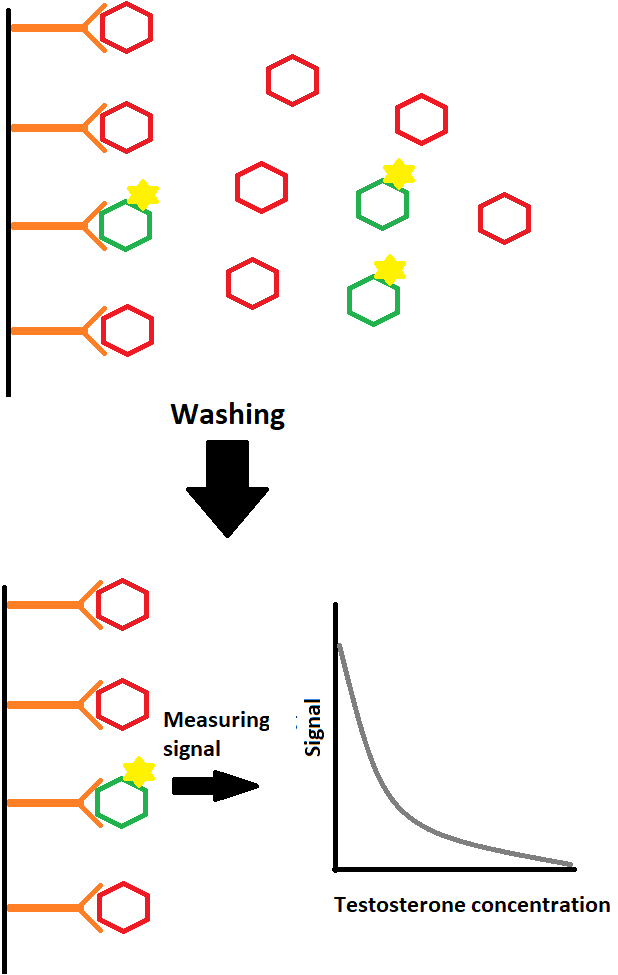
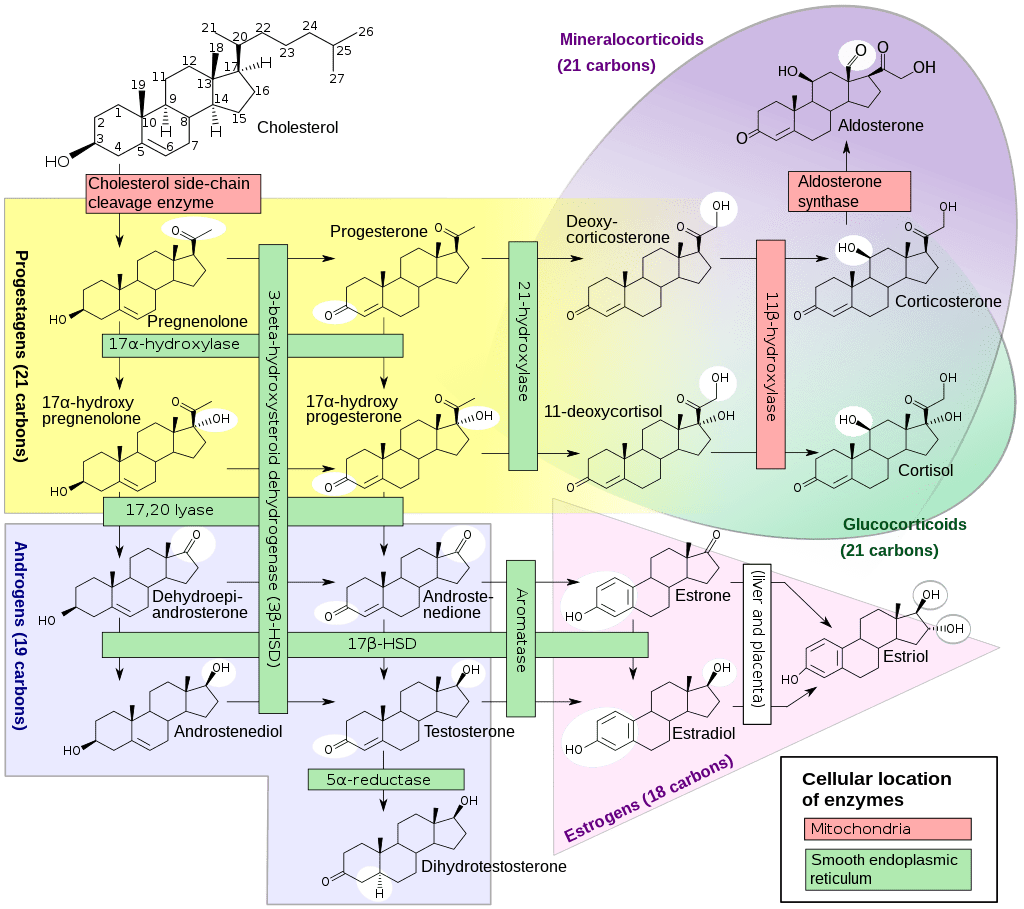
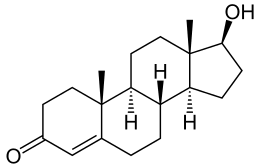
Introduzione:

A causa della natura catabolica del Cortisolo e del desiderio viscerale di molti bodybuilder di mantenere uno stato di anabolismo muscolare costante si è speso molto tempo per cercare di contenere il rilascio di Cortisolo, soprattutto quando non necessario. Gli allenamenti sono stati ridotti in volume e intensità nella sciocca speranza di tenere sotto controllo il Cortisolo nei momenti cruciali della sua funzione fisiologica.
Tuttavia, questa visione cortisolocentrica e del suo impatto in acuto è sia riduttiva che controproducente. Essa non tiene conto della differenza tra gli aumenti acuti e cronici del corticosteroide in questione.
Detto ciò, approfondiamo il ruolo dell’ormone Cortisolo nel processo di ipertrofia muscolare.
Caratteristiche principali del Cortisolo:
Il Cortisolo è un ormone steroideo, appartenente alla classe degli ormoni glucocorticoidi. Quando viene utilizzato come farmaco, è noto come Idrocortisone.
Viene sintetizzato in molti animali, principalmente dalla zona fascicolata della corteccia surrenale nella ghiandola surrenale.[1][2] Viene prodotto in altri tessuti in quantità inferiori.[3] Viene rilasciato con un ciclo diurno e il suo rilascio aumenta in risposta allo stress e a una bassa concentrazione di glucosio nel sangue. Funziona per aumentare la glicemia ematica attraverso la gluconeogenesi, per sopprimere il sistema immunitario e per coadiuvare il metabolismo di grassi, proteine e carboidrati.[4] Diminuisce anche la formazione delle ossa.[5] Molte di queste funzioni sono svolte dal Cortisolo che si lega ai recettori dei glucocorticoidi o dei mineralocorticoidi all’interno della cellula, che poi si legano al DNA per influenzare l’espressione genica.[6][7]
Grazie alle proprietà immunoregolatrici del Cortisone, i derivati farmaceutici del Cortisolo, come il Prednisone, sono utilizzati per controllare forti reazioni allergiche, artrite e altre condizioni infiammatorie. I pericoli di un aumento cronico del Cortisolo sono evidenti nel modo attento in cui questi farmaci vengono dosati e nella breve durata dei trattamenti che li utilizzano.
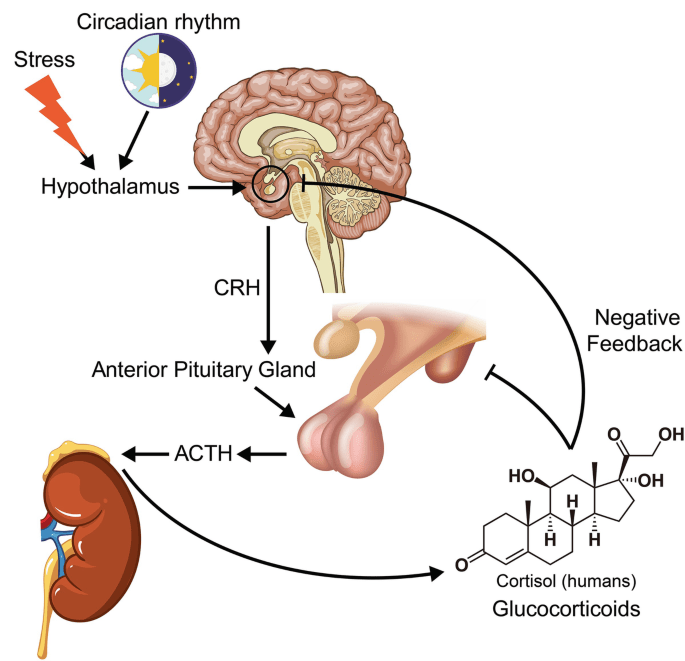
Il Cortisolo è sintetizzato a partire dal Colesterolo. Come già accennato, la sua sintesi avviene nella zona fascicolata della corteccia surrenale (il nome Cortisolo deriva da corteccia). Sebbene la corteccia surrenale produca anche Aldosterone (nella zona glomerulosa) e alcuni ormoni sessuali (nella zona reticolare), il Cortisolo è la sua secrezione principale nell’uomo e in molte altre specie. La midollare della ghiandola surrenale si trova sotto la corteccia e secerne principalmente le catecolamine Adrenalina (Epinefrina) e Noradrenalina (Norepinefrina) sotto stimolazione simpatica.

La sintesi di Cortisolo nella ghiandola surrenale è stimolata dal lobo anteriore dell’ipofisi con l’ACTH; la produzione di ACTH è a sua volta stimolata dal CRH, rilasciato dall’ipotalamo. L’ACTH aumenta la concentrazione di Colesterolo nella membrana mitocondriale interna, attraverso la regolazione della proteina regolatrice steroidogenica acuta. Stimola inoltre la principale fase limitante della sintesi del Cortisolo, in cui il Colesterolo viene convertito in Pregnenolone e catalizzato dal citocromo P450SCC (enzima di scissione della catena laterale).[8]
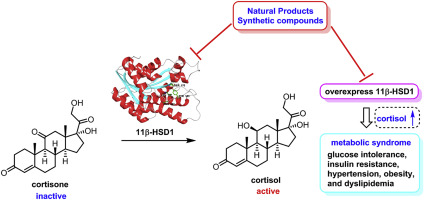
Il Cortisolo viene metabolizzato reversibilmente a Cortisone[9-89] dal sistema dell’11-beta idrossisteroide deidrogenasi (11-beta HSD), che consiste in due enzimi:11-beta HSD1 e 11-beta HSD2. Il metabolismo del Cortisolo a Cortisone comporta l’ossidazione del gruppo ossidrilico in posizione 11-beta.[10]
- L’11-beta HSD1 utilizza il cofattore NADPH per convertire il Cortisone biologicamente inerte in Cortisolo biologicamente attivo.
- L’11-beta HSD2 utilizza il cofattore NAD+ per convertire il Cortisolo in Cortisone.
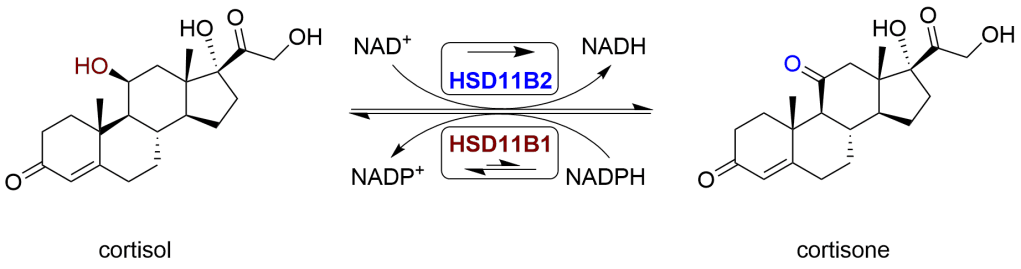
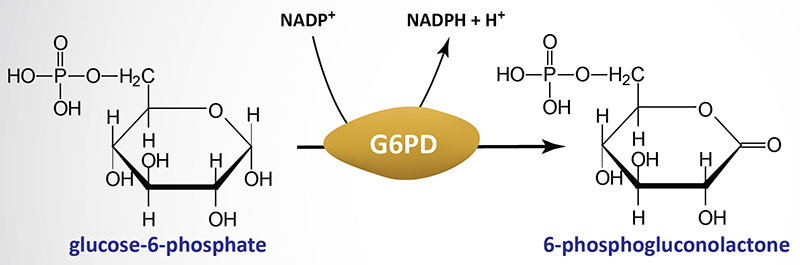
Nel complesso, l’effetto netto è che l’11-beta HSD1 serve ad aumentare le concentrazioni locali di Cortisolo biologicamente attivo in un dato tessuto; l’11-beta HSD2 serve a diminuire le concentrazioni locali di Cortisolo biologicamente attivo. Se è presente l’esoso-6-fosfato deidrogenasi (H6PDH), l’equilibrio può favorire l’attività dell’11-beta HSD1. L’H6PDH rigenera NADPH, aumentando l’attività dell’11-beta HSD1 e diminuendo quella dell’11-beta HSD2.[11]
È stato ipotizzato che un’alterazione dell’11-beta HSD1 svolga un ruolo nella patogenesi dell’obesità, dell’ipertensione e dell’insulino-resistenza, note come sindrome metabolica.[12]
Un’alterazione dell’11-beta HSD2 è stata implicata nell’ipertensione essenziale ed è nota per portare alla sindrome da eccesso apparente di mineralcorticoidi (SAME).
A breve termine, l’aumento del Cortisolo è associato a una diminuzione della sintesi proteica. Il motivo è che una delle azioni del Cortisolo è quella di fornire substrati energetici alternativi all’organismo quando non c’è abbastanza glucosio. Ciò si verifica durante la restrizione calorica o il digiuno, ma anche durante l’esercizio fisico intenso. Il Cortisolo media la degradazione muscolare in modo che gli aminoacidi presenti nel tessuto muscolare possano essere utilizzati per creare glucosio, attraverso la gluconeogenesi.
Il Cortisolo svolge anche un ruolo importante, ma indiretto, nella glicogenolisi epatica e muscolare (la scissione del glicogeno in glucosio-1-fosfato e glucosio) che si verifica in seguito all’azione del Glucagone e dell’Adrenalina. Inoltre, il Cortisolo facilita l’attivazione della glicogeno fosforilasi, necessaria affinché l’Adrenalina abbia effetto sulla glicogenolisi.[13][14]

È paradossale che il Cortisolo promuova non solo la gluconeogenesi nel fegato, ma anche la glicogenesi: è quindi meglio pensare che il Cortisolo stimoli il turnover di glucosio/glicogeno nel fegato. [Questo è in contrasto con l’effetto del cortisolo nel muscolo scheletrico, dove la glicogenolisi è promossa indirettamente attraverso le catecolamine.[15] In questo modo, il Cortisolo e le catecolamine lavorano sinergicamente per promuovere la scissione del glicogeno muscolare in glucosio, che viene poi utilizzato da altri tessuti.
Il Cortisolo aumenta anche i livelli di glucosio nel sangue riducendo l’assorbimento del glucosio nei tessuti muscolari e adiposi, diminuendo la sintesi proteica e aumentando la scomposizione dei trigliceridi di deposito in grassi acidi liberi (lipolisi). Tutte queste modifiche metaboliche hanno l’effetto netto di aumentare i livelli di glucosio nel sangue, che alimentano il cervello e altri tessuti durante la risposta di lotta o fuga [16] … e i workout…
Livelli elevati di Cortisolo, se prolungati, quindi elevati in cronico, possono portare alla proteolisi (disgregazione delle proteine) protratta e al deperimento muscolare.[17] La ragione della proteolisi è quella di fornire ai tessuti interessati una materia prima per la gluconeogenesi; si vedano gli aminoacidi glucogenici.[18] Gli effetti del Cortisolo sul metabolismo lipidico sono più complicati, poiché la lipogenesi è osservata in pazienti con livelli cronici elevati di glucocorticoidi circolanti,[18] mentre un aumento acuto del Cortisolo circolante promuove la lipolisi. La spiegazione abituale di questa apparente discrepanza è anche l’aumento della concentrazione di glucosio nel sangue (per azione del Cortisolo) stimola il rilascio di Insulina. L’Insulina stimola la lipogenesi, quindi questa è una conseguenza indiretta dell’aumento della concentrazione di cortisolo nel sangue, ma si verifica solo su una scala temporale più lunga. Stiamo parlando sempre di condizioni croniche e non in range fisiologici.
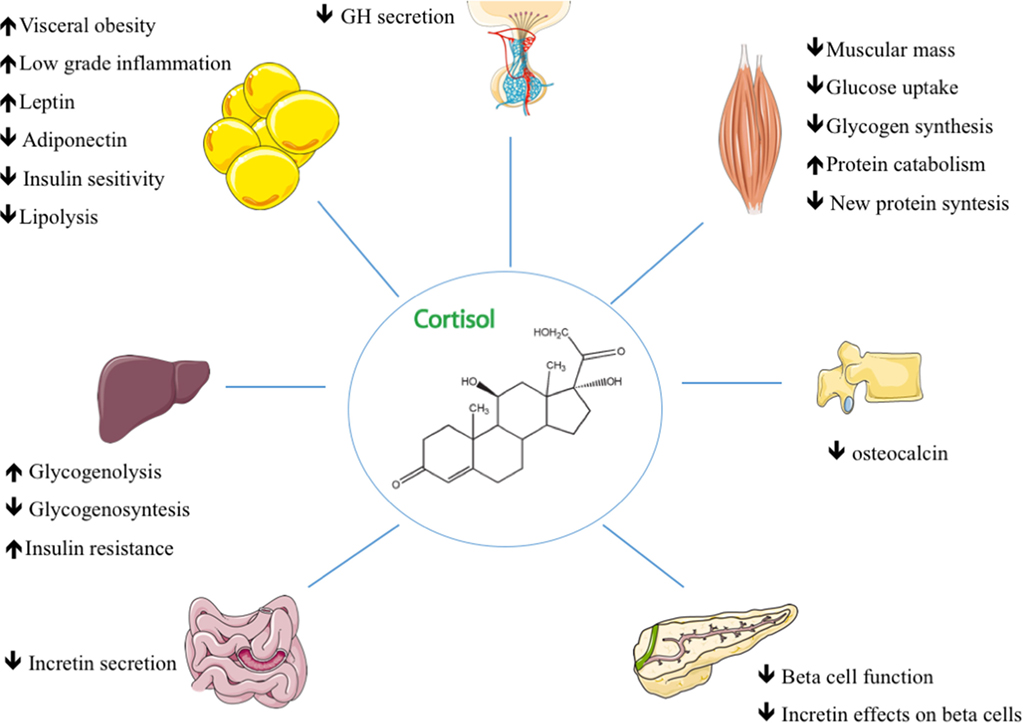
Il Cortisolo è un ormone controinsulinare, contribuisce quindi all’iperglicemia stimolando la gluconeogenesi[19] e inibisce l’utilizzo periferico del glucosio (insulino-resistenza)[19] diminuendo la traslocazione dei trasportatori del glucosio (in particolare GLUT4) sulla membrana cellulare. Il Cortisolo aumenta anche la sintesi di glicogeno (glicogenesi) nel fegato, immagazzinando il glucosio in forma facilmente accessibile.[20] L’effetto permissivo del Cortisolo sull’azione dell’Insulina nella glicogenesi epatica è stato osservato in coltura di epatociti in laboratorio, anche se il meccanismo di questo fenomeno è sconosciuto.

Il Cortisolo aumenta gli aminoacidi liberi nel siero inibendo la formazione di Collagene, diminuendo l’assorbimento di aminoacidi da parte del muscolo e inibendo la sintesi proteica.[21] Il Cortisolo (sotto forma di Opticortinolo) può inibire inversamente le cellule precursori delle IgA nell’intestino dei vitelli.[22] Il Cortisolo inibisce anche le IgA nel siero, come le IgM; tuttavia, non è dimostrato che inibisca le IgE.[23]

Il Cortisolo diminuisce la velocità di filtrazione glomerulare e il flusso plasmatico renale dai reni, aumentando così l’escrezione di fosfati e aumentando la ritenzione di Sodio e acqua e l’escrezione di Potassio agendo sui recettori dei mineralocorticoidi. Aumenta inoltre l’assorbimento di Sodio e acqua e l’escrezione di Potassio nell’intestino.[24]
Il Cortisolo favorisce l’assorbimento del Sodio attraverso l’intestino tenue dei mammiferi.[25] La deplezione di Sodio, tuttavia, non influisce sui livelli di Cortisolo[26] e quindi questo ormone non può essere utilizzato per regolare il Sodio sierico.
Un carico di Sodio aumenta l’intensa escrezione di Potassio da parte del Cortisolo. In questo caso, il Corticosterone è paragonabile al Cortisolo.[27] Affinché il Potassio esca dalla cellula, il Cortisolo sposta un numero uguale di ioni Sodio all’interno della cellula.[28] Ciò dovrebbe facilitare la regolazione del pH (a differenza della normale situazione di carenza di Potassio, in cui due ioni Sodio si spostano all’interno per ogni tre ioni Potassio che si spostano all’esterno, il che si avvicina all’effetto del Desossicorticosterone).
Cortisolo e workout:
Nell’articolo del 1998 “Stress-Related Cortisol Secretion in Men: Relationships with Abdominal Obesity and Endocrine, Metabolic, and Hemodynamic Abnormalities”, i ricercatori del Sahlgrenska University Hospital in Svezia hanno dato diversi contributi preziosi alla nostra comprensione del Cortisolo e delle sue attività differenti in acuto e in cronico. Innanzitutto, le singole letture dei livelli di Cortisolo di un soggetto “non sono altamente informative, perché il Cortisolo viene secreto in modo molto irregolare”.
I livelli di Cortisolo in genere salgono e scendono nel corso della giornata e un livello elevato in un determinato momento non è indicativo di un problema. Al contrario, livelli di Cortisolo variabili, flessibili e reattivi riflettono un sistema endocrino sano. Se il corpo perdesse la capacità di rispondere ai fattori di stress e di regolare in modo appropriato i livelli di Cortisolo, sarebbe un problema.
Un secondo punto che lo studio svedese fornisce riguarda un aspetto che molte persone sbagliano nella loro ricerca di una body fat ridotta, soprattutto addominale. Il Cortisolo viene spesso definito “l’ormone del grasso della pancia”, ma la verità è che il Cortisolo ha il suo maggiore impatto sul grasso viscerale, che è il grasso che circonda gli organi, non il grasso sottocutaneo che copre gli addominali. Se la body fat rende poco visibile il retto addominale, il problema principale non è il Cortisolo.
Nel 2006, Stephen Bird ha pubblicato una serie di articoli che tracciano un buon quadro dei cambiamenti ormonali che si verificano in seguito al sollevamento pesi e di come i diversi interventi nutrizionali influiscano su tali cambiamenti. Nel loro insieme, questi lavori forniscono un quadro della differenza tra i cambiamenti ormonali a breve termine, ad esempio durante o dopo una sessione di allenamento, e quelli a lungo termine.
Nello studio di Bird erano presenti quattro gruppi di soggetti, suddivisi in base a ciò che potevano bere durante gli allenamenti: acqua, aminoacidi essenziali, carboidrati o aminoacidi essenziali più carboidrati. Nell’arco di 12 settimane, tutti i gruppi hanno perso all’incirca la stessa quantità di grasso corporeo, mentre il gruppo che aveva una supplementazione più completa durante l’allenamento (EAA + carboidrati) ha guadagnato più muscoli.
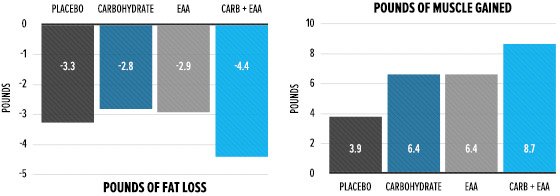
Esaminiamo ora i cambiamenti acuti che hanno accompagnato questa differente risposta. I ricercatori hanno misurato l’aminoacido 3-metil-istidina nelle urine come marcatore della degradazione muscolare. Come mostra il grafico sottostante, il gruppo che ha bevuto solo acqua (il placebo) ha registrato un aumento della disgregazione muscolare 48 ore dopo la sessione di allenamento. I gruppi che hanno assunto aminoacidi essenziali o carboidrati non hanno subito variazioni. Il gruppo che ha assunto la bevanda combinata per l’allenamento ha registrato una diminuzione dei livelli di 3-metil-istidina dopo l’allenamento.
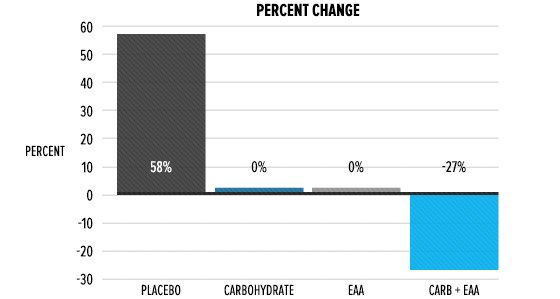
Ciò che è successo, molto banalmente, è che i substrati ingeriti con la bevanda intra-workout hanno tamponato l’uso delle proteine strutturali.
cosa è successo al Cortisolo? Come si può vedere qui sotto, i livelli di Cortisolo 30 minuti dopo l’esercizio fisico sono aumentati di oltre il 50% nel gruppo che ha bevuto acqua, mentre sono rimasti praticamente invariati nel gruppo EAA. Il Cortisolo è diminuito in entrambi i gruppi che hanno assunto Carboidrati come parte dell’alimentazione peri-workout.

Pensate alla gluconeogenesi, il processo che nel fegato crea glucosio da fonti non glucidiche per fornire energia alle cellule del corpo che ne hanno essenziale bisogno. L’organismo non ha bisogno di generare glucosio – un processo ad alto costo metabolico – quando nel flusso ematico c’è glucosio extra dato da una bevanda sportiva. Pertanto, non si è verificato alcun aumento sensibile del Cortisolo in presenza di carboidrati.
Il catabolismo muscolare a breve termine e il picco di Cortisolo in acuto per il gruppo che beveva acqua possono sembrare significativi, ma non bisogna dimenticare che tutti i gruppi hanno guadagnato massa muscolare nel corso dello studio. Il gruppo che ha bevuto solo acqua ha aggiunto quasi due chili di massa muscolare in 12 settimane! Adesso cominciate ad avere chiara la differenza tra effetto in acuto e effetto in cronico?… Il catabolismo è propedeutico all’anabolismo! Eventi in acuto sono largamente compensati dai processi di recupero, in fisiologia.
I picchi di Cortisolo decrescono nel breve termine!
Quindi le persone con la più alta risposta catabolica in acuto hanno comunque guadagnato muscoli? Certo che si! Ed è piuttosto semplice, in realtà: oltre all’attività propedeutica del catabolismo per avviare i processi anabolici, la fisiologia dei soggetti osservati si è adattata allo stimolo dell’allenamento contro-resistenza nel corso del tempo e ha rilasciato sempre meno Cortisolo, anche senza alcun intervento nutrizionale. Nel gruppo che beveva acqua, i livelli di Cortisolo post-esercizio sono diminuiti del 28% nel corso delle 12 settimane dello studio.

Sì, è emerso che i livelli di Cortisolo e la degradazione muscolare acuta non hanno affatto un impatto negativo sull’aumento dei muscoli o sulla perdita di grasso per un periodo di 12 settimane.
I ricercatori della McMaster hanno analizzato la relazione tra i livelli di Cortisolo post-allenamento e i cambiamenti nella forza, nella massa magra e nella sezione trasversale delle fibre muscolari. Hanno scoperto che dopo 12 settimane di allenamento contro-resistenza, alti livelli di Cortisolo post-allenamento erano correlati (anche se debolmente) con l’aumento della massa magra e con le variazioni delle dimensioni delle fibre muscolari di tipo II.
È bene ripeterlo: Le persone con livelli di Cortisolo più elevati in acuto erano quelle che avevano maggiori probabilità di guadagnare più muscoli nel corso dello studio. Tutto il contrario di quello che i limitati detrattori del Cortisolo si sarebbero aspettati!
Conclusioni:
In definitiva, i dati della ricerca sottolineano che l’interruzione di un allenamento contro-resistenza e/o l’abbassamento del intensità e del volume per paura che i livelli di Cortisolo post-allenamento impennassero, è potenzialmente controproducente ai fini ipertrofici. Lo studio della McMaster ha lasciato intendere che potrebbe addirittura esistere una correlazione tra l’innalzamento acuto del Cortisolo e la crescita muscolare a lungo termine.
Detto ciò potreste chiedervi: “Se gli innalzamenti acuti del Cortisolo riflettono una buona sessione di allenamento, allora dovrei smettere di usare i protocolli nutrizionali che riducono il Cortisolo?”. Direi di no, non è assolutamente necessaria l’eliminazione del intra-workout. Le proteine e i carboidrati assunti prima, durante e dopo l’allenamento sono comunque importanti per avviare il processo di recupero.
In queste situazioni, il Cortisolo elevato è semplicemente un indicatore di un allenamento produttivo. E, per non dimenticare, nello studio iniziale di 12 settimane il gruppo che ha assunto aminoacidi e carboidrati ha guadagnato più del doppio dei muscoli rispetto a chi ha bevuto solo acqua; per ovvie ragioni di substrati disponibili e migliore prestazione data dal consumo di CHO.
Un ultimo dubbio: se dobbiamo ignorare i livelli di cortisolo post-allenamento, questo significa che dobbiamo dimenticarci del tutto del Cortisolo e ignorare qualsiasi cambiamento a lungo termine nei nostri livelli?
Assolutamente no!
I cambiamenti a lungo termine del Cortisolo e la diminuzione della sua flessibilità circadiana dovrebbero essere monitorati. Gli effetti sistemici di questo ormone catabolico devono essere presi in considerazione quando si guarda al quadro generale dell’allenamento, della alimentazione e dello stile di vita in generale.
Sonno adeguato, calorie e attenzione al recupero sono i tre fattori più importanti su cui abbiamo il controllo quotidiano. Oltre a questi fattori, è stato suggerito l’uso di integratori come il SAMe o l’Ashwagandha per favorire l’adattamento allo stress e prevenire ulteriormente l’aumento cronico del Cortisolo, o, se atleti “enhanced” farmaci come il Trilostano o l’Aminoglutettimide che sono inibitori della biosintesi steroidea.
Indipendentemente dalla forza e dalla forma fisica, gli elevati livelli di Cortisolo indotti dallo stress cronico possono compromettere il benessere psicofisico. Aumentano il rischio di ipertensione e di malattie cardiovascolari e aggravano qualsiasi altro problema di cui si possa soffrire. Ricordate, tuttavia, di mantenere le cose in prospettiva e di guardare al lungo termine.
Gabriel Bellizzi [CEO BioGenTech]
Approfondimenti supplementari:
- Liquid carbohydrate/essential amino acid ingestion during a short-term bout of resistance exercise suppresses myofibrillar protein degradation.
- Effects of liquid carbohydrate/essential amino acid ingestion on acute hormonal response during a single bout of resistance exercise in untrained men.
- Independent and combined effects of liquid carbohydrate/essential amino acid ingestion on hormonal and muscular adaptations following resistance training in untrained men.
- Stress-related cortisol secretion in men: relationships with abdominal obesity and endocrine, metabolic and hemodynamic abnormalities.
- Associations of exercise-induced hormone profiles and gains in strength and hypertrophy in a large cohort after weight training.
Riferimenti:
- Lightman SL, Birnie MT, Conway-Campbell BL (June 2020). “Dynamics of ACTH and Cortisol Secretion and Implications for Disease”. Endocr Rev.
- Jump up to: Scott E (22 September 2011). “Cortisol and Stress: How to Stay Healthy”. About.com. Retrieved 29 November 2011.
- Taves MD, Gomez-Sanchez CE, Soma KK (July 2011). “Extra-adrenal glucocorticoids and mineralocorticoids: evidence for local synthesis, regulation, and function”. American Journal of Physiology. Endocrinology and Metabolism.
- Hoehn K, Marieb EN (2010). Human Anatomy & Physiology. San Francisco: Benjamin Cummings.
- Jump up to:a b Chyun YS, Kream BE, Raisz LG (February 1984). “Cortisol decreases bone formation by inhibiting periosteal cell proliferation”. Endocrinology.
- Lightman SL, Birnie MT, Conway-Campbell BL (June 2020). “Dynamics of ACTH and Cortisol Secretion and Implications for Disease”. Endocrine Reviews.
- DeRijk RH, Schaaf M, de Kloet ER (June 2002). “Glucocorticoid receptor variants: clinical implications”. The Journal of Steroid Biochemistry and Molecular Biology.
- Margioris AN, Tsatsanis C (2011). “ACTH Action on the Adrenal”. In Chrousos G (ed.). Adrenal physiology and diseases. Endotext.org. Archived from the original on 29 November 2011. Retrieved 5 June 2012.
- ^ Jump up to:a b Finken MJ, Andrews RC, Andrew R, Walker BR (September 1999). “Cortisol metabolism in healthy young adults: sexual dimorphism in activities of A-ring reductases, but not 11beta-hydroxysteroid dehydrogenases”. The Journal of Clinical Endocrinology and Metabolism. 84 (9): 3316–3321. doi:10.1210/jcem.84.9.6009. PMID 10487705.
- ^ Dammann C, Stapelfeld C, Maser E (April 2019). “Expression and activity of the cortisol-activating enzyme 11β-hydroxysteroid dehydrogenase type 1 is tissue and species-specific”. Chemico-Biological Interactions. 303: 57–61. doi:10.1016/j.cbi.2019.02.018. PMID 30796905. S2CID 73467693.
- ^ Atanasov AG, Nashev LG, Schweizer RA, Frick C, Odermatt A (July 2004). “Hexose-6-phosphate dehydrogenase determines the reaction direction of 11beta-hydroxysteroid dehydrogenase type 1 as an oxoreductase”. FEBS Letters. 571 (1–3): 129–133. doi:10.1016/j.febslet.2004.06.065. PMID 15280030. S2CID 6360244.
- ^ Tomlinson JW, Walker EA, Bujalska IJ, Draper N, Lavery GG, Cooper MS, Hewison M, Stewart PM (October 2004). “11beta-hydroxysteroid dehydrogenase type 1: a tissue-specific regulator of glucocorticoid response”. Endocrine Reviews.
- Martin PA, Crump MH (2003). “The adrenal gland”. In Dooley MP, Pineda MH (eds.). McDonald’s veterinary endocrinology and reproduction (5th ed.). Ames, Iowa: Iowa State Press. ISBN 978-0-8138-1106-2.
- ^ Coderre L, Srivastava AK, Chiasson JL (June 1991). “Role of glucocorticoid in the regulation of glycogen metabolism in skeletal muscle”. The American Journal of Physiology. 260 (6 Pt 1): E927–32.
- Kuo T, McQueen A, Chen TC, Wang JC (2015). “Regulation of Glucose Homeostasis by Glucocorticoids”. In Wang JC, Harris C (eds.). Glucocorticoid Signaling: From Molecules to Mice to Man. Advances in Experimental Medicine and Biology. Vol. 872. Springer. pp. 99–126.
- Khani S, Tayek JA (December 2001). “Cortisol increases gluconeogenesis in humans: its role in the metabolic syndrome”. Clin Sci (Lond).
- Simmons PS, Miles JM, Gerich JE, Haymond MW (February 1984). “Increased proteolysis. An effect of increases in plasma cortisol within the physiologic range”. The Journal of Clinical Investigation.
- Laycock JF (2013). Integrated endocrinology. Meeran, Karim. Chichester, West Sussex, UK: Wiley-Blackwell.
- Brown DF, Brown DD (2003). USMLE Step 1 Secrets: Questions You Will Be Asked on USMLE Step 1. Philadelphia: Hanley & Belfus. p. 63.
- Baynes J, Dominiczak M (2009). Medical biochemistry. Mosby Elsevier.
- Manchester, KL (1964). “Sites of Hormonal Regulation of Protein Metabolism”. In Allison, NH; Munro JB (eds.). Mammalian Protein Metabolism. New York: Academic Press.
- Husband AJ, Brandon MR, Lascelles AK (October 1973). “The effect of corticosteroid on absorption and endogenous production of immunoglobulins in calves”. The Australian Journal of Experimental Biology and Medical Science.
- ^ Posey WC, Nelson HS, Branch B, Pearlman DS (December 1978). “The effects of acute corticosteroid therapy for asthma on serum immunoglobulin levels”. The Journal of Allergy and Clinical Immunology.
- ^ McKay LI, Cidlowski JA (2003). “Physiologic and Pharmacologic Effects of Corticosteroids”. In Kure DW, Pollock RE, Weichselbaum RR, Bast RC, Ganglier TS, Holland JF, Frei E (eds.). Holland-Frei Cancer Medicine (6th ed.). Hamilton, Ontario: Decker.
- ^ Sandle GI, Keir MJ, Record CO (1981). “The effect of hydrocortisone on the transport of water, sodium, and glucose in the jejunum. Perfusion studies in normal subjects and patients with coeliac disease”. Scandinavian Journal of Gastroenterology.
- ^ Mason PA, Fraser R, Morton JJ, Semple PF, Wilson A (August 1977). “The effect of sodium deprivation and of angiotensin II infusion on the peripheral plasma concentrations of 18-hydroxycorticosterone, aldosterone and other corticosteroids in man”. Journal of Steroid Biochemistry.
- Muller AF, Oconnor CM (1958). An International Symposium on Aldosterone. Little Brown & Co. p. 58.
- Knight RP, Kornfeld DS, Glaser GH, Bondy PK (February 1955). “Effects of intravenous hydrocortisone on electrolytes of serum and urine in man”. The Journal of Clinical Endocrinology and Metabolism.