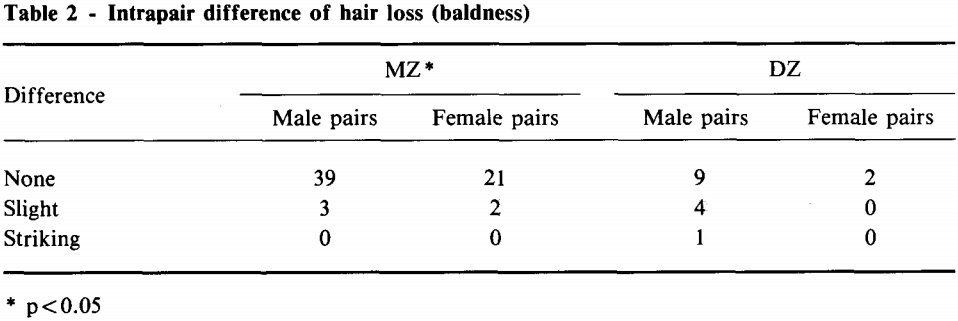Con l’avanzare dell’età, molti uomini sviluppano una perdita di capelli con un andamento caratteristico. Può iniziare dal cuoio capelluto frontale, spostandosi verso l’alto ai lati (“bitemporale”), oppure dalla parte posteriore/superiore della testa in un’area chiamata corona (anche “vertice”). Questa perdita di capelli continua progressivamente fino al cuoio capelluto medio. I capelli ai lati della testa e sotto la corona nella parte posteriore sono risparmiati (la “regione occipitale”). Questa forma di perdita di capelli è nota come calvizie maschile o alopecia androgenetica.
L’alopecia androgenetica ha la parola “androgeno” al suo interno, e per una buona ragione. Un articolo di rilievo pubblicato nel 1942, intitolato “Male hormone stimulation is prerequisite and an incitant in common baldness” (La stimolazione degli ormoni maschili è un prerequisito e un incitante nella calvizie comune) [1], affronta in modo eloquente il legame tra androgeni e alopecia androgenetica. Come avrete capito dal nome dell’articolo, la condizione era ancora chiamata “calvizie comune”. L’autore fece alcune osservazioni interessanti. In primo luogo, l’autore osservò che gli eunuchi e gli uomini castrati in età prepuberale non sviluppavano l’alopecia. Scrive: “Anche la recessione della linea di capelli sulle tempie e sulla fronte, che si osserva nella maggior parte degli uomini normali, non è comparsa”. In secondo luogo, quando è stato somministrato il Testosterone, alcuni uomini hanno sviluppato una perdita di capelli. È interessante notare che quando due di questi uomini hanno interrotto la terapia con Testosterone per un anno, la calvizie ha smesso di progredire. Tuttavia, quando sono stati rimessi in terapia con il Testosterone, la calvizie ha ripreso a progredire. In terzo luogo, ha osservato che gli uomini che hanno sviluppato l’alopecia dopo il trattamento androgenico appartenevano a famiglie in cui i membri maschi adulti normali tendono a essere calvi. Allo stesso modo, quelli che non hanno sviluppato l’alopecia appartenevano a famiglie senza tendenze pronunciate alla calvizie tra gli uomini normali. Si sospettò quindi una predisposizione genetica all’alopecia.
Un articolo pubblicato sulla rivista “Science” nel 1974 ha dimostrato il ruolo centrale del Diidrotestosterone (DHT) nel mediare l’effetto del Testosterone su questa condizione [2]. Il DHT è un metabolita del Testosterone sintetizzato per interazione un membro della famiglia degli enzimi 5α-reduttasi, la 5α-reduttasi di tipo 2. Il DHT è un androgeno più potente del Testosterone e quindi l’effetto del Testosterone è amplificato nei tessuti che esprimono questi enzimi. È stato riscontrato che i soggetti con un deficit di 5α-reduttasi non sviluppano la recessione dell’attaccatura dei capelli. In effetti, in un successivo articolo di review si legge che l’alopecia androgenetica non è mai stata osservata in persone con questa condizione [3].
L’importante ruolo della genetica nello sviluppo dell’alopecia androgenetica è sottolineato da uno studio sui gemelli che esamina l’invecchiamento fisico e la longevità [4]. Hanno partecipato 76 coppie di gemelli identici (gemelli monozigoti), di cui 65 (42 maschi, 23 femmine) sono stati inclusi nell’analisi della perdita di capelli. Un campione più piccolo di 21 coppie di gemelli non identici (gemelli dizigoti) ha partecipato all’analisi della perdita di capelli, di cui 16 sono state incluse nell’analisi (14 maschi, 2 femmine). I ricercatori hanno quindi classificato la differenza di perdita di capelli (calvizie) tra le coppie come “nessuna”, “lieve” o “notevole”. I risultati sono stati i seguenti:
Abbreviazioni: MZ, monozigote; DZ, dizigote.
Come si può vedere nella tabella, la differenza era praticamente inesistente in quasi tutte le coppie monozigoti, mentre lievi differenze potevano essere osservate più frequentemente nei gemelli dizigoti, e persino una differenza eclatante in una coppia. Per esprimere questo dato in numeri, gli autori hanno calcolato il tasso di concordanza intracoppia. Si tratta di un termine elegante per indicare la percentuale di identità di un tratto tra i gemelli. Il tasso di concordanza intracoppia sulla perdita di capelli è stato del 92,3% nelle coppie monozigoti e del 68,7% nelle coppie dizigoti. Poiché le coppie monozigoti hanno geni praticamente identici, mentre le coppie dizigoti condividono solo il 50%, ciò implica una notevole componente genetica nello sviluppo della caduta dei capelli. Diciamo che il termine androgenetico in alopecia androgenetica è giustificato.
Meccanismo d’azione della caduta dei capelli Androgeno-correlata:
Per rispondere a questa domanda, dobbiamo vedere come funziona la crescita dei capelli. I capelli crescono in cicli: “cicli del follicolo pilifero” [5, 6]. Questi cicli possono essere suddivisi in tre fasi o stadi:
Crescita (fase anagen)
Involuzione/regressione (fase catagen)
Riposo (fase telogen)
La fase anagen determina la lunghezza di una ciocca di capelli. Durante questa fase, cresce una nuova ciocca di capelli. La crescita avviene grazie a un’ampia proliferazione delle cosiddette cellule della matrice del capello. Si tratta di un gruppo di cellule che si trovano proprio sopra la papilla dermica che si trova alla base del follicolo pilifero. La papilla dermica ha un ricco apporto di sangue che fornisce le sostanze nutritive necessarie per questo processo di ampia proliferazione cellulare. In altre parole, alla base del follicolo pilifero c’è una popolazione di cellule che si divide continuamente, aggiungendosi alla ciocca di capelli in crescita e spingendola verso l’alto. All’inizio della fase anagen, spinge fuori la vecchia ciocca di capelli (se ancora presente). La fase anagen completa dura di solito alcuni anni per i capelli sulla sommità del capo.
Alla fase anagen segue la fase catagen. Nella parte inferiore del follicolo pilifero molte cellule muoiono per apoptosi e la ciocca si separa dalle cellule della matrice pilifera. La parte inferiore della ciocca di capelli forma una struttura arrotondata chiamata “clava del capello”. Da questo momento in poi il capello non può più crescere e aspetta solo di cadere. O con un po’ di forza, o durante la successiva fase anagen, quando una nuova ciocca di capelli spingerà fuori quella vecchia. Questa fase dura circa due settimane.
Infine, il follicolo entra nella fase telogen o di riposo. A questo punto, non succede sostanzialmente nulla fino a quando la fase anagen non ricomincia. Normalmente questa fase dura circa 3 mesi.
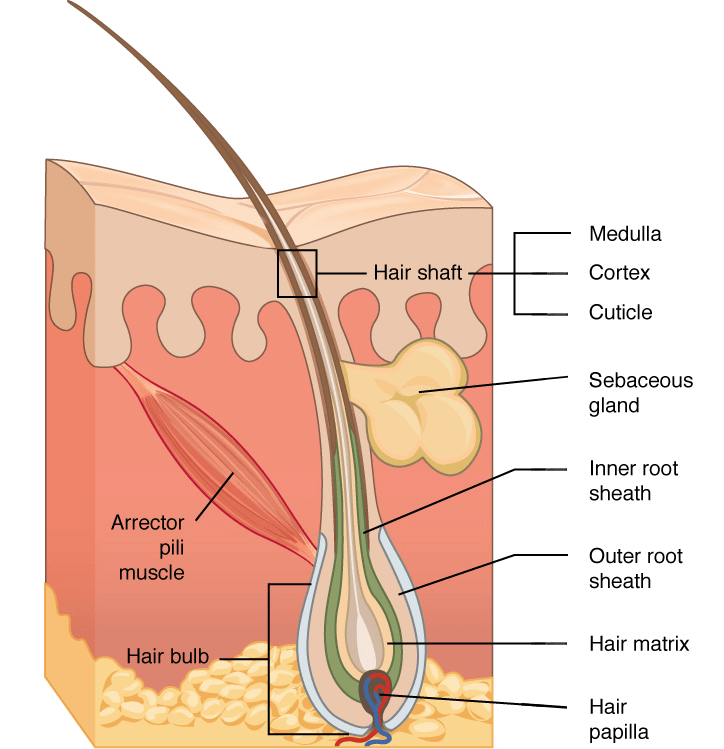
Un’immagine (vedi sopra) rende probabilmente più facile seguire questo insieme di parole. Ripercorriamola brevemente. In basso si vede la papilla pilifera, o dermica. Essa ha un ricco apporto di sangue che le consente di coprire il fabbisogno di nutrienti per lo sviluppo del pelo, che avviene continuamente durante la fase anagen da parte della matrice del pelo. Una popolazione di cellule che si trova proprio sopra di essa. Esse producono le cellule della ciocca di capelli, che accumuleranno molta cheratina dura e alla fine moriranno, e le cellule che circondano la ciocca di capelli (la guaina radicolare interna ed esterna). Inoltre, in alto a destra è visibile la ghiandola sebacea che aggiunge il sebo, che si muoverà fino alla superficie della pelle. A sinistra si vede un piccolo muscolo che può tirare un pelo in alto (cosa che accade con la “pelle d’oca”). L’area in cui questo muscolo si attacca è chiamata rigonfiamento. Durante la fase catagenica, alla base del follicolo pilifero, proprio sotto il rigonfiamento, si verifica un processo chiamato “involuzione”, durante il quale un gruppo di cellule di sostegno muore (apoptosi) e la ciocca di capelli si stacca dalla matrice pilifera.
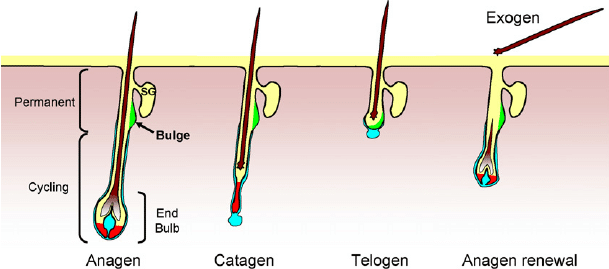
Ecco un’altra immagine che descrive approssimativamente queste fasi:
Ora che abbiamo affrontato tutte queste nozioni di base, qual è l’effetto degli AAS su questo aspetto? Senza parlare delle vie di segnalazione (e sono tante): diminuiscono la lunghezza della fase anagen e aumentano la lunghezza della fase telogen in follicoli piliferi selezionati [7]. A ogni ciclo successivo del capello, la fase anagen continua a diminuire in lunghezza e la fase telogen continua ad aumentare in lunghezza. Di conseguenza, in qualsiasi momento, ci saranno più follicoli piliferi in fase telogen, con capelli facilmente eliminabili, e meno follicoli piliferi in fase anagen. Pertanto, in quest’area le ciocche di capelli che fuoriescono dal cuoio capelluto sono meno numerose. A un certo punto, la fase anagen può diventare così breve che una nuova ciocca di capelli raggiunge a malapena la superficie della pelle.
Inoltre, gli androgeni inducono un fenomeno chiamato miniaturizzazione [8]. Il follicolo pilifero diventa progressivamente più piccolo, così come il fusto del capello e la ciocca che ne deriva. I capelli terminali (che sono i normali capelli sulla sommità del capo) si trasformano in capelli vellutati. I capelli vellutati sono capelli molto corti, morbidi e privi di pigmento.
Le opzioni terapeutiche mirano ad arrestare, o preferibilmente a invertire, questa progressione di miniaturizzazione e interruzione del ciclo del follicolo pilifero. Ad oggi, solo due farmaci per l’alopecia androgenetica sono stati approvati dalla Food and Drug Administration (FDA). Il primo farmaco approvato è stato il Minoxidil topico, nel 1988. Questo farmaco (o almeno la sua versione orale) non è stato sviluppato specificamente per questa indicazione. La fortuna è stata semplicemente quella di notare che molti pazienti ipertesi sviluppavano ipertricosi (crescita anormale dei capelli) e che molti mostravano un’inversione della calvizie [9].
Il secondo farmaco approvato colpisce gli enzimi (5α-reduttasi) responsabili della conversione del Testosterone nel più potente androgeno DHT. Si tratta di un farmaco chiamato Finasteride, attualmente approvato solo per uso orale. È stato approvato per il trattamento dell’alopecia androgenetica nel 1997.
Struttura molecolare del modulatore della via Wnt SM04554
Ora starete pensando: il più recente farmaco approvato per l’alopecia androgenetica è stato approvato nel 1997? Già. Da allora sono state acquisite molte conoscenze sul ciclo del follicolo pilifero e sullo sviluppo dell’alopecia androgenetica, ma nessun nuovo farmaco è stato immesso sul mercato. Eppure. Tuttavia, le aziende farmaceutiche hanno in cantiere alcune opzioni terapeutiche. Alcune meno innovative (Finasteride topica) di altre (ad esempio SM04554, un modulatore della via Wnt). Con un po’ di fortuna vedremo alcuni di questi nuovi farmaci ottenere l’approvazione della FDA nel prossimo futuro.
Nel prossimo articolo tratterò in modo più dettagliato le due modalità di trattamento approvate dalla FDA, oltre ad altre modalità di trattamento in via di definizione.
Continua…
Riferimenti:
J. B. Hamilton. Male hormone stimulation is prerequisite and an incitant in common baldness. American Journal of Anatomy, 71(3):451–480, 1942.
Imperato-McGinley, Julianne, et al. “Steroid 5α-reductase deficiency in man: an inherited form of male pseudohermaphroditism.” Science 186.4170 (1974): 1213-1215.
Imperato-McGinley, Jullianne, and Y-S. Zhu. “Androgens and male physiology the syndrome of 5α-reductase-2 deficiency.” Molecular and cellular endocrinology 198.1-2 (2002): 51-59.
Hayakawa, K., et al. “Intrapair differences of physical aging and longevity in identical twins.” Acta geneticae medicae et gemellologiae: twin research 41.2-3 (1992): 177-185.
Paus, Ralf, and George Cotsarelis. “The biology of hair follicles.” New England journal of medicine 341.7 (1999): 491-497.
Alonso, Laura, and Elaine Fuchs. “The hair cycle.” Journal of cell science 119.3 (2006): 391-393.
Lolli, Francesca, et al. “Androgenetic alopecia: a review.” Endocrine 57.1 (2017): 9-17.
Whiting, David A. “Possible mechanisms of miniaturization during androgenetic alopecia or pattern hair loss.” Journal of the American Academy of Dermatology 45.3 (2001): S81-S86.
Zappacosta, Anthony R. “Reversal of baldness in patient receiving minoxidil for hypertension.” The New England journal of medicine 303.25 (1980): 1480-1481.
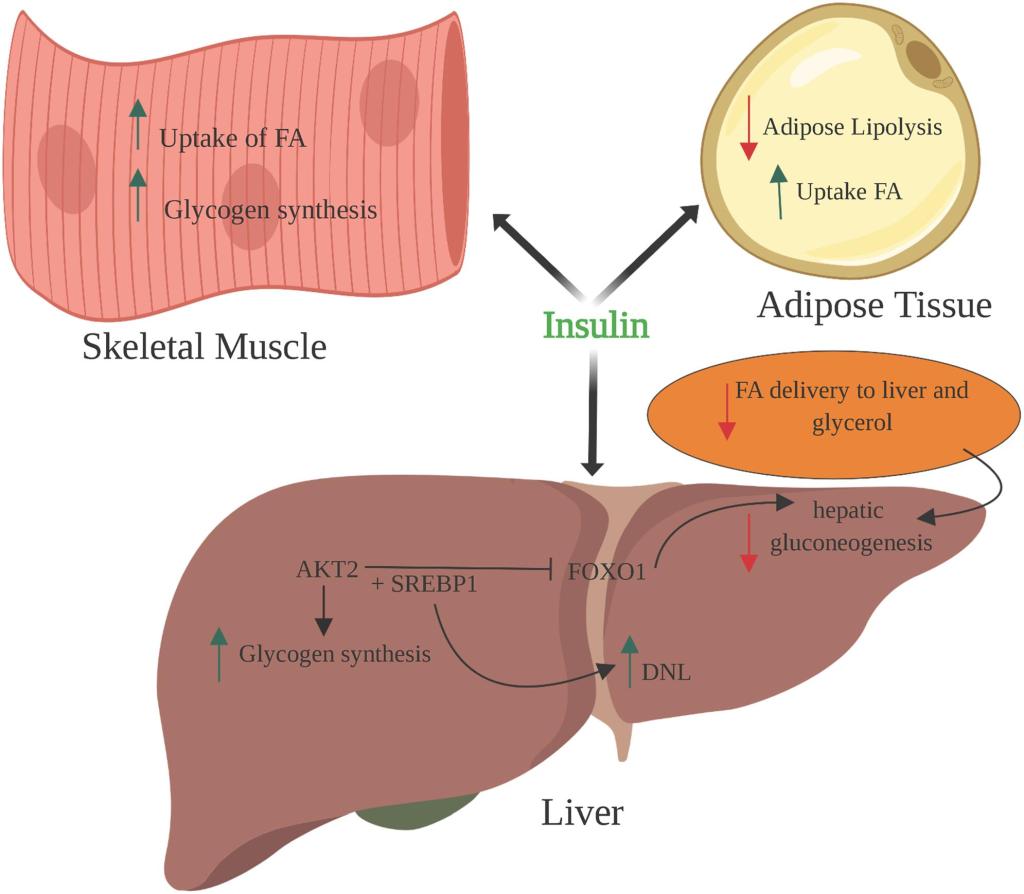
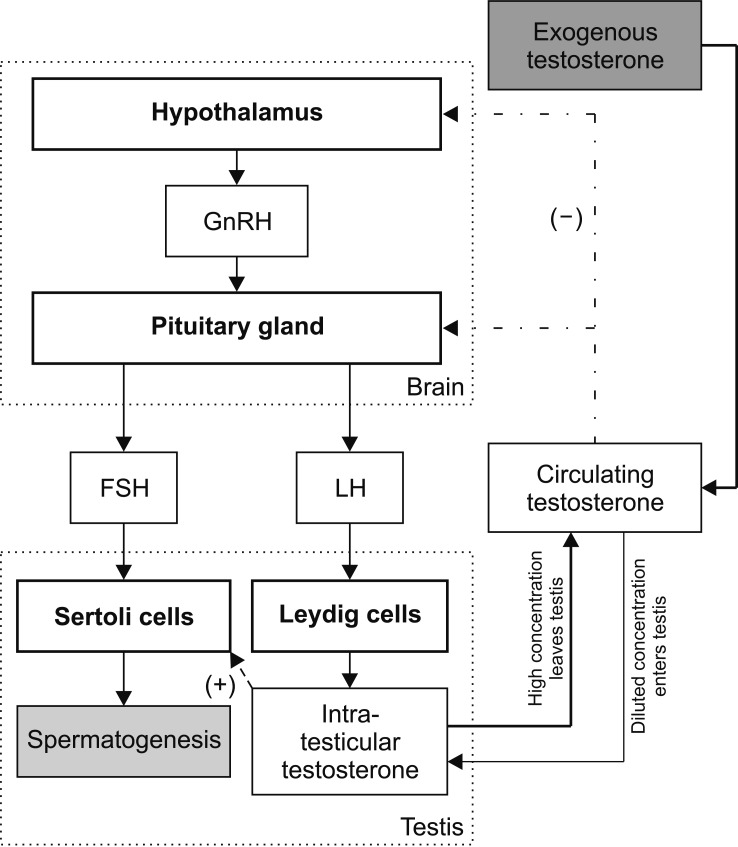
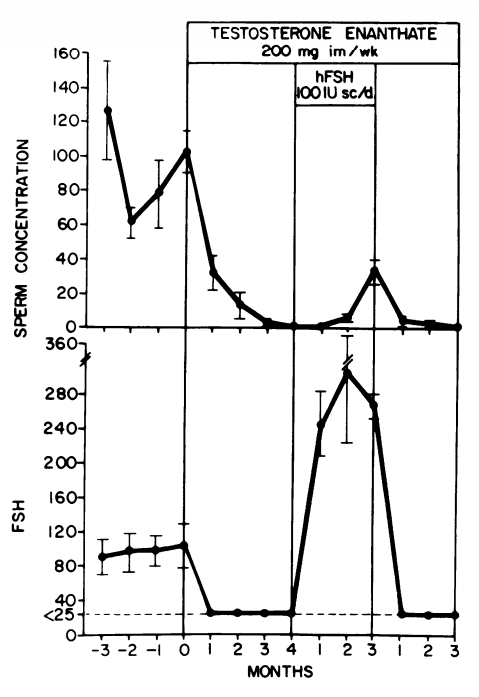
Attualmente gli unici trattamenti approvati per l’ipogonadismo o la carenza di Testosterone sono la Terapia Sostitutiva con Testosterone (TRT) e la terapia con gonadotropina corionica umana (hCG). Tra le due, la TRT è sicuramente quella più comunemente prescritta. Uno dei motivi è rappresentato dal fatto che l’hCG è inefficace nell’ipogonadismo primario, un tipo di ipogonadismo in cui la causa è l’insufficienza testicolare. Questo esclude circa il 15% dei casi di carenza di Testosterone [1]. Altri motivi possono essere il fatto che l’hCG richiede iniezioni frequenti (di solito tre volte alla settimana) ed è più costoso di alcune alternative alla TRT.
Un problema centrale della TRT non affiancata dall’uso regolare di hCG (o abbinamento dell’hCG con l’hMG) è che sopprime la spermatogenesi e quindi porta all’infertilità in un numero considerevole di uomini. Inoltre, le dimensioni dei testicoli diminuiscono. Per gli uomini che desiderano preservare la fertilità e le dimensioni dei testicoli, la TRT in modalità priva di hCG è ovviamente un candidato non ideale. Sebbene questo aspetto sia meno importante per gli uomini più anziani che possono beneficiare della TRT, in quanto è meno probabile che abbiano in programma di avere figli, è un problema importante per gli uomini giovani che desiderano trattare l’ipogonadismo.
Come discusso nel mio precedente articolo sulla fertilità durante l’uso di AAS o in TRT, il Testosterone sopprime la secrezione di LH e FSH, con conseguente inibizione della spermatogenesi. Parte di questa soppressione è mediata dalla conversione del Testosterone in Estradiolo. Si potrebbero quindi aumentare i livelli di Testosterone annullando l’effetto soppressivo dell’Estradiolo sull’ipotalamo e, conseguentemente, sull’ipofisi. In effetti, l’uso di modulatori selettivi dei recettori degli estrogeni (SERM) – che esercitano un’azione antagonista sui recettori degli estrogeni nell’ipotalamo e nell’ipofisi – porta a un forte aumento di LH, FSH e Testosterone negli uomini con ipogonadismo secondario [2]. Allo stesso modo, l’uso di inibitori dell’Aromatasi – che impediscono al Testosterone di essere convertito in Estradiolo dall’azione dell’Enzima Aromatasi – porta a un aumento di LH, FSH e Testosterone negli uomini trattati [3]. Una conseguenza di ciò è che la spermatogenesi può essere preservata sebbene l’uso di AI sia maggiormente deleterio per il profilo lipidico ematico.
Enclomifene nell’Ipogonadismo Secondario:
Per inserire tra le opzioni terapeutiche per il trattamento dell’ipogonadismo secondario l’Enclomifene, un’azienda farmaceutica, la Repros Therapeutics Inc. ha tentato di farlo approvare dalla FDA. Prima di continuare a parlare di come si è svolto il processo, vorrei fornire alcune informazioni sul SERM in questione: l’Enclomifene Citrato (nome commerciale Androxal, successivamente ribattezzato EnCyzix).
Negli anni ’60 è stato scoperto che un farmaco chiamato clomifene citrato induce l’ovulazione. In quanto tale, poteva essere utilizzato come modalità di trattamento per promuovere la fertilità in caso di anovulazione o oligovulazione. Già all’epoca si sapeva che il clomifene agisce aumentando il rilascio di gonadotropine (LH e FSH) [4]. Per questo motivo, i ricercatori hanno iniziato a valutarne l’effetto anche negli uomini sulla spermatogenesi e sul testosterone. Nei decenni successivi, numerosi studi hanno dimostrato la sua efficacia nello stimolare la produzione di testosterone negli uomini ipogonadici. Tuttavia, il clomifene non è stato approvato dalla FDA per il trattamento dell’ipogonadismo. Tuttavia, viene prescritto off-label per questa indicazione e la linea guida 2018 per la valutazione e la gestione della carenza di testosterone dell’American Urological Association ne sostiene condizionatamente l’uso come alternativa alla TRT [5].
Un problema legato al trattamento con Clomifene è che, nonostante il significativo aumento dei livelli di Testosterone, i dati sul suo effetto sulla riduzione dei sintomi dell’ipogonadismo sono contrastanti [6]. Studi su larga scala e di buona qualità potrebbero chiarire questi aspetti e forse fare luce su quali pazienti potrebbero trarre i maggiori benefici dal suo utilizzo. Poiché il brevetto del farmaco è scaduto da tempo e vengono prodotti farmaci generici, le aziende farmaceutiche non sono molto attratte dagli investimenti. Pertanto, questi studi potrebbero non venir mai realizzati.
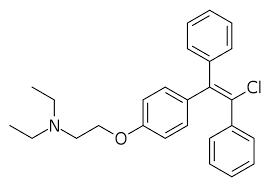
Come accade per molti altri farmaci, anche il Clomifene è una miscela racemica. Ciò significa che è costituito da una molecola di tipo “levogiro” e una di tipo “destrogiro”. In genere solo uno di questi stereoisomeri, come vengono chiamati, è il composto attivo. E ciò dà come risultato che si adatta meglio al recettore su cui agisce. Come un guanto si adatta solo a una mano e non all’altra, il tipo “levogiro” è più efficace nel legarsi a un recettore “levogiro” rispetto allo stereoisomero “destrogiro”. Il Clomifene è costituito dagli stereoisomeri Zuclomifene (nell’immagine sotto a sinistra) e, come alcuni di voi già sapranno, l’Enclomifene (nell’immagine sotto a destra):
Da sinistra: Zuclomifene e Enclomifene.
In generale, lo Zuclomifene è considerato un agonista del recettore degli estrogeni, mentre l’Enclomifene è considerato un potente antagonista degli estrogeni [7]. L’Enclomifene può quindi essere considerato lo stereoisomero attivo del Clomifene. L’idea dell’Enclomifene privo dello stereoisomero Zuclomifene, quindi, è quella di avere qualcosa di più efficace e sicuro del Clomifene. Tuttavia, l’aspetto più importante è che Repros Therapeutics Inc. potrebbe brevettarne l’uso terapeutico per il trattamento dell’ipogonadismo maschile.
Per richiedere l’approvazione della FDA, l’azienda farmaceutica ha dovuto condurre alcuni studi clinici. Il primo studio pubblicato comprendeva solo 12 uomini e non era in cieco [8]. In altre parole, sia i partecipanti che i ricercatori sapevano quale trattamento stavano ricevendo gli uomini. I partecipanti erano uomini con ipogonadismo secondario trattati in precedenza con Testosterone topico. Sono stati randomizzati a ricevere nuovamente Testosterone topico o Enclomifene (25mg al giorno).
Dopo sei mesi di trattamento, i livelli di Testosterone erano praticamente gli stessi tra i gruppi: 545ng/dL (18,9nmol/L) nel gruppo che riceveva il gel e 525ng/dL (18,2nmol/L) nel gruppo che riceveva l’Enclomifene. Anche i livelli di Testosterone libero sono aumentati e sono rimasti praticamente invariati tra i gruppi. Inoltre, e naturalmente, il numero di spermatozoi è stato ridotto negli uomini che ricevevano Testosterone, con numeri intorno ai 20milioni/mL. Inoltre, come previsto, il numero di spermatozoi è aumentato negli uomini che hanno ricevuto l’Enclomifene, con una media di circa 150milioni/mL.
Sono stati condotti un paio di studi clinici successivi. Forse il più interessante è stato quello pubblicato nel 2016, rivolto a uomini ipogonadici obesi [9]. Il documento comprende due studi paralleli randomizzati, in doppio cieco, a doppio braccio e controllati con placebo. Si tratta di un’affermazione che lascia a bocca aperta e credo che il termine “doppio cieco” richieda qualche spiegazione. Nel precedente studio di cui mi sono occupato, ho detto che era di natura non cieca. Quindi i partecipanti e i ricercatori sapevano quale trattamento stava ricevendo ciascun soggetto. Di solito, quando si confrontano due farmaci diversi, si possono semplicemente mettere in cieco i soggetti (e i ricercatori) dando ai gruppi capsule, o pastiglie, o altro identici. Tuttavia, il gel di Testosterone è un gel, mentre l’Enclomifene è una compressa da inghiottire. Quindi non è possibile farlo. Per poter effettuare uno studio come questo in cieco, è necessario somministrare a entrambi i gruppi sia le pastiglie che il gel. Quindi un gruppo riceve un gel placebo e l’Enclomifene, mentre l’altro gruppo riceve un gel di Testosterone e una compressa placebo. Ovvero, doppio braccio. (E poiché lo studio era controllato con placebo, un gruppo ha ricevuto un gel e una compressa placebo).
I due studi descritti in questo articolo hanno utilizzato lo stesso protocollo e l’aspetto forse più interessante è stata la dimensione del campione: 256 soggetti in totale! Finalmente si è capito qualcosa. L’intervento è durato 16 settimane e i soggetti del gruppo Enclomifene hanno ricevuto 12,5mg al giorno e sono stati trattati fino a 25mg al giorno se i livelli di Testosterone non erano aumentati ad almeno 450ng/dL (15,6nmol/L) alla quarta settimana. La dose è stata aumentata per la metà dei soggetti che ricevevano l’Enclomifene. A questo punto le cose iniziano a farsi interessanti: sebbene metà dei soggetti sia stata modificata nel dosaggio alla quarta settimana, non è successo assolutamente nulla con la concentrazione media di Testosterone:
E, in effetti, alla fine dell’intervento, la media del gruppo era appena al di sotto del valore limite di 450ng/dL (15,6nmol/L) per l’up-titration. Infine, 29 degli 85 uomini del gruppo Enclomifene non hanno visto il loro Testosterone aumentare al di sopra del valore limite di ipogonadismo di 300ng/dL (10,4nmol/L) dopo 16 settimane di trattamento. Inoltre, i ricercatori hanno fatto un LAVORO ORRIBILE nel trattare correttamente il gruppo che utilizzava il gel di Testosterone, come si può vedere dalla concentrazione media di Testosterone di quel gruppo. Quasi come se l’avessero fatto apposta per far sì che il gruppo Enclomifene facesse meglio in alcune misurazioni… (anche se si tratta di uno studio a doppio braccio, è comunque possibile istruire i pazienti in modo scorretto con l’applicazione del gel).
È importante notare che gli unici endpoint erano i livelli di Testosterone, LH e FSH e la concentrazione di sperma. Non sono stati analizzati endpoint clinicamente rilevanti, come il desiderio sessuale, la funzione erettile, la stanchezza/vitalità, ecc. A quanto pare, nemmeno negli altri studi (pubblicati). O, forse, sono stati analizzati, ma semplicemente non sono stati riportati nei risultati dello studio perché erano deludenti. E penso che potrebbe essere stata la seconda ipotesi, visto che la FDA non ha approvato il farmaco per il trattamento dell’ipogonadismo secondario, a causa della mancanza di un miglioramento sintomatico misurabile [10]. Anche l’equivalente della FDA nell’UE, l’EMA, ha rifiutato l’autorizzazione all’immissione in commercio dell’Enclomifene qualche tempo dopo, con preoccupazioni simili:
“Il CHMP [Comitato per i Medicinali per Uso Umano] ha osservato che, sebbene gli studi abbiano mostrato un aumento dei livelli di Testosterone con EnCyzix [Enclomifene], non hanno esaminato se EnCyzix migliorasse sintomi quali la densità e resistenza ossea, l’aumento di peso, l’impotenza e la libido. Inoltre, il farmaco comporta un rischio di tromboembolismo venoso (problemi dovuti alla formazione di coaguli di sangue nelle vene)”.
E il Clomifene mostra in realtà risultati molto simili, anche mg per mg, a quelli dell’Enclomifene. Non riassumerò qui l’intera letteratura sul Clomifene, ma prendiamo ad esempio uno studio di Katz et al. in cui 86 giovani uomini ipogonadici hanno ricevuto il Clomifene Citrato a 25mg o 50mg a giorni alterni per una media di 19 mesi e hanno visto aumentare il Testosterone totale del 152% (da 192 ng/dL a 485 ng/dL) [11]. In particolare, il Testosterone libero è aumentato di ben il 332%. Se consideriamo un altro studio condotto su uomini obesi, il Testosterone è aumentato del 98% (da 303ng/dL a 599ng/dL) con 25mg al giorno [12]. In termini di aumento del Testosterone, l’Enclomifene non sembra avere un vantaggio rispetto al Clomifene (non sono riuscito a trovare uno studio di confronto testa a testa).
Conclusioni:
Quindi, per concludere, purtroppo non esiste ancora un’alternativa approvata dalla FDA oltre all’hCG o alla TRT per il trattamento dell’ipogonadismo. E con ciò, gli uomini ipogonadici che cercano un trattamento saranno vincolati alle iniezioni di hCG ( e spesso anche hMG) se desiderano preservare la fertilità durante la TRT. Forse i SERM (attuali) sono solo un vicolo cieco, poiché il loro antagonismo con gli estrogeni contrasta anche gli effetti positivi. Infatti, come hanno dimostrato elegantemente Finkelstein et al., l’aggiunta di un inibitore dell’Aromatasi a un gel di Testosterone ha un impatto negativo sul grasso corporeo e sulla funzione sessuale [13]. Avrebbero dovuto inserire anche l’aumento della neurotossicità e cardiotossicità da carenza di Estradiolo, oltre a stati depressivi e condizioni annesse.
Gabriel Bellizzi
Riferimenti:
Tajar, Abdelouahid, et al. “Characteristics of secondary, primary, and compensated hypogonadism in aging men: evidence from the European Male Ageing Study.” The Journal of Clinical Endocrinology & Metabolism 95.4 (2010): 1810-1818.
Wheeler, Karen M., et al. “Clomiphene citrate for the treatment of hypogonadism.” Sexual medicine reviews 7.2 (2019): 272-276.
De Ronde, Willem, and Frank H. de Jong. “Aromatase inhibitors in men: effects and therapeutic options.” Reproductive Biology and Endocrinology 9.1 (2011): 1-7.
Jungck, Edwin C., et al. “Effect of clomiphene citrate on spermatogenesis in the human: a preliminary report.” Obstetrical & Gynecological Survey 19.3 (1964): 520.
Mulhall, John P., et al. “Evaluation and management of testosterone deficiency: AUA guideline.” The Journal of urology 200.2 (2018): 423-432.
Scovell, Jason M., and Mohit Khera. “Testosterone replacement therapy versus clomiphene citrate in the young hypogonadal male.” European urology focus 4.3 (2018): 321-323.
Fontenot, Gregory K., Ronald D. Wiehle, and Joseph S. Podolski. “Differential effects of isomers of clomiphene citrate on reproductive tissues in male mice.” BJU Int 117.2 (2016): 344-50.
Kaminetsky, Jed, et al. “Oral enclomiphene citrate stimulates the endogenous production of testosterone and sperm counts in men with low testosterone: comparison with testosterone gel.” The journal of sexual medicine 10.6 (2013): 1628-1635.
Kim, Edward D., Andrew McCullough, and Jed Kaminetsky. “Oral enclomiphene citrate raises testosterone and preserves sperm counts in obese hypogonadal men, unlike topical testosterone: restoration instead of replacement.” BJU international 117.4 (2016): 677-685.
Earl, Joshua A., and Edward D. Kim. “Enclomiphene citrate: A treatment that maintains fertility in men with secondary hypogonadism.” Expert review of endocrinology & metabolism 14.3 (2019): 157-165.
Katz, Darren J., et al. “Outcomes of clomiphene citrate treatment in young hypogonadal men.” BJU International-British Journal of Urology 110.4 (2012): 573.
Pelusi, Carla, et al. “Clomiphene citrate effect in obese men with low serum testosterone treated with metformin due to dysmetabolic disorders: a randomized, double-blind, placebo-controlled study.” PLoS One 12.9 (2017): e0183369.
Finkelstein, Joel S., et al. “Gonadal steroids and body composition, strength, and sexual function in men.” New England Journal of Medicine 369.11 (2013): 1011-1022.
Il BodyBuilding si differenzia dagli sport di prestazione perché il giorno della gara gli atleti vengono giudicati in base all’aspetto piuttosto che alle capacità atletiche. I bodybuilder posano sul palco dove vengono giudicati per la muscolatura, la definizione e la simmetria. Nel corso di una stagione, i bodybuilder attraversano tre fasi diverse: la fase di crescita muscolare (Off-Season), la dieta per la competizione (preparazione alla gara) e la gara stessa. La maggior parte della letteratura riguarda la fase di dieta pre-gara e la peak week.[1]
Tuttavia, la letteratura scientifica sulle raccomandazioni alimentari per i bodybuilder durante la Off-Season è carente. Si tratta di una lacuna importante, poiché la maggior parte della carriera di un bodybuilder si svolge in questa fase, in cui l’obiettivo è aumentare la massa muscolare riducendo al minimo l’aumento eccessivo della massa grassa. I bodybuilder sono noti per avere atteggiamenti rigidi nei confronti della selezione degli alimenti, della frequenza dei pasti, dei tempi di alimentazione e dell’integrazione [2]. Storicamente, le informazioni sull’alimentazione e l’integrazione sono state trasmesse dalle riviste di bodybuilding e dai concorrenti di successo, ma recentemente sono emerse più informazioni attraverso Internet e i forum [3,4]. Di conseguenza, molte delle strategie alimentari utilizzate dai bodybuilder non hanno un solido supporto scientifico e la letteratura scientifica dimostra che alcune di queste strategie, tra cui l’uso massiccio di farmaci, ma anche di integratori più in generale, possono essere ovviamente dannosi per la salute [5,6,7].
Poiché i bodybuilder trascorrono la maggior parte del loro tempo in Off-Season, è evidente la necessità di raccomandazioni nutrizionali e di supplementazione, sia OTC che PEDs, il più possibile “sicure” e basate sull’evidenza per questa popolazione. È stato inoltre dimostrato che alcuni bodybuilder, e non soltanto i concorrenti di alto livello nel bodybuilding “Natural”, potrebbero essere interessati a informazioni basate sull’evidenza [8]. Con il supporto della review realizzata e pubblicata da Juma Iraki et al. che tratta del Off-Season a livello alimentare e integrativo, lo scopo di questo articolo sarà quello di riportare quanto evidenziato dalla letteratura scientifica sugli argomenti relativi all’alimentazione e all’integrazione alimentare e supplementazione PEDs rilevanti per i bodybuilder nella Off-Season e di fornire raccomandazioni pratiche sull’assunzione di energia, macronutrienti, frequenza dei pasti, tempistica dei nutrienti, integratori alimentari e PEDs .
Transizione dalla dieta pre-gara/peak week alla dieta in Off-Season – Reverse Diet Vs. Recovery Diet:
Il primo step che il bodybuilder si trova davanti è la gestione del passaggio da una dieta ipocalorica ad una ipercalorica. Ed è in questo frangente che emergono due strategie simili all’apparenza ma in realtà diverse: la “Recovery Diet” e la “Reverse Diet”.
Ora, molto semplicemente, la “Recovery Diet” consiste in un graduale aumento calorico ma di consistenza tale che l’atleta esca dalla condizione di ipocalorica nel giro di due settimane circa. Con la “Reverse Diet”, invece, abbiamo sempre un graduale aumento calorico ma caratterizzato da una ridotta consistenza dello stesso (si parla di circa 100Kcal/die a settimana). In questo caso specifico, il bodybuilder rimarrebbe in ipocalorica per diverse settimane con possibile emersione di problemi psicofisici legati al protrarsi dello stato stressorio.
Quindi, con il termine “Recovery Diet” ci riferiamo ad uno schema alimentare avente l’obiettivo generale di RECUPERARE da un periodo di dieta cronica sperimentato durante la preparazione alla gara. La “Recovery Diet” incoraggia i bodybuilder a guadagnare il 5-10% del loro peso di gara nelle prime 4-8 settimane successive all’evento. Questo con l’intento di accelerare l’aumento di grasso corporeo e far rientrare il soggetto in un range di grasso corporeo “sano”, fisiologico, il prima possibile. In seguito, si consiglia agli atleti di rallentare il ritmo di aumento del peso e di mantenere un surplus controllato, con un aumento medio dello 0,5-1% del peso corporeo al mese passando pienamente nella Off-Season. Questo fino a quando non raggiungono un punto in cui un ulteriore aumento di peso è considerato improduttivo. Con il termine “Reverse Diet” ci si riferisce ad una strategia la quale può ancora essere attuata con discreti vantaggi per aiutare un agonista a recuperare dopo il contest. Tuttavia, se rispettata e seguita correttamente, piccoli aumenti di cibo di ~100 Kcal/die a settimana potrebbero comunque protrarre il deficit calorico del soggetto, prolungando così il periodo di dieta ipocalorica. Sebbene questa possa essere una strategia utile in alcune circostanze, ad esempio durante l’avvicinamento alla competizione, le modalità di applicazione non permettono un recupero di una bf salubre in tempi ottimali. È risaputo che un bodybuilder in condizioni di picco non è necessariamente al massimo della salute, e questo è in gran parte correlato al livello di grasso corporeo. Accettare un certo aumento di grasso avrà effetti positivi su tutti gli aspetti della Off-Season come le prestazioni in allenamento, i marcatori ormonali, la disponibilità di energia, la qualità del sonno e, inoltre, sarà vantaggioso sulla longevità complessiva dello sport praticato.
In definitiva, se si parte da body fat estremamente basse, tipiche da gara, allora la “Recovery Diet” è la scelta migliore per shiftare dal regime ipocalorico che ha caratterizzato il periodo di preparazione alla gara a quello ipercalorico del Off-Season. Discorso diverso se ci troviamo di fronte ad un soggetto amatoriale, con una body fat del 8-10% arrivato al termine del percorso di “Cut”. In questo caso la “Reverse Diet” è la scelta più funzionale permettendo un controllo migliore degli incrementi calorici evitando che la massa grassa sfori eccessivamente e che il lavoro precedentemente svolto in “Cut” venga facilmente e totalmente compromesso. Anche “ibridazioni” con aumenti settimanali di 45-50g di CHO die possono essere applicati con buoni risultati.
Energia:
Durante la Off-Season, l’obiettivo principale di un bodybuilder è quello di aumentare la massa muscolare riducendo al minimo l’aumento della massa grassa attraverso l’uso di allenamenti contro-resistenza e il mantenimento di un bilancio energetico positivo. Per valutare con precisione il fabbisogno energetico dei bodybuilder durante la bassa stagione, è necessario considerare il volume, la frequenza e l’intensità dell’allenamento. Durante la fase off-season, è stato riportato che i bodybuilder si allenano alla resistenza 5-6 volte a settimana, esercitando ogni gruppo muscolare 1-2 volte a settimana [9]. È stato inoltre riferito che seguono una routine di allenamento ad alto volume con 4-5 esercizi per gruppo muscolare, eseguendo 3-6 serie per esercizio, 7-12 ripetizioni massime (RM) per ogni serie con 1-2 minuti di riposo tra le serie. La durata della sessione di allenamento è stata indicata in ~40-90 minuti. Tuttavia, i piani di allenamento possono variare notevolmente da atleta ad atleta. È necessario valutare anche l’apporto calorico medio dei bodybuilder. Nella fase off-season, l’apporto energetico è di solito sostanzialmente più elevato rispetto alla fase di dieta: tra i bodybuilder maschi è stato riportato un apporto medio di ~3800 kcal/giorno durante la fase off-season e di ~2400 kcal/giorno durante la fase di dieta [2].
Bilancio energetico positivo:
È stato dimostrato che un bilancio energetico positivo ha un importante effetto anabolico, anche in assenza di allenamento contro-resistenza [10]. Tuttavia, la combinazione di un bilancio energetico positivo con l’allenamento contro-resistenza rappresenta il metodo più efficace per garantire che gli effetti anabolici siano diretti all’aumento della massa muscolo-scheletrica [11,12]. L’entità del surplus energetico ideale per guadagnare massa muscolare limitando l’accumulo di tessuto adiposo può variare in base allo stato di allenamento. Nei soggetti non allenati, è stato dimostrato che un surplus energetico sostanziale di circa 2.000 kcal, combinato con l’allenamento contro-resistenza, fornisce un robusto aumento di peso, in cui il contributo della massa magra (LBM) può raggiungere il 100% [12]. Tuttavia, nei soggetti allenati, un surplus energetico sostanziale potrebbe non essere necessario o vantaggioso. Uno studio condotto su atleti d’élite ha esaminato l’effetto delle indicazioni dietetiche sui cambiamenti della composizione corporea tra gli atleti d’élite quando l’allenamento contro-resistenza è stato combinato con diverse entità di surplus energetico. Un gruppo con un peso corporeo medio di 75kg ha consumato energia ad libitum (2964 kcal) per raggiungere un surplus molto ridotto, mentre un secondo gruppo con un peso corporeo medio di 71kg ha ricevuto una consulenza dietetica e ha consumato ~600 kcal in più rispetto al gruppo ad libitum [13].
Entrambi i gruppi hanno seguito lo stesso programma di allenamento contro-resistenza di 4 giorni alla settimana per un periodo di 8-12 settimane. I ricercatori hanno ipotizzato che il gruppo ipercalorico avrebbe avuto un aumento maggiore del peso corporeo e della LBM. Sebbene il gruppo ipercalorico abbia ottenuto un aumento maggiore della LBM rispetto a quelli che mangiavano ad libitum, questo non ha raggiunto la significatività statistica (1,7kg contro 1,2kg, rispettivamente). Inoltre, rispetto al gruppo che mangiava a sazietà, hanno registrato un aumento significativamente maggiore della massa grassa (1,1kg contro 0,2kg, rispettivamente). I ricercatori hanno concluso che un surplus di 200-300 kcal al giorno negli atleti altamente allenati potrebbe essere più appropriato di 500 kcal per minimizzare il rischio di inutili aumenti di grasso corporeo. I soggetti non allenati, più lontani dal loro tetto genetico di massa muscolare, possono essere in grado di aumentare i muscoli a un ritmo più veloce rispetto agli individui allenati.
Il tasso di crescita muscolare può rallentare con l’avanzare dell’età [14]. Pertanto, un maggiore surplus energetico può essere più vantaggioso per i bodybuilder alle prime armi, mentre i bodybuilder avanzati potrebbero trarre maggiore beneficio da diete ipercaloriche conservative per limitare inutili aumenti di grasso corporeo. Studi precedenti hanno raccomandato ai bodybuilder di consumare una dieta leggermente ipercalorica, con un aumento dell’apporto energetico di circa il 15% rispetto al mantenimento nella Off-Season [15]. Tuttavia, ciò non tiene conto della storia di allenamento e del livello di esperienza del singolo bodybuilder. Poiché la capacità di aumentare la massa muscolare è limitata, un surplus aggressivo può portare a un inutile aumento del grasso corporeo, che aumenterebbe la durata o la gravità dei successivi periodi di preparazione alle gare, aumentando di conseguenza la durata o la gravità della scarsa disponibilità energetica. Pertanto, il numero di calorie che un bodybuilder consuma al di sopra del livello di mantenimento può essere stabilito in base al livello di esperienza e poi regolato in base al tasso di aumento di peso e ai cambiamenti nella composizione corporea. Dato che i bodybuilder spesso aumentano rapidamente di peso dopo una gara, potrebbe essere utile avere un obiettivo di aumento di peso per settimana e regolarsi di conseguenza [16,17].
Tuttavia, come detto precedentemente, inizialmente, dopo la gara, potrebbe essere utile un aumento di peso più rapido per aiutare a riportare il concorrente a uno stato di salute sia psicologico che fisiologico, prima che il tasso di aumento di peso venga rallentato per limitare l’accumulo eccessivo di tessuto adiposo. Nella letteratura scientifica si raccomanda di puntare a un aumento di peso di circa 0,25-0,5 kg a settimana per cercare di aumentare la LBM e ridurre al minimo l’aumento della massa grassa [14,18]. Per un bodybuilder avanzato, un potenziale aumento di 2kg di peso corporeo su base mensile potrebbe essere eccessivo e comportare un’inutile accumulazione di grasso corporeo; pertanto, questo tasso dovrebbe essere considerato con cautela. Sulla base delle prove attuali, potrebbe essere opportuno raccomandare ai bodybuilder di consumare una dieta leggermente ipercalorica (~10-20% sopra le calorie di mantenimento) nella Off-Season e raccomandare ai bodybuilder avanzati di puntare all’estremità inferiore di questa raccomandazione, o addirittura di essere più conservativi se si verificano aumenti sostanziali della massa grassa. Dato che i bodybuilder consumano in media 45 kcal/kg durante la bassa stagione, il surplus raccomandato equivale a circa 42-48 kcal/kg [2]. Potrebbe essere utile puntare a un aumento di peso di circa 0,25-0,5% del peso corporeo a settimana, regolando al contempo l’apporto energetico in base alle variazioni della composizione corporea. Inoltre, potrebbe essere più appropriato considerare le variazioni di peso medie settimanali basate su pesate giornaliere (o più volte alla settimana) per limitare gli errori delle fluttuazioni giornaliere del peso che possono verificarsi durante la settimana. Una volta determinato il surplus calorico, il passo successivo sarà quello di distribuire le calorie tra proteine, grassi e carboidrati.
Proteine:
Il turnover proteico del muscolo scheletrico è il rapporto tra la sintesi proteica muscolare (MPS) e la degradazione proteica muscolare (MPB). L’ipertrofia del muscolo scheletrico richiede un equilibrio netto in cui la MPS supera la MPB. L’esercizio contro-resistenza fornisce lo stimolo di tensione iniziale che induce l’ipertrofia risultante dall’aumento cumulativo della MPS dopo l’esercizio cronico [19]; tuttavia, l’aumento della massa grassa (FFM) può essere limitato se l’apporto proteico giornaliero è insufficiente [20]. Oltre alla quantità totale consumata al giorno, i ricercatori hanno ipotizzato che la qualità delle proteine possa aumentare il guadagno muscolare indotto dall’allenamento contro-resistenza [21]. Pertanto, entrambi questi argomenti saranno discussi nelle sezioni seguenti.
Introito proteico giornaliero:
Mentre l’attuale RDA per le proteine negli individui sani sedentari è di 0,8 g/kg, in una meta-analisi del 2018 di Morton e colleghi [22] è stato osservato che il doppio di questa quantità massimizza l’ipertrofia indotta dall’allenamento contro-resistenza. Inoltre, gli autori hanno osservato che “potrebbe essere prudente raccomandare ~2,2g di proteine/kg/die per coloro che cercano di massimizzare i guadagni di FFM indotti dall’allenamento contro-resistenza”, poiché 2,2g/kg era l’estremità superiore del limite di confidenza [22] e le differenze individuali impongono che alcuni atleti abbiano un fabbisogno proteico più elevato di altri [23]. Inoltre, la raccomandazione “meglio prevenire che curare” è probabilmente sicura, vista l’assenza di danni apparenti in studi di 1-2 anni tra i sollevatori che consumavano apporti proteici di almeno 2,2 g/kg [24,25]. Infine, la media e il limite superiore di confidenza del 95% per il fabbisogno proteico utilizzando la tecnica di ossidazione degli aminoacidi con indicatore tra i bodybuilder maschi nei giorni di non allenamento sono stati riportati rispettivamente come 1,7 e 2,2g/kg [26], che è simile al fabbisogno tra le donne quando è normalizzato alla FFM [27].
Tuttavia, è stato riportato che i bodybuilder consumano fino a 4,3g/kg di proteine al giorno tra i soggetti di sesso maschile e 2,8g/kg tra quelli di sesso femminile, superando di gran lunga queste raccomandazioni [2]. Le linee guida precedentemente fornite per i bodybuilder nella Off-Season erano di consumare il 25-30% del loro apporto energetico dalle proteine [15]. Potrebbe essere ragionevole opporsi all’indicazione di raccomandazioni basate su percentuali dell’apporto energetico totale, poiché un individuo con un peso non particolarmente elevato ma con un alto fabbisogno energetico potrebbe finire per consumare proteine che superano di gran lunga quelle necessarie e quindi richieste. Inoltre, questo può portare a un’assunzione insufficiente di carboidrati e grassi se l’atleta mira a un apporto calorico specifico. Pertanto, potrebbe essere più appropriato raccomandare un fabbisogno proteico basato sul peso corporeo. Pertanto, i bodybuilder dovrebbero consumare un minimo di 1,6g/kg di proteine nella Off-Season, anche se un obiettivo più vicino a 2,2 g/kg potrebbe garantire una risposta ottimizzata in modo più coerente in una maggiore percentuale di atleti.
E per i “Doped”? Dovremo ormai sapere che la fisiologia di base è la medesima per ogni individuo con le consuete variabili. Detto ciò, l’uso di PEDs va si ad alterare la fisiologia ma in questo specifico ambito, ossia introito proteico per massimizzare lo stimolo ipertrofico, hanno una azione di perfezionamento dell'”economia proteica cellulare”: in parole più semplici, sembra che l’uso di AAS porti ad una migliore resa nell’utilizzo degli amminoacidi scissi e assorbiti dalle proteine alimentari. Di conseguenza, a parità di apporto proteico, la veicolazione degli amminoacidi a scopo plastico è maggiore come minore è l’attività catabolica. Ciò significa che abusare delle proteine, in special modo durante una fase ipercalorica, perchè si è sotto AAS potrebbe risultare più inutile di quanto non lo sia in contesto “Natural”.
Infine, ed è necessario sottolinearlo, tra i bodybuilder che lottano con la fame in Off-Season e che di conseguenza assumono quantità caloriche che portano a un aumento di peso più rapido e all’accumulo di grasso in eccesso, un apporto proteico più elevato può essere utile (se non controindicato per motivi clinici). In uno studio condotto da Antonio e colleghi, i partecipanti ad allenamenti contro-resistenza che consumavano più proteine (4,4g/kg al giorno) e più calorie hanno guadagnato una quantità simile di FFM, ma non hanno guadagnato ulteriore grasso corporeo rispetto al gruppo che consumava meno proteine e meno calorie [28]. Allo stesso modo, in uno studio di follow-up, un gruppo che consumava 3,4g/kg di proteine al giorno ha guadagnato una quantità simile di FFM, ma ha perso una percentuale maggiore di grasso corporeo rispetto a un gruppo a basso contenuto proteico, ancora una volta, nonostante un apporto energetico più elevato [29]. Gli autori di questi studi sulla “vita libera” hanno ipotizzato che i loro risultati fossero dovuti a un aumento della termogenesi indotta dalla dieta attraverso protocolli alimentari ad alto contenuto proteico. Tuttavia, ciò è in contrasto con uno studio di Bray e colleghi del 2012 sul reparto metabolico, più strettamente controllato, in cui il contenuto proteico della dieta influenzava la percentuale di massa corporea acquisita, mentre la massa corporea totale era dettata dal solo contenuto energetico della dieta [30].
Pertanto, mentre la termogenesi indotta dalla dieta potrebbe essere significativamente più elevata con assunzioni di proteine nell’intervallo di 3 g/kg o superiore, la perdita di grasso o la mancanza di aumento di peso osservata da Antonio e colleghi, nonostante un apporto energetico più elevato, potrebbe con più probabilità riflettere l’effetto saziante di assunzioni proteiche molto elevate che diminuiscono l’assunzione calorica effettiva, piuttosto che un aumento della sola termogenesi.
Qualità delle Proteine:
Gli aminoacidi essenziali (EAA) sono gli unici aminoacidi necessari per stimolare il processo di MPS [31]. Sebbene tutti gli aminoacidi forniscano i “mattoni” necessari per la sintesi di nuovi tessuti, l’aminoacido Leucina in particolare sembra essere particolarmente importante come “innesco metabolico” della MPS [32]. È stato suggerito che una concentrazione sufficiente di Leucina è necessaria per raggiungere una “soglia di Leucina” che è richiesta per stimolare al massimo la MPS [33]. In breve, dal punto di vista della costruzione muscolare, le fonti proteiche che innescano una consistente risposta della MPS (quantità sufficiente di Leucina) e forniscono i mattoni essenziali per la costruzione di nuovo tessuto muscolare (contengono l’intero spettro di aminoacidi essenziali in abbondanza) possono essere considerate di “qualità superiore”.
Sebbene l’effetto meccanicistico della Leucina sulle MPS esuli dallo scopo di questo articolo, si invitano i lettori a leggere una rassegna che tratta questo argomento in dettaglio [34]. In generale, su una base di grammo per grammo, le fonti proteiche di origine animale contengono in genere più Leucina ed EAA, anche se ci sono eccezioni degne di nota. Le proteine della soia, uno dei più comuni integratori proteici di origine vegetale, contengono tutti gli EAA, ma in una quantità inferiore per grammo rispetto alle proteine del latte e quindi, in uno studio, hanno prodotto un aumento minore delle MPS rispetto al siero di latte dopo un’ingestione acuta [35]. È interessante notare che in questo stesso studio la soia ha prodotto un aumento maggiore delle MPS rispetto alla caseina, anch’essa una proteina casearia di “alta qualità”, presumibilmente a causa della più lenta velocità di digestione della caseina [35]. Rammentate sempre la differenza tra risposta “acuta” e “cronica”. Per l’appunto, ciò significa che, sebbene il contenuto di Leucina e di EAA di una fonte proteica debba essere preso in considerazione, la risposta acuta alla MPS non è l’unica variabile legata all’ipertrofia a lungo termine. Infatti, una proteina di alta qualità ma “lenta” come la caseina produce inizialmente una risposta MPS di minore ampiezza. Tuttavia, la caseina (e altre proteine a lenta digestione) può produrre un’area MPS sotto la curva simile o maggiore se osservata longitudinalmente rispetto a una fonte proteica “veloce” come il siero di latte, che determina un aumento iniziale maggiore e poi una brusca riduzione [36].
Inoltre, la risposta acuta della MPS a un determinato tipo di proteina non deve essere vista in una prospettiva riduzionista. Nel mondo reale si consumano quotidianamente più porzioni di varie fonti proteiche, rendendo probabilmente superflue alcune di queste distinzioni nel profilo aminoacidico e nella cinetica di digestione. Infatti, in una meta-analisi che ha confrontato i cambiamenti longitudinali della composizione corporea con diversi tipi di integratori proteici, non sono state riscontrate differenze significative tra i partecipanti che consumavano soia rispetto al siero di latte, ad altre proteine del latte o alle proteine isolate del manzo [37].
Come dimostrato in uno studio che ha messo a confronto gruppi che consumavano proteine dopo l’allenamento (in aggiunta a una dieta già composta dal 25% di proteine), sia che venissero forniti 48g di proteine del siero del latte (contenenti 5,5g di Leucina), sia che venissero forniti 48g di proteine del riso (contenenti 3,8g di Leucina), non è stato osservato alcun impatto sui cambiamenti della composizione corporea tra i gruppi dopo otto settimane [38]. Pertanto, se consumate in quantità sufficienti (soprattutto se si considera l’apporto proteico totale giornaliero), la qualità delle proteine di un singolo pasto è meno preoccupante. Tuttavia, se si volesse consumare una dieta dominata da fonti proteiche di origine vegetale, esistono alternative alla soia e al riso. Ad esempio, le proteine isolate del pisello sono ricche di EAA e di Leucina. In uno studio di 12 settimane, un gruppo che consumava 50g di proteine isolate di pisello al giorno ha registrato un aumento maggiore dello spessore muscolare indotto dall’allenamento di resistenza rispetto al placebo, non significativamente diverso da un gruppo che consumava 50g di siero di latte [39].
Pertanto, nel contesto delle indicazioni di questo articolo, la qualità delle proteine può essere un problema solo se si utilizza la fascia bassa delle linee guida sulle proteine (1,6g/kg) o se si consuma una dieta a base prevalentemente vegetale. In entrambi i casi, potrebbe essere utile integrare con fonti proteiche ricche di Leucina e di EAA, a seconda delle preferenze alimentari (ad esempio, proteine del latte o del pisello se si è vegani), per garantire la risposta attesa della MPS all’assunzione di proteine.
Grassi:
Il grasso è un nutriente fondamentale per molte funzioni dell’organismo. Tuttavia, non si sa molto dell’effetto dei grassi alimentari sull’ipertrofia del muscolo scheletrico. È stato riportato che l’assunzione di grassi alimentari tra i bodybuilder varia dall’8 al 33% delle calorie totali [2]. Sebbene i trigliceridi intramuscolari possano fungere da substrato energetico durante l’allenamento di resistenza, non sono un fattore limitante poiché i substrati derivano principalmente da processi anaerobici [40]. Di interesse per il bodybuilder, è dimostrato che negli atleti allenati contro-resistenza [41] e nei giocatori di hockey [42] le diete a basso contenuto di carboidrati (30-45% dell’energia o meno) possono influire sul rapporto Testosterone libero/Cortisolo (fTC), il che potrebbe avere un impatto negativo sul recupero. D’altra parte, la riduzione dei grassi alimentari nelle diete isocaloriche da ~30-40% a ~15-25% ha portato a riduzioni significative ma modeste dei livelli di Testosterone [43,44,45,46].
Tuttavia, non è chiaro se le variazioni di Testosterone all’interno di intervalli normali influenzino in modo significativo l’aumento della massa muscolare [47]. Nonostante la possibilità che i livelli di testosterone possano essere più elevati quando si consuma una percentuale maggiore di energia proveniente dai grassi alimentari, i cambiamenti effettivi nella massa muscolare durante gli studi longitudinali di individui allenati alla resistenza che seguono diete “chetogeniche” ad alto contenuto di grassi sono stati costantemente inferiori rispetto ad approcci moderati o a basso contenuto di grassi con ampi carboidrati [48,49,50,51]. Non è ancora stato chiarito se ciò sia dovuto a cambiamenti nella capacità di esercizio, ad alterazioni del rapporto fTC o a qualche altro meccanismo legato alla componente ad alto contenuto di grassi o a basso contenuto di carboidrati della dieta.
Tuttavia, ciò indica che forse si dovrebbe consumare una proporzione più moderata di grassi nella dieta, piuttosto che un apporto basso o alto. In letteratura sono state proposte raccomandazioni del 15-20% e del 20-30% delle calorie provenienti dai grassi alimentari [15,52]. Tuttavia, sono necessarie ulteriori ricerche per stabilire l’effetto e la quantità ottimale di grassi alimentari per favorire l’ipertrofia muscolare.
Sulla base delle evidenze attuali, può essere prudente raccomandare che i grassi alimentari rappresentino il 20-35% delle calorie, in linea con le raccomandazioni dell’American College of Sports Medicine per gli atleti [53], che nella maggior parte dei casi corrispondono a circa 0,5-1,5 g/kg/giorno. Inoltre, va notato che un apporto sufficiente di proteine e carboidrati non deve essere compromesso da un’elevata assunzione di grassi nella dieta.
Anche la qualità dei grassi, come gli essenziali omega 3 e gli omega 6, potrebbe essere importante per i bodybuilder. Se l’apporto di questi acidi grassi è sufficiente, non è necessario integrarli con una dieta di alta qualità contenente buone fonti di acidi grassi. Tuttavia, per alcuni potrebbe essere difficile assumere le quantità ottimali. Per questo motivo, l’argomento verrà trattato in modo più approfondito nella sezione dedicata agli integratori alimentari.
Carboidrati:
A differenza delle proteine e dei grassi, i carboidrati sono considerati non essenziali per la dieta umana perché l’organismo è in grado di produrre il glucosio necessario ai tessuti attraverso la gluconeogenesi [54]. Tuttavia, l’assunzione di carboidrati ha un ruolo importante nella dieta del bodybuilder come regolatore degli ormoni tiroidei e come contributo al fabbisogno di micronutrienti [55,56]. Inoltre, una dieta a basso contenuto di carboidrati potrebbe limitare la rigenerazione dell’adenosina trifosfato (ATP) e limitare la capacità dei muscoli di contrarsi con una forza elevata [57,58]. Durante l’esercizio ad alta intensità, il glicogeno muscolare è il principale contributore di substrato energetico ed è stato dimostrato che la glicolisi fornisce circa l’80% del fabbisogno di ATP di una serie di flessioni del gomito se portata al cedimento muscolare [59]. Nonostante ciò, parte del glicogeno utilizzato durante questo tipo di esercizio può essere risintetizzato dal lattato, il che potrebbe ridurre il fabbisogno di carboidrati. È stato inoltre dimostrato che l’allenamento contro-resistenza riduce il glicogeno muscolare del 24-40% in una singola sessione [59,60].
La quantità esaurita può variare in base alla durata, all’intensità e al lavoro svolto, ma l’allenamento tipico del bodybuilding con ripetizioni più elevate e carichi moderati sembra causare la maggiore riduzione delle scorte di glicogeno muscolare [61]. Inoltre, è stato suggerito che quando le scorte di glicogeno sono troppo basse (~70 mmol/kg), ciò può inibire il rilascio di calcio e accelerare l’insorgenza della fatica muscolare [62]. Un basso livello di glicogeno muscolare riduce significativamente il numero di ripetizioni eseguite quando si eseguono tre serie di Squat all’80% di 1RM [57].
Tuttavia, è stato dimostrato che il consumo di una dieta contenente 7,7 g/kg/die di carboidrati per 48 ore prima di una sessione di allenamento non ha un effetto maggiore sulle prestazioni rispetto a 0,37g/kg/die quando si eseguono 15 serie a 15RM di esercizi per la parte inferiore del corpo [63]. Analogamente, un altro studio ha rilevato che una dieta con il 70% di carboidrati rispetto a una dieta con il 50% di carboidrati non ha un effetto maggiore sulle prestazioni durante l’esercizio sopramassimale; tuttavia, una dieta composta dal 25% di carboidrati ha ridotto significativamente le prestazioni [64].
Inoltre, visti gli effetti negativi a lungo termine sulla massa muscolare osservati di recente in studi su popolazioni allenate alla resistenza che seguono diete chetogeniche [49,51], potrebbe essere prudente per i bodybuilder assicurarsi semplicemente un apporto sufficiente di carboidrati, visti questi risultati disparati. Pertanto, mentre le diete a moderato e alto contenuto di carboidrati sono probabilmente appropriate per il bodybuilding, le diete a bassissimo contenuto di carboidrati possono essere dannose per l’allenamento.
Nei bodybuilder maschi, sono stati riportati apporti medi di carboidrati pari a 5,3g/kg/giorno durante la Off-Season [2]. Tuttavia, non sono state stabilite le quantità ottimali di carboidrati per i bodybuilder. In letteratura sono state proposte raccomandazioni per gli sport di forza, tra cui il bodybuilding, con assunzioni di 4-7g/kg/giorno e 5-6g/kg [15,65]. I carboidrati sembrano essere importanti per il bodybuilder, ma per ottenere benefici possono essere necessarie solo quantità moderate. Pertanto, dopo aver destinato le calorie alle proteine (1,6-2,2g/kg/die) e ai grassi (0,5-1,5g/kg/die), le restanti calorie dovrebbero essere destinate ai carboidrati. Tuttavia, sulla base delle prove attuali, potrebbe essere ragionevole consumare quantità sufficienti di carboidrati nell’intervallo ≥3-5g/kg/giorno, se possibile.
Sono necessarie ulteriori ricerche tra i bodybuilder per stabilire se l’assunzione abituale di carboidrati, superiore o inferiore a quella osservata, possa produrre ulteriori benefici. La Tabella sottostante riassume le raccomandazioni per le calorie e i macronutrienti.
Raccomandazioni dietetiche per i bodybuilder in Off-Season.
Distribuzione e timing dei nutrienti:
Si dice che i bodybuilder consumino in media sei pasti al giorno [66]; tuttavia, non esistono studi che esaminino specificamente quale possa essere la frequenza ottimale dei pasti per questa popolazione [65]. Questa elevata frequenza dei pasti si basa sulla convinzione di un maggiore stato di anabolismo e persino di un migliore utilizzo dei nutrienti durante il giorno, che potrebbe tradursi in un miglioramento della composizione corporea.
Il concetto di temporizzazione dell’assunzione di proteine per massimizzare l’ipertrofia comprende diverse strategie di dosaggio. La prima a comparire in letteratura è stata il consumo di proteine in prossimità dell’allenamento contro-resistenza. I picchi di MPS sono più elevati in questo periodo quando si consumano proteine; pertanto, questa strategia è stata proposta per migliorare l’efficienza della riparazione e del rimodellamento del muscolo scheletrico [31]. Inoltre, a causa dell'”effetto muscolo pieno”, per cui un ulteriore apporto di proteine non aumenta la MPS finché non è trascorso un tempo sufficiente, distribuire uniformemente l’assunzione di proteine tra più pasti è un’altra strategia studiata per massimizzare la MPS totale giornaliera [67]. Infine, il consumo prima di andare a letto di proteine a lenta digestione (come la caseina) per evitare periodi catabolici prolungati durante il sonno è la strategia proposta più di recente per migliorare il bilancio proteico netto giornaliero [68], sebbene si sia dimostrata inutile nel perseguire il fine o, per lo meno, non molto diversa dalla risultante di una assunzione di isolate in un contesto alimentare con parità nel totale proteico giornaliero. Ciascuna di queste tre strategie sarà discussa in seguito.
Dosaggio proteico:
Il periodo post-allenamento consente un picco della MPS più elevato quando si consumano proteine [31] e per raggiungere il picco di MPS può essere necessaria un’adeguata dose di Leucina “soglia” [32]. Diversi studi hanno esaminato il dosaggio proteico necessario per massimizzare la MPS dopo l’allenamento [69,70,71]. In uno studio sono stati consumati 0, 5, 10, 20 o 40g di proteine d’uovo intere dopo l’esercizio contro-resistenza della parte inferiore del corpo, con 20g che stimolavano al massimo la MPS [69]. Risultati simili sono stati riscontrati anche in un altro studio, in cui 20 g di siero di latte sono stati sufficienti a stimolare al massimo i tassi post-assorbitivi di MPS sia a riposo che dopo un lavoro unilaterale delle gambe all’80% del 1RM [70]. Inoltre, 40g di siero di latte non hanno prodotto ulteriori aumenti di MPS in questo studio e hanno portato all’ossidazione amminoacidica e alla produzione di urea.
Tuttavia, uno studio recente ha rilevato che, durante l’esecuzione di esercizi contro-resistenza per tutto il corpo al 75% del 1RM, 40g di siero di latte hanno prodotto una risposta MPS significativamente più elevata rispetto a 20g [71]. Esiste quindi una relazione tra il volume di tessuto muscolare danneggiato e stimolato e l’assunzione adeguata di proteine. È interessante notare che gli autori di una meta-analisi del 2013 hanno osservato che, nonostante gli studi con traccianti a breve termine mostrassero risposte nella MPS maggiori quando le proteine venivano consumate nella “finestra anabolica” post-allenamento, negli studi longitudinali sull’allenamento non è stato riscontrato alcun effetto significativo sull’ipertrofia quando si controllava l’apporto proteico totale giornaliero, indipendentemente dal fatto che le proteine fossero consumate all’interno della “finestra anabolica” o al di fuori di essa [72].
Nutrient Timing:
Analogamente, i ricercatori di uno studio tracciante a breve termine che ha esaminato il dosaggio delle proteine nel corso di 12 ore hanno riportato una maggiore area sotto la curva della MPS quando sono state consumate quattro dosi di proteine del siero di latte da 20g ogni tre ore rispetto a due dosi da 40g a distanza di sei ore e otto dosi da 10g ogni ora e mezza [73]. In teoria, data la soglia oltre la quale le proteine supplementari consumate in una singola seduta non contribuiscono ulteriormente alla MPS [69] e a causa del “periodo refrattario” postprandiale durante il quale la MPS non può essere nuovamente stimolata al massimo [67], si potrebbe concludere che un bodybuilder dovrebbe raggiungere, ma non superare, questa dose soglia ogni poche ore per massimizzare l’ipertrofia a lungo termine. Tuttavia, gli autori di una review sistematica del 2018 sugli integratori proteici, comprendente 34 studi randomizzati e controllati, hanno riportato guadagni di massa magra simili tra i gruppi che utilizzavano un programma di dosaggio con i pasti (che comportava un minor numero di dosi di proteine di entità elevata) e tra i pasti (che comportava un maggior numero di dosi di proteine di entità moderata) [74].
È interessante notare che i dati che esaminano l’alimentazione proteica notturna mostrano uno distacco simile tra gli studi meccanicistici a breve termine e gli interventi di allenamento a lungo termine. Nel 2012 è stata condotta la prima ricerca che esaminava la risposta acuta all’alimentazione notturna con caseina [68]. Gli autori hanno riportato che 40g di caseina consumati prima di andare a letto sono stati digeriti, assorbiti e hanno stimolato la MPS e migliorato l’equilibrio proteico dell’intero corpo durante il periodo notturno in misura maggiore rispetto al placebo. Negli anni successivi sono stati pubblicati altri studi in acuto che hanno confermato [75] e riconfermato questi risultati in una popolazione più anziana [76]. Nel 2015, gli autori del primo studio longitudinale hanno riportato un aumento della forza e dell’ipertrofia in un gruppo a cui era stato somministrato un supplemento proteico notturno rispetto a un gruppo placebo [77].
Tuttavia, la quantità totale di proteine giornaliere non è stata equiparata, in quanto il gruppo con proteine notturne ha consumato 1,9g/kg/giorno, mentre il gruppo placebo ha consumato solo 1,3g/kg. È importante notare che in entrambi gli unici studi longitudinali con corrispondenza proteica che hanno confrontato l’integrazione notturna di caseina con i gruppi che hanno assunto l’integrazione prima, non sono state riportate differenze significative nell’aumento della FFM tra i gruppi [78,79]. Pertanto, la domanda è la stessa per ogni strategia di distribuzione: perché ci sono ripetuti distacchi tra gli studi meccanicistici a breve termine sulle MPS e le ricerche a lungo termine che esaminano l’effettiva ipertrofia? La risposta potrebbe risiedere nei metodi utilizzati negli studi sulla MPS, in quanto i partecipanti sono a digiuno, ricevono solo proteine in polvere in isolamento, spesso viene loro somministrato del siero di latte (che viene digerito molto rapidamente) e vengono osservati per brevi periodi. Questi contesti di laboratorio determinano tempi di digestione e cinetiche degli aminoacidi diversi da quelli che si verificano nel “mondo reale”. In particolare, in queste condizioni di laboratorio i livelli di base degli aminoacidi nel corpo sono più bassi del normale e la digestione e il successivo apporto di aminoacidi al muscolo sono più rapidi.
In condizioni di vita libera, le proteine vengono consumate principalmente da fonti alimentari intere, più volte al giorno e insieme ad altri alimenti, il che ritarda lo svuotamento gastrico. Per questi motivi, gli aminoacidi vengono titolati nel flusso sanguigno in modo più lento e costante; pertanto, in condizioni normali, le scorte sono quasi sempre prontamente disponibili [80]. Pertanto, l’efficacia della “finestra anabolica” e persino delle strategie di distribuzione delle proteine potrebbe non tradursi nella pratica. Inoltre, le limitazioni specifiche del laboratorio si estendono anche agli studi sull’alimentazione notturna. Si consideri, ad esempio, che 26g di proteine provenienti da una bistecca magra determinano un aumento sostenuto della MPS che dura almeno sei ore (l’intero periodo di tempo studiato) [81].
Inoltre, 26g sono solo il ~37% della dose di proteine contenuta in media in una cena americana [82], che richiederebbe più tempo per essere digerita a causa della maggiore porzione di proteine e dell’aggiunta di fibre, lipidi e altri nutrienti che ritarderebbero ulteriormente la digestione [80]. Pertanto, il tipico pasto finale potrebbe già soddisfare lo scopo di un frullato di caseina. Detto questo, nonostante queste discrepanze tra MPS e risultati della composizione corporea, non c’è nulla di male nel tentare queste strategie, soprattutto se attuate in modo pragmatico e senza introdurre ulteriori oneri logistici nel proprio programma quotidiano.
Pertanto, potrebbe essere prudente consigliare ai bodybuilder di suddividere l’assunzione giornaliera di 1,6-2,2 g/kg di proteine in più pasti contenenti ciascuno ~0,40-0,55g/kg [80] e di fare in modo che uno di questi pasti avvenga entro 1-2 ore prima o dopo l’allenamento, mentre un’alimentazione costituita da una fonte proteica e non proteica venga consumata 1-2 ore prima di dormire. Ad esempio, un bodybuilder di 90 kg potrebbe consumare 40-50g di proteine alle 8-9 del mattino per la colazione, allenarsi alle 11, consumare 40-50g di proteine alle 12-13 per il pranzo/post-allenamento, 40-50g di proteine a cena tra le 17-18, e poi un pasto finale di 40-50g di proteine non contenenti fonti proteiche grasse alle 21-10 prima di andare a letto entro le 23.
I carboidrati consumati prima dell’allenamento sono spesso una strategia utilizzata dagli atleti per migliorare le prestazioni negli esercizi ad alta intensità. La completa risintesi del glicogeno può essere raggiunta entro 24 ore da un allenamento che depaupera il glicogeno se si consumano quantità sufficienti di carboidrati [83]. Tuttavia, solo il 24-40% del glicogeno muscolare viene esaurito dopo un allenamento contro-resistenza [59,60]. Pertanto, una quantità di ≥3-5g/kg di carboidrati al giorno sarebbe probabilmente sufficiente per la risintesi del glicogeno. Questo elevato apporto giornaliero di carboidrati probabilmente riduce anche l’impatto della tempistica dei carboidrati pre-allenamento sulle prestazioni dell’esercizio.
Inoltre, per i bodybuilder che non hanno bisogno di enfatizzare il rifornimento di glicogeno, le proteine aumentano la MPS post-allenamento a livelli massimi anche senza l’aggiunta di carboidrati [86,87]. Anche se il consumo di carboidrati nel post-allenamento non è certo dannoso, è improbabile che questo favorisca l’ipertrofia a lungo termine, come discusso in precedenti review [1,88]. Pertanto, è meglio concentrarsi sul consumo di un’adeguata quantità di carboidrati giornalieri e basare la distribuzione dei carboidrati intorno all’allenamento sulle preferenze personali.
Supplementazione OTC:
In un recente sondaggio condotto tra i bodybuilder, è stato riportato che tutti i partecipanti assumevano integratori alimentari [9]. Gli integratori alimentari più comuni erano: integratori di proteine (86%), creatina (68%), aminoacidi a catena ramificata (67%), glutammina (42%), vitamine (40%), olio di pesce (37%) e prodotti contenenti caffeina/efedrina (24%).
Sebbene gli integratori proteici siano molto popolari tra i bodybuilder, vengono utilizzati prevalentemente come gli alimenti interi per raggiungere gli obiettivi proteici. Pertanto, non verranno discussi in dettaglio. I lettori sono invitati a leggere la posizione dell’ISSN su questo argomento [89]. Inoltre, la trattazione di tutti gli integratori comunemente utilizzati dai bodybuilder esula dallo scopo di questo articolo. L’attenzione si concentrerà piuttosto sugli integratori alimentari che potrebbero potenzialmente produrre un effetto ergogenico e sugli integratori che possono garantire un apporto sufficiente di micronutrienti e acidi grassi essenziali.
Creatina Monoidrato:
La Creatin-fosfato si trova in alte concentrazioni nel muscolo scheletrico e cardiaco, dove agisce come fonte di energia [90]. La Creatina può essere ottenuta anche attraverso la dieta nei soggetti che consumano carne; tuttavia, le concentrazioni di Creatina nella carne si riducono con la cottura [91].
Numerosi studi hanno osservato un aumento della massa e della forza muscolare in seguito a fasi di carico di Creatina, in genere di 20g al giorno per circa una settimana, spesso seguite da fasi di mantenimento di 2-3g di Creatina al giorno [92]. Tuttavia, la fase di carico potrebbe non essere necessaria. È stato dimostrato che la saturazione della Creatina muscolare dopo un’integrazione di 3g di Creatina Monoidrato per 28 giorni è simile al consumo di Creatina Monoidrato dopo la tipica fase di carico [93].
La maggior parte degli individui non raggiunge i 3g giornalieri con la dieta e può essere necessaria un’integrazione. Esistono numerose forme di Creatina negli integratori in commercio, tra le quali la Creatina Monoidrato è la più studiata. Le versioni più recenti di Creatina, come la kre-alkalyn [94] e la Creatina etil-estere [95], non si sono dimostrate superiori alla Creatina Monoidrato, nonostante abbiano in genere un prezzo più elevato. Pertanto, si raccomanda il consumo di 3-5g di Creatina Monoidrato al giorno. La tempistica di assunzione della Creatina non sembra avere importanza, poiché la saturazione delle riserve di Creatin-fosfato richiede circa 28 giorni per raggiungere le concentrazioni massime quando si consumano 3g al giorno e non ha un effetto in acuto [93].
Caffeina:
Uno degli integratori alimentari più utilizzati dai bodybuilder sono gli stimolanti, in particolare la Caffeina [9]. Oltre ad aumentare l’eccitazione [96], la Caffeina può ridurre il dolore e lo sforzo percepito durante l’esercizio [97] e migliora la gestione del Calcio, aumentando la potenza [98]. Studi sull’allenamento contro-resistenza hanno rilevato che la Caffeina riduce la fatica e aumenta la forza [99,100]. Tuttavia, non tutti gli studi hanno dimostrato un effetto ergogenico sull’allenamento contro-resistenza [101]. Gli studi che hanno dimostrato un effetto ergogenico hanno utilizzato dosaggi elevati di caffeina (5-6 mg/kg), che sono al limite superiore di quello che è considerato un dosaggio sicuro [99,100]. Tuttavia, può essere consigliabile consumare il dosaggio minimo efficace per individuo, poiché l’assunzione regolare può generare tolleranza [102]. A causa dell’effetto acuto della Caffeina, è consigliabile assumerla circa 1 ora prima dell’esercizio fisico [99]. Tuttavia, l’emivita della Caffeina è di circa 3-9 ore; pertanto, può essere consigliabile consumare la Caffeina all’inizio della giornata per favorire un sonno sano se l’esercizio fisico viene svolto più tardi nel corso della giornata [103]. Sono necessarie ulteriori ricerche per trovare un consenso sull’uso della Caffeina nell’allenamento contro-resistenza, ma sulla base delle prove attuali un dosaggio di 5-6 mg/kg consumato prima dell’esercizio potrebbe produrre un effetto ergogenico sulle prestazioni nell’allenamento contro-resistenza.
Beta-Alanina:
È stato dimostrato che l’ingestione di 4-6 g di beta-alanina aumenta i livelli di carnosina muscolare [104]. La carnosina agisce come tampone del pH nel muscolo scheletrico e può ritardare l’inizio dell’affaticamento muscolare durante l’esercizio ad alta intensità [105]. Una meta-analisi ha concluso che la beta-alanina potrebbe produrre effetti ergogenici durante l’esercizio ad alta intensità della durata di 60-240 secondi [104]. Inoltre, non sono stati riscontrati effetti benefici negli esercizi di durata inferiore a 60 secondi. La maggior parte degli studi inclusi nella meta-analisi riguardava l’esercizio di resistenza.
Tuttavia, è dimostrato che l’integrazione di beta-alanina può migliorare la resistenza muscolare negli atleti allenati alla resistenza [105] e può migliorare la composizione corporea [106]. Sono necessari ulteriori studi per esaminare l’effetto ergogenico della beta-alanina sulla composizione corporea e sulle prestazioni. Tuttavia, dato che i bodybuilder si allenano spesso con più di 10 ripetizioni per serie e spesso includono tecniche di intensità come drop set, pause di riposo, myo reps e altre, la beta-alanina potrebbe apportare un beneficio alla resistenza di queste serie [9].
Pertanto, potrebbe essere ragionevole per un bodybuilder consumare 3-5 g di beta alanina al giorno durante le fasi di allenamento ad alte ripetizioni o nelle fasi di allenamento in cui si incorporano diverse tecniche di intensità che prolungano la durata di un set. Come la creatina monoidrato, la beta-alanina non ha un effetto acuto, in quanto le concentrazioni di carnosina muscolare richiedono circa 4 settimane per raggiungere concentrazioni tali da produrre un effetto ergogenico, a condizione che se ne consumi una quantità sufficiente al giorno [104].
Citrullina Malato:
Recentemente, la Citrullina Malato ha guadagnato popolarità tra i bodybuilder. Il potenziale effetto ergogenico è dovuto all’aumento del flusso ematico al muscolo, alla produzione di ATP e alla potenziale capacità della Citrullina Malato di agire come agente tampone [107]. È stato dimostrato che il consumo di 8g di Citrullina Malato aumenta le ripetizioni fino al cedimento del 50% [107,108,109,110], riduce l’indolenzimento muscolare del 40% [107] e migliora la forza massimale e la potenza anaerobica [111].
Tuttavia, non tutti gli studi hanno osservato effetti ergogenici del consumo di Citrullina Malato. Due studi recenti non hanno mostrato un miglioramento delle prestazioni, un aumento della risposta del gonfiore muscolare dovuto all’allenamento, un’attenuazione della fatica o un aumento dell’attenzione e dell’energia in seguito all’integrazione di Citrullina Malato in uomini allenati contro-resistenza a livello amatoriale [112,113].
Una recente meta-analisi di Trexler et al. ha analizzato 12 studi sullla CM per le prestazioni di forza e potenza [114]. Sebbene abbiano riscontrato solo una piccola dimensione dell’effetto (0,20), hanno concluso che questo potrebbe essere rilevante per gli atleti di alto livello in cui i risultati delle competizioni si decidono su margini ridotti, come i culturisti agonisti di alto livello. Si consiglia di assumere la Citrullina Malato circa 60 minuti prima dell’esercizio fisico per consentire un assorbimento sufficiente.
Sono necessarie ulteriori ricerche per determinare l’efficacia della Citrullina Malato nell’esercizio contro-resistenza. Allo stato attuale, i dati indicano un effetto benefico o neutro sulle prestazioni. Pertanto, sulla base delle prove attuali, 8g al giorno di Citrullina Malato consumati prima dell’esercizio potrebbero avere dei benefici interessanti per i bodybuilder.
Alfa-GPC:
L’Alfa-GPC (alfa-glicerofosfocolina o colina alfoscerato) è un fosfolipide contenente colina. Quando viene ingerita, l’Alfa-GPC viene metabolizzata in colina e glicerolo-1-fosfato. La colina è un precursore dell’acetilcolina, un neurotrasmettitore coinvolto nella memoria, nell’attenzione e nella contrazione dei muscoli scheletrici. Il glicerolo-1-fosfato serve a sostenere le membrane cellulari.[https://pubmed.ncbi.nlm.]
L’Alfa-GPC sembra attraversare facilmente la barriera emato-encefalica e viene assorbito rapidamente. Attualmente è il miglior colinergico per aumentare i livelli plasmatici e cerebrali di colina.[https://pubmed.ncbi.nlm.]
L’integrazione orale di Alfa-GPC è interessante soprattutto per scopi nootropici o di potenziamento cognitivo. Esistono numerosi studi sui roditori che supportano questo effetto, ma non è ancora stato dimostrato negli esseri umani altrimenti sani. Negli anziani affetti da demenza lieve o moderata – che comporta un’alterazione della neurotrasmissione colinergica – l’Alfa-GPC migliora i sintomi cognitivi (ad esempio, disturbi della memoria e dell’attenzione).[https://pubmed.ncbi.nlm] L’Alfa-GPC può anche migliorare l’efficacia degli inibitori dell’acetilcolinesterasi (cioè i farmaci che aumentano la disponibilità di acetilcolina rallentandone la degradazione), utilizzati per il trattamento della malattia di Alzheimer.[https://pubmed.ncbi.nlm.]
Gli atleti sono un’altra popolazione che può trarre beneficio dall’integrazione di Alfa-GPC. Prove preliminari suggeriscono che l’alfa-GPC aumenta la potenza del salto verticale.[https://jissn.biomedcentral.com][https://pubmed.ncbi.nlm.] Inoltre, uno studio pilota ha riportato che l’Alfa-GPC ha aumentato il picco di forza nella panca, ma non la potenza di picco o il tasso di sviluppo della forza.[Ziegenfuss T, Landis J, Hofheins JJ Int Soc Sports Nutr.] Attualmente non è chiaro se l’Alfa-GPC aumenti la forza isometrica, ma i dati empirici e aneddotici sono incoraggianti [https://pubmed.ncbi.nlm.]
L’integrazione di un dosaggio pari a 600mg di Alpha-GPC prima di un test di potenza (spinte su panca) ha riportato un miglioramento della potenza del 14% rispetto al placebo quando assunta 45 minuti prima dell’attività; si trattava di uno studio pilota.[http://www.jissn.com] In media si è notato che il dosaggio di Alfa-GPC efficacie per trarre miglioramenti nella forza è nel range dei 300-600mg 45-30 minuti prima della seduta allenante.
Multi Vitaminico-Multi Minerale:
Storicamente, i bodybuilder hanno utilizzato diete restrittive che eliminano alimenti o interi gruppi di alimenti. Di conseguenza, sono comuni numerose carenze di vitamine e minerali. Nei bodybuilder a dieta sono state osservate carenze di Calcio, vitamina D, Zinco, Ferro e altre ancora [115,116,117]. Tuttavia, la maggior parte della letteratura sulle pratiche alimentari dei bodybuilder risale agli anni ’80 e ’90; pertanto, sono necessari dati più recenti [2].
Più di recente, le pratiche alimentari dei bodybuilder che seguono una dieta tradizionale restrittiva sono state confrontate con quelle degli agonisti che utilizzano un approccio dietetico basato sui macronutrienti, in cui nessun alimento o gruppo alimentare è off limits [118]. Non sorprende che i concorrenti che utilizzano un approccio dietetico più flessibile presentino meno carenze di micronutrienti. In particolare, la vitamina E, la vitamina K e le proteine sono risultate significativamente inferiori nelle donne che utilizzavano approcci dietetici rigidi rispetto a quelle che utilizzavano approcci più flessibili. Nel presente articolo, specie se si parla di Off-Season, si raccomanda di utilizzare un approccio dietetico flessibile, in cui nessun alimento o gruppo viene eliminato dalla dieta.
In questo modo, è meno probabile che si verifichino carenze di micronutrienti, soprattutto se si considera che le atlete in Off-Season hanno a disposizione una maggiore quantità di calorie rispetto a quelle a dieta per un contest, il che dovrebbe consentire loro di incorporare una maggiore varietà di alimenti.
Ciononostante, può essere consigliabile raccomandare un integratore multivitaminico/minerale a basso dosaggio (≤100% RDA) come misura di sicurezza per prevenire eventuali carenze di micronutrienti, sottolineando al contempo il consumo di una buona varietà di alimenti al giorno per soddisfare il fabbisogno di micronutrienti.
Omega 3 (EPA-DHA):
Gli acidi grassi polinsaturi con un doppio legame a tre atomi di distanza dal gruppo metilico terminale sono noti come ω-3 o acidi grassi omega-3 (O3). Un basso apporto di O3 nelle diete occidentali rispetto ad altre fonti di grassi alimentari (come gli acidi grassi omega-6) è associato a un peggioramento della salute multispettrale negli studi epidemiologici [119]. Pertanto, è interessante concentrarsi specificamente sulle modifiche della dieta per fornire acidi eicosapentaenoici e docosaesaenoici (EPA e DHA) – la carenza alimentare più comune nel mondo occidentale; ma vale la pena notare che la misurazione, l’interazione e l’effetto di O3 e acidi grassi omega-6 in relazione alla salute non sono chiari e vanno oltre lo scopo di questo articolo. Per una rassegna si rimanda ad altra pubblicazione [120].
Oltre alla salute, c’è interesse per i potenziali effetti anabolici degli integratori di EPA e DHA [121], che di solito vengono forniti attraverso l’olio di pesce o, in alcuni casi, l’olio di alghe. Tuttavia, ci sono dati contrastanti sulla capacità dell’olio di pesce di aumentare la risposta della sintesi proteica muscolare all’ingestione di proteine. Mentre un articolo di revisione del 2014 ha evidenziato una serie di studi secondo cui l’olio di pesce può aumentare la risposta [122], uno studio recente non ha rilevato alcun effetto sulla risposta della MPS a una sessione di allenamento contro-resistenza e all’ingestione di proteine dopo l’allenamento [123]. Inoltre, i dati sull’ipertrofia longitudinale sono pochi [124] e gli studi sulle prestazioni dell’allenamento contro-resistenza sono contrastanti [125] e in gran parte non applicabili o difficili da valutare a causa dell’uso di partecipanti non allenati o di allenamenti non standardizzati ed ecologicamente non realistici rispetto al bodybuilding.
In una recente review che affronta specificamente la questione se gli integratori di O3 possano o meno aumentare l’ipertrofia [126], gli autori hanno concluso che attualmente non ci sono prove sufficienti per fare tale affermazione. Sebbene siano necessarie ulteriori ricerche prima di poter raccomandare l’integrazione di O3 (o di alterazioni della dieta) a fini di costruzione muscolare, i benefici per la salute dell’integrazione di O3 sono degni di nota. Ad esempio, recenti meta-analisi hanno riportato che l’integrazione di olio di pesce riduce i sintomi della depressione [127], diminuisce il rischio di morte cardiaca [128], riduce la pressione sanguigna [129] e diminuisce la circonferenza vita [130]. Pertanto, gli atleti estetici possono prendere in considerazione l’integrazione giornaliera di olio di pesce (o di alghe) (1.5-2.5g di EPA/DHA) per la salute generale e multi spettro, ma sono necessari studi futuri per formulare raccomandazioni relative alle prestazioni nel bodybuilding.
Acido Arachidonico (AA):
L’Acido Arachidonico (AA) è l’acido grasso omega-6 più rilevante dal punto di vista biologico e, nella membrana lipidica di una cellula, è l’acido grasso che viene confrontato con i due acidi grassi dell’olio di pesce (EPA e DHA) nella costituzione di un rapporto omega-3:6. Dati recenti suggeriscono un’assunzione giornaliera di 50-250mg di Acido Arachidonico[https://www.ncbi.nlm.][https://www.ncbi.nlm.][https://www.ncbi.nlm.][https://www.ncbi.nlm.] con alcune fonti che stimano livelli fino a 500mg al giorno;[https://www.ncbi.nlm.] l’assunzione di Acido Arachidonico sembra essere inferiore nei vegetariani[https://www.ncbi.nlm.].
Si ritiene che l’Acido Arachidonico sia importante per il metabolismo del muscolo scheletrico, poiché si pensa che i fosfolipidi della membrana del sarcoplasma riflettano la dieta,[https://www.ncbi.nlm.][https://www.ncbi.nlm.] l’allenamento stesso sembra alterare il contenuto di fosfolipidi del muscolo (indipendentemente dalla composizione delle fibre muscolari[https://www.ncbi.nlm.] e associato a un rapporto omega 6:3 più basso[https://www.ncbi.nlm.][https://www.ncbi.nlm.]) e gli eicosanoidi dell’Acido Arachidonico interagiscono con la sintesi proteica muscolare attraverso i loro recettori.
L’Acido Arachidonico segnala la sintesi proteica muscolare attraverso una via dipendente dalla COX-2 (che suggerisce il coinvolgimento delle prostaglandine)[https://www.ncbi.nlm.] che è associata ad aumenti sia della prostaglandina E2 (PGE2) che del PGF(2α),[https://www.ncbi.nlm.][https://www.ncbi.nlm.][https://www.ncbi.nlm.][https://www.ncbi.nlm.] anche se l’incubazione con PGE2 o PGF(2α) isolati non sembra replicare pienamente gli effetti ipertrofici dell’Acido Arachidonico. [https://www.ncbi.nlm.] PGE2 e PGF(2α) sono indotti anche dall’esercizio fisico (nello specifico, dallo stiramento delle cellule muscolari in vitro[https://www.ncbi.nlm.]) ed è stato osservato sia nel siero[https://pubmed.ncbi.nlm.][https://www.ncbi.nlm.] che a livello intramuscolare (quadruplicato, da 0,95+/-0,26ng/mL a 3,97+/-0. La capacità del riflesso da stiramento di aumentare le concentrazioni di PGE2 e PGF(2α)[https://www.ncbi.nlm.] potrebbe essere dovuta semplicemente al fatto che lo stiramento aumenta l’attività delle COX2.[https://www.ncbi.nlm.][https://www.ncbi.nlm.]
Va notato che l’integrazione di 1.500mg di Acido Arachidonico (rispetto a una dieta di controllo contenente 200mg dello stesso) per 49 giorni ha aumentato la secrezione di PGE2 da parte di cellule immunitarie stimolate (del 50-100%) in giovani uomini altrimenti sani,[https://www.ncbi.nlm.] ma la rilevanza di questo studio per il muscolo scheletrico non è nota. Questo studio ha anche osservato che, senza stimolazione, non c’erano differenze significative tra i gruppi.[https://www.ncbi.nlm.] Altrove, è stata osservata una tendenza all’aumento delle concentrazioni sieriche di PGE2 a riposo in uomini allenati a cui sono stati somministrati 1.000mg di Acido Arachidonico per 50 giorni.[https://www.ncbi.nlm.]
L’Acido Arachidonico, attraverso gli eicosanoidi noti come PGF(2α) e PGE2, stimola la sintesi proteica muscolare. Sono prodotti a partire dall’Acido Arachidonico, ma normalmente non formano i rispettivi eicosanoidi per la costruzione del muscolo finché la cellula non viene stimolata da un fattore di stress (come il riflesso di stiramento di una cellula muscolare) che ne induce la produzione.
Il recettore per il PGF(2α) (recettore FP) sembra essere sovraregolato dagli inibitori della COX1 (l’acetaminofene utilizzato in questo studio)[https://www.ncbi.nlm.] e si ritiene che una maggiore segnalazione del PGF(2α) sia alla base del miglioramento della sintesi proteica muscolare osservato nei soggetti anziani con farmaci antinfiammatori. La supplementazione di Acido Arachidonico non sembra influenzare la quantità di recettori FP nei giovani;[https://www.ncbi.nlm.] mentre l’esercizio fisico stesso può aumentare il contenuto di recettori EP3, né gli inibitori della COX1[https://www.ncbi.nlm.] né l’Acido Arachidonico[https://www.ncbi.nlm.] sembrano influenzarlo ulteriormente.
Tuttavia, è stato riscontrato che l’uso di inibitori della COX2 (nei giovani) sopprime l’aumento di PGF(2α) indotto dall’esercizio fisico (Ibuprofene e Acetaminofene)[https://www.ncbi.nlm.][https://www.ncbi.nlm.] e di PGE2,[https://www.ncbi.nlm.] il che si pensa sia dovuto al fatto che la conversione da PGH2 in questi metaboliti dipende dall’attività della COX2.
Poiché la produzione di questi eicosanoidi dipende dall’enzima COX2, si ritiene che l’inibizione di questo enzima riduca gli effetti anabolizzanti dell’esercizio fisico se assunto prima dello stesso.
L’acido arachidonico (così come l’EPA dall’olio di pesce) non ha compromesso l’assorbimento del glucosio nelle cellule muscolari isolate e 10μM di acido grasso sono in grado di attenuare la resistenza all’Insulina indotta dai grassi saturi; [https://pubmed.ncbi.nlm.] un fenomeno osservato con i grassi saturi a 18 o più catene di carbonio[https://www.ncbi.nlm.] che non sembra applicarsi agli acidi grassi polinsaturi di uguale lunghezza di catena[https://www.ncbi.nlm.][https://www.ncbi.nlm.] ed è probabilmente legato all’aumento delle ceramidi intracellulari[https://www.ncbi.nlm.] che compromettono la segnalazione di Akt[https://www.ncbi.nlm.][https://www.ncbi.nlm.] e riducono l’assorbimento di glucosio mediato da GLUT4 con l’Insulina.[https://www.ncbi.nlm.]
L’Acido Arachidonico e i grassi polinsaturi omega-3 sono entrambi associati a una migliore sensibilità all’Insulina delle cellule muscolari, che potrebbe essere secondaria alla riduzione dei livelli di grassi saturi nella membrana lipidica e quindi alla riduzione delle concentrazioni intracellulari di ceramidi. È possibile che ciò non sia correlato agli eicosanoidi o al rapporto omega-3:6.
In 31 uomini allenati, sottoposti a un programma di sollevamento pesi e a una dieta standardizzata (500kcal in eccesso con 2g/kg di proteine) con 1g di Acido Arachidonico al giorno o placebo, l’integrazione per 50 giorni è sembrata aumentare la potenza di picco (7,1%) e la potenza media (3,6%) al test di Wingate, ma non è riuscita a influenzare positivamente la massa muscolare o le misure di potenza del sollevamento pesi (bench press e leg press).[https://www.ncbi.nlm.]
Attualmente non ci sono prove sufficienti per raccomandare una dose ideale di integrazione di Acido Arachidonico, ma aneddoticamente si usa un dosaggio di circa 1.500 mg da assumere 45 minuti prima dell’allenamento per un periodo medio di 8 settimane. Non è certo che si tratti di una dose ottimale o che sia necessaria la tempistica.
Va inoltre notato che per le persone affette da patologie infiammatorie croniche, come l’artrite reumatoide o le malattie infiammatorie intestinali, la dose ideale di Acido Arachidonico può essere in realtà una sua restrizione dietetica. Nei casi di malattie infiammatorie, l’integrazione di Acido Arachidonico è probabilmente controindicata.
Raccomandazioni per gli integratori alimentari e il dosaggio per i bodybuilder in Off-Season:
Creatina Monoidrato= 3-5g/die;
Beta-Alanina= 3-5g/die;
Citrullina Malato= 8g/pre-workout;
Alfa-GPC= 300-600mg/pre-workout;
Caffeina= 5-6mg/Kg/pre-workout (media standard tra 200 e 600mg/die);
Multi Vitaminico – Multi Minerale= ≤100% RDA/die;
Omega 3 (EPA-DHA)= 1.5-2.5g/die;
Acido Arachidonico= 1.5g/pre-workout.
Supplementazione PEDs:
Una cosa occorre premettere prima di procedere con la descrizione delle molecole più utilizzate nel contesto della Off-Season: non esistono PEDs esclusivamente confinabili in uno dei contesti della programmazione di un bodybuilder. Esiste il grado di versatilità il quale sta ad indicare quanto una molecola possa essere gestita con facilità in situazioni preparatorie differenti. Esistono molecole che per caratteristiche possono dare vantaggi maggiori in Off-Season/Bulk per via di alcune loro caratteristiche che in altro contesto, per esempio il pre-contest, risulterebbero più complesse da gestire. Ma questo non significa che tali molecole siano generalemnte da considerarsi “off-limitz” in un altra fase della preparazione annuale.
Premesso ciò, l’attenzione in questo paragrafo si concentrerà sui principali PEDs usati in Off-Season.
Tra tutti gli AAS, il Testosterone è quello che non ha bisogno di particolari presentazioni. Si tratta dell’ormone sessuale maschile per antonomasia. Nell’uomo, il Testosterone svolge un ruolo fondamentale nello sviluppo dei tessuti riproduttivi maschili, come i testicoli e la prostata, oltre a promuovere le caratteristiche sessuali secondarie, come l’aumento della massa muscolare e ossea e la crescita dei peli. Inoltre, in entrambi i sessi, il Testosterone è coinvolto nella salute e nel benessere, compresi gli stati d’animo, il comportamento e la prevenzione dell’osteoporosi in cooperazione con l’Estradiolo. Livelli insufficienti di Testosterone negli uomini possono portare ad anomalie, tra cui la fragilità e la perdita ossea.
In generale, il Testosterone promuove la sintesi proteica e quindi la crescita dei tessuti dotati di recettori per gli androgeni. Il Testosterone può essere descritto come avente effetti virilizzanti e anabolizzanti (anche se queste descrizioni categoriali sono in qualche modo arbitrarie, poiché vi è una grande sovrapposizione reciproca tra di essi).
Gli effetti anabolizzanti comprendono la crescita della massa e della forza muscolare, l’aumento della densità e della resistenza ossea e la stimolazione della crescita lineare e della maturazione ossea.
Gli effetti androgeni comprendono la maturazione degli organi sessuali, in particolare del pene, e la formazione dello scroto nel feto, e dopo la nascita (di solito nella pubertà) l’approfondimento della voce, la crescita dei peli del viso (come la barba) e dei peli ascellari. Molti di questi effetti rientrano nella categoria dei caratteri sessuali secondari maschili.
Al principio degli anni 30 del novecento avvenne la sintesi chimica del Testosterone, quando Butenandt e G. Hanisch pubblicarono un articolo che descriveva “Un metodo per preparare il Testosterone dal colesterolo”. Solo una settimana dopo, il terzo gruppo, Ruzicka e A. Wettstein, annunciò una domanda di brevetto in un documento “Sulla preparazione artificiale dell’ormone testicolare Testosterone (Androsten-3-one-17-ol).” Ruzicka e Butenandt ricevettero il premio Nobel per la chimica nel 1939 per il loro lavoro.
Gli studi clinici sull’uomo, che prevedevano dosi PO (per via orale) di Methyltestosterone o iniezioni di Testosterone Propionato, iniziarono già nel 1937. Il Testosterone Propionato è menzionato in una lettera all’editore della rivista Strength and Health nel 1938; questo è il primo riferimento noto a un AAS in una rivista statunitense di sollevamento pesi o Bodybuilding.
Lo sviluppo delle proprietà di costruzione muscolare del Testosterone proseguì negli anni ’40, in Unione Sovietica e nei paesi del blocco orientale come la Germania dell’Est, dove sono stati utilizzati programmi di AAS per migliorare le prestazioni dei sollevatori di pesi olimpici e di altri dilettanti già prima degli anni ’50. In risposta al successo dei sollevatori di pesi russi, il medico della squadra olimpica statunitense John Ziegler lavorò con un equipe di chimici per sviluppare un AAS con effetti androgeni ridotti. Ma questa è un altra storia.
L’uso del Testosterone nello sport si diffuse tra gli anni ’50 e gli anni ’60. Le forme utilizzate nei primi tempi erano il Testosterone in sospensione e il Testosterone Propionato, che rappresentano con il Methyltestosterone (Testosterone metilato in C-17) le forme più datate dell’ormone in questione (1935).
In ambito culturistico, il Testosterone rappresenta un AAS sufficientemente versatile in maniera dose-dipendente e sensibilità-dipendente dal momento che il dosaggio dovrebbe essere tarato in base alle risposte metaboliche soggettive alle quali è soggetto l’ormone (vedi, ad esempio, aromatizzazione in estrogeni). Questo ultimo punto è di estrema importanza al fine di evitare l’uso/abuso di AI (Inibitori dell’Aromatasi) e/o SERM (Modulatori Selettivi del Recettore degli Estrogeni). Oltre a peggiorare potenzialmente il quadro lipidico, sommandosi all’azione degli AAS utilizzati, essi riducono l’espressione epatica di IGF-1 cosa che può ridurre la risposta anabolizzante del protocollo PEDs. Nei soggetti caratterizzati da una elevata sensibilità all’attività estrogenica, le procedure applicate vedono: 1) l’uso di Raloxifene o Tamoxifene (SERM) a dosi sufficienti a impedire la comparsa o il peggioramento di una ginecomastia in stadio iniziale già presente e non ancora asportata chirurgicamente 2) l’uso di dosi fisiologiche di Testosterone come base onde evitare la comparsa di stati letargici, affaticabilità, disfunzioni sessuali ecc 3) l’uso di un “mix” composto da Testosterone e Boldenone (vedi in seguito) tale da poter usufruire della bassa e diversa sensibilità all’azione dell’Enzima Aromatasi su quest’ultimo riuscendo ad avere un controllo estrogenico teoricamente migliore (Testosterone e Boldenone mostrano qualità anabolizzanti intrinseche simili).
In un contesto Off-Season, quindi, vista l’importanza della presenza di un buon livello di Estradiolo sia sul complesso degli effetti anabolizzanti ricercati sia per la sua attività sessuale e cerebrale, il Testosterone andrebbe inizialmente calibrato sul soggetto e nel caso affiancato da dosi altrettanto ben tarate di SERM la dove ne risultasse un reale bisogno.
L’uso di un estere che garantisca un rilascio graduale della molecola (vedi Enantato o Cypionato) risulta la scelta migliore al fine di creare una soglia ematica stabile e esente da picchi e cali che possono risultare controproducenti a livello psicofisico. Tenere sempre in considerazione l’emivita di una molecola è uno dei punti fondamentali per sfruttarla al meglio. Nel caso degli esteri sopra citati, una divisione del dosaggio settimanale in due somministrazioni uguali distanziate da quattro-cinque giorni l’una dall’altra risulta una pratica ottimale allo scopo di creare una soglia ematica stabile.
I dosaggi comunemente utilizzati, parlando di molecole esterificate, vanno da 200mg ad 1g a settimana. Per quanto riguarda il Testosterone in sospensione, le dosi comunemente utilizzate vanno dai 175mg ai 700mg a settimana.
Il Boldenone [1,4-androstadiene-3-one,17b-ol], commercializzato con il nome di Equipoise, Ganabol, Equigan, Ultragan, e Boldane, è uno steroide anabolizzante-androgeno spesso legato all’estere Undecylenato. Strutturalmente molto simile al Testosterone, il Boldenone differisce da questo per il doppio legame tra C1 e C2.
La Ciba brevettò il Boldenone nel 1949. Successivamente, negli anni ’50 e ’60, sviluppò diversi esteri sperimentali del farmaco. Uno di questi era il Boldenone Undecilenato, che fu introdotto per uso clinico con il marchio Parenabol e fu utilizzato alla fine degli anni ’60 e all’inizio degli anni ’70. Tuttavia, fu sospeso prima della fine degli anni ’70. Ad oggi l’uso del Boldenone è legale in alcuni paesi in campo veterinario.
Se qualcuno volesse usare 500mg di Testosterone, ma non potrebbe usare un tale dosaggio dal momento che presenta particolare difficoltà nella gestione estrogenica in specie senza l’uso di AI come Exemestane o Anastrozolo, una conclusione a cui molti superficialmente sono giunti è che si potrebbe semplicemente usare il Boldenone al dosaggio sopra citato per ridurre della metà l’attività estrogenica, ma comunque supportare un’adeguata produzione di Estradiolo. Ma quando si approfondisci l’ipotesi e la si testa sul campo, è davvero così che stanno le cose? In realtà no, o, comunque, la media delle variabili di risposta spinge a confermare una maggiore validità nel “mixare” Testosterone e Boldenone coprendo la dose base calcolata in precedenza, e con variazione di percentuale T:B ratio da 1:1 a 2:1.
Comunque, oltre a rappresentare genericamente una discreta molecola sia in in preparazione alla gara che in Off-Season, I dosaggi utilizzati si settano nel range tra i 200mg ed i 500mg a settimana, spesso abbinato ad una dose variabile (vedi sopra) di Testosterone.
Il Nandrolone, noto anche come 19-nortestosterone, è uno Steroide Androgeno Anabolizzante (AAS) utilizzato sotto forma di molecola legata a esteri come quello Decanoato (nome commerciale Deca-Durabolin) e il Fenilpropionato (nome commerciale Durabolin). Gli esteri del Nandrolone sono utilizzati nel trattamento di anemie, cachessia (sindrome da deperimento), osteoporosi, cancro al seno e per altre indicazioni mediche.
Il Nandrolone è stato sintetizzato per la prima volta nel 1950. È stato introdotto per la prima volta nel mercato farmaceutico, come Nandrolone Fenilpropionato, nel 1959, e poi come Nandrolone Decanoato nel 1962, seguito da ulteriori esteri.
Il Nandrolone ha una bassa affinità di interazione con l’Enzima Aromatasi convertendo in Estrone, un estrogeno molto meno potente dell’Estradiolo, circa 10 volte meno attivo, e, come tale, è un estrogeno relativamente debole. In una condizione di somministrazione del Nandrolone senza una base di Testosterone, i livelli di Estradiolo calerebbero marcatamente a favore di un aumento del Estrone il quale non potrebbe però sostituire nelle diverse attività tissutali il prima citato E2. Le conseguenze negative si verificherebbero dall’attività sessuale all’attività neurosteroidea.
Infatti, un effetto da non sottovalutare con l’uso di Nandrolone è il suo impatto sul SNC. L’impatto del Nandrolone sul Sistema Nervoso Centrale è stato osservato scientificamente. Nello studio intitolato “The Impact of Nandrolone Decanoate on the Central Nervous System” vengono descritti chiaramente i numerosi effetti psicologici di questa molecola. Essi comprendono e influenzano:
1- Aggressività 2- Ansia, paura e stress 3- Ricompensa e dipendenza 4- Apprendimento, memoria e capacità di lavoro 5- Locomozione e attività fisica 6- Effetti sulla HPAA (Asse Ipotalamo-Pituitaria-Surrene) 7- Effetto sui neurotrasmettitori: Recettore Acido γ-Aminobutirrico Tipo A (GABAA); Recettori 5-idrossitriptamina (5-HT) e 5-HT; Recettori della Dopamina e Recettori Oppioidi.
Questo effetto, unito alla modesta potenzialità anabolizzante se confrontata con altre molecole anche della stessa famiglia, fa pendere l’ago della bilancia verso gli svantaggi d’uso piuttosto che i vantaggi. Sebbene vi sia un rapporto tra Testosterone e Nandrolone finalizzato a ridurre la comparsa di questi effetti avversi (ratio T:N = 2:1) su un buon numero di soggetti risulta dare comunque problemi rilevanti.
Il suo uso principale in Off-Season comprende dosaggi medi tra i 200mg ed i 400mg a settimana, con un adeguato rapporto con il Testosterone. Se utilizzato a fini di recupero articolare viene usato a dosaggi di 100mg a settimana, e con tali dosaggi difficilmente emergono i problemi sopra elencati a patto che ci sia una base di Testosterone.
Il Drostanolone, noto anche come 2α-metil-5α-diidrotestosterone (2α-metil-DHT) o come 2α-metil-5α-androstan-17β-ol-3-one, è uno steroide androstano sintetico e un derivato del DHT. Si tratta nello specifico di DHT con un gruppo metile in posizione C2α. La forma esterificata Drostanolone Propionato è stata usata in passato nel trattamento del cancro al seno nelle donne per via della sua attività antiestrogenica. Questa azione il Drostanolone la esplica sia agendo come antagonista del recettore degli estrogeni e sia come inibitore dell’Enzima Aromatasi. Ed è proprio per questo motivo che una molecola generalmente relegata all’uso in “Cut” o pre-gara trova un suo uso funzionale in Off-Season. La sua attività AI è comunque moderata ma sufficiente in un buon numero di soggetti per evitare l’aggiunta di SERM e/o AI di altro genere. L’attività AI moderata sembra non incidere negativamente in modo sensibile sull’Asse GH/IGF1.
L’effetto miotrofico risulta simile a quello osservato con il Methenolone, in generale moderatamente inferiore al Testosterone. I dosaggi utilizzati in Off-Season per il controllo estrogenico sono nel range dei 200-400mg a settimana (diviso in due iniezioni distanziate da 4-5 giorni) per l’estere Enantato, mentre per il Propionato 150-350mg a settimana (dosi a giorni alterni o giornaliere).
Il Trenbolone, noto anche come 19-nor-δ9,11-testosterone o come estra-4,9,11-trien-17β-ol-3-one, è uno steroide sintetico e un derivato del Nandrolone (19-nortestosterone) sintetizzato per la prima volta nel 1963. Si tratta nello specifico di Nandrolone con due doppi legami aggiuntivi nel nucleo steroideo. Gli esteri del Trenbolone, che hanno un estere in posizione C17β, includono il Trenbolone Acetato, il Trenbolone Enantato, Il Trenbolone Hexahydrobenzylcarbonato e il Trenbolone Undecanoato. Il Trenbolone Acetato (marchi Finajet, Finaplix, e altri) e il Trenbolone Hexahydrobenzylcarbonato (marchi Parabolan, Hexabolan), sono o sono stati commercializzati per uso veterinario e clinico nell’uomo. Il Trenbolone Acetato è utilizzato in medicina veterinaria nel bestiame per aumentare la crescita muscolare e l’appetito degli animali, mentre il Trenbolone Hexahydrobenzylcarbonato è stato utilizzato in passato a livello clinico nell’uomo, ma ora non è più commercializzato.
Si tratta di uno degli AAS più versatili in assoluto, con un ottima resa tanto in preparazione alla gara quanto in Off-Season. L’enorme potenziale anabolizzante del Trenbolone, così come dei suoi analoghi, è stato riportato fin dagli anni ’60. La sua diffusione nel Bodybuilding è iniziata circa negli anni ’80 del secolo scorso. La sua elevata potenzialità miotrofica, lipolitica e di spinta mentale lo resero in poco tempo estremamente popolare tra i culturisti.
In Off-Season viene utilizzato nelle sue forme eseterificate Enantato e Hexahydrobenzylcarbonato a dosaggi nell’ordine dei 100-400mg a settimana (divisa in due somministrazioni distanziate l’una dall’altra da 4-5 giorni), sebbene il trend d’oltre oceano è arrivato a dosaggi decisamente eccessivi e nell’ordine del grammo. Per l’esetere Acetato i dosaggi medi vanno da 150mg a 350mg a settimana con dosaggi a giorni alterni o giornalieri.
E’ necessario ricordare ai lettori che gli effetti collaterali a livello del SNC possono verificarsi in alcuni punti come nel caso del Nandrolone sebbene i vantaggi rendano il Trenbolone più bilanciato tra sides e vantaggi.
Il Trestolone, noto anche come 7α-metil-19-nortestosterone (MENT) o come 7α-metilestr-4-en-17β-ol-3-one, è uno steroide sintetico e un derivato del Nandrolone (19-nortestosterone). È una forma modificata del Nandrolone con un gruppo metile in posizione C7α. Tra gli AAS strettamente correlati vi sono il 7α-metil-19-norandrostenedione (MENT dione, trestione), un pro-ormone androgeno del Trestolone, e il Dimetandrolone (7α, 11β-dimetil-19-nortestosterone), il derivato metilato C11β del Trestolone, nonché il Mibolerone (7α,17α-dimetil-19-nortestosterone) e il Dimetiltrienolone (7α,17α-dimetil-δ9,11-19-nortestosterone). Anche il progestinico Tibolone (7α-metil-17α-etinil-δ5(10)-19-nortestosterone) è strettamente correlato al Trestolone.
Il Trestolone è stato descritto per la prima volta nel 1963. Tuttavia, non è stato successivamente studiato fino al 1990. Lo sviluppo del Trestolone per un potenziale uso nella contraccezione ormonale maschile e nella terapia sostitutiva degli androgeni è stato avviato nel 1993 ed è proseguito in seguito. Non sembra che siano stati condotti ulteriori sviluppi dal 2013. Il Trestolone è stato sviluppato dal Population Council, un’organizzazione non governativa senza scopo di lucro dedicata alla salute riproduttiva.
Come AAS, il Trestolone è un agonista del recettore degli androgeni (AR), analogamente agli androgeni come il Testosterone e il Diidrotestosterone (DHT). Questo AAS presenta spiccate proprietà anticortisolemiche sia attraverso l’inibizione enzimatica sia per attività antagonista recettoriale. Il Trestolone non è un substrato per la 5α-reduttasi e quindi non è potenziato o inattivato nei cosiddetti tessuti “androgeni” come la pelle, i follicoli piliferi e la ghiandola prostatica. Come tale, ha un elevato rapporto tra attività anabolica e androgena, analogamente ad altri derivati del Nandrolone. Il Trestolone è un substrato per l’Aromatasi e quindi produce come metabolita l’estrogeno 7α-metilestradiolo. Tuttavia, il Trestolone ha solo una debole attività estrogenica e una quantità che sembrerebbe essere insufficiente per scopi terapici sostitutivi, come evidenziato dalla diminuzione della densità minerale ossea negli uomini trattati con esso per l’ipogonadismo.
Il potenziale anabolizzante del Trestolone ha mostrato un grado di superiorità miotrofica rispetto al Trenbolone. Le sue caratteristiche ne fanno prediligere l’uso in Off-Season/Bulk. I dosaggi utilizzati con la forma Acetato sono nell’ordine dei 150-350mg a settimana con una cadenza nelle somministrazioni a giorni alterni. Sebbene sia più rara da reperire, la forma Enantato è utilizzato nel range dei 200-400mg a settimana divisi in somministrazioni ogni 4-5 giorni.
L’Oxymetholone, noto anche come 2-idrossimetilene-17α-metil-4,5α-diidrotestosterone (2-idrossimetilene-17α-metil-DHT) o come 2-idrossimetilene-17α-metil-5α-androstan-17β-olo-3-one, è uno steroide androstanico sintetico e un derivato 17α-alchilato del DHT. L’Oxymetholone è stato descritto per la prima volta in un articolo del 1959 da scienziati della Syntex. È stato introdotto per uso medico dalla Syntex e dalla Imperial Chemical Industries nel Regno Unito con il marchio Anapolon nel 1961. L’Oxymetholone è stato introdotto anche con i marchi Adroyd (Parke-Davis) nel 1961 e Anadrol (Syntex) nel 1962. Il farmaco è stato commercializzato negli Stati Uniti nei primi anni ’60.
Come altri AAS, l’Oxymetholone è un agonista del recettore degli androgeni (AR). Non è un substrato per la 5α-reduttasi (dal momento che è già 5α-ridotto) ed è uno substrato scarso per il 3α-idrossisteroide deidrogenasi (3α-HSD), e quindi mostra un alto rapporto di attività anabolizzante rispetto all’effetto androgenico.
Data la sua derivanza dal DHT, l’Oxymetholone non è un substrato per l’Enzima Aromatasi e quindi non può essere aromatizzato in metaboliti estrogenici. Tuttavia, caratteristica unica tra i derivati del DHT, l’Oxymetholone è comunque associato a un’estrogenicità relativamente elevata ed è noto per avere il potenziale di produrre effetti collaterali estrogenici come ginecomastia (anche se non comune) e ritenzione idrica. È stato suggerito che questo può essere una conseguenza del legame diretto a l’attivazione del recettore degli estrogeni da parte dell’Oxymetholone (estrogenicità intrinseca). L’Oxymetholone non possiede alcuna attività progestinica significativa. Per via della caratteristica attività estrogenica intrinseca, con l’uso di Oxymetholone è spesso necessario l’uso di un SERM onde avere un controllo sulla aumentata attività estrogenica.
A causa della sua struttura 17α-alchilata, l’Oxymetholone è epatotossico. L’uso a lungo termine del farmaco può causare una varietà di disturbi gravi, tra cui l’epatite, il cancro al fegato e la cirrosi; pertanto si raccomandano test periodici di funzionalità epatica per coloro che assumono l’Oxymetholone a fini terapeutici. Questa molecola ha ottenuto, infatti, la nomea di essere uno tra gli AAS più epatotossici. Ciò deriva da i dosaggi comunemente, ed erroneamente, utilizzati in contesto culturistico. Si parla di dosaggi che facilmente sforano i 150mg/die.
Osservazioni e esaminazione di diversi referti di esami ematici hanno evidenziato una soglia di “vantaggio/svantaggio” a favore del primo con un dosaggio calcolato con la formula 1mg/Kg. Genericamente, però, il dosaggio standard e conservativo si attesta nel range dei 50-100mg/die per non più di 28 giorni consecutivi, al fine di ridurre l’impatto negativo sul fegato e lipidemia.
Il Methandrostenolone, noto anche come 17α-metil-δ1-testosterone o come 17α-metilandrost-1,4-dien-17β-ol-3-one, è uno steroide androstanico sintetico e un derivato 17α-alchilato del Testosterone. È una modifica del Testosterone con un gruppo metile in posizione C17α e un doppio legame aggiuntivo tra le posizioni C1 e C2. Il farmaco è anche il derivato 17α-metilato del Boldenone (δ1-testosterone) e l’analogo δ1 del Methyltestosterone (17α-metiltestosterone).
Il Methandrostenolone è stato descritto per la prima volta nel 1955. È stato sintetizzato dai ricercatori dei laboratori CIBA di Basilea, in Svizzera. La CIBA depositò un brevetto statunitense nel 1957 e iniziò a commercializzare il farmaco sotto il nome di Dianabol nel 1958 negli Stati Uniti. Inizialmente veniva prescritto alle vittime di ustioni e agli anziani. Tra i primi utilizzatori vi furono i giocatori dell’Oklahoma University e l’allenatore dei San Diego Chargers Sid Gillman, che somministrò il Dianabol alla sua squadra a partire dal 1963.
Anche se il primo a somministrare il Methandrostenolone agli atleti fu il Dr. John Ziegler, personaggio che ebbe non poca importanza nella storia dell’uso degli AAS negli Stati Uniti. Ziegler contribuì a facilitare l’adozione degli AAS in generale, e del Dianabol in particolare, da parte degli atleti americani. Ziegler fu la prima persona a somministrare il Dianabol agli atleti competitivi poco dopo la sua introduzione da parte della CIBA nel 1958. Ebbe accesso al laboratorio CIBA a Summit (New Jersey) nel corso degli anni 50’ e somministrava già ai pesisti il Testosterone Propionato per “scopi di ricerca”. Da li il passo fu breve per la diffusione a macchia d’olio di questo AAS tra i culturisti.
Data la sua principale modifica strutturale, ossia la metilazione in C-17, il Methandrostenolone mostra un aumentata stabilità del legame recettoriale aumentando così l’affinità sia al AR sia, successivamente all’aromatizzazione nel suo metabolita 17-Methylestradiolo, per i recettori estrogenici rendendo il composto molto più estrogenico del Testosterone. Tale caratteristiche migliora però il potenziale proliferativo dei AR e l’influenza positiva sulla sintesi di IGF-1. Da non dimenticare è il suo significativo impatto anticortisolemico.
Trattandosi di una molecola con una discreta tendenza all’aromatizzazione, il suo uso tipico la vede inserita nelle fasi Off-Season. Il calcolo del dosaggio, per via dati aneddotici e osservativi raccolti, lo si ottiene con la formula 5mg/12Kg di peso corporeo. Trattandosi di un composto orale metilato in C-17 se ne scoraggia l’utilizzo oltre i 28 giorni consecutivi onde ridurre l’impatto negativo su fegato e lipidemia. Data la sua emivita di circa 4h, il dosaggio giornaliero dovrebbe essere diviso in più assunzioni distribuite durante l’arco della giornata.
L’Ormone della Crescita (GH) o Somatotropina, noto anche come Ormone della Crescita Umano (hGH o HGH), è un ormone peptidico che stimola la crescita, la riproduzione e la rigenerazione cellulare nell’uomo e in altri animali. È quindi importante per lo sviluppo umano. Il GH stimola anche la produzione di IGF-1 e aumenta la concentrazione di glucosio e acidi grassi liberi nel sangue. È un tipo di mitogeno specifico solo per i recettori di alcuni tipi di cellule. Il GH è un polipeptide a catena singola di 191 aminoacidi che viene sintetizzato, immagazzinato e secreto dalle cellule somatotrope nelle ali laterali dell’ipofisi anteriore.
Una forma ricombinante di hGH, chiamata Somatropina, viene utilizzata come farmaco da prescrizione per il trattamento dei disturbi della crescita nei bambini e della carenza di Ormone della Crescita negli adulti. Molte delle funzioni dell’hGH rimangono sconosciute.
Nel suo ruolo di agente anabolizzante, l’hGH è stato utilizzato dagli sportivi agonisti almeno dal 1982, quando la sola forma disponibile era quella derivata dall’Ipofisi dei cadaveri, ed è stato vietato dal CIO e dall’NCAA. L’analisi tradizionale delle urine non è in grado di rilevare il doping con HGH, pertanto il divieto è stato applicato solo all’inizio degli anni 2000, quando sono stati sviluppati test del sangue in grado di distinguere tra hGH naturale e artificiale.
In ambiente bodybuilding, l’hGH viene utilizzato in Off-Season (dai soggetti meglio informati) a dosaggi nel range delle 4-8UI al giorno o 8-16UI a giorni alterni. La somministrazione in concomitanza con l’uso di Insulina ha mostrato effetti sinergici molto evidenti che trovano la loro origine nel miglioramento della sintesi di IGF-1 e della sua frazione libera quindi attiva. Ricordo inoltre che l’uso di hGH può causare una sottoregolazione della funzionalità tiroidea per via del feedback negativo causato da un aumento della conversione del T4 in T3 per azione del GH. L’uso di T4, nel periodo d’uso in Off-Season, è in alcuni casi una necessità.
Il Fattore di Crescita Insulino-Simile 1 (IGF-1), chiamato anche Somatomedina C, è un ormone dalla struttura molecolare simile a quella dell’insulina che svolge un ruolo importante nella crescita infantile e ha effetti anabolici negli adulti. L’IGF-1 è costituito da 70 aminoacidi in una singola catena con tre ponti disolfuro intramolecolari.
L’IGF-1 è prodotto principalmente dal fegato. La produzione è stimolata dall’Ormone della Crescita (GH). La maggior parte dell’IGF-1 è legata a una delle 6 proteine di legame (IGF-BP). L’IGFBP-1 è regolato dall’Insulina. L’IGF-1 viene prodotto durante tutta la vita; i tassi più alti di produzione di IGF-1 si verificano durante la crescita puberale. I livelli più bassi si verificano nell’infanzia e nella vecchiaia.
L’IGF-1 lega e attiva il proprio recettore, l’IGF-1R, attraverso l’espressione sulla superficie cellulare delle tirosin-chinasi recettoriali (RTK) e segnala ulteriormente attraverso molteplici cascate di trasduzione intracellulare. L’IGF-1R è l’induttore che svolge un ruolo critico nella modulazione degli effetti metabolici dell’IGF-1 per la senescenza e la sopravvivenza cellulare. L’IGF-1 è responsabile di stimolare la crescita di tutti i tipi di cellule e di provocare effetti metabolici significativi. Un importante effetto metabolico dell’IGF-1 è la sua capacità di segnalare alle cellule che sono disponibili nutrienti sufficienti per l’ipertrofia e la divisione cellulare. Questi segnali consentono inoltre all’IGF-1 di inibire l’apoptosi cellulare e di aumentare la produzione di proteine cellulari. I recettori dell’IGF-1 sono ubiquitari, il che consente che i cambiamenti metabolici causati dall’IGF-1 si verifichino in tutti i tipi di cellule. Gli effetti metabolici dell’IGF-1 sono di vasta portata e possono coordinare il metabolismo delle proteine, dei carboidrati e dei grassi in una varietà di tipi di cellule diverse. La regolazione degli effetti metabolici dell’IGF-1 sui tessuti bersaglio è coordinata anche con altri ormoni, come l’Ormone della Crescita e l’Insulina.
L’IGF-1 da DNA ricombinante è disponibile principalmente in due diversi formati/varianti, lr3 e DES. È importante ricordare che, a prescindere dalla variante, tutti funzionano a livello sistemico nell’organismo e che, nonostante la somministrazione dell’ormone per via intramuscolare direttamente in un muscolo specifico, non genererà una crescita localizzata misurabile.
Ovviamente tralascerò di descrivere l’IGF-1 bioidentico commercializzato come Mecasermina dal momento che la sua farmacocinetica è identica a quella del IGF-1 endogeno. Dirò soltanto che mediamente viene utilizzato in dosi giornaliere nel range tra 60-1.000mcg post-workout. L’emivita di questa forma di IGF-1 è di circa 5.8h.
IGF-1 LR3: Questa forma è la variante di IGF-1 più comune e molto popolare sul mercato e utilizzata da bodybuilder e atleti di altre discipline. Contiene IGF-1 bioidentico costituito dalla catena originale di 70 aminoacidi, ma con 13 aminoacidi in più all’estremità N, per un totale di 83 aminoacidi. Possiede anche una seconda modifica, in cui un’Arginina si trova in 3a posizione invece dell’Acido Glutammico originale. Il risultato di queste modifiche è che l’IGF-1 continua a svolgere la sua attività originaria sul recettore dell’IGF-1 nei tessuti corporei e ha un’affinità di legame molto bassa per le proteine leganti l’IGF menzionate in precedenza. Inoltre, presenta una vita attiva significativamente più lunga, di circa 20-30 ore, rispetto a quella dell’IGF-1 di 12-15 ore. L’insieme di questi fattori ha dimostrato che l’LR3 ha un’efficacia circa tre volte superiore a quella dell’IGF-1.
I dosaggi medi utilizzati per questa forma sono nel range dei 40-80mcg/die. A causa della sua lunga vita attiva nell’organismo, la variante LR3 non dovrebbe essere somministrata più di una volta al giorno per il semplice fatto che non risulta necessario. Nei giorni di allenamento, il dosaggio di IGF-1 è solitamente somministrato subito dopo l’allenamento. La scelta è a discrezione dell’utilizzatore, in quanto può essere benissimo somministrato sia prima che dopo (solo prima dell’allenamento o solo dopo l’allenamento). E’ possibile comunque dividere il dosaggio giornaliero in due somministrazioni nell’arco della giornata, il dosaggio giornaliero completo può essere diviso quindi a metà tra i due (ad esempio, 20mcg prima dell’allenamento e 20mcg dopo l’allenamento, per un totale di 40mcg al giorno). Nei giorni di non allenamento, può essere somministrato in qualsiasi momento della giornata.
IGF-1 DES: Conosciuto anche come DES(1-3)IGF-1, questa è la forma di IGF-1 comunemente conosciuta come ad azione molto rapida e di solito è la meno preferita tra le due. Le sue modifiche rispetto alla molecola originale di IGF-1 sono tali da farle mancare i primi 3 aminoacidi all’N terminale, il che conferisce all’IGF-1 DES un totale di 67 aminoacidi nella sua catena rispetto ai 70 originali. Questa modifica garantisce all’IGF-1 DES una ridotta affinità di legame per le proteine leganti l’IGF menzionate in precedenza, oltre a una maggiore forza di legame e potenziale miotrofico, circa dieci volte superiore a quella dell’IGF-1 originale e cinque volte superiore a quella dell’IGF-1 LR3. A differenza dell’IGF-1 LR3, l’IGF-1 DES ha un’emivita molto più breve, di circa 20-30 minuti. Grazie alla sua attività più rapida e alla maggiore forza/potenza, la variante DES dell’IGF-1 è comunemente ritenuta in grado di ottenere una crescita muscolare localizzata nel sito in cui viene iniettata. Sebbene ciò sia in parte vero, gli studi hanno dimostrato che, come l’IGF-1 in generale, agisce a livello sistemico una volta raggiunti i capillari e il flusso sanguigno. Quindi l’effetto localizzato è minimo e non significativamente differente dall’effetto sistemico.
Il dosaggio della variante DES è leggermente più variabile rispetto a quello del LR3. Per l’IGF-1 DES, il dosaggio varia da 50 a 150 mcg al giorno. A causa della sua emivita molto più breve rispetto alla variante LR3, è possibile utilizzare dosaggi più elevati con una ipotetica riduzione del rischio di effetti a lungo termine sull’organismo, anche se è necessario usare comunque cautela. Può essere utilizzato nello stesso modo dell’IGF-1 LR3 post-workout, ed è infatti comunemente usato in questo modo a causa della sua breve emivita.
Entrambe le forme di IGF-1 possono essere somministrate per via intramuscolare o sottocutanea. L’uso di una delle due forme non deve superare la durata di 30 giorni prima di una pausa di almeno 2 settimane, anche se fare pause più lunghe di 2 settimane tra un ciclo di IGF-1 e l’altro è l’opzione migliore. Questo non solo per ridurre il rischio di effetti sulla salute a lungo termine, ma anche per garantire che i recettori dell’IGF-1 tornino ad un grado di sensibilità ottimale e, quindi, a “rispondere” correttamente dopo un ciclo.
L’insulina è un ormone peptidico prodotto dalle cellule beta delle isole pancreatiche. Regola il metabolismo dei carboidrati, dei grassi e delle proteine promuovendo l’assorbimento del glucosio dal sangue nelle cellule del fegato, dei grassi e dei muscoli scheletrici. In questi tessuti il glucosio assorbito viene convertito in glicogeno attraverso la glicogenesi o in grassi (trigliceridi) attraverso la lipogenesi o, nel caso del fegato, in entrambi. La produzione e la secrezione di glucosio da parte del fegato sono fortemente inibite da alte concentrazioni di Insulina nel sangue. L’Insulina circolante influisce anche sulla sintesi di proteine in un’ampia varietà di tessuti. È quindi un ormone anabolico e anticatabolico, che promuove la conversione di piccole molecole nel sangue in grandi molecole all’interno delle cellule. Bassi livelli di Insulina nel sangue hanno l’effetto opposto, favorendo un diffuso catabolismo, soprattutto del grasso corporeo di riserva.
La maggior parte dei bodybuilder utilizza una sola forma di Insulina (ad azione rapida o ultra-rapida), anche se alcuni utilizzano anche un’Insulina a lunga durata d’azione o in monoterapia insulinica o in conbinazione con le forme ad azione rapida o ultra-rapida.
L’Humalog® (Insulina Lispro) è senza dubbio la forma di Insulina più diffusa tra i bodybuilder insieme all’Humulin-R. L’Humalog è un analogo a breve durata d’azione dell’Insulina umana, in particolare l’analogo Lys(B28) Pro(B29) dell’Insulina che si crea quando gli aminoacidi in posizione 28 e 29 sono invertiti. È considerata equipotente all’Insulina solubile normale su base unitaria, ma con un’attività più rapida. L’inizio dell’azione del farmaco in seguito alla somministrazione sottocutanea è di circa 10-15 minuti e il suo picco d’effetto viene raggiunto in 30-90 minuti. La durata d’azione totale è compresa tra 3-5 ore. L’Insulina lispro viene solitamente utilizzata come supplemento a un prodotto a base di Insulina a più lunga durata d’azione, fornendo un farmaco ad azione rapida che può essere assunto prima o subito dopo i pasti per imitare la secrezione insulinica naturale dell’organismo. Molti atleti ritengono che la sua breve finestra d’effetto la renda un farmaco insulinico ideale per scopi dopanti, in quanto la maggior parte dell’azione può essere concentrata nel periodo successivo all’allenamento sfruttando l’assimilazione dei nutrienti durante la così detta “finestra anabolica”. Proprio al fine di potenziare la “finestra anabolica”, l’Humalog viene usata in concomitanza del GH il quale viene somministrato in una tempistica tale che i due picchi di rilascio (curva ematica massima) si “incrocino” andando a creare un affetto additivo di potenziamento della sintesi epatica di IGF-1 e della sua attività per via della riduzione dei trasportatori IGFBP.
Tuttavia, l’uso di una base insulinica composta da Insuline Glargine (Lantus), con una vita attiva di 24-26.5h, la quale sembra avere effetti di maggiore affinità di legame per il recettore del IGF-1 rispetto all’Insulina umana regolare o uno dei qualsiasi altri analoghi, viene da alcuni inserita nei protocolli Off-Season.
I dosaggi di Insulina non andrebbero calcolati in modo distaccato dal piano alimentare e dal suo contenuto glucidico. Se il margine di “sicurezza” indica un assunzione di 10-15g di Carboidrati per UI di Insulina, questi non dovrebbero essere addizionati al piano alimentare già tarata in surplus calorico. Il calcolo delle unità dovrebbe essere tarato sul quantitativo glucidico della dieta e sul rapporto con il peso corporeo dell’atleta. Facciamo un semplice esempio: Soggetto di 90Kg = formula 1UI ogni 10Kg di peso = 9UI massime somministrabili per pasto e in base alla vita attiva della forma utilizzata = assicurarsi che il pasto appena successivo alla somministrazione dell’Insulina a questo dosaggio sia pari o superiore ai 90g di Carboidrati. Il monitoraggio della glicemia attraverso un glucometro è ovviamente d’obbligo in un protocollo di Insulina.
Nota: tali informazioni esposte non rappresentano in nessun modo un parere medico ne tanto meno una prescrizione e/o incentivo all’uso di sostanze dopanti e illegali. Le descrizioni presentate per i PEDs solitamente più utilizzati in Off-Season sono sintetiche sia per motivi di “Off Topic” sia per ragioni legate alla loro descrizione approfondita in altri articoli presenti nel database di questo sito. In queste pubblicazioni potrete trovare informazioni inerenti anche agli affetti collaterali connessi ad un uso/abuso “off-label” dei diversi PEDs.
Conclusioni:
Per concludere e fare una sintesi delle nozioni esposte in questo articolo, dobbiamo ricordarci che i bodybuilder in Off-Season dovrebbero concentrarsi sul consumo di una dieta leggermente ipercalorica (~10-20% sopra le calorie di mantenimento) con l’obiettivo di guadagnare ~0,25-0,5% del peso corporeo a settimana per un “Natural”, mentre nel caso di un “Doped” la soglia può spostarsi tra l’1-2% con variabili connesse a risposte genetiche differenziali e anzianità nella carriera culturistica (principiante, intermedio e avanzato). In ogni caso, in una fetta maggioritaria di praticanti, ai bodybuilder avanzati si consiglia di essere più prudenti con il surplus calorico e il tasso di aumento di peso settimanale. L’assunzione di proteine nella dieta è raccomandata a 1,6-2,2 g/kg/giorno, con particolare attenzione a una quantità sufficiente di proteine a ogni pasto (0,40-0,55 g/kg/pasto) e a una distribuzione uniforme nell’arco della giornata (3-6 pasti). Per i “Doped”, in alcuni casi, l’introito proteico può essere portato, con minimi vantaggi in contesto ipercalorico, a 2,5g/Kg con le medesime linee guida di suddivisione per numero di pasti. I grassi alimentari devono essere consumati a livelli moderati, né troppo bassi né troppo alti (0,5-1,5 g/kg/die), per evitare un rapporto fTC sfavorevole e per prevenire riduzioni dei livelli di testosterone. Nei “Doped” l’obbiettivo con i lipidi è principalmente quello di assumerne una dose necessaria, e altamente qualitativa, al fine di assimilare vitamine liposolubili, per substrato strutturale, per sintesi di eicosanoidi (vedi assunzione EPA, DHA e AA), protezioni epidermide e capelli; di conseguenza attenersi ad un dosaggio medio pari a 35-50g/die. Dopo che le calorie sono state distribuite tra Proteine e Grassi, le restanti calorie dovrebbero provenire dai Carboidrati, assicurandosi di consumarne una quantità sufficiente (≥3-5 g/kg/giorno). Si possono ottenere benefici maggiori consumando proteine (0,40-0,55 g/kg/pasto) in prossimità delle sessioni di allenamento (1-2 ore prima dell’esercizio ed entro 1-2 ore dopo l’esercizio). È opportuno prendere in considerazione la Creatina Monoidrato (3-5 g/giorno) e la Caffeina (5-6 mg/kg), in quanto possono produrre effetti ergogenici per i bodybuilder. Inoltre, Beta-Alanina (3-5 g/die) e Citrullina Malato (8 g/die) sono integratori alimentari che possono essere presi in considerazione in quanto potenzialmente utili per i bodybuilder, a seconda dei regimi di allenamento individuali. I bodybuilder che non sono in grado di assumere un apporto sufficiente di micronutrienti e acidi grassi essenziali nella loro dieta dovrebbero prendere in considerazione l’integrazione di questi nutrienti per evitare carenze. Il limite principale di questo articolo è la mancanza di studi su larga scala e a lungo termine sui bodybuilder durante la Off-Season. Sono necessarie ulteriori ricerche su questa popolazione per ottimizzare la nutrizione e le raccomandazioni sugli integratori alimentari.
4. Philen R.M., Ortiz D.I., Auerbach S.B., Falk H. Survey of Advertising for Nutritional Supplements in Health and Bodybuilding Magazines. JAMA. 1992;268:1008. doi: 10.1001/jama.1992.03490080082029. [PubMed] [CrossRef] [Google Scholar]
5. Giampreti A., Lonati D., Locatelli C., Rocchi L., Campailla M.T. Acute neurotoxicity after yohimbine ingestion by a bodybuilder. [(accessed on 25 March 2019)];Clin. Toxicol. 2009 47:827–829. doi: 10.1080/15563650903081601. Available online: https://www.ncbi.nlm.nih.gov/pubmed/19640235 [PubMed] [CrossRef] [Google Scholar]
7. Della Guardia L., Cavallaro M., Cena H. The risks of self-made diets: The case of an amateur bodybuilder. J. Int. Soc. Sports Nutr. 2015;12:5. doi: 10.1186/s12970-015-0077-8. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
9. Hackett D.A., Johnson N.A., Chow C.-M. Training Practices and Ergogenic Aids Used by Male Bodybuilders. J. Strength Cond. Res. 2013;27:1609–1617. doi: 10.1519/JSC.0b013e318271272a. [PubMed] [CrossRef] [Google Scholar]
10. Forbes G.B., Brown M.R., Welle S.L., Lipinski B.A. Deliberate overfeeding in women and men: Energy cost and composition of the weight gain. Br. J. Nutr. 1986;56:1–9. doi: 10.1079/BJN19860080. [PubMed] [CrossRef] [Google Scholar]
11. Kreider R.B., Klesges R., Harmon K., Ramsey L., Bullen D., Wood L., Almada A., Grindstaff P., Li Y. Effects of Ingesting Supplements Designed to Promote Lean Tissue Accretion on Body Composition during Resistance Training. Int. J. Sport Nutr. 1996;6:234–246. doi: 10.1123/ijsn.6.3.234. [PubMed] [CrossRef] [Google Scholar]
12. Rozenek R., Ward P., Long S., Garhammer J. Effects of high-calorie supplements on body composition and muscular strength following resistance training. J. Sports Med. Phys. Fit. 2002;42:340–347. [PubMed] [Google Scholar]
13. Garthe I., Raastad T., Refsnes P.E., Sundgot-Borgen J. Effect of nutritional intervention on body composition and performance in elite athletes. Eur. J. Sport Sci. 2013;13:295–303. doi: 10.1080/17461391.2011.643923. [PubMed] [CrossRef] [Google Scholar]
14. American College og Sports Medicine American College of Sports Medicine position stand. Progression models in resistance training for healthy adults. [(accessed on 25 March 2019)];Med. Sci. Sport. Exerc. 2009 41:687–708. doi: 10.1249/MSS.0b013e3181915670. Available online: https://www.ncbi.nlm.nih.gov/pubmed/19204579 [PubMed] [CrossRef] [Google Scholar]
15. Lambert C.P., Frank L.L., Evans W.J., Lambert D.C.P. Macronutrient Considerations for the Sport of Bodybuilding. Sports Med. 2004;34:317–327. doi: 10.2165/00007256-200434050-00004. [PubMed] [CrossRef] [Google Scholar]
16. Walberg-Rankin J., Edmonds C.E., Gwazdauskas F.C. Diet and Weight Changes of Female Bodybuilders Before and After Competition. Int. J. Sport Nutr. 1993;3:87–102. doi: 10.1123/ijsn.3.1.87. [PubMed] [CrossRef] [Google Scholar]
17. Lamar-Hildebrand N., Saldanha L., Endres J. Dietary and exercise practices of college-aged female bodybuilders. J. Am. Diet. Assoc. 1989;89:1308–1310. [PubMed] [Google Scholar]
18. Houston M.E. Gaining Weight: The Scientific Basis of Increasing Skeletal Muscle Mass. Can. J. Appl. Physiol. 1999;24:305–316. doi: 10.1139/h99-024. [PubMed] [CrossRef] [Google Scholar]
19. Phillips S.M. A Brief Review of Critical Processes in Exercise-Induced Muscular Hypertrophy. Sports Med. 2014;44:71–77. doi: 10.1007/s40279-014-0152-3. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
20. Campbell B.I., Aguilar D., Conlin L., Vargas A., Schoenfeld B.J., Corson A., Gai C., Best S., Galvan E., Couvillion K. Effects of High Versus Low Protein Intake on Body Composition and Maximal Strength in Aspiring Female Physique Athletes Engaging in an 8-Week Resistance Training Program. Int. J. Sport Nutr. Exerc. Metab. 2018;28:580–585. doi: 10.1123/ijsnem.2017-0389. [PubMed] [CrossRef] [Google Scholar]
22. Morton R.W., Murphy K.T., McKellar S.E., Schoenfeld B.J., Henselmans M., Helms E., Aragon A.A., Devries M.C., Banfield L., Krieger J.W., et al. A systematic review, meta-analysis and meta-regression of the effect of protein supplementation on resistance training-induced gains in muscle mass and strength in healthy adults. [(accessed on 25 March 2019)];Br. J. Sports Med. 2018 52:376–384. doi: 10.1136/bjsports-2017-097608. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28698222 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
23. Houltham S.D., Rowlands D.S. A snapshot of nitrogen balance in endurance-trained women. Appl. Physiol. Nutr. Metab. 2014;39:219–225. doi: 10.1139/apnm-2013-0182. [PubMed] [CrossRef] [Google Scholar]
25. Antonio J., Ellerbroek A., Silver T., Vargas L., Peacock C. The effects of a high protein diet on indices of health and body composition—A crossover trial in resistance-trained men. J. Int. Soc. Sports Nutr. 2016;13:8. doi: 10.1186/s12970-016-0114-2. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
26. Bandegan A., Courtney-Martin G., Rafii M., Pencharz P.B., Lemon P.W. Indicator Amino Acid–Derived Estimate of Dietary Protein Requirement for Male Bodybuilders on a Nontraining Day Is Several-Fold Greater than the Current Recommended Dietary Allowance. J. Nutr. 2017;147:850–857. doi: 10.3945/jn.116.236331. [PubMed] [CrossRef] [Google Scholar]
27. Malowany J.M., West D.W.D., Williamson E., Volterman K.A., Sawan S.A., Mazzulla M., Moore D.R. Protein to Maximize Whole-Body Anabolism in Resistance-trained Females after Exercise. Med. Sci. Sports Exerc. 2019;51:798–804. doi: 10.1249/MSS.0000000000001832. [PubMed] [CrossRef] [Google Scholar]
28. Antonio J., Peacock C.A., Ellerbroek A., Fromhoff B., Silver T. The effects of consuming a high protein diet (4.4 g/kg/d) on body composition in resistance-trained individuals. J. Int. Soc. Sports Nutr. 2014;11:19. doi: 10.1186/1550-2783-11-19. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
29. Antonio J., Ellerbroek A., Silver T., Orris S., Scheiner M., Gonzalez A., Peacock C.A. A high protein diet (3.4 g/kg/d) combined with a heavy resistance training program improves body composition in healthy trained men and women—A follow-up investigation. J. Int. Soc. Sports Nutr. 2015;12:39. doi: 10.1186/s12970-015-0100-0. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
30. Bray G.A., Smith S.R., de Jonge L., Xie H., Rood J., Martin C.K., Most M., Brock C., Mancuso S., Redman L.M. Effect of dietary protein content on weight gain, energy expenditure, and body composition during overeating: A randomized controlled trial. [(accessed on 25 March 2019)];JAMA. 2012 307:47–55. doi: 10.1001/jama.2011.1918. Available online: https://www.ncbi.nlm.nih.gov/pubmed/22215165 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
31. Tipton K.D., Ferrando A.A., Phillips S.M., Doyle D., Wolfe R.R. Postexercise net protein synthesis in human muscle from orally administered amino acids. Am. J. Physiol. Metab. 1999;276:628–634. doi: 10.1152/ajpendo.1999.276.4.E628. [PubMed] [CrossRef] [Google Scholar]
32. Rieu I., Balage M., Sornet C., Giraudet C., Pujos E., Grizard J., Mosoni L., Dardevet D. Leucine supplementation improves muscle protein synthesis in elderly men independently of hyperaminoacidaemia. J. Physiol. 2006;575:305–315. doi: 10.1113/jphysiol.2006.110742. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
33. Burd N.A., Tang J.E., Moore D.R., Phillips S.M. Exercise training and protein metabolism: Influences of contraction, protein intake, and sex-based differences. [(accessed on 25 March 2019)];J. Appl. Physiol. 2008 106:1692–1701. doi: 10.1152/japplphysiol.91351.2008. Available online: https://www.ncbi.nlm.nih.gov/pubmed/19036897 [PubMed] [CrossRef] [Google Scholar]
34. Drummond M.J., Dreyer H.C., Fry C.S., Glynn E.L., Rasmussen B.B. Nutritional and contractile regulation of human skeletal muscle protein synthesis and mTORC1 signaling. J. Appl. Physiol. 2009;106:1374–1384. doi: 10.1152/japplphysiol.91397.2008. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
35. Tang J.E., Moore D.R., Kujbida G.W., Tarnopolsky M.A., Phillips S.M. Ingestion of whey hydrolysate, casein, or soy protein isolate: Effects on mixed muscle protein synthesis at rest and following resistance exercise in young men. J. Appl. Physiol. 2009;107:987–992. doi: 10.1152/japplphysiol.00076.2009. [PubMed] [CrossRef] [Google Scholar]
36. Kanda A., Nakayama K., Sanbongi C., Nagata M., Ikegami S., Itoh H. Effects of Whey, Caseinate, or Milk Protein Ingestion on Muscle Protein Synthesis after Exercise. Nutrients. 2016;8:339. doi: 10.3390/nu8060339. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
37. Messina M., Lynch H., Dickinson J.M., Reed K.E. No Difference Between the Effects of Supplementing With Soy Protein Versus Animal Protein on Gains in Muscle Mass and Strength in Response to Resistance Exercise. Int. J. Sport Nutr. Exerc. Metab. 2018;28:674–685. doi: 10.1123/ijsnem.2018-0071. [PubMed] [CrossRef] [Google Scholar]
38. Joy J.M., Lowery R.P., Wilson J.M., Purpura M., De Souza E.O., Mc Wilson S., Kalman D.S., Dudeck J.E., Jäger R. The effects of 8 weeks of whey or rice protein supplementation on body composition and exercise performance. Nutr. J. 2013;12:86. doi: 10.1186/1475-2891-12-86. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
39. Babault N., Paizis C., Deley G., Guérin-Deremaux L., Saniez M.-H., Lefranc-Millot C., Allaert F.A. Pea proteins oral supplementation promotes muscle thickness gains during resistance training: A double-blind, randomized, Placebo-controlled clinical trial vs. Whey protein. J. Int. Soc. Sports Nutr. 2015;12:1692. doi: 10.1186/s12970-014-0064-5. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
40. Tesch P.A. Glycogen and triglyceride utilization in relation to muscle metabolic characteristics in men performing heavy-resistance exercise. Graefe’s Arch. Clin. Exp. Ophthalmol. 1990;61:5–10. [PubMed] [Google Scholar]
41. Lane A.R., Duke J.W., Hackney A.C. Influence of dietary carbohydrate intake on the free testosterone: Cortisol ratio responses to short-term intensive exercise training. [(accessed on 25 March 2019)];Eur. J. Appl. Physiol. 2010 108:1125–1131. doi: 10.1007/s00421-009-1220-5. Available online: https://www.ncbi.nlm.nih.gov/pubmed/20091182 [PubMed] [CrossRef] [Google Scholar]
42. Tegelman R., Aberg T., Pousette A., Carlström K. Effects of a diet regimen on pituitary and steroid hormones in male ice hockey players. [(accessed on 25 March 2019)];Int. J. Sports Med. 1992 13:420–430. doi: 10.1055/s-2007-1021292. Available online: https://www.ncbi.nlm.nih.gov/pubmed/1387870 [PubMed] [CrossRef] [Google Scholar]
43. Dorgan J.F., Judd J.T., Longcope C., Brown C., Schatzkin A., Clevidence B.A., Campbell W.S., Nair P.P., Franz C., Kahle L., et al. Effects of dietary fat and fiber on plasma and urine androgens and estrogens in men: A controlled feeding study. Am. J. Clin. Nutr. 1996;64:850–855. doi: 10.1093/ajcn/64.6.850. [PubMed] [CrossRef] [Google Scholar]
44. Hämäläinen E., Adlercreutz H., Puska P., Pietinen P. Decrease of serum total and free testosterone during a low-fat high-fibre diet. J. Steroid Biochem. 1983;18:369–370. doi: 10.1016/0022-4731(83)90117-6. [PubMed] [CrossRef] [Google Scholar]
45. Hämäläinen E., Adlercreutz H., Puska P., Pietinen P. Diet and serum sex hormones in healthy men. J. Steroid Biochem. 1984;20:459–464. doi: 10.1016/0022-4731(84)90254-1. [PubMed] [CrossRef] [Google Scholar]
46. Wang C., Catlin D.H., Starcevic B., Heber D., Ambler C., Berman N., Lucas G., Leung A., Schramm K., Lee P.W.N., et al. Low-Fat High-Fiber Diet Decreased Serum and Urine Androgens in Men. J. Clin. Endocrinol. Metab. 2005;90:3550–3559. doi: 10.1210/jc.2004-1530. [PubMed] [CrossRef] [Google Scholar]
47. Morton R.W., Sato K., Gallaugher M.P.B., Oikawa S.Y., McNicholas P.D., Fujita S., Phillips S.M. Muscle Androgen Receptor Content but Not Systemic Hormones Is Associated With Resistance Training-Induced Skeletal Muscle Hypertrophy in Healthy, Young Men. Front. Physiol. 2018;9:9. doi: 10.3389/fphys.2018.01373. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
48. Tinsley G.M., Willoughby D.S. Fat-Free Mass Changes During Ketogenic Diets and the Potential Role of Resistance Training. Int. J. Sport Nutr. Exerc. Metab. 2016;26:78–92. doi: 10.1123/ijsnem.2015-0070. [PubMed] [CrossRef] [Google Scholar]
49. Vargas S., Romance R., Petro J.L., Bonilla D.A., Galancho I., Espinar S., Kreider R.B., Benítez-Porres J. Efficacy of ketogenic diet on body composition during resistance training in trained men: A randomized controlled trial. J. Int. Soc. Sports Nutr. 2018;15:31. doi: 10.1186/s12970-018-0236-9. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
50. Kephart W.C., Pledge C.D., Roberson P.A., Mumford P.W., Romero M.A., Mobley C.B., Martin J.S., Young K.C., Lowery R.P., Wilson J.M., et al. The Three-Month Effects of a Ketogenic Diet on Body Composition, Blood Parameters, and Performance Metrics in CrossFit Trainees: A Pilot Study. Sports. 2018;6:1. doi: 10.3390/sports6010001. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
51. Greene D.A., Varley B.J., Hartwig T.B., Chapman P., Rigney M. A Low-Carbohydrate Ketogenic Diet Reduces Body Mass Without Compromising Performance in Powerlifting and Olympic Weightlifting Athletes. [(accessed on 26 March 2019)];J. Strength Cond. Res. 2018 32:3373–3382. Available online: https://www.ncbi.nlm.nih.gov/pubmed/30335720 [PubMed] [Google Scholar]
52. Bird S. Strength Nutrition: Maximizing Your Anabolic Potential. Strength Cond. J. 2010;32:80–86. doi: 10.1519/SSC.0b013e3181d5284e. [CrossRef] [Google Scholar]
53. American Dietetic Association. Dietitians of Canada. American College of Sports Medicine. Rodriguez N.R., Di Marco N.M., Langley S. American College of Sports Medicine position stand. Nutrition and athletic performance. [(accessed on 26 March 2019)];Med. Sci. Sports Exerc. 2009 41:709–731. Available online: https://www.ncbi.nlm.nih.gov/pubmed/19225360 [PubMed] [Google Scholar]
54. Chung S.T., Chacko S.K., Sunehag A.L., Haymond M.W. Measurements of Gluconeogenesis and Glycogenolysis: A Methodological Review. Diabetes. 2015;64:3996–4010. doi: 10.2337/db15-0640. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
55. Azizi F. Effect of dietary composition on fasting-induced changes in serum thyroid hormones and thyrotropin. Metabolism. 1978;27:935–942. doi: 10.1016/0026-0495(78)90137-3. [PubMed] [CrossRef] [Google Scholar]
56. Mathieson R.A., Walberg J.L., Gwazdauskas F.C., Hinkle D.E., Gregg J.M. The effect of varying carbohydrate content of a very-low-caloric diet on resting metabolic rate and thyroid hormones. Metabolism. 1986;35:394–398. doi: 10.1016/0026-0495(86)90126-5. [PubMed] [CrossRef] [Google Scholar]
57. Leveritt M., Abernethy P.J. Effects of Carbohydrate Restriction on Strength Performance. J. Strength Cond. Res. 1999;13:52–57. [Google Scholar]
58. Jacobs I., Kaiser P., Tesch P. Muscle strength and fatigue after selective glycogen depletion in human skeletal muscle fibers. Graefe’s Arch. Clin. Exp. Ophthalmol. 1981;46:47–53. doi: 10.1007/BF00422176. [PubMed] [CrossRef] [Google Scholar]
59. Ray S., Sale D.G., Lee P., Garner S., MacDougall J.D., McCartney N. Muscle Substrate Utilization and Lactate Production During Weightlifting. Can. J. Appl. Physiol. 1999;24:209–215. [PubMed] [Google Scholar]
60. Tesch P.A., Colliander E.B., Kaiser P. Muscle metabolism during intense, heavy-resistance exercise. Graefe’s Arch. Clin. Exp. Ophthalmol. 1986;55:362–366. doi: 10.1007/BF00422734. [PubMed] [CrossRef] [Google Scholar]
63. Mitchell J.B., DiLauro P.C., Pizza F.X., Cavender D.L. The Effect of Preexercise Carbohydrate Status on Resistance Exercise Performance. Int. J. Sport Nutr. 1997;7:185–196. doi: 10.1123/ijsn.7.3.185. [PubMed] [CrossRef] [Google Scholar]
64. Lima-Silva A.E., Silva-Cavalcante M.D., Oliveira R.S., Kiss M.A., Pires F.O., Bertuzzi R., Bishop D. Effects of a low- or a high-carbohydrate diet on performance, energy system contribution, and metabolic responses during supramaximal exercise. Appl. Physiol. Nutr. Metab. 2013;38:928–934. doi: 10.1139/apnm-2012-0467. [PubMed] [CrossRef] [Google Scholar]
65. Vega F., Jackson R. Dietary habits of bodybuilders and other regular exercisers. Nutr. Res. 1996;16:3–10. doi: 10.1016/0271-5317(95)02054-3. [CrossRef] [Google Scholar]
66. Chappell A.J., Simper T., Barker M.E. Nutritional strategies of high level natural bodybuilders during competition preparation. J. Int. Soc. Sports Nutr. 2018;15:4. doi: 10.1186/s12970-018-0209-z. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
67. Atherton P.J., Etheridge T., Watt P.W., Wilkinson D., Selby A., Rankin D., Smith K., Rennie M.J. Muscle full effect after oral protein: Time-dependent concordance and discordance between human muscle protein synthesis and mTORC1 signaling. Am. J. Clin. Nutr. 2010;92:1080–1088. doi: 10.3945/ajcn.2010.29819. [PubMed] [CrossRef] [Google Scholar]
68. Res P.T., Groen B., Pennings B., Beelen M., Wallis G.A., Gijsen A.P., Senden J.M., Van Loon L.J. Protein ingestion before sleep improves postexercise overnight recovery. [(accessed on 25 March 2019)];Med. Sci. Sports Exerc. 2012 44:1560–1569. doi: 10.1249/MSS.0b013e31824cc363. Available online: https://www.ncbi.nlm.nih.gov/pubmed/22330017 [PubMed] [CrossRef] [Google Scholar]
69. Moore D.R., Robinson M.J., Fry J.L., Tang J.E., Glover E.I., Wilkinson S.B., Prior T., Tarnopolsky M.A., Phillips S.M. Ingested protein dose response of muscle and albumin protein synthesis after resistance exercise in young men. [(accessed on 25 March 2019)];Am. J. Clin. Nutr. 2009 89:161–168. doi: 10.3945/ajcn.2008.26401. Available online: https://www.ncbi.nlm.nih.gov/pubmed/19056590 [PubMed] [CrossRef] [Google Scholar]
70. Witard O.C., Jackman S.R., Breen L., Smith K., Selby A., Tipton K.D. Muscle protein synthesis rates subsequent to a meal in response to increasing doses of whey protein at rest and after. [(accessed on 25 March 2019)];Am. J. Clin. Nutr. 2014 99:86–95. doi: 10.3945/ajcn.112.055517. Available online: https://www.ncbi.nlm.nih.gov/pubmed/24257722 [PubMed] [CrossRef] [Google Scholar]
71. Macnaughton L.S., Wardle S.L., Witard O.C., McGlory C., Hamilton D.L., Jeromson S., Lawrence C.E., Wallis G.A., Tipton K.D. The response of muscle protein synthesis following whole-body resistance exercise is greater following 40 g than 20 g of ingested whey protein. Physiol. Rep. 2016;4:e12893. doi: 10.14814/phy2.12893. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
72. Schoenfeld B.J., Aragon A.A., Krieger J.W. The effect of protein timing on muscle strength and hypertrophy: A meta-analysis. J. Int. Soc. Sports Nutr. 2013;10:53. doi: 10.1186/1550-2783-10-53. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
73. Areta J.L., Burke L.M., Ross M.L., Camera D.M., West D.W.D., Broad E.M., Jeacocke N.A., Moore D.R., Stellingwerff T., Phillips S.M., et al. Timing and distribution of protein ingestion during prolonged recovery from resistance exercise alters myofibrillar protein synthesis. J. Physiol. 2013;591:2319–2331. doi: 10.1113/jphysiol.2012.244897. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
74. Hudson J.L., Bergia R.E., Campbell W.W. Effects of protein supplements consumed with meals, versus between meals, on resistance training–induced body composition changes in adults: A systematic review. Nutr. Rev. 2018;76:461–468. doi: 10.1093/nutrit/nuy012. [PubMed] [CrossRef] [Google Scholar]
75. Trommelen J., Kouw I.W.K., Holwerda A.M., Snijders T., Halson S.L., Rollo I., Verdijk L.B., Van Loon L.J.C. Pre-sleep dietary protein-derived amino acids are incorporated in myofibrillar protein during post-exercise overnight recovery. [(accessed on 25 March 2019)];Am. J. Physiol. Metab. 2018 1:457–467. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28536184 [Google Scholar]
76. Kouw I.W., Holwerda A.M., Trommelen J., Kramer I.F., Bastiaanse J., Halson S.L., Wodzig W.K., Verdijk L.B., Van Loon L.J. Protein Ingestion before Sleep Increases Overnight Muscle Protein Synthesis Rates in Healthy Older Men: A Randomized Controlled Trial. [(accessed on 25 March 2019)];J. Nutr. 2017 147:2252–2261. doi: 10.3945/jn.117.254532. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28855419 [PubMed] [CrossRef] [Google Scholar]
77. Snijders T., Res P.T., Smeets J.S., Van Vliet S., Van Kranenburg J., Maase K., Kies A.K., Verdijk L.B., Van Loon L.J. Protein ingestion before sleep increases muscle mass and strength gains during prolonged resistance-type exercise training in healthy young men. [(accessed on 25 March 2019)];J. Nutr. 2015 145:1178–1184. doi: 10.3945/jn.114.208371. Available online: https://www.ncbi.nlm.nih.gov/pubmed/25926415 [PubMed] [CrossRef] [Google Scholar]
78. Joy J.M., Vogel R.M., Broughton K.S., Kudla U., Kerr N.Y., Davison J.M., Wildman R.E.C., DiMarco N.M. Daytime and nighttime casein supplements similarly increase muscle size and strength in response to resistance training earlier in the day: A preliminary investigation. J. Int. Soc. Sports Nutr. 2018;15:24. doi: 10.1186/s12970-018-0228-9. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
79. Antonio J., Ellerbroek A., Peacock C., Silver T. Casein Protein Supplementation in Trained Men and Women: Morning versus Evening. Int. J. Exerc. Sci. 2017;10:479–486. [PMC free article] [PubMed] [Google Scholar]
80. Schoenfeld B.J., Aragon A.A. How much protein can the body use in a single meal for muscle-building? Implications for daily protein distribution. J. Int. Soc. Sports Nutr. 2018;15:10. doi: 10.1186/s12970-018-0215-1. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
81. Pennings B., Groen B.B., Van Dijk J.-W., De Lange A., Kiskini A., Kuklinski M., Senden J.M., Van Loon L.J. Minced beef is more rapidly digested and absorbed than beef steak, resulting in greater postprandial protein retention in older men. Am. J. Clin. Nutr. 2013;98:121–128. doi: 10.3945/ajcn.112.051201. [PubMed] [CrossRef] [Google Scholar]
82. Kim I.Y., Schutzler S., Schrader A., Spencer H.J., Azhar G., Ferrando A.A., Wolfe R.R. The anabolic response to a meal containing different amounts of protein is not limited by the maximal stimulation of protein synthesis in healthy young adults. [(accessed on 25 March 2019)];Am. J. Physiol. Metab. 2016 310:73–80. doi: 10.1152/ajpendo.00365.2015. Available online: https://www.ncbi.nlm.nih.gov/pubmed/26530155 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
83. Jentjens R., Jeukendrup A.E. Determinants of Post-Exercise Glycogen Synthesis During Short-Term Recovery. Sports Med. 2003;33:117–144. doi: 10.2165/00007256-200333020-00004. [PubMed] [CrossRef] [Google Scholar]
84. Biolo G., Williams B.D., Fleming R.Y., Wolfe R.R. Insulin action on muscle protein kinetics and amino acid transport during recovery after resistance exercise. Diabetes. 1999;48:949–957. doi: 10.2337/diabetes.48.5.949. [PubMed] [CrossRef] [Google Scholar]
85. Greenhaff P.L., Karagounis L.G., Peirce N., Simpson E.J., Hazell M., Layfield R., Wackerhage H., Smith K., Atherton P., Selby A., et al. Disassociation between the effects of amino acids and insulin on signaling, ubiquitin ligases, and protein turnover in human muscle. Am. J. Physiol. Metab. 2008;295:E595–E604. doi: 10.1152/ajpendo.90411.2008. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
86. Glynn E.L., Fry C.S., Timmerman K.L., Drummond M.J., Volpi E., Rasmussen B.B., Leroy J.L., Gadsden P., De Cossío T.G., Gertler P. Addition of Carbohydrate or Alanine to an Essential Amino Acid Mixture Does Not Enhance Human Skeletal Muscle Protein Anabolism123. J. Nutr. 2013;143:307–314. doi: 10.3945/jn.112.168203. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
87. Koopman R., Beelen M., Stellingwerff T., Pennings B., Saris W.H.M., Kies A.K., Kuipers H., Van Loon L.J.C. Coingestion of carbohydrate with protein does not further augment postexercise muscle protein synthesis. Am. J. Physiol. Metab. 2007;293:E833–E842. doi: 10.1152/ajpendo.00135.2007. [PubMed] [CrossRef] [Google Scholar]
88. Aragon A.A., Schoenfeld B.J. Nutrient timing revisited: Is there a post-exercise anabolic window? J. Int. Soc. Sports Nutr. 2013;10:5. doi: 10.1186/1550-2783-10-5. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
89. Jäger R., Kerksick C.M., Campbell B.I., Cribb P.J., Wells S.D., Skwiat T.M., Purpura M., Ziegenfuss T.N., Ferrando A.A., Arent S.M., et al. International Society of Sports Nutrition position stand: Protein and exercise. [(accessed on 25 March 2019)];J. Int. Soc. Sport. Nutr. 2017 4:20. doi: 10.1186/s12970-017-0177-8. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28642676 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
90. Darrabie M.D., Arciniegas A.J.L., Mishra R., Bowles D.E., Jacobs D.O., Santacruz L. AMPK and substrate availability regulate creatine transport in cultured cardiomyocytes. Am. J. Physiol. Metab. 2011;300:870–876. doi: 10.1152/ajpendo.00554.2010. [PubMed] [CrossRef] [Google Scholar]
91. Purchas R., Busboom J., Wilkinson B. Changes in the forms of iron and in concentrations of taurine, carnosine, coenzyme Q10, and creatine in beef longissimus muscle with cooking and simulated stomach and duodenal digestion. Meat Sci. 2006;74:443–449. doi: 10.1016/j.meatsci.2006.03.015. [PubMed] [CrossRef] [Google Scholar]
92. Branch J.D. Effect of Creatine Supplementation on Body Composition and Performance: A Meta-analysis. Int. J. Sport Nutr. Exerc. Metab. 2003;13:198–226. doi: 10.1123/ijsnem.13.2.198. [PubMed] [CrossRef] [Google Scholar]
93. Hultman E., Söderlund K., Timmons J.A., Cederblad G., Greenhaff P.L. Muscle creatine loading in men. [(accessed on 25 March 2019)];J. Appl. Physiol. Soc. 1996 81:232–237. doi: 10.1152/jappl.1996.81.1.232. Available online: https://www.ncbi.nlm.nih.gov/pubmed/8828669 [PubMed] [CrossRef] [Google Scholar]
94. Jagim A.R., Oliver J.M., Sanchez A., Galvan E., Fluckey J., Riechman S., Greenwood M., Kelly K., Meininger C., Rasmussen C., et al. A buffered form of creatine does not promote greater changes in muscle creatine content, body composition, or training adaptations than creatine monohydrate. J. Int. Soc. Sports Nutr. 2012;9:43. doi: 10.1186/1550-2783-9-43. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
95. Spillane M., Schoch R., Cooke M., Harvey T., Greenwood M., Kreider R., Willoughby D.S., Cooke M. The effects of creatine ethyl ester supplementation combined with heavy resistance training on body composition, muscle performance, and serum and muscle creatine levels. J. Int. Soc. Sports Nutr. 2009;6:6. doi: 10.1186/1550-2783-6-6. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
96. Childs E., De Wit H., Wit H. Subjective, behavioral, and physiological effects of acute caffeine in light, nondependent caffeine users. Psychopharmacology. 2006;185:514–523. doi: 10.1007/s00213-006-0341-3. [PubMed] [CrossRef] [Google Scholar]
97. Bellar D., Kamimori G.H., Glickman E.L. The Effects of Low-Dose Caffeine on Perceived Pain During a Grip to Exhaustion Task. J. Strength Cond. Res. 2011;25:1225–1228. doi: 10.1519/JSC.0b013e3181d9901f. [PubMed] [CrossRef] [Google Scholar]
98. Davis J.K., Green J.M. Caffeine and anaerobic performance: Ergogenic value and mechanisms of action. [(accessed on 25 March 2019)];Sport. Med. 2009 39:813–832. doi: 10.2165/11317770-000000000-00000. Available online: https://www.ncbi.nlm.nih.gov/pubmed/19757860 [PubMed] [CrossRef] [Google Scholar]
99. Wickwire P.J., McLester J.R., Gendle S., Hudson G., Pritchett R.C., Laurent C.M., Green J.M. Effects of Caffeine on Repetitions to Failure and Ratings of Perceived Exertion during Resistance Training. Int. J. Sports Physiol. Perform. 2007;2:250–259. [PubMed] [Google Scholar]
100. Duncan M.J., Oxford S.W. The effect of caffeine ingestion on mood state and bench press performance to failure. [(accessed on 25 March 2019)];J. Strength Cond. Res. 2001 25:178–185. doi: 10.1519/JSC.0b013e318201bddb. Available online: https://www.ncbi.nlm.nih.gov/pubmed/22124354 [PubMed] [CrossRef] [Google Scholar]
101. Williams A.D., Cribb P.J., Cooke M.B., Hayes A. The Effect of Ephedra and Caffeine on Maximal Strength and Power in Resistance-Trained Athletes. J. Strength Cond. Res. 2008;22:464–470. doi: 10.1519/JSC.0b013e3181660320. [PubMed] [CrossRef] [Google Scholar]
102. Tarnopolsky M.A., Atkinson S.A., MacDougall J.D., Sale D.G., Sutton J.R. Physiological responses to caffeine during endurance running in habitual caffeine users. Med. Sci. Sports Exerc. 1989;21:418–424. doi: 10.1249/00005768-198908000-00013. [PubMed] [CrossRef] [Google Scholar]
103. Blanchard J., Sawers S.J.A. The absolute bioavailability of caffeine in man. Eur. J. Clin. Pharmacol. 1983;24:93–98. doi: 10.1007/BF00613933. [PubMed] [CrossRef] [Google Scholar]
104. Hobson R.M., Saunders B., Ball G., Harris R.C., Sale C. Effects of β-alanine supplementation on exercise performance: A meta-analysis. Amino Acids. 2012;43:25–37. doi: 10.1007/s00726-011-1200-z. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
105. Hoffman J., Ratamess N.A., Ross R., Kang J., Magrelli J., Neese K., Faigenbaum A.D., Wise J.A. Beta-alanine and the hormonal response to exercise. [(accessed on 25 March 2019)];Int. J. Sports Med. 2008 29:952–958. doi: 10.1055/s-2008-1038678. Available online: https://www.ncbi.nlm.nih.gov/pubmed/18548362 [PubMed] [CrossRef] [Google Scholar]
106. Hoffman J., Ratamess N., Kang J., Mangine G., Faigenbaum A., Stout J. Effect of creatine and β-alanine supplementation on performance and endocrine responses in strength/power athletes. [(accessed on 25 March 2019)];Int. J. Sport Nutr. Exerc. Metab. 2006 16:430–446. doi: 10.1123/ijsnem.16.4.430. Available online: https://www.ncbi.nlm.nih.gov/pubmed/17136944 [PubMed] [CrossRef] [Google Scholar]
108. Wax B., Kavazis A.N., Weldon K., Sperlak J. Effects of Supplemental Citrulline Malate Ingestion During Repeated Bouts of Lower-Body Exercise in Advanced Weightlifters. J. Strength Cond. Res. 2015;29:786–792. doi: 10.1519/JSC.0000000000000670. [PubMed] [CrossRef] [Google Scholar]
109. Wax B., Kavazis A.N., Luckett W. Effects of Supplemental Citrulline-Malate Ingestion on Blood Lactate, Cardiovascular Dynamics and Resistance Exercise Performance in Trained Males. [(accessed on 25 March 2019)];J. Diet. 2016 13:269–282. doi: 10.3109/19390211.2015.1008615. Available online: https://www.ncbi.nlm.nih.gov/pubmed/25674699 [PubMed] [CrossRef] [Google Scholar]
110. Glenn J.M., Gray M., Wethington L.N., Stone M.S., Stewart R.W., Jr., Moyen N.E. Acute citrulline malate supplementation improves upper- and lower-body submaximal weightlifting exercise performance in resistance-trained females. [(accessed on 25 March 2019)];Eur. J. Nutr. 2017 56:775–784. doi: 10.1007/s00394-015-1124-6. Available online: https://www.ncbi.nlm.nih.gov/pubmed/26658899 [PubMed] [CrossRef] [Google Scholar]
111. Glenn J.M., Gray M., Jensen A., Stone M.S., Vincenzo J.L. Acute citrulline-malate supplementation improves maximal strength and anaerobic power in female, masters athletes tennis players. Eur. J. Sport Sci. 2016;16:1–9. doi: 10.1080/17461391.2016.1158321. [PubMed] [CrossRef] [Google Scholar]
112. Gonzalez A.M., Spitz R.W., Ghigiarelli J.J., Sell K.M., Mangine G.T. Acute Effect of Citrulline Malate Supplementation on Upper-Body Resistance Exercise Performance in Recreationally Resistance-Trained Men. J. Strength Cond. Res. 2018;32:3088–3094. doi: 10.1519/JSC.0000000000002373. [PubMed] [CrossRef] [Google Scholar]
113. Farney T.M., Bliss M.V., Hearon C.M., Salazar D.A. The Effect of Citrulline Malate Supplementation On Muscle Fatigue Among Healthy Participants. J. Strength Cond. Res. 2017:1. doi: 10.1519/JSC.0000000000002356. [PubMed] [CrossRef] [Google Scholar]
114. Trexler E.T., Persky A.M., Ryan E.D., Schwartz T.A., Stoner L., Smith-Ryan A.E. Acute Effects of Citrulline Supplementation on High-Intensity Strength and Power Performance: A Systematic Review and Meta-Analysis. Sports Med. 2019;49:707–718. doi: 10.1007/s40279-019-01091-z. [PubMed] [CrossRef] [Google Scholar]
115. Kleiner S.M., Bazzarre T.L., Litchford M.D. Metabolic profiles, diet, and health practices of championship male and female bodybuilders. J. Am. Diet. Assoc. 1990;90:962–967. [PubMed] [Google Scholar]
116. Kleiner S.M., Bazzarre T.L., Ainsworth B.E. Nutritional Status of Nationally Ranked Elite Bodybuilders. Int. J. Sport Nutr. 1994;4:54–69. doi: 10.1123/ijsn.4.1.54. [PubMed] [CrossRef] [Google Scholar]
117. Sandoval W.M., Heyward V.H. Food Selection Patterns of Bodybuilders. Int. J. Sport Nutr. 1991;1:61–68. doi: 10.1123/ijsn.1.1.61. [PubMed] [CrossRef] [Google Scholar]
118. Ismaeel A., Weems S., Willoughby D.S. A Comparison of the Nutrient Intakes of Macronutrient-Based Dieting and Strict Dieting Bodybuilders. Int. J. Sport Nutr. Exerc. Metab. 2018;28:502–508. doi: 10.1123/ijsnem.2017-0323. [PubMed] [CrossRef] [Google Scholar]
119. Nelson J.R., Raskin S. The eicosapentaenoic acid:arachidonic acid ratio and its clinical utility in cardiovascular disease. Postgrad. Med. 2019;131:268–277. doi: 10.1080/00325481.2019.1607414. [PubMed] [CrossRef] [Google Scholar]
120. Harris W.S. The Omega-6: Omega-3 ratio: A critical appraisal and possible successor. [(accessed on 15 June 2019)];Prostaglandins Leukot Essent Fatty Acids. 2018 132:34–40. doi: 10.1016/j.plefa.2018.03.003. Available online: https://www.ncbi.nlm.nih.gov/m/pubmed/29599053/ [PubMed] [CrossRef] [Google Scholar]
121. Tachtsis B., Camera D., Lacham-Kaplan O. Potential Roles of n-3 PUFAs during Skeletal Muscle Growth and Regeneration. Nutrients. 2018;10:309. doi: 10.3390/nu10030309. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
122. Di Girolamo F.G., Situlin R., Mazzucco S., Valentini R., Toigo G., Biolo G. Omega-3 fatty acids and protein metabolism: Enhancement of anabolic interventions for sarcopenia. [(accessed on 15 June 2019)];Curr. Opin. Clin. Nutr. Metab Care. 2014 17:145–150. doi: 10.1097/MCO.0000000000000032. Available online: https://www.ncbi.nlm.nih.gov/pubmed/24500439 [PubMed] [CrossRef] [Google Scholar]
123. McGlory C., Wardle S.L., Macnaughton L.S., Witard O.C., Scott F., Dick J., Bell J.G., Phillips S.M., Galloway S.D.R., Hamilton D.L., et al. Fish oil supplementation suppresses resistance exercise and feeding-induced increases in anabolic signaling without affecting myofibrillar protein synthesis in young men. Physiol. Rep. 2016;4:e12715. doi: 10.14814/phy2.12715. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
124. Crestani D.M., Bonin E.F.R., Barbieri R.A., Zagatto A.M., Higino W.P., Milion F. Chronic supplementation of omega-3 can improve body composition and maximal strength, but does not change the resistance to neuromuscular fatigue. [(accessed on 15 June 2019)];Sport Sci. Health. 2017 13:259–265. doi: 10.1007/s11332-016-0322-9. Available online: https://link.springer.com/article/10.1007/s11332-016-0322-9 [CrossRef] [Google Scholar]
125. Lewis E.J.H., Radonic P.W., Wolever T.M.S., Wells G.D. 21 days of mammalian omega-3 fatty acid supplementation improves aspects of neuromuscular function and performance in male athletes compared to olive oil placebo. J. Int. Soc. Sports Nutr. 2015;12:28. doi: 10.1186/s12970-015-0089-4. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
126. Rossato L.T., Schoenfeld B.J., De Oliveira E.P. Is there sufficient evidence to supplement omega-3 fatty acids to increase muscle mass and strength in young and older adults? Clin. Nutr. 2019 doi: 10.1016/j.clnu.2019.01.001. [PubMed] [CrossRef] [Google Scholar]
127. Mocking R.J.T., Harmsen I., Assies J., Koeter M.W.J., Ruhé H.G., Schene A.H. Meta-analysis and meta-regression of omega-3 polyunsaturated fatty acid supplementation for major depressive disorder. Transl. Psychiatry. 2016;6:e756. doi: 10.1038/tp.2016.29. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
128. Maki K.C., Palacios O.M., Bell M., Toth P.P. Use of supplemental long-chain omega-3 fatty acids and risk for cardiac death: An updated meta-analysis and review of research gaps. J. Clin. Lipidol. 2017;11:1152–1160.e2. doi: 10.1016/j.jacl.2017.07.010. [PubMed] [CrossRef] [Google Scholar]
129. Miller P.E., Van Elswyk M., Alexander D.D. Long-Chain Omega-3 Fatty Acids Eicosapentaenoic Acid and Docosahexaenoic Acid and Blood Pressure: A Meta-Analysis of Randomized Controlled Trials. Am. J. Hypertens. 2014;27:885–896. doi: 10.1093/ajh/hpu024. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
130. Du S., Jin J., Fang W., Su Q. Does Fish Oil Have an Anti-Obesity Effect in Overweight/Obese Adults? A Meta-Analysis of Randomized Controlled Trials. PLoS ONE. 2015;10:e0142652. doi: 10.1371/journal.pone.0142652. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
Per accedere alle precedenti parti, dalla prima alla terza, clicca qui, qui e qui.
Disclaimer: quelle che seguono sono informazioni provenienti da casi studio [tranne dove diversamente specificato] e testimonianze di Bodybuilder sull’uso dell’Insulina, alcune delle quali sono palesemente sbagliate. Non prendete assolutamente queste opinioni come consigli.
Insulina – dall’uso clinico al Bodybuinding:
Come abbiamo visto nella prima parte, inizialmente l’Insulina farmaceutica era di origine animale. In questo caso, l’Insulina viene estratta dal pancreas di suino o di mucca (o di entrambi) e preparata per uso medico. Queste preparazioni sono ulteriormente suddivise nelle categorie “standard” e “purificate”, a seconda del livello di purezza e del contenuto non insulinico della soluzione. Con questi preparati c’è sempre la possibilità che contaminanti pancreatici siano presenti nel farmaco.
Nel 1977, Herbert Boyer e i suoi collaboratori Keiichi Itakura e Arthur Riggs al City of Hope National Medical Center descrisse la prima sintesi ed espressione di un gene codificante per un peptide. Nell’agosto del 1978, Boyer produsse Insulina sintetica utilizzando i suoi nuovi batteri transgenici geneticamente modificati, seguita nel 1979 dall’Ormone della Crescita. La Tecnologia del DNA ricombinante faceva il suo debutto e cambiava la storia dell’Insulina per uso medico.
L’Insulina umana biosintetica (insulina umana rDNA), attualmente e maggiormente utilizzata per uso clinico, è prodotta con la tecnologia del DNA ricombinante. L’Insulina umana biosintetica ha una maggiore purezza rispetto all’Insulina animale estrattiva e riduce la formazione di anticorpi. I ricercatori sono riusciti a introdurre il gene dell’Insulina umana nelle piante come un altro metodo per produrre Insulina (“biopharming”) nel cartamo. Si prevede che questa tecnica ridurrà i costi di produzione.
Sono disponibili diversi analoghi dell’Insulina umana. Questi analoghi dell’Insulina sono strettamente correlati alla struttura dell’Insulina umana e sono stati sviluppati per aspetti specifici del controllo glicemico in termini di azione rapida (insuline prandiali) e azione prolungata (insuline basali). Il primo analogo biosintetico dell’insulina è stato sviluppato per l’uso clinico al momento del pasto (insulina prandiale), Humalog (Insulina lispro), è assorbita più rapidamente dopo l’iniezione sottocutanea rispetto all’Insulina normale, con un effetto a 15 minuti dopo l’iniezione. Altri analoghi ad azione rapida sono NovoRapid e Apidra, con profili simili. Tutti vengono assorbiti rapidamente grazie a sequenze aminoacidiche che riducono la formazione di dimeri ed esameri (le insuline monomeriche vengono assorbite più rapidamente). Le insuline ad azione rapida non richiedono l’intervallo iniezione-pasto precedentemente raccomandato per l’Insulina umana e le insuline animali. L’altro tipo è l’Insulina a lunga durata d’azione; la prima di queste è stata Lantus (Insulina glargine). Queste hanno un effetto costante per un periodo prolungato, da 18 a 24 ore. Allo stesso modo, un altro analogo dell’Insulina a lunga durata d’azione (Levemir) si basa su un approccio di acilazione degli acidi grassi. A questo analogo è legata una molecola di acido miristico, che associa la molecola di Insulina all’abbondante albumina sierica, prolungando così l’effetto e riducendo il rischio di ipoglicemia. Entrambi gli analoghi ad azione prolungata devono essere assunti una sola volta al giorno e sono utilizzati nei diabetici di tipo 1 come Insulina basale. È disponibile anche una combinazione di un’Insulina ad azione rapida e di un’Insulina protratta, che consente ai pazienti di ottenere un profilo insulinico simile a quello del rilascio di Insulina da parte dell’organismo. L’Insulina viene utilizzata anche in molte linee cellulari, come CHO-s, HEK 293 o Sf9, per la produzione di anticorpi monoclonali, vaccini virali e prodotti per la terapia genica.
L’Insulina viene solitamente somministrata sotto forma di iniezioni sottocutanee tramite siringhe monouso con aghi, tramite un microinfusore di Insulina o tramite penne da insulina a uso ripetuto con aghi monouso. Sul mercato statunitense è disponibile anche l’Insulina per inalazione.
A differenza di molti farmaci, l’Insulina non può essere assunta per bocca perché, come quasi tutte le altre proteine introdotte nel tratto gastrointestinale, si riduce in frammenti amminoacidici, perdendo tutto il suo potenziale di attività. Sono state condotte alcune ricerche su come proteggere l’Insulina dal tratto digestivo, in modo da poterla somministrare per via orale o sublinguale.
Nel 2021, l’Organizzazione Mondiale della Sanità ha aggiunto l’Insulina al suo modello di elenco di farmaci essenziali.
Complice la descrizione iniziale allettante riportata in letteratura riguardante l’azione dell’Insulina sul metabolismo glucidico e proteico, dove tale peptide veniva descritto come l'”ormone più anabolico”, unita alla maggiore disponibilità di approvvigionamento del farmaco data dalla Tecnologia del DNA ricombinante, nel mondo della cultura fisica di alto livello non mancarono i primi pionieri del suo utilizzo “”off-label”.
Si può stimare che nel giro di 40 anni, vale a dire dagli anni 80 ad oggi, l’uso dell’Insulina nel Bodybuilding abbia subito sia un abuso pratico che teorico. Quello che spero di fare con questa mia piccola opera divulgativa è proprio quello di cambiare questa situazione. Come uomo di scienza con anni di ricerca alle spalle sono qualificato per giudicare le conoscenze dei Bodybuilder. Molti di loro conoscono molto meno di me il funzionamento e l’uso dell’Insulina. Quello che porto sul tavolo sono anni di ricerca nella comunità del bodybuilding e una corposo serie di prospettive diverse di culturisti sull’uso dell’Insulina, raccolte da 20 interviste con utilizzatori di Insulina con un’esperienza che va da mesi a decenni. Tranquilli però, fortunatamente non sono un “classico camicie bianco”, ma uno che analizza con attenzione è sa ammettere quando gli atleti hanno ragione e la ricerca scientifica si sbaglia. È ora quindi di trasformare la “broscience” dell’Insulina in scienza vera e propria e di correggere alcuni miti potenzialmente pericolosi.
Sto scrivendo questo articolo non solo per dimostrare le diverse, e persino contraddittorie, opinioni che i bodybuilder hanno sull’insulina, ma anche per ispirarvi a riflettere lucidamente.
I culturisti, quelli con doti intellettive un minimo sopra la media dei loro colleghi, sanno cose che i medici non potrebbero mai sapere perché hanno un obiettivo diverso e priorità diverse. Ma una cosa è certa, i bodybuilder accorti vogliono praticare il bodybuilding in modo scientificamente informato.
Non saranno presenti i nomi dei culturisti intervistati, dei quali sono stati esaminati i video e di cui sono state raccolte le affermazioni nei forum, per un principio etico che prevede di non rivelare l’identità delle persone che hanno contribuito alla presente ricerca, per evitare qualsiasi rischio di danno alla loro persona e immagine. Pertanto, tutti i nomi sono pseudonimi. È sufficiente dire che sono stati intervistati alcuni individui riconosciuti come esperti mondiali nell’uso dell’Insulina per il bodybuilding. sono stati inclusi anche i bodybuilder medi.
L’Insulina vista dai BodyBuilder:
I bodybuilder hanno tra loro visioni piuttosto diverse riguardo all’Insulina e ai suoi effetti. Per esempio, essi non sono d’accordo all’unanimità su quanto sia anabolizzante l’Insulina. Una minoranza afferma che l’Insulina esogena non è direttamente anabolizzante. Alcuni suggeriscono che l’Insulina induce l’anabolismo solo aumentando l’appetito. Ma, come sappiamo bene, di per se, quest’ultima affermazione ha ben poco senso.
Una piccolissima minoranza di bodybuilder sostiene che l’impatto dell’Insulina esogena sia principalmente, o puramente, cosmetico, in quanto l’Insulina fa apparire il muscolo più “pieno” (piuttosto che aumentare effettivamente le dimensioni del muscolo) aumentando la ritenzione idrica intracellulare.
Al contrario, molti bodybuilder sostengono che l’Insulina sia l’ormone più anabolico.
Alcuni partecipanti hanno descritto l’uso dell’Insulina come il risultato di un aumento muscolare di 3-6kg a settimana rispetto a quello che si potrebbe ottenere con il solo uso di Steroidi Androgeni Anabolizzanti (AAS) o con l’uso combinato di AAS e Ormone della Crescita (GH). Tuttavia, queste affermazioni sono state contestate da alcuni. Vi sono bodybuilder che ritengono che i benefici dell’Insulina non siano sufficienti e non hanno intenzione di utilizzare nuovamente questo peptide. Uno di questi non raccomanda più l’uso dell’Insulina ai suoi clienti.
Mentre alcuni bodybuilder sostengono che l’Insulina da sola sia anabolizzante, altri suggeriscono che essa sia significativamente anabolizzante solo in sinergia con AAS e hGH.
La maggior parte dei bodybuilder concorda sul funzionamento dell’Insulina, anche se le loro spiegazioni sono più o meno scientifiche.
In genere i bodybuilder descrivono l’Insulina come una “navetta” o un “mezzo” che trasporta i nutrienti nelle cellule muscolari. L’Insulina viene descritta come una “chiave” che apre le porte delle cellule o come un “autobus” che trasporta i nutrienti. Alcuni bodybuilder citano la letteratura scientifica quando descrivono l’azione dell’Insulina:
Sembra che l’Insulina abbia almeno un effetto permissivo sulla sintesi proteica, tanto che i suoi livelli basali sono necessari per la normale sintesi proteica miofibrillare (MPS), ma l’aumento dell’Insulina dopo un pasto potrebbe non aumentare la MPS (Greenhaff et al., 2008). Tuttavia, l’Insulina promuove l’anabolismo muscolare (bilancio proteico proattivo) attraverso il suo effetto inibitorio sulla degradazione delle proteine muscolari (MPB) (Deutz e Wolf 2013). Inoltre, l’Insulina può aumentare l’MPS attraverso un maggiore assorbimento di aminoacidi (essenziali) nel muscolo scheletrico, provocato da un aumento del flusso sanguigno associato alla vasodilatazione (Biolo et al., 1995; Fujita et al., 2006; Timmerman et al., 2010). [Chad via e-mail].
Molti concordano sul fatto che:
l’Insulina è molto efficace nel trasportare i nutrienti nelle cellule muscolari, ma anche nelle cellule adipose.
l’Insulina è anabolizzante grazie al suo ruolo nella ripartizione dei nutrienti, in quanto lavora di concerto con GH e IGF-I.
l’Insulina promuove l’anabolismo muscolare grazie al suo effetto inibitorio sulla degradazione delle proteine muscolari.
l’Insulina (così come gli AAS e il GH) promuove la sintesi proteica muscolare solo in presenza di un adeguato apporto di aminoacidi.
l’Insulina svolge un ruolo nel controllo fisiologico della riproduzione, agendo sulla secrezione dell’Ormone di Rilascio delle Gonadotropine (GnRH)/luteinizzante (LH).
L’Insulina è anche descritta da alcuni bodybuilder come anti-catabolica.
Un bodybuilder di alto livello ha dichiarato che l’Insulina dovrebbe essere usata solo se un individuo è carente di Insulina:
A volte è una buona idea prendere l’Insulina solo per aiutare il pancreas a fare il suo lavoro. … se il vostro corpo producesse abbastanza da solo, perché avreste bisogno di assumere Insulina esogena? In altre parole, non apporta alcun beneficio. È utile solo se non si produce abbastanza Insulina. Quindi bisogna innanzitutto stabilire se non si produce abbastanza Insulina. Procuratevi un glucometro e controllate la glicemia. … Quindi l’Insulina dovrebbe essere usata solo se si ha una carenza di insulina, perché si sta usando molto GH o perché si sta mangiando una quantità esorbitante di carboidrati.
Definirei questa ipotesi come “patologica indotta/deficitaria”. Ricordatevi sempre che l’omeostasi organica è regolata da feedback. Ciò significa che l’uso di Insulina esogena causerà una sottoregolazione/inibizione della biosintesi endogena di Insiluna. Di conseguenza, parlare di “pancreas ipoattivi” o “supporto pancreatico” non è in definitiva corretto. In tal caso si parla di una vera e propria terapia ormonale sostitutiva dell’Insulina.
Un bodybuilder ha anche affermato che gli effetti anabolici dell’Insulina sono dovuti alla sua azione osmotica e alla sua capacità di aprire “tutti i recettori del corpo”. Descrizione alquanto particolare ma che può rendere una certa idea di uno degli effetti dell’Insulina.
Mentre alcuni bodybuilder mettono in guardia dall’uso dell’Insulina perché può causare ipoglicemia con conseguente coma ipoglicemico potenzialmente letale, la maggior parte ritiene che i rischi dell’Insulina siano stati sopravvalutati e alcuni suggeriscono che la morte dovuta all’uso di Insulina è estremamente improbabile. Molti pensano che “bisogna essere una testa di cazzo per uccidersi con l’Insulina”.
Sebbene la morte di diversi culturisti di alto livello nel corso della mia ricerca sia stata inizialmente suggerita da membri della comunità dei culturisti come correlata all’Insulina, l’Insulina non è stata implicata nelle cause ufficiali dei loro decessi. Non mi sono imbattuto in un caso confermato di morte o di danni significativi causati dall’uso di Insulina per il bodybuilding, anche se alcuni bodybuilder hanno dichiarato di conoscere qualcuno che è morto a causa dell’uso di Insulina (e uno di loro ha avuto un grave incidente d’auto a causa di una ipoglicemia avuta in autostrada, ma fortunatamente nessuno è rimasto ferito). Nella letteratura medica sono riportati due casi di bodybuilder in coma ipoglicemico (Heidet et al., 2019; Petrovic et al., 2015). Non credo che la mancanza di decessi confermati sia dovuta al fatto che l’Insulina non sia pericolosa, ma più probabilmente perché non è comunemente testata o difficile da rilevare.
Un bodybuilder ha suggerito che le morti premature dovute all’uso di Insulina potrebbero non essere dovute solo all’ipoglicemia, ma ha suggerito che l’aumento dei livelli di Insulina nel corso della vita accorcia la durata della stessa e che quindi i bodybuilder si mettono a rischio in questo senso.
Alcuni bodybuilder sostengono che l’Insulina sia uno dei farmaci più sicuri del loro arsenale, in particolare rispetto al DNP e al Trenbolone. Alcuni suggeriscono addirittura che l’Insulina sia più sicura di qualsiasi AAS.
Mentre tutti i bodybuilder hanno descritto almeno lievi sintomi di ipoglicemia in alcuni momenti del loro utilizzo di Insulina, la maggior parte degli episodi di ipoglicemia si sono verificati durante le prime fasi di utilizzo, quando stavano elaborando il dosaggio dell’Insulina, o sono stati attribuiti alla loro stupidità (ad esempio, dimenticando di mangiare). Tutti i bodybuilder hanno dichiarato che l’ipoglicemia era facilmente gestibile consumando zuccheri.
Molti suggeriscono che per essere competitivi come bodybuilder professionisti è necessario utilizzare l’Insulina. Tuttavia, altri suggeriscono che non è necessario.
L’Insulina fa ormai parte del bodybuilding, ne è parte integrante. È come i denti sbiancati. Tutti sbiancano i denti, tutti hanno denti bianchi e splendenti. Se c’è uno che non li ha, gli si chiede: “Cosa c’è che non va in te?”. … Se non fai l’Insulina, cosa che alcuni professionisti non fanno, alcuni non ne hanno bisogno, allora il tuo aspetto è un po’ diverso da quello degli altri bodybuilder e potresti distinguerti in modo negativo.
Molti attribuiscono il significativo aumento della muscolatura dei mostri di massa all’Insulina e/o all’Ormone della Crescita. Alcuni suggeriscono che l’Insulina abbia rovinato il bodybuilding, poiché l’attenzione si è spostata dall’estetica alle dimensioni a scapito dell’estetica. Altri criticano ulteriormente il look dell’Insulina:
Prima dell’arrivo dell’Insulina, tornando ai primi tempi di Bertil Fox, Tom Platts, Arnold, Sergio, i loro muscoli avevano questo aspetto duro e granitico, sembravano scolpiti nella pietra. Ora ci sono questi ragazzi, certo grandi e stravaganti come i Ramy e tutti questi ragazzi, sono grandi ma non hanno quell’aspetto duro e denso.
Ma altri suggeriscono che Dorian Yates è stato il primo a portare sul palcoscenico del Olympia un fisico potenziato dall’Insulina e viene spesso descritto come se avesse un aspetto granitico.
Alcuni suggeriscono che l’Insulina (e/o l’Ormone della Crescita) provochi la “bolla intestinale” o il “palumboismo” [vedi anche “GH Gut”], e una fonte ha affermato che l’Insulina ha causato l’organomegalia. Per questi motivi alcuni affermano che l’Insulina ha rovinato l’estetica del bodybuilding.
Il dosaggio dell’insulina è molto vario tra i bodybuilder. Ho parlato con culturisti che usano un massimo di 4UI al giorno, e altri che hanno usato un massimo di 360UI al giorno! Anche se lo considero ben poco credibile. Tuttavia, in linea con le precedenti ricerche accademiche che riportavano dosaggi compresi tra 10 e 20 unità al giorno (Dawson e Harrison 1997; Evans 1997; Hildebrandt et al., 2007) e con i sondaggi condotti all’interno della comunità, ho scoperto che la maggior parte dei bodybuilder utilizza dosi che si collocano all’estremità inferiore dello spettro. Tuttavia, una buona parte dei bodybuilder tende a usare più di quanto riportato in precedenza nella letteratura accademica e nella comunità, con una dose giornaliera mediana di 40 unità e una dose mediana di 0,39 unità per chilogrammo di peso corporeo.
La maggior parte dei bodybuilder utilizza una sola forma di insulina (ad azione rapida o ultra-rapida), anche se un quarto degli intervistati (n=20) utilizza anche un’Insulina a lunga durata d’azione.
Ma vediamo nel dettaglio i tipi di Insulina utilizzati:
Humalog® (Insulina Lispro): Humalog® è un analogo a breve durata d’azione dell’Insulina umana, in particolare l’analogo Lys(B28) Pro(B29) dell’Insulina che si crea quando gli aminoacidi in posizione 28 e 29 sono invertiti. È considerata equipotente all’Insulina solubile normale su base unitaria, ma con un’attività più rapida. L’inizio dell’azione del farmaco in seguito alla somministrazione sottocutanea è di circa 10-15 minuti e il suo picco d’effetto viene raggiunto in 30-90 minuti. La durata d’azione totale è compresa tra 3-5 ore. L’Insulina lispro viene solitamente utilizzata come supplemento a un prodotto a base di Insulina a più lunga durata d’azione, fornendo un farmaco ad azione rapida che può essere assunto prima o subito dopo i pasti per imitare la secrezione insulinica naturale dell’organismo. Molti atleti ritengono che la sua breve finestra d’effetto la renda un farmaco insulinico ideale per scopi dopanti, in quanto la maggior parte dell’azione può essere concentrata nel periodo successivo all’allenamento sfruttando l’assimilazione dei nutrienti durante la così detta “finestra anabolica”.
Novolog® (Insulina Aspart):Novolog è un analogo a breve durata d’azione dell’Insulina umana, creato quando l’aminoacido prolina in posizione B28 viene sostituito con l’acido aspartico. L’inizio dell’azione del farmaco dopo la somministrazione sottocutanea è di circa 15 minuti e l’effetto di picco si raggiunge in 1-3 ore. La durata d’azione totale è compresa tra le 3 e le 5 ore. L’Insulina Aspart viene solitamente utilizzata come supporto a un prodotto contenente insulina a più lunga durata d’azione, fornendo un farmaco a rapida azione che può essere assunto prima o subito dopo i pasti per imitare la risposta insulinica dell’organismo. Molti atleti ritengono che la sua breve finestra di effetto la renda ideale per scopi dopanti, tanto quanto la Lispro, in quanto la maggior parte della sua azione si può concentra nel periodo successivo all’allenamento durante la “finestra anabolica”.
Humulin®-R “Regular” (insulina Inj): Identica all’Insulina umana. Venduta in alcuni mercati anche come Humulin-S® (Solubile), questo prodotto è costituito da cristalli di zinco-insulina disciolti in un liquido chiaro. Non viene aggiunto nulla per rallentare il rilascio di questo prodotto, per cui viene genericamente indicato come insulina umana solubile. Questo farmaco agisce rapidamente e ha una breve durata d’azione. L’inizio dell’azione del farmaco dopo la somministrazione sottocutanea è di 20-30 minuti, e il suo picco d’effetto si raggiunge in 1-3 ore. Ha una durata d’azione totale tra le 5 e le 8 ore. Insieme a Humalog, queste due forme di Insulina sono le scelte più popolari tra gli atleti e i culturisti per scopi dopanti.
Humulin®-N, NPH (Insulina Isofana): Una sospensione cristallina di Insulina con protamina e zinco per ritardarne il rilascio e prolungarne l’azione. L’Insulina Isofana è considerata un’Insulina di lunghezza intermedia. L’inizio dell’azione del farmaco dopo la somministrazione sottocutanea è di circa 1-2 ore e il picco d’effetto si raggiunge in 4-10 ore. La durata totale dell’attività è superiore a 14 ore. Questo tipo di Insulina non è comunemente usata come agente dopante.
Humulin®-L, Lente (sospensione media di Zinco): Una sospensione cristallina di Insulina con zinco per ritardarne il rilascio e prolungarne l’azione. Humulin-L è considerata un’insulina di lunghezza d’azione intermedia. L’inizio dell’azione del farmaco dopo somministrazione sottocutanea è di circa 1-3 ore e l’effetto di picco viene raggiunto in 6-14 ore. Ha una durata totale di attività superiore alle 20 ore. Questo tipo di Insulina non è comunemente usato per scopi dopanti.
Humulin®-U, Ultralente (sospensione prolungata di Zinco): Una sospensione cristallina di Insulina con zinco per ritardarne il rilascio e prolungarne l’azione. Humulin-U è considerata un’Insulina a lunga durata d’azione. L’inizio dell’azione del farmaco dopo somministrazione sottocutanea è di circa 6 ore, e l’effetto di picco viene raggiunto in 14-18 ore. La durata totale dell’attività è di 18-24 ore. Questo tipo di insulina non è comunemente usato per scopi dopanti.
Lantus (Insulina Glargine): Analogo a lunga durata d’azione dell’Insulina umana. L’Insulina Glargine viene creata quando l’aminoacido asparagina in posizione A21 viene sostituito con la glicina e vengono aggiunte due arginine al C-terminale della catena B dell’Insulina. L’inizio dell’azione del farmaco dopo la somministrazione sottocutanea è di circa 1-2 ore, e il farmaco è considerato privo di un picco significativo (è stato progettato per un modello di rilascio molto stabile per tutta la durata dell’attività). L’Insulina Glargine ha una durata d’azione compresa tra 20-24 ore nell’organismo dopo l’iniezione sottocutanea. Questo tipo di Insulina non è comunemente usato per scopi dopanti.
Humulin® (Miscele) : Sono miscele di Insulina solubile normale per un’azione rapida, e di un’Insulina a lunga durata d’azione o ad azione intermetizzata per un effetto prolungato. Queste miscele sono etichettate con la percentuale di miscela, di solito 10/90, 20/80, 30/70, 40/60 e 50/50. Sono anche disponibili le miscele che utilizzano Humalog come Insulina ad azione rapida.
Indipendentemente dal tipo, tutte le insuline forniscono gli stessi effetti di base, e la durata di azione è la differenziazione primaria. Il trasporto dei nutrienti, l’aumento della sintesi proteica, la diminuzione del catabolismo proteico, l’aumento del IGF-1, l’aumento della biodisponibilità del IGF-1, e una maggiore vasodilatazioni sono i vantaggi più noti.
Non è da molto tempo che un tipo di Insulina precedentemente marginale a fini dopanti è diventata di moda tra alcuni culturisti. Sto parlando della Lantus (Insulina Glargine), appunto.
Di tutte le diverse insuline disponibili, la Lantus è probabilmente quella meno utilizzata anche perchè e paradossalmente la più complessa da gestire. Il suo scarso utilizzo nel bodybuilding ha portato, come ovvia conseguenza, ad una scarsità delle informazioni disponibili su di essa.
A differenza delle insuline a breve durata d’azione, che forniscono i benefici di cui sopra per poche ore al giorno, la Lantus continuerà il trasporto dei nutrienti, l’aumento della sintesi proteica, il miglioramento della vasodilatazioni, ecc, per tutto il giorno, anche mentre dormiamo, ed è questo ultimo punto a renderla di non facile gestione. Ma uno degli svantaggi principali della Lantus risiede nella sua possibilità di essere utilizzata solo per brevi periodi di tempo, in quanto l’esposizione continua a livelli elevati di Insulina esogena, e lo sappiamo bene, porterà ad una riduzione della sensibilità all’insulina, la successiva sotto-regolazione dei trasportatori GLUT-4, cose che si vorrebbero evitare sia da un punto di vista della saluta che della crescita muscolare. Così, mentre la Lantus può essere superiore per lo stimolo della crescita muscolare nel breve termine, troviamo che le cose cominciano a pareggiarsi nel lungo periodo, e protocolli come quelli pre-allenamento più comunemente impiegati possono invece essere utilizzati a tempo indeterminato, senza danneggiare eccessivamente la sensibilità all’Insulina a qualsiasi grado significativo. Questo rende la Lantus ideale per dei “blitz”, in cui l’atleta vuole mettere su muscoli il più rapidamente possibile, ma non è adatta per un uso prolungato.
Differenza nella curva di rilascio tra Humulin N e Lantus in pazienti con Diabete di Tipo I.
Una caratteristica decisamente interessante della Lantus è il suo effetto sul IGF-1 ed i suoi recettori. In diversi studi universitari, la Lantus ha dimostrato una maggiore affinità di legame per il recettore del IGF-1 rispetto all’Insulina umana regolare o uno dei qualsiasi altri analoghi. È interessante notare che Levemir, l’unico altro analogo dell’Insulina ad azione prolungata sul mercato, mostra una ridotta affinità di legame ai recettori del IGF-1 umani. Questo mette la Lantus e la Levemir alle estremità opposte dello spettro in termini di affinità di legame. Mentre un aumento di IGF-1 vincolante è visto generalmente come una cosa positiva per la crescita muscolare, è stato il punto focale del dibattito in corso nella comunità medica per parecchi anni, per il fatto che alcuni studi hanno mostrato un aumento del rischio di cancro quando si usa la Lantus. Da allora, altri studi hanno confutato questa nozione, ma il dibattito continua, con la comunità medica riluttante a prendere una posizione in un modo o nell’altro.
Via di segnalazione del Recettore dell’Insulina (IR) e del Recettore del Fattore di Crescita Insulino-Simile 1 (IGF1R). L’Insulina e l’IGF1 si legano ai loro recettori, inducendo un cambiamento conformazionale e l’autofosforilazione della subunità beta di IR e IGF1R. Successivamente, le proteine substrato del recettore dell’Insulina (IRS) o Shc vengono reclutate e fosforilate. Shc attiva la via della mitogen-activated protein kinase-extracellular signal regulated kinase (MAPK-ERK) e le proteine IRS inducono prevalentemente l’attivazione della via della fosfoinositide 3-chinasi (PI3K)-AKT. In questo caso, l’attivazione di PI3K causa la conversione del fosfatidilinositolo 4,5-bisfosfato (PIP 2 ) in fosfatidilinositolo (3,4,5)-trifosfato (PIP 3 ) e l’attivazione e la fosforilazione di AKT da parte della proteina chinasi 1 dipendente dal fosfoinositide. La regolazione dipendente da AKT della forkhead box O (FoxO), del mammalian target of rapamycin complex 1 (mTORC1) e della glicogeno sintasi chinasi 3b (GSK3b) regola la crescita degli assoni, la trascrizione genica, la sintesi proteica e la plasticità neuronale. MEK, MAPK/ERK chinasi; PDK1, proteina chinasi 1 fosfoinositide-dipendente; SOS, son-of-sevenless. [Adattato da Servier Medical Art di Servier, con licenza Creative CommonsAttribuzione 3.0 Unported].
In definitiva, la Lantus viene solitamente usata come “base” di un protocollo di Insulina affiancata dall’uso del Humalog nei protocolli di Insulina e GH pre o post workout.
UI:CHO ratio e timing di somministrazione
Sappiamo tutti che il rapporto più frequentemente citato tra carboidrati e insulina (UI:CHO ratio) è di 10-15g di carboidrati per 1UI di Insulina. Ma non tutti i bodybuilder si attengono a questo rapporto. Diversi culturisti non hanno stabilito un rapporto fisso tra carboidrati e Insulina, mentre altri hanno utilizzato una gamma di rapporti (5-20g per unità di Insulina) con una media di 9-10g di carboidrati per ogni UI di Insulina.
La maggior parte dei bodybuilder ha assunto l’Insulina durante i pasti, ma considerando che i bodybuilder mangiano spesso, questo non ci dice molto. C’è stato un grande dibattito sul momento più efficace per l’uso dell’Insulina: alcuni suggeriscono che il momento più efficace sia il pre-workout, mentre altri affermano che si tratta di un uso irresponsabile, in quanto è difficile determinare quanti carboidrati verranno ossidati durante l’allenamento e quindi l’uso dell’Insulina nel pre-allenamento potrebbe essere pericoloso. Alcuni bodybuilder usano l’Insulina prima e dopo l’allenamento, altri solo dopo.
Alcuni bodybuilder sostengono che l’uso di Insulina a scopo ricreativo espone i bodybuilder al rischio di sviluppare il diabete. Altri bodybuilder sostengono che l’uso dell’Insulina riduce il rischio di diabete in quanto diminuisce l’impatto delle diete per il bodybuilding. Lasciatemi dire che l’ultima affermazione non ha alcun senso. Il corpo mantiene una condizione di omeostasi attraverso elaborati feedback di controllo. La somministrazione di Insulina esogena causerà un feedback negativo a livello della secrezione endogena pancreatica, e l’Insulina esogena somministrata avrà il medesimo effetto a livello centrale e periferico in un regime alimentare ipercalorico (vedi “dieta per il bodybuilding”) dell’Insulina endogena! E le affermazioni secondo le quali l’uso dell’Insulina esogena sortirebbe un effetto di protezione all’affaticamento pancreatico beh, è un affermazione che non ha basi di riscontro.
La follia del protocollo “No Fat Gain Insulin Program”:
Diversi anni fa riportai un protocollo d’uso dell’Insulina denominato “No Fat Gain Insulin Program”. Questo protocollo “alternativo” fu ideato da Mike Zumpano e Oliver Starr i quali si chiesero se ci poteva essere una strategia che permettesse ai bodybuilder di non aumentare eccessivamente di bf durante l’uso di Insulina. In realtà, la motivazione di base per la quale molti culturisti diventano più grassi che grossi quando nelle loro preparazioni inseriscono l’Insulina è fondamentalmente la “la paura” dell’ipoglicemia. La maggior parte del guadagno di grasso è causato dal consumo eccessivo di carboidrati durante l’uso di Insulina. Una regola di “sicurezza” diffusa con l’uso di Insulina dice che bisogna consumare un minimo di 10-15g di carboidrati per ogni UI di Insulina utilizzata (distribuiti nell’arco di tempo d’azione dell’Insulina utilizzata). Un altro errore commesso da molti bodybuilder e che porta ad un eccesso calorico addizionale è che essi non calcolano le UI in base ai CHO della dieta ma calcolano le UI in base al peso e di conseguenza aggiungono i carboidrati di “sicurezza” a quelli già presenti nel loro programma alimentare. Comunque sia, un Bodybuilder che utilizza 8UI di Insulina 2 volte al giorno, in aggiunta al suo normale apporto di carboidrati, proteine e grassi andrà (con il metodo standard) a consumere una quota addizionale di carboidrati pari a 160g. Difficilmente ci si aspetta che 160g in più di carboidrati, o 640Kcal in più al giorno facciano una differenza significativa su un soggetto che magari mangia 5000 o più calorie al giorno.
Anche se la quantità di carboidrati supplementari (10gXUI) comunemente applicata non sembra terribilmente eccessiva, alcuni “pionieri” dei PEDs alla fine degli anni ‘90 erano certi che fosse il motivo principale per cui gli utilizzatori di Insulina guadagnavano quantità sproporzionate di grasso.
Il plasma umano contiene solo circa 5g di carboidrati in uno specifico momento. I diabetici che hanno preso troppa Insulina di solito possono riportare i loro livelli glicemici nel sangue nel range di normalità consumando cinque grammi (solo 20 calorie!) di Destrosio.
Comunque sia, il metodo “alternativo” lo trovarono e fu ribattezzato, come precedentemente accennato, protocollo “No Fat Gain”. Il trucco, se così possiamo definirlo, sarebbe quello di assumere l’Insulina, ma seguendo un dieta Low-Carb. Proprio così, Low. Con un contenuto glucidico di circa 50g al giorno. Oliver Starr, con rudimentali conoscenze in biochimica e fisiologia umana, si chiese se ci fosse qualche altro modo per mantenere la glicemia nel sangue moderata con un alto grado di stabilità. La sua (riduttiva) risposta è stata la gluconeogenesi. Se si guarda su un grafico dei processi biochimici, si può chiaramente vedere che, quando le riserve di glicogeno epatico e muscolare sono esaurite, ma prima che il soggetto vada in chetosi, il corpo comincia a convertire aminoacidi in glucosio per mantenere stabili i livelli di glucosio nel sangue. Questo processo è noto come gluconeogenesi. Come risaputo, seguire una dieta molto povera di carboidrati provoca un esaurimento delle riserve di glicogeno epatico e muscolare. Questo provoca un sovra-regolazione degli enzimi necessari per la conversione rapida ed efficace degli aminoacidi in glucosio. La parola gluconeogenesi significa letteralmente “la nascita di nuovo glucosio.”
La seconda metà dell’ipotesi applicata, ovviamente, è il contenuto proteico della dieta. Se non si mangiano molti carboidrati, l’unico modo con cui il corpo può produrre glucosio è principalmente quello di convertire gli aminoacidi in glucosio. Questo accade in una certa misura ogni volta che si mangiano proteine, tuttavia, quando si mangia una grande quantità di proteine, si viene a creare ancora più glucosio. E’ il livello di glucosio creato dall’eccesso di proteine che dovrebbe impedire il verificarsi di uno stato ipoglicemico in un contesto nel quale si utilizza Insulina esogena con una dieta a basso contenuto di carboidrati.
Per questo protocollo è necessaria l’assunzione di proteine in polvere, perché non c’è modo di riuscire a essere in grado di mangiare la quantità di proteine che si richiedono da cibi interi. Per sostenere il livello di gluconeogenesi supposto per coprire le necessità durante l’utilizzo di Insulina si è proposto che la migliore strategia è quella in cui si consumano 600g di proteine da una combinazione di proteine del siero di latte e caseina, più un pasto solido che contiene da 50 a 100g di proteine, più alcune verdure fibrose a foglia verde. Il resto delle calorie devono provenire da fonti di grassi con scarsissimo o nullo contenuto di carboidrati.
Si può suddividere l’assunzione consumando una bevanda proteica ogni 30 minuti o un’ora, mescolando in un contenitore 3 litri con 100g di proteine e mantenendo una lista di quante volte si svuota ogni giorno.
Bisogna ricordare, però, che in questo protocollo, l’unica cosa che dovrebbe salvaguardare l’atleta (letteralmente) è l’assunzione di proteine. Se si utilizza l’Insulina con tali modalità e non si mantiene un adeguato apporto di proteine, le conseguenze saranno più che spiacevoli, saranno gravissime.
L’approccio teorico all’uso di Insulina in questo protocollo “No Fat Gains” è:
Giorni da 1 a 3: la deplezione di carboidrati. È necessario diminuire i carboidrati al di sotto dei 100g al giorno. Si suggerisce di arrivare a 50g di carboidrati il giorno 3. L’assunzione proteica deve aumentare a 450g al giorno.
Giorni da 4 a 30: le proteine devono essere pari o superiore a 600g al giorno. I carboidrati devono essere mantenuti tra i 100 e i 50g al giorno (50g è meglio) e si dovrebbero utilizzare i grassi affinché si compensi l’equilibrio delle proprie esigenze caloriche giornaliere. Come già detto, si raccomanda l’uso di proteine in polvere di composizione mista (siero di latte e caseina), anche se è possibile utilizzare alcuni cibi interi, se lo si desidera. (Basta tenere a mente che 600g grammi di proteine corrispondono all’incirca a più di 2.5Kg di petto di pollo o di tacchino al giorno)
Partire da una piccola dose di Insulina (4 UI) per poi aumentarla gradualmente (fino anche a 12UI x 3 volte al giorno).
Monitorare regolarmente la glicemia durante il giorno e in specie nel periodo di massima azione dell’Insulina. Se si inizia a perdere la capacità di rimanere svegli, prendere una zolletta di zucchero.
Si, questo protocollo non è soltanto folle ma, cosa fondamentale, è basato su una conoscenza superficiale e con enormi lacune della fisiologia umana. Perchè? Perchè l’Insulina è un regolatore in negativo della gluconeogenesi!
Il ruolo dell’Insulina nella regolazione della produzione epatica di glucosio è ampiamente accettato. Negli individui sani, l’iperinsulinemia fisiologica sopprime la gluconeogenesi del 20%, mentre la glicogenolisi è completamente soppressa. L’iperglicemia da sola sopprime la glicogenolisi epatica con effetti minimi sull’immagazzinamento del glicogeno. Solo la combinazione di iperglicemia e iperinsulinemia ha un effetto significativo sulla sintesi epatica di glicogeno. Pertanto, l’Insulina svolge un ruolo cruciale nel metabolismo epatico del glucosio.
Effetti dell’Insulina sul metabolismo del glucosio e dei lipidi.
Il meccanismo dominante della regolazione insulino-mediata della gluconeogenesi epatica non è però chiaro. L’Insulina esercita un controllo diretto della gluconeogenesi agendo sul fegato, ma influisce anche indirettamente sulla gluconeogenesi agendo su altri tessuti. L’effetto diretto dell’Insulina è stato dimostrato nei cani a digiuno, dove l’Insulina plasmatica portale ha soppresso la produzione epatica di glucosio, anche in assenza di variazioni del glucagone o dei precursori gluconeogenici. Tuttavia, nei modelli murini, l’Insulina è risultata avere effetti più potenti sulla produzione epatica di glucosio in vivo piuttosto che in vitro. Inoltre, è stato dimostrato che gli effetti indiretti dell’Insulina sui tessuti extraepatici sono sufficienti a mantenere il normale metabolismo del glucosio, suggerendo un ruolo importante per la regolazione indiretta della gluconeogenesi da parte dell’Insulina.
Regolazione dell’espressione genica della gluconeogenesi da parte del segnale insulinico epatico. L’azione dell’Insulina regola l’attività dei fattori di trascrizione che controllano l’espressione dei geni gluconeogenici. La fosforilazione mediata da AKT porta all’esportazione nucleare di FOXO1. La fosforilazione inibitoria di CBP/p300 blocca la formazione del complesso di trascrizione di CREB. La modifica di PGC-1α mediante acetilazione mediata da GCN5 o fosforilazione mediata da AKT/CLK2 riduce l’attività trascrizionale di PGC-1α.
Un sistema di segnalazione insulinica intatto è fondamentale per mantenere i livelli di glucosio nel sangue all’interno di un range glicemico normale e ristretto durante i periodi di digiuno o di eccesso di disponibilità di nutrienti, e questo si ottiene in particolare attraverso la regolazione del flusso metabolico attraverso la via gluconeogenica epatica. Un importante nodo di controllo coinvolge la regolazione trascrizionale dell’espressione dei geni chiave della gluconeogenesi epatica Pck1 e G6pc, che avviene principalmente attraverso il fattore di trascrizione FOXO1 e il recettore nucleare HNF4α e il loro coattivatore trascrizionale PGC-1α. La comprensione di queste vie di regolazione è di estrema importanza per comprendere come un massivo consumo proteico in regime low carb non possa assolutamente garantire una stabilità glicemica in presenza di trattamento con Insulina esogena!
Ma si ingrassa di meno con questo protocollo? Direi di no o, comunque, la differenza è irrisoria… Vorrei ricordare in tal sede che, per quanto dispendioso in termini energetici, in un contesto di eccesso calorico (essenziale in regimi “Bulk”) dove si consumano più proteine del necessario, l’organismo utilizza gli AA che le compongono o come come fonte energetica di scarsa resa [3,3Kcal per 1lt di Ossigeno] o li converte in acidi grassi! Certo, per necessità di deficienza nutrizionale la gluconeogenesi degli AA garantisce una stabilità glicemica, ma in fisiologia e non quando quest’ultima viene marcatamente alterata dall’uso di Insulina esogena!
Per concludere questo paragrafo, è corretto sottolineare che la gluconeogenesi si verifica dopo circa 8 ore di digiuno o scarso apporto glucidico, quando le scorte di glicogeno del fegato iniziano a esaurirsi ed è necessaria una fonte alternativa di glucosio. Inoltre è un processo biochimico piuttosto lento e assolutamente non garante della ben che minima sicurezza in un regime d’uso di Insulina in contesto low carb.
L’uso dell’Insulina pre-workout:
I dibattiti sulla reale efficacia dell’Insulina come agente anabolizzante ha spinto atleti e preparatori a sperimentare protocolli diversi. L’unico di questi che si è avvicinato maggiormente ad una logica d’insieme è ““The Ultimate Insulin Protocol” di Mike Arnold.
Ciò che sta alla base di questo protocollo non è altro che una versione “arricchita” di ciò che sta alla base dell’integrazione “intra-workout”.
Spesso si ragiona sul fatto che durante l’allenamento ci si trova in uno stato fondamentalmente catabolico e dopo in uno stato fondamentalmente anabolico. In realtà però, la sintesi proteica ed i meccanismi di anabolismo e catabolismo sono sempre attivi, con diverse prevalenze, quindi i processi di riparazione tissutale iniziano già nel momento in cui il muscolo viene danneggiato e durante il danneggiamento. Questo è ancor più vero nel momento in cui il workout sarà incentrato su più gruppi muscolari. La presenza di una concentrazione di Insulina esogena in circolo addizionata all’introduzione di macronutrienti ben calibrati (vedi integrazione intra-workout) renderebbe maggiormente incisivo il processo.
Il pump e la pienezza muscolare che si può raggiungere seguendo questo protocollo sono, a detta dei “tester”, impressionanti. Il programma trova la sua “magia” nella sua tempistica e nella sinergia degli ingredienti utilizzati.
Qui di seguito è riportato il protocollo nella sua interezza:
60 minuti pre-workout: *** optional (Uno qualsiasi dei supplementi NO stimolanti sul mercato. Gaspari Nutrition “Vasotropin” è un ottimo prodotto).
45 minuti pre-workout: 15UI di Humulin R.
20 minuti pre-workout: 50g di carboidrati ad alto peso molecolare (ex: Vitargo, Karbolyn, etc). 20g di proteine idrolizzate (es: Hydrowhey, Carnivore). 20g di Glicerolo Monostearato. 3g di Leucina. 5g di Creatina Monoidrata Micronizata. 2g grammi di Beta-Alanina. 10g di Glutammina. 3g di Taurina. 500mg di Potassio. 1g di Vitamina C.
75 minuti dopo il 1° shake: 50g di carboidrati ad alto peso molecolare. 20g di protein idrolizzate (es: Hydrowhey, Carnivore). 10g di Glicerolo Monostearato. 3g di Leucina. 5g di Creatina Monoidrata Micronizzata. 2g Beta-Alanina. 10g di Glutammina. 3g di Taurina.
75 minuti dopo il 2° shake: 50g di carboidrati ad alto peso molecolare. 20g di protein idrolizzate (es: Hydrowhey, Carnivore). 3g di Leucina. 5g di Glutammina.
Proteine totali: 60g (esclusi gli amminoacidi in forma libera aggiunti)
Carboidrati totali: 150g (escluse le trace di carboidrati contenute nelle polveri proteiche).
Prima di tutto, nel formulare il rapporto macros/Insulina sopra esposto, Arnold ha aumentato la quantità di carboidrati-proteine al di sopra di ciò che è tipicamente necessario per una UI di Insulina, al fine di tenere conto degli utilizzatori che dimostrano un grado superiore alla media di sensibilità all’Insulina. La maggior parte degli utilizzatori di Insulina, richiedono circa 8g di carboidrati-proteine per UI di Insulina, al fine di chiudere in pareggio e mantenere la normale soglia di glucosio nel sangue. Questo protocollo utilizza un rapporto 14:1 (macros/Insulina), che dovrebbe permettere a praticamente chiunque di utilizzare questo programma mantenendo il glucosio nel sangue all’interno di un range di normalità.
Va notato che questo programma è stato progettato per essere seguito “come è scritto”, soprattutto per quanto riguarda i tempi di assunzione dei nutrienti e le loro quantità. Per gli utilizzatori di Insulina già “navigati” che sanno quali rapporti sono ideali per loro, essi hanno la libertà di ridurre la quantità di macros consumati per UI, se necessario, come determinato dalla valutazione della loro risposta metabolica. Per gli utilizzatori inesperti, la componente nutrizionale del programma dovrebbe essere rispettata come scritta per almeno 2 settimane, a questo punto l’utilizzatore può quindi iniziare a personalizzare il suo rapporto macros/Insulina, se necessario.
La base di questo programma poggia sul tipo specifico di macros utilizzati. Senza di loro, ogni altro componente/aspetto del programma è influenzato negativamente e in alcuni casi rende il tutto sensibilmente limitato negli effetti. Carboidrati ad alto peso molecolare, come il Vitargo o le Ciclodestrine Altamente Ramificate, hanno dimostrato di essere superiori ad altre forme di carboidrati in diversi modi, per esempio un rapido e costante rilascio di glucosio nel sangue e una bassissima osmolaritá in soluzione.
Passando alla componente proteica; le proteine idrolizzate sono molto più velocemente assorbite rispetto ad altri tipi di proteine e sono l’unica proteina che può essere consumata insieme ai carboidrati ad alto peso molecolare senza compromettere il loro assorbimento.
In questo protocollo vi è anche la possibilità di un aggiunta di uno stimolatore del NO a scelta. L’aggiunta di questi stimolatori (vedi, ad esempio, la Citrullina Malato), pur non “necessario”, aumenterà ulteriormente la circolazione e il trasporto dei nutrienti ai muscoli che lavorano, così come contribuiscono ad aumentare il pumping sperimentato durante e dopo l’allenamento. Il Glicerolo monostearato è incluso anche tra gli ingredienti per il suo ruolo di volumizzante muscolare. Questo composto viene spesso utilizzato appena prima della gara, al fine di contribuire al raggiungimento di uno aspetto pieno e asciutto durante l’esibizione. Sono presenti anche dei volumizatori tradizionali, come la Glutammina, la Taurina, la Creatina, e il Potassio.
Al fine di promuovere un maggiore recupero e una maggiore risposta per una crescita muscolare, la temporizzazione dell’assunzione degli shake è stata messa a punto per mantenere un flusso costante dei nutrienti per tutta la vita attiva dell’Insulina. L’Humulin R è stata appositamente scelta per questo scopo, dal momento che con la sua emivita permetterà all’utilizzatore di sfruttare le “finestre” sia intra che post-allenamento. L’Humulin R offre anche un picco di Insulina meno pronunciato, che risulta più facile da gestire per una buona parte degli utilizzatori rispetto ad una versione di Insulina ad azione più rapida, come ad esempio l’Humalog che, però, viene in alcuni casi sostituita alla scelta classica.
Per via del tempo di esposizione all’Insulina limitato con la pratica di questo protocollo, la sensibilità all’Insulina è solo moderatamente influenzata in modo diretto quando si utilizza il programma per circa 5-6 volte alla settimana. Per gli individui che scelgono di utilizzare il presente protocollo per 3-4 volte alla settimana, le alterazioni dirette sulla sensibilità all’Insulina non risulta essere un problema. Per coloro che eseguono il protocollo per 5-6 volte a settimana, possono intervenire in due modi per assicurare una pienamente ottimale sensibilità all’insulina:
L’utilizzatore può fare 2 settimane “off” ogni 4 settimane “on”.
L’individuo può aggiungere Metformina nel suo programma di 3-4 volte a settimana ad un dosaggio di 750mg-1g/die.
Per gli utilizzatori che si apprestano all’uso di questo protocollo per la prima volta, mentre il rapporto macros / Insulina di cui sopra è sufficiente, l’autore consiglia sempre di iniziare con un dosaggio ridotto e aumentarlo poco a poco fino al raggiungimento del pieno dosaggio. Per uno novizio, un dosaggio di 6-8UI è ideale. Questo può essere seguito da una seconda iniezione da 8-10UI, per poi passare ad una terza iniezione di 10-12UI prima di arrivare al dosaggio massimo di 15UI.
Il protocollo in questione non contempla il solo uso di “shake e insulina” ma anche di GH (nel pre-workout a distanza dall’Insulina) e del IGF-1lr3 (nel post workout).
Il protocollo inizia con un’assunzione di GH, circa 30 minuti prima del workout per far si che i livelli plasmatici siano ragionevolmente alti, prima di aggiungere la dose di Insulina. L’idea alla base di questo, è assicurarsi che il GH passi attraverso il fegato mentre si ha una notevole quantità di Insulina in circolo. Questo è il modo in cui produciamo grandi picchi di IGF-1. Dopo l’allenamento, si somministra l’IGF-1LR3.
Il protocollo esemplificativo è il seguente:
30 minuti pre-workout: 6-10UI di GH subq
15 minuti pre-workout: 6-16UI di Novalog subq
10 minuti pre-workout: assumere lo shake #1
Dopo ogni set eseguito: sorseggiare lo shake #2, e terminarlo entro la fine dell’allenamento.
Andare a casa
Somministrare la dose di 100mcg di IGF-1lr3 (per i suoi effetti sulla sensibilità all’Insulina
Assumere lo shake #3
Formulazione degli shake:
Shake 1: 10-20g di EAA (Amino Acidi Essenziali) o PeptoPro, 40-60g Vitargo, 5g Creatina Monoidrata Micronizata, 200mg di Caffeina (migliora la resintesi di glicogeno); anche in questo caso la dose di carboidrati per ogni UI si aggira in media sui 7-10g.
Shake 2: 10-20g EAA o PeptoPro, 50-100g Vitargo, 5g di Carnitina Monoidrata Micronizata.
Shake 3: 2 tazze di albume pastorizzato, 1 tazza di avena istantanea, 1 banana o 1 tazza di mirtilli, Splenda o Stevia.
La somministrazione di Insulina pre-workout trova la sua motivazione d’essere nel fatto che durante l’allenamento con i pesi si viene a creare uno stress meccanico che a sua volta crea una sovra regolazione dello stimolo anabolico. Più comunemente, questo stato iper-anabolizzante viene indicato come “finestra anabolica”; un determinato periodo di tempo che dura dall’inizio dell’allenamento fino a poche ore dopo. Un modo in cui il corpo reagisce all’allenamento con i pesi è attraverso un aumento della sensibilità all’Insulina. Questo accade quando i recettori per l’Insulina, che risiedono sulla superficie della cellula, rispondono al segnale dell’Insulina in modo più efficiente, cosa che ci permette un migliore assorbimento dei nutrienti all’interno della cellula. Oltre a ciò, avviene un aumento della proliferazione dei GLUT-4 sulla superficie cellulare in maniera insulino-indipendente, caratteristica che richiede una minore quantità di Insulina per avere i massimi effetti nell’uptake cellulare.
Infatti, l’allenamento promuove anche il recupero e la crescita a livello intracellulare aumentando la sintesi proteica, la glicogeno sintasi, dei GLUT4 e l’espressione del trasportatore degli aminoacidi, e diminuendo i livelli di Miostatina. In combinazione con un aumento della sensibilità all’Insulina, queste cose non solo si traducono in una crescita accelerata, ma forniscono un effetto di ri-partizionamento dei nutrienti, in cui il cibo che assumiamo e i macronutrienti ivi contenuti vengono indirizzati maggiormente verso il miocita (cellule muscolari), piuttosto che immagazzinati come grasso.
L’Insulina è il complemento perfetto per questa “finestra anabolica” durante l’allenamento, in quanto non solo i nutrienti vengono trasportati alle cellule muscolari, permettendo così al corpo di approfittare di questo stato anabolizzante intensificato, ma l’Insulina agisce anche per molti dei processi di costruzione muscolare, fornendo uno stimolo ipertrofico raddoppiato nel momento in cui il corpo è più sensibile alla risposta del segnale dell’Insulina.
Come anti-catabolico, e lo abbiamo anche visto nella review presente nella terza parte di questa serie di articoli, l’Insulina è decisamente efficace, riducendo sensibilmente la degradazione del tessuto muscolare che si somma attivamente con la ripartizione delle proteine muscolari, e il tempo di recupero risulta ridotto. Questo ha un duplice effetto:
alterazione in positivo del rapporto catabolismo:anabolismo e maggiore supercompensazione (crescita muscolare);
il recupero più rapido si traduce in un aumento della possibile frequenza di allenamento, permettendo ai muscoli di essere stimolati più volte entro un determinato periodo di tempo e, in definitiva, di crescere più rapidamente;
infine, le concentrazioni di Insulina sovrafisiologiche aumentano in modo significativo il volume ematico all’interno nel tessuto muscolare, aggiungendo un pump muscolare generale che di per se stimola la crescita.
Tuttavia, affinché il corpo possa mettere a frutto tutto questo, sono necessari lo stimolo allenante adeguato e i nutrienti giusti che devono essere presenti al momento giusto. A questo punto entra in gioco la nutrizione intra-allenamento presentata in precedenza.
Nonostante questo protocollo abbia rappresentato la migliore applicazione dell’uso di Insulina nel Bodybuilding, esso presenta delle lacune oltre che dei problemi tecnici che riducono la pienezza del potenziale e il margine di “sicurezza”.
Alcuni atleti hanno sperimentato una maggiore tendenza all’ipoglicemia durante i loro workout con protocolli di Insulina pre workout. Questo imprevisto è limitante oltre che decisamente pericoloso, specie durante un workout. E’ ovvio che il controllo della glicemia e il rispetto dei punti base del protocollo riducono le possibilità del verificarsi di casi ipoglicemici. Ma il rischio è comunque maggiore rispetto ad una condizione di trattamento base (assunzione classica ai pasti).
Inoltre, tiene poco in considerazione le reale farmacocinetica dei PEDs utilizzati riducendo le piene ed ottimali interazioni ed effetti additivi.
GH/Insulin Protocol – pre e post-workout:
In risposta alle lacune del protocollo di Arnold che abbiamo appena visto, ho realizzato una versione perfezionata denominata “GH/Insulin Protocol”.
Tale perfezionamento prende in considerazione in modo preciso sia la farmacocinetica che la farmacodinamica incrociata dei componenti Insulina e GH.
Ora, sappiamo che la curva di rilascio del hGH somministrato per via sottocutanea raggiunge un picco iniziale dopo 30 minuti post iniezione per poi attestarsi a 2-3h e subire un calo significativo dopo 4h dalla somministrazione. Lo strascico di IGF-1 perdura per circa 24h sopra il basale.
Sappiamo inoltre che l’Insulina aumenta la sensibilità epatica del GH con risposta massiva nella sintesi e rilascio di IGF-1riduzione del IGFBP-1 e IGFBP-2 con conseguente aumento della frazione libera e bioattiva di IGF-1;3) l’aumento della sensibilità del GH a livello epatico porta anche ad una riduzione della IGF-1/IGFBP-3 ratio con ulteriore incremento della frazione libera e bioattiva di IGF-1.
Interazioni biologiche a livello ipofisario ed epatico tra Insulina, Ormone della Crescita (GH), Fattore di Crescita Insulino-Simile-I (IGF-I) e Proteine Leganti il Fattore di Crescita Insulino-Simile (IGFBPs). Le frecce aperte indicano la stimolazione e le linee nere sottili l’inibizione. Livelli elevati di Insulina (a destra) possono aumentare indirettamente la biodisponibilità di IGF-I (cerchi pieni) sopprimendo la produzione di IGFBP-1 e, in misura minore, di IGFBP-2 (simboli ombreggiati). A sua volta, l’aumento della biodisponibilità di IGF-I può aumentare l’effetto di feedback negativo sul GH (quadrato aperto), portando a una riduzione della secrezione di GH e a una minore produzione epatica di IGF-I e IGFBP-3. Tuttavia, livelli elevati di Insulina possono anche aumentare il numero e l’attività dei recettori epatici del GH (barra aperta), riflessi da un aumento dei livelli di Proteina Legante l’Ormone della Crescita (GHBP) circolante. Questo effetto può portare a un aumento della produzione epatica di IGF-I e IGFBP-3 regolata dal GH, con un aumento maggiore dei livelli di IGF-I circolante rispetto a quelli di IGFBP-3. Pertanto, insieme a fattori genetici, ormonali e ambientali, l’entità relativa di questi due effetti opposti dell’Insulina sulla produzione di IGF-I potrebbe determinare i livelli di IGF-I circolante. Nel tempo, un’eccessiva biodisponibilità di IGF-I potrebbe aumentare il rischio di cancro del colon-retto, favorendo la sopravvivenza di cellule trasformate e mutate che normalmente andrebbero incontro ad apoptosi.
Quindi, dal momento che l’obbiettivo è quello di creare un ambiente non fisiologicamente riproducibile senza alterazioni iatrogene al fine di ottenere la massima risposta anabolica dal protocollo, il primo punto di congiunzione, o meglio la chiave di volta del protocollo, deve essere l’incrocio del picco di hGH con quello dell’Insulina. Per fare ciò le modalità di somministrazione dovrebbero contemplare primariamente l’iniezione di hGH pre-workout e quella di Insulina (preferibilmente Humalog per via del picco raggiunto entro 15 minuti dalla somministrazione) nel post-workout. Un vantaggio aggiuntivo di questa modifica è la riduzione del rischio ipoglicemico durante la sessione di allenamento.
E no, il vantaggio dello shake intra-workout non viene perso dal momento che la ripartizione calorica è di per se ottimale per via di meccanismi insulino-indipendenti dati dall’attività contro-resistenza (vedi aumento dei GLUT-4 sulla superficie cellulare in seguito all’attività muscolare). Si veda anche l’assorbimento dilazionato dei composti facenti parte della soluzione ingerita e costituente lo shake intra-workout.
I punti chiave del protocollo sono i seguenti:
somministrazione di hGH pre-workout [UI utilizzate dalle 4 alle 8, in questo ultimo caso divise in una dose da 4UI pre workout e due da 2UI appena sveglio e prima di dormire, a secondo della modalità d’uso del hGH; se somministrato giornalmente o a giorni alterni. Vedi a tal proposito l’articolo dedicato alla somministrazione di hGH a giorni alterni];
1h pre-workout possibilità di assumere tra i 25 ed i 50mg di Sildenafil seguiti a 30 minuti dall’inizio del workout dall’assunzione di 8g di Citrullina Malato;
consumare uno shake intra-workout contenente di base 0,5-1g di carboidrati ad alto peso molecolare, 0,25g di proteine idrolizzate e 5g di Creatina Monoidrato;
somministrazione dell’Insulina (Humalog) post-workout [le UI vanno calibrate in base ai CHO consumati nel pasto post-workout; in linea di massima 1UI ogni 10Kg di peso]. L’Humalog può essere sostituita con l’Humulin-R se per la risposta del soggetto in quanto a tolleranza è migliore.
Schema esemplificativo dell’azione incrociata tra Insulina e GH a livello epatico e sue consequenziali principali aree di influenza ricercate.
Esiste una variante del suddetto protocollo la quale contempla l’uso di due tipi di Insulina: una base (Lantus) e una post-workout (Humalog):
calcolare la dose totale di Insulina giornaliera con la formula 1UI ogni 10Kg di peso;
dividere il totale della dose a metà tra Insulina Glargine (Lantus) e Insulina lispro (Humalog) o Humulin-R;
somministrazione di Insulina glargina (Lantus) dividendo la dose giornaliera in 2: la prima, pari al 65% della dose totale, al mattino in concomitanza con il primo pasto e la seconda, pari al 35% della dose totale, 12h dopo la prima somministrazione;
somministrazione di hGH pre-workout [UI utilizzate dalle 4 alle 8 a secondo della modalità d’uso del hGH];
somministrare la dose di Insulina lispro o Humulin-R nel post-workout.
L’uso di una base di Insulina rappresentata dalla Insulina glargina ha il potenziale di aumentare ulteriormente l’espressione del IGF-1 e, di conseguenza, la risultante anabolizzante del protocollo. Ma, ovviamente, i rischi di ipoglicemia con questa modifica protocollare sono maggiori.
Grafico esemplificativo delle curve ematiche di Insulina, hGH e IGF-1 nel protocollo con Insulina Glargine di base.
Il monitoraggio regolare della glicemia e il rispetto dei punti base del protocollo evitano con un buon margine il verificarsi di casi ipoglicemici. Quindi, il glucometro, che sia classico o con sensore, è essenziale in questi casi.
Repetita iuvant: QUESTO PROTOCOLLO COME I PRECEDENTEMENTE PRESENTATI, E L’USO DI INSULINA, SONO APPANNAGGIO DI ATLETI AVANZATI E MONITORATI DA PERSONALE QUALIFICATO! NULLA DI CIO’ CHE E’ STATO DESCRITTO DEVE ESSERE PRESO COME UNA PRESCRIZIONE MEDICA O UN CONSIGLIO!
Conclusioni “dopo un lungo viaggio”:
Siamo ora giunti alla conclusione di questa serie di articoli dedicati all’Insulina e al suo centesimo anniversario.
Durante questo lungo percorso abbiamo imparato a conoscere meglio questo affascinante e mal compreso peptide. Abbiamo visto come è stato scoperto, abbiamo compreso l’enorme passo avanti nella medicina e nella tutela della vita umana che la sua scoperta ha rappresentato, abbiamo compreso come essa sia fisiologicamente regolata e quali sono le sue reali azioni a livello sistemico ed abbiamo imparato a separare i luoghi comuni che vi aleggiano intorno dai fatti.
Con la terza parte molti sono rimasti delusi nel constatare che in fisiologia l’Insulina abbia un effetto prettamente anticatabolico, effetto preminente che mantiene anche se somministrata esogenamente entro i range fisiologici [<1.200ng/dl]. Altresì gli umori sono migliorati quando siamo venuti a conoscenza del fatto che non solo il dosaggio fa la differenza tra preminenza anticatabolica e anabolica ma anche la sensibilità.
Con la recente constatazione che il risultato dell’equazione “Insulina/anabolismo” cambi drasticamente in positivo se vi si aggiunge la variabile del hGH, abbiamo imparato che l’unico uso minimamente sensato dell’Insulina per il miglioramento della massa muscolo-scheletrica è in associazione con il peptide ipofisario con le ultime due modalità esposte.
Ci tengo però a precisare che l’uso dell’Insulina “off-label” dovrebbe rappresentare la componente più marginale nella carriera di un culturista. Vale a dire che se ne può benissimo fare a meno, in specie quando si è semplici amatori o agonisti di piccoli o medi circuiti competitivi. Non complicatevi la vita.
Fate tesoro delle nozioni che vi ho esposto affinché il confine della conoscenza si espanda e prevalga su quello dell’ignoranza.
Uno dei farmaci più incompresi e discussi nel BodyBuilding è sicuramente l’Insulina. Ciò è dovuto dal fatto che non esiste una vera scienza che funga da base per le modalità in cui i bodybuilder possano utilizzarla con criterio. Questo fa sì che tutte le conoscenze in possesso della maggior parte dei culturisti sull’uso dell’Insulina siano nulla più che “broscience”. Usando il termine “broscience” non intendo screditare una certa forma di conoscenza esperienziale. Infatti essa, se correttamente intesa nei suoi limiti, ha una certa importanza tanto che a volte capita che alcuni intuitivi atleti siano in grado di scoprire dettagli prima che questi vengano catalogati dalla letteratura scientifica e possono avere ragione anche quando la ricierca scientifica pecca nel design degli studi in cui vuole dimostrare una tesi (Holt 2009). Ma spesso e volentieri quello che i bodybuilder dicono sull’Insulina è una vera e propria stronzata. La pratica dell’uso di Insulina da parte dei bodybuilder si basa su un mucchio di studi mal intesi e su un mucchio di dicerie da guru che parlano di spiegazioni dal sapore pseudo-scientifico. Pochi di questi soggetti hanno una formazione scientifica o medica, per non parlare della competenza in endocrinologia. Alcuni di loro non hanno la minima idea di cosa stiano parlando, ma si comportano come se l’avessero. Come si fa a sapere a chi dare retta? Semplice! Conoscendo l’Insulina dalle basi alla pratica!
Ho quindi deciso, visto anche il centenario della sua scoperta, di scrivere una serie di articoli attraverso i quali vi accompagnerò lungo un secolo di storia dell’Insulina, dal suo isolamento alla sua applicazione medica passando, infine, al suo uso nel BodyBuilding.
In questa prima parte vedremo il lato accademico dell’Insulina…
Tanto tempo fa, tra due continenti…:
Nel 1869, studiando la struttura del pancreas al microscopio, Paul Langerhans, studente di medicina a Berlino, identificò alcuni ammassi di tessuto precedentemente inosservati, sparsi nella maggior parte del pancreas.[1] La funzione di questi “mucchietti di cellule”, in seguito noti come isolotti di Langerhans, rimase inizialmente sconosciuta, ma Édouard Laguesse suggerì in seguito che potessero produrre secrezioni che svolgono un ruolo regolatore nella digestione.[2] Anche il figlio di Paul Langerhans, Archibald, contribuì a comprendere questo ruolo regolatore.
Paul Langerhans (25 luglio 1847 – 20 luglio 1888) è stato un patologo, fisiologo e biologo tedesco, a cui si deve la scoperta delle cellule che secernono Insulina, che da lui prendono il nome di isole di Langerhans.
Nel 1889, il medico Oskar Minkowski, in collaborazione con Joseph von Mering, rimosse il pancreas da un cane sano per verificare il suo presunto ruolo nella digestione. Analizzando l’urina, trovarono dello zucchero, stabilendo per la prima volta una relazione tra il pancreas e il diabete. Nel 1901, un altro passo importante fu compiuto dal medico e scienziato americano Eugene Lindsay Opie, quando isolò il ruolo del pancreas alle isole di Langerhans: “Il diabete mellito, quando è il risultato di una lesione del pancreas, è causato dalla distruzione delle isole di Langerhans e si verifica solo quando questi corpi sono in parte o completamente distrutti”.[3][4][5]
Oskar Minkowski (13 gennaio 1858 – 18 luglio 1931) è stato un medico e fisiologo tedesco, titolare di una cattedra all’Università di Breslau e famoso soprattutto per le sue ricerche sul diabete. Era fratello del matematico Hermann Minkowski e padre dell’astrofisico Rudolph Minkowski.
Nei due decenni successivi i ricercatori fecero diversi tentativi di isolare le secrezioni delle isole pancreatiche. Nel 1906 George Ludwig Zuelzer ottenne un parziale successo nel trattamento di cani con estratti pancreatici, ma non fu in grado di continuare il suo lavoro. Tra il 1911 e il 1912, E.L. Scott dell’Università di Chicago sperimentò estratti acquosi di pancreas e notò “una leggera diminuzione della glicosuria”, ma non riuscì a convincere il suo direttore del valore del suo lavoro, che venne interrotto. Israel Kleiner dimostrò effetti simili alla Rockefeller University nel 1915, ma la Prima Guerra Mondiale interruppe il suo lavoro e non lo riprese.[6]
Georg Ludwig Zülzer (10 aprile 1870 a Berlino;16 ottobre 1949 a New York) è stato un medico internista tedesco che ha condotto ricerche nel campo del trattamento del diabete mellito. Sulla base della scoperta di Oskar Minkowski, alla fine del XIX secolo, che l’asportazione del pancreas nei cani scatenava il diabete mellito di tipo I, all’inizio del XX secolo Georg Ludwig Zülzer condusse esperimenti sull’uso di estratti di pancreas per il trattamento del diabete.
Nel 1916, Nicolae Paulescu sviluppò un estratto acquoso di pancreas che, iniettato in un cane diabetico, aveva un effetto normalizzante sui livelli di zucchero nel sangue. Dovette interrompere i suoi esperimenti a causa della Prima Guerra Mondiale e nel 1921 scrisse quattro articoli sul suo lavoro svolto a Bucarest e sui suoi test su un cane diabetico. Più tardi, nello stesso anno, pubblicò “Research on the Role of the Pancreas in Food Assimilation”.[7][8]
Nicolae Constantin Paulescu (30 ottobre 1869 (O.S.) – 17 luglio 1931) è stato un fisiologo, professore di medicina e politico rumeno, famoso soprattutto per i suoi lavori sul diabete, tra cui il brevetto della pancreina (un estratto pancreatico contenente Insulina). La “pancreina” era un estratto di pancreas bovino in soluzione salina, dopo di che alcune impurità venivano rimosse con acido cloridrico e idrossido di sodio. Paulescu fu anche cofondatore, insieme ad A. C. Cuza, dell’Unione Nazionale Cristiana e successivamente della Lega di Difesa Nazionale Cristiana in Romania. È stato anche un membro di spicco della Guardia di Ferro.
Il nome “Insulin” fu coniato da Edward Albert Sharpey-Schafer nel 1916 per un’ipotetica molecola prodotta dalle isole pancreatiche di Langerhans (in latino insula per isolotto o isola) che controlla il metabolismo del glucosio. All’insaputa di Sharpey-Schafer, Jean de Meyer aveva introdotto il termine molto simile “Insulina” nel 1909 per la stessa molecola.[9][10]
Sir Edward Albert Sharpey-Schafer (2 giugno 1850 – 29 marzo 1935) è stato un fisiologo inglese. È considerato un fondatore dell’endocrinologia: nel 1894 scoprì e dimostrò l’esistenza dell’adrenalina insieme a George Oliver e coniò il termine “endocrino” per le secrezioni delle ghiandole non duttili. Il metodo di respirazione artificiale di Schafer prende il nome da lui. Schafer coniò il termine “insulin” dopo aver teorizzato che l’assenza di una singola sostanza prodotta dal pancreas fosse responsabile del diabete mellito.
Nell’ottobre del 1920, il canadese Frederick Banting giunse alla conclusione che le secrezioni digestive studiate originariamente da Minkowski stavano disgregando il secreto delle isole, rendendone impossibile l’estrazione. Chirurgo di formazione, Banting sapeva che l’ostruzione del dotto pancreatico avrebbe portato all’atrofia della maggior parte del pancreas, lasciando intatte le isole di Langerhans. Pensò che si sarebbe potuto ricavare un estratto relativamente puro dalle isole una volta che la maggior parte del resto del pancreas fosse stata eliminata. Si appuntò una nota: “Legare i dotti pancreatici del cane. Mantenere i cani in vita finché gli acini non degenerano lasciando gli isolotti. Cercare di isolare la secrezione interna di questi ultimi e alleviare la glicosuria.”[11][12]
Sir Frederick Grant Banting (14 novembre 1891 – 21 febbraio 1941), scienziato, medico, pittore e premio Nobel noto come co-scopritore dell’Insulina e del suo potenziale terapeutico.
Nella primavera del 1921, Banting si recò a Toronto per spiegare la sua idea a J.J.R. Macleod, professore di fisiologia all’Università di Toronto. Macleod era inizialmente scettico, poiché Banting non aveva un background di ricerca e non conosceva la letteratura più recente, ma accettò di mettere a disposizione di Banting uno spazio di laboratorio per testare le sue idee. Macleod fece anche in modo che due studenti universitari fossero gli assistenti di laboratorio di Banting quell’estate, ma Banting aveva bisogno di un solo assistente di laboratorio. Charles Best e Clark Noble lanciarono una moneta; Best vinse il lancio e prese il primo turno. Ciò si rivelò sfortunato per Noble, poiché Banting tenne Best per tutta l’estate e alla fine divise con Best metà del premio Nobel e il merito della scoperta.[13] Il 30 luglio 1921, Banting e Best riuscirono a isolare con successo un estratto (“isleton”) dalle isole di un cane e lo iniettarono in un cane diabetico, scoprendo che l’estratto riduceva la glicemia del 40% in 1 ora.[14][12]
Charles Herbert Best (27 febbraio 1899 – 31 marzo 1978) è stato uno scienziato medico americano-canadese, uno dei co-scopritori dell’insulina insieme al collega Banting.
Banting e Best presentarono i loro risultati a Macleod al suo ritorno a Toronto nell’autunno del 1921, ma Macleod sottolineò i difetti del disegno sperimentale e suggerì di ripetere gli esperimenti con un maggior numero di cani e con attrezzature migliori. Trasferì Banting e Best in un laboratorio migliore e iniziò a pagare a Banting uno stipendio con le sue borse di ricerca. Alcune settimane dopo, anche la seconda serie di esperimenti fu un successo e Macleod contribuì a pubblicare i risultati privatamente a Toronto nel novembre dello stesso anno. Bloccato dal lungo compito di legare i cani ai condotti pancreatici e di aspettare diverse settimane per estrarre l’Insulina, Banting ebbe l’idea di estrarre l’Insulina dal pancreas di un vitello fetale, che non aveva ancora sviluppato le ghiandole digestive. A dicembre, riuscirono a estrarre l’insulina anche dal pancreas di una mucca adulta. Macleod interruppe tutte le altre ricerche nel suo laboratorio per concentrarsi sulla purificazione dell’Insulina. Invitò il biochimico James Collip ad aiutarlo in questo compito e il team si sentì pronto per un test clinico entro un mese.[12]
John James Rickard Macleod (6 settembre 1876 – 16 marzo 1935) è stato un biochimico e fisiologo britannico. Ha dedicato la sua carriera a diversi argomenti di fisiologia e biochimica, ma si è interessato soprattutto al metabolismo dei carboidrati. È noto per il suo ruolo nella scoperta e nell’isolamento dell’Insulina durante il suo incarico di docente all’Università di Toronto, per il quale ricevette, insieme a Frederick Banting, il premio Nobel per la fisiologia o la medicina nel 1923. L’assegnazione del premio a Macleod fu all’epoca controversa, perché secondo la versione dei fatti di Banting, il ruolo di Macleod nella scoperta era trascurabile. Solo decenni dopo gli eventi, una revisione indipendente ha riconosciuto un ruolo molto più importante di quello attribuitogli all’inizio.
L’11 gennaio 1922, Leonard Thompson, un quattordicenne diabetico che giaceva in fin di vita al Toronto General Hospital, ricevette la prima iniezione di insulina.[15][16][17][18] Tuttavia, l’estratto era così impuro che Thompson ebbe una grave reazione allergica e le ulteriori iniezioni furono annullate. Nei 12 giorni successivi, Collip lavorò giorno e notte per migliorare l’estratto di pancreas di bue. Una seconda dose fu iniettata il 23 gennaio, eliminando la glicosuria tipica del diabete senza causare effetti collaterali evidenti. La prima paziente americana fu Elizabeth Hughes, figlia del Segretario di Stato americano Charles Evans Hughes.[19][20] Il primo paziente trattato negli Stati Uniti fu il futuro artista di xilografie James D. Havens;[21] il dottor John Ralston Williams importò l’Insulina da Toronto a Rochester, New York, per trattare Havens.[22]
Leonard Thompson (17 luglio 1908 – 20 aprile 1935) è la prima persona ad aver ricevuto un’iniezione di Insulina come trattamento per il diabete di tipo I.
Banting e Best non lavorarono mai bene con Collip, considerandolo una specie di intruso, e Collip lasciò il progetto poco dopo. Nella primavera del 1922, Best riuscì a migliorare le sue tecniche al punto da poter estrarre grandi quantità di Insulina su richiesta, ma la preparazione rimase impura. L’azienda farmaceutica Eli Lilly and Company aveva offerto assistenza non molto tempo dopo le prime pubblicazioni del 1921, e in aprile accettò l’offerta della Lilly. A novembre, il capo chimico della Lilly, George B. Walden, scoprì la precipitazione isoelettrica e fu in grado di produrre grandi quantità di Insulina altamente purificata. Poco dopo, l’Insulina fu messa in vendita al pubblico.
Cartella per Elizabeth Hughes Autore: Hughes, Elizabeth Evans Luogo/Data: [Toronto], 16 agosto 1922 Descrizione fisica: 1 carta 28 x 22 cm. Scopo e contenuto: Si tratta di una tabella utilizzata per tenere traccia del sangue, delle urine, della dieta in grammi e delle prescrizioni dietetiche in grammi. Si tratta di una pagina compilata a mano. Il grafico mostra che il 3 settembre la Hughes aveva preso 9 chili rispetto alla prima iniezione di Insulina del 17 agosto. Raccolta: Banting Posizione: MS. COLL. 76 (Banting), Box 8A, Folder 25B Fonte del titolo: Titolo basato sul contenuto della carta. Nota generale: le annotazioni sono di mano di Elizabeth Hughes. Si tratta di un campione dei moduli utilizzati per annotare le sue condizioni mediche da quando le fu diagnosticato il diabete. Informazioni sui diritti: Nessuna restrizione di accesso nota Deposito: Thomas Fisher Rare Book Library, Università di Toronto, Toronto, Ontario Canada, M5S 1A5, library.utoronto.ca/fisher Collezione: Parte della collezione Discovery and Early Development of Insulin link.library.utoronto.ca/insulin/
Verso la fine del gennaio 1922, le tensioni tra i quattro “co-scopritori” dell’insulina aumentarono e Collip minacciò brevemente di brevettare separatamente il suo processo di purificazione. John G. FitzGerald, direttore dell’istituzione sanitaria pubblica non commerciale Connaught Laboratories, intervenne quindi come paciere. L’accordo del 25 gennaio 1922 stabilì due condizioni fondamentali: 1) i collaboratori avrebbero firmato un contratto in cui si impegnavano a non sottoscrivere un brevetto con un’azienda farmaceutica commerciale durante un periodo iniziale di lavoro con Connaught; e 2) non sarebbero stati permessi cambiamenti nella politica di ricerca se non prima discussi tra FitzGerald e i quattro collaboratori.[23] Ciò contribuì a contenere il disaccordo e a vincolare la ricerca al mandato pubblico di Connaught.
John Gerald “Gerry” FitzGerald (9 dicembre 1882 a Drayton, Ontario – 20 giugno 1940) è stato un medico canadese e specialista della salute pubblica che ha contribuito in modo determinante al controllo della difterite, prima producendo e distribuendo gratuitamente l’antitossina e poi, nel 1924, utilizzando la produzione di massa per consentire l’uso diffuso del vaccino ideato da Gaston Ramon.
Inizialmente, Macleod e Banting erano particolarmente riluttanti a brevettare il loro processo per l’Insulina per motivi di etica medica. Tuttavia, rimaneva il timore che un terzo privato potesse dirottare e monopolizzare la ricerca (come aveva lasciato intendere Eli Lilly and Company[24]) e che sarebbe stato difficile garantire una distribuzione sicura senza una capacità di controllo della qualità. A tal fine, Edward Calvin Kendall fornì preziosi consigli. Egli aveva isolato la Tiroxina presso la Mayo Clinic nel 1914 e aveva brevettato il processo attraverso un accordo tra lui, i fratelli Mayo e l’Università del Minnesota, trasferendo il brevetto all’università pubblica.[25] Il 12 aprile, Banting, Best, Collip, Macleod e FitzGerald scrissero congiuntamente al presidente dell’Università di Toronto per proporre un accordo simile con l’obiettivo di assegnare un brevetto al Board of Governors dell’università.[26] La lettera sottolineava che:[27] Il brevetto non sarebbe stato utilizzato per nessun altro scopo se non quello di impedire il conseguimento di un brevetto da parte di altre persone. Quando i dettagli del metodo di preparazione saranno pubblicati, chiunque sarà libero di preparare l’estratto, ma nessuno potrà assicurarsi un monopolio redditizio.
Edward Calvin Kendall (8 marzo 1886 – 4 maggio 1972) è stato un chimico americano. Nel 1950, Kendall ricevette il Premio Nobel per la Fisiologia o la Medicina insieme al chimico svizzero Tadeusz Reichstein e al medico della Mayo Clinic Philip S. Hench, per il loro lavoro sugli ormoni della ghiandola surrenale. Kendall non si concentrò solo sulle ghiandole surrenali, ma fu anche responsabile dell’isolamento della Tiroxina, un ormone della ghiandola tiroidea, e collaborò con il team che cristallizzò il Glutatione e ne identificò la struttura chimica.
La cessione al Consiglio superiore dell’Università di Toronto fu completata il 15 gennaio 1923, con il pagamento simbolico di 1 dollaro.[28] L’accordo è stato giudicato da The World’s Work del 1923 come “un passo avanti nell’etica medica”.[29] Ha ricevuto molta attenzione da parte dei media anche negli anni 2010 per quanto riguarda la questione dell’assistenza sanitaria e dell’accessibilità dei farmaci.
A seguito di ulteriori preoccupazioni riguardanti i tentativi di Eli Lilly di brevettare separatamente parti del processo di produzione, il vicedirettore di Connaught e capo della divisione Insulina Robert Defries ha stabilito una politica di pooling dei brevetti che avrebbe richiesto ai produttori di condividere liberamente qualsiasi miglioramento del processo di produzione senza compromettere l’accessibilità dei farmaci.[30]
Nel 1923 il comitato del Premio Nobel attribuì l’estrazione pratica dell’Insulina a un team dell’Università di Toronto e assegnò il Premio Nobel a due uomini: Frederick Banting e J.J.R. Macleod.[31] Essi ricevettero il Premio Nobel per la Fisiologia o la Medicina nel 1923 per la scoperta dell’Insulina. Banting, incredulo per la mancata menzione di Best,[32] condivise il premio con lui, mentre Macleod condivise immediatamente il suo con James Collip. Il brevetto dell’Insulina fu venduto all’Università di Toronto per un dollaro.
Altri due premi Nobel sono stati assegnati per lavori sull’Insulina. Il biologo molecolare britannico Frederick Sanger, che nel 1955 determinò la struttura primaria dell’Insulina, ricevette il Premio Nobel per la Chimica nel 1958.[33] Rosalyn Sussman Yalow ricevette il Premio Nobel per la Medicina nel 1977 per lo sviluppo del test radioimmunologico dell’Insulina.
Diversi premi Nobel hanno anche un legame indiretto con l’Insulina. George Minot, co-ricevente del Premio Nobel 1934 per lo sviluppo del primo trattamento efficace per l’anemia perniciosa, era affetto da diabete mellito di tipo I. Il dottor William Castle ha osservato che la scoperta dell’Insulina nel 1921, arrivata in tempo per mantenere in vita Minot, era quindi anche responsabile della scoperta di una cura per l’anemia perniciosa.[34] Dorothy Hodgkin ha ricevuto il Premio Nobel per la Chimica nel 1964 per lo sviluppo della cristallografia, la tecnica che ha utilizzato per decifrare la struttura molecolare completa dell’Insulina nel 1969.[35]
Dorothy Mary Crowfoot Hodgkin (nata Crowfoot; 12 maggio 1910 – 29 luglio 1994) è stata una chimica britannica vincitrice del premio Nobel che ha fatto progredire la tecnica della cristallografia a raggi X per determinare la struttura delle biomolecole, divenuta essenziale per la biologia strutturale.
Il lavoro pubblicato da Banting, Best, Collip e Macleod rappresentava la preparazione di un estratto purificato di Insulina adatto all’uso su pazienti umani.[36] Sebbene Paulescu avesse scoperto i principi del trattamento, il suo estratto salino non poteva essere usato sugli esseri umani; non fu menzionato nel Premio Nobel del 1923. Il professor Ian Murray fu particolarmente attivo nel lavorare per correggere “l’errore storico” contro Nicolae Paulescu. Murray era professore di fisiologia presso l’Anderson College of Medicine di Glasgow, in Scozia, capo del dipartimento di Malattie Metaboliche di un importante ospedale di Glasgow, vicepresidente della British Association of Diabetes e membro fondatore della International Diabetes Federation. Murray ha scritto:
Non è stato dato sufficiente riconoscimento a Paulescu, l’illustre scienziato rumeno, che all’epoca in cui l’équipe di Toronto stava iniziando le sue ricerche era già riuscito a estrarre l’ormone antidiabetico del pancreas e a dimostrarne l’efficacia nel ridurre l’iperglicemia nei cani diabetici.[37]
In una comunicazione privata, il professor Arne Tiselius, ex capo dell’Istituto Nobel, espresse la sua personale opinione che Paulescu fosse ugualmente degno del premio nel 1923.[38]
Arne Wilhelm Kaurin Tiselius (10 agosto 1902 – 29 ottobre 1971) è stato un biochimico svedese che ha vinto il Premio Nobel per la Chimica nel 1948 “per le sue ricerche sull’elettroforesi e sull’analisi di adsorbimento, in particolare per le sue scoperte sulla natura complessa delle proteine del siero”.
Analisi strutturale e sintesi di laboratorio:
L’Insulina purificata di origine animale era inizialmente l’unico tipo di Insulina disponibile per gli esperimenti e i diabetici. John Jacob Abel fu il primo a produrre la forma cristallizzata nel 1926.[39] La prova della natura proteica fu fornita per la prima volta da Michael Somogyi, Edward A. Doisy e Philip A. Shaffer nel 1924.[40] Fu pienamente dimostrata quando Hans Jensen e Earl A. Evans Jr. isolarono gli aminoacidi fenilalanina e prolina nel 1935.[41]
Da sinistra: il Dr. Michael Somogyi (7 marzo 1883 – 21 luglio 1971), professore ungherese-americano di biochimica presso la Washington University e l’ospedale ebraico di Saint Louis e Edward Adelbert Doisy (13 novembre 1893 – 23 ottobre 1986), biochimico americano.
La struttura aminoacidica dell’Insulina fu caratterizzata per la prima volta nel 1951 da Frederick Sanger,[42] e la prima Insulina sintetica fu prodotta simultaneamente nei laboratori di Panayotis Katsoyannis dell’Università di Pittsburgh e di Helmut Zahn dell’Università RWTH di Aquisgrana a metà degli anni Sessanta. [43][44][45][46][47] L’Insulina bovina cristallina sintetica è stata ottenuta da ricercatori cinesi nel 1965.[48] La struttura tridimensionale completa dell’Insulina è stata determinata mediante cristallografia a raggi X nel laboratorio di Dorothy Hodgkin nel 1969.[49]
Frederick Sanger (13 agosto 1918 – 19 novembre 2013), biochimico inglese che ha vinto due volte il Premio Nobel per la Chimica. Nel 1958 gli è stato assegnato il Premio Nobel per la Chimica “per il suo lavoro sulla struttura delle proteine, in particolare quella dell’Insulina”.
Il dottor Hans E. Weber scoprì la preproinsulina mentre lavorava come ricercatore presso l’Università della California Los Angeles nel 1974. Nel 1973-1974, Weber imparò le tecniche per isolare, purificare e tradurre l’RNA messaggero. Per studiare ulteriormente l’Insulina, ottenne tessuti pancreatici da un macello di Los Angeles e successivamente da animali dell’UCLA. Isolò e purificò l’RNA messaggero totale dalle cellule dell’isoletta pancreatica, che fu poi tradotto in oociti di Xenopus laevis e precipitato usando anticorpi anti-insulina. Quando la proteina totale tradotta è stata sottoposta a elettroforesi su gel di SDS-poliacrilammide e gradiente di saccarosio, sono stati isolati i picchi corrispondenti all’Insulina e alla proinsulina. Tuttavia, con sorpresa del Dr. Weber, è stato isolato un terzo picco corrispondente a una molecola più grande della proinsulina. Dopo aver riprodotto l’esperimento diverse volte, ha notato costantemente questo grande picco prima della proinsulina, che ha stabilito essere una molecola precursore più grande a monte della proinsulina. Nel maggio 1975, in occasione del meeting dell’American Diabetes Association a New York, Weber presentò oralmente il suo lavoro[50-146] e fu il primo a chiamare questa molecola precursore “preproinsulina”. In seguito a questa presentazione orale, Weber fu invitato a cena dal dottor Donald Steiner, un ricercatore che aveva contribuito alla caratterizzazione della proinsulina, per discutere del suo lavoro e delle sue scoperte. Un anno dopo, nell’aprile 1976, questa molecola fu ulteriormente caratterizzata e sequenziata da Steiner, facendo riferimento al lavoro e alla scoperta di Hans Weber.[51] La preproinsulina divenne una molecola importante per studiare il processo di trascrizione e traduzione.
La prima Insulina “umana” geneticamente ingegnerizzata e sintetica è stata prodotta con l’E. coli nel 1978 da Arthur Riggs e Keiichi Itakura presso il Beckman Research Institute della Città della Speranza in collaborazione con Herbert Boyer della Genentech.[52][53] La Genentech, fondata da Swanson, Boyer e Eli Lilly and Company, ha continuato nel 1982 a vendere la prima Insulina umana biosintetica disponibile in commercio con il marchio Humulin [La stragrande maggioranza dell’Insulina utilizzata in tutto il mondo è Insulina “umana” biosintetica o suoi analoghi].[54] Recentemente, un altro approccio è stato utilizzato da un gruppo pionieristico di ricercatori canadesi, che ha utilizzato una pianta di cartamo facilmente coltivabile, per la produzione di Insulina molto più economica.[55]
Herbert Wayne “Herb” Boyer (nato il 10 luglio 1936), biotecnologo americano, ricercatore e imprenditore nel campo delle biotecnologie. Insieme a Stanley N. Cohen e Paul Berg ha scoperto un metodo per indurre i batteri a produrre proteine estranee, dando così il via al campo dell’ingegneria genetica [tecnologia del DNA ricombinante].
L’Insulina ricombinante viene prodotta nel lievito (di solito Saccharomyces cerevisiae) o in E. coli.[56] Nel lievito, l’Insulina può essere ingegnerizzata come una proteina a catena singola con un sito di endoproteasi KexII (un omologo del PCI/PCII del lievito) che separa la catena A dell’Insulina da una catena B dell’Insulina troncata C-terminalmente. Una coda C-terminale sintetizzata chimicamente viene quindi innestata sull’Insulina mediante proteolisi inversa utilizzando la proteasi tripsina, poco costosa; in genere la lisina sulla coda C-terminale è protetta con un gruppo protettivo chimico per impedire la proteolisi. La facilità della sintesi modulare e la relativa sicurezza delle modifiche in quella regione spiega i comuni analoghi dell’Insulina con modifiche C-terminali (ad esempio lispro, aspart, glulisine). La sintesi Genentech e le sintesi completamente chimiche come quella di Bruce Merrifield non sono preferibili perché l’efficienza della ricombinazione delle due catene di Insulina è bassa, soprattutto a causa della competizione con la precipitazione della catena B dell’Insulina.
Da sinistra: diagramma di Richardson di un monomero di Insulina suina, che mostra la sua caratteristica struttura secondaria. Questa è la forma biologicamente attiva dell’insulina. A destra, il diagramma di Richardson di un esamero di Insulina suina. La sfera al centro è un atomo di zinco stabilizzante, circondato da residui di istidina coordinati. Questa è la forma in cui l’Insulina viene immagazzinata nelle cellule beta.
Caratteristiche dell’Insulina:
Grazie ad annali ricerche oggi sappiamo che l’Insulina è un ormone peptidico prodotto dalle cellule beta delle isole pancreatiche, codificato nell’uomo dal gene INS. È considerato il principale ormone anabolico dell’organismo sebbene la sua attività prevalente sia diretta alla riduzione del catabolismo.[57] Regola il metabolismo dei carboidrati, dei grassi e delle proteine promuovendo l’assorbimento del glucosio dal sangue nelle cellule epatiche, lipidiche e del muscolo-scheletrico [In questi tessuti il glucosio assorbito viene convertito in glicogeno attraverso la glicogenesi o in alcuni casi in grassi (trigliceridi) attraverso la lipogenesi o, nel caso del fegato, in entrambi].[58] La produzione e la secrezione di glucosio da parte del fegato sono fortemente inibite da alte concentrazioni di Insulina nel sangue.[59] L’Insulina circolante influisce anche sulla sintesi di proteine in un’ampia varietà di tessuti. È quindi un ormone anabolico, che promuove la conversione di piccole molecole nel sangue in grandi molecole all’interno delle cellule. Bassi livelli di Insulina nel sangue hanno l’effetto opposto, favorendo un diffuso catabolismo, soprattutto del grasso corporeo di riserva.
Le cellule beta sono sensibili ai livelli della glicemia nel sangue, per cui secernono Insulina nel sangue in risposta a livelli elevati di glucosio e inibiscono la secrezione di Insulina quando i livelli di glucosio sono bassi.[60] L’Insulina aumenta l’assorbimento e il metabolismo del glucosio nelle cellule, riducendo così il livello della glicemia ematica. Le cellule alfa vicine, prendendo spunto dalle cellule beta,[60] secernono Glucagone nel sangue in modo opposto: aumento della secrezione quando il glucosio nel sangue è basso e diminuzione della secrezione quando le concentrazioni di glucosio sono elevate. Il Glucagone aumenta il livello di glucosio nel sangue stimolando la glicogenolisi e la gluconeogenesi nel fegato.[58][60] La secrezione di Insulina e Glucagone nel sangue in risposta alla concentrazione di glucosio nel sangue è il meccanismo principale dell’omeostasi del glucosio.[60]
Schema della regolazione dell’Insulina in caso di glicemia elevata.
L’insulina è quindi prodotta esclusivamente nelle cellule beta delle isole pancreatiche nei mammiferi e nel corpo di Brockmann in alcuni pesci. L’Insulina umana è prodotta dal gene INS, situato sul cromosoma 11.[61] I roditori hanno due geni funzionali dell’Insulina: uno è l’omologo della maggior parte dei geni dei mammiferi (Ins2) e l’altro è una copia retroposta che include la sequenza del promotore ma che manca di un introne (Ins1) [La trascrizione del gene dell’Insulina aumenta in risposta all’aumento del glucosio nel sangue].[62] Ciò è controllato principalmente da fattori di trascrizione che legano sequenze enhancer nelle circa 400 paia di basi prima del sito di inizio della trascrizione del gene.[61][62]
I principali fattori di trascrizione che influenzano la secrezione insulinica sono PDX1, NeuroD1 e MafA.[63][64][65][66]
L’Insulina subisce un’ampia modificazione post-traslazionale lungo la via di produzione. La produzione e la secrezione sono ampiamente indipendenti; l’Insulina sintetizzata viene immagazzinata in attesa della secrezione. Sia il C-peptide che l’Insulina matura sono biologicamente attivi. I componenti cellulari e le proteine di questa immagine non sono in scala.
In uno stato di basso livello di glucosio, PDX1 (pancreatic and duodenal homeobox protein 1) si trova nella periferia nucleare in seguito all’interazione con HDAC1 e 2,[67] il che determina una sottoregolazione della secrezione insulinica.[68] Un aumento dei livelli di glucosio nel sangue provoca la fosforilazione di PDX1, che subisce una traslocazione nucleare e si lega all’elemento A3 all’interno del promotore dell’Insulina.[69] Dopo la traslocazione interagisce con i coattivatori HAT p300 e SETD7. PDX1 influisce sulle modificazioni degli istoni attraverso l’acetilazione, la deacetilazione e la metilazione. Si dice anche che sopprima il glucagone.[70]
NeuroD1, noto anche come β2, regola l’esocitosi dell’Insulina nelle cellule β pancreatiche inducendo direttamente l’espressione di geni coinvolti nell’esocitosi.[71] È localizzato nel citosol, ma in risposta all’elevato livello di glucosio viene glicosilato da OGT e/o fosforilato da ERK, il che provoca la traslocazione nel nucleo. Nel nucleo β2 eterodimerizza con E47, si lega all’elemento E1 del promotore dell’insulina e recluta il co-attivatore p300 che acetilerà β2. È in grado di interagire anche con altri fattori di trascrizione nell’attivazione del gene dell’Insulina.[71]
MafA viene degradato dai proteasomi quando i livelli di glucosio nel sangue sono bassi. L’aumento dei livelli di glucosio rende glicosilata una proteina sconosciuta. Questa proteina funziona come fattore di trascrizione per MafA in modo sconosciuto e MafA viene trasportata fuori dalla cellula. MafA viene poi traslocata di nuovo nel nucleo dove lega l’elemento C1 del promotore dell’insulina.[72][73]
Questi fattori di trascrizione lavorano in modo sinergico e complesso. L’aumento del glucosio nel sangue può, dopo un po’, distruggere le capacità di legame di queste proteine e quindi ridurre la quantità di Insulina secreta, causando il diabete. La diminuzione delle attività di legame può essere mediata dallo stress ossidativo indotto dal glucosio e si ritiene che gli antiossidanti prevengano la diminuzione della secrezione di Insulina nelle cellule β pancreatiche glucotossiche. Le molecole di segnalazione dello stress e le specie reattive dell’ossigeno inibiscono il gene dell’Insulina interferendo con i cofattori che legano i fattori di trascrizione e con i fattori di trascrizione stessi.[74]
Diverse sequenze regolatrici nella regione del promotore del gene dell’Insulina umana si legano ai fattori di trascrizione. In generale, le A-box si legano ai fattori Pdx1, le E-box a NeuroD, le C-box a MafA e gli elementi di risposta al cAMP a CREB. Esistono anche dei silenziatori che inibiscono la trascrizione.
L’insulina viene sintetizzata come molecola precursore inattiva, una proteina di 110 aminoacidi chiamata “preproinsulina”. La preproinsulina viene tradotta direttamente nel reticolo endoplasmatico ruvido (RER), dove il suo peptide segnale viene rimosso dalla peptidasi segnale per formare la “proinsulina”.[60] Durante il ripiegamento della proinsulina, le estremità opposte della proteina, chiamate “catena A” e “catena B”, vengono fuse insieme con tre legami disolfuro.[60] La proinsulina ripiegata passa quindi attraverso l’apparato di Golgi e viene impacchettata in vescicole secretorie specializzate [Nel granulo, la proinsulina viene scissa dalla proproteina convertasi 1/3 e dalla proproteina convertasi 2, rimuovendo la parte centrale della proteina, chiamata “peptide C”].[60] Infine, la carbossipeptidasi E rimuove due coppie di aminoacidi dalle estremità della proteina, dando origine all’Insulina attiva – le catene A e B dell’insulina, ora collegate da due legami disolfuro.[60]
Struttura primaria della preproinsulina.
L’Insulina matura risultante è impacchettata all’interno di granuli maturi in attesa di segnali metabolici (come leucina, arginina, glucosio e mannosio) e della stimolazione del nervo vagale per essere esocitata dalla cellula nella circolazione.[75]
È stato dimostrato che l’Insulina e le proteine ad essa correlate sono prodotte all’interno del cervello e che livelli ridotti di queste proteine sono collegati alla malattia di Alzheimer.[76][77][78]
Il rilascio di Insulina è stimolato anche dalla stimolazione del recettore beta-2 e inibito dalla stimolazione del recettore alfa-1. Inoltre, il Cortisolo, il Glucagone e l’Ormone della Crescita antagonizzano le azioni dell’Insulina nei periodi di stress. L’Insulina inibisce anche il rilascio di acidi grassi da parte della lipasi ormonosensibile nel tessuto adiposo.[79]
Contrariamente alla convinzione iniziale che gli ormoni fossero generalmente molecole chimiche di piccole dimensioni, l’Insulina, primo ormone peptidico di cui si conosce la struttura, si è rivelata piuttosto grande.[80] Una singola proteina (monomero) di Insulina umana è composta da 51 aminoacidi e ha una massa molecolare di 5808 Da. La formula molecolare dell’Insulina umana è C257H383N65O77S6.[81-44] Si tratta di una combinazione di due catene peptidiche (dimeri) denominate catena A e catena B, legate tra loro da due legami disolfuro. La catena A è composta da 21 aminoacidi, mentre la catena B è composta da 30 residui. I legami disolfuro di collegamento (intercatena) si formano sui residui di cisteina tra le posizioni A7-B7 e A20-B19. Esiste un ulteriore legame disolfuro (intracatena) all’interno della catena A tra i residui di cisteina nelle posizioni A6 e A11. La catena A presenta due regioni α-eliche in corrispondenza di A1-A8 e A12-A19 che sono antiparallele; mentre la catena B presenta un’α-elica centrale (che copre i residui B9-B19) affiancata dal legame disolfuro su entrambi i lati e da due foglietti β (che coprono B7-B10 e B20-B23).[80][82-45]
La struttura dell’Insulina. Il lato sinistro è un modello di riempimento dello spazio del monomero dell’insulina, ritenuto biologicamente attivo. Il carbonio è verde, l’idrogeno bianco, l’ossigeno rosso e l’azoto blu. A destra c’è un diagramma a nastro dell’esamero dell’insulina, che si ritiene essere la forma immagazzinata. Un’unità monomerica è evidenziata con la catena A in blu e la catena B in ciano. Il giallo indica i legami disolfuro e le sfere magenta sono ioni di zinco.
La sequenza aminoacidica dell’insulina è fortemente conservata e varia solo leggermente tra le specie. L’insulina bovina differisce da quella umana solo per tre residui aminoacidici e quella suina per uno. Anche l’insulina di alcune specie di pesci è abbastanza simile a quella umana da essere clinicamente efficace nell’uomo. L’insulina di alcuni invertebrati ha una sequenza molto simile a quella dell’insulina umana e ha effetti fisiologici simili. Il C-peptide della proinsulina, tuttavia, differisce molto di più tra le specie; è anch’esso un ormone, ma secondario.[82]
L’Insulina viene prodotta e immagazzinata nell’organismo sotto forma di esamero (un’unità di sei molecole di insulina), mentre la forma attiva è il monomero. L’esamero ha una dimensione di circa 36000 Da. Le sei molecole sono legate insieme come tre unità dimeriche per formare una molecola simmetrica. Una caratteristica importante è la presenza di atomi di zinco (Zn2+) sull’asse di simmetria, che sono circondati da tre molecole d’acqua e da tre residui di istidina in posizione B10.[68][82]
L’esamero è una forma inattiva con stabilità a lungo termine, che serve a mantenere l’insulina altamente reattiva protetta, ma prontamente disponibile. La conversione esamero-monomero è uno degli aspetti centrali delle formulazioni di insulina per iniezione. L’esamero è molto più stabile del monomero, il che è auspicabile per motivi pratici; tuttavia, il monomero è un farmaco che reagisce molto più rapidamente, poiché la velocità di diffusione è inversamente correlata alla dimensione delle particelle. Un farmaco a reazione rapida significa che le iniezioni di insulina non devono precedere di ore i pasti, il che a sua volta offre alle persone con diabete una maggiore flessibilità negli orari giornalieri.[83] L’Insulina può aggregarsi e formare foglietti beta fibrillari interdigitati. Ciò può causare amiloidosi da iniezione e impedisce la conservazione dell’insulina per lunghi periodi.[84]
Le cellule beta delle isole di Langerhans rilasciano insulina in due fasi. Il rilascio della prima fase avviene rapidamente in risposta all’aumento dei livelli di glucosio nel sangue e dura circa 10 minuti. La seconda fase è un rilascio lento e prolungato di vescicole di nuova formazione, innescato indipendentemente dallo zucchero, che raggiunge il suo picco tra le 2 e le 3 ore. Le due fasi del rilascio di insulina suggeriscono che i granuli di insulina sono presenti in diverse popolazioni dichiarate o “pool”. Durante la prima fase dell’esocitosi dell’insulina, la maggior parte dei granuli predisposti all’esocitosi viene rilasciata dopo l’internalizzazione del calcio. Questo pool è noto come Readily Releasable Pool (RRP). I granuli RRP rappresentano lo 0,3-0,7% della popolazione totale di granuli contenenti insulina e si trovano immediatamente adiacenti alla membrana plasmatica. Durante la seconda fase dell’esocitosi, i granuli di insulina richiedono la mobilizzazione dei granuli verso la membrana plasmatica e una precedente preparazione per essere rilasciati.[85] Pertanto, la seconda fase del rilascio di insulina è regolata dalla velocità con cui i granuli si preparano al rilascio. Questo pool è noto come pool di riserva (RP). L’RP viene rilasciato più lentamente dell’RRP (RRP: 18 granuli/min; RP: 6 granuli/min).[86] Un ridotto rilascio di insulina nella prima fase può essere il primo difetto rilevabile delle cellule beta che predice l’insorgenza del diabete di tipo 2.[87] Il rilascio nella prima fase e la sensibilità all’insulina sono predittori indipendenti del diabete.[88]
La descrizione del rilascio della prima fase è la seguente:
Il glucosio entra nelle β-cellule attraverso il trasportatore del glucosio, GLUT 2. A bassi livelli di zucchero nel sangue poco glucosio entra nelle β-cellule; ad alte concentrazioni di glucosio nel sangue grandi quantità di glucosio entrano in queste cellule.[89]
Il glucosio che entra nella β-cellula viene fosforilato a glucosio-6-fosfato (G-6-P) dalla glucochinasi (esochinasi IV) che non è inibita dal G-6-P come le esochinasi di altri tessuti (esochinasi I-III). Ciò significa che la concentrazione intracellulare di G-6-P rimane proporzionale alla concentrazione di zucchero nel sangue.[89]
Il glucosio-6-fosfato entra nella via glicolitica e poi, attraverso la reazione della piruvato deidrogenasi, nel ciclo di Krebs, dove vengono prodotte più molecole di ATP ad alta energia dall’ossidazione dell’acetil CoA (substrato del ciclo di Krebs), con conseguente aumento del rapporto ATP:ADP all’interno della cellula.[90]
Un aumento del rapporto ATP:ADP intracellulare chiude il canale del potassio SUR1/Kir6.2 sensibile all’ATP (vedi recettore delle sulfoniluree). Questo impedisce agli ioni potassio (K+) di lasciare la cellula per diffusione facilitata, portando a un accumulo di ioni potassio intracellulare. Di conseguenza, l’interno della cellula diventa meno negativo rispetto all’esterno, portando alla depolarizzazione della membrana della superficie cellulare.
In seguito alla depolarizzazione, si aprono i canali degli ioni calcio (Ca2+) voltaggio-gati, consentendo agli ioni calcio di spostarsi nella cellula per diffusione facilitata.
La concentrazione citosolica di ioni calcio può anche essere aumentata dal rilascio di calcio dai depositi intracellulari attraverso l’attivazione dei recettori rianodinici.[91]
La concentrazione di ioni calcio nel citosol delle cellule beta può essere aumentata anche, o in aggiunta, attraverso l’attivazione della fosfolipasi C derivante dal legame di un ligando extracellulare (ormone o neurotrasmettitore) a un recettore di membrana accoppiato a proteine G. La fosfolipasi C scinde il fosfolipide di membrana, il fosfatidil inositolo 4,5-bisfosfato, in inositolo 1,4,5-trifosfato e diacilglicerolo. L’inositolo 1,4,5-trisfosfato (IP3) si lega quindi a proteine recettoriali nella membrana plasmatica del reticolo endoplasmatico (ER). Ciò consente il rilascio di ioni Ca2+ dall’ER attraverso canali IP3-gated, che aumentano la concentrazione citosolica di ioni calcio indipendentemente dagli effetti di un’elevata concentrazione di glucosio nel sangue. La stimolazione parasimpatica delle isole pancreatiche opera attraverso questa via per aumentare la secrezione di insulina nel sangue.[92]
L’aumento significativo della quantità di ioni calcio nel citoplasma delle cellule provoca il rilascio nel sangue dell’Insulina precedentemente sintetizzata e immagazzinata nelle vescicole secretorie intracellulari.
Questo è il meccanismo principale di rilascio dell’insulina. Altre sostanze note per stimolare il rilascio di insulina sono gli aminoacidi arginina e leucina, il rilascio parasimpatico di acetilcolina (che agisce attraverso la via della fosfolipasi C), le sulfoniluree, la colecistochinina (CCK, anch’essa attraverso la fosfolipasi C),[93-56] e le incretine di derivazione gastrointestinale, come il peptide glucagone-simile-1 (GLP-1) e il peptide insulinotropico glucosio-dipendente (GIP).
Il polipeptide insulinotropico glucosio-dipendente (GIP), noto anche come polipeptide inibitore gastrico o peptide inibitore gastrico (abbreviato anche in GIP), è un ormone inibitore della famiglia delle secretine. Pur essendo un debole inibitore della secrezione acida gastrica, il suo ruolo principale è quello di stimolare la secrezione di Insulina. La GIP, insieme al peptide glucagone-simile-1 (GLP-1), appartiene a una classe di molecole denominate incretine.
Il rilascio di insulina è fortemente inibito dalla noradrenalina, che porta a un aumento dei livelli di glucosio nel sangue durante lo stress. Sembra che il rilascio di catecolamine da parte del sistema nervoso simpatico abbia influenze contrastanti sul rilascio di insulina da parte delle cellule beta, perché il rilascio di Insulina è inibito dai recettori α2-adrenergici[94] e stimolato dai recettori β2-adrenergici.[95] L’effetto netto della noradrenalina dai nervi simpatici e dell’epinefrina dalle ghiandole surrenali sul rilascio di insulina è l’inibizione dovuta alla dominanza dei recettori α-adrenergici.[96]
Quando il livello di glucosio scende al valore fisiologico abituale, il rilascio di insulina da parte delle cellule β rallenta o si arresta. Se il livello di glucosio nel sangue scende al di sotto di questo valore, soprattutto a livelli pericolosamente bassi, il rilascio di ormoni iperglicemizzanti (in particolare il glucagone dalle cellule alfa dell’isolotto di Langerhans) forza il rilascio di glucosio nel sangue dalle scorte di glicogeno del fegato, integrato dalla gluconeogenesi se le scorte di glicogeno si esauriscono. Aumentando il glucosio nel sangue, gli ormoni iperglicemizzanti prevengono o correggono l’ipoglicemia pericolosa per la vita.
L’evidenza di un alterato rilascio di insulina nella prima fase può essere osservata nel test di tolleranza al glucosio, dimostrato da un livello di glucosio nel sangue sostanzialmente elevato a 30 minuti dall’ingestione di un carico di glucosio (75 o 100 g di glucosio), seguito da un lento calo nei 100 minuti successivi, per rimanere al di sopra di 120 mg/100 ml dopo due ore dall’inizio del test. In una persona normale il livello di glucosio nel sangue è corretto (e può anche essere leggermente sovracorretto) alla fine del test. Il picco insulinico è una “prima risposta” all’aumento del glucosio nel sangue; questa risposta è individuale e specifica per la dose, anche se in passato si è sempre ritenuto che fosse specifica solo per il tipo di alimento.
Anche durante la digestione, in genere una o due ore dopo un pasto, il rilascio di insulina da parte del pancreas non è continuo, ma oscilla con un periodo di 3-6 minuti, passando dal generare una concentrazione di insulina nel sangue superiore a circa 800 pmol/l a meno di 100 pmol/l (nei ratti).[97] Si pensa che questo avvenga per evitare la sottoregolazione dei recettori dell’Insulina nelle cellule bersaglio e per aiutare il fegato a estrarre l’insulina dal sangue [Questa oscillazione è importante da considerare quando si somministrano farmaci insulino-stimolanti, poiché idealmente si dovrebbe ottenere una concentrazione ematica oscillante del rilascio di insulina, e non una concentrazione elevata costante].[97] Ciò può essere ottenuto somministrando l’insulina in modo ritmico nella vena porta, con una somministrazione attivata dalla luce o con il trapianto di cellule dell’isoletta nel fegato.[98][99][100]
Il livello di Insulina nel sangue può essere misurato in unità internazionali, come µIU/mL o in concentrazione molare, come pmol/L, dove 1 µIU/mL equivale a 6,945 pmol/L.[101] Un livello ematico tipico tra i pasti è di 8-11 μIU/mL (57-79 pmol/L).[102]
Gli effetti dell’insulina sono avviati dal suo legame con un recettore, il recettore dell’insulina (IR), presente nella membrana cellulare. La molecola del recettore contiene una subunità α e una subunità β. Due molecole si uniscono per formare il cosiddetto omodimero. L’insulina si lega alla subunità α dell’omodimero, che è rivolta verso il lato extracellulare delle cellule. Le subunità β hanno un’attività enzimatica tirosin-chinasica che viene attivata dal legame con l’insulina. Questa attività provoca l’autofosforilazione delle subunità β e successivamente la fosforilazione di proteine all’interno della cellula, note come substrati del recettore dell’insulina (IRS). La fosforilazione dell’IRS attiva una cascata di trasduzione del segnale che porta all’attivazione di altre chinasi e di fattori di trascrizione che mediano gli effetti intracellulari dell’insulina.[103]
Recettore dell’Insulina (IR).
La cascata che porta all’inserimento dei trasportatori di glucosio GLUT4 nelle membrane cellulari delle cellule muscolari e adipose e alla sintesi di glicogeno nel fegato e nel tessuto muscolare, nonché alla conversione del glucosio in trigliceridi nel fegato, nell’adipe e nel tessuto della ghiandola mammaria in allattamento, opera attraverso l’attivazione, da parte dell’IRS-1, della fosfoinositolo 3 chinasi (PI3K). Questo enzima converte un fosfolipide della membrana cellulare, il fosfatidilinositolo 4,5-bisfosfato (PIP2), in fosfatidilinositolo 3,4,5-trifosfato (PIP3), che a sua volta attiva la protein chinasi B (PKB). La PKB attivata facilita la fusione degli endosomi contenenti GLUT4 con la membrana cellulare, con conseguente aumento dei trasportatori GLUT4 nella membrana plasmatica.[104] La PKB fosforila anche la glicogeno sintasi chinasi (GSK), inattivando così questo enzima.[104] Ciò significa che il suo substrato, la glicogeno sintasi (GS), non può essere fosforilato e rimane de-fosforilato, e quindi attivo. L’enzima attivo, la glicogeno sintasi (GS), catalizza la fase limitante della sintesi del glicogeno dal glucosio. Defosforilazioni simili interessano gli enzimi che controllano il tasso di glicolisi che porta alla sintesi dei grassi attraverso il malonil-CoA nei tessuti che possono generare trigliceridi, nonché gli enzimi che controllano il tasso di gluconeogenesi nel fegato. L’effetto complessivo di queste de-fosforilazioni enzimatiche finali è che, nei tessuti in grado di effettuare queste reazioni, viene stimolata la sintesi di glicogeno e di grassi a partire dal glucosio, mentre viene inibita la produzione di glucosio da parte del fegato attraverso la glicogenolisi e la gluconeogenesi.[105] Anche la scomposizione dei trigliceridi da parte del tessuto adiposo in acidi grassi liberi e glicerolo viene inibita.[104]
Struttura del GLUT4. Il GLUT4 contiene anche un dominio UBX. Si tratta di regioni regolatrici dell’ubiquitina che possono contribuire alla segnalazione cellulare.
Una volta prodotto il segnale intracellulare derivante dal legame dell’insulina con il suo recettore, è necessario interrompere la segnalazione. Come menzionato di seguito nella sezione sulla degradazione, l’endocitosi e la degradazione del recettore legato all’insulina è un meccanismo principale per terminare la segnalazione.[106] Inoltre, la via di segnalazione viene terminata anche dalla de-fosforilazione dei residui di tirosina nelle varie vie di segnalazione da parte delle tirosina fosfatasi. Le serina/treonina chinasi sono anche note per ridurre l’attività dell’insulina.
La struttura del complesso insulina-recettore dell’insulina è stata determinata con le tecniche della cristallografia a raggi X.[107]
Una volta che la molecola di Insulina si è agganciata al recettore e ha svolto la sua azione, può essere rilasciata nell’ambiente extracellulare o essere degradata dalla cellula. I due siti principali per l’eliminazione dell’Insulina sono il fegato e il rene.[108] Viene scomposta dall’enzima proteina-disolfuro reduttasi (Glutatione),[109] che rompe i legami disolfuro tra le catene A e B. Il fegato elimina la maggior parte dell’Insulina durante il transito di primo passaggio, mentre il rene elimina la maggior parte dell’Insulina nella circolazione sistemica. La degradazione comporta normalmente l’endocitosi del complesso insulino-recettore, seguita dall’azione di enzimi degradanti l’Insulina. Si stima che una molecola di Insulina prodotta endogenamente dalle cellule beta venga degradata entro circa un’ora dal suo rilascio iniziale in circolo (emivita dell’Insulina ~ 4-6 minuti).[109][110]
Struttura del Glutatione.
Le azioni dell’Insulina a livello del metabolismo umano globale comprendono:
Aumento dell’assorbimento di alcune sostanze da parte delle cellule, in particolare del glucosio nei muscoli e nel tessuto adiposo (circa i due terzi delle cellule del corpo)[111]
Aumento della replicazione del DNA e della sintesi proteica attraverso il controllo dell’assorbimento degli aminoacidi.
Modifica dell’attività di numerosi enzimi.
Le azioni dell’Insulina (indirette e dirette) sulle cellule comprendono:
Stimola l’assorbimento del glucosio – L’Insulina diminuisce la concentrazione di glucosio nel sangue inducendo l’assunzione di glucosio da parte delle cellule. Ciò è possibile perché l’insulina provoca l’inserimento del trasportatore GLUT4 nelle membrane cellulari dei tessuti muscolari e adiposi, permettendo al glucosio di entrare nella cellula.[112]
Aumento della sintesi dei grassi – l’insulina costringe le cellule grasse ad accogliere il glucosio nel sangue, che viene convertito in trigliceridi; la diminuzione dell’insulina provoca l’inverso.[111]
Aumento dell’esterificazione degli acidi grassi – costringe il tessuto adiposo a produrre grassi neutri (cioè trigliceridi) dagli acidi grassi; la diminuzione dell’insulina provoca l’inverso.[111]
Diminuzione della lipolisi – costringe a ridurre la conversione dei depositi di lipidi delle cellule adipose in acidi grassi e glicerolo nel sangue; la diminuzione dell’insulina provoca l’effetto inverso.[111]
Sintesi indotta di glicogeno – Quando i livelli di glucosio sono elevati, l’insulina induce la formazione di glicogeno attraverso l’attivazione dell’enzima esochinasi, che aggiunge un gruppo fosfato al glucosio, ottenendo così una molecola che non può uscire dalla cellula. Allo stesso tempo, l’insulina inibisce l’enzima glucosio-6-fosfatasi, che rimuove il gruppo fosfato. Questi due enzimi sono fondamentali per la formazione del glicogeno. Inoltre, l’insulina attiva gli enzimi fosfofruttochinasi e glicogeno sintasi, responsabili della sintesi del glicogeno.[113]
Diminuzione della gluconeogenesi e della glicogenolisi – diminuisce la produzione di glucosio da substrati non glucidici, principalmente nel fegato (la maggior parte dell’insulina endogena che arriva al fegato non lascia mai il fegato); la diminuzione dell’insulina causa la produzione di glucosio da parte del fegato a partire da substrati diversi.[111]
Diminuzione della proteolisi – diminuzione della scomposizione delle proteine[111]
Diminuzione dell’autofagia – diminuzione del livello di degradazione degli organelli danneggiati. I livelli postprandiali inibiscono completamente l’autofagia[114].
Aumento dell’assorbimento di aminoacidi – costringe le cellule ad assorbire gli aminoacidi circolanti; la diminuzione dell’insulina inibisce l’assorbimento.[111]
Tono muscolare arterioso – costringe i muscoli della parete arteriosa a rilassarsi, aumentando il flusso sanguigno, soprattutto nelle microarterie; la diminuzione dell’Insulina riduce il flusso permettendo a questi muscoli di contrarsi.[115]
Aumento della secrezione di acido cloridrico da parte delle cellule parietali dello stomaco.[citazione necessaria]
Aumento dell’assorbimento di potassio – costringe le cellule che sintetizzano glicogeno (una sostanza molto spugnosa e “umida”, che aumenta il contenuto di acqua intracellulare e i relativi ioni K+)[116] ad assorbire il potassio dai fluidi extracellulari; la mancanza di insulina inibisce l’assorbimento. L’aumento dell’assorbimento cellulare di potassio da parte dell’insulina abbassa i livelli di potassio nel plasma sanguigno. Ciò potrebbe avvenire attraverso la traslocazione indotta dall’insulina della Na+/K+-ATPasi sulla superficie delle cellule muscolari scheletriche.[117][118]
Diminuzione dell’escrezione renale di sodio.[119]
L’Insulina influenza anche altre funzioni corporee, come la compliance vascolare e la cognizione. Una volta che l’Insulina entra nel cervello umano, migliora l’apprendimento e la memoria, in particolare la memoria verbale.[120] Il potenziamento della segnalazione cerebrale dell’Insulina mediante la somministrazione intranasale di insulina migliora anche la risposta termoregolatoria e glucoregolatoria acuta all’assunzione di cibo, suggerendo che l’insulina a livello nervoso centrale contribuisce al coordinamento di un’ampia varietà di processi omeostatici o regolatori nel corpo umano. [121] L’insulina ha anche effetti stimolanti sull’ormone di rilascio delle gonadotropine dall’ipotalamo, favorendo così la fertilità.[122]
Una nota interessante riguarda il fatto che l’Insulina è un importante regolatore del metabolismo degli endocannabinoidi (EC) e il trattamento con insulina ha dimostrato di ridurre gli EC intracellulari, il 2-arachidonoilglicerolo (2-AG) e l’anandamide (AEA), che corrispondono a cambiamenti di espressione sensibili all’insulina negli enzimi del metabolismo degli EC. Negli adipociti insulino-resistenti, i modelli di espressione degli enzimi indotti dall’insulina sono disturbati in modo coerente con un’elevata sintesi di EC e una ridotta degradazione di EC. I risultati suggeriscono che gli adipociti insulino-resistenti non riescono a regolare il metabolismo delle EC e diminuiscono i livelli intracellulari di EC in risposta alla stimolazione insulinica, per cui gli individui obesi insulino-resistenti presentano un aumento delle concentrazioni di EC.[123][124] Questa disregolazione contribuisce all’eccessivo accumulo di grasso viscerale e al ridotto rilascio di adiponectina dal tessuto adiposo addominale, nonché all’insorgenza di diversi fattori di rischio cardiometabolici associati all’obesità e al diabete di tipo II.[125]
Opie EL (1901). “Diabetes Mellitus Associated with Hyaline Degeneration of the islands of Langerhans of the Pancreas”. Bulletin of the Johns Hopkins Hospital. 12 (125): 263–64. hdl:2027/coo.31924069247447.
The American Institute of Nutrition (1967). “Proceedings of the Thirty-first Annual Meeting of the American Institute of Nutrition”. Journal of Nutrition. 92 (4): 509. doi:10.1093/jn/92.4.507.
de Leiva A, Brugués E, de Leiva-Pérez A (2011). “The discovery of insulin: Continued controversies after ninety years”. Endocrinología y Nutrición (English Edition). 58 (9): 449–456. doi:10.1016/j.endoen.2011.10.001.
Zuger A (October 4, 2010). “Rediscovering the First Miracle Drug”. The New York Times. Retrieved 2010-10-06. Elizabeth Hughes was a cheerful, pretty little girl, five feet tall, with straight brown hair and a consuming interest in birds. On Dr. Allen’s diet her weight fell to 65 pounds, then 52 pounds, and then, after an episode of diarrhea that almost killed her in the spring of 1922, 45 pounds. By then she had survived three years, far longer than expected. And then her mother heard the news: Insulin had finally been isolated in Canada.
Bliss M (2007). The discovery of insulin (25th anniversary ed.). Chicago: University of Chicago Press. p. 132. ISBN9780226058993. OCLC74987867. The Lilly company would be delighted to work with Toronto, Clowes wrote, and hinted, perhaps intentionally, perhaps not, that Toronto could be bypassed: “I have thus far refrained from starting work in our laboratories on the field of this question as I was anxious to avoid in any way intruding on the field of yourself and your associates until you had published your results. I feel, however, that the matter is now one of such immediate importance that we should take up the experimental end of the question without delay, preferably cooperating with you and your associates…”
Tsou CL (2015). 对人工合成结晶牛胰岛素的回忆 [Memory on the research of synthesizing bovine insulin]. 生命科学 [Chinese Bulletin of Life Science] (in Simplified Chinese). 27 (6): 777–79.
Katsoyannis PG, Fukuda K, Tometsko A, Suzuki K, Tilak M (1964). “Insulin Peptides. X. The Synthesis of the B-Chain of Insulin and Its Combination with Natural or Synthetis A-Chin to Generate Insulin Activity”. Journal of the American Chemical Society. 86 (5): 930–32. doi:10.1021/ja01059a043.
Kung YT, Du YC, Huang WT, Chen CC, Ke LT (November 1965). “Total synthesis of crystalline bovine insulin”. Scientia Sinica. 14 (11): 1710–6. PMID5881570.
Marglin A, Merrifield RB (November 1966). “The synthesis of bovine insulin by the solid phase method”. Journal of the American Chemical Society. 88 (21): 5051–2. doi:10.1021/ja00973a068. PMID5978833.
Tsou CL (2015). 对人工合成结晶牛胰岛素的回忆 [Memory on the research of synthesizing bovine insulin]. 生命科学 [Chinese Bulletin of Life Science] (in Simplified Chinese). 27 (6): 777–79.
Iwase H, Kobayashi M, Nakajima M, Takatori T (January 2001). “The ratio of insulin to C-peptide can be used to make a forensic diagnosis of exogenous insulin overdosage”. Forensic Science International. 115 (1–2): 123–127. doi:10.1016/S0379-0738(00)00298-X. PMID11056282.
Dimitriadis G, Mitrou P, Lambadiari V, Maratou E, Raptis SA (August 2011). “Insulin effects in muscle and adipose tissue”. Diabetes Research and Clinical Practice. 93 (Suppl 1): S52–59.
Bergamini E, Cavallini G, Donati A, Gori Z (October 2007). “The role of autophagy in aging: its essential part in the anti-aging mechanism of caloric restriction”. Annals of the New York Academy of Sciences. 1114 (1): 69–78. Bibcode:2007NYASA1114…69B. doi:10.1196/annals.1396.020. PMID17934054. S2CID21011988.
Non è per me raro discutere dell’argomento fertilità negli utilizzatori di AAS o nei soggetti in TRT. Complice una classe medica non sempre aggiornata, molti sono spinti a credere che una condizione di sterilità sia ineluttabile, tanto nei soggetti utilizzatori di dosi sovrafisiologiche di AAS quanto in quelli sottoposti a Terapia Sostitutiva del Testosterone [TRT].
A sottolineare questa eventualità ci ha pensato uno studio pubblicato nel 2019 nel quale veniva riportato che “la terapia con Testosterone è un contraccettivo, anche se di scarsa efficacia. Gli uomini in età riproduttiva con Testosterone basso devono essere informati degli effetti negativi della TRT sulla fertilità. Se la TRT viene prescritta a uomini interessati a preservare la fertilità, è opportuno proporre un’analisi del liquido seminale e l’eventuale crioconservazione dello sperma. Opzioni come il Clomifene Citrato e l’hCG, insieme al rinvio a un urologo della riproduzione, dovrebbero essere prese in considerazione per aumentare naturalmente i livelli di Testosterone negli uomini con testosterone basso che vogliono evitare la TRT.”
Immagine che spiega l’effetto contraccettivo del Testosterone esogeno. In sintesi, agisce attraverso due meccanismi: la diminuzione del Testosterone intratesticolare e l’inibizione della spermatogenesi. La maggior parte del Testosterone intra-testicolare è prodotto dalle cellule di Leydig nel testicolo. In presenza di Testosterone esogeno, esso inibisce la produzione di Ormone di Rilascio delle Gonadotropine (GnRH), che a sua volta inibisce la produzione di Ormone Luteinizzante (LH) e diminuisce la produzione endogena di Testosterone da parte delle cellule di Leydig, diminuendo la concentrazione di Testosterone intra-testicolare. L’inibizione della produzione di GnRH inibisce anche il rilascio dell’Ormone Follicolo-Stimolante (FSH), che compromette la spermatogenesi nelle cellule del Sertoli.
Quindi possiamo chiudere qui e liquidare la questione con un “si, anche in TRT si è destinati ad una condizione di sterilità”? Assolutamente no! Per quanto corretta nei punti espositivi, la conclusione di Amir Shahreza Patel et al. è incompleta. Per quale motivo? Ve lo spiegherò in questo articolo…
Breve panoramica sulla spermatogenesi:
Gli Steroidi Anabolizzanti Androgeni non influiscono solo sulla produzione endogena di Testosterone, ma anche sulla produzione di sperma, un processo chiamato spermatogenesi.
La spermatogenesi è strettamente regolata dalle cellule di Leydig e Sertoli del testicolo. Le cellule di Leydig producono Testosterone in risposta all’attivazione del recettore LHCG (LHCGR). Questo recettore è attivato dal legame con l’Ormone Luteinizzante (LH). Il Testosterone, a sua volta, agisce sulle cellule vicine, comprese le cellule del Sertoli, per controllare la spermatogenesi. L’attivazione del recettore dell’FSH (FSHR) sulle cellule del Sertoli controlla direttamente la spermatogenesi.
La produzione di spermatozoi avviene nei tubuli seminiferi e può essere suddivisa approssimativamente in tre fasi, come illustrato di seguito:
Le diverse fasi della spermatogenesi a partire da uno spermatogonio.
Tutte queste fasi si svolgono nei tubuli seminiferi. Durante la prima fase, gli spermatogoni migrano tra le cellule del Sertoli verso il lume dei tubuli. Mentre migrano lungo le cellule del Sertoli, questi spermatogoni si dividono lentamente e si differenziano in cellule spermatiche mature. In primo luogo, subiscono la mitosi, ossia la divisione in due cellule figlie identiche. Alcune di queste cellule figlie subiranno ulteriori modifiche e ingrandimenti, un processo noto come spermatocitogenesi, che darà origine agli spermatociti primari. Queste cellule, a loro volta, subiranno la meiosi. In questo caso, si verificano due divisioni cellulari consecutive, che danno origine a un totale di quattro cellule figlie. Ognuna di queste cellule avrà la metà del numero di cromosomi della cellula madre. Dopo la prima divisione cellulare chiamiamo queste cellule spermatociti secondari, mentre dopo la seconda divisione meiotica le chiamiamo spermatidi. Infine, gli spermatidi si differenziano in spermatozoi (spermatozoi maturi) durante la spermiogenesi.
L’intero processo di spermatogenesi richiede circa 74 giorni per essere completato [1]. Dopodiché, ci vorranno altri 1-21 giorni prima che gli spermatozoi finiscano nell’eiaculato [2]. Di conseguenza, quando la spermatogenesi si interrompe e riprende, ci vorrà un po’ di tempo prima che ciò si rifletta in un’analisi del liquido seminale.
La spermatogenesi dipende in larga misura dalla concentrazione di Testosterone intratesticolare (ITT). Poiché l’LH stimola i testicoli a produrre Testosterone e quindi è responsabile della concentrazione di ITT, l’LH è importante per la spermatogenesi. Normalmente, la concentrazione di ITT è circa 100 volte superiore a quella del sangue [3]. La somministrazione settimanale di 200mg di Testosterone Enantato la riduce notevolmente, fino a circa il 2% dei livelli basali. Sebbene non sia mai stato studiato nell’uomo, il limite inferiore della concentrazione di ITT necessaria per una spermatogenesi quantitativamente normale nei ratti è circa il 20% del livello basale [4]. Una volta scesi al di sotto di questo valore, esiste una chiara relazione tra il calo della concentrazione di ITT e la conta spermatica.
Breve parentesi su Estrogeni e fertilità:
Il Recettore α degli Estrogeni (ERα) è essenziale per la fertilità maschile. La sua attività è responsabile del mantenimento della citoarchitettura epiteliale nei dotti efferenti e del riassorbimento del liquido per la concentrazione degli spermatozoi nella testa dell’epididimo. Queste e altre scoperte hanno contribuito a stabilire il ruolo bisessuale degli estrogeni nell’importanza riproduttiva. È stato dimostrato che gli Estrogeni regolano l’espressione dello scambiatore Na+/H+-3 (NHE3) e la velocità di trasporto del 22Na+, sensibile a un inibitore di NHE3. Pertanto, nel maschio, gli estrogeni regolano uno dei più importanti trasportatori epiteliali di ioni e mantengono la differenziazione morfologica epiteliale nei dotti efferenti del maschio, indipendentemente dalla regolazione del trasporto di Na+.[https://www.pnas.org/]
17 β-estradiolo (E2) legato a ERα (giallo) e ERβ (blu). Solo due residui, cioè L384/M336 e M421/I373 (Erα/ERβ), differiscono nelle tasche di legame di ERα e ERβ. Non sorprende che l’E2 si leghi ai sottotipi in modo leggermente diverso.
Così come una concentrazione ottimale di E2 porta ad un miglioramento dei quadri della fertilità, livelli elevati di Estradiolo sono correlati all’infertilità maschile. Le cause dell’iperestrogenismo includono malattie della corteccia surrenale, del testicolo o uso di farmaci che influenzano l’asse ipotalamo-ipofisi-gonadi.[https://www.nature.com/]
Da notare che i dati raccolti hanno sollevato la possibilità di puntare sul ERα nello sviluppo di un contraccettivo per l’uomo.
AAS è soppressione della spermatogenesi:
E’ un dato di fatto che l’uso di AAS sopprime la produzione endogena di Testosterone. Lo fa attraverso un feedback negativo a livello dell’ipotalamo e dell’ipofisi. In breve, l’ipotalamo secerne un ormone chiamato Ormone di Rilascio delle Gonadotropine (GnRH) che viene rilasciato nel sistema portale ipofisario. Attraverso questo sistema, può raggiungere l’ipofisi anteriore. Qui, si legherà al suo recettore cognitivo che porterà alla secrezione di gonadotropine da parte dell’ipofisi anteriore. Queste gonadotropine, l’Ormone Luteinizzante (LH) e l’Ormone Follicolo-Stimolante (FSH), raggiungono la circolazione sistemica che le trasporta all’organo bersaglio: i testicoli. Il legame dell’LH al suo recettore specifico porta alla produzione di Testosterone. Il legame dell’FSH con il suo recettore specifico svolge un ruolo importante nella spermatogenesi. E, come descritto in precedenza, anche il Testosterone prodotto è fondamentale nella spermatogenesi.
Gli AAS inibiscono la secrezione di GnRH da parte dell’ipotalamo e la secrezione di gonadotropine da parte dell’ipofisi. Di conseguenza, sia la produzione di Testosterone che quella di spermatozoi vengono soppresse. Questo può portare a una condizione chiamata azoospermia, in cui non si trovano spermatozoi in un campione di sperma. Oppure può portare all’oligozoospermia, in cui la concentrazione di spermatozoi è molto bassa (inferiore a 15 milioni per mL o 39 milioni per eiaculato).[5]
Ad esempio, in uno studio, il 65% degli uomini è diventato azoospermico entro 6 mesi dalla somministrazione di Testosterone Enantato a 200mg settimanali [6]. Poiché l’LH e l’FSH non sono stati completamente soppressi (rispettivamente -66,7 e -62,5%), si può ipotizzare che un numero maggiore di uomini sarebbe diventato azoospermico con un dosaggio più elevato e più soppressivo. In effetti, in combinazione con un progestinico (che porterebbe a una più forte soppressione di LH e FSH), si registrano generalmente tassi di azoospermia di quasi il 90% [7]. Tuttavia, uno studio prospettico osservazionale (lo studio HAARLEM) che ha seguito 100 consumatori di AAS prima, durante e in due momenti successivi al ciclo di AAS, ha visto risultati simili a quelli dello studio in cui il 65% degli uomini è diventato azoospermico [8]. I dati relativi all’analisi dello sperma erano disponibili per 91 utilizzatori al termine del ciclo. Nonostante la soppressione praticamente totale di LH e FSH in quasi tutti gli utilizzatori, la concentrazione di spermatozoi era inferiore a 15 milioni per mL nel 68% degli utilizzatori (la conta totale degli spermatozoi era inferiore a 40 milioni nel 77%). Una differenza fondamentale in questo caso potrebbe essere il tempo di soppressione, in quanto l’altro studio ha mostrato il tasso cumulativo di azoospermia fino a 6 mesi, mentre gli utilizzatori di AAS si sono sottoposti a somministrazioni per periodi di tempo variabili, con una durata mediana di 13 settimane. Inoltre, alcuni dei consumatori di AAS hanno utilizzato l’hCG durante il ciclo, che potrebbe aver stimolato in qualche misura la spermatogenesi (tornerò su questo punto più avanti). Anche se gli autori scrivono: “(…) l’uso di hCG non ha avuto effetti rilevabili sulle dimensioni dei testicoli o sulla spermatogenesi”. Questo potrebbe essere attribuito a un sottodosaggio di hCG, a un uso non corretto o forse, in qualche misura, alla mancanza di potenza statistica. Infine, alti dosaggi di AAS – in modo del tutto casuale – potrebbero stimolare la spermatogenesi sostituendo una parte dell’attività androgena endogena mancante, come spiegato nella sezione precedente.
In ogni caso, è chiaro che l’uso di AAS di per se compromette in modo significativo la spermatogenesi.
Uso di AAS e atrofia testicolare: I testicoli comprendono il compartimento interstiziale, che ospita le cellule di Leydig, e il compartimento dei tubuli seminiferi, che ospita la spermatogenesi. Quest’ultimo è responsabile della maggior parte del volume del testicolo, con valori che in letteratura variano dal 60 al 90% [9, 10]. Gran parte di questo volume è costituito da cellule spermatiche in via di sviluppo. Di conseguenza, quando la spermatogenesi è compromessa, i testicoli diminuiscono di dimensioni. Ad esempio, lo studio citato in precedenza, in cui il 65% degli uomini è diventato azoospermico entro 6 mesi dalla somministrazione di Testosterone, ha visto una diminuzione del volume testicolare del 16,5% [6]. Uno studio in cui il Testosterone è stato combinato con un dosaggio molto basso di un progestinico orale (Levonorgestrel) per ottenere una soppressione più forte ha registrato una riduzione maggiore del volume testicolare, pari a circa il 30% [11]. Lo studio HAARLEM, citato in precedenza, ha registrato una riduzione del 24%. È interessante notare che i consumatori di AAS hanno visto il loro volume testicolare tornare a quello che era 3 mesi dopo la cessazione dell’uso (c’era solo una differenza del -4% a quel punto).
La terapia con gonadotropine (hCG e hMG/FSH) può preservare la spermatogenesi: L’effetto dell’hCG e dell’FSH o dell’hMG sulla spermatogenesi è forse dimostrato in modo più elegante da una serie di esperimenti di Matsumoto et al. [12]. In primo luogo, soggetti maschi sani hanno ricevuto 5000 UI di hCG due volte alla settimana per 7 mesi. Questo stimola fortemente la produzione di Testosterone da solo e di conseguenza l’FSH viene completamente soppresso. Ciononostante, è stata mantenuta una certa produzione di spermatozoi, la cui concentrazione è stata ridotta da 88 milioni/mL a 22 milioni/mL dopo 4 mesi. Dopo questi 7 mesi, il Testosterone Enantato (200mg settimanali) è stato aggiunto all’hCG per altri 6 mesi in questi uomini. Le concentrazioni di sperma sono rimaste praticamente inalterate (26 milioni/mL negli ultimi 3 mesi).
Dopo questo periodo, 4 soggetti hanno continuato l’hCG per altri 3 mesi senza Testosterone. Successivamente, in due dei soggetti è stato aggiunto l’FSH (100 UI al giorno) e negli altri due l’hMG (75 UI al giorno). L’aggiunta di FSH o hMG ha portato a un forte aumento della produzione di spermatozoi, raggiungendo una media di 103 milioni/mL negli ultimi 2 mesi:
Allo stesso modo, l’FSH da solo può preservare una parte della spermatogenesi durante la soppressione della terapia con testosterone, come illustrato nella figura seguente [13]:
Ciò che si può dedurre da questi risultati è che sia l’FSH che l’hCG possono preservare una certa spermatogenesi durante la soppressione delle gonadotropine da parte del Testosterone, ma che entrambi sono necessari per una spermatogenesi quantitativamente normale. Va notato, tuttavia, che ci sono state marcate differenze interindividuali. Nel precedente studio con hCG, un uomo è diventato azoospermico durante il trattamento con hCG.
Un piccolo studio retrospettivo suggerisce che l’hCG da solo, al dosaggio di 500 UI a giorni alterni, può preservare completamente la spermatogenesi in associazione alla Terapia Sostitutiva del Testosterone [14]. Forse in questi uomini c’era una secrezione residua di FSH sufficiente a consentire la piena conservazione della spermatogenesi. Inoltre, la natura retrospettiva dello studio potrebbe aver portato a una distorsione dei risultati.
Differenze tra hCG, LH e FSH.
Questo mi porta a un altro aspetto che vorrei discutere: il dosaggio. Uno studio ha rilevato che iniettando hCG al dosaggio di 250 UI a giorni alterni si ottiene una concentrazione di Testosterone intratesticolare praticamente uguale a quella del basale [15]. Dato il ruolo centrale del Testosterone intratesticolare nella spermatogenesi, si potrebbe sostenere che questo basso dosaggio dovrebbe essere sufficiente per preservare la spermatogenesi durante l’uso di AAS. Tuttavia, questo aspetto non è stato studiato direttamente in uno studio controllato.
L’hMG (chiamata anche Menotropina o Gonadotropina Umana della Menopausa – human Menopausal Gonadotropin), commercializzato in Italia sotto il nome di MENOGON ®, è un principio attivo per il trattamento dei disordini della fertilità. Si compone di gonadotropine che vengono estratte dalle urine di donne in post-menopausa, gonadotropine che sono solitamente l’Ormone Luteinizzante (LH) e l’Ormone Follicolo-Stimolante (FSH). Spesso, contiene anche Gonadotropina Corionica umana (hCG).
Un dosaggio più elevato, ma comunque inferiore a quello utilizzato negli studi di Matsumoto, ha dimostrato la conservazione di una certa spermatogenesi in pazienti con ipogonadismo secondario con hCG dosato a 500-2500 UI due volte alla settimana [16]. I dosaggi sono stati titolati in base ai livelli di Testosterone raggiunti. Per ottenere una spermatogenesi quantitativamente normale era necessaria l’aggiunta di FSH (3x 150 UI hMG settimanali). Anche in questo caso, però, si trattava di uno studio retrospettivo.
Infine, sono state sollevate alcune perplessità sull’effetto dell’hCG sulla morfologia degli spermatozoi. Uno studio finlandese suggerisce che l’uso concomitante di hCG e AAS ad alti dosaggi può avere un impatto negativo sulla morfologia dello sperma [17]. Lo studio ha seguito 18 atleti di forza amatoriali, 16 dei quali hanno utilizzato l’hCG insieme ad alti dosaggi di AAS. I campioni di sperma sono stati prelevati alla fine del ciclo di AAS, circa 1,5 mesi dopo il ciclo e circa 6 mesi dopo il ciclo. Naturalmente, la produzione di sperma era compromessa, con una conta media di 33 milioni di spermatozoi/mL alla fine del ciclo di AAS. Un soggetto è diventato azoospermico (e lo è rimasto per tutto il successivo periodo di sospensione). Ciò sembra dimostrare che l’uso di hCG può preservare una certa spermatogenesi durante l’uso di AAS. La morfologia dello sperma, tuttavia, era solo del 15% rispetto a una media del 40% di una coorte finlandese di donatori di banche del seme. Inoltre, hanno trovato una correlazione tra la dose totale di hCG utilizzata e gli spermatozoi morfologicamente anormali.
Quando hanno stratificato gli utilizzatori in due gruppi: un gruppo ad alta dose di hCG (>12.000 UI totali) e un gruppo a bassa dose (<12.000 UI totali), hanno notato che c’era una differenza significativa nella morfologia dello sperma tra i due. In media, il 22% era normale nel gruppo ad alto dosaggio e il 72% nel gruppo a basso dosaggio alla fine del ciclo di AAS. Ma come? Se la media del gruppo è del 15%, come può essere più alta sia nel gruppo ad alta dose che in quello a bassa dose? C’è qualcosa di sbagliato nei dati. Questo è un problema dello studio in questione. Da notare che, poiché il gruppo ad alta dose aveva una concentrazione di spermatozoi quasi cinque volte superiore, la quantità assoluta di spermatozoi morfologicamente normali era maggiore nel gruppo ad alta dose.
Si potrebbe obiettare che potrebbe essere l’assenza di FSH, piuttosto che l’hCG di per sé, ad avere un impatto sulla morfologia. Infatti, è stato riscontrato che dosi elevate di hCG migliorano la motilità degli spermatozoi e la morfologia normale in uomini subfertili con livelli normali di FSH [17]. Inoltre, si potrebbe sostenere che l’AAS stesso potrebbe avere un effetto negativo diretto sulla morfologia degli spermatozoi a dosi elevate [18]. Questo potrebbe non manifestarsi se vengono prodotte solo piccole quantità di spermatozoi, come nel caso del gruppo a basso dosaggio. Anche Matsumoto et al. hanno dimostrato che l’hCG (3x 5000 UI settimanali) non ha alcun effetto sulla morfologia degli spermatozoi in associazione al testosterone in un piccolo studio [19]. Infine, anche l’abuso di altre sostanze non dichiarate potrebbe aver avuto un impatto.
Conclusioni:
Ricapitolando, la spermatogenesi è strettamente regolata da LH e FSH. Quando si somministrano AAS, la secrezione di questi due ormoni viene fortemente ridotta. Di conseguenza, anche la spermatogenesi viene fortemente ridotta. Nella maggior parte degli uomini questo porta all’azoospermia. È stato riscontrato che l’uso di hCG mantiene una certa spermatogenesi, anche se a un livello inferiore al normale. L’aggiunta di FSH (direttamente o come parte di hMG) è necessaria per preservare completamente la spermatogenesi. Il dosaggio necessario per mantenere in modo ottimale la spermatogenesi con il solo hCG durante un ciclo AAS non è del tutto chiaro. Dato l’importante ruolo del Testosterone intratesticolare nel mantenimento della spermatogenesi, si potrebbe sostenere che un dosaggio che sostenga questo aspetto sostenga in modo ottimale anche la spermatogenesi. Si potrebbe quindi arrivare a un dosaggio di circa 250 UI a giorni alterni. Tuttavia, gli studi clinici (controllati) che hanno valutato direttamente l’impatto sulla spermatogenesi con la soppressione delle gonadotropine hanno tutti utilizzato dosaggi nettamente superiori. I dati di studi retrospettivi suggeriscono che potrebbero essere sufficienti da 500 a 2500 UI due volte alla settimana. L’ideale sarebbe testare il proprio sperma per capire quale sia il dosaggio più adatto. Si tenga presente che l’intero processo di spermatogenesi e la successiva comparsa di spermatozoi nell’eiaculato possono richiedere fino a circa 3 mesi. I cambiamenti nella terapia potrebbero quindi richiedere almeno 3 mesi prima che i loro effetti si riflettano nell’analisi dello sperma.
Dopo le informazioni fin qui riportate, non ci si stupisce del fatto che nello studio citato nell’introduzione la TRT fosse stata classificata come un “contraccettivo di bassa efficacia”. Se infatti togliamo dall’equazione la somministrazione esogena di hCG e/o FSH [o in alternativa hMG], la condizione di azoospermia è praticamente una certezza. Da considerarsi anche i dosaggi di questi ancillari della TRT. Dosaggi che devono tenere conto della risposta terapeutica soggettiva. Ciò significa che i dosaggi standard per l’hCG, per esempio, rappresentano per la maggior parte dei soggetti solo un punto di partenza che dovrà essere riconsiderato alla luce di esami specifici [vedi spermiogramma].
Un protocollo di fertilità nel quale mi sono imbattuto spesso parlando con atleti o preparatori d’oltre oceano è costituito da una hCG, hMG e Clomifene Citrato. L'”invenzione” di questo protocollo si attribuisce a Dave Palumbo. Non propriamente un luminare dell’endocrinologia ma sicuramente un vagliatore di tester non da poco.
Il protocollo è il seguente:
hCG – 2000 UI a giorni alterni hMG – 75 UI a giorni alterni Clomifene Citrato – 50mg al giorno
Clomifene Citrato
In alternativa all’uso di Clomifene Citrato si opta per Enclomifene Citrato, l’isomero trans del Clomifene Citrato. Ma di lui parlerò in un articolo apposito. Rimane comunque il dubbio di una loro sufficiente efficacia additiva.
Encolimfene Citrato
Ho avuto l’opportunità di raccogliere molte testimonianze di utilizzatori. Alcuni di loro hanno trascorso più di un decennio tra cicli, bridge e fasi in TRT. Anche nei casi più estremi, quando è arrivato il momento di avere un figlio, una parte consistente di quelli che hanno seguito questo semplice protocollo sono riusciti a ingravidare la propria moglie/fidanzata:
La sospensione temporanea della TRT per seguire un protocollo di ristabilizzazione dell’Asse HPT e aumentare il numero di spermatozoi con il protocollo di fertilità, non rappresentava una costante ma una eventualità che poteva interessare alcuni individui .
Ovviamente, quanto detto non rappresenta assolutamente una prescrizione medica o un consiglio terapeutico! Si tratta, come sempre, di pura divulgazione scientifica volta alla formazione di una cultura di base utile alla tutela della propria e altrui salute.
Gabriel Bellizzi [CEO BioGenTech]
Riferimenti:
“Book on Steroids” di Peter Bond [capitolo 6 – Side effects and managing them – sezione 6.12. – Low/undetectable sperm count (oligo-/azoospermia)].
Amann, Rupert P. “The cycle of the seminiferous epithelium in humans: a need to revisit?.” Journal of andrology 29.5 (2008): 469-487.
Rowley, Mavis J., Florence Teshima, and Carl G. Heller. “Duration of transit of spermatozoa through the human male ductular system.” Fertility and sterility 21.5 (1970): 390-396.
McLachlan, Robert I., et al. “Effects of testosterone plus medroxyprogesterone acetate on semen quality, reproductive hormones, and germ cell populations in normal young men.” The Journal of Clinical Endocrinology & Metabolism 87.2 (2002): 546-556.
Zirkin, Barry R., et al. “Maintenance of advanced spermatogenic cells in the adult rat testis: quantitative relationship to testosterone concentration within the testis.” Endocrinology 124.6 (1989): 3043-3049.
T. G. Cooper, E. Noonan, S. Von Eckardstein, J. Auger, H. Baker, H. M. Behre, T. B. Haugen, T. Kruger, C. Wang, M. T. Mbizvo, et al. World health organization reference values for human semen characteristics. Human reproduction update, 16(3):231–245, 2010.
W. H. O. T. F. on Methods for the Regulation of Male Fertility. Contraceptive efficacy of testosterone-induced azoospermia in normal men. The Lancet, 336(8721):955–959, 1990.
Page, Stephanie T., John K. Amory, and William J. Bremner. “Advances in male contraception.” Endocrine reviews 29.4 (2008): 465-493.
Smit, D. L., et al. “Disruption and recovery of testicular function during and after androgen abuse: the HAARLEM study.” Human Reproduction 36.4 (2021): 880-890.
S. Melmed. Williams textbook of endocrinology. 13th edition. Elsevier Health Sciences, 2016.
M. Simoni and I. T. Huhtaniemi. Endocrinology of the Testis and Male Reproduction. Springer, 2017.
Anawalt, Bradley D., et al. “Intramuscular testosterone enanthate plus very low dosage oral levonorgestrel suppresses spermatogenesis without causing weight gain in normal young men: a randomized clinical trial.” Journal of andrology 26.3 (2005): 405-413.
Matsumoto, Alvin M., Anthony E. Karpas, and William J. Bremner. “Chronic human chorionic gonadotropin administration in normal men: evidence that follicle-stimulating hormone is necessary for the maintenance of quantitatively normal spermatogenesis in man.” The Journal of Clinical Endocrinology & Metabolism 62.6 (1986): 1184-1192.
Matsumoto, Alvin M., et al. “Reinitiation of sperm production in gonadotropin-suppressed normal men by administration of follicle-stimulating hormone.” The Journal of clinical investigation 72.3 (1983): 1005-1015.
Hsieh, Tung-Chin, et al. “Concomitant intramuscular human chorionic gonadotropin preserves spermatogenesis in men undergoing testosterone replacement therapy.” The Journal of urology 189.2 (2013): 647-650.
Coviello, Andrea D., et al. “Low-dose human chorionic gonadotropin maintains intratesticular testosterone in normal men with testosterone-induced gonadotropin suppression.” The Journal of Clinical Endocrinology & Metabolism 90.5 (2005): 2595-2602.
Depenbusch, Marion, et al. “Maintenance of spermatogenesis in hypogonadotropic hypogonadal men with human chorionic gonadotropin alone.” European journal of endocrinology 147.5 (2002): 617-624.
Homonnai, Z. T., M. Peled, and G. F. Paz. “Changes in semen quality and fertility in response to endocrine treatment of subfertile men.” Gynecologic and obstetric investigation 9.5 (1978): 244-255.
Torres-Calleja, J., et al. “Effect of androgenic anabolic steroids on sperm quality and serum hormone levels in adult male bodybuilders.” Life sciences 68.15 (2001): 1769-1774.
Matsumoto, Alvin M., et al. “Human chorionic gonadotropin and testicular function: stimulation of testosterone, testosterone precursors, and sperm production despite high estradiol levels.” The Journal of Clinical Endocrinology & Metabolism 56.4 (1983): 720-728.
Negli ultimi anni il mondo del Bodybuilding e del Fitness è stato letteralmente invaso da affermazioni sensazionalistiche di stampo pubblicitario su una classe di molecole steroidee provenienti dal mondo animale e vegetale: gli Ecdysteroidi. Tra questi sono emersi agli onori della cronaca il 20-hydroxyecdysone e il Turkesterone. Quest’ultimo, in particolare, è stato fortemente pubblicizzato da “influencer” con nozioni di biochimica pari o prossime allo 0 e da venditori con sete speculativa mista ad ignoranza e malafede.
In base a quanto sinteticamente esposto, ho deciso di riportare le cose come sono alla luce delle attuali evidenze scientifiche e della lucida osservazione empirica e dei dati aneddotici.
Introduzione agli Ecdysteroidi:
Gli Ecdysteroidi sono ormoni steroidei degli artropodi responsabili principalmente della muta, dello sviluppo e, in misura minore, della riproduzione;[1][2][3] esempi di Ecdysteroidi includono Ecdysone, Ecdysterone, Turkesterone e 2-deossiecdysone. [4] Questi composti sono sintetizzati negli artropodi a partire dal colesterolo alimentare attraverso la fase metabolica influenzata dalla famiglia dei citocromi P450.[5] I Fitodisteroidi sono presenti anche in molte piante, per lo più come agenti protettivi (tossine o antifeedanti) contro gli insetti erbivori.[6][7]
Infatti, gli Ecdysteroidi sono presenti in molte piante (circa il 6% delle piante esistenti)[8], anche se a livelli solitamente considerati insufficienti per l’estrazione o l’attività biologica. Alcune piante che presentano una maggiore quantità di Ecdysteroidi bioattivi sono:
Asparagus Filicinus[9]
Spinacia oleracea (Spinaci, fonte di 20-idrossidisone)[10]
Quinoa, soprattutto nella crusca, che contiene principalmente 2-idrossidisone e makisteroni[11] e varia da 450-1300mcg/g di ecdysone equivalente[12].
Ignami[13]
Funghi a bottone bianco[11]
Ajuga Turkestanica, fonte del “Turkesterone” idrossilato C-11[14]
Rhaponticum carthamoides
Silene Praemixta (2-deossiecdysterone e 2-deossi-alfa-ecdysone)
Vitex Scabra, con l’1,8% di ecdisteroidi in peso[15] e altre specie di Vitex[16] come cymosa[17] e canescens[18].
Gli Ecdysteroidi prendono il loro nome dal fatto di avere uno scheletro carbossilico steroideo (sterone) e dall’essere associati al processo di muta, altrimenti noto come ecdisi. La ragione della loro esistenza nelle piante (essendo un ormone degli insetti), come precedentemente accennato, è che proteggono le piante dagli insetti non adattati alla loro tolleranza, e quindi sono una fitoalessina.[19-13]
Gli Ecdysteroidi sono composti ormonali coinvolti nel comportamento sessuale degli insetti, nella muta e nella metamorfosi. Gli Ecdysteroidi presentano una somiglianza strutturale con il Testosterone e sono considerati il composto simile al Testosterone più attivo negli insetti.
Di seguito è riportata la struttura generale della famiglia degli Ecdysteroidi:
Sebbene dal 2001 siano noti oltre 200 Ecdysteroidi,[8] e ne siano stati registrati fino a 463[19], la maggior parte di essi non è bioattiva se ingerita per via orale.[20] Tra quelli più comuni, sia nella ricerca che nell’assunzione per via orale, vi sono:
Ecdysone
Ecdysterone e beta-ecdysterone
20-idrossiecdysone
Turkesterone
Integristerone A
24(28)-deidramakisterone A
Viticosterone E
Sileneoside A e C
Ponasterone A
Citasterone
Farmacologia [biodisponibilità, farmacocinetica, interazione cellulare/recettoriale e metabolismo]:
In uno studio, utilizzando Ecdysteroidi da 0,2mg/kg di peso corporeo (come ecdysone e 20-idrossidysone), l’Ecdysone sembrava avere un’emivita di eliminazione di 4 ore e il 20-idrossidysone un’emivita di eliminazione di 9 ore nell’uomo.[20] Non è nota un’emivita attiva nell’uomo.
Tuttavia, i modelli murini mostrano un’emivita di 8,15 minuti per il 20-idrossidysone quando viene iniettato alla dose di 50mg/kg di peso corporeo nella vena caudale[21] e risultati simili sono stati replicati con il 20-idrossidysone altrove.[22] È stata notata anche un’emivita di 48 minuti (per l’Ecdysteroide Ponasterone A) quando viene iniettato alla dose di 750g.[23]
Nella drosofilia è stato clonato un recettore citoplasmatico, denominato DopEcR, che si lega agli Ecdysteroni e alla Dopamina.[24] È stato teorizzato che alcuni dei meccanismi d’azione avvengano attraverso questo recettore e siano di natura non genomica (non influenzano il nucleo della cellula).[25][26] Tra i possibili effetti non genomici vi è l’afflusso di ioni calcio che inducono la fosforilazione di Akt, di cui parlerò più avanti.
Si ipotizzano anche recettori nucleari (nei mammiferi) della superfamiglia dei recettori nucleari[27]. Il recettore dell’Ecdysterone si dimerizza con i recettori Ultraspiracle (USP) negli insetti per influenzare i geni, ma negli esseri umani deve dimerizzare con il recettore RXR.[20] Sebbene negli insetti l’USP possa dimerizzare con un’ampia varietà di recettori nucleari, nei mammiferi deve formarsi un complesso EcR:RXR perché si verifichino gli effetti. [28] Il legame dell’EcR con recettori non RXR non produce effetti genetici nei vertebrati.[29] È stato tuttavia osservato che l’RXR è un partner “riluttante” per l’EcR e che per la segnalazione genetica tramite EcR:RXR è necessario un relativo eccesso di RXR; questo è stato menzionato in uno studio[20] in relazione a un altro che ha indagato un modello in vitro su una linea cellulare di ovaio di criceto cinese.[30]
Il gambero bruno Crangon crangon possiede molteplici isoforme del recettore degli ecdisteroidi (CrcEcR) e del recettore dei retinoidi-X (CrcRXR). La troncatura dei recettori CrcEcR e CrcRXR non compromette ma altera l’attività transattiva. La modellazione in silico prevede che il tributilstagno (TBT) si adatti alla tasca di legame del ligando di CrcRXR. ► L’esposizione in vitro mostra che la TBT influenza la transattivazione degli Ecdysteroidi. ► La TBT porta a un’alterazione dell’espressione in vivo di CrcEcR e CrcRXR, soprattutto nelle ovaie.
Gli Ecdysteroidi, in particolare il 20-idrossiecdysone e il Pinnatasterone, sono stati chiamati in causa come inibitori delle pompe di efflusso della P-glicoproteina e possono interagire con altri farmaci che vengono metabolizzati ampiamente dalla P-glicoproteina, come la Berberina o l’Icariina.[31]
Pinnatasterone
Nei topi (e nell’uomo) l’escrezione avviene sia per via fecale che per via urinaria. Alcuni studi suggeriscono che la via fecale sia favorita, in quanto gli Ecdysteroidi vengono raccolti dal fegato e poi espulsi negli acidi biliari[32][33], tuttavia almeno uno studio osserva che entrambe le vie possono essere ugualmente importanti,[21] sebbene quest’ultimo studio abbia utilizzato un’iniezione di Ecdysteroidi da 50mg/kg di peso corporeo.
Nell’esaminare i metaboliti fecali, sono stati notati il 4-deossicedisone e composti con un anello B completamente ridotto.[34] In una review è stato osservato che questo metabolismo “ricorda la riduzione epatica del 4-en-3-one sull’anello-A degli ormoni steroidei dei vertebrati”.[20] Quando la scissione della catena laterale avviene tra il C20 e il C22, possono risultare i metaboliti Poststerone e 12-deossicedisone (dal 20-idrossicedysone). [Nei ratti è stato osservato anche un nuovo metabolita, il 2β,3β,6α,22R,25-pentaidrossi-5β-colest-8(14)-ene.[22] Infine, il metabolita 14-deossi-20-ecdysone (osservato come metabolita primario nelle urine umane) può avere interazioni con la microflora intestinale, poiché è noto che la microflora causa la deidrossilazione dei composti steroidei.[35]
Nell’uomo, l’escrezione urinaria di Ecdysterone tende a risultare in uno dei tre composti: l’Ecdysterone in forma invariata, il 2-deossiacidysterone o il Deossiacidysone. Il principale metabolita urinario, con una percentuale del 99,34%, è il Desossiacidysone a 21 ore dall’ingestione di 20mg di Ecdysterone.[36] Un’escrezione urinaria bifasica del composto progenitore è stata notata anche con l’analisi delle urine a 68 ore.[36] Questi metaboliti si trovano anche nelle urine dei ratti.[37]
In realtà non ci sono molte informazioni su questo argomento che possano essere considerate “conclusive”. Sembra che ci sia un’ampia varietà di metaboliti che non sono stati studiati e che il 20-HE persista più a lungo negli esseri umani che nei topi (4,1 ore contro 8,15 m) o che la sua risposta sia dipendente dalla dose. Al momento non si hanno risposte a proposito.
Interazioni neurologiche:
L’Ecdysterone è in grado di aumentare l’induzione enzimatica sia dell’acetilcolinesterasi[38] che della glutammato decarbossilasi.[39] Questi effetti sono a valle della capacità degli Ecdysteroidi di aumentare la sintesi proteica, in quanto l’aumento della sintesi proteica (attraverso l’aumento dell’efficacia dell’mRNA, come ipotizzato da Uchiyama & Otaka[40][41]) si applica ai tessuti proteici (muscolo scheletrico, proteine degli organi) e agli enzimi. L’aumento del glutammato decarbossilato è stato misurato al 25-30% in vivo dopo 2,5-50ug/kg di peso corporeo, anche se la dipendenza dalla dose non era chiara.[39]
Da sinistra: Acetilcolinesterasi e Glutammato Decarbossilasi
Esercita inoltre alcuni effetti protettivi contro le tossine neurologiche.[42][43]
Salute cardiovascolare:
I Fitoecdysoni (la famiglia di cui fa parte l’Ecdysterone) sono promettenti come agenti di riduzione del Colesterolo[44], probabilmente attraverso una maggiore conversione del colesterolo in acidi biliari. Questi effetti sono stati riscontrati a 2,5mg/kg di peso corporeo.
L’Ecdysterone (termine intercambiabile con 20-idrossiecdysterone, o 20-HE) sembra essere in grado di sopprimere la formazione epatica di glucosio e quindi di abbassare i livelli di zucchero nel sangue indipendentemente dalla secrezione di Insulina e dai livelli sierici.[45] La soppressione del metabolismo del glucosio sembra provenire dalla fosfoenolpiruvato carbossichinasi e dalla glucosio-6-fosfatasi, oltre a indurre la fosforilazione di Akt nelle cellule epatiche.[46]
Quando viene somministrato ai ratti alla dose di 10mg/kg di peso corporeo, il composto correlato 20-idrossidysone è in grado di esercitare effetti antidiabetici e antiobesogeni in modelli di obesità animale, suggerendo che potrebbe esercitare questi stessi effetti nell’uomo.[46] Questi cambiamenti hanno anche portato a una maggiore secrezione di Adiponectina da parte degli adipociti di ratto.[46] È stato dimostrato che in altri modelli passati esercita proprietà antidiabetiche simili, indipendentemente dal metodo di ingestione/iniezione.[47][48]
Muscolo-scheletrico e prestazioni sportive:
La somministrazione di Ecdysterone (per via sottocutanea o endovenosa), a circa 5mg/kg di peso corporeo, sembra essere in grado di indurre la sintesi proteica in organi animali come il fegato[49][50] o il muscolo scheletrico.[51] Ciò è probabilmente dovuto a un aumento dell’efficienza di traduzione dell’mRNA piuttosto che a un aumento della sintesi dell’mRNA (trascrizione).[45] Inoltre, gli Ecdysteroidi possono essere in grado di aumentare l’incorporazione della leucina nelle cellule a una dose di 5mg/kg di peso corporeo (lo studio ha riguardato il fegato).[52]
Studi in vitro su cellule muscolari (con 20-idrossiecdysone e Turkesterone) hanno rilevato miglioramenti statisticamente significativi nella sintesi proteica in modo dipendente dalla dose a partire da 0,08nM, con un picco a 0,1nM con una sintesi proteica superiore del 110-120% rispetto al controllo e un plateau a concentrazioni comprese tra 1 e 10nM. [Il metabolita del 20-idrossisterone, il Rubrosterone, sembra essere altrettanto potente se si considera il fegato di topo.[49]
In studi comparativi diretti, l’Ecdysteroide chiamato “Turkesterone” sembra essere più potente rispetto agli altri Ecdysteroidi studiati[53][15], seguito dal Cyasterone e poi dal 20-idrossisterone.[54][22]
Cyasterone
Rispetto al controllo, il Turkesterone ha aumentato la crescita dei ratti (sulla base di mg/die) del 63,5%, l’Ecdysterone del 51,9%, il 2-deossiecdysterone del 21,2% e l’alfa-ecdystone del 19,2%. Questo studio ha utilizzato il Metilandrostenediolo (51,9%) e il Nerobol (57,7%) come composti di confronto, anche se gli effetti del Nerobol [Methandienone] erano più localizzati al muscolo scheletrico, mentre gli Ecdysteroidi avevano un aumento della sintesi proteica sistemica (organo e muscolo).[15] Gli Ecdysteroidi in questo studio non hanno soppresso né causato lo sviluppo della prostata o delle vescicole seminali, e non hanno avuto effetti uterotropi nei ratti femmina; il peso del timo è aumentato del 23-35%, mentre è diminuito del 20% con il Nerobol. Le dosi utilizzate in questo studio sono state di 5mg/kg di peso corporeo per tutti gli Ecdysteroidi e di 10mg/kg di peso corporeo per il Metilandrostenediolo e il Nerobol.[15]
Per quanto riguarda i meccanismi d’azione, gli Ecdysteroidi sembrano in grado di provocare un rapido afflusso di Ca2+ nei miociti che porta alla fosforilazione di Akt e quindi alla sintesi proteica. [Questo effetto si verifica dopo 10 minuti di incubazione ed è inibito dagli inibitori della PI3K, come già visto in altri studi, ma anche dagli inibitori della GPRC e della PLC; inoltre, quando le cellule vengono private del calcio intracellulare, Akt non viene fosforilato e il legame del calcio libero con l’EGTA abbassa la sintesi proteica dal 16% all’8%.[55] Il calcio di per sé può essere un importante mediatore di Akt e della sintesi proteica[56-51], e gli Ecdysteroidi sembrano funzionare in modo vicario attraverso il Ca2+ e Akt.[57]
L’afflusso di calcio aumenta la fosforilazione di Akt di oltre 3 volte a una concentrazione di 0,1uM, con una dose-risposta decrescente fino a 5 volte a 1-10uM.[55] L’effetto di 1uM di 20-idrossiecdysterone su Akt ha raggiunto il picco a 2-4 ore, ma è rimasto superiore al valore di base fino a 24 ore.[55]
Nella discussione di uno studio[24] è stato anche notato che la “coda” degli Ecdysteroidi (γ-idrossi-γ-metilpentanoato), se separata dallo scheletro carbossilico steroideo, assomiglia al metabolita anabolizzante della Leucina HMB (beta-idrossi-metilbutirrato).
Studi in vivo hanno rilevato un aumento dell’anabolismo in un’ampia varietà di animali, come ratti e topi,[50][58][59] suini[60] e quaglie.[61] Gli effetti sul miglioramento della forza sembrano essere indipendenti dall’attività, come valutato dal tempo di nuoto forzato nei ratti che migliora senza un allenamento costante. Alcuni studi passati (precedenti al 2000) suggeriscono che possa aumentare la sintesi proteica anche negli esseri umani.[62] Anche nei ratti sono stati rilevati miglioramenti delle prestazioni.[63]
Nelle pecore, una dose orale di 0,02mcg/kg di Ecdysteroidi al giorno è stata in grado di aumentare il tasso di crescita corporea e la produzione di lana[22] ed è stata più evidente con un apporto nutritivo più scarso. Una dose altrettanto ridotta di 0,4 mg/kg di peso corporeo è stata in grado di aumentare la ritenzione di azoto e preservare la massa magra (al 112-116% del controllo) quando l’assunzione di cibo è stata ridotta dell’11-17% nei suini.[64]
L’aumento dell’attività della fosfatasi alcalina indotta dall’Ecdysterone sembra avvenire attraverso il recettore degli estrogeni.[65] Attraverso questo recettore, anche l’attività dei geni reporter degli estrogeni viene aumentata dall’Ecdysterone.
L’aumento dell’attività osservato nell’espressione del collagene di tipo I, dell’osteocalcina e di Runx2 non sembra avvenire attraverso il recettore degli estrogeni.[66]
Interazioni ormonali:
Al momento è stato condotto un solo studio sull’uomo con l’Ecdysterone. Dosato a 200 mg al giorno, non è stato osservato alcun risultato nei maschi che si allenavano alla resistenza per quanto riguarda il testosterone totale e libero o i cambiamenti nella composizione corporea rispetto al placebo.[64] Quando è stato testato il legame con il recettore degli androgeni, il 20-idrossiecdysterone non sembra avere alcuna affinità di legame e quindi non può attivare il recettore degli androgeni.[67]
Detto questo, nonostante l’assenza di influenza sul Testosterone stesso, l’Ecdysterone potrebbe essere in grado di esercitare effetti simili al testosterone attraverso le vie di trasduzione del segnale (anche se il meccanismo esatto non è ancora noto); un’azione che in definitiva ha lo stesso significato biologico del testosterone.[68]
Non ci sono molte prove, oltre a quelle in vitro, che suggeriscano l’utilità dell’Ecdysterone per la sintesi proteica muscolare o per l’aumento della forza.[69]
Quando è stato testato in vitro in C2C12, 1µM di 20-idrossiecdysone (20-HE) ha aumentato il diametro dei miotubi in modo indipendente dal recettore degli androgeni; sia i corticosteroidi che i bloccanti del recettore degli estrogeni hanno impedito al 20-HE di promuovere la crescita muscolare. [52] In seguito a ulteriori test, il 20-HE sembra attivare sia la variante alfa del recettore degli estrogeni (ERα; EC50 di 25,7nM) sia la variante beta (ERβ; EC50 13nM) e si è riscontrato che 10nM di 20-HE promuove la crescita muscolare attraverso ERβ.[52]
Da sinistra: Recettore Estrogeno α [ERα] e Recettore Estrogeno β [ERβ].
Quando è stato testato in vitro, il 20-idrossiecdysone sembra promuovere l’ipertrofia delle cellule muscolari agendo sul recettore beta degli estrogeni. Questa molecola sembra agire anche sul recettore alfa, e quando entrambi i recettori agiscono contemporaneamente la cellula muscolare sperimenta comunque l’ipertrofia.
Interazioni con i processi ossidativi:
L’Ecdysterone esercita anche effetti protettivi contro la perossidazione lipidica da parte dei radicali liberi, ottenendo uno status di antiossidante.[70] Questo effetto è stato osservato a una dose molto bassa di 0,1mg/kg di peso corporeo ed è risultato più potente della vitamina D su base molecolare.
Meccanismo della sostituzione radicalica che porta alla perossidazione lipidica.
Sistemi organici periferici:
L’Ecdysterone, alla dose di 5mg/kg di peso corporeo, è in grado di ripristinare la normale velocità di filtrazione glomerulare e di sopprimere l’albuminuria nei ratti trattati con una miscela nefrotossica[46] e può alleviare i sintomi dell’uremia associata al danno epatico.[58]
Come discusso nella sezione sul metabolismo dei grassi, l’Ecdysterone è in grado di aumentare il tasso di secrezione biliare e di migliorare la rigenerazione del fegato dopo un danno da tossina (Eliotrina).[46]
Eliotrina
Per quanto riguarda la pelle, l’Ecdysterone sembra essere in grado di promuovere la differenziazione dei cheratinociti e di accelerare piccole ferite e ustioni quando viene applicato esternamente.[46]
Longevità e aspettativa di vita:
Gli Ecdysteroidi sono uno dei due ormoni degli insetti (l’altro è l’ormone giovanile) che sembrano essere coinvolti nella durata della vita di questi animali, con l’Ecdysterone come agente che aumenta la durata della vita.[71][72] La trasfezione di Drosophilia con un recettore per l’Ecdysone aumenta la durata della vita.[72] Tuttavia, gli studi sull’uomo sono inesistenti e gli altri modelli animali molto preliminari.
Drosophila Melanogaster [moscerino della frutta]
Sicurezza e tossicità:
Gli Ecdysteroidi, nel complesso, sono abbastanza sicuri per l’ingestione. I benefici sembrano manifestarsi a dosi intorno ai 10mg/kg di peso corporeo, mentre la tossicità accertata nei mammiferi (roditori) è di 6400mg/kg di peso corporeo se iniettati e >9000mg/kg di peso corporeo se consumati per via orale.[42][41]
Tuttavia, i dati sull’uomo sono pochi e non sempre aventi un design ottimale al fine di poter fare una analisi oggettiva dei dati in essi riportati. Al momento però, sono maggiori le segnalazioni di mancanza totale di effetti piuttosto che di conseguenze negative dopo la loro somministrazione orale o transdermica. Gli effetti avversi negativi denunciati da alcuni utilizzatori sono stati dolori gastro-intestinali, reflusso e dissenteria comparse dopo l’inizio della somministrazione orale e cessate poco dopo l’interruzione della stessa. In alcuni casi, la somministrazione topica ha comportato la comparsa di rush cutanei nella zona di applicazione.
Bisogna comunque ricordare che ad ogni azione corrisponde una reazione uguale e contraria, nel mondo fisico e umano. Indi per cui, pretendere modifiche dell’omeostasi senza potenziali conseguenze indesiderate è da idioti illusi.
Conclusioni:
Se avete compreso bene quanto riportate, vi sarete resi conto che mancano interventi validi sull’uomo. Quindi, parlare di assenza di effetti collaterali o efficacia certa è solamente espressione di becera ignoranza venduta ad un pubblico in perenne ricerca della “pillola magica”. E, a dirla tutta, dopo anni di dati raccolti, vi posso dire che non è proprio il caso degli Ecdysteroidi l’essere la panacea di tutti i mali.
Vorrei che consideraste il fatto che il 20-idrossiecdysone (20HE), per fare un esempio, è risultato limitato nella segnalazione muscolo-scheletrica e epatica della protein chinasi B/Akt-mTOR1 nei topi. Anche la biodisponibilità del 20HE, consumato da solo o con la Leucina, è rimasta bassa a tutte le dosi ingerite.[https://pubmed.ncbi.nlm.nih.gov/26584207/]
Schema semplificato della via di segnalazione PI3K/AKT/mTOR nel Condrocita.
Sebbene esista uno studio svolto su 46 soggetti (principianti) affermi che la somministrazione di Ecdysterone ha portato ad osservare un aumento significativo della massa muscolare e aumenti significativamente più pronunciati nelle prestazioni di panca ad una ripetizione. Come se non bastasse, i ricercatori hanno affermato che non è stato rilevato alcun aumento dei biomarcatori di tossicità epatica o renale. La loro conclusione? Ché i dati raccolti sottolineano l’efficacia di un’integrazione di Ecdysterone rispetto alle prestazioni sportive, suggerendo fortemente l’inclusione dell’Ecdysterone nell’elenco delle sostanze e dei metodi proibiti nello sport nella classe S1.2 “altri agenti anabolizzanti”. Lo studio in questione è del 2019, ma rappresenta un eccezione in una letteratura contraria o neutra. Basta leggere bene lo studio per trovarne le “crepe di design” le quali lo rendono ottimo per i venditori ma pessimo per chiarire oggettivamente le cose.
La questione recettoriale mette ancora di più in difficoltà il valutare le possibili e reali potenzialità ipertrofiche negli esseri umani. Sebbene la struttura degli Ecdysteroidi sia in qualche modo simile a quella degli ormoni steroidei dei vertebrati, esistono diverse differenze strutturali tra i due gruppi di steroidi. Nonostante queste differenze strutturali essenziali, gli Ecdysteroidi esercitano nei vertebrati numerosi effetti simili a quelli degli steroidi ormonali dei vertebrati e possono servire come efficaci agenti anabolizzanti, epatoprotettivi, immunoprotettivi, antiossidanti e ipoglicemizzanti. Gli Ecdysteroidi non si legano ai recettori steroidei citosolici, ma è probabile che influenzino le vie di trasduzione del segnale, come gli steroidi anabolizzanti, probabilmente attraverso recettori legati alla membrana. L’applicazione di fitoecdisteroidi sarebbe anche una promettente alternativa all’uso di steroidi anabolizzanti-androgeni per l’apparente mancanza di effetti avversi, se non fosse che le conoscenze in nostro possesso sui reali effetti quando somministrati agli uomini siano molto limitate. Chiunque affermi con certezza che l’applicazione dei fitoecdisteroidi porti all’aumento delle dimensioni muscolari dovrebbe sapere che per poter affermare con certezza ciò è necessaria una ricerca rigorosa, poiché non è ancora disponibile una spiegazione citologica adeguata oltre a prove incontrovertibili.
E’ ormai risaputo che gli estrogeni hanno una serie di effetti metabolici sul muscolo scheletrico. Quando gli individui di sesso femminile perdono gli estrogeni attraverso l’ovariectomia, la funzione mitocondriale, la microviscosità della membrana e le attività del complesso I e I+III diminuiscono (Torres et al., 2018). La perdita di estrogeni provoca anche un aumento della produzione mitocondriale di H2O2 (Valencia et al., 2016), una diminuzione dei livelli di proteine antiossidanti come la glutatione perossidasi, la catalasi e la superossido dismutasi (Baltgalvis et al., 2010; Valencia et al., 2016) e un’alterata sensibilità all’insulina (Torres et al., 2018). Questi effetti sono dovuti alla perdita di estrogeni, poiché il ripristino di livelli normali di questi ormoni ripristina la redox cellulare e l’omeostasi del glucosio nel muscolo scheletrico (Spangenburg et al., 2012; Camporez et al., 2013; Torres et al., 2018).
Al di là dei ruoli metabolici, gli estrogeni sono chiaramente benefici per la massa e la forza muscolare, almeno nei modelli animali (McClung et al., 2006; Kitajima e Ono, 2016). Si ipotizza, quindi, che in assenza di estrogeni, il muscolo è più soggetto a lesioni e questo limita la ricrescita (McClung et al., 2006). Sulla base di questi e altri dati, Enns e Tiidus (2010) hanno proposto che gli estrogeni potrebbero stabilizzare la matrice extracellulare o agire come antiossidanti per ridurre le lesioni muscolari; tuttavia, l’effetto degli estrogeni sul muscolo umano non è stato definito con altrettanta chiarezza perché le variazioni di estrogeni sono transitorie o associate a differenze confondenti di età, livello di forma fisica e tipo e intensità di esercizio (Enns e Tiidus, 2010). Infine, molti studi che cercano di capire il ruolo degli estrogeni sulla funzione muscolare si concentrano sulle differenze di sesso, che vanno ben oltre le semplici variazioni dei livelli ormonali.
Una delle differenze muscolo-scheletriche meglio caratterizzate tra uomini e donne è il tasso di rottura del legamento crociato anteriore (ACL). Le rotture del legamento crociato anteriore si verificano con una frequenza da 2 a 8 volte maggiore tra le atlete rispetto agli uomini (Arendt e Dick, 1995; Adachi et al., 2008). Quando la concentrazione di estrogeni aumenta durante il ciclo mestruale, aumenta anche la lassità del ginocchio (Shultz et al., 2010, 2011, 2012a). Infatti, è stato riscontrato che la lassità del ginocchio è aumentata tra 1 e 5 mm tra il primo giorno delle mestruazioni e il giorno successivo all’ovulazione, a seconda dei livelli di estrogeni. Infine, Park et al. hanno riscontrato una diminuzione del 17% della rigidità del ginocchio durante la fase ovulatoria, con una variazione della lassità del ginocchio da 13,35 ± 2,53 mm durante la fase follicolare a 14,43 ± 2,60 mm durante l’ovulazione (Park et al., 2009). Al contrario, Carcia et al. (2004) non hanno riscontrato variazioni nello spostamento del ginocchio in relazione al ciclo; tuttavia, è importante notare che questi autori hanno utilizzato la lunghezza del ciclo auto-riferita per stimare la fase mestruale, mentre gli altri studi hanno misurato direttamente i livelli di estrogeni in concerto con la lassità del ginocchio. Poiché Myer et al. (2008) hanno dimostrato che per ogni aumento di 1,3 mm dello spostamento del ginocchio, il rischio di lesione del legamento crociato anteriore aumenta di 4 volte, l’aumento della lassità del ginocchio riportato da Deie, Park e Shultz potrebbe spiegare il tasso di rottura del legamento crociato anteriore da 2 a 8 volte superiore nelle donne (Arendt e Dick, 1995; Adachi et al., 2008). Quindi, se gli estrogeni diminuiscono l’attività della lisil-ossidasi nei tendini, ci si aspetterebbe che questo diminuisca la rigidità dei tendini e quindi l’incidenza di lesioni ai muscoli associati. In effetti, come già detto, le donne subiscono meno lesioni muscolari degli uomini (Hägglund et al., 2009; Edouard et al., 2016).
È chiaro che gli estrogeni hanno un effetto significativo sulla funzione muscolo-scheletrica. In passato, gran parte della ricerca si è concentrata sulla forte connessione tra estrogeni e ossa. Tuttavia, recentemente l’effetto degli estrogeni su altri tessuti muscolo-scheletrici, come muscoli, tendini e legamenti, è diventato oggetto di maggiori ricerche. Questi studi chiariscono che gli estrogeni migliorano la proteostasi muscolare e aumentano il contenuto di collagene dei tendini; tuttavia, i benefici sull’osso e sul muscolo avvengono al prezzo di una minore rigidità del tessuto connettivo. Tuttavia, con l’aumento delle donne che praticano sport, è chiaro che questi effetti fisiologici degli estrogeni contribuiscono a diminuire la potenza e le prestazioni e rendono le donne più inclini a subire infortuni catastrofici ai legamenti.
Ma perchè ho riportato tutto ciò? Semplicemente per farvi capire alcuni punti essenziali:
La mancanza di interazione con i AR da parte degli Ecdysteroidi e la loro affinità nei mammiferi a carico dei ERα e ERβ;
tale affinità ha il potenziale di promuove la crescita muscolare attraverso il ERβ;
tale interazione, però, non è mai stata accuratamente monitorata e quantificata nell’uomo, di conseguenza non si ha alcuna certezza che il potenziale sia statisticamente significativo;
date tali caratteristiche sembrerebbe più probabile che l’assunzione di Ecdysteroidi possa avere un qualche effetto protettivo su tendini e legamenti oltre che sulla matrice ossea;
la scarsa biodisponibilità che caratterizza gli Ecdysteroidi assunti per via orale renderebbero molto difficile il mantenimento di una soglia ematica efficace;
notando le caratteristiche strutturali degli Ecdysteroidi, sebbene abbiano mostrato una emivita di 8h circa in media, non suggerisce un potenziale di legame ormone-recettore elevato con conseguente ulteriore dubbio del grado di efficacia;
l’affinità con i ER potrebbe, se si dovessero raggiungere soglie ematiche sufficienti, indurre un feedback negativo a livello ipotalamico-ipofisario con conseguente calo della secrezione di LH e FSH e alterazioni consequenziali di Testosterone, DHT ed Estradiolo [vedi alterazioni nel comportamento sessuale, depressione, letargia ecc…];
si è proposta l’ipotesi che l’interazione con i ER a livello ipotalamico-ipofisario possa dare un effetto simile a quello riscontrato con i SERM. Tuttavia tale ipotesi risulta ad oggi indimostrata;
l’affinità con i ER potrebbe, se si dovessero raggiungere soglie ematiche sufficienti, causare la comparsa di effetti collaterali estrogeno-dipendenti, sebbene l’iterazione con il ERβ, in specie l’isoforma ERβ1, abbia mostrato un attività protettiva antitumorale che, secondo alcuni, potrebbe avere una valenza preventiva sullo sviluppo di ginecomastia. Anche questa rimane una ipotesi senza dimostrazione;
possibile interazione ad esito sconosciuto con l’asse GH/IGF-1 mediata dal legame con il ERα e ERβ a livello epatico.
Siete confusi? Non dovreste. Infondo, adesso, avete gli strumenti di logica per valutare quanto possa valere la pena acquistare un integratore contenente Ecdysteroidi. Si tratta di un azzardo che potrebbe non limitarsi solo alla componente economica visti i dosaggi necessari per poter sperare (forse) di osservare miglioramenti nella composizione corporea e/o nelle prestazioni atletiche.
Gabriel Bellizzi [CEO BioGenTech]
Riferimenti:
de Loof A (2006). “Ecdysteroids: the overlooked sex steroids of insects? Males: the black box”. Insect Science.
Krishnakumaran A, Schneiderman HA (December 1970). “Control of molting in mandibulate and chelicerate arthropods by ecdysones”. The Biological Bulletin.
Margam VM, Gelman DB, Palli SR (June 2006). “Ecdysteroid titers and developmental expression of ecdysteroid-regulated genes during metamorphosis of the yellow fever mosquito, Aedes aegypti (Diptera: Culicidae)”. Journal of Insect Physiology.
Continua la disamina dei principali PEDs utilizzati e del confine che delimita l’uso dall’abuso. In questo terzo articolo della serie tratteremo due ormoni, o meglio un precursore poco attivo [T4] ed il suo derivato molto attivo [T3], che non rientrano pienamente nella categoria PEDs, ma che, volenti o nolenti, si sono diffusi da decenni nel mondo del BodyBuilding, in ambo i sessi. Inutile dire che l’abuso con questa classe di farmaci è alquanto facile e spesso praticato.
Tanto per ribadirlo, questo articolo non rappresenta ne un incitamento all’uso di farmaci fuori dalla prescrizione medica ne tantomeno un indicazione medica. Si tratta di divulgazione scientifica.
Introduzione agli ormoni tiroidei [T4 e T3]:
Gli ormoni tiroidei sono ormoni secreti dalla tiroide. La tiroide è una ghiandola endocrina situata nella parte anteriore del collo, direttamente sotto la laringe (pomo d’Adamo), ed ha un peso di circa 20g. I due principali ormoni tiroidei che secerne sono la Triiodotironina (T3) e la Tiroxina (T4). Quest’ultima ha soprattutto una attività da pro-ormone, poiché la maggior parte dei suoi effetti dipende dalla conversione in T3. Questa conversione da T4 a T3, chiamata anche deiodinazione dell’anello esterno, avviene principalmente al di fuori della tiroide, nei tessuti periferici. Complessivamente, ciò porta a una produzione giornaliera di circa 88mcg (113 nmol) di T4 e 28mcg (43 nmol) di T3 [2]. Circa un quinto della T3 deriva dalla tiroide, mentre gli altri quattro quinti sono prodotti dalla conversione extratiroidea di T4 in T3 [3].
Come nel caso degli steroidi anabolizzanti, gli ormoni tiroidei sono trasportati nel flusso sanguigno da proteine trasportatrici. La maggior parte è legata alla globulina legante la tiroxina (TBG), mentre la parte restante è legata alla transtiretina, all’albumina e ad alcune lipoproteine. Nel complesso, esse legano oltre il 99% degli ormoni tiroidei in circolazione. Si ritiene che la frazione non legata sia disponibile per i tessuti per l’assorbimento e sia responsabile dei suoi effetti [4]. Sebbene vi siano alcune riserve sulle prove a sostegno di questa tesi, non intendo addentrarmi in una discussione sull’ipotesi dell’ormone libero (se non ricordare che nella sua forma più rigorosa è sbagliata, ma le misurazioni dell’ormone tiroideo libero sono comunque utili).
Una volta che raggiunge i tessuti periferici e attraversa la membrana plasmatica di una cellula, esso esplica la sua attività. Nel caso del T4, deve prima essere convertito in T3, come già detto, in quanto il T4 può essere considerato un pro-ormone. Questa conversione avviene all’interno della cellula, o vicino alla membrana plasmatica (dopo di che si equilibra rapidamente con il plasma sanguigno), o vicino al nucleo della cellula, il sito d’azione [5].Il T3, invece, può continuare direttamente il suo viaggio entrando nel nucleo della cellula. Il nucleo cellulare è l’organello della cellula dove avviene la trascrizione dei geni. Proprio come gli steroidi anabolizzanti, gli ormoni tiroidei esercitano i loro effetti principalmente attraverso la modulazione della trascrizione genica. Lo fanno legandosi ai recettori degli ormoni tiroidei che si trovano principalmente all’interno del nucleo cellulare, legati al DNA.
Attività di legame T3-recettore.
Gli ormoni tiroidei agiscono su una vasta gamma di tessuti e hanno un’infinità di effetti, ma in questo articolo mi concentrerò sull’effetto che essi hanno sul metabolismo energetico e sul turnover delle proteine (muscolo scheletrico). Con tutta probabilità sono i due aspetti che più interessano le persone che leggono questo articolo per quanto riguarda la sua efficacia.
Effetto sul metabolismo energetico (Parte 1):
Quando è presente una quantità insufficiente di ormoni tiroidei, si parla di ipotiroidismo. Una delle caratteristiche dell’ipotiroidismo è l’aumento di peso. Al contrario, quando la quantità di ormoni tiroidei è eccessiva, si parla di ipertiroidismo. Una delle sue caratteristiche è la perdita di peso. Queste variazioni di peso sono probabilmente il risultato di cambiamenti nel tasso metabolico basale. È noto che gli ormoni tiroidei aumentano il dispendio energetico.
Sono stati proposti alcuni meccanismi che spiegano come gli ormoni tiroidei riescano a ottenere questo risultato. In questo articolo tratterò i tre più interessanti (o forse semplicemente quelli che si incontrano di più nella letteratura scientifica). I primi due meccanismi si basano sull’energia necessaria per mantenere i gradienti ionici all’interno della cellula. Ad esempio, le cellule mantengono una bassa concentrazione intracellulare di sodio e un’alta concentrazione intracellulare di potassio rispetto all’esterno della cellula. Il mantenimento di questa condizione è assicurato da pompe incorporate nella membrana plasmatica, che richiedono energia per funzionare. Esse pompano ioni sodio fuori dalla cellula e ioni potassio dentro la cellula. Queste pompe sono note come Na+/K+-ATPasi, o semplicemente pompe sodio-potassio. L’energia necessaria al funzionamento di queste pompe deriva dalla molecola portatrice di energia adenosina trifosfato (ATP). L’ATP è utilizzato da molti processi cellulari per alimentare il proprio fabbisogno energetico e l’energia contenuta in queste molecole deriva dai macronutrienti che mangiamo: carboidrati, acidi grassi e proteine (aminoacidi). Ed ora d’obbligo descrivere il modo principale in cui le cellule producono queste molecole di ATP attraverso un processo chiamato fosforilazione ossidativa.
Uno dei modi in cui gli ormoni tiroidei potrebbero aumentare il dispendio energetico è simile al modo in cui il famoso DNP ottiene questo risultato: “sabotando” la fosforilazione ossidativa.
Fosforilazione ossidativa: ottenere energia dal passaggio degli elettroni.
È inutile ribadire che è sempre un piacere per me trattare di biochimica in un articolo. Ritengo che questi principi di base tolgano un po’ di magia agli effetti dei farmaci, e forniscano quindi un quadro più chiaro di come funzionano le cose. Con un po’ di fortuna potrei anche, forse, interessare qualcuno di voi che sta leggendo questo articolo ad approfondire l’argomento. La biochimica e la biologia cellulare sono campi di studio estremamente interessanti.
Le cellule del vostro corpo svolgono continuamente ogni sorta di funzione per, essenzialmente, mantenervi in vita. Molti di questi processi consumano energia. Questa energia deriva, in ultima analisi, dagli alimenti che mangiamo. Carboidrati, grassi e proteine, persino l’alcol, hanno tutti energia immagazzinata nei loro legami chimici. È compito dell’organismo estrarre questa energia e trasformarla in qualcosa di utile. Come il motore della vostra auto non funziona con il petrolio grezzo, questi processi cellulari non funzionano direttamente con i macronutrienti. Al contrario, la maggior parte di questi processi richiede energia da una molecola chiamata adenosina trifosfato (ATP), proprio come il motore di un’automobile richiede specificamente la benzina.
Vediamo come funziona per una molecola di glucosio, un carboidrato. Quando una molecola di glucosio viene utilizzata da una cellula per produrre ATP, subisce prima un processo chiamato glicolisi. La glicolisi è un processo composto da varie fasi enzimatiche che scindono la molecola di glucosio in 2 molecole di piruvato e producono 2 molecole di ATP (oltre ad altre molecole). In poche parole:
glucosio -> 2 piruvato + 2 ATP
Tuttavia, un processo chiamato fosforilazione ossidativa estrarrà molta più energia, cioè molecole di ATP, dalle 2 molecole di piruvato risultanti.
La fosforilazione ossidativa è un processo che avviene nei mitocondri. Quindi è qui che il piruvato è diretto. I mitocondri sono organelli della cellula che si occupano principalmente della produzione di energia. Sono piccole fabbriche di energia di dimensioni microscopiche. Sono costituiti da una membrana esterna e da una membrana interna. Lo spazio tra la membrana esterna e quella interna è chiamato spazio intermembrana. Lo spazio incapsulato dalla membrana interna è chiamato matrice mitocondriale. La membrana interna è ripiegata in modo caratteristico. Queste pieghe sono chiamate cristae. L’aspetto è questo:
1) Crista, 2) membrana esterna, 3) spazio intermembrana e 4) matrice mitocondriale.
Quando il piruvato si trova all’interno della matrice mitocondriale, viene convertito in acetil-CoA e successivamente subisce una serie di reazioni che vengono chiamate collettivamente ciclo dell’acido citrico o ciclo di Krebs. Durante questo processo, tutta l’energia viene estratta da quella che in origine era una molecola di piruvato. Viene ossidata. Tuttavia, l’energia non si è ancora trasformata in ATP. Prima viene trasferita ai vettori energetici NAD e FAD (e al GTP, ma non ne parlerò). I vettori energetici NAD e FAD parteciperanno al processo chiamato fosforilazione ossidativa che segue il ciclo dell’acido citrico.
L’energia viene immagazzinata in coppie di elettroni che vengono donati a NAD e FAD. Questo processo riduce queste molecole, come viene chiamato, producendo rispettivamente NADH e FADH2. Successivamente, NADH e FADH2 cedono la coppia di elettroni a grandi complessi proteici incorporati nella membrana interna. Questa è la prima fase della fosforilazione ossidativa. Quando queste coppie di elettroni vengono cedute a tali complessi proteici, parte dell’energia in essi immagazzinata viene utilizzata per pompare un protone (H+) fuori dalla matrice mitocondriale nello spazio intermembrana. Si tratta di un aspetto estremamente cruciale, di cui si capirà presto il motivo.
Successivamente, le coppie di elettroni vengono trasferite un paio di volte da un complesso all’altro, staccando ogni volta un po’ dell’energia in esse contenuta e utilizzandola per pompare fuori un protone. A ogni passaggio, gli elettroni raggiungono uno stato energetico inferiore. (Non vengono trasferiti direttamente da un complesso all’altro, ci sono alcune proteine/molecole intermedie che li trasportano tra questi complessi proteici che pompano protoni). E ogni volta una parte dell’energia sottratta viene sfruttata per pompare fuori un protone. Se si utilizza una ruota idraulica, l’aspetto è simile a questo:
Immagine di Peter Bond
La destinazione finale degli elettroni è quella di combinarsi con l’idrogeno e l’ossigeno per formare H2O, ovvero l’acqua. Il processo di fosforilazione ossidativa ha stabilito un gradiente elettrochimico di protoni. La concentrazione di protoni nella matrice mitocondriale sarà inferiore rispetto allo spazio intermembrana. Questo gradiente contiene energia potenziale. Proprio come una ruota idraulica ruota con l’acqua che si muove in discesa, un macchinario molecolare chiamato ATP sintasi inizia a ruotare con i protoni che si muovono lungo il loro gradiente elettrochimico dallo spazio intermembrana alla matrice mitocondriale. Questa energia viene poi sfruttata per generare ATP combinando l’ADP con un gruppo fosfato inorganico. E voilà, l’intero processo di passaggio degli elettroni, di sottrazione di energia per pompare fuori i protoni e di successivo utilizzo del gradiente protonico stabilito per sintetizzare ATP, è chiamato fosforilazione ossidativa.
Per ricapitolare ciò che è stato trattato, e che non è poco:
Il glucosio viene scisso in due molecole di piruvato dalla glicolisi.
il piruvato viene trasportato nella matrice mitocondriale per essere convertito in acetil-CoA
L’acetil-CoA viene ossidato, trasferendo la sua energia nei vettori energetici NAD e FAD nelle loro forme ridotte NADH e FADH2, accettando una coppia di elettroni.
Queste molecole di NADH e FADH2 donano le loro coppie di elettroni a un grande complesso proteico incorporato nella membrana interna, che poi viene trasferito in continuazione fino a combinarsi con idrogeno e ossigeno per formare acqua. Con questi trasferimenti, parte dell’energia viene sfruttata per pompare protoni (H+) fuori dalla matrice mitocondriale. Si stabilisce così un gradiente elettrochimico: bassa concentrazione di protoni all’interno della matrice mitocondriale, alta concentrazione di protoni all’esterno della matrice mitocondriale.
Il flusso di protoni lungo il gradiente di concentrazione fornisce energia all’ATP sintasi per svolgere il suo lavoro e generare ATP.
Effetto sul metabolismo energetico (Parte 2):
Dopo questa dovuta parentesi, torniamo alle pompe sodio-potassio. Alcune prove suggeriscono che gli ormoni tiroidei aumentano la permeabilità della membrana plasmatica agli ioni sodio e potassio [6]. Ciò significa che una quantità maggiore di questi ioni fuoriesce lungo il gradiente di concentrazione. Pertanto, gli ioni potassio fuoriescono dalla cellula e gli ioni sodio vi entrano. Di conseguenza, le pompe sodio-potassio devono pompare maggiormente per mantenere le concentrazioni intracellulari desiderate di questi ioni e questo costa energia. Alcuni studi suggeriscono addirittura che tutti i tessuti dei mammiferi mostrano un aumento dell’attività della pompa sodio-potassio in risposta alla T3 [7].
Qualcosa di simile è stato suggerito per quanto riguarda gli ioni calcio nelle cellule muscolari [8]. Le cellule muscolari sono cellule piuttosto speciali sotto molti aspetti. Uno di questi è che contengono un organello chiamato reticolo sarcoplasmatico. Si tratta di una forma specializzata del reticolo endoplasmatico presente nelle cellule normali. Una delle caratteristiche che lo rendono speciale è che funziona come sito di stoccaggio degli ioni calcio. Questi ioni di calcio svolgono un ruolo fondamentale nella contrazione muscolare, poiché lo scarico di questi ioni di calcio dal reticolo sarcoplasmatico al resto della cellula porta alla contrazione muscolare. Quando la contrazione deve cessare, questi ioni vengono nuovamente pompati nel reticolo sarcoplasmatico. Anche questo processo, ovviamente, consuma energia. E qui viene il bello: si è visto che gli ormoni tiroidei regolano l’espressione di queste pompe del calcio in modelli animali. Inoltre, aumentano l’attività di un certo tipo di recettore nel tessuto muscolare che stimola lo scarico di questi ioni nel citosol [9]. Questo è un altro elemento che indica un potenziale aumento del dispendio energetico come risultato del mantenimento dell’accumulo di ioni calcio nel reticolo endoplasmatico.
Infine, ci sono buone prove che indicano che “sabota” la fosforilazione ossidativa. Come detto sopra, ma questa volta in breve, la fosforilazione ossidativa avviene in un organello cellulare chiamato mitocondrio. I macronutrienti che mangiamo vengono ulteriormente scomposti in componenti più piccoli e in questo processo viene rilasciata energia sotto forma di coppie di elettroni. Un complesso gioco molecolare nei mitocondri tra varie molecole e complessi proteici estrae l’energia da queste coppie di elettroni, utilizzandola essenzialmente per pompare protoni (H+). Questi protoni vengono pompati all’esterno del nucleo dei mitocondri, chiamato matrice mitocondriale, e nello spazio intermembrana – lo spazio tra la membrana mitocondriale interna e quella esterna (i mitocondri hanno due membrane, una che avvolge l’altra). Questo crea un gradiente protonico, con un’alta concentrazione di protoni nello spazio intermembrana e una concentrazione relativamente bassa nella matrice mitocondriale. Proprio come l’acqua che scorre dall’alto verso il basso, da cui possiamo estrarre energia con una turbina ad acqua, le cellule possono estrarre energia da questi protoni che scendono lungo il loro gradiente di concentrazione guidando questo flusso attraverso un fantastico macchinario proteico chiamato ATP sintasi. È questo che alimenta la sintesi di ATP.
Ok, torniamo al modo in cui gli ormoni tiroidei influiscono su questo aspetto: aumentano l’espressione delle proteine di disaccoppiamento [10, 11]. Si tratta di proteine incorporate nella membrana interna dei mitocondri che lasciano fuoriuscire i protoni lungo il loro gradiente di concentrazione. I protoni passano quindi dallo spazio intermembrana alla matrice mitocondriale, senza passare per l’ATP sintasi. In questo modo, l’energia viene rilasciata come calore anziché essere destinata alla produzione di ATP.
Gli ormoni tiroidei influenzano il turnover delle proteine:
Sembrerà strano, ma non è così raro sentire qualcuno che dice di assumere T3 in Bulk nel tentativo aumentare il turnover proteico. Ma è una buona idea? No. Mentre il turnover proteico aumenta, si verifica un contemporaneo aumento sia della sintesi proteica sia della degradazione proteica, quest’ultima supera il tasso di sintesi. Di conseguenza, si verifica una degradazione netta delle proteine.
In uno studio in cui i soggetti hanno ricevuto 150mcg di T3 al giorno per 7 giorni, la degradazione proteica è aumentata notevolmente [12]. L’escrezione di azoto (un indicatore della degradazione delle proteine) è aumentata del 45% e l’ossidazione della leucina del 74%. È stato riscontrato anche un piccolo aumento della sintesi proteica corporea, ma l’entità era inferiore all’aumento della degradazione proteica. Un altro studio, nel quale è stata usata una dose di 100mcg di T3 al giorno per 2 settimane, ha ottenuto risultati simili [13]. La sintesi proteica corporea a digiuno è aumentata del 9%, anche se in modo non statisticamente significativo, mentre la degradazione proteica e l’ossidazione della leucina hanno mostrato un aumento statisticamente significativo, rispettivamente del 12 e del 24%.
L’aspetto forse più interessante è che i ricercatori hanno anche prelevato biopsie muscolari dal muscolo gastrocnemio. Hanno misurato una serie di elementi, tra cui l’area della sezione trasversale (CSA) delle fibre muscolari. I risultati sono stati i seguenti:
Si tratta di una situazione piuttosto drastica per sole 2 settimane. (Si noti anche il cambiamento del tipo di fibra indotto da uno stato di ipertiroidismo).
In un altro studio, sei partecipanti hanno ricevuto 2mcg/kg di peso corporeo di T4 al giorno per 6 settimane, insieme a 1mcg/kg di peso corporeo di T3 al giorno per le ultime 2 settimane [14]. Questo (le prime 4 settimane) è un po’ più alto di un dosaggio completo di ormoni tiroidei. In effetti, il TSH è stato soppresso da 1,8 a 0,3 mIU/L e sia il T4 che il T3 sono aumentati in modo significativo. La successiva aggiunta di T3 ha reso i livelli di TSH non rilevabili e ha aumentato ulteriormente i livelli di T3. In questo studio non è stata misurata la cinetica delle proteine muscolari. È stata misurata la sintesi e la degradazione delle proteine nell’intero corpo nello stato di post-assorbimento. L’integrazione di ormoni tiroidei ha portato a un aumento di entrambi, ma con un aumento sostanziale della degradazione. Sarebbe ragionevole ipotizzare che questo rifletta anche ciò che accade nel tessuto muscolare.
Infine, vale la pena sottolineare un altro studio di lunga durata, con un dosaggio relativamente basso rispetto agli altri studi. Lovejoy et al. hanno somministrato T3 per 2 mesi a un piccolo gruppo di uomini [15]. Il dosaggio è iniziato con 75 mcg di T3 al giorno, ma è stato ridotto a 50 o 62,5 mcg al giorno quando i livelli di T3 nel siero superavano i 4,6 nmol/L. Cosa che, in effetti, si è verificata per 5 dei 7 uomini partecipanti. Il bilancio dell’azoto è risultato significativamente ridotto rispetto al basale nella seconda e terza settimana, ma in seguito tendeva a tornare verso lo zero. Questo fa pensare a un meccanismo di risparmio proteico che entra in funzione dopo le prime settimane. Inoltre, hanno riscontrato una diminuzione significativa della massa magra (-1,5 kg) e della massa grassa (-2,7 kg) dopo 6 settimane. Alla 9a settimana, la massa magra non è diminuita ulteriormente (-0,1 kg rispetto alla 6a settimana), mentre la massa grassa è sembrata continuare a diminuire (-0,6 kg), anche se non si tratta di una differenza statisticamente significativa rispetto alla 6a settimana. Non sono state riscontrate differenze statisticamente significative nelle misure del turnover proteico, ma questo è stato probabilmente il risultato delle ridotte dimensioni del campione: un errore statistico di tipo 2.
Conclusioni:
Gli agenti anabolizzanti, che essi siano SARM steroidei o non steroidei, possono annullare gli effetti catabolici degli ormoni tiroidei? Dai dati aneddotici ed empirici raccolti sul campo sembrerebbe molto probabile, in una certa misura, ma non ci sono dati clinici al riguardo. La variabile di picco nella questione è il dosaggio. Si è potuto osservare che gli atleti con maggiori vantaggi dalla somministrazione di T3 in regimi ipocalorici protratti li ottenevano con dosaggi nel range tra 25 e 50mcg/die massimo! Tale dosaggio, con riscontro per via esami ematici, permette all’atleta di mantenere livelli tiroidei da normo o ipercalorica, senza sforare il range di riferimento fisiologico, nonostante la forte restrizione alimentare. Ovviamente, questi atleti sono sottoposti ad una preparazione complessa comprendente l’uso di uno o più PEDs.
I dosaggi da 100-150mcg/die di T3 o 200mcg/die di T4 sono del tutto controproducenti, a meno che per il modesto aumento del dispendio energetico (poche centinaia di kcal, con un aumento del 10%-15% del tasso metabolico a riposo) siate disposti a ritrovarvi ipertiroidei e fortemente catabolici.
Oltretutto, in ipocalorica, il T4 subisce comunque una riduzione della conversione in T3. L’uso concomitante di GH può migliorare questa risposta.
In conclusione, ricordiamo gli effetti collaterali legati ad uno stato di ipertiroidismo:
accelerazione della frequenza cardiaca;
palpitazioni;
possibili aritmie;
forte calo di peso e perdita di massa muscolare;
insonnia;
ansia;
tremori;
sudorazione;
debolezza muscolare;
aumento del reverse T3 [legato ad abuso di farmaci contenenti T3 e/o T4].
Riflettete e traete le corrette conclusioni… la conoscenza per farlo ora non vi manca. Per la capacità beh, miracoli non ne faccio…
Gabriel Bellizzi
Riferimenti:
Carlé, Allan, Anne Krejbjerg, and Peter Laurberg. “Epidemiology of nodular goitre. Influence of iodine intake.” Best practice & research Clinical endocrinology & metabolism 28.4 (2014): 465-479.
Nicoloff, John T., et al. “Simultaneous measurement of thyroxine and triiodothyronine peripheral turnover kinetics in man.” The Journal of clinical investigation 51.3 (1972): 473-483.
Bianco, Antonio C., et al. “Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases.” Endocrine reviews 23.1 (2002): 38-89.
Mendel, Carl M. “The free hormone hypothesis: a physiologically based mathematical model.” Endocrine reviews 10.3 (1989): 232-274.
Gereben, Balázs, et al. “Cellular and molecular basis of deiodinase-regulated thyroid hormone signaling.” Endocrine reviews 29.7 (2008): 898-938.
Silva, J. Enrique. “Thermogenic mechanisms and their hormonal regulation.” Physiological reviews 86.2 (2006): 435-464.
Ismail-Beigi, Faramarz. “Thyroid hormone regulation of Na, K-ATPase expression.” Trends in Endocrinology & Metabolism 4.5 (1993): 152-155.
Everts, M. E. “Effects of thyroid hormones on contractility and cation transport in skeletal muscle.” Acta Physiologica Scandinavica 156.3 (1996): 325-333.
Mullur, Rashmi, Yan-Yun Liu, and Gregory A. Brent. “Thyroid hormone regulation of metabolism.” Physiological reviews 94.2 (2014): 355-382.
Barbe, Pierre, et al. “Triiodothyronine‐mediated upregulation of UCP2 and UCP3 mRNA expression in human skeletal muscle without coordinated induction of mitochondrial respiratory chain genes.” The FASEB Journal 15.1 (2001): 13-15.
de Lange, Pieter, et al. “Uncoupling protein-3 is a molecular determinant for the regulation of resting metabolic rate by thyroid hormone.” Endocrinology 142.8 (2001): 3414-3420.
Gelfand, Robert A., et al. “Catabolic effects of thyroid hormone excess: the contribution of adrenergic activity to hypermetabolism and protein breakdown.” Metabolism 36.6 (1987): 562-569.
Martin, WH 3rd, et al. “Mechanisms of impaired exercise capacity in short duration experimental hyperthyroidism.” The Journal of clinical investigation 88.6 (1991): 2047-2053.
Tauveron, I. G. O. R., et al. “Response of leucine metabolism to hyperinsulinemia under amino acid replacement in experimental hyperthyroidism.” American Journal of Physiology-Endocrinology and Metabolism 269.3 (1995): E499-E507.
Lovejoy, Jennifer C., et al. “A paradigm of experimentally induced mild hyperthyroidism: effects on nitrogen balance, body composition, and energy expenditure in healthy young men.” The Journal of Clinical Endocrinology & Metabolism 82.3 (1997): 765-770.
L’ACE-031 può rientrare a pieno titolo nel “club” delle molecole PEDs semisconosciute. Un peptide praticamente unico nel panorama “doped”, sicuramente promettente, specie nel BodyBuilding, ma del quale se ne parla poco.
Nel 2013 sembrava che la ricerca sul ACE-031 fosse stata definitivamente interrotta, nonostante funzionasse piuttosto bene.
Le aziende farmaceutiche Acceleron Pharma e Shire misero in pausa la ricerca sull’inibitore della Miostatina ACE-031 [Acceleronpharma.com 2 maggio 2013]. E questo evento risultò piuttosto strano. In un comunicato stampa congiunto rilasciato qualche tempo dopo il sopra citato annuncio, Muscle & Nerve aveva pubblicato uno studio che dimostrava che l’ACE-031 è un composto che un culturista supplementato farmacologicamente aggiungerebbe volentieri al suo “arsenale”.
L’ACE-031 iniettabile è un recettore sintetico dell’Attivina di Tipo IIB. Anche le cellule muscolari hanno questo recettore. È destinato a proteine come la Miostatina, il GDF11 e l’Attivina A e B. Se la Miostatina si lega al recettore dell’Attivina di Tipo IIB, la crescita delle fibre muscolari si riduce. Nelle circostanze “giuste” la Miostatina arriva addirittura a degradare il muscolo-scheletrico.
Se si somministra l’ACE-031, questo non accade o, comunque, l’effetto viene marcatamente ridotto. Il recettore sintetico dell’Attivina di Tipo IIB si lega con il tristemente noto peptide Miostatina impedendo a quest’ultimo di legarsi al sito recettore della cellula e compiere la sua attività di riduzione ipertrofica e degradazione del tessuto muscolo-scheletrico.
ACE-031 e “recettori esca”:
Come accennato pocanzi, l’ACE-031 non è altro che un “recettore esca”. Un recettore esca è un recettore in grado di riconoscere e legare in modo efficiente specifici fattori di crescita o citochine, ma non è strutturalmente in grado di segnalare o attivare il complesso recettoriale previsto. Agisce come un inibitore, legando un ligando e impedendogli di legarsi al suo recettore abituale. I recettori esca partecipano a un metodo comune di inibizione del segnale e sono anche abbondanti nei tessuti maligni, costituendo un argomento significativo nella ricerca sul cancro.[1]
“Recettori esca”: si legano ai ligandi e inibiscono la segnalazione attraverso i recettori veri e propri.
IL1R2 è stato uno dei primi recettori esca identificati.[2] [3] Lega IL1A e IL1B e inibisce il loro legame con IL1R1, impedendo la risposta infiammatoria che è generalmente promossa dal legame delle interleuchine di tipo 1 con il recettore 1 dell’interleuchina di tipo I.[4]
Un altro membro di questa categoria è il recettore DcR3, conosciuto anche come TNFRSF6, che si trova principalmente nei tessuti maligni umani.[5] Agisce come recettore esca per i membri delle citochine TNF: FasL, LIGHT e TL1A, inibendo la capacità delle citochine di segnalare la morte cellulare o l’apoptosi.
TNFRSF6
Il VEGFR-1 è una tirosin-chinasi recettoriale che modula negativamente l’angiogenesi agendo come recettore esca.[6] La caratteristica di “esca” del VEGFR-1 è necessaria per lo sviluppo e l’angiogenesi normali. Il VEGFR-1 inibisce l’attività del VEGFR-2 sequestrando il VEGF, impedendo così al VEGFR-2 di legarsi al VEGF.
Quindi eccoci di nuovo con ACE-031. Esso è stato studiato in quanto è un recettore esca ingegnerizzato con attività inibitoria della Miostatina potenzialmente utile nel tentativo di trattare i bambini affetti da distrofia muscolare di Duchenne (DMD). Il recettore ACE-031 circola al di fuori della membrana della fibra muscolare. Poiché questo recettore si lega alla Miostatina, riduce la quantità di questo peptide che può legarsi al recettore nativo nella membrana (ActRIIB), impedendo alla Miostatina di fornire il segnale che limita la crescita muscolare e ne promuove il catabolismo.[7]
I principali studi su ACE-031:
Nel 2007 Acceleron Pharma aveva grandi aspettative su ACE-031. All’epoca l’azienda aveva condotto solo studi sugli animali. Tuttavia, nel marzo 2013 AP ha pubblicato uno studio sull’uomo in cui 48 donne sane di età compresa tra 45 e 75 anni hanno ricevuto una singola iniezione con 0.02, 0.05, 0.1, 0.3, 1 o 3 mg di ACE-031 per kg di peso corporeo. Il composto ha circolato per alcune settimane nell’organismo dei soggetti trattati. L’emivita è stata stimata essere di 10-15 giorni. Tuttavia, questa singola iniezione ha prodotto una crescita muscolare. La dose di 3mg/kg ha mostrato un aumento del volume muscolare del 5%. La massa magra è aumentata del 3% [poco più di un chilo] e sembra anche diminuire la massa grassa.
L’iniezione ha ridotto la Leptina e aumentato la concentrazione di Adiponectina. Ciò suggerisce che l’ACE-031 riduce la massa grassa.
Inoltre, è aumentato l’inibitore della Miostatina, i livelli di fosfatasi alcalina specifica per le ossa [BSAP] nel sangue e si è ridotto quello del telopeptide C-terminale del collagene di tipo 1 [CTX]. Ciò suggerisce che l’ACE-031 rende le ossa più forti. Negli studi sugli animali con RAP-031, la versione per topi di ACE-031, Acceleron è riuscita a dimostrare questi effetti. [Endocrinology. 2010 Sep; 151 (9) :4289-300].
Se si legge lo studio su Muscle & Nerve, ci si chiede perché mai la Acceleron abbia interrotto lo sviluppo di ACE-031. E perché non agisce legalmente contro tutti gli store online che si puliscono le terga con i brevetti di Acceleron e vendono l’ACE-031 a un prezzo al quale una normale azienda farmaceutica non può trarre alcun profitto.[Muscle Nerve. 2013 Mar; 47 (3) :416-23.]
La risposta si trova in un messaggio sul sito web dell’Associazione per la Distrofia Muscolare. [Quest.mda.org 2 maggio 2013] In esso si legge che nel 2011, durante uno studio [NCT01099761] in cui i ricercatori somministravano l’ACE 031 a bambini affetti da malattie muscolari, sono emersi effetti collaterali che hanno costretto i ricercatori a interrompere lo studio.
“Gli eventi avversi che i partecipanti alla sperimentazione hanno subito – piccoli sanguinamenti del naso e delle gengive e dilatazione dei vasi sanguigni della pelle – non sono stati considerati di per sé pericolosi. Tuttavia, le aziende e le agenzie regolatorie coinvolte affermano di aver bisogno di comprendere appieno questi eventi prima di continuare gli studi clinici sull’ACE-031”. “
Un altro strano effetto collaterale è stato rivelato nello studio pubblicato su Muscle & Nerve. È emerso che la somministrazione di ACE-031 abbia ridotto fortemente la concentrazione di FSH nelle donne partecipanti. I ricercatori non ne conoscono la causa e le possibili conseguenze.
Sembrava che ACE-031 fosse stato definitivamente accantonato dalla ricerca fino alla pubblicazione nel 2017 di uno studio sul recettore esca , sempre su Muscle Nerve [Myostatin inhibitor ACE-031 treatment of ambulatory boys with Duchenne muscular dystrophy: Results of a randomized, placebo-controlled clinical trial]. L’ACE-031 è stato somministrato per via sottocutanea ogni 2-4 settimane a ragazzi affetti da DMD [distrofia muscolare di Duchenne] in uno studio randomizzato, in doppio cieco, controllato con placebo, a dose crescente. L’obiettivo primario era la valutazione della sicurezza. Gli obiettivi secondari comprendevano la caratterizzazione della farmacocinetica e della farmacodinamica.
L’ACE-031, durante lo studio, non è stato associato a eventi avversi gravi o molto gravi. Lo studio è stato interrotto dopo il secondo regime di dosaggio a causa di potenziali problemi di sicurezza legati a epistassi e teleangectasie. È stata rilevata una tendenza al mantenimento della distanza del test del cammino di 6 minuti (6MWT) nei gruppi ACE-031 rispetto al calo osservato nel gruppo placebo (non statisticamente significativo), nonché una tendenza all’aumento della massa magra e della densità minerale ossea (BMD) e alla riduzione della massa grassa.
Anche in questo studio, l’uso dell’ACE-031 ha dimostrato tendenze per gli effetti farmacodinamici sulla massa magra, sulla massa grassa, sulla BMD e sul 6MWT (6-minute walk test). Ma, come successo in precedenza, gli eventi avversi non correlati ai muscoli hanno contribuito alla decisione di interrompere lo studio. Nonostante l’inibizione della Miostatina è un approccio terapeutico promettente per la DMD.
Neanche lo studio su MYO-029, il miostatinblokker della Wyeth, ha avuto successo. Nel 2008 uno studio deludente ha dimostrato che gli adulti con distrofia muscolare, dopo la somministrazione di MYO-029, non sono diventati più forti. [Ann Neurol. 2008 May, 63 (5) :561-71] e la Wyeth ha interrotto lo sviluppo del MYO-029.
Uso nel BodyBuilding e conclusioni:
Ora sappiamo che questo “recettore esca” può favorire lo sviluppo del muscolo-scheletrico legandosi alla Miostatina ed impedendo a questa di esercitare la sua azione di controllo e catabolismo muscolare. Sappiamo inoltre che gli studi effettuati su esseri umani sono stati promettenti ma non sufficientemente sicuri da permetterne uno sviluppo completo. I casi di epistassi e teleangectasie hanno spinto i ricercatori ad interrompere la ricerca. Ma come spesso accade, ogni qualvolta nel panorama scientifico si affaccia una molecola potenzialmente vantaggiosa per lo sportivo, e per il BodyBuilder in particolare, anche se la ricerca si interrompe non si può dire lo stesso per quella svolta illegalmente da improvvisate cavie umane. E questo evento si è verificato anche per l’ACE-031.
Partendo dalle prove emerse durante gli studi, sappiamo che una dose di 3mg/Kg ha comportato un aumento del volume muscolare del 5%, un aumento della massa muscolare del 3% e sembra portare anche a una riduzione della massa grassa. La molecola sembra ridurre la concentrazione di Leptina, condizione che potrebbe portare ad uno scompenso nella regolazione fame/sazietà, ed un aumento dell’Adiponectina, la quale è correlata ad un miglioramento della sensibilità all’Insulina.
Prove sul campo raccolte negli ultimi anni, hanno permesso di quantificare i dosaggi mediamente efficaci per un Bodybuilder e i tempi di somministrazione: 1-3mg per chilogrammo di peso corporeo ogni 15 giorni è risultato essere il range standard per ottenere i migliori risultati possibili. Per quanto concerne la lunghezza del trattamento, si presume che l’uso debba essere circoscritto in un arco temporale di circa 5-6 settimane, limite di conservazione che non dovrebbe essere superato.
Ricordo che il principale effetto collaterale di ACE-031 è la dilatazione dei vasi sanguigni. Tuttavia, questo effetto collaterale, se contenuto, non sembra avere svantaggi. Inoltre, l’uso di ACE-031 può causare epistassi e gengive sanguinanti. Non sono noti altri effetti collaterali. I soggetti emofiliaci sono a forte rischio emorragico potenziale con l’uso di ACE-031.
Anche se dovrebbe essere scontato, ribadisco il fatto che nessuno sta invitando all’uso sperimentale ed illegale di una molecola della quale, oltretutto, si sa poco. Le informazioni ivi presenti sono a puro scopo divulgativo e non rappresentano in alcun modo prescrizioni mediche e affini.
Gabriel Bellizzi
Riferimenti:
Decoy Receptor”. Encyclopedia of Cancer. Springer Berlin Heidelberg. 2012. p. 1070.
Come ben sappiamo, la maggior parte dei farmaci con potenziale sulla perdita di peso agiscono sul aumento della lipolisi e/o della termogenesi, ma anche sulla soppressione dell’appetito che può essere presente insieme alle prima citate reazioni iatrogene nella medesima molecola. Ma esiste un farmaco che si differenzia di molto dalle molecole classicamente utilizzate per la riduzione del peso/grasso. Questo farmaco non è molto conosciuto e fino a poco tempo fa era in fase di test su scimmie rhesus obese: si dice che “uccida” le cellule adipose. I ricercatori dell’Università del Texas pensavano che il farmaco potesse un giorno aiutare a combattere l’obesità negli esseri umani.
Infatti, basti pensare che nel giro di soli 20 anni (dal 1990 al 2010), si è verificato un drammatico aumento dell’obesità negli Stati Uniti e i tassi rimangono alti. Nel 2010, nessuno stato degli Stati Uniti aveva una prevalenza di obesità inferiore al 20%. Circa un adulto su tre e un bambino su sei sono obesi. L’obesità è oggi epidemica negli Stati Uniti e una delle principali cause di morte, attribuibile a malattie cardiache, cancro e diabete. L’Europa non se la passa sicuramente bene. Sulla base dell’indice di massa corporea, nel 2019 il 45% degli adulti europei era normopeso, mentre il 53% era in sovrappeso, con un 17% in condizione di obesità.
Nonostante gli sforzi significativi nell’ultimo decennio, pochissimi farmaci sono stati sviluppati con successo per il trattamento dei pazienti obesi. Attualmente, solo due farmaci approvati dalla Food and Drug Administration (FDA) per la perdita di peso sono disponibili negli Stati Uniti: il soppressore dell’appetito Fentermina e l’inibitore della digestione e assorbimento dei grassi Orlistat. L’Orlistat (Xenical) è un farmaco per la perdita di peso a lungo termine. Questo farmaco riduce la digestione e l’assorbimento dei grassi alimentari nello stomaco e nell’intestino. Altri tentativi di trattare l’obesità si sono concentrati prevalentemente su farmaci volti a sopprimere l’appetito o ad aumentare il metabolismo, ma questi sforzi sono stati ostacolati dai loro effetti collaterali. Sfortunatamente, per una persona nella media è comune riprendere peso indipendentemente dai metodi di trattamento dell’obesità applicati.
Un gruppo di ricercatori ha progettato un farmaco, il peptidomimetico ligando-diretto CKGGRAKDC-GG-D(KLAKLAK)2 (chiamato Adipotide), che è un peptide sintetico che innesca la morte del adipocita. Il farmaco agisce sul tessuto adiposo bianco. Il tessuto adiposo bianco è, per fare un esempio, il tipo di grasso malsano che si accumula sottocute e a livello viscerale.
Caratteristiche del Adipotide:
Più nello specifcio, sto parlando del Prohibitin-targeting peptide 1 (noto anche come prohibitin-TP01 e TP01; nome commerciale Adipotide), un peptidomimetico con sequenza CKGGRAKDC-GG-D(KLAKLAK)2. È un farmaco sperimentale proapoptotico[1] che ha dimostrato di causare una rapida perdita di peso nei topi[2] e nelle scimmie rhesus. [3] Il suo meccanismo d’azione è quello di colpire i vasi sanguigni specifici che riforniscono di sangue il tessuto adiposo, causare il restringimento dei vasi e l’apoptosi delle cellule adipose alimentate da quei vasi.[4] Il TP01 è progettato per legarsi a due recettori, il ANXA2 e quello della prohibitina, che sono specifici dei vasi sanguigni che riforniscono il tessuto adiposo bianco.[5]
In precedenti ricerche precliniche, i topi obesi hanno perso circa il 30% del loro peso corporeo con questo peptidomimetico.[6] Scimmie di tre specie diverse hanno mostrato cambiamenti prevedibili e reversibili nella funzione del tubulo prossimale renale.[6] I livelli di grasso corporeo complessivo e addominale sono scesi, con effetti collaterali reversibili nel Peso, BMI e circonferenza addominale che hanno continuato a scendere per tre settimane dopo la fine del trattamento prima di iniziare lentamente a invertire il trend durante la quarta settimana del periodo di follow-up. Le scimmie negli studi non hanno mostrato segni di nausea o di evitamento del cibo. L’effetto renale era dose-dipendente, prevedibile e reversibile. Questa è una scoperta potenzialmente importante poiché gli effetti collaterali spiacevoli hanno limitato l’uso di farmaci approvati che riducono l’assorbimento dei grassi nell’intestino.
Nota: le barre nere sono in riferimento ai topi trattati con Adipotide.
Nel complesso, questi dati nei primati stabiliscono che l’Adipotide avrebbe potuto divenire un prototipo di una nuova classe di farmaci candidati che possono essere utili per trattare l’obesità negli esseri umani.
Comunque sia l’Adipotide risulta funziona prendendo di mira le cellule che si trovano nel tessuto adiposo bianco, come affermato da Steven Reinberg a USA Today. L’Adipotide uccide il grasso “interagendo con recettori specifici nei vasi sanguigni degli adipociti e innescando l’espressione di una proteina sintetica che fa morire le cellule. In seguito, quelle cellule morte vengono riassorbite dal corpo e metabolizzate.
Le scimmie trattate con questo peptide sono risultate più magre, almeno. In sole quattro settimane, le scimmie obese hanno perso l’11% del loro peso corporeo.[7] Le scimmie hanno anche perso il 27% del loro grasso addominale, come affermato da Tim Barribeau a io9. Attenzione però: le scimmie che erano già magre non hanno perso nemmeno un chilo, il che significa che la molecola potrebbe mirare solo al grasso extra, o subisce una riduzione nell’attività recettoriale (es. riduzione del numero e densità dei recettori target), senza intaccare la messa grassa essenziale alla sopravvivenza.
I test sono risultati senza dubbio promettenti per l’uso negli esseri umani. Di solito, i farmaci “bruciagrassi” sono testati sui topi. I ricercatori credono che questa ricerca sia particolarmente “rilevante perché è stata fatta con i primati”, ha affermato Jennifer Booton a Fox Business. Inoltre, le scimmie più grasse nello studio erano diventate corpulente grazie al loro stesso eccesso di cibo e alla mancanza di esercizio; proprio come molti umani obesi.
Come accennato in precedenza, uno studio su animali ha mostrato che l’Adipotide può portare a una significativa e rapida perdita di peso distruggendo l’apporto di sangue alle cellule adipose. Il farmaco in questione ha aiutato le scimmie rhesus obese a perdere in media l’11% del loro peso corporeo dopo quattro settimane di trattamento. Il farmaco, che funziona sulla base di un trattamento del cancro, mira alle proteine sulla superficie dei vasi sanguigni che alimentano gli adipociti bianchi e li distrugge rilasciando una molecola di sintesi che innesca un processo naturale di morte cellulare. I ricercatori guidati da scienziati dell’Università del Texas hanno prima studiato l’efficacia del nuovo farmaco su topi obesi che ha causato una diminuzione del 30% del loro peso corporeo. La prova successiva è stata effettuata su 15 scimmie in quanto le loro somiglianze con l’uomo li rendono un buon modello per prevedere la possibile efficacia e gli effetti collaterali di un farmaco nell’uomo, anche se sempre in maniera marginale. Dopo quattro settimane, i 10 primati che hanno ricevuto un’iniezione quotidiana di Adipotide hanno perso in media il 38,7% del loro grasso corporeo totale, rispetto al 14,8% degli altri cinque esemplari che sono stati trattati con placebo. Le scimmie trattate hanno anche perso il 27% del loro grasso addominale, come riportato dagli scienziati nella rivista Science Translational Medicine. [8] Anche l’indice di massa corporea (BMI) e la circonferenza addominale (giro vita) sono stati ridotti, mentre tutte e tre le misure erano invariate nelle scimmie del gruppo di controllo non trattate. Le scimmie macaco Rhesus sono state selezionate dalla colonia per lo studio in base alla loro condizione di obesità, contribuendo a fornire un modello di prova perfetto per l’obesità umana e di trattamento del diabete di tipo II.[4] Da notare, a proposito, è che il trattamento con Adipotide ha anche portato ad una migliore sensibilità all’insulina.[9]
A. Mostra la variazione del fabbisogno di insulina (area sotto la curva) per i gruppi trattati (rosso) e di controllo (blu). L’AUC è stata calcolata da un test IVGTT. B. Mostra l’indice insulinogenico prima e dopo nei gruppi di trattamento (rosso) e di controllo (blu). I gruppi trattati mostrano una drastica riduzione della secrezione di insulina. C. Variazione del consumo di cibo nei gruppi trattati (rosso) e di controllo (blu).
“Lo sviluppo di questo composto per uso umano fornirebbe un modo non chirurgico per ridurre effettivamente il tessuto adiposo bianco accumulato, in contrasto con gli attuali farmaci per la perdita di peso che tentano di controllare l’appetito o prevenire l’assorbimento del grasso alimentare”, ha affermato Renata Pasqualini, co-autore senior dello studio. I precedenti tentativi di trattare l’obesità si sono concentrati principalmente su farmaci volti a sopprimere l’appetito o a causare un aumento del metabolismo, ma questi sforzi sono stati ostacolati dai loro effetti collaterali. Il nuovo farmaco progettato dal gruppo MD Anderson include un agente che si lega a una proteina sulla superficie dei vasi sanguigni che supportano il grasso bianco e un peptide sintetico che innesca la morte delle cellule adipose, non appena il loro approvvigionamento di sangue cessa, le cellule adipose vengono riassorbite e metabolizzate. Il professor Wadih Arap, co-autore senior, ha affermato: “L’obesità è un importante fattore di rischio per lo sviluppo del cancro, più o meno l’equivalente dell’uso del tabacco, ed entrambi sono potenzialmente reversibili”.
Dallo studio su scimmie rhesus, la risonanza magnetica conferma che la perdita di peso deriva da una marcata diminuzione del volume del tessuto adiposo bianco. (A) La variazione percentuale del volume di grasso è stata determinata quantificando il volume con immagini di risonanza magnetica assiale T1-pesata. La variazione è rappresentata come variazione percentuale rispetto al basale (giorno 1) ed è significativamente diminuita alla fine del trattamento e alla fine del recupero (test di Mann-Whitney-Wilcoxon, P = 0,02 e P = 0,04, rispettivamente). Le barre di errore indicano il SEM (controllo, n = 3; trattato, n = 6). (B) Un modello a effetti misti dei dati nel tempo indica la significatività della diminuzione della percentuale di grasso per i gruppi trattati rispetto a quelli di controllo (P < 0,0001). (C) Immagini sagittali e assiali pesate in T1 rappresentative di uno degli animali trattati. L’intervallo del livello di finestra è indicato dalla barra colorata sulla destra. Le immagini assiali sono prese in corrispondenza della sezione trasversale indicata dalla linea bianca tratteggiata nell’immagine sagittale. Una diminuzione del contenuto di grasso è rappresentata da una diminuzione del livello della finestra (cioè dell’intensità della visualizzazione dell’immagine).
Come risultato delle sfide nello sviluppo di farmaci per la perdita di peso, attualmente c’è solo un farmaco per l’obesità approvato dalla FDA (e non solo) sul mercato, Alli, che riduce la digestione e l’assorbimento dei grassi alimentari. “Non ci può essere alcun dubbio sulla necessità di nuove strategie in merito”, ha detto Wadih Arap. “E questo rappresenta un salto di qualità in termini di una nuova strategia per il trattamento dell’obesità”. Pasqualini e il Dr. Wadih Arap, suo marito e anche un ricercatore del M.D. Anderson, sono stati in grado di sviluppare il farmaco per l’obesità dopo aver ideato una tecnica per “mappare” le varie reti di vasi sanguigni nel corpo umano. Durante più di un decennio di ricerca, hanno identificato i piccoli pezzi di proteina che si legano con le varie reti di vasi sanguigni nel corpo. In sostanza, quindi, hanno identificato lo “ZIP codes” per ciascuno di questi tipi di vasi sanguigni, e hanno sintetizzato agenti con “ZIP codes” per i vasi sanguigni delle cellule adipose che possono spegnerli. Il loro lavoro ha dimostrato che le diverse cellule hanno dei vasi sanguigni con “firme molecolari” distinte che i ricercatori paragonano ai codici postali. I ricercatori hanno teorizzato di poter privare i tumori del loro approvvigionamento di sangue combinando una terapia letale con una molecola che ha individuato il CAP dei vasi sanguinei in determinate cellule cancerose bloccandone il rifornimento di ossigeno e substrati energetici. Dopo aver identificato lo “ZIP codes” che pensavano potessero funzionare nel cancro alla prostata, si sono interrogati sulla possibilità di colpire i vasi che alimentano gli adipociti bianchi. [10]
Se iniettato su base giornaliera, i ricercatori ritengono che l’Adipodide potrebbe aiutare le persone a perdere il 40% del loro grasso corporeo in sole quattro settimane. Il team americano dietro il nuovo farmaco affermò che la loro formulazione fosse più sicura dei precedenti farmaci dietetici, che sono stati vietati per timori di sicurezza negli ultimi anni, poiché lavora direttamente sul corpo piuttosto che sul SNC.
Nonostante i riscontri positivi avuti su topi e scimmie, la ricerca sul Adipotide è stata interrotta nel 2019.[11] Tale decisione può essere riconducibile al potenziale effetto collaterale a carico dei reni, sebbene tale effetto fosse stato ridimensionato dalle dichiarazioni dei ricercatori per via della sua facile reversibilità.
Conclusioni:
Ora, sappiamo che l’Adipotide agisce sui vasi sanguinei degli adipociti bianchi causando una cessazione del flusso sanguineo e, di conseguenza, del rifornimento cellulare di ossigeno e substrati energetici: il risultato è l’apoptosi cellulare. Ma, come ho precedentemente riportato, sebbene la sua somministrazione in scimmie abbia portato ad una perdita del’11% del peso corporeo totale e il 27% della massa grassa addominale, la sua sperimentazione è stata praticamente interrotta nel 2019, nonostante l’espressione degli effetti collaterali fosse stata descritta come facilmente reversibile e non preoccupante (vedi funzione renale).
L’Adipotide non è un peptide sconosciuto agli atleti, soprattutto nella sottocultura del BodyBuilding. Sono circa 11 anni che se ne parla, anche se la discussione è sempre stata di nicchia rispetto ad altre molecole. Ed è proprio perchè se ne sa poco che bisogna fare dei dovuti chiarimenti.
Se si analizza con attenzione lo studio svolto su scimmie rhesus se ne può notare l’esatto significato dei dati, e su come questi potrebbero darci un idea su eventuali vantaggi e svantaggi di utilizzo.
Ad una coorte di scimmie (n = 15) sono stati somministrati tre livelli di dose di Adipotide (0,25, 0,43 e 0,75 mg/kg) al giorno per 28 giorni. Le scimmie rhesus magre che hanno ricevuto Adipotide (0,25 e 0,43 mg/kg) non hanno perso peso. Le scimmie del gruppo con la dose più alta hanno mantenuto il peso precedente allo studio o hanno mostrato una lieve perdita di peso.
Nelle scimmie sottoposte a necroscopia 24 ore dopo la dose finale di Adipotide, sono state osservate lesioni associate al rene che sono risultate dipendenti dalla dose; tali lesioni non erano presenti nel gruppo di controllo . Le lesioni osservate sono state classificate da minime a lievi nel gruppo a bassa dose, da minime a lievi nella maggior parte delle scimmie a dose media e da minime a moderate nel gruppo ad alta dose. Le lesioni primarie sono state classificate come degenerative/necrotiche (necrosi monocellulare) e reattive/rigenerative. Nelle scimmie sottoposte a necrosi alla fine del periodo di recupero, è stata osservata una degenerazione tubulare minima con poche cellule degenerate in una scimmia del gruppo a dose media e in due scimmie del gruppo ad alta dose. La rigenerazione tubulare e la necrosi tubulare (singola cellula con poche cellule necrotiche) erano minime in tutte le scimmie dopo il recupero. Pertanto, l’effetto collaterale principale dell’Adipotide è un danno renale relativamente lieve, prevedibile e reversibile e un’alterazione della funzione tubulare. L’accumulo anomalo di lipidi (compresa la steatosi epatica) non è stato osservato in nessuna delle scimmie che hanno ricevuto Adipotide.
E’ emerso, valutando dose-risposta ed effetti collaterali, che la dose ottimale era di 0.43mg/Kg peso per le scimmie rhesus, e non per l’uomo! La dose conservativa per l’uomo non è nota. Se dovessimo rapportare il dosaggio usato per le scimmie ad un dosaggio per l’uomo, utilizzando l’apposita formula, esso risulterebbe pari a circa 0.14mg/Kg per 28 giorni.
Valutazione antropometrica di scimmie rhesus obese trattate a dose fissa (0,43 mg/kg, sottocutaneo al giorno) di Adipotide. (Da A a C) La variazione percentuale media rispetto al basale del peso corporeo, della circonferenza addominale e dell’IMC è stata calcolata settimanalmente durante gli intervalli di trattamento (28 giorni) e di recupero (28 giorni) per ogni animale che ha ricevuto Adipotide o soluzione salina. Nel gruppo di trattamento, è stata osservata una marcata diminuzione (A) del peso corporeo medio (10,6%), (B) dell’IMC (10,0%) e (C) della circonferenza addominale (8,4%) rispetto alle misurazioni di base. Le barre di errore indicano il SEM (controllo, n = 5; trattato, n = 10). (Da D a F) Questi risultati erano statisticamente significativi (modello a effetti misti, P < 0,0001 per ogni variabile). Durante un periodo di recupero di 4 settimane, la diminuzione del peso corporeo, della circonferenza addominale e del BMI ha iniziato a invertirsi lentamente.
Va notato, inoltre, che l’effetto sensibile di perdita di grasso si è notato solo nelle scimmie in sovrappeso, mentre in quelle magre la differenza è stata irrisoria. Ciò significa che, se tale peptide venisse usato da un soggetto in sovrappeso o obeso l’effetto potrebbe essere decisamente più significativo sul totale della body fat presente alla fine della terapia rispetto all’inizio paragonato a, per esempio, un bodybuilder con il 10% di bf. Sto parlando del PARAGONE DEL TOTALE DELLA BODY FAT ALL’INIZIO E ALLA FINE DELLA TERAPIA INDIPENDENTEMENTE DALLA PERCENTUALE. in soldoni, è ovvio che uno con il 20% di bf avrà una perdita ponderale maggiore di uno con il 10%, partendo con più grasso… ecco, non mi sto riferendo a questo.
Questa differenza di risposta può essere spiegata attraverso meccanismi di controllo recettoriali che portano ad una sottoregolazione maggiore quanto più la percentuale di grasso e bassa.
Ovviamente non sto consigliando a nessuno di diventare una cavia da esperimenti, d’altronde non sappiamo praticamente nulla sugli effetti collaterali nell’uomo e sul loro grado anche qualora combaciassero in buona parte con quelli osservati nelle scimmie.
Kolonin MG, Saha PK, Chan L, Pasqualini R, Arap W (June 2004). “Reversal of obesity by targeted ablation of adipose tissue”. Nature Medicine. Nature Publishing Group. 10 (6): 625–32.