Come si potrà facilmente capire dalle prime righe di questo articolo, la Rauwolscina è una molecola molto simile nella sua struttura alla Yohimbina e probabilmente condivide simili effetti; ipoteticamente potrebbe essere più potente, ma esistono prove limitate.
Dal momento che mi è capitato molto spesso di ricevere domande su questa forma di Yohimbina, ho deciso di scrivere alcune righe in merio trattando gli effetti maggiormente ricercati dall’utilizzatore medio dell’alcaloide in questione.
Rauwolscina: caratteristiche proprie e similitudini con la Yohimbina
La Rauwolscina, nota anche come Isoyohimbina, α-yohimbina e Corynanthidina, è un alcaloide presente in varie specie del genere Rauvolfia e Pausinystalia (precedentemente noto come Corynanthe).[1] È uno stereoisomero della Yohimbina. [1] la Rauwolscina è uno stimolante del sistema nervoso centrale, un anestetico locale e possiede un certo potenziale afrodisiaco.[1] La Rauwolscina agisce prevalentemente come antagonista del recettore α2-adrenergico.[2][3] È stato anche dimostrato che agisce come agonista parziale del recettore 5-HT1A e antagonista del recettore 5-HT2A e 5-HT2B. [4] [5] [6]
la Rauwolscina, o {3H}Rauwolscina, come già accennato, è un antagonista dei recettori adrenergici alfa-1 e alfa-2 (con una maggiore selettività per quest’ultimo).[7] La Yohimbina condivide il medesimo meccanismo d’azione recettoriale.
Come ormai risaputo, la Yohimbina agisce sul sistema dei recettori adrenergici delle cellule adipose, che regolano la termogenesi. Le subunità beta dei recettori adrenergici (bersagli, per esempio, dell’Efedrina) possono essere viste come stimolanti per la perdita di grasso poiché aumentano l’attività dell’enzima adenil ciclasi e successivamente dei livelli di cAMP (principalmente attraverso le subunità b1 e b2; con la b3 che è meno attiva negli umani).[8][9] Le subunità alfa sono soppressive del metabolismo lipidico, e attraverso la loro attivazione si riduce l’attività dell’adenil ciclasi e si riducono i livelli di cAMP (in particolare per opera degli alfa-2). La Yohimbina, come la Rauwolscina, è un antagonista selettivo del recettore alfa-2 adrenergico (inattivatore), che inibisce l’attivazione del set di recettori soppressivi e preserva l’attività dell’adenil ciclasi e gli effetti mediati dai recettori beta.[10]
Osservando le interazioni della Yohimbina a livello del recettore, la molecola è risultata essere un antagonista alfa2-adrenergico selettivo con un’affinità 44 volte maggiore per la subunità alfa2 rispetto alla subunità alfa1 quando testata su ratti anoccigei e dotti deferenti; questo differisce dal relativo composto Corinantino e Rauwolscina che sono selettivi per il recettore alfa1 (33 volte) e per lo più non selettivi (3,3 volte); rispettivamente.[11] Questi valori sono stati derivati da sperimentazioni in vitro e un secondo test nel quale è stato osservato il legame competitivo in campioni di cervello notando che la selettività era ridotta da 45 a 5,7. Quando si osserva il recettore alfa2 stesso, la Yohimbina sembra avere ulteriore selettività per la subunità alfa2C piuttosto che per la A o la B; nell’intervallo di 4-15 volte la selettività,[12] mentre la Rauwolscina sembra essere non selettiva tra queste tre subunità.[13][12] La Rauwolscina sembra essere efficace a livello del recettore quanto la Yohimbina,[14] con la Coynantina che presenta la minore entità di efficacia.[11]
Oltre a ciò, la stessa Yohimbina può potenzialmente indurre la perdita di grasso per via indiretta attraverso il rilascio di Adrenalina; l’Adrenalina stessa è un attivatore dei recettori beta-adrenergici.[15] Tuttavia, questo aumento di Adrenalina può svanire con il tempo raggiungendo l’irrilevanza statistica 2 settimane dopo l’inizio dell’ingestione giornaliera.[16] L’aumento degli acidi grassi liberi plasmatici e la densità dei recettori alfa2-adrenergici rimangono simili in entrambi i momenti, suggerendo che la Yohimbina perde selettivamente il picco di Adrenalina ma non gli effetti diretti sulla sovra-regolazione della lipolisi recettore-dipendente.
La Yohimbina è stata inizialmente studiata per il suo ruolo nella riduzione degli accumuli adiposi localizzati grazie al suo utilizzo come crema topica (potendo scegliere dove applicare la Yohimbina)[17][18] ma anche secondariamente al suo utilizzo per ridurre l’adipe nelle cosce delle donne, poiché livelli elevati di Estrogeni aumentano l’attività del recettore alfa2-adrenergico.[19] A causa dell’aumento dell’attività alfa2-adrenergica nel tessuto adiposo sito nelle cosce delle donne, si pensava che l’antagonismo di questi recettori riducesse l’adiposità in modo selettivo; i risultati con la soluzione topica sono ad oggi vaghi, con uno studio con esito positivo [18] e l’altro nel quale si sono notati benefici sia con la Forskolina (da Coleus Forskohlii) che con l’Aminofillina ma non con la Yohimbina. [17]
La chetogenesi, o la produzione di corpi chetonici, è potenziata dalla presenza di Noradrenalina in condizioni normali. Il blocco dei recettori alfa adrenergici, tramite l’antagonismo della Yohimbina (e della Rauwolscina) sul recettore alfa-2 adrenergico, aumenta gli effetti chetogenici della Noradrenalina. [20]
Uno studio nel quale si è osservato un aumento della ossidazione lipidica (aumento dei biomarcatori di NEFA e glicerolo) ha anche notato che questo aumento è stato soppresso durante lo stato di alimentazione.[21] È stato ipotizzato che questa interazione con lo stato di digiuno così come gli effetti apparentemente additivi/sinergici della Yohimbina e l’esercizio sulla lipolisi[21] potrebbero portare a tempistiche di assunzione ideale della Yohimbina prima dell’esercizio mattutino.[22] Se abbinata al cibo in modo acuto, la Yohimbina può effettivamente aumentare il rilascio di Insulina indotto dal glucosio attraverso lo stimolo delle cellule pancreatiche[23][24] ma non si verifica a digiuno dopo il consumo orale di 0,2g/kg.[25][21]
Se il meccanismo avviene tramite la stimolazione del rilascio di Insulina che riduce la lipolisi, è teorico che un pasto privo di carboidrati e a basso contenuto proteico (per ridurre la stimolazione indotta dagli amminoacidi) potrebbe essere simile allo stato di digiuno; questo non è stato però studiato.
Uno studio è stato condotto con la somministrazione di Yohimbina a giocatori di calcio d’élite che assumevano 10mg della molecola due volte al giorno (20 mg in totale) per un periodo di 21 giorni. Durante lo studio si è osservato che, in seguito al controllo della dieta, la percentuale di grasso era diminuita dallo 9,3 +/- 1,1% allo 7,1 +/- 2,2% (valutato tramite calibro), mentre nel gruppo placebo è stato registrato un aumento non significativo.[26] La dose di 0,2mg/kg di Yohimbina in uomini altrimenti sani sembra aumentare gli effetti sul miglioramento del metabolismo lipidico e della beta-ossidazione della Noradrenalina endogena e sembra essere più efficace durante i periodi di esercizio e attenuarsi se somministrati dei beta-bloccanti;[21] un altro studio ha rilevato che questa attenuazione deve essere misurata al 70%.[25] Tuttavia, almeno uno studio ha rilevato risultati nulli, in quanto la Yohimbina non ha fatto diminuire il peso nei volontari sani.[27]
La {3H} Rauwolscina è meno potente della Yohimbina nel proteggere dagli aumenti della pressione sanguigna indotti dall’Adrenalina (entrambi meno potenti della Corinantina e più potenti della 3-epi-alfa-yohimbina), questo effetto sembra correlato alla loro affinità per gli alfa -2 adrenorecettori.[28]
Come la Yohimbina, la Rauwolscina è un agonista dei recettori 5-HT1a/b e induce effetti simili alla Serotonina. Mentre la Yohimbina ha più affinità per tale recettore, la Rauwolscina ha un valore IC50 più basso (il che significa che può saturare più recettori alla stessa dose) e può essere considerata leggermente più potente nell’attività serotoninergica.[29][30]
Non mi dilungherò oltre, dal momento che gli argomenti trattati fino a questo momento sono di maggiore interesse per la valutazione di supplementi contenenti Rauwolscina e del loro possibile utilizzo.
Conclusione:
Ora, sappiamo che le caratteristiche di entrambe le forme di Yohimbina sono pressoché identiche sebbene la selettività recettoriale della Rauwolscina sembri minore di quella della Yohimbina. La caratteristica che sembra spostare l’interesse sulla Rauwolscina è la sua più lunga emivita ( Yohimbine 0.25-2.5h; Rauwolscina 0.5-5h ipotetiche). Se però valutiamo quest’aspetto alla luce della sua bassa selettività e alto potenziale di saturazione recettoriale, viene facile intuire che ciò possa influire negativamente sulla percentuale di emersione di effetti collaterali tipici dell’alcaloide (tremori, insonnia, emicrania, tachicardia, ecc…). Con molta probabilità il dosaggio “ideale” con un certo margine di “sicurezza” può essere attestato a 0.1mg/Kg/die.
Gabriel Bellizzi
Riferimenti:
KOHLI JD, DE NN (June 1956). “Pharmacological action of rauwolscine”. Nature. 177 (4521): 1182. doi:10.1038/1771182a0. PMID13334509.
Perry BD, U’Prichard DC (December 1981). “[3H]rauwolscine (alpha-yohimbine): a specific antagonist radioligand for brain alpha 2-adrenergic receptors”. European Journal of Pharmacology. 76 (4): 461–4. doi:10.1016/0014-2999(81)90123-0. PMID6276200.
Arthur JM, Casañas SJ, Raymond JR (June 1993). “Partial agonist properties of rauwolscine and yohimbine for the inhibition of adenylyl cyclase by recombinant human 5-HT1A receptors”. Biochemical Pharmacology. 45 (11): 2337–41. doi:10.1016/0006-2952(93)90208-E. PMID8517875.
Kaumann AJ (June 1983). “Yohimbine and rauwolscine inhibit 5-hydroxytryptamine-induced contraction of large coronary arteries of calf through blockade of 5 HT2 receptors”. Naunyn-Schmiedeberg’s Archives of Pharmacology. 323 (2): 149–54. doi:10.1007/BF00634263. PMID6136920.
DISCLAIMER: Il presente articolo è a solo scopo educativo, di intrattenimento e informativo. Non rappresenta in alcun modo una forma di incitamento all’uso/abuso di sostanze dopanti. L’autore ed il sito, per tanto, è esentato da qualsiasi responsabilità dipendente dalla libera scelta individuale.
Introduzione ad un dilemma…
Chiunque frequenti l’ambiente del Bodybuilding e del Fitness avrà letto o sentito almeno una volta nella vita espressioni del genere “se mi dopassi sarei anche io così [indicando Flex Wheeler]” o “ho provato di tutto e senza farmaci non riuscirò ad ottenere risultati”. Andando poi ad approfondire la storia di ognuno di questi soggetti si scopre in percentuale quasi assoluta che si tratta di individui nella norma (o al di sotto) frustrati e/o con personalità deboli, speranzosi omini che attendono placidamente che accada una svolta miracolosa nella loro banale e piatta esistenza e, cosa molto importante, con il minimo dello sforzo (meglio se nessuno).
Nella mia esperienza come ricercatore e operatore nel campo della cultura fisica in qualità di Preparatore Atletico, ho assistito a innumerevoli casi in cui un soggetto aspirava al miglioramento della propria composizione corporea trascurando, consciamente o inconsciamente, le basi fondamentali rappresentate da Nutrizione e Allenamento baipassandole in vista della possibile prescrizione di una pillola miracolosa capace di renderlo/a possessore della forma fisica ambita.
Tralasciando l’ovvio ragionamento che spinge ogni essere umano dotato di un minimo d’intelletto verso la comprensione che la genetica è il blocco d’argilla sul quale si va ad operare, ma le sue qualità e difetti sono presenti in modo eterogeneo nella popolazione mondiale, e ciò non è modificabile nemmeno con la farmacologia più oculata, quando ci si trova davanti al bivio tra “pillola rossa” (PEDs) e “pillola blu” (drug free) bisogna essere pienamente consapevoli non solo del fattore illegalità ma del fattore conoscitivo. Purtroppo, la politica del terrore ha operato in modo fallimentare nel goffo intento di allontanare dalla scelta “rossa”, e ciò si è tradotto in un numero sensibile di soggetti abusatori con tutte le conseguenze cliniche derivanti.
Se un individuo non ha raggiunto un livello di maturità sportiva tale da conferirgli una gestione corretta della nutrizione e della periodizzazione allenante (gestione delle variabili volume, intensità, densità ecc…), è molto meglio per lui/lei rivedere i suoi programmi e scegliere ancora la “pillola blu”. Capita, a volte, di incontrare persone decise ad intraprendere la via del “lato oscuro” che, dopo una approfondita chiacchierata sulla gestione dei suddetti fattori, rivede le proprie posizioni.
Per tutti coloro i quali sono immersi nel dilemma della scelta, vi espongo alcuni punti per rendere l’eventuale decisione meno rischiosa anche se pur sempre illegale nel “bel paese”…
“Pillola blu o pillola rossa?” I punti da tenere in considerazione per una scelta consapevole:
#1 Raggiungere una adeguata maturità sportiva
Per “maturità sportiva”, in particolare riferimento al BodyBuilding, si intende la capacità del atleta di sapersi alimentare e allenare correttamente con piena gestione delle proprie potenzialità fisiologiche/genetiche. Questa è la base, se viene a mancare ciò non solo la vostra esperienza finirà per deludervi e rendervi ancora di più dei frustrati, ma potrebbe rovinosamente portarvi ad un abuso cronico a senso inesorabilmente negativo…
#2 I PEDs non faranno miracoli
Una cosa da tenere bene a mente, e questo non dovrebbe interessare solo gli aspiranti “doped”, è che l’uso di PEDs non renderà diversi da ciò che rientra nelle potenzialità espressive del proprio patrimonio genetico. Certamente le caratteristiche genetiche verranno “iperespresse”, nel bene e nel male, dall’uso di PEDs ma non vi sarà nessun miracolo! Migliorerete ma non sarete ne più ne meno di ciò che potete essere!
Un esempio per capire come la base genetica faccia la differenza anche con protocolli che, ad oggi, spesso non raggiungono nemmeno i livelli del “bridge” più soft..
#3 Ridurre la percentuale di grasso corporeo
Il tessuto adiposo rappresenta uno dei siti dove il Testosterone, ed altri AAS soggetti all’aromatizzazione, viene convertito in Estradiolo. Soggetti con percentuali di grasso corporeo elevate vedrebbero una alterazione marcata della Testosterone:Estradiolo ratio a favore della componente estrogenica, con conseguenze quali alterazione del comportamento sessuale (impotenza, difficoltà nel raggiungere e/o mantenere l’erezione), ritenzione idrica, accumulo di grasso con modello femminile e ginecomastia. E no, l’uso di DHT derivati o di SARM non steroidei senza una base di Testosterone non risolverebbe il problema o, per lo meno, porterebbe ad altre conseguenze negative, che pur non comprendendo, per esempio, ritenzione idrica e ginecomastia, interesserebbero l’attività sessuale e la condizione psichica del soggetto trattato. [1]
Schema esemplificato del processo di aromatizzazione degli androgeni aumentati in un soggetto con percentuale di grasso corporeo alta.
Allo stesso tempo, i rischi cardiovascolari della somministrazione di AAS- come il possibile aumento esponenziale del Ematocrito, l’aumento del LDL e Trigliceridi a discapito di una riduzione del HDL, e l’aumento della pressione sanguigna – sarebbero già presenti in certa misura quando la body fat è già alta e sarebbero quindi soggetti ad un repentino aggravamento.
Se la percentuale di grasso è relativamente alta, si dovrebbe prima di tutto considerare di migliorare la composizione corporea con una adeguata routine alimentare e allenante (senza farmaci) prima di iniziare solo a pensare all’uso di AAS. Sicuramente ciò renderà la scelta più efficace e meno rischiosa.
Nel caso fosse necessario sottolinearlo, no, non è saggio nemmeno utilizzare agenti PEDs a fini lipolitici e/o antiadipogenici e/o termogenici (compresi gli Ormoni Tiroidei). A meno che non siate affetti da ipotiroidismo, e in questo caso la terapia vi dovrebbe essere stilata dal vostro medico, per ridurre in modo sensibile la body fat non sono necessari i farmaci!
#4 Controllare se si ha una storia familiare di trombosi (o qualsiasi altra malattia cardiovascolare)
Molte malattie cardiovascolari hanno una componente di base genetica. Uno stile di vita sano può ridurne sensibilmente la loro insorgenza, ma l’uso di AAS può causare l’attivazione di specifici geni implicati nella comparsa di malattie cardio-circolatorie. Caratteristico dell’interazione tra AAS e geni specifici è un caso studio ben documentato che ricercatori americani hanno pubblicato sul “Blood Coagulation & Fibrinolysis”.[2]
Trombosi venosa
Oltre all’attivazione genica diretta dagli AAS, e nociva per il sistema cardio-circolatorio, vi sono altre condizioni negative innescate dall’uso/abuso di Steroidi Anabolizzanti, e di altri PEDs, come, per esempio, l’aumento del tasso di coagulazione, l’incremento eccessivo dell’Ematocrito con aumento pressorio, rigidità dell’endotelio vascolare con perdita di efficienza strutturale e aumento della pressione ematica con incremento delle possibilità di danno strutturale dei componenti del sistema interessato.
#5 Inserire delle sedute di allenamento cardio prima, durante e dopo l’uso di PEDs
Un moderato allenamento cardiovascolare è sicuramente una delle migliori strategie preventive contro la comparsa di malattie cardio-circolatorie. Tale tipologia di allenamento può portare un miglioramento e/o riduzione delle alterazioni lipidiche ematiche del praticante, fornendo un, seppur minimo, tampone all’azione negativa degli AAS e SARM non steroidei sui livelli di LDL (aumento), Trigliceridi (aumento) e HDL (diminuzione). Secondo quanto riportato da una interessante review del 2013, l’abbinamento di sedute cardio e in sala pesi possono avere una azione additiva benefica sui livelli di LDL, Trigliceridi e HDL.[3]
Risulta interessante anche quanto emerso da alcuni studi su animali a seguito dei quali si è osservato un significativo grado di protezione dato dall’allenamento cardio negli esemplari trattati con AAS.[4]
#6 Assicurarsi di rimanere ben idratati
Oltre ad agevolare il mantenimento di un Ematocrito migliore, una buona idratazione risulta positiva sulla pressione di lavoro renale nel filtraggio del sangue. Diversi AAS come il Trenbolone e i metilati in C-17 presentano una particolare resistenza metabolica che, oltre a causare un aumentato stress epatico, può portare ad una sofferenza renale sfociabile nel patologico. Si è osservato come una combinazione di AAS, dieta iperproteica e supplementazione di Creatina possa aumentare l’incidenza di problemi renali.[5] In un soggetto in fisiologia, la sola dieta ad altro contenuto proteico e la supplementazione di Creatina non hanno mostrato nessun grado di pericolosità, soprattutto sul breve/medio termine.
#7 Non usare “droghe ricreative”
A livello globale, il numero di decessi tra gli abusatori di AAS è in aumento. Alcuni, troppo superficialmente, dicono che questo sia dovuto al fatto che sempre più uomini e donne usano AAS, ma questa è solo una spiegazione dozzinale e limitata. Il sospetto ricade soprattutto sulle modalità di approccio dei consumatori di AAS: i dosaggi sono drammaticamente aumentati e un numero crescente di individui combina PEDs con “droghe ricreative”. Ed è su questi due ultimi punti che risiede la spiegazione principale dell’aumento statistico prima menzionato. Soprattutto la combinazione di PEDs e le così dette “droghe ricreative” risulta essere probabilmente un fattore significativo, come evidenziato alcuni anni fa da ricercatori australiani. Nel loro studio sono state analizzate tutte le morti documentate tra i consumatori di AAS a Sydney tra il 1997 e il 2012, scoprendo che le droghe ricreative come la cocaina avevano avuto un ruolo nella schiacciante molteplicità dei casi. Dagli studi sugli animali ora sappiamo della possibilità che la co-assunzione di un AAS come il Nandrolone con la cocaina vede moltiplicati gli effetti cardiotossici rispetto ai singoli composti.[6] E secondo studi in vitro la combinazione di Testosterone e cocaina aumenterebbe la possibilità di formazione di coaguli nel flusso ematico. [7]
#8 Corretta modalità di iniezione e herpes labiale
Gli utilizzatori di AAS a volte sviluppano ascessi, ma non sempre dovuti alla bassa qualità dei prodotti utilizzati.
Alcuni medici ritengono che gli utilizzatori di AAS dovrebbero effettivamente ricevere una formazione sulle tecniche di iniezione corrette, onde evitare embolie oleose o ascessi per cattiva gestione igienica della procedura. [8]
Molti utilizzatori ancora non sanno che disturbi come l’herpes labiale rendono le iniezioni ancora più rischiose. Perchè? Il virus che causa l’herpes labiale, come altri patogeni, riduce l’efficienza del sistema immunitario, fornendo così terreno fertile per infezioni batteriche i cui microorganismi scatenanti vengono inoculati nel corpo del soggetto attraverso l’iniezione in modo diretto o indiretto.
#9 Non fare affidamento sugli integratori
Secondo un buon numero di studi svolti su animali, alcuni integratori proteggono dagli effetti collaterali degli AAS. Secondo alcune ricerche, la Taurina, la Vitamina C ed E proteggono i testicoli durante un ciclo e la vitamina C e il cacao proteggono la prostata.
L’utilità dei risultati provenienti da questi studi è limitata per tre motivi:
A. gli animali da laboratorio non sono esseri umani, e
B. le dosi utilizzate e rapportate ad un essere umano sono quasi sempre molto inferiori rispetto a quelle utilizzate dai “doped”, e
C. la ricerca in campo psicologico mostra che l’uso di integratori stimola comportamenti rischiosi e malsani. I supplementi fanno pensare agli utilizzatori di essere invulnerabili e di non dover comportarsi in modo sano ed attento.[9]
Gli integratori possono aiutare a creare una mentalità che non si dovrebbe avere da utilizzatore consapevole di AAS.
Ovviamente, alcuni supplementi “protettivi” utilizzati dai soggetti meglio informati hanno un potenziale di “tamponare” in modo discreto alcune alterazioni legate all’uso di AAS e SARM come, ma non limitato a, Riso Rosso fermantato (controllo lipidico) [10], Silimarina (epatoprotezione), NAC (epatoprotezione) [11], Niacina (controllo lipidico) ecc…
#10 Ridurre al minimo (se non eliminare) il consumo di alcolici
Potrebbe sembrare un indicazione superflua ma non lo è.
L’abuso di alcol è indubbiamente uno dei problemi sociali più diffusi. Uno dei problemi correlati all’abuso di alcol e l’epatopatia alcolica. Questo stato patologico è derivante da un processo infiammatorio progressivo ai danni del fegato legato al consumo eccessivo di alcolici. È una malattia a più stadi. La steatosi provoca un ingrossamento del fegato causato da un accumulo di trigliceridi, spesso senza sintomi per molto tempo. I rischi correlati sono la steatosi (fegato grasso), l’epatite alcolica e la cirrosi epatica. Il rapporto con l’alcolismo è complesso. Non tutti i bevitori, infatti, hanno danni al fegato, anche se sono altamente probabili. La causa è da rinvenire in una trasformazione dell’alcol (etanolo) in sostanze tossiche che danneggiano il fegato in maniera irreversibile e cronica, con un rischio elevato di insufficienza epatica e di cancro, fino alla necessità di un trapianto di fegato.
In acuto, invece, l’alcol può essere una causa di alterazione delle transaminasi ma non si può sapere se e con quale modalità si potrebbero innalzare: dipende molto dalla risposta individuale dell’organismo. In caso di stress preesistente, di causa iatrogena e/o alimentare, si può presentare una alterazione significativa. [12]
Il primo caso è una consequenziale possibile se eventi stressori concomitanti si presentano in cronico. Ed è semplice giungere alla conclusione che l’uso di AAS, specie se metilati, possa comportare un aumentato stress epatico che potrebbe degenerare in peliosi epatica, cirrosi ecc…
Che siate “doped” o “natural”, per ragioni legate e non, dovreste evitare di consumare più di 25g per gli uomini, o 12,5g per le donne, di Etanolo al giorno.
#11 Sottoporsi a regolari controlli medici pre, intra e post utilizzo
Il monitoraggio della salute dovrebbe essere la base fondante del comportamento del utilizzatore consapevole e minimamente attento ai potenziali rischi nei quali potrebbe imbattersi.
Gli esami di controllo sono i seguenti:
Esami ematici e delle urine (comprendenti il quadro ormonale secondo necessità);
Elettrocardiogramma ogni 6 mesi circa;
Elettrocardiogramma sotto sforzo (prima di iniziare);
Ecocardiogramma ogni 6 mesi circa;
Coronarografia ogni 6 mesi circa;
Monitoraggio della pressione ematica;
TAC addome completa ogni 6 mesi circa.
Ovviamente, ogni accertamento , al di la degli esami ematici, deve essere gestito in base alle esigenze soggettive, caratteristiche e tipo di PEDs utilizzati.
#12 Essere seguiti da personale qualificato
Fin troppa gente è stata salutisticamente deturpata da gorilla di spogliatoio a mala pena consapevoli dell’esistenza dei macronutrienti e che, nonostante ciò, si sono improvvisati farmacisti. Donne divenuti uomini e uomini divenuti simili a cagne in calore per via di orrende ginecomastie. Evitate il fai da te e l’affidarsi a semianalfabeti … la somaticità sopra la norma è cosa diversa dall’intelligenza e alla competenza in biologia, biochimica e farmacologia… senza offesa per tutti quelli che “io mi facevo e ho vinto! Senzia scienzia!” …
#13 Pensare seriamente al post ciclo prima del ciclo
Molti aspiranti “doped” non considerano il fattore post ciclo. La maggior parte di loro è convinta che la PCT sarà una facile soluzione alla sottoregolazione dell’Asse HPT, ma in realtà non è proprio così. Esistono diversi casi studio che mostrano come gli ex utilizzatori abbiano spesso livelli di Testosterone inferiori rispetto al pre-utilizzo anche a distanza di anni dal cessato uso di AAS. Sembra che i fattori che aumentano le possibilità e il grado di tale effetto sul lungo termine siano:
Tempo di somministrazione;
Età
Molecole utilizzate (con maggiore impatto negativo dato dai19-norsteroidi come il Nandrolone per via della lunga permanenza dei metaboliti nel sistema).
Tutto ciò è indipendente dalla qualità della PCT, anche se essa può avere dei riscontri positivi specie nel primo periodo di stacco dagli AAS. Le alterazioni ormonali legate ad una alterazione dell’Asse HPT comprendono depressione, ansia, bassa libido, difficoltà nel raggiungere e mantenere l’erezione, stanchezza cronica ecc…
Per questa ragione molti scelgono di entrare in TRT (Terapia Sostitutiva del Testosterone) dopo il primo ciclo.
Quale conclusione?…
Se mai non dovesse bastare il disclaimer, questo articolo non rappresenta in alcun modo un consiglio e, ne tanto meno, un incitamento all’uso di sostanze dopanti! E’ semplicemente a fine divulgativo con l’obbiettivo di far comprendere a più persone possibili che la scelta di intraprendere coscientemente certe pratiche (illegali) necessita di una sufficiente (e veritiera) conoscenza del argomento.
Quindi? Leggete e comprendete correttamente ciò che ho riportato in sintesi fruibile ad un largo pubblico… Pensate prima di tutto ad alimentarvi e allenarvi in modo ottimale!
La conoscenza della Verità rende liberi dalla cattiva informazione, dagli strumenti commerciali e dal relativismo… Negarla è semplice e pericolosa manifestazione di profonda ignoranza… di VERO NEGAZIONISMO!
Se avete una buona conoscenza della lingua inglese e volete approfondire l’argomento PEDs e Sport, potete leggere il libro ANABOLICS 11th Edition di William Llewellyn
“Per la prima volta, le persone possono ottenere attraverso i farmaci ciò che era possibile solo attraverso un intervento chirurgico per dimagrire “, è l’affermazione sensazionalistica estratta da un comunicato stampa della endocrinologa e professoressa britannica Rachel Batterham.[1] Il farmaco di cui parla la Batterham è il Semaglutide. In uno studio pubblicato sul New England Journal of Medicine, è stato riportato che i soggetti obesi hanno perso in media 15 libbre (circa 6,80Kg) quando trattati con Semaglutide.[2]
Semaglutide? Una spiegazione d’obbligo…
Semaglutide
Il Semaglutide, venduto con i marchi Ozempic e Rybelsus, è un farmaco antidiabetico utilizzato nello specifico per il trattamento del diabete di tipo 2. [3][4]
Il Semaglutide agisce come il peptide Glucagone simile-1 umano (GLP-1) in modo che aumenti la secrezione di Insulina, migliorando di conseguenza il metabolismo glucidico cellulare. È distribuito sotto forma di soluzione iniettabile da praticarsi sottocute all’interno di una penna predosata. Uno dei suoi vantaggi rispetto ad altri farmaci antidiabetici è che ha una lunga durata d’azione, quindi è sufficiente solo un’iniezione una volta alla settimana. [5]
Una versione iniettabile (Ozempic) è stata approvata per uso medico negli Stati Uniti nel dicembre 2017,[6] e nell’Unione Europea,[7] Canada,[8] e Giappone nel 2018. Una versione che viene assunta oralmente (Rybelsus) è stata approvata per uso medico negli Stati Uniti nel settembre 2019,[9] e nell’Unione Europea nell’aprile 2020. [10] E’ il primo trattamento a base di una proteina con affinità per il recettore del peptide glucagone simile 1 approvato per l’uso negli Stati Uniti che non ha bisogno di essere iniettato, ed è stato prodotto dalla Novo Nordisk.
Il Semaglutide è chimicamente simile al GLP-1 umano, con una somiglianza del 94%. Le uniche differenze sono due sostituzioni di amminoacidi nelle posizioni 8 e 34, dove l’Alanina e la Lisina sono sostituiti rispettivamente dall’acido 2-amminoisobutirrico e dall’Arginina. [11] La sostituzione degli amminoacidi nella posizione 8 previene la rottura chimica da parte di un enzima dipeptidile peptidasi-4. Inoltre, la Lisina in posizione 26 è nella sua forma derivata (acilata con diacid stearico). L’acilazione con un distanziale e una catena di diacidi grassi C-18 aumenta il legame del farmaco con la proteina del sangue albumina, il che consente una vita attiva più lunga della molecola. La sua emivita è di circa 7 giorni (165-184 ore), quindi è sufficiente un’iniezione una volta alla settimana. [5][12]
Come precedentemente accennato, il Semaglutide è un agonista del recettore del peptide Glucagone simile-1. Aumenta la sintesi di Insulina, che ovviamente abbassa il livello della glicemia ematica. [13] Sembra anche aumentare la crescita delle cellule β nel pancreas, che sono i siti di sintesi del Insulina. [12] Dall’altra parte inibisce il Glucagone, ormone con effetto iperglicemizzante. Inoltre riduce l’assunzione di cibo abbassando l’appetito e rallentando la digestione gastrica. [14] In questo modo può agire come agente per la riduzione del grasso corporeo. [15]
In sintesi, nel corpo, il GLP-1 viene rilasciato quando il cibo raggiunge la fine dell’intestino tenue e i livelli di glucosio aumentano. Nel cervello il GLP-1 inibisce l’appetito, e nel pancreas stimola il rilascio di Insulina. Il GLP-1 somministrato per via esogena viene rapidamente eliminato dal corpo, ma il Semaglutide presentando le prima citate modifiche strutturali, rimane in circolo per un periodo di tempo prolungato. Gli enzimi che scompongono il GLP-1 in singoli amminoacidi hanno difficoltà nella lisi del Semaglutide.
Studio:
Tornando a parlare nello specifico dello studio citato al principio di questo articolo, la Batterham ei suoi colleghi hanno reclutato 1.961 persone di età superiore ai 18 anni con un BMI ≥30 sottoponendole al loro studio per 68 settimane. Una parte dei soggetti è stata trattata con un placebo, mentre un’altra parte di essi ha ricevuto una iniezione settimanale di Semaglutide.
La dose iniziale per iniezione era di 0,25mg di Semaglutide, ma la dose è aumentata gradualmente fino a 2,4mg nelle prime 16 settimane dello studio. Alcuni soggetti non sono stati in grado di tollerare questa dose di 2,4mg e hanno continuato a usarne una quantità inferiore.
I ricercatori hanno utilizzato un prodotto Novo-Nordisk. Come sappiamo ormai, la Novo-Nordisk produce Semaglutide sia in forma orale che iniettabile. Diabetologi ed endocrinologi somministrano la molecola in questione a una dose inferiore alle persone con diabete di tipo 2.
Nota: lo studio potrebbe godere di credibilità limitata visto che è statpo sponsorizzato dalla Novo-Nordisk.
Risultati dello studio:
Il soggetto medio del gruppo sperimentale ha perso poco più di 15 libbre (circa 6.80Kg). Ciò è principalmente dovuto al fatto che il Semiglutide riduce l’appetito.
La somministrazione di Semaglutide ha anche comportato una certa perdita di massa corporea magra (questa legata alla mancanza di una adeguata routine allenante contro-resistenza). Tuttavia, poiché i soggetti hanno perso significativamente più massa grassa rispetto alla massa corporea magra, il Semaglutide ha migliorato la composizione corporea anche grazie al miglioramento della ripartizione calorica (vedi anche miglioramento del insulino-sensibilità che ne è alla base). I soggetti nel gruppo sperimentale hanno sperimentato una significativa riduzione del girovita. Inoltre, il loro equilibrio del colesterolo è ovviamente migliorato. Infine, i soggetti hanno anche segnalato un aumento della loro qualità di vita.
Effetti collaterali riscontrati con il Semaglutide:
Una piccola percentuale dei soggetti nel gruppo sperimentale si è ritirata a causa di effetti collaterali come nausea, costipazione, diarrea e vomito. Questo non è sorprendente, perché gli analoghi del GLP-1 rallentano la funzione intestinale. Secondo lo studio del NEJM, il Semaglutide è relativamente sicuro.
Bisognerebbe anche tenere presente che il Semaglutide è un nuovo farmaco, i cui effetti collaterali non sono stati ancora del tutto identificati. Sappiamo di altri agonisti del GLP-1 più vecchi, che potrebbero avere effetti collaterali che gli utilizzatori dovrebbero prendere seriamente in considerazione. Ci sono alcune indicazioni che i vecchi analoghi del GLP-1 possono danneggiare i reni in alcuni soggetti. [16] Inoltre, la FDA statunitense è a conoscenza di alcuni casi in cui gli utilizzatori di analoghi del GLP-1 hanno sviluppato un’infiammazione del pancreas. Alcuni di questi casi sono stati fatali.[17]
Breve conclusione sul Semaglutide nel BodyBuilding:
Molti di voi si staranno chiedendo come questo farmaco possa (e in quale circostanza) essere utilizzato per migliorare la composizione corporea. A riguardo, tenendo in considerazione quanto pocanzi esposto, si può essere spinti a pensare che tale peptide possa trovare uso funzionale in fasi ipercaloriche permettendo il mantenimento di un miglior partizionamento calorico anche se, a dire il vero, la soppressione dell’appetito e il rallentamento dello svuotamento gastrico non sono le migliori condizioni da avere in un momento come il “Bulk”. Potrebbe quindi essere funzionale in una leggera ipocalorica o isocalorica seguente una fase “Bulk” con finalità di miglioramento marcato della sensibilità insulinica, della ripartizione calorica ed un effetto sulla soppressione dell’appetito che può tornare utile in tali contesti.
In conclusione? Lasciatelo (almeno per il momento) ai diabetici e obesi…
In rete sono consultabili articoli a tema DNP, scritti da improvvisati biochimici o da qualche “guru” del settore Fitness e BodyBuilding, nei quali si espongono “consigli” sulla riduzione degli effetti collaterali legati all’uso di questo disaccoppiante della fosforilazione ossidativa. Alcuni di questi sono incentrati sulla caratteristica del DNP di aumentare massivamente i radicali liberi. Per ovviare a ciò, si riportano mix integrativi caratterizzati da antiossidanti come la Vitamina C, il NAC o direttamente il Glutatione. Ma tale pratica svolge un reale effetto riduttivo sui potenziali danni di una concentrazione elevata di radicali liberi DNP dipendente? Se si legge lo studio svolto su animali che i biologi finlandesi hanno pubblicato su “Comparative Biochemistry and Physiology Part C”, la risposta a questa specifica domanda potrebbe essere negativa o, comunque, l’effetto potrebbe essere poco rilevante nel contesto del tentativo di ridurre gli effetti collaterali del nitrocomposto in questione.[1] Lo so, stiamo parlando di uno studio su animali, ma ci offre comunque una visione d’insieme abbastanza concreta per porre ipotesi sull’efficacia di questa supplementazione sull’impatto negativo correlato all’uso di DNP.
2,4-dinitrofenolo
Dettagli dello studio:
Antoine Stier dell’Università di Turku in Finlandia ha svolto il suo esperimento utilizzando i fringuelli zebra. Gli animali sono stati divisi in due gruppi: al primo era stata somministrata acqua potabile per 4 anni mentre all’altro gruppo ha ricevuto acqua potabile contenente DNP. Ai fringuelli sono stati somministrati 4mg di DNP per chilo di peso corporeo al giorno. Se il dosaggio usato nei fringuelli fosse stato trasposto agli esseri umani, essi avrebbero ricevuto solo una frazione di quella dose. Il metabolismo dei fringuelli zebra è molto più elevato di quello dell’uomo.
Fringuelli zebra
Risultati dello studio:
I fringuelli trattati con il DNP, ovviamente, risposero alla somministrazione con un calo del peso e della massa grassa. Allo stesso tempo, i fringuelli del gruppo DNP vivevano il 21% in meno rispetto ai fringuelli del gruppo di controllo [solo acqua potabile].
Di per sé, la durata della vita più breve dei fringuelli zebra trattati con DNP non sorprende. Il DNP è un dissipatore di ATP sotto forma di calore, il che causa una produzione massiva di radicali liberi nei mitocondri. Da qui il consiglio di assumere quantità significative di antiossidanti con il DNP . Ma, stranamente, la concentrazione del perossido di idrogeno (uno dei due radicali liberi più conosciuti a contenuto d’ossigeno [ROS da Reacting Oxygen Species]) non è aumentata nei fringuelli zebra del gruppo DNP.
Conclusione:
Lo studio evidenzia che, anche a una dose moderata […], un trattamento cronico con DNP può abbreviare la durata della vita. Il DNP promuove il flusso di protoni non solo attraverso la membrana mitocondriale, ma anche attraverso la membrana plasmatica. Questo potrebbe essere un elemento chiave che spiega l’impatto negativo del DNP sulla durata della vita, ma potrebbe essere potenzialmente risolto utilizzando disaccoppiatori di prossima generazione (ad esempio BAM15) specifici per la membrana mitocondriale. Ulteriori studi che indagano i percorsi molecolari e fisiologici attraverso i quali il DNP riduce la durata della vita nei fringuelli zebra sarebbero utili per consentire indagini mirate di effetti deleteri subletali in altri modelli animali e potenzialmente nell’uomo. Il presente studio dovrebbe essere un potenziale segnale di avvertimento per gli attuali utilizzatori di DNP e sollevare domande per gli scienziati che indagano sull’uso del DNP come medicinale.
Quindi è inutile una supplementazione a base di antiossidanti per attenuare gli effetti collaterali cellulari legati alla somministrazione di DNP? Beh, dai risultati sembrerebbe che la questione radicali liberi sia addirittura molto secondaria nella lista degli effetti collaterali DNP-dipendenti ma, ed è da tenere in considerazione, parliamo di uno studio effettuato utilizzando una specie con caratteristiche nettamente differenti da quella umana e, oltretutto, lo studio è stato svolto con dosaggi di DNP somministrati in cronico (4 anni). Ovviamente, gli utilizzatori di DNP usano tale composto solo per brevi periodi di tempo; parliamo di una media di 2 settimane consecutive. Detto ciò, l’assunzione di antiossidanti non è da considerarsi (al momento) del tutto inutile visto il tasso di stressor aggiuntivi ai quali il soggetto in trattamento con DNP viene sottoposto.
Nota:l’effetto collaterale predominante e incisivo (non che letale) del DNP rimane lo shock termico da dose letale la quale è molto vicina alla dose comunemente utilizzata a scopi termogenici.
Questione ormai conosciuta è l’effetto delle proteine sulla sazietà percepita. E’ infatti risaputo che, dopo il consumo di proteine (ma anche di grassi) nel duodeno e nella prima parte dell’intestino si libera un ormone, la Colecistochinina (CCK), che segnala al cervello di smettere di mangiare. Inoltre, nell’intestino e nel colon, le proteine ingerite determinano la produzione di un altro ormone saziante, il PYY. Il suo livello si alza dopo 1-2 ore dal pasto e rimane alto per circa 6 ore, limitando così l’insorgenza dell’appetito in questo periodo di tempo. Quindi, le proteine, da un lato, grazie al CCK e all’effetto del PYY, favoriscono la sazietà. Altrettanto conosciuto è il maggiore effetto sulla TID (Termogenesi Indotta dalla Dieta) dato dall’azione dinamica specifica delle proteine le quali, con variabili date dalla fonte, per essere digerite ed assimilate richiedono un dispendio energetico tra il 10 ed il 35% (media del 22,5%).
Da quanto riportato in uno studio pubblicato sette anni fa (2013) sul Journal of Nutrition [1], sembrerebbe che l’aggiunta di Capsaicina, uno degli alcaloidi responsabili della maggior parte della “piccantezza” dei peperoncini, ad una dieta con alto apporto proteico possa migliorarne l’effetto, migliorando ulteriormente il tempo e grado di efficacia di una dieta ipocalorica.
Dettagli dello studio
I ricercatori che hanno svolto lo studio in questione, hanno reclutato 28 soggetti sani facendoli permanere per otto periodi di 24 ore in una camera di respirazione, dove potevano essere osservati e misurate le quantità esatte di calorie ossidate dagli individui in osservazione. In ogni occasione i soggetti hanno ricevuto pasti diversi.
Ai soggetti è stato somministrato il 100% della quantità calorica ossidata giornalmente. Il 10% della quota calorica dei pasti proveniva dalle proteine.
Ai soggetti è stato somministrato l’80% della quantità calorica ossidata giornalmente. Il 10% della quota calorica dei pasti proveniva dalle proteine.
Ai soggetti è stato somministrato il 100% della quantità calorica ossidata giornalmente. Il 10% della quota calorica dei pasti proveniva dalle proteine. Inoltre ai soggetti sono state somministrate 2 capsule contenenti 40.000 unità di calore Scoville dal pepe ad ogni pasto (Capsaicina).
La sostanza bioattiva più importante del pepe è la Capsaicina. I nutrizionisti hanno utilizzato un prodotto realizzato dalla Solaray. [solarayuk.co.uk]
Ai soggetti è stato somministrato l’80% della quantità calorica ossidata giornalmente. Il 10% della quota calorica dei pasti proveniva dalle proteine. Inoltre, i soggetti hanno assunto 2 capsule contenenti 40.000 unità di calore Scoville dal pepe ad ogni pasto.
Ai soggetti è stato somministrato il 100% della quantità calorica ossidata giornalmente. Il 25% dell’introito calorico dei pasti proveniva dalle proteine. I ricercatori hanno ridotto la quantità di carboidrati sostituendola con proteine.
Ai soggetti è stato somministrato l’80% della quantità calorica ossidata giornalmente. Il 25% dell’introito calorico dei pasti proveniva dalle proteine.
Ai soggetti è stato somministrato il 100% della quantità calorica ossidata giornalmente. Il 25% dell’introito calorici dei pasti proveniva dalle proteine. Inoltre i soggetti hanno assunto 2 capsule contenenti 40.000 unità di calore Scoville dal pepe ad ogni pasto (Capsaicina).
Ai soggetti è stato somministrato l’80% della quantità calorica ossidata giornalmente. Il 25% dell’introito calorico dei pasti proveniva dalle proteine. Inoltre, i soggetti hanno assunto 2 capsule contenenti 40.000 unità di calore Scoville dal pepe ad ogni pasto (Capsaicina).
Risultato dello studio
Come mostrato nella figura seguente, il ridotto apporto calorico ha portato ad una riduzione del 5% del dispendio energetico dei soggetti osservati. La riduzione del dispendio energetico a seguito della riduzione dell’apporto calorico non si è verificata quando i soggetti assumevano la Capsaicina o aumentavano il loro apporto proteico. La combinazione di una dieta ricca di proteine con la componente supplementare di Capsaicina ha portato a migliori risultati.
Colonne chiare: apporto calorico = 100% delle calorie ossidate; Colonne scure = apporto calorico = 80% delle calorie ossidate .
La Capsaicina integrativa di per se ha mostrato effetti anoressizzanti causando, consequenzialmente, una migliore compliance del protocollo alimentare. I ricercatori hanno riportato risultati migliori nei soggetti che seguivano una dieta ricca di proteine combinata con l’integrazione di Capsaicina. Indi, si è verificato un effetto additivo sulla soppressione della fame.
Sembrerebbe, quindi, che una combinazione di Capsaicina con la sostituzione di una parte dell’introito calorico giornaliero proveniente dai Carboidrati con Proteine, ed un apporto calorico pari al 20% in meno del totale di mantenimento, possa portare ad un maggiore dispendio energetico e sazietà rispetto a una dieta di controllo del solo bilancio energetico.
In conclusione, i ricercatori sottolineano che l’efficacia della combinazione di Capsaicina e Proteine dovrebbe essere ulteriormente valutata in studi sulla perdita di peso ben progettati su individui in sovrappeso e obesi.
Nota: è utile aggiungere che se si è intenzionati a testare l’effetto della Capsaicina sulla perdita di peso, bisogna tenere in considerazione la tollerabilità individuale alla molecola. Infatti, alle dosi di Capsaicina utilizzate dai ricercatori, specie sul lungo termine, possono causare problemi gastrointestinali (stomaco e tratto digerente).[2]
Sono ormai diversi anni che in nutrizione si discute della questione “dolcificanti artificiali” e se essi siano o meno deleteri nel contesto dell’alimentazione umana. Molti studi hanno “assolto” dalla loro presunta pericolosità dolcificati ipocalorici molto diffusi come l’Aspartame, con le corrette modalità d’uso ovviamente (vedi dosaggio totale giornaliero). Mentre altri dolcificanti artificiali sono decisamente posizionati nella “zona grigia”, come l’Acesulfame-K. Il peggiore, secondo quanto emerso dalle ultime ricerche, sembrerebbe essere il Sucralosio. Il Sucralosio, un dolcificante sintetico mille volte più dolce dello zucchero da cucina (Saccarosio), sembra che possa causare sintomi pre-diabetici nelle persone sane. I ricercatori dell’Università di Yale hanno riportato della comparsa di questi sintomi in un articolo comparso recentemente su “Cell Metabolism”.[1] Sebbene i soggetti dello studio non fossero effettivamente patologici, i risultati sono stati così preoccupanti che l’università ha consigliato ai ricercatori di interrompere lo studio.
Caratteristiche del Sucralosio:
La maggior parte del Sucralosio (E-955) ingerito non viene enzimaticamente scomposto, quindi non apporta calorie. [2] È prodotto dalla clorurazione del saccarosio. Il Sucralosio è da 320 a 1.000 volte più dolce del Saccarosio [3], tre volte più dolce dell’Aspartame e dell’Acesulfame-K, e due volte più dolce della Saccarina Sodica.
Sebbene il Sucralosio è ampiamente considerato stabile e sicuro per l’uso a temperature elevate (come nei prodotti da forno), ci sono alcune prove che mostrano un iniziale degradazione a temperature superiori a 119 gradi Celsius. [4][5] Il successo commerciale dei prodotti a base di Sucralosio deriva semplicemente dal confronto favorevole con altri dolcificanti ipocalorici in termini di gusto, stabilità e sicurezza nelle prima citate circostanze.[6]
Lo studio in questione e risultati emersi…
Per lo svolgimento dello studio che qui andiamo trattando, i ricercatori hanno diviso 45 soggetti sani in tre gruppi. Ogni gruppo si recava al laboratorio di controllo sette volte durante un periodo di due settimane. Li, ai soggetti veniva data una bevanda analcolica da 355ml.
Il contenuto della suddetta bevanda differiva nei tre gruppi esaminati come segue:
Contenuto 1° gruppo [LCS]: 60mg di Sucralosio;
Contenuto 2° gruppo [Sugar]: 30g di Saccarosio [normale zucchero da tavola];
Contenuto 3° gruppo [Combi]: 60mg di Sucralosio + 31g di Maltodestrine.
Come già accennato, la struttura chimica del Sucralosio è molto simile a quella del Saccarosio. In tre punti, tuttavia, il Sucralosio presenta gruppi cloro che mancano nel Saccarosio. A causa di questi gruppi cloro, secondo alcuni studi, il Sucralosio è mille volte più dolce del Saccarosio.
Le bevande analcoliche assunte dai partecipanti dei gruppi 1 e 2 non hanno avuto alcun effetto sulla farmacocinetica del glucosio negli individui esaminati. Quando i ricercatori hanno somministrato a questi soggetti un lotto di glucosio dopo 2 settimane, la glicemia ematica si è ridotta con la stessa velocità osservata prima del periodo di due settimane dello studio. A questo proposito, le bevande analcoliche erano sicure.
Il quadro è cambiato quando i ricercatori hanno esaminato la quantità di insulina che era presente nel sangue dei soggetti dopo la somministrazione del glucosio. Questa quantità era significativamente maggiore nei soggetti che avevano ricevuto bevande analcoliche contenenti Maltodestrine più Sucralosio.
Ciò implica che la combinazione di Sucralosio con un carboidrato ad assorbimento altera il metabolismo glucidico peggiorando, sebbene in acuto, la sensibilità all’Insulina.
I ricercatori hanno anche osservato che in un certo numero di soggetti, la combinazione di Sucralosio e un carboidrato a rapido assorbimento portava ad un aumento dell’Insulina basale, misurata al mattino prima che i soggetti consumassero il loro primo pasto della giornata. Ciò suggerisce anche una possibile ridotta sensibilità all’insulina in cronico.
In bocca, nell’intestino e in altri punti del corpo, i dolcificanti come il Sucralosio interagiscono con i recettori del dolce T1R2 / T1R3. Questi recettori sono in realtà destinati al glucosio e ad altri zuccheri naturali. Regolano l’assorbimento degli zuccheri da parte dell’intestino tenue.
I ricercatori ipotizzano che, tramite questi recettori, il Sucralosio possa indurre il corpo ad assorbire rapidamente i carboidrati assimilandoli ancora più velocemente, interrompendo l’equilibrio tra glucosio e insulina e riducendo la sensibilità all’insulina non solo in acuto ma anche, potenzialmente, in cronico.
I ricercatori hanno scritto che questi risultati suggeriscono che il consumo di Sucralosio altera il metabolismo del glucosio consumato simultaneamente producendo rapidamente effetti deleteri sulla salute metabolica.
Durate di esposizione simili quasi certamente si verificano negli esseri umani nella quotidianità, soprattutto se si considera il consumo di una bevanda dietetica insieme ad un pasto. Ciò solleva la possibilità che l’effetto combinato possa essere un importante contributo all’aumento dell’incidenza del diabete di tipo 2 e l’obesità, in senso indiretto o induttivo.
In tal caso, l’aggiunta di dolcificanti a basso contenuto calorico per aumentare la dolcezza di cibi e bevande già contenenti carboidrati dovrebbe essere scoraggiato e il consumo di bevande dietetiche durante i pasti dovrebbe essere sconsigliato.
Nota:Il Sucralosio risulta particolarmente deleterio anche sul microbiota intestinale. Il primo studio che ha valutato il Sucralosio sul microbiota intestinale è stato eseguito nel 2008 con l’uso di campioni fecali di ratti Sprague-Dawley che hanno ricevuto il dolcificante per 12 settimane. Il consumo di Sucralosio ha ridotto il numero totale di batteri anaerobici e aerobici, bifidobatteri, lattobacilli, Bacteroides e Clostridium.(7) La somministrazione di 15mg di Sucralosio/kg ha influenzato l’abbondanza relativa del Clostridium cluster XIVa nei topi.(8) Più recentemente, la somministrazione di Sucralosio nei topi ha prodotto modifiche nel microbiota intestinale a 14 diversi livelli tassonomici, tra cui Turicibacteraceae, Lachnospiraceae, Ruminococcaceae, Verrucomicrobiaceae, Staphylococcaceae, Streptococcaceae, Dehalobacteriaceae, Dehalobacterium, Lachnospiraceae, Lachnospiraceae ordine Bacillales e cambiamenti nella sintesi e regolazione degli amminoacidi. Queste variazioni erano correlate all’infiammazione nell’ospite.(9)
Nonostante lo studio sia di piccole dimensioni e non sia controllato (non vi è sicurezza nel comportamento alimentare seguito dai soggetti esaminati al di fuori di quanto emergesse durante i controlli), esso rappresenta un forte incentivo verso la ricerca sugli effettivi vantaggi e svantaggi del consumo di dolcificanti in soggetti sani e non.
Nota:Mancano ad oggi prove di un possibile beneficio per la perdita di peso a lungo termine con alcuni dati che supporto il rischio di un aumento di peso e di sviluppo di malattie cardiache con l’uso di questo dolcificante.[10]
Splenda alters gut microflora and increases intestinal p-glycoprotein and cytochrome p-450 in male rats.Abou-Donia MB, El-Masry EM, Abdel-Rahman AA, McLendon RE, Schiffman SSJ Toxicol Environ Health A. 2008; 71(21):1415-29.
Effects of Consuming Xylitol on Gut Microbiota and Lipid Metabolism in Mice.Uebanso T, Kano S, Yoshimoto A, Naito C, Shimohata T, Mawatari K, Takahashi ANutrients. 2017 Jul 14; 9(7):.
Gut Microbiome Response to Sucralose and Its Potential Role in Inducing Liver Inflammation in Mice.Bian X, Chi L, Gao B, Tu P, Ru H, Lu KFront Physiol. 2017; 8():487.
La Niacina è largamente utilizzata dagli atleti supplementati chimicamente, in special modo da coloro i quali usano molecole con un potenziale negativo marcato sui lipidi ematici. Ma come spesso capita, gli utilizzatori non conoscono a sufficienza le caratteristiche di ciò che assumono, e questa essenziale vitamina del gruppo B (B3) non è da meno. Per la maggior parte degli individui tanto basta sapere che una sua integrazione si traduce in livelli migliorati di Colesterolo e Trigliceridi. Purtroppo, però, si trascurano caratteristiche importanti la cui conoscenza può fare la differenza tra un uso più o meno funzionale per la salute sistemica. Infatti, un effetto collaterale dell’integrazione di Niacina è un peggioramento della resistenza all’insulina, cosa che limita i benefici di tale supplementazione sulla salute cardiovascolare se non vengono prese adeguate precauzioni.
Prima di correre a defenestrare in preda al panico la vostra Niacina, leggete con attenzione (e comprendete) le informazioni che seguono…
Introduzione alla Niacina (vitamina B3)
Niacina
La Niacina, nota anche come Acido Nicotinico, è un composto organico e una forma di vitamina B3, un micronutriente essenziale per l’essere umano. [1] La Niacina ha formula bruta C6H5NO2 e appartiene al gruppo dell’acido piridinecarbossilico.[1] Come precursore di NAD e NADP, la Niacina è coinvolta nella riparazione del DNA.[2] La Niacina viene assunta attraverso la dieta da una varietà di alimenti interi e trasformati, con il più alto contenuto in alimenti confezionati fortificati, carne, pollame, pesce rosso come tonno e salmone, con minori quantità nelle noci, legumi e semi. [1] [3] La Niacina come integratore alimentare viene anche utilizzata per trattare la pellagra, una malattia causata da una sua carenza. Segni e sintomi includono lesioni della pelle e della bocca, anemia, mal di testa e stanchezza.[4] Molti paesi richiedono la sua aggiunta alla farina di grano o ad altri cereali, riducendo così il rischio di pellagra.[1][5] Come vitamina, le raccomandazioni di dosaggio giornaliero indicate in diversi paesi sono 14-18mg/die per gli adulti, quota sufficiente per soddisfare le esigenze delle persone sane. [6] [7] [8]
Sebbene la Niacina e la Nicotinamide (Niacinamide) siano identiche nella loro attività vitaminica, la Nicotinamide non ha gli stessi effetti farmacologici, modificanti i lipidi o gli effetti collaterali della Niacina, cioè quando la Niacina assume il gruppo -amide, non riduce il Colesterolo né causa vampate di calore.[9][10] La Nicotinamide è raccomandata come trattamento per la carenza di Niacina poiché può essere somministrata in quantità correttive senza causare l’effetto negativo del rossore.[11]
La Niacina è anche un farmaco di prescrizione. Quantità molto superiori all’assunzione dietetica raccomandata per le funzioni vitaminiche ridurranno i Trigliceridi nel sangue e le lipoproteine a bassa densità (LDL-C) e aumenteranno le lipoproteine ad alta densità (HDL-C). Ne esistono due forme: Niacina a rilascio immediato e a rilascio prolungato. Le quantità iniziali di prescrizione sono di 500mg/die, con possibilità di essere aumentate nel tempo fino a raggiungere l’effetto terapeutico ricercato. Le dosi a rilascio immediato possono arrivare fino a 3g/die; quelle a rilascio prolungato fino a 2g/die. [12] Nonostante i comprovati cambiamenti lipidici, la Niacina non è stata trovata utile per ridurre il rischio di malattie cardiovascolari nei soggetti già in trattamento con statine. [13] Una review del 2010 aveva concluso che l’efficacia della Niacina si osservava in mono-terapia, [14] ma una review del 2017 che incorporava il doppio del numero degli studi ha concluso che la Niacina su prescrizione, pur influenzando i livelli lipidici, non riduceva la mortalità per tutte le cause, la mortalità cardiovascolare, gli infarti del miocardio, né ictus fatali o non fatali. [15] È stato dimostrato che la Niacina da prescrizione provoca epatotossicità [16] e aumenta il rischio di diabete di tipo 2. [17] [18] Le prescrizioni di Niacina negli Stati Uniti avevano raggiunto il picco nel 2009, a 9,4 milioni, in calo a 1,3 milioni entro il 2017.[19]
Niacina, flusso ematico, pressione e vasodilatazione
Uno studio sulla supplementazione di Niacina che ha valutato il flusso sanguigno dell’avambraccio non è riuscito a trovare un effetto significativo fino a 1g al giorno somministrati nel corso di due settimane in soggetti altrimenti sani, [20] e 1.5g di Niacina a rilascio prolungato negli uomini con sindrome metabolica non sono riusciti a influenzare la dilatazione flusso- mediata (FMD). [21] Un altro studio non è riuscito a trovare un effetto significativo in un intero gruppo di pazienti affetti da afta epizootica, mentre in un gruppo di pazienti con malattia coronarica ha riscontrato un miglioramento in un sottogruppo con bassi livelli HDL-C. [22]
In soggetti con bassi livelli di HDL-C, è stato osservato che 1g di Niacina a rilascio prolungato per una settimana aumenta il flusso sanguigno (via FMD) del 4,5%; questo meccanismo non era correlato alle Prostaglandini, poiché il Laropiprant (un inibitore della Prostaglandine D2) non ha influenzato l’effetto. [23] Questo effetto ha anche coinciso con un aumento della bilirubina indiretta (ma non totale) del 62%. [23] Poiché la bilirubina del acido biliare è un antiossidante endoteliale, [24] e poiché i benefici della niacina sulla funzione endoteliale in questo studio sono stati ritenuti dipendenti dall’ossido nitrico, [23] è stato ipotizzato che un effetto conservativo della bilirubina sulla biodisponibilità dell’ossido nitrico sia alla base della beneficio osservato. Sia l’aumento della bilirubina che il miglioramento del flusso sanguigno si sono dissipati una settimana dopo l’interruzione della Niacina.[23]
Laropiprant
I soggetti che in precedenza avevano subito infarto del miocardio, a seguito del trattamento con Niacina (con Laropiprant) hanno riscontrato un aumento del flusso sanguigno dipendente dall’ossido nitrico (FMD) dopo dodici settimane di terapia insieme a un miglioramento della vasodilatazione indotta da nitroglicerina, entrambe non correlate con alterazioni dei trigliceridi. [25] Miglioramenti simili nel flusso sanguigno sono stati osservati in pazienti con infezione da HIV e con bassi livelli di HDL-C trattati con la sola Niacina. [26]
Prostaglandine D2 (PGD2)
È noto che la Niacina influenza il diametro dei vasi sanguigni, in particolare per via della sua reazione vasodilatativa cutanea (allargamento dei vasi nella pelle), che ha portato a ipotizzare che potrebbe influenzare la pressione sanguigna aumentando il diametro delle arterie e vene. Tuttavia, una review [27] ha notato che un possibile effetto di riduzione della pressione arteriosa della Niacina è indipendente dalla Prostaglandine che media il rossore, nota come PGD2.
È stato osservato che le infusioni di Niacina riducono acutamente la pressione sanguigna negli ipertesi senza alcun effetto nei soggetti con pressione sanguigna normale ed è stata associata ad un aumento della gittata cardiaca e della frequenza cardiaca che era simile in entrambi i gruppi. [28] Un altro studio ha confermato questo risultato, scoprendo che la pressione arteriosa ambulatoriale di 24 ore non sembra essere influenzata da un supplemento di Niacina fino a 1g nell’arco di due settimane in soggetti altrimenti sani. [20]
In termini di effetti della Niacina in cronico sulla pressione sanguigna, una review [27] che ha valutato gli studi che hanno misurato la pressione sanguigna negli ipertesi [29] [30] [31] [32] non ha notato alcun effetto statisticamente significativo nella riduzione della pressione sanguigna associata alla supplementazione di Niacina, sebbene questi studi in quanto a metodologie di misurazione sulle variazioni della pressione sanguigna non fossero ideali secondo gli autori della review. Tuttavia, la review ha osservato che in un ampio studio (il Coronary Drug Project), che inizialmente non è riuscito a trovare alcuna influenza della terapia con Niacina sulla pressione arteriosa, [32] ha osservato variazioni sensibili soltanto sui soggetti con sindrome metabolica. Questi presentavano un lieve riduzione di 2,2mmHg della pressione arteriosa sistolica con una moderata riduzione di 2,9mmHg della pressione diastolica. [33] Un’analisi post-hoc di un altro studio clinico [34] ha rilevato che la pressione arteriosa sistolica è stata abbassata di 2,2mmHg e la pressione sistolica di 2,7 rispetto al placebo nei pazienti dislipidemici trattati per 24 settimane. [35]
Niacina, Trigliceridi, Colesterolo e Aterosclerosi
Apolipoproteina B
La Niacina sembra abbassare i trigliceridi nel sangue inibendo sia la sintesi degli acidi grassi sia la loro esterificazione epatica per formare i trigliceridi, il che aumenta il tasso di degradazione dell’apolipoproteina B riducendo la sua secrezione dalle cellule epatiche. [36] Un meccanismo con cui la Niacina fa questo è attraverso l’inibizione diretta e non competitiva della diacilglicerolo aciltransferasi 2 (DGAT2), l’enzima finale nella sintesi dei trigliceridi nelle cellule epatiche, senza inibizione della DGAT1. [37]
Si è visto che gli effetti della Niacina sulla sintesi dei trigliceridi influenzano i livelli sierici di lipoproteine a densità molto bassa (vLDL-C), dove la terapia con Niacina per 16 settimane in soggetti con malattia del fegato grasso non alcolica (NAFLD) sembra ridurre le vLDL-C nel siero così come i complessi con trigliceridi (vLDL-TG) e apolipoproteina B (vLDL-ApoB) rispetto al placebo e con una potenza paragonabile al fenofibrato. [38] La Niacina lo fa riducendo la secrezione epatica di vLDL-C, sebbene ciò non aumenti la quantità di trigliceridi nel fegato anche nello stato di NAFLD. [38]
Oltre ai suoi effetti sul fegato, la Niacina può anche sopprimere il rilascio di acidi grassi liberi dal tessuto adiposo [39] che normalmente verrebbero reesterificati come trigliceridi nel fegato e quindi secreti via vLDL. [40] Tuttavia, questo meccanismo specifico, che è mediato dal recettore HM74A, [39] non sembra essere rilevante per le proprietà riducenti dei trigliceridi della Niacina. [41]
I benefici sui livelli di trigliceridi possono verificarsi entro una settimana dall’inizio della supplementazione con Niacina a rilascio prolungato (1g), sebbene in misura minore di circa il 4%. [23]
L’integrazione di 1.5-2g di Niacina a rilascio prolungato per due anni con follow-up di un anno nelle persone in terapia con statine caratterizzate da bassi livelli di HDL-C ha mostrato una riduzione dei trigliceridi del 28,6% (statina da sola dell’8,1%). [42]
Esiste un fenomeno noto come “rimbalzo degli acidi grassi” associato alla supplementazione di Niacina, in quanto l’azione iniziale del composto sul suo recettore (HM74A) nel tessuto adiposo può determinare una minore lipolisi e una minore secrezione di acidi grassi non esterificati (NEFA) nel sangue [43] e una migliore conservazione adiposa; [44] si tratta di fenomeni prontamente reversibili in quanto in un giorno di esposizione continua vi è un aumento netto del NEFA piuttosto che la sua soppressione [45] [46] [47] e alterazioni nel NEFA possono non riflette alterazioni dei trigliceridi.
Il primo meccanismo pensato per spiegare il miglioramento del profilo sierico di colesterolo in seguito alla supplementazione di Niacina è stato attraverso la riduzione del rilascio di acidi grassi non esterificati (NEFA) dai tessuti, che non è più considerato un probabile meccanismo in quanto l’integrazione di niacina in cronico è associata ad un aumento, piuttosto che alla soppressione, di NEFA mentre il recettore HM74A appare superfluo in termini di effetti della Niacina nei topi con altri ligandi del HM74A (Acipimox [48] e MK-0354 [49]) che si sono mostrati rispettivamente meno efficaci o inefficaci sul colesterolo. Attualmente si ritiene che l’influenza della Niacina sui NEFA nel siero non sia un fattore determinante nel modo in cui influenza i livelli di colesterolo nel corpo, con le teorie attuali che ipotizzano che il fattore sia determinato dalla sua sintesi e dal suo tasso di catabolismo.
Il primo potenziale meccanismo prevede la sintesi di HDL-C nel fegato attraverso l’aumento della trascrizione del gene ABCA1 (che dipende dal legame LXRα alla regione del promotore DR4 di questo gene). [50] L’attività di ABCA1 promuove la “lipidazione” della principale proteina dell’HDL nota come apolipoproteina AI (ApoAI) aumentando il tasso che associa ai fosfolipidi e al colesterolo, [51] [52] un passaggio obbligatorio nella sintesi dell’HDL-C che è aumentato di 500-1000µM con Niacina in vitro. [50] Questo meccanismo non è stato confermato, poiché mentre l’ApoAI può essere aumentato parallelamente all’aumento dell’HDL-C in soggetti trattati con Niacina e con livelli di HDL-C bassi di base, [53] LXRα sembra richiedere un coattivatore (PPARγ) per esercitare questi effetti, [54] che è attivato dal recettore della Niacina. [55] Tuttavia, l’attività del recettore della Niacina non è stata richiesta per i suoi effetti sui livelli di colesterolo, suggerendo che altri meccanismi potrebbero essere rilevanti.
PPARγ
L’altra teoria relativa alla sintesi di HDL dalla Niacina afferma che ciò dipenda dalla proteina di trasferimento dell’estere del colesterolo (CETP) nonostante la riduzione del colesterolo totale e dei trigliceridi non richieda per entrambe questa proteina. [56] [57] CETP è una proteina che facilita il trasferimento di lipidi tra diverse lipoproteine (generalmente donando un trigliceride da vLDL a HDL e prendendo un estere di colesterolo in un processo noto come trasporto inverso di colesterolo. [58]) La Niacina riduce l’espressione di CETP nel fegato e la sua attività nel sangue dei topi; [56] una riduzione del CETP aumenta la quantità di HDL-C nel sangue poiché i tassi di catabolismo dell’HDL / LDL riflettono l’attività del trasporto inverso del colesterolo e raggiungono rapidamente l’equilibrio, [59] e se il CETP è ridotto allora sarebbe necessario più HDL per normalizzare i tassi di trasporto inverso del colesterolo. Questo meccanismo può anche essere correlato a LXRα, poiché mentre un eteromero di LXRα con il recettore nucleare di vitamina A (RXR) attiva l’elemento DR4 aumenta la CETP [60] la Niacina agevola l’eterodimerizzazione di LXRα e PPARγ che attiva ancora DR4, ma in un modo che promuove l’efflusso di colesterolo. [61-44] Questa eterodimerizzazione competitiva [62] non è stata ancora dimostrata sperimentalmente, e lo studio che ha utilizzato dosi di Niacina da 2g nell’uomo non è riuscito a trovare un’influenza sull’attività del CETP nel siero nonostante un aumento dell’HDL. [63]
L’ultimo potenziale meccanismo per l’aumento dell’HDL non consiste nel suo incremento di sintesi ma piuttosto nel preservare il colesterolo HDL già sintetizzato arricchito con apoAI, riducendo il tasso in cui la lipoproteina viene assunta nelle cellule epatiche nonostante la donazione di colesterolo dall’HDL a queste cellule sia inalterata a causa della riduzione dell’espressione del recettore (catena beta sintasi ATP) che normalmente trasporta l’HDL nella cellula. [64] Questa ipotesi funziona meglio con le osservazioni che suggeriscono che il ridotto catabolismo dell’HDL è il principale fattore determinante dei suoi livelli più elevati, [65] e influenza anche l’apoA1 poiché la sua clearance dal sangue e l’assorbimento da parte dei reni sono ridotti. [66]
Una supplementazione di Niacina a rilascio prolungato (1g) della durata di una settimana in soggetti con bassi livelli di HDL-C non sembra essere sufficiente da aumentare sensibilmente i livelli totali di HDL-C, sebbene sia stata notata una riduzione della dimensione media delle particelle; [23] le variazioni di HDL -C possono mediare un miglioramento della vasodilatazione dipendente dall’ossido nitrico, sebbene sia stato anche osservato un aumento della bilirubina indiretta. [23]
L’integrazione prolungata di Niacina nei diabetici è associata ad un aumento della quantità e delle dimensioni particellari dell’HDL-C (32,7%) mentre le particelle di dimensioni più piccole sono diminuite (8,2%). [67]
È stato osservato che la Niacina conferisce un effetto protettivo sulla mortalità cardiovascolare poiché una metanalisi [68] ha osservato che negli studi su soggetti con malattia coronarica la terapia con Niacina era associata a un minor rischio di rivascolarizzazione dell’arteria coronarica (RR di 0,31; IC al 95% di 0,15-0,63), infarto miocardico non fatale (RR di 0,72; IC al 95% di 0,60-0,86) e attacco ischemico transitorio (RR di 0,76; IC al 95% di 0,61-0,94) mentre la riduzione della mortalità complessiva non è riuscita a raggiungere significatività statistica (RR 0,883; IC 95% di 0,773-1,008). I sette studi inclusi in questa meta-analisi [32] [29] [31] [30] (e un follow-up [69]) hanno totalizzato 5137 pazienti che utilizzavano anche vari prodotti farmaceutici della classe di statine e fibrati .
In uno studio i cui partecipanti erano in terapia con statine e avevano bassi livelli di colesterolo HDL è stato rilevato che 1.5-2g di Niacina a rilascio prolungato sono stati in grado di fornire benefici additivi nel miglioramento dell’HDL-C (20%) e nella riduzione dell’LDL-C (17%) rispetto al placebo, sebbene per quanto riguarda l’endpoint clinico predeterminato (morte o ricovero in ospedale) sia la Niacina che il placebo avevano una uguale quantità di responder. [70] Questo studio ha rilevato un’alta percentuale di pazienti con sindrome metabolica (80%) e commenti [71] hanno suggerito che a causa di una possibile capacità della Niacina a rilascio prolungato di deteriorare l’insulino-resistenza [72] che i suoi benefici potrebbero essere compensati da questo effetto avverso, mentre lo studio stesso ha suggerito che i benefici delle statine hanno sostituito i benefici della Niacina.
Mentre uno studio precedente che utilizzava alte dosi di Niacina a rilascio immediato (3g) ha riscontrato una riduzione della morte del 14% rispetto al placebo insieme a una riduzioni del colesterolo totale, [32] ed è stato osservato che questa riduzione è simile per grandezza agli studi che combinano statine con placebo.
Studi in vitro suggeriscono che la Niacina potrebbe in teoria prevenire la formazione di placche aterosclerotiche riducendo l’infiammazione e il danno alla parete endoteliale attraverso diversi meccanismi. Limitate ricerche su animali hanno mostrato che la Niacina nella dieta, a concentrazioni paragonabili a quelle utilizzate per ridurre il colesterolo, riduce la deposizione della placca sulla parete dell’arteria e ritarda l’aterosclerosi.[73][74][75][76][77][78][79][80]
Niacina e sue interazioni con il metabolismo del glucosio
L’assunzione prolungata di Niacina è stata osservata causare una riduzione della sensibilità all’insulina, causando un aumento compensativo della produzione di insulina da parte delle cellule β del pancreas per mantenere i livelli di glucosio nel sangue. [81] La Niacina non sembra avere effetti diretti sulle cellule β pancreatiche, tuttavia, poiché la perfusione negli isolotti pancreatici (isole di Langerhans) di ratto isolati con Niacina in vitro non ha influenzato la secrezione di insulina. [82] Ciò indica che la Niacina aumenta la produzione di insulina mediante un meccanismo indiretto, secondario a causare insulino-resistenza periferica. È stato osservato che la supplementazione induce resistenza all’insulina a dosi comprese tra 500mg e 1g, che rientrano nell’intervallo di dosaggio che conferisce effetti di riduzione del colesterolo. [83]
In particolare, sembra che sia necessaria una supplementazione cronica di Niacina per aumentare la produzione di Insulina, poiché in uno studio è stato dimostrato che la supplementazione acuta riduce i livelli di questo peptide in soggetti altrimenti sani prima di un picco dopo un giorno, [84] mentre altri studi in acuto hanno notato un effetto minimo o nullo sui livelli di Insulina. [85] [86] [87] [88]
Gli effetti dell’integrazione cronica di Niacina sui livelli di Insulina possono anche dipendere dalla popolazione. È stato osservato che la Niacina provoca iperinsulinemia in soggetti che invecchiano altrimenti sani [83] (1g / die) ed è stato dimostrato che quasi raddoppiano i livelli di Insulina nei soggetti con NAFLD (2g / giorno [89] [90]). Nei pazienti con sindrome metabolica, l’integrazione di Niacina a 6 settimane di somministrazione alla dose di 1.5g / die ha aumentato i livelli di Insulina del 30%. [91]
Nei soggetti obesi con malattia del fegato grasso non alcolico (NAFLD), l’integrazione giornaliera di Niacina a rilascio prolungato (titolata fino a 2g) per 16 settimane sembrava aumentare lo stato di resistenza all’insulina nel fegato, nei muscoli e nel tessuto adiposo [89] con un effetto inibitorio sulle azioni dell’Insulina nel fegato notate negli uomini non diabetici con dislipidemia. [92]
Adiponectina
Negli uomini adulti con sindrome metabolica, è stato osservato che la Niacina a rilascio prolungato alla dose di 1.5g ostacola in modo significativo la sensibilità all’Insulina, valutata dall’HOMA-IR (42%), che è stata associata ad un aumento dell’Insulina sierica nonostante un aumento dell’Adiponectina sierica. [91] Questo è stato notato anche in un altro studio (aumento del 22% dell’HOMA-IR), in cui l’Aspirina assunta insieme alla Niacina non ha impedito la comparsa di una ridotta sensibilità all’Insulina. [93]
Questo effetto può persistere in soggetti altrimenti sani, poiché i soggetti trattati con 1g di Niacina per due settimane a cui veniva somministrato un clamp iperinsulinaemico-euglicemico richiedono meno glucosio per mantenere l’omeostasi, il che è indicativo di una riduzione dell’assorbimento del glucosio (attraverso un aumento dell’Insulino-resistenza). [94]
La resistenza all’Insulina indotta dalla Niacina è stata inizialmente attribuita a un effetto di rebound nel tessuto adiposo in cui un aumento del rilascio di acidi grassi non esterificati (NEFA) da parte della Niacina compromette gli effetti della segnalazione dell’Insulina. [95] [96] Ciò è plausibile, poiché la resistenza all’Insulina può essere indotta con infusione di NEFA in 24 ore nei roditori. [97] Altre fonti suggeriscono che la resistenza all’Insulina non è associata al rebound del NEFA, poiché i soggetti con NAFLD che sperimentano resistenza all’Insulina dalla terapia con Niacina non hanno necessariamente un aumento del NEFA nel siero. [89].
Modello ipotetico per i ruoli intracellulari del DGAT1 e DGAT2.
Un’altra possibile opzione è che la Niacina può inibire in modo non competitivo l’enzima noto come diacilglicerolo aciltransferasi 2 (DGAT2) con un IC50 di 100 µM (potenza simile a circa 300 µM). [98] L’inibizione di questo enzima non causa di per sé resistenza all’insulina con la somministrazione di Niacina, [92] ma poiché il DGAT catalizza il primo stadio della sintesi dei trigliceridi, la sua inibizione può favorire l’accumulo di diacilglicerolo (DAG) che è la molecola che si ritiene spieghi parzialmente la resistenza all’insulina data dalla Niacina. [92] Poiché l’aumento del DAG nelle cellule del fegato sopprime la segnalazione dell’Insulina, [99-162] l’inibizione mediata dalla Niacina del DGAT2 provoca insulino-resistenza, [98] [89] ostacolando così la capacità dell’Insulina di sopprimere la sintesi di glucosio e promuovendo indirettamente uno stato di iperglicemia.
Sebbene l’integrazione cronica di alte dosi di Niacina riduca la sensibilità all’Insulina, ciò non è associato a variazioni dei livelli di glucosio a digiuno. [90] Ciò può essere spiegato da un aumento compensativo della sintesi di Insulina che contrasta la resistenza alla stessa, lasciando sostanzialmente invariati i livelli di glucosio nel sangue. [81]
L’attivazione del recettore della Niacina (HM74A) da parte di alcuni altri agonisti sembra ridurre rapidamente il glucosio sierico nei diabetici migliorando la sensibilità all’Insulina [100] o comunque migliorando i tassi di smaltimento del glucosio. [101] Ciò indica che lo stesso recettore della Niacina può avere effetti benefici sul metabolismo del glucosio e che la resistenza all’Insulina indotta dalla Niacina non si verifica tramite l’attivazione del HM74A.
Quando si osserva il muscolo scheletrico, è stato dimostrato che la terapia con Niacina induce resistenza all’Insulina in questo tessuto in soggetti obesi con NAFLD (2g al giorno nel corso di 16 settimane). Uno studio svolto su ratti a digiuno (il digiuno aumenta la concentrazione plasmatica di acidi grassi non esterificati (NEFA), similmente alla somministrazione di Niacina [102-135] e diminuisce il glicogeno del muscolo scheletrico [103]) in cui sono stati accuratamente somministrati 20mg/kg di Niacina ha mostrato che il glicogeno nel soleo era ridotto mentre il gastrocnemius e il fegato non sono stati influenzati. [103]
Metilgliossale
Quando il processo di glicazione è testato in vitro, la Niacina ha avuto solo effetti inibitori minori sulla glicazione dell’albumina sierica bovina da un noto agente glicante (Metilgliossale [104]) nonostante altri antiossidanti testati come lo Zinco (10-25 µg / mL) avessero più potenti benefici. [105]
È importante sottolineare che qualsiasi effetto della Niacina sulla glicazione in vitro deve essere interpretato con l’avvertenza che la Niacina riduce la sensibilità all’Insulina. Mentre la resistenza all’Insulina indotta dalla Niacina è ben compensata in soggetti sani giovani, lasciando sostanzialmente invariati i livelli di glucosio nel sangue, [81] la compensazione delle cellule β del pancreas negli individui più anziani o in quelli con ridotta tolleranza al glucosio era incompleta in uno studio, [83] causando aumenti nei livelli ematici di glucosio. Pertanto, la misura in cui la Niacina possa influenzare la glicazione in vivo non è chiara e probabilmente dipendente dalla popolazione.
Obesità e massa grassa
L’Adiponectina, un’adipochina nota per migliorare la sensibilità all’Insulina, per essere cardioprotettiva e ritenuta anche antiobesogena, [106] è aumentata in risposta all’attivazione mediata dalla Niacina del recettore HM74A nei topi. [107] La produzione di Adiponectina indotta dalla Niacina è stata rapida in questo studio, aumentando i livelli di questa adipochina del 37% entro 10 minuti da una dose di 30mg / kg per iniezione. I livelli sierici hanno raggiunto il picco dopo 60 minuti e sono rimasti elevati al di sopra del basale fino a 24 ore dopo la somministrazione. [107]
Leptina
È noto anche che la Leptina è aumentata in seguito alla somministrazione di Niacina nell’uomo [91], il che si ritiene si verifichi tramite un meccanismo simile poiché l’agonista farmaceutico HM74A Acipimox induce anch’esso la secrezione di Leptina dal tessuto adiposo in vitro [108] e in vivo. [109]
È stato osservato che la supplementazione di Niacina nel corso di sei settimane negli uomini obesi aumenta l’Adiponectina sierica del 43-56%, con circa metà dell’aumento rappresentato dalla forma ad alto peso molecolare [93] [91] insieme a un aumento del 26,8% della Leptina [91 ] senza cambiamenti osservabili nella Resistina. [91] L’Adiponectina è stata osservata aumentare di circa il 30% in soggetti obesi con NAFLD in risposta alla terapia con Niacina (fino a 2g al giorno), che era correlata con un aumento dell’Insulino-resistenza, [90] portando all’ipotesi che i due meccanismi siano intrecciati, forse come risposta adattativa. [90]
Resistina
Lo “spillover” degli acidi grassi risultante da una conservazione inefficiente del grasso dopo un pasto aumenta i lipidi sierici non esterificati (NEFA), [110] che influenzano negativamente la sensibilità all’Insulina epatica, aumentando la produzione di VLDL e potenzialmente svolgono un ruolo causale nella steatosi epatica. [111] [112] La somministrazione in acuto di Niacina (285 mg per via endovenosa) nell’uomo durante l’alimentazione ha dimostrato di ridurre lo spillover degli acidi grassi, promuovendo l’assorbimento del grasso alimentare nel tessuto adiposo e riducendo i Trigliceridi sierici e i NEFA. [113]
Al contrario, è stato osservato che un trattamento prolungato con Niacina, noto per favorire la resistenza all’Insulina nell’uomo, induce la resistenza all’Insulina adipocitaria, [114] che favorirebbe lo spillover degli acidi grassi, aumentando i livelli sierici di NEFA.[115]
Glucosio-6-fosfato deidrogenasi (G6PD)
È stato osservato che la Nicotinamide sopprime la differenziazione degli adipociti 3T3-L1 in modo dipendente dalla concentrazione con un range superiore a 10mM (il valore ED50), raggiungendo la soppressione completa a 20mM dopo nove giorni. [116] Si ritiene che ciò sia correlato a un effetto inibitorio sulla poli (ADP-ribosio) sintetasi, [116] che la Nicotinamide inibisce a 50µM mentre la Niacina non lo fa. [117] Quando aggiunta dopo differenziazione e in condizioni di glucosio elevato, la Nicotinamide sembra inibire il glucosio-6-fosfato deidrogenasi (G6PD) e prevenire il normale stress ossidativo. [118]
Il recettore dell’Acido Nicotinico è espresso negli dipociti in cui la sua attivazione sopprime l’adenilato ciclasi. [119] Questo effetto sembra essere circa il 30% più efficace negli adipociti rispetto ad altre linee cellulari (milza). [120] Poiché l’attivazione di questo recettore inibisce l’adenilato ciclasi, [119] e i fenolici che agiscono su di esso riducono anch’essi i tassi di lipolisi, [35] l’effetto complessivo dell’Acido Nicotinico sarebbe quello di ridurre la lipolisi negli adipociti, almeno a breve termine.
A lungo termine, tuttavia, il recettore dell’Acido Nicotinico può essere desensibilizzato con esposizione cronica a un agonista [121] e uno studio sui topi ha evidenziato che gli adipociti che sono diventati insulino-resistenti dopo la terapia con Niacina hanno mostrato una maggiore reattività dei recettori adrenergici (β1 e β2) all’aumentare dei livelli di cAMP nella cellula adiposa, [114] (il cAMP viene normalmente soppresso dalla Niacina che agisce sul recettore GRP109A [119]). Ciò potrebbe essere stato correlato alla sottoregolazione dei geni mediata dalla Niacina nella via di segnalazione dell’Insulina incluso il PDE3B, che normalmente degrada il cAMP, [114] una potenziale risposta adattativa nelle cellule adipose che è stata osservata avere la funzione di normalizzare i tassi di lipolisi (nei ratti sotto l’infusione di Niacina) . [97]
Un piccolo studio su sette partecipanti altrimenti sani che assumevano Niacina a 500mg/die, e aumentando la dose a 2g nel corso di due settimane ha mostrato una riduzione dei tassi di ossidazione dei grassi. [94] Tuttavia, a causa di un aumento dei tassi di ossidazione dei carboidrati, non vi era alcuna differenza netta nel tasso metabolico tra Niacina e placebo [94].
I topi privi di PARP-1 sembrano avere tassi metabolici più alti e una minore massa grassa; in assenza di PARP, aumentano le concentrazioni di NAD +, attivando le SIRT1 che quindi lavorano per deacetilare varie proteine (PGC-1α e FOXO1) per promuovere il dispendio energetico attraverso un metabolismo ossidativo aumentato e un incremento dei mitocondri.[122]
La SIRT2 e la SIRT3 non sono influenzate dalla bassa attività del PARP-1, [122] e l’inibizione della ribosilazione dell’ADP con altri mezzi come il knockdown NMNAT-1 sembra conferire anche effetti antiobesità nei roditori. [182] L’alimentazione aumenta acutamente l’attività del PARP-1 nei topi e ostacola transitoriamente l’attività della SIRT1, [122] che si pensa sia correlata al PARP-1 che ha la priorità per l’uso dei donatori di NAD +.
La supplementazione orale di Nicotinamide Riboside a 400mg/kg nel topo sembra aumentare il contenuto di NAD + nel muscolo scheletrico similmente a quanto avviene alla stessa dose di Niacina (Nicotinamide Mononucleotide inefficace in questo organo) [123] e sembra aumentare il dispendio energetico nei topi nutriti con un alto contenuto di grassi insieme all’aumento dell’attività dei geni bersaglio di FOXO1, suggerendo che l’integrazione orale è efficace. [123]
Esistono prove limitate nell’uomo che valutano gli effetti della Niacina sul tasso metabolico, sebbene l’estremità inferiore del dosaggio farmacologico della niacina (1g) in soggetti altrimenti sani non sia riuscito ad aumentare il tasso metabolico rispetto al placebo. [94]
Niacina, muscolo scheletrico e prestazioni fisiche
La somministrazione di Niacina nell’uomo ha dimostrato di aumentare l’espressione dei fattori di trascrizione PPARδ e PPARγ coactivator-1α (PGC-1α) nel muscolo scheletrico. [124] Poiché questi fattori di trascrizione sono importanti regolatori del metabolismo ossidativo e della biogenesi mitocondriale, [125] [126] questo suggerisce che l’integrazione con Niacina può svolgere un ruolo nella resistenza dei muscoli scheletrici.
Gli studi sugli animali hanno supportato questa idea, in cui è stato dimostrato che l’integrazione di Niacina provoca una transizione di fibre muscolari dal tipo II (contrazione rapida) al tipo I (contrazione lenta), aumentando anche il numero complessivo di fibre di tipo I nei muscoli scheletrici nello Zucker (ratto) obeso [127] e suini in crescita [128] (750mg di Niacina/kg di dieta) e pecore (1g di Niacina al giorno). [129] Questo effetto può essere limitato a determinati modelli animali, tuttavia, poiché studi su ratti sani hanno dimostrato che la Niacina ha un effetto trascurabile sulla distribuzione del tipo di fibra muscolare o sul fenotipo metabolico. [130] Inoltre, nonostante la Niacina aumenti l’espressione dei fattori di trascrizione pro-ossidativa nell’uomo, [124] fino ad oggi nessuno studio ha dimostrato che migliora le prestazioni o la capacità di resistenza del muscolo scheletrico.
Tuttavia, come substrato per la sintesi di NAD +, un’adeguata presenza di Niacina può supportare indirettamente il metabolismo ossidativo e la resistenza muscolare. In soggetti altrimenti sani, un lieve esercizio fisico sembra essere associato ad un aumento delle concentrazioni di NAD + nel sangue rispetto a uno stato di riposo (indipendente da qualsiasi integrazione [131]) mentre, quando testato in un esercizio lieve nei roditori, portava anche ad un aumento del NAD + nel sangue prima che diminuisse durante un esercizio ad esaurimento, [131] che è stato notato anche nel muscolo scheletrico. [132] A questo livello di esaurimento c’è un concomitante aumento del contenuto di NADH nel muscolo scheletrico [133] [134] che è stato proposto [135] indicativo di una riduzione del trasferimento di elettroni dal NADH alla sintesi di ATP.
È stato inoltre proposto [135] che da quando l’esercizio aumenta l’ossidazione nei tessuti stimolati e i fattori di stress ossidativo sono noti per alterare l’attività del ciclo di Kreb (TCA) [136] e la catena di trasporto degli elettroni (compresa la NADH deidrogenasi [137]) che forniscono antiossidanti aumenterebbe la resistenza secondaria alla conservazione della cinetica intramuscolare di NAD + / NADH. Quando si forniscono 36mg di picnogenolo [135] come antiossidante durante l’esercizio fisico fino all’esaurimento, sembra che la diminuzione del NAD + nel sangue sia stata invertita in un aumento con gli effetti (sia la diminuzione che l’aumento in attesa di integrazione) più marcati negli atleti allenati. [135]
Impatto organico della Niacina e principali effetti collaterali
Indometacina
In uno studio svolto sui ratti, la Nicotinamide ad un dosaggio di 20mg/kg somministrata un’ora prima di una dose di Indometacina che induceva ulcerazioni allo stomaco ha impedito l’ulcerazione a un livello paragonabile sia al controllo (nessuna induzione di ulcere) sia al farmaco di riferimento di 400mg/kg di sucralfato, che agisce localmente per forma una superficie protettiva per lo stomaco. [138] Questo effetto si è verificato insieme alla conservazione dell’attività del glutatione, alla riduzione della perossidazione lipidica e al miglioramento del muco gastrico. [138] Simili effetti protettivi contro l’ulcerazione indotta da etanolo e stress sono stati notati altrove, con il metabolita primario della Nicotinamide (1-metilnicotinamide; MNA). [139] Questo effetto gastroprotettivo è stato associato con un aumento dell’attività delle prostaglandine, in particolare la PGI2, [139] e nicotinamide, nonché il suo metabolita MNA sono stati implicati nell’aumento del flusso sanguigno gastrico [139] e nella riduzione della permeabilità microvascolare [138] dopo l’ulcerazione.
Interleuchina 10 (IL-10)
Nel colon dei topi, il recettore della Niacina (GPR109A) è necessario per la proliferazione ottimale delle cellule T CD4 + e la produzione di IL-10, che si traduce in un effetto antiinfiammatorio. [140] Questo effetto antinfiammatorio guidato dal GPR109A è mediato dal butirrato, l’acido grasso a catena corta del colon [140], che è un agonista del GPR109A ed è prodotto attraverso la fermentazione della fibra alimentare da parte dei batteri nel colon. [141] [142]
L’effetto riducente dei Trigliceridi dato dalla Niacina sembra da doversi ricondurre al fegato, dove la secrezione di lipoproteine a bassissima densità (vLDL) è ridotta; poiché le vLDL normalmente trasportano i Trigliceridi dal fegato ad altri tessuti, riducendo la secrezione di vLDL ciò si traduce in un livello sierico di Trigliceridi inferiori. [89] La diminuzione della secrezione di vLDL può essere secondaria all’inibizione della lipolisi nel tessuto adiposo, poiché l’aumento cronico di acidi grassi liberi nel siero può regolare negativamente la secrezione di vLDL. [143]
Sembra che l’integrazione di Niacina in acuto (che riduce gli acidi grassi liberi nel siero) sopprime anche la produzione di vLDL e la sua complessazione con i trigliceridi. [144] Ciò suggerisce un altro possibile meccanismo, che può verificarsi attraverso la soppressione acuta del PGC-1β, [145] una proteina nota per promuovere la secrezione di Trigliceridi dal fegato in risposta all’ingestione di grassi nella dieta. [146] In accordo con quest’ultimo meccanismo, la somministrazione di Niacina con un pasto ad alto contenuto di grassi sembra ridurre il picco normale dei trigliceridi postprandiali. [147]
Regioni regolatorie, fattori di trascrizione e molecole di segnalazione (citochine, fattori di crescita, metaboliti, farmaci) che modulano l’espressione del gene ABCA1 nei macrofagi e in altri tessuti. Le frecce e le linee di blocco indicano rispettivamente l’attivazione e la repressione.
Non è confermato come la Niacina riduca le vLDL-C, ma la sua capacità di stimolare l’attività del gene ABCA1 e aumentare il suo contenuto proteico nelle cellule del fegato è alla base dell’aumento dell’HDL-C, [148] che è noto anche per sopprimere la secrezione di vLDL-C. [57] La Niacina (2g per 16 settimane), nonostante riduca le vLDL-C e il complesso con Trigliceridi, non sembra aumentare significativamente il contenuto di trigliceridi intraepatici in soggetti con malattia del fegato grasso non alcolica (NAFLD). [149]
La Niacina sembra anche agire sulle cellule del fegato per promuovere l’accumulo di diacilglicerolo (DAG), che è associato all’insulino-resistenza localizzata. [92] La resistenza all’insulina nelle cellule del fegato riduce l’effetto soppressivo dell’insulina sulla sintesi del glucosio, con conseguente aumento dell’efflusso di glucosio dal fegato nel sangue. [150] Poiché gli stadi iniziali dell’insulino-resistenza indotta dalla Niacina (prima degli aumenti dell’insulina basale e del glucosio) sono stati associati a un fabbisogno ridotto di glucosio per bilanciare un morsetto euglicemico iperinsulinaemico, [109] questo suggerisce che l’inizio dell’insulino-resistenza avviene a livello del fegato. Il ruolo preciso del DAG in questo processo è tuttavia incerto. Mentre il DAG promuove la resistenza all’insulina attraverso l’attivazione di PKCε, [151] l’attivazione di questa proteina non è stata osservata nelle cellule del fegato che sono diventate insulino-resistenti con la Niacina. [92]
TRANSAMINASI. Enzimi intracellulari prodotti principalmente dagli epatociti. normalmente presenti in circolo a bassi livelli nel sangue. Aumentano in caso di danno cellulare. Indici molto sensibili ma moderatamente specifici di danno epatico. ALT è un marker più specifico di danno epatocellulare. (localizzazione citoplasmatica e più lunga emivita)
Nota: La Niacina in dosi terapeutiche può causare aumenti modesti delle transaminasi sieriche e della bilirubina non coniugata, entrambi biomarcatori del danno epatico. Le modifiche vengono invertite se il trattamento farmacologico viene interrotto e di solito si risolvono anche quando si continua l’assunzione. [152] [153] [154] Tuttavia, meno comunemente, la forma di rilascio prolungato del farmaco può portare a gravi epatotossicità, con insorgenza in giorni o settimane. I primi sintomi di gravi danni al fegato includono nausea, vomito e dolore addominale, seguiti da ittero e prurito. Si ritiene che il meccanismo sia una tossicità diretta della Niacina sierica elevata. Abbassare la dose o passare alla forma a rilascio immediato può risolvere i sintomi. In rari casi la lesione è grave e progredisce fino a insufficienza epatica. [152]
È noto che la Niacina rende le cellule β pancreatiche (un tipo di cellula specializzata che secerne insulina in risposta al glucosio) meno sensibile al glucosio sierico. [81] Inoltre, la normale riduzione della sensibilità al glucosio delle cellule β del pancreas associata all’invecchiamento può essere ulteriormente esacerbata dalla supplementazione di Niacina (500mg-1g due volte al giorno). [83] Anche se sembra esserci un aumento compensativo della secrezione di insulina nella risposta alla Niacina [83], in un modello di primati con diabete di tipo I, [155] questo non è stato sufficiente a ridurre la glicemia a livelli normali, con conseguente lieve iperglicemia e iperinsulinemia dopo due settimane di integrazione.[83]
Va notato che una linea cellulare coinvolta nel rossore cutaneo tipico della Niacina, nota come Langerhans, [156] [157] è diversa dall’area del pancreas nota come “Isole di Langerhans”.
Nota: il rossore dato dalla Niacina – una dilatazione a breve termine delle arteriole della pelle, che causa il colore della pelle rossastra – di solito dura circa 15-30 minuti, anche se a volte può persistere per settimane con uso cronico e di forme a lento rilascio. In genere, il viso è maggiormente interessato, ma la reazione può estendersi al collo e alla parte superiore del torace. La causa è la dilatazione dei vasi sanguigni [158] [93] dovuta all’aumento della prostaglandina GD2 (PGD2) e serotonina. [159] [160] [161] [162] Si pensava spesso che il rossore riguardasse l’istamina, ma è stato dimostrato che l’istamina non è coinvolta nella reazione. [159] Il rossore a volte è accompagnato da una sensazione di prurito, in particolare, nelle aree coperte da indumenti. [93]
Acido Acetilsalicilico (Aspirina)
La prevenzione del rossore richiede l’alterazione o il blocco della via mediata dalle prostaglandine. [93] [163] L’Aspirina [165-325mg] assunta mezz’ora prima della Niacina riduce fortemente il rossore, così come l’Ibuprofene [200mg] (una riduzione della frequenza e intensità del rossore fino al 50%). L’assunzione di Niacina ai pasti aiuta anche a ridurre questo effetto collaterale. [93] La tolleranza acquisita aiuterà a ridurre l’effetto; dopo diverse settimane a dosaggio costante, la maggior parte delle persone non ha più esperienza di vampate di calore. [93] Sono state sviluppate forme di Niacina a rilascio lento o “prolungato” per ridurre questi effetti collaterali. [164] [165]
Conclusioni sulla supplementazione di Niacina
Le informazioni che abbiamo a disposizione sulla Niacina e la sua supplementazione, ci presentano un composto senz’altro utile per il controllo o riassesto del quadro lipidico ma allo stesso tempo questa molecola risulta di una complessità d’azione biochimica non trascurabile. Il suo peggiorare l’insulino-resistenza in cronico ma con un maggior picco in acuto, picco che sembra venire controbilanciato da altri fattori come l’aumento della Adiponectina. Lo stesso effetto sulla riduzione della lipolisi può destare preoccupazione nell’atleta, specie se questo si trova in una fase ipocalorica con il principale intento di ridurre la massa grassa. Anche in questo caso sembrerebbe che l’effetto si manifesti in acuto per poi rientrare in condizioni pre-utilizzo. Ciò che è quasi certo, è che le osservazioni sul campo non hanno fatto emergere grosse differenze nell’alterazione della composizione corporea, sia con l’uso della Niacina in regimi ipercalorici che ipocalorici. L’utilizzo di GDA (in specie Berberina) anche alle dosi base efficaci potrebbe essere un “tampone” sufficienti a compensare almeno in parte i possibili peggioramenti dei parametri dell’insulino resistenza. I controlli della glicemia basale e della insulinemia a digiuno sono indicatori da tenere sotto controllo durante l’uso di Niacina. Non è da trascurare la possibilità che la Niacina possa influenzare lo “shift” dalle fibre muscolari di tipo II a quelle di tipo I, cosa non auspicabile per un Bodybuilder o altro atleta di forza.
In definitiva, considerando i dosaggi efficaci e la migliore azione in combinazione con statine (vedi Monacolina K), l’assunzione di Niacina può essere mantenuta con un certo margine di sicurezza tra i 500mg ed 1g/die, ovviamente tarando il dosaggio in risposta agli esami ematici di controllo.
Gabriel Bellizzi
Riferimenti:
“Niacin”. Micronutrient Information Center, Linus Pauling Institute, Oregon State University, Corvallis, OR. 8 October 2018. Retrieved 16 September 2019.
Hegyi J, Schwartz RA, Hegyi V (January 2004). “Pellagra: dermatitis, dementia, and diarrhea”. International Journal of Dermatology. 43 (1): 1–5.
“Why fortify?”. Food Fortification Initiative. 2017. Retrieved 4 April 2017.
Institute of Medicine (1998). “Niacin”. Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline. Washington, DC: The National Academies Press. pp. 123–149.
Bruckert E, Labreuche J, Amarenco P (June 2010). “Meta-analysis of the effect of nicotinic acid alone or in combination on cardiovascular events and atherosclerosis”. Atherosclerosis. 210 (2): 353–61.
“Niacin”. IN: LiverTox: Clinical and Research Information on Drug-Induced Liver Injury (Internet). Bethesda, MD: National Institute of Diabetes and Digestive and Kidney Diseases. February 2014.
Ricercatori australiani presso la University of New South Wales stanno testando un nuovo farmaco per la perdita di peso che agisce similmente al DNP (2,4-dinitrofenolo), ma senza i numerosi effetti collaterali connessi a quest’ultimo. Il loro lavoro è stato recentemente pubblicato su Nature Communications.[1]
BAM15?
Finché l’epidemia di obesità continuerà a crescere, la ricerca di farmaci per la perdita di peso continuerà. Parte di questa ricerca si svolge tenendo come riferimento il pericoloso ma estremamente efficace DNP. Sarebbe possibile sintetizzare una molecola che sia efficace come DNP, ma non così pericolosa?
Il DNP è un disaccoppiatore della fosforilizzazione ossidativa: interferisce con la resintesi di ATP nel mitocondrio causando una significativa dispersione di energia sotto forma di calore.
Ricercatori americani presso la Yale University stanno studiando un analogo metilato del DNP, il DNPME.[2]
La giapponese Otsuka Pharmaceutical sta conducendo esperimenti con OPC-163493.[3]
I ricercatori australiani che hanno pubblicato la ricerca che qui si sta trattando, stanno svolgendo esperimenti sul N5, N6-bis (2-fluorofenil) [1,2,5] oxadiazolo [3,4-b] pirazina-5,6-diammina. In breve: BAM15 .
Effetti del BAM15 nei topi
I ricercatori hanno somministrato ai topi dosi orali di BAM15 e hanno osservato che nelle ore successive alla somministrazione, il consumo di ossigeno degli animali – e quindi il loro consumo calorico – era aumentato di alcune decine di punti percentuale. L’effetto è stato temporaneo. Questo perché l’emivita del BAM15 nei topi è di sole 1,7 ore.
In altri esperimenti, in cui i topi sono stati alimentati con cibo contenente BAM15, i ricercatori hanno scoperto che la molecola aumentava il dispendio calorico solo di notte. Ciò non sorprende, perché i topi sono animali notturni e quindi preferiscono mangiare quando è buio. Il BAM15 non ha aumentato la quantità di attività fisica svolta.
Il dosaggio utilizzato nei topi trasposto in un essere umano adulto corrisponderebbe a circa 1g/die di BAM15.
In un altro esperimento, i ricercatori hanno nutrito i topi con zucchero e grasso addizionali [WD], rendendo gli animali più grassi. Se i topi venivano trattati con il BAM15, la loro massa grassa aumentava di meno, o per nulla, o addirittura diminuiva. L’equivalente umano delle dosi alle quali si sono verificati questi effetti è stato rispettivamente di 500mg, 1g e 1,5g di BAM15 al giorno.
Il BAM15 non ha aumentato la temperatura corporea dei topi, non ha ridotto la massa magra o aumentato l’attività dei radicali liberi. Per quanto i ricercatori hanno potuto scoprire, il BAM15 aumenta anche la sensibilità all’Insulina.
Conclusioni
Concludendo, i ricercatori riportano che il BAM15 rappresenta un raro disaccoppiatore mitocondriale che previene e inverte l’obesità senza influire sull’assunzione di cibo o sulla massa magra. Una limitazione del BAM15 è la bassa solubilità acquosa, ma questa proprietà non ha influito sulla biodisponibilità orale e infatti la bassa solubilità acquosa è un parametro importante che consente al BAM15 di penetrare nelle membrane cellulari ed entrare nei mitocondri. Un’altra limitazione del BAM15 è l’emivita di 1,7 ore e le direzioni future esamineranno le strategie di formulazione per migliorare l’esposizione. Collettivamente, i dati qui presentati supportano l’ulteriore sviluppo del BAM15 come potenziale terapeutico per l’obesità e le malattie metaboliche.
Negli ultimi anni il Cannabidiolo (noto anche con la sigla CBD) ha subito una crescente popolarità data sia dai promettenti risultati ipoteticamente applicabili e replicabili nel trattamento di varie condizioni patologiche (vedi, ad esimpio, SLA) provenienti dagli studi in vitro e su animali da laboratorio e, soprattutto, dalla sua relativamente facile reperibilità, essendo legale in molti paesi, che rendeva (e rende) il composto testabile dal singolo senza rischi legali. Il suo uso ha iniziato a diffondersi anche tra gli sportivi, ed i culturisti in particolar modo ne sono stati attratti per le sue potenzialità nella riduzione della percezione dello stress, come agente anoressizzante e come trattamento del dolore.
Ma quanto c’è di concretamente e fruttuosamente applicabile del Cannabidiolo in ambito medico e anche sportivo? Nel seguente articolo verranno riportate le informazioni necessarie per una risposta scientificamente onestà a questa domanda.
La storia del CBD
Cannabidiolo (CBD)
Il Cannabidiolo (CBD) è un fitocannabinoide scoperto nel 1960. È uno dei 113 cannabinoidi identificati nelle piante di cannabis e rappresenta fino al 40% dell’estratto della pianta.[1] I cannabinoidi nella cannabis sono stati studiati dagli anni ’40,
[2], con le strutture di THC (delta-9-tetraidrocannabinolo) e CBD scoperte negli anni ’60. La ricerca di questi composti ha subito un picco negli anni ’90, quando sono stati trovati i recettori dei cannabinoidi nel corpo [3], e in seguito è stato scoperto che sono stati attivati anche da composti endogeni (prodotti nel corpo) nel 1992.[4]
La popolarità del CBD è esplosa negli anni 2000, soprattutto dopo che i genitori di bambini epilettici hanno iniziato a sperimentare l’effetto del CBD sui loro figli, come opzione di trattamento per convulsioni resistenti ai farmaci nella rara e grave condizione di epilessia chiamata Sindrome di Dravet.[5]
Delta-9-tetraidrocannabinolo (THC)
I prodotti a base di CBD che mantengono una percentuale di THC “low-to-zero” (inferiore allo 0,3%) si trovano attualmente in un’area grigia della legalità negli Stati Uniti, ma con una legalità rafforzata dopo l’approvazione del Farm Act nel dicembre 2018, sebbene la FDA richiede ancora tecnicamente che gli integratori commercializzati debbano essere approvati prima dalla stessa. [6] La FDA ha inviato lettere di avvertimento nell’aprile del 2019 a tre società che ritenevano stessero formulando dichiarazioni sanitarie significative, tra cui: [7]
“Il CBD ha bloccato con successo le cellule tumorali in diverse varietà di cancro cervicale.”
“Il CBD ha anche ridotto la crescita e la diffusione delle cellule di glioma umano, suggerendo così un possibile ruolo del CBD come agente antitumorale.”
“Per i pazienti con Alzheimer, il CBD è un’opzione di trattamento che sta rallentando la progressione di quella malattia.”
“La fibromialgia è concepita come uno stato di sensibilizzazione centrale con iperalgesia secondaria. Il CBD ha dimostrato la capacità di bloccare i meccanismi spinali, periferici e gastrointestinali responsabili del dolore associato a emicrania, fibromialgia, IBS e altri disturbi correlati. “
“Il cannabidiolo può essere efficace nel trattamento dei disturbi da uso di sostanze.”
“Il CBD ha ridotto gli effetti gratificanti della morfina e ha ridotto la ricerca dell’eroina.”
“Il CBD può essere utilizzato per evitare o ridurre i sintomi di astinenza.”
Presumibilmente, la reazione della FDA è stata dettata dalle scarse prove che si hanno in questo momento per il trattamento della maggior parte delle condizioni al di fuori della grave epilessia, e poiché queste affermazioni non includono tale dichiarazione di non responsabilità, i pazienti potrebbero presumere che il livello di evidenza sia più forte di quanto non sia in realtà. Pertanto, la FDA afferma che “non tollereremo questo tipo di marketing ingannevole per i pazienti vulnerabili”. L’interazione tra la regolamentazione FDA di alimenti, droghe e integratori (che possono essere tutti applicabili alla cannabis), insieme a diverse normative sulla cannabis in diversi stati, rende complesse le questioni legali relative al CBD e ad altri prodotti a base di cannabinoidi. [8]
Nota: in Italia la vendita della cannabis light è legale dal 2016, e ciò in virtù di una legge (numero 242) pienamente in vigore. La vendita è consentita per cannabis che abbia THC (i principi attivi che generano effetti psicotropi) fino e non oltre la percentuale dello 0,2. Ma il venditore di cannabis legale non è responsabile penalmente fino alla soglia dello 0,6 per cento, oltre la quale la marijuana viene sequestrata da parte dell’autorità giudiziaria e si apre un fascicolo che poi arriverà in tribunale. Queste due limitazioni aprono certo le porte a diverse interpretazioni e smagliature della legge. Molte profonde incertezze attualmente coinvolgono il CBD legale in Italia: questo a seguito della lacunosa sentenza emessa il 30 Maggio 2019 da parte della Corte di Cassazione e che ha visto “rivoluzionare”, secondo l’accezione meno positiva del termine, quanto invece stabilito dalla Legge 242. La sentenza emessa 30 Maggio 2019 circa la cannabis light e il CBD fino ad allora legale, ha stabilito che in Italia non sarebbe più concessa la vendita o la cessione a qualunque titolo di tutti i prodotti “derivati dalla coltivazione della cannabis”, quali l’olio al CBD, così come foglie, le infiorescenze e la resina, questo secondo la massima provvisoria emessa che recita testualmente -“Integrano il reato previsto dal Testo unico sulle droghe (articolo 73, commi 1 e 4, dpr 309/1990) le condotte di cessione, di vendita, e, in genere, la commercializzazione al pubblico, a qualsiasi titolo, dei prodotti derivati dalla coltivazione della cannabis sativa light, salvo che tali prodotti siano in concreto privi di efficacia drogante”. Ma, e c’è un ma, la legge italiana sull’utilizzo e sulla vendita del CBD esplica tutto ciò che concerne la filiera della canapa in Italia, dalla coltivazione al consumo. Nella stessa si legge che vi è una promozione alla coltivazione, trasformazione, impiego della stessa per lo sviluppo del territorio, produzione di alimenti, cosmetici, opere di bioingegneria, bonifica dei terreni, ricerca.
Le varietà di canapa che possono essere seminate devono essere presenti nel catalogo europeo delle sementi, in queste sono riportate solo quelle tipologie che hanno dimostrato di possedere un THC inferiore allo 0.2%.
Per questo è possibile affermare che i prodotti a base di CBD possono essere venduti in quanto rientrano nella tipologia ad uso tecnico, ciò vuol dire che nella legge non si fa esplicito riferimento alla possibilità di fumare la cannabis light, tuttavia questo non è neanche vietato e per questo l’utilizzo finale è a discrezione di chi la compra. La canapa legale è la principale fonte di approvvigionamento di CBD legale in Italia.
I negozi, online o fisici, possono commercializzare tisane, bevande, torte, vestiti e anche bustine di marijuana light. Ad essere illegale infatti è un valore elevato di THC ma non il CBD che ha esclusivamente potenziali proprietà rilassanti e antiossidanti e quindi, come dimostrano i test svolti in laboratorio, non ha alcun effetto ‘ludico’ comparabile all’utilizzo di droghe.
Per la serie “leggi ambigue e dove trovarle”…
Fonti del CBD
Il CBD viene in genere estratto dalla pianta di canapa. Ecco una rapida panoramica dei termini: la cannabis è un genere di pianta, che si presenta in due tipi botanici principali: la cannabis indica e la cannabis sativa. La marijuana (che contiene sia CBD che THC) può essere di entrambi i tipi, mentre la canapa è solo di tipo sativa.
Si noti che entrambe appartengono allo stesso genere e specie e sono piuttosto distinti i loro livello di THC. La canapa contiene un massimo dello 0,3% di THC, mentre la marijuana varia in genere tra il 5-20% di THC. [9] La marijuana viene spesso coltivata per uso ricreativo o misto ricreativo/medicinale, mentre la canapa viene coltivata per una più ampia varietà di usi (abbigliamento di canapa, olio di canapa, fibra di canapa, CBD isolato e dozzine di altri derivati).
CBD e THC non sono gli unici cannabinoidi presenti sul mercato. Ad esempio, il CBG (cannabigerolo) si trova in quantità minori nella cannabis, ma è stato studiato per le malattie infiammatorie in un modello animale.[10]
Attività biologica (farmacodinamica)
Poiché il sistema endocannabinoide ha un impatto su così tante sfaccettature della vita di base di un individuo (ad es. Appetito, funzione immunitaria, riproduzione e gestione del dolore), esistono una serie di possibilità di azione per il CBD (e altri cannabinoidi) che incidono sulla salute.[11] Il CBD può impedire l’attivazione eccessiva dei recettori CB1 e CB2, riducendo gli effetti dello stress mediato da tali recettori.[12] Altri ruoli includono l’antiossidazione: sia il CBD che il THC hanno agito come antiossidanti neuroprotettivi nei ratti.[13] Mentre il THC agisce principalmente sui recettori CB1 e CB2, il CBD agisce su altri recettori tra cui il canale del calcio TRPV1, attivandolo e rapidamente desensibilizzandolo, risultando in definitiva in meno potenziale di ipereccitazione.[14]
Nota: Il CBD non sembra avere effetti psicotropi (“alti”) come quelli causati dal ∆9-THC nella marijuana, ma, come già detto, è attualmente in fase di ricerca preliminare per i suoi possibili effetti anti-ansia e anti-psicotici legati ad un suo possibile effetto psicotropo. Mentre si sviluppa la comprensione delle differenze nei cannabinoidi medici, gli esperti stanno lavorando per distinguere la “marijuana medica” (con vari gradi di effetti psicotropi e deficit nella funzione esecutiva) dalle “terapie mediche con CBD” che comunemente presenterebbero avere un profilo degli effetti collaterali ridotto o non psicoattivo. Il CBD sembrerebbe non causare dipendenza.
Farmacocinetica
Il CBD assunto per via orale (in particolare in riferimento al farmaco liquido Epidiolex) impiega tra le 2,5 e le 5 ore per raggiungere la massima concentrazione ematica. L’emivita è tra le 56 e le 61 ore.[15] Sia nell’uomo che nei ratti, essere in stato di alimentazione sembra aumentare i livelli plasmatici di CBD.[16][17] La farmacocinetica del CBD può variare abbastanza ampiamente tra persone e persona [18], con risposte che possono essere aggravate da variazioni da lotto a lotto ed imprecisioni di etichettatura in alcuni prodotti a base di CBD.
Potenziali effetti terapeutici del CBD
In questo momento, c’è solo un accenno di promessa per il CBD per il miglioramento delle condizioni della pelle, ma nessuna basata sulla ricerca randomizzata.[19] Ad esempio, il CBD in uno studio preclinico in vitro ha mostrato effetti antiinfiammatori , antiproliferativi e lipostatici che suggeriscono che potrebbe potenzialmente avere una certa efficacia nei trattamenti di condizioni della pelle come l’acne vulgaris.[20]
Un caso studio ha riferito che una dose giornaliera di 2x300mg di CBD era associata al miglioramento della disfagia da SLA. Mentre diciotto mesi dopo l’esordio, il progresso della disfagia subivano un calo, e la debolezza degli arti, il fascicolazione e l’atrofia peggioravano relativamente meno. Gli autori teorizzano che la progressione di alcuni ma non tutti i sintomi della malattia dei motoneuroni può essere rallentata con il CBD.[21]
Il disturbo dello spettro autistico (ASD) probabilmente comporta alterazioni in alcuni percorsi chimici neurologici, come il glutammato e il GABA. È con questa idea che uno studio ha testato se il CBD potesse alterare questi percorsi in un modo che potesse aiutare il trattamento dell’autismo, con l’utilizzo della spettroscopia di risonanza magnetica. I ricercatori hanno scoperto che il CBD non era efficace per questo scopo.[22] Tuttavia, una serie di casi retrospettivi hanno mostrato un comportamento migliorato nel 61% di un campione di bambini con ASD. Tuttavia, non vi era alcun gruppo di controllo.[23]
Il CBD non sembra nemmeno migliorare i sintomi legati al movimento nel Parkinson, sebbene possa migliorare i problemi del sonno derivanti dalla condizione, in particolare la parassitnia con incubi e la perdita di atonia muscolare durante il sonno REM.[24][25]
In uno studio esplorativo, 2000mg/die di CBD hanno ridotto i sintomi psicotici della schizofrenia e i pazienti trattati con CBD avevano maggiori probabilità di essere valutati come migliorati dal medico curante.[26] Uno studio randomizzato nel quale si è testato l’effetto del CBD ha scoperto che sia esso che l’amisulpride hanno migliorato i sintomi della patologia in questione, ma il CBD ha avuto un migliore profilo degli effetti collaterali.[27] Tuttavia, in uno studio randomizzato nel quale si è utilizzata una dose di 600mg/die di CBD, non sono stati osservati miglioramenti nei sintomi.[28] Uno studio non controllato non ha riscontrato alcun beneficio del CBD con dosi aumentate da 40mg/die a 1280mg/die in 35 giorni su pazienti con schizofrenia resistente al trattamento.[29]
In sei studi randomizzati con oltre 500 pazienti con convulsioni gravi (in genere bambini piccoli), il CBD migliora significativamente i sintomi convulsivi.[30] In particolare, la percentuale di pazienti che ha avuto una riduzione di oltre il 50% delle crisi è diminuita, così come il numero di persone che hanno smesso di avere crisi epilettiche. Mentre gli studi randomizzati hanno in genere esaminato i pazienti con sindrome di Dravet e Lennox-Gastaut, studi estesi non randomizzati (studi sull’uso compassionevole) hanno anche mostrato efficacia nel trattamento del disturbo da carenza di CDKL5 e nelle sindromi di Aicardi, Doose e Dup15q.[31] Una serie di casi di tre pazienti ha anche suggerito che il CBD può aiutare con l’epilessia correlata al tumore al cervello.[32]
Studi a più lungo termine confermano che il CBD continua ad essere efficace e relativamente sicuro in questi pazienti [33][34][35], sebbene gli effetti collaterali inizialmente comuni come la riduzione dell’appetito e la diarrea persistessero in uno studio.[36] Il CBD sembra avere un effetto di dimensioni simili sull’epilessia pediatrica grave come altri farmaci antiepilettici.[37] Un piccolo studio di coorte suggerisce che anche i pediatri hanno ritenuto che la salute generale dei loro bambini con epilessia grave fosse migliorata.[38]
Il motivo per cui il CBD può aiutare con le convulsioni ha a che fare con le vie di segnalazione degli endocannabinoidi. Questi sono alterati nel disturbo convulsivo e THC e CBD agiscono in diversi modi per aiutare potenzialmente a marginare questo disturbo. Il THC probabilmente aiuta attivando il recettore CB1 [39], mentre i meccanismi del CBD sono meno conosciuti e possono includere una varietà di altri recettori, come il GPR55 e il TRPV1.[40][41][42]
Mentre il potenziale per il CBD di aiutare ad alleviuare alcuni tipi di dolore cronico è elevato, le prove di base sono molto piccole, con solo uno studio randomizzato e due studi non randomizzati a sostegno.
In uno studio randomizzato sono stati osservati dolore e spasticità in pazienti con sclerosi multipla, lesioni del midollo spinale e altre condizioni con questi sintomi. Sebbene diversi pazienti non abbiano completato lo studio, il controllo del dolore è migliorato nel gruppo CBD (mentre lo spasmo e altri sintomi non hanno subito variazioni).
Due studi non randomizzati e non controllati hanno esaminato il CBD al fine di valutarne il potenziale nell’alleviare il dolore. Uno studio ha osservato il dolore nelle ragazze giovani con dolore cronico da vaccino HPV. Si è riscontrato una riduzione del dolore corporeo e un migliore funzionamento con l’uso del CBD.[43] Uno studio molto piccolo ha esaminato il dolore nei pazienti con trapianto di rene e ha scoperto che due su sette pazienti avevano un sollievo totale dal dolore, mentre quattro avevano un sollievo non particolarmente percepibile.[44]
In uno studio, una singola dose di THC sembrava aumentare l’ansia rispetto al placebo, mentre una singola dose di CBD è risultata simile al placebo come stato ansioso.[45] Mancano studi randomizzati a lungo termine, ma studi randomizzati a breve termine e studi di serie di casi a lungo termine hanno esplorato i potenziali effetti benefici del CBD sull’ansia.
In una serie di casi di 72 pazienti ambulatoriali psichiatrici, il CBD (con la maggior parte dei pazienti che ne assumevano 25mg/die) ha migliorato rapidamente i punteggi di ansia in circa l’80% dei pazienti, con un effetto generalmente prolungato nel corso di tre mesi.[46]
Tre prove hanno esaminato gli effetti del CBD sull’ansia indotta sperimentalmente dal parlare in pubblico. Uno studio precoce non randomizzato nel 1993 ha mostrato che 300 mg di CBD hanno migliorato lo stato ansioso dopo il test di parlare in pubblico, ma non prima.[47] Uno studio randomizzato del 2017 dello stesso autore ha anche mostrato una riduzione dell’ansia alla stessa dose di 300mg, dopo il test del parlato, con 100mg e 900mg che non hanno mostrato alcun effetto.[48] Un altro gruppo ha studiato l’effetto di 600mg di CBD riscontrando una significativa riduzione dell’ansia durante il test di conversazione pubblica.[49]
Gli studi non controllati forniscono prove più deboli, ma estendono tali prove ad altre popolazioni. Una serie di casi retrospettivi hanno mostrato che quasi l’80% dei pazienti per i quali il CBD era utilizzato in una clinica psichiatrica per trattare l’ansia aveva migliorato questa condizione, e la maggior parte dei pazienti hanno mantenuto i benefici dopo il primo mese.[46]
Un caso studio ha suggerito che la somministrazione di CBD per 10 giorni ha ridotto i sintomi di astinenza da cannabis in un pesante abusatore, come ansia, perdita di appetito e irritabilità.[50]
Prove preliminari hanno suggerito che il CBD ha ridotto il consumo di fumo di sigaretta (nonostante non abbia influito sulla brama di sigarette)[51], e ha ridotto la piacevolezza legata alla sigaretta dopo una notte di astinenza.[52] Purtroppo, nei fumatori di sigarette che mirano all’astinenza, il CBD non ha migliorato la memoria di lavoro verbale o spaziale o l’impulsività.[53]
Un piccolo studio pilota condotto su individui dipendenti da oppioidi ha mostrato che il CBD in dosi di 400 o 800mg riduceva la brama dopo che i soggetti erano stati fatti astenere dagli oppioidi per sette giorni (dopo una sessione video per indurre la brama).
[54] Tuttavia, la maggior parte delle prove per i potenziali benefici del CBD sulla sospensione degli oppioidi sono di origine preclinica e basate su studi molto piccoli.[55]
Una serie di casi ha riportato una riduzione dei sintomi del PTSD (Post Traumatic Stress Disorder) in 10 di 11 pazienti con l’uso di CBD, dopo otto settimane di trattamento, in cui il CBD è stato ben tollerato.[56]
La prima citata serie di casi di 72 pazienti ambulatoriali psichiatrici ha anche scoperto che il CBD (sempore con la maggior parte dei pazienti che ne assumevano 25mg/die) non ha migliorato i punteggi del sonno nel corso di tre mesi.[46]
D’altro canto, uno studio crossover in doppio cieco ha scoperto che il CBD non ha interrotto il ciclo sonno-veglia dei pazienti che assumevano una dose relativamente alta intesa a ridurre clinicamente l’ansia (300 mg).[57]
Quando si parla di cancro e CBD, gli studi affidabili sull’uomo mancano totalmente.
Un caso studio ha riportato di un paziente con carcinoma polmonare che ha avuto una “risposta sorprendente” con evidente riduzione del tumore dopo auto-somministrazione di CBD.[58] Naturalmente, questo è un caso clinico, ed è una pura supposizione che il CBD sia l’elemento causale. Ma è comunque uno spunto di riflessione.
Parlando di casi clinici, un altro ha suggerito che il CBD potrebbe aver migliorato la chemioradioterapia in due casi di gliomi di alto grado.[59] Una serie di casi retrospettivi ha riferito che il 92% dei 119 casi di cancro esaminati aveva una riduzione delle cellule tumorali circolanti e talvolta una riduzione delle dimensioni del tumore, quando veniva assunto il CBD di livello farmaceutico.[106-60] Ancora una volta, questa è una prova di basso livello, ma pur sempre qualcosa che merita approfondimenti.
In uno studio nel quale sono stati utiulizzati 10mg di CBD, due volte al giorno, al fine di valkutarne gli effetti sul Morbo di Crohn, non sono stati riscontrati effetti benefici. Gli autori ipotizzano che ciò potrebbe essere stato influenzato dalla bassa dose o dal piccolo numero di pazienti nello studio o dalla mancanza di altri cannabinoidi sinergici (ovvero l’effetto entourage).[61]
Uno studio di prova (randomizzato, con un gruppo placebo) ha testato l’effetto di un estratto botanico ricco di CBD sulla Colite Ulcerosa, e non ha riscontrato differenze di remissione rispetto al placebo, ma un miglioramento di alcuni sintomi.[62]
Status del CBD nello Sport
Il CBD è stato utilizzato da atleti professionisti e dilettanti in diverse discipline e paesi, con l’Agenzia mondiale antidoping che ha rimosso il CBD dall’elenco delle sostanze vietate. L’Agenzia antidoping degli Stati Uniti e l’Agenzia antidoping del Regno Unito non hanno politiche anti-CBD, con quest’ultima che afferma che “il CBD non è attualmente nell’elenco delle sostanze proibite dell’Agenzia mondiale antidoping. Di conseguenza, è consentito l’uso di esso nello sport. Tutti gli altri cannabinoidi (inclusi ma non limitati a cannabis, hashish, marijuana e THC) sono vietati in competizione. L’intenzione del regolamento è proibire i cannabinoidi che attivano gli stessi recettori nel cervello come attivati dal THC. “[63] [64] Nel 2019, il principale produttore di prodotti a base di cannabis, Canopy Growth, ha acquisito la proprietà di maggioranza di BioSteel Sports Nutrition, che sta sviluppando prodotti a base di CBD con l’approvazione di numerosi atleti professionisti. [65] La National Hockey League Alumni Association ha avviato un progetto con Canopy Growth per determinare se il CBD o altri prodotti a base di cannabis potrebbero migliorare i sintomi neurologici e la qualità della vita dei giocatori feriti alla testa.[65] Numerosi atleti professionisti usano il CBD, principalmente per il trattamento del dolore.[65][66][67]
Interazioni alimentari e farmacologiche del CBD
CYP2D6
Il CBD sembra interagire con i farmaci antiepleettici comunemente usati, modificando significativamente i loro livelli sierici. Questi includono clobazam, rufinamide, topiramato, zonisamide ed eslicarbazepina.[68]
Il CBD può anche inibire un enzima chiamato CYP2D6, che è preso di mira da farmaci comuni tra cui omeprazolo e risperidone. Può anche inibire l’enzima CYP2C9, che ridurrebbe la metabolizzazione di warfarin e diclofenac.[69] Non è noto se le inibizioni in vitro si tradurranno in inibizioni in vivo negli esseri umani, e sono necessari ulteriori studi a riguardo.[70] In uno studio nel quale sono stati utilizzati 6x100mg/die di CBD, ha aumentato la biodisponibilità e l’emivita di eliminazione dell’esobarbitale.[71]
D’altro canto, è anche possibile che altri farmaci influenzino la metabolizzazione del CBD. Ad esempio, la rifampicina antibiotica riduce le concentrazioni di picco del CBD nel sangue a causa dell’induzione dell’enzima CYP3A4, mentre il ketoconazolo, l’inibitore del CYP3A4, raddoppia quasi il picco della concentrazione di CBD.[72]
Effetti collaterali del CBD e possibili contaminazioni dei prodotti che lo contengono
(±)-Amisulpride
Per trovare eventi avversi ed effetti collaterali, è necessario un campione relativamente grande. Quindi i principali studi per valutare questi in riferimento al CBD erano gli studi più ampi su giovani pazienti con gravi disturbi convulsivi, vale a dire la sindrome di Dravet e la sindrome di Lennox-Gastaut.
Gli effetti che si sono verificati più frequentemente con paragone tra CBD e placebo includono: sonnolenza, diarrea, affaticamento, vomito, febbre e letargia. Alcuni pazienti avevano anche livelli aumentati di aminotransferasi epatica.[73][5]
Due studi hanno esaminato i potenziali effetti collaterali emodinamici (come variazioni della frequenza cardiaca, della frequenza respiratoria o della pressione sanguigna) e non hanno trovato effetti significativi quando è stato assunto il CBD.[74][75]
Una review completa ha rilevato che dosi fino a 1.500mg/die sembrano essere abbastanza ben tollerate [76], sebbene non sia affatto sicuro presumere che la dose sia sicura per un determinato individuo (a causa della relativa mancanza di informazioni sulla sicurezza a lungo termine e della varietà di condizioni/farmaci/ecc. Che potrebbero teoricamente interagire con il CBD).
Gli studi sugli animali hanno riportato alcuni effetti collaterali teorici non osservati negli studi sull’uomo esistenti, come la ridotta capacità di fertilizzazione e dell’inibizione attraverso il metabolismo epatico del farmaco.[77][78]
Il CBD è risultato promettente come metodo per ridurre gli effetti collaterali quando vengono assunti altri farmaci, come osservato nel già citato studio sul CBD somministrato a pazienti con schizofrenia che assumevano amisulpride.[79]
I contaminanti da piante di cannabis coltivate, raccolte e confezionate con pratiche meno controllate potrebbero teoricamente comparire anche in prodotti a base di CBD, inclusi pesticidi, particelle metalliche, cannabinoidi sintetici, metalli pesanti, muffe, batteri e aflatossine.[80][81] I solventi residui del processo di produzione possono anche comparire in estratti di cannabis, vale a dire esano, etanolo, alcool isopropilico, toluene, benzene, xilene e acetone.[82] Questa ricerca riguarda tuttavia tutti gli estratti generali della cannabis; la ricerca sui contaminanti specificamente nei prodotti isolati di CBD deve ancora essere svolta.
Conclusioni sul CBD
Cosa concludere riguardo al CBD dopo l’esposizione dei dati precedentemente riportati? Sicuramente che la ricerca sull’uomo è molto limitata e non ci permette ad oggi di esporre un giudizio sufficientemente fondato sulla reale portata terapeutica del CBD. Si tratta ovviamente di una conclusione dettata dalla scarsità dei dati e dall’impatto degli studi esistenti. Possiamo comunque scorgere dei potenziali sul trattamento dell’ansia e, ipoteticamente, sullo stress percepito e il trattamento del dolore. Tale caratteristica rende il CBD sperimentabile come aggiunta ad una preparazione sportiva particolarmente intensa come durante il picco di preparazione per una competizione o un contest di BodyBuilding. La sua possibile influenza sull’appetito (effetto anoressizzante) potrebbe tornare senz’altro molto utile nei periodi di restrizione calorica. Alcuni speculeranno su un suo possibile impatto indiretto significativo sui livelli di Cortisolo dato, per l’appunto, da una ridotta percezione dello stress, ma i dati certi al momento non sussistono. Il fatto che si tratti di un composto dagli effetti non del tutto accertati non invoglia particolarmente a investire denaro per testarne le potenzialità. Se poi consideriamo il fatto che i più non conoscono ne le modalità di utilizzo ne le dosi graduate da applicare con criterio crescente in caso di risposta positiva con dosaggi più blandi, non c’è da stupirsi se spesso ci si imbatte in testimonianze molto contrastanti provenienti dagli utilizzatori che scoraggiano i lettori meno preparati in materia.
Quindi, vale la pena investire denaro per testare l’efficacia del CBD? Considerando che il costo di prodotti di alta qualità non è propriamente economico, e che i dati in letteratura non sono molto impattanti a livello dimostrativo (almeno ad oggi), a meno che non abbiate una adegiuata conoscenza per testarne e valutarne gli effetti, per il momento si può saggiamente investire su altro… per adesso…
Gabriel Bellizzi
Riferimenti:
1- Campos AC, Moreira FA, Gomes FV, Del Bel EA, Guimarães FS (December 2012). “Multiple mechanisms involved in the large-spectrum therapeutic potential of cannabidiol in psychiatric disorders”. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences (Review). 367 (1607): 3364–78.
2- LOEWE S. Studies on the pharmacology and acute toxicity of compounds with marihuana activity. J Pharmacol Exp Ther (1946)
3- Howlett AC, et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev (2002)
4- Devane WA, et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science (1992)
5- Devinsky O, Cross JH, Wright S. Trial of Cannabidiol for Drug-Resistant Seizures in the Dravet Syndrome.
N Engl J Med (2017)
6- Statement from FDA Commissioner Scott Gottlieb, M.D., on signing of the Agriculture Improvement Act and the agency’s regulation of products containing cannabis and cannabis-derived compounds.
7- Statement from FDA Commissioner Scott Gottlieb, M.D., on new steps to advance agency’s continued evaluation of potential regulatory pathways for cannabis-containing and cannabis-derived products.
8- O’Connor SM, Lietzan E. The Surprising Reach of FDA Regulation of Cannabis, Even After Descheduling. Am Univ Law Rev. (2019)
9- Hilderbrand RL. Hemp & Cannabidiol: What is a Medicine?. Mo Med. (2018)
10- Borrelli F, et al. Beneficial effect of the non-psychotropic plant cannabinoid cannabigerol on experimental inflammatory bowel disease. Biochem Pharmacol. (2013)
11- Battista N, et al. The endocannabinoid system: an overview. Front Behav Neurosci. (2012)
12- Laprairie RB, et al. Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. Br J Pharmacol. (2015)
13- Hampson AJ, et al. Cannabidiol and (-)Delta9-tetrahydrocannabinol are neuroprotective antioxidants. Proc Natl Acad Sci U S A. (1998)
14- Iannotti FA, et al. Nonpsychotropic plant cannabinoids, cannabidivarin (CBDV) and cannabidiol (CBD), activate and desensitize transient receptor potential vanilloid 1 (TRPV1) channels in vitro: potential for the treatment of neuronal hyperexcitability. ACS Chem Neurosci. (2014)
16- a b Zgair A, et al. Dietary fats and pharmaceutical lipid excipients increase systemic exposure to orally administered cannabis and cannabis-based medicines. Am J Transl Res. (2016)
17- Winter H, et al. Effect of a high-calorie, high-fat meal on the bioavailability and pharmacokinetics of PA-824 in healthy adult subjects. Antimicrob Agents Chemother. (2013)
18- Huestis MA. Human cannabinoid pharmacokinetics. Chem Biodivers. (2007)
19- Eagleston LRM, et al. Cannabinoids in dermatology: a scoping review. Dermatol Online J. (2018)
20- Oláh A, et al. Cannabidiol exerts sebostatic and antiinflammatory effects on human sebocytes. J Clin Invest. (2014)
21- Co-medication with Cannabidiol May Slow Down the Progression of Motor Neuron Disease: A Case Report; 2017..
22- Pretzsch CM, et al. Effects of cannabidiol on brain excitation and inhibition systems; a randomised placebo-controlled single dose trial during magnetic resonance spectroscopy in adults with and without autism spectrum disorder. Neuropsychopharmacology. (2019)
23- Aran A, et al. Brief Report: Cannabidiol-Rich Cannabis in Children with Autism Spectrum Disorder and Severe Behavioral Problems-A Retrospective Feasibility Study. J Autism Dev Disord. (2019)
24- Chagas MH, et al. Effects of cannabidiol in the treatment of patients with Parkinson’s disease: an exploratory double-blind trial. J Psychopharmacol. (2014)
25- Peres FF, et al. Cannabidiol as a Promising Strategy to Treat and Prevent Movement Disorders?. Front Pharmacol. (2018)
26- McGuire P, et al. Cannabidiol (CBD) as an Adjunctive Therapy in Schizophrenia: A Multicenter Randomized Controlled Trial. Am J Psychiatry. (2018)
27- Leweke FM, et al. Cannabidiol enhances anandamide signaling and alleviates psychotic symptoms of schizophrenia. Transl Psychiatry. (2012)
28- Boggs DL, et al. The effects of cannabidiol (CBD) on cognition and symptoms in outpatients with chronic schizophrenia a randomized placebo controlled trial. Psychopharmacology (Berl). (2018)
29- Zuardi AW, et al. Cannabidiol monotherapy for treatment-resistant schizophrenia. J Psychopharmacol. (2006)
30- Stockings E, et al. Evidence for cannabis and cannabinoids for epilepsy: a systematic review of controlled and observational evidence. J Neurol Neurosurg Psychiatry. (2018)
31- Devinsky O, et al. Open-label use of highly purified CBD (Epidiolex®) in patients with CDKL5 deficiency disorder and Aicardi, Dup15q, and Doose syndromes. Epilepsy Behav. (2018)
32- Warren PP, et al. The use of cannabidiol for seizure management in patients with brain tumor-related epilepsy. Neurocase. (2017)
33- Thiele E, et al. Cannabidiol in patients with Lennox-Gastaut syndrome: Interim analysis of an open-label extension study. Epilepsia. (2019)
34- Szaflarski JP, et al. Cannabidiol improves frequency and severity of seizures and reduces adverse events in an open-label add-on prospective study. Epilepsy Behav. (2018)
35- Szaflarski JP, et al. Long-term safety and treatment effects of cannabidiol in children and adults with treatment-resistant epilepsies: Expanded access program results. Epilepsia. (2018)
36- Sands TT, et al. Long-Term Safety, Tolerability, and Efficacy of Cannabidiol in Children with Refractory Epilepsy: Results from an Expanded Access Program in the US. CNS Drugs. (2019)
37- Ali S, Scheffer IE, Sadleir LG. Efficacy of cannabinoids in paediatric epilepsy. Dev Med Child Neurol. (2019)
38- Chen KA, et al. Cannabidiol for treating drug-resistant epilepsy in children: the New South Wales experience. Med J Aust. (2018)
39- Reddy DS, Golub VM. The Pharmacological Basis of Cannabis Therapy for Epilepsy. J Pharmacol Exp Ther. (2016)
40- Rosenberg EC, Patra PH, Whalley BJ. Therapeutic effects of cannabinoids in animal models of seizures, epilepsy, epileptogenesis, and epilepsy-related neuroprotection. Epilepsy Behav. (2017)
41- Bisogno T, et al. Molecular targets for cannabidiol and its synthetic analogues: effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br J Pharmacol. (2001)
42- Bialer M, et al. Progress report on new antiepileptic drugs: A summary of the Thirteenth Eilat Conference on New Antiepileptic Drugs and Devices (EILAT XIII). Epilepsia. (2017)
43- Palmieri B, Laurino C, Vadalà M. Short-Term Efficacy of CBD-Enriched Hemp Oil in Girls with Dysautonomic Syndrome after Human Papillomavirus Vaccination. Isr Med Assoc J. (2017)
44- Cuñetti L, et al. Chronic Pain Treatment With Cannabidiol in Kidney Transplant Patients in Uruguay. Transplant Proc. (2018)
45- Martin-Santos R, et al. Acute effects of a single, oral dose of d9-tetrahydrocannabinol (THC) and cannabidiol (CBD) administration in healthy volunteers. Curr Pharm Des. (2012)
46- a b c Shannon S, et al. Cannabidiol in Anxiety and Sleep: A Large Case Series. Perm J. (2019)
47- Zuardi AW, et al. Effects of ipsapirone and cannabidiol on human experimental anxiety. J Psychopharmacol. (1993)
48- Zuardi AW, et al. Inverted U-Shaped Dose-Response Curve of the Anxiolytic Effect of Cannabidiol during Public Speaking in Real Life. Front Pharmacol. (2017)
49- Bergamaschi MM, et al. Cannabidiol reduces the anxiety induced by simulated public speaking in treatment-naïve social phobia patients. Neuropsychopharmacology. (2011)
50- Crippa JA, et al. Cannabidiol for the treatment of cannabis withdrawal syndrome: a case report. J Clin Pharm Ther. (2013)
51- Morgan CJ, et al. Cannabidiol reduces cigarette consumption in tobacco smokers: preliminary findings. Addict Behav. (2013)
52- Hindocha C, et al. Cannabidiol reverses attentional bias to cigarette cues in a human experimental model of tobacco withdrawal. Addiction. (2018)
53- Hindocha C, et al. The effects of cannabidiol on impulsivity and memory during abstinence in cigarette dependent smokers. Sci Rep. (2018)
54- Ren Y, et al. Cannabidiol, a nonpsychotropic component of cannabis, inhibits cue-induced heroin seeking and normalizes discrete mesolimbic neuronal disturbances. J Neurosci. (2009)
55- Hurd YL, et al. Early Phase in the Development of Cannabidiol as a Treatment for Addiction: Opioid Relapse Takes Initial Center Stage. Neurotherapeutics. (2015)
56- Elms L, et al. Cannabidiol in the Treatment of Post-Traumatic Stress Disorder: A Case Series. J Altern Complement Med. (2019)
57- Linares IMP, et al. No Acute Effects of Cannabidiol on the Sleep-Wake Cycle of Healthy Subjects: A Randomized, Double-Blind, Placebo-Controlled, Crossover Study. Front Pharmacol. (2018)
58- Sulé-Suso J, et al. Striking lung cancer response to self-administration of cannabidiol: A case report and literature review. SAGE Open Med Case Rep. (2019)
59- Dall’Stella PB, et al. Case Report: Clinical Outcome and Image Response of Two Patients With Secondary High-Grade Glioma Treated With Chemoradiation, PCV, and Cannabidiol. Front Oncol. (2019)
60- Kenyon J, Liu W, Dalgleish A. Report of Objective Clinical Responses of Cancer Patients to Pharmaceutical-grade Synthetic Cannabidiol. Anticancer Res. (2018)
61- Naftali T, et al. Low-Dose Cannabidiol Is Safe but Not Effective in the Treatment for Crohn’s Disease, a Randomized Controlled Trial. Dig Dis Sci. (2017)
62- Irving PM, et al. A Randomized, Double-blind, Placebo-controlled, Parallel-group, Pilot Study of Cannabidiol-rich Botanical Extract in the Symptomatic Treatment of Ulcerative Colitis. Inflamm Bowel Dis. (2018)
63- “Athlete Advisory Note: Cannabidiol (CBD)”. United Kingdom Anti-Doping Agency. Retrieved October 9, 2019.
64- “Athletes: 6 things to know about cannabidiol”. US Anti-Doping Agency. October 23, 2018. Retrieved February 17, 2020.
65- a b c Donna Spencer (October 2, 2019). “Canopy cannabis company buys ex-NHL player’s sports nutrition business”. CTV Business. The Canadian Press. Retrieved February 17, 2020. BioSteel’s brand ambassadors also include well-known athletes across major sports leagues in North America, which could be beneficial as the company’s attempt to push regulated CBD nutrition products into the mainstream health and wellness segments
66- Amanda Loudin (December 7, 2019). “As more pro athletes use cannabis for aches and pain, the more they run afoul of rules”. The Washington Post. Retrieved February 17, 2020.
67- Sara Harrison (September 28, 2019). “Lots of athletes say CBD Is a better painkiller. Is It?”. Wired. Retrieved February 17, 2020.
68- Gaston TE, et al. Interactions between cannabidiol and commonly used antiepileptic drugs. Epilepsia. (2017)
69- Ujváry I, Hanuš L. Human Metabolites of Cannabidiol: A Review on Their Formation, Biological Activity, and Relevance in Therapy. Cannabis Cannabinoid Res. (2016)
70- Welty TE, Luebke A, Gidal BE. Cannabidiol: promise and pitfalls. Epilepsy Curr. (2014)
71- Brzozowska N, et al. ABC transporters P-gp and Bcrp do not limit the brain uptake of the novel antipsychotic and anticonvulsant drug cannabidiol in mice. PeerJ. (2016)
72- Stott C, et al. A Phase I, open-label, randomized, crossover study in three parallel groups to evaluate the effect of Rifampicin, Ketoconazole, and Omeprazole on the pharmacokinetics of THC/CBD oromucosal spray in healthy volunteers. Springerplus. (2013)
73- Devinsky O, et al. Effect of Cannabidiol on Drop Seizures in the Lennox-Gastaut Syndrome. N Engl J Med. (2018)
74- Manini AF, et al. Safety and pharmacokinetics of oral cannabidiol when administered concomitantly with intravenous fentanyl in humans. J Addict Med. (2015)
75- Arndt DL, de Wit H. Cannabidiol Does Not Dampen Responses to Emotional Stimuli in Healthy Adults. Cannabis Cannabinoid Res. (2017)
76- Bergamaschi MM, et al. Safety and side effects of cannabidiol, a Cannabis sativa constituent. Curr Drug Saf. (2011)
77- Schuel H, et al. Cannabinoids inhibit fertilization in sea urchins by reducing the fertilizing capacity of sperm. Pharmacol Biochem Behav. (1991)
78- Stout SM, Cimino NM. Exogenous cannabinoids as substrates, inhibitors, and inducers of human drug metabolizing enzymes: a systematic review. Drug Metab Rev. (2014)
79- Jara-Oseguera A, Simon SA, Rosenbaum T. TRPV1: on the road to pain relief. Curr Mol Pharmacol. (2008)
80- Russo EB. Current Therapeutic Cannabis Controversies and Clinical Trial Design Issues. Front Pharmacol. (2016)
81- Busse FP, et al. Lead poisoning due to adulterated marijuana in leipzig. Dtsch Arztebl Int. (2008)
82- Pavlovic R, et al. Quality Traits of “Cannabidiol Oils”: Cannabinoids Content, Terpene Fingerprint and Oxidation Stability of European Commercially Available Preparations. Molecules. (2018)
Androgenico: 78-254
Anabolico: 107 (valore sovrastimato)
Standard: Testosterone
Nome Chimico: 17α-Methyl-4,5α-dihydrotestosterone
Attività estrogenica: nessuna
Attività Progestinica: nessuna
Aromatizzazione: no
Il Mestanolone, noto anche come 17α-methyl-4,5α-Diidrotestosterone (17α-methyl-DHT) o 17α-methyl-5α-androstan-17β-ol-3-one, è uno steroide androgeno anabolizzante (AAS), commercializzato in forma orale sotto il nome di Androstalone e Ermalone, ad oggi per lo più in disuso in ambito medico.[1][2][3][4]
Il Mestanolone è uno steroide androstano sintetico e un derivato del Diidrotestosterone (DHT) metilato in posizione C17α. [1][4] Infatti, differisce dal DHT solo per la presenza del gruppo metile nella posizione C17α.[1][4] Stretti parenti sintetici del Mestanolone includono Mesterolone (1α-Methyl-4,5α-dihydrotestosterone), Oxandrolone (2-oxa-17α-methyl-DHT), Oxymetholone (2-idrossimetilene-17α-methyl-DHT) e Stanozololo (un derivato del 17α-methyl-DHT (Mestanolone) con un anello pirazolico fuso con l’anello A.)[1][4]
Differenze nella struttura dello scheletro carbossilico tra Mestanolone (metilazione in C17α) e Mesterolone (metilazione in C1α).
Il Mestanolone fu sintetizzato per la prima volta nel 1935 insieme al Methyltestosterone e al Methandriolo.[5][6] È stato sviluppato dalla Roussel negli anni ’50 ed è stato introdotto per uso medico, con i marchi Androstalone ed Ermalone, intorno al 1960.[4][10][8] Venne inizialmente commercializzato in Germania.[4] Inizialmente si pensava che il farmaco fosse un potente agente anabolizzante, ma le ricerche successive hanno dimostrato che in realtà ha effetti anabolici relativamente deboli ed esplica principalmente azione androgena.[4] Il Mestanolone, insieme al molto più conosciuto 4-Chlorodehydromethyltestosterone (Oral Turinabol) è stato utilizzato come agente dopante negli atleti olimpionici della Germania Orientale all’interno di un programma di doping sponsorizzato dallo stato negli anni ’70 e ’80.[4] Il motivo del suo uso, come precedentemente accennato, era giustificato maggiormente dal suo valore androgeno piuttosto che su quello anabolizzante. Il suo uso garantiva un ottima risposta neuro steroidea con conseguente stimolazione del Sistema Nervoso Centrale e migliorata interazione neuromuscolare, con vantaggi nella velocità, nella forza, nell’aggressività, nella concentrazione, nella resistenza fisica e allo stress mentale.[4] Oggi, l’uso del Mestanolone è stato per lo più sospeso in medicina, sebbene sia ancora disponibile in Giappone.[2][3][4] Il Mestanolone era comunemente disponibile sotto forma di compresse sublinguali da 25mg (marchio Ermalone).[7]
Il Mestanolone è un AAS con effetti molto simili all’Androstanolone (diidrotestosterone; DHT) essendo praticamente una versione orale di quest’ultimo.[4] A causa dell’inattivazione da parte della 3α-idrossisteroide deidrogenasi (3α-HSD) nel muscolo scheletrico, il Mestanolone, sebbene dotato di una metilazione in posizione C-17 la quale ne migliora la stabilità del legame recettoriale, è descritto come un agente anabolizzante molto scarso, analogamente all’Androstanolone e al Mesterolone.[4] Poiché il Mestanolone è un composto 5α ridotto, non è un substrato soggetto all’enzima aromatasi e. quindi, non convertendo in estrogeno oltre a non possedere attività estrogenica intrinseca.[4] Inoltre, il farmaco non ha attività progestinica.[4] Come per gli altri AAS 17α-alchilati, il Mestanolone presenta un certo grado di epatotossicità.[4]
Differenze nella struttura dello scheletro carbossilico tra Diidrotestosterone e Mestanolone (aggiunta metilazione in C17α).
Come risaputo, gli AAS, a diverso grado di impatto, possono avere effetti deleteri sul colesterolo sierico. Il Mestanolone non è da meno presentando la tendenza a causare una riduzione delle concentrazioni di colesterolo HDL (“buono”) e un aumento delle concentrazioni di colesterolo LDL (“cattivo”), cosa che comporta uno sbilanciamento dell’equilibrio HDL/LDL che si traduce in un rischio maggiore di sviluppare arteriosclerosi. L’impatto relativo all’assunzione di un AAS nei confronti dei lipidi ematici dipende dalla dose, dalla via di somministrazione (per via orale o iniettabile), dal tipo di steroide (aromatizzabile o non aromatizzabile), e dal livello di resistenza al metabolismo epatico.
Essendo il Mestanolone un AAS con consistente attività androgenica, la soglia dei possibili forti effetti collaterali androgenici è generalmente alta ed è paragonabile a quella riscontrabile con altri composti come il Mesterolone. Per questa ragione, il suo uso in ambito femminile non è stato molto diffuso dal momento che poteva essere causa di severi effetti virilizzanti.
Quando l’uso del Mestanolone veniva applicato in campo sportivo, i dosaggi comunemente utilizzati erano mediamente tra i 10 ed i 20mg al giorno con un timing di somministrazione tra una dose e la successiva di 12 ore. I tempi di utilizzo rimanevano entro le 6-8 settimane onde evitare di creare un eccessivo stress epatico. Tra i bodybuilder, il Mestanolone era spesso inserito durante la preparazione alla gara vista la sua facile gestibilità non causando ritenzione idrica, non essendo soggetto ad aromatizzazione, esercitando una blanda azione anti-estrogenica (sia recettoriale che come ligando inibitorio dell’enzima Aromatasi) e possedendo una buona azione lipolitica (legame con i AR adipocitari).
Come già detto in precedenza, l’uso del Mestanolone è stato per lo più sospeso in ambito medico anche se rimane disponibile in Giappone.[2][3][4] Nel mercato nero è raramente reperibile per via della sua attuale e pressoché assente richiesta tra gli atleti.
Gabriel Bellizzi
Riferimenti:
1- J. Elks (14 November 2014). The Dictionary of Drugs: Chemical Data: Chemical Data, Structures and Bibliographies. Springer. pp. 775.
2- Index Nominum 2000: International Drug Directory. Taylor & Francis. 2000. pp. 655.
4- William Llewellyn (2017). Anabolics 11th. Molecular Nutrition Llc. p. 284-285-286.
5- Schänzer W (1996). “Metabolism of anabolic androgenic steroids”. Clin. Chem. 42 (7): 1001–20.
6- Ruzicka, L.; Goldberg, M. W.; Rosenberg, H. R. (1935). “Sexualhormone X. Herstellung des 17-Methyl-testosterons und anderer Androsten- und Androstanderivate. Zusammenhänge zwischen chemischer Konstitution und männlicher Hormonwirkung”. Helvetica Chimica Acta. 18 (1): 1487–1498.
7- H.-L. Krüskemper (22 October 2013). Anabolic Steroids. Elsevier. pp. 196.

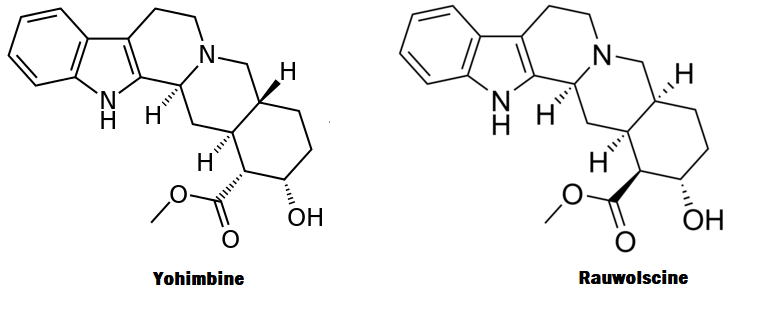
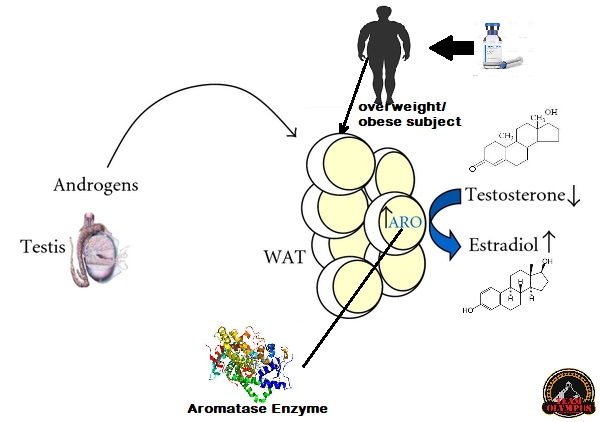
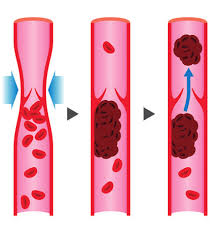



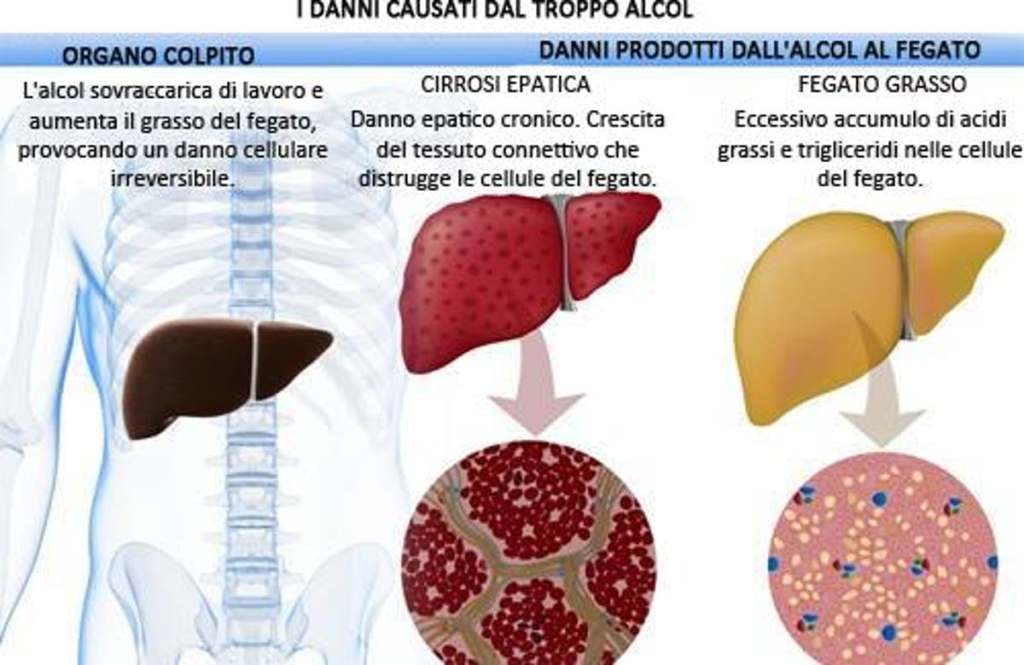











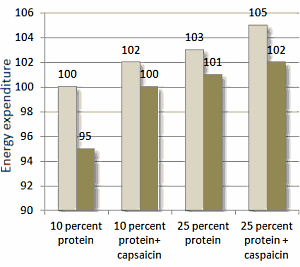
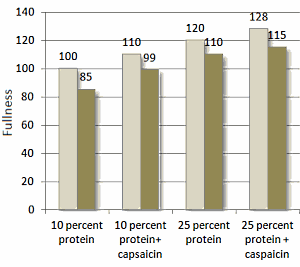




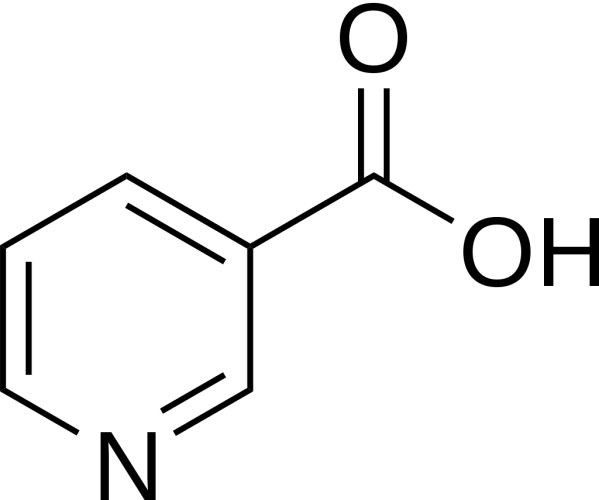
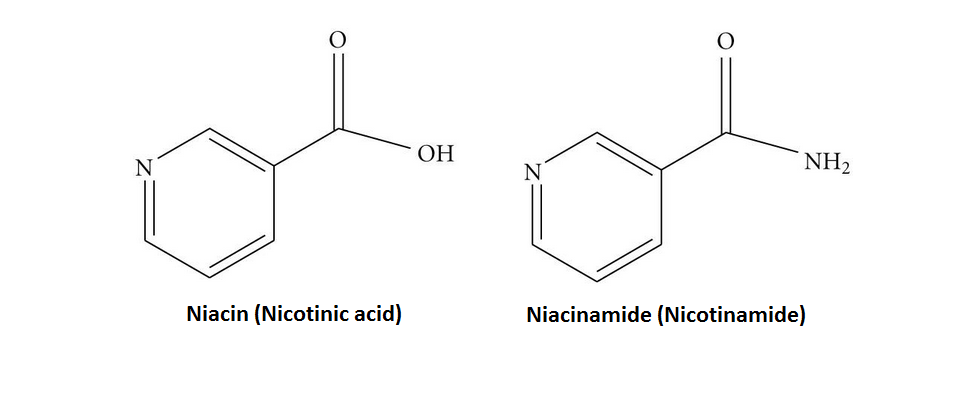

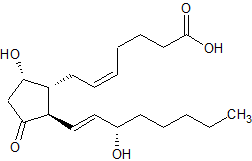

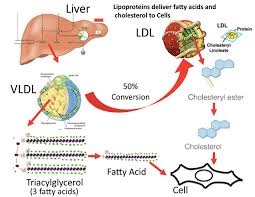
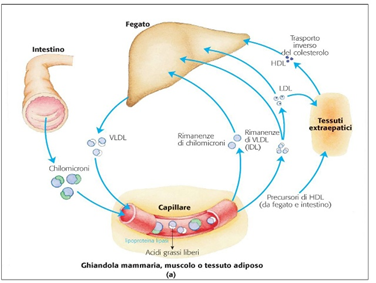
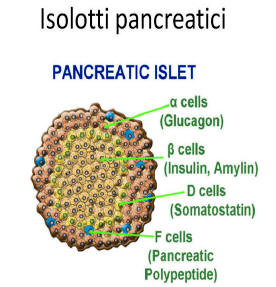
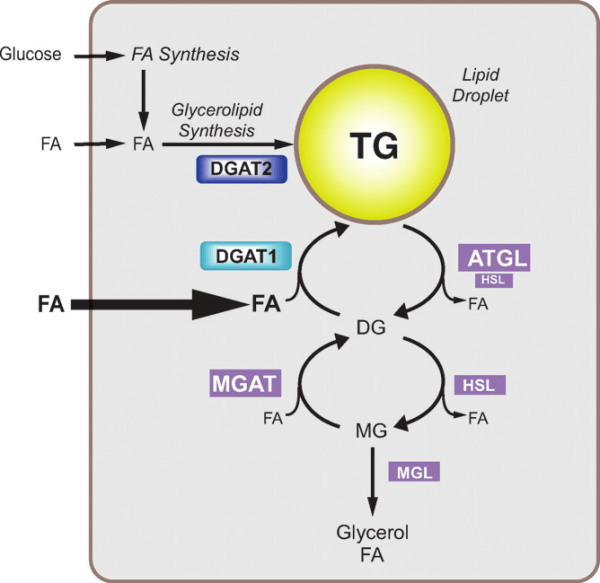
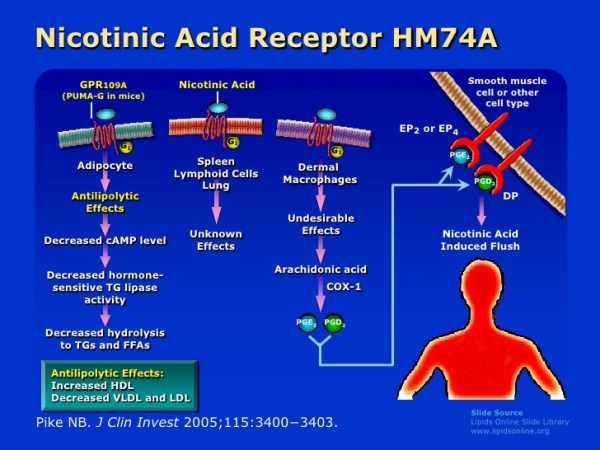
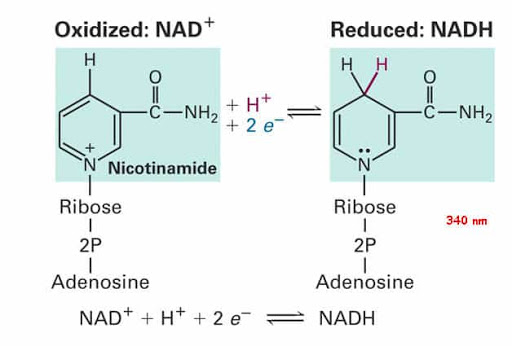

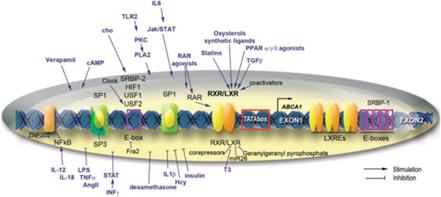

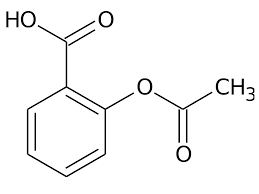
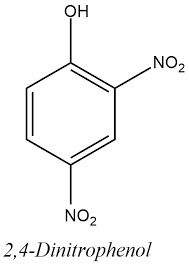

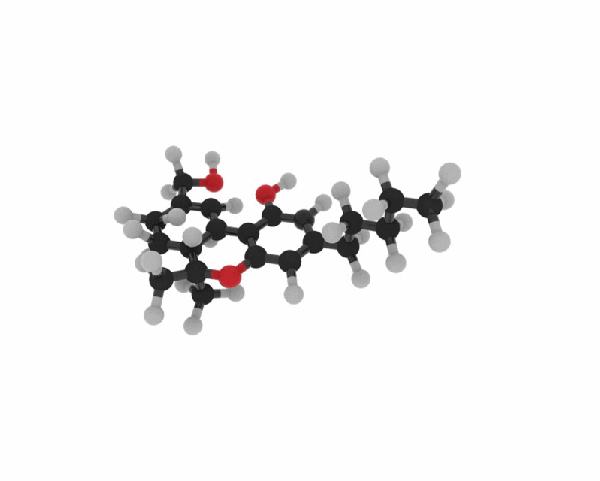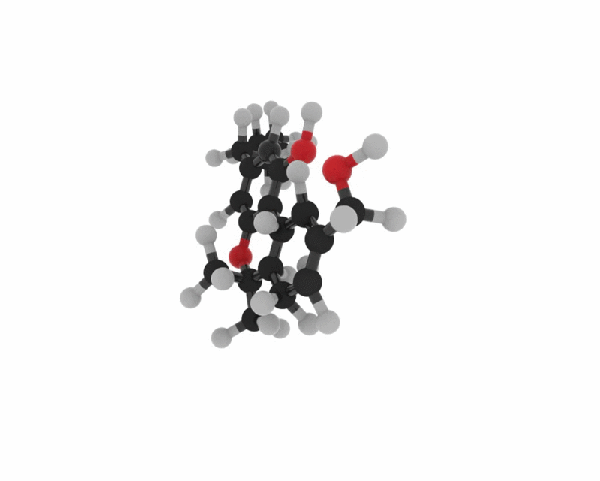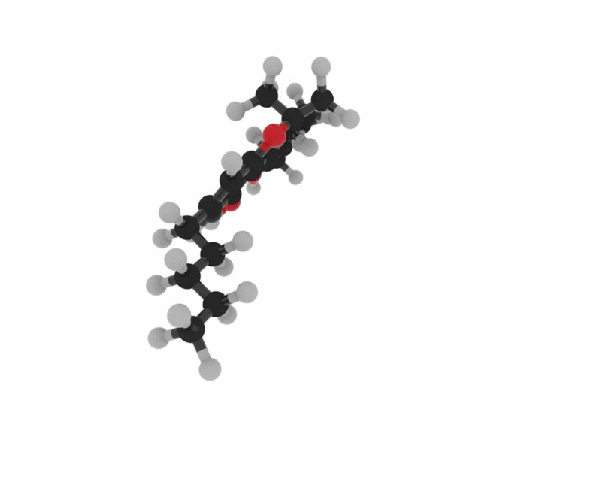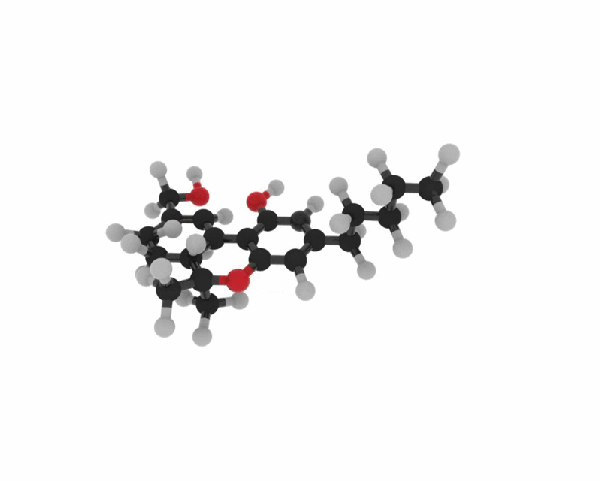


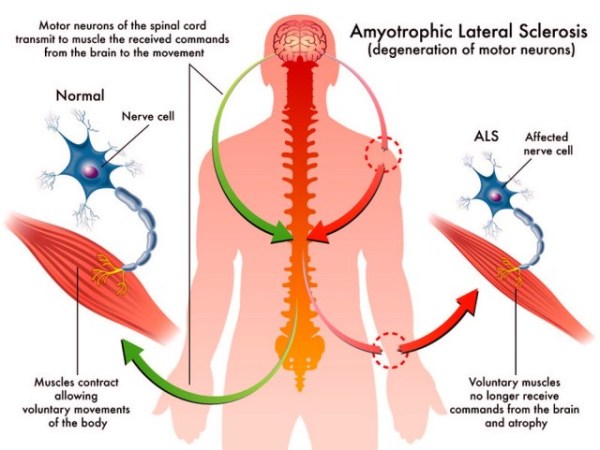

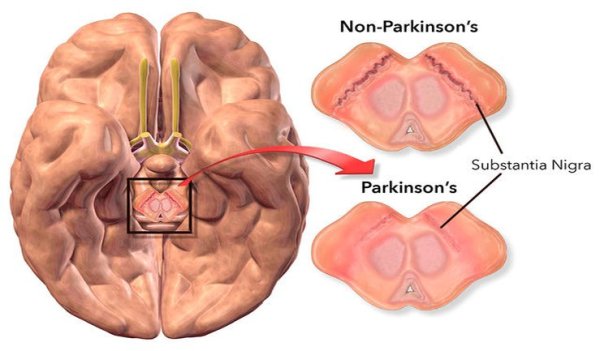
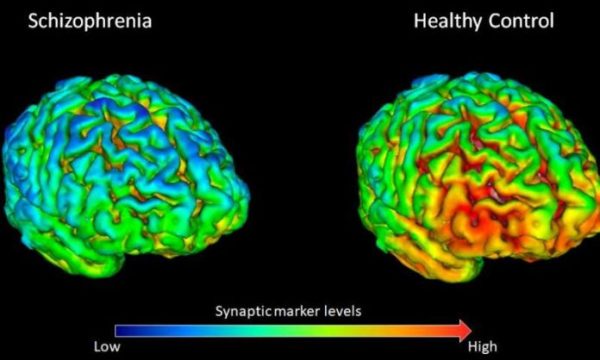


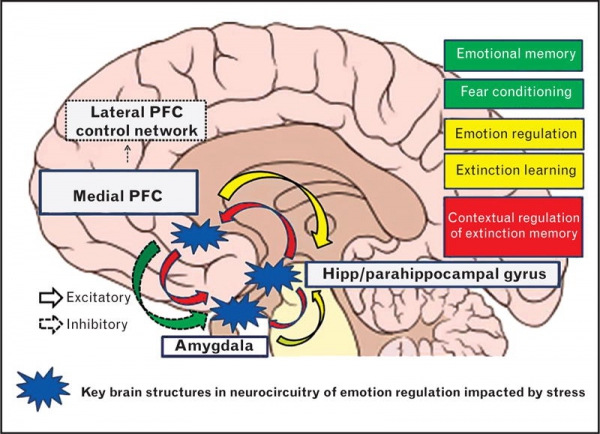





 to come agente dopante negli atleti olimpionici della Germania Orientale all’interno di un programma di doping sponsorizzato dallo stato negli anni ’70 e ’80.[4] Il motivo del suo uso, come precedentemente accennato, era giustificato maggiormente dal suo valore androgeno piuttosto che su quello anabolizzante. Il suo uso garantiva un ottima risposta neuro steroidea con conseguente stimolazione del Sistema Nervoso Centrale e migliorata interazione neuromuscolare, con vantaggi nella velocità, nella forza, nell’aggressività, nella concentrazione, nella resistenza fisica e allo stress mentale.[4] Oggi, l’uso del Mestanolone è stato per lo più sospeso in medicina, sebbene sia ancora disponibile in Giappone.[2][3][4] Il Mestanolone era comunemente disponibile sotto forma di compresse sublinguali da 25mg (marchio Ermalone).[7]
to come agente dopante negli atleti olimpionici della Germania Orientale all’interno di un programma di doping sponsorizzato dallo stato negli anni ’70 e ’80.[4] Il motivo del suo uso, come precedentemente accennato, era giustificato maggiormente dal suo valore androgeno piuttosto che su quello anabolizzante. Il suo uso garantiva un ottima risposta neuro steroidea con conseguente stimolazione del Sistema Nervoso Centrale e migliorata interazione neuromuscolare, con vantaggi nella velocità, nella forza, nell’aggressività, nella concentrazione, nella resistenza fisica e allo stress mentale.[4] Oggi, l’uso del Mestanolone è stato per lo più sospeso in medicina, sebbene sia ancora disponibile in Giappone.[2][3][4] Il Mestanolone era comunemente disponibile sotto forma di compresse sublinguali da 25mg (marchio Ermalone).[7]