convertendo primariamente nel poco attivo Estrone.
Le sostituzioni alchiliche 7-alfa rendono il Testosterone meno suscettibile alla 5-α riduzione e ne aumentano la selettività tissutale rispetto alla Prostata. Pertanto, il 7-alfa metil, 19-nortestosterone ha attività anabolica teoricamente superiore all’attività androgena, sebbene i test fatti sono stati svolti su topi attraverso il ben poco affidabile se rapportato all’uomo “test di Hershberger” (per approfondimenti clicca qui). Comunque, altre molecole di questa serie con gruppi alchilici variabili sono state studiate per la loro attività anabolica.
Il Testosterone viene eliminato rapidamente dalla circolazione e ha una breve emivita. L’esterificazione del gruppo ossidrile 17-β rende la molecola più idrofoba; più lunga è la catena laterale dell’estere, maggiore è l’idrofobicità. Quando gli esteri idrossilici 17-β del Testosterone vengono somministrati attraverso un iniezione intramuscolare in una sospensione oleosa, vengono rilasciati lentamente dal deposito oleoso nella circolazione. Il lento rilascio di esteri idrossilici 17-β dal deposito oleoso estende la loro durata d’azione. Tuttavia, la de-esterificazione degli esteri di Testosterone non limita la velocità della metabolizzazione molecolare; in breve, l’emivita del Testosterone Enantato nel plasma non è significativamente diversa da quella del Testosterone non esterificato una volta scissa l’esterificazione. Allo stesso modo, l’esterificazione del Nandrolone per formare il Nandrolone Decanoato aumenta la sua emivita.
Molecola di Testosterone legata ad un estere Enantato.
L’Oxandrolone è un AAS orale derivato dal DHT che ha un sostituente metilico 17-alfa. La sostituzione del secondo carbonio con l’ossigeno aumenta la stabilità del 3-cheto gruppo e ne aumenta l’attività anabolizzante. Non aromatizza in estrogeno e ha mostrato una bassa attività androgena. Indi, esso è un altro esempio di SARM steroideo.
Struttura molecolare del Oxandrolone
SARM non-steroidei
Gli sforzi pionieristici degli scienziati della Ligand Pharmaceuticals e dell’Università del Tennessee hanno fornito le prime basi della scoperta dei SARM non-steroidei. Da allora, sono state esplorate una serie di categorie strutturali di SARM farmacofori: aril-propionamide (GTX, Inc.), idantoina biciclica (BMS), chinolinoni (Ligand Pharmaceuticals), analoghi della tetraidrochinolina (Kaken Pharmaceuticals, Inc.), benizimidazolo, imidazolopirazolo. , indolo e derivati pirazolina (Johnson e Johnson), derivati azasteroidali (Merck) e derivati anilina, diaril anilina e bezoxazepinoni (GSK) (vedi figura seguente). Poiché è stata pubblicata solo una parte della ricerca sulla scoperta, è probabile che esistano categorie strutturali aggiuntive. Una recente review di Narayanan et al fornisce un eccellente trattato delle strutture dei SARM (28).
Le modifiche strutturali degli analoghi dell’aril propionammide bicalutamide e idrossiflutamide hanno portato alla scoperta della prima generazione di SARM. I composti S1 e S4 in questa serie si legano al AR con elevata affinità e dimostrano selettività tissutale nel impreciso test di Hershberger che utilizza un modello di ratto castrato (30, 31). In questo modello di ratto castrato, sia S1 che S4 hanno prevenuto l’atrofia indotta dalla castrazione del muscolo levat ani e hanno agito come deboli agonisti nella Prostata (30, 31, 32). Alla dose di 3 mg/kg/die, S4 ha parzialmente ripristinato il peso della prostata a < 20% di quello intatto, ma ha ripristinato completamente il peso del levator ani, la forza dei muscoli scheletrici, la densità minerale ossea, la forza ossea e la massa corporea magra e ha soppresso LH e FSH (33, 34). S4 ha anche prevenuto la perdita ossea indotta dall’ovariectomia nel modello di osteoporosi femminile di ratto (35). La capacità dei SARM di promuovere sia la forza muscolare che la forza meccanica ossea costituisce un vantaggio unico rispetto ad altre terapie per l’osteoporosi che aumentano solo la densità ossea.
S1 e S4 sono agonisti parziali; quindi, in ratti maschi intatti (31), S1 e S4 competono con gli androgeni endogeni (o esogeni) e agiscono come antagonisti nella Prostata, tali SARM con attività antagonista o bassa attività intrinseca nella Prostata potrebbero essere utili nel trattamento dell’IPB o del cancro alla Prostata. Gli effetti soppressivi di questa classe di SARM sulla secrezione di gonadotropine nei ratti suggeriscono una potenziale applicazione per la contraccezione maschile.(31)
SARM non-steroideo S4 (Andarina)
Il legame etereo e la sostituzione della posizione-para dell’anello B sono fondamentali per l’attività agonista dei SARM aril propionammidi (30). Sulla base delle strutture cristalline, i composti con legame etereo sembrano adattare una conformazione più compatta rispetto alla bicalutamide a causa della formazione di un legame H intramolecolare, consentendo all’anello B di evitare il conflitto sterico con la catena laterale di W741 nel AR e potenzialmente spiegando l’attività agonista.(36)
I derivati dell’idantoina, sviluppati dal gruppo BMS (37), hanno una struttura ad anello A simile a quella della bicalutamide. Il gruppo ciano o nitro di queste molecole interagisce con Q711 e R752 (38, 39). L’anello benzenico o gruppo naftile, insieme all’anello idantoico, si sovrappone al piano steroideo, mentre l’azoto dell’anello idantoinico forma un legame H con N705. BMS-564929 lega al AR con alta affinità e alta specificità. BMS-564929 ha dimostrato attività anabolizzante nel muscolo levator ani e un alto grado di selettività tissutale, come indicato da una ED50 sostanzialmente più elevata per la Prostata. I derivati dell’idantoina sono potenti soppressori dell’LH. BMS-564929 è disponibile per via orale nell’uomo, con un’emivita di 8-14 ore. L’emivita prolungata di questi ligandi nei ratti può spiegare la dose più bassa necessaria per ottenere effetti farmacologici; differenze nelle attività in vivo di SARM che condividono affinità di legame e attività in vitro simili possono essere correlate alle differenze nella farmacocinetica e nell’esposizione al farmaco.(40)
Hanada et al (41) della Kaken Pharmaceutical Co. hanno riportato una serie di derivati della tetraidrochinolina come agonisti dell’AR nell’osso. Sebbene questi composti mostrino un’elevata affinità per l’AR e una forte attività agonista nella Prostata e nel levator ani, hanno dimostrato una scarsa selettività tra i tessuti androgeni e anabolici (41). Una significativa attività farmacologica in vivo è stata osservata solo ad alte dosi sottocutanee.(28, 41)
I composti ligandi LGD2226 e LGD 2941 che sono derivati biciclici del 6-anilino chinolinone hanno mostrato attività anabolica sul muscolo levator ani, nonché sulla massa ossea e sulla forza, pur avendo scarso effetto sulla dimensione della Prostata in un modello preclinico di roditori (42, 43, 44). È stato anche dimostrato che LGD2226 mantiene il comportamento riproduttivo maschile nel modello di roditore castrato (42). Gli scienziati della Johnson e Johnson hanno sostituito il legante propionammidico con elementi ciclici come pirazoli, benzimidazoli, indoli e mimetici propionanilidi ciclici (45). Gli scienziati della Merck hanno sviluppato una serie di derivati 4-azasteroidali e butanammidi (28). Ulteriori composti sono stati sviluppati da altre aziende farmaceutiche, ma una discussione dettagliata di ciascun composto esula dallo scopo di questo articolo.
Meccanismi di selettività tissutale dei SARM
Narayanan et al hanno confrontato le vie attivate da un aril propionamide SARM, S-22, con quelle attivate dal DHT (46) e hanno scoperto che S-22 e DHT attivavano diverse vie di segnalazione distinte. S-22 e DHT differivano significativamente nel reclutamento del AR e dei suoi co-regolatori come potenziatore del PSA. L’S-22 differiva anche dal DHT nell’induzione della rapida fosforilazione di diverse chinasi (46). Tuttavia, i meccanismi che contribuiscono all’attivazione trascrizionale tessuto-specifica e alla selettività degli effetti biologici dei SARM rimangono poco compresi. Sono state proposte tre ipotesi generali, anche se queste ipotesi non si escludono a vicenda. L’ipotesi del co-attivatore presuppone che il repertorio di proteine co-regolatrici che si associa al AR legato al SARM differisce da quello associato al AR legato al Testosterone che porta all’attivazione trascrizionale di un insieme di geni regolati in modo differenziale.
Antigene Prostatico Specifico (Prostate Specific Antigen, PSA)
L’ipotesi conformazionale afferma che le differenze funzionali nelle classi di ligandi (agonisti, antagonisti e SARM) si riflettono in stati conformazionalmente distinti con partizionamento termodinamico distinto. Il legame con il ligando induce specifici cambiamenti conformazionali nel dominio di legame del ligando, che potrebbe modulare la topologia di superficie e le successive interazioni proteina-proteina tra AR e altri co-regolatori coinvolti nell’attivazione trascrizionale genomica o proteine citosoliche coinvolte nella segnalazione non genomica. Le differenze nella conformazione del recettore ligando-specifico e le interazioni proteina-proteina potrebbero portare a una regolazione genica tessuto-specifica, a causa di potenziali cambiamenti nelle interazioni con ARE, co-regolatori o fattori di trascrizione. Le interazioni proteina-proteina indotte dal ligando contribuiscono alle interazioni tra le estremità amminiche e carbossiliche del AR (cioè l’interazione N/C) e il reclutamento di co-attivatori (47). Entrambe le interazioni sono mediate dall’interazione tra la regione AF2 del AR ed i motivi di legame FXXLF o LXXLL (48). Il solco idrofobo presente nella regione AF2 del AR LBD sembra essere più favorevole per il legame della fenilalanina, il che suggerisce che l’interazione N/C è preferita. Sebbene la conformazione AR-LBD legata al SARM non steroideo non sia stata ben caratterizzata, Sathya et al (49) hanno riportato che alcuni SARM steroidei che hanno attività agonista in vitro inducono un cambiamento conformazionale attivante senza facilitare le interazioni N/C. Questi dati suggeriscono che il cambiamento conformazionale specifico del ligando è ottenibile con ligandi sintetici.
(A) Il gene AR consiste di 8 esoni che codificano per il recettore degli androgeni con un prodotto genico della dimensione tipica di 919 amminoacidi. Il AR è composto da un dominio N-terminale (NTD), un dominio di legame al DNA centrale (DBD), una regione a cerniera corta e un LBD C-terminale. (B) LBD comprende una struttura elicoidale 12 che racchiude una tasca centrale di legame dell’ormone (HBP), un secondo dominio della funzione di attivazione (AF2) che si trova all’estremità carbossi-terminale dell’LBD e un sito di legame scoperto di recente, funzione di legame 3 (BF3). La conformazione adottata dell’H12 è inequivocabilmente associata al meccanismo d’azione molecolare dei ligandi legati all’HBP. (C) Come mostrato nella struttura complessa di Diidrotestosterone (DHT) e AR-LBD, l’AR HBP è composto principalmente da residui idrofobici (palla verde) che possono formare forti interazioni non polari con il DHT. L’ancoraggio proteina-ligando può essere ulteriormente stabilizzato da una rete di legami idrogeno (linea tratteggiata blu) che coinvolge i residui polari R752, Q711, N705 e T877.[fonte immagine https://www.researchgate.net/%5D
Bohl et al (36) hanno riportato che la bicalutamide adotta una conformazione molto piegata nel AR. Sebbene l’anello A e il legame ammidico della molecola di bicalutamide si sovrappongano al piano steroideo, l’anello B della bilcautammide si piega lontano dal piano, puntando verso la parte superiore della tasca di legame del ligando (LBP), che costituisce una caratteristica strutturale unica di questo classe di leganti (36). Il gruppo ciano dell’anello A forma legami H con Q711 e R752, simile al 3-cheto gruppo nel 5α-DHT (36). Il gruppo idrossile chirale forma legami H con L704 e N705, imitando l’anello C e il gruppo 17β-OH nel 5α-DHT (36). Queste interazioni di legame H sono fondamentali per un’elevata affinità di legame. Lievi modifiche strutturali possono cambiare il ligando da antagonista AR ad agonista. Il legame idrogeno favorevole tra il ligando e la catena laterale T877, le caratteristiche strutturali che imitano il 3-cheto gruppo del Testosterone e le interazioni idrofobiche sono fondamentali affinché il ligando si leghi con alta affinità e stimoli l’azione del AR. La struttura cristallina a raggi X del AR legato a S-1 ha rivelato che la catena laterale W741 è spostata dall’anello B per espandere la tasca di legame in modo che il composto si orienti verso la regione AF2 (50). Il ripiegamento proteico del AR legato al SARM è lo stesso che si tratti di un SARM steroideo e non steroideo (50). Non è chiaro come l’interazione ligando-recettore determini l’attività agonista o antagonista del ligando.
La selettività tissutale dei SARM potrebbe anche essere correlata a differenze nella loro distribuzione tissutale, potenziali interazioni con la 5α-reduttasi o l’aromatasi CYP19, o l’espressione tessuto-specifica di co-regolatori (51). Tuttavia, studi di autoradiografia con derivati di bicalutamide e idantoina (52) hanno mostrato che non si accumulano preferenzialmente nei tessuti “anabolizzanti”. L’azione del Testosterone in alcuni tessuti androgeni è amplificata dalla sua conversione in 5α-DHT (53); i SARM non steroidei non fungono da substrati per la 5α-reduttasi. La selettività tissutale dei SARM potrebbe essere correlata all’espressione tessuto-specifica delle proteine co-regolatorie. Allo stesso modo, alcune differenze delle azioni dei SARM rispetto al Testosterone potrebbero essere correlate all’incapacità dei SARM non steroidei di subire l’aromatizzazione.
Esperienza di studi preclinici e clinici con i SARM di prima generazione
Un gran numero di SARM candidati sono stati sottoposti a studi preclinici di verifica teorica e tossicologici e sono entrati in studi clinici di fase I e II (27, 28). Gli studi preclinici hanno rivelato una promettente selettività dei tessuti; tuttavia, poiché molti di questi dati generati dalle aziende farmaceutiche sono rimasti inediti, i confronti della potenza relativa e della selettività dei tessuti tra i diversi SARM sono difficili da convalidare.
Un certo numero di SARM di prima generazione sono stati testati in prove di fase I. Questi composti sono stati posizionati per studi di efficacia precoci per il trattamento dell’osteoporosi, la fragilità ossea, la cachessia del cancro e le limitazioni funzionali associate all’invecchiamento. Inoltre, i SARM che inibiscono potentemente le gonadotropine, ma risparmiano l’attività a livello della Prostata, hanno suscitato una certa attrattiva come candidati per la contraccezione maschile. È stato proposto l’uso di SARM per il trattamento delle sindromi da carenza di androgeni negli uomini; i vantaggi relativi ai SARM rispetto al Testosterone per questa indicazione non sono immediatamente evidenti e risultano limitati. Molte funzioni biologiche del Testosterone, in particolare i suoi effetti sulla libido e sul comportamento, sulle ossa e sui lipidi plasmatici, richiedono la sua aromatizzazione in estrogeni; poiché i SARM attualmente disponibili non sono né aromatizzabili né 5-alfa riducibili, questi composti risultano fortemente limitati come base terapica di sostituzione androgena in andropausa e dovrebbero affrontare una barra normativa in salita per l’approvazione in quanto sarebbero tenuti a dimostrare efficacia e sicurezza in molti più domini di azione degli androgeni rispetto a quanto richiesto dalle formulazioni di Testosterone la quale si conosce per effetti diretti ed indiretti in condizione terapeutica sostitutiva degli androgeni endogeni.
Alle dosi che sono state testate, i SARM di prima generazione inducono modesti guadagni di massa corporea magra in volontari sani, che non sono affatto vicini ai guadagni molto maggiori nella massa muscolare scheletrica riportati con dosi sovrafisiologiche di Testosterone. I modesti guadagni da 1,0 a 1,5 kg di massa magra con i SARM di prima generazione in 4-6 settimane dovrebbero essere confrontati con i guadagni di 5-7 kg di massa magra con dosi da 300 e 600mg di Testosterone Enantato (pari approssimativamente a 216mg e 432mg di Testosterone effettivo rispettivamente). Tuttavia, è possibile che la prossima generazione di molecole SARM avrà maggiore potenza e selettività rispetto ai SARM di prima generazione, ma ad oggi non sussiste ancora dimostrazione a riguardo.
Raggiungimento della selettività e spiegazione dei meccanismi d’azione
Un altro approccio per ottenere la selettività d’azione è chiarire i meccanismi dell’azione del Testosterone sulla Prostata e identificare le molecole a valle associate all’attivazione della segnalazione AR nel muscolo scheletrico, ma non nella Prostata. Attraverso la comprensione di questi meccanismi, potrebbe essere possibile identificare molecole candidate che prendono di mira aspetti specifici della cascata di segnalazione AR.
Le analisi delle biopsie muscolari di uomini trattati con dosi graduate di testosterone hanno rivelato che la somministrazione di testosterone induce ipertrofia delle fibre muscolari sia di tipo I che di tipo II (54, 55); I cambiamenti nelle aree trasversali di entrambe le fibre di tipo I e II sono correlati alla dose di Testosterone e alle concentrazioni di Testosterone totale e libero (54). Tuttavia, né il numero assoluto né la proporzione relativa delle fibre di tipo I e II cambiano durante la somministrazione di Testosterone.
Poiché le cellule satellite muscolari sono state implicate nell’ipertrofia del muscolo scheletrico e nell’aumento del numero mionucleare (56), sono state quantificate le cellule satellite e il numero mionucleare mediante microscopia elettronica, utilizzando metodi di conteggio diretto e orientamento spaziale nelle biopsie del vasto laterale ottenute al basale e dopo 20- settimane di trattamento con un agonista del GnRH e dosi graduate di Testosterone Enantato. Il numero assoluto e percentuale di cellule satellite a 20 settimane era significativamente maggiore del basale negli uomini che ricevevano dosi sovrafisiologiche di Testosterone (57). La variazione del numero di cellule satellite era correlata alle variazioni dei livelli di Testosterone totale e libero (57). Quindi, l’ipertrofia delle fibre muscolari indotta dal Testosterone è associata ad un aumento delle cellule satellite e del numero di mionuclei.
Il Testosterone e il DHT promuovono la differenziazione delle cellule staminali mesenchimali multipotenti in linea miogenica e inibiscono la loro differenziazione in linea adipogenica (58, 59). Il Testosterone inibisce anche la differenziazione dei pre-adipociti in adipociti (59, 60). Altri hanno suggerito che l’ipertrofia indotta dal Testosterone sia causata dalla stimolazione della sintesi proteica e dall’inibizione della degradazione proteica (61, 62). Testosterone e DHT promuovono l’associazione del ligando AR con il suo co-attivatore, β-catenina; questa interazione stabilizza la β-catenina, promuove la sua traslocazione nel nucleo e l’associazione con TCF-4, e l’attivazione trascrizionale di un certo numero di geni bersaglio Wnt (63). La β-catenina svolge un ruolo essenziale nel mediare gli effetti del Testosterone sulla differenziazione miogenica. Il Testosterone sovra-regola l’espressione della Follistatina in vivo e in vitro (63); l’infusione della proteina Follistatina ricombinante aumenta la massa muscolare e diminuisce la massa grassa nei topi castrati. Il Testosterone sovra-regola l’SMAD 7 e sotto-regola la segnalazione del SMAD mediata dal TGFβ e i geni bersaglio del TGFβ (63). La Follistatina inibisce l’azione di diversi membri della famiglia del TGFβ. Questi studi supportano l’ipotesi che gli effetti del Testosterone siano trasmessi in modo incrociato dalla via Wnt alla via TGFβ-SMAD attraverso la Follistatina. Pertanto, è possibile che molecole candidate come la Follistatina che sono a valle del AR e β-catenina e che mediano gli effetti del Testosterone sul muscolo possano fornire la selettività desiderata degli effetti anabolici. La via di segnalazione mediata dal AR a valle della β-catenina può essere un interessante serbatoio di bersagli candidati per lo sviluppo di farmaci anabolizzanti selettivi.
Molecola di Follistatina
Ostacoli normativi allo sviluppo dei SARM
Negli studi di fase I e II, i SARM di prima generazione hanno mostrato riduzioni significative delle concentrazioni di colesterolo HDL e SHBG e lievi aumenti transitori di AST e ALT. Non è chiaro se gli aumenti delle transaminasi riflettano la tossicità epatica di primo passaggio tipica degli androgeni somministrati per via orale o un effetto di classe sulla trascrizione del gene AST. Allo stesso modo, la soppressione del colesterolo HDL potrebbe riflettere gli effetti combinati della via di somministrazione orale e la mancanza di aromatizzazione. È possibile che una via di somministrazione sistemica – transdermica o intramuscolare – possa attenuare il potenziale di aumento delle transaminasi e riduzioni di HDL-C.
Globulina Legante gli Ormoni Sessuali (in inglese sex hormone-binding globulin o SHBG)
Mentre il percorso normativo per l’approvazione dei farmaci per l’osteoporosi è stato ben delineato a causa della precedenza stabilita dai farmaci precedentemente approvati, il percorso per l’approvazione delle terapie anabolizzanti che promuovono la suddetta funzione non è stato chiaramente stabilito. Sono in corso sforzi considerevoli per generare un consenso su indicazioni, risultati di efficacia negli studi cardine e differenze clinicamente importanti minime nei risultati di efficacia chiave; questi sforzi dovrebbero facilitare le prove di efficacia delle molecole candidate. Ma il risultato, ad oggi, non è molto promettente.
Allora i SARM non-steroidei sono tessuto-selettivi?
Ammetto che quanto esposto fino ad ora non è propriamente “masticabile” da tutti, ed è per questo che vi renderò la comprensione più facile.
Allora, un modo per ottenere la selettività tissutale è tramite un fapping molecolare che implica l’attivazione del recettore degli androgeni (AR) specificamente nel tessuto muscolare. Mentre l’AR è lo stesso in tutti i tessuti, il contesto cellulare è diverso: puoi immaginare che il contenuto di una cellula muscolare sia abbastanza diverso da quello di una cellula della ghiandola sebacea. Quando l’AR viene attivato per indurre la trascrizione genica, che alla fine porterà ai guadagni muscolari, entrano in gioco molte altre proteine. Queste proteine coinvolte nella trascrizione sono i cosiddetti coregolatori trascrizionali. Chiamiamoli cofattori in breve. Questi possono aiutare nella trascrizione (coattivatori) o reprimerla (corepressori). Quei cofattori, e le loro proporzioni, che vengono reclutati da un AR attivato, possono variare da un tessuto all’altro. Questo dipende, in parte, da quale molecola è legata all’AR. In quanto tale, un SARM potrebbe essere in grado di reclutare un gruppo di cofattori che porteranno a una trascrizione genica minima o nulla nel tessuto A (Prostata), mentre portano alla trascrizione genica completa nel tessuto B (Muscolo).
Quanto detto sopra sembra comunque piuttosto complesso, e lo è, ma non mi è possibile comunicare a gesti per spiegarvi una cosa che è di base complessa. Comunque sia, come si fa a sapere quale tipo di ligando per l’AR interagisce con quali cofattori e in che misura? Non lo fa, si dovrebbero eseguire test quasi infiniti sul composto in questione per determinarlo effettivamente. E questo processo sembra richiedere molto tempo. Tuttavia, questo è attualmente pubblicizzato come uno dei motivi per cui i SARM – in sostanza avendolo scoperto per “caso” – esercitano i loro effetti specifici sui tessuti. Ad esempio, è stato dimostrato che l’antiandrogeno steroideo TSAA-291 esercita un’attività tessuto-specifica che coincide con profili di reclutamento di coregolatori differenziali rispetto al Diidrotestosterone (DHT) [64]. Tuttavia, poiché non hanno confrontato altri AAS, potrebbe anche essere che avrebbero visto diversi profili di reclutamento di coregolatori con altri AAS. Pertanto, è difficile vedere quanto sia effettivamente rilevante per le proprietà specifiche dei SARM. Dopotutto, la correlazione non implica la causalità.
Oxendolone (TSAA-291)
Andando avanti con la semplificazione pratica del concetto di selettività specifica, un altro modo in cui un SARM potrebbe esercitare tale specificità tissutale è attraverso il la sua via di metabolizzazione. Una molecola viene metabolizzata dall’azione degli enzimi. E la presenza di tali enzimi metabolizzanti può differire da un tessuto all’altro. Ad esempio, questo è molto evidente con la metabolizzazione del Testosterone. Il Testosterone è suscettibile di metabolizzazione per riduzione sul suo quinto atomo di carbonio. Questa riduzione è catalizzata dall’enzima 5α-reduttasi. Il risultato di questa riduzione è il più potente androgeno Diidrotestosterone (DHT). Pertanto, l’effetto del testosterone viene amplificato nei tessuti che esprimono questo enzima. Sfortunatamente, il muscolo scheletrico non è uno di quei tessuti. E, in effetti, il DHT viene degradato nel molto debole androgeno 3α-Androstanediolo dall’enzima 3α-HSD nel muscolo [65], diminuendo così il suo effetto in loco.
3α-idrossisteroide deidrogenasi ( 3α-HSD o aldo-cheto reduttasi famiglia 1 membro C4)
Tuttavia, questo aspetto è leggermente diverso per i SARM. Gli enzimi steroidogeni, come la 5α-reduttasi e la 3α-HSD, non hanno effetto sui SARM non steroidei. Gli enzimi che metabolizzano i SARM variano da una classe di SARM all’altra. Come tale, deve essere studiato per ogni SARM, analizzandone il modo in cui viene metabolizzato e con quale velocità ciò si verifica nei vari tessuti. Questo risulta essere più banale per la maggior parte degli AAS sui quali possiamo ampiamente prevederlo. Contrariamente, risulta difficile per lo sviluppo dei SARM non steroidei.
3α-Androstanediolo
Infine, è noto che gli AAS possono esercitare anche effetti non genomici [66]. Come suggerisce il nome, questi sono effetti che non sono mediati dalla trascrizione genica. Pertanto, questi effetti si verificano molto rapidamente (entro secondi/minuti dopo l’esposizione di una cellula ad essa). Alcune ricerche indicano che il recettore degli androgeni localizzato nella membrana plasmatica, così come altri recettori legati alla membrana, mediano questi effetti. Ipoteticamente è possibile che AAS – e per estensione SARM – siano in grado di influenzare le vie di segnalazione a seconda del contesto cellulare, cioè gli effetti potrebbero differire da una cellula all’altra: specificità del tessuto.
Più di 20 anni di ricerca sui SARM ma nessuna approvazione clinica
Sapere queste cose è interessante e utile per comprendere l’attività di tali molecole, ma tali attività ci mostrano di essere ben lungi (ancora) dal possedere la chiave di volta nello sviluppo di SARM terapeuticamente e pienamente efficaci. Ma almeno abbiamo una base attraverso la quale i SARM potrebbero effettivamente funzionare. Tuttavia, dopo oltre 2 decenni di ricerca sui SARM [67], nessuno è stato approvato dalla Food and Drug Administration (FDA). E no, non c’entra “bIg PhaRma”, complottaro da tastiera.
Parte del motivo per cui ciò avviene può essere ricondotto al modo in cui i ricercatori hanno esaminato i potenziali SARM. Come ho riportato in un mio precedente articolo, la anabolico:androgeno ratio, come valutato dal test di Hershberger, è pressoché inutile. Eppure questo test è stato utilizzato dalle aziende farmaceutiche per decidere se perseguire o meno la ricerca su determinati SARM di particolare interesse, queste aziende includono la GTx, Inc. con lo sviluppo del Enobosarm (GTx-024) [68], la GlaxoSmithKline con lo sviluppo del GSK2881078 [69 ], la Takeda Pharmaceutical Company con lo sviluppo del SARM-2f [70], la Aska Pharmaceuticals con lo sviluppo del S42 [71], e la Merck & Co, Inc con lo sviluppo del MK-4541 [72], ecc.
Non si sono forse già visti risultati ridicolmente buoni con AAS convenzionali in passato utilizzando questi test? Si, e non per una molecola. Ad esempio, si dice che lo Stanozololo abbia un rapporto anabolico/androgeno circa 10 volte superiore a quello del Testosterone, mentre il Methyldrostanolone ha circa un rapporto anabolico/androgeno 20 volte superiore [73]. Tuttavia, come sappiamo, queste molecole non sono considerate SARM sito-specifici e non sono scevre da eventuali effetti androgenizzanti. Perché? Perchè uno studio con molteplici variabili svolto su roditori non può essere rapportato correttamente all’uomo, come ho spiegato nell’articolo dedicato alla anabolico:androgeno ratio.
Un ulteriore problema con la ricerca sui SARM emerge quando si esaminano gli studi clinici. Poiché i SARM vengono sviluppati per superare gli AAS convenzionali, non ci si aspetterebbe forse che essi vengano confrontati con gli AAS convenzionali negli studi clinici? Per qualche ragione, in tutti gli studi clinici con i SARM, questi vengono confrontati con un placebo. Se si vuole valutare l’efficacia reale di una molecola rispetto ad un altra, non lo si fa confrontandola solo ad un placebo, o forse solo inizialmente lo si farebbe, come in una sperimentazione pilota per risparmiare sui costi, e per valutare se ne vale la pena o meno. Questi studi mostrano comunemente guadagni marginali (nell’ordine di 1kg) di LBM in un periodo di diverse settimane/mesi con una corrispondente buona tollerabilità. Anche gli AAS convenzionali sono generalmente ben tollerati e aumentano marginalmente l’LBM quando vengono somministrati a basso dosaggio, niente di sconvolgente in questo. La Ligand Pharmaceuticals ha persino trovato la necessità di menzionare quanto segue nella conclusione del loro abstract di studio che copre gli effetti del loro SARM LGD-4033: “LGD-4033 era sicuro, aveva un profilo farmacocinetico favorevole e un aumento della massa corporea magra anche durante questo breve periodo senza cambiamento nell’antigene prostatico specifico”. Cosa si aspettavano in poche settimane di trattamento con il loro SARM? Anche 600mg di Testosterone Enantato a settimana per 20 settimane non aumentano l’antigene prostatico specifico (PSA) negli uomini giovani [74, 75] o negli uomini più anziani [76].
LGD-4033
Se l’unico requisito ricercato è che un SARM non steroideo sia più efficace di un placebo pur essendo ben tollerato, ce l’hanno fatta. Ma praticamente tutti gli AAS convenzionali sono anche più efficaci di un placebo pur essendo ben tollerati. Superare il placebo non è mai stato l’obiettivo dello sviluppo dei SARM, quindi perché gli studi testa a testa sono ancora gravemente carenti? Forse perchè non vi è superiorità ne negli effetti benefici e nel rapporto tra benefici e rischi sistemici? …
Conclusioni:
I SARM si basano sulla selettività dei tessuti per esercitare i loro effetti anabolici (costruzione muscolare), mantenendo gli effetti collaterali al minimo assoluto. Dopotutto, gli effetti collaterali si riducono in gran parte, ma non totalmente, all’azione androgena nei tessuti diversi dai muscoli. I SARM possono esercitare questi effetti tessuto-specifici attraverso circa tre diversi meccanismi. Uno sfrutta le differenze nelle molecole tra i diversi tipi di cellule che “aiutano” un SARM ad avviare la trascrizione genica. Un altro si basa su enzimi di espressione tessuto-specifici che metabolizzano il SARM. Un terzo si basa sugli effetti non genomici che potrebbero essere mediati da un SARM che, ancora una volta, potrebbe variare da un tipo di cellula all’altro.
Poiché questi processi biochimici sono estremamente difficile da prevedere in anticipo, le aziende farmaceutiche devono esaminare molte molecole per vedere quale potrebbe essere la soluzione migliore. Nessun SARM è stato ancora approvato e credo che ciò sia in parte dovuto a questo processo di screening che si basa su metodi obsoleti e imperfetti come il test di Hershberger e all’incapacità di sopperire all’attività fisiologica del DHT e dell’Estradiolo, i quali subiscono una marcata soppressione consequenziale al abbassamento dei livelli di Testosterone endogeno. Questo punto deve essere sicuramente migliorato. Ed è quindi questa la strada che dovrebbe intraprendere la ricerca sui SARM.
Negli sport, ed in particolare nel Bodybuilding, l’uso dei SARM non steroidei, dopo l’iniziale eccitazione per le promesse commerciali affiancate al loro uso da parte dei rivenditori e brand, sono caduti in un uso più che altro amatoriale, da parte di persone poco informate in materia e dalla mente facilmente manipolabile dalla pubblicità e informazioni incomplete se non del tutto errate.
L’unico ambito in cui i SARM non steroidei hanno visto un certo potenziale è nel culturismo femminile. In questa circostanza, le molecole più testate, prima su tutte l’Ostarina, ha mostrato un certo vantaggio se l’obbiettivo era quello di aumenti contenuti del tessuto muscolare e la mancanza di possibili effetti mascolinizzanti alle dosi comprese tra 5 e 10mg/die.
Nell’uso maschile i SARM hanno lasciato una serie di delusioni e promesse non mantenute. In monoterapia il loro uso ha portato ad atleti con problemi non indifferenti nella sfera sessuale, con difficoltà di raggiungimento e mantenimento dell’erezione, letargia, stanchezza cronica, affaticabilità, depressione e stati ansiosi. Tutti sintomi legati ad un calo significativo del DHT e del Estradiolo, con conseguente riduzione o mancanza della loro, per esempio, attività a livello cerebrale (neurosteroideo).
Di conseguenza, utilizzare uno o più SARM senza una base esogena di Testosterone (o, per lo meno, di hCG) è una totale pazzia! E, comunque, l’uso dei SARM come aggiunte ad un ciclo di classici AAS iniettabili non risulta quasi mai all’altezza delle aspettative di risposta ipertrofica rispetto all’uso, per esempio, di AAS orali come starter e/o finisher. Ovviamente la valutazione si basa anche e soprattutto sul rapporto effetti collaterali:benefici in contesto preparatorio correttamente impostato.
Inoltre, gli effetti collaterali a livello epatico e della lipidemia ematica non sono estranei all’uso di SARM non steroidei, sebbene essi si mostrino a diverso grado di entità molecola-dipendente e dose-dipendente. La stessa Ostarina aveva mostrato lievi alterazioni di ALT e AST con riduzione del HDL al dosaggio di 3mg in studi clinici; la molecola in ambito “physique” viene assunta ad un dosaggio nel range di 10-20mg/die, e l’impatto sulle transaminasi, colesterolo totale, LDL e HDL osservato attraverso esami ematici mostrano variazioni significative e variabili in misura soggettiva.
Il SARM non steroideo con il più alto carico di effetti collaterali è risultato essere LGD4033, il quale, in diversi casi studio, ha mostrato di poter causare forte stress epatico oltre che alterare sensibilmente la lipidemia ematica. Nel caso di questa molecola, si è osservato anche una perdita della selettività con possibile comparsa di effetti androgenicizzanti. Complice di questi riscontri è soprattutto l’abuso che se ne fa della molecola, sforando i dosaggi efficaci e contenitivi (2-8mg/die) a favore di somministrazioni elevate (≥10mg/die).
Anche il RAD140 sembra non essere privo di effetti collaterali significativi a livello epatico, nonostante il suo potenziale effetto protettivo sulla Prostata che, a dosaggi minimi (5mg/die) potrebbe avere un riscontro terapeutico preventivo per l’ipertrofia prostatica.
SARM non-steroideo RAD140
Lascerei perdere discorsi ipotetici su altri SARM comunemente utilizzati dagli atleti (specialmente amatori) ma che alle spalle sono privi di studi clinici (vedi, per esempio, l’S23) e, quindi, di dati oggettivi sulle possibili attività nell’uomo. L’unica eccezione tra questi la fa, forse, il SARM steroideo YK11, il quale sembra essere gestibile a dosi di 5-10mg/die con un buon rapporto tra benefici ed alterazioni dei marker ematici.
SARM steroideo YK11
Per concludere, mi sembra di avervi dato sufficienti informazioni per valutare correttamente i SARM e deporli con cognizione logica dall'”altarino” di innocuità sul quale brand e venditori li hanno posti e dove una parte di voi continua a tenerli.
Gabriel Bellizzi
Riferimenti:
Mohler ML, Bohl CE, Jones A, Coss CC, Narayanan R, He Y, Hwang DJ, Dalton JT, Miller DD (June 2009). “Nonsteroidal selective androgen receptor modulators (SARMs): dissociating the anabolic and androgenic activities of the androgen receptor for therapeutic benefit”. Journal of Medicinal Chemistry. 52(12): 3597–617.
Yin D, Gao W, Kearbey JD, Xu H, Chung K, He Y, Marhefka CA, Veverka KA, Miller DD, Dalton JT (March 2003). “Pharmacodynamics of selective androgen receptor modulators”. The Journal of Pharmacology and Experimental Therapeutics. 304 (3): 1334–40.
Manfredi MC, Bi Y, Nirschl AA, Sutton JC, Seethala R, Golla R, Beehler BC, Sleph PG, Grover GJ, Ostrowski J, Hamann LG (August 2007). “Synthesis and SAR of tetrahydropyrrolo[1,2-b][1,2,5]thiadiazol-2(3H)-one 1,1-dioxide analogues as highly potent selective androgen receptor modulators”. Bioorganic & Medicinal Chemistry Letters. 17 (16): 4487–90.
Zhang X, Li X, Allan GF, Sbriscia T, Linton O, Lundeen SG, Sui Z (August 2007). “Design, synthesis, and in vivo SAR of a novel series of pyrazolines as potent selective androgen receptor modulators”. Journal of Medicinal Chemistry. 50 (16): 3857–69.
Long YO, Higuchi RI, Caferro TR, Lau TL, Wu M, Cummings ML, Martinborough EA, Marschke KB, Chang WY, López FJ, Karanewsky DS, Zhi L (May 2008). “Selective androgen receptor modulators based on a series of 7H-[1,4]oxazino[3,2-g]quinolin-7-ones with improved in vivo activity”. Bioorganic & Medicinal Chemistry Letters. 18 (9): 2967–71.
Bhasin S, Cunningham GR, Hayes FJ, Matsumoto AM, Snyder PJ, Swerdloff RS, Montori VM. Testosterone therapy in adult men with androgen deficiency syndromes: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2006;91:1995–2010.
Baumgartner RN. Body composition in healthy aging. Annals of the New York Academy of Sciences. 2000;904:437–448.
Bassey EJ, Fiatarone MA, O’Neill EF, Kelly M, Evans WJ, Lipsitz LA. Leg extensor power and functional performance in very old men and women. Clin Sci (Lond) 1992;82:321–327.
. Lexell J, Downham D, Sjostrom M. Distribution of different fibre types in human skeletal muscles. A statistical and computational study of the fibre type arrangement in m. vastus lateralis of young, healthy males. Journal of the neurological sciences. 1984;65:353–365.
Orwoll E, Lambert LC, Marshall LM, Blank J, Barrett-Connor E, Cauley J, Ensrud K, Cummings SR. Endogenous testosterone levels, physical performance, and fall risk in older men. Arch Intern Med. 2006;166:2124–2131.
Bhasin S, Storer TW, Berman N, Yarasheski KE, Clevenger B, Phillips J, Lee WP, Bunnell TJ, Casaburi R. Testosterone replacement increases fat-free mass and muscle size in hypogonadal men. J Clin Endocrinol Metab. 1997;82:407–413.
Snyder PJ, Peachey H, Berlin JA, Hannoush P, Haddad G, Dlewati A, Santanna J, Loh L, Lenrow DA, Holmes JH, Kapoor SC, Atkinson LE, Strom BL. Effects of testosterone replacement in hypogonadal men. J Clin Endocrinol Metab. 2000;85:2670–2677.
. Bhasin S, Storer TW, Berman N, Callegari C, Clevenger B, Phillips J, Bunnell TJ, Tricker R, Shirazi A, Casaburi R. The effects of supraphysiologic doses of testosterone on muscle size and strength in normal men. N Engl J Med. 1996;335:1–7.
. Bhasin S, Woodhouse L, Casaburi R, Singh AB, Bhasin D, Berman N, Chen X, Yarasheski KE, Magliano L, Dzekov C, Dzekov J, Bross R, Phillips J, Sinha-Hikim I, Shen R, Storer TW. Testosterone dose-response relationships in healthy young men. Am J Physiol Endocrinol Metab. 2001;281:E1172–1181.
Bhasin S, Woodhouse L, Casaburi R, Singh AB, Mac RP, Lee M, Yarasheski KE, Sinha-Hikim I, Dzekov C, Dzekov J, Magliano L, Storer TW. Older men are as responsive as young men to the anabolic effects of graded doses of testosterone on the skeletal muscle. J Clin Endocrinol Metab. 2005;90:678–688.
Casaburi R, Bhasin S, Cosentino L, Porszasz J, Somfay A, Lewis MI, Fournier M, Storer TW. Effects of testosterone and resistance training in men with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2004;170:870–878.
Johansen KL, Mulligan K, Schambelan M. Anabolic effects of nandrolone decanoate in patients receiving dialysis: a randomized controlled trial. Jama. 1999;281:1275–1281.
Woodhouse LJ, Reisz-Porszasz S, Javanbakht M, Storer TW, Lee M, Zerounian H, Bhasin S. Development of models to predict anabolic response to testosterone administration in healthy young men. Am J Physiol Endocrinol Metab. 2003;284:E1009–1017.
Storer TW, Magliano L, Woodhouse L, Lee ML, Dzekov C, Dzekov J, Casaburi R, Bhasin S. Testosterone dose-dependently increases maximal voluntary strength and leg power, but does not affect fatigability or specific tension. J Clin Endocrinol Metab. 2003;88:1478–1485.
Calof O, Singh AB, Lee ML, Urban RJ, Kenny AM, Tenover JL, Bhasin S. Adverse events associated with testosterone supplementation of odler men. J Greontol Med Sci. 2005 in press.
Bhasin S, Calof OM, Storer TW, Lee ML, Mazer NA, Jasuja R, Montori VM, Gao W, Dalton JT. Drug insight: Testosterone and selective androgen receptor modulators as anabolic therapies for chronic illness and aging. Nature Clinical Practice Endocrinology & Metabolism. 2006;2:146–159.
Narayanan R, Mohler ML, Bohl CE, Miller DD, Dalton JT. Selective androgen receptor modulators in preclinical and clinical development. Nuclear receptor signaling. 2008;6:e010. An excellent treatise of SARM chemistry and structure-activity relationships.
Negro-Vilar A. Selective androgen receptor modulators (SARMs): a novel approach to androgen therapy for the new millennium. J Clin Endocrinol Metab. 1999;84:3459–3462.
Yin D, He Y, Perera MA, Hong SS, Marhefka C, Stourman N, Kirkovsky L, Miller DD, Dalton JT. Key structural features of nonsteroidal ligands for binding and activation of the androgen receptor. Mol Pharmacol. 2003;63:211–223.
Gao W, Kearbey JD, Nair VA, Chung K, Parlow AF, Miller DD, Dalton JT. Comparison of the pharmacological effects of a novel selective androgen receptor modulator, the 5alpha-reductase inhibitor finasteride, and the antiandrogen hydroxyflutamide in intact rats: new approach for benign prostate hyperplasia. Endocrinology. 2004;145:5420–5428.
Gao W, Reiser PJ, Coss CC, Phelps MA, Kearbey JD, Miller DD, Dalton JT. Selective Androgen Receptor Modulator (SARM) Treatment Improves Muscle Strength and Body Composition, and Prevents Bone Loss in Orchidectomized Rats. Endocrinology 2005
Gao W, Reiser PJ, Coss CC, Phelps MA, Kearbey JD, Miller DD, Dalton JT. Selective androgen receptor modulator treatment improves muscle strength and body composition and prevents bone loss in orchidectomized rats. Endocrinology. 2005;146:4887–4897.
Gao W, Reiser PJ, Kearbey JD, Phelps MA, Coss CC, Miller DD, Dalton JT. Effects of Novel Selective Androgen Receptor Modulator (SARM) on Skeletal Muscle Mass and Strength in Castrated Male Rats. The Endocrine Society; New Orleans: 2004.
Kearbey JD, Gao W, Narayanan R, Fisher SJ, Wu D, Miller DD, Dalton JT. Selective Androgen Receptor Modulator (SARM) treatment prevents bone loss and reduces body fat in ovariectomized rats. Pharmaceutical research. 2007;24:328–335.
Bohl CE, Gao W, Miller DD, Bell CE, Dalton JT. Structural basis for antagonism and resistance of bicalutamide in prostate cancer. Proc Natl Acad Sci U S A. 2005;102:6201–6206. An important paper that describes the structural basis of antagonism of bicalutamide based on the crystal structure.
Hamann LG, Manfredi MC, Sun C, Krystek SR, Jr, Huang Y, Bi Y, Augeri DJ, Wang T, Zou Y, Betebenner DA, Fura A, Seethala R, Golla R, Kuhns JE, Lupisella JA, Darienzo CJ, Custer LL, Price JL, Johnson JM, Biller SA, Zahler R, Ostrowski J. Tandem optimization of target activity and elimination of mutagenic potential in a potent series of N-aryl bicyclic hydantoin-based selective androgen receptor modulators. Bioorganic & medicinal chemistry letters. 2007;17:1860–1864.
Manfredi MC, Bi Y, Nirschl AA, Sutton JC, Seethala R, Golla R, Beehler BC, Sleph PG, Grover GJ, Ostrowski J, Hamann LG. Synthesis and SAR of tetrahydropyrrolo[1,2-b][1,2,5]thiadiazol-2(3H)-one 1,1-dioxide analogues as highly potent selective androgen receptor modulators. Bioorganic & medicinal chemistry letters. 2007;17:4487–4490.
Ostrowski J, Kuhns JE, Lupisella JA, Manfredi MC, Beehler BC, Krystek SR, Jr, Bi Y, Sun C, Seethala R, Golla R, Sleph PG, Fura A, An Y, Kish KF, Sack JS, Mookhtiar KA, Grover GJ, Hamann LG. Pharmacological and x-ray structural characterization of a novel selective androgen receptor modulator: potent hyperanabolic stimulation of skeletal muscle with hypostimulation of prostate in rats. Endocrinology. 2007;148:4–12.
Kim J, Wu D, Hwang DJ, Miller DD, Dalton JT. The para substituent of S-3-(phenoxy)-2-hydroxy-2-methyl-N-(4-nitro-3-trifluoromethyl-phenyl)-prop ionamides is a major structural determinant of in vivo disposition and activity of selective androgen receptor modulators. The Journal of pharmacology and experimental therapeutics. 2005;315:230–239.
Hanada K, Furuya K, Yamamoto N, Nejishima H, Ichikawa K, Nakamura T, Miyakawa M, Amano S, Sumita Y, Oguro N. Bone anabolic effects of S-40503, a novel nonsteroidal selective androgen receptor modulator (SARM), in rat models of osteoporosis. Biol Pharm Bull. 2003;26:1563–1569.
Miner JN, Chang W, Chapman MS, Finn PD, Hong MH, Lopez FJ, Marschke KB, Rosen J, Schrader W, Turner R, van Oeveren A, Viveros H, Zhi L, Negro-Vilar A. An orally active selective androgen receptor modulator is efficacious on bone, muscle, and sex function with reduced impact on prostate. Endocrinology. 2007;148:363–373.
van Oeveren A, Motamedi M, Mani NS, Marschke KB, Lopez FJ, Schrader WT, Negro-Vilar A, Zhi L. Discovery of 6-N,N-bis(2,2,2-trifluoroethyl)amino-4-trifluoromethylquinolin-2(1H)-one as a novel selective androgen receptor modulator. Journal of medicinal chemistry. 2006;49:6143–6146.
van Oeveren A, Motamedi M, Martinborough E, Zhao S, Shen Y, West S, Chang W, Kallel A, Marschke KB, Lopez FJ, Negro-Vilar A, Zhi L. Novel selective androgen receptor modulators: SAR studies on 6-bisalkylamino-2-quinolinones. Bioorganic & medicinal chemistry letters. 2007;17:1527–1531.
Ng RA, Lanter JC, Alford VC, Allan GF, Sbriscia T, Lundeen SG, Sui Z. Synthesis of potent and tissue-selective androgen receptor modulators (SARMs): 2-(2,2,2)-Trifluoroethyl-benzimidazole scaffold. Bioorganic & medicinal chemistry letters. 2007;17:1784–1787.
Narayanan R, Coss CC, Yepuru M, Kearbey JD, Miller DD, Dalton JT. Steroidal androgens and nonsteroidal, tissue-selective androgen receptor modulator, S-22, regulate androgen receptor function through distinct genomic and nongenomic signaling pathways. Mol Endocrinol. 2008;22:2448–2465. This paper showed that DHT and SARMs activate distinct signaling pathways.
Masiello D, Chen SY, Xu Y, Verhoeven MC, Choi E, Hollenberg AN, Balk SP. Recruitment of beta-catenin by wild-type or mutant androgen receptors correlates with ligand-stimulated growth of prostate cancer cells. Mol Endocrinol. 2004;18:2388–2401.
Song LN, Herrell R, Byers S, Shah S, Wilson EM, Gelmann EP. Beta-catenin binds to the activation function 2 region of the androgen receptor and modulates the effects of the N-terminal domain and TIF2 on ligand-dependent transcription. Mol Cell Biol. 2003;23:1674–1687.
Sathya G, Chang CY, Kazmin D, Cook CE, McDonnell DP. Pharmacological uncoupling of androgen receptor-mediated prostate cancer cell proliferation and prostate-specific antigen secretion. Cancer Res. 2003;63:8029–8036.
Sathya G, Chang CY, Kazmin D, Cook CE, McDonnell DP. Pharmacological uncoupling of androgen receptor-mediated prostate cancer cell proliferation and prostate-specific antigen secretion. Cancer Res. 2003;63:8029–8036.
Bohl CE, Wu Z, Miller DD, Bell CE, Dalton JT. Crystal structure of the T877A human androgen receptor ligand-binding domain complexed to cyproterone acetate provides insight for ligand-induced conformational changes and structure-based drug design. J Biol Chem. 2007;282:13648–13655.
Heinlein CA, Chang C. Androgen receptor (AR) coregulators: an overview. Endocr Rev. 2002;23:175–200.
Hamann LG. Discovery and preclinical profile of a highly potent and muscle selective androgen receptor modulator (SARM). 227th National Meeting of the American Chemical Society Medicinal Chemistry Division.2004.
Sinha-Hikim I, Artaza J, Woodhouse L, Gonzalez-Cadavid N, Singh AB, Lee MI, Storer TW, Casaburi R, Shen R, Bhasin S. Testosterone-induced increase in muscle size in healthy young men is associated with muscle fiber hypertrophy. Am J Physiol Endocrinol Metab. 2002;283:E154–164.
Kadi F, Eriksson A, Holmner S, Thornell LE. Effects of anabolic steroids on the muscle cells of strength-trained athletes. Medicine and science in sports and exercise. 1999;31:1528–1534.
Sinha-Hikim I, Roth SM, Lee MI, Bhasin S. Testosterone-induced muscle hypertrophy is associated with an increase in satellite cell number in healthy, young men. Am J Physiol Endocrinol Metab. 2003;285:E197–205.
Singh R, Artaza JN, Taylor WE, Gonzalez-Cadavid NF, Bhasin S. Androgens stimulate myogenic differentiation and inhibit adipogenesis in C3H 10T1/2 pluripotent cells through an androgen receptor-mediated pathway. Endocrinology. 2003;144:5081–5088. This paper was the first to report that androgens regulate myogenic differentiation of mesenchymal multipotent cells.
Gupta V, Bhasin S, Guo W, Singh R, Miki R, Chauhan P, Choong K, Tchkonia T, Lebrasseur NK, Flanagan JN, Hamilton JA, Viereck JC, Narula NS, Kirkland JL, Jasuja R. Effects of dihydrotestosterone on differentiation and proliferation of human mesenchymal stem cells and preadipocytes. Molecular and cellular endocrinology. 2008;296:32–40.
Singh R, Artaza JN, Taylor WE, Braga M, Yuan X, Gonzalez-Cadavid NF, Bhasin S. Testosterone inhibits adipogenic differentiation in 3T3-L1 cells: nuclear translocation of androgen receptor complex with beta-catenin and T-cell factor 4 may bypass canonical Wnt signaling to down-regulate adipogenic transcription factors. Endocrinology. 2006;147:141–154.
Brodsky IG, Balagopal P, Nair KS. Effects of testosterone replacement on muscle mass and muscle protein synthesis in hypogonadal men–a clinical research center study. J Clin Endocrinol Metab. 1996;81:3469–3475.
Ferrando AA, Sheffield-Moore M, Yeckel CW, Gilkison C, Jiang J, Achacosa A, Lieberman SA, Tipton K, Wolfe RR, Urban RJ. Testosterone administration to older men improves muscle function: molecular and physiological mechanisms. Am J Physiol Endocrinol Metab. 2002;282:E601–607.
Singh R, Bhasin S, Braga M, Artaza JN, Pervin S, Taylor WE, Krishnan V, Sinha SK, Rajavashisth TB, Jasuja R. Regulation of Myogenic Differentiation by Androgens: Cross-Talk between Androgen Receptor/{beta}-Catenin and Follistatin/TGF-{beta} Signaling Pathways. Endocrinology. 2008 This paper describes the important role of beta-catenin/Wnt pathway in mediating the effects of testosterone on myogenic differentiation and the role of follistatin in cross-communicating the signal from Wnt to TGFbeta/SMAD pathway.
Hikichi, Yukiko, et al. “Selective androgen receptor modulator activity of a steroidal antiandrogen TSAA-291 and its cofactor recruitment profile.” European journal of pharmacology 765 (2015): 322-331.
Becker, H., et al. “In vivo uptake and metabolism of 3H-testosterone and 3H-5α-dihydrotestosterone by human benign prostatic hypertrophy.” European Journal of Endocrinology 71.3 (1972): 589-599.
Foradori, C. D., M. J. Weiser, and R. J. Handa. “Non-genomic actions of androgens.” Frontiers in neuroendocrinology 29.2 (2008): 169-181.
Negro-Vilar, Andres. “Selective androgen receptor modulators (SARMs): a novel approach to androgen therapy for the new millennium.” The Journal of Clinical Endocrinology & Metabolism 84.10 (1999): 3459-3462.
Kim, Juhyun, et al. “The para substituent of S-3-(phenoxy)-2-hydroxy-2-methyl-N-(4-nitro-3-trifluoromethyl-phenyl)-propionamides is a major structural determinant of in vivo disposition and activity of selective androgen receptor modulators.” Journal of Pharmacology and Experimental Therapeutics 315.1 (2005): 230-239.
Neil, David, et al. “GSK2881078, a SARM, produces dose-dependent increases in lean mass in healthy older men and women.” The Journal of Clinical Endocrinology & Metabolism 103.9 (2018): 3215-3224.
Aikawa, Katsuji, et al. “Synthesis and biological evaluation of novel selective androgen receptor modulators (SARMs) Part III: Discovery of 4-(5-oxopyrrolidine-1-yl) benzonitrile derivative 2f as a clinical candidate.” Bioorganic & medicinal chemistry 25.13 (2017): 3330-3349.
Min, Liu, et al. “A novel synthetic androgen receptor ligand, S42, works as a selective androgen receptor modulator and possesses metabolic effects with little impact on the prostate.” Endocrinology 150.12 (2009): 5606-5616.
Schmidt, Azriel, et al. “Identification of an anabolic selective androgen receptor modulator that actively induces death of androgen-independent prostate cancer cells.” The Journal of steroid biochemistry and molecular biology 143 (2014): 29-39.
Basaria, Shehzad, et al. “The safety, pharmacokinetics, and effects of LGD-4033, a novel nonsteroidal oral, selective androgen receptor modulator, in healthy young men.” Journals of Gerontology Series A: Biomedical Sciences and Medical Sciences 68.1 (2013): 87-95.
Bhasin, Shalender, et al. “Testosterone dose-response relationships in healthy young men.” American Journal of Physiology-Endocrinology And Metabolism (2001).
Bhasin, Shalender, et al. “Effect of testosterone supplementation with and without a dual 5α-reductase inhibitor on fat-free mass in men with suppressed testosterone production: a randomized controlled trial.” Jama 307.9 (2012): 931-939.
Bhasin, Shalender, et al. “Older men are as responsive as young men to the anabolic effects of graded doses of testosterone on the skeletal muscle.” The Journal of Clinical Endocrinology & Metabolism 90.2 (2005): 678-688.
Di norma il Nandrolone viene considerato un composto con una bassa androgenicità pari a circa il 37% di quella del Testosterone. Questa sua caratteristica è stata osservata e documentata fin dai primi anni di sperimentazione clinica. Però, la questione della sua androgenicità vista in questo modo risulta incompleta e, quindi, limitante per la sua reale e totale valutazione.
Sono decenni che atleti, soprattutto di sesso femminile, optano per il Nandrolone dal momento che i suoi effetti androgeni sono rari e generalmente lievi. Gli uomini che soffrono di alopecia androgenetica e che desiderano ridurre al minimo la perdita di capelli con l’uso di AAS hanno optato per anni per l’uso del Nandrolone come parte della base dei loro cicli.
E allora qual è il problema? Beh, esso risiede nelle abitudini di trattamento iatrogeno che gli atleti con alopecia androgenetica abbinano alle loro preparazioni contenenti Nandrolone al fine di arrestare la perdita di capelli…
Ma vediamo di cosa si tratta…
C’è “androgenicità” e “androgenicità”…
Il motivo per cui il Nandrolone è abbastanza sicuro per i capelli è la sua relativa mancanza di androgenicità nel corpo.
Rispetto agli ormoni di base che normalmente vengono considerati come punto di riferimento per la caduta dei capelli (vedi Testosterone e DHT), il Nandrolone è un’opzione superiore. Tuttavia, questo non avviene perché il Nandrolone è meno androgeno del DHT ma non del Testosterone. In effetti, il Nandrolone è più androgeno del Testosterone, sebbene risulti comunque meno androgeno del DHT.
La bassa attività androgena è dovuta esclusivamente all’interazione del Nandrolone con la 5α-reduttasi. Come ben sappiamo, la 5α-riduttasi è l’enzima principalmente responsabile per la sintesi di Diidrotestosterone (DHT) nel corpo. Il Testosterone viene in parte convertito tramite la 5α-reduttasi nel suo metabolita più androgeno, il DHT. Questo stesso processo enzimatico converte anche in parte il Nandrolone in un metabolita noto come Diidronandrolone (DHN).
Il DHN è il meno androgeno dei quattro ormoni sopra citati.
Se dovessimo elencare questi quattro ormoni in ordine dal più al meno androgeno, verrebbe fuori una graduatoria così come segue:
Diidrotestosterone (DHT) – maggior attività androgena
Nandrolone
Testosterone
Diidronandrolone (DHN) – minore attività androgena.
5α-Dihydronandrolone [DHN]
Poiché la 5α-reduttasi è significativamente espressa nel cuoio capelluto, in realtà si finisce per restare in una situazione di bassa nocività del Nandrolone a livello del bulbo del capello attraverso i processi enzimatici endogeni naturali.
In effetti, questo è anche il motivo per cui l’inibizione di questo processo enzimatico attraverso l’uso degli inibitori della 5α-reduttasi può diventare problematico per gli utilizzatori di AAS.
Coloro che utilizzano il Nandrolone con Finasteride o Dutasteride vanno ad inibire effettivamente questo processo enzimatico della 5α-reduttasi che converte il Nandrolone nel metabolita DHN, molto più sicuro per i capelli. Pertanto, quando si combina il Nandrolone con Finasteride o Dutasteride, si verifica l’effetto opposto rispetto a quanto accade con il Testosterone.[1]
Effetto degli inibitori della 5α-reduttasi sulla attività androgena di Testosterone, Nandrolone e Trestolone in ratti castrati.
Come si può vedere dal grafico sopra esposto, sebbene vada considerato con un certo margine di fallibilità parziale dal momento che si tratta di uno studio svolto su animali (ratti), l’uso di un inibitore della 5α-reduttasi ha parzialmente inibito la conversione del Testosterone in DHT. Questo è il motivo per cui la stimolazione ventrale della prostata (legato all’attività androgena come alla caduta dei capelli) è diminuita così significativamente nel gruppo trattato con l’inibitore della 5α-reduttasi.
Nei gruppi trattati con Nandrolone, l’uso di un inibitore della 5α-reduttasi ha parzialmente inibito la conversione del Nandrolone in DHN. Poiché il Nandrolone è molto più androgeno del DHN, l’inibizione di questo processo enzimatico ha mantenuto concentrazioni sieriche più elevate di Nandrolone nei tessuti dove la 5α-reduttasi è maggiormente espressa e la stimolazione ventrale della prostata è aumentata in modo significativo.
La maggior parte degli AAS utilizzati nel BodyBuilding e nel PowerLifting non sono potenti substrati per la 5α-reduttasi o non lo sono affatto. Ciò significa che Finasteride e Dutasteride non aiuteranno assolutamente a prevenire la caduta dei capelli se assunti durante un protocollo contenente AAS come Methandrostenolone, Trenbolone, Oxymetholone, Stanozololo, Drostanolone, Boldenone, Trestolone, Mesterolone, Methenolone, DHB ecc… .
Inoltre, non aiuterà a prevenire la caduta dei capelli con l’uso di SARM non steroidei androgeni come l’S23.
S-23
L’S-23 è un Modulatore Selettivo del Recettore degli Androgeni (SARM) sperimentale sviluppato da GTX come potenziale contraccettivo ormonale maschile. Si lega al recettore degli androgeni in modo più forte rispetto ai farmaci più vecchi come l’Andarina con un Ki di 1,7nM, è possiede una potenziale attività androgena superiore alla maggior parte dei SARM non steroidei.
Alla fine della giostra, l’unico AAS degno di nota che può essere attenuato nei suoi effetti androgeni a livello del cuoio capelluto mediante l’uso di Finasteride o Dutasteride è il Testosterone. E anche con il DHT sostanzialmente inibito con Finasteride, o quasi completamente eliminato con Dutasteride, il Testosterone ha ancora la sua androgenicità intrinseca e atrofizzerà i follicoli piliferi, soltanto in misura molto minore e più lenta.
Questo è il motivo per cui alcuni individui utilizzatori di Dutasteride con alopecia androgenetica aggressiva perdono ancora i capelli anche con zero DHT in circolo.
La soluzione in caso di uso di Nandrolone da parte di soggetti predisposti alla alopecia androgenetica, risulta essere l’uso del RU58841.
RU-58841
Il RU-58841, noto anche come PSK-3841 o HMR-3841, è un antiandrogeno non steroideo (NSAA) inizialmente sviluppato negli anni ’80 da Roussel Uclaf, l’azienda farmaceutica francese da cui ha preso il nome. In precedenza era oggetto di indagine da parte della ProStrakan (precedentemente ProSkelia e Strakan) per un potenziale utilizzo come trattamento topico per condizioni androgeno-dipendenti tra cui acne, perdita di capelli,[2] e crescita eccessiva dei capelli.[3][4][5][6]
Il composto è simile nella struttura all’NSAA RU-58642 ma contiene una catena laterale diversa.[7] Questi composti sono simili nella struttura chimica alla Nilutamide,[8] che è correlata a Flutamide, Bicalutamide ed Enzalutamide, che sono tutte NSAA.[9]
Il RU-58841 può essere sintetizzato sia sintetizzando la porzione idantoina che mediante accoppiamento arilico con 5,5-dimetilidantoina.[10] Il RU-58841 produce Cianonilutamide (RU-56279) e RU-59416 come metaboliti negli animali.[11]
La Cianonilutamide ha un’affinità relativamente bassa per il recettore degli androgeni, ma mostra una significativa attività antiandrogena negli animali.[11] Mentre il RU-59416 ha un’affinità molto bassa per il recettore degli androgeni.[11]
Fortunatamente, sembra che il RU58841 abbia un’affinità di legame equivalente o maggiore di quella del Testosterone per l’AR, quindi in realtà è abbastanza efficace nell’inibire l’attività androgena sia del debole DHN che del Testosterone e delle quantità residue di Nandrolone nel cuoio capelluto.
Conclusioni:
Mi sembra ovvio che il messaggio principale da portarsi a casa sia quello di evitare , la dove si voglia tamponare la perdita di capelli legata alla alopecia androgenetica, l’abbinamento di inibitori della 5α-reduttasi (vedi Finasteride o Dutasteride) quando nel protocollo di supplementazione farmacologica è presente il Nandrolone.
Se si desidera utilizzare un trattamento più efficace per arginare l’alopecia androgenetica durante un ciclo contenente Nandrolone, sarebbe opportuno considerare l’uso di un anti-androgeno topico di forte affinità di legame all’AR come il RU58841.
Battmann T, Bonfils A, Branche C, Humbert J, Goubet F, Teutsch G, Philibert D (January 1994). “RU 58841, a new specific topical antiandrogen: a candidate of choice for the treatment of acne, androgenetic alopecia and hirsutism”. J. Steroid Biochem. Mol. Biol.
Van Dort ME, Jung YW (April 2001). “Synthesis and structure-activity studies of side-chain derivatized arylhydantoins for investigation as androgen receptor radioligands”. Bioorg. Med. Chem. Lett. 11 (8): 1045–7.
Poulos GA, Mirmirani P (February 2005). “Investigational medications in the treatment of alopecia”. Expert Opin Investig Drugs. 14 (2): 177–84.
Leonard, Matthew J.; Lingham, Anthony R.; Niere, Julie O.; Jackson, Neale R. C.; McKay, Peter G.; Hügel, Helmut M. (2014). “Alternative synthesis of the anti-baldness compound RU58841”. RSC Adv. 4 (27): 14143–14148.
Jump up to:abc Cousty-Berlin D, Bergaud B, Bruyant MC, Battmann T, Branche C, Philibert D (October 1994). “Preliminary pharmacokinetics and metabolism of novel non-steroidal antiandrogens in the rat: relation of their systemic activity to the formation of a common metabolite”. J. Steroid Biochem. Mol. Biol.
Chi segue il sito e legge con attenzione i miei lavori, si ricorderà certamente che l’argomento della Anabolico:Androgeno ratio era già stato toccato nell’articolo di analisi dettagliata sul Methenolone. Visto che la questione alzò una non indifferente reazione da parte degli “irriducibili” del “ribattere con banalità”, e che la lettura di certi validi testi in lingua inglese sembra per i più ostica, ho deciso di trattare con minuzia di dettagli questo tanto dibattuto argomento. Ovviamente, chi vive di convinzioni basate sul nulla difficilmente potrà accettare quanto mi accingerò a riportare. Per tutti gli altri sarà un altra occasione per imparare qualcosa di nuovo e potenzialmente utile.
Ma andiamo avanti…
Tutti noi sappiamo che gli Steroidi Androgeni Anabolizzanti (AAS) hanno proprietà anaboliche e androgene, da cui il nome. In generale, per proprietà anaboliche si indica l’effetto di costruzione muscolare e l’effetto stimolante sulla densità minerale ossea (BMD). Gli altri effetti sono considerati effetti Androgeni, come l’impatto sulla ipertrofia prostatica, sullo stimolo del midollo osseo, sul cuore, sull’ipotalamo e sull’ipofisi, ecc. In generale, questi effetti sono considerati indesiderati. Ad esempio, una meta-analisi ha rilevato che un aumento dell’ematocrito (la % del volume di sangue occupato dagli eritrociti) è l’effetto avverso più frequente associato alla Terapia Sostitutiva del Testosterone (TRT).[1] Un altro problema degli androgeni è che sono in grado di indurre, in particolari circostanze, la crescita del cancro alla prostata. In quanto tale, una delle terapie utilizzate per il trattamento del cancro alla prostata è la terapia di deprivazione androgenica. Tuttavia, è importante notare che esiste un limite alla capacità degli androgeni di stimolare la crescita del cancro alla prostata. Ciò significa che, fino a una certa concentrazione, gli androgeni ne stimoleranno la crescita, ma al di sopra di essa avranno poco o nessun ulteriore effetto. Da qui sono nate alcune ipotesi tra le quali quella del “modello di saturazione dei recettori degli androgeni” [2], un modello dibattuto e tutt’altro che dimostrato. Comunque sia, questo effetto sulla riduzione dell’attività ipertrofica prostatica è in realtà qualcosa che sembra già avvenire a basse concentrazioni di Testosterone, nel intervallo basso classico del soggetto ipogonadico. Avremo comunque tempo di ritornare nuovamente su questo punto più tardi, quando parlerò di un rinomato test che viene utilizzato per valutare il rapporto tra potenza anabolica e androgena.
Lo scopo della Androgeno:Anabolico ratio è quello di fornire dati numerici al fine di dividere i diversi AAS in termini di potenza anabolica e androgena. Quindi, ad esempio, si potrebbe prendere il Testosterone come AAS di “paragone”, assegnandogli una Anabolico/Androgeno ratio di 100 e 100 (o solo 1). Quindi, attraverso alcuni esperimenti, viene determinato che un altro AAS ha un rapporto pari a 400:200 (o solo 2) Ciò implicherebbe che questo AAS è 4 volte più anabolico del Testosterone, pur essendo solo due volte più androgena.
Se le cose stessero in questo modo, allora, volendo ridurre al minimo il rischio di effetti collaterali androgeni, si potrebbe semplicemente scegliere un AAS con una Anabolico-Androgeno ratio molto favorevole e il problema non sussisterebbe. Tuttavia, ci sono così tanti problemi e variabili sia con il concetto stesso di Anabolico:Androgeno, ratio sia con il modo in cui esso è determinato sperimentalmente, che tutti questi rapporti che si trovano online o in letteratura sono praticamente inutili.
Il test di Hershberger
Un test molto comune utilizzato per determinare la Anabolico:Androgeno ratio è il cosiddetto test di Hershberger. Il test è stato descritto per la prima volta nel 1953 da Hershberger e dai suoi colleghi dell’Università del Wisconsin.[3] Come già accennato nell’articolo sul Methenolone, il test funziona come segue. Si prendono dei ratti e li si castra. La castrazione assicura che si abbia pochissimo Testosterone endogeno nell’animale e che ciò possa influenzare i risultati del test. Successivamente, si somministra l’AAS di cui si vuole conoscere il rapporto tra potenza anabolico e androgena all’animale. Successivamente si attende un po’ di tempo (8 giorni nel caso del originale test di Hershberger) e si procede con l’uccisione dei ratti trattati per sezionarli e pesarne il muscolo levator ani, la prostata ventrale e le vescicole seminali. L’aumento di peso de levator ani sarebbe quindi indicativo dell’attività anabolica dell’AAS, mentre quello della prostata ventrale e delle vescicole seminali sarebbe indicativo della sua attività androgena.
LA: levator ani.
Anche se questo test può sembra un metodo ragionevolmente valido, in esso vi sono una serie di problemi. Un primo punto su cui vorrei soffermarmi riguarda il muscolo levator ani, che, appunto, è il muscolo bulbocavernoso dorsale.[4] È un muscolo che fa parte del sistema riproduttivo maschile e quindi non dovrebbe essere considerato in alcun modo rappresentativo del muscolo scheletrico. È un muscolo fortemente androgeno-dipendente e dopo la castrazione subisce un tasso di diminuzione del peso simile a quello dell’atrofia da denervazione nei muscoli scheletrici.[5] Proprio questa informazione da sola garantisce già che il lato anabolico dell'”equazione” sia imperfetto. Un altro problema è che il muscolo bulbocavernoso dorsale e le vescicole seminali rispondono in modo diverso a una diminuzione della concentrazione di AAS all’interno del range fisiologico.[6] Di conseguenza, il rapporto determinato sperimentalmente dipenderà dalla dose utilizzata e dal momento in cui vengono effettuate le misurazioni. Ciò è ben illustrato nella figura sottostante tratta da una pubblicazione di van der Vies.[6] Durante i primi 3 giorni, la concentrazione di AAS (Nandrolone in questo caso) è abbastanza alta da stimolare la crescita sia delle vescicole seminali che del muscolo bulbocavernoso dorsale. Tuttavia, dopo tre giorni la concentrazione non è abbastanza elevata da sostenere questa crescita per le vescicole seminali, che diminuiscono nuovamente di dimensioni. Tuttavia, il muscolo bulbocavernoso è ancora sufficientemente stimolato per continuare a crescere di dimensioni. Pertanto, se determinassi il rapporto anabolico/androgeno il giorno 3, esso sarebbe molto diverso rispetto al risultato che rileverei se la misurazione venisse fatta il giorno 7, nonostante sia utilizzato lo stesso composto.
Scomparsa del Nandrolone Fenilpropionato dal deposito intramuscolare ed effetti sui pesi del muscolo bulbocavernoso dorsale e delle vescicole seminali. Figura tratta da van der Vies [6].
Ciò evidenzia anche che i diversi organi rispondono semplicemente in modo diverso a seconda della concentrazione della molecola. E anche se ci fosse un modo accurato per determinare una anabolico:androgeno ratio, estrapolarlo oltre le concentrazioni fisiologiche sarebbe completamente errato.
Un altro difetto è l’ipotesi che la crescita della prostata ventrale o delle vescicole seminali sia rappresentativa di tutti gli altri effetti androgeni. Non ci sono mai state prove a sostegno di questa ipotesi. I tessuti androgeni variano molto in risposta l’uno dall’altro e non ci si deve assolutamente aspettare che un tessuto risponda nella stessa misura di un altro. In effetti, ricordate quanto menzionato nell’introduzione sulla prostata? Gli androgeni sembrano già smettere di stimolare ulteriormente la crescita della prostata oltre l’intervallo ipogonadico basso. Infatti, il volume della prostata rimane invariato quando a uomini sani vengono somministrati 600mg di Testosterone Enantato (pari a 432mg di Testosterone) settimanalmente per 20 settimane.[7] Eppure sappiamo per certo che altri effetti collaterali androgeni iniziano a comparire quando il dosaggio comincia ad essere elevato! Comunque, questo mette in luce anche la questione del concetto stesso di rapporto tra potenza anabolica e androgena. Un singolo rapporto non è mai in grado di catturare le risposte differenziali dei vari tessuti sensibili agli androgeni o la complessità della risposta androgena all’interno di un tessuto specifico, per essere di valore. Diversi tessuti rispondono in modo diverso ad un AAS, come sarà mai possibile rappresentarlo con un singolo numero?
Naturalmente, il test di Hershberger viene eseguito sui ratti, non sugli esseri umani. È un altro errore è quello di presumere che i tessuti omologhi nell’uomo rispondano allo stesso modo osservato in un ratto trattato con AAS. L’intero test di Hershberger è semplicemente pieno di falle, e nonostante ciò viene attualmente utilizzato per lo screening di potenziali Modulatori Selettivi del Recettore degli Androgeni (SARM). Ad esempio, GlaxoSmithKline ha valutato la selettività tissutale del proprio SARM GSK2881078 utilizzando il classico test di Hershberger.[8]
GSK2881078
Test di affinità di legame relativo (RBA)
Mentre il test di Hershberger viene eseguito in un organismo vivente, i saggi di affinità di legame relativa (RBA) vengono eseguiti in una sorta di piastra di Petri. Viene quindi esaminata l’affinità di legame dei composti per il Recettore degli Androgeni (AR). In questo contesto, l’affinità di legame si riferisce alla forza con cui un AAS si lega all’AR. L’RBA mostra quindi quanto fortemente un AAS si lega all’AR rispetto ad un altro. O in altre parole: relativamente l’uno all’altro.
Il principio alla base è abbastanza semplice. Si prende un AAS di riferimento, comunemente il Methyltrienolone (R1811), e si misura la sua affinità di legame. Successivamente si misura l’affinità di legame di altri AAS e si esprimono i dati relativi all’AAS di riferimento. Quindi al Methyltrienolone viene assegnato un RBA di 1, essendo il composto di riferimento, e quindi se qualche altra molecola si lega con una potenza maggiore di due volte gli viene assegnato un RBA di 2. Allo stesso modo, se un’altra molecola si lega due volte più debolmente le viene assegnato un RBA di 0,5. Si potrebbero fare queste misurazioni in diversi tipi di cellule. Una che rappresenta il muscolo scheletrico e un altra che rappresenta in qualche modo i suoi effetti androgeni (ad esempio le cellule della prostata). Come tale, anche in questo caso si possono porre alcune delle stesse obiezioni del test Hershberger descritte sopra.
Ad ogni modo, l’immagine seguente raccoglie gli RBA di una selezione di AAS popolari misurati nei tessuti di ratto e coniglio.[9] Se dovessimo ottenere questi risultati, il Testosterone avrebbe un rapporto anabolico-androgeno più favorevole rispetto al Nandrolone. È un po’ l’opposto di quello che si osserva nei test di Hershberger. È anche un po’ sorprendente, dato che gli effetti androgeni del Testosterone sono amplificati nei tessuti che esprimono la 5α-reduttasi, a causa della conversione all’androgenicamente più potente DHT. Al contrario, l’azione androgena del Nandrolone è indebolita nei tessuti che esprimono questo enzima, a causa della conversione al meno potente androgeno Dihydronandrolone (DHN).[10]
Gli RBA di una selezione di AAS presi dal lavoro di Saartok et al. [9]. Il Methyltrienolone è servito come steroide di riferimento.
Un’altra cosa che questi dati rivelano in modo appropriato sono le differenze interspecie dei valori RBA. Nel muscolo di ratto, l’1α-methyl DHT si lega all’AR circa 3 volte più debolmente del Testosterone. Se si osservano i dati provenienti dall’analisi del muscolo di coniglio, fondamentalmente si vede un risultato contrario: 1α-methyl-DHT si lega con una forza all’incirca 3 volte maggiore all’AR come il Testosterone. L’estrapolazione da una specie animale a un’altra è (altamente) problematica, e quindi anche l’estrapolazione dal ratto, coniglio o qualsiasi altro animale, all’uomo.
Nel caso ti stia chiedendo perché il DHT dimostri un RBA così basso nel muscolo di coniglio e ratto, questo è probabilmente dovuto alla sua rapida degradazione nel tessuto muscolare. Il DHT costituisce un eccellente substrato per l’enzima 3α-HSD. Questo enzima lo scompone in 3α-androstanediolo, il quale si lega molto debolmente all’AR.[11] Questo accade anche negli esseri umani [12], e questo è uno dei motivi per cui non si vedono protocolli basati sull’uso di DHT.
Un ultimo punto che deve essere evidenziato è che l’affinità di legame non determina la potenza del AAS nel modulare anche l’espressione genica. Che è alla fine ciò che più interessa. Tuttavia, questo è possibilmente valutabile per via sperimentale. Si tratta del test del gene reporter responsivo agli androgeni (dosaggi biologici AR). Questi test biologici, per quanto ne so, sono stati inizialmente utilizzati per lo screening di nuovi androgeni di design nei campioni di urina per contrastare l’uso di doping. Un test biologico AR è essenzialmente in grado di dimostrare se un campione contiene qualcosa che riesce ad attivare il recettore degli androgeni e avviare la trascrizione genica. Per contrastare il doping, questo è molto utile. Dopotutto, puoi dimostrare che un campione di urina contiene qualcosa che attiva l’AR senza conoscere la struttura chimica del composto utilizzato.
Ad ogni modo, uno di questi test è stato sviluppato da un team di scienziati olandesi. [13] I ricercatori hanno utilizzato un test chiamato test biologico della LUciferasi attivata da sostanze chimiche reattive agli androgeni (AR CALUX). Hanno preso una linea cellulare di osteosarcoma umano e l’hanno co-trasfettata con l’AR umano e un gene reporter della luciferasi che è sotto il controllo trascrizionale degli elementi di risposta agli androgeni (ARE). Ciò significa che quando l’AR viene attivato, l’enzima luciferasi arriva all’espressione. Questo enzima produce bioluminescenza, o per dirla semplicemente: luce. E la luce può essere misurata. Quindi il grado di bioluminescenza è il grado in cui avviene l’attivazione del recettore degli androgeni.
I ricercatori hanno quindi proceduto a testare una varietà di AAS noti con il test biologico AR CALUX. Simile all’RBA, con esso si può calcolare la potenza relativa in termini di attivazione del recettore (REP). E non è solo stato fatto per l’AR, ma lo hanno fatto anche per il recettore del progesterone (PR), entrambe le isoforme del recettore degli estrogeni (ERα e ERβ) e il recettore dei glucocorticoidi (GR). Nella tabella seguente sono elencati gli REP di alcuni (popolari) AAS.
Gli REP di una selezione di AAS tratti dal lavoro di Houtman et al. [13]. Il DHT è servito come steroide di riferimento per il AR, ORG-2058 per il PR, l’Estradiolo per ERα/β e il Desametasone per GR.
Permettetemi di evidenziare il REP del Testosterone e del DHT per l’AR. Il REP del Testosterone è circa 5 volte inferiore al REP del DHT. Cosa ci dice questo? Semplicemente che quel DHT non è stato degradato enzimaticamente nella linea cellulare che hanno usato come sarebbe successo nel mondo reale se si fosse legato al AR del muscolo scheletrico. Il metabolismo che di solito avviene nel muscolo scheletrico non sembra quindi avvenire in questa linea cellulare. Questo problema invalida i risultati di questo test biologico per quegli AAS che sono metabolizzati nel muscolo scheletrico, come il DHT, ma probabilmente anche il Methenolone (Primobolan). Un altro problema è che l’espressione genica è complessa (e dirlo è un eufemismo). L’AR regola un vasto numero di geni. Se due composti aumentano la trascrizione genica di un determinato gene in misura simile, non significa necessariamente che questi due composti modulino in modo comparabile la trascrizione genica di altri geni. Sicuramente non sarebbe sorprendente se ci fosse una correlazione in un modo o nell’altro, ma questi test biologici dipingono solo un quadro approssimativo. Anche se è probabile che questa immagine approssimativa sia più accurata di quella degli RBA. Tuttavia, fino ad oggi, i biotest AR non sono stati eseguiti in più linee cellulari di vari tessuti (sensibili agli androgeni) per fornirci “nuovi” rapporti tra il potenziale anabolico e androgeno.
Conclusioni
Sia il test di Hershberger che gli studi che valutano gli RBA dell’AAS in vari tessuti sono irrimediabilmente inaffidabili. Inoltre, il concetto di numero che cattura la complessità delle proprietà anabolizzanti e di quelle androgene dovrebbe essere abbandonato. Un singolo rapporto è semplicemente incapace di descrivere le risposte differenziali di vari tessuti agli androgeni, così come la complessità della risposta androgena all’interno di un tessuto specifico, per essere di valore. Forse sarebbe più appropriato un “profilo di attività” che descriva l’azione androgenica su base tissutale. Qualcosa di simile è stato proposto per descrivere come dovrebbe essere un SARM ideale per il trattamento di una condizione specifica. Tuttavia, è estremamente difficile quantificare l’azione androgena per tessuto, se non impossibile. Forse i biotest AR eseguiti su linee cellulari di tessuti di interesse potrebbero avere un valore clinico predittivo. Alla fine, dopo tutto, sono necessari studi clinici per dimostrare (il grado di) eventi avversi che si verificano con l’uso di un determinato composto.
Ovviamente, e lo dico per i tordi che affermano “e allora perchè con l’Oxandrolone gli effetti androgeni sono bassi come descritto dalla sua anabolico:androgeno ratio?” Bambino caro, l’Oxandrolone, come altri composti steroidei testati nel corso degli ultimi sessant’anni, hanno passato trial clinici dove gli effetti sono stati documentati anche nel caso di trattamento di donne in pre e post-menopausa. Ma non sono mai stati effettuati confronti di potenziale ed estrapolate ratio anabolico:androgeno. Semplicemente hanno osservato il miglioramento clinico dei pazienti trattati per svariate patologie e condizioni come, ad esempio, soggetti ustionati o gravemente sottopeso (vedi malati di HIV). Sono state effettuate biopsie, controlli della ritenzione d’azoto ma nessun test comparativo. Così facendo, puoi basarti sul grado di trofismo indotto dalla molecola e dal grado di espressione dei caratteri androgeni, ma, lo ribadisco, nessuna ratio di confronto!
Ricordate, inoltre, che per i soggetti sensibili l’espressione degli effetti androgeno-correlati avviene anche con le molecole con la ratio più bassa… Fatevene una ragione…
Gabriel Bellizzi
Riferimenti:
Calof, Olga M., et al. “Adverse events associated with testosterone replacement in middle-aged and older men: a meta-analysis of randomized, placebo-controlled trials.” The Journals of Gerontology Series A: Biological Sciences and Medical Sciences 60.11 (2005): 1451-1457.
Morgentaler, Abraham, and Abdulmaged M. Traish. “Shifting the paradigm of testosterone and prostate cancer: the saturation model and the limits of androgen-dependent growth.” European urology 55.2 (2009): 310-321.
Hershberger, L. G., Elva G. Shipley, and Roland K. Meyer. “Myotrophic activity of 19-nortestosterone and other steroids determined by modified levator ani muscle method.” Proceedings of the Society for Experimental Biology and Medicine 83.1 (1953): 175-180.
Hayes, Keith J. “The so-called ‘levator ani’ of the rat.” European Journal of Endocrinology 48.3 (1965): 337-347.
Gori, Zina, C. Pellegrino, and Maria Pollera. “The castration atrophy of the dorsal bulbocavernosus muscle of rat: an electron microscopic study.” Experimental and molecular pathology 6.2 (1967): 172-198.
Van der Vies, J. “Implications of basic pharmacology in the therapy with esters of nandrolone.” European Journal of Endocrinology 110.3_Suppla (1985): S38-S44.
Bhasin, Shalender, et al. “Effect of testosterone supplementation with and without a dual 5α-reductase inhibitor on fat-free mass in men with suppressed testosterone production: a randomized controlled trial.” Jama 307.9 (2012): 931-939.
Neil, David, et al. “GSK2881078, a SARM, produces dose-dependent increases in lean mass in healthy older men and women.” The Journal of Clinical Endocrinology & Metabolism 103.9 (2018): 3215-3224.
Saartok, Tönu, Erik Dahlberg, and JAN-ÅKE GUSTAFSSON. “Relative binding affinity of anabolic-androgenic steroids: comparison of the binding to the androgen receptors in skeletal muscle and in prostate, as well as to sex hormone-binding globulin.” Endocrinology 114.6 (1984): 2100-2106.
Bergink, E. W., et al. “Comparison of the receptor binding properties of nandrolone and testosterone under in vitro and in vivo conditions.” Journal of steroid biochemistry 22.6 (1985): 831-836.
Jin, Yi, and Trevor M. Penning. “Steroid 5α-reductases and 3α-hydroxysteroid dehydrogenases: key enzymes in androgen metabolism.” Best Practice & Research Clinical Endocrinology & Metabolism 15.1 (2001): 79-94.
Becker, H., et al. “In vivo uptake and metabolism of 3H-testosterone and 3H-5α-dihydrotestosterone by human benign prostatic hypertrophy.” European Journal of Endocrinology 71.3 (1972): 589-599.
Houtman, Corine J., et al. “Detection of anabolic androgenic steroid abuse in doping control using mammalian reporter gene bioassays.” Analytica chimica acta 637.1-2 (2009): 247-258.
Torno a parlare dell’argomento AAS e sistema immunitario con questa piccola parentesi incentrata sulla risposta alle possibilità di sviluppo di eventi avversi in seguito a contaminazione virale, soprattutto da ceppi delle famiglia dei coronaviridae della cui famiglia fa parte l’ormai famoso SARS-CoV-2.
Come ben sapete, io ed altri divulgatori d’oltre oceano abbiamo sottolineato come l’uso di dosi sovrafisiologiche di AAS possano portare ad uno stato di lieve immunosoppressione, tale da poter aumentare il rischio di sviluppare sintomatologia da contagio virale. Secondo recenti studi scientifici, probabilmente avevamo ragione.[1]
Attività androgenica e SARS-CoV-2 (e coronaviridaee)
Le donne con la PCOS hanno livelli elevati di androgeni. Se queste donne sono infettate dal SARS-CoV-2, come da altri ceppi della stessa famiglia, e non solo, i sintomi sembrano essere più gravi rispetto alle donne senza la PCOS.[2]
Gli inibitori della 5α-reduttasi riducono fortemente la conversione del Testosterone nel più androgeno DHT. Se si trattano gli uomini con inibitori della 5α-reduttasi, sembra che l’infezione da SARS-CoV-2 risulti meno grave nella sintomatologia.[3,4]
Una minore attività androgenica nei soggetti di sesso maschile affetti da SARS-CoV-2 sembrano quindi manifestare una sintomatologia lieve. Alcuni ricercatori sospettano che ciò sia dovuto al fatto che gli androgeni attivano l’enzima TMPRSS2. Questo enzima fa sì che le cellule presentino un numero maggiore di recettori ACE2 sulla superficie della membrana fosfolipidica. Come ormai noto, i recettori ACE2 sono il bersaglio di legame per le proteine spike del SARS-CoV-2. Grazie al legame con tali proteine di membrana il virus in questione può penetrare all’interno della cellula e dare avvio ai processi di replicazione.
Meccanismi di infezione cellulare (cardiomiocita) da parte del SARS-CoV-2. Come si può notare, vi è connessione di attività tra l’enzima TMPRSS2 e il recettore ACE2, il legame della proteine spike del virus.
Il caso studio “Oxandrolone e Proxalutamide”
In base a quanto detto in precedenza, significa anche che gli atleti che usano AAS hanno maggiori probabilità di sviluppare una sintomatologia maggiore in seguito ad infezione da SARS-CoV-2? Sembrerebbe di sì, e gli endocrinologi brasiliani dell’Università federale di San Paolo hanno riportato una loro osservazione su BMJ Case Reports.
Quindi, secondo quanto emerso dalle loro osservazioni, gli utilizzatori di AAS sono più vulnerabili all’infezione e sviluppo di sintomatologia acuta da SARS-CoV-2.
I ricercatori si basano sul caso di un bodybuilder di 28 anni con una grave infezione da SARS-CoV-2. Il caso è stato notevole, perché l’uomo non aveva di base fattori di rischio. I genitori del soggetto avevano contratto il virus, ma la sintomatologia non è stata grave. Perché allora il bodybuilder si è ammalato gravemente? Quando gli endocrinologi lo hanno esaminato, è emerso che aveva assunto 40mg di Oxandrolone al giorno per un mese.
I medici hanno convinto l’uomo a interrompere l’assunzione di Oxandrolone prescrivendogli una terapia a base dell’anti-androgeno Proxalutamide. Il Proxalutamide è anche un farmaco sperimentale per il trattamento dell’infezione da SARS-CoV-2 negli uomini.[5]
I medici hanno inizialmente somministrato al bodybuilder una singola dose di 600mg di Proxalutamide. Quindi al soggetto è stata prescritta l’assunzione di 200mg/die del anti-androgeno per una settimana.
Già 24 ore dopo la prima somministrazione, il bodybuilder ha riferito di sentirsi significativamente meglio. Allo stesso tempo, vari marker ematici erano visibilmente migliorati in quel breve periodo di tempo.
L’immagine schematica descrive il ruolo del enzima TMPRSS2 nell’infezione da SARS-CoV-2 e l’espressione androgeno-mediata di ACE2 e di TMPRSS2 che potrebbero potenzialmente essere modulati con l’uso di molecole anti-androgene.
Conclusione
La più alta percentuale di infezioni e manifestazione di sintomatologia severa possibilmente riscontrabile degli utilizzatori di AAS era cosa già conosciuta e constata nel mio articolo dedicato. Detto ciò, adesso abbiamo la conferma che la manifestazione della sintomatologia conseguente all’infezione da SARS-CoV-2 può essere maggiore negli utilizzatori di AAS. E, di certo, questo interessa tutti i virus che possono dare una risposta sintomatologica di varia entità, non solo il SARS-CoV-2 o altri coronaviridaee.
DISCLAIMER: Il presente articolo è a solo scopo educativo, di intrattenimento e informativo. Non rappresenta in alcun modo una forma di incitamento all’uso/abuso di sostanze dopanti. L’autore ed il sito, per tanto, è esentato da qualsiasi responsabilità dipendente dalla libera scelta individuale.
Introduzione ad un dilemma…
Chiunque frequenti l’ambiente del Bodybuilding e del Fitness avrà letto o sentito almeno una volta nella vita espressioni del genere “se mi dopassi sarei anche io così [indicando Flex Wheeler]” o “ho provato di tutto e senza farmaci non riuscirò ad ottenere risultati”. Andando poi ad approfondire la storia di ognuno di questi soggetti si scopre in percentuale quasi assoluta che si tratta di individui nella norma (o al di sotto) frustrati e/o con personalità deboli, speranzosi omini che attendono placidamente che accada una svolta miracolosa nella loro banale e piatta esistenza e, cosa molto importante, con il minimo dello sforzo (meglio se nessuno).
Nella mia esperienza come ricercatore e operatore nel campo della cultura fisica in qualità di Preparatore Atletico, ho assistito a innumerevoli casi in cui un soggetto aspirava al miglioramento della propria composizione corporea trascurando, consciamente o inconsciamente, le basi fondamentali rappresentate da Nutrizione e Allenamento baipassandole in vista della possibile prescrizione di una pillola miracolosa capace di renderlo/a possessore della forma fisica ambita.
Tralasciando l’ovvio ragionamento che spinge ogni essere umano dotato di un minimo d’intelletto verso la comprensione che la genetica è il blocco d’argilla sul quale si va ad operare, ma le sue qualità e difetti sono presenti in modo eterogeneo nella popolazione mondiale, e ciò non è modificabile nemmeno con la farmacologia più oculata, quando ci si trova davanti al bivio tra “pillola rossa” (PEDs) e “pillola blu” (drug free) bisogna essere pienamente consapevoli non solo del fattore illegalità ma del fattore conoscitivo. Purtroppo, la politica del terrore ha operato in modo fallimentare nel goffo intento di allontanare dalla scelta “rossa”, e ciò si è tradotto in un numero sensibile di soggetti abusatori con tutte le conseguenze cliniche derivanti.
Se un individuo non ha raggiunto un livello di maturità sportiva tale da conferirgli una gestione corretta della nutrizione e della periodizzazione allenante (gestione delle variabili volume, intensità, densità ecc…), è molto meglio per lui/lei rivedere i suoi programmi e scegliere ancora la “pillola blu”. Capita, a volte, di incontrare persone decise ad intraprendere la via del “lato oscuro” che, dopo una approfondita chiacchierata sulla gestione dei suddetti fattori, rivede le proprie posizioni.
Per tutti coloro i quali sono immersi nel dilemma della scelta, vi espongo alcuni punti per rendere l’eventuale decisione meno rischiosa anche se pur sempre illegale nel “bel paese”…
“Pillola blu o pillola rossa?” I punti da tenere in considerazione per una scelta consapevole:
#1 Raggiungere una adeguata maturità sportiva
Per “maturità sportiva”, in particolare riferimento al BodyBuilding, si intende la capacità del atleta di sapersi alimentare e allenare correttamente con piena gestione delle proprie potenzialità fisiologiche/genetiche. Questa è la base, se viene a mancare ciò non solo la vostra esperienza finirà per deludervi e rendervi ancora di più dei frustrati, ma potrebbe rovinosamente portarvi ad un abuso cronico a senso inesorabilmente negativo…
#2 I PEDs non faranno miracoli
Una cosa da tenere bene a mente, e questo non dovrebbe interessare solo gli aspiranti “doped”, è che l’uso di PEDs non renderà diversi da ciò che rientra nelle potenzialità espressive del proprio patrimonio genetico. Certamente le caratteristiche genetiche verranno “iperespresse”, nel bene e nel male, dall’uso di PEDs ma non vi sarà nessun miracolo! Migliorerete ma non sarete ne più ne meno di ciò che potete essere!
Un esempio per capire come la base genetica faccia la differenza anche con protocolli che, ad oggi, spesso non raggiungono nemmeno i livelli del “bridge” più soft..
#3 Ridurre la percentuale di grasso corporeo
Il tessuto adiposo rappresenta uno dei siti dove il Testosterone, ed altri AAS soggetti all’aromatizzazione, viene convertito in Estradiolo. Soggetti con percentuali di grasso corporeo elevate vedrebbero una alterazione marcata della Testosterone:Estradiolo ratio a favore della componente estrogenica, con conseguenze quali alterazione del comportamento sessuale (impotenza, difficoltà nel raggiungere e/o mantenere l’erezione), ritenzione idrica, accumulo di grasso con modello femminile e ginecomastia. E no, l’uso di DHT derivati o di SARM non steroidei senza una base di Testosterone non risolverebbe il problema o, per lo meno, porterebbe ad altre conseguenze negative, che pur non comprendendo, per esempio, ritenzione idrica e ginecomastia, interesserebbero l’attività sessuale e la condizione psichica del soggetto trattato. [1]
Schema esemplificato del processo di aromatizzazione degli androgeni aumentati in un soggetto con percentuale di grasso corporeo alta.
Allo stesso tempo, i rischi cardiovascolari della somministrazione di AAS- come il possibile aumento esponenziale del Ematocrito, l’aumento del LDL e Trigliceridi a discapito di una riduzione del HDL, e l’aumento della pressione sanguigna – sarebbero già presenti in certa misura quando la body fat è già alta e sarebbero quindi soggetti ad un repentino aggravamento.
Se la percentuale di grasso è relativamente alta, si dovrebbe prima di tutto considerare di migliorare la composizione corporea con una adeguata routine alimentare e allenante (senza farmaci) prima di iniziare solo a pensare all’uso di AAS. Sicuramente ciò renderà la scelta più efficace e meno rischiosa.
Nel caso fosse necessario sottolinearlo, no, non è saggio nemmeno utilizzare agenti PEDs a fini lipolitici e/o antiadipogenici e/o termogenici (compresi gli Ormoni Tiroidei). A meno che non siate affetti da ipotiroidismo, e in questo caso la terapia vi dovrebbe essere stilata dal vostro medico, per ridurre in modo sensibile la body fat non sono necessari i farmaci!
#4 Controllare se si ha una storia familiare di trombosi (o qualsiasi altra malattia cardiovascolare)
Molte malattie cardiovascolari hanno una componente di base genetica. Uno stile di vita sano può ridurne sensibilmente la loro insorgenza, ma l’uso di AAS può causare l’attivazione di specifici geni implicati nella comparsa di malattie cardio-circolatorie. Caratteristico dell’interazione tra AAS e geni specifici è un caso studio ben documentato che ricercatori americani hanno pubblicato sul “Blood Coagulation & Fibrinolysis”.[2]
Trombosi venosa
Oltre all’attivazione genica diretta dagli AAS, e nociva per il sistema cardio-circolatorio, vi sono altre condizioni negative innescate dall’uso/abuso di Steroidi Anabolizzanti, e di altri PEDs, come, per esempio, l’aumento del tasso di coagulazione, l’incremento eccessivo dell’Ematocrito con aumento pressorio, rigidità dell’endotelio vascolare con perdita di efficienza strutturale e aumento della pressione ematica con incremento delle possibilità di danno strutturale dei componenti del sistema interessato.
#5 Inserire delle sedute di allenamento cardio prima, durante e dopo l’uso di PEDs
Un moderato allenamento cardiovascolare è sicuramente una delle migliori strategie preventive contro la comparsa di malattie cardio-circolatorie. Tale tipologia di allenamento può portare un miglioramento e/o riduzione delle alterazioni lipidiche ematiche del praticante, fornendo un, seppur minimo, tampone all’azione negativa degli AAS e SARM non steroidei sui livelli di LDL (aumento), Trigliceridi (aumento) e HDL (diminuzione). Secondo quanto riportato da una interessante review del 2013, l’abbinamento di sedute cardio e in sala pesi possono avere una azione additiva benefica sui livelli di LDL, Trigliceridi e HDL.[3]
Risulta interessante anche quanto emerso da alcuni studi su animali a seguito dei quali si è osservato un significativo grado di protezione dato dall’allenamento cardio negli esemplari trattati con AAS.[4]
#6 Assicurarsi di rimanere ben idratati
Oltre ad agevolare il mantenimento di un Ematocrito migliore, una buona idratazione risulta positiva sulla pressione di lavoro renale nel filtraggio del sangue. Diversi AAS come il Trenbolone e i metilati in C-17 presentano una particolare resistenza metabolica che, oltre a causare un aumentato stress epatico, può portare ad una sofferenza renale sfociabile nel patologico. Si è osservato come una combinazione di AAS, dieta iperproteica e supplementazione di Creatina possa aumentare l’incidenza di problemi renali.[5] In un soggetto in fisiologia, la sola dieta ad altro contenuto proteico e la supplementazione di Creatina non hanno mostrato nessun grado di pericolosità, soprattutto sul breve/medio termine.
#7 Non usare “droghe ricreative”
A livello globale, il numero di decessi tra gli abusatori di AAS è in aumento. Alcuni, troppo superficialmente, dicono che questo sia dovuto al fatto che sempre più uomini e donne usano AAS, ma questa è solo una spiegazione dozzinale e limitata. Il sospetto ricade soprattutto sulle modalità di approccio dei consumatori di AAS: i dosaggi sono drammaticamente aumentati e un numero crescente di individui combina PEDs con “droghe ricreative”. Ed è su questi due ultimi punti che risiede la spiegazione principale dell’aumento statistico prima menzionato. Soprattutto la combinazione di PEDs e le così dette “droghe ricreative” risulta essere probabilmente un fattore significativo, come evidenziato alcuni anni fa da ricercatori australiani. Nel loro studio sono state analizzate tutte le morti documentate tra i consumatori di AAS a Sydney tra il 1997 e il 2012, scoprendo che le droghe ricreative come la cocaina avevano avuto un ruolo nella schiacciante molteplicità dei casi. Dagli studi sugli animali ora sappiamo della possibilità che la co-assunzione di un AAS come il Nandrolone con la cocaina vede moltiplicati gli effetti cardiotossici rispetto ai singoli composti.[6] E secondo studi in vitro la combinazione di Testosterone e cocaina aumenterebbe la possibilità di formazione di coaguli nel flusso ematico. [7]
#8 Corretta modalità di iniezione e herpes labiale
Gli utilizzatori di AAS a volte sviluppano ascessi, ma non sempre dovuti alla bassa qualità dei prodotti utilizzati.
Alcuni medici ritengono che gli utilizzatori di AAS dovrebbero effettivamente ricevere una formazione sulle tecniche di iniezione corrette, onde evitare embolie oleose o ascessi per cattiva gestione igienica della procedura. [8]
Molti utilizzatori ancora non sanno che disturbi come l’herpes labiale rendono le iniezioni ancora più rischiose. Perchè? Il virus che causa l’herpes labiale, come altri patogeni, riduce l’efficienza del sistema immunitario, fornendo così terreno fertile per infezioni batteriche i cui microorganismi scatenanti vengono inoculati nel corpo del soggetto attraverso l’iniezione in modo diretto o indiretto.
#9 Non fare affidamento sugli integratori
Secondo un buon numero di studi svolti su animali, alcuni integratori proteggono dagli effetti collaterali degli AAS. Secondo alcune ricerche, la Taurina, la Vitamina C ed E proteggono i testicoli durante un ciclo e la vitamina C e il cacao proteggono la prostata.
L’utilità dei risultati provenienti da questi studi è limitata per tre motivi:
A. gli animali da laboratorio non sono esseri umani, e
B. le dosi utilizzate e rapportate ad un essere umano sono quasi sempre molto inferiori rispetto a quelle utilizzate dai “doped”, e
C. la ricerca in campo psicologico mostra che l’uso di integratori stimola comportamenti rischiosi e malsani. I supplementi fanno pensare agli utilizzatori di essere invulnerabili e di non dover comportarsi in modo sano ed attento.[9]
Gli integratori possono aiutare a creare una mentalità che non si dovrebbe avere da utilizzatore consapevole di AAS.
Ovviamente, alcuni supplementi “protettivi” utilizzati dai soggetti meglio informati hanno un potenziale di “tamponare” in modo discreto alcune alterazioni legate all’uso di AAS e SARM come, ma non limitato a, Riso Rosso fermantato (controllo lipidico) [10], Silimarina (epatoprotezione), NAC (epatoprotezione) [11], Niacina (controllo lipidico) ecc…
#10 Ridurre al minimo (se non eliminare) il consumo di alcolici
Potrebbe sembrare un indicazione superflua ma non lo è.
L’abuso di alcol è indubbiamente uno dei problemi sociali più diffusi. Uno dei problemi correlati all’abuso di alcol e l’epatopatia alcolica. Questo stato patologico è derivante da un processo infiammatorio progressivo ai danni del fegato legato al consumo eccessivo di alcolici. È una malattia a più stadi. La steatosi provoca un ingrossamento del fegato causato da un accumulo di trigliceridi, spesso senza sintomi per molto tempo. I rischi correlati sono la steatosi (fegato grasso), l’epatite alcolica e la cirrosi epatica. Il rapporto con l’alcolismo è complesso. Non tutti i bevitori, infatti, hanno danni al fegato, anche se sono altamente probabili. La causa è da rinvenire in una trasformazione dell’alcol (etanolo) in sostanze tossiche che danneggiano il fegato in maniera irreversibile e cronica, con un rischio elevato di insufficienza epatica e di cancro, fino alla necessità di un trapianto di fegato.
In acuto, invece, l’alcol può essere una causa di alterazione delle transaminasi ma non si può sapere se e con quale modalità si potrebbero innalzare: dipende molto dalla risposta individuale dell’organismo. In caso di stress preesistente, di causa iatrogena e/o alimentare, si può presentare una alterazione significativa. [12]
Il primo caso è una consequenziale possibile se eventi stressori concomitanti si presentano in cronico. Ed è semplice giungere alla conclusione che l’uso di AAS, specie se metilati, possa comportare un aumentato stress epatico che potrebbe degenerare in peliosi epatica, cirrosi ecc…
Che siate “doped” o “natural”, per ragioni legate e non, dovreste evitare di consumare più di 25g per gli uomini, o 12,5g per le donne, di Etanolo al giorno.
#11 Sottoporsi a regolari controlli medici pre, intra e post utilizzo
Il monitoraggio della salute dovrebbe essere la base fondante del comportamento del utilizzatore consapevole e minimamente attento ai potenziali rischi nei quali potrebbe imbattersi.
Gli esami di controllo sono i seguenti:
Esami ematici e delle urine (comprendenti il quadro ormonale secondo necessità);
Elettrocardiogramma ogni 6 mesi circa;
Elettrocardiogramma sotto sforzo (prima di iniziare);
Ecocardiogramma ogni 6 mesi circa;
Coronarografia ogni 6 mesi circa;
Monitoraggio della pressione ematica;
TAC addome completa ogni 6 mesi circa.
Ovviamente, ogni accertamento , al di la degli esami ematici, deve essere gestito in base alle esigenze soggettive, caratteristiche e tipo di PEDs utilizzati.
#12 Essere seguiti da personale qualificato
Fin troppa gente è stata salutisticamente deturpata da gorilla di spogliatoio a mala pena consapevoli dell’esistenza dei macronutrienti e che, nonostante ciò, si sono improvvisati farmacisti. Donne divenuti uomini e uomini divenuti simili a cagne in calore per via di orrende ginecomastie. Evitate il fai da te e l’affidarsi a semianalfabeti … la somaticità sopra la norma è cosa diversa dall’intelligenza e alla competenza in biologia, biochimica e farmacologia… senza offesa per tutti quelli che “io mi facevo e ho vinto! Senzia scienzia!” …
#13 Pensare seriamente al post ciclo prima del ciclo
Molti aspiranti “doped” non considerano il fattore post ciclo. La maggior parte di loro è convinta che la PCT sarà una facile soluzione alla sottoregolazione dell’Asse HPT, ma in realtà non è proprio così. Esistono diversi casi studio che mostrano come gli ex utilizzatori abbiano spesso livelli di Testosterone inferiori rispetto al pre-utilizzo anche a distanza di anni dal cessato uso di AAS. Sembra che i fattori che aumentano le possibilità e il grado di tale effetto sul lungo termine siano:
Tempo di somministrazione;
Età
Molecole utilizzate (con maggiore impatto negativo dato dai19-norsteroidi come il Nandrolone per via della lunga permanenza dei metaboliti nel sistema).
Tutto ciò è indipendente dalla qualità della PCT, anche se essa può avere dei riscontri positivi specie nel primo periodo di stacco dagli AAS. Le alterazioni ormonali legate ad una alterazione dell’Asse HPT comprendono depressione, ansia, bassa libido, difficoltà nel raggiungere e mantenere l’erezione, stanchezza cronica ecc…
Per questa ragione molti scelgono di entrare in TRT (Terapia Sostitutiva del Testosterone) dopo il primo ciclo.
Quale conclusione?…
Se mai non dovesse bastare il disclaimer, questo articolo non rappresenta in alcun modo un consiglio e, ne tanto meno, un incitamento all’uso di sostanze dopanti! E’ semplicemente a fine divulgativo con l’obbiettivo di far comprendere a più persone possibili che la scelta di intraprendere coscientemente certe pratiche (illegali) necessita di una sufficiente (e veritiera) conoscenza del argomento.
Quindi? Leggete e comprendete correttamente ciò che ho riportato in sintesi fruibile ad un largo pubblico… Pensate prima di tutto ad alimentarvi e allenarvi in modo ottimale!
La conoscenza della Verità rende liberi dalla cattiva informazione, dagli strumenti commerciali e dal relativismo… Negarla è semplice e pericolosa manifestazione di profonda ignoranza… di VERO NEGAZIONISMO!
Se avete una buona conoscenza della lingua inglese e volete approfondire l’argomento PEDs e Sport, potete leggere il libro ANABOLICS 11th Edition di William Llewellyn
Sfogliando superficialmente la letteratura medica si può giungere all’erronea conclusione secondo cui gli effetti collaterali consequenziali all’uso/abuso di AAS si limitino principalmente alla sfera fisica e, quindi, organica e sistemica. Ma andando ad approfondire la ricerca in merito ci si imbatte in non pochi casi studio i quali non riportano solamente i classici effetti collaterali correlati agli AAS: aumentato rischio di malattie cardiovascolari, disturbi endocrinologici e, se si parla soprattutto di AAS orali metilati in C-17, anomalie epatiche. Un altro effetto collaterale dovrebbe essere preso in considerazione, anche perchè potrebbe peggiorare, e non di poco, la qualità della vita dell’utilizzatore; sto parlando degli effetti collaterali di natura psicologica. Nel 2019, i ricercatori dell’Università di Oslo hanno pubblicato i risultati di una loro ricerca nella quale si evidenziava che gli effetti collaterali più comuni degli AAS non sono fisici, ma di natura psicologica.[1]
Prima di trattare lo studio, però, è necessario comprendere la correlazione tra cervello e attività steroidea.
I Neurosteroidi
I neurosteroidi, noti anche come steroidi neuroattivi, sono steroidi endogeni o esogeni che alterano rapidamente l’eccitabilità neuronale attraverso l’interazione con canali ionici ligando-dipendenti e altri recettori della superficie cellulare. [2] [3] Il termine neurosteroide è stato coniato dal fisiologo francese Étienne-Émile Baulieu e si riferisce agli steroidi sintetizzati nel cervello. [4] [5] Per essere più precisi, il termine steroide neuroattivo si riferisce agli steroidi che possono essere sintetizzati nel cervello, o sono sintetizzati da una ghiandola endocrina, partendo dalla quale raggiungono il cervello attraverso il flusso sanguigno e hanno effetti sulla funzione cerebrale. [6] Il termine steroidi neuroattivi è stato coniato per la prima volta nel 1992 da Steven Paul e Robert Purdy. Oltre alle loro azioni sui recettori della membrana neuronale, alcuni di questi steroidi possono anche esercitare effetti sull’espressione genica attraverso i recettori degli ormoni steroidei nucleari. I neurosteroidi hanno una vasta gamma di potenziali applicazioni cliniche dalla sedazione al trattamento dell’epilessia [7] e delle lesioni cerebrali traumatiche. [8] [9] Il Ganaxolone, un analogo sintetico del neurosteroide endogeno Allopregnanolone, è in fase di studio per il trattamento dell’epilessia. [10]
Dr. Étienne-Émile Baulieu
I neurosteroidi sono sintetizzati ovviamente dal colesterolo, che viene convertito in Pregnenolone e poi in tutti gli altri steroidi endogeni. I neurosteroidi sono prodotti nel cervello dopo la sintesi locale o per conversione di steroidi surrenali di derivazione periferica o steroidi gonadici. Si accumulano soprattutto nelle cellule gliali mieliniche, dal colesterolo o dai precursori steroidei importati da fonti periferiche. [11] [12] La 5α-reduttasi di tipo I e la 3α-idrossisteroide deidrogenasi sono coinvolte nella biosintesi dei neurosteroidi inibitori, mentre la 3β-idrossisteroide deidrogenasi e l’idrossisteroide sulfotransferasi sono coinvolte nella produzione di neurosteroidi eccitatori. [4]
3α-Idrossisteroide deidrogenasi (3α-HSDoaldo-keto reductasi family 1 member C4)
Sulla base delle differenze di attività e struttura, i neurosteroidi possono essere ampiamente classificati in diversi gruppi principali. [4]
Neurosteroidi inibitori
Questi neurosteroidi esercitano azioni inibitorie sulla neurotrasmissione. Agiscono come modulatori allosterici positivi del recettore GABAA (in particolare isoforme contenenti subunità δ) e possiedono, in nessun ordine particolare, attività antidepressiva, ansiolitica, riducente lo stress, gratificante, [13] prosociale, [14] antiaggressiva, [15] prosessuale, [13] sedativa, pro-sonno, [16] cognitiva e con disturbi della memoria, analgesica, [17] effetti anestetici, anticonvulsivanti, neuroprotettivi e neurogeni. [4] I principali esempi includono il Tetraidrodeossicorticosterone (THDOC), l’Androstano 3α-androstandiolo, il Colestano colesterolo e il Pregnanolone (Eltanolone), Allopregnanolone (3α, 5α-THP). [18] [19]
Tetrahydrodeoxycorticosterone
Neurosteroidi eccitatori
Questi neurosteroidi hanno effetti eccitatori sulla neurotrasmissione. Agiscono come potenti modulatori allosterici negativi del recettore GABAA, modulatori allosterici deboli positivi del recettore NMDA e / o agonisti del recettore σ1 e per lo più hanno effetti antidepressivi, ansiogeni, cognitivi e di potenziamento della memoria, convulsivi, neuroprotettivi e neurogenici . [4] I principali esempi includono il Pregnenolon Solfato (PS), l’Epipregnanolone e l’Isopregnanolone (Sepranolone), gli androstani Deidroepiandrosterone (DHEA; Prasterone) e il Deidroepiandrosterone Solfato (DHEA-S; Prasterone Solfato) e il Colestano 24S-Hydroxycholesterol (Sidrossicol) (Selettivo per il recettore NMDA; molto potente). [20]
Sintesi del 24S-hydroxycholesterol dal colesterolo, catalizzato dall’enzima CYP46A1.
Feromoni
I feromoni sono neurosteroidi che influenzano l’attività cerebrale, in particolare la funzione ipotalamica, tramite l’attivazione delle cellule del recettore vomeronasale. [21] [22] [23] Includono androstani Androstadienol, Androstadienone, Androstenol e Androstenone e Estrane Estratetraenolo.
Androstenone (5α-androst-16-en-3-one)
Altri neurosteroidi
Alcuni altri steroidi endogeni, come il Pregnenolone, [24] Progesterone, [25] [26] Estradiolo, [6] e il Corticosterone sono anch’essi neurosteroidi. Tuttavia, a differenza di quelli sopra elencati, questi neurosteroidi non modulano i recettori GABAA o NMDA e invece influenzano vari altri recettori della superficie cellulare e bersagli non genomici. Inoltre, molti steroidi endogeni, tra cui Pregnenolone, Progesterone, Corticosterone, Desossicorticosterone, DHEA e Testosterone, vengono metabolizzati in (altri) neurosteroidi, funzionando efficacemente come cosiddetti proneurosteroidi.
Progesterone
AAS derivati dal 19-nortestosterone/progestinici
In questa categoria vengono inseriti tutti gli AAS con struttura simile al Progesterone e che interagiscono a diverso grado con i recettori dello stesso, anche a livello neuronale. Non solo il Nandrolone è stato osservato avere effetti significativi a livello cerebrale e sul SNC ma anche altri composti della stessa famiglia (derivati del 19-nortestosterone), come il Trenbolone, hanno mostrato effetti di tipo misto eccitatorio-depressivo. Tali effetti, che si manifestano con maggior enfasi se la molecola in questione va a creare uno squilibrio endocrino maggiormente in contrapposizione con quello mediamente mantenuto in fisiologia (vedi anche rapporto Testosterone:Progestinici), possono essere i seguenti [27]:
1- Aggressività 2- Ansia, paura e stress 3- Ricompensa e dipendenza 4- Alterazione dell’apprendimento, memoria e capacità di lavoro 5- Alterazione della locomozione e attività fisica 6- Effetti sulla HPAA (Asse Ipotalamo-Pituitaria-Surrene) 7- Effetto sui neurotrasmettitori: Recettore Acido γ-Aminobutirrico Tipo A (GABAA); Recettori 5-idrossitriptamina (5-HT) e 5-HT; Recettori della Dopamina e Recettori Oppioidi.
Trenbolone
Alcune delle principali funzioni biologiche note dei neurosteroidi includono la modulazione della plasticità neurale, [28] processi di apprendimento e memoria, [29] comportamento, [30] [31] e suscettibilità alle crisi [32], nonché risposte a stress, ansia e depressione . [33] I neurosteroidi sembrano anche svolgere un ruolo importante in vari comportamenti sessualmente dimorfici e risposte emotive. [34]
Lo stress acuto aumenta i livelli di neurosteroidi inibitori come l’Allopregnanolone e questi neurosteroidi sono noti per contrastare molti degli effetti dello stress. [35] Questo è simile al caso delle endorfine, che vengono rilasciate in risposta allo stress e al dolore fisico e contrastano gli effetti soggettivi negativi di tali stati. Pertanto, è stato suggerito che una delle funzioni biologiche di questi neuromodulatori potrebbe essere quella di aiutare a mantenere l’omeostasi emotiva. [30] [36] Lo stress cronico è stato associato a livelli ridotti di Allopregnanolone e reattività allo stress di Allopregnanolone alterata, disturbi psichiatrici e disregolazione dell’asse ipotalamo-ipofisi-surrene. [33] [35]
Si ritiene che le fluttuazioni dei livelli di neurosteroidi inibitori durante il ciclo mestruale e la gravidanza svolgano un ruolo importante in una varietà di condizioni femminili, tra cui sindrome premestruale (PMS), disturbo disforico premestruale (PMDD), depressione postpartum (PPD), psicosi postpartum e l’epilessia catameniale. [37] [38] [39] Inoltre, si pensa che i cambiamenti nei livelli di neurosteroidi possano essere coinvolti nei cambiamenti di umore, ansia e desiderio sessuale che si verificano durante la pubertà in entrambi i sessi e durante la menopausa nelle donne. [4] [40] [41]
Livelli elevati di neurosteroidi inibitori, vale a dire Allopregnanolone, possono produrre effetti paradossali, come umore negativo, ansia, irritabilità e aggressività. [42] [43] [44] [45] Ciò sembra essere dovuto al fatto che questi neurosteroidi, come altri modulatori allosterici positivi del recettore GABAA come le benzodiazepine, i barbiturici e l’etanolo, [36] [44] possiedono azioni bifasiche a forma di U – livelli moderati (nell’intervallo di 1,5– Alloprogesterone totale 2 nM / L, che sono approssimativamente equivalenti ai livelli della fase luteale) inibiscono l’attività del recettore GABAA, mentre concentrazioni più basse e più alte facilitano l’attività del recettore. [42] [43]
E’ interessante notare come alcuni farmaci antidepressivi come la Fluoxetina e la Fluvoxamina, che si ritiene generalmente influenzino la depressione agendo come inibitori selettivi della ricaptazione della serotonina (SSRI), sono stati osservati agire come normalizzatori dei livelli di alcuni neurosteroidi (che sono frequentemente carenti nei pazienti depressi) a dosi nelle quali non presentano attività nell’influenzare la ricaptazione della Serotonina. Ciò suggerisce che anche altre azioni che coinvolgono i neurosteroidi possono essere in gioco nell’efficacia di questi farmaci contro la depressione. [45] [46]
Fluoxetina
Inoltre, le benzodiazepine possono influenzare il metabolismo dei neurosteroidi in virtù della loro azione sulla proteina traslocatrice (TSPO; “recettore periferico delle benzodiazepine”). [47] Le azioni farmacologiche delle benzodiazepine a livello del recettore GABAA sono simili a quelle dei neurosteroidi. I fattori che influenzano la capacità delle singole benzodiazepine di alterare i livelli di neurosteroidi possono dipendere dal fatto che il singolo farmaco benzodiazepinico interagisca con TSPO. Alcune benzodiazepine possono anche inibire gli enzimi neurosteroidogenici riducendo la sintesi dei neurosteroidi. [48]
Adesso che avete una panoramica di cosa sia un Neurosteroide e come esso possa agire in base al tipo di molecola, passiamo allo studio prima citato…
Dettagli dello studio
I ricercatori hanno utilizzato i dati di 232 utilizzatori di AAS e 60 loro amici e familiari. Queste persone, hanno contattato il progetto Steroid del governo norvegese nel 2015-2019. Il progetto Steroid mira a indirizzare gli utilizzatore di AAS con problemi medici verso specialisti al fine di cercare di consentire loro di cessarne l’uso.
In questo studio, apparso su Substance Abuse Treatment, Prevention, and Policy nel 2019, i ricercatori hanno fatto il punto della situazione sui problemi degli utilizzatori che li hanno contattati.
Risultati dello studio
Il 77,2% degli utilizzatori ha contattato lo Steroid Project e ha deciso di smettere di usarli. Quando i ricercatori hanno confrontato i soggetti che avevano cessato l’utilizzo con gli utilizzatori che volevano continuare a percorrere la strada della supplementazione farmacologica, hanno scoperto che i primi erano più grandi, avevano maggiori probabilità di avere figli e hanno anche segnalato problemi medici più spesso. Bene, questo ha senso.
La figura seguente mostra i problemi segnalati più frequentemente dagli utilizzatori. Erano di natura psicologica piuttosto che fisica. Depressione, cambiamento di comportamento e ansia sono stati i tre effetti collaterali più frequentemente riportati.
Gli amici e i familiari hanno visto principalmente effetti collaterali come cambiamento comportamentale, aggressività e depressione tra gli utenti.
Conclusioni
I ricercatori concludono scrivendo che i problemi contro cui gli utilizzatori di AAS stanno lottando sono piuttosto complessi. I programmi di riduzione del danno, o programmi che vogliono incoraggiare gli utilizzatori a smettere, devono tenerne conto.
I problemi della sfera psicologica si possono presentare sia durante il ciclo (a diverso grado) che nel post ciclo, sebbene una corretta PCT possa smorzarne il grado almeno fin tano che essa persiste (il vero recupero avviene nei mesi successivi alla PCT, ed è qui che gli effetti di una omeostasi ancora alterata si fanno percepire; vedi fase OCT).
Quindi, anche voi lettori, che siate utilizzatori o intenzionati a farlo, dovreste mettere sul piatto della bilancia anche l’impatto psicologico AAS-dipendente/AAS-correlato…
Nota: anche l’uso di SARM, attraverso il feedback negativo AR-dipendente a livello ipotalamico, possono causare i medesimi squilibri nella sintesi ed attività dei neurosteroidi.
12-^ Mellon SH, Griffin LD (2002). “Neurosteroids: biochemistry and clinical significance”. Trends in Endocrinology and Metabolism. 13(1): 35–43.
13- Rougé-Pont F, Mayo W, Marinelli M, Gingras M, Le Moal M, Piazza PV (July 2002). “The neurosteroid allopregnanolone increases dopamine release and dopaminergic response to morphine in the rat nucleus accumbens”. The European Journal of Neuroscience. 16 (1): 169–73. doi:10.1046/j.1460-9568.2002.02084.x. PMID12153544. S2CID9953445.
16- ^ Terán-Pérez G, Arana-Lechuga Y, Esqueda-León E, Santana-Miranda R, Rojas-Zamorano JÁ, Velázquez Moctezuma J (October 2012). “Steroid hormones and sleep regulation”. Mini Reviews in Medicinal Chemistry. 12 (11): 1040–8. doi:10.2174/138955712802762167. PMID23092405.
17- ^ Patte-Mensah C, Meyer L, Taleb O, Mensah-Nyagan AG (February 2014). “Potential role of allopregnanolone for a safe and effective therapy of neuropathic pain”. Progress in Neurobiology. 113: 70–8. doi:10.1016/j.pneurobio.2013.07.004. PMID23948490. S2CID207407077.
23- ^ Baulieu E, Schumacher M (2000). “Progesterone as a neuroactive neurosteroid, with special reference to the effect of progesterone on myelination”. Steroids. 65 (10–11): 605–12. doi:10.1016/s0039-128x(00)00173-2. PMID11108866. S2CID14952168.
33- ^ Jump up to:ab Bäckström T, Andersson A, Andreé L, Birzniece V, Bixo M, Björn I, Haage D, Isaksson M, Johansson IM, Lindblad C, Lundgren P, Nyberg S, Odmark IS, Strömberg J, Sundström-Poromaa I, Turkmen S, Wahlström G, Wang M, Wihlbäck AC, Zhu D, Zingmark E (December 2003). “Pathogenesis in menstrual cycle-linked CNS disorders”. Annals of the New York Academy of Sciences. 1007 (1): 42–53. Bibcode:2003NYASA1007…42B. doi:10.1196/annals.1286.005. PMID14993039. S2CID20995334.
38- ^ Andréen L, Sundström-Poromaa I, Bixo M, Nyberg S, Bäckström T (August 2006). “Allopregnanolone concentration and mood–a bimodal association in postmenopausal women treated with oral progesterone”. Psychopharmacology. 187 (2): 209–21.
39- Bäckström T, Haage D, Löfgren M, Johansson IM, Strömberg J, Nyberg S, Andréen L, Ossewaarde L, van Wingen GA, Turkmen S, Bengtsson SK (September 2011). “Paradoxical effects of GABA-A modulators may explain sex steroid induced negative mood symptoms in some persons”. Neuroscience. 191: 46–54. doi:10.1016/j.neuroscience.2011.03.061. PMID21600269. S2CID38928854.
40- ^ Jump up to:ab Andréen L, Nyberg S, Turkmen S, van Wingen G, Fernández G, Bäckström T (September 2009). “Sex steroid induced negative mood may be explained by the paradoxical effect mediated by GABAA modulators”. Psychoneuroendocrinology. 34 (8): 1121–32. doi:10.1016/j.psyneuen.2009.02.003. PMID19272715. S2CID22259026.
41- ^ Jump up to:ab Bäckström T, Bixo M, Johansson M, Nyberg S, Ossewaarde L, Ragagnin G, Savic I, Strömberg J, Timby E, van Broekhoven F, van Wingen G (February 2014). “Allopregnanolone and mood disorders”. Progress in Neurobiology. 113: 88–94.
44-^ Pinna G, Costa E, Guidotti A (June 2006). “Fluoxetine and norfluoxetine stereospecifically and selectively increase brain neurosteroid content at doses that are inactive on 5-HT reuptake”. Psychopharmacology. 186 (3): 362–72. doi:10.1007/s00213-005-0213-2. PMID16432684. S2CID7799814.


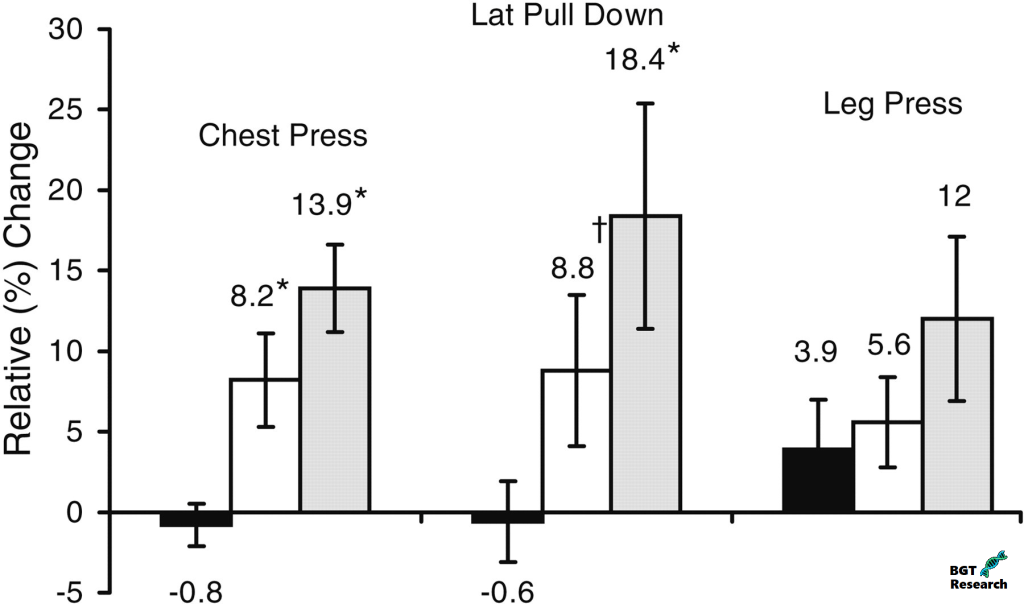

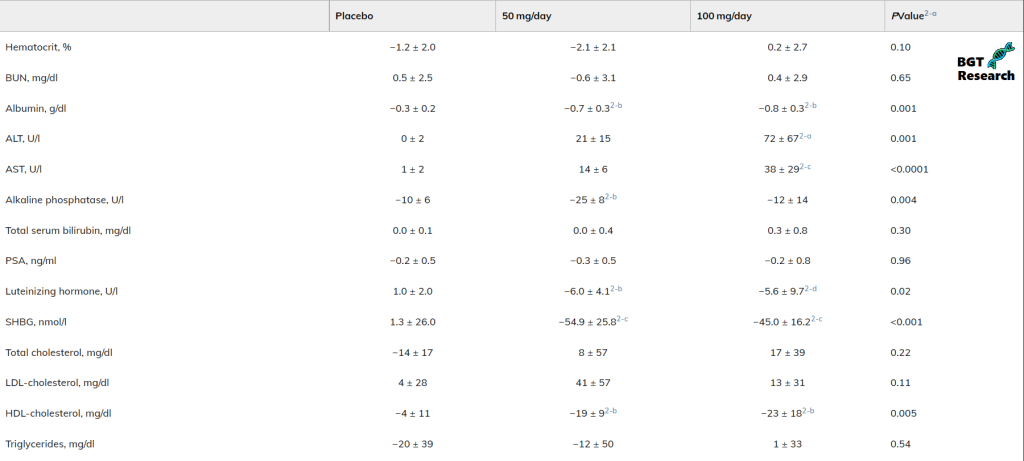

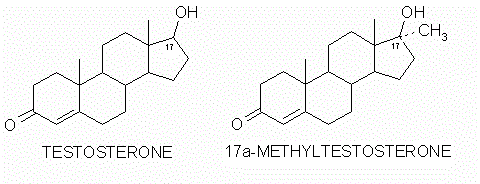
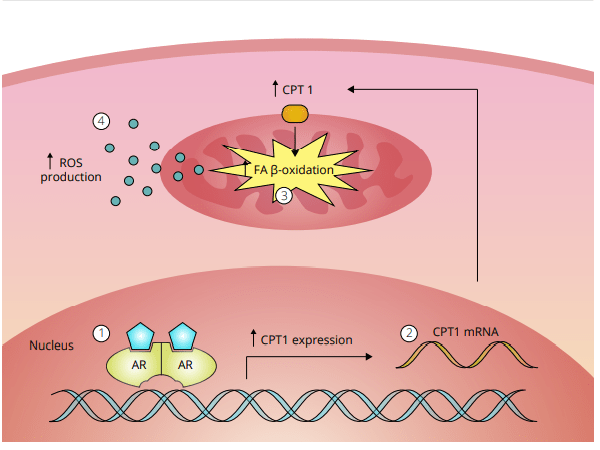


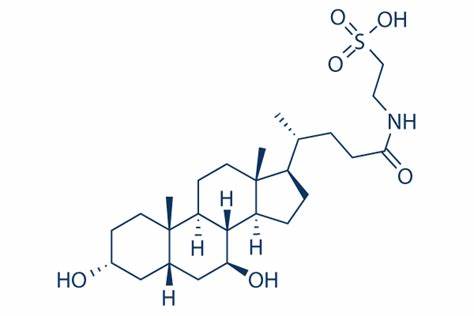
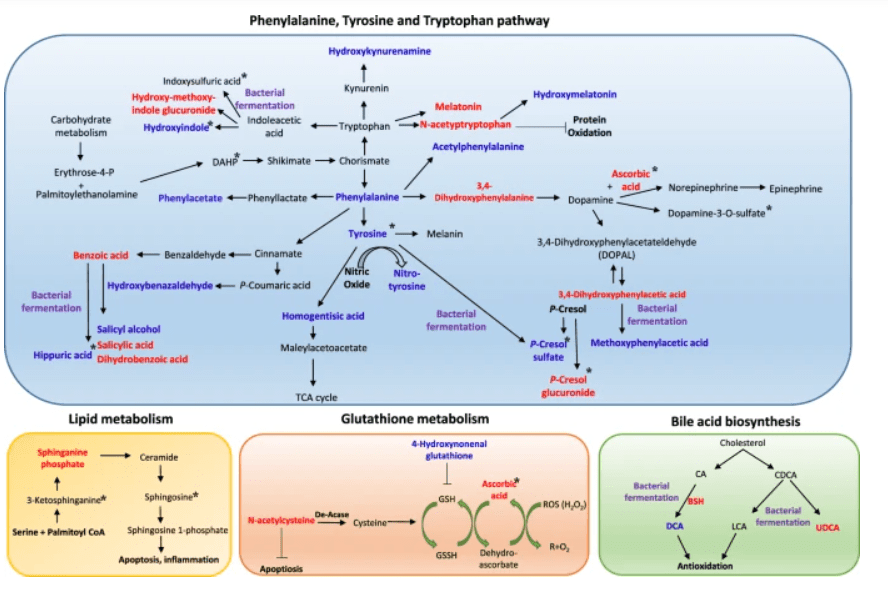































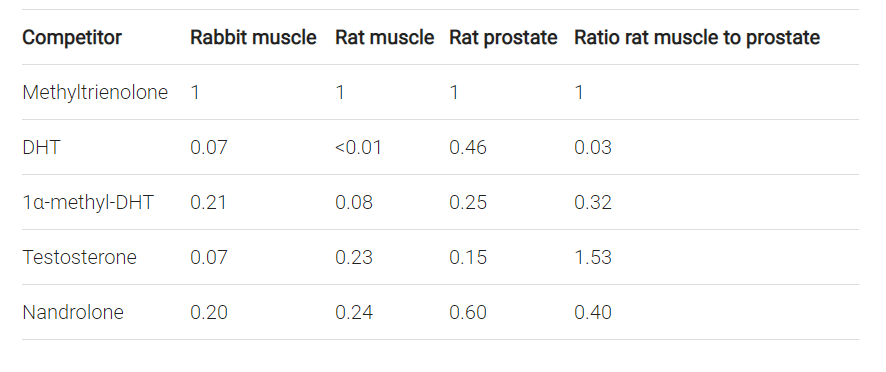
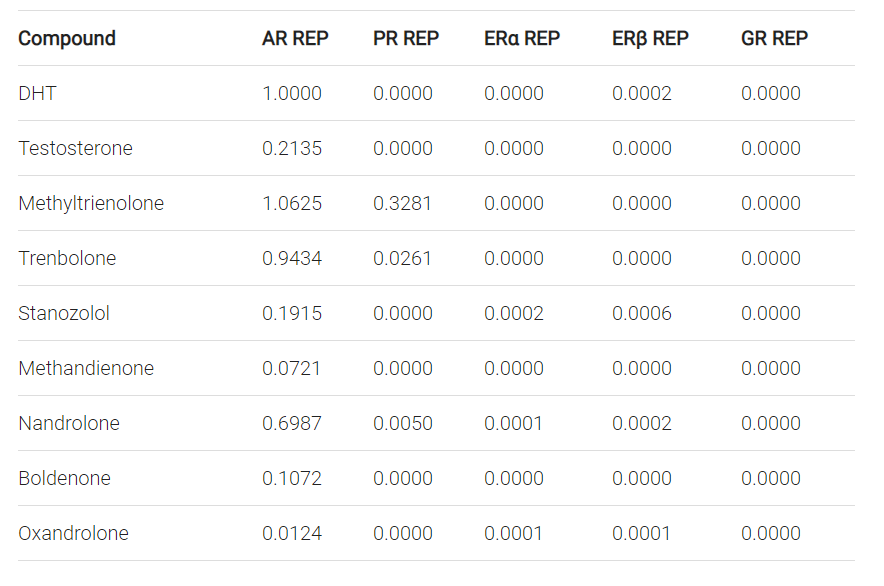

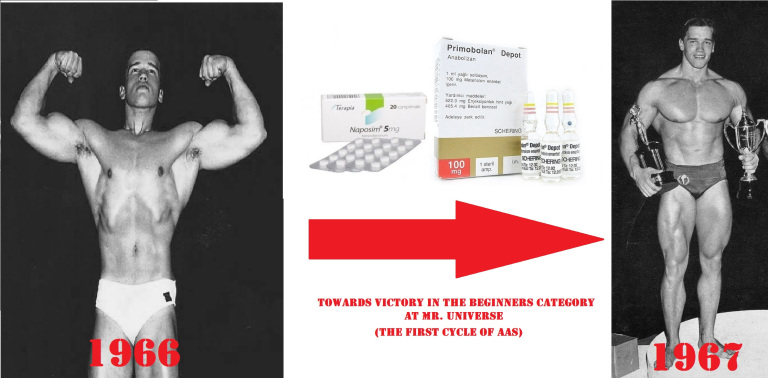
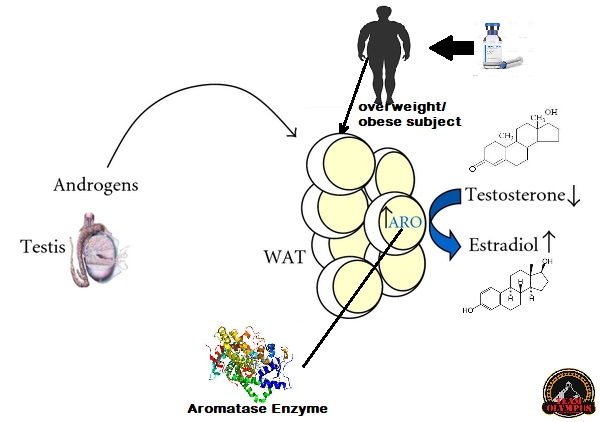
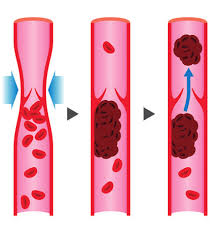



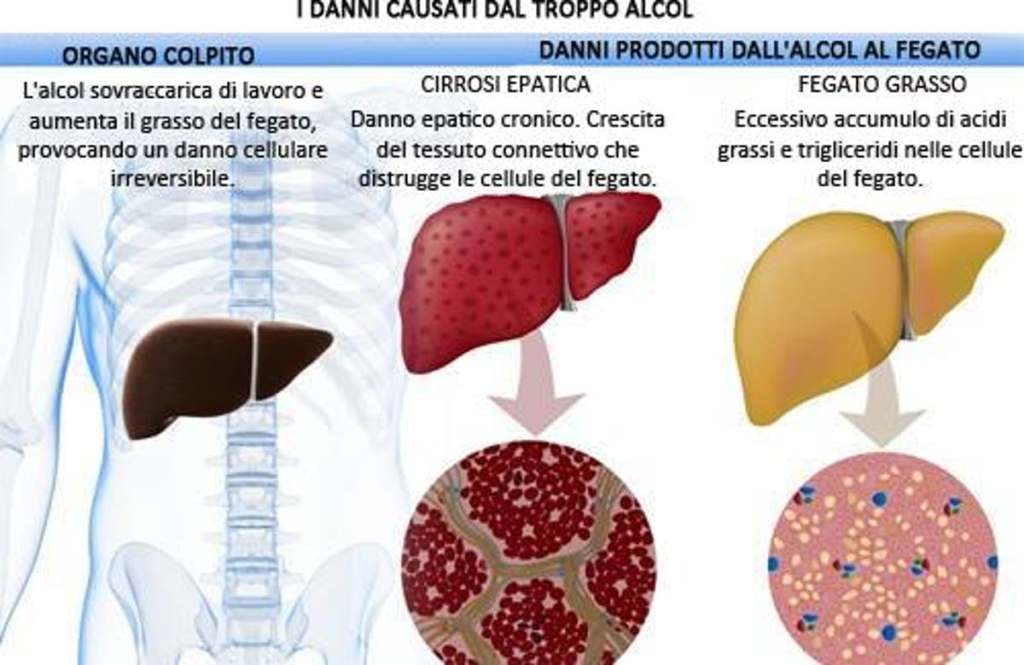














{kind=link}