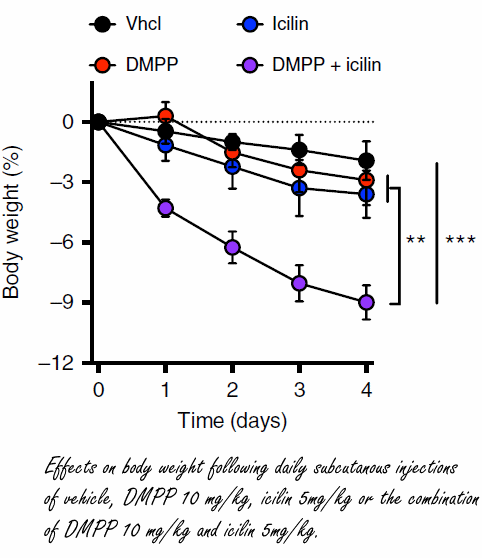
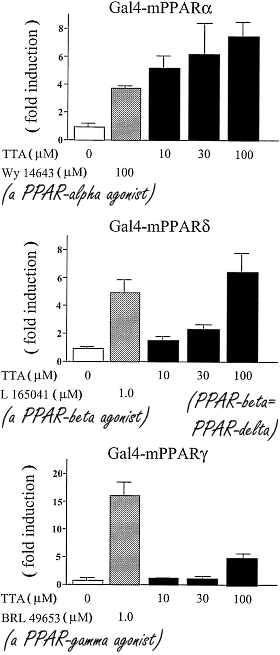
Scienziati danesi potrebbero essersi imbattuti in una strategia farmacologica completamente nuova per indurre la perdita di grasso. Essi hanno scoperto che uno stimolo simultaneo dei recettori dell’Icilina e del DMPP porta a una rapida perdita di grasso nei topi. (1)
Il recettore TRPM8 si trova negli strati esterni della pelle, dove si percepisce il freddo. Se il TRMP8 viene attivato, l’attività del metabolismo lipidico aumenta a causa, tra le altre cose, di un incremento dell’ossidazione lipidica nel tessuto adiposi marrone. Per esempio, il Mentolo stimola il recettore TRMP8.
I ricercatori hanno scoperto un composto sintetico che stimola il TRMP8 e che è quasi 200 volte più potente del Mentolo e 2,5 volte più efficace: l’Icilina. Questo composto è stato iniettato a dosaggi differenti in topi sovrappeso permettendo ai ricercatori di osservare una perdita di grasso da parte degli animali dipendente da un aumento del loro dispendio energetico.
Tuttavia, il primo autore dello studio che qui si sta trattando, Christoffer Clemmensen, affiliato all’Università di Copenhagen, ha affermato che il recettore TRMP8 non è presente nel tessuto adiposo marrone. (2) Sembra che il recettore del freddo [TRMP8] sulla superficie della pelle mandi un segnale al cervello che successivamente attiva il tessuto adiposo marrone tramite i connettori nervosi.
I topi hanno subito una riduzione della percentuale del grasso corporeo quando trattati con Icilina per via di un aumento del loro turnover energetico. Tuttavia, l’effetto riscontrato non era sufficientemente incisivo da portare ad un effetto reale su ipotetici pazienti, anche la dove l’azione della molecola venisse ottimizzata. Un punto fondamentale che i ricercatori danesi non hanno tralasciato, è che se si desidera modificare il peso corporeo di un soggetto, non è sufficiente prendere di mira solamente il turnover energetico. Come affermato da Christoffer Clemmensen, per creare davvero un bilancio energetico negativo, è necessario anche fare in modo che il soggetto mangi di meno.
E’ noto che i fumatori mangiano meno delle persone che non fumano, e ciò è dovuto in parte perché la Nicotina attiva il sottotipo del recettore Nicotinico (nAChR) alfa3beta4. I ricercatori hanno scoperto che la sostanza sintetica Dimetilfenilpiperazinio [DMPP] funziona allo stesso modo. E così ne hanno sperimentato l’effetto iniettandola nei topi sovrappeso.
Come conseguenza, gli animali trattati mangiavano di meno e perdevano peso.
Il DMPP non solo sopprime l’appetito, ma ha anche un enorme effetto positivo sul metabolismo glucidico rispetto alla Nicotina, la quale ha un effetto negativo sul grasso epatico e sulla sensibilità all’insulina.
I ricercatori hanno cosomministrato ai topi l’Icilina e il DMPP ottenendo un effetto sinergico sulla perdita del peso corporeo. Presi singolarmente, i due composti non causano effetti particolarmente significativi sulla perdita di peso, ma una loro assunzione combinata ha mostrato di poter causare una marcata perdita di peso.
I ricercatori non sono certi che tale trattamento possa essere pienamente efficace e sicuro nell’uomo. Questo studio, come affermato anche dagli stessi autori, rappresenta semplicemente una prova preliminare.
Nel febbraio di quest’anno ho scritto un articolo nel quale riportavo alcuni studi svolti sul SARM GSK20881078. Tra gli studi citati ve ne era uno svolto sull’uomo (studio di fase 1).(1) Attualmente i ricercatori della GlaxoSmithKline stanno proseguendo i test sugli esseri umani. Un recente studio che ha preso in esame gli effetti del GSK20881078 sugli esseri umani, il quale verrà a breve pubblicato sul Journal of Clinical Endocrinology & Metabolism, ha mostrato, per la prima volta, il potenziale anabolizzante di questo SARM nell’uomo.(2) Tuttavia, lo studio suggerisce anche che il GSK20881078 potrebbe avere degli effetti collaterali non di entità non trascurabile.
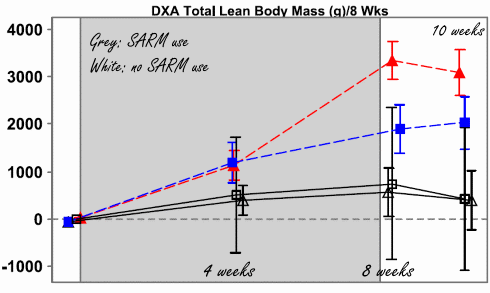
I ricercatori hanno somministrato il GSK20881078 a 100 persone sane di età superiore ai 50 per 8 settimane. I soggetti presi in esame erano sia maschi che femmine e sono stati trattati con dosaggi differenti.
I soggetti di sesso maschile ai quali è stata somministrata una dose di 4mg/die di GSK20881078 hanno avuto un guadagno di 1,5Kg di massa magra in 8 settimane di trattamento. Le donne alle quali è stata somministrata una dose di 1,5mg/die di GSK2088078, hanno avuto un guadagnato 3Kg di massa magra.
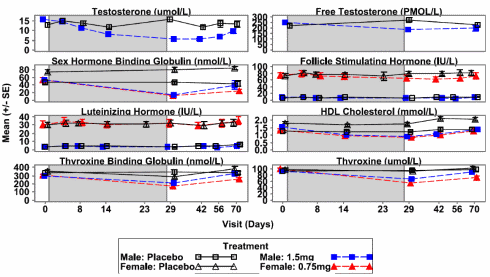
Ovviamente, come per altri SARM o PED in generale, l’uso del GSK20881078 può portare ad alcuni effetti collaterali. Il livello di Testosterone totale negli uomini si è ridotto di due terzi durante il trattamento con il SARM. Due settimane dopo la fine del trattamento, i livelli di Testosterone non erano ancora tornati in soglia basale (nei range della fascia d’età dei soggetti esaminati).
Anche gli effetti sui livelli di HDL non sono da sottovalutare. Infatti, si è verificato un calo del 30-45% delle lipoproteine ad alta densità.
Gli effetti collaterali di cui sopra sono stati rilevati a metà del periodo di somministrazione. Quale entità abbiano gli effetti collaterali dopo 8 settimane di trattamento non è dato saperlo dal momento che i ricercatori, stranamente e con non pochi dubbi, non lo hanno verificato. Per risolvere questo “mistero d’omissione”, o ,per lo meno, per farsi un idea plausibile sulle reali cause di ciò, basta conoscere i finanziatori dello studio (GlaxoSmithKline). Un altro dato mancante è rappresentato dall’impatto della molecola a livello epatico.
I ricercatori concludono affermando che è il trattamento con GSK20881078 può portare ad aumenti potenzialmente significativi a livello clinico della massa magra con una risposta differenziale tra i sessi. I cambiamenti nella chimica clinica sono stati coerenti con quelli precedentemente segnalati per altri SARM ed erano relativamente miti, monitorabili e reversibili. Ulteriori ricerche sono ora in programma per analizzare gli effetti di aumento della massa magra osservati.
Sebbene le risposte nell’aumento della massa magra possano sembrare promettenti ai dosaggi indicati, specie negli individui di sesso femminile, l’entità dei possibili effetti collaterali rende questo composto decisamente meno interessante per gli atleti; specie se paragonato con altri PED attualmente in uso ed il loro rapporto tra possibili benefici ed effetti collaterali. Un calo del HDL del 35-40% con una dose di 4mg/die per 4 settimane non lascia spazio a rosee previsioni sull’effetto riscontrabile con l’uso di dosi più elevate per lo stesso lasso di tempo o per periodi più lunghi.
Una nota interessante, che va oltre lo studio che qui è stato trattato, è data dal fatto che l’antidoping ha già sviluppato un test per la rilevazione del GSK20881078.(3)
A seguito di uno studio in vitro svolto da tossicologi dell’Università di Basilea e pubblicato nel 2012, è emerso che il Fluoxymesterone può causare un aumento significativo dei livelli di Cortisolo. (1) Argomento da me accennato nell’articolo dedicato alla molecola in questione.
Come risaputo, il Fluoxymesterone è un AAS orale metilato in C-17, con un potere androgeno elevato e non soggetto all’enzima aromatasi.
Nonostante quest’ultimo punto, la casa produttrice (Pfizer) riporta nelle avvertenze del prodotto una caratteristica che non ci si aspetta da una molecola priva di attività estrogenica diretta e indiretta: “L’edema, con o senza insufficienza cardiaca congestizia, può essere una seria complicanza in pazienti con preesistente malattia cardiaca, renale o epatica”.(2)
I ricercatori hanno scoperto il meccanismo attraverso il quale il Fluoxymesterone può causare edema e quindi aggravare ulteriormente la sua influenza sulla salute del sistema cardiovascolare. Il Fluoxymesterone si lega all’enzima 11-beta-HSD2, enzima preposto alla conversione del Cortisolo in Cortisone (inattivo). Di conseguenza il gruppo 11-idrossile del Fluoxymesterone viene convertito in un gruppo 11-oxo. Poiché l’attività dell’11-beta-HSD2 subisce una riduzione, si osserva un aumento della concentrazione di Cortisolo.
I ricercatori hanno esteso la loro ricerca ad altri composti al fine di valutarne una possibile azione sui meccanismi di conversione del Cortisolo in Cortisone. L’esito è stata la scoperta della marcata attività inibitoria dell’11-beta-HSD2 da parte del Fluoxymesterone. Durante l’esperimento è stato constatato che il Fluoxymesterone esplica una potenza maggiore sull’alterazione dei livelli di Cortisolo dell’Acido Glicirretico, sostanza presente nella liquirizia che causa un aumentano della produzione endogena di corticosteroidi, attraverso l’inibizione degli enzimi 4 e 5-beta-reduttasi che inattivano gli steroidi.(3)
L’Oxymesterone – o 4-idrossi-17-metil-testosterone – e, in misura minore, l’Oxymetholone inibiscono l’11-beta-HSD2 quasi quanto il Fluoxymesterone.
I ricercatori hanno scoperto anche che il Fluoxymesterone non può interagire direttamente con i Recettori del Cortisolo. Ma, le concentrazioni aumentate di Cortisolo portano, ovviamente, ad un consequenziale aumento dell’attività di quest’ultimo.
Un eccesso di Cortisolo altera l’equilibrio elettrolitico data l’interazione dello steroide con i Recettori Mineralocorticoidi. Induce il corpo a trattenere più sodio e quindi aumenta la ritenzione idrica extracellulare. Ciò significa che la quantità di plasma nel sangue aumenta e di conseguenza aumenta la pressione sanguigna. Oltre a ciò, un livello elevato di Cortisolo ha un effetto restringente sui vasi sanguigni, cosa che, a sua volta, causa un aumento della pressione sanguigna. Infine, un livello elevato di Cortisolo rende i vasi sanguigni più suscettibili ai danni causati dall’accumulo di colesterolo nelle loro pareti.
Pertanto, gli AAS con azione inibitoria nei confronti dell’11-beta-HSD2, come il Fluoxymesterone, possono avere un azione avversa maggiore nel causare effetti cardiovascolari avversi.
Si è ipotizzata anche una differenza nell’impatto sull’11-beta-HSD2 e i livelli di Cortisolo tra assunzione orale e somministrazione per iniezione con maggiore influenza data da quest’ultima. La cosa potrebbe con molta probabilità essere legata alle modifiche che la farmacocinetica subisce con la somministrazione tramite iniezione rispetto alla classica somministrazione orale.
Per avere una visione d’insieme più completa riguardo al Fluoxymesterone vi rimando all’artico ad esso dedicato.
La supplementazione con estratto di semi d’uva sembra poter fornire una protezione a cuore e vasi sanguigni durante l’uso di AAS. Questa possibile azione è emersa da uno studio svolto su animali che i ricercatori dell’Università di Tanta (Egitto) hanno pubblicato sul Oxidative Medicine and Cellular Longevity.(1)
I ricercatori hanno svolto l’esperimento dividendo ratti da laboratorio maschi in 4 gruppi. Durante le otto settimane di durata dell’esperimento, i ricercatori non hanno dato alcuna sostanza attiva al primo gruppo di ratti [Controllo].
Procianidina C1 (membro della famiglia delle Proantocianidine presenti nei semi d’uva)
Il secondo gruppo di ratti ha ricevuto una dose consistente di un estratto di semi d’uva purificato due volte alla settimana tramite un sondino gastrico. Questo estratto, prodotto dalla Merck, conteneva Proantocianidine [GSPE]. Se i ratti fossero stati esseri umani, avrebbero ricevuto una dose media di 700mg di Estratto di semi d’uva due volte a settimana.
Il terzo gruppo di animali esaminati è stato trattato con un iniezione settimanale di Boldenone. Infine, Il quarto gruppo è stato trattato con un iniezione settimanale di Boldenone insieme alla supplementazione con estratto di semi d’uva.
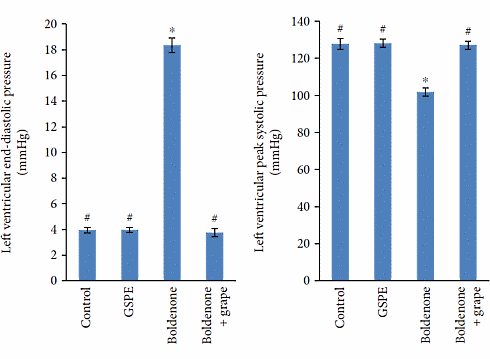
I ratti trattati con Boldenone hanno sviluppato ipertrofia cardiaca. L’aggiunta dell’estratto di semi d’uva al trattamento con Boldenone ha annullato tale effetto.
La figura riportata di seguito mostra come il Boldenone ha indotto l’ipertrofia cardiaca. Se il ventricolo sinistro del cuore – la parte del muscolo cardiaco che pompa sangue ricco di ossigeno nel corpo – si era contratto, il sangue era ancora sotto un elevata pressione nei vasi sanguigni degli animali esaminati a cui era stato somministrato il Boldenone [Sinistra].
Questo indicava che il cuore era sottoposto ad un lavoro maggiore legato alla condizione ipertrofica. Ciò indicava anche che la salute dei vasi sanguigni non era ottimale.
Ancora una volta, l’estratto di semi d’uva ha annullato questo effetto.
Le figure sottostanti mostrano cosa è successo esattamente ai vasi sanguigni degli animali trattati. Il Boldenone aveva attivato gli enzimi NOX. Gli enzimi NOX producono radicali liberi. Le cellule immunitarie, ma anche le cellule che formano i vasi sanguigni, producono enzimi NOX. Questi enzimi sono utili quando il corpo sta combattendo degli agenti patogeni, ma se questi enzimi si attivano senza una buona ragione, possono causare rigidità dei vasi sanguigni e, di conseguenza, danneggiarli.
Il NOX2 è prodotto dalle cellule endoteliali nelle pareti dei vasi sanguigni, il NOX4 nel muscolo cardiaco.
I ricercatori concludono affermando che, queste nuove scoperte sull’attività antiossidante delle Proantocianidine contenute nell’estratto di semi d’uva dovrebbero servire come base per lo sviluppo di migliori strategie chemiopreventive o terapeutiche per la tossicità cardiaca indotta dal Boldenone.
E’ necessario però fare alcune precisazioni…
La dose di Boldenone usata dai ricercatori era tutto sommato contenuta: 5mg/Kg a settimana. Se questo non è un errore di battitura, l’equivalente umano della dose utilizzata dai ricercatori è di circa 70mg a settimana. Gli utilizzatori di AAS assumono dosi settimanali minime di 200-250mg dello steroide preso in esame, e alcuni arrivano anche al grammo. Ovviamente, insieme al Boldenone vengono generalmente somministrati altri AAS. Quest’ultimo punto riduce ulteriormente la validità protettiva del supplemento esaminato nel presente studio in un contesto di utilizzo di AAS a scopo dopante.
In conclusione, è assai improbabile che una supplementazione con estratto di semi d’uva possa fornire una protezione verso l’ipertrofia cardiaca o il danno endoteliale negli utilizzatori di AAS.
Con il termine Antiestrogeni ci si riferisce genericamente ad una classe di farmaci aventi azione diretta o indiretta sull’attività tissutale e/o concentrazione ematica degli estrogeni. Agiscono bloccando il recettore dell’estrogeno (ER) e/o riducendo o sopprimendo la sintesi estrogenica.(1)(2) Una recente categoria di agenti facenti parte di questa classe di farmaci, i SERD (Selective Estrogen Receptor Degrader), esplicano la loro azione antiestrogena degradando/sottoregolando il recettore dell’estrogeno. Gli Antiestrogeni sono una delle tre classi di farmaci antagonisti dell’ormone sessuale, insieme agli Antiandrogeni e agli Antiprogestinici.(3)
Largamente utilizzati in ambito sportivo, in special modo nell’ambiente culturistico, con il fine di controllare l’attività estrogenica durante l’uso di AAS aromatizzabili, o aventi attività estrogenica intrinseca, e durante la PCT con lo scopo aggiunto di stimolare la ripresa dell’HPTA, questi farmaci hanno un discreto carico di effetti collaterali tra i quali, quelli che destano maggior preoccupazione nell’atleta previdente, vi sono la dislipidemia (aumento dell’LDL, dei Trigliceridi, riduzione del HDL e alterazione delle loro ratio), l’atralgia (dolore articolare), il calo della libido/disfunzione erettile e l’affaticamento/letargia. Non sono di certo da meno le preoccupazioni legate all’alterazione dell’Asse GH/IGF-1 o la riduzione delle potenzialità di induzione ipertrofica di un ciclo in seguito ad una eccessiva soppressione dell’attività e/o delle concentrazioni estrogeniche. Ma esiste un’altra preoccupazione legata all’uso di composti antiestrogeni, ed è la possibilità che si verifichi un rebound estrogenico in seguito al l’oro uso. Purtroppo, la letteratura a disposizione è al quanto scarsa e poco chiara nella specifica del problema. E’ possibile, però, fare maggiore chiarezza sulla questione analizzando le caratteristiche dei composti antiestrogeni e il loro impatto, passando in rassegna tutti i componenti dell’addizione (Recettori Estrogeni e enzima Aromatasi). In questo articolo cercherò di esporre un ragionamento logico grazie al quale, seppur non avendo una risposta definitiva, sarà possibile avere un idea, la più concreta possibile, sul binomio antiestrogeni/rebound estrogenico.
Una analisi della questione…
Recettori dell’Estrogeno, SERM e “rebound estrogenico”
Un dimero della regione legame-ligando del ERa.
I Recettori degli Estrogeni (ER) sono un gruppo di proteine presenti all’interno delle cellule. Sono recettori attivati dall’ormone estrogeno (con maggiore attività del 17β-estradiolo).(4) Esistono due classi di ER: i Recettori degli Estrogeni nucleari (ERα e ERβ), che sono membri della famiglia dei recettori nucleari e dei recettori intracellulari, ed i Recettori degli Estrogeni di Membrana (MR) (GPER (GPR30), ER-X e Gq-mER), che sono per lo più recettori accoppiati alla proteina G. In questa sede ci si riferirà ai primi (ER).
Una volta attivato dall’estrogeno, l’ER è in grado di traslocare nel nucleo e legarsi al DNA per regolare l’attività di diversi geni (ciò significa che è un fattore di trascrizione del DNA). Tuttavia, ha anche funzioni aggiuntive indipendenti dal legame con il DNA.(5)
Poiché l’estrogeno è un ormone steroideo, può passare attraverso le membrane fosfolipidiche della cellula, e pertanto i recettori non hanno bisogno di essere legati alla membrana per potersi legare a loro volta con l’estrogeno.
L’estrogeno esplica la sua attività cellulare attraverso un azione Genomica e Non-Genomica.
• Genomica
In assenza di ormoni, i ER si trovano in gran parte nel citosol. Il legame dell’ormone al recettore innesca un numero di eventi che iniziano con la migrazione del recettore dal citosol nel nucleo, la dimerizzazione del recettore e il successivo legame del dimero del recettore a specifiche sequenze di DNA conosciute come elementi di risposta ormonale. Il complesso DNA / recettore quindi recluta altre proteine che sono responsabili della trascrizione del DNA a valle in mRNA e, infine, in una proteina la quale porta a dei cambiamenti nella funzione cellulare. I recettori degli estrogeni si trovano anche all’interno del nucleo della cellula, ed entrambi i sottotipi del recettore dell’estrogeno hanno un dominio di legame con il DNA e possono funzionare come fattori di trascrizione per regolare la produzione di proteine.
Il recettore interagisce anche con la proteina attivatore 1 e Sp-1 per promuovere la trascrizione, attraverso diversi coattivatori come il PELP-1.(6)
L’acetilazione diretta del recettore alfa dell’estrogeno ai residui della lisina nella regione cerniera mediante il p300 regola la transattivazione e la sensibilità ormonale.(7)
• Non-Genomica
Alcuni recettori per gli estrogeni sono presenti nelle membrana della superficie della cellula e possono essere rapidamente attivati dall’esposizione di questa agli estrogeni.(8)(9)
Inoltre, alcuni ER possono associarsi alle membrane cellulari legandole alla caveolina-1 e formarmando complessi con la proteine G, striatina, tirosina chinasi del recettore (es. EGFR e IGF-1) e tirosina chinasi non recettoriale (es. Src). (6)(8) Attraverso la striatina, alcuni di questi ER legati alla membrana possono portare a livelli aumentati di Ca2 + e ossido nitrico (NO).(10) Attraverso il recettore tirosin chinasi, i segnali vengono inviati al nucleo attraverso la via della proteina chinasi attivata dal mitogeno (MAPK / ERK) e la via del fosfoinositide 3-chinasi (Pl3K / AKT).(11) La glicogeno sintasi chinasi-3 (GSK) -3β inibisce la trascrizione dal ER nucleare inibendo la fosforilazione della serina 118 dell’ERa nucleare. La fosforilazione di GSK-3β rimuove il suo effetto inibitorio, e questo può essere ottenuto tramite il pathway PI3K / AKT e il pathway MAPK / ERK, tramite rsk.
Il 17β-estradiolo ha dimostrato di attivare il recettore GPR30 accoppiato alla proteina G.(12) Tuttavia, la localizzazione subcellulare e il ruolo di questo recettore sono ancora oggetto di controversie.(13)
ERb.
Gli estrogeni e gli ER sono implicati nel cancro al seno, nel carcinoma ovarico, nel cancro del colon, nel cancro alla prostata e nel cancro dell’endometrio. Il carcinoma del colon avanzato è associato a una perdita di ERβ, l’ER predominante nel tessuto del colon, e il tumore del colon è trattato con agonisti specifici per ERβ.(14)
Sappiamo che i recettori degli estrogeni sono sovraespressi in circa il 70% dei casi di cancro al seno, indicati come “ER-positivi”, e possono essere dimostrati in tali tessuti mediante l’immunoistochimica.(15) E’ ipotizzabile ,quindi, che gli atleti più sensibili agli effetti estrogenici presentino un espressione dei ER più elevata del normale, cosa che li porta a sviluppare con maggiore facilità effetti avversi dati da un eccesso dei livelli estrogenici e/o da un aumento della loro attività dato dalla cosomministrazione con progestinici (es. Nandrolone e Trenbolone).
I Modulatori Selettivi del Recettore dell’Estrogeno (SERM) sono composti antiestrogenici che agiscono a livello del ER. (16) Una caratteristica che distingue queste sostanze dagli agonisti e antagonisti ER puri (cioè agonisti completi e antagonisti silenti) è che la loro azione è diversa nei vari tessuti, garantendo in tal modo la possibilità di inibire selettivamente o stimolare l’azione estrogenica in diversi tessuti.
I SERM sono agonisti parziali competitivi del ER.(17) Tessuti diversi presentano differenti gradi di sensibilità all’attività degli estrogeni, pertanto i SERM esplicano effetti estrogenici o antiestrogeni a seconda del tessuto specifico con il quale interagiscono e della percentuale di attività intrinseca (IA) del composto in questione.(18) Un esempio di SERM con alta IA, e quindi di effetti prevalentemente estrogenici, è rappresentato dal Clorotrianisene, mentre un esempio di SERM con bassa IA, e quindi avente per lo più attività antiestrogenica, è rappresentato dall’Ethamoxytrifetolo. SERM come il Clomifene e il Tamoxifene, largamente utilizzati in ambito sportivo, sono considerabili come composti con valore IA intermedio essendo molecole con una azione bilanciata tra effetti estrogenici e antiestrogenici. Il Raloxifene è un SERM che presenta una azione antiestrogenica maggiore del Tamoxifene; entrambi hanno una attività estrogenica (sebbene differente) a livello osseo, ma il Raloxifene presenta una attività antiestrogenica nell’utero mentre il Tamoxifene ha un azione estrogenica nel tessuto dell’utero.(18)
Tamoxifene
Il Tamoxifene è un farmaco di prima linea per il trattamento del carcinoma mammario metastatico ER-positivo. È usato per la riduzione delle possibilità di sviluppo del cancro al seno nelle donne ad alto rischio, come trattamento adiuvante del nodo ascellare negativo e positivo, e nel carcinoma duttale in situ.(19)(20)
Il Tamoxifene è classificabile come un profarmaco, dal momento che la sua affinità per la proteina bersaglio (ER) è limitata. Il Tamoxifene viene metabolizzato nel fegato dall’isoforma del citocromo CYP2D6 e CYP3A4 in metaboliti attivi come l’Afimoxifene (4-idrossitamoxifene; 4-OHT) e l’Endoxifene (N-desmetil-4-idrossitamoxifene) (21) che presentano una affinità da 30 a 100 volte maggiore per il ER rispetto al Tamoxifene. (22) Questi metaboliti attivi competono con gli estrogeni per il legame con il recettore. Nel tessuto mammario, il 4-OHT agisce come un antagonista del ER in modo da inibire la trascrizione dei geni che reagiscono agli estrogeni. (23) Il Tamoxifene ha rispettivamente il 7% e il 6% dell’affinità dell’Estradiolo per il ERα e il ERβ, mentre il 4-OHT ha il 178% e il 338% dell’affinità dell’Estradiolo per il ERα e il ERβ.(24)
Afimoxifene (4-OHT)
Il 4-OHT si lega al ER, il complesso ER/Tamoxifene recluta altre proteine note come co-repressori e quindi si lega al DNA per modulare l’espressione genica. Alcune di queste proteine includono la NCoR e la SMRT. (25) La funzione del Tamoxifene può essere regolata da una serie di variabili diverse, compresi i fattori di crescita.(26) Il Tamoxifene deve bloccare le proteine del fattore di crescita come ErbB2/HER2 (27) perché è stato dimostrato che livelli elevati di ErbB2 si manifestano nei tumori resistenti al Tamoxifene.(28) Il Tamoxifene sembra richiedere una proteina PAX2 affinché possa esplicare il suo pieno effetto antitumorale. (27)(29) In presenza di un elevata espressione della PAX2, il complesso Tamoxifene/ER è in grado di sopprimere l’espressione della proteina pro-proliferativa del ERBB2. Al contrario, quando l’espressione del AIB-1 è superiore alla PAX2, il complesso di Tamoxifene/ER aumenta l’espressione del ERBB2 con conseguente stimolazione della crescita del cancro al seno. (27)(30)
Il 4-OHT si lega al ER in modo competitivo (rispetto all’estrogeno agonista) nelle cellule tumorali e in altri bersagli tissutali, producendo un complesso nucleare che riduce la sintesi del DNA e inibisce gli effetti degli estrogeni. È un agente non steroideo con potenti proprietà antiestrogeniche che competono con gli estrogeni per i siti di legame nel seno e in altri tessuti. Il Tamoxifene fa sì che le cellule rimangano nelle fasi G0 e G1 del ciclo cellulare. Poiché impedisce alle cellule (pre) cancerose di dividersi ma non provoca la morte cellulare, il Tamoxifene è citostatico piuttosto che citocida.
La letteratura scientifica riguardante l’attività del Tamoxifene è a dir poco complessa ed occorre prestare particolare attenzione ai dati disponibili per stabilire se il Tamoxifene, o il suo metabolita 4-idrossi, abbiano il maggiore impatto complessivo.
Norendoxifene
Il Norendoxifene (N, N-didesmetil-4-idrossitamoxifene), un altro metabolita attivo del Tamoxifene, è stato osservato agire come un potente inibitore dell’aromatasi competitivo (IC50 = 90 nM), cosa che a sua volta può amplificare l’attività antiestrogenica complessiva del Tamoxifene.(31)
Come già accennato in precedenza, e come molti sapranno, il Tamoxifene è largamente utilizzato in ambito sportivo, sia da solo che in abbinamento con altri SERM come il Clomifene ( in PCT) o con AI (“on-cycle” e/o in PCT). La sua applicazione all’interno di una preparazione che contempla l’uso di AAS aromatizzabili, alla luce di quanto esposto pocanzi, lo vede come agente preventivo o di trattamento dell’attività estrogenica a livello tissutale, in specie per quanto concerne l’attività estrogenica nel tessuto mammario al fine di evitare (o “tamponare”) la comparsa della ginecomastia. In un contesto PCT tale composto, oltre ad esercitare la funzione di regolazione dell’attività estrogenica appena esposta, agendo a livello ipotalamico stimola il rilascio di GnRH e, consequenzialmente, di LH ed FSH dall’Ipofisi che a loro volta stimoleranno la sintesi di Testosterone e la spermatogenesi.
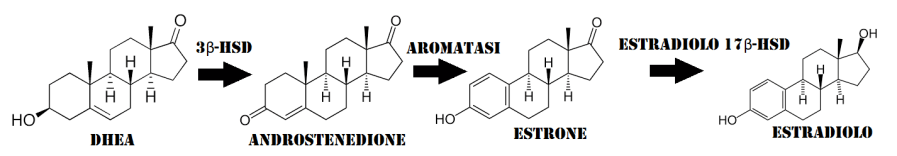
Il suo utilizzo massivo e cronico è stato spesso collegato aneddoticamente a rebound estrogenico. Ora, conoscendo la complessità d’azione che questo composto (ed i suoi metaboliti) ha sul controllo dell’attività estrogenica, si può facilmente ipotizzare che un suo uso protratto (legato anche alla dose e, quindi, al suo impatto sulla attività estrogenica sistemica) possa innescare degli adattamenti reattivi con conseguente aumento dell’attività estrogenica attraverso l’incremento dei livelli serici di Estradiolo e dell’attività non-genomica dello steroide (ipotizzabile anche un aumento del numero dei ER). Nel corso degli anni sono state esposte diverse ipotesi volte a spiegare i meccanismi attraverso i quali un abuso di Tamoxifene possa portare ad un rebound estrogenico. Una di queste ipotesi venne riportata all’inizio del secolo dal compianto A.L. Rea il quale affermava che la causa andasse ricercata nell’aumento del rilascio di DHEA da parte delle ghiandole surrenali e dalla sua successiva (e aumentata) conversione in Androstenedione e, attraverso l’intervento dell’enzima aromatasi che lo converte in Estrone e la successiva azione del estradiolo 17beta-deidrogenasi, Estradiolo.(32) In breve, secondo questa teoria i processi innescati causerebbero l’instaurarsi di livelli di E2 cronicamente alti con conseguente impossibilità del SERM di esplicare la sua azione. Questa teoria seppur, in parte, possa dare una spiegazione logica dei possibili meccanismi implicati manca di alcuni tasselli. Il principale “tallone d’Achille” è rappresentato dai livelli di E2 che, una volta aumentati, diventano dei competitor recettoriali più aggressivi rispetto al 4-OHT (che ricordiamo avere il 178% dell’affinità dell’Estradiolo per il ERα). Ciò potrebbe avvenire in situazioni di calo delle concentrazioni di 4-OHT seguenti alla riduzione del dosaggio del farmaco o alla sua cessazione, quindi, in questo ultimo caso, esplicabili in crescendo nei 7-14 giorni successivi all’interruzione della somministrazione e con una durata indeterminata. Di conseguenza, sembra più plausibile che l’aumento delle concentrazioni di E2, durante l’uso del Tamoxifene, si affianchi ad un consequenziale incremento dell’attività Non-Genomica dell’ormone e da un aumentato numero di ER. Seguendo questa logica, una volta interrotto l’uso del Tamoxifene, queste condizioni tenderanno ad aggravarsi come gli effetti avversi a loro legati.
Consultando la bibliografia scientifica disponibile, non si trovano accenni su un possibile rebound estrogenico in seguito all’uso di Tamoxifene, ma si parla nello specifico di “resistenza al Tamoxifene” o “sottoregolazione degli ER”.(33)(34) Nel caso della “resistenza al Tamoxifene” sembra che l’aumento dell’espressione del gene MACROD2 porti ad una risposta negativa all’azione del SERM con conseguente proliferazione delle cellule cancerose estradiolo-dipendenti. La sovra espressione di tale gene sembra essere di base genetica anche se non si esclude una risposta di adattamento in seguito ad uso cronico del composto in questione.
Raloxifene
Il Raloxifene, un altro SERM discretamente utilizzato nella pratica sportiva, è un agonista-antagonista misto del ER.(35)(36)(37) Ha effetti estrogenici a livello osseo ed epatico con effetti antiestrogenici nei seni e nell’utero. Le azioni biologiche del Raloxifene sono quindi ampiamente mediate dal legame con i ER. Questo legame determina l’attivazione di percorsi estrogenici in alcuni tessuti (agonismo) e il blocco di questi in altri (antagonismo). Le sue caratteristiche d’azione similari a quelle del Tamoxifene, sembrano poter far pensare ad un medesimo e ipotetico meccanismo che possa portare ad un rebound estrogenico. Questa volta la letteratura scientifica sembra dare alcune conferme. In un caso studio (38), una paziente di 66 anni si è presentata con recidiva metastatica acuta estrogeno-positiva e progesterone-positiva, carcinoma mammario Her-2 / neu-negativo, lesioni ossee (colonna lombare, bacino), noduli polmonari, metastasi epatiche, antigene tumorale elevato 15 e enzimi epatici, dispepsia e diarrea. La paziente aveva assunto Raloxifene per circa 8 anni. Dopo la sospensione del farmaco, parametri e sintomi clinici sono migliorati rapidamente senza terapia oncologica o altre forme di trattamento. Tre mesi dopo la sospensione del Raloxifene, l’oncologo ha prescritto alla paziente l’uso della Capecitabina dato che non riteneva plausibile un effetto di rebound estrogenico (anti-estrogen withdrawal effect – AEWE). Tuttavia, la regressione duratura è stata più indicativa di un effetto rebound dato dal Raloxifene rispetto alla chemioterapia o ad altri interventi. In seguito la paziente si è mostrata asintomatica con un buono stato di prestazione. La regressione metastatica epatica è stata confermata, senza alcun trattamento oncologico somministrato negli ultimi 16 mesi e circa 23 mesi dopo il termine d’uso del Raloxifene. Questo caso evidenzia la necessità di esaminare pazienti con carcinoma mammario per la possibilità di un AEWE con l’uso di Raloxifene o con altri SERM . Ovviamente, il caso presentato non è molto comparabile, soprattutto per quanto riguarda i tempi di somministrazione, ad un BodyBuilder supplementato chimicamente nella “media” ma, ciò nonostante, ci offre un indizio sulla probabilità che si possa manifestare un rebound estrogenico con l’uso di SERM.
Enzima Aromatasi, Inibitori della Aromatasi e “rebound estrogenico”
Enzima Aromatasi
L’Enzima Aromatasi, chiamato anche estrogeno sintetasi o estrogeno sintasi, è un enzima responsabile del processo fondamentale della biosintesi degli Estrogeni. Denominato CYP19A1, questo enzima è un membro della superfamiglia del citocromo P450 (EC 1.14.14.1), che sono monoossigenasi che catalizzano molte reazioni coinvolte nella steroidogenesi. In particolare, l’Aromatasi è responsabile dell’aromatizzazione degli Androgeni in Estrogeni. L’enzima Aromatasi è sintetizzato in molti tessuti tra cui le gonadi (cellule della granulosa), cervello, tessuto adiposo, placenta, vasi sanguigni, pelle e ossa, nonché nei tessuti dell’endometriosi, dei fibromi uterini, del cancro al seno e del cancro dell’endometrio.
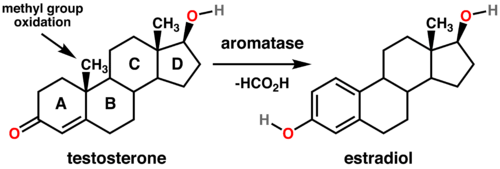
L’Aromatasi è localizzato nel reticolo endoplasmatico dove è regolato da promotori tissutali che sono a loro volta controllati da ormoni, citochine e altri fattori. Catalizza gli ultimi passaggi della biosintesi degli estrogeni dagli androgeni (in particolare, converte l’Androstenedione in Estrone e il Testosterone in Estradiolo). Queste fasi comprendono tre idrossilazioni successive del gruppo 19-metilico degli androgeni, seguite dall’eliminazione simultanea del gruppo metilico come formiato e aromatizzazione dell’anello A.
Reazioni generali per la conversione del Testosterone in Estradiolo catalizzata dall’Aromatasi. Gli Steroidi sono formati da quattro anelli fusi (A-B-C-D). L’Enzima Aromatasi converte l’anello “A” in uno stato aromatico.
Il gene esprime due varianti di trascrizione. (39) Nell’uomo, il gene CYP19, situato sul cromosoma 15q21.1, codifica per l’Enzima Aromatasi. (40) Il gene ha nove esoni codificanti e un numero di primi esoni non codificanti alternativi che regolano l’espressione specifica del tessuto. (41)
Il CYP19 è presente in un cordato precoce divergente, l’anfiosso cefalocordato (il Florida lancelet, Branchiostoma floridae), ma non nel precedente tunicato divergente Ciona intestinalis. Pertanto, gli evoluzionisti ipotizzano che il gene Aromatasi si sia evoluto precocemente nell’evoluzione dei cordati e non sembra essere presente negli invertebrati non-cordati (ad esempio insetti, molluschi, echinodermi, spugne, coralli). Tuttavia, gli Estrogeni possono essere sintetizzati in alcuni di questi organismi, attraverso altri percorsi sconosciuti.
I fattori noti che aumentano l’attività dell’Aromatasi includono l’età, l’obesità, l’Insulina, le gonadotropine e l’alcol. L’attività dell’Aromatasi risulta diminuita dalla Prolattina, dall’ormone anti-Mülleriano e dal glifosato , un comune erbicida.(42) L’attività dell’Aromatasi sembra essere migliorata in alcuni tessuti estrogeno-dipendenti come il tessuto mammario, nel carcinoma dell’endometrio, nell’endometriosi e nei fibromi uterini.
Gli Inibitori dell’Aromatasi (AI) sono una gruppo di farmaci usati nel trattamento del carcinoma mammario nelle donne in postmenopausa e nella ginecomastia negli uomini. Come i SERM, trovano un largo uso off-label in ambito sportivo durante la somministrazione di AAS aromatizzabili o durante la PCT. Possono anche essere utilizzati per la chemioprevenzione in donne ad alto rischio.
Esistono due tipi di Inibitori dell’Aromatasi approvati per il trattamento del carcinoma mammario e, quindi, diffusi anche per l’uso off-label: (43)
– Gli inibitori steroidei irreversibili, come l’Exemestano (nome commerciale Aromasin), formano un legame permanente e disattivante con l’Enzima Aromatasi.
– Gli inibitori non steroidei, come l’Anastrozolo (nome commerciale Arimidex) e il Letrozolo (nome commerciale Femara), inibiscono la sintesi degli Estrogeni attraverso la competizione reversibile per l’Enzima Aromatasi.
Gli inibitori dell’Aromatasi disponibili (AI) includono:
– Non selettivi:
• L’Aminoglutetimide, il quale però inibisce l’enzima P450scc agendo come inibitore della biosintesi di tutti gli ormoni steroidei (aprirò una nota a riguardo più avanti).
• Testolactone (nome commerciale Teslac) – Selettivi:
• Anastrozolo (Arimidex)
• Letrozolo (Femara)
• Exemestano (Aromasin)
• Vorozolo (Rivizor)
• Formestano (Lentaron)
• Fadrozolo (Afema)
– Non classificati:
• 1,4,6-Androstatrien-3,17-dione (ATD)
• 4-Androstene-3,6,17-trione (“6-OXO”)
Oltre agli AI farmaceutici, alcuni composti naturali hanno mostrato effetti di inibizione dell’Aromatasi, come le foglie di damiana. Il loro impatto non è stato pienamente chiarito sull’uomo.
Gli Inibitori dell’Aromatasi agiscono, proprio come suggerisce il nome, inibendo l’azione dell’enzima Aromatasi, che converte gli Androgeni in Estrogeni mediante un processo chiamato aromatizzazione. Poiché il tessuto mammario è stimolato dagli Estrogeni, diminuirne la produzione è un modo per sopprimere la recidiva del tessuto tumorale del seno. La principale fonte di Estrogeni è rappresentata dalle ovaie nelle donne in premenopausa, mentre nelle donne in post-menopausa la maggior parte degli Estrogeni del corpo viene prodotta nei tessuti periferici (al di fuori del SNC) e anche in alcuni siti del SNC in varie regioni del cervello. L’Estrogeno viene prodotto e agisce localmente in questi tessuti, ma qualsiasi estrogeno circolante, che esercita effetti estrogenici sistemici in uomini e donne, è il risultato dell’Estrogeno che sfugge al metabolismo locale e si diffonde nel sistema circolatorio.(44)
Come già accennato pocanzi, i composti AI sono anch’essi, al pari dei SERM, largamente utilizzati in ambito sportivo, sia come agenti di controllo dei livelli estrogenici durante l’uso di AAS aromatizzabili (uso preventivo della comparsa di effetti estrogenici), in caso di ginecomastia (spesso in combinazione con un SERM, specie se l’AI utilizzato è l’Exemestano) o in combinazione con i SERM in ambito PCT (specie nella fase preliminare dove viene utilizzata l’hCG).
Più che con i SERM, il rebound estrogenico è stato riportato, soprattutto aneddoticamente, con l’uso di AI, specialmente quelli reversibili (vedi Anastrozolo e Letrozolo).
L’Anastrozolo ed il Letrozolo agiscono legandosi in modo reversibile all’Enzima Aromatasi (unità eme del citocromo P450) e, attraverso l’inibizione competitiva, blocca la conversione degli Androgeni in Estrogeni nei tessuti periferici (extragonali).
Il Letrozolo ha dimostrato, attraverso studi clinici, di poter abbassare rapidamente il livello degli estrogeni fino al 65%. Il motivo principale è probabilmente legato alla capacità che la molecola ha di abbassare drasticamente gli estrogeni attraverso un legame competitivo reversibile al gruppo eme della relativa unità del citocromo P450. L’Anastrozolo, il quale agisce similmente al Letrozolo, ha mostrato una riduzione del livello estrogenico in soggetti di sesso maschile del 50%.(45) Il problema di un possibile rebound estrogenico con questi composti nasce proprio dalla loro natura “reversibile”.
Rebound estrogenici sono stati riportati sia con l’uso di Letrozolo che con l’uso di Anastrozolo, sebbene il Letrozolo, avendo un azione inibitoria più marcata, sembra causare rebound di intensità maggiore dopo la sua interruzione. La causa del rebound estrogenico indotto da cessazione d’uso di Letrozolo o di Anastrozolo è proprio legata al comportamento che queste due molecole esplicano nei confronti dell’Enzima aromatasi. Il legame tra la molecola di Letrozolo o di Anastrozolo con l’Enzima Aromatasi è solo temporanea e non decreta la completa de-attivazione dell’enzima responsabile della conversione degli Androgeni in Estrogeni. Una volta interrotta l’assunzione del composto, i livelli di Aromatasi possono salire significativamente con la possibile comparsa di un rebound estrogenico. Una pratica per evitare che ciò si verifichi consiste nell’uso limitato dei due composti e in una loro graduale sospensione. Con questi farmaci, il rebound estrogenico può essere “multifattoriale” derivando non solo dalla cessazione del farmaco in questione ma anche da un incremento dell’espressione dell’Enzima Aromatasi come risposta adattativa all’uso (specie nel lungo termine). Ciò significa che, anche durate un utilizzo cronico, i livelli di E2 possono mostrare degli aumenti, aumenti che diverranno maggiormente significativi una volta cessato l’uso del farmaco. Cessata l’azione del composto non solo viene a mancare un controllo dell’aromatizzazione ma questa risulta anche incrementata rispetto ai tassi pre-utilizzo (l’aumento dell’espressione dell’aromatasi è un comportamento adattativo che si può manifestare anche durante cicli particolarmente lunghi). Prendendo in considerazione la vita attiva del Letrozolo e dell’Anastrozolo, il possibile rebound estrogenico potrebbe manifestarsi in crescendo dopo 64-120h circa dall’ultima assunzione.
Exemestano
L’Exemestano, invece, è un inibitore dell’Aromatasi steroideo irreversibile di tipo I, strutturalmente correlato al substrato naturale 4-androstenedione. Agisce come un falso substrato per l’Enzima Aromatasi e viene trasformato in un intermedio che si lega irreversibilmente al sito attivo dell’enzima causandone l’inattivazione, un effetto noto anche come “inibizione suicida”. Essendo strutturalmente simile agli obiettivi dell’enzima, l’Exemestano si lega in modo permanente a quest’ultimo, impedendo la sua azione di conversione degli Androgeni in Estrogeni. Il tasso di soppressione degli Estrogeni da parte dell’Exemestano varia dal 35% per l’Estradiolo (E2) al 70% per l’Estrone (E1).(46)
Grazie alla sua caratteristica di “inibitore selettivo”, l’Exemestano sembra non causare un rebound estrogenico dopo la sua cessazione. Nonostante ciò, un suo uso temporalmente protratto potrebbe (teoricamente) causare, similmente a quanto accade con l’uso di Letrozolo e Anastrozolo, un aumento dell’espressione dell’Enzima Aromatasi nonché un aumento del numero di ER come risposta adattativa.
Ovviamente, questa possibilità può interessare tutti gli AI con legame irreversibile (es. Formestano).
Queste sono semplici ipotesi nate da una riflessione sulle possibili cause e meccanismi che potrebbero (teoricamente) portare al manifestarsi di un rebound estrogenico con l’uso di tali composti. La letteratura scientifica, purtroppo, non ci aiuta a fare molta chiarezza sulla connessione AI/rebound estrogenico, sebbene esistono alcuni studi nei quali la cosa viene accennata.(47)
Aminoglutetimide
*Nota sull’Aminoglutetimide: inibendo l’enzima P450scc e agendo, di conseguenza, come inibitore della biosintesi di tutti gli ormoni steroidei, l’abuso di Aminoglutetimide può potenzialmente causare non solo un rebound estrogenico ma anche un rebound dei livelli di cortisolo. Lo stesso vale per il farmaco Trylostano.
Conclusioni
Basarsi per la maggior parte sui dati aneddotici è un azzardo, anche perché la maggior parte delle variabili soggettive in gioco rimangono celate. Banalmente, alcuni lamentano rebound estrogenici che alla fine non risultano legati all’uso del SERM o del AI ma alla loro (o del Preparatore) ignoranza, come quando cessano l’utilizzo di AAS, e di SERM e/o AI, senza preoccuparsi di svolgere un adeguata PCT convinti, magari, che un po’ di Mesterolone (Proviron) risolvi tutto. Infatti, la maggior parte dei casi di presunti rebound estrogenici SERM o AI dipendenti sono causati da una repentina cessazione d’uso di questi e di AAS, oppure da una PCT mal pianificata e/o che non ha dato i risultati sperati (vedi anche alterazione della Testosterone:Estradiolo ratio). Queste condizioni sono legate più che altro ad una alterazione del HPTA data dall’uso di AAS e non ad una presunta azione diretta del SERM e/o AI precedentemente utilizzati.
In conclusione, l’uso ponderato e consapevole è l’unica vera arma che l’atleta supplementato chimicamente (o il Preparatore che lo segue) ha per far si che ipotetici rebound non si manifestino.
Gabriel Bellizzi
Riferimenti:
1- “Definition of antiestrogen – NCI Dictionary of Cancer Terms, Definition of antiestrogen – NCI Dictionary of Cancer Terms”.,
2- Jump up ^ “antiestrogen” at Dorland’s Medical Dictionary
3- Jump up ^ Judi Lindsley Nath (2006). Using Medical Terminology: A Practical Approach. Lippincott Williams & Wilkins. pp. 977–. ISBN 978-0-7817-4868-1.
4- Dahlman-Wright K, Cavailles V, Fuqua SA, Jordan VC, Katzenellenbogen JA, Korach KS, Maggi A, Muramatsu M, Parker MG, Gustafsson JA (Dec 2006). “International Union of Pharmacology. LXIV. Estrogen receptors”. Pharmacological Reviews. 58 (4): 773–81. doi:10.1124/pr.58.4.8. PMID 17132854.
5- Levin ER (Aug 2005). “Integration of the extranuclear and nuclear actions of estrogen”. Molecular Endocrinology. 19 (8): 1951–9. doi:10.1210/me.2004-0390. PMC 1249516 . PMID 15705661.
6- Levin ER (Aug 2005). “Integration of the extranuclear and nuclear actions of estrogen”. Molecular Endocrinology. 19 (8): 1951–9. doi:10.1210/me.2004-0390. PMC 1249516 . PMID 15705661.
7- Wang C, Fu M, Angeletti RH, Siconolfi-Baez L, Reutens AT, Albanese C, Lisanti MP, Katzenellenbogen BS, Kato S, Hopp T, Fuqua SA, Lopez GN, Kushner PJ, Pestell RG (May 2001). “Direct acetylation of the estrogen receptor alpha hinge region by p300 regulates transactivation and hormone sensitivity”. The Journal of Biological Chemistry. 276 (21): 18375–83. doi:10.1074/jbc.M100800200. PMID 11279135.
8- Zivadinovic D, Gametchu B, Watson CS (2005). “Membrane estrogen receptor-alpha levels in MCF-7 breast cancer cells predict cAMP and proliferation responses”. Breast Cancer Research. 7 (1): R101–12. doi:10.1186/bcr958. PMC 1064104 . PMID 15642158.
9- Björnström L, Sjöberg M (Jun 2004). “Estrogen receptor-dependent activation of AP-1 via non-genomic signalling”. Nuclear Receptor. 2 (1): 3. doi:10.1186/1478-1336-2-3. PMC 434532 . PMID 15196329.
10- Lu Q, Pallas DC, Surks HK, Baur WE, Mendelsohn ME, Karas RH (Dec 2004). “Striatin assembles a membrane signaling complex necessary for rapid, nongenomic activation of endothelial NO synthase by estrogen receptor alpha”. Proceedings of the National Academy of Sciences of the United States of America. 101 (49): 17126–31. doi:10.1073/pnas.0407492101. PMC 534607 . PMID 15569929.
11- Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, Masushige S, Gotoh Y, Nishida E, Kawashima H, Metzger D, Chambon P (Dec 1995). “Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase”. Science. 270 (5241): 1491–4. doi:10.1126/science.270.5241.1491. PMID 7491495.
12- Prossnitz ER, Arterburn JB, Sklar LA (Feb 2007). “GPR30: A G protein-coupled receptor for estrogen”. Molecular and Cellular Endocrinology. 265-266: 138–42. doi:10.1016/j.mce.2006.12.010. PMC 1847610 . PMID 17222505.
13- Otto C, Rohde-Schulz B, Schwarz G, Fuchs I, Klewer M, Brittain D, Langer G, Bader B, Prelle K, Nubbemeyer R, Fritzemeier KH (Oct 2008). “G protein-coupled receptor 30 localizes to the endoplasmic reticulum and is not activated by estradiol”. Endocrinology. 149 (10): 4846–56. doi:10.1210/en.2008-0269. PMID 18566127.
14- Harris HA, Albert LM, Leathurby Y, Malamas MS, Mewshaw RE, Miller CP, Kharode YP, Marzolf J, Komm BS, Winneker RC, Frail DE, Henderson RA, Zhu Y, Keith JC (Oct 2003). “Evaluation of an estrogen receptor-beta agonist in animal models of human disease”. Endocrinology. 144 (10): 4241–9. doi:10.1210/en.2003-0550. PMID 14500559.
15- Deroo BJ, Korach KS (Mar 2006). “Estrogen receptors and human disease”. The Journal of Clinical Investigation. 116 (3): 561–70. doi:10.1172/JCI27987. PMC 2373424 . PMID 16511588.
16- Riggs BL, Hartmann LC (Feb 2003). “Selective estrogen-receptor modulators — mechanisms of action and application to clinical practice”. The New England Journal of Medicine. 348 (7): 618–29. doi:10.1056/NEJMra022219. PMID 12584371.
17- Cameron JL, Cameron AM (20 November 2013). Current Surgical Therapy. Elsevier Health Sciences. pp. 582–. ISBN 978-0-323-22511-3.
18- Huang X, Aslanian RG (19 April 2012). Case Studies in Modern Drug Discovery and Development. John Wiley & Sons. pp. 392–394. ISBN 978-1-118-21967-6.
19- Pickar JH, Komm BS (Sep 2015). “Selective estrogen receptor modulators and the combination therapy conjugated estrogens/bazedoxifene: A review of effects on the breast”. Post Reproductive Health. 21 (3): 112–21. doi:10.1177/2053369115599090. PMID 26289836.
20- Mirkin S, Pickar JH (Jan 2015). “Selective estrogen receptor modulators (SERMs): a review of clinical data”. Maturitas. 80 (1): 52–7. doi:10.1016/j.maturitas.2014.10.010. PMID 25466304.
21- Desta Z, Ward BA, Soukhova NV, Flockhart DA (Sep 2004). “Comprehensive evaluation of tamoxifen sequential biotransformation by the human cytochrome P450 system in vitro: prominent roles for CYP3A and CYP2D6”. The Journal of Pharmacology and Experimental Therapeutics. 310 (3): 1062–75. doi:10.1124/jpet.104.065607. PMID 15159443.
22- Ahmad A, Shahabuddin S, Sheikh S, Kale P, Krishnappa M, Rane RC, Ahmad I (December 2010). “Endoxifen, a new cornerstone of breast cancer therapy: demonstration of safety, tolerability, and systemic bioavailability in healthy human subjects”. Clinical Pharmacology and Therapeutics. 88 (6): 814–7. doi:10.1038/clpt.2010.196. PMID 20981001.
23- Wang DY, Fulthorpe R, Liss SN, Edwards EA (Feb 2004). “Identification of estrogen-responsive genes by complementary deoxyribonucleic acid microarray and characterization of a novel early estrogen-induced gene: EEIG1”. Molecular Endocrinology. 18 (2): 402–11. doi:10.1210/me.2003-0202. PMID 14605097.
24- Kuhl H (2005). “Pharmacology of estrogens and progestogens: influence of different routes of administration”. Climacteric. 8 Suppl 1: 3–63. doi:10.1080/13697130500148875. PMID 16112947.
25- Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M (Dec 2000). “Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription”. Cell. 103 (6): 843–52. doi:10.1016/S0092-8674(00)00188-4. PMID 11136970.
26- Massarweh S, Osborne CK, Creighton CJ, Qin L, Tsimelzon A, Huang S, Weiss H, Rimawi M, Schiff R (Feb 2008). “Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function”. Cancer Research. 68 (3): 826–33. doi:10.1158/0008-5472.CAN-07-2707. PMID 18245484.
27- Hurtado A, Holmes KA, Geistlinger TR, Hutcheson IR, Nicholson RI, Brown M, Jiang J, Howat WJ, Ali S, Carroll JS (Dec 2008). “Regulation of ERBB2 by oestrogen receptor-PAX2 determines response to tamoxifen”. Nature. 456 (7222): 663–6. Bibcode:2008Natur.456..663H. doi:10.1038/nature07483. PMC 2920208 . PMID 19005469.
28- Osborne CK, Bardou V, Hopp TA, Chamness GC, Hilsenbeck SG, Fuqua SA, Wong J, Allred DC, Clark GM, Schiff R (Mar 2003). “Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer”. Journal of the National Cancer Institute. 95 (5): 353–61. doi:10.1093/jnci/95.5.353. PMID 12618500.
29- “New Mechanism Predicts Tamoxifen Response: PAX2 gene implicated in tamoxifen-induced inhibition of ERBB2/HER2-mediated tumor growth”. http://www.modernmedicine.com. 2008-11-13. Archived from the original on 2011-07-14. Retrieved 2008-11-14.
30- “Study sheds new light on tamoxifen resistance”. News. CORDIS News. Archived from the original on 2009-02-20. Retrieved 2008-11-14.
31- Liu J, Flockhart PJ, Lu D, Lv W, Lu WJ, Han X, Cushman M, Flockhart DA (2013). “Inhibition of cytochrome p450 enzymes by the e- and z-isomers of norendoxifen”. Drug Metab. Dispos. 41 (9): 1715–20. doi:10.1124/dmd.113.052506. PMC 3876808 . PMID 23824607.
32- Chemical muscle enhancement. Report. B.B. desk reference. di Author L. Rea. Pag. 106.
33- https://www.rdmag.com/article/2014/12/journal-watch-tamoxifen-news-again
34- https://www.ncbi.nlm.nih.gov/pubmed/14687597
35- Bryant HU (2001). “Mechanism of action and preclinical profile of raloxifene, a selective estrogen receptor modulation”. Rev Endocr Metab Disord. 2 (1): 129–38. PMID 11704975.
36- Thiebaud D, Secrest RJ (2001). “Selective estrogen receptor modulators: mechanism of action and clinical experience. Focus on raloxifene”. Reprod. Fertil. Dev. 13 (4): 331–6. PMID 11800172.
37- Gizzo S, Saccardi C, Patrelli TS, Berretta R, Capobianco G, Di Gangi S, Vacilotto A, Bertocco A, Noventa M, Ancona E, D’Antona D, Nardelli GB (2013). “Update on raloxifene: mechanism of action, clinical efficacy, adverse effects, and contraindications”. Obstet Gynecol Surv. 68 (6): 467–81. doi:10.1097/OGX.0b013e31828baef9. PMID 23942473.
38- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5739193/
39- “Entrez Gene: CYP19A1 cytochrome P450, family 19, subfamily A, polypeptide 1”.
40- Toda K, Shizuta Y (April 1993). “Molecular cloning of a cDNA showing alternative splicing of the 5′-untranslated sequence of mRNA for human aromatase P-450”. European Journal of Biochemistry. 213 (1): 383–9. doi:10.1111/j.1432-1033.1993.tb17772.x. PMID 8477708.
41- Czajka-Oraniec I, Simpson ER (2010). “Aromatase research and its clinical significance”. Endokrynologia Polska. 61 (1): 126–34. PMID 20205115.
42- Gasnier C, Dumont C, Benachour N, Clair E, Chagnon MC, Séralini GE (August 2009). “Glyphosate-based herbicides are toxic and endocrine disruptors in human cell lines”. Toxicology. 262 (3): 184–91. doi:10.1016/j.tox.2009.06.006. PMID 19539684.
43- Mokbel K (2002). “The evolving role of aromatase inhibitors in breast cancer”. Int. J. Clin. Oncol. 7 (5): 279–83. doi:10.1007/s101470200040 (inactive 2017-01-15). PMID 12402060.
44- Simpson ER (2003). “Sources of estrogen and their importance”. J. Steroid Biochem. Mol. Biol. 86 (3–5): 225–30. doi:10.1016/S0960-0760(03)00360-1. PMID 14623515.
45- Leder BZ, Rohrer JL, Rubin SD, Gallo J, Longcope C (March 2004). “Effects of aromatase inhibition in elderly men with low or borderline-low serum testosterone levels”. J. Clin. Endocrinol. Metab. 89 (3): 1174–80. doi:10.1210/jc.2003-031467. PMID 15001605.
46- Mauras, N; Lima, J; Patel, D; Rini, A; Di Salle, E; Kwok, A; Lippe, B (2003). “Pharmacokinetics and Dose Finding of a Potent Aromatase Inhibitor, Aromasin (Exemestane), in Young Males”. The Journal of Clinical Endocrinology & Metabolism. 88 (12): 5951–6. doi:10.1210/jc.2003-031279. PMID 14671195.
47- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3263690/
L’uso della Gonadotropina Corionica Umana (hCG) è largamente diffuso nell’ambiente culturistico. Usata principalmente per ripristinare la funzionalità gonadale in seguito all’uso di AAS, questo peptide vede la sua applicazione anche durante l’uso di questa classe di farmaci (ciclo, Bridge o TRT), o di altri composti causanti un ciclo di feedback negativo dell’HPTA (vedi SARM), al fine di prevenire l’istaurarsi di una disfunzione testicolare. Fin dai primi anni della sua applicazione su soggetti di sesso maschile, l’hCG è stato oggetto di speculazioni riguardo la possibilità o meno che il suo uso possa portare ad una desensibilizzazione delle cellule di Leydig con conseguente sviluppo di ipogonadismo ipergonadotropo. Il seguente articolo è volto a riportare le caratteristiche del hCG, le sue possibili applicazioni e, in modo approfondito, fare maggiore chiarezza sulla questione legata alla possibile desensibilizzazione hCG-dipendente.
hCG: storia, usi clinici e off-label
Gonadotropina Corionica Umana
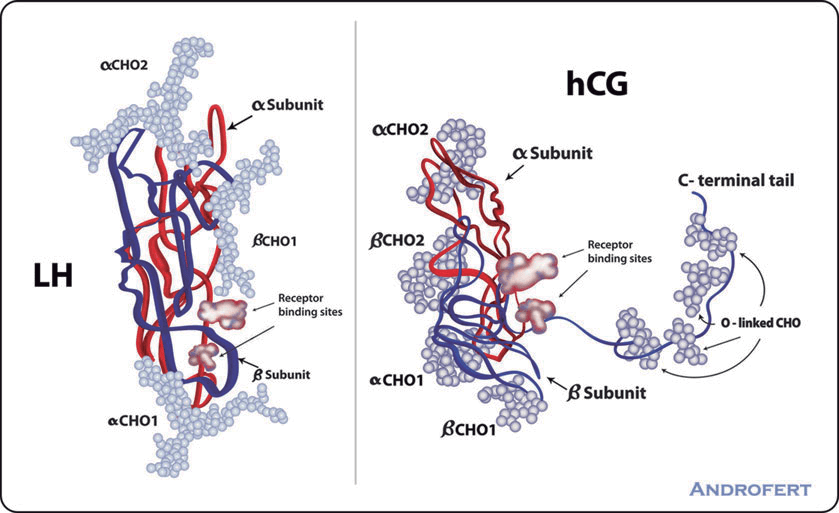
L’hCG (Human chorionic gonadotropin) o Gonadotropina Corionica è un ormone polipeptidico prodotto dall’embrione all’inizio della seconda settimana di sviluppo, in particolare dalle cellule del sinciziotrofoblasto, un tessuto epiteliale monostratificato posto nella porzione profonda del cito-sinciziotrofoblasto, subito dopo l’impianto nell’endometrio. La molecola di hCG è un eterodimero, composto da due subunità (α e β). La subunità α ha struttura identica a quella delle altre gonadotropine (LH e FSH), mentre la subunità β è specifica di ciascun ormone. Per questo motivo, i metodi di dosaggio dell’hCG utilizzano anticorpi diretti contro la subunità β dell’hCG.
Più specificatamente, la Gonadotropina Corionica è una glicoproteina oligosaccaridica composta da 244 aminoacidi. La subunità α è lunga 92 aminoacidi ed è identica a quella dell’Ormone Luteinizzante (LH), dell’Ormone Follicolo-Stimolante (FSH) e dell’Ormone Stimolante la Tiroide (TSH). Come già accennato, la subunità beta è unica per l’hCG.
L’hCG è quindi un analogo del LH, l’ormone prodotto dall’ipofisi che stimola la produzione di ormoni sessuali nei testicoli o nelle ovaie. L’hCG si lega e attiva lo stesso recettore dell’LH ed è ugualmente efficace nello stimolare la produzione di Testosterone negli uomini e di Estrogeni nelle donne.
La Gonadotropina Corionica venne isolata ed identificata per la prima volta nel 1920 (1) venendo in seguito classificata come un ormone della gravidanza circa otto anni dopo.(2) La prima preparazione farmaceutica contenente Gonadotropina Corionica si presentava sotto forma di estratto pituitario animale, il quale venne sviluppato come prodotto commerciale dalla Organon. La Organon introdusse nel mercato l’estratto nel 1931, con il nome commerciale di Pregnon. Una controversia sui marchi obbligò la compagnia a cambiare il nome Pregnyl, che raggiunse il mercato nel 1932. Il Pregnyl è attualmente venduto dalla MSD–Organon, anche se il principio attivo non è più estratto dalla pituitaria animale. Nel 1940 furono introdotte tecniche di produzione che consentivano di ottenere l’ormone filtrando e purificando l’urina delle donne incinta, e alla fine degli anni ’60 questa tecnica di produzione fu adottata da tutti i produttori che avevano usato precedentemente gli estratti animali. Nel corso degli anni i processi di produzione sono stati perfezionati, ma l’hCG è ottenuta essenzialmente nello stesso modo oggi come lo era decenni fa. Nonostante i preparati moderni siano di origine biologica, si afferma che i rischi di contaminanti biologici siano bassi (sebbene non possano essere completamente esclusi).
Al principio della sua applicazione clinica, gli usi indicati per le preparazioni a base di Gonadotropina Corionica erano molto più ampi di quanto non lo siano attualmente. La letteratura inerente al composto degli anni ’50 e ’60 raccomandava l’uso di questo farmaco per, tra le altre cose, il trattamento del sanguinamento uterino e dell’amenorrea, la sindrome di Froehlich, il criptochismo, la sterilità femminile, l’obesità, la depressione e l’impotenza maschile. Un buon esempio degli ampi usi della Gonadotropina Corionica è illustrato nel preparato Glukor, che fu descritto nel 1958 come “Tre volte più efficace del Testosterone. Per i giovani stanchi dal climaterio maschile. Per vecchi stanchi dalla senilità maschile. Benefici nell’impotenza, angina e malattia coronarica, neuropsicosi, prostatite, [e] miocardite.” Tali raccomandazioni, tuttavia, riflettono un’era meno strettamente regolata dall’agenzia governativa e meno dipendente da studi cliniche comprovati. Oggi, le indicazioni approvate dalla FDA per l’uso del hCG sono limitate al trattamento dell’ipogonadotropismo ipogonadico e del criptocridismo negli uomini e alla sterilità anovulatoria nelle donne.
Dr. A.T.W. Simeons
L’hCG non ha alcuna attività significativa di stimolo della tiroide. Questo necessita di essere specificato dato che l’hCG è stata ampiamente usata in passato per il trattamento dell’obesità. Questa applicazione d’uso sembra che sia divenuta popolare nel 1954, dopo la pubblicazione di un articolo del Dr. A.T.W. Simeons nel quale sosteneva che la Gonadotropina Corionica era un’aggiunta efficace alla dieta. Secondo lo studio, i pazienti sono stati in grado di sopprimere efficacemente l’appetito seguendo una dieta con marcata restrizione calorica abbinata alla somministrazione di hCG. Soprannominata la dieta Simeons, le persone in tutti gli Stati Uniti si sottoposero presto a severe restrizioni caloriche (500 Kcal al giorno) e iniezioni di hCG. Poco dopo, l’ormone stesso divenne il coadiuvante principale per la perdita di grasso. Infatti, nel 1957 si diceva che l’hCG era il farmaco più comunemente prescritto per la perdita di peso. Indagini più recenti e complete, tuttavia, confutano l’esistenza di qualsiasi vantaggio anoressizzante o metabolico dato dall’uso di hCG.(3) Nel 1962, il Journal of American Medical Association aveva già avvertito i consumatori circa la dieta Simeons inclusiva di hCG, affermando che la grave restrizione calorica tipica di tale protocollo dimagrante (che si rifletteva in un accentuato catabolismo del tessuto magro) era più pericolosa dell’obesità stessa. Nel 1974, la FDA aveva raccolto abbastanza dichiarazioni sull’uso del hCG per la perdita di grasso che fece inserire una dichiarazione in merito nel bugiardino dei prodotti contenenti l’ormone, nella quale affermava che non vi erano dimostrazioni sulla presunta efficacia nella perdita di peso data dalla somministrazione di hCG in concomitanza con regimi alimentari ipocalorici. Questo avvertimento è tutt’oggi presente su tutti prodotto venduti negli Stati Uniti. Nonostante questo avvertimento e prove che confutano l’efficace di tale pratica, alcune cliniche promuovono ancora l’uso di hCG per la perdita di peso.
La Gonadotropina Corionica Umana è oggi una preparazione farmaceutica molto popolare, poiché rimane una parte indispensabile della terapia di ovulazione per molti casi di infertilità femminile. Sebbene la forma di hCG sintetizzata tramite la tecnica del DNA ricombinante sia stata introdotta sul mercato negli ultimi anni, l’ampia offerta e il basso costo dell’hCG biologico continuano a renderlo un prodotto di base per gli usi clinici e off-label.
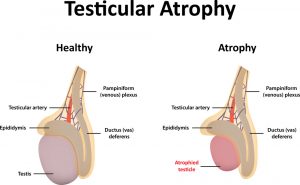
Quando vengono somministrati AAS (o SARM), i livelli di LH diminuiscono rapidamente. Il calo o l’assenza del rilascio ipofisario di LH, e suo consequenziale segnale, induce un calo o interruzione dell’attività testicolare (la quale, ovviamente, si riflette negativamente sulla sintesi di Testosterone) che causa la rapida insorgenza dell’atrofia testicolare. Questa degenerazione testicolare inizia con una riduzione del volume delle cellule di Leydig, seguita da una riduzioni rapida del Testosterone Intra-Testicolare (ITT), dei perossisomi e del fattore insulino-simile 3 (INSL3) – Tutti bio-marcatori e fattori importanti per una corretta funzione testicolare e biosintesi di Testosterone.
Tuttavia, questa degenerazione testicolare viene trattata dai Bodybuilder supplementari chimicamente con la somministrazione di hCG, in special modo all’uscita di un ciclo e per il periodo iniziale della PCT.
Tutte, o quasi tutte, le esperienze pratiche con questo farmaco nel Bodybuilding avvengono con l’uso del hCG biologico (estratto dalle urine di donne gravide), che viene generalmente venduto in vial contenenti polvere liofilizzata da ricombinare con acqua fisiologica o batteriostatica, con un contenuto che va dalle 250 alle 10.000UI per vial.
Il dosaggio clinico di hCG per trattare i casi di ipogonadismo ipogonadotropo è stato tradizionalmente di 5000UI per iniezione. Prima del 1998, la dose tipicamente utilizzata nel bodybuilding per il ripristino della funzione testicolare era la medesima. Di conseguenza, trattandosi di un quantitativo molto elevato, è stato per molto tempo considerato un farmaco di non facile gestione e dagli effetti collaterali, presunti o tali, che destavano non poca preoccupazione (vedi desensibilizzazione delle cellule di Leydig che tratterò più avanti).
Successivamente, venne introdotto l’uso di un dosaggio più basso con un limite di 1500UI per ogni singola iniezione, con una preferenza di dosaggio non superiore alle 1000UI, e con l’uso consigliato di un dosaggio pari a 500UI a somministrazione.
Molti Preparatori danno come raccomandazione quella di non superare le 500UI per ogni somministrazione, poiché non è stato riscontrato alcun vantaggio aggiuntivo nell’utilizzare un dosaggio singolo superiore a questo, a condizione che le iniezioni siano ragionevolmente frequenti (ogni 2-4 giorni).
L’intervallo di dosaggio settimanale comunemente consigliato è compreso tra circa le 700 e le 1750UI. Le dosi di esempio sono 100-250UI al giorno, 250-500 UI a giorni alterni o 250-500UI da tre volte a settimana a somministrazioni distanziate l’una dall’altra da quattro giorni.
Con tali dosaggi sono stati seguiti un numero molto elevato di individui per diversi anni e con eccellenti risultati, e la ricerca scientifica sembra aver convalidato l’utilità del mantenersi all’interno di queste dosi. Come misurato dai livelli intratesticolari di testosterone, questo livello di dosaggio massimizza i risultati. Semplicemente non risulta conveniente la somministrazione di dosi maggiori.
Si raccomandano generalmente iniezioni multiple settimanali dal momento che l’emivita del hCG è di circa 36 ore. Iniezioni meno frequenti comportano uno scarso mantenimento dei livelli ematici.
Prima del 1996, l’uso tradizionale del hCG era quello di inserirla post-ciclo con lo scopo di ripristinare una funzionalità testicolare ottimale. Ma tale pratica non risulta pienamente ottimale dal momento che rallenta comunque i processi di recupero dell’HPTA. Infatti, il tempo medio di recupero della funzionalità testicolare con l’uso del hCG risulta essere in media di 4-8 settimane. Di conseguenza, la scelta migliore, in contesti nei quali i cicli durano più di quattro settimane e/o quando il ciclo viene seguito da un “Bridge” o TRT, l’uso del hCG durante il ciclo permette di conservare una buona attività testicolare permettendo, per esempio, all’atleta in uscita da un ciclo di accelerare i processi di recupero dell’HPTA dal momento che, così facendo, evita quel periodo transitorio (e potenzialmente controproducente) tra la fine del ciclo ed il ripristino di una corretta funzionalità testicolare.
Nei contesti sopra citati, la hCG viene somministrata durante il ciclo con varianti temporali che vanno dalla seconda settimana alla quarta (dipendente dalla durata complessiva del ciclo e da ciò che l’atleta farà nel post ciclo). I dosaggi mediamente utilizzati sono 100 UI al giorno, 200 UI a giorni alterni o 250UI da 3 volte a settimana a ogni 4 giorni.
Un’altra pratica d’uso del hCG è quella di inserirla durante i cicli che non contemplano l’uso di AAS soggetti ad aromatizzazione. Con il solo uso di AAS non aromatizzabili, i livelli di estrogeni diminuiscono in modo anomalo in seguito alla sottoregolazione/soppressione del Testosterone endogeno e la consequenziale diminuzione dei substrati soggetti all’aromatizzazione. Questa condizione interferisce con l’anabolismo, la libido, l’umore, la funzione articolare e, sul lungo termine, la salute cardiovascolare. Un modo ovvio per risolvere questo problema è quello di includere almeno una piccola quantità di uno AAS aromatizzabile (vedi base terapeutica di Testosterone). In questo caso i dosaggi di hCG tipicamente utilizzati sono compresi nella fascia altra d’intervallo del dosaggio efficace suggerito (500UI a giorni alterni). La risultante sarà una sintesi di Testosterone endogeno e Estradiolo.
Terminate le dovute precisazioni sul hCG adesso possiamo trattare l’argomento centrale di questo articolo…
hCG e possibile desensibilizzazione (?)
La questione sulla possibilità secondo cui l’uso prolungato di hCG possa portare ad una condizione di ipogonadismo ipergonadotropo è tutt’ora dibattuta. L’utilizzatore deve comunque tenere a mente che il dosaggio di tale composto deve essere attentamente calibrato in specie con somministrazioni prolungate, poiché alti livelli di hCG possono anche causare un aumento dell’espressione dell’aromatasi testicolare (con conseguente innalzamento dei livelli di estrogeni), (4). Esistono studi piuttosto datati, e svolti per la maggior parte sui ratti, che riportano il verificarsi della desensibilizzazione testicolare al LH in seguito a somministrazione di alti dosaggi e per lunghi periodi di tempo.(5) Il farmaco in questione può effettivamente avere il potenziale di indurre ipogonadismo primario se usato impropriamente, peggiorando notevolmente, non migliorando, la funzionalità testicolare.
I protocolli d’uso di hCG che contemplano la somministrazione di dosi pari a 250UI per via sottocutanea ogni 3 o 4 giorni con una dose massima di 500UI, sviluppati dal Dr. John Crisler, una figura ben nota nel campo dell’Anti-Aging e della terapia ormonale sostitutiva, vengono spesso utilizzati dai soggetti in Terapia Sostitutiva del Testosterone (TRT). L’atrofia testicolare per i pazienti in TRT è un disturbo cosmetico comune. Il programma di somministrazione di hCG del Dr. Crisler è progettato per risolvere questo problema con un uso a lungo termine senza causare l’ipotetica desensibilizzazione. Coloro i quali sono interessati a gestire il timing di somministrazione del hCG con precisione in relazione ad una TRT, il dott. Crisler raccomanda quanto segue: “… i miei pazienti in TRT con Testosterone Cypionato ora somministrano la loro dose di hCG di 250IU nei due giorni precedenti l’iniezione intramuscolare (Testosterone Cypionato NdR.). Tutti i pazienti somministrano la loro dose di hCG per via sottocutanea e il dosaggio può essere aggiustato secondo necessità (devo ancora vedere una necessità di dosaggio superiore alle 350 UI per somministrazione) … Quei pazienti in TRT che preferiscono usare un Testosterone transdermico, o anche Testosterone orale (sebbene io non sia favorevole a ciò) , somministrano la loro dose di hCG ogni tre giorni. ”
Il Dr. John Crisler afferma che è importante non somministrare più di 500UI di hCG in un dato giorno. Egli infatti afferma che vi è solo una quantità massima di stimolazione, e il superamento di questo dosaggio non solo è uno spreco, ma ha conseguenze negative importanti. Dosi più elevate stimolano eccessivamente l’aromatasi testicolare, che aumenta in modo inappropriato i livelli di estrogeni portando alla comparsa di effetti collaterali tipici del iperestrogenemia. Il Dr. Crisler continua dicendo che dosi superiori a quella sopra indicata (500UI) causino anche la desensibilizzazione delle cellule di Leydig verso LH inducendo quindi all’ipogonadismo primario. Egli ribadisce che 250IU ogni 2-4 giorni sia una dose efficace e sicura. Dopotutto, stiamo semplicemente sostituendo ciò che è stato inibito.
Il Dr. Scalley, dal canto suo, critica la posizione del Dr. John Crisler affermando che, la somministrazione dell’hCG per due giorni consecutivi non ha senso, inoltre la dose è omeopatica (inutile). Inoltre, il Dr. Scalley ritiene che, nonostante il Dr. Crisler qualifichi le sue affermazione ricollegandosi a determinati studi, l’errore sta nel considerare come assodato che le dosi più elevate di quelle che consiglia causino la desensibilizzazione. Il Dr. Crisler sembra mancare di una comprensione corretta della letteratura.
Scalley riporta che la desensibilizzazione hCG-dipendente si può potenzialmente verificare in caso di somministrazione prolungata di 5.000UI (cinquemila). Ma, anche in questo caso l’incidenza non è universalmente osservata. C’è anche da aggiungere che il problema della desensibilizzazione non è quasi mai stato osservato nella pratica clinica.
Gli studi solitamente menzionati non danno in realtà alcun supporto a dimostrazione che la desensibilizzazione si verifichi con dosi superiori alle 500UI o che l’uso di 250 UI X2 volte a settimana sia una terapia utile. Se ci si pensaun attimo, qual è lo scopo dell’uso di hCG per due giorni di seguito? Questa pratica risulta completamente bizzarra. Come prima cosa, sfido chiunque a riportare la letteratura (articolo o citazioni) a sostegno del suo trattamento (del Dr. Crisler). Se Crisler è così sicuro di sé, perché non cita alcuna pubblicazione a supporto della sua terapia o, meglio, pubblichi i risultati del trattamento.
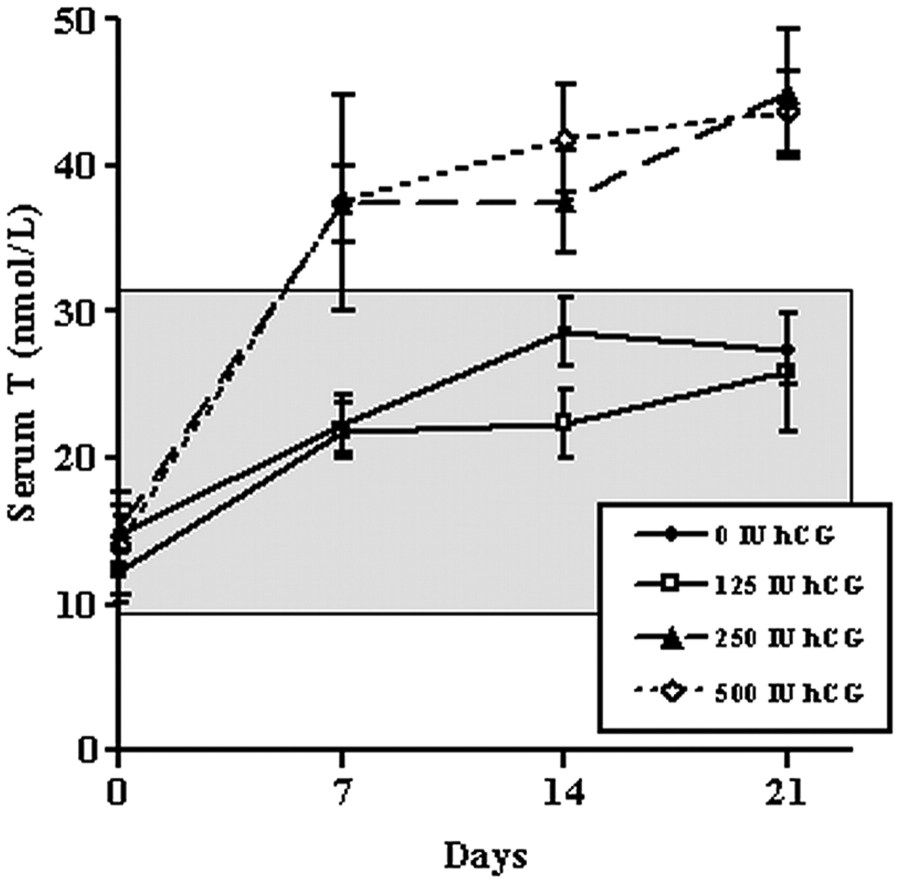
Innanzitutto, lo studio che spesso viene citato a sostegno delle tesi del Dr. Crisler (6) valuta il Testosterone Intratesticolare (ITT) e questo, di per se, non è di poca importanza. I partecipanti a questo studio sono stati trattati con Testosterone Enantato (TE), 200 mg alla settimana, per la soppressione rapida della gonadotropina in combinazione con una dose variabile di hCG, somministrata sottocute ogni 2 giorni per 3 settimane: 0 (placebo salino), 125, 250 o 500 UI hCG. Il gruppo placebo è servito come gruppo di controllo. [Nota: la differenza sostanziale è che, anche se lo studio supporta Crisler, il dosaggio è molto diverso da quello da lui raccomandato.]
Quindi, quello che lo studio ci offre sono soggetti di sesso maschile con elevati livelli di Testosterone per via di iniezioni settimanali di 200mg di Testosterone Enantato. La loro produzione endogena di Testosterone è completamente soppressa (teoricamente) come le loro gonadotropine. Il ITT risulta quindi soppresso a causa dell’inibizione delle gonadotropine date dalla somministrazione di Testosterone Enantato. I ricercatori hanno scoperto che ogni dose di hCG (125, 250 e 500 UI) riportava la concentrazione di ITT alla normalità. Si da il caso però che in un maschio normale con un normale livello di Testosterone serico il suo ITT sarà normale. Tutto questo studio è stato semplicemente prendere un maschio normale e sostituire il suo Testosterone con del Testosterone esogeno per poi somministrargli hCG come sostituto del suo LH.
L’unica cosa che può essere salvata di questo studio, è che può essere istruttiva per chi usa hCG a basse dosi in on-cicle, in “Bridge” o in TRT. Più precisamente ci dice qualcosa sulla terapia con hCG mentre si usa un dosaggio “simil-TRT”.
Nello studio risulta interessante esaminare i dati sulle variazioni seriche di Testosterone con ciascuna dose di hCG. I soggetti presi in esame hanno usato una dose contenuta (seppur fisiologicamente alta) di Testosterone Enantato, creando una situazione che per certi versi riproduce quella di un individuo che usa hCG in TRT. Il risultato è stato che la dose di hCG da 125UI a giorni alterni non ha avuto effetti sul Testosterone serico. Le due dosi più elevate (250-500UI) hanno alzato i livelli di Testosterone nel siero al di sopra del normale.
Non ci sono dati individuali (sempre motivo di sospetto quando si esamina la letteratura) e non sono riportati livelli significativi. L’analisi del grafico dello studio riportato di seguito, tuttavia, mostra che il livello di Testosterone del siero non era significativamente diverso dal controllo fino al giorno 21[altra nota a discredito delle affermazioni del Dr. Crisler].
Ci sono quindi molti possibili errori nell’analisi dello studio appena discusso. Dal momento che non ci mostra un analisi sufficientemente accurata tale da permetterci di identificare una soglia di dosaggio che porti alla desensibilizzazione delle cellule di Leydig.
Si può ipotizzare che la modalità di somministrazione dell’hCG nei due giorni precedenti l’iniezione settimanale di Testosterone (come indicato nel protocollo del Dr Crisler) serva da teorico “supporto” al calo della soglia ematica di quest’ultimo. Se si legge la letteratura disponibile sugli effetti dell’hCG, il rialzo dei livelli di Testosterone serico si manifestano in modo significativo a circa 48-72 ore dopo la somministrazione del peptide. Questo dosaggio concentrato in due giorni non da reali vantaggi sulla funzionalità testicolare. Quindi, fino a dimostrazione contraria, le ipotesi del Dr. Crisler sulla somministrazione ottimale di hCG, per effetti e sicurezza riguardo la desensibilizzazione testicolari, non sono altro che opinabili speculazioni.
Per tutti gli scopi pratici, la desensibilizzazione delle cellule di Leydig hCG-dipendente praticamente non sussiste all’interno del quadro clinico, sebbene rimanga una possibilità con l’uso di dosi elevate e per un lungo periodo di tempo (>5000UI)
17 alfa-hydroxyprogesterone
Esiste uno studio che, seppur “isolato”, ritengo sia interessante per farsi un idea delle variabili e della differenza tra possibilità universalmente riscontrate e possibilità di bassa o scarsa incidenza. Si tratta di uno studio nel quale si è osservato che la somministrazione di Tamoxifene in maschi sani ha causato una riduzione dell’accumulo del 17 α-hydroxyprogesterone hCG-indotto.(7)
In questo studio, la somministrazione per via intramuscolare di 1500 UI di hCG al giorno per 3 giorni ha indotto un accumulo transitorio di 17 α-hydroxyprogesterone (17 OHP) rispetto al Testosterone (T) in uomini normali, raggiungendo il massimo nelle 24 ore successive la prima iniezione (rapporto 17 OHP / T, 1,7 +/- 0,3 volte il basale, P <0,01). La somministrazione simultanea di hCG e Tamoxifene (20 mg due volte al giorno) ha quasi completamente soppresso il blocco steroidogenico indotto dal hCG e osservato con il 17 OHP:T ratio (rapporto 17 OHP-T a 24 ore, 1,1 +/- 0,1 volte il basale; 0,01 vs hCG da solo). Questi dati suggeriscono indirettamente che, nell’uomo, la lesione steroidogenica indotta dal hCG potrebbe essere mediata attraverso il suo effetto stimolante sugli estrogeni.
Un altro studio svolto sulla falsariga del precedente, ma ad una distanza di undici anni, ha osservato l’effetto del Tamoxifene sulla risposta testicolare al hCG. (8) Se si legge con attenzione il presente studio, anche alla luce di quanto affermato pocanzi, si riesce ad avere un quadro molto più chiaro sulla questione.
Tamoxifene
In questo studio è stato osservato l’effetto del Tamoxifene (Tx) in concomitanza con la somministrazione acuta e cronica di hCG in pazienti con ipogonadismo ipogonadotropo (HH) e in uomini normali. Un test con hCG (5000 UI hCG) è stato svolto prima e dopo due mesi di somministrazione di hCG (2000 UI di hCG tre volte a settimana) e dopo due mesi di hCG + Tx (2000 UI hCG tre volte a settimana più 20 mg/die di Tamoxifene). I campioni di sangue sono stati prelevati 24 e 72 ore prima e dopo ogni test per determinare i livelli di Testosterone , Estradiolo, 17OHP e SHBG. Il Testosterone è aumentato solo nel gruppo HH con entrambi i trattamenti (X +/- SEM: basale: 97,9 +/- 19,7; hCG: 237,7 +/- 43,2; hCG +/- Tx: 204,7 +/- 10,7 ng / 100 ml). Il 17OHP è aumentato con la somministrazione di hCG da solo, ma non con hCG + Tx in entrambi i gruppi. Il rapporto Estradiolo, SHBG e 17OHP / Testosterone non è cambiato dopo i trattamenti. In risposta al hCG il Testosterone è aumentato 24 ore dopo la somministrazione in ogni test. Il rapporto 17OHP / Testosterone è salito dopo 24 ore nel primo e nel secondo test, ma nel terzo test non è cambiato. Questi risultati supportano il ruolo del Estradiolo nella desensibilizzazione delle cellule di Leydig indotto da una somministrazione acuta di hCG. Tuttavia, l’associazione di Tx non migliora i livelli serici di Testosterone, suggerendo che l’Estradiolo potrebbe non essere il fattore unico coinvolto nei meccanismi di desensibilizzazione testicolare.
Sembrerebbe, quindi, che il fattore determinante legato ad una possibile desensibilizzazione o sottoregolazione della funzionalità testicolare (in particolar modo in riferimento alle cellule di Leydig) sia la dose in acuto e, soprattutto, in cronico. Per ciò che concerne la dose utile questa è invece determinata dal limite fisiologico di stimolo della secrezione di Testosterone che, negli uomini sani con una sensibilità testicolare normale, si è visto corrispondere ad una dose di sole 250UI, con ulteriori aumenti minimi ottenuti con 500UI a 5000UI.
Conclusioni
Sebbene, come già accennato, la questione non sia del tutto chiarita, le informazioni riportate in questo articolo possono senz’altro permettere una pratica d’uso del hCG “sicura” e, soprattutto, intelligente nel contesto di una preparazione farmacologica. Risulta abbastanza chiaro che iniezione intramuscolare o sottocutanee di hCG a dosi di 100-200UI al giorno, 200-250 UI a giorni alterni o 250-500UI da tre volte a settimana a una somministrazione ogni 4 giorni, risultano pienamente efficace per evitare la disfunzione e seguente atrofia testicolare durante l’uso di AAS (o SARM) mantenendone una buona funzionalità in mancanza di stimoli dati dal LH. Dosi superiori a quelle riportate non offrono ulteriori vantaggi. Si tenga inoltre bene a mente che la desensibilizzazione delle cellule di Leydig può manifestarsi con maggiore facilità in una situazione di mancanza di segnale dato dall’LH, condizione che viene spesso osservata in quegli atleti che non usano hCG durante i cicli. Tale desensibilizzazione, però, risulta più semplice da trattare rispetto ad una desensibilizzazione indotta da uno stimolo eccessivo delle cellule di Leydig che, in casi cronici, obbliga il soggetto colpito a doversi sottoporre a trattamento con Testosterone esogeno (TRT).
Quindi, in definitiva, le nozioni base da tenere bene a mente sono:
Uso di dosi e tempi di somministrazione utili allo scopo prefissato (evitare la disfunzione testicolare e avere un ottimale stimolo della biosintesi di Testosterone)
Iniziare la somministrazione di hCG durante il ciclo (tempo variabile dalle 2 alle 4 settimane dall’inizio del ciclo e determinato dalle scelte future al ciclo [PCT, Bridge o TRT]
Non eccedere le 500UI a giorni alterni (principalmente perché dosi più elevate non portano vantaggi considerevoli)
Un’altra nota che mi sento di aggiungere è in riferimento alla ricombinazione del contenuto delle vial e delle procedure per la sterilità del prodotto. Le vial di hCG dovrebbero essere ricostituite con una quantità di soluzione acquosa (sterile o batteriostatica) basata sul quantitativo effettivo in UI della vial. Ad esempio, una vial da 5000UI può essere convenientemente ricombinata con 2,5ml d’acqua. Ciò fornisce una soluzione di 2000IU/ml , che consente un facile calcolo del dosaggio necessario. Ad esempio, una dose di 200 UI richiederebbe quindi l’aspirazione di 0,1mL di soluzione, che sarebbe contrassegnata con “10 UI” su una siringa da insulina.
Se la capacità della vial lo consente, è possibile aggiungere 5,0ml di acqua in una vial da 5000UI. La soluzione risultante sarebbe ovviamente di 1000IU/mL, consentendo un calcolo ancora più semplice del dosaggio necessario.
L’iniezione può essere eseguita intramuscolarmente o sottocute in base alle preferenze personali.
Le vial di hCG non ricostituite devono essere conservate in frigo. Sebbene possano essere spedite a temperatura ambiente. Le vial ricostituite devono sempre essere conservate in frigo; tuttavia, se una vial viene accidentalmente lasciata a temperatura ambiente per un giorno, il principio attivo non subirà alcun deterioramento.
Cosa molto importante quando si manipola l’hCG è quella di impiegare corrette procedure per mantenere la sterilità della vial e della soluzione ivi contenuta. La membrana di gomma deve sempre essere pulita accuratamente con alcool e l’ago deve essere sterile. Il peptide acquoso, o in questo caso le soluzioni glicopeptidiche possono supportare la crescita batterica molto più di quanto possano fare le soluzioni oleose, quindi è raccomandata la massima cura della sterilità del prodotto. Se si nota un intorpidimento della soluzione è consigliabile che il prodotto non venga utilizzato.
Gabriel Bellizzi
Riferimenti:
Exogenous stimulation of corpus luteum formation in the rabbit; influence of extracts of human placenta, decidua, fetus, hydatid mole and corpus luteum on the rabbit gonad. Hirose T 1920 J Jpn Gynecol Sot 16:1055.
Die Schwangerschaftsdiagnose ausdem Ham durch Nachweis des Hypophysenvorderlappen-hormone. II. Pracktishe und theoretische Ergebnisse aus den hamuntersuchungen. Ascheim S, Zondek B 1928 Klin Wochenschr 7:1453-1457.
Un maggior utilizzo di AAS da parte di un atleta si traduce in una maggiore probabilità di avere problemi sessuali durante i periodi di non utilizzo. E no, la PCT non sembra rendere esenti da tali possibili problemi. I ricercatori del American Mayo hospital hanno osservato tale effetto in seguito ad uno studio svolto su diverse centinaia di Bodybuilder supplementati farmacologicamente.(1)
I ricercatori hanno contattato 231 utilizzatori di AAS maschi, attraverso nove forum di bodybuilding, i quali erano pronti a rispondere a domande online sulla loro salute sessuale e sul loro uso di AAS. Per rilevare eventuali disfunzioni erettili, i ricercatori hanno utilizzato una versione abbreviata del questionario IEEF-5. Potete trovarne una copia qui. Gli uomini che totalizzano un punteggio pari o maggiore di 22 non hanno nulla di cui lamentarsi sessualmente parlando, mentre gli uomini che ottengono un punteggio di 17-21 soffrono di un lieve disturbo erettile.
I partecipanti hanno ottenuto un punteggio leggermente superiore sull’IEEF-5 se erano sottoposti ad iniezioni superiori ai 600mg di Testosterone a settimana, se usavano anche un AAS orale o un anti-estrogeno e se erano in buone condizioni di salute. Non molto sorprendente come risultato.
127 utilizzatori hanno affermato che nei loro periodi “off” la libido diminuiva. Questo però non è stato riportato da altri 94 utilizzatori. Quando i ricercatori hanno messo a confronto questi due gruppi, hanno notato che il rischio che si verifichi una riduzione della libido aumentava in modo significativo qualora gli utilizzatori avessero seguito protocolli di AAS per molte settimane all’anno e avessero una lunga storia di utilizzo di questa classe di farmaci.
Lo svolgimento di una PCT era pratica diffusa nel gruppo in cui la libido tra i cicli non diminuiva rispetto al gruppo in cui la libido diminuiva. Tuttavia, questa differenza non era statisticamente significativa.
L’attuale studio rappresenta ad oggi la più grande valutazione sull’impatto dell’utilizzo di AAS ad alto dosaggio e nel lungo periodo sulla funzione sessuale. I risultati dimostrano che l’aumento della durata e della frequenza di utilizzo degli AAS sono associate a più alti tassi di disfunzione erettile de novo e diminuzione della libido dopo l’interruzione d’uso del/i composto/i.
Gli uomini con disfunzione erettile de novo avevano anche maggiori probabilità di riportare altri sintomi legati a bassi livelli di Testosterone, come riduzione della libido, diminuzione dell’energia, depressione, riduzione soggettiva della massa muscolare e aumento soggettivo della massa grassa. Diversamente a ciò, durante l’uso di un dosaggio più alto di Testosterone e l’uso (con tutta probabilità ponderato) di anti-estrogeni si sono osservati punteggi più alti sul questionario IEEF-5.
Se non avete ancora letto le precedenti parti componenti questa serie di articoli vi invito a farlo prima di procedere con la lettura di questa quarta ed ultima parte: 1° Parte – 2° Parte – 3° Parte.
Farmacocinetica, Farmacodinamica e Feedback Negativi
Come già detto, il fegato rappresenta il principale bersaglio del GH, il quale è il principale regolatore della sintesi epatica di IGF-1. Per causare tale effetto, il GH si lega con i GHR localizzati nel dominio extracellulare degli epatociti stimolando successivamente la produzione di IGF-1 endocrino tramite la trascrizione genica, utilizzando la via di segnalazione JAK-STAT. Inoltre, è stato dimostrato che la somministrazione di GH causa una rapida sovraregolazione dell’mRNA del IGF-1 nel fegato.[338]
Aumenti dei livelli serici di IGF-1 si verificano molto rapidamente anche in presenza di un grande bolo di rHGH. Incrementi significativi di IGF-1 sono già osservabili dopo 6-12h dall’iniezione.[339] Questi livelli serici di IGF-1 continuano ad aumentare fino a raggiungere il loro punto di saturazione dose-dipendente entro 4-7 giorni, anche quando si utilizzano dosi estremamente elevate che ammontano a 20-30UI al giorno di rHGH.[340] In particolare, il punto di saturazione si è rivelato essere compreso nell’intervallo dei 700-800 ng/mL e sembra suggerire che i livelli endocrini di IGF-1 hanno un tetto massimo negli adulti sani. I meccanismi esatti devono ancora essere chiariti, ma sono probabilmente il risultato dei complessi meccanismi di controllo intrinseci all’Asse GH/IGF-1. Coloro i quali desiderano elevare i livelli endocrini di IGF-1 al fine di ottenerne un vantaggio sull’ipertrofia dovrebbe tenerlo a mente, in quanto vi è un punto in cui l’uso di dosi maggiori di rHGH semplicemente non si traducono in elevati livelli serici di IGF-1. Qui di seguito ho riportato il grafico dello studio di Tanaka il quale mostra la relazione tra l’rhGH e i livelli serici di IGF-:
Ora, vorrei dedicarmi brevemente all’analisi dell’azione del IGF-1autocrino e del perché esso rappresenti un mediatore cruciale del processo ipertrofico, prima di tornare nuovamente a discutere su questioni inerenti alla farmacodinamica e farmacocinetica. La segnalazione recettoriale del IGF-1 è unica nel suo genere, e questo lo si deve al fatto che utilizza due percorsi distinti per stimolare la proliferazione o la differenziazione.[341-343] Questo è un comportamento abbastanza interessante, poiché nessun altro membro della famiglia dei fattori di crescita ha dimostrato di agire in tal modo. Poiché la proliferazione e la differenziazione sono processi opposti, inizialmente era difficile per i ricercatori capire come un singolo fattore di crescita, attraverso un singolo recettore, potesse inviare un segnale che attivasse entrambi.[294] Da quando sono state fatte queste prime scoperte, è stato ulteriormente chiarito che l’IGF-1 non svolge simultaneamente queste azioni. Test su varie linee di coltura cellulare hanno dimostrato che gli effetti proliferativi arrivano prima, durando tra le 24 e le 36 ore. È solo dopo questa fase proliferativa iniziale che si verifica la differenziazione miogenica.[344]
Gli effetti proliferativi mediati dall’IGF-1 sui mioblasti sono noti sin dagli anni ’70, quando vennero osservati per la prima volta nelle cellule epatiche di ratto.[345] Questa stimolazione proliferativa del IGF-1 si traduce in un aumento del numero di cellule, nei livelli di proteine, nella sintesi del DNA, nell’assorbimento di aminoacidico, nell’assorbimento del glucosio e nella soppressione della proteolisi.[346] Nelle colture cellulari umane, l’IGF-1 ha anche dimostrato di aumentare la dimensione dei miotubi indipendentemente dal fatto che i mioblasti proliferino attivamente o che la proliferazione sia cessata. Regola la dimensione dei miotubi attivando la sintesi proteica, inibendo la degradazione proteica e inducendo la fusione delle cellule di riserva.[347-348] La capacità dell’IGF di sopprimere la proteolisi nel muscolo scheletrico, la scomposizione delle proteine in aminoacidi, è stata dimostrata innumerevoli volte nel corso degli anni.[349-352] È stato anche dimostrato che l’IGF-1 induce la proliferazione e la differenziazione delle cellule satelliti in miociti maturi, come determinato da un aumento del numero di miofibre nucleate a livello centrale rispetto a quelle periferiche.[148,353-354]
La capacità dell’IGF-1 autocrino di causare la differenziazione dei mioblasti è stata in realtà una scoperta che potremmo definire quasi “ibrida” dal momento che degli studi svolti negli anni ’60 avevano mostrato che questo effetto si verifica con alti livelli di Insulina.[355] Successivamente è stato dimostrato che gli IGF sono stimolatori molto più potenti nella differenziazione miogenica rispetto all’Insulina e si è concluso che la stessa Insulina agisce realmente come un analogo dell’IGF-1 in questo sistema.[356-357] Gli effetti di differenziazione dati dall’IGF-1 autocrino sono bifasici, con basse concentrazioni che stimolano progressivamente la differenziazione dei mioblasti mentre concentrazioni molto elevate mostrano una cessazione dell’attività di differenziazione. Il limite massimo per la differenziazione sembra attestarsi a circa 100ng/mL per l’IGF-1 e 300ng/mL per IGF-2.[358] Questo effetto non è legato alla proliferazione, poiché non si osservano ulteriori aumenti nel numero complessivo delle cellule.[294] È possibile che le molecole di segnalazione coinvolte nella regolazione negativa del sistema miogenico siano aumentate, ma questa è una affermazione puramente speculativa.[359-360]
La somministrazione di rHGH eleva l’espressione dell’mRNA dell’IGF-1 nel muscolo scheletrico in numerosi modelli cellulari, umani e animali.[127,150,361-364] Ciò avviene abbastanza rapidamente, entro 60 minuti dall’iniezione sottocutanea di rHGH ed i picchi sono segnalati tra le 6 e le 12 ore post iniezione.[363] In questo particolare modello animale citato, il raddoppio della dose di GH non ha portato ad ulteriori aumentati dei livelli di mRNA dell’IGF-1, il che suggerisce che esiste un sistema di regolazione che determina quanto GH sia necessario per stimolare al massimo l’espressione locale dell’IGF-1 nel muscolo scheletrico. In precedenza si è potuto appurare che la differenziazione dei miociti mediata dall’IGF-1 si arresta quando le concentrazioni locali raggiungono circa i 100ng/mL, ma quanto GH è necessario per raggiungere il punto di saturazione dell’espressione dell’mRNA dell’IGF-1?
Gli studi sui miociti umani mostrano che il GH aumenta l’espressione dell’mRNA dell’IGF-1 entro 30-60 minuti con picchi molto più rapidi rispetto a quelli osservati negli studi sugli animali, entro 1-2 ore, usando la via di segnalazione JAK / STAT5b.[365] Questi livelli elevati di mRNA hanno dimostrato di durare fino a 48 ore dopo una singola esposizione al GH. La quantità di GH necessaria per stimolare al massimo l’espressione dell’mRNA dell’IGF-1 è risultata essere una dose compresa tra i 7,5ng/mL e 30ng/ml [366], con una dose media efficace che si attesta a 3ng/ml. Questi numeri sono in linea con gli intervalli di dose fisiologica osservati negli animali, che sono effettivamente compresi tra i 2-100 ng/mL.[367] Inoltre, si collocano esattamente in linea con quanto si osserva endogenamente nell’uomo, con concentrazioni normali di picco comprese tra i 22,4 e 32,4ng/mL.[368-369,436] Ci sono stati casi in cui gli uomini presi in esame hanno mostrato concentrazioni di picco leggermente più alte, ma questi devono essere considerati valori anomali.[370] In ogni caso, ciò che questi dati tendono a suggerire è che il corpo umano è particolarmente adatto a gestire livelli naturali di picco della secrezioni di GH endogeno. Cercare di incidere ulteriormente il sistema elevando i livelli di GH oltre quelli endogeni, unicamente per tentare di potenziare i processi ipertrofici, potrebbe in realtà non tradursi nell’effetto desiderato.
Gli studi che mettono a confronto le infusioni locali con le infusioni sistemiche di GH o IGF-1 sono un po’ più difficili da trovare di quanto si vorrebbe. Le poche sperimentazioni sugli animali che sono riuscito a trovare indicano che l’infusione diretta di GH o IGF-1 nei tessuti bersaglio determina un aumento della massa muscolare. Questo aumento dell’ipertrofia si verifica anche senza che il muscolo bersaglio sia stato sottoposto ad attività motoria.[371-372] Gli studi dimostrano anche che le iniezioni locali di GH portano a livelli sostanzialmente più alti nell’espressione dell’mRNA dell’ IGF-1 locale rispetto alle iniezioni locali di IGF-1, di un fattore di oltre venti.[127] Sono riuscito a trovare uno studio nel quale si confrontavano le risposte dei ratti (attivi e non) all’infusione locale di IGF-1. Il gruppo “IGF-1 plus training” ha mostrato un aumento sia della massa muscolare che della forza locale maggiore rispetto al semplice trattamento in isolamento.[373] Quindi, anche se limitata, la letteratura disponibile è apparentemente in grado di dimostrare che le iniezione locali di GH o IGF-1 hanno effettivamente valore.
Ne ho già parlato diverse volte ma, nel tentativo di imprimere ulteriormente questo concetto, è necessario ricordarsi che i livelli autocrini di IGF-1 sembrano essere molto più importanti dei livelli endocrini di IGF-1 in relazione alla regolazione della massa muscolare. Oltre a questo punto, la sovraespressione dell’IGF-1 autocrino nel muscolo provoca l’ipertrofia delle fibre.[374] La sovraespressione dell’IGF-1 autocrino ha anche mostrato effetti anti-catabolici, con modelli animali tendenti a mostrare una resistenza generale all’atrofia muscolare normalmente osservata con l’invecchiamento.[375] L’IGF-1 localizzato fornisce anche capacità rigenerative indipendenti dall’età nelle cellule muscolari.[376]
Vi sono anche alcune prove convincenti che suggeriscono che l’IGF-1 endocrino agisce direttamente come un regolatore di feedback negativo sulla produzione di IGF-1 autocrino. Questo meccanismo di feedback negativo è dipendente dal pathway PI3K/Akt [377-378]. Inoltre, elevati livelli di IGF-1 endocrino possono anche agire indirettamente per sopprimere la produzione di IGF-1 autocrino. Quindi, in altre parole, non solo l’IGF-1 endocrino ha un impatto diretto minore sulla regolazione della massa muscolare, ma può anche sopprimere l’IGF-1 autocrino che ha impatti maggiori sull’ipertrofia.
Elevati livelli di IGF-1 circolante e, nello specifico, di IGF-1 libero elevati agiscono in modo negativo sul GH determinando un tasso di soppressione della produzione di IGF-1 autocrino a valle.[379] Non è del tutto chiaro, tuttavia, se la regolazione negativa dell’IGF-1 modifichi l’emivita dell’mRNA dell’IGF-1 o influenzi direttamente l’espressione del gene IGF-1. Oltre a questo, è stato anche dimostrato che l’espressione dell’IGF-1 autocrino è sottoregolata nelle cellule muscolari dopo trattamento con IGF-1.[366] È stato anche dimostrato che l’espressione epatica dell’mRNA dell’IGF-1 è sottoregolata dall’esposizione acuta all’IGF-1.[127] Quindi, mantenere livelli endocrini il più possibile soppressi con rispettiva dose di rHGH, elevando contemporaneamente i livelli autocrini, dovrebbe essere un fattore prioritario in un protocollo di GH volto all’ipertrofia.
Il GH è pulsatile per natura sia nell’uomo che nelle specie animali. Quindi, sarebbe logico pensare che molti dei processi intrinseci del corpo saranno tarati in modo tale da rispondere in maniera ottimale all’esposizione al GH in modo simile. In accordo con questa affermazione è stato dimostrato che solo la somministrazione di GH pulsatile, e non l’infusione continua, ha la capacità di stimolare massimamente l’espressione dell’mRNA dell’IGF-1 nel muscolo scheletrico.[366,380-381] È stato anche dimostrato che la somministrazione pulsatile porta ad un aumento del potenziale di crescita postnatale complessivo rispetto all’infusione continua.[89,382] La somministrazione pulsatile può anche portare a livelli endocrini di IGF-1 serici comparabili, o addirittura diminuiti [383], il che è vantaggioso a causa delle potenziali capacità di regolazione negativa che possiede sull’espressione dell’IGF-1 autocrino e che sono state discusse in precedenza. L’evidenza suggerisce anche che il picco stesso, e non necessariamente il numero di picchi, potrebbe essere della massima importanza per i tessuti bersaglio.[384] Per la massima crescita e potenziale ipertrofico, l’evidenza tende a suggerire che creare picchi di GH elevati, e quindi tornare ai livelli basali più volte al giorno, può essere preferibile rispetto a mantenerli elevati per periodi di tempo più lunghi. Questo pratica permette di riprodurre gli schemi secretori in vivo.
I pathways del GH coinvolti nell’anabolismo sono anche suscettibili alla desensibilizzazione, che è parte della fisiologia del GH endogeno.[385] A causa della natura intrinsecamente pulsatile del GH in vivo, l’attività dei recettori e dei pathways sono regolati da un impulso seguito da un periodo di inattività.[386] L’esposizione continua o ripetuta al GH senza un adeguato lasso di tempo refrattario comporterà livelli di attività fortemente soppressi. In effetti, nel corso degli anni numerosi studi hanno dimostrato che tale effetto si verifica. Le cellule ed il tessuto muscolare richiedono un periodo refrattario piuttosto lungo prima che la loro piena risposta al GH venga recuperata. Dopo l’esposizione al GH, le cellule muscolari non sono nemmeno in grado di rispondere alle successive dosi di GH. In realtà, occorrono due ore complete per riprendere parzialmente la reattività nei modelli cellulari, con un totale di 6-8 ore di astinenza dall’uso di GH necessarie per ripristinare la piena sensibilità.[366] Viceversa, quando il GH è micro-dosato in impulsi di dieci minuti, seguiti da intervalli di otto ore, è stato mostrato aumentare progressivamente l’mRNA dell’IGF-1 con ogni impulso successivo.[386]
Questo fenomeno è potenzialmente il risultato di una desensibilizzazione complessiva all’interno della via JAK-STAT5, poiché è stato dimostrato che l’esposizione al GH negli studi sulle cellule epatiche causa resistenza alla successiva attivazione della via STAT5 per 4-8 ore.[387-388] Questo lasso di tempo è sufficiente per sincronizzarsi abbastanza bene con ciò che è stato visto nei modelli di cellule miocitarie citati in precedenza. Nei modelli di cellule epatiche, il GH ha stimolato un significativo aumento dell’espressione del SOCS3, che è un potente inibitore dell’azione del GH.[389]. Poiché il GH non ha avuto alcun effetto sull’espressione del SOCS3 nelle cellule muscolari, questo deve essere un altro meccanismo causante il periodo refrattario. Questo meccanismo può essere dipeso dalla sottoregolazione dei GHR, dall’inibizione mediata da un’altra proteina SOCS, o dall’induzione di una tirosina fosfatasi che semplicemente inattiva la via JAK / STAT.[390] La via JAK-STAT5b, che come ricorderete è intimamente associata al muscolo scheletrico e all’espressione dell’IGF-1, è di natura transitoria – con attivazione massima raggiunta entro 10-30 minuti, seguita da un prolungato periodo di inattivazione.
Una scoperta piuttosto nuova di Xu et al. [391] ha dimostrato che anche distanziare le esposizioni al GH di cinque ore lasciava entrambi i percorsi a valle MEK1/2 e ERK1/2 significativamente soppressi rispetto a tutti i percorsi a monte, a causa di una potenziale disconnessione nella trasduzione del segnale . Ciò è di particolare interesse in quanto questi stessi due percorsi a valle sono stati coinvolti in modo significativo sia nella crescita sia nella proliferazione.[392-393] È stato anche scoperto che l’attivazione indotta da GH di STAT1 e STAT3 è stata desensibilizzata, ma l’esposizione all’Insulina inverte la desensibilizzazione osservata in tutti i percorsi interessati. Anche se non sto per trattare approfonditamente l’Insulina, ci sono un paio di importanti punti da dovere prendere in considerazione. Bisogna comprendere innanzitutto che ci sono molti obiettivi a valle del recettore del GH e molti di questi hanno il potenziale per essere desensibilizzati dopo l’esposizione al GH. Bisogna comprendi anche che l’Insulina possiede l’abilità unica di risensibilizzare molti di questi percorsi. Ciò ha un senso vista la relazione tipo yin-yang tra i due composti. È noto che il GH e l’Insulina possiedono una relazione anabolica sinergica a causa di molti effetti che esercitano l’uno sull’altro. Questo sembra essere soltanto un’anteprima di uno di questi effetti.
Asse GH/IGF-1 – Relazione con altri ormoni
Prima di passare alle note conclusive, vorrei trattare brevemente alcuni altri ormoni connessi a diverso grado con l’Asse GH/IGF-1. Per prima cosa, voglio trattare brevemente l’Asse Tiroideo dal momento che l’inserimento di composti tiroidei insieme al GH è una pratica comune anche durante i protocolli di massa.
Il muscolo scheletrico è il principale bersaglio di segnalazione dell’ormone tiroideo, con trasportatori degli ormoni tiroidei e enzimi di conversione espressi localmente.[394] È ben noto che il GH potenzia la deiodinazione periferica che converte il T4 in T3, riducendo così il T4 e il reverse T3, aumentando contemporaneamente i livelli di T3.[395-398] Ciò che molte persone non riescono a capire è che questo è un effetto transitorio, e studi a lungo termine sembrano indicare che gli effetti mediati dal GH sulla conversione periferica si stabilizzino con il tempo.[399-402]
Via ubiquitina/proteasoma
Invece di proseguire ulteriormente su questo, avendo già trattato la questione nel dettaglio in un mio vecchio articolo, preferirei concentrarmi su alcune pubblicazioni relative alla tiroide che non vengono discusse abbastanza spesso. Gli ormoni tiroidei, per loro natura, sono composti tendenzialmente catabolici in quanto stimolano la disgregazione proteica dell’intero corpo in misura maggiore rispetto alla sintesi proteica.[403] A livello locale, nel muscolo scheletrico stimolano un aumento dell’attività all’interno della via ubiquitina/proteasoma, che è ampiamente coinvolta nella proteolisi.[404-406] Il risultato di questo è un tasso accelerato del turnover proteico e una perdita netta complessiva degli aminoacidi situati all’interno dei muscoli scheletrici.
Inoltre, negli esseri umani, sia gli stati di ipertiroidismo che di ipotiroidismo sono stati associati a livelli di IGF-1 soppressi con una tendenza alla normalizzazione quando viene ristabilita una condizione di eutiroidismo. L’ipertiroidismo è anche associato ad una bassa attività di legame recettoriale del GH, che si ipotizza essere il risultato di una ridotta capacità di elaborazione dei recettori del GH.[407] E’ stato anche ipotizzato che l’ipertiroidismo sia in grado anche di accelerare la clearance del GH urinario.[408] Inoltre, studi su animali hanno dimostrato che gli ormoni tiroidei possono avere importanti effetti soppressivi sulla sintesi di IGF-1 stimolata con il GH.[409] Ovviamente, a causa della complessa relazione che l’Asse Tiroideo ha con l’Asse GH/IGF-1, raggruppando tutte le interazioni che hanno tra loro in pochi paragrafi, trattare l’argomento diventerebbe poco pratico. Tuttavia, quando il corpo della letteratura scientifica viene esaminato nella sua interezza, ci sono molte prove che suggeriscono che la supplementazione con composti tiroidei esogeni potrebbe non essere l’ideale quando l’obiettivo di un individuo è l’ipertrofia, anche se la regolazione del dosaggio dei tiroidei in tale contesto rimane la misura di “sicurezza” più intelligente visto l’impatto negativo sulla funzionalità tiroidea dato dal GH. Comunque, per tutti coloro che sono interessati ad approfondire questo argomento, consiglio di iniziare con la review nella nota seguente.[410]
Miostatina
Mi piacerebbe trattare anche la Miostatin, che rappresenta un argomento molto discusso nei vari forum di BodyBuilding presenti in rete. La sua fama proviene dai risultati ipertrofici espressi dai bovini privi per mutazione del gene della Miostatina, i quali mostrano una massa muscolare significativamente maggiore rispetto ai loro simili non mutati.[411] La Miostatina, un fattore di crescita e differenziazione appartenente alla superfamiglia dei TGF-beta, ha dimostrato di inibire selettivamente la miogenesi, in gran parte tramite il suo effetto soppressivo sulla proliferazione dei mioblasti.[412] È espressa e secreta prevalentemente dal muscolo scheletrico. Come molti sanno, se riesci a sopprimere o inibire la Miostatina, di conseguenza il potenziale ipertrofico aumenta significativamente.
Le mutazioni della Miostatina sono state osservate sia negli animali che nell’uomo. Queste mutazioni del gene della Miostatina portano ad un fenotipo ipertrofico negli animali, come accennato in precedenza.[413-415] L’Asse GH/IGF-1 e la Miostatina sembrano avere una relazione regolativa diretta tra loro, come osservato nei pazienti affetti contemporaneamente da GHD e HIV che mostrano marcati aumenti nell’espressione dell’mRNA della Miostatina.[416] Quindi, è possibile che attraverso una supplementazione di dosi sovrafisiologiche di rHGH si possa indurre una diminuzione dell’mRNA della Miostatina [209,417-419]? Sfortunatamente, nonostante la presenza di alcuni casi studio selezionati, non credo che si abbiano abbastanza dati in questo momento per sapere se ciò possa dare risultati apprezzabili in seguito alla sua applicazione.
Quello che sappiamo è che aumenti dell’espressione dell’mRNA dell’IGF-1 e le concentrazioni circolanti di IGF-1 sono state osservati dopo inibizione della Miostatina.[419-421] Sappiamo anche che l’inibizione della Miostatina tende a causare l’ipertrofia attraverso molte delle stesse modalità osservate con l’IGF-1 autocrino, cioè l’aumento della sintesi proteica e l’attivazione delle cellule satelliti.[422-425] E sappiamo anche che l’ipertrofia indotta dalla sovraespressione dell’IGF-1 o dall’inibizione della Miostatina utilizza la stessa identica via – PI3K/Akt/mTOR.[426-428] Tuttavia, l’IGF-1 non è un requisito per l’ipertrofia indotta dalla Follistatina, tranne nel caso di livelli di Insulina estremamente bassi – come ben sappiamo, la Follistatina è un inibitore della Miostatina [429]. E l’esposizione cronica al GH può in realtà portare ad un’espressione sovrastimolata della Miostatina e del suo recettore.[209]
Quindi quello che possiamo dire, con certezza, è che l’espressione della Miostatina non sarà un fattore diretto o indiretto per quanto riguarda il potenziamento dei processi ipertrofici, né dell’attività contrattile, nei muscoli scheletrici umani.[430] Proprio per questo motivo, non ritengo che sia un fattore sul quale gli atleti debbano eccessivamente concentrarsi, al di fuori di un utile arricchimento delle proprie conoscenze in materia.
Applicazioni pratiche e pensieri conclusivi
Arrivati a questo punto è mia intenzione unire tutto ciò che è stato esposto in questa serie di articoli e esporlo sotto forma di alcuni suggerimenti pratici rivolti a tutti coloro i quali vogliono semplicemente massimizzare la loro capacità ipertrofica.
Ora è chiaro che il GH possiede pochissimi, se non nulli, effetti diretti sull’ipertrofia. Pertanto, qualsiasi protocollo di massa che contempli il suo uso dovrà tenere in considerazione questo punto includendo gli AAS, i quali, per l’appunto, hanno anche una proficua sinergia con il GH. Sia la letteratura scientifica che i dati aneddotici dimostrano chiaramente che l’uso combinato di entrambi i composti ha un massimale ipertrofico significativamente più alto rispetto all’uso singolo. Personalmente, penso che i BodyBuilder dovrebbero sempre optare per l’utilizzo di una base di Testosterone e Boldenone (correttamente rapportati in base a contesto e alle caratteristiche individuali) anche in una fase “Bulk” nella quale viene inserito l’uso del GH. Il Trenbolone può essere considerato come parte “accessoria” di un protocollo di massa, a causa della sua intrinseca difficoltà gestazionale come sostanza anabolizzante. Dovrebbe essere usato con parsimonia e con cautela poiché, insieme ai suoi numerosi punti di forza come composto anabolizzante, presenta alcune limitazioni. Queste limitazioni derivano per lo più, come già accennato, dalla difficile gestione del composto in quanto esso non è facilmente tollerato da una buona parte degli individui. Quindi, se viene usato il Trenbolone, dovrebbe essere inserito calcolando con attenzione il dosaggio e la tolleranza individuale, anche per quanto concerne la tolleranza temporale individuale all’uso di tale composto.
Dopo un periodo d’uso prolungato di dosi sovrafisiologico di AAS (Ciclo+Bridge), è buona cosa procedere con l’interruzione o con una marcata riduzione del numero e degli AAS utilizzati. Detta in parole semplici, questa interruzione può contemplare una completa astinenza dagli AAS, svolgendo un adeguata PCT al fine di ristabilire una omeostasi ormonale fisiologica, o una transizione ad una TRT, metodologia comunemente chiamata “blast and cruise”. La struttura del protocollo di supplementazione farmacologica dovrebbe sempre seguire i principi del dosaggio minimo efficace con aumenti nei dosaggi degli AAS solo nel caso si sia raggiunto il limite di crescita con il precedente dosaggio, assicurandosi che tutte le altre variabili nello stile di vita siano correttamente regolate. L’utilizzo di questo approccio limita il rischio che si sviluppino effetti collaterali indesiderati sul lungo termine.
Quando si decide di usare il GH, la dove ce ne sia la possibilità economica, esso dovrebbe provenire da uno dei prodotti presenti nel mercato farmaceutico. Questi prodotti approvati devono superare anni di studi strettamente controllati per dimostrare la loro sicurezza, purezza ed efficacia su soggetti umani. I progressi tecnologici nel corso degli anni hanno reso molto più facile la produzione di rHGH. Per questo motivo, i produttori ora provengono da tutto il mondo. Spesso questi produttori realizzano ciò che viene definito “GH generico” sui forum, ma tale termine non mi piace molto. Definire qualcosa come “generico” implica che sia una replica perfetta di prodotti legati a specifici marchi farmaceutici approvati dell’agenzia del farmaco che hanno perso il cui brevetto è scaduto, il che non è il caso del GH. Infatti, a causa della natura estremamente complessa del processo di produzione del rHGH, per esempio, la FDA non consente nemmeno l’uso del termine “generico” quando si tratta di rHGH e utilizza invece il termine ” follow-on protein product ” o FOPP.
Spesso questi marchi off-label sono venduti ad un costo molto ridotto, ed è qui che sta il dilemma, in quanto questo può essere molto allettante. Tuttavia, con questo costo ridotto per il consumatore, non ci sarà nemmeno la garanzia del produttore su cosa ci sia realmente nella fiala o persino su come è stato prodotto. Il problema di fondo è che il processo di produzione del rHGH è estremamente complessa, ed è molto facile che nelle fasi di questo processo si commettano errori con conseguenti variazioni nella catena proteica che potenzialmente portano ad effetti indesiderati, o anche a risposte autoimmuni.
Spesso gli atleti si affidano semplicemente ai test serici per misurare i livelli di GH e/o IGF-1 al fine di concludere che un prodotto contenete GH sia “buono”, ma dobbiamo ricordarci che raggiungere livelli ematici ormonali elevati è la parte relativamente facile. Anche le molecole di GH che sono state alterate o danneggiate durante la produzione possono dare questo esito. Tuttavia, queste stesse molecole di GH danneggiate o mutate possono spesso stimolare risposte autoimmuni. Ciò potrebbe indurre il corpo ad avere una risposta recettoriale degradata, che può anche riflettersi sulla secrezione endogena nel tempo. [431-432] Rimane poi il problema del reale contenuto della vial o fiala, la quale può non presentare nessun principio attivo al suo interno.
Il GH dovrebbe essere usato in modo pulsatile, per mimare le condizioni in vivo. Tra queste iniezioni, deve esserci un periodo di refrattarietà o si deve consumare un pasto che abbia un buon stimolo sull’Insulina. Può anche essere utilizzata l’Insulina esogena al fine di bypassare molte delle limitazioni del periodo refrattario, ma questo va oltre lo scopo di questo articolo. Anche se il cumulo delle dosi giornaliere dovrebbe essere sovrafisiologico, le dosi individuali non hanno bisogno di essere ad alto dosaggio, poiché la massima stimolazione dell’IGF-1 autocrino nel tessuto muscolo scheletrico si verifica ben all’interno delle concentrazioni fisiologiche di GH. Aneddoticamente, sembra anche esserci un limite con il quale l’uso di rHGH diventa additivo in presenza di AAS. Potrebbe essere necessario un po’ di ponderata (e supportata da personale qualificato) auto-sperimentazione per scoprire dove si trova questa singola dose di saturazione, ma la maggior parte dei soggetti troverà questo limite tra le 4 e le 8 UI/die. Oltre questo dosaggio, la maggior parte degli utilizzatori tenderà a scoprire che la giustificazione dei costi e il rapporto rischio /beneficio tendono a diminuire rapidamente.
Non bisognerebbe passare troppo tempo a riflettere sul tempo delle iniezioni di GH, dal momento che gli aumenti dei livelli di IGF-1 autocrino avvengono rapidamente e possono rimanere elevati per giorni. Bisognerebbe concentrati invece sul programma di iniezione che si addice meglio al contesto della giornata, tenendo contemporaneamente presenti le linee guida per il periodo di refrattarietà del GH. Si possono anche prendere in considerazione piccole o grandi iniezioni, poiché alcuni potrebbero trovare più pratiche e funzionali iniezioni con un dosaggio inferiore e somministrate più frequenti mentre altri potrebbero preferire iniezioni con un dosaggio maggiore e somministrazioni meno frequenti. Naturalmente, maggiore è il contenuto dell’iniezione, maggiore è la probabilità che si superi la soglia massima di espressione dell’IGF-1 autocrino.
Massimizzare l’espressione dell’IGF-1 autocrino, mentre contemporaneamente si sopprimono i livelli di IGF-1 endocrino, sarà una priorità. Esistono prove a sostegno dell’ipotesi secondo cui un iniezione locale di GH possa aiutare a raggiungere questo obiettivo, con una conseguente minore possibilità di feedback negativo. Sono stati osservati aumenti significativi della massa muscolare in appena due settimane di iniezioni locali di IGF-1.[441]
La dove cause di forza maggiore non lo impediscano, è consigliabile evitare o comunque regolare attentamente l’uso di tutti quei composti che possono avere interazioni negative con l’uso del GH a fini ipertrofici. Inibitori della Aromatasi, Modulatori Selettivi del Recettore degli Estrogeni e T3 hanno tutti dimostrato di avere un potenziale effetto negativo sul processo globale ipertrofico legato al GH e per tale motivo dovrebbero essere usati con parsimonia, se non omessi del tutto dove possibile (vedi in particolare il T3).
Questo non dovrebbe sorprendere nessuno e non dovrebbe nemmeno essere troppo difficile da tenere a mente: allenarsi duramente, allenarsi in modo intelligente e allenarsi in modo coerente. Sebbene non sia stato affrontato direttamente nell’articolo, bisogna capire che l’allenamento contro resistenza ha impatti unici e additivi sull’ipertrofia. In effetti, alcuni di questi meccanismi non sono nemmeno mediati dall’Asse AR e/o GH/IGF-1. [433] Bisogna anche comprendere che non esiste una “magica” routine allenante universalmente applicabile, la chiave di volta sarà la coerenza nel garantire un carico di lavoro adeguato, con elementi di sovraccarico progressivo nel tempo assicurando un adeguato stimolo meccanico gestendo al meglio le variabili allenanti (intensità, volume, densità e intensità percepita). La logica composizione del piano allenante servirà a garantire lo stimolo ipertrofico di base che sarà coadiuvato dall’azione dei composti utilizzati.
Nonostante la mole e l’importanza delle informazioni presentate in questa serie di articoli, è necessario ricordare che i meccanismi d’azione ormonali sono governati da innumerevoli fattori. Anche esaminando l’intero corpo della letteratura scientifica equivarrebbe a poco più che accumulare una serie di utili nozioni garanti di assicurare una solida base conoscitiva sull’argomento la quale rappresenterà un punto di partenza intelligente per l’applicazione pratica, rimanendo sempre soggetti alle variabili di risposta individuale. Seguendo questa linea, i migliori risultati nella pratica spesso provengono da coloro i quali posseggono una buona conoscenza dei principi scientifici e la capacità innata di saperli applicare non solo su se stessi ma, soprattutto, su terzi. Infatti, molto raramente due persone rispondono in modo identico alla supplementazione di ormoni esogeni (e non solo), quindi, non bisogna assolutamente pensare che basti semplicemente applicare su se stessi o su terzi un protocollo che ha portato benefici realmente apprezzabili su un soggetto per ottenere la medesima risposta.
A tal fine, invito atleti e Preparatori a utilizzare queste pubblicazioni come punto di partenza per essere in grado di gestire l’applicazione pratica in modo più consapevole e produttivo. Inoltre, chi ne fosse in grado, può consultare il vasto numero di riferimenti riportati nel corso di queste pubblicazioni è tentare di ragionare sulle mie conclusioni. Ad ogni citazione presente in questa serie di articoli, assicuratevi che il riferimento elencato supporti effettivamente le affermazioni fatte. Mantenere sempre una mente aperta ma con i giusti “filtri”, e cercare di non credere per partito preso ad una singola opinione, specialmente di fronte alle nuove evidenze scientifiche. Infine, è buona cosa verificare sempre la veridicità di quanto è stato affermato.
Punti conclusivi per un corretto utilizzo della chimica e del GH a fini ipertrofici:
Usare il GH in combinazione con gli AAS
Usare GH e AAS di grado farmaceutico la dove ciò è possibile
Assicurarsi una base di Testosterone correttamente rapportata al Boldenone aggiungendo (in base a maturità e tolleranza) il Trenbolone
Iniettare il GH in modo pulsatile, considerare l’opzione delle iniezioni locali nei gruppi carenti
Mantenere un dosaggio complessivo ottimale di GH il quale si attesta tra le 4 e le 8UI/die
Evitare o regolare attentamente l’uso dei composti che possono interagire negativamente con i processi ipertrofici legati al uso di GH (vedi AI, SERM e T3)
Dopo un periodo di tempo (variabile) nel quale si è stati sottoposti a dosaggi ormonali sovrafisiologici optare per una PCT o per una TRT (a seconda delle proprie necessità e priorità)
Essere a conoscenza dei potenziali effetti collaterali legati all’auso/abuso di GH (nausea, vomito, cefalea, ritenzione idrica e sodica, edemi, parestesie, sindrome del tunnel carpale, rigidità articolare, dolori articolari, artrite, dolori muscolari, ipertensione, insulino-resistenza, diabete di tipo II, acromegalia, dilatazione addominale, ipertrofia cardiaca ecc…)
Svolgere regolarmente esami del sangue; sia durante i periodi di picco nell’uso della farmacologia sia nel periodo successivo (vedi PCT/OCT o TRT)
Gestire al meglio le variabili legate agli stressor ambientali, all’allenamento, all’alimentazione e al sonno.
Gabriel Bellizzi
Riferimenti:
338. Mathews LS, Norstedt G, Palmiter RD. Regulation of insulin-like growth factor I gene expression by growth hormone. Proc Natl Acad Sci U S A. 1986 Dec;83(24):9343-7.
339. Keller A, Wu Z, Kratzsch J, Keller E, Blum WF, Kniess A, Preiss R, Teichert J, Strasburger CJ, Bidlingmaier M. Pharmacokinetics and pharmacodynamics of GH: dependence on route and dosage of administration. Eur J Endocrinol. 2007 Jun;156(6):647-53.
340. Tanaka T, Seino Y, Fujieda K, Igarashi Y, Yokoya S, Tachibana K, Ogawa Y. Pharmacokinetics and metabolic effects of high-dose growth hormone administration in healthy adult men. Endocr J. 1999 Aug;46(4):605-12.
341. Quinn LS, Steinmetz B, Maas A, Ong L, Kaleko M. Type-1 insulin-like growth factor receptor overexpression produces dual effects on myoblast proliferation and differentiation. J Cell Physiol. 1994 Jun;159(3):387-98.
342. Coolican SA, Samuel DS, Ewton DZ, McWade FJ, Florini JR. The mitogenic and myogenic actions of insulin-like growth factors utilize distinct signaling pathways. J Biol Chem. 1997 Mar 7;272(10):6653-62.
343. Foulstone EJ, Huser C, Crown AL, Holly JM, Stewart CE. Differential signalling mechanisms predisposing primary human skeletal muscle cells to altered proliferation and differentiation: roles of IGF-I and TNFalpha. Exp Cell Res. 2004 Mar 10;294(1):223-35.
344. Ewton DZ, Roof SL, Magri KA, McWade FJ, Florini JR. IGF-II is more active than IGF-I in stimulating L6A1 myogenesis: greater mitogenic actions of IGF-I delay differentiation. J Cell Physiol. 1994 Nov;161(2):277-84.
345. Florini JR, Nicholson ML, Dulak NC. Effects of peptide anabolic hormones on growth of myoblasts in culture. Endocrinology. 1977 Jul;101(1):32-41.
346. Laviola L, Natalicchio A, Giorgino F. The IGF-I signaling pathway. Curr Pharm Des. 2007;13(7):663-9. Review.
347. Jacquemin V, Furling D, Bigot A, Butler-Browne GS, Mouly V. IGF-1 induces human myotube hypertrophy by increasing cell recruitment. Exp Cell Res. 2004 Sep 10;299(1):148-58.
348. Jacquemin V, Butler-Browne GS, Furling D, Mouly V. IL-13 mediates the recruitment of reserve cells for fusion during IGF-1-induced hypertrophy of human myotubes. J Cell Sci. 2007 Feb 15;120(Pt 4):670-81. Epub 2007 Jan 30.
349. Ballard FJ, Francis GL. Effects of anabolic agents on protein breakdown in L6 myoblasts. Biochem J. 1983 Jan 15;210(1):243-9.
350. Ewton DZ, Falen SL, Florini JR. The type II insulin-like growth factor (IGF) receptor has low affinity for IGF-I analogs: pleiotypic actions of IGFs on myoblasts are apparently mediated by the type I receptor. Endocrinology. 1987 Jan;120(1):115-23.
351. Hembree JR, Hathaway MR, Dayton WR. Isolation and culture of fetal porcine myogenic cells and the effect of insulin, IGF-I, and sera on protein turnover in porcine myotube cultures. J Anim Sci. 1991 Aug;69(8):3241-50.
352. Hong D, Forsberg NE. Effects of serum and insulin-like growth factor I on protein degradation and protease gene expression in rat L8 myotubes. J Anim Sci. 1994 Sep;72(9):2279-88.
353. Florini JR, Ewton DZ, Roof SL. Insulin-like growth factor-I stimulates terminal myogenic differentiation by induction of myogenin gene expression. Mol Endocrinol. 1991 May;5(5):718-24.
354. Musarò A, McCullagh KJ, Naya FJ, Olson EN, Rosenthal N. IGF-1 induces skeletal myocyte hypertrophy through calcineurin in association with GATA-2 and NF-ATc1. Nature. 1999 Aug 5;400(6744):581-5.
355. Haba GDL, Cooper GW, Elting V. HORMONAL REQUIREMENTS FOR MYOGENESIS OF STRIATED MUSCLE IN VITRO: INSULIN AND SOMATOTROPIN. Proceedings of the National Academy of Sciences of the United States of America. 1966;56(6):1719-1723.
356. Florini JR, Ewton DZ. Insulin acts as a somatomedin analog in stimulating myoblast growth in serum-free medium. In Vitro. 1981 Sep;17(9):763-8.
357. Schmid C, Steiner T, Froesch ER. Preferential enhancement of myoblast differentiation by insulin-like growth factors (IGF I and IGF II) in primary cultures of chicken embryonic cells. FEBS Lett. 1983 Sep 5;161(1):117-21.
358. Florini JR, Ewton DZ, Falen SL, Van Wyk JJ. Biphasic concentration dependency of stimulation of myoblast differentiation by somatomedins. Am J Physiol. 1986 May;250(5 Pt 1):C771-8.
359. Quinn LS, Ehsan M, Steinmetz B, Kaleko M. Ligand-dependent inhibition of myoblast differentiation by overexpression of the type-1 insulin-like growth factor receptor. J Cell Physiol. 1993 Sep;156(3):453-61.
360. Olson EN. Signal transduction pathways that regulate skeletal muscle gene expression. Mol Endocrinol. 1993 Nov;7(11):1369-78. Review.
361. Murphy LJ, Bell GI, Friesen HG. Growth hormone stimulates sequential induction of c-myc and insulin-like growth factor I expression in vivo. Endocrinology. 1987 May;120(5):1806-12.
362. Turner JD, Rotwein P, Novakofski J, Bechtel PJ. Induction of mRNA for IGF-I and -II during growth hormone-stimulated muscle hypertrophy. Am J Physiol. 1988 Oct;255(4 Pt 1):E513-7.
363. Isgaard J, Nilsson A, Vikman K, Isaksson OG. Growth hormone regulates the level of insulin-like growth factor-I mRNA in rat skeletal muscle. J Endocrinol. 1989 Jan;120(1):107-12.
364. Bichell DP, Kikuchi K, Rotwein P. Growth hormone rapidly activates insulin-like growth factor I gene transcription in vivo. Mol Endocrinol. 1992 Nov;6(11):1899-908.
365. Sadowski CL, Wheeler TT, Wang LH, Sadowski HB. GH regulation of IGF-I and suppressor of cytokine signaling gene expression in C2C12 skeletal muscle cells. Endocrinology. 2001 Sep;142(9):3890-900.
366. Frost RA, Nystrom GJ, Lang CH. Regulation of IGF-I mRNA and signal transducers and activators of transcription-3 and -5 (Stat-3 and -5) by GH in C2C12 myoblasts. Endocrinology. 2002 Feb;143(2):492-503.
367. MacLeod JN, Pampori NA, Shapiro BH. Sex differences in the ultradian pattern of plasma growth hormone concentrations in mice. J Endocrinol. 1991 Dec;131(3):395-9.
368. Rochiccioli P, Messina A, Tauber MT, Enjaume C. Correlation of the parameters of 24-hour growth hormone secretion with growth velocity in 93 children of varying height. Horm Res. 1989;31(3):115-8.
369. Hansen TK, Gravholt CH, ØRskov H, Rasmussen MH, Christiansen JS, Jørgensen JO. Dose dependency of the pharmacokinetics and acute lipolytic actions of growth hormone. J Clin Endocrinol Metab. 2002 Oct;87(10):4691-8.
370. Baum WF, Klöditz E, Hesse V, Jahreis G, Schneyer U, Giebler H. [Increase in spontaneous growth hormone secretion in asthmatic children–a symptom of atopic disposition?]. Kinderarztl Prax. 1993 Nov;61(9):323-8.
371. Adams GR, McCue SA. Localized infusion of IGF-I results in skeletal muscle hypertrophy in rats. J Appl Physiol (1985). 1998 May;84(5):1716-22.
372. Alzghoul MB, Gerrard D, Watkins BA, Hannon K. Ectopic expression of IGF-I and Shh by skeletal muscle inhibits disuse-mediated skeletal muscle atrophy and bone osteopenia in vivo. FASEB J. 2004 Jan;18(1):221-3. Epub 2003 Nov 3.
373. Lee S, Barton ER, Sweeney HL, Farrar RP. Viral expression of insulin-like growth factor-I enhances muscle hypertrophy in resistance-trained rats. J Appl Physiol (1985). 2004 Mar;96(3):1097-104. Erratum in: J Appl Physiol. 2004 Jun;96(6):2343.
374. Coleman ME, DeMayo F, Yin KC, Lee HM, Geske R, Montgomery C, Schwartz RJ. Myogenic vector expression of insulin-like growth factor I stimulates muscle cell differentiation and myofiber hypertrophy in transgenic mice. J Biol Chem. 1995 May 19;270(20):12109-16.
375. Barton-Davis ER, Shoturma DI, Musaro A, Rosenthal N, Sweeney HL. Viral mediated expression of insulin-like growth factor I blocks the aging-related loss of skeletal muscle function. Proc Natl Acad Sci U S A. 1998 Dec 22;95(26):15603-7.
376. Musarò A, McCullagh K, Paul A, Houghton L, Dobrowolny G, Molinaro M, Barton ER, Sweeney HL, Rosenthal N. Localized Igf-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat Genet. 2001 Feb;27(2):195-200
377. Lewis MI, Bulut Y, Biring MS, Da X, Fournier M. (1999) IGF-I administration prevents corticosteroids-induced diaphragm atrophy in emphysema . Am J Respir Crit Care Med 159:A580
378. Fournier M, Huang ZS, Cercek B, Li H, Bykhovskaya I, Lewis MI. (2000) Administration of insulin-like growth factor-1 (IGF-I) and corticosteroids in emphysematous hamsters: influences on diaphragm IGF-I . Am J Respir Crit Care Med 161:A18
379. Shavlakadze T, Grounds M. Of bears, frogs, meat, mice and men: complexity of factors affecting skeletal muscle mass and fat. Bioessays. 2006 Oct;28(10):994-1009. Review.
380. Maiter D, Underwood LE, Maes M, Davenport ML, Ketelslegers JM. Different effects of intermittent and continuous growth hormone (GH) administration on serum somatomedin-C/insulin-like growth factor I and liver GH receptors in hypophysectomized rats. Endocrinology. 1988 Aug;123(2):1053-9.
381. Isgaard J, Carlsson L, Isaksson OG, Jansson JO. Pulsatile intravenous growth hormone (GH) infusion to hypophysectomized rats increases insulin-like growth factor I messenger ribonucleic acid in skeletal tissues more effectively than continuous GH infusion. Endocrinology. 1988 Dec;123(6):2605-10.
382. Clark RG, Jansson JO, Isaksson O, Robinson IC. Intravenous growth hormone: growth responses to patterned infusions in hypophysectomized rats. J Endocrinol. 1985 Jan;104(1):53-61.
383. Bick T, Hochberg Z, Amit T, Isaksson OG, Jansson JO. Roles of pulsatility and continuity of growth hormone (GH) administration in the regulation of hepatic GH-receptors, and circulating GH-binding protein and insulin-like growth factor-I. Endocrinology. 1992 Jul;131(1):423-9.
384. Weltman A, Weltman JY, Schurrer R, Evans WS, Veldhuis JD, Rogol AD. Endurance training amplifies the pulsatile release of growth hormone: effects of training intensity. J Appl Physiol (1985). 1992 Jun;72(6):2188-96.
385. Flores-Morales A, Greenhalgh CJ, Norstedt G, Rico-Bautista E. Negative regulation of growth hormone receptor signaling. Mol Endocrinol. 2006 Feb;20(2):241-53. Epub 2005 Jul 21. Review.
386. Hartman ML, Veldhuis JD, Thorner MO. Normal control of growth hormone secretion. Horm Res. 1993;40(1-3):37-47. Review.
387. Fernández L, Flores-Morales A, Lahuna O, Sliva D, Norstedt G, Haldosén LA, Mode A, Gustafsson JA. Desensitization of the growth hormone-induced Janus kinase 2 (Jak 2)/signal transducer and activator of transcription 5 (Stat5)-signaling pathway requires protein synthesis and phospholipase C. Endocrinology. 1998 Apr;139(4):1815-24.
388. Gebert CA, Park SH, Waxman DJ. Termination of growth hormone pulse-induced STAT5b signaling. Mol Endocrinol. 1999 Jan;13(1):38-56.
389. Ram PA, Waxman DJ. SOCS/CIS protein inhibition of growth hormone-stimulated STAT5 signaling by multiple mechanisms. J Biol Chem. 1999 Dec 10;274(50):35553-61.
390. Ram PA, Waxman DJ. Role of the cytokine-inducible SH2 protein CIS in desensitization of STAT5b signaling by continuous growth hormone. J Biol Chem.2000 Dec 15;275(50):39487-96.
391. Xu J, Keeton AB, Franklin JL, Li X, Venable DY, Frank SJ, Messina JL. Insulin enhances growth hormone induction of the MEK/ERK signaling pathway. J Biol Chem. 2006 Jan 13;281(2):982-92. Epub 2005 Nov 4.
392. Lewis TS, Shapiro PS, Ahn NG. Signal transduction through MAP kinase cascades. Adv Cancer Res. 1998;74:49-139. Review.
394. Mebis L, Paletta D, Debaveye Y, Ellger B, Langouche L, D’Hoore A, Darras VM, Visser TJ, Van den Berghe G. Expression of thyroid hormone transporters during critical illness. Eur J Endocrinol. 2009 Aug;161(2):243-50.
395. Jørgensen JO, Pedersen SA, Laurberg P, Weeke J, Skakkebaek NE, Christiansen JS. Effects of growth hormone therapy on thyroid function of growth hormone-deficient adults with and without concomitant thyroxine-substituted central hypothyroidism. J Clin Endocrinol Metab. 1989 Dec;69(6):1127-32.
396. Jørgensen JO, Pedersen SB, Børglum J, Møller N, Schmitz O, Christiansen JS, Richelsen B. Fuel metabolism, energy expenditure, and thyroid function in growth hormone-treated obese women: a double-blind placebo-controlled study. Metabolism. 1994 Jul;43(7):872-7.
397. Wolthers T, Grøftne T, Møller N, Christiansen JS, Orskov H, Weeke J, Jørgensen JO. Calorigenic effects of growth hormone: the role of thyroid hormones. J Clin Endocrinol Metab. 1996 Apr;81(4):1416-9.
398. Feldt-Rasmussen U. Interactions between growth hormone and the thyroid gland — with special reference to biochemical diagnosis. Curr Med Chem. 2007;14(26):2783-8. Review.
399. Kalina-Faska B, Kalina M, Koehler B. Effects of recombinant growth hormone therapy on thyroid hormone concentrations. Int J Clin Pharmacol Ther. 2004 Jan;42(1):30-4.
400. Hubina E, Mersebach H, Rasmussen AK, Juul A, Sneppen SB, Góth MI, Feldt-Rasmussen U. Effect of growth hormone replacement therapy on pituitary hormone secretion and hormone replacement therapies in GHD adults. Horm Res. 2004;61(5):211-7. Epub 2004 Jan 30.
401. Seminara S, Stagi S, Candura L, Scrivano M, Lenzi L, Nanni L, Pagliai F, Chiarelli F. Changes of thyroid function during long-term hGH therapy in GHD children. A possible relationship with catch-up growth? Horm Metab Res. 2005 Dec;37(12):751-6.
402. Losa M, Scavini M, Gatti E, Rossini A, Madaschi S, Formenti I, Caumo A, Stidley CA, Lanzi R. Long-term effects of growth hormone replacement therapy on thyroid function in adults with growth hormone deficiency. Thyroid. 2008 Dec;18(12):1249-54.
403. Müller MJ, Seitz HJ. Thyroid hormone action on intermediary metabolism. Part III. Protein metabolism in hyper- and hypothyroidism. Klin Wochenschr. 1984 Feb 1;62(3):97-102.
404. Tawa NE Jr, Odessey R, Goldberg AL. Inhibitors of the proteasome reduce the accelerated proteolysis in atrophying rat skeletal muscles. J Clin Invest. 1997 Jul 1;100(1):197-203. PubMed PMID: 9202072
405. Dace A, Zhao L, Park KS, et al. Hormone binding induces rapid proteasome-mediated degradation of thyroid hormone receptors. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(16):8985-8990.
406. Clément K, Viguerie N, Diehn M, Alizadeh A, Barbe P, Thalamas C, Storey JD, Brown PO, Barsh GS, Langin D. In vivo regulation of human skeletal muscle gene expression by thyroid hormone. Genome Res. 2002 Feb;12(2):281-91.
407. Miell JP, Taylor AM, Zini M, Maheshwari HG, Ross RJ, Valcavi R. Effects of hypothyroidism and hyperthyroidism on insulin-like growth factors (IGFs) and growth hormone- and IGF-binding proteins. J Clin Endocrinol Metab. 1993 Apr;76(4):950-5.
408. Murao K, Takahara J, Sato M, Tamaki M, Niimi M, Ishida T. Relationship between thyroid functions and urinary growth hormone secretion in patients with hyper- and hypothyroidism. Endocr J. 1994 Oct;41(5):517-22.
409. Wolf M, Ingbar SH, Moses AC. Thyroid hormone and growth hormone interact to regulate insulin-like growth factor-I messenger ribonucleic acid and circulating levels in the rat. Endocrinology. 1989 Dec;125(6):2905-14.
410. Laron Z. Interactions between the thyroid hormones and the hormones of the growth hormone axis. Pediatr Endocrinol Rev. 2003 Dec;1 Suppl 2:244-9-discussion 250. Review.
411. Fiems LO. Double Muscling in Cattle: Genes, Husbandry, Carcasses and Meat. Animals : an Open Access Journal from MDPI. 2012;2(3):472-506.
412. Langley B, Thomas M, Bishop A, Sharma M, Gilmour S, Kambadur R. Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J Biol Chem. 2002 Dec 20;277(51):49831-40. Epub 2002 Sep 18.
413. McPherron AC, Lee SJ. Double muscling in cattle due to mutations in the myostatin gene. Proc Natl Acad Sci U S A. 1997 Nov 11;94(23):12457-61.
414. Schuelke M, Wagner KR, Stolz LE, Hübner C, Riebel T, Kömen W, Braun T, Tobin JF, Lee SJ. Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med. 2004 Jun 24;350(26):2682-8.
415. Clop A, Marcq F, Takeda H, Pirottin D, Tordoir X, Bibé B, Bouix J, Caiment F, Elsen JM, Eychenne F, Larzul C, Laville E, Meish F, Milenkovic D, Tobin J, Charlier C, Georges M. A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat Genet. 2006 Jul;38(7):813-8.
416. Gonzalez-Cadavid NF, Taylor WE, Yarasheski K, Sinha-Hikim I, Ma K, Ezzat S, Shen R, Lalani R, Asa S, Mamita M, Nair G, Arver S, Bhasin S. Organization of the human myostatin gene and expression in healthy men and HIV-infected men with muscle wasting. Proc Natl Acad Sci U S A. 1998 Dec 8;95(25):14938-43.
417. Liu W, Thomas SG, Asa SL, Gonzalez-Cadavid N, Bhasin S, Ezzat S. Myostatin is a skeletal muscle target of growth hormone anabolic action. J Clin Endocrinol Metab. 2003 Nov;88(11):5490-6.
418. Oldham JM, Osepchook CC, Jeanplong F, Falconer SJ, Matthews KG, Conaglen JV, Gerrard DF, Smith HK, Wilkins RJ, Bass JJ, McMahon CD. The decrease in mature myostatin protein in male skeletal muscle is developmentally regulated by growth hormone. J Physiol. 2009 Feb 1;587(3):669-77.
419. Williams NG, Interlichia JP, Jackson MF, Hwang D, Cohen P, Rodgers BD. Endocrine actions of myostatin: systemic regulation of the IGF and IGF binding protein axis. Endocrinology. 2011 Jan;152(1):172-80.
420. Winbanks CE, Weeks KL, Thomson RE, Sepulveda PV, Beyer C, Qian H, Chen JL, Allen JM, Lancaster GI, Febbraio MA, Harrison CA, McMullen JR, Chamberlain JS, Gregorevic P. Follistatin-mediated skeletal muscle hypertrophy is regulated by Smad3 and mTOR independently of myostatin. J Cell Biol. 2012 Jun 25;197(7):997-1008.
421. Lach-Trifilieff E, Minetti GC, Sheppard K, Ibebunjo C, Feige JN, Hartmann S, Brachat S, Rivet H, Koelbing C, Morvan F, Hatakeyama S, Glass DJ. An antibody blocking activin type II receptors induces strong skeletal muscle hypertrophy and protects from atrophy. Mol Cell Biol. 2014 Feb;34(4):606-18.
422. Bark TH, McNurlan MA, Lang CH, Garlick PJ. Increased protein synthesis after acute IGF-I or insulin infusion is localized to muscle in mice. Am J Physiol. 1998 Jul;275(1 Pt 1):E118-23.
423. Barton-Davis ER, Shoturma DI, Sweeney HL. Contribution of satellite cells to IGF-I induced hypertrophy of skeletal muscle. Acta Physiol Scand. 1999 Dec;167(4):301-5.
424. Suryawan A, Frank JW, Nguyen HV, Davis TA. Expression of the TGF-beta family of ligands is developmentally regulated in skeletal muscle of neonatal rats. Pediatr Res. 2006 Feb;59(2):175-9.
425. Gilson H, Schakman O, Kalista S, Lause P, Tsuchida K, Thissen JP. Follistatin induces muscle hypertrophy through satellite cell proliferation and inhibition of both myostatin and activin. Am J Physiol Endocrinol Metab. 2009 Jul;297(1):E157-64.
426. Bodine SC, Stitt TN, Gonzalez M, Kline WO, Stover GL, Bauerlein R, Zlotchenko E, Scrimgeour A, Lawrence JC, Glass DJ, Yancopoulos GD. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol. 2001 Nov;3(11):1014-9.
428. Kalista S, Schakman O, Gilson H, Lause P, Demeulder B, Bertrand L, Pende M, Thissen JP. The type 1 insulin-like growth factor receptor (IGF-IR) pathway is mandatory for the follistatin-induced skeletal muscle hypertrophy. Endocrinology. 2012 Jan;153(1):241-53.
429. Barbé C, Kalista S, Loumaye A, Ritvos O, Lause P, Ferracin B, Thissen JP. Role of IGF-I in follistatin-induced skeletal muscle hypertrophy. Am J Physiol Endocrinol Metab. 2015 Sep 15;309(6):E557-67. doi: 10.1152/ajpendo.00098.2015. Epub 2015 Jul 28.
430. Coffey VG, Shield A, Canny BJ, Carey KA, Cameron-Smith D, Hawley JA. Interaction of contractile activity and training history on mRNA abundance in skeletal muscle from trained athletes. Am J Physiol Endocrinol Metab. 2006 May;290(5):E849-55.
431. Moore WV, Leppert P. Role of aggregated human growth hormone (hGH) in development of antibodies to hGH. J Clin Endocrinol Metab. 1980 Oct;51(4):691-7
432. Dannies PS. Protein folding and deficiencies caused by dominant-negative mutants of hormones. Vitam Horm. 2000;58:1-26. Review.
433. DeVol DL, Rotwein P, Sadow JL, Novakofski J, Bechtel PJ. Activation of insulin-like growth factor gene expression during work-induced skeletal muscle growth. Am J Physiol. 1990 Jul;259(1 Pt 1):E89-95.
434. Hermansen K, Bengtsen M, Kjær M, Vestergaard P, Jørgensen JOL. Impact of GH administration on athletic performance in healthy young adults: A systematic review and meta-analysis of placebo-controlled trials. Growth Horm IGF Res. 2017 Jun;34:38-44.
435. de Souza GL, Hallak J. Anabolic steroids and male infertility: a comprehensive review. BJU Int. 2011 Dec;108(11):1860-5.
436. Kraemer WJ, Marchitelli L, Gordon SE, Harman E, Dziados JE, Mello R, Frykman P, McCurry D, Fleck SJ. Hormonal and growth factor responses to heavy resistance exercise protocols. J Appl Physiol (1985). 1990 Oct;69(4):1442-50.
437. Pfeffer LA, Brisson BK, Lei H, Barton ER. The insulin-like growth factor (IGF)-I E-peptides modulate cell entry of the mature IGF-I protein. Mol Biol Cell. 2009 Sep;20(17):3810-7.
438. Mills P, Dominique JC, Lafrenière JF, Bouchentouf M, Tremblay JP. A synthetic mechano growth factor E Peptide enhances myogenic precursor cell transplantation success. Am J Transplant. 2007 Oct;7(10):2247-59.
439. Brisson BK, Barton ER. Insulin-like growth factor-I E-peptide activity is dependent on the IGF-I receptor. PLoS One. 2012;7(9):e45588.
440. Brisson BK, Spinazzola J, Park S, Barton ER. Viral expression of insulin-like growth factor I E-peptides increases skeletal muscle mass but at the expense of strength. Am J Physiol Endocrinol Metab. 2014 Apr 15;306(8):E965-74.
441. Goldspink G, Harridge S. Mechanism for adaptation in skeletal muscle In: Komi P, editor. Strength and power in sport: Olympic encyclopedia of sports medicine. Oxford: Blackwell; 2002. p. 231–51.
442. Janssen JA, Hofland LJ, Strasburger CJ, van den Dungen ES, Thevis M. Potency of Full-Length MGF to Induce Maximal Activation of the IGF-I R Is Similar to Recombinant Human IGF-I at High Equimolar Concentrations. PLoS One. 2016 Mar 18;11(3):e0150453.
Nei primi anni del XXI secolo, negli scaffali dei negozi di integratori (fisici o online) emersero alcuni nuovi supplementi e tra questi emerse l’Acido Tetradeciltioacetico [TTA]. In quel periodo questo particolare composto era uno dei principi attivi, insieme a molte altre sostanze, di alcuni controversi supplementi per la perdita di grasso. La maggior parte di questi prodotti è scomparsa dal mercato, ma l’Acido Tetradeciltiocetico è stato recentemente reinserito nel mercato. Ora, spesso in concentrazioni significativamente più elevate rispetto a un decennio fa, il TTA è spesso l’unico ingrediente attivo presente nei supplementi che lo contengono. E alcuni produttori di integratori vendono tale composto sotto forma di polvere. È quindi interessante approfondire le caratteristiche del composto e di cosa è ipoteticamente in grado di fare.
L’Acido Tetradeciltioacetico è un acido grasso sintetico che non può essere convertito in energia.
Negli anni ’90 gli scienziati scoprirono che l’Acido Tetradeciltioacetico stimolava il metabolismo dei grassi attivando i PPAR-α. I ricercatori hanno scoperto anche che, attraverso lo stesso meccanismo, la sostanza era in grado di inibire l’infiammazione, la crescita delle cellule tumorali e lo sviluppo/peggioramento dell’insulino-resistenza.
Esistono tre tipi di recettori PPAR: α, γ e δ. Il recettore α è espresso a livello epatico. Quando viene attivato, il fegato utilizza più acidi grassi per produrre energia. Il recettore γ è espresso nelle cellule adipose. Se viene stimolato, la cellula adiposa immagazzina acidi grassi. Il recettore δ è espresso a livello muscolare. Se il recettore δ viene stimolato, i muscoli utilizzano maggiormente gli acidi grassi per produrre energia.
Nel 2002, scienziati norvegesi hanno riferito che gli animali presi in esame, e che avevano ricevuto una dose significativa di Acido Tetradeciltioacetico per via orale, non hanno mostrato un aumento della percentuale di grasso corporeo in concomitanza con un regime alimentare ipercalorico.(1) La suddetta pubblicazione ha anche riportato che l’Acido Tetradeciltioacetico non solo stimolava il PPAR- α, ma anche il PPAR- γ e, in misura minore, il PPAR-δ.
Nel 2009 nutrizionisti affiliati all’Università di Oslo, hanno pubblicato un altro studio svolto su animali. In questo studio, i norvegesi hanno fatto ingrassare i ratti dando loro un mangime che conteneva una elevata percentuale di grasso [Lard] per 7 settimane.
La metà dei ratti presi in esame ha ricevuto anche Acido Tetradeciltioacetico [Lard Plus TTA]. Se i topi fossero stati esseri umani adulti, avrebbero consumato giornalmente circa 1,5g di Acido Tetradeciltioacetico.
L’integrazione con TTA ha significativamente inibito l’aumento del peso corporeo degli animali trattati. Tuttavia, però, la supplementazione ha indotto gli animali a consumare più cibo. Tale comportamento è strano, dal momento che la maggior parte degli agonisti PPAR-α diminuiscono l’appetito interferendo con la biosintesi di alcuni neuropeptidi ipotalamici.
Al termine delle 7 settimane del test, i ratti trattati con Acido Tetradeciltioacetico non erano solo più magri dei ratti non trattati. La loro composizione corporea differiva anche dai ratti del gruppo “Lard”: presentavano una miglior massa muscolare e una ridotta percentuale di grasso.
Di seguito viene mostrato come l’Acido Tetradeciltioacetico stimola il metabolismo nei ratti. Nel fegato, l’attività del gene UCP3, che potenzia il metabolismo, ha subito un incremento di non meno di un fattore di duemila.
I ricercatori scrivono che i loro dati sono in accordo nel riconoscere l’attivazione del PPAR- α come mediatore principale degli effetti dell’Acido Tetradeciltiocetico sull’espressione del gene epatico. L’effetto dell’Acido Tetradeciltiocetico sull’assunzione di cibo è risultato opposto a quanto riportato per altri ligandi il PPAR- α e può essere correlato all’attivazione concomitante del PPAR- γ, alla riduzione della segnalazione della Leptina o alla riduzione della sensibilità ipotalamica del malonil-CoA.
Per chiarire ulteriormente gli effetti dell’Acido Tetradeciltioacetico sull’assunzione di cibo e sull’efficienza del cibo, continuano i ricercatori, l’uso di gabbie metaboliche potrebbe chiarire laquestione. Inoltre, dovrebbero essere prese in considerazione le indagini sull’eventualità che l’Acido Tetradeciltioacetico si accumuli nell’ipotalamo e promuova […] i livelli di sazietà regolati dai neuropeptidi.
Quando i primi supplementi per la perdita di grasso contenenti Acido Tetradeciltioacetico sono comparsi nel mercato degli integratori nel primo decennio del XXI secolo, i produttori hanno cercato di prevenire l’aumento dell’appetito combinando il TTA con composti aventi azione anoressizzante l’Oleoietanolamide.
Se quei supplementi funzionassero davvero è un altro discorso. C’è da considerare anche che la quantità di Acido Tetradeciltioacetico in essi contenuta era bassa. Tuttavia, con i nuovi prodotti presenti sul mercato, le dosi di TTA sono molto più alte.
Sebbene in numero limitato, esistono degli studi svolti sull’uomo che hanno osservato l’effetto del TTA… e i risultati ottenuti non sono così promettenti come quelli osservati sugli animali.
Nel 2004, i ricercatori del Rikshospitalet di Oslo hanno somministrato a 10 pazienti affetti da HIV 1g di TTA al giorno per 4 settimane.(2) L’integrazione non ha avuto alcun effetto sulle cellule T, ma ha ridotto la concentrazione di TNF-α, LDL e Trigliceridi nel sangue. Ma la perdita di peso? Non si è verificata.
Nel 2008, i ricercatori norvegesi, affiliati all’Università di Bergen, hanno somministrato a 18 volontari 200mg, 600mg o 1g di TTA al giorno durante uno studio di fase 1 per 7 giorni. Non sono emersi effetti collaterali degni di nota.(3) Per quanto possibile dedurre da un test della ristretta durata di 7 giorni, il TTA è risultato essere sicuro.
Nel 2009, i ricercatori norvegesi dell’Università di Bergen hanno pubblicato un piccolo studio svolto su esseri umani nel quale hanno somministrato a 16 uomini con diabete di tipo II 1g di TTA ogni giorno per 4 settimane. Gli uomini seguivano una dieta apposita o usavano farmaci per il trattamento del diabete. Alcuni di loro usavano anche statine.
Ovviamente, i livelli di colesterolo e trigliceridi degli uomini sono migliorati (effetto da attribuirsi maggiormente ai farmaci utilizzati), ma non hanno mostrato alcuna perdita di peso.
Nella stessa pubblicazione, i ricercatori parlano anche di esperimenti con cellule muscolari umane. In concentrazioni di diverse decine di micromoli, il TTA ha aumentato l’ossidazione degli acidi grassi.
Nelle cellule muscolari la TTA ha aumentato la produzione di Carnitina Palmitoiltransferasi-I, un enzima coinvolto nell’ossidazione degli acidi grassi, e di CD36, una proteina che è coinvolta nell’assorbimento cellulare degli acidi grassi.
Tuttavia, questi effetti apparentemente non sono abbastanza incisivi da causare una perdita di peso. Forse sono annullati da un aumento dell’appetito, che è stato dimostrato negli studi svolti su animali precedentemente esposti.
Come accennato al principio di questo articolo, il TTA è un acido grasso sintetico, che non si trova in natura. Necessita di essere consumato nel ordine dei grammi [una capsula generalmente può contenerne 0,5g]. Il TTA non viene utilizzato come fonte energetica dall’organismo. Inoltre, il TTA è una sostanza con attività farmacologica. È a tutti gli effetti un farmaco sperimentale, la cui sicurezza non è stata ancora studiata adeguatamente. E negli studi sull’uomo, non sembra funzionare.
In seguito a quanto esposto, non considero una supplementazione con TTA una buona idea sia per la probabile inefficacia in termini di perdita di grasso e sia per le incognite di un suo uso sul lungo termine.
Se non avete ancora letto la prima e la seconda parte di questa serie di articoli vi invito a farlo prima di procedere con la lettura di questa terza parte: 1° Parte – 2° Parte.
Effetti diretti di GH e IGF-1 sull’ipertrofia muscolare
Andiamo dritti al punto – di per sé, il GH non causa direttamente ipertrofia muscolare. Questa caratteristica è stata ampiamente osservata per decenni e, finora, nessuno studio attendibile è stato in grado di mostrare un chiaro effetto della somministrazione di rHGH a medio-lungo termine sull’ipertrofia – anche in dosi sovrafisiologiche somministrate ad atleti di alto livello sottoposti ad intensi allenamenti contro resistenza.
Il fatto che questa correlazione non sia stata dimostrata non è certamente dovuto alla mancanza di tentativi in tal senso. Diversi gruppi di ricerca nel corso degli anni hanno tentato di identificare un ipotetica ipertrofia GH-mediata in soggetti adulti sani [48,195-199] o in soggetti anziani [188,198,200-203] senza successo, o in modo inconcludente. Inoltre, è stato dimostrato che gli aumenti della secrezione di GH indotta dall’esercizio in acuto non hanno prodotto cambiamenti nella MPS o nell’ipertrofia [204-205]. È interessante notare che, sebbene la quantità di studi presenti in letteratura siano significativamente meno comuni di quelli in cui è stato somministrato GH, anche la somministrazione sistemica di rhIGF-1 non ha prodotto alcun effetto ipertrofico misurabile sia in soggetti giovani [63,206] che negli anziani [207- 208].
Vi sono prove recenti che suggeriscono che l’esposizione cronica al GH aumenta l’espressione delle vie intramuscolari responsabili dell’atrofia.[209] È ovvio che molte delle caratteristiche anaboliche dimostrate dal GH possono essere compensate da questa maggiore espressione del catabolismo, che potrebbe essere il motivo per cui l’esposizione cronica al GH non porta all’ipertrofia. Potrebbe anche essere responsabile del perché l’esposizione cronica al GH può produrre muscoli meno efficienti e più deboli.[210] Molto semplicemente, questo potrebbe essere ancora un altro fattore nella lunga serie di effetti regolatori negativi intrinseci nell’Asse GH/IGF-1, ma ulteriori studi dovranno essere condotti per chiarire maggiormente questa ipotesi.
Per quelli che difficilmente accettano le informazioni riportate in questa serie di articoli, e vogliono addentrarsi loro stessi nella letteratura scientifica sperando di trovare informazioni che confutino quanto da me riportato, voglio esporre un concetto molto importante da me già espresso nella prima parte di questa serie di articoli. La maggior parte degli studi che hanno preso in esame il GH segnalano un aumento della massa magra nei soggetti dei gruppi trattati con tale ormone. Quindi, ad un lettore inesperto, potrebbero risultare infondate le affermazioni sulla mancanza di un effetto ipertrofico diretto del GH. Bisogna ricordare, però, che il GH causa una non indifferente ritenzione idrica, oltre ad aumentare la massa dei tessuti molli della quale parlerò brevemente. Nello specifico, il GH aumenta la ritenzione di sodio e, di conseguenza, dell’acqua extracellulare, in modo dose-dipendente, attraverso i suoi effetti sul sistema renina-angiotensina.[211] Questi incrementi nella ritenzione idrica e sodica sono stati osservati anche con la somministrazione di IGF-1, poiché l’IGF-1 stesso sembra essere un regolatore chiave del tasso di escrezione renale di sodio.[83,212-213] Quindi il punto della questione è che bisogna stare molto attenti nel trarre conclusioni quando non si ha ben chiara la differenza tra aumento della massa magra e crescita effettiva del tessuto muscolare.
Effetti indiretti di GH e IGF-1 sull’ipertrofia muscolare
Nella sezione precedente si è dimostrato che il GH e l’IGF-1, da soli, non hanno alcun impatto diretto sull’ipertrofia. Tuttavia, questo non significa che questi peptidi non abbiano un ruolo nei processi ipertrofici. L’obiettivo principale di questa sezione è quello di spiegare alcuni dei molti meccanismi che interessano in modo indiretto il GH e l’IGF-1 nei processi ipertrofici, molti dei quali saranno fattori importanti per gli atleti interessati a massimizzare il loro potenziale ipertrofico.
L’Ormone della Crescita è un potente stimolatore della sintesi di collagene sia nei tendini che nei muscoli. Questo effetto è probabilmente mediato dalla capacità dell’IGF-1 autocrino di stimolare i fibroblasti per sintetizzarlo.[214-215] In realtà, questo processo avviene senza che il peptide influenzi la sintesi proteica muscolare, nonostante i livelli di IGF-1 circolante e di quello locale siano significativamente più alti. Questo effetto è anche indotto indipendentemente dall’allenamento contro resistenza ed è stato persino osservato in soggetti immobilizzati hai quali è stato somministrato il GH.[216] La componente connettiva del muscolo scheletrico è vitale per la trasmissione della forza, che è prodotta dalle fibre muscolari, ai tendini e alle ossa affinché si verifichi il movimento. In particolare, il collagene è un importante componente nella trasmissione della forza della matrice extracellulare, che viene continuamente sottoposta a carichi intesi durante i movimenti.
A causa dei potenti effetti del GH sui componenti della matrice extracellulare, si può chiaramente iniziare a capire il perché gli aneddoti nel corso degli anni suggerivano che l’aggiunta del GH in una preparazione farmacologica produceva impatti positivi sulla riduzione del dolore tendineo-articolare. D’altra parte però, questo potrebbe anche essere un fattore che contribuisce in primo luogo al motivo per cui vari effetti collaterali sono segnalati dagli utilizzatori di GH come l’edema dei tessuti molli, il dolore articolare e la sindrome del tunnel carpale.[217-219] Ci sono anche molti che credono che il GH possa accelerare i tempi di recupero dagli infortuni, ma questo è un argomento complesso che verrà discusso in futuro.
Gli impatti del GH sulla sintesi di collagene potrebbero anche essere di grande interesse per gli atleti di forza che non sono necessariamente interessati all’ipertrofia, ma il cui obiettivo principale è la creazione di una condizione favorevole allo spostamento del carico massimale. Stimolare la sintesi del collagene potrebbe aiutare a rafforzare l’intero sistema di supporto dei muscoli scheletrici. Ora, vale la pena aggiungere una piccola nota di chiarimento. Nonostante questo suoni positivo in linea di principio, la supplementazione di GH non ha mai portato direttamente a guadagni di forza in nessuno degli studi svolti su soggetti sani, coprendo vari gruppi.[48,50,184,196-198,200-201,203,220-221] Naturalmente, se il GH fosse usato insieme ad un composto con la capacità di aumentare direttamente la forza, non è difficile supporre che in questo caso l’effetto addizionale potrebbe fare del GH un prezioso componente della preparazione.
Decorina
La Decorina è una proteina strutturale, che risiede principalmente nella matrice extracellulare del muscolo scheletrico e il cui ruolo è correlato alla crescita e alla riparazione dei muscoli.[222-223] Qualche decennio fa è stato dimostrato per la prima volta che la somministrazione di GH potrebbe aumentare direttamente l’espressione del gene della Decorina negli animali [224]. Recentemente, è stato dimostrato che questo effetto si manifesta anche in soggetti umani sottoposti ad allenamenti ricreativi.[225] Nello studio più recente, i livelli di Decorina sono fortemente correlati a quelli di PIIINP, che induce la stimolazione del Decorina GH-mediata che può essere coinvolta nel processo di assemblaggio della matrice di collagene osseo. Questo effetto è più pronunciato negli uomini rispetto alle donne e può essere un sottoprodotto dei livelli più alti di IGF-1 osservati negli uomini, sebbene questa affermazione sia speculativa. L’aumento dell’espressione di Decorina non è stato alterato dall’aggiunta di Testosterone, quindi questo effetto è indipendente dagli androgeni. Dopo aver visto gli effetti che il GH ha sulla sintesi di collagene e Decorina, sta diventando piuttosto chiaro che l’asse GH/IGF-1 è molto più importante per rafforzare la matrice extracellulare di supporto piuttosto che contribuire direttamente alla crescita del tessuto muscolare.
Adenosina Trifosfato (ATP)
La somministrazione acuta di GH in soggetti sani ha anche dimostrato di causare una maggiore produzione di ATP mitocondriale e una maggiore attività della citrato sintasi nel muscolo scheletrico, con una maggiore abbondanza di mRNA muscolari codificanti l’IGF-1.[226] Non solo questo potrebbe essere un fattore che contribuisce al motivo per cui il GH potrebbe avere la capacità di promuovere un aumento dei tassi di spesa energetica giornaliera ma potrebbe anche essere coinvolto nello spostamento verso la preferenza dei lipidi come substrato energetico. Anche se, come abbiamo visto in precedenza, il corpo della letteratura non supporta questa pratica, l’aumento della produzione di ATP mitocondriale potrebbe avere un ruolo nella capacità aerobica.[227]
È stato dimostrato che il GH promuove la fusione dei mioblasti con i miotubi in modelli cellulari [84], un effetto completamente indipendente dalla sovraregolazione locale del IGF-1. Per capire perché questo possa essere importante, bisogna approfondire maggiormente la questione dei fattori cellulari coinvolti nell’ipertrofia del muscolo scheletrico. L’ipertrofia dei muscoli scheletrici negli esseri umani si basa sulle cellule satelliti, che sono cellule dormienti situate all’interno delle miofibre, proprio sotto lo strato di lamina basale nella matrice extracellulare.[148] Una volta attivate queste cellule satellite, spesso con l’esercizio fisico o il danno muscolare, proliferano. Dopo la proliferazione, queste cellule satellite migrano verso siti ove si trova il danno differenziandosi e fondendosi con le miofibre esistenti che forniscono nuovi nuclei per l’ipertrofia e la riparazione. [228] Non è ancora del tutto chiaro se il GH abbia un effetto diretto sulla proliferazione e la differenziazione delle cellule satelliti.[229-230] Vale anche la pena di affermare che ciò che si osserva nelle colture cellulari potrebbe non essere del tutto indicativo di ciò che accade in vivo a causa di vari fattori esterni che non possono essere spiegati in condizioni di laboratorio.
Ci sono due fasi distinte in questa fusione dei mioblasti che si verifica.[85] Il primo sarebbe lo stadio iniziale della differenziazione in cui un sottoinsieme di cellule mononucleate si fondono per formare miotubi nascenti (fusione mioblasto/mioblasto). Questo è seguito dal secondo stadio che coinvolge ulteriori cellule disponibili che si fondono con questi miotubi nascenti e dove avviene la crescita muscolare effettiva (fusione mioblasto/miotubo). È all’interno di quest’ultimo stadio in cui il GH esercita i suoi effetti.
Questa è una scoperta piuttosto interessante, ma ancora una volta, numerosi studi sugli esseri umani non sono riusciti a dimostrare un effetto ipertrofico del GH in condizioni reali. Quindi, possiamo probabilmente dedurre da ciò che gli effetti che il GH ha sulla fusione dei nascenti miotubi non si traducono direttamente nell’ipertrofia. Tuttavia, cosa accadrebbe se si aggiungesse un’altra variabile nell’equazione in grado di creare un ambiente in cui esistessero numeri di celle satelliti potenziati, creando più materiale sul quale l’azione del GH possa manifestarsi?[231]
Introduzione degli AAS
A differenza del GH e del IGF, l’uso degli AAS ha mostrato di avere un impatto pronunciato su ipertrofia e forza. Questo è stato ben noto per decenni anche perché sono stati usati e abusati da atleti fin dalla loro creazione negli anni ’30 (con buona pace dei “puritani” della “old school”).[232] La famiglia degli AAS consiste in un gruppo di potenti composti sintetici che sono simili nella struttura chimica al Testosterone e/o al suo derivato 5α-ridotto (DHT). Vari tipi di singoli composti AAS sono stati creati nel corso degli anni usando come base la molecola naturale di Testosterone manipolandola, quindi, attraverso l’aggiunta di un gruppo etile, metile, idrossile o benzile in uno o più siti lungo la sua struttura.[233-234 ] Alcune delle varianti AAS più facilmente riconoscibili includono i composti metilati in C-17, i quali sono notoriamente dotati di un elevata biodisponibilità orale, e le varianti del 19-nortestosterone nelle quali viene rimosso il gruppo 19-metilico dalla molecola di Testosterone nel tentativo di aumentare la sua attività anabolica diminuendo al contempo la sua androgenicità e tendenza all’aromatizzazione. Conosciuti anche come “19-norsteroidi”, questa famiglia include ben note varianti di AAS come il Nandrolone ed il Trenbolone.
Molte di questi composti sono nati come risultato di un desiderio di aumentare le caratteristiche anaboliche del Testosterone a livello muscolare, abbassando allo stesso tempo gli effetti collaterali androgenici intrinsecamente inerenti alla molecola di Testosterone.[235] In generale, i rischi complessivi degli effetti collaterali derivanti dall’uso cronico di AAS sembrano essere relativamente bassi rispetto a molte sostanze socialmente accettate come l’alcol, il tabacco e vari farmaci da prescrizione.[233,236] Ovviamente, l’uso e l’abuso sono termini che si escludono a vicenda e abusare di qualsiasi sostanza tende a creare un ambiente a più alto rischio di effetti collaterali. A meno che non sia diversamente specificato, da questo punto in poi, parlerò specificamente del Testosterone in quanto è l’ormone sessuale endogeno maschile nativo, nonché l’androgeno più approfonditamente studiato in letteratura. Tanto per avere un riferimento, i maschi adulti sani producono una media di 14-77mg/settimana di Testosterone endogeno. [435]
Testosterone
Il Testosterone è un noto regolatore della massa muscolare, e gli aumenti della massa muscolare mediati dal Testosterone sono associati all’ipertrofia delle fibre, così come ad un aumento delle cellule satelliti e del numero mio nucleare.[231,237-240] Il muscolo scheletrico sembra essere uno dei tessuti più reattivi al Testosterone e si stima che i livelli circolanti di Testosterone rappresentino il fattore causale significativo (incidenza del 40-75%) dei guadagni nella massa muscolare osservati in studi di controllo randomizzati. Se ricordate ciò che è stato affermato e dimostrato nella precedente sezione, il GH possiede un’abilità molto particolare in quanto può aumentare la fusione dei mioblasti durante il processo ipertrofico. La massimizzazione di questa capacità si basa sull’avere un numero adeguato di cellule satellitari disponibili.
I risultati mostrano costantemente che i trattamenti con Testosterone determinano aumenti dose-dipendenti della massa e della forza del muscolo scheletrico, indipendentemente dal fatto che i soggetti presi in esame siano maschi più giovani o più anziani.[241-243] Al contrario, negli studi con soggetti sani giovani in cui il Testosterone endogeno è stato soppresso artificialmente, la forza e la composizione corporea hanno entrambi subito peggioramenti significativi.[244] Per ribadire il nostro precedente punto, gli aumenti della massa muscolare mediati dal Testosterone sono basati sull’ipertrofia e non sono il risultato di una transizione della fibra (cambiando le fibre di tipo I in fibre di tipo II o viceversa) o di una sua scissione.[245] A causa dei loro meccanismi anabolici unici e non sovrapposti, la somministrazione di Testosterone e l’allenamento contro resistenza hanno anche mostrato effetti sinergici e additivi l’uno sull’altro per quanto riguarda lo stimolo dell’aumento della massa muscolare.[246]
Recettore degli Androgeni (AR)
Gli Androgeni mediano principalmente i loro effetti attraverso il gene del recettore degli androgeni (AR) che è espresso in mioblasti, miofibre e cellule satelliti.[247-248] I AR sono stati rilevati anche nelle cellule che supportano i muscoli, come i fibroblasti e le cellule endoteliali. La densità dei AR sembra essere specifica per i gruppi muscolari, con l’allenamento contro resistenza e l’uso di AAS che hanno la capacità di influenzare il numero di AR presenti in questi gruppi muscolari. Oltre ai suoi effetti sulla densità dei AR, l’uso di AAS ha anche dimostrato la capacità di influenzare i livelli di attività dei AR sia in modo acuto che sul lungo termine.[249-250] Questi sono fattori piuttosto importanti da considerare quando ci si imbatte in individui che sostengono che il precedente utilizzo di AAS non offre necessariamente un vantaggio competitivo permanente.
A causa della complessità dell’argomento, ci sono state diverse ipotesi riguardo ai meccanismi con cui l’AAS esercita le sue azioni anaboliche sul muscolo scheletrico.[251] È stato dimostrato che i trattamenti con Testosterone aumentano i tassi di sintesi proteica muscolare (MPS) [252], riducono i tassi di degradazione delle proteine [253] facendo persino in modo che il corpo utilizzi più efficientemente gli amminoacidi prontamente disponibili. Quindi, ancora una volta, questo è un sistema abbastanza complesso che può anche essere semplificato ricordando che gli AAS promuovono l’anabolismo muscolare attraverso la loro capacità di incidere positivamente sull’equilibrio degli aminoacidi.
Via Wnt/B-catenina
È generalmente accettato che gli AAS esercitino i loro effetti anabolizzanti attraverso il legame e attivazione del AR che successivamente attiva cascate di segnalazione a valle che coinvolgono la via Wnt/β-catenina.[254-256]. I Wingless-INT (Wnt) sono una famiglia di glicoproteine secrete che regolano la proliferazione e la differenziazione cellulare.[257-258] I modelli cellulari hanno mostrato che il AR forma un complesso con la beta-catenina che si potenzia in presenza di AAS. [259-260] Una volta attivato questo complesso, migra nei nuclei in cui regola l’espressione dei geni target e la differenziazione delle cellule satelliti.[261-262] Questo è anche il pathway degli AAS in gran parte responsabile della miogenesi, la formazione del tessuto muscolare.[263-265]
Vale la pena notare che l’AAS possiede anche caratteristiche non genomiche che possono influenzare rapidamente numerosi processi ormonali e metabolici al di fuori del classico legame con il recettore. In letteratura sono stati riportati casi di maschi adulti con disturbi di insensibilità agli androgeni, causati da mutazioni del AR, che hanno risposto al Testosterone in modo molto simile ai soggetti sani. Questi casi studio rafforzano l’ipotesi che gli effetti anabolici dell’AAS possano essere mediati indipendentemente dal AR.[266] Le azioni non genomiche degli Androgeni possono in realtà essere un argomento piuttosto affascinante, ma che va oltre lo scopo previsto per questo articolo. Per coloro che vogliono approfondire l’argomento, consiglio di iniziare con le seguenti note di riferimento.[267-268]
AAS e potenziale sinergico con l’Asse GH/IGF-1
Ho riportato molte informazioni utili fino a questo punto, ma è qui che le cose cominciano davvero a farsi interessanti. A questo punto una domanda logica sarebbe: esistono studi svolti su soggetti sani nei quali si sono confrontate le differenze tra trattamenti singoli con GH o Androgeni e trattamenti combinati? Fortunatamente per noi, la risposta è “sì” in quanto vi sono stati alcuni studi, principalmente utilizzando soggetti anziani, sia maschi che femmine. I risultati di ognuno di questi studi dimostrano chiaramente che il GH ha un effetto additivo sui benefici consolidati che la terapia con ormoni sessuali fornisce: l’ipertrofia, la lipolisi, la sintesi del collagene, la funzione fisica, la qualità della vita e altri vari indicatori di prestazione.[187-188,269-270] Dato che è abbastanza chiaro che esiste un effetto additivo, vediamo se è possibile approfondire ulteriormente l’argomento al fine di scoprire alcuni dei meccanismi sottostanti che operano nella sinergia tra Androgeni e GH.
GHRH
Deve essere chiaro che il Testosterone, di per sé, ha un effetto additivo sull’intero Asse GH/IGF-1. Questo è stato osservato sia in modelli umani che animali, con la somministrazione di Testosterone che porta ad un aumento dei livelli circolanti di GH e IGF-1.[241,271-276] Viceversa, il deficit di Testosterone è comunemente associato a livelli significativamente ridotti di IGF-1.[277] L’effetto stimolante del Testosterone sull’Asse GH/IGF-1 sembra essere mediato a livello ipotalamico da una promozione della funzionalità del GHRH.[278]
Inoltre, e questo è bene ricordarlo, gli AAS non aromatizzabili sembrano non possedere lo stesso effetto stimolante sull’Asse GH/IGF-1.[279] Gli inibitori dell’aromatasi (AI), progettati per sopprimere il processo di aromatizzazione degli Androgeni, hanno dimostrato di attenuare direttamente la stimolazione del GH seguente la somministrazione di Testosterone. Questi indizi forniscono prove abbastanza convincenti sul fatto che gli estrogeni locali, derivanti dall’aromatizzazione, svolgono un ruolo fondamentale nella regolazione della secrezione di GH nei maschi.[280-281] Poiché l’aromatasi non è espressa nel fegato, gli AI non influenzano l’azione epatica del GH, mentre influenzano la sua azione a livello del sistema centrale.[282-283] Tuttavia i modulatori selettivi del recettore degli estrogeni (SERM) sono ancora più soppressivi in quanto agiscono ad entrambi i livelli a causa del loro meccanismo d’azione.[284-285]
Anche gli Androgeni che aumentano i livelli serici di Estrogeni, come il Nandrolone (Nor-Estrogeni), mostrano un effetto minimo sui livelli sistemici di GH e IGF-1 rispetto al Testosterone.[286] Ciò è legato ipoteticamente sia al fatto che il Nandrolone è poco soggetto all’aromatizzazione e sia al fatto che la forma estrogenica derivata (Nor-Estrogeno) abbia un ridotto potenziale di attività.[287] Ricordiamoci che l’aromatizzazione sembra essere il passo più cruciale nella stimolazione ipotalamica mediata dagli Androgeni. Ora, è ovvio che un utilizzatore di rHGH deve preoccuparsi parzialmente di questo effetto rispetto ad un non utilizzatore, considerando il fatto che, in tal caso, i livelli ormonali sono controllati quasi esclusivamente da mezzi esogeni. Detto questo, è sempre utile analizzare il quadro generale, soprattutto se l’obiettivo è massimizzare l’ipertrofia.
GHR
Un altro potenziale motivo legato agli aumenti dei livelli di GH e IGF osservati durante i trattamenti con Testosterone, è rappresentato dagli effetti diretti di quest’ultimo sui GHR. Sia gli studi sull’uomo che sugli animali hanno fornito prove del fatto che il Testosterone modifica i GHR sia nel fegato che nei tessuti periferici, migliorandone l’espressione.[288-289] Inoltre, soggetti umani ipopituitari e ipogonadici sottoposti a trattamenti con GH hanno mostrato una aumentata risposta sia all’IGF-1 locale che all’espressione del gene del recettore degli androgeni quando sottoposti anche a trattamento con Testosterone.[187,290-291] Inoltre, i maschi ipopituitari sottoposti a trattamenti con Testosterone hanno mostrato effetti notevoli sull’anabolismo proteico in presenza di GH, con il sito primario di interazione ormonale a livello epatico.[292] Quindi anche quando i livelli ormonali sono carenti, c’è ancora un’interazione molto importante tra il Testosterone e l’Asse GH/IGF-1.
Come accennato in precedenza, il GHR è espresso in quasi tutti i principali tipi di tessuto. Vale la pena sottolineare che il GHR è espresso in quantità molto bassa nel muscolo scheletrico – solo circa il 4-33% dei livelli osservati in altri tessuti. D’altra parte, il recettore del IGF-1 è espresso in maniera molto più elevata nei muscoli scheletrici, proprio come nel tessuto epatico.[293-294] Detto questo, l’aumento della sensibilità del GHR in un ambiente con livelli sovra fisiologici di GH andrà certamente a beneficio del BodyBuilder. I BodyBuilder cercano continuamente di utilizzare quantità elevate di rHGH nel tentativo di massimizzare i processi ipertrofici, e se il GH vede potenziata la sua azione e, quindi, il suo stimolo diretto sulla sintesi e rilascio del IGF-1, il quale opera direttamente sul tessuto muscolare, venendosi a creare quindi una vantaggiosa alterazione dell’Asse GH/IGF-1, l’effetto ipertrofico potenziato sarà indubbio.
IGFBP-4
Gli Androgeni hanno mostrato di avere la capacità di aumentare l’espressione del mRNA del IGF-1 locale nel muscolo scheletrico. Possiamo pertanto ipotizzare che gli Androgeni, in particolare a dosi più elevate, creino un ambiente all’interno del muscolo scheletrico estremamente favorevole alla gestione di livelli più alti di IGF-1 seguenti alla somministrazione di dosi sovrafisioogiche di rHGH. Ci sono persino stati studi sull’uomo che hanno mostrato livelli ridotti di IGFBP-4 locale nei campioni di tessuto muscolare, oltre a maggiori livelli di mRNA del IGF-1. Ciò potrebbe indicare che i cambiamenti hanno avuto luogo in quei muscoli per liberare più IGF-1 locale affinché esso possa legarsi ai suoi recettori.[277,295] Non ho molto approfondito la questione delle proteine leganti l’IGF, ma l’IGFBP-4 inibisce l’azione dell’IGF-1 e, quindi, minore è il livello della proteina legante, maggiori saranno i livelli di IGF-1 libero e attivo.[149,296]
È stato anche dimostrato che il Testosterone promuove l’ipertrofia negli stati deficitari di GH/IGF-1. [297-298] Questo è interessante in quanto dimostra che il Testosterone possiede sia vie metaboliche IGF-indipendenti che IGF-indipendenti nel tessuto muscolare.[299] Inoltre, i modelli cellulari hanno dimostrato che il Testosterone può sovraregolare l’espressione delle varie isoforme di IGF nei muscoli scheletrici, anche in assenza di GH/IGF-1.[298] E, anche se questo è stato dimostrato nei fibroblasti, il Testosterone ha mostrato di aumentare l’espressione del IGFBP-3 – un effetto che è stato ulteriormente migliorato dalla somministrazione di IGF-1.[300] È abbastanza chiaro, comunque, che il Testosterone eserciti effetti sinergici e additivi sull’anabolismo mediato da GH/IGF-1.
Fino ad ora mi sono concentrato sul Testosterone, tuttavia, però, sono stati osservati comportamenti leggermente diversi in relazione alle varianti androgene e al loro impatto sull’espressione sistemica e locale del IGF-1. Volevo analizzare un paio di specifici composti che sono spesso presenti nei protocolli farmacologici: sto parlando del Trenbolone e del Nandrolone. Salvo diverse indicazione, i risultati in seguito esposti provengono da studi svolti su animali.
Nandrolone
La somministrazione di Nandrolone ha costantemente dimostrato di non causare cambiamenti nei livelli di IGF-1 endocrino, nonostante produca simultaneamente un’espressione del IGF-1 muscolare significativamente più alta e un aumento della CSA delle fibre muscolari.[286,301-302] Inoltre, i livelli di IGFBP-3 locali sono stati riportati come significativamente più alti mentre i livelli di IGFBP-4 sono stati significativamente soppressi, il che ci rimanda a quanto esposto in precedenza – cioè ad un fattore che determina un aumento dei livelli di IGF-1 locale libero. In tutti gli studi, la somministrazione di Nandrolone ha portato direttamente ad un aumento dell’ipertrofia, nonostante non abbia avuto alcun impatto sui livelli di IGF-1 sistemici. Ciò rafforza ulteriormente l’ipotesi secondo cui l’IGF-1 endocrino non sia un fattore primario nell’ipertrofia dei muscoli scheletrici e quindi i livelli elevati non sono un prerequisito per l’aumento della massa muscolare.[303-305]
Trenbolone
È stato inoltre universalmente dimostrato che il Trenbolone aumenta i tassi di crescita muscolare in tutte le varie specie nelle quali è stato testato. A differenza del Testosterone e del Nandrolone, non si converte in estrogeno ed è stato suggerito fin dagli anni ’70 che l’aggiunta di Estradiolo al Trenbolone sembra migliorare gli effetti anabolici del composto.[306-307] Ci sono stati anche effetti potenziati sull’ipertrofia quando il Trenbolone è stato somministrato insieme ad un fattore di rilascio dell’Ormone della Crescita (GHRF).[308] Come accennato in precedenza, l’aumento di GH/IGF-1 necessita di livelli adeguati di estrogeni (derivanti principalmente dall’aromatizzazione) affinché questi esercitino uno stimolo dell’asse GH/IGF-1. Poiché la somministrazione di Trenbolone diminuisce intrinsecamente i livelli di Estradiolo, mediante inibizione a feedback negativo del Testosterone attraverso l’Asse Ipotalamo-Ipofisi-Gonadi (HPG) [309], la somministrazione di Estradiolo, tecnicamente, dovrebbe migliorare la funzionalità dell’asse GH/IGF-1. Questo dovrebbe quindi favorire ulteriormente la sinergia anabolica che avrebbe con l’androgeno. Questa ipotesi è in linea con ciò che vari studi hanno dimostrato nel corso degli anni.
Nelle colture cellulari, è stato anche dimostrato che gli Estrogeni alterano direttamente i tassi di MPS e MPB del Trenbolone attraverso meccanismi che coinvolgono sia il recettore estrogenico che il recettore del IGF-1.[310-311] In effetti, in linea di massima, i trattamenti con solo Trenbolone non mostrano aumenti significativi dei livelli endocrini o autocrini del IGF-1. Tuttavia, la co-somministrazione con Estradiolo ha tradizionalmente mostrato aumenti nei livelli di IGF-1 autocrini, similmente a quanto osservato con il Testosterone.[312-314] Questa è solo un’ulteriore prova che suggerisce che l’Estrogeno, sia di origine sistemica che derivata dall’aromatasi, è un componente chiave sia della massima stimolazione dell’Asse GH/IGF-1 che delle capacità anaboliche massimali degli Androgeni.
Proprio come il suo “cugino” 19-nor, il Trenbolone ha anche mostrato una maggiore espressione del fattore di crescita nei tessuti muscolo scheletrici, oltre ad una maggiore reattività dei muscoli a tali fattori di crescita.[315] Il Trenbolone ha anche mostrato di operare un aumento dell’attivazione e della proliferazione delle cellule satelliti in varie specie, in misura simile al Testosterone.[316-317] Ora, essendo a conoscenza delle interazioni legate al GH, si può facilmente giungere alla conclusione che entrambi questi effetti sarebbero vantaggiosi in un protocollo che includesse entrambi i composti.
PIIINP
Tornando al Testosterone, sia il GH che questo Androgeno aumentano i marker di sintesi del collagene come il PIIINP. Inoltre, il Testosterone ha anche dimostrato di potenziare le capacità del GH di aumentare la sintesi di collagene sia nei muscoli che nei tendini.[318] A sostegno di ciò, la somministrazione concomitante di GH e Testosterone in soggetti umani allenati (a livello amatoriale) ha causato aumenti significativi sia dei marker del IGF-1 che del collagene.[319] E’ interessante notare come alcuni di questi stessi marker del collagene dei quali si sta discutendo sono indicatori esatti che vengono esaminati come parte dei test antidoping per la rilevazione del GH.[320-321]
In precedenza ho solo brevemente toccato il percorso JAK-STAT ma, arrivati a questo punto, è necessario trattare l’argomento in maniera leggermente più approfondita. La via JAK-STAT è un componente critico del GH e riguarda sia la trascrizione del gene del IGF-1 che la crescita postnatale. Una delle proteine STAT in particolare, la STAT5, sembra essere intimamente coinvolta anche nella regolazione del muscolo scheletrico.[322] Ci sono due sottoproteine nella famiglia STAT5 e sono indicate come STAT5a e STAT5b. Sebbene siano identiche al 96%, è la variante STAT5b che è abbondante nei muscoli e nel tessuto epatico ed è quindi la proteina specifica su cui mi concentrerò da questo punto in avanti.[323-324]
Una revisione completa del percorso di segnalazione renderebbe questo articolo estremamente prolisso, ma ritengo importante che si comprenda che il percorso JAK/STAT5b è stato ripetutamente dimostrato avere una relazione diretta con l’espressione dell’IGF-1 locale nel tessuto muscolare e sul’ipertrofia.[248,325-332] Per questo motivo, se ci fossero modi per migliorare o ottimizzare questo specifico percorso, sembrerebbe che ciò si possa tradurre non solo in un aumento dell’attivazione del gene del IGF-1 [333-335] ma anche in un maggiore potenziale ipertrofico.
Fortunatamente, alcuni nuovi studi svolti su animali hanno già mostrato come i percorsi AR e JAK-STAT siano intimamente correlati.[248,336] Per essere precisi, il percorso STAT5a/b è a monte e l’AR è un bersaglio diretto a valle tramite la regolazione dell’espressione genica del AR. Studi su esseri umani hanno anche dimostrato che questo si verifica anche nell’uomo, con l’attività della STAT5 correlata positivamente all’espressione AR nelle linee cellulari del cancro alla prostata. [337]
Nella prossima parte di questa serie di articoli, parlerò dei modi in cui è possibile tentare di garantire che la via JAK-STAT5b-AR sia massimamente sensibilizzata, assicurando così che il potenziale ipertrofico sia potenziato quando Androgeni e GH vengono co-somministrati. Inoltre tratterò le applicazioni pratiche dell’uso del GH a fini ipertrofici esponendo infine le mie conclusioni in merito.
Stay tuned!
Gabriel Bellizzi
Riferimenti:
Yarasheski KE, Campbell JA, Smith K, Rennie MJ, Holloszy JO, Bier DM. Effect of growth hormone and resistance exercise on muscle growth in young men. Am J Physiol. 1992 Mar;262(3 Pt 1):E261-7.
Yarasheski KE, Zachwieja JJ, Campbell JA, Bier DM. Effect of growth hormone and resistance exercise on muscle growth and strength in older men. Am J Physiol. 1995 Feb;268(2 Pt 1):E268-76.
Fryburg DA. Insulin-like growth factor I exerts growth hormone- and insulin-like actions on human muscle protein metabolism. Am J Physiol. 1994 Aug;267(2 Pt 1):E331-6.
Ohlsson C, Mohan S, Sjögren K, Tivesten A, Isgaard J, Isaksson O, Jansson JO, Svensson J. The role of liver-derived insulin-like growth factor-I. Endocr Rev. 2009 Aug;30(5):494-535.
Wakelam MJ. The fusion of myoblasts. Biochem J. 1985 May 15;228(1):1-12. Review.
Crist DM, Peake GT, Egan PA, Waters DL. Body composition response to exogenous GH during training in highly conditioned adults. J Appl Physiol (1985). 1988 Aug;65(2):579-84.
Deyssig R, Frisch H, Blum WF, Waldhör T. Effect of growth hormone treatment on hormonal parameters, body composition and strength in athletes. Acta Endocrinol (Copenh). 1993 Apr;128(4):313-8.
Yarasheski KE, Zachweija JJ, Angelopoulos TJ, Bier DM. Short-term growth hormone treatment does not increase muscle protein synthesis in experienced weight lifters. J Appl Physiol (1985). 1993 Jun;74(6):3073-6.
Lange KH, Andersen JL, Beyer N, Isaksson F, Larsson B, Rasmussen MH, Juul A, Bülow J, Kjaer M. GH administration changes myosin heavy chain isoforms in skeletal muscle but does not augment muscle strength or hypertrophy, either alone or combined with resistance exercise training in healthy elderly men. J Clin Endocrinol Metab. 2002 Feb;87(2):513-23.
Ehrnborg C, Ellegård L, Bosaeus I, Bengtsson BA, Rosén T. Supraphysiological growth hormone: less fat, more extracellular fluid but uncertain effects on muscles in healthy, active young adults. Clin Endocrinol (Oxf). 2005 Apr;62(4):449-57.
Rudman D, Feller AG, Nagraj HS, Gergans GA, Lalitha PY, Goldberg AF, Schlenker RA, Cohn L, Rudman IW, Mattson DE. Effects of human growth hormone in men over 60 years old. N Engl J Med. 1990 Jul 5;323(1):1-6.
Taaffe DR, Pruitt L, Reim J, Hintz RL, Butterfield G, Hoffman AR, Marcus R. Effect of recombinant human growth hormone on the muscle strength response to resistance exercise in elderly men. J Clin Endocrinol Metab. 1994 Nov;79(5):1361-6.
Taaffe DR, Jin IH, Vu TH, Hoffman AR, Marcus R. Lack of effect of recombinant human growth hormone (GH) on muscle morphology and GH-insulin-like growth factor expression in resistance-trained elderly men. J Clin Endocrinol Metab. 1996 Jan;81(1):421-5.
Hennessey JV, Chromiak JA, DellaVentura S, Reinert SE, Puhl J, Kiel DP, Rosen CJ, Vandenburgh H, MacLean DB. Growth hormone administration and exercise effects on muscle fiber type and diameter in moderately frail older people. J Am Geriatr Soc. 2001 Jul;49(7):852-8.
West DW, Kujbida GW, Moore DR, Atherton P, Burd NA, Padzik JP, De Lisio M, Tang JE, Parise G, Rennie MJ, Baker SK, Phillips SM. Resistance exercise-induced increases in putative anabolic hormones do not enhance muscle protein synthesis or intracellular signalling in young men. J Physiol. 2009 Nov 1;587(Pt 21):5239-47.
West DW, Burd NA, Tang JE, Moore DR, Staples AW, Holwerda AM, Baker SK, Phillips SM. Elevations in ostensibly anabolic hormones with resistance exercise enhance neither training-induced muscle hypertrophy nor strength of the elbow flexors. J Appl Physiol (1985). 2010 Jan;108(1):60-7.
Fryburg DA, Jahn LA, Hill SA, Oliveras DM, Barrett EJ. Insulin and insulin-like growth factor-I enhance human skeletal muscle protein anabolism during hyperaminoacidemia by different mechanisms. J Clin Invest. 1995 Oct;96(4):1722-9
Butterfield GE, Thompson J, Rennie MJ, Marcus R, Hintz RL, Hoffman AR. Effect of rhGH and rhIGF-I treatment on protein utilization in elderly women. Am J Physiol. 1997 Jan;272(1 Pt 1):E94-9.
Friedlander AL, Butterfield GE, Moynihan S, Grillo J, Pollack M, Holloway L, Friedman L, Yesavage J, Matthias D, Lee S, Marcus R, Hoffman AR. One year of insulin-like growth factor I treatment does not affect bone density, body composition, or psychological measures in postmenopausal women. J Clin Endocrinol Metab. 2001 Apr;86(4):1496-503.
Consitt LA, Saneda A, Saxena G, List EO, Kopchick JJ. Mice overexpressing growth hormone exhibit increased skeletal muscle myostatin and MuRF1 with attenuation of muscle mass. Skelet Muscle. 2017 Sep 4;7(1):17.
Wolf E, Wanke R, Schenck E, Hermanns W, Brem G. Effects of growth hormone overproduction on grip strength of transgenic mice. Eur J Endocrinol. 1995 Dec;133(6):735-40.
Ho KY, Weissberger AJ. The antinatriuretic action of biosynthetic human growth hormone in man involves activation of the renin-angiotensin system. Metabolism. 1990 Feb;39(2):133-7.
Blazer-Yost BL, Cox M. Insulin-like growth factor 1 stimulates renal epithelial Na+ transport. Am J Physiol. 1988 Sep;255(3 Pt 1):C413-7.
Giordano M, DeFronzo RA. Acute effect of human recombinant insulin-like growth factor I on renal function in humans. Nephron. 1995;71(1):10-5.
Ehrnborg C, Lange KH, Dall R, Christiansen JS, Lundberg PA, Baxter RC, Boroujerdi MA, Bengtsson BA, Healey ML, Pentecost C, Longobardi S, Napoli R, Rosén T; GH-2000 Study Group. The growth hormone/insulin-like growth factor-I axis hormones and bone markers in elite athletes in response to a maximum exercise test. J Clin Endocrinol Metab. 2003 Jan;88(1):394-401.
Doessing S, Heinemeier KM, Holm L, Mackey AL, Schjerling P, Rennie M, Smith K, Reitelseder S, Kappelgaard AM, Rasmussen MH, Flyvbjerg A, Kjaer M. Growth hormone stimulates the collagen synthesis in human tendon and skeletal muscle without affecting myofibrillar protein synthesis. J Physiol. 2010 Jan 15;588(Pt 2):341-51.
Boesen AP, Dideriksen K, Couppé C, Magnusson SP, Schjerling P, Boesen M, Kjaer M, Langberg H. Tendon and skeletal muscle matrix gene expression and functional responses to immobilisation and rehabilitation in young males: effect of growth hormone administration. J Physiol. 2013 Dec 1;591(23):6039-52.
Cohn L, Feller AG, Draper MW, Rudman IW, Rudman D. Carpal tunnel syndrome and gynaecomastia during growth hormone treatment of elderly men with low circulating IGF-I concentrations. Clin Endocrinol (Oxf). 1993 Oct;39(4):417-25.
Sullivan DH, Carter WJ, Warr WR, Williams LH. Side effects resulting from the use of growth hormone and insulin-like growth factor-I as combined therapy to frail elderly patients. J Gerontol A Biol Sci Med Sci. 1998 May;53(3):M183-7.
Dickerman RD, Douglas JA, East JW. Bilateral median neuropathy and growth hormone use: a case report. Arch Phys Med Rehabil. 2000 Dec;81(12):1594-5.
Papadakis MA, Grady D, Black D, Tierney MJ, Gooding GA, Schambelan M, Grunfeld C. Growth hormone replacement in healthy older men improves body composition but not functional ability. Ann Intern Med. 1996 Apr 15;124(8):708-16.
Zachwieja JJ, Yarasheski KE. Does growth hormone therapy in conjunction with resistance exercise increase muscle force production and muscle mass in men and women aged 60 years or older? Phys Ther. 1999 Jan;79(1):76-82. Review.
Kishioka Y, Thomas M, Wakamatsu J, Hattori A, Sharma M, Kambadur R, Nishimura T. Decorin enhances the proliferation and differentiation of myogenic cells through suppressing myostatin activity. J Cell Physiol. 2008 Jun;215(3):856-67.
Kanzleiter T, Rath M, Görgens SW, Jensen J, Tangen DS, Kolnes AJ, Kolnes KJ, Lee S, Eckel J, Schürmann A, Eckardt K. The myokine decorin is regulated by contraction and involved in muscle hypertrophy. Biochem Biophys Res Commun. 2014 Jul 25;450(2):1089-94.
Zhang CZ, Li H, Bartold PM, Young WG, Waters MJ. Effect of growth hormone on the distribution of decorin and biglycan during odontogenesis in the rat incisor. J Dent Res. 1995 Oct;74(10):1636-43.
Bahl N, Stone G, McLean M, Ho KKY, Birzniece V. Decorin, a growth hormone regulated protein in humans. Eur J Endocrinol. 2017 Nov 14. pii: EJE-17-0844.
Short KR, Moller N, Bigelow ML, Coenen-Schimke J, Nair KS. Enhancement of muscle mitochondrial function by growth hormone. J Clin Endocrinol Metab. 2008 Feb;93(2):597-604. Epub 2007 Nov 13.
Lange KH, Isaksson F, Juul A, Rasmussen MH, Bülow J, Kjaer M. Growth hormone enhances effects of endurance training on oxidative muscle metabolism in elderly women. Am J Physiol Endocrinol Metab. 2000 Nov;279(5):E989-96.
Chargé SB, Rudnicki MA. Cellular and molecular regulation of muscle regeneration. Physiol Rev. 2004 Jan;84(1):209-38. Review.
Halevy O, Hodik V, Mett A. The effects of growth hormone on avian skeletal muscle satellite cell proliferation and differentiation. Gen Comp Endocrinol. 1996 Jan;101(1):43-52.
Kim H, Barton E, Muja N, Yakar S, Pennisi P, Leroith D. Intact insulin and insulin-like growth factor-I receptor signaling is required for growth hormone effects on skeletal muscle growth and function in vivo. Endocrinology. 2005 Apr;146(4):1772-9. Epub 2004 Dec 23.
Sinha-Hikim I, Roth SM, Lee MI, Bhasin S. Testosterone-induced muscle hypertrophy is associated with an increase in satellite cell number in healthy, young men. Am J Physiol Endocrinol Metab. 2003 Jul;285(1):E197-205. Epub 2003 Apr 1.
Ruzicka, L., Wettstein, A. and Kägi, H. (1935), Sexualhormone VIII. Darstellung von Testosteron unter Anwendung gemischter Ester. HCA, 18: 1478–1482.
Haupt HA, Rovere GD. Anabolic steroids: a review of the literature. Am J Sports Med. 1984 Nov-Dec;12(6):469-84. Review.
Shahidi NT. A review of the chemistry, biological action, and clinical applications of anabolic-androgenic steroids. Clin Ther. 2001 Sep;23(9):1355-90. Review.
Calof OM, Singh AB, Lee ML, Kenny AM, Urban RJ, Tenover JL, Bhasin S. Adverse events associated with testosterone replacement in middle-aged and older men: a meta-analysis of randomized, placebo-controlled trials. J Gerontol A Biol Sci Med Sci. 2005 Nov;60(11):1451-7
Hartgens F, Kuipers H. Effects of androgenic-anabolic steroids in athletes. Sports Med. 2004;34(8):513-54. Review.
Kadi F, Eriksson A, Holmner S, Thornell LE. Effects of anabolic steroids on the muscle cells of strength-trained athletes. Med Sci Sports Exerc. 1999 Nov;31(11):1528-34.
Herbst KL, Bhasin S. Testosterone action on skeletal muscle. Curr Opin Clin Nutr Metab Care. 2004 May;7(3):271-7. Review.
Eriksson A, Kadi F, Malm C, Thornell LE. Skeletal muscle morphology in power-lifters with and without anabolic steroids. Histochem Cell Biol. 2005 Aug;124(2):167-75.
Kadi F. Cellular and molecular mechanisms responsible for the action of testosterone on human skeletal muscle. A basis for illegal performance enhancement. Br J Pharmacol. 2008 Jun;154(3):522-8. Epub 2008 Apr 14. Review.
Bhasin S, Woodhouse L, Casaburi R, Singh AB, Bhasin D, Berman N, Chen X, Yarasheski KE, Magliano L, Dzekov C, Dzekov J, Bross R, Phillips J, Sinha-Hikim I, Shen R, Storer TW. Testosterone dose-response relationships in healthy young men. Am J Physiol Endocrinol Metab. 2001 Dec;281(6):E1172-81.
Woodhouse LJ, Reisz-Porszasz S, Javanbakht M, Storer TW, Lee M, Zerounian H, Bhasin S. Development of models to predict anabolic response to testosterone administration in healthy young men. Am J Physiol Endocrinol Metab. 2003 May;284(5):E1009-17.
Bhasin S, Woodhouse L, Casaburi R, Singh AB, Mac RP, Lee M, Yarasheski KE, Sinha-Hikim I, Dzekov C, Dzekov J, Magliano L, Storer TW. Older men are as responsive as young men to the anabolic effects of graded doses of testosterone on the skeletal muscle. J Clin Endocrinol Metab. 2005 Feb;90(2):678-88. Epub 2004 Nov 23.
Kvorning T, Andersen M, Brixen K, Madsen K. Suppression of endogenous testosterone production attenuates the response to strength training: a randomized, placebo-controlled, and blinded intervention study. Am J Physiol Endocrinol Metab. 2006 Dec;291(6):E1325-32. Epub 2006 Jul 25.
Sinha-Hikim I, Artaza J, Woodhouse L, Gonzalez-Cadavid N, Singh AB, Lee MI, Storer TW, Casaburi R, Shen R, Bhasin S. Testosterone-induced increase in muscle size in healthy young men is associated with muscle fiber hypertrophy. Am J Physiol Endocrinol Metab. 2002 Jul;283(1):E154-64.
Bhasin S, Storer TW, Berman N, Callegari C, Clevenger B, Phillips J, Bunnell TJ, Tricker R, Shirazi A, Casaburi R. The effects of supraphysiologic doses of testosterone on muscle size and strength in normal men. N Engl J Med. 1996 Jul 4;335(1):1-7.
Sinha-Hikim I, Taylor WE, Gonzalez-Cadavid NF, Zheng W, Bhasin S. Androgen receptor in human skeletal muscle and cultured muscle satellite cells: up-regulation by androgen treatment. J Clin Endocrinol Metab. 2004 Oct;89(10):5245-55.
Klover P, Chen W, Zhu BM, Hennighausen L. Skeletal muscle growth and fiber composition in mice are regulated through the transcription factors STAT5a/b: linking growth hormone to the androgen receptor. FASEB J. 2009 Sep;23(9):3140-8.
Sheffield-Moore M, Urban RJ, Wolf SE, Jiang J, Catlin DH, Herndon DN, Wolfe RR, Ferrando AA. Short-term oxandrolone administration stimulates net muscle protein synthesis in young men. J Clin Endocrinol Metab. 1999 Aug;84(8):2705-11.
Kadi F, Bonnerud P, Eriksson A, Thornell LE. The expression of androgen receptors in human neck and limb muscles: effects of training and self-administration of androgenic-anabolic steroids. Histochem Cell Biol. 2000 Jan;113(1):25-9.
de Rooy C, Grossmann M, Zajac JD, Cheung AS. Targeting muscle signaling pathways to minimize adverse effects of androgen deprivation. Endocr Relat Cancer. 2016 Jan;23(1):R15-26.
Brodsky IG, Balagopal P, Nair KS. Effects of testosterone replacement on muscle mass and muscle protein synthesis in hypogonadal men–a clinical research center study. J Clin Endocrinol Metab. 1996 Oct;81(10):3469-75.
Ferrando AA, Sheffield-Moore M, Paddon-Jones D, Wolfe RR, Urban RJ. Differential anabolic effects of testosterone and amino acid feeding in older men. J Clin Endocrinol Metab. 2003 Jan;88(1):358-62.
Bauer ER, Daxenberger A, Petri T, Sauerwein H, Meyer HH. Characterisation of the affinity of different anabolics and synthetic hormones to the human androgen receptor, human sex hormone binding globulin and to the bovine progestin receptor. APMIS. 2000 Dec;108(12):838-46.
Singh R, Artaza JN, Taylor WE, Gonzalez-Cadavid NF, Bhasin S. Androgens stimulate myogenic differentiation and inhibit adipogenesis in C3H 10T1/2 pluripotent cells through an androgen receptor-mediated pathway. Endocrinology. 2003 Nov;144(11):5081-8. Epub 2003 Jul 24.
Yarrow JF, McCoy SC, Borst SE. Tissue selectivity and potential clinical applications of trenbolone (17beta-hydroxyestra-4,9,11-trien-3-one): A potent anabolic steroid with reduced androgenic and estrogenic activity. Steroids. 2010 Jun;75(6):377-89.
Mulholland DJ, Dedhar S, Coetzee GA, Nelson CC. Interaction of nuclear receptors with the Wnt/beta-catenin/Tcf signaling axis: Wnt you like to know? Endocr Rev. 2005 Dec;26(7):898-915. Epub 2005 Aug 26. Review.
Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006 Nov 3;127(3):469-80. Review.
Singh R, Bhasin S, Braga M, Artaza JN, Pervin S, Taylor WE, Krishnan V, Sinha SK, Rajavashisth TB, Jasuja R. Regulation of myogenic differentiation by androgens: cross talk between androgen receptor/ beta-catenin and follistatin/transforming growth factor-beta signaling pathways. Endocrinology. 2009 Mar;150(3):1259-68.
Zhao JX, Hu J, Zhu MJ, Du M. Trenbolone enhances myogenic differentiation by enhancing β-catenin signaling in muscle-derived stem cells of cattle. Domest Anim Endocrinol. 2011 May;40(4):222-9.
Armstrong DD, Esser KA. Wnt/beta-catenin signaling activates growth-control genes during overload-induced skeletal muscle hypertrophy. Am J Physiol Cell Physiol. 2005 Oct;289(4):C853-9. Epub 2005 May 11.
Takada I, Kouzmenko AP, Kato S. Wnt and PPARgamma signaling in osteoblastogenesis and adipogenesis. Nat Rev Rheumatol. 2009 Aug;5(8):442-7.
Cossu G, Borello U. Wnt signaling and the activation of myogenesis in mammals. EMBO J. 1999 Dec 15;18(24):6867-72. Review.
Buckingham M. Skeletal muscle formation in vertebrates. Curr Opin Genet Dev. 2001 Aug;11(4):440-8. Review.
Polesskaya A, Seale P, Rudnicki MA. Wnt signaling induces the myogenic specification of resident CD45+ adult stem cells during muscle regeneration. Cell. 2003 Jun 27;113(7):841-52.
Tincello DG, Saunders PT, Hodgins MB, Simpson NB, Edwards CR, Hargreaves TB, Wu FC. Correlation of clinical, endocrine and molecular abnormalities with in vivo responses to high-dose testosterone in patients with partial androgen insensitivity syndrome. Clin Endocrinol (Oxf). 1997 Apr;46(4):497-506.
Foradori CD, Weiser MJ, Handa RJ. Non-genomic Actions of Androgens. Frontiers in neuroendocrinology. 2008;29(2):169-181.
Lucas-Herald AK, Alves-Lopes R, Montezano AC, Ahmed SF, Touyz RM. Genomic and non-genomic effects of androgens in the cardiovascular system: clinical implications. Clin Sci (Lond). 2017 Jul 1;131(13):1405-1418.
Münzer T, Harman SM, Hees P, Shapiro E, Christmas C, Bellantoni MF, Stevens TE, O’Connor KG, Pabst KM, St Clair C, Sorkin JD, Blackman MR. Effects of GH and/or sex steroid administration on abdominal subcutaneous and visceral fat in healthy aged women and men. J Clin Endocrinol Metab. 2001 Aug;86(8):3604-10.
Sattler FR, Castaneda-Sceppa C, Binder EF, Schroeder ET, Wang Y, Bhasin S, Kawakubo M, Stewart Y, Yarasheski KE, Ulloor J, Colletti P, Roubenoff R, Azen SP. Testosterone and growth hormone improve body composition and muscle performance in older men. J Clin Endocrinol Metab. 2009 Jun;94(6):1991-2001.
Illig R, Prader A. Effect of testosterone on growth hormone secretion in patients with anorchia and delayed puberty. J Clin Endocrinol Metab. 1970 May;30(5):615-8.
Pfeilschifter J, Scheidt-Nave C, Leidig-Bruckner G, Woitge HW, Blum WF, Wüster C, Haack D, Ziegler R. Relationship between circulating insulin-like growth factor components and sex hormones in a population-based sample of 50- to 80-year-old men and women. J Clin Endocrinol Metab. 1996 Jul;81(7):2534-40.
Erfurth EM, Hagmar LE, Sääf M, Hall K. Serum levels of insulin-like growth factor I and insulin-like growth factor-binding protein 1 correlate with serum free testosterone and sex hormone binding globulin levels in healthy young and middle-aged men. Clin Endocrinol (Oxf). 1996 Jun;44(6):659-64.
van Kesteren P, Lips P, Deville W, Popp-Snijders C, Asscheman H, Megens J, Gooren L. The effect of one-year cross-sex hormonal treatment on bone metabolism and serum insulin-like growth factor-1 in transsexuals. J Clin Endocrinol Metab. 1996 Jun;81(6):2227-32.
Veldhuis JD, Keenan DM, Mielke K, Miles JM, Bowers CY. Testosterone supplementation in healthy older men drives GH and IGF-I secretion without potentiating peptidyl secretagogue efficacy. Eur J Endocrinol. 2005 Oct;153(4):577-86.
Lewis MI, Fournier M, Storer TW, Bhasin S, Porszasz J, Ren SG, Da X, Casaburi R. Skeletal muscle adaptations to testosterone and resistance training in men with COPD. J Appl Physiol (1985). 2007 Oct;103(4):1299-310.
Mauras N, Hayes V, Welch S, Rini A, Helgeson K, Dokler M, Veldhuis JD, Urban RJ. Testosterone deficiency in young men: marked alterations in whole body protein kinetics, strength, and adiposity. J Clin Endocrinol Metab. 1998 Jun;83(6):1886-92.
Bondanelli M, Ambrosio MR, Margutti A, Franceschetti P, Zatelli MC, degli Uberti EC. Activation of the somatotropic axis by testosterone in adult men: evidence for a role of hypothalamic growth hormone-releasing hormone. Neuroendocrinology. 2003 Jun;77(6):380-7.
Veldhuis JD, Metzger DL, Martha PM Jr, Mauras N, Kerrigan JR, Keenan B, Rogol AD, Pincus SM. Estrogen and testosterone, but not a nonaromatizable androgen, direct network integration of the hypothalamo-somatotrope (growth hormone)-insulin-like growth factor I axis in the human: evidence from pubertal pathophysiology and sex-steroid hormone replacement. J Clin Endocrinol Metab. 1997 Oct;82(10):3414-20.
Weissberger AJ, Ho KK. Activation of the somatotropic axis by testosterone in adult males: evidence for the role of aromatization. J Clin Endocrinol Metab. 1993 Jun;76(6):1407-12.
Veldhuis JD, Mielke KL, Cosma M, Soares-Welch C, Paulo R, Miles JM, Bowers CY. Aromatase and 5alpha-reductase inhibition during an exogenous testosterone clamp unveils selective sex steroid modulation of somatostatin and growth hormone secretagogue actions in healthy older men. J Clin Endocrinol Metab. 2009 Mar;94(3):973-81.
Yamamoto T, Sakai C, Yamaki J, Takamori K, Yoshiji S, Kitawaki J, Fujii M, Yasuda J, Honjo H, Okada H. Estrogen biosynthesis in human liver–a comparison of aromatase activity for C-19 steroids in fetal liver, adult liver and hepatoma tissues of human subjects. Endocrinol Jpn. 1984 Jun;31(3):277-81.
Hata S, Miki Y, Saito R, Ishida K, Watanabe M, Sasano H. Aromatase in human liver and its diseases. Cancer Med. 2013 Jun;2(3):305-15.
Riggs BL, Hartmann LC. Selective estrogen-receptor modulators — mechanisms of action and application to clinical practice. N Engl J Med. 2003 Feb 13;348(7):618-29. Review. Erratum in: N Engl J Med. 2003 Mar 20;348(12):1192.
Löfgren L, Wallberg B, Wilking N, Fornander T, Rutqvist LE, Carlström K, von Schoultz B, von Schoultz E. Tamoxifen and megestrol acetate for postmenopausal breast cancer: diverging effects on liver proteins, androgens, and glucocorticoids. Med Oncol. 2004;21(4):309-18.
Centrella M, McCarthy TL, Chang WZ, Labaree DC, Hochberg RB. Estren (4-estren-3alpha,17beta-diol) is a prohormone that regulates both androgenic and estrogenic transcriptional effects through the androgen receptor. Mol Endocrinol. 2004 May;18(5):1120-30.
Yu YM, Domené HM, Sztein J, Counts DR, Cassorla F. Developmental changes and differential regulation by testosterone and estradiol of growth hormone receptor expression in the rabbit. Eur J Endocrinol. 1996 Nov;135(5):583-90.
Zung A, Phillip M, Chalew SA, Palese T, Kowarski AA, Zadik Z. Testosterone effect on growth and growth mediators of the GH-IGF-I axis in the liver and epiphyseal growth plate of juvenile rats. J Mol Endocrinol. 1999 Oct;23(2):209-21.
Hayes VY, Urban RJ, Jiang J, Marcell TJ, Helgeson K, Mauras N. Recombinant human growth hormone and recombinant human insulin-like growth factor I diminish the catabolic effects of hypogonadism in man: metabolic and molecular effects. J Clin Endocrinol Metab. 2001 May;86(5):2211-9.
Sculthorpe N, Solomon AM, Sinanan AC, Bouloux PM, Grace F, Lewis MP. Androgens affect myogenesis in vitro and increase local IGF-1 expression. Med Sci Sports Exerc. 2012 Apr;44(4):610-5.
Birzniece V, Meinhardt UJ, Umpleby MA, Handelsman DJ, Ho KK. Interaction between testosterone and growth hormone on whole-body protein anabolism occurs in the liver. J Clin Endocrinol Metab. 2011 Apr;96(4):1060-7.
Mertani HC, Delehaye-Zervas MC, Martini JF, Postel-Vinay MC, Morel G. Localization of growth hormone receptor messenger RNA in human tissues. Endocrine. 1995 Feb;3(2):135-42.
Florini JR, Ewton DZ, Coolican SA. Growth hormone and the insulin-like growth factor system in myogenesis. Endocr Rev. 1996 Oct;17(5):481-517. Review.
Urban RJ, Bodenburg YH, Gilkison C, Foxworth J, Coggan AR, Wolfe RR, Ferrando A. Testosterone administration to elderly men increases skeletal muscle strength and protein synthesis. Am J Physiol. 1995 Nov;269(5 Pt 1):E820-6.
Ewton DZ, Coolican SA, Mohan S, Chernausek SD, Florini JR. Modulation of insulin-like growth factor actions in L6A1 myoblasts by insulin-like growth factor binding protein (IGFBP)-4 and IGFBP-5: a dual role for IGFBP-5. J Cell Physiol. 1998 Oct;177(1):47-57.
Venken K, Movérare-Skrtic S, Kopchick JJ, Coschigano KT, Ohlsson C, Boonen S, Bouillon R, Vanderschueren D. Impact of androgens, growth hormone, and IGF-I on bone and muscle in male mice during puberty. J Bone Miner Res. 2007 Jan;22(1):72-82.
Serra C, Bhasin S, Tangherlini F, Barton ER, Ganno M, Zhang A, Shansky J, Vandenburgh HH, Travison TG, Jasuja R, Morris C. The role of GH and IGF-I in mediating anabolic effects of testosterone on androgen-responsive muscle. Endocrinology. 2011 Jan;152(1):193-206.
Spangenburg EE, Le Roith D, Ward CW, Bodine SC. A functional insulin-like growth factor receptor is not necessary for load-induced skeletal muscle hypertrophy. J Physiol. 2008 Jan 1;586(1):283-91.
Yoshizawa A, Clemmons DR. Testosterone and insulin-like growth factor (IGF) I interact in controlling IGF-binding protein production in androgen-responsive foreskin fibroblasts. J Clin Endocrinol Metab. 2000 Apr;85(4):1627-33.
Gayan-Ramirez G, Rollier H, Vanderhoydonc F, Verhoeven G, Gosselink R, Decramer M. Nandrolone decanoate does not enhance training effects but increases IGF-I mRNA in rat diaphragm. J Appl Physiol (1985). 2000 Jan;88(1):26-34.
Lewis MI, Horvitz GD, Clemmons DR, Fournier M. Role of IGF-I and IGF-binding proteins within diaphragm muscle in modulating the effects of nandrolone. Am J Physiol Endocrinol Metab. 2002 Feb;282(2):E483-90
Salmons S. Myotrophic effects of an anabolic steroid in rabbit limb muscles. Muscle Nerve. 1992 Jul;15(7):806-12.
Bisschop A, Gayan-Ramirez G, Rollier H, Dekhuijzen PN, Dom R, de Bock V, Decramer M. Effects of nandrolone decanoate on respiratory and peripheral muscles in male and female rats. J Appl Physiol (1985). 1997 Apr;82(4):1112-8
Lewis MI, Fournier M, Yeh AY, Micevych PE, Sieck GC. Alterations in diaphragm contractility after nandrolone administration: an analysis of potential mechanisms. J Appl Physiol (1985). 1999 Mar;86(3):985-92.
Heitzman RJ. The effectiveness of anabolic agents in increasing rate of growth in farm animals; report on experiments in cattle. Environ Qual Saf Suppl. 1976;(5):89-98. Review.
Buttery, P., Vernon, B., & Pearson, J. (1978). Anabolic agents—some thoughts on their mode of action. Proceedings of the Nutrition Society, 37(3), 311-315.
Hongerholt DD, Crooker BA, Wheaton JE, Carlson KM, Jorgenson DM. Effects of a growth hormone-releasing factor analogue and an estradiol-trenbolone acetate implant on somatotropin, insulin-like growth factor I, and metabolite profiles in growing Hereford steers. J Anim Sci. 1992 May;70(5):1439-48.
Tan RS, Scally MC. Anabolic steroid-induced hypogonadism–towards a unified hypothesis of anabolic steroid action. Med Hypotheses. 2009 Jun;72(6):723-8.
Kamanga-Sollo E, White ME, Hathaway MR, Weber WJ, Dayton WR. Effect of Estradiol-17beta on protein synthesis and degradation rates in fused bovine satellite cell cultures. Domest Anim Endocrinol. 2010 Jul;39(1):54-62.
Kamanga-Sollo E, Thornton KJ, White ME, Dayton WR. Role of G protein-coupled estrogen receptor-1, matrix metalloproteinases 2 and 9, and heparin binding epidermal growth factor-like growth factor in estradiol-17β-stimulated bovine satellite cell proliferation. Domest Anim Endocrinol. 2014 Oct;49:20-6.
Dunn JD, Johnson BJ, Kayser JP, Waylan AT, Sissom EK, Drouillard JS. Effects of flax supplementation and a combined trenbolone acetate and estradiol implant on circulating insulin-like growth factor-I and muscle insulin-like growth factor-I messenger RNA levels in beef cattle. J Anim Sci. 2003 Dec;81(12):3028-34.
Pampusch MS, Johnson BJ, White ME, Hathaway MR, Dunn JD, Waylan AT, Dayton WR. Time course of changes in growth factor mRNA levels in muscle of steroid-implanted and nonimplanted steers. J Anim Sci. 2003 Nov;81(11):2733-40.
Pampusch MS, White ME, Hathaway MR, Baxa TJ, Chung KY, Parr SL, Johnson BJ, Weber WJ, Dayton WR. Effects of implants of trenbolone acetate, estradiol, or both, on muscle insulin-like growth factor-I, insulin-like growth factor-I receptor, estrogen receptor-{alpha}, and androgen receptor messenger ribonucleic acid levels in feedlot steers. J Anim Sci. 2008 Dec;86(12):3418-23.
Thompson SH, Boxhorn LK, Kong WY, Allen RE. Trenbolone alters the responsiveness of skeletal muscle satellite cells to fibroblast growth factor and insulin-like growth factor I. Endocrinology. 1989 May;124(5):2110-7.
Johnson BJ, Halstead N, White ME, Hathaway MR, DiCostanzo A, Dayton WR. Activation state of muscle satellite cells isolated from steers implanted with a combined trenbolone acetate and estradiol implant. J Anim Sci. 1998 Nov;76(11):2779-86.
Dalbo VJ, Roberts MD, Mobley CB, Ballmann C, Kephart WC, Fox CD, Santucci VA, Conover CF, Beggs LA, Balaez A, Hoerr FJ, Yarrow JF, Borst SE, Beck DT. Testosterone and trenbolone enanthate increase mature myostatin protein expression despite increasing skeletal muscle hypertrophy and satellite cell number in rodent muscle. Andrologia. 2017 Apr;49(3).
Bhasin S, He EJ, Kawakubo M, Schroeder ET, Yarasheski K, Opiteck GJ, Reicin A, Chen F, Lam R, Tsou JA, Castaneda-Sceppa C, Binder EF, Azen SP, Sattler FR. N-terminal propeptide of type III procollagen as a biomarker of anabolic response to recombinant human GH and testosterone. J Clin Endocrinol Metab. 2009 Nov;94(11):4224-33.
Nelson AE, Meinhardt U, Hansen JL, Walker IH, Stone G, Howe CJ, Leung KC, Seibel MJ, Baxter RC, Handelsman DJ, Kazlauskas R, Ho KK. Pharmacodynamics of growth hormone abuse biomarkers and the influence of gender and testosterone: a randomized double-blind placebo-controlled study in young recreational athletes. J Clin Endocrinol Metab. 2008 Jun;93(6):2213-22.
Holt RI. Detecting growth hormone misuse in athletes. Indian J Endocrinol Metab. 2013 Oct;17(Suppl 1):S18-22.
Tan SH, Lee A, Pascovici D, Care N, Birzniece V, Ho K, Molloy MP, Khan A. Plasma biomarker proteins for detection of human growth hormone administration in athletes. Sci Rep. 2017 Aug 30;7(1):10039.
Jørgensen JO, Jessen N, Pedersen SB, Vestergaard E, Gormsen L, Lund SA, Billestrup N. GH receptor signaling in skeletal muscle and adipose tissue in human subjects following exposure to an intravenous GH bolus. Am J Physiol Endocrinol Metab. 2006 Nov;291(5):E899-905.
Liu X, Robinson GW, Gouilleux F, Groner B, Hennighausen L. Cloning and expression of Stat5 and an additional homologue (Stat5b) involved in prolactin signal transduction in mouse mammary tissue. Proc Natl Acad Sci U S A. 1995 Sep 12;92(19):8831-5.
Hennighausen L, Robinson GW. Interpretation of cytokine signaling through the transcription factors STAT5A and STAT5B. Genes Dev. 2008 Mar 15;22(6):711-21.
Eshet R, Laron Z, Pertzelan A, Arnon R, Dintzman M. Defect of human growth hormone receptors in the liver of two patients with Laron-type dwarfism. Isr J Med Sci. 1984 Jan;20(1):8-11.
Teglund S, McKay C, Schuetz E, van Deursen JM, Stravopodis D, Wang D, Brown M, Bodner S, Grosveld G, Ihle JN. Stat5a and Stat5b proteins have essential and nonessential, or redundant, roles in cytokine responses. Cell. 1998 May 29;93(5):841-50.
Hwa V, Little B, Adiyaman P, Kofoed EM, Pratt KL, Ocal G, Berberoglu M, Rosenfeld RG. Severe growth hormone insensitivity resulting from total absence of signal transducer and activator of transcription 5b. J Clin Endocrinol Metab. 2005 Jul;90(7):4260-6. Epub 2005 Apr 12.
Rowland JE, Lichanska AM, Kerr LM, White M, d’Aniello EM, Maher SL, Brown R, Teasdale RD, Noakes PG, Waters MJ. In vivo analysis of growth hormone receptor signaling domains and their associated transcripts. Mol Cell Biol. 2005 Jan;25(1):66-77. Erratum in: Mol Cell Biol. 2005 Mar;25(5):2072.
Klover P, Hennighausen L. Postnatal body growth is dependent on the transcription factors signal transducers and activators of transcription 5a/b in muscle: a role for autocrine/paracrine insulin-like growth factor I. Endocrinology. 2007 Apr;148(4):1489-97.
Barclay JL, Kerr LM, Arthur L, Rowland JE, Nelson CN, Ishikawa M, d’Aniello EM, White M, Noakes PG, Waters MJ. In vivo targeting of the growth hormone receptor (GHR) Box1 sequence demonstrates that the GHR does not signal exclusively through JAK2. Mol Endocrinol. 2010 Jan;24(1):204-17.
Hwa V, Nadeau K, Wit JM, Rosenfeld RG. STAT5b deficiency: lessons from STAT5b gene mutations. Best Pract Res Clin Endocrinol Metab. 2011 Feb;25(1):61-75.
Varco-Merth B, Feigerlová E, Shinde U, Rosenfeld RG, Hwa V, Rotwein P. Severe growth deficiency is associated with STAT5b mutations that disrupt protein folding and activity. Mol Endocrinol. 2013 Jan;27(1):150-61.
Davey HW, Xie T, McLachlan MJ, Wilkins RJ, Waxman DJ, Grattan DR. STAT5b is required for GH-induced liver IGF-I gene expression. Endocrinology. 2001 Sep;142(9):3836-41.
Woelfle J, Chia DJ, Rotwein P. Mechanisms of growth hormone (GH) action. Identification of conserved Stat5 binding sites that mediate GH-induced insulin-like growth factor-I gene activation. J Biol Chem. 2003 Dec 19;278(51):51261-6. Epub 2003 Oct 7.
Woelfle J, Billiard J, Rotwein P. Acute control of insulin-like growth factor-I gene transcription by growth hormone through Stat5b. J Biol Chem. 2003 Jun 20;278(25):22696-702. Epub 2003 Apr 7.
MacLean HE, Chiu WS, Notini AJ, Axell AM, Davey RA, McManus JF, Ma C, Plant DR, Lynch GS, Zajac JD. Impaired skeletal muscle development and function in male, but not female, genomic androgen receptor knockout mice. FASEB J. 2008 Aug;22(8):2676-89.
Tan SH, Dagvadorj A, Shen F, Gu L, Liao Z, Abdulghani J, Zhang Y, Gelmann EP, Zellweger T, Culig Z, Visakorpi T, Bubendorf L, Kirken RA, Karras J, Nevalainen MT. Transcription factor Stat5 synergizes with androgen receptor in prostate cancer cells. Cancer Res. 2008 Jan 1;68(1):236-48.
de Souza GL, Hallak J. Anabolic steroids and male infertility: a comprehensive review. BJU Int. 2011 Dec;108(11):1860-5.