
Introduzione:
Per i più informati, è ormai appurato il fatto che la soppressione dell’attività dell’Asse HPT, per via dell’uso di AAS, è determinata da:
- L’origine del AAS
- Il tasso di conversione del AAS ad estrogeno, attraverso l’enzima Aromatasi in alcuni tessuti (adiposo, mammario)
- Dose e tempo d’uso/abuso del AAS
- Attività androgena del AAS.
Di conseguenza, dovrebbe essere chiaro che anche farmaci puramente androgeni o essenzialmente anabolizzanti e con forte potenziale di legame con il AR [vedi SARM non steroidei] possono causare una sotto-regolazione della funzionalità dell’Asse HPT, quindi con meccanismi indipendenti dalla aromatizzazione della molecola.
Infatti, gli AAS [ed i SARM non steroidei] attraversano la barriera ematoencefalica e si legano ai recettori Ipotalamici. Ciò comporterà una marcata soppressione dell’HPTA per via di intermediari quali i peptidi oppioidi endogeni.
Da qualche tempo, e per i risultati ottenuti nel trattamento di soggetti dipendenti da droghe e alcool, è emerso il potenziale degli antagonisti dei recettori oppiacei, come il Naltrexone, di bloccare l’attività dei peptidi oppioidi endogeni sull’Ipotalamo e, consequenzialmente, impedire una riduzione del rilascio di GnRH e, di conseguenza, di LH e FSH.
Questo indubbio potenziale ha attirato l’interesse degli utilizzatori di AAS, sempre in cerca, e a ragione, di un modo per ridurre gli effetti collaterali in largo spetro dato dall’uso di PEDs.
In questo articolo descriverò come i recettori oppioidi situati nel Ipotalamo agiscano causando una riduzione gonadotropica e, di rimando, androgena. Tratterò in dettaglio il farmaco Naltrexone e dove questo potrebbe realmente trovare applicazione.
Ma prima un breve ripasso sul HPTA…
Dettaglia sul HPTA:
Con Asse-Ipotalamo-Ipofisi-Testicoli (HPTA) ci si riferisce alla connessione tra ipotalamo, ghiandola pituitaria e testicoli come se queste singole ghiandole endocrine fossero una singola entità. Poiché queste ghiandole spesso agiscono in concerto, i fisiologi e gli endocrinologi ritengono conveniente e descrittivo parlare di esse come di un unico sistema.
L’asse HPTA svolge una parte critica nello sviluppo e nella regolazione di un certo numero di sistemi del corpo, come i sistemi riproduttivi e immunitari. Le fluttuazioni di questo asse causano variazioni negli ormoni prodotti da ciascuna ghiandola e hanno diversi effetti locali e sistemici nel corpo.
In breve, l’asse HPT rappresenta un sistema di stimolazione/inibizione degli ormoni prodotti dalle rispettiva strutture:
- Ipotalamo: GnRH (ormone di rilascio delle gonadotropine; in inglese Gonadotropin-releasing hormone).
- Ipofisi (o ghiandola Pituitaria): dalle cellule beta e gamma rispettivamente l’ormone follicolo-stimolante (FSH) e l’ormone luteinizzante (LH).
- Testicoli: Testosterone, Androstenedione, DHEAS, Inibina.
Come ben sappiamo, diversi AAS sono derivati sintetici del Testosterone, il principale androgeno nei maschi. Il Testosterone sopprime marcatamente l’HPTA, mentre altri derivati lo fanno in misura maggiore o minore.
In questo specifico caso ci concentreremo sulla soppressione/sottoregolazione del HPTA Androgeno-dipendente.
Come accennato pocanzi, l’effetto sotto-regolatore dato dagli androgeni a livello Ipotalamico è mediato dai peptidi oppioidi endogeni. Ma come si verifica la disfunzione endocrina derivata dalla attivazione dei recettori oppioidi?
Oppioidi e disfunzione endocrina:
Gli oppioidi sono stati usati per secoli come metodo principale per alleviare il dolore intenso, in particolare il dolore acuto e il dolore da cancro avanzato. Gli oppioidi somministrati regolarmente 24 ore su 24 continuano a essere il cardine nella gestione del dolore associato alle condizioni di fine vita e sono sanciti nella scala analgesica dell’Organizzazione Mondiale della Sanità. Cinquemila anni fa il Papaver somniferumfu veniva coltivato dai Sumeri. Da allora si è intrecciato con l’esperienza umana di molte culture successive, fungendo da benedizione ma anche da maledizione, come analgesico e ansiolitico, stupefacente e musa. Su di esso sono state combattute guerre; qualcuno potrebbe dire che questo continua ad essere perpetrato. Nel 1804 Friedrich Sertürner isolò la morfina e nel 1843 Alexander Wood utilizzò la prima forma iniettabile di questa.
Le proprietà analgesiche degli oppioidi hanno alleviato la sofferenza di innumerevoli persone e il loro ruolo nella gestione del dolore acuto e del dolore oncologico è ben consolidato. Tuttavia, circa il 50% dei riceventi con dolore non oncologico soffre di effetti avversi e nel 20% di questi ciò porta all’interruzione della terapia. (1) Gli effetti avversi, ben noti sia agli operatori sanitari che ai non addetti, comprendono sonnolenza, costipazione, nausea, prurito e depressione respiratoria. Gli effetti avversi meno noti includono instabilità cardiovascolare, mal di testa e spasmi muscolari sia della muscolatura liscia che striata, che portano a ritenzione urinaria, rallentamento della motilità intestinale e scatti mioclonici. (2) Meno ancora saranno a conoscenza della potenziale compromissione della funzione immunitaria, nota per essere specifica per gli oppioidi.(3) I medici del dolore probabilmente conosceranno l’iperalgesia indotta da oppioidi, anche se raramente viene diagnosticata.
Gli effetti a lungo termine meno conosciuti sono la soppressione della funzione endocrina e l’effetto sulla funzione cognitiva.(3) Con l’invecchiamento della popolazione, l’aumento dei tassi di sopravvivenza al cancro e l’uso crescente di oppioidi per il dolore persistente non oncologico, il numero di prescrizioni di oppioidi nella comunità è aumentato da 6 milioni a 15 milioni nel Regno Unito tra il 1999 e il 2008 (NHS Information Center). Gli Stati Uniti hanno il 5% della popolazione mondiale ma consumano l’80% dell’offerta globale di oppioidi. Il numero di overdose fatali accidentali correlate a oppioidi da prescrizione negli Stati Uniti è ora superiore a quello per l’uso ricreativo di eroina e cocaina combinati. (4)
È per ciò fondamentale che tutti coloro che gestiscono e prescrivono oppioidi per il dolore persistente siano consapevoli degli effetti a lungo termine degli oppioidi sulla funzione endocrina e siano in grado di diagnosticare carenze, monitorare i livelli ormonali, comprendere e spiegare al paziente le possibili conseguenze di queste carenze, sforzarsi di ridurre e sospendere gli oppioidi quando appropriato e collaborare con altri medici per sostituire le carenze ormonali.
- Effetti endocrini degli oppioidi:
Nel 1895 il reverendo RH Graves (5) notò come “l’oppio mangiava la virilità dell’individuo” e nel 1925 il chirurgo generale HS Cumming (6) affermò che “l’oppio rende effeminato un uomo”. Katz (7) ha già citato Charles Bruce, che, nel 1839, definì i tossicodipendenti da oppio in Assam “più effeminati delle donne”. (8)
È stato riportato in molte occasioni che gli oppioidi, somministrati per qualsiasi via, sopprimono l’asse ipotalamo-ipofisi-gonadi e hanno un impatto misurabile sulla funzione gonadica. (7)
L’ipotalamo, come sappiamo, è fondamentale per la regolazione degli ormoni sessuali. Esercita il suo controllo attraverso la secrezione dell’ormone di rilascio delle gonadotropine (GnRH) dall’area preottica nella circolazione portale ipofisaria all’eminenza mediana. Il GnRH stimola il rilascio dell’ormone follicolo-stimolante (FSH) e dell’ormone luteinizzante (LH) dall’ipofisi anteriore attivando il proprio recettore del GnRH, che, attraverso l’aumento dei livelli di calcio e proteina chinasi C, porta alla formazione e secrezione di FSH e LH (Figura seguente).
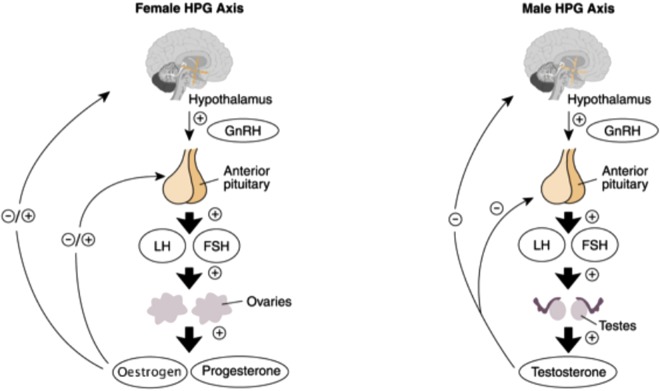
(9) .
(Riprodotto con il permesso di NIAAA).
Il GnRH viene normalmente rilasciato a impulsi in tutti i vertebrati studiati. Questi impulsi producono picchi circadiani essenziali per il corretto funzionamento del sistema riproduttivo determinando quando ciascun ormone viene rilasciato. La secrezione non pulsatile di GnRH provoca una sottoregolazione dell’ipofisi e porta a una secrezione di LH deficitaria. 7
L’FSH è responsabile della crescita precoce dei follicoli ovarici nelle femmine e del mantenimento dell’epitelio spermatogeno nei maschi. L’LH è responsabile della maturazione finale dei follicoli e della loro secrezione di estrogeni, nonché dell’ovulazione e della formazione iniziale del corpo luteo nella femmina. Nel maschio stimola le cellule di Leydig a secernere testosterone. 10 Questi ormoni sono necessari anche per lo sviluppo appropriato dell’essere umano – fisiologicamente, fisicamente e socialmente.
L’asse ipotalamo-ipofisario è costantemente sotto l’effetto di molteplici sostanze tra cui neurotrasmettitori, ormoni steroidei e oppioidi endogeni. Gli oppioidi esogeni esercitano un effetto sugli stessi recettori degli oppioidi endogeni e hanno dimostrato di interferire con il rilascio (compresa la sua natura pulsatile) di GnRH. 7 , 10 , 11 Il naloxone ha mostrato un aumento dei livelli di GnRH, e quindi un aumento della concentrazione di LH e della frequenza del polso, che ha suggerito un livello basale di inibizione della secrezione di LH a base di oppioidi. È stato suggerito che la morfina inibisca la biosintesi del GnRH. 11 Gli oppioidi riducono anche il feedback negativo degli steroidi sessuali sull’ipofisi anteriore, così come la sua risposta al GnRH. 7Al contrario, la secrezione di FSH non è, o solo in minima parte, influenzata.
Con la riduzione dei livelli di LH, il testosterone e l’estradiolo si abbassano proporzionalmente. Li Shizhen scrisse dell’oppio nel suo Compendium of Materia Medica (1578) che “le persone lo usano per l’arte del sesso, in particolare per “arrestare l’emissione seminale”” 11 . Gli studi sugli animali hanno aggiunto credito all’osservazione di Shizhen, dimostrando una ricettività sessuale inibita in entrambi i sessi di ratti con oppioidi e il contrario con naloxone. 12 Ciò sembra essere dovuto alla diminuzione dell’eccitazione piuttosto che all’impotenza. In modo allarmante, studi correlati hanno mostrato che l’esposizione prepuberale agli oppioidi inibiva la maturazione sessuale. 13 Nell’uomo, in seguito al trattamento con oppioidi per via orale e intratecale, si sono verificate irregolarità mestruali, inclusa l’amenorrea. Daniele14 ha notato che c’era anche una diminuzione degli androgeni surrenali, e quindi spiegando la diminuzione della libido e delle prestazioni sessuali così spesso incontrate in coloro che sono esposti a oppioidi a lungo termine. Sembra che la diminuzione del comportamento sessuale possa anche essere dovuta all’azione diretta degli oppioidi sui recettori µ e ∂ nell’ipotalamo. 15 , 16
Il termine “ormoni sessuali” comprende gli steroidi sessuali (testosterone ed estradiolo) e gli ormoni non steroidei (GnRH, FSH e LH).
Il testosterone è il principale ormone dei testicoli, sintetizzato dal colesterolo nelle cellule di Leydig e dall’androstenedione nella corteccia surrenale. La sua secrezione è controllata da LH a 4–9 mg/die. Piccole quantità sono secrete dalle ovaie e forse dalla corteccia surrenale nelle donne. È legato per il 98% alle proteine (globulina e albumina leganti gli steroidi gonadici) nel plasma. Solo il testosterone libero e debolmente legato all’albumina è disponibile per agire sui recettori degli androgeni. Alcune cellule bersaglio convertono il testosterone in diidrotestosterone, che forma complessi ormone-recettore più stabili. Nei maschi il testosterone ha un ritmo circadiano, con i livelli più alti al mattino. La variazione massima può essere dell’ordine del 35%.
Il testosterone impartisce un feedback negativo sul rilascio di LH dall’ipofisi. Sviluppa e mantiene le caratteristiche sessuali secondarie maschili e incoraggia i comportamenti sessuali maschili. È anabolico, aumenta il tasso di crescita e, insieme all’FSH, promuove la spermatogenesi. Fa tutto questo legandosi ai recettori intracellulari e formando complessi che si legano al DNA, facilitando così una certa espressione genica.
Gli estrogeni (17ß-estradiolo, estrene ed estriolo) sono i principali steroidi sessuali femminili e sono biosintetizzati dal testosterone e dall’androstenedione. Sono secreti dalle cellule della granulosa nei follicoli ovarici. Il novantotto per cento degli estrogeni è legato alle proteine. Sono metabolizzati dal fegato ed escreti nelle urine. Gli estrogeni facilitano lo sviluppo dei follicoli ovarici, aiutano nella regolazione del ciclo mestruale e nei necessari cambiamenti nell’anatomia interna femminile e hanno effetti anabolici sull’utero e sulle tube di Falloppio. Diminuiscono i livelli di FSH e alterano i livelli di LH. Sono responsabili dei comportamenti sessuali femminili e della libido e sono in gran parte responsabili dello sviluppo del seno. Questi effetti, combinati con l’assenza di androgeni, portano a caratteristiche sessuali secondarie femminili.
In sintesi, gli oppioidi portano a una diminuzione della secrezione di GnRH, che a sua volta porta a livelli ridotti di LH. Ciò si traduce in una diminuzione della secrezione di testosterone ed estradiolo, che porta ai segni e sintomi elencati nell’immagine sottostante. È importante notare che questi cambiamenti si sviluppano nel corso di settimane o anni.

Diagnosi di ipogonadismo nel maschio:
La diagnosi di ipogonadismo dipende dall’anamnesi e dall’esame insieme ai test di laboratorio. Nel maschio postpuberale le potenziali cause di ipogonadismo primario menzionate nella tabella sottostante possono causare difficoltà diagnostiche, soprattutto per quanto riguarda la loro graduale insorgenza. Se si sospetta l’ipogonadismo, è quindi importante differenziare l’insufficienza gonadica primaria da quella dell’asse ipotalamo-ipofisario.

Per la seconda consultazione internazionale dell’Organizzazione mondiale della sanità sulla disfunzione erettile, che ha considerato il ritmo circadiano e la natura pulsante della secrezione di Testosterone, dovrebbero essere prelevati due campioni di sangue tra le 8:00 e le 11:00 (quando si presume che i livelli di Testosterone siano al massimo, sebbene il ritmo circadiano può diminuire con l’aumentare dell’età e con variazioni tra sportivi e sedentari). I campioni devono essere inviati per la misurazione del testosterone sierico, della globulina legante gli ormoni sessuali (SHBG), della prolattina, dei livelli di FSH e LH.
Livelli di FSH/LH da normali ad alti potrebbero indicare un ipogonadismo primario (vedi tabella sopra). È importante misurare il livello di FSH poiché ha un’emivita più lunga e dimostra una minore variabilità rispetto all’LH. Ipogonadismo secondario (Tabella seguente) è indicato da bassi livelli di testosterone e livelli di FSH/LH da normali a bassi ( Riquadro seguante).

Con l’invecchiamento si riduce la fluttuazione diurna del testosterone sierico (le variabili vi sono nella popolazione sportivamente attiva). Il livello scende notevolmente durante il giorno, rafforzando l’importanza del campionamento mattutino. Nell’ipogonadismo in questa fascia di età i livelli di testosterone totale possono essere normali se i livelli di globulina legante gli ormoni sessuali (SHBG) sono aumentati. I livelli di SHBG aumentano con l’età e quindi riducono la biodisponibilità del testosterone.
I dati recenti del Massachusetts Male Aging Study (MMAS) forniscono prospettive sui normali intervalli di androgeni, come mostrato nella tabella seguente.

Diagnosi di ipogonadismo nella femmina:
Allo stesso modo, l’ipogonadismo nelle femmine può essere dovuto a un asse ipotalamo-ipofisario o a un difetto gonadico primario. I segni ei sintomi sono strettamente legati al ciclo mestruale negli anni postmenarca e premenopausale. I segni comuni includono oligomenorrea, amenorrea e mancato concepimento. Possono verificarsi sintomi più sottili come vampate di calore e ansia, raramente con cambiamenti nella distribuzione dei peli pubici e nella dimensione del seno 21 . Sebbene gli effetti clinici possano essere gravi per le donne come per gli uomini, i cambiamenti esteriori o visibili non sono così evidenti e non sono stati studiati in dettaglio, specialmente nelle donne in postmenopausa.
Sebbene sia stato dimostrato che la carenza di androgeni è sintomatica nelle donne che utilizzano la terapia con oppioidi intratecali e in quelle in trattamento con metadone di mantenimento, i livelli di testosterone non sono stati misurati di routine. Si ritiene che il diidroepiandrosterone (DHEA) sia abbassato ed è noto che i livelli di LH sono notevolmente ridotti. Il DHEA è un marker della produzione di androgeni surrenali e circa il 50% degli androgeni prodotti nella femmina sono di origine surrenale. Vi è una scarsità di dati per quanto riguarda i livelli ormonali nelle donne e sono necessari ulteriori studi per quantificare il significato clinico di questo aspetto potenzialmente molto interessante della carenza di androgeni indotta da oppioidi (OPIAD).
Effetto degli oppioidi sugli ormoni surrenali:
Il dolore acuto e cronico, come risposte fisiologiche, determina un aumento della secrezione dell’ormone adrenocorticotropo (ACTH) e del cortisolo. Tuttavia, in diversi studi è stato riscontrato che l’uso cronico di oppioidi esogeni riduce i livelli di ACTH e cortisolo e le risposte del cortisolo alle sfide dell’adrenocorticotropina. 19 Gli oppioidi influenzano anche i ritmi circadiani della secrezione di cortisolo, determinando un aumento persistente dei livelli di ACTH e cortisolo e alla fine attenuando la risposta allo stress. 20 Anche i livelli di deidroepiandrosterone solfato (DHEAS), un precursore degli androgeni surrenali, sono stati notevolmente ridotti nei consumatori cronici di oppioidi sia maschi che femmine. 14 , 18Occasionalmente, la somministrazione di oppioidi può causare insufficienza surrenalica franca, ma i fattori di rischio per questo sono attualmente sconosciuti. 15
Prolattina:
La somministrazione acuta di oppioidi stimola il rilascio di prolattina dall’ipofisi anteriore attraverso un effetto a livello dell’ipotalamo. 15 Questo effetto può essere bloccato dalla metoclopramide, suggerendo che sia mediato da meccanismi dopaminergici. L’effetto della somministrazione a lungo termine di oppioidi sulla prolattina è meno chiaro. C’è un aumento occasionale del rilascio di prolattina, probabilmente dipendente dal tipo di oppioide. Il significato clinico di questo è sconosciuto, ma può causare galattorrea.
Ormoni tiroidei:
In generale, gli oppioidi non sembrano alterare in modo significativo la funzione tiroidea, 19 sebbene possano stimolare l’ormone stimolante la tiroide (TSH) attraverso l’ipotalamo. 15 Questo non è importante negli individui con tiroxina libera normale, ma gli individui con ipotiroidismo possono avere risposte prolungate ed esagerate agli oppioidi. 22
Ormone della crescita:
La somministrazione acuta di oppioidi porta ad un aumento della secrezione dell’ormone della crescita (GH), attraverso meccanismi che coinvolgono i recettori degli oppioidi, i livelli di feedback e la trascrizione genica. La dose minima richiesta è di circa 15 mg di morfina. 15 Abs et al. 23 hanno riscontrato una carenza di GH in circa il 15% dei pazienti che ricevevano oppioidi intratecali a lungo termine, ma non in tutti i pazienti. È stato dimostrato che il naloxone inibisce il GH nei soggetti sani, ma lo aumenta nelle donne obese. L’effetto della somministrazione cronica di oppioidi sul GH è complesso e attualmente poco conosciuto, ma sembra essere correlato agli ormoni sessuali, alla composizione corporea e al grado di insulino-resistenza. 15
Vasopressina:
È stato scoperto che il tramadolo causa iponatriemia attraverso il rilascio di vasopressina indotto dalla serotonina. 24 L’effetto degli oppioidi sull’ipofisi posteriore non è chiaro: sono stati riscontrati livelli di vasopressina sia aumentati che diminuiti, a seconda dello stato di idratazione. 22
Ossitocina:
Studi su donne in gravidanza hanno dimostrato che la morfina inibisce la produzione di ossitocina nelle prime fasi del travaglio e durante l’allattamento al seno dopo il parto. 25
Obesità e diabete:
Un numero crescente di dati suggerisce che gli oppioidi svolgono un ruolo nella regolazione dell’assunzione di cibo e della scelta del cibo, e forse la ricompensa associata ai cibi di buon gusto, attraverso meccanismi centrali. 15 L’uso cronico di oppioidi è associato ad aumento di peso, iperglicemia e peggioramento del diabete. Questa può essere un’azione centrale attraverso il sistema nervoso simpatico e una ridotta secrezione di insulina. 26 L’ipogonadismo è associato ad un aumento della resistenza all’insulina e al rischio di diabete mellito, 15 un rischio che è migliorato dalla sostituzione del testosterone. 27
Metabolismo delle catecolamine:
La terapia con oppioidi aumenta la secrezione di catecolamine attraverso l’ipotalamo e il tronco cerebrale. I pazienti che assumono oppioidi a lungo termine devono essere sottoposti a screening per l’ipertensione. 22
Metabolismo osseo:
Esistono molti fattori di rischio per la diminuzione della densità minerale ossea e l’osteoporosi nei pazienti trattati con oppioidi, tra cui possibile scarso stato nutrizionale, ipogonadismo, inibizione degli osteoblasti, ridotta sintesi di osteocalcina, calcio anormale e ormone paratiroideo e aumento del riassorbimento osseo, mediato dall’interleuchina 1. rappresenta un aumento del rischio di frattura ossea nei pazienti che assumono oppioidi. 3
- Via di somministrazione:
Gli oppioidi per uso cronico vengono somministrati per via orale o intratecale. Occasionalmente possono essere utilizzate altre vie, ad esempio l’iniezione ripetuta, sebbene questa pratica non sia raccomandata. 28
Cambiamenti ormonali che si verificano dopo la somministrazione intratecale sono stati riportati da diversi autori. 23 , 29 , 30
Si stima che il 90% dei pazienti che assumono oppioidi intratecali svilupperà ipogonadismo. Gli oppioidi orali hanno lo stesso effetto, sebbene l’inizio dell’azione possa essere più lento.
- Tipo e dose di oppioide:
Le classi di oppioidi differiscono nel loro effetto sulla soppressione delle gonadi. Tramadolo e buprenorfina non alterano significativamente i livelli di testosterone negli animali e nell’uomo e la buprenorfina non sopprime il cortisolo sierico.
L’incidenza di ipogonadismo era maggiore nei sopravvissuti al cancro che ricevevano una dose equivalente o superiore a 200 mg di morfina al giorno per almeno 1 anno rispetto ai sopravvissuti di pari età non in terapia con oppioidi, suggerendo che gli effetti sono correlati alla dose. 31 Anche la durata della terapia con oppioidi sembra aumentare la possibilità di soppressione ormonale, sebbene ciò debba essere studiato ulteriormente.
Esiste una possibile differenza di genere negli effetti endocrini, tendente a una maggiore sintomatologia nelle donne, ma sono necessari ulteriori studi. 22
Quando e cosa si misura prima di iniziare una terapia con oppioidi:
Prima di iniziare la terapia cronica con oppioidi si raccomanda di misurare quanto segue:
- pressione sanguigna;
- elettroliti (soprattutto se si utilizza tramadolo);
- livelli di glucosio a digiuno;
- funzione tiroidea (per escludere l’ipotiroidismo);
- livelli di testosterone, globulina legante l’ormone sessuale, LH/FSH ed estradiolo; e
- densità ossea (in un gruppo “a rischio”).
- Monitoraggio:
Non ci sono standard accettati, ma sembra ragionevole ripetere i test di cui sopra ogni 6 mesi.
- Considera i livelli di cortisolo nel sangue, DHEA, ACTH e GH a digiuno mattutino. (Ricorda che un livello di cortisolo nel sangue a digiuno anormalmente alto può rappresentare la perdita della variazione diurna e dovresti chiedere consiglio.)
- Ripetere la densità ossea ogni anno nel gruppo “a rischio”.
- Misurare i livelli di prolattina se c’è galattorrea.
- Terapia sostitutiva:
Non esistono standard accettati per la gestione della disfunzione endocrina indotta da oppioidi.
L’opzione migliore è ridurre gradualmente e ritirare gli oppioidi e monitorare la risposta per un periodo di alcuni mesi, se appropriato. Non è noto se il cambio di oppioidi sia di qualche beneficio. La buprenorfina sembra avere un effetto minore sugli ormoni surrenali ma ha un effetto maggiore sul TSH rispetto alla morfina. La risposta a diversi oppioidi è in gran parte sconosciuta al momento della scrittura.
Se non è possibile ottenere l’astinenza da oppiacei e il paziente presenta sintomi definiti di disfunzione endocrina, si raccomanda la sostituzione ormonale, con il monitoraggio da parte di un endocrinologo.
Il testosterone può essere sostituito, sia negli uomini che nelle donne, come cerotto o gel transdermico o per iniezione. È necessario un attento monitoraggio poiché gli effetti collaterali includono reazioni in sede, policitemia e aumento del rischio di cancro alla prostata negli uomini e irregolarità mestruali e irsutismo nelle donne.
La terapia sostitutiva con estrogeni è meglio monitorata da un ginecologo.
Cosa estrapolare di utile per un utilizzatore di PEDs da quanto detto?
Innanzi tutto bisogna sapere che l’attività di soppressione/sottoregolazione dell’Asse HPT androgeno-dipendente ha come intermediari i peptidi oppioidi endogeni, con attività principale da parte della Beta-Endorfina, delle Encefaline e Dinorfine attraverso il legame con i recettori oppioidi μ.
Quindi, di conseguenza, comprendere i meccanismi e gli effetti dell’uso di oppioidi sulla omeostasi ormonale, soprattutto riguardante l’HPTA, ci permette di capire meglio il legame tra attività AR di una molecola e suoi effetti mediati a livello ipotalamico che causano una sottoregolazione/soppressione della funzione del Asse HPT, anche se la molecola non è aromatizzabile e non possiede attività estrogenica e/o progestinica.
Adesso possiamo approfondire il discorso passando all’analisi del antagonisti degli oppioidi Naltrexone.
Caratteristiche del Naltrexone:
Il Naltrexone, venduto con tra gli altri con i nomi commerciali di ReVia e Vivitrol, è un farmaco utilizzato principalmente per gestire l’abuso di alcol o il disturbo da uso di oppioidi, riducendo le voglie e le sensazioni di euforia associate al disturbo da uso di sostanze “compensative”.[32] È stato anche osservato essere efficace nel trattamento di altre dipendenze e può essere utilizzato per loro in modalità off-label. [33] Una persona dipendente da oppioidi non dovrebbe ricevere il Naltrexone prima della disintossicazione.[4] Viene assunto per bocca o tramite iniezione intra-muscolo.[32] Gli effetti iniziano entro 30 minuti dalla somministrazione.[32] Una diminuzione del desiderio di oppioidi può richiedere alcune settimane per verificarsi.[32]
Il Naltrexone è un antagonista degli oppioidi e funziona bloccando gli effetti degli oppioidi, sia quelli endogeni che esogeni.[32]
Il Naltrexone è stato prodotto per la prima volta nel 1965 ed è stato approvato per uso medico negli Stati Uniti nel 1984.[32][34] Il Naltrexone, come Naltrexone/bupropione (nome commerciale Contrave), è anche usato per trattare l’obesità.[35]
Il Naltrexone, noto anche come N-ciclopropil-metilnorossimorfone, è un derivato dell’Ossimorfone (14-idrossi-diidromorfone). È specificamente il derivato dell’Ossimorfone in cui il sostituto metilico dell’ammina terziaria è sostituito con il Metilciclopropano.
Il farmaco strettamente correlato, il Metilnaltrexone (N-metilnaltrexone), è usato per trattare la costipazione indotta dagli oppioidi, ma non tratta la dipendenza in quanto non attraversa la barriera emato-encefalica e, quindi, non è di nostro interesse per l’ipotetico utilizzo in ambito di PEDs. Il Nalmefene (6-desossi-6-metilenaltrexone) è simile al Naltrexone ed è usato per gli stessi scopi. Il Naltrexone non deve essere confuso con il Naloxone (N-allylnoroxymorphone), che è usato in casi di emergenza di overdose da oppioidi. Altri antagonisti degli oppioidi correlati al Naltrexone includono 6β-naltrexol (6β-idrossinaltrexone), Samidorphan (3-carbossamido-4-idrossinaltrexone), β-funaltrexamine (Naltrexone Fumarato Metil Estere), Nalodeina (N-allylnorcodeine), Nalorphine (N-allylnormorphine), e Nalbuphine (N-cyclobutylmethyl-14-hydroxydihydronormorphine).
- Farmacocinetica del Naltrexone:
L’assorbimento del Naltrexone con la somministrazione orale è rapido e quasi completo (96%).[36] La biodisponibilità del Naltrexone con la somministrazione orale è dal 5 al 60% a causa di un esteso metabolismo di primo passaggio.[37][38] Le concentrazioni di picco del naltrexone sono da 19 a 44 μg/L dopo una singola dose orale di 100 mg e il tempo per le concentrazioni di picco del naltrexone e del 6β-naltrexol (metabolita) è entro 1 ora. [37][38][36] Aumenti lineari nelle concentrazioni circolanti di naltrexone e 6β-naltrexolo in un intervallo di dosi orali da 50 a 200 mg.[37] Il naltrexone non sembra essere accumulato con la somministrazione orale ripetuta una volta al giorno e non vi è alcun cambiamento nel tempo di picco delle concentrazioni con la somministrazione ripetuta.[37]

Il legame alle proteine plasmatiche del naltrexone è di circa il 20% in un intervallo di concentrazione di naltrexone da 0,1 a 500 μg/L.[37][36] Il suo volume apparente di distribuzione a 100 mg per via orale è di 16,1 L/kg dopo una singola dose e 14,2 L/kg con dosi ripetute.[37]
Il naltrexone viene metabolizzato nel fegato principalmente dalle diidrodiol deidrogenasi in 6β-naltrexolo (6β-idrossinaltrexone).[37][38] I livelli di 6β-naltrexolo sono da 10 a 30 volte superiori a quelli del naltrexone con la somministrazione orale a causa del vasto metabolismo di primo passaggio. [39] Al contrario, l’esposizione al 6β-naltrexolo è solo circa 2 volte superiore a quella del naltrexone con l’iniezione intramuscolare di naltrexone in microsfere (nome commerciale Vivitrol).[40] Il 6β-Naltrexolo è un antagonista dei recettori degli oppioidi in modo simile al naltrexone e mostra un profilo di legame comparabile ai recettori degli oppioidi. [41] Tuttavia, il 6β-naltrexolo è selettivo a livello periferico e attraversa il cervello molto meno facilmente del naltrexone.[41] In ogni caso, il 6β-naltrexolo mostra ancora una certa attività centrale e può contribuire significativamente alle azioni centrali del naltrexone orale. [41][37] Altri metaboliti del naltrexone includono il 2-idrossi-3-metossi-6β-naltrexolo e il 2-idrossi-3-metossinaltrexone.[37] Dopo la loro formazione, i metaboliti del naltrexone sono ulteriormente metabolizzati per coniugazione con l’acido glucuronico per formare glucuronidi.[37] Il Naltrexone non è metabolizzato dal sistema del citocromo P450 e ha un basso potenziale di interazioni farmacologiche.[42]
L’eliminazione del naltrexone è biesponenziale e rapida nelle prime 24 ore seguita da un terzo declino estremamente lento dopo 24 ore.[37] Le emivite di eliminazione rapida del naltrexone e del suo metabolita 6β-naltrexolo sono rispettivamente di circa 4 ore e 13 ore.[43] Nelle compresse orali Contrave, che contengono anche bupropione e sono descritte come a rilascio prolungato, l’emivita del naltrexone è di 5 ore. [44] L’emivita di eliminazione lenta in fase terminale del naltrexone è di circa 96 ore.[45] Come microsfere di Naltrexone per iniezione intramuscolare (Vivitrol), le emivite di eliminazione del naltrexone e del 6β-naltrexolo sono entrambe di 5-10 giorni.[46] Mentre il naltrexone orale viene somministrato quotidianamente, il naltrexone in microsfere per iniezione intramuscolare è adatto alla somministrazione una volta ogni 4 settimane o una volta al mese.[46]


Il Naltrexone e i suoi metaboliti sono escreti nelle urine.[36]
- Farmacodinamica del Naltrexone:
Il Naltrexone e il suo metabolita attivo 6β-naltrexolo sono antagonisti competitivi dei recettori oppioidi.[47][48] Il naltrexone è specificamente un antagonista preferenzialmente del recettore μ-opioide (MOR), in misura minore del recettore κ-opioide (KOR), e in misura molto minore del recettore δ-opioide (DOR). [47] Tuttavia, il naltrexone non è in realtà un antagonista silenzioso di questi recettori, ma agisce invece come un debole agonista parziale, con valori di Emax dal 14 al 29% al MOR, dal 16 al 39% al KOR, e dal 14 al 25% al DOR in diversi studi.[48][49][50] In accordo con il suo agonismo parziale, sebbene il naltrexone sia descritto come un puro antagonista dei recettori oppioidi, ha mostrato alcune prove di deboli effetti oppioidi in studi clinici e preclinici.[37]

Da solo, il naltrexone agisce come un antagonista o un debole agonista parziale dei recettori oppioidi.[48] In combinazione con agonisti del MOR come la morfina, tuttavia, il naltrexone sembra diventare un agonista inverso del MOR.[48] Al contrario, il naltrexone rimane un antagonista neutro (o un debole agonista parziale) del KOR e DOR. [In contrasto con il naltrexone, il 6β-naltrexolo è puramente un antagonista neutro dei recettori oppioidi.[51] L’agonismo inverso MOR del naltrexone quando è co-presente con agonisti MOR può in parte spiegare la sua capacità di far precipitare l’astinenza in individui dipendenti da oppioidi.[51][48] Ciò può essere dovuto alla soppressione della segnalazione MOR basale attraverso l’agonismo inverso.[51][48]
L’occupazione dei recettori degli oppioidi nel cervello da parte del Naltrexone è stata studiata utilizzando la tomografia a emissione di positroni (PET).[52][53] Il Naltrexone alla dose di 50 mg/giorno è risultato occupare circa il 90-95% dei MOR del cervello e il 20-35% dei DOR del cervello. [52] Il Naltrexone alla dose di 100 mg/giorno è stato osservato raggiungere l’87% e il 92% di occupazione cerebrale del KOR in diversi studi.[54][53][55] Per simulazione, una dose più bassa di Naltrexone di 25 mg/giorno potrebbe essere prevista per raggiungere circa il 60% di occupazione cerebrale del KOR ma ancora vicino al 90% di occupazione del MOR. [53] In uno studio sulla durata del blocco del MOR con il Naltrexone, il farmaco con una singola dose da 50mg ha mostrato un blocco del 91% del legame cerebrale al [11C]carfentanil (un ligando selettivo del MOR) a 48 ore (2 giorni), un blocco dell’80% a 72 ore (3 giorni), un blocco del 46% a 120 ore (5 giorni) e un blocco del 30% a 168 ore (7 giorni). [8][9] Il tempo di dimezzamento del blocco MOR del cervello da parte del Naltrexone in questo studio era da 72 a 108 ore (3. 0 a 4,5 giorni).[56][45] Sulla base di questi risultati, dosi di Naltrexone anche inferiori a 50mg/giorno dovrebbero raggiungere un’occupazione dei MOR cerebrali praticamente completa.[8][9] Il blocco dei MOR cerebrali con Naltrexone è molto più duraturo rispetto ad altri antagonisti oppioidi come il Naloxone (tempo di dimezzamento di ~ 1,7 ore per via intranasale) o il Nalmefene (tempo di dimezzamento di ~ 29 ore).[56][57][58]

L’emivita di occupazione dei MOR cerebrali e la durata dell’effetto clinico del Naltrexone sono molto più lunghe di quanto suggerito dalla sua emivita di eliminazione plasmatica.[56][59][45] Una singola dose orale di 50mg di Naltrexone ha mostrato di bloccare i MOR cerebrali e gli effetti oppioidi per almeno 48-72 ore. [59][45][60] L’emivita del blocco dei MOR cerebrali da parte del Naltrexone (72-108 ore) è molto più lunga della componente di clearance plasmatica rapida del Naltrexone e del 6β-naltrexolo (~4-12 ore), ma è stato riportato che corrisponde bene alla fase terminale più lunga della clearance plasmatica del Naltrexone (96 ore). [56][45][61] Come possibilità alternativa, la prolungata occupazione cerebrale dei MOR da parte degli antagonisti oppioidi come il Naltrexone e il Nalmefene può essere dovuta alla lenta dissociazione dai MOR conseguente alla loro affinità MOR molto alta (<1,0 nM).[58][62]
Il Naltrexone blocca gli effetti degli agonisti MOR come la Morfina, l’Eroina e l’Idromorfone nell’uomo attraverso il suo antagonismo MOR.[37][42] Dopo una singola dose di 100mg di Naltrexone, gli effetti soggettivi e oggettivi dell’Eroina sono stati bloccati del 90% a 24 ore, con un blocco che è diminuito fino a 72 ore.[1] Allo stesso modo, da 20 a 200mg di Naltrexone hanno antagonizzato in modo dose-dipendente gli effetti dell’Eroina fino a 72 ore. [37] Il Naltrexone blocca anche gli effetti degli agonisti KOR come la Salvinorina A, la Pentazocina e il Butorfanolo negli esseri umani attraverso il suo antagonismo KOR.[63][64][65][66] Oltre agli oppioidi, il Naltrexone è stato osservato bloccare o ridurre gli effetti gratificanti e altri effetti di altre droghe euforizzanti tra cui l’alcol,[67] la Nicotina,[68] e le Anfetamine.[69]
Come visto in precedenza, i recettori degli oppioidi sono coinvolti nella regolazione neuroendocrina.[37] Gli agonisti MOR producono aumenti dei livelli di Prolattina e diminuzioni dei livelli di Ormone Luteinizzante (LH) e Testosterone. [37] Dosi di Naltrexone da 25 a 150mg/giorno sono state osservate produrre aumenti significativi nei livelli di β-endorfina, Cortisolo e LH, cambiamenti equivoci nei livelli di Prolattina e Testosterone, e nessun cambiamento significativo nei livelli di ormone Adrenocorticotropico (ACTH) o Ormone Follicolo-Stimolante (FSH). [37] Il Naltrexone influenza l’Asse Ipotalamo-Ipofisi-Surrene (Asse HPT) probabilmente attraverso l’interferenza con la segnalazione del recettore degli oppioidi da parte delle endorfine.
Si pensa che il blocco dei MOR sia il meccanismo d’azione del Naltrexone nella gestione della dipendenza da oppioidi – blocca o attenua reversibilmente gli effetti degli oppioidi. Si pensa anche che sia coinvolto nell’efficacia del Naltrexone nella dipendenza da alcol riducendo gli effetti euforici di quest’ultimo. Il ruolo della modulazione del KOR da parte del Naltrexone nella sua efficacia per la dipendenza da alcol non è chiaro, ma questa azione potrebbe anche essere coinvolta in base alla teoria e agli studi sugli animali.[70][71]
Oltre ai recettori degli oppioidi, il Naltrexone si lega e agisce come antagonista del recettore del fattore di crescita degli oppioidi (OGFR) e del recettore toll-like 4 (TLR4) e interagisce con siti di legame ad alta e bassa affinità nella filamina-A (FLNA).[72][73][74][75] Si dice che dosi molto basse di Naltrexone (<0. 001-1 mg/die) interagiscono con FLNA, basse dosi (da 1 a 5 mg/die) producono antagonismo di TLR4, e dosi cliniche standard (da 50 a 100 mg/die) esercitano antagonismo del recettore degli oppioidi e OGFR.[72][74] Le interazioni del Naltrexone con FLNA e TLR4 sono ritenute coinvolte negli effetti terapeutici del Naltrexone a basse dosi.[72]
- Farmacogenetica del Naltrexone:
Prove provvisorie suggeriscono che la storia familiare e la presenza del polimorfismo Asn40Asp predice l’efficacia del Naltrexone.[76][77]
Effetti collaterali del Naltrexone:
Gli effetti collaterali più comuni riportati con il Naltrexone sono disturbi gastrointestinali come diarrea e crampi addominali.[36] Questi effetti avversi sono analoghi ai sintomi dell’astinenza da oppioidi, poiché il blocco del recettore μ-opioide aumenterà la motilità gastrointestinale.
Gli effetti collaterali del naltrexone per incidenza sono i seguenti:[36]
- Più del 10%: difficoltà a dormire, ansia, nervosismo, dolori addominali/crampi, nausea e/o vomito, poca energia, dolori articolari/muscolari e mal di testa.[36]
- Meno del 10%: perdita di appetito, diarrea, costipazione, sete, aumento di energia, sensazione di depressione, irritabilità, vertigini, eruzione cutanea, eiaculazione ritardata, disfunzione erettile e brividi.[36]
- Una varietà di altri eventi avversi sono stati riportati anche con un’incidenza inferiore all’1%.[36] È stato segnalato che il Naltrexone può causare danni al fegato se somministrato a dosi più elevate di quelle raccomandate.[52] È oggetto di un avvertimento della FDA per questo raro effetto collaterale.
Ipotesi d’uso del Naltrexone in contesto PEDs:
Ora, abbiamo tutte le informazioni necessarie per poter comprendere come un antagonista dei recettori oppioidi possa portare a dei possibili vantaggi durante l’uso di PEDs o, meglio, di alcuni farmaci rientranti in tale categoria.
Riassumendo in breve:
- Sappiamo che la sottoregolazione/soppressione del Asse HPT avviene anche in modo androgeno-dipendente: quindi, anche nel caso di molecole non aromatizzabili e non aventi attività estrogeniche intrinseche o progestiniche;
- Sappiamo che i peptidi oppioidi endogeni agiscono da intermediari dell’attività androgena ipotalamica: attività che vede una maggiore interazione da parte delle Encefaline, Beta-Endorfine e Dinorfine con i recettori oppioidi mu (μ) (MOR OP3 (I));
- Sappiamo che il Naltrexone può bloccare l’attività dei delle Encefaline, Beta-Endorfine e Dinorfine con i recettori oppioidi mu (μ) (MOR OP3 (I));
- Sappiamo che tale blocco recettoriale può tradursi in una mancata (o per lo meno marcata e significativa) sottoregolazione/soppressione androgeno-dipendente del Asse HPT.
Se a ciò aggiungiamo che tale potenziale sarebbe al quanto insignificante in un contesto d’uso di AAS aromatizzabili e/o aventi attività estrogenica intrinseca e/o progestinica, anche volendoci abbinare un SERM il gioco non varrebbe la candela, l’unica possibilità d’uso con ritorno “positivo” si verificherebbe senza dubbio con l’uso di soli SARM non steroidei o, tutt’al più, con l’uso di DHT derivati senza la sopramenzionata caratteristica di attività estrogenica intrinseca (quindi niente Oxymetholone).
Per quanto rimanga un idiozia per un atleta di sesso maschile intraprendere protocolli di soli SARM non steroidei o AAS non-aromatizzabili, o progestinici, senza una base per lo meno fisiologica di Testosterone o l’uso di hCG onde garantire almeno un sufficiente “equilibrio” etsrogenico e di DHT, rappresenti il culmine dell’ignoranza e dell’incompetenza (se parliamo di “preparatori”), per i “testofobici” e gli “agofobici” l’uso del Naltrexone potrebbe (e, sottolineo, il condizionale “potrebbe”) garantire una conservazione sufficiente dell’attività del Asse HPT.
Nonostante vi siano scarsi lavori che hanno osservato l’attività cerebrale dei SARM non steroidei, e che uno in particolare abbia affermato che l’Ostarina non passi in modo significativo la barriera ematoencefalica, questa categoria di PEDs è per lo più strutturata su modifiche della molecola antiandrogena Bicalutamide la quale oltrepassa la barriera ematoencefalica.
Sebbene le modifiche strutturali determinino, per ovvie ragioni, una diversa farmacodinamica, il fatto che la sottoregolazione del HPTA, misurata anche a dosi di 3mg/die di Ostarina, sia con tutta probabilità androgeno-dipendente a livello ipotalamico [androgeno dipendente in senso di legame con i AR non nel senso di effetto androgeno genico] mi spinge ad ipotizzare una potenziale utilità del Naltrexone. Senza considerare che SARM non steroidei come LGD 4033, nonostante abbiano caratteristiche strutturali diverse dall’Ostarina o dalla Andarina, causino sottoregolazioni molto più marcate dei SARM non steroidei con struttura base del Bicalutamide.
Conclusioni:
Questo articolo è un esposizione accurata di una ipotesi e nulla più! Per quanto sensata possa essa essere, non abbiamo attualmente documentazione scientifica che dimostri inconfutabilmente che tale pratica sia funzionale e vantaggiosa o tantomeno sicura.
Il Naltrexone è un farmaco con una serie di potenziali effetti avversi (vedi sopra). Alcuni di essi potrebbero, in alcuni soggetti, vanificare i vantaggi sulla sfera sessuale ottenuti da un ipotetica conservazione del attività del HPTA. Da considerare anche la cura della dose somministrata per possibile danno epatico da sovradosaggio.
Gabriel Bellizzi
Riferimenti:
- Moore AR, McQuay HJ. Prevalenza di eventi avversi da oppioidi nel dolore cronico non maligno: revisione sistematica di studi randomizzati di oppioidi orali . Artrite Res Ther 2005; 7 : 1046–1051. [ Articolo gratuito PMC ] [ PubMed ] [ Google Scholar ]2.
- Furlan AD, Sandoval JA, Mailis-Gagnon A, et al. Oppioidi per il dolore cronico non oncologico: una meta-analisi di efficacia ed effetti collaterali . Can Med Assoc J 2006; 174 : 1589–1594. [ Articolo gratuito PMC ] [ PubMed ] [ Google Scholar ]3.
- Raghavan S, Harvey AD, Humble SR. Nuovi effetti collaterali e implicazioni per gli oppioidi per la terapia a lungo termine . Tendenze Anaesth Crit Care 2011; 1 : 18–21. [ Google Scholar ]4.
- Okie S. Un’ondata di oppioidi, una marea crescente di morti . N inglese J Med 2010; 363 ( 21 ): 1981–1983. [ PubMed ] [ Google Scholar ]5.
- Graves RH. Quarant’anni in Cina, Baltimora: Woodward , 1895. [ Google Scholar ]6.
- Cumming HS, Control of DrugAddiction Mainly a Police Problem, The American City Magazine (novembre 1925, fascicolo 126, riquadro 3, sottoserie 1, serie 3.7.
- Katz N, Mazer NA. L’impatto degli oppioidi sul sistema endocrino . Clin J Dolore 2009; 25 ( 2 ): 170–175. [ PubMed ] [ Google Scholar ]8.
- Bruce CA. Relazione sulla produzione del tè e sull’estensione e la produzione delle piantagioni di tè nell’Assam . Calcutta: : Bishop’s College Press, 1839. [ Google Scholar ]9.
- Hiller-Sturmhofel S, Bartke A. Il sistema endocrino: una panoramica . Alcol Health Res World 1998; 22 ( 3 ): 153–164. [ Articolo gratuito PMC ] [ PubMed ] [ Google Scholar ]10.
- Ganong WF. Rassegna di fisiologia medica . USA: Lange/McGraw-Hill, 2005. [ Google Scholar ]11.
- Luo Xiwen tr. Bencao Gangmu: Compendio di Materia Medica . Foreign Languages Press, 2003. Pubblicato per la prima volta nel 157812.
- Van Vugt DA, Baby N, Stewart M, Reid RL. L’effetto stimolante paradossale della morfina sulla secrezione di LH è dose-dipendente e reversibile al naloxone . Neuroendocrinologia 1989; 50 : 109–116. [ PubMed ] [ Google Scholar ]13.
- De la Rosa RE, Hennessey JV. Ipogonadismo e metadone: ipogonadismo ipotalamico dopo l’uso a lungo termine di metadone ad alte dosi . Pratica Endocr 1996; 2 :4–7. [ PubMed ] [ Google Scholar ]14.
- Daniel HW. Deficit di DHEAS durante il consumo di oppioidi prescritti ad azione prolungata: evidenza dell’inibizione indotta da oppioidi della produzione di androgeni surrenali . J Dolore 2006; 7 ( 12 ): 901–907. [ PubMed ] [ Google Scholar ]15.
- Vuong C, Van Uum SHM, O’Dell L, et al. Gli effetti degli oppioidi e degli analoghi degli oppioidi sui sistemi endocrini animali e umani . Endocr Rev 2010; 31 ( 1 ): 98–132. [ Articolo gratuito PMC ] [ PubMed ] [ Google Scholar ]16.
- Van Furth WR, van Emst MG, van Ree JM. Oppioidi e comportamento sessuale dei ratti maschi: coinvolgimento dell’area preottica mediale . Comportamento Neurosci 1995; 109 ; 123–134. [ PubMed ] [ Google Scholar ]17.
- Daniell Harry W. Le linee guida cliniche della Endocrine Society sulla terapia con testosterone negli uomini adulti con sindromi da carenza di androgeni . J Clin Metab endocrinolo 2010; 95 ( 6 ): 2536–2559. [ PubMed ] [ Google Scholar ]18.
- O’Donne AB, Araujo AB, McKinlay JB. La salute degli uomini che invecchiano normalmente: The Massachusetts Male Aging Study (1987–2004) . Scad. Gerontolo 2004; ( 7 ): 975–984. [ PubMed ] [ Google Scholar ]19.
- Katz N. L’impatto degli oppioidi sul sistema endocrino . Round di gestione del dolore 2005; 1 ( 9 ). www.painmanagementrounds.org (visitato il 5 febbraio 2012) [ Google Scholar ]20.
- Faccinetti F, Grasso A, Petraglia F, et al. Ridotta ritmicità circadiana di β-lipotropina, β-endorfina e ACTH nei tossicodipendenti . Acta endocrinolo 1984; 105 ( 2 ): 149–155. [ PubMed ] [ Google Scholar ]21.
- Daniel HW. Endocrinopatia da oppioidi nelle donne che consumano oppioidi ad azione prolungata prescritti per il controllo del dolore non maligno . J Dolore 2008; 9 ( 1 ): 28–36. [ PubMed ] [ Google Scholar ]22.
- Thosani S, Jiminez C. Alterazioni biochimiche dell’asse neuroendocrino indotte da oppioidi . Esperto Rev Endocrinol Metab 2011; 6 ( 5 ): 705–713. [ Google Scholar ]23.
- Abs R, Verhelst J, Maeyaert J, et al. Conseguenze endocrine della somministrazione intratecale a lungo termine di oppioidi . J Clin Endo Metab 2000; 85 ( 6 ): 2215–2222. [ PubMed ] [ Google Scholar ]24.
- Sarret D, Le Berre JP, Zemraoui N. Iponatriemia indotta da Tramadol . Am J Kidney Dis 2008; 52 ( 5 ): 1026. [ PubMed ] [ Google Scholar ]25.
- Lindow SW, Hendricks MS, Nugent FA, et al. La morfina sopprime la risposta all’ossitocina nelle donne che allattano . Gynecol Obstet Invest 1999; 48 ( 1 ): 33–37. [ PubMed ] [ Google Scholar ]26.
- Giugliano D. Morfina, peptidi oppioidi e funzione delle isole pancreatiche . Cura del diabete 1984; 7 : 92–98. [ PubMed ] [ Google Scholar ]27.
- Kapoor D, Goodwin E, Channer KS, Jones TH. La terapia sostitutiva del testosterone migliora la resistenza all’insulina, il controllo glicemico, l’adiposità viscerale e l’ipercolesterolemia negli uomini ipogonadici con diabete di tipo 2 . Eur J Endocrinolo 2006; 154 : 899–906. [ PubMed ] [ Google Scholar ]28. Oppiacei per il dolore persistente; buona pratica . 2010.
- The British Pain Society, Facoltà di Medicina del Dolore del Royal College of Anesthetists, Royal College of General Practitioners e Facoltà di Medicina delle Dipendenze del Royal College of Psychiatrists . Disponibile su: http://www.britishpainsociety.org/book_opioid_main.pdf (visitato il 5 febbraio 2012)
29. Roberts LJ, Finch PM, Pullan PT, et al. Soppressione dell’ormone sessuale da parte degli oppioidi intratecali: uno studio prospettico . Clin J Dolore 2002; 18 : 144–148. [ PubMed ] [ Google Scholar ]
30.Thimineur MA, Kravitz E, Vodapally MS. Trattamento intratecale con oppioidi per il dolore cronico non maligno: uno studio prospettico di 3 anni . Dolore 2004; 109 : 242–249. [ PubMed ] [ Google Scholar ]
31.Rajagopal A, Vasilopoulou-Sellin R, Palmer JL, et al. Ipogonadismo sintomatico nei sopravvissuti maschi al cancro con esposizione cronica agli oppioidi . Cancro 2004; 100 ( 4 ): 851–858. [ PubMed ] [ Google Scholar ]
32. “Naltrexone Monograph for Professionals – Drugs.com”. Drugs.com. American Society of Health-System Pharmacists. Archived from the original on 9 November 2017. Retrieved 9 November 2017.
33. Aboujaoude E, Salame WO (August 2016). “Naltrexone: A Pan-Addiction Treatment?”. CNS Drugs. 30 (8): 719–33.
34. Sadock BJ, Sadock VA, Sussman N (2012). Kaplan & Sadock’s Pocket Handbook of Psychiatric Drug Treatment. Lippincott Williams & Wilkins. p. 265. ISBN 9781451154467. Archived from the original on 2017-12-05.
35. “Naltrexone/bupropion for obesity”. Drug and Therapeutics Bulletin. 55 (11): 126–129. November 2017.
36.https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/018932s017lbl.pdf
37. Gonzalez JP, Brogden RN (March 1988). “Naltrexone. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic efficacy in the management of opioid dependence”. Drugs. 35 (3): 192–213.
38. Lee MW, Fujioka K (August 2009). “Naltrexone for the treatment of obesity: review and update”. Expert Opin Pharmacother. 10 (11): 1841–5.
39. Davis MP, Glare PA, Hardy J (28 May 2009). Opioids in Cancer Pain. Oxford University Press. pp. 41–.
40. Dunbar JL, Turncliff RZ, Dong Q, Silverman BL, Ehrich EW, Lasseter KC (March 2006). “Single- and multiple-dose pharmacokinetics of long-acting injectable naltrexone”. Alcohol Clin Exp Res. 30 (3): 480–90.
41.Hipkin, R. William; Dolle, Roland E. (2010). “Opioid Receptor Antagonists for Gastrointestinal Dysfunction”. Annual Reports in Medicinal Chemistry. Vol. 45. Elsevier. pp. 142–155.
42.Sevarino, Kevin A.; Kosten, Thomas R. (2009). “Naltrexone for Initiation and Maintenance of Opiate Abstinence”. Opiate Receptors and Antagonists. Humana Press. pp. 227–245.
43.http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/018932s017lbl.pdf
44.https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/200063s020lbl.pdf
45.Lee MC, Wagner HN, Tanada S, Frost JJ, Bice AN, Dannals RF (July 1988). “Duration of occupancy of opiate receptors by naltrexone”. J Nucl Med. 29 (7): 1207–11.
46.https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/021897s015lbl.pdf
47.Niciu MJ, Arias AJ (October 2013). “Targeted opioid receptor antagonists in the treatment of alcohol use disorders”. CNS Drugs. 27 (10): 777–87.
48.Wang D, Sun X, Sadee W (May 2007). “Different effects of opioid antagonists on mu-, delta-, and kappa-opioid receptors with and without agonist pretreatment”. J Pharmacol Exp Ther. 321 (2): 544–52.
49.Wentland MP, Lou R, Lu Q, Bu Y, Denhardt C, Jin J, Ganorkar R, VanAlstine MA, Guo C, Cohen DJ, Bidlack JM (April 2009). “Syntheses of novel high affinity ligands for opioid receptors”. Bioorg Med Chem Lett. 19 (8): 2289–94.
50.Linda P. Dwoskin, ed. (29 January 2014). Emerging Targets and Therapeutics in the Treatment of Psychostimulant Abuse. Academic Press. pp. 398–.
51.Sadée W, Wang D, Bilsky EJ (February 2005). “Basal opioid receptor activity, neutral antagonists, and therapeutic opportunities”. Life Sci. 76 (13): 1427–37.
52.Ray LA, Chin PF, Miotto K (March 2010). “Naltrexone for the treatment of alcoholism: clinical findings, mechanisms of action, and pharmacogenetics”. CNS Neurol Disord Drug Targets. 9 (1): 13–22.
53.de Laat B, Nabulsi N, Huang Y, O’Malley SS, Froehlich JC, Morris ED, Krishnan-Sarin S (June 2020). “Occupancy of the kappa opioid receptor by naltrexone predicts reduction in drinking and craving”. Mol Psychiatry. 26 (9): 5053–5060.
54.Placzek MS (August 2021). “Imaging Kappa Opioid Receptors in the Living Brain with Positron Emission Tomography”. Handb Exp Pharmacol. Handbook of Experimental Pharmacology. 271: 547–577.
55.Vijay, AIshwarya; Morris, Evan; Goldberg, Alissa; Petrulli, Joseph; Liu, Heather; Huang, Yiyun; Krishnan-Sarin, Suchitra (1 April 2017). “Naltrexone occupancy at kappa opioid receptors investigated in alcoholics by PET occupancy at kappa opioid receptors investigated in alcoholics by PET”. Journal of Nuclear Medicine. 58 (Supplement 1): 1297. ISSN 0161-5505. Retrieved 29 October 2021.
56.Colasanti, Alessandro; Lingford-Hughes, Anne; Nutt, David (2013). “Opioids Neuroimaging”. In Miller, Peter M. (ed.). Biological Research on Addiction. Comprehensive Addictive Behaviors and Disorders. Vol. 2. Elsevier. pp. 675–687.
57.van Waarde, Aren; Absalom, Anthony R.; Visser, Anniek K. D.; Dierckx, Rudi A. J. O. (30 September 2020). “Positron Emission Tomography (PET) Imaging of Opioid Receptors”. PET and SPECT of Neurobiological Systems. Springer International Publishing. pp. 749–807.
58.Soyka M, Rösner S (November 2010). “Nalmefene for treatment of alcohol dependence”. Expert Opin Investig Drugs. 19 (11): 1451–9.
59.Mannelli P, Peindl KS, Wu LT (June 2011). “Pharmacological enhancement of naltrexone treatment for opioid dependence: a review”. Subst Abuse Rehabil. 2011 (2): 113–123.
60. Narcotic Antagonists: Naltrexone Pharmacochemistry and Sustained-release Preparations. Department of Health and Human Services, Public Health Service, Alcohol, Drug Abuse, and Mental Health Administration, National Institute on Drug Abuse, Division of Research. 1981. pp. 148–.
61.Miotto K, McCann M, Basch J, Rawson R, Ling W (2002). “Naltrexone and dysphoria: fact or myth?”. Am J Addict. 11 (2): 151–60.
62.Ingman K, Hagelberg N, Aalto S, Någren K, Juhakoski A, Karhuvaara S, Kallio A, Oikonen V, Hietala J, Scheinin H (December 2005). “Prolonged central mu-opioid receptor occupancy after single and repeated nalmefene dosing”. Neuropsychopharmacology. 30 (12): 2245–53.
63.Maqueda AE, Valle M, Addy PH, Antonijoan RM, Puntes M, Coimbra J, Ballester MR, Garrido M, González M, Claramunt J, Barker S, Lomnicka I, Waguespack M, Johnson MW, Griffiths RR, Riba J (July 2016). “Naltrexone but Not Ketanserin Antagonizes the Subjective, Cardiovascular, and Neuroendocrine Effects of Salvinorin-A in Humans”. Int J Neuropsychopharmacol. 19 (7): pyw016.
64.Walsh SL, Chausmer AE, Strain EC, Bigelow GE (January 2008). “Evaluation of the mu and kappa opioid actions of butorphanol in humans through differential naltrexone blockade”. Psychopharmacology (Berl). 196 (1): 143–55.
65.Preston KL, Bigelow GE (February 1993). “Differential naltrexone antagonism of hydromorphone and pentazocine effects in human volunteers”. J Pharmacol Exp Ther. 264 (2): 813–23.
66.Clark SD, Abi-Dargham A (October 2019). “The Role of Dynorphin and the Kappa Opioid Receptor in the Symptomatology of Schizophrenia: A Review of the Evidence”. Biol Psychiatry. 86 (7): 502–511.
67.Unterwald EM (September 2008). “Naltrexone in the treatment of alcohol dependence”. J Addict Med. 2 (3): 121–7.
68.Modesto-Lowe V, Van Kirk J (August 2002). “Clinical uses of naltrexone: a review of the evidence”. Exp Clin Psychopharmacol. 10 (3): 213–27.
69.Lam L, Anand S, Li X, Tse ML, Zhao JX, Chan EW (April 2019). “Efficacy and safety of naltrexone for amfetamine and methamfetamine use disorder: a systematic review of randomized controlled trials”. Clin Toxicol (Phila). 57 (4): 225–233.
70.Soyka M, Friede M, Schnitker J (March 2016). “Comparing Nalmefene and Naltrexone in Alcohol Dependence: Are there any Differences? Results from an Indirect Meta-Analysis”. Pharmacopsychiatry. 49 (2): 66–75.
71.Koob GF, Volkow ND (August 2016). “Neurobiology of addiction: a neurocircuitry analysis”. The Lancet. Psychiatry. 3 (8): 760–73.
72.Toljan K, Vrooman B (September 2018). “Low-Dose Naltrexone (LDN)-Review of Therapeutic Utilization”. Med Sci (Basel). 6 (4): 82.
73.Bachtell R, Hutchinson MR, Wang X, Rice KC, Maier SF, Watkins LR (2015). “Targeting the Toll of Drug Abuse: The Translational Potential of Toll-Like Receptor 4”. CNS Neurol Disord Drug Targets. 14 (6): 692–9.
74. Lee B, Elston DM (June 2019). “The uses of naltrexone in dermatologic conditions”. J Am Acad Dermatol. 80 (6): 1746–1752.
75.Zagon IS, Verderame MF, McLaughlin PJ (February 2002). “The biology of the opioid growth factor receptor (OGFr)”. Brain Res Brain Res Rev. 38 (3): 351–76.
76.Ray LA, Barr CS, Blendy JA, Oslin D, Goldman D, Anton RF (March 2012). “The role of the Asn40Asp polymorphism of the mu opioid receptor gene (OPRM1) on alcoholism etiology and treatment: a critical review”. Alcoholism, Clinical and Experimental Research. 36 (3): 385–94.
77.Garbutt JC, Greenblatt AM, West SL, Morgan LC, Kampov-Polevoy A, Jordan HS, Bobashev GV (August 2014). “Clinical and biological moderators of response to naltrexone in alcohol dependence: a systematic review of the evidence”. Addiction. 109 (8): 1274–84.
