Introduzione:

La pressione alta, nota anche come ipertensione, è una delle cause più frequenti (che contribuiscono) di morte e complicazioni cardiovascolari nel mondo. Quando viene misurata, si divide in pressione sistolica (il numero superiore) e pressione diastolica (il numero inferiore). La pressione arteriosa sistolica è la pressione più alta raggiunta durante la contrazione del cuore, mentre la pressione diastolica è la pressione più bassa raggiunta durante il rilassamento del cuore. Una tipica lettura della pressione arteriosa potrebbe essere 120/80 mmHg, ovvero una pressione sistolica di 120 mmHg e una pressione diastolica di 80 mmHg. (L’unità di misura, millimetri di mercurio [Hg], risale a quando la pressione sanguigna veniva ancora misurata con manometri contenenti mercurio).
Per quantificare quanto sia grave l’ipertensione, diamo un’occhiata a un documento storico pubblicato su Lancet nel 2002 che, secondo Google Scholar, è stato citato ben 12.000 volte [1]. In questo lavoro, i ricercatori hanno riunito i dati dei singoli pazienti provenienti da 61 studi prospettici osservazionali. Questo studio comprendeva circa un milione di adulti senza precedenti malattie vascolari al basale. Per questo motivo, hanno avuto a disposizione dati davvero straordinari su cui lavorare e da cui trarre conclusioni.
Che cosa hanno dimostrato i dati? Hanno dimostrato che la mortalità per malattie coronariche e ictus aumenta con una pressione sistolica superiore a 115 mmHg e una pressione diastolica superiore a 75 mmHg. Ogni aumento di 20 mmHg della pressione arteriosa sistolica e di 10 mmHg della pressione arteriosa diastolica oltre questi valori raddoppia la mortalità per coronaropatia e ictus. In altre parole, chi ha una pressione arteriosa sistolica di 135 mmHg ha il doppio del rischio di morire per malattia coronarica o ictus rispetto a chi ha una pressione arteriosa sistolica di 115 mmHg. Si tratta di una differenza notevole. Questa relazione tra pressione arteriosa e mortalità, ad esempio per ictus, è illustrata nell’immagine sottostante:

Si noti che anche la probabilità di morire per ictus aumenta fortemente con l’aumentare dell’età. Il che ha senso, ovviamente. Sebbene non siano molte le persone che muoiono di ictus a 40 anni, è molto più comune negli anziani. Pertanto, l’aumento del rischio relativo di ipertensione diventa più rilevante con l’aumentare dell’età, poiché il rischio assoluto è molto più elevato.
Oltre a questo evidente aumento della mortalità a causa di eventi cardiovascolari, l’ipertensione provoca alterazioni strutturali e funzionali di diversi organi, danneggiandoli. Il danno agli organi bersaglio comprende, oltre al cuore e alla vascolarizzazione, il cervello, gli occhi e i reni. Il danno agli organi bersaglio può manifestarsi, oltre che con eventi cardiovascolari fatali e non fatali, con retinopatia, demenza, ischemia, albuminuria, glomerulopatia e ipertrofia ventricolare sinistra [2].
È chiaro che la pressione arteriosa elevata è dannosa per la salute.
Come influiscono gli steroidi anabolizzanti sulla pressione sanguigna?
Per rispondere alla domanda su come gli steroidi anabolizzanti influenzino la pressione arteriosa, si possono effettuare due tipi di studi. Un tipo di studio è costituito dagli studi prospettici interventistici. Questi, in sostanza, sono i più affidabili. Si prende un gruppo di persone, si somministra loro uno steroide anabolizzante e le si segue nel tempo per vedere cosa succede alla loro pressione sanguigna. Inoltre, si può includere un gruppo di controllo/placebo con cui confrontare i risultati (e se si randomizzano i soggetti si ottiene uno studio randomizzato-controllato). Sebbene questi studi siano sicuramente i migliori in termini di qualità delle prove, soffrono di un grosso inconveniente: non imitano correttamente l’uso reale, poiché i dosaggi sono inferiori a quelli utilizzati dalla maggior parte delle persone che fanno uso di steroidi anabolizzanti in modo illecito.
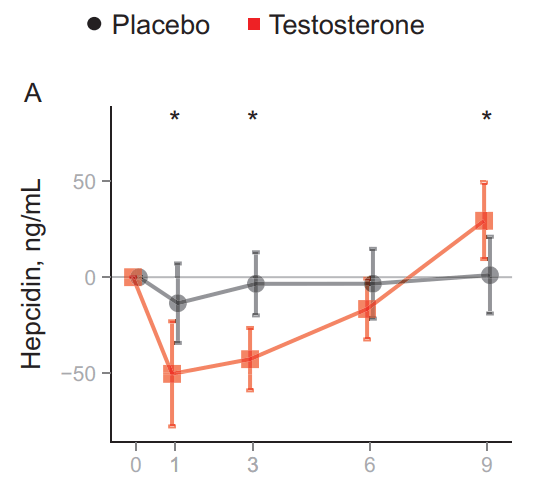
Detto questo, diamo un’occhiata a queste prove. Li ho riassunti nella tabella sottostante per fornire una buona panoramica:
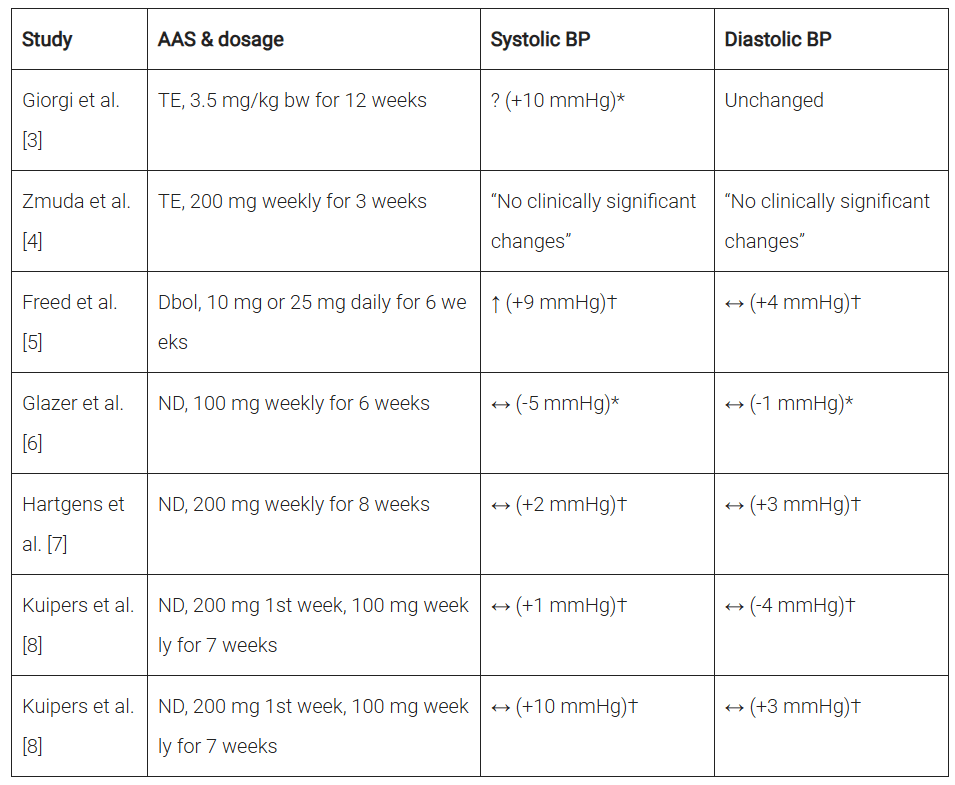
Effetto degli steroidi anabolizzanti sulla pressione arteriosa in studi prospettici interventistici. ↑ significa un aumento statisticamente significativo, ? significa che non sono stati eseguiti test statistici, * significa rispetto al basale, † significa rispetto alla variazione nel gruppo placebo. Abbreviazioni: BP, pressione sanguigna; TE, testosterone enantato; Dbol, metandienone; ND, nandrolone decanoato.
L’unico studio che ha dimostrato un aumento statisticamente significativo della pressione arteriosa (sistolica) è stato quello di Freed et al [5]. In questo caso, sollevatori di pesi esperti hanno ricevuto 10mg o 25mg di Methandienone (Dianabol) al giorno per 6 settimane in doppio cieco controllato con placebo. La pressione arteriosa sistolica è aumentata significativamente di circa 9 mmHg. La pressione diastolica ha mostrato un leggero aumento di circa 4 mmHg, ma non è stato statisticamente significativo.
Gli altri studi non hanno eseguito test statistici [3,4] o non hanno rilevato cambiamenti statisticamente significativi rispetto al basale [6] o rispetto al cambiamento nel gruppo placebo [7, 8].
Come si può notare anche osservando i dosaggi, questi erano piuttosto bassi e non possono essere considerati rappresentativi dell’uso di steroidi anabolizzanti che si fa regolarmente in ambito del culturismo e simili. Per avere un’idea più precisa, si potrebbe ricorrere a studi prospettici osservazionali. In questi studi gli utilizzatori di AAS vengono seguiti nel tempo autosomministrando il proprio ciclo di AAS. Naturalmente, questi studi presentano anche degli inconvenienti. Uno di questi è il “policonsumo”. Non tutti gli steroidi anabolizzanti possono influire allo stesso modo sulla pressione arteriosa e, quando i consumatori di AAS li cumulano, è difficile dire quale steroide anabolizzante possa esserne responsabile. Per non parlare del fatto che è molto probabile che un consumatore di AAS stia somministrando steroidi anabolizzanti diversi da quelli che pensa di somministrare a causa di un’etichettatura errata. [9]. Inoltre, i consumatori di AAS potrebbero associarli a diversi altri tipi di farmaci, come l’rhGH, tiroidei, beta-agonisti e, al giorno d’oggi, la vasta gamma di farmaci sperimentali per il miglioramento delle prestazioni, il cui uso è in aumento.
Consideriamo anche brevemente alcuni studi prospettici osservazionali. Hartgens et al. hanno osservato gli effetti degli AAS autosomministrati per un periodo di 8 settimane in un piccolo gruppo di atleti di forza [10]. Prima dello studio, i soggetti, in media, non avevano fatto uso di AAS per quasi 8 mesi. Il dosaggio medio era relativamente basso, circa 400mg a settimana, il che mi fa pensare a quanto sia stato accurato. In ogni caso, la pressione arteriosa sistolica è aumentata da 131 a 139 mmHg. Il gruppo di controllo ha registrato un aumento da 129 a 134 mmHg. Quindi la variazione media rispetto al gruppo di controllo è stata di +3 mmHg. È stato osservato un piccolo aumento di 2 mmHg della pressione arteriosa diastolica, mentre il gruppo di controllo non ha registrato alcuna variazione. In ogni caso, le differenze non erano statisticamente significative.
Se si parte dal presupposto che gli AAS possono influenzare la pressione sanguigna e che questo fenomeno è completamente reversibile dopo la cessazione dell’uso, è possibile utilizzare un disegno di studio leggermente diverso. In altre parole, si potrebbe prendere un gruppo di utilizzatori mentre fanno uso di AAS, misurare la loro pressione sanguigna e poi misurarla di nuovo dopo un certo periodo di tempo, quando hanno smesso di usare gli AAS. Questo è esattamente il tipo di approccio che altri due gruppi hanno utilizzato [11, 12].
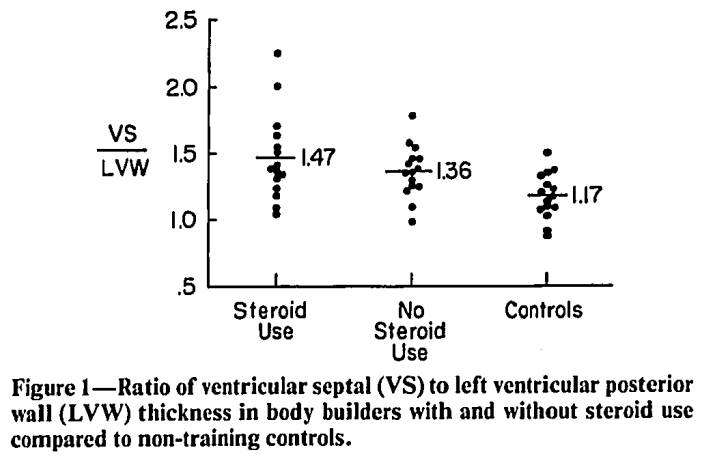
Uno di questi ha valutato tre gruppi: soggetti sedentari, bodybuilder che non fanno uso di AAS e bodybuilder che ne fanno uso [11]. I cicli di AAS duravano in media 8 settimane e, purtroppo, i dosaggi non possono essere ricavati con precisione dallo studio. Ciononostante, sembrano essere bassi. Subito dopo i cicli, la pressione sanguigna misurava 141/84 mmHg e 9 settimane dopo la cessazione dell’uso era 140/83 mmHg. Come riferimento, i bodybuilder che non ne facevano uso avevano una pressione sanguigna di 136/87 e i soggetti sedentari di 139/85 mmHg.
Palatini et al. hanno effettuato misurazioni della pressione arteriosa nelle 24 ore in un piccolo gruppo di consumatori di AAS [12]. I cicli duravano in media 9 settimane e il dosaggio era di circa 500mg settimanali. La pressione arteriosa era di 128/83 mmHg alla fine dei cicli e di 129/84 mmHg circa 12 settimane dopo la cessazione.
Nello studio HAARLEM, 100 consumatori di steroidi anabolizzanti sono stati seguiti nel tempo mentre si autosomministravano AAS [9]. Il dosaggio medio, basato sulle informazioni riportate sull’etichetta, era di 898mg a settimana, rendendo così il loro ciclo di AAS abbastanza rappresentativo dell’uso comune da parte dei bodybuilder. Le misurazioni sono state effettuate prima, durante, 3 mesi dopo la fine del ciclo e 1 anno dopo l’inizio del ciclo. I dati non pubblicati di questo studio hanno dimostrato un aumento di 7 mmHg della pressione sanguigna sistolica e di 3 mmHg della pressione sanguigna diastolica durante l’uso di steroidi anabolizzanti rispetto al basale [DL Smit, comunicazione personale]. Queste misurazioni sono tornate al valore basale dopo il ciclo. Data la dimensione relativamente ampia del campione di 100 utilizzatori di AAS e la natura osservazionale prospettica di questo studio, questa è attualmente la migliore stima della misura in cui gli AAS potrebbero influenzare la pressione sanguigna ai dosaggi comunemente utilizzati dai bodybuilder.
Nel complesso, si può concludere con cautela che i dosaggi sovrafisiologici di AAS possono aumentare transitoriamente la pressione arteriosa sistolica di circa 5-10 mmHg durante l’uso. È difficile dire in che misura questo aggravi il rischio cardiovascolare. Ma potremmo trarre qualche indizio da altri dati presenti nella letteratura scientifica. Ne parlerò nel prossimo articolo, in cui tratterò dei farmaci per abbassare la pressione sanguigna.ù
[altri] PEDs e pressione arteriosa:
Le evidenze nella ricerca e i dati aneddotici hanno mostrato un effetto ipertensivo legato all’uso di β-Agonisti sia non selettivi che selettivi. In particolare, è stato osservato che l’allele Gly16 del recettore adrenergico β-2 AR associato all’ipertensione. Questo effetto è stato osservato sia in trattamento con Salbutamolo che con Clenbuterolo, ed è responsivo ad alterazioni maggiori in base al dosaggio utilizzato [https://pubmed.ncbi.nlm.nih.gov/10373227/].
Anche l’Insulina può aumentare la pressione arteriosa attraverso diversi meccanismi. Per esempio, portando all’aumento del riassorbimento renale di sodio, all’attivazione del sistema nervoso simpatico, all’alterazione del trasporto ionico transmembrana e all’ipertrofia dei vasi di resistenza. Ovviamente si tratta di casi emersi, o possibili, in condizione di IR o alterazione subclinica del metabolismo glucidico e dell’attività biochimica dell’Insulina. Non è raro che bodybuilder in fase di Off-Season, con abuso di Insulina esogena e/o GH, ma anche in situazioni di non uso del peptide, presentino alterazioni pressorie correlati a sensibili aumenti di peso: tale causa vede anche la condizione di IR come co-fattore peggiorativo [https://pubmed.ncbi.nlm.nih.gov/].

In letteratura viene riportato che in seguito a somministrazione di rhGH si manifesti ritenzione idrica, e che essa sia un effetto collaterale concreto e documentato. Infatti, la maggior parte dei dati indicano che i pazienti adulti con deficit di hGH sono disidratati, cioè non hanno un volume d’acqua positivo nel corpo, e presentano una bassa concentrazione di acqua extracellulare nel plasma. Quando viene avviata la terapia sostitutiva del GH in questi pazienti i loro fluidi corporei vengono ripristinati alla normalità. La capacità di ritenzione dei fluidi del GH dovrebbe quindi essere considerata in ambito clinico come una normalizzazione fisiologica desiderabile dell’omeostasi dei liquidi corporei piuttosto che un effetto collaterale sgradevole [https://www.ncbi.nlm.nih.gov/pubmed/10592455]. Ovviamente, nel Bodybuilding le cose cambiano nettamente come l’incidenza quantitativa e anche indiretta sulla pressione vascolare di questo effetto sulla ritenzione idrica dato dall’uso di rhGH.
Si è ipotizzato che l’ormone alterato dall’uso di rhGH e che causa la ritenzione idrica possa essere l’Aldosterone. Nel qual caso, un diuretico antagonista come lo Spironolattone aiuterebbe. Il problema può essere risolto con del Lasix, il Furosemide (non è un consiglio!) , ma dal momento che l’esperienza sul campo non ha mostrato risoluzione al problema con queste pratiche, la domanda non ha così trovato una risposta chiara.
Un altra ipotesi indica una correlazione tra ritenzione idrica da rhGH e un aumento dell’ADH (Ormone Antidiuretico, conosciuto anche come vasopressina). Uno studio giunge alla conclusione che il hGH aumenta l’ADH [https://www.ncbi.nlm.nih.gov/pubmed/2405233], come effetto che trova la sua causa nella attivazione del sistema renina-angiotensina.

Il GH esogeno aumenta la Somatostatina, e dato che il rene possiede recettori specifici per la Somatostatina questi possono attivare il sistema renina-angiotensina [https://www.ncbi.nlm.nih.gov/pubmed/2405233]. Ciò può causare la ritenzione idrica da GH che può essere inibita da un ACE-inibitore.
I ricercatori, per vederci più chiaro, hanno studiato gli effetti di un preparato biosintetico autentico del hGH (bio-hGH) sul metabolismo del sodio e l’attività del sistema renina-angiotensina. Questa preparazione è stata somministrata a 6 giovani uomini ad un dosaggio di 0,2 U / kg / die per via sottocutanea per cinque giorni consecutivi. E’ stata effettuata la raccolta delle urine nelle ventiquattro ore per la misurazione dell’escrezione di sodio e l’osmolalità, ed è stato prelevato il sangue per quantificare i cambiamenti del sodio, dell’osmolalità, dell’attività della renina plasmatica (PRA), dell’aldosterone, e delle concentrazioni della arginina vasopressina (AVP). La somministrazione di Bio-hGH ha determinato un calo nelle 24 ore dell’escrezione urinaria di sodio (197 +/- 38 a 42 +/- 20 mmol, media +/- SD, P meno di 0,005), una riduzione del volume delle urine (1.652 + / – 182-848 +/- 348 mL, P inferiore a 0,05), ma non l’osmolalità. Il PRA è aumentato in modo significativo di 1.118 +/- 73 a 3.608 +/- 1.841 fmol angiotensina 1 L / s (P meno di 0,005), il che è stato associato con un aumento di sette volte nella concentrazione plasmatica (52 +/- 12-402 + / – 99 pg / mL, P meno di 0,001). L’osmolalità del plasma e le concentrazioni di AVP non sono cambiate in modo significativo. I risultati mostrano che la ritenzione di sodio indotta dal Bio-GH comporta l’attivazione del sistema renina-angiotensina. Questo meccanismo può spiegare in parte l’insorgenza dell’espansione del volume plasmatico e l’ipertensione e suggerisce un rischio di ritenzione di liquidi e, eventualmente, l’ipertensione nei soggetti trattati con dosi sovrafisiologiche di bio-hGH per il trattamento della bassa statura [http://www.dtic.mil/dtic/tr/fulltext/u2/611818.pdf].
Quindi, il rhGH provoca un rialzo del ADH(Ormone Antidiuretico). L’ADH è un costrittore coronarico molto potente che costringente vascolare/venoso. dato questo aumento del ADH, abbiamo la ritenzione idrica aumentata, e ciò provoca un aumento della pressione sanguinea. L’aumento del ADH causato dal rhGH è dose-dipendente.

L’uso della Clonidina è indicato se si soffre di pressione alta causata dal ADH.
Ovviamente, la somministrazione di rhGH, soprattutto se in concomitanza con AAS fortemente aromatizzabili peggiora considerevolmente la situazione collegandosi anche all’Insulino Resistenza e all’azione di alcuni AAS con i recettori mineralocorticoidi e sul danno endoteliale di questi.
La Clondina viene consigliata principalmente perché agisce direttamente sul ADH è la ritenzione idrica causata da questo ormone. Tuttavia, alcuni assumono il Furosemide (una molecola tutt’altro che sicura) per via della sua maggior potenza; ma non sembra lavorare tanto efficacemente nel complesso. Anche lo Spironolattone mostra una sua efficacia in tale circostanza con il suo effetto di diminuzione dell’attività dell’Aldosterone; riducendo quindi l’edema.
Ma la Clonidina risulta essere la prima scelta per contrastare l’effetto di un aumento del ADH dovuto alla somministrazione di rhGH esogeno. La sua somministrazione è solitamente indicata prima di dormire.
Come misurare la pressione arteriosa
In primo luogo, naturalmente, è necessario un dispositivo per misurare la pressione sanguigna a casa. Consiglio vivamente di utilizzare un apparecchio elettronico automatico che la misuri a livello della parte superiore del braccio. Questi dispositivi sono affidabili e richiedono la minima abilità, per cui c’è la minima possibilità di sbagliare la misurazione. Sono facilmente reperibili e costano circa 50 dollari. Valgono bene l’investimento. La maggior parte dei miei clienti utilizza dispositivi Omron, ma sono certo che esistono molte altre marche che producono dispositivi eccellenti.
Inoltre, assicuratevi che il misuratore di pressione sia dotato di un bracciale di dimensioni adeguate al vostro braccio. Di solito, i dispositivi per la misurazione della pressione arteriosa sono dotati di un bracciale di misura M, adatto a braccia con una circonferenza massima di 31-33 cm. Naturalmente, questa non è la circonferenza del braccio per i bicipiti flessi, ma la circonferenza quando il braccio è leggermente piegato senza essere flesso. La maggior parte degli utilizzatori di AAS ha braccia più grandi. Nella maggior parte dei casi è appropriato un bracciale di taglia L, che si adatta a braccia con circonferenza fino a 41-43 cm. Se siete molto grandi, potreste aver bisogno della taglia XL, che si adatta a braccia con circonferenza fino a circa 51-53 cm.
Un bracciale di dimensioni adeguate è importante perché, se troppo piccolo, potrebbe sovrastimare la pressione arteriosa. Ciò è ben illustrato in uno studio che ha esaminato le differenze di pressione arteriosa tra un bracciale di taglia M e uno di taglia L in 193 bodybuilder che partecipavano al Campionato Nazionale Messicano di Bodybuilding e Fitness [13]. Coloro che avevano braccia troppo grandi per il bracciale di taglia M (>33 cm) avevano una pressione sistolica più alta di 8,2 mmHg con questo bracciale rispetto al bracciale di taglia L. La pressione diastolica era più alta di 1,6 mmHg.
Anche un altro studio, condotto su individui obesi, ha sottolineato l’importanza di un bracciale di dimensioni adeguate [14]. Per ogni aumento di 5 cm della circonferenza del braccio oltre i 35 cm, si registrava un aumento di 2-5 mmHg della pressione arteriosa sistolica e di 1-3 mmHg della pressione arteriosa diastolica.
Ora che siete pronti a misurarla, come dovete fare? La Società Internazionale dell’Ipertensione ha un’ottima immagine che lo illustra [15], diamo un’occhiata:
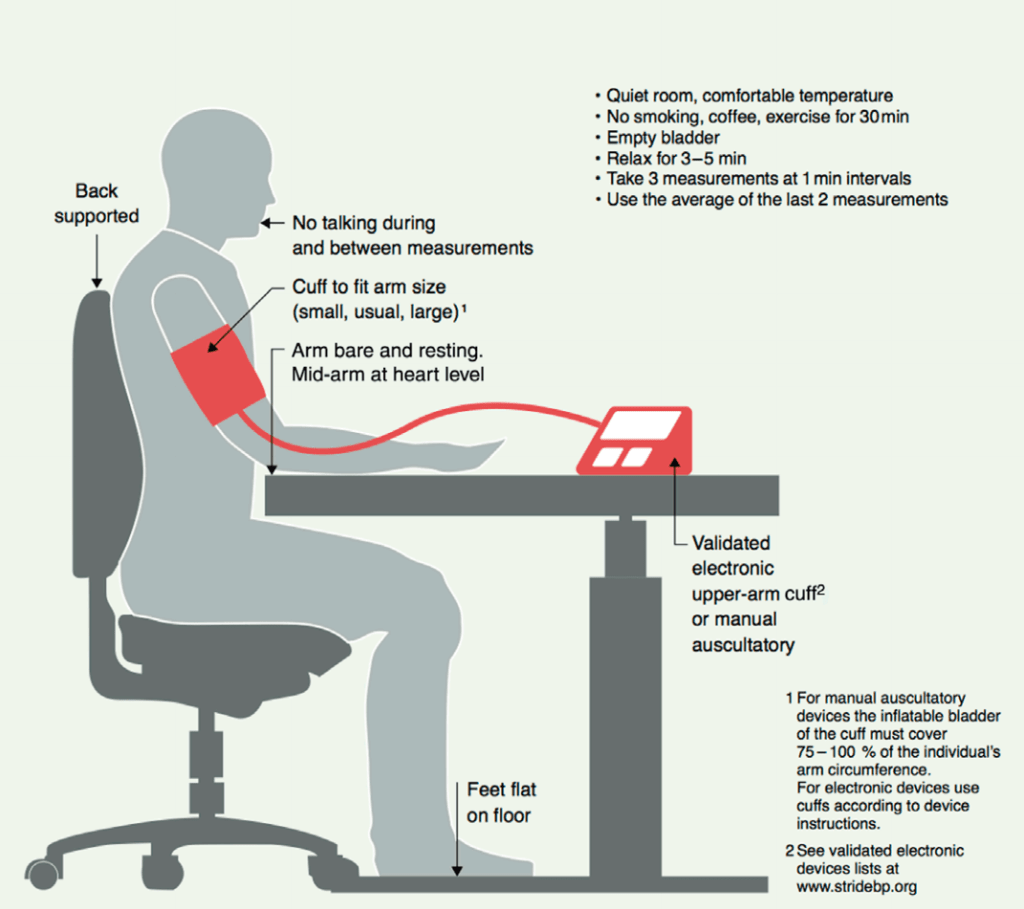
Assicuratevi di non dover fare pipì, di non fumare (ovviamente non fumate, giusto?), di non aver bevuto caffè/caffeina o di aver fatto esercizio fisico 30 minuti prima della misurazione e di mettervi in una stanza tranquilla e confortevole per rilassarvi un paio di minuti. Siete seduti su una sedia che sostiene adeguatamente la vostra schiena dietro una scrivania. Si indossa il bracciale e si appoggia il braccio sulla scrivania, appoggiandolo completamente con la parte centrale del braccio all’altezza del cuore. Si appoggiano i piedi sul pavimento, non si accavallano le gambe e si batte il più forte possibile per far sì che il dispositivo misuri la pressione sanguigna. Ripetete la misurazione due volte con una piccola pausa tra l’una e l’altra e voilà. Prendete la media delle ultime due misurazioni e annotatela da qualche parte (la maggior parte degli apparecchi elettronici per la misurazione della pressione arteriosa ha anche una funzione di memoria, quindi potete evitare di scriverla).
Con quale frequenza si dovrebbero effettuare queste misurazioni? Le linee guida della Società Europea dell’Ipertensione per il monitoraggio domiciliare della pressione arteriosa raccomandano di farlo inizialmente almeno 3 e preferibilmente 7 giorni prima di considerare il trattamento della pressione arteriosa [16]. Le misurazioni dovrebbero essere effettuate sia al mattino che alla sera. Dopo questo periodo iniziale, è sufficiente misurarla circa una volta alla settimana.
Quando iniziare a trattare la pressione arteriosa
Dopo aver letto quanto fino a questo punto esposto, potreste pensare di dover trattare la pressione arteriosa quando è superiore a 115/75 mmHg. Tuttavia, una recente revisione sistematica e meta-analisi ha rilevato che, nella prevenzione primaria, l’abbassamento della pressione arteriosa riduce la mortalità e il rischio di malattie cardiovascolari solo se la pressione sistolica al basale è pari o superiore a 140 mmHg [17]. Se era inferiore a quella al basale, gli autori non sono riusciti a trovare alcun beneficio per quanto riguarda la mortalità o il rischio di malattie cardiovascolari. In effetti, questo è anche il motivo per cui la Società Europea dell’Ipertensione classifica l’ipertensione come una pressione arteriosa sistolica in ufficio pari o superiore a 140 mmHg e/o una pressione arteriosa diastolica pari o superiore a 90 mmHg [16]. L’ipertensione è definita come il livello di pressione arteriosa al quale i benefici del trattamento (con interventi sullo stile di vita o con farmaci) superano inequivocabilmente i rischi del trattamento, come documentato da studi clinici.
Va sottolineato che questa soglia di 140/90 mmHg riguarda le misurazioni della pressione arteriosa effettuate in ufficio. Queste sono di solito leggermente più alte rispetto alle misurazioni della pressione sanguigna effettuate a casa. Pertanto, la soglia per le misurazioni domiciliari della pressione arteriosa è definita come un valore medio di 135/85 mmHg [16].
Il trattamento dell’ipertensione produce chiari benefici clinici. Una meta-analisi mostra che ogni riduzione di 10 mmHg della pressione arteriosa sistolica riduce il rischio di eventi cardiovascolari maggiori del 20%, di malattia coronarica del 17%, di ictus del 27%, di insufficienza cardiaca del 28% e di mortalità per tutte le cause del 13% [18]. Sfortunatamente, il trattamento dell’ipertensione non annulla completamente tutti i rischi osservati nei grandi studi osservazionali. Due probabili ragioni sono: 1) l’ipertensione per periodi prolungati può danneggiare in modo irreversibile alcuni organi, e il trattamento non può annullare i danni subiti; 2) l’ipertensione spesso è associata a diverse altre comorbilità che possono influire sull’esito (ad esempio, l’obesità). Il primo motivo può essere affrontato iniziando il trattamento il prima possibile quando necessario, mentre il secondo non è particolarmente applicabile a un aumento della pressione sanguigna indotto da farmaci (come nel caso degli AAS). Tuttavia, non bisogna assolutamente dimenticare che, al di là del modesto aumento della pressione arteriosa, gli AAS hanno un impatto negativo sulla salute. Quindi, la correzione della pressione arteriosa, ovviamente, non annulla completamente i rischi per la salute degli AAS, così come non risolve i rischi per la salute di altre comorbidità che spesso vanno di pari passo con l’ipertensione.
Trattamento dell’ipertensione: modifiche allo stile di vita

Proprio come nella popolazione generale, ci possono essere alcuni cambiamenti nello stile di vita da adottare per ridurre la pressione sanguigna prima di ricorrere ai farmaci per abbassarla. Tuttavia, in alcuni casi i farmaci devono essere utilizzati immediatamente insieme ai cambiamenti dello stile di vita. La Società Europea dell’Ipertensione raccomanda di iniziare immediatamente il trattamento farmacologico nei soggetti con un rischio elevato o molto elevato di malattie cardiovascolari, malattie renali o danni agli organi mediati dall’ipertensione. Raccomanda inoltre un trattamento farmacologico immediato in tutti i pazienti che hanno una pressione arteriosa pari o superiore a 160/100 mmHg. In questi casi, vi invito a non usare steroidi anabolizzanti e a rivolgervi al vostro medico per un trattamento. Sconsiglio vivamente l’uso di steroidi anabolizzanti se questo è il vostro caso.
Detto questo, ecco alcuni cambiamenti nello stile di vita. Uno ovvio è quello di smettere di fumare, se lo fate. Non tanto per abbassare la pressione sanguigna, ma semplicemente perché il fumo aumenta enormemente il rischio di malattie cardiovascolari. Probabilmente la lettura di questo articolo non vi farà smettere di fumare (se solo fosse così facile, no?), ma volevo solo informarvi.
Una strategia efficace consiste nel ridurre il sodio alimentare, cioè il sale. Diverse linee di evidenza hanno costantemente implicato l’assunzione di sale nella dieta con il rischio cardiovascolare [19]. Una meta-analisi Cochrane del 2013 di 34 studi randomizzati e controllati ha dimostrato una riduzione della pressione arteriosa di 4,2/2,1 mmHg per ogni riduzione di 4,4 g/die di assunzione di sale (=1,8 g di sodio) [20]. Di conseguenza, la Società Europea dell’Ipertensione raccomanda di limitare l’assunzione di sale a 5 g al giorno (= 2 g di sodio) [16]. Tuttavia, l’assunzione di sale con la dieta mostra una curva a U per quanto riguarda il rischio di eventi cardiovascolari e di morte. Ciò significa che, mentre la riduzione dell’assunzione di sale diminuisce questo rischio, esso ricomincia ad aumentare al di sotto di una certa dose giornaliera. Una recente meta-analisi ha rilevato che, rispetto a un’assunzione di 7 o più g di sodio al giorno, 4-5 g al giorno comportano un rischio inferiore di eventi cardiovascolari e morte [21]. Allo stesso modo, anche un’assunzione di meno di 3 g al giorno mostrava un rischio maggiore rispetto a un’assunzione di 4-5 g di sodio al giorno. (Gli studi hanno esaminato l’escrezione urinaria di sodio come proxy dell’assunzione di sodio. Questa è eccellente come proxy, quindi in questo articolo faccio finta che siano la stessa cosa). Non è del tutto chiaro quale sia la causa, poiché 3 g di sodio al giorno sono sufficienti a coprire il fabbisogno giornaliero. La Società Europea dell’Ipertensione si aggrappa all’effetto di abbassamento della pressione arteriosa come decisivo per ridurla ulteriormente. Sentitevi liberi di farlo, ma credo che sia più pragmatico attenersi ai 4-5 g al giorno, a meno che non abbiate già un apporto inferiore, e lasciarlo così. E poi realizzare un’ulteriore riduzione della pressione arteriosa, se necessario, con ulteriori interventi sullo stile di vita o con farmaci. Infine, una cosa che non potrò mai sottolineare abbastanza: controllate il contenuto di sale di tutto ciò che mangiate. Potreste rimanere sorpresi dal contenuto di sale di alcuni prodotti che consumate.
Se bevete molto alcol, è ovviamente consigliabile moderare il consumo di alcol (o astenersi dal farlo). L’effetto di abbassamento della pressione sanguigna è molto modesto (riduzione di ~1,2/0,7 mmHg [22]), ma l’alcol fa male alla salute (cardiovascolare) a prescindere. La raccomandazione è di limitare il consumo a 14 unità a settimana per gli uomini (8 a settimana per le donne; 1 unità equivale a 125 ml di vino o 250 ml di birra) [16]. A parte questo, aggiungete un po’ di esercizio aerobico alla vostra routine, se non l’avete già fatto, e assicuratevi di non ingrassare. Anche questo aiuta.
Trattamento dell’ipertensione: i farmaci

Per il trattamento dell’ipertensione sono disponibili diversi farmaci. Esistono cinque classi principali di farmaci raccomandati per questo scopo: ACE-inibitori, bloccanti del recettore dell’angiotensina (ARB), beta-bloccanti, calcio-antagonisti (CCB) e diuretici tiazidici. Esistono alcune piccole differenze per quanto riguarda gli esiti specifici per causa tra questi farmaci. Tuttavia, gli esiti cardiovascolari maggiori e la mortalità sono complessivamente simili e pertanto tutti sono raccomandati dalla Società Europea dell’Ipertensione come trattamento di prima linea [16]. Anche la Società Internazionale dell’Ipertensione raccomanda questi farmaci come trattamento di prima linea, ad eccezione dei beta-bloccanti [15]. Ogni classe di farmaci ha le proprie controindicazioni. Ad esempio, gli atleti e i pazienti fisicamente attivi sono indicati come possibile controindicazione all’uso dei beta-bloccanti, mentre una frequenza cardiaca inferiore a 60 bpm è indicata come controindicazione assoluta sia per i beta-bloccanti che per alcuni calcio-antagonisti (le non diidropiridine) [16]. I beta-bloccanti vengono generalmente aggiunti al trattamento quando esiste un’indicazione specifica per il loro utilizzo. Inoltre, i diuretici tiazidici sono preferiti ai tiazidici. In definitiva, la terapia si riduce ai calcio-antagonisti diidropiridinici, agli ACE-inibitori, agli ARB e ai diuretici tiazidici. Secondo la mia esperienza, i consumatori di AAS hanno un accesso relativamente più facile a questi ultimi tre, ma non ai calcio-antagonisti. (Naturalmente, a meno che non vengano prescritti dal medico, ma in quel caso si fa quello che prescrive il medico). Pertanto, mi concentrerò su queste tre modalità di trattamento.
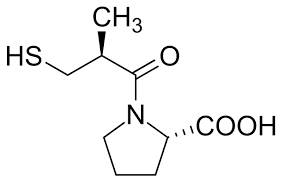
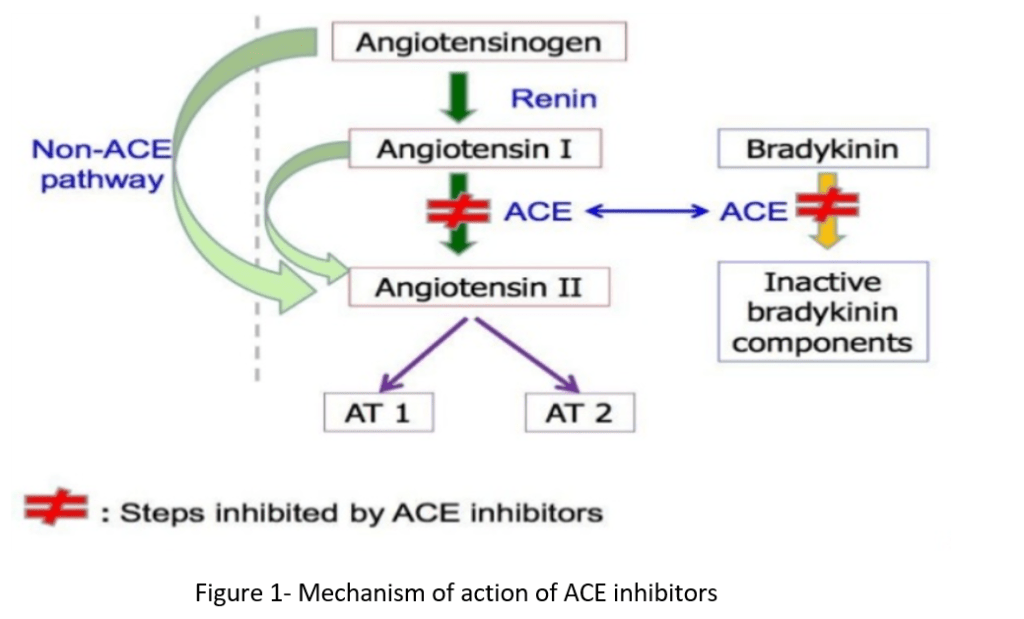
Sia gli ACE-inibitori che gli ARB si agganciano al cosiddetto sistema renina-angiotensina-aldosterone (RAAS). Si tratta di un sistema ormonale che svolge un ruolo estremamente importante nella regolazione del volume del sangue, degli elettroliti e della resistenza vascolare sistemica. Come tale, costituisce un bersaglio molto interessante per il trattamento dell’ipertensione. Questo sistema ormonale funziona come segue. I reni rilasciano un enzima chiamato renina ogni volta che rilevano un calo della pressione sanguigna. Questo enzima, a sua volta, converte una proteina prodotta dalla leva, l’angiotensinogeno, in angiotensina I. Si tratta di un piccolo peptide che costituisce il substrato per l’enzima di conversione dell’angiotensina (ACE), che taglia altri due aminoacidi da questo peptide producendo angiotensina II. L’angiotensina II è responsabile della vasocostrizione, soprattutto nelle arteriole. Di conseguenza, aumenta la pressione arteriosa, chiudendo così il circuito avviato dal rilevamento di una diminuzione della pressione arteriosa da parte dei reni. Inoltre, l’angiotensina II inibisce il processo di escrezione di acqua (diuresi) e sodio (natriuresi) da parte dei reni. Questo effetto si ottiene in parte evocando il rilascio di aldosterone da parte dei surreni. L’aldosterone è un ligando per i recettori dei mineralocorticoidi (MR) situati nei reni. L’attivazione del MR provoca la ritenzione di acqua e sodio e la secrezione di potassio. Il RAAS è schematizzato di seguito:
Ora che sapete come funziona il RAAS, sapete anche come funzionano gli ACE-inibitori e gli ARB. Gli ACE inibitori inibiscono l’enzima ACE (naturalmente, è scritto nel nome). In questo modo inibiscono la formazione di angiotensina II a partire dall’angiotensina I. Allo stesso modo, gli ARB – bloccanti del recettore dell’angiotensina – assicurano che l’angiotensina II non possa svolgere la sua azione bloccando il recettore a cui l’angiotensina II dovrebbe legarsi.
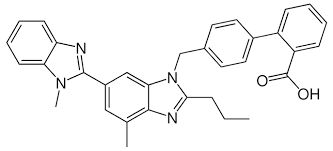
Sia gli ACE-inibitori che gli ARB hanno effetti simili sulla riduzione della pressione sanguigna. Le meta-analisi Cochrane hanno rilevato una riduzione della pressione arteriosa sistolica di 8 mmHg e della pressione arteriosa diastolica di 5 mmHg nel trattamento dell’ipertensione primaria in entrambi [23, 24]. La metà della dose massima giornaliera raccomandata dal produttore ha ottenuto un effetto di abbassamento della pressione sanguigna pari al 90% della dose massima nel caso degli ACE-inibitori e all’80% della dose massima nel caso degli ARB. Di conseguenza, l’aumento del dosaggio di questi farmaci di solito porta solo a riduzioni molto modeste della pressione arteriosa. Diventa quindi più interessante associarli a un diuretico tiazidico. Tuttavia, vorrei sottolineare una cosa: se avete bisogno di più farmaci per abbassare sufficientemente la pressione sanguigna, vi esorto a farlo sotto la supervisione di un medico.

Sebbene entrambi i farmaci siano abbastanza sicuri e ben tollerati in generale, come ogni farmaco possono avere effetti collaterali. Questi includono ipotensione/vertigini di prima dose, insufficienza renale acuta, iperkaliemia, tosse, eruzioni cutanee, disturbi del gusto (disgeusia), epatotossicità e angioedema per gli ACE-inibitori [25]. L’ipotensione da prima dose si riferisce all’improvviso calo della pressione sanguigna che può verificarsi nelle prime fasi del trattamento. Tenetene conto in caso di situazioni come la guida, ecc. Questo effetto è esacerbato quando si è disidratati (per la competizione o quando si utilizza un diuretico per qualsiasi motivo). Non utilizzare il farmaco in questi casi. L’insufficienza renale acuta si verifica in alcuni pazienti, ma di solito non comporta alcun segno clinico. Non è permanente: una volta smesso, la funzione renale torna normale. Anche in questo caso la disidratazione è un ulteriore fattore di rischio. A questo scopo, è necessario misurare la creatinina sierica nel tempo (o altri marcatori, forse più affidabili, utilizzati per stimare la velocità di filtrazione glomerulare [GFR]).
L’iperkaliemia (eccesso di potassio nel sangue) è abbastanza rara da sviluppare se questo è l’unico farmaco in uso, ma la combinazione con altri farmaci risparmiatori di potassio, un apporto molto elevato di potassio dalla dieta o un’insufficienza renale esistente possono aumentare il rischio. Per questo motivo, oltre alla misurazione della funzionalità renale, è necessario misurare anche gli elettroliti sierici. Ridurre il dosaggio (se possibile) se si è iperkaliemici, o passare a diuretici tiazidici.
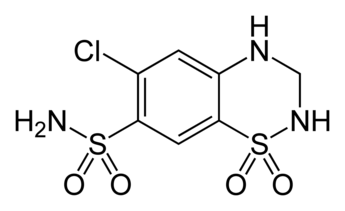
L’effetto collaterale più caratteristico degli ACE-inibitori è forse la tosse secca e irritante. Si verifica all’incirca in 1 persona su 10 [26], e sembra essere nettamente maggiore negli asiatici [27]. Occasionalmente, alcune persone sviluppano anche un’eruzione cutanea a causa degli ACE-inibitori [28]. Anche in questo caso, è necessario ridurre il dosaggio e talvolta il passaggio da un ACE-inibitore a un altro risolve il problema (in particolare il passaggio dal captopril). In alcuni casi molto rari, sembrano verificarsi colestasi, epatite colestatica o lesioni epatocellulari [29]. Ma in letteratura esistono solo alcuni limitati casi di questo tipo.
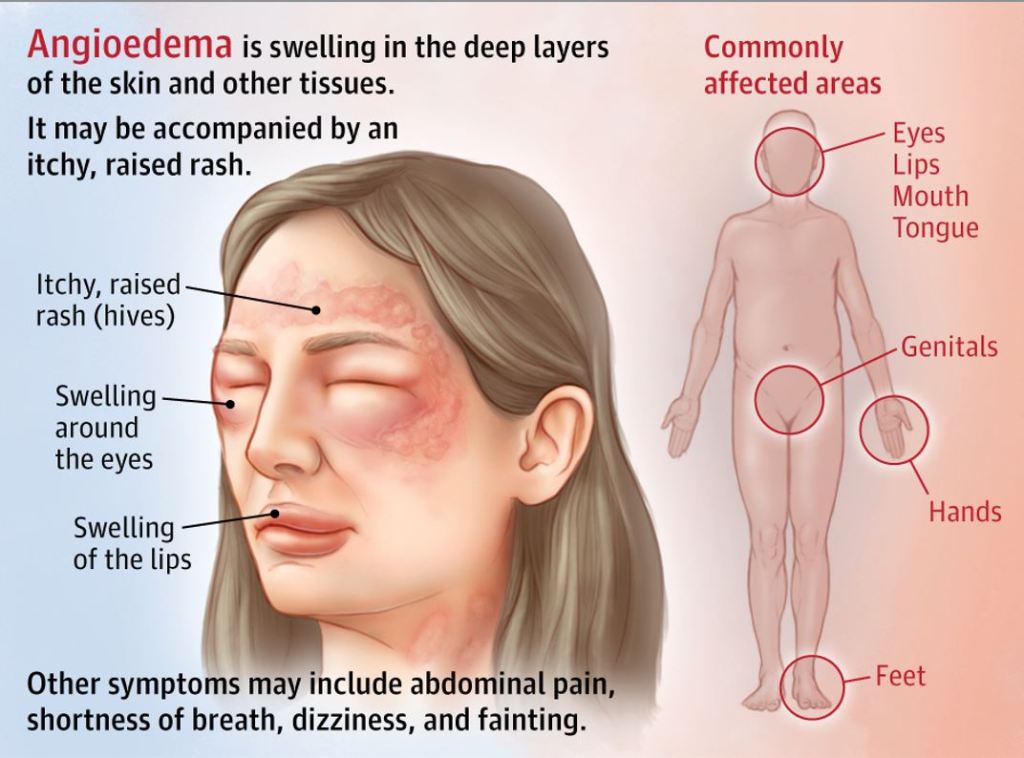
Un ultimo effetto collaterale che vorrei sottolineare è l’angioedema: l’accumulo di liquido sotto la pelle o le membrane mucose. Questo può includere il viso, la mucosa orale, la lingua, le labbra e anche la faringe e la laringe [30]. A seconda del luogo in cui si verifica, può causare una situazione di pericolo di vita bloccando le vie respiratorie. L’incidenza di questo fenomeno, tuttavia, è piuttosto bassa. Una meta-analisi ha rilevato un’incidenza dello 0,3% rispetto allo 0,07% del placebo [31]. Questo effetto collaterale non deve necessariamente verificarsi nelle fasi iniziali di utilizzo, ma può talvolta manifestarsi anche dopo anni di utilizzo. La raccomandazione è di interrompere completamente l’uso di qualsiasi ACE-inibitore quando si verifica l’angioedema e di non utilizzarlo mai più.
L’aspetto positivo degli ARB è che non causano tosse come gli ACE-inibitori [32] e non sembrano aumentare il rischio di angioedema [31]. Tuttavia, rimane l’aumento del rischio di ipotensione, iperkaliemia e disfunzione renale [32]. In ogni caso, il suo tasso di aderenza è superiore a quello di qualsiasi altra classe di farmaci antipertensivi [33]. Alla luce di ciò e del fatto che una recente meta-analisi non ha riscontrato differenze tra ACE-inibitori e ARB in termini di riduzione della pressione arteriosa, eventi fatali per qualsiasi causa e cause cardiovascolari, infarti miocardici fatali e non fatali e ictus [34], gli ARB sembrano la scelta più probabile tra i due, se disponibili.
Gli ACE-inibitori comunemente prescritti sono Benazepril, Captopril, Enalapril, Fosinopril, Lisinopril, Perindopril, Quinapril, Ramipril e Zofenopril. Gli ARB comunemente prescritti sono Candesartan, Eprosartan, Irbesartan, Losartan, Olmesartan, Telmisartan e Valsartan. Le revisioni Cochrane che ho citato in precedenza non hanno rilevato alcun ACE-inibitore con prestazioni migliori o peggiori rispetto agli altri, e lo stesso vale per gli ARB. Potrebbero esserci delle eccezioni in alcune popolazioni (ad esempio i diabetici), ma in generale si tratta di quello che si riesce a reperire. Gli ACE-inibitori generalmente più prescritti sono il Captopril, l’Enalapril e il Lisinopril, mentre gli ARB più prescritti sono il Valsartan, il Candesartan, il Telmisartan e il Losartan. In generale, la metà della dose massima raccomandata dal produttore è una buona dose iniziale. Eseguire un esame del sangue prima e un mese dopo l’inizio (o dopo un aumento della dose) e, se tutto risulta normale, ogni sei mesi. Includere creatinina, eGFR ed elettroliti. (Esistono molte linee guida sul monitoraggio, ma non c’è molto consenso al riguardo [35]). Tenete presente che sono necessarie circa 4-6 settimane per ottenere l’effetto completo del trattamento. L’ideale è raggiungere una pressione arteriosa inferiore o uguale a 130/80 mmHg (ma superiore a 120 mmHg).
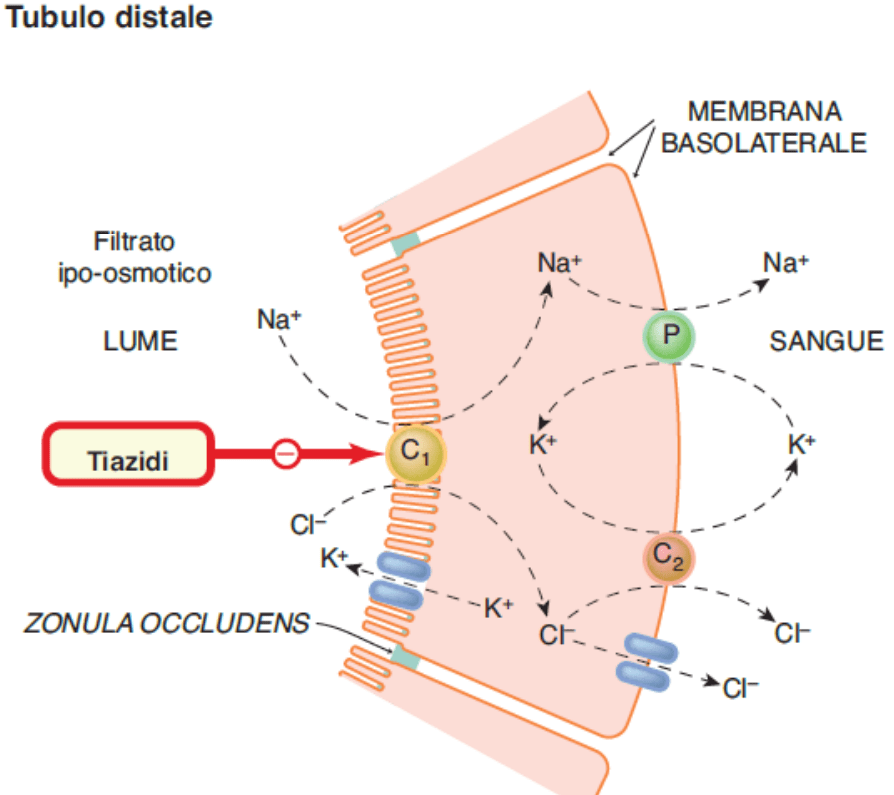
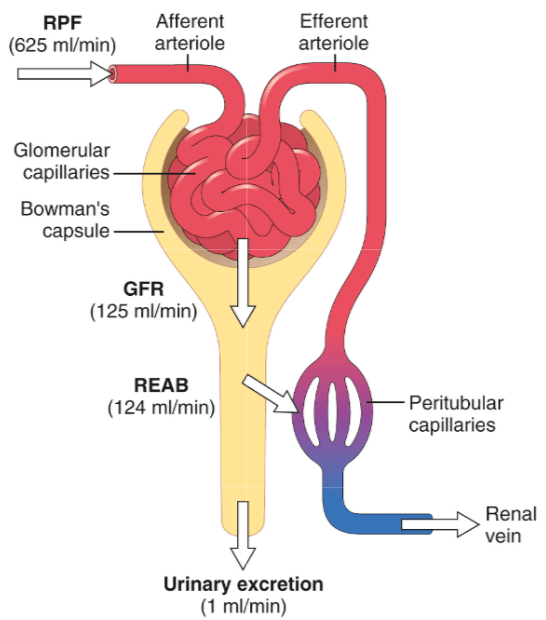
I diuretici tiazidici inibiscono l’azione dei simpatizzanti sodio-cloruro nel lume del tubulo distale dei nefroni. I nefroni sono gli elementi costitutivi dei reni, l’unità di base del funzionamento. Ognuno di essi (e i reni ne contengono diverse centinaia di migliaia) contribuisce in minima parte alla funzione di filtraggio cumulativa dei reni. Guardate l’immagine qui sotto per avere un’idea di come si presenta.
Il sangue viene filtrato attraverso un gruppo di capillari “specializzati” chiamati glomeruli e il filtrato viene poi catturato in un sacco simile a una tazza che lo circonda, chiamato capsula di Bowman. Il filtrato entra quindi nel tubulo renale per essere trasformato in urina. Durante questo percorso, varie sostanze vengono riassorbite dal filtrato nel sangue e secrete dal sangue nel filtrato.
I simpaticatori su cui agiscono i diuretici tiazidici trasportano il sodio e il cloruro fuori dal filtrato. Pertanto, bloccando questo simpatizzante, nel filtrato rimane più cloruro di sodio e quindi viene espulsa più acqua. In altre parole, i diuretici tiazidici devono la loro azione diuretica (escrezione di acqua) all’azione natriuretica (escrezione di sodio).
Come nel caso di qualsiasi diuretico che porti a un aumento delle concentrazioni di sodio nella parte distale del tubulo distale, si verifica una perdita di potassio. Questo accade perché il sodio viene assorbito in misura maggiore e finisce per essere scambiato con il potassio (che viene quindi secreto nel filtrato). Pertanto, contrariamente a quanto avviene con gli ARB e gli ACE-inibitori, può verificarsi un’ipopotassiemia (un livello troppo basso di potassio nel sangue). Se si sviluppa un’ipopotassiemia, di solito è lieve, ma evidenzia la necessità di analizzare gli elettroliti. Un’ipokaliemia lieve (3,0-3,5 mmol/L) raramente provoca sintomi. Quindi non la noterete. Tuttavia, se diventa più grave (<2,5-3,0 mmol/L), si possono sviluppare sintomi come debolezza generalizzata, affaticamento e costipazione [36]. Livelli di potassio molto bassi possono anche evocare aritmie cardiache. Nel contesto dei bodybuilder che fanno uso di PED, diversi altri composti possono contribuire allo sviluppo dell’ipokaliemia. Tra questi vi sono i beta-agonisti, giàcitati in precedenza, come il clenbuterolo, ma anche dosi elevate di insulina. Ciò può causare uno spostamento transitorio di potassio dal compartimento extracellulare a quello intracellulare. Sebbene tale spostamento duri solo un paio d’ore, l’effetto può essere molto drastico nel contesto di un’ipopotassiemia esistente. Inoltre, anche l’assunzione di caffeina può contribuire profondamente. Per questo motivo, è bene controllare anche gli integratori pre-allenamento che si utilizzano, poiché si può osservare una diminuzione di 0,26 mmol/L 4 ore dopo una dose di 180 mg di caffeina e un aumento ancora maggiore di 0,44 mmol/L dopo 360 mg [37]. In ogni caso, se i risultati del sangue mostrano una lieve ipokaliemia, è possibile aumentare l’apporto di potassio con la dieta.
Come con qualsiasi natriuretico, si può sviluppare iponatriemia (bassi livelli di sodio). Il rischio è basso, ma può essere, ovviamente, molto pericoloso [38, 39]. Bisogna fare attenzione a nausea, mal di testa, crampi muscolari, affaticamento, disturbi dell’andatura, vomito, sensazione di confusione e difficoltà di pensiero. Anche bere molta acqua può contribuire a sviluppare l’iponatriemia. Non bevete quindi di proposito litri e litri di acqua. In alcuni casi possono verificarsi anche ipomagnesiemia e ipercalcemia.
Misurare creatinina, eGFR ed elettroliti prima e una settimana dopo l’inizio del trattamento. Se tutto risulta normale, effettuare una seconda misurazione entro 4-8 settimane, dopodiché è possibile ripeterla ogni 6-12 mesi [40].
Utilizzare preferibilmente Clortalidone o Indapamide e iniziare con un dosaggio basso, ad esempio 12,5 mg di Clortalidone al giorno o 1,25 mg di Indapamide al giorno (anche se di solito sono disponibili in formato da 2,5 mg senza interruzione). Dopo che la seconda misurazione del sangue è risultata normale e la pressione arteriosa non è inferiore a 130/80 mmHg, si può prendere in considerazione un ulteriore aumento della dose (si tratta di raddoppiare la dose a 25 mg e 2,5 mg per Clortalidone e Indapamide, rispettivamente). Ripetere gli esami del sangue una settimana dopo l’aumento della dose.
Conclusioni:
La pressione alta è un killer silenzioso. La maggior parte delle persone non la sente, ma aumenta drasticamente la mortalità dovuta a malattie coronariche o ictus. Ogni aumento di 20 mmHg della pressione arteriosa sistolica oltre i 115 mmHg e ogni aumento di 10 mmHg della pressione arteriosa diastolica oltre i 75 mmHg è associato a un raddoppio della mortalità per queste cause. Inoltre, l’ipertensione per un periodo di tempo prolungato provoca danni agli organi bersaglio. Questo non riguarda solo il cuore e la vascolarizzazione, ma anche altri organi come il cervello, gli occhi e i reni. I dati della letteratura indicano che l’uso di steroidi anabolizzanti ad alti dosaggi può potenzialmente aumentare leggermente la pressione sanguigna. Una cifra indicativa è di circa 5-10 mmHg per la pressione arteriosa sistolica e la metà per quella diastolica. Naturalmente, le persone possono discostarsi da questi intervalli. Ciò può dipendere o meno dal dosaggio, dai tipi di AAS utilizzati e dall’uso concomitante di farmaci ausiliari che possono influire. Fortunatamente, questi aumenti sono transitori e reversibili dopo la cessazione dell’uso.
Ora sapete come misurare correttamente la pressione arteriosa, quando è necessario trattarla e come trattarla. Il trattamento va effettuato quando la pressione è superiore a 135/85mmHg, misurata a casa, e va effettuato con ARB, ACE-inibitori o diuretici tiazidici sotto stretto controllo medico. In generale, opterei per gli ARB, che di solito sono meglio tollerati. Come nota finale di questo articolo, vi invito ancora una volta a chiedere al vostro medico di curare la vostra pressione arteriosa piuttosto che farlo da soli.
Gabriel Bellizzi [CEO BioGenTech]
Riferimenti:
- Prospective Studies Collaboration. “Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies.” The Lancet 360.9349 (2002): 1903-1913.
- Cohuet, G., and H. Struijker-Boudier. “Mechanisms of target organ damage caused by hypertension: therapeutic potential.” Pharmacology & therapeutics 111.1 (2006): 81-98.
- Giorgi, Anthony, Robert P. Weatherby, and Peter W. Murphy. “Muscular strength, body composition and health responses to the use of testosterone enanthate: a double blind study.” Journal of science and medicine in sport 2.4 (1999): 341-355.
- Zmuda, Joseph M., et al. “The effect of testosterone aromatization on high-density lipoprotein cholesterol level and postheparin lipolytic activity.” Metabolism 42.4 (1993): 446-450.
- Freed, D. L., et al. “Anabolic steroids in athletics: crossover double-blind trial on weightlifters.” Br Med J 2.5969 (1975): 471-473.
- Glazer, Gary, and Anthony L. Suchman. “Lack of demonstrated effect of nandrolone on serum lipids.” Metabolism 43.2 (1994): 204-210.
- Hartgens, F., E. C. Cheriex, and H. Kuipers. “Prospective echocardiographic assessment of androgenic-anabolic steroids effects on cardiac structure and function in strength athletes.” International journal of sports medicine 24.05 (2003): 344-351.
- Kuipers, H., et al. “Influence of anabolic steroids on body composition, blood pressure, lipid profile and liver functions in body builders.” International journal of sports medicine 12.04 (1991): 413-418.
- Smit, Diederik L., et al. “Baseline characteristics of the HAARLEM study: 100 male amateur athletes using anabolic androgenic steroids.” Scandinavian journal of medicine & science in sports 30.3 (2020): 531-539.
- Hartgens, F., E. C. Cheriex, and H. Kuipers. “Prospective echocardiographic assessment of androgenic-anabolic steroids effects on cardiac structure and function in strength athletes.” International journal of sports medicine 24.05 (2003): 344-351.
- De Piccoli, B., et al. “Anabolic steroid use in body builders: an echocardiographic study of left ventricle morphology and function.” International journal of sports medicine 12.04 (1991): 408-412.
- Palatini, Paolo, et al. “Cardiovascular effects of anabolic steroids in weight‐trained subjects.” The Journal of Clinical Pharmacology 36.12 (1996): 1132-1140.
- Prospective Studies Collaboration. “Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies.” The Lancet 360.9349 (2002): 1903-1913.
- Fonseca-Reyes, Salvador, et al. “Differences and effects of medium and large adult cuffs on blood pressure readings in individuals with muscular arms.” Blood pressure monitoring 14.4 (2009): 166-171.
- Fonseca-Reyes, Salvador, et al. “Effect of standard cuff on blood pressure readings in patients with obese arms. How frequent are arms of a ‘large circumference’?.” Blood pressure monitoring 8.3 (2003): 101-106.
- Unger, Thomas, et al. “2020 International Society of Hypertension global hypertension practice guidelines.” Hypertension 75.6 (2020): 1334-1357.
- Parati, Gianfranco, et al. “European Society of Hypertension practice guidelines for home blood pressure monitoring.” Journal of human hypertension 24.12 (2010): 779-785.
- Brunström, Mattias, and Bo Carlberg. “Association of blood pressure lowering with mortality and cardiovascular disease across blood pressure levels: a systematic review and meta-analysis.” JAMA internal medicine 178.1 (2018): 28-36.
- Ettehad, Dena, et al. “Blood pressure lowering for prevention of cardiovascular disease and death: a systematic review and meta-analysis.” The Lancet 387.10022 (2016): 957-967.
- He, Feng J., and Graham A. MacGregor. “Salt, blood pressure and cardiovascular disease.” Current opinion in cardiology 22.4 (2007): 298-305.
- He, Feng J., Jiafu Li, and Graham A. MacGregor. “Effect of longer‐term modest salt reduction on blood pressure.” Cochrane database of systematic reviews 4 (2013).
- Mente, Andrew, et al. “Associations of urinary sodium excretion with cardiovascular events in individuals with and without hypertension: a pooled analysis of data from four studies.” The Lancet 388.10043 (2016): 465-475.
- Cushman, William C., et al. “Prevention and Treatment of Hypertension Study (PATHS): effects of an alcohol treatment program on blood pressure.” Archives of internal medicine 158.11 (1998): 1197-1207.
- Heran, Balraj S., et al. “Blood pressure lowering efficacy of angiotensin converting enzyme (ACE) inhibitors for primary hypertension.” Cochrane Database of Systematic Reviews 4 (2008).
- Heran, Balraj S., et al. “Blood pressure lowering efficacy of angiotensin receptor blockers for primary hypertension.” Cochrane Database of Systematic Reviews 4 (2008).
- Alderman, Christopher P. “Adverse effects of the angiotensin-converting enzyme inhibitors.” Annals of Pharmacotherapy 30.1 (1996): 55-61.
- Bangalore, Sripal, Sunil Kumar, and Franz H. Messerli. “Angiotensin-converting enzyme inhibitor associated cough: deceptive information from the Physicians’ Desk Reference.” The American journal of medicine 123.11 (2010): 1016-1030.
- Tseng, Daniel S., et al. “Angiotensin-converting enzyme-related cough among Chinese-Americans.” The American journal of medicine 123.2 (2010): 183-e11.
- Weber, Michael A. “Safety issues during antihypertensive treatment with angiotensin converting enzyme inhibitors.” The American journal of medicine 84 (1988): 16-23.
- Chitturi, Shivakumar, and Jacob George. “Hepatotoxicity of commonly used drugs: nonsteroidal anti-inflammatory drugs, antihypertensives, antidiabetic agents, anticonvulsants, lipid-lowering agents, psychotropic drugs.” Seminars in liver disease. Vol. 22. No. 02. Copyright© 2002 by Thieme Medical Publishers, Inc., 333 Seventh Avenue, New York, NY 10001, USA. Tel.:+ 1 (212) 584-4662, 2002.
- Campo, Paloma, et al. “Angioedema induced by angiotensin-converting enzyme inhibitors.” Current opinion in allergy and clinical immunology 13.4 (2013): 337-344.
- Makani, Harikrishna, et al. “Meta-analysis of randomized trials of angioedema as an adverse event of renin–angiotensin system inhibitors.” The American journal of cardiology 110.3 (2012): 383-391.
- Caldeira, Daniel, Cláudio David, and Cristina Sampaio. “Tolerability of angiotensin-receptor blockers in patients with intolerance to angiotensin-converting enzyme inhibitors.” American Journal of Cardiovascular Drugs 12.4 (2012): 263-277.
- Kronish, Ian M., et al. “Meta-analysis: impact of drug class on adherence to antihypertensives.” Circulation 123.15 (2011): 1611-1621.
- Dimou, Chrisa, et al. “A systematic review and network meta-analysis of the comparative efficacy of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers in hypertension.” Journal of human hypertension 33.3 (2019): 188-201.
- McDowell, Sarah E., et al. “A practical guide to monitoring for adverse drug reactions during antihypertensive drug therapy.” Journal of the Royal Society of Medicine 106.3 (2013): 87-119.
- Gennari, F. John. “Hypokalemia.” New England Journal of Medicine 339.7 (1998): 451-458.
- Passmore, A. P., G. B. Kondowe, and G. D. Johnston. “Caffeine and hypokalemia.” Annals of internal medicine 105.3 (1986): 468-468.
- Rodenburg, Eline M., et al. “Thiazide-associated hyponatremia: a population-based study.” American journal of kidney diseases 62.1 (2013): 67-72.
- Spital, Aaron. “Diuretic-induced hyponatremia.” American journal of nephrology 19.4 (1999): 447-452.