Introduzione:
Come intuibile dal titolo, questa volta il mio intento non sarà quello di esporre una semplice descrizione “commerciale” del Methenolone, ma ben si una sua dettagliata descrizione molecolare e bio-attiva a livello muscolare.
Per molti professionisti e profani, il Methenolone è un AAS “debole” con una capacità miotrofica poco inferiore a quella del Testosterone. Questo effetto è generalmente attribuito al metabolismo di questa molecola. In particolare, la cosiddetta 3α-riduzione che è nota avvenire nel muscolo scheletrico, e che rende AAS come il Mesterolone, soggetto a questa riduzione, con un valore miotrofico dipendente dal legame AR molto scarso. Ma, dal momento che si sta comunque parlando di una 3α-riduzione, vorrei anche sottolineare che questo è probabilmente il motivo per cui l’AAS Methenolone ha proprietà di stimolo ipertrofico della massa muscolare così relativamente deboli. Si lega all’AR tanto quanto il Testosterone e in uno studio dove veniva misurata l’attività androgena, la sua potenza relativa è persino superiore a quella del Testosterone.[1] Allora come si può considerare questo AAS come una molecola debole? Bene, questo studio è stato svolto utilizzando cellule del osteosarcoma, e non cellule muscolo-scheletriche. Le cellule muscolo-scheletriche esprimono l’enzima 3α-HSD e infatti i metaboliti primari del Methenolone sono 3α-ridotti. Questi metaboliti 3α ridotti sono il risultato di una singola reazione chimica, catalizzata dal 3α-HSD. Questo praticamente non avviene con il Testosterone, il quale è un cattivo substrato per questo enzima. Questi risultati suggeriscono che il Methenolone subisce modifiche strutturali in misura apprezzabile nel muscolo scheletrico, rendendolo meno potente in questo tessuto. Quindi, la sua attività nel muscolo scheletrico (e in altri tessuti che esprimono il 3α-HSD) subisce una riduzione, mentre ciò non si verifica nei tessuti che ne sono privi, rendendolo quindi una cattiva scelta per chi cerca AAS con un effetto miotrofico rilevante.
No, l’articolo non finisce qui… siamo solo all’introduzione…
In questo articolo cercherò di presentare le prove disponibili in merito alle affermazioni sopra esposte in modo equilibrato. Esaminerò ciò che si sa sul metabolismo del Methenolone, le insidie che possono esserci nei test degli androgeni e alcuni degli studi clinici che hanno utilizzato il Methenolone.
Methenolone – caratteristiche molecolari e metabolismo – :
Prima di entrare nel dettaglio del metabolismo del Methenolone, è utile esaminare brevemente la sua formula strutturale. Dopo tutto, il suo metabolismo è il risultato della sua struttura chimica.
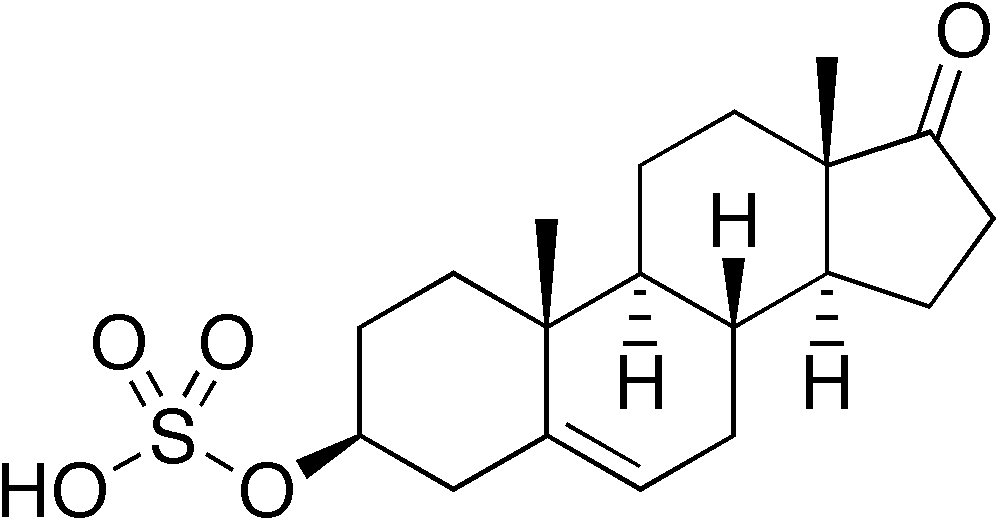
Il Methenolone [17beta-Hydroxy-1-methyl-5alpha-androst-1-en-3-one] è stato descritto per la prima volta nel 1960.(2) Squibb introdusse il farmaco (nella forma orale e iniettabile) negli Stati Uniti nel 1962.(3) Si trattava e, ovviamente, si tratta di un AAS derivato dal DHT con valore androgeno/anabolizzante pari a 44-57:88 . Si tratta, inoltre, di una molecola presente in natura, sebbene essa si trovi all’interno delle ghiandole surrenali dei felini domestici in gravidanza.(4)
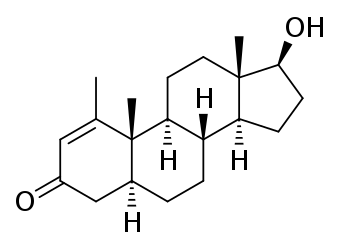
Nota: Il Methenolone, essendo una molecola 5-alfa ridotta (in quanto derivato del DHT) non subisce alcuna aromatizzazione (5) e non presenta alcuna attività estrogenica misurabile.
Le caratteristiche chimiche che sono importanti sono il gruppo cheto (= O) al carbonio 3, l’atomo di idrogeno α-orientato (-H) al carbonio 5, il gruppo metile (-CH3) attaccato al carbonio 1 e il doppio legame (C = C) tra gli atomi di carbonio 1 e 2.
Il gruppo cheto al carbonio 3 e l’atomo di idrogeno α-orientato al carbonio 5 sono presenti anche nel Diidrotestosterone (DHT). Gli steroidi con queste due caratteristiche chimiche generalmente formano buoni substrati per enzimi che riducono il gruppo cheto al carbonio 3. Questi enzimi sono indicati come 3α / 3β-idrossisteroide deidrogenasi. Il risultato è un gruppo idrossile (-OH). Il gruppo 3-cheto è molto importante in termini di affinità di legame per il recettore degli androgeni. Quasi tutti gli steroidi anabolizzanti comuni hanno un gruppo 3-cheto: Testosterone, Nandrolone, Trenbolone, Boldenone, Drostanolone, Oxandrolone, Fluoxymesterone, Oxymetholone, Methandienone, 4-Chlorodehydromethyltestosterone, ecc. La ragione di ciò è che l’atomo di ossigeno può funzionare come un accettore di legami idrogeno per gli amminoacidi carichi del recettore degli androgeni che si trovano nelle immediate vicinanze dell’anello A quando si legano [6]. Se riduci questo atomo di ossigeno, non ci sono coppie di elettroni che funzionino come accettori di legami idrogeno, e quindi l’affinità di legame diminuisce enormemente. Lo Stanozololo potrebbe essere un’eccezione strutturale, poiché non ha un gruppo 3-cheto (né un gruppo idrossile li), ma accade la stessa cosa. Il secondo atomo di azoto dell’anello pirazolico dello Stanozololo può funzionare come accettore di legami idrogeno. Il messaggio da portare a casa qui è che, una volta che uno steroide vede ridotto il suo gruppo 3-cheto, è effettivamente reso inutile.

Gli esseri umani esprimono 4 diversi cosiddetti isoenzimi che possono catalizzare la riduzione 3α [7]. Questi isoenzimi sono denominati da AKR1C1 a AKR1C4. Tutti sono in grado di catalizzare la riduzione 3α, ma differiscono per la distribuzione tissutale e la specificità del substrato. Questo è in netto contrasto nel ratto, che esprime solo un singolo enzima in grado di ridurre il 3α. A causa della presenza di più enzimi in grado di catalizzare la riduzione del 3α, nonché del fatto che il singolo nei ratti non è identico a nessuno dei 4 isoenzimi nell’uomo, è necessario prestare molta attenzione quando si estrapola un dato dai ratti e lo si trasferisce all’uomo.
Gli isoenzimi AKR1C2 e AKR1C4 sono quelli che sembrano dimostrare la maggiore attività di 3α-riduzione. AKR1C4 è espresso in gran parte nel fegato, ma solo in misura minore nel polmone, nella prostata, nella ghiandola mammaria, nel cervello, nell’intestino tenue e nei testicoli.[7] In quanto tale, AKR1C4 sembra essere una forma specifica del fegato. AKR1C2 d’altra parte, oltre ad essere espresso nel fegato, è anche espresso in larga misura nei polmoni e nella prostata. Alcune espressioni più piccole, ma significative, si verificano anche in altri tessuti.
“E i muscoli-scheletrici?”. Sfortunatamente, lo studio che ho menzionato sopra non ha verificato questo punto. Un altro studio ha verificato la presenza nel muscolo scheletrico, ma lo ha fatto solo per AKR1C1 e AKR1C4.[8] Entrambi non sono stati rilevati nel muscolo scheletrico. Ancora un altro studio ha scoperto che AKR1C3 è espresso nei muscoli e che la sua espressione attraverso l’mRNA è sovraregolata con la terapia con Testosterone (250 mg di Sustanon ogni due settimane). [9] Non sono riuscito a trovare nessuno studio che esamini l’espressione del AKR1C2 nel muscolo scheletrico.
Finora abbiamo stabilito che almeno uno, e forse due, degli isoenzimi che possono ridurre gli steroidi 3α sono espressi nel muscolo scheletrico. Esistono quindi anche studi che dimostrano un’attività significativa di questi enzimi nel muscolo scheletrico? Un vecchio lavoro tedesco lo ha dimostrato elegantemente iniettando per via endovenosa DHT tracciato a tre partecipanti al fine di monitorare il suo destino metabolico [10]. I campioni di tessuto muscolare sono stati raccolti da 20 a 60 minuti dopo l’iniezione. Il 29,4-51,0% dello steroide marcato è stato identificato come 3α-androstandiolo (il metabolita 3α-ridotto del DHT) in questi campioni. Sono stati identificati anche un paio di altri metaboliti e solo dal 18,1 al 26,2% di DHT era rimasto integro. Ciò dimostra infatti una significativa modifica del DHT che si verifica nel tessuto muscolo scheletrico come risultato della riduzione del suo gruppo 3-cheto. Come nota a margine, hanno fatto la stessa cosa con il Testosterone marcato, di cui il 69,5-79,8% dello steroide marcato era ancora identificato come Testosterone nei campioni di tessuto muscolare.
E il Methenolone? Come accennato all’inizio di questa sezione dell’articolo, esso presenta un gruppo cheto al carbonio 3 e un atomo di idrogeno α-orientato al carbonio 5. Il motivo per cui non segue necessariamente lo stesso destino del DHT, è a causa delle altre 2 alterazioni : il gruppo metile attaccato al carbonio 1 e il doppio legame tra il carbonio 1 e 2. Cosa si sa del metabolismo del Methenolone? La tabella seguente proviene da uno studio condotto dal produttore del Methenolone (Schering, oggi Bayer). [11]

Le parole “stark gehemmt” sono tedesche e stanno per “fortemente inibito”. Come tale, questa tabella suggerisce che la riduzione 3α del Methenolone è fortemente inibita. Ciò è quanto seguirebbe dalla tabella 5 dello stesso documento. La Tabella 5 elenca 2 potenziali metaboliti 3α-ridotti del Methenolone che hanno controllato. Infatti, uno di loro, 1- (α) -Methyl-androstan-3α-ol-17-on (Methylandrosterone), è stato elencato come non rilevabile. L’altro, androstan-3α-ol-17-on (androsterone), è stato elencato come (+), il che significa che ne hanno trovato una traccia. Lo stesso Methenolone è stato recuperato nella misura massima, elencato come +++ nella tabella. (Indubbiamente questo è stato coniugato, poiché la ricerca successiva non è stata in grado di recuperare il Methenolone immodificato dall’urina dopo l’iniezione [12].)
Il Methylandrosterone è una forma 3α ridotta di Methenolone che manca del doppio legame tra il carbonio 1 e 2. L’altro metabolita, Androsterone, manca inoltre del gruppo 1α-metile. Ma questi non sono gli unici potenziali metaboliti 3α-ridotti del Methenolone. In effetti, ricerche successive hanno dimostrato che dopo la somministrazione orale, il metabolita principale del Methenolone era il 3α-idrossi-1-metilen-5a-andorstan-17-one [13]. In effetti, è stato escreto in misura maggiore del 50% circa rispetto allo stesso Methenolone (coniugato). In quanto tale, la nota che la riduzione 3α è “fortemente inibita” nella tabella sopra si basa su uno screening incompleto dei metaboliti e dovrebbe essere scartata come falsa data la ricerca che è emersa in seguito.
Tuttavia, si nota che hanno estratto l’1,62-1,65% del Methenolone ingerito come immodificato nelle urine e il 2,4% della dose di Methenolone escreta come metabolita 3α-ridotto (3α-idrossi-1-metilen-5α-andorstan-17-one ) entro 7 giorni dall’ingestione. Non menzionano la % di altri metaboliti, ma si può dedurre che cumulativamente costituissero non più dell’1% della dose di Methenolone. Tutto sommato, circa il 95% della dose ingerita rimane dispersa. Ciò può essere spiegato da due fattori:
- Mancano metaboliti aggiuntivi
- Escrezione attraverso altre vie (feci attraverso il riciclo entroepatico).
Ricerche svolte successivamente. e che utilizzano una tecnica all’avanguardia (LiQuiD ChRoMaToGrApHy QuAdRuPoLe TiMe-Of-FliGhT mAsS sPeCtRoMeTry aka LC-QTOFMS), hanno effettivamente trovato diversi metaboliti aggiuntivi dopo l’iniezione di Methenolone [14]. Tuttavia, non hanno eseguito un’analisi quantitativa, quindi non può essere derivata la parte % mancante della dose di Methenolone. È mia opinione che la maggior parte finisca nelle feci piuttosto che i metaboliti non rilevati costituiscano la maggior parte della dose non quantificabile. Inoltre, lo studio [12] non ha trovato Methenolone letteralmente invariato nelle urine (hanno trovato Methenolone coniugato senza altre alterazioni chimiche). Ciò significa che il Methenolone immodificato riportato in altri studi deve essere stato un miscuglio con un metabolita diverso, oppure i gruppi sulfo o acido glucuronico sono stati rimossi dopo la raccolta del campione di urina. In ogni caso, il messaggio da portare a casa è che il Methenolone si 3α-riduce significativamente, ma in misura minore rispetto al DHT. Le domande senza risposta sono in che misura ciò avvenga nel muscolo scheletrico (dati i diversi isoenzimi) e con quale velocità. Queste domande sono senza risposta con i dati attuali.
Approfondimenti da non trascurare:
Dato che il Methenolone si 3α-riduce e il muscolo scheletrico esprime enzimi che possono catalizzare questa reazione, si può sostenere che l’effetto del Methenolone nel muscolo scheletrico diminuisce. Non è chiaro in che misura ciò accada. Inoltre, altri tessuti sensibili agli androgeni che esprimono enzimi in grado di farlo diminuiranno anche il suo effetto in questi. Si può quindi sostenere che in alcuni tessuti sensibili agli androgeni anche il suo effetto sarà ridotto. Quello che sto cercando di dire qui è che da un lato può essere dannoso per la suo cosiddetta Anabolizzante/Androgeno ratio, mentre dall’altro potrebbe essere utile, a seconda del tessuto.
Quantificare la anabolico-androgeno ratio per un determinato AAS è qualcosa che alcuni test hanno cercato di fare. L’esempio principale di tale analisi è il dosaggio Hershberger. Questo test è stato descritto per la prima volta da Hershberger e dai suoi colleghi nel 1953 [15]. Per lo svolgimento si prendono dei ratti e li si castra per sbarazzarsi del Testosterone endogeno prodotto che potrebbe interferire con i risultati. Quindi si inietta loro l’AAS sotto ricerca e si aspetta un certo periodo di tempo (generalmente 8 giorni) prima di uccidere i ratti e sezionarli. Si sezionano per pesare il muscolo levator ani, la prostata ventrale e le vescicole seminali. Si suppone che l’aumento di peso del muscolo levator ani rifletta l’attività anabolica del composto e si suppone invece che l’aumento di peso della prostata ventrale e delle vescicole seminali rifletta la sua attività androgena.
I problemi con questo test sono numerosi. Innanzitutto, scegliere il muscolo levator ani come sostituto del muscolo scheletrico è un non lieve difetto di forma per l’esecuzione di uno studio con variabili accettabili, non per nulla ha ricevuto alcune critiche dalla comunità scientifica. Questo muscolo, a quanto pare, non è un normale muscolo scheletrico. È un muscolo fortemente dipendente dagli androgeni che fa parte del sistema riproduttivo del ratto (in realtà è il muscolo bulbocavernoso). Un articolo di Keith Hayes copre ampiamente questo aspetto [16]. In un certo senso, si potrebbe quindi sostenere che un aumento di peso del muscolo bulbocavernoso è rappresentativo dell’attività androgenica piuttosto che dell’attività anabolica. Un altro problema è che il muscolo bulbocavernoso e le vescicole seminali rispondono in modo diverso alle diverse concentrazioni di un androgeno. Come tale, il rapporto misurato sarà diverso a seconda della dose utilizzata e del momento in cui sono state effettuate le misurazioni. Ciò è ben illustrato nella figura seguente presa da van der Vies dopo l’iniezione di Nandrolone [17].
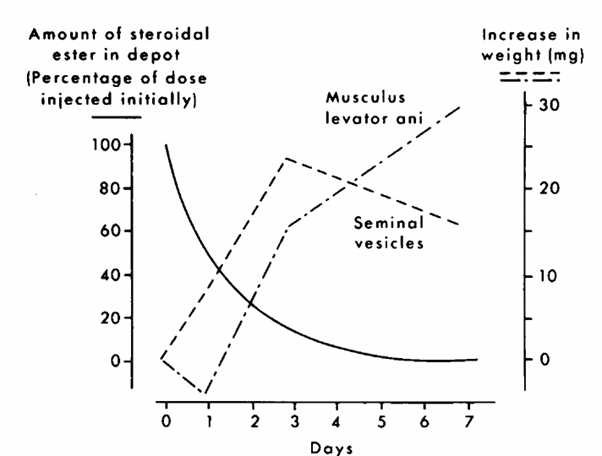
Durante i primi 3 giorni, la concentrazione è sufficientemente elevata da stimolare la crescita sia delle vescicole seminali che del muscolo bulbocavernoso. Tuttavia, dopo tre giorni la concentrazione non è abbastanza alta da mantenere questa crescita per le vescicole seminali e le loro dimensioni diminuiscono nuovamente. Tuttavia, il muscolo bulbocavernoso è ancora sufficientemente stimolato per continuare a crescere di dimensioni. In quanto tale, la androgeno/anabolico ratio misurata il giorno 3 sarà molto diversa da quella che si misurerà il giorno 7, nonostante si stia utilizzando lo stesso composto. Ciò evidenzia anche che organi diversi rispondono semplicemente in modo diverso a seconda della concentrazione del composto. Anche se ci fosse un modo accurato per determinare una androgeno/anabolico ratio, estrapolarlo da concentrazioni fisiologiche a concentrazioni sovrafisiologiche sarebbe fortemente soggetto ad errori interpretativi, se non completamente sbagliato!
Continuando con questo ragionamento logico, è un presupposto errato che la crescita della prostata ventrale o delle vescicole seminali sia rappresentativa di tutti gli altri effetti androgeni. Non ci sono prove a sostegno di questa ipotesi (anzi, le prove esistenti sono contrarie).
Infine, ovviamente, i ratti non sono esseri umani. È un altro enorme presupposto che i tessuti omologhi negli esseri umani risponderebbero allo stesso modo ad un AAS come osservato nel ratto. Nessun valore deve essere attribuito ai rapporti provenienti dal dosaggio Hershberger. Un altro metodo consiste nel misurare l’affinità di legame relativa di uno steroide in vari tessuti. Saartok et al. ha utilizzato questo metodo determinando l’affinità di legame relativa del Methenolone nel muscolo del coniglio, nel muscolo del ratto e nella prostata del ratto, rispetto al composto di riferimento (Methyltrienolone) [18]. Gli autori erano consapevoli dei problemi con il muscolo bulbocavernoso e, di conseguenza, sembra che abbiano utilizzato i muscoli soleo e gastrocnemio. I risultati sono stati i seguenti:
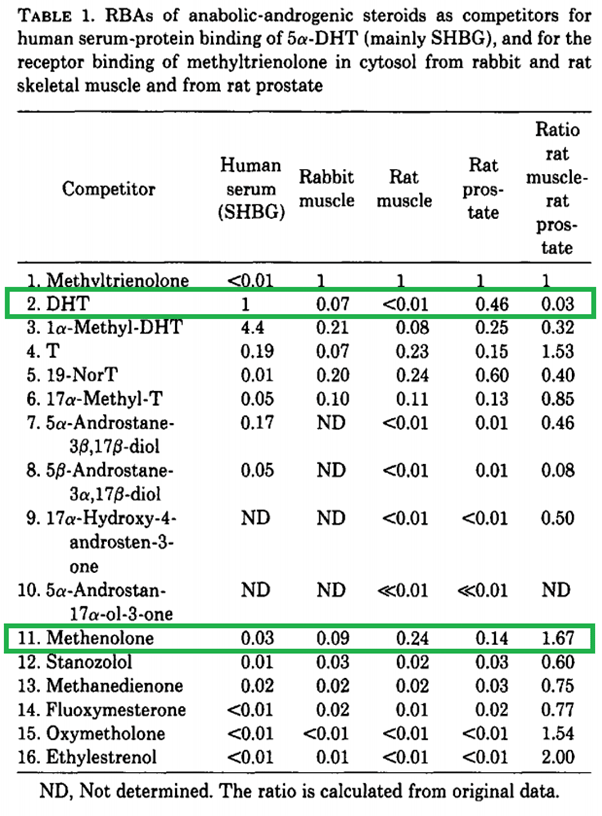
Diamo prima un’occhiata al DHT e confrontiamolo con Testosterone. Nel muscolo di ratto, l’affinità di legame è molto inferiore a quella del Testosterone. Ciò è molto probabilmente dovuto alla rapida metabolizzazione attraverso la 3α-riduzione nel muscolo scheletrico (come ipotizzato anche dagli autori). Tuttavia, l’affinità di legame è simile a quella del Testosterone nel muscolo del coniglio. Ciò evidenzia una notevole differenza tra le due specie animali. Apparentemente si verifica una 3α-riduzione del DHT significativamente inferiore nel muscolo scheletrico del coniglio. Ora, se guardiamo al Methenolone, le affinità di legame sono nello stesso campo di applicazione di quelle del Testosterone. Questo significa che non si 3α-riduce negli esseri umani? Assolutamente no. Come accennato in precedenza in questo articolo, ci sono diversi enzimi che agiscono in tal senso e che differiscono nella loro specificità del substrato rispetto ai ratti, e questo studio da solo mostra già una notevole differenza tra due specie animali quando si guarda al DHT.
L’inutilità di questo test per avere un’idea della androgeno/anabolico ratio (o della sola attività anabolica) negli esseri umani è ulteriormente evidenziata dalle affinità di legame ridicolmente basse di alcuni degli altri AAS elencati.
Un test finale di cui vorrei discutere è il dosaggio CALUX del recettore degli androgeni (AR) [19]. Il test è stato sviluppato per rilevare nuove sostanze dopanti negli atleti piuttosto che essere utilizzato per derivare questi rapporti o l’attività anabolica di un composto. Allo scopo di rilevare nuove sostanze dopanti, è perfetto. Fondamentalmente hanno modificato una cellula in modo tale che emani una bioluminescenza quando viene attivato il recettore degli androgeni. Lo hanno fatto co-trasfettando la cellula con un gene della luciferasi che è sotto il controllo trascrizionale del recettore degli androgeni. A questo scopo, hanno utilizzato cellule di osteosarcoma. Tratterò questo a breve, perché è uno dei problemi principali riguardanti il Methenolone dal momento che queste cellule chiaramente non hanno alcuna significativa attività nella 3α-riduzione. Ciò è dimostrato dal fatto che il DHT si presenta come un AAS estremamente potente in questo test. Quindi, si può anche leggere quello che hanno misurato con il Methenolone, ma ciò non dirà nulla su quello che realmente accade nel muscolo scheletrico con questo AAS.
Studi clinici:
In fine, quello che si vorrebbe sapere è come il Methenolone si sovrappone a un altro AAS in termini di effetti collaterali e crescita muscolare in uno studio clinico. Esistono studi del genere? Sì, ma effettuati in pazienti con cancro al seno e in assenza di misurazioni della crescita muscolare in un contesto clinico, praticamente irrilevante per i bodybuilder. Un’altra cosa che è possibile fare per dare risposta al quesito iniziale, è quella di confrontare ciò che è stato riportato negli studi clinici senza un confronto diretto tra AAS e mettere in rapporto ad altri studi che hanno utilizzato altri composti in funzione di confronto potenziale. Certo, questo è già un tentativo fallito in quanto il confronto tra i dati, specialmente con campioni di dimensioni inferiori, è molto problematico.
C’è uno studio che avrebbe potuto essere fantastico. Si tratta di una ricerca svolta reclutando 47 uomini i quali sono stati randomizzati a ricevere un placebo, Methenolone Acetato, placebo+ Methenolone Acetato e Methenolone Acetato + Allenamento [20]. Lo studio è durato ben 16 settimane. Hanno eseguito misurazioni della forza e del peso dei soggetti. Allora perché non c’è da entusiasmarsi? Perché hanno dato il Methenolone (20mg al giorno) per via orale invece che tramite iniezioni. Sebbene abbia una certa biodisponibilità orale, non è sicuramente all’altezza di quella di un AAS 17a metilato (la biodisponibilità del Methenolone Acetato in forma orale è molto simile a quella del Mesterolone assunto per la medesima via [3% circa]). In quanto tale, viene metabolizzato troppo rapidamente per esercitare un effetto apprezzabile a quel dosaggio. In effetti, gli autori non hanno riscontrato differenze tra i gruppi in nessuna delle misurazioni.
Un altro studio ha esaminato l’effetto di 100mg di Methenolone Enantato mediante iniezione intramuscolare una volta alla settimana per 6 settimane [19]. Tuttavia, è stato fatto insieme alla terapia riabilitativa in pazienti con emiplegia 1-8 mesi dopo l’ictus. Cioè, pazienti con paralisi completa di metà del corpo (ovviamente la terapia riabilitativa mirata alla metà del corpo non paralizzata.). Lo studio non era in cieco e non randomizzato ed i partecipanti ai gruppi placebo e Methenolone erano anziani (in media dai 62 ai 67 anni di età). Le misurazioni della forza erano aumentate, così come l’area della sezione trasversale muscolare delle cosce. È difficile, se non impossibile, dire come si comporta rispetto ad altri AAS. La “colpevolezza” del Methenolone non è tanto nel “non funzionare”: perchè, in effetti, funziona. Il colpevole è che la sua attività nel muscolo scheletrico potrebbe diminuire, rendendo non ottimale per acutizzare l’ipertrofia muscolare (anche se, in una certa misura, lo fa).
Esiste uno studio nel quale pazienti con cancro al seno (donne) hanno ricevuto Methenolone Enantato tramite iniezione intramuscolare [21]. Le donne hanno ricevuto prima 3x 400mg a settimana, dopodiché il dosaggio è stato ridotto a 3x 200mg o 3x 300mg a settimana. La terapia su queste donne è durata, in media, 7,7 mesi. C’è poco che può essere derivato da questo studio, a parte il fatto che il Methenolone causa un aumento del colesterolo totale in modo abbastanza significativo. Questo è qualcosa di condiviso con altri AAS, specie se DHT derivati, non soggetti ad aromatizzazione e orali metilati in C-17. E forse questo effetto potrebbe essere più pronunciato nelle donne, ma non è chiaro. Ad ogni modo, ho trovato uno studio in cui hanno fatto un confronto tra AAS in pazienti con carcinoma mammario avanzato [22]. Un gruppo ha ricevuto un dosaggio molto elevato di Methenolone (3x 400mg a settimana) e l’altro ha ricevuto un alto dosaggio di Testosterone Propionato (3x 100mg a settimana). Il 48% dei pazienti nel gruppo Methenolone ha avuto un miglioramento oggettivo del cancro al seno (e chiaramente un tempo di sopravvivenza più lungo), mentre nessuno ha mostrato miglioramenti nel gruppo Testosterone. È tutto molto interessante, certo, ma questo non ci porta oltre per quanto riguarda il confronto con altri composti in termini di ipertrofia muscolare. Se qualcuno è a conoscenza di qualche vecchio e oscuro studio clinico di confronto tra AAS, con presenza del Methenolone rispetto ad un altro AAS in persone sane, in cui è stata misurata la composizione corporea o la forza, per favore mi contattati.
Conclusioni:
Finora abbiamo stabilito alcune cose sul Methenolone.
Il Methenolone viene modificato in misura apprezzabile mediante la 3α-riduzione, come evidenziato dal suo metabolita primario 3α-idrossi-1-metilen-5a-andorstan-17-one. Una volta 3α-ridotto, la sua attività è praticamente nulla. Ci sono 4 isoenzimi nell’uomo che sono distinti dal singolo enzima nei ratti che catalizza questa reazione e variano l’uno dall’altro in termini di specificità del substrato e distribuzione tissutale. Ciò rende l’estrapolazione dei dati dai ratti (o da qualsiasi altro animale per questa materia) non attendibile. Di questi 4 isoenzimi, almeno 1 è espresso nel muscolo scheletrico, e possibilmente 2. Mentre il Methenolone si 3α-riduce, lo fa in misura minore rispetto al DHT. Non è noto in che misura ciò avvenga nel muscolo scheletrico e con quale velocità. I dati attuali non sono sufficienti per rispondere a queste importanti domande. I dosaggi degli androgeni sono essenzialmente buoni, ma hanno tutti difetti significativi, rendendoli notoriamente inaffidabili per farci affidamento e per dire se un determinato composto è più anabolico o androgeno di un altro nella pratica. Pertanto, da questi studi non è possibile ricavare informazioni affidabili riguardo il Methenolone.
Ci sono alcuni studi clinici svolti con utilizzo del Methenolone, e uno in particolare sarebbe stato estremamente interessante; se non avessero somministrato il Methenolone per via orale invece di iniettarlo. La mancanza di prove di confronto tra AAS con partecipanti sani in cui viene misurata la composizione corporea o la forza muscolare rende impossibile rispondere a qualsiasi domanda su come il Methenolone si relazioni ad altri AAS.
Comunque sia, la maggior parte delle persone mi ha descritto il Methenolone come un AAS relativamente mite. Ciò implica che “devono” abbinarlo con altri AAS, o dosarlo massivamente, per un effetto apprezzabile sull’ipertrofia muscolare. Questa è stata la ragione principale per cui ho ricollegato il possibile “effetto mite” alla 3α-riduzione: dal momento che i feedback riportatimi descrivevano il Methenolone come un AAS lieve, e dato che la molecola è soggetta alla 3α-riduzione nel muscolo scheletrico, la spiegazione più probabile si trova in questa caratteristica. Tuttavia, sono sicuro che altre persone non saranno d’accordo con questa descrizione sull’effetto del Methenolone e / o con quello che ne ho ricavato. Infine, che vi piaccia oppure no, stanno crescendo le prove che qualunque AAS sia denunciato sull’etichetta della fiala o multidose acquistato nel mercato nero, con una discreta probabilmente non la conterrà (a meno che tu non l’abbia preso direttamente da una farmacia, o da un seller con collegamenti esteri “sicuri”, ma questa non è la regola nel torbido mondo delle UGL).
Un recente studio condotto dalla clinica ambulatoriale per utilizzatori di steroidi anabolizzanti nei Paesi Bassi ha esaminato 272 campioni di AAS di 46 diversi marchi forniti loro da i soggetti che si sono arruolati nella loro sperimentazione clinica [23]. I risultati possono essere trovati nella tabella tratta dal documento qui sotto:
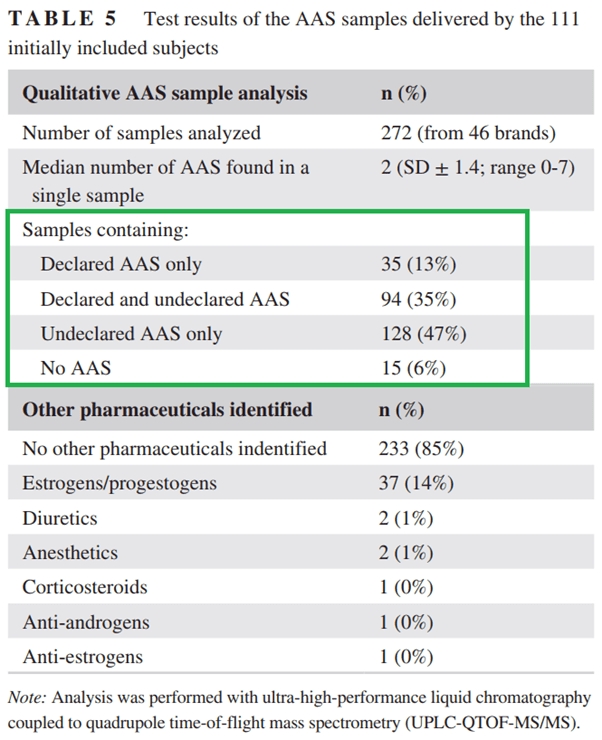
Sorprendentemente, solo il 13% degli AAS forniti conteneva solo la molecola dichiarata. Attenzione però, questa era un’analisi qualitativa. Quindi questo non significa nemmeno che questi AAS siano stati dosati come dichiarato sull’etichetta! Vi sento dire: “Sì, ma quelli sono fake, il mio è reale, ho un fornitore affidabile”. Guarda caso, tutti i soggetti della ricerca in questione erano estremamente convinti che il loro AAS fosse buono come descritto sull’etichetta. Per loro era impensabile che i prodotti in loro possesso non fossero quelli riportati. Avevano il miglior fornitore sulla piazza. Tuttavia, si sbagliavano. Gli autori dell’articolo menzionano anche questo: “Il tentativo di aggirare prodotti AAS inaffidabili acquistando da un marchio o rivenditore apparentemente” affidabile “- un argomento sempre interessante per gli utilizzatori di AAS – sembra insignificante poiché nessun marchio ha mostrato un’affidabilità costante”. Risultati simili sono stati pubblicati nel 2005 dall’autorità antidoping olandese dopo aver esaminato 336 prodotti [24] e in Svezia, quando hanno analizzato oltre 1.000 campioni di prodotti dopanti sequestrati al confine [25]. L’ombra del mercato nero degli AAS (che continuerà ad esistere fin tanto che la conoscenza verrà oscurata dalle ideologie di sistema) si aggiunge all’inaffidabilità dei resoconti personali sull’utilizzo di AAS, al di là degli ovvi inconvenienti legati ai dati aneddotici. La maggior parte degli utilizzatori di AAS semplicemente non assume ciò che pensa di assumere.
Nota riflessiva: Durante un intervista dove si trattava l’argomento su quali AAS i bodybuilder della “Golden Era” assumevano, Steve Davis ha affermato:
- Steve Davis: Sai, nella mia epoca, era consuetudine assumere 3 Dianabol al giorno e una iniezione a settimana.
- Ric Drasin: È esattamente così.
- Steve Davis: E abbiamo sentito che alcuni austriaci (vedi Arnold Schwarzenegger) che assumevano 4 Dianabol al giorno e una iniezione alla settimana (Methenolone NdR).
- Ric Drasin: Sì.
[link all’intervista integrale https://www.youtube.com/watch?v=31-kQbjtxDc ]

Allora, visto che il Methenolone Enantato è stato commercializzato fin dal suo ingresso nel mercato farmaceutico in fiale da 100mg/ml, e che il Methandrostenolone (Dianabol) era comunemente venduto in compresse da 5mg, un tipico ciclo di Schwarzenegger avrebbe potuto essere composto da 100mg/week di Methenolone Enantato e 20mg/die di Methandrostenolone…
Lo stesso Arnold, nella sua autobiografia “Total Recall – My Unbelievably True Life Story”, scrive che la sua scoperta degli AAS risale a prima della competizione Mr.Universe del 1967 mentre faceva ricerche sui metodi di allenamento dei tedeschi dell’est e dei sovietici. Correva voce che stessero usando farmaci per migliorare le prestazioni e per ottenere risultati superiori dai loro sollevatori di pesi, lanciatori e nuotatori e presto scoprì che gli AAS erano i farmaci che stavano usando.
Arnold continua raccontando di quando si recò dal medico per chiedere una prescrizione di AAS. Ha semplicemente chiesto:
- Puoi farmi provare?
- Il dottore acconsentì…. e gli fu poi prescritta un’iniezione ogni due settimane e delle pillole da prendere giornalmente.

Tenete presente che questo è stato il primo ciclo di AAS di Arnold in assoluto.
I suoi dosaggi potrebbero essere aumentati nei cicli successivi e probabilmente ha sperimentato diversi anabolizzanti ad un certo punto almeno una o due volte.
Sebbene Arnold non abbia precisato esattamente ciò che gli era stato prescritto, è possibile ipotizzarlo con un certo margine di attendibilità utilizzando alcune informazioni utili sulla pratica di somministrazione di ciascun composto (orale e iniettabile) e quale tipo di farmaci rientrerebbero con molta probabilità in queste due categorie in quel preciso momento storico, senza tralasciare tutte le altre informazioni provenienti da interviste di altri campioni della “Golden Era”. A questo punto, sappiamo che stava usando un composto iniettabile e un composto orale.
In un’altra intervista Arnold più o meno sottintende di aver usato 3 Dianabol (15mg ca.) al giorno. Ed è noto nella comunità del bodybuilding che Arnold era un utilizzatore di Dianabol nel corso della giornata.

Dedicato ai buon temponi sognatori che “mi inietto un pò di genetica”…
Gabriel Bellizzi
Riferimenti:
1- Book on Steroids di Peter Bond
2- Wiechert R. et al. Chem Ber. 93 (1960):1710.
3- Methenolone acetate, Summary of information for clinical investigators, New Brunswick, NJ.The Squibb Institute for Medical Research, May 30, 1962. Methenolone enanthate, Summary of information for clinical investigators, New Brunswick, NJ. The Squibb Institute for Medical Research, April 15, 1962.
4- https://en.wikipedia.org/wiki/Metenolone
- 6- Pereira de Jésus-Tran, Karine, et al. “Comparison of crystal structures of human androgen receptor ligand‐binding domain complexed with various agonists reveals molecular determinants responsible for binding affinity.” Protein Science 15.5 (2006): 987-999.
- 7- Penning, Trevor M., et al. “Human 3α-hydroxysteroid dehydrogenase isoforms (AKR1C1–AKR1C4) of the aldo-keto reductase superfamily: functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones.” Biochemical journal 351.1 (2000): 67-77.
- 8- O’CONNOR, Tania, et al. “Major differences exist in the function and tissue-specific expression of human aflatoxin B1 aldehyde reductase and the principal human aldo-keto reductase AKR1 family members.” Biochemical journal 343.2 (1999): 487-504.
- 9- Gharahdaghi, Nima, et al. “Testosterone therapy induces molecular programming augmenting physiological adaptations to resistance exercise in older men.” Journal of Cachexia, Sarcopenia and Muscle 10.6 (2019): 1276-1294.
- 10-Becker, H., et al. “In vivo uptake and metabolism of 3H-testosterone and 3H-5α-dihydrotestosterone by human benign prostatic hypertrophy.” European Journal of Endocrinology 71.3 (1972): 589-599.
- 11-Langecker, H. “BEZIEHUNGEN ZWISCHEN SUBSTITUTION IM RING A UND ABBAU IM STOFFWECHSEL BEI VERWANDTEN DES TESTOSTERONS.” European Journal of Endocrinology 41.4 (1962): 494-506.
- 12-Goudreault, Danielle, and Robert Massé. “Studies on anabolic steroids—4. Identification of new urinary metabolites of methenolone acetate (primobolan®) in human by gas chromatography/mass spectrometry.” The Journal of steroid biochemistry and molecular biology 37.1 (1990): 137-154.
- 13-He, Genye, et al. “New long term metabolite in human urine for metenolone misuse by liquid chromatography quadrupole time-of-flight mass spectrometry.” Steroids 105 (2016): 1-11.
- 14-Hershberger, L. G., Elva G. Shipley, and Roland K. Meyer. “Myotrophic activity of 19-nortestosterone and other steroids determined by modified levator ani muscle method.” Proceedings of the Society for Experimental Biology and Medicine 83.1 (1953): 175-180.
- 15-Hayes, Keith J. “The so-called ‘levator ani’ of the rat.” European Journal of Endocrinology 48.3 (1965): 337-347.
- 16-Van der Vies, J. “Implications of basic pharmacology in the therapy with esters of nandrolone.” European Journal of Endocrinology 110.3_Suppla (1985): S38-S44.
- 17-Saartok, Tönu, Erik Dahlberg, and Jan-Åke Gustafsson. “Relative binding affinity of anabolic-androgenic steroids: comparison of the binding to the androgen receptors in skeletal muscle and in prostate, as well as to sex hormone-binding globulin.” Endocrinology 114.6 (1984): 2100-2106.
- 18-Houtman, Corine J., et al. “Detection of anabolic androgenic steroid abuse in doping control using mammalian reporter gene bioassays.” Analytica chimica acta 637.1-2 (2009): 247-258.
- 19-Fowler JR, William M., Gerald W. Gardner, and Glen H. Egstrom. “Effect of an anabolic steroid on physical performance of young men.” Journal of Applied Physiology 20.5 (1965): 1038-1040.
- 20-Shimodozono, Megumi, et al. “Addition of an anabolic steroid to strength training promotes muscle strength in the nonparetic lower limb of poststroke hemiplegia patients.” International Journal of Neuroscience 120.9 (2010): 617-624.
- 21-Garbrecht, M., et al. “Hyperlipoproteinämie bei additiver Behandlung des metastasierenden Mammakarzinoms mit Metenolonönanthat.” DMW-Deutsche Medizinische Wochenschrift 106.13 (1981): 400-403.
- 22-Kennedy, B. J., and John W. Yarbro. “Effect of methenolone enanthate (NSC‐64967) in advanced cancer of the breast.” Cancer 21.2 (1968): 197-201.
- 23-Smit, Diederik L., et al. “Baseline characteristics of the HAARLEM study: 100 male amateur athletes using anabolic androgenic steroids.” Scandinavian Journal of Medicine & Science in Sports 30.3 (2020): 531-539.
- 24-https://www.dopingautoriteit.nl/media/files/documenten/Kwaliteit%20Illegale%20Dopingmiddelen.pdf (Retrieved 17-09-2020)
- 25-Weber, Christina, et al. “Qualitative and semiquantitative analysis of doping products seized at the Swiss border.” Substance use & misuse 52.6 (2017): 742-753.