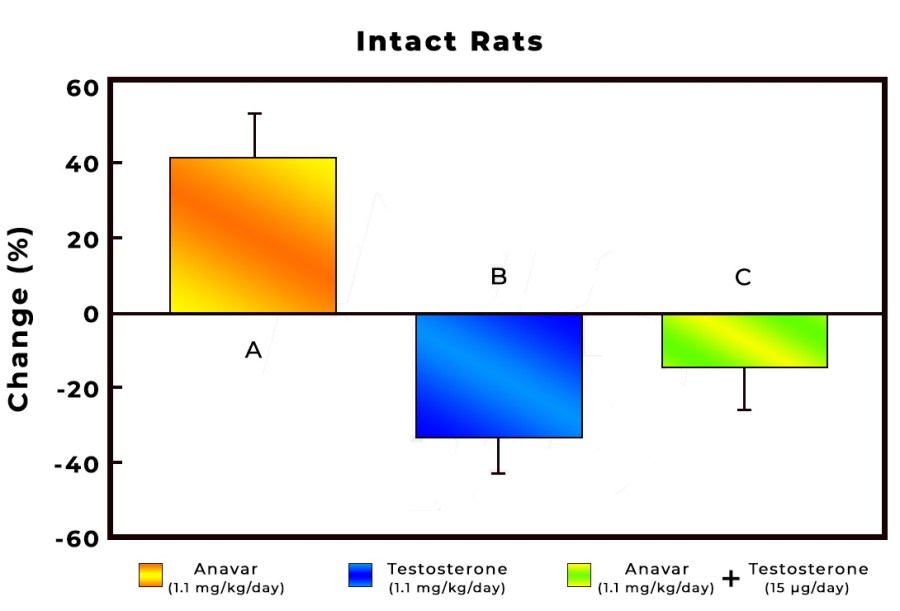
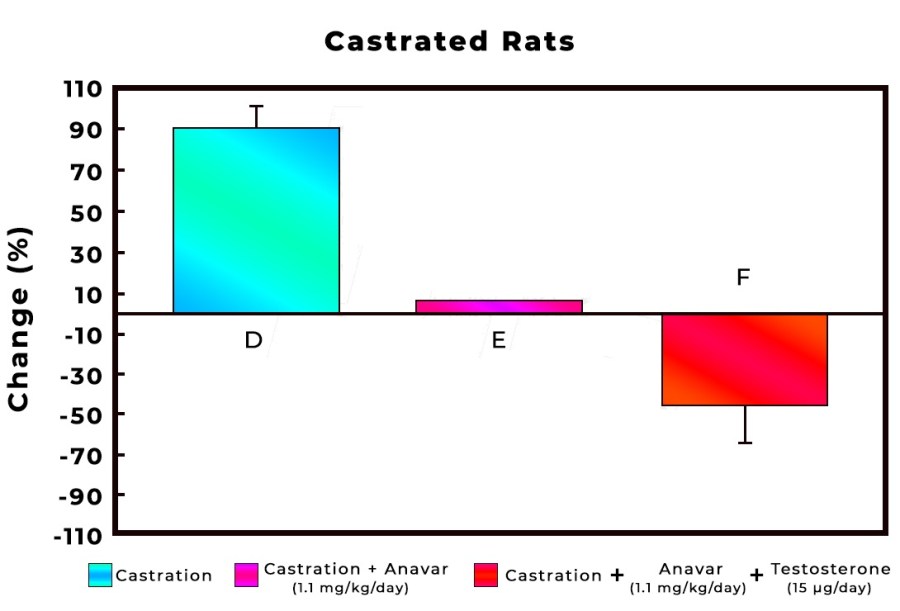
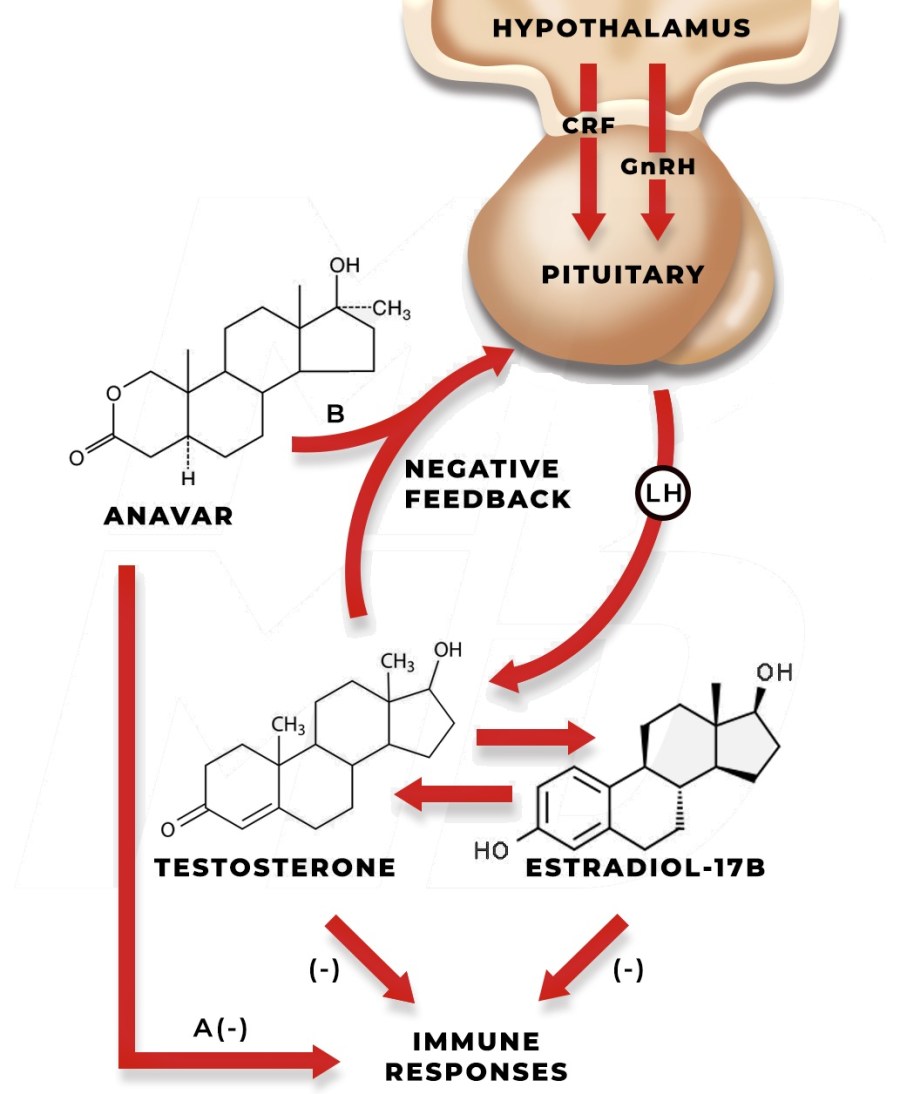
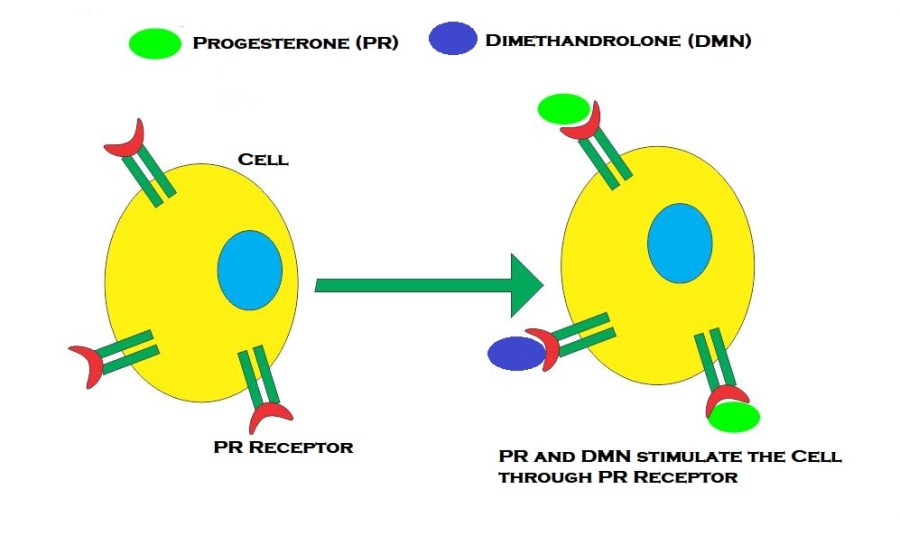
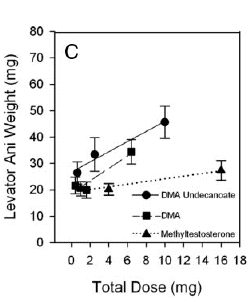
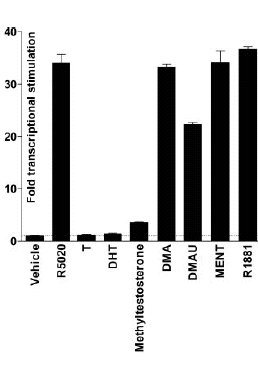
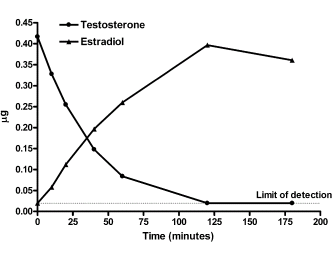
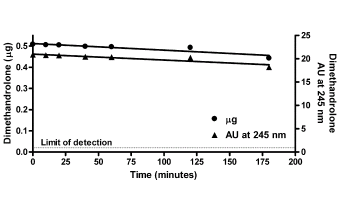
La Niacina è largamente utilizzata dagli atleti supplementati chimicamente, in special modo da coloro i quali usano molecole con un potenziale negativo marcato sui lipidi ematici. Ma come spesso capita, gli utilizzatori non conoscono a sufficienza le caratteristiche di ciò che assumono, e questa essenziale vitamina del gruppo B (B3) non è da meno. Per la maggior parte degli individui tanto basta sapere che una sua integrazione si traduce in livelli migliorati di Colesterolo e Trigliceridi. Purtroppo, però, si trascurano caratteristiche importanti la cui conoscenza può fare la differenza tra un uso più o meno funzionale per la salute sistemica. Infatti, un effetto collaterale dell’integrazione di Niacina è un peggioramento della resistenza all’insulina, cosa che limita i benefici di tale supplementazione sulla salute cardiovascolare se non vengono prese adeguate precauzioni.
Prima di correre a defenestrare in preda al panico la vostra Niacina, leggete con attenzione (e comprendete) le informazioni che seguono…
Introduzione alla Niacina (vitamina B3)

La Niacina, nota anche come Acido Nicotinico, è un composto organico e una forma di vitamina B3, un micronutriente essenziale per l’essere umano. [1] La Niacina ha formula bruta C6H5NO2 e appartiene al gruppo dell’acido piridinecarbossilico.[1] Come precursore di NAD e NADP, la Niacina è coinvolta nella riparazione del DNA.[2] La Niacina viene assunta attraverso la dieta da una varietà di alimenti interi e trasformati, con il più alto contenuto in alimenti confezionati fortificati, carne, pollame, pesce rosso come tonno e salmone, con minori quantità nelle noci, legumi e semi. [1] [3] La Niacina come integratore alimentare viene anche utilizzata per trattare la pellagra, una malattia causata da una sua carenza. Segni e sintomi includono lesioni della pelle e della bocca, anemia, mal di testa e stanchezza.[4] Molti paesi richiedono la sua aggiunta alla farina di grano o ad altri cereali, riducendo così il rischio di pellagra.[1][5] Come vitamina, le raccomandazioni di dosaggio giornaliero indicate in diversi paesi sono 14-18mg/die per gli adulti, quota sufficiente per soddisfare le esigenze delle persone sane. [6] [7] [8]

Sebbene la Niacina e la Nicotinamide (Niacinamide) siano identiche nella loro attività vitaminica, la Nicotinamide non ha gli stessi effetti farmacologici, modificanti i lipidi o gli effetti collaterali della Niacina, cioè quando la Niacina assume il gruppo -amide, non riduce il Colesterolo né causa vampate di calore.[9][10] La Nicotinamide è raccomandata come trattamento per la carenza di Niacina poiché può essere somministrata in quantità correttive senza causare l’effetto negativo del rossore.[11]

La Niacina è anche un farmaco di prescrizione. Quantità molto superiori all’assunzione dietetica raccomandata per le funzioni vitaminiche ridurranno i Trigliceridi nel sangue e le lipoproteine a bassa densità (LDL-C) e aumenteranno le lipoproteine ad alta densità (HDL-C). Ne esistono due forme: Niacina a rilascio immediato e a rilascio prolungato. Le quantità iniziali di prescrizione sono di 500mg/die, con possibilità di essere aumentate nel tempo fino a raggiungere l’effetto terapeutico ricercato. Le dosi a rilascio immediato possono arrivare fino a 3g/die; quelle a rilascio prolungato fino a 2g/die. [12] Nonostante i comprovati cambiamenti lipidici, la Niacina non è stata trovata utile per ridurre il rischio di malattie cardiovascolari nei soggetti già in trattamento con statine. [13] Una review del 2010 aveva concluso che l’efficacia della Niacina si osservava in mono-terapia, [14] ma una review del 2017 che incorporava il doppio del numero degli studi ha concluso che la Niacina su prescrizione, pur influenzando i livelli lipidici, non riduceva la mortalità per tutte le cause, la mortalità cardiovascolare, gli infarti del miocardio, né ictus fatali o non fatali. [15] È stato dimostrato che la Niacina da prescrizione provoca epatotossicità [16] e aumenta il rischio di diabete di tipo 2. [17] [18] Le prescrizioni di Niacina negli Stati Uniti avevano raggiunto il picco nel 2009, a 9,4 milioni, in calo a 1,3 milioni entro il 2017.[19]
Niacina, flusso ematico, pressione e vasodilatazione

Uno studio sulla supplementazione di Niacina che ha valutato il flusso sanguigno dell’avambraccio non è riuscito a trovare un effetto significativo fino a 1g al giorno somministrati nel corso di due settimane in soggetti altrimenti sani, [20] e 1.5g di Niacina a rilascio prolungato negli uomini con sindrome metabolica non sono riusciti a influenzare la dilatazione flusso- mediata (FMD). [21] Un altro studio non è riuscito a trovare un effetto significativo in un intero gruppo di pazienti affetti da afta epizootica, mentre in un gruppo di pazienti con malattia coronarica ha riscontrato un miglioramento in un sottogruppo con bassi livelli HDL-C. [22]
In soggetti con bassi livelli di HDL-C, è stato osservato che 1g di Niacina a rilascio prolungato per una settimana aumenta il flusso sanguigno (via FMD) del 4,5%; questo meccanismo non era correlato alle Prostaglandini, poiché il Laropiprant (un inibitore della Prostaglandine D2) non ha influenzato l’effetto. [23] Questo effetto ha anche coinciso con un aumento della bilirubina indiretta (ma non totale) del 62%. [23] Poiché la bilirubina del acido biliare è un antiossidante endoteliale, [24] e poiché i benefici della niacina sulla funzione endoteliale in questo studio sono stati ritenuti dipendenti dall’ossido nitrico, [23] è stato ipotizzato che un effetto conservativo della bilirubina sulla biodisponibilità dell’ossido nitrico sia alla base della beneficio osservato. Sia l’aumento della bilirubina che il miglioramento del flusso sanguigno si sono dissipati una settimana dopo l’interruzione della Niacina.[23]

I soggetti che in precedenza avevano subito infarto del miocardio, a seguito del trattamento con Niacina (con Laropiprant) hanno riscontrato un aumento del flusso sanguigno dipendente dall’ossido nitrico (FMD) dopo dodici settimane di terapia insieme a un miglioramento della vasodilatazione indotta da nitroglicerina, entrambe non correlate con alterazioni dei trigliceridi. [25] Miglioramenti simili nel flusso sanguigno sono stati osservati in pazienti con infezione da HIV e con bassi livelli di HDL-C trattati con la sola Niacina. [26]

È noto che la Niacina influenza il diametro dei vasi sanguigni, in particolare per via della sua reazione vasodilatativa cutanea (allargamento dei vasi nella pelle), che ha portato a ipotizzare che potrebbe influenzare la pressione sanguigna aumentando il diametro delle arterie e vene. Tuttavia, una review [27] ha notato che un possibile effetto di riduzione della pressione arteriosa della Niacina è indipendente dalla Prostaglandine che media il rossore, nota come PGD2.
È stato osservato che le infusioni di Niacina riducono acutamente la pressione sanguigna negli ipertesi senza alcun effetto nei soggetti con pressione sanguigna normale ed è stata associata ad un aumento della gittata cardiaca e della frequenza cardiaca che era simile in entrambi i gruppi. [28] Un altro studio ha confermato questo risultato, scoprendo che la pressione arteriosa ambulatoriale di 24 ore non sembra essere influenzata da un supplemento di Niacina fino a 1g nell’arco di due settimane in soggetti altrimenti sani. [20]
In termini di effetti della Niacina in cronico sulla pressione sanguigna, una review [27] che ha valutato gli studi che hanno misurato la pressione sanguigna negli ipertesi [29] [30] [31] [32] non ha notato alcun effetto statisticamente significativo nella riduzione della pressione sanguigna associata alla supplementazione di Niacina, sebbene questi studi in quanto a metodologie di misurazione sulle variazioni della pressione sanguigna non fossero ideali secondo gli autori della review. Tuttavia, la review ha osservato che in un ampio studio (il Coronary Drug Project), che inizialmente non è riuscito a trovare alcuna influenza della terapia con Niacina sulla pressione arteriosa, [32] ha osservato variazioni sensibili soltanto sui soggetti con sindrome metabolica. Questi presentavano un lieve riduzione di 2,2mmHg della pressione arteriosa sistolica con una moderata riduzione di 2,9mmHg della pressione diastolica. [33] Un’analisi post-hoc di un altro studio clinico [34] ha rilevato che la pressione arteriosa sistolica è stata abbassata di 2,2mmHg e la pressione sistolica di 2,7 rispetto al placebo nei pazienti dislipidemici trattati per 24 settimane. [35]
Niacina, Trigliceridi, Colesterolo e Aterosclerosi

La Niacina sembra abbassare i trigliceridi nel sangue inibendo sia la sintesi degli acidi grassi sia la loro esterificazione epatica per formare i trigliceridi, il che aumenta il tasso di degradazione dell’apolipoproteina B riducendo la sua secrezione dalle cellule epatiche. [36] Un meccanismo con cui la Niacina fa questo è attraverso l’inibizione diretta e non competitiva della diacilglicerolo aciltransferasi 2 (DGAT2), l’enzima finale nella sintesi dei trigliceridi nelle cellule epatiche, senza inibizione della DGAT1. [37]
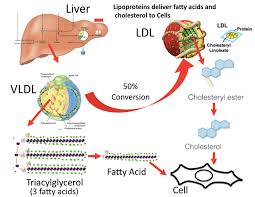
Si è visto che gli effetti della Niacina sulla sintesi dei trigliceridi influenzano i livelli sierici di lipoproteine a densità molto bassa (vLDL-C), dove la terapia con Niacina per 16 settimane in soggetti con malattia del fegato grasso non alcolica (NAFLD) sembra ridurre le vLDL-C nel siero così come i complessi con trigliceridi (vLDL-TG) e apolipoproteina B (vLDL-ApoB) rispetto al placebo e con una potenza paragonabile al fenofibrato. [38] La Niacina lo fa riducendo la secrezione epatica di vLDL-C, sebbene ciò non aumenti la quantità di trigliceridi nel fegato anche nello stato di NAFLD. [38]

Oltre ai suoi effetti sul fegato, la Niacina può anche sopprimere il rilascio di acidi grassi liberi dal tessuto adiposo [39] che normalmente verrebbero reesterificati come trigliceridi nel fegato e quindi secreti via vLDL. [40] Tuttavia, questo meccanismo specifico, che è mediato dal recettore HM74A, [39] non sembra essere rilevante per le proprietà riducenti dei trigliceridi della Niacina. [41]
I benefici sui livelli di trigliceridi possono verificarsi entro una settimana dall’inizio della supplementazione con Niacina a rilascio prolungato (1g), sebbene in misura minore di circa il 4%. [23]
L’integrazione di 1.5-2g di Niacina a rilascio prolungato per due anni con follow-up di un anno nelle persone in terapia con statine caratterizzate da bassi livelli di HDL-C ha mostrato una riduzione dei trigliceridi del 28,6% (statina da sola dell’8,1%). [42]
Esiste un fenomeno noto come “rimbalzo degli acidi grassi” associato alla supplementazione di Niacina, in quanto l’azione iniziale del composto sul suo recettore (HM74A) nel tessuto adiposo può determinare una minore lipolisi e una minore secrezione di acidi grassi non esterificati (NEFA) nel sangue [43] e una migliore conservazione adiposa; [44] si tratta di fenomeni prontamente reversibili in quanto in un giorno di esposizione continua vi è un aumento netto del NEFA piuttosto che la sua soppressione [45] [46] [47] e alterazioni nel NEFA possono non riflette alterazioni dei trigliceridi.
Il primo meccanismo pensato per spiegare il miglioramento del profilo sierico di colesterolo in seguito alla supplementazione di Niacina è stato attraverso la riduzione del rilascio di acidi grassi non esterificati (NEFA) dai tessuti, che non è più considerato un probabile meccanismo in quanto l’integrazione di niacina in cronico è associata ad un aumento, piuttosto che alla soppressione, di NEFA mentre il recettore HM74A appare superfluo in termini di effetti della Niacina nei topi con altri ligandi del HM74A (Acipimox [48] e MK-0354 [49]) che si sono mostrati rispettivamente meno efficaci o inefficaci sul colesterolo. Attualmente si ritiene che l’influenza della Niacina sui NEFA nel siero non sia un fattore determinante nel modo in cui influenza i livelli di colesterolo nel corpo, con le teorie attuali che ipotizzano che il fattore sia determinato dalla sua sintesi e dal suo tasso di catabolismo.
Il primo potenziale meccanismo prevede la sintesi di HDL-C nel fegato attraverso l’aumento della trascrizione del gene ABCA1 (che dipende dal legame LXRα alla regione del promotore DR4 di questo gene). [50] L’attività di ABCA1 promuove la “lipidazione” della principale proteina dell’HDL nota come apolipoproteina AI (ApoAI) aumentando il tasso che associa ai fosfolipidi e al colesterolo, [51] [52] un passaggio obbligatorio nella sintesi dell’HDL-C che è aumentato di 500-1000µM con Niacina in vitro. [50] Questo meccanismo non è stato confermato, poiché mentre l’ApoAI può essere aumentato parallelamente all’aumento dell’HDL-C in soggetti trattati con Niacina e con livelli di HDL-C bassi di base, [53] LXRα sembra richiedere un coattivatore (PPARγ) per esercitare questi effetti, [54] che è attivato dal recettore della Niacina. [55] Tuttavia, l’attività del recettore della Niacina non è stata richiesta per i suoi effetti sui livelli di colesterolo, suggerendo che altri meccanismi potrebbero essere rilevanti.
L’altra teoria relativa alla sintesi di HDL dalla Niacina afferma che ciò dipenda dalla proteina di trasferimento dell’estere del colesterolo (CETP) nonostante la riduzione del colesterolo totale e dei trigliceridi non richieda per entrambe questa proteina. [56] [57] CETP è una proteina che facilita il trasferimento di lipidi tra diverse lipoproteine (generalmente donando un trigliceride da vLDL a HDL e prendendo un estere di colesterolo in un processo noto come trasporto inverso di colesterolo. [58]) La Niacina riduce l’espressione di CETP nel fegato e la sua attività nel sangue dei topi; [56] una riduzione del CETP aumenta la quantità di HDL-C nel sangue poiché i tassi di catabolismo dell’HDL / LDL riflettono l’attività del trasporto inverso del colesterolo e raggiungono rapidamente l’equilibrio, [59] e se il CETP è ridotto allora sarebbe necessario più HDL per normalizzare i tassi di trasporto inverso del colesterolo. Questo meccanismo può anche essere correlato a LXRα, poiché mentre un eteromero di LXRα con il recettore nucleare di vitamina A (RXR) attiva l’elemento DR4 aumenta la CETP [60] la Niacina agevola l’eterodimerizzazione di LXRα e PPARγ che attiva ancora DR4, ma in un modo che promuove l’efflusso di colesterolo. [61-44] Questa eterodimerizzazione competitiva [62] non è stata ancora dimostrata sperimentalmente, e lo studio che ha utilizzato dosi di Niacina da 2g nell’uomo non è riuscito a trovare un’influenza sull’attività del CETP nel siero nonostante un aumento dell’HDL. [63]
L’ultimo potenziale meccanismo per l’aumento dell’HDL non consiste nel suo incremento di sintesi ma piuttosto nel preservare il colesterolo HDL già sintetizzato arricchito con apoAI, riducendo il tasso in cui la lipoproteina viene assunta nelle cellule epatiche nonostante la donazione di colesterolo dall’HDL a queste cellule sia inalterata a causa della riduzione dell’espressione del recettore (catena beta sintasi ATP) che normalmente trasporta l’HDL nella cellula. [64] Questa ipotesi funziona meglio con le osservazioni che suggeriscono che il ridotto catabolismo dell’HDL è il principale fattore determinante dei suoi livelli più elevati, [65] e influenza anche l’apoA1 poiché la sua clearance dal sangue e l’assorbimento da parte dei reni sono ridotti. [66]
Una supplementazione di Niacina a rilascio prolungato (1g) della durata di una settimana in soggetti con bassi livelli di HDL-C non sembra essere sufficiente da aumentare sensibilmente i livelli totali di HDL-C, sebbene sia stata notata una riduzione della dimensione media delle particelle; [23] le variazioni di HDL -C possono mediare un miglioramento della vasodilatazione dipendente dall’ossido nitrico, sebbene sia stato anche osservato un aumento della bilirubina indiretta. [23]
L’integrazione prolungata di Niacina nei diabetici è associata ad un aumento della quantità e delle dimensioni particellari dell’HDL-C (32,7%) mentre le particelle di dimensioni più piccole sono diminuite (8,2%). [67]
È stato osservato che la Niacina conferisce un effetto protettivo sulla mortalità cardiovascolare poiché una metanalisi [68] ha osservato che negli studi su soggetti con malattia coronarica la terapia con Niacina era associata a un minor rischio di rivascolarizzazione dell’arteria coronarica (RR di 0,31; IC al 95% di 0,15-0,63), infarto miocardico non fatale (RR di 0,72; IC al 95% di 0,60-0,86) e attacco ischemico transitorio (RR di 0,76; IC al 95% di 0,61-0,94) mentre la riduzione della mortalità complessiva non è riuscita a raggiungere significatività statistica (RR 0,883; IC 95% di 0,773-1,008). I sette studi inclusi in questa meta-analisi [32] [29] [31] [30] (e un follow-up [69]) hanno totalizzato 5137 pazienti che utilizzavano anche vari prodotti farmaceutici della classe di statine e fibrati .
In uno studio i cui partecipanti erano in terapia con statine e avevano bassi livelli di colesterolo HDL è stato rilevato che 1.5-2g di Niacina a rilascio prolungato sono stati in grado di fornire benefici additivi nel miglioramento dell’HDL-C (20%) e nella riduzione dell’LDL-C (17%) rispetto al placebo, sebbene per quanto riguarda l’endpoint clinico predeterminato (morte o ricovero in ospedale) sia la Niacina che il placebo avevano una uguale quantità di responder. [70] Questo studio ha rilevato un’alta percentuale di pazienti con sindrome metabolica (80%) e commenti [71] hanno suggerito che a causa di una possibile capacità della Niacina a rilascio prolungato di deteriorare l’insulino-resistenza [72] che i suoi benefici potrebbero essere compensati da questo effetto avverso, mentre lo studio stesso ha suggerito che i benefici delle statine hanno sostituito i benefici della Niacina.
Mentre uno studio precedente che utilizzava alte dosi di Niacina a rilascio immediato (3g) ha riscontrato una riduzione della morte del 14% rispetto al placebo insieme a una riduzioni del colesterolo totale, [32] ed è stato osservato che questa riduzione è simile per grandezza agli studi che combinano statine con placebo.

Studi in vitro suggeriscono che la Niacina potrebbe in teoria prevenire la formazione di placche aterosclerotiche riducendo l’infiammazione e il danno alla parete endoteliale attraverso diversi meccanismi. Limitate ricerche su animali hanno mostrato che la Niacina nella dieta, a concentrazioni paragonabili a quelle utilizzate per ridurre il colesterolo, riduce la deposizione della placca sulla parete dell’arteria e ritarda l’aterosclerosi.[73][74][75][76][77][78][79][80]
Niacina e sue interazioni con il metabolismo del glucosio

L’assunzione prolungata di Niacina è stata osservata causare una riduzione della sensibilità all’insulina, causando un aumento compensativo della produzione di insulina da parte delle cellule β del pancreas per mantenere i livelli di glucosio nel sangue. [81] La Niacina non sembra avere effetti diretti sulle cellule β pancreatiche, tuttavia, poiché la perfusione negli isolotti pancreatici (isole di Langerhans) di ratto isolati con Niacina in vitro non ha influenzato la secrezione di insulina. [82] Ciò indica che la Niacina aumenta la produzione di insulina mediante un meccanismo indiretto, secondario a causare insulino-resistenza periferica. È stato osservato che la supplementazione induce resistenza all’insulina a dosi comprese tra 500mg e 1g, che rientrano nell’intervallo di dosaggio che conferisce effetti di riduzione del colesterolo. [83]
In particolare, sembra che sia necessaria una supplementazione cronica di Niacina per aumentare la produzione di Insulina, poiché in uno studio è stato dimostrato che la supplementazione acuta riduce i livelli di questo peptide in soggetti altrimenti sani prima di un picco dopo un giorno, [84] mentre altri studi in acuto hanno notato un effetto minimo o nullo sui livelli di Insulina. [85] [86] [87] [88]

Gli effetti dell’integrazione cronica di Niacina sui livelli di Insulina possono anche dipendere dalla popolazione. È stato osservato che la Niacina provoca iperinsulinemia in soggetti che invecchiano altrimenti sani [83] (1g / die) ed è stato dimostrato che quasi raddoppiano i livelli di Insulina nei soggetti con NAFLD (2g / giorno [89] [90]). Nei pazienti con sindrome metabolica, l’integrazione di Niacina a 6 settimane di somministrazione alla dose di 1.5g / die ha aumentato i livelli di Insulina del 30%. [91]
Nei soggetti obesi con malattia del fegato grasso non alcolico (NAFLD), l’integrazione giornaliera di Niacina a rilascio prolungato (titolata fino a 2g) per 16 settimane sembrava aumentare lo stato di resistenza all’insulina nel fegato, nei muscoli e nel tessuto adiposo [89] con un effetto inibitorio sulle azioni dell’Insulina nel fegato notate negli uomini non diabetici con dislipidemia. [92]

Negli uomini adulti con sindrome metabolica, è stato osservato che la Niacina a rilascio prolungato alla dose di 1.5g ostacola in modo significativo la sensibilità all’Insulina, valutata dall’HOMA-IR (42%), che è stata associata ad un aumento dell’Insulina sierica nonostante un aumento dell’Adiponectina sierica. [91] Questo è stato notato anche in un altro studio (aumento del 22% dell’HOMA-IR), in cui l’Aspirina assunta insieme alla Niacina non ha impedito la comparsa di una ridotta sensibilità all’Insulina. [93]
Questo effetto può persistere in soggetti altrimenti sani, poiché i soggetti trattati con 1g di Niacina per due settimane a cui veniva somministrato un clamp iperinsulinaemico-euglicemico richiedono meno glucosio per mantenere l’omeostasi, il che è indicativo di una riduzione dell’assorbimento del glucosio (attraverso un aumento dell’Insulino-resistenza). [94]
La resistenza all’Insulina indotta dalla Niacina è stata inizialmente attribuita a un effetto di rebound nel tessuto adiposo in cui un aumento del rilascio di acidi grassi non esterificati (NEFA) da parte della Niacina compromette gli effetti della segnalazione dell’Insulina. [95] [96] Ciò è plausibile, poiché la resistenza all’Insulina può essere indotta con infusione di NEFA in 24 ore nei roditori. [97] Altre fonti suggeriscono che la resistenza all’Insulina non è associata al rebound del NEFA, poiché i soggetti con NAFLD che sperimentano resistenza all’Insulina dalla terapia con Niacina non hanno necessariamente un aumento del NEFA nel siero. [89].

Un’altra possibile opzione è che la Niacina può inibire in modo non competitivo l’enzima noto come diacilglicerolo aciltransferasi 2 (DGAT2) con un IC50 di 100 µM (potenza simile a circa 300 µM). [98] L’inibizione di questo enzima non causa di per sé resistenza all’insulina con la somministrazione di Niacina, [92] ma poiché il DGAT catalizza il primo stadio della sintesi dei trigliceridi, la sua inibizione può favorire l’accumulo di diacilglicerolo (DAG) che è la molecola che si ritiene spieghi parzialmente la resistenza all’insulina data dalla Niacina. [92] Poiché l’aumento del DAG nelle cellule del fegato sopprime la segnalazione dell’Insulina, [99-162] l’inibizione mediata dalla Niacina del DGAT2 provoca insulino-resistenza, [98] [89] ostacolando così la capacità dell’Insulina di sopprimere la sintesi di glucosio e promuovendo indirettamente uno stato di iperglicemia.
Sebbene l’integrazione cronica di alte dosi di Niacina riduca la sensibilità all’Insulina, ciò non è associato a variazioni dei livelli di glucosio a digiuno. [90] Ciò può essere spiegato da un aumento compensativo della sintesi di Insulina che contrasta la resistenza alla stessa, lasciando sostanzialmente invariati i livelli di glucosio nel sangue. [81]
L’attivazione del recettore della Niacina (HM74A) da parte di alcuni altri agonisti sembra ridurre rapidamente il glucosio sierico nei diabetici migliorando la sensibilità all’Insulina [100] o comunque migliorando i tassi di smaltimento del glucosio. [101] Ciò indica che lo stesso recettore della Niacina può avere effetti benefici sul metabolismo del glucosio e che la resistenza all’Insulina indotta dalla Niacina non si verifica tramite l’attivazione del HM74A.
Quando si osserva il muscolo scheletrico, è stato dimostrato che la terapia con Niacina induce resistenza all’Insulina in questo tessuto in soggetti obesi con NAFLD (2g al giorno nel corso di 16 settimane). Uno studio svolto su ratti a digiuno (il digiuno aumenta la concentrazione plasmatica di acidi grassi non esterificati (NEFA), similmente alla somministrazione di Niacina [102-135] e diminuisce il glicogeno del muscolo scheletrico [103]) in cui sono stati accuratamente somministrati 20mg/kg di Niacina ha mostrato che il glicogeno nel soleo era ridotto mentre il gastrocnemius e il fegato non sono stati influenzati. [103]

Quando il processo di glicazione è testato in vitro, la Niacina ha avuto solo effetti inibitori minori sulla glicazione dell’albumina sierica bovina da un noto agente glicante (Metilgliossale [104]) nonostante altri antiossidanti testati come lo Zinco (10-25 µg / mL) avessero più potenti benefici. [105]
È importante sottolineare che qualsiasi effetto della Niacina sulla glicazione in vitro deve essere interpretato con l’avvertenza che la Niacina riduce la sensibilità all’Insulina. Mentre la resistenza all’Insulina indotta dalla Niacina è ben compensata in soggetti sani giovani, lasciando sostanzialmente invariati i livelli di glucosio nel sangue, [81] la compensazione delle cellule β del pancreas negli individui più anziani o in quelli con ridotta tolleranza al glucosio era incompleta in uno studio, [83] causando aumenti nei livelli ematici di glucosio. Pertanto, la misura in cui la Niacina possa influenzare la glicazione in vivo non è chiara e probabilmente dipendente dalla popolazione.
Obesità e massa grassa

L’Adiponectina, un’adipochina nota per migliorare la sensibilità all’Insulina, per essere cardioprotettiva e ritenuta anche antiobesogena, [106] è aumentata in risposta all’attivazione mediata dalla Niacina del recettore HM74A nei topi. [107] La produzione di Adiponectina indotta dalla Niacina è stata rapida in questo studio, aumentando i livelli di questa adipochina del 37% entro 10 minuti da una dose di 30mg / kg per iniezione. I livelli sierici hanno raggiunto il picco dopo 60 minuti e sono rimasti elevati al di sopra del basale fino a 24 ore dopo la somministrazione. [107]

È noto anche che la Leptina è aumentata in seguito alla somministrazione di Niacina nell’uomo [91], il che si ritiene si verifichi tramite un meccanismo simile poiché l’agonista farmaceutico HM74A Acipimox induce anch’esso la secrezione di Leptina dal tessuto adiposo in vitro [108] e in vivo. [109]
È stato osservato che la supplementazione di Niacina nel corso di sei settimane negli uomini obesi aumenta l’Adiponectina sierica del 43-56%, con circa metà dell’aumento rappresentato dalla forma ad alto peso molecolare [93] [91] insieme a un aumento del 26,8% della Leptina [91 ] senza cambiamenti osservabili nella Resistina. [91] L’Adiponectina è stata osservata aumentare di circa il 30% in soggetti obesi con NAFLD in risposta alla terapia con Niacina (fino a 2g al giorno), che era correlata con un aumento dell’Insulino-resistenza, [90] portando all’ipotesi che i due meccanismi siano intrecciati, forse come risposta adattativa. [90]

Lo “spillover” degli acidi grassi risultante da una conservazione inefficiente del grasso dopo un pasto aumenta i lipidi sierici non esterificati (NEFA), [110] che influenzano negativamente la sensibilità all’Insulina epatica, aumentando la produzione di VLDL e potenzialmente svolgono un ruolo causale nella steatosi epatica. [111] [112] La somministrazione in acuto di Niacina (285 mg per via endovenosa) nell’uomo durante l’alimentazione ha dimostrato di ridurre lo spillover degli acidi grassi, promuovendo l’assorbimento del grasso alimentare nel tessuto adiposo e riducendo i Trigliceridi sierici e i NEFA. [113]
Al contrario, è stato osservato che un trattamento prolungato con Niacina, noto per favorire la resistenza all’Insulina nell’uomo, induce la resistenza all’Insulina adipocitaria, [114] che favorirebbe lo spillover degli acidi grassi, aumentando i livelli sierici di NEFA.[115]

È stato osservato che la Nicotinamide sopprime la differenziazione degli adipociti 3T3-L1 in modo dipendente dalla concentrazione con un range superiore a 10mM (il valore ED50), raggiungendo la soppressione completa a 20mM dopo nove giorni. [116] Si ritiene che ciò sia correlato a un effetto inibitorio sulla poli (ADP-ribosio) sintetasi, [116] che la Nicotinamide inibisce a 50µM mentre la Niacina non lo fa. [117] Quando aggiunta dopo differenziazione e in condizioni di glucosio elevato, la Nicotinamide sembra inibire il glucosio-6-fosfato deidrogenasi (G6PD) e prevenire il normale stress ossidativo. [118]
Il recettore dell’Acido Nicotinico è espresso negli dipociti in cui la sua attivazione sopprime l’adenilato ciclasi. [119] Questo effetto sembra essere circa il 30% più efficace negli adipociti rispetto ad altre linee cellulari (milza). [120] Poiché l’attivazione di questo recettore inibisce l’adenilato ciclasi, [119] e i fenolici che agiscono su di esso riducono anch’essi i tassi di lipolisi, [35] l’effetto complessivo dell’Acido Nicotinico sarebbe quello di ridurre la lipolisi negli adipociti, almeno a breve termine.

A lungo termine, tuttavia, il recettore dell’Acido Nicotinico può essere desensibilizzato con esposizione cronica a un agonista [121] e uno studio sui topi ha evidenziato che gli adipociti che sono diventati insulino-resistenti dopo la terapia con Niacina hanno mostrato una maggiore reattività dei recettori adrenergici (β1 e β2) all’aumentare dei livelli di cAMP nella cellula adiposa, [114] (il cAMP viene normalmente soppresso dalla Niacina che agisce sul recettore GRP109A [119]). Ciò potrebbe essere stato correlato alla sottoregolazione dei geni mediata dalla Niacina nella via di segnalazione dell’Insulina incluso il PDE3B, che normalmente degrada il cAMP, [114] una potenziale risposta adattativa nelle cellule adipose che è stata osservata avere la funzione di normalizzare i tassi di lipolisi (nei ratti sotto l’infusione di Niacina) . [97]
Un piccolo studio su sette partecipanti altrimenti sani che assumevano Niacina a 500mg/die, e aumentando la dose a 2g nel corso di due settimane ha mostrato una riduzione dei tassi di ossidazione dei grassi. [94] Tuttavia, a causa di un aumento dei tassi di ossidazione dei carboidrati, non vi era alcuna differenza netta nel tasso metabolico tra Niacina e placebo [94].

I topi privi di PARP-1 sembrano avere tassi metabolici più alti e una minore massa grassa; in assenza di PARP, aumentano le concentrazioni di NAD +, attivando le SIRT1 che quindi lavorano per deacetilare varie proteine (PGC-1α e FOXO1) per promuovere il dispendio energetico attraverso un metabolismo ossidativo aumentato e un incremento dei mitocondri.[122]
La SIRT2 e la SIRT3 non sono influenzate dalla bassa attività del PARP-1, [122] e l’inibizione della ribosilazione dell’ADP con altri mezzi come il knockdown NMNAT-1 sembra conferire anche effetti antiobesità nei roditori. [182] L’alimentazione aumenta acutamente l’attività del PARP-1 nei topi e ostacola transitoriamente l’attività della SIRT1, [122] che si pensa sia correlata al PARP-1 che ha la priorità per l’uso dei donatori di NAD +.

La supplementazione orale di Nicotinamide Riboside a 400mg/kg nel topo sembra aumentare il contenuto di NAD + nel muscolo scheletrico similmente a quanto avviene alla stessa dose di Niacina (Nicotinamide Mononucleotide inefficace in questo organo) [123] e sembra aumentare il dispendio energetico nei topi nutriti con un alto contenuto di grassi insieme all’aumento dell’attività dei geni bersaglio di FOXO1, suggerendo che l’integrazione orale è efficace. [123]
Esistono prove limitate nell’uomo che valutano gli effetti della Niacina sul tasso metabolico, sebbene l’estremità inferiore del dosaggio farmacologico della niacina (1g) in soggetti altrimenti sani non sia riuscito ad aumentare il tasso metabolico rispetto al placebo. [94]
Niacina, muscolo scheletrico e prestazioni fisiche

La somministrazione di Niacina nell’uomo ha dimostrato di aumentare l’espressione dei fattori di trascrizione PPARδ e PPARγ coactivator-1α (PGC-1α) nel muscolo scheletrico. [124] Poiché questi fattori di trascrizione sono importanti regolatori del metabolismo ossidativo e della biogenesi mitocondriale, [125] [126] questo suggerisce che l’integrazione con Niacina può svolgere un ruolo nella resistenza dei muscoli scheletrici.
Gli studi sugli animali hanno supportato questa idea, in cui è stato dimostrato che l’integrazione di Niacina provoca una transizione di fibre muscolari dal tipo II (contrazione rapida) al tipo I (contrazione lenta), aumentando anche il numero complessivo di fibre di tipo I nei muscoli scheletrici nello Zucker (ratto) obeso [127] e suini in crescita [128] (750mg di Niacina/kg di dieta) e pecore (1g di Niacina al giorno). [129] Questo effetto può essere limitato a determinati modelli animali, tuttavia, poiché studi su ratti sani hanno dimostrato che la Niacina ha un effetto trascurabile sulla distribuzione del tipo di fibra muscolare o sul fenotipo metabolico. [130] Inoltre, nonostante la Niacina aumenti l’espressione dei fattori di trascrizione pro-ossidativa nell’uomo, [124] fino ad oggi nessuno studio ha dimostrato che migliora le prestazioni o la capacità di resistenza del muscolo scheletrico.
Tuttavia, come substrato per la sintesi di NAD +, un’adeguata presenza di Niacina può supportare indirettamente il metabolismo ossidativo e la resistenza muscolare. In soggetti altrimenti sani, un lieve esercizio fisico sembra essere associato ad un aumento delle concentrazioni di NAD + nel sangue rispetto a uno stato di riposo (indipendente da qualsiasi integrazione [131]) mentre, quando testato in un esercizio lieve nei roditori, portava anche ad un aumento del NAD + nel sangue prima che diminuisse durante un esercizio ad esaurimento, [131] che è stato notato anche nel muscolo scheletrico. [132] A questo livello di esaurimento c’è un concomitante aumento del contenuto di NADH nel muscolo scheletrico [133] [134] che è stato proposto [135] indicativo di una riduzione del trasferimento di elettroni dal NADH alla sintesi di ATP.

È stato inoltre proposto [135] che da quando l’esercizio aumenta l’ossidazione nei tessuti stimolati e i fattori di stress ossidativo sono noti per alterare l’attività del ciclo di Kreb (TCA) [136] e la catena di trasporto degli elettroni (compresa la NADH deidrogenasi [137]) che forniscono antiossidanti aumenterebbe la resistenza secondaria alla conservazione della cinetica intramuscolare di NAD + / NADH. Quando si forniscono 36mg di picnogenolo [135] come antiossidante durante l’esercizio fisico fino all’esaurimento, sembra che la diminuzione del NAD + nel sangue sia stata invertita in un aumento con gli effetti (sia la diminuzione che l’aumento in attesa di integrazione) più marcati negli atleti allenati. [135]
Impatto organico della Niacina e principali effetti collaterali

In uno studio svolto sui ratti, la Nicotinamide ad un dosaggio di 20mg/kg somministrata un’ora prima di una dose di Indometacina che induceva ulcerazioni allo stomaco ha impedito l’ulcerazione a un livello paragonabile sia al controllo (nessuna induzione di ulcere) sia al farmaco di riferimento di 400mg/kg di sucralfato, che agisce localmente per forma una superficie protettiva per lo stomaco. [138] Questo effetto si è verificato insieme alla conservazione dell’attività del glutatione, alla riduzione della perossidazione lipidica e al miglioramento del muco gastrico. [138] Simili effetti protettivi contro l’ulcerazione indotta da etanolo e stress sono stati notati altrove, con il metabolita primario della Nicotinamide (1-metilnicotinamide; MNA). [139] Questo effetto gastroprotettivo è stato associato con un aumento dell’attività delle prostaglandine, in particolare la PGI2, [139] e nicotinamide, nonché il suo metabolita MNA sono stati implicati nell’aumento del flusso sanguigno gastrico [139] e nella riduzione della permeabilità microvascolare [138] dopo l’ulcerazione.

Nel colon dei topi, il recettore della Niacina (GPR109A) è necessario per la proliferazione ottimale delle cellule T CD4 + e la produzione di IL-10, che si traduce in un effetto antiinfiammatorio. [140] Questo effetto antinfiammatorio guidato dal GPR109A è mediato dal butirrato, l’acido grasso a catena corta del colon [140], che è un agonista del GPR109A ed è prodotto attraverso la fermentazione della fibra alimentare da parte dei batteri nel colon. [141] [142]
L’effetto riducente dei Trigliceridi dato dalla Niacina sembra da doversi ricondurre al fegato, dove la secrezione di lipoproteine a bassissima densità (vLDL) è ridotta; poiché le vLDL normalmente trasportano i Trigliceridi dal fegato ad altri tessuti, riducendo la secrezione di vLDL ciò si traduce in un livello sierico di Trigliceridi inferiori. [89] La diminuzione della secrezione di vLDL può essere secondaria all’inibizione della lipolisi nel tessuto adiposo, poiché l’aumento cronico di acidi grassi liberi nel siero può regolare negativamente la secrezione di vLDL. [143]
Sembra che l’integrazione di Niacina in acuto (che riduce gli acidi grassi liberi nel siero) sopprime anche la produzione di vLDL e la sua complessazione con i trigliceridi. [144] Ciò suggerisce un altro possibile meccanismo, che può verificarsi attraverso la soppressione acuta del PGC-1β, [145] una proteina nota per promuovere la secrezione di Trigliceridi dal fegato in risposta all’ingestione di grassi nella dieta. [146] In accordo con quest’ultimo meccanismo, la somministrazione di Niacina con un pasto ad alto contenuto di grassi sembra ridurre il picco normale dei trigliceridi postprandiali. [147]

Non è confermato come la Niacina riduca le vLDL-C, ma la sua capacità di stimolare l’attività del gene ABCA1 e aumentare il suo contenuto proteico nelle cellule del fegato è alla base dell’aumento dell’HDL-C, [148] che è noto anche per sopprimere la secrezione di vLDL-C. [57] La Niacina (2g per 16 settimane), nonostante riduca le vLDL-C e il complesso con Trigliceridi, non sembra aumentare significativamente il contenuto di trigliceridi intraepatici in soggetti con malattia del fegato grasso non alcolica (NAFLD). [149]
La Niacina sembra anche agire sulle cellule del fegato per promuovere l’accumulo di diacilglicerolo (DAG), che è associato all’insulino-resistenza localizzata. [92] La resistenza all’insulina nelle cellule del fegato riduce l’effetto soppressivo dell’insulina sulla sintesi del glucosio, con conseguente aumento dell’efflusso di glucosio dal fegato nel sangue. [150] Poiché gli stadi iniziali dell’insulino-resistenza indotta dalla Niacina (prima degli aumenti dell’insulina basale e del glucosio) sono stati associati a un fabbisogno ridotto di glucosio per bilanciare un morsetto euglicemico iperinsulinaemico, [109] questo suggerisce che l’inizio dell’insulino-resistenza avviene a livello del fegato. Il ruolo preciso del DAG in questo processo è tuttavia incerto. Mentre il DAG promuove la resistenza all’insulina attraverso l’attivazione di PKCε, [151] l’attivazione di questa proteina non è stata osservata nelle cellule del fegato che sono diventate insulino-resistenti con la Niacina. [92]

Nota: La Niacina in dosi terapeutiche può causare aumenti modesti delle transaminasi sieriche e della bilirubina non coniugata, entrambi biomarcatori del danno epatico. Le modifiche vengono invertite se il trattamento farmacologico viene interrotto e di solito si risolvono anche quando si continua l’assunzione. [152] [153] [154] Tuttavia, meno comunemente, la forma di rilascio prolungato del farmaco può portare a gravi epatotossicità, con insorgenza in giorni o settimane. I primi sintomi di gravi danni al fegato includono nausea, vomito e dolore addominale, seguiti da ittero e prurito. Si ritiene che il meccanismo sia una tossicità diretta della Niacina sierica elevata. Abbassare la dose o passare alla forma a rilascio immediato può risolvere i sintomi. In rari casi la lesione è grave e progredisce fino a insufficienza epatica. [152]
È noto che la Niacina rende le cellule β pancreatiche (un tipo di cellula specializzata che secerne insulina in risposta al glucosio) meno sensibile al glucosio sierico. [81] Inoltre, la normale riduzione della sensibilità al glucosio delle cellule β del pancreas associata all’invecchiamento può essere ulteriormente esacerbata dalla supplementazione di Niacina (500mg-1g due volte al giorno). [83] Anche se sembra esserci un aumento compensativo della secrezione di insulina nella risposta alla Niacina [83], in un modello di primati con diabete di tipo I, [155] questo non è stato sufficiente a ridurre la glicemia a livelli normali, con conseguente lieve iperglicemia e iperinsulinemia dopo due settimane di integrazione.[83]
Va notato che una linea cellulare coinvolta nel rossore cutaneo tipico della Niacina, nota come Langerhans, [156] [157] è diversa dall’area del pancreas nota come “Isole di Langerhans”.
Nota: il rossore dato dalla Niacina – una dilatazione a breve termine delle arteriole della pelle, che causa il colore della pelle rossastra – di solito dura circa 15-30 minuti, anche se a volte può persistere per settimane con uso cronico e di forme a lento rilascio. In genere, il viso è maggiormente interessato, ma la reazione può estendersi al collo e alla parte superiore del torace. La causa è la dilatazione dei vasi sanguigni [158] [93] dovuta all’aumento della prostaglandina GD2 (PGD2) e serotonina. [159] [160] [161] [162] Si pensava spesso che il rossore riguardasse l’istamina, ma è stato dimostrato che l’istamina non è coinvolta nella reazione. [159] Il rossore a volte è accompagnato da una sensazione di prurito, in particolare, nelle aree coperte da indumenti. [93]

La prevenzione del rossore richiede l’alterazione o il blocco della via mediata dalle prostaglandine. [93] [163] L’Aspirina [165-325mg] assunta mezz’ora prima della Niacina riduce fortemente il rossore, così come l’Ibuprofene [200mg] (una riduzione della frequenza e intensità del rossore fino al 50%). L’assunzione di Niacina ai pasti aiuta anche a ridurre questo effetto collaterale. [93] La tolleranza acquisita aiuterà a ridurre l’effetto; dopo diverse settimane a dosaggio costante, la maggior parte delle persone non ha più esperienza di vampate di calore. [93] Sono state sviluppate forme di Niacina a rilascio lento o “prolungato” per ridurre questi effetti collaterali. [164] [165]
Conclusioni sulla supplementazione di Niacina
Le informazioni che abbiamo a disposizione sulla Niacina e la sua supplementazione, ci presentano un composto senz’altro utile per il controllo o riassesto del quadro lipidico ma allo stesso tempo questa molecola risulta di una complessità d’azione biochimica non trascurabile. Il suo peggiorare l’insulino-resistenza in cronico ma con un maggior picco in acuto, picco che sembra venire controbilanciato da altri fattori come l’aumento della Adiponectina. Lo stesso effetto sulla riduzione della lipolisi può destare preoccupazione nell’atleta, specie se questo si trova in una fase ipocalorica con il principale intento di ridurre la massa grassa. Anche in questo caso sembrerebbe che l’effetto si manifesti in acuto per poi rientrare in condizioni pre-utilizzo. Ciò che è quasi certo, è che le osservazioni sul campo non hanno fatto emergere grosse differenze nell’alterazione della composizione corporea, sia con l’uso della Niacina in regimi ipercalorici che ipocalorici. L’utilizzo di GDA (in specie Berberina) anche alle dosi base efficaci potrebbe essere un “tampone” sufficienti a compensare almeno in parte i possibili peggioramenti dei parametri dell’insulino resistenza. I controlli della glicemia basale e della insulinemia a digiuno sono indicatori da tenere sotto controllo durante l’uso di Niacina. Non è da trascurare la possibilità che la Niacina possa influenzare lo “shift” dalle fibre muscolari di tipo II a quelle di tipo I, cosa non auspicabile per un Bodybuilder o altro atleta di forza.
In definitiva, considerando i dosaggi efficaci e la migliore azione in combinazione con statine (vedi Monacolina K), l’assunzione di Niacina può essere mantenuta con un certo margine di sicurezza tra i 500mg ed 1g/die, ovviamente tarando il dosaggio in risposta agli esami ematici di controllo.
Gabriel Bellizzi
Riferimenti:
- “Niacin”. Micronutrient Information Center, Linus Pauling Institute, Oregon State University, Corvallis, OR. 8 October 2018. Retrieved 16 September 2019.
- Kennedy DO (January 2016). “B Vitamins and the Brain: Mechanisms, Dose and Efficacy—A Review”. Nutrients. 8 (2): 68.
- “Niacin Fact Sheet for Health Professionals”. Office of Dietary Supplements, US National Institutes of Health. 3 June 2020. Retrieved 29 June 2020.
- Hegyi J, Schwartz RA, Hegyi V (January 2004). “Pellagra: dermatitis, dementia, and diarrhea”. International Journal of Dermatology. 43 (1): 1–5.
- “Why fortify?”. Food Fortification Initiative. 2017. Retrieved 4 April 2017.
- Institute of Medicine (1998). “Niacin”. Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline. Washington, DC: The National Academies Press. pp. 123–149.
- “Overview on Dietary Reference Values for the EU population as derived by the EFSA Panel on Dietetic Products, Nutrition and Allergies” (PDF). 2017.
- “Nutrient reference values for Australia and New Zealand”(PDF). National Health and Medical Research Council. 9 September 2005. Archived from the original (PDF) on 21 January 2017. Retrieved 19 June 2018.
- Jaconello P (October 1992). “Niacin versus niacinamide”. CMAJ. 147 (7): 990.
- Kirkland JB (May 2012). “Niacin requirements for genomic stability”. Mutation Research. 733 (1–2): 14–20.
- World Health Organization (2000). “Pellagra And Its Prevention And Control In Major Emergencies”. World Health Organization (WHO).
- “Niacin”. Drugs.com. 16 March 2019. Retrieved 27 April 2020.
- Keene D, Price C, Shun-Shin MJ, Francis DP (July 2014). “Effect on cardiovascular risk of high density lipoprotein targeted drug treatments niacin, fibrates, and CETP inhibitors: meta-analysis of randomised controlled trials including 117,411 patients”. BMJ. 349: g4379.
- Bruckert E, Labreuche J, Amarenco P (June 2010). “Meta-analysis of the effect of nicotinic acid alone or in combination on cardiovascular events and atherosclerosis”. Atherosclerosis. 210 (2): 353–61.
- Schandelmaier S, Briel M, Saccilotto R, Olu KK, Arpagaus A, Hemkens LG, Nordmann AJ (June 2017). “Niacin for primary and secondary prevention of cardiovascular events”. The Cochrane Database of Systematic Reviews. 6: CD009744.
- “Niacin”. IN: LiverTox: Clinical and Research Information on Drug-Induced Liver Injury (Internet). Bethesda, MD: National Institute of Diabetes and Digestive and Kidney Diseases. February 2014.
- Ong KL, Barter PJ, Waters DD (April 2014). “Cardiovascular drugs that increase the risk of new-onset diabetes”. Am. Heart J. 167 (4): 421–8.
- Goldie C, Taylor AJ, Nguyen P, McCoy C, Zhao XQ, Preiss D (February 2016). “Niacin therapy and the risk of new-onset diabetes: a meta-analysis of randomised controlled trials”. Heart. 102 (3): 198–203.
- “Niacin – Drug Usage Statistics”. ClinCalc. Retrieved 11 April2020.
- Kelly JJ1, et al. Effects of nicotinic acid on insulin sensitivity and blood pressure in healthy subjects. J Hum Hypertens. (2000)
- Westphal S1, et al. Extended-release niacin raises adiponectin and leptin. Atherosclerosis. (2007)
- Warnholtz A1, et al. Effects of oral niacin on endothelial dysfunction in patients with coronary artery disease: results of the randomized, double-blind, placebo-controlled INEF study. Atherosclerosis. (2009)
- Nasser Figueiredo V1, et al. Short-term effects of extended-release niacin with and without the addition of laropiprant on endothelial function in individuals with low HDL-C: a randomized, controlled crossover trial. Clin Ther. (2014)
- Stocker R. Antioxidant activities of bile pigments. Antioxid Redox Signal. (2004)
- Bregar U1, et al. Extended-release niacin/laropiprant improves endothelial function in patients after myocardial infarction. Heart Vessels. (2014)
- Chow DC1, et al. Short-term effects of extended-release niacin on endothelial function in HIV-infected patients on stable antiretroviral therapy. AIDS. (2010)
- Bays HE1, Rader DJ. Does nicotinic acid (niacin) lower blood pressure. Int J Clin Pract. (2009)
- Gadegbeku CA1, et al. Hemodynamic effects of nicotinic acid infusion in normotensive and hypertensive subjects. Am J Hypertens. (2003)
- Brown BG1, et al. Simvastatin and niacin, antioxidant vitamins, or the combination for the prevention of coronary disease. N Engl J Med. (2001)
- Carlson LA1, Rosenhamer G. Reduction of mortality in the Stockholm Ischaemic Heart Disease Secondary Prevention Study by combined treatment with clofibrate and nicotinic acid. Acta Med Scand. (1988)
- Whitney EJ1, et al. A randomized trial of a strategy for increasing high-density lipoprotein cholesterol levels: effects on progression of coronary heart disease and clinical events. Ann Intern Med. (2005)
- [No authors listed. Clofibrate and niacin in coronary heart disease. JAMA. (1975)
- Canner PL1, Furberg CD, McGovern ME. Benefits of niacin in patients with versus without the metabolic syndrome and healed myocardial infarction (from the Coronary Drug Project). Am J Cardiol. (2006)
- Maccubbin D1, et al. Lipid-modifying efficacy and tolerability of extended-release niacin/laropiprant in patients with primary hypercholesterolaemia or mixed dyslipidaemia. Int J Clin Pract. (2008)
- Bays HE1, et al. Blood pressure-lowering effects of extended-release niacin alone and extended-release niacin/laropiprant combination: a post hoc analysis of a 24-week, placebo-controlled trial in dyslipidemic patients. Clin Ther. (2009)
- Ganji SH1, et al. Niacin noncompetitively inhibits DGAT2 but not DGAT1 activity in HepG2 cells. J Lipid Res. (2004)
- Fabbrini E1, et al. Effect of fenofibrate and niacin on intrahepatic triglyceride content, very low-density lipoprotein kinetics, and insulin action in obese subjects with nonalcoholic fatty liver disease. J Clin Endocrinol Metab. (2010)
- Tunaru S1, et al. PUMA-G and HM74 are receptors for nicotinic acid and mediate its anti-lipolytic effect. Nat Med. (2003)
- Eaton RP, Berman M, Steinberg D. Kinetic studies of plasma free fatty acid and triglyceride metabolism in man. J Clin Invest. (1969)
- Lauring B1, et al. Niacin lipid efficacy is independent of both the niacin receptor GPR109A and free fatty acid suppression. Sci Transl Med. (2012)
- AIM-HIGH Investigators, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. (2011)
- Wang W1, et al. Effects of nicotinic acid on fatty acid kinetics, fuel selection, and pathways of glucose production in women. Am J Physiol Endocrinol Metab. (2000)
- Nelson RH1, et al. Intravenous niacin acutely improves the efficiency of dietary fat storage in lean and obese humans. Diabetes. (2012)
- Ahlström C1, et al. Feedback modeling of non-esterified fatty acids in obese Zucker rats after nicotinic acid infusions. J Pharmacokinet Pharmacodyn. (2013)
- Ahlström C1, et al. Feedback modeling of non-esterified fatty acids in rats after nicotinic acid infusions. J Pharmacokinet Pharmacodyn. (2011)
- Oh YT1, et al. Continuous 24-h nicotinic acid infusion in rats causes FFA rebound and insulin resistance by altering gene expression and basal lipolysis in adipose tissue. Am J Physiol Endocrinol Metab. (2011)
- O’Kane MJ1, et al. A comparison of acipimox and nicotinic acid in type 2b hyperlipidaemia. Br J Clin Pharmacol. (1992)
- Lai E1, et al. Effects of a niacin receptor partial agonist, MK-0354, on plasma free fatty acids, lipids, and cutaneous flushing in humans. J Clin Lipidol. (2008)
- van der Hoorn JW1, et al. Niacin increases HDL by reducing hepatic expression and plasma levels of cholesteryl ester transfer protein in APOE-3Leiden.CETP mice. Arterioscler Thromb Vasc Biol. (2008)
- Zhang LH1, et al. Niacin increases HDL biogenesis by enhancing DR4-dependent transcription of ABCA1 and lipidation of apolipoprotein A-I in HepG2 cells. J Lipid Res. (2012)
- Lee JY1, Parks JS. ATP-binding cassette transporter AI and its role in HDL formation. Curr Opin Lipidol. (2005)
- Sahoo D1, et al. ABCA1-dependent lipid efflux to apolipoprotein A-I mediates HDL particle formation and decreases VLDL secretion from murine hepatocytes. J Lipid Res. (2004)
- Sakai T1, Kamanna VS, Kashyap ML. Niacin, but not gemfibrozil, selectively increases LP-AI, a cardioprotective subfraction of HDL, in patients with low HDL cholesterol. Arterioscler Thromb Vasc Biol. (2001)
- Wu ZH1, Zhao SP. Niacin promotes cholesterol efflux through stimulation of the PPARgamma-LXRalpha-ABCA1 pathway in 3T3-L1 adipocytes. Pharmacology. (2009)
- Knowles HJ1, et al. Niacin induces PPARgamma expression and transcriptional activation in macrophages via HM74 and HM74a-mediated induction of prostaglandin synthesis pathways. Biochem Pharmacol. (2006)
- Hernandez M1, Wright SD, Cai TQ. Critical role of cholesterol ester transfer protein in nicotinic acid-mediated HDL elevation in mice. Biochem Biophys Res Commun. (2007)
- Barter PJ1, et al. Cholesteryl ester transfer protein: a novel target for raising HDL and inhibiting atherosclerosis. Arterioscler Thromb Vasc Biol. (2003)
- Barter PJ, Hopkins GJ, Calvert GD. Transfers and exchanges of esterified cholesterol between plasma lipoproteins. Biochem J. (1982)
- Luo Y1, Tall AR. Sterol upregulation of human CETP expression in vitro and in transgenic mice by an LXR element. J Clin Invest. (2000)
- Zhang LH1, et al. Niacin increases HDL biogenesis by enhancing DR4-dependent transcription of ABCA1 and lipidation of apolipoprotein A-I in HepG2 cells. J Lipid Res. (2012)
- Zhang LH1, et al. Niacin increases HDL biogenesis by enhancing DR4-dependent transcription of ABCA1 and lipidation of apolipoprotein A-I in HepG2 cells. J Lipid Res. (2012)
- Cinquin O1, Page KM. Generalized, switch-like competitive heterodimerization networks. Bull Math Biol. (2007)
- Lamon-Fava S1, et al. Extended-release niacin alters the metabolism of plasma apolipoprotein (Apo) A-I and ApoB-containing lipoproteins. Arterioscler Thromb Vasc Biol. (2008)
- Zhang LH1, et al. Niacin inhibits surface expression of ATP synthase beta chain in HepG2 cells: implications for raising HDL. J Lipid Res. (2008)
- Blum CB, et al. High density lipoprotein metabolism in man. J Clin Invest. (1977)
- van der Hoorn JW1, et al. Niacin increases HDL by reducing hepatic expression and plasma levels of cholesteryl ester transfer protein in APOE-3Leiden.CETP mice. Arterioscler Thromb Vasc Biol. (2008)
- Bays H1, et al. Extended-release niacin/laropiprant effects on lipoprotein subfractions in patients with type 2 diabetes mellitus. Metab Syndr Relat Disord. (2012)
- Duggal JK1, et al. Effect of niacin therapy on cardiovascular outcomes in patients with coronary artery disease. J Cardiovasc Pharmacol Ther. (2010)
- Cashin-Hemphill L1, et al. Beneficial effects of colestipol-niacin on coronary atherosclerosis. A 4-year follow-up. JAMA. (1990)
- AIM-HIGH Investigators, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. (2011)
- Brouwers MC, Stehouwer CD. Niacin in cardiovascular patients receiving statins. N Engl J Med. (2012)
- Fabbrini E1, et al. Effect of fenofibrate and niacin on intrahepatic triglyceride content, very low-density lipoprotein kinetics, and insulin action in obese subjects with nonalcoholic fatty liver disease. J Clin Endocrinol Metab. (2010)
- Si Y1, et al. Niacin inhibits vascular inflammation via downregulating nuclear transcription factor-κB signaling pathway. Mediators Inflamm. (2014)
- Lipszyc PS1, et al. Niacin Modulates Pro-inflammatory Cytokine Secretion. A Potential Mechanism Involved in its Anti-atherosclerotic Effect. Open Cardiovasc Med J. (2013)
- Shashkin P1, Dragulev B, Ley K. Macrophage differentiation to foam cells. Curr Pharm Des. (2005)
- Rubic T1, Trottmann M, Lorenz RL. Stimulation of CD36 and the key effector of reverse cholesterol transport ATP-binding cassette A1 in monocytoid cells by niacin. Biochem Pharmacol. (2004)
- Tontonoz P1, et al. PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell. (1998)
- Chinetti G1, et al. PPAR-alpha and PPAR-gamma activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nat Med. (2001)
- Bortnick AE1, et al. The correlation of ATP-binding cassette 1 mRNA levels with cholesterol efflux from various cell lines. J Biol Chem. (2000)
- Zhang F, et al. Niacin-induced enhancement of lysosomal cholesterol efflux in macrophages through CD38 – NAADP signaling pathway: implication in reduced foam cell formation (671.6). FASEB J. (2014)
- Kahn SE1, et al. Increased beta-cell secretory capacity as mechanism for islet adaptation to nicotinic acid-induced insulin resistance. Diabetes. (1989)
- ^ Loffler, Trautschold. Influence of nicotinic acid on insulin secretion in vivo and in vitro. In Metabolic Effects of Nicotinic Acid and its Derivatives. Gey KF, Carlson LA, editors.. Bern: Hanss Huber Publishers; pp. 487–496.. (1971)
- Chang AM1, et al. Impaired beta-cell function in human aging: response to nicotinic acid-induced insulin resistance. J Clin Endocrinol Metab. (2006)
- Chen X1, Iqbal N, Boden G. The effects of free fatty acids on gluconeogenesis and glycogenolysis in normal subjects. J Clin Invest. (1999)
- Gross RC, Carlson LA. Metabolic effects of nicotinic acid in acute insulin deficiency in the rat. Diabetes. (1968)
- Davidson MB, Bernstein JM. The effect of nicotinic acid on growth hormone-induced lipolysis and glucose intolerance. J Lab Clin Med. (1973)
- Reaven GM1, Chang H, Hoffman BB. Additive hypoglycemic effects of drugs that modify free-fatty acid metabolism by different mechanisms in rats with streptozocin-induced diabetes. Diabetes. (1988)
- Kelly JJ1, et al. Effects of nicotinic acid on insulin sensitivity and blood pressure in healthy subjects. J Hum Hypertens. (2000)
- Fabbrini E1, et al. Effect of fenofibrate and niacin on intrahepatic triglyceride content, very low-density lipoprotein kinetics, and insulin action in obese subjects with nonalcoholic fatty liver disease. J Clin Endocrinol Metab. (2010)
- Fraterrigo G1, et al. Relationship between Changes in Plasma Adiponectin Concentration and Insulin Sensitivity after Niacin Therapy. Cardiorenal Med. (2012)
- Westphal S1, et al. Extended-release niacin raises adiponectin and leptin. Atherosclerosis. (2007)
- Blond E1, et al. Nicotinic acid effects on insulin sensitivity and hepatic lipid metabolism: an in vivo to in vitro study. Horm Metab Res. (2014)
- Plaisance EP1, et al. Increased total and high-molecular weight adiponectin after extended-release niacin. Metabolism. (2008)
- Kelly JJ1, et al. Effects of nicotinic acid on insulin sensitivity and blood pressure in healthy subjects. J Hum Hypertens. (2000)
- Alvarsson M1, Grill V. Impact of nicotinic acid treatment on insulin secretion and insulin sensitivity in low and high insulin responders. Scand J Clin Lab Invest. (1996)
- Poynten AM1, et al. Nicotinic acid-induced insulin resistance is related to increased circulating fatty acids and fat oxidation but not muscle lipid content. Metabolism. (2003)
- Oh YT1, et al. Continuous 24-h nicotinic acid infusion in rats causes FFA rebound and insulin resistance by altering gene expression and basal lipolysis in adipose tissue. Am J Physiol Endocrinol Metab. (2011)
- Ganji SH1, et al. Niacin noncompetitively inhibits DGAT2 but not DGAT1 activity in HepG2 cells. J Lipid Res. (2004)
- Mithieux G. Brain, liver, intestine: a triumvirate to coordinate insulin sensitivity of endogenous glucose production. Diabetes Metab. (2010)
- Dobbins RL1, et al. GSK256073, a selective agonist of G-protein coupled receptor 109A (GPR109A) reduces serum glucose in subjects with type 2 diabetes mellitus. Diabetes Obes Metab. (2013)
- Fulcher GR1, et al. Acipimox increases glucose disposal in normal man independent of changes in plasma nonesterified fatty acid concentration and whole-body lipid oxidation rate. Metabolism. (1993)
- Oh YT1, et al. Continuous 24-h nicotinic acid infusion in rats causes FFA rebound and insulin resistance by altering gene expression and basal lipolysis in adipose tissue. Am J Physiol Endocrinol Metab. (2011)
- Kokubun E1, et al. Changes of glycogen content in liver, skeletal muscle, and heart from fasted rats. Cell Biochem Funct. (2009)
- Beránek M1, et al. Glycation and advanced glycation end-products in laboratory experiments in vivo and in vitro. Acta Medica (Hradec Kralove). (2006)
- Tarwadi KV1, Agte VV. Effect of micronutrients on methylglyoxal-mediated in vitro glycation of albumin. Biol Trace Elem Res. (2011)
- Parker-Duffen JL1, Walsh K2. Cardiometabolic effects of adiponectin. Best Pract Res Clin Endocrinol Metab. (2014)
- Plaisance EP1, et al. Niacin stimulates adiponectin secretion through the GPR109A receptor. Am J Physiol Endocrinol Metab. (2009)
- Wang-Fisher YL1, Han J, Guo W. Acipimox stimulates leptin production from isolated rat adipocytes. J Endocrinol. (2002)
- Worm D1, et al. The nicotinic acid analogue acipimox increases plasma leptin and decreases free fatty acids in type 2 diabetic patients. Eur J Endocrinol. (2000)
- Miles JM1, et al. Systemic and forearm triglyceride metabolism: fate of lipoprotein lipase-generated glycerol and free fatty acids. Diabetes. (2004)
- [No authors listed. Baron Theodore Rose. Lancet. (1978)
- Deivanayagam S1, et al. Nonalcoholic fatty liver disease is associated with hepatic and skeletal muscle insulin resistance in overweight adolescents. Am J Clin Nutr. (2008)
- Nelson RH1, et al. Intravenous niacin acutely improves the efficiency of dietary fat storage in lean and obese humans. Diabetes. (2012)
- Heemskerk MM1, et al. Long-term niacin treatment induces insulin resistance and adrenergic responsiveness in adipocytes by adaptive downregulation of phosphodiesterase 3B. Am J Physiol Endocrinol Metab. (2014)
- Vega GL1, et al. Influence of extended-release nicotinic acid on nonesterified fatty acid flux in the metabolic syndrome with atherogenic dyslipidemia. Am J Cardiol. (2005)
- Lewis JE, Shimizu Y, Shimizu N. Nicotinamide inhibits adipocyte differentiation of 3T3-L1 cells. FEBS Lett. (1982)
- Purnell MR, Whish WJ. Novel inhibitors of poly(ADP-ribose) synthetase. Biochem J. (1980)
- Torres-Ramírez N1, et al. Nicotinamide, a glucose-6-phosphate dehydrogenase non-competitive mixed inhibitor, modifies redox balance and lipid accumulation in 3T3-L1 cells. Life Sci. (2013)
- Aktories K, Jakobs KH, Schultz G. Nicotinic acid inhibits adipocyte adenylate cyclase in a hormone–like manner. FEBS Lett. (1980)
- Soudijn W1, van Wijngaarden I, Ijzerman AP. Nicotinic acid receptor subtypes and their ligands. Med Res Rev. (2007)
- Aktories K, Jakobs KH. In vivo and in vitro desensitization of nicotinic acid-induced adipocyte adenylate cyclase inhibition. Naunyn Schmiedebergs Arch Pharmacol. (1982)
- Bai P1, et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. (2011)
- Cantó C1, et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. (2012)
- Wang YX1, et al. Regulation of muscle fiber type and running endurance by PPARdelta. PLoS Biol. (2004)
- Schuler M1, et al. PGC1alpha expression is controlled in skeletal muscles by PPARbeta, whose ablation results in fiber-type switching, obesity, and type 2 diabetes. Cell Metab. (2006)
- Ringseis R1, et al. Supplementing obese Zucker rats with niacin induces the transition of glycolytic to oxidative skeletal muscle fibers. J Nutr. (2013)
- Khan M1, et al. Niacin supplementation increases the number of oxidative type I fibers in skeletal muscle of growing pigs. BMC Vet Res. (2013)
- Khan M1, et al. Niacin supplementation induces type II to type I muscle fiber transition in skeletal muscle of sheep. Acta Vet Scand. (2013)
- Scholz K1, et al. Supplementing healthy rats with a high-niacin dose has no effect on muscle fiber distribution and muscle metabolic phenotype. Eur J Nutr. (2014)
- Fukuwatari T1, et al. Elevation of blood NAD level after moderate exercise in young women and mice. J Nutr Sci Vitaminol (Tokyo). (2001)
- Graham T, et al. NAD in muscle of man at rest and during exercise. Pflugers Arch. (1978)
- Sahlin K. NADH in human skeletal muscle during short-term intense exercise. Pflugers Arch. (1985)
- Sahlin K1, Katz A, Henriksson J. Redox state and lactate accumulation in human skeletal muscle during dynamic exercise. Biochem J. (1987)
- Mach J1, et al. The effect of antioxidant supplementation on fatigue during exercise: potential role for NAD+(H). Nutrients. (2010)
- Zhang SJ1, et al. Activation of aconitase in mouse fast-twitch skeletal muscle during contraction-mediated oxidative stress. Am J Physiol Cell Physiol. (2007)
- Cardoso SM1, Pereira C, Oliveira R. Mitochondrial function is differentially affected upon oxidative stress. Free Radic Biol Med. (1999)
- Zandi-Nejad K1, et al. The role of HCA2 (GPR109A) in regulating macrophage function. FASEB J. (2013)

















































 to come agente dopante negli atleti olimpionici della Germania Orientale all’interno di un programma di doping sponsorizzato dallo stato negli anni ’70 e ’80.[4] Il motivo del suo uso, come precedentemente accennato, era giustificato maggiormente dal suo valore androgeno piuttosto che su quello anabolizzante. Il suo uso garantiva un ottima risposta neuro steroidea con conseguente stimolazione del Sistema Nervoso Centrale e migliorata interazione neuromuscolare, con vantaggi nella velocità, nella forza, nell’aggressività, nella concentrazione, nella resistenza fisica e allo stress mentale.[4] Oggi, l’uso del Mestanolone è stato per lo più sospeso in medicina, sebbene sia ancora disponibile in Giappone.[2][3][4] Il Mestanolone era comunemente disponibile sotto forma di compresse sublinguali da 25mg (marchio Ermalone).[7]
to come agente dopante negli atleti olimpionici della Germania Orientale all’interno di un programma di doping sponsorizzato dallo stato negli anni ’70 e ’80.[4] Il motivo del suo uso, come precedentemente accennato, era giustificato maggiormente dal suo valore androgeno piuttosto che su quello anabolizzante. Il suo uso garantiva un ottima risposta neuro steroidea con conseguente stimolazione del Sistema Nervoso Centrale e migliorata interazione neuromuscolare, con vantaggi nella velocità, nella forza, nell’aggressività, nella concentrazione, nella resistenza fisica e allo stress mentale.[4] Oggi, l’uso del Mestanolone è stato per lo più sospeso in medicina, sebbene sia ancora disponibile in Giappone.[2][3][4] Il Mestanolone era comunemente disponibile sotto forma di compresse sublinguali da 25mg (marchio Ermalone).[7]