BPC-157 è il termine usato per riferirsi a un pentadecapeptide, una proteina composta da una catena di 15 amminoacidi. BPC è l’acronimo di “Body Protection Compounds” e si riferisce a “peptidi comprendenti 8-15 residui di amminoacidi con un peso molecolare di 900-1.600 dalton” secondo il brevetto per il BPC-157[1], sebbene un altro studio affermi che BPC si riferisce a una proteina gastroprotettiva utilizzata per isolare il BPC-157.[2] Questa particolare sequenza non condivide l’omologia con altri peptidi gastrici noti,[2] con almeno uno studio che rileva che questa sequenza non è stata registrata nel database Protein BLAST (a partire dal 2016[3]). Ci sono alcuni studi nei quali questo peptide è indicato anche come PL 14736, PL-10,[4] e Bepecin[3]. In questo articolo si utilizzerà esclusivamente l’acronimo BPC-157.
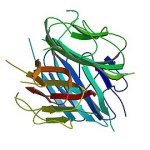
Struttura molecolare del BPC-157
Il BPC-157 è liberamente solubile in acqua con un valore pH normale.[5] La sequenza del pentadecapeptide è Gly-Glu-Pro-Pro-Pro-Gly-Lys-Pro-Ala-Asp-Asp-Ala-Gly-Leu-Val[5] e si dice che sia abbastanza stabile rispetto ad altri peptidi non degradandosi nell’acido dello stomaco (ex vivo) per almeno 24 ore.[2][6] È stato dimostrato che è moderatamente stabile nel plasma ex vivo, con il 36% del peptide intatto che permane dopo 60 minuti.[3]
Caratteristiche farmacodinamiche del BPC-157:
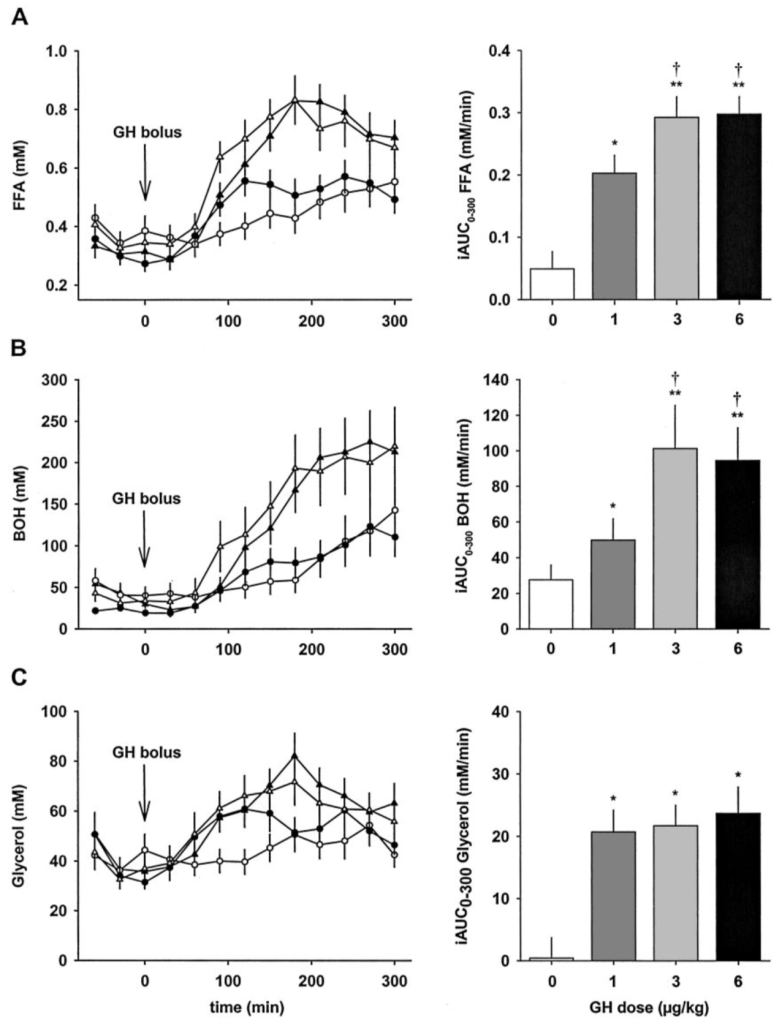
Quando i ricercatori hanno testato il BPC-157 in un test CAM (embrione di pulcino), sembrava essere in grado di aumentare il processo di angiogenesi (produzione di vasi sanguigni) del 129 +/- 7% e del 152 +/- 14% se somministrato a dosi di 0,01 μg e 0,1 μg, rispettivamente. Questo effetto è stato successivamente confermato negli HUVEC, dove concentrazioni di 0,1 μg/mL e 1 μg/mL hanno aumentato la formazione di vasi sanguinei perfetti del 119+/-9% e del 147 +/- 7% nelle 24 ore di incubazione (con 1 μg/ ml essendo determinata essere la concentrazione ottimale in HUVEC).[7] Questa osservazione è stata confermata anche nei ratti con danni agli arti. Dopo una settimana di trattamento con BPC-157, sembravano esserci più vasi sanguigni nell’arto danneggiato rispetto al controllo.[7]
Angiogenesi: processo multifasico che genera nuovi vasi sanguigni dal pre-esistente letto vascolare.
Un aumento dell’espressione di VEGFR2 è stato notato nei ratti con un arto ferito a cui era stato somministrato BPC-157 rispetto al controllo, che si pensava fosse alla base dell’aumento della produzione di vasi sanguigni. Quando sono stati ulteriormente testati, i ricercatori hanno scoperto che il VEGF-A è completamente inalterato alla concentrazione di 1μg/mL, mentre il VEGFR2 è aumentato in modo dipendente dal tempo di esposizione all’interno della cellula e quindi ha proceduto all’attivazione della via VEGFR2-Akt-eNOS (una via importante all’angiogenesi).[7] Quando è stato introdotto il Dynasore, un inibitore del VEGFR2,[8], l’intera via non è stata più attivata e la formazione del vaso sanguineo non si è più verificata in vitro.[7]
È stato anche scoperto che il BPC-157 stimola l’mRNA del fattore di crescita EGR-1 nelle cellule intestinali (Caco-2) a 10-100μM, con la massima efficacia a 50μM. Anche una proteina correlata, l’mRNA NAB2, è stata aumentata poco dopo. Entrambi questi effetti sono paralleli agli effetti del PDGF-BB (un fattore di crescita endogeno) sebbene richiedano concentrazioni molto più elevate. Anche il contenuto di proteine EGF-1 sembrava essere aumentato.[4]
Quando incubato nel plasma ex vivo, sembra che una grande quantità del peptide rilevato venga registrata come ‘metaboliti’ (79+/-2%) del composto originario entro 60 minuti, anche se poi sembra stabilizzarsi, con il rimanente peptide intatto che permane fino a 240 minuti.[3]
È stato menzionato indirettamente dall’autore di molti studi che con il BPC-157 non è stato trovato alcun legame noto con i recettori della dopamina, sebbene non sia stata fornita alcuna citazione per questa particolare affermazione.[9] Quando somministrato a 10ng/kg o 10μg/kg, il BPC-157 somministrato contemporaneamente all’anfetamina ha mostrato che solo la dose più elevata era in grado di attenuare alcuni effetti osservabili dell’anfetamina (comportamenti dei ratti come annusare, leccare e rosicchiare). Anche la somministrazione del BPC-157 un’ora dopo l’anfetamina ha mostrato alcuni benefici.[2] Quando ai ratti è stato precedentemente somministrato Aloperidolo (che rende i ratti successivamente più sensibili agli effetti dell’anfetamina[10]), la co-somministrazione di BPC-157 sembrava mitigare la prevista sensibilità indotta dall’Aloperidolo.[2] Questo effetto apparentemente antagonista può anche applicarsi cronicamente, il che significa che una singola dose di BPC-157 (10μg/kg IP; 10ng/kg inefficace) somministrata prima della somministrazione cronica di anfetamine sembrava attenuare gli effetti comportamentali dell’anfetamina nei ratti durante il periodo di osservazione.[11]
Aloperidolo
Il BPC-157 è stato studiato per il suo coinvolgimento nel sistema serotoninergico dovuto alla sua influenza nella salute dell’intestino, ed i ricercatori suggeriscono un possibile asse cervello-intestino a monte degli effetti del BPC-157 in entrambe queste aree.[12] Per quanto riguarda una connessione tra il cervello e l’intestino, la Serotonina è un probabile giocatore a causa della sua alta prevalenza nell’organo.[13]
I ricercatori hanno scoperto che i ratti trattati con 10μg/kg (iniezione sottocutanea) di BPC-157 sperimentano acutamente un aumento della sintesi della Serotonina dopo 40 minuti in diverse regioni del cervello, tra cui la substantia nigra reticulata e il nucleo olfattivo anteriore mediale, mentre contemporaneamente sperimentano una diminuzione nell’Ipotalamo, nell’Ippocampo (ventrale e dorsale), e nel talamo (dorsale ma non ventrale).[14] Quando questa dose veniva somministrata per una settimana, persisteva l’aumento della sintesi di Serotonina nella substantia nigra (che si verificava sia nella reticulata che nella compacta) mentre le diminuzioni della sintesi di Serotonina osservate con una singola dose non persistevano più.[14]
Asse Cervello-Intestino
Il BPC-157 sembra avere effetti protettivi sul tessuto cerebrale quando somministrato ai ratti (sia somministrato tramite l’acqua da bere che attraverso le iniezioni) insieme alla tossina Cuprizone, riducendo la quantità di cellule danneggiate in numerose regioni del cervello, compreso l’Ippocampo.[15] Il Cuprizone[15] è una tossina utilizzata per simulare i danni osservati nella sclerosi multipla[16] e potenzialmente nella schizofrenia.[17]
L’ingestione orale di BPC-157 a una quantità stimata di 10 μg/kg (0,16 μg/mL in acqua) è stata altrettanto efficace delle iniezioni di 10ng/kg e 10μg/kg[15], sebbene sia noto che il Cuprizone è una tossina che può indurre danno neuronale (in particolare demielinizzazione) senza necessariamente raggiungere il cervello.[18]
Cuprizone
Nelle femmine di ratto sottoposte a test di nuoto forzato (test di Porsolt), il BPC-157 (somministrazione intraperitoneale) alle dosi sia di 10ng/kg che di 10μg/kg sembra funzionare in misura statisticamente uguale ai controlli attivi sia del Imipramina (15 mg e 30 mg) che della Nialamide (30 mg e 40 mg), e hanno tutti superato il gruppo di controllo.[5] Il BPC-157 è apparso anche efficace nell’assistere questi ratti in un modello di stress cronico imprevedibile simile a 30mg di Imipramina.[5]
A concentrazioni di 2μg/mL nei fibroblasti tendinei che sono stati poi espiantati, le cellule trattate con il BPC-157 sembravano crescere più velocemente dei fibroblasti non trattati con BPC-157 entro due giorni, raggiungendo una quantità significativamente maggiore dopo una settimana. Questo effetto è stato associato sia ad una maggiore resistenza ossidativa al perossido di idrogeno che ad un aumento dipendente dalla concentrazione delle proteine FAK e Paxillina non osservate nel gruppo di controllo.[19] Anche la formazione di F-actina, importante per il processo di diffusione dei fibroblasti tendinei,[20] sembra essere notevolmente aumentata con il BPC-157 rispetto al controllo[19] ed è correlata alle azioni delle suddette proteine (FAK e Paxillina).[ 21] Questo studio ha anche scoperto che i fibroblasti tendinei ex vivo in isolamento non erano influenzati dal BPC-157, solo quelli espiantati nei ratti,[19] un effetto notato anche altrove quando i Tendociti coltivati non erano influenzati dal solo BPC-157.[22] Tuttavia, l’effetto inibitorio della crescita del 4-idrossinonenale (HNE) è stato negato dalla presenza del BPC-157 in queste cellule.[22]
Paxillina
I ricercatori hanno osservato benefici quando il BPC-157 veniva messo su una spugna durante l’intervento chirurgico, dove sembrava migliorare il tasso di riformazione del collagene, inizialmente superando il fattore di crescita delle piastrine dopo quattro giorni, ma alla fine risultando equipotente dopo otto giorni[4]. Sono stati osservati benefici nei ratti sottoposti a iniezioni intraperitoneali dopo una lesione del tallone d’Achille, dove il tasso di guarigione della lesione è stato confermato visivamente con dimensioni e profondità del taglio inferiori.[22]
È stato riscontrato che gli effetti protettivi del BPC-157 sulle ulcere vengono prevenuti nei ratti attraverso la somministrazione concomitante di Aloperidolo (antagonista dei recettori alfa-1A e dopaminergico), Fentolamina (antagonista alfa adrenergico, non selettivo) e Clonidina (antagonista alfa-2A adrenergico, simile all’Agmatina) ma non è stato influenzato dalla Prazosina, dal Domperidone o dalla Yohimbina.[23]
Il BPC-157 ha mostrato effetti protettivi contro vari agenti che inducono ulcere gastriche, come la Ciclofosfamide[24] e l’Aloperidolo.[25]
Quando si tratta di infiammazione, il BPC-157 ha mostrato benefici nei ratti contro le tossine Acido Trinitrobenzensolfonico (TNBS)[6] e Cisteamina,[26][27][15] dove sono stati ridotti sia i biomarker dell’infiammazione che i marker visivi di danno quando il BPC-157 è stato somministrato insieme alle tossine. Il BPC-157 non è unico in questo senso d’azione, poiché altri composti attivi controllati come la Ranitidina e l’Omeprazolo hanno mostrato efficacia nello stesso modello di infiammazione intestinale,[27] sebbene sia stato menzionato in una review degli autori[28] che il BPC-157 può essere più pratico a causa dei comprovati benefici in altre complicanze della malattia intestinale: guarigione dell’anastomosi, sindrome dell’intestino corto e fistole.
Omeprazolo
Un’anastomosi è una connessione tra due cose che normalmente non sono collegate, con una fistola che è un tipo anormale comunemente osservato durante le malattie intestinali. Numerosi studi hanno dimostrato che le iniezioni di BPC-157 nei ratti hanno proprietà riparatrici sull’anastomosi in numerose regioni del corpo, comprese quelle aortiche[29] ed esofagogastriche.[30] Negli studi che hanno valutato l’intestino, sono stati dimostrati benefici per le fistole colo-vescicali,[31] retto-vaginali,[32] colon-colon,[15] e ileoileali[33]. Questo particolare beneficio può essere correlato alla segnalazione dell’Ossido Nitrico (potenzialmente la via VEGFR2-Akt-eNOS influenzata dal BPC-157[7]) poiché L-NAME, un inibitore della sintasi dell’Ossido Nitrico, peggiora la guarigione dell’anastomosi che viene migliorato dal BPC-157.[30]
Ossido Nitrico
Anche gli studi che valutano il BPC-157 in modelli sperimentali di sindrome dell’intestino corto riportano benefici, con iniezioni di BPC-157 che migliorano questo stato[34][35] anche quando lo stato è peggiorato con l’aggiunta di L-NAME e Diclofenac.[35]
In particolare, è stato riscontrato un beneficio per la guarigione dell’anastomosi (esofagogastrica) nei ratti trattati con BPC-157 nell’acqua di abbeveramento (circa 10ng/kg o 10 μg/kg al giorno) senza iniezione, senza differenze significative nell’efficacia tra le due dosi ed efficacia statisticamente simile alle iniezioni di 10ng/kg e 10μg/kg.[30]
Uno studio sui ratti che utilizzava la tossina MPTP (che induce danni simili a quelli osservati nel morbo di Parkinson nei roditori), la somministrazione di BPC-157 per via intraperitoneale sembrava mitigare alcuni dei danni causati dall’MPTP.[36]
Nei roditori a cui è stato somministrato Cuprizone (per indurre danni simili a quelli osservati nella sclerosi multipla[16]) quelli a cui è stato somministrato il BPC-157 insieme al Cuprizone (0,16 ng/mL o 0,16 μg/mL in acqua potabile per quattro giorni o 10ng/kg o 10μg/kg per via intragastrica nell’ultimo giorno) sembravano mostrare danni cerebrali e anomalie cliniche significativamente inferiori dal Cuprizone rispetto ai ratti di controllo a cui non era stato somministrato il BPC-157.[15]
Conclusioni:
Come abbiamo visto, i ricercatori hanno condotto numerosi studi sui roditori utilizzando il BPC-157 il quale ha mostrato di avere effetti protettivi che si estendono oltre lo stomaco e il tratto intestinale. È stato dimostrato che il BPC-157 favorisce la guarigione delle ulcere nello stomaco, dei danni intestinali come fistole e disturbi infiammatori, la guarigione di ossa e articolazioni e i tassi di crescita e danni agli organi. Ha anche alcune influenze sul cervello.
I ricercatori hanno osservato effetti protettivi marcati quando il BPC-157 viene somministrato ai ratti insieme a una tossina utilizzata nella ricerca o a una procedura chirurgica dannosa. Sono necessarie ulteriori ricerche per chiarire se il BPC-157 ha molteplici meccanismi d’azione, ma la ricerca attuale suggerisce che questo pentadecapeptide influenza diversi fattori di crescita solitamente coinvolti nell’angiogenesi (la produzione di vasi sanguigni) e altri fattori coinvolti nella rigenerazione a seguito di un danno.
Il BPC-157 è sicuramente promettente, ma sono necessari studi sull’uomo per dimostrare che questi benefici si estendono oltre gli animali da ricerca. La maggior parte degli studi sul BPC-157 sono condotti su ratti sottoposti a iniezioni del supplemento. Nonostante il BPC-157 sia un peptide temporalmente stabile a livello gastrico, i peptidi sono un gruppo di composti che normalmente sono scarsamente assorbiti dopo l’integrazione orale, specie in forme oltre la tripeptide, quindi i ricercatori usano prevalentemente le iniezioni negli studi sui roditori. Inoltre, non ci sono prove d’efficacia accademicamente documentata del BPC-157 sugli esseri umani e la maggior parte della ricerca è stata condotta da un singolo gruppo di ricerca. A causa della sua natura sintetica, potrebbero esserci problemi legali associati alla vendita di questo composto in alcune regioni e potrebbe essere vietato da alcune organizzazioni sportive.
Tornando sulla questione dell’assunzione orale del BPC-157, vorrei ricordare che la stabilità gastrica non si traduce in un assorbimento intestinale di una catena composta da 15 amminoacidi. La Pepsina dello stomaco e le proteasi pancreatiche scompongono tutte le proteine/peptidi in amminoacidi, dipeptidi e tripeptidi, i quali vengono assorbiti a livello intestinale attraverso specifici trasportatori. Quindi, l’assunzione orale può portare benefici a livello gastrointestinale e, per connessione cerebrale attraverso il sistema nervoso enterico, benefici a livello mentale. Le proprietà (supposte anche nell’uomo) a livello delle articolazioni e tendini sono ben poco probabili con l’assunzione orale mentre sono una potenziale risultante dal trattamento per iniezione.
Digestione proteica
“Io ho usato la forma orale e ho recuperato più velocemente da una infiammazione alla spalla!” Si, sei proprio sicuro che sia dovuto alla supplementazione con il BPC-157? Oppure è la conseguenza di una combinazione di effetti sul recupero dati dai PEDs che stai utilizzando e il miglioramento dello stato psicologico consequenziale all’impatto a livello intestinale del peptide in questione? Prima di “gridare al miracolo” assicuratevi che lo sia…
Detto ciò, la dose orale più vicina possibile alla logica di trasposizione tra test su roditori ed esseri umani si basa su studi sui ratti in cui tale metodo di somministrazione ha mostrato benefici, poiché la maggior parte degli studi, come già detto, somministra il supplemento tramite iniezione. Si stima che la dose orale efficace nei ratti, 10μg/kg, sia equivalente nell’uomo a 1,6μg/kg, ovvero:
96mcg per una persona di 60Kg;
112mcg per una persona di 70Kg;
128mcg per una persona di 80Kg.
Attualmente, per ovvie ragioni, non ci sono studi di farmacocinetica umana per valutare le differenze di specie.
I dosaggi per la forma iniettabile si attestano tra i 200 ed i 300mcg/die per via sottocutanea o intramuscolare (non direttamente nell’articolazione) per un periodo di tempo variabile tra le 2 e le 4 settimane.
Sebbene il peptide BPC-157 non sia attualmente incluso nell’elenco delle sostanze vietate dell’Agenzia Mondiale Antidoping (WADA), è importante che gli atleti sappiano che questa sostanza non è approvata per l’uso clinico umano. È stato sviluppato e pubblicato un test antidoping per la rilevazione del BPC-157 nelle urine. Nonostante la WADA abbia chiarito che al momento il BPC-157 non è una sostanza proibita, questo potrebbe cambiare in futuro se si determinasse di soddisfare almeno due dei tre criteri di inclusione per l’elenco delle sostanze vietate dalla WADA.
Poiché il BPC-157 non è stato ampiamente studiato negli esseri umani, nessuno sa se esiste una dose sicura o se esiste un metodo per utilizzare questo composto con un buon grado di sicurezza per trattare condizioni mediche specifiche.
Dai dati empirici provenienti dagli utilizzatori “off-label” sono emersi effetti avversi quali dolore e arrossamento nel sito di iniezione, così come con qualsiasi iniezione, mal di testa, vertigini e nausea.
Se si domandasse al bodybuilder nella media quale sia l’oggetto di maggiore interesse nella sua programmazione per il “Bulk” quasi sicuramente, dopo aver detto “raggiungere la massima ipertrofia”, direbbe “ridurre al minimo l’aumento della massa grassa”. Ed è più che logico visto che l’atleta con un minimo di senso logico sa che un peggioramento marcato della “bf” in una fase ipercalorica si tradurrà mediamente in un tempo generalmente più lungo in ipocalorica con restrizioni caloriche (o output calorici) più elevate ed un rischio relativamente aumentato di veder persa più massa contrattile o, peggio ancora, il dover calcare con i PEDs per rientrare nei tempi della preparazione al contest (o a qualsiasi evento dove il soggetto vuole essere nella sua “Top Condition”).
Tralasciando la pratica della “Break Diet”, altamente funzionale se gestita correttamente, esiste una strategia alimentare piuttosto datata ma, complici anche “neo-trafilettari” impomatati, riemersa recentemente, ovvero seguire un regime alimentare “High Fat” nel periodo di “Bulk”.
Siamo soliti collegare le diete “Low Carbs” e “High Fat” nel periodo di restrizione calorica, sapendo benissimo che il vantaggio assoluto esiste solo e soltanto con pratiche supplementative mirate (PEDs), parlando sempre di atleti e non di soggetti in sovrappeso o obesi che inizialmente giovano nel seguire una dieta Chetogenica o iperproteica in quanto a compliance insulino-resistenza correlata. Ma in ipercalorica? Possiamo veramente aspettarci una qualità migliore in quanto aumento della massa muscolare ed il peso guadagnato rispetto ad una dieta con prevalenza glucidica?
Con questo articolo cercherò di dare una risposta la più oggettiva possibile avvalendomi della conoscenza scientifica in nostro possesso che riguarda il metabolismo lipidico, l’impatto dei lipidi sulla massa muscolo-scheletrica, sulle prestazioni e i raffronti con il metabolismo glucidico partendo però dalla storia pratica della metodica alimentare qui trattata…
Back to the Future: Mauro Di Pasquale e la sua “Soluzione Anabolica”
Chi è che non ricorda Mauro Di Pasquale e il suo (a detta dell’autore) “Santo Graal” delle diete, “La Dieta Metabolica”? E la sua versione per i Bodybuilder ” La Soluzione Anabolica”? Immagino che molti di voi conosceranno entrambi i libri e i concetti viziati da bias in essi contenuti.
Visto l’argomento partirò proprio da qui…
Per chi non lo sapesse, Mauro G. Di Pasquale è un Powerlifter campione del mondo, autore di articoli sul bodybuilding e opinionista. Di Pasquale è stato assistente professore all’Università di Toronto dal 1988 al 1998. Ha tenuto conferenze e fatto ricerche sulle prestazioni atletiche, sugli integratori alimentari e sull’uso di farmaci nello sport. Ha conseguito una laurea con lode in scienze biologiche, specializzandosi in biochimica molecolare (1968) e ha una laurea in medicina (1971) – entrambe presso l’Università di Toronto. Di Pasquale è certificato come Medical Review Officer (MRO) dal Medical Review Officer Certification Council (MROCC). Era il MRO per la National Association for Stock Car Auto Racing (NASCAR). Dal 1997 al 1999 Di Pasquale si è occupato di redazione, ricerca e sviluppo di prodotti per le Scienze Sperimentali e Applicate (EAS). Come autore di bodybuilding, Di Pasquale ha scritto migliaia di articoli per molte grandi riviste di bodybuilding e fitness come Muscle & Fitness e Iron Man;[1-4] I suoi articoli e libri sono stati tradotti in lingua italiana e pubblicati in Italia da Sandro Ciccarelli per la rivista Olympian’s News.
Di Pasquale divenne anche un oppositore all’uso del doping pubblicizzando i suoi regimi alimentari e integrativi come “sostitutivi salutari” dei farmaci per il miglioramento delle prestazioni.
Da destra: Mauro di Pasquale insieme al bodybuilder professionista Eddie Robinson.
Nonostante la titolatura sopra esposta, Di Pasquale divulgò alcune teorie decisamente opinabili abbracciando la filosofia delle diete “Low Carb” come soluzione universale per perdere grasso e aumentare la massa muscolare. Egli rappresenta una sorta di “paladino” per la fazione dei sostenitori della così detta “ipotesi dell’Insulina”, tra l’altro smentita scientificamente, la quale ipotizza che non sia l’eccesso energetico (calorie) a causare l’aumento di peso/grasso ma i Carboidrati ed il loro stimolo sull’Insulina. Peccato però che la termodinamica ci dimostri il contrario, ovvero che è l’eccesso energetico ha causare una conservazione dello stesso e non il semplice stimolo ormonale (sul quale, tra l’altro, ci sarebbe molto da dire). Oltretutto, alcune fonti proteiche (vedi prodotti lattiero caseari) hanno un impatto insulinico maggiore del pane bianco, sebbene questo venga regolato da una concomitante risposta del Glucagone. Comunque sia, di quest’ultimo punto, il Di Pasquale sembra non curarsene più di tanto, similmente a quanto fa della risposta insulinica data dalla coingestione di grassi e proteine preoccupandosi del carico glucidico da ciclicizzare (cosa, quest’ultima, condivisibile ma da contestualizzare).
Ma arriviamo al dunque sul suo approccio “High Fat” in Bulk…
Nel 1995 pubblica il libro “The Anabolic Diet“, pubblicato poi in Italia con il nome di “La Soluzione Anabolica”.
Il libro in questione espone le vedute dell’autore, accuratamente servite per vendere il prodotto e convincere il lettore, dividendo in sei fasi la preparazione:
Fase di Inizio;
Fase di Massa;
Fase di Forza;
Fase di Definizione;
Fase Pregara;
Fase di Recupero.
Tralasciando le fasi 1, 3,4,5 e 6 che, per argomento trattato nel presente articolo, non ci interessano, concentriamoci sulla “Fase di Massa“.
Dopo aver trovato la quota calorica di “mantenimento”, procedendo con i primi 12 giorni sostituendo le kcal tipicamente consumate dai Carboidrati con Grassi e Proteine riducendo significativamente i primi (es. 1g/CHO-Pro = 4Kcal; 1g Fat = 9Kcal; 400g di CHO/120g di Pro/50g di Fat = circa 30g di CHO, 220g/die di Pro e 134g/die di Grassi), si dovrebbe passare alla prima “ricarica di Carboidrati” seguita a sua volta da 5-6 giorni “High Fat” e 1 o 2 giorni “High Carbs” per 3-4 settimane.
A questo punto l’autore consiglia di permettere al peso un aumento del 15% oltre il peso ideale (es. Peso ideale = 98Kg, peso da raggiungere = 113Kg [+15%]) premurandosi di avvisare il lettore che non deve commettere l’errore di mangiare eccessivamente e vanificare il potenziale della sua strategia di farvi aumentare di massa magra con poco incremento della massa grassa…
Quindi, il bodybuilder dovrebbe seguire lo schema “High Fat” nei giorni feriali ed effettuare le “ricariche” glucidiche nel fine settimana, con un monte calorico indicativo pari a 55Kcal per Kg di peso corporeo desiderato (es. 90Kg = 4950Kcal/die). E le differenze di risposta delle vie metaboliche? Capacità di gestione delle Kcal connesse alle richieste prestative e al livello dell’atleta?… Punti non pervenuti… Per lo meno, il Di Pasquale dice che se il soggetto presenta difficoltà nell’ingestione di grossi quantitativi calorici giornalieri può far diventare il totale calorico da raggiungere un obbiettivo settimanale, abbandonando saggiamente il concetto forviante di “evento in acuto”.
Nelle indicazioni viene riportata anche la soglia del grasso corporeo da non superare. La quota riportata è del 10%… Ora, fatta eccezione per soggetti con un set point adipocitario sensibilmente basso, arrivare a pesare il 15% in più del peso ideale mantenendo una “bf” del 10% è cosa non proprio semplice, leggermente di più se parliamo di atleti “Natural”. Molto più logico sarebbe stato indicare una soglia tra il 12 ed il 15%. Ma l’autore scrive che la Fase di Massa deve essere interrotta quando si raggiunge il peso desiderato o il 10% di “bf”… Se siete agonisti dovete interromperla se arrivate al limite delle 12 settimane dalla gara.
Per quanto concerne l’obbiettivo di incremento settimanale di peso, viene indicato come “ideale” 1Kg a settimana.
Ma quanti glucidi dovrebbero essere consumati durante il/i giorno/i di “ricarica”? Di Pasquale afferma che si possono raggiungere “quantità enormi” di CHO e Kcal nel week and, anche fino a 12.000Kcal tanto il sabato quanto la domenica. Inoltre sia la quota di CHO giornaliera che il numero di giorni con rialzo glucidico possono essere aumentati in base alle risposte. E già questo punto denota una mancanza di funzionalità di base del suo piano alimentare “High Fat” per una “massa pulita” che però va oltre il concetto di “pulito” arrivando al “nodo metabolico”: il vantaggio indiscutibile del substrato glucidico per sostenere l’attività fisica in uno sport contro-resistenza che sfrutta prevalentemente un metabolismo anaerobico glicolitico.
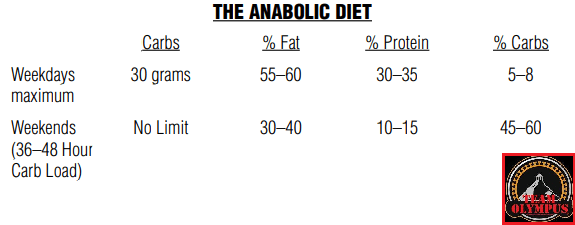
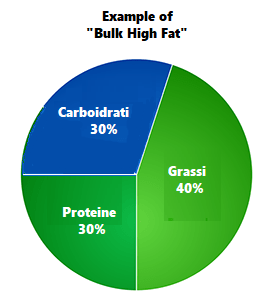
Percentuali di ripartizione dei macronutrienti durante i giorni “High Fat” e le “ricariche di Carboidrati”. (fonte immagine “THE ANABOLIC DIET”, pag. 30 Capitolo 3).
Lascerò da parte la sfilza di integratori marca “MetabolicDiet”, che per lo più altro non sono che Creatina e Proteine in polvere, e le riduttive, ridicole e confutate chiacchiere sulla manipolazione ormonale “simil-doped”, riflettendo direttamente su quanto detto fino a questo punto.
Ora, abbiamo una dieta per il “Bulk” a tutti gli effetti “High Fat” per 3-4/5 della settimana con delle “ricariche glucidiche” di base di 1 o 2 giorni ma incrementabili a bisogno. Ricordo che questa dieta ha la bellezza di 26 anni, ed il sottoscritto, ai tempi in cui ero fortemente tentato a prestar orecchio più alla “carne ed al sangue” piuttosto che alla logica e al buon senso, la conosce e osserva i suoi effetti sugli atleti che l’hanno voluta sperimentare da circa 15 anni. Beh, posso assicurarvi che i risultati con questo tipo di alimentazione in “Bulk” erano apprezzabili (questo non vuol dire che erano spettacolari o che riflettevano le promesse dell’autore) principalmente in due contesti:
Alteti “doped” con un set point adipocitario basso e una media di 3 copiose “ricariche” a settimana;
Atleti “natural” con un set point adipocitario basso e una media di 3 copiose “ricariche” a settimana e un minimo di CHO giornalieri pari a 2-3g/Kg.;
Atleti di sesso femminile con variabili sul numero di “ricariche” e tempi di svolgimento delle medesime.
Ed i casi osservati con risultanti apprezzabili, comunque, sono pochi rispetto ad una maggioranza di individui non significativamente avvantaggiati dal protocollo.
Come potete vedere, il concetto di base della dieta crolla inesorabilmente sotto il peso di dati empirici raccolti direttamente ed indirettamente nel corso degli anni.
“Mah Gabriel! Esistono altre metodologie “High Fat” per una “massa pulita” e funzionano!” Alt, non ti agitare piccolo “troll”, perchè con il termine “High Fat” mi stai dicendo “tutto e niente”, un pò come succede quando si parla di “dieta iperproteica”.
Si possono considerare “High Fat” tutte le diete che in ipercalorica superano il 20-25% delle calorie giornaliere dai grassi! E’ ovvio che possono esistere degli abissi programmatici tra uno schema alla Di Pasquale e un altro che presenta linee di percentuali macro-caloriche nei range sopra indicati.
Prima di proseguire con esempi di schemi alimentari “High Fat” ben più logici rispetto a quanto presentato ne “La Soluzione Anabolica”, e considerare le argomentazioni a favore di questo tipo di dieta ipercalorica, vediamo come i diversi acidi grassi influenzano la massa muscolare. Passiamo a un pò di teoria per tornare successivamente alla pratica con maggiori conoscenze…
La modulazione nella massa e funzione del muscolo-scheletrico dei lipidi
È importante iniziare il discorso sottolineando che vi sono prove crescenti che supportano un ruolo degli acidi grassi e dei loro intermedi lipidici derivati nella regolazione della massa e della funzione del muscolo scheletrico. E’ mia intenzione quindi discutere le prove relative a quei percorsi che sono coinvolti nella riduzione, aumento e/o conservazione della massa muscolare scheletrica da parte dei lipidi.
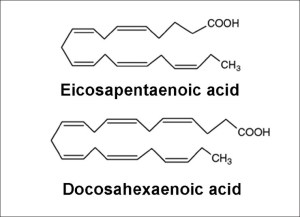
L’evidenza di diversi studi suggerisce che gli acidi grassi saturi e insaturi possono agire per regolare in modo differenziale la massa e la funzione del muscolo scheletrico. Ad esempio, è stato dimostrato che l’esposizione dei miotubi C2C12 al palmitato (C16:0), l’acido grasso saturo circolante più abbondante, riduce il diametro del miotubo e sopprime la segnalazione dell’Insulina. [2] In accordo con questo, è stato riportato che la fornitura di palmitato nelle cellule muscolari induce l’espressione di geni pro-atrofici come atrogin-1/MAFbx, in concomitanza con una maggiore localizzazione nucleare del suo regolatore trascrizionale FoxO3. [3] Al contrario, l’applicazione di acido docosaesaenoico (DHA), un acido grasso polinsaturo omega-3 (PUFA), non ha alterato la morfologia dei miotubi quando applicato da solo ed è stato dimostrato che contromodula l’atrofia indotta dal palmitato nei miotubi C2C12. [2] Coerentemente con ciò, uno studio separato ha riportato il miglioramento della degradazione proteica indotta dal palmitato nei miotubi C2C12 dopo il co-trattamento con DHA. [3] In particolare, ciò ha coinciso con la capacità del DHA di mitigare la localizzazione nucleare potenziata di FoxO3 e l’espressione genica di atrogin-1/MAFbx in risposta alla fornitura di palmitato. [3]
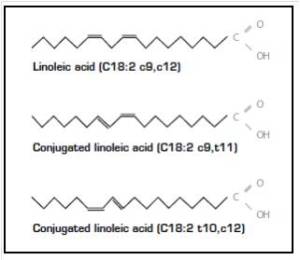
In accordo con questi risultati nelle cellule muscolari di coltura, diversi studi in vivo hanno anche riportato la capacità degli acidi grassi insaturi di trasmettere risposte benefiche che agiscono per prevenire l’atrofia muscolare. Ad esempio, l’alimentazione di topi portatori dell’adenocarcinoma del colon-26, un modello animale di cachessia tumorale, con una dieta integrata con acido linoleico coniugato, ha dimostrato di preservare la massa muscolare del gastrocnemio. [4] In particolare, questo effetto protettivo ha coinciso con una riduzione dell’espressione del recettore TNF-α del muscolo scheletrico, suggerendo che i PUFA possono agire per prevenire l’atrofia muscolare, almeno in parte, riducendo le azioni cataboliche della citochina TNF-α. [4 , 5] In uno studio separato, l’integrazione alimentare con acido eicosapentaenoico (EPA; C20:5 (n-3)) ha attenuato la degradazione proteica nel muscolo gastrocnemio di topi portatori del tumore MAC16 che induce la cachessia. [6] Il trattamento con EPA è stato riportato anche per evitare riduzioni dell’artrite indotta e aumento del peso del muscolo gastrocnemio nei ratti dopo somministrazione di adiuvante di Freund, concomitante con la normalizzazione dell’espressione genica di atrogin-1 / MAFbx e MuRF1. [7] Inoltre, i criceti distrofici alimentati con una dieta arricchita in acido α-linolenico PUFA (ALA) (C18:3(n-6)) hanno mostrato miglioramenti nella morfologia e nella funzione muscolare, compreso l’ingrossamento delle miofibre . [8] In accordo con questi risultati, è stato anche dimostrato che i PUFA omega-3 e omega-6 aumentano la fosforilazione di p70S6K1 a Thr389, indicativo della sua maggiore attività, durante la differenziazione miogenica dei miociti L6. [9] Insieme, questi studi supportano l’idea che gli acidi grassi insaturi possono fornire protezione contro l’atrofia muscolare in risposta a varie condizioni patologiche e potenzialmente migliorare le condizione trofiche in soggetti sani. Inoltre, questi risultati evidenziano le risposte distinte che gli acidi grassi saturi e insaturi inducono rispettivamente per promuovere o contrastare l’atrofia muscolare e la degradazione proteica.
Un certo numero di differenti vie di segnalazione e/o intermedi sono stati implicati come potenziali mediatori della atrofia muscolare, che a loro volta possono essere regolati in risposta alla assunzione di acidi grassi (vedi Figura seguente). Ad esempio, è noto che il palmitato agisce come un potente repressore della segnalazione diretta di PKB/Akt nel muscolo scheletrico, almeno in parte attraverso la sua capacità di indurre l’accumulo di intermedi lipidici tossici come la Ceramide. [10 , 11] Infatti, tali sfingolipidi possono agire stimolando le isoforme della proteina fosfatasi 2A (PP2A) o della proteina chinasi C (PKC) atipica (PKCζ) per inibire PKB/Akt. [12] In accordo con ciò, è stato riportato che l’atrofia del miotubo C2C12 indotta da TNF-α coincide con livelli elevati di Ceramide intracellulare, [13] mentre è stato dimostrato che il blocco della sintesi di Ceramide attenua l’atrofia muscolare indotta dal TNF-α nei miotubi L6, oltre a proteggere i topi contro l’atrofia del muscolo scheletrico tumore indotto ( via impianto di carcinoma C26) in vivo. [13] In particolare, queste risposte benefiche hanno contribuito a una maggiore sintesi proteica e a una diminuzione della proteolisi, in concomitanza con una ridotta espressione del gene atrogin-1/MAFbx tramite la funzione Foxo3 soppressa, nonché una maggiore abbondanza di mediatori chiave della sintesi proteica tra cui S6K1 e PKB/Akt. [13] Inoltre, è stato riportato che la fornitura esogena di Ceramide nelle cellule muscolari L6 riduce i livelli proteici del fattore di trascrizione miogenico miogenina attraverso l’ inibizione della fosfolipasi D, mentre l’inibizione della sintesi di Ceramide migliora l’espressione della miogenina e accelera la formazione di miotubi. [14]Uno studio di Turpin e colleghi ha anche dimostrato un aumento del contenuto di Ceramide muscolare dopo l’infusione acuta (5 h) di intralipid®, che ha coinciso con l’attivazione della segnalazione pro-apoptotica, come dimostrato dall’aumento dell’attività della caspasi-3 nel muscolo gastrocnemio. [15] Tuttavia, il ruolo della Ceramide nel promuovere questo aumento dell’apoptosi muscolare guidato dai lipidi non è stato studiato, ad esempio mediante la co-somministrazione di inibitori della sintesi della Ceramide. In alternativa, livelli elevati di Ceramide associati all’iperlipidemia possono anche agire per sopprimere la sintesi proteica inducendo l’espressione e/o l’attività di repressori chiave della segnalazione mTORC1-S6K come Regulated in Development e DNA Damage 1 (REDD1). [16 , 17]In particolare, va anche evidenziato che il ganglioside GM3 (trisialotetrahexosylganglioside), un glicosfingolipide contenente acido sialico derivato dalla Ceramide, è stato anche implicato come regolatore negativo della crescita e/o differenziazione del muscolo scheletrico, in concomitanza con la sua capacità segnalata di modificare l’azione dell’insulina alterando la funzione del recettore specifico (Recettore dell’Insulina). [18 , 19 , 20 , 21] Inoltre, è stato dimostrato che un altro lipide derivato dalla Ceramide, ceramide-1-fosfato, stimola la proliferazione dei mioblasti C2C12 attraverso un meccanismo che coinvolge l’attivazione di Akt, mTOR e ERK1/2. [22] In effetti, un ulteriore lavoro che utilizza topi carenti di GM3 sintasi, l’enzima responsabile della sintesi di GM3, potrebbe far luce sul ruolo di questo ganglioside nel controllo della massa muscolare scheletrica, ad esempio in risposta all’obesità e/o all’invecchiamento.
Riassunto delle vie che mediano l’atrofia muscolare da acidi grassi saturi. L’esposizione delle cellule muscolari ad acidi grassi saturi come il palmitato (C16:0) provoca l’accumulo intracellulare di intermedi lipidici tossici come Ceramide e Diacilglicerolo. (A) L’aumento dei livelli di Ceramide può portare all’inibizione della proteina chinasi B/Akt attraverso l’attivazione di isoforme atipiche della proteina chinasi C (ξ/λ) e/o della proteina fosfatasi 2A. Inoltre, la Ceramide agisce come precursore per la sintesi del glicosfingolipide GM3 che ha dimostrato di compromettere la funzione del recettore dell’Insulina. Inoltre, la Ceramide può anche agire per modulare l’assorbimento dei nutrienti, ad esempio reprimendo l’espressione del trasportatore amminoacidico neutro SNAT2, riducendo così l’apporto cellulare di amminoacidi. (B) È stato dimostrato che la stimolazione indotta da diacilglicerolo della protein chinasi Cθ promuove la fosforilazione della serina dell’IRS-1, con conseguente sua funzione compromessa. La risultante inibizione della protein chinasi B/Akt a sua volta può portare alla repressione della sintesi proteica attraverso la soppressione del segnale meccanicistico bersaglio della rapamicina (mTOR)/p70-S6 chinasi 1-dipendente (C), l’attivazione del forkhead box O (FoxO ) fattori di trascrizione e induzione dei loro geni atrofici bersaglio (D) e/o l’attivazione della proteolisi dipendente dalla caspasi (E). Inoltre, la stimolazione della segnalazione proinfiammatoria da parte degli acidi grassi saturi a catena lunga può portare alla sovraregolazione dei geni atrofici dipendente dal fattore nucleare kappa B (F).
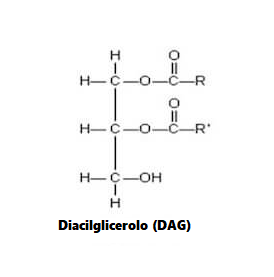
Oltre agli sfingolipidi, i diacilgliceroli (DAG) sono una classe alternativa di lipidi che possono essere generati in risposta alla ingestione di acidi grassi. In particolare, l’aumento dei livelli di DAG è stato associato allo sviluppo dell’insulino-resistenza, fattore di per se limitante sul corretto ripartizionamento calorico a favore del miocita e, quindi, sulla sintesi proteica . [23] sono stati rilevati Inoltre, un aumento dei livelli di DAG muscolare dopo infusione di lipidi nei topi, con concomitante aumento della attività nel muscolo gastrocnemio della caspasi-3 .[15] Sebbene si sappia poco sul ruolo dei DAG nella regolazione della massa muscolare scheletrica, è stato riportato che l’attivazione meccanica ex-vivo della DAG chinasiζ (DGKζ), un enzima che catalizza la conversione di DAG in acido fosfatidico (PA), favorisce un aumento Segnalazione dipendente da mTOR e ipertrofia associata nel muscolo estensore lungo delle dita (EDL) di topo isolato, in concomitanza con la capacità riportata di PA di legare e attivare direttamente mTOR. [24 , 25] In accordo con ciò, è stato anche dimostrato che la sovraespressione cardiaca specifica di DGKζ migliora l’atrofia miocardica nei topi diabetici indotti da streptozotocina. [26] Pertanto, questi risultati suggeriscono che l’attivazione e/o la sovraespressione di DGKζ può fornire un mezzo per stimolare i tassi di sintesi proteica e le risposte ipertrofiche, e quindi migliorare le perdite di massa muscolare, sia riducendo i livelli cellulari di DAG e/o aumentando l’attivazione della segnalazione mTOR indotta dal PA. È importante sottolineare che il lavoro futuro potrebbe comportare lo studio dei potenziali effetti benefici della sovraespressione di DGKζ nel muscolo come mezzo per contrastare l’atrofia muscolare indotta dall’età e/o dalla dieta. Inoltre, modelli animali che mostrano livelli elevati di DAG nel muscolo scheletrico, compresi i topi che sono carenti di lipasi ormone-sensibile (HSL), [27] possono anche essere utili per chiarire il ruolo di DAG nell’atrofia del muscolo scheletrico.
Un’altra considerazione importante riguarda la possibilità che specie di DAG distinte possano avere un impatto diverso sulle vie coinvolte nella regolazione della massa muscolare, ad esempio come determinato dalla composizione dei gruppi acilici grassi che si esterificano a livello sn-1,2, sn‐ 1,3, o le posizioni sn-2,3 della base di glicerolo del DAG. [28 , 29] Infatti, è stato dimostrato che il trattamento dei miotubi L6 di ratto con palmitato porta ad aumenti significativi dei livelli cellulari di alcune specie di DAG, nonché del contenuto totale di DAG cellulare. [30] Inoltre, è stato dimostrato che il co-trattamento con l’acido grasso monoinsaturo (MUFA) palmitoleato (C16:1) sopprime selettivamente gli aumenti indotti dal palmitato nei livelli di specie DAG contenenti porzioni di acidi grassi saturi C18:0 e C20:0, in coincidenza con l’azione antinfiammatoria del MUFA. [30] Sebbene non determinati in questo studio, stereoisomeri distinti di DAG possono anche regolare in modo differenziale la segnalazione anabolica/catabolica muscolare. Per supportare questa nozione, è stato riportato che gli stereoisomeri sn-1,2 DAG (rispetto agli isomeri sn-1,3) sono più potenti nell’attivare le vie di segnalazione legate all’insulino-resistenza, inclusa l’attivazione della PKC. [31] Insieme, questi studi forniscono prove emergenti che alcune molecole/isomeri DAG possono svolgere un ruolo più importante nello sviluppo dell’atrofia muscolare, ad esempio promuovendo la resistenza all’insulina e/o aumentando la spinta proinfiammatoria. Tuttavia, sarà necessario un ulteriore lavoro per determinare quali di queste molecole di DAG, se presenti, sono responsabili delle azioni di atrofia muscolare. Nel tentativo di affrontare questo problema, studi futuri potrebbero comportare il trattamento di cellule muscolari in coltura con diverse molecole/stereoisomeri DAG al fine di determinare i loro effetti sulla miogenesi e/o sull’atrofia muscolare. In alternativa, un ulteriore lavoro può anche incorporare un’analisi lipidomica dettagliata di varie specie di DAG intramuscolari in tessuti isolati da modelli animali di atrofia muscolare, oltre a monitorare potenziali cambiamenti nella loro abbondanza a seguito di interventi noti per aumentare la massa muscolare (ad es. apporto dietetico di PUFA o aumento dell’attività fisica). Infatti, se tali studi dovessero rivelare un ruolo chiave per l’accumulo di DAG nello sviluppo dell’atrofia del muscolo scheletrico, il lavoro successivo potrebbe implicare la determinazione dell’origine di tali specie di DAG, ad esempio inibendo l’attività degli enzimi implicati nella formazione del triacilglicerolo dal DAG ( sintesi di TAG) (es. glicerolo fosfato transferasi (GPAT), acilglicerolfosfato aciltransferasi (AGPAT) e lipina), o alterando l’attività di enzimi implicati nell’idrolisi di TAG e/o DAG (es. lipasi dei trigliceridi adiposa (ATGL) o HSL). A questo fine, i lavori di Badin e collaboratori hanno riportato un’elevata abbondanza di proteine ATGL nel muscolo scheletrico di individui diabetici di tipo 2 rispetto a soggetti di controllo magri, nonché una ridotta espressione di HSL muscolare in individui obesi.[32] Inoltre, gli autori dello stesso studio hanno ulteriormente dimostrato che la sovraespressione di ATGL o l’inibizione dell’attività dell’HSL nei miotubi primari umani determinava l’accumulo di DAG cellulare e una compromissione associata nella segnalazione dell’Insulina. Tuttavia, in questo studio non è stato determinato se questi cambiamenti nei livelli di DAG siano collegati all’atrofia muscolare.
Oltre a modulare la segnalazione diretta di PKB/Akt e/o mTORC1, gli acidi grassi e/oi loro lipidi derivati possono ulteriormente contribuire alla perdita muscolare modulando il trasporto dei nutrienti (aminoacidi) e/o la segnalazione associata. Ad esempio, lavori svolti da diversi gruppi di ricerca hanno dimostrato la capacità della Ceramide di sottoregolare l’espressione e/o l’attività dei principali trasportatori di nutrienti, incluso il trasportatore di amminoacidi neutri SNAT2 (SLC38A2). [33 , 34] In tal modo, gli acidi grassi che agiscono attraverso tali intermedi lipidici possono agire per compromettere l’assorbimento degli aminoacidi, contribuendo così a una perdita di massa muscolare. È interessante notare che in uno studio separato, è stato dimostrato che l’incubazione di miotubi L6 di ratto con acido linoleico (C18:2) limita la sovraregolazione adattativa dell’espressione e dell’attività di SNAT2 in risposta alla carenza di aminoacidi. [35] In particolare, questa riduzione indotta da acidi grassi nell’attività di trasporto del Sistema A è stata mediata da una maggiore ubiquitinazione e degradazione proteasomica della proteina SNAT2. [35] Al contrario, in uno studio separato di Li e collaboratori, è stato riportato che l’espressione dell’mRNA dei transcettori di aminoacidi LAT1 (un trasportatore di aminoacidi di tipo L) e SNAT2 è sovra-regolata nel longissimus dorsi di suini alimentati con diete n. -6 e n-3 PUFA.[36] Quindi, è possibile che gli acidi grassi e/o i loro lipidi derivati possano funzionare per modulare le strategie adattative che vengono utilizzate da tessuti come il muscolo scheletrico, al fine di massimizzare o minimizzare l’assorbimento di nutrienti durante condizioni di digiuno o privazione di nutrienti e, presumibilmente, tale meccanismo può subire alterazioni durante stati di sovra-alimentazione.
È importante sottolineare che la letteratura attuale descrive prove che suggeriscono che gli acidi grassi insaturi possono agire per contrastare i mediatori pro-atrofici, compresi quelli attivati in seguito all’esposizione agli acidi grassi saturi. Ad esempio, è stato riportato che MUFA e PUFA prevengono le riduzioni indotte dal palmitato della sensibilità all’Insulina e veicolano effetti antinfiammatori nelle cellule del muscolo scheletrico. [30 , 37, 38] In effetti, la regolazione trascrizionale dipendente da NF-kB è stata implicata nel promuovere l’atrofia muscolare da disuso nel muscolo soleo di ratto aumentando l’attivazione mediata da FoxO del promotore MuRF1. [39] Inoltre, uno studio recente ha dimostrato che la diminuzione del segnale anabolico nel muscolo scheletrico di topi anziani coincideva con l’accumulo di Ceramide intramuscolare e DAG, nonché con una maggiore abbondanza di mRNA di TNF-α. [40] È interessante notare che la somministrazione di olio di pesce ai suinetti in fase di svezzamento, che ha portato all’arricchimento di EPA, DHA e contenuto totale di PUFA omega-3 all’interno del muscolo gastrocnemio, ha coinciso con una riduzione dei livelli di TNF-α muscolare e una ridotta espressione del recettore Toll-like 4 (TLR4), un recettore bersaglio per gli acidi grassi saturi che stimola la segnalazione proinfiammatoria in risposta alla sua attivazione. [41] In particolare, è stato riportato che la stimolazione del TLR4 da parte del suo ligando lipopolisaccaride induce il catabolismo muscolare nei miotubi C2C12 attraverso l’attivazione delle vie ubiquitina-proteasoma e autofagia-lisosoma. [42] Inoltre, è stato dimostrato che il trattamento con DHA in cellule muscolari umane co-coltivate con macrofagi attenua il contenuto proteico indotto dai macrofagi di Fn14, un modulatore positivo dell’espressione di MuRF-1. [43 , 44] Pertanto, sulla base di questi risultati, è concepibile che le azioni antinfiammatorie riportate degli acidi grassi insaturi nelle cellule muscolari scheletriche possano contribuire, almeno in parte, alla loro capacità di preservare la massa e/o la funzione muscolare.
In particolare, queste azioni protettive possono essere collegate a miglioramenti della funzione mitocondriale, la cui compromissione è stata suggerita dal tipo di dieta e/o all’atrofia muscolare indotta dall’età. [45 , 46] Ad esempio, un recente studio di Roseno e colleghi ha riportato che una dieta ricca di grassi a breve termine (3 settimane) ha aumentato l’atrofia muscolare da denervazione nei topi inducendo la degradazione proteica nel soleo ricco di mitocondri, ma non nel muscolo EDL glicolitico. In particolare, la denervazione di 14 giorni ha indotto una perdita del contenuto proteico mitocondriale nel soleo ma non nell’EDL, indipendentemente dalla dieta. Pertanto, questi risultati suggeriscono che la perdita di mitocondri indotta dalla denervazione e la compromissione della funzione mitocondriale indotta da una dieta ricca di grassi possono combinarsi per promuovere l’atrofia del muscolo scheletrico. Al contrario, uno studio indipendente di Tardif e collaboratori ha dimostrato che i ratti anziani alimentati con una dieta arricchita di oleati mostrano notevoli miglioramenti nella sensibilità all’insulina e un aumento della sintesi proteica muscolare, in concomitanza con una maggiore espressione di geni implicati nella stimolazione della ossidazione mitocondriale, compreso il recettore attivato dal proliferatore dei perossisomi PPARα e PPARβ, nonché CPT-1β. [47 , 48] Inoltre, è stato dimostrato che i miotubi C2C12 trattati con PUFA acido linolenico e ALA mostrano una maggiore attivazione del AMPK, un altro regolatore positivo chiave della -ossidazione mitocondriale. [49] Inoltre, è stato riportato che il DHA inibisce la degradazione proteica nei miotubi C2C12 attraverso una via PPARγ-dipendente. [50] Infatti, la capacità ossidativa mitocondriale potenziata e/o preservata, come precedentemente riportato in risposta alla sola o co-fornitura di acidi grassi insaturi, può anche aiutare a prevenire l’accumulo intramuscolare di intermedi lipotossici come la Ceramide che sono stati implicati nella promozione dell’atrofia muscolare. [51 , 52] Inoltre, è possibile che l’integrazione di PUFA possa agire per alterare le proprietà contrattili e metaboliche muscolari, ad esempio promuovendo un passaggio da fibre glicolitiche veloci a fibre lente (ossidative), cosa non propriamente positiva per un atleta di potenza. A sostegno di questa idea, lavori precedenti hanno dimostrato che l’alimentazione di ratti Wistar con una dieta arricchita in PUFA n-3 porta alla sovraregolazione delle proteine implicate nell’attivazione del metabolismo ossidativo (ad es. nel muscolo EDL, un tessuto muscolare dominante di tipo veloce). [53] È interessante notare che questo cambiamento metabolico mediato da PUFA ha coinciso anche con livelli ridotti di proteine dell’isoforma di tipo veloce MyHC-2b (catena pesante della miosina 2b) nel muscolo EDL. Pertanto, è concepibile che uno spostamento mediato dai PUFA verso un tipo di fibra muscolare a lenta ossidazione possa contribuire, almeno in parte, a guadagni benefici nella massa muscolare e/o nella funzione metabolica.
In alternativa, la regolazione della massa muscolare da parte dei lipidi può anche comportare la modulazione dell’autofagia, un meccanismo omeostatico che facilita la degradazione e il riciclaggio di proteine e organelli attraverso il macchinario lisosomiale. [54] In particolare, è stato riportato che un aumento della degradazione autofagica coincide con l’atrofia muscolare in varie condizioni e/o patologie tra cui cancro, [55] denervazione, [56] e invecchiamento. [55 , 57] Inoltre, è stato dimostrato che un’alimentazione ricca di grassi a breve termine (3 settimane) aumenta l’abbondanza di marcatori autofagosomiali nel soleo denervato dei topi. In accordo con ciò, Yuzefovych e collaboratori hanno dimostrato un aumento dell’autofagia nei miotubi L6 a seguito della somministrazione di palmitato. Pertanto, sebbene non sia ancora stato stabilito un collegamento diretto in vivo , è ipotizzabile che il turnover proteico alterato tramite l’ autofagia possa, almeno in parte, mediare le alterazioni indotte dai lipidi nella massa muscolare.
Va inoltre evidenziato che alcuni acidi grassi insaturi possono alterare il tasso di proliferazione delle cellule satelliti che funzionano come cellule progenitrici miogeniche necessarie per la crescita e la rigenerazione muscolare. Ad esempio, è stato dimostrato che DHA ed EPA inibiscono la proliferazione dei mioblasti C2C12 e delle cellule satelliti isolate dal muscolo di tacchino. [58 , 59] In particolare, queste azioni di soppressione della crescita sono state collegate a livelli ridotti di ciclina E e CDK2, proteine che svolgono un ruolo critico nella progressione del ciclo cellulare, nonché all’attivazione soppressa di ERK1/2, una proteina chinasi attivata da mitogeni implicata nel promuovere la crescita e la divisione cellulare. [59 , 60] Al contrario, è stato dimostrato che l’alimentazione di criceti distrofici carenti di δ-sarcoglicano con una dieta arricchita in ALA (un PUFA omega-3) aumenta la proliferazione e la differenziazione delle cellule satellite nel muscolo EDL, in concomitanza con un miglioramento dell’istologia muscolare. In particolare, queste risposte benefiche hanno coinciso con la capacità dell’ALA di aumentare la proporzione di miofibre α-MHC positive nel muscolo scheletrico dei criceti distrofici, insieme a una riduzione dell’espressione di β-MHC, contribuendo così alla conservazione di un α/β più fisiologico. Rapporto MHC. Inoltre, nello stesso studio, è stato anche dimostrato che l’integrazione di ALA alimentare previene l’accumulo aberrante citoplasmatico di proteine di membrana chiave nei muscoli adduttori dei criceti distrofici, inclusa la caveolina-3, una proteina coinvolta nella regolazione dell’adesione cellulare e della riparazione della membrana, nonché essendo implicato nel controllo della differenziazione muscolare e della segnalazione indotta dall’insulina. [61 , 62] Infatti, dato che le aberrazioni nella funzione e/o localizzazione della caveolina-3 sono state associate a vari fenotipi di malattie del muscolo scheletrico, [63 , 64 , 65 , 66 , 67] è plausibile che gli acidi grassi e/o i loro derivati lipidici possano influenzare la proliferazione delle cellule satellite e/o la differenziazione muscolare, almeno in parte, alterando la funzione e/o la localizzazione subcellulare delle isoforme della caveolina, nonché di altri componenti chiave della membrana strutturale.
Gli effetti degli acidi grassi sulla massa muscolare e sulla differenziazione possono essere mediati anche da una serie di metaboliti lipidici derivati. Ad esempio, è stato dimostrato che la capacità dell’acido arachidonico PUFA (C20; 4n-6) di aumentare le dimensioni, il contenuto mionucleare e il contenuto proteico dei miotubi C2C12 è mediata dall’attività della cicloossigenasi-2 (COX-2), implicando dipendenza da sintesi delle prostaglandine a valle. [68] In accordo con ciò, è stato riportato che la crescita indotta dall’acido arachidonico dei miociti C2C12 coincide con l’aumento della secrezione degli eicosanoidi PGF2α e PGE. [68]È interessante notare che diversi studi hanno anche documentato il ruolo positivo che le prostaglandine svolgono nel promuovere eventi precoci sulla superficie cellulare, inclusa l’adesione cellula-cellula, che successivamente mediano la fusione dei mioblasti nei miotubi. [69 , 70] Infatti, importanti studi di follow-up possono comportare la determinazione dell’esatta identità dei bersagli molecolari attraverso i quali le prostaglandine mediano le loro azioni, ad esempio agendo sui recettori prostanoidi legati alla proteina G (es. EP1). [69 , 71] Al contrario, è stato recentemente riportato che un altro metabolita lipidico derivato dall’acido arachidonico noto come 2-arachidonoilglicerolo (2-AG), un ligando lipidico endogeno chiave del sistema endocannabinoide, inibisce la differenziazione delle cellule satellite primarie umane e dei mioblasti murini C2C12 prendendo di mira il recettore-G ‐ 1 dei cannabinoidi accoppiati a proteine e sua successiva inibizione dei canali Kv7.4. [72] Pertanto, non si può escludere il possibile coinvolgimento di tali intermedi lipidici nella regolazione del catabolismo muscolare in risposta a determinate condizioni patologiche o alimentari.
L’aumento dell’adiposità osservato nell’invecchiamento umano è stato collegato a risposte sintetiche proteiche muscolari alterate negli individui anziani. [73] Pertanto, è ipotizzabile che i cambiamenti nei livelli e/o nella composizione dei lipidi possano contribuire all’atrofia muscolare in queste condizioni ma non limitatamente ad esse. A sostegno di questa idea, ci sono prove che suggeriscono che la modifica della composizione alimentare può avere un impatto sulla massa muscolare e/o sulla sua funzione negli esseri umani. Ad esempio, uno studio di McGlory e collaboratori ha riportato che il consumo di olio di pesce ha aumentato il contenuto di PUFA omega-3 del muscolo ( Vastus lateralis) in individui maschi sani, che ha coinciso con un’elevata espressione di proteine di segnalazione anabolizzanti incluso mTOR. [74] Inoltre, è stato riportato che l’inclusione di MUFA e PUFA nella dieta riduce l’espressione di geni lipogenici nel muscolo scheletrico di soggetti insulino-resistenti, in concomitanza con ridotti tassi di sintesi frazionaria di DAG intramuscolare e triacilgliceroli. [75] In particolare, queste risposte benefiche possono essere collegate a miglioramenti nella sensibilità all’insulina veicolati dall’integrazione alimentare di PUFA omega-3 negli esseri umani. [76 , 77 , 78]
E’ stato riportato che l’integrazione alimentare con acidi grassi insaturi può anche agire per migliorare le prestazioni fisiche e/o aumentare gli effetti metabolici benefici associati all’esercizio, in particolare in individui sedentari o non allenati . [79] A sostegno di questa idea, è stato dimostrato che un basso apporto alimentare di olio di tonno promuove la resistenza all’affaticamento muscolare nei ratti, in concomitanza con un aumento selettivo del contenuto di fosfolipidi della membrana e DHA all’interno del muscolo gastrocnemio. [80 , 81] Pertanto, un approccio più integrato che coinvolga modifiche al consumo di grassi nella dieta e un aumento dell’esercizio fisico può fornire una strategia più efficace per alleviare gli effetti deleteri associati all’atrofia muscolare. Ma stiamo comunque parlando di effetti apprezzabili su soggetti sedentari e non allenati, non su culturisti intermedi o avanzati.
Tirando le somme di quanto fino qui esposto, c’è un certo interesse giustificato sul ruolo importante che gli acidi grassi e/oi loro derivati lipidici possono svolgere nella modulazione della massa muscolare e della sua funzione. Nel complesso, le prove presentate indicano che gli acidi grassi saturi agiscono per trasmettere effetti dannosi sulla funzione muscolare, ad esempio alterando o riducendo la sintesi proteica e accentuando il catabolismo. Al contrario, è stato dimostrato che diversi acidi grassi insaturi contrastano molte delle azioni pro-cataboliche associate alla assunzione di acidi grassi saturi. Tuttavia, sarà necessari ulteriori studi per delineare le vie e i processi alla base dell’atrofia muscolare indotta da acidi grassi, nonché quelli che mediano miglioramenti nella funzione muscolare in risposta alla fornitura di acidi grassi insaturi (cioè aumento della sintesi proteica, ridotta atrofia, miglioramento della funzione metabolica). A tal fine, strategie volte ad alterare il contenuto e/o la composizione lipidica intramuscolare in quelle condizioni che possono favorire l’atrofia muscolare (es. aumento dell’obesità, invecchiamento, inattività fisica), ad esempio sopprimendo l’accumulo di mediatori lipidici come le Ceramidi (es. de novosintesi di ceramide) o prostaglandine (ad es. inibendo l’attività della COX-2), possono fornire informazioni utili sul ruolo che diverse classi di lipidi svolgono nella modulazione della massa e della funzione muscolare. È importante sottolineare che tale lavoro potrebbe implicare l’uso di modelli animali pertinenti o soggetti umani che richiederebbero di prendere in considerazione fattori come il background genetico, la composizione della dieta e l’apporto calorico. Inoltre, questi studi possono anche comportare la determinazione di potenziali alterazioni indotte dai lipidi dell’architettura muscolare e della composizione del tipo di fibra che possono influenzare la forza muscolare, nonché il monitoraggio dei cambiamenti nella segnalazione intramuscolare e nei metaboliti all’interno di specifici tipi di fibre muscolari. Insieme a questo, ulteriori lavori che esplorano il ruolo degli acidi grassi e degli intermedi lipidici nella regolazione della proliferazione, sarebbe necessario eseguire la differenziazione e/o la funzione delle cellule satellite derivate da muscoli umani e dei miotubi primari al fine di effettuare confronti appropriati con i dati ottenuti in altri modelli sperimentali (ad es. miotubi C2C12), che hanno dimostrato di mostrare differenze funzionali (ad es. grado di maturazione).[82] Nel complesso , i dati ottenuti da tali studi possono portare allo sviluppo di nuove strategie terapeutiche per contrastare l’atrofia muscolare e/o migliorare la capacità rigenerativa a seguito di lesioni o malattie.
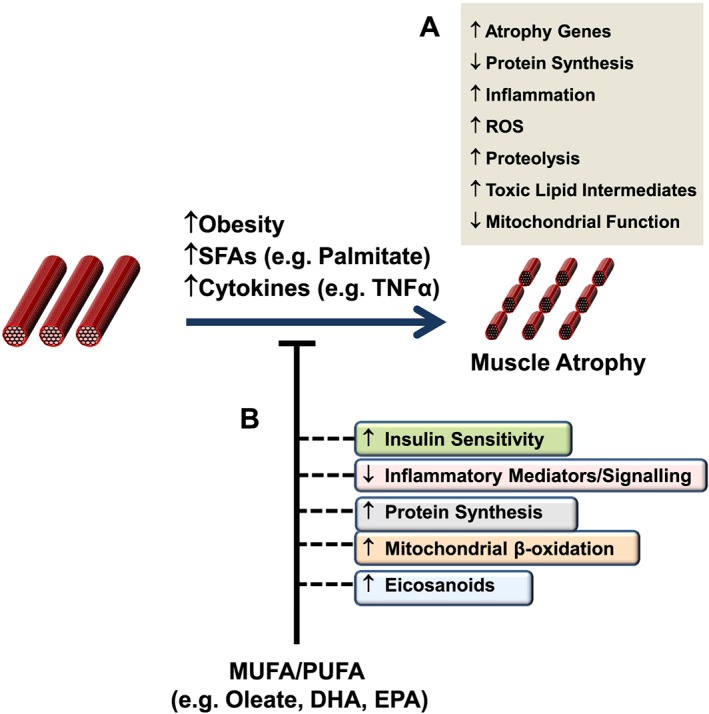
Potenziali meccanismi attraverso i quali gli acidi grassi insaturi possono contrastare l’obesità e/o l’atrofia del muscolo scheletrico indotta da acidi grassi saturi. Livelli elevati di acidi grassi saturi e/o citochine proinfiammatorie, ad esempio durante l’obesità e ipoteticamente anche nel sovrappeso marcato, possono favorire lo sviluppo dell’atrofia del muscolo scheletrico attraverso varie vie e processi come indicato (A). È importante sottolineare che gli acidi grassi monoinsaturi e polinsaturi hanno dimostrato di contrastare queste azioni pro-atrofiche, che possono essere mediate dalla loro capacità di aumentare la sensibilità all’insulina e la produzione di eicosanoidi protettivi, oltre a migliorare la capacità ossidativa mitocondriale e la sintesi proteica, mentre riducono contemporaneamente la spinta proinfiammatoria (B).
Torniamo alla pratica: tesi, antitesi e sintesi del “Bulk High Fat”.
Quindi, cosa concludere da tutto ciò che ho esposto proveniente dalla letteratura scientifica se parliamo di Bodybuilding e alimentazione “High Fat” in ipercalorica? Quanto segue:
La cosa certa, e risaputa in tutti i contesti dietetici, è quella di preoccuparsi di ridurre l’assunzione di acidi grassi saturi a circa e non oltre il 10% del totale calorico giornaliero (soprattutto l’AcidoPalmitico, Miristico e Laurico);
Concentrare l’assunzione di acidi grassi utilizzando fonti principalmente di Omega-3 (con particolare attenzione ad assumere EPA e DHA in un range tra 1 e 5g/die)
Assumere una buona dose di GLA (Omega-6) la quale può portare ad una migliore risposta nella biosintesi di prostaglandine anti-infiammatorie;
Possibilità di assumere Acido Arachidonico (Omega-6) in un range di dosaggio tra i 100mg ed 1.5g/die (quest’ultimo dosaggio va calibrato con un apporto di EPA/DHA pari ad un minimo di 2,5g/die) che può portare ad un miglioramento della biosintesi di prostaglandine anti-infiammatorie e con attività anabolizzante (vedi, per esempio, la PGF2-α responsabile con la PGE2 della regolazione della proteolisi e della sintesi proteica; così come della proliferazione, differenziazione e fusione delle celle satellite);
Uno schema di ripartizionamento dei macronutrienti potrebbe essere il seguente: 30-40% di Grassi, 30-20% di Carboidrati e 40-30% di Proteine.
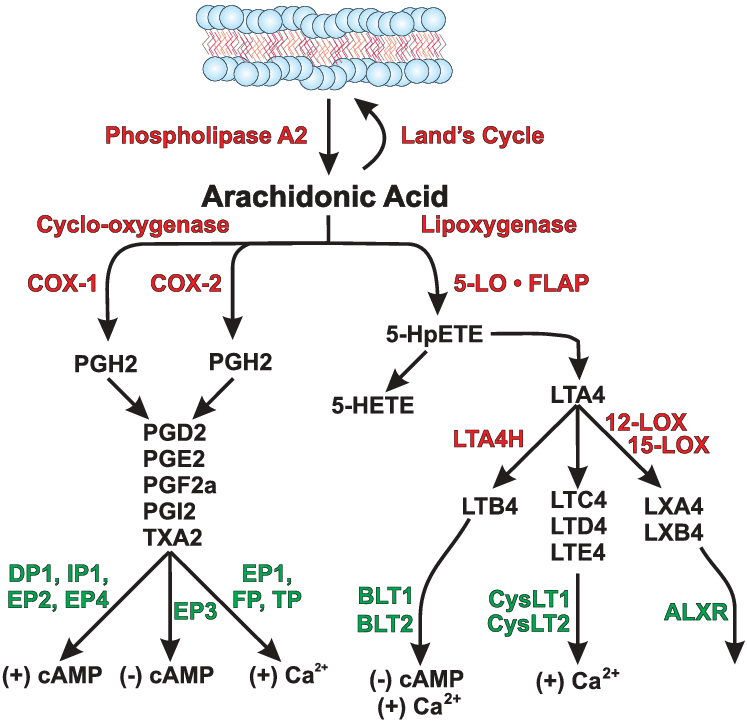
Metabolismo e vie di segnalazione dell’Acido Arachidonico. Sono riassunte 3 delle 4 principali vie metaboliche dell’Acido Arachidonico: il ciclo di Terra, la conversione in prostaglandine che richiede l’attività della cicloossigenasi e la conversione in leucotrieni e lipossine tramite l’attività della 5-lipossigenasi. Non è mostrato il metabolismo del citocromo P-450 dell’Acido Arachidonico. I processi enzimatici sono mostrati in rosso, i substrati ei prodotti sono mostrati in nero e i recettori di superficie della proteina G delle cellule sono mostrati in verde. Gli effetti noti dell’attivazione del recettore sull’adenosina monofosfato ciclico (cAMP) a valle o sui livelli di calcio intracellulare sono mostrati in basso. COX-1, cicloossigenasi-1; COX-2, cicloossigenasi-2; 5-LO, 5-lipossigenasi; FLAP, proteina attivante la 5-lipossigenasi; PGH2, PGD2, PGE2, PGF2a e PGI2, sono prostaglandine H2, D2, E2, F2a e I2, rispettivamente; TXA2: trombossano A2; acido 5-HpETE, 5S-idroperossi-6E,8Z,11Z,14Z-eicosatetraenoico; acido 5-HETE, 5S-idrossi6E,8Z,11Z,14Z-eicosatetraenoico; LTA4, LTB4, LTC4, LTD4 e LTE4 sono i leucotrieni A4, B4, C4, D4 ed E4, rispettivamente; LXA4 e LXB4, lipossine A4 e B4; DP1, EP1-EP4, FP e IP, recettori delle prostaglandine; TP, recettore del trombossano A2; BLT1 e BLT2, recettore dei leucotrieni B4; CysLT1 e CysLT2: recettori dei cisteinil leucotrieni; ALXR: recettore delle lipossine.
Partendo da questi punti si hanno delle “linee guida” finalizzate alla qualità degli acidi grassi e alla quantità media percentuale.
Ora, mettiamo il caso che un soggetto per caratteristiche genetiche non riesca a gestire grosse quantità di glucidi e abbia un buon metabolismo lipidico sul quale poter puntare per eventuali surplus calorici.
Il soggetto in questione si trova bene con un quantitativo di CHO pari a 3g/Kg, e pesando 80Kg la suo quota glucidica giornaliera ammonta a 240g. L’assunzione proteica giornaliera è pari a 2,5g/Kg, per un totale di 200g.
Il suo TDEE di mantenimento è di circa 2.500Kcal. Punta ad un surplus calorico giornaliero di 500Kcal (obbiettivo 3000Kcal/die). Dal momento che sa già quanti CHO e proteine dovrà assumere e a quante calorie ammontano (1760Kcal totali da CHO e Pro) il restante lo andrà ad assumere dai Grassi (1240Kcal = circa 138g di Fat).
Totale macronutrienti del soggetto:
Carboidrati: 240g
Proteine: 200g
Grassi: 138g (di cui il 10% di Saturi ed il restante 90% di insaturi, polinsaturi e monoinsaturi; presenza di 5g/die di EPA e DHA, 300mg/die di GLA e 1.5g/die di Acido Arachidonico per 8-12 settimane).
Il soggetto in questione seguirà questo schema per circa 4 settimana senza effettuare alcuna giornata con un aumento dei CHO e una riduzione della componente proteica e lipidica. In base alle risposte ottenute rimodulerà a bisogno il suo schema alimentare.
Ecco, questo è un banalissimo esempio di come una dieta “Bulk High Fat” possa essere per lo meno impostata in modo logico, anche per quanto riguarda il tipo ipotetico di soggetto menzionato. Nonostante ciò, e vi parlo da persona che ha analizzato i risultati di svariati schemi similari a questo, un vantaggio vero e proprio sulla qualità dell’aumento di peso non è praticamente mai emerso, o per lo meno mai in modo sufficientemente apprezzabile.
A questo punto ci sarebbe anche da riportare quanto affermato in uno studio dal Dr. Jose Antonio, PhD, ricercatore della ISSN, che ha affermato come non vi siano aumenti maggiori di grasso corporeo tra una dieta ipercalorica “High Fat” e una “High Carbs” dopo circa 14 giorni.[https://italia-podcast.it/] Questa affermazione va comunque presa come una ipotesi nata da osservazione di alcuni studi. Sebbene tra la teoria e la pratica spesso c’è differenza, quando si parla di surplus calorico e aumento del grasso corporeo sappiamo tutti quanto gli acidi grassi alimentari vengano facilmente stoccati negli adipociti sotto forma di trigliceridi di deposito quando in surplus calorico e ingeriti in quantità significative con carboidrati. Questi ultimi, o meglio il glucosio, in eccesso, in un soggetto in salute vengono in gran parte dissipati in calore e solo un 5-10% è coinvolto nei processi di De Novo Lipogenesi (DNL).
Alcuni sostenitori delle diete “High Fat” in “Bulk” affermano che tali strategie alimentari portino ad un miglioramento della “durezza muscolare”, anche grazie ad un basso stimolo insulinico e consequenziale riduzione della ritenzione idrica, e della pienezza dei ventri data dai Trigliceridi intra-muscolari. Peccato che, Insulina o meno, gli acidi grassi in eccesso vengono captati e depositati negli adipociti per intervento, tra l’altro, della Proteina Stimolante l’Acilazione (ASP), la quale ha azione insulino-indipendente. I Trigliceridi intra-muscolari (IMTG) danno si un effetto di pienezza muscolare dal momento che legano in piccola parte acqua, ma parliamo di un effetto additivo al ben maggiore volume dato dal glicogeno muscolare e apprezzabile a “bf” basse, da pre-contest per intenderci.
Come molti di voi già sapranno, la sintesi proteica aumenta principalmente:
In seguito ad adattamenti in risposta a sedute di allenamento contro-resistenza correttamente svolte generando tensione meccanica, stress metabolico e danno muscolare;
Mangiando sufficienti calorie;
Assumendo un quantitativo adeguato di proteine (media 1,5-2,5g/Kg).
Raggiungere il primo punto, però, diventa molto più difficile se si consuma una quota glucidica bassa. Le diete ricche di grassi possono fornire molte calorie e proteine, ma risultano per la stragrande maggioranza dei soggetti disfunzionali alle prestazioni in sala pesi. Ricordo, per l’ennesima volta, che un bodybuilder è un atleta che sfrutta primariamente il metabolismo glucidico al fine della massima prestazione essendo praticante di uno sport anaerobico alattacido e lattacido.
Durante l’esercizio intenso, i muscoli si basano principalmente sul metabolismo glucidico. E come certamente saprete, il glicogeno è un polimero di glucosio immagazzinato nel muscolo-scheletrico e nel fegato, ed è scomposto nelle sue unità di glucosio e utilizzato come substrato energetico durante l’esercizio nel distretto muscolare di deposito oppure venendo rilasciato nel flusso ematico dal deposito epatico.
Le richieste di carboidrati nella dieta variano da soggetto a soggetto, ma un culturista che si allena seriamente richiede in media una consistente quantità di glucidi per mantenere le sue riserve di glicogeno e sostenere una buona prestazione, motivo per cui le diete ricche di grassi e povere di carboidrati riducono significativamente i livelli di questo polimero. Ed i problemi annessi emergono soprattutto quando ci si allena con pesi consistenti.
Fare solo 6-9 serie può ridurre i livelli di glicogeno muscolare di circa il 40% [83] e, se ci si allena con livelli già bassi, è ragionevole presumere che si farà fatica a sollevare più peso.
Ma cosa mostrano gli studi sugli atleti? La maggior parte degli studi dimostrano che le diete ricche di grassi e povere di carboidrati riducono le prestazioni atletiche [84, 85, 86] mentre altri affermano che non vi è alcuna differenza [87]e persino alcuni che mostrano un aumento delle prestazioni.[88] Tuttavia, ci sono altri motivi per pensare che le diete ad alto contenuto di carboidrati potrebbero essere superiori per l’aumento della massa muscolare.
La ricerca mostra che la disponibilità di glicogeno influenza direttamente la sintesi proteica ei tassi di degradazione.[89] In poche parole: le diete ricche di grassi e povere di carboidrati comportano livelli inferiori di sintesi proteica rispetto a quelle ad alto contenuto di carboidrati.
Un altro modo in cui i carboidrati influenzano favorevolmente l’equilibrio proteico muscolare ha a che fare con la produzione di Insulina.
L’insulina è un ormone peptidico rilasciato dal pancreas che agevola l’assorbimento dei nutrienti dal sangue alle cellule. Possiede, in fisiologia, proprietà prettamente anticataboliche, il che significa che quando i livelli di Insulina sono elevati, viene soppressa la lisi delle proteine muscolari.[90]
Ora, poiché la produzione di Insulina è stimolata ingerendo cibo, e mangiando carboidrati in particolare, ma anche proteine con stimoli differenti dati dalla composizione amminoacidica, non sorprende che le persone che seguono una dieta ricca di carboidrati abbiano generalmente livelli ottimali di Insulina rispetto alle persone che seguono una dieta povera di carboidrati e ricca di grassi.
La formazione del complesso Insulina (di colore rosso) – Recettore dell’Insulina (di colore blu) determina una cascata di segnali per l’esplicazione della funzione dell’ormone. Ispirato da discussioni e cifre in Siddle (2011). L’immagine del complesso Recettore Insulina-Insulina è adattata da Goodsell (2015).
I carboidrati sono principalmente energetici e se non si è molto attivi, non si necessita di loro quantità elevate. Tuttavia, livelli ottimali di insulina (e di sensibilità insulinica) sono altamente desiderabili se si sta cercando di costruire massa muscolare, semplicemente perché si viene a creare un ambiente più anabolico in cui i muscoli possono vedere agevolata la loro crescita. E questa non è solo teoria.
La ricerca condotta dagli scienziati della Ball State University ha scoperto che bassi livelli di glicogeno muscolare (che è inevitabile con una dieta povera di carboidrati e ricca di grassi) compromettono la segnalazione cellulare post-allenamento relativa alla crescita muscolare.[91]
Un altro studio condotto da ricercatori dell’Università della Carolina del Nord ha scoperto che, se combinata con l’esercizio quotidiano, una dieta povera di carboidrati e ricca di grassi aumenta i livelli di Cortisolo a riposo e diminuisce i livelli di Testosterone libero.[92] Sicuramente una situazione compromettente soprattutto per un “Natural”. Quando si tratta di costruire massa muscolare, ciò che si vuole sono bassi livelli di Cortisolo a riposo e alti livelli di Testosterone libero, l’esatto opposto di ciò che porta una dieta a basso contenuto di carboidrati.
Quanto detto aiuta a spiegare i risultati di altri studi sulla questione dei carboidrati e della composizione corporea e delle prestazioni.
Ad esempio, uno studio condotto da ricercatori dell’Università del Rhode Island ha esaminato come l’assunzione di carboidrati a basso e ad alto contenuto di carboidrati influenzasse il danno muscolare indotto dall’esercizio, il recupero della forza e il metabolismo proteico di tutto il corpo dopo un intenso allenamento. Quello che hanno scoperto è che i soggetti con una dieta a basso contenuto di carboidrati hanno perso più forza, si sono ripresi più lentamente e hanno mostrato livelli più bassi di sintesi proteica. Vale anche la pena notare che il gruppo “a basso contenuto di carboidrati” (e quindi ad alto contenuto di grassi) non era poi così “Low Carbs”. Stavano ingerendo circa 220g di carboidrati al giorno contro i 350g assunti dal gruppo con dieta ad alto contenuto di carboidrati.[93] E questi effetti diventano ancora più pronunciati man mano che l’assunzione di carboidrati diminuisce.
Ancora un altro studio degno di nota è stato condotto dai ricercatori della McMaster University, che ha confrontato la dieta ad alto e basso contenuto di carboidrati con soggetti che eseguono allenamenti quotidiani per le gambe. I soggetti che seguono una dieta a basso contenuto di carboidrati (30% delle calorie giornaliere) hanno mostrato tassi più elevati di degradazione proteica e tassi più bassi di sintesi proteica rispetto ai soggetti a dieta ricca di carboidrati (60% delle calorie giornaliere), con conseguente minore crescita muscolare complessiva.[94]
Conclusionie ultime indicazioni:
La linea di fondo è questa: Alcuni studi dimostrano che le diete ricche di grassi possono funzionare per la costruzione muscolare, ma un approccio più ideale e redditizio sembra rimanere quello di seguire una dieta ricca di carboidrati secondo capacità individuali.
I dati empirici ci suggeriscono mediamente la stessa medesima cosa, senza però cadere nell’errore di valutare le innumerevoli variabili soggettive in un unico macro-gruppo di risposta positiva ai regimi prevalentemente glucidici.
Questo significa, in poche parole, che la soggettività genetica non va mai sottovalutata o peggio rinnegata per via di futili credenze pseudoscientifiche o convinzioni personali.
Ciò non toglie, però, che i regimi ipercalorici “High Fat” non portano a sostanziali benefici se non ad un ristretto numero di individui caratterizzati da ipotetiche alterazioni metaboliche rispetto alla media della popolazione. Ma anche in questi casi il “tetto favorevole” del “High Fat” si attesta mediamente al 40% delle calorie totali giornaliere con un totale glucidico del 30-25%.
Ciò che dobbiamo ricordare è che i test alimentari soggettivi con modulazioni macro-caloriche sono utili al fine di attestare una quota singolarmente funzionale per ogni macronutriente. Solo in questo modo, e con una valutazione oggettiva, possiamo capire fin dove possiamo spingerci in quanto a quantità di macronutrienti al fine di ottenerne il massimo dei vantaggi prestativi ed estetici. Questo può significare differenze non da poco in un piano ipercalorico tra Grassi e Carboidrati.
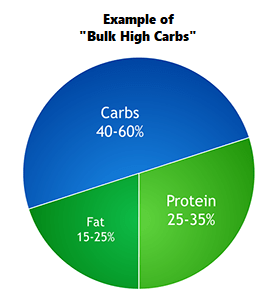
Altri tre esempi di variazione nella percentuale dei macronutrienti in funzione delle risposte soggettive.
Personalmente, ritengo che il range della quota lipidica dovrebbe coprire un arco di grammatura da 0,6 a 2g/Kg. Sempre su base soggettiva e rapporto funzionale tra carboidrati e grassi.
In regimi ipercalorici, con l’obbiettivo di non perdere la flessibilità metabolica, si possono utilizzare, oltre alla “C:G ratio” di Ludovico Lemme, uno schema d’esempio di rapporto come segue:
CHO 4-5g/Kg= Fat 0,6g/Kg
CHO 5-6g/Kg= Fat 0,8g/Kg
CHO 6-7g/Kg= Fat 1g/Kg
CHO 7-8g/Kg= Fat 1,5/Kg
CHO 8-10g/Kg= Fat 2g/Kg
E se volessi tarare il quantitativo lipidico per un regime “Bulk High Fat”? Le linee indicative sarebbero:
CHO 3-4g/Kg= Fat 1-1,2g/Kg
CHO 2.5-3g/Kg= Fat 1,5-1,6g/Kg
CHO 2-2.5g/Kg= Fat 1,8-2g/Kg
CHO 0,5-2g/Kg= Fat 2-2,5g/Kg
Queste sono semplici indicazioni generali estrapolate da dati raccolti empiricamente, nulla di scolpito nella roccia o inconfutabilmente dimostrato.
Prima di chiudere con il presente articolo, è logico che vi dica di fare attenzioni alle fonti lipidiche che assumete o che andrete ad assumere (cosa già detta in precedenza). Nessuno vi sta dicendo di evitare la carne rossa (tagli magri) ma di concentrarvi soprattutto su fonti lipidiche quali tuorlo d’uovo, Olio Extravergine di Oliva, EPA, DHA e MCT (controllandone la composizione ed evitando l’olio di Cocco ricco di Acido Laurico). Fonti proteiche quali salmone selvatico, sgombro, pesce spada e carni da allevamenti “Grass Feed” sono pienamente consigliate, se alla propria portata economica.
Gabriel Bellizzi
Riferimenti:
Dr. Mauro Di Pasquale, bodybuilding.com, accessed February 21, 2007. His articles and books have been translated into Italian language and published in Italy by Sandro Ciccarelli Olympian’s News magazine.
Woodworth‐Hobbs ME, Hudson MB, Rahnert JA, Zheng B, Franch HA, Price SR. Docosahexaenoic acid prevents palmitate‐induced activation of proteolytic systems in C2C12 myotubes. J Nutr Biochem 2014;25:868–74.
Graves E, Hitt A, Pariza MW, Cook ME, McCarthy DO. Conjugated linoleic acid preserves gastrocnemius muscle mass in mice bearing the colon‐26 adenocarcinoma. Res Nurs Health 2005;28:48–55.
Langen RC, Schols AM, Kelders MC, van der Velden JL, Wouters EF, Janssen‐Heininger YM. Muscle wasting and impaired muscle regeneration in a murine model of chronic pulmonary inflammation. Am J Respir Cell Mol Biol 2006;35:689–96.
Whitehouse AS, Smith HJ, Drake JL, Tisdale MJ. Mechanism of attenuation of skeletal muscle protein catabolism in cancer cachexia by eicosapentaenoic acid. Cancer Res 2001;61:3604–9.
Castillero E, Martin AI, Lopez‐Menduina M, Villanua MA, Lopez‐Calderon A. Eicosapentaenoic acid attenuates arthritis‐induced muscle wasting acting on atrogin‐1 and on myogenic regulatory factors. Am J Physiol Regul Integr Comp Physiol 2009;297:R1322–31.
Fiaccavento R, Carotenuto F, Vecchini A, Binaglia L, Forte G, Capucci E, et al. An omega‐3 fatty acid‐enriched diet prevents skeletal muscle lesions in a hamster model of dystrophy. Am J Pathol 2010;177:2176–84.
Briolay A, Jaafar R, Nemoz G, Bessueille L. Myogenic differentiation and lipid‐raft composition of L6 skeletal muscle cells are modulated by PUFAs. Biochim Biophys Acta 2013;1828:602–13.
Watson ML, Coghlan M, Hundal HS. Modulating serine palmitoyl transferase (SPT) expression and activity unveils a crucial role in lipid‐induced insulin resistance in rat skeletal muscle cells. Biochem J 2009;417:791–801.
. Yuzefovych L, Wilson G, Rachek L. Different effects of oleate vs. palmitate on mitochondrial function, apoptosis, and insulin signaling in L6 skeletal muscle cells: role of oxidative stress. Am J Physiol Endocrinol Metab 2010;299:E1096–105.
Mahfouz R, Khoury R, Blachnio‐Zabielska A, Turban S, Loiseau N, Lipina C, et al. Characterising the inhibitory actions of ceramide upon insulin signaling in different skeletal muscle cell models: a mechanistic insight. PLoS One 2014;9:e101865.
De Larichaudy J, Zufferli A, Serra F, Isidori AM, Naro F, Dessalle K, et al. TNF‐alpha‐ and tumor‐induced skeletal muscle atrophy involves sphingolipid metabolism. Skelet Muscle 2012;2:2.
Mebarek S, Komati H, Naro F, Zeiller C, Alvisi M, Lagarde M, et al. Inhibition of de novo ceramide synthesis upregulates phospholipase D and enhances myogenic differentiation. J Cell Sci 2007;120:407–16.
Turpin SM, Ryall JG, Southgate R, Darby I, Hevener AL, Febbraio MA, et al. Examination of ‘lipotoxicity’ in skeletal muscle of high‐fat fed and ob/ob mice. J Physiol 2009;587:1593–605.
Williamson DL, Dungan CM, Jadhav KS. High lipid concentrations regulate skeletal muscle REDD1. FASEB J 2013;27:Supplement 942.1.
Gordon BS, Williamson DL, Lang CH, Jefferson LS, Kimball SR. Nutrient‐induced stimulation of protein synthesis in mouse skeletal muscle is limited by the mTORC1 repressor REDD1. J Nutr 2015;145:708–13.
Leskawa KC, Erwin RE, Buse PE, Hogan EL. Glycosphingolipid biosynthesis during myogenesis of rat L6 cells in vitro. Mol Cell Biochem 1988;83:47–54.
Anastasia L, Papini N, Colazzo F, Palazzolo G, Tringali C, Dileo L, et al. NEU3 sialidase strictly modulates GM3 levels in skeletal myoblasts C2C12 thus favoring their differentiation and protecting them from apoptosis. J Biol Chem 2008;283:36265–71.
Papini N, Anastasia L, Tringali C, Dileo L, Carubelli I, Sampaolesi M, et al. MmNEU3 sialidase over‐expression in C2C12 myoblasts delays differentiation and induces hypertrophic myotube formation. J Cell Biochem 2012;113:2967–78.
Lipina C, Hundal HS. Ganglioside GM3 as a gatekeeper of obesity‐associated insulin resistance: evidence and mechanisms. FEBS Lett 2015;589:3221–7.
Gangoiti P, Bernacchioni C, Donati C, Cencetti F, Ouro A, Gomez‐Munoz A, et al. Ceramide 1‐phosphate stimulates proliferation of C2C12 myoblasts. Biochimie 2012;94:597–607.
Szendroedi J, Yoshimura T, Phielix E, Koliaki C, Marcucci M, Zhang D, et al. Role of diacylglycerol activation of PKCtheta in lipid‐induced muscle insulin resistance in humans. Proc Natl Acad Sci U S A 2014;111:9597–602.
You JS, Lincoln HC, Kim CR, Frey JW, Goodman CA, Zhong XP, et al. The role of diacylglycerol kinase zeta and phosphatidic acid in the mechanical activation of mammalian target of rapamycin (mTOR) signaling and skeletal muscle hypertrophy. J Biol Chem 2014;289:1551–63.
Shad BJ, Smeuninx B, Atherton PJ, Breen L. The mechanistic and ergogenic effects of phosphatidic acid in skeletal muscle. Appl Physiol Nutr Metab 2015;40:1233–41.
Bilim O, Takeishi Y, Kitahara T, Arimoto T, Niizeki T, Sasaki T, et al. Diacylglycerol kinase zeta inhibits myocardial atrophy and restores cardiac dysfunction in streptozotocin‐induced diabetes mellitus. Cardiovasc Diabetol 2008;7:2.
Haemmerle G, Zimmermann R, Hayn M, Theussl C, Waeg G, Wagner E, et al. Hormone‐sensitive lipase deficiency in mice causes diglyceride accumulation in adipose tissue, muscle, and testis. J Biol Chem 2002;277:4806–15.
Marignani PA, Epand RM, Sebaldt RJ. Acyl chain dependence of diacylglycerol activation of protein kinase C activity in vitro. Biochem Biophys Res Commun 1996;225:469–73.
Eichmann TO, Kumari M, Haas JT, Farese RV Jr, Zimmermann R, Lass A, et al. Studies on the substrate and stereo/regioselectivity of adipose triglyceride lipase, hormone‐sensitive lipase, and diacylglycerol‐O‐acyltransferases. J Biol Chem 2012;287:41446–57.
Macrae K, Stretton C, Lipina C, Blachnio‐Zabielska A, Baranowski M, Gorski J, et al. Defining the role of DAG, mitochondrial function, and lipid deposition in palmitate‐induced proinflammatory signaling and its counter‐modulation by palmitoleate. J Lipid Res 2013;54:2366–78.
Rando RR, Young N. The stereospecific activation of protein kinase C. Biochem Biophys Res Commun 1984;122:818–23.
Badin PM, Louche K, Mairal A, Liebisch G, Schmitz G, Rustan AC, et al. Altered skeletal muscle lipase expression and activity contribute to insulin resistance in humans. Diabetes 2011;60:1734–42.
Hyde R, Hajduch E, Powell DJ, Taylor PM, Hundal HS. Ceramide down‐regulates System A amino acid transport and protein synthesis in rat skeletal muscle cells. FASEB J 2005;19:461–3.
Guenther GG, Peralta ER, Rosales KR, Wong SY, Siskind LJ, Edinger AL. Ceramide starves cells to death by downregulating nutrient transporter proteins. Proc Natl Acad Sci U S A 2008;105:17402–7.
Nardi F, Hoffmann TM, Stretton C, Cwiklinski E, Taylor PM, Hundal HS. Proteasomal modulation of cellular SNAT2 (SLC38A2) abundance and function by unsaturated fatty acid availability. J Biol Chem 2015;290:8173–84.
Li F, Duan Y, Li Y, Tang Y, Geng M, Oladele OA, et al. Effects of dietary n‐6:n‐3 PUFA ratio on fatty acid composition, free amino acid profile and gene expression of transporters in finishing pigs. Br J Nutr 2015;113:739–48.
Coll T, Eyre E, Rodriguez‐Calvo R, Palomer X, Sanchez RM, Merlos M, et al. Oleate reverses palmitate‐induced insulin resistance and inflammation in skeletal muscle cells. J Biol Chem 2008;283:11107–16.
Salvado L, Coll T, Gomez‐Foix AM, Salmeron E, Barroso E, Palomer X, et al. Oleate prevents saturated‐fatty‐acid‐induced ER stress, inflammation and insulin resistance in skeletal muscle cells through an AMPK‐dependent mechanism. Diabetologia 2013;56:1372–82.
Wu CL, Cornwell EW, Jackman RW, Kandarian SC. NF‐kappaB but not FoxO sites in the MuRF1 promoter are required for transcriptional activation in disuse muscle atrophy. Am J Physiol Cell Physiol 2014;306:C762–7.
Rivas DA, McDonald DJ, Rice NP, Haran PH, Dolnikowski GG, Fielding RA. Diminished anabolic signaling response to insulin induced by intramuscular lipid accumulation is associated with inflammation in aging but not obesity. Am J Physiol Regul Integr Comp Physiol 2016;310:R561–9.
Liu Y, Chen F, Odle J, Lin X, Zhu H, Shi H, et al. Fish oil increases muscle protein mass and modulates Akt/FOXO, TLR4, and NOD signaling in weanling piglets after lipopolysaccharide challenge. J Nutr 2013;143:1331–9.
Doyle A, Zhang G, Abdel Fattah EA, Eissa NT, Li YP. Toll‐like receptor 4 mediates lipopolysaccharide‐induced muscle catabolism via coordinate activation of ubiquitin–proteasome and autophagy–lysosome pathways. FASEB J 2011;25:99–110.
Mittal A, Bhatnagar S, Kumar A, Lach‐Trifilieff E, Wauters S, Li H, et al. The TWEAK‐Fn14 system is a critical regulator of denervation‐induced skeletal muscle atrophy in mice. J Cell Biol 2010;188:833–49.
Finlin BS, Varma V, Nolen GT, Dube J, Starnes CP, Rasouli N, et al. DHA reduces the atrophy‐associated Fn14 protein in differentiated myotubes during coculture with macrophages. J Nutr Biochem 2012;23:885–91.
Jang YC, Lustgarten MS, Liu Y, Muller FL, Bhattacharya A, Liang H, et al. Increased superoxide in vivo accelerates age‐associated muscle atrophy through mitochondrial dysfunction and neuromuscular junction degeneration. FASEB J 2010;24:1376–90.
Gouspillou G, Sgarioto N, Kapchinsky S, Purves‐Smith F, Norris B, Pion CH, et al. Increased sensitivity to mitochondrial permeability transition and myonuclear translocation of endonuclease G in atrophied muscle of physically active older humans. FASEB J 2014;28:1621–33.
Muoio DM, Way JM, Tanner CJ, Winegar DA, Kliewer SA, Houmard JA, et al. Peroxisome proliferator‐activated receptor‐alpha regulates fatty acid utilization in primary human skeletal muscle cells. Diabetes 2002;51:901–9.
Tardif N, Salles J, Landrier JF, Mothe‐Satney I, Guillet C, Boue‐Vaysse C, et al. Oleate‐enriched diet improves insulin sensitivity and restores muscle protein synthesis in old rats. Clin Nutr 2011;30:799–806.
Park SY, Kim MH, Ahn JH, Lee SJ, Lee JH, Eum WS, et al. The stimulatory effect of essential fatty acids on glucose uptake involves both Akt and AMPK activation in C2C12 skeletal muscle cells. Korean J Physiol Pharmacol: Off J Korean Phys Soc Korean Soc Pharm 2014;18:255–61.
Wang Y, Lin QW, Zheng PP, Zhang JS, Huang FR. DHA inhibits protein degradation more efficiently than EPA by regulating the PPARgamma/NFkappaB pathway in C2C12 myotubes. BioMed Res Int 2013;2013:318981.
Lanza IR, Blachnio‐Zabielska A, Johnson ML, Schimke JM, Jakaitis DR, Lebrasseur NK, et al. Influence of fish oil on skeletal muscle mitochondrial energetics and lipid metabolites during high‐fat diet. Am J Physiol Endocrinol Metab 2013;304:E1391–403.
Lim JH, Gerhart‐Hines Z, Dominy JE, Lee Y, Kim S, Tabata M, et al. Oleic acid stimulates complete oxidation of fatty acids through protein kinase A‐dependent activation of SIRT1‐PGC1alpha complex. Nutr Metab 2013;288:7117–26.
Mizunoya W, Iwamoto Y, Shirouchi B, Sato M, Komiya Y, Razin FR, et al. Dietary fat influences the expression of contractile and metabolic genes in rat skeletal muscle. PLoS One 2013;8:e80152.
Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011;147:728–41.
Penna F, Costamagna D, Pin F, Camperi A, Fanzani A, Chiarpotto EM, et al. Autophagic degradation contributes to muscle wasting in cancer cachexia. Am J Pathol 2013;182:1367–78.
O’Leary MF, Vainshtein A, Carter HN, Zhang Y, Hood DA. Denervation‐induced mitochondrial dysfunction and autophagy in skeletal muscle of apoptosis‐deficient animals. Am J Physiol Cell Physiol 2012;303:C447–54.
Wenz T, Rossi SG, Rotundo RL, Spiegelman BM, Moraes CT. Increased muscle PGC‐1alpha expression protects from sarcopenia and metabolic disease during aging. Proc Natl Acad Sci U S A 2009;106:20405–10.
McFarland DC, Velleman SG, Pesall JE, Coy CS. Effect of lipids on avian satellite cell proliferation, differentiation and heparan sulfate proteoglycan expression. Comp Biochem Physiol A Mol Integr Physiol 2011;159:188–95.
Peng Y, Zheng Y, Zhang Y, Zhao J, Chang F, Lu T, et al. Different effects of omega‐3 fatty acids on the cell cycle in C2C12 myoblast proliferation. Mol Cell Biochem 2012;367:165–73.
Schevzov G, Kee AJ, Wang B, Sequeira VB, Hook J, Coombes JD, et al. Regulation of cell proliferation by ERK and signal‐dependent nuclear translocation of ERK is dependent on Tm5NM1‐containing actin filaments. Mol Biol Cell 2015;26:2475–90, doi:10.1091/mbc.E14-10-1453.
Parton RG, Way M, Zorzi N, Stang E. Caveolin‐3 associates with developing T‐tubules during muscle differentiation. J Cell Biol 1997;136:137–54.
Oshikawa J, Otsu K, Toya Y, Tsunematsu T, Hankins R, Kawabe J, et al. Insulin resistance in skeletal muscles of caveolin‐3‐null mice. Proc Natl Acad Sci U S A 2004;101:12670–5.
Galbiati F, Volonte D, Minetti C, Chu JB, Lisanti MP. Phenotypic behavior of caveolin‐3 mutations that cause autosomal dominant limb girdle muscular dystrophy (LGMD‐1C). Retention of LGMD‐1C caveolin‐3 mutants within the golgi complex. J Biol Chem 1999;274:25632–41.
Minetti C, Bado M, Broda P, Sotgia F, Bruno C, Galbiati F, et al. Impairment of caveolae formation and T‐system disorganization in human muscular dystrophy with caveolin‐3 deficiency. Am J Pathol 2002;160:265–70.
Park DS, Woodman SE, Schubert W, Cohen AW, Frank PG, Chandra M, et al. Caveolin‐1/3 double‐knockout mice are viable, but lack both muscle and non‐muscle caveolae, and develop a severe cardiomyopathic phenotype. Am J Pathol 2002;160:2207–17.
Capozza F, Combs TP, Cohen AW, Cho YR, Park SY, Schubert W, et al. Caveolin‐3 knockout mice show increased adiposity and whole body insulin resistance, with ligand‐induced insulin receptor instability in skeletal muscle. Am J Physiol Cell Physiol 2005;288:C1317–31.
Gazzerro E, Sotgia F, Bruno C, Lisanti MP, Minetti C. Caveolinopathies: from the biology of caveolin‐3 to human diseases. Eur J Hum Genet: EJHG 2010;18:137–45.
Markworth JF, Cameron‐Smith D. Arachidonic acid supplementation enhances in vitro skeletal muscle cell growth via a COX‐2‐dependent pathway. Am J Physiol Cell Physiol 2013;304:C56–67.
Hausman RE, Velleman SG. Prostaglandin E1 receptors on chick embryo myoblasts. Biochem Biophys Res Commun 1981;103:213–8.
Santini MT, Indovina PL, Hausman RE. Prostaglandin dependence of membrane order changes during myogenesis in vitro. Biochim Biophys Acta 1988;938:489–92.
Hausman RE, elGendy H, Craft F. Requirement for G protein activity at a specific time during embryonic chick myogenesis. Cell Differ Dev: The Official J Int Soc Dev Biol 1990;29:13–20.
Iannotti FA, Silvestri C, Mazzarella E, Martella A, Calvigioni D, Piscitelli F, et al. The endocannabinoid 2‐AG controls skeletal muscle cell differentiation via CB1 receptor‐dependent inhibition of Kv7 channels. Proc Natl Acad Sci U S A 2014;111:E2472–81.
Murton AJ, Marimuthu K, Mallinson JE, Selby AL, Smith K, Rennie MJ, et al. Obesity appears to be associated with altered muscle protein synthetic and breakdown responses to increased nutrient delivery in older men, but not reduced muscle mass or contractile function. Diabetes 2015;64:3160–71.
McGlory C, Galloway SD, Hamilton DL, McClintock C, Breen L, Dick JR, et al. Temporal changes in human skeletal muscle and blood lipid composition with fish oil supplementation. Prostaglandins Leukot Essent Fatty Acids 2014;90:199–206.
Jans A, van Hees AM, Gjelstad IM, Sparks LM, Tierney AC, Riserus U, et al. Impact of dietary fat quantity and quality on skeletal muscle fatty acid metabolism in subjects with the metabolic syndrome. Metabolism 2012;61:1554–65.
Rasic‐Milutinovic Z, Perunicic G, Pljesa S, Gluvic Z, Sobajic S, Djuric I, et al. Effects of N‐3 PUFAs supplementation on insulin resistance and inflammatory biomarkers in hemodialysis patients. Ren Fail 2007;29:321–9.
Dangardt F, Chen Y, Gronowitz E, Dahlgren J, Friberg P, Strandvik B. High physiological omega‐3 fatty acid supplementation affects muscle fatty acid composition and glucose and insulin homeostasis in obese adolescents. J Nutr Metab 2012;2012:395757.
Jimenez‐Gomez Y, Cruz‐Teno C, Rangel‐Zuniga OA, Peinado JR, Perez‐Martinez P, Delgado‐Lista J, et al. Effect of dietary fat modification on subcutaneous white adipose tissue insulin sensitivity in patients with metabolic syndrome. Mol Nutr Food Res 2014;58:2177–88.
Kawabata F, Neya M, Hamazaki K, Watanabe Y, Kobayashi S, Tsuji T. Supplementation with eicosapentaenoic acid‐rich fish oil improves exercise economy and reduces perceived exertion during submaximal steady‐state exercise in normal healthy untrained men. Biosci Biotechnol Biochem 2014;78:2081–8.
Henry R, Peoples GE, McLennan PL. Muscle fatigue resistance in the rat hindlimb in vivo from low dietary intakes of tuna fish oil that selectively increase phospholipid n‐3 docosahexaenoic acid according to muscle fibre type. Br J Nutr 2015;114:873–84.
Peoples GE, McLennan PL. Long‐chain n‐3 DHA reduces the extent of skeletal muscle fatigue in the rat in vivo hindlimb model. Br J Nutr 2014;111:996–1003.
Langelaan ML, Boonen KJ, Rosaria‐Chak KY, van der Schaft DW, Post MJ, Baaijens FP. Advanced maturation by electrical stimulation: differences in response between C2C12 and primary muscle progenitor cells. J Tissue Eng Regen Med 2011;5:529–39.
Siamo pienamente a conoscenza dell’impatto che una condizione di stress cronicamente protratto, sia fisico che psichico, ha sulla attivazione dell’Asse Ipotalamo-Ipofisi-Surrene (nota anche nella denominazione inglese di Hypothalamic–Pituitary–Adrenal Axis; abbreviato in HPA Axis) e la sintesi di Cortisolo con conseguenze negative anche a livello fisico con un accentuato stato catabolico del tessuto proteico del muscolo-scheletrico. Ma lo stress cronico potrebbe compromettere maggiormente la capacità di sviluppo della massa contrattile rispetto a quanto normalmente potremmo essere indotti a pensare.
Asse Ipotalamo-Ipofisi-Surrene (HPA). Sperimentare un fattore di stress ambientale, come percepito dal cervello, provoca l’attivazione dell’Asse HPA. L’Ipotalamo secernerà quindi l’Ormone di Rilascio della Corticotropina (CRH). Nel lobo anteriore dell’Ipofisi, il CRH stimola la secrezione dell’Ormone Adrenocorticotropo (ACTH). La corteccia delle ghiandole surrenali produrrà quindi glucocorticoidi (Cortisolo nell’uomo) in risposta all’ACTH. Il Cortisolo genererà quindi una risposta allo stress.
In un piccolo studio svolto su topi presso la University of Colorado e pubblicato nel settembre 2010 sul American Journal of Physiology, è stato riportato che lo stress causa una iper-espressione del gene della Miostatina.[1]
Miostatina
Sappiamo benissimo che la Miostatina, espressa nell’uomo dal gene MSTN, è un peptide che nel muscolo maturo inibisce l’Akt, una chinasi sufficiente a causare l’ipertrofia muscolare, in parte attraverso l’attivazione della sintesi proteica mentre stimola la produzione di ubiquitina ligasi, proteine che regolano la disgregazione proteica muscolare. Tuttavia, l’Akt non è responsabile di tutti gli effetti ipertrofici muscolari osservati che sono mediati dall’inibizione della miostatina[2] Pertanto la Miostatina agisce in due modi: inibendo la sintesi proteica indotta dal Akt e stimolando la degradazione proteica regolata dall’ubiquitina.
Caratteristiche dello studio
I ricercatori hanno fatto la loro scoperta quando hanno posto dei topi in una gabbia diversa ogni giorno per una settimana [CS], o li hanno messi per un breve periodo di tempo ogni giorno in una piccola “camera restrittiva” [RS]. Quest’ultimo trattamento risulta essere particolarmente doloroso per questi animali.
Dopo 7 giorni la massa muscolare di entrambi i gruppi era diminuita, ma in modo maggiore dei topi RS.
La sigla “TA” sta per muscolo del polpaccio tibiale anteriore, “SOL” per soleo. BC = massa muscolare all’inizio dell’esperimento, HC = massa muscolare in un gruppo di controllo di topi che sono stati pesati ogni giorno, ma non hanno ricevuto stimoli stressanti.
I ricercatori notarono che i muscoli dei topi sottoposti a stress iniziavano a produrre più Miostatina il giorno 1, in particolare i topi RS.
Poiché volevano sapere se la Miostatina svolgesse davvero un ruolo così importante nello stress psicologico, i ricercatori hanno ripetuto il loro esperimento con topi geneticamente modificati che non producevano Miostatina [MSTN KO]. Questi sono i topi massivamente ipertrofici le cui foto sono reperibili in rete. Essi provengono dal laboratorio dell’esperto di Miostatina Se-Jin Lee.
I topi non modificati sono topi WT.
Conclusioni
I ricercatori hanno concluso che lo stress psicologico può portare all’obesità e all’indebolimento muscolare.
Le diminuzioni della massa muscolare possono contribuire ovviamente a un cambiamento nella composizione corporea che può favorire l’obesità. Una perdita di massa muscolare magra riduce la quantità di tessuto metabolicamente attivo disponibile per la degradazione ossidativa dei substrati energetici.
Inoltre, una diminuzione della massa muscolo-scheletrica in risposta allo stress psicologico può anche predisporre il muscolo scheletrico a una maggiore probabilità o gravità di lesioni.
Ma nell’uomo le conseguenze dello stress psicofisico possono essere le medesime sui livelli aumentati di Miostatina osservati nei topi? Non possiamo ancora sapere con certezza quanto e come ciò possa influire sull’espressione genica MSTN nell’uomo, ma possiamo ipotizzare che ciò sia potenzialmente verificabile. Inoltre ciò non dovrebbe nemmeno stupire. Un organismo sottoposto a stress cronico tende a ricercare una omeostasi anche attraverso la riduzione del dispendio energetico tramite una riduzione del tessuto metabolicamente attivo. Gli esempi non mancano in letteratura. Pensate per esempio ai soggetti denutriti o stanziati in ambienti inospitali.
2- Sartori R, Gregorevic P, Sandri M (September 2014). “TGFβ and BMP signaling in skeletal muscle: potential significance for muscle-related disease”. Trends in Endocrinology and Metabolism.
Il più delle volte risulta inutile ribadire banalità largamente conosciute, ma in questo caso era d’obbligo sottolinearne il fatto dal momento che tornerà utile anche al fine di comprendere come la maggior parte dei (presunti) preparatori agisca quando si parla di PEDs.
Il GH, in modo particolare, è avvolto da dicerie di ogni genere. Dalle convinzioni sul momento in cui “inizia a funzionare” e la leggenda (fuori da ogni base fisiologica e attività espressiva iper-indotta iatrogenamente) del “assottiglia la pelle”. Ma la cosa di cui vorrei parlare riguarda i dosaggi. Nell’agonismo se ne sentono e vedono di tutti i tipi, dosaggi mostruosi da 10-20UI/die portatori di neuropatie croniche e probabili stati diabetici subclinici o patologici. La cosa triste è che tali dosaggi vengono indicati come “funzionali” solo perchè danno l’effetto ricercato che spesso e volentieri si confonde con quello indotto dallo svariato numero di farmaci cosomministrati e dalla additività tra questi. In poche parole: l’effetto lo avrebbero raggiunto comunque e nello stesso arco temporale utilizzando 1/10 della dose somministrata.
Si, mi dispiace infrangere le vostre fantasie ma il GH ha un limite nello stimolo massimo della lipolisi molto sottile…
GH e suo limite di dosaggio per il massimo stimolo lipolitico:
In effetti è stato calcolato un tetto massimo alla lipolisi indotta da una singola somministrazione di GH, ed è molto più bassa di quanto la maggior parte delle persone pensi.
Come riportato nello studio “Pharmacokinetics and acute lipolytic actions of growth hormone. Impact of age, body composition, binding proteins, and other hormonesvedere” nel quale si è valutata la farmacocinetica e l’azione lipolitica acuta del GH, si verificano significativi effetti dose-risposta confrontando l’area incrementale sotto la curva sia degli acidi grassi liberi che del 3-idrossi-butirrato nel sangue dopo la somministrazione di 0, 1 , e 3mcg/kg di GH, mentre non si osservano differenze tra le risposte dopo 3 e 6mcg/kg di GH.[1]
Questi dosaggi sono stati somministrati per via endovenosa.
1mg di Somatropina corrisponde a 3UI (Unità Internazionali) della medesima, come si può anche leggere nelle informazioni riferite al Norditropin presenti di seguito.[2]
Quindi, questo significa che per un soggetto di 100kg, la lipolisi è stimolata al massimo con circa 300mcg di Somatropina somministrata endovena, che equivale a 0,9UI di GH di grado farmaceutico IV (endovena).
Occorre quindi calcolare la dose sottocutanea corrispondente in base ai dati di confronto data dalla biodisponibilità e bioattività che abbiamo sugli esseri umani a cui è stato somministrato hGH.
Ovviamente i dosaggi somministrati endovena non sono rappresentativi di quali sarebbero i dosaggi ideali tramite il metodo di somministrazione molto più realistico e tollerabile quale è quello sottocutaneo. In uno studio, la disponibilità media stimata di hGH iniettato per via sottocutanea ha dimostrato di essere del 63% di quella di hGH somministrato per via endovenosa. dopo aver corretto le differenze nella dose di GH. [3] Un altro studio ha mostrato che la disponibilità di hGH iniettato per via sottocutanea è circa il 70% di quella di hGH somministrato per via endovenosa.[4]
Quindi, per un uomo di 100kg, i benefici della perdita di grasso sarebbero massimizzati per ogni somministrazione iniettando circa 1,35UI di GH (arrotondabili a massimo 2UI). Esiste infatti un periodo di refrattarietà durante il quale le cellule non danno risposta ad altro impulso lipolitico dato dal GH esogeno.
Conclusioni pratiche:
Di conseguenza, la risposta massima dello stimolo della lipolisi per somministrazione si aggira intorno a circa 1.5-2UI (3-4UI/die totali) in un individuo di 100Kg. Questo rispecchia in parte quanto osservato aneddoticamente ed annotato empiricamente con la somministrazione giornaliera di GH ad un dosaggio di 4UI/die divise in due somministrazioni uguali durante periodi di “Cut” in molti atleti di diverse categorie. Ciò vuole anche dire, però, che la maggior parte dei soggetti potrebbero avere risposte ottimali con la somministrazione di appena 2UI/die di GH. Parlo, ovviamente, di GH di grado farmaceutico, cioè equivalente al mcg al contenuto riportato in etichetta, non così abbondante nel mercato nero.
Le risposte temporali alla riduzione del grasso corporeo per azione del GH non sono dovute al dosaggio ma alla percentuale di grasso di partenza e alla distanza tra questo ed il punto percentuale prefissato da raggiungere oltre che dalla additività con altre molecole aventi effetti lipolitici e/o termogenici: più è breve e meno tempo di somministrazione sarà necessario. Ovviamente, i tempi di risposta sono maggiori rispetto ad atleti non trattati, mi pare ovvio e scontato. E, lo ripeto, vanno anche considerate nella somma dell’effetto le altre molecole co-somministrate e aventi attività lipolitica (diretta o indiretta) e/o termogenica.
Ma questo vale anche per le donne? Di questo me ne occuperò in un altro articolo ma vi anticipo già che in termini assoluti il dosaggio non ha variabili significative ma circostanze gestionali diverse.
DISCLAIMER: Il presente articolo è a solo scopo educativo, di intrattenimento e informativo. Non rappresenta in alcun modo una forma di incitamento all’uso/abuso di sostanze dopanti. L’autore ed il sito, per tanto, è esentato da qualsiasi responsabilità dipendente dalla libera scelta individuale.
Introduzione ad un dilemma…
Chiunque frequenti l’ambiente del Bodybuilding e del Fitness avrà letto o sentito almeno una volta nella vita espressioni del genere “se mi dopassi sarei anche io così [indicando Flex Wheeler]” o “ho provato di tutto e senza farmaci non riuscirò ad ottenere risultati”. Andando poi ad approfondire la storia di ognuno di questi soggetti si scopre in percentuale quasi assoluta che si tratta di individui nella norma (o al di sotto) frustrati e/o con personalità deboli, speranzosi omini che attendono placidamente che accada una svolta miracolosa nella loro banale e piatta esistenza e, cosa molto importante, con il minimo dello sforzo (meglio se nessuno).
Nella mia esperienza come ricercatore e operatore nel campo della cultura fisica in qualità di Preparatore Atletico, ho assistito a innumerevoli casi in cui un soggetto aspirava al miglioramento della propria composizione corporea trascurando, consciamente o inconsciamente, le basi fondamentali rappresentate da Nutrizione e Allenamento baipassandole in vista della possibile prescrizione di una pillola miracolosa capace di renderlo/a possessore della forma fisica ambita.
Tralasciando l’ovvio ragionamento che spinge ogni essere umano dotato di un minimo d’intelletto verso la comprensione che la genetica è il blocco d’argilla sul quale si va ad operare, ma le sue qualità e difetti sono presenti in modo eterogeneo nella popolazione mondiale, e ciò non è modificabile nemmeno con la farmacologia più oculata, quando ci si trova davanti al bivio tra “pillola rossa” (PEDs) e “pillola blu” (drug free) bisogna essere pienamente consapevoli non solo del fattore illegalità ma del fattore conoscitivo. Purtroppo, la politica del terrore ha operato in modo fallimentare nel goffo intento di allontanare dalla scelta “rossa”, e ciò si è tradotto in un numero sensibile di soggetti abusatori con tutte le conseguenze cliniche derivanti.
Se un individuo non ha raggiunto un livello di maturità sportiva tale da conferirgli una gestione corretta della nutrizione e della periodizzazione allenante (gestione delle variabili volume, intensità, densità ecc…), è molto meglio per lui/lei rivedere i suoi programmi e scegliere ancora la “pillola blu”. Capita, a volte, di incontrare persone decise ad intraprendere la via del “lato oscuro” che, dopo una approfondita chiacchierata sulla gestione dei suddetti fattori, rivede le proprie posizioni.
Per tutti coloro i quali sono immersi nel dilemma della scelta, vi espongo alcuni punti per rendere l’eventuale decisione meno rischiosa anche se pur sempre illegale nel “bel paese”…
“Pillola blu o pillola rossa?” I punti da tenere in considerazione per una scelta consapevole:
#1 Raggiungere una adeguata maturità sportiva
Per “maturità sportiva”, in particolare riferimento al BodyBuilding, si intende la capacità del atleta di sapersi alimentare e allenare correttamente con piena gestione delle proprie potenzialità fisiologiche/genetiche. Questa è la base, se viene a mancare ciò non solo la vostra esperienza finirà per deludervi e rendervi ancora di più dei frustrati, ma potrebbe rovinosamente portarvi ad un abuso cronico a senso inesorabilmente negativo…
#2 I PEDs non faranno miracoli
Una cosa da tenere bene a mente, e questo non dovrebbe interessare solo gli aspiranti “doped”, è che l’uso di PEDs non renderà diversi da ciò che rientra nelle potenzialità espressive del proprio patrimonio genetico. Certamente le caratteristiche genetiche verranno “iperespresse”, nel bene e nel male, dall’uso di PEDs ma non vi sarà nessun miracolo! Migliorerete ma non sarete ne più ne meno di ciò che potete essere!
Un esempio per capire come la base genetica faccia la differenza anche con protocolli che, ad oggi, spesso non raggiungono nemmeno i livelli del “bridge” più soft..
#3 Ridurre la percentuale di grasso corporeo
Il tessuto adiposo rappresenta uno dei siti dove il Testosterone, ed altri AAS soggetti all’aromatizzazione, viene convertito in Estradiolo. Soggetti con percentuali di grasso corporeo elevate vedrebbero una alterazione marcata della Testosterone:Estradiolo ratio a favore della componente estrogenica, con conseguenze quali alterazione del comportamento sessuale (impotenza, difficoltà nel raggiungere e/o mantenere l’erezione), ritenzione idrica, accumulo di grasso con modello femminile e ginecomastia. E no, l’uso di DHT derivati o di SARM non steroidei senza una base di Testosterone non risolverebbe il problema o, per lo meno, porterebbe ad altre conseguenze negative, che pur non comprendendo, per esempio, ritenzione idrica e ginecomastia, interesserebbero l’attività sessuale e la condizione psichica del soggetto trattato. [1]
Schema esemplificato del processo di aromatizzazione degli androgeni aumentati in un soggetto con percentuale di grasso corporeo alta.
Allo stesso tempo, i rischi cardiovascolari della somministrazione di AAS- come il possibile aumento esponenziale del Ematocrito, l’aumento del LDL e Trigliceridi a discapito di una riduzione del HDL, e l’aumento della pressione sanguigna – sarebbero già presenti in certa misura quando la body fat è già alta e sarebbero quindi soggetti ad un repentino aggravamento.
Se la percentuale di grasso è relativamente alta, si dovrebbe prima di tutto considerare di migliorare la composizione corporea con una adeguata routine alimentare e allenante (senza farmaci) prima di iniziare solo a pensare all’uso di AAS. Sicuramente ciò renderà la scelta più efficace e meno rischiosa.
Nel caso fosse necessario sottolinearlo, no, non è saggio nemmeno utilizzare agenti PEDs a fini lipolitici e/o antiadipogenici e/o termogenici (compresi gli Ormoni Tiroidei). A meno che non siate affetti da ipotiroidismo, e in questo caso la terapia vi dovrebbe essere stilata dal vostro medico, per ridurre in modo sensibile la body fat non sono necessari i farmaci!
#4 Controllare se si ha una storia familiare di trombosi (o qualsiasi altra malattia cardiovascolare)
Molte malattie cardiovascolari hanno una componente di base genetica. Uno stile di vita sano può ridurne sensibilmente la loro insorgenza, ma l’uso di AAS può causare l’attivazione di specifici geni implicati nella comparsa di malattie cardio-circolatorie. Caratteristico dell’interazione tra AAS e geni specifici è un caso studio ben documentato che ricercatori americani hanno pubblicato sul “Blood Coagulation & Fibrinolysis”.[2]
Trombosi venosa
Oltre all’attivazione genica diretta dagli AAS, e nociva per il sistema cardio-circolatorio, vi sono altre condizioni negative innescate dall’uso/abuso di Steroidi Anabolizzanti, e di altri PEDs, come, per esempio, l’aumento del tasso di coagulazione, l’incremento eccessivo dell’Ematocrito con aumento pressorio, rigidità dell’endotelio vascolare con perdita di efficienza strutturale e aumento della pressione ematica con incremento delle possibilità di danno strutturale dei componenti del sistema interessato.
#5 Inserire delle sedute di allenamento cardio prima, durante e dopo l’uso di PEDs
Un moderato allenamento cardiovascolare è sicuramente una delle migliori strategie preventive contro la comparsa di malattie cardio-circolatorie. Tale tipologia di allenamento può portare un miglioramento e/o riduzione delle alterazioni lipidiche ematiche del praticante, fornendo un, seppur minimo, tampone all’azione negativa degli AAS e SARM non steroidei sui livelli di LDL (aumento), Trigliceridi (aumento) e HDL (diminuzione). Secondo quanto riportato da una interessante review del 2013, l’abbinamento di sedute cardio e in sala pesi possono avere una azione additiva benefica sui livelli di LDL, Trigliceridi e HDL.[3]
Risulta interessante anche quanto emerso da alcuni studi su animali a seguito dei quali si è osservato un significativo grado di protezione dato dall’allenamento cardio negli esemplari trattati con AAS.[4]
#6 Assicurarsi di rimanere ben idratati
Oltre ad agevolare il mantenimento di un Ematocrito migliore, una buona idratazione risulta positiva sulla pressione di lavoro renale nel filtraggio del sangue. Diversi AAS come il Trenbolone e i metilati in C-17 presentano una particolare resistenza metabolica che, oltre a causare un aumentato stress epatico, può portare ad una sofferenza renale sfociabile nel patologico. Si è osservato come una combinazione di AAS, dieta iperproteica e supplementazione di Creatina possa aumentare l’incidenza di problemi renali.[5] In un soggetto in fisiologia, la sola dieta ad altro contenuto proteico e la supplementazione di Creatina non hanno mostrato nessun grado di pericolosità, soprattutto sul breve/medio termine.
#7 Non usare “droghe ricreative”
A livello globale, il numero di decessi tra gli abusatori di AAS è in aumento. Alcuni, troppo superficialmente, dicono che questo sia dovuto al fatto che sempre più uomini e donne usano AAS, ma questa è solo una spiegazione dozzinale e limitata. Il sospetto ricade soprattutto sulle modalità di approccio dei consumatori di AAS: i dosaggi sono drammaticamente aumentati e un numero crescente di individui combina PEDs con “droghe ricreative”. Ed è su questi due ultimi punti che risiede la spiegazione principale dell’aumento statistico prima menzionato. Soprattutto la combinazione di PEDs e le così dette “droghe ricreative” risulta essere probabilmente un fattore significativo, come evidenziato alcuni anni fa da ricercatori australiani. Nel loro studio sono state analizzate tutte le morti documentate tra i consumatori di AAS a Sydney tra il 1997 e il 2012, scoprendo che le droghe ricreative come la cocaina avevano avuto un ruolo nella schiacciante molteplicità dei casi. Dagli studi sugli animali ora sappiamo della possibilità che la co-assunzione di un AAS come il Nandrolone con la cocaina vede moltiplicati gli effetti cardiotossici rispetto ai singoli composti.[6] E secondo studi in vitro la combinazione di Testosterone e cocaina aumenterebbe la possibilità di formazione di coaguli nel flusso ematico. [7]
#8 Corretta modalità di iniezione e herpes labiale
Gli utilizzatori di AAS a volte sviluppano ascessi, ma non sempre dovuti alla bassa qualità dei prodotti utilizzati.
Alcuni medici ritengono che gli utilizzatori di AAS dovrebbero effettivamente ricevere una formazione sulle tecniche di iniezione corrette, onde evitare embolie oleose o ascessi per cattiva gestione igienica della procedura. [8]
Molti utilizzatori ancora non sanno che disturbi come l’herpes labiale rendono le iniezioni ancora più rischiose. Perchè? Il virus che causa l’herpes labiale, come altri patogeni, riduce l’efficienza del sistema immunitario, fornendo così terreno fertile per infezioni batteriche i cui microorganismi scatenanti vengono inoculati nel corpo del soggetto attraverso l’iniezione in modo diretto o indiretto.
#9 Non fare affidamento sugli integratori
Secondo un buon numero di studi svolti su animali, alcuni integratori proteggono dagli effetti collaterali degli AAS. Secondo alcune ricerche, la Taurina, la Vitamina C ed E proteggono i testicoli durante un ciclo e la vitamina C e il cacao proteggono la prostata.
L’utilità dei risultati provenienti da questi studi è limitata per tre motivi:
A. gli animali da laboratorio non sono esseri umani, e
B. le dosi utilizzate e rapportate ad un essere umano sono quasi sempre molto inferiori rispetto a quelle utilizzate dai “doped”, e
C. la ricerca in campo psicologico mostra che l’uso di integratori stimola comportamenti rischiosi e malsani. I supplementi fanno pensare agli utilizzatori di essere invulnerabili e di non dover comportarsi in modo sano ed attento.[9]
Gli integratori possono aiutare a creare una mentalità che non si dovrebbe avere da utilizzatore consapevole di AAS.
Ovviamente, alcuni supplementi “protettivi” utilizzati dai soggetti meglio informati hanno un potenziale di “tamponare” in modo discreto alcune alterazioni legate all’uso di AAS e SARM come, ma non limitato a, Riso Rosso fermantato (controllo lipidico) [10], Silimarina (epatoprotezione), NAC (epatoprotezione) [11], Niacina (controllo lipidico) ecc…
#10 Ridurre al minimo (se non eliminare) il consumo di alcolici
Potrebbe sembrare un indicazione superflua ma non lo è.
L’abuso di alcol è indubbiamente uno dei problemi sociali più diffusi. Uno dei problemi correlati all’abuso di alcol e l’epatopatia alcolica. Questo stato patologico è derivante da un processo infiammatorio progressivo ai danni del fegato legato al consumo eccessivo di alcolici. È una malattia a più stadi. La steatosi provoca un ingrossamento del fegato causato da un accumulo di trigliceridi, spesso senza sintomi per molto tempo. I rischi correlati sono la steatosi (fegato grasso), l’epatite alcolica e la cirrosi epatica. Il rapporto con l’alcolismo è complesso. Non tutti i bevitori, infatti, hanno danni al fegato, anche se sono altamente probabili. La causa è da rinvenire in una trasformazione dell’alcol (etanolo) in sostanze tossiche che danneggiano il fegato in maniera irreversibile e cronica, con un rischio elevato di insufficienza epatica e di cancro, fino alla necessità di un trapianto di fegato.
In acuto, invece, l’alcol può essere una causa di alterazione delle transaminasi ma non si può sapere se e con quale modalità si potrebbero innalzare: dipende molto dalla risposta individuale dell’organismo. In caso di stress preesistente, di causa iatrogena e/o alimentare, si può presentare una alterazione significativa. [12]
Il primo caso è una consequenziale possibile se eventi stressori concomitanti si presentano in cronico. Ed è semplice giungere alla conclusione che l’uso di AAS, specie se metilati, possa comportare un aumentato stress epatico che potrebbe degenerare in peliosi epatica, cirrosi ecc…
Che siate “doped” o “natural”, per ragioni legate e non, dovreste evitare di consumare più di 25g per gli uomini, o 12,5g per le donne, di Etanolo al giorno.
#11 Sottoporsi a regolari controlli medici pre, intra e post utilizzo
Il monitoraggio della salute dovrebbe essere la base fondante del comportamento del utilizzatore consapevole e minimamente attento ai potenziali rischi nei quali potrebbe imbattersi.
Gli esami di controllo sono i seguenti:
Esami ematici e delle urine (comprendenti il quadro ormonale secondo necessità);
Elettrocardiogramma ogni 6 mesi circa;
Elettrocardiogramma sotto sforzo (prima di iniziare);
Ecocardiogramma ogni 6 mesi circa;
Coronarografia ogni 6 mesi circa;
Monitoraggio della pressione ematica;
TAC addome completa ogni 6 mesi circa.
Ovviamente, ogni accertamento , al di la degli esami ematici, deve essere gestito in base alle esigenze soggettive, caratteristiche e tipo di PEDs utilizzati.
#12 Essere seguiti da personale qualificato
Fin troppa gente è stata salutisticamente deturpata da gorilla di spogliatoio a mala pena consapevoli dell’esistenza dei macronutrienti e che, nonostante ciò, si sono improvvisati farmacisti. Donne divenuti uomini e uomini divenuti simili a cagne in calore per via di orrende ginecomastie. Evitate il fai da te e l’affidarsi a semianalfabeti … la somaticità sopra la norma è cosa diversa dall’intelligenza e alla competenza in biologia, biochimica e farmacologia… senza offesa per tutti quelli che “io mi facevo e ho vinto! Senzia scienzia!” …
#13 Pensare seriamente al post ciclo prima del ciclo
Molti aspiranti “doped” non considerano il fattore post ciclo. La maggior parte di loro è convinta che la PCT sarà una facile soluzione alla sottoregolazione dell’Asse HPT, ma in realtà non è proprio così. Esistono diversi casi studio che mostrano come gli ex utilizzatori abbiano spesso livelli di Testosterone inferiori rispetto al pre-utilizzo anche a distanza di anni dal cessato uso di AAS. Sembra che i fattori che aumentano le possibilità e il grado di tale effetto sul lungo termine siano:
Tempo di somministrazione;
Età
Molecole utilizzate (con maggiore impatto negativo dato dai19-norsteroidi come il Nandrolone per via della lunga permanenza dei metaboliti nel sistema).
Tutto ciò è indipendente dalla qualità della PCT, anche se essa può avere dei riscontri positivi specie nel primo periodo di stacco dagli AAS. Le alterazioni ormonali legate ad una alterazione dell’Asse HPT comprendono depressione, ansia, bassa libido, difficoltà nel raggiungere e mantenere l’erezione, stanchezza cronica ecc…
Per questa ragione molti scelgono di entrare in TRT (Terapia Sostitutiva del Testosterone) dopo il primo ciclo.
Quale conclusione?…
Se mai non dovesse bastare il disclaimer, questo articolo non rappresenta in alcun modo un consiglio e, ne tanto meno, un incitamento all’uso di sostanze dopanti! E’ semplicemente a fine divulgativo con l’obbiettivo di far comprendere a più persone possibili che la scelta di intraprendere coscientemente certe pratiche (illegali) necessita di una sufficiente (e veritiera) conoscenza del argomento.
Quindi? Leggete e comprendete correttamente ciò che ho riportato in sintesi fruibile ad un largo pubblico… Pensate prima di tutto ad alimentarvi e allenarvi in modo ottimale!
La conoscenza della Verità rende liberi dalla cattiva informazione, dagli strumenti commerciali e dal relativismo… Negarla è semplice e pericolosa manifestazione di profonda ignoranza… di VERO NEGAZIONISMO!
Se avete una buona conoscenza della lingua inglese e volete approfondire l’argomento PEDs e Sport, potete leggere il libro ANABOLICS 11th Edition di William Llewellyn
Anni dopo l’interruzione dell’uso di AAS da parte di soggetti di sesso maschile questi ultimi potrebbero riscontrare ancora una condizione di ipo-funzionamento testicolare. La concentrazione ematica del fattor isulino simile 3 (INSL3), un peptide del quale si sa ancora poco, rende chiaro quanto possa durare l’impatto endocrinologico dato dall’uso di AAS. Ciò risulta particolarmente evidente dai risultati di uno studio danese, al quale hanno partecipato 132 bodybuilder.[1]
Fattore Insulino Simile-3 (INSL3)
Dettagli dello studio
I ricercatori, un gruppo di endocrinologi dell’Università di Copenaghen, hanno osservato 132 uomini di età compresa tra 18 e 50 anni che si allenavano con i pesi. Questi uomini sono stati divisi in 3 gruppi formati all’incirca dallo stesso numero di individui:
Un gruppo non aveva mai usato AAS;
un secondo gruppo era sotto ciclo di AAS durante lo studio;
infine, il terzo gruppo aveva usato AAS in passato.
Il partecipante medio allo studio in quest’ultimo gruppo aveva smesso di usare AAS 32 mesi prima. I ricercatori hanno misurato la concentrazione di INSL3 nel sangue dei soggetti partecipanti allo studio. Quell’ormone, che di per sé ha anche proprietà anabolizzanti, è prodotto dai testicoli. Gli endocrinologi sospettano che i testicoli siano più sani in quanto producono più INSL3 e pensano persino che questo peptide sia un marker di riferimento maggiormente importante per la valutazione della vitalità dei testicoli rispetto al Testosterone.
Risultati dello studio
Dei tre gruppi sotto osservazione, i soggetti che utilizzavano AAS nel periodo dello studio avevano la più bassa concentrazione ematica di INSL3. Tuttavia, i soggetti che avevano usato AAS in passato avevano anch’essi meno INSL3 sierico rispetto a coloro che non avevano mai utilizzato AAS:
I non utilizzatori avevano una media di 0,59mcg di INSL3 per litro;
gli ex utenti 0,39mcg per litro.
Più a lungo gli ex utilizzatori erano stati sotto AAS in passato, minore era il INSL3 rilevato attraverso esame ematico.
I dati riportati nella figura sopra sono di notevole interesse. Si guardi all’associazione tra il passare del tempo dopo l’ultima somministrazione di AAS da una parte e la concentrazione sierica di INSL3, dall’altro. Bene, si nota una consequenziale en significativa risposta…
Contrariamente, è ovvio che se non ci fosse stato alcun legame significativo il INSL3 non avrebbe alcuna valenza come marker per la valutazione dello stato di salute dei testicoli.
Conclusioni
Il principale ricercatore dello studio, Jon Jarlov Rasmussen, in un comunicato stampa ha affermato che è ancora dibattuto se l’uso off-label di AAS provochi una carenza di Testosterone sul lungo termine. I loro risultati suggeriscono, però, la presenza di una capacità gonadica compromessa sul lungo termine nei precedenti utilizzatori di AAS. I risultati sollevano la questione se alcuni precedenti utilizzatori di AAS debbano ricevere una terapia di stimolazione medica per aumentare la capacità funzionale delle cellule di Leydig nei testicoli.[2]
Rasmussen ipotizza come base di trattamento inibitori dell’Aromatasi e SERM.
Nonostante il terrorismo diffuso da alcuni scientisti riguardo alla “malvagità assoluta” del IGF-1 e della sua correlazione con il cancro (esistente ma contestualizzabile), la terapia sostitutiva dell’Ormone della Crescita (GH) sembra non aumentare il rischio di cancro; sembrerebbe che, addirittura, ne riduca il rischio. Ciò è stato evidenziato in una meta-analisi pubblicata da endocrinologi cinesi della Zhejiang University College of Medicine nell’Open Journal of Endocrine and Metabolic Diseases.[1]
I ricercatori hanno esaminato 10 studi pubblicati in precedenza che hanno seguito adulti trattati con Ormone della Crescita per diversi anni. Ai soggetti è stato diagnosticato un deficit dell’ormone della crescita negli adulti. Ovviamente, e mi riferisco al lettore nella media che troppo velocemente trae conclusioni inesatte, gli studi potrebbero non dire molto sugli effetti che gli atleti supplementati farmacologicamente possano incorrere nel contesto dell’incidenza di sviluppo del cancro.
Nella tabella sottostante troverete maggiori informazioni sugli studi utilizzati.
Negli studi esaminati, i soggetti trattati con il GH non hanno sviluppato il cancro in maniera maggiore rispetto ai soggetti dei gruppi di controllo. Da quanto è emerso, per lo meno dagli studi presi in esame, l’Ormone della Crescita ha persino ridotto il rischio di cancro.
I ricercatori hanno però trovato presenza di bias. Ciò significa che sembra che non siano stati pubblicati studi con risultati meno interessanti. Tuttavia, il bias sembrava essere modesto ed i ricercatori sospettano che l’inclusione degli studi potenzialmente mancanti nella loro meta-analisi non ne altererebbe realmente il risultato.
I ricercatori concludono dicendo che la loro analisi corrobora le prove di studi precedenti che dimostrano che la terapia sostitutiva dell’Ormone della Crescita nei pazienti con deficit di questo ormone in età adulta non vedrebbero aumentare il loro rischio di sviluppare il cancro; invece, potrebbe addirittura diminuire il rischio. I risultati hanno suggerito che la terapia sostitutiva dell’Ormone della Crescita nei pazienti con deficit dell’ormone in età adulta era sicura.
Nota: i dosaggi terapici variano nettamente a seconda dell’età e delle finalità della terapia. Sono generalmente dosaggi inferiori a quelli utilizzati a scopo estetico-prestativo. Pertanto, il presente articolo non ha assolutamente lo scopo di far passare un dato che ad oggi non è statisticamente comprovato.
A differenza del trattamento pediatrico con GH, spesso dosato in microgrammi / chilogrammi di peso corporeo / giorno, il dosaggio sostitutivo del GH attualmente raccomandato negli adulti è individualizzato indipendentemente dal peso, tenendo conto dell’età del paziente, del sesso e dello stato degli estrogeni (Johannsson et al., 1997a; Hoffman et al., 2004b). L’inizio della terapia a basse dosi (dose totale 0,2-0,4 mg / die SC) riduce la probabilità di sviluppare effetti collaterali comuni come rigidità articolare, artralgie, mialgie, parestesie ed edema periferico, con ritenzione di liquidi. La dose deve essere titolata a intervalli di 6-8 settimane in base alla risposta clinica, evitando effetti collaterali e monitorando i livelli sierici di IGF-I. Si desidera raggiungere un obiettivo nella metà superiore dell’intervallo normale del IGF-I corretto per l’età del paziente. È ragionevole iniziare con dosi più elevate (0,4-0,5 mg / die) nei pazienti di età inferiore a 30 anni, ma i pazienti più anziani (di età superiore a 60 anni) dovrebbero iniziare con dosi più basse (0,1-0,2 mg / die) e titolato più lentamente per ridurre al minimo il verificarsi di effetti collaterali. Alcuni pazienti con AGHD ad esordio infantile con IGF-I pretrattamento molto basso possono sviluppare effetti collaterali da GH ma non raggiungere livelli medi normali nonostante dosi elevate di quest’ultimo.
Le donne che assumono una terapia sostitutiva degli estrogeni per via orale possono richiedere dosi più elevate per la GHRT, presumibilmente perché l’estrogeno orale inibisce la produzione e la secrezione di IGF-I nel fegato con effetto di primo passaggio (Weissberger et al., 1991). Di solito non è necessario alcun aggiustamento della dose nei pazienti che assumono dosi moderate di estrogeni transdermici.
Da qualche tempo stanno circolando teorie secondo le quali l’Ibutamoren (comunemente noto come MK-677) può causare danni cerebrali per via dello stimolo cronico dei recettori della Grelina (bersaglio della molecola in questione).
Ma è mai stato osservato un effetto simile sull’uomo negli studi presenti svolti su esseri umani? No, e non risulta nulla di simile nemmeno dai dati aneddotici provenienti dagli utilizzatori, almeno per il momento.
Allora da cosa nasce la teoria? Nasce dai dati provenienti da test di laboratorio svolti su topi. Ed in effetti, i dati provenienti da questi test mostrano collegamenti tra esposizione cronica allo stimolo dei recettori della Grelina e esiti negativi a livello cerebrale.
Ma approfondiamo la questione…
MK-677 e aumento della paura nei ratti.
Uno studio sui ratti ha valutato se l’MK-677 possa causare un aumento dei livelli di paura.[1]
Esistono prove a sostegno del fatto che uno dei modi in cui la Grelina viene modulata è attraverso l’esposizione allo stress.
Ciò che questo studio intendeva valutare specificamente era se i ratti avessero una maggiore probabilità di sperimentare la paura e il disturbo da stress post-traumatico (PTSD) con livelli cronicamente alti di Grelina, situazione “mimata” artificialmente 24/7 con l’utilizzo di un agonista del recettore della Grelina (MK-677).
Ai ratti veniva somministrato in cronico l’MK-677 e nello stesso tempo venivano costantemente spaventati. I ricercatori valutavano quindi la loro differenza di risposta (gruppo trattato con MK-677 Vs. gruppo non trattato).
È stato quindi scoperto che i ratti del gruppo trattato con MK-677 avevano hanno migliorato la memoria della paura rispetto al basale.
Come ho già detto, ci sono prove a sostegno del fatto che uno dei modi in cui la Grelina viene modulata è attraverso l’esposizione allo stress, e questo studio è stato in grado di suscitare un po’ di controversie nella sottocultura del BodyBuilding con i suoi risultati che mostrano un effetto palesemente negativo sull’amigdala in cui gli effetti di una sovrastimolazione del recettore della grelina porta ad un della paura.
La Grelina e l’Ormone della Crescita agiscono insieme nell’amigdala per aumentare la percezione della paura, almeno come osservato in questo modello di roditori.
Si noti che in questo studio, i ratti sono stati trattati con iniezioni intraperitoneale (I.P.) di 1ml/kg.
Nel caso non lo sapeste, una iniezione intraperitoneale consiste nell’iniezione di una sostanza nel peritoneo (cavità corporea).Il peritoneo è la membrana sierosa che forma il rivestimento della cavità addominale. Ed è qui che l’MK-677 è stato somministrato per la prima volta per valutare se una maggiore attivazione del recettore della Grelina è sufficiente per migliorare la memoria della paura.
Dopo aver stabilito che l’attivazione del recettore della Grelina è effettivamente sufficiente per migliorare la memoria della paura, hanno quindi continuato a infondere MK-677 direttamente nel complesso basolaterale dell’amigdala per determinare se l’attivazione ripetuta del recettore della Grelina nel complesso basolaterale dell’amigdala sia sufficiente per migliorare la memoria della paura.
Ripetute infusioni intra-amigdala miglioravano la memoria della paura.[2]
Ora, mentre tutto ciò dipinge un quadro piuttosto negativo dell’MK-677, bisognerebbe tener presente che questo studio ha anche dimostrato che una singola iniezione di un agonista del recettore della Grelina non era sufficiente per aumentare la paura, e persino l’agonismo cronico del recettore della Grelina non alterare la locomozione, l’ansia innata o l’espressione di ricordi di paura acquisiti in precedenza.[2]
Quanto possono interessare l’essere umano i risultati ottenuti?
Negli studi effettuati sull’uomo, l’MK-677 è stato osservato essere ben tollerato per oltre un anno consecutivo di utilizzo di una dose pari a 25mg/die assunta per via orale.
Gli utilizzatori ovviamente non si somministrano cronicamente il composto nella cavità peritoneale o nel cervello.
Questa scoperta non è stata ancora replicata nelle prove umane, e nemmeno nei resoconti aneddotici fino ad oggi.
Detto ciò, la ricerca sugli esseri umani sta indagando su questo particolare effetto?
Probabilmente no, e ovviamente quelli che usano l’MK-677 a livello ricreativo non vengono sottoposti di norma a scansioni cerebrali prima e dopo l’utilizzo del composto.
Molto probabilmente il problema potrebbe emergere se l’uso fosse protratto nel cronico, per anni. Non per nulla esistono meccanismi di autoregolazione nel corpo per una ragione ben specifica.
Anche se non vi fosse alcun rischio neurologico, bisogna ricordare che l’MK-677 causa un peggioramento della sensibilità all’insulina, fattore di certo non da trascurare e da gestire con cicli temporalmente non eccessivi e l’arginamento di tale conseguenza iatrogena con “composti tampone” quali Berberina o Metformina. L’effetto sulla sensibilità all’insulina è certamente una preoccupazione più immediata di qualsiasi altra affermazione estrapolata da studi sui roditori che mostrano i suoi possibili effetti sulla salute del cervello.
Uno degli argomenti che spesso vengono dibattuti in ambito sportivo, e soprattutto culturistico, è il senso, non ché l’efficacia, dell’uso del solo GH per il miglioramento delle prestazioni. Ormai dovrebbe essere di dominio pubblico il fatto che il GH, di per se, non è una molecola anabolizzante (se non, e per un certo grado diretto/indiretto, a carico delle fibre di tipo I), e che anche l’aumento del solo IGF-1, legato al picco del precedente peptide, non è garanzia di incrementi significativi nelle dimensioni del tessuto contrattile se non accompagnato da altri fattori come l’incremento concomitante di AAS circolanti. Quindi, è facile giungere alla conclusione che protocolli farmacologici volti all’ipertrofia, o al mantenimento della stessa, basati esclusivamente sul GH sono fallimentari, e, alle dosi consuete o minimamente efficaci, con un carico di possibili effetti avversi sicuramente maggiore rispetto ai possibili benefici (vedi, ad esempio, peggioramento dell’insulino resistenza e quello consequenziale dello stato metabolico glucidico/lipidico e della composizione corporea). I benefici sulle articolazioni, se qualcuno se lo chiedesse, possono essere raggiunti con dosi decisamente inferiori a quelle comunemente utilizzate dai BodyBuilder e, con l’avvento di peptidi specifici per tali trattamenti, oggi l’applicazione del GH in monoterapia è quasi del tutto in disuso anche nell’ambiente “doped” (quello minimamente aggiornato). Comunque sia, l’uso del solo GH per il miglioramento delle prestazioni potrebbe non risultare sempre inutile…se si è atleti di sport caratterizzati da sprint. Questa è la conclusione raggiunta dall’esperto dell’Ormone della Crescita Ken Ho presso la University of New South Wales, in una review pubblicata su Archives of Endocrinology and Metabolism.(1)
Ken Ho ha voluto trovare una risposta al quesito pocanzi discusso valutando quanto forti fossero le evidenze scientifiche che avvalorassero il potenziale del GH sul miglioramento delle prestazione. Per fare ciò ha raccolto tutti gli studi sull’uomo che prendevano in esame soggetti sani e in forma.
In quasi tutti gli studi raccolti, i soggetti avevano ricevuto solo l’Ormone della Crescita. Come ben sappiamo, nella pratica sportiva, tali pratiche monoterapiche sono praticamente inesistenti. Gli atleti supplemenmtati farmacologicamente usano quasi sempre il GH in combinazione con altri composti che migliorano le prestazioni come gli AAS.
Negli studi raccolti da Ho, i soggetti presi in esame sono stati trattati con dosi di GH dai 15 ai 180mcg per Kg di peso corporeo al giorno per un periodo massimo di 12 settimane.
Diversi studi non hanno riportato alcuna prova che l’Ormone della Crescita possa aumentare la forza [2] o la resistenza aerobica [3] degli atleti trattati con tale peptide.
Tuttavia, esiste uno studio in cui la somministrazione dell’Ormone della Crescita ha portato ad un aumento della capacità negli esercizi anaerobici degli atleti pari al 3,8%. [4] La dose ricevuta dagli atleti in questione era di 2mg di GH al giorno. Questa dose è di circa 2-3 volte superiore alla capacità di sintesi endogena giornaliera di un soggetto in giovane età.
La somministrazione in questo specifico caso è durata 8 settimane.
Secondo quanto riportato dai ricercatori, se tradotto in riduzione proporzionale del tempo, il 3,8% potrebbe equivalere a un miglioramento di 0,4 secondi in uno sprint di 10 secondi, di 1,2 secondi nei 100 metri e di 30 secondi nel attraversamento di una vasca olimpionica di 50 metri.
I ricercatori concludono che, Nei soggetti in forma, il GH in dosi utilizzate negli studi eticamente controllati non influisce sulla forza muscolare o sulla capacità aerobica, ma migliora la capacità anaerobica. Le prove suggeriscono che è improbabile che l’Ormone della Crescita avvantaggi negli sport di potenza o di resistenza ma che probabilmente benefici eventi di breve durata, vedi, appunto, gli sprint.
L’Adiponectina (denominata anche come GBP-28, apM1, AdipoQ, Acrp30 e Liponectine[1]) è un ormone proteico coinvolto nella regolazione dei livelli di glucosio e nella scomposizione degli acidi grassi. Nell’uomo è codificato dal gene ADIPOQ ed è prodotto nel tessuto adiposo. [2]
Nel 1995, l’Adiponectina è stata inizialmente osservata esercitare una azione di differenziazione degli adipociti 3T3-L1 (Scherer PE et al.).[3] Essa venne infatti scoperta nel 1995 da quattro diversi gruppi di ricerca che lavoravano indipendentemente l’uno dall’altro.[4] Nel 1996 è stato osservato che nei topi l’Adiponectina è la trascrizione dell’mRNA più espressa negli adipociti.[2] Nel 2007, l’Adiponectina è stata osservata essere una trascrizione altamente espressa nei preadipociti [5] (precursori delle cellule adipose) che si differenziano in adipociti.[5][6]
L’omologo umano è stato identificato come la trascrizione più abbondante nel tessuto adiposo. Contrariamente alle aspettative, nonostante venga sintetizzata nel tessuto adiposo, l’Adiponectina risultata essere sottoregolata nei soggetti obesi.[7][8][9] Questa sottoregolazione non è stata completamente spiegata. Il gene è stato localizzato nel cromosoma 3q27, una regione evidenziata per avere una certa influenza nella suscettibilità genetica al diabete di tipo 2 e all’obesità. Il trattamento con diverse forme di Adiponectina è stato in grado di migliorare il controllo dell’Insulina, della glicemia e dei livelli di Trigliceridi nei modelli murini.
Il gene è stato studiato per le variabili che predispongono al diabete di tipo 2.[9][5][10][11][12][13]Diversi polimorfismi a singolo nucleotide nella regione codificante e nella sequenza circostante sono stati identificati in diverse popolazioni, con prevalenze, gradi di associazione e forza di effetto variabili sul diabete di tipo 2. La Berberina, un alcaloide isochinolina, ha dimostrato di aumentare l’espressione del Adiponectina [14], il che spiega, in parte, i suoi effetti benefici sui disturbi metabolici. Topi nutriti con gli acidi grassi Omega-3 Acido Eicosapentaenoico (EPA) e Acido Docosaesaenoico (DHA) hanno mostrato un aumento dell’Adiponectina plasmatica.[15] Anche la Curcumina, la Capsaicina, il Gingerolo e le Catechine hanno mostrato di poter aumentare l’espressione dell’Adiponectina.[16]
La distribuzione filogenetica include l’espressione negli uccelli [17] e nei pesci.[18]
L’Adiponectina è un polipeptide (proteina) composto da una catena di 247 aminoacidi. Esistono quattro regioni distinte nella struttura molecolare della Adiponectina. La prima regione è formata da una breve sequenza di segnali che interessa l’ormone nella secrezione all’esterno della cellula; la seconda regione varia tra le specie; la terza è una regione composta di 65 aminoacidi con somiglianza con le proteine del Collagene; l’ultima è un dominio globale. Nel complesso questa proteina mostra una somiglianza con i fattori del complemento 1Q (C1Q). Tuttavia, quando è stata determinata la struttura tridimensionale della regione globulare, è stata osservata una sorprendente somiglianza con il TNFα (Fattore di Necrosi Tumorale α), nonostante sequenze proteiche non correlate.[19]
Come già accennato, l’Adiponectina è un ormone proteico che modula una serie di processi metabolici, tra i quali la regolazione del glucosio e l’ossidazione degli acidi grassi.[7] L’Adiponectina viene secreta dal tessuto adiposo (e anche dalla placenta in gravidanza [20]) nel flusso ematico ed è molto abbondante nel plasma rispetto a molti altri ormoni. Molti studi hanno scoperto che l’Adiponectina è inversamente correlata all’indice di massa corporea nelle popolazioni di pazienti.[8] Tuttavia, una meta analisi non è stata in grado di confermare questa associazione negli adulti sani.[21] Le concentrazioni circolanti di Adiponectina aumentano durante la restrizione calorica negli animali e nell’uomo, come nei pazienti con anoressia nervosa. Questa osservazione è senza dubbio sorprendente, dato che l’Adiponectina è sintetizzata nel tessuto adiposo. Tuttavia, un recente studio suggerisce che il tessuto adiposo nel midollo osseo, che aumenta durante la restrizione calorica, contribuisce alle elevate concentrazioni ematiche di Adiponectina in tale contesto.[22]
Mitocondrio
Topi transgenici con elevati livelli di Adiponectina mostrano una ridotta differenziazione degli adipociti e un aumento del dispendio energetico associato al disaccoppiamento mitocondriale.[23] L’ormone in questione svolge un ruolo nella soppressione delle alterazioni metaboliche correlate al diabete di tipo 2, [8] obesità, aterosclerosi, [7] epatopatia adiposa non alcolica (NAFLD) e un fattore di rischio indipendente per la sindrome metabolica.[24] L’Adiponectina in associazione con la Leptina ha dimostrato di invertire completamente lo stato di insulino-resistenza nei topi.[25]
L’Adiponectina viene secreta nel flusso sanguigno dove rappresenta circa lo 0,01% di tutte le proteine plasmatiche ad un dosaggio di circa 5-10μg/mL (mg/L). Negli adulti, le concentrazioni plasmatiche sono più elevate nelle femmine rispetto ai maschi e sono ridotte nei diabetici rispetto ai non diabetici. La riduzione del peso ne aumenta significativamente le concentrazioni circolanti.[26]
L’Adiponectina si auto-associa automaticamente in strutture più grandi. Inizialmente, tre molecole di Adiponectina si legano insieme per formare un omotrimero. I trimeri continuano ad auto-associarsi e formano esameri o dodecameri. Come la concentrazione plasmatica, i livelli relativi delle strutture di ordine superiore sono sessualmente dimorfici, dove le femmine hanno livelli maggiori delle forme ad alto peso molecolare. Studi recenti hanno dimostrato che la forma ad alto peso molecolare può essere quella biologicamente più attiva per quanto riguarda l’omeostasi del glucosio.[27] L’Adiponectina ad alto peso molecolare è stata inoltre osservata associarsi ad un minor rischio di diabete con un’entità di associazione simile all’Adiponectina totale.[28] Tuttavia, si è scoperto che la malattia coronarica è associata positivamente con l’Adiponectina ad alto peso molecolare, ma non con l’Adiponectina a basso peso molecolare.[29]
Leptina
L’Adiponectina esercita alcuni dei suoi effetti di riduzione del peso attraverso il cervello. Questa azione è simile a quella esercitata dalla Leptina[9]; l’Adiponectina e la Leptina possono agire sinergicamente.
L’Adiponectina non si lega ad un solo recettore. Finora, sono stati identificati due recettori con l’omologia dei recettori accoppiati a proteine G (GPCRs) e un recettore simile alla famiglia delle Caderine [30][31]:
Recettore 1 dell’adiponectina (AdipoR1)
Recettore 2 dell’adiponectina (AdipoR2)
T-caderina – CDH13
Questi recettori hanno specificità tissutali distinte all’interno del corpo e hanno affinità diverse con le varie forme di Adiponectina. I recettori influenzano a valle l’AMP chinasi, un importante punto di controllo del tasso metabolico cellulare. L’espressione dei recettori è correlata ai livelli di Insulina, cosa osservata nei modelli murini diabetici, in particolare nel muscolo scheletrico e nel tessuto adiposo.[32][33] Nel 2016 l’Università di Tokyo annunciò l’avvio di un’indagine, spinta dalle richieste fatte in modo anonimo, sulla presunta falsificazione dei dati rilasciati sull’identificazione dei recettori AdipoR1 e AdipoR2.[34]
Sia AdipoR1 che AdipoR2 sono orientati in modo opposto ai GPCR nella membrana (cioè N-terminale citoplasmatico, C-terminale extracellulare) e non si legano alle proteine G di memnrana.[35] I recettori dell’Adiponectina, AdipoR1 e AdipoR2, fungono da recettori per l’Adiponectina globulare e integrale e mediano un aumento delle attività dei ligandi AMPK e PPAR-α, nonché l’ossidazione degli acidi grassi e l’assorbimento del glucosio per attività dell’Adiponectina.[35][36]
Gli effetti legati all’attività della Adiponectina sono:
Regolazione del flusso del glucosio ematico
riduzione della gluconeogenesi
Aumento dell’assorbimento cellulare di glucosio [7][9][11]
2. Catabolismo lipidico
β-ossidazione [9]
Liberazione dei trigliceridi [9]
3. Protezione dalla disfunzione endoteliale (aspetto importante della formazione aterosclerotica)
4. Miglioramento della Sensibilità all’Insulina
5. Perdita di peso
6. Controllo del metabolismo energetico [11]
7. Sovraregolazione delle proteine disaccoppianti (UCP) [23]
8. Riduzione del TNF-α.
Regolazione dell’Adiponectina:
L’obesità è associata alla riduzione dell’Adiponectina.
L’esatto meccanismo di regolazione non è noto, ma l’Adiponectina potrebbe essere regolata da meccanismi post-trasduzionali nelle cellule.[37]
Bassi livelli di Adiponectina sono associati all’ADHD negli adulti.[38]
È stato scoperto che i livelli di Adiponectina sono aumentati nei pazienti con artrite reumatoide che rispondono alla terapia con DMARD o inibitori del TNF. [39]
Il rilascio di Adiponectina indotto dall’esercizio fisico ha aumentato la crescita dell’ippocampo e ha portato a risposte antidepressive nei topi.[40]
Un basso livello di Adiponectina è un fattore di rischio indipendente per lo sviluppo di:
Sindrome metabolica [24]
Diabete mellito di tipo II [9] [5] [10] [12] [13]
I livelli circolanti di Adiponectina possono essere indirettamente aumentati attraverso modifiche dello stile di vita e alcuni farmaci come le Statine.[41]
Esistono dei composti sintetici che interagiscono con i recettori dell’Adiponectina come l’AdipoRon, un agonista selettivo, attivo per via orale, del recettore 1 (AdipoR1) e del recettore 2 dell’Adiponectina (AdipoR2) (Kd = 1,8 μM e 3,1 μM, rispettivamente).[42] L’Università di Tokyo che nel 2016 annunciò l’avvio dell’indagine, spinta dalle richieste fatte in modo anonimo, sulla presunta falsificazione dei dati rilasciati sull’identificazione dei recettori AdipoR1 e AdipoR2, si è occupata anche di questo agonista selettivo dei recettori per l’Adiponectina.[34]
È stato riportato che estratti di patate dolci aumentano i livelli di Adiponectina portando, quindi, ad un miglioramento del controllo glicemico nell’uomo.[43] Tuttavia, una review sistematica ha concluso che non vi sono prove sufficienti a supporto del consumo di patate dolci per il trattamento del diabete mellito di tipo 2.[44]
L’Adiponectina è apparentemente in grado di attraversare la barriera emato-encefalica [45] sebbene esistano dati contrastanti a riguardo.[46] L’Adiponectina ha un’emivita di 2,5 ore nell’uomo.[47]
AdipoRon
Ho accennato pocanzi al AdipoRon, agonista sintetico dei recettori per l’Adiponectina AdipoR1 e AdipoR2.[42] Analogamente all’Adiponectina, questa molecola attiva la segnalazione del AMPK e del PPARα causando un miglioramento dell’insulino sensibilità, della dislipidemia e dell’intolleranza al glucosio nei topi db/db (un modello animale per il diabete di tipo II e l’obesità).[42][48] Inoltre, è stato scoperto che AdipoRon estende la durata della vita dei topi db/db alimentati con una dieta ricca di grassi, oltre a migliorarne la resistenza all’esercizio fisico. [42] [48] [49] La molecola è stata scoperta da ricercatori giapponesi nel 2013 attraverso la revisione della letteratura disponibile, ed è il primo agonista dei recettori per l’Adiponectina attivo per via orale ad essere stato identificato.[42][48]
Gli agonisti del recettore dell’Adiponectina come AdipoRon hanno attirato l’interesse come potenziali terapie per il trattamento dell’obesità, del diabete, delle malattie cardiovascolari, della malattia del fegato grasso non alcolico e una panoplia di altre condizioni.[42][48] Inoltre, è stato recentemente chiarito il ruolo di mediatore dell’adiponectina sugli effetti antidepressivi, ansiolitici e neurogenici indottidall’esercizio fisico. [50][51][52] E’ interessante notare che l’aumento dei livelli di Adiponectina dopo una seduta di esercizio fisico moderato perdura per per 24 a 72 ore. La disregolazione dell’espressione dell’Adiponectina è stata anche implicata nella patologia dei disturbi dell’umore, dei disturbi d’ansia, dei disturbi alimentari, dei disturbi neurodegenerativi e di vari altri disturbi neuropsichiatrici.[53] Inoltre, è stato determinato che l’esercizio fisico migliora la resistenza all’insulina attraverso l’attivazione del recettore AdipoR1.[54] Come tale, gli agonisti del recettore dell’Adiponectina sono un target terapeutico molto interessante per il trattamento di una varietà di condizioni diverse.[42][48][52][53] Inoltre, è stato suggerito che potrebbero essere potenzialmente utilizzati come sostituti dell’esercizio fisico per ottenere benefici simili sulla salute fisica e mentale.[42][48][52][55] Questa opzione è da prendere in considerazione solo e soltanto in quei soggetti impossibilitati a svolgere attività fisica “significativa”.
A causa delle limitazioni nella produzione di Adiponectina biologicamente attiva, gli agonisti degli AdipoRs adiponectino-mimetici sono stati suggeriti come possibili alternative per espandere l’opportunità di sviluppare agenti anti-diabetici. Basandosi sulla struttura cristallina del AdipoR1, i ricercatori hanno progettato gli agonisti dei peptidi del AdipoR1 usando la simulazione del docking proteico-peptidico analizzando le loro capacità di legame per i recettori e le funzioni biologiche attraverso la risonanza plasmonica di superficie (SPR) e l’analisi biologica. Sono stati selezionati e confermati tre peptidi candidati, BHD1028, BHD43 e BHD44 per attivare le vie del segnale mediate da AdipoR1. Al fine di migliorare la stabilità e la solubilità degli agonisti peptidici, i peptidi candidati sono stati PEGilati. Il BHD1028 PEGilato ha mostrato la sua attività biologica alla concentrazione nano-molare e potrebbe essere un potenziale agente terapeutico per il trattamento del diabete. Inoltre, l’SPR e tecniche di screening virtuale possono essere potenzialmente applicate ad altri processi di screening di farmaci peptidici per le proteine del recettore di membrana.[56]
Arctiina
Altre forme di agonisti dei recettori per l’Adiponectina sono i peptidi ADP-355 e ADP-399 [57], i non-peptidi (–)-Arctigenina, Arctiina, Gramina, Matairesinol, Deoxyschizandrina, Parthenolide,
Syringing e Taxifoliol.[58] L’ADP-400 è invece un peptide antagonista del recettore per l’Adiponectina. [58]
Date le potenzialità legate all’Adiponectina, l’uso e la diffusione degli agonisti sintetici dei suoi recettori nella sottocultura del BodyBuilding, in particolare, e del Fitness, in generale, non sarà di certo un evento improbabile nel prossimo futuro. In attesa di questo evento, diversi divulgatori d’oltre oceano, più o meno autorevoli, hanno incominciato a cercare soluzioni OTC per incrementare la sintesi endogena di Adipnectina.
Come già detto all’inizio di questo articolo, i ricercatori hanno scoperto che l’assunzione di grassi monoinsaturi presenti nell’olio di pesce (vedi in particolare EPA e DHA), causa un aumento dei livelli di Adiponectina dal 14 al 60%. Anche l’olio di Cartamo ha dimostrato di aumentare la sintesi di Adiponectina. .[15] Per tale ragione alcuni dei prima citati divulgatori consiglia di assumere 4g/die di CLA derivato dall’olio di Cartamo.
I ricercatori hanno scoperto che l’aggiunta di adeguate quantità di fibre alla dieta causa un aumento dei livelli di Adiponectina tra il 60 ed il 115%.[59] Un motivo in più, se mai ce ne fosse stato il bisogno, di consumare la quantità raccomandata di fibre pari a 30g al giorno.[60]
Il consumo regolare di caffè è stato correlato ad un aumento dei livelli di Adiponectina e ad una riduzione delle citochine pro-infiammatorie, che potrebbero aiutare ad aumentare la perdita di peso e ridurre i livelli di infiammazione.[61]
Curcumina
Si è ipotizzato, con ben poche evidenze, che la Curcumina aumenti la sintesi di Adiponectina. La funzione verrebbe esercitata tramite la riduzione della sintesi di sostanza pro-infiammatorie nell’adipocita con un conseguente aumento della sintesi di Adiponectina.[62]
Resveratrolo
Uno studio del 2011, condotto da ricercatori dell’Università del Texas Health Science Center di San Antonio, pubblicato sul Journal of Biological Chemistry, ha riportato che il Resveratrolo stimola anche l’espressione dell’Adiponectina. [63]
Secondo uno studio del 2012, completato all’Università di Gerusalemme, una dieta sperimentale con carboidrati consumata principalmente a cena, piuttosto che durante il giorno, sembra avvantaggiare le persone che soffrono di obesità grave e morbosa. Questa dieta sembra influenzare i modelli di secrezione degli ormoni responsabili della fame e della sazietà, nonché gli ormoni associati alla sindrome metabolica, compreso un aumento della produzione di Adiponectina durante il giorno.[64]
In uno studio pubblicato sull’Iranian Journal of Diabetes and Obesity nel giugno 2012, è stato osservato un aumento significativo dei livelli di Adiponectina nei soggetti che avevano assunto 50mg di Zinco rispetto a al gruppo di controllo.[65] Un altro studio simile, nel quale soggetti diabetici di tipo II sono stati trattati con 30mg/die di Zinco, è stato osservato un aumento significativo della Adiponectina rispetto al basale ma non rispetto al gruppo di controllo (e il 53.3% presentava un insufficienza di Zinco al basale).[66]
Cianidina 3-glucoside
L’antocianina C3G (cianadina 3-glucoside), se assunta in quantità sufficiente in forma di integratore, sembra migliorare la composizione corporea. Si è sempre pensato che questo effetto fosse causato dagli effetti positivi diretti C3G sulla sensibilità all’Insulina. Infatti, è noto da tempo che la C3G possa aumentare la lipolisi stimolando la sintesi di adipocitochine (proteine di segnalazione cellulare) come l’Adiponectina, che regola appunto i livelli di glucosio e l’ossidazione degli acidi grassi. Recentemente sono emerse alcune ricerche secondo cui questo aumento di Adiponectina mediato dal C3G potrebbe anche contribuire alla crescita muscolare.[67]
L’Agaricus blazei Murrill è un fungo medicinale non facente parte della Medicina Tradizionale Cinese (MTC), ma che ha suscitato grande interessa per la sua peculiare capacità di regolare la risposta immunitaria, ha mostrato di poter aumentare la concentrazione plasmatica di Adiponectina del 20%.[68] Favorendo un aumento di concentrazione di Adiponectina, l’AbM risulterebbe molto utile in caso di steatosi epatica non alcolica e insulino-resistenza. C’è da considerare però il fatto che, nello studio citato, l’estratto del AbM era somministrata in concomitanza con Metformina e Gliclazide.
Non fatevi troppe illusioni però, è molto probabile che se vi metteste a provare una o più di queste ipotetiche metodiche per aumentare i livelli di Adiponetina, il risultato che ne ricavereste, nel migliore delle ipotesi, non sarebbe da attribuirsi ad altra cosa se non al effetto placebo. Ma, da ricercatore quale sono, non biasimerò di certo chi vorrà testare e cercare di comprendere, eliminando per quanto possibile le variabili in gioco, se l’effetto ottenuto (se se ne è ottenuto qualcuno) è attribuibile ad un incremento dell’Adiponectina… risultante comunque molto speculativa vista la mancanza per la stragrande maggioranza delle persone di un laboratorio ove sottoporsi a periodiche analisi al fine di quantificare i (se mai ci fossero) incrementi del peptide in questione.
Maeda K, Okubo K, Shimomura I, Funahashi T, Matsuzawa Y, Matsubara K (April 1996). “cDNA cloning and expression of a novel adipose specific collagen-like factor, apM1 (AdiPose Most abundant Gene transcript 1)”. Biochemical and Biophysical Research Communications. 221 (2): 286–9. doi:10.1006/bbrc.1996.0587
Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF (November 1995). “A novel serum protein similar to C1q, produced exclusively in adipocytes”. The Journal of Biological Chemistry. 270 (45): 26746–9. doi:10.1074/jbc.270.45.26746
Lara-Castro C, Fu Y, Chung BH, Garvey WT (June 2007). “Adiponectin and the metabolic syndrome: mechanisms mediating risk for metabolic and cardiovascular disease”. Current Opinion in Lipidology. 18 (3): 263–70.
Matsuzawa Y, Funahashi T, Kihara S, Shimomura I (January 2004). “Adiponectin and metabolic syndrome”. Arteriosclerosis, Thrombosis, and Vascular Biology. 24 (1): 29–33.
Díez JJ, Iglesias P (March 2003). “The role of the novel adipocyte-derived hormone adiponectin in human disease”. European Journal of Endocrinology. 148 (3): 293–300.
Ukkola O, Santaniemi M (November 2002). “Adiponectin: a link between excess adiposity and associated comorbidities?”. Journal of Molecular Medicine. 80 (11): 696–702.
Hara K, Yamauchi T, Kadowaki T (April 2005). “Adiponectin: an adipokine linking adipocytes and type 2 diabetes in humans”. Current Diabetes Reports. 5 (2): 136–40.
Vasseur F, Leprêtre F, Lacquemant C, Froguel P (April 2003). “The genetics of adiponectin”. Current Diabetes Reports. 3 (2): 151–8.
Vasseur F, Meyre D, Froguel P (November 2006). “Adiponectin, type 2 diabetes and the metabolic syndrome: lessons from human genetic studies”. Expert Reviews in Molecular Medicine. 8 (27): 1–12.
Grimshaw CE, Matthews DA, Varughese KI, Skinner M, Xuong NH, Bray T, Hoch J, Whiteley JM (August 1992). “Characterization and nucleotide binding properties of a mutant dihydropteridine reductase containing an aspartate 37-isoleucine replacement”. The Journal of Biological Chemistry. 267 (22): 15334–9.
Yuan J, Liu W, Liu ZL, Li N (2006). “cDNA cloning, genomic structure, chromosomal mapping and expression analysis of ADIPOQ (adiponectin) in chicken”. Cytogenetic and Genome Research. 112 (1–2): 148–51.
Nishio S, Gibert Y, Bernard L, Brunet F, Triqueneaux G, Laudet V (June 2008). “Adiponectin and adiponectin receptor genes are coexpressed during zebrafish embryogenesis and regulated by food deprivation”. Developmental Dynamics. 237 (6): 1682–90.
Shapiro L, Scherer PE (March 1998). “The crystal structure of a complement-1q family protein suggests an evolutionary link to tumor necrosis factor”. Current Biology. 8 (6): 335–8.
Chen J, Tan B, Karteris E, Zervou S, Digby J, Hillhouse EW, Vatish M, Randeva HS (June 2006). “Secretion of adiponectin by human placenta: differential modulation of adiponectin and its receptors by cytokines”. Diabetologia. 49 (6): 1292–302.
Kuo SM, Halpern MM (December 2011). “Lack of association between body mass index and plasma adiponectin levels in healthy adults”. International Journal of Obesity. 35 (12): 1487–94.
Bauche IB, El Mkadem SA, Pottier AM, Senou M, Many MC, Rezsohazy R, Penicaud L, Maeda N, Funahashi T, Brichard SM (April 2007). “Overexpression of adiponectin targeted to adipose tissue in transgenic mice: impaired adipocyte differentiation”. Endocrinology. 148 (4): 1539–49
Renaldi O, Pramono B, Sinorita H, Purnomo LB, Asdie RH, Asdie AH (January 2009). “Hypoadiponectinemia: a risk factor for metabolic syndrome”. Acta Medica Indonesiana. 41 (1): 20–4.
Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, Mori Y, Ide T, Murakami K, Tsuboyama-Kasaoka N, Ezaki O, Akanuma Y, Gavrilova O, Vinson C, Reitman ML, Kagechika H, Shudo K, Yoda M, Nakano Y, Tobe K, Nagai R, Kimura S, Tomita M, Froguel P, Kadowaki T (August 2001). “The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity”. Nature Medicine. 7 (8): 941–6.
Coppola A, Marfella R, Coppola L, Tagliamonte E, Fontana D, Liguori E, Cirillo T, Cafiero M, Natale S, Astarita C (May 2009). “Effect of weight loss on coronary circulation and adiponectin levels in obese women”. International Journal of Cardiology. 134 (3): 414–6.
Oh DK, Ciaraldi T, Henry RR (May 2007). “Adiponectin in health and disease”. Diabetes, Obesity & Metabolism. 9 (3): 282–9.
Rizza S, Gigli F, Galli A, Micchelini B, Lauro D, Lauro R, Federici M (April 2010). “Adiponectin isoforms in elderly patients with or without coronary artery disease”. Journal of the American Geriatrics Society. 58 (4): 702–6.
Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, Sugiyama T, Miyagishi M, Hara K, Tsunoda M, Murakami K, Ohteki T, Uchida S, Takekawa S, Waki H, Tsuno NH, Shibata Y, Terauchi Y, Froguel P, Tobe K, Koyasu S, Taira K, Kitamura T, Shimizu T, Nagai R, Kadowaki T (June 2003). “Cloning of adiponectin receptors that mediate antidiabetic metabolic effects”. Nature. 423 (6941): 762–9.
Fang X, Sweeney G (November 2006). “Mechanisms regulating energy metabolism by adiponectin in obesity and diabetes”. Biochemical Society Transactions. 34 (Pt 5): 798–801.
Bonnard C, Durand A, Vidal H, Rieusset J (February 2008). “Changes in adiponectin, its receptors and AMPK activity in tissues of diet-induced diabetic mice”. Diabetes & Metabolism. 34 (1): 52–61.
Lim S, Quon MJ, Koh KK (April 2014). “Modulation of adiponectin as a potential therapeutic strategy”. Atherosclerosis. 233 (2): 721–8.
Okada-Iwabu M, Yamauchi T, Iwabu M, Honma T, Hamagami K, Matsuda K, Yamaguchi M, Tanabe H, Kimura-Someya T, Shirouzu M, Ogata H, Tokuyama K, Ueki K, Nagano T, Tanaka A, Yokoyama S, Kadowaki T (November 2013). “A small-molecule AdipoR agonist for type 2 diabetes and short life in obesity”. Nature. 503 (7477): 493–9.
Ludvik B, Hanefeld M, Pacini G (July 2008). “Improved metabolic control by Ipomoea batatas (Caiapo) is associated with increased adiponectin and decreased fibrinogen levels in type 2 diabetic subjects”. Diabetes, Obesity & Metabolism. 10 (7): 586–92.
Ooi CP, Loke SC (September 2013). “Sweet potato for type 2 diabetes mellitus”. The Cochrane Database of Systematic Reviews. 9 (9): CD009128.
Spranger J, Verma S, Göhring I, Bobbert T, Seifert J, Sindler AL, Pfeiffer A, Hileman SM, Tschöp M, Banks WA (January 2006). “Adiponectin does not cross the blood-brain barrier but modifies cytokine expression of brain endothelial cells”. Diabetes. 55 (1): 141–7.
Hoffstedt J, Arvidsson E, Sjölin E, Wåhlén K, Arner P (March 2004). “Adipose tissue adiponectin production and adiponectin serum concentration in human obesity and insulin resistance”. The Journal of Clinical Endocrinology and Metabolism. 89 (3): 1391–6.
Nicolas S, Veyssière J, Gandin C, Zsürger N, Pietri M, Heurteaux C, Glaichenhaus N, Petit-Paitel A, Chabry J (2015). “Neurogenesis-independent antidepressant-like effects of enriched environment is dependent on adiponectin”. Psychoneuroendocrinology. 57: 72–83.
Li A, Yau SY, Machado S, Yuan TF, So KF (2015). “Adult neurogenic and antidepressant effects of adiponectin: a potential replacement for exercise?”. CNS Neurol Disord Drug Targets. 14 (9): 1129–1144.
Cho JK, Kim S, Hong HR, Yoon JH, Kang H (2015). “Exercise Training Improves Whole Body Insulin Resistance via Adiponectin Receptor 1”. Int J Sports Med. 36 (13): e24–e30.
Gorman, Katherina, et al, “Adiponectin is necessary for exercise training-induced muscular hypertrophy and vascular adaptation.” The FASEB Journal, April 2016. Vol. 30 no. 1 Supplement 1240.23.