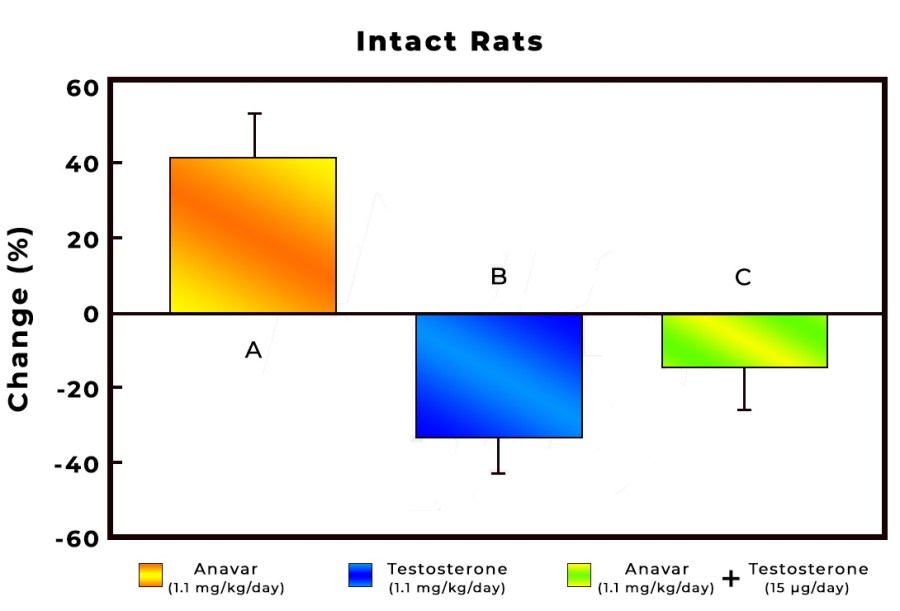
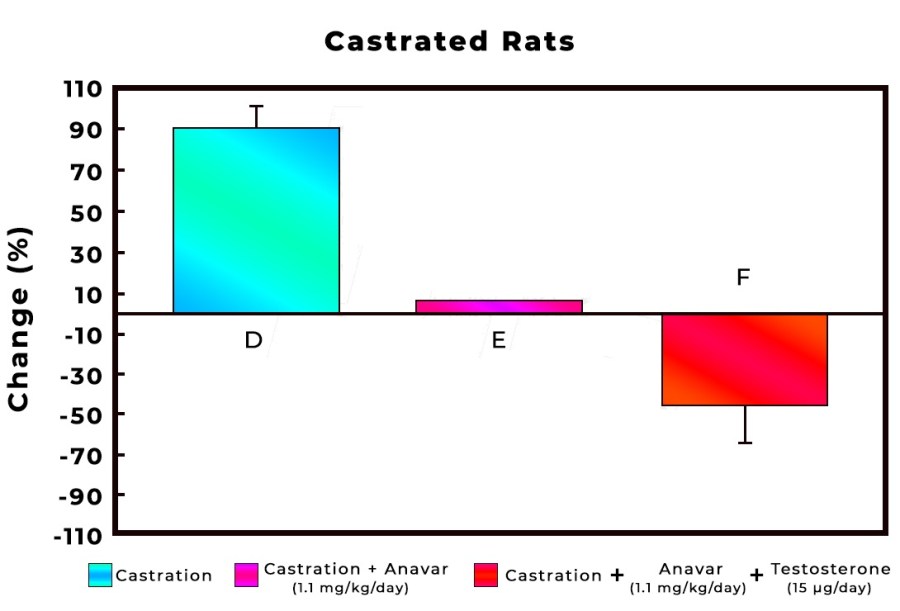
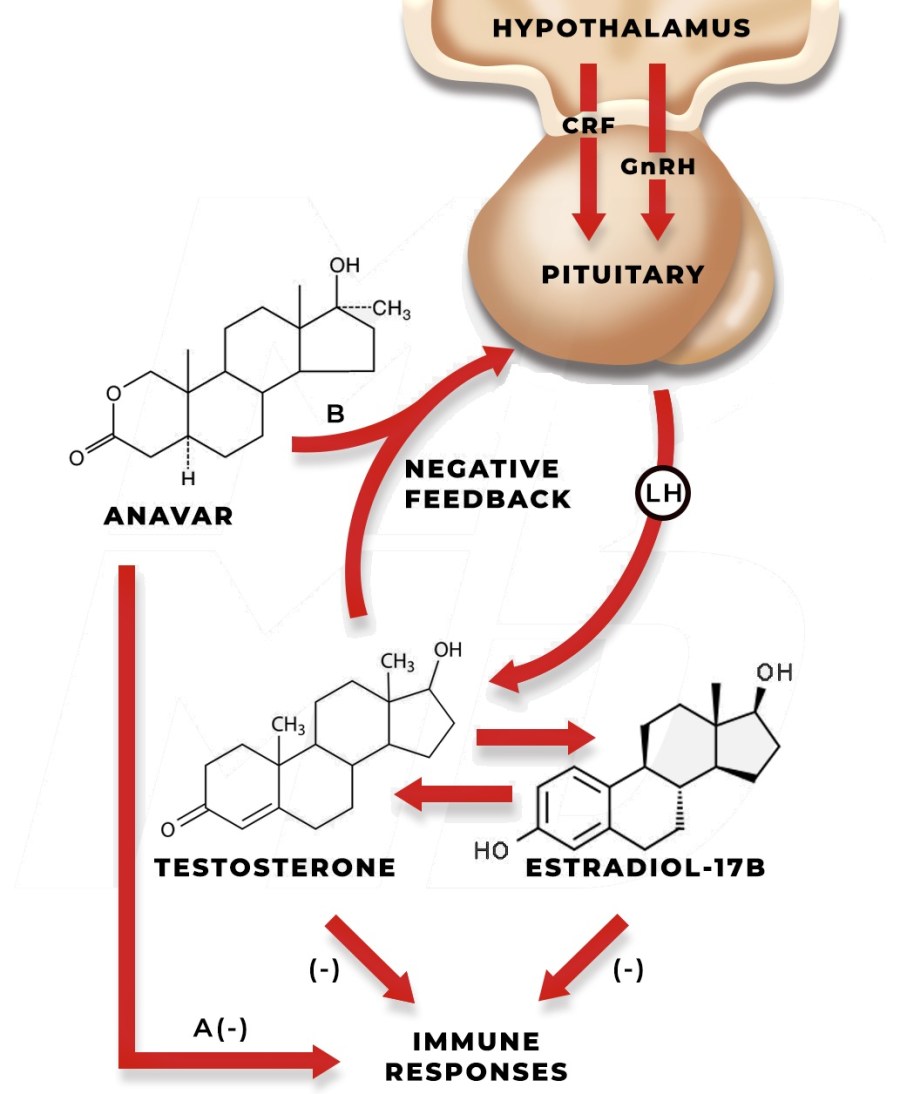
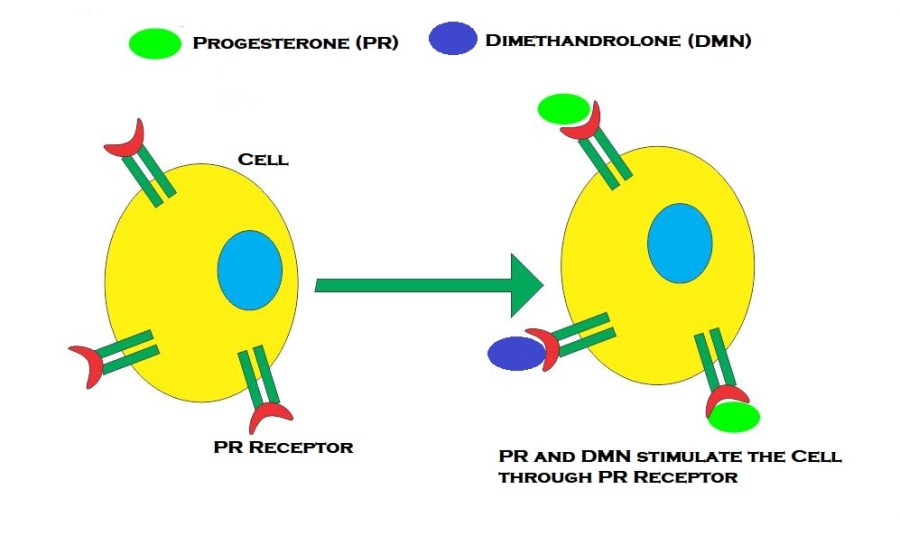
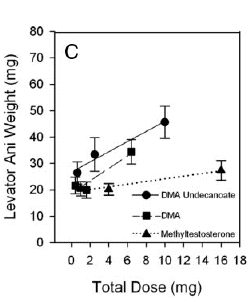
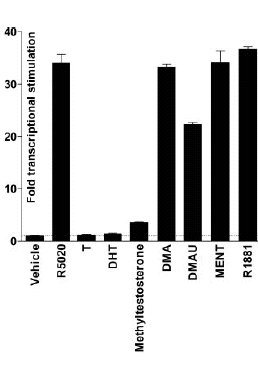
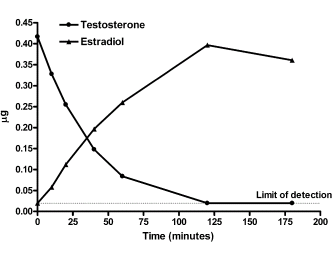
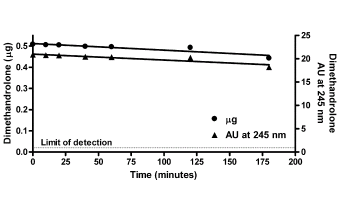
Introduzione
Negli ultimi anni il Cannabidiolo (noto anche con la sigla CBD) ha subito una crescente popolarità data sia dai promettenti risultati ipoteticamente applicabili e replicabili nel trattamento di varie condizioni patologiche (vedi, ad esimpio, SLA) provenienti dagli studi in vitro e su animali da laboratorio e, soprattutto, dalla sua relativamente facile reperibilità, essendo legale in molti paesi, che rendeva (e rende) il composto testabile dal singolo senza rischi legali. Il suo uso ha iniziato a diffondersi anche tra gli sportivi, ed i culturisti in particolar modo ne sono stati attratti per le sue potenzialità nella riduzione della percezione dello stress, come agente anoressizzante e come trattamento del dolore.
Ma quanto c’è di concretamente e fruttuosamente applicabile del Cannabidiolo in ambito medico e anche sportivo? Nel seguente articolo verranno riportate le informazioni necessarie per una risposta scientificamente onestà a questa domanda.
La storia del CBD

Il Cannabidiolo (CBD) è un fitocannabinoide scoperto nel 1960. È uno dei 113 cannabinoidi identificati nelle piante di cannabis e rappresenta fino al 40% dell’estratto della pianta.[1] I cannabinoidi nella cannabis sono stati studiati dagli anni ’40,
[2], con le strutture di THC (delta-9-tetraidrocannabinolo) e CBD scoperte negli anni ’60. La ricerca di questi composti ha subito un picco negli anni ’90, quando sono stati trovati i recettori dei cannabinoidi nel corpo [3], e in seguito è stato scoperto che sono stati attivati anche da composti endogeni (prodotti nel corpo) nel 1992.[4]
La popolarità del CBD è esplosa negli anni 2000, soprattutto dopo che i genitori di bambini epilettici hanno iniziato a sperimentare l’effetto del CBD sui loro figli, come opzione di trattamento per convulsioni resistenti ai farmaci nella rara e grave condizione di epilessia chiamata Sindrome di Dravet.[5]

I prodotti a base di CBD che mantengono una percentuale di THC “low-to-zero” (inferiore allo 0,3%) si trovano attualmente in un’area grigia della legalità negli Stati Uniti, ma con una legalità rafforzata dopo l’approvazione del Farm Act nel dicembre 2018, sebbene la FDA richiede ancora tecnicamente che gli integratori commercializzati debbano essere approvati prima dalla stessa. [6] La FDA ha inviato lettere di avvertimento nell’aprile del 2019 a tre società che ritenevano stessero formulando dichiarazioni sanitarie significative, tra cui: [7]
- “Il CBD ha bloccato con successo le cellule tumorali in diverse varietà di cancro cervicale.”
- “Il CBD ha anche ridotto la crescita e la diffusione delle cellule di glioma umano, suggerendo così un possibile ruolo del CBD come agente antitumorale.”
- “Per i pazienti con Alzheimer, il CBD è un’opzione di trattamento che sta rallentando la progressione di quella malattia.”
- “La fibromialgia è concepita come uno stato di sensibilizzazione centrale con iperalgesia secondaria. Il CBD ha dimostrato la capacità di bloccare i meccanismi spinali, periferici e gastrointestinali responsabili del dolore associato a emicrania, fibromialgia, IBS e altri disturbi correlati. “
- “Il cannabidiolo può essere efficace nel trattamento dei disturbi da uso di sostanze.”
- “Il CBD ha ridotto gli effetti gratificanti della morfina e ha ridotto la ricerca dell’eroina.”
- “Il CBD può essere utilizzato per evitare o ridurre i sintomi di astinenza.”
Presumibilmente, la reazione della FDA è stata dettata dalle scarse prove che si hanno in questo momento per il trattamento della maggior parte delle condizioni al di fuori della grave epilessia, e poiché queste affermazioni non includono tale dichiarazione di non responsabilità, i pazienti potrebbero presumere che il livello di evidenza sia più forte di quanto non sia in realtà. Pertanto, la FDA afferma che “non tollereremo questo tipo di marketing ingannevole per i pazienti vulnerabili”. L’interazione tra la regolamentazione FDA di alimenti, droghe e integratori (che possono essere tutti applicabili alla cannabis), insieme a diverse normative sulla cannabis in diversi stati, rende complesse le questioni legali relative al CBD e ad altri prodotti a base di cannabinoidi. [8]

Nota: in Italia la vendita della cannabis light è legale dal 2016, e ciò in virtù di una legge (numero 242) pienamente in vigore. La vendita è consentita per cannabis che abbia THC (i principi attivi che generano effetti psicotropi) fino e non oltre la percentuale dello 0,2. Ma il venditore di cannabis legale non è responsabile penalmente fino alla soglia dello 0,6 per cento, oltre la quale la marijuana viene sequestrata da parte dell’autorità giudiziaria e si apre un fascicolo che poi arriverà in tribunale. Queste due limitazioni aprono certo le porte a diverse interpretazioni e smagliature della legge. Molte profonde incertezze attualmente coinvolgono il CBD legale in Italia: questo a seguito della lacunosa sentenza emessa il 30 Maggio 2019 da parte della Corte di Cassazione e che ha visto “rivoluzionare”, secondo l’accezione meno positiva del termine, quanto invece stabilito dalla Legge 242. La sentenza emessa 30 Maggio 2019 circa la cannabis light e il CBD fino ad allora legale, ha stabilito che in Italia non sarebbe più concessa la vendita o la cessione a qualunque titolo di tutti i prodotti “derivati dalla coltivazione della cannabis”, quali l’olio al CBD, così come foglie, le infiorescenze e la resina, questo secondo la massima provvisoria emessa che recita testualmente -“Integrano il reato previsto dal Testo unico sulle droghe (articolo 73, commi 1 e 4, dpr 309/1990) le condotte di cessione, di vendita, e, in genere, la commercializzazione al pubblico, a qualsiasi titolo, dei prodotti derivati dalla coltivazione della cannabis sativa light, salvo che tali prodotti siano in concreto privi di efficacia drogante”. Ma, e c’è un ma, la legge italiana sull’utilizzo e sulla vendita del CBD esplica tutto ciò che concerne la filiera della canapa in Italia, dalla coltivazione al consumo. Nella stessa si legge che vi è una promozione alla coltivazione, trasformazione, impiego della stessa per lo sviluppo del territorio, produzione di alimenti, cosmetici, opere di bioingegneria, bonifica dei terreni, ricerca.
Le varietà di canapa che possono essere seminate devono essere presenti nel catalogo europeo delle sementi, in queste sono riportate solo quelle tipologie che hanno dimostrato di possedere un THC inferiore allo 0.2%.
Per questo è possibile affermare che i prodotti a base di CBD possono essere venduti in quanto rientrano nella tipologia ad uso tecnico, ciò vuol dire che nella legge non si fa esplicito riferimento alla possibilità di fumare la cannabis light, tuttavia questo non è neanche vietato e per questo l’utilizzo finale è a discrezione di chi la compra. La canapa legale è la principale fonte di approvvigionamento di CBD legale in Italia.
I negozi, online o fisici, possono commercializzare tisane, bevande, torte, vestiti e anche bustine di marijuana light. Ad essere illegale infatti è un valore elevato di THC ma non il CBD che ha esclusivamente potenziali proprietà rilassanti e antiossidanti e quindi, come dimostrano i test svolti in laboratorio, non ha alcun effetto ‘ludico’ comparabile all’utilizzo di droghe.
Per la serie “leggi ambigue e dove trovarle”…
Fonti del CBD
Il CBD viene in genere estratto dalla pianta di canapa. Ecco una rapida panoramica dei termini: la cannabis è un genere di pianta, che si presenta in due tipi botanici principali: la cannabis indica e la cannabis sativa. La marijuana (che contiene sia CBD che THC) può essere di entrambi i tipi, mentre la canapa è solo di tipo sativa.

Si noti che entrambe appartengono allo stesso genere e specie e sono piuttosto distinti i loro livello di THC. La canapa contiene un massimo dello 0,3% di THC, mentre la marijuana varia in genere tra il 5-20% di THC. [9] La marijuana viene spesso coltivata per uso ricreativo o misto ricreativo/medicinale, mentre la canapa viene coltivata per una più ampia varietà di usi (abbigliamento di canapa, olio di canapa, fibra di canapa, CBD isolato e dozzine di altri derivati).

CBD e THC non sono gli unici cannabinoidi presenti sul mercato. Ad esempio, il CBG (cannabigerolo) si trova in quantità minori nella cannabis, ma è stato studiato per le malattie infiammatorie in un modello animale.[10]
Attività biologica (farmacodinamica)

Poiché il sistema endocannabinoide ha un impatto su così tante sfaccettature della vita di base di un individuo (ad es. Appetito, funzione immunitaria, riproduzione e gestione del dolore), esistono una serie di possibilità di azione per il CBD (e altri cannabinoidi) che incidono sulla salute.[11] Il CBD può impedire l’attivazione eccessiva dei recettori CB1 e CB2, riducendo gli effetti dello stress mediato da tali recettori.[12] Altri ruoli includono l’antiossidazione: sia il CBD che il THC hanno agito come antiossidanti neuroprotettivi nei ratti.[13] Mentre il THC agisce principalmente sui recettori CB1 e CB2, il CBD agisce su altri recettori tra cui il canale del calcio TRPV1, attivandolo e rapidamente desensibilizzandolo, risultando in definitiva in meno potenziale di ipereccitazione.[14]
Nota: Il CBD non sembra avere effetti psicotropi (“alti”) come quelli causati dal ∆9-THC nella marijuana, ma, come già detto, è attualmente in fase di ricerca preliminare per i suoi possibili effetti anti-ansia e anti-psicotici legati ad un suo possibile effetto psicotropo. Mentre si sviluppa la comprensione delle differenze nei cannabinoidi medici, gli esperti stanno lavorando per distinguere la “marijuana medica” (con vari gradi di effetti psicotropi e deficit nella funzione esecutiva) dalle “terapie mediche con CBD” che comunemente presenterebbero avere un profilo degli effetti collaterali ridotto o non psicoattivo. Il CBD sembrerebbe non causare dipendenza.
Farmacocinetica

Il CBD assunto per via orale (in particolare in riferimento al farmaco liquido Epidiolex) impiega tra le 2,5 e le 5 ore per raggiungere la massima concentrazione ematica. L’emivita è tra le 56 e le 61 ore.[15] Sia nell’uomo che nei ratti, essere in stato di alimentazione sembra aumentare i livelli plasmatici di CBD.[16][17] La farmacocinetica del CBD può variare abbastanza ampiamente tra persone e persona [18], con risposte che possono essere aggravate da variazioni da lotto a lotto ed imprecisioni di etichettatura in alcuni prodotti a base di CBD.
Potenziali effetti terapeutici del CBD

In questo momento, c’è solo un accenno di promessa per il CBD per il miglioramento delle condizioni della pelle, ma nessuna basata sulla ricerca randomizzata.[19] Ad esempio, il CBD in uno studio preclinico in vitro ha mostrato effetti antiinfiammatori , antiproliferativi e lipostatici che suggeriscono che potrebbe potenzialmente avere una certa efficacia nei trattamenti di condizioni della pelle come l’acne vulgaris.[20]

Un caso studio ha riferito che una dose giornaliera di 2x300mg di CBD era associata al miglioramento della disfagia da SLA. Mentre diciotto mesi dopo l’esordio, il progresso della disfagia subivano un calo, e la debolezza degli arti, il fascicolazione e l’atrofia peggioravano relativamente meno. Gli autori teorizzano che la progressione di alcuni ma non tutti i sintomi della malattia dei motoneuroni può essere rallentata con il CBD.[21]

Il disturbo dello spettro autistico (ASD) probabilmente comporta alterazioni in alcuni percorsi chimici neurologici, come il glutammato e il GABA. È con questa idea che uno studio ha testato se il CBD potesse alterare questi percorsi in un modo che potesse aiutare il trattamento dell’autismo, con l’utilizzo della spettroscopia di risonanza magnetica. I ricercatori hanno scoperto che il CBD non era efficace per questo scopo.[22] Tuttavia, una serie di casi retrospettivi hanno mostrato un comportamento migliorato nel 61% di un campione di bambini con ASD. Tuttavia, non vi era alcun gruppo di controllo.[23]

Il CBD non sembra nemmeno migliorare i sintomi legati al movimento nel Parkinson, sebbene possa migliorare i problemi del sonno derivanti dalla condizione, in particolare la parassitnia con incubi e la perdita di atonia muscolare durante il sonno REM.[24][25]

In uno studio esplorativo, 2000mg/die di CBD hanno ridotto i sintomi psicotici della schizofrenia e i pazienti trattati con CBD avevano maggiori probabilità di essere valutati come migliorati dal medico curante.[26] Uno studio randomizzato nel quale si è testato l’effetto del CBD ha scoperto che sia esso che l’amisulpride hanno migliorato i sintomi della patologia in questione, ma il CBD ha avuto un migliore profilo degli effetti collaterali.[27] Tuttavia, in uno studio randomizzato nel quale si è utilizzata una dose di 600mg/die di CBD, non sono stati osservati miglioramenti nei sintomi.[28] Uno studio non controllato non ha riscontrato alcun beneficio del CBD con dosi aumentate da 40mg/die a 1280mg/die in 35 giorni su pazienti con schizofrenia resistente al trattamento.[29]

In sei studi randomizzati con oltre 500 pazienti con convulsioni gravi (in genere bambini piccoli), il CBD migliora significativamente i sintomi convulsivi.[30] In particolare, la percentuale di pazienti che ha avuto una riduzione di oltre il 50% delle crisi è diminuita, così come il numero di persone che hanno smesso di avere crisi epilettiche. Mentre gli studi randomizzati hanno in genere esaminato i pazienti con sindrome di Dravet e Lennox-Gastaut, studi estesi non randomizzati (studi sull’uso compassionevole) hanno anche mostrato efficacia nel trattamento del disturbo da carenza di CDKL5 e nelle sindromi di Aicardi, Doose e Dup15q.[31] Una serie di casi di tre pazienti ha anche suggerito che il CBD può aiutare con l’epilessia correlata al tumore al cervello.[32]
Studi a più lungo termine confermano che il CBD continua ad essere efficace e relativamente sicuro in questi pazienti [33][34][35], sebbene gli effetti collaterali inizialmente comuni come la riduzione dell’appetito e la diarrea persistessero in uno studio.[36] Il CBD sembra avere un effetto di dimensioni simili sull’epilessia pediatrica grave come altri farmaci antiepilettici.[37] Un piccolo studio di coorte suggerisce che anche i pediatri hanno ritenuto che la salute generale dei loro bambini con epilessia grave fosse migliorata.[38]
Il motivo per cui il CBD può aiutare con le convulsioni ha a che fare con le vie di segnalazione degli endocannabinoidi. Questi sono alterati nel disturbo convulsivo e THC e CBD agiscono in diversi modi per aiutare potenzialmente a marginare questo disturbo. Il THC probabilmente aiuta attivando il recettore CB1 [39], mentre i meccanismi del CBD sono meno conosciuti e possono includere una varietà di altri recettori, come il GPR55 e il TRPV1.[40][41][42]

Mentre il potenziale per il CBD di aiutare ad alleviuare alcuni tipi di dolore cronico è elevato, le prove di base sono molto piccole, con solo uno studio randomizzato e due studi non randomizzati a sostegno.
In uno studio randomizzato sono stati osservati dolore e spasticità in pazienti con sclerosi multipla, lesioni del midollo spinale e altre condizioni con questi sintomi. Sebbene diversi pazienti non abbiano completato lo studio, il controllo del dolore è migliorato nel gruppo CBD (mentre lo spasmo e altri sintomi non hanno subito variazioni).
Due studi non randomizzati e non controllati hanno esaminato il CBD al fine di valutarne il potenziale nell’alleviare il dolore. Uno studio ha osservato il dolore nelle ragazze giovani con dolore cronico da vaccino HPV. Si è riscontrato una riduzione del dolore corporeo e un migliore funzionamento con l’uso del CBD.[43] Uno studio molto piccolo ha esaminato il dolore nei pazienti con trapianto di rene e ha scoperto che due su sette pazienti avevano un sollievo totale dal dolore, mentre quattro avevano un sollievo non particolarmente percepibile.[44]

In uno studio, una singola dose di THC sembrava aumentare l’ansia rispetto al placebo, mentre una singola dose di CBD è risultata simile al placebo come stato ansioso.[45] Mancano studi randomizzati a lungo termine, ma studi randomizzati a breve termine e studi di serie di casi a lungo termine hanno esplorato i potenziali effetti benefici del CBD sull’ansia.
In una serie di casi di 72 pazienti ambulatoriali psichiatrici, il CBD (con la maggior parte dei pazienti che ne assumevano 25mg/die) ha migliorato rapidamente i punteggi di ansia in circa l’80% dei pazienti, con un effetto generalmente prolungato nel corso di tre mesi.[46]
Tre prove hanno esaminato gli effetti del CBD sull’ansia indotta sperimentalmente dal parlare in pubblico. Uno studio precoce non randomizzato nel 1993 ha mostrato che 300 mg di CBD hanno migliorato lo stato ansioso dopo il test di parlare in pubblico, ma non prima.[47] Uno studio randomizzato del 2017 dello stesso autore ha anche mostrato una riduzione dell’ansia alla stessa dose di 300mg, dopo il test del parlato, con 100mg e 900mg che non hanno mostrato alcun effetto.[48] Un altro gruppo ha studiato l’effetto di 600mg di CBD riscontrando una significativa riduzione dell’ansia durante il test di conversazione pubblica.[49]
Gli studi non controllati forniscono prove più deboli, ma estendono tali prove ad altre popolazioni. Una serie di casi retrospettivi hanno mostrato che quasi l’80% dei pazienti per i quali il CBD era utilizzato in una clinica psichiatrica per trattare l’ansia aveva migliorato questa condizione, e la maggior parte dei pazienti hanno mantenuto i benefici dopo il primo mese.[46]

Un caso studio ha suggerito che la somministrazione di CBD per 10 giorni ha ridotto i sintomi di astinenza da cannabis in un pesante abusatore, come ansia, perdita di appetito e irritabilità.[50]
Prove preliminari hanno suggerito che il CBD ha ridotto il consumo di fumo di sigaretta (nonostante non abbia influito sulla brama di sigarette)[51], e ha ridotto la piacevolezza legata alla sigaretta dopo una notte di astinenza.[52] Purtroppo, nei fumatori di sigarette che mirano all’astinenza, il CBD non ha migliorato la memoria di lavoro verbale o spaziale o l’impulsività.[53]
Un piccolo studio pilota condotto su individui dipendenti da oppioidi ha mostrato che il CBD in dosi di 400 o 800mg riduceva la brama dopo che i soggetti erano stati fatti astenere dagli oppioidi per sette giorni (dopo una sessione video per indurre la brama).
[54] Tuttavia, la maggior parte delle prove per i potenziali benefici del CBD sulla sospensione degli oppioidi sono di origine preclinica e basate su studi molto piccoli.[55]
Una serie di casi ha riportato una riduzione dei sintomi del PTSD (Post Traumatic Stress Disorder) in 10 di 11 pazienti con l’uso di CBD, dopo otto settimane di trattamento, in cui il CBD è stato ben tollerato.[56]

La prima citata serie di casi di 72 pazienti ambulatoriali psichiatrici ha anche scoperto che il CBD (sempore con la maggior parte dei pazienti che ne assumevano 25mg/die) non ha migliorato i punteggi del sonno nel corso di tre mesi.[46]
D’altro canto, uno studio crossover in doppio cieco ha scoperto che il CBD non ha interrotto il ciclo sonno-veglia dei pazienti che assumevano una dose relativamente alta intesa a ridurre clinicamente l’ansia (300 mg).[57]
Quando si parla di cancro e CBD, gli studi affidabili sull’uomo mancano totalmente.
Un caso studio ha riportato di un paziente con carcinoma polmonare che ha avuto una “risposta sorprendente” con evidente riduzione del tumore dopo auto-somministrazione di CBD.[58] Naturalmente, questo è un caso clinico, ed è una pura supposizione che il CBD sia l’elemento causale. Ma è comunque uno spunto di riflessione.
Parlando di casi clinici, un altro ha suggerito che il CBD potrebbe aver migliorato la chemioradioterapia in due casi di gliomi di alto grado.[59] Una serie di casi retrospettivi ha riferito che il 92% dei 119 casi di cancro esaminati aveva una riduzione delle cellule tumorali circolanti e talvolta una riduzione delle dimensioni del tumore, quando veniva assunto il CBD di livello farmaceutico.[106-60] Ancora una volta, questa è una prova di basso livello, ma pur sempre qualcosa che merita approfondimenti.
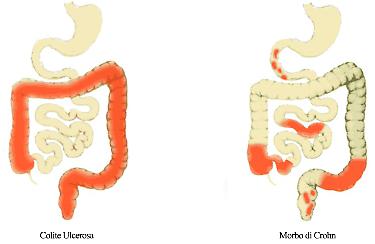
In uno studio nel quale sono stati utiulizzati 10mg di CBD, due volte al giorno, al fine di valkutarne gli effetti sul Morbo di Crohn, non sono stati riscontrati effetti benefici. Gli autori ipotizzano che ciò potrebbe essere stato influenzato dalla bassa dose o dal piccolo numero di pazienti nello studio o dalla mancanza di altri cannabinoidi sinergici (ovvero l’effetto entourage).[61]
Uno studio di prova (randomizzato, con un gruppo placebo) ha testato l’effetto di un estratto botanico ricco di CBD sulla Colite Ulcerosa, e non ha riscontrato differenze di remissione rispetto al placebo, ma un miglioramento di alcuni sintomi.[62]
Status del CBD nello Sport

Il CBD è stato utilizzato da atleti professionisti e dilettanti in diverse discipline e paesi, con l’Agenzia mondiale antidoping che ha rimosso il CBD dall’elenco delle sostanze vietate. L’Agenzia antidoping degli Stati Uniti e l’Agenzia antidoping del Regno Unito non hanno politiche anti-CBD, con quest’ultima che afferma che “il CBD non è attualmente nell’elenco delle sostanze proibite dell’Agenzia mondiale antidoping. Di conseguenza, è consentito l’uso di esso nello sport. Tutti gli altri cannabinoidi (inclusi ma non limitati a cannabis, hashish, marijuana e THC) sono vietati in competizione. L’intenzione del regolamento è proibire i cannabinoidi che attivano gli stessi recettori nel cervello come attivati dal THC. “[63] [64] Nel 2019, il principale produttore di prodotti a base di cannabis, Canopy Growth, ha acquisito la proprietà di maggioranza di BioSteel Sports Nutrition, che sta sviluppando prodotti a base di CBD con l’approvazione di numerosi atleti professionisti. [65] La National Hockey League Alumni Association ha avviato un progetto con Canopy Growth per determinare se il CBD o altri prodotti a base di cannabis potrebbero migliorare i sintomi neurologici e la qualità della vita dei giocatori feriti alla testa.[65] Numerosi atleti professionisti usano il CBD, principalmente per il trattamento del dolore.[65][66][67]
Interazioni alimentari e farmacologiche del CBD

Il CBD sembra interagire con i farmaci antiepleettici comunemente usati, modificando significativamente i loro livelli sierici. Questi includono clobazam, rufinamide, topiramato, zonisamide ed eslicarbazepina.[68]
Il CBD può anche inibire un enzima chiamato CYP2D6, che è preso di mira da farmaci comuni tra cui omeprazolo e risperidone. Può anche inibire l’enzima CYP2C9, che ridurrebbe la metabolizzazione di warfarin e diclofenac.[69] Non è noto se le inibizioni in vitro si tradurranno in inibizioni in vivo negli esseri umani, e sono necessari ulteriori studi a riguardo.[70] In uno studio nel quale sono stati utilizzati 6x100mg/die di CBD, ha aumentato la biodisponibilità e l’emivita di eliminazione dell’esobarbitale.[71]
D’altro canto, è anche possibile che altri farmaci influenzino la metabolizzazione del CBD. Ad esempio, la rifampicina antibiotica riduce le concentrazioni di picco del CBD nel sangue a causa dell’induzione dell’enzima CYP3A4, mentre il ketoconazolo, l’inibitore del CYP3A4, raddoppia quasi il picco della concentrazione di CBD.[72]
Effetti collaterali del CBD e possibili contaminazioni dei prodotti che lo contengono

Per trovare eventi avversi ed effetti collaterali, è necessario un campione relativamente grande. Quindi i principali studi per valutare questi in riferimento al CBD erano gli studi più ampi su giovani pazienti con gravi disturbi convulsivi, vale a dire la sindrome di Dravet e la sindrome di Lennox-Gastaut.
Gli effetti che si sono verificati più frequentemente con paragone tra CBD e placebo includono: sonnolenza, diarrea, affaticamento, vomito, febbre e letargia. Alcuni pazienti avevano anche livelli aumentati di aminotransferasi epatica.[73][5]
Due studi hanno esaminato i potenziali effetti collaterali emodinamici (come variazioni della frequenza cardiaca, della frequenza respiratoria o della pressione sanguigna) e non hanno trovato effetti significativi quando è stato assunto il CBD.[74][75]
Una review completa ha rilevato che dosi fino a 1.500mg/die sembrano essere abbastanza ben tollerate [76], sebbene non sia affatto sicuro presumere che la dose sia sicura per un determinato individuo (a causa della relativa mancanza di informazioni sulla sicurezza a lungo termine e della varietà di condizioni/farmaci/ecc. Che potrebbero teoricamente interagire con il CBD).
Gli studi sugli animali hanno riportato alcuni effetti collaterali teorici non osservati negli studi sull’uomo esistenti, come la ridotta capacità di fertilizzazione e dell’inibizione attraverso il metabolismo epatico del farmaco.[77][78]
Il CBD è risultato promettente come metodo per ridurre gli effetti collaterali quando vengono assunti altri farmaci, come osservato nel già citato studio sul CBD somministrato a pazienti con schizofrenia che assumevano amisulpride.[79]
I contaminanti da piante di cannabis coltivate, raccolte e confezionate con pratiche meno controllate potrebbero teoricamente comparire anche in prodotti a base di CBD, inclusi pesticidi, particelle metalliche, cannabinoidi sintetici, metalli pesanti, muffe, batteri e aflatossine.[80][81] I solventi residui del processo di produzione possono anche comparire in estratti di cannabis, vale a dire esano, etanolo, alcool isopropilico, toluene, benzene, xilene e acetone.[82] Questa ricerca riguarda tuttavia tutti gli estratti generali della cannabis; la ricerca sui contaminanti specificamente nei prodotti isolati di CBD deve ancora essere svolta.
Conclusioni sul CBD

Cosa concludere riguardo al CBD dopo l’esposizione dei dati precedentemente riportati? Sicuramente che la ricerca sull’uomo è molto limitata e non ci permette ad oggi di esporre un giudizio sufficientemente fondato sulla reale portata terapeutica del CBD. Si tratta ovviamente di una conclusione dettata dalla scarsità dei dati e dall’impatto degli studi esistenti. Possiamo comunque scorgere dei potenziali sul trattamento dell’ansia e, ipoteticamente, sullo stress percepito e il trattamento del dolore. Tale caratteristica rende il CBD sperimentabile come aggiunta ad una preparazione sportiva particolarmente intensa come durante il picco di preparazione per una competizione o un contest di BodyBuilding. La sua possibile influenza sull’appetito (effetto anoressizzante) potrebbe tornare senz’altro molto utile nei periodi di restrizione calorica. Alcuni speculeranno su un suo possibile impatto indiretto significativo sui livelli di Cortisolo dato, per l’appunto, da una ridotta percezione dello stress, ma i dati certi al momento non sussistono. Il fatto che si tratti di un composto dagli effetti non del tutto accertati non invoglia particolarmente a investire denaro per testarne le potenzialità. Se poi consideriamo il fatto che i più non conoscono ne le modalità di utilizzo ne le dosi graduate da applicare con criterio crescente in caso di risposta positiva con dosaggi più blandi, non c’è da stupirsi se spesso ci si imbatte in testimonianze molto contrastanti provenienti dagli utilizzatori che scoraggiano i lettori meno preparati in materia.
Quindi, vale la pena investire denaro per testare l’efficacia del CBD? Considerando che il costo di prodotti di alta qualità non è propriamente economico, e che i dati in letteratura non sono molto impattanti a livello dimostrativo (almeno ad oggi), a meno che non abbiate una adegiuata conoscenza per testarne e valutarne gli effetti, per il momento si può saggiamente investire su altro… per adesso…
Gabriel Bellizzi
Riferimenti:
1- Campos AC, Moreira FA, Gomes FV, Del Bel EA, Guimarães FS (December 2012). “Multiple mechanisms involved in the large-spectrum therapeutic potential of cannabidiol in psychiatric disorders”. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences (Review). 367 (1607): 3364–78.
2- LOEWE S. Studies on the pharmacology and acute toxicity of compounds with marihuana activity. J Pharmacol Exp Ther (1946)
3- Howlett AC, et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev (2002)
4- Devane WA, et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science (1992)
5- Devinsky O, Cross JH, Wright S. Trial of Cannabidiol for Drug-Resistant Seizures in the Dravet Syndrome.
N Engl J Med (2017)
6- Statement from FDA Commissioner Scott Gottlieb, M.D., on signing of the Agriculture Improvement Act and the agency’s regulation of products containing cannabis and cannabis-derived compounds.
7- Statement from FDA Commissioner Scott Gottlieb, M.D., on new steps to advance agency’s continued evaluation of potential regulatory pathways for cannabis-containing and cannabis-derived products.
8- O’Connor SM, Lietzan E. The Surprising Reach of FDA Regulation of Cannabis, Even After Descheduling. Am Univ Law Rev. (2019)
9- Hilderbrand RL. Hemp & Cannabidiol: What is a Medicine?. Mo Med. (2018)
10- Borrelli F, et al. Beneficial effect of the non-psychotropic plant cannabinoid cannabigerol on experimental inflammatory bowel disease. Biochem Pharmacol. (2013)
11- Battista N, et al. The endocannabinoid system: an overview. Front Behav Neurosci. (2012)
12- Laprairie RB, et al. Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. Br J Pharmacol. (2015)
13- Hampson AJ, et al. Cannabidiol and (-)Delta9-tetrahydrocannabinol are neuroprotective antioxidants. Proc Natl Acad Sci U S A. (1998)
14- Iannotti FA, et al. Nonpsychotropic plant cannabinoids, cannabidivarin (CBDV) and cannabidiol (CBD), activate and desensitize transient receptor potential vanilloid 1 (TRPV1) channels in vitro: potential for the treatment of neuronal hyperexcitability. ACS Chem Neurosci. (2014)
15- Epidiolex(Cannabidiol) prescribing information. Carlsbad,CA: Greenwich Biosciences, Inc.; 2018..
16- a b Zgair A, et al. Dietary fats and pharmaceutical lipid excipients increase systemic exposure to orally administered cannabis and cannabis-based medicines. Am J Transl Res. (2016)
17- Winter H, et al. Effect of a high-calorie, high-fat meal on the bioavailability and pharmacokinetics of PA-824 in healthy adult subjects. Antimicrob Agents Chemother. (2013)
18- Huestis MA. Human cannabinoid pharmacokinetics. Chem Biodivers. (2007)
19- Eagleston LRM, et al. Cannabinoids in dermatology: a scoping review. Dermatol Online J. (2018)
20- Oláh A, et al. Cannabidiol exerts sebostatic and antiinflammatory effects on human sebocytes. J Clin Invest. (2014)
21- Co-medication with Cannabidiol May Slow Down the Progression of Motor Neuron Disease: A Case Report; 2017..
22- Pretzsch CM, et al. Effects of cannabidiol on brain excitation and inhibition systems; a randomised placebo-controlled single dose trial during magnetic resonance spectroscopy in adults with and without autism spectrum disorder. Neuropsychopharmacology. (2019)
23- Aran A, et al. Brief Report: Cannabidiol-Rich Cannabis in Children with Autism Spectrum Disorder and Severe Behavioral Problems-A Retrospective Feasibility Study. J Autism Dev Disord. (2019)
24- Chagas MH, et al. Effects of cannabidiol in the treatment of patients with Parkinson’s disease: an exploratory double-blind trial. J Psychopharmacol. (2014)
25- Peres FF, et al. Cannabidiol as a Promising Strategy to Treat and Prevent Movement Disorders?. Front Pharmacol. (2018)
26- McGuire P, et al. Cannabidiol (CBD) as an Adjunctive Therapy in Schizophrenia: A Multicenter Randomized Controlled Trial. Am J Psychiatry. (2018)
27- Leweke FM, et al. Cannabidiol enhances anandamide signaling and alleviates psychotic symptoms of schizophrenia. Transl Psychiatry. (2012)
28- Boggs DL, et al. The effects of cannabidiol (CBD) on cognition and symptoms in outpatients with chronic schizophrenia a randomized placebo controlled trial. Psychopharmacology (Berl). (2018)
29- Zuardi AW, et al. Cannabidiol monotherapy for treatment-resistant schizophrenia. J Psychopharmacol. (2006)
30- Stockings E, et al. Evidence for cannabis and cannabinoids for epilepsy: a systematic review of controlled and observational evidence. J Neurol Neurosurg Psychiatry. (2018)
31- Devinsky O, et al. Open-label use of highly purified CBD (Epidiolex®) in patients with CDKL5 deficiency disorder and Aicardi, Dup15q, and Doose syndromes. Epilepsy Behav. (2018)
32- Warren PP, et al. The use of cannabidiol for seizure management in patients with brain tumor-related epilepsy. Neurocase. (2017)
33- Thiele E, et al. Cannabidiol in patients with Lennox-Gastaut syndrome: Interim analysis of an open-label extension study. Epilepsia. (2019)
34- Szaflarski JP, et al. Cannabidiol improves frequency and severity of seizures and reduces adverse events in an open-label add-on prospective study. Epilepsy Behav. (2018)
35- Szaflarski JP, et al. Long-term safety and treatment effects of cannabidiol in children and adults with treatment-resistant epilepsies: Expanded access program results. Epilepsia. (2018)
36- Sands TT, et al. Long-Term Safety, Tolerability, and Efficacy of Cannabidiol in Children with Refractory Epilepsy: Results from an Expanded Access Program in the US. CNS Drugs. (2019)
37- Ali S, Scheffer IE, Sadleir LG. Efficacy of cannabinoids in paediatric epilepsy. Dev Med Child Neurol. (2019)
38- Chen KA, et al. Cannabidiol for treating drug-resistant epilepsy in children: the New South Wales experience. Med J Aust. (2018)
39- Reddy DS, Golub VM. The Pharmacological Basis of Cannabis Therapy for Epilepsy. J Pharmacol Exp Ther. (2016)
40- Rosenberg EC, Patra PH, Whalley BJ. Therapeutic effects of cannabinoids in animal models of seizures, epilepsy, epileptogenesis, and epilepsy-related neuroprotection. Epilepsy Behav. (2017)
41- Bisogno T, et al. Molecular targets for cannabidiol and its synthetic analogues: effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br J Pharmacol. (2001)
42- Bialer M, et al. Progress report on new antiepileptic drugs: A summary of the Thirteenth Eilat Conference on New Antiepileptic Drugs and Devices (EILAT XIII). Epilepsia. (2017)
43- Palmieri B, Laurino C, Vadalà M. Short-Term Efficacy of CBD-Enriched Hemp Oil in Girls with Dysautonomic Syndrome after Human Papillomavirus Vaccination. Isr Med Assoc J. (2017)
44- Cuñetti L, et al. Chronic Pain Treatment With Cannabidiol in Kidney Transplant Patients in Uruguay. Transplant Proc. (2018)
45- Martin-Santos R, et al. Acute effects of a single, oral dose of d9-tetrahydrocannabinol (THC) and cannabidiol (CBD) administration in healthy volunteers. Curr Pharm Des. (2012)
46- a b c Shannon S, et al. Cannabidiol in Anxiety and Sleep: A Large Case Series. Perm J. (2019)
47- Zuardi AW, et al. Effects of ipsapirone and cannabidiol on human experimental anxiety. J Psychopharmacol. (1993)
48- Zuardi AW, et al. Inverted U-Shaped Dose-Response Curve of the Anxiolytic Effect of Cannabidiol during Public Speaking in Real Life. Front Pharmacol. (2017)
49- Bergamaschi MM, et al. Cannabidiol reduces the anxiety induced by simulated public speaking in treatment-naïve social phobia patients. Neuropsychopharmacology. (2011)
50- Crippa JA, et al. Cannabidiol for the treatment of cannabis withdrawal syndrome: a case report. J Clin Pharm Ther. (2013)
51- Morgan CJ, et al. Cannabidiol reduces cigarette consumption in tobacco smokers: preliminary findings. Addict Behav. (2013)
52- Hindocha C, et al. Cannabidiol reverses attentional bias to cigarette cues in a human experimental model of tobacco withdrawal. Addiction. (2018)
53- Hindocha C, et al. The effects of cannabidiol on impulsivity and memory during abstinence in cigarette dependent smokers. Sci Rep. (2018)
54- Ren Y, et al. Cannabidiol, a nonpsychotropic component of cannabis, inhibits cue-induced heroin seeking and normalizes discrete mesolimbic neuronal disturbances. J Neurosci. (2009)
55- Hurd YL, et al. Early Phase in the Development of Cannabidiol as a Treatment for Addiction: Opioid Relapse Takes Initial Center Stage. Neurotherapeutics. (2015)
56- Elms L, et al. Cannabidiol in the Treatment of Post-Traumatic Stress Disorder: A Case Series. J Altern Complement Med. (2019)
57- Linares IMP, et al. No Acute Effects of Cannabidiol on the Sleep-Wake Cycle of Healthy Subjects: A Randomized, Double-Blind, Placebo-Controlled, Crossover Study. Front Pharmacol. (2018)
58- Sulé-Suso J, et al. Striking lung cancer response to self-administration of cannabidiol: A case report and literature review. SAGE Open Med Case Rep. (2019)
59- Dall’Stella PB, et al. Case Report: Clinical Outcome and Image Response of Two Patients With Secondary High-Grade Glioma Treated With Chemoradiation, PCV, and Cannabidiol. Front Oncol. (2019)
60- Kenyon J, Liu W, Dalgleish A. Report of Objective Clinical Responses of Cancer Patients to Pharmaceutical-grade Synthetic Cannabidiol. Anticancer Res. (2018)
61- Naftali T, et al. Low-Dose Cannabidiol Is Safe but Not Effective in the Treatment for Crohn’s Disease, a Randomized Controlled Trial. Dig Dis Sci. (2017)
62- Irving PM, et al. A Randomized, Double-blind, Placebo-controlled, Parallel-group, Pilot Study of Cannabidiol-rich Botanical Extract in the Symptomatic Treatment of Ulcerative Colitis. Inflamm Bowel Dis. (2018)
63- “Athlete Advisory Note: Cannabidiol (CBD)”. United Kingdom Anti-Doping Agency. Retrieved October 9, 2019.
64- “Athletes: 6 things to know about cannabidiol”. US Anti-Doping Agency. October 23, 2018. Retrieved February 17, 2020.
65- a b c Donna Spencer (October 2, 2019). “Canopy cannabis company buys ex-NHL player’s sports nutrition business”. CTV Business. The Canadian Press. Retrieved February 17, 2020. BioSteel’s brand ambassadors also include well-known athletes across major sports leagues in North America, which could be beneficial as the company’s attempt to push regulated CBD nutrition products into the mainstream health and wellness segments
66- Amanda Loudin (December 7, 2019). “As more pro athletes use cannabis for aches and pain, the more they run afoul of rules”. The Washington Post. Retrieved February 17, 2020.
67- Sara Harrison (September 28, 2019). “Lots of athletes say CBD Is a better painkiller. Is It?”. Wired. Retrieved February 17, 2020.
68- Gaston TE, et al. Interactions between cannabidiol and commonly used antiepileptic drugs. Epilepsia. (2017)
69- Ujváry I, Hanuš L. Human Metabolites of Cannabidiol: A Review on Their Formation, Biological Activity, and Relevance in Therapy. Cannabis Cannabinoid Res. (2016)
70- Welty TE, Luebke A, Gidal BE. Cannabidiol: promise and pitfalls. Epilepsy Curr. (2014)
71- Brzozowska N, et al. ABC transporters P-gp and Bcrp do not limit the brain uptake of the novel antipsychotic and anticonvulsant drug cannabidiol in mice. PeerJ. (2016)
72- Stott C, et al. A Phase I, open-label, randomized, crossover study in three parallel groups to evaluate the effect of Rifampicin, Ketoconazole, and Omeprazole on the pharmacokinetics of THC/CBD oromucosal spray in healthy volunteers. Springerplus. (2013)
73- Devinsky O, et al. Effect of Cannabidiol on Drop Seizures in the Lennox-Gastaut Syndrome. N Engl J Med. (2018)
74- Manini AF, et al. Safety and pharmacokinetics of oral cannabidiol when administered concomitantly with intravenous fentanyl in humans. J Addict Med. (2015)
75- Arndt DL, de Wit H. Cannabidiol Does Not Dampen Responses to Emotional Stimuli in Healthy Adults. Cannabis Cannabinoid Res. (2017)
76- Bergamaschi MM, et al. Safety and side effects of cannabidiol, a Cannabis sativa constituent. Curr Drug Saf. (2011)
77- Schuel H, et al. Cannabinoids inhibit fertilization in sea urchins by reducing the fertilizing capacity of sperm. Pharmacol Biochem Behav. (1991)
78- Stout SM, Cimino NM. Exogenous cannabinoids as substrates, inhibitors, and inducers of human drug metabolizing enzymes: a systematic review. Drug Metab Rev. (2014)
79- Jara-Oseguera A, Simon SA, Rosenbaum T. TRPV1: on the road to pain relief. Curr Mol Pharmacol. (2008)
80- Russo EB. Current Therapeutic Cannabis Controversies and Clinical Trial Design Issues. Front Pharmacol. (2016)
81- Busse FP, et al. Lead poisoning due to adulterated marijuana in leipzig. Dtsch Arztebl Int. (2008)
82- Pavlovic R, et al. Quality Traits of “Cannabidiol Oils”: Cannabinoids Content, Terpene Fingerprint and Oxidation Stability of European Commercially Available Preparations. Molecules. (2018)













 to come agente dopante negli atleti olimpionici della Germania Orientale all’interno di un programma di doping sponsorizzato dallo stato negli anni ’70 e ’80.[4] Il motivo del suo uso, come precedentemente accennato, era giustificato maggiormente dal suo valore androgeno piuttosto che su quello anabolizzante. Il suo uso garantiva un ottima risposta neuro steroidea con conseguente stimolazione del Sistema Nervoso Centrale e migliorata interazione neuromuscolare, con vantaggi nella velocità, nella forza, nell’aggressività, nella concentrazione, nella resistenza fisica e allo stress mentale.[4] Oggi, l’uso del Mestanolone è stato per lo più sospeso in medicina, sebbene sia ancora disponibile in Giappone.[2][3][4] Il Mestanolone era comunemente disponibile sotto forma di compresse sublinguali da 25mg (marchio Ermalone).[7]
to come agente dopante negli atleti olimpionici della Germania Orientale all’interno di un programma di doping sponsorizzato dallo stato negli anni ’70 e ’80.[4] Il motivo del suo uso, come precedentemente accennato, era giustificato maggiormente dal suo valore androgeno piuttosto che su quello anabolizzante. Il suo uso garantiva un ottima risposta neuro steroidea con conseguente stimolazione del Sistema Nervoso Centrale e migliorata interazione neuromuscolare, con vantaggi nella velocità, nella forza, nell’aggressività, nella concentrazione, nella resistenza fisica e allo stress mentale.[4] Oggi, l’uso del Mestanolone è stato per lo più sospeso in medicina, sebbene sia ancora disponibile in Giappone.[2][3][4] Il Mestanolone era comunemente disponibile sotto forma di compresse sublinguali da 25mg (marchio Ermalone).[7]