
Introduzione:
In passato mi sono già cimentato nella trattazione del Colesterolo, soprattutto in ambito Enhanced, esplicando in modo chiaro i meccanismi che portano l’utilizzatore di AAS in una condizione dislipidemica/ipercolesterolemica potenzialmente negativa specie se mantenuta nel tempo. In questo artricolo, invece, è mia intenzione approfondire le caratteristiche di questa molecola tanto vituperata quando mal intesa al fine di far conoscere al lettore non semplicemente le basi di biochimica che reggono la sua omeostasi ma, soprattutto, quali sono ad oggi i trattamenti farmacologici applicati per intervenire sulle alterazione del Colesterolo ematico.
Il Colesterolo, un lipide strutturale e di segnalazione indispensabile, è fondamentale per l’integrità delle membrane cellulari, la steroidogenesi e le vie metaboliche dei morfogeni durante lo sviluppo. La sua omeostasi dipende dal preciso coordinamento di quattro moduli metabolici interdipendenti: biosintesi de novo, assorbimento intestinale, conversione enzimatica ed eliminazione sistemica. In questo articolo tratteremo i meccanismi molecolari che regolano questi processi, dalle vie di sintesi di Bloch/Kandutsch-Russell e dall’assorbimento di colesterolo mediato da NPC1L1 (Niemann-Pick C1-like 1) alla sintesi degli acidi biliari guidata dalla colesterolo 7α-idrossilasi (CYP7A1) e al trasporto inverso dipendente dalle HDL. Vengono inoltre approfonditi i molteplici ruoli del colesterolo nell’assemblaggio dei lipid raft, nella trasduzione del segnale Hedgehog e nella produzione di vitamina D e ormoni.
La disregolazione del flusso di Colesterolo è alla base della patogenesi dell’aterosclerosi, della steatosi epatica associata a disfunzione metabolica (MAFLD), delle malattie neurodegenerative e dell’oncogenesi, con cascate di sintesi, efflusso o esterificazione alterate che fungono da fattori chiave. Le strategie terapeutiche emergenti vanno oltre le statine convenzionali e gli inibitori della proproteina convertasi subtilisina/kexina di tipo 9 (PCSK9) per includere modalità innovative: l’editing genetico in vivo basato su CRISPR (ad esempio, VERVE-101 che ha come bersaglio PCSK9), le terapie con RNA interferente (siRNA) (inclisiran) e gli interventi mirati al microbiota. Approcci pionieristici contro bersagli quali l’angiopoietina-simile 3 (ANGPTL3), la lipoproteina(a) [Lp(a)] e il recettore 1 dell’asialoglicoproteina (ASGR1), insieme ad agenti naturali riproposti (berberina, probiotici), offrono promettenti soluzioni per mitigare il rischio cardiovascolare residuo e promuovere la medicina cardiometabolica di precisione. Integrando le conoscenze sui meccanismi d’azione con i progressi clinici, andrà sottolineata la transizione dalle terapie ad ampio spettro a regimi personalizzati multi-bersaglio, offrendo una tabella di marcia per la mitigazione delle malattie correlate al Colesterolo nell’era della medicina genomica e metabolica.
Questa disamina si dividerà in due parti al fine di dare la possibilità al lettore di assimilare le nozioni e poter essere preparato alle successive presenti della seconda parte.
Regolazione dell’omeostasi del Colesterolo
L’omeostasi del colesterolo è meticolosamente orchestrata attraverso l’integrazione di quattro moduli metabolici chiave: sintesi, assorbimento, conversione ed eliminazione (vedi la figura seguente). Questo ciclo metabolico completo garantisce che il Colesterolo svolga i suoi ruoli fondamentali nella costruzione delle membrane, nella trasduzione del segnale e nell’omeostasi sistemica.
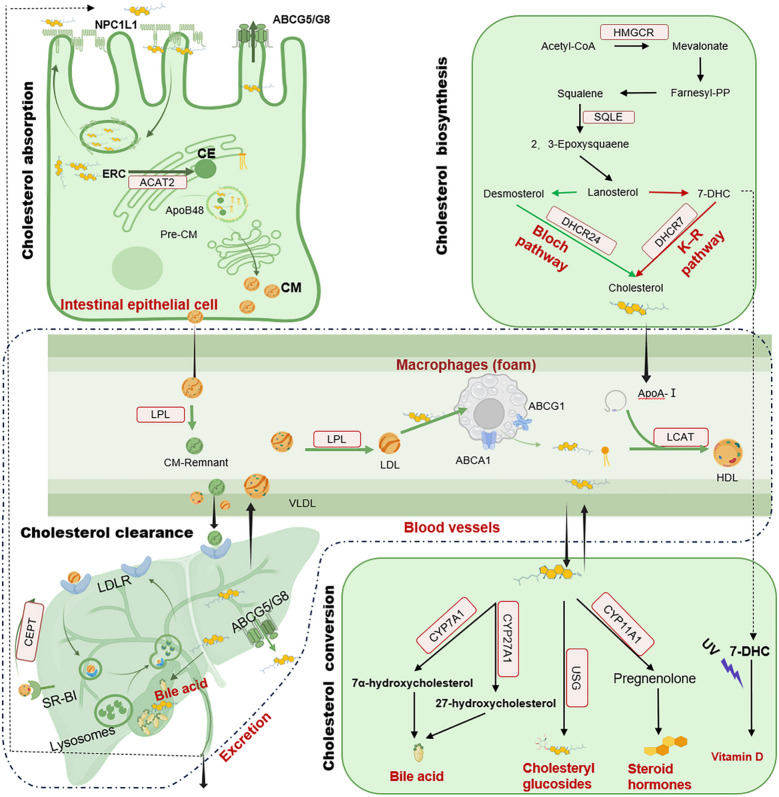
- Biosintesi del Colesterolo
La sintesi de novo del Colesterolo nei vertebrati avviene prevalentemente nel fegato (80%), con contributi minori dai tessuti extraepatici (20%). Questo processo è metabolicamente impegnativo, consumando 18 molecole di adenosina trifosfato (ATP) e 16 molecole di nicotinammide adenina dinucleotide (NAD) in forma ridotta (NADPH), oltre ad acetil-CoA e ossigeno, per ogni molecola di colesterolo prodotta. Procede attraverso due vie metaboliche conservate nel design: la via canonica di Bloch (> 90% della produzione totale), chiarita dagli studi premiati con il Nobel da Konrad Bloch negli anni ’50, e la via di Kandutsch-Russell (K-R), scoperta negli anni ’60. La via K-R è dominante in condizioni di ipossia o stress da raggi ultravioletti (UV) in tessuti come la pelle e le gonadi. Entrambi i percorsi metabolici condividono le stesse fasi iniziali, a partire dalla condensazione delle molecole di acetil-CoA. La tiolasi combina due molecole di acetil-CoA per formare acetoacetil-CoA, al quale la 3-idrossi-3-metilglutaril-CoA (HMG-CoA) sintasi aggiunge una terza molecola di acetil-CoA, producendo HMG-CoA. L’enzima limitante la velocità di reazione, l’HMG-CoA reduttasi (HMGCR), riduce quindi l’HMG-CoA a Mevalonato (MVA) utilizzando NADPH. L’MVA subisce fosforilazione e isomerizzazioni sequenziali per produrre farnesil pirofosfato (FPP). La squalene sintasi (SQS) dimerizza l’FPP in squalene, che viene successivamente ossidato dal secondo enzima limitante la velocità di reazione, la squalene epossidasi (SQLE), per formare 2,3-ossidosqualene. La lanosterolo sintasi (LSS) ciclizza il 2,3-ossidosqualene in lanosterolo, il primo intermedio steroideo. Oltre il lanosterolo, la via di Bloch e la via di Krebs-Ribbent-Robinson (K-R) divergono. Nella via di Bloch, il lanosterolo subisce demetilazione, desaturazione e riduzione per produrre infine Colesterolo. Nella via di K-R, il lanosterolo viene convertito sequenzialmente in 24,25-diidrolanosterolo e 7-deidrocolesterolo (7-DHC), seguito dalla riduzione del 7-DHC a Colesterolo tramite la 7-DHC reduttasi (DHCR7). Sebbene meno efficiente, la via di K-R evita le fasi dipendenti dall’ossigeno, fornendo un vantaggio fisiologico in ambienti con limitata disponibilità di ossigeno.
- Assorbimento del Colesterolo
L’assorbimento del Colesterolo, che avviene principalmente nel duodeno e nel digiuno prossimale, è un processo strettamente regolato, fondamentale per il mantenimento dell’omeostasi lipidica sistemica, garantendo un assorbimento efficiente del Colesterolo alimentare e biliare e prevenendone l’accumulo eccessivo. Il principale mediatore dell’assorbimento intestinale del colesterolo è la proteina di membrana degli enterociti niemann-pick C1-like 1 (NPC1L1), che si sposta tra la superficie cellulare e i compartimenti di riciclo endocitico (ERC) per facilitarne l’assorbimento. NPC1L1 contiene cinque domini transmembrana, tra cui un dominio di rilevamento degli steroli che rileva i livelli di Colesterolo . Un elevato livello di colesterolo luminale induce l’integrazione del colesterolo libero nella membrana degli enterociti, dove NPC1L1 lo lega e ne facilita l’internalizzazione. Questo avviene principalmente tramite endocitosi mediata da clatrina/AP2, trasportando il Colesterolo lungo i filamenti di actina verso gli ERC per l’immagazzinamento. Studi di microscopia crioelettronica rivelano un meccanismo aggiuntivo: il legame del Colesterolo induce un cambiamento conformazionale in NPC1L1, formando un tunnel di trasporto transmembrana che facilita direttamente l’assorbimento indipendentemente dall’endocitosi. In condizioni di basso Colesterolo, NPC1L1 viene riciclato verso la membrana per riprendere l’assorbimento.
Una volta internalizzato, il Colesterolo subisce esterificazione o efflusso per bilanciare i livelli cellulari. Il colesterolo libero si sposta nel reticolo endoplasmatico, dove l’acil-CoA:colesterolo aciltransferasi 2 (ACAT2) lo esterifica con acidi grassi in esteri del Colesterolo. Questi esteri idrofobici vengono impacchettati nei chilomicroni, trasportati all’apparato di Golgi per essere elaborati e infine entrano nella circolazione sistemica attraverso il sistema linfatico (dotto toracico) per essere distribuiti ai tessuti periferici. Nel frattempo, il Colesterolo libero in eccesso viene attivamente pompato di nuovo nel lume intestinale dai trasportatori eterodimerici ATP-binding cassette (ABC) G5/G8 (ABCG5/G8). Questo meccanismo di efflusso limita in modo critico l’assorbimento netto e protegge dal sovraccarico cellulare.
- Conversione del Colesterolo
All’interno delle cellule, il Colesterolo funge da precursore versatile per la sintesi di composti biologicamente essenziali, tra cui acidi biliari, glucosidi del Colesterolo, vitamina D e vari ormoni steroidei come Androgeni, Estrogeni, Progesterone, Glucocorticoidi e Mineralcorticoidi.
- Sintesi degli Acidi Biliari
Nel fegato, la maggior parte del Colesterolo viene convertita in Acidi Biliari, costituiti principalmente da Acidi Biliari primari come l’Acido Colico (CA) e l’Acido Chenodesossicolico (CDCA), insieme ad acidi biliari secondari come l’Acido Desossicolico (DCA) e tracce di Acido Litocolico (LCA), attraverso due percorsi distinti. Il percorso classico, responsabile di oltre il 90% della produzione di acidi biliari, inizia con la Colesterolo 7α-idrossilasi (CYP7A1) che idrossila il Colesterolo in posizione 7α. Questa fase limitante la velocità produce 7α-idrossicolesterolo, che viene poi convertito in 7α-idrossi-4-colesten-3-one (C4) dalla 3β-idrossi-Δ5-C27-steroide deidrogenasi (3β-HSD). In particolare, il C4 funge da precursore comune sia per il CA che per il CDCA ed è spesso utilizzato come biomarcatore sierico per la velocità di sintesi degli acidi biliari. Al contrario, la via alternativa agisce come un meccanismo compensatorio essenziale durante l’alterazione della via classica o lo stress metabolico, inizia con la sterolo 27-idrossilasi mitocondriale (CYP27A1) che converte il Colesterolo in 27-idrossicolesterolo (27-OHC), seguita dall’ossisterolo 7α-idrossilasi (CYP7B1) che catalizza un’ulteriore idrossilazione per generare acido 3β,7α-diidrossi-5-colestenoico, che subisce ossidazione della catena laterale e accorciamento tramite 3β-HSD tipo 7 (HSD3B7) per produrre CDCA. Sebbene questa via alternativa rappresenti solo circa il 10% della sintesi degli acidi biliari in condizioni normali, diventa di fondamentale importanza negli stati patologici.
- Glucosidi del Colesterolo (CG)
I CG sono glicosidi sterolici in cui una porzione di Glucosio è esterificata al gruppo idrossilico del Colesterolo. I CG contribuiscono alla stabilità della membrana alterando l’impacchettamento e la fluidità dei lipidi, in particolare nei raft lipidici. I CG possono anche agire come molecole di segnalazione nelle risposte immunitarie. Ad esempio, sono implicati nell’attivazione dei macrofagi e nella produzione di citochine. Nelle piante, i CG difendono dai patogeni microbici, suggerendo ruoli analoghi nell’immunità innata dei mammiferi. Nei mammiferi, i CG vengono sintetizzati tramite glicosilazione enzimatica del colesterolo principalmente nell’apparato di Golgi o nel reticolo endoplasmatico, dove il colesterolo e l’UDP-glucosio sono accessibili. L’UDP-glucosio:sterolo glucosiltransferasi (USG) catalizza il trasferimento del glucosio dall’UDP-glucosio al gruppo 3β-idrossilico del colesterolo. La produzione di CG è regolata dalla disponibilità di colesterolo e dallo stress cellulare; in condizioni di sovraccarico di colesterolo o stress ossidativo, la sintesi di CG può aumentare per modulare la fluidità della membrana o sequestrare il Colesterolo in eccesso. La disregolazione della sintesi di CG è collegata a disturbi dell’accumulo di lipidi e malattie infiammatorie. Ad esempio, livelli elevati di CG si osservano nella malattia di Niemann-Pick di tipo C, una malattia da accumulo lisosomiale. È interessante notare che alcuni organismi patogeni, come Helicobacter pylori, sono anche in grado di sintetizzare glucosidi del colesterolo, sebbene i loro percorsi biosintetici differiscano fondamentalmente da quelli dei mammiferi.
- Sintesi della Vitamina D
La vitamina D, un secosteroide fondamentale per l’omeostasi del Calcio e la regolazione immunitaria, viene sintetizzata a partire dal 7-DHC attraverso un percorso dipendente dai raggi UV e successive modifiche enzimatiche. Nello strato basale e spinoso dell’epidermide, il 7-DHC, un intermedio nella sintesi del Colesterolo, viene convertito in pre-vitamina D3 in seguito all’esposizione alle radiazioni UVB (290-315 nm). Questa reazione non enzimatica avviene spontaneamente. La pre-vitamina D3 subisce un’isomerizzazione dipendente dalla temperatura in colecalciferolo (vitamina D3) nell’arco di circa 48 ore. La vitamina D3 viene idrossilata in due fasi sequenziali per acquisire l’attività biologica. Nel fegato, il citocromo P450 2R1 (CYP2R1) converte la vitamina D3 in 25-idrossivitamina D3 [25(OH)D3], la principale forma circolante. Nel rene, il citocromo P450 27B1 (CYP27B1) idrossila la 25(OH)D3 in 1,25-diidrossivitamina D3 [1,25(OH)2D3], che aumenta l’assorbimento intestinale di Calcio e fosfato e promuove il riassorbimento renale.
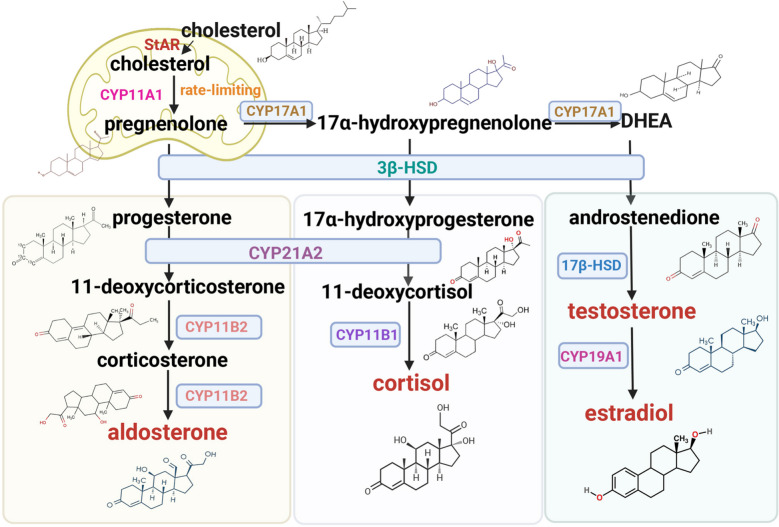
- Biosintesi steroidea
Il Colesterolo, precursore universale di tutti gli ormoni steroidei, inclusi mineralcorticoidi, glucocorticoidi e ormoni sessuali, subisce una biotrasformazione tessuto-specifica all’interno della corteccia surrenale. Questo sito di sintesi primario è organizzato in tre zone: la zona glomerulare, che produce mineralcorticoidi (ad esempio, Aldosterone) per regolare l’equilibrio elettrolitico e la pressione sanguigna; la zona fascicolata, che sintetizza glucocorticoidi (ad esempio, Cortisolo) che regolano la risposta allo stress e il metabolismo; e la zona reticolare, che genera steroidi sessuali (ad esempio, Androgeni) essenziali per la fisiologia riproduttiva.
Gli ormoni sessuali (androgeni, estrogeni e progestinici) sono regolatori essenziali dello sviluppo riproduttivo, delle caratteristiche sessuali secondarie e del dimorfismo fisiologico. L’attività 17,20-liasi del CYP17A1 converte il 17α-idrossipregnenolone in deidroepiandrosterone (DHEA), precursore universale degli steroidi gonadici. Il metabolismo del DHEA diverge attraverso l’ossidazione mediata da 3β-HSD-1 ad androstenedione o la riduzione dipendente da 17β-HSD-1 a 5-androstenediolo, entrambe convergenti nel testosterone. La successiva elaborazione enzimatica da parte dell’aromatasi (CYP19A1/P450aro) o della 5α-reduttasi produce rispettivamente 17β-estradiolo o 5α-diidrotestosterone (5α-DHT).
- Eliminazione del Colesterolo
L’omeostasi del Colesterolo nei mammiferi si basa su meccanismi di eliminazione strettamente regolati per prevenire l’accumulo patologico nei tessuti. Due principali vie complementari mediate dalle lipoproteine, tra cui il trasporto inverso del Colesterolo (RCT) guidato dalle lipoproteine ad alta densità (HDL) e l’assorbimento epatico dipendente dalle lipoproteine a bassa densità (LDL), sono essenziali per il mantenimento dell’omeostasi del Colesterolo. Le HDL rimuovono il Colesterolo in eccesso dai tessuti periferici, mentre le LDL e il loro recettore LDLR assicurano un efficiente assorbimento epatico del colesterolo circolante. Le strategie terapeutiche che prendono di mira queste vie, come gli agenti che potenziano LDLR o HDL, sono promettenti per il trattamento della dislipidemia e dell’aterosclerosi.
- Trasporto del Colesterolo mediato dalle HDL
Il trasporto inverso del colesterolo (RCT) è un processo critico attraverso il quale il colesterolo viene trasportato dai tessuti periferici al fegato per essere escreto tramite la bile o le feci. Un componente principale del percorso RCT è l’HDL. Le particelle HDL nascenti hanno una forma discoidale e sono composte da apolipoproteina A1 (ApoA1) e fosfolipidi [24]. Le particelle HDL acquisiscono colesterolo libero dalle cellule schiumose periferiche, come i macrofagi o le cellule muscolari lisce vascolari, tramite i trasportatori ABC A1 (ABCA1) e G1 (ABCG1). ABCA1 interagisce con l’apoA1 povera di lipidi, mentre ABCG1 facilita l’efflusso di colesterolo verso l’HDL matura. La lecitina-colesterolo aciltransferasi (LCAT) esterifica il colesterolo libero all’interno dell’HDL, convertendolo in esteri del colesterolo. Questo nucleo idrofobico trasforma l’HDL in particelle sferiche. L’HDL matura trasporta il colesterolo al fegato attraverso l’assorbimento selettivo mediato dal recettore scavenger di classe B tipo I (SR-BI) o il trasferimento facilitato dalla proteina di trasferimento degli esteri del colesterolo (CETP). L’SR-BI estrae selettivamente gli esteri del colesterolo dall’HDL senza degradare l’intera particella, mentre la CETP trasporta gli esteri del colesterolo dall’HDL alle lipoproteine contenenti apolipoproteina B (ApoB) (ad esempio, LDL, lipoproteine a bassissima densità (VLDL)), che vengono successivamente eliminate tramite LDLR epatico.
- Eliminazione del Colesterolo mediata dalle LDL
Le particelle LDL, che trasportano esteri del Colesterolo e apolipoproteina B-100 (ApoB-100), vengono eliminate prevalentemente dal fegato. I recettori LDLR, altamente espressi negli epatociti, legano l’ApoB-100 presente sulle particelle LDL tramite interazioni elettrostatiche. I complessi LDL-LDLR vengono internalizzati in vescicole rivestite di clatrina, che si fondono con i lisosomi. All’interno dei lisosomi, gli esteri del colesterolo vengono idrolizzati a colesterolo libero, mentre i recettori LDLR vengono riciclati sulla superficie cellulare. In alcuni casi, le particelle HDL che esprimono apolipoproteina E (ApoE) possono legarsi ai recettori LDLR o alla proteina 1 correlata al recettore LDL (LRP1), consentendo la loro internalizzazione e degradazione epatica. Questa via metabolica fornisce un percorso ausiliario per l’eliminazione del Colesterolo HDL.
La proproteina convertasi subtilisina/kexina di tipo 9 (PCSK9) è un membro della famiglia delle serina proteasi, nota per il suo ruolo critico nell’attivazione proteolitica, nella modificazione e nella degradazione delle proteine secrete. Gli LDLR epatici fungono da recettori per la PCSK9 circolante. La PCSK9 si lega al dominio EGF-A (epidermal growth factor-like repeat A) degli LDLR tramite il suo dominio catalitico, promuovendo l’endocitosi degli LDLR negli endosomi. Il pH acido all’interno degli endosomi aumenta l’interazione PCSK9-LDLR di 150 volte, impedendo il riciclo degli LDLR. Di conseguenza, il complesso PCSK9-LDLR viene indirizzato alla degradazione lisosomiale, riducendo la densità degli LDLR sulla superficie degli epatociti, diminuendo così la clearance delle particelle di colesterolo LDL (LDL-C) epatiche e aumentando i livelli circolanti di LDL-C. L’inibizione di PCSK9 migliora la clearance del Colesterolo LDL, una strategia terapeutica per l’ipercolesterolemia.
Funzione biologica del Colesterolo
Il Colesterolo è una molecola multifunzionale con un’ampia gamma di ruoli biologici. Le sue funzioni spaziano dal mantenimento dell’integrità e della fluidità delle membrane cellulari al ruolo di precursore di ormoni essenziali e acidi biliari. Inoltre, il Colesterolo svolge un ruolo cruciale nella regolazione del metabolismo lipidico, nella modulazione delle vie di trasduzione del segnale e nell’influenza sulla funzione immunitaria. Comprendere i diversi ruoli del Colesterolo è fondamentale per apprezzarne l’importanza nel mantenimento della salute generale e per sviluppare strategie terapeutiche volte a contrastare i disturbi correlati all’alterazione del Colesterolo.
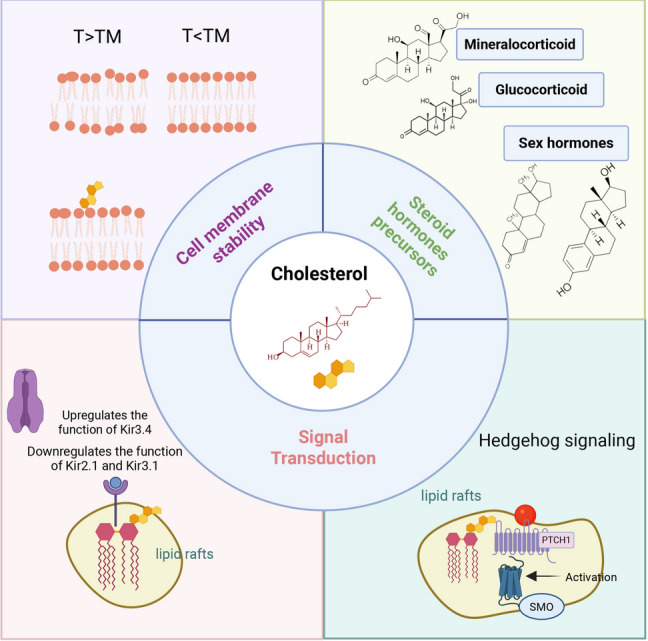
Tra le principali funzioni biologiche del Colesterolo troviamo:
- Colesterolo e stabilità della membrana cellulare
Il Colesterolo è un componente chiave delle membrane cellulari eucariotiche, rappresentando circa il 20-30% dei lipidi di membrana. Un doppio strato fosfolipidico puro esiste in una fase gel (rigida) al di sotto della sua temperatura di transizione (TM) e in una fase liquido-cristallina (fluida) al di sopra di Tₘ . Intercalandosi tra le catene fosfolipidiche, il colesterolo può smussare il punto di flesso netto della transizione di fase e contribuire a tamponare il cambiamento di fase, stabilizzando così la fase liquido-cristallina in un intervallo di temperature più ampio. Pertanto, quando l’ambiente è vicino o al di sopra della temperatura di transizione, il Colesterolo limita i movimenti vigorosi, in particolare i movimenti laterali, delle catene fosfolipidiche, prevenendo così un’eccessiva fluidità della membrana. Al contrario, a temperature più basse, l’anello sterolico del colesterolo ostacola la disposizione ordinata delle catene fosfolipidiche, impedendo la formazione di uno stato gel rigido.
L’architettura anfifilica del Colesterolo consente una duplice funzione regolatrice. Da un lato, l’anello steroideo idrofobico si inserisce nelle code idrofobiche dei fosfolipidi, associandosi alle catene di acidi grassi saturi per aumentare la densità della membrana. Riempiendo gli spazi tra le catene fosfolipidiche, il colesterolo riduce i difetti della membrana e ne migliora la resistenza meccanica. Dall’altro lato, la porzione flessibile della catena alchilica del colesterolo può interagire con le code idrofobiche delle catene di acidi grassi insaturi, diminuendo l’impacchettamento disordinato causato dalle loro pieghe e prevenendo un’eccessiva fluidità.
Inoltre, il Colesterolo collabora con gli sfingolipidi per formare i domini lipidici (lipid raft), microdomini spazialmente segregati caratterizzati da un elevato grado di ordine pur mantenendo un certo livello di fluidità, noti come fase liquido-ordinata (Lo). Al contrario, le regioni circostanti, ricche di fosfolipidi insaturi, presentano un’elevata fluidità che facilita la diffusione delle sostanze e la deformazione della membrana, e sono note come fase liquido-disordinata (Ld). Regolando l’assemblaggio e il disassemblaggio dei lipid raft, il Colesterolo influenza indirettamente la fluidità delle regioni non-raft. Le variazioni di fluidità di questi lipid raft possono anche attivare proteine di membrana come le tirosin chinasi recettoriali, influenzando così l’efficienza della trasduzione del segnale.
- Modulazione dei canali ionici tramite interazioni con il Colesterolo
Il Colesterolo regola bidirezionalmente la funzione dei canali ionici attraverso meccanismi strutturali e allosterici. Le alterazioni dei livelli di Colesterolo possono influenzare la funzione dei canali ionici. Ad esempio, è noto che i canali del potassio a rettificazione interna (Kir) sono influenzati da tali cambiamenti. Nello specifico, il Colesterolo aumenta la funzione di Kir3.4 mentre riduce le funzioni di Kir2.1 e Kir3.1. Un fattore che contribuisce a queste differenze è l’alterazione della distribuzione del Colesterolo. Rispetto a Kir2.1, il potenziale sito di legame del Colesterolo di Kir3.4 ha subito delle alterazioni. L’accumulo preferenziale di colesterolo nell’elica transmembrana distale è accoppiato allostericamente con la dinamica conformazionale a livello del filtro di selettività. Questo accoppiamento allosterico tra la funzione del canale e il legame dei lipidi è un meccanismo necessario per l’attivazione dei canali Kir mediata da PIP [109].
- Il Colesterolo come precursore universale della sintesi degli ormoni steroidei
Come già accennato in precedenza, il Colesterolo funge da precursore fondamentale per tutti gli ormoni steroidei, operando come impalcatura molecolare per questi regolatori critici dei processi di sviluppo, dell’omeostasi metabolica e dell’adattamento allo stress. La steroidogenesi impiega due sistemi enzimatici conservati nei tessuti endocrini: le ossidasi del citocromo P450 (CYP) e le idrossisteroide deidrogenasi (HSD), nonostante i modelli di espressione specifici delle ghiandole. Gli enzimi CYP contengono domini di legame all’eme conservati, mentre le HSD sono prive di eme ma richiedono i cofattori NAD e NADP. La cascata steroidogenica inizia con la fase limitante: la traslocazione del colesterolo alle membrane interne mitocondriali mediata dalla proteina regolatrice acuta steroidogenica (StAR). All’interno dei mitocondri, l’enzima di scissione della catena laterale del citocromo P450 (CYP11A1/P450scc) catalizza la conversione del colesterolo in pregnenolone, il precursore universale degli ormoni steroidei.
- Colesterolo e mTOR
Una funzione biologica poco conosciuta che interessa il Colesterolo è che esso rappresenta un elemento essenziale per l’attivazione della via metabolica di mTOR. La presenza di Colesterolo libero a livello delle membrane cellulari e il suo corretto trasporto intracellulare sono infatti necessari per permettere l’assemblaggio e l’attivazione del complesso mTORC1, fondamentale per la crescita e la sintesi proteica.
L’mTORC1 è un regolatore primario dei processi anabolici. Aumenta i livelli di nSREBP2 fosforilando e impedendo l’ingresso nucleare di Lipin 1. Viceversa, il fattore di trascrizione lipogenico ChREBP (proteina legante l’elemento di risposta ai carboidrati) promuove l’ubiquitinazione e la degradazione proteasomica di nSREBP2 attraverso un meccanismo sconosciuto. Il digiuno attiva la deacetilazione di SREBP2 mediata da SIRT1, arrestando il processo di biosintesi del colesterolo, che consuma energia, in condizioni di carenza di nutrienti. Oltre all’acetilazione, nSREBP2 può subire fosforilazione da parte di ERK e AMPK, portando rispettivamente a un aumento o a una diminuzione dell’attività trascrizionale. Anche la sumoilazione di nSREBP2 riduce la sua attività trascrizionale. AMPK è stata identificata come una chinasi a monte di LXR. L’AMPK attivata (AMPK fosforilata) inibisce la produzione di ligandi LXR endogeni, riducendo così l’espressione di LXR e bloccando la regolazione trascrizionale mediata da LXR.
L’mTOR agisce come un regolatore di molteplici stimoli cellulari. I principali attivatori includono:
1- Nutrienti: Aminoacidi, in particolare la leucina, e il glucosio.
2- Segnali ormonali: Insulina e fattori di crescita (come l’IGF-1).
3- Stimoli meccanici: Sforzi fisici intensi e allenamento contro resistenza (ipertrofia).
Malattie associate al metabolismo del Colesterolo
Come abbiamo visto, il metabolismo del Colesterolo è caratterizzato da un complesso equilibrio dinamico. La disregolazione dell’omeostasi del Colesterolo, caratterizzata da squilibri nella sintesi, nell’assorbimento, nel trasporto o nell’escrezione, rappresenta un fattore patogenetico critico per una serie di malattie croniche e degenerative. Queste condizioni interessano i sistemi cardiovascolare, epatico, neurologico e oncologico, riflettendo l’impatto sistemico del dismetabolismo del Colesterolo.
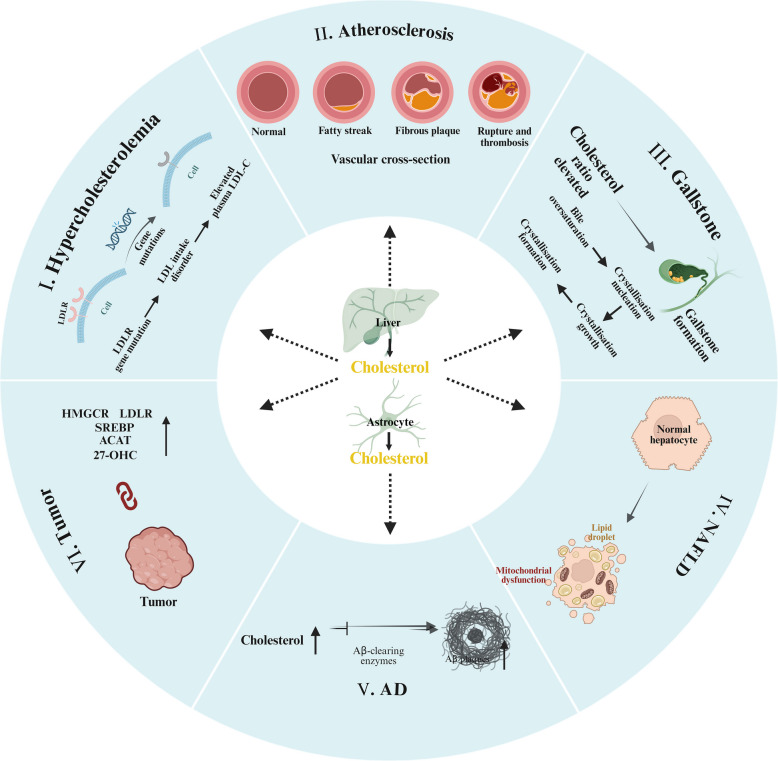
- Ipercolesterolemia
L’ipercolesterolemia, una dislipidemia clinicamente significativa caratterizzata da livelli cronicamente elevati di Colesterolo totale plasmatico (≥ 5,2 mmol/L [200 mg/dL]) e/o livelli di LDL-C, costituisce un importante fattore di rischio modificabile per le malattie cardiovascolari aterosclerotiche. Classificata secondo le Linee guida cinesi del 2024 e 2026 per la gestione dei lipidi nel sangue e allineata con i criteri dell’American College of Cardiology, questa patologia metabolica si manifesta in due forme eziologiche principali: primaria (ereditaria) e secondaria. L’ipercolesterolemia primaria deriva prevalentemente da difetti genetici, in particolare dall’ipercolesterolemia familiare (FH), una condizione autosomica dominante guidata principalmente da varianti patogene nel gene LDLR. Queste mutazioni compromettono la clearance dell’LDL-C, con conseguente accumulo patologico di LDL-C. L’ipercolesterolemia secondaria deriva da fattori acquisiti, tra cui comorbilità (ad esempio, coronaropatia, diabete mellito), farmaci (ad esempio, diuretici, beta-bloccanti, glucocorticoidi, AAS) e fattori legati allo stile di vita. In particolare, l’obesità amplifica il rischio attraverso la disregolazione del metabolismo lipidico e l’iperlipidemia, mentre l’eccessivo consumo di grassi saturi, la sedentarietà, il consumo cronico di alcol e lo stress psicosociale aggravano ulteriormente l’alterazione dell’omeostasi del Colesterolo.
Data la sua natura multifattoriale, l’ipercolesterolemia può essere inizialmente gestita attraverso interventi sullo stile di vita mirati a fattori estrinseci modificabili. Le attuali strategie di intervento includono: 1) la modifica della composizione e della struttura della dieta, limitando il consumo di cibi ad alto contenuto di grassi e riducendo l’assunzione di Colesterolo esogeno. La ricerca indica che la dieta DASH, specificamente progettata per prevenire e gestire l’ipertensione, è efficace nel ridurre i livelli di LDL-C. Inoltre, i risultati di Luiza et al. suggeriscono che l’adesione alla dieta mediterranea può anche migliorare la dislipidemia negli individui affetti. 2) Impegnarsi in un’attività fisica regolare per migliorare i processi metabolici, migliorando così il metabolismo lipidico anomalo; e 3) Praticare un consumo moderato di alcol per mitigare l’assunzione complessiva di alcol. Quando le modifiche dello stile di vita non producono una risposta sufficiente, diventa imperativo intensificare la farmacoterapia. Gli agenti di prima linea includono statine, inibitori dell’assorbimento del Colesterolo, inibitori di PCSK9, sequestranti degli acidi biliari e fibrati. Queste modalità terapeutiche, con i loro distinti meccanismi e applicazioni cliniche, saranno analizzate in modo esaustivo nelle sezioni successive.
- Aterosclerosi
L’aterosclerosi (AS) è una malattia immunoinfiammatoria cronica del sistema arterioso, caratterizzata patologicamente dall’accumulo di placche ricche di lipidi all’interno delle pareti dei vasi, che porta a stenosi luminale e ridotta compliance vascolare. Colpisce principalmente le arterie di grandi e medie dimensioni ed è guidata da una varietà di fattori di rischio, tra cui la dislipidemia, in particolare i livelli elevati di LDL, costituisce il principale determinante patogeno. Le particelle di LDL circolanti migrano attraverso l’endotelio e subiscono una modificazione ossidativa (ad esempio, coniugazione con malondialdeide), formando LDL ossidate pro-infiammatorie (oxLDL). Le oxLDL attivano i recettori scavenger dei macrofagi (ad esempio, CD36, LOX-1), dando inizio a un ciclo autosostenuto di formazione di cellule schiumose. I macrofagi internalizzano le oxLDL ma non possono esportare efficacemente il colesterolo a causa della disfunzione delle HDL. In condizioni di ipercolesterolemia, il sistema RCT, mediato dal legame dell’HDL ai recettori epatici SR-B1, viene sovraccaricato. Ciò promuove lo sviluppo di un nucleo necrotico e di una capsula fibrosa, che sono i tratti distintivi delle placche vulnerabili. Pertanto, il metabolismo del Colesterolo disregolato, in particolare la ridotta clearance lipidica dovuta a livelli elevati di LDL e a una disfunzione dell’HDL, funge da meccanismo centrale nella patogenesi dell’AS, guidando l’intero processo dal danno endoteliale alla formazione della placca. Sebbene l’ipercolesterolemia rimanga il principale fattore di rischio modificabile, altri fattori contribuenti includono ipertensione, diabete mellito, obesità e fattori legati allo stile di vita come il fumo.
- Malattia del fegato grasso associata a disfunzione metabolica (MAFLD)
La MAFLD è una condizione epatica cronica caratterizzata dall’accumulo di lipidi nel fegato (steatosi) in assenza di altre cause identificabili, come disturbi genetici o consumo eccessivo di alcol. Lo spettro della malattia spazia dalla semplice steatosi epatica (FL), alla steatoepatite non alcolica (NASH), alla fibrosi e alla cirrosi. La sua progressione è guidata da molteplici fattori interagenti, tra cui predisposizione genetica, influenze ambientali, disbiosi del microbiota intestinale, stress ossidativo, insulino-resistenza, infiammazione e dislipidemia, che possono agire in parallelo o in sequenza nelle diverse fasi della malattia. Il Colesterolo è riconosciuto come il principale agente lipotossico nella patogenesi della MAFLD, e la sua disregolazione gioca un ruolo centrale nella progressione della MAFLD verso la NASH e la fibrosi. Un’eccessiva assunzione e accumulo di Colesterolo promuovono la progressione della malattia e le risposte infiammatorie indotte dal Colesterolo sono state identificate come un fattore determinante in queste condizioni.
La gestione della MAFLD comprende strategie sia non farmacologiche che farmacologiche, adattate ai profili di rischio individuali. Modifiche dietetiche, come la dieta mediterranea [143] e la dieta chetogenica, sono opzioni terapeutiche efficaci. Anche l’esercizio fisico regolare dimostra effetti benefici nel trattamento della MAFLD. L’asse intestino-fegato gioca un ruolo importante, poiché la salute intestinale influenza significativamente la funzione epatica, rendendo il microbiota intestinale un potenziale bersaglio terapeutico. Ad esempio, i probiotici possono modulare la composizione del microbiota intestinale ed esercitare effetti benefici sulla MAFLD, e alcune caratteristiche della flora potrebbero fungere da biomarcatori diagnostici o prognostici [147]. Tuttavia, uno studio del 2021 condotto da Nor et al. ha suggerito che sei mesi di integrazione probiotica non hanno prodotto un miglioramento clinico significativo, indicando che i probiotici potrebbero essere più adatti come terapia aggiuntiva piuttosto che come terapia autonoma. Il primo trattamento farmacologico per la NASH è Rezdiffra, un agonista orale del THR-β che attiva selettivamente il recettore epatico dell’ormone tiroideo β. Modula il metabolismo lipidico, promuove il dispendio energetico e riduce il grasso epatico e l’infiammazione. Altri approcci farmacologici mirano alla patogenesi sottostante, tra cui agenti ipolipemizzanti (ad esempio, statine), agonisti del PPARα (ad esempio, acido obeticolico) e agonisti dell’FXR (ad esempio, cilofexor ed elafibranor).
- Calcoli biliari
I calcoli biliari sono depositi cristallini solidi che si formano nella cistifellea o nelle vie biliari e rappresentano una comune patologia gastrointestinale. Si distinguono principalmente in due tipi: calcoli di colesterolo e calcoli pigmentati. La patogenesi della calcolosi biliare è multifattoriale e coinvolge una complessa interazione di predisposizioni genetiche, fattori legati allo stile di vita e altre influenze. Il meccanismo predominante alla base della formazione dei calcoli biliari è la sovrasaturazione di colesterolo nella bile, che favorisce lo sviluppo dei calcoli di colesterolo. In condizioni fisiologiche, il colesterolo è solubilizzato all’interno di micelle e vescicole formate da acidi biliari e lecitina. Tuttavia, quando i livelli di Colesterolo superano la capacità di solubilizzazione di questi composti, si verifica la sovrasaturazione e la conseguente cristallizzazione. In particolare, studi recenti suggeriscono che la sovrasaturazione di colesterolo nei pazienti con calcoli biliari potrebbe essere dovuta principalmente a una carenza di acidi biliari piuttosto che a un’eccessiva produzione di colesterolo.
Le strategie di trattamento per i calcoli biliari includono interventi farmacologici e procedure chirurgiche. L’Acido Ursodesossicolico (UDCA) è un farmaco standard per i calcoli biliari di Colesterolo, poiché promuove la dissoluzione dei calcoli aumentando la secrezione di acidi biliari e sopprimendo la sintesi epatica di Colesterolo, riducendo così la saturazione di Colesterolo nella bile. Anche le Statine sono emerse come potenziale trattamento grazie alla loro capacità di modulare il metabolismo del Colesterolo e diminuire la formazione di calcoli biliari. Secondo Georgescu et al., le statine possono inoltre agire attraverso la modulazione del microbiota intestinale. La medicina tradizionale cinese contribuisce ulteriormente alla gestione dei calcoli biliari. Ad esempio, Huang et al. hanno riportato che i polisaccaridi di Ganoderma lucidum alleviano la formazione di calcoli biliari di Colesterolo attraverso la regolazione dipendente da FXR del metabolismo del Colesterolo e degli acidi biliari. Altre formulazioni a base di erbe, come i granuli Shugan Lidan Xiaoshi (SLXG), hanno anche dimostrato un potenziale terapeutico prendendo di mira geni tra cui HMGCR, SOAT2 e UGT1A1, modulando così l’omeostasi del Colesterolo. Inoltre, prove sempre più numerose indicano il ruolo del microbiota intestinale nella prevenzione e nel trattamento dei calcoli biliari di colesterolo. Ad esempio, Wang et al. hanno dimostrato che l’integrazione di lattobacilli ha ridotto l’incidenza e la gravità dei calcoli biliari indotti da una dieta ricca di grassi, indicando una nuova promettente direzione terapeutica.
- Malattie neurodegenerative
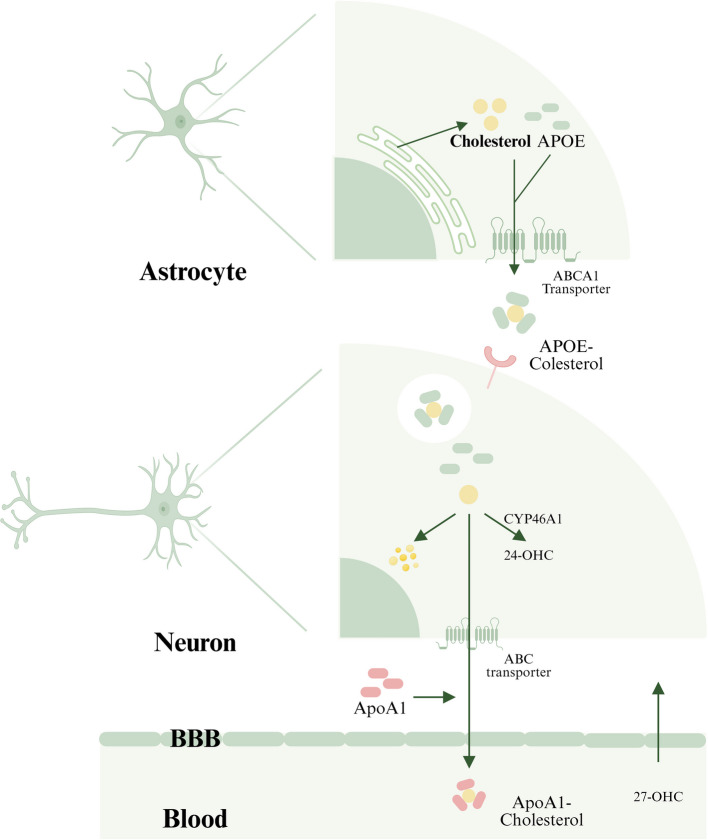
Il cervello umano contiene la più alta concentrazione di Colesterolo tra tutti gli organi, rappresentando circa il 25% del Colesterolo totale del corpo. Questo pool strettamente regolato è essenziale per il mantenimento dell’architettura neuronale, della plasticità sinaptica e della neurotrasmissione, con prove emergenti che collegano il metabolismo del Colesterolo disregolato alle malattie neurodegenerative. Separato dalla circolazione sistemica dalla barriera emato-encefalica (BBB), il Colesterolo cerebrale opera come un sistema metabolico autonomo. Gli astrociti sono la fonte primaria di Colesterolo cerebrale, producendolo attraverso la sintesi de novo e consegnandolo ai neuroni tramite il trasporto mediato da ApoE. Il Colesterolo in eccesso subisce tre destini regolatori: (1) accumulo come goccioline lipidiche citoplasmatiche, (2) efflusso mediato dai trasportatori ABC tramite particelle legate ad ApoA1, o (3) conversione enzimatica in ossisteroli (24-idrossicolesterolo [24-OHC] e 27-OHC) per l’eliminazione attraverso la BBB.
La malattia di Alzheimer (AD), una patologia neurodegenerativa prototipica, è caratterizzata dalla deposizione di placche di beta-amiloide (Aβ) e da grovigli neurofibrillari. L’aumento dei livelli di Colesterolo nelle membrane neuronali è correlato alla progressione dell’AD [160, 161]. L’ipercolesterolemia promuove la produzione di Aβ facilitando la processazione della proteina precursore dell’amiloide (APP) all’interno dei lipid raft ricchi di colesterolo tramite le β- e γ-secretasi. L’allele ApoE4 (ε4), il più forte fattore di rischio genetico per l’AD ad esordio tardivo, compromette il trasporto del Colesterolo e la clearance dell’Aβ. In particolare, le dinamiche degli ossisteroli mostrano ruoli opposti: il 27-OHC aggrava la produzione di Aβ e la progressione dell’AD, mentre il 24-OHC promuove l’attività dell’α-secretasi per ridurre il carico di Aβ, esercitando effetti neuroprotettivi [159].
Il limitato successo clinico delle attuali terapie mirate all’Aβ ha spostato l’attenzione verso interventi sul metabolismo del Colesterolo. Le statine, tradizionalmente utilizzate per la dislipidemia, mostrano un potenziale nella gestione dell’AD. Una meta-analisi di 55 studi osservazionali ha indicato che la terapia con statine è associata a una riduzione del 14% del rischio di demenza e a una diminuzione del 18% dell’incidenza dell’AD. Questi benefici possono derivare dall’inibizione della sintesi del Colesterolo e dalla ridotta aggregazione di Aβ, in particolare con statine lipofile che attraversano la barriera emato-encefalica come la Simvastatina. Prove emergenti evidenziano la rilevanza terapeutica del CYP46A1, un enzima neuronale specifico che converte il colesterolo in 24-OHC. La sovraespressione di CYP46A1 migliora la cognizione nei modelli murini e la sua modulazione farmacologica si dimostra promettente: ad esempio, KaiXinSan regola positivamente CYP46A1 per alleviare i deficit cognitivi indotti da 27-OHC, mentre il farmaco anti-HIV efavirenz agisce come modulatore dipendente dalla concentrazione di CYP46A1. Nuovi analoghi di efavirenz progettati specificamente per colpire l’attività di CYP46A1 offrono un ulteriore potenziale per il trattamento dell’AD . Insieme, questi risultati stabiliscono la regolazione del metabolismo del colesterolo come una promettente frontiera terapeutica nella patogenesi dell’AD.
- Tumori
Prove emergenti identificano il metabolismo del Colesterolo disregolato come un segno distintivo metabolico critico del cancro. Rispetto alle cellule normali, le cellule maligne mostrano profonde alterazioni nell’omeostasi del Colesterolo, caratterizzate da quattro modifiche chiave: (1) iperattivazione della biosintesi de novo del Colesterolo, (2) aumento dell’assorbimento mediato da LDLR, (3) alterazione dell’efflusso di colesterolo e (4) accumulo patologico di derivati del Colesterolo. Studi comparativi indicano che i tessuti tumorali presentano livelli di colesterolo significativamente più elevati rispetto ai tessuti normali adiacenti, il che è correlato a una maggiore aggressività e a un maggiore potenziale metastatico. Ciò è supportato da una marcata sovraregolazione dei geni coinvolti nella biosintesi del Colesterolo, come SREBP2, HMGCR, SQS, OSC e SQLE, insieme a un’elevata espressione di LDLR e a una maggiore esterificazione del Colesterolo.
Le attuali strategie antitumorali che prendono di mira il metabolismo del Colesterolo si concentrano principalmente sull’inibizione della biosintesi, del trasporto e dell’esterificazione. Le Statine, inibitori classici dell’HMGCR, mostrano un’attività antitumorale ad ampio spettro nei tumori della prostata, dello stomaco, dell’esofago e della mammella. Agiscono riducendo i livelli di Colesterolo cellulare e sopprimendo la sintesi degli isoprenoidi (ad esempio, geranilgeranil pirofosfato (GGPP) e FPP), interrompendo così la funzione GTPasi nelle cellule tumorali. Tuttavia, l’uso a lungo termine delle Statine (> 4 anni) è stato associato a un aumento del rischio di diversi tumori, tra cui quelli del colon, della vescica e del polmone. Oltre agli effetti ipocolesterolemizzanti, le Statine esercitano un’azione antitumorale attraverso molteplici meccanismi: inducono l’apoptosi tramite l’attivazione di FOXO3a e l’inibizione dell’autofagia nei tumori del cavo orale e del colon, innescano la ferroptosi tramite l’interruzione della via del mevalonato, promuovono la piroptosi e modulano il microambiente tumorale. Clinicamente, le statine mostrano effetti sinergici se combinate con le terapie convenzionali, migliorando gli esiti nei pazienti con carcinoma squamocellulare dell’esofago sottoposti a chemioradioterapia e superando la resistenza ai farmaci nel carcinoma polmonare a piccole cellule recidivante. Per ridurre gli effetti collaterali come la miopatia, sono in fase di sviluppo Statine nanoformulate con un targeting tumorale migliorato.
Il fattore di trascrizione SREBP2 è un regolatore critico della sintesi del Colesterolo. La sua attivazione tramite isomerizzazione mediata dalla prolil isomerasi fosfato-dipendente (PIN1) promuove l’accumulo di Colesterolo oncogenico e il silenziamento di PIN1 riduce i livelli di colesterolo cellulare, mostrando un potenziale terapeutico nel cancro alla vescica. Anche l’inibizione del trasporto del Colesterolo attraverso il blocco del rilascio lisosomiale di Colesterolo o il targeting di NPC1 con agenti come l’itraconazolo sopprime efficacemente la crescita tumorale. Inoltre, l’esterificazione del Colesterolo attraverso ACAT/SOAT1 rappresenta un nodo vulnerabile, con inibitori come l’Avasimibe che esercitano potenti effetti antitumorali in modelli di melanoma. Le evidenze cliniche mostrano un accumulo patologico di esteri del colesterolo nel cancro al pancreas, al colon-retto e alla prostata metastatica, dove l’inibizione di ACAT1 riduce sostanzialmente la progressione e le metastasi.
Sebbene il targeting del metabolismo del Colesterolo abbia un ampio potenziale antitumorale, sempre più evidenze suggeriscono la necessità di approcci terapeutici specifici per tipo di cancro. Le strategie attuali includono l’inibizione della sintesi, l’interruzione del trasporto e il blocco dell’esterificazione, ognuna delle quali richiede un’applicazione personalizzata in base alle dipendenze metaboliche del tumore. Le prospettive future prevedono lo sviluppo di terapie di precisione che combinino il targeting metabolico con trattamenti convenzionali e sistemi di somministrazione avanzati come la nanotecnologia.
AAS e dislipidemia
Gli steroidi anabolizzanti-androgeni (AAS) causano, a diverso grado dipendente dalla natura strutturale di sintesi della molecola, una significativa dislipidemia aterogena alterando il metabolismo del Colesterolo. In genere, riducono sensibilmente l’HDL-C dal 20% al 70% e aumentano l’LDL-C dal 30% al 50%, oltre a causare un aumento dei Trigliceridi ematici. Queste anomalie delle lipoproteine possono aumentare il rischio di cardiopatia coronarica da tre a sei volte.
- AAS e riduzione dell’HDL
Diversi studi interventistici hanno esaminato l’effetto dell’uso di AAS sul colesterolo. Peter Bond ha fatto un piccolo riassunto di questi studi nel suo libro “Book on Steroids” il quale riporto nella tabella sottostante. Sebbene non tutti gli studi abbiano riscontrato una diminuzione statisticamente significativa del colesterolo HDL (↔️), molti lo fanno e nel complesso mostrano inequivocabilmente una diminuzione. Ciò è particolarmente vero per gli AAS orali, che sembrano avere l’effetto maggiore sul colesterolo HDL.
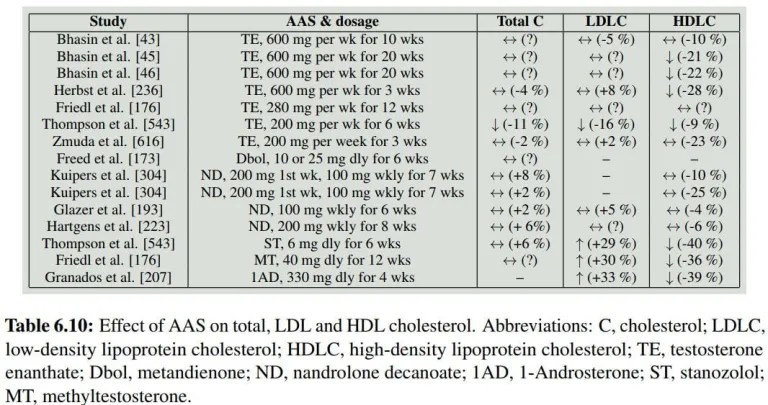
Si ritiene che gli AAS riducano l’HDL aumentando l’attività di un enzima chiamato lipasi epatica. Si tratta di un enzima prodotto principalmente dal fegato. Essendo una lipasi, catalizza le reazioni di idrolisi dei lipidi. In particolare, scinde gli acidi grassi dal triacilglicerolo (Trigliceride) e i fosfolipidi dalle particelle lipoproteiche, come l’HDL. Idrolizzando il triacilglicerolo e i fosfolipidi dall’HDL, riduce le dimensioni di queste particelle. Queste particelle più piccole vengono catabolizzate a un ritmo più elevato.
Thompson et al. hanno esaminato queste sottofrazioni di colesterolo HDL che differiscono per dimensioni. Hanno misurato i livelli di colesterolo HDL2 e HDL3: le particelle di colesterolo HDL2 sono più grandi e di densità inferiore rispetto a quelle HDL3. Gli uomini partecipanti hanno ricevuto 200mg di Testosterone Enantato alla settimana o 6mg di Stanozololo orale (Winstrol) al giorno per 6 settimane in un design crossover. I risultati sono stati i seguenti:

Come si può notare, la maggiore diminuzione relativa è stata osservata nella frazione HDL2 più grande a seguito del trattamento con Stanozololo. Al contrario, il Testosterone non ha mostrato una diminuzione statisticamente significativa nella frazione HDL2, ma ha fatto altrettanto nella frazione HDL3 più piccola. Non è del tutto chiaro cosa provochi la diminuzione di questa frazione.
Quando dei bodybuilder sono stati randomizzati a ricevere 200mg di Nandrolone Decanoato alla settimana o un placebo. Non sono stati riscontrati cambiamenti statisticamente significativi nel colesterolo totale, nel colesterolo LDL e nel colesterolo HDL. Analogamente, non sono stati riscontrati cambiamenti significativi nelle sottofrazioni di colesterolo HDL2 e HDL3. In particolare, nella stessa pubblicazione, gli autori riferiscono anche di uno studio in cui hanno seguito un gruppo di atleti di forza che si autosomministravano steroidi anabolizzanti. Sono stati utilizzati diversi composti in vari dosaggi, ma vale la pena sottolineare che la maggior parte di essi comprendeva anche uno steroide anabolizzante orale (soprattutto Stanozololo). In questo caso, il colesterolo HDL è sceso in picchiata: da 1,08 mmol/L a 0,43 mmol/L dopo 8 settimane. La sottofrazione di colesterolo HDL2 è scesa da 0,21 a 0,05 e la sottofrazione di colesterolo HDL3 è scesa da 0,87 a 0,40 mmol/L.
- AAS e funzione dell’HDL
Dato il legame tra l’effetto di un farmaco sui livelli di HDL e il rischio di malattie cardiovascolari, la ricerca ha iniziato a concentrarsi sulla funzione del HDL. L’HDL è il protagonista di un processo chiamato trasporto inverso del colesterolo. Nell’aterosclerosi, il colesterolo si accumula nelle cellule del sistema immunitario (macrofagi) e nelle cellule muscolari lisce che circondano i vasi sanguigni. Queste cellule, a loro volta, diventano le cosiddette cellule schiumose, che segnano il punto di partenza dell’aterosclerosi. Le particelle di HDL sono in grado di raccogliere il colesterolo da queste cellule – efflusso di Colesterolo. L’efflusso del colesterolo dalle cellule schiumose nelle particelle di HDL è uno dei modi in cui si ritiene che il HDL eserciti i suoi effetti protettivi sulle arterie. Il HDL raccolto può poi essere riportato al fegato, che lo incorpora nella bile e può quindi essere secreto nelle feci. Allo stesso modo, le particelle di HDL possono trasferire parte del loro contenuto alle particelle LDL, che possono finire nuovamente nelle cellule schiumose o essere assorbite dal fegato.
Esistono metodi per misurare la capacità di efflusso del HDL e l’idea attuale è che la sua modulazione possa influire sul rischio di malattie cardiovascolari, contrariamente ai livelli di HDL in sé. Esistono diversi modi in cui l’HDL può assorbire il Colesterolo dalle cellule schiumose. Uno di questi coinvolge un trasportatore chiamato ATP-binding casette transporter A1 (ABCA1), che si ritiene sia il più importante. Contribuiscono anche altri trasportatori, come ABCG1 e il recettore scavenger B1, oltre alla diffusione semplice.
In uno studio (non controllato), uomini anziani ipogonadici sono stati randomizzati alla TRT con o senza Dutasteride (un inibitore della 5a-reduttasi). Dopo 3 mesi, la TRT era riuscita a riportare i livelli di Testosterone di questi uomini all’interno del range di normalità. L’HDL e la capacità di efflusso del HDL sono rimasti inalterati.
Un altro studio, randomizzato e controllato, ha applicato un approccio leggermente diverso. Uomini sani, di età compresa tra i 19 e i 55 anni, sono stati castrati medicalmente per sopprimere completamente la loro produzione endogena. In seguito, hanno ricevuto un placebo, una TRT a basso dosaggio, una TRT sostitutiva completa o una TRT sostitutiva completa con Letrozolo, un inibitore dell’Aromatasi che inibisce la conversione del Testosterone in Estradiolo. L’HDL è aumentato leggermente nel gruppo placebo e in quello a basso dosaggio, mentre è rimasto inalterato nei due gruppi che hanno ricevuto una dose sostitutiva completa. Inoltre, mentre è stata riscontrata una piccola diminuzione della capacità di efflusso di ABCA1 nel gruppo che ha ricevuto anche il Letrozolo, non sono stati osservati cambiamenti negli altri tre gruppi. A causa delle dimensioni ridotte dei gruppi, è possibile che un piccolo effetto non sia stato notato.
Esiste un solo studio che ha esaminato questo aspetto, ed era di natura trasversale (si tratta di misurazioni effettuate in un solo momento, il che rende impossibile/difficile trarre conclusioni). I ricercatori hanno confrontato le misurazioni di un gruppo di utilizzatori di AAS con quelle di non utilizzatori e controlli sedentari, che avevano un’età corrispondente. I consumatori di AAS erano forti utilizzatori, avendo fatto uso di AAS in media per circa 8 anni con un dosaggio medio di (apparentemente) 2,5g settimanali. La capacità delle HDL di effluire il Colesterolo dai macrofagi è risultata inferiore del 13% nei consumatori di AAS rispetto ai non consumatori con allenamento della forza. Anche in questo caso, a causa della natura trasversale dello studio, è difficile dire se questo sia causale.
Effetti clinici principali:
- Riduzione dell’HDL: le riduzioni possono essere estreme, soprattutto con gli AAS non aromatizzabili come lo Stanozololo o l’Oxandrolone.
- Aumento dell’LDL: dosi soprafisiologiche di AAS sovrastimolano le lipasi epatiche, alterando la sintesi delle apolipoproteine e accelerando la circolazione delle particelle di LDL.
- Alterazione del eflusso del efflusso del HDL: dosi soprafisiologiche di AAS alterano l’efflusso del HDL contribuendo nel lungo termine al deposito di placche aterosclerotiche.
- Trigliceridi: livelli elevati sono comunemente osservati anche durante l’uso attivo di AAS.
Insorgenza e reversibilità:
- Insorgenza: Le alterazioni del profilo lipidico si manifestano rapidamente, spesso entro le prime 9 settimane di somministrazione di AAS, anche se con l’uso di alcuni AAS orali metilati in C-17 l’insorgenza di una alterazione lipidica può manifestarsi entro 3-4 settimane.
- Reversibilità: Gli studi dimostrano che i profili lipidici sono in gran parte reversibili e tendono a normalizzarsi entro 2,5-5 mesi dalla completa interruzione dell’uso di AAS.
Conseguenze a lungo termine:
- Sebbene i marcatori lipidici in genere tornino ai valori basali al termine dei cicli, l’uso cronico e ripetuto accelera l’accumulo di placche aterosclerotiche, aumentando il rischio di infarto miocardico precoce e ictus ischemico.
- I medici spesso raccomandano pannelli lipidici a digiuno completi, test di funzionalità epatica e monitoraggio cardiovascolare per le persone con una storia di abuso di AAS.
Fine I° parte…
Gabriel Bellizzi [CEO BioGenTech]
Riferimenti:

Una risposta a "Colesterolo – dalla biosintesi ai trattamenti della ipercolesterolemia – I° Parte."