Continua la disamina dei principali PEDs utilizzati e del confine che delimita l’uso dall’abuso. Nel primo articolo della serie abbiamo analizzato l’Oxymetholone e come questa molecola sia soggetta a facile abuso da un numero considerevole di atleti di diverse categorie. In questo secondo articolo analizzeremo il Trenbolone, una molecola “must” nel BodyBuilding.
Caratteristiche della molecola di Trenbolone:
Il Trenbolone, noto anche come 19-nor-δ9,11-testosterone o come estra-4,9,11-trien-17β-ol-3-one, è un 19 nor- steroide derivato dal Nandrolone, e condivide con il suo precursore la modifica molecolare in C-19. Nello specifico, il Trenbolone è Nandrolone con due doppi legami aggiuntivi nel nucleo steroideo. Le differenze strutturali tra la molecola di Trenbolone e quella di Nandrolone, quindi, comprendono il doppio legame presente in C9– C10, che inibisce totalmente l’aromatizzazione e aumenta la resistenza al passaggio epatico, e quello in C11-C12 che aumenta l’affinità per il recettore androgeno, rendendo il Trenbolone uno degli anabolizzante con la più forte affinità AR.[1]
Differenze nella struttura molecolare tra Nandrolone e Trenbolone.
Gli esteri del Trenbolone, che presentano un estere in posizione C17β, includono il Trenbolone Acetato, il Trenbolone Enantato, il Trenbolone Hexahydrobenzylcarbonate e il Trenbolone Undecanoato.[2][3][4][5]
Il Trenbolone ha sia effetti anabolizzanti che androgeni.[4] Una volta metabolizzato, il Trenbolone causa un aumento dell’assorbimento di ioni ammonio da parte del tessuto muscolare, effetto che porta ad un aumento del tasso di sintesi proteica. Può anche avere effetti secondari di stimolo dell’appetito (effetto oressizzante) e di diminuzione del tasso catabolico, con quest’ultima caratteristica condivisa in diversa misura con tutti gli AAS.[6] Almeno uno studio svolto sui ratti ha dimostrato che il Trenbolone può indurre l’espressione genica del recettore degli androgeni (AR) almeno allo stesso livello del Diidrotestosterone (DHT). Questa caratteristica sta a indicare che il Trenbolone può causare un aumento delle caratteristiche sessuali secondarie maschili senza la necessità che esso converta in un androgeno più potente.[7]
Gli studi sul metabolismo del Trenbolone sono quanto meno eterogenei, con alcuni studi che mostrano che è metabolizzato dall’enzima Aromatasi o 5α-reduttasi in composti estrogenici e androgenici, nonostante le sue caratteristiche escludano questa potenzialità.[8][9]
Il Trenbolone si lega con alta affinità anche al recettore del Progesterone,[4][10][11][12] e al recettore dei glucocorticoidi.[11]
Per prolungare la sua emivita, il Trenbolone viene somministrato sotto forma di molecola coniugata ad un estere come i precedentemente citati Trenbolone Acetato, Trenbolone Enantato o Trenbolone Hexahydrobenzylcarbonate.[2][3][13][4] Le Lipasi plasmatiche poi scindono il gruppo estere nel flusso sanguigno rendendo libero, e quindi attivo, il Trenbolone.
Dosaggio di Trenbolone per il bestiame Vs dosaggio per esseri umani – :
Il Trenbolone Acetato è stato ed è utilizzato in medicina veterinaria nel bestiame per aumentarne la crescita muscolare e l’appetito, mentre Il Trenbolone Hexahydrobenzylcarbonate in passato è stato utilizzato clinicamente negli esseri umani, ma dalla fine degli anni 90 non è più commercializzato legalmente per tale scopo.[2][3][13][4]
Se si dà un’occhiata alla pagina ufficiale del sito che presenta il prodotto veterinario Finaplix-H, che consiste in pellet da impianto contenenti Trenbolone Acetato e utilizzati ancora oggi nel bestiame, si può notare come il dosaggio somministrato ad una mucca per indurne un aumento di peso è pari a 200mg. Avete capito bene, sono 200mg per capo di bestiame! Ok, sono bovini, hanno caratteristiche diverse tra metabolismo di escrezione, sensibilità recettoriale e risposta genica, ma già questo punto dovrebbe farvi cominciare a riflettere.
E’ altresì utile sottolineare che la somministrazione dei 10 pellet da 200mg di Trenbolone Acetato l’uno avviene per ogni singolo capo di bestiame nel giro di 63 giorni. Ciò significa un totale di 2g di Trenbolone Acetato (1.740mg di Trenbolone effettivo) per capo di bestiame nelle ultime 9 settimane circa prima della macellazione.
E l’uso clinico del Trenbolone negli esseri umani?
Come alcuni di voi già sapranno, nonostante la mancanza di prove cliniche su esseri umani, negli anni 70, in Francia, venne immesso sul mercato farmaceutico il Trenbolone per uso umano, sotto il nome commerciale di Parabolan (Trenbolone Hexahydrobenzylcarbonate), prodotto dalla Negma Laboratories.
Il Parabolan era usato clinicamente come agente anabolizzante per il risparmio proteico in caso di cachessia (deperimento della massa magra) e nella malnutrizione, oltre che per combattere alcune forme di osteoporosi. Le sue linee guida di prescrizione includevano raccomandazioni per il trattamento delle popolazioni androgeno-sensibile, come le donne e gli anziani. Grazie alle sue marcate proprietà androgene, tuttavia, il farmaco è stato controindicato per l’uso nei bambini, e soprattutto nelle giovani donne. Il Parabolan è rimasto sul mercato francese per un tempo molto lungo, anche se è stato interrotto (volontariamente) dalla Negma nel 1997. Da allora, nessun altro preparato a base di Trenbolone è stato approvato per uso umano.
Il Parabolan era generalmente somministrato ad un dosaggio clinico pari a 3 fiale al mese (228mg totali). La terapia veniva avviata il primo mese con tutte e 3 fiale somministrate nel corso dei primi 15 giorni. Durante i successivi 3 mesi, veniva somministrata una iniezione (76 mg) ogni 10 giorni. 76mg di Trenbolone Hexahydrobenzylcarbonate equivalgono a circa 50mg di Trenbolone attivo dopo la scissione del legame molecolare con l’estere.
Il protocollo di dosaggio clinico di Trenbolone negli esseri umani con utilizzo del Parabolan (Negma Laboratories) era il seguente:
114mg Trenbolone Hexahydrobenzylcarbonate (75 mg di ormone attivo) a settimana per le prime due settimane (totale 228mg di Trenbolone Hexahydrobenzylcarbonate);
Successivamente 76mg di Trenbolone Hexahydrobenzylcarbonate (50mg di ormone attivo) ogni 10 giorni.
Si sta ovviamente parlando di dosaggi basati su protocolli clinici, ma sono pienamente considerabili dosaggi più che efficaci specie per un atleta al suo primo utilizzo della suddetta molecola legata al suddetto estere. Parliamo comunque di 75mg di Trenbolone effettivo a settimana. Paragonandolo poi al dosaggio utilizzato per il bestiame (200mg di Trenbolone Acetato equivalente a 174mg di Trenbolone effettivo), si capisce che l’uso di dosaggi nell’ordine di 400-600mg a settimana di Trenbolone Hexahydrobenzylcarbonate (circa 300-400mg di Trenbolone effettivo) o 700mg di Trenbolone Acetato (609mg di Trenbolone effettivo) siano palesemente e a tutti gli effetti un abuso della molecola. Tralasciando certe abitudini yankee che consistono nella somministrazione massiva di Trenbolone a dosaggi fino ad 1g a settimana! Certo, 1g legato all’estere, ma se fate un po’ i conti l’abuso persiste indipendentemente dall’estere, con una variabile discreta tra estere Acetato e estere Enantato o Hexahydrobenzylcarbonate.
Per quanto possa urtare la convinzione di alcuni, usare il Trenbolone a dosaggi più alti di quelli somministrati ad un bovino di 720-1100Kg non è esattamente una pratica sostenibile.
Ad oggi non abbiamo informazioni certe sull’effetto che questo farmaco possa avere negli esseri umani, quindi sarebbe saggio evitarne quantomeno l’abuso. E questo è un dato di fatto.
La questione degli effetti collaterali a carico della sfera cognitiva:
Chi mi segue e legge i miei lavori conoscerà la mia posizioni in merito al Nandrolone e allo sbilanciamento tra “benefici e rischi”, a favore di questi ultimi, dati dal suo utilizzo, in specie a scopi dopanti. Infatti, l’uso del Nandrolone ha un significativo impatto sul SNC, con particolari differenze nel grado di manifestazione rispetto ad altri AAS non progestinici. Questo effetto del Nandrolone sul Sistema Nervoso Centrale è stato osservato scientificamente. Nello studio intitolato “The Impact of Nandrolone Decanoate on the Central Nervous System” vengono descritti chiaramente i numerosi effetti psicologici di questa molecola. Essi comprendono e influenzano:
1- Aggressività 2- Ansia, paura e stress 3- Ricompensa e dipendenza 4- Apprendimento, memoria e capacità di lavoro 5- Locomozione e attività fisica 6- Effetti sulla HPAA (Asse Ipotalamo-Pituitaria-Surrene) 7- Effetto sui neurotrasmettitori: Recettore Acido γ-Aminobutirrico Tipo A (GABAA); Recettori 5-idrossitriptamina (5-HT) e 5-HT; Recettori della Dopamina e Recettori Oppioidi.
Questi effetti sono potenzialmente riconducibili a tutti i progestinici, compreso il Trenbolone. Sebbene vi siano differenze sia nel grado di manifestazione dell’influenza che nel rapporto tra “vantaggi e svantaggi”, senza dubbio a favore del Trenbolone, questi aspetti neurologici non dovrebbero essere comunque tralasciati.
I neuroni nel cervello sono in grado di conservare le informazioni e di elaborarle perché creano continuamente nuove connessioni tra loro. La Proteina precorritrice della beta-amiloide [APP] svolge un ruolo importante in questo processo. Per essere precisi: la APP è una proteina di trans-membrana di tipo 1.
Proteina Precorritrice della Beta-Amiloide [APP]
Gli enzimi scompongono la APP in pezzi e se questo processo si svolge correttamente le cellule cerebrali funzionano in modo normale. Ma se gli enzimi iniziano a non agire come dovrebbero – a causa di geni difettosi o fattori ambientali pericolosi – si formano frammenti proteici tossici. Il più rischioso di questi è l’amiloide-beta-42, che si accumula nel cervello, formando placche e uccidendo infine i neuroni. Il cervello delle persone decedute a causa dell’Alzheimer contengono grandi quantità di amiloidi-beta-42, per cui la maggior parte dei neurologi pensa che l’amiloide-beta-42 sia la causa dell’Alzheimer e di forme legate alla demenza.
Il Testosterone, l’Estradiolo e il DHT offrono protezione contro l’Alzheimer, con influenza incrociata e interdipendente. Ecco perché Ma e Lui si sono chiesti quale effetto avrebbe avuto il Trenbolone sulla formazione di amiloide-beta-42. Così, hanno somministrato a dei topi 5 iniezioni di Trenbolone in un periodo di 48 ore. L’equivalente umano delle dosi utilizzate dai ricercatori sarebbe di circa 0,85mg per kg di peso corporeo.
L’amiloide-beta-42 si è accumulata nel cervello dei ratti maschi trattati con Trenbolone. Il grafico seguente mostra i risultati emersi nel periodo di 48 ore nel quale sono state distribuite le 5 iniezioni di Trenbolone.
Il grafico qui sopra (barre grigie), riporta i dati estrapolati dagli esperimenti in vitro che i ricercatori hanno inoltre svolto con le cellule cerebrali esponendole per 48 ore a 100 nanomoli di Trenbolone [TB]. L’aggiunta di un antiandrogeno come la Flutamina [Flu] ha ridotto l’accumulo di amiloide-beta-42. Così è emerso che i danni cerebrali causati dal Trenbolone sono dovuti agli effetti androgeni di questa molecola.
Una combinazione di Trenbolone e DHT ha mostrato di aumentare l’accumulo di amiloide-beta-42.
In definitiva, per quanto emerso dallo studio, dal momento che i danni ai neuroni possono verificarsi molto prima dei sintomi clinici dei disturbi neurodegenerativi, l’esposizione al Trenbolone dovrebbe essere considerata come un fattore ambientale ad alto rischio per lo sviluppo della malattia di Alzheimer. Ciò nonostante, si parla ancora di ipotesi le quali dovrebbero essere accertate ed avvalorate da ulteriori ricerche.
Siamo più o meno tutti consapevoli del fatto che l’aumento dell’aggressività riscontrabile in diversi bodybuilder “doped” sia una costante con variabili di intensità tra soggetto e soggetto. Ma è piuttosto facile osservare maggiori problemi di instabilità mentale negli utilizzatori di Trenbolone rispetto ad altri AAS.
Generalmente, se un soggetto ha una personalità già di base aggressiva, con l’aumento degli androgeni circolanti tale caratteristica subisce una significativa esacerbazione.
Non stupisce, quindi, che i modelli animali trattati con Trenbolone mostrino effettivamente un degrado cognitivo.
Conclusioni sul Trenbolone:
Grazie ai molti dati enpirici ed aneddotici da me raccolti negli anni riguardo al Trenbolone e al suo utilizzo da parte dei BodyBuilder e PowerLifter, vi esorto innanzi tutto ad essere molto cauti con un suo possibile uso, soprattutto quando si è consapevoli del fatto che i dati clinici a riguardo sono scarsi e basati su modelli animali.
Nonostante alcuni di voi saranno sicuramente sorpresi del fatto che il Trenbolone non è un farmaco che ha avuto studi sugli esseri umani, la verità è questa.
Ci sono molte persone convinte del fatto che, dato l’utilizzo diffuso e annoso nella comunità del Bodybuilding di questa molecola, e dato che è stato precedentemente prescritto clinicamente per il trattamento di malattie degenerative muscolo-scheletriche nell’uomo, il Trenbolone possa essere somministrato con una certa sicurezza anche a dosaggi elevati. Peccato però che di “dati di sicurezza” nell’uso umano non vi sia la minima traccia. Ci si basa solo e soltanto su dati empirici e aneddotici!
Paradossalmente, esistono più dati clinici su esseri umani di molecole relativamente recenti rispetto al Trenbolone che, tanto per delucidazioni temporali, è stato sintetizzato per la prima volta nel 1963.
Qualora una persona decidesse di testare il Trenbolone dovrebbe tenere conto di alcuni punti “conservativi”:
Non superare un dosaggio iniziale pari a 76-100mg di Trenbolone Hexahydrobenzylcarbonato o di 100mg di Trenbolone Enantato a settimana. Una dose massima iniziale può essere pari a 150mg di Trenbolone Acetato a settimana;
Successivamente, calibrare il dosaggio attraverso la formula 2mg/Kg (Trenbolone/Kg di peso corporeo) a settimana (es. 90Kg = 180mg);
Ridurre l’uso della molecola a massimo 1-2 volte l’anno.
ATTENZIONE! Non si tratta di consigli ma di divulgazione preventiva! Non per nulla, questi articoli nascono come mezzo per evitare l’abuso sconsiderato di PEDs!
Continuare ad usare dosaggi molto al di sopra del margine utile e conservativo non porterà a nessun reale vantaggio. E ciò non interessa soltanto gli individui con una genetica nella media, in parte convinti che basti usare un dosaggio maggiore e crescente per essere quello che non si è, ma anche e soprattutto i tendenti “freak”, soggetti che come la mal erba crescono con dosaggi tutt’altro che facenti parte della posologia dei doped “millennial”.
Per il resto, ad ognuno la propria riflessione in merito e il dialogo con la propria coscienza.
Yarrow, Joshua F.; McCoy, Sean C.; Borst, Stephen E. (2010). “Tissue selectivity and potential clinical applications of trenbolone (17β-hydroxyestra-4,9,11-trien-3-one): A potent anabolic steroid with reduced androgenic and estrogenic activity”. Steroids. 75 (6): 377–89. doi:10.1016/j.steroids.2010.01.019. PMID20138077. S2CID205253265.
Gettys, TW; d’Occhio, MJ; Henricks, DM; Schanbacher, BD (1984). “Suppression of LH secretion by oestradiol, dihydrotestosterone and trenbolone acetate in the acutely castrated bull”. The Journal of Endocrinology. 100 (1): 107–12. doi:10.1677/joe.0.1000107. PMID6361192.
Nonostante decenni di “lotta al doping” esso rimane assai diffuso, e non solo nelle competizioni di alto livello. L’errore alla base di questa campagna mediatico-salutistica è stata la generalizzazione; ossia fornire informazioni imprecise, accentuando i possibili sides senza però premurarsi di una vera e propria informativa preventiva chiara, veritiera ed efficace. In poche parole, quello che non si è fatto è dire: “l’uso di PEDs ha una serie di possibili effetti collaterali di gravità dipendente dal tipo di molecola, dal tempo e dalle modalità di assunzione”. Tutto ciò accompagnato da un manuale scientificamente corretto e di facile comprensione, contenente informazioni utili riguardanti la materia PEDs tale da permettere una migliore comprensione della questione che, a sua volta, renda possibile una più consapevole scelta individuale. Ma ciò non è stato fatto. Con l’unica eccezione di alcuni esperti indipendenti che nel corso degli anni hanno pubblicato libri e scritto articoli di una certa utilità.
Lo scopo di questa serie di articoli sarà quello di arginare il fenomeno dell’abuso dei PEDs, cosa che sta degenerando e che sta mostrando i suoi peggiori effetti su atleti di ambo i sessi.
Per la prima pubblicazione di questa nuova serie iniziamo con l’Oxymetholone…
Una (sempre utile) introduzione alla molecola di Oxymetholone:
L’Oxymetholone, noto anche come 2-idrossimetilene-17α-metil-4,5α-diidrotestosterone (2-idrossimetilene-17α-metil-DHT) o come 2-idrossimetilene-17α-metil-5α-androstan-17β-ol-3-one, è uno steroide androstano sintetico e un derivato 17α-alchilato del DHT.[1][2][3]
Le informazioni disponibili sulla farmacocinetica di questo AAS sono limitate.[4] Sembra essere ben assorbito con la somministrazione orale.[4] L’Oxymetholone ha affinità molto bassa per le globuline leganti gli ormoni sessuali nel siero umano (SHBG), meno del 5% di quella del Testosterone e meno dell’1% di quella del DHT. [5] Il farmaco viene metabolizzato nel fegato tramite ossidazione in posizione C2, riduzione in posizione C3, idrossilazione in posizione C17 e coniugazione. [4][6] Il gruppo C2 idrossimetilene del Oxymetholone può essere scisso per formare il Mestanolone (17α-metil-DHT), che può contribuire agli effetti della molecola precursore.[3] L’emivita del Oxymetholone è sconosciuta sebbene vi siano alcune ipotesi a riguardo.[6] L’Oxymetholone e suoi metaboliti vengono eliminati attraverso le urine.[5][6]
Come altri AAS, l’Oxymetholone è un agonista del recettore degli androgeni (AR).[3] Non è un substrato per la 5α-reduttasi (dal momento che è già 5α-ridotto) ed è uno substrato scarso per il 3α-idrossisteroide deidrogenasi (3α-HSD), e quindi mostra un alto rapporto di attività anabolizzante rispetto all’effetto androgenico.[3]
Data la sua derivanza dal DHT, l’Oxymetholone non è un substrato per l’enzima Aromatasi e quindi non può essere aromatizzato in metaboliti estrogenici.[3] Tuttavia, caratteristica unica tra i derivati del DHT, l’Oxymetholone è comunque associato a un’estrogenicità relativamente elevata ed è noto per avere il potenziale di produrre effetti collaterali estrogenici come ginecomastia (raramente) e ritenzione idrica. [3][7][8][9] È stato suggerito che questo può essere una conseguenza del legame diretto a l’attivazione del recettore degli estrogeni da parte dell’Oxymetholone (estrogenicità intrinseca).[3] L’Oxymetholone non possiede alcuna attività progestinica significativa.[3]
A causa della sua struttura 17α-alchilata, l’Oxymetholone è epatotossico.[3] L’uso a lungo termine del farmaco può causare una varietà di disturbi gravi, tra cui l’epatite, il cancro al fegato e la cirrosi; pertanto si raccomandano test periodici di funzionalità epatica per coloro che assumono l’Oxymetholone a fini terapeutici.[10] Questa molecola ha ottenuto, infatti, la nomea di essere uno tra gli AAS più epatotossici. Ciò deriva da i dosaggi comunemente, ed erroneamente, utilizzati in contesto culturistico. Si parla di dosaggi che facilmente sforano i 100-150mg/die. Ma tali dosaggi sono realmente vantaggiosi in termini di guadagni ipertrofici specie se messi in rapporto con gli effetti collaterali possibilmente verificabili? Questa domanda può ottenere una risposta sufficientemente esaustiva attraverso i risultati di uno studio che ha messo a confronto gli effetti di una dose di Oxymetholone da 50mg/die e una da 100mg/die.[11]
Oxymetholone – 50mg Vs. 100mg:
In questo studio, possiamo vedere i cambiamenti nel peso corporeo, nella massa magra, e la perdita di grasso in risposta a un dosaggio moderato e alto di Oxymetholone (50 mg vs 100 mg).
I cambiamenti nella composizione corporea sono mostrati per i gruppi placebo (barre nere), 50mg di Oxymetholone al giorno (barre bianche) e 100mg al giorno (barre grigie). I numeri sopra le barre rappresentano i cambiamenti assoluti medi e le barre di errore sono ± 1 SE. Per la massa corporea magra totale (LBM) e il grasso totale, le differenze tra i 3 gruppi erano significative (P <0,0001, ANOVA a una via). * Differenze significative rispetto al placebo, P ≤ 0,001.
Come ci si aspetterebbe, il gruppo placebo non ha guadagnato massa magra, né ha perso grasso corporeo.
Il gruppo trattato con 50mg di Oxymetholone ha guadagnato 3,3Kg di massa magra e ha perso 2,6kg di grasso.
Il gruppo trattato con 100mg di Oxymetholone ha guadagnato 4,2Kg di massa magra e ha perso 2,5kg di grasso.
I cambiamenti nella composizione regionale (n = 16) sono mostrati per i gruppi placebo, 50mg/die e 100mg/die. A: i numeri sopra le barre rappresentano i cambiamenti assoluti medi per il grasso del tronco mediante assorbimetria a raggi X a doppia energia (DEXA). B: le barre rappresentano i cambiamenti assoluti medi (kg) per la LBM dell’arto superiore (braccio destro più braccio sinistro) mediante DEXA. C: area della sezione trasversale del muscolo totale prossimale (barre grigie) e posteriore (barre nere) dei muscoli della coscia tramite risonanza magnetica. Le barre di errore sono ± 1 SE. * Differenza significativa rispetto al placebo, P ≤ 0,005. .
Guardando la massa corporea magra, è possibile vedere che quando si confrontano i due gruppi di dosaggio, il gruppo da 100mg ha guadagnato solo 0,9kg di massa corporea magra in più rispetto al gruppo da 50mg.
Questo dopo tre mesi di esposizione al doppio della quantità di farmaco.
Se si confrontano i biomarcatori tra i due gruppi, è possibile vedere che l’effetto di 100mg di Oxymetholone ha avuto sui livelli di ALT e AST era molto più deleterio rispetto al gruppo di 50 mg.
Caratteristiche di base della popolazione dello studio
Come molti di voi già sapranno, l’alanina aminotransferasi (ALT) e l’aspartato aminotransferasi (AST) sono biomarcatori comunemente usati per valutare i danni al fegato.
La somministrazione di un dosaggio di Oxymetholone doppio rispetto al basale di 50mg ha prodotto un ulteriore 27% di crescita muscolare relativa (la massa magra non è composta solo dal muscolo scheletrico!), ma ha provocato un picco 3.4x più alto di ALT e un picco 2.7x più alto nei livelli di AST.
Il calo del HDL è stato simile in entrambi i gruppi 50mg/die e 100mg/die.
Quelli sono solo biomarcatori con valore diagnostico per un eventuale danno epatico ma non sono indicativi di ciò che comporta la variabile del dosaggio sull’ipertrofia ventricolare, o altri fattori comunemente trascurati che dovrebbero essere utilizzati per valutare la salute cardiovascolare.
Anche se è possibile che gli aumenti di massa magra misurati dalla DEXA fossero legati in buona parte alla ritenzione idrica causata dalla terapia con Oxymetholone, i notevoli aumenti di forza muscolare misurati con il metodo 1-RM nei gruppi da 50 e 100mg/die (8,2-18,4%) suggeriscono che gli aumenti di massa magra erano probabilmente dovuti all’accrescimento di proteine miofibrillari oltre che alla semplice massa magra totale, poiché la forza è in una certa misura legata alle dimensioni dei muscoli. Inoltre, i membri del gruppo di ricerca hanno riferito che i cambiamenti nella massa magra appendicolare tramite DEXA sono quantitativamente correlati ai cambiamenti nella forza muscolare scheletrica in risposta a stimoli anabolici. In effetti, nel presente studio, sono stati in grado di corroborare questa relazione dimostrando che gli aumenti significativi del tessuto magro della parte superiore del corpo mediante scansione DEXA appendicolare erano altamente correlati con i cambiamenti nella forza della parte superiore del corpo come valutato da esercizi di Chest Press e Lat Pull-Down. Inoltre, i cambiamenti nella forza muscolare massima volontaria per gli esercizi della parte superiore del corpo hanno mostrato una risposta legata alla dose.
I cambiamenti relativi (%) nella forza sono mostrati per i gruppi placebo (barre nere), 50mg/giorno Oxymetholone (barre bianche) e 100mg/giorno Oxymetholone (barre grigie). I numeri sopra le barre rappresentano il cambiamento relativo (%) dal basale alla settimana 12 per le prove di forza massima a 1 ripetizione. Le barre di errore rappresentano ± 1 SE dalla media. * Differenza significativa rispetto al placebo, P < 0,05; † differenza significativa rispetto al placebo con il test di Wilcoxon, P < 0,02.
Al contrario, c’erano guadagni non significativi tra i tre gruppi di trattamento per la forza degli arti inferiori (3,9-12,0%), coerentemente con la mancanza di un aumento significativo della massa magra degli arti inferiori mediante scansione DEXA. Tuttavia, c’era una differenza quasi significativa (P = 0,052) tra i gruppi per il cambiamento del area della sezione trasversale del muscolo (CSA) dei muscoli della coscia tramite la risonanza magnetica, suggerendo che la terapia dello studio può aver influenzato positivamente i muscoli degli arti inferiori. È possibile che i test di forza di gruppi muscolari multipli e di grandi dimensioni, come quelli utilizzati con l’esercizio Leg Press, siano meno sensibili ai modesti cambiamenti nella massa muscolare, e lo studio potrebbe non aver avuto sufficiente potenza per rilevare piccoli ma significativi guadagni nelle estremità inferiori. Si ipotizza che ciò sia dovuto al fatto che i grandi muscoli delle gambe sono abitualmente utilizzati più frequentemente per sostenere il carico (ad esempio, camminare, alzarsi da una sedia) rispetto ai muscoli dell’estremità superiore negli adulti più anziani. Piccoli ma significativi guadagni nella forza e nella massa muscolare della parte inferiore del corpo possono essere meno dimostrabili che per i muscoli della parte superiore del corpo, che possono essere utilizzati meno per il lavoro ad alto volume e più inclini alla sarcopenia nelle persone anziane. Inoltre, i muscoli degli arti superiori, rispetto ai muscoli degli arti inferiori, hanno proporzioni maggiori di fibre a contrazione rapida di tipo II, che possono essere perse preferibilmente con l’invecchiamento. Inoltre, uno studio longitudinale in uomini anziani ha mostrato che le fibre di tipo I sono state perse principalmente nel vasto laterale della gamba, portando all’ipotesi che ci potrebbe essere una maggiore perdita di fibre di tipo II nelle braccia con l’invecchiamento. Così la risposta agli stimoli anabolici può essere più facilmente dimostrabile nelle estremità superiori di questa popolazione.
C’erano anche significative ma simili diminuzioni del grasso corporeo totale di 2,6 ± 1,2 e 2,5 ± 1,6 kg nei gruppi di 50 e 100mg al giorno, rispettivamente. Una parte importante del miglioramento dell’adiposità riguardava la diminuzione del grasso del tronco (1,7 ± 1,0 e 2,2 ± 0,9 kg nei due rispettivi gruppi di trattamento attivo). Una riduzione significativa del grasso del tronco potrebbe influenzare favorevolmente i fattori di rischio per le malattie cardiovascolari. Anche se ci aspetteremmo che la riduzione del grasso addominale si rifletta in una migliore sensibilità all’insulina, le misure indirette (HOMA-IR e QUICKI) potrebbero non essere state abbastanza sensibili. È anche possibile che ci fossero troppo pochi soggetti in ogni gruppo per rilevare cambiamenti piccoli ma significativi.
Ci sono ragioni teoriche per temere che l’eccesso di androgeni possa provocare o essere associato all’insulino-resistenza, anche se questa relazione è stata dimostrata solo in donne con sindrome dell’ovaio policistico. Non è stata misurata direttamente la sensibilità all’insulina né con il clamp euglicemico iperinsulinemico né con test di tolleranza al glucosio endovena a campionamento frequente. Tuttavia, le misure indirette della sensibilità insulinica (insulina a digiuno, HOMA-IR, QUICKI) non hanno mostrato prove di resistenza insulinica.
Cosa estrapolare?
Questo studio però presenta alcune limitazioni che possono averne influenzato i risultati. In primo luogo, la piccola dimensione del campione di meno di una dozzina di soggetti per gruppo può aver limitato la capacità di rilevare piccoli ma importanti cambiamenti in variabili come la massa magra (LBM) delle estremità inferiori e il CSA della muscolatura della coscia. Allo stesso modo, è possibile che le differenze osservate per i cambiamenti nella LBM totale e nella forza avrebbero potuto essere significative tra i gruppi di trattamento con dimensioni del campione maggiori. Quest’ultimo avrebbe fornito ulteriore supporto alla nostra supposizione di una risposta dose-dipendente con l’Oxymetholone. In secondo luogo, la popolazione rappresentava uomini adulti più anziani, che sono stati caratterizzati come a rischio di sarcopenia legata all’età sulla base dei rapporti che mostrano la perdita di massa e forza muscolare con l’invecchiamento. Tuttavia, i soggetti non sono stati reclutati per la perdita di peso, la fragilità o l’ipogonadismo palese di per sé, dal momento che è stato dimostrato che gli uomini più giovani con concentrazioni di Testosterone normali possono ottenere aumenti apprezzabili della massa muscolare e della forza dopo l’integrazione di androgeni. Inoltre, ci sono prove che la sintesi proteica miofibrillare nelle persone anziane può essere significativamente aumentata a livelli paragonabili a quelli raggiunti nelle persone più giovani in risposta a un potente stimolo anabolico. Infine, poiché l’Oxymetholone è un AAS 17-metilato che provoca un elevato effetto di primo passaggio nel fegato, e che nel presente studio non sono state prese misure di contenimento per l’epatotossicità potenziale, i risultati di AST e ALT ottenuti rappresentano solamente modelli privi di ancillari volti ad una epatoprotezione.
Conclusioni sul dosaggio “ottimale” di Oxymetholone:
Evidenziati i limiti dello studio, pur prendendo i dati ivi riportati universalmente rapportabili al basale d’uso della molecola (es. vedi epatotossicità), possiamo giungere, grazie all’ausilio di dati empirici raccolti negli anni attraverso indagini svolte sulle preparazioni di svariati atleti di medio e alto livello, ad identificare un dosaggio con una ratio “efficacia:rischio (E:R)” favorevole per l’atleta.
Un dato è emerso preponderante nel corso delle indagini svolte: quale fosse il peso dell’atleta e il suo condizionamento atletico, nonché l’utilizzo di una adeguata epatoprotezione e controllo della dislipidemia, il margine della ratio E:R diveniva evidentemente sfavorevole oltre i 150mg/die. Indi per cui, i dosaggi elevati raggiunti da certi atleti, arrivando a picchi di 200-300mg/die, sono risultati inutili al miglioramento delle risposte anabolizzanti complessive e inficianti per il corretto svolgimento della stessa preparazione (vedi, ad esempio, marcata inappetenza e nausea).
Dosaggi standard per un atleta di sesso maschile non dovrebbero discostarsi dal range 50-100mg/die, considerando che la taratura del “dosaggio ideale” si è ottenuta calcolando la dose individuale con la formula 1mg/Kg di peso corporeo. Ovviamente, l’assicurarsi una adeguata protezione epatica e lipidica è il punto parallelo da raggiungere.
Nelle atlete, invece, vista la loro maggiore sensibilità agli aumenti degli androgeni circolanti, la “dose ideale” si è attestata a 25mg/die con punte massime (anche se non necessarie) di 50mg/die. A tal proposito, vorrei ricordare che l’Oxymetholone è risultato essere una molecola più vantaggiosa nel controllo degli effetti collaterali androgenizzanti rispetto a composti quali Methenolone e Boldenone.
La linea tra abuso e uso è spesso molto sottile, ma nel caso del Oxymetholone essa si mostra sufficientemente marcata…
Pavlatos AM, Fultz O, Monberg MJ, Vootkur A (June 2001). “Review of oxymetholone: a 17alpha-alkylated anabolic-androgenic steroid”. Clinical Therapeutics. 23 (6): 789–801, discussion 771.
Saartok T, Dahlberg E, Gustafsson JA (June 1984). “Relative binding affinity of anabolic-androgenic steroids: comparison of the binding to the androgen receptors in skeletal muscle and in prostate, as well as to sex hormone-binding globulin”. Endocrinology. 114 (6): 2100–6.
Hengge UR, Stocks K, Wiehler H, Faulkner S, Esser S, Lorenz C, et al. (March 2003). “Double-blind, randomized, placebo-controlled phase III trial of oxymetholone for the treatment of HIV wasting”. AIDS. 17 (5): 699–710.
Cortesgallegos V, Castaneda G, Alonso R, Perezpasten E, Reyeslugo V, Barron C, Mondragon L, Villalpando S (January 1982). “Spontaneous and Oxymetholone-Induced Gynecomastia”. Journal of Andrology. C/O Allen Press, Inc Po Box 368, Lawrence, Ks 66044: Amer Soc Andrology, Inc. 3 (1): 33.
Villalpando S, Mondragon L, Barron C, Reyeslugo U, Perezpasten E, Alonso R, Castaneda G, Gallegos V (January 1982). “5-Alpha Reductase Blockade May Be Responsible for Spontaneous and Oxymetholone-Induced Gynecomastia”. Archivos de Investigacion Medica. Social Apdo Postal 73-032, Mexico Df 03020, Mexico: Inst Mexicano Seguro. 13 (2): s13.
Il fegato è un organo importante ed è vitale per la sopravvivenza del soggetto. È responsabile di diverse e importanti funzioni nel corpo umano. Produce acidi biliari e proteine plasmatiche, immagazzina glicogeno e produce glucosio attraverso la gluconeogenesi, gioca un ruolo nel sistema immunitario, metabolizza un numero elevato di molecole, ecc. Quindi, si, avete capito bene: è importante. Quando qualcosa risulta dannosa per il fegato, essa si indica come epatotossico (dal greco hêpar-atos, fegato). Un chiaro esempio è l’alcol. Gli alcolisti tendono a sviluppare una malattia del fegato a un certo punto della loro vita. Tuttavia, molti farmaci da prescrizione, o anche over-the-counter, possono essere epatotossici, come l’Acetaminofene. E, come è ben dimostrato, anche gli AAS possono essere epatotossici, anche se specifici. Come sembra, solo quelli con una specifica alterazione chimica sembrano essere maggiormente epatotossici – in particolare, quelli che presentano una metilazione in pozione C-17α.
Modifica della struttura carbossilica del Testosterone (sinistra) in posizione C-17α (destra).
In questo articolo tratterò principalmente ciò che sembra causare questa epatotossicità indotta da AAS. L’effetto epatotossico può essere riscontrato attraverso l’osservazione dei cambiamenti nei marcatori ematici del danno epatico, come Alanina Transaminasi (ALAT), Aspartato Transaminasi (ASAT), γ-glutamiltransferasi (GGT) e la Fosfatasi Alcalina (ALP). Una nota di cautela deve essere presa in considerazione quando si interpretano gli aumenti di ALAT e ASAT, poiché entrambi aumenteranno anche a causa del intyenso lavoro muscolare [1]. È bene sapere che in questi casi, ASAT sarà di solito più alto del ALAT, mantenendo un rapporto ASAT/ALAT superiore a 1. Quindi, quando questi aumentano con un rapporto inferiore a 1, si può essere più sicuri che il danno muscolare non è il colpevole dell’alterazione. Idealmente, nessun esercizio (contro-resistenza) viene svolto 1-2 settimane prima dell’esame del sangue per escludere il danno muscolare muscolare come causa dell’innalzamento, sebbene ciò dipenda anche dall’intensità del allenamento. In rari casi, il danno al fegato potrebbe avanzare clinicamente fino allo sviluppo di ittero colestatico [2]. In questo caso, un prodotto della degradazione dei globuli rossi (bilirubina) si accumula nel corpo. L’ittero può essere osservato visivamente (tono giallo della pelle e della sclera degli occhi), e si possono sviluppare sintomi come nausea, vomito, dolore allo stomaco e prurito. Inoltre, alcuni rari casi di peliosis hepatis (Peliosi Epatica) sono stati segnalati verificarsi come risultato dell’uso di AAS orali ad alte dosi [3]. Questa è una condizione nella quale si vengono a formare cisti piene di sangue nel fegato. La sospensione dell’AAS in questione è solitamente sufficiente e porterà alla scomparsa di queste caratteristiche cliniche entro pochi mesi. In casi più gravi, tuttavia, potrebbero richiedere un intervento chirurgico. Infine, alcuni casi in letteratura hanno riportato un’associazione tra uso di AAS e carcinoma epatico [4] e adenoma [5].
Ho già trattato in passato tale problematica legata all’uso di AAS, ma questa volta voglio trattare la questione più nello specifico, analizzando le due ipotesi che ruotano intorno all’epatotossicità AAS-dipendente: “ipotesi dello stress ossidativo” e “ipotesi di coniugazione dell’anello D”.
L’ipotesi dello stress ossidativo:
L’ipotesi dello stress ossidativo che tratterò qui si basa su un documento che William Llewellyn, Peter Van Mol e Peter Bond hanno pubblicato [6]. Lo stress ossidativo è qualcosa che si pensa possa risultare nell’epatotossicità osservata con l’uso di AAS, e se l’ipotesi è vera, dà qualche opportunità per contrastarla in modo migliore. Quindi, cominciamo con spiegare quello che è lo stress ossidativo. Lo stress ossidativo è descritto da Helmut Sies come un disturbo nell’equilibrio pro-ossidante-antiossidante a favore del primo [7], che si riduce a molecole contenenti ossigeno, che sono altamente reattive (specie reattive dell’ossigeno [ROS]), sopraffacendo il sistema antiossidante. Poiché le ROS sono così altamente reattive, possono reagire con molecole come lipidi, proteine, carboidrati e acidi nucleici (elementi costitutivi del DNA). Quando si dice “reagire con queste molecole”, si intende che danneggia queste molecole (estremamente semplificato, ma è sufficiente per far comprendere il processo). Questi ROS provengono da varie reazioni catalizzate da enzimi come la respirazione cellulare (l’ossidazione dei macronutrienti per fornire energia), altri processi metabolici e radiazioni. La fonte primaria di ROS all’interno di una cellula sono i mitocondri, il che non è sorprendente dato che i mitocondri sono le “centrali energetiche” della cellula. È il posto nella cellula dove i carboidrati alimentari, gli acidi grassi e le proteine (o, meglio, gli amminoacidi che le compongono) finiscono per essere ossidate per produrre energia in un processo chiamato fosforilazione ossidativa. Come suggerisce il nome, la fosforilazione ossidativa ossida e richiede ossigeno per farlo. Questo processo, tuttavia, non è perfetto. Per non complicare troppo le cose al lettore, non mi addentrerò nelle complessità delle reazioni chimiche, ma fondamentalmente, questo processo può produrre ROS come sottoprodotto (superossido in particolare). Le cellule del corpo sono dotate di meccanismi per tenere a bada questi ROS generati (la parte antiossidante dell’equazione). In circostanze normali questo porta ad un sottile equilibrio tra i due. Avere qualche ROS qua e là nelle cellule è normale. Essi giocano un ruolo essenziale nel normale funzionamento di vari processi vitali [8]. Tuttavia, il problema nasce quando questo equilibrio si altera a favore della parte proossidante dell’equazione: lo stress ossidativo. Questo è il momento in cui i ROS prendono il sopravvento, per così dire, e possono iniziare a creare il caos nella cellula. Quanto sopra è un quadro un po’ troppo semplificato. Ci sono diversi tipi di ROS (radicali liberi e non radicali). Ciò che conta è dove si trovano questi ROS nella cellula e come evolvono nel tempo. Inoltre, questo interagisce con il sistema antiossidativo delle cellule, il che complica ulteriormente il quadro. Ma credo che quanto sopra sia sufficiente per dare una buona comprensione di tutto questo. Ciò che conta è che l’epatotossicità indotta da AAS è stata ripetutamente dimostrata essere associata allo stress ossidativo nelle cellule epatiche (fegato) di modelli animali [9]. Questo fa sorgere la domanda: è solo un’associazione, o c’è una relazione causale con l’epatotossicità indotta da AAS? Dopo aver scavato nella letteratura, sono emersi alcuni studi che sembrano sostenere una relazione causale. Uno studio svolto su un carcinoma prostatico umano epiteliale (22Rv1) ha collegato l’attivazione del recettore degli androgeni (AR) a un aumento dei ROS basali [10]. Più tardi, lo stesso gruppo ha pubblicato una ricerca applicando un disegno di studio simile. Questo studio ha confermato i precedenti risultati e ha anche dimostrato che l’aumento dei ROS è dovuto a un aumento indotto dall’AAS nella β-ossidazione mitocondriale degli acidi grassi [11]. Quindi, l’attivazione di l’AR porta a una maggiore ossidazione degli acidi grassi nei mitocondri, con conseguente maggiore produzione di ROS come sottoprodotto. Da notare che questo studio ha anche trovato un aumento dell’mRNA della carnitina palmitoiltransferasi (CPT1). Tutto quello che dovete sapere è che la CPT1 è considerata essere l’enzima che regola la velocità nel processo di ossidazione mitocondriale degli acidi grassi. Quindi, se si aumenta la CPT1, si aumenta l’ossidazione mitocondriale degli acidi grassi. Ora, le cellule del cancro alla prostata non sono cellule del fegato, ovviamente. Ma ciò che è interessante è che l’AAS 17α-alchilato Fluoxymesterone e Metilandrostanolone hanno dimostrato di aumentare l’attività del CPT1 nel fegato di ratto [12]. Inoltre, se si guardano agli epatociti di ratto (cellule epatiche) trattati con AAS 17α-alchilati, si vedrà il gonfiore dei mitocondri e solo cristae leggermente definite [13]. (Le criste sono quelle pieghe caratteristiche della membrana interna dei mitocondri). Infatti, la produzione di ROS è una causa nota di gonfiore mitocondriale, e il gonfiore è un fattore importante che porta alla successiva morte cellulare [14]. Quindi, apparentemente, suggerisce un potenziale ruolo dello stress ossidativo. Questo non vuol dire che qualsiasi aumento nella produzione di energia di una cellula sia negativo. Usando i muscoli aumenta anche la produzione di energia nelle cellule muscolari. Di conseguenza, più ROS vengono prodotti anche in queste cellule. In contrasto con l’aumento di ROS indotto dall’AAS nelle cellule del fegato, questi aumenti sono transitori invece che continui. Inoltre, le cellule muscolari differiscono nei loro meccanismi antiossidanti per gestire questa condizione. Quindi, normalmente, questo non è assolutamente un problema. Tuttavia, l’esercizio intenso e prolungato può anche provocare danni ossidativi alle molecole delle cellule muscolari [15].
L’ipotesi dello stress ossidativo nella epatotossicità indotta da AAS come descritto da Bond et al. [49]. 1 Un androgeno si lega a, e attiva, il recettore degli androgeni (AR) nelle cellule epatiche. Questo porta a 2 la sovra-regolazione della Carnitina Palmitoiltransferasi 1 (CPT1), l’enzima che regola il tasso di β-ossidazione degli acidi grassi (FA). Si pensa che questo porti a 3 un aumento della β-ossidazione degli acidi grassi nei mitocondri. Di conseguenza, 4 la produzione di specie reattive dell’ossigeno (ROS) è aumentata. L’aumento dei ROS poi danneggia i mitocondri, il che sembra essere alla base dell’epatotossicità indotta dall’AAS.
Ora, se si integrassero gli antiossidanti (mitocondriali), si allevierebbe questo danno? Può darsi. Mentre non c’è un trial di buona qualità che valuti questo, uno studio osservazionale su 320 atleti dimostra qualcosa del genere [16]. In breve, gli utilizzatori di AAS che hanno preso un supplemento contenente alcuni composti antiossidanti non ha mostrato alcun aumento dei marcatori di danno epatico dopo il ciclo rispetto a quelli che non hanno assunto quel supplemento. Ancora una volta, questo sarebbe in linea con lo stress ossidativo che gioca un ruolo causale nell’epatotossicità indotta da AAS. Infine, sembra che l’epatotossicità indotta da AAS potrebbe essere legata all’attivazione del AR nelle cellule epatiche. In un vecchio studio del 1964, Marquardt et al. non sono riusciti a dimostrare che l’AAS non 17α-alchilato produce test di funzionalità epatica anormali [17]. Infatti, gli AAS 17α-alchilati mostrano segni di epatotossicità in diversi studi, mentre non si vede questo con AAS non-17αalchilati, nemmeno con un alto dosaggio di 600 mg di Testosterone Enantato settimanale [18]. La 17α-alchilazione sembra quasi necessaria per rendere epatotossico un AAS, probabilmente perché è l’unica alterazione che lo rende sufficientemente biodisponibile per via orale. E, di conseguenza, porta ad alte concentrazioni del composto nel fegato. Ma possiamo individuare le differenze tra i vari AAS 17α-alchilati che riguardano la loro capacità di attivare l’AR? Certamente sembra così. In generale, sembra che sia vero quanto segue:
Epatotossicità = resistenza alla decomposizione epatica×potenza di attivazione del AR
Quindi, facciamo un esempio. Il Methyltrienolone (R1881) ha un’affinità molto alta per l’AR, ha un’alta potenza per la transattivazione dell’AR [19], ed è fortemente resistente al metabolismo epatico. Come tale, è un composto ideale per un saggio dei siti di legame agli androgeni [20]. Infatti, un studio clinico che impiega un basso dosaggio dello steroide (≤1 mg al giorno) ha dimostrato un significativo aumento dei marcatori di danno epatico entro due settimane [21]. Gli autori lo hanno definito “(…) attualmente lo steroide più epatotossico”. Lo steroide 17α-alchilato meno epatotossico è solitamente considerato l’Oxandrolone. Anche con alti dosaggi fino a 80mg al giorno, mostra solo deboli segni di epatotossicità [22]. Mentre lo steroide è abbastanza resistente al metabolismo epatico [23], ha una bassa affinità per il AR [23]. La sua potenza relativa in termini di transattivazione AR è anche quasi 100 volte inferiore a quella del Methyltrienolone [19]. Allo stesso modo, anche l’Oxymetholone ha una bassa affinità per l’AR [23] e la sua potenza in termini di transattivazione AR è molto simile a quella dell’Oxandrolone [19]. Non sorprende che mostri segni di epatotossicità solo in una minoranza di pazienti, nonostante gli alti dosaggi (100-150 mg al giorno) [24].
L’ipotesi di coniugazione dell’anello D:
Avete mai sfogliato il libro Doping in Sports di Thieme e Hemmersbach? [25] In questo libro gli autori notano che non c’è correlazione tra la tossicità epatica e gli effetti farmacologici primari (cioè gli effetti anabolizzanti) – il che è sufficientemente ovvio perché gli AAS non 17α-alchilati sono rapidamente metabolizzati nel fegato, quindi la loro concentrazione in loco non sarebbe come quella dei 17α-alchilati. Naturalmente, non si troverà una correlazione se si guarda solo a questo fattore. Bisogna anche prendere in considerazione la sua resistenza al metabolismo epatico come è stato fatto con l’ipotesi dello stress ossidativo descritta sopra.
In ogni caso, questo ha portato gli autori a formulare un’alternativa ipotesi di ciò che causa l’epatotossicità indotta da AAS. E sembrava essere l’unica. Essi suggeriscono che l’epatotossicità è probabilmente dovuta alla coniugazione dell’anello D con l’acido glucuronico. Questo processo è chiamato glucuronidazione ed è una cosiddetta comune reazione di fase 2 nel metabolismo del farmaco. Rende la molecola madre più solubile in acqua, facilitando così la sua escrezione nelle urine.
Il gruppo 17β-glucuronide (in blu) attaccato al anello D di uno steroide 17α-metilato (gruppo 17α-metilico in rosso).
È semplicemente l’attaccamento (coniugazione) dell’acido glucuronico alla molecola madre (vedi figura sopra). Quando il Testosterone con un gruppo 17β-glucuronide (così come diversi estrogeni con questa modifica) viene iniettato nel ratto, il flusso biliare è inibito [521]. Presumibilmente, perché questi composti condividono somiglianze strutturali con gli acidi biliari, questi composti competono con gli acidi biliari per legarsi a certi recettori. Tuttavia, a parte questo, non c’è molta sostanza per sostenere questa ipotesi come la ragione per l’epatotossicità indotta da AAS, soprattutto perché molti degli AAS non 17α-alchilati, compreso il Testosterone, subiscono la glucuronizzazione del loro gruppo 17β-idrossi. Eppure questi non sono sensibilmente epatotossici. Infatti, la 17βglucuronidazione è stata identificata solo per alcuni AAS 17α-alchilati, e sembra che essi subiscono questo processo solo in piccola misura [26]. Così, ironicamente, se questa ipotesi fosse vera, o significativa, ci si aspetterebbe l’epatotossicità con il Testosterone ma non con gli AAS 17α-alchilati.
Conclusioni sulle ipotesi esposte:
Non è sicuramente una novità per l’utilizzatore medio, ma anche per il semplice soggetto interessato all’argomento PEDs, che gli AAS metilati in C-17 (17α-alchilati) abbiano un effetto epatotossico con lievi variabili tra molecole aventi la stessa modifica strutturale. E non è nemmeno una rivelazione che la supplementazione con antiossidanti (vedi NAC e Silimarina) possa ridurre tale effetto. Di conseguenza, l’ipotesi dello stress ossidativo sembra essere la principale causa del epatotossicità AAS-indotta. Ma non l’unico fattore.
Nell’ultimo decennio si è aggiunto ai classici composti antiossidanti l’uso di acidi biliari come l’Acido Ursodesossicolico e l’Acido Tauroursodesossicolico assunti oralmente.
L’Acido Ursodesossicolico è un acido biliare secondario che deriva dal metabolismo dell’acido colico da parte del microbiota umano intestinale. Il suo nome deriva dal fatto che è il principale acido biliare negli orsi (dal latino ursus). In biologia e biochimica lo si etichetta con l’acronimo UDCA. Il nome completo del UDCA è Acido 3α,7β-diidrossi-5β-colanoico.[27]
Acido Ursodesossicolico (UDCA)
L’Acido Tauroursodesossicolico (TUDCA) è un acido biliare ambifilico. È la forma coniugata di Taurina ed il precedentemente citato Acido Ursodeossicolico (UDCA). Il nome completo del TUDCA è 2-{(4R)-4-[(1R,3aS,3bR,4S,5aS,7R,9aS,9bS,11aR)-4,7-Dihydroxy-9a,11a-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-1-yl]pentanamido} acido etan-1-sulfonico.[28]
Acido Tauroursodesossicolico (TUDCA)
l’UDCA è approvato per il trattamento della cirrosi biliare primaria.[1][2] Di conseguenza, l’Acido Ursodesossicolico (UDCA) ha mostrato effetti epatoprotettivi. Tuttavia, i suoi meccanismi molecolari sottostanti rimangono poco chiari. Per tale motivazione, sono stati condotti alcuni studi come quello di Da Jung Kim et al. nel quale è stato osservato l’effetto epatoprotettivo dell’UDCA e della vitamina E utilizzando la metabolomica e l’analisi metagenomica. In questo studio, sono stati analizzati campioni di sangue e urine di pazienti con obesità e disfunzione epatica. Nove pazienti sono stati assegnati in modo casuale a ricevere UDCA (300 mg due volte al giorno), e 10 soggetti hanno ricevuto la vitamina E (400 UI due volte al giorno) per 8 settimane. L’UDCA ha migliorato significativamente i punteggi della funzionalità epatica dopo 4 settimane di trattamento e ha ridotto efficacemente i livelli epatici di acido Desossicolico e di microRNA-122 nel siero. Per comprendere meglio il suo meccanismo protettivo, è stato condotto uno studio di metabolomica globale ed è stato scoperto che l’UDCA ha regolato le tossine uremiche (acido ippurico, solfato di p-cresolo e metaboliti derivati dall’indolo), gli antiossidanti (solfato di ascorbato e N-acetil-L-cisteina) e il percorso fenilalanina/tirosina. Inoltre, il coinvolgimento del microbioma, in particolare di Lactobacillus e Bifidobacterium, è stato dimostrato attraverso l’analisi metagenomica delle vescicole extracellulari derivate dai batteri. Nel frattempo, il trattamento con vitamina E non ha portato a tali alterazioni, tranne che ha ridotto le tossine uremiche e la disfunzione epatica. I nostri risultati hanno suggerito che entrambi i trattamenti erano efficaci nel migliorare la funzione epatica, anche se attraverso meccanismi diversi.
Schema dei potenziali meccanismi terapeutici del trattamento con UDCA. L’analisi metabolomica ha rivelato che l’UDCA riduce i principali composti nei percorsi fenilalanina/tirosina e triptofano, tra cui fenilalanina, fenilacetato, acetilfenilalanina, aldeide 3,4-idrossifenilacetato, dopamina-3-O-solfato, idrossibenzaldeide, p-cresolo solfato, idrossicynurenamina, idrossindolo e acido ippurico, nel plasma e nelle urine. I metaboliti intermedi degli aminoacidi aromatici come l’idrossimelatonina, l’acido benzoico e l’acido salicilico sono stati aumentati. I forti antiossidanti come l’ascorbato, l’acetiltriptofano e la N-acetil-L-cisteina erano elevati. Inoltre, la disintossicazione delle tossine uremiche tramite glucuronidazione (idrossimetossiindolo glucuronide e p-cresolo glucuronide) è stata osservata dopo il trattamento UDCA. Tuttavia, la vitamina E ha ridotto l’acido indolo-propionico, il solfato di indoxile, la 3-ketosphinganina e la sfingosina, che non sono stati regolati dall’UDCA. Il colore blu indica una diminuzione del livello del metabolita, e il colore rosso indica un aumento del livello del metabolita dopo il trattamento UDCA. I metaboliti che sono cambiati dopo il trattamento con vitamina E sono contrassegnati da un asterisco (*). I metaboliti che sono stati possibilmente regolati da modifiche batteriche sono contrassegnati da un colore viola.
Inoltre, si sa che l’UDCA a livello epatico stimola la secrezione di ATP da parte degli epatociti[29]; sebbene il significato di quest’azione non è ancora noto. Si sa però che interagisce col sistema dei citocromi P450 e che riduce la Glicuronazione degli estrogeni sintetici e non solo.[30] Vi ricorda qualcosa? Esatto! L’ipotesi di coniugazione dell’anello D e la sua potenzialità di essere parte dell’effetto epatotossico AAS-indotto! Se a ciò aggiungiamo che l’UDCA possiede la capacità di attivare direttamente il recettore per i glucocorticoidi, che contribuirebbe ad allargare i meccanismi della sua azione anticolestatica ed antinfiammatoria sul parenchima epatico [31], e che stimola la sintesi del glutatione (GSH), potente antiossidante endogeno, attraverso l’intervento delle chinasi dipendenti dai fosfoinositidi (PI-3K e PKB) [32], ciò fa si che l’UDCA risulti la chiave di volta nella protezione epatica durante l’uso di AAS con marcata resistenza al metabolismo epatico in abbinamento ai largamente utilizzati NAC (precursone ad alta biodisponibilità del Glutatione) e Silimarina.
Quanto detto non rappresenta ne un consiglio medico ne una scusa per abusare di AAS di qualsiasi tipo! Si tratta semplicemente della divulgazione di informazioni che la seria ricerca scientifica ha permesso di estrapolare, per il momento…
Gabriel Bellizzi
Riferimenti:
W. J. Meyer, A. Webb, C. A. Stuart, J. W. Finkelstein, B. Lawrence, and P. A. Walker. Physical and hormonal evaluation of transsexual patients: a longitudinal study. Archives of sexual behavior, 15(2):121–138, 1986.
A. M. Elsharkawy, S. McPherson, S. Masson, A. D. Burt, R. T. Dawson, and M. Hudson. Cholestasis secondary to anabolic steroid use in young men. Bmj, 344, 2012.
J. Nadell and J. Kosek. Peliosis hepatis. twelve cases associated with oral androgen therapy. Archives of pathology & laboratory medicine, 101(8):405–410, 1977.
F. L. Johnson, K. Lerner, M. Siegel, J. Feagler, P. Majerus, J. Hartmann, and E. D. Thomas. Association of androgenic-anabolic steroid therapy with development of hepatocellular carcinoma. The Lancet, 300(7790):1273–1276, 1972.
L. Hernandez-Nieto, M. Bruguera, J. A. Bombi, L. Camacho, and C. Rozman. Benign liver-cell adenom associated with long-term administration of an androgenic-anabolic steroid (methandienone). Cancer,40(4):1761–1764, 1977.
P. Bond, W. Llewellyn, and P. Van Mol. Anabolic androgenic steroid-induced hepatotoxicity. Medical Hypotheses, 93:150–153, 2016.
H. Sies et al. Oxidative stress: introductory remarks. Oxidative stress, 501:1–8, 1985.
K. Brieger, S. Schiavone, F. J. Miller Jr, and K.-H. Krause. Reactive oxygen species: from health to disease. Swiss medical weekly, 142:w13659, 2012.
S. P. Frankenfeld, L. P. Oliveira, V. H. Ortenzi, I. C. Rego-Monteiro, E. A. Chaves, A. C. Ferreira, A. C. Leitáo, D. P. Carvalho, and R. S. Fortunato. The anabolic androgenic steroid nandrolone decanoate disrupts redox homeostasis in liver, heart and kidney of male wistar rats. PloS one, 9(9):e102699, 2014.
J. H. Pinthus, I. Bryskin, J. Trachtenberg, J.-P. Luz, G. Singh, E. Fridman, and B. C. Wilson. Androgen induces adaptation to oxidative stress in prostate cancer: implications for treatment with radiation therapy. Neoplasia, 9(1):68–80, 2007.
H. Lin, J.-P. Lu, P. Laflamme, S. Qiao, B. Shayegan, I. Bryskin, L. Monardo, B. C. Wilson, G. Singh, and J. H. Pinthus. Inter-related in vitro effects of androgens, fatty acids and oxidative stress in prostate cancer: a mechanistic model supporting prevention strategies. International journal of oncology, 37(4):761–766, 2010.
M. Guzmán, A. Saborido, J. Castro, F. Molano, and A. Megias. Treatment with anabolic steroids increases the activity of the mitochondrial outer carnitine palmitoyltransferase in rat liver and fast-twitch muscle. Biochemical pharmacology, 41(5):833–835, 1991.
R. Gragera, A. Saborido, F. Molano, L. Jimenez, E. Muñiz, and A. Megias. Ultrastructural changes induced by anabolic steroids in liver of trained rats. Histology and histopathology, 1993.
X. Chapa-Dubocq, V. Makarov, and S. Javadov. Simple kinetic model of mitochondrial swelling in cardiac cells. Journal of cellular physiology, 233(7):5310–5321, 2018.
S. K. Powers, L. L. Ji, A. N. Kavazis, and M. J. Jackson. Reactive oxygen species: impact on skeletal muscle. Comprehensive Physiology, 1(2):941–969, 2011.
T. A. Pagonis, G. N. Koukoulis, C. S. Hadjichristodoulou, P. N. Toli, and N. V. Angelopoulos. Multivitamins and phospholipids complex protects the hepatic cells from androgenic-anabolic-steroids-induced toxicity. Clinical Toxicology, 46(1):57–66, 2008.
G. H. Marquardt, C. E. Logan, W. G. Tomhave, and R. M. Dowben. Failure of non-17-alkylated anabolic steroids to produce abnormal liver function tests. The Journal of Clinical Endocrinology & Metabolism, 24(12):1334–1336, 1964.
S. Bhasin, L. Woodhouse, R. Casaburi, A. B. Singh, D. Bhasin, N. Berman, X. Chen, K. E. Yarasheski, L. Magliano, C. Dzekov, et al. Testosterone dose-response relationships in healthy young men. American Journal of Physiology-Endocrinology And Metabolism, 281(6):E1172–E1181, 2001.
C. J. Houtman, S. S. Sterk, M. P. Van de Heijning, A. Brouwer, R. W. Stephany, B. Van der Burg, and E. Sonneveld. Detection of anabolic androgenic steroid abuse in doping control using mammalian reporter gene bioassays. Analytica chimica acta, 637(1-2):247–258, 2009.
C. Bonne and J.-P. Raynaud. Assay of androgen binding sites by exchange with methyltrienolone (r 1881). Steroids, 27(4):497–507, 1976.
H. L. Krüskemper and G. Noell. Liver toxicity of a new anabolic agent: methyltrienolone (17α-methyl-4, 9, 11-estratriene-17β-ol-3-one). Steroids, 8(1):13–24, 1966.
C. Grunfeld, D. P. Kotler, A. Dobs, M. Glesby, S. Bhasin, O. S. Group, et al. Oxandrolone in the treatment of hiv-associated weight loss in men: a randomized, double-blind, placebo-controlled study. JAIDS Journal of Acquired Immune Deficiency Syndromes, 41(3):304–314, 2006.
J. A. Kemppainen, E. Langley, C.-i. Wong, K. Bobseine, W. R. Kelce, and E. M. Wilson. Distinguishing androgen receptor agonists and antagonists: distinct mechanisms of activation by medroxyprogesterone acetate and dihydrotestosterone. Molecular Endocrinology, 13(3):440–454, 1999.
U. R. Hengge, K. Stocks, S. Faulkner, H. Wiehler, C. Lorenz, W. Jentzen, D. Hengge, and G. Ringham. Oxymetholone for the treatment of hiv-wasting: a double-blind, randomized, placebo-controlled phase iii trial in eugonadal men and women. HIV clinical trials, 4:150–163, 2003.
A. Sansone, F. Romanelli, M. Sansone, A. Lenzi, and L. Di Luigi. Gynecomastia and hormones. Endocrine, 55(1):37–44, 2017.
W. Schänzer. Metabolism of anabolic androgenic steroids. Clinical chemistry, 42(7):1001–1020, 1996.
Boatright, Jeffrey H.; Nickerson, John M.; Moring, Anisha G.; Pardue, Machelle T. (2009). “Bile acids in treatment of ocular disease”. Journal of Ocular Biology, Diseases, and Informatics. 2 (3): 149–159.
Nathanson MH et al. Stimulation of ATP secretion in the liver by therapeutic bile acids. Biochem J. 2001; 358(Pt 1):1-5.
Weitzel C et al. Ursodeoxycholic acid induced activation of the glucocorticoid receptor in primary rat hepatocytes. Eur J Gastroenterol Hepatol. 2005 Feb; 17(2):169-77.
Sanchez Pozzi EJ et al. Ursodeoxycholate reduces ethinylestradiol glucuronidation in the rat: role in prevention of estrogen-induced cholestasis. J Pharmacol Exp Ther. 2003 Jul; 306(1):279-86.
Arisawa S et al. Ursodeoxycholic acid induces glutathione synthesis through activation of PI3K/Akt pathway in HepG2 cells. Biochem Pharmacol. 2009 Mar 1;77(5):858-66.
Come di mia consuetudine, mi servirò della letteratura scientifica ad oggi disponibile per trattare nel modo più accurato ed esaustivo, rimanendo pur sempre comprensibile da chi non avvezzo alla biochimica e all’endocrinologia, il tema annoso della cadenza di somministrazione del GH.
Iniziamo subito andando ad esaminare la “genesi del dibattito” …
La genesi del dibattito in uno studio:
Il trattamento dei bambini con bassa statura idiopatica mediante iniezioni giornaliere di GH umano (hGH) è seguito, dopo la sua sospensione, da una decelerazione della crescita con livelli sierici normali di GH e IGF-I.
Il studio ivi riportato [1] è stato progettato per capire e prevenire la decelerazione della crescita. I ricercatori hanno ipotizzato che questo fenomeno sia dovuto alla tolleranza a livello dell’organo bersaglio, che la tolleranza si sviluppi in risposta alla farmacocinetica non fisiologica dell’hGH iniettato quotidianamente, e che la terapia con hGH a giorni alterni lo prevenga.
Trentotto bambini prepuberi con bassa statura idiopatica, di età 3.3-9.0 anni, sono stati esaminati. Le loro altezze erano meno di -2 SD score, il tasso di crescita era superiore al 10 ° percentile per l’età, l’età ossea era inferiore al 75% dell’età cronologica, e la concentrazione sierica stimolata di GH era maggiore di 10 μg/litro.
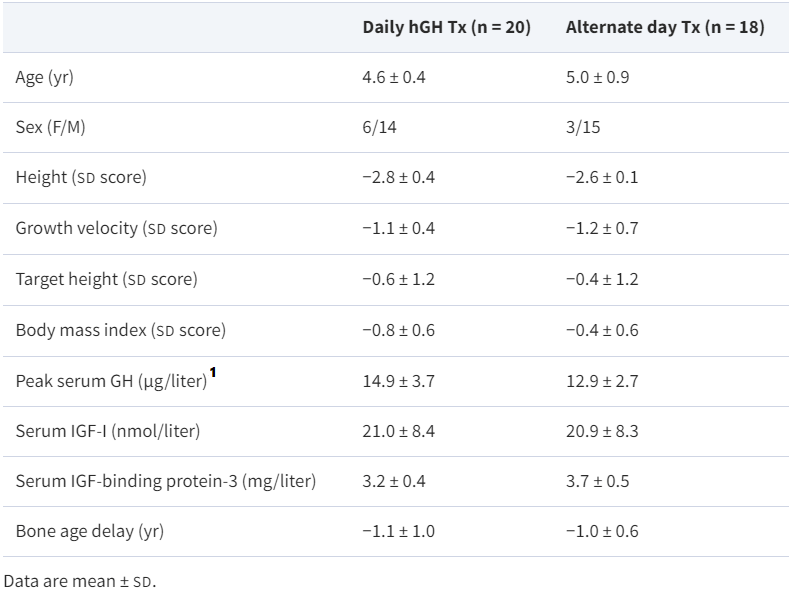
I bambini sono stati abbinati per sesso, altezza e punteggio SD della velocità di crescita per ricevere hGH giornaliero o a giorni alterni alla stessa dose settimanale di 6 mg/m2 per un periodo di 2 anni. Le velocità di crescita medie del 1° e 2° anno erano rispettivamente 3.4 e 2.3 SD score per il gruppo di terapia giornaliera e 3.0 e 2.0 SD score per il gruppo a giorni alterni (P = NS).
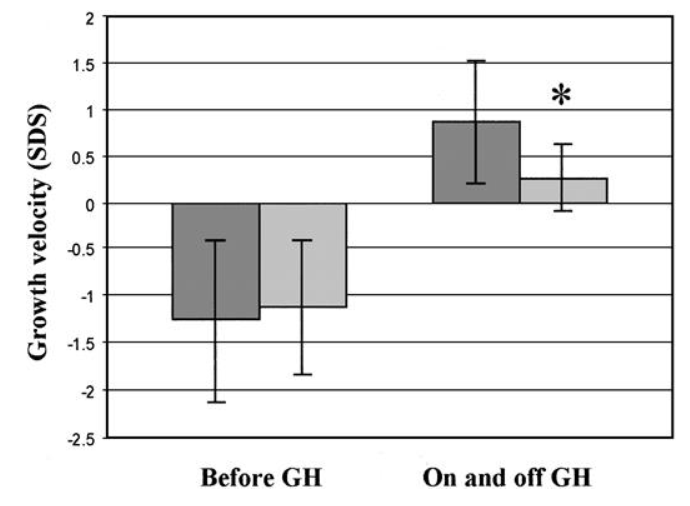
Velocità di crescita dei bambini trattati con GH a giorni alterni (▨) o con un regime giornaliero di GH prima, durante e 2 anni dopo l’interruzione della terapia. I valori sono la media ± SD. *, P < 0,05; **, P < 0,01.
Nei 6 mesi iniziali dopo la sospensione della terapia, la velocità di crescita è decelerata fino a un nadir di -3,9 SD score nel gruppo di terapia giornaliera, mentre è decelerata nel gruppo del giorno alternato a solo -0,2 SD score (P < 0,01).
Velocità di crescita pre-trattamento e cumulativa a 4 anni dei bambini trattati con GH a giorni alterni (▨) o con un regime giornaliero di GH . I valori sono la media ± SD. *, P < 0.002.
Durante tutti i 2 anni di interruzione della terapia, quest’ultimo gruppo ha mantenuto tassi di crescita medi da -0,2 a -1,2 SD score, simili alle loro velocità di pretrattamento. Il gruppo giornaliero ha recuperato lentamente per riprendere il loro tasso medio di pretrattamento solo alla quarta valutazione semestrale fuori dalla terapia.
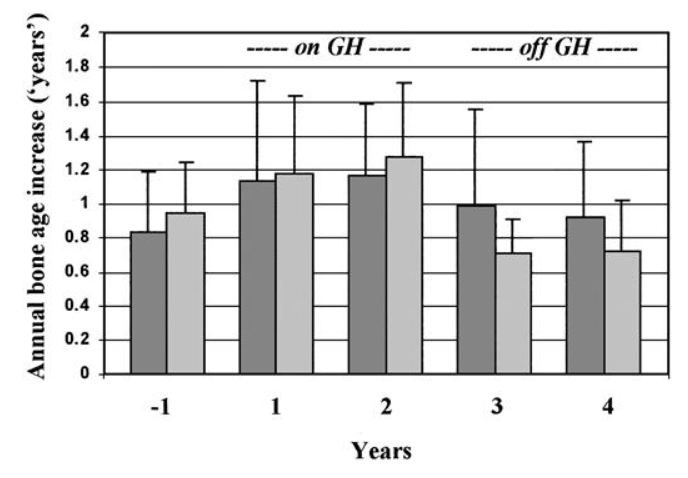
Avanzamento annuale della crescita ossea nei bambini trattati con GH a giorni alterni (▨) o con un regime giornaliero di GH prima, durante e 2 anni dopo l’interruzione della terapia. I valori sono la media ± SD.
La velocità di crescita cumulativa a 4 anni (2 anni con e 2 anni senza terapia) del gruppo a giorni alterni era maggiore di quella del gruppo a terapia giornaliera (media, 0,9 contro 0,3 SD score; P < 0,002). Alla fine del periodo di terapia di 4 anni, la previsione di altezza da adulto del gruppo a giorni alterni era maggiore di quella del gruppo giornaliero di una media di 6,5 cm (P = 0,06).
Punteggio SD dell’altezza dei bambini trattati con GH a giorni alterni (▨) o con un regime giornaliero di GH prima, durante e 2 anni dopo l’interruzione della terapia. I valori sono la media ± SD. *, P < 0,05; **, P < 0,01. Caratteristiche cliniche di 20 pazienti che hanno ricevuto iniezioni giornaliere di hGH, rispetto a 18 pazienti che hanno ricevuto una terapia di GH a giorni alterni a una dose settimanale identica per metro quadrato di superficie corporea. 1= Test di stimolazione dell’Arginina.
Discussione oggettiva sui dati appresi:
Si tratta senza dubbio di uno studio molto approfondito e ben controllato, durato quattro anni e pubblicato sul The Journal of Clinical Endocrinology & Metabolism. Esso mostra chiaramente che le iniezioni di hGH a giorni alterni (EOD) sono molto più vantaggiose a lungo termine delle iniezioni quotidiane.
Le iniezioni quotidiane sembrano abbassare drasticamente la sensibilità del corpo alla propria secrezione di GH, e al GH esogeno. Lo studio comprendeva bambini con bassa statura idiopatica, ma i risultati possono essere estrapolati e trasposti, almeno in buona parte, a soggetti in fisiologia, e cioè non carenti di hGH e che possono utilizzare hGH esogeno periodicamente per Anti-Aging e Bodybuilding, per esempio.
Come abbiamo visto, i 38 bambini sono stati divisi in due gruppi:
Gruppo I: ha ricevuto iniezioni giornaliere di hGH;
Gruppo II: ha ricevuto iniezioni di hGH a giorni alterni.
È importante notare che il dosaggio settimanale totale di hGH era lo stesso per entrambi i gruppi. Entrambi i gruppi hanno ricevuto la terapia di hGH in modo contiguo per due anni. La loro crescita naturale è stata seguita per altri due anni dopo la fine della terapia hGH.
Sono stati tutti misurati a intervalli di tre mesi durante il periodo di quattro anni – due anni con la terapia di hGH e due anni dopo. Il GH sierico è stato misurato con un kit RIA a doppio anticorpo.
Durante la terapia con hGH, entrambi i gruppi hanno accelerato la loro crescita in modo sostanziale:
Gruppo I: ricevendo le iniezioni giornaliere di hGH nel primo e secondo anno la velocità di crescita era di 3.4 e 2.3 SD;
Gruppo II: ricevendo le iniezioni di hGH a giorni alterni aveva un tasso nella velocità di crescita di 3.0 e 2.0 SD per il primo e il secondo anno, rispettivamente.
Nel corso dei sei mesi iniziali dopo il termine della terapia, la velocità di crescita è decelerata ad un basso nadir pari a -3.9 SD di punteggio per il gruppo di terapia a somministrazione giornaliera, mentre è decelerato nel gruppo di terapia a giorni alterni di solo -0.2 SD di punteggio.
Durante i 2 anni seguenti la fine della terapia, quest’ultimo gruppo al quale sono state somministrate iniezioni EOD ha mantenuto tassi di crescita da -0.2 a -1.2 di punteggio SD, che è simile al loro punteggio SD prima del trattamento con hGH esogeno. Il gruppo giornaliero ha anch’esso mostrato un recuperato, seppur molto lentamente, alla quarta valutazione semestrale dopo la conclusione della terapia. La velocità di crescita cumulativa di 4 anni – 2 anni con e 2 anni senza terapia – del gruppo a giorni alterni era maggiore di quella del gruppo con terapia giornaliera: media, 0.9 contro 0.3 SD score.
Alla fine del periodo di terapia di 4 anni, la previsione dell’altezza adulta del gruppo a giorni alterni era maggiore di quella del gruppo giornaliero di una media di 6,5 cm – che è più di 2,5“ in altezza.
Per dirlo il più semplicemente possibile, per tradurre ciò che può significare tutto ciò per un bodybuilder, l’uso giornaliero di hGH darà solo trascurabilmente migliori risultati a breve termine. Tuttavia, l’uso di hGH a giorni alterni darà risultati radicalmente migliori a lungo termine e un recupero molto migliore. Ciò significa che il corpo può tornare all’omeostasi molto più velocemente.
I due gruppi hanno ottenuto lo stesso dosaggio settimanale totale di hGH, così che il gruppo “EOD” è stato trattato con iniezioni che comprendevano il totale del giorno successivo (es. 4UI/die e 8UI/EOD), ovvero il doppio di UI del gruppo trattato ogni giorno, ma con un totale settimanale identico! I ricercatori hanno riportato che la dose era di minore importanza rispetto al programma delle iniezioni. La terapia di hGH quotidiana per 3 anni ha causato una crescita subnormale che persiste per 1,5 anni (molto male).
Può essere che il problema non sia legato tanto ai livelli di secrezione di hGH o IGF-1, ma piuttosto alla diminuita sensibilità del corpo ad esso. La parte interessante è che i livelli sierici di GH e i livelli sierici di IGF-I e IGF-binding protein sono rimasti inalterati, o relativamente mutati.
La secrezione endogena di GH del corpo riprende in pochi giorni, anche dopo una terapia di hGH a lungo termine.
L’ipotesi dei ricercatori è che la tolleranza può essere insita nella trasduzione del segnale del GH in organi bersaglio selettivi in risposta alla scomparsa del modello unico pulsatile di GH sierico durante la terapia con GH esogeno. Ciò è dovuto al fatto che il GH assunto tramite iniezioni SubQ (sottocutanea) non corrisponde alla pulsatilità di rilascio del GH del corpo.
Pulsatilità circadiana del GH negli uomini (in altro) e nelle donne (in basso).
La somministrazione giornaliera SubQ di GH si traduce in un profilo di GH sierico non fisiologico, con livelli di picco a 3-4 ore e un lento declino nel corso delle successive 12-24 ore. Questo modello può essere considerato come una somministrazione continua, piuttosto che i naturali impulsi di GH fisiologici del corpo con una frequenza di circa otto impulsi al giorno.
Farmacocinetica GH esogeno somministrato per via parenterale sottocutanea.
Supponendo che la sindrome da astinenza sia legata alla tolleranza che potrebbe essersi sviluppata verso l’hGH o l’IGF-I, si è cercato di prevenirla con un trattamento a giorni alterni. Inoltre, le dosi di hGH utilizzate in terapia spesso stimolano l’IGF-I a livelli sierici sovrafisiologici, suggerendo che i tessuti bersaglio del IGF-I possono ovviamente essere sovrastimolati rispetto al normale. Il meccanismo sembra, quindi, risiedere nell’azione del hGH e del IGF-I nei confronti di loro tessuti bersaglio. E’ stato dimostrata, fino a prova contraria, quindi, che la terapia a giorni alterni con hGH nei bambini con un asse GH-IGF-I intatto impedisce la sindrome da astinenza.
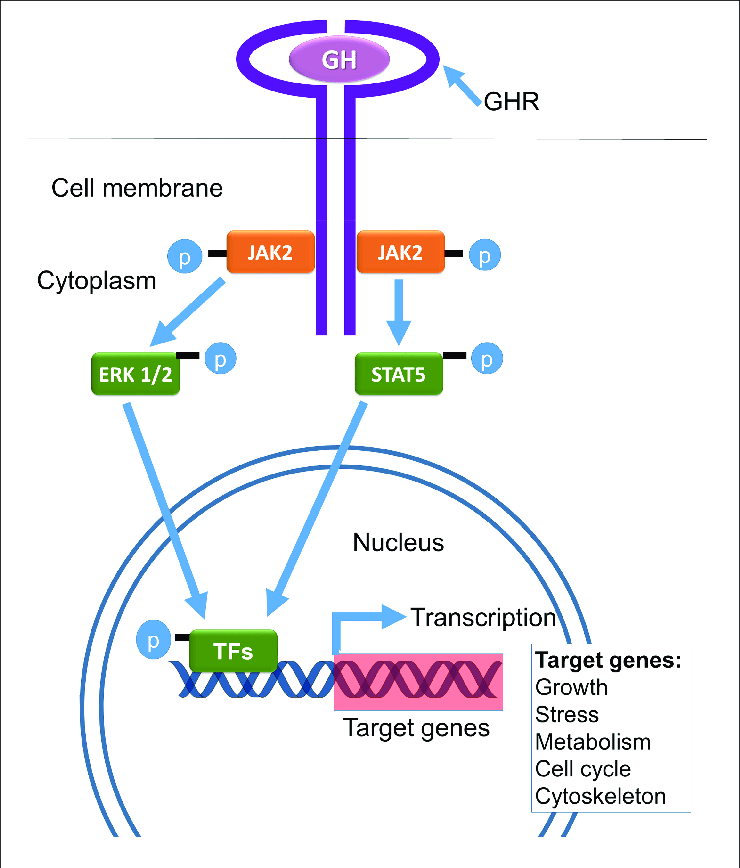
Legame GH-GHR (Recettore del GH) e seguenti pathways.
I ricercatori collegano l’analogia con un’altra sindrome di tolleranza e astinenza endocrina: “la terapia a giorni alterni con glucocorticosteroidi previene la tolleranza a quell’ormone in misura sostanziale. È interessante notare che la sindrome da astinenza da glucocorticoidi può verificarsi anche mentre l’asse ipotalamo-ipofisi-surrene è intatto, indicando che la tolleranza ai glucocorticoidi si è sviluppata a livello dell’organo bersaglio”.
Conclusioni:
Adesso sappiamo che le iniezioni giornaliere di GH abbassano drasticamente la sensibilità del corpo all’attività dell’ormone a livello dei tessuti bersaglio, sia durante l’uso di GH esogeno sia post utilizzo (bassa risposta ai propri impulsi di GH endogeno).
Come abbiamo potuto constatare, la desensibilizzazione si è verificata, a parità di dosaggio settimanale, in risposta alla somministrazione quotidiana, a differenza del protocollo EOD.
Lo stesso GH ha una breve emivita quando viene iniettato per via endovenosa, la via di somministrazione ottimale, ma l’iniezione IM o subQ porta a un rilascio lento e prolungato e a un’elevazione al di sopra dei livelli basali per 12-24 ore, che comporta una stimolazione cronica dei recettori. Questo porta a una drammatica desensibilizzazione del tessuto bersaglio che può persiste per un lungo periodi di tempo.
Per maggiori benefici, la somministrazione di hGH in ambito Bodybuilding, che sia per la crescita muscolare, la lipolisi e l’antiaging dovrebbe aderire al dosaggio a giorni alterni per massimizzare i risultati e prevenire la tolleranza nei recettori dei tessuti bersaglio. Il dosaggio EOD per ridurre la tolleranza – mantenendo una maggiore sensibilità sia all’HGH esogeno che alla produzione endogena del corpo – ha dimostrato di produrre risultati a lungo termine molto migliori rispetto alla somministrazione quotidiana.
Repetita iuvant: La somministrazione EOD mantiene una maggiore sensibilità sia all’HGH esogeno che alla produzione endogena dell’organismo post utilizzo rispetto alle iniezioni quotidiane, mentre il dosaggio settimanale rimane lo stesso.
Praticamente, il doppio dosaggio di HGH dovrebbe essere somministrato in un giorno con un intervallo di circa 8 ore. Ad esempio al mattino e alla sera e il giorno successivo dovrebbe essere omesso, e così via. Questa somministrazione previene la tolleranza nei recettori del GH e massimizza i risultati a lungo termine. Si prega di notare che il dosaggio settimanale rimane lo stesso.
Un esempio di somministrazione “EOD” potrebbe essere il seguente:
L’hGH assunto per 12-16 settimane o più a 8 UI ogni due giorni, diviso in 4 UI a digiuno subito dopo il risveglio e altre 4 UI prese otto ore dopo. Questo approccio è abbastanza conservativo e può essere ottimale. La dose può essere ulteriormente suddivisa, se lo si desidera, per ridurre il totale delle UI iniettate in qualsiasi momento (es. 2UI appena sveglio – 2UI pre-workout – 2UI 4h dopo – 2UI prima di andare a dormire).
Ovviamente, si può estendere oltre i quattro mesi, e prendere più UI al giorno. L’approccio sopra esposto è di 8UI EOD, quindi è equivalente ad una assunzione giornaliera di 4UI, che è la media utilizzata dalla maggior parte degli utilizzatori di PEDs.
Bisogna però mettere da parte gli assolutismi, dal momento che lo studio in questione ha preso in considerazione l’altezza negli adolescenti, non la massa magra in culturisti adulti, o gli effetti Anti-Aging in adulti di mezza età, quindi è ancora una questione di sperimentazione sul campo ed estrapolazione se i risultati possono essere applicati a questi sottogruppi di utilizzatori. Comunque sia, è vero che i bodybuilder non sono bambini, né carenti di hGH idiopatico, ma la risposta sottoregolativa dei recettori del GH sono una possibilità. Vi ricordo che la “GH resistenza” esiste.
Poiché i dosaggi settimanali rimangono gli stessi, così come la durata dell’uso di hGH, il solo cambiamento del protocollo “die” a quello “EOD” varrebbe la pena di essere testato, dato che sembra statisticamente una pratica migliore rispetto al protocollo ordinario/giornaliero.
Vorrei concludere con il rendere noto che “l’ho usato tutti i giorni per mesi e mi sono tirato!” è una affermazione vuota di significato reale e realmente applicabile al discorso qui trattato: vantaggio di una somministrazione a giorni alterni di GH! Oltretutto, caro il mio bongo, dubito fortemente che tu stessi utilizzando solo GH, e che le altre molecole da te cosomministrate non abbiano avuto, a diverso grado, un impatto sulla massa grassa! Inoltre, dato ciò, non puoi affermare né uno svantaggio né una parità d’effetto delle due metodiche di cadenza nella somministrazione… a meno che tu non abbia testato tale pratica su un numero sufficiente di persone, dividendole in due gruppi trattati con una o l’altra modalità e, con la minore presenza possibile di bias, tu abbia potuto valutare oggettivamente i risultati…
OPK-88004 è un nuovo SARM non steroideo sviluppato dalla Transition Therapeutics, e che è stato acquistato dalla OKPO nel 2016.
Struttura molecolare del OPK-88004
Uno studio di recente pubblicazione svolto su questo SARM sembra aver mostrato che causa un aumento dose-dipendente della massa muscolare, che diminuisce la massa grassa e aumenta anche la quantità di Testosterone libero. Prima di eccitarvi troppo sull’ultimo punto, vediamo nel dettaglio lo studio. Caratteristiche dello studio:
In questo studio controllato con placebo, randomizzato, in doppio cieco, 114 uomini, di età ≥19 anni, che avevano subito una prostatectomia radicale per un cancro alla prostata di basso grado e localizzato all’organo, PSA non rilevabile (<0,1 ng/mL) per ≥2 anni dopo la prostatectomia radicale e carenza di Testosterone sono stati randomizzati per gradi a placebo [0mg] o 1, 5, o 15mg/die di OPK-88004 per 12 settimane. I risultati includevano la recidiva del PSA, l’attività sessuale, il desiderio sessuale, la funzione erettile, la composizione corporea, la forza muscolare e le misure della funzione fisica, l’umore, la fatica e i marker ossei.
Risultati dello studio:
I partecipanti avevano un’età media di 67,5 anni e una grave disfunzione sessuale (punteggi medi della funzione erettile e del dominio del desiderio sessuale 7,3 e 14,6, rispettivamente). Nessun partecipante ha avuto recidive di PSA o eritrocitosi. OPK-88004 è stato associato a un aumento correlato alla dose della massa magra [non specificatamente muscolare] (P <0,001) e appendicolare (P <0,001) e a una diminuzione significativamente maggiore della percentuale di grasso corporeo (P <0,001) e della fosfatasi alcalina nel siero (P <0,001) rispetto al placebo. I cambiamenti nell’attività sessuale, il desiderio sessuale, la funzione erettile, l’umore, l’affaticamento, le prestazioni fisiche e i marker ossei non differiscono tra i gruppi (P = 0,73).
Risultati dei test per valutare il miglioramento delle prestazioni fisiche.Variabili ormonali riscontrate durante lo studio.
Conclusioni sul OPK-88004:
La somministrazione di OPK-88004 è stata sicura e non è stata associata alla recidiva del PSA in uomini con deficit di androgeni che erano stati sottoposti a prostatectomia radicale per cancro alla prostata confinato all’organo. OPK-88004 ha aumentato la massa corporea magra e diminuito la massa grassa, ma non ha migliorato i sintomi sessuali o le prestazioni fisiche.
In conseguenza dei dati estrapolati dallo studio ivi esposto, e nonostante i dati di sicurezza a breve termine siano rassicuranti, questo SARM non steroideo mostra i difetti dei suoi predecessori:
Nonostante vi sia un aumento del Testosterone libero che potrebbe interessare maggiormente il pubblico rispetto al Testosterone totale, non bisogna dimenticarsi del fatto che gli Androgeni possono interagire con le attività cellulari anche attraverso interazioni non genomiche (non mediate direttamente dal recettore androgeno), le quali avvengono anche con l’ormone legato all’albumina (trasportatore ematico che lega l’ormone sessuale; pari circa al 55-35% del Testosterone). Inoltre, anche SARM non steroidei “datati” come l’Ostarina hanno mostrato le medesime caratteristiche sui livelli di Testosterone.
La diminuzione dell’Estradiolo (E2) può causare, in misura dipendente dall’entità del calo e dalla sensibilità individuale, molteplici problemi come depressione, letargia, affaticabilità, ansia, o disfuzione erettile (o difficolta a raggiungere l’erezione e/o a mantenerla) e riduzione della libido.
Problemi legati ai precedenti sono riscontrabili dalla riduzione del DHT, in maniera sempre dipendente dall’entità del calo e dalla sensibilità individuale. A tal proposito si veda la Testosterone:Estradiolo ratio o la più approfondita DHT:Estradiolo ratio.
Non è un caso che il trattamento con il suddetto SARM non abbia portato ad un aumento della funzione sessuale.
La mancanza di miglioramento nelle prestazioni fisiche e dei marker ossei, non lo rende molto allettante.
L’unico punto interessante rimane la riduzione della massa grassa, essendo l’aumento della massa magra estremamente generico e non inducibile a specifiche miotrofiche. Nonostante ciò, i possibili svantaggi superano di netto il suddetto vantaggio che, oltretutto, è riscontrabile in maniera accentuata in un “vecchio” SARM non steroideo, L’Andarina (S4).
Ora, come si può vedere, analizzando con logica tutti i dati in nostro possesso, possiamo valutare concretamente questo nuovo SARM non steroideo e rilegarlo in una posizione di basso interesse sia per uso terapeutico che “off-label”. Ma la ricerca effettuata non è letteralmente da buttare. I dati raccolti devono spingere la ricerca a migliorare queste molecole, sviluppando nuovi SARM che migliorino non solo il trofismo muscolo-scheletrico ma che portino ad una ottimale funzione sessuale e delle prestazioni psicofisiche.
L’Anamorelina (INN) (nomi in codice di sviluppo ONO-7643, RC-1291, ST-1291), nota anche come Anamorelina Cloridrato (USAN, JAN), è un agonista selettivo non peptidico, attivo per via orale, penetrante-centrale, selettivo del recettore della grelina / un secretagogo dell’Ormone della Crescita (GHSR) con effetti anabolizzanti e di stimolo dell’appetito, sviluppato dalla Helsinn Healthcare SA per il trattamento della cachessia e dell’anoressia da cancro.[2][3][4]
Struttura molecolare della Anamorelina
L’Anamorelina aumenta significativamente i livelli plasmatici dell’Ormone della Crescita (GH), del Fattore di Crescita Insulino-Simile 1 (IGF-1) e della proteina legante il Fattore di Crescita Insulino-Simile 3 (IGFBP-3) nell’uomo, senza influenzare i livelli plasmatici di Prolattina, Cortisolo, Insulina, Glucosio (in acuto), Ormone Adrenocorticotropo (ACTH), Ormone Luteinizzante (LH), Ormone Follicolo-Stimolante (FSH), e dell’Ormone Stimolante la Tiroide (TSH). [3][5] Inoltre, l’Anamorelina aumenta significativamente l’appetito, il peso corporeo complessivo, la massa magra e la forza muscolare,[4][5] con aumenti di peso corporeo direttamente correlati ad aumenti dei livelli plasmatici di IGF-1.[3]
Vie di azione della Anamorelina (e del Ibutamoren).
Un interessante studio sulla Anamorelina del 2018:
Oncologi giapponesi, affiliati all’Institute of Biomedical Research and Innovation, hanno pubblicato uno studio sulla rivista scientifica “Cancer” nel 2018. In questo studio, i ricercatori hanno diviso 174 persone con cancro al polmone avanzato in 2 gruppi.
Un gruppo ha ricevuto un placebo per 12 settimane, l’altro ha assunto giornalmente 2 compresse contenente ognuna 50mg di Anamorelina. I soggetti hanno quindi assunto 100mg di Anamorelina al giorno.
Rispetto al gruppo placebo, la somministrazione di Anamorelina ha portato a un aumento della massa corporea magra di poco meno di 1.5Kg. Anche se la sperimentazione è durata 12 settimane, questo aumento si è verificato completamente durante le prime 3 settimane dello studio. Una classica risposta da adattamento omeostatico.
Si può vedere nella figura sopra il perché l’Anamorelina perde il suo effetto dopo 3 settimane. Durante le prime 3 settimane di prova, le concentrazioni sia di IGF-1 che di IGF-BP3 nel sangue erano aumentate, dopo di che si è verificato un calo. La IGF-BP3 è una proteina legante che, per l’appunto, lega l’IGF-1 proteggendolo dalla degradazione enzimatica, ma non ne riduce l’attività biologica sulla matrice ossea e il muscolo scheletrico.
Un altro studio del 2019 dimostra la riduzione d’efficacia dopo 3 settimane:
Naturalmente bisogna sempre stare attenti a prendere per affidabili le affermazioni riportate in un singolo studio. Ma, seppur non rappresenti un enorme differenza, esiste un altro studio, in qualche misura comparabile con il precedente, ed è sempre uno studio giapponese, condotto tra persone con cancro colorettale, allo stomaco o al pancreas in stadio avanzato, dove la somministrazione di 100mg di Anamorelina ha stimolato il guadagno di massa magra solo per le prime 3 settimane.
Stato legale della Amorelina:
Fino a poco tempo, l’Anamorelina faceva parte della corposa compagine di molecole sperimentali (vedi SARM-non steroidei, SPPARM, ecc…) venduti dal mercato “grigio” dei siti UK.
Infatti, mentre le ricerche sul Ibutamoren sono state interrotte, quelle sulla Anamorelina, d’altra parte, sono proseguite ed essa è stata approvato per uso medico, per lo meno in Giappone.
L’Anamorelina è disponibile nel paese del sol levante sotto il nome di Adlumiz dall’aprile 2021. Il farmaco è destinato al trattamento di persone che stanno perdendo massa muscolare e forza a causa di una condizione di cancro avanzato.
Confezione di Adlumiz venduta in Giappone
A partire dal febbraio del 2016, l’Anamorelina ha completato gli studi clinici di fase III per il trattamento della cachessia e dell’anoressia associate al carcinoma polmonare non a piccole cellule.[6][7]
Il 18 maggio 2017, l’Agenzia europea per i medicinali ha raccomandato il rifiuto dell’autorizzazione all’immissione in commercio del medicinale, destinato al trattamento dell’anoressia, della cachessia o della perdita di peso involontaria in pazienti con carcinoma polmonare non a piccole cellule. La Helsinn ha chiesto un riesame del parere iniziale. Dopo aver considerato le motivazioni di questa richiesta, l’Agenzia europea per i medicinali ha riesaminato il parere, e ha confermato il rifiuto dell’autorizzazione all’immissione in commercio il 14 settembre 2017.[8] L’Agenzia europea per i medicinali ha concluso che gli studi mostrano un effetto marginale dell’Anamorelina sulla massa corporea magra e nessun effetto dimostrato sulla forza della presa delle mani o sulla qualità della vita dei pazienti. Inoltre, a seguito di un’ispezione presso i siti degli studi clinici, l’agenzia ha ritenuto che i dati sulla sicurezza del farmaco non fossero stati registrati adeguatamente. Pertanto, l’agenzia era del parere che i benefici dell’Anamorelina non superassero i suoi rischi.[9]
Conclusioni:
Nonostante la sua autorizzazione in Giappone per l’uso nel trattamento di pazienti umani affetti da cancro in stato avanzato, gli studi non mostrano reali vantaggi terapeutici dal momento che l’effetto del farmaco sembra limitato a tre settimane di uso giornaliero.
Anche se alcuni bodybuilder hanno utilizzato la molecola come sostituto del Ibutamoren, quest’ultimo sembra dimostrare un efficacia prolungata rispetto alla Anamorelina.
Ricordo, inoltre, che secondo legislazione italiana, anche l’acquisto e il possesso di SARM-non steroidei, SPPARM (vedi Cardarina) ed altri è illegale soggetto a sequestro e ammenda dipendenti dalla quantità di materiale in possesso.
Currow DC, Abernethy AP (April 2014). “Anamorelin hydrochloride in the treatment of cancer anorexia-cachexia syndrome”. Future Oncology. 10 (5): 789–802.
Garcia JM, Polvino WJ (June 2009). “Pharmacodynamic hormonal effects of anamorelin, a novel oral ghrelin mimetic and growth hormone secretagogue in healthy volunteers”. Growth Hormone & IGF Research. 19 (3): 267–73.
Garcia JM, Boccia RV, Graham CD, Yan Y, Duus EM, Allen S, Friend J (January 2015). “Anamorelin for patients with cancer cachexia: an integrated analysis of two phase 2, randomised, placebo-controlled, double-blind trials”. The Lancet. Oncology.
Garcia JM, Friend J, Allen S (January 2013). “Therapeutic potential of anamorelin, a novel, oral ghrelin mimetic, in patients with cancer-related cachexia: a multicenter, randomized, double-blind, crossover, pilot study”. Supportive Care in Cancer. 21 (1): 129–37.
Temel JS, Abernethy AP, Currow DC, Friend J, Duus EM, Yan Y, Fearon KC (April 2016). “Anamorelin in patients with non-small-cell lung cancer and cachexia (ROMANA 1 and ROMANA 2): results from two randomised, double-blind, phase 3 trials”. The Lancet. Oncology. 17 (4): 519–531.
Con il nuovo anno riprendo la pubblicazione degli articoli e lo faccio trattando un argomento che spesso, direttamente o indirettamente, è emerso nelle discussioni tra clienti e colleghi.
Il Testosterone è senza dubbio l’ormone simbolo per l’uomo della strada, preso dalla frenesia del mondo moderno e dal raggiungimento di obbiettivi tanto futili quanto irrealistici. Lo so che ve lo state domandando e la risposta è “si”. Questa entrata filosofica è perfetta per introdurre una questione legata al Androgeno per eccellenza.
Tanto per fare un esempio: quanti rimedi da banco vi sono stati proposti per migliorare i livelli plasmatici di Testosterone? Tra Tribulus Terrestris, Maca e Boro il conto è presto perso. E quanti di questi supplementi OTC hanno dato reali risultati? Misurabili, quantificabili con i livelli di partenza e che si sono tradotti in significativi miglioramenti della composizione corporea? …
La necessità di un ottimale apporto di Zinco, Vitamina D e altri macro e microelementi implicati nella biosintesi androgena, nella Testosterone:Estradiolo ratio ecc… non sono di certo messi in dubbio. Ad esserlo è il marketing, è l’affermazione sensazionalistica che va sempre con cura soppesata e valutata in concreto.
Ma, ipotizzando un miglioramento dei livelli di Testosterone endogeno rispetto al basale di partenza, ed entro l’intervallo di riferimento standard (es. per gli uomini dai 240 ai 950ng/dl dopo i 18 anni), garantite da trattamenti iatrogeni, quanto può incidere ciò nel miglioramento della composizione corporea?…
L’articolo che segue si basa sulle informazioni raccolte nella Research Review di James Krieger.
Iniziamo dalla letteratura scientifica
È assodato che l’uso AAS, che comporta la somministrazione di dosi sovrafisiologiche di Testosterone o ormoni correlati, provoca marcati aumenti delle dimensioni muscolari, ben oltre ciò che può essere ottenuto di base fisiologica dal soggetto, anche se questi guadagni addizionali sono limitati geneticamente. Nonostante ciò, mentre è assodato che dosi sovrafisiologiche di Testosterone, suoi derivati e analoghi aumenteranno significativamente i potenziali guadagni ipertrofici, questo non ci dice se le variazioni del Testosterone all’interno di un normale intervallo fisiologico possano avere qualche impatto. C’è una vasta gamma di livelli ematici di Testosterone da un uomo all’altro. Ad esempio, in uno studio nel quale sono stati presi in esame 456 uomini sani e non obesi di età compresa tra 19 e 39 anni, l’intervallo delle concentrazioni di Testosterone nel sangue (misurato al mattino dopo un digiuno notturno) era il seguente:
Lo studio di cui sopra è stato eseguito su un campione di individui della Framingham Heart Study Generation 3. Tuttavia, gli intervalli di concentrazione di Testosterone possono variare a seconda della popolazione e del dosaggio utilizzato per misurare il Testosterone. Anche i laboratori variano molto nei loro intervalli di riferimento. Travison et al. hanno estrapolato i dati da quattro importanti studi di coorte e hanno utilizzato modelli statistici per stabilire intervalli di riferimento che potrebbero essere applicati in diversi laboratori. Ecco la gamma di concentrazioni di Testosterone nel sangue che hanno stabilito:
Indipendentemente dall’intervallo di riferimento utilizzato, non c’è dubbio che vi sia un’ampia variazione nei livelli di Testosterone tra gli uomini, anche tra gli uomini sani e non obesi (poiché, come ben sappiamo, l’obesità è associata a un livello di Testosterone inferiore). Ciò solleva la questione se le variazioni in un intervallo normale possano influire sensibilmente sui guadagni muscolari. Un uomo con livelli di Testosterone naturalmente più alti riesce ad avere un maggior margine ipertrofico muscolare rispetto ad un uomo con livelli più bassi, anche se entrambi gli uomini sono all’interno di un intervallo normale? La risposta a questa domanda può avere particolare rilevanza per gli uomini che invecchiano. Il Testosterone diminuisce con l’età, ed è un altro dato di fatto, sebbene l’attività contro resistenza e una alimentazione sana possono rallentarne il declino. Ad esempio, ecco i dati dello studio French Telecom, che mostra il calo del Testosterone in tutti i percentili con l’età degli uomini.
Percentili di distribuzione plasmatica del Testosterone in un campione di 1.408 uomini caucasici dello studio Telecom, Parigi, Francia, 1985-1987
Ancora una volta, si può vedere l’ampia variazione nei livelli fisiologici di Testosterone, che vanno da 350-400ng/dL nel 5° percentile 850-1000ng/dL nel 95° percentile. Il declino continua negli anni ’60, ’70 e oltre. Ecco i dati che mostrano i livelli medi di Testosterone nei decenni di durata della vita; questi dati sono tratti da sei studi:
Testosterone totale (ng/ml) per fascia di età (moltiplicare per 100 per ottenere ng/dL); dati da 6 diversi studi.
Poiché anche la massa muscolare diminuisce con l’età e poiché gli uomini con bassi livelli di Testosterone mostrano tassi di perdita muscolare più rapidi rispetto agli uomini con livelli più alti, potremmo ipotizzare che gli uomini più anziani potrebbero trarre beneficio dal portare il Testosterone nell’intervallo fisiologico medio-alto.
Pertanto, tutti questi dati sollevano una serie di domande importanti:
I livelli di Testosterone di base sono correlati alla risposta all’allenamento?
Il Testosterone estremamente basso compromette la massa muscolare e i guadagni muscolari?
Le variazioni all’interno del normale range fisiologico influiscono sulla massa muscolare?
Se i livelli di Testosterone sono bassi o al limite, portare i livelli fino alla fascia media o superiore aiuta a migliorare la massa muscolare?
Se le variazioni nel normale range fisiologico hanno un impatto sulle condizioni muscolari negli uomini, hanno lo stesso impatto anche nelle donne?
Diamo un’occhiata alla ricerca per poter cercare di dare una risposta a queste domande.
I livelli basali di Testosterone sono correlati con la risposta all’allenamento?
Un modo per esaminare se esiste una relazione tra Testosterone in range fisiologico e guadagni muscolari è quello di guardare le risposte all’allenamento di un insieme di individui e vedere se i livelli di Testosterone di base sono correlati alla quantità di muscoli guadagnata da ciascuna persona. McCall et al. non hanno trovato alcuna correlazione tra i livelli basali di Testosterone e i cambiamenti nella dimensione muscolare in giovani uomini allenati a livello amatoriale. Tuttavia, Ahtiainen et al. hanno trovato una forte correlazione tra i livelli di Testosterone di base e il miglioramento della forza isometrica massima in 21 settimane. Ma la correlazione con l’ipertrofia non è stata affrontata.
Data la relazione tra l’ipertrofia e l’espressione di forza isometrica, potremmo ipotizzare che ci fosse una relazione tra il Testosterone di base e l’ipertrofia in questo studio, ma non è possibile saperlo con certezza.
In uno studio di Bhasin et al., uomini con infezione da HIV con Testosterone basso (<349ng/dL) sono stati assegnati in modo casuale a gruppo placebo, solo allenamento contro-resistenza, solo iniezioni di Testosterone o Testosterone e allenamento contro-resistenza combinati.
I livelli di Testosterone al basale non erano correlati con la variazione della massa magra (FFM) e non c’erano differenze significative nel guadagno assoluto di FFM tra uomini che avevano livelli di Testosterone <275ng/dL e uomini che avevano livelli di 275-350ng/dL .
E’ possibile anche confrontare i guadagni muscolari tra maschi e femmine, poiché gli uomini hanno 10 volte più Testosterone delle donne. Se i livelli di Testosterone di base fossero correlati con la risposta all’allenamento, ci aspetteremmo che gli uomini abbiano maggiori guadagni rispetto alle donne. Tuttavia, quando uomini e donne vengono sottoposti a programmi di allenamento contro-resistenza, mentre i guadagni muscolari assoluti sono maggiori negli uomini, i guadagni muscolari relativi (cioè i guadagni percentuali) sono per lo più simili.
Nel complesso, questi dati limitati suggerirebbero che i livelli di Testosterone non influiscono realmente sui guadagni. Tuttavia, si tratta di dati trasversali e non sono realmente progettati per affrontare la questione se le variazioni del Testosterone fisiologico abbiano un impatto sui guadagni di massa muscolare.
Pertanto, è necessario esaminare alcune ricerche in cui i livelli di Testosterone vengono direttamente manipolati.
Livelli di Testosterone estremamente bassi compromettono la massa muscolare e i guadagni muscolari?
Un modo per esaminare l’impatto del Testosterone sui guadagni muscolari è vedere cosa succede quando si sopprime la produzione di Testosterone. Maura et al. ha somministrato a giovani uomini il Lupron, un farmaco antiandrogeno che sopprime la produzione naturale di Testosterone. I livelli di Testosterone sono scesi da 535ng/dL a 31ng/dL dopo 10 settimane. Pertanto, la media dei soggetti trattati con Lupron aveva livelli di Testosterone simili a quelli di una donna.
La massa magra è diminuita di 2,1 kg e la sintesi proteica dell’intero corpo è diminuita del 13%. Naturalmente, non c’era alcun tipo di allenamento in questo studio. Forse l’allenamento con i pesi potrebbe interagire con questa risposta.
Kvorning et al. hanno somministrato a giovani uomini il Goserelin, che sopprime la produzione naturale di Testosterone, o un placebo. Gli uomini, che avevano una minima esperienza di allenamento contro-resistenza, si sono impegnati in un programma di allenamento della forza di 8 settimane. I livelli di Testosterone sono scesi da 651ng/dL a 57ng/dL, e poi 31ng/dL nel gruppo che ha ricevuto il Goserelina.
La soppressione del Testosterone non ha compromesso i miglioramenti nelle prestazioni del 10-RM rispetto al placebo. Tuttavia, i miglioramenti nella forza isometrica erano significativamente inferiori con il Goserelina.
I miglioramenti nella massa magra delle gambe erano significativamente inferiori per il gruppo Goserelina e anche la massa corporea magra totale tendeva verso quella direzione (valore P di 0,07, dove 0,05 è considerato significativo). L’aumento medio della massa magra è stato di 1kg maggiore nel gruppo placebo rispetto al gruppo Goserelina. La differenza nella massa magra della gamba era di 0,2kg.
Pertanto, questo studio ha dimostrato che la soppressione della produzione di Testosterone ha compromesso i guadagni di massa magra, ma la differenza non era marcata, pari a circa 1kg di differenza complessiva nei guadagni di massa magra in 8 settimane.
Non sono state eseguite misurazioni dirette della dimensione muscolare, sebbene le grandi differenze nei guadagni di forza isometrica probabilmente indichino che i guadagni muscolari erano inferiori con la soppressione del Testosterone. Quindi, questi dati suggerirebbero che c’è un impatto del Testosterone sul guadagno muscolare, pur essendo di piccola entità.
Molecola di Goserelina. La Goserelina è un agonista delle gonadotropine iniettabile (agonista GnRH), conosciuta anche come agonista dell’Ormone di Rilascio dell’Ormone Luteinizzante (LHRH).
Quindi le variazioni all’interno dell’intervallo fisiologico normale influiscono sulla massa muscolare?
Nessuna delle ricerche discusse finora può davvero dirci se le variazioni all’interno del normale range fisiologico possono avere un impatto sulla massa muscolare. Ci sono tre modi in cui è possibile rispondere a questa domanda. Il primo modo è guardare ai dati trasversali. Più semplicemente si tratta di prendere grandi gruppi di uomini e di dividerli in categorie in base ai loro livelli di Testosterone. Quindi si osserva se la massa muscolare differisce tra gli uomini in diverse categorie o se i livelli di Testosterone sono correlati ai livelli di massa muscolare.
He et al. hanno esaminato 270 uomini sedentari dell’HERITAGE Family Study. Dopo aver controllato per età e ascendenza, il Testosterone non era correlato alla massa magra. È interessante notare, tuttavia, che era correlato negativamente con l’indice di massa magra (FFM diviso per altezza al quadrato, simile all’IMC), il che significa che le persone con un indice FFM più elevato avevano livelli più bassi di Testosterone. Questo significa che avere più Testosterone significa in realtà avere meno muscoli? No! Questi dati sono confusi dal fatto che alcuni degli uomini erano obesi e che l’indice di massa corporea più elevato e le percentuali di grasso corporeo più elevate erano associate a un livello di Testosterone più basso. Ecco i livelli di testosterone per quartili di BMI; è possibile notare che i livelli di Testosterone diminuiscono all’aumentare dell’IMC.
Poiché gli uomini obesi hanno anche più FFM, questo può far credere che ci sia una relazione negativa tra FFM e livelli di Testosterone. Ciò di cui si ha bisogno per comprendere la questione è una ricerca che esamini la relazione negli individui non obesi.
Testosterone per quartili di BMI negli uomini. 1 nmol/L = 0,0347 ng/dL
Poiché gli uomini obesi hanno anche più FFM, questo può far sembrare che ci sia una relazione negativa tra FFM e livelli di Testosterone. Ciò di cui si necessita per comprendere la questione è una ricerca che esamini la relazione negli individui non obesi.
Van Den Beld et al. non hanno trovata alcuna relazione tra Testosterone e massa magra negli uomini anziani (età 73-94 anni).
Mouser et al. hanno raccolto dati sul Testosterone e sulla composizione corporea di 252 uomini nel National Health And Nutrition Examination Survey (NHANES) del 1999-2000 di età compresa tra 18 e 85 anni. Uomini che non rientravano nell’intervallo normale per il Testosterone (da 240 a 950ng/dL). ) non sono stati inclusi nell’analisi. Gli uomini sono stati suddivisi in quartili in base ai loro livelli di Testosterone. Gli uomini hanno mostrato quantità progressivamente più elevate di massa magra nella parte inferiore del corpo con livelli crescenti di Testosterone, anche dopo aver aggiustato la media per età, razza, presenza di diabete, partecipazione auto-riferita all’attività fisica, proteina C-reattiva e assunzione di proteine nella dieta. I quartili 3 e 4 erano statisticamente significativi rispetto al quartile 1. Un modello in qualche modo simile è emerso per la parte superiore del corpo, sebbene non vi fosse alcuna differenza tra il quartile 3 e 4.
Questi dati hanno mostrato che gli uomini nel 3° quartile avevano il 14,2% in più di massa magra nell’area inferiore e il 5,6% in più di massa magra in quella superiore rispetto agli uomini nel 1° quartile. Gli uomini del 4° quartile avevano il 22,1% in più di massa magra nell’area inferiore e il 5,6% in più di massa magra in quella superiore rispetto agli uomini del 1° quartile. Se si prendesse un ipotetico uomo nel 1° quartile con 17kg di massa magra nell’area inferiore, si potrebbe prevedere che un uomo nel 3° quartile possa avere 19,4kg e un uomo nel 4° quartile 20,6kg. Pertanto, questi dati hanno mostrato che gli uomini nell’estremità superiore dell’intervallo fisiologico del Testosterone avevano una massa corporea magra maggiore rispetto agli uomini nell’estremità inferiore, anche tenendo conto di altre variabili che potrebbero influenzare il Testosterone.
I dati trasversali di Mouser indicano che esiste potenzialmente una relazione tra i livelli di Testosterone nell’intervallo fisiologico e la massa magra di cui si dispone. Tuttavia, un problema con i dati trasversali è che non possono stabilire causa ed effetto. Un altro modo in cui è possibile affrontare la questione se le variazioni all’interno di un intervallo fisiologico influiscano sulla massa muscolare è sopprimere la produzione naturale di Testosterone usando farmaci, quindi somministrare dosi diverse di Testosterone e osservare se c’è un effetto dose-risposta. Ci sono quattro studi che hanno fatto questo.
Shalendar Bhasin et al. hanno somministrato a giovani uomini sani un agonista dell’ormone di rilascio delle gonadotropine (GnRH) per sopprimere la secrezione endogena di Testosterone. Hanno quindi somministrato agli uomini iniezioni settimanali di 25, 50, 125, 300 o 600mg di Testosterone Enantato per 20 settimane. Ecco i livelli ematici medi di Testosterone per le diverse dosi; come prevedibile, i livelli ematici sono aumentati con l’aumentare delle dosi e le dosi da 300 e 600mg hanno ovviamente portato a livelli di Testosterone al di sopra del normale intervallo fisiologico.
C’è stato un aumento dose-dipendente della massa magra; maggiori livelli ematici di Testosterone hanno portato a maggiori aumenti della FFM.
Anche il volume muscolare della coscia è aumentato in modo dose-dipendente.
La variazione della massa magra e la variazione del volume muscolare del quadricipite erano significativamente correlate con i livelli ematici di Testosterone.
Nel complesso, questo studio ha mostrato un effetto dose-risposta del Testosterone sulla dimensione muscolare, anche all’interno dell’intervallo fisiologico. Infatti, il solo passaggio dalla fascia bassa del fisiologico (306ng/dL) alla fascia media (542ng/dL) ha comportato un aumento della massa magra di 2,8 kg.
Bhasin ha ripetuto lo stesso esperimento in uomini più anziani di età compresa tra 60 e 75 anni. I risultati erano molto simili; i grafici seguenti mostrano gli effetti dose-risposta negli uomini più anziani.
Un terzo studio di Shalendar Bhasin ha coinvolto un design simile. La secrezione naturale di Testosterone è stata soppressa utilizzando il Lupron in uomini sani di età compresa tra 18 e 50 anni. Agli uomini sono state quindi somministrate dosi di 50, 125, 300 o 600 mg/settimana di Testosterone Enatnato, con o senza un inibitore della 5α-reduttasi (un farmaco che blocca la conversione del Testosterone in Diidrotestosterone [DHT]). I risultati sono stati ancora una volta simili, con una maggiore massa magra all’aumentare dei livelli ematici di Testosterone.
Finkelstein et al. hanno somministrato la Goserelina a 198 uomini sani di età compresa tra 20 e 50 anni per sopprimere i loro livelli di Testosterone. Sono stati quindi assegnati in modo casuale a ricevere giornalmente un gel placebo, o 1,25g, 2,5g, 5g o 10g di un gel contenente Testosterone per 16 settimane. Altri 202 uomini sono stati sottoposti allo stesso protocollo, tranne per il fatto che hanno ricevuto anche un inibitore dell’Aromatasi (Anastrozolo) per sopprimere la conversione del Testosterone in Estradiolo. C’è stato un effetto dose-risposta delle diverse dosi di Testosterone sui livelli ematici del ormone in questione, che vanno da al di sotto dell’intervallo normale fisiologico per le dosi di 0 e 1,25g, fino all’estremità superiore dell’intervallo fisiologico per la dose di 10g. Le barre nere rappresentano il gruppo trattato con Anastrozolo, mentre le barre rosse rappresentano il gruppo non trattato con Anastrozolo.
Livelli di Testosterone nel sangue con diverse dosi di un gel contenente Testosterone, dopo la soppressione del Testosterone endogeno con Goserelina. Le barre rosse rappresentano un gruppo che ha ricevuto Anastrozolo, un inibitore dell’aromatasi, per ridurre la conversione del Testosterone in Estradiolo. Dati da Finkelstein et al., NEJM, 2013
I cambiamenti nella massa magra e nell’area muscolare della coscia hanno mostrato un po’ di effetto dose-risposta, anche se non così chiaro come gli quanto osservato negli articoli di Bhasin che hanno utilizzato somministrazione per iniezioni. Nessuna dose di Testosterone ha provocato una significativa perdita di massa magra, mentre la dose più alta ha portato al guadagno maggiore di questa, sebbene molte delle differenze non fossero statisticamente significative. I numeri uguali non indicano differenze statisticamente significative rispetto ad altre barre.
C’era un’enorme quantità di variazione nel modo in cui gli individui rispondevano al Testosterone, come si può vedere in questo grafico a dispersione.
Un terzo modo per esaminare se le variazioni in un intervallo fisiologico influiscono sui guadagni muscolari è vedere se portare il +stosterone al limite molto superiore dell’intervallo normale (come quello che si verifica negli studi sui contraccettivi maschili di Testosterone) influisce sulla massa magra. Herbst et al. ha studiato l’impatto del testosterone esogeno (100 mg di testosterone enathnato a settimana) su uomini sani con normali livelli di testosterone. I livelli di testosterone sono aumentati da 570 ng/dL a 734 ng/dL (il livello subito prima dell’iniezione successiva), con un picco di 1196 ng/dL (24 ore dopo l’iniezione). Pertanto, il livello di picco era al limite molto superiore del range di normalità e il minimo era nella parte superiore del normale. Massa magra aumentata di 2,5 kg. Nel complesso, questi tre corpi di prove (dati trasversali, dati sulla risposta alla dose e dati sui contraccettivi maschili) indicano che le variazioni all’interno dell’intervallo fisiologicamente normale influiscono sulla massa magra che si trasporta. Ora, qui c’è una differenza tra quanta massa magra porti e quanto guadagnerai da un programma di allenamento (ne parleremo più avanti), ma sembra esserci un effetto. Il che ci porta alla nostra prossima domanda…
Se i livelli di Testosterone sono bassi o al limite del limite basso, portare i livelli fino al livello medio o superiore aiuta a migliorare la condizione della massa muscolare?
Partendo dal precedente quesito, cosa succede se si prendono delle persone con bassi livelli di Testosterone e li si aumenta i livelli con iniezioni di Testosterone esogeno? Fortunatamente c’è la ricerca a darci una risposta.
Urban et al. hanno reclutato 6 uomini sani e anziani con un’età media di 67 anni. I loro livelli di Testosterone erano di 480ng/dL o meno e sono stati somministrati loro iniezioni di Testosterone per 4 settimane per raggiungere livelli simili a quelli degli uomini più giovani. Sia la forza muscolare che la sintesi proteica muscolare sono migliorate, suggerendo che aumentare i livelli all’interno dell’intervallo fisiologico può aiutare a migliorare le condizioni della massa e la forza muscolare. Una limitazione è che la massa muscolare non è stata direttamente misurata; sono state determinate solo la sintesi proteica muscolare e la forza.
Sullivan et al. hanno reclutato 71 uomini di età compresa tra 65 e 93 anni e li hanno assegnati in modo casuale a uno di 4 gruppi:
Esercizio a bassa resistenza (3 x 8 con 20% del 1-RM) + Placebo
Esercizio a bassa resistenza + 100 mg/settimana di Testosterone
Esercizio di resistenza ad alta intensità (3 x 8 all’80% del 1-RM) + Placebo
Esercizio di resistenza ad alta intensità + 100 mg/settimana di Testosterone
Le iniezioni di Testosterone hanno più che raddoppiato i livelli del ormone rispetto al placebo, portando i livelli alla fascia alta del normale (804 ng/dL contro 304 ng/dL). Anche i guadagni nell’area della sezione trasversale dei muscoli a metà coscia sono stati più del doppio con le iniezioni di Testosterone rispetto al placebo. I guadagni di forza erano molto più alti nella condizione di esercizio a bassa resistenza quando veniva somministrato Testosterone rispetto al placebo. Tuttavia, quando l’allenamento era ad alta intensità, non c’era più un beneficio significativo del Testosterone, indicando che il carico di allenamento aveva un impatto maggiore sulla forza rispetto al Testosterone in questo studio.
Bhasin et al. hanno trattato uomini ipogonadici (età 19 – 47 anni) con 100mg di Testosterone Enantato a settimana per 10 settimane. I livelli medi di Testosterone al basale sono aumentati da 72ng/dL (leggermente al di sopra dell’intervallo per una donna media) a 767ng/dL alla settimana 10. La massa magra è aumentata di 5kg, la dimensione del tricipite è aumentata del 12% e la dimensione del quadricipite è aumentata del 8%.
Bhasin et al. hanno reclutato uomini con infezione da HIV con bassi livelli di Testosterone e li hanno trattati con una crema topica di Testosterone per 12 settimane. I livelli di Testosterone sono migliorati da 258ng/dL a 367ng/dL. La massa magra è aumentata di 1,4kg.
In un altro studio del Dr. Bhasin, uomini con infezione da HIV con bassi livelli di Testosterone (<349 ng/dL) sono stati assegnati in modo casuale a uno dei 4 seguenti gruppi:
Placebo
Testosterone Enatnato (100 mg/settimana)
Allenamento di resistenza
Testosterone + allenamento di resistenza
Il trattamento con Testosterone ha aumentato i livelli ematici da una media di 201 – 205ng/dL a 311 – 337ng/dL. La massa magra è aumentata di 4kg nel gruppo solo Testosterone, 2kg nel gruppo solo allenamento e 1,6kg nel gruppo allenamento + Testosterone. Il volume muscolare della coscia è aumentato di 40cm³ nel gruppo solo Testosterone, 62cm³ nel gruppo solo allenamento e 44cm³ nel gruppo combinato. Non è chiaro il motivo per cui non vi è stato alcun effetto combinato nel gruppo allenamento + Testosterone.
Sattler e colleghi hanno somministrato a uomini anziani (età media 71 anni) 5 o 10g al giorno di Testosterone transdermico (formulazione per somministrazione sulla pelle). Gli uomini trattati avevano livelli ematici di Testosterone di 550ng/dL o meno (la concentrazione media era 385 nel gruppo 5g/die e 350 nel gruppo 10g/die). Le concentrazioni medie di Testosterone sono aumentate di 150ng/dl nel gruppo 5g/die (aumentando i livelli a circa 535ng/dl) e 500ng/dl nel gruppo 10g (aumentando i livelli a circa 850ng/dl). La massa corporea magra è aumentata di 1kg nel gruppo 5g/die e di 1,6kg nel gruppo 10g/die.
Basaria et al. hanno reclutato uomini con Testosterone basso (<350 ng/dL) a causa dell’abuso di oppiacei e hanno somministrato loro un 5g/die di gel contenente Testosterone. Il testosterone medio è aumentato da 243ng/dL a 790ng/dL. La massa magra aumentata di 1kg.
Storer et al. hanno reclutati uomini di età superiore ai 59 anni con livelli di Testosterone tra 100 e 400ng/dL. Agli uomini è stato somministrato 7,5g di un gel contenente Testosterone o un placebo al giorno per 3 anni. Il Testosterone ematico è aumentato da 307 ng/dL a 567 ng/dL nel gruppo gel. La massa corporea magra è aumentata di 0,7kg.
Brodsky e colleghi hanno osservato gli effetti della somministrazione di Testosterone negli uomini con livelli di questo ormone inferiori a 200 ng/dL. I livelli di Testosterone sono aumentati gradualmente da 106 ng/dL a 576 ng/dL in 4 mesi. E da 432 ng/dL in 6 mesi. Queste erano le concentrazioni più basse osservate prima di ogni iniezione. Le iniezioni sono state somministrate ogni 2 settimane; l’ultimo livello di Testosterone misurato è stato una settimana dopo l’ultima iniezione ed era di 1277ng/dL, appena al di sopra del range fisiologico. La massa magra è aumentata di 8,7kg. La sintesi proteica muscolare mista è aumentata del 56% e la sintesi proteica miofibrillare è aumentata del 46%. La sintesi proteica muscolare totale in tutto il corpo è aumentata del 71-87% (da circa 2,4 grammi all’ora a 4,3 grammi all’ora).
Snyder et al. hanno somministrato a uomini con Testosterone basso (a causa di una malattia) un cerotto cutaneo con Testosterone per 3 anni. I livelli di Testosterone sono aumentati da 78ng/dL a 407ng/dL. La massa magra è aumentata di 3,1kg.
Wang e colleghi hanno somministrato a uomini con bassi livelli di Testosterone un cerotto o uno dei due diversi gel (50mg o 100 mg/giorno) per 90 giorni. Il Testosterone è aumentato da 236 ng/dL a 417 ng/dL nel gruppo cerotto, da 236 ng/dL a 552 ng/dL nel gruppo del gel da 50mg e da 248 ng/dL a 791 ng/dL nel gruppo del gel da 100mg. La massa corporea magra è aumentata rispettivamente di 1,2 kg, 1,3 kg e 2,7 kg in questi gruppi.
In un altro studio di Wang, agli uomini con bassi livelli di Testosterone (<300 ng/dL) sono state somministrate varie dosi di gel di Testosterone per un massimo di 42 mesi. I livelli totali sono aumentati di circa 260 ng/dL e sono rimasti nell’intervallo medio-basso normale per la durata dello studio. La massa magra è aumentata di 2,9kg.
Tenover ha reclutato uomini di età compresa tra 57 e 76 anni, con livelli di Testosterone inferiori a 400 ng/dL, e ha iniettato loro 100 mg di Testosterone Enantato a settimana. I livelli ematici medi sono aumentati da 334 ng/dl a 568 ng/dl. La massa magra è aumentata di 1,8kg.
Snyder e colleghi hanno assegnato casualmente a uomini di età superiore ai 65 anni un cerotto di Testosterone o a un placebo. I livelli di Testosterone sono aumentati da 367 ng/dL a 625 ng/dL in 6 mesi. La massa magra è aumentata di 1,6 kg in 6 mesi.
Ferrando et al. hanno reclutato uomini di età pari o superiore a 60 anni e con livelli di Testosterone nel sangue inferiori a 480 ng/dL somministrando loro Testosterone Enatnato o un placebo su base settimanale per 6 mesi. Il livello medio basale di Testosterone era 363 ng/dL. Le iniezioni di Testosterone sono state regolate individualmente per cercare di mantenere un livello ematico tra 490 e 807 ng/dL, sebbene ciò non abbia avuto un successo totale e molti individui abbiano riscontrato livelli leggermente superiori a tale obiettivo. Il livello medio a 6 mesi era di 882 ng/dL. La massa magra è aumentata di 4,2 kg, mentre è diminuita di 2 kg nel gruppo placebo. Il volume muscolare delle gambe è aumentato di 488 ml, mentre è diminuito di 96 ml nel gruppo placebo. Il Testosterone ha anche determinato un aumento dell’equilibrio netto delle proteine muscolari, a causa di una diminuzione del catabolismo delle proteine muscolari.
Dias et al. hanno reclutato uomini di età compresa tra 65-82 anni e livelli di Testosterone <350 ng/dL assegnandoli in modo casuale a un placebo, Anastrozolo (un inibitore dell’Aromatasi) o un gel di Testosterone. Inibendo l’enzima Aromatasi, l’enzima che converte il Testosterone in Estradiolo, è possibile aumentare efficacemente i livelli di Testosterone, ed è una cosa largamente risaputa. L’Anastrozolo ha aumentato il Testosterone da 272 ng/dL a circa 500 ng/dL a 6 mesi; la massa magra è aumentata di 1,5kg. Il Testosterone somministrato attraverso il gel ha aumentato i livelli dell’ormone da 300 ng/dL a circa 650 ng/dL e la massa magra non ha raggiunto un aumento statisticamente significativo (0,9 kg).
Magnusson e colleghi hanno randomizzato soggetti diabetici di tipo 2 di età compresa tra 50 e 70 anni trattandoli con un Testosterone gel o un placebo per 6 mesi. I livelli di Testosterone sono aumentati da 205 ng/dL a 637 ng/dL e la massa magra è aumentata di 1,9 kg.
Ribeiro e Abucham hanno somministrato a uomini ipogonadici Clomifene Citrato, il quale causa un aumento del Testosterone endogeno legandosi ai recettori degli estrogeni ipotalamici. Legandosi ai recettori degli estrogeni, induce il cervello a percepire che non ci siano così tanti estrogeni nel corpo per garantire l’omeostasi. Questo porta ad un aumento di GnRH seguito da LH e FSH. L’LH (Ormone Luteinizzante), stimola le cellule di Leydig nei testicoli a sintetizzare più Testosterone. I livelli di Testosterone nei “responder” sono aumentati da 201 ng/dL a 435 ng/dL dopo 3 mesi e la massa magra è aumentata di 1 kg.
Liu et al. hanno reclutato uomini più anziani con bassi livelli di Testosterone e somministrato loro iniezioni di gonadotropina corionica umana (HCG). L’HCG, mimando l’LH, stimola i testicoli a sintetizzare Testosterone. Il Testosterone è aumentato da 320 ng/dL a circa 720 ng/dL e la massa magra è aumentata di 2 kg.
Bayram et al. hanno somministrato a uomini ipogonadici iniezioni di HCG. Il Testosterone è aumentato da 39 ng/dl (nell’intervallo) a 512 ng/dl, con un aumento di 473 ng/dl. La massa magra è aumentata di 2,8 kg.
In uno studio di Casaburi et al., uomini con BPCO e basso livello di Testosterone (≤400 ng/dL) sono stati assegnati in modo casuale a uno dei 4 gruppi:
-Placebo -Testosterone Enantato (100 mg/week) -Resistance Training -Testosterone + Resistance Training
Le iniezioni di Testosterone hanno aumentato i livelli ematici da 302 ng/dL nel gruppo senza allenamento a 595 ng/dL e da 408 ng/dL a 656 ng/dL nel gruppo con allenamento. La massa magra è aumentata di 2,3 kg nel gruppo solo Testosterone, 0,2 kg nel gruppo solo resistance training e 3,29 kg nel gruppo Testosterone + resistance training. Va notato che l’allenamento è stato eseguito solo nella parte inferiore del corpo. Se si osservano i guadagni di massa magra delle gambe, erano 1,07kg nel gruppo solo Testosterone, 0,49 kg nel gruppo solo resistance training e 1,41 kg nel gruppo combinato.
Ecco un riassunto di tutti questi studi appena discussi, in cui i livelli bassi o al limite del livello di Testosterone sono stati aumentati in un intervallo fisiologico utilizzando iniezioni o sistemi di somministrazione transdermica. Puoi vedere che tutti hanno mostrato impatti positivi sulla massa magra. Alcuni hanno avuto aumenti relativamente piccoli del Testosterone (come circa 100-250 ng/dL) e hanno mostrato aumenti significativi della massa magra di circa 1-2 kg. In alcuni di questi studi, i soggetti si trovavano nella fascia più bassa del range di normalità (piuttosto che al di sotto del range di riferimento), e anche con quei soggetti, portare i livelli fino alla fascia medio-alta del range fisiologico ha avuto benefici positivamente apprezzabili.
E’ possibile vedere che i guadagni di massa magra con le iniezioni ( righe arancioni) tendono ad essere maggiori rispetto alla somministrazione transdermica ( righe grigie). Ciò è probabilmente legato al fatto che le iniezioni causano un picco iniziale di Testosterone che può essere all’estremità superiore dell’intervallo fisiologico, se non superare leggermente l’intervallo fisiologico.
Tipicamente, in questi studi, il Testosterone viene misurato 1-2 settimane dopo l’iniezione, rappresentando il minimo o il livello più basso di Testosterone. Pertanto, i livelli finali di Testosterone non rappresentano i livelli di picco raggiunti. Si può vedere questa differenza quando si guarda lo studio di Brodsky et al, dove il livello di picco, misurato 1 settimana dopo l’iniezione, era tre volte superiore al livello più basso, misurato 2 settimane dopo l’iniezione. Pertanto, tutti questi studi sulla terapia sostitutiva del Testosterone (TRT) dimostrano un beneficio nella massa magra dallo spostamento al di sotto dell’intervallo fisiologico, o dall’estremità inferiore dell’intervallo fisiologico, all’intervallo fisiologico medio o alto. Pertanto, sembra che anche il passaggio da una fascia bassa della gamma fisiologica a quella superiore abbia un vantaggio.
Testosterone più alto = Muscolo basale più alto, solo tassi di guadagno leggermente maggiori.
Nel complesso, i dati trasversali, gli studi dose-risposta, gli studi sui contraccettivi maschili e gli studi su TRT (compresi quelli in cui il Testosterone al basale era ancora normale) mostrano che le variazioni del Testosterone all’interno dell’intervallo fisiologico hanno un impatto sulla massa magra e muscolare. Questo significa che qualcuno con un livello di Testosterone più alto guadagnerà più velocemente di qualcuno con un livello più basso? Non proprio. Quando si guarda il corpo delle prove, il maggiore impatto del Testosterone sembra essere sul mantenimento di un certo livello di base della massa muscolare, piuttosto che sul tasso di guadagno muscolare. Ad esempio, i malati di cancro alla prostata sono spesso sottoposti a terapia di deprivazione di androgeni, in cui i loro livelli di Testosterone sono stati soppressi. In questo studio, il Testosterone medio era 45,7 ng/dL (all’interno dell’intervallo di una donna), rispetto a 430 ng/dL per i controlli. La sintesi proteica muscolare a riposo e a stomaco pieno era più bassa nei pazienti deprivati di androgeni. Tuttavia, quando l’alimentazione è stata combinata con l’allenamento contro-resistenza, la risposta alla sintesi proteica muscolare non era statisticamente diversa dai controlli (sebbene la media grezza fosse ancora leggermente inferiore).
Tassi di sintesi proteica muscolare in soggetti di controllo rispetto a pazienti in terapia di deprivazione androgenica (ADT). La sintesi proteica muscolare è significativamente più bassa a riposo e anche dopo un pasto (FED). Tuttavia, dopo l’allenamento contro-resistenza, la sintesi proteica muscolare non è significativamente diversa dai controlli dopo un pasto (EX-FED).
Mentre il guadagno percentuale nello studio di cui sopra era leggermente favorito negli uomini, non lo era di molto. Tuttavia, il guadagno assoluto è stato quasi il doppio di quello negli uomini rispetto alle donne. Pertanto, un aumento del 15% della massa muscolare negli uomini sarà generalmente maggiore su base assoluta rispetto alle donne, poiché gli uomini hanno una linea di base più ampia.
Una terza linea di supporto a questo concetto viene dal famoso studio Bhasin del 1996 sugli steroidi anabolizzanti. In questo studio di 10 settimane, uomini normali sono stati assegnati in modo casuale a uno dei quattro gruppi:
Placebo senza allenamento
Testosterone senza allenamento
Placebo con allenamento contro-resistenza
Testosterone con allenamento contro-resistenza.
Il testosterone è stato somministrato in dosi sovrafisiologiche (600 mg/settimana). I livelli di testosterone sono stati elevati a 2828 – 3244 ng/dL con le iniezioni, rispetto ai livelli normali di 453 – 667 ng/dL nel gruppo placebo. L’iniezione di testosterone, senza allenamento, ha comportato un aumento della massa magra di 3,2 kg. Il solo allenamento ha comportato un aumento della massa magra di 2 kg. Quando l’allenamento è stato combinato con l’iniezione di testosterone, l’aumento di massa magra è stato di 6,1 kg. La dimensione del muscolo quadricipite è aumentata in modo simile nel gruppo testosterone + nessun allenamento e nel gruppo solo allenamento, mentre i guadagni sono stati raddoppiati nel gruppo testosterone + allenamento.
La cosa interessante qui è che, quando guardi questi dati, puoi vedere che c’era principalmente un effetto additivo, piuttosto che sinergico, del testosterone e dell’allenamento. La FFM è aumentata di 3,2 kg con il solo testosterone. È aumentato di 2 kg solo con l’allenamento. Quando sommi queste due quantità, ottieni 3,2 + 2 = 5,2 kg, che è ragionevolmente vicino al guadagno di 6,1 kg osservato nel gruppo combinato. In altre parole, solo circa 0,9 kg potrebbero essere spiegati da un effetto sinergico tra testosterone e allenamento. Puoi anche vedere un effetto simile per l’aumento delle dimensioni del quadricipite. Il cambiamento nella dimensione del quadricipite era più o meno lo stesso nei gruppi solo testosterone e solo allenamento, ed era per lo più additivo nel gruppo combinato. Ancora una volta, solo una piccola parte del cambiamento nella dimensione del quadricipite potrebbe essere spiegata da un effetto sinergico tra testosterone e allenamento.
Questo è simile ai dati osservati quando osserviamo uomini e donne. I maggiori livelli di testosterone negli uomini non aumentano molto il tasso di guadagno rispetto alle donne; è solo che dà agli uomini una linea di base più alta per cominciare, e quindi i guadagni assoluti sono maggiori. Se ci fosse un forte effetto sinergico tra testosterone e allenamento, allora i guadagni relativi negli uomini sarebbero significativamente maggiori rispetto alle donne, ma ovviamente non è così.
Una quarta evidenza viene dallo studio di Casaburi e colleghi sugli uomini con BPCO, di cui si è brevemente accennato in precedenza. In questo studio è stato eseguito solo l’allenamento delle gambe. I guadagni di massa magra delle gambe nel gruppo combinato allenamento+testosterone erano 1,41 kg, che è vicino alla somma dei guadagni sperimentati dal gruppo solo testosterone (1,07 kg) e il gruppo solo allenamento (0,49 kg). Questo suggerisce ancora una volta che gli effetti del testosterone e dell’allenamento di resistenza sono per lo più additivi e non sinergici.
Pertanto, i tuoi livelli di testosterone influenzano la quantità di muscoli che porti in giro, indipendentemente dal fatto che ti alleni o meno. Quindi, quando inizi ad allenarti, la tua reattività all’allenamento è per lo più simile indipendentemente dal fatto che tu abbia livelli di testosterone bassi o alti. Potrebbe essere un po’ meno con un testosterone più basso, ma l’impatto maggiore è sulla tua linea di base.
Ad esempio, supponiamo che tu abbia una massa magra di base di 50 kg e che tu abbia un livello di testosterone nella fascia bassa (diciamo circa 300 ng/dL). Guadagni il 10% in 6 mesi, ovvero 5 kg.
Ora, prendi la stessa situazione, ma la persona ha un testosterone di base a 600 ng/dL. La tua massa magra di base ora potrebbe essere di 52 kg. Guadagni ancora il 10% in 6 mesi, ovvero 5,2 kg. Pertanto, il guadagno relativo è simile. Tuttavia, il punto di partenza e il guadagno assoluto sono maggiori a causa del testosterone più alto.
Consideriamo un altro esempio. Diciamo che una persona segue un qualche tipo di sostituzione del testosterone, aumentando il testosterone da 250 ng/dL a 500 o 600 ng/dL. Quella persona inizialmente sperimenterà alcuni guadagni relativi superiori al normale, mentre si muove verso la sua nuova linea di base per il suo nuovo livello di testosterone. Ti sembrerà di guadagnare da “principiante”. Tuttavia, una volta che quella persona ha raggiunto la sua nuova linea di base, i suoi guadagni relativi saranno simili a quando aveva un testosterone più basso.
Quantificare l’impatto delle variazioni del Testosterone fisiologico sulla massa magra Quindi sappiamo che le variazioni del testosterone fisiologico influiscono sulla quantità di muscoli che hai. Ma quanto? Se passi da 300 ng/dL a 600 ng/dL, quanta massa magra in più puoi aspettarti di avere?
Per rispondere a questa domanda, torniamo agli studi dose-risposta di Shalendar Bhasin di cui abbiamo discusso in precedenza. Possiamo prendere i dati dai tre studi ed eseguire una regressione su di essi per vedere come cambia la massa magra al variare dei livelli di testosterone all’interno dell’intervallo fisiologico.
Ecco la linea di regressione per i dati di Bhasin et al. 2001, 2005 e 2012. Ho usato solo punti dati in cui il testosterone si trovava all’interno di un intervallo fisiologico o appena al di fuori di esso (da 176 ng/dL a 1345 ng/dL). Sono 11 punti dati. Per ogni aumento di 100 ng/dL di testosterone, la massa magra aumenta di 0,6 kg. L’R al quadrato per la vestibilità del modello era 0,85, il che è molto buono.
Variazione della massa magra in relazione al cambiamento del Testosterone rispetto al basale, entro un intervallo di variazione da -340 ng/dL a +691 ng/dL. Pendenza della linea = 0,006, il che significa un ulteriore 0,6 kg in FFM per ogni aumento di 100ng/dL del Testosterone. R-Quadrato = 0,85. Dati di Bhasin et al. 2001, 2005 e 2012.
Il valore di 0,6 kg di FFM per ogni aumento di 100 ng/dL è in accordo con uno studio dose-risposta di Huang e colleghi su donne isterectomizzate. Hanno anche scoperto che la FFM aumenta di 0,6 kg per ogni aumento di 100 ng/dL di testosterone.
Possiamo anche eseguire una regressione sui dati che ho discusso in precedenza da Finkelstein e colleghi, in cui i livelli di testosterone sono stati soppressi e quindi ai soggetti sono state somministrate diverse dosi di un gel di testosterone. Sebbene i numeri effettivi della massa magra non siano stati riportati da questo studio, possiamo stimarli dai valori di base riportati e dalle variazioni percentuali. Per ogni aumento di 100 ng/dL di testosterone, la massa magra aumenta di 0,3 kg. L’R al quadrato per la vestibilità del modello è 0,70, il che è buono.
Cambiamento nella massa magra in relazione al cambiamento nel Testosterone dal basale. Pendenza della linea = 0,003, il che significa un ulteriore 0,3 kg in FFM per ogni aumento di 100ng/dL del Testosterone. R-Quadrato = 0,70. Dati da Finkelstein et al. 2013.
Mentre potremmo anche provare a eseguire una regressione su alcuni degli studi discussi in cui i livelli di testosterone erano bassi e portati in un intervallo normale fisiologico, il problema con questi è che tutti usavano diversi metodi di somministrazione per il testosterone (iniezione vs gel vs. . patch), che possono avere dinamiche diverse in termini di come vengono modificati i livelli ematici. Inoltre, il problema con il tentativo di aggregare diversi studi di iniezione è che variano nel tempo in cui misurano il testosterone e variano anche in termini di frequenza delle iniezioni. Puoi avere una misurazione del testosterone molto diversa se misuri 1 settimana dopo un’iniezione, rispetto a 2 settimane.
Tuttavia, possiamo dare un’occhiata a studi in cui è stato somministrato un farmaco che ha stimolato la produzione naturale di testosterone; tali studi possono imitare meglio il modo in cui la massa magra risponde alle variazioni dei livelli di testosterone endogeno. Dias et al. dato agli uomini Anastrozolo, un inibitore dell’Aromatasi. Inibendo l’Aromatasi, l’enzima che converte il Testosterone in Estrogeno, puoi aumentare efficacemente i livelli di Testosterone. In teoria, questi livelli di Testosterone sarebbero relativamente stabili, poiché rappresenterebbero livelli di testosterone endogeno piuttosto che testosterone esogeno da iniezione o somministrazione transdermica. L’Anastrozolo ha aumentato il testosterone da 272 ng/dL a circa 500 ng/dL a 6 mesi e la massa magra è aumentata di 1,5 kg. Sono circa 0,6 kg di FFM per ogni aumento di 100 ng/dL, il che è in accordo con le nostre analisi precedenti. Ribeiro e Abucham hanno somministrato agli uomini ipogonadici Clomifene Citrato, che aumenta il Testosterone endogeno (i livelli all’interno del corpo, rispetto a quello esogeno come da un’iniezione o da una crema) legandosi ai recettori degli ipotalamici degli Estrogeni. Legandosi ai recettori degli estrogeni, induce il cervello a pensare che non ci siano così tanti estrogeni nel corpo. Questo porta il cervello a pompare più ormone Luteinizzante (LH), che poi stimola i testicoli a produrre più testosterone. I livelli di testosterone nei soggetti responsivi sono aumentati da 201 ng/dL a 435 ng/dL dopo 3 mesi e la massa magra è aumentata di 1 kg. Sono 0,4 kg per ogni aumento di 100 ng/dL di testosterone. Liu et al. iniettato HCG in uomini che avevano bassi livelli di testosterone al limite; L’HCG è un ormone che stimola i testicoli a produrre più testosterone. Il testosterone è aumentato da 320 ng/dL a circa 720 ng/dL, un aumento di 400 ng/dL. Massa magra aumentata di 2 kg. Sono 0,5 kg di massa magra per ogni 100 ng/dL di aumento del testosterone, che è ancora una volta in accordo con il range che abbiamo stabilito. Infine, Bayram et al. uomini ipogonadici iniettati con HCG. Il testosterone è aumentato da 39 ng/dL a 512 ng/dL, un aumento di 473 ng/dL. Massa magra aumentata di 2,8 kg. Sono 0,6 kg per ogni aumento di 100 ng/dL. Pertanto, questi 4 studi suggeriscono un aumento di 0,5 – 0,6 kg di massa magra per ogni aumento di 100 ng/dL di testosterone, che è in accordo con le regressioni di Bhasin e Huang.
Mettendo insieme tutto questo, i dati suggeriscono che la massa magra aumenterà di 0,7 – 1,3 libbre (0,3 – 0,6 kg) per ogni aumento di 100 ng/dL dei livelli ematici di testosterone all’interno dell’intervallo fisiologico. Quindi, se passassi da 300 ng/dL a 600 ng/dL, questo sarebbe 0,9 – 1,8 kg o circa 2,1 – 4 libbre.
Ora, tieni presente che ci sono dei limiti a questa analisi. In primo luogo, si basa su medie; i risultati individuali possono essere diversi. Ad esempio, come accennato in precedenza, Finkelstein et al. hanno mostrato un’ampia variazione nel modo in cui i soggetti hanno risposto a diversi livelli di testosterone. In secondo luogo, si basa su analisi tra soggetti; ciò che accade all’interno delle persone può essere diverso da ciò che si osserva tra le persone. Terzo, parte di esso si basa su iniezioni di testosterone esogeno. Il problema è che, con le iniezioni, i livelli medi di testosterone nel sangue saranno superiori a quelli misurati. Questo perché, quando inietti il testosterone, ottieni un grande picco nei livelli ematici e poi decade lentamente nell’arco di 1-2 settimane. I ricercatori di solito misurano il testosterone alla depressione del decadimento dopo l’iniezione, di solito 1-2 settimane dopo. Questa limitazione diventa evidente quando si osservano alcuni dei dati dose-risposta di Bhasin. Ad esempio, nello studio Bhasin 2001, gli uomini che hanno ricevuto 125 mg di testosterone iniettato hanno guadagnato 3,4 kg di massa magra, ma il livello di testosterone nel sangue misurato di 542 ng/dL era simile al livello di base naturale dei soggetti prima che avessero il loro testosterone livelli soppressi. Pertanto, i loro livelli medi di testosterone erano probabilmente molto più alti di 542 ng/dL.
Sebbene questo sia certamente un grosso limite nell’analisi, va anche ricordato che, con la regressione, stiamo valutando principalmente le differenze nella massa magra tra diversi livelli di testosterone, piuttosto che la relazione con un particolare livello assoluto. In altre parole, stiamo osservando come la massa magra cambia per un cambiamento di 100 ng/dl nel testosterone, piuttosto che come la massa magra si riferisce, ad esempio, a un livello ematico di 500 ng/dl. Quindi, anche se i livelli di testosterone sono dovuti a iniezioni esogene, e anche se i livelli medi sono molto più alti di quelli misurati alla depressione, il rapporto tra i livelli non dovrebbe cambiare drasticamente. Va anche notato che il rapporto di 0,6 kg/100 ng/dL riscontrato negli studi Bhasin ha retto nelle donne a cui sono state somministrate dosi molto più basse. Tuttavia, è ancora una limitazione che deve essere considerata.
Una cosa interessante da notare è che le variazioni all’interno di un intervallo fisiologico possono avere un impatto maggiore rispetto alle variazioni al di fuori dell’intervallo fisiologico. In altre parole, la massa magra non aumenta in modo lineare con l’aumento dei livelli di testosterone. Quando superi l’intervallo fisiologico, la pendenza della relazione diminuisce e il testosterone non ha lo stesso impatto. Ciò diventa evidente quando guardiamo di nuovo alla nostra regressione dei tre studi Bhasin, ma questa volta includiamo i dati che sono ben al di sopra dell’intervallo normale fisiologico.
Puoi vedere che la curva si adatta meglio ai dati rispetto alla linea retta. La pendenza per la linea retta è 0,002, il che significa che la massa magra è aumentata di 0,2 kg per ogni 100 ng/dL di testosterone, che è inferiore agli 0,6 kg che abbiamo osservato con gli stessi dati in precedenza. Anche l’adattamento del modello non è altrettanto buono (R-quadrato = 0,77). Questo perché i livelli estremamente elevati di testosterone attenuano la relazione. Ogni aumento di 100 ng/dL di testosterone non ha un effetto così forte a intervalli soprafisiologici rispetto al normale intervallo fisiologico. Questo è supportato anche quando diamo un’occhiata allo studio Bhasin del 1996 sul testosterone ad alte dosi che ho menzionato prima. Il testosterone nel sangue è aumentato di circa 2326 ng/dL nel gruppo con solo testosterone e la massa magra è aumentata di 3,2 kg. Questo è un aumento di 0,13 kg per ogni aumento di 100 ng/dL di testosterone, che non è lontano dall’aumento di 0,2 kg menzionato in precedenza quando abbiamo incluso alte dosi di testosterone.
E le donne?
Le donne hanno livelli di Testosterone molto più bassi rispetto agli uomini e ci sono dati limitati per stabilire intervalli di riferimento sulle donne. Uno dei problemi è che alcuni test di laboratorio tradizionali per il Testosterone, come i radioimmunodosaggi (RIA), non sono abbastanza sensibili da misurare con precisione il Testosterone nelle donne. Misurazioni accurate del testosterone nelle donne richiedono tecniche sensibili come la cromatografia liquida-spettrometria di massa tandem (LC-MS/MS). Esistono alcuni dati che stabiliscono intervalli di riferimento utilizzando questa tecnica. Ecco i dati di Haring e colleghi che mostrano i percentili più bassi e più alti per le donne in premenopausa di età compresa tra 20 e 49 anni (si noti che sto mescolando alcuni dei dati di distribuzione effettivi per ottenere il 25° e il 75° percentile, con i loro modelli di regressione quantile per ottenere il 2,5° e 97,5° percentile, ma per i nostri scopi va benissimo).
Simile agli uomini, il testosterone diminuisce con l’età.
Testosterone misurato da GC-MS / MS in 985 donne. Dati da Haring et al., J Clin Endocrinol Metab, 2012. 1 nmol/L = 0,0347 ng/dL
È stato anche riscontrato che le donne che assumevano contraccettivi orali o terapia ormonale sostitutiva avevano in media livelli di testosterone più bassi, sebbene l’intervallo percentile superiore fosse più alto. Il 25° percentile per queste donne era 10 ng/dL (vs 13) e il 75° percentile era 56 ng/dL (vs 47).
Testosterone per età nelle donne, confrontando le donne trattate con contraccettivi orali o HRT a quelle che non lo sono. Dati da Haring et al., J Clin Endocrinol Metab, 2012. 1 nmol/L = 0,0347 ng/dL
Rari et al. non ha riscontrato alcuna relazione tra testosterone totale e massa magra nelle donne anziane (età 67-94 anni), ma ha osservato una relazione significativa tra testosterone libero e massa magra (il testosterone libero è la forma che non è legata ad alcuna proteina, da qui il termine “libero”).
Possiamo anche esaminare i dati in cui alle donne con bassi livelli di testosterone è stato somministrato testosterone esogeno. Anche in questo caso, i dati sono limitati, ma ci sono alcuni studi.
Gruber e colleghi hanno somministrato alle donne in postmenopausa (età media di 51 anni) testosterone topico o un placebo per 6 mesi. Ora, va notato che in questo studio, le donne non avevano bassi livelli di testosterone (i livelli medi erano 29 ng/dL, che rientra in un intervallo normale). Il testosterone è aumentato appena al di sopra dell’intervallo normale (72 ng/dL) e non vi è stato alcun impatto sulla massa magra.
Cambiamento nella massa magra con diverse dosi di Testosterone nelle donne in menopausa isterectomizzate. Dati da Huang et al, Menopausa, 2014. Solo la dose di 25mg ha determinato un aumento statisticamente significativo. La massa magra è aumentata di 0,6kg per ogni aumento di 100ng/dL del Testosterone, che è al di fuori del normale intervallo fisiologico per le donne (13 – 56 ng/dL).
Questi dati suggeriscono che è necessario aumentare i livelli ematici di testosterone ben al di fuori di un intervallo normale fisiologico (verso la fascia molto bassa di un maschio) nelle donne in post-menopausa per ottenere aumenti misurabili della massa magra e della funzione sessuale. Nel complesso, questi dati indicano che le variazioni del testosterone nelle donne, all’interno del normale range fisiologico, hanno un impatto misurabile molto piccolo, se non nullo, sul muscolo. Ciò non sorprende se consideriamo l’aumento di 0,3 – 0,6 kg per ogni aumento di 100 ng/dL di testosterone di cui abbiamo discusso. L’intervallo normale per le femmine va da 13 a 56 ng/dL, un intervallo di soli 43 ng/dL. Ciò equivale solo a un quarto di chilogrammo (mezza libbra) o meno nelle donne.
Tiriamo le somme
Quando esaminiamo l’intero corpo di prove, è chiaro che le variazioni nei livelli fisiologici di Testosterone influiscono sulla quantità di muscoli che il soggetto ha, ma avranno un impatto minimo sui guadagni relativi (%). Riassumendo: I dati trasversali, gli studi dose-risposta e gli studi TRT supportano tutti variazioni all’interno dell’intervallo normale in quanto hanno un impatto sulla massa magra e sui muscoli-scheletrici. Le variazioni nei livelli ematici di Testosterone influiscono sul livello “base” dei muscoli, ma hanno un impatto minimo sui guadagni relativi (%). Quindi, avere livelli di Testosterone più alti significa avere un livello base più alto di massa muscolare. Mentre i guadagni relativi saranno per lo più simili, i guadagni assoluti saranno più alti a causa della linea di base più elevata. Gli impatti del Testosterone e dell’allenamento contro-resistenza sulla massa magra e sui muscoli-scheletrici sono principalmente additivi piuttosto che sinergici. La massa magra al basale aumenta di circa 0,7 – 1,3 libbre o 0,3 – 0,6 kg per ogni aumento di 100 ng/dL del Testosterone fisiologico; questo si basa su medie e dati tra soggetti, quindi i risultati individuali possono variare considerevolmente. Ci sono anche limitazioni a questa analisi, come il fatto che parte di essa si basa su dati dose-risposta provenienti da iniezioni, che potrebbero non riflettere accuratamente i cambiamenti nei livelli endogeni. L’impatto del Testosterone sulla FFM è attenuato a livelli sovrafisiologici (>1500 ng/dL); La FFM di base aumenta di circa 0,1-0,2 kg per ogni 100 ng/dL per quei livelli. Le variazioni del Testosterone nelle donne, all’interno del normale range fisiologico, hanno un impatto minimo o nullo sul muscolo-scheletrico. Quindi, sì, i soggetti di sesso maschile con un livello di Testosterone più alto hanno un vantaggio in termini assoluti sulla massa muscolare. Questi dati indicano anche che gli uomini che invecchiano, con livelli di Testosterone al limite o bassi, possono ottenere un beneficio nella costruzione muscolare da terapie progettate per aumentare i livelli di Testosterone in range fisiologici, sia da fonti esogene (come iniezioni o gel) sia da fonti che stimolano la produzione di Testosterone (come il Clomifene). , HCG o inibitori dell’Aromatasi), anche se si allenano già con i pesi.
Però, attenzione a fare comparazioni fuori luogo: la fisiologia e quello che può manifestare non è paragonabile alle sue alterazioni fuori range… nel bene e nel male…
Chi mi conosce sa come io prenda con estrema cautela qualsiasi affermazione sensazionalistica nei confronti di derivati erboristici et similari, ma non solo. Ogni qual volta mi capita di leggere qualche studio o serie di dati aneddotici sono solito indagare tutto lo scindibile riguardante l’oggetto che si ritiene causa primaria di un dato evento migliorativo nella composizione corporea e/o nelle prestazioni. Non di rado le mie ricerche mi hanno portato a conclusioni nettamente negative che liquidavano le affermazioni fatte da taluni come “placebo” o “non riconducibili alla molecola in questione. Mi capitò nei primi anni di ricerca con la Carnitina e il suo presunto effetto nel miglioramento del trasporto degli acidi grassi nel mitocondrio (cosa strettamente regolata e non sovraesprimibile con integrazione della medesima), o con il Tribulus Terrestris, la Maca e altri presunti “Testo-booster”. La lista è lunga.
E’ solo di recente che la mia attenzione è stata attirata verso due molecole, un precursore e il suo derivato, contenute in significative concentrazioni (in particolare riferimento al precursore) nelle crucifere (Broccoli, Cavoli ecc…), le quali presentano una interessante, sebbene contenuta, letteratura che ne sottolinea il potenziale di azioni biochimiche tra le quali spicca quella sul metabolismo degli estrogeni. Sto parlando del Indolo-3-Carbinolo (I3C) e del suo derivato 3,3′-Diindolylmethano (DIM).
E’ mia intenzione, quindi, esporre le loro caratteristiche e la possibile portata attualmente ipotizzata dalla loro assunzione.
I3C e DIM- loro caratteristiche molecolari e attività biochimica:
L’Indolo-3-Carbinolo (C9H9NO) è prodotto dalla scomposizione del Glucosinolato Glucobrassicina, che può essere trovato a livelli relativamente alti nelle verdure crocifere come Broccoli, Cavoli, Cavolfiori, Cavolini di Bruxelle ecc… .[1] È disponibile anche sotto forma di integratore alimentare.[2] L’Indolo-3-Carbinolo è oggetto di continua ricerca biomedica sui suoi possibili effetti anticancerogeni,[3] antiossidanti e anti-aterogeni.[4] La ricerca sull’Indolo-3-Carbinolo è stata condotta principalmente utilizzando animali da laboratorio e cellule coltivate in vitro.[5] Sono stati riportati studi umani limitati e per ora inconcludenti. Una recente review della letteratura sulla ricerca biomedica ha rilevato che “l’evidenza di un’associazione inversa tra l’assunzione di verdure crocifere e il cancro al seno o alla prostata negli esseri umani è limitata e incoerente” e “sono necessari studi controllati randomizzati più ampi” per determinare se l’Indolo-3-Carbinolo supplementare ha benefici per la salute.[6]
Lo studio dei meccanismi attraverso i quali il consumo di Indolo-3-carbinolo potrebbe influenzare l’incidenza del cancro si concentra sulla sua capacità di alterare il metabolismo degli estrogeni e altri effetti cellulari. Sono stati condotti studi controllati su animali come ratti, topi e trote arcobaleno, introducendo vari livelli controllati di agenti cancerogeni e livelli di Indolo-3-Carbinolo nella loro dieta quotidiana. I risultati hanno mostrato diminuzioni dose-correlate della suscettibilità al tumore dovute all’Indolo-3-Carbinolo (indotto dalla diminuzione del legame aflatossina-DNA). La prima prova diretta dell’attività anti-iniziale pura di un anticancerogeno naturale (indolo-3-carbinolo) presente nella dieta umana è stata rivendicata da Dashwood et al. nel 1989.[7]
L’Indolo-3-Carbinolo (I3C) agisce principalmente attraverso il suo principale metabolita, il Diindolylmethano (DIM) (può comprendere fino a un terzo dei derivati del I3C[8]) e alcuni altri metaboliti che possono essere prodotti spontaneamente dall’instabile I3C (come l’indolo {3,2-b}carbazolo,[9] un costituente minore[8]). La formazione precisa di questi metaboliti implica la catalizzazione del I3C per formare indoli reattivi che poi si combinano tra loro per “costruire” una molecola più grande ma stabile, essendo il DIM il risultato della formazione di due di questi indoli.[8]
Il Diindolylmethano (DIM), come già accennato, è il principale metabolita derivato dall’acido farmaceuticamente attivo dell’Indolo-3-Carbinolo (I3C) il quale si trova in molte verdure Brassica attraverso il composto madre glucobrassicina.[10][11][12] La glucobrassicina ingerita viene catalizzata tramite l’enzima Mirosinasi (contenuto nei vegetali) convertendo in Indolo-3-Carbinolo, il quale viene rapidamente metabolizzato sia in DIM che in vari altri metaboliti nello stomaco umano tramite reazioni di condensazione acido-mediate.[8][13]
Le fonti di glucosinolati (in generale) sono elencate di seguito, con qualsiasi fonte che citi il Diindolylmethano o il suo precursore (Indole-3-Carbinolo) specificatamente menzionata in grassetto:
Cavoletti di Bruxelles, 104mg per 44 g (mezza tazza)[14];
Crescione da giardino, 98mg per 25g (mezza tazza)[14];
Senape, 79mg per 28g (mezza tazza, tritata)[14];
Rapa, 60mg per 65g (mezza tazza, cubetti)[14]
Cavolo Verza, 35mg per 45g (mezza tazza, tritato)[14]
Cavolo riccio, 67mg per 67g (1 tazza, tritato)[14];
Crescione, 32mg per 34g (1 tazza, tritato)[14];
Cavolo rapa, 31mg per 67g (mezza tazza, tritato)[14];
Cavolo rosso, 29mg per 45g (mezza tazza, tritato)[14];
Broccoli, 27mg per 44g (mezza tazza, tritati)[14];
Rafano, 24mg per 15g (cucchiaio)[14];
Cavolfiore, 22mg per 50g (mezza tazza tritata)[14];
Bok Choy, 19mg per 35g (mezza tazza, tritato)[14].
Poiché la glucobrassicina si degrada in I3C per azione dell’enzima Mirosinasi contenuto nella pianta, la disattivazione di questo enzima mediante trattamento termico (cottura) può ridurre la biodisponibilità orale di qualsiasi glucosinolato incluso DIM.[15][16] Tuttavia, una certa biodisponibilità viene conservata a causa dell’espressione della Mirosinasi anche nell’intestino umano.[17]
Tioglucosidasi (Mirosinasi)
L’ebollizione[18] e il microonde (750-900 watt)[19][20] sembrano i maggiori sospettati per la riduzione della biodisponibilità del glucosinolato; il primo a causa dell’eccesso di acqua che assorbe i composti bioattivi solubili in acqua dal cibo. In questo senso, i metodi di cottura che utilizzano meno acqua trattengono più glucosinolati rispetto a quelli che utilizzano molta acqua.[21]
È stato dimostrato che il DIM attiva la segnalazione del Fattore Nucleare Kappa-Beta (NF-kB), l’attivazione della caspasi, l’attivazione del citocromo P450 (in particolare CYP1A1, CYP1A2 e CYP19), la riparazione del DNA, il recettore degli idrocarburi arilici (AHR) e varie protein chinasi.[22][23][24]
Fattore Nucleare Kappa-Beta
L’Indolo-3-Carbinolo alimentare o integrativo, tramite il metabolita DIM, si ritiene che possa aumentare il peso del fegato come riflesso di un aumento generale della produzione dell’enzima P450;[25] questa risposta organica sembra essere dose dipendente tra basse concentrazioni nella dieta (250 ppm ) fino a quelli molto elevati (5.000 ppm) con la 2-idrossilazione degli estrogeni in aumento in relazione al peso complessivo del fegato.[25]
Uno studio che utilizzava Indole-3-Carbinol ha rilevato che le iniezioni giornaliere di 5mg nell’intestino sono state in grado di attenuare l’aumento previsto di grasso corporeo associato a una dieta ricca di grassi/calorie.[26]
Se si rapporta questa dose utilizzata in topi da laboratorio in una adatta per un essere umano adulto di 80kg si arriverebbe a circa 30mg al giorno. Se fosse somministrato per via orale probabilmente si avrebbe bisogno di una dose teoricamente più alta per ipotizzarne una qualche efficacia in tal senso.
È stato notato che il recettore degli idrocarburi arilici (AhR) ha un ruolo in alcune cellule immunitarie e nelle cellule natural killer (NK) l’attivazione di questo recettore (osservata con 10µM di 3,3′-diindolilmetano[27]) può aumentare la produzione di IFN-γ e funzione effettrice, aumentando così la loro inibizione della crescita delle cellule tumorali.[27]
Cellule Natural Killer (NK)
È stato notato che il 3,3′-Diindolylmethano (DIM) attiva sia il sottoinsieme alfa del recettore degli estrogeni (ERα)[28] che il sottoinsieme beta (ERβ),[29][30] con promozione da parte della molecola della crescita cellulare tramite ERα[ 28] non essendo un ligando diretto[31] mentre anche l’aumento della segnalazione tramite ERβ (15μM) sembra essere mediato indirettamente.[29][30] L’attivazione di ERα può dipendere dal tipo di cellula, poiché concentrazioni simili (10-15 μM; la concentrazione più bassa proposta per essere raggiunta tramite una dieta ricca di crocifere[32]) hanno mostrato efficacia nell’agire su questo recettore nel cancro al seno MCF7 e T47D cellule [28] ma non cellule MDA-MB-231 o HeLa,[29] o può essere dovuto alla sensibilità, poiché anche nelle cellule reattive concentrazioni più elevate (50μM) non riescono a causare una risposta.[28] È noto che l’attivazione indiretta è mediata prevalentemente dall’attivazione di PKA[29][31] che poi attiva MAPK e CREB.[31]
Recettore degli Estrogeni alfa (ERα), noto anche come NR3A1 (sottofamiglia del recettore nucleare 3, gruppo A, membro 1).
La maggiore concentrazione di DIM sembra indurre geni sensibili ad AhR nelle cellule del cancro al seno (CYP1A1 e CYP1B1[28-21]) suggerendo un diverso meccanismo dipendente dalla concentrazione. L’attivazione dell’AhR di per sé induce la produzione di alcuni di questi enzimi di fase I[33] che è un meccanismo di estrogenicità (attraverso l’aumento dell’attività dell’Aromatasi) osservato con pochi estrogeni ambientali[34] ma a causa della minore affinità del DIM verso l’AhR rispetto alla selezionare degli estrogeni ambientali (PCB, diossine e PAH) la combinazione dei due può comportare una minore estrogenicità relativa rispetto ai soli estrogeni ambientali.[35][36][37]
Il DIM è stato implicato nella modifica degli estrogeni preesistenti in altri metaboliti. Il processo di 2-idrossilazione, probabilmente secondario all’attivazione di AhR,[38] può aumentare il rapporto tra 2-idrossiestrone e 16α-idrossiestrone, che si pensa sia un profilo meno estrogenico dato dagli estrogeni.[39] I processi di 4-idrossilazione e 16-idrossilazione non sembrano significativamente influenzati.[40] È stato osservato che l’Indolo-3-Carbinolo induce la formazione di 2-idrossiestrone secondario ad un aumento del processo di 2-idrossilazione[41] e l’integrazione orale di DIM (108mg) nelle donne con anamnesi di carcinoma mammario in fase iniziale aumenta l’incremento delle vie urinarie. concentrazioni di 2-idrossiestrone (insieme a un aumento non significativo del rapporto tra 2-idrossiestrone e 16α-idrossiestrone.[42] Nei ratti trattati con I3C nella dieta per un periodo di tempo prolungato 200-1.000ppm sembravano essere efficaci nell’aumentare la 2-idrossilazione dell’Estradiolo con l’efficacia raggiunta quasi al doppio di circa 600-1.000ppm (17,6-36,3mg/kg),[32] traducendosi in circa 3-6mg/kg in un essere umano adulto.
2-Idrossiestrone
Le iniezioni di DIM nei ratti per due settimane prima dell’irradiazione corporea totale hanno fatto notare miglioramenti dose-dipendenti della sopravvivenza (fino al 60% da 75 mg/kg), e mentre 7,5mg/kg erano inefficaci se somministrati in questo periodo di tempo mentre una singola dose un giorno prima della irradiazione è sembrato conferire il 55% di sopravvivenza.[43] Si pensava che questo effetto protettivo fosse dovuto all’attivazione dell’atassia-teleangectasia mutata (ATM), un enzima riparatore che aumenta l’attività in risposta al danno genetico,[44] osservato con DIM 300nM ritenuto secondario all’inibizione di PP2A (MRE11 e BRCA1 anche richiesto);[43] PP2A normalmente si complessa con ATM mantenendolo in uno stato inattivo e la sua inibizione consente ad ATM di diventare iperattivo in risposta al danno genetico.[48]
Nel tessuto normale, il DIM (300nM) può attivare la via di riparazione genetica ATM in risposta al danno da irradiazione in modo dipendente da BRCA1 (uno dei suoi bersagli[43]) senza aumentare la sopravvivenza delle cellule del cancro al seno (MDA-MB-231[43]); ci sono alterazioni note in questo percorso in alcuni tumori al seno in cui BRCA1 è ridotto mentre l’ATM stesso sembra essere iperattivo ed è stato notato che l’integrazione orale di 300mg di DIM aumenta i livelli di mRNA di BRCA1 dopo 4-6 settimane di integrazione (misurata nei globuli bianchi) nelle donne che avevano una mutazione a bassa attività.[49] Alcuni studi sugli animali (usando DIM o il suo precursore I3C) che trovano effetti antitumorali sulle cellule del cancro al seno notano che questi cambiamenti si verificano insieme all’aumento della 2-idrossilazione dell’Estradiolo,[50] che sembra essere dose-dipendente fino a dosi orali molto grandi (5.000ppm nei topi o oltre 10g/kg rispetto al peso corporeo).[50]
Idrossilazione dell’Estradiolo
Nei ratti, l’ingestione orale di Indolo-3-Carbinolo (I3C) per una settimana prima dell’induzione del cancro mammario tramite DMBA ha ridotto significativamente l’incidenza (70-90%) e la molteplicità (91-96%) rispetto al controllo cancerogeno,[50] dimostrando efficacia anche sul cancerogeno ad azione diretta N-Nitroso-N-metilurea ma in misura minore (riduzione del 65% della molteplicità).[50] Anche la crescita tumorale spontanea piuttosto che indotta da tossine sembra essere appena dimezzata in uno studio (della durata di 250 giorni) in ratti alimentati con 64-128mg/kg di I3C nella dieta (l’assunzione stimata rispetto al peso corporeo è di 4,8-9,6g/kg) rispetto al controllo, con anche la molteplicità in qualche modo ridotta.[50]
Nei ratti predisposti al cancro dell’endometrio (ratti Donryu) trattati con livelli dietetici di Indolo-3-Carbinolo (I3C; 200-1.000ppm) e valutati per un periodo sperimentale prolungato, i tassi di neoplasie spontanee nell’utero dopo 660 giorni erano significativamente più alti nei controlli (38%) piuttosto che negli esemplari trattati a bassa dose di I3C (25%) con 600-1.000ppm con prestazioni uguali (14-16%);[32] questo effetto è stato osservato insieme all’aumento della 2-idrossilazione dell’Estradiolo.[32]
È stato notato che il DIM antagonizza gli effetti del Diidrotestosterone (DHT) nelle cellule del cancro prostatico (LNCaP e PC-3) di oltre il 50% a una concentrazione di 1μM in modo dipendente dal Recettore degli Androgeni, sembrava essere un antagonista diretto al recettore con affinità simile a Casodex (Bicalutamide).[51] Gli effetti antitumorali del DIM a livello della cellula prostatica non sembrano essere completamente dipendenti da questo recettore sebbene non siano dipendenti da p53 (cellule DU145[42]) e possono indurre l’arresto cellulare in un modo dipendente dall’induzione di p27 (Kip1 ) tramite Sp1 (10μM),[52] due proteine che tendono ad avere una minore attività nelle cellule della prostata androgeno-indipendenti.[53] Questa era l’attivazione di p38 a valle[52] nota che si verifica con DIM anche in altre cellule tumorali.[53]
Bicalutamide
Conclusioni sul uso di I3C o DIM per il controllo estrogenico:
Nel tessuto mammario, ma anche in altri tessuti come quello adiposo, il CYP19 (Aromatasi) catalizza le fasi finali della conversione degli androgeni (Testosterone o Androstenedione) in estrogeni (rispettivamente 17β-Estradiolo o Estrone). Ora sappiamo che il I3C, maggiormente per via della sua conversione in DIM, riduce l’espressione di CYP19 nelle cellule mammarie non tumorali e tumorigeniche estrogeno-responsive (ER+), mentre l’espressione di CYP19 è aumentata nelle cellule mammarie tumorigeniche estrogeno-indipendenti (ER-) trattate con I3C/DIM [54]. Tale effetto potrebbe verificarsi a livello sistemico il che potrebbe comportare un uso di integratori di I3C o DIM come mezzo di controllo estrogenico in quei soggetti nei quali il CYP19 viene espresso in maniera maggiore anche in situazioni di terapia ormonale sostitutiva (vedi TRT).
Ruolo dell’Aromatasi nella sintesi degli Estrogeni.
Come abbiamo visto, gli enzimi metabolizzanti di fase I, CYP1A1, CYP1A2 e CYP1B1, sono stati coinvolti nel metabolismo ossidativo degli estrogeni. Il 17β-Estradiolo può essere convertito in 2-idrossiestradiolo (2HE2) e 4-idrossiestradiolo (4HE2) rispettivamente da CYP1A1/2 e CYP1B1. 2HE2 e 4HE2 sono ulteriormente metabolizzati a 2- e 4-metossimetaboliti dall’enzima di fase II, catecol-O-metiltransferasi (COMT) [55]. Il 2HE2 è un agente non cancerogeno con un potenziale estrogenico più debole del 17β-estradiolo, mentre il 4-HE2 può essere convertito in radicali liberi che possono formare addotti del DNA e promuovere la carcinogenesi [56-57]. In diverse linee cellulari di cancro al seno, è stato dimostrato che I3C e DIM, in particolare, sovraregolano l’espressione di CYP1A1, CYP1A2 e CYP1B1 a livello di trascritto (mRNA) ma non a livello di proteina [58]. Inoltre, gli estrogeni endogeni 17β-Estradiolo ed Estrone possono essere metabolizzati irreversibilmente a 16a-idrossiestrone (16HE1) [59]. A differenza del 2-idrossiestrone (2HE1), il 16HE1 è altamente estrogenico ed è stato scoperto che stimola la proliferazione di diverse linee cellulari tumorali sensibili agli estrogeni [60-61]. È stato ipotizzato che spostare il metabolismo del 17β-Estradiolo verso 2HE1 e lontano da 16HE1, potrebbe ridurre il rischio di tumori sensibili agli estrogeni, come il cancro al seno [62]. Negli studi clinici controllati, l’integrazione orale con I3C o DIM ha costantemente aumentato le concentrazioni urinarie di 2HE1 oi rapporti urinari 2HE1:16HE1 nelle donne [63-64]. Tuttavia, ampi studi caso-controllo e prospettici di coorte non sono riusciti a trovare associazioni significative tra i rapporti urinari 2HE1:16HE1 e il rischio di cancro al seno e all’endometrio [65-66].
16a-idrossiestrone (16HE1)
Gli estrogeni endogeni, compreso il 17β-Estradiolo, esercitano i loro effetti estrogenici legandosi a specifici recettori nucleari chiamati Recettori per gli Estrogeni (ER). All’interno del nucleo, gli ER attivati dagli estrogeni possono legarsi a specifiche sequenze di DNA, note come Elementi di Risposta agli Estrogeni (ERE), nei promotori dei geni che rispondono agli estrogeni. I complessi estrogeno-ER legati all’ERE agiscono come fattori di trascrizione reclutando proteine coattivatrici e fattori di rimodellamento della cromatina nei promotori, innescando così la trascrizione dei geni bersaglio [67]. Come sappiamo, esistono due principali sottotipi di ER, ERα ed ERβ, codificati rispettivamente da due geni separati ESR1 e ESR2. Il ERα è il principale driver dell’effetto proliferativo degli estrogeni, mentre l’espressione del ERβ è stata inversamente associata alla tumorigenesi della ghiandola mammaria [68]. Livelli elevati di ERα promuovono la proliferazione cellulare nel seno e nell’utero, aumentando probabilmente il rischio di sviluppare tumori sensibili agli estrogeni [69].
Nelle cellule del cancro al seno umano sensibili agli estrogeni fatte interagire con il 17β-Estradiolo, è stato scoperto che l’I3C inibisce la trascrizione dei geni sensibili agli estrogeni senza legarsi né al ERβ né al ERα [70-71]. In effetti, è stato dimostrato che il legame di I3C ad AhR innesca la degradazione dipendente dal proteasoma di ERα [72]. La perdita del ERα indotta da I3C ha portato alla sotto-regolazione dei prodotti genici che rispondono al ERα come il fattore di trascrizione GATA3. Poiché GATA3 regola la trascrizione del gene codificante ERα ESR1, l’I3C ha impedito la sintesi di nuove trascrizioni e proteine ERα, sopprimendo infine la via di segnalazione ERα. L’interruzione dell’anello cross-regolatorio GATA3/ERα da parte del I3C ha infine arrestato la proliferazione cellulare ERα-dipendente [73]. I prodotti di condensazione acida del I3C che legano e attivano AhR possono anche inibire la trascrizione dei geni sensibili agli estrogeni competendo per i co-attivatori o aumentando la degradazione del ERα [74]. Il trattamento con I3C ha anche influenzato l’espressione di altri geni ERα-responsivi, compresi quelli che codificano per il Recettore del Fattore di Crescita Insulino-Simile-1 (IGFR1) e il substrato del recettore dell’Insulina-1 (IRS-1), coinvolti nella proliferazione cellulare e deregolati nel cancro al seno ( Figura seguente) [75].
Recettore del Fattore di Crescita Insulino-Simile-1 (IGFR1)
In base alle informazioni riportate in letteratura, sebbene limitate, possiamo ipotizzare che una supplementazione di I3C o DIM possa essere funzionale ad un controllo estrogenico in soggetti trattati con terapia sostitutiva del Testosterone (TRT) che presentano superiori espressioni dell’enzima Aromatasi legate a fattori non controllabili attraverso la semplice dieta e l’allenamento (vedi riduzione della massa grassa). Parliamo quindi di condizioni di lieve iperestrogenemia o alterata estrogenemia soggetto-sensibile (cioè non quantificabile con l’intervallo di riferimento standard ma solo con analisi dei sintomi legati ad una aumentata attività estrogenica). La sua efficacia di controllo estrogenico potrebbe però non essere sufficiente in contesti di uso di dosi sovrafisiologiche di AAS aromatizzabili, specie se queste superano i 180mg di Testosterone (netto) a settimana [dati raccolti aneddoticamente].
L’I3C è disponibile come prodotto da banco senza prescrizione medica anche in Italia, da solo o in combinazione con altre molecole. Il dosaggio varia tra 200 mg/die e 800 mg/die [76]. L’integrazione di I3C ha aumentato le concentrazioni urinarie di 2HE1 negli adulti a dosi da 300 a 400 mg/die [77]. Dosi di I3C di 200 mg/die o 400 mg/die hanno migliorato la regressione della neoplasia intraepiteliale cervicale (CIN) in uno studio clinico preliminare [78]. L’I3C in dosi fino a 400 mg/die è stato usato per trattare la papillomatosi respiratoria ricorrente (vedi Trattamento della malattia) [79-80]. In caso di lieve iperestrogenemia o alterata estrogenemia soggetto-sensibile, il dosaggio di 400mg/die ha portato benefici apprezzabili, sebbene con risposte soggettive, nel giro di 7-14 giorni di somministrazione continua [dati raccolti aneddoticamente].
Il DIM è anch’esso disponibile senza prescrizione medica come integratore alimentare da banco, nonostante sia più difficile da trovare, da solo o in combinazione con altre molecole. In un piccolo studio clinico, l’integrazione di DIM alla dose di 108mg/die per 30 giorni ha aumentato l’escrezione urinaria di 2HE1 nelle donne in postmenopausa con anamnesi di cancro al seno [81]. Dosaggi di 100-200mg/die si sono dimostrati discretamente efficaci in caso di lieve iperestrogenemia o alterata estrogenemia soggetto-sensibile in individui in terapia sostitutiva del Testosterone [dati raccolti aneddoticamente].
Leggeri aumenti delle concentrazioni sieriche dell’enzima epatico, alanina aminotransferasi (ALT) sono stati osservati in due donne che hanno assunto dosi non specificate di integratori di I3C per quattro settimane [64]. Una persona ha riportato un’eruzione cutanea durante l’assunzione di 375 mg/die di I3C [82]. Alte dosi di I3C (800 mg/die) sono state associate a sintomi di squilibrio e tremore, che si sono risolti quando la dose è stata ridotta [83]. In uno studio di fase I in donne ad alto rischio di cancro al seno, 5 partecipanti su 20 hanno manifestato sintomi gastrointestinali con dosi singole ≥600 mg, sebbene altri non abbiano avuto effetti avversi con dosi singole fino a 1.200mg [84]. Non sono stati segnalati effetti avversi con il consumo giornaliero di 400mg di I3C per quattro settimane [84]. In alcuni modelli animali, è stato scoperto che l’integrazione di I3C migliora lo sviluppo del cancro indotto dal cancerogeno quando somministrato cronicamente dopo il cancerogeno [85-86]. Quando somministrato prima o contemporaneamente al cancerogeno, l’I3C orale ha inibito la tumorigenesi in modelli animali di tumori della ghiandola mammaria [87-88], dell’utero [89], dello stomaco [90], del colon [91-92], del polmone [93] e fegato [94-95]. Sebbene non siano noti gli effetti a lungo termine dell’integrazione di I3C sul rischio di cancro nell’uomo, i risultati contraddittori degli studi sugli animali hanno portato diversi esperti a mettere in guardia contro l’uso diffuso di integratori di I3C e DIM negli esseri umani fino a quando i loro potenziali rischi e benefici non saranno meglio compresi [86-96-97]. La sicurezza degli integratori contenenti I3C o DIM durante la gravidanza o l’allattamento non è stata stabilita [98].
Non sono state segnalate interazioni farmacologiche con l’integrazione di I3C o DIM nell’uomo. Tuttavia, l’evidenza preliminare che I3C e DIM possono aumentare l’attività del CYP1A2 [99-100] suggerisce che l’integrazione con I3C o DIM può ridurre le concentrazioni sieriche dei farmaci metabolizzati dal CYP1A2 [101]. Sia I3C che DIM aumentano modestamente l’attività del CYP3A4 nei ratti quando somministrati cronicamente [102]. Questa osservazione aumenta il potenziale di interazioni farmacologiche avverse nell’uomo poiché il CYP3A4 è coinvolto nel metabolismo di circa il 60% dei farmaci terapeutici. L’ambiente acido dello stomaco consente alle molecole I3C di condensare e generare un numero di oligomeri I3C biologicamente attivi. I farmaci che bloccano la produzione di acidi dello stomaco, come gli antiacidi, gli antagonisti del recettore dell’istamina2 (H2) e gli inibitori della pompa protonica, probabilmente impedirebbero la generazione di DIM e ICZ. Tuttavia, non è noto se questi farmaci limitino le attività biologiche attribuite all’I3C e ai suoi derivati [98].
Si esorta il lettore ad avere cautela nell’uso delle summenzionate molecole. A causa del loro effetto sui livelli di Estrogeni (ricordo che gli estrogeni hanno, tra le altre cose, un impatto significativo sulla funzione cerebrale, metabolismo osseo e comportamento/attività sessuale).[103][104] Prima di procedere con il trattamento assicurarsi, per via di analisi specifiche e consulto di specialisti, che i livelli estrogenici e/o la loro attività tissutale necessitino di un controllo per via di trattamento con molecole esogene.
Park, N. I.; Kim, J. K.; Park, W. T.; Cho, J. W.; Lim, Y. P.; Park, S. U. (2010). “An efficient protocol for genetic transformation of watercress (Nasturtium officinale) using Agrobacterium rhizogenes”. Molecular Biology Reports. 38(8): 4947–4953.
“indole-3-methanol (CHEBI:24814)”. Chemical Entities of Biological Interest (ChEBI). European Bioinformatics Institute. Retrieved 2016-03-25.
Dashwood, R. H.; Arbogast, D. N.; Fong, A. T.; Pereira, C.; Hendricks, J. D.; Bailey, G. S. (1989). “Quantitative inter-relationships between aflatoxin B1 carcinogen dose, indole-3-carbinol anti-carcinogen dose, target organ DNA adduction and final tumor response”. Carcinogenesis. 10 (1): 175–181.
Belous AR, Hachey DL, Dawling S, Roodi N, Parl FF. Cytochrome P450 1B1-mediated estrogen metabolism results in estrogen-deoxyribonucleoside adduct formation. Cancer Res. 2007;67(2):812-817.
Jefcoate CR, Liehr JG, Santen RJ, et al. Tissue-specific synthesis and oxidative metabolism of estrogens. J Natl Cancer Inst Monogr. 2000(27):95-112.
Kwon YJ, Baek HS, Ye DJ, Shin S, Kim D, Chun YJ. CYP1B1 enhances cell proliferation and metastasis through induction of EMT and activation of Wnt/beta-catenin signaling via Sp1 upregulation. PLoS One. 2016;11(3):e0151598.
Park SA, Lee MH, Na HK, Surh YJ. 4-Hydroxyestradiol induces mammary epithelial cell transformation through Nrf2-mediated heme oxygenase-1 overexpression. Oncotarget. 2016;8(1):164-178.
Szaefer H, Licznerska B, Krajka-Kuzniak V, Bartoszek A, Baer-Dubowska W. Modulation of CYP1A1, CYP1A2 and CYP1B1 expression by cabbage juices and indoles in human breast cell lines. Nutr Cancer. 2012;64(6):879-888.
Ziegler RG, Fuhrman BJ, Moore SC, Matthews CE. Epidemiologic studies of estrogen metabolism and breast cancer. Steroids. 2015;99(Pt A):67-75.
Telang NT, Suto A, Wong GY, Osborne MP, Bradlow HL. Induction by estrogen metabolite 16 alpha-hydroxyestrone of genotoxic damage and aberrant proliferation in mouse mammary epithelial cells. J Natl Cancer Inst. 1992;84(8):634-638.
Yuan F, Chen DZ, Liu K, Sepkovic DW, Bradlow HL, Auborn K. Anti-estrogenic activities of indole-3-carbinol in cervical cells: implication for prevention of cervical cancer. Anticancer Res. 1999;19(3A):1673-1680.
Bradlow HL, Michnovicz JJ, Halper M, Miller DG, Wong GY, Osborne MP. Long-term responses of women to indole-3-carbinol or a high fiber diet. Cancer Epidemiol Biomarkers Prev. 1994;3(7):591-595.
Wong GY, Bradlow L, Sepkovic D, Mehl S, Mailman J, Osborne MP. Dose-ranging study of indole-3-carbinol for breast cancer prevention. J Cell Biochem Suppl. 1997;28-29:111-116.
Arslan AA, Shore RE, Afanasyeva Y, Koenig KL, Toniolo P, Zeleniuch-Jacquotte A. Circulating estrogen metabolites and risk for breast cancer in premenopausal women. Cancer Epidemiol Biomarkers Prev. 2009;18(8):2273-2279.
Zeleniuch-Jacquotte A, Shore RE, Afanasyeva Y, et al. Postmenopausal circulating levels of 2- and 16alpha-hydroxyestrone and risk of endometrial cancer. Br J Cancer. 2011;105(9):1458-1464.
Liehr JG. Is estradiol a genotoxic mutagenic carcinogen? Endocr Rev. 2000;21(1):40-54.
Ashok BT, Chen Y, Liu X, Bradlow HL, Mittelman A, Tiwari RK. Abrogation of estrogen-mediated cellular and biochemical effects by indole-3-carbinol. Nutr Cancer. 2001;41(1-2):180-187.
Meng Q, Yuan F, Goldberg ID, Rosen EM, Auborn K, Fan S. Indole-3-carbinol is a negative regulator of estrogen receptor-alpha signaling in human tumor cells. J Nutr. 2000;130(12):2927-2931.
Marconett CN, Sundar SN, Poindexter KM, Stueve TR, Bjeldanes LF, Firestone GL. Indole-3-carbinol triggers aryl hydrocarbon receptor-dependent estrogen receptor (ER)alpha protein degradation in breast cancer cells disrupting an ERalpha-GATA3 transcriptional cross-regulatory loop. Mol Biol Cell. 2010;21(7):1166-1177.
Chen I, McDougal A, Wang F, Safe S. Aryl hydrocarbon receptor-mediated antiestrogenic and antitumorigenic activity of diindolylmethane. Carcinogenesis. 1998;19(9):1631-1639.
Marconett CN, Singhal AK, Sundar SN, Firestone GL. Indole-3-carbinol disrupts estrogen receptor-alpha dependent expression of insulin-like growth factor-1 receptor and insulin receptor substrate-1 and proliferation of human breast cancer cells. Mol Cell Endocrinol. 2012;363(1-2):74-84.
Stewart ZA, Westfall MD, Pietenpol JA. Cell-cycle dysregulation and anticancer therapy. Trends Pharmacol Sci. 2003;24(3):139-145. (PubMed)
Mao CG, Tao ZZ, Chen Z, Chen C, Chen SM, Wan LJ. Indole-3-carbinol inhibits nasopharyngeal carcinoma cell growth in vivo and in vitro through inhibition of the PI3K/Akt pathway. Exp Ther Med. 2014;8(1):207-212. (PubMed)
Leem SH, Li XJ, Park MH, Park BH, Kim SM. Genome-wide transcriptome analysis reveals inactivation of Wnt/beta-catenin by 3,3′-diindolylmethane inhibiting proliferation of colon cancer cells. Int J Oncol. 2015;47(3):918-926. (PubMed)
Wong GY, Bradlow L, Sepkovic D, Mehl S, Mailman J, Osborne MP. Dose-ranging study of indole-3-carbinol for breast cancer prevention. J Cell Biochem Suppl. 1997;28-29:111-116.
Bell MC, Crowley-Nowick P, Bradlow HL, et al. Placebo-controlled trial of indole-3-carbinol in the treatment of CIN. Gynecol Oncol. 2000;78(2):123-129.
Rosen CA, Woodson GE, Thompson JW, Hengesteg AP, Bradlow HL. Preliminary results of the use of indole-3-carbinol for recurrent respiratory papillomatosis. Otolaryngol Head Neck Surg. 1998;118(6):810-815.
Rosen CA, Bryson PC. Indole-3-carbinol for recurrent respiratory papillomatosis: long-term results. J Voice. 2004;18(2):248-253.
Dalessandri KM, Firestone GL, Fitch MD, Bradlow HL, Bjeldanes LF. Pilot study: effect of 3,3′-diindolylmethane supplements on urinary hormone metabolites in postmenopausal women with a history of early-stage breast cancer. Nutr Cancer. 2004;50(2):161-167. (PubMed)
McAlindon TE, Gulin J, Chen T, Klug T, Lahita R, Nuite M. Indole-3-carbinol in women with SLE: effect on estrogen metabolism and disease activity. Lupus. 2001;10(11):779-783.
Rosen CA, Woodson GE, Thompson JW, Hengesteg AP, Bradlow HL. Preliminary results of the use of indole-3-carbinol for recurrent respiratory papillomatosis. Otolaryngol Head Neck Surg. 1998;118(6):810-815.
Reed GA, Arneson DW, Putnam WC, et al. Single-dose and multiple-dose administration of indole-3-carbinol to women: pharmacokinetics based on 3,3′-diindolylmethane. Cancer Epidemiol Biomarkers Prev. 2006;15(12):2477-2481.
Kim DJ, Han BS, Ahn B, et al. Enhancement by indole-3-carbinol of liver and thyroid gland neoplastic development in a rat medium-term multiorgan carcinogenesis model. Carcinogenesis. 1997;18(2):377-381.
Stoner G, Casto B, Ralston S, Roebuck B, Pereira C, Bailey G. Development of a multi-organ rat model for evaluating chemopreventive agents: efficacy of indole-3-carbinol. Carcinogenesis. 2002;23(2):265-272.
Grubbs CJ, Steele VE, Casebolt T, et al. Chemoprevention of chemically-induced mammary carcinogenesis by indole-3-carbinol. Anticancer Res. 1995;15(3):709-716.
Bradlow HL, Michnovicz J, Telang NT, Osborne MP. Effects of dietary indole-3-carbinol on estradiol metabolism and spontaneous mammary tumors in mice. Carcinogenesis. 1991;12(9):1571-1574.
Kojima T, Tanaka T, Mori H. Chemoprevention of spontaneous endometrial cancer in female Donryu rats by dietary indole-3-carbinol. Cancer Res. 1994;54(6):1446-1449.
Wattenberg LW, Loub WD. Inhibition of polycyclic aromatic hydrocarbon-induced neoplasia by naturally occurring indoles. Cancer Res. 1978;38(5):1410-1413.
Wargovich MJ, Chen CD, Jimenez A, et al. Aberrant crypts as a biomarker for colon cancer: evaluation of potential chemopreventive agents in the rat. Cancer Epidemiol Biomarkers Prev. 1996;5(5):355-360.
Guo D, Schut HA, Davis CD, Snyderwine EG, Bailey GS, Dashwood RH. Protection by chlorophyllin and indole-3-carbinol against 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP)-induced DNA adducts and colonic aberrant crypts in the F344 rat. Carcinogenesis. 1995;16(12):2931-2937.
Morse MA, LaGreca SD, Amin SG, Chung FL. Effects of indole-3-carbinol on lung tumorigenesis and DNA methylation induced by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) and on the metabolism and disposition of NNK in A/J mice. Cancer Res. 1990;50(9):2613-2617.
Dashwood RH, Arbogast DN, Fong AT, Hendricks JD, Bailey GS. Mechanisms of anti-carcinogenesis by indole-3-carbinol: detailed in vivo DNA binding dose-response studies after dietary administration with aflatoxin B1. Carcinogenesis. 1988;9(3):427-432.
Oganesian A, Hendricks JD, Williams DE. Long term dietary indole-3-carbinol inhibits diethylnitrosamine-initiated hepatocarcinogenesis in the infant mouse model. Cancer Lett. 1997;118(1):87-94.
Dashwood RH. Indole-3-carbinol: anticarcinogen or tumor promoter in brassica vegetables? Chem Biol Interact. 1998;110(1-2):1-5.
Lee BM, Park KK. Beneficial and adverse effects of chemopreventive agents. Mutat Res. 2003;523-524:265-278.
He YH, Friesen MD, Ruch RJ, Schut HA. Indole-3-carbinol as a chemopreventive agent in 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) carcinogenesis: inhibition of PhIP-DNA adduct formation, acceleration of PhIP metabolism, and induction of cytochrome P450 in female F344 rats. Food Chem Toxicol. 2000;38(1):15-23.
Lake BG, Tredger JM, Renwick AB, Barton PT, Price RJ. 3,3′-Diindolylmethane induces CYP1A2 in cultured precision-cut human liver slices. Xenobiotica. 1998;28(8):803-811.
Natural Medicines. Professional monograph: Indole-3-carbinol/Interactions with drugs; 2016.
Leibelt DA, Hedstrom OR, Fischer KA, Pereira CB, Williams DE. Evaluation of chronic dietary exposure to indole-3-carbinol and absorption-enhanced 3,3′-diindolylmethane in Sprague-Dawley rats. Toxicol Sci. 2003;74(1):10-21.
Introduzione al “GH Gut” – tra ipotesi e conclusioni affrettate – :
Non molto tempo fa parlai di “piaga della ginecomastia” nel Bodybuilding agonistico e non, evidenziando quanta poca cura nella gestione estrogenica vi fosse (e vi sia) nel ambiente culturistico, sia da parte dei singoli atleti che (cosa assai più importante e grave) dei preparatori o presunti tali. Ora, però, in linea di coerenza con la denuncia di una così grave deturpazione estetico-salutistica, mi accingo a trattare un altro problema, frutto dell’ignoranza, che affligge il Bodybuilding agonistico, mi riferisco alla così detta “GH Gut” o “Palumboismo” (definizione nata dal nome del famoso bodybuilder Dave Palumbo che fu uno dei primi a mostrare tale deturpazione estetica).
Dave Palumbo (da sinistra a destra): anni 90 (pre-“GH Gut”) Vs primi anni 2000 .
Come molti di voi già sapranno, con il termine “GH Gut” ci si riferisce ad una condizione caratterizzata da una pronunciata dilatazione addominale in un soggetto (bodybuilder) con una “bf” sensibilmente bassa .
Le ipotesi sulla causa scatenante tale condizione si sono susseguite nel tempo comprendendo assurdità e letture conclusioni semplicistiche.
Una di queste ipotesi venne diffusa anche grazie ai libri del compianto A.L.Rea, il quale affermava già nel suo libro “Chemichal Muscle Enhancement” , senza esternazione di validità di ipotesi, che la causa del “GH Gut” fosse l’ipertrofia gastrointestinale. Argomentava la sua affermazione dicendo che nel tratto gastrointestinale vi fosse un numero maggiore di recettori per l’IGF-1 rispetto al tessuto muscolo-scheletrico. Peccato però che la regolazione dell’ipertrofia/iperplasia gastrointestinale sia strettamente controllata, e che ogni sovra-stimolazione rientra in breve tempo attraverso meccanismi omeostatici come la sotto-regolazione dei recettori per l’IGF-1.[1] Inoltre, una crescita così spropositata della componente viscerale causerebbe gravi problemi di funzionalità organica (per esempio, occlusione intestinale data dall’alterata motilità gastrica) molto prima che l’atleta riesca a mostrare alterazioni visivamente così accentuate e calcare il palco.
Un altra ipotesi fantasiosa, e tutta “nostrana”, afferma che la suddetta condizione è conseguenza di un accumulo massivo di grasso viscerale il quale, notoriamente, è correlato all’insulino–resistenza (IR). Purtroppo però tale tesi è viziata da un errore di computo ed esso risiede nella reale correlazione tra soggetti sottoposti a somministrazione di PEDs i quali possono ridurre l’accumulo di grasso viscerale e aumentarne la sua mobilitazione anche in condizione di non ottimale IR, e il raggiungimento di un tale deposito di grasso intra-organico (oltretutto in ipocalorica) degno di un grande obeso (IMC maggiore di 40 kg/m²) capace di causare una così prominente dilatazione addominale.[2] Oltretutto, le analisi DEXA (il gold standard per la valutazione della composizione corporea) non hanno mai mostrato una presenza così invasiva di grasso viscerale in questo tipo di atleti.
Ma allora quale è la causa del “GH Gut” nel Bodybuilding? Diciamo che per rispondere a questa domanda bisogna dire che è cosa semplice trovare “l’arma del delitto” (dieta ipercalorica in cronico, abuso di GH e Insulina) mentre il capire come si è arrivati al risultato finale è più difficile da individuare… ma ve lo spiegherò…
Le cause del “GH Gut”:
La causa del “GH Gut” nel Bodybuilding, tralasciando ovviamente il discorso del gonfiore addominale ansia-correlato, è di triplice natura:
Alimentazione ipercalorica;
Abuso cronico di GH;
Abuso cronico di Insulina.
Questi tre fattori, anche se presi singolarmente, possono causare una condizione di insulino-resistenza che, se protratta e cronicizzata, può sfociare in una condizione patologica denominata “gastroparesi”.[3]
La gastroparesi è una complicanza cronica del diabete, o marcata IR correlata anche da abuso di GH, espressione della presenza di una neuropatia che provoca un rallentato svuotamento gastrico dopo un pasto solido, in assenza di cause meccaniche ostruttive. In pratica, i muscoli dello stomaco non funzionano in modo corretto.
Quindi, l’abuso di GH e Insulina possono anche causare indirettamente il “GH Gut”, ma il meccanismo con il quale lo provocano è molto diverso da quello che la maggior parte di voi crede.
Non è raro, infatti, che un bodybuilder di grandi dimensioni assuma in “Bulk” più di 8000Kcal nel tentativo di aumentare la massa muscolare. Nel frattempo, lo stesso bodybuilder potrebbe assumere GH e/o Insulina creando, nel medio/lungo termine un peggioramento sostanziale della resistenza all’insulina con comparsa di sintomi pre-diabetici.
Inoltre, quando l’intestino è sottoposto ad una sovrabbondanza di cibo, può svilupparsi un accumulo di batteri nell’intestino tenue e la salute dell’organo viene compromessa mentre il corpo tenta di rimediare alla situazione. Una sovrabbondanza di batteri nell’intestino tenue porta a gonfiore e gas significativi e ad una consequenziale ed evidente distensione addominale. A differenza dell’intestino crasso (colon), l’intestino tenue non ha solitamente un gran numero di batteri. E, come già detto in precedenza, quando un bodybuilder consuma troppe calorie, può iniziare a diventare resistente all’insulina e l’abuso di GH e Insulina contribuiscono all’aggravarsi di ciò acutizzando lo stato di iperglicemia e alterazione del metabolismo glucidico e lipidico.
Quando si instaura una condizione di insulino-resistenza e vi sono livelli di glicemia ematica cronicamente elevati, lo svuotamento gastrico viene ritardato e l’intestino inizia a perdere la capacità di contrarsi in modo altrettanto efficace. Di conseguenza, il transito intestinale è gravemente compromesso.
Se la velocità del transito intestinale è influenzata negativamente dall’instaurarsi di un insulino-resistenza marcata, può verificarsi un riflusso batterico dal colon nell’intestino tenue, dove viene colonizzato da batteri del colon. Ciò si traduce in una eccessiva proliferazione batterica nell’intestino tenue e, in definitiva, in un peggioramento del “GH Gut”. Man mano che la resistenza all’insulina peggiora, la motilità intestinale viene parallelamente ostacolata, aggravando il problema.
Normalmente, forti contrazioni muscolari autonome (quindi non percepibili) spingono il cibo ingerito attraverso il tratto digestivo. In caso di gastroparesi, i muscoli della parete dello stomaco lavorano poco o niente; ciò impedisce allo stomaco di svuotarsi correttamente e completamente interferendo con i processi della digestione e causando un marcato gonfiore addominale.
La gastroparesi si associa in genere a scarso controllo dei valori della glicemia, neuropatia, retinopatia e nefropatia.
Non è sempre chiaro che cosa determini la comparsa di gastroparesi. Sembrano essere coinvolti molti meccanismi e/o eventuali interazioni tra di essi: fluttuazioni croniche dei valori della glicemia, neuropatia, anomalie in alcune cellule interstiziali poste tra lo stomaco e l’intestino (Cellule interstiziali di Cajal), utilizzo di farmaci incretino-mimetici per normalizzare i picchi glicemici postprandiali e forse – secondo alcuni autori – fattori psicosomatici. In molti casi si pensa che la gastroparesi sia causata da un danno a un nervo (neuropatia) che controlla i muscoli dello stomaco (nervo vago). Il nervo vago consente di gestire i complessi processi del tratto digestivo. Un nervo vago danneggiato non riesce a inviare i segnali ai muscoli dello stomaco. Ciò può far sì che il cibo rimanga nello stomaco più a lungo, invece di muoversi normalmente verso l’intestino tenue per essere digerito. Il nervo vago può essere danneggiato da malattie, come il diabete (neuropatia diabetica), abuso di GH e/o Insulina, o da un intervento chirurgico allo stomaco.
Locazione e innervazioni del Nervo Vago.
I medici usano diversi test per diagnosticare la gastroparesi ed escludere condizioni che possano causare sintomi simili (diagnosi differenziale). I test possono includere:
La misurazione dello svuotamento gastrico. Ci sono diverse metodiche per valutarla, dirette ed indirette.
L’endoscopia del tratto gastrointestinale superiore. Un’endoscopia può aiutare a escludere altre condizioni che possano causare un ritardo dello svuotamento gastrico.
Quindi, per trattare la gastroparesi è necessario prima di tutto identificare la condizione di base che l’ha provocata. Per esempio, se il diabete o la cronicizzazione subclinica del IR è la causa della gastroparesi, il medico darà indicazioni per controllare Insulina basale, Glicemia a digiuno e dopo i pasti, Emoglobina Glicata e test della curva del Glucosio e Insulina.[4][5] La terapia della gastroparesi ha come obiettivo il controllo dei sintomi e il mantenimento di un adeguato stato nutrizionale; purtroppo questa finalità appare spesso difficile e insoddisfacente in termini di risultati. Molti culturisti con gastroparesi hanno un introito calorico inferiore rispetto a quello di mantenimento e/o un deficit sia di macro che di micronutrienti. L’introito calorico necessario in questi casi può essere calcolato moltiplicando 33 kcal con il peso corporeo attuale in chilogrammi. Il trattamento di fondo consiste nell’assunzione di piccoli pasti a basso contenuto di fibre, con l’aggiunta, se necessario, di farmaci procinetici o farmaci antiemetici.
Due delle più note molecole appartenenti alla classe dei farmaci procinetici (Betanecolo) e antiemetici (Clorpromazina).
Un dietista/nutrizionista potrà selezionare tutti gli alimenti che siano più facili da digerire, a seconda dei casi.[6] Da tenere presente che grassi e fibre tendono a ritardare lo svuotamento gastrico per questo potrebbe essere utile limitarne il consumo in base alle esigenze pertanto che la condizione perdura. Lo specialista potrebbe consigliare anche alcuni comportamenti idonei, per esempio:
mangiare piccoli pasti, a piccoli intervalli;
consumare poche fibre;
scegliere alimenti a basso contenuto di grassi;
evitare frutta e verdura fibrosa, come le arance e i broccoli, che possono provocare bezoari;
provare a frullare i cibi e a consumare più zuppe;
bere acqua durante ogni pasto;
muoversi dopo aver mangiato.
Un bezoario è un agglomerato compatto di materiale parzialmente digerito o non digerito, che si verifica in genere nello stomaco. I bezoari gastrici possono verificarsi in tutte le fasce di età e spesso si verificano in pazienti con disturbi del comportamento, svuotamento gastrico anormale o alterazione dell’anatomia gastrointestinale. Molti bezoari sono asintomatici, ma alcuni causano l’insorgenza di sintomi. Alcuni bezoari possono essere sciolti chimicamente, altri richiedono la rimozione endoscopica e alcuni altri richiedono anche l’intervento chirurgico.
È imperativo cercare di ottimizzare il controllo dei valori glicemici per minimizzare i sintomi acuti della gastroparesi e migliorare lo svuotamento gastrico così da influenzare positivamente la regressione della condizione. L’iperglicemia ritarda lo svuotamento gastrico anche in assenza di neuropatia o miopatia; inoltre, può inibire l’effetto di accelerazione dei farmaci procinetici. Per cui è importante mettere in atto insieme al proprio specialista di riferimento delle strategie d’intervento per minimizzare i picchi iperglicemici postprandiali.
I farmaci per il trattamento della gastroparesi[7]possono includere:
Farmaci per controllare la nausea e il vomito (antiemetici)
Farmaci per stimolare i muscoli dello stomaco (procinetici). Gli effetti collaterali di questi farmaci sono importanti e questo andrà considerato insieme al medico.
In caso di inefficacia di questi farmaci, come ulteriore terapia sono stati proposti dei trattamenti sperimentali, per esempio l’impiego della tossina botulinica, al fine di ridurre il tono neuromuscolare e di conseguenza lo spasmo del piloro, o degli analoghi della somatostatina, per ridurre l’entità della secrezione gastrica e altre molecole.
Altri farmaci procinetici sono in corso di studio: agonisti della motilina, agonisti della grelina, nuovi agonisti 5-HT4. Nei casi gravi, che non rispondono alla terapia medica, può rendersi necessario il ricorso a terapie più invasive, come la nutrizione enterale mediante digiunostomia endoscopica, la gastrectomia, la digiunostomia o altri tipi di intervento chirurgico.
Strutture chimiche degli agenti procinetici (agonisti dei recettori 5-HT4). (A) Velusetrag, un agonista del recettore 5-HT4, aumenta significativamente il transito intestinale e del colon. (B) La Prucalopride, un derivato diidro-benzofurancarbossammide di prima classe, è un agonista altamente selettivo del recettore 5-HT4. (C) Tegaserod, il primo agonista del recettore 5-HT4 per il trattamento IBS-C a breve termine nelle donne. (D) Naronapride, un agonista del recettore 5-HT4 altamente selettivo, accelera significativamente il transito globale del colon.
Un trattamento non farmacologico alternativo alla chirurgia, recentemente proposto per la terapia della gastroparesi, è rappresentato dalla gastrostimolazione elettrica (GES) a mezzo di elettrodi posizionati sulla parete muscolare dello stomaco, i cui risultati appaiono molto incoraggianti. La GES migliora nausea, vomito, qualità della vita e stato nutrizionale nei pazienti con gastroparesi refrattaria.
La gastroparesi può causare diverse complicazioni, per esempio:
Perdita di peso e malnutrizione. La gastroparesi può rendere difficile assorbire e digerire in modo corretto le sostanze nutrienti.
Crescita eccessiva di batteri nello stomaco. Il residuo alimentare che rimane nello stomaco può iniziare a fermentare e a rompere l’equilibrio locale tra batteri buoni (microbiota) e cattivi.
Frazioni di cibo non digerito che formano masse solide (bezoari) e rimangono nello stomaco. I bezoari possono causare nausea e vomito e possono anche essere pericolosi.
Fluttuazioni della glicemia. La gastroparesi anche se non causa il diabete, può determinare ed essere suscettibile alle variazioni nei livelli di zucchero nel sangue.
Alterazioni del microbiota intestinale, specie se marcate, oltre a causare disturbi gastrointestinali come gonfiore addominale da fenomeni fermentativi, può portare a disturbi di natura psicologica i quali possono peggiorare in forma di risposta somatica la dilatazione addominale.
Un altro dato che dimostra la reale infondatezza del mito dell’ipertrofia/iperplasia gastrointestinale è rappresentato anche dalla piuttosto semplice reversibilità della condizione, un fattore che molte persone che predicano questo mito sembra non prendere in considerazione nonostante ci siano stati molti Bodybuilder professionisti di successo che hanno sfoggiato una prominente dilatazione addominale sul palco per poi tornare successivamente con linee decisamente più armoniose. I loro organi si sono improvvisamente rimpiccioliti e riorganizzati? Con tutta probabilità, la risposta è no. Il loro addome non sporgeva perchè le viscere lo spingevano dall’interno verso l’esterno, era semplicemente una conseguenza che rispecchia una salute intestinale compromessa (che causava gonfiore) e distensione da gastroparesi.
Nota: anche condizioni di forte ansia possono peggiorare o causare una sensibile dilatazione addominale durante la Peak Week e il giorno del contest. Il controllo dello stress è essenziale per evitare che ciò si presenti.
Ben Pakulski è un ottimo esempio da utilizzare per sfatare la teoria della crescita intestinale attribuita alla condizione del “GH Gut”. Per questo atleta è stato sufficiente abbandonare l’obbiettivo di un aumento drastico dell’ipertrofia ed è stato in grado di tenere sotto controllo la sua salute intestinale, risolvendo completamente il suo precedentemente mostrato “GH Gut”. Se i suoi organi fossero davvero cresciuti, non sarebbe stato in grado di liberarsene così facilmente e rapidamente.
Ben Pakulski “Reverse GH Gut”
Ben Pakulski ha evidentemente perso un po’ di massa muscolare tra i suoi spettacoli precedenti, durante i quali mostrava una marcata dilatazione addominale, e i suoi spettacoli più recenti nei quali aveva una fantastica posa in vacuum e nessun problema di dilatazione, e questo può essere attribuito esclusivamente alla sua decisione di non perseguire più volumi muscolari enormi mangiando un monte calorico estremamente alto e abusando di GH e/o Insulina. Questo è quello che stava facendo in precedenza quando aveva orribili problemi di dilatazione addominale, mentre cercava di rimanere competitivo con i colleghi “freak”.
Roelly Winklaar è un altro ottimo esempio di bodybuilder di alto livello che ha risolto la sua condizione di “GH Gut” ed è stato in grado di continuare a fare progressi. Può essere attribuito al suo uso di un bustino? Decisamente no. La mia ipotesi è che abbia più a che fare con i cambiamenti nella dieta e nella supplementazione farmacologica. Se date un’occhiata ai vecchi spettacoli di Roelly, potrete vedere chiaramente che era uno dei peggiori casi di “GH Gut”. Ma, ciò nonostante, ora può quasi riuscire a fare il vacuum sul palco.
Roelly Winklaar “Reverse GH Gut” (da sinistra a destra: forma presentata nel 2015 e nel 2018).
E’ corretto specificare che un bodybuilder può letteralmente passare dall’avere la possibilità di esibire un ottimo vacuum all’avere una estreme dilatazione addominale / GH Gut durante la notte semplicemente consumando quantità eccessive di carboidrati, con conseguente effetto fermentativo il quale è legato ad una compromissione della salute intestinale prima di salire sul palco, senza contare l’effetto dell’iperglicemia sulla gastroparesi prima esposto. Questo è il motivo per cui si vedono bodybuilder presentarsi sul palco con un enorme dilatazione addominale, e poi una settimana dopo presentarsi in un altro contest con il problema completamente risolto.
Ci sono molti altri casi di “GH Gut” nel mondo del Bodybuilding, ma quello di Phil Heath ha attirato più attenzione di tutti a causa delle sue vittorie all’Olympia che molti pensavano di non aver meritato.
Non credo che l’ernia di Phil Heath sia stata la causa dei suoi problemi di dilatazione addominale. Probabilmente l’ernia ha giocato un ruolo sul suo controllo addominale, ma resta il fatto che il suo addome è dilatato da alcuni anni e credo che la radice del problema sia la salute intestinale compromessa e metabolica. Credo anche che Phil Heath stia mostrando i primi segni di insulino-resistenza cronica, chiamata anche “palumboismo”.
Phil Heath al Mr. Olympia nel 2012 (a sinistra) e nel 2018 (a destra).
Se confrontate Phil Heath nel 2012 con Phil Heath nel 2018, l’unica differenza evidente è il suo addome. Il risultato di ciò è con molta probabilità l’insieme dell’abuso di GH e/o Insulina e il consumo eccessivo di cibo per raggiungere maggiori dimensioni muscolari per rimanere in cima alla competizione, o per lo meno questa è l’ipotesi che considero più probabile.
Phil Heath nel 2018 e nel 2020.
Conclusioni sul problema:
Da quanto detto fino a questo punto, sappiamo che la condizione denominata “GH Gut” non è legata ad una massiva crescita viscerale data dagli aumenti di IGF-1, e non è nemmeno causata da un drastico accumulo di grasso viscerale. Sappiamo infatti che la condizione è legata ad alterazioni della salute intestinale e da uno stato di gastroparesi legata al peggioramento del IR correlata ad iperalimentazione e abuso di GH e/o Insulina.
Il GH, l’MK-677, IGF-1 e suo varianti non causano di per se la condizione detta “GH Gut”, ma possono contribuire al suo instaurarsi se usate in cronico e, soprattutto, in concomitanza di regimi alimentari ipercalorici.
La prevenzione del “GH Gut” si basa, quindi, su una alimentazione ben calibrata e non estremamente elevata in carboidrati (specie nei periodi di refeed pre-contest), nell’evitamento dell’abuso di GH e/o Insulina e il controllo della glicemia ematica e dello stato del IR tramite rapporto glicemia basale:Insulina basale (a digiuno) e, in aggiunta, anche un test della tolleranza al glucosio con curva insulinica.
La conoscenza di questi fattori dovrebbe essere sufficiente a far desistere coloro i quali, pur non potendo raggiungere dimensioni da “freak”, per esempio, si ostinano a voler abusare pesantemente della farmacologia pensando che essa sia “la chiave”… Per tutti coloro che soffrono già di questa condizione, possono usufruire di questo articolo come input per uscirne e tornare a preparazioni generalmente più salubri ed esteticamente considerabili affini alla cultura fisica.
Krishnasamy S, Abell TL – Diabetic Gastroparesis: Principles and Current Trends in Management. Diabetes Ther. 2018 Jun 22. doi: 10.1007/s13300-018-0454-9.
BPC-157 è il termine usato per riferirsi a un pentadecapeptide, una proteina composta da una catena di 15 amminoacidi. BPC è l’acronimo di “Body Protection Compounds” e si riferisce a “peptidi comprendenti 8-15 residui di amminoacidi con un peso molecolare di 900-1.600 dalton” secondo il brevetto per il BPC-157[1], sebbene un altro studio affermi che BPC si riferisce a una proteina gastroprotettiva utilizzata per isolare il BPC-157.[2] Questa particolare sequenza non condivide l’omologia con altri peptidi gastrici noti,[2] con almeno uno studio che rileva che questa sequenza non è stata registrata nel database Protein BLAST (a partire dal 2016[3]). Ci sono alcuni studi nei quali questo peptide è indicato anche come PL 14736, PL-10,[4] e Bepecin[3]. In questo articolo si utilizzerà esclusivamente l’acronimo BPC-157.
Struttura molecolare del BPC-157
Il BPC-157 è liberamente solubile in acqua con un valore pH normale.[5] La sequenza del pentadecapeptide è Gly-Glu-Pro-Pro-Pro-Gly-Lys-Pro-Ala-Asp-Asp-Ala-Gly-Leu-Val[5] e si dice che sia abbastanza stabile rispetto ad altri peptidi non degradandosi nell’acido dello stomaco (ex vivo) per almeno 24 ore.[2][6] È stato dimostrato che è moderatamente stabile nel plasma ex vivo, con il 36% del peptide intatto che permane dopo 60 minuti.[3]
Caratteristiche farmacodinamiche del BPC-157:
Quando i ricercatori hanno testato il BPC-157 in un test CAM (embrione di pulcino), sembrava essere in grado di aumentare il processo di angiogenesi (produzione di vasi sanguigni) del 129 +/- 7% e del 152 +/- 14% se somministrato a dosi di 0,01 μg e 0,1 μg, rispettivamente. Questo effetto è stato successivamente confermato negli HUVEC, dove concentrazioni di 0,1 μg/mL e 1 μg/mL hanno aumentato la formazione di vasi sanguinei perfetti del 119+/-9% e del 147 +/- 7% nelle 24 ore di incubazione (con 1 μg/ ml essendo determinata essere la concentrazione ottimale in HUVEC).[7] Questa osservazione è stata confermata anche nei ratti con danni agli arti. Dopo una settimana di trattamento con BPC-157, sembravano esserci più vasi sanguigni nell’arto danneggiato rispetto al controllo.[7]
Angiogenesi: processo multifasico che genera nuovi vasi sanguigni dal pre-esistente letto vascolare.
Un aumento dell’espressione di VEGFR2 è stato notato nei ratti con un arto ferito a cui era stato somministrato BPC-157 rispetto al controllo, che si pensava fosse alla base dell’aumento della produzione di vasi sanguigni. Quando sono stati ulteriormente testati, i ricercatori hanno scoperto che il VEGF-A è completamente inalterato alla concentrazione di 1μg/mL, mentre il VEGFR2 è aumentato in modo dipendente dal tempo di esposizione all’interno della cellula e quindi ha proceduto all’attivazione della via VEGFR2-Akt-eNOS (una via importante all’angiogenesi).[7] Quando è stato introdotto il Dynasore, un inibitore del VEGFR2,[8], l’intera via non è stata più attivata e la formazione del vaso sanguineo non si è più verificata in vitro.[7]
È stato anche scoperto che il BPC-157 stimola l’mRNA del fattore di crescita EGR-1 nelle cellule intestinali (Caco-2) a 10-100μM, con la massima efficacia a 50μM. Anche una proteina correlata, l’mRNA NAB2, è stata aumentata poco dopo. Entrambi questi effetti sono paralleli agli effetti del PDGF-BB (un fattore di crescita endogeno) sebbene richiedano concentrazioni molto più elevate. Anche il contenuto di proteine EGF-1 sembrava essere aumentato.[4]
Quando incubato nel plasma ex vivo, sembra che una grande quantità del peptide rilevato venga registrata come ‘metaboliti’ (79+/-2%) del composto originario entro 60 minuti, anche se poi sembra stabilizzarsi, con il rimanente peptide intatto che permane fino a 240 minuti.[3]
È stato menzionato indirettamente dall’autore di molti studi che con il BPC-157 non è stato trovato alcun legame noto con i recettori della dopamina, sebbene non sia stata fornita alcuna citazione per questa particolare affermazione.[9] Quando somministrato a 10ng/kg o 10μg/kg, il BPC-157 somministrato contemporaneamente all’anfetamina ha mostrato che solo la dose più elevata era in grado di attenuare alcuni effetti osservabili dell’anfetamina (comportamenti dei ratti come annusare, leccare e rosicchiare). Anche la somministrazione del BPC-157 un’ora dopo l’anfetamina ha mostrato alcuni benefici.[2] Quando ai ratti è stato precedentemente somministrato Aloperidolo (che rende i ratti successivamente più sensibili agli effetti dell’anfetamina[10]), la co-somministrazione di BPC-157 sembrava mitigare la prevista sensibilità indotta dall’Aloperidolo.[2] Questo effetto apparentemente antagonista può anche applicarsi cronicamente, il che significa che una singola dose di BPC-157 (10μg/kg IP; 10ng/kg inefficace) somministrata prima della somministrazione cronica di anfetamine sembrava attenuare gli effetti comportamentali dell’anfetamina nei ratti durante il periodo di osservazione.[11]
Aloperidolo
Il BPC-157 è stato studiato per il suo coinvolgimento nel sistema serotoninergico dovuto alla sua influenza nella salute dell’intestino, ed i ricercatori suggeriscono un possibile asse cervello-intestino a monte degli effetti del BPC-157 in entrambe queste aree.[12] Per quanto riguarda una connessione tra il cervello e l’intestino, la Serotonina è un probabile giocatore a causa della sua alta prevalenza nell’organo.[13]
I ricercatori hanno scoperto che i ratti trattati con 10μg/kg (iniezione sottocutanea) di BPC-157 sperimentano acutamente un aumento della sintesi della Serotonina dopo 40 minuti in diverse regioni del cervello, tra cui la substantia nigra reticulata e il nucleo olfattivo anteriore mediale, mentre contemporaneamente sperimentano una diminuzione nell’Ipotalamo, nell’Ippocampo (ventrale e dorsale), e nel talamo (dorsale ma non ventrale).[14] Quando questa dose veniva somministrata per una settimana, persisteva l’aumento della sintesi di Serotonina nella substantia nigra (che si verificava sia nella reticulata che nella compacta) mentre le diminuzioni della sintesi di Serotonina osservate con una singola dose non persistevano più.[14]
Asse Cervello-Intestino
Il BPC-157 sembra avere effetti protettivi sul tessuto cerebrale quando somministrato ai ratti (sia somministrato tramite l’acqua da bere che attraverso le iniezioni) insieme alla tossina Cuprizone, riducendo la quantità di cellule danneggiate in numerose regioni del cervello, compreso l’Ippocampo.[15] Il Cuprizone[15] è una tossina utilizzata per simulare i danni osservati nella sclerosi multipla[16] e potenzialmente nella schizofrenia.[17]
L’ingestione orale di BPC-157 a una quantità stimata di 10 μg/kg (0,16 μg/mL in acqua) è stata altrettanto efficace delle iniezioni di 10ng/kg e 10μg/kg[15], sebbene sia noto che il Cuprizone è una tossina che può indurre danno neuronale (in particolare demielinizzazione) senza necessariamente raggiungere il cervello.[18]
Cuprizone
Nelle femmine di ratto sottoposte a test di nuoto forzato (test di Porsolt), il BPC-157 (somministrazione intraperitoneale) alle dosi sia di 10ng/kg che di 10μg/kg sembra funzionare in misura statisticamente uguale ai controlli attivi sia del Imipramina (15 mg e 30 mg) che della Nialamide (30 mg e 40 mg), e hanno tutti superato il gruppo di controllo.[5] Il BPC-157 è apparso anche efficace nell’assistere questi ratti in un modello di stress cronico imprevedibile simile a 30mg di Imipramina.[5]
A concentrazioni di 2μg/mL nei fibroblasti tendinei che sono stati poi espiantati, le cellule trattate con il BPC-157 sembravano crescere più velocemente dei fibroblasti non trattati con BPC-157 entro due giorni, raggiungendo una quantità significativamente maggiore dopo una settimana. Questo effetto è stato associato sia ad una maggiore resistenza ossidativa al perossido di idrogeno che ad un aumento dipendente dalla concentrazione delle proteine FAK e Paxillina non osservate nel gruppo di controllo.[19] Anche la formazione di F-actina, importante per il processo di diffusione dei fibroblasti tendinei,[20] sembra essere notevolmente aumentata con il BPC-157 rispetto al controllo[19] ed è correlata alle azioni delle suddette proteine (FAK e Paxillina).[ 21] Questo studio ha anche scoperto che i fibroblasti tendinei ex vivo in isolamento non erano influenzati dal BPC-157, solo quelli espiantati nei ratti,[19] un effetto notato anche altrove quando i Tendociti coltivati non erano influenzati dal solo BPC-157.[22] Tuttavia, l’effetto inibitorio della crescita del 4-idrossinonenale (HNE) è stato negato dalla presenza del BPC-157 in queste cellule.[22]
Paxillina
I ricercatori hanno osservato benefici quando il BPC-157 veniva messo su una spugna durante l’intervento chirurgico, dove sembrava migliorare il tasso di riformazione del collagene, inizialmente superando il fattore di crescita delle piastrine dopo quattro giorni, ma alla fine risultando equipotente dopo otto giorni[4]. Sono stati osservati benefici nei ratti sottoposti a iniezioni intraperitoneali dopo una lesione del tallone d’Achille, dove il tasso di guarigione della lesione è stato confermato visivamente con dimensioni e profondità del taglio inferiori.[22]
È stato riscontrato che gli effetti protettivi del BPC-157 sulle ulcere vengono prevenuti nei ratti attraverso la somministrazione concomitante di Aloperidolo (antagonista dei recettori alfa-1A e dopaminergico), Fentolamina (antagonista alfa adrenergico, non selettivo) e Clonidina (antagonista alfa-2A adrenergico, simile all’Agmatina) ma non è stato influenzato dalla Prazosina, dal Domperidone o dalla Yohimbina.[23]
Il BPC-157 ha mostrato effetti protettivi contro vari agenti che inducono ulcere gastriche, come la Ciclofosfamide[24] e l’Aloperidolo.[25]
Quando si tratta di infiammazione, il BPC-157 ha mostrato benefici nei ratti contro le tossine Acido Trinitrobenzensolfonico (TNBS)[6] e Cisteamina,[26][27][15] dove sono stati ridotti sia i biomarker dell’infiammazione che i marker visivi di danno quando il BPC-157 è stato somministrato insieme alle tossine. Il BPC-157 non è unico in questo senso d’azione, poiché altri composti attivi controllati come la Ranitidina e l’Omeprazolo hanno mostrato efficacia nello stesso modello di infiammazione intestinale,[27] sebbene sia stato menzionato in una review degli autori[28] che il BPC-157 può essere più pratico a causa dei comprovati benefici in altre complicanze della malattia intestinale: guarigione dell’anastomosi, sindrome dell’intestino corto e fistole.
Omeprazolo
Un’anastomosi è una connessione tra due cose che normalmente non sono collegate, con una fistola che è un tipo anormale comunemente osservato durante le malattie intestinali. Numerosi studi hanno dimostrato che le iniezioni di BPC-157 nei ratti hanno proprietà riparatrici sull’anastomosi in numerose regioni del corpo, comprese quelle aortiche[29] ed esofagogastriche.[30] Negli studi che hanno valutato l’intestino, sono stati dimostrati benefici per le fistole colo-vescicali,[31] retto-vaginali,[32] colon-colon,[15] e ileoileali[33]. Questo particolare beneficio può essere correlato alla segnalazione dell’Ossido Nitrico (potenzialmente la via VEGFR2-Akt-eNOS influenzata dal BPC-157[7]) poiché L-NAME, un inibitore della sintasi dell’Ossido Nitrico, peggiora la guarigione dell’anastomosi che viene migliorato dal BPC-157.[30]
Ossido Nitrico
Anche gli studi che valutano il BPC-157 in modelli sperimentali di sindrome dell’intestino corto riportano benefici, con iniezioni di BPC-157 che migliorano questo stato[34][35] anche quando lo stato è peggiorato con l’aggiunta di L-NAME e Diclofenac.[35]
In particolare, è stato riscontrato un beneficio per la guarigione dell’anastomosi (esofagogastrica) nei ratti trattati con BPC-157 nell’acqua di abbeveramento (circa 10ng/kg o 10 μg/kg al giorno) senza iniezione, senza differenze significative nell’efficacia tra le due dosi ed efficacia statisticamente simile alle iniezioni di 10ng/kg e 10μg/kg.[30]
Uno studio sui ratti che utilizzava la tossina MPTP (che induce danni simili a quelli osservati nel morbo di Parkinson nei roditori), la somministrazione di BPC-157 per via intraperitoneale sembrava mitigare alcuni dei danni causati dall’MPTP.[36]
Nei roditori a cui è stato somministrato Cuprizone (per indurre danni simili a quelli osservati nella sclerosi multipla[16]) quelli a cui è stato somministrato il BPC-157 insieme al Cuprizone (0,16 ng/mL o 0,16 μg/mL in acqua potabile per quattro giorni o 10ng/kg o 10μg/kg per via intragastrica nell’ultimo giorno) sembravano mostrare danni cerebrali e anomalie cliniche significativamente inferiori dal Cuprizone rispetto ai ratti di controllo a cui non era stato somministrato il BPC-157.[15]
Conclusioni:
Come abbiamo visto, i ricercatori hanno condotto numerosi studi sui roditori utilizzando il BPC-157 il quale ha mostrato di avere effetti protettivi che si estendono oltre lo stomaco e il tratto intestinale. È stato dimostrato che il BPC-157 favorisce la guarigione delle ulcere nello stomaco, dei danni intestinali come fistole e disturbi infiammatori, la guarigione di ossa e articolazioni e i tassi di crescita e danni agli organi. Ha anche alcune influenze sul cervello.
I ricercatori hanno osservato effetti protettivi marcati quando il BPC-157 viene somministrato ai ratti insieme a una tossina utilizzata nella ricerca o a una procedura chirurgica dannosa. Sono necessarie ulteriori ricerche per chiarire se il BPC-157 ha molteplici meccanismi d’azione, ma la ricerca attuale suggerisce che questo pentadecapeptide influenza diversi fattori di crescita solitamente coinvolti nell’angiogenesi (la produzione di vasi sanguigni) e altri fattori coinvolti nella rigenerazione a seguito di un danno.
Il BPC-157 è sicuramente promettente, ma sono necessari studi sull’uomo per dimostrare che questi benefici si estendono oltre gli animali da ricerca. La maggior parte degli studi sul BPC-157 sono condotti su ratti sottoposti a iniezioni del supplemento. Nonostante il BPC-157 sia un peptide temporalmente stabile a livello gastrico, i peptidi sono un gruppo di composti che normalmente sono scarsamente assorbiti dopo l’integrazione orale, specie in forme oltre la tripeptide, quindi i ricercatori usano prevalentemente le iniezioni negli studi sui roditori. Inoltre, non ci sono prove d’efficacia accademicamente documentata del BPC-157 sugli esseri umani e la maggior parte della ricerca è stata condotta da un singolo gruppo di ricerca. A causa della sua natura sintetica, potrebbero esserci problemi legali associati alla vendita di questo composto in alcune regioni e potrebbe essere vietato da alcune organizzazioni sportive.
Tornando sulla questione dell’assunzione orale del BPC-157, vorrei ricordare che la stabilità gastrica non si traduce in un assorbimento intestinale di una catena composta da 15 amminoacidi. La Pepsina dello stomaco e le proteasi pancreatiche scompongono tutte le proteine/peptidi in amminoacidi, dipeptidi e tripeptidi, i quali vengono assorbiti a livello intestinale attraverso specifici trasportatori. Quindi, l’assunzione orale può portare benefici a livello gastrointestinale e, per connessione cerebrale attraverso il sistema nervoso enterico, benefici a livello mentale. Le proprietà (supposte anche nell’uomo) a livello delle articolazioni e tendini sono ben poco probabili con l’assunzione orale mentre sono una potenziale risultante dal trattamento per iniezione.
Digestione proteica
“Io ho usato la forma orale e ho recuperato più velocemente da una infiammazione alla spalla!” Si, sei proprio sicuro che sia dovuto alla supplementazione con il BPC-157? Oppure è la conseguenza di una combinazione di effetti sul recupero dati dai PEDs che stai utilizzando e il miglioramento dello stato psicologico consequenziale all’impatto a livello intestinale del peptide in questione? Prima di “gridare al miracolo” assicuratevi che lo sia…
Detto ciò, la dose orale più vicina possibile alla logica di trasposizione tra test su roditori ed esseri umani si basa su studi sui ratti in cui tale metodo di somministrazione ha mostrato benefici, poiché la maggior parte degli studi, come già detto, somministra il supplemento tramite iniezione. Si stima che la dose orale efficace nei ratti, 10μg/kg, sia equivalente nell’uomo a 1,6μg/kg, ovvero:
96mcg per una persona di 60Kg;
112mcg per una persona di 70Kg;
128mcg per una persona di 80Kg.
Attualmente, per ovvie ragioni, non ci sono studi di farmacocinetica umana per valutare le differenze di specie.
I dosaggi per la forma iniettabile si attestano tra i 200 ed i 300mcg/die per via sottocutanea o intramuscolare (non direttamente nell’articolazione) per un periodo di tempo variabile tra le 2 e le 4 settimane.
Sebbene il peptide BPC-157 non sia attualmente incluso nell’elenco delle sostanze vietate dell’Agenzia Mondiale Antidoping (WADA), è importante che gli atleti sappiano che questa sostanza non è approvata per l’uso clinico umano. È stato sviluppato e pubblicato un test antidoping per la rilevazione del BPC-157 nelle urine. Nonostante la WADA abbia chiarito che al momento il BPC-157 non è una sostanza proibita, questo potrebbe cambiare in futuro se si determinasse di soddisfare almeno due dei tre criteri di inclusione per l’elenco delle sostanze vietate dalla WADA.
Poiché il BPC-157 non è stato ampiamente studiato negli esseri umani, nessuno sa se esiste una dose sicura o se esiste un metodo per utilizzare questo composto con un buon grado di sicurezza per trattare condizioni mediche specifiche.
Dai dati empirici provenienti dagli utilizzatori “off-label” sono emersi effetti avversi quali dolore e arrossamento nel sito di iniezione, così come con qualsiasi iniezione, mal di testa, vertigini e nausea.

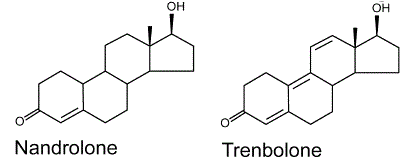
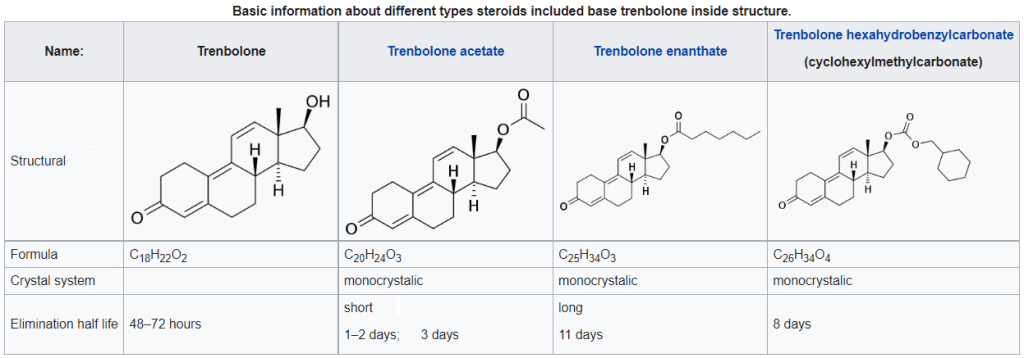







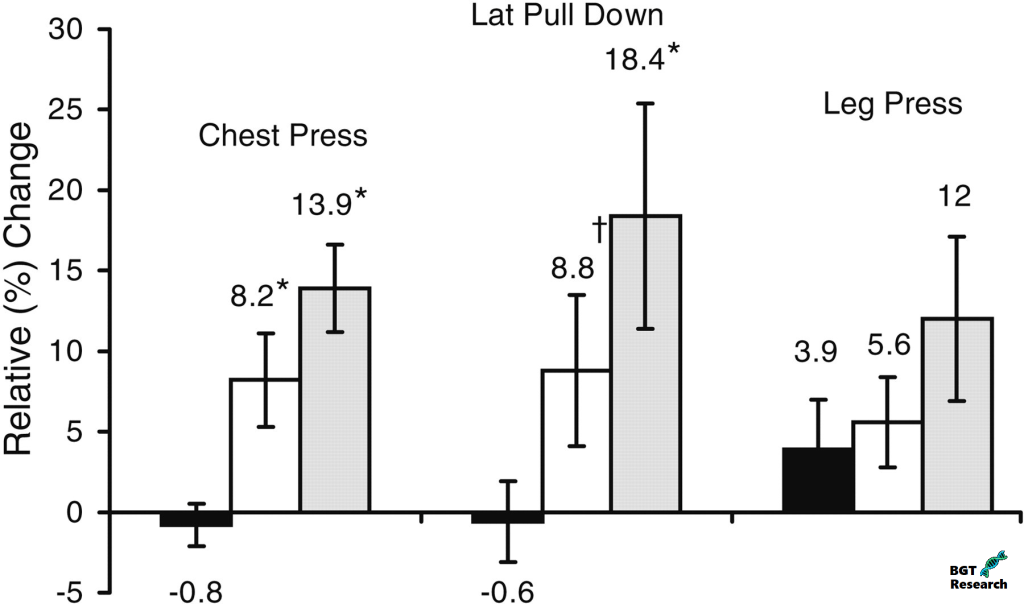

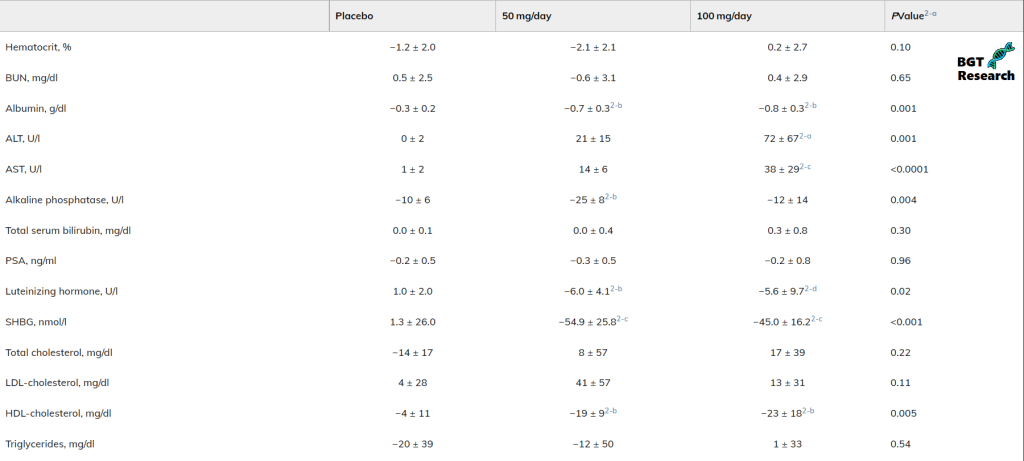

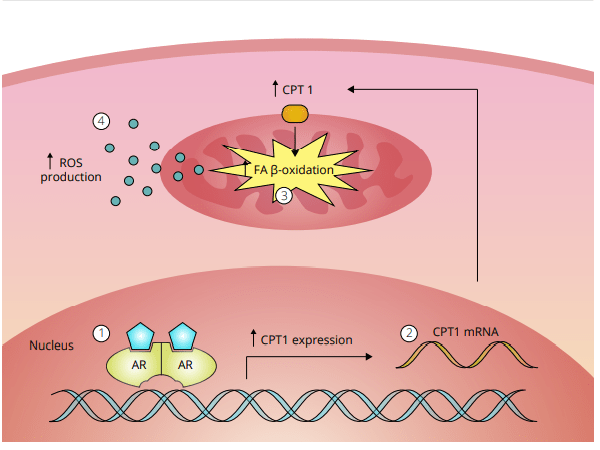


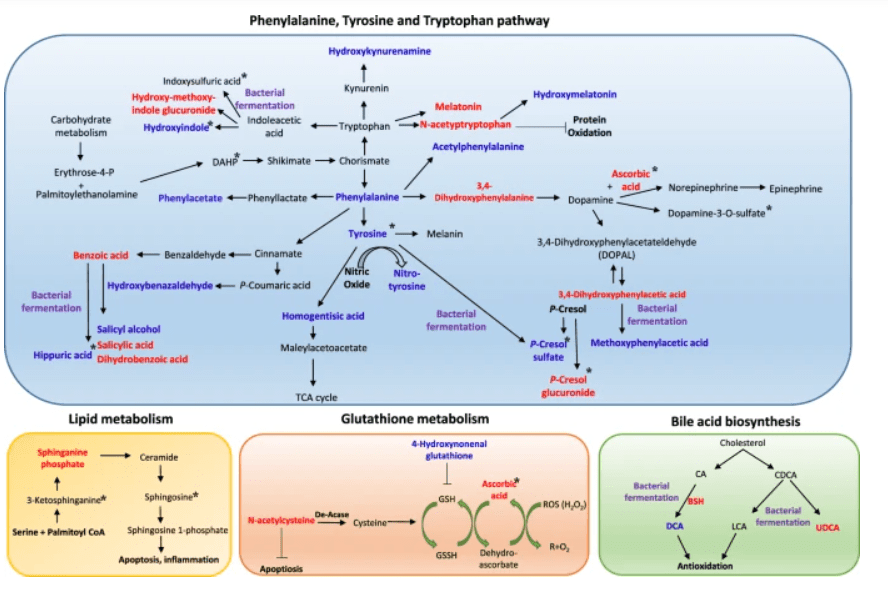

 prima, durante e 2 anni dopo l’interruzione della terapia. I valori sono la media ± SD. *, P < 0,05; **, P < 0,01.
prima, durante e 2 anni dopo l’interruzione della terapia. I valori sono la media ± SD. *, P < 0,05; **, P < 0,01.