
Introduzione

Con il termine Antiestrogeni ci si riferisce genericamente ad una classe di farmaci aventi azione diretta o indiretta sull’attività tissutale e/o concentrazione ematica degli estrogeni. Agiscono bloccando il recettore dell’estrogeno (ER) e/o riducendo o sopprimendo la sintesi estrogenica.(1)(2) Una recente categoria di agenti facenti parte di questa classe di farmaci, i SERD (Selective Estrogen Receptor Degrader), esplicano la loro azione antiestrogena degradando/sottoregolando il recettore dell’estrogeno. Gli Antiestrogeni sono una delle tre classi di farmaci antagonisti dell’ormone sessuale, insieme agli Antiandrogeni e agli Antiprogestinici.(3)
Largamente utilizzati in ambito sportivo, in special modo nell’ambiente culturistico, con il fine di controllare l’attività estrogenica durante l’uso di AAS aromatizzabili, o aventi attività estrogenica intrinseca, e durante la PCT con lo scopo aggiunto di stimolare la ripresa dell’HPTA, questi farmaci hanno un discreto carico di effetti collaterali tra i quali, quelli che destano maggior preoccupazione nell’atleta previdente, vi sono la dislipidemia (aumento dell’LDL, dei Trigliceridi, riduzione del HDL e alterazione delle loro ratio), l’atralgia (dolore articolare), il calo della libido/disfunzione erettile e l’affaticamento/letargia. Non sono di certo da meno le preoccupazioni legate all’alterazione dell’Asse GH/IGF-1 o la riduzione delle potenzialità di induzione ipertrofica di un ciclo in seguito ad una eccessiva soppressione dell’attività e/o delle concentrazioni estrogeniche. Ma esiste un’altra preoccupazione legata all’uso di composti antiestrogeni, ed è la possibilità che si verifichi un rebound estrogenico in seguito al l’oro uso. Purtroppo, la letteratura a disposizione è al quanto scarsa e poco chiara nella specifica del problema. E’ possibile, però, fare maggiore chiarezza sulla questione analizzando le caratteristiche dei composti antiestrogeni e il loro impatto, passando in rassegna tutti i componenti dell’addizione (Recettori Estrogeni e enzima Aromatasi). In questo articolo cercherò di esporre un ragionamento logico grazie al quale, seppur non avendo una risposta definitiva, sarà possibile avere un idea, la più concreta possibile, sul binomio antiestrogeni/rebound estrogenico.
Una analisi della questione…
- Recettori dell’Estrogeno, SERM e “rebound estrogenico”

I Recettori degli Estrogeni (ER) sono un gruppo di proteine presenti all’interno delle cellule. Sono recettori attivati dall’ormone estrogeno (con maggiore attività del 17β-estradiolo).(4) Esistono due classi di ER: i Recettori degli Estrogeni nucleari (ERα e ERβ), che sono membri della famiglia dei recettori nucleari e dei recettori intracellulari, ed i Recettori degli Estrogeni di Membrana (MR) (GPER (GPR30), ER-X e Gq-mER), che sono per lo più recettori accoppiati alla proteina G. In questa sede ci si riferirà ai primi (ER).
Una volta attivato dall’estrogeno, l’ER è in grado di traslocare nel nucleo e legarsi al DNA per regolare l’attività di diversi geni (ciò significa che è un fattore di trascrizione del DNA). Tuttavia, ha anche funzioni aggiuntive indipendenti dal legame con il DNA.(5)
Poiché l’estrogeno è un ormone steroideo, può passare attraverso le membrane fosfolipidiche della cellula, e pertanto i recettori non hanno bisogno di essere legati alla membrana per potersi legare a loro volta con l’estrogeno.
L’estrogeno esplica la sua attività cellulare attraverso un azione Genomica e Non-Genomica.
• Genomica
In assenza di ormoni, i ER si trovano in gran parte nel citosol. Il legame dell’ormone al recettore innesca un numero di eventi che iniziano con la migrazione del recettore dal citosol nel nucleo, la dimerizzazione del recettore e il successivo legame del dimero del recettore a specifiche sequenze di DNA conosciute come elementi di risposta ormonale. Il complesso DNA / recettore quindi recluta altre proteine che sono responsabili della trascrizione del DNA a valle in mRNA e, infine, in una proteina la quale porta a dei cambiamenti nella funzione cellulare. I recettori degli estrogeni si trovano anche all’interno del nucleo della cellula, ed entrambi i sottotipi del recettore dell’estrogeno hanno un dominio di legame con il DNA e possono funzionare come fattori di trascrizione per regolare la produzione di proteine.
Il recettore interagisce anche con la proteina attivatore 1 e Sp-1 per promuovere la trascrizione, attraverso diversi coattivatori come il PELP-1.(6)
L’acetilazione diretta del recettore alfa dell’estrogeno ai residui della lisina nella regione cerniera mediante il p300 regola la transattivazione e la sensibilità ormonale.(7)
• Non-Genomica
Alcuni recettori per gli estrogeni sono presenti nelle membrana della superficie della cellula e possono essere rapidamente attivati dall’esposizione di questa agli estrogeni.(8)(9)
Inoltre, alcuni ER possono associarsi alle membrane cellulari legandole alla caveolina-1 e formarmando complessi con la proteine G, striatina, tirosina chinasi del recettore (es. EGFR e IGF-1) e tirosina chinasi non recettoriale (es. Src). (6)(8) Attraverso la striatina, alcuni di questi ER legati alla membrana possono portare a livelli aumentati di Ca2 + e ossido nitrico (NO).(10) Attraverso il recettore tirosin chinasi, i segnali vengono inviati al nucleo attraverso la via della proteina chinasi attivata dal mitogeno (MAPK / ERK) e la via del fosfoinositide 3-chinasi (Pl3K / AKT).(11) La glicogeno sintasi chinasi-3 (GSK) -3β inibisce la trascrizione dal ER nucleare inibendo la fosforilazione della serina 118 dell’ERa nucleare. La fosforilazione di GSK-3β rimuove il suo effetto inibitorio, e questo può essere ottenuto tramite il pathway PI3K / AKT e il pathway MAPK / ERK, tramite rsk.
Il 17β-estradiolo ha dimostrato di attivare il recettore GPR30 accoppiato alla proteina G.(12) Tuttavia, la localizzazione subcellulare e il ruolo di questo recettore sono ancora oggetto di controversie.(13)

Gli estrogeni e gli ER sono implicati nel cancro al seno, nel carcinoma ovarico, nel cancro del colon, nel cancro alla prostata e nel cancro dell’endometrio. Il carcinoma del colon avanzato è associato a una perdita di ERβ, l’ER predominante nel tessuto del colon, e il tumore del colon è trattato con agonisti specifici per ERβ.(14)
Sappiamo che i recettori degli estrogeni sono sovraespressi in circa il 70% dei casi di cancro al seno, indicati come “ER-positivi”, e possono essere dimostrati in tali tessuti mediante l’immunoistochimica.(15) E’ ipotizzabile ,quindi, che gli atleti più sensibili agli effetti estrogenici presentino un espressione dei ER più elevata del normale, cosa che li porta a sviluppare con maggiore facilità effetti avversi dati da un eccesso dei livelli estrogenici e/o da un aumento della loro attività dato dalla cosomministrazione con progestinici (es. Nandrolone e Trenbolone).

I Modulatori Selettivi del Recettore dell’Estrogeno (SERM) sono composti antiestrogenici che agiscono a livello del ER. (16) Una caratteristica che distingue queste sostanze dagli agonisti e antagonisti ER puri (cioè agonisti completi e antagonisti silenti) è che la loro azione è diversa nei vari tessuti, garantendo in tal modo la possibilità di inibire selettivamente o stimolare l’azione estrogenica in diversi tessuti.
I SERM sono agonisti parziali competitivi del ER.(17) Tessuti diversi presentano differenti gradi di sensibilità all’attività degli estrogeni, pertanto i SERM esplicano effetti estrogenici o antiestrogeni a seconda del tessuto specifico con il quale interagiscono e della percentuale di attività intrinseca (IA) del composto in questione.(18) Un esempio di SERM con alta IA, e quindi di effetti prevalentemente estrogenici, è rappresentato dal Clorotrianisene, mentre un esempio di SERM con bassa IA, e quindi avente per lo più attività antiestrogenica, è rappresentato dall’Ethamoxytrifetolo. SERM come il Clomifene e il Tamoxifene, largamente utilizzati in ambito sportivo, sono considerabili come composti con valore IA intermedio essendo molecole con una azione bilanciata tra effetti estrogenici e antiestrogenici. Il Raloxifene è un SERM che presenta una azione antiestrogenica maggiore del Tamoxifene; entrambi hanno una attività estrogenica (sebbene differente) a livello osseo, ma il Raloxifene presenta una attività antiestrogenica nell’utero mentre il Tamoxifene ha un azione estrogenica nel tessuto dell’utero.(18)

Il Tamoxifene è un farmaco di prima linea per il trattamento del carcinoma mammario metastatico ER-positivo. È usato per la riduzione delle possibilità di sviluppo del cancro al seno nelle donne ad alto rischio, come trattamento adiuvante del nodo ascellare negativo e positivo, e nel carcinoma duttale in situ.(19)(20)
Il Tamoxifene è classificabile come un profarmaco, dal momento che la sua affinità per la proteina bersaglio (ER) è limitata. Il Tamoxifene viene metabolizzato nel fegato dall’isoforma del citocromo CYP2D6 e CYP3A4 in metaboliti attivi come l’Afimoxifene (4-idrossitamoxifene; 4-OHT) e l’Endoxifene (N-desmetil-4-idrossitamoxifene) (21) che presentano una affinità da 30 a 100 volte maggiore per il ER rispetto al Tamoxifene. (22) Questi metaboliti attivi competono con gli estrogeni per il legame con il recettore. Nel tessuto mammario, il 4-OHT agisce come un antagonista del ER in modo da inibire la trascrizione dei geni che reagiscono agli estrogeni. (23) Il Tamoxifene ha rispettivamente il 7% e il 6% dell’affinità dell’Estradiolo per il ERα e il ERβ, mentre il 4-OHT ha il 178% e il 338% dell’affinità dell’Estradiolo per il ERα e il ERβ.(24)

Il 4-OHT si lega al ER, il complesso ER/Tamoxifene recluta altre proteine note come co-repressori e quindi si lega al DNA per modulare l’espressione genica. Alcune di queste proteine includono la NCoR e la SMRT. (25) La funzione del Tamoxifene può essere regolata da una serie di variabili diverse, compresi i fattori di crescita.(26) Il Tamoxifene deve bloccare le proteine del fattore di crescita come ErbB2/HER2 (27) perché è stato dimostrato che livelli elevati di ErbB2 si manifestano nei tumori resistenti al Tamoxifene.(28) Il Tamoxifene sembra richiedere una proteina PAX2 affinché possa esplicare il suo pieno effetto antitumorale. (27)(29) In presenza di un elevata espressione della PAX2, il complesso Tamoxifene/ER è in grado di sopprimere l’espressione della proteina pro-proliferativa del ERBB2. Al contrario, quando l’espressione del AIB-1 è superiore alla PAX2, il complesso di Tamoxifene/ER aumenta l’espressione del ERBB2 con conseguente stimolazione della crescita del cancro al seno. (27)(30)
Il 4-OHT si lega al ER in modo competitivo (rispetto all’estrogeno agonista) nelle cellule tumorali e in altri bersagli tissutali, producendo un complesso nucleare che riduce la sintesi del DNA e inibisce gli effetti degli estrogeni. È un agente non steroideo con potenti proprietà antiestrogeniche che competono con gli estrogeni per i siti di legame nel seno e in altri tessuti. Il Tamoxifene fa sì che le cellule rimangano nelle fasi G0 e G1 del ciclo cellulare. Poiché impedisce alle cellule (pre) cancerose di dividersi ma non provoca la morte cellulare, il Tamoxifene è citostatico piuttosto che citocida.
La letteratura scientifica riguardante l’attività del Tamoxifene è a dir poco complessa ed occorre prestare particolare attenzione ai dati disponibili per stabilire se il Tamoxifene, o il suo metabolita 4-idrossi, abbiano il maggiore impatto complessivo.

Il Norendoxifene (N, N-didesmetil-4-idrossitamoxifene), un altro metabolita attivo del Tamoxifene, è stato osservato agire come un potente inibitore dell’aromatasi competitivo (IC50 = 90 nM), cosa che a sua volta può amplificare l’attività antiestrogenica complessiva del Tamoxifene.(31)
Come già accennato in precedenza, e come molti sapranno, il Tamoxifene è largamente utilizzato in ambito sportivo, sia da solo che in abbinamento con altri SERM come il Clomifene ( in PCT) o con AI (“on-cycle” e/o in PCT). La sua applicazione all’interno di una preparazione che contempla l’uso di AAS aromatizzabili, alla luce di quanto esposto pocanzi, lo vede come agente preventivo o di trattamento dell’attività estrogenica a livello tissutale, in specie per quanto concerne l’attività estrogenica nel tessuto mammario al fine di evitare (o “tamponare”) la comparsa della ginecomastia. In un contesto PCT tale composto, oltre ad esercitare la funzione di regolazione dell’attività estrogenica appena esposta, agendo a livello ipotalamico stimola il rilascio di GnRH e, consequenzialmente, di LH ed FSH dall’Ipofisi che a loro volta stimoleranno la sintesi di Testosterone e la spermatogenesi.
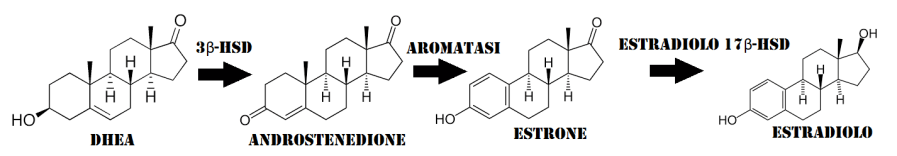
Il suo utilizzo massivo e cronico è stato spesso collegato aneddoticamente a rebound estrogenico. Ora, conoscendo la complessità d’azione che questo composto (ed i suoi metaboliti) ha sul controllo dell’attività estrogenica, si può facilmente ipotizzare che un suo uso protratto (legato anche alla dose e, quindi, al suo impatto sulla attività estrogenica sistemica) possa innescare degli adattamenti reattivi con conseguente aumento dell’attività estrogenica attraverso l’incremento dei livelli serici di Estradiolo e dell’attività non-genomica dello steroide (ipotizzabile anche un aumento del numero dei ER). Nel corso degli anni sono state esposte diverse ipotesi volte a spiegare i meccanismi attraverso i quali un abuso di Tamoxifene possa portare ad un rebound estrogenico. Una di queste ipotesi venne riportata all’inizio del secolo dal compianto A.L. Rea il quale affermava che la causa andasse ricercata nell’aumento del rilascio di DHEA da parte delle ghiandole surrenali e dalla sua successiva (e aumentata) conversione in Androstenedione e, attraverso l’intervento dell’enzima aromatasi che lo converte in Estrone e la successiva azione del estradiolo 17beta-deidrogenasi, Estradiolo.(32) In breve, secondo questa teoria i processi innescati causerebbero l’instaurarsi di livelli di E2 cronicamente alti con conseguente impossibilità del SERM di esplicare la sua azione. Questa teoria seppur, in parte, possa dare una spiegazione logica dei possibili meccanismi implicati manca di alcuni tasselli. Il principale “tallone d’Achille” è rappresentato dai livelli di E2 che, una volta aumentati, diventano dei competitor recettoriali più aggressivi rispetto al 4-OHT (che ricordiamo avere il 178% dell’affinità dell’Estradiolo per il ERα). Ciò potrebbe avvenire in situazioni di calo delle concentrazioni di 4-OHT seguenti alla riduzione del dosaggio del farmaco o alla sua cessazione, quindi, in questo ultimo caso, esplicabili in crescendo nei 7-14 giorni successivi all’interruzione della somministrazione e con una durata indeterminata. Di conseguenza, sembra più plausibile che l’aumento delle concentrazioni di E2, durante l’uso del Tamoxifene, si affianchi ad un consequenziale incremento dell’attività Non-Genomica dell’ormone e da un aumentato numero di ER. Seguendo questa logica, una volta interrotto l’uso del Tamoxifene, queste condizioni tenderanno ad aggravarsi come gli effetti avversi a loro legati.
Consultando la bibliografia scientifica disponibile, non si trovano accenni su un possibile rebound estrogenico in seguito all’uso di Tamoxifene, ma si parla nello specifico di “resistenza al Tamoxifene” o “sottoregolazione degli ER”.(33)(34) Nel caso della “resistenza al Tamoxifene” sembra che l’aumento dell’espressione del gene MACROD2 porti ad una risposta negativa all’azione del SERM con conseguente proliferazione delle cellule cancerose estradiolo-dipendenti. La sovra espressione di tale gene sembra essere di base genetica anche se non si esclude una risposta di adattamento in seguito ad uso cronico del composto in questione.
Il Raloxifene, un altro SERM discretamente utilizzato nella pratica sportiva, è un agonista-antagonista misto del ER.(35)(36)(37) Ha effetti estrogenici a livello osseo ed epatico con effetti antiestrogenici nei seni e nell’utero. Le azioni biologiche del Raloxifene sono quindi ampiamente mediate dal legame con i ER. Questo legame determina l’attivazione di percorsi estrogenici in alcuni tessuti (agonismo) e il blocco di questi in altri (antagonismo). Le sue caratteristiche d’azione similari a quelle del Tamoxifene, sembrano poter far pensare ad un medesimo e ipotetico meccanismo che possa portare ad un rebound estrogenico. Questa volta la letteratura scientifica sembra dare alcune conferme. In un caso studio (38), una paziente di 66 anni si è presentata con recidiva metastatica acuta estrogeno-positiva e progesterone-positiva, carcinoma mammario Her-2 / neu-negativo, lesioni ossee (colonna lombare, bacino), noduli polmonari, metastasi epatiche, antigene tumorale elevato 15 e enzimi epatici, dispepsia e diarrea. La paziente aveva assunto Raloxifene per circa 8 anni. Dopo la sospensione del farmaco, parametri e sintomi clinici sono migliorati rapidamente senza terapia oncologica o altre forme di trattamento. Tre mesi dopo la sospensione del Raloxifene, l’oncologo ha prescritto alla paziente l’uso della Capecitabina dato che non riteneva plausibile un effetto di rebound estrogenico (anti-estrogen withdrawal effect – AEWE). Tuttavia, la regressione duratura è stata più indicativa di un effetto rebound dato dal Raloxifene rispetto alla chemioterapia o ad altri interventi. In seguito la paziente si è mostrata asintomatica con un buono stato di prestazione. La regressione metastatica epatica è stata confermata, senza alcun trattamento oncologico somministrato negli ultimi 16 mesi e circa 23 mesi dopo il termine d’uso del Raloxifene. Questo caso evidenzia la necessità di esaminare pazienti con carcinoma mammario per la possibilità di un AEWE con l’uso di Raloxifene o con altri SERM . Ovviamente, il caso presentato non è molto comparabile, soprattutto per quanto riguarda i tempi di somministrazione, ad un BodyBuilder supplementato chimicamente nella “media” ma, ciò nonostante, ci offre un indizio sulla probabilità che si possa manifestare un rebound estrogenico con l’uso di SERM.
- Enzima Aromatasi, Inibitori della Aromatasi e “rebound estrogenico”

L’Enzima Aromatasi, chiamato anche estrogeno sintetasi o estrogeno sintasi, è un enzima responsabile del processo fondamentale della biosintesi degli Estrogeni. Denominato CYP19A1, questo enzima è un membro della superfamiglia del citocromo P450 (EC 1.14.14.1), che sono monoossigenasi che catalizzano molte reazioni coinvolte nella steroidogenesi. In particolare, l’Aromatasi è responsabile dell’aromatizzazione degli Androgeni in Estrogeni. L’enzima Aromatasi è sintetizzato in molti tessuti tra cui le gonadi (cellule della granulosa), cervello, tessuto adiposo, placenta, vasi sanguigni, pelle e ossa, nonché nei tessuti dell’endometriosi, dei fibromi uterini, del cancro al seno e del cancro dell’endometrio.
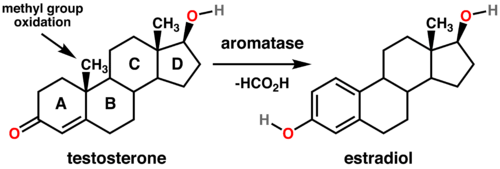
L’Aromatasi è localizzato nel reticolo endoplasmatico dove è regolato da promotori tissutali che sono a loro volta controllati da ormoni, citochine e altri fattori. Catalizza gli ultimi passaggi della biosintesi degli estrogeni dagli androgeni (in particolare, converte l’Androstenedione in Estrone e il Testosterone in Estradiolo). Queste fasi comprendono tre idrossilazioni successive del gruppo 19-metilico degli androgeni, seguite dall’eliminazione simultanea del gruppo metilico come formiato e aromatizzazione dell’anello A.
Androstenedione + 3O2 + 3NADPH + 3H+  Estrone + Formiato + 4H2O + 3NADP+
Estrone + Formiato + 4H2O + 3NADP+
Testosterone + 3O2 + 3NADPH + 3H+ 17β-estradiol + Formiato + 4H2O + 3NADP+
Il gene esprime due varianti di trascrizione. (39) Nell’uomo, il gene CYP19, situato sul cromosoma 15q21.1, codifica per l’Enzima Aromatasi. (40) Il gene ha nove esoni codificanti e un numero di primi esoni non codificanti alternativi che regolano l’espressione specifica del tessuto. (41)
Il CYP19 è presente in un cordato precoce divergente, l’anfiosso cefalocordato (il Florida lancelet, Branchiostoma floridae), ma non nel precedente tunicato divergente Ciona intestinalis. Pertanto, gli evoluzionisti ipotizzano che il gene Aromatasi si sia evoluto precocemente nell’evoluzione dei cordati e non sembra essere presente negli invertebrati non-cordati (ad esempio insetti, molluschi, echinodermi, spugne, coralli). Tuttavia, gli Estrogeni possono essere sintetizzati in alcuni di questi organismi, attraverso altri percorsi sconosciuti.
I fattori noti che aumentano l’attività dell’Aromatasi includono l’età, l’obesità, l’Insulina, le gonadotropine e l’alcol. L’attività dell’Aromatasi risulta diminuita dalla Prolattina, dall’ormone anti-Mülleriano e dal glifosato , un comune erbicida.(42) L’attività dell’Aromatasi sembra essere migliorata in alcuni tessuti estrogeno-dipendenti come il tessuto mammario, nel carcinoma dell’endometrio, nell’endometriosi e nei fibromi uterini.

Gli Inibitori dell’Aromatasi (AI) sono una gruppo di farmaci usati nel trattamento del carcinoma mammario nelle donne in postmenopausa e nella ginecomastia negli uomini. Come i SERM, trovano un largo uso off-label in ambito sportivo durante la somministrazione di AAS aromatizzabili o durante la PCT. Possono anche essere utilizzati per la chemioprevenzione in donne ad alto rischio.
Esistono due tipi di Inibitori dell’Aromatasi approvati per il trattamento del carcinoma mammario e, quindi, diffusi anche per l’uso off-label: (43)
– Gli inibitori steroidei irreversibili, come l’Exemestano (nome commerciale Aromasin), formano un legame permanente e disattivante con l’Enzima Aromatasi.
– Gli inibitori non steroidei, come l’Anastrozolo (nome commerciale Arimidex) e il Letrozolo (nome commerciale Femara), inibiscono la sintesi degli Estrogeni attraverso la competizione reversibile per l’Enzima Aromatasi.
Gli inibitori dell’Aromatasi disponibili (AI) includono:
– Non selettivi:
• L’Aminoglutetimide, il quale però inibisce l’enzima P450scc agendo come inibitore della biosintesi di tutti gli ormoni steroidei (aprirò una nota a riguardo più avanti).
• Testolactone (nome commerciale Teslac)
– Selettivi:
• Anastrozolo (Arimidex)
• Letrozolo (Femara)
• Exemestano (Aromasin)
• Vorozolo (Rivizor)
• Formestano (Lentaron)
• Fadrozolo (Afema)
– Non classificati:
• 1,4,6-Androstatrien-3,17-dione (ATD)
• 4-Androstene-3,6,17-trione (“6-OXO”)
Oltre agli AI farmaceutici, alcuni composti naturali hanno mostrato effetti di inibizione dell’Aromatasi, come le foglie di damiana. Il loro impatto non è stato pienamente chiarito sull’uomo.
Gli Inibitori dell’Aromatasi agiscono, proprio come suggerisce il nome, inibendo l’azione dell’enzima Aromatasi, che converte gli Androgeni in Estrogeni mediante un processo chiamato aromatizzazione. Poiché il tessuto mammario è stimolato dagli Estrogeni, diminuirne la produzione è un modo per sopprimere la recidiva del tessuto tumorale del seno. La principale fonte di Estrogeni è rappresentata dalle ovaie nelle donne in premenopausa, mentre nelle donne in post-menopausa la maggior parte degli Estrogeni del corpo viene prodotta nei tessuti periferici (al di fuori del SNC) e anche in alcuni siti del SNC in varie regioni del cervello. L’Estrogeno viene prodotto e agisce localmente in questi tessuti, ma qualsiasi estrogeno circolante, che esercita effetti estrogenici sistemici in uomini e donne, è il risultato dell’Estrogeno che sfugge al metabolismo locale e si diffonde nel sistema circolatorio.(44)
Come già accennato pocanzi, i composti AI sono anch’essi, al pari dei SERM, largamente utilizzati in ambito sportivo, sia come agenti di controllo dei livelli estrogenici durante l’uso di AAS aromatizzabili (uso preventivo della comparsa di effetti estrogenici), in caso di ginecomastia (spesso in combinazione con un SERM, specie se l’AI utilizzato è l’Exemestano) o in combinazione con i SERM in ambito PCT (specie nella fase preliminare dove viene utilizzata l’hCG).
Più che con i SERM, il rebound estrogenico è stato riportato, soprattutto aneddoticamente, con l’uso di AI, specialmente quelli reversibili (vedi Anastrozolo e Letrozolo).
L’Anastrozolo ed il Letrozolo agiscono legandosi in modo reversibile all’Enzima Aromatasi (unità eme del citocromo P450) e, attraverso l’inibizione competitiva, blocca la conversione degli Androgeni in Estrogeni nei tessuti periferici (extragonali).

Il Letrozolo ha dimostrato, attraverso studi clinici, di poter abbassare rapidamente il livello degli estrogeni fino al 65%. Il motivo principale è probabilmente legato alla capacità che la molecola ha di abbassare drasticamente gli estrogeni attraverso un legame competitivo reversibile al gruppo eme della relativa unità del citocromo P450. L’Anastrozolo, il quale agisce similmente al Letrozolo, ha mostrato una riduzione del livello estrogenico in soggetti di sesso maschile del 50%.(45) Il problema di un possibile rebound estrogenico con questi composti nasce proprio dalla loro natura “reversibile”.
Rebound estrogenici sono stati riportati sia con l’uso di Letrozolo che con l’uso di Anastrozolo, sebbene il Letrozolo, avendo un azione inibitoria più marcata, sembra causare rebound di intensità maggiore dopo la sua interruzione. La causa del rebound estrogenico indotto da cessazione d’uso di Letrozolo o di Anastrozolo è proprio legata al comportamento che queste due molecole esplicano nei confronti dell’Enzima aromatasi. Il legame tra la molecola di Letrozolo o di Anastrozolo con l’Enzima Aromatasi è solo temporanea e non decreta la completa de-attivazione dell’enzima responsabile della conversione degli Androgeni in Estrogeni. Una volta interrotta l’assunzione del composto, i livelli di Aromatasi possono salire significativamente con la possibile comparsa di un rebound estrogenico. Una pratica per evitare che ciò si verifichi consiste nell’uso limitato dei due composti e in una loro graduale sospensione. Con questi farmaci, il rebound estrogenico può essere “multifattoriale” derivando non solo dalla cessazione del farmaco in questione ma anche da un incremento dell’espressione dell’Enzima Aromatasi come risposta adattativa all’uso (specie nel lungo termine). Ciò significa che, anche durate un utilizzo cronico, i livelli di E2 possono mostrare degli aumenti, aumenti che diverranno maggiormente significativi una volta cessato l’uso del farmaco. Cessata l’azione del composto non solo viene a mancare un controllo dell’aromatizzazione ma questa risulta anche incrementata rispetto ai tassi pre-utilizzo (l’aumento dell’espressione dell’aromatasi è un comportamento adattativo che si può manifestare anche durante cicli particolarmente lunghi). Prendendo in considerazione la vita attiva del Letrozolo e dell’Anastrozolo, il possibile rebound estrogenico potrebbe manifestarsi in crescendo dopo 64-120h circa dall’ultima assunzione.

L’Exemestano, invece, è un inibitore dell’Aromatasi steroideo irreversibile di tipo I, strutturalmente correlato al substrato naturale 4-androstenedione. Agisce come un falso substrato per l’Enzima Aromatasi e viene trasformato in un intermedio che si lega irreversibilmente al sito attivo dell’enzima causandone l’inattivazione, un effetto noto anche come “inibizione suicida”. Essendo strutturalmente simile agli obiettivi dell’enzima, l’Exemestano si lega in modo permanente a quest’ultimo, impedendo la sua azione di conversione degli Androgeni in Estrogeni. Il tasso di soppressione degli Estrogeni da parte dell’Exemestano varia dal 35% per l’Estradiolo (E2) al 70% per l’Estrone (E1).(46)
Grazie alla sua caratteristica di “inibitore selettivo”, l’Exemestano sembra non causare un rebound estrogenico dopo la sua cessazione. Nonostante ciò, un suo uso temporalmente protratto potrebbe (teoricamente) causare, similmente a quanto accade con l’uso di Letrozolo e Anastrozolo, un aumento dell’espressione dell’Enzima Aromatasi nonché un aumento del numero di ER come risposta adattativa.
Ovviamente, questa possibilità può interessare tutti gli AI con legame irreversibile (es. Formestano).
Queste sono semplici ipotesi nate da una riflessione sulle possibili cause e meccanismi che potrebbero (teoricamente) portare al manifestarsi di un rebound estrogenico con l’uso di tali composti. La letteratura scientifica, purtroppo, non ci aiuta a fare molta chiarezza sulla connessione AI/rebound estrogenico, sebbene esistono alcuni studi nei quali la cosa viene accennata.(47)

*Nota sull’Aminoglutetimide: inibendo l’enzima P450scc e agendo, di conseguenza, come inibitore della biosintesi di tutti gli ormoni steroidei, l’abuso di Aminoglutetimide può potenzialmente causare non solo un rebound estrogenico ma anche un rebound dei livelli di cortisolo. Lo stesso vale per il farmaco Trylostano.
Conclusioni
Basarsi per la maggior parte sui dati aneddotici è un azzardo, anche perché la maggior parte delle variabili soggettive in gioco rimangono celate. Banalmente, alcuni lamentano rebound estrogenici che alla fine non risultano legati all’uso del SERM o del AI ma alla loro (o del Preparatore) ignoranza, come quando cessano l’utilizzo di AAS, e di SERM e/o AI, senza preoccuparsi di svolgere un adeguata PCT convinti, magari, che un po’ di Mesterolone (Proviron) risolvi tutto. Infatti, la maggior parte dei casi di presunti rebound estrogenici SERM o AI dipendenti sono causati da una repentina cessazione d’uso di questi e di AAS, oppure da una PCT mal pianificata e/o che non ha dato i risultati sperati (vedi anche alterazione della Testosterone:Estradiolo ratio). Queste condizioni sono legate più che altro ad una alterazione del HPTA data dall’uso di AAS e non ad una presunta azione diretta del SERM e/o AI precedentemente utilizzati.
In conclusione, l’uso ponderato e consapevole è l’unica vera arma che l’atleta supplementato chimicamente (o il Preparatore che lo segue) ha per far si che ipotetici rebound non si manifestino.
Gabriel Bellizzi
Riferimenti:
1- “Definition of antiestrogen – NCI Dictionary of Cancer Terms, Definition of antiestrogen – NCI Dictionary of Cancer Terms”.,
2- Jump up ^ “antiestrogen” at Dorland’s Medical Dictionary
3- Jump up ^ Judi Lindsley Nath (2006). Using Medical Terminology: A Practical Approach. Lippincott Williams & Wilkins. pp. 977–. ISBN 978-0-7817-4868-1.
4- Dahlman-Wright K, Cavailles V, Fuqua SA, Jordan VC, Katzenellenbogen JA, Korach KS, Maggi A, Muramatsu M, Parker MG, Gustafsson JA (Dec 2006). “International Union of Pharmacology. LXIV. Estrogen receptors”. Pharmacological Reviews. 58 (4): 773–81. doi:10.1124/pr.58.4.8. PMID 17132854.
5- Levin ER (Aug 2005). “Integration of the extranuclear and nuclear actions of estrogen”. Molecular Endocrinology. 19 (8): 1951–9. doi:10.1210/me.2004-0390. PMC 1249516 . PMID 15705661.
6- Levin ER (Aug 2005). “Integration of the extranuclear and nuclear actions of estrogen”. Molecular Endocrinology. 19 (8): 1951–9. doi:10.1210/me.2004-0390. PMC 1249516 . PMID 15705661.
7- Wang C, Fu M, Angeletti RH, Siconolfi-Baez L, Reutens AT, Albanese C, Lisanti MP, Katzenellenbogen BS, Kato S, Hopp T, Fuqua SA, Lopez GN, Kushner PJ, Pestell RG (May 2001). “Direct acetylation of the estrogen receptor alpha hinge region by p300 regulates transactivation and hormone sensitivity”. The Journal of Biological Chemistry. 276 (21): 18375–83. doi:10.1074/jbc.M100800200. PMID 11279135.
8- Zivadinovic D, Gametchu B, Watson CS (2005). “Membrane estrogen receptor-alpha levels in MCF-7 breast cancer cells predict cAMP and proliferation responses”. Breast Cancer Research. 7 (1): R101–12. doi:10.1186/bcr958. PMC 1064104 . PMID 15642158.
9- Björnström L, Sjöberg M (Jun 2004). “Estrogen receptor-dependent activation of AP-1 via non-genomic signalling”. Nuclear Receptor. 2 (1): 3. doi:10.1186/1478-1336-2-3. PMC 434532 . PMID 15196329.
10- Lu Q, Pallas DC, Surks HK, Baur WE, Mendelsohn ME, Karas RH (Dec 2004). “Striatin assembles a membrane signaling complex necessary for rapid, nongenomic activation of endothelial NO synthase by estrogen receptor alpha”. Proceedings of the National Academy of Sciences of the United States of America. 101 (49): 17126–31. doi:10.1073/pnas.0407492101. PMC 534607 . PMID 15569929.
11- Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, Masushige S, Gotoh Y, Nishida E, Kawashima H, Metzger D, Chambon P (Dec 1995). “Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase”. Science. 270 (5241): 1491–4. doi:10.1126/science.270.5241.1491. PMID 7491495.
12- Prossnitz ER, Arterburn JB, Sklar LA (Feb 2007). “GPR30: A G protein-coupled receptor for estrogen”. Molecular and Cellular Endocrinology. 265-266: 138–42. doi:10.1016/j.mce.2006.12.010. PMC 1847610 . PMID 17222505.
13- Otto C, Rohde-Schulz B, Schwarz G, Fuchs I, Klewer M, Brittain D, Langer G, Bader B, Prelle K, Nubbemeyer R, Fritzemeier KH (Oct 2008). “G protein-coupled receptor 30 localizes to the endoplasmic reticulum and is not activated by estradiol”. Endocrinology. 149 (10): 4846–56. doi:10.1210/en.2008-0269. PMID 18566127.
14- Harris HA, Albert LM, Leathurby Y, Malamas MS, Mewshaw RE, Miller CP, Kharode YP, Marzolf J, Komm BS, Winneker RC, Frail DE, Henderson RA, Zhu Y, Keith JC (Oct 2003). “Evaluation of an estrogen receptor-beta agonist in animal models of human disease”. Endocrinology. 144 (10): 4241–9. doi:10.1210/en.2003-0550. PMID 14500559.
15- Deroo BJ, Korach KS (Mar 2006). “Estrogen receptors and human disease”. The Journal of Clinical Investigation. 116 (3): 561–70. doi:10.1172/JCI27987. PMC 2373424 . PMID 16511588.
16- Riggs BL, Hartmann LC (Feb 2003). “Selective estrogen-receptor modulators — mechanisms of action and application to clinical practice”. The New England Journal of Medicine. 348 (7): 618–29. doi:10.1056/NEJMra022219. PMID 12584371.
17- Cameron JL, Cameron AM (20 November 2013). Current Surgical Therapy. Elsevier Health Sciences. pp. 582–. ISBN 978-0-323-22511-3.
18- Huang X, Aslanian RG (19 April 2012). Case Studies in Modern Drug Discovery and Development. John Wiley & Sons. pp. 392–394. ISBN 978-1-118-21967-6.
19- Pickar JH, Komm BS (Sep 2015). “Selective estrogen receptor modulators and the combination therapy conjugated estrogens/bazedoxifene: A review of effects on the breast”. Post Reproductive Health. 21 (3): 112–21. doi:10.1177/2053369115599090. PMID 26289836.
20- Mirkin S, Pickar JH (Jan 2015). “Selective estrogen receptor modulators (SERMs): a review of clinical data”. Maturitas. 80 (1): 52–7. doi:10.1016/j.maturitas.2014.10.010. PMID 25466304.
21- Desta Z, Ward BA, Soukhova NV, Flockhart DA (Sep 2004). “Comprehensive evaluation of tamoxifen sequential biotransformation by the human cytochrome P450 system in vitro: prominent roles for CYP3A and CYP2D6”. The Journal of Pharmacology and Experimental Therapeutics. 310 (3): 1062–75. doi:10.1124/jpet.104.065607. PMID 15159443.
22- Ahmad A, Shahabuddin S, Sheikh S, Kale P, Krishnappa M, Rane RC, Ahmad I (December 2010). “Endoxifen, a new cornerstone of breast cancer therapy: demonstration of safety, tolerability, and systemic bioavailability in healthy human subjects”. Clinical Pharmacology and Therapeutics. 88 (6): 814–7. doi:10.1038/clpt.2010.196. PMID 20981001.
23- Wang DY, Fulthorpe R, Liss SN, Edwards EA (Feb 2004). “Identification of estrogen-responsive genes by complementary deoxyribonucleic acid microarray and characterization of a novel early estrogen-induced gene: EEIG1”. Molecular Endocrinology. 18 (2): 402–11. doi:10.1210/me.2003-0202. PMID 14605097.
24- Kuhl H (2005). “Pharmacology of estrogens and progestogens: influence of different routes of administration”. Climacteric. 8 Suppl 1: 3–63. doi:10.1080/13697130500148875. PMID 16112947.
25- Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M (Dec 2000). “Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription”. Cell. 103 (6): 843–52. doi:10.1016/S0092-8674(00)00188-4. PMID 11136970.
26- Massarweh S, Osborne CK, Creighton CJ, Qin L, Tsimelzon A, Huang S, Weiss H, Rimawi M, Schiff R (Feb 2008). “Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function”. Cancer Research. 68 (3): 826–33. doi:10.1158/0008-5472.CAN-07-2707. PMID 18245484.
27- Hurtado A, Holmes KA, Geistlinger TR, Hutcheson IR, Nicholson RI, Brown M, Jiang J, Howat WJ, Ali S, Carroll JS (Dec 2008). “Regulation of ERBB2 by oestrogen receptor-PAX2 determines response to tamoxifen”. Nature. 456 (7222): 663–6. Bibcode:2008Natur.456..663H. doi:10.1038/nature07483. PMC 2920208 . PMID 19005469.
28- Osborne CK, Bardou V, Hopp TA, Chamness GC, Hilsenbeck SG, Fuqua SA, Wong J, Allred DC, Clark GM, Schiff R (Mar 2003). “Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer”. Journal of the National Cancer Institute. 95 (5): 353–61. doi:10.1093/jnci/95.5.353. PMID 12618500.
29- “New Mechanism Predicts Tamoxifen Response: PAX2 gene implicated in tamoxifen-induced inhibition of ERBB2/HER2-mediated tumor growth”. http://www.modernmedicine.com. 2008-11-13. Archived from the original on 2011-07-14. Retrieved 2008-11-14.
30- “Study sheds new light on tamoxifen resistance”. News. CORDIS News. Archived from the original on 2009-02-20. Retrieved 2008-11-14.
31- Liu J, Flockhart PJ, Lu D, Lv W, Lu WJ, Han X, Cushman M, Flockhart DA (2013). “Inhibition of cytochrome p450 enzymes by the e- and z-isomers of norendoxifen”. Drug Metab. Dispos. 41 (9): 1715–20. doi:10.1124/dmd.113.052506. PMC 3876808 . PMID 23824607.
32- Chemical muscle enhancement. Report. B.B. desk reference. di Author L. Rea. Pag. 106.
33- https://www.rdmag.com/article/2014/12/journal-watch-tamoxifen-news-again
34- https://www.ncbi.nlm.nih.gov/pubmed/14687597
35- Bryant HU (2001). “Mechanism of action and preclinical profile of raloxifene, a selective estrogen receptor modulation”. Rev Endocr Metab Disord. 2 (1): 129–38. PMID 11704975.
36- Thiebaud D, Secrest RJ (2001). “Selective estrogen receptor modulators: mechanism of action and clinical experience. Focus on raloxifene”. Reprod. Fertil. Dev. 13 (4): 331–6. PMID 11800172.
37- Gizzo S, Saccardi C, Patrelli TS, Berretta R, Capobianco G, Di Gangi S, Vacilotto A, Bertocco A, Noventa M, Ancona E, D’Antona D, Nardelli GB (2013). “Update on raloxifene: mechanism of action, clinical efficacy, adverse effects, and contraindications”. Obstet Gynecol Surv. 68 (6): 467–81. doi:10.1097/OGX.0b013e31828baef9. PMID 23942473.
38- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5739193/
39- “Entrez Gene: CYP19A1 cytochrome P450, family 19, subfamily A, polypeptide 1”.
40- Toda K, Shizuta Y (April 1993). “Molecular cloning of a cDNA showing alternative splicing of the 5′-untranslated sequence of mRNA for human aromatase P-450”. European Journal of Biochemistry. 213 (1): 383–9. doi:10.1111/j.1432-1033.1993.tb17772.x. PMID 8477708.
41- Czajka-Oraniec I, Simpson ER (2010). “Aromatase research and its clinical significance”. Endokrynologia Polska. 61 (1): 126–34. PMID 20205115.
42- Gasnier C, Dumont C, Benachour N, Clair E, Chagnon MC, Séralini GE (August 2009). “Glyphosate-based herbicides are toxic and endocrine disruptors in human cell lines”. Toxicology. 262 (3): 184–91. doi:10.1016/j.tox.2009.06.006. PMID 19539684.
43- Mokbel K (2002). “The evolving role of aromatase inhibitors in breast cancer”. Int. J. Clin. Oncol. 7 (5): 279–83. doi:10.1007/s101470200040 (inactive 2017-01-15). PMID 12402060.
44- Simpson ER (2003). “Sources of estrogen and their importance”. J. Steroid Biochem. Mol. Biol. 86 (3–5): 225–30. doi:10.1016/S0960-0760(03)00360-1. PMID 14623515.
45- Leder BZ, Rohrer JL, Rubin SD, Gallo J, Longcope C (March 2004). “Effects of aromatase inhibition in elderly men with low or borderline-low serum testosterone levels”. J. Clin. Endocrinol. Metab. 89 (3): 1174–80. doi:10.1210/jc.2003-031467. PMID 15001605.
46- Mauras, N; Lima, J; Patel, D; Rini, A; Di Salle, E; Kwok, A; Lippe, B (2003). “Pharmacokinetics and Dose Finding of a Potent Aromatase Inhibitor, Aromasin (Exemestane), in Young Males”. The Journal of Clinical Endocrinology & Metabolism. 88 (12): 5951–6. doi:10.1210/jc.2003-031279. PMID 14671195.
47- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3263690/
