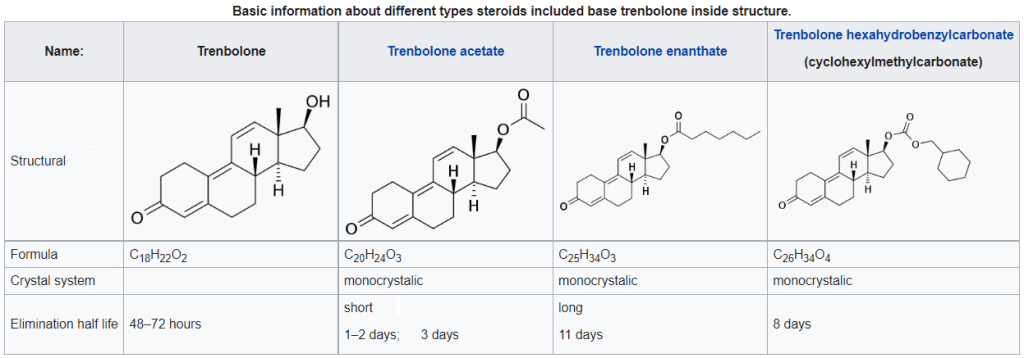
Introduzione:
Uno dei farmaci più incompresi e discussi nel BodyBuilding è sicuramente l’Insulina. Ciò è dovuto dal fatto che non esiste una vera scienza che funga da base per le modalità in cui i bodybuilder possano utilizzarla con criterio. Questo fa sì che tutte le conoscenze in possesso della maggior parte dei culturisti sull’uso dell’Insulina siano nulla più che “broscience”. Usando il termine “broscience” non intendo screditare una certa forma di conoscenza esperienziale. Infatti essa, se correttamente intesa nei suoi limiti, ha una certa importanza tanto che a volte capita che alcuni intuitivi atleti siano in grado di scoprire dettagli prima che questi vengano catalogati dalla letteratura scientifica e possono avere ragione anche quando la ricierca scientifica pecca nel design degli studi in cui vuole dimostrare una tesi (Holt 2009). Ma spesso e volentieri quello che i bodybuilder dicono sull’Insulina è una vera e propria stronzata. La pratica dell’uso di Insulina da parte dei bodybuilder si basa su un mucchio di studi mal intesi e su un mucchio di dicerie da guru che parlano di spiegazioni dal sapore pseudo-scientifico. Pochi di questi soggetti hanno una formazione scientifica o medica, per non parlare della competenza in endocrinologia. Alcuni di loro non hanno la minima idea di cosa stiano parlando, ma si comportano come se l’avessero. Come si fa a sapere a chi dare retta? Semplice! Conoscendo l’Insulina dalle basi alla pratica!
Ho quindi deciso, visto anche il centenario della sua scoperta, di scrivere una serie di articoli attraverso i quali vi accompagnerò lungo un secolo di storia dell’Insulina, dal suo isolamento alla sua applicazione medica passando, infine, al suo uso nel BodyBuilding.
In questa prima parte vedremo il lato accademico dell’Insulina…
Tanto tempo fa, tra due continenti…:
Nel 1869, studiando la struttura del pancreas al microscopio, Paul Langerhans, studente di medicina a Berlino, identificò alcuni ammassi di tessuto precedentemente inosservati, sparsi nella maggior parte del pancreas.[1] La funzione di questi “mucchietti di cellule”, in seguito noti come isolotti di Langerhans, rimase inizialmente sconosciuta, ma Édouard Laguesse suggerì in seguito che potessero produrre secrezioni che svolgono un ruolo regolatore nella digestione.[2] Anche il figlio di Paul Langerhans, Archibald, contribuì a comprendere questo ruolo regolatore.

Nel 1889, il medico Oskar Minkowski, in collaborazione con Joseph von Mering, rimosse il pancreas da un cane sano per verificare il suo presunto ruolo nella digestione. Analizzando l’urina, trovarono dello zucchero, stabilendo per la prima volta una relazione tra il pancreas e il diabete. Nel 1901, un altro passo importante fu compiuto dal medico e scienziato americano Eugene Lindsay Opie, quando isolò il ruolo del pancreas alle isole di Langerhans: “Il diabete mellito, quando è il risultato di una lesione del pancreas, è causato dalla distruzione delle isole di Langerhans e si verifica solo quando questi corpi sono in parte o completamente distrutti”.[3][4][5]

Nei due decenni successivi i ricercatori fecero diversi tentativi di isolare le secrezioni delle isole pancreatiche. Nel 1906 George Ludwig Zuelzer ottenne un parziale successo nel trattamento di cani con estratti pancreatici, ma non fu in grado di continuare il suo lavoro. Tra il 1911 e il 1912, E.L. Scott dell’Università di Chicago sperimentò estratti acquosi di pancreas e notò “una leggera diminuzione della glicosuria”, ma non riuscì a convincere il suo direttore del valore del suo lavoro, che venne interrotto. Israel Kleiner dimostrò effetti simili alla Rockefeller University nel 1915, ma la Prima Guerra Mondiale interruppe il suo lavoro e non lo riprese.[6]
Nel 1916, Nicolae Paulescu sviluppò un estratto acquoso di pancreas che, iniettato in un cane diabetico, aveva un effetto normalizzante sui livelli di zucchero nel sangue. Dovette interrompere i suoi esperimenti a causa della Prima Guerra Mondiale e nel 1921 scrisse quattro articoli sul suo lavoro svolto a Bucarest e sui suoi test su un cane diabetico. Più tardi, nello stesso anno, pubblicò “Research on the Role of the Pancreas in Food Assimilation”.[7][8]

Il nome “Insulin” fu coniato da Edward Albert Sharpey-Schafer nel 1916 per un’ipotetica molecola prodotta dalle isole pancreatiche di Langerhans (in latino insula per isolotto o isola) che controlla il metabolismo del glucosio. All’insaputa di Sharpey-Schafer, Jean de Meyer aveva introdotto il termine molto simile “Insulina” nel 1909 per la stessa molecola.[9][10]

Schafer coniò il termine “insulin” dopo aver teorizzato che l’assenza di una singola sostanza prodotta dal pancreas fosse responsabile del diabete mellito.
Nell’ottobre del 1920, il canadese Frederick Banting giunse alla conclusione che le secrezioni digestive studiate originariamente da Minkowski stavano disgregando il secreto delle isole, rendendone impossibile l’estrazione. Chirurgo di formazione, Banting sapeva che l’ostruzione del dotto pancreatico avrebbe portato all’atrofia della maggior parte del pancreas, lasciando intatte le isole di Langerhans. Pensò che si sarebbe potuto ricavare un estratto relativamente puro dalle isole una volta che la maggior parte del resto del pancreas fosse stata eliminata. Si appuntò una nota: “Legare i dotti pancreatici del cane. Mantenere i cani in vita finché gli acini non degenerano lasciando gli isolotti. Cercare di isolare la secrezione interna di questi ultimi e alleviare la glicosuria.”[11][12]

Nella primavera del 1921, Banting si recò a Toronto per spiegare la sua idea a J.J.R. Macleod, professore di fisiologia all’Università di Toronto. Macleod era inizialmente scettico, poiché Banting non aveva un background di ricerca e non conosceva la letteratura più recente, ma accettò di mettere a disposizione di Banting uno spazio di laboratorio per testare le sue idee. Macleod fece anche in modo che due studenti universitari fossero gli assistenti di laboratorio di Banting quell’estate, ma Banting aveva bisogno di un solo assistente di laboratorio. Charles Best e Clark Noble lanciarono una moneta; Best vinse il lancio e prese il primo turno. Ciò si rivelò sfortunato per Noble, poiché Banting tenne Best per tutta l’estate e alla fine divise con Best metà del premio Nobel e il merito della scoperta.[13] Il 30 luglio 1921, Banting e Best riuscirono a isolare con successo un estratto (“isleton”) dalle isole di un cane e lo iniettarono in un cane diabetico, scoprendo che l’estratto riduceva la glicemia del 40% in 1 ora.[14][12]

Banting e Best presentarono i loro risultati a Macleod al suo ritorno a Toronto nell’autunno del 1921, ma Macleod sottolineò i difetti del disegno sperimentale e suggerì di ripetere gli esperimenti con un maggior numero di cani e con attrezzature migliori. Trasferì Banting e Best in un laboratorio migliore e iniziò a pagare a Banting uno stipendio con le sue borse di ricerca. Alcune settimane dopo, anche la seconda serie di esperimenti fu un successo e Macleod contribuì a pubblicare i risultati privatamente a Toronto nel novembre dello stesso anno. Bloccato dal lungo compito di legare i cani ai condotti pancreatici e di aspettare diverse settimane per estrarre l’Insulina, Banting ebbe l’idea di estrarre l’Insulina dal pancreas di un vitello fetale, che non aveva ancora sviluppato le ghiandole digestive. A dicembre, riuscirono a estrarre l’insulina anche dal pancreas di una mucca adulta. Macleod interruppe tutte le altre ricerche nel suo laboratorio per concentrarsi sulla purificazione dell’Insulina. Invitò il biochimico James Collip ad aiutarlo in questo compito e il team si sentì pronto per un test clinico entro un mese.[12]

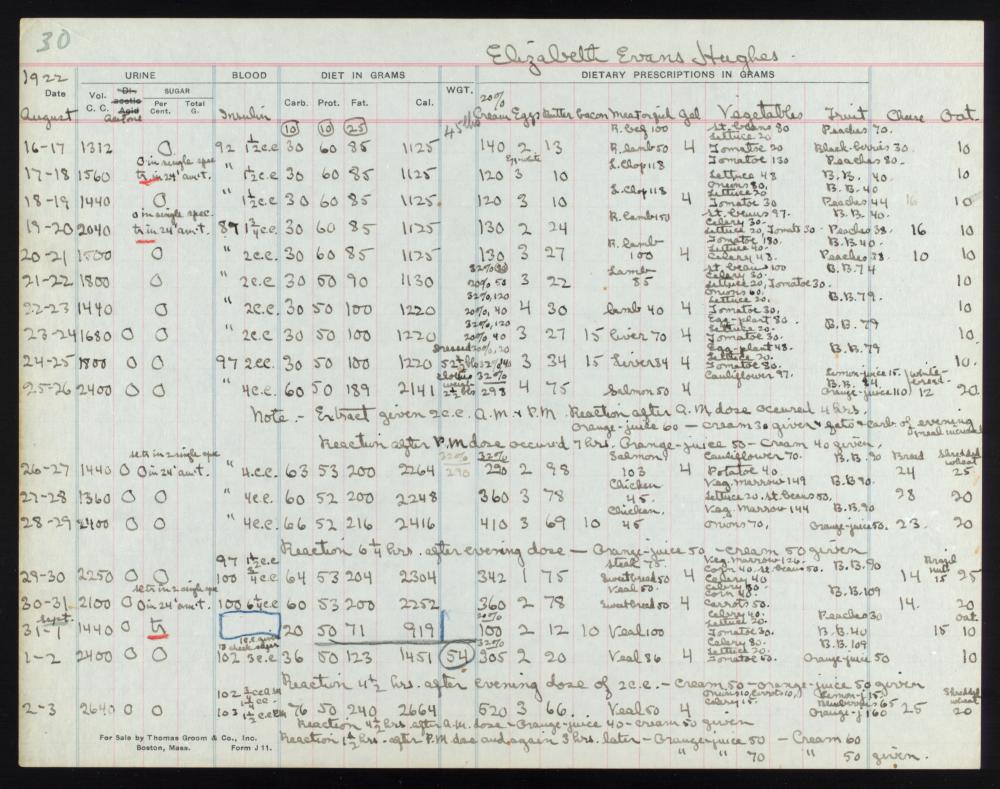
L’11 gennaio 1922, Leonard Thompson, un quattordicenne diabetico che giaceva in fin di vita al Toronto General Hospital, ricevette la prima iniezione di insulina.[15][16][17][18] Tuttavia, l’estratto era così impuro che Thompson ebbe una grave reazione allergica e le ulteriori iniezioni furono annullate. Nei 12 giorni successivi, Collip lavorò giorno e notte per migliorare l’estratto di pancreas di bue. Una seconda dose fu iniettata il 23 gennaio, eliminando la glicosuria tipica del diabete senza causare effetti collaterali evidenti. La prima paziente americana fu Elizabeth Hughes, figlia del Segretario di Stato americano Charles Evans Hughes.[19][20] Il primo paziente trattato negli Stati Uniti fu il futuro artista di xilografie James D. Havens;[21] il dottor John Ralston Williams importò l’Insulina da Toronto a Rochester, New York, per trattare Havens.[22]

Banting e Best non lavorarono mai bene con Collip, considerandolo una specie di intruso, e Collip lasciò il progetto poco dopo. Nella primavera del 1922, Best riuscì a migliorare le sue tecniche al punto da poter estrarre grandi quantità di Insulina su richiesta, ma la preparazione rimase impura. L’azienda farmaceutica Eli Lilly and Company aveva offerto assistenza non molto tempo dopo le prime pubblicazioni del 1921, e in aprile accettò l’offerta della Lilly. A novembre, il capo chimico della Lilly, George B. Walden, scoprì la precipitazione isoelettrica e fu in grado di produrre grandi quantità di Insulina altamente purificata. Poco dopo, l’Insulina fu messa in vendita al pubblico.
Verso la fine del gennaio 1922, le tensioni tra i quattro “co-scopritori” dell’insulina aumentarono e Collip minacciò brevemente di brevettare separatamente il suo processo di purificazione. John G. FitzGerald, direttore dell’istituzione sanitaria pubblica non commerciale Connaught Laboratories, intervenne quindi come paciere. L’accordo del 25 gennaio 1922 stabilì due condizioni fondamentali: 1) i collaboratori avrebbero firmato un contratto in cui si impegnavano a non sottoscrivere un brevetto con un’azienda farmaceutica commerciale durante un periodo iniziale di lavoro con Connaught; e 2) non sarebbero stati permessi cambiamenti nella politica di ricerca se non prima discussi tra FitzGerald e i quattro collaboratori.[23] Ciò contribuì a contenere il disaccordo e a vincolare la ricerca al mandato pubblico di Connaught.

Inizialmente, Macleod e Banting erano particolarmente riluttanti a brevettare il loro processo per l’Insulina per motivi di etica medica. Tuttavia, rimaneva il timore che un terzo privato potesse dirottare e monopolizzare la ricerca (come aveva lasciato intendere Eli Lilly and Company[24]) e che sarebbe stato difficile garantire una distribuzione sicura senza una capacità di controllo della qualità. A tal fine, Edward Calvin Kendall fornì preziosi consigli. Egli aveva isolato la Tiroxina presso la Mayo Clinic nel 1914 e aveva brevettato il processo attraverso un accordo tra lui, i fratelli Mayo e l’Università del Minnesota, trasferendo il brevetto all’università pubblica.[25] Il 12 aprile, Banting, Best, Collip, Macleod e FitzGerald scrissero congiuntamente al presidente dell’Università di Toronto per proporre un accordo simile con l’obiettivo di assegnare un brevetto al Board of Governors dell’università.[26] La lettera sottolineava che:[27]
Il brevetto non sarebbe stato utilizzato per nessun altro scopo se non quello di impedire il conseguimento di un brevetto da parte di altre persone. Quando i dettagli del metodo di preparazione saranno pubblicati, chiunque sarà libero di preparare l’estratto, ma nessuno potrà assicurarsi un monopolio redditizio.

La cessione al Consiglio superiore dell’Università di Toronto fu completata il 15 gennaio 1923, con il pagamento simbolico di 1 dollaro.[28] L’accordo è stato giudicato da The World’s Work del 1923 come “un passo avanti nell’etica medica”.[29] Ha ricevuto molta attenzione da parte dei media anche negli anni 2010 per quanto riguarda la questione dell’assistenza sanitaria e dell’accessibilità dei farmaci.
A seguito di ulteriori preoccupazioni riguardanti i tentativi di Eli Lilly di brevettare separatamente parti del processo di produzione, il vicedirettore di Connaught e capo della divisione Insulina Robert Defries ha stabilito una politica di pooling dei brevetti che avrebbe richiesto ai produttori di condividere liberamente qualsiasi miglioramento del processo di produzione senza compromettere l’accessibilità dei farmaci.[30]
Nel 1923 il comitato del Premio Nobel attribuì l’estrazione pratica dell’Insulina a un team dell’Università di Toronto e assegnò il Premio Nobel a due uomini: Frederick Banting e J.J.R. Macleod.[31] Essi ricevettero il Premio Nobel per la Fisiologia o la Medicina nel 1923 per la scoperta dell’Insulina. Banting, incredulo per la mancata menzione di Best,[32] condivise il premio con lui, mentre Macleod condivise immediatamente il suo con James Collip. Il brevetto dell’Insulina fu venduto all’Università di Toronto per un dollaro.
Altri due premi Nobel sono stati assegnati per lavori sull’Insulina. Il biologo molecolare britannico Frederick Sanger, che nel 1955 determinò la struttura primaria dell’Insulina, ricevette il Premio Nobel per la Chimica nel 1958.[33] Rosalyn Sussman Yalow ricevette il Premio Nobel per la Medicina nel 1977 per lo sviluppo del test radioimmunologico dell’Insulina.
Diversi premi Nobel hanno anche un legame indiretto con l’Insulina. George Minot, co-ricevente del Premio Nobel 1934 per lo sviluppo del primo trattamento efficace per l’anemia perniciosa, era affetto da diabete mellito di tipo I. Il dottor William Castle ha osservato che la scoperta dell’Insulina nel 1921, arrivata in tempo per mantenere in vita Minot, era quindi anche responsabile della scoperta di una cura per l’anemia perniciosa.[34] Dorothy Hodgkin ha ricevuto il Premio Nobel per la Chimica nel 1964 per lo sviluppo della cristallografia, la tecnica che ha utilizzato per decifrare la struttura molecolare completa dell’Insulina nel 1969.[35]

Il lavoro pubblicato da Banting, Best, Collip e Macleod rappresentava la preparazione di un estratto purificato di Insulina adatto all’uso su pazienti umani.[36] Sebbene Paulescu avesse scoperto i principi del trattamento, il suo estratto salino non poteva essere usato sugli esseri umani; non fu menzionato nel Premio Nobel del 1923. Il professor Ian Murray fu particolarmente attivo nel lavorare per correggere “l’errore storico” contro Nicolae Paulescu. Murray era professore di fisiologia presso l’Anderson College of Medicine di Glasgow, in Scozia, capo del dipartimento di Malattie Metaboliche di un importante ospedale di Glasgow, vicepresidente della British Association of Diabetes e membro fondatore della International Diabetes Federation. Murray ha scritto:
Non è stato dato sufficiente riconoscimento a Paulescu, l’illustre scienziato rumeno, che all’epoca in cui l’équipe di Toronto stava iniziando le sue ricerche era già riuscito a estrarre l’ormone antidiabetico del pancreas e a dimostrarne l’efficacia nel ridurre l’iperglicemia nei cani diabetici.[37]
In una comunicazione privata, il professor Arne Tiselius, ex capo dell’Istituto Nobel, espresse la sua personale opinione che Paulescu fosse ugualmente degno del premio nel 1923.[38]
Analisi strutturale e sintesi di laboratorio:
L’Insulina purificata di origine animale era inizialmente l’unico tipo di Insulina disponibile per gli esperimenti e i diabetici. John Jacob Abel fu il primo a produrre la forma cristallizzata nel 1926.[39] La prova della natura proteica fu fornita per la prima volta da Michael Somogyi, Edward A. Doisy e Philip A. Shaffer nel 1924.[40] Fu pienamente dimostrata quando Hans Jensen e Earl A. Evans Jr. isolarono gli aminoacidi fenilalanina e prolina nel 1935.[41]

La struttura aminoacidica dell’Insulina fu caratterizzata per la prima volta nel 1951 da Frederick Sanger,[42] e la prima Insulina sintetica fu prodotta simultaneamente nei laboratori di Panayotis Katsoyannis dell’Università di Pittsburgh e di Helmut Zahn dell’Università RWTH di Aquisgrana a metà degli anni Sessanta. [43][44][45][46][47] L’Insulina bovina cristallina sintetica è stata ottenuta da ricercatori cinesi nel 1965.[48] La struttura tridimensionale completa dell’Insulina è stata determinata mediante cristallografia a raggi X nel laboratorio di Dorothy Hodgkin nel 1969.[49]

Il dottor Hans E. Weber scoprì la preproinsulina mentre lavorava come ricercatore presso l’Università della California Los Angeles nel 1974. Nel 1973-1974, Weber imparò le tecniche per isolare, purificare e tradurre l’RNA messaggero. Per studiare ulteriormente l’Insulina, ottenne tessuti pancreatici da un macello di Los Angeles e successivamente da animali dell’UCLA. Isolò e purificò l’RNA messaggero totale dalle cellule dell’isoletta pancreatica, che fu poi tradotto in oociti di Xenopus laevis e precipitato usando anticorpi anti-insulina. Quando la proteina totale tradotta è stata sottoposta a elettroforesi su gel di SDS-poliacrilammide e gradiente di saccarosio, sono stati isolati i picchi corrispondenti all’Insulina e alla proinsulina. Tuttavia, con sorpresa del Dr. Weber, è stato isolato un terzo picco corrispondente a una molecola più grande della proinsulina. Dopo aver riprodotto l’esperimento diverse volte, ha notato costantemente questo grande picco prima della proinsulina, che ha stabilito essere una molecola precursore più grande a monte della proinsulina. Nel maggio 1975, in occasione del meeting dell’American Diabetes Association a New York, Weber presentò oralmente il suo lavoro[50-146] e fu il primo a chiamare questa molecola precursore “preproinsulina”. In seguito a questa presentazione orale, Weber fu invitato a cena dal dottor Donald Steiner, un ricercatore che aveva contribuito alla caratterizzazione della proinsulina, per discutere del suo lavoro e delle sue scoperte. Un anno dopo, nell’aprile 1976, questa molecola fu ulteriormente caratterizzata e sequenziata da Steiner, facendo riferimento al lavoro e alla scoperta di Hans Weber.[51] La preproinsulina divenne una molecola importante per studiare il processo di trascrizione e traduzione.
La prima Insulina “umana” geneticamente ingegnerizzata e sintetica è stata prodotta con l’E. coli nel 1978 da Arthur Riggs e Keiichi Itakura presso il Beckman Research Institute della Città della Speranza in collaborazione con Herbert Boyer della Genentech.[52][53] La Genentech, fondata da Swanson, Boyer e Eli Lilly and Company, ha continuato nel 1982 a vendere la prima Insulina umana biosintetica disponibile in commercio con il marchio Humulin [La stragrande maggioranza dell’Insulina utilizzata in tutto il mondo è Insulina “umana” biosintetica o suoi analoghi].[54] Recentemente, un altro approccio è stato utilizzato da un gruppo pionieristico di ricercatori canadesi, che ha utilizzato una pianta di cartamo facilmente coltivabile, per la produzione di Insulina molto più economica.[55]

L’Insulina ricombinante viene prodotta nel lievito (di solito Saccharomyces cerevisiae) o in E. coli.[56] Nel lievito, l’Insulina può essere ingegnerizzata come una proteina a catena singola con un sito di endoproteasi KexII (un omologo del PCI/PCII del lievito) che separa la catena A dell’Insulina da una catena B dell’Insulina troncata C-terminalmente. Una coda C-terminale sintetizzata chimicamente viene quindi innestata sull’Insulina mediante proteolisi inversa utilizzando la proteasi tripsina, poco costosa; in genere la lisina sulla coda C-terminale è protetta con un gruppo protettivo chimico per impedire la proteolisi. La facilità della sintesi modulare e la relativa sicurezza delle modifiche in quella regione spiega i comuni analoghi dell’Insulina con modifiche C-terminali (ad esempio lispro, aspart, glulisine). La sintesi Genentech e le sintesi completamente chimiche come quella di Bruce Merrifield non sono preferibili perché l’efficienza della ricombinazione delle due catene di Insulina è bassa, soprattutto a causa della competizione con la precipitazione della catena B dell’Insulina.
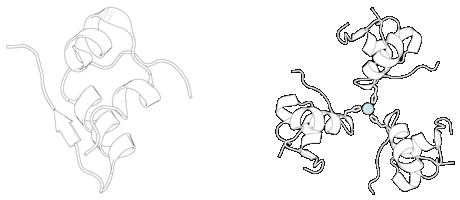
Caratteristiche dell’Insulina:
Grazie ad annali ricerche oggi sappiamo che l’Insulina è un ormone peptidico prodotto dalle cellule beta delle isole pancreatiche, codificato nell’uomo dal gene INS. È considerato il principale ormone anabolico dell’organismo sebbene la sua attività prevalente sia diretta alla riduzione del catabolismo.[57] Regola il metabolismo dei carboidrati, dei grassi e delle proteine promuovendo l’assorbimento del glucosio dal sangue nelle cellule epatiche, lipidiche e del muscolo-scheletrico [In questi tessuti il glucosio assorbito viene convertito in glicogeno attraverso la glicogenesi o in alcuni casi in grassi (trigliceridi) attraverso la lipogenesi o, nel caso del fegato, in entrambi].[58] La produzione e la secrezione di glucosio da parte del fegato sono fortemente inibite da alte concentrazioni di Insulina nel sangue.[59] L’Insulina circolante influisce anche sulla sintesi di proteine in un’ampia varietà di tessuti. È quindi un ormone anabolico, che promuove la conversione di piccole molecole nel sangue in grandi molecole all’interno delle cellule. Bassi livelli di Insulina nel sangue hanno l’effetto opposto, favorendo un diffuso catabolismo, soprattutto del grasso corporeo di riserva.
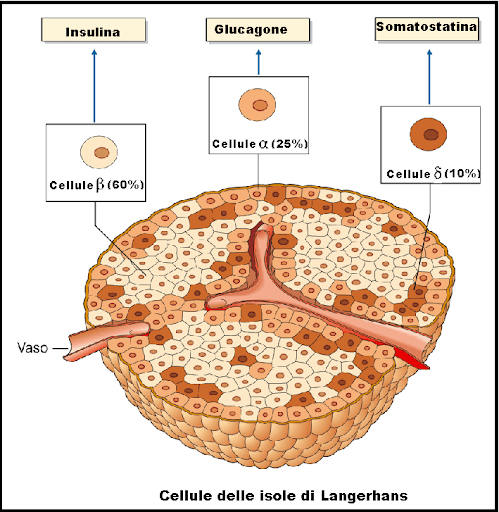
Le cellule beta sono sensibili ai livelli della glicemia nel sangue, per cui secernono Insulina nel sangue in risposta a livelli elevati di glucosio e inibiscono la secrezione di Insulina quando i livelli di glucosio sono bassi.[60] L’Insulina aumenta l’assorbimento e il metabolismo del glucosio nelle cellule, riducendo così il livello della glicemia ematica. Le cellule alfa vicine, prendendo spunto dalle cellule beta,[60] secernono Glucagone nel sangue in modo opposto: aumento della secrezione quando il glucosio nel sangue è basso e diminuzione della secrezione quando le concentrazioni di glucosio sono elevate. Il Glucagone aumenta il livello di glucosio nel sangue stimolando la glicogenolisi e la gluconeogenesi nel fegato.[58][60] La secrezione di Insulina e Glucagone nel sangue in risposta alla concentrazione di glucosio nel sangue è il meccanismo principale dell’omeostasi del glucosio.[60]
L’insulina è quindi prodotta esclusivamente nelle cellule beta delle isole pancreatiche nei mammiferi e nel corpo di Brockmann in alcuni pesci. L’Insulina umana è prodotta dal gene INS, situato sul cromosoma 11.[61] I roditori hanno due geni funzionali dell’Insulina: uno è l’omologo della maggior parte dei geni dei mammiferi (Ins2) e l’altro è una copia retroposta che include la sequenza del promotore ma che manca di un introne (Ins1) [La trascrizione del gene dell’Insulina aumenta in risposta all’aumento del glucosio nel sangue].[62] Ciò è controllato principalmente da fattori di trascrizione che legano sequenze enhancer nelle circa 400 paia di basi prima del sito di inizio della trascrizione del gene.[61][62]
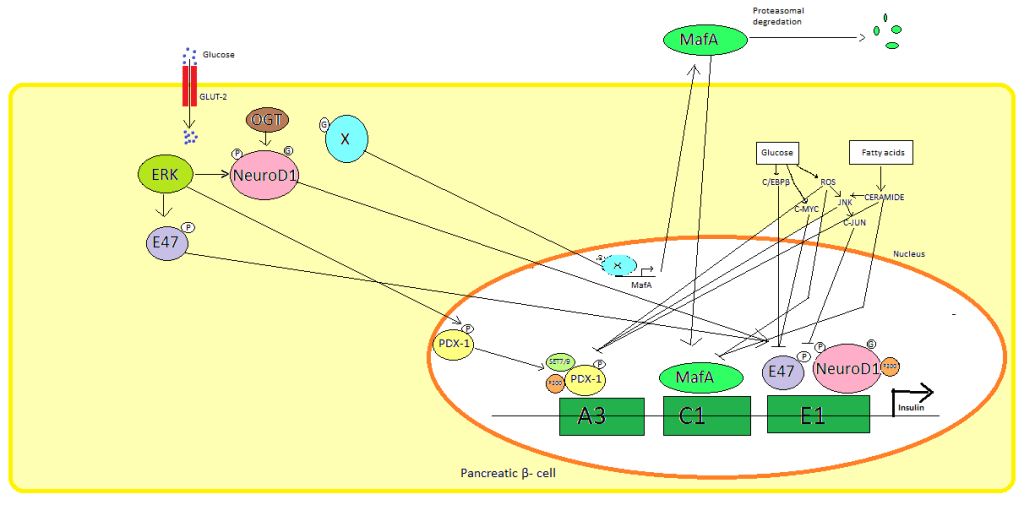
I principali fattori di trascrizione che influenzano la secrezione insulinica sono PDX1, NeuroD1 e MafA.[63][64][65][66]
In uno stato di basso livello di glucosio, PDX1 (pancreatic and duodenal homeobox protein 1) si trova nella periferia nucleare in seguito all’interazione con HDAC1 e 2,[67] il che determina una sottoregolazione della secrezione insulinica.[68] Un aumento dei livelli di glucosio nel sangue provoca la fosforilazione di PDX1, che subisce una traslocazione nucleare e si lega all’elemento A3 all’interno del promotore dell’Insulina.[69] Dopo la traslocazione interagisce con i coattivatori HAT p300 e SETD7. PDX1 influisce sulle modificazioni degli istoni attraverso l’acetilazione, la deacetilazione e la metilazione. Si dice anche che sopprima il glucagone.[70]
NeuroD1, noto anche come β2, regola l’esocitosi dell’Insulina nelle cellule β pancreatiche inducendo direttamente l’espressione di geni coinvolti nell’esocitosi.[71] È localizzato nel citosol, ma in risposta all’elevato livello di glucosio viene glicosilato da OGT e/o fosforilato da ERK, il che provoca la traslocazione nel nucleo. Nel nucleo β2 eterodimerizza con E47, si lega all’elemento E1 del promotore dell’insulina e recluta il co-attivatore p300 che acetilerà β2. È in grado di interagire anche con altri fattori di trascrizione nell’attivazione del gene dell’Insulina.[71]
MafA viene degradato dai proteasomi quando i livelli di glucosio nel sangue sono bassi. L’aumento dei livelli di glucosio rende glicosilata una proteina sconosciuta. Questa proteina funziona come fattore di trascrizione per MafA in modo sconosciuto e MafA viene trasportata fuori dalla cellula. MafA viene poi traslocata di nuovo nel nucleo dove lega l’elemento C1 del promotore dell’insulina.[72][73]
Questi fattori di trascrizione lavorano in modo sinergico e complesso. L’aumento del glucosio nel sangue può, dopo un po’, distruggere le capacità di legame di queste proteine e quindi ridurre la quantità di Insulina secreta, causando il diabete. La diminuzione delle attività di legame può essere mediata dallo stress ossidativo indotto dal glucosio e si ritiene che gli antiossidanti prevengano la diminuzione della secrezione di Insulina nelle cellule β pancreatiche glucotossiche. Le molecole di segnalazione dello stress e le specie reattive dell’ossigeno inibiscono il gene dell’Insulina interferendo con i cofattori che legano i fattori di trascrizione e con i fattori di trascrizione stessi.[74]
Diverse sequenze regolatrici nella regione del promotore del gene dell’Insulina umana si legano ai fattori di trascrizione. In generale, le A-box si legano ai fattori Pdx1, le E-box a NeuroD, le C-box a MafA e gli elementi di risposta al cAMP a CREB. Esistono anche dei silenziatori che inibiscono la trascrizione.
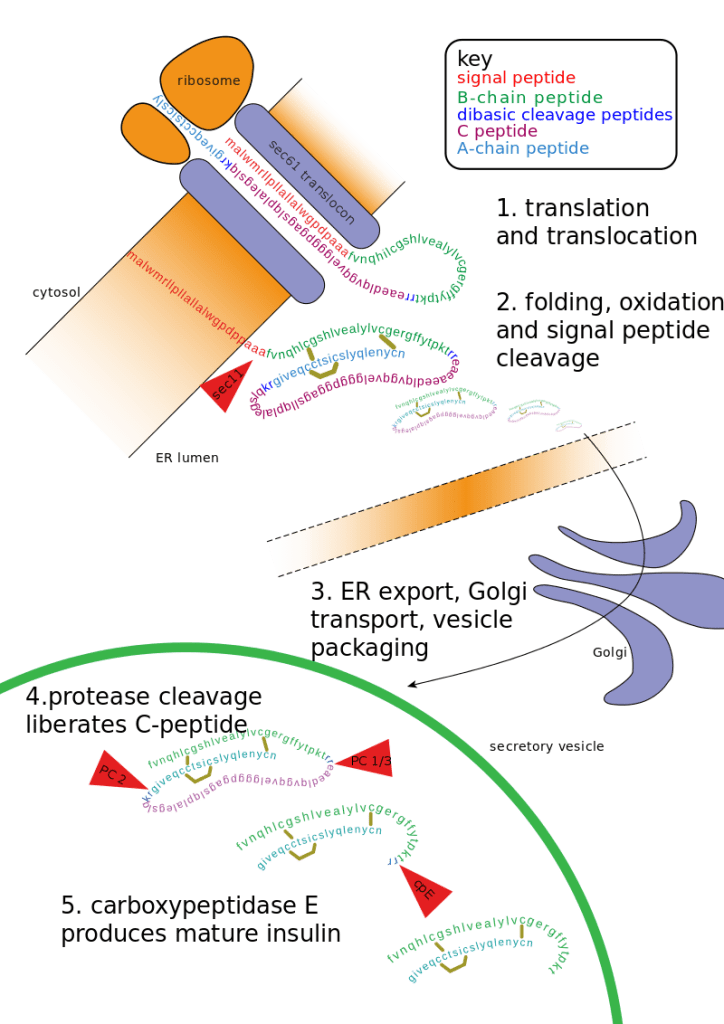
L’insulina viene sintetizzata come molecola precursore inattiva, una proteina di 110 aminoacidi chiamata “preproinsulina”. La preproinsulina viene tradotta direttamente nel reticolo endoplasmatico ruvido (RER), dove il suo peptide segnale viene rimosso dalla peptidasi segnale per formare la “proinsulina”.[60] Durante il ripiegamento della proinsulina, le estremità opposte della proteina, chiamate “catena A” e “catena B”, vengono fuse insieme con tre legami disolfuro.[60] La proinsulina ripiegata passa quindi attraverso l’apparato di Golgi e viene impacchettata in vescicole secretorie specializzate [Nel granulo, la proinsulina viene scissa dalla proproteina convertasi 1/3 e dalla proproteina convertasi 2, rimuovendo la parte centrale della proteina, chiamata “peptide C”].[60] Infine, la carbossipeptidasi E rimuove due coppie di aminoacidi dalle estremità della proteina, dando origine all’Insulina attiva – le catene A e B dell’insulina, ora collegate da due legami disolfuro.[60]
L’Insulina matura risultante è impacchettata all’interno di granuli maturi in attesa di segnali metabolici (come leucina, arginina, glucosio e mannosio) e della stimolazione del nervo vagale per essere esocitata dalla cellula nella circolazione.[75]
È stato dimostrato che l’Insulina e le proteine ad essa correlate sono prodotte all’interno del cervello e che livelli ridotti di queste proteine sono collegati alla malattia di Alzheimer.[76][77][78]
Il rilascio di Insulina è stimolato anche dalla stimolazione del recettore beta-2 e inibito dalla stimolazione del recettore alfa-1. Inoltre, il Cortisolo, il Glucagone e l’Ormone della Crescita antagonizzano le azioni dell’Insulina nei periodi di stress. L’Insulina inibisce anche il rilascio di acidi grassi da parte della lipasi ormonosensibile nel tessuto adiposo.[79]

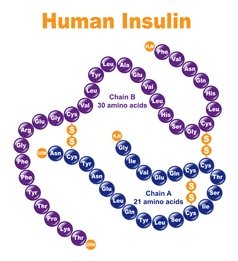
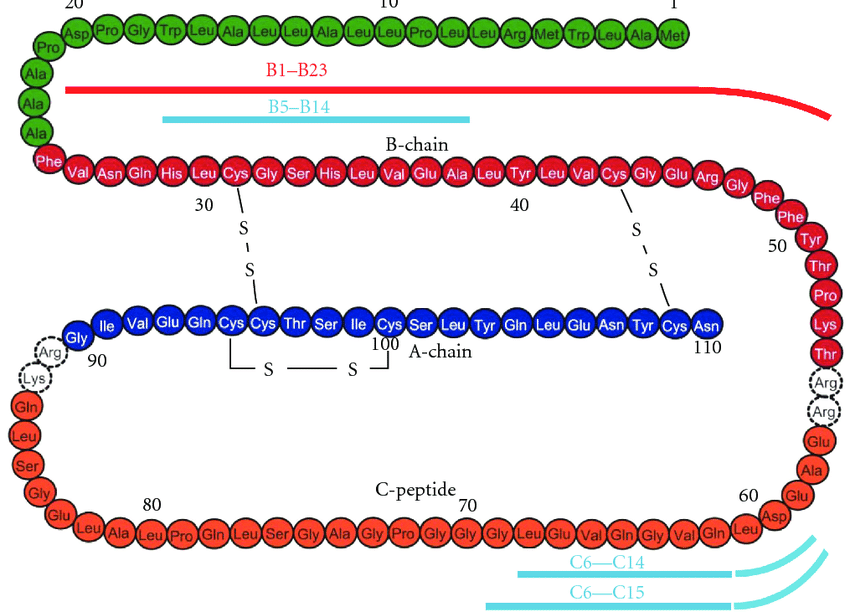
Contrariamente alla convinzione iniziale che gli ormoni fossero generalmente molecole chimiche di piccole dimensioni, l’Insulina, primo ormone peptidico di cui si conosce la struttura, si è rivelata piuttosto grande.[80] Una singola proteina (monomero) di Insulina umana è composta da 51 aminoacidi e ha una massa molecolare di 5808 Da. La formula molecolare dell’Insulina umana è C257H383N65O77S6.[81-44] Si tratta di una combinazione di due catene peptidiche (dimeri) denominate catena A e catena B, legate tra loro da due legami disolfuro. La catena A è composta da 21 aminoacidi, mentre la catena B è composta da 30 residui. I legami disolfuro di collegamento (intercatena) si formano sui residui di cisteina tra le posizioni A7-B7 e A20-B19. Esiste un ulteriore legame disolfuro (intracatena) all’interno della catena A tra i residui di cisteina nelle posizioni A6 e A11. La catena A presenta due regioni α-eliche in corrispondenza di A1-A8 e A12-A19 che sono antiparallele; mentre la catena B presenta un’α-elica centrale (che copre i residui B9-B19) affiancata dal legame disolfuro su entrambi i lati e da due foglietti β (che coprono B7-B10 e B20-B23).[80][82-45]
La sequenza aminoacidica dell’insulina è fortemente conservata e varia solo leggermente tra le specie. L’insulina bovina differisce da quella umana solo per tre residui aminoacidici e quella suina per uno. Anche l’insulina di alcune specie di pesci è abbastanza simile a quella umana da essere clinicamente efficace nell’uomo. L’insulina di alcuni invertebrati ha una sequenza molto simile a quella dell’insulina umana e ha effetti fisiologici simili. Il C-peptide della proinsulina, tuttavia, differisce molto di più tra le specie; è anch’esso un ormone, ma secondario.[82]
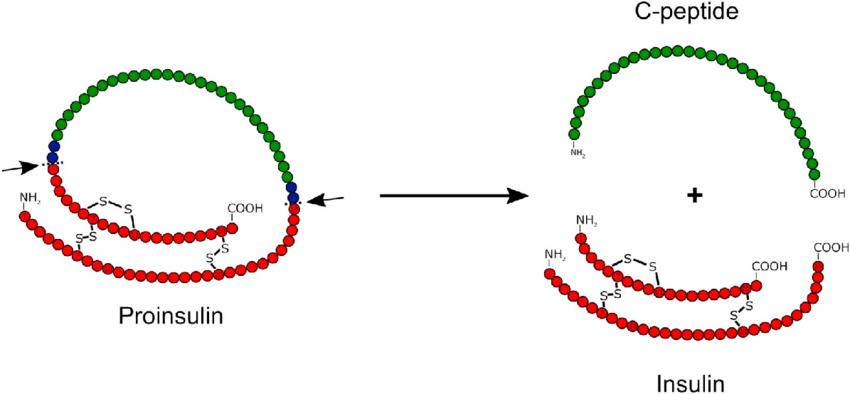
L’Insulina viene prodotta e immagazzinata nell’organismo sotto forma di esamero (un’unità di sei molecole di insulina), mentre la forma attiva è il monomero. L’esamero ha una dimensione di circa 36000 Da. Le sei molecole sono legate insieme come tre unità dimeriche per formare una molecola simmetrica. Una caratteristica importante è la presenza di atomi di zinco (Zn2+) sull’asse di simmetria, che sono circondati da tre molecole d’acqua e da tre residui di istidina in posizione B10.[68][82]
L’esamero è una forma inattiva con stabilità a lungo termine, che serve a mantenere l’insulina altamente reattiva protetta, ma prontamente disponibile. La conversione esamero-monomero è uno degli aspetti centrali delle formulazioni di insulina per iniezione. L’esamero è molto più stabile del monomero, il che è auspicabile per motivi pratici; tuttavia, il monomero è un farmaco che reagisce molto più rapidamente, poiché la velocità di diffusione è inversamente correlata alla dimensione delle particelle. Un farmaco a reazione rapida significa che le iniezioni di insulina non devono precedere di ore i pasti, il che a sua volta offre alle persone con diabete una maggiore flessibilità negli orari giornalieri.[83] L’Insulina può aggregarsi e formare foglietti beta fibrillari interdigitati. Ciò può causare amiloidosi da iniezione e impedisce la conservazione dell’insulina per lunghi periodi.[84]
Le cellule beta delle isole di Langerhans rilasciano insulina in due fasi. Il rilascio della prima fase avviene rapidamente in risposta all’aumento dei livelli di glucosio nel sangue e dura circa 10 minuti. La seconda fase è un rilascio lento e prolungato di vescicole di nuova formazione, innescato indipendentemente dallo zucchero, che raggiunge il suo picco tra le 2 e le 3 ore. Le due fasi del rilascio di insulina suggeriscono che i granuli di insulina sono presenti in diverse popolazioni dichiarate o “pool”. Durante la prima fase dell’esocitosi dell’insulina, la maggior parte dei granuli predisposti all’esocitosi viene rilasciata dopo l’internalizzazione del calcio. Questo pool è noto come Readily Releasable Pool (RRP). I granuli RRP rappresentano lo 0,3-0,7% della popolazione totale di granuli contenenti insulina e si trovano immediatamente adiacenti alla membrana plasmatica. Durante la seconda fase dell’esocitosi, i granuli di insulina richiedono la mobilizzazione dei granuli verso la membrana plasmatica e una precedente preparazione per essere rilasciati.[85] Pertanto, la seconda fase del rilascio di insulina è regolata dalla velocità con cui i granuli si preparano al rilascio. Questo pool è noto come pool di riserva (RP). L’RP viene rilasciato più lentamente dell’RRP (RRP: 18 granuli/min; RP: 6 granuli/min).[86] Un ridotto rilascio di insulina nella prima fase può essere il primo difetto rilevabile delle cellule beta che predice l’insorgenza del diabete di tipo 2.[87] Il rilascio nella prima fase e la sensibilità all’insulina sono predittori indipendenti del diabete.[88]
La descrizione del rilascio della prima fase è la seguente:
- Il glucosio entra nelle β-cellule attraverso il trasportatore del glucosio, GLUT 2. A bassi livelli di zucchero nel sangue poco glucosio entra nelle β-cellule; ad alte concentrazioni di glucosio nel sangue grandi quantità di glucosio entrano in queste cellule.[89]
- Il glucosio che entra nella β-cellula viene fosforilato a glucosio-6-fosfato (G-6-P) dalla glucochinasi (esochinasi IV) che non è inibita dal G-6-P come le esochinasi di altri tessuti (esochinasi I-III). Ciò significa che la concentrazione intracellulare di G-6-P rimane proporzionale alla concentrazione di zucchero nel sangue.[89]
- Il glucosio-6-fosfato entra nella via glicolitica e poi, attraverso la reazione della piruvato deidrogenasi, nel ciclo di Krebs, dove vengono prodotte più molecole di ATP ad alta energia dall’ossidazione dell’acetil CoA (substrato del ciclo di Krebs), con conseguente aumento del rapporto ATP:ADP all’interno della cellula.[90]
- Un aumento del rapporto ATP:ADP intracellulare chiude il canale del potassio SUR1/Kir6.2 sensibile all’ATP (vedi recettore delle sulfoniluree). Questo impedisce agli ioni potassio (K+) di lasciare la cellula per diffusione facilitata, portando a un accumulo di ioni potassio intracellulare. Di conseguenza, l’interno della cellula diventa meno negativo rispetto all’esterno, portando alla depolarizzazione della membrana della superficie cellulare.
- In seguito alla depolarizzazione, si aprono i canali degli ioni calcio (Ca2+) voltaggio-gati, consentendo agli ioni calcio di spostarsi nella cellula per diffusione facilitata.
- La concentrazione citosolica di ioni calcio può anche essere aumentata dal rilascio di calcio dai depositi intracellulari attraverso l’attivazione dei recettori rianodinici.[91]
- La concentrazione di ioni calcio nel citosol delle cellule beta può essere aumentata anche, o in aggiunta, attraverso l’attivazione della fosfolipasi C derivante dal legame di un ligando extracellulare (ormone o neurotrasmettitore) a un recettore di membrana accoppiato a proteine G. La fosfolipasi C scinde il fosfolipide di membrana, il fosfatidil inositolo 4,5-bisfosfato, in inositolo 1,4,5-trifosfato e diacilglicerolo. L’inositolo 1,4,5-trisfosfato (IP3) si lega quindi a proteine recettoriali nella membrana plasmatica del reticolo endoplasmatico (ER). Ciò consente il rilascio di ioni Ca2+ dall’ER attraverso canali IP3-gated, che aumentano la concentrazione citosolica di ioni calcio indipendentemente dagli effetti di un’elevata concentrazione di glucosio nel sangue. La stimolazione parasimpatica delle isole pancreatiche opera attraverso questa via per aumentare la secrezione di insulina nel sangue.[92]
- L’aumento significativo della quantità di ioni calcio nel citoplasma delle cellule provoca il rilascio nel sangue dell’Insulina precedentemente sintetizzata e immagazzinata nelle vescicole secretorie intracellulari.
Questo è il meccanismo principale di rilascio dell’insulina. Altre sostanze note per stimolare il rilascio di insulina sono gli aminoacidi arginina e leucina, il rilascio parasimpatico di acetilcolina (che agisce attraverso la via della fosfolipasi C), le sulfoniluree, la colecistochinina (CCK, anch’essa attraverso la fosfolipasi C),[93-56] e le incretine di derivazione gastrointestinale, come il peptide glucagone-simile-1 (GLP-1) e il peptide insulinotropico glucosio-dipendente (GIP).

Il rilascio di insulina è fortemente inibito dalla noradrenalina, che porta a un aumento dei livelli di glucosio nel sangue durante lo stress. Sembra che il rilascio di catecolamine da parte del sistema nervoso simpatico abbia influenze contrastanti sul rilascio di insulina da parte delle cellule beta, perché il rilascio di Insulina è inibito dai recettori α2-adrenergici[94] e stimolato dai recettori β2-adrenergici.[95] L’effetto netto della noradrenalina dai nervi simpatici e dell’epinefrina dalle ghiandole surrenali sul rilascio di insulina è l’inibizione dovuta alla dominanza dei recettori α-adrenergici.[96]
Quando il livello di glucosio scende al valore fisiologico abituale, il rilascio di insulina da parte delle cellule β rallenta o si arresta. Se il livello di glucosio nel sangue scende al di sotto di questo valore, soprattutto a livelli pericolosamente bassi, il rilascio di ormoni iperglicemizzanti (in particolare il glucagone dalle cellule alfa dell’isolotto di Langerhans) forza il rilascio di glucosio nel sangue dalle scorte di glicogeno del fegato, integrato dalla gluconeogenesi se le scorte di glicogeno si esauriscono. Aumentando il glucosio nel sangue, gli ormoni iperglicemizzanti prevengono o correggono l’ipoglicemia pericolosa per la vita.
L’evidenza di un alterato rilascio di insulina nella prima fase può essere osservata nel test di tolleranza al glucosio, dimostrato da un livello di glucosio nel sangue sostanzialmente elevato a 30 minuti dall’ingestione di un carico di glucosio (75 o 100 g di glucosio), seguito da un lento calo nei 100 minuti successivi, per rimanere al di sopra di 120 mg/100 ml dopo due ore dall’inizio del test. In una persona normale il livello di glucosio nel sangue è corretto (e può anche essere leggermente sovracorretto) alla fine del test. Il picco insulinico è una “prima risposta” all’aumento del glucosio nel sangue; questa risposta è individuale e specifica per la dose, anche se in passato si è sempre ritenuto che fosse specifica solo per il tipo di alimento.
Anche durante la digestione, in genere una o due ore dopo un pasto, il rilascio di insulina da parte del pancreas non è continuo, ma oscilla con un periodo di 3-6 minuti, passando dal generare una concentrazione di insulina nel sangue superiore a circa 800 pmol/l a meno di 100 pmol/l (nei ratti).[97] Si pensa che questo avvenga per evitare la sottoregolazione dei recettori dell’Insulina nelle cellule bersaglio e per aiutare il fegato a estrarre l’insulina dal sangue [Questa oscillazione è importante da considerare quando si somministrano farmaci insulino-stimolanti, poiché idealmente si dovrebbe ottenere una concentrazione ematica oscillante del rilascio di insulina, e non una concentrazione elevata costante].[97] Ciò può essere ottenuto somministrando l’insulina in modo ritmico nella vena porta, con una somministrazione attivata dalla luce o con il trapianto di cellule dell’isoletta nel fegato.[98][99][100]
Il livello di Insulina nel sangue può essere misurato in unità internazionali, come µIU/mL o in concentrazione molare, come pmol/L, dove 1 µIU/mL equivale a 6,945 pmol/L.[101] Un livello ematico tipico tra i pasti è di 8-11 μIU/mL (57-79 pmol/L).[102]
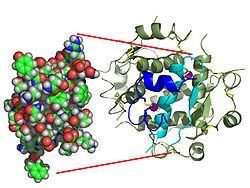
Gli effetti dell’insulina sono avviati dal suo legame con un recettore, il recettore dell’insulina (IR), presente nella membrana cellulare. La molecola del recettore contiene una subunità α e una subunità β. Due molecole si uniscono per formare il cosiddetto omodimero. L’insulina si lega alla subunità α dell’omodimero, che è rivolta verso il lato extracellulare delle cellule. Le subunità β hanno un’attività enzimatica tirosin-chinasica che viene attivata dal legame con l’insulina. Questa attività provoca l’autofosforilazione delle subunità β e successivamente la fosforilazione di proteine all’interno della cellula, note come substrati del recettore dell’insulina (IRS). La fosforilazione dell’IRS attiva una cascata di trasduzione del segnale che porta all’attivazione di altre chinasi e di fattori di trascrizione che mediano gli effetti intracellulari dell’insulina.[103]
La cascata che porta all’inserimento dei trasportatori di glucosio GLUT4 nelle membrane cellulari delle cellule muscolari e adipose e alla sintesi di glicogeno nel fegato e nel tessuto muscolare, nonché alla conversione del glucosio in trigliceridi nel fegato, nell’adipe e nel tessuto della ghiandola mammaria in allattamento, opera attraverso l’attivazione, da parte dell’IRS-1, della fosfoinositolo 3 chinasi (PI3K). Questo enzima converte un fosfolipide della membrana cellulare, il fosfatidilinositolo 4,5-bisfosfato (PIP2), in fosfatidilinositolo 3,4,5-trifosfato (PIP3), che a sua volta attiva la protein chinasi B (PKB). La PKB attivata facilita la fusione degli endosomi contenenti GLUT4 con la membrana cellulare, con conseguente aumento dei trasportatori GLUT4 nella membrana plasmatica.[104] La PKB fosforila anche la glicogeno sintasi chinasi (GSK), inattivando così questo enzima.[104] Ciò significa che il suo substrato, la glicogeno sintasi (GS), non può essere fosforilato e rimane de-fosforilato, e quindi attivo. L’enzima attivo, la glicogeno sintasi (GS), catalizza la fase limitante della sintesi del glicogeno dal glucosio. Defosforilazioni simili interessano gli enzimi che controllano il tasso di glicolisi che porta alla sintesi dei grassi attraverso il malonil-CoA nei tessuti che possono generare trigliceridi, nonché gli enzimi che controllano il tasso di gluconeogenesi nel fegato. L’effetto complessivo di queste de-fosforilazioni enzimatiche finali è che, nei tessuti in grado di effettuare queste reazioni, viene stimolata la sintesi di glicogeno e di grassi a partire dal glucosio, mentre viene inibita la produzione di glucosio da parte del fegato attraverso la glicogenolisi e la gluconeogenesi.[105] Anche la scomposizione dei trigliceridi da parte del tessuto adiposo in acidi grassi liberi e glicerolo viene inibita.[104]
Una volta prodotto il segnale intracellulare derivante dal legame dell’insulina con il suo recettore, è necessario interrompere la segnalazione. Come menzionato di seguito nella sezione sulla degradazione, l’endocitosi e la degradazione del recettore legato all’insulina è un meccanismo principale per terminare la segnalazione.[106] Inoltre, la via di segnalazione viene terminata anche dalla de-fosforilazione dei residui di tirosina nelle varie vie di segnalazione da parte delle tirosina fosfatasi. Le serina/treonina chinasi sono anche note per ridurre l’attività dell’insulina.
La struttura del complesso insulina-recettore dell’insulina è stata determinata con le tecniche della cristallografia a raggi X.[107]
Una volta che la molecola di Insulina si è agganciata al recettore e ha svolto la sua azione, può essere rilasciata nell’ambiente extracellulare o essere degradata dalla cellula. I due siti principali per l’eliminazione dell’Insulina sono il fegato e il rene.[108] Viene scomposta dall’enzima proteina-disolfuro reduttasi (Glutatione),[109] che rompe i legami disolfuro tra le catene A e B. Il fegato elimina la maggior parte dell’Insulina durante il transito di primo passaggio, mentre il rene elimina la maggior parte dell’Insulina nella circolazione sistemica. La degradazione comporta normalmente l’endocitosi del complesso insulino-recettore, seguita dall’azione di enzimi degradanti l’Insulina. Si stima che una molecola di Insulina prodotta endogenamente dalle cellule beta venga degradata entro circa un’ora dal suo rilascio iniziale in circolo (emivita dell’Insulina ~ 4-6 minuti).[109][110]
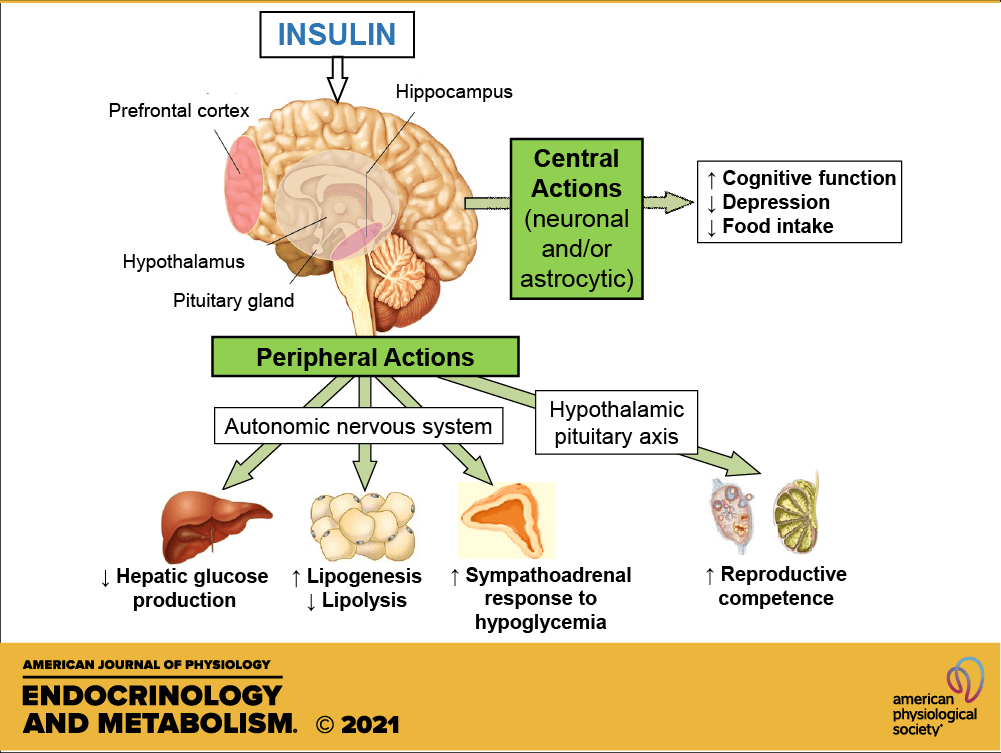
Le azioni dell’Insulina a livello del metabolismo umano globale comprendono:
- Aumento dell’assorbimento di alcune sostanze da parte delle cellule, in particolare del glucosio nei muscoli e nel tessuto adiposo (circa i due terzi delle cellule del corpo)[111]
- Aumento della replicazione del DNA e della sintesi proteica attraverso il controllo dell’assorbimento degli aminoacidi.
- Modifica dell’attività di numerosi enzimi.
Le azioni dell’Insulina (indirette e dirette) sulle cellule comprendono:
- Stimola l’assorbimento del glucosio – L’Insulina diminuisce la concentrazione di glucosio nel sangue inducendo l’assunzione di glucosio da parte delle cellule. Ciò è possibile perché l’insulina provoca l’inserimento del trasportatore GLUT4 nelle membrane cellulari dei tessuti muscolari e adiposi, permettendo al glucosio di entrare nella cellula.[112]
- Aumento della sintesi dei grassi – l’insulina costringe le cellule grasse ad accogliere il glucosio nel sangue, che viene convertito in trigliceridi; la diminuzione dell’insulina provoca l’inverso.[111]
- Aumento dell’esterificazione degli acidi grassi – costringe il tessuto adiposo a produrre grassi neutri (cioè trigliceridi) dagli acidi grassi; la diminuzione dell’insulina provoca l’inverso.[111]
- Diminuzione della lipolisi – costringe a ridurre la conversione dei depositi di lipidi delle cellule adipose in acidi grassi e glicerolo nel sangue; la diminuzione dell’insulina provoca l’effetto inverso.[111]
- Sintesi indotta di glicogeno – Quando i livelli di glucosio sono elevati, l’insulina induce la formazione di glicogeno attraverso l’attivazione dell’enzima esochinasi, che aggiunge un gruppo fosfato al glucosio, ottenendo così una molecola che non può uscire dalla cellula. Allo stesso tempo, l’insulina inibisce l’enzima glucosio-6-fosfatasi, che rimuove il gruppo fosfato. Questi due enzimi sono fondamentali per la formazione del glicogeno. Inoltre, l’insulina attiva gli enzimi fosfofruttochinasi e glicogeno sintasi, responsabili della sintesi del glicogeno.[113]
- Diminuzione della gluconeogenesi e della glicogenolisi – diminuisce la produzione di glucosio da substrati non glucidici, principalmente nel fegato (la maggior parte dell’insulina endogena che arriva al fegato non lascia mai il fegato); la diminuzione dell’insulina causa la produzione di glucosio da parte del fegato a partire da substrati diversi.[111]
- Diminuzione della proteolisi – diminuzione della scomposizione delle proteine[111]
- Diminuzione dell’autofagia – diminuzione del livello di degradazione degli organelli danneggiati. I livelli postprandiali inibiscono completamente l’autofagia[114].
- Aumento dell’assorbimento di aminoacidi – costringe le cellule ad assorbire gli aminoacidi circolanti; la diminuzione dell’insulina inibisce l’assorbimento.[111]
- Tono muscolare arterioso – costringe i muscoli della parete arteriosa a rilassarsi, aumentando il flusso sanguigno, soprattutto nelle microarterie; la diminuzione dell’Insulina riduce il flusso permettendo a questi muscoli di contrarsi.[115]
- Aumento della secrezione di acido cloridrico da parte delle cellule parietali dello stomaco.[citazione necessaria]
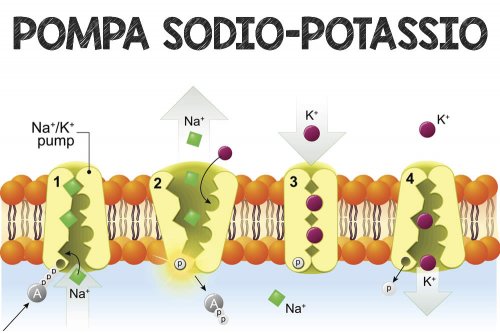
- Aumento dell’assorbimento di potassio – costringe le cellule che sintetizzano glicogeno (una sostanza molto spugnosa e “umida”, che aumenta il contenuto di acqua intracellulare e i relativi ioni K+)[116] ad assorbire il potassio dai fluidi extracellulari; la mancanza di insulina inibisce l’assorbimento. L’aumento dell’assorbimento cellulare di potassio da parte dell’insulina abbassa i livelli di potassio nel plasma sanguigno. Ciò potrebbe avvenire attraverso la traslocazione indotta dall’insulina della Na+/K+-ATPasi sulla superficie delle cellule muscolari scheletriche.[117][118]
- Diminuzione dell’escrezione renale di sodio.[119]
L’Insulina influenza anche altre funzioni corporee, come la compliance vascolare e la cognizione. Una volta che l’Insulina entra nel cervello umano, migliora l’apprendimento e la memoria, in particolare la memoria verbale.[120] Il potenziamento della segnalazione cerebrale dell’Insulina mediante la somministrazione intranasale di insulina migliora anche la risposta termoregolatoria e glucoregolatoria acuta all’assunzione di cibo, suggerendo che l’insulina a livello nervoso centrale contribuisce al coordinamento di un’ampia varietà di processi omeostatici o regolatori nel corpo umano. [121] L’insulina ha anche effetti stimolanti sull’ormone di rilascio delle gonadotropine dall’ipotalamo, favorendo così la fertilità.[122]
Una nota interessante riguarda il fatto che l’Insulina è un importante regolatore del metabolismo degli endocannabinoidi (EC) e il trattamento con insulina ha dimostrato di ridurre gli EC intracellulari, il 2-arachidonoilglicerolo (2-AG) e l’anandamide (AEA), che corrispondono a cambiamenti di espressione sensibili all’insulina negli enzimi del metabolismo degli EC. Negli adipociti insulino-resistenti, i modelli di espressione degli enzimi indotti dall’insulina sono disturbati in modo coerente con un’elevata sintesi di EC e una ridotta degradazione di EC. I risultati suggeriscono che gli adipociti insulino-resistenti non riescono a regolare il metabolismo delle EC e diminuiscono i livelli intracellulari di EC in risposta alla stimolazione insulinica, per cui gli individui obesi insulino-resistenti presentano un aumento delle concentrazioni di EC.[123][124] Questa disregolazione contribuisce all’eccessivo accumulo di grasso viscerale e al ridotto rilascio di adiponectina dal tessuto adiposo addominale, nonché all’insorgenza di diversi fattori di rischio cardiometabolici associati all’obesità e al diabete di tipo II.[125]
Continua…
Gabriel Bellizzi [CEO BioGenTech]
Riferimenti:
- Sharma NC (2021-10-01). “WHO adds new drugs to its essential medicines’ list”. mint. Retrieved 2021-10-09.
- Sakula A (July 1988). “Paul Langerhans (1847-1888): a centenary tribute”. Journal of the Royal Society of Medicine. 81 (7): 414–5. doi:10.1177/014107688808100718. PMC 1291675. PMID 3045317.
- Petit H. “Edouard Laguesse (1861–1927)”. Museum of the Regional Hospital of Lille (in French). Retrieved 25 July 2018.
- Opie EL (1901). “Diabetes Mellitus Associated with Hyaline Degeneration of the islands of Langerhans of the Pancreas”. Bulletin of the Johns Hopkins Hospital. 12 (125): 263–64. hdl:2027/coo.31924069247447.
- Opie EL (1901). “On the Relation of Chronic Interstitial Pancreatitis to the Islands of Langerhans and to Diabetes Mellitus”. Journal of Experimental Medicine. 5 (4): 397–428. doi:10.1084/jem.5.4.397. PMC 2118050. PMID 19866952.
- Opie EL (1901). “The Relation of Diabetes Mellitus to Lesions of the Pancreas. Hyaline Degeneration of the Islands of Langerhans”. Journal of Experimental Medicine. 5 (5): 527–40. doi:10.1084/jem.5.5.527. PMC 2118021. PMID 19866956.
- The American Institute of Nutrition (1967). “Proceedings of the Thirty-first Annual Meeting of the American Institute of Nutrition”. Journal of Nutrition. 92 (4): 509. doi:10.1093/jn/92.4.507.
- Paulesco NC (August 31, 1921). “Recherche sur le rôle du pancréas dans l’assimilation nutritive”. Archives Internationales de Physiologie. 17: 85–109.
- Lestradet H (1997). “Le 75e anniversaire de la découverte de l’insuline”. Diabetes & Metabolism. 23 (1): 112.
- de Leiva A, Brugués E, de Leiva-Pérez A (2011). “The discovery of insulin: Continued controversies after ninety years”. Endocrinología y Nutrición (English Edition). 58 (9): 449–456. doi:10.1016/j.endoen.2011.10.001.
- Vecchio I, Tornali C, Bragazzi NL, Martini M (2018-10-23). “The Discovery of Insulin: An Important Milestone in the History of Medicine”. Frontiers in Endocrinology. 9: 613. doi:10.3389/fendo.2018.00613. PMC 6205949. PMID 30405529.
- Banting FG (31 October 1920). “Note dated Oct 31/20 from loose leaf notebook 1920/21”. University of Toronto Libraries.
- Jump up to:a b c Rosenfeld L (December 2002). “Insulin: discovery and controversy”. Clinical Chemistry. 48 (12): 2270–88. doi:10.1093/clinchem/48.12.2270. PMID 12446492.
- Wright JR (December 2002). “Almost famous: E. Clark Noble, the common thread in the discovery of insulin and vinblastine”. CMAJ. 167 (12): 1391–96. PMC 137361. PMID 12473641.
- Krishnamurthy K (2002). Pioneers in scientific discoveries. Mittal Publications. p. 266. ISBN 978-81-7099-844-0. Retrieved 26 July 2011.
- Bliss M (July 1993). “Rewriting medical history: Charles Best and the Banting and Best myth” (PDF). Journal of the History of Medicine and Allied Sciences. 48 (3): 253–74. doi:10.1093/jhmas/48.3.253. PMID 8409364.
- Toronto star weekly (14 Jan 1922). “Work on diabetes shows progress against disease”. University of Toronto Libraries.
- Fletcher AA (November 1962). “Early clinical experiences with insulin”. Canadian Medical Association Journal. 87: 1052–5. PMC 1849803. PMID 13945508.
- Banting FG (Dec 1921 – Jan 1922). “Patient records for Leonard Thompson”. University of Toronto Libraries.
- Zuger A (October 4, 2010). “Rediscovering the First Miracle Drug”. The New York Times. Retrieved 2010-10-06.
Elizabeth Hughes was a cheerful, pretty little girl, five feet tall, with straight brown hair and a consuming interest in birds. On Dr. Allen’s diet her weight fell to 65 pounds, then 52 pounds, and then, after an episode of diarrhea that almost killed her in the spring of 1922, 45 pounds. By then she had survived three years, far longer than expected. And then her mother heard the news: Insulin had finally been isolated in Canada.
- Banting FG (16 August 1922). “Chart for Elizabeth Hughes”. University of Toronto Libraries.
- Woodbury DO (February 1963). “Please save my son!”. University of Toronto Libraries.
- Marcotte B (November 22, 2010). “Rochester’s John Williams a man of scientific talents”. Democrat and Chronicle. Rochester, New York. Gannett Company. pp. 1B, 4B. Archived from the original on November 23, 2010. Retrieved November 22, 2010.
- University of Toronto Board of Governors Insulin Committee (25 Jan 1922). “Memorandum in reference to the co-operation of the Connaught Antitoxin Laboratories in the researches conducted by Dr. Banting, Mr. Best and Dr. Collip under the general direction of Professor J.J.R. Macleod to obtain an extract of pancreas having a specific effect on blood sugar concentration”. University of Toronto Libraries.
- Bliss M (2007). The discovery of insulin (25th anniversary ed.). Chicago: University of Chicago Press. p. 132. ISBN 9780226058993. OCLC 74987867.
The Lilly company would be delighted to work with Toronto, Clowes wrote, and hinted, perhaps intentionally, perhaps not, that Toronto could be bypassed: “I have thus far refrained from starting work in our laboratories on the field of this question as I was anxious to avoid in any way intruding on the field of yourself and your associates until you had published your results. I feel, however, that the matter is now one of such immediate importance that we should take up the experimental end of the question without delay, preferably cooperating with you and your associates…”
- Kendall EC (10 April 1922). “Letter to Dr. J. J. R. Macleod 10/04/1922”. University of Toronto Libraries: Discovery and Early Development of Insulin.
- Macleod JJ (28 April 1924). “Statement read by J. J. R. Macleod at the Insulin Committee meeting regarding patents and royalties 28/04/1924”. University of Toronto Libraries: The Discovery and Early Development of Insulin.
- Bliss M (2007). The discovery of insulin (25th anniversary ed.). Chicago: University of Chicago Press. pp. 131–133. ISBN 9780226058993. OCLC 74987867.
- Banting FG, Best C, Collip JS (15 January 1923). “Assignment to the Governors of the University of Toronto”. University of Toronto Libraries: Discovery and Early Development of Insulin.
- “Copy of the article: A step forward in medical ethics”. University of Toronto Libraries: The Discovery and Early Development of Insulin. The World’s Work. February 1923.
- Kjeldsen T (September 2000). “Yeast secretory expression of insulin precursors” (PDF). Applied Microbiology and Biotechnology. 54 (3): 277–86. doi:10.1007/s002530000402. PMID 11030562. S2CID 9246671. Archived from the original (PDF) on 2017-09-27.
- “The Nobel Prize in Physiology or Medicine 1923”. The Nobel Foundation.
- Stretton AO (October 2002). “The first sequence. Fred Sanger and insulin”. Genetics. 162 (2): 527–32.
- Felman A (22 November 2018). “Who discovered insulin?”. Medical News Today.
- Tsou CL (2015). 对人工合成结晶牛胰岛素的回忆 [Memory on the research of synthesizing bovine insulin]. 生命科学 [Chinese Bulletin of Life Science] (in Simplified Chinese). 27 (6): 777–79.
- Castle WB (1962). “The Gordon Wilson Lecture. A Century of Curiosity About Pernicious Anemia”. Transactions of the American Clinical and Climatological Association. 73: 54–80. PMC 2249021. PMID 21408623.
- Banting FG, Best CH, Collip JB, Campbell WR, Fletcher AA (March 1922). “Pancreatic Extracts in the Treatment of Diabetes Mellitus”. Canadian Medical Association Journal. 12 (3): 141–46. PMC 1524425. PMID 20314060.
- Drury MI (July 1972). “The golden jubile of insulin”. Journal of the Irish Medical Association.
- Bliss M (2007). The discovery of insulin (25th anniversary ed.). Chicago: University of Chicago Press. p. 181. ISBN 9780226058993. OCLC 74987867.
- Abel JJ (February 1926). “Crystalline Insulin”. Proceedings of the National Academy of Sciences of the United States of America. 12 (2): 132–6. Bibcode:1926PNAS…12..132A. doi:10.1073/pnas.12.2.132. PMC 1084434. PMID 16587069.
- Somogyi M, Doisy EA, Shaffer PA (May 1924). “On the Preparation of Insulin” (PDF). Journal of Biological Chemistry. 60 (1): 31–58. doi:10.1016/S0021-9258(18)85220-6.
- Jensen H, Evans EA (1935-01-01). “Studies on Crystalline Insulin Xviii. the Nature of the Free Amino Groups in Insulin and the Isolation of Phenylalanine and Proline from Crystalline Insulin” (PDF). Journal of Biological Chemistry. 108 (1): 1–9. doi:10.1016/S0021-9258(18)75301-5.
- Sanger F, Tuppy H (September 1951). “The amino-acid sequence in the phenylalanyl chain of insulin. I. The identification of lower peptides from partial hydrolysates”. The Biochemical Journal. 49 (4): 463–81. doi:10.1042/bj0490463. PMC 1197535. PMID 14886310.; Sanger F, Tuppy H (September 1951). “The amino-acid sequence in the phenylalanyl chain of insulin. 2. The investigation of peptides from enzymic hydrolysates”. The Biochemical Journal. 49 (4): 481–90. doi:10.1042/bj0490481. PMC 1197536. PMID 14886311.; Sanger F, Thompson EO (February 1953). “The amino-acid sequence in the glycyl chain of insulin. I. The identification of lower peptides from partial hydrolysates”. The Biochemical Journal. 53 (3): 353–66. doi:10.1042/bj0530353. PMC 1198157. PMID 13032078.; Sanger F, Thompson EO (February 1953). “The amino-acid sequence in the glycyl chain of insulin. II. The investigation of peptides from enzymic hydrolysates”. The Biochemical Journal. 53 (3): 366–74. doi:10.1042/bj0530366. PMC 1198158. PMID 13032079.
- Katsoyannis PG, Fukuda K, Tometsko A, Suzuki K, Tilak M (1964). “Insulin Peptides. X. The Synthesis of the B-Chain of Insulin and Its Combination with Natural or Synthetis A-Chin to Generate Insulin Activity”. Journal of the American Chemical Society. 86 (5): 930–32. doi:10.1021/ja01059a043.
- Kung YT, Du YC, Huang WT, Chen CC, Ke LT (November 1965). “Total synthesis of crystalline bovine insulin”. Scientia Sinica. 14 (11): 1710–6. PMID 5881570.
- Marglin A, Merrifield RB (November 1966). “The synthesis of bovine insulin by the solid phase method”. Journal of the American Chemical Society. 88 (21): 5051–2. doi:10.1021/ja00973a068. PMID 5978833.
- Costin GE (January 2004). “What is the advantage of having melanin in parts of the central nervous system (e.g. substantia nigra)?”. IUBMB Life. Time Inc. 56 (1): 47–9. doi:10.1080/15216540310001659029. PMID 14992380.
- Wollmer A, Dieken ML, Federwisch M, De Meyts P (2002). Insulin & related proteins structure to function and pharmacology. Boston: Kluwer Academic Publishers. ISBN 978-1-4020-0655-5.
- Tsou CL (2015). 对人工合成结晶牛胰岛素的回忆 [Memory on the research of synthesizing bovine insulin]. 生命科学 [Chinese Bulletin of Life Science] (in Simplified Chinese). 27 (6): 777–79.
- Jump up to:a b Blundell TL, Cutfield JF, Cutfield SM, Dodson EJ, Dodson GG, Hodgkin DC, et al. (June 1971). “Atomic positions in rhombohedral 2-zinc insulin crystals”. Nature. 231 (5304): 506–11. Bibcode:1971Natur.231..506B. doi:10.1038/231506a0. PMID 4932997. S2CID 4158731.
- Weber, H.E. (1975) Diabetes 24, 405. (see figure)
- “First Successful Laboratory Production of Human Insulin Announced”. News Release. Genentech. 1978-09-06. Archived from the original on 2016-09-27. Retrieved 2016-09-26.
- Jump up to:a b c Tof I (1994). “Recombinant DNA technology in the synthesis of human insulin”. Little Tree Publishing. Retrieved 2009-11-03.
- Jump up to:a b Aggarwal SR (December 2012). “What’s fueling the biotech engine-2011 to 2012”. Nature Biotechnology. 30 (12): 1191–7.
- Chan SJ, Keim P, Steiner DF. Cell-free synthesis of rat preproinsulins: Characterization and partial amino acid sequence determination. Proc Natl Acad Sci. USA 1976;73:1964-1968.
- “Safflowers may provide new insulin source | CTV News”.
- GRCh38: Ensembl release 89: ENSG00000254647 – Ensembl, May 2017
- Jump up to:a b c GRCm38: Ensembl release 89: ENSMUSG00000000215 – Ensembl, May 2017
- “Human PubMed Reference:”. National Center for Biotechnology Information, U.S. National Library of Medicine.
- “Mouse PubMed Reference:”. National Center for Biotechnology Information, U.S. National Library of Medicine.
- “Insulin | Meaning of Insulin by Lexico”. Lexico Dictionaries | English.
- Fu Z, Gilbert ER, Liu D (January 2013). “Regulation of insulin synthesis and secretion and pancreatic Beta-cell dysfunction in diabetes”. Curr Diabetes Rev. 9 (1): 25–53. doi:10.2174/157339913804143225. PMC 3934755. PMID 22974359.
- Bernardo AS, Hay CW, Docherty K (November 2008). “Pancreatic transcription factors and their role in the birth, life and survival of the pancreatic beta cell” (PDF). review. Molecular and Cellular Endocrinology. 294 (1–2): 1–9. doi:10.1016/j.mce.2008.07.006. PMID 18687378. S2CID 28027796.
- Rutter GA, Pullen TJ, Hodson DJ, Martinez-Sanchez A (March 2015). “Pancreatic β-cell identity, glucose sensing and the control of insulin secretion”. review. The Biochemical Journal. 466 (2): 203–18. doi:10.1042/BJ20141384. PMID 25697093. S2CID 2193329.
- Rutter GA, Tavaré JM, Palmer DG (June 2000). “Regulation of Mammalian Gene Expression by Glucose”. review. News in Physiological Sciences. 15 (3): 149–54. doi:10.1152/physiologyonline.2000.15.3.149. PMID 11390898.
- Poitout V, Hagman D, Stein R, Artner I, Robertson RP, Harmon JS (April 2006). “Regulation of the insulin gene by glucose and d acids”. review. The Journal of Nutrition. 136 (4): 873–76. doi:10.1093/jn/136.4.873. PMC 1853259. PMID 16549443.
- Vaulont S, Vasseur-Cognet M, Kahn A (October 2000). “Glucose regulation of gene transcription”. review. The Journal of Biological Chemistry. 275 (41): 31555–58. doi:10.1074/jbc.R000016200. PMID 10934218.
- Christensen DP, Dahllöf M, Lundh M, Rasmussen DN, Nielsen MD, Billestrup N, Grunnet LG, Mandrup-Poulsen T (2011). “Histone deacetylase (HDAC) inhibition as a novel treatment for diabetes mellitus”. Molecular Medicine. 17 (5–6): 378–90. doi:10.2119/molmed.2011.00021. PMC 3105132. PMID 21274504.
- Wang W, Shi Q, Guo T, Yang Z, Jia Z, Chen P, Zhou C (June 2016). “PDX1 and ISL1 differentially coordinate with epigenetic modifications to regulate insulin gene expression in varied glucose concentrations”. Molecular and Cellular Endocrinology. 428: 38–48. doi:10.1016/j.mce.2016.03.019. PMID 26994512.
- Wang X, Wei X, Pang Q, Yi F (August 2012). “Histone deacetylases and their inhibitors: molecular mechanisms and therapeutic implications in diabetes mellitus”. Acta Pharmaceutica Sinica B. 2 (4): 387–95. doi:10.1016/j.apsb.2012.06.005.
- Jump up to:a b Andrali SS, Sampley ML, Vanderford NL, Ozcan S (October 2008). “Glucose regulation of insulin gene expression in pancreatic beta-cells”. review. The Biochemical Journal. 415 (1): 1–10. doi:10.1042/BJ20081029. PMID 18778246.
- Kaneto H, Matsuoka TA, Kawashima S, Yamamoto K, Kato K, Miyatsuka T, Katakami N, Matsuhisa M (July 2009). “Role of MafA in pancreatic beta-cells”. Advanced Drug Delivery Reviews. 61 (7–8): 489–96. doi:10.1016/j.addr.2008.12.015. PMID 19393272.
- Aramata S, Han SI, Kataoka K (December 2007). “Roles and regulation of transcription factor MafA in islet beta-cells”. Endocrine Journal. 54 (5): 659–66. doi:10.1507/endocrj.KR-101. PMID 17785922.
- Kaneto H, Matsuoka TA (October 2012). “Involvement of oxidative stress in suppression of insulin biosynthesis under diabetic conditions”. International Journal of Molecular Sciences. 13 (10): 13680–90. doi:10.3390/ijms131013680. PMC 3497347. PMID 23202973.
- Jump up to:a b Najjar S (2001). “Insulin Action: Molecular Basis of Diabetes”. Encyclopedia of Life Sciences. John Wiley & Sons. doi:10.1038/npg.els.0001402. ISBN 978-0470016176.
- Gustin N (2005-03-07). “Researchers discover link between insulin and Alzheimer’s”. EurekAlert!. American Association for the Advancement of Science. Retrieved 2009-01-01.
- de la Monte SM, Wands JR (February 2005). “Review of insulin and insulin-like growth factor expression, signaling, and malfunction in the central nervous system: relevance to Alzheimer’s disease” (PDF). Journal of Alzheimer’s Disease. 7 (1): 45–61. doi:10.3233/JAD-2005-7106. PMID 15750214.
- Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, Xu XJ, Wands JR, de la Monte SM (February 2005). “Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease—is this type 3 diabetes?” (PDF). Journal of Alzheimer’s Disease. 7 (1): 63–80.
- Stryer L (1995). Biochemistry (Fourth ed.). New York: W.H. Freeman and Company. pp. 773–74. ISBN 0-7167-2009-4.
- Weiss M, Steiner DF, Philipson LH (2000). “Insulin Biosynthesis, Secretion, Structure, and Structure-Activity Relationships”. In Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, et al. (eds.). Endotext. MDText.com, Inc. PMID 25905258. Retrieved 2020-02-18.
- “Insulin human”. PubChem. Retrieved 26 February 2019.
- Jump up to:a b c Fu Z, Gilbert ER, Liu D (January 2013). “Regulation of insulin synthesis and secretion and pancreatic Beta-cell dysfunction in diabetes”. Current Diabetes Reviews. 9 (1): 25–53. doi:10.2174/157339913804143225. PMC 3934755. PMID 22974359.
- Dunn MF (August 2005). “Zinc-ligand interactions modulate assembly and stability of the insulin hexamer — a review”. Biometals. 18 (4): 295–303. doi:10.1007/s10534-005-3685-y. PMID 16158220. S2CID 8857694.
- Ivanova MI, Sievers SA, Sawaya MR, Wall JS, Eisenberg D (November 2009). “Molecular basis for insulin fibril assembly”. Proceedings of the National Academy of Sciences of the United States of America. 106 (45): 18990–5. Bibcode:2009PNAS..10618990I. doi:10.1073/pnas.0910080106. PMC 2776439. PMID 19864624.
- Omar-Hmeadi M, Idevall-Hagren O (March 2021). “Insulin granule biogenesis and exocytosis”. Cellular and Molecular Life Sciences. 78 (5): 1957–1970. doi:10.1007/s00018-020-03688-4. PMC 7966131. PMID 33146746.
- Bratanova-Tochkova TK, Cheng H, Daniel S, Gunawardana S, Liu YJ, Mulvaney-Musa J, et al. (February 2002). “Triggering and augmentation mechanisms, granule pools, and biphasic insulin secretion”. Diabetes. 51 (Suppl 1): S83–S90. doi:10.2337/diabetes.51.2007.S83. PMID 11815463.
- Gerich JE (February 2002). “Is reduced first-phase insulin release the earliest detectable abnormality in individuals destined to develop type 2 diabetes?”. Diabetes. 51 (Suppl 1): S117–S121. doi:10.2337/diabetes.51.2007.s117. PMID 11815469.
- Lorenzo C, Wagenknecht LE, Rewers MJ, Karter AJ, Bergman RN, Hanley AJ, Haffner SM (September 2010). “Disposition index, glucose effectiveness, and conversion to type 2 diabetes: the Insulin Resistance Atherosclerosis Study (IRAS)”. Diabetes Care. 33 (9): 2098–2103. doi:10.2337/dc10-0165. PMC 2928371. PMID 20805282.
- Jump up to:a b Schuit F, Moens K, Heimberg H, Pipeleers D (November 1999). “Cellular origin of hexokinase in pancreatic islets”. The Journal of Biological Chemistry (published 1999). 274 (46): 32803–09. doi:10.1074/jbc.274.46.32803. PMID 10551841.
- Schuit F, De Vos A, Farfari S, Moens K, Pipeleers D, Brun T, Prentki M (July 1997). “Metabolic fate of glucose in purified islet cells. Glucose-regulated anaplerosis in beta cells”. The Journal of Biological Chemistry (published 1997). 272 (30): 18572–79. doi:10.1074/jbc.272.30.18572. PMID 9228023.
- Santulli G, Pagano G, Sardu C, Xie W, Reiken S, D’Ascia SL, Cannone M, Marziliano N, Trimarco B, Guise TA, Lacampagne A, Marks AR (May 2015). “Calcium release channel RyR2 regulates insulin release and glucose homeostasis”. The Journal of Clinical Investigation. 125 (5): 1968–78. doi:10.1172/JCI79273. PMC 4463204. PMID 25844899.
- Stryer L (1995). Biochemistry (Fourth ed.). New York: W.H. Freeman and Company. pp. 343–44. ISBN 0-7167-2009-4.
- Cawston EE, Miller LJ (March 2010). “Therapeutic potential for novel drugs targeting the type 1 cholecystokinin receptor”. British Journal of Pharmacology. 159 (5): 1009–21. doi:10.1111/j.1476-5381.2009.00489.x. PMC 2839260. PMID 19922535.
- Nakaki T, Nakadate T, Kato R (August 1980). “Alpha 2-adrenoceptors modulating insulin release from isolated pancreatic islets”. Naunyn-Schmiedeberg’s Archives of Pharmacology. 313 (2): 151–53. doi:10.1007/BF00498572. PMID 6252481. S2CID 30091529.
- Layden BT, Durai V, Lowe WL Jr (2010). “G-Protein-Coupled Receptors, Pancreatic Islets, and Diabetes”. Nature Education. 3 (9): 13.
- Sircar S (2007). Medical Physiology. Stuttgart: Thieme Publishing Group. pp. 537–38.
- Hellman B, Gylfe E, Grapengiesser E, Dansk H, Salehi A (2007). “[Insulin oscillations—clinically important rhythm. Antidiabetics should increase the pulsative component of the insulin release]”. Läkartidningen (in Swedish).
- Sarode BR, Kover K, Tong PY, Zhang C, Friedman SH (November 2016). “Light Control of Insulin Release and Blood Glucose Using an Injectable Photoactivated Depot”. Molecular Pharmaceutics. 13 (11): 3835–3841. doi:10.1021/acs.molpharmaceut.6b00633. PMC 5101575. PMID 27653828.
- Jain PK, Karunakaran D, Friedman SH (January 2013). “Construction of a photoactivated insulin depot” (PDF). Angewandte Chemie. 52 (5): 1404–9. doi:10.1002/anie.201207264. PMID 23208858. Archived from the original (PDF) on 2019-11-02. Retrieved 2019-11-03.
- Rowlett R (13 June 2001). “A Dictionary of Units of Measurement”. The University of North Carolina at Chapel Hill. Archived from the original on 2013-10-28.
- Iwase H, Kobayashi M, Nakajima M, Takatori T (January 2001). “The ratio of insulin to C-peptide can be used to make a forensic diagnosis of exogenous insulin overdosage”. Forensic Science International. 115 (1–2): 123–127. doi:10.1016/S0379-0738(00)00298-X. PMID 11056282.
- Jump up to:a b “Handbook of Diabetes, 4th Edition, Excerpt #4: Normal Physiology of Insulin Secretion and Action”. Diabetes In Control. A free weekly diabetes newsletter for Medical Professionals. 2014-07-28. Retrieved 2017-06-01.
- McManus EJ, Sakamoto K, Armit LJ, Ronaldson L, Shpiro N, Marquez R, Alessi DR (April 2005). “Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis”. The EMBO Journal. 24 (8): 1571–83. doi:10.1038/sj.emboj.7600633. PMC 1142569. PMID 15791206.
- Fang X, Yu SX, Lu Y, Bast RC, Woodgett JR, Mills GB (October 2000). “Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A”. Proceedings of the National Academy of Sciences of the United States of America. 97 (22): 11960–75. Bibcode:2000PNAS…9711960F. doi:10.1073/pnas.220413597. PMC 17277. PMID 11035810.
- Jump up to:a b Stryer L (1995). Biochemistry (Fourth ed.). New York: W.H. Freeman and Company. pp. 351–56, 494–95, 505, 605–06, 773–75.
- Najjar S (2001). “Insulin Action: Molecular Basis of Diabetes”. Encyclopedia of Life Sciences. John Wiley & Sons.
- Menting JG, Whittaker J, Margetts MB, Whittaker LJ, Kong GK, Smith BJ, Watson CJ, Záková L, Kletvíková E, Jiráček J, Chan SJ, Steiner DF, Dodson GG, Brzozowski AM, Weiss MA, Ward CW, Lawrence MC (January 2013).
- Koh HE, Cao C, Mittendorfer B (January 2022). “Insulin Clearance in Obesity and Type 2 Diabetes”. International Journal of Molecular Sciences. 23 (2): 596. doi:10.3390/ijms23020596. PMC 8776220. PMID 35054781.
- “EC 1.8.4.2”. iubmb.qmul.ac.uk. Retrieved 25 July 2022.
- Duckworth WC, Bennett RG, Hamel FG (October 1998). “Insulin degradation: progress and potential”. Endocrine Reviews. 19 (5): 608–24. doi:10.1210/edrv.19.5.0349. PMID 9793760.
- Palmer BF, Henrich WL. “Carbohydrate and insulin metabolism in chronic kidney disease”. UpToDate, Inc.
- Dimitriadis G, Mitrou P, Lambadiari V, Maratou E, Raptis SA (August 2011). “Insulin effects in muscle and adipose tissue”. Diabetes Research and Clinical Practice. 93 (Suppl 1): S52–59.
- “Handbook of Diabetes, 4th Edition, Excerpt #4: Normal Physiology of Insulin Secretion and Action”. Diabetes In Control. A free weekly diabetes newsletter for Medical Professionals. 2014-07-28. Retrieved 2017-06-01.
- “Physiologic Effects of Insulin”. http://www.vivo.colostate.edu. Retrieved 2017-06-01.
- Bergamini E, Cavallini G, Donati A, Gori Z (October 2007). “The role of autophagy in aging: its essential part in the anti-aging mechanism of caloric restriction”. Annals of the New York Academy of Sciences. 1114 (1): 69–78. Bibcode:2007NYASA1114…69B. doi:10.1196/annals.1396.020. PMID 17934054. S2CID 21011988.
- Zheng C, Liu Z (June 2015). “Vascular function, insulin action, and exercise: an intricate interplay”. Trends in Endocrinology and Metabolism. 26 (6): 297–304. doi:10.1016/j.tem.2015.02.002. PMC 4450131. PMID 25735473.
- Kreitzman SN, Coxon AY, Szaz KF (July 1992). “Glycogen storage: illusions of easy weight loss, excessive weight regain, and distortions in estimates of body composition” (PDF). The American Journal of Clinical Nutrition. 56 (Suppl 1): 292S–93S. doi:10.1093/ajcn/56.1.292S. PMID 1615908. Archived from the original (PDF) on 2012-10-18.
- Benziane B, Chibalin AV (September 2008). “Frontiers: skeletal muscle sodium pump regulation: a translocation paradigm”. American Journal of Physiology. Endocrinology and Metabolism. 295 (3): E553–58. doi:10.1152/ajpendo.90261.2008. PMID 18430962. S2CID 10153197.
- Clausen T (September 2008). “Regulatory role of translocation of Na+-K+ pumps in skeletal muscle: hypothesis or reality?”. American Journal of Physiology. Endocrinology and Metabolism. 295 (3): E727–28, author reply 729. doi:10.1152/ajpendo.90494.2008. PMID 18775888. S2CID 13410719.
- Gupta AK, Clark RV, Kirchner KA (January 1992). “Effects of insulin on renal sodium excretion”. Hypertension. 19 (Suppl 1): I78–82. doi:10.1161/01.HYP.19.1_Suppl.I78. PMID 1730458.
- Benedict C, Hallschmid M, Hatke A, Schultes B, Fehm HL, Born J, Kern W (November 2004). “Intranasal insulin improves memory in humans” (PDF). Psychoneuroendocrinology. 29 (10): 1326–1334. doi:10.1016/j.psyneuen.2004.04.003. PMID 15288712. S2CID 20321892.
- Benedict C, Brede S, Schiöth HB, Lehnert H, Schultes B, Born J, Hallschmid M (January 2011). “Intranasal insulin enhances postprandial thermogenesis and lowers postprandial serum insulin levels in healthy men”. Diabetes.
- D’Eon TM, Pierce KA, Roix JJ, Tyler A, Chen H, Teixeira SR (May 2008). “The role of adipocyte insulin resistance in the pathogenesis of obesity-related elevations in endocannabinoids”. Diabetes. 57 (5): 1262–68. doi:10.2337/db07-1186. PMID 18276766.
- Gatta-Cherifi B, Cota D (February 2016). “New insights on the role of the endocannabinoid system in the regulation of energy balance”. International Journal of Obesity. 40 (2): 210–19. doi:10.1038/ijo.2015.179. PMID 26374449. S2CID 20740277.
- Di Marzo V (August 2008). “The endocannabinoid system in obesity and type 2 diabetes”. Diabetologia. 51 (8): 1356–67.