La Luteolina, un flavonoide contenuto nel rosmarino, timo, prezzemolo e negli agrumi, sembra essere un efficace composto anti-aromatase, almeno secondo uno studio svolto su animali e pubblicato da ricercatori del Centro di Scienze Sanitarie dell’Università di Pechino nel Journal of Pharmacology and Experimental Therapeutics.(1) A differenza dei composti anti-aromatasi sintetici, la Luteolina sembra avere il potenziale di migliorare anche l’equilibrio del Colesterolo.
Già nel 2013, in uno studio in vitro svolto da farmacologi dell’Istituto di biologia di Chengdu, dove si erano prese in esame oltre 100 sostanze al fine di valutarne il potenziale antiestrogenico, la Luteolina risultò la più interessante a tal fine.(2)
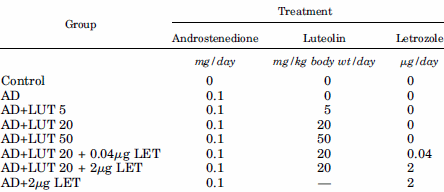
Nello studio del quale ho introdotto brevemente i risultati emersi all’inizio di questo articolo, i ricercatori hanno usato topi di sesso femminile le cui ovaie sono state rimosse chirurgicamente come pratica preliminare per lo svolgimento dell’esperimento. Ad un certo numero di animali sono state somministrate iniezioni giornaliere di Androstenedione [AD]. Come ben sappiamo, l’enzima aromatasi converte l’Androstenedione in Estrone e, attraverso l’azione dell’Estradiolo 17beta-deidrogenasi, in Estradiolo.
Ad alcuni animali è stata anche somministrata per via orale la Luteolina [LUT]. Le dosi utilizzate sono mostrate nella figura riportata di seguito. L’equivalente umano delle dosi utilizzate varia dai 45mg ai 450mg di Luteolina al giorno. Alcuni topi sono stati trattati con iniezioni di Letrozolo [LET], conosciutissimo e potente inibitore dell’aromatasi non steroideo di terza generazione.
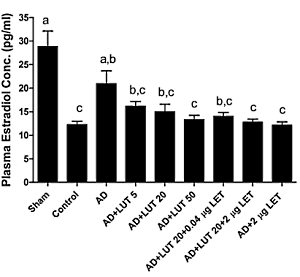
Dopo 12 settimane, i ricercatori hanno misurato le concentrazioni di Estradiolo nel sangue dei topi. La figura seguente mostra che l’effetto anti-aromatasi dato dalla dose più alta di Luteolina era uguale a quello ottenuto con l’uso del Letrozolo.
I ricercatori avevano impiantato nei topi cellule di cancro al seno estradiolo-sensibili. Sia la Luteolina che il Letrozolo hanno inibito la crescita tumorale, ma il Letrozolo ha ottenuto risultati leggermente migliori rispetto alla Luteolina.
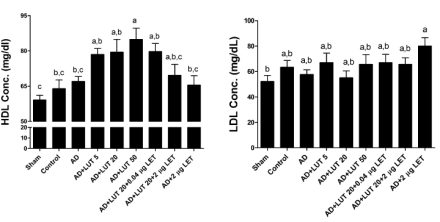
L’uso degli inibitori dell’aromatasi ha tra i suoi effetti collaterali quello di poter causare uno squilibrio delle lipoproteine. Le concentrazioni di LDL aumentano mentre quelle di HDL diminuiscono. La Luteolina, pur agendo attraverso l’inibizione dell’enzima aromatasi, mostra l’effetto opposto sull’equilibrio del Colesterolo. La Luteolina mostra di causare un abbassamento delle concentrazioni di LDL e aumento delle concentrazioni di HDL.
La figura sopra riportata mostra che la Luteolina può anche annullare l’effetto negativo del Letrozolo sui livelli di Colesterolo.
La Luteolina non ha avuto alcun effetto sui livelli di Trigliceridi.
I ricercatori sottolineano il fatto che, sebbene la Luteolina sia ampiamente presenti in diversi alimenti vegetali, i dosaggi utilizzati nello studio qui riportato erano superiori ai livelli normalmente consumati dagli esseri umani. Tuttavia, questo studio potrebbe fornire le basi scientifiche per lo sviluppo nutraceutico o farmacologico di questo flavone.
In fine, i ricercatori concludono che, dato il potenziale di alterazione del rapporto LDL/HDL solitamente associato all’uso a lungo termine degli inibitori dell’aromatasi, la somministrazione di Luteolina può essere un potenziale meccanismo di compensazione senza compromissioni sull’aromatasi.
Se non avete ancora letto le precedenti parti componenti questa serie di articoli vi invito a farlo prima di procedere con la lettura di questa quarta ed ultima parte: 1° Parte – 2° Parte – 3° Parte.
Farmacocinetica, Farmacodinamica e Feedback Negativi
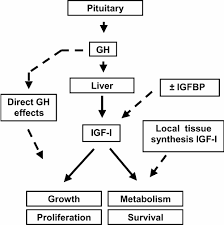
Come già detto, il fegato rappresenta il principale bersaglio del GH, il quale è il principale regolatore della sintesi epatica di IGF-1. Per causare tale effetto, il GH si lega con i GHR localizzati nel dominio extracellulare degli epatociti stimolando successivamente la produzione di IGF-1 endocrino tramite la trascrizione genica, utilizzando la via di segnalazione JAK-STAT. Inoltre, è stato dimostrato che la somministrazione di GH causa una rapida sovraregolazione dell’mRNA del IGF-1 nel fegato.[338]
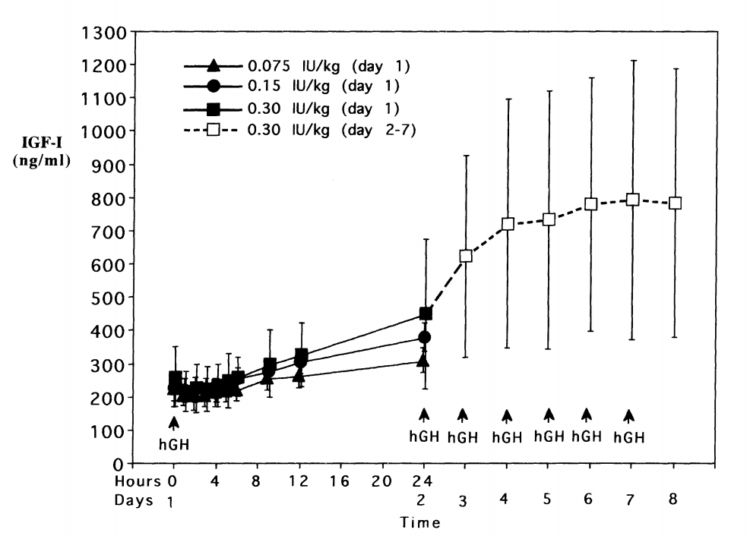
Aumenti dei livelli serici di IGF-1 si verificano molto rapidamente anche in presenza di un grande bolo di rHGH. Incrementi significativi di IGF-1 sono già osservabili dopo 6-12h dall’iniezione.[339] Questi livelli serici di IGF-1 continuano ad aumentare fino a raggiungere il loro punto di saturazione dose-dipendente entro 4-7 giorni, anche quando si utilizzano dosi estremamente elevate che ammontano a 20-30UI al giorno di rHGH.[340] In particolare, il punto di saturazione si è rivelato essere compreso nell’intervallo dei 700-800 ng/mL e sembra suggerire che i livelli endocrini di IGF-1 hanno un tetto massimo negli adulti sani. I meccanismi esatti devono ancora essere chiariti, ma sono probabilmente il risultato dei complessi meccanismi di controllo intrinseci all’Asse GH/IGF-1. Coloro i quali desiderano elevare i livelli endocrini di IGF-1 al fine di ottenerne un vantaggio sull’ipertrofia dovrebbe tenerlo a mente, in quanto vi è un punto in cui l’uso di dosi maggiori di rHGH semplicemente non si traducono in elevati livelli serici di IGF-1. Qui di seguito ho riportato il grafico dello studio di Tanaka il quale mostra la relazione tra l’rhGH e i livelli serici di IGF-:
Ora, vorrei dedicarmi brevemente all’analisi dell’azione del IGF-1autocrino e del perché esso rappresenti un mediatore cruciale del processo ipertrofico, prima di tornare nuovamente a discutere su questioni inerenti alla farmacodinamica e farmacocinetica. La segnalazione recettoriale del IGF-1 è unica nel suo genere, e questo lo si deve al fatto che utilizza due percorsi distinti per stimolare la proliferazione o la differenziazione.[341-343] Questo è un comportamento abbastanza interessante, poiché nessun altro membro della famiglia dei fattori di crescita ha dimostrato di agire in tal modo. Poiché la proliferazione e la differenziazione sono processi opposti, inizialmente era difficile per i ricercatori capire come un singolo fattore di crescita, attraverso un singolo recettore, potesse inviare un segnale che attivasse entrambi.[294] Da quando sono state fatte queste prime scoperte, è stato ulteriormente chiarito che l’IGF-1 non svolge simultaneamente queste azioni. Test su varie linee di coltura cellulare hanno dimostrato che gli effetti proliferativi arrivano prima, durando tra le 24 e le 36 ore. È solo dopo questa fase proliferativa iniziale che si verifica la differenziazione miogenica.[344]
Gli effetti proliferativi mediati dall’IGF-1 sui mioblasti sono noti sin dagli anni ’70, quando vennero osservati per la prima volta nelle cellule epatiche di ratto.[345] Questa stimolazione proliferativa del IGF-1 si traduce in un aumento del numero di cellule, nei livelli di proteine, nella sintesi del DNA, nell’assorbimento di aminoacidico, nell’assorbimento del glucosio e nella soppressione della proteolisi.[346] Nelle colture cellulari umane, l’IGF-1 ha anche dimostrato di aumentare la dimensione dei miotubi indipendentemente dal fatto che i mioblasti proliferino attivamente o che la proliferazione sia cessata. Regola la dimensione dei miotubi attivando la sintesi proteica, inibendo la degradazione proteica e inducendo la fusione delle cellule di riserva.[347-348] La capacità dell’IGF di sopprimere la proteolisi nel muscolo scheletrico, la scomposizione delle proteine in aminoacidi, è stata dimostrata innumerevoli volte nel corso degli anni.[349-352] È stato anche dimostrato che l’IGF-1 induce la proliferazione e la differenziazione delle cellule satelliti in miociti maturi, come determinato da un aumento del numero di miofibre nucleate a livello centrale rispetto a quelle periferiche.[148,353-354]
La capacità dell’IGF-1 autocrino di causare la differenziazione dei mioblasti è stata in realtà una scoperta che potremmo definire quasi “ibrida” dal momento che degli studi svolti negli anni ’60 avevano mostrato che questo effetto si verifica con alti livelli di Insulina.[355] Successivamente è stato dimostrato che gli IGF sono stimolatori molto più potenti nella differenziazione miogenica rispetto all’Insulina e si è concluso che la stessa Insulina agisce realmente come un analogo dell’IGF-1 in questo sistema.[356-357] Gli effetti di differenziazione dati dall’IGF-1 autocrino sono bifasici, con basse concentrazioni che stimolano progressivamente la differenziazione dei mioblasti mentre concentrazioni molto elevate mostrano una cessazione dell’attività di differenziazione. Il limite massimo per la differenziazione sembra attestarsi a circa 100ng/mL per l’IGF-1 e 300ng/mL per IGF-2.[358] Questo effetto non è legato alla proliferazione, poiché non si osservano ulteriori aumenti nel numero complessivo delle cellule.[294] È possibile che le molecole di segnalazione coinvolte nella regolazione negativa del sistema miogenico siano aumentate, ma questa è una affermazione puramente speculativa.[359-360]
La somministrazione di rHGH eleva l’espressione dell’mRNA dell’IGF-1 nel muscolo scheletrico in numerosi modelli cellulari, umani e animali.[127,150,361-364] Ciò avviene abbastanza rapidamente, entro 60 minuti dall’iniezione sottocutanea di rHGH ed i picchi sono segnalati tra le 6 e le 12 ore post iniezione.[363] In questo particolare modello animale citato, il raddoppio della dose di GH non ha portato ad ulteriori aumentati dei livelli di mRNA dell’IGF-1, il che suggerisce che esiste un sistema di regolazione che determina quanto GH sia necessario per stimolare al massimo l’espressione locale dell’IGF-1 nel muscolo scheletrico. In precedenza si è potuto appurare che la differenziazione dei miociti mediata dall’IGF-1 si arresta quando le concentrazioni locali raggiungono circa i 100ng/mL, ma quanto GH è necessario per raggiungere il punto di saturazione dell’espressione dell’mRNA dell’IGF-1?
Gli studi sui miociti umani mostrano che il GH aumenta l’espressione dell’mRNA dell’IGF-1 entro 30-60 minuti con picchi molto più rapidi rispetto a quelli osservati negli studi sugli animali, entro 1-2 ore, usando la via di segnalazione JAK / STAT5b.[365] Questi livelli elevati di mRNA hanno dimostrato di durare fino a 48 ore dopo una singola esposizione al GH. La quantità di GH necessaria per stimolare al massimo l’espressione dell’mRNA dell’IGF-1 è risultata essere una dose compresa tra i 7,5ng/mL e 30ng/ml [366], con una dose media efficace che si attesta a 3ng/ml. Questi numeri sono in linea con gli intervalli di dose fisiologica osservati negli animali, che sono effettivamente compresi tra i 2-100 ng/mL.[367] Inoltre, si collocano esattamente in linea con quanto si osserva endogenamente nell’uomo, con concentrazioni normali di picco comprese tra i 22,4 e 32,4ng/mL.[368-369,436] Ci sono stati casi in cui gli uomini presi in esame hanno mostrato concentrazioni di picco leggermente più alte, ma questi devono essere considerati valori anomali.[370] In ogni caso, ciò che questi dati tendono a suggerire è che il corpo umano è particolarmente adatto a gestire livelli naturali di picco della secrezioni di GH endogeno. Cercare di incidere ulteriormente il sistema elevando i livelli di GH oltre quelli endogeni, unicamente per tentare di potenziare i processi ipertrofici, potrebbe in realtà non tradursi nell’effetto desiderato.
Gli studi che mettono a confronto le infusioni locali con le infusioni sistemiche di GH o IGF-1 sono un po’ più difficili da trovare di quanto si vorrebbe. Le poche sperimentazioni sugli animali che sono riuscito a trovare indicano che l’infusione diretta di GH o IGF-1 nei tessuti bersaglio determina un aumento della massa muscolare. Questo aumento dell’ipertrofia si verifica anche senza che il muscolo bersaglio sia stato sottoposto ad attività motoria.[371-372] Gli studi dimostrano anche che le iniezioni locali di GH portano a livelli sostanzialmente più alti nell’espressione dell’mRNA dell’ IGF-1 locale rispetto alle iniezioni locali di IGF-1, di un fattore di oltre venti.[127] Sono riuscito a trovare uno studio nel quale si confrontavano le risposte dei ratti (attivi e non) all’infusione locale di IGF-1. Il gruppo “IGF-1 plus training” ha mostrato un aumento sia della massa muscolare che della forza locale maggiore rispetto al semplice trattamento in isolamento.[373] Quindi, anche se limitata, la letteratura disponibile è apparentemente in grado di dimostrare che le iniezione locali di GH o IGF-1 hanno effettivamente valore.
Ne ho già parlato diverse volte ma, nel tentativo di imprimere ulteriormente questo concetto, è necessario ricordarsi che i livelli autocrini di IGF-1 sembrano essere molto più importanti dei livelli endocrini di IGF-1 in relazione alla regolazione della massa muscolare. Oltre a questo punto, la sovraespressione dell’IGF-1 autocrino nel muscolo provoca l’ipertrofia delle fibre.[374] La sovraespressione dell’IGF-1 autocrino ha anche mostrato effetti anti-catabolici, con modelli animali tendenti a mostrare una resistenza generale all’atrofia muscolare normalmente osservata con l’invecchiamento.[375] L’IGF-1 localizzato fornisce anche capacità rigenerative indipendenti dall’età nelle cellule muscolari.[376]
Vi sono anche alcune prove convincenti che suggeriscono che l’IGF-1 endocrino agisce direttamente come un regolatore di feedback negativo sulla produzione di IGF-1 autocrino. Questo meccanismo di feedback negativo è dipendente dal pathway PI3K/Akt [377-378]. Inoltre, elevati livelli di IGF-1 endocrino possono anche agire indirettamente per sopprimere la produzione di IGF-1 autocrino. Quindi, in altre parole, non solo l’IGF-1 endocrino ha un impatto diretto minore sulla regolazione della massa muscolare, ma può anche sopprimere l’IGF-1 autocrino che ha impatti maggiori sull’ipertrofia.
Elevati livelli di IGF-1 circolante e, nello specifico, di IGF-1 libero elevati agiscono in modo negativo sul GH determinando un tasso di soppressione della produzione di IGF-1 autocrino a valle.[379] Non è del tutto chiaro, tuttavia, se la regolazione negativa dell’IGF-1 modifichi l’emivita dell’mRNA dell’IGF-1 o influenzi direttamente l’espressione del gene IGF-1. Oltre a questo, è stato anche dimostrato che l’espressione dell’IGF-1 autocrino è sottoregolata nelle cellule muscolari dopo trattamento con IGF-1.[366] È stato anche dimostrato che l’espressione epatica dell’mRNA dell’IGF-1 è sottoregolata dall’esposizione acuta all’IGF-1.[127] Quindi, mantenere livelli endocrini il più possibile soppressi con rispettiva dose di rHGH, elevando contemporaneamente i livelli autocrini, dovrebbe essere un fattore prioritario in un protocollo di GH volto all’ipertrofia.
Il GH è pulsatile per natura sia nell’uomo che nelle specie animali. Quindi, sarebbe logico pensare che molti dei processi intrinseci del corpo saranno tarati in modo tale da rispondere in maniera ottimale all’esposizione al GH in modo simile. In accordo con questa affermazione è stato dimostrato che solo la somministrazione di GH pulsatile, e non l’infusione continua, ha la capacità di stimolare massimamente l’espressione dell’mRNA dell’IGF-1 nel muscolo scheletrico.[366,380-381] È stato anche dimostrato che la somministrazione pulsatile porta ad un aumento del potenziale di crescita postnatale complessivo rispetto all’infusione continua.[89,382] La somministrazione pulsatile può anche portare a livelli endocrini di IGF-1 serici comparabili, o addirittura diminuiti [383], il che è vantaggioso a causa delle potenziali capacità di regolazione negativa che possiede sull’espressione dell’IGF-1 autocrino e che sono state discusse in precedenza. L’evidenza suggerisce anche che il picco stesso, e non necessariamente il numero di picchi, potrebbe essere della massima importanza per i tessuti bersaglio.[384] Per la massima crescita e potenziale ipertrofico, l’evidenza tende a suggerire che creare picchi di GH elevati, e quindi tornare ai livelli basali più volte al giorno, può essere preferibile rispetto a mantenerli elevati per periodi di tempo più lunghi. Questo pratica permette di riprodurre gli schemi secretori in vivo.
I pathways del GH coinvolti nell’anabolismo sono anche suscettibili alla desensibilizzazione, che è parte della fisiologia del GH endogeno.[385] A causa della natura intrinsecamente pulsatile del GH in vivo, l’attività dei recettori e dei pathways sono regolati da un impulso seguito da un periodo di inattività.[386] L’esposizione continua o ripetuta al GH senza un adeguato lasso di tempo refrattario comporterà livelli di attività fortemente soppressi. In effetti, nel corso degli anni numerosi studi hanno dimostrato che tale effetto si verifica. Le cellule ed il tessuto muscolare richiedono un periodo refrattario piuttosto lungo prima che la loro piena risposta al GH venga recuperata. Dopo l’esposizione al GH, le cellule muscolari non sono nemmeno in grado di rispondere alle successive dosi di GH. In realtà, occorrono due ore complete per riprendere parzialmente la reattività nei modelli cellulari, con un totale di 6-8 ore di astinenza dall’uso di GH necessarie per ripristinare la piena sensibilità.[366] Viceversa, quando il GH è micro-dosato in impulsi di dieci minuti, seguiti da intervalli di otto ore, è stato mostrato aumentare progressivamente l’mRNA dell’IGF-1 con ogni impulso successivo.[386]
Questo fenomeno è potenzialmente il risultato di una desensibilizzazione complessiva all’interno della via JAK-STAT5, poiché è stato dimostrato che l’esposizione al GH negli studi sulle cellule epatiche causa resistenza alla successiva attivazione della via STAT5 per 4-8 ore.[387-388] Questo lasso di tempo è sufficiente per sincronizzarsi abbastanza bene con ciò che è stato visto nei modelli di cellule miocitarie citati in precedenza. Nei modelli di cellule epatiche, il GH ha stimolato un significativo aumento dell’espressione del SOCS3, che è un potente inibitore dell’azione del GH.[389]. Poiché il GH non ha avuto alcun effetto sull’espressione del SOCS3 nelle cellule muscolari, questo deve essere un altro meccanismo causante il periodo refrattario. Questo meccanismo può essere dipeso dalla sottoregolazione dei GHR, dall’inibizione mediata da un’altra proteina SOCS, o dall’induzione di una tirosina fosfatasi che semplicemente inattiva la via JAK / STAT.[390] La via JAK-STAT5b, che come ricorderete è intimamente associata al muscolo scheletrico e all’espressione dell’IGF-1, è di natura transitoria – con attivazione massima raggiunta entro 10-30 minuti, seguita da un prolungato periodo di inattivazione.
Una scoperta piuttosto nuova di Xu et al. [391] ha dimostrato che anche distanziare le esposizioni al GH di cinque ore lasciava entrambi i percorsi a valle MEK1/2 e ERK1/2 significativamente soppressi rispetto a tutti i percorsi a monte, a causa di una potenziale disconnessione nella trasduzione del segnale . Ciò è di particolare interesse in quanto questi stessi due percorsi a valle sono stati coinvolti in modo significativo sia nella crescita sia nella proliferazione.[392-393] È stato anche scoperto che l’attivazione indotta da GH di STAT1 e STAT3 è stata desensibilizzata, ma l’esposizione all’Insulina inverte la desensibilizzazione osservata in tutti i percorsi interessati. Anche se non sto per trattare approfonditamente l’Insulina, ci sono un paio di importanti punti da dovere prendere in considerazione. Bisogna comprendere innanzitutto che ci sono molti obiettivi a valle del recettore del GH e molti di questi hanno il potenziale per essere desensibilizzati dopo l’esposizione al GH. Bisogna comprendi anche che l’Insulina possiede l’abilità unica di risensibilizzare molti di questi percorsi. Ciò ha un senso vista la relazione tipo yin-yang tra i due composti. È noto che il GH e l’Insulina possiedono una relazione anabolica sinergica a causa di molti effetti che esercitano l’uno sull’altro. Questo sembra essere soltanto un’anteprima di uno di questi effetti.
Asse GH/IGF-1 – Relazione con altri ormoni
Prima di passare alle note conclusive, vorrei trattare brevemente alcuni altri ormoni connessi a diverso grado con l’Asse GH/IGF-1. Per prima cosa, voglio trattare brevemente l’Asse Tiroideo dal momento che l’inserimento di composti tiroidei insieme al GH è una pratica comune anche durante i protocolli di massa.
Il muscolo scheletrico è il principale bersaglio di segnalazione dell’ormone tiroideo, con trasportatori degli ormoni tiroidei e enzimi di conversione espressi localmente.[394] È ben noto che il GH potenzia la deiodinazione periferica che converte il T4 in T3, riducendo così il T4 e il reverse T3, aumentando contemporaneamente i livelli di T3.[395-398] Ciò che molte persone non riescono a capire è che questo è un effetto transitorio, e studi a lungo termine sembrano indicare che gli effetti mediati dal GH sulla conversione periferica si stabilizzino con il tempo.[399-402]
Via ubiquitina/proteasoma
Invece di proseguire ulteriormente su questo, avendo già trattato la questione nel dettaglio in un mio vecchio articolo, preferirei concentrarmi su alcune pubblicazioni relative alla tiroide che non vengono discusse abbastanza spesso. Gli ormoni tiroidei, per loro natura, sono composti tendenzialmente catabolici in quanto stimolano la disgregazione proteica dell’intero corpo in misura maggiore rispetto alla sintesi proteica.[403] A livello locale, nel muscolo scheletrico stimolano un aumento dell’attività all’interno della via ubiquitina/proteasoma, che è ampiamente coinvolta nella proteolisi.[404-406] Il risultato di questo è un tasso accelerato del turnover proteico e una perdita netta complessiva degli aminoacidi situati all’interno dei muscoli scheletrici.
Inoltre, negli esseri umani, sia gli stati di ipertiroidismo che di ipotiroidismo sono stati associati a livelli di IGF-1 soppressi con una tendenza alla normalizzazione quando viene ristabilita una condizione di eutiroidismo. L’ipertiroidismo è anche associato ad una bassa attività di legame recettoriale del GH, che si ipotizza essere il risultato di una ridotta capacità di elaborazione dei recettori del GH.[407] E’ stato anche ipotizzato che l’ipertiroidismo sia in grado anche di accelerare la clearance del GH urinario.[408] Inoltre, studi su animali hanno dimostrato che gli ormoni tiroidei possono avere importanti effetti soppressivi sulla sintesi di IGF-1 stimolata con il GH.[409] Ovviamente, a causa della complessa relazione che l’Asse Tiroideo ha con l’Asse GH/IGF-1, raggruppando tutte le interazioni che hanno tra loro in pochi paragrafi, trattare l’argomento diventerebbe poco pratico. Tuttavia, quando il corpo della letteratura scientifica viene esaminato nella sua interezza, ci sono molte prove che suggeriscono che la supplementazione con composti tiroidei esogeni potrebbe non essere l’ideale quando l’obiettivo di un individuo è l’ipertrofia, anche se la regolazione del dosaggio dei tiroidei in tale contesto rimane la misura di “sicurezza” più intelligente visto l’impatto negativo sulla funzionalità tiroidea dato dal GH. Comunque, per tutti coloro che sono interessati ad approfondire questo argomento, consiglio di iniziare con la review nella nota seguente.[410]
Miostatina
Mi piacerebbe trattare anche la Miostatin, che rappresenta un argomento molto discusso nei vari forum di BodyBuilding presenti in rete. La sua fama proviene dai risultati ipertrofici espressi dai bovini privi per mutazione del gene della Miostatina, i quali mostrano una massa muscolare significativamente maggiore rispetto ai loro simili non mutati.[411] La Miostatina, un fattore di crescita e differenziazione appartenente alla superfamiglia dei TGF-beta, ha dimostrato di inibire selettivamente la miogenesi, in gran parte tramite il suo effetto soppressivo sulla proliferazione dei mioblasti.[412] È espressa e secreta prevalentemente dal muscolo scheletrico. Come molti sanno, se riesci a sopprimere o inibire la Miostatina, di conseguenza il potenziale ipertrofico aumenta significativamente.
Le mutazioni della Miostatina sono state osservate sia negli animali che nell’uomo. Queste mutazioni del gene della Miostatina portano ad un fenotipo ipertrofico negli animali, come accennato in precedenza.[413-415] L’Asse GH/IGF-1 e la Miostatina sembrano avere una relazione regolativa diretta tra loro, come osservato nei pazienti affetti contemporaneamente da GHD e HIV che mostrano marcati aumenti nell’espressione dell’mRNA della Miostatina.[416] Quindi, è possibile che attraverso una supplementazione di dosi sovrafisiologiche di rHGH si possa indurre una diminuzione dell’mRNA della Miostatina [209,417-419]? Sfortunatamente, nonostante la presenza di alcuni casi studio selezionati, non credo che si abbiano abbastanza dati in questo momento per sapere se ciò possa dare risultati apprezzabili in seguito alla sua applicazione.
Quello che sappiamo è che aumenti dell’espressione dell’mRNA dell’IGF-1 e le concentrazioni circolanti di IGF-1 sono state osservati dopo inibizione della Miostatina.[419-421] Sappiamo anche che l’inibizione della Miostatina tende a causare l’ipertrofia attraverso molte delle stesse modalità osservate con l’IGF-1 autocrino, cioè l’aumento della sintesi proteica e l’attivazione delle cellule satelliti.[422-425] E sappiamo anche che l’ipertrofia indotta dalla sovraespressione dell’IGF-1 o dall’inibizione della Miostatina utilizza la stessa identica via – PI3K/Akt/mTOR.[426-428] Tuttavia, l’IGF-1 non è un requisito per l’ipertrofia indotta dalla Follistatina, tranne nel caso di livelli di Insulina estremamente bassi – come ben sappiamo, la Follistatina è un inibitore della Miostatina [429]. E l’esposizione cronica al GH può in realtà portare ad un’espressione sovrastimolata della Miostatina e del suo recettore.[209]
Quindi quello che possiamo dire, con certezza, è che l’espressione della Miostatina non sarà un fattore diretto o indiretto per quanto riguarda il potenziamento dei processi ipertrofici, né dell’attività contrattile, nei muscoli scheletrici umani.[430] Proprio per questo motivo, non ritengo che sia un fattore sul quale gli atleti debbano eccessivamente concentrarsi, al di fuori di un utile arricchimento delle proprie conoscenze in materia.
Applicazioni pratiche e pensieri conclusivi
Arrivati a questo punto è mia intenzione unire tutto ciò che è stato esposto in questa serie di articoli e esporlo sotto forma di alcuni suggerimenti pratici rivolti a tutti coloro i quali vogliono semplicemente massimizzare la loro capacità ipertrofica.
Ora è chiaro che il GH possiede pochissimi, se non nulli, effetti diretti sull’ipertrofia. Pertanto, qualsiasi protocollo di massa che contempli il suo uso dovrà tenere in considerazione questo punto includendo gli AAS, i quali, per l’appunto, hanno anche una proficua sinergia con il GH. Sia la letteratura scientifica che i dati aneddotici dimostrano chiaramente che l’uso combinato di entrambi i composti ha un massimale ipertrofico significativamente più alto rispetto all’uso singolo. Personalmente, penso che i BodyBuilder dovrebbero sempre optare per l’utilizzo di una base di Testosterone e Boldenone (correttamente rapportati in base a contesto e alle caratteristiche individuali) anche in una fase “Bulk” nella quale viene inserito l’uso del GH. Il Trenbolone può essere considerato come parte “accessoria” di un protocollo di massa, a causa della sua intrinseca difficoltà gestazionale come sostanza anabolizzante. Dovrebbe essere usato con parsimonia e con cautela poiché, insieme ai suoi numerosi punti di forza come composto anabolizzante, presenta alcune limitazioni. Queste limitazioni derivano per lo più, come già accennato, dalla difficile gestione del composto in quanto esso non è facilmente tollerato da una buona parte degli individui. Quindi, se viene usato il Trenbolone, dovrebbe essere inserito calcolando con attenzione il dosaggio e la tolleranza individuale, anche per quanto concerne la tolleranza temporale individuale all’uso di tale composto.
Dopo un periodo d’uso prolungato di dosi sovrafisiologico di AAS (Ciclo+Bridge), è buona cosa procedere con l’interruzione o con una marcata riduzione del numero e degli AAS utilizzati. Detta in parole semplici, questa interruzione può contemplare una completa astinenza dagli AAS, svolgendo un adeguata PCT al fine di ristabilire una omeostasi ormonale fisiologica, o una transizione ad una TRT, metodologia comunemente chiamata “blast and cruise”. La struttura del protocollo di supplementazione farmacologica dovrebbe sempre seguire i principi del dosaggio minimo efficace con aumenti nei dosaggi degli AAS solo nel caso si sia raggiunto il limite di crescita con il precedente dosaggio, assicurandosi che tutte le altre variabili nello stile di vita siano correttamente regolate. L’utilizzo di questo approccio limita il rischio che si sviluppino effetti collaterali indesiderati sul lungo termine.
Quando si decide di usare il GH, la dove ce ne sia la possibilità economica, esso dovrebbe provenire da uno dei prodotti presenti nel mercato farmaceutico. Questi prodotti approvati devono superare anni di studi strettamente controllati per dimostrare la loro sicurezza, purezza ed efficacia su soggetti umani. I progressi tecnologici nel corso degli anni hanno reso molto più facile la produzione di rHGH. Per questo motivo, i produttori ora provengono da tutto il mondo. Spesso questi produttori realizzano ciò che viene definito “GH generico” sui forum, ma tale termine non mi piace molto. Definire qualcosa come “generico” implica che sia una replica perfetta di prodotti legati a specifici marchi farmaceutici approvati dell’agenzia del farmaco che hanno perso il cui brevetto è scaduto, il che non è il caso del GH. Infatti, a causa della natura estremamente complessa del processo di produzione del rHGH, per esempio, la FDA non consente nemmeno l’uso del termine “generico” quando si tratta di rHGH e utilizza invece il termine ” follow-on protein product ” o FOPP.
Spesso questi marchi off-label sono venduti ad un costo molto ridotto, ed è qui che sta il dilemma, in quanto questo può essere molto allettante. Tuttavia, con questo costo ridotto per il consumatore, non ci sarà nemmeno la garanzia del produttore su cosa ci sia realmente nella fiala o persino su come è stato prodotto. Il problema di fondo è che il processo di produzione del rHGH è estremamente complessa, ed è molto facile che nelle fasi di questo processo si commettano errori con conseguenti variazioni nella catena proteica che potenzialmente portano ad effetti indesiderati, o anche a risposte autoimmuni.
Spesso gli atleti si affidano semplicemente ai test serici per misurare i livelli di GH e/o IGF-1 al fine di concludere che un prodotto contenete GH sia “buono”, ma dobbiamo ricordarci che raggiungere livelli ematici ormonali elevati è la parte relativamente facile. Anche le molecole di GH che sono state alterate o danneggiate durante la produzione possono dare questo esito. Tuttavia, queste stesse molecole di GH danneggiate o mutate possono spesso stimolare risposte autoimmuni. Ciò potrebbe indurre il corpo ad avere una risposta recettoriale degradata, che può anche riflettersi sulla secrezione endogena nel tempo. [431-432] Rimane poi il problema del reale contenuto della vial o fiala, la quale può non presentare nessun principio attivo al suo interno.
Il GH dovrebbe essere usato in modo pulsatile, per mimare le condizioni in vivo. Tra queste iniezioni, deve esserci un periodo di refrattarietà o si deve consumare un pasto che abbia un buon stimolo sull’Insulina. Può anche essere utilizzata l’Insulina esogena al fine di bypassare molte delle limitazioni del periodo refrattario, ma questo va oltre lo scopo di questo articolo. Anche se il cumulo delle dosi giornaliere dovrebbe essere sovrafisiologico, le dosi individuali non hanno bisogno di essere ad alto dosaggio, poiché la massima stimolazione dell’IGF-1 autocrino nel tessuto muscolo scheletrico si verifica ben all’interno delle concentrazioni fisiologiche di GH. Aneddoticamente, sembra anche esserci un limite con il quale l’uso di rHGH diventa additivo in presenza di AAS. Potrebbe essere necessario un po’ di ponderata (e supportata da personale qualificato) auto-sperimentazione per scoprire dove si trova questa singola dose di saturazione, ma la maggior parte dei soggetti troverà questo limite tra le 4 e le 8 UI/die. Oltre questo dosaggio, la maggior parte degli utilizzatori tenderà a scoprire che la giustificazione dei costi e il rapporto rischio /beneficio tendono a diminuire rapidamente.
Non bisognerebbe passare troppo tempo a riflettere sul tempo delle iniezioni di GH, dal momento che gli aumenti dei livelli di IGF-1 autocrino avvengono rapidamente e possono rimanere elevati per giorni. Bisognerebbe concentrati invece sul programma di iniezione che si addice meglio al contesto della giornata, tenendo contemporaneamente presenti le linee guida per il periodo di refrattarietà del GH. Si possono anche prendere in considerazione piccole o grandi iniezioni, poiché alcuni potrebbero trovare più pratiche e funzionali iniezioni con un dosaggio inferiore e somministrate più frequenti mentre altri potrebbero preferire iniezioni con un dosaggio maggiore e somministrazioni meno frequenti. Naturalmente, maggiore è il contenuto dell’iniezione, maggiore è la probabilità che si superi la soglia massima di espressione dell’IGF-1 autocrino.
Massimizzare l’espressione dell’IGF-1 autocrino, mentre contemporaneamente si sopprimono i livelli di IGF-1 endocrino, sarà una priorità. Esistono prove a sostegno dell’ipotesi secondo cui un iniezione locale di GH possa aiutare a raggiungere questo obiettivo, con una conseguente minore possibilità di feedback negativo. Sono stati osservati aumenti significativi della massa muscolare in appena due settimane di iniezioni locali di IGF-1.[441]
La dove cause di forza maggiore non lo impediscano, è consigliabile evitare o comunque regolare attentamente l’uso di tutti quei composti che possono avere interazioni negative con l’uso del GH a fini ipertrofici. Inibitori della Aromatasi, Modulatori Selettivi del Recettore degli Estrogeni e T3 hanno tutti dimostrato di avere un potenziale effetto negativo sul processo globale ipertrofico legato al GH e per tale motivo dovrebbero essere usati con parsimonia, se non omessi del tutto dove possibile (vedi in particolare il T3).
Questo non dovrebbe sorprendere nessuno e non dovrebbe nemmeno essere troppo difficile da tenere a mente: allenarsi duramente, allenarsi in modo intelligente e allenarsi in modo coerente. Sebbene non sia stato affrontato direttamente nell’articolo, bisogna capire che l’allenamento contro resistenza ha impatti unici e additivi sull’ipertrofia. In effetti, alcuni di questi meccanismi non sono nemmeno mediati dall’Asse AR e/o GH/IGF-1. [433] Bisogna anche comprendere che non esiste una “magica” routine allenante universalmente applicabile, la chiave di volta sarà la coerenza nel garantire un carico di lavoro adeguato, con elementi di sovraccarico progressivo nel tempo assicurando un adeguato stimolo meccanico gestendo al meglio le variabili allenanti (intensità, volume, densità e intensità percepita). La logica composizione del piano allenante servirà a garantire lo stimolo ipertrofico di base che sarà coadiuvato dall’azione dei composti utilizzati.
Nonostante la mole e l’importanza delle informazioni presentate in questa serie di articoli, è necessario ricordare che i meccanismi d’azione ormonali sono governati da innumerevoli fattori. Anche esaminando l’intero corpo della letteratura scientifica equivarrebbe a poco più che accumulare una serie di utili nozioni garanti di assicurare una solida base conoscitiva sull’argomento la quale rappresenterà un punto di partenza intelligente per l’applicazione pratica, rimanendo sempre soggetti alle variabili di risposta individuale. Seguendo questa linea, i migliori risultati nella pratica spesso provengono da coloro i quali posseggono una buona conoscenza dei principi scientifici e la capacità innata di saperli applicare non solo su se stessi ma, soprattutto, su terzi. Infatti, molto raramente due persone rispondono in modo identico alla supplementazione di ormoni esogeni (e non solo), quindi, non bisogna assolutamente pensare che basti semplicemente applicare su se stessi o su terzi un protocollo che ha portato benefici realmente apprezzabili su un soggetto per ottenere la medesima risposta.
A tal fine, invito atleti e Preparatori a utilizzare queste pubblicazioni come punto di partenza per essere in grado di gestire l’applicazione pratica in modo più consapevole e produttivo. Inoltre, chi ne fosse in grado, può consultare il vasto numero di riferimenti riportati nel corso di queste pubblicazioni è tentare di ragionare sulle mie conclusioni. Ad ogni citazione presente in questa serie di articoli, assicuratevi che il riferimento elencato supporti effettivamente le affermazioni fatte. Mantenere sempre una mente aperta ma con i giusti “filtri”, e cercare di non credere per partito preso ad una singola opinione, specialmente di fronte alle nuove evidenze scientifiche. Infine, è buona cosa verificare sempre la veridicità di quanto è stato affermato.
Punti conclusivi per un corretto utilizzo della chimica e del GH a fini ipertrofici:
Usare il GH in combinazione con gli AAS
Usare GH e AAS di grado farmaceutico la dove ciò è possibile
Assicurarsi una base di Testosterone correttamente rapportata al Boldenone aggiungendo (in base a maturità e tolleranza) il Trenbolone
Iniettare il GH in modo pulsatile, considerare l’opzione delle iniezioni locali nei gruppi carenti
Mantenere un dosaggio complessivo ottimale di GH il quale si attesta tra le 4 e le 8UI/die
Evitare o regolare attentamente l’uso dei composti che possono interagire negativamente con i processi ipertrofici legati al uso di GH (vedi AI, SERM e T3)
Dopo un periodo di tempo (variabile) nel quale si è stati sottoposti a dosaggi ormonali sovrafisiologici optare per una PCT o per una TRT (a seconda delle proprie necessità e priorità)
Essere a conoscenza dei potenziali effetti collaterali legati all’auso/abuso di GH (nausea, vomito, cefalea, ritenzione idrica e sodica, edemi, parestesie, sindrome del tunnel carpale, rigidità articolare, dolori articolari, artrite, dolori muscolari, ipertensione, insulino-resistenza, diabete di tipo II, acromegalia, dilatazione addominale, ipertrofia cardiaca ecc…)
Svolgere regolarmente esami del sangue; sia durante i periodi di picco nell’uso della farmacologia sia nel periodo successivo (vedi PCT/OCT o TRT)
Gestire al meglio le variabili legate agli stressor ambientali, all’allenamento, all’alimentazione e al sonno.
Gabriel Bellizzi
Riferimenti:
338. Mathews LS, Norstedt G, Palmiter RD. Regulation of insulin-like growth factor I gene expression by growth hormone. Proc Natl Acad Sci U S A. 1986 Dec;83(24):9343-7.
339. Keller A, Wu Z, Kratzsch J, Keller E, Blum WF, Kniess A, Preiss R, Teichert J, Strasburger CJ, Bidlingmaier M. Pharmacokinetics and pharmacodynamics of GH: dependence on route and dosage of administration. Eur J Endocrinol. 2007 Jun;156(6):647-53.
340. Tanaka T, Seino Y, Fujieda K, Igarashi Y, Yokoya S, Tachibana K, Ogawa Y. Pharmacokinetics and metabolic effects of high-dose growth hormone administration in healthy adult men. Endocr J. 1999 Aug;46(4):605-12.
341. Quinn LS, Steinmetz B, Maas A, Ong L, Kaleko M. Type-1 insulin-like growth factor receptor overexpression produces dual effects on myoblast proliferation and differentiation. J Cell Physiol. 1994 Jun;159(3):387-98.
342. Coolican SA, Samuel DS, Ewton DZ, McWade FJ, Florini JR. The mitogenic and myogenic actions of insulin-like growth factors utilize distinct signaling pathways. J Biol Chem. 1997 Mar 7;272(10):6653-62.
343. Foulstone EJ, Huser C, Crown AL, Holly JM, Stewart CE. Differential signalling mechanisms predisposing primary human skeletal muscle cells to altered proliferation and differentiation: roles of IGF-I and TNFalpha. Exp Cell Res. 2004 Mar 10;294(1):223-35.
344. Ewton DZ, Roof SL, Magri KA, McWade FJ, Florini JR. IGF-II is more active than IGF-I in stimulating L6A1 myogenesis: greater mitogenic actions of IGF-I delay differentiation. J Cell Physiol. 1994 Nov;161(2):277-84.
345. Florini JR, Nicholson ML, Dulak NC. Effects of peptide anabolic hormones on growth of myoblasts in culture. Endocrinology. 1977 Jul;101(1):32-41.
346. Laviola L, Natalicchio A, Giorgino F. The IGF-I signaling pathway. Curr Pharm Des. 2007;13(7):663-9. Review.
347. Jacquemin V, Furling D, Bigot A, Butler-Browne GS, Mouly V. IGF-1 induces human myotube hypertrophy by increasing cell recruitment. Exp Cell Res. 2004 Sep 10;299(1):148-58.
348. Jacquemin V, Butler-Browne GS, Furling D, Mouly V. IL-13 mediates the recruitment of reserve cells for fusion during IGF-1-induced hypertrophy of human myotubes. J Cell Sci. 2007 Feb 15;120(Pt 4):670-81. Epub 2007 Jan 30.
349. Ballard FJ, Francis GL. Effects of anabolic agents on protein breakdown in L6 myoblasts. Biochem J. 1983 Jan 15;210(1):243-9.
350. Ewton DZ, Falen SL, Florini JR. The type II insulin-like growth factor (IGF) receptor has low affinity for IGF-I analogs: pleiotypic actions of IGFs on myoblasts are apparently mediated by the type I receptor. Endocrinology. 1987 Jan;120(1):115-23.
351. Hembree JR, Hathaway MR, Dayton WR. Isolation and culture of fetal porcine myogenic cells and the effect of insulin, IGF-I, and sera on protein turnover in porcine myotube cultures. J Anim Sci. 1991 Aug;69(8):3241-50.
352. Hong D, Forsberg NE. Effects of serum and insulin-like growth factor I on protein degradation and protease gene expression in rat L8 myotubes. J Anim Sci. 1994 Sep;72(9):2279-88.
353. Florini JR, Ewton DZ, Roof SL. Insulin-like growth factor-I stimulates terminal myogenic differentiation by induction of myogenin gene expression. Mol Endocrinol. 1991 May;5(5):718-24.
354. Musarò A, McCullagh KJ, Naya FJ, Olson EN, Rosenthal N. IGF-1 induces skeletal myocyte hypertrophy through calcineurin in association with GATA-2 and NF-ATc1. Nature. 1999 Aug 5;400(6744):581-5.
355. Haba GDL, Cooper GW, Elting V. HORMONAL REQUIREMENTS FOR MYOGENESIS OF STRIATED MUSCLE IN VITRO: INSULIN AND SOMATOTROPIN. Proceedings of the National Academy of Sciences of the United States of America. 1966;56(6):1719-1723.
356. Florini JR, Ewton DZ. Insulin acts as a somatomedin analog in stimulating myoblast growth in serum-free medium. In Vitro. 1981 Sep;17(9):763-8.
357. Schmid C, Steiner T, Froesch ER. Preferential enhancement of myoblast differentiation by insulin-like growth factors (IGF I and IGF II) in primary cultures of chicken embryonic cells. FEBS Lett. 1983 Sep 5;161(1):117-21.
358. Florini JR, Ewton DZ, Falen SL, Van Wyk JJ. Biphasic concentration dependency of stimulation of myoblast differentiation by somatomedins. Am J Physiol. 1986 May;250(5 Pt 1):C771-8.
359. Quinn LS, Ehsan M, Steinmetz B, Kaleko M. Ligand-dependent inhibition of myoblast differentiation by overexpression of the type-1 insulin-like growth factor receptor. J Cell Physiol. 1993 Sep;156(3):453-61.
360. Olson EN. Signal transduction pathways that regulate skeletal muscle gene expression. Mol Endocrinol. 1993 Nov;7(11):1369-78. Review.
361. Murphy LJ, Bell GI, Friesen HG. Growth hormone stimulates sequential induction of c-myc and insulin-like growth factor I expression in vivo. Endocrinology. 1987 May;120(5):1806-12.
362. Turner JD, Rotwein P, Novakofski J, Bechtel PJ. Induction of mRNA for IGF-I and -II during growth hormone-stimulated muscle hypertrophy. Am J Physiol. 1988 Oct;255(4 Pt 1):E513-7.
363. Isgaard J, Nilsson A, Vikman K, Isaksson OG. Growth hormone regulates the level of insulin-like growth factor-I mRNA in rat skeletal muscle. J Endocrinol. 1989 Jan;120(1):107-12.
364. Bichell DP, Kikuchi K, Rotwein P. Growth hormone rapidly activates insulin-like growth factor I gene transcription in vivo. Mol Endocrinol. 1992 Nov;6(11):1899-908.
365. Sadowski CL, Wheeler TT, Wang LH, Sadowski HB. GH regulation of IGF-I and suppressor of cytokine signaling gene expression in C2C12 skeletal muscle cells. Endocrinology. 2001 Sep;142(9):3890-900.
366. Frost RA, Nystrom GJ, Lang CH. Regulation of IGF-I mRNA and signal transducers and activators of transcription-3 and -5 (Stat-3 and -5) by GH in C2C12 myoblasts. Endocrinology. 2002 Feb;143(2):492-503.
367. MacLeod JN, Pampori NA, Shapiro BH. Sex differences in the ultradian pattern of plasma growth hormone concentrations in mice. J Endocrinol. 1991 Dec;131(3):395-9.
368. Rochiccioli P, Messina A, Tauber MT, Enjaume C. Correlation of the parameters of 24-hour growth hormone secretion with growth velocity in 93 children of varying height. Horm Res. 1989;31(3):115-8.
369. Hansen TK, Gravholt CH, ØRskov H, Rasmussen MH, Christiansen JS, Jørgensen JO. Dose dependency of the pharmacokinetics and acute lipolytic actions of growth hormone. J Clin Endocrinol Metab. 2002 Oct;87(10):4691-8.
370. Baum WF, Klöditz E, Hesse V, Jahreis G, Schneyer U, Giebler H. [Increase in spontaneous growth hormone secretion in asthmatic children–a symptom of atopic disposition?]. Kinderarztl Prax. 1993 Nov;61(9):323-8.
371. Adams GR, McCue SA. Localized infusion of IGF-I results in skeletal muscle hypertrophy in rats. J Appl Physiol (1985). 1998 May;84(5):1716-22.
372. Alzghoul MB, Gerrard D, Watkins BA, Hannon K. Ectopic expression of IGF-I and Shh by skeletal muscle inhibits disuse-mediated skeletal muscle atrophy and bone osteopenia in vivo. FASEB J. 2004 Jan;18(1):221-3. Epub 2003 Nov 3.
373. Lee S, Barton ER, Sweeney HL, Farrar RP. Viral expression of insulin-like growth factor-I enhances muscle hypertrophy in resistance-trained rats. J Appl Physiol (1985). 2004 Mar;96(3):1097-104. Erratum in: J Appl Physiol. 2004 Jun;96(6):2343.
374. Coleman ME, DeMayo F, Yin KC, Lee HM, Geske R, Montgomery C, Schwartz RJ. Myogenic vector expression of insulin-like growth factor I stimulates muscle cell differentiation and myofiber hypertrophy in transgenic mice. J Biol Chem. 1995 May 19;270(20):12109-16.
375. Barton-Davis ER, Shoturma DI, Musaro A, Rosenthal N, Sweeney HL. Viral mediated expression of insulin-like growth factor I blocks the aging-related loss of skeletal muscle function. Proc Natl Acad Sci U S A. 1998 Dec 22;95(26):15603-7.
376. Musarò A, McCullagh K, Paul A, Houghton L, Dobrowolny G, Molinaro M, Barton ER, Sweeney HL, Rosenthal N. Localized Igf-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat Genet. 2001 Feb;27(2):195-200
377. Lewis MI, Bulut Y, Biring MS, Da X, Fournier M. (1999) IGF-I administration prevents corticosteroids-induced diaphragm atrophy in emphysema . Am J Respir Crit Care Med 159:A580
378. Fournier M, Huang ZS, Cercek B, Li H, Bykhovskaya I, Lewis MI. (2000) Administration of insulin-like growth factor-1 (IGF-I) and corticosteroids in emphysematous hamsters: influences on diaphragm IGF-I . Am J Respir Crit Care Med 161:A18
379. Shavlakadze T, Grounds M. Of bears, frogs, meat, mice and men: complexity of factors affecting skeletal muscle mass and fat. Bioessays. 2006 Oct;28(10):994-1009. Review.
380. Maiter D, Underwood LE, Maes M, Davenport ML, Ketelslegers JM. Different effects of intermittent and continuous growth hormone (GH) administration on serum somatomedin-C/insulin-like growth factor I and liver GH receptors in hypophysectomized rats. Endocrinology. 1988 Aug;123(2):1053-9.
381. Isgaard J, Carlsson L, Isaksson OG, Jansson JO. Pulsatile intravenous growth hormone (GH) infusion to hypophysectomized rats increases insulin-like growth factor I messenger ribonucleic acid in skeletal tissues more effectively than continuous GH infusion. Endocrinology. 1988 Dec;123(6):2605-10.
382. Clark RG, Jansson JO, Isaksson O, Robinson IC. Intravenous growth hormone: growth responses to patterned infusions in hypophysectomized rats. J Endocrinol. 1985 Jan;104(1):53-61.
383. Bick T, Hochberg Z, Amit T, Isaksson OG, Jansson JO. Roles of pulsatility and continuity of growth hormone (GH) administration in the regulation of hepatic GH-receptors, and circulating GH-binding protein and insulin-like growth factor-I. Endocrinology. 1992 Jul;131(1):423-9.
384. Weltman A, Weltman JY, Schurrer R, Evans WS, Veldhuis JD, Rogol AD. Endurance training amplifies the pulsatile release of growth hormone: effects of training intensity. J Appl Physiol (1985). 1992 Jun;72(6):2188-96.
385. Flores-Morales A, Greenhalgh CJ, Norstedt G, Rico-Bautista E. Negative regulation of growth hormone receptor signaling. Mol Endocrinol. 2006 Feb;20(2):241-53. Epub 2005 Jul 21. Review.
386. Hartman ML, Veldhuis JD, Thorner MO. Normal control of growth hormone secretion. Horm Res. 1993;40(1-3):37-47. Review.
387. Fernández L, Flores-Morales A, Lahuna O, Sliva D, Norstedt G, Haldosén LA, Mode A, Gustafsson JA. Desensitization of the growth hormone-induced Janus kinase 2 (Jak 2)/signal transducer and activator of transcription 5 (Stat5)-signaling pathway requires protein synthesis and phospholipase C. Endocrinology. 1998 Apr;139(4):1815-24.
388. Gebert CA, Park SH, Waxman DJ. Termination of growth hormone pulse-induced STAT5b signaling. Mol Endocrinol. 1999 Jan;13(1):38-56.
389. Ram PA, Waxman DJ. SOCS/CIS protein inhibition of growth hormone-stimulated STAT5 signaling by multiple mechanisms. J Biol Chem. 1999 Dec 10;274(50):35553-61.
390. Ram PA, Waxman DJ. Role of the cytokine-inducible SH2 protein CIS in desensitization of STAT5b signaling by continuous growth hormone. J Biol Chem.2000 Dec 15;275(50):39487-96.
391. Xu J, Keeton AB, Franklin JL, Li X, Venable DY, Frank SJ, Messina JL. Insulin enhances growth hormone induction of the MEK/ERK signaling pathway. J Biol Chem. 2006 Jan 13;281(2):982-92. Epub 2005 Nov 4.
392. Lewis TS, Shapiro PS, Ahn NG. Signal transduction through MAP kinase cascades. Adv Cancer Res. 1998;74:49-139. Review.
394. Mebis L, Paletta D, Debaveye Y, Ellger B, Langouche L, D’Hoore A, Darras VM, Visser TJ, Van den Berghe G. Expression of thyroid hormone transporters during critical illness. Eur J Endocrinol. 2009 Aug;161(2):243-50.
395. Jørgensen JO, Pedersen SA, Laurberg P, Weeke J, Skakkebaek NE, Christiansen JS. Effects of growth hormone therapy on thyroid function of growth hormone-deficient adults with and without concomitant thyroxine-substituted central hypothyroidism. J Clin Endocrinol Metab. 1989 Dec;69(6):1127-32.
396. Jørgensen JO, Pedersen SB, Børglum J, Møller N, Schmitz O, Christiansen JS, Richelsen B. Fuel metabolism, energy expenditure, and thyroid function in growth hormone-treated obese women: a double-blind placebo-controlled study. Metabolism. 1994 Jul;43(7):872-7.
397. Wolthers T, Grøftne T, Møller N, Christiansen JS, Orskov H, Weeke J, Jørgensen JO. Calorigenic effects of growth hormone: the role of thyroid hormones. J Clin Endocrinol Metab. 1996 Apr;81(4):1416-9.
398. Feldt-Rasmussen U. Interactions between growth hormone and the thyroid gland — with special reference to biochemical diagnosis. Curr Med Chem. 2007;14(26):2783-8. Review.
399. Kalina-Faska B, Kalina M, Koehler B. Effects of recombinant growth hormone therapy on thyroid hormone concentrations. Int J Clin Pharmacol Ther. 2004 Jan;42(1):30-4.
400. Hubina E, Mersebach H, Rasmussen AK, Juul A, Sneppen SB, Góth MI, Feldt-Rasmussen U. Effect of growth hormone replacement therapy on pituitary hormone secretion and hormone replacement therapies in GHD adults. Horm Res. 2004;61(5):211-7. Epub 2004 Jan 30.
401. Seminara S, Stagi S, Candura L, Scrivano M, Lenzi L, Nanni L, Pagliai F, Chiarelli F. Changes of thyroid function during long-term hGH therapy in GHD children. A possible relationship with catch-up growth? Horm Metab Res. 2005 Dec;37(12):751-6.
402. Losa M, Scavini M, Gatti E, Rossini A, Madaschi S, Formenti I, Caumo A, Stidley CA, Lanzi R. Long-term effects of growth hormone replacement therapy on thyroid function in adults with growth hormone deficiency. Thyroid. 2008 Dec;18(12):1249-54.
403. Müller MJ, Seitz HJ. Thyroid hormone action on intermediary metabolism. Part III. Protein metabolism in hyper- and hypothyroidism. Klin Wochenschr. 1984 Feb 1;62(3):97-102.
404. Tawa NE Jr, Odessey R, Goldberg AL. Inhibitors of the proteasome reduce the accelerated proteolysis in atrophying rat skeletal muscles. J Clin Invest. 1997 Jul 1;100(1):197-203. PubMed PMID: 9202072
405. Dace A, Zhao L, Park KS, et al. Hormone binding induces rapid proteasome-mediated degradation of thyroid hormone receptors. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(16):8985-8990.
406. Clément K, Viguerie N, Diehn M, Alizadeh A, Barbe P, Thalamas C, Storey JD, Brown PO, Barsh GS, Langin D. In vivo regulation of human skeletal muscle gene expression by thyroid hormone. Genome Res. 2002 Feb;12(2):281-91.
407. Miell JP, Taylor AM, Zini M, Maheshwari HG, Ross RJ, Valcavi R. Effects of hypothyroidism and hyperthyroidism on insulin-like growth factors (IGFs) and growth hormone- and IGF-binding proteins. J Clin Endocrinol Metab. 1993 Apr;76(4):950-5.
408. Murao K, Takahara J, Sato M, Tamaki M, Niimi M, Ishida T. Relationship between thyroid functions and urinary growth hormone secretion in patients with hyper- and hypothyroidism. Endocr J. 1994 Oct;41(5):517-22.
409. Wolf M, Ingbar SH, Moses AC. Thyroid hormone and growth hormone interact to regulate insulin-like growth factor-I messenger ribonucleic acid and circulating levels in the rat. Endocrinology. 1989 Dec;125(6):2905-14.
410. Laron Z. Interactions between the thyroid hormones and the hormones of the growth hormone axis. Pediatr Endocrinol Rev. 2003 Dec;1 Suppl 2:244-9-discussion 250. Review.
411. Fiems LO. Double Muscling in Cattle: Genes, Husbandry, Carcasses and Meat. Animals : an Open Access Journal from MDPI. 2012;2(3):472-506.
412. Langley B, Thomas M, Bishop A, Sharma M, Gilmour S, Kambadur R. Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J Biol Chem. 2002 Dec 20;277(51):49831-40. Epub 2002 Sep 18.
413. McPherron AC, Lee SJ. Double muscling in cattle due to mutations in the myostatin gene. Proc Natl Acad Sci U S A. 1997 Nov 11;94(23):12457-61.
414. Schuelke M, Wagner KR, Stolz LE, Hübner C, Riebel T, Kömen W, Braun T, Tobin JF, Lee SJ. Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med. 2004 Jun 24;350(26):2682-8.
415. Clop A, Marcq F, Takeda H, Pirottin D, Tordoir X, Bibé B, Bouix J, Caiment F, Elsen JM, Eychenne F, Larzul C, Laville E, Meish F, Milenkovic D, Tobin J, Charlier C, Georges M. A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat Genet. 2006 Jul;38(7):813-8.
416. Gonzalez-Cadavid NF, Taylor WE, Yarasheski K, Sinha-Hikim I, Ma K, Ezzat S, Shen R, Lalani R, Asa S, Mamita M, Nair G, Arver S, Bhasin S. Organization of the human myostatin gene and expression in healthy men and HIV-infected men with muscle wasting. Proc Natl Acad Sci U S A. 1998 Dec 8;95(25):14938-43.
417. Liu W, Thomas SG, Asa SL, Gonzalez-Cadavid N, Bhasin S, Ezzat S. Myostatin is a skeletal muscle target of growth hormone anabolic action. J Clin Endocrinol Metab. 2003 Nov;88(11):5490-6.
418. Oldham JM, Osepchook CC, Jeanplong F, Falconer SJ, Matthews KG, Conaglen JV, Gerrard DF, Smith HK, Wilkins RJ, Bass JJ, McMahon CD. The decrease in mature myostatin protein in male skeletal muscle is developmentally regulated by growth hormone. J Physiol. 2009 Feb 1;587(3):669-77.
419. Williams NG, Interlichia JP, Jackson MF, Hwang D, Cohen P, Rodgers BD. Endocrine actions of myostatin: systemic regulation of the IGF and IGF binding protein axis. Endocrinology. 2011 Jan;152(1):172-80.
420. Winbanks CE, Weeks KL, Thomson RE, Sepulveda PV, Beyer C, Qian H, Chen JL, Allen JM, Lancaster GI, Febbraio MA, Harrison CA, McMullen JR, Chamberlain JS, Gregorevic P. Follistatin-mediated skeletal muscle hypertrophy is regulated by Smad3 and mTOR independently of myostatin. J Cell Biol. 2012 Jun 25;197(7):997-1008.
421. Lach-Trifilieff E, Minetti GC, Sheppard K, Ibebunjo C, Feige JN, Hartmann S, Brachat S, Rivet H, Koelbing C, Morvan F, Hatakeyama S, Glass DJ. An antibody blocking activin type II receptors induces strong skeletal muscle hypertrophy and protects from atrophy. Mol Cell Biol. 2014 Feb;34(4):606-18.
422. Bark TH, McNurlan MA, Lang CH, Garlick PJ. Increased protein synthesis after acute IGF-I or insulin infusion is localized to muscle in mice. Am J Physiol. 1998 Jul;275(1 Pt 1):E118-23.
423. Barton-Davis ER, Shoturma DI, Sweeney HL. Contribution of satellite cells to IGF-I induced hypertrophy of skeletal muscle. Acta Physiol Scand. 1999 Dec;167(4):301-5.
424. Suryawan A, Frank JW, Nguyen HV, Davis TA. Expression of the TGF-beta family of ligands is developmentally regulated in skeletal muscle of neonatal rats. Pediatr Res. 2006 Feb;59(2):175-9.
425. Gilson H, Schakman O, Kalista S, Lause P, Tsuchida K, Thissen JP. Follistatin induces muscle hypertrophy through satellite cell proliferation and inhibition of both myostatin and activin. Am J Physiol Endocrinol Metab. 2009 Jul;297(1):E157-64.
426. Bodine SC, Stitt TN, Gonzalez M, Kline WO, Stover GL, Bauerlein R, Zlotchenko E, Scrimgeour A, Lawrence JC, Glass DJ, Yancopoulos GD. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol. 2001 Nov;3(11):1014-9.
428. Kalista S, Schakman O, Gilson H, Lause P, Demeulder B, Bertrand L, Pende M, Thissen JP. The type 1 insulin-like growth factor receptor (IGF-IR) pathway is mandatory for the follistatin-induced skeletal muscle hypertrophy. Endocrinology. 2012 Jan;153(1):241-53.
429. Barbé C, Kalista S, Loumaye A, Ritvos O, Lause P, Ferracin B, Thissen JP. Role of IGF-I in follistatin-induced skeletal muscle hypertrophy. Am J Physiol Endocrinol Metab. 2015 Sep 15;309(6):E557-67. doi: 10.1152/ajpendo.00098.2015. Epub 2015 Jul 28.
430. Coffey VG, Shield A, Canny BJ, Carey KA, Cameron-Smith D, Hawley JA. Interaction of contractile activity and training history on mRNA abundance in skeletal muscle from trained athletes. Am J Physiol Endocrinol Metab. 2006 May;290(5):E849-55.
431. Moore WV, Leppert P. Role of aggregated human growth hormone (hGH) in development of antibodies to hGH. J Clin Endocrinol Metab. 1980 Oct;51(4):691-7
432. Dannies PS. Protein folding and deficiencies caused by dominant-negative mutants of hormones. Vitam Horm. 2000;58:1-26. Review.
433. DeVol DL, Rotwein P, Sadow JL, Novakofski J, Bechtel PJ. Activation of insulin-like growth factor gene expression during work-induced skeletal muscle growth. Am J Physiol. 1990 Jul;259(1 Pt 1):E89-95.
434. Hermansen K, Bengtsen M, Kjær M, Vestergaard P, Jørgensen JOL. Impact of GH administration on athletic performance in healthy young adults: A systematic review and meta-analysis of placebo-controlled trials. Growth Horm IGF Res. 2017 Jun;34:38-44.
435. de Souza GL, Hallak J. Anabolic steroids and male infertility: a comprehensive review. BJU Int. 2011 Dec;108(11):1860-5.
436. Kraemer WJ, Marchitelli L, Gordon SE, Harman E, Dziados JE, Mello R, Frykman P, McCurry D, Fleck SJ. Hormonal and growth factor responses to heavy resistance exercise protocols. J Appl Physiol (1985). 1990 Oct;69(4):1442-50.
437. Pfeffer LA, Brisson BK, Lei H, Barton ER. The insulin-like growth factor (IGF)-I E-peptides modulate cell entry of the mature IGF-I protein. Mol Biol Cell. 2009 Sep;20(17):3810-7.
438. Mills P, Dominique JC, Lafrenière JF, Bouchentouf M, Tremblay JP. A synthetic mechano growth factor E Peptide enhances myogenic precursor cell transplantation success. Am J Transplant. 2007 Oct;7(10):2247-59.
439. Brisson BK, Barton ER. Insulin-like growth factor-I E-peptide activity is dependent on the IGF-I receptor. PLoS One. 2012;7(9):e45588.
440. Brisson BK, Spinazzola J, Park S, Barton ER. Viral expression of insulin-like growth factor I E-peptides increases skeletal muscle mass but at the expense of strength. Am J Physiol Endocrinol Metab. 2014 Apr 15;306(8):E965-74.
441. Goldspink G, Harridge S. Mechanism for adaptation in skeletal muscle In: Komi P, editor. Strength and power in sport: Olympic encyclopedia of sports medicine. Oxford: Blackwell; 2002. p. 231–51.
442. Janssen JA, Hofland LJ, Strasburger CJ, van den Dungen ES, Thevis M. Potency of Full-Length MGF to Induce Maximal Activation of the IGF-I R Is Similar to Recombinant Human IGF-I at High Equimolar Concentrations. PLoS One. 2016 Mar 18;11(3):e0150453.
Se non avete ancora letto la prima e la seconda parte di questa serie di articoli vi invito a farlo prima di procedere con la lettura di questa terza parte: 1° Parte – 2° Parte.
Effetti diretti di GH e IGF-1 sull’ipertrofia muscolare
Andiamo dritti al punto – di per sé, il GH non causa direttamente ipertrofia muscolare. Questa caratteristica è stata ampiamente osservata per decenni e, finora, nessuno studio attendibile è stato in grado di mostrare un chiaro effetto della somministrazione di rHGH a medio-lungo termine sull’ipertrofia – anche in dosi sovrafisiologiche somministrate ad atleti di alto livello sottoposti ad intensi allenamenti contro resistenza.
Il fatto che questa correlazione non sia stata dimostrata non è certamente dovuto alla mancanza di tentativi in tal senso. Diversi gruppi di ricerca nel corso degli anni hanno tentato di identificare un ipotetica ipertrofia GH-mediata in soggetti adulti sani [48,195-199] o in soggetti anziani [188,198,200-203] senza successo, o in modo inconcludente. Inoltre, è stato dimostrato che gli aumenti della secrezione di GH indotta dall’esercizio in acuto non hanno prodotto cambiamenti nella MPS o nell’ipertrofia [204-205]. È interessante notare che, sebbene la quantità di studi presenti in letteratura siano significativamente meno comuni di quelli in cui è stato somministrato GH, anche la somministrazione sistemica di rhIGF-1 non ha prodotto alcun effetto ipertrofico misurabile sia in soggetti giovani [63,206] che negli anziani [207- 208].
Vi sono prove recenti che suggeriscono che l’esposizione cronica al GH aumenta l’espressione delle vie intramuscolari responsabili dell’atrofia.[209] È ovvio che molte delle caratteristiche anaboliche dimostrate dal GH possono essere compensate da questa maggiore espressione del catabolismo, che potrebbe essere il motivo per cui l’esposizione cronica al GH non porta all’ipertrofia. Potrebbe anche essere responsabile del perché l’esposizione cronica al GH può produrre muscoli meno efficienti e più deboli.[210] Molto semplicemente, questo potrebbe essere ancora un altro fattore nella lunga serie di effetti regolatori negativi intrinseci nell’Asse GH/IGF-1, ma ulteriori studi dovranno essere condotti per chiarire maggiormente questa ipotesi.
Per quelli che difficilmente accettano le informazioni riportate in questa serie di articoli, e vogliono addentrarsi loro stessi nella letteratura scientifica sperando di trovare informazioni che confutino quanto da me riportato, voglio esporre un concetto molto importante da me già espresso nella prima parte di questa serie di articoli. La maggior parte degli studi che hanno preso in esame il GH segnalano un aumento della massa magra nei soggetti dei gruppi trattati con tale ormone. Quindi, ad un lettore inesperto, potrebbero risultare infondate le affermazioni sulla mancanza di un effetto ipertrofico diretto del GH. Bisogna ricordare, però, che il GH causa una non indifferente ritenzione idrica, oltre ad aumentare la massa dei tessuti molli della quale parlerò brevemente. Nello specifico, il GH aumenta la ritenzione di sodio e, di conseguenza, dell’acqua extracellulare, in modo dose-dipendente, attraverso i suoi effetti sul sistema renina-angiotensina.[211] Questi incrementi nella ritenzione idrica e sodica sono stati osservati anche con la somministrazione di IGF-1, poiché l’IGF-1 stesso sembra essere un regolatore chiave del tasso di escrezione renale di sodio.[83,212-213] Quindi il punto della questione è che bisogna stare molto attenti nel trarre conclusioni quando non si ha ben chiara la differenza tra aumento della massa magra e crescita effettiva del tessuto muscolare.
Effetti indiretti di GH e IGF-1 sull’ipertrofia muscolare
Nella sezione precedente si è dimostrato che il GH e l’IGF-1, da soli, non hanno alcun impatto diretto sull’ipertrofia. Tuttavia, questo non significa che questi peptidi non abbiano un ruolo nei processi ipertrofici. L’obiettivo principale di questa sezione è quello di spiegare alcuni dei molti meccanismi che interessano in modo indiretto il GH e l’IGF-1 nei processi ipertrofici, molti dei quali saranno fattori importanti per gli atleti interessati a massimizzare il loro potenziale ipertrofico.
L’Ormone della Crescita è un potente stimolatore della sintesi di collagene sia nei tendini che nei muscoli. Questo effetto è probabilmente mediato dalla capacità dell’IGF-1 autocrino di stimolare i fibroblasti per sintetizzarlo.[214-215] In realtà, questo processo avviene senza che il peptide influenzi la sintesi proteica muscolare, nonostante i livelli di IGF-1 circolante e di quello locale siano significativamente più alti. Questo effetto è anche indotto indipendentemente dall’allenamento contro resistenza ed è stato persino osservato in soggetti immobilizzati hai quali è stato somministrato il GH.[216] La componente connettiva del muscolo scheletrico è vitale per la trasmissione della forza, che è prodotta dalle fibre muscolari, ai tendini e alle ossa affinché si verifichi il movimento. In particolare, il collagene è un importante componente nella trasmissione della forza della matrice extracellulare, che viene continuamente sottoposta a carichi intesi durante i movimenti.
A causa dei potenti effetti del GH sui componenti della matrice extracellulare, si può chiaramente iniziare a capire il perché gli aneddoti nel corso degli anni suggerivano che l’aggiunta del GH in una preparazione farmacologica produceva impatti positivi sulla riduzione del dolore tendineo-articolare. D’altra parte però, questo potrebbe anche essere un fattore che contribuisce in primo luogo al motivo per cui vari effetti collaterali sono segnalati dagli utilizzatori di GH come l’edema dei tessuti molli, il dolore articolare e la sindrome del tunnel carpale.[217-219] Ci sono anche molti che credono che il GH possa accelerare i tempi di recupero dagli infortuni, ma questo è un argomento complesso che verrà discusso in futuro.
Gli impatti del GH sulla sintesi di collagene potrebbero anche essere di grande interesse per gli atleti di forza che non sono necessariamente interessati all’ipertrofia, ma il cui obiettivo principale è la creazione di una condizione favorevole allo spostamento del carico massimale. Stimolare la sintesi del collagene potrebbe aiutare a rafforzare l’intero sistema di supporto dei muscoli scheletrici. Ora, vale la pena aggiungere una piccola nota di chiarimento. Nonostante questo suoni positivo in linea di principio, la supplementazione di GH non ha mai portato direttamente a guadagni di forza in nessuno degli studi svolti su soggetti sani, coprendo vari gruppi.[48,50,184,196-198,200-201,203,220-221] Naturalmente, se il GH fosse usato insieme ad un composto con la capacità di aumentare direttamente la forza, non è difficile supporre che in questo caso l’effetto addizionale potrebbe fare del GH un prezioso componente della preparazione.
Decorina
La Decorina è una proteina strutturale, che risiede principalmente nella matrice extracellulare del muscolo scheletrico e il cui ruolo è correlato alla crescita e alla riparazione dei muscoli.[222-223] Qualche decennio fa è stato dimostrato per la prima volta che la somministrazione di GH potrebbe aumentare direttamente l’espressione del gene della Decorina negli animali [224]. Recentemente, è stato dimostrato che questo effetto si manifesta anche in soggetti umani sottoposti ad allenamenti ricreativi.[225] Nello studio più recente, i livelli di Decorina sono fortemente correlati a quelli di PIIINP, che induce la stimolazione del Decorina GH-mediata che può essere coinvolta nel processo di assemblaggio della matrice di collagene osseo. Questo effetto è più pronunciato negli uomini rispetto alle donne e può essere un sottoprodotto dei livelli più alti di IGF-1 osservati negli uomini, sebbene questa affermazione sia speculativa. L’aumento dell’espressione di Decorina non è stato alterato dall’aggiunta di Testosterone, quindi questo effetto è indipendente dagli androgeni. Dopo aver visto gli effetti che il GH ha sulla sintesi di collagene e Decorina, sta diventando piuttosto chiaro che l’asse GH/IGF-1 è molto più importante per rafforzare la matrice extracellulare di supporto piuttosto che contribuire direttamente alla crescita del tessuto muscolare.
Adenosina Trifosfato (ATP)
La somministrazione acuta di GH in soggetti sani ha anche dimostrato di causare una maggiore produzione di ATP mitocondriale e una maggiore attività della citrato sintasi nel muscolo scheletrico, con una maggiore abbondanza di mRNA muscolari codificanti l’IGF-1.[226] Non solo questo potrebbe essere un fattore che contribuisce al motivo per cui il GH potrebbe avere la capacità di promuovere un aumento dei tassi di spesa energetica giornaliera ma potrebbe anche essere coinvolto nello spostamento verso la preferenza dei lipidi come substrato energetico. Anche se, come abbiamo visto in precedenza, il corpo della letteratura non supporta questa pratica, l’aumento della produzione di ATP mitocondriale potrebbe avere un ruolo nella capacità aerobica.[227]
È stato dimostrato che il GH promuove la fusione dei mioblasti con i miotubi in modelli cellulari [84], un effetto completamente indipendente dalla sovraregolazione locale del IGF-1. Per capire perché questo possa essere importante, bisogna approfondire maggiormente la questione dei fattori cellulari coinvolti nell’ipertrofia del muscolo scheletrico. L’ipertrofia dei muscoli scheletrici negli esseri umani si basa sulle cellule satelliti, che sono cellule dormienti situate all’interno delle miofibre, proprio sotto lo strato di lamina basale nella matrice extracellulare.[148] Una volta attivate queste cellule satellite, spesso con l’esercizio fisico o il danno muscolare, proliferano. Dopo la proliferazione, queste cellule satellite migrano verso siti ove si trova il danno differenziandosi e fondendosi con le miofibre esistenti che forniscono nuovi nuclei per l’ipertrofia e la riparazione. [228] Non è ancora del tutto chiaro se il GH abbia un effetto diretto sulla proliferazione e la differenziazione delle cellule satelliti.[229-230] Vale anche la pena di affermare che ciò che si osserva nelle colture cellulari potrebbe non essere del tutto indicativo di ciò che accade in vivo a causa di vari fattori esterni che non possono essere spiegati in condizioni di laboratorio.
Ci sono due fasi distinte in questa fusione dei mioblasti che si verifica.[85] Il primo sarebbe lo stadio iniziale della differenziazione in cui un sottoinsieme di cellule mononucleate si fondono per formare miotubi nascenti (fusione mioblasto/mioblasto). Questo è seguito dal secondo stadio che coinvolge ulteriori cellule disponibili che si fondono con questi miotubi nascenti e dove avviene la crescita muscolare effettiva (fusione mioblasto/miotubo). È all’interno di quest’ultimo stadio in cui il GH esercita i suoi effetti.
Questa è una scoperta piuttosto interessante, ma ancora una volta, numerosi studi sugli esseri umani non sono riusciti a dimostrare un effetto ipertrofico del GH in condizioni reali. Quindi, possiamo probabilmente dedurre da ciò che gli effetti che il GH ha sulla fusione dei nascenti miotubi non si traducono direttamente nell’ipertrofia. Tuttavia, cosa accadrebbe se si aggiungesse un’altra variabile nell’equazione in grado di creare un ambiente in cui esistessero numeri di celle satelliti potenziati, creando più materiale sul quale l’azione del GH possa manifestarsi?[231]
Introduzione degli AAS
A differenza del GH e del IGF, l’uso degli AAS ha mostrato di avere un impatto pronunciato su ipertrofia e forza. Questo è stato ben noto per decenni anche perché sono stati usati e abusati da atleti fin dalla loro creazione negli anni ’30 (con buona pace dei “puritani” della “old school”).[232] La famiglia degli AAS consiste in un gruppo di potenti composti sintetici che sono simili nella struttura chimica al Testosterone e/o al suo derivato 5α-ridotto (DHT). Vari tipi di singoli composti AAS sono stati creati nel corso degli anni usando come base la molecola naturale di Testosterone manipolandola, quindi, attraverso l’aggiunta di un gruppo etile, metile, idrossile o benzile in uno o più siti lungo la sua struttura.[233-234 ] Alcune delle varianti AAS più facilmente riconoscibili includono i composti metilati in C-17, i quali sono notoriamente dotati di un elevata biodisponibilità orale, e le varianti del 19-nortestosterone nelle quali viene rimosso il gruppo 19-metilico dalla molecola di Testosterone nel tentativo di aumentare la sua attività anabolica diminuendo al contempo la sua androgenicità e tendenza all’aromatizzazione. Conosciuti anche come “19-norsteroidi”, questa famiglia include ben note varianti di AAS come il Nandrolone ed il Trenbolone.
Molte di questi composti sono nati come risultato di un desiderio di aumentare le caratteristiche anaboliche del Testosterone a livello muscolare, abbassando allo stesso tempo gli effetti collaterali androgenici intrinsecamente inerenti alla molecola di Testosterone.[235] In generale, i rischi complessivi degli effetti collaterali derivanti dall’uso cronico di AAS sembrano essere relativamente bassi rispetto a molte sostanze socialmente accettate come l’alcol, il tabacco e vari farmaci da prescrizione.[233,236] Ovviamente, l’uso e l’abuso sono termini che si escludono a vicenda e abusare di qualsiasi sostanza tende a creare un ambiente a più alto rischio di effetti collaterali. A meno che non sia diversamente specificato, da questo punto in poi, parlerò specificamente del Testosterone in quanto è l’ormone sessuale endogeno maschile nativo, nonché l’androgeno più approfonditamente studiato in letteratura. Tanto per avere un riferimento, i maschi adulti sani producono una media di 14-77mg/settimana di Testosterone endogeno. [435]
Testosterone
Il Testosterone è un noto regolatore della massa muscolare, e gli aumenti della massa muscolare mediati dal Testosterone sono associati all’ipertrofia delle fibre, così come ad un aumento delle cellule satelliti e del numero mio nucleare.[231,237-240] Il muscolo scheletrico sembra essere uno dei tessuti più reattivi al Testosterone e si stima che i livelli circolanti di Testosterone rappresentino il fattore causale significativo (incidenza del 40-75%) dei guadagni nella massa muscolare osservati in studi di controllo randomizzati. Se ricordate ciò che è stato affermato e dimostrato nella precedente sezione, il GH possiede un’abilità molto particolare in quanto può aumentare la fusione dei mioblasti durante il processo ipertrofico. La massimizzazione di questa capacità si basa sull’avere un numero adeguato di cellule satellitari disponibili.
I risultati mostrano costantemente che i trattamenti con Testosterone determinano aumenti dose-dipendenti della massa e della forza del muscolo scheletrico, indipendentemente dal fatto che i soggetti presi in esame siano maschi più giovani o più anziani.[241-243] Al contrario, negli studi con soggetti sani giovani in cui il Testosterone endogeno è stato soppresso artificialmente, la forza e la composizione corporea hanno entrambi subito peggioramenti significativi.[244] Per ribadire il nostro precedente punto, gli aumenti della massa muscolare mediati dal Testosterone sono basati sull’ipertrofia e non sono il risultato di una transizione della fibra (cambiando le fibre di tipo I in fibre di tipo II o viceversa) o di una sua scissione.[245] A causa dei loro meccanismi anabolici unici e non sovrapposti, la somministrazione di Testosterone e l’allenamento contro resistenza hanno anche mostrato effetti sinergici e additivi l’uno sull’altro per quanto riguarda lo stimolo dell’aumento della massa muscolare.[246]
Recettore degli Androgeni (AR)
Gli Androgeni mediano principalmente i loro effetti attraverso il gene del recettore degli androgeni (AR) che è espresso in mioblasti, miofibre e cellule satelliti.[247-248] I AR sono stati rilevati anche nelle cellule che supportano i muscoli, come i fibroblasti e le cellule endoteliali. La densità dei AR sembra essere specifica per i gruppi muscolari, con l’allenamento contro resistenza e l’uso di AAS che hanno la capacità di influenzare il numero di AR presenti in questi gruppi muscolari. Oltre ai suoi effetti sulla densità dei AR, l’uso di AAS ha anche dimostrato la capacità di influenzare i livelli di attività dei AR sia in modo acuto che sul lungo termine.[249-250] Questi sono fattori piuttosto importanti da considerare quando ci si imbatte in individui che sostengono che il precedente utilizzo di AAS non offre necessariamente un vantaggio competitivo permanente.
A causa della complessità dell’argomento, ci sono state diverse ipotesi riguardo ai meccanismi con cui l’AAS esercita le sue azioni anaboliche sul muscolo scheletrico.[251] È stato dimostrato che i trattamenti con Testosterone aumentano i tassi di sintesi proteica muscolare (MPS) [252], riducono i tassi di degradazione delle proteine [253] facendo persino in modo che il corpo utilizzi più efficientemente gli amminoacidi prontamente disponibili. Quindi, ancora una volta, questo è un sistema abbastanza complesso che può anche essere semplificato ricordando che gli AAS promuovono l’anabolismo muscolare attraverso la loro capacità di incidere positivamente sull’equilibrio degli aminoacidi.
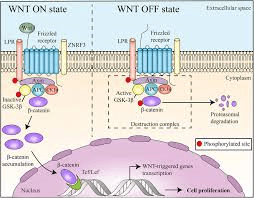
Via Wnt/B-catenina
È generalmente accettato che gli AAS esercitino i loro effetti anabolizzanti attraverso il legame e attivazione del AR che successivamente attiva cascate di segnalazione a valle che coinvolgono la via Wnt/β-catenina.[254-256]. I Wingless-INT (Wnt) sono una famiglia di glicoproteine secrete che regolano la proliferazione e la differenziazione cellulare.[257-258] I modelli cellulari hanno mostrato che il AR forma un complesso con la beta-catenina che si potenzia in presenza di AAS. [259-260] Una volta attivato questo complesso, migra nei nuclei in cui regola l’espressione dei geni target e la differenziazione delle cellule satelliti.[261-262] Questo è anche il pathway degli AAS in gran parte responsabile della miogenesi, la formazione del tessuto muscolare.[263-265]
Vale la pena notare che l’AAS possiede anche caratteristiche non genomiche che possono influenzare rapidamente numerosi processi ormonali e metabolici al di fuori del classico legame con il recettore. In letteratura sono stati riportati casi di maschi adulti con disturbi di insensibilità agli androgeni, causati da mutazioni del AR, che hanno risposto al Testosterone in modo molto simile ai soggetti sani. Questi casi studio rafforzano l’ipotesi che gli effetti anabolici dell’AAS possano essere mediati indipendentemente dal AR.[266] Le azioni non genomiche degli Androgeni possono in realtà essere un argomento piuttosto affascinante, ma che va oltre lo scopo previsto per questo articolo. Per coloro che vogliono approfondire l’argomento, consiglio di iniziare con le seguenti note di riferimento.[267-268]
AAS e potenziale sinergico con l’Asse GH/IGF-1
Ho riportato molte informazioni utili fino a questo punto, ma è qui che le cose cominciano davvero a farsi interessanti. A questo punto una domanda logica sarebbe: esistono studi svolti su soggetti sani nei quali si sono confrontate le differenze tra trattamenti singoli con GH o Androgeni e trattamenti combinati? Fortunatamente per noi, la risposta è “sì” in quanto vi sono stati alcuni studi, principalmente utilizzando soggetti anziani, sia maschi che femmine. I risultati di ognuno di questi studi dimostrano chiaramente che il GH ha un effetto additivo sui benefici consolidati che la terapia con ormoni sessuali fornisce: l’ipertrofia, la lipolisi, la sintesi del collagene, la funzione fisica, la qualità della vita e altri vari indicatori di prestazione.[187-188,269-270] Dato che è abbastanza chiaro che esiste un effetto additivo, vediamo se è possibile approfondire ulteriormente l’argomento al fine di scoprire alcuni dei meccanismi sottostanti che operano nella sinergia tra Androgeni e GH.
GHRH
Deve essere chiaro che il Testosterone, di per sé, ha un effetto additivo sull’intero Asse GH/IGF-1. Questo è stato osservato sia in modelli umani che animali, con la somministrazione di Testosterone che porta ad un aumento dei livelli circolanti di GH e IGF-1.[241,271-276] Viceversa, il deficit di Testosterone è comunemente associato a livelli significativamente ridotti di IGF-1.[277] L’effetto stimolante del Testosterone sull’Asse GH/IGF-1 sembra essere mediato a livello ipotalamico da una promozione della funzionalità del GHRH.[278]
Inoltre, e questo è bene ricordarlo, gli AAS non aromatizzabili sembrano non possedere lo stesso effetto stimolante sull’Asse GH/IGF-1.[279] Gli inibitori dell’aromatasi (AI), progettati per sopprimere il processo di aromatizzazione degli Androgeni, hanno dimostrato di attenuare direttamente la stimolazione del GH seguente la somministrazione di Testosterone. Questi indizi forniscono prove abbastanza convincenti sul fatto che gli estrogeni locali, derivanti dall’aromatizzazione, svolgono un ruolo fondamentale nella regolazione della secrezione di GH nei maschi.[280-281] Poiché l’aromatasi non è espressa nel fegato, gli AI non influenzano l’azione epatica del GH, mentre influenzano la sua azione a livello del sistema centrale.[282-283] Tuttavia i modulatori selettivi del recettore degli estrogeni (SERM) sono ancora più soppressivi in quanto agiscono ad entrambi i livelli a causa del loro meccanismo d’azione.[284-285]
Anche gli Androgeni che aumentano i livelli serici di Estrogeni, come il Nandrolone (Nor-Estrogeni), mostrano un effetto minimo sui livelli sistemici di GH e IGF-1 rispetto al Testosterone.[286] Ciò è legato ipoteticamente sia al fatto che il Nandrolone è poco soggetto all’aromatizzazione e sia al fatto che la forma estrogenica derivata (Nor-Estrogeno) abbia un ridotto potenziale di attività.[287] Ricordiamoci che l’aromatizzazione sembra essere il passo più cruciale nella stimolazione ipotalamica mediata dagli Androgeni. Ora, è ovvio che un utilizzatore di rHGH deve preoccuparsi parzialmente di questo effetto rispetto ad un non utilizzatore, considerando il fatto che, in tal caso, i livelli ormonali sono controllati quasi esclusivamente da mezzi esogeni. Detto questo, è sempre utile analizzare il quadro generale, soprattutto se l’obiettivo è massimizzare l’ipertrofia.
GHR
Un altro potenziale motivo legato agli aumenti dei livelli di GH e IGF osservati durante i trattamenti con Testosterone, è rappresentato dagli effetti diretti di quest’ultimo sui GHR. Sia gli studi sull’uomo che sugli animali hanno fornito prove del fatto che il Testosterone modifica i GHR sia nel fegato che nei tessuti periferici, migliorandone l’espressione.[288-289] Inoltre, soggetti umani ipopituitari e ipogonadici sottoposti a trattamenti con GH hanno mostrato una aumentata risposta sia all’IGF-1 locale che all’espressione del gene del recettore degli androgeni quando sottoposti anche a trattamento con Testosterone.[187,290-291] Inoltre, i maschi ipopituitari sottoposti a trattamenti con Testosterone hanno mostrato effetti notevoli sull’anabolismo proteico in presenza di GH, con il sito primario di interazione ormonale a livello epatico.[292] Quindi anche quando i livelli ormonali sono carenti, c’è ancora un’interazione molto importante tra il Testosterone e l’Asse GH/IGF-1.
Come accennato in precedenza, il GHR è espresso in quasi tutti i principali tipi di tessuto. Vale la pena sottolineare che il GHR è espresso in quantità molto bassa nel muscolo scheletrico – solo circa il 4-33% dei livelli osservati in altri tessuti. D’altra parte, il recettore del IGF-1 è espresso in maniera molto più elevata nei muscoli scheletrici, proprio come nel tessuto epatico.[293-294] Detto questo, l’aumento della sensibilità del GHR in un ambiente con livelli sovra fisiologici di GH andrà certamente a beneficio del BodyBuilder. I BodyBuilder cercano continuamente di utilizzare quantità elevate di rHGH nel tentativo di massimizzare i processi ipertrofici, e se il GH vede potenziata la sua azione e, quindi, il suo stimolo diretto sulla sintesi e rilascio del IGF-1, il quale opera direttamente sul tessuto muscolare, venendosi a creare quindi una vantaggiosa alterazione dell’Asse GH/IGF-1, l’effetto ipertrofico potenziato sarà indubbio.
IGFBP-4
Gli Androgeni hanno mostrato di avere la capacità di aumentare l’espressione del mRNA del IGF-1 locale nel muscolo scheletrico. Possiamo pertanto ipotizzare che gli Androgeni, in particolare a dosi più elevate, creino un ambiente all’interno del muscolo scheletrico estremamente favorevole alla gestione di livelli più alti di IGF-1 seguenti alla somministrazione di dosi sovrafisioogiche di rHGH. Ci sono persino stati studi sull’uomo che hanno mostrato livelli ridotti di IGFBP-4 locale nei campioni di tessuto muscolare, oltre a maggiori livelli di mRNA del IGF-1. Ciò potrebbe indicare che i cambiamenti hanno avuto luogo in quei muscoli per liberare più IGF-1 locale affinché esso possa legarsi ai suoi recettori.[277,295] Non ho molto approfondito la questione delle proteine leganti l’IGF, ma l’IGFBP-4 inibisce l’azione dell’IGF-1 e, quindi, minore è il livello della proteina legante, maggiori saranno i livelli di IGF-1 libero e attivo.[149,296]
È stato anche dimostrato che il Testosterone promuove l’ipertrofia negli stati deficitari di GH/IGF-1. [297-298] Questo è interessante in quanto dimostra che il Testosterone possiede sia vie metaboliche IGF-indipendenti che IGF-indipendenti nel tessuto muscolare.[299] Inoltre, i modelli cellulari hanno dimostrato che il Testosterone può sovraregolare l’espressione delle varie isoforme di IGF nei muscoli scheletrici, anche in assenza di GH/IGF-1.[298] E, anche se questo è stato dimostrato nei fibroblasti, il Testosterone ha mostrato di aumentare l’espressione del IGFBP-3 – un effetto che è stato ulteriormente migliorato dalla somministrazione di IGF-1.[300] È abbastanza chiaro, comunque, che il Testosterone eserciti effetti sinergici e additivi sull’anabolismo mediato da GH/IGF-1.
Fino ad ora mi sono concentrato sul Testosterone, tuttavia, però, sono stati osservati comportamenti leggermente diversi in relazione alle varianti androgene e al loro impatto sull’espressione sistemica e locale del IGF-1. Volevo analizzare un paio di specifici composti che sono spesso presenti nei protocolli farmacologici: sto parlando del Trenbolone e del Nandrolone. Salvo diverse indicazione, i risultati in seguito esposti provengono da studi svolti su animali.
Nandrolone
La somministrazione di Nandrolone ha costantemente dimostrato di non causare cambiamenti nei livelli di IGF-1 endocrino, nonostante produca simultaneamente un’espressione del IGF-1 muscolare significativamente più alta e un aumento della CSA delle fibre muscolari.[286,301-302] Inoltre, i livelli di IGFBP-3 locali sono stati riportati come significativamente più alti mentre i livelli di IGFBP-4 sono stati significativamente soppressi, il che ci rimanda a quanto esposto in precedenza – cioè ad un fattore che determina un aumento dei livelli di IGF-1 locale libero. In tutti gli studi, la somministrazione di Nandrolone ha portato direttamente ad un aumento dell’ipertrofia, nonostante non abbia avuto alcun impatto sui livelli di IGF-1 sistemici. Ciò rafforza ulteriormente l’ipotesi secondo cui l’IGF-1 endocrino non sia un fattore primario nell’ipertrofia dei muscoli scheletrici e quindi i livelli elevati non sono un prerequisito per l’aumento della massa muscolare.[303-305]
Trenbolone
È stato inoltre universalmente dimostrato che il Trenbolone aumenta i tassi di crescita muscolare in tutte le varie specie nelle quali è stato testato. A differenza del Testosterone e del Nandrolone, non si converte in estrogeno ed è stato suggerito fin dagli anni ’70 che l’aggiunta di Estradiolo al Trenbolone sembra migliorare gli effetti anabolici del composto.[306-307] Ci sono stati anche effetti potenziati sull’ipertrofia quando il Trenbolone è stato somministrato insieme ad un fattore di rilascio dell’Ormone della Crescita (GHRF).[308] Come accennato in precedenza, l’aumento di GH/IGF-1 necessita di livelli adeguati di estrogeni (derivanti principalmente dall’aromatizzazione) affinché questi esercitino uno stimolo dell’asse GH/IGF-1. Poiché la somministrazione di Trenbolone diminuisce intrinsecamente i livelli di Estradiolo, mediante inibizione a feedback negativo del Testosterone attraverso l’Asse Ipotalamo-Ipofisi-Gonadi (HPG) [309], la somministrazione di Estradiolo, tecnicamente, dovrebbe migliorare la funzionalità dell’asse GH/IGF-1. Questo dovrebbe quindi favorire ulteriormente la sinergia anabolica che avrebbe con l’androgeno. Questa ipotesi è in linea con ciò che vari studi hanno dimostrato nel corso degli anni.
Nelle colture cellulari, è stato anche dimostrato che gli Estrogeni alterano direttamente i tassi di MPS e MPB del Trenbolone attraverso meccanismi che coinvolgono sia il recettore estrogenico che il recettore del IGF-1.[310-311] In effetti, in linea di massima, i trattamenti con solo Trenbolone non mostrano aumenti significativi dei livelli endocrini o autocrini del IGF-1. Tuttavia, la co-somministrazione con Estradiolo ha tradizionalmente mostrato aumenti nei livelli di IGF-1 autocrini, similmente a quanto osservato con il Testosterone.[312-314] Questa è solo un’ulteriore prova che suggerisce che l’Estrogeno, sia di origine sistemica che derivata dall’aromatasi, è un componente chiave sia della massima stimolazione dell’Asse GH/IGF-1 che delle capacità anaboliche massimali degli Androgeni.
Proprio come il suo “cugino” 19-nor, il Trenbolone ha anche mostrato una maggiore espressione del fattore di crescita nei tessuti muscolo scheletrici, oltre ad una maggiore reattività dei muscoli a tali fattori di crescita.[315] Il Trenbolone ha anche mostrato di operare un aumento dell’attivazione e della proliferazione delle cellule satelliti in varie specie, in misura simile al Testosterone.[316-317] Ora, essendo a conoscenza delle interazioni legate al GH, si può facilmente giungere alla conclusione che entrambi questi effetti sarebbero vantaggiosi in un protocollo che includesse entrambi i composti.
PIIINP
Tornando al Testosterone, sia il GH che questo Androgeno aumentano i marker di sintesi del collagene come il PIIINP. Inoltre, il Testosterone ha anche dimostrato di potenziare le capacità del GH di aumentare la sintesi di collagene sia nei muscoli che nei tendini.[318] A sostegno di ciò, la somministrazione concomitante di GH e Testosterone in soggetti umani allenati (a livello amatoriale) ha causato aumenti significativi sia dei marker del IGF-1 che del collagene.[319] E’ interessante notare come alcuni di questi stessi marker del collagene dei quali si sta discutendo sono indicatori esatti che vengono esaminati come parte dei test antidoping per la rilevazione del GH.[320-321]
In precedenza ho solo brevemente toccato il percorso JAK-STAT ma, arrivati a questo punto, è necessario trattare l’argomento in maniera leggermente più approfondita. La via JAK-STAT è un componente critico del GH e riguarda sia la trascrizione del gene del IGF-1 che la crescita postnatale. Una delle proteine STAT in particolare, la STAT5, sembra essere intimamente coinvolta anche nella regolazione del muscolo scheletrico.[322] Ci sono due sottoproteine nella famiglia STAT5 e sono indicate come STAT5a e STAT5b. Sebbene siano identiche al 96%, è la variante STAT5b che è abbondante nei muscoli e nel tessuto epatico ed è quindi la proteina specifica su cui mi concentrerò da questo punto in avanti.[323-324]
Una revisione completa del percorso di segnalazione renderebbe questo articolo estremamente prolisso, ma ritengo importante che si comprenda che il percorso JAK/STAT5b è stato ripetutamente dimostrato avere una relazione diretta con l’espressione dell’IGF-1 locale nel tessuto muscolare e sul’ipertrofia.[248,325-332] Per questo motivo, se ci fossero modi per migliorare o ottimizzare questo specifico percorso, sembrerebbe che ciò si possa tradurre non solo in un aumento dell’attivazione del gene del IGF-1 [333-335] ma anche in un maggiore potenziale ipertrofico.
Fortunatamente, alcuni nuovi studi svolti su animali hanno già mostrato come i percorsi AR e JAK-STAT siano intimamente correlati.[248,336] Per essere precisi, il percorso STAT5a/b è a monte e l’AR è un bersaglio diretto a valle tramite la regolazione dell’espressione genica del AR. Studi su esseri umani hanno anche dimostrato che questo si verifica anche nell’uomo, con l’attività della STAT5 correlata positivamente all’espressione AR nelle linee cellulari del cancro alla prostata. [337]
Nella prossima parte di questa serie di articoli, parlerò dei modi in cui è possibile tentare di garantire che la via JAK-STAT5b-AR sia massimamente sensibilizzata, assicurando così che il potenziale ipertrofico sia potenziato quando Androgeni e GH vengono co-somministrati. Inoltre tratterò le applicazioni pratiche dell’uso del GH a fini ipertrofici esponendo infine le mie conclusioni in merito.
Stay tuned!
Gabriel Bellizzi
Riferimenti:
Yarasheski KE, Campbell JA, Smith K, Rennie MJ, Holloszy JO, Bier DM. Effect of growth hormone and resistance exercise on muscle growth in young men. Am J Physiol. 1992 Mar;262(3 Pt 1):E261-7.
Yarasheski KE, Zachwieja JJ, Campbell JA, Bier DM. Effect of growth hormone and resistance exercise on muscle growth and strength in older men. Am J Physiol. 1995 Feb;268(2 Pt 1):E268-76.
Fryburg DA. Insulin-like growth factor I exerts growth hormone- and insulin-like actions on human muscle protein metabolism. Am J Physiol. 1994 Aug;267(2 Pt 1):E331-6.
Ohlsson C, Mohan S, Sjögren K, Tivesten A, Isgaard J, Isaksson O, Jansson JO, Svensson J. The role of liver-derived insulin-like growth factor-I. Endocr Rev. 2009 Aug;30(5):494-535.
Wakelam MJ. The fusion of myoblasts. Biochem J. 1985 May 15;228(1):1-12. Review.
Crist DM, Peake GT, Egan PA, Waters DL. Body composition response to exogenous GH during training in highly conditioned adults. J Appl Physiol (1985). 1988 Aug;65(2):579-84.
Deyssig R, Frisch H, Blum WF, Waldhör T. Effect of growth hormone treatment on hormonal parameters, body composition and strength in athletes. Acta Endocrinol (Copenh). 1993 Apr;128(4):313-8.
Yarasheski KE, Zachweija JJ, Angelopoulos TJ, Bier DM. Short-term growth hormone treatment does not increase muscle protein synthesis in experienced weight lifters. J Appl Physiol (1985). 1993 Jun;74(6):3073-6.
Lange KH, Andersen JL, Beyer N, Isaksson F, Larsson B, Rasmussen MH, Juul A, Bülow J, Kjaer M. GH administration changes myosin heavy chain isoforms in skeletal muscle but does not augment muscle strength or hypertrophy, either alone or combined with resistance exercise training in healthy elderly men. J Clin Endocrinol Metab. 2002 Feb;87(2):513-23.
Ehrnborg C, Ellegård L, Bosaeus I, Bengtsson BA, Rosén T. Supraphysiological growth hormone: less fat, more extracellular fluid but uncertain effects on muscles in healthy, active young adults. Clin Endocrinol (Oxf). 2005 Apr;62(4):449-57.
Rudman D, Feller AG, Nagraj HS, Gergans GA, Lalitha PY, Goldberg AF, Schlenker RA, Cohn L, Rudman IW, Mattson DE. Effects of human growth hormone in men over 60 years old. N Engl J Med. 1990 Jul 5;323(1):1-6.
Taaffe DR, Pruitt L, Reim J, Hintz RL, Butterfield G, Hoffman AR, Marcus R. Effect of recombinant human growth hormone on the muscle strength response to resistance exercise in elderly men. J Clin Endocrinol Metab. 1994 Nov;79(5):1361-6.
Taaffe DR, Jin IH, Vu TH, Hoffman AR, Marcus R. Lack of effect of recombinant human growth hormone (GH) on muscle morphology and GH-insulin-like growth factor expression in resistance-trained elderly men. J Clin Endocrinol Metab. 1996 Jan;81(1):421-5.
Hennessey JV, Chromiak JA, DellaVentura S, Reinert SE, Puhl J, Kiel DP, Rosen CJ, Vandenburgh H, MacLean DB. Growth hormone administration and exercise effects on muscle fiber type and diameter in moderately frail older people. J Am Geriatr Soc. 2001 Jul;49(7):852-8.
West DW, Kujbida GW, Moore DR, Atherton P, Burd NA, Padzik JP, De Lisio M, Tang JE, Parise G, Rennie MJ, Baker SK, Phillips SM. Resistance exercise-induced increases in putative anabolic hormones do not enhance muscle protein synthesis or intracellular signalling in young men. J Physiol. 2009 Nov 1;587(Pt 21):5239-47.
West DW, Burd NA, Tang JE, Moore DR, Staples AW, Holwerda AM, Baker SK, Phillips SM. Elevations in ostensibly anabolic hormones with resistance exercise enhance neither training-induced muscle hypertrophy nor strength of the elbow flexors. J Appl Physiol (1985). 2010 Jan;108(1):60-7.
Fryburg DA, Jahn LA, Hill SA, Oliveras DM, Barrett EJ. Insulin and insulin-like growth factor-I enhance human skeletal muscle protein anabolism during hyperaminoacidemia by different mechanisms. J Clin Invest. 1995 Oct;96(4):1722-9
Butterfield GE, Thompson J, Rennie MJ, Marcus R, Hintz RL, Hoffman AR. Effect of rhGH and rhIGF-I treatment on protein utilization in elderly women. Am J Physiol. 1997 Jan;272(1 Pt 1):E94-9.
Friedlander AL, Butterfield GE, Moynihan S, Grillo J, Pollack M, Holloway L, Friedman L, Yesavage J, Matthias D, Lee S, Marcus R, Hoffman AR. One year of insulin-like growth factor I treatment does not affect bone density, body composition, or psychological measures in postmenopausal women. J Clin Endocrinol Metab. 2001 Apr;86(4):1496-503.
Consitt LA, Saneda A, Saxena G, List EO, Kopchick JJ. Mice overexpressing growth hormone exhibit increased skeletal muscle myostatin and MuRF1 with attenuation of muscle mass. Skelet Muscle. 2017 Sep 4;7(1):17.
Wolf E, Wanke R, Schenck E, Hermanns W, Brem G. Effects of growth hormone overproduction on grip strength of transgenic mice. Eur J Endocrinol. 1995 Dec;133(6):735-40.
Ho KY, Weissberger AJ. The antinatriuretic action of biosynthetic human growth hormone in man involves activation of the renin-angiotensin system. Metabolism. 1990 Feb;39(2):133-7.
Blazer-Yost BL, Cox M. Insulin-like growth factor 1 stimulates renal epithelial Na+ transport. Am J Physiol. 1988 Sep;255(3 Pt 1):C413-7.
Giordano M, DeFronzo RA. Acute effect of human recombinant insulin-like growth factor I on renal function in humans. Nephron. 1995;71(1):10-5.
Ehrnborg C, Lange KH, Dall R, Christiansen JS, Lundberg PA, Baxter RC, Boroujerdi MA, Bengtsson BA, Healey ML, Pentecost C, Longobardi S, Napoli R, Rosén T; GH-2000 Study Group. The growth hormone/insulin-like growth factor-I axis hormones and bone markers in elite athletes in response to a maximum exercise test. J Clin Endocrinol Metab. 2003 Jan;88(1):394-401.
Doessing S, Heinemeier KM, Holm L, Mackey AL, Schjerling P, Rennie M, Smith K, Reitelseder S, Kappelgaard AM, Rasmussen MH, Flyvbjerg A, Kjaer M. Growth hormone stimulates the collagen synthesis in human tendon and skeletal muscle without affecting myofibrillar protein synthesis. J Physiol. 2010 Jan 15;588(Pt 2):341-51.
Boesen AP, Dideriksen K, Couppé C, Magnusson SP, Schjerling P, Boesen M, Kjaer M, Langberg H. Tendon and skeletal muscle matrix gene expression and functional responses to immobilisation and rehabilitation in young males: effect of growth hormone administration. J Physiol. 2013 Dec 1;591(23):6039-52.
Cohn L, Feller AG, Draper MW, Rudman IW, Rudman D. Carpal tunnel syndrome and gynaecomastia during growth hormone treatment of elderly men with low circulating IGF-I concentrations. Clin Endocrinol (Oxf). 1993 Oct;39(4):417-25.
Sullivan DH, Carter WJ, Warr WR, Williams LH. Side effects resulting from the use of growth hormone and insulin-like growth factor-I as combined therapy to frail elderly patients. J Gerontol A Biol Sci Med Sci. 1998 May;53(3):M183-7.
Dickerman RD, Douglas JA, East JW. Bilateral median neuropathy and growth hormone use: a case report. Arch Phys Med Rehabil. 2000 Dec;81(12):1594-5.
Papadakis MA, Grady D, Black D, Tierney MJ, Gooding GA, Schambelan M, Grunfeld C. Growth hormone replacement in healthy older men improves body composition but not functional ability. Ann Intern Med. 1996 Apr 15;124(8):708-16.
Zachwieja JJ, Yarasheski KE. Does growth hormone therapy in conjunction with resistance exercise increase muscle force production and muscle mass in men and women aged 60 years or older? Phys Ther. 1999 Jan;79(1):76-82. Review.
Kishioka Y, Thomas M, Wakamatsu J, Hattori A, Sharma M, Kambadur R, Nishimura T. Decorin enhances the proliferation and differentiation of myogenic cells through suppressing myostatin activity. J Cell Physiol. 2008 Jun;215(3):856-67.
Kanzleiter T, Rath M, Görgens SW, Jensen J, Tangen DS, Kolnes AJ, Kolnes KJ, Lee S, Eckel J, Schürmann A, Eckardt K. The myokine decorin is regulated by contraction and involved in muscle hypertrophy. Biochem Biophys Res Commun. 2014 Jul 25;450(2):1089-94.
Zhang CZ, Li H, Bartold PM, Young WG, Waters MJ. Effect of growth hormone on the distribution of decorin and biglycan during odontogenesis in the rat incisor. J Dent Res. 1995 Oct;74(10):1636-43.
Bahl N, Stone G, McLean M, Ho KKY, Birzniece V. Decorin, a growth hormone regulated protein in humans. Eur J Endocrinol. 2017 Nov 14. pii: EJE-17-0844.
Short KR, Moller N, Bigelow ML, Coenen-Schimke J, Nair KS. Enhancement of muscle mitochondrial function by growth hormone. J Clin Endocrinol Metab. 2008 Feb;93(2):597-604. Epub 2007 Nov 13.
Lange KH, Isaksson F, Juul A, Rasmussen MH, Bülow J, Kjaer M. Growth hormone enhances effects of endurance training on oxidative muscle metabolism in elderly women. Am J Physiol Endocrinol Metab. 2000 Nov;279(5):E989-96.
Chargé SB, Rudnicki MA. Cellular and molecular regulation of muscle regeneration. Physiol Rev. 2004 Jan;84(1):209-38. Review.
Halevy O, Hodik V, Mett A. The effects of growth hormone on avian skeletal muscle satellite cell proliferation and differentiation. Gen Comp Endocrinol. 1996 Jan;101(1):43-52.
Kim H, Barton E, Muja N, Yakar S, Pennisi P, Leroith D. Intact insulin and insulin-like growth factor-I receptor signaling is required for growth hormone effects on skeletal muscle growth and function in vivo. Endocrinology. 2005 Apr;146(4):1772-9. Epub 2004 Dec 23.
Sinha-Hikim I, Roth SM, Lee MI, Bhasin S. Testosterone-induced muscle hypertrophy is associated with an increase in satellite cell number in healthy, young men. Am J Physiol Endocrinol Metab. 2003 Jul;285(1):E197-205. Epub 2003 Apr 1.
Ruzicka, L., Wettstein, A. and Kägi, H. (1935), Sexualhormone VIII. Darstellung von Testosteron unter Anwendung gemischter Ester. HCA, 18: 1478–1482.
Haupt HA, Rovere GD. Anabolic steroids: a review of the literature. Am J Sports Med. 1984 Nov-Dec;12(6):469-84. Review.
Shahidi NT. A review of the chemistry, biological action, and clinical applications of anabolic-androgenic steroids. Clin Ther. 2001 Sep;23(9):1355-90. Review.
Calof OM, Singh AB, Lee ML, Kenny AM, Urban RJ, Tenover JL, Bhasin S. Adverse events associated with testosterone replacement in middle-aged and older men: a meta-analysis of randomized, placebo-controlled trials. J Gerontol A Biol Sci Med Sci. 2005 Nov;60(11):1451-7
Hartgens F, Kuipers H. Effects of androgenic-anabolic steroids in athletes. Sports Med. 2004;34(8):513-54. Review.
Kadi F, Eriksson A, Holmner S, Thornell LE. Effects of anabolic steroids on the muscle cells of strength-trained athletes. Med Sci Sports Exerc. 1999 Nov;31(11):1528-34.
Herbst KL, Bhasin S. Testosterone action on skeletal muscle. Curr Opin Clin Nutr Metab Care. 2004 May;7(3):271-7. Review.
Eriksson A, Kadi F, Malm C, Thornell LE. Skeletal muscle morphology in power-lifters with and without anabolic steroids. Histochem Cell Biol. 2005 Aug;124(2):167-75.
Kadi F. Cellular and molecular mechanisms responsible for the action of testosterone on human skeletal muscle. A basis for illegal performance enhancement. Br J Pharmacol. 2008 Jun;154(3):522-8. Epub 2008 Apr 14. Review.
Bhasin S, Woodhouse L, Casaburi R, Singh AB, Bhasin D, Berman N, Chen X, Yarasheski KE, Magliano L, Dzekov C, Dzekov J, Bross R, Phillips J, Sinha-Hikim I, Shen R, Storer TW. Testosterone dose-response relationships in healthy young men. Am J Physiol Endocrinol Metab. 2001 Dec;281(6):E1172-81.
Woodhouse LJ, Reisz-Porszasz S, Javanbakht M, Storer TW, Lee M, Zerounian H, Bhasin S. Development of models to predict anabolic response to testosterone administration in healthy young men. Am J Physiol Endocrinol Metab. 2003 May;284(5):E1009-17.
Bhasin S, Woodhouse L, Casaburi R, Singh AB, Mac RP, Lee M, Yarasheski KE, Sinha-Hikim I, Dzekov C, Dzekov J, Magliano L, Storer TW. Older men are as responsive as young men to the anabolic effects of graded doses of testosterone on the skeletal muscle. J Clin Endocrinol Metab. 2005 Feb;90(2):678-88. Epub 2004 Nov 23.
Kvorning T, Andersen M, Brixen K, Madsen K. Suppression of endogenous testosterone production attenuates the response to strength training: a randomized, placebo-controlled, and blinded intervention study. Am J Physiol Endocrinol Metab. 2006 Dec;291(6):E1325-32. Epub 2006 Jul 25.
Sinha-Hikim I, Artaza J, Woodhouse L, Gonzalez-Cadavid N, Singh AB, Lee MI, Storer TW, Casaburi R, Shen R, Bhasin S. Testosterone-induced increase in muscle size in healthy young men is associated with muscle fiber hypertrophy. Am J Physiol Endocrinol Metab. 2002 Jul;283(1):E154-64.
Bhasin S, Storer TW, Berman N, Callegari C, Clevenger B, Phillips J, Bunnell TJ, Tricker R, Shirazi A, Casaburi R. The effects of supraphysiologic doses of testosterone on muscle size and strength in normal men. N Engl J Med. 1996 Jul 4;335(1):1-7.
Sinha-Hikim I, Taylor WE, Gonzalez-Cadavid NF, Zheng W, Bhasin S. Androgen receptor in human skeletal muscle and cultured muscle satellite cells: up-regulation by androgen treatment. J Clin Endocrinol Metab. 2004 Oct;89(10):5245-55.
Klover P, Chen W, Zhu BM, Hennighausen L. Skeletal muscle growth and fiber composition in mice are regulated through the transcription factors STAT5a/b: linking growth hormone to the androgen receptor. FASEB J. 2009 Sep;23(9):3140-8.
Sheffield-Moore M, Urban RJ, Wolf SE, Jiang J, Catlin DH, Herndon DN, Wolfe RR, Ferrando AA. Short-term oxandrolone administration stimulates net muscle protein synthesis in young men. J Clin Endocrinol Metab. 1999 Aug;84(8):2705-11.
Kadi F, Bonnerud P, Eriksson A, Thornell LE. The expression of androgen receptors in human neck and limb muscles: effects of training and self-administration of androgenic-anabolic steroids. Histochem Cell Biol. 2000 Jan;113(1):25-9.
de Rooy C, Grossmann M, Zajac JD, Cheung AS. Targeting muscle signaling pathways to minimize adverse effects of androgen deprivation. Endocr Relat Cancer. 2016 Jan;23(1):R15-26.
Brodsky IG, Balagopal P, Nair KS. Effects of testosterone replacement on muscle mass and muscle protein synthesis in hypogonadal men–a clinical research center study. J Clin Endocrinol Metab. 1996 Oct;81(10):3469-75.
Ferrando AA, Sheffield-Moore M, Paddon-Jones D, Wolfe RR, Urban RJ. Differential anabolic effects of testosterone and amino acid feeding in older men. J Clin Endocrinol Metab. 2003 Jan;88(1):358-62.
Bauer ER, Daxenberger A, Petri T, Sauerwein H, Meyer HH. Characterisation of the affinity of different anabolics and synthetic hormones to the human androgen receptor, human sex hormone binding globulin and to the bovine progestin receptor. APMIS. 2000 Dec;108(12):838-46.
Singh R, Artaza JN, Taylor WE, Gonzalez-Cadavid NF, Bhasin S. Androgens stimulate myogenic differentiation and inhibit adipogenesis in C3H 10T1/2 pluripotent cells through an androgen receptor-mediated pathway. Endocrinology. 2003 Nov;144(11):5081-8. Epub 2003 Jul 24.
Yarrow JF, McCoy SC, Borst SE. Tissue selectivity and potential clinical applications of trenbolone (17beta-hydroxyestra-4,9,11-trien-3-one): A potent anabolic steroid with reduced androgenic and estrogenic activity. Steroids. 2010 Jun;75(6):377-89.
Mulholland DJ, Dedhar S, Coetzee GA, Nelson CC. Interaction of nuclear receptors with the Wnt/beta-catenin/Tcf signaling axis: Wnt you like to know? Endocr Rev. 2005 Dec;26(7):898-915. Epub 2005 Aug 26. Review.
Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006 Nov 3;127(3):469-80. Review.
Singh R, Bhasin S, Braga M, Artaza JN, Pervin S, Taylor WE, Krishnan V, Sinha SK, Rajavashisth TB, Jasuja R. Regulation of myogenic differentiation by androgens: cross talk between androgen receptor/ beta-catenin and follistatin/transforming growth factor-beta signaling pathways. Endocrinology. 2009 Mar;150(3):1259-68.
Zhao JX, Hu J, Zhu MJ, Du M. Trenbolone enhances myogenic differentiation by enhancing β-catenin signaling in muscle-derived stem cells of cattle. Domest Anim Endocrinol. 2011 May;40(4):222-9.
Armstrong DD, Esser KA. Wnt/beta-catenin signaling activates growth-control genes during overload-induced skeletal muscle hypertrophy. Am J Physiol Cell Physiol. 2005 Oct;289(4):C853-9. Epub 2005 May 11.
Takada I, Kouzmenko AP, Kato S. Wnt and PPARgamma signaling in osteoblastogenesis and adipogenesis. Nat Rev Rheumatol. 2009 Aug;5(8):442-7.
Cossu G, Borello U. Wnt signaling and the activation of myogenesis in mammals. EMBO J. 1999 Dec 15;18(24):6867-72. Review.
Buckingham M. Skeletal muscle formation in vertebrates. Curr Opin Genet Dev. 2001 Aug;11(4):440-8. Review.
Polesskaya A, Seale P, Rudnicki MA. Wnt signaling induces the myogenic specification of resident CD45+ adult stem cells during muscle regeneration. Cell. 2003 Jun 27;113(7):841-52.
Tincello DG, Saunders PT, Hodgins MB, Simpson NB, Edwards CR, Hargreaves TB, Wu FC. Correlation of clinical, endocrine and molecular abnormalities with in vivo responses to high-dose testosterone in patients with partial androgen insensitivity syndrome. Clin Endocrinol (Oxf). 1997 Apr;46(4):497-506.
Foradori CD, Weiser MJ, Handa RJ. Non-genomic Actions of Androgens. Frontiers in neuroendocrinology. 2008;29(2):169-181.
Lucas-Herald AK, Alves-Lopes R, Montezano AC, Ahmed SF, Touyz RM. Genomic and non-genomic effects of androgens in the cardiovascular system: clinical implications. Clin Sci (Lond). 2017 Jul 1;131(13):1405-1418.
Münzer T, Harman SM, Hees P, Shapiro E, Christmas C, Bellantoni MF, Stevens TE, O’Connor KG, Pabst KM, St Clair C, Sorkin JD, Blackman MR. Effects of GH and/or sex steroid administration on abdominal subcutaneous and visceral fat in healthy aged women and men. J Clin Endocrinol Metab. 2001 Aug;86(8):3604-10.
Sattler FR, Castaneda-Sceppa C, Binder EF, Schroeder ET, Wang Y, Bhasin S, Kawakubo M, Stewart Y, Yarasheski KE, Ulloor J, Colletti P, Roubenoff R, Azen SP. Testosterone and growth hormone improve body composition and muscle performance in older men. J Clin Endocrinol Metab. 2009 Jun;94(6):1991-2001.
Illig R, Prader A. Effect of testosterone on growth hormone secretion in patients with anorchia and delayed puberty. J Clin Endocrinol Metab. 1970 May;30(5):615-8.
Pfeilschifter J, Scheidt-Nave C, Leidig-Bruckner G, Woitge HW, Blum WF, Wüster C, Haack D, Ziegler R. Relationship between circulating insulin-like growth factor components and sex hormones in a population-based sample of 50- to 80-year-old men and women. J Clin Endocrinol Metab. 1996 Jul;81(7):2534-40.
Erfurth EM, Hagmar LE, Sääf M, Hall K. Serum levels of insulin-like growth factor I and insulin-like growth factor-binding protein 1 correlate with serum free testosterone and sex hormone binding globulin levels in healthy young and middle-aged men. Clin Endocrinol (Oxf). 1996 Jun;44(6):659-64.
van Kesteren P, Lips P, Deville W, Popp-Snijders C, Asscheman H, Megens J, Gooren L. The effect of one-year cross-sex hormonal treatment on bone metabolism and serum insulin-like growth factor-1 in transsexuals. J Clin Endocrinol Metab. 1996 Jun;81(6):2227-32.
Veldhuis JD, Keenan DM, Mielke K, Miles JM, Bowers CY. Testosterone supplementation in healthy older men drives GH and IGF-I secretion without potentiating peptidyl secretagogue efficacy. Eur J Endocrinol. 2005 Oct;153(4):577-86.
Lewis MI, Fournier M, Storer TW, Bhasin S, Porszasz J, Ren SG, Da X, Casaburi R. Skeletal muscle adaptations to testosterone and resistance training in men with COPD. J Appl Physiol (1985). 2007 Oct;103(4):1299-310.
Mauras N, Hayes V, Welch S, Rini A, Helgeson K, Dokler M, Veldhuis JD, Urban RJ. Testosterone deficiency in young men: marked alterations in whole body protein kinetics, strength, and adiposity. J Clin Endocrinol Metab. 1998 Jun;83(6):1886-92.
Bondanelli M, Ambrosio MR, Margutti A, Franceschetti P, Zatelli MC, degli Uberti EC. Activation of the somatotropic axis by testosterone in adult men: evidence for a role of hypothalamic growth hormone-releasing hormone. Neuroendocrinology. 2003 Jun;77(6):380-7.
Veldhuis JD, Metzger DL, Martha PM Jr, Mauras N, Kerrigan JR, Keenan B, Rogol AD, Pincus SM. Estrogen and testosterone, but not a nonaromatizable androgen, direct network integration of the hypothalamo-somatotrope (growth hormone)-insulin-like growth factor I axis in the human: evidence from pubertal pathophysiology and sex-steroid hormone replacement. J Clin Endocrinol Metab. 1997 Oct;82(10):3414-20.
Weissberger AJ, Ho KK. Activation of the somatotropic axis by testosterone in adult males: evidence for the role of aromatization. J Clin Endocrinol Metab. 1993 Jun;76(6):1407-12.
Veldhuis JD, Mielke KL, Cosma M, Soares-Welch C, Paulo R, Miles JM, Bowers CY. Aromatase and 5alpha-reductase inhibition during an exogenous testosterone clamp unveils selective sex steroid modulation of somatostatin and growth hormone secretagogue actions in healthy older men. J Clin Endocrinol Metab. 2009 Mar;94(3):973-81.
Yamamoto T, Sakai C, Yamaki J, Takamori K, Yoshiji S, Kitawaki J, Fujii M, Yasuda J, Honjo H, Okada H. Estrogen biosynthesis in human liver–a comparison of aromatase activity for C-19 steroids in fetal liver, adult liver and hepatoma tissues of human subjects. Endocrinol Jpn. 1984 Jun;31(3):277-81.
Hata S, Miki Y, Saito R, Ishida K, Watanabe M, Sasano H. Aromatase in human liver and its diseases. Cancer Med. 2013 Jun;2(3):305-15.
Riggs BL, Hartmann LC. Selective estrogen-receptor modulators — mechanisms of action and application to clinical practice. N Engl J Med. 2003 Feb 13;348(7):618-29. Review. Erratum in: N Engl J Med. 2003 Mar 20;348(12):1192.
Löfgren L, Wallberg B, Wilking N, Fornander T, Rutqvist LE, Carlström K, von Schoultz B, von Schoultz E. Tamoxifen and megestrol acetate for postmenopausal breast cancer: diverging effects on liver proteins, androgens, and glucocorticoids. Med Oncol. 2004;21(4):309-18.
Centrella M, McCarthy TL, Chang WZ, Labaree DC, Hochberg RB. Estren (4-estren-3alpha,17beta-diol) is a prohormone that regulates both androgenic and estrogenic transcriptional effects through the androgen receptor. Mol Endocrinol. 2004 May;18(5):1120-30.
Yu YM, Domené HM, Sztein J, Counts DR, Cassorla F. Developmental changes and differential regulation by testosterone and estradiol of growth hormone receptor expression in the rabbit. Eur J Endocrinol. 1996 Nov;135(5):583-90.
Zung A, Phillip M, Chalew SA, Palese T, Kowarski AA, Zadik Z. Testosterone effect on growth and growth mediators of the GH-IGF-I axis in the liver and epiphyseal growth plate of juvenile rats. J Mol Endocrinol. 1999 Oct;23(2):209-21.
Hayes VY, Urban RJ, Jiang J, Marcell TJ, Helgeson K, Mauras N. Recombinant human growth hormone and recombinant human insulin-like growth factor I diminish the catabolic effects of hypogonadism in man: metabolic and molecular effects. J Clin Endocrinol Metab. 2001 May;86(5):2211-9.
Sculthorpe N, Solomon AM, Sinanan AC, Bouloux PM, Grace F, Lewis MP. Androgens affect myogenesis in vitro and increase local IGF-1 expression. Med Sci Sports Exerc. 2012 Apr;44(4):610-5.
Birzniece V, Meinhardt UJ, Umpleby MA, Handelsman DJ, Ho KK. Interaction between testosterone and growth hormone on whole-body protein anabolism occurs in the liver. J Clin Endocrinol Metab. 2011 Apr;96(4):1060-7.
Mertani HC, Delehaye-Zervas MC, Martini JF, Postel-Vinay MC, Morel G. Localization of growth hormone receptor messenger RNA in human tissues. Endocrine. 1995 Feb;3(2):135-42.
Florini JR, Ewton DZ, Coolican SA. Growth hormone and the insulin-like growth factor system in myogenesis. Endocr Rev. 1996 Oct;17(5):481-517. Review.
Urban RJ, Bodenburg YH, Gilkison C, Foxworth J, Coggan AR, Wolfe RR, Ferrando A. Testosterone administration to elderly men increases skeletal muscle strength and protein synthesis. Am J Physiol. 1995 Nov;269(5 Pt 1):E820-6.
Ewton DZ, Coolican SA, Mohan S, Chernausek SD, Florini JR. Modulation of insulin-like growth factor actions in L6A1 myoblasts by insulin-like growth factor binding protein (IGFBP)-4 and IGFBP-5: a dual role for IGFBP-5. J Cell Physiol. 1998 Oct;177(1):47-57.
Venken K, Movérare-Skrtic S, Kopchick JJ, Coschigano KT, Ohlsson C, Boonen S, Bouillon R, Vanderschueren D. Impact of androgens, growth hormone, and IGF-I on bone and muscle in male mice during puberty. J Bone Miner Res. 2007 Jan;22(1):72-82.
Serra C, Bhasin S, Tangherlini F, Barton ER, Ganno M, Zhang A, Shansky J, Vandenburgh HH, Travison TG, Jasuja R, Morris C. The role of GH and IGF-I in mediating anabolic effects of testosterone on androgen-responsive muscle. Endocrinology. 2011 Jan;152(1):193-206.
Spangenburg EE, Le Roith D, Ward CW, Bodine SC. A functional insulin-like growth factor receptor is not necessary for load-induced skeletal muscle hypertrophy. J Physiol. 2008 Jan 1;586(1):283-91.
Yoshizawa A, Clemmons DR. Testosterone and insulin-like growth factor (IGF) I interact in controlling IGF-binding protein production in androgen-responsive foreskin fibroblasts. J Clin Endocrinol Metab. 2000 Apr;85(4):1627-33.
Gayan-Ramirez G, Rollier H, Vanderhoydonc F, Verhoeven G, Gosselink R, Decramer M. Nandrolone decanoate does not enhance training effects but increases IGF-I mRNA in rat diaphragm. J Appl Physiol (1985). 2000 Jan;88(1):26-34.
Lewis MI, Horvitz GD, Clemmons DR, Fournier M. Role of IGF-I and IGF-binding proteins within diaphragm muscle in modulating the effects of nandrolone. Am J Physiol Endocrinol Metab. 2002 Feb;282(2):E483-90
Salmons S. Myotrophic effects of an anabolic steroid in rabbit limb muscles. Muscle Nerve. 1992 Jul;15(7):806-12.
Bisschop A, Gayan-Ramirez G, Rollier H, Dekhuijzen PN, Dom R, de Bock V, Decramer M. Effects of nandrolone decanoate on respiratory and peripheral muscles in male and female rats. J Appl Physiol (1985). 1997 Apr;82(4):1112-8
Lewis MI, Fournier M, Yeh AY, Micevych PE, Sieck GC. Alterations in diaphragm contractility after nandrolone administration: an analysis of potential mechanisms. J Appl Physiol (1985). 1999 Mar;86(3):985-92.
Heitzman RJ. The effectiveness of anabolic agents in increasing rate of growth in farm animals; report on experiments in cattle. Environ Qual Saf Suppl. 1976;(5):89-98. Review.
Buttery, P., Vernon, B., & Pearson, J. (1978). Anabolic agents—some thoughts on their mode of action. Proceedings of the Nutrition Society, 37(3), 311-315.
Hongerholt DD, Crooker BA, Wheaton JE, Carlson KM, Jorgenson DM. Effects of a growth hormone-releasing factor analogue and an estradiol-trenbolone acetate implant on somatotropin, insulin-like growth factor I, and metabolite profiles in growing Hereford steers. J Anim Sci. 1992 May;70(5):1439-48.
Tan RS, Scally MC. Anabolic steroid-induced hypogonadism–towards a unified hypothesis of anabolic steroid action. Med Hypotheses. 2009 Jun;72(6):723-8.
Kamanga-Sollo E, White ME, Hathaway MR, Weber WJ, Dayton WR. Effect of Estradiol-17beta on protein synthesis and degradation rates in fused bovine satellite cell cultures. Domest Anim Endocrinol. 2010 Jul;39(1):54-62.
Kamanga-Sollo E, Thornton KJ, White ME, Dayton WR. Role of G protein-coupled estrogen receptor-1, matrix metalloproteinases 2 and 9, and heparin binding epidermal growth factor-like growth factor in estradiol-17β-stimulated bovine satellite cell proliferation. Domest Anim Endocrinol. 2014 Oct;49:20-6.
Dunn JD, Johnson BJ, Kayser JP, Waylan AT, Sissom EK, Drouillard JS. Effects of flax supplementation and a combined trenbolone acetate and estradiol implant on circulating insulin-like growth factor-I and muscle insulin-like growth factor-I messenger RNA levels in beef cattle. J Anim Sci. 2003 Dec;81(12):3028-34.
Pampusch MS, Johnson BJ, White ME, Hathaway MR, Dunn JD, Waylan AT, Dayton WR. Time course of changes in growth factor mRNA levels in muscle of steroid-implanted and nonimplanted steers. J Anim Sci. 2003 Nov;81(11):2733-40.
Pampusch MS, White ME, Hathaway MR, Baxa TJ, Chung KY, Parr SL, Johnson BJ, Weber WJ, Dayton WR. Effects of implants of trenbolone acetate, estradiol, or both, on muscle insulin-like growth factor-I, insulin-like growth factor-I receptor, estrogen receptor-{alpha}, and androgen receptor messenger ribonucleic acid levels in feedlot steers. J Anim Sci. 2008 Dec;86(12):3418-23.
Thompson SH, Boxhorn LK, Kong WY, Allen RE. Trenbolone alters the responsiveness of skeletal muscle satellite cells to fibroblast growth factor and insulin-like growth factor I. Endocrinology. 1989 May;124(5):2110-7.
Johnson BJ, Halstead N, White ME, Hathaway MR, DiCostanzo A, Dayton WR. Activation state of muscle satellite cells isolated from steers implanted with a combined trenbolone acetate and estradiol implant. J Anim Sci. 1998 Nov;76(11):2779-86.
Dalbo VJ, Roberts MD, Mobley CB, Ballmann C, Kephart WC, Fox CD, Santucci VA, Conover CF, Beggs LA, Balaez A, Hoerr FJ, Yarrow JF, Borst SE, Beck DT. Testosterone and trenbolone enanthate increase mature myostatin protein expression despite increasing skeletal muscle hypertrophy and satellite cell number in rodent muscle. Andrologia. 2017 Apr;49(3).
Bhasin S, He EJ, Kawakubo M, Schroeder ET, Yarasheski K, Opiteck GJ, Reicin A, Chen F, Lam R, Tsou JA, Castaneda-Sceppa C, Binder EF, Azen SP, Sattler FR. N-terminal propeptide of type III procollagen as a biomarker of anabolic response to recombinant human GH and testosterone. J Clin Endocrinol Metab. 2009 Nov;94(11):4224-33.
Nelson AE, Meinhardt U, Hansen JL, Walker IH, Stone G, Howe CJ, Leung KC, Seibel MJ, Baxter RC, Handelsman DJ, Kazlauskas R, Ho KK. Pharmacodynamics of growth hormone abuse biomarkers and the influence of gender and testosterone: a randomized double-blind placebo-controlled study in young recreational athletes. J Clin Endocrinol Metab. 2008 Jun;93(6):2213-22.
Holt RI. Detecting growth hormone misuse in athletes. Indian J Endocrinol Metab. 2013 Oct;17(Suppl 1):S18-22.
Tan SH, Lee A, Pascovici D, Care N, Birzniece V, Ho K, Molloy MP, Khan A. Plasma biomarker proteins for detection of human growth hormone administration in athletes. Sci Rep. 2017 Aug 30;7(1):10039.
Jørgensen JO, Jessen N, Pedersen SB, Vestergaard E, Gormsen L, Lund SA, Billestrup N. GH receptor signaling in skeletal muscle and adipose tissue in human subjects following exposure to an intravenous GH bolus. Am J Physiol Endocrinol Metab. 2006 Nov;291(5):E899-905.
Liu X, Robinson GW, Gouilleux F, Groner B, Hennighausen L. Cloning and expression of Stat5 and an additional homologue (Stat5b) involved in prolactin signal transduction in mouse mammary tissue. Proc Natl Acad Sci U S A. 1995 Sep 12;92(19):8831-5.
Hennighausen L, Robinson GW. Interpretation of cytokine signaling through the transcription factors STAT5A and STAT5B. Genes Dev. 2008 Mar 15;22(6):711-21.
Eshet R, Laron Z, Pertzelan A, Arnon R, Dintzman M. Defect of human growth hormone receptors in the liver of two patients with Laron-type dwarfism. Isr J Med Sci. 1984 Jan;20(1):8-11.
Teglund S, McKay C, Schuetz E, van Deursen JM, Stravopodis D, Wang D, Brown M, Bodner S, Grosveld G, Ihle JN. Stat5a and Stat5b proteins have essential and nonessential, or redundant, roles in cytokine responses. Cell. 1998 May 29;93(5):841-50.
Hwa V, Little B, Adiyaman P, Kofoed EM, Pratt KL, Ocal G, Berberoglu M, Rosenfeld RG. Severe growth hormone insensitivity resulting from total absence of signal transducer and activator of transcription 5b. J Clin Endocrinol Metab. 2005 Jul;90(7):4260-6. Epub 2005 Apr 12.
Rowland JE, Lichanska AM, Kerr LM, White M, d’Aniello EM, Maher SL, Brown R, Teasdale RD, Noakes PG, Waters MJ. In vivo analysis of growth hormone receptor signaling domains and their associated transcripts. Mol Cell Biol. 2005 Jan;25(1):66-77. Erratum in: Mol Cell Biol. 2005 Mar;25(5):2072.
Klover P, Hennighausen L. Postnatal body growth is dependent on the transcription factors signal transducers and activators of transcription 5a/b in muscle: a role for autocrine/paracrine insulin-like growth factor I. Endocrinology. 2007 Apr;148(4):1489-97.
Barclay JL, Kerr LM, Arthur L, Rowland JE, Nelson CN, Ishikawa M, d’Aniello EM, White M, Noakes PG, Waters MJ. In vivo targeting of the growth hormone receptor (GHR) Box1 sequence demonstrates that the GHR does not signal exclusively through JAK2. Mol Endocrinol. 2010 Jan;24(1):204-17.
Hwa V, Nadeau K, Wit JM, Rosenfeld RG. STAT5b deficiency: lessons from STAT5b gene mutations. Best Pract Res Clin Endocrinol Metab. 2011 Feb;25(1):61-75.
Varco-Merth B, Feigerlová E, Shinde U, Rosenfeld RG, Hwa V, Rotwein P. Severe growth deficiency is associated with STAT5b mutations that disrupt protein folding and activity. Mol Endocrinol. 2013 Jan;27(1):150-61.
Davey HW, Xie T, McLachlan MJ, Wilkins RJ, Waxman DJ, Grattan DR. STAT5b is required for GH-induced liver IGF-I gene expression. Endocrinology. 2001 Sep;142(9):3836-41.
Woelfle J, Chia DJ, Rotwein P. Mechanisms of growth hormone (GH) action. Identification of conserved Stat5 binding sites that mediate GH-induced insulin-like growth factor-I gene activation. J Biol Chem. 2003 Dec 19;278(51):51261-6. Epub 2003 Oct 7.
Woelfle J, Billiard J, Rotwein P. Acute control of insulin-like growth factor-I gene transcription by growth hormone through Stat5b. J Biol Chem. 2003 Jun 20;278(25):22696-702. Epub 2003 Apr 7.
MacLean HE, Chiu WS, Notini AJ, Axell AM, Davey RA, McManus JF, Ma C, Plant DR, Lynch GS, Zajac JD. Impaired skeletal muscle development and function in male, but not female, genomic androgen receptor knockout mice. FASEB J. 2008 Aug;22(8):2676-89.
Tan SH, Dagvadorj A, Shen F, Gu L, Liao Z, Abdulghani J, Zhang Y, Gelmann EP, Zellweger T, Culig Z, Visakorpi T, Bubendorf L, Kirken RA, Karras J, Nevalainen MT. Transcription factor Stat5 synergizes with androgen receptor in prostate cancer cells. Cancer Res. 2008 Jan 1;68(1):236-48.
de Souza GL, Hallak J. Anabolic steroids and male infertility: a comprehensive review. BJU Int. 2011 Dec;108(11):1860-5.
Se non avete ancora letto la prima parte di questa serie di articoli vi invito a farlo prima di procedere con la lettura di questa seconda parte: 1° Parte.
Informazioni base sull’IGF-1
IGF-1
Nella prima parte di questa serie di articoli ho esposto la storia e cronologia legate alla scoperta del GH e dll’IGF-1. Tuttavia, vorrei approfondire l’argomento riguardante i membri della famiglia dei fattori di crescita insulino-similimi, esponendo le loro caratteristiche e i loro ruoli nel processo ipertrofico. Gli IGF sono una famiglia di peptidi, in gran parte dipendenti dal GH, che mediano molte delle azioni di stimolo della crescita date dal GH.[121] Il fegato è il principale responsabile di tutta la produzione endocrina di IGF-1, con circa il 75% della sintesi a carico epatico sotto la regolazione del GH.[83,122-123] Ciò presuppone che ci sia un apporto macro-calorico sufficiente e livelli elevati di Insulina portale.[124-125] La sintesi autocrina di IGF-1 è anche regolata dal GH, in aggiunta ad altri fattori autocrini dipendenti dal tessuto.[126-128]
IGF-2
Alla famiglia degli IGF appartengono oltre dieci proteine strutturalmente simili tra cui IGF-1, IGF-2, Insulina, Relaxina e Pro-Insulina.[129] Sono tutti altamente omologhi sia nella struttura che nella funzione e gli effetti metabolici dell’IGF-1 sono stati definiti “insulino-simili” proprio a causa delle somiglianze e dei percorsi che condividono l’uno con l’altro. L’IGF-1 ha un’omologia di sequenza aminoacidica superiore al 50% con l’Insulina e il recettore del IGF-1 ha un’omologia della sequenza aminoacidica del 60% con il recettore dell’Insulina.[121,130-131] Gli evoluzionisti suppongono che la somiglianze nella struttura di questi peptidi sia dovuta al fatto che essi si siano evoluti da una singola molecola precursore trovata in vertebrati di oltre 60 milioni di anni fa.[132] Sia il rilascio di IGF-1 che quello di Insulina sono stimolati dall’assunzione di cibo, rilascio che viene inibito in condizioni di digiuno.[83]
Recettore del IGF-1
A causa di queste somiglianze strutturali, i membri della famiglia IGF possono spesso legarsi con i recettori nativi in modo “incrociato”.[133] Per riassumere brevemente queste relazioni di legame, la molecola di IGF-1 si lega con il recettore del IGF-1 con un elevata affinità, tuttavia sia l’IGF-2 che l’Insulina possono legarsi al recettore del IGF-1, ma con una affinità significativamente inferiori. L’IGF-2 si lega al recettore del IGF-2 con una elevata affinità, e l’IGF-1 si lega a questo recettore con un’affinità inferiore mentre l’Insulina non presenta alcun legame con esso.
Adipocita
La famiglia dei recettori IGF ha densità che variano significativamente in base ai tipi di cellule in cui sono presenti.[132] Questo è uno dei motivi per cui l’Insulina e l’IGF-1 possono avere diverse azioni metaboliche nonostante siano strutturalmente simili. Cellule come gli epatociti e gli adipociti hanno molti più recettori dell’Insulina rispetto ai recettori del IGF-1. Al contrario, le cellule muscolari lisce vascolari situate nei vasi sanguigni hanno un numero significativamente più elevato di recettori del IGF-1 rispetto ai recettori dell’Insulina.
Poiché ho già fatto un’analisi approfondita in precedenza sulle basi chimiche che si verificano durante l’attivazione del GHR, non mi ripeterò su tale questione. Però, è necessario comprendere che la famiglia dei recettori IGF è anche attivata dalla Tirosina Chinasi che, come ora sappiamo, porta alla fosforilazione dei substrati, all’attivazione delle vie cellulari e infine all’espressione genica e alla sintesi proteica.[121] L’attivazione del recettore del IGF-1 sembra essere indipendente dall’isoforma da cui è stato prodotto l’IGF-1. Inoltre, si noti che entrambi i tipi di recettori IGF sono stati trovati nelle cellule muscolari umane.[134]
I livelli serici di IGF-1 sono stabili negli adulti sani e vi sono poche variazioni da un giorno all’altro, o anche da una settimana all’altra. In effetti, osservare i livelli serici di IGF-1 di un individuo può essere un indicatore abbastanza affidabile di eventuali problemi di sensibilità al GH in relazione ad intervalli ben definiti, corretti per età e sesso.[135] Naturalmente, quando si cerca di decidere se esistono reali problemi di sensibilità, è necessario prendere in considerazione aspetti come lo stato nutrizionale generale dell’individuo e la salute epatica.
IGFBP-1
Nel flusso sanguineo, l’IGF-1 esiste principalmente in forma legata a proteine leganti l’IGF (IGFBP). La superfamiglia IGFBP comprende sei proteine ad alta affinità che vanno dal IGFBP-1 al IGFBP-6, nonché un certo numero di proteine a bassa affinità denominate proteine legate all’IGFBP.[136] Quasi il 95% di tutto l’IGF-1 circolante esiste in forma legata, con circa il 75% legato specificamente con l’IGFBP-3.[137] Una piccola frazione di IGF-1 (normalmente inferiore al 5%) può anche esistere in forma libera, e queste molecole non legate agiscono come regolatore negativo della secrezione di GH.[104] Gli IGFBP possono legarsi con l’IGF-1 e l’IGF-2, ma non con l’insulina. [138]
Azioni regolatorie delle proteine leganti l’IGF-1
L’IGF-1 legato esiste più comunemente in un complesso ternario da 150-kDa mentre è nel circolo ematico. Questo complesso ternario è costituito da una molecola di IGF-1, dal IGFBP-3 e dalla subunità labile acida (ALS) – sebbene possa esistere in un complesso binario con altri IGFBP.[139-140] Questi complessi servono a scopi come l’aumento della biodisponibilità degli IGF circolanti, estendendo la loro emivita serica, trasportando gli IGF alle cellule bersaglio e modulando l’interazione degli IGF con i loro rispettivi recettori di membrana posti sulla superficie delle cellule.[141-144] Ad esempio, nel plasma, il complesso ternario stabilizza IGF-1, aumentando significativamente la sua emivita da meno di 5 minuti a oltre 16 ore in alcuni casi.[137]
Gli IGFB sembrano normalmente inibire l’azione degli IGF, e questo perché competono con i recettori IGF per l’affinità di legame con gli IGF.[145] Tuttavia, non è sempre così, poiché gli IGFBP sono anche in grado di potenziare le azioni dell’IGF, potenzialmente facilitando la consegna dell’IGF al recettore.[146] Sebbene esista un’interazione piuttosto complessa, basti ricordare che il ruolo principale degli IGFBP è quello di trasportare gli IGF dal flusso ematico ai tessuti periferici. Una volta che ciò è avvenuto, gli IGFBP vengono rilasciati dai complessi binari e ternari mediante proteolisi o tramite legame alla matrice extracellulare del recettore del IGF-1.[147] Una volta rilasciate, le molecole di IGF-1 diventano libere, attive e possono quindi esplicare la loro azione.[137,143]
Una volta nei tessuti, gli IGFBP modulano le azioni dell’IGF in quanto hanno una maggiore affinità per il sito recettore rispetto all’IGF stesso [148], tuttavia essi possono anche esercitare effetti indipendenti dall’IGF.[149] Alcuni degli effetti diretti del IGFBP che sono già stati chiariti includono l’inibizione della crescita, l’induzione diretta dell’apoptosi e la modulazione degli effetti dei fattori di crescita non-IGF.[121]
È anche noto che lo splicing alternativo del gene IGF-1 produce tre isoforme distinte nell’uomo che hanno sia azioni dirette che indirette che contribuiscono agli effetti di promozione della crescita del IGF-1.[150-151] Sebbene non siano richieste per la secrezione di IGF-1, queste isoforme possono aumentare la reale biodisponibilità del IGF-1 al suo sito recettore.[437] Le tre isoforme sono denominate IGF-1Ea, IGF-1Eb e IGF-1Ec. E’ corretto sottolineare il fatto che roditori e pesci posseggono solo due isoforme, e in questo articolo ci si riferirà solo alle isoforme umane, a meno che non sia chiaramente indicato diversamente, per spera in questo modo di creare meno confusione al lettore.
L’IGF-1Ea è simile all’isoforma IGF principale espressa dagli epatociti e ha l’esone 4 del gene IGF-1 maturo giuntato direttamente all’esone 6.[152] Si pensa che l’IGF-1Eb sia prevalentemente espresso nel fegato, ma il suo ruolo nei muscoli non è ancora del tutto chiaro.[153] Si estende più a valle dell’esone 5, ma solo i primi 17 aminoacidi di questa isoforma sono identici a quelli della variante finale dell’isoforma che tratterò a momenti[154]. Si ritiene che questa isoforma sia unica per i primati, e per l’uomo, in quanto non è stata trovata nei roditori o nei pesci.[155]
MGF
L’IGF-1Ec è anche chiamato Fattore di Crescita Maccanico (MGF) ed è chiamato così perché è espresso in risposta a tensione meccanica e stress.[156-157] Nella prima parte di questa serie di articoli ho riportato in breve come questi siano due dei principali meccanismi alla base del processo ipertrofico all’interno del muscolo scheletrico. Tratterò più nel dettaglio dell’MGF in seguito. Questa isoforma contiene parte dell’esone 5 giuntata all’esone 6 che si traduce in un frame-shift e questo dal mRNA è tradotto in una isoforma con una alternativa sequenza di 25 aminoacidi al C-terminale.[152] Nei roditore l’IGF-1Eb condivide un’alta omologia con l’MGF umano ed entrambi sono spesso usati in modo intercambiabile in letteratura.[158] Riporto ciò solamente perché può diventare forviante quando si esamina la letteratura scientifica trattante questa isoforma, specialmente quando si passa da modelli animali a umani.
L’MGF ha mostrato in modelli di colture cellulari di aumentare la proliferazione e la migrazione dei mioblasti, oltre ad essere coinvolto nell’attivazione delle cellule satelliti. Comunque sia, se ciò si verifichi in vivo è a tutt’oggi una fonte di contesa. Questo comportamento è stato visto anche in presenza dell’inattivazione del IGF-1, il che suggerisce che l’MGF abbia la capacità di operare indipendentemente dall’IGF-1 maturo.[438] Detto questo, tutte le isoforme di IGF-1 richiedono un recettore del IGF-1 funzionale per produrre effettivamente ipertrofia muscolare poiché tale recettore non viene influenzato in assenza di IGF-1 maturo.[439-440] La risposta effettiva al MGF si riscontra in un ambiente con un pool attivo di cellule satelliti, poiché i tessuti muscolari invecchiati sono normalmente in uno stato di dormienza. Infine, sebbene l’MGF iniettabile sia quasi una chimera nel ambiente del BodyBuilding, osservando la totalità del suo effetto sembra avere una minore attività muscolare rispetto all’IGF-1 maturo, quindi il suo valore intrinseco per i Bodybuilder può effettivamente essere stato sovrastimato.[442]
Somatopausa
Lo studio di soggetti anziani offre una prospettiva alquanto singolare dal momento che è noto come la sarcopenia, termine che sta ad indicare una degenerazione del tessuto muscolare, si manifesti con l’avanzare dell’età. È anche noto che i livelli di GH secreto e IGF-1 circolante diminuiscono gradualmente nel corso della vita dopo il picco della pubertà.[159-162] Il declino dei livelli ormonali è piuttosto marcato, con una diminuzione della secrezione di GH del 10-15% ogni decennio dopo i 20 anni.[163] Molti anni fa è stato suggerito che questi cambiamenti senescenti nella composizione corporea e nelle funzioni metaboliche sono direttamente correlati alla diminuzione dei livelli ormonali all’interno dell’Asse GH/IGF-1. La comunità scientifica ha coniato il termine “somatopausa” per descrivere questo fenomeno.[159,164]
In sintesi, i processi che caratterizzano l’”ipotesi della Somatopausa” [128] sono:
I cambiamenti nello stile di vita e le predisposizioni genetiche promuovono l’accumulo di grasso corporeo con l’avanzare dell’età;
Questa maggiore massa grassa aumenta la disponibilità di FFA e quindi induce insulino-resistenza;
Livelli elevati di Insulina sopprimono l’IGFBP-1 determinando un aumento relativo dei livelli di IGF-1 liberi;
Aumenti sistemici degli FFA, dell’Insulina e una soppressione del rilascio ipofisario di GH dato dal aumento del IGF-1, portano ad un ulteriore aumento della massa grassa;
Il GH endogeno viene eliminato più rapidamente nei soggetti con maggiore massa grassa
Come si può vedere, il grasso corporeo aumenta con l’avanzare dell’età, di conseguenza si verifica un peggioramento dell’insulino-resistenza seguita da un aumento dei livelli di Insulina che, attraverso la soppressione del IGFBP-1 e all’aumento del IGF-1 libero, porta alla soppressione della secrezione di GH, che a sua volta peggiora la composizione corporea (vedi aumento della percentuale di grasso). È piuttosto interessante vedere che la frequenza degli impulsi di GH rimane essenzialmente intatta. L’attenuazione legata all’età è in realtà solo una marcata riduzione dell’ampiezza dell’impulso insieme ad un aumento della secrezione di SRIF.[165]
I cambiamenti associati alla somatopausa sono molto simili a quelli osservati negi giovani adulti con deficit dell’Ormone della Crescita (GHD). Sebbene simili, le persone anziane non sono normalmente così gravemente colpite da questa condizione rispetto agli individui con GHD e la somatopause non è considerata uno stato patologico.[160,166-167] Esempi di alcuni dei cambiamenti associati alla somatopausa includono la riduzione della massa muscolare e ossea, riduzione della forza, diminuzione dell’attività fisica e della capacità cardiaca, aumento del grasso corporeo (in particolare nella regione viscerale) e deterioramento cognitivo.
A causa del desiderio di invertire i molti effetti dannosi legati all’invecchiamento, vi è una diffusa ipotesi secondo cui la somministrazione di GH possa essere d’aiuto come parte di un programma di terapia ormonale sostitutiva completa (HRT). Una revisione completa riguardante HRT e anziani va oltre lo scopo di questo articolo, ma quelli che sono interessati possono trovare una discussione recente sull’argomento qui.[168]
L’ormone della crescita migliora le prestazioni atletiche?
Ben Johnson alle Olimpiadi di Seul del 1988.
L’emergere del GH come farmaco per il miglioramento delle prestazioni (PED), al di fuori dei circoli ristretti del BodyBuilding, è in gran parte attribuito al rilascio del famigerato “Underground Steroid Handbook” nei primi anni ’80.[169] Successivamente, il GH ha colpito l’opinione pubblica quando il vincitore della medaglia d’oro delle Olimpiadi del 1988, Ben Johnson, ammise di usarlo insieme agli AAS dopo essere stato privato del suo titolo in seguito alla positività del test anti-doping al quale era stato sottoposto.[170] Durante quel periodo, si credeva comunemente che l’uso del GH avrebbe aumentato la massa muscolare e contemporaneamente migliorato gli aspetti della prestazione atletica.[171] E, come risposta a questa convinzione, nel 1989 il CIO ha vietato l’uso del GH definendolo un PED e inserendolo come parte di una nuova classe di farmaci dopanti denominata “ormoni peptidici e analoghi”. Ha vietato l’uso del GH nonostante in quel momento vi fosse la mancanza di un test legittimo per la rilevazione del rHGH.[172] Nonostante tutto ciò, la domanda rimane ancora: anche con le prove che suggeriscono l’uso decennale del GH nell’atletica competitiva, questo ormone apporta veramente un significativo miglioramento delle prestazioni?
Ci sono state in realtà più revisioni sistematiche che hanno tentato di rispondere a questa domanda, ma sfortunatamente sono state tutt’altro che conclusive per quanto riguarda gli effetti ergogenici del GH.[173-175] E nonostante vari scandali nel corso degli anni, così come la prevalenza d’uso del GH da parte di atleti professionisti, ci sono ancora pochissime prove cliniche che suggeriscono che il GH da solo abbia un impatto significativo sull’aumento delle prestazioni sia negli adulti sani che nei soggetti giovani.[173,176- 179]
Sono stati svolti soltanto pochi studi strettamente controllati che più direttamente hanno tentato di cercare il reale impatto del GH sulle prestazioni fisiche in soggetti sani e allenati. Probabilmente il più interessante di questi ha dimostrato che dosi sovrafisiologiche di GH abbinate agli AAS non hanno apportato miglioramenti significativi del VO2max, nella forza o nella potenza esplosiva misurata attraverso l’altezza del salto.[180] E’ stato notato un leggero miglioramento nella capacità di sprint anaerobico, che era più evidente negli uomini e specialmente in quelli trattati con GH e AAS. Considerando ciò, in un evento sportivo nel quale le frazioni di secondo potrebbero significare la differenza tra vincere e perdere, questo dato ha una certa importanza.
Passando in rassegna il corpo della letteratura scientifica, non si osservano miglioramenti delle prestazioni aerobiche tramite l’uso di GH. Ciò si riscontra sia nel caso in cui il GH viene somministrato a dosi fisiologiche a soggetti sani [181-182] sia nel caso in cui viene somministrato in dosi sovrafisiologiche.[180,183-184] La capacità aerobica non è inoltre influenzata dalla somministrazione acuta di GH prima dell’allenamento.[185] Tutti gli aumenti delle prestazioni aerobiche del GH sembrano essere mediati tramite gli Androgeni, e questo è ulteriormente supportato dai risultati di uno studio che dimostrano come gli ex utilizzatori di AAS esprimano un aumento del VO2max. Sebbene fossero passati alcuni mesi dal loro ultimo utilizzo di AAS, tale lasso di tempo non era sufficiente affinchè l’effetto “di coda” dell’uso di questa categoria di farmaci fosse esaurito.[186]
Soggetti anziani sani a cui erano state somministrate dosi combinate di Testosterone e GH, calibrate affinchè si raggiungesse una soglia ematica nel range dell’età giovanile, hanno sperimentato in qualche misura il miglioramento dell’equilibrio e delle prestazioni fisiche.[187] Questi miglioramenti delle prestazioni sembrano essere più pronunciati negli uomini e, comunque, sono marginali, anche con i trattamenti combinati.[188] Come è stato discusso in precedenza, le dosi soprafisiologiche di GH possono aumentare la capacità anaerobica.[180] Questo è un effetto osservato anche in alcune occasioni in cui i soggetti GHD sono stati trattati con GH, ripristinando i “normali” intervalli dell’ormone.[182,189]
L’Asse GH/IGF-1 può anche svolgere un ruolo nella regolazione del tono vascolare, o nel grado di costrizione rispetto allo stato di massima dilatazione dei vasi sanguigni, regolando in tal modo la resistenza periferico.[190-191] L’IGF-1 è stato identificato come un potente vasodilatatore, un effetto parzialmente mediato dall’aumento dell’emissione di ossido nitrico dall’endotelio, il tessuto che forma uno strato di cellule che rivestono organi come il cuore e i vasi linfatici.[192-194] Questo potenziale di aumento della capacità del flusso sanguigno ha una miriade di benefici ipotetici sugli sforzi atletici.
Per concludere, nonostante la mancanza di prove convincenti in letteratura [434], gli atleti usano spesso dosi e protocolli che non vengono replicati durante gli studi. Detto ciò, si dovrebbe comunque essere cauti prima di liquidare completamente il GH come composto per il miglioramento delle prestazioni basandosi semplicemente su ciò che, al momento, il corpo della letteratura scientifica ci suggerisce. È molto probabile che eserciti un contributo significativo negli atleti di alto livello, e potrebbe farlo attraverso mezzi diretti o indiretti.
Nella terza parte di questa serie di articoli mi occuperò degli effetti diretti e indiretti del GH e del IGF-1 nell’ipertrofia muscolare, dell’uso degli AAS in combinazione con GH e IGF-1 analizzando, infine, il loro potenziale sinergico con l’Asse GH/IGF-1.
Stay tuned!
Gabriel Bellizzi
Riferimenti:
Cohen P. Overview of the IGF-I system. Horm Res. 2006;65 Suppl 1:3-8. Epub 2006 Mar 2. Review.
Yakar S, Liu JL, Stannard B, Butler A, Accili D, Sauer B, LeRoith D. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc Natl Acad Sci U S A. 1999 Jun 22;96(13):7324-9.
Laron Z. Insulin-like growth factor 1 (IGF-1): a growth hormone. Molecular Pathology. 2001;54(5):311-316.
Wurzburger MI, Prelevic GM, Sönksen PH, Balint-Peric LA, Wheeler M. The effect of recombinant human growth hormone on regulation of growth hormone secretion and blood glucose in insulin-dependent diabetes. J Clin Endocrinol Metab. 1993 Jul;77(1):267-72.
Yakar S, Rosen CJ, Beamer WG, Ackert-Bicknell CL, Wu Y, Liu JL, Ooi GT, Setser J, Frystyk J, Boisclair YR, LeRoith D. Circulating levels of IGF-1 directly regulate bone growth and density. J Clin Invest. 2002 Sep;110(6):771-81.
D’Ercole AJ, Stiles AD, Underwood LE. Tissue concentrations of somatomedin C: further evidence for multiple sites of synthesis and paracrine or autocrine mechanisms of action. Proc Natl Acad Sci U S A. 1984 Feb;81(3):935-9.
Gosteli-Peter MA, Winterhalter KH, Schmid C, Froesch ER, Zapf J. Expression and regulation of insulin-like growth factor-I (IGF-I) and IGF-binding protein messenger ribonucleic acid levels in tissues of hypophysectomized rats infused with IGF-I and growth hormone. Endocrinology. 1994 Dec;135(6):2558-67.
Gunawardane K, Krarup Hansen T, Sandahl Christiansen J, et al. Normal Physiology of Growth Hormone in Adults. [Updated 2015 Nov 12]. In: De Groot LJ, Chrousos G, Dungan K, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.
Lu C, Lam HN, Menon RK. New members of the insulin family: regulators of metabolism, growth and now … reproduction. Pediatr Res. 2005 May;57(5 Pt 2):70R-73R.
Yakar S, Sun H, Zhao H, Pennisi P, Toyoshima Y, Setser J, Stannard B, Scavo L, Leroith D. Metabolic effects of IGF-I deficiency: lessons from mouse models. Pediatr Endocrinol Rev. 2005 Sep;3(1):11-9. Review.
Samani AA, Yakar S, LeRoith D, Brodt P. The role of the IGF system in cancer growth and metastasis: overview and recent insights. Endocr Rev. 2007 Feb;28(1):20-47. Epub 2006 Aug 24. Review.
Clemmons DR. Metabolic actions of insulin-like growth factor-I in normal physiology and diabetes. Endocrinol Metab Clin North Am. 2012 Jun;41(2):425-43, vii-viii.
Kim JJ, Accili D. Signalling through IGF-I and insulin receptors: where is the specificity? Growth Horm IGF Res. 2002 Apr;12(2):84-90. Review.
Shimizu M, Webster C, Morgan DO, Blau HM, Roth RA. Insulin and insulin-like growth factor receptors and responses in cultured human muscle cells. Am J Physiol. 1986 Nov;251(5 Pt 1):E611-5.
Buckway CK, Guevara-Aguirre J, Pratt KL, Burren CP, Rosenfeld RG. The IGF-I generation test revisited: a marker of GH sensitivity. J Clin Endocrinol Metab. 2001 Nov;86(11):5176-83.
Hwa V, Oh Y, Rosenfeld RG. The insulin-like growth factor-binding protein (IGFBP) superfamily. Endocr Rev. 1999 Dec;20(6):761-87. Review.
Firth SM, Baxter RC. Cellular actions of the insulin-like growth factor binding proteins. Endocr Rev. 2002 Dec;23(6):824-54. Review.
Bach LA, Hsieh S, Sakano K, Fujiwara H, Perdue JF, Rechler MM. Binding of mutants of human insulin-like growth factor II to insulin-like growth factor binding proteins 1-6. J Biol Chem. 1993 May 5;268(13):9246-54.
Boisclair YR, Rhoads RP, Ueki I, Wang J, Ooi GT. The acid-labile subunit (ALS) of the 150 kDa IGF-binding protein complex: an important but forgotten component of the circulating IGF system. J Endocrinol. 2001 Jul;170(1):63-70. Review.
Duan C. Specifying the cellular responses to IGF signals: roles of IGF-binding proteins. J Endocrinol. 2002 Oct;175(1):41-54. Review.
LeRoith D. Insulin-like growth factor receptors and binding proteins. Baillieres Clin Endocrinol Metab. 1996 Jan;10(1):49-73. Review.
Rajaram S, Baylink DJ, Mohan S. Insulin-like growth factor-binding proteins in serum and other biological fluids: regulation and functions. Endocr Rev. 1997 Dec;18(6):801-31. Review.
Monzavi R, Cohen P. IGFs and IGFBPs: role in health and disease. Best Pract Res Clin Endocrinol Metab. 2002 Sep;16(3):433-47. Review.
Giustina A, Mazziotti G, Canalis E. Growth hormone, insulin-like growth factors, and the skeleton. Endocr Rev. 2008 Aug;29(5):535-59. Epub 2008 Apr 24. Review.
Collett-Solberg PF, Cohen P. Genetics, chemistry, and function of the IGF/IGFBP system. Endocrine. 2000 Apr;12(2):121-36. Review.
Wetterau LA, Moore MG, Lee KW, Shim ML, Cohen P. Novel aspects of the insulin-like growth factor binding proteins. Mol Genet Metab. 1999 Oct;68(2):161-81. Review.
Parker A, Rees C, Clarke J, Busby WH Jr, Clemmons DR. Binding of insulin-like growth factor (IGF)-binding protein-5 to smooth-muscle cell extracellular matrix is a major determinant of the cellular response to IGF-I. Mol Biol Cell. 1998 Sep;9(9):2383-92.
Velloso CP. Regulation of muscle mass by growth hormone and IGF-I. Br J Pharmacol. 2008 Jun;154(3):557-68. Review.
Jones JI, Clemmons DR. Insulin-like growth factors and their binding proteins: biological actions. Endocr Rev. 1995 Feb;16(1):3-34. Review.
Hameed M, Lange KH, Andersen JL, Schjerling P, Kjaer M, Harridge SD, Goldspink G. The effect of recombinant human growth hormone and resistance training on IGF-I mRNA expression in the muscles of elderly men. J Physiol. 2004 Feb 15;555(Pt 1):231-40. Epub 2003 Oct 17.
Pfeffer LA, Brisson BK, Lei H, Barton ER. The insulin-like growth factor (IGF)-I E-peptides modulate cell entry of the mature IGF-I protein. Mol Biol Cell. 2009 Sep;20(17):3810-7.
Zabłocka B, Goldspink PH, Goldspink G, Górecki DC. Mechano-Growth Factor: an important cog or a loose screw in the repair machinery? Front Endocrinol (Lausanne). 2012 Nov 1;3:131.
Rotwein P. Two insulin-like growth factor I messenger RNAs are expressed in human liver. Proc Natl Acad Sci U S A. 1986 Jan;83(1):77-81.
Siegfried JM, Kasprzyk PG, Treston AM, Mulshine JL, Quinn KA, Cuttitta F. A mitogenic peptide amide encoded within the E peptide domain of the insulin-like growth factor IB prohormone. Proc Natl Acad Sci U S A. 1992 Sep 1;89(17):8107-11.
Barton ER, DeMeo J, Lei H. The insulin-like growth factor (IGF)-I E-peptides are required for isoform-specific gene expression and muscle hypertrophy after local IGF-I production. J Appl Physiol (1985). 2010 May;108(5):1069-76.
Yang S, Alnaqeeb M, Simpson H, Goldspink G. Cloning and characterization of an IGF-1 isoform expressed in skeletal muscle subjected to stretch. J Muscle Res Cell Motil. 1996 Aug;17(4):487-95.
McKoy G, Ashley W, Mander J, Yang SY, Williams N, Russell B, Goldspink G. Expression of insulin growth factor-1 splice variants and structural genes in rabbit skeletal muscle induced by stretch and stimulation. J Physiol. 1999 Apr 15;516 (Pt 2):583-92.
Yang SY, Goldspink G. Different roles of the IGF-I Ec peptide (MGF) and mature IGF-I in myoblast proliferation and differentiation. FEBS Lett. 2002 Jul 3;522(1-3):156-60. Erratum in: FEBS Lett. 2006 May 1;580(10):2530.
Rudman D, Kutner MH, Rogers CM, Lubin MF, Fleming GA, Bain RP. Impaired growth hormone secretion in the adult population: relation to age and adiposity. J Clin Invest. 1981 May;67(5):1361-9.
Corpas E, Harman SM, Blackman MR. Human growth hormone and human aging. Endocr Rev. 1993 Feb;14(1):20-39. Review.
Veldhuis JD, Liem AY, South S, Weltman A, Weltman J, Clemmons DA, Abbott R, Mulligan T, Johnson ML, Pincus S, et al. Differential impact of age, sex steroid hormones, and obesity on basal versus pulsatile growth hormone secretion in men as assessed in an ultrasensitive chemiluminescence assay. J Clin Endocrinol Metab. 1995 Nov;80(11):3209-22.
Veldhuis JD, Iranmanesh A, Weltman A. Elements in the pathophysiology of diminished growth hormone (GH) secretion in aging humans. Endocrine. 1997 Aug;7(1):41-8. Review.
Iranmanesh A, Lizarralde G, Veldhuis JD. Age and relative adiposity are specific negative determinants of the frequency and amplitude of growth hormone (GH) secretory bursts and the half-life of endogenous GH in healthy men. J Clin Endocrinol Metab. 1991 Nov;73(5):1081-8
Rudman D. Growth hormone, body composition, and aging. J Am Geriatr Soc. 1985 Nov;33(11):800-7. Review.
Russell-Aulet M, Jaffe CA, Demott-Friberg R, Barkan AL. In vivo semiquantification of hypothalamic growth hormone-releasing hormone (GHRH) output in humans: evidence for relative GHRH deficiency in aging. J Clin Endocrinol Metab. 1999 Oct;84(10):3490-7.
Martin FC, Yeo AL, Sonksen PH. Growth hormone secretion in the elderly: aging and the somatopause. Baillieres Clin Endocrinol Metab. 1997 Jul;11(2):223-50. Review.
Chertman LS, Merriam GR, Kargi AY. Growth Hormone in Aging. [Updated 2015 May 4]. In: De Groot LJ, Chrousos G, Dungan K, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.
Sattler FR. Growth hormone in the aging male. Best Pract Res Clin Endocrinol Metab. 2013 Aug;27(4):541-55.
Duchaine D. Underground steroid handbook. 1. California: HLR Technical Books; 1983. p. 84
Erotokritou-Mulligan I, Holt RI, Sönksen PH. Growth hormone doping: a review. Open Access Journal of Sports Medicine. 2011;2:99-111.
Liu H, Bravata DM, Olkin I, Friedlander A, Liu V, Roberts B, Bendavid E, Saynina O, Salpeter SR, Garber AM, Hoffman AR. Systematic review: the effects of growth hormone on athletic performance. Ann Intern Med. 2008 May 20;148(10):747-58. Epub 2008 Mar 17. Review.
Birzniece V, Nelson AE, Ho KK. Growth hormone and physical performance. Trends Endocrinol Metab. 2011 May;22(5):171-8. Mar 17. Review.
Baumann GP. Growth hormone doping in sports: a critical review of use and detection strategies. Endocr Rev. 2012 Apr;33(2):155-86. Epub 2012 Feb 24. Review.
Gibney J, Healy ML, Sönksen PH. The growth hormone/insulin-like growth factor-I axis in exercise and sport. Endocr Rev. 2007 Oct;28(6):603-24. Epub 2007 Sep 4. Review.
Holt RI, Sönksen PH. Growth hormone, IGF-I and insulin and their abuse in sport. Br J Pharmacol. 2008 Jun;154(3):542-56. Epub 2008 Mar 31. Review.
Barroso O, Mazzoni I, Rabin O. Hormone abuse in sports: the antidoping perspective. Asian J Androl. 2008 May;10(3):391-402.
Frystyk J. Exercise and the growth hormone-insulin-like growth factor axis. Med Sci Sports Exerc. 2010 Jan;42(1):58-66.
Meinhardt U, Nelson AE, Hansen JL, Birzniece V, Clifford D, Leung KC, Graham K, Ho KK. The effects of growth hormone on body composition and physical performance in recreational athletes: a randomized trial. Ann Intern Med. 2010 May 4;152(9):568-77.
Wallace JD, Cuneo RC, Baxter R, Orskov H, Keay N, Pentecost C, Dall R, Rosén T, Jørgensen JO, Cittadini A, Longobardi S, Sacca L, Christiansen JS, Bengtsson BA, Sönksen PH. Responses of the growth hormone (GH) and insulin-like growth factor axis to exercise, GH administration, and GH withdrawal in trained adult males: a potential test for GH abuse in sport. J Clin Endocrinol Metab. 1999 Oct;84(10):3591-601.
Chikani V, Ho KK. Action of GH on skeletal muscle function: molecular and metabolic mechanisms. J Mol Endocrinol. 2013 Dec 19;52(1):R107-23.
Lange KH, Larsson B, Flyvbjerg A, Dall R, Bennekou M, Rasmussen MH, Ørskov H, Kjaer M. Acute growth hormone administration causes exaggerated increases in plasma lactate and glycerol during moderate to high intensity bicycling in trained young men. J Clin Endocrinol Metab. 2002 Nov;87(11):4966-75.
Berggren A, Ehrnborg C, Rosén T, Ellegård L, Bengtsson BA, Caidahl K. Short-term administration of supraphysiological recombinant human growth hormone (GH) does not increase maximum endurance exercise capacity in healthy, active young men and women with normal GH-insulin-like growth factor I axes. J Clin Endocrinol Metab. 2005 Jun;90(6):3268-73. Epub 2005 Mar 22.
Irving BA, Patrie JT, Anderson SM, Watson-Winfield DD, Frick KI, Evans WS, Veldhuis JD, Weltman A. The effects of time following acute growth hormone administration on metabolic and power output measures during acute exercise. J Clin Endocrinol Metab. 2004 Sep;89(9):4298-305. Epub 2004 Aug 24.
Graham MR, Baker JS, Evans P, Kicman A, Cowan D, Hullin D, Davies B. Evidence for a decrease in cardiovascular risk factors following recombinant growth hormone administration in abstinent anabolic-androgenic steroid users. Growth Horm IGF Res. 2007 Jun;17(3):201-9. Epub 2007 Feb 26.
Brill KT, Weltman AL, Gentili A, Patrie JT, Fryburg DA, Hanks JB, Urban RJ, Veldhuis JD. Single and combined effects of growth hormone and testosterone administration on measures of body composition, physical performance, mood, sexual function, bone turnover, and muscle gene expression in healthy older men. J Clin Endocrinol Metab. 2002 Dec;87(12):5649-57.
Blackman MR, Sorkin JD, Münzer T, Bellantoni MF, Busby-Whitehead J, Stevens TE, Jayme J, O’Connor KG, Christmas C, Tobin JD, Stewart KJ, Cottrell E, St Clair C, Pabst KM, Harman SM. Growth hormone and sex steroid administration in healthy aged women and men: a randomized controlled trial. JAMA. 2002 Nov 13;288(18):2282-92.
Chikani V, Cuneo RC, Hickman I, Ho KK. Growth hormone (GH) enhances anaerobic capacity: impact on physical function and quality of life in adults with GH deficiency. Clin Endocrinol (Oxf). 2016 Oct;85(4):660-8.
Saccà L, Cittadini A, Fazio S. Growth hormone and the heart. Endocr Rev. 1994 Oct;15(5):555-73. Review.
Svensson J, Tivesten A, Isgaard J. Growth hormone and the cardiovascular function. Minerva Endocrinol. 2005 Mar;30(1):1-13. Review.
Copeland KC, Nair KS. Recombinant human insulin-like growth factor-I increases forearm blood flow. J Clin Endocrinol Metab. 1994 Jul;79(1):230-2.
Pete G, Hu Y, Walsh M, Sowers J, Dunbar JC. Insulin-like growth factor-I decreases mean blood pressure and selectively increases regional blood flow in normal rats. Proc Soc Exp Biol Med. 1996 Nov;213(2):187-92.
Walsh MF, Barazi M, Pete G, Muniyappa R, Dunbar JC, Sowers JR. Insulin-like growth factor I diminishes in vivo and in vitro vascular contractility: role of vascular nitric oxide. Endocrinology. 1996 May;137(5):1798-803.
Hermansen K, Bengtsen M, Kjær M, Vestergaard P, Jørgensen JOL. Impact of GH administration on athletic performance in healthy young adults: A systematic review and meta-analysis of placebo-controlled trials. Growth Horm IGF Res. 2017 Jun;34:38-44.
Quello che verrà riportato in questo e negli altri articoli della serie che seguiranno, è il frutto di un lavoro di ricerca ed estrapolazione di informazioni che trovano la loro origine nel ottimo lavoro di Chest Rockwell sull’uso del Ormone della Crescita a fini ipertrofici intitolato “The Most Effective Growth Hormone Protocol for Hypertrophy”.
Nota introduttiva
Se volete un metodo altamente efficace per spingere un Bodybuilder a guardarvi prima con confusione e, in seguito, con totale irritazione, basta che usiate qualsiasi variazione della seguente affermazione:
L’Ormone della Crescita determina un aumento significativo della massa corporea magra, è altamente anabolico, ma non causa la crescita del tessuto muscolare…
Nel corso di questi articoli, spiegherò come questa affermazione sia precisa al 100% nonostante sembri un po’ assurdo ad una prima lettura. Per impostare alcuni parametri in anticipo, si parlerà di AAS e della loro relazione sinergica con il GH, ma ci saranno solo alcune menzioni sull’Insulina. Questa serie di articoli rappresenta già di suo un’impresa enorme. Penso che per impostare in modo corretto l’argomento, il tema GH/Insulina meriti un articolo a parte (cosa parzialmente già fatta) a causa delle immense complessità che esistono nella loro correlazione. Non ho nemmeno intenzione di toccare l’argomento dei secretagoghi del GH, dei peptidi sperimentali o di altri analoghi in modo che ci si possa concentrare principalmente sui fondamentali della questione GH ed Ipertrofia. Se non diversamente specificato nell’articolo, si parlerà esplicitamente dell’Ormone della Crescita di origine endogena o ricombinante (esogeno).
E, infine, questi articoli sono rivolti esclusivamente agli uomini. Mi dispiace per le atlete, ma i maschi sono molto più sensibili agli effetti anabolici della supplementazione con GH rispetto alle donne, data la spiccata natura sessualmente dimorfica del GH.(1) Quindi, a meno che non dichiari specificatamente il contrario, le informazioni che seguiranno saranno orientate verso l’uomo. In futuro è probabile che realizzerò un articolo in merito dedicato alle donne.
Introduzione all’Anabolismo e alla Crescita Muscolare
La prima cosa da fare è conoscere concisamente le corrette definizioni di termini quali anabolismo, ipertrofia e iperplasia. Li vedo spesso usati quasi in modo intercambiabile, ma ci sono alcune differenze chiave. Ad esempio, solo perché qualcosa è anabolico non significa automaticamente che causerà la crescita del tessuto muscolare. Viceversa solo perché qualcosa è catabolico non significa automaticamente che non possa contribuire alla crescita del tessuto muscolare in modo positivo.
Schema dei processi anabolici
L’anabolismo può essere definito come qualsiasi stato in cui la ritenzione d’azoto nella massa magra è positiva, sia attraverso la stimolazione della sintesi proteica che attraverso la soppressione dei tassi di proteolisi.(2) Il primo chiarimento da fare in riferimento alla mia precedente affermazione è che la misurazione della massa corporea magra non include soltanto la massa muscolare ma, per esempio, anche gli organi, le ossa e l’acqua ivi contenuta (3), fattore quest’ultimo soggetto all’azione dell’Ormone della Crescita. Quindi, leggere uno studio nel quale si afferma che la massa corporea magra è aumentata in seguito al trattamento con GH, non vuol dire che ci si riferisca ad un aumento della massa muscolare.
L’apparato muscolare è un tessuto plastico molto complesso e in grado di adattarsi alle mutevoli esigenze funzionali alle quali viene sottoposto. Quando parliamo di aumento della massa muscolare, stiamo parlando principalmente di un evento che si verifica attraverso due meccanismi primari – ipertrofia e iperplasia.
L’ipertrofia è il processo attraverso il quale l’aumento della massa muscolare si verifica tramite l’aumento delle dimensioni dell’area della sezione trasversa di una fibra muscolare esistente (CSA). Il processo ipertrofico è mediato da molti fattori come con l’ipertrofia indotta dall’esercizio, di particolare interesse per i Bodybuilder, essendo essa mediata da una combinazione di tensione meccanica, danno muscolare e stress metabolico.(4)
L’iperplasia, ivece, è il processo mediante il quale l’aumento della massa muscolare viene raggiunto attraverso un incremento del numero effettivo di fibre muscolari. È generalmente accettato che, negli esseri umani, il numero di fibre all’interno del muscolo sia geneticamente predeterminato e fissato durante il periodo perinatale.(5) Esistono alcuni studi svolti su animali i quali hanno dimostrato che l’iperplasia può verificarsi (6-7), spesso in condizioni di test unici, ma il tentativo di dedurre da ciò che essa si possa verificare negli esseri umani (8-9) è altamente speculativo nella migliore delle ipotesi. Anche se l’iperplasia si verifica nell’apparato muscolare umano, è molto probabile che sia solo un fattore minore che determina la massa complessiva. Tuttavia, a causa di una cattiva comprensione (se così la vogliamo chiamare) degli studi prima citati spesso vengono formulate affermazioni definitive secondo cui il GH causa iperplasia. Vale quindi la pena di ribadire che questi tipi di affermazioni dovrebbero essere considerate nient’altro che speculative.
La scoperta dell'”Ormone della Crescita”
Ormone della Crescita
Sono trascorsi oltre 100 anni da quando Harvey Cushing suppose per la prima volta l’esistenza di un “Ormone della Crescita” (10), e questo ormone venne successivamente isolato, identificato ed estratto dall’ipofisi umana negli anni ’40.(11) Nel decennio successivo, si fece largo un’ipotesi cardine secondo cui la “crescita” non era direttamente attribuibile al GH, ma piuttosto ad un gruppo di fattori serici regolati dall’espressione del GH.(12) Questi fattori serici sono stati in seguito chiamati fattori di solfatazione, termine volto ad indicare sostanze controllate dal GH che stimolavano l’assorbimento di solfato nella cartilagine e nei tessuti. Questa ipotesi ha cercato di aiutare i ricercatori a conciliare meglio il modo in cui la crescita somatica veniva regolata da una sostanza secreta dall’Ipofisi, mentre allo stesso tempo rinforzava il fatto che questa sostanza ipofisaria non agiva direttamente sui suoi tessuti bersaglio per promuovere la crescita.(13-14)
Nei decenni successivi, molti esperimenti hanno dimostrato l’ampia varietà di funzioni possedute da questa famiglia di fattori di solfatazione e il termine “somatomedina” è stato proposto per includere tutti i membri di questa famiglia.(15) È interessante notare che l’ipotesi originale che descrive la regolazione di questi fattori di solfatazione mediati dal GH è ancora oggi generalmente indicata come “l’ipotesi della somatomedina”.(16) L’ipotesi originale affermava che la crescita somatica è causata dall’azione del GH a livello epatico, dove stimola la sintesi di IGF-1 che viene successivamente rilasciato agendo sui tessuti bersaglio con modello endocrino. Questa ipotesi è rimasta il modello accettato per decenni fino a quando non sono stati identificati i ruoli autocrini del IGF (17-18) insieme agli effetti diretti che il GH ha nella crescita ossea.(19-20)Per riconciliare queste nuove scoperte, l’ipotesi originaria è stata leggermente modificata prendendo il nome di “Teoria del Doppio Effetto”, che ha definito sia un ruolo autocrino che endocrino del IGF-1.(21)
Tornando indietro di qualche tempo, verso la fine degli anni ’70, l’identità chimica di questi fattori di solfatazione era più intimamente riconosciuta come la manifestazione di due peptidi con un’elevatissima somiglianza con l’Insulina e quindi ribattezzati “fattori di crescita isulino-simili (IGF)”.(22-24) Fu all’incirca nello stesso periodo nel quale vennero identificate anche le proteine leganti l’IGF (IGFBP), e di conseguenza la conoscenza della biologia dell’IGF-1 decollò ad un tasso esponenziale.(25)
Nel frattempo, gli estratti ipofisari umani diventarono disponibili tramite l’estrazione dai cadaveri e gli esperimenti iniziali svolti su animali ed esseri umani mostrarono quanto complesse fossero realmente le azioni del GH umano. (26-33) La pratica dell’uso di questi estratti ipofisari venne interrotta dai medici in seguito alla comparsa della malattia di Creutzfeldt-Jakob (CJD) in pazienti a cui era stato precedentemente somministrato il GH contenuto nel Ipofisi estratta dai cadaveri.(34) La CJD è una malattia cerebrale particolarmente sgradevole e universalmente fatale, con la stragrande maggioranza dei soggetti affetti che muoiono poco dopo la sua diagnosi.
Fortunatamente, nel 1985, nello stesso periodo in cui questi casi di CJD cominciarono a fare notizia, la FDA approvò il primo GH ricombinante sintetico (rHGH) per l’utilizzo su soggetti con deficit dell’Ormone della Crescita. Questa versione di rHGH è stata prodotta dalla Genentech e denominata Protropin.(35-36) Vale la pena notare che, analizzando la composizione dei vecchi farmaci contenenti GH si può osservare che erano costituiti da una catena di 192 amminoacidi (met-hGH) rispetto al rHGH che consiste in una catena di 191 amminoacidi.(37) In ogni caso, con l’rHGH, ora di facile disponibilità, è stata inaugurata un’era completamente nuova, con condizioni cliniche molto più sicure, per il numero in continua crescita di pazienti trattati.
Effetti dell’Ormone della Crescita sulla sintesi proteica
All’inizio dell’articolo, ho definito a grandi linee cos’è l’anabolismo ed ho anche affermato che l’Ormone della Crescita possiede una natura anabolica. Adesso, descriverò ciò che rende anabolizzante l’Ormone della Crescita e approfondirò parte della letteratura scientifica esistente sull’argomento.
Nel corso degli anni, il GH è stato ampiamente studiato in diversi modi. La maggior parte degli studi svolti, quando considerati nel loro insieme, suggeriscono che il GH sia anabolico. Più specificamente, il GH è anabolico perché stimola la sintesi proteica di tutto il corpo con o nessun effetto, o un effetto soppressivo, sui tassi di degradazione proteica. [38] Tuttavia, quando si approfondisce l’argomento, le cose diventano un po’ meno chiare dal momento che i risultati degli studi tendono ad essere diversi. I diversi risultati sono un riflesso diretto dell’ immensa complessità del GH.
Il GH esplica i suoi effetti sulla sintesi proteica legandosi prima con il suo recettore specifico (GHR) e successivamente aumentando la trascrizione del gene muscolare attraverso i percorsi di segnalazione a valle, in definitiva attivando la segnalazione del mTOR. [39-40] Questi effetti si manifestano in acuto, spesso si verificano in pochi minuti e sono di natura simile all’Insulina, usando molte delle stesse vie anaboliche. [41-44] La rapida comparsa di questi cambiamenti metabolici legati alle proteine suggerisce che essi siano direttamente causati dal GH e non secondariamente mediati tramite l’IGF-1 [45]. L’impatto del GH sulla proteolisi, d’altro canto, è molto probabilmente di natura indiretta. A detta di tutti, ciò ha più a che fare con i suoi effetti inibitori sull’Insulina, che è stata vista avere effetti diretti sulla proteolisi.[46]
Ora, poiché sarete interessati principalmente alla crescita muscolare, riporterò come il GH influenza specificamente i tassi di sintesi delle proteine muscolari (MPS). Vi sono numerosi studi in letteratura nei quali il GH è stato somministrato a soggetti adulti sani senza aver mostrato alcun impatto sui tassi di MPS. [45,47-52] Ciò che risulta affascinante di questi risultati è che un paio di studi includevano anche una allenamento di resistenza, sebbene, nella fattispecie, non sono stati comunque riscontrati aumenti nei tassi di MPS locali. Al contrario, ci sono una serie di studi che hanno riportato di aumenti nei tassi di MPS senza cambiamenti significativi nei tassi di sintesi proteica dell’intero corpo. [53-55]
Ci sono molte ragioni per cui questi risultati appaiono non del tutto coerenti all’interno del corpo della letteratura scientifica. Uno dei motivi principali è rappresentato dal modo in cui gli studi sono stati svolti per quanto riguarda il tipo di somministrazione del GH (ad esempio, le concentrazione di dosaggio, se somministrato localmente o in modo sistemico, nonché se l’ormone è stato somministrato in modo pulsatile o costante). Alcune delle altre ragioni includono come è stata misurata la sintesi proteica, se i soggetti esaminati erano a digiuno o nutriti, che tipo di muscolo scheletrico è stato esaminato e, anche, per quanto tempo è durato lo studio. Come menzionato in precedenza, molti degli effetti che il GH ha sul metabolismo proteico avvengono in acuto.
Sebbene questa serie di articoli è principalmente incentrata sull’ipertrofia, ritengo comunque che valga la pena di discutere le differenze che il GH ha sul metabolismo delle proteine in condizioni di digiuno e nutrizione. Ciò aiuterà ad avere un quadro generale più chiaro di come il comportamento di questo ormone sia spesso il risultato diretto dell’ambiente nel quale è introdotto. Come ho spiegato dettagliatamente nell’articolo dedicato a GH e lipolisi, la secrezione di GH subisce un aumento durante periodi prolungati di digiuno. Questo è un meccanismo di sopravvivenza, con l’obiettivo principale di conservare preziosi pool amminoacidici immagazzinati prevenendo la degradazione delle proteine. [56] Questo stesso comportamento di risparmio di proteine può essere visto, in misura minore, in soggetti trattati con GH e sottoposti a severe restrizioni dietetiche [57], soggetti obesi sottoposti a vari tipi di dieta ipocalorica [58-61] e soggetti privati di proteine dietetiche [ 62].
IGF-1
È stato dimostrato che l’IGF-1 inibisce allo stesso modo la disgregazione proteica a livello sistemico [63], il che avrebbe senso a causa della stretta co-relazione con il GH. Quando gli amminoacidi e l’Insulina vengono somministrati ai soggetti esaminati, già sottoposti a somministrazione di IGF-1, è stato dimostrato, sia negli uomini che negli animali, che i tassi di sintesi proteica aumentano a livello sistemico [64-65]. Vale la pena notare che l’IGF-1 è bifasico nel senso che quando è somministrato ad alto dosaggio e, di conseguenza, i livelli serici diventano elevati, il suo comportamento cambia passando da un azione “GH-simile” ad una “insulino-simile”. Tratterò nel dettaglio l’argomento in seguito.
Per riassumere, il GH è molto adatto per prevenire la degradazione proteica, e lo fa in una vasta gamma di condizioni alimentari a ristretto apporto calorico. Tuttavia, in presenza di un apporto energetico sufficiente, il suo comportamento cambia. L’effetto principale del GH sul metabolismo proteico è volto dapprima a creare un ambiente con una ossidazione amminoacidica ridotta [47,66] e successivamente ad aumentare la sintesi proteica sistemica. [67]
Il ruolo di GH e IGF-1 nella crescita postnatale
È noto che il GH regola la crescita postnatale e che questi effetti di promozione della crescita sono principalmente mediati dall’IGF-1. [68-69] Tuttavia, giusto per ribadirlo, è necessario chiarire che questi effetti di induzione della crescita non sono esclusivi dell’ipertrofia muscolare. La crescita lineare di un organismo include cambiamenti nell’apparato scheletrico, negli organi e nei muscoli.
Per riportare esempi di un certo rilievo sull’importanza che GH e IGF-1 hanno sulla crescita postnatale, basta osservare gli individui con mutazioni o disturbi correlati all’Asse GH/IGF. Una revisione dettagliata su questo tema va oltre lo scopo di questo articolo ma, in generale, e penso sia abbastanza risaputo, i disturbi che sopprimono/inibiscono l’Asse GH/IGF in soggetti in giovane età si caratterizzano in una bassa statura di questi mentre i disturbi che stimolano l’Asse GH/IGF si caratterizzano nel gigantismo. [70-71]
La stragrande maggioranza degli effetti del GH sulla promozione della crescita sono mediati tramite l’IGF-1, tuttavia ci sono una serie di processi che sono GH-mediati o IGF-indipendenti. L’esempio più eloquente di questo è rappresentato nei modelli animali in cui i topi “knockout” mutati e deficitari di GH e IGF-1 hanno mostrato una maggiore gravità nel ritardo della crescita rispetto ai “knockout” GH-deficienti o IGF-deficienti [72]. Ma osserviamo nel dettaglio alcuni degli effetti più specifici che sono stati identificati nei modelli umani.
Il primo è legato alla steatosi epatica, nota anche come “malattia del fegato grasso” [73]. È stato dimostrato sia negli esseri umani con la sindrome di Laron che negli animali con soppressione del GHR che questa condizione può ancora verificarsi in presenza di livelli di IGF-1 soppressi[74]. La sindrome di Laron fornisce alcune intuizioni piuttosto uniche sul sistema GH/IGF-1 a causa del fatto che è causata da una mutazione nel GHR che si traduce in livelli significativamente bassi di IGF-1 endocrino, dal momento che al GH viene impedito di legarsi al suo sito recettore e di stimolare efficacemente la produzione di IGF-1.
Un’altra azione specifica del GH è legata alla sua capacità di migliorare lo sviluppo del follicolo preantrale ovarico. In effetti, il GH è stato anche recentemente oggetto di studi per i suoi potenziali miglioramenti sulla fertilità femminile. I risultati indicano che alcuni gruppi che utilizzano la fecondazione in vitro (FIV) sembrano beneficiare della somministrazione di GH. [75-76] Anche i modelli animali con deficit di GHR hanno mostrato un numero inferiore di follicoli primari preantrali e antrali rispetto ai loro simili di controllo.
Una delle prime e più interessanti scoperte riguardanti l’azione del GH , è stata il ruolo specifico del GH nel promuovere la crescita ossea longitudinale attraverso i suoi effetti sulla generazione dei condrociti (cellule cartilaginee) nella regione delle placche di crescita epifisaria. [19-20,77-80]. Il GH ha in realtà due ruoli nella promozione della crescita ossea longitudinale; sia attraverso i suddetti effetti sulla generazione dei condrociti nella piastra di crescita che attraverso un ruolo mediato dall’IGF-1 nella promozione dell’ipertrofia dei condrociti.[81] Vale la pena notare che i livelli endocrini di IGF-1 completamente intatti non sono nemmeno una necessità quando si tratta di crescita ossea postnatale. Infatti, fino a quando sono presenti almeno il 10-20% dei livelli di IGF-1 endocrino circolante, la combinazione di IGF-1 e GH autocrini può comunque garantire la normale crescita ossea postnatale. [82] Ciò è probabilmente dovuto ai ruoli sovrapposti di IGF-1 autocrino ed endocrino in relazione alla crescita ossea longitudinale. [83]
Il GH promuove un aumento dei tassi di fusione delle cellule muscolari tardive che possono avere la capacità di aumentare la dimensione delle fibre muscolari in un modo completamente indipendente dalla sovraregolazione dell’IGF-1.[84] Utilizzando una nuova tecnica di sperimentazione su cellule animali, i ricercatori sono stati in grado di dimostrare che il GH promuove la fusione dei mioblasti con i miotubi nascenti senza un aumento dell’espressione effettiva dell’mRNA del IGF-1. I miotubi nascenti sono presenti negli stadi successivi della fusione delle cellule muscolari [85] e il GH ha dimostrato di aumentare il numero di nuclei per miotubo.
La relazione tra secrezione di GH e IGF-1
L’Ormone della Crescita è noto per aumentare i livelli di IGF-1 circolante così come la sintesi locale di IGF-1, in modo autocrino. Entrambe queste azioni giocano un ruolo fondamentale nella regolazione della massa muscolare e, quindi, in conseguenza di ciò, risulta utile comprendere meglio come la secrezione di GH porta ad un aumento dei livelli endocrino e autocrino di IGF-1.
La stragrande maggioranza dell’Ormone della Crescita negli adulti sani è secreto dalla ghiandola pituitaria e, più specificamente, dalle cellule somatotrope nel lobo anteriore mediate dal fattore di trascrizione Prophet of Pit-1 (PROP1).[86-87] Il GH può anche essere sintetizzato localmente in molti tessuti come il cervello, le cellule immunitarie, il tessuto mammario, i denti e la placenta che sono tutti al di fuori della regolazione dell’ipofisi. [88] Questo supporta l’idea secondo cui il GH abbia ruoli autocrini oltre ai suoi già consolidati ruoli endocrini.
Il GH è secreto in modo pulsatile in tutte le specie [89-90] e questo modello secretorio gioca un ruolo fisiologico importante in tutte le sue caratteristiche, da quelle dimorfiche sessuali all’RNA del IGF-1, di cui scopriremo di più in seguito. I giovani maschi adulti sani secernono tra 0,4-0,5mg di GH ogni 24 ore e molte di queste secrezioni si verificano come “impulsi all’interno di impulsi”. [91] Normalmente, ci sono circa 10-12 impulsi secretorie ogni giorno con gli uomini che presentano un modello di impulso significativamente più regolare rispetto alle donne. Nei maschi, il GH viene secreto in modo episodico, con la ben nota grande ondata serale che si verifica in prossimità dell’inizio del sonno ad onde lente. I maschi hanno anche secrezioni più contenute che si verificano poche ore dopo aver consumato i pasti.[92-94] Le femmine hanno livelli ematici inter-secretori più elevati, in particolare nella fase follicolare del ciclo mestruale, con impulsi di GH più frequenti durante il giorno e un impulso notturno significativamente più basso rispetto ai maschi [95-96]. Non è del tutto chiaro perché esista questo schema secretivo sessualmente dimorfico.
GHRH
La secrezione di GH è regolata in un modo molto complesso che coinvolge la partecipazione di diversi neurotrasmettitori, così come di feedback sia ormonali che metabolici. È regolato principalmente in modo positivo dal GHRH (Ormone di Rilascio della Somatotropina o Somatorelina) [97], e in modo negativo dalla SRIF (Somatostatina). Entrambi questi peptidi sono prodotti all’interno dell’ipotalamo. Infatti, oltre al suo ruolo di base nella produzione ormonale, l’ipotalamo monitora costantemente il rapporto GHRH/SRIF e di conseguenza opera un controllo sulla secrezione ipofisaria. [98]
Oltre alla sua funzione primaria di stimolazione della secrezione di GH, il GHRH svolge anche un ruolo essenziale nella proliferazione e nello sviluppo delle suddette cellule somatotrope. Infatti, in un sistema con GHRH alterato o assente, è stata osservata ipoplasia anteriore dell’ipofisi che è probabilmente il risultato di un alterato sviluppo somatotropo.[99-100] Gli esseri umani con il GHRH soppresso per azione di un antagonista hanno mostrato un rilascio pulsatile di GH gravemente compromesso e una risposta soppressa del GH al GHRH [101], quindi è chiaramente una componente vitale all’interno dell’Asse GH/IGF-1.
SRIF
La SRIF, il principale regolatore negativo della secrezione di GH, sopprime il TSH. Inoltre, in misura minore, sopprime sia la Prolattina che l’Ormone Adrenocorticotropo [97]. Tutti questi ormoni sono noti per avere strette relazioni con l’Asse GH/IGF-1. La SRIF ha un’emivita breve di circa 2-3 minuti nel siero e viene quindi rapidamente inattivata dalle peptidasi tissutali. Durante la sua vita attiva, sopprime non solo il rilascio spontaneo di GH, ma anche la risposta del GH a tutti gli stimoli esterni incluso il GHRH, l’ipoglicemia, l’Arginina e l’esercizio fisico solo per nominarne alcuni. I suoi effetti soppressivi apparentemente sono limitati all’ampiezza della liberazione basale e pulsatile di GH, poiché non è stato dimostrato che alteri la frequenza degli impulsi di GH.[102]
Il GH circolante è in gran parte legato alle proteine di trasporto chiamate GHBP o proteine leganti l’ormone della crescita. Queste proteine carrier sono essenzialmente una forma solubile e troncata del dominio extracellulare del GHR – GHR circolanti mobili che non si trovano all’interno delle membrane cellulari.[103] Il GH nel flusso ematico può anche esistere in forma libera o non legata e il rapporto tra frazione legata e non legata dipende dal modello pulsatile della sua secrezione. [104] I complessi di GH circolante nell’uomo possono essere costituiti da una delle due distinte forme di GH (22-kDa e 20-kDa), con circa il 90% rappresentato dalla forma molecolare da 22-kDa nonostante le prime stime abbiano ridotto tale numero.[105-106] Le moderne metodologie utilizzate nei test antidoping per la rilevazione indiretta del GH possono effettivamente sfruttare i rapporti in tempo reale delle molecole di GH circolanti all’interno del sistema di un atleta per determinare se questo ha usato rHGH nelle ultime 24-36 ore prima del test. La molecola dell’Ormone della Crescita, nella sua forma corretta da 22-kDa, è raffigurata di seguito. [107]:
Molecola dell’Ormone della Crescita nella sua forma corretta da 22kDa
In definitiva, questo GH circolante si lega al GHR, recettore dell’Ormone della Crescita appartenente alla superfamiglia delle proteine transmembrana presenti in tutte le cellule del corpo e che include il recettore della Prolattina e un certo numero di recettori delle citochine [108-110]. I livelli sulla superficie cellulare, o la densità recettoriale, dei GHR sono il determinante principale dell’affinità di legame del GH con le cellule. La traslocazione del GHR, cioè il recettore che si sposta dal nucleo di una cellula alla sua membrana esterna, è direttamente inibita dall’IGF-1 – che è uno dei molti meccanismi di feedback che esistono tra questi ormoni strettamente correlati. Mediante l’inibizione della traslocazione dei GHR, l’IGF-1 contribuisce direttamente ad abbassare la reattività di queste cellule a uno stimolo esterno di GH. [111]
Una analisi profonda dell’attivazione della Tirosina Chinasi, delle vie di segnalazione a valle e dell’espressione genica va oltre lo scopo di questo articolo. Quindi, mi limiterò a trattarlo superficialmente. E’ necessario comunque toccare alcuni dei punti più importanti della segnalazione intracellulare per comprendere veramente le basi del processo ipertrofico e del perché esiste sia sinergie composte che potenziali strategie ottimizzate per massimizzare il processo.
Come già accennato, i GHR esistono sulle membrane cellulari come omodimeri preformati e inattivi. Questo significa che il GHR ha due dimeri identici del recettore della proteina, e questi omodimeri saranno sempre accoppiati al JAK2 quando sono privi di attività enzimatica. Questo accoppiamento al JAK2 provoca un’azione inibitoria complessiva sul recettore. [112-113] In altre parole, il GHR rimane dormiente finché non viene attivato come parte del processo di legame GH/GHR. Quando una molecola di GH si lega al GHR, si verifica un cambiamento strutturale all’interno del GHR che si traduce in movimento effettivo dei domini intracellulari del recettore separatamente l’uno dall’altro. Questo smorza quell’azione inibitoria data dal JAK2 e consente loro di attivarsi l’un l’altro.[114-116]
Successivamente, la molecola di GH si lega sequenzialmente a uno dei due omodimeri di GHR e il completamento di questo processo di legame facilita le interazioni con il secondo omodimero. Dopo questo, i domini intracellulari di questo dimero-GHR appena formato subiscono una rotazione effettiva. La rotazione del nuovo dimero-GHR consente ai domini chinasi del JAK2 di essere in contatto l’uno con l’altro, consentendo loro di transactivare e ciascuno successivamente si lega a una molecola di JAK2. [117-118] Ciascuna molecola di JAK2 eseguirà quindi la fosforilazione incrociata (attivazione) dei residui di Tirosina, e sono proprio questi residui che formano “punti di attracco” per molte delle diverse molecole di segnalazione che costituiscono le vie di segnalazione a valle, e alla fine portano all’espressione genica. [114,118 -120] Uno dei più importanti percorsi a valle di maggior interesse per l’argomento specifico che qui vado trattando è il percorso JAK-STAT. Questo percorso è di vitale importanza sia per la trascrizione epatica di IGF-1 dal GH che per molti dei processi anabolici mediati dal GH all’interno del tessuto muscolare.
Nella seconda parte di questa serie di articoli mi occuperò più nel dettaglio del IGF-1, della “somatopausa” e della reale efficacia dell’uso del GH nello sport.
Stay tuned!
Gabriel Bellizzi
Riferimenti:
The Most Effective Growth Hormone Protocol for Hypertrophy – by Chest Rockwell
Dall R, Longobardi S, Ehrnborg C, Keay N, Rosén T, Jørgensen JO, Cuneo RC,Boroujerdi MA, Cittadini A, Napoli R, Christiansen JS, Bengtsson BA, Sacca L,Baxter RC, Basset EE, Sönksen PH. The effect of four weeks of supraphysiological growth hormone administration on the insulin-like growth factor axis in women and men. GH-2000 Study Group. J Clin Endocrinol Metab. 2000 Nov;85(11):4193-200.
Kuhn CM. Anabolic steroids. Recent Prog Horm Res. 2002;57:411-34. Review.
Ribeiro SML, Kehayias JJ. Sarcopenia and the Analysis of Body Composition. Advances in Nutrition. 2014;5(3):260-267.
Schoenfeld BJ. The mechanisms of muscle hypertrophy and their application to resistance training. J Strength Cond Res. 2010 Oct;24 (10):2857-72.
Stickland NC. Muscle development in the human fetus as exemplified by m.sartorius: a quantitative study. J Anat. 1981 Jun;132(Pt 4):557-79.
Antonio J, Gonyea WJ. Skeletal muscle fiber hyperplasia. Med Sci Sports Exerc. 1993 Dec;25(12):1333-45. Review.
Fernández AM, Dupont J, Farrar RP, Lee S, Stannard B, Le Roith D.Muscle-specific inactivation of the IGF-I receptor induces compensatory hyperplasia in skeletal muscle. J Clin Invest. 2002 Feb;109(3):347-55.
Kadi F, Thornell LE. Training affects myosin heavy chain phenotype in the trapezius muscle of women. Histochem Cell Biol. 1999 Jul;112(1):73-8.
D’Antona G, Lanfranconi F, Pellegrino MA, Brocca L, Adami R, Rossi R, Moro G, Miotti D, Canepari M, Bottinelli R.Skeletal muscle hypertrophy and structure and function of skeletal muscle fibres in male body builders. J Physiol. 2006 Feb 1;570(Pt 3):611-27.
Cohen-Gadol AA, Liu JK, Laws ER Jr. Cushing’s first case of transsphenoidal surgery: the launch of the pituitary surgery era. J Neurosurg. 2005 Sep;103(3):570-4.
Li CH, Evans HM. THE ISOLATION OF PITUITARY GROWTH HORMONE. Science. 1944 Mar 3;99(2566):183-4.
SALMON WD Jr, DAUGHADAY WH. A hormonally controlled serum factor which stimulates sulfate incorporation by cartilage in vitro. J Lab Clin Med. 1957 Jun;49(6):825-36.
Daughaday WH, Reeder C. Synchronous activation of DNA synthesis in hypophysectomized rat cartilage by growth hormone. J Lab Clin Med. 1966 Sep;68(3):357-68.
Garland JT, Lottes ME, Kozak S, Daughaday WH. Stimulation of DNA synthesis in isolated chondrocytes by sulfation factor. Endocrinology. 1972 Apr;90(4):1086-90.
Daughaday WH, Hall K, Raben MS, Salmon WD Jr, van den Brande JL, van Wyk JJ.Somatomedin: proposed designation for sulphation factor. Nature. 1972 Jan 14;235(5333):107.
Le Roith D, Bondy C, Yakar S, Liu JL, Butler A. The somatomedin hypothesis: 2001. Endocr Rev. 2001 Feb;22(1):53-74. Review.
D’Ercole AJ, Applewhite GT, Underwood LE. Evidence that somatomedin is synthesized by multiple tissues in the fetus. Dev Biol. 1980 Mar 15;75(2):315-28.
Han VK, Lund PK, Lee DC, D’Ercole AJ. Expression of somatomedin/insulin-like growth factor messenger ribonucleic acids in the human fetus: identification, characterization, and tissue distribution. J Clin Endocrinol Metab. 1988 Feb;66(2):422-9.
Isaksson OG, Jansson JO, Gause IA. Growth hormone stimulates longitudinal bone growth directly. Science. 1982 Jun 11;216 (4551):1237-9.
Isaksson OG, Lindahl A, Nilsson A, Isgaard J. Mechanism of the stimulatory effect of growth hormone on longitudinal bone growth. Endocr Rev. 1987 Nov;8(4):426-38. Review.
Green H, Morikawa M, Nixon T. A dual effector theory of growth-hormone action. Differentiation. 1985;29(3):195-8. Review.
Rinderknecht E, Humbel RE. The amino acid sequence of human insulin-like growth factor I and its structural homology with proinsulin. J Biol Chem. 1978 Apr 25;253(8):2769-76.
Klapper DG, Svoboda ME, Van Wyk JJ. Sequence analysis of somatomedin-C: confirmation of identity with insulin-like growth factor I. Endocrinology. 1983 Jun;112(6):2215-7.
Daughaday WH, Rotwein P. Insulin-like growth factors I and II. Peptide, messenger ribonucleic acid and gene structures, serum, and tissue concentrations. Endocr Rev. 1989 Feb;10(1):68-91. Review.
Hintz RL, Liu F. Demonstration of specific plasma protein binding sites for somatomedin. J Clin Endocrinol Metab. 1977 Nov;45(5):988-95.
BECK JC, McGARRY EE, DYRENFURTH I, VENNING EH. The metabolic effects of human and monkey growth hormone in man. Ann Intern Med. 1958 Nov;49(5):1090-105.
IKKOS D, LUFT R, GEMZELL CA. The effect of human growth hormone in man. Lancet. 1958 Apr 5;1(7023):720-1.
RABEN MS, HOLLENBERG CH. Effect of growth hormone on plasma fatty acids. J Clin Invest. 1959 Mar;38(3):484-8. PubMed PMID: 13641397
RABEN MS. Growth hormone. 1. Physiologic aspects. N Engl J Med. 1962 Jan 4;266:31-5.
RABEN MS. Growth hormone. 2. Clinical use of human growth hormone. N Engl J Med. 1962 Jan 11;266:82-6 concl.
ZIERLER KL, RABINOWITZ D. ROLES OF INSULIN AND GROWTH HORMONE, BASED ON STUDIES OF FOREARM METABOLISM IN MAN. Medicine (Baltimore). 1963 Nov;42:385-402.
RABINOWITZ D, KLASSEN GA, ZIERLER KL. EFFECT OF HUMAN GROWTH HORMONE ON MUSCLE AND ADIPOSE TISSUE METABOLISM IN THE FOREARM OF MAN. J Clin Invest. 1965 Jan;44:51-61.
Fineberg SE, Merimee TJ. Acute metabolic effects of human growth hormone. Diabetes. 1974 Jun;23(6):499-504.
Appleby BS, Lu M, Bizzi A, et al. Iatrogenic Creutzfeldt-Jakob Disease from Commercial Cadaveric Human Growth Hormone. Emerging Infectious Diseases. 2013;19(4):682-684. doi:10.3201/eid1904.121504.
Flodh H. Human growth hormone produced with recombinant DNA technology: development and production. Acta Paediatr Scand Suppl. 1986;325:1-9.
Crist DM, Peake GT, Egan PA, Waters DL. Body composition response to exogenous GH during training in highly conditioned adults. J Appl Physiol (1985). 1988 Aug;65(2):579-84.
Møller N, Copeland KC, Nair KS. Growth hormone effects on protein metabolism. Endocrinol Metab Clin North Am. 2007 Mar;36(1):89-100. Review.
Argetsinger LS, Carter-Su C. Mechanism of signaling by growth hormone receptor. Physiol Rev. 1996 Oct;76(4):1089-107. Review.
Hayashi AA, Proud CG. The rapid activation of protein synthesis by growth hormone requires signaling through mTOR. Am J Physiol Endocrinol Metab. 2007 Jun;292(6):E1647-55.
Kostyo JL. Rapid effects of growth hormone on amino acid transport and protein synthesis. Ann N Y Acad Sci. 1968 Feb 5;148(2):389-407.
Cameron CM, Kostyo JL, Adamafio NA, Brostedt P, Roos P, Skottner A, Forsman A, Fryklund L, Skoog B. The acute effects of growth hormone on amino acid transport and protein synthesis are due to its insulin-like action. Endocrinology. 1988 Feb;122(2):471-4.
Vanderkuur JA, Butch ER, Waters SB, Pessin JE, Guan KL, Carter-Su C. Signaling molecules involved in coupling growth hormone receptor to mitogen-activated protein kinase activation. Endocrinology. 1997 Oct;138(10):4301-7.
Costoya JA, Finidori J, Moutoussamy S, Seãris R, Devesa J, Arce VM. Activation of growth hormone receptor delivers an antiapoptotic signal: evidence for a role of Akt in this pathway. Endocrinology. 1999 Dec;140(12):5937-43.
Copeland KC, Nair KS. Acute growth hormone effects on amino acid and lipid metabolism. J Clin Endocrinol Metab. 1994 May;78(5):1040-7.
Umpleby AM, Boroujerdi MA, Brown PM, Carson ER, Sönksen PH. The effect of metabolic control on leucine metabolism in type 1 (insulin-dependent) diabetic patients. Diabetologia. 1986 Mar;29(3):131-41.
Horber FF, Haymond MW. Human growth hormone prevents the protein catabolic side effects of prednisone in humans. J Clin Invest. 1990 Jul;86(1):265-72.
Yarasheski KE, Campbell JA, Smith K, Rennie MJ, Holloszy JO, Bier DM. Effect of growth hormone and resistance exercise on muscle growth in young men. Am J Physiol. 1992 Mar;262(3 Pt 1):E261-7.
Zachwieja JJ, Bier DM, Yarasheski KE. Growth hormone administration in older adults: effects on albumin synthesis. Am J Physiol. 1994 Jun;266(6 Pt 1):E840-4.
Yarasheski KE, Zachwieja JJ, Campbell JA, Bier DM. Effect of growth hormone and resistance exercise on muscle growth and strength in older men. Am J Physiol. 1995 Feb;268(2 Pt 1):E268-76.
Healy ML, Gibney J, Russell-Jones DL, Pentecost C, Croos P, Sönksen PH, Umpleby AM. High dose growth hormone exerts an anabolic effect at rest and during exercise in endurance-trained athletes. J Clin Endocrinol Metab. 2003 Nov;88(11):5221-6.
Giannoulis MG, Jackson N, Shojaee-Moradie F, Nair KS, Sonksen PH, Martin FC, Umpleby AM. The effects of growth hormone and/or testosterone on whole body protein kinetics and skeletal muscle gene expression in healthy elderly men: a randomized controlled trial. J Clin Endocrinol Metab. 2008 Aug;93(8):3066-74.
Fryburg DA, Gelfand RA, Barrett EJ. Growth hormone acutely stimulates forearm muscle protein synthesis in normal humans. Am J Physiol. 1991 Mar;260(3 Pt 1):E499-504.
Fryburg DA, Louard RJ, Gerow KE, Gelfand RA, Barrett EJ. Growth hormone stimulates skeletal muscle protein synthesis and antagonizes insulin’s antiproteolytic action in humans. Diabetes. 1992 Apr;41(4):424-9.
Fryburg DA, Barrett EJ. Growth hormone acutely stimulates skeletal muscle but not whole-body protein synthesis in humans. Metabolism. 1993 Sep;42(9):1223-7.
Nørrelund H, Nair KS, Jørgensen JO, Christiansen JS, Møller N. The protein-retaining effects of growth hormone during fasting involve inhibition of muscle-protein breakdown. Diabetes. 2001 Jan;50(1):96-104.
Manson JM, Wilmore DW. Positive nitrogen balance with human growth hormone and hypocaloric intravenous feeding. Surgery. 1986 Aug;100(2):188-97.
Clemmons DR, Snyder DK, Williams R, Underwood LE. Growth hormone administration conserves lean body mass during dietary restriction in obese subjects. J Clin Endocrinol Metab. 1987 May;64(5):878-83.
Snyder DK, Clemmons DR, Underwood LE. Treatment of obese, diet-restricted subjects with growth hormone for 11 weeks: effects on anabolism, lipolysis, and body composition. J Clin Endocrinol Metab. 1988 Jul;67(1):54-61.
Tagliaferri M, Scacchi M, Pincelli AI, Berselli ME, Silvestri P, Montesano A, Ortolani S, Dubini A, Cavagnini F. Metabolic effects of biosynthetic growth hormone treatment in severely energy-restricted obese women. Int J Obes Relat Metab Disord. 1998 Sep;22(9):836-41.
Nørrelund H, Børglum J, Jørgensen JO, Richelsen B, Møller N, Nair KS, Christiansen JS. Effects of growth hormone administration on protein dynamics andsubstrate metabolism during 4 weeks of dietary restriction in obese women. Clin Endocrinol (Oxf). 2000 Mar;52(3):305-12.
Lundeberg S, Belfrage M, Wernerman J, von der Decken A, Thunell S, Vinnars E. Growth hormone improves muscle protein metabolism and whole body nitrogen economy in man during a hyponitrogenous diet. Metabolism. 1991 Mar;40(3):315-22
Fryburg DA. Insulin-like growth factor I exerts growth hormone- and insulin-like actions on human muscle protein metabolism. Am J Physiol. 1994 Aug;267(2 Pt 1):E331-6.
Russell-Jones DL, Umpleby AM, Hennessy TR, Bowes SB, Shojaee-Moradie F, Hopkins KD, Jackson NC, Kelly JM, Jones RH, Sönksen PH. Use of a leucine clamp to demonstrate that IGF-I actively stimulates protein synthesis in normal humans. Am J Physiol. 1994 Oct;267(4 Pt 1):E591-8.
Jacob R, Hu X, Niederstock D, Hasan S, McNulty PH, Sherwin RS, Young LH. IGF-I stimulation of muscle protein synthesis in the awake rat: permissive role of insulin and amino acids. Am J Physiol. 1996 Jan;270(1 Pt 1):E60-6.
Buijs MM, Romijn JA, Burggraaf J, De Kam ML, Cohen AF, Frölich M, Stellaard F, Meinders AE, Pijl H. Growth hormone blunts protein oxidation and promotes protein turnover to a similar extent in abdominally obese and normal-weight women. J Clin Endocrinol Metab. 2002 Dec;87(12):5668-74.
Gibney J, Wolthers T, Johannsson G, Umpleby AM, Ho KK. Growth hormone and testosterone interact positively to enhance protein and energy metabolism in hypopituitary men. Am J Physiol Endocrinol Metab. 2005 Aug;289(2):E266-71.
Le Roith D, Scavo L, Butler A. What is the role of circulating IGF-I? Trends Endocrinol Metab. 2001 Mar;12(2):48-52. Review.
Mauras N, Haymond MW. Are the metabolic effects of GH and IGF-I separable? Growth Horm IGF Res. 2005 Feb;15(1):19-27. Review.
Laron Z. Laron syndrome (primary growth hormone resistance or insensitivity): the personal experience 1958-2003. J Clin Endocrinol Metab. 2004 Mar;89(3):1031-44.
Muhammad A, van der Lely AJ, Neggers SJ. Review of current and emerging treatment options in acromegaly. Neth J Med. 2015 Oct;73(8):362-7. Review.
Lupu F, Terwilliger JD, Lee K, Segre GV, Efstratiadis A. Roles of growth hormone and insulin-like growth factor 1 in mouse postnatal growth. Dev Biol. 2001 Jan 1;229(1):141-62.
Waters MJ. The growth hormone receptor. Growth Horm IGF Res. 2016 Jun;28:6-10.
Laron Z, Ginsberg S, Webb M. Nonalcoholic fatty liver in patients with Laron syndrome and GH gene deletion – preliminary report. Growth Horm IGF Res. 2008 Oct;18(5):434-8.
Sharara FI. Value of growth hormone in ovulation induction? Fertil Steril. 1996 Jun;65(6):1259-61.
Kolibianakis EM, Venetis CA, Diedrich K, Tarlatzis BC, Griesinger G. Addition of growth hormone to gonadotrophins in ovarian stimulation of poor responders treated by in-vitro fertilization: a systematic review and meta-analysis. Hum Reprod Update. 2009 Nov-Dec;15(6):613-22.
Russell SM, Spencer EM. Local injections of human or rat growth hormone or of purified human somatomedin-C stimulate unilateral tibial epiphyseal growth in hypophysectomized rats. Endocrinology. 1985 Jun;116(6):2563-7.
Schlechter NL, Russell SM, Greenberg S, Spencer EM, Nicoll CS. A direct growth effect of growth hormone in rat hindlimb shown by arterial infusion. Am J Physiol. 1986 Mar;250(3 Pt 1):E231-5.
Isaksson OG, Ohlsson C, Nilsson A, Isgaard J, Lindahl A. Regulation of cartilage growth by growth hormone and insulin-like growth factor I. Pediatr Nephrol. 1991 Jul;5(4):451-3. Review.
Wang J, Zhou J, Cheng CM, Kopchick JJ, Bondy CA. Evidence supporting dual, IGF-I-independent and IGF-I-dependent, roles for GH in promoting longitudinal bone growth. J Endocrinol. 2004 Feb;180(2):247-55.
Yakar S, Rosen CJ, Bouxsein ML, Sun H, Mejia W, Kawashima Y, Wu Y, Emerton K, Williams V, Jepsen K, Schaffler MB, Majeska RJ, Gavrilova O, Gutierrez M, Hwang D, Pennisi P, Frystyk J, Boisclair Y, Pintar J, Jasper H, Domene H, Cohen P, Clemmons D, LeRoith D. Serum complexes of insulin-like growth factor-1 modulate skeletal integrity and carbohydrate metabolism. FASEB J. 2009 Mar;23(3):709-19.
Ohlsson C, Mohan S, Sjögren K, Tivesten A, Isgaard J, Isaksson O, Jansson JO, Svensson J. The role of liver-derived insulin-like growth factor-I. Endocr Rev. 2009 Aug;30(5):494-535.
Sotiropoulos A, Ohanna M, Kedzia C, Menon RK, Kopchick JJ, Kelly PA, Pende M. Growth hormone promotes skeletal muscle cell fusion independent of insulin-like growth factor 1 up-regulation. Proc Natl Acad Sci U S A. 2006 May 9;103(19):7315-20.
Wakelam MJ. The fusion of myoblasts. Biochem J. 1985 May 15;228(1):1-12. Review.
Pfäffle RW, Blankenstein O, Wüller S, Kentrup H. Combined pituitary hormone deficiency: role of Pit-1 and Prop-1. Acta Paediatr Suppl. 1999 Dec;88(433):33-41. Review.
Hemchand K, Anuradha K, Neeti S, Vaman K, Roland P, Werner B, Sharmila B. Entire prophet of Pit-1 (PROP-1) gene deletion in an Indian girl with combined pituitary hormone deficiencies. J Pediatr Endocrinol Metab. 2011;24(7-8):579-80.
Waters MJ, Shang CA, Behncken SN, Tam SP, Li H, Shen B, Lobie PE. Growth hormone as a cytokine. Clin Exp Pharmacol Physiol. 1999 Oct;26(10):760-4. Review.
Jansson JO, Edén S, Isaksson O. Sexual dimorphism in the control of growth hormone secretion. Endocr Rev. 1985 Spring;6(2):128-50. Review.
Giustina A, Veldhuis JD. Pathophysiology of the neuroregulation of growth hormone secretion in experimental animals and the human. Endocr Rev. 1998 Dec;19(6):717-97. Review.
Hartman ML, Faria AC, Vance ML, Johnson ML, Thorner MO, Veldhuis JD. Temporal structure of in vivo growth hormone secretory events in humans. Am J Physiol. 1991 Jan;260(1 Pt 1):E101-10.
Takahashi Y, Kipnis DM, Daughaday WH. Growth hormone secretion during sleep. J Clin Invest. 1968 Sep;47(9):2079-90.
Ho KY, Veldhuis JD, Johnson ML, Furlanetto R, Evans WS, Alberti KG, Thorner MO. Fasting enhances growth hormone secretion and amplifies the complex rhythms of growth hormone secretion in man. J Clin Invest. 1988 Apr;81(4):968-75.
Jaffe CA, Ocampo-Lim B, Guo W, Krueger K, Sugahara I, DeMott-Friberg R, Bermann M, Barkan AL. Regulatory mechanisms of growth hormone secretion are sexually dimorphic. J Clin Invest. 1998 Jul 1;102(1):153-64.
Jessup SK, Dimaraki EV, Symons KV, Barkan AL. Sexual dimorphism of growth hormone (GH) regulation in humans: endogenous GH-releasing hormone maintains basal GH in women but not in men. J Clin Endocrinol Metab. 2003 Oct;88(10):4776-80.
Goldenberg N, Barkan A. Factors regulating growth hormone secretion in humans. Endocrinol Metab Clin North Am. 2007 Mar;36(1):37-55. Review.
Müller EE, Locatelli V, Cocchi D. Neuroendocrine control of growth hormone secretion. Physiol Rev. 1999 Apr;79(2):511-607. Review.
Murray RA, Maheshwari HG, Russell EJ, Baumann G. Pituitary hypoplasia in patients with a mutation in the growth hormone-releasing hormone receptor gene. AJNR Am J Neuroradiol. 2000 Apr;21(4):685-9.
Murray PG, Higham CE, Clayton PE. 60 YEARS OF NEUROENDOCRINOLOGY: The hypothalamo-GH axis: the past 60 years. J Endocrinol. 2015 Aug;226(2):T123-40.
Russell-Aulet M, Dimaraki EV, Jaffe CA, DeMott-Friberg R, Barkan AL. Aging-related growth hormone (GH) decrease is a selective hypothalamic GH-releasing hormone pulse amplitude mediated phenomenon. J Gerontol A Biol Sci Med Sci. 2001 Feb;56(2):M124-9
Dimaraki EV, Jaffe CA, Demott-Friberg R, Russell-Aulet M, Bowers CY, Marbach P, Barkan AL. Generation of growth hormone pulsatility in women: evidence against somatostatin withdrawal as pulse initiator. Am J Physiol Endocrinol Metab. 2001 Mar;280(3):E489-95.
Baumann G. Growth hormone heterogeneity: genes, isohormones, variants, and binding proteins. Endocr Rev. 1991 Nov;12(4):424-49. Review.
Vijayakumar A, Novosyadlyy R, Wu Y, Yakar S, LeRoith D. Biological effects of growth hormone on carbohydrate and lipid metabolism. Growth Horm IGF Res. 2010 Feb;20(1):1-7. doi: 10.1016/j.ghir.2009.09.002. Epub 2009 Oct 1. Review.
Herrington J, Carter-Su C. Signaling pathways activated by the growth hormone receptor. Trends Endocrinol Metab. 2001 Aug;12(6):252-7. Review.
Baumann G. Growth hormone binding protein. The soluble growth hormone receptor. Minerva Endocrinol. 2002 Dec;27(4):265-76. Review.
Bairoch A, Apweiler R. The SWISS-PROT protein sequence data bank and its new supplement TREMBL. Nucleic Acids Res. 1996 Jan 1;24(1):21-5.
Kelly PA, Djiane J, Postel-Vinay MC, Edery M. The prolactin/growth hormone receptor family. Endocr Rev. 1991 Aug;12(3):235-51. Review.
Vikman K, Carlsson B, Billig H, Edén S. Expression and regulation of growth hormone (GH) receptor messenger ribonucleic acid (mRNA) in rat adipose tissue, adipocytes, and adipocyte precursor cells: GH regulation of GH receptor mRNA. Endocrinology. 1991 Sep;129(3):1155-61.
Zou L, Menon RK, Sperling MA. Induction of mRNAs for the growth hormone receptor gene during mouse 3T3-L1 preadipocyte differentiation. Metabolism. 1997 Jan;46(1):114-8.
Leung KC, Waters MJ, Markus I, Baumbach WR, Ho KK. Insulin and insulin-like growth factor-I acutely inhibit surface translocation of growth hormone receptors in osteoblasts: a novel mechanism of growth hormone receptor regulation. Proc Natl Acad Sci U S A. 1997 Oct 14;94(21):11381-6.
Birzniece V, Sata A, Ho KK. Growth hormone receptor modulators. Rev Endocr Metab Disord. 2009 Jun;10(2):145-56.
Sawada T, Arai D, Jing X, Miyajima M, Frank SJ, Sakaguchi K. Molecular interactions of EphA4, growth hormone receptor, Janus kinase 2, and signal transducer and activator of transcription 5B. PLoS One. 2017 Jul 7;12(7):e0180785.
Brooks AJ, Dai W, O’Mara ML, Abankwa D, Chhabra Y, Pelekanos RA, Gardon O, Tunny KA, Blucher KM, Morton CJ, Parker MW, Sierecki E, Gambin Y, Gomez GA, Alexandrov K, Wilson IA, Doxastakis M, Mark AE, Waters MJ. Mechanism of activation of protein kinase JAK2 by the growth hormone receptor. Science. 2014 May 16;344(6185):1249783.
Liu Y, Berry PA, Zhang Y, Jiang J, Lobie PE, Paulmurugan R, Langenheim JF, Chen WY, Zinn KR, Frank SJ. Dynamic analysis of GH receptor conformational changes by split luciferase complementation. Mol Endocrinol. 2014 Nov;28(11):1807-19.
Waters MJ, Brooks AJ. JAK2 activation by growth hormone and other cytokines. Biochem J. 2015 Feb 15;466(1):1-11.
Brown RJ, Adams JJ, Pelekanos RA, Wan Y, McKinstry WJ, Palethorpe K, Seeber RM, Monks TA, Eidne KA, Parker MW, Waters MJ. Model for growth hormone receptor activation based on subunit rotation within a receptor dimer. Nat Struct Mol Biol. 2005 Sep;12(9):814-21. Epub 2005 Aug 21.
Lanning NJ, Carter-Su C. Recent advances in growth hormone signaling. Rev Endocr Metab Disord. 2006 Dec;7(4):225-35. Review.
Carter-Su C, Schwartz J, Smit LS. Molecular mechanism of growth hormone action. Annu Rev Physiol. 1996;58:187-207. Review.
Brooks AJ, Waters MJ. The growth hormone receptor: mechanism of activation and clinical implications. Nat Rev Endocrinol. 2010 Sep;6(9):515-25.
La questione degli Estrogeni nell’ambito delle preparazioni comprendenti farmaci per il miglioramento delle prestazioni, in special modo nel Bodybuilding, è generalmente mal compresa e, di conseguenza, mal gestita. Complice di questa “mala gestione” è l’ignoranza sia specifica, cioè portata da una scarsa disponibilità di testi di facile accesso (e comprensione) che trattano l’argomento in modo esaustivo, sia indotta dal comportamento della maggior parte dei preparatori o degli atleti “solisti” che ignorano totalmente la necessità di un aggiornamento continuo in tutte le componenti della preparazione (allenamento, nutrizione e supplementazione). Con questa pubblicazione è mia intenzione fornire una buona base di conoscenza sugli Estrogeni (loro biosintesi, azioni ecc…) e sulla migliore gestione di questi nel contesto di una preparazione.
Iniziamo per gradi…
Estrogeni e loro caratteristiche/azioni principali (nella donna e nell’uomo)
Estradiolo
Gli Estrogeni sono i principali ormoni sessuali femminili. Si tratta di ormoni steroidei responsabili dello sviluppo e della regolazione del sistema riproduttivo femminile e delle caratteristiche sessuali secondarie. Con il termine estrogeno ci si può anche riferire a qualsiasi sostanza, naturale o sintetica, che mima gli effetti dell’ormone naturale.(1) L’estrogeno prevalente in quantità e potenza è l’Estradiolo, anche se diversi metaboliti dell’estradiolo hanno anche una marcata attività ormonale estrogenica. Versioni sintetiche degli estrogeni vengono utilizzate come principio attivo di farmaci contraccettivi orali, nella terapia ormonale sostitutiva per le donne in postmenopausa, nelle donne ipogonadiche e nei transgender, oltre che nel trattamento di alcuni tumori ormone-sensibili come il cancro alla prostata e il cancro al seno. Gli Estrogeni sono una delle tre classi di ormoni sessuali, insieme agli Steroidi Androgeni/Anabolizzanti come il Testosterone e i progestinici come il Progesterone.
Gli Estrogeni sono sintetizzati in tutti i vertebrati (2) così come in alcuni insetti.(3) Le tre principali forme di estrogeno presenti naturalmente nelle donne sono l’Estrone (E1), l’Estradiolo (E2) e l’Estriolo (E3). Un altro tipo di Estrogeno chiamato Estetrolo (E4) viene prodotto solo durante la gravidanza. Quantitativamente, gli Estrogeni circolano a livelli inferiori rispetto agli Androgeni sia negli uomini che nelle donne.(4) Mentre i livelli di Estrogeni sono significativamente più bassi nei maschi rispetto alle femmine, gli Estrogeni, tuttavia, hanno anche importanti ruoli fisiologici nei soggetti di sesso maschile.(5)
Come tutti gli ormoni steroidei, gli Estrogeni penetrano facilmente attraverso la membrana cellulare. Una volta all’interno della cellula, si legano e attivano i recettori degli estrogeni (ER) che a loro volta modulano l’espressione di molti geni.(6) Inoltre, gli Estrogeni si legano e attivano rapidamente i recettori degli estrogeni di membrana (mER), (7)(8) come i GPER (GPR30).(9)
Come già precedentemente accennato, i tre principali Estrogeni presenti naturalmente nelle donne sono l’Estrone (E1), l’Estradiolo (E2) e l’Estriolo (E3). L’Estradiolo è l’estrogeno predominante negli anni riproduttivi sia in termini assoluti nel siero che in termini di attività estrogenica. Durante la menopausa, l’Estrone è l’Estrogeno circolante predominante mentre durante la gravidanza è l’Estriolo ad essere l’Estrogeno circolante predominante in termini di livelli sierici. Anche se l’Estriolo è il più abbondante dei tre estrogeni è anche il più debole, mentre l’Estradiolo è il più forte con una potenza di circa 80 volte quella dell’Estriolo.(10) Quindi, l’Estradiolo è l’Estrogeno più importante nelle donne non in gravidanza che si trovano tra le fasi del menarca e della menopausa.
Estetrolo
Tuttavia, durante la gravidanza questo ruolo passa all’Estriolo e nelle donne in postmenopausa l’Estrone diventa la forma primaria di Estrogeno nel corpo. Come già accennato, un altro tipo di Estrogeno chiamato Estetrolo (E4) viene prodotto solo durante la gravidanza. Oltre che dai follicoli ovarici, tutte le diverse forme di estrogeni sono sintetizzate a partire dagli Androgeni, in particolare dal Testosterone e dal Androstenedione, da parte dell’enzima aromatasi.
Altri substrati utilizzati dal corpo per la sintesi estrogenica, senza il coinvolgimento primario dell’enzima aromatasi, sono il 27-idrossicholesterolo, il Deidroepiandrosterone (DHEA), il 7-oxo-DHEA, il 7α-idrossi-DHEA, il 16α-idrossi-DHEA, il 7β-idrossiepiandrosterone, l’Androstenedione (A4), l’Androstenediolo (A5), il 3α-androstanediolo e il 3β-androstanediolo. Alcuni metaboliti estrogenici, come i catecol-estrogeni 2-idrossiestradiolo, 2-idrossestrone, 4-idrossiestradiolo e 4-idrossestrone, nonché il 16α-idrossiestrone, sono composti estrogenici con vari gradi di attività. L’importanza biologica di questi estrogeni minori non è del tutto chiara. Tutti questi composti possono avere importanti funzioni estrogeniche nel corpo.
Steroidogenesi; gli Estrogeni sono rappresentati in basso a destra.
Recettore degli Estrogeni alfa (ERα)
Le azioni degli Estrogeni sono mediate dal Recettore Estrogeno (ER), una proteina nucleare dimerica che si lega al DNA e controlla l’espressione genica. Come altri ormoni steroidei, l’Estrogeno entra passivamente nella cellula nella quale si lega e attiva il Recettore dell’Estrogeno. Il complesso ER si lega a specifiche sequenze di DNA chiamate “elemento di risposta ormonale” per attivare la trascrizione di geni bersaglio: in uno studio nel quale si è utilizzate una linea di cellule del cancro al seno estrogeno-dipendente come modello, sono stati identificati 89 di questi geni.(11) Poiché l’Estrogeno entra in tutte le cellule, le sue azioni dipendono dalla presenza del ER nella cellula. L’ER è espresso in tessuti specifici tra cui l’ovaio, l’utero e il seno. Gli effetti metabolici degli Estrogeni nelle donne in postmenopausa sono stati legati al polimorfismo genetico dell’ER. (12)
Mentre gli Estrogeni sono presenti sia negli uomini che nelle donne, sono solitamente presenti a livelli significativamente più alti nelle donne in età riproduttiva. Essi promuovono lo sviluppo delle caratteristiche sessuali secondarie femminili, come i seni, e sono anche coinvolti nell’ispessimento dell’endometrio e altri aspetti della regolazione del ciclo mestruale. Nei maschi, l’Estrogeno regola alcune funzioni del sistema riproduttivo importanti per la maturazione dello sperma (13)(14)(15) e sembra essere necessario per una sana libido.(16) Inoltre, vi sono molti altri cambiamenti strutturali indotti dall’Estrogeno oltre ad altre funzioni.
Gli Estrogeni, nelle femmine, sono prodotti principalmente dalle ovaie e, durante la gravidanza, dalla placenta.(17) L’Ormone Follicolo Stimolante (FSH) stimola la produzione ovarica di Estrogeni da parte delle cellule della granulosa dei follicoli ovarici e dei corpi lutei. Alcuni Estrogeni vengono anche prodotti in quantità minori in altri tessuti come il fegato, le ghiandole surrenali e i seni. Queste fonti secondarie di Estrogeni sono particolarmente importanti nelle donne in postmenopausa. Anche nelle cellule adipose vengono sintetizzati Estrogeni (vedi azione dell’Enzima Aromatasi)(18)
Nelle femmine, la sintesi degli Estrogeni inizia nelle cellule della teca follicolare interna nell’ovaio, dalla sintesi del Androstenedione a partire dal Colesterolo. L’Androstenedione è uno steroide con debole attività androgenica il cui scopo è prevalentemente quello di fungere da precursore di più potenti androgeni come il Testosterone così come dell’Estrogeno. Questo steroide attraversa la membrana basale nelle cellule della granulosa circostanti, dove o viene immediatamente convertito in Estrone, o in Testosterone e poi successivamente in Estradiolo. La conversione del Androstenedione in Testosterone è catalizzata dal 17β-idrossisteroide deidrogenasi (17β-HSD), mentre la conversione del Androstenedione e del Testosterone in Estrone ed Estradiolo è rispettivamente catalizzata dal aromatasi, entrambi enzimi che sono espressi nelle cellule della granulosa. Al contrario, le cellule della granulosa non dispongono di 17α-idrossilasi e 17,20-liasi, mentre le cellule della teca follicolare interna esprimono questi enzimi e il 17β-HSD, ma non l’aromatasi. Quindi sia le cellule della granulosa che le cellule della teca follicolare interna sono essenziali per la produzione di Estrogeni nelle ovaie.
I livelli di Estrogeni variano durante il ciclo mestruale, con livelli più alti vicino alla fine della fase follicolare e poco prima dell’ovulazione.
Intervalli di riferimento nelle concentrazioni di Estradiolo, il principale Estrogeno circolante, durante il ciclo mestruale. CYP1A1
Gli Estrogeni sono metabolizzati tramite idrossilazione da enzimi citocromo P450 quali CYP1A1 e CYP3A4 e tramite coniugazione del estrogeno sulfotransferasi (sulfato) e del UDP-glucuroniltransferasi (glucuronidazione). Inoltre, l’Estradiolo viene deidrogenato dal 17β-idrossiesteroide deidrogenasi nel molto meno potente Estrone. Queste reazioni si verificano principalmente nel fegato, ma avvengono anche in altri tessuti.
Gli estrogeni sono implicati nel rilascio di GH e IGF-1 sia nell’uomo che nella donna. Le donne hanno mediamente livelli di GH più elevati sebbene sembri che ne siano meno sensibili (vedi percentuale di grasso corporeo femminile).
Quindi, per riepilogare, gli Estrogeni nelle femmine regolano la maturazione sessuale intervenendo nello sviluppo dell’apparato genitale.
La loro massiccia secrezione in epoca puberale induce la chiusura delle cartilagini di coniugazione delle ossa lunghe, terminando di fatto, la fase di accrescimento staturale.
Gli estrogeni stimolano lo sviluppo stromale della mammella e il mantenimento delle caratteristiche femminili secondarie (crescita delle mammelle, distribuzione dei peli, voce, statura, ossatura, distribuzione del grasso).
Permettono la fecondazione e la gravidanza, intervenendo nella regolazione del ciclo mestruale.
Regolano la distribuzione del grasso corporeo, favorendone il deposito nelle anche, nelle natiche, nelle cosce e nell’addome al di sotto dell’ombelico.
Mantengono il trofismo osseo ed hanno quindi azione protettiva nei confronti dell’osteoporosi
Stimolano la sintesi di trigliceridi e l’aumento delle lipoproteine ad alta densità (HDL) proteggendo le pareti vasali dal danno arteriosclerotico.
Stimolano la lipolisi nel tessuto muscolare ed adiposo. Per questo motivo gli estrogeni migliorano la prestazione degli sport di durata risparmiando il glicogeno muscolare a scapito degli acidi grassi
Regolano molte funzioni cerebrali fra cui l’attenzione e la memoria.
Stimolano la sintesi epatica di numerosi enzimi e proteine (SHBG, angiotensinogeno).
Enzima Aromatasi
Nell’uomo, invece, gli estrogeni circolanti derivano per la maggior parte dall’aromatizzazione degli Androgeni circolanti (19). Il complesso enzimatico conosciuto come aromatasi è responsabile dell’aromatizzazione dell’anello A degli Androgeni che comporta la trasformazione in sostanze ad attività estrogenica. (20) (21) (22)
Recettore degli Estrogeni beta (ERβ)
La trascrizione del gene per l’aromatasi e l’espressione dell’enzima aromatasi avviene in un ampio numero di tessuti, come i testicoli, il tessuto adiposo, il muscolo, il fegato, il cervello, i follicoli piliferi, i fibroblasti etc. (20). L’azione degli Estrogeni, come già accennato, si esplica attraverso il loro legame a recettori specifici, che appartengono alla ‘superfamiglia’ dei recettori nucleari (23). Finora sono stati caratterizzati due tipi di recettori per gli Estrogeni (ER): l’ER classico, ora denominato ERα (24) (25) (26), ed un altro sottotipo denominato ERβ (27). Entrambi sono recettori intranucleari che, una volta attivati, modulano a livello genomico l’attività di trascrizione genica. Di recente è stata documentata anche una via non-genomica dell’azione degli Estrogeni, che prevede un’interazione plasma-membrana del recettore estrogenico (28) (29) (30).
Conversione del Testosterone in Estradiolo
L’Estradiolo (E2) nel maschio è sintetizzato dal Testosterone attraverso l’enzima aromatasi o dall’Estrone attraverso la 17β-idrossisteroido deidrogenasi (17β-HSD) (31). L’E2 prodotto giornalmente nel maschio è stimato essere circa 35-45 μg (0.130- 0.165 μmol), dei quali il 15-20% è direttamente prodotto dal testicolo (32) (19), un 60% è prodotto dall’aromatizzazione periferica del Testosterone circolante ed il restante 20% dalla conversione periferica dell’Estrone. L’Estrone (E1) può anche essere direttamente secreto dai surreni (33) o derivare dall’aromatizzazione periferica dell’Androstenedione, quest’ultimo prodotto in parte dal surrene ed in parte derivante dalla conversione periferica del Testosterone. I testicoli comunque contribuiscono maggiormente, rispetto ai surreni, alla totale produzione di Estradiolo circolante e la via enzimatica di sintesi maggiormente coinvolta nella secrezione di Estrogeni nel maschio è quella aromatasica, mentre la via della 17β-HSD ha una minore rilevanza “in vivo”. Solo il 2-3% dell’Estradiolo circolante è libero, la maggior parte dell’ormone è legato all’albumina o alla globulina legante gli ormoni sessuali (SHBG) (34) e la frazione non legata alla SHBG è considerata bioattiva a livello dei tessuti bersaglio.
SHBG
I progressi negli studi effettuati negli ultimi anni hanno infatti dimostrato in maniera inequivocabile il ruolo fisiologico che gli Estrogeni hanno anche nell’uomo. Gli Estrogeni svolgono un ruolo chiave sul sistema immunitario, a livello del timo, agiscono a livello ipofisario e cardiovascolare, osseo e del metabolismo glucidico e lipidico.
Un eccesso estrogenico nella donna favorisce l’accumulo di tessuto adiposo e la comparsa di ritenzione idrica, oltre ad esporre la donna ad un elevato rischio di sviluppare alcune forme di cancro come quello alla mammella, l’insulinoresistenza, l’infertilità e l’ovaiopolicistico. Nell’uomo l’eccesso estrogenico è legato ad effetti collaterali spiacevoli tra i quali troviamo la ginecomastia, la diminuzione del desiderio sessuale, problemi di erezione (che si verifica anche con un calo estrogenico eccessivo), e diminuzione della fertilità.
Ricordiamoci però che, tanto nella donna come nell’uomo, un livello estremamente basso di Estrogeni comporta diversi problemi tra i quali emergono: elevati livelli sierici di Trigliceridi, LDL e colesterolo totale con ridotti livelli di HDL (35)(36)(37)(38)(39), ridotti livelli di LDL, HDL e colesterolo totale con Trigliceridi normali (40)(41), osteoporosi, dolore osseo, e diabete mellito di tipo II (37).
Si noti che la descrizione di pazienti maschi affetti da deficit congenito di Estrogeni (42) (43) (44) (45) (46) (47) (48) (49) (50) (51) ha fornito un modello in vivo unico che ha permesso di stabilire che la completa ossificazione delle cartilagini epifisarie, anche nel maschio, non può avvenire senza l’azione degli Estrogeni. Inoltre, è stato dimostrato che gli Androgeni da soli non sono sufficienti per garantire una normale mineralizzazione dello scheletro in età adulta, dal momento che un quadro di osteopenia e/o osteoporosi è stato documentato in tutti i pazienti con deficit congenito di Estrogeni (42) (43) (44) (45) (46) (47) (48) (49) (50) (51). Ad oggi è stato chiarito che gli effetti degli steroidi sessuali sul processo di mineralizzazione ossea nel maschio sono solo in parte da ascrivere agli Androgeni di origine testicolare, mentre l’azione degli Estrogeni, che derivano dall’aromatizzazione degli androgeni stessi, sembra giocare un ruolo di maggiore importanza (52) (53). Infatti in letteratura vi sono evidenze riguardo l’associazione tra ipogonadismo e riduzione della massa ossea in entrambi i sessi (54) (55) (56) (57) (58) (59). Inoltre, il concetto generale che gli Androgeni mantengono la massa ossea nell’uomo, così come gli Estrogeni la mantengono nella donna, continua ad essere il punto di vista prevalente tra i medici, sebbene recenti studi suggeriscano il contrario (42) (43) (44) (45) (60) (61). Nel 1997, infatti, è stata descritta l’efficacia della terapia Estrogenica nell’ottenere sia la completa ossificazione delle epifisi che l’aumento di densità minerale ossea (BMD) in un paziente maschio affetto da deficit congenito di aromatasi (44).
Fonti degli estrogeni circolanti nel maschio ed organi e tessuti bersaglio dell’azione degli estrogeni. La figura indica la produzione in µmol/L.(modificata da Baird et al., 1969a e Saez et al., 1972)
Una funzione interessante dell’Estradiolo nel maschio è rappresentata dal suo impatto sulla funzione sessuale. Come già accennato, l‘Estradiolo negli uomini è essenziale per modulare la libido, la funzione erettile e la spermatogenesi. I recettori degli Estrogeni, così come l’aromatasi, sono abbondanti nel cervello, nel pene e nei testicoli, organi importanti per la funzione sessuale. Nel cervello, la sintesi dell’Estradiolo è elevata nelle aree correlate all’auda sessuale. Inoltre, nel pene, i recettori degli Estrogeni si trovano in tutto il corpus cavernosum con elevata concentrazione intorno ai fasci neurovascolari. Un livello basso di Testosterone e elevato di Estrogeni aumenta l’incidenza della disfunzione erettile indipendentemente l’uno dall’altro. Nei testicoli, la spermatogenesi è modulata a tutti i livelli dagli Estrogeni, a partire dall’asse ipotalamo-ipofisi-gonadi, seguita dalle cellule di Leydig, Sertoli e germinali e terminando con l’epitelio ductale, l’epididimo e lo sperma maturo. La regolazione delle cellule testicolari mediante l’Estradiolo mostra sia un’influenza inibitoria che una stimolatoria, indicando un intricata sinfonia di modulazione dose-dipendente e temporalmente sensibile.(62)
Adesso che abbiamo una buona conoscenza di base sugli Estrogeni e le loro azioni sia nell’uomo che nella donna, possiamo passare a trattare la questione della gestione funzionale degli Estrogeni durante la preparazione.
AAS aromatizzabili e gestione estrogenica
Come ormai ben sappiamo, il Testosterone è il substrato primario usato nell’uomo per la sintesi dell’Estradiolo, principale ormone sessuale femminile. Con una leggera alterazione strutturale del Testosterone dovuta all’azione dell’enzima aromatasi, l’Estradiolo viene prodotto nel maschio. L’attività dell’enzima aromatasi nel maschio si verifica in diverse regioni del corpo , compreso il tessuto adiposo, il fegato, le gonadi, il Sistema Nervoso Centrale e il tessuto muscolo scheletrico.(63) (64) (65) (66) (67) In un soggetto sano di sesso maschile , la quantità di Estrogeni prodotta non è generalmente molto significativa ed è basata sulle richieste e caratteristiche fisiologiche divenendo anche un vantaggio, come precedentemente visto, nella qualità e quantità dei valori di colesterolo e nella mineralizzazione ossea. Nei casi in cui le concentrazioni di Estrogeni (in particolare dell’Estradiolo) raggiungono livelli ematici elevati, che ciò avvenga come conseguenza non patologica (vedi obesità e cattiva alimentazione) o patologica (vedi neoplasia al testicolo), o come conseguenza del uso di AAS aromatizzabili e dell’alterazione omeostatica dovuta all’uso di tali composti, il soggetto sperimenta effetti avversi tra i quali vi sono la comparsa di ritenzione idrica, l’accumulo adiposo con modello femminile e la ginecomastia. Per questo motivo, molti atleti si concentrano sulla riduzione dell’incremento e/o dell’attività degli estrogeni nel corpo con l’uso di inibitori dell’aromatasi (es. Anastrozolo, Letrozolo e Exemestane), di Modulatori Selettivi del Recettore degli Estrogeni (SERM) (es. Tamoxifene e Raloxifene) o di inibitori della biosintesi steroidea (es. Aminoglutettimide e Trylostano), in specie quando il ciclo che stanno seguendo è composto da molecole altamente aromatizzabili (es. Testosterone e Methandrostenolone) o quando l’atleta desidera avere un maggior controllo estrogenico in un contesto nel quale si cerca di aumentare la definizione muscolare.
Oxymetholone
Nota:Con l’uso del Oxymetholone, il quale possiede attività estrogenica intrinseca, la gestione del fattore estrogenico (in particolare con la presenza di substrati soggetti all’aromatizzazione) assume caratteristiche leggermente più “complesse”. L’Oxymetholone è infatti un AAS avente azione altamente estrogenica. Tale azione, per l’appunto, non è dipendente dall’aromatizzazione del composto in estrogeno – essendo un derivato del DHT non può essere aromatizzato. L’uso di inibitori dell’aromatasi come l’Anastrozolo o l’Exemestane non influiscone sulla estrogenicità relativa di questo AAS. Alcuni hanno suggerito che l’alto livello di attività estrogenica del Oxymetholone sia in realtà dovuto alla presunta azione progestinica del composto, simile al Nandrolone. Gli effetti collaterali estrogenici e progestinici possono essere molto simili (anche perché l’azione dei due steroidi è co-attiva), il che ha reso plausibile questa ipotesi. Tuttavia, esiste uno studio nel quale è stata esaminata l’attività progestinica del Oxymetholone. Lo studio in questione stabilì che non vi era alcuna attività progestinica derivante dal composto.(68) Con tali risultati, sembra plausibile che l’Oxymetholone possa attivare il recettore degli estrogeni similmente a, ma più profondamente, dell’androgeno estrogenico Methandriolo. In definitiva, l’attività estrogenica di questo composto può essere gestita solamente con l’uso di SERM come il Tamoxifene o il Raloxifene dal momento che, per i motivi sopra detti, composti anti aromatasi e gli inibitori della biosintesi steroidea non hanno alcuna incidenza sull’attività estrogenica di questo AAS.
Anche alla luce di quanto accennato in precedenza, non dobbiamo tuttavia essere indotti a pensare che gli Estrogeni non diano nessun beneficio prestativo. Livelli di Estradiolo mantenuti nei limiti del range di riferimento offrono diversi vantaggi.
Gli atleti sono da anni a conoscenza del fatto che gli AAS aromatizzabili presentano un vantaggio nell’anabolismo muscolare, ma è solo di recente che siamo arrivati finalmente a capire i meccanismi che stanno alla base di tale effetto. Sembra che i motivi vadano oltre l’aumento di peso e di forza che si attribuiscono alla ritenzione idrica data dall’attività estrogenica, comprendendo che questo ormone (Estradiolo) possiede effettivamente un effetto diretto sui processi anabolici. Ciò si manifesta attraverso l’aumento dell’utilizzo del Glucosio, la secrezione dell’Ormone della Crescita (e di IGF-1) e la proliferazione dei recettori degli Androgeni.
Estrogeni e utilizzo del Glucosio
G6PD
Gli Estrogeni possono giocare un ruolo molto importante nella promozione di uno stato anabolico influenzando l’utilizzo del Glucosio nel tessuto muscolare. Ciò avviene attraverso un’alterazione del livello di glucosio 6-fosfato deidrogenasi disponibile, un enzima direttamente legato all’uso del glucosio per la crescita e il recupero del tessuto muscolare.(69) (70) Più specificamente, il G6PD è l’enzima che catalizza la prima reazione della via dei pentoso fosfati (definita anche Shunt dell’Esosomonofosfato [HMP shunt] o PPP da Pentose phosphate pathway), un processo metabolico citoplasmatico, parallelo alla glicolisi, in grado di generare NADPH e zuccheri pentosi (a 5 atomi di carbonio). Durante il periodo di rigenerazione tissutale seguente il danno muscolare, i livelli di G6PD aumentano considerevolmente, il che è ritenuto essere un meccanismo che il corpo attua per migliorare il recupero quando necessario. Sorprendentemente, scopriamo che l’Estrogeno è direttamente legato al livello di G6PD che deve essere messo a disposizione delle cellule in questa fase di recupero. In sintesi, gli Estrogeni svolgono anche una azione metabolica accelerando la sintesi degli acidi nucleici, delle proteine e del glicogeno.
Il legame tra Estrogeno e G6PD è stato stabilito attraverso uno studio nel quale è stato dimostrato che i livelli di questo enzima deidrogenasi aumentano dopo la somministrazione di Testosterone Propionato. Lo studio ha inoltre mostrato che era l’aromatizzazione del Testosterone in Estradiolo ad essere direttamente responsabile di questo aumento e non l’azione Androgena del AAS.(71) In questo studio sono stati presi in esame anche due composti non-aromatizzabili, il Dihydrotestosterone e il Fluoxymesterone, ma l’effetto avuto con l’uso di Testosterone Propionato non è stato replicato. Inoltre, l’effetto positivo del Testosterone Propionato è stato bloccato quando è stato aggiunto l’inibitore dell’aromatasi 4-idrossiandrostenedione (Formestano), mentre la somministrazione di 17-beta Estradiolo da solo ha causato un aumento di G6PD simile a quello osservato con il Testosterone Propionato. Il 17-alfa Estradiolo, isomero ormonalmente inattivo del 17-beta Estradiolo, che non può legarsi al recettore estrogenico, non ha avuto alcun effetto sul G6PD. Ulteriori prove sono state fatte utilizzando il Testosterone Propionato con l’antiandrogeno Flutamide dimostrando che anche quest’ultimo farmaco non ha alterato negativamente le concentrazioni di G6PD, sottolineando l’effetto indipendente dall’azione del Testosterone con il recettore androgeno.
Estrogeni e GH/IGF-1
L’Estrogeno può anche svolgere un ruolo importante nella produzione dell’Ormone della Crescita e del IGF-1. L’IGF-1 (Fattore di Crescita Insulino-Simile) è un ormone anabolizzante rilasciato nel fegato e in vari tessuti periferici attraverso lo stimolo dell’Ormone della Crescita. L’IGF-1 è il principale responsabile dell’attività anabolica dell’Ormone della Crescita, attraverso una maggiore ritenzione di azoto / sintesi proteica e iperplasia cellulare (proliferazione). Uno dei primi studi da portare all’attenzione riguardante la questione qui trattata ha esaminato gli effetti del SERM Tamoxifene sui livelli di IGF-1, dimostrando che il composto esaminato ha un effetto soppressivo.(72) Un secondo studio, forse più degno di nota, svolto nel 1993, ha esaminato gli effetti della terapia sostitutiva del Testosterone sui soli livelli di GH e IGF-1 e li ha confrontati con gli effetti del Testosterone combinato nuovamente con il Tamoxifene. (73) Quando è stato somministrato Tamoxifene, i livelli di GH e IGF-1 hanno subito un notevole calo, mentre entrambi i valori sono stati elevati con la somministrazione di solo Testosterone Enantato. Un altro studio ha mostrato come la somministrazione settimanale di 300mg di Testosterone Enantato causi un lieve aumento del IGF-1 in uomini normali. Questi 300mg di Testosterone Enantato hanno causato un aumento dei livelli di Estradiolo, cosa normale con tale dose. Questo è stato confrontato con l’effetto portato con lo stesso dosaggio di Nandrolone Decanoato; tuttavia, questo AAS non è riuscito a produrre lo stesso aumento nei livelli di IGF-1. Questo risultato è molto interessante, specialmente quando notiamo che i livelli di Estrogeni sono stati effettivamente ridotti quando questo AAS è stato somministrato (ricordiamoci che il Nandrolone aromatizza il 20% del Testosterone). (74) Un altro studio ha dimostrato che la secrezione di GH e IGF-1 è aumentata con la somministrazione di Testosterone in maschi con pubertà ritardata, mentre il Dihydrotestosterone (non aromatizzabile) sembra sopprimere la secrezione di GH e IGF-1. (75)
Estrogeni e Recettori degli Androgeni
Recettore degli Androgeni
È stato anche dimostrato che l’Estrogeno può aumentare la concentrazione dei recettori degli androgeni in alcuni tessuti. Ciò è stato dimostrato in studi svolti su ratti che hanno esaminato gli effetti degli Estrogeni sui Recettori degli Androgeni cellulari in animali sottoposti ad orchiectomia (rimozione dei testicoli, spesso effettuata per diminuire la produzione endogena di Androgeni). Secondo lo studio, la somministrazione di Estrogeni ha determinato un aumento del legame recettoriale nel muscolo levator ani del Metribolone pari al 480%. (76) La spiegazione per tale evento suggerita è che l’Estrogeno stimola direttamente la proliferazione dei Recettori Androgeni, o forse ne diminuisce il tasso di danneggiamento. Anche se la crescita del muscolo levator ani è comunemente usata come riferimento per determinare l’attività anabolica dei composti steroidei, esso è un muscolo dell’organo sessuale ed è diverso dal tessuto muscolo scheletrico in quanto possiede una concentrazione molto più alta di Recettori Androgeni. Questo studio, tuttavia, ha esaminato l’effetto sui AR dato dagli Estrogeni nei tessuti musco scheletrici veloci (tibialis anterior e extensor digitorum longus), ma senza notare lo stesso aumento visto nel levator ani. Anche se scoraggiante come notizia, il fatto che l’Estrogeno possa aumentare il legame del Recettore degli Androgeni in qualsiasi tessuto rimane una scoperta estremamente significativa, soprattutto alla luce del fatto che ora sappiamo che gli Androgeni hanno alcuni effetti positivi sulla crescita muscolare mediati al di fuori del tessuto muscolare.
Estrogeni e fatica/stanchezza
La così detta “Steroid Fatigue” è una frase comunemente utilizzata in riferimento ad un’altra importante funzione dell’Estrogeno sia nel corpo maschile che femminile, vale a dire la sua capacità di promuovere uno stato mentale di vigilanza. Data la larga disponibilità di potenti inibitori dell’aromatasi di terza generazione, i Bodybuilder odierni raggiungono (a volte) un livello di soppressione degli Estrogeni molto più marcata di quanto fosse stato possibile in passato. Spesso associati a questa marcata soppressione si manifestano stati di stanchezza. In tali condizioni, l’atleta, anche se sta seguendo un ciclo correttamente formulato, potrebbe non essere in grado di massimizzare i propri risultati di miglioramento della condizione fisica a causa di un’incapacità di allenarsi con pieno vigore. Questo effetto è talvolta anche soprannominato “letargia steroidea”. La ragione principale per cui ciò accade è legata all’importante azione di supporto all’attività della Serotonina data dall’Estrogeno. La Serotonina è uno dei principali neurotrasmettitori del corpo, di essenziale importanza per un adeguata lucidità mentale e un regolare ciclo sonno / veglia. (77) (78)L’alterazione di questo neurotrasmettitore è associata anche alla sindrome da affaticamento cronico, (79) (80) e ciò ci fa comprendere quanto possa essere incisiva in particolare per la stanchezza. L’abbassamento dei livelli estrogenici nella menopausa è stata associata anche alla stanchezza, (81) così come l’uso clinico di inibitori dell’aromatasi più recenti (e più potenti) come l’Anastrozolo, (82) il Letrozolo, (83) l’Exemestane, (84) e il Fadrozolo (85) in alcuni pazienti. Questo effetto deve essere preso in importante considerazione quando si pianifica un ciclo. Anche se non tutti notano questo problema quando il livello estrogenico è basso, per coloro i quali lo subiscono, l’aggiunta di un po’ di Testosterone o di altro AAS soggetto all’aromatizzazione (oltre che l’abbassamento del composto AI) può aiutare a correggere di molto questo sintomo. È anche da notare che l’uso di AAS non aromatizzabili a volte causa questo effetto, probabilmente dovuto alla soppressione della produzione naturale di Testosterone (abbassando la disponibilità del principale substrato usato dal corpo maschile per sintetizzare Estradiolo).
Methylestradiolo
*Nota:gli AAS metilati in C-17, soggetti ad aromatizzazione (vedi Methyltestosterone e Mathandrostenolone), convertono nel potente Methylestradiolo, o 17α-methylestradiolo (17α-ME). A causa della presenza del gruppo 17α-metile, il Methylestradiolo non può essere disattivato per ossidazione del gruppo 17β-idrossile, con conseguente migliorata stabilità metabolica e potenza di legame recettoriale (e conseguente attività estrogenica) rispetto all’Estradiolo. (86)(87)(88)
Quindi, come gestire il fattore estrogenico durante la preparazione?
Dave Bautista mostra un classico esempio di cattiva gestione estrogenica.
Arrivati a questo punto, quale significato ha quanto è stato trattato per un Bodybuilder che cerca di ottenere una forma ottimale? Fondamentalmente penso che il punto chiave sia quello di mantenere un approccio prudente all’uso di farmaci per il controllo estrogenico, un approccio che deve essere dettato dalle reali esigenze verificate attraverso appositi esami ematici. Ovviamente, è chiaro che l’approccio all’uso dei composti per il controllo estrogenico debba variare a seconda dell’obbiettivo (vale a dire se l’obbiettivo primario è l’aumento della massa muscolare o se è l’aumento della definizione muscolare). Ripeto, è ovvio che l’utilizzo di composti volti al controllo estrogenico è una chiara necessità qualora si manifestassero effetti collaterali estrogenici, come lo è, nel caso, il calo dei composti soggetti all’azione dell’enzima aromatasi, ma gli esami ematici sono sempre di primaria importanza ai fini di una gestione ottimale del problema. La ginecomastia è certamente un problema indesiderato per l’utilizzatore di AAS, come lo è la ritenzione idrica ed un eccessivo accumulo di massa grassa. Ma se tali problemi non emergono, o se dagli esami ematici non emerge la reale necessità, l’aggiunta di composti per il controllo estrogenico (che siano AI, SERM o inibitori della biosintesi steroidea) può inficiare i risultati ottenibili in un contesto “Bulk”.
Quindi, concludendo, in un contesto “Bulk”, al fine di mantenere i benefici estrogenici senza incappare negli effetti collaterali estrogeno-dipendenti, la soglia ematica di Estradiolo dovrebbe mantenersi all’interno del range medio di riferimento; ciò significa che, in teoria, non dovrebbe ne scendere al di sotto dei 30pg/pg ne salire al di sopra dei 60 pg/ml. In un contesto “Cut” o “Pre-Gara”, ovviamente, le cose cambiano. In tali contesti, considerando gli effetti estrogenici anche sullo spessore della pelle, la cosa migliore sarebbe optare per il mantenimento di un livello di estrogeni basso-normale (20-30pg/ml) per tutta la fase “Cut” della preparazione per poi, se si è in preparazione alla gara, calare i livelli a 10pg o meno nell’ultimo paio di settimane prima del contest.
Per le atlete il discorso è simile anche se diverso sotto alcuni aspetti. In ambito femminile il controllo dei livelli e dell’attività estrogenica è di importanza anche maggiore per il raggiungimento di una forma da gara ottimale (vedere ad esempio “Nolvadex (Tamoxifene Citrato)”e “RALOXIFENE E COMPOSIZIONE CORPOREA NELLE DONNE”).
In definitiva, come logica conclusione, il metodo d’approccio per la gestione estrogenica deve basarsi sulle reali necessità, su dati oggettivi, e non su un fallace e molto poco scientifico “passa parola” dettato dall'”esperienza” di qual si voglia agonista o ex tale…
2- Ryan KJ (August 1982). “Biochemistry of aromatase: significance to female reproductive physiology”. Cancer Res. 42 (8 Suppl): 3342s–3344s. PMID7083198.
5- Lombardi G, Zarrilli S, Colao A, Paesano L, Di Somma C, Rossi F, De Rosa M (2001). “Estrogens and health in males”. Molecular and Cellular Endocrinology. 178 (1–2): 51–5. doi:10.1016/S0303-7207(01)00420-8. PMID11403894.
7- Soltysik K, Czekaj P (April 2013). “Membrane estrogen receptors — is it an alternative way of estrogen action?”. J. Physiol. Pharmacol. 64 (2): 129–42. PMID23756388.
12- Darabi M, Ani M, Panjehpour M, Rabbani M, Movahedian A, Zarean E (2011). “Effect of estrogen receptor β A1730G polymorphism on ABCA1 gene expression response to postmenopausal hormone replacement therapy”. Genet Test Mol Biomarkers. 15 (1–2): 11–5. doi:10.1089/gtmb.2010.0106. PMID21117950.
14- Hess RA, Bunick D, Lee KH, Bahr J, Taylor JA, Korach KS, Lubahn DB (1997). “A role for estrogens in the male reproductive system”. Nature. 390 (6659): 447–8. doi:10.1038/37352. PMID9393999.
16- Hill RA, Pompolo S, Jones ME, Simpson ER, Boon WC (2004). “Estrogen deficiency leads to apoptosis in dopaminergic neurons in the medial preoptic area and arcuate nucleus of male mice”. Mol. Cell. Neurosci. 27 (4): 466–76. doi:10.1016/j.mcn.2004.04.012. PMID15555924.
18- Nelson LR, Bulun SE (September 2001). “Estrogen production and action”. J. Am. Acad. Dermatol. 45 (3 Suppl): S116–24. doi:10.1067/mjd.2001.117432. PMID11511861.
19- MacDonald PC, Madden JD, Brenner PF, Wilson JD, Siiteri PK: Origin of estrogen in normal men and in women with testicular feminization. J Clin Endocrinol Metab 49:905-916, 1979
20- Simpson ER, Mahendroo MS, Means GD, Kilgore MW, Hinshelwood MM, Graham-Lorence S, Amarneh B, Ito Y, Fisher CR, Michael MD, et al.: Aromatase cytochrome P450, the enzyme responsible for estrogen biosyn- thesis. Endocr Rev 15:342-355, 1994
21- Simpson ER, Zhao Y, Agarwal VR, Michael MD, Bulun SE, Hinshelwood MM, Graham-Lorence S, Sun T, Fisher CR, Qin K, Mendelson CR: Aromatase expression in health and disease. Recent Prog Horm Res 52:185-213; discus- sion 213-184, 1997
22- Simpson ER: Aromatase: biologic relevance of tissue-specific expression. Semin Reprod Med 22:11-23, 2004
23- Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM: The nuclear recep- tor superfamily: the second decade. Cell 83:835-839, 1995
24- Green S, Walter P, Kumar V, Krust A, Bornert JM, Argos P, Chambon P: Human oestrogen receptor cDNA: sequence, expression and homology to v-erb-A. Nature 320:134-139, 1986
25- Greene GL, Gilna P, Waterfield M, Baker A, Hort Y, Shine J: Sequence and expression of human estrogen receptor complementary DNA. Science 231:1150-1154, 1986
26- Kuiper GG, Carlsson B, Grandien K, Enmark E, Haggblad J, Nilsson S, Gustafsson JA: Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology 138:863-870, 1997
27- Enmark E, Gustafsson JA: Oestrogen receptors – an overview. J Intern Med 246:133-138, 1999
28- Levin ER: Cellular Functions of the Plasma Membrane Estrogen Receptor. Trends Endocrinol Metab 10:374-377, 1999
29- Pedram A, Razandi M, Levin ER: Nature of functional estrogen receptors at the plasma membrane. Mol Endocrinol 20:1996-2009, 2006
30- Filardo E, Quinn J, Pang Y, Graeber C, Shaw S, Dong J, Thomas P: Activation of the novel estrogen receptor G protein-coupled receptor 30 (GPR30) at the plasma membrane. Endocrinology 148:3236-3245, 2007
31- de Ronde W, Pols HA, van Leeuwen JP, de Jong FH: The importance of oestrogens in males. Clin Endocrinol (Oxf) 58:529-542, 2003
32- Baird DT, Horton R, Longcope C, Tait JF: Steroid dynamics under steady- state conditions. Recent Prog Horm Res 25:611-664, 1969
33- Baird DT, Uno A, Melby JC: Adrenal secretion of androgens and oestrogens. J Endocrinol 45:135-136, 1969
34- Dunn JF, Nisula BC, Rodbard D: Transport of steroid hormones: bind- ing of 21 endogenous steroids to both testosterone-binding globulin and corticosteroid-binding globulin in human plasma. J Clin Endocrinol Metab 53:58-68, 1981
35- Carr BR: Disorders of the ovaries and female reproductive tract. Williams Textbook of Endocrinology. 11th ed. W.B. Saunders Company ed. Polonsky KS, Melmed S, Kronenberg HM, Larsen PR, Eds. Philadelphia, 2008
36- Weinbauer GF, Nieschlag E: The role of testosterone in spermatogene- sis. Springer-Verlag ed. Nieschlag E, Behre H, Eds. Berlin Heidelberg, Testosterone action deficiency substitution, 1990, p. 23-50
37- Zondek B: Mass excretion of oestrogenic hormone in the urine of the stallion. Nature 33:209-210, 1934
38- Heard RD, Jellinck PH, O’Donnell VJ: Biogenesis of the estrogens: the con- version of testosterone-4-C14 to estrone in the pregnant mare. Endocrinology 57:200-204, 1955.
39- West CD, Damast BL, Sarro SD, Pearson OH: Conversion of testosterone to estrogens in castrated, adrenalectomized human females. J Biol Chem 218:409-418, 1956.
40- Davis JW, Gut M, Lemon HM, Wotiz HH: Studies in steroid metabolism. V. The conversion of testosterone-4-C14 to estrogens by human ovarian tissue. J Biol Chem 222:487-495, 1956
41- Baggett B, Dorfman RI, Engel LL, Savard K: The conversion of testoster- one-3-C14 to C14-estradiol-17beta by human ovarian tissue. J Biol Chem 221:931-941, 1956 8. Baggett B, Engel LL, Balderas L, Lanman G: Conversion of C14-testosterone to C14-estrogenic steroids by endocrine tissues. Endocrinology 64:600-608, 1959
42- Smith EP, Boyd J, Frank GR, Takahashi H, Cohen RM, Specker B, Williams TC, Lubahn DB, Korach KS: Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. N Engl J Med 331:1056-1061, 1994
43- Morishima A, Grumbach MM, Simpson ER, Fisher C, Qin K: Aromatase deficiency in male and female siblings caused by a novel mutation and the physiological role of estrogens. J Clin Endocrinol Metab 80:3689-3698, 1995
44- Carani C, Qin K, Simoni M, Faustini-Fustini M, Serpente S, Boyd J, Korach KS, Simpson ER: Effect of testosterone and estradiol in a man with aromata- se deficiency. N Engl J Med 337:91-95, 1997
45- Bilezikian JP, Morishima A, Bell J, Grumbach MM: Increased bone mass as a result of estrogen therapy in a man with aromatase deficiency. N Engl J Med 339:599-603, 1998
46- Herrmann BL, Saller B, Janssen OE, Gocke P, Bockisch A, Sperling H, Mann K, Broecker M: Impact of estrogen replacement therapy in a male with con- genital aromatase deficiency caused by a novel mutation in the CYP19 gene. J Clin Endocrinol Metab 87:5476-5484, 2002
47- Pura M, Mittre H, Carreau S, Kottler ML: Clinical findings in an adult man with a novel mutation in the aromatase gene. In Program of the 85th Annual Meeting of the Endocrine Society, Abstract Book Philadelphia, PA, 2003
48- Maffei L, Murata Y, Rochira V, Tubert G, Aranda C, Vazquez M, Clyne CD, Davis S, Simpson ER, Carani C: Dysmetabolic syndrome in a man with a novel mutation of the aromatase gene: effects of testosterone, alendronate, and estradiol treatment. J Clin Endocrinol Metab 89:61-70, 2004
49- Maffei L, Rochira V, Zirilli L, Antunez P, Aranda C, Fabre B, Simone ML, Pignatti E, Simpson ER, Houssami S, Clyne CD, Carani C: A novel com- pound heterozygous mutation of the aromatase gene in an adult man: reinforced evidence on the relationship between congenital oestrogen deficiency, adiposity and the metabolic syndrome. Clin Endocrinol (Oxf) 67:218-224, 2007
50- Luberto A, Rochira V, Zirilli L, Pignatti E, Simpson ER, Maffei L, Carani C: A novel compound heterozygous mutation of the aromatase gene in an adult man: a reinforced estrogen deficincy, adiposity and the metabolic syndrome. J Endocrinol Invest 30 Suppl 4:50, 2007
51- Bouillon R, Bex M, Vanderschueren D, Boonen S: Estrogens are essen- tial for male pubertal periosteal bone expansion. J Clin Endocrinol Metab 89:6025-6029, 2004
52- Vanderschueren D, Boonen S, Bouillon R: Action of androgens versus estrogens in male skeletal homeostasis. Bone 23:391-394, 1998
53- Vanderschueren D, Venken K, Ophoff J, Bouillon R, Boonen S: Clinical Review: Sex steroids and the periosteum–reconsidering the roles of androgens and estrogens in periosteal expansion. J Clin Endocrinol Metab 91:378-382, 2006
54- Horsman A, Gallagher JC, Simpson M, Nordin BE: Prospective trial of oestrogen and calcium in postmenopausal women. Br Med J 2:789-792, 1977
55- Lindsay R, Hart DM, Forrest C, Baird C: Prevention of spinal osteoporosis in oophorectomised women. Lancet 2:1151-1154, 1980
56- Christiansen C, Christensen MS, Transbol I: Bone mass in postmenopausal women after withdrawal of oestrogen/gestagen replacement therapy. Lancet 1:459-461, 1981
57- Finkelstein JS, Neer RM, Biller BM, Crawford JD, Klibanski A: Osteopenia in men with a history of delayed puberty. N Engl J Med 326:600-604, 1992
58- Speroff L, Rowan J, Symons J, Genant H, Wilborn W: The comparative effect on bone density, endometrium, and lipids of continuous hormones as replacement therapy (CHART study). A randomized controlled trial. Jama 276:1397-1403, 1996
59- Wang C, Eyre DR, Clark R, Kleinberg D, Newman C, Iranmanesh A, Veldhuis J, Dudley RE, Berman N, Davidson T, Barstow TJ, Sinow R, Alexander G, Swerdloff RS: Sublingual testosterone replacement improves muscle mass and strength, decreases bone resorption, and increases bone formation markers in hypogonadal men–a clinical research center study. J Clin Endocrinol Metab 81:3654-3662, 1996
60- Faustini-Fustini M, Rochira V, Carani C: Oestrogen deficiency in men: where are we today? Eur J Endocrinol 140:111-129, 1999
61- Vanderschueren D, Venken K, Ophoff J, Bouillon R, Boonen S: Clinical Review: Sex steroids and the periosteum–reconsidering the roles of androgens and estrogens in periosteal expansion. J Clin Endocrinol Metab 91:378-382, 2006
69- Pentose Cycle Activity in Muscle from Fetal, Neonatal and Infant Rhesus Monkeys. Arch Biochem Biophys 117:275-81 1966
70- The pentose phosphate pathway in regenerating skeletal muscle. Biochem J 170: 17 1978
71- Aromatization of androgens to estrogens mediates increased activity of glucose 6-phosphate dehydrogenase in rat levator ani muscle. Endocrinol 106(2):440-43 1980
72- Influence of tamoxifen, aminoglutethimide and goserelin on human plasma IGF-1 levels in breast cancer patients. J steroid Biochem Mol Bio 41:541- 3,1992
73- Activation of the somatotropic axis by testosterone in adult males: Evidence for the role of aromatization. J Clin. Endocrinol Metab 76:1407-12 1993
74- Testosterone administration increases insulin-like growth factor-I levels in normal men. J Clin Endocrinol Metab 77(3):776-9 1993
80- Association between serotonin transporter gene polymorphism and chronic fatigue syndrome. Narita M et al. Biochem Biophys Res Commun 2003 Nov 14;311(2)264-6
81- Premenstrual Syndrome. Dickerson LM et al. Am Fam Physician 2003 Apr 15;67(8):1743-52
82- Phase II trial of anastrozole in women with asymptomatic mullerian cancer. Gynecol Oncol. 2003 Dec;91(3):596-602.
83- Letrozole. A review of its use in postmenopausal women with advanced breast cancer. Drugs. 1998 Dec;56(6):1125-40. Review.
84- Exemestane: a review of its clinical efficacy and safety. Breast. 2001 Jun;10(3):198-208.
85- A study of fadrozole, a new aromatase inhibitor, in postmenopausal women with advanced metastatic breast cancer. J Clin Oncol. 1992 Jan;10(1):111-6.
87- Feenstra A, Vaalburg W, Nolten GM, Reiffers S, Talma AG, Wiegman T, van der Molen HD, Woldring MG (1983). “Estrogen receptor binding radiopharmaceuticals: II. Tissue distribution of 17 alpha-methylestradiol in normal and tumor-bearing rats”. J. Nucl. Med. 24 (6): 522–8. PMID6406650.
In questa terza parte della serie di articoli dedicati all’approfondimento sul Trenbolone descriverò nel dettaglio le proprietà anabolizzanti della molecola. Vale sempre la pena ribadire che tutti gli studi relativi agli argomenti qui trattati provengono da studi svolti sugli animali. Quindi, il mio obiettivo è quello di riportare ciò che è disponibile nella letteratura scientifica discutendo su ciò che è potenzialmente rilevante per l’essere umano e, più nel dettaglio, per il BodyBuilder. Molti meccanismi biologici riportati di seguito sono simili tra i mammiferi e l’uomo, e farò del mio meglio per sottolineare quando questo sussiste o meno.
Nota: se non avete letto la prima e la seconda parte vi invito a farlo prima di procedere con la lettura del presente articolo.
IX. Anabolismo
Prima di entrare nel dettaglio, è importante sottolineare una cosa ovvia ma che ad alcuni sfugge, e cioè che vi sono differenze tra gli esseri umani e gli animali più comunemente usati negli studi che hanno preso in esame il Trenbolone (ad esempio pecore, topi, mucche, ecc.). Mentre ci si addentra nell’analisi della documentazione scientifica disponibile, è utile che teniate a mente queste differenze, in quanto possono sicuramente avere un impatto rilevante per gli esseri umani nella loro differenza di risposta.
Levator ani
La maggior parte dell’apparato muscolo-scheletrico dei roditori possiede una percentuale molto bassa di nuclei AR positivi. Un esempio è dato dalle caratteristiche del extensor digitorum longus, situato vicino alla parte anteriore della zampa posteriore, il quale presenta solo il 7% di mionuclei AR positivi.(1) Questo però non è universalmente vero, poiché il complesso muscolare levator ani / bulbocavernosus (LABC) (situato vicino alla pelvi) presenta il 70-75% di mionuclei AR-positivi e, tale caratteristica, si traduce in una marcata risposta miotropica alla somministrazione di androgeni.(2)(3)(4) Quindi, se si prendono in esame diversi studi svolti sui roditori, nei quali sono stati analizzati i muscoli sopra riportati in modo separato, i risultati saranno nettamente differenti.
In alto un esemplare di Toro, in basso un esemplare di Manzo.
Viceversa, i bovini sono generalmente molto sensibili agli stimoli indotti dagli androgeni a causa delle alte concentrazioni di AR nel loro apparato muscolo-scheletrico e nelle loro cellule satelliti.(5)(6)(7) E’ necessario capire anche che, quando si parla di tori, si parla di bovini di sesso maschile maturi e intatti, mentre quando si parla di manzi, si parla di bovini di sesso maschile i quali hanno subito castrazione prima di raggiungere la maturità sessuale. La stragrande maggioranza degli studi esistenti è stata svolta sui manzo sottoposti ad impianti di Trenbolone, dal momento che tali impianti non presentano effetti rilevanti sui tori. Probabilmente questi animali hanno raggiunto il loro massimo potenziale di crescita muscolare data dai loro livelli elevati di endogeni endogeni, comunque sia sono necessari impianti che combinano TBA ed E2 per produrre la massima crescita ed efficienza alimentare nei bovini castrati.(8) Le giovenche sono giovani bovini di sesso femminile che non hanno mai partorito; sono usate occasionalmente per testare gli impianti.
I tori sintetizzano livelli molto elevati di Testosterone. Oltre ad avere una risposta molto scarsa agli impianti, generalmente hanno anche fibre muscolari più grandi rispetto ai manzi.(9) I tori tendono inoltre ad avere una percentuale più elevata di fibre glicolitiche-ossidanti a contrazione rapida combinate con una percentuale inferiore di fibre glicolitiche a contrazione rapida nei muscoli longissimus dorsi (LD) rispetto ai manzi.(10) È per questi motivi che i tori producono un totale di carne più elevate, anche se generalmente di qualità inferiore. I manzi tendono ad avere una percentuale di grasso più elevata ma, tuttavia, sono compensati da una minore velocità di aumento di peso e una minore efficienza alimentare. Quindi, nel tentativo di ottenere maggiori rese con carni di qualità superiore, i ricercatori hanno iniziato a studiare l’effetto degli impianti per verificare se fosse possibile ottenere il meglio di entrambe le caratteristiche che caratterizzano i tori ed i manzi .
Infine, una breve nota a margine: noi esseri umani siamo molto simili ai bovini per quanto concerne la risposta agli androgeni, in quanto rispondiamo in modo marcato agli stimoli androgenici a causa delle alte percentuali di mionuclei AR-positivi.(11)
Affinità per il Recettore degli Androgeni
È stato dimostrato che il Trenbolone si lega con il AR umano, e con il AR di varie specie animali, con una affinità approssimativa pari a tre volte superiore a quella del Testosterone, o approssimativamente uguale a quella del DHT.(12)(13)(14)(15)(16) Nei AR umani, il metabolita attivo 17β-TbOH ha mostrato un’affinità di legame del 109% rispetto al DHT, mentre il metabolita inattivo 17α-TbOH ha mostrato una affinità di legame del 4,5%.(13) Detto questo, gli studi sul legame recettoriale dovrebbero essere visti come uno strumento per una rapida valutazione iniziale di un ligando, non prendendo in considerazione aspetti come la successiva attivazione della trascrizione genica, ecc. In altre parole, anche se il Trenbolone presenta una affinità di legame al AR tre volte superiore rispetto al Testosterone, questo non significa letteralmente che produrrà un ipertrofia tre volte maggiore.
Oltre a questo, negli studi comparativi è stato dimostrato che il Trenbolone produce una crescita uguale o leggermente superiore nel complesso muscolare del LABC rispetto al Testosterone.(14)(17)(18)(19)(20)(21)(22) Il LABC è un tessuto androgeno reattivo che manca dell’enzima 5α-reduttasi. Mentre il Testosterone esercita effetti potenziati nei tessuti che esprimono l’enzima 5α-reduttasi, il Trenbolone esercita effetti uguali in questi tessuti rispetto a quelli che non causano una anabolico:androgeno ratio favorevole in comparazione al Testosterone.(23) Approfondirò questo argomento quando tratterò dei rischi di sviluppare cancro alla prostata.
X. Ipertrofia
Inizialmente avevo in programma di esporre un’analisi approfondita dei meccanismi alla base dell’ipertrofia, ma trattandosi di un argomento decisamente complesso occorrerebbe un articolo a parte esclusivamente dedicato a questa tematica. Avrò comunque bisogno di esporre alcuni elementi fondamentali dell’ipertrofia, altrimenti molto di quanto tratterò in seguito potrebbe risultare più complicato di quanto dovrebbe. Quindi, non mi dilungherò troppo nella descrizione dei percorsi di segnalazione intracellulare, poiché questo renderebbe il presente articolo inutilmente prolisso. Comunque, se l’argomento dovesse suscitare abbastanza interesse, forse in futuro realizzerò un articolo dedicato esclusivamente all’ipertrofia.
Fondamenti di ipertrofia
Prima di tutto è necessario parlare un po ‘di cos’è l’ipertrofia e di come essa si manifesta nei mammiferi. Questo è un passaggio volto a fornire al lettore nozioni base sull’argomento, affinché i termini utilizzati in seguito vengano meglio compresi.
Il numero di fibre muscolari nei mammiferi è essenzialmente fissato alla nascita, quindi la crescita muscolare postnatale è la risultante dell’ipertrofia delle fibre muscolari esistenti. Questa ipertrofia delle fibre richiede un aumento del numero dei mionuclei presenti all’interno di esse; tuttavia i nuclei presenti nelle fibre muscolari non sono in grado di dividersi, quindi, essi, devono provenire dall’esterno della fibra.(24) La fonte dei nuclei necessari per supportare l’ipertrofia delle fibre è rappresentata da un gruppo di cellule miogeniche mononucleate (cellule satelliti) situate tra la lamina basale e la membrana plasmatica (sarcolemma) delle fibre muscolari.(25) Esiste una forte correlazione tra i tassi di crescita postnatale e i tassi con cui le cellule satellite si accumulano all’interno dei tessuti muscolari. Ciò sembrerebbe avere un senso in quanto saranno disponibili più meccanismi generali per alimentare il processo ipertrofico.(26)
Queste cellule satelliti svolgono un ruolo cruciale nella crescita muscolare postnatale fondendosi con le fibre muscolari esistenti, fornendo i nuclei necessari per la crescita delle fibre postnatali. Negli animali appena nati, si riscontra una percentuale molto più alta di nuclei muscolari legati alle cellule satelliti, ma questa percentuale diminuisce significativamente con l’età.(27) Non solo c’è una riduzione del numero di cellule satelliti, ma quelle cellule ancora presenti si ritirano dallo stato proliferativo del ciclo cellulare e entrano in uno stato di quiescenza, che di conseguenza porta ad un plateau della crescita. Quindi, trovare modi per superare questi limiti fisiologici può ipoteticamente portare a tassi di crescita postnatale superiori.
Per garantire che ci sia un numero adeguato di cellule satelliti utilizzabili, devono prima essere attivate per consentire loro di progredire attraverso il ciclo cellulare e infine di contribuire con il DNA della fibra muscolare esistente. Dopo che queste cellule satelliti dormienti sono state attivate, vi è successivamente la necessità di un rilascio dei fattori di crescita in grado di stimolare la proliferazione e la differenziazione delle cellule satelliti. Sia l’IGF-1 che il Fattore di Crescita dei Fibroblasti 2 (FGF-2) sono esempi di potenti stimolatori della proliferazione delle cellule satelliti.(28)(29) L’IGF-1 è unico in quanto promuove la differenziazione delle cellule muscolari nei muscoli-scheletrici, mentre l’FGF-2 inibisce la differenziazione [30]. Tra poco parlerò più nel dettaglio della relazione tra Trenbolone e IGF-1.
Quindi, facendo un leggero passo indietro, quando si verifica un evento di attivazione dell’ipertrofia (ad esempio esercizio o danno muscolare), questo porta anche alla proliferazione delle cellule satelliti. Questa proliferazione delle cellule satelliti induce queste a fondersi con le fibre muscolari esistenti, fornendo nuovi nuclei per l’ipertrofia e la riparazione e per supportare l’aumentata sintesi proteica. Un modo semplice per comprendere quanto appena detto è il seguente: le cellule satellite possono essere attivate per proliferare (dividere) e donare il loro DNA (nuclei) alle fibre muscolari esistenti (differenziazione).
Questo DNA donato porta le fibre muscolari a generare la fusione di mioblasti (cellule proliferanti) in fibre muscolari multinucleate denominate miotubi. Questi miotubi possono fondersi con le miofibre esistenti, o anche l’uno con l’altro, generando direttamente nuove fibre muscolari. Per il momento questo è l’approfondimento necessario per tale l’argomento.
Effetti di stimolo della crescita
Gli effetti di stimolo della crescita dati dal Trenbolone sono ben noti e sono stati studiati dai ricercatori per decenni. L’obiettivo è sempre stato quello di trovare metodiche per promuovere maggiori rese di carne insieme ad un prodotto finito di qualità superiore. Ci concentreremo principalmente sulle rese di carne per ora, poiché la qualità della carne tende spesso a coincidere con la quantità di grassi intramuscolari ivi contenuti. Questo rientra maggiormente nel campo della lipolisi, che tratteremo nel prossimo articoli.
È stato dimostrato che il Trenbolone aumenta la crescita totale della massa corporea e della massa muscolo-scheletrica in vari studi svolti su animali quando somministrato da solo (3)(14)(17) (19)(31)(32)(33)(34)(35)(36)(37)(38)(39)(40)(41)(42)(43)(44), in combinazione con Estradiolo (45) (46) (47)(48)(49)(50)(51)(52)(53)(54)(55)(56)(57)(58)(59)(60)(61)(62)(63)(64)(65)(66), in combinazione con Testosterone ed Estradiolo (67), nonché in combinazione con Estradiolo e Ormone della Crescita.(68) Questo potenziale ipertrofico è stato universalmente osservato attraverso molteplici metodi di somministrazione in diverse specie animali.
È interessante notare che numerosi studi hanno dimostrato che un impianto contenente la combinazione TBA / E2 è più efficace rispetto ad un impianto contenente soltanto TBA o E2 nello stimolare la crescita dei manzi all’ingrasso.(8)(45)(52)(54)(55)(69)(70)(71)(72)(73)(74) L’ipotesi secondo la quale l’Estradiolo aumenterebbe gli effetti anabolizzanti del Trenbolone è emersa intorno agli anni ’70 (75)(76). Un fatto ancora più interessante è che il trattamento combinato (TBA/E2) aumenta il potenziale di ipertrofia nonostante il fatto che tale metodo si traduce in livelli di Trenbolone serici effettivamente inferiori, di circa la metà (8).
Asse GH/IGF-1
Uno dei motivi che mi spinge a credere nella concretezza di un effetto potenziale nella cosomministrazione di Trenbolone ed Estradiolo, in particolare nei manzi , è il fatto che gli impianti contenenti solo Trenbolone portano alla soppressione dei livelli di Estradiolo endogeno a causa del impatto del Trenbolone sull’asse HPG limitandone la crescita potenziale. L’Estrogeno e, più specificamente, l’attività dell’aromatasi sono un potente stimolatore dell’asse GH/IGF-1. A supporto di questa ipotesi, gli impianti con solo Trenbolone hanno dimostrato di abbassare i livelli serici di GH.(8,68,69,70,71) Al contrario, è stato dimostrato che i manzi impiantati con E2 hanno aumentato le concentrazioni ematiche sia di GH che di IGF-1.(77-78) Gli impianti contenenti la combinazione TBA / E2 probabilmente determinano un aumento dei livelli di GH e possono persino alterare il numero e/o l’affinità dei GHR nei tessuti come quello epatico.(79) Come abbiamo appreso in precedenza, avere livelli adeguati di fattori di crescita per stimolare la proliferazione e la differenziazione delle cellule satelliti è un fattore significativo nel processo ipertrofico.
TBA/E2 ratio ottimale
Poiché i trattamenti combinati sembrano avere caratteristiche anaboliche migliori, molti studi nel corso degli anni hanno cercato di rispondere alla domanda del rapporto ottimale tra TBA / E2 per ottenere il massimo rendimento di crescita. Ce ne sono stati alcuni che hanno affermato che la risposta a tale quesito era un rapporto di 5: 1 e 8: 1 (52,54), tuttavia i risultati hanno subito delle leggere variazioni nel corso degli anni. Infatti, in uno studio, i tassi medi di crescita giornaliera (ADG) erano abbastanza simili nei manzi impiantati con una combinazione di 120mg TBA+25mg E2 o una combinazione di 120mg TBA+24mg E2 (80).
Un altro studio ha dimostrato che 120mg di TBA+24mg E2 hanno aumentato i tassi medi di crescita del 20-25% e l’efficienza dell’alimentazione del 15-20% (55). Infatti, è stato dimostrato che i trattamenti combinati portano al 50% in più nella proliferazione delle cellule satelliti nel muscolo semimembranoso (tendine del ginocchio) dei manzi. (81) Come detto in precedenza, la proliferazione delle cellule satelliti è un fattore significativo nel processo ipertrofico. Altri studi hanno mostrato un aumento indotto dal Trenbolone sia nell’attivazione che nella proliferazione delle cellule satelliti. (82-83) Sembra che il Trenbolone e il Testosterone aumentino il numero di cellule satelliti per fibra muscolare in misura simile (22), quindi questo non è un effetto dato solo dal Trenbolone. I suoi effetti sulle cellule satelliti possono essere, almeno in parte, mediati attraverso il recettore del IGF-1, come l’inibizione di diversi target a valle del IGF-1 (es. MAPK, MEK / ERK, PI3K / Akt) e la successiva soppressione della proliferazione delle cellule satelliti Trenbolone-indotta osservata nelle colture cellulari.(7)
Nel 2007 la FDA ha approvato la commercializzazione del Revalor-XS il quale contiene una combinazione di 200mg di TBA + 40mg di E2, e che è stato progettato per avere un lento rilascio degli ormoni in esso contenuti dovuto all’azione di un rivestimento polimerico presente su sei dei dieci pellet componenti il prodotto. Ciò è utile in quanto i metodi di impianto tradizionali richiedono più impianti con il potenziale di aggiungere stress che potrebbe influire negativamente sul rendimento del bestiame. Gran parte delle variazione dei risultati riscontrate negli studi nel corso degli anni potrebbero benissimo essere correlate a questo fattore. Le studi condotti sul Revalor-XS hanno mostrato che la dose più elevata di TBA / E2 migliora il rendimento dei manzi sono alimentati per periodi più lunghi. (65,140) Nonostante la speculazione sul fatto che più impianti possano causare stress aggiunto ai manzi, il pattern di lento rilascio del Revalor-XS in realtà non fornisce effetti unici sul rendimento delle carni, o della qualità, se confrontato con una strategia di impianto multiplo di pari dosaggio di TBA + E2.
Effetti del IGF-1
Molecola di IGF-1 umano
Gli impianti contenenti TBA / E2 hanno dimostrato di aumentare significativamente i livelli di IGF-1. Questi trattamenti combinati hanno portato a un aumento dei livelli serici di IGF-1 (59,84,85,86), aumenti dell’espressione del mRNA del IGF-1 epatico (56) e aumenti dell’espressione del mRNA del IGF-1 nei tessuti muscolo-scheletrici. (59,61,81)
Anche gli impianti contenenti solo TBA hanno dimostrato di aumentare i livelli di IGF-1 in varie specie, tuttavia, però, il grado di aumento dei livelli di IGF-1 non è stato paragonabile a quello riscontrato con impianti contenenti la combinazione TBA/E2.(87,88,89) In effetti, il Trenbolone non aumenta significativamente l’IGF-1 autocrino o endocrino in maniera maggiore rispetto al Testosterone. Uno studio ha anche dimostrato che il Testosterone aumenta i livelli di IGF-1 autocrino in maniera leggermente più elevata rispetto al TBA.(4) L’evidenza sembra suggerire che qualsiasi effetto sull’IGF-1 possa essere mediato attraverso l’estradiolo e possa anche essere stimolato tramite distinti meccanismi dei recettori degli androgeni e degli estrogeni, che includono il coinvolgimento del recettore accoppiato alla proteina G (GPR30).(90) In particolare, uno studio ha rilevato che l’aumento dell’espressione autocrina del IGF-1 nel muscolo scheletrico richiede la presenza di estrogeni e che gli impianti con solo TBA non hanno determinato aumenti dei livelli di mRNA del IGF-1 muscolare.(91) È certamente ragionevole ipotizzare che possa esserci una soglia da rispettare, nonostante il fatto che essa potrebbe non essere realisticamente rapportabile dagli studi sugli animali e l’uomo.
Studi in vitro svolti utilizzando cellule satelliti bovine (BSC) hanno mostrato una relazione dose-dipendente tra Trenbolone e livelli del mRNA del IGF-1. Nelle cellule trattate con 1nM o 10nM di Trenbolone per 48 ore, i livelli del mRNA del IGF-1 erano 1,7 volte più alti, tuttavia i livelli di mRNA non erano influenzati dai trattamenti con 0,001, 0,01 o 0,1 nM. (92) È anche emerso che gli effetti sono stati almeno parzialmente AR-mediati in quanto il co-trattamento con Flutamide (inibitore AR) ha completamente annullato l’aumento dell’espressione di IGF-1 osservata in queste colture cellulari.(90)
Non ci vuole molto tempo per osservare un aumento dei livelli di IGF-1 dopo l’applicazione di un impianto. In uno studio, agnelli impiantati con Revalor-S (120mg TBA / 24mg E2) hanno mostrato un aumento dei livelli serici di IGF-1 entro il 3° e il 10 ° giorno rispettivamente del 43% e del 62% (56). Questo aumento del IGF-1 è stato mantenuto per tutti i 24 giorni dello studio e i livelli del mRNA del IGF-1 epatico erano risultati del 150% più alti negli agnelli impiantati rispetto a quelli di controllo, suggerendo che il fegato è probabilmente una fonte primaria dalla quale provengono gli aumenti di IGF-1 circolante. I livelli del mRNA del IGF-1 autocrino erano del 68% più alti nei muscoli longissimus degli agnelli impiantati rispetto a quelli osservati negli animali di controllo. Il dosaggio di TBA ed E2, per chilogrammo di peso corporeo, era circa tre volte superiore a quello approvato per l’uso nei manzi. A causa delle differenze tra le specie e il dosaggio, si dovrebbe usare cautela quando si tenta di prendere questi risultati e applicarli ai manzi.
L’uso di questa stessa dose nei manzi ha dimostrato di produrre livelli di IGF-1 serici più elevati rispetto ai bovini non impiantati, entro 6-42 giorni dall’impianto.(93) In sole 48 ore, i manzi impiantati hanno avuto un aumento del 13,4% delle concentrazioni seriche di IGF-1.(84) Nei giorni 21 e 40, i manzi impiantati presentavano livelli di IGF-1 superiori del 16% e del 22% rispetto agli animali di controllo. La cosa interessante è che i livelli di IGF-1 raggiunsero il picco durante questo periodo di tempo e successivamente iniziarono a calare fino al giorno 115 dello studio, dove finirono per assomigliare ai valori del giorno 1. Detto questo, i manzi di controllo avevano ancora livelli IGF-1 più bassi rispetto al primo giorno. Quindi, anche se gli aumenti dei livelli di IGF-1 appaiono transitori, gli impianti sembrano ancora fornire un effetto additivo complessivo, anche con un uso a lungo termine.
Altri studi sul bestiame hanno mostrato campioni muscolari con un più alto livello del mRNA del IGF-1 entro 30-40 giorni dall’applicazione dell’impianto. (56,61) Questi animali impiantati mostravano anche cellule satelliti più proliferanti rispetto ai manzi non impiantati suggerendo che gli aumenti della massa muscolare osservati con la combinazione TBA / E2 possono essere almeno in parte ricondotti all’aumento del IGF-1. Come abbiamo discusso in precedenza, è ben noto che la crescita muscolare postnatale dipende dalla fusione delle cellule satelliti con le fibre esistenti per fornire i mionuclei necessari a sostenere la crescita.(24) Questo aumento è importante anche perché solo un piccolo numero di cellule satelliti sono presenti nei bovini di un anno e molte delle cellule esistenti sono diventate inattive o hanno lasciato il ciclo cellulare. Vale anche la pena ricordare che la sovraespressione del IGF-1 estende la durata della vita replicativa delle cellule satelliti, almeno nelle colture cellulari. (94) Pertanto, sembra ragionevole ipotizzare che l’aumentata espressione del IGF-I muscolare giochi un ruolo nell’aumento indotto dall’AAS nel numero delle cellule satelliti muscolari.
In un altro studio, i livelli del mRNA del IGF-1 allo stato stazionario sono stati mostrati essere del 69% più alti nei manzi impiantati, suggerendo ancora una volta che il fegato può essere un fattore importante nell’aumento dell’IGF-1 circolante negli animali sottoposti ad impiantati.(61) Si prega di notare che c’è stato almeno uno studio, di cui sono a conoscenza, che non ha mostrato differenze nelle concentrazioni di IGF-1 tra manzi impiantati e di controllo.(95) Comunque, questo risultato può benissimo essere considerato come l’eccezione che conferma la regola.
Un elemento di risposta agli androgeni (ARE) è stato identificato nella regione del promotore del gene IGF-1, suggerendo che il complesso recettore-ligando androgeno possa interagire con questo ARE per stimolare la trascrizione del gene IGF-1. Gli androgeni tendono ad agire attraverso meccanismi multipli sui muscoli, e l’estrogeno tende ad agire sull’ipotalamo / pituitaria anteriore per stimolare l’asse GH / IGF-1.(96) La relazione tra l’estrogeno e l’asse GH / IGF-1 ha dimostrato di essere additiva.(97-98)
Elementi sugli Estrogeni
Dato che l’impatto degli estrogeni è stato parzialmente trattato, è corretto concentrarsi sul loro ruolo nel contesto di cui stiamo trattando prima di andare avanti.
Studi in vitro hanno dimostrato che il trattamento di colture di cellule satelliti bovine con E2 per 48 ore porta ad un aumento significativo dell’espressione del mRNA del IGF-1.(92) Ciò è in linea con quello che già sappiamo sull’E2, poiché è stato dimostrato che stimola l’espressione dell’mRNA dell’IGF-1 in un certo numero di tessuti. (99-100) È interessante notare che il co-trattamento con ICI (antagonista del recettore dell’estrogeno) non ha soppresso l’espressione del IGF-1 stimolata con l’E2. Ciò sembra suggerire che il meccanismo attraverso il quale l’E2 stimola l’espressione dell’mRNA del IGF-I nelle BSC può essere diverso dal meccanismo che si attua in altri tessuti che sono stati esaminati fino ad oggi.
Anche se il gene del IGF-1 non contiene un tradizionale elemento di risposta agli estrogeni (ERE) nella sua regione di regolazione, la stimolazione da parte dell’E2 dell’espressione dell’mRNA del IGF-1 può avvenire tramite un percorso che coinvolge il fattore di trascrizione AP-1.(101) Come accennato in precedenza, oltre ai recettori degli estrogeni classici, il recettore GRP30) può svolgere un ruolo nella mediazione delle azioni degli estrogeni.(102,103,104) Questo è rilevante per i nostri interessi in quanto il tessuto muscolare contiene l’mRNA del GPR30 e gli studi immunoistochimici hanno localizzato la proteina del recettore GPR30 all’interno delle cellule muscolo-scheletriche.(105) Inoltre, gli effetti dell’agonista / antagonista GPR indicano fortemente che il recettore GPR30 è coinvolto nell’aumento dello stimolo del E2 sul mRNA del IGF-1 osservato nelle colture di cellule satelliti bovine.(90)
Effetti sulle fibre muscolari
(Fonte immagine “Project Invictus”)
Gli impianti contenenti TBA / E2 aumentano l’area della sezione trasversale delle fibre muscolari (CSA) a causa di un iniziale aumento della trascrizione del DNA seguito da un aumento dei nuclei all’interno delle fibre muscolari che supportano l’ipertrofia.(106) Il TBA (da solo o in combinazione con E2) ha dimostrato di aumentare il CSA delle fibre muscolari di tipo I ma non di quelle di tipo II (9,107). È stato anche dimostrato che l’impianto nei manzi combinato con iperalimentazione aumenta il CSA nei muscoli LM nelle fibre di tipo I e IIA.(110) Ci sono state comunque delle eccezioni, dal momento che è stato dimostrato che uno studio ha aumentato le fibre di tipo IIB senza alcun impatto sulla dimensione o sul numero delle fibre di tipo I.(57) Questi studi, se considerati nel loro complesso, sembrano suggerire che il Trenbolone induca la conversione delle fibre muscolari da glicolitiche a ossidative, il che indica un aumento della capacità ossidativa delle fibre del muscolo-scheletrico.
Tornando alle potenziali differenze osservate tra le diverse specie animali, nonostante l’aumento del peso muscolare e della dimensione delle fibre muscolari, il numero dei mionuclei per fibra non ha subito miglioramenti nei ratti ai quali è stato somministrato Trenbolone o Testosterone.(22) Ciò contraddice i risultati di uno studio precedente il quale, però, ha visto la somministrazione di Testosterone all’inizio della pubertà, una fase caratterizzata da una crescita rapida del muscolo LABC rispetto a quanto osservabile negli esemplari maturi dello studio precedente.(108) È altamente probabile che l’ipertrofia indotta dagli androgeni nei ratti adulti senza stimoli fisici non richieda l’aggiunta mionucleare (109), il che va contro senso rispetto a ciò di cui abbiamo parlato nell’intero articolo. Ma queste sono le differenze da tenere in considerazione quando si osservano studi su animali e si cerca di stabilire modelli che potrebbero potenzialmente essere tradotti negli esseri umani.
Effetti dei Recettori degli Androgeni
Recettore degli Androgeni, modello molecolare.
Ci sono stati più esperimenti in vitro che hanno indicano una sovra regolazione dell’espressione del mRNA del AR (111-112). Tuttavia, sembra che vi sia un limite massimo oltre il quale anche con dosi più elevate non si riesce a modificare i livelli di mRNA in misura relativa a quelli presenti nelle colture di controllo non trattate.(92)
Tuttavia, ciò non è stato universalmente dimostrato negli studi, in quanto alcuni non hanno mostrato effetti mediati dal Trenbolone sull’espressione del mRNA del AR.(4,91) Questa discrepanza può essere dovuta al fatto che l’espressione elevata del AR si verifica in un punto temporale precedente rispetto alla raccolta dei dati effettuata in questi studi, ma ciò è speculativo. L’evidenza in vitro indica anche che il Trenbolone induce la traslocazione del AR umano nel nucleo cellulare in modo dose-dipendente e stimola anche la trascrizione del gene almeno allo stesso grado del DHT.(14)
XI. Atrofia/Anti-Catabolismo
La reputazione del Trenbolone come AAS fortemente anti-catabolico è in realtà ben meritata. Anche in questo caso, è utile trattare brevemente le basi dell’atrofia muscolo-scheletrica prima di proseguire con l’esposizione della letteratura associata al Trenbolone.
Il Proteosoma spezza i substrati ubiquitinilati in corti peptidi, riciclando le molecole di Ubiquitina.
Durante diversi stati catabolici, la via dell’ubiquitina-proteasoma aumenta la disgregazione proteica che porta all’atrofia muscolare. Nello specifico, due ubiquitin ligasi, MuRF1 e MAFbx (anche denominate Atrogin-1) fungono da marker dell’atrofia muscolare in diverse condizioni cataboliche come il digiuno, il cancro, l’insufficienza renale e il diabete.(113,114,115) È stato dimostrato che il Trenbolone riduce significativamente l’espressione del mRNA del MuRF1 e del Atrogin-1 nei tessuti muscolo-scheletrici di un fattore 3 nei ratti castrati. I tassi di Atrogina-1 sono stati soppressi in questi animali ad un livello ancora maggiore rispetto a quanto osservato con la somministrazione di Testosterone.(4)
Glucocorticoidi
Cortisolo, il principale Glucocorticoide sintetizzato dalla Corteccia Surrenale.
I Glucocorticoidi sono ormoni steroidei che aiutano a regolare l’omeostasi metabolica dell’intero organismo. Essi esercitano anche la loro influenza sul muscolo-scheletrico, infatti una elevata esposizione del tessuto muscolare a questi ormoni può potenzialmente portare all’atrofia dei tessuti. I membri principali della famiglia dei Glucocorticoidi sono il Cortisolo, il Corticosterone e il Cortisone. Si legano con il recettore Glucocorticoidi intracellulare (GRα) attivandolo ed esercitando i loro effetti. Vale la pena ricordare che il Cortisolo può legarsi sia al recettore glucocorticoide (GRα) che al recettore mineralcorticoide (MR), tuttavia un approfondimento in tal senso va oltre lo scopo di questa serie di articoli.
È stato dimostrato che il Trenbolone riduce la capacità di legame dei Glucocorticoidi causando una riduzione nel numero di GRα nei tessuti muscolo-scheletrici. (36,116) Studi in vitro hanno dimostrato che il Trenbolone agisce come un antagonista del recettore glucocorticoide [14] nonostante il 17β-TbOH possegga solo un’affinità di legame relativa del 9,4% per il recettore glucocorticoide bovino rispetto al cortisolo [13]. Altri studi hanno dimostrato che il Trenbolone riduce la capacità del Cortisolo di legarsi ai GR nel muscolo-scheletrico e sottoregola l’espressione complessiva dei GR. (117-118) Infatti, il Trenbolone sopprime l’espressione dei GRα del 50% in più rispetto al Testosterone.(4) E le sue azioni anti-glucocorticoidi probabilmente aiutano a creare un’inibizione significativamente più marcata della degradazione delle proteine muscolari (MPB) rispetto al Testosterone, che riduce solo leggermente l’MPB aumentando contemporaneamente l’MPS. (119)
È stato dimostrato che il Trenbolone riduce le concentrazioni circolanti di Corticosterone nei roditori (37,39,116,120) e del Cortisolo nei bovini impiantati.(50) L’evidenza suggerisce che il Trenbolone agisca a livello delle ghiandole surrenali sopprimendo la sintesi del Cortisolo ACTH-stimolata e sopprimendo il rilascio di Cortisolo.(121)
Ora possiamo provare ad estrapolare un po’ di informazioni correlate alle possibilità che si presentano in un contesto dove i livelli di glucocorticoidi sono abbassati. Ad esempio, i glucocorticoidi inibiscono l’assorbimento del glucosio e aiutano a stimolare la disgregazione del glicogeno immagazzinato nel muscolo-scheletrico attenuando la traslocazione dei GLUT4 nelle membrane cellulari indotta dall’insulina.(122) La segnalazione dell’insulina nei tessuti muscolari è essenzialmente soppressa dai glucocorticoidi.(123) Detto questo, è possibile che la somministrazione di Trenbolone possa portare ad un maggiore utilizzo del glucosio? Abbiamo già visto la capacità del Trenbolone di aumentare la sensibilità all’insulina nei ratti, ma cosa succederebbe se lo si co-somministrasse con l’insulina esogena?
I Glucocorticoidi tendono anche ad aumentare i livelli di trigliceridi intramuscolari.(124) È quindi ragionevole ipotizzare che gli effetti cosmetici tradizionalmente attribuiti al Trenbolone possano avere qualcosa a che fare con questo? Se il Trenbolone riduce ì i livelli di trigliceridi intramuscolari, allora questo potrebbe essere un fattore primario dietro all’aspetto muscolare poco “voluminoso” (nonostante la presenza di un carico glicemico pienamente sufficiente) che molti tendono ad avere? Cercherò di tornare su queste domande nelle mie osservazioni conclusive di questa serie di articoli.
Effetti sulla sintesi e degradazione proteica
Una delle caratteristiche più paradossali riscontrate sul Trenbolone è rappresentata da un tasso di sintesi proteica muscolare (MPS) diminuito in seguito alla somministrazione di questo AAS. Ciò è stato dimostrato in studi nei quali si sono osservati gli effetti del trattamento con impianti contenenti TBA o TBA+E2.(17,32,48) Molti si chiedono come sia possibile che una molecola notoriamente molto anabolizzante come il Trenbolone riduca i tassi di MPS. La chiave per comprendere ciò risiede nell’impatto del Trenbolone sul MPB, in quanto il Trenbolone possiede un azione riduttiva sui tassi di MPB maggiore rispetto a quelli sul MPS, il che si traduce in uno stato nettamente anabolico.
Infatti, nonostante la riduzione dei tassi di MPS, è stato dimostrato che il Trenbolone aumenta la ritenzione di azoto di tutto il corpo in varie specie animali. (32,125,126,127) Ancora una volta, questo ha molto a che fare con l’impatto del Trenbolone sui tassi di MPB. È stato dimostrato che causa una riduzione significativa dei tassi di MPB totale e miofibrillare in varie specie animali.(32,34,36,120,128)
Vale la pena notare che studi in vitro hanno effettivamente mostrato aumenti dipendenti dalla concentrazione indotti dal Trenbolone nei tassi di MPS. Possono essere significativi, con un aumento fino a 1,7 volte utilizzando la dose più alta di 10 nM [129]. Quindi, similmente a quello che abbiamo visto prima con l’espressione del IGF-1, potrebbe esserci una soglia oltre la quale il Trenbolone smette di sopprimere la MPS e inizia ad aumentarla. È probabile che questa soglia si estenda oltre i realistici casi d’uso del mondo reale, dato che studi in vivo su vari animali non mostrano questo effetto. A livello cellulare, anche le percentuali di degradazione delle proteine sono state abbassate, con la dose massima di TBA che causa un tasso di degradazione pari al 70% in meno di quello mostrato nelle colture senza TBA. Questo è stato un effetto parzialmente AR-mediato in quanto il Flutamide sopprime la capacità del Trenbolone di stimolare la sintesi proteica e di sopprimere i tassi di degradazione delle proteine. Anche il trattamento delle colture cellulari con JB1 (inibitore del IGF-1) influisce sugli effetti del Trenbolone sulla sintesi / degradazione delle proteine, quindi è altamente probabile che questi effetti richiedano in una certa misura l’attivazione dei recettori degli androgeni e del IGF-1.
È stato anche dimostrato che il Trenbolone sopprime la degradazione degli amminoacidi nel fegato.(37,130) Questo può anche essere un fattore chiave per gli effetti complessivi sul MPB, in quanto il primo passo nella degradazione degli aminoacidi avviene a livello epatico – la rimozione dell’azoto. In effetti, il principale sito di degradazione degli amminoacidi nei mammiferi è il fegato.
Effetti sull’osso
L’ipogonadismo legato all’età è un fattore importante che contribuisce alla perdita di tessuto osseo negli uomini anziani.(131) Come abbiamo discusso in precedenza, il trattamento di fatto per l’ipogonadismo è la terapia sostitutiva del Testosterone (TRT). Il problema è che con le TRT si vengono a creare solo modesti miglioramenti della densità minerale ossea nei soggetti trattati. (132-133) Al contrario, le dosi sovrafisiologiche di Testosterone offrono una protezione completa dalla perdita ossea ma, tuttavia, a tali dosaggi si manifestano molti effetti collaterali indesiderati. (134,135,136) Quindi, sono tornato a trattare un punto già esposto nella prima parte di questa serie di articoli, e cioè cercare di ottenere con l’uso del Trenbolone gli effetti protettivi dati dalla somministrazione di dosi sovra fisiologiche di Testosterone, ma senza gli effetti indesiderati.
Le prime indicazioni in questo senso sono promettenti in quanto gli studi sui roditori dimostrano che il Trenbolone impedisce la perdita di tessuto osseo indotta dal ipogonadismo in ratti castrati a un livello uguale a quello rilevato con dosi sovra fisiologiche di Testosterone, ma senza causare ipertrofia prostatica o aumenti dell’emoglobina che sono frequentemente osservati con trattamenti a base di Testosterone.(3,20)
Corticosterone
Il Trenbolone potenzialmente esercita parte della sua influenza sull’osso attraverso la riduzione del Corticosterone circolante, attraverso la sua attività anti-glucocorticoide.(14,39) E, nonostante l’azione soppressiva del Trenbolone sui livelli estrogenici, questa molecola possiede ancora caratteristiche di conservazione del tessuto osseo simili al Testosterone. Questo è similare a quanto osservato con il DHT, quindi sembra che gli androgeni non aromatizzabili siano in grado di proteggere le ossa direttamente attraverso percorsi AR-mediati.(137-138) Ci sono ancora persone convinte che un lieve grado di aromatizzazione del testosterone in E2 a livello scheletrico sia essenziale per garantire la protezione delle ossa nei maschi.(139) Quindi, prima di poter trarre conclusioni, dovranno essere condotti studi sul Trenbolone a lungo termine.
Anche per questa terza parte siamo arrivati al termine. Nella quarta ed ultima parte di questa serie di articoli tratterò della lipolisi, dei potenziali rischi legati all’uso del Trenbolone ed esporrò le mie osservazioni e raccomandazioni riguardo a quanto trattato.
Stay tuned!
Gabriel Bellizzi
Riferimenti:
Monks DA, O’Bryant EL, Jordan CL. Androgen receptor immunoreactivity in skeletal muscle: enrichment at the neuromuscular junction. J Comp Neurol. 2004 May 17;473(1):59-72.
Monks DA, Kopachik W, Breedlove SM, Jordan CL. Anabolic responsiveness of skeletal muscles correlates with androgen receptor protein but not mRNA. Can J Physiol Pharmacol. 2006 Feb;84(2):273-7
McCoy SC, Yarrow JF, Conover CF, Borsa PA, Tillman MD, Conrad BP, Pingel JE, Wronski TJ, Johnson SE, Kristinsson HG, Ye F, Borst SE. 17β-Hydroxyestra-4,9,11-trien-3-one (Trenbolone) preserves bone mineral density in skeletally mature orchiectomized rats without prostate enlargement. Bone. 2012 Oct;51(4):667-73.
Ye F, McCoy SC, Ross HH, Bernardo JA, Beharry AW, Senf SM, Judge AR, Beck DT, Conover CF, Cannady DF, Smith BK, Yarrow JF, Borst SE. Transcriptional regulation of myotrophic actions by testosterone and trenbolone on androgen-responsive muscle. Steroids. 2014 Sep;87:59-66.
Sauerwein H, Meyer HH. Androgen and estrogen receptors in bovine skeletal muscle: relation to steroid-induced allometric muscle growth. J Anim Sci. 1989 Jan;67(1):206-12.
Brandstetter AM, Pfaffl MW, Hocquette JF, Gerrard DE, Picard B, Geay Y, Sauerwein H. Effects of muscle type, castration, age, and compensatory growth rate on androgen receptor mRNA expression in bovine skeletal muscle. J Anim Sci. 2000 Mar;78(3):629-37.
Kamanga-Sollo E, White ME, Hathaway MR, Chung KY, Johnson BJ, Dayton WR. Roles of IGF-I and the estrogen, androgen and IGF-I receptors in estradiol-17beta- and trenbolone acetate-stimulated proliferation of cultured bovine satellite cells. Domest Anim Endocrinol. 2008 Jul;35(1):88-97.
Hunt DW, Henricks DM, Skelley GC, Grimes LW. Use of trenbolone acetate and estradiol in intact and castrate male cattle: effects on growth, serum hormones, and carcass characteristics. J Anim Sci. 1991 Jun;69(6):2452-62.
Clancy MJ, Lester JM, Roche JF. The effects of anabolic agents and breed on the fibers of the longissimus muscle of male cattle. J Anim Sci. 1986 Jul;63(1):83-91.
Young OA, Bass JJ. Effect of castration on bovine muscle composition. Meat Sci. 1984;11(2):139-56.
Sinha-Hikim I, Taylor WE, Gonzalez-Cadavid NF, Zheng W, Bhasin S. Androgen receptor in human skeletal muscle and cultured muscle satellite cells: up-regulation by androgen treatment. J Clin Endocrinol Metab. 2004 Oct;89(10):5245-55.
Sinnett-Smith PA, Palmer CA, Buttery PJ. Androgen receptors in skeletal muscle cytosol from sheep treated with trenbolone acetate. Horm Metab Res. 1987 Mar;19(3):110-4.
Bauer ER, Daxenberger A, Petri T, Sauerwein H, Meyer HH. Characterisation of the affinity of different anabolics and synthetic hormones to the human androgen receptor, human sex hormone binding globulin and to the bovine progestin receptor. APMIS. 2000 Dec;108(12):838-46.
Wilson VS, Lambright C, Ostby J, Gray LE Jr. In vitro and in vivo effects of 17beta-trenbolone: a feedlot effluent contaminant. Toxicol Sci. 2002 Dec;70(2):202-11.
Ankley GT, Defoe DL, Kahl MD, Jensen KM, Makynen EA, Miracle A, Hartig P, Gray LE, Cardon M, Wilson V. Evaluation of the model anti-androgen flutamide for assessing the mechanistic basis of responses to an androgen in the fathead minnow (Pimephales promelas). Environ Sci Technol. 2004 Dec 1;38(23):6322-7.
Lee HS, Jung DW, Han S, Kang HS, Suh JH, Oh HS, Hwang MS, Moon G, Park Y, Hong JH, Koo YE. Veterinary drug, 17β-trenbolone promotes the proliferation of human prostate cancer cell line through the Akt/AR signaling pathway. Chemosphere. 2018 May;198:364-369.
Vernon BG, Buttery PJ. Protein turnover in rats treated with trienbolone acetate. Br J Nutr. 1976 Nov;36(3):575-9.
Freyberger A, Ellinger-Ziegelbauer H, Krötlinger F. Evaluation of the rodent Hershberger bioassay: testing of coded chemicals and supplementary molecular-biological and biochemical investigations. Toxicology. 2007 Sep 24;239(1-2):77-88.
Hotchkiss AK, Nelson RJ. An environmental androgen, 17beta-trenbolone, affects delayed-type hypersensitivity and reproductive tissues in male mice. J Toxicol Environ Health A. 2007 Jan 15;70(2):138-40.
Yarrow JF, Conover CF, McCoy SC, Lipinska JA, Santillana CA, Hance JM, Cannady DF, VanPelt TD, Sanchez J, Conrad BP, Pingel JE, Wronski TJ, Borst SE. 17β-Hydroxyestra-4,9,11-trien-3-one (trenbolone) exhibits tissue selective anabolic activity: effects on muscle, bone, adiposity, hemoglobin, and prostate. Am J Physiol Endocrinol Metab. 2011 Apr;300(4):E650-60.
Beck DT, Yarrow JF, Beggs LA, Otzel DM, Ye F, Conover CF, Miller JR, Balaez A, Combs SM, Leeper AM, Williams AA, Lachacz SA, Zheng N, Wronski TJ, Borst SE. Influence of aromatase inhibition on the bone-protective effects of testosterone. J Bone Miner Res. 2014 Nov;29(11):2405-13.
Dalbo VJ, Roberts MD, Mobley CB, Ballmann C, Kephart WC, Fox CD, Santucci VA, Conover CF, Beggs LA, Balaez A, Hoerr FJ, Yarrow JF, Borst SE, Beck DT. Testosterone and trenbolone enanthate increase mature myostatin protein expression despite increasing skeletal muscle hypertrophy and satellite cell number in rodent muscle. Andrologia. 2017 Apr;49(3).
Yarrow JF, McCoy SC, Borst SE. Tissue selectivity and potential clinical applications of trenbolone (17beta-hydroxyestra-4,9,11-trien-3-one): A potent anabolic steroid with reduced androgenic and estrogenic activity. Steroids. 2010 Jun;75(6):377-89.
Campion DR. The muscle satellite cell: a review. Int Rev Cytol. 1984;87:225-51. Review.
Moss FP, Leblond CP. Satellite cells as the source of nuclei in muscles of growing rats. Anat Rec. 1971 Aug;170(4):421-35.
Allen Trenkle, D. L. DeWitt, David G. Topel; Influence of Age, Nutrition and Genotype on Carcass Traits and Cellular Development of the M. Longissimus of Cattle, Journal of Animal Science, Volume 46, Issue 6, 1 June 1978, Pages 1597–1603
Cardasis CA, Cooper GW. A method for the chemical isolation of individual muscle fibers and its application to a study of the effect of denervation on the number of nuclei per muscle fiber. J Exp Zool. 1975 Mar;191(3):333-46.
Johnson SE, Allen RE. The effects of bFGF, IGF-I, and TGF-beta on RMo skeletal muscle cell proliferation and differentiation. Exp Cell Res. 1990 Apr;187(2):250-4.
Allen RE, Rankin LL. Regulation of satellite cells during skeletal muscle growth and development. Proc Soc Exp Biol Med. 1990 Jun;194(2):81-6. Review.
Allen RE, Boxhorn LK. Regulation of skeletal muscle satellite cell proliferation and differentiation by transforming growth factor-beta, insulin-like growth factor I, and fibroblast growth factor. J Cell Physiol. 1989 Feb;138(2):311-5.
Best JM. The use of trienbolone acetate implants in heifer beef production at pasture. Vet Rec. 1972 Dec 16;91(25):624-6
Vernon BG, Buttery PJ. The effect of trenbolone acetate with time on the various responses of protein synthesis of the rat. Br J Nutr. 1978 Nov;40(3):563-72.
Galbraith H. Effect of trenbolone acetate on growth, blood metabolites and hormones of cull beef cows. Vet Rec. 1980 Dec 13;107(24):559-60.
Ranaweera KN, Wise DR. The effects of trienbolone acetate on carcass composition, conformation and skeletal growth of turkeys. Br Poult Sci. 1981 Mar;22(2):105-14.
Henricks DM, Edwards RL, Champe KA, Gettys TW, Skelley GC Jr, Gimenez T. Trenbolone, estradiol-17 beta and estrone levels in plasma and tissues and live weight gains of heifers implanted with trenbolone acetate. J Anim Sci. 1982 Nov;55(5):1048-56.
Santidrian S, Thompson JR. Effect of sex, testosterone propionate and trienbolone acetate on the rate of growth and myofibrillar protein degradation in growing young rats. Reproduccion. 1982 Jan-Mar;6(1):33-41.
Thomas KM, Rodway RG. Effects of trenbolone acetate on adrenal function and hepatic enzyme activities in female rats. J Endocrinol. 1983 Jul;98(1):121-7.
Hunter RA, Vercoe JE. Reduction of energy requirements of steers fed on low-quality-roughage diets using trenbolone acetate. Br J Nutr. 1987 Nov;58(3):477-83.
Sillence MN, Rodway RG. Effects of trenbolone acetate and testosterone on growth and on plasma concentrations of corticosterone and ACTH in rats. J Endocrinol. 1990 Sep;126(3):461-6.
Castaldo DJ, Jones JE, Maurice DV. Growth and carcass composition of female turkeys implanted with anabolic agents and fed high-protein and low-protein diets. Arch Tierernahr. 1990 Aug;40(8):703-12.
Apple JK, Dikeman ME, Simms DD, Kuhl G. Effects of synthetic hormone implants, singularly or in combinations, on performance, carcass traits, and longissimus muscle palatability of Holstein steers. J Anim Sci. 1991 Nov;69(11):4437-48.
Freyberger A, Hartmann E, Krötlinger F. Evaluation of the rodent Hershberger bioassay using three reference (anti)androgens. Arh Hig Rada Toksikol. 2005 Jun;56(2):131-9.
Donner DG, Beck BR, Bulmer AC, Lam AK, Du Toit EF. Improvements in body composition, cardiometabolic risk factors and insulin sensitivity with trenbolone in normogonadic rats. Steroids. 2015 Feb;94:60-9.
Galbraith H, Watson HB. Performance, blood and carcase characteristics of finishing steers treated with trenbolone acetate and hexoestrol. Vet Rec. 1978 Jul 8;103(2):28-31.
Galbraith H, Dempster DG. Effect of hexoestrol on the response of finishing steers to treatment with trenbolone acetate. Vet Rec. 1979 Sep 22;105(12):283-4.
Wise DR, Ranaweera KN. The effects of trienbolone acetate and other anabolic agents in growing turkeys. Br Poult Sci. 1981 Mar;22(2):93-104.
Sinnett-Smith PA, Dumelow NW, Buttery PJ. Effects of trenbolone acetate and zeranol on protein metabolism in male castrate and female lambs. Br J Nutr. 1983 Sep;50(2):225-34.
Renaville R, Burny A, Sneyers M, Rochart S, Portetelle D, Théwis A. Effects of an anabolic treatment before puberty with trenbolone acetate-oestradiol or oestradiol alone on growth rate, testicular development and luteinizing hormone and testosterone plasma concentrations. Theriogenology. 1988 Feb;29(2):461-76.
Lee CY, Henricks DM, Skelley GC, Grimes LW. Growth and hormonal response of intact and castrate male cattle to trenbolone acetate and estradiol. J Anim Sci. 1990 Sep;68(9):2682-9.
Perry TC, Fox DG, Beermann DH. Effect of an implant of trenbolone acetate and estradiol on growth, feed efficiency, and carcass composition of Holstein and beef steers. J Anim Sci. 1991 Dec;69(12):4696-702.
Bartle SJ, Preston RL, Brown RE, Grant RJ. Trenbolone acetate/estradiol combinations in feedlot steers: dose-response and implant carrier effects. J Anim Sci. 1992 May;70(5):1326-32.
Cecava MJ, Hancock DL. Effects of anabolic steroids on nitrogen metabolism and growth of steers fed corn silage and corn-based diets supplemented with urea or combinations of soybean meal and feathermeal. J Anim Sci. 1994 Feb;72(2):515-22.
Herschler RC, Olmsted AW, Edwards AJ, Hale RL, Montgomery T, Preston RL, Bartle SJ, Sheldon JJ. Production responses to various doses and ratios of estradiol benzoate and trenbolone acetate implants in steers and heifers. J Anim Sci. 1995 Oct;73(10):2873-81.
Johnson BJ, Anderson PT, Meiske JC, Dayton WR. Effect of a combined trenbolone acetate and estradiol implant on feedlot performance, carcass characteristics, and carcass composition of feedlot steers. J Anim Sci. 1996 Feb;74(2):363-71.
Johnson BJ, White ME, Hathaway MR, Christians CJ, Dayton WR. Effect of a combined trenbolone acetate and estradiol implant on steady-state IGF-I mRNA concentrations in the liver of wethers and the longissimus muscle of steers. J Anim Sci. 1998 Feb;76(2):491-7.
Fritsche S, Solomon MB, Paroczay EW, Rumsey TS. Effects of growth-promoting implants on morphology of Longissimus and Semitendinosus muscles in finishing steers. Meat Sci. 2000 Nov;56(3):229-37.
McClure KE, Solomont MB, Loerch SC. Body weight and tissue gain in lambs fed an all-concentrate diet and implanted with trenbolone acetate or grazed on alfalfa. J Anim Sci. 2000 May;78(5):1117-24.
Dunn JD, Johnson BJ, Kayser JP, Waylan AT, Sissom EK, Drouillard JS. Effects of flax supplementation and a combined trenbolone acetate and estradiol implant on circulating insulin-like growth factor-I and muscle insulin-like growth factor-I messenger RNA levels in beef cattle. J Anim Sci. 2003 Dec;81(12):3028-34.
Scheffler JM, Buskirk DD, Rust SR, Cowley JD, Doumit ME. Effect of repeated administration of combination trenbolone acetate and estradiol implants on growth, carcass traits, and beef quality of long-fed Holstein steers. J Anim Sci. 2003 Oct;81(10):2395-400.
White ME, Johnson BJ, Hathaway MR, Dayton WR. Growth factor messenger RNA levels in muscle and liver of steroid-implanted and nonimplanted steers. J Anim Sci. 2003 Apr;81(4):965-72.
Kreikemeier WM, Mader TL. Effects of growth-promoting agents and season on yearling feedlot heifer performance. J Anim Sci. 2004 Aug;82(8):2481-8.
Lefebvre B, Malouin F, Roy G, Giguère K, Diarra MS. Growth performance and shedding of some pathogenic bacteria in feedlot cattle treated with different growth-promoting agents. J Food Prot. 2006 Jun;69(6):1256-64.
Mader TL, Kreikemeier WM. Effects of growth-promoting agents and season on blood metabolites and body temperature in heifers. J Anim Sci. 2006 Apr;84(4):1030-7.
Parr SL, Chung KY, Hutcheson JP, Nichols WT, Yates DA, Streeter MN, Swingle RS, Galyean ML, Johnson BJ. Dose and release pattern of anabolic implants affects growth of finishing beef steers across days on feed. J Anim Sci. 2011 Mar;89(3):863-73.
Parr SL, Chung KY, Galyean ML, Hutcheson JP, DiLorenzo N, Hales KE, May ML, Quinn MJ, Smith DR, Johnson BJ. Performance of finishing beef steers in response to anabolic implant and zilpaterol hydrochloride supplementation. J Anim Sci. 2011 Feb;89(2):560-70.
Cranwell CD, Unruh JA, Brethour JR, Simms DD, Campbell RE. Influence of steroid implants and concentrate feeding on performance and carcass composition of cull beef cows. J Anim Sci. 1996 Aug;74(8):1770-6.
Hongerholt DD, Crooker BA, Wheaton JE, Carlson KM, Jorgenson DM. Effects of a growth hormone-releasing factor analogue and an estradiol-trenbolone acetate implant on somatotropin, insulin-like growth factor I, and metabolite profiles in growing Hereford steers. J Anim Sci. 1992 May;70(5):1439-48.
Heitzman RJ, Harwood DJ, Kay RM, Little W, Mallinson CB, Reynolds IP. Effects of implanting prepuberal dairy heifers with anabolic steroids on hormonal status, puberty and parturition. J Anim Sci. 1979 Apr;48(4):859-66.
Hancock, D. L., J. F. Wagner, and D. B. Anderson 1991. Effects of estrogens and androgens on animal growth. Pages 255–297 in Growth Regulation in Farm Animals. Advances in Meat Research. Vol. 7. A. M. Pearson and T. R. Dutson ed. Elsevier Applied Science, New York, NY.
Hayden JM, Bergen WG, Merkel RA. Skeletal muscle protein metabolism and serum growth hormone, insulin, and cortisol concentrations in growing steers implanted with estradiol-17 beta, trenbolone acetate, or estradiol-17 beta plus trenbolone acetate. J Anim Sci. 1992 Jul;70(7):2109-19.
Henricks DM, Brandt RT Jr, Titgemeyer EC, Milton CT. Serum concentrations of trenbolone-17 beta and estradiol-17 beta and performance of heifers treated with trenbolone acetate, melengestrol acetate, or estradiol-17 beta. J Anim Sci. 1997 Oct;75(10):2627-33.
Schneider BA, Tatum JD, Engle TE, Bryant TC. Effects of heifer finishing implants on beef carcass traits and longissimus tenderness. J Anim Sci. 2007 Aug;85(8):2019-30.
Heitzman RJ. The effectiveness of anabolic agents in increasing rate of growth in farm animals; report on experiments in cattle. Environ Qual Saf Suppl. 1976;(5):89-98. Review.
Buttery, P., Vernon, B., & Pearson, J. (1978). Anabolic agents—some thoughts on their mode of action. Proceedings of the Nutrition Society, 37(3), 311-315
Grigsby ME, Trenkle 1986 Plasma growth hormone, insulin, glucocorticoids and thyroid hormones in large, medium and small breeds of steers with and without an estradiol implant Domestic Animal Endocrinology p.261-267
Breier, B. H., P. D. Gluckman, and J. J. Bass. 1988. Influence of nutritional status and oestradiol-17b on plasma growth hormone, insulin-like growth factors-I and -II and the response to exogenous growth hormone in young steers. J. Endocrinol. 118:243
Breier BH, Gluckman PD, Bass JJ. The somatotrophic axis in young steers: influence of nutritional status and oestradiol-17 beta on hepatic high- and low-affinity somatotrophic binding sites. J Endocrinol. 1988 Feb;116(2):169-77.
Chung KY, Baxa TJ, Parr SL, Luqué LD, Johnson BJ. Administration of estradiol, trenbolone acetate, and trenbolone acetate/estradiol implants alters adipogenic and myogenic gene expression in bovine skeletal muscle. J Anim Sci. 2012 May;90(5):1421-7.
Johnson BJ, Halstead N, White ME, Hathaway MR, DiCostanzo A, Dayton WR. Activation state of muscle satellite cells isolated from steers implanted with a combined trenbolone acetate and estradiol implant. J Anim Sci. 1998 Nov;76(11):2779-86.
Thompson SH, Boxhorn LK, Kong WY, Allen RE. Trenbolone alters the responsiveness of skeletal muscle satellite cells to fibroblast growth factor and insulin-like growth factor I. Endocrinology. 1989 May;124(5):2110-7.
Johnson BJ, Chung KY. Alterations in the physiology of growth of cattle with growth-enhancing compounds. Vet Clin North Am Food Anim Pract. 2007 Jul;23(2):321-32, viii. Review.
Johnson BJ, Hathaway MR, Anderson PT, Meiske JC, Dayton WR. Stimulation of circulating insulin-like growth factor I (IGF-I) and insulin-like growth factor binding proteins (IGFBP) due to administration of a combined trenbolone acetate and estradiol implant in feedlot cattle. J Anim Sci. 1996 Feb;74(2):372-9.
Schoonmaker JP, Loerch SC, Fluharty FL, Turner TB, Moeller SJ, Rossi JE, Dayton WR, Hathaway MR, Wulf DM. Effect of an accelerated finishing program on performance, carcass characteristics, and circulating insulin-like growth factor concentration of early-weaned bulls and steers. J Anim Sci. 2002 Apr;80(4):900-10.
Pampusch MS, Johnson BJ, White ME, Hathaway MR, Dunn JD, Waylan AT, Dayton WR. Time course of changes in growth factor mRNA levels in muscle of steroid-implanted and nonimplanted steers. J Anim Sci. 2003 Nov;81(11):2733-40.
Lee CY, Lee HP, Jeong JH, Baik KH, Jin SK, Lee JH, Sohnt SH. Effects of restricted feeding, low-energy diet, and implantation of trenbolone acetate plus estradiol on growth, carcass traits, and circulating concentrations of insulin-like growth factor (IGF)-I and IGF-binding protein-3 in finishing barrows. J Anim Sci. 2002 Jan;80(1):84-93.
Walker DK, Titgemeyer EC, Sissom EK, Brown KR, Higgins JJ, Andrews GA, Johnson BJ. Effects of steroidal implantation and ractopamine-HCl on nitrogen retention, blood metabolites and skeletal muscle gene expression in Holstein steers. J Anim Physiol Anim Nutr (Berl). 2007 Oct;91(9-10):439-47.
Winterholler SJ, Parsons GL, Walker DK, Quinn MJ, Drouillard JS, Johnson BJ. Effect of feedlot management system on response to ractopamine-HCl in yearling steers. J Anim Sci. 2008 Sep;86(9):2401-14.
Kamanga-Sollo E, White ME, Chung KY, Johnson BJ, Dayton WR. Potential role of G-protein-coupled receptor 30 (GPR30) in estradiol-17beta-stimulated IGF-I mRNA expression in bovine satellite cell cultures. Domest Anim Endocrinol. 2008 Oct;35(3):254-62.
Pampusch MS, White ME, Hathaway MR, Baxa TJ, Chung KY, Parr SL, Johnson BJ, Weber WJ, Dayton WR. Effects of implants of trenbolone acetate, estradiol, or both, on muscle insulin-like growth factor-I, insulin-like growth factor-I receptor, estrogen receptor-{alpha}, and androgen receptor messenger ribonucleic acid levels in feedlot steers. J Anim Sci. 2008 Dec;86(12):3418-23.
Kamanga-Sollo E, Pampusch MS, Xi G, White ME, Hathaway MR, Dayton WR. IGF-I mRNA levels in bovine satellite cell cultures: effects of fusion and anabolic steroid treatment. J Cell Physiol. 2004 Nov;201(2):181-9.
Bryant TC, Engle TE, Galyean ML, Wagner JJ, Tatum JD, Anthony RV, Laudert SB. Effects of ractopamine and trenbolone acetate implants with or without estradiol on growth performance, carcass characteristics, adipogenic enzyme activity, and blood metabolites in feedlot steers and heifers. J Anim Sci. 2010 Dec;88(12):4102-19.
Barton-Davis ER, Shoturma DI, Sweeney HL. Contribution of satellite cells to IGF-I induced hypertrophy of skeletal muscle. Acta Physiol Scand. 1999 Dec;167(4):301-5.
Reinhardt CD, Lee TL, Thomson DU, Mamedova LK, Bradford BJ. Restricted nutrient intake does not alter serum-mediated measures of implant response in cell culture. J Anim Sci Biotechnol. 2013 Nov 19;4(1):45.
Wu Y, Zhao W, Zhao J, Pan J, Wu Q, Zhang Y, Bauman WA, Cardozo CP. Identification of androgen response elements in the insulin-like growth factor I upstream promoter. Endocrinology. 2007 Jun;148(6):2984-93.
Preston RL, Bartle SJ, Kasser TR, Day JW, Veenhuizen JJ, Baile CA. Comparative effectiveness of somatotropin and anabolic steroids in feedlot steers. J Anim Sci. 1995 Apr;73(4):1038-47.
Elsasser TH, Rumsey TS, Kahl S, Czerwinski SM, Moseley WM, Ono Y, Solomon MB, Harris F, Fagan JM. Effects of Synovex-S and recombinant bovine growth hormone (Somavubove) on growth responses of steers: III. Muscle growth and protein responses. J Anim Sci. 1998 Sep;76(9):2346-53.
Martin MB, Stoica A. Insulin-like growth factor-I and estrogen interactions in breast cancer. J Nutr. 2002 Dec;132(12):3799S-3801S.
Venken K, Schuit F, Van Lommel L, Tsukamoto K, Kopchick JJ, Coschigano K, Ohlsson C, Movérare S, Boonen S, Bouillon R, Vanderschueren D. Growth without growth hormone receptor: estradiol is a major growth hormone-independent regulator of hepatic IGF-I synthesis. J Bone Miner Res. 2005 Dec;20(12):2138-49.
Umayahara Y, Kawamori R, Watada H, Imano E, Iwama N, Morishima T, Yamasaki Y, Kajimoto Y, Kamada T. Estrogen regulation of the insulin-like growth factor I gene transcription involves an AP-1 enhancer. J Biol Chem. 1994 Jun 10;269(23):16433-42.
Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005 Mar 11;307(5715):1625-30.
Prossnitz ER, Arterburn JB, Sklar LA. GPR30: A G protein-coupled receptor for estrogen. Mol Cell Endocrinol. 2007 Feb;265-266:138-42.
Filardo E, Quinn J, Pang Y, Graeber C, Shaw S, Dong J, Thomas P. Activation of the novel estrogen receptor G protein-coupled receptor 30 (GPR30) at the plasma membrane. Endocrinology. 2007 Jul;148(7):3236-45.
Prossnitz ER, Arterburn JB, Smith HO, Oprea TI, Sklar LA, Hathaway HJ. Estrogen signaling through the transmembrane G protein-coupled receptor GPR30. Annu Rev Physiol. 2008;70:165-90.
Kellermeier JD, Tittor AW, Brooks JC, Galyean ML, Yates DA, Hutcheson JP, Nichols WT, Streeter MN, Johnson BJ, Miller MF. Effects of zilpaterol hydrochloride with or without an estrogen-trenbolone acetate terminal implant on carcass traits, retail cutout, tenderness, and muscle fiber diameter in finishing steers. J Anim Sci. 2009 Nov;87(11):3702-11.
Gonzalez JM, Carter JN, Johnson DD, Ouellette SE, Johnson SE. Effect of ractopamine-hydrochloride and trenbolone acetate on longissimus muscle fiber area, diameter, and satellite cell numbers in cull beef cows. J Anim Sci. 2007 Aug;85(8):1893-901.
Joubert Y, Tobin C, Lebart MC. Testosterone-induced masculinization of the rat levator ani muscle during puberty. Dev Biol. 1994 Mar;162(1):104-10.
McCarthy JJ, Mula J, Miyazaki M, Erfani R, Garrison K, Farooqui AB, Srikuea R, Lawson BA, Grimes B, Keller C, Van Zant G, Campbell KS, Esser KA, Dupont-Versteegden EE, Peterson CA. Effective fiber hypertrophy in satellite cell-depleted skeletal muscle. Development. 2011 Sep;138(17):3657-66.
Hughes, N. J., G. T. Schelling, M. J. Garber, J. S. Eastridge, M. B. Solomon, and R. A. Roeder 1998. Skeletal muscle morphology alterations due to Posilac® and Revalor S® treatments alone or in combination in feedlot steers. J. Anim. Sci. 49(Proceedings Western Section):90–93
Sone K, Hinago M, Itamoto M, Katsu Y, Watanabe H, Urushitani H, Tooi O, Guillette LJ Jr, Iguchi T. Effects of an androgenic growth promoter 17beta-trenbolone on masculinization of Mosquitofish (Gambusia affinis affinis). Gen Comp Endocrinol. 2005 Sep 1;143(2):151-60.
Zhao JX, Hu J, Zhu MJ, Du M. Trenbolone enhances myogenic differentiation by enhancing β-catenin signaling in muscle-derived stem cells of cattle. Domest Anim Endocrinol. 2011 May;40(4):222-9.
Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001 Nov 23;294(5547):1704-8.
Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc Natl Acad Sci U S A. 2001 Dec 4;98(25):14440-5.
Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, Price SR, Mitch WE, Goldberg AL. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004 Jan;18(1):39-51.
Danhaive PA, Rousseau GG. Evidence for sex-dependent anabolic response to androgenic steroids mediated by muscle glucocorticoid receptors in the rat. J Steroid Biochem. 1988 Jun;29(6):575-81.
Sharpe PM, Buttery PJ, Haynes NB. The effect of manipulating growth in sheep by diet or anabolic agents on plasma cortisol and muscle glucocorticoid receptors. Br J Nutr. 1986 Jul;56(1):289-304.
Reiter M, Walf VM, Christians A, Pfaffl MW, Meyer HH. Modification of mRNA expression after treatment with anabolic agents and the usefulness for gene expression-biomarkers. Anal Chim Acta. 2007 Mar 14;586(1-2):73-81.
Sheffield-Moore M. Androgens and the control of skeletal muscle protein synthesis. Ann Med. 2000 Apr;32(3):181-6. Review.
Santidrián S, Thompson JR, Young VR. Effect of trienbolone acetate on the rate of myofibrillar protein breakdown in young adrenalectomized male rate treated with corticosterone. Arch Farmacol Toxicol. 1981 Dec;7(3):333-40.
Isaacson WK, Jones SJ, Krueger RJ. Testosterone, Dihydrotestosterone, trenbolone acetate, and zeranol alter the synthesis of cortisol in bovine adrenocortical cells. J Anim Sci. 1993 Jul;71(7):1771-7.
Morgan SA, Sherlock M, Gathercole LL, Lavery GG, Lenaghan C, Bujalska IJ, Laber D, Yu A, Convey G, Mayers R, Hegyi K, Sethi JK, Stewart PM, Smith DM, Tomlinson JW. 11beta-hydroxysteroid dehydrogenase type 1 regulates glucocorticoid-induced insulin resistance in skeletal muscle. Diabetes. 2009 Nov;58(11):2506-15.
Coderre L, Srivastava AK, Chiasson JL. Role of glucocorticoid in the regulation of glycogen metabolism in skeletal muscle. Am J Physiol. 1991 Jun;260
Gounarides JS, Korach-André M, Killary K, Argentieri G, Turner O, Laurent D. Effect of dexamethasone on glucose tolerance and fat metabolism in a diet-induced obesity mouse model. Endocrinology. 2008 Feb;149(2):758-66.
Chan KH, Heitzman RJ, Kitchenham BA. Digestibility and N-balance studies on growing heifers implanted with trienbolone acetate. Br Vet J. 1975 Mar-Apr;131(2):170-4.
van Weerden EJ, Grandadam JA. The effect of an anabolic agent on N deposition, growth, and slaughter quality in growing castrated male pigs. Environ Qual Saf Suppl. 1976;(5):115-22.
Lobley GE, Connell A, Mollison GS, Brewer A, Harris CI, Buchan V, Galbraith H. The effects of a combined implant of trenbolone acetate and oestradiol-17 beta on protein and energy metabolism in growing beef steers. Br J Nutr. 1985 Nov;54(3):681-94.
Kerth CR, Montgomery JL, Morrow KJ, Galyean ML, Miller MF. Protein turnover and sensory traits of longissimus muscle from implanted and nonimplanted heifers. J Anim Sci. 2003 Jul;81(7):1728-35.
Kamanga-Sollo E, White ME, Hathaway MR, Weber WJ, Dayton WR. Effect of trenbolone acetate on protein synthesis and degradation rates in fused bovine satellite cell cultures. Domest Anim Endocrinol. 2011 Jan;40(1):60-6.
Rodway RG, Galbraith H. Effects of anabolic steroids on hepatic enzymes of amino acid catabolism. Horm Metab Res. 1979 Aug;11(8):489-90.
Compston JE. Sex steroids and bone. Physiol Rev. 2001 Jan;81(1):419-447. Review.
Leifke E, Körner HC, Link TM, Behre HM, Peters PE, Nieschlag E. Effects of testosterone replacement therapy on cortical and trabecular bone mineral density, vertebral body area and paraspinal muscle area in hypogonadal men. Eur J Endocrinol. 1998 Jan;138(1):51-8.
Martin AC. Osteoporosis in men: a review of endogenous sex hormones and testosterone replacement therapy. J Pharm Pract. 2011 Jun;24(3):307-15.
Bhasin S, Tenover JS. Age-associated sarcopenia–issues in the use of testosterone as an anabolic agent in older men. J Clin Endocrinol Metab. 1997 Jun;82(6):1659-60.
Calof OM, Singh AB, Lee ML, Kenny AM, Urban RJ, Tenover JL, Bhasin S. Adverse events associated with testosterone replacement in middle-aged and older men: a meta-analysis of randomized, placebo-controlled trials. J Gerontol A Biol Sci Med Sci. 2005 Nov;60(11):1451-7.
Bhasin S, Cunningham GR, Hayes FJ, Matsumoto AM, Snyder PJ, Swerdloff RS, Montori VM; Task Force, Endocrine Society. Testosterone therapy in men with androgen deficiency syndromes: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2010 Jun;95(6):2536-59.
Kasperk CH, Wakley GK, Hierl T, Ziegler R. Gonadal and adrenal androgens are potent regulators of human bone cell metabolism in vitro. J Bone Miner Res. 1997 Mar;12(3):464-71.
Wiren KM, Zhang X-W, Olson DA, Turner RT, Iwaniec UT. Androgen prevents hypogonadal bone loss via inhibition of resorption mediated by mature osteoblasts/osteocytes. Bone. 2012;51(5):835-846.
Vandenput L, Ohlsson C. Estrogens as regulators of bone health in men. Nat Rev Endocrinol. 2009 Aug;5(8):437-43.
Smith ZK, Thompson AJ, Hutcheson JP, Nichols WT, Johnson BJ. Evaluation of Coated Steroidal Implants Containing Trenbolone Acetate and Estradiol-17β on Live Performance, Carcass Traits, and Sera Metabolites in Finishing Steers. J Anim Sci. 2018 Mar 9.
Nella prima parte di questa serie di articoli, ho esposto diverse informazioni riguardanti principalmente le caratteristiche metaboliche del Trenbolone nei mammiferi. Se non avete già letto la prima parte, vi esorto a farlo prima di proseguire con la lettura della seconda parte, poiché alcuni degli argomenti trattati in precedenza verranno approfonditi nel presente articolo.
VI. Effetti sull’Asse HPG
Nei vertebrati, l’Asse Ipotalamo-Ipofisi-Gonadi (HPG), conosciuto nell’uomo anche come HPTA, controlla i processi riproduttivi attraverso una varietà di ormoni che agiscono sui tessuti bersaglio direttamente o indirettamente. Ad alti livelli, nei maschi, l’Ormone Rilasciante la Gonadotropina (GnRH) secreto dall’ipotalamo stimola l’ipofisi a rilasciare l’Ormone Luteinizzante (LH) e l’Ormone Follicolo-Stimolante (FSH). Questi, a loro volta, stimolano il rilascio di ormoni sessuali dai testicoli (l’FSH incrementa la risposta all’LH attraverso l’up- regulation dei recettori nelle cellule di Leydig.).(1)(2) La forte connessione tra i diversi componenti del sistema che caratterizza l’Asse HPG si traduce nel fatto che nessuno di questi funzioni in maniera isolata. Entrando in circolo, sia gli ormoni androgenici che quelli estrogenici sono in grado di attraversare la barriera emato-encefalica e di esercitare un feedback negativo sull’ipofisi e sull’ipotalamo, sottoregolando quindi il rilascio del GnRH e, di conseguenza, causando una sottoregolazione/soppressione del funzionamento dell’intero asse.(3)
La somministrazione di Trenbolone è associata a numerosi tipi di alterazioni dell’Asse HPG, il che è in linea con ciò che è stato osservato con vari altri trattamenti androgeni nel corso degli anni. (4) Alcune delle alterazioni indotte dal Trenbolone osservate negli anni includono livelli ridotti di LH serico (5)(6)(7)(8)(9)(10), ridotti livelli di FSH serico (11), ridotti livelli di Testosterone serico (5)(6) (7)(8)(9)(10)(12)(13)(14)(15)(16), ridotti livelli di DHT serico (11), ridotti livelli di estradiolo serico (13)(15), atrofia testicolare (7)(17)(18) e un principio di ritardato della pubertà (19). Questi effetti si verificano abbastanza rapidamente poiché, in uno studio svolto su ratti, sono stati registrati entro dieci giorni dalla somministrazione di Trenbolone Enantato tassi di soppressione del Testosterone serico pari all’80% e tassi di soppressione del DHT serico pari al 70% rispetto agli animali di controllo. (20) Vale la pena notare che essendo il Trenbolone Enantato (TBE) una variante a lunga durata d’azione, con l’uso del Trenbolone Acetato (TBA) gli effetti registrati si sarebbero potenzialmente verificati in tempi più rapidi.
Non è del tutto chiaro quali siano i meccanismi attraverso i quali si esplicano gli effetti di soppressione dati dal Trenbolone sull’Asse HPG, tuttavia ci sono state certamente alcune prove nel corso degli anni che forniscono indizi in merito. Un’ipotesi diffusa prevede l’inibizione diretta del feedback ipotalamico, come evidenziato dalla ridotta trascrizione di GnRH osservata nel cervello dei modelli animali (pesci). Questo può essere additivo ai suoi effetti diretti sulla biosintesi degli steroidi testicolari, come sostenuto dalla sottoregolazione dell’espressione del CYP17 testicolare.(21) Il CYP17 è un enzima molto importante nella biosintesi degli steroidi e catalizza sequenzialmente due reazioni chiave nella produzione di steroidi sessuali nei maschi.
È anche interessante notare che qualunque sia il meccanismo, non sembra essere dipendente dal Recettore degli Androgeni (AR).(22)(23) Sostenendo ulteriormente questa linea di pensiero, nelle colture di tessuto ovarico di pesce, androgeni non aromatizzabili come il Trenbolone hanno mostrato effetti inibitori diretti e non genomici, anti-androgeno-insensibili, sulla sintesi di estrogeni.(24) È altamente probabile che i meccanismi di feedback alla base dell’alterazione dell’Asse HPG da parte del Trenbolone siano del tutto simili ad altri androgeni, con conseguente inibizione dei livelli di GnRH e, in definitiva, la sottoregolazione/inibizione della produzione di FSH e LH.(25)
Un altro potenziale gene candidato coinvolto nelle diminuite concentrazioni di steroidi sessuali, osservate in esperimenti su pesci i quali erano stati esposti al forte androgeno 17-trenbolone, è l’idrossisteroide (17β) deidrogenasi 12a (hsd17b12a). L’Hsd17b12a catalizza la conversione del Androstenedione in Testosterone che, a sua volta, viene convertito in 17β-estradiolo dall’enzima aromatasi. Pertanto, la sottoregolazione del hsd17b12a, come si può osservare nei pesci esposti al Trenbolone, ha come prevedibile risposta un declino sia nei livelli di Testosterone che di Estradiolo.(14)
VII. Effetti sui Pathways Anabolici
Come già detto nella prima parte, il Trenbolone è considerabile quale SARM con il suo indice terapeutico pari a circa 3,4. Vorrei utilizzare questa sezione per discutere parte delle caratteristiche che fanno del Trenbolone un composto SARM-simile.
5α-Reduttasi
Nonostante abbia una somiglianza strutturale con il Testosterone, il Trenbolone non è soggetto alla 5α-riduzione a causa della presenza di una struttura a 3-ossotrieni che impedisce la riduzione dell’anello A.(26) Questo è il percorso enzimatico utilizzato per la conversione del Testosterone nel suo metabolita fortemente androgeno Dihydrotestosterone (DHT). Poiché il Trenbolone non è un substrato soggetto all’azione del enzima 5α-reduttasi, ha dimostrato di stimolare effetti androgenici meno pronunciati rispetto al Testosterone nei tessuti sensibili agli androgeni che esprimono l’enzima 5α-reduttasi, compresi gli organi sessuali accessori e la prostata.(27)(28)(29)(30)(31)(32). Per rendere meglio l’idea di ciò che si è appena affermato, il Testosterone ha una potenza circa tre volte superiore nei tessuti androgenici che esprimono l’enzima 5α-reduttasi nonostante abbia un’affinità di legame ai AR significativamente più bassa rispetto al Trenbolone.(33) Discuteremo di come questa caratteristica influenzi il potenziale ipertrofico in questi tessuti in seguito.
Come presumibile, un motivo particolare per cui il Trenbolone sta iniziando a prendere piede nella comunità scientifica è dovuto al suo potenziale di ridurre i rischi associati al cancro alla prostata nei pazienti trattati per l’ipogonadismo. L’attuale strategia per il trattamento degli individui ipogonadici consiste nella somministrazione di Testosterone al fine di ripristinare i livelli fisiologici dell’ormone. Tuttavia, nei maschi adulti, la crescita benigna e maligna del tessuto prostatico ghiandolare è ampiamente regolata dagli ormoni sessuali. Inoltre, è stato dimostrato che anche aumenti moderati del Testosterone circolante si traducono direttamente in pronunciati effetti iperplastici nei tessuti prostatici, mediati dalla sua 5α riduzione in DHT.(34)(35) Più avanti, indagheremo più a fondo nella letteratura disponibile per constatare se il potenziale attribuito al Trenbolone di abbassare il rischio di cancro alla prostata sia una realtà concreta.
Enzima Aromatasi
Enzima Aromatasi
Stranamente, uno dei quesiti sul Trenbolone maggiormente posti è se questo AAS abbia o meno effetti sui livelli degli estrogeni serici, e se possa aromatizzare o meno come il Testosterone. E’ un fatto pienamente riconosciuto nella comunità scientifica che il Trenbolone, ed altri composti C19 nor-steroidi, non sia soggetto all’azione dell’enzima aromatasi.(36)(37) Detto questo, è necessario che voi capiate che la precedente affermazione non sta a significare che i composti C19 nor-steroidi non possano essere convertiti in estrogeni o non possano indurre effetti estrogenici. (38)(39)
Il Trenbolone è in gran parte ritenuto non avere alcuna attività estrogenica (40)(41) e ci sono stati numerosi studi sugli animali che hanno dimostrato che il suo uso causa una riduzione delle concentrazioni ematiche di Estradiolo. (13)(14)(15)(42)(43)(44)Tenete presente che ci sono stati alcuni studi che non hanno mostrato questo effetto soppressivo sui livelli estrogenici (10)(45)(46) ma, nel complesso, il corpo della letteratura nel suo complesso supporta l’ipotesi che il Trenbolone possieda effetti anti-estrogenici.
Sulla base di ciò che ora sappiamo sull’Asse HPG, avrebbe sicuramente senso pensare che gli effetti anti-estrogenici causati dalla somministrazione di Trenbolone siano probabilmente legati al suo feedback negativo sull’Asse. Questo feedback negativo, causando l’inibizione della produzione endogena di Testosterone, porta ad una riduzione dei substrati soggetti all’azione dell’enzima aromatasi, il quale è necessario per la biosintesi degli estrogeni endogeni nei maschi. Questo impatto sull’Asse HPG causerebbe un tasso di inibizione estrogenica più marcato rispetto a qualsiasi potenziale effetto diretto del Trenbolone sui recettori degli estrogeni e / o l’enzima aromatasi.(5)(6)(8)(21)(47) L’impatto di questo AAS sui livelli estrogenici e la loro attività potrebbe contare anche un meccanismo secondario legato alla capacità del Trenbolone di sottoregolare l’espressione di entrambi i recettori estrogenici α e β (REα/Reβ).(48)
Vi sono state altre scoperte interessanti riguardo ai meccanismi alla base della relazione del Trenbolone con gli estrogeni, così come le risposte compensatorie associate ai livelli dell’ormone soppresso. È stato dimostrato che il Trenbolone riduce le concentrazioni tissutali e l’espressione genica della VTG (Vitellogenina), una proteina positivamente associata all’esposizione a composti estrogenici. (13)(14)(21)(41)(47)(49)(50)(51)(52)(53)(54) E’ stata anche dimostrata una sottoregolazione a livello cerebrale del CYP19B (aromatasi B) e una sovra regolazione gonadica del CYP19A (aromatasi A) in pesci femmina, anche se la cosa non è risultata interessante nei maschi. (14)(54)
Flutamide
L’impatto del Trenbolone sugli estrogeni non sembra essere AR-dipendente, poiché gli studi hanno dimostrato che la co-somministrazione con un antagonista dei AR (Flutamide) ha portato alla stessa attività anti-estrogenica nei pesci.(13) È interessante notare che c’è stato un altro studio sui pesci il quale ha riportato che il Trenbolone possiede una bassa affinità per il recettore degli estrogeni ma può potenzialmente attivarlo.(44) Che questa caratteristica sia specifica o meno per la specie presa in esame è questione di dibattito, poiché in altri studi che ho esaminato non ho riscontrato che tale effetto si verifichi. Tuttavia, esperimenti di coltura cellulare e analisi biologiche dimostrano che il Trenbolone ed i suoi metaboliti hanno un’affinità di legame molto bassa con i recettori degli estrogeni, circa il 20% dell’affinità dell’Estradiolo.(40)
Quindi, il Trenbolone può aromatizzare? Date le informazioni sopra esposte (soppressione dei livelli di E2) e la presenza di un doppio legame inserito in C9– C10 che esclude teoricamente tale possibilità, al momento non ho trovato nulla che suggerisca in via definitiva che ciò possa verificarsi. Esiste però un’ipotesi esposta da Holland et al. (55) che trovo abbastanza interessante da includere nella sua interezza:
“In precedenza abbiamo riportato che il Trenbolone Enantato riduce la massa grassa viscerale in animali giovani e anziani ORX, indicando che la perdita di grasso si verifica in risposta alla somministrazione di androgeni, anche in assenza di un substrato androgenico soggetto all’azione dell’enzima aromatasi. Tuttavia, il nostro precedente lavoro non ha tenuto conto della possibilità che l’Androstenedione (derivato dal Deidroepiandrosterone) possa essere aromatizzato in Estrone e, successivamente, convertito in E2 per azione del 17β-idrossisteroide deidrogenasi nei tessuti, come il grasso, esprimendo gli enzimi richiesti.”
Progesterone/SHBG
Dimero SHBG umano.
È stato dimostrato che il Trenbolone possiede un’alta affinità per il recettore progestinico dei bovini e si presume che abbia un’affinità simile allo stesso Progesterone verso tale recettore.(56) L’analisi in vitro ha rivelato che la relativa affinità di legame con il recettore del progesterone bovino, rispetto al progesterone stesso, era del 137,4% per 17β-TbOH e del 2,1% per 17α-TbOH (57). Infine, la relativa affinità di legame tra Trenbolone e SHBG umano, rispetto al DHT, è del 29,4% per il 17β-TbOH e del 94,8% per il 17α-TbOH.
VIII. Effetti sui marker della salute metabolica
Uno dei motivi principali per cui i detrattori del Trenbolone dispensano ammonimenti contro il suo uso è legato alla severità con cui il composto incide apparentemente sui marker della salute. Speravo di avere qualche riferimento effettivo su esami ematici da aggiungere a questo articolo, ma sfortunatamente i miei sforzi di crowdsourcing non hanno avuto successo poiché non molte persone usano il Trenbolone da solo. Quindi quello che farò in questa sezione è proseguire con l’esposizione della letteratura scientifica disponibile (studi su animali) che espone l’impatto del Trenbolone su diversi marker della salute.
Asse Ipotalamo-Ipofisi-Tiroide (Asse Tiroideo)
Il rapporto tra il Trenbolone e l’Asse Tiroideo è decisamente dibattuto nel mondo del BodyBuilding. Sebbene gli effetti osservati siano stati un po’ incoerenti, sembra esserci un pattern che suggerisce che il Trenbolone abbia un effetto soppressivo complessivo sull’Asse Tiroideo. In uno studio, il Trenbolone ha ridotto il T4 nelle giovenche, il Trenbolone e l’Estradiolo hanno ridotto il T4 nei manzi, mentre non è stato osservato alcun impatto sul uptake del T3.(58) In un altro studio, il Trenbolone e l’Estradiolo hanno effettivamente aumentato il T3, mentre il Trenbolone da solo ha ridotto sia il T3 che il T4.(59) Bisogna ricordare, però, che l’Estradiolo stimola l’asse GH / IGF, il che aumenta acutamente la conversione del T4 in T3. Ciò può aiutare a spiegare il perché la co-somministrazione con estradiolo può portare a livelli più elevati di T3 mentre la somministrazione di solo Trenbolone ha come conseguenza l’effetto opposto, poiché i livelli di Estradiolo sono marcatamente soppressi. Anche negli studi in cui i livelli tiroidei non sono risultati significativamente differenti, il Trenbolone ha ridotto i tassi metabolici a digiuno, portando ad un minor fabbisogno calorico per creare un surplus energetico.(60)
Pertanto, può essere ragionevole ipotizzare che l’aumentata efficienza alimentare vista in numerosi studi nel corso degli anni potrebbe essere correlata ad una alterazione metabolica trenbolone-mediata. Naturalmente, va notato che i bovini più magri tendono a crescere più velocemente e ad utilizzare i mangimi in modo più efficiente, quindi potrebbe anche essere solo un sottoprodotto di questo.(61) Prima di concludere questa serie di articoli, parlerò un po’ più approfonditamente della gestione pratica di questo possibile impatto dato dal Trenbolone.
Colesterolo
In generale, esiste una forte correlazione tra la perdita di grasso e i cambiamenti favorevoli nei livelli serici dei lipidi, in particolare negli uomini. (62)(63) Pertanto, poiché il Trenbolone ha dimostrato costantemente di migliorare la composizione corporea, è ragionevole ipotizzare che possa avere un impatto favorevole sui marker lipidici. Quindi, vediamo quali prove esistono a riguardo analizzando le informazioni provenienti dagli studi svolti sugli animali.
Studi sui ratti hanno dimostrato che sia il Testosterone che il Trenbolone possiedono capacità protettive simili contro il colesterolo elevato nonostante la capacità del Trenbolone di provocare la perdita del grasso viscerale sia maggiore. Ciò suggerisce che i livelli serici di colesterolo possono essere regolati principalmente dalla composizione corporea complessiva, indipendentemente dai cambiamenti nelle riserve adipose viscerali. In uno studio, il colesterolo totale serico, l’HDL e l’LDL erano tutti significativamente più bassi nei ratti trattati con Trenbolone rispetto ai ratti di controllo (- 62%, – 57% e – 78% rispettivamente). Il sottoprodotto di questo era che i ratti trattati avevano un rapporto HDL:LDL maggiore. Anche i Trigliceridi serici erano diminuiti di un significativo 51% rispetto ai ratti di controllo.(15) In un altro studio, sia il Testosterone che il Trenbolone hanno ridotto il colesterolo circolante nei ratti alimentati con una dieta ricca di grassi e zuccheri, ma solo il Trenbolone ha ridotto i livelli di Trigliceridi circolanti.(64) Si consiglia vivamente di tenere d’occhio i livelli di colesterolo quando si usano dosi sovrafisiologiche di androgeni, in quanto tendono ad avere la capacità di aumentare lo stimolo delle catecolamine sull’ormone lipasi sensibile (HSL) nel tessuto epatico e cardiaco.(65)(66) Questa aumentata attività della HSL tende a determinare un aumento del tasso di mobilitazione dei trigliceridi e un aumento della degradazione del HDL. Avere livelli di HDL cronicamente bassi rappresenta un fattore di rischio cardiovascolare indipendente, quindi, un uso prolungato di AAS che portano ad una soppressione marcata del HDL (vedi, per esempio, composti metilati in C-17) dovrebbe essere attentamente monitorato.(67)
Marker epatici
Enzimi epatocellulari (ALT e AST) sono immagazzinati all’interno delle cellule epatiche e gli enzimi epatici colestatici (es. ALP e GGT) sono associati a dotti biliari esterni alle cellule epatiche.
Ci sono alcuni marker epatici comunemente utilizzati per valutare la funzionalità e la salute del fegato, così come il danno a quest’organo. L’albumina è un marker indicativo della funzione epatica generale mentre AST, ALT, ALP sono tutti marker generali del danno epatico.
Recenti studi svolti sui roditori hanno dimostrato che il Trenbolone non sembra indurre danni significativi al tessuto epatico. In uno studio, campioni di tessuto epatico di ratti trattati con Trenbolone hanno mostrato una morfologia simile a quella dei ratti di controllo. AST, ALT, ALP e albumina erano tutti a livello simile tra i ratti trattati con Trenbolone ed i ratti del gruppo di controllo.(15) In un altro studio, sono stati osservati valori simili negli enzimi epatici con ratti alimentati con una dieta ricca di grassi e zuccheri in tutti i gruppi di trattamento, compresi i gruppi trattati con Testosterone e Trenbolone.(64) Questo steroide possiede comunque un forte livello di resistenza alla disattivazione epatica, e una significativa tossicità epatica è stata osservato nei body builder che abusano del Trenbolone.(68) Sebbene non sia una costante, l’epatotossicità non può essere completamente esclusa, in particolare con alte dosi.
Insulina
L’Insulina è un altro ormone che tende ad avere una correlazione diretta con il grasso corporeo, e in particolare con i livelli di grasso viscerale. L’accumulo di grasso viscerale e l’aumento dei livelli di trigliceridi circolanti sono entrambi associati con il peggioramento dell’insulino-resistenza. (69) Al contrario, la restrizione calorica e la consequenziale perdita di peso nei soggetti non diabetici visceralmente obesi ha indotto miglioramenti significativi nella sensibilità all’insulina.(70) Oltre all’obesità, esistono anche prove convincenti che dimostrano come bassi livelli di androgeni promuovano il peggioramento dell’insulino-rsistenza. (71) È stato ipotizzato che il trattamento con Trenbolone in modelli animali possa promuovere effetti di miglioramento della sensibilità all’insulina attraverso meccanismi simili a quelli ottenuti dalla restrizione calorica nei soggetti umani di sesso maschile.
Uno studio ha dimostrato che l’Insulina serica è significativamente più bassa nei ratti trattati con Trenbolone (riduzione del 38%) rispetto ai ratti di controllo, e che ciò si è tradotto in un valore HOMA-IR (indice utilizzato per valutare l’insulino resistenza) significativamente più basso.(15) I ratti che sono stati sottoposti a regimi alimentari ad alto contenuto di grassi e zuccheri presentavano livelli di Insulina serica significativamente elevati e che sono stati ripristinati solo parzialmente con la somministrazione di Testosterone, mentre con la somministrazione di Trenbolone i livelli insulinici si sono ridotti significativamente.(64) In effetti, il gruppo trattato con Trenbolone era l’unico a mostrare valori di HOMA-IR ridotti, indicando un aumento della funzionalità delle cellule beta e una riduzione della resistenza all’insulina. Quindi, anche se limitata, l’evidenza suggerisce che il Trenbolone abbia effetti sulla sensibilità all’Insulina superiore al Testosterone.
L’adiponectina è un’adipochina sensibilizzante dell’insulina da 30 kDa che viene principalmente secreta dal tessuto adiposo viscerale.(72)(73) In generale, i livelli serici di adiponectina sono inversamente proporzionali alla massa grassa (74). Il Testosterone ed il Trenbolone tendono a ridurre i livelli totali di adiponectina in misura simile nei ratti (75).
Eritropoiesi
L’eritropoiesi è il processo di formazione dei globuli rossi (RBC) attraverso una serie di elementi cellulari immaturi (serie eritroblastica). Uno degli effetti collaterali più comunemente riportati durante le TRT è l’aumento dei livelli di ematocrito ed emoglobina. Nello specifico, la deprivazione androgenica riduce sia l’ematocrito che l’emoglobina mentre la somministrazione di Testosterone determina un aumento dose-dipendente di entrambi.(76)(77)
Eritropoietina (EPO)
I meccanismi mediante i quali gli androgeni aumentano la produzione di RBC possono essere direttamente correlati alla stimolazione della secrezione di eritropoietina renale o persino del midollo osseo.(78) E sulla base delle prove esistenti, sembrerebbe che gli androgeni elevino direttamente l’eritropoiesi attraverso meccanismi AR-mediati.(11) Non sembra che l’aromatizzazione del Testosterone sia necessaria per l’eritropoiesi poiché la somministrazione di DHT ne causa un analogo aumento nei soggetti di sesso maschile.(79) Inoltre, è stato dimostrato che ciò si verifica anche in soggetti di sesso maschile con carenza dell’enzima aromatasi.(80) Allo stesso modo, per l’eritropoiesi non sembra essere necessaria una riduzione della 5α riduzione del Testosterone poiché la co-somministrazione di Testosterone e Finasteride (inibitore dell’enzima 5α-riduttasi) ha portato ad un aumento sia dell’ematocrito che dell’emoglobina nella stessa misura data dalla sola somministrazione di Testosterone nonostante una riduzione nei livelli di DHT del 65% nel gruppo Finasteride .(81)
Se il Trenbolone non causasse l’innalzamento dell’ematocrito e dell’emoglobina osservati con le tradizionali TRT, allora questo potrebbe essere un altro potenziale motivo che renderebbe questa molecola un candidato interessante per le HTR.
Prove preliminari indicano che il Trenbolone aumenta l’emoglobina nei roditori maschi in modo dose-dipendente, e in misura leggermente superiore rispetto a dosi sovrafisiologiche di Testosterone (8-10%), nonostante il DHT sia soppresso di oltre il 70% dopo la somministrazione.(20) In un altro studio, a dosi somministrate che erano sette volte superiori a quelle del Testosterone, i ratti trattati con Trenbolone avevano livelli di emoglobina quasi identici, sebbene entrambi fossero significativamente elevati rispetto al gruppo di controllo.(82)
Penso che le informazioni esposte fino a questo punto siano più che sufficienti per concludere questa seconda parte. Nella terza parte, inizieremo ad approfondire il potenziale ipertrofico del Trenbolone.
Stay tuned!
Gabriel Bellizzi
Riferimenti:
Simoni M, Weinbauer GF, Gromoll J, Nieschlag E. Role of FSH in male gonadal function. Ann Endocrinol (Paris). 1999 Jul;60(2):102-6. Review.
Liu PY, Iranmanesh A, Nehra AX, Keenan DM, Veldhuis JD. Mechanisms of hypoandrogenemia in healthy aging men. Endocrinol Metab Clin North Am. 2005 Dec;34(4):935-55, ix. Review.
Kumar TR, Low MJ. Hormonal regulation of human follicle-stimulating hormone-beta subunit gene expression: GnRH stimulation and GnRH-independent androgen inhibition. Neuroendocrinology. 1995 Jun;61(6):628-37.
Tan RS, Scally MC. Anabolic steroid-induced hypogonadism–towards a unified hypothesis of anabolic steroid action. Med Hypotheses. 2009 Jun;72(6):723-8.
Fabry J, Renaville R, Halleux V, Burny A. Plasma testosterone and LH responses to LHRH in double-muscled bulls treated with trenbolone acetate and zeranol. J Anim Sci. 1983 Nov;57(5):1138-45.
Gettys TW, D’Occhio MJ, Henricks DM, Schanbacher BD. Suppression of LH secretion by oestradiol, dihydrotestosterone and trenbolone acetate in the acutely castrated bull. J Endocrinol. 1984 Jan;100(1):107-12.
Silcox RW, Keeton JT, Johnson BH. Effects of zeranol and trenbolone acetate on testis function, live weight gain and carcass traits of bulls. J Anim Sci. 1986 Aug;63(2):358-68.
Renaville R, Burny A, Sneyers M, Rochart S, Portetelle D, Théwis A. Effects of an anabolic treatment before puberty with trenbolone acetate-oestradiol or oestradiol alone on growth rate, testicular development and luteinizing hormone and testosterone plasma concentrations. Theriogenology. 1988 Feb;29(2):461-76.
Lee CY, Henricks DM, Skelley GC, Grimes LW. Growth and hormonal response of intact and castrate male cattle to trenbolone acetate and estradiol. J Anim Sci. 1990 Sep;68(9):2682-9.
Hunt DW, Henricks DM, Skelley GC, Grimes LW. Use of trenbolone acetate and estradiol in intact and castrate male cattle: effects on growth, serum hormones, and carcass characteristics. J Anim Sci. 1991 Jun;69(6):2452-62.
Yarrow JF, McCoy SC, Borst SE. Tissue selectivity and potential clinical applications of trenbolone (17beta-hydroxyestra-4,9,11-trien-3-one): A potent anabolic steroid with reduced androgenic and estrogenic activity. Steroids. 2010 Jun;75(6):377-89.
Galbraith, H. (1982). Growth, hormonal and metabolic response of post-pubertal entire male cattle to trenbolone acetate and hexoestrol. Animal Science, 35(2), 269-276.
Ankley GT, Defoe DL, Kahl MD, Jensen KM, Makynen EA, Miracle A, Hartig P, Gray LE, Cardon M, Wilson V. Evaluation of the model anti-androgen flutamide for assessing the mechanistic basis of responses to an androgen in the fathead minnow (Pimephales promelas). Environ Sci Technol. 2004 Dec 1;38(23):6322-7.
Dorts J, Richter CA, Wright-Osment MK, Ellersieck MR, Carter BJ, Tillitt DE. The genomic transcriptional response of female fathead minnows (Pimephales promelas) to an acute exposure to the androgen, 17beta-trenbolone. Aquat Toxicol. 2009 Jan 18;91(1):44-53.
Donner DG, Beck BR, Bulmer AC, Lam AK, Du Toit EF. Improvements in body composition, cardiometabolic risk factors and insulin sensitivity with trenbolone in normogonadic rats. Steroids. 2015 Feb;94:60-9.
Ma F, Liu D. 17β-trenbolone, an anabolic-androgenic steroid as well as an environmental hormone, contributes to neurodegeneration. Toxicol Appl Pharmacol. 2015 Jan 1;282(1):68-76.
O’Lamhna M, Roche JF. Effect of repeated implantation with anabolic agents on growth rate, carcase weight, testicular size and behaviour of bulls. Vet Rec. 1983 Dec 3;113(23):531-4.
López-Bote C, Sancho G, Martínez M, Ventanas J, Gázquez A, Roncero V. Trenbolone acetate induced changes in the genital tract of male pigs. Zentralbl Veterinarmed B. 1994 Mar;41(1):42-8.
Moran C, Prendiville DJ, Quirke JF, Roche JF. Effects of oestradiol, zeranol or trenbolone acetate implants on puberty, reproduction and fertility in heifers. J Reprod Fertil. 1990 Jul;89(2):527-36.
Yarrow JF, Conover CF, McCoy SC, Lipinska JA, Santillana CA, Hance JM, Cannady DF, VanPelt TD, Sanchez J, Conrad BP, Pingel JE, Wronski TJ, Borst SE. 17β-Hydroxyestra-4,9,11-trien-3-one (trenbolone) exhibits tissue selective anabolic activity: effects on muscle, bone, adiposity, hemoglobin, and prostate. Am J Physiol Endocrinol Metab. 2011 Apr;300(4):E650-60.
Zhang X, Hecker M, Park JW, Tompsett AR, Jones PD, Newsted J, Au DW, Kong R, Wu RS, Giesy JP. Time-dependent transcriptional profiles of genes of the hypothalamic-pituitary-gonadal axis in medaka (Oryzias latipes) exposed to fadrozole and 17beta-trenbolone. Environ Toxicol Chem. 2008 Dec;27(12):2504-11.
Heinlein CA, Chang C. The roles of androgen receptors and androgen-binding proteins in nongenomic androgen actions. Mol Endocrinol. 2002 Oct;16(10):2181-7. Review.
Martinović D, Blake LS, Durhan EJ, Greene KJ, Kahl MD, Jensen KM, Makynen EA, Villeneuve DL, Ankley GT. Reproductive toxicity of vinclozolin in the fathead minnow: confirming an anti-androgenic mode of action. Environ Toxicol Chem. 2008 Feb;27(2):478-88.
Braun AM, Thomas P. Androgens inhibit estradiol-17beta synthesis in Atlantic croaker (Micropogonias undulatus) ovaries by a nongenomic mechanism initiated at the cell surface. Biol Reprod. 2003 Nov;69(5):1642-50.
MacIndoe JH, Perry PJ, Yates WR, Holman TL, Ellingrod VL, Scott SD. Testosterone suppression of the HPT axis. J Investig Med. 1997 Oct;45(8):441-7.
Pottier J, Cousty C, Heitzman RJ, Reynolds IP. Differences in the biotransformation of a 17 beta-hydroxylated steroid, trenbolone acetate, in rat and cow. Xenobiotica. 1981 Jul;11(7):489-500.
Wilson VS, Lambright C, Ostby J, Gray LE Jr. In vitro and in vivo effects of 17beta-trenbolone: a feedlot effluent contaminant. Toxicol Sci. 2002 Dec;70(2):202-11.
Ashby J, Lefevre PA, Tinwell H, Odum J, Owens W. Testosterone-stimulated weanlings as an alternative to castrated male rats in the Hershberger anti-androgen assay. Regul Toxicol Pharmacol. 2004 Apr;39(2):229-38.
Freyberger A, Hartmann E, Krötlinger F. Evaluation of the rodent Hershberger bioassay using three reference (anti)androgens. Arh Hig Rada Toksikol. 2005 Jun;56(2):131-9.
Freyberger A, Ellinger-Ziegelbauer H, Krötlinger F. Evaluation of the rodent Hershberger bioassay: testing of coded chemicals and supplementary molecular-biological and biochemical investigations. Toxicology. 2007 Sep 24;239(1-2):77-88.
Owens W, Gray LE, Zeiger E, Walker M, Yamasaki K, Ashby J, Jacob E. The OECD program to validate the rat Hershberger bioassay to screen compounds for in vivo androgen and antiandrogen responses: phase 2 dose-response studies. Environ Health Perspect. 2007 May;115(5):671-8.
Moon HJ, Kang TS, Kim TS, Kang IH, Ki HY, Kim SH, Han SY. OECD validation of phase 3 Hershberger assay in Korea using surgically castrated male rats with coded chemicals. J Appl Toxicol. 2009 May;29(4):350-5.
Wilson JD. The role of 5alpha-reduction in steroid hormone physiology. Reprod Fertil Dev. 2001;13(7-8):673-8. Review.
Borst SE, Lee JH, Conover CF. Inhibition of 5alpha-reductase blocks prostate effects of testosterone without blocking anabolic effects. Am J Physiol Endocrinol Metab. 2005 Jan;288(1):E222-7.
Vargas RA, Oliveira LP, Frankenfeld S, Souza DB, Costa WS, Favorito LA, Sampaio FJ. The prostate after administration of anabolic androgenic steroids: a morphometrical study in rats. Int Braz J Urol. 2013 Sep-Oct;39(5):675-82.
Donaldson IA, Hart IC, Heitzman RJ. Growth hormone, insulin, prolactin and total thyroxine in the plasma of sheep implanted with the anabolic steroid trenbolone acetate alone or with oestradiol. Res Vet Sci. 1981 Jan;30(1):7-13.
Quinn MJ Jr, Lavoie ET, Ottinger MA. Reproductive toxicity of trenbolone acetate in embryonically exposed Japanese quail. Chemosphere. 2007 Jan;66(7):1191-6.
Kuhl H, Wiegratz I. Can 19-nortestosterone derivatives be aromatized in the liver of adult humans? Are there clinical implications? Climacteric. 2007 Aug;10(4):344-53. Review.
Attardi BJ, Pham TC, Radler LC, Burgenson J, Hild SA, Reel JR. Dimethandrolone (7alpha,11beta-dimethyl-19-nortestosterone) and 11beta-methyl-19-nortestosterone are not converted to aromatic A-ring products in the presence of recombinant human aromatase. J Steroid Biochem Mol Biol. 2008 Jun;110(3-5):214-22.
Le Guevel R, Pakdel F. Assessment of oestrogenic potency of chemicals used as growth promoter by in-vitro methods. Hum Reprod. 2001 May;16(5):1030-6.
Hemmer MJ, Cripe GM, Hemmer BL, Goodman LR, Salinas KA, Fournie JW, Walker CC. Comparison of estrogen-responsive plasma protein biomarkers and reproductive endpoints in sheepshead minnows exposed to 17beta-trenbolone. Aquat Toxicol. 2008 Jun 23;88(2):128-36.
Neumann F. Pharmacological and endocrinological studies on anabolic agents. Environ Qual Saf Suppl. 1976;(5):253-64. Review.
Hunt DW, Henricks DM, Skelley GC, Grimes LW. Use of trenbolone acetate and estradiol in intact and castrate male cattle: effects on growth, serum hormones, and carcass characteristics. J Anim Sci. 1991 Jun;69(6):2452-62.
Ankley GT, Jensen KM, Makynen EA, Kahl MD, Korte JJ, Hornung MW, Henry TR, Denny JS, Leino RL, Wilson VS, Cardon MC, Hartig PC, Gray LE. Effects of the androgenic growth promoter 17-beta-trenbolone on fecundity and reproductive endocrinology of the fathead minnow. Environ Toxicol Chem. 2003 Jun;22(6):1350-60.
Henricks DM, Edwards RL, Champe KA, Gettys TW, Skelley GC Jr, Gimenez T. Trenbolone, estradiol-17 beta and estrone levels in plasma and tissues and live weight gains of heifers implanted with trenbolone acetate. J Anim Sci. 1982 Nov;55(5):1048-56.
Henricks DM, Brandt RT Jr, Titgemeyer EC, Milton CT. Serum concentrations of trenbolone-17 beta and estradiol-17 beta and performance of heifers treated with trenbolone acetate, melengestrol acetate, or estradiol-17 beta. J Anim Sci. 1997 Oct;75(10):2627-33.
Zhang X, Hecker M, Park JW, Tompsett AR, Jones PD, Newsted J, Au DW, Kong R, Wu RS, Giesy JP. Time-dependent transcriptional profiles of genes of the hypothalamic-pituitary-gonadal axis in medaka (Oryzias latipes) exposed to fadrozole and 17beta-trenbolone. Environ Toxicol Chem. 2008 Dec;27(12):2504-11.
Reiter M, Walf VM, Christians A, Pfaffl MW, Meyer HH. Modification of mRNA expression after treatment with anabolic agents and the usefulness for gene expression-biomarkers. Anal Chim Acta. 2007 Mar 14;586(1-2):73-81.
Seki M, Fujishima S, Nozaka T, Maeda M, Kobayashi K. Comparison of response to 17 beta-estradiol and 17 beta-trenbolone among three small fish species. Environ Toxicol Chem. 2006 Oct;25(10):2742-52.
Jensen KM, Ankley GT. Evaluation of a commercial kit for measuring vitellogenin in the fathead minnow (Pimephales promelas). Ecotoxicol Environ Saf. 2006 Jun;64(2):101-5. Epub 2006 Apr 17.
Holbech H, Kinnberg K, Petersen GI, Jackson P, Hylland K, Norrgren L, Bjerregaard P. Detection of endocrine disrupters: evaluation of a Fish Sexual Development Test (FSDT). Comp Biochem Physiol C Toxicol Pharmacol. 2006 Sep;144(1):57-66.
Orn S, Yamani S, Norrgren L. Comparison of vitellogenin induction, sex ratio, and gonad morphology between zebrafish and Japanese medaka after exposure to 17alpha-ethinylestradiol and 17beta-trenbolone. Arch Environ Contam Toxicol. 2006 Aug;51(2):237-43.
Park JW, Tompsett A, Zhang X, Newsted JL, Jones PD, Au D, Kong R, Wu RS, Giesy JP, Hecker M. Fluorescence in situ hybridization techniques (FISH) to detect changes in CYP19a gene expression of Japanese medaka (Oryzias latipes). Toxicol Appl Pharmacol. 2008 Oct 15;232(2):226-35.
Zhang X, Hecker M, Park JW, Tompsett AR, Newsted J, Nakayama K, Jones PD, Au D, Kong R, Wu RS, Giesy JP. Real-time PCR array to study effects of chemicals on the Hypothalamic-Pituitary-Gonadal axis of the Japanese medaka. Aquat Toxicol. 2008 Jul 7;88(3):173-82.
Holland AM, Roberts MD, Mumford PW, Mobley CB, Kephart WC, Conover CF, Beggs LA, Balaez A, Otzel DM, Yarrow JF, Borst SE, Beck DT. Testosterone inhibits expression of lipogenic genes in visceral fat by an estrogen-dependent mechanism. J Appl Physiol (1985). 2016 Sep 1;121(3):792-805.
H. D. MEYER and M. RAPP Reversible binding of the anabolic steroid trenbolone to steroid receptors Acta Endocrinol 110 S129-S130
Bauer ER, Daxenberger A, Petri T, Sauerwein H, Meyer HH. Characterisation of the affinity of different anabolics and synthetic hormones to the human androgen receptor, human sex hormone binding globulin and to the bovine progestin receptor. APMIS. 2000 Dec;108(12):838-46.
Heitzman RJ, Donaldson IA, Hart IC. Effect of anabolic steroids on plasma thyroid hormones in steers and heifers. Br Vet J. 1980 Mar-Apr;136(2):168-74.
Mader TL, Kreikemeier WM. Effects of growth-promoting agents and season on blood metabolites and body temperature in heifers. J Anim Sci. 2006 Apr;84(4):1030-7.
Hunter RA, Vercoe JE. Reduction of energy requirements of steers fed on low-quality-roughage diets using trenbolone acetate. Br J Nutr. 1987 Nov;58(3):477-83.
Field RA. Effect of castration on meat quality and quantity. J Anim Sci. 1971 May;32(5):849-58.
Leenen R, van der Kooy K, Droop A, Seidell JC, Deurenberg P, Weststrate JA, Hautvast JG. Visceral fat loss measured by magnetic resonance imaging in relation to changes in serum lipid levels of obese men and women. Arterioscler Thromb. 1993 Apr;13(4):487-94.
Leenen R, van der Kooy K, Meyboom S, Seidell JC, Deurenberg P, Weststrate JA. Relative effects of weight loss and dietary fat modification on serum lipid levels in the dietary treatment of obesity. J Lipid Res. 1993 Dec;34(12):2183-91.
Donner DG, Elliott GE, Beck BR, Bulmer AC, Lam AK, Headrick JP, Du Toit EF. Trenbolone Improves Cardiometabolic Risk Factors and Myocardial Tolerance to Ischemia-Reperfusion in Male Rats With Testosterone-Deficient Metabolic Syndrome. Endocrinology. 2016 Jan;157(1):368-81.
Rebuffé-Scrive M, Mårin P, Björntorp P. Effect of testosterone on abdominal adipose tissue in men. Int J Obes. 1991 Nov;15(11):791-5.
Langfort J, Jagsz S, Dobrzyn P, Brzezinska Z, Klapcinska B, Galbo H, Gorski J. Testosterone affects hormone-sensitive lipase (HSL) activity and lipid metabolism in the left ventricle. Biochem Biophys Res Commun. 2010 Sep 3;399(4):670-6.
Goldbourt U, Yaari S, Medalie JH. Isolated low HDL cholesterol as a risk factor for coronary heart disease mortality. A 21-year follow-up of 8000 men. Arterioscler Thromb Vasc Biol. 1997 Jan;17(1):107-13.
Cholestasis induced by Parabolan successfully treated with the molecular adsorbent recirculating system. Anand JS et al. ASAIO 2006. JanFeb;52(1):117-8.
Katsuki A, Sumida Y, Urakawa H, Gabazza EC, Murashima S, Maruyama N, Morioka K, Nakatani K, Yano Y, Adachi Y. Increased visceral fat and serum levels of triglyceride are associated with insulin resistance in Japanese metabolically obese, normal weight subjects with normal glucose tolerance. Diabetes Care. 2003 Aug;26(8):2341-4.
Bruun JM, Verdich C, Toubro S, Astrup A, Richelsen B. Association between measures of insulin sensitivity and circulating levels of interleukin-8, interleukin-6 and tumor necrosis factor-alpha. Effect of weight loss in obese men. Eur J Endocrinol. 2003 May;148(5):535-42.
Kapoor D, Malkin CJ, Channer KS, Jones TH. Androgens, insulin resistance and vascular disease in men. Clin Endocrinol (Oxf). 2005 Sep;63(3):239-50. Review.
Motoshima H, Wu X, Sinha MK, Hardy VE, Rosato EL, Barbot DJ, Rosato FE, Goldstein BJ. Differential regulation of adiponectin secretion from cultured human omental and subcutaneous adipocytes: effects of insulin and rosiglitazone. J Clin Endocrinol Metab. 2002 Dec;87(12):5662-7.
Swarbrick MM, Havel PJ. Physiological, pharmacological, and nutritional regulation of circulating adiponectin concentrations in humans. Metab Syndr Relat Disord. 2008 Jun;6(2):87-102.
Gavrila A, Chan JL, Yiannakouris N, Kontogianni M, Miller LC, Orlova C, Mantzoros CS. Serum adiponectin levels are inversely associated with overall and central fat distribution but are not directly regulated by acute fasting or leptin administration in humans: cross-sectional and interventional studies. J Clin Endocrinol Metab. 2003 Oct;88(10):4823-31.
Yarrow JF, Beggs LA, Conover CF, McCoy SC, Beck DT, Borst SE. Influence of Androgens on Circulating Adiponectin in Male and Female Rodents. Lobaccaro J-MA, ed. PLoS ONE. 2012;7(10):e47315.
Calof OM, Singh AB, Lee ML, Kenny AM, Urban RJ, Tenover JL, Bhasin S. Adverse events associated with testosterone replacement in middle-aged and older men: a meta-analysis of randomized, placebo-controlled trials. J Gerontol A Biol Sci Med Sci. 2005 Nov;60(11):1451-7.
Coviello AD, Kaplan B, Lakshman KM, Chen T, Singh AB, Bhasin S. Effects of graded doses of testosterone on erythropoiesis in healthy young and older men. J Clin Endocrinol Metab. 2008 Mar;93(3):914-9.
Shahani S, Braga-Basaria M, Maggio M, Basaria S. Androgens and erythropoiesis: past and present. J Endocrinol Invest. 2009 Sep;32(8):704-16.
Sakhri S, Gooren LJ. Safety aspects of androgen treatment with 5alpha-dihydrotestosterone. Andrologia. 2007 Dec;39(6):216-22. Review.
Rochira V, Zirilli L, Madeo B, Maffei L, Carani C. Testosterone action on erythropoiesis does not require its aromatization to estrogen: Insights from the testosterone and estrogen treatment of two aromatase-deficient men. J Steroid Biochem Mol Biol. 2009 Feb;113(3-5):189-94.
Amory JK, Watts NB, Easley KA, Sutton PR, Anawalt BD, Matsumoto AM, Bremner WJ, Tenover JL. Exogenous testosterone or testosterone with finasteride increases bone mineral density in older men with low serum testosterone. J Clin Endocrinol Metab. 2004 Feb;89(2):503-10.
McCoy SC, Yarrow JF, Conover CF, Borsa PA, Tillman MD, Conrad BP, Pingel JE, Wronski TJ, Johnson SE, Kristinsson HG, Ye F, Borst SE. 17β-Hydroxyestra-4,9,11-trien-3-one (Trenbolone) preserves bone mineral density in skeletally mature orchiectomized rats without prostate enlargement. Bone. 2012 Oct;51(4):667-73.
Alcuni di voi potrebbero essersi già imbattuti nell’articolo di Chest Rockwell intitolato “The Science of Trenbolone” il quale si trova da qualche tempo su alcuni siti dedicati al BodyBuilding. L’intento dell’autore è stato quello di creare un articolo approfondito sul Trenbolone. Tale articolo è stato progettato per essere una guida di riferimento rapida volta a rispondere a molte domande riguardanti questo AAS. La mole di informazioni presenti in esso ha reso questo articolo un punto di riferimento per comprendere le caratteristiche e le potenziali applicazioni del Trenbolone. Il mio intento è quello di realizzare una “guida” sul Trenbolone, sul modello di quella realizzata da Chest Rockwell, in grado di fornire una serie di importanti informazioni riguardanti questo AAS e utilizzabili da atleti e preparatori.
II. Introduzione
Il Trenbolone è senza dubbio l’AAS con una reputazione quasi mitica all’interno del mondo del BodyBuilding. Poiché i dati sugli esseri umani sono molto limitati, spesso dobbiamo fare affidamento sugli aneddoti nel tentativo di formulare ipotesi su di esso. Come si può leggere in quasi tutti i forum dedicati al BodyBuilding, le esperienze con il Trenbolone variano ampiamente – con alcuni utenti che adorano assolutamente il composto mentre altri ne consigliano estrema cautela nell’uso o dicono agli altri utenti di evitarlo a tutti i costi. Nonostante questa grande divergenza di opinioni, non si può contestare la sua popolarità poiché numerosi studi hanno dimostrato negli anni che tale molecola è uno dei composti anabolizzanti più frequentemente usati, con una percentuale compresa tra il 20 e il 25% dei culturisti supplementari chimicamente che hanno riferito di averlo usato nell’arco degli ultimi dodici mesi. (1)(2)(3)
Il mio obiettivo con questo articolo sarà quello di utilizzare quante più informazioni disponibili per cercare di formulare alcune solide conclusioni riguardo al funzionamento e al potenziale utilizzo del composto. Allo stesso tempo spero di contribuire a dissipare alcuni miti che vengono ancora propagandati troppo spesso.
Come precedentemente accennato, esistono solo un paio di studi sull’uomo di cui sono a conoscenza, quindi la maggior parte del materiale citato proverrà da studi svolti su animali o in vitro. La domanda che dovrebbe essere posta è la seguente: possiamo prendere questi dati e applicarli concretamente sui BodyBuilder? Personalmente, ritengo che ci sono dei dati molto concreti e universalmente applicabili sugli esseri umani e poi ce ne sono altri che potrebbero richiedere un disclaimer. Cercherò di fare del mio meglio per indicarli nel corso di questo articolo.
III. Nozioni di base sul Trenbolone
Trenbolone
Il Trenbolone è considerabile quale Modulatore Selettivo del Recettore degli Androgeni (SARM), non progettato per l’uso umano (4), anche se venne commercializzato come farmaco da prescrizione per uso umano dalla Negma Laboratoires in Francia sotto il nome di Parabolan (Trenbolone Hexahydrobenzylcarbonato). Nonostante la sua primaria designazione, questo AAS continua ad essere pesantemente utilizzato dai Bodybuilder per promuovere la crescita muscolare, la riduzione del grasso e, di conseguenza, migliorare la composizione corporea.(5)(6) Quando sentiamo parlare di SARM colleghiamo tale termine ad una classe di agenti anabolizzanti non steroidei (vedi Ostarina, Andarina, LGD4033, ecc…). Però, tale definizione “particolaristica” non è del tutto corretta. Infatti, tutti gli AAS aventi un indice terapeutico (dato dalla Anabolico:Androgeno ratio) superiore a “1” sono considerabili quali SARM (l’indice terapeutico del Trenbolone è di circa 3,4). Si sa perfettamente che i SARM sono analoghi modificati degli ormoni sessuali maschili che normalmente esibiscono attività anabolica favorevole mentre, contemporaneamente, presentano una attività androgenica da moderata a minima in vivo rispetto agli androgeni endogeni.(7)(8)(9) Sono composti in fase di sviluppo da parte di molte aziende farmaceutiche nel tentativo di creare mezzi alternativi per il trattamento di condizioni come l’ipogonadismo e gli stati di deperimento muscolare e osseo. In sostanza, l’obiettivo è quello di ricreare gli aspetti positivi dati da dosi sovrafisiologiche di testosterone eliminando al contempo il rischio di eventi avversi che tendono a verificarsi quando si utilizzano tali dosaggi.(10) Questo obbiettivo lo si ritrova, appunto, concretizzato in tutti gli AAS che presentano modifiche dello scheletro carbossilico tali da permettere tale effetto.
La maggior parte dei SARM steroidei hanno come base di partenza una molecola di Testosterone. La struttura chimica della molecola di Testosterone viene quindi tradizionalmente modificata in uno dei seguenti tre modi (11)(12):
Esterificazione del gruppo 17β-idrossile che aumenta l’idrofobicità o la probabilità che la molecola possa essere respinta da una massa d’acqua;
Alchilazione in posizione 7α la quale riduce l’affinità di legame con l’enzima 5α-reduttasi;
Modifica strategica dei legami di carbonio in C1, C2, C9, C11 o C19 per ottenere una vasta gamma di effetti terapeutici.
Il Trenbolone è un C19-norsteroide (19-nor), derivato dal Nandrolone (Nortestosterone). La rimozione del gruppo metilico nella posizione C19 dello scheletro carbossilico steroideo riduce significativamente la suscettibilità dei 19-norsteroidi all’azione dell’enzima aromatasi e a quella dell’enzima 5α-reduttasi.(4) Entreremo più nel dettaglio dei meccanismi sopracitati in seguito ma, per ora, è necessario che si comprenda semplicemente come le sottili modifiche allo scheletro carbossilico della molecola di Testosterone possano tradursi direttamente in cambiamenti significativi del comportamento della nuova molecola derivata. Alcuni di questi cambiamenti possono includere l’affinità di legame del composto per il recettore degli androgeni e la sua affinità di legame con numerosi enzimi in grado di convertirlo in altri steroidi.(13)
Il Trenbolone differisce dal suo precursore (Nandrolone) per:
1- Il doppio legame inserito in C9– C10, che inibisce totalmente l’aromatizzazione e aumenta la resistenza al passaggio epatico;
2- l’insaturazione in C11-C12 che aumenta l’affinità per il recettore androgeno, rendendo il Trenbolone uno degli anabolizzante con la più forte affinità AR. (14)
Il valore Anabolico/Androgeno del Trenbolone rispetto al Testosterone (100/100) è pari a 625:185.
Quindi, il Trenbolone ha proprietà SARM-simili in quanto presenta una affinità significativamente minore ai percorsi metabolici ai quali è soggetto il Testosterone. Ma di questo ne parleremo in seguito.
IV. Storia del Trenbolone
Metribolone
L’enorme potenziale anabolizzante del Trenbolone, così come dei suoi analoghi, è stato riportato fin dagli anni ’60. Come molti di voi già sapranno, venne sintetizzata anche una versione orale denominata Metribolone (conosciuta anche come Methyltrienolone o Methyltrenbolone), tuttavia non è mai stata commercializzata come agente anabolizzante a causa della sua tossicità epatica estrema – causando colestasi intraepatica a quantità somministrate per via orale pari a 1 mg/giorno.(15)
A parte la parentesi francese del Parabolan (Trenbolone Hexahydrobenzylcarbonato), commercializzato dalla Negma fino al 1997, il Trenbolone non è mai stato approvato per l’uso umano e il suo utilizzo è stato (ed è) principalmente quello di agente per la promozione della crescita nel bestiame.(16)(17) Per tale scopo viene usato sia singolarmente che in combinazione con Estradiolo (E2).(18) L’uso di impianti per il bestiame contenenti la combinazione di Trenbolone ed Estradiolo è stata approvata dalla FDA nel 1992 (19), e ora circa il 90% dei bovini da carne negli Stati Uniti viene trattato con un mix di estrogeni, androgeni e/o progestinici volto a promuoverne la crescita.(20) Gli impianti per il bestiame sono un grande business con fino a 20 milioni di bovini all’anno impiantati con Trenbolone e un reddito annuo probabilmente superiore a un miliardo di dollari.(21)
Trenbolone Acetato
Nonostante l’approvazione da parte della FDA, sussistono ancora problemi di sicurezza poiché il Trenbolone Acetato (TBA), estere utilizzato per trattare il bestiame, ei suoi metaboliti sono stati identificati come potenziali sostanze chimiche dannose per il sistema endocrino (EDC). Gli EDC sono molecole esogene che possono imitare o inibire l’azione dei recettori ormonali come i recettori degli estrogeni, degli androgeni e degli ormoni tiroidei. Questi EDC possono anche alterare la sintesi, l’azione, il metabolismo e la secrezione di ormoni endogeni la quale può portare a problemi gravi come obesità, diabete e persino il cancro. (22)(23)
Meccanismi d’azione degli EDC. A: Interazione diretta del EDC con un recettore nucleare dell’ormone che porta alla stimolazione (agonismo) o inibizione (antagonismo) della sua attività trascrizionale. B: Stimolazione o inibizione della biosintesi degli ormoni endogeni. C: Stimolazione o inibizione della degradazione degli ormoni endogeni. D: Stimolazione o inibizione della proteina legante gli ormoni endogeni che porta ad un aumento o diminuzione della disponibilità degli ormoni circolanti.
A causa della potenziale gravità degli EDC, negli ultimi due decenni si è registrata una maggiore attenzione internazionale sull’esposizione ambientale e sugli effetti degli EDC nell’uomo e nella fauna selvatica.(24)(25) Come precedentemente accennato, il TBA ei suoi metaboliti sono stati identificati come EDC attraverso numerosi studi, possono essere diffusi in ambienti agricoli e sono associati a tossicità riproduttiva. (26)(27)(28) Il problema è che è necessaria soltanto un’esposizione a concentrazioni molto basse per causare potenziali problemi, come è stato dimostrato negli animali come i pesci, i quali hanno mostrato disordini nel comportamento sessuale e una diminuzione della fertilità. (29)
Sarà anche importante essere in grado di distinguere i vari tipi di impianti contenenti TBA, dal momento che molti degli studi che esamineremo in seguito utilizzeranno tipi diversi sugli animali presi in esame. Quello che segue è un elenco dei tipi di impianti comuni usati negli Stati Uniti, con specificazione delle loro concentrazioni ormonali:
Revalor-XS (200mg TBA / 40mg E2)
Revalor-200 (200mg TBA / 20mg E2)
Revalor-H (140mg TBA / 14mg E2)
Revalor-S (120mg TBA / 24mg E2)
Revalor-IS (80mg TBA / 16mg E2)
Revalor-IH (80mg TBA / 8mg E2)
Revalor-G (40mg TBA / 8mg E2)
Synovex PLUS (200mg TBA / 28mg E2)
Synovex-C (100mg Progesterone / 10mg E2)
Synovex-ONE Grass (150mg TBA / 15mg E2)
Synovex-S (200mg Progesterone / 20mg E2)
Synovex-H (200mg Testosterone / 20mg E2)
V. Metabolismo e fisiologia
Precedentemente ho fatto breve menzione ai metaboliti del TBA, quindi, ora, per completezza è giusto entrare nei dettagli. E’ corretto rammentare che la maggior parte delle informazioni riguardanti il metabolismo del Trenbolone in vivo provengono da osservazioni su bestiame e roditori (30)(31)(32). È fondamentale anche comprendere che vi sono differenze marcate nella quantità di vari metaboliti osservati nei modelli di ratti e mucche, i due mammiferi più intensamente studiati. (33) Torneremo a trattare questo argomento dopo aver esaminato alcuni dei principi fondamentali.
Trendione
Il nome chimico del TBA è 17ß-idrossi-estra-4,9,11-trien-3-one-17-acetato, talvolta abbreviato in 17β-TBOH-acetato. Dopo un’iniezione intramuscolare, viene rapidamente idrolizzato nel metabolita biologicamente attivo noto come 17β-idrossi-estra-4,9,11-trien-3-one o 17β-TBOH.(34) Da lì viene ulteriormente convertita in metaboliti inclusi i glucuronidi (per esempio Trendione / TBO) e altri cinque metaboliti idrossilati polari.(35) La serie di questi processi metabolici può essere riassunta come segue:
Il 17β-TBOH ha una maggiore affinità per gli AR rispetto a qualsiasi altro dei suoi metaboliti primari suggerendo che la biotrasformazione del Trenbolone riduce l’attività biologica dello steroide.(26)(27)(35) Per mettere questo in prospettiva, in uno studio l’elevata affinità del 17β-TBOH nei confronti del recettore degli androgeni umani e del recettore del progesterone bovino è stata ridotta dopo che la molecola è stata metabolizzata in 17α-TBOH e TBO a meno di 1/24 della dose originale del composto.(36) Questo comportamento è in netto contrasto con quello osservato nel Testosterone la cui conversione in DHT e in estrogeni porta a composti più potenti in relazione all’affinità del legame con il recettore bersaglio.(37)(38) Tuttavia, il comportamento del TBA è simile per natura ad altri composti 19-norsteroidei (come il Nandrolone), la cui affinità AR diminuisce una volta 5α-ridotta.(39)
Come accennato in precedenza, vi è una certa variazione nel metabolismo del 17-TBOH tra i mammiferi poiché i metaboliti primari sono il 17ß-idrossi-estra-4,9,11-trien-3-one e l’Estra-4,9,11-triene- 3,17-dione con, a loro volta, metaboliti 16α e 16ß-idrossilati nel ratto. Nella mucca questi metaboliti sono trascurabili e il 17α-TBOH è il prodotto principale insieme a piccole quantità di 16α e 16ß-idrossi-17α-TBOH (30)(31). Segue un grafico dettagliato che confronta le differenze tra gli animali:
Confronto dei metaboliti biliari del 17beta-trenbolone acetato nel ratto e nella mucca.
Fortunatamente per noi, esiste uno studio su esseri umani che ci può aiutare a chiarire come l’uomo metabolizzi il Trenbolone – almeno dopo che questo è stato ingerito per via orale. (35) Lo studio è stato progettato per indagare sugli effetti dell’ingestione di cibo contaminato e quindi il team di ricerca ha iniettato il 17β-TBOH in un pezzo di hamburger fritto da 5g, con una dose di 0,04 mg/kg di peso corporeo. Dopo un singolo consumo, il 63% della dose somministrata è stata escreta tramite l’urina entro 72 ore; a 24 ore il 50% della dose somministrata è stata osservata nei campioni di urina.
I risultati hanno anche rivelato che, negli esseri umani, il 17β-TBOH ingerito è principalmente escreto intatto come 17β-TBOH, come Epitrenbolone (17α-TBOH) o come Trendione (TBO) – con la maggior parte in forma 17α-TBOH. In questo senso, la biotrasformazione del 17ß-TBOH negli esseri umani assomiglia più da vicino a quella delle mucche che a quella dei roditori. Inoltre, nell’urina umana sono stati rilevati numerosi metaboliti polari di 17β-TBOH, sebbene in concentrazione molto inferiore rispetto a quei metaboliti precedentemente menzionati. (40)
Il 17β-TBOH presenta una bassa biodisponibilità orale dal momento che non presenta una metilazione in posizione 17α. I risultati di due saggi di Hershberger dimostrano che il Trenbolone è circa 80-100 volte meno efficace assunto per via orale rispetto a somministrazione tramite iniezione (comportamento legato appunto alla biodisponibilità).(26) Nonostante questo, il TBA e, ovviamente, 17β-TBOH hanno ancora dimostrato di alterare il sistema riproduttivo di umani, maiali, topi, ratti e altre specie di mammiferi a livelli di dosaggio relativamente bassi quando somministrati per via orale.
Nella prossima parte di questa serie di articoli, esploreremo l’impatto del Trenbolone su vari percorsi anabolici e marker della salute metabolica. Vedremo anche come il Trenbolone influenzi la produzione endogena di ormoni e inizieremo ad analizzare in maniera approfondita i suoi effetti su anabolismo e ipertrofia.
Stay tuned!
Gabriel Bellizzi
Riferimenti:
The Science of Trenbolone – By Chest Rockwell
Perry PJ, Lund BC, Deninger MJ, Kutscher EC, Schneider J. Anabolic steroid use in weightlifters and bodybuilders: an internet survey of drug utilization. Clin J Sport Med. 2005 Sep;15(5):326-30.
Parkinson AB, Evans NA. Anabolic androgenic steroids: a survey of 500 users. Med Sci Sports Exerc. 2006 Apr;38(4):644-51.
Ip EJ, Barnett MJ, Tenerowicz MJ, Perry PJ. The Anabolic 500 survey: characteristics of male users versus nonusers of anabolic-androgenic steroids for strength training. Pharmacotherapy. 2011 Aug;31(8):757-66.
Donner DG, Beck BR, Bulmer AC, Lam AK, Du Toit EF. Improvements in body composition, cardiometabolic risk factors and insulin sensitivity with trenbolone in normogonadic rats. Steroids. 2015 Feb;94:60-9.
Daniels JM, van Westerloo DJ, de Hon OM, Frissen PH. [Rhabdomyolysis in a bodybuilder using steroids]. Ned Tijdschr Geneeskd. 2006 May 13;150(19):1077-80. Dutch.
Geraci MJ, Cole M, Davis P. New onset diabetes associated with bovine growth hormone and testosterone abuse in a young body builder. Hum Exp Toxicol. 2011 Dec;30(12):2007-12.
Omwancha J, Brown TR. Selective androgen receptor modulators: in pursuit of tissue-selective androgens. Curr Opin Investig Drugs. 2006 Oct;7(10):873-81. Review.
Gao W, Dalton JT. Expanding the therapeutic use of androgens via selective androgen receptor modulators (SARMs). Drug Discov Today. 2007 Mar;12(5-6):241-8. Review.
Thevis M, Schänzer W. Synthetic anabolic agents: steroids and nonsteroidal selective androgen receptor modulators. Handb Exp Pharmacol. 2010;(195):99-126.
Calof OM, Singh AB, Lee ML, Kenny AM, Urban RJ, Tenover JL, Bhasin S. Adverse events associated with testosterone replacement in middle-aged and older men: a meta-analysis of randomized, placebo-controlled trials. J Gerontol A Biol Sci Med Sci. 2005 Nov;60(11):1451-7.
Kicman AT. Pharmacology of anabolic steroids. British Journal of Pharmacology. 2008;154(3):502-521.
Haendler B, Cleve A. Recent developments in antiandrogens and selective androgen receptor modulators. Mol Cell Endocrinol. 2012 Apr 16;352(1-2):79-91.
Fragkaki AG, Angelis YS, Koupparis M, Tsantili-Kakoulidou A, Kokotos G, Georgakopoulos C. Structural characteristics of anabolic androgenic steroids contributing to binding to the androgen receptor and to their anabolic and androgenic activities. Applied modifications in the steroidal structure. Steroids. 2009 Feb;74(2):172-97.
Krüskemper HL, Noell G. Liver toxicity of a new anabolic agent: methyltrienolone (17-alpha-methyl-4,9,11-estratriene-17 beta-ol-3-one). Steroids. 1966 Jul;8(1):13-24.
Hunt DW, Henricks DM, Skelley GC, Grimes LW. Use of trenbolone acetate and estradiol in intact and castrate male cattle: effects on growth, serum hormones, and carcass characteristics. J Anim Sci. 1991 Jun;69(6):2452-62.
Chung KY, Johnson BJ. Application of cellular mechanisms to growth and development of food producing animals. J Anim Sci. 2008 Apr;86(14 Suppl):E226-35.Epub 2007 Oct 26. Review.
Metzler M, Pfeiffer E. Genotoxic potential of xenobiotic growth promoters and their metabolites. APMIS. 2001 Feb;109(2):89-95. Review.
Chung KY, Baxa TJ, Parr SL, Luqué LD, Johnson BJ. Administration of estradiol, trenbolone acetate, and trenbolone acetate/estradiol implants alters adipogenic and myogenic gene expression in bovine skeletal muscle. J Anim Sci. 2012 May;90(5):1421-7.
Balter M. Scientific cross-claims fly in continuing beef war. Science. 1999 May 28;284(5419):1453, 1455.
Lawrence JD, Ibarburu MA. Proceedings of the NCCC-134 Conference on Applied Commodity Price Analysis, Forecasting, and Market Risk Management; 16 and 17 April 2007; Chicago.
Diamanti-Kandarakis E, Bourguignon JP, Giudice LC, Hauser R, Prins GS, Soto AM, Zoeller RT, Gore AC. Endocrine-disrupting chemicals: an Endocrine Society scientific statement. Endocr Rev. 2009 Jun;30(4):293-342. Review.
Rachoń D. Endocrine disrupting chemicals (EDCs) and female cancer: Informing the patients. Rev Endocr Metab Disord. 2015 Dec;16(4):359-64.
Birnbaum LS. State of the science of endocrine disruptors. Environ Health Perspect. 2013 Apr;121(4):A107.
Baynes RE, Dedonder K, Kissell L, Mzyk D, Marmulak T, Smith G, Tell L, Gehring R, Davis J, Riviere JE. Health concerns and management of select veterinary drug residues. Food Chem Toxicol. 2016 Feb;88:112-22.
Wilson VS, Lambright C, Ostby J, Gray LE Jr. In vitro and in vivo effects of 17beta-trenbolone: a feedlot effluent contaminant. Toxicol Sci. 2002 Dec;70(2):202-11.
Durhan EJ, Lambright CS, Makynen EA, Lazorchak J, Hartig PC, Wilson VS, Gray LE, Ankley GT. Identification of metabolites of trenbolone acetate in androgenic runoff from a beef feedlot. Environ Health Perspect. 2006 Apr;114 Suppl 1:65-8.
Gall HE, Sassman SA, Lee LS, Jafvert CT. Hormone discharges from a midwest tile-drained agroecosystem receiving animal wastes. Environ Sci Technol. 2011 Oct 15;45(20):8755-64.
Jensen KM, Makynen EA, Kahl MD, Ankley GT. Effects of the feedlot contaminant 17alpha-trenbolone on reproductive endocrinology of the fathead minnow. Environ Sci Technol. 2006 May 1;40(9):3112-7.
Pottier J, Busigny M, Grandadam JA. Plasma kinetics, excretion in mild and tissue levels in the cow following implantation of trenbolone acetate. J Anim Sci. 1975 Sep;41(3):962-8.
Pottier J, Cousty C, Heitzman RJ, Reynolds IP. Differences in the biotransformation of a 17 beta-hydroxylated steroid, trenbolone acetate, in rat and cow. Xenobiotica. 1981 Jul;11(7):489-500.
Evrard P, Maghuin-Rogister G, Rico AG. Fate and residues of trenbolone acetate in edible tissues from sheep amd calves implanted with tritium-labeled trenbolone acetate. J Anim Sci. 1989 Jun;67(6):1489-96
Metzler M. Metabolism of some anabolic agents: toxicological and analytical aspects. J Chromatogr. 1989 Apr 7;489(1):11-21.
Dorts J, Richter CA, Wright-Osment MK, Ellersieck MR, Carter BJ, Tillitt DE. The genomic transcriptional response of female fathead minnows (Pimephales promelas) to an acute exposure to the androgen, 17beta-trenbolone. Aquat Toxicol.2009 Jan 18;91(1):44-53.
Spranger B, Metzler M. Disposition of 17 beta-trenbolone in humans. J Chromatogr. 1991 Apr 5;564(2):485-92.
Bauer ER, Daxenberger A, Petri T, Sauerwein H, Meyer HH. Characterisation of the affinity of different anabolics and synthetic hormones to the human androgen receptor, human sex hormone binding globulin and to the bovine progestin receptor. APMIS. 2000 Dec;108(12):838-46.
Wilson JD. The role of 5alpha-reduction in steroid hormone physiology. Reprod Fertil Dev. 2001;13(7-8):673-8. Review.
Santen RJ, Brodie H, Simpson ER, Siiteri PK, Brodie A. History of aromatase: saga of an important biological mediator and therapeutic target. Endocr Rev. 2009 Jun;30(4):343-75.
Sundaram K, Kumar N, Monder C, Bardin CW. Different patterns of metabolism determine the relative anabolic activity of 19-norandrogens. J Steroid Biochem Mol Biol. 1995 Jun;53(1-6):253-7.
Yarrow JF, McCoy SC, Borst SE. Tissue selectivity and potential clinical applications of trenbolone (17beta-hydroxyestra-4,9,11-trien-3-one): A potent anabolic steroid with reduced androgenic and estrogenic activity. Steroids. 2010 Jun;75(6):377-89.
Una delle singolarità che maggiormente caratterizzano l’Ormone della Crescita (GH) è rappresentata dall’aumento della sua secrezione correlata al sonno, che si verifica vicino all’inizio del così detto sonno a onde lente (fase tre) [1-2]. Questa secrezione notturna rappresenta quasi il 70% dell’intera sintesi giornaliera di GH secreta negli individui di sesso maschile. L’impulso notturno è di natura sessualmente dimorfica e significativamente meno pronunciato nelle donne [3-4].
Poiché questo è un evento secretorio endogeno così sostanziale, molte persone, sia atleti che preparatori, basandosi anche su speculazioni frutto di teorie più o meno plausibili esposte su alcuni libri del settore (errore in cui caddi anche io), nel tentativo di massimizzare l’efficacia dell’utilizzo di GH esogeno spesso si convincono del fatto che un particolare protocollo di temporizzazione possa essere usato per preservare questa secrezione notturna. Ma, questa ipotesi ha un reale riscontro nella pratica?
Come in tutti gli altri sistemi endocrini, la secrezione di GH è regolata da molteplici cicli di feedback negativo che sono tradizionalmente suddivisi in tre categorie: feedback ultracorto, corto, e lungo [5]. Il ciclo di feedback ultracorto è rappresentato dall’azione del GHRH (Growth hormone releasing hormone; Ormone di Rilascio della Somatotropina o Somatorelina) – che inibisce acutamente la secrezione di GH – e della Somatostatina (SRIF) – che sopprime il rilascio di GH [6-8].
Il ciclo di feedback corto è rappresentato dall’azione dei livelli sierici di GH elevati che agiscono direttamente sull’ipofisi per inibire l’ulteriore il rilascio di GH attraverso la soppressione del GHRH. Infatti, gli elevati livelli sierici di GH possono anche indurre l’inibizione del SRIF e del GHRH all’interno dell’ipotalamo. Sembra però che tale processo richieda un certo lasso di tempo per manifestarsi, poiché alcune prove hanno dimostrato che questo particolare circuito di feedback può richiedere da due a quattro ore per impostarsi [9-14]. Questo ciclo di feedback è anche quello sul quale prestiamo maggiore interesse.
Per completezza d’informazioni, il ciclo di feedback lungo si verifica quando elevati livelli sierici di IGF-1 agiscono sull’ipofisi riducendo consequenzialmente la secrezione di GH [15-16].
Fortunatamente, esistono due studi sull’uomo che possono fare ulteriore chiarezza in merito al quesito posto in questa sede. Il primo studio ha preso in esame soggetti sani di sesso maschile e femminile sottoponendoli ha somministrazioni di GH esogeno pari a 2UI/die divise in due iniezioni: una prima iniezione AM (08:00) e una seconda iniezione PM (17:00) [17]. Nonostante l’ultima somministrazione di GH sia avvenuta più di sei ore prima del sonno, il gruppo di ricercatori ha notato una completa soppressione delle secrezioni endogene notturna dell’ormone nonostante i livelli elevati di GH fossero stati eliminati da tempo dal sistema. Ciò sembrerebbe implicare l’azione di un meccanismo di feedback oltre a quello legato agli effetti diretti del GH.
Secrezione dell’Ormone della Crescita correlata al sonno dopo somministrazione di GH esogeno o soluzione salina – ore dopo l’inizio del sonno.
Il secondo studio [18] ha preso in esame soggetti sani di sesso maschile sottoponendoli alla somministrazione di una singola dose sottocutanea di 20kDa GH alle 21:00. La dose era variabile ed i soggetti sono stati divisi in quattro gruppi di dosaggio: 0.01, 0.025, 0.05 o 0.1mg/kg. Con uno scarto di alcune ore, tutti i gruppi di dosaggio hanno mostrato una completa soppressione delle secrezioni endogena di GH per un lasso di tempo pari a 16-24 ore. Infatti, eliminando due valori anomali, i soggetti rimanenti hanno mostrato una completa soppressione per tutte le 24 ore del monitoraggio. È stato interessante notare che uno dei valori anomali era nel gruppo ad alto dosaggio.
Secrezione dell’Ormone della Crescita correlata al sonno dopo somministrazione di GH esogeno o soluzione salina – tempo dopo la somministrazione.
Basandomi su quanto esposto, penso che il vero punto da tenere presente sia che gli utilizzatori di GH esogeno non dovrebbero concentrarsi sulla secrezione endogena di GH nelle ore notturne nel vano tentativo di preservare tale impulso. Come si è potuto vedere, sembrerebbe che qualsiasi dose significativa di GH esogeno aumenti la probabilità che tale impulso venga soppresso a prescindere. Infatti, a meno che non si utilizzi il GH esclusivamente per scopi lipolitici, l’impulso serale probabilmente non fornirà comunque un effetto additivo semplicemente a causa di come esso è strutturato.
Gabriel Bellizzi
Riferimenti:
Exogenous Growth Hormone and Nocturnal Secretions – By Chest Rockwell
Holl RW, Hartman ML, Veldhuis JD, Taylor WM, Thorner MO. Thirty-second sampling of plasma growth hormone in man: correlation with sleep stages. J Clin Endocrinol Metab. 1991 Apr;72(4):854-61
Obal F Jr, Krueger JM. GHRH and sleep. Sleep Med Rev. 2004 Oct;8(5):367-77. Review Van Cauter E, Plat L, Copinschi G. Interrelations between sleep and the somatotropic axis. Sleep. 1998 Sep 15;21(6):553-66. Review.
Jaffe CA, Ocampo-Lim B, Guo W, Krueger K, Sugahara I, DeMott-Friberg R, Bermann M, Barkan AL. Regulatory mechanisms of growth hormone secretion are sexually dimorphic. J Clin Invest. 1998 Jul 1;102(1):153-64.
Farhy LS, Straume M, Johnson ML, Kovatchev B, Veldhuis JD. Unequal autonegative feedback by GH models the sexual dimorphism in GH secretory dynamics. Am J Physiol Regul Integr Comp Physiol. 2002 Mar;282(3):R753-64.
Lumpkin MD, McDonald JK. Blockade of growth hormone-releasing factor (GRF) activity in the pituitary and hypothalamus of the conscious rat with a peptidic GRF antagonist. Endocrinology. 1989 Mar;124(3):1522-31.
Lumpkin MD, Mulroney SE, Haramati A. Inhibition of pulsatile growth hormone (GH) secretion and somatic growth in immature rats with a synthetic GH-releasing factor antagonist. Endocrinology. 1989 Mar;124(3):1154-9.
Berelowitz M, Firestone SL, Frohman LA. Effects of growth hormone excess and deficiency on hypothalamic somatostatin content and release and on tissue somatostatin distribution. Endocrinology. 1981 Sep;109(3):714-9.
Chomczynski P, Downs TR, Frohman LA. Feedback regulation of growth hormone (GH)-releasing hormone gene expression by GH in rat hypothalamus. Mol Endocrinol. 1988 Mar;2(3):236-41
Frohman MA, Downs TR, Chomczynski P, Frohman LA. Cloning and characterization of mouse growth hormone-releasing hormone (GRH) complementary DNA: increased GRH messenger RNA levels in the growth hormone-deficient lit/lit mouse. Mol Endocrinol. 1989 Oct;3(10):1529-36.
Rosenthal SM, Kaplan SL, Grumbach MM. Short term continuous intravenous infusion of growth hormone (GH) inhibits GH-releasing hormone-induced GH secretion: a time-dependent effect. J Clin Endocrinol Metab. 1989 Jun;68(6):1101-5.
Yamauchi N, Shibasaki T, Ling N, Demura H. In vitro release of growth hormone-releasing factor (GRF) from the hypothalamus: somatostatin inhibits GRF release. Regul Pept. 1991 Mar 26;33(1):71-8.
Pontiroli AE, Lanzi R, Monti LD, Sandoli E, Pozza G. Growth hormone (GH) autofeedback on GH response to GH-releasing hormone. Role of free fatty acids and somatostatin. J Clin Endocrinol Metab. 1991 Feb;72(2):492-5.
Hashimoto Y, Kamioka T, Hosaka M, Mabuchi K, Mizuchi A, Shimazaki Y, Tsunoo M, Tanaka T. Exogenous 20K growth hormone (GH) suppresses endogenous 22K GH secretion in normal men. J Clin Endocrinol Metab. 2000 Feb;85(2):601-6.