Al principio del mese di giugno di quest’anno ho riportato alcuni casi studio i quali facevano emergere il potenziale effetto epatotossico dato dall’uso dei SARM RAD-140 e LGD-4033. Il caso studio riguardante LGD-4033 non era di per sé convincente, poiché il Bodybuilder in questione era solito consumare discrete quantità di alcol. Di recente, i medici del Baylor College of Medicine negli Stati Uniti hanno segnalato un altro caso di danno epatico legato all’uso di LGD-4033.[1] E in questo nuovo caso studio, non ci sono altri fattori esplicativi del problema.
Il soggetto protagonista del caso studio è un Bodybuilder di 32 anni che ha riferito ai medici di aver usato 10mg/die di LGD-4033 in forma liquida per 15 giorni. Dopo di che aveva cominciato a lamentare malessere e interruppe il suo ciclo con il suddetto SARM. L’uomo aveva dolori di stomaco e nausea, oltre a prurito e ittero. Le sue feci erano grigie, e aveva perso l’appetito. Quando si è rivolto ai medici, l’uomo aveva già perso 18Kg.
Questi sono classici sintomi da danno epatico. Infatti, quando i medici hanno scansionato la cavità addominale del Bodybuilder, hanno notato che il fegato dell’uomo era più grande del normale. Una biopsia ha mostrato che il fegato del aveva cicatrici in alcuni punti. I dotti biliari, che trasportano i sali biliari all’intestino, erano ostruiti.
Nelle settimane successive, i medici hanno monitorato quattro marker del danno epatico nel sangue del Bodybuilder. La figura seguente mostra che le condizioni del fegato dell’uomo sono lentamente migliorate.
Come avevo già riportato nell’articolo di giugno, secondo uno studio del 2012 condotto dai produttori del LGD-4033, questo SARM non è significativamente dannoso per il fegato. Ma in questo studio, i soggetti non hanno ricevuto più di 1mg/die di LGD-4033.[2]
Le aziende che vendono SARM online e alcuni guru del bodybuilding raccomandano dosi significativamente più elevate di 1mg/die. Ad esempio, il paziente del quale si è parlato in questo articolo ha assunto dieci volte la dose più alta testata nello studio del 2012. Con tutta probabilità, un dosaggio di LGD-4033 di tale entità o superiore rappresenta uno stress epatico eccessivo, in specie se l’utilizzatore presenta una marcata sensibilità e manca di una efficace epatoprotezione (comunque non garante di immunità da effetti collaterali a livello epatico).
Alcuni utilizzatori di LGD-4033 hanno pubblicato il loro esame ematico sui forum presenti in rete. Sembra che non mostrino segni di danno epatico, ma l’affidabilità di certi dati è assai scarsa.
Forse il Bodybuilder in questione stava usando un prodotto contaminato o fake. Non tutti i SARM negli store online sono prodotti con le giuste misure di controllo, come riportato da una recente ricerca inglese e americana.[3]
E’ anche possibile che il Bodybuilder del caso studio stava usando altre sostanze oltre al solo LGD-4033 e non ne ha fatto menzione ai medici che lo hanno preso in cura. Le possibilità sono diverse ma ciò che è sufficientemente certo è che l’uso di LGD-4033 ad alte dosi, per vie metaboliche intrinseche, è un potenziale fattore causale per stress e danno epatico.
Nonostante il terrorismo diffuso da alcuni scientisti riguardo alla “malvagità assoluta” del IGF-1 e della sua correlazione con il cancro (esistente ma contestualizzabile), la terapia sostitutiva dell’Ormone della Crescita (GH) sembra non aumentare il rischio di cancro; sembrerebbe che, addirittura, ne riduca il rischio. Ciò è stato evidenziato in una meta-analisi pubblicata da endocrinologi cinesi della Zhejiang University College of Medicine nell’Open Journal of Endocrine and Metabolic Diseases.[1]
I ricercatori hanno esaminato 10 studi pubblicati in precedenza che hanno seguito adulti trattati con Ormone della Crescita per diversi anni. Ai soggetti è stato diagnosticato un deficit dell’ormone della crescita negli adulti. Ovviamente, e mi riferisco al lettore nella media che troppo velocemente trae conclusioni inesatte, gli studi potrebbero non dire molto sugli effetti che gli atleti supplementati farmacologicamente possano incorrere nel contesto dell’incidenza di sviluppo del cancro.
Nella tabella sottostante troverete maggiori informazioni sugli studi utilizzati.
Negli studi esaminati, i soggetti trattati con il GH non hanno sviluppato il cancro in maniera maggiore rispetto ai soggetti dei gruppi di controllo. Da quanto è emerso, per lo meno dagli studi presi in esame, l’Ormone della Crescita ha persino ridotto il rischio di cancro.
I ricercatori hanno però trovato presenza di bias. Ciò significa che sembra che non siano stati pubblicati studi con risultati meno interessanti. Tuttavia, il bias sembrava essere modesto ed i ricercatori sospettano che l’inclusione degli studi potenzialmente mancanti nella loro meta-analisi non ne altererebbe realmente il risultato.
I ricercatori concludono dicendo che la loro analisi corrobora le prove di studi precedenti che dimostrano che la terapia sostitutiva dell’Ormone della Crescita nei pazienti con deficit di questo ormone in età adulta non vedrebbero aumentare il loro rischio di sviluppare il cancro; invece, potrebbe addirittura diminuire il rischio. I risultati hanno suggerito che la terapia sostitutiva dell’Ormone della Crescita nei pazienti con deficit dell’ormone in età adulta era sicura.
Nota: i dosaggi terapici variano nettamente a seconda dell’età e delle finalità della terapia. Sono generalmente dosaggi inferiori a quelli utilizzati a scopo estetico-prestativo. Pertanto, il presente articolo non ha assolutamente lo scopo di far passare un dato che ad oggi non è statisticamente comprovato.
A differenza del trattamento pediatrico con GH, spesso dosato in microgrammi / chilogrammi di peso corporeo / giorno, il dosaggio sostitutivo del GH attualmente raccomandato negli adulti è individualizzato indipendentemente dal peso, tenendo conto dell’età del paziente, del sesso e dello stato degli estrogeni (Johannsson et al., 1997a; Hoffman et al., 2004b). L’inizio della terapia a basse dosi (dose totale 0,2-0,4 mg / die SC) riduce la probabilità di sviluppare effetti collaterali comuni come rigidità articolare, artralgie, mialgie, parestesie ed edema periferico, con ritenzione di liquidi. La dose deve essere titolata a intervalli di 6-8 settimane in base alla risposta clinica, evitando effetti collaterali e monitorando i livelli sierici di IGF-I. Si desidera raggiungere un obiettivo nella metà superiore dell’intervallo normale del IGF-I corretto per l’età del paziente. È ragionevole iniziare con dosi più elevate (0,4-0,5 mg / die) nei pazienti di età inferiore a 30 anni, ma i pazienti più anziani (di età superiore a 60 anni) dovrebbero iniziare con dosi più basse (0,1-0,2 mg / die) e titolato più lentamente per ridurre al minimo il verificarsi di effetti collaterali. Alcuni pazienti con AGHD ad esordio infantile con IGF-I pretrattamento molto basso possono sviluppare effetti collaterali da GH ma non raggiungere livelli medi normali nonostante dosi elevate di quest’ultimo.
Le donne che assumono una terapia sostitutiva degli estrogeni per via orale possono richiedere dosi più elevate per la GHRT, presumibilmente perché l’estrogeno orale inibisce la produzione e la secrezione di IGF-I nel fegato con effetto di primo passaggio (Weissberger et al., 1991). Di solito non è necessario alcun aggiustamento della dose nei pazienti che assumono dosi moderate di estrogeni transdermici.
Ricercatori australiani presso la University of New South Wales stanno testando un nuovo farmaco per la perdita di peso che agisce similmente al DNP (2,4-dinitrofenolo), ma senza i numerosi effetti collaterali connessi a quest’ultimo. Il loro lavoro è stato recentemente pubblicato su Nature Communications.[1]
BAM15?
Finché l’epidemia di obesità continuerà a crescere, la ricerca di farmaci per la perdita di peso continuerà. Parte di questa ricerca si svolge tenendo come riferimento il pericoloso ma estremamente efficace DNP. Sarebbe possibile sintetizzare una molecola che sia efficace come DNP, ma non così pericolosa?
Il DNP è un disaccoppiatore della fosforilizzazione ossidativa: interferisce con la resintesi di ATP nel mitocondrio causando una significativa dispersione di energia sotto forma di calore.
Ricercatori americani presso la Yale University stanno studiando un analogo metilato del DNP, il DNPME.[2]
La giapponese Otsuka Pharmaceutical sta conducendo esperimenti con OPC-163493.[3]
I ricercatori australiani che hanno pubblicato la ricerca che qui si sta trattando, stanno svolgendo esperimenti sul N5, N6-bis (2-fluorofenil) [1,2,5] oxadiazolo [3,4-b] pirazina-5,6-diammina. In breve: BAM15 .
Effetti del BAM15 nei topi
I ricercatori hanno somministrato ai topi dosi orali di BAM15 e hanno osservato che nelle ore successive alla somministrazione, il consumo di ossigeno degli animali – e quindi il loro consumo calorico – era aumentato di alcune decine di punti percentuale. L’effetto è stato temporaneo. Questo perché l’emivita del BAM15 nei topi è di sole 1,7 ore.
In altri esperimenti, in cui i topi sono stati alimentati con cibo contenente BAM15, i ricercatori hanno scoperto che la molecola aumentava il dispendio calorico solo di notte. Ciò non sorprende, perché i topi sono animali notturni e quindi preferiscono mangiare quando è buio. Il BAM15 non ha aumentato la quantità di attività fisica svolta.
Il dosaggio utilizzato nei topi trasposto in un essere umano adulto corrisponderebbe a circa 1g/die di BAM15.
In un altro esperimento, i ricercatori hanno nutrito i topi con zucchero e grasso addizionali [WD], rendendo gli animali più grassi. Se i topi venivano trattati con il BAM15, la loro massa grassa aumentava di meno, o per nulla, o addirittura diminuiva. L’equivalente umano delle dosi alle quali si sono verificati questi effetti è stato rispettivamente di 500mg, 1g e 1,5g di BAM15 al giorno.
Il BAM15 non ha aumentato la temperatura corporea dei topi, non ha ridotto la massa magra o aumentato l’attività dei radicali liberi. Per quanto i ricercatori hanno potuto scoprire, il BAM15 aumenta anche la sensibilità all’Insulina.
Conclusioni
Concludendo, i ricercatori riportano che il BAM15 rappresenta un raro disaccoppiatore mitocondriale che previene e inverte l’obesità senza influire sull’assunzione di cibo o sulla massa magra. Una limitazione del BAM15 è la bassa solubilità acquosa, ma questa proprietà non ha influito sulla biodisponibilità orale e infatti la bassa solubilità acquosa è un parametro importante che consente al BAM15 di penetrare nelle membrane cellulari ed entrare nei mitocondri. Un’altra limitazione del BAM15 è l’emivita di 1,7 ore e le direzioni future esamineranno le strategie di formulazione per migliorare l’esposizione. Collettivamente, i dati qui presentati supportano l’ulteriore sviluppo del BAM15 come potenziale terapeutico per l’obesità e le malattie metaboliche.
Chi si interessa in modo approfondito di supplementazione farmacologica nello sport, penserà di avere una conoscenza discretamente completa su una delle pratiche più conosciute nell’ambiente, vale a dire la PCT (Post Cycle Therapy). Questo tentativo di recupero della propria funzionalità dell’Asse HPT ha subito perfezionamenti nel corso degli ultimi decenni. Si è passati da una illogica accozzaglia di farmaci tra i quali spiccavano il Mesterolone e l’Oxandrolone insieme ai classici SERM e hCG, ad una logica sequenza di composti strutturata sugli andamenti della curva ematica delle molecole utilizzate durante il ciclo e all’azione sinergica e ordinata di hCG seguito da Tamoxifene Citrato e Clomifene Citrato, con la recente aggiunta di Inibitori dell’Aromatasi (AI). Vedi PCT Scally.
Da qualche tempo, però, circola la voce secondo la quale il piano di recupero ormonale dell’HPTA può essere migliorato con l’inserimento di un altra classe di farmaci. Questa classe di farmaci è quella degli Antiandrogeni.
Prima di svelarvi il nesso che ha spinto qualche mente speculatrice a partorire tale idea, è corretto darvi una base di cultura generale sugli Antiandrogeni per concludere con la spiegazione del perchè un loro possibile inserimento in una PCT possa essere favorevole…forse…
Una panoramica sugli Antiandrogeni
Gli Antiandrogeni, noti anche come antagonisti degli androgeni o bloccanti del Testosterone, sono una classe di farmaci che impediscono agli androgeni come il Testosterone e il Dihydrotestosterone (DHT) di mediare i loro effetti biologici nel corpo. Agiscono bloccando il Recettore degli Androgeni (AR) e/o inibendo o sopprimendo la produzione di androgeni.[1][2] Possono essere pensati come gli opposti funzionali degli agonisti AR, come ad esempio gli Steroidi Anabolizzanti Androgeni (AAS) e i Modulatori Selettivi del Recettore degli Androgeni (SARM). Gli antiandrogeni sono uno dei tre tipi di antagonisti degli ormoni sessuali, gli altri sono antiestrogeni e antiprogestinici.[3]
Gli Antiandrogeni sono usati per trattare una serie di condizioni androgeno-dipendenti. [4] Nei maschi, gli Antiandrogeni sono usati nel trattamento del cancro alla prostata, ipertrofia prostatica, perdita di capelli, desiderio sessuale eccessivamente elevato, impulsi sessuali insoliti e problematici e pubertà precoce.[4][5] Nelle donne, gli antiandrogeni sono usati per trattare l’acne, la seborrea, l’eccessiva crescita dei peli, la perdita dei capelli e gli alti livelli di androgeni, come quelli che si verificano nella sindrome dell’ovaio policistico (PCOS).[4] Gli antiandrogeni sono anche usati come componente della terapia ormonale femminizzante per i transgender e come bloccanti della pubertà nelle ragazze transgender.[4]
Ciproterone Acetato
Le Antigonadotropine come gli Estrogeni e i progestinici furono entrambe introdotte per la prima volta negli anni ’30. [6] Gli effetti benefici della deprivazione di androgeni attraverso la castrazione chirurgica o la terapia con estrogeni ad alte dosi sul cancro alla prostata furono scoperti nel 1941. [7][8] antagonisti del AR furono scoperti per la prima volta nei primi anni ’60.[9] Il Ciproterone Acetato è un antiandrogeno steroideo scoperto nel 1961 e introdotto nel 1973 ed è spesso descritto come il primo antiandrogeno commercializzato. [10] [11] Tuttavia, lo Spironolattone fu introdotto nel 1959, [12] [13], sebbene i suoi effetti antiandrogeni non fossero stati riconosciuti o sfruttati fin da subito e fossero originariamente considerati un’azione indesiderata fuori bersaglio del farmaco.[14] Oltre allo Spironolattone, il Clormadinone Acetato e il Megestrolo Acetato sono antiandrogeni steroidei che sono più deboli del Ciproterone Acetato ma sono stati introdotti precedentemente, negli anni ’60. [15] [16] [17] Altri primi antiandrogeni steroidei che sono stati sviluppati in questo periodo ma che non sono mai stati commercializzati includono il Benorterone (SKF-7690; 17α-metil-B-Nortestosterone), BOMT (Ro 7-2340), il Ciproterone (SH-80881) e il Trimetiltrienolone (R- 2956).[18][19]
Flutamide
La Flutamide è un antiandrogena non steroideo descritto per la prima volta nel 1967. [20] Fu introdotto sul mercato nel 1983 ed è stato il primo antiandrogeno non steroideo commercializzato. [21] [22] Un altro antiandrogeno precoce non steroideo, [23] DIMP (Ro 7-8117), che è strutturalmente correlato alla Talidomide [24] ed è un antiandrogeno relativamente debole, [25] [26] fu descritto per la prima volta nel 1973 e non fu mai commercializzato. [27] La Flutamide è stata seguita dalla Nilutamide nel 1989 e dalla Bicalutamide nel 1995. [28] Oltre a questi tre farmaci, che sono stati considerati antiandrogeni non steroidei di prima generazione, gli antiandrogeni non steroidei di seconda generazione Enzalutamide e Apalutamide sono stati introdotti rispettivamente nel 2012 e nel 2018. [29] [30] [31] Differiscono dai precedenti antiandrogeni non steroidei, in particolar modo per il fatto che sono molto più efficaci.[30]
Aminoglutetimide
Gli inibitori della sintesi androgena Aminoglutetimide e Ketoconazolo furono commercializzati per la prima volta rispettivamente nel 1960 e nel 1977 [32] [33] e il più recente farmaco Abiraterone Acetato è stato introdotto nel sul mercato nel 2011. [34] I modulatori del GnRH furono introdotti per la prima volta negli anni ’80. [35] Gli inibitori della 5α-reduttasi Finasteride e Dutasteride sono stati introdotti sul mercato rispettivamente nel 1992 e nel 2002.[36] [37] L’Elagolix, il primo modulatore GnRH attivo per via orale ad essere commercializzato, è stato introdotto sul mercato nel 2018. [38]
Abiraterone Acetato
Quindi, gli antiandrogeni possono essere suddivisi in diversi tipi in base alla struttura chimica, inclusi antiandrogeni steroidei, antiandrogeni non steroidei e peptidi. Gli antiandrogeni steroidei comprendono composti come il Ciproterone Acetato, lo Spironolattone, l’Estradiolo, l’Abiraterone Acetato e la Finasteride; antiandrogeni non steroidei includono composti come il Bicalutamide, l’Elagolix, il Dietilstilbestrolo, l’Aminoglutetimide e Ketoconazolo; e i peptidi includono analoghi del GnRH come Leuprorelina e il Cetrorelix.
Gli Antiandrogeni si dividono in cinque gruppi principali: [39]
Antagonisti del recettore degli androgeni: farmaci che si legano direttamente al AR bloccando il legame con l’ormone bersaglio.[40][41] Questi farmaci comprendono gli antiandrogeni steroidei Ciproterone Acetato, Megestrolo Acetato, Clormadinone Acetato, Spironolattone, Oxendolone e Osaterone Acetato (veterinario) e gli antiandrogeni non steroidei Flutamide, Bicalutamide, Nilutamide, Topilutamide, Enzalutamide e Apalutamide. [41][40] ] [42] A parte il Ciproterone Acetato e il Clormadinone Acetato, alcuni altri progestinici usati nei contraccettivi orali e / o nella TOS in menopausa tra cui Dienogest, Drospirenone, Medrogestone, Nomegestrolo Acetato, Promegestone e Trimegestone hanno anche vari gradi di attività AR-antagonista. [43] [44] [45]
Inibitori della sintesi degli Androgeni: farmaci che inibiscono direttamente la biosintesi enzimatica di androgeni come Testosterone e/o DHT. [46] [47] Gli esempi includono gli inibitori del CYP17A1 Ketoconazolo, Abiraterone Acetato e Seviteronel, [46] l’inibitore del CYP11A1 (P450scc) Aminoglutetimidico , [46] e gli inibitori della 5α-reduttasi Finasteride, Dutasteride, Epristeride, Alfatradiolo e il blando Saw Palmetto (Palmetto Seghettato).[88] Numerosi altri antiandrogeni, tra cui Ciproterone Acetato, Spironolattone, Medrogestone, Flutamide, Nilutamide e Bifluranolo, sono anche noti per inibire debolmente la sintesi degli Androgeni.
Antigonadotropici: farmaci che sopprimono il rilascio di gonadotropine indotto dall’ormone di rilascio delle gonadotropine (GnRH) e conseguente attivazione della produzione di androgeni gonadici. [2] [48] Gli esempi includono modulatori del GnRH come Leuprorelina (un agonista del GnRH) e Cetrorelix (un antagonista del GnRH), [90] progestinici come Allilestrenolo, Clormadinone Acetato, Ciproterone Acetato, Gestonorone Caproato, Idrossiprogesterone Caproato, Medroxyprogesterone Acetato, Megestrol Acetato, Osaterone Acetato (veterinario), e Oxendolone, [49] [50] ed estrogeni come Estradiolo, esteri dell’Estradiolo, Etinilestradiolo, Estrogeni coniugati e Dietilstilbestrolo. [2] [49]
Miscellanei: farmaci che si oppongono agli effetti degli androgeni con mezzi diversi da quelli sopra indicati. Esempi includono Estrogeni, in particolare sintetici orali (ad esempio Etinilestradiolo, Dietilstilbestrolo), che stimolano la produzione di globulina legante gli ormoni sessuali (SHBG) nel fegato e quindi diminuiscono i livelli liberi e quindi bioattivi di Testosterone e DHT; anticorticotropine come i glucocorticoidi, che sopprimono la produzione indotta dall’ormone adrenocorticotropo (ACTH) di androgeni surrenali; e immunogeni e vaccini contro l’Androstenedione come l’albumina Ovandrotone e l’albumina Androstenedione, che riducono i livelli di androgeni attraverso la generazione di anticorpi contro il precursore androgeno androstenedione (usato solo in medicina veterinaria).
Come si è potuto vedere, alcuni antiandrogeni combinano molti dei meccanismi di cui sopra. [39] [51] Un esempio è l’antiandrogeno steroideo Ciproterone Acetato, che è un potente antagonista AR, un potente progestinico e quindi antigonadotropico, un glucocorticoide debole e quindi anticorticotropo e un inibitore debole della sintesi degli androgeni. [39] [51] [52] [53]
Per ovvie ragioni di sintesi, la lista sopra include antagonisti AR, inibitori della sintesi degli androgeni e progestinici commercializzati per l’uso o ampiamente usati come antiandrogeni, ma non include specificatamente agonisti del GnRH, antagonisti del GnRH, inibitori della 5α-reduttasi o Estrogeni.
La classe degli Antagonisti del Recettore degli Androgeni è di nostro particolare interesse…
Gli antagonisti del AR agiscono legandosi direttamente e sostituendo in modo competitivo gli androgeni come il Testosterone e il DHT dal AR, impedendo così loro di attivare il recettore e mediare i loro effetti biologici. [40] [41] Gli antagonisti del AR, come abbiamo già visto, sono classificati in due tipi, in base alla struttura chimica: steroidei e non steroidei. [54] [42] [40] [41] [55] Gli antagonisti di AR steroide sono strutturalmente correlati agli ormoni steroidei come Testosterone e Progesterone, mentre gli antagonisti del AR non steroidei non sono steroidi e sono strutturalmente distinti. Gli antagonisti del AR steroidei tendono ad avere azioni ormonali fuori bersaglio a causa della loro somiglianza strutturale con altri ormoni steroidei. [55] Al contrario, gli antagonisti del AR non steroidei sono selettivi per l’AR e non hanno attività ormonale fuori bersaglio. [55] Per questo motivo, a volte sono descritti come antiandrogeni “puri”. [55]
Spironolattone
Sebbene siano descritti come antiandrogeni e in effetti mostrano solo tali effetti in generale, la maggior parte o tutti gli antagonisti AR steroidei non sono in realtà antagonisti inattivi del AR ma piuttosto sono agonisti parziali deboli e sono in grado di attivare il recettore in assenza di agonisti AR più potenti come Testosterone e DHT. [40] [47] [55] [56] Ciò può avere implicazioni cliniche nel contesto specifico del trattamento del cancro alla prostata. [40] [55] Ad esempio, gli antagonisti del AR steroidei sono in grado di aumentare il peso della prostata e accelerare la crescita delle cellule tumorali della prostata in assenza di più potenti agonisti dell’AR, [40] [55] e lo Spironolattone ha dimostrato di accelerare la progressione del cancro alla prostata nei casi clinici [57] [58] Inoltre, mentre il Ciproterone Acetato produce genitali ambigui attraverso la femminilizzazione nei feti maschi quando somministrato ad animali in gravidanza, [59] è stato osservato che causa la mascolinizzazione dei genitali dei feti femminili di animali in gravidanza. [40] A differenza degli antagonisti AR steroidei, gli antagonisti AR non steroidei sono antagonisti inattivi del AR e, quindi, non attivano il recettore. [60] [47] [61] [55] Questo potrebbe essere il motivo per cui hanno una maggiore efficacia rispetto agli antagonisti del AR steroidei nel trattamento del cancro alla prostata ed è un motivo importante per cui li hanno ampiamente sostituiti per questa indicazione in medicina. [60] [47] [61] [55]
Bicalutamide
Gli antiandrogeni non steroidei hanno un’affinità relativamente bassa per il AR rispetto ai ligandi AR steroidei. [47] [61] [62] Ad esempio, la Bicalutamide ha circa il 2% dell’affinità di DHT per il AR e circa il 20% dell’affinità del CPA per il AR. [62] Nonostante la loro bassa affinità con il AR, tuttavia, la mancanza di un’attività agonista parziale debole degli NSAA sembra migliorare la loro potenza rispetto agli antiandrogeni steroidei. [62] [63] Ad esempio, sebbene la Flutamide abbia un’affinità circa 10 volte inferiore per il AR rispetto al CPA, mostra una potenza pari o leggermente maggiore al CPA come antiandrogeno nei biotest. [62] [63] Inoltre, le concentrazioni terapeutiche circolanti di antiandrogeni non steroidei sono molto elevate, nell’ordine di migliaia di volte superiori a quelle di Testosterone e DHT, e ciò consente loro di competere efficacemente e bloccare la segnalazione del AR. [64]
Gli antagonisti del AR non possono legarsi o bloccare i recettori degli androgeni di membrana (mARs), che sono distinti dal AR nucleare classico. [65] [66] [67] Tuttavia, le mARs non sembrano essere coinvolte nella mascolinizzazione. Ciò è evidenziato dal fenotipo perfettamente femminile di donne con sindrome da insensibilità agli androgeni completa. [68] [69] Queste donne hanno un cariotipo 46, XY (cioè geneticamente “maschio”) e alti livelli di androgeni ma possiedono un AR difettoso e per questo motivo non mascolinizzano mai. [68] [69] Sono descritti come altamente femminili, sia fisicamente che mentalmente e comportamentalmente. [70] [71] [72]
Perchè questo interesse per gli Antagonisti del Recettore degli Androgeni?
Asse Ipotalamo-Ipofisi-Testicoli
Piccolo ripasso sul controllo omeostatico ormonale riferito all’Asse HPT.
Con Asse Ipotalamo-Ipofisi-Testicoli (HPTA) ci si riferisce alla connessione tra ipotalamo, ghiandola pituitaria e testicoli come se queste singole ghiandole endocrine fossero una singola entità. Poiché queste ghiandole spesso agiscono in concerto, i fisiologi e gli endocrinologi ritengono conveniente e descrittivo parlare di esse come di un unico sistema.
L’asse HPTA svolge una parte critica nello sviluppo e nella regolazione di un certo numero di sistemi del corpo, come i sistemi riproduttivi e immunitari. Le fluttuazioni di questo asse causano variazioni negli ormoni prodotti da ciascuna ghiandola e hanno diversi effetti locali e sistemici nel corpo.
In breve, l’asse HPTA rappresenta un sistema di stimolazione/inibizione degli ormoni prodotti dalle rispettiva strutture:
Ipotalamo: GnRH (ormone di rilascio delle gonadotropine; in inglese Gonadotropin-releasing hormone).
Ipofisi (o ghiandola Pituitaria): dalle cellule beta e gamma rispettivamente l’ormone follicolo-stimolante (FSH) e l’ormone luteinizzante (LH).
Come ben sappiamo, diversi AAS sono derivati sintetici del Testosterone, il principale androgeno nei maschi. Il Testosterone sopprime marcatamente l’HPTA, mentre altri derivati lo fanno in misura maggiore o minore.
I fattori che contribuiscono alla soppressione dell’HPTA sono:
L’origine del AAS
Il tasso di conversione del AAS ad estrogeno, attraverso l’Enzima Aromatasi in alcuni tessuti (adiposo, mammario)
Dose e tempo d’uso/abuso del AAS
Attività androgena del AAS
Bingo! Ci siete arrivati adesso? In ogni caso andiamo avanti…
Conosciamo tutti il feedback negativo indotto dagli estrogeni a livello ipotalamico.
Estradiolo
Gli estrogeni (principalmente E2-beta Estradiolo) causano un feedback negativo sull’ipotalamo per la produzione di GnRH, che a sua volta stimola LH che stimola la sintesi di Testosterone nelle cellule Leydig nei testicoli. Pertanto, gli AAS fortemente soggetti all’aromatizzazione o che posseggono una attività estrogenica intrinseca (Oxymetholone, Methyltestosterone, Testosterone, Methandienone ecc…) influenzano marcatamente la funzione dell’HPTA.
Ed ecco perchè l’uso di SERM causa un incremento del GnRH, e consequenzialmente del LH e FSH, bloccando il legame recettoriale estrogenico ipotalamico inducendo un feedback positivo.
Gli AAS con alta affinità con il AR si legano fortemente ad esso. Gli AAS attraversano la barriera ematoencefalica e si legano ai recettori sull’ipotalamo. Ciò comporterà una marcata soppressione dell’HPTA. L’attività androgena si traduce nelle caratteristiche sessuali secondarie (crescita dei peli e della barba, allargamento delle spalle e il rafforzarsi dei muscoli, l’ingrandimento del pene, dei testicoli e della prostata.)
E qui entrano in gioco gli Antagonisti del Recettore Androgeno che, agendo similmente ai SERM, causano un incremento della secrezione di LH. Tale incremento è stato osservato in diversi studi tra i quali uno svolto su animali nel 1989, nel quale si era utilizzata la Flutamide.[73] L’effetto indotto è quindi progonadotropico.[74]
Ed è da ciò che è nata l’idea di inserire piccole quantità per un breve lasso di tempo di Antiandrogeni (nello specifico Antagonisti del Recettore degli Androgeni non steroidei) nel protocollo PCT al fine di potenziarne gli affetti.
Nota: Gli effetti collaterali degli antiandrogeni variano a seconda del tipo di antiandrogeno – ovvero se si tratta di un antagonista AR selettivo o un inibitore della biosintesi androgena – nonché dalla presenza di attività fuori bersaglio terapico dell’antiandrogeno in questione. [74][75] Ad esempio, mentre gli antiandrogeni antigonadotropici come i modulatori del GnRH e il Ciproterone Acetato sono associati a disfunzione sessuale pronunciata e osteoporosi negli uomini, gli antagonisti selettivi del AR come la Bicalutamide non sono associati all’osteoporosi e sono stati correlati solo a una disfunzione sessuale minima. [74] [76] [77] Queste differenze sono ritenute una conseguenza del fatto che le antigonadotropine sopprimono i livelli di androgeni e, per estensione, dei livelli dei metaboliti bioattivi degli androgeni come estrogeni e neurosteroidi, mentre gli antagonisti selettivi del AR neutralizzano gli effetti degli androgeni ma lasciano intatti i livelli degli stessi (e di fatto i loro metaboliti) potendo persino aumentarli a causa dei loro effetti progonadotropici.[74] Come altro esempio, gli antiandrogeni steroidei Ciproterone Acetato e Spironolattone possiedono azioni off-target tra cui attività progestinica, antimineralocorticoide e / o glucocorticoide in aggiunta alla loro attività antiandrogena, e queste attività off-target possono provocare ulteriori effetti collaterali.[75]
Nei maschi, i principali effetti collaterali degli antiandrogeni sono la demasculinizzazione e la femminilizzazione.[78] Questi effetti collaterali includono dolore al seno / lipomastia e ginecomastia (sviluppo del seno / ingrossamento), riduzione della crescita / densità dei peli corporei, riduzione della massa e della forza muscolare, cambiamenti femminili nella massa e nella distribuzione del grasso e riduzione della lunghezza del pene e delle dimensioni dei testicoli. [78] I tassi di ginecomastia negli uomini con monoterapia antagonista selettiva del AR sono stati stimati tra il 30 e l’85%. [79] Inoltre, gli antiandrogeni possono causare infertilità, osteoporosi, vampate di calore, disfunzione sessuale (inclusa perdita di libido e disfunzione erettile), depressione, affaticamento, anemia e riduzione del volume spermatico / eiaculato nei maschi.[78] Al contrario, gli effetti collaterali degli antagonisti selettivi del AR nelle donne sono minimi. [80] [81] Tuttavia, gli antiandrogeni antigonadotropici come il Ciproterone Acetato possono produrre ipoestrogenismo, amenorrea e osteoporosi nelle donne in premenopausa, tra gli altri effetti collaterali. [82] [83] [84]
Numerosi antiandrogeni sono stati associati a epatotossicità. [85] Questi includono, in varia misura, Ciproterone Acetato, Flutamide, Nilutamide, Bicalutamide, Aminoglutetimide e Ketoconazolo. [85] Al contrario, Spironolattone, Enzalutamide, [86] e altri antiandrogeni non sono associati a epatotossicità. Tuttavia, sebbene non presentino un rischio di epatotossicità, lo Spironolattone ha un rischio di causare iperkaliemia e l’Enzalutamide ha un rischio di causare convulsioni.
Conclusioni
E’ ovvio che si sta parlando di pura teoria, lungi dall’essere dimostrata come terapeuticamente valida. Ma, per amor di conoscenza, ho ritenuto utile trattare l’argomento in modo tale che meno persone si facessero strane e confuse idee a riguardo, magari dopo essersi imbattuti nel “bongo” da spogliatoio che, con atteggiamento del primate dominante, dispensa consigli applicativi di un qualcosa per lui difficilmente comprensibile.
Gabriel Bellizzi
Riferimenti:
Mowszowicz I (1989). “Antiandrogens. Mechanisms and paradoxical effects”. Ann. Endocrinol. Paris. 50 (3): 50(3):189–99.
Brueggemeier, Robert W. (2006). “Sex Hormones (Male): Analogs and Antagonists”. Encyclopedia of Molecular Cell Biology and Molecular Medicine.
Student S, Hejmo T, Poterała-Hejmo A, Leśniak A, Bułdak R (January 2020). “Anti-androgen hormonal therapy for cancer and other diseases”. Eur. J. Pharmacol. 866: 172783.
Gillatt D (2006). “Antiandrogen treatments in locally advanced prostate cancer: are they all the same?”. J Cancer Res Clin Oncol. 1: S17-26.
William Figg; Cindy H. Chau; Eric J. Small (14 September 2010). Drug Management of Prostate Cancer. Springer Science & Business Media. pp. 71–72, 75, 91–96.
Benno Clemens Runnebaum; Thomas Rabe; Ludwig Kiesel (6 December 2012). Female Contraception: Update and Trends. Springer Science & Business Media. pp. 136–.
Liu, Bo; Su, Lei; Geng, Jingkun; Liu, Junjie; Zhao, Guisen (2010). “Developments in Nonsteroidal Antiandrogens Targeting the Androgen Receptor”. ChemMedChem. 5 (10): 1651–1661.
Heyns, W.; G., Verhoeven; De Moor, P. (1976). “Androgen binding in rat uterus cytosol. Study of the specificity”. Journal of Steroid Biochemistry. 7 (5): 335–343.
Boris, A.; Scott, J. W.; DeMartino, L.; Cox, D. C. (1973). “Endocrine Profile of a Nonsteroidal Antiandrogen N-(3,5-Dimethyl-4-Isoxazolylmethyl)Phthalimide (Dimp)”. European Journal of Endocrinology. 72 (3): 604–614.
Menon MP, Higano CS (2013). “Enzalutamide, a second generation androgen receptor antagonist: development and clinical applications in prostate cancer”. Curr Oncol Rep. 15 (2): 69–75.
Kolvenbag, Geert J. C. M.; Furr, Barrington J. A. (2009). “Nonsteroidal Antiandrogens”. In V. Craig Jordan; Barrington J. A. Furr (eds.). Hormone Therapy in Breast and Prostate Cancer. Humana Press. pp. 347–368.
Shen, Howard C.; Taplin, Mary-Ellen; Balk, Steven P. (2010). “Androgen Receptor Antagonists”. Drug Management of Prostate Cancer: 71–81.
Kolvenbag, Geert J. C. M.; Furr, Barrington J. A. (2009). “Nonsteroidal Antiandrogens”. In V. Craig Jordan; Barrington J. A. Furr (eds.). Hormone Therapy in Breast and Prostate Cancer. Humana Press. pp. 347–368.
William Figg; Cindy H. Chau; Eric J. Small (14 September 2010). Drug Management of Prostate Cancer. Springer Science & Business Media. pp. 71–72, 75, 91–96.
Peter B. Farmer; John M. Walker (6 December 2012). The Molecular Basis of Cancer. Springer Science & Business Media. pp. 232–.
de Lignières B, Silberstein S (April 2000). “Pharmacodynamics of oestrogens and progestogens”. Cephalalgia: An International Journal of Headache. 20 (3): 200–7.
^William Ledger; William D. Schlaff; Thierry G. Vancaillie (11 December 2014). Chronic Pelvic Pain. Cambridge University Press. pp. 55–.
Louise Hanna; Tom Crosby; Fergus Macbeth (19 November 2015). Practical Clinical Oncology. Cambridge University Press. pp. 37–.
Mahler C, Verhelst J, Denis L (May 1998). “Clinical pharmacokinetics of the antiandrogens and their efficacy in prostate cancer”. Clin Pharmacokinet. 34 (5): 405–17.
Schröder, Fritz H.; Radlmaier, Albert (2009). “Steroidal Antiandrogens”. In V. Craig Jordan; Barrington J. A. Furr (eds.). Hormone Therapy in Breast and Prostate Cancer. Humana Press. pp. 325–346.
Poyet P, Labrie F (October 1985). “Comparison of the antiandrogenic/androgenic activities of flutamide, cyproterone acetate and megestrol acetate”. Molecular and Cellular Endocrinology. 42 (3): 283–8.
Luthy IA, Begin DJ, Labrie F (1988). “Androgenic activity of synthetic progestins and spironolactone in androgen-sensitive mouse mammary carcinoma (Shionogi) cells in culture”. Journal of Steroid Biochemistry. 31 (5): 845–52.
Flynn T, Guancial EA, Kilari M, Kilari D (2016). “Case Report: Spironolactone Withdrawal Associated With a Dramatic Response in a Patient With Metastatic Castrate-Resistant Prostate Cancer”. Clin Genitourin Cancer. 15 (1): e95–e97.
Caubet JF, Tosteson TD, Dong EW, Naylon EM, Whiting GW, Ernstoff MS, Ross SD (1997). “Maximum androgen blockade in advanced prostate cancer: a meta-analysis of published randomized controlled trials using nonsteroidal antiandrogens”. Urology. 49 (1): 71–8. Because steroidal antiandrogens such as cyproterone acetate have intrinsic androgenic activity and lower antiandrogenic activity than the NSAAs such as flutamide and nilutamide,39–43 it is not surprising that the two classes of antiandrogens may have different efficacies.
Singh SM, Gauthier S, Labrie F (February 2000). “Androgen receptor antagonists (antiandrogens): structure-activity relationships”. Current Medicinal Chemistry. 7 (2): 211–47.
Ayub M, Levell MJ (August 1989). “The effect of ketoconazole related imidazole drugs and antiandrogens on [3H] R 1881 binding to the prostatic androgen receptor and [3H]5 alpha-dihydrotestosterone and [3H]cortisol binding to plasma proteins”. J. Steroid Biochem. 33 (2): 251–5.
Yamasaki K, Sawaki M, Noda S, Muroi T, Takakura S, Mitoma H, Sakamoto S, Nakai M, Yakabe Y (2004). “Comparison of the Hershberger assay and androgen receptor binding assay of twelve chemicals”. Toxicology. 195 (2–3): 177–86.
William B. Pratt (1994). The Anticancer Drugs. Oxford University Press. pp. 220–. In patients receiving flutamide at the usual dosage of 250 mg every 8 hours, the minimal plasma concentration of hydroxyflutamide is about 5 uM, which is 5,000 times the plasma concentration of testosterone (1 nM) in patients treated with an LHRH agonist.127 As hydroxyflutamide is only one percent as potent as testosterone in competing for binding to the androgen receptor,126 a plasma level of 5 uM hydroxyflutamide is required to ensure effective competition.127 […] Both cyproterone acetate and flutamide have been demonstrated to be effective therapy (roughly equivalent to an estrogen) when used alone in the treatment of carcinoma of the prostate.123
Bennett NC, Gardiner RA, Hooper JD, Johnson DW, Gobe GC (2010). “Molecular cell biology of androgen receptor signalling”. Int. J. Biochem. Cell Biol. 42 (6): 813–27.
Iversen P, Melezinek I, Schmidt A (2001). “Nonsteroidal antiandrogens: a therapeutic option for patients with advanced prostate cancer who wish to retain sexual interest and function”. BJU Int. 87 (1): 47–56.
Higano CS (2003). “Side effects of androgen deprivation therapy: monitoring and minimizing toxicity”. Urology. 61 (2 Suppl 1): 32–8.
Di Lorenzo G, Autorino R, Perdonà S, De Placido S (December 2005). “Management of gynaecomastia in patients with prostate cancer: a systematic review”. Lancet Oncol. 6 (12): 972–9.
Erem C (2013). “Update on idiopathic hirsutism: diagnosis and treatment”. Acta Clin Belg. 68 (4): 268–74.
W. Futterweit (6 December 2012). Polycystic Ovarian Disease. Springer Science & Business Media. pp. 282–.
Katsambas AD, Dessinioti C (2010). “Hormonal therapy for acne: why not as first line therapy? facts and controversies”. Clin. Dermatol. 28 (1): 17–23.
Thole Z, Manso G, Salgueiro E, Revuelta P, Hidalgo A (2004). “Hepatotoxicity induced by antiandrogens: a review of the literature”. Urol. Int. 73 (4): 289–95.
Keating GM (March 2015). “Enzalutamide: a review of its use in chemotherapy-naïve metastatic castration-resistant prostate cancer”. Drugs & Aging. 32 (3): 243–9.
Nel comune pensare dell’uomo (e dell’atleta) medio, il Dihydrotestosterone (DHT) è, al pari degli Estrogeni, visto come un ormone tendenzialmente negativo, da ridurre il più possibile. Ovviamente questa visione è a dir poco ristretta dal momento che valuta l’attività del suddetto metabolita del Testosterone solamente in quelle circostanze dove un suo consistente livello può causare, specie nei soggetti predisposti o in determinate circostanze multifattoriali, acne, perdita accelerata dei capelli e ipertrofia prostatica (ovviamente parliamo di soggetti di sesso maschile). Inoltre, il DHT è considerato un metabolita pressoché insignificante nel miglioramento delle prestazioni, soprattutto per quanto concerne l’ipertrofia muscolare. Ma è veramente così limitato il suo impatto per un atleta? ..
Per rispondere a questo quesito nel presente articolo, in modo simile a quanto già feci nell’articolo dedicato agli Estrogeni, esporrò una panoramica dettagliata di tutto ciò che concerne il Dihydrotestosterone e le sue caratteristiche anche alla luce di recenti ed interessanti studi.
Cos’è il DHT?
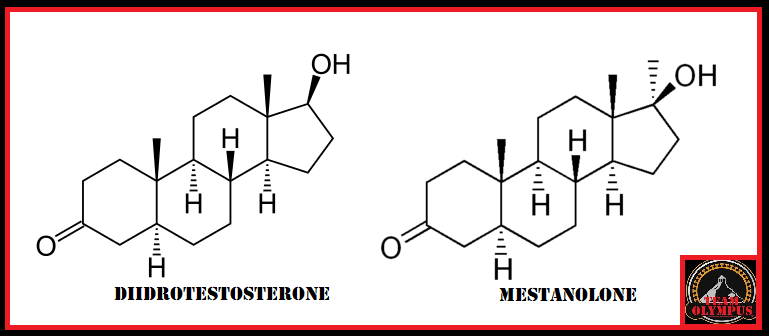
Il Dihydrotestosterone (DHT, 5α-dihydrotestosterone, 5α-DHT, Androstanolone o Stanolone) è uno steroide con caratteristiche fortemente androgene, principalmente ottenuto dalla 5α-riduzione del Testosterone. Infatti, l’enzima 5α-reduttasi catalizza la formazione di DHT dal Testosterone in alcuni tessuti tra cui la ghiandola prostatica, le vescicole seminali, le epididimidi, la pelle, i follicoli piliferi, il fegato e il cervello. Questo enzima media la riduzione del doppio legame C4-5 del Testosterone. Rispetto al Testosterone, il DHT è considerevolmente più potente come agonista del recettore degli androgeni (AR), seppure limitato da percorsi enzimatici.
Oltre al suo ruolo di ormone naturale, il DHT è stato usato come farmaco, ad esempio nel trattamento di bassi livelli di Androgeni negli uomini (vedi Androstanolone).
Il DHT nella Storia
Adolf Friedrich Johann Butenandt (24 marzo 1903-18 gennaio 1995) fu un biochimico tedesco. Nel 1939 gli fu assegnato il premio Nobel per la chimica per il suo “lavoro sugli ormoni sessuali”. Inizialmente respinse il premio a causa della politica nazional-socialista, accettandolo solo nel 1949 dopo la seconda guerra mondiale.
Il DHT fu sintetizzato per la prima volta da Adolf Butenandt e dai suoi colleghi nel 1935. [1][2] Venne ottenuto mediante idrogenazione del Testosterone [3], che era stato scoperto all’inizio di quell’anno.[4] Il DHT è stato introdotto per uso medico come AAS nel 1953 ed è stato inizialmente notato per essere più potente del Testosterone ma con maggiore androgenicità.[5][6][7] Ma il suo potenziale androgeno non fu chiaro fino al 1956, quando venne dimostrato che veniva sintetizzato dal Testosterone negli omogenati di fegato di ratto.[2][8] Inoltre, l’importanza biologica del DHT non è stata realizzata fino agli inizi degli anni ’60, quando si è scoperto che era prodotto dalla 5α-riduzione del Testosterone circolante nei tessuti bersaglio come la ghiandola prostatica e le vescicole seminali risultando più potente del Testosterone in test biologici.[9][10][11][12] Le funzioni biologiche del DHT nell’uomo sono state definite in modo molto più chiaro alla scoperta e alla caratterizzazione del deficit di 5α-reduttasi di tipo II nel 1974.[13] Il DHT è stato l’ultimo importante ormone sessuale, gli altri sono Testosterone, Estradiolo e Progesterone, ad essere scoperto, ed è unico in quanto risulta essere il solo ormone sessuale principale che agisce fondamentalmente come ormone intracrino e paracrino piuttosto che come ormone endocrino.[12]
Biosintesi e distribuzione
Il DHT, noto anche come 5α-androstan-17β-ol-3-one, è uno steroide androstano presente in natura con un gruppo chetonico nella posizione C3 e un gruppo idrossile nella posizione C17β. È il derivato del Testosterone in cui il doppio legame tra le posizioni C4 e C5 è stato ridotto o idrogenato.
Differenze strutturali tra Testosterone e DHT
Il DHT è sintetizzato irreversibilmente dal Testosterone dall’enzima 5α-reduttasi. [14] [15] Ciò si verifica in vari tessuti tra cui i genitali (pene, scroto, clitoride, grandi labbra), [16] prostata, pelle, follicoli piliferi, fegato e cervello. [14] Circa il 5-7% del Testosterone subisce una 5α-riduzione in DHT [17] [18], e circa 200-300μg di DHT vengono sintetizzati giornalmente nel corpo. La maggior parte del DHT è prodotta nei tessuti periferici come la pelle e il fegato, mentre la maggior parte del DHT circolante proviene specificamente dal fegato. I testicoli e la ghiandola prostatica contribuiscono relativamente poco alle concentrazioni di DHT nel circolo ematico.[14]
Ghiandola sebacea
Esistono due isoforme principali di 5α-reduttasi, la SRD5A1 (tipo I) e la SRD5A2 (tipo II), quest’ultimo isoenzima ha una maggiore importanza biologica.[14] Esiste anche una terza forma di 5α-reduttasi: SRD5A3. [19] L’SRD5A2 è maggiormente espressa nei genitali, nella ghiandola prostatica, nelle epididimidi, nelle vescicole seminali, nella pelle genitale, nei follicoli piliferi del viso, del torace [20][21] e nel fegato, mentre si osserva un’espressione più bassa in alcune aree del cervello, pelle non genitale / follicoli piliferi, testicoli e reni. L’SRD5A1 è maggiormente espressa nei follicoli non genitali della pelle / dei capelli, nel fegato e in alcune aree del cervello, mentre sono presenti livelli più bassi nella prostata, nelle epididimidi, nelle vescicole seminali, nella pelle genitale, nei testicoli, nelle ghiandole surrenali e nei reni.[14] Nella pelle, la 5α-reduttasi è espressa in ghiandole sebacee, ghiandole sudoripare, cellule epidermiche e follicoli piliferi.[20][21] Entrambi gli isoenzimi sono espressi nei follicoli piliferi del cuoio capelluto [22], sebbene l’SRD5A2 predomina in queste cellule.[21] Il sottotipo SRD5A2 è l’isoforma quasi esclusivamente espressa nella ghiandola prostatica.[23][24]
Globuline leganti gli ormoni sessuali (SHBG)
Il legame del DHT con le proteine plasmatiche è superiore al 99%. Negli uomini, circa lo 0,88% del DHT non è legato e quindi libero, mentre nelle donne in premenopausa, circa lo 0,47-0,48% non è legato. Negli uomini, il DHT è legato per il 49,7% alla globulina legante gli ormoni sessuali (SHBG), il 39,2% per l’albumina e lo 0,22% per la globulina legante i corticosteroidi (CBG), mentre nelle donne in premenopausa il DHT è legato per il 78,1-78,4% alle SHBG, 21,0-21,3% all’albumina e lo 0,12% al CBG. Nella tarda gravidanza, solo lo 0,07% del DHT non è legato nelle donne; Il 97,8% è legato alle SHBG mentre il 2,15% è legato all’albumina e lo 0,04% è legato al CBG. [25][26] Il DHT ha un’affinità maggiore per le SHBG rispetto al Testosterone, all’Estradiolo o qualsiasi altro ormone steroideo.[27][26]
Funzioni e attività biologiche del DHT
Il DHT è biologicamente importante per la differenziazione sessuale dei genitali maschili durante l’embriogenesi, la maturazione del pene e dello scroto durante la pubertà, la crescita dei peli nel viso, nel corpo e dei peli pubici e lo sviluppo e il mantenimento della ghiandola prostatica e delle vescicole seminali. Come già accennato, è principalmente sintetizzato per via della 5α-riduzione del Testosterone in alcuni tessuti ed è il principale androgeno nei genitali, nella ghiandola prostatica, nelle vescicole seminali, nella pelle e nei follicoli piliferi. [28]
3α-Hydroxysteroide dehydrogenasi (3α-HSD)
Il DHT esplica una segnalazione principalmente in maniera intracrina e paracrina nei tessuti in cui viene sintetizzato, svolgendo un ruolo secondario, sebbene non trascurabile, come ormone endocrino circolante.[29][30][31] I livelli circolanti di DHT sono 1/10 e 1/20 di quelli del Testosterone in termini di concentrazioni totali e libere, rispettivamente [32], mentre i livelli locali di DHT possono essere fino a 10 volte quelli del Testosterone nei tessuti con alta espressione del 5α-reduttasi come la prostata.[33] Inoltre, a differenza del Testosterone, il DHT viene inattivato dalla 3α-idrossisteroide deidrogenasi (3α-HSD) nell’androgeno 3α-androstanediolo molto debole in vari tessuti come quello muscolare, adiposo e epatico, tra gli altri [31][34][35], e in relazione a questo, è generalmente stato riportato che il DHT è un agente anabolico molto scarso quando somministrato esogenamente come farmaco. [36] Ma su questo ci torneremo più avanti.
Progressione della alopecia androgenetica
Oltre alle funzioni biologiche di base, il DHT svolge anche un importante ruolo causale in una serie di condizioni dipendenti dagli androgeni, tra cui le condizioni inerenti alla crescita della peluria come l’irsutismo (eccessiva crescita dei peli sul viso / corpo) e anche la perdita di capelli (alopecia androgenetica o calvizie) e malattie della prostata come l’iperplasia prostatica benigna (IPB) e il carcinoma prostatico.[28] Gli inibitori della 5α-reduttasi, che impediscono la sintesi di DHT, sono efficaci nella prevenzione e nel trattamento di queste condizioni, sebbene siano accompagnati da pesanti effetti collaterali.[37][38][39][40] Inoltre, il DHT può svolgere una funzione nel reclutamento e nella funzione del trasportatore di aminoacidi nel muscolo scheletrico.[41] Ed anche su questo punto torneremo tra poco.
Recettore Estrogeno beta (ERβ)
È stato scoperto che i metaboliti del DHT agiscono come neurosteroidi con la propria attività biologica indipendente dall’AR.[42] Il 3α-Androstanediol è un potente modulatore allosterico positivo del recettore GABAA, mentre il 3β-androstanediol è un potente e selettivo agonista del sottotipo ERβ del Recettore degli Estrogeni (ER).[42] Questi metaboliti possono svolgere un ruolo importante negli effetti centrali del DHT e per estensione del Testosterone, inclusi i loro effetti antidepressivi, ansiolitici, gratificanti / edonici, antistress e pro-cognitivi.[42][43] Ed è soprattutto grazie all’azione neurosteroidea dei metaboliti del DHT a conferire a questa molecola i suoi benefici sull’aumento della forza muscolare e del focus mentale, entrambe caratteristiche ricercate negli sport di potenza e propedeutiche ad un migliore stimolo ipertrofico indotto dall’allenamento contro-resistenza.
Il DHT è un potente agonista dell’AR ed è in effetti il ligando endogeno più potente conosciuto per questo recettore. Ha un’affinità (Kd) compresa tra 0,25 e 0,5 nM per la RA umana, che è circa 2-3 volte superiore a quella del Testosterone (Kd = 0,4 a 1,0 nM) [44] e 15-30 volte superiore a quella degli androgeni surrenali.[45] Inoltre, il tasso di dissociazione del DHT dall’AR è 5 volte più lento di quello del Testosterone.[46 L’EC50 del DHT per l’attivazione dell’AR è 0,13 nM, che è circa 5 volte più forte di quello del Testosterone (EC50 = 0,66 nM).[47] Nei biotest, il DHT è risultato essere da 2,5 a 10 volte più potente del Testosterone.[44]
L’emivita di eliminazione del DHT nel corpo (53 minuti) è più lunga di quella del Testosterone (34 minuti), e ciò potrebbe spiegare alcune delle differenze nella loro potenza.[48] Uno studio sul trattamento transdermico con DHT e Testosterone ha riportato emivite terminali rispettivamente di 2,83 ore e 1,29 ore.[49]
A differenza di altri androgeni come il Testosterone, il DHT non può essere convertito dall’enzima aromatasi in estrogeno come l’Estradiolo. Pertanto, viene spesso utilizzato in contesti di ricerca per distinguere tra gli effetti del testosterone causati dal legame con l’AR e quelli causati dalla conversione del Testosterone in Estradiolo e il successivo legame e attivazione del ER.[50] Sebbene il DHT non possa essere aromatizzato, viene comunque trasformato in metaboliti con significativa affinità e attività ER. Questi sono 3α-androstanediolo e 3β-androstanediolo, che sono agonisti predominanti dell’ERβ.[51] Determinano l’effetto anti-estrogenico attribuito al DHT.
I livelli sierici di DHT sono circa il 10% di quelli del Testosterone, ma i livelli nella ghiandola prostatica sono da 5 a 10 volte superiori a quelli del Testosterone a causa di una conversione di oltre il 90% di quest’ultimo in DHT da parte della 5α-reduttasi espressa localmente.[33] Per questo motivo, e oltre al fatto che il DHT è molto più potente come agonista dell’AR rispetto al Testosterone [44], il DHT è considerato il principale androgeno della ghiandola prostatica.[33]
3α-androstanediol
Il DHT è inattivato nel fegato e nei tessuti extraepatici come la pelle in 3α-androstanediol dall’enzima 3α-idrossistoidea deidrogenasi, e in 3β-androstanediol dall’enzimi 3β-idrossisteroidide deidrogenasi.[34][52]Questi metaboliti vengono a loro volta convertiti, rispettivamente, in Androsterone ed Epiandrosterone, quindi coniugati (tramite glucuronidazione e/o solfatazione), rilasciati in circolazione ed escreti nelle urine.[34]
Come già detto, a differenza del Testosterone, il DHT non può essere aromatizzato in estrogeno come l’Estradiolo e, per questo motivo, non ha propensione ad esercitare effetti estrogenici.[53] Quindi, il DHT viene escreto nelle urine sotto forma di metaboliti, come i coniugati di 3α-androstanediol e Androsterone.[54][34]
Uso del DHT in medicina
Il DHT è disponibile in formulazioni farmaceutiche per uso medico come steroide anabolizzante androgeno (AAS) con finalità prettamente androgene.[55] È usato come ancillare principalmente nel trattamento dell’ipogonadismo maschile.[56] Quando usato come farmaco, il DHT viene chiamato Androstanolone (INN) o Stanolone (BAN) [55] [57] [58], e viene venduto sotto nomi commerciali differenti come Andractim. [55] [57] [58] [56] [59] La disponibilità di DHT farmaceutica è limitata; non è disponibile negli Stati Uniti o in Canada, [60] [61] ma è disponibile in alcuni paesi europei. [58] [56] Le formulazioni disponibili di DHT includono compresse orali o sublinguali, gel topici e, come esteri in olio, iniettabili come Androstanolone propionato e Androstanolone Valerato.[55] [56] [59]
L’Androstanolone è disponibile in formulazioni farmaceutiche per uso medico come androgeno.[4] È usato principalmente come forma ancillare nella terapia sostitutiva degli androgeni nel trattamento dell’ipogonadismo maschile ed è specificamente approvato per questa indicazione in alcuni paesi.[62] [13] [63] [64] [65] [66] [67] Non è più raccomandato come solo farmaco nelle terapie sostitutive degli androgeni a causa delle differenze biologiche con il Testosterone come la mancanza di effetti Estrogenici e effetti androgeni parziali.[68] L’Androstanolone topico è utile nel trattamento della ginecomastia.[69] Allo stesso modo, l’Androstanolone Enantato tramite iniezione intramuscolare è risultato efficace nel trattamento della ginecomastia puberale persistente.[70] Il farmaco è stato anche usato come gel topico per il trattamento del pene piccolo nei ragazzi pre e peripubertali con sindrome da insensibilità agli androgeni lieve o parziale.[71] [72] [73]
Drostanolone Propionato
L’Androstanolone è risultato efficace nel trattamento del carcinoma mammario in fase avanzata nelle donne negli anni ’50, sebbene fosse utilizzato in dosi molto elevate e causasse una grave virilizzazione.[74] [75] [76] È stato usato in sospensione acquosa microcristallina mediante iniezione intramuscolare.[77] [78] [79] Poco dopo, il Drostanolone Propionato (2α-Methylandrostanolone Propionato) fu sviluppato per questo uso al fine di sostituire l’Androstanolone a causa della sua superiore farmacodinamica e fu introdotto per questa indicazione negli Stati Uniti e in Europa nei primi anni ’60.[80] [81] [82] [83]
L’Androstanolone è stato usato alla dose di 25mg per via sublinguale da due a tre volte al giorno nella terapia sostitutiva con androgeni per gli uomini.[84] Questo è anche il dosaggio di Androstanolone comunemente utilizzato nel trattamento di individui si sesso maschile.[84]
La questione DHT e ipertrofia muscolare
La sarcopenia, caratterizzata da una perdita di massa muscolare, ossea, forza e resistenza, si verifica con l’invecchiamento e disturbi medici cronici come l’infezione da virus dell’immunodeficienza umana (HIV) e la terapia a lungo termine con glucocorticoidi sistemici (Gcc). D’altra parte, la somministrazione di Testosterone negli uomini più anziani e negli uomini con infezione da HIV con perdita di peso che hanno basse concentrazioni di Testosterone (Bhasin et al. 2001), nonché gli uomini che richiedono un trattamento sistemico a lungo termine di Gcc (Truhan e Ahmed 1989) aumentano il grasso corporeo- massa magra e forza muscolare.
Recettore degli Androgeni
Il muscolo scheletrico è uno dei tessuti bersaglio per l’azione anabolica degli androgeni. Recettori degli Androgeni (AR), localizzati nelle cellule muscolari e adipose, cellule nervose e pluripotenti mesenchimali che risiedono nel tessuto muscolare, probabilmente mediano gli effetti degli androgeni aumentando la massa muscolare, la sintesi proteica, il contenuto ribosomiale, le aree mitocondriali, il numero mioonucleare, il numero di cellule satellite e la miogenesi delle cellule mesenchimali pluripotenti riducendo la degradazione delle proteine e l’adipogenesi delle cellule mesenchimali pluripotenti (Herbst & Bhasin 2004). Sul ligando che si lega all’AR intracellulare, il complesso androgeno-AR viene traslocato nel nucleo e si lega a sequenze specifiche di DNA, elementi di risposta agli androgeni, con conseguente trascrizione di geni specifici (Michel & Baulieu 1980, Simental et al. 1991). Gli androgeni hanno anche azioni rapide non genomiche nel muscolo (Estrada et al. 2000, 2003), tra cui il recettore di membrana accoppiato alla proteina G, il recettore dell’inositolo 1,4,5-trisfosfato (IP3), lo ione calcio (Ca2 +) e la cascata della fosforilazione della proteina chinasi mitogeno-attivata (MAPK) / proteina chinasi regolata da segnali extracellulari (ERK). Oltre al suo ruolo nella contrazione muscolare, si ritiene che Ca2+ intracellulare regola l’espressione genica nel muscolo scheletrico (Estrada et al. 2001, Araya et al. 2003). Pertanto, le azioni genomiche e non genomiche degli androgeni sono responsabili della trascrizione dei geni sensibili agli androgeni (ARG).
Tuttavia, i meccanismi molecolari dell’effetto anabolico degli androgeni nel muscolo scheletrico sono mal compresi. Con l’avvento dell’analisi seriale dell’espressione genica (SAGE) (Velculescu et al. 1995), sono sorte nuove possibilità per l’analisi del trascrittoma su larga scala. Usando questo metodo, si sono precedentemente studiati i meccanismi molecolari responsabili dell’atrofia muscolare causata dall’immobilizzazione nei ratti (St-Amand et al. 2001), nonché il profilo di espressione genica degli uomini allenati per la resistenza (Yoshioka et al. 2003). In un interessante studio del 2006 [85], si sono studiati gli effetti della castrazione (GDX) e del DHT sull’espressione genica globale nel muscolo scheletrico dei topi maschi usando la strategia SAGE. Le trascrizioni modulate DHT sono coinvolte nel rilascio di Ca2 +, nella segnalazione cellulare, nella proliferazione cellulare, nella sintesi di mRNA e proteine e nel metabolismo energetico. Questi risultati costituiscono un primo passo verso una comprensione precisa dei meccanismi molecolari coinvolti negli effetti fisiologici degli androgeni nel muscolo scheletrico.
Topo C57BL6
Nello studio, è stato asportato il muscolo gastrocnemio destro dai topi C57BL6 di età compresa tra 12 e 14 settimane. Gli animali sono stati tenuti con luci accese da 0715 a 1915h, e hanno avuto accesso all’acqua ad libitum. Nessun trattamento è stato eseguito su 26 topi intatti. Il GDX è stato eseguito 7 giorni prima della raccolta di organi in ciascuno dei 14 topi dai gruppi GDX e DHT. I topi del gruppo GDX hanno ricevuto un i.p. della soluzione del veicolo (0,4% (p / v) Methocel A15 LV Premium / 5% etanolo; Dow Chemicals Co, Laval, Quebec, Canada) 24 ore prima della morte, mentre una dose fisiologica di DHT (0,1 mg / topo) è stato iniettato 1, 3, 6 e 24 ore prima della loro uccisione (gruppi DHT 1 h, DHT 3 h, DHT 6 he DHT 24 h). Il muscolo gastrocnemio destro è stato campionato da ciascun topo e messo insieme per l’analisi dello stesso gruppo per eliminare le variazioni inter-individuali ed estrarre quantità sufficienti di mRNA. I tessuti sono stati conservati a -80 ° C fino all’estrazione dell’RNA.
Poliammina Spermina
Gli ormoni anabolizzanti stimolano la crescita muscolare principalmente aumentando la sintesi proteica (Rooyackers & Nair 1997). In questo studio, la condizione GDX ha represso l’espressione del membro della famiglia delle proteine da shock termico 7 (Hspb7), mentre l’iniezione di DHT ha regolato verso l’alto cinque geni che codificano proteine ribosomiali e chaperoni (Mrpl51, Rpl34, Rps20, Cct8 e Cabc1) entro 3 ore e modulando altre tre trascrizioni (Fxr2h, Rps24 e Rps27) a 24 h. Oltre alla sintesi proteica, Rpl34 e Rps20 sono implicati nella biosintesi delle poliammine (Panagiotidis et al. 1995). La proteina Cct8 la cui espressione è fortemente dipendente dalla crescita cellulare (Yokota et al. 1999) piega le proteine appena sintetizzate, compresi i componenti cellulari necessari per la crescita cellulare (Thulasiraman et al. 1999), oltre a comportarsi come proteina associata ai microtubuli (Roobol et al. 1999). Mrpl51 è codificato dal DNA mitocondriale e Cabc1 codifica una proteina mitocondriale essenziale per la corretta conformazione e funzionamento dei complessi proteici nella catena respiratoria (Iiizumi et al. 2002). In effetti, le espressioni di 18 trascrizioni relative alla produzione di OxPhos e ATP sono state sovra-regolate dal DHT in questo studio. Questi dati suggeriscono che il DHT aumenta la sintesi proteica e la stabilizzazione in parallelo con la crescita cellulare entro 3 ore nei topi in vivo.
Le trascrizioni modulate 24h dopo il trattamento con DHT nel presente studio suggeriscono quanto segue:
l’aumento del trasporto di mRNA tra citoplasma e nucleolo da parte delle proteine fragili correlate all’X (FMRP) (Tamanini et al. 1999);
la soppressione della degradazione dell’mRNA da parte della proteina ribosomiale S27 (Revenkova et al. 1999);
la diminuzione della sintesi proteica poiché la proteina ribosomiale S24 è strettamente coinvolta sia nei processi di iniziazione che di allungamento durante la sintesi proteica (Bommer et al. 1988).
Il fatto che il tasso di sintesi proteica diminuisca drasticamente durante la mitosi nelle cellule di mammifero potrebbe spiegare i dati.
La degradazione delle proteine da parte del proteasoma 26S è essenziale per la progressione del ciclo cellulare, il metabolismo delle poliammine e la presentazione della catena pesante di classe I del maggiore complesso di istocompatibilità (MHC) sulla superficie cellulare. Il DHT ha indotto Psmc3 e Psmc5 le cui proteine sono i componenti integrali della subunità normativa 19S del proteasoma 26S, che potrebbe riflettere l’induzione della proliferazione cellulare e la modulazione dell’immunità da DHT.
La regolazione trascrizionale è un punto di controllo essenziale per diverse funzioni cellulari come la proliferazione cellulare, la differenziazione, la trasformazione e l’apoptosi. Il DHT ha sovra-regolato cinque fattori trascrizionali (Ogt, Pttg1, Psmc3, Psmc5 e Smyd2) entro 3 ore dopo il trattamento e il Fxr2h a 24h. L’attivazione della cascata MAPK provoca la traslocazione della proteina citoplasmatica Pttg1 nel nucleo (Pei 2000) dove la proteina Pttg1 transattiva i geni bersaglio che promuovono la proliferazione cellulare (Pei 2001). Le proteine di Psmc3 e Psmc5 suggeriscono ruoli nella transattivazione del recettore dell’ormone tiroideo (Ishizuka et al. 2001). La condizione GDX ha anche ridotto il livello di espressione del Ttr, il cui prodotto è una proteina plasmatica omotetramericana che trasporta tiroxina e retinolo. Sebbene gli ormoni tiroidei siano essenziali durante la crescita, sia un eccesso che una carenza causano un degrado muscolare da meccanismi sconosciuti (Rooyackers & Nair 1997). La proteina Fxr2h mostra una forte attivazione della trascrizione (Hillman & Gecz 2001). La glicosilazione delle proteine nucleari e citoplasmatiche è una modifica post-traduzionale diffusa e reversibile nelle cellule eucariotiche. La glicosilazione intracellulare dei residui di serina e treonina è catalizzata dalla proteina di Ogt, che regola un numero di funzioni cellulari tra cui l’attivazione trascrizionale (geni bersaglio p53) / repressione (RNA polimerasi II) e l’attivazione traslazionale (Wells et al. 2003). La presenza nel gene Smyd2 di domini SET e MYND sarebbe in accordo con gli effetti rispettivamente sulla deacetilazione e metilazione dell’istone (Sims et al. 2002). Pertanto, i risultati suggeriscono che almeno alcune delle azioni del DHT si verificano attraverso l’attivazione o la repressione dei regolatori trascrizionali.
Via di segnalazione MAPK
Nel muscolo scheletrico, il Ca2+ svolge un ruolo chiave nella contrazione e nel rilassamento. Il presente studio ha dimostrato che il trattamento con DHT ha aumentato l’espressione della Pvalb, una proteina di legame Ca2+ ad alta affinità che agisce come fattore di rilassamento muscolare dopo la contrazione, e Trdn che forma un complesso quaternario con il recettore della ryanodina, junctina e calsequestrina nel lume del reticolo sarcoplasmatico ( SR) per il buffering passivo di Ca2+ luminale SR, nonché un rilascio Ca2+ attivo dal processo SR durante l’accoppiamento eccitazione-contrazione. Topi transgenici che sovraesprimono Trdn1 nel cuore mostrano ipertrofia cardiaca con rilassamento alterato e contrattilità attenuata (Kirchhefer et al. 2001). Pertanto, l’induzione di entrambi i fattori di rilassamento muscolare e di contrazione potrebbe contribuire a una generazione di energia generalmente osservata negli atleti che assumono steroidi anabolizzanti.
Fosfolipasi C
La depolarizzazione delle cellule muscolari provoca anche un rilascio transitorio lento di Ca2 +, che è mediato dalla fosfolipasi C (PLC) e IP3 tramite i recettori IP3 (Estrada et al. 2001, Powell et al. 2001) e porta alla fosforilazione di ERK1 / 2 (Powell et al. 2001). Negli osteoblasti, il DHT attiva la proteina Gβ4 accoppiata al PLC-β2 che aumenta la formazione di IP3 e diacilglicerolo (DAG) e innesca il rilascio di Ca2 + intracellulare dal reticolo endoplasmatico (Zagar et al. 2004). Gli aumenti dei livelli di DAG e Ca2 + regolano l’attività della proteina chinasi C (PKC) che stimola ERK1 / 2 attraverso l’attivazione di MAPK chinasi 1/2 (Zagar et al. 2004). I risultati hanno mostrato le induzioni di Camk2g, Dusp1 e Hint1 entro 3 ore dall’iniezione di DHT. Ca2 + multifunzionale / proteinodinasi dipendente dalla calmodulina (CaMKII) media le risposte cellulari alla Ca2 + intracellulare ed è implicato nel controllo di funzioni essenziali quali trasmissione sinaptica, canali ionici, trascrizione genica e progressione del ciclo cellulare (Santella 1998, Anderson 2005). Le cellule proliferanti (Tombes & Krystal 1997) e il cuore ipertrofico con maggiore contrattilità (Colomer et al. 2003) esprimono l’isoforma CaMKIIγ codificata da Camk2g. Dusp1 (chiamato anche CL100 o MAPK fosfatasi 1) è stato originariamente identificato come un gene precoce immediato indotto da mitogeni (Charles et al. 1992, Keyse & Emslie 1992), e il suo livello di trascrizione riflette l’attivazione di ERK1 / 2 (Camps et al. 2000).
Proteina Hint1
La Hint1, una proteina che interagisce con la PKC che originariamente si pensava inibisse quest’ultima, può svolgere un ruolo di soppressore del tumore (Su et al. 2003). Abbiamo osservato la sovra-regolazione di Mpp6, Oaz, Psmc3 e Psmc5 entro 3 ore dal trattamento con DHT. La funzione di Mpp6, un membro della sottofamiglia guanilato chinasi (MAGUK) associata alla membrana p55, non è ancora nota. Tuttavia, il MAGUK interagisce con i recettori del glutammato e vari canali ionici (Godreau et al. 2004). Le poliammine (spermine, spermidina e putrescina) interagiscono anche con alcuni canali ionici e controllano i livelli intracellulari di Ca2 + (Williams 1997). La biosintesi della poliammina nelle cellule di mammifero inizia con una produzione di putrescina da parte dell’ornitina decarbossilasi (ODC). Quando i livelli di poliammina intracellulare aumentano, l’antizima ODC, codificato da Oaz, si lega all’OCD e facilita il suo rapido degrado da parte del proteosoma 26S (Thomas & Thomas 2003). Pertanto, le induzioni di Oaz così come Psmc3 e Psmc5, che sono componenti integranti del proteasoma 26S, possono riflettere il livello aumentato di poliammine intracellulari. Inoltre, le modulazioni di queste trascrizioni e di Mpp6 entro 3 ore, lo stesso decorso di Camk2g, suggeriscono la loro partecipazione al controllo di Ca2 + intracellulare. Inoltre, il percorso Ras / MAPK controlla la trascrizione di Cct8 (Yamazaki et al. 2003) che è stata indotta a 1 ora dopo il trattamento con DHT nel presente studio. Nel loro insieme, il secondo messaggero, ovvero Ca2 + intracellulare, e le sue cascate a valle tra cui PKC e MAPK, che sono essenziali per la regolazione della crescita cellulare, sembrano essere modulati dal DHT.
Le cellule satellite / mioblasti all’interno del tessuto muscolo-scheletrico proliferano in seguito all’esposizione a fattori di crescita e a seguito di lesioni muscolari, ma smettono di dividersi quando si fondono con fibre muscolari preesistenti. La fusione è generalmente accoppiata con l’inizio della proliferazione cellulare. Nel presente studio, la trascrizioni sovra-regolate da parte del DHT e correlate all’entrata in fase S (Pttg1) (Nasmyth et al. 2000), assemblaggio di microtubuli (Cct8) (Roobol et al. 1999), formazione del fuso bipolare (Tctex1) (Vaisberg et al 1993), uguale segregazione cromosomica (Pttg1) (Nasmyth et al. 2000), accatastamento di Golgi cisternae (Gorasp2) (Shorter et al. 1999) e disintossicazione dei metaboliti reattivi prodotti durante la proliferazione cellulare (Akr1a4) (Barski et al. 2004 ), suggeriscono un’induzione della proliferazione cellulare da parte del DHT. D’altra parte, il DHT ha sotto-regola un fattore cistostatico, Lgals1, che mantiene G0 e controlla la traversata G2 (Wells & Mallucci 1991). Il fuso mitotico richiede il montaggio / smontaggio di icrotubuli e l’azione di complessi motori come il dynein (Vaisberg et al. 1993). La proteina Tctex1 è una catena leggera del complesso motorio dynein (Tai et al. 1998). Il Cct8 aumenta durante la transizione G1 / S attraverso la prima fase S (Yokota et al. 1999). Pttg1, protezione umana, si accumula all’inizio della fase S con picchi nelle fasi G2 – M, e previene l’attivazione prematura delle separine durante la mitosi (Nasmyth et al. 2000).
Il DHT sovra-regola Psmc3, Dusp1, Gadd45g e Pttg1 nelle presenti condizioni. La sovraespressione di Psmc3 aumenta le proteine p53 e p21 (Pollice et al. 2004). In risposta al danno al DNA e ad altri stress, il soppressore del tumore p53 induce l’arresto del ciclo cellulare o l’apoptosi a seconda dei contesti cellulari specifici (Yu & Zhang 2005). In risposta al danno al DNA, p53 promuove la riparazione del DNA influenzando il percorso di riparazione dell’escissione del DNA e arrestando le cellule in G1 attraverso l’induzione di p21 che contribuiscono a fornire più tempo per la riparazione (Smith & Seo 2002), mentre anche l’arresto G1 mediato da p53 si verifica per induzione di Dusp1 in assenza di danni al DNA (Li et al. 2003). Le proteine codificate da Gadd45g interagiscono con p21 e sopprimono la crescita cellulare senza alcuna evidenza di apoptosi (Nakayama et al. 1999). L’arresto della crescita mediato dagli inibitori del ciclo cellulare p21 e Gadd45 inibisce la risposta apoptotica indotta da bersagli apoptotici di p53 (Yu & Zhang 2005). Inoltre, la protezione codificata da Pttg1 inibisce la capacità di p53 di indurre la morte cellulare (Bernal et al. 2002). Nel loro insieme, il DHT potrebbe promuovere l’arresto di G1 senza indurre l’apoptosi, almeno secondo ciò che è emerso dal presente studio.
S-adenosilmetionina decarbossilasi (SAMDC)
Il presente studio riporta l’induzione di Amd1 che codifica per la S-adenosilmetionina decarbossilasi (SAMDC), Oaz, Rps20 e Rpl34, nonché Psmc3 e Psmc5 dopo l’iniezione di DHT. D’altra parte, il DHT sotto-regolato Sbp il cui prodotto è correlato all’accumulo di spermatozoi (Moruzzi et al. 1982). I composti policristici sintetizzati dagli enzimi che limitano la velocità, ODC e SAMDC, sono cruciali per la crescita e la proliferazione delle cellule di mammifero. L’antizima ODC codificato da Oaz, così come le proteine ribosomiali L34 e S20, inibiscono le decarbossilasi di arginina e ODC (Panagiotidis et al. 1995). ODC e SAMDC sono degradati dal proteasoma 26S (Yerlikaya e Stanley 2004) che è codificato da Psmc3 e Psmc5. L’antizima ODC inibisce anche l’assorbimento della poliammina e stimola l’escrezione (Sakata et al. 2000). Inoltre, l’assorbimento della poliammina è inibito dal PKC ed è stimolato dalla sua inibizione (Dot et al. 2000). Per coincidenza, Hint1, che inibisce la PKC, aveva un modello di modulazione simile a quello di Oaz con il trattamento a base di DHT. Nel loro insieme, le modulazioni di Pttg1, Cct8, Tctex1, Gorasp2, Akr1a4, Lgals1, Amd1, Oaz, Rpl34, Rps20, Psmc3, Psmc5 e Sbp mediante dal DHT nel presente studio potrebbero riflettere la proliferazione di cellule satelliti / mioblasti nel muscolo scheletrico .
Inoltre, la proteina Pttg1 induce angiogenesi sia in vitro che in vivo (Ishikawa et al. 2001). Nel presente studio, il DHT ha sovra-regolato Pttg1 e Asb5, che è una nuova proteina implicata nell’inizio dell’arteriogenesi (Boengler et al. 2003). La vascolarizzazione è un importante fattore determinante dell’approvvigionamento energetico e della rimozione dei rifiuti durante la contrazione muscolare e la sua stimolazione da parte del DHT in questo studio è quindi in accordo con altri dati presentati.
Lpin1
Nel metabolismo lipidico, la condizione GDX ha spento la Apina2 e il DHT a sovra-regolato la Lpin1. L’apolipoproteina A-II codificata da Apoa2, la seconda proteina più abbondante delle particelle di lipoproteine ad alta densità (HDL), esercita un marcato effetto sul legame HDL e sull’assorbimento selettivo dei lipidi da parte dei recettori scavenger di classe B. Nel topo, l’espressione migliorata di Lpin1 nel muscolo scheletrico promuove l’obesità diminuendo il dispendio energetico dell’intero corpo e l’utilizzo dei grassi, nonché inducendo resistenza all’insulina (Phan & Reue 2005). Contrariamente a quanto accade nel muscolo, la sovraespressione di Lpin1 nel tessuto adiposo provoca obesità senza insulino-resistenza (Phan & Reue 2005). Ho precedentemente riportato che il livello di espressione di Lpin1 nel tessuto adiposo rimane inalterato con il trattamento a base di DHT (Bolduc et al. 2004). L’induzione di Lpin1 solo nel muscolo potrebbe suggerire che il carboidrato fosse usato per aumentare la produzione di OxPhos e ATP. Ulteriori studi sono necessari per chiarire questo intrigante meccanismo.
Le prime risposte all’iniezione di DHT (DHT 1, 3 e 6 h) sono l’induzione sia dei fattori di rilassamento muscolare (Pvalb) che di contrazione (Trdn) che modulano i livelli intracellulari di Ca2+ . Il DHT ha anche indotto le trascrizioni relative alla segnalazione cellulare come Ca2 + (Camk2g), PKC (Hint1) e percorsi MAPK (Dusp1) nonché la biosintesi della poliammina (Amd1, Oaz, Psmc3, Rps20 e Rpl34), proliferazione cellulare (Akr1a4, Cct8 Pttg1 e Tctex1), arresto del ciclo cellulare (Gadd45g), p53 (Cabc1, Dusp1 e Pttg1) e angiogenesi (Asb5 e Pttg1). L’induzione di mRNA correlati alla trascrizione (Ogt, Psmc3 e Psmc5), sintesi proteica (Mrpl51, Ogt, Rps20 e Rpl34), modifica (Cabc1, Cct8 e Ogt) e degradazione (Psmc3 e Psmc5), fosforilazione ossidativa (Cyc1, MtCo1, in questi punti sono stati osservati anche MtCo2, Cyp27a1, Ndufa5 e Ndufb2), produzione di ATP (Atp5j2 e EST Atpaf1), metabolismo lipidico (Lpin1) e immunità (Cd59a e Ga17). Tuttavia, l’induzione di trascrizioni relative alla segnalazione di Ca2+, MAPK, arresto del ciclo cellulare, p53, sintesi proteica e angiogenesi non è più significativa dopo 24 ore dall’iniezione di DHT mentre le trascrizioni relative alla progressione del ciclo cellulare sono ancora sovraregolate. Inoltre, le trascrizioni relative alla sintesi proteica (Rps24 e Rps27) erano sotto-regolate. Questi risultati indicano che l’iniezione di DHT induce la generazione di energia, la sintesi proteica, la funzione mitocondriale e la proliferazione di cellule satellite / mioblasti a livello trascrizionale in vivo, supportando precedenti risultati di un’azione anabolica del composto in questione.
Panoramica degli ARG nel muscolo scheletrico. Le trascrizioni sovra e sotto regolate dal trattamento con DHT sono mostrate rispettivamente in rosa e in blu. Le trascrizioni che mostrano solo una risposta precoce (DHT 1 ora, 3 ore e 6 ore) o tardiva (DHT 24 ore) sono indicate rispettivamente come (E) o (L). Le linee continue e tratteggiate rappresentano rispettivamente attivazione / induzione e inibizione. Akr1a4, aldo-keto reductasi famiglia 1 membro A4; Amd1, S-adenosilmetionina decarbossilasi 1; Asb5, ripetizione di ankyrin e proteina 5 contenente scatola SOCs; Atp5j2, ATP sintasi subunità mitocondriale F0 complessa f isoforma 2; Atp6, ATP sintasi 6; Atpaf1, fattore di assemblaggio complesso mitocondriale F1 sintasi ATP 1; B2m, microglobulina β-2; Cabc1, attività di accompagnatore ABC1 del complesso bc1 simile; Camk2g, protein chinasi IIγ calcio / calmodulina-dipendente; Cct8, subunità chaperonin 8; Cd59a, antigene CD59a; Col1a2, procollagene tipo I α2; Cyc1, citocromo c-1; Cyp27a1, citocromo P450 famiglia 27 sottofamiglia un polipeptide 1; DAG, diacilglicerolo; Dusp1, fosfatasi 1 a doppia specificità; EST, tag di sequenza espressi; Fxr2h, fragile 2 ritardo mentale X gene 2; Ga17, proteina cellulare dendritica GA17; Gadd45g, arresto della crescita e DNA inducibile 45γ; Gorasp2, Golgi riassemblando la proteina 2; Suggerimento 1, proteina 1 di legame alla triade di nucleotidi di istidina; IP3, inositolo 1,4,5-trisfosfato; Lgals1, lectina legante il galattosio solubile 1; Lpin1, lipina 1; MAPK, protein chinasi attivata dal mitogeno; Mrpl51, proteina ribosomiale L51; MtCo1, citocromo c ossidasi 1; MtCo2, citocromo c ossidasi 2; MtCo3, citocromo c ossidasi 3; MtNd2, NADH deidrogenasi 2; MtNd3, NADH deidrogenasi 3; MtNd4, NADH deidrogenasi 4; Ndufa5, NADH deidrogenasi 1α sottocomplex 5; Ndufb2, NADH deidrogenasi 1β sottocomplex 2; Ndufb9, NADH deidrogenasi 1β sottocomplex 9; Oaz, ornitina decarbossilasi antizima; Ogt, N-acetilglucosamina transferasi legata all’O; PKC, proteina chinasi C; PLC, fosfolipasi C; Psmc, subunità ATPase 26S proteasoma; Pvalb, parvalbumina; Rp, proteina ribosomiale; Pttg1, trasformazione del tumore pituitario 1; Sbp, proteina legante lo sperminozoo; Tctex1, testicolo complesso t espresso 1; TPO1, trasportatore di poliammina; Trdn, triadin.
Un altro studio interessante del 2011 e pubblicato sul “The Journal of Physiology” [86] ci offre ulteriori indizzi sul potenziale del DHT nell’ipertrofia muscolare.
Il gruppo di ricerca che ha svolto questo studio ne aveva in precedenza condotto un altro attraverso il quale avevano dimostrato che il Dihydrotestosterone (DHT), ma non il Testosterone, aumenta la produzione di forza nei muscoli a contrazione rapida e la diminuisce in quelli a contrazione lenta. Questi risultati hanno loro suggerito che il DHT poteva essere un androgeno con capacità ben superiori a quelle comunemente attribuiteli. Nel presente studio, i ricercatori hanno esaminato gli effetti di questi ormoni sul trasporto degli aminoacidi nei fasci muscolari dell’apparato muscolo-scheletrico a rapida contrazione del topo. I risultati mostrano che il DHT aumenta la sintesi proteica e l’aumento delle proteine che trasportano aminoacidi essenziali in fasci muscolari a contrazione rapida. Questo non è stato osservato con il Testosterone.
E’ stato osservato che il DHT esercita azioni acute/non genomiche nel muscolo-scheletrico di mammiferi adulti le cui funzioni fisiologiche sono state capite di recente, pertanto l’obiettivo primario di questo studio era di osservare gli effetti acuti / non genomici del DHT sul uptacke di aminoacidi e sugli eventi di trasduzione del segnale cellulare alla base di queste azioni in fasci muscolari scheletrici a contrazione rapida e lenta di topo. Gli aminoacidi marcati con 14C sono stati usati per studiare gli effetti del DHT e del Testosterone (T) sull’assorbimento degli aminoacidi e sono stati usati interventi farmacologici per determinare gli eventi di trasduzione del segnale cellulare che mediano queste azioni. Mentre il T non ha avuto alcun effetto sull’assorbimento di isoleucina (Ile) e acido α-metilamminoisobutirrico (MeAIB) in entrambi i tipi di fibre, il DHT ha aumentato il loro assorbimento nei fasci di fibre a contrazione rapida. Questo effetto è stato invertito dagli inibitori della traslazione proteica, del recettore del prodotto per la crescita epidemica (EGFR), del sistema A, del sistema L, del mTOR e del MEK. Tuttavia, è stato relativamente correlato agli inibitori della trascrizione, dei recettori degli androgeni e dell’IP3K / Akt. In aggiunta, il trattamento con DHT ha aumentato l’espressione della LAT2 e l’insufflazione fosforilata del GRFR, mentre il miscuglio rapido-twitch si mescola e in entrambi i tipi di ERK1 / 2, RSK1 / 2 e ATF2. Inoltre, ha diminuito la fosforilazione di eEF2 e ha aumentato l’incorporazione di proteine di ferro in entrambi i tipi di fibre. La maggior parte di questi effetti è stata superata dagli inibitori di EGFR e MEK. Da questi risultati si ipotizza che un’altra funzione fisiologica delle azioni acute/non genomiche del DHT nelle fibre muscolari isolate dei mammiferi è quella di stimolare l’assorbimento di aminoacidi. Questo effetto è mediato dall’EGFR e comporta l’attivazione della via MAPK e un aumento dell’espressione LAT2.
IGF-1
Un risultato chiave nel presente studio è stato l’osservazione che il trattamento di piccoli fasci di fibre muscolari scheletriche isolati dall’EDL e dal soleo di topi femmine adulti con concentrazioni fisiologiche (630pgml − 1) di DHT, per 1 ora, significativamente (P = 0,001) ha aumentato l’assorbimento di Ile e MeAiB nei fasci di fibre isolati dall’EDL ma non in quelli isolati dal soleo. Sebbene la somministrazione acuta di ormoni come l’insulina (Biolo et al. 1995), il fattore di crescita insulino-simile 1 (IGF-1) (Fryburg et al. 1995) e l’ormone della crescita (Fryburg et al. 1995) hanno dimostrato di aumentare la sintesi proteica e per promuovere l’assorbimento di aminoacidi nel muscolo scheletrico umano, è la prima volta che è stato dimostrato un aumento dell’assorbimento di aminoacidi in risposta alla somministrazione acuta di uno steroide anabolizzante-androgeno nel muscolo scheletrico dei mammiferi adulti. Solo altri due studi hanno precedentemente osservato gli effetti dell’assorbimento di aminoacidi. Entrambi gli studi hanno utilizzato soggetti umani e non sono stati in grado di dimostrare alcun cambiamento nell’assorbimento degli aminoacidi in gruppi muscolari interi (Bhasin et al. 1997; Ferrando et al. 1998). Sebbene nel presente studio siano state utilizzate piccole fibre muscolari e il T sia stata applicato direttamente ai fasci di fibre, è importante notare che questo ormone non ha avuto alcun effetto sull’assorbimento degli aminoacidi in entrambi i tipi di fibre. Al contrario, il trattamento dei fasci di fibre con DHT ha comportato un marcato aumento dell’assorbimento di Ile e MeAIB solo nei fasci di fibre a contrazione rapida (Fig. 1). In precedenza, abbiamo anche dimostrato che il T non ha effetti acuti sulla produzione di forza in fasci muscolari di topo isolati intatti, mentre il DHT ha aumentato la produzione di forza nei fasci di fibre a contrazione rapida, ma l’ha diminuita in quelli a contrazione lenta (Hamdi & Mutungi, 2010). Nel loro insieme, questi risultati suggeriscono che il T potrebbe non avere effetti acuti / non genomici nelle fasce muscolari scheletriche dei mammiferi adulti.
Come già detto, nella maggior parte dei tessuti il T viene convertito in DHT dall’enzima 5 α-reduttasi. Pertanto, è probabile che tutti gli effetti acuti del T osservati in precedenza nei miociti in coltura (Estrada et al. 2003) potrebbero essere stati esercitati dal DHT. In effetti, è stato precedentemente suggerito che gli effetti anabolici del T nei muscoli scheletrici umani possano essere indiretti o secondari al rilascio di un altro ormone come IGF-1 (Ferrando et al. 1998). Con la presente si precisa che oltre all’IGF-1, gli effetti del T nel muscolo scheletrico dei mammiferi possono anche essere esercitati attraverso il rilascio di DHT. I ricercatori suggeriscono anche che il DHT è il principale steroide anabolizzante-androgeno nei muscoli scheletrici dei mammiferi adulti.
Nonostante la maggior parte degli studi è stata effettuata su fasci muscolari di topi femmina, in precedenza era già stato dimostrato che il DHT ha effetti simili sulla produzione di forza nei fasci muscolari scheletrici di topi maschi e femmine (Hamdi & Mutungi, 2010).
I risultati riportati in questo studio (vedi figura seguente) mostrano che sebbene il DHT aumenti l’attività del SNAT2, ciò non influisce sulla sua espressione. Al contrario, aumenta l’attività e l’espressione del LAT2 (vedi Fig. 3). Inoltre, il trattamemto del fascio di fibre muscolari con il classico sistema di inibitori A e L-typeaminoacid ha portato a una marcata riduzione dell’assorbimento basale di L- [U-14C] Ile in entrambi i tipi di fibre e ha completamente soppresso l’aumento indotto da DHT nell’assorbimento di Ile.
Gli effetti del DHT sull’assorbimento di aminoacidi nei fasci di fibre muscolari di topo sono parzialmente mediati attraverso un trasportatore di aminoacidi del sistema A e sono mediati attraverso LAT2.
Effetti simili sono stati osservati anche quando i fasci di fibre sono stati trattati con la soluzione di Ringer contenente anticorpi contro il LAT2, suggerendo che gli effetti del DHT sull’Ile sono mediati attraverso questo trasportatore. Da queste osservazioni si ipotizza che i due trasportatori (LAT2, SNAT2) siano in qualche modo collegati. Pertanto, modulando l’espressione e l’attività di LAT2, anche il DHT sembra regolare indirettamente l’attività di SNAT2.
Recettore del Fattore di Crescita dell’Epidermide (EGFR)
In precedenza, era stato dimostrato che le azioni acute/ non genomiche del DHT sulla produzione di forza nei fasci muscolo-scheletrici di topi adulti sono mediate attraverso il Recettore del Fattore di Crescita dell’Epidermide (EGFR) e comportano l’attivazione di ERK1 / 2 (Hamdi & Mutungi, 2010). È interessante notare che i risultati qui riportati suggeriscono che le azioni acute/non genomiche del DHT sull’assorbimento di aminoacidi nelle fibre muscolari dei mammiferi adulti sono mediate attraverso lo stesso recettore e lo stesso percorso. Inoltre, il pretrattamento dei fasci di fibre con Ciproterone o Flutamide in modo significativo (P <0,05) ha attenuato gli effetti del DHT sull’assorbimento dell’Ile senza sopprimerlo completamente. In precedenza, è stato suggerito che il Testosterone può attivare la via MAPK tramite un recettore degli androgeni accoppiato con proteina G (Estrada et al. 2003). Tuttavia, se questo meccanismo, comunemente indicato come transattivazione, è quello che media gli effetti acuti/non genomici del DHT nelle fibre muscolari dei mammiferi adulti è incerto e sono necessarie ulteriori ricerche per chiarirlo.
Sistema regolatorio di segnalazione ERK1/2.
Dopo l’attivazione, ERK1 / 2 può rimanere nel citosol o traslare nel nucleo dove svolge un ruolo critico nella regolazione dell’espressione genica e della replicazione del DNA (Brunet et al. 1999). Nel nucleo, ERK1 / 2 fosforila una serie di target, inclusi molti fattori di trascrizione e una famiglia di chinasi correlate a RSK, le chinasi proteiche attivate dallo stress e dal mitogeno (MSK) (Deaketal.1998). Anche se nel presente studio è stato esaminato un gran numero di target a valle di ERK1 / 2, il trattamento con DHT ha aumentato soltanto la fosforilazione di RSK1 / 2 e ATF2 . Inoltre, il DHT non ha avuto alcun effetto sulla fosforilazione degli altri modulatori MAPK. Per i ricercatori, questi risultati suggeriscono che le azioni acute / non genomiche del DHT nei muscoli scheletrici dei mammiferi adulti sono esercitate principalmente attraverso l’EGFR e comportano l’attivazione della modulazione ERK1 / 2 del percorso MAPK. L’ipotesi è che il DHT, direttamente o indirettamente, attivi l’EGFR che a sua volta attiva la via MAPK che porta ad un aumento del trasporto di aminoacidi nelle fibre aumentando anche la sintesi proteica in esse. Inoltre, il DHT sembra avere un attività principale sulle fibre a contrazione rapida.
Negli animali adulti, un aumento della massa muscolo scheletrica (ipertrofia) si verifica principalmente a causa di un aumento delle dimensioni piuttosto che del numero di fibre muscolari ed è generalmente ritenuto che sia regolata dal percorso Akt / mTOR (Glass, 2003). Il percorso Akt / mTOR è attivato da molti stimoli tra cui fattori di crescita, altri ormoni e sostanze nutritive. Inoltre, la sua attivazione culmina nel blocco del apoptosi, induzione della sintesi proteica, trascrizione genica e proliferazione cellulare (Dann etal.2007). Pertanto, è stato stimato che il DHT e il T potessero attivare questo percorso. L’assorbimento di amminoacidi indotto dal DHT nelle fibre a contrazione rapida senza la sua completa soppressione, suggerisce che le azioni acute / non genomiche del DHT non sono mediate attraverso Akt. Invece, i risultati che vengono presentati nel presente studio suggeriscono che il DHT aumenta la sintesi proteica regolando la traduzione dell’mRNA già presente nelle cellule. In effetti, il trattamento dei fasci di fibre muscolari con DHT ha portato a una marcata riduzione della fosforilazione di eEF2 e ad un moderato aumento (∼50%) della sintesi proteica nei fasci di fibre a contrazione rapida.
È anche degno di nota il fatto che il pretrattamento dei fasci di fibre con l’inibitore specifico del mTOR non solo ha ridotto l’assorbimento basale di Ile, ma ha anche soppresso completamente l’aumento indotto dal DHT dell’assorbimento di Ile nei gruppi di fibre a contrazione rapida. Per i ricercatori questi risultati suggeriscono che alcuni degli effetti acuti / non genomici del DHT sono probabilmente mediati attraverso mTOR. In effetti, numerosi studi hanno suggerito che mTOR è il legame tra la disponibilità di aminoacidi e una maggiore sintesi proteica (Beugnet et al. 2003; Avruch et al. 2009). Tuttavia, la rapamicina può anche indurre l’autofagia (Ravikumar et al. 2004). Pertanto, un’altra possibilità è che i suoi effetti siano dovuti all’accumulo di aminoacidi nelle fibre muscolari.
Via di segnalazione cellulare che media gli effetti del DHT sul trasporto di aminoacidi nelle fibre muscolo-scheletriche dei mammiferi. Un diagramma schematico che mostra la via di segnalazione cellulare ipotizzata che media gli effetti acuti del DHT sull’amminoacido nei fasci muscolari dei mammiferi. L’ipotesi è che il DHT, attraverso un meccanismo sconosciuto, attiva l’EGFR e questo porta all’attivazione di RSK1 / 2 da parte di ERK1 / 2. L’RSK1 / 2 attivato aumenta quindi l’attività e l’espressione di LAT2 (cerchio blu con una croce), che a sua volta aumenta il trasporto di Ile nei fasci muscolari. Inoltre, RSK1 / 2 migliora la traduzione dell’mRNA in proteine. Si noti che il trasporto di Ile è accoppiato a quello di piccoli aminoacidi neutri come la glutammina (Gln) che vengono trasportati nella cellula dai trasportatori di aminoacidi del sistema A come SNAT2 (cerchio rosso con una croce). Pertanto, un aumento dell’attività di LAT2 aumenta indirettamente l’attività di SNAT2. Gli eventuali effetti di tutti questi processi sono di aumentare la sintesi proteica e quindi la massa muscolare, specialmente nelle fibre muscolari a contrazione rapida.
In conclusione, ci sono state prove che dimostrano che un’altra funzione fisiologica delle azioni acute / non genomiche del DHT nelle fibre muscolari dei mammiferi adulti è quella di aumentare l’assorbimento di aminoacidi essenziali. Sebbene non si possa escludere completamente il coinvolgimento del recettore degli androgeni, i principali risultati suggeriscono che l’aumento dell’assorbimento di aminoacidi e la sintesi proteica sono mediati attraverso l’EGFR e comportano l’attivazione del modulo ERK1 / 2 del pathway MAPK che a sua volta attiva RSK1 / 2 portando ad un aumento dell’espressione del trasportatore di aminoacidi di tipo L LAT2.
Piccola parentesi sul Mesterolone
Arrivati a questo punto, prima di trattare le conclusioni sul DHT, vorrei riportarvi due interessanti studi nei quali si è osservata l’azione di una forma di DHT metilata in C-1, il noto Mesterolone, sulla massa muscolare.
Senza dilungarsi troppo in preamboli, come ben sappiamo, il Mesterolone (nome commerciale Proviron) [1 alpha-methyl-17 beta- hydroxy-5 alpha-androstan-3-one] è un AAS derivato dal Dihydrotestosterone (DHT). A differenza del DHT il Mesterolone presenta l’aggiunta di un gruppo metilico in C1 (similmente al Metenolone) cosa che ne aumenta discretamente la biodisponibilità orale, senza però apportare eccessivo stress epatico. Questa alterazione però non aumenta la stabilità del C3 chetogruppo, essenziale per l’attività anabolizzante a livello muscolare mediata dal legame recettoriale, come il suo precursore.
Ora, sappiamo anche che ci sono frequenti segnalazioni di abuso di Mesterolone negli sport umani ed equini per il miglioramento della prestazione. Tuttavia, sono disponibili informazioni limitate su come questo farmaco esercita i suoi effetti sul muscolo scheletrico.
Le cellule satellite (SC) sono cellule staminali miogeniche mononucleari che contribuiscono alla crescita e alla riparazione dei muscoli postnatali. Poiché l’attivazione delle SC e la successiva differenziazione nei nuovi myonuclei sono un evento importante durante l’ipertrofia muscolare. In uno studio pubblicato nel maggio del 2012 [87], si è osservata l’influenza del Mesterolone sulla distribuzione delle SC all’interno del muscolo pettorale dei polli (Gallus gallus ). Nello specifico, questo studio ha testato le ipotesi secondo cui che il Mesterolone induca l’ipertrofia del muscolo scheletrico aviario e che aumenti anche il numero di SC nel muscolo scheletrico aviario. Sono state utilizzate solide tecniche immunocitochimiche e analisi morfometriche per calcolare il numero di SC e myonuclei. Inoltre, sono stati misurati i livelli di concentrazione di DNA e proteine Pax7 per confermare i risultati immunocitochimici. Il Mesterolone ha aumentato significativamente la massa pettorale e le dimensioni delle fibre. Tutti gli indici delle SC e il numero di myonuclei sono aumentati significativamente con la somministrazione di Mesterolone. Inoltre, negli uccelli trattati con Mesterolone sono state riscontrate una maggiore concentrazione di DNA ed espressione della proteina Pax7. Questo studio indica che il Mesterolone può indurre ipertrofia del muscolo scheletrico aviario e che questo è correlato con un aumento del numero di SC. I ricercatori suggeriscono che le SC siano intermediari cellulari chiave per l’ipertrofia muscolare indotta da Mesterolone. L’ipotesi, che necessità di ulteriori ricerche, è che tale effetto possa riscontrarsi anche in altre specie compreso l’uomo.
Identificazione immunofluorescente di SC nelle sezioni trasversali dei muscoli pettorali ottenuti da polli di controllo e trattati con Mesterolone. (A) e (C) rappresentano una sezione ottenuta dal muscolo pettorale di un esemplare di controllo. (B) e (D) rappresentano una sezione ottenuta dal muscolo pettorale di un esemplare trattato con Mesterolone. (A) e (B) mostrano tutti i nuclei in blu (colorazione DAPI) e le lamine basali delle fibre muscolari in rosso (colorazione anti-laminina). (C) e (D) mostrano SC in verde (etichettatura anti-Pax7) e le lamine basali in rosso. Barre di scala = 50 μm.
In un altro studio pubblicato nel 2010 [88], la microscopia ottica ed elettronica e la morfometria quantitativa sono state utilizzate per determinare gli effetti dell’esercizio e del Mesterolone sul muscolo soleo dei topi. Sia l’esercizio fisico che il Mesterolone hanno causato una significativa ipertrofia delle fibre muscolari extrafusali. L’ipertrofia delle fibre di tipo I era maggiore di quella delle fibre di tipo II. Non c’era iperplasia. I mitocondri erano più numerosi e più grandi rispetto a quanto osservato nei muscoli degli animali sedentari. La capillarità è aumentata e sono comparse piccole fibre muscolari nucleate centralmente, di solito in piccoli gruppi e molto spesso nei muscoli degli animali esposti al Mesterolone. Una piccola percentuale di cellule satelliti mostrava segni di attivazione ma nei muscoli degli animali trattati con Mesterolone c’era una massa muscolare maggiore dopo l’esercizio. I muscoli di animali che erano stati entrambi fatti esercitati e trattati con Mesterolone presentavano i maggiori cambiamenti: la massa muscolare e l’ipertrofia delle fibre muscolari erano maggiori rispetto a tutti gli altri gruppi di animali, la capillarità era maggiore e> il 30% di tutte le cellule satellitari riconosciute mostrava segni di attivazione. Gruppi di piccole fibre muscolari nucleate centralmente sono state comunemente osservate in questi muscoli. Sembravano essere il risultato di lacerazioni rigenerate in fibre muscolari esistenti. Sia con l’esercizio fisico sia con il Mesterolone, da solo o in combinazione, si è verificato un aumento della percentuale di fibre muscolari di tipo I e una diminuzione della proporzione di tipo II.
Entrambi gli studi esposti, sebbene non siano di grande impatto, fanno sicuramente nascere il dubbio in coloro i quali hanno sempre valutato le possibili azioni di una molecola in modo ristretto, sia per mancanza di una conoscenza sufficienza in materia sia per limitatezza nel formulare ipotesi estrapolate dall’osservazione.
Ora, non sto sicuramente dicendo che un culturista trattato con solo Mesterolone possa ambire a grandi cambiamenti nella composizione corporea. Sto semplicemente ipotizzando, che le nozioni in merito alle attività degli AAS sono datate e parziali, anche perchè si basano per lo più, tanto quanto gli studi esposti, sulla capacità di legame degli AAS testati osservata nella prostata e nel levator ani dei topi. Inoltre, il Mesterolone ha visto una riscoperta come farmaco ancillare nella TRT o come anti-depressivo funzionale in alcune tipologie di depressione.
In fondo, il peggior difetto del Mesterolone è dipeso dalla sua consuetudinaria via di somministrazione…
Nota: la biodisponibilità orale del Mesterolone è stata stimata al 3%.
Tiriamo le somme…con possibili critiche…
Già sento i soliti limitati in intelletto e pazienza, sempre pronti a parlare senza cognizione di causa, urlare al “esiste lo studio X che afferma che l’aggiunta di Dutasteride con dosi sovrafisiologiche di Testosterone non cambia il risultato!” … Lo conosco anche io capre!… A proposito, vediamo di cosa si tratta…
Testosterone Enantato
Il principale studio è quello pubblicato su JAMA nel 2012.[89] Si tratta di uno studio in doppio cieco, randomizzato, controllato con placebo. Lo studio è stato diretto dal dott. Bhasin e colleghi, i quali hanno reclutato 139 uomini sani, di età compresa tra 18 e 50 anni, con livelli normali di Testosterone.
Ai pazienti è stata assegnata 1 su 4 dosi di Testosterone Enantato (50, 125, 300 o 600 mg / settimana) più 2,5mg/die di Dutasteride o sono stati assegnati a 1 su 4 gruppi a cui è stato somministrato 1 di 4 dosi di Testosterone Enantato (50, 125, 300 o 600 mg / settimana) più placebo per 20 settimane.
Dutasteride
Il cambiamento nella massa magra rispetto al basale, misurata dall’assorbtiometria a raggi X a doppia energia, è stato il risultato primario. I ricercatori hanno anche misurato i livelli di Testosterone, la forza muscolare, la funzione sessuale, la produzione di sebo e l’acne.
La forza nel Leg Press e del petto sono state misurate con il metodo 1 ripetizione massimale con l’uso di macchine Keizer.
La funzione sessuale è stata valutata con l’indice internazionale della funzione erettile e il questionario sulla salute sessuale maschile per una valutazione più completa del desiderio sessuale.
Il volume della prostata è stato misurato con la risonanza magnetica 1.5-Tesla.
La produzione di sebo è stata misurata con l’uso di nastri di sebo. La scala Palatzi è stata utilizzata per valutare l’acne.
139 uomini furono assegnati in modo casuale; 102 (54 nei gruppi placebo e 48 nei gruppi dutasteride) hanno completato l’intervento di 20 settimane.
Gli uomini assegnati alla Dutasteride erano simili al basale degli uomini assegnati al placebo. Inoltre, i livelli di Testosterone totale e libero non differivano tra i due gruppi.
L’aumento della massa magra ottenuta dai gruppi Dutasteride+Testosterone era di 0,6 kg (IC 95%, da -0,1 a 1,2 kg) quando ricevevano 50 mg/settimana di Testosterone Enantato, 2,6 kg (IC 95%, 0,9 – 4,3 kg) per 125mg/settimana, 5,8 kg (IC 95%, 4,8 – 6,9 kg) per 300mg/settimana e 7,1kg (IC 95%, 6,0 – 8,2 kg) per 600 mg/settimana.
La massa magra ottenuta dai gruppi placebo+Testosterone era di 0,8 kg (IC 95%, da -0,1 a 1,7 kg) quando ricevevano 50mg/settimana di Testosterone Enantato, 3,5 kg (IC 95%, 2,1-4,8 kg) per 125 mg/settimana, 5,7 kg (IC 95%, 4,8 – 6,5 kg) per 300mg/settimana e 8,1 kg (IC 95%, 6,7 – 9,5 kg) per 600mg/settimana.
Le differenze dose-dipendente tra i gruppi Dutasteride e placebo per la massa magra non erano significative (P = .18).
I cambiamenti nella massa grassa, nella forza muscolare, nella funzione sessuale, nel volume della prostata, nella produzione di sebo e nei livelli di ematocrito e lipidi non differivano tra i gruppi.
La frequenza complessiva di eventi avversi, inclusa la frequenza di disfunzione sessuale, acne, vampate di calore e dolorabilità mammaria era simile nei gruppi placebo e dutasteride.
Misure in riferimento alla composizione corporea e alla forza muscolare raccolte nello studio trattato. Nei grafici a sinistra, i marker indicati hanno un margine di errore con intervalli di confidenza al 95%. Nei grafici a destra (variazione dei livelli di Testosterone), le scale dell’asse orizzontale sono state spostate (offset) di 1000ng/dL e quindi è stata applicata una trasformazione della radice quadrata. Il livellamento semiparametrico è stato ottenuto utilizzando modelli di additivi generalizzati. Le aree ombreggiate indicano intervalli di confidenza al 95%.
Bene…possiamo cestinare tutto quanto ipotizzato in precedenza? Visto anche il tipo di studio esposto? Non esattamente …
Ora, da notare la tipologia di soggetti arruolati e il loro grado di “maturità sportiva”, più specificatamente negli allenamenti contro resistenza. Essa non è chiaramente specificata e la misurazione dell’espressione della forza in soggetti poco o per nulla allenati non è un grande parametro sebbene lo scopo dello studio fosse quello di constatare l’impatto nella forza e ipertrofia dato da un protocollo a dosaggio differenziato di Testosterone Enantato con o senza un inibitore della 5α-reduttasi (Dutasteride).
Secondo punto da notare, la mancanza di misurazione specifica della massa muscolare. Infatti, la composizione corporea dei partecipanti è stata valutata per i parametri di massa grassa e massa magra. Come risaputo, quando si parla di massa magra si prende anche in considerazione l’acqua. Sebbene la differenza sia bassa, probabilmente essa è da ricollegare anche al fatto che le dosi di Testosterone utilizzate erano differenti e con limite a 600mg di Testosterone Enantato a settimana (pari a 432mg di Testosterone), e che tale dosaggio non sempre crea iperestrogenemia marcata (considerando anche un lasso di tempo contenuto) tale da causare accumuli sensibili di acqua extracellulare. Considerando poi che uno degli effetti collaterali collegati all’uso di inibitore della 5α-reduttasi sia proprio un aumento degli estrogeni circolanti (per aumento del substrato di sintesi) con ripercussioni a carico di (e non solo limitate a) ritenzione idrica, accumulo di grasso con modello ginoide e ginecomastia, i dubbi sulle metodiche di analisi dei partecipanti allo studio diventano senz’altro dubbie.
Da notare che il Dutasteride è stato osservato ridurre il DHT sierico del >90% e quello intraprostatico del 94% alla dose di 0.5mg/die.[90] Nello studio presentato, i partecipanti assegnati ai gruppi Dutasteride e Dutasteride+Testosterone erano trattati con una dose di Dutasteride pari a 2,5mg/die.
I ricercatori volevano condurre uno studio meticolosamente progettato ed eseguito per rispondere a una domanda vecchia di quasi 5 decenni: il Testosterone, il DHT o entrambi a livelli fisiologici e soprafisiologici sono importanti per il guadagno della massa magra? Purtroppo sembra che non siano stati sufficientemente meticolosi…
Non è di certo mia intenzione far valere di più uno studio svolto in vitro o su animali rispetto ad uno svolto su esseri umani. E’ invece mia intenzione far riflettere il lettore sul design di uno studio, ed in base al livello di questo poter valutare quanto in esso riportato.
Un altra critica riduttiva che potrebbe essere mossa contro le nuove ipotesi di attività del DHT potrebbe essere riferita all’enzima 3α-Hydroxysteroide dehydrogenasi (3α-HSD). Si dimentica però che tale enzima è un “limitatore” dell’attività del DHT a livello di tessuti come quello muscolare e adiposo. Infatti, la sua azione non esclude assolutamente la potenziale attività in acuto e non genomica del DHT nei tessuti dove l’enzima 3α-HSD viene espresso. E’ possibile che la cooperatività del DHT con il Testosterone sia di tipo sequenziale: attività prettamente recettoriale e in cronico (sebbene sotto regolazione organica) data dal Testosterone preceduta e seguita da una attività acuta e non genomica data dal DHT.
Inoltre, ed è una cosa molto importante da tenere in considerazione, la ricerca è tutt’ora in essere. Esistono ipotesi diverse con il loro bagaglio di ricerca. Ciò che il lettore preparato può fare è semplicemente soppesare le informazioni raccolte al fine di farsi un idea il più concreta possibile della questione.
Sebbene la ricerca scientifica abbia fatto enormi passi avanti nell’ultimo secolo, siamo ancora lungi dal conoscere tutto… anche quando si parla di ormoni e la loro attività diretta e indiretta…
Nota conclusiva: è ovvio che non si mette in dubbio l’utilità del controllo terapeutico del DHT in condizioni patologiche (vedi iperplasia prostatica maligna) o preventive in particolari soggetti. Negli individui in fisiologia e senza particolari predisposizioni, l’uso di inibitori della 5α-reduttasi potrebbe risultare una scelta controproducente sotto molteplici aspetti.
Gabriel Bellizzi
Riferimenti:
Schnitzer R (1 January 1967). Experimental Chemotherapy. Elsevier Science. pp. 156–. ISBN 978-0-323-14611-1.
Krüskemper H (22 October 2013). Anabolic Steroids. Elsevier. pp. 12–. ISBN 978-1-4832-6504-9.
Taylor WN (16 January 2002). Anabolic Steroids and the Athlete, 2d ed. McFarland. pp. 178–. ISBN 978-0-7864-1128-3.
William Andrew Publishing (2007). Pharmaceutical Manufacturing Encyclopedia. William Andrew Pub. ISBN 978-0-8155-1526-5.
Newsweek. Newsweek. 1953.
New and Nonofficial Drugs. Lippincott. 1958.
Rubin BL, Dorfman RI (1956). “In vitro conversion of testosterone to 17beta-hydroxyandrostan-3-one”. Proc. Soc. Exp. Biol. Med. 91 (4): 585–6. doi:10.3181/00379727-91-22337. PMID 13323010.
Agmo A (18 April 2011). Functional and Dysfunctional Sexual Behavior: A Synthesis of Neuroscience and Comparative Psychology. Academic Press. pp. 196–. ISBN 978-0-08-054938-5.
Oreopoulos DG, Michelis M, Herschorn S (6 December 2012). Nephrology and Urology in the Aged Patient. Springer Science & Business Media. pp. 495–. ISBN 978-94-011-1822-4.
Webster GF, Rawlings AV (17 May 2007). Acne and Its Therapy. CRC Press. pp. 168–. ISBN 978-1-4200-1841-7.
Smith LB, Mitchell RT, McEwan IJ (1 October 2013). Testosterone: From Basic Research to Clinical Applications. Springer Science & Business Media. pp. 5–. ISBN 978-1-4614-8978-8.
Azzouni F, Mohler J (2012). “Role of 5α-reductase inhibitors in benign prostatic diseases”. Prostate Cancer Prostatic Dis. 15 (3): 222–30.
Melmed S (2016). Williams Textbook of Endocrinology. Elsevier Health Sciences. pp. 621, 711.
Blume-Peytavi U, Whiting DA, Trüeb RM (26 June 2008). Hair Growth and Disorders. Springer Science & Business Media. pp. 161–162.
Rhoades RA, Bell DR (18 January 2012). Medical Phisiology: Principles for Clinical Medicine. Lippincott Williams & Wilkins. pp. 690–.
Rakel D (12 April 2012). Integrative Medicine E-Book. Elsevier Health Sciences. pp. 321–.
Morrison MF (4 May 2000). Hormones, Gender and the Aging Brain: The Endocrine Basis of Geriatric Psychiatry. Cambridge University Press. pp. 17–.
Azzouni F, Godoy A, Li Y, Mohler J (2012). “The 5 alpha-reductase isozyme family: a review of basic biology and their role in human diseases”. Advances in Urology. 2012: 1–18.
Zouboulis CC, Chen WC, Thornton MJ, Qin K, Rosenfield R (2007). “Sexual hormones in human skin”. Horm. Metab. Res. 39 (2): 85–95.
Bolognia JL, Jorizzo JL, Schaffer JV (8 June 2012). Dermatology E-Book. Elsevier Health Sciences. pp. 1094–. ISBN 978-0-7020-5182-1.
Murphy MJ (24 March 2011). Molecular Diagnostics in Dermatology and Dermatopathology. Springer Science & Business Media. pp. 373–.
eam SJ, Scott LJ (2008). “Dutasteride: a review of its use in the management of prostate disorders”. Drugs. 68 (4): 463–85.
Heesakkers J, Chapple C, Ridder DD, Farag F (24 February 2016). Practical Functional Urology. Springer. pp. 280–.
Nieschlag E, Behre HM, Nieschlag S (26 July 2012). Testosterone: Action, Deficiency, Substitution. Cambridge University Press. pp. 61–.
Dunn JF, Nisula BC, Rodbard D (July 1981). “Transport of steroid hormones: binding of 21 endogenous steroids to both testosterone-binding globulin and corticosteroid-binding globulin in human plasma”. J. Clin. Endocrinol. Metab. 53 (1): 58–68.
Williams DA, Foye WO, Lemke TL (2002). Foye’s Principles of Medicinal Chemistry. Lippincott Williams & Wilkins. pp. 707–.
Coutts, S. B.; Kicman, A. T.; Hurst, D. T.; Cowan, D. A. (1997-11-01). “Intramuscular administration of 5α-dihydrotestosterone heptanoate: changes in urinary hormone profile”. Clinical Chemistry. 43 (11): 2091–2098.
Marks LS (2004). “5α-reductase: history and clinical importance”. Rev Urol. 6 Suppl 9: S11–21.
Horton R (1992). “Dihydrotestosterone is a peripheral paracrine hormone”. J. Androl. 13 (1): 23–7. doi:10.1002/j.1939-4640.1992.tb01621.x (inactive 2020-03-09).
Wilson JD (1996). “Role of dihydrotestosterone in androgen action”. Prostate Suppl. 6: 88–92.
Swerdloff RS, Dudley RE, Page ST, Wang C, Salameh WA (2017). “Dihydrotestosterone: Biochemistry, Physiology, and Clinical Implications of Elevated Blood Levels”. Endocr. Rev. 38 (3): 220–254.
Bhasin S (13 February 1996). Pharmacology, Biology, and Clinical Applications of Androgens: Current Status and Future Prospects. John Wiley & Sons. pp. 72–.
Hay ID, Wass JA (26 January 2009). Clinical Endocrine Oncology. John Wiley & Sons. pp. 37–.
Melmed S (2016). Williams Textbook of Endocrinology. Elsevier Health Sciences. pp. 621, 711.
Jin Y, Penning TM (2001). “Steroid 5alpha-reductases and 3alpha-hydroxysteroid dehydrogenases: key enzymes in androgen metabolism”. Best Pract. Res. Clin. Endocrinol. Metab. 15 (1): 79–94.
Llewellyn W (2009). Anabolics. Molecular Nutrition Llc. pp. 19, 163.
Blume-Peytavi U, Whiting DA, Trüeb RM (26 June 2008). Hair Growth and Disorders. Springer Science & Business Media. pp. 161–162.
Azzouni F, Mohler J (2012). “Role of 5α-reductase inhibitors in benign prostatic diseases”. Prostate Cancer Prostatic Dis. 15 (3): 222–30.
Azzouni F, Mohler J (2012). “Role of 5α-reductase inhibitors in prostate cancer prevention and treatment”. Urology. 79 (6): 1197–205.
Lotti F, Maggi M (28 April 2015). “Hormonal Treatment for Skin Androgen-Related Disorders”. In Katsambas A, Lotti T, Dessinioti C, D’Erme AM (eds.). European Handbook of Dermatological Treatments. Springer. pp. 1451–1464.
Wendowski, Oskar; Redshaw, Zoe; Mutungi, Gabriel (February 2017). “Dihydrotestosterone treatment rescues the decline in protein synthesis as a result of sarcopenia in isolated mouse skeletal muscle fibres”. Journal of Cachexia, Sarcopenia and Muscle. 8 (1): 48–56.
Kohtz AS, Frye CA (2012). Dissociating behavioral, autonomic, and neuroendocrine effects of androgen steroids in animal models. Methods Mol. Biol. Methods in Molecular Biology. 829. pp. 397–431.
Mozayani A, Raymon L (18 September 2011). Handbook of Drug Interactions: A Clinical and Forensic Guide. Springer Science & Business Media. pp. 656–.
Hemat RA (2004). Principles Of Orthomolecularism. Urotext. p. 426.
Grino PB, Griffin JE, Wilson JD (February 1990). “Testosterone at high concentrations interacts with the human androgen receptor similarly to dihydrotestosterone”. Endocrinology. 126 (2): 1165–72.
Wilderer PA (1 September 2010). “Bioassays for Estrogenic and Androgenic Effects of Water Constituents”. Treatise on Water Science, Four-Volume Set. Newnes. pp. 1805–.
Diamanti-Kandarakis E (1999). “Current aspects of antiandrogen therapy in women”. Current Pharmaceutical Design. 5 (9): 707–23. PMID 10495361.
von Deutsch DA, Abukhalaf IK, Lapu-Bula R (15 October 2003). “Anabolic Doping Agents”. In Mozayani A, Raymon L (eds.). Handbook of Drug Interactions: A Clinical and Forensic Guide. Springer Science & Business Media. pp. 510–.
Swerdloff RS, Wang C (October 1998). “Dihydrotestosterone: a rationale for its use as a non-aromatizable androgen replacement therapeutic agent”. Baillière’s Clinical Endocrinology and Metabolism. 12 (3): 501–6.
Kohtz AS, Frye CA (2012). Dissociating behavioral, autonomic, and neuroendocrine effects of androgen steroids in animal models. Methods Mol. Biol. Methods in Molecular Biology. 829. pp. 397–431.
Rizner TL, Lin HK, Peehl DM, Steckelbroeck S, Bauman DR, Penning TM (July 2003). “Human type 3 3alpha-hydroxysteroid dehydrogenase (aldo-keto reductase 1C2) and androgen metabolism in prostate cells”. Endocrinology. 144 (7): 2922–32.
Weiner IB, Gallagher M (2003). Handbook of Psychology, Biological Psychology. John Wiley & Sons. pp. 333–.
Schill W, Comhaire FH, Hargreave TB (26 August 2006). Andrology for the Clinician. Springer Science & Business Media. pp. 243–.
Hyde TE, Gengenbach MS (2007). Conservative Management of Sports Injuries. Jones & Bartlett Learning. pp. 1100–.
“Androstanolone Drug Profile”. Adis Insight. 4 December 2006.
Elks J (14 November 2014). The Dictionary of Drugs: Chemical Data: Chemical Data, Structures and Bibliographies. Springer. pp. 640–.
Index Nominum 2000: International Drug Directory. Taylor & Francis. January 2000. pp. 63–.
List PH, Hörhammer L (12 March 2013). Chemikalien und Drogen: Teil B: R, S. Springer-Verlag. pp. 523–.
“Drugs@FDA: FDA Approved Drug Products”. United States Food and Drug Administration. Retrieved 16 November 2016.
“Drug Product Database – Health Canada”. Health Canada. Retrieved 13 November 2016.
Blume-Peytavi U, Whiting DA, Trüeb RM (26 June 2008). Hair Growth and Disorders. Springer Science & Business Media. pp. 161–162.
Azzouni F, Mohler J (2012). “Role of 5α-reductase inhibitors in prostate cancer prevention and treatment”. Urology. 79 (6): 1197–205.
Lotti F, Maggi M (28 April 2015). “Hormonal Treatment for Skin Androgen-Related Disorders”. In Katsambas A, Lotti T, Dessinioti C, D’Erme AM (eds.). European Handbook of Dermatological Treatments. Springer.
Wendowski, Oskar; Redshaw, Zoe; Mutungi, Gabriel (February 2017). “Dihydrotestosterone treatment rescues the decline in protein synthesis as a result of sarcopenia in isolated mouse skeletal muscle fibres”. Journal of Cachexia, Sarcopenia and Muscle. 8 (1): 48–56.
Kohtz AS, Frye CA (2012). Dissociating behavioral, autonomic, and neuroendocrine effects of androgen steroids in animal models. Methods Mol. Biol. Methods in Molecular Biology. 829. pp. 397–431.
Llewellyn W (2009). Anabolics. Molecular Nutrition Llc. pp. 19, 163.
Okeigwe I, Kuohung W (2014). “5-Alpha reductase deficiency: a 40-year retrospective review”. Curr Opin Endocrinol Diabetes Obes. 21 (6): 483–7.
Heesakkers J, Chapple C, Ridder DD, Farag F (24 February 2016). Practical Functional Urology. Springer. pp. 280–.
Imperato-McGinley J, Zhu YS (2002). “Androgens and male physiology the syndrome of 5alpha-reductase-2 deficiency”. Mol. Cell. Endocrinol. 198 (1–2): 51–9.
Coutts, S. B.; Kicman, A. T.; Hurst, D. T.; Cowan, D. A. (1997-11-01). “Intramuscular administration of 5α-dihydrotestosterone heptanoate: changes in urinary hormone profile”. Clinical Chemistry. 43 (11): 2091–2098.
Imperato-McGinley J, Peterson RE, Gautier T, Sturla E (1979). “Androgens and the evolution of male-gender identity among male pseudohermaphrodites with 5alpha-reductase deficiency”. N. Engl. J. Med. 300 (22): 1233–7.
Kang HJ, Imperato-McGinley J, Zhu YS, Rosenwaks Z (2014). “The effect of 5α-reductase-2 deficiency on human fertility”. Fertil. Steril. 101 (2): 310–6.
Katz MD, Cai LQ, Zhu YS, Herrera C, DeFillo-Ricart M, Shackleton CH, Imperato-McGinley J (1995). “The biochemical and phenotypic characterization of females homozygous for 5 alpha-reductase-2 deficiency”. J. Clin. Endocrinol. Metab. 80 (11): 3160–7.
Cilotti A, Danza G, Serio M (2001). “Clinical application of 5alpha-reductase inhibitors”. J. Endocrinol. Invest. 24 (3): 199–203.
Bradbury R (30 January 2007). Cancer. Springer Science & Business Media. pp. 49–.Burchum J, Rosenthal L (2 December 2014). Lehne’s Pharmacology for Nursing Care. Elsevier Health Sciences. pp. 803–.
Bostwick DG, Cheng L (24 January 2014). Urologic Surgical Pathology. Elsevier Health Sciences. pp. 492–.
Sun J, Xiang H, Yang LL, Chen JB (2011). “A review on steroidal 5α-reductase inhibitors for treatment of benign prostatic hyperplasia”. Curr. Med. Chem. 18 (23): 3576–89.
Torres F (2015). Androgenetic, diffuse and senescent alopecia in men: practical evaluation and management. Curr. Probl. Dermatol. Current Problems in Dermatology. 47. pp. 33–44.
Check JH, Cohen R (2015). “An update on the treatment of female alopecia and the introduction of a potential novel therapy”. Clin Exp Obstet Gynecol. 42 (4): 411–5.
Blume-Peytavi U, Whiting DA, Trüeb RM (26 June 2008). Hair Growth and Disorders. Springer Science & Business Media. pp. 182, 369.
Check JH, Cohen R (2015). “An update on the treatment of female alopecia and the introduction of a potential novel therapy”. Clin Exp Obstet Gynecol. 42 (4): 411–5.
Blume-Peytavi U, Whiting DA, Trüeb RM (26 June 2008). Hair Growth and Disorders. Springer Science & Business Media. pp. 182, 369.
Negli ultimi anni il Cannabidiolo (noto anche con la sigla CBD) ha subito una crescente popolarità data sia dai promettenti risultati ipoteticamente applicabili e replicabili nel trattamento di varie condizioni patologiche (vedi, ad esimpio, SLA) provenienti dagli studi in vitro e su animali da laboratorio e, soprattutto, dalla sua relativamente facile reperibilità, essendo legale in molti paesi, che rendeva (e rende) il composto testabile dal singolo senza rischi legali. Il suo uso ha iniziato a diffondersi anche tra gli sportivi, ed i culturisti in particolar modo ne sono stati attratti per le sue potenzialità nella riduzione della percezione dello stress, come agente anoressizzante e come trattamento del dolore.
Ma quanto c’è di concretamente e fruttuosamente applicabile del Cannabidiolo in ambito medico e anche sportivo? Nel seguente articolo verranno riportate le informazioni necessarie per una risposta scientificamente onestà a questa domanda.
La storia del CBD
Cannabidiolo (CBD)
Il Cannabidiolo (CBD) è un fitocannabinoide scoperto nel 1960. È uno dei 113 cannabinoidi identificati nelle piante di cannabis e rappresenta fino al 40% dell’estratto della pianta.[1] I cannabinoidi nella cannabis sono stati studiati dagli anni ’40,
[2], con le strutture di THC (delta-9-tetraidrocannabinolo) e CBD scoperte negli anni ’60. La ricerca di questi composti ha subito un picco negli anni ’90, quando sono stati trovati i recettori dei cannabinoidi nel corpo [3], e in seguito è stato scoperto che sono stati attivati anche da composti endogeni (prodotti nel corpo) nel 1992.[4]
La popolarità del CBD è esplosa negli anni 2000, soprattutto dopo che i genitori di bambini epilettici hanno iniziato a sperimentare l’effetto del CBD sui loro figli, come opzione di trattamento per convulsioni resistenti ai farmaci nella rara e grave condizione di epilessia chiamata Sindrome di Dravet.[5]
Delta-9-tetraidrocannabinolo (THC)
I prodotti a base di CBD che mantengono una percentuale di THC “low-to-zero” (inferiore allo 0,3%) si trovano attualmente in un’area grigia della legalità negli Stati Uniti, ma con una legalità rafforzata dopo l’approvazione del Farm Act nel dicembre 2018, sebbene la FDA richiede ancora tecnicamente che gli integratori commercializzati debbano essere approvati prima dalla stessa. [6] La FDA ha inviato lettere di avvertimento nell’aprile del 2019 a tre società che ritenevano stessero formulando dichiarazioni sanitarie significative, tra cui: [7]
“Il CBD ha bloccato con successo le cellule tumorali in diverse varietà di cancro cervicale.”
“Il CBD ha anche ridotto la crescita e la diffusione delle cellule di glioma umano, suggerendo così un possibile ruolo del CBD come agente antitumorale.”
“Per i pazienti con Alzheimer, il CBD è un’opzione di trattamento che sta rallentando la progressione di quella malattia.”
“La fibromialgia è concepita come uno stato di sensibilizzazione centrale con iperalgesia secondaria. Il CBD ha dimostrato la capacità di bloccare i meccanismi spinali, periferici e gastrointestinali responsabili del dolore associato a emicrania, fibromialgia, IBS e altri disturbi correlati. “
“Il cannabidiolo può essere efficace nel trattamento dei disturbi da uso di sostanze.”
“Il CBD ha ridotto gli effetti gratificanti della morfina e ha ridotto la ricerca dell’eroina.”
“Il CBD può essere utilizzato per evitare o ridurre i sintomi di astinenza.”
Presumibilmente, la reazione della FDA è stata dettata dalle scarse prove che si hanno in questo momento per il trattamento della maggior parte delle condizioni al di fuori della grave epilessia, e poiché queste affermazioni non includono tale dichiarazione di non responsabilità, i pazienti potrebbero presumere che il livello di evidenza sia più forte di quanto non sia in realtà. Pertanto, la FDA afferma che “non tollereremo questo tipo di marketing ingannevole per i pazienti vulnerabili”. L’interazione tra la regolamentazione FDA di alimenti, droghe e integratori (che possono essere tutti applicabili alla cannabis), insieme a diverse normative sulla cannabis in diversi stati, rende complesse le questioni legali relative al CBD e ad altri prodotti a base di cannabinoidi. [8]
Nota: in Italia la vendita della cannabis light è legale dal 2016, e ciò in virtù di una legge (numero 242) pienamente in vigore. La vendita è consentita per cannabis che abbia THC (i principi attivi che generano effetti psicotropi) fino e non oltre la percentuale dello 0,2. Ma il venditore di cannabis legale non è responsabile penalmente fino alla soglia dello 0,6 per cento, oltre la quale la marijuana viene sequestrata da parte dell’autorità giudiziaria e si apre un fascicolo che poi arriverà in tribunale. Queste due limitazioni aprono certo le porte a diverse interpretazioni e smagliature della legge. Molte profonde incertezze attualmente coinvolgono il CBD legale in Italia: questo a seguito della lacunosa sentenza emessa il 30 Maggio 2019 da parte della Corte di Cassazione e che ha visto “rivoluzionare”, secondo l’accezione meno positiva del termine, quanto invece stabilito dalla Legge 242. La sentenza emessa 30 Maggio 2019 circa la cannabis light e il CBD fino ad allora legale, ha stabilito che in Italia non sarebbe più concessa la vendita o la cessione a qualunque titolo di tutti i prodotti “derivati dalla coltivazione della cannabis”, quali l’olio al CBD, così come foglie, le infiorescenze e la resina, questo secondo la massima provvisoria emessa che recita testualmente -“Integrano il reato previsto dal Testo unico sulle droghe (articolo 73, commi 1 e 4, dpr 309/1990) le condotte di cessione, di vendita, e, in genere, la commercializzazione al pubblico, a qualsiasi titolo, dei prodotti derivati dalla coltivazione della cannabis sativa light, salvo che tali prodotti siano in concreto privi di efficacia drogante”. Ma, e c’è un ma, la legge italiana sull’utilizzo e sulla vendita del CBD esplica tutto ciò che concerne la filiera della canapa in Italia, dalla coltivazione al consumo. Nella stessa si legge che vi è una promozione alla coltivazione, trasformazione, impiego della stessa per lo sviluppo del territorio, produzione di alimenti, cosmetici, opere di bioingegneria, bonifica dei terreni, ricerca.
Le varietà di canapa che possono essere seminate devono essere presenti nel catalogo europeo delle sementi, in queste sono riportate solo quelle tipologie che hanno dimostrato di possedere un THC inferiore allo 0.2%.
Per questo è possibile affermare che i prodotti a base di CBD possono essere venduti in quanto rientrano nella tipologia ad uso tecnico, ciò vuol dire che nella legge non si fa esplicito riferimento alla possibilità di fumare la cannabis light, tuttavia questo non è neanche vietato e per questo l’utilizzo finale è a discrezione di chi la compra. La canapa legale è la principale fonte di approvvigionamento di CBD legale in Italia.
I negozi, online o fisici, possono commercializzare tisane, bevande, torte, vestiti e anche bustine di marijuana light. Ad essere illegale infatti è un valore elevato di THC ma non il CBD che ha esclusivamente potenziali proprietà rilassanti e antiossidanti e quindi, come dimostrano i test svolti in laboratorio, non ha alcun effetto ‘ludico’ comparabile all’utilizzo di droghe.
Per la serie “leggi ambigue e dove trovarle”…
Fonti del CBD
Il CBD viene in genere estratto dalla pianta di canapa. Ecco una rapida panoramica dei termini: la cannabis è un genere di pianta, che si presenta in due tipi botanici principali: la cannabis indica e la cannabis sativa. La marijuana (che contiene sia CBD che THC) può essere di entrambi i tipi, mentre la canapa è solo di tipo sativa.
Si noti che entrambe appartengono allo stesso genere e specie e sono piuttosto distinti i loro livello di THC. La canapa contiene un massimo dello 0,3% di THC, mentre la marijuana varia in genere tra il 5-20% di THC. [9] La marijuana viene spesso coltivata per uso ricreativo o misto ricreativo/medicinale, mentre la canapa viene coltivata per una più ampia varietà di usi (abbigliamento di canapa, olio di canapa, fibra di canapa, CBD isolato e dozzine di altri derivati).
CBD e THC non sono gli unici cannabinoidi presenti sul mercato. Ad esempio, il CBG (cannabigerolo) si trova in quantità minori nella cannabis, ma è stato studiato per le malattie infiammatorie in un modello animale.[10]
Attività biologica (farmacodinamica)
Poiché il sistema endocannabinoide ha un impatto su così tante sfaccettature della vita di base di un individuo (ad es. Appetito, funzione immunitaria, riproduzione e gestione del dolore), esistono una serie di possibilità di azione per il CBD (e altri cannabinoidi) che incidono sulla salute.[11] Il CBD può impedire l’attivazione eccessiva dei recettori CB1 e CB2, riducendo gli effetti dello stress mediato da tali recettori.[12] Altri ruoli includono l’antiossidazione: sia il CBD che il THC hanno agito come antiossidanti neuroprotettivi nei ratti.[13] Mentre il THC agisce principalmente sui recettori CB1 e CB2, il CBD agisce su altri recettori tra cui il canale del calcio TRPV1, attivandolo e rapidamente desensibilizzandolo, risultando in definitiva in meno potenziale di ipereccitazione.[14]
Nota: Il CBD non sembra avere effetti psicotropi (“alti”) come quelli causati dal ∆9-THC nella marijuana, ma, come già detto, è attualmente in fase di ricerca preliminare per i suoi possibili effetti anti-ansia e anti-psicotici legati ad un suo possibile effetto psicotropo. Mentre si sviluppa la comprensione delle differenze nei cannabinoidi medici, gli esperti stanno lavorando per distinguere la “marijuana medica” (con vari gradi di effetti psicotropi e deficit nella funzione esecutiva) dalle “terapie mediche con CBD” che comunemente presenterebbero avere un profilo degli effetti collaterali ridotto o non psicoattivo. Il CBD sembrerebbe non causare dipendenza.
Farmacocinetica
Il CBD assunto per via orale (in particolare in riferimento al farmaco liquido Epidiolex) impiega tra le 2,5 e le 5 ore per raggiungere la massima concentrazione ematica. L’emivita è tra le 56 e le 61 ore.[15] Sia nell’uomo che nei ratti, essere in stato di alimentazione sembra aumentare i livelli plasmatici di CBD.[16][17] La farmacocinetica del CBD può variare abbastanza ampiamente tra persone e persona [18], con risposte che possono essere aggravate da variazioni da lotto a lotto ed imprecisioni di etichettatura in alcuni prodotti a base di CBD.
Potenziali effetti terapeutici del CBD
In questo momento, c’è solo un accenno di promessa per il CBD per il miglioramento delle condizioni della pelle, ma nessuna basata sulla ricerca randomizzata.[19] Ad esempio, il CBD in uno studio preclinico in vitro ha mostrato effetti antiinfiammatori , antiproliferativi e lipostatici che suggeriscono che potrebbe potenzialmente avere una certa efficacia nei trattamenti di condizioni della pelle come l’acne vulgaris.[20]
Un caso studio ha riferito che una dose giornaliera di 2x300mg di CBD era associata al miglioramento della disfagia da SLA. Mentre diciotto mesi dopo l’esordio, il progresso della disfagia subivano un calo, e la debolezza degli arti, il fascicolazione e l’atrofia peggioravano relativamente meno. Gli autori teorizzano che la progressione di alcuni ma non tutti i sintomi della malattia dei motoneuroni può essere rallentata con il CBD.[21]
Il disturbo dello spettro autistico (ASD) probabilmente comporta alterazioni in alcuni percorsi chimici neurologici, come il glutammato e il GABA. È con questa idea che uno studio ha testato se il CBD potesse alterare questi percorsi in un modo che potesse aiutare il trattamento dell’autismo, con l’utilizzo della spettroscopia di risonanza magnetica. I ricercatori hanno scoperto che il CBD non era efficace per questo scopo.[22] Tuttavia, una serie di casi retrospettivi hanno mostrato un comportamento migliorato nel 61% di un campione di bambini con ASD. Tuttavia, non vi era alcun gruppo di controllo.[23]
Il CBD non sembra nemmeno migliorare i sintomi legati al movimento nel Parkinson, sebbene possa migliorare i problemi del sonno derivanti dalla condizione, in particolare la parassitnia con incubi e la perdita di atonia muscolare durante il sonno REM.[24][25]
In uno studio esplorativo, 2000mg/die di CBD hanno ridotto i sintomi psicotici della schizofrenia e i pazienti trattati con CBD avevano maggiori probabilità di essere valutati come migliorati dal medico curante.[26] Uno studio randomizzato nel quale si è testato l’effetto del CBD ha scoperto che sia esso che l’amisulpride hanno migliorato i sintomi della patologia in questione, ma il CBD ha avuto un migliore profilo degli effetti collaterali.[27] Tuttavia, in uno studio randomizzato nel quale si è utilizzata una dose di 600mg/die di CBD, non sono stati osservati miglioramenti nei sintomi.[28] Uno studio non controllato non ha riscontrato alcun beneficio del CBD con dosi aumentate da 40mg/die a 1280mg/die in 35 giorni su pazienti con schizofrenia resistente al trattamento.[29]
In sei studi randomizzati con oltre 500 pazienti con convulsioni gravi (in genere bambini piccoli), il CBD migliora significativamente i sintomi convulsivi.[30] In particolare, la percentuale di pazienti che ha avuto una riduzione di oltre il 50% delle crisi è diminuita, così come il numero di persone che hanno smesso di avere crisi epilettiche. Mentre gli studi randomizzati hanno in genere esaminato i pazienti con sindrome di Dravet e Lennox-Gastaut, studi estesi non randomizzati (studi sull’uso compassionevole) hanno anche mostrato efficacia nel trattamento del disturbo da carenza di CDKL5 e nelle sindromi di Aicardi, Doose e Dup15q.[31] Una serie di casi di tre pazienti ha anche suggerito che il CBD può aiutare con l’epilessia correlata al tumore al cervello.[32]
Studi a più lungo termine confermano che il CBD continua ad essere efficace e relativamente sicuro in questi pazienti [33][34][35], sebbene gli effetti collaterali inizialmente comuni come la riduzione dell’appetito e la diarrea persistessero in uno studio.[36] Il CBD sembra avere un effetto di dimensioni simili sull’epilessia pediatrica grave come altri farmaci antiepilettici.[37] Un piccolo studio di coorte suggerisce che anche i pediatri hanno ritenuto che la salute generale dei loro bambini con epilessia grave fosse migliorata.[38]
Il motivo per cui il CBD può aiutare con le convulsioni ha a che fare con le vie di segnalazione degli endocannabinoidi. Questi sono alterati nel disturbo convulsivo e THC e CBD agiscono in diversi modi per aiutare potenzialmente a marginare questo disturbo. Il THC probabilmente aiuta attivando il recettore CB1 [39], mentre i meccanismi del CBD sono meno conosciuti e possono includere una varietà di altri recettori, come il GPR55 e il TRPV1.[40][41][42]
Mentre il potenziale per il CBD di aiutare ad alleviuare alcuni tipi di dolore cronico è elevato, le prove di base sono molto piccole, con solo uno studio randomizzato e due studi non randomizzati a sostegno.
In uno studio randomizzato sono stati osservati dolore e spasticità in pazienti con sclerosi multipla, lesioni del midollo spinale e altre condizioni con questi sintomi. Sebbene diversi pazienti non abbiano completato lo studio, il controllo del dolore è migliorato nel gruppo CBD (mentre lo spasmo e altri sintomi non hanno subito variazioni).
Due studi non randomizzati e non controllati hanno esaminato il CBD al fine di valutarne il potenziale nell’alleviare il dolore. Uno studio ha osservato il dolore nelle ragazze giovani con dolore cronico da vaccino HPV. Si è riscontrato una riduzione del dolore corporeo e un migliore funzionamento con l’uso del CBD.[43] Uno studio molto piccolo ha esaminato il dolore nei pazienti con trapianto di rene e ha scoperto che due su sette pazienti avevano un sollievo totale dal dolore, mentre quattro avevano un sollievo non particolarmente percepibile.[44]
In uno studio, una singola dose di THC sembrava aumentare l’ansia rispetto al placebo, mentre una singola dose di CBD è risultata simile al placebo come stato ansioso.[45] Mancano studi randomizzati a lungo termine, ma studi randomizzati a breve termine e studi di serie di casi a lungo termine hanno esplorato i potenziali effetti benefici del CBD sull’ansia.
In una serie di casi di 72 pazienti ambulatoriali psichiatrici, il CBD (con la maggior parte dei pazienti che ne assumevano 25mg/die) ha migliorato rapidamente i punteggi di ansia in circa l’80% dei pazienti, con un effetto generalmente prolungato nel corso di tre mesi.[46]
Tre prove hanno esaminato gli effetti del CBD sull’ansia indotta sperimentalmente dal parlare in pubblico. Uno studio precoce non randomizzato nel 1993 ha mostrato che 300 mg di CBD hanno migliorato lo stato ansioso dopo il test di parlare in pubblico, ma non prima.[47] Uno studio randomizzato del 2017 dello stesso autore ha anche mostrato una riduzione dell’ansia alla stessa dose di 300mg, dopo il test del parlato, con 100mg e 900mg che non hanno mostrato alcun effetto.[48] Un altro gruppo ha studiato l’effetto di 600mg di CBD riscontrando una significativa riduzione dell’ansia durante il test di conversazione pubblica.[49]
Gli studi non controllati forniscono prove più deboli, ma estendono tali prove ad altre popolazioni. Una serie di casi retrospettivi hanno mostrato che quasi l’80% dei pazienti per i quali il CBD era utilizzato in una clinica psichiatrica per trattare l’ansia aveva migliorato questa condizione, e la maggior parte dei pazienti hanno mantenuto i benefici dopo il primo mese.[46]
Un caso studio ha suggerito che la somministrazione di CBD per 10 giorni ha ridotto i sintomi di astinenza da cannabis in un pesante abusatore, come ansia, perdita di appetito e irritabilità.[50]
Prove preliminari hanno suggerito che il CBD ha ridotto il consumo di fumo di sigaretta (nonostante non abbia influito sulla brama di sigarette)[51], e ha ridotto la piacevolezza legata alla sigaretta dopo una notte di astinenza.[52] Purtroppo, nei fumatori di sigarette che mirano all’astinenza, il CBD non ha migliorato la memoria di lavoro verbale o spaziale o l’impulsività.[53]
Un piccolo studio pilota condotto su individui dipendenti da oppioidi ha mostrato che il CBD in dosi di 400 o 800mg riduceva la brama dopo che i soggetti erano stati fatti astenere dagli oppioidi per sette giorni (dopo una sessione video per indurre la brama).
[54] Tuttavia, la maggior parte delle prove per i potenziali benefici del CBD sulla sospensione degli oppioidi sono di origine preclinica e basate su studi molto piccoli.[55]
Una serie di casi ha riportato una riduzione dei sintomi del PTSD (Post Traumatic Stress Disorder) in 10 di 11 pazienti con l’uso di CBD, dopo otto settimane di trattamento, in cui il CBD è stato ben tollerato.[56]
La prima citata serie di casi di 72 pazienti ambulatoriali psichiatrici ha anche scoperto che il CBD (sempore con la maggior parte dei pazienti che ne assumevano 25mg/die) non ha migliorato i punteggi del sonno nel corso di tre mesi.[46]
D’altro canto, uno studio crossover in doppio cieco ha scoperto che il CBD non ha interrotto il ciclo sonno-veglia dei pazienti che assumevano una dose relativamente alta intesa a ridurre clinicamente l’ansia (300 mg).[57]
Quando si parla di cancro e CBD, gli studi affidabili sull’uomo mancano totalmente.
Un caso studio ha riportato di un paziente con carcinoma polmonare che ha avuto una “risposta sorprendente” con evidente riduzione del tumore dopo auto-somministrazione di CBD.[58] Naturalmente, questo è un caso clinico, ed è una pura supposizione che il CBD sia l’elemento causale. Ma è comunque uno spunto di riflessione.
Parlando di casi clinici, un altro ha suggerito che il CBD potrebbe aver migliorato la chemioradioterapia in due casi di gliomi di alto grado.[59] Una serie di casi retrospettivi ha riferito che il 92% dei 119 casi di cancro esaminati aveva una riduzione delle cellule tumorali circolanti e talvolta una riduzione delle dimensioni del tumore, quando veniva assunto il CBD di livello farmaceutico.[106-60] Ancora una volta, questa è una prova di basso livello, ma pur sempre qualcosa che merita approfondimenti.
In uno studio nel quale sono stati utiulizzati 10mg di CBD, due volte al giorno, al fine di valkutarne gli effetti sul Morbo di Crohn, non sono stati riscontrati effetti benefici. Gli autori ipotizzano che ciò potrebbe essere stato influenzato dalla bassa dose o dal piccolo numero di pazienti nello studio o dalla mancanza di altri cannabinoidi sinergici (ovvero l’effetto entourage).[61]
Uno studio di prova (randomizzato, con un gruppo placebo) ha testato l’effetto di un estratto botanico ricco di CBD sulla Colite Ulcerosa, e non ha riscontrato differenze di remissione rispetto al placebo, ma un miglioramento di alcuni sintomi.[62]
Status del CBD nello Sport
Il CBD è stato utilizzato da atleti professionisti e dilettanti in diverse discipline e paesi, con l’Agenzia mondiale antidoping che ha rimosso il CBD dall’elenco delle sostanze vietate. L’Agenzia antidoping degli Stati Uniti e l’Agenzia antidoping del Regno Unito non hanno politiche anti-CBD, con quest’ultima che afferma che “il CBD non è attualmente nell’elenco delle sostanze proibite dell’Agenzia mondiale antidoping. Di conseguenza, è consentito l’uso di esso nello sport. Tutti gli altri cannabinoidi (inclusi ma non limitati a cannabis, hashish, marijuana e THC) sono vietati in competizione. L’intenzione del regolamento è proibire i cannabinoidi che attivano gli stessi recettori nel cervello come attivati dal THC. “[63] [64] Nel 2019, il principale produttore di prodotti a base di cannabis, Canopy Growth, ha acquisito la proprietà di maggioranza di BioSteel Sports Nutrition, che sta sviluppando prodotti a base di CBD con l’approvazione di numerosi atleti professionisti. [65] La National Hockey League Alumni Association ha avviato un progetto con Canopy Growth per determinare se il CBD o altri prodotti a base di cannabis potrebbero migliorare i sintomi neurologici e la qualità della vita dei giocatori feriti alla testa.[65] Numerosi atleti professionisti usano il CBD, principalmente per il trattamento del dolore.[65][66][67]
Interazioni alimentari e farmacologiche del CBD
CYP2D6
Il CBD sembra interagire con i farmaci antiepleettici comunemente usati, modificando significativamente i loro livelli sierici. Questi includono clobazam, rufinamide, topiramato, zonisamide ed eslicarbazepina.[68]
Il CBD può anche inibire un enzima chiamato CYP2D6, che è preso di mira da farmaci comuni tra cui omeprazolo e risperidone. Può anche inibire l’enzima CYP2C9, che ridurrebbe la metabolizzazione di warfarin e diclofenac.[69] Non è noto se le inibizioni in vitro si tradurranno in inibizioni in vivo negli esseri umani, e sono necessari ulteriori studi a riguardo.[70] In uno studio nel quale sono stati utilizzati 6x100mg/die di CBD, ha aumentato la biodisponibilità e l’emivita di eliminazione dell’esobarbitale.[71]
D’altro canto, è anche possibile che altri farmaci influenzino la metabolizzazione del CBD. Ad esempio, la rifampicina antibiotica riduce le concentrazioni di picco del CBD nel sangue a causa dell’induzione dell’enzima CYP3A4, mentre il ketoconazolo, l’inibitore del CYP3A4, raddoppia quasi il picco della concentrazione di CBD.[72]
Effetti collaterali del CBD e possibili contaminazioni dei prodotti che lo contengono
(±)-Amisulpride
Per trovare eventi avversi ed effetti collaterali, è necessario un campione relativamente grande. Quindi i principali studi per valutare questi in riferimento al CBD erano gli studi più ampi su giovani pazienti con gravi disturbi convulsivi, vale a dire la sindrome di Dravet e la sindrome di Lennox-Gastaut.
Gli effetti che si sono verificati più frequentemente con paragone tra CBD e placebo includono: sonnolenza, diarrea, affaticamento, vomito, febbre e letargia. Alcuni pazienti avevano anche livelli aumentati di aminotransferasi epatica.[73][5]
Due studi hanno esaminato i potenziali effetti collaterali emodinamici (come variazioni della frequenza cardiaca, della frequenza respiratoria o della pressione sanguigna) e non hanno trovato effetti significativi quando è stato assunto il CBD.[74][75]
Una review completa ha rilevato che dosi fino a 1.500mg/die sembrano essere abbastanza ben tollerate [76], sebbene non sia affatto sicuro presumere che la dose sia sicura per un determinato individuo (a causa della relativa mancanza di informazioni sulla sicurezza a lungo termine e della varietà di condizioni/farmaci/ecc. Che potrebbero teoricamente interagire con il CBD).
Gli studi sugli animali hanno riportato alcuni effetti collaterali teorici non osservati negli studi sull’uomo esistenti, come la ridotta capacità di fertilizzazione e dell’inibizione attraverso il metabolismo epatico del farmaco.[77][78]
Il CBD è risultato promettente come metodo per ridurre gli effetti collaterali quando vengono assunti altri farmaci, come osservato nel già citato studio sul CBD somministrato a pazienti con schizofrenia che assumevano amisulpride.[79]
I contaminanti da piante di cannabis coltivate, raccolte e confezionate con pratiche meno controllate potrebbero teoricamente comparire anche in prodotti a base di CBD, inclusi pesticidi, particelle metalliche, cannabinoidi sintetici, metalli pesanti, muffe, batteri e aflatossine.[80][81] I solventi residui del processo di produzione possono anche comparire in estratti di cannabis, vale a dire esano, etanolo, alcool isopropilico, toluene, benzene, xilene e acetone.[82] Questa ricerca riguarda tuttavia tutti gli estratti generali della cannabis; la ricerca sui contaminanti specificamente nei prodotti isolati di CBD deve ancora essere svolta.
Conclusioni sul CBD
Cosa concludere riguardo al CBD dopo l’esposizione dei dati precedentemente riportati? Sicuramente che la ricerca sull’uomo è molto limitata e non ci permette ad oggi di esporre un giudizio sufficientemente fondato sulla reale portata terapeutica del CBD. Si tratta ovviamente di una conclusione dettata dalla scarsità dei dati e dall’impatto degli studi esistenti. Possiamo comunque scorgere dei potenziali sul trattamento dell’ansia e, ipoteticamente, sullo stress percepito e il trattamento del dolore. Tale caratteristica rende il CBD sperimentabile come aggiunta ad una preparazione sportiva particolarmente intensa come durante il picco di preparazione per una competizione o un contest di BodyBuilding. La sua possibile influenza sull’appetito (effetto anoressizzante) potrebbe tornare senz’altro molto utile nei periodi di restrizione calorica. Alcuni speculeranno su un suo possibile impatto indiretto significativo sui livelli di Cortisolo dato, per l’appunto, da una ridotta percezione dello stress, ma i dati certi al momento non sussistono. Il fatto che si tratti di un composto dagli effetti non del tutto accertati non invoglia particolarmente a investire denaro per testarne le potenzialità. Se poi consideriamo il fatto che i più non conoscono ne le modalità di utilizzo ne le dosi graduate da applicare con criterio crescente in caso di risposta positiva con dosaggi più blandi, non c’è da stupirsi se spesso ci si imbatte in testimonianze molto contrastanti provenienti dagli utilizzatori che scoraggiano i lettori meno preparati in materia.
Quindi, vale la pena investire denaro per testare l’efficacia del CBD? Considerando che il costo di prodotti di alta qualità non è propriamente economico, e che i dati in letteratura non sono molto impattanti a livello dimostrativo (almeno ad oggi), a meno che non abbiate una adegiuata conoscenza per testarne e valutarne gli effetti, per il momento si può saggiamente investire su altro… per adesso…
Gabriel Bellizzi
Riferimenti:
1- Campos AC, Moreira FA, Gomes FV, Del Bel EA, Guimarães FS (December 2012). “Multiple mechanisms involved in the large-spectrum therapeutic potential of cannabidiol in psychiatric disorders”. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences (Review). 367 (1607): 3364–78.
2- LOEWE S. Studies on the pharmacology and acute toxicity of compounds with marihuana activity. J Pharmacol Exp Ther (1946)
3- Howlett AC, et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev (2002)
4- Devane WA, et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science (1992)
5- Devinsky O, Cross JH, Wright S. Trial of Cannabidiol for Drug-Resistant Seizures in the Dravet Syndrome.
N Engl J Med (2017)
6- Statement from FDA Commissioner Scott Gottlieb, M.D., on signing of the Agriculture Improvement Act and the agency’s regulation of products containing cannabis and cannabis-derived compounds.
7- Statement from FDA Commissioner Scott Gottlieb, M.D., on new steps to advance agency’s continued evaluation of potential regulatory pathways for cannabis-containing and cannabis-derived products.
8- O’Connor SM, Lietzan E. The Surprising Reach of FDA Regulation of Cannabis, Even After Descheduling. Am Univ Law Rev. (2019)
9- Hilderbrand RL. Hemp & Cannabidiol: What is a Medicine?. Mo Med. (2018)
10- Borrelli F, et al. Beneficial effect of the non-psychotropic plant cannabinoid cannabigerol on experimental inflammatory bowel disease. Biochem Pharmacol. (2013)
11- Battista N, et al. The endocannabinoid system: an overview. Front Behav Neurosci. (2012)
12- Laprairie RB, et al. Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. Br J Pharmacol. (2015)
13- Hampson AJ, et al. Cannabidiol and (-)Delta9-tetrahydrocannabinol are neuroprotective antioxidants. Proc Natl Acad Sci U S A. (1998)
14- Iannotti FA, et al. Nonpsychotropic plant cannabinoids, cannabidivarin (CBDV) and cannabidiol (CBD), activate and desensitize transient receptor potential vanilloid 1 (TRPV1) channels in vitro: potential for the treatment of neuronal hyperexcitability. ACS Chem Neurosci. (2014)
16- a b Zgair A, et al. Dietary fats and pharmaceutical lipid excipients increase systemic exposure to orally administered cannabis and cannabis-based medicines. Am J Transl Res. (2016)
17- Winter H, et al. Effect of a high-calorie, high-fat meal on the bioavailability and pharmacokinetics of PA-824 in healthy adult subjects. Antimicrob Agents Chemother. (2013)
18- Huestis MA. Human cannabinoid pharmacokinetics. Chem Biodivers. (2007)
19- Eagleston LRM, et al. Cannabinoids in dermatology: a scoping review. Dermatol Online J. (2018)
20- Oláh A, et al. Cannabidiol exerts sebostatic and antiinflammatory effects on human sebocytes. J Clin Invest. (2014)
21- Co-medication with Cannabidiol May Slow Down the Progression of Motor Neuron Disease: A Case Report; 2017..
22- Pretzsch CM, et al. Effects of cannabidiol on brain excitation and inhibition systems; a randomised placebo-controlled single dose trial during magnetic resonance spectroscopy in adults with and without autism spectrum disorder. Neuropsychopharmacology. (2019)
23- Aran A, et al. Brief Report: Cannabidiol-Rich Cannabis in Children with Autism Spectrum Disorder and Severe Behavioral Problems-A Retrospective Feasibility Study. J Autism Dev Disord. (2019)
24- Chagas MH, et al. Effects of cannabidiol in the treatment of patients with Parkinson’s disease: an exploratory double-blind trial. J Psychopharmacol. (2014)
25- Peres FF, et al. Cannabidiol as a Promising Strategy to Treat and Prevent Movement Disorders?. Front Pharmacol. (2018)
26- McGuire P, et al. Cannabidiol (CBD) as an Adjunctive Therapy in Schizophrenia: A Multicenter Randomized Controlled Trial. Am J Psychiatry. (2018)
27- Leweke FM, et al. Cannabidiol enhances anandamide signaling and alleviates psychotic symptoms of schizophrenia. Transl Psychiatry. (2012)
28- Boggs DL, et al. The effects of cannabidiol (CBD) on cognition and symptoms in outpatients with chronic schizophrenia a randomized placebo controlled trial. Psychopharmacology (Berl). (2018)
29- Zuardi AW, et al. Cannabidiol monotherapy for treatment-resistant schizophrenia. J Psychopharmacol. (2006)
30- Stockings E, et al. Evidence for cannabis and cannabinoids for epilepsy: a systematic review of controlled and observational evidence. J Neurol Neurosurg Psychiatry. (2018)
31- Devinsky O, et al. Open-label use of highly purified CBD (Epidiolex®) in patients with CDKL5 deficiency disorder and Aicardi, Dup15q, and Doose syndromes. Epilepsy Behav. (2018)
32- Warren PP, et al. The use of cannabidiol for seizure management in patients with brain tumor-related epilepsy. Neurocase. (2017)
33- Thiele E, et al. Cannabidiol in patients with Lennox-Gastaut syndrome: Interim analysis of an open-label extension study. Epilepsia. (2019)
34- Szaflarski JP, et al. Cannabidiol improves frequency and severity of seizures and reduces adverse events in an open-label add-on prospective study. Epilepsy Behav. (2018)
35- Szaflarski JP, et al. Long-term safety and treatment effects of cannabidiol in children and adults with treatment-resistant epilepsies: Expanded access program results. Epilepsia. (2018)
36- Sands TT, et al. Long-Term Safety, Tolerability, and Efficacy of Cannabidiol in Children with Refractory Epilepsy: Results from an Expanded Access Program in the US. CNS Drugs. (2019)
37- Ali S, Scheffer IE, Sadleir LG. Efficacy of cannabinoids in paediatric epilepsy. Dev Med Child Neurol. (2019)
38- Chen KA, et al. Cannabidiol for treating drug-resistant epilepsy in children: the New South Wales experience. Med J Aust. (2018)
39- Reddy DS, Golub VM. The Pharmacological Basis of Cannabis Therapy for Epilepsy. J Pharmacol Exp Ther. (2016)
40- Rosenberg EC, Patra PH, Whalley BJ. Therapeutic effects of cannabinoids in animal models of seizures, epilepsy, epileptogenesis, and epilepsy-related neuroprotection. Epilepsy Behav. (2017)
41- Bisogno T, et al. Molecular targets for cannabidiol and its synthetic analogues: effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br J Pharmacol. (2001)
42- Bialer M, et al. Progress report on new antiepileptic drugs: A summary of the Thirteenth Eilat Conference on New Antiepileptic Drugs and Devices (EILAT XIII). Epilepsia. (2017)
43- Palmieri B, Laurino C, Vadalà M. Short-Term Efficacy of CBD-Enriched Hemp Oil in Girls with Dysautonomic Syndrome after Human Papillomavirus Vaccination. Isr Med Assoc J. (2017)
44- Cuñetti L, et al. Chronic Pain Treatment With Cannabidiol in Kidney Transplant Patients in Uruguay. Transplant Proc. (2018)
45- Martin-Santos R, et al. Acute effects of a single, oral dose of d9-tetrahydrocannabinol (THC) and cannabidiol (CBD) administration in healthy volunteers. Curr Pharm Des. (2012)
46- a b c Shannon S, et al. Cannabidiol in Anxiety and Sleep: A Large Case Series. Perm J. (2019)
47- Zuardi AW, et al. Effects of ipsapirone and cannabidiol on human experimental anxiety. J Psychopharmacol. (1993)
48- Zuardi AW, et al. Inverted U-Shaped Dose-Response Curve of the Anxiolytic Effect of Cannabidiol during Public Speaking in Real Life. Front Pharmacol. (2017)
49- Bergamaschi MM, et al. Cannabidiol reduces the anxiety induced by simulated public speaking in treatment-naïve social phobia patients. Neuropsychopharmacology. (2011)
50- Crippa JA, et al. Cannabidiol for the treatment of cannabis withdrawal syndrome: a case report. J Clin Pharm Ther. (2013)
51- Morgan CJ, et al. Cannabidiol reduces cigarette consumption in tobacco smokers: preliminary findings. Addict Behav. (2013)
52- Hindocha C, et al. Cannabidiol reverses attentional bias to cigarette cues in a human experimental model of tobacco withdrawal. Addiction. (2018)
53- Hindocha C, et al. The effects of cannabidiol on impulsivity and memory during abstinence in cigarette dependent smokers. Sci Rep. (2018)
54- Ren Y, et al. Cannabidiol, a nonpsychotropic component of cannabis, inhibits cue-induced heroin seeking and normalizes discrete mesolimbic neuronal disturbances. J Neurosci. (2009)
55- Hurd YL, et al. Early Phase in the Development of Cannabidiol as a Treatment for Addiction: Opioid Relapse Takes Initial Center Stage. Neurotherapeutics. (2015)
56- Elms L, et al. Cannabidiol in the Treatment of Post-Traumatic Stress Disorder: A Case Series. J Altern Complement Med. (2019)
57- Linares IMP, et al. No Acute Effects of Cannabidiol on the Sleep-Wake Cycle of Healthy Subjects: A Randomized, Double-Blind, Placebo-Controlled, Crossover Study. Front Pharmacol. (2018)
58- Sulé-Suso J, et al. Striking lung cancer response to self-administration of cannabidiol: A case report and literature review. SAGE Open Med Case Rep. (2019)
59- Dall’Stella PB, et al. Case Report: Clinical Outcome and Image Response of Two Patients With Secondary High-Grade Glioma Treated With Chemoradiation, PCV, and Cannabidiol. Front Oncol. (2019)
60- Kenyon J, Liu W, Dalgleish A. Report of Objective Clinical Responses of Cancer Patients to Pharmaceutical-grade Synthetic Cannabidiol. Anticancer Res. (2018)
61- Naftali T, et al. Low-Dose Cannabidiol Is Safe but Not Effective in the Treatment for Crohn’s Disease, a Randomized Controlled Trial. Dig Dis Sci. (2017)
62- Irving PM, et al. A Randomized, Double-blind, Placebo-controlled, Parallel-group, Pilot Study of Cannabidiol-rich Botanical Extract in the Symptomatic Treatment of Ulcerative Colitis. Inflamm Bowel Dis. (2018)
63- “Athlete Advisory Note: Cannabidiol (CBD)”. United Kingdom Anti-Doping Agency. Retrieved October 9, 2019.
64- “Athletes: 6 things to know about cannabidiol”. US Anti-Doping Agency. October 23, 2018. Retrieved February 17, 2020.
65- a b c Donna Spencer (October 2, 2019). “Canopy cannabis company buys ex-NHL player’s sports nutrition business”. CTV Business. The Canadian Press. Retrieved February 17, 2020. BioSteel’s brand ambassadors also include well-known athletes across major sports leagues in North America, which could be beneficial as the company’s attempt to push regulated CBD nutrition products into the mainstream health and wellness segments
66- Amanda Loudin (December 7, 2019). “As more pro athletes use cannabis for aches and pain, the more they run afoul of rules”. The Washington Post. Retrieved February 17, 2020.
67- Sara Harrison (September 28, 2019). “Lots of athletes say CBD Is a better painkiller. Is It?”. Wired. Retrieved February 17, 2020.
68- Gaston TE, et al. Interactions between cannabidiol and commonly used antiepileptic drugs. Epilepsia. (2017)
69- Ujváry I, Hanuš L. Human Metabolites of Cannabidiol: A Review on Their Formation, Biological Activity, and Relevance in Therapy. Cannabis Cannabinoid Res. (2016)
70- Welty TE, Luebke A, Gidal BE. Cannabidiol: promise and pitfalls. Epilepsy Curr. (2014)
71- Brzozowska N, et al. ABC transporters P-gp and Bcrp do not limit the brain uptake of the novel antipsychotic and anticonvulsant drug cannabidiol in mice. PeerJ. (2016)
72- Stott C, et al. A Phase I, open-label, randomized, crossover study in three parallel groups to evaluate the effect of Rifampicin, Ketoconazole, and Omeprazole on the pharmacokinetics of THC/CBD oromucosal spray in healthy volunteers. Springerplus. (2013)
73- Devinsky O, et al. Effect of Cannabidiol on Drop Seizures in the Lennox-Gastaut Syndrome. N Engl J Med. (2018)
74- Manini AF, et al. Safety and pharmacokinetics of oral cannabidiol when administered concomitantly with intravenous fentanyl in humans. J Addict Med. (2015)
75- Arndt DL, de Wit H. Cannabidiol Does Not Dampen Responses to Emotional Stimuli in Healthy Adults. Cannabis Cannabinoid Res. (2017)
76- Bergamaschi MM, et al. Safety and side effects of cannabidiol, a Cannabis sativa constituent. Curr Drug Saf. (2011)
77- Schuel H, et al. Cannabinoids inhibit fertilization in sea urchins by reducing the fertilizing capacity of sperm. Pharmacol Biochem Behav. (1991)
78- Stout SM, Cimino NM. Exogenous cannabinoids as substrates, inhibitors, and inducers of human drug metabolizing enzymes: a systematic review. Drug Metab Rev. (2014)
79- Jara-Oseguera A, Simon SA, Rosenbaum T. TRPV1: on the road to pain relief. Curr Mol Pharmacol. (2008)
80- Russo EB. Current Therapeutic Cannabis Controversies and Clinical Trial Design Issues. Front Pharmacol. (2016)
81- Busse FP, et al. Lead poisoning due to adulterated marijuana in leipzig. Dtsch Arztebl Int. (2008)
82- Pavlovic R, et al. Quality Traits of “Cannabidiol Oils”: Cannabinoids Content, Terpene Fingerprint and Oxidation Stability of European Commercially Available Preparations. Molecules. (2018)
Fin dalla loro comparsa nel mercato della supplementazione sportiva “border line” , i SARM hanno goduto fin da subito di una enorme popolarità portata più dalle promesse che i produttori elencavano nelle descrizioni dei loro prodotti che da veri e propri dati di fatto. La “principale promessa” connessa a questa classe di molecole (classe più complessa di quanto comunemente inteso e non del tutto separata dagli AAS) era quella di apportare tutti i benefici degli AAS senza il carico di effetti collaterali ad essi legati. Questa è risultata una affermazione non vera, almeno in buona parte. Personalmente, ho svolto ricerche e studi per molto tempo, un tempo sufficiente per constatare tramite letteratura e sperimentazione che gli effetti collaterali tipici degli AAS continuano a persistere anche nei SARM. Non solo, paradossalmente il loro uso in monoterapia è assai più problematico, per certi versi, di quello che potrebbe essere con l’uso del vecchi Testosterone. Ma il tema di questo articolo è più specifico e si riferisce alla epatotossicità potenziale di due SARM, il RAD-140 ed LGD-4033. Recentemente è stato pubblicato su Epatology Communications un articolo nel quale un equipe australiana ha descritto due casi di danno epatico riscontrati in uomini che avevano usato RAD 140 e LGD4033.[1] Purtroppo, i casi esaminati presentano più variabili che ne impediscono una corretta valutazione del potenziale negativo a livello epatico dei composti in questione.
Ma andiamo per ordine ….
Casi studio e approfondimenti sulla questione
RAD-140
Il primo caso riguarda un uomo di 49 anni che aveva assunto RAD 140 per 5 settimane consecutive. I ricercatori hanno analizzato il contenuto del “supplemento” nel loro laboratorio constatando un effettivo contenuto di RAD 140. Non sono stati trovati contaminanti. L’uomo era sottoposto a terapia antidepressiva a base di anche usato la Venlafaxina, un antidepressivo SSRI che può interferire con la funzionalità epatica in alcune soggetti particolarmente sensibili.[2]
Dopo che l’uomo ha sviluppato ittero e prurito, ha cercato assistenza medica. L’analisi del sangue ha mostrato che la concentrazione dei marker ematici, legati alla valutazione della salute epatica, era sensibilmente aumentata. L’analisi ha suggerito che l’uomo soffriva di colestasi, un classico effetto collaterale da abuso di AAS orali metilati in C-17.
L’esame dei campioni di tessuto epatico ha mostrato che i tubi che drenano i sali biliari nell’intestino erano gravemente compromessi. Il corpo si libera dei prodotti di scarto liposolubili attraverso i sali biliari. Inoltre, i campioni di tessuto epatico erano infiammati e danneggiati.
I dottori hanno impedito all’uomo di assumere le sue medicine e gli hanno somministrato Ursodiolo e Colestiramina. Il suo fegato si riprese nei mesi seguenti. Un anno dopo, tutti i suoi valori erano tornati alla normalità.
LGD-4033
Il secondo caso descritto dagli australiani è quello di un uomo di 24 anni che aveva assunto LGD-4033 per 9 settimane. I ricercatori hanno anche analizzato anche questo “supplemento” trovando effettivamente LGD-4033, e nessun contaminante.
L’uomo si era ammalato una settimana dopo la fine del suo ciclo, ma solo dopo 3 settimane si recò dal medico. Aveva l’ittero, aveva la nausea, mangiava a malapena e aveva già perso 5 chili.
L’uomo non stava usando nessun altro farmaco. Tuttavia, una volta al mese era solito abbandonare lo stile di vita controllato e consumava molto alcol. Nel suo sangue, i ricercatori hanno trovato una maggiore concentrazione di enzimi che indicano danni al fegato. Il fegato dell’uomo aveva dimensioni più grandi della norma, ma secondo una scansione, non c’erano prove di un ridotto drenaggio dei sali biliari.
I ricercatori hanno deciso di aspettare e hanno visto che il fegato si era ripreso 4 mesi dopo.
I ricercatori concludono il loro articolo affermando che la lesione epatica in entrambi i casi rientra nello spettro descritto della lesione epatica associata agli steroidi anabolizzanti. Questi effetti off-target mettono in discussione la cosiddetta selettività tissutale dei SARM, che è stato il loro principale punto di forza.
Resta da vedere se questi casi rappresentino la punta dell’iceberg, ma data la crescente popolarità dell’uso dei SARM, è necessaria una maggiore vigilanza e segnalazione di potenziali casi.
Come abbiamo visto, in entrambi i casi sono presenti delle variabili che non permettono una piena valutazione del potenziale epatotossico delle due molecole. Nel primo caso la variabile era rappresentata dalla Venlafaxina, mentre nel secondo caso era l’abuso di alcool.
Ciò non significa che i due SARM non abbiano un possibile impatto epatico negativo. Studi controllati hanno mostrato infatti una epatotossicità potenziale del RAD-140 dose-dipendente.[3] Mentre con LGD-4033, a dose entro 1mg/die, non hanno osservato alterazioni significative i ALT e AST, sebbene, anche in questo caso, il problema aumenta in modo dose-dipendente.[4] Giova sottolineare che in ambito sportivo, è abbastanza comune per i soggetti di sesso maschile utilizzare dai 10 ai 20mg/die di LGD-4033, mentre le atlete rimangono nel range tra 5 e 10mg/die.
Questi dosaggi sono stati determinati dagli utilizzatori sulla base di informazioni aneddotiche e sperimentazione personale e non rappresentano in alcun modo delle linee guida che determinano un uso corretto o errato della molecola in questione.
Nota: il dosaggio più elevato di LGD-4033 utilizzato negli studi clinici di fase 2 è stato di 2mg al giorno a fini terapeutici.[5]
Come prendere questi dati? In modo costruttivo, ovviamente. Considerate i SARM come un qualsiasi altro farmaco, valutando concretamente il lato “positivo” ed il lato “negativo”, e non come una sorta di “pillola rossa” in grado di darvi la visione e concretizzazione della soluzione definitiva alle vostre smanie estetiche.
Da qualche tempo stanno circolando teorie secondo le quali l’Ibutamoren (comunemente noto come MK-677) può causare danni cerebrali per via dello stimolo cronico dei recettori della Grelina (bersaglio della molecola in questione).
Ma è mai stato osservato un effetto simile sull’uomo negli studi presenti svolti su esseri umani? No, e non risulta nulla di simile nemmeno dai dati aneddotici provenienti dagli utilizzatori, almeno per il momento.
Allora da cosa nasce la teoria? Nasce dai dati provenienti da test di laboratorio svolti su topi. Ed in effetti, i dati provenienti da questi test mostrano collegamenti tra esposizione cronica allo stimolo dei recettori della Grelina e esiti negativi a livello cerebrale.
Ma approfondiamo la questione…
MK-677 e aumento della paura nei ratti.
Uno studio sui ratti ha valutato se l’MK-677 possa causare un aumento dei livelli di paura.[1]
Esistono prove a sostegno del fatto che uno dei modi in cui la Grelina viene modulata è attraverso l’esposizione allo stress.
Ciò che questo studio intendeva valutare specificamente era se i ratti avessero una maggiore probabilità di sperimentare la paura e il disturbo da stress post-traumatico (PTSD) con livelli cronicamente alti di Grelina, situazione “mimata” artificialmente 24/7 con l’utilizzo di un agonista del recettore della Grelina (MK-677).
Ai ratti veniva somministrato in cronico l’MK-677 e nello stesso tempo venivano costantemente spaventati. I ricercatori valutavano quindi la loro differenza di risposta (gruppo trattato con MK-677 Vs. gruppo non trattato).
È stato quindi scoperto che i ratti del gruppo trattato con MK-677 avevano hanno migliorato la memoria della paura rispetto al basale.
Come ho già detto, ci sono prove a sostegno del fatto che uno dei modi in cui la Grelina viene modulata è attraverso l’esposizione allo stress, e questo studio è stato in grado di suscitare un po’ di controversie nella sottocultura del BodyBuilding con i suoi risultati che mostrano un effetto palesemente negativo sull’amigdala in cui gli effetti di una sovrastimolazione del recettore della grelina porta ad un della paura.
La Grelina e l’Ormone della Crescita agiscono insieme nell’amigdala per aumentare la percezione della paura, almeno come osservato in questo modello di roditori.
Si noti che in questo studio, i ratti sono stati trattati con iniezioni intraperitoneale (I.P.) di 1ml/kg.
Nel caso non lo sapeste, una iniezione intraperitoneale consiste nell’iniezione di una sostanza nel peritoneo (cavità corporea).Il peritoneo è la membrana sierosa che forma il rivestimento della cavità addominale. Ed è qui che l’MK-677 è stato somministrato per la prima volta per valutare se una maggiore attivazione del recettore della Grelina è sufficiente per migliorare la memoria della paura.
Dopo aver stabilito che l’attivazione del recettore della Grelina è effettivamente sufficiente per migliorare la memoria della paura, hanno quindi continuato a infondere MK-677 direttamente nel complesso basolaterale dell’amigdala per determinare se l’attivazione ripetuta del recettore della Grelina nel complesso basolaterale dell’amigdala sia sufficiente per migliorare la memoria della paura.
Ripetute infusioni intra-amigdala miglioravano la memoria della paura.[2]
Ora, mentre tutto ciò dipinge un quadro piuttosto negativo dell’MK-677, bisognerebbe tener presente che questo studio ha anche dimostrato che una singola iniezione di un agonista del recettore della Grelina non era sufficiente per aumentare la paura, e persino l’agonismo cronico del recettore della Grelina non alterare la locomozione, l’ansia innata o l’espressione di ricordi di paura acquisiti in precedenza.[2]
Quanto possono interessare l’essere umano i risultati ottenuti?
Negli studi effettuati sull’uomo, l’MK-677 è stato osservato essere ben tollerato per oltre un anno consecutivo di utilizzo di una dose pari a 25mg/die assunta per via orale.
Gli utilizzatori ovviamente non si somministrano cronicamente il composto nella cavità peritoneale o nel cervello.
Questa scoperta non è stata ancora replicata nelle prove umane, e nemmeno nei resoconti aneddotici fino ad oggi.
Detto ciò, la ricerca sugli esseri umani sta indagando su questo particolare effetto?
Probabilmente no, e ovviamente quelli che usano l’MK-677 a livello ricreativo non vengono sottoposti di norma a scansioni cerebrali prima e dopo l’utilizzo del composto.
Molto probabilmente il problema potrebbe emergere se l’uso fosse protratto nel cronico, per anni. Non per nulla esistono meccanismi di autoregolazione nel corpo per una ragione ben specifica.
Anche se non vi fosse alcun rischio neurologico, bisogna ricordare che l’MK-677 causa un peggioramento della sensibilità all’insulina, fattore di certo non da trascurare e da gestire con cicli temporalmente non eccessivi e l’arginamento di tale conseguenza iatrogena con “composti tampone” quali Berberina o Metformina. L’effetto sulla sensibilità all’insulina è certamente una preoccupazione più immediata di qualsiasi altra affermazione estrapolata da studi sui roditori che mostrano i suoi possibili effetti sulla salute del cervello.
Androgenico: 22-55
Anabolico: 100-200
Standard: Methyltestosterone (orale)
Nome Chimico: 17α-Ethylestr-4-en-17β-ol-3-one; 17α-Ethyl-19-norandrost-4-en-17β-ol-3-one.
Attività estrogenica: alta
Attività Progestinica: alta
Aromatizzazione: si
Il Norethandrolone, noto anche come 17α-Ethyl-19-nortestosterone o 17α-Ethylestr-4-en-17β-ol-3-one; 17α-Ethyl-19-norandrost-4-en-17β-ol-3-one, conosciuto con i nomi commerciali di Nilevar e Pronabol, è uno steroide androgene anabolizzante (AAS), disponibile in forma orale, utilizzato in campo clinico come agente per la promozione della crescita muscolare, per curare gravi ustioni, traumi fisici e per il trattamento dell’anemia aplastica, anche se attualmente non è quasi più utilizzato in medicina.[1][2][3] Tuttavia, è ancora disponibile in Francia.[2][3]
Il Norethandrolone è uno steroide estrano sintetico e un derivato metilato in C17 del 19-nortestosterone (Nandrolone).[1][2] È strettamente correlato al Normethandrone (17α-metil-19-nortestosterone) e all’Etilestrenolo (3-deketo-17α-etil-19-nortestosterone).[1][2] In letteratura sono state pubblicate le vie di sintesi chimica del Norethandrolone.[4]
Il Norethandrolone è un AAS con un alto rapporto tra attività anabolica e androgena.[2] Analogamente al caso del nandrolone e del 5α-diidronandrolone, e del 5α-diidronoretandrolone, il metabolita 5α-ridotto del Norethandrolone, mostra una ridotta affinità per il recettore degli androgeni rispetto alla molecola progenitrice.[2][5] Ciò è probabilmente correlato all’alto rapporto tra attività anabolica e androgena osservata con il Norethandrolone.[2][5] Il Norethandrolone ha un’attività estrogenica relativamente elevata per via della sua conversione ad opera dell’enzima Aromatasi in Etilestradiolo.[2] Ha anche una forte attività progestinica.[2] La potenza progestinica del Norethandrolone è simile a quella del Noretisterone in termini di alterazioni dell’endometrio nelle donne.[6-11] Inoltre, il Norethandrolone è epatotossico per via della metilazione in C17.[2]
Il Norethandrolone è stato uno dei primi AAS orali disponibili negli Stati Uniti. Era essenzialmente la risposta della Searles all’AAS di nuova creazione da parte della svizzera Ciba, il Methandrostenolone (Dianabol), che venne rilasciato sul mercato lo stesso anno. In effetti, spesso venne usato il Methandrostenolone come paragone per valutare l’impatto del Norethandrolone sull’aumento di peso, sull’anabolismo e sulla ritenzione idrica.
Sette anni prima del rilascio del Norethandrolone, la Mayo Clinic aveva annunciato l’efficacia drammatica del Cortisone nel trattamento dell’artrite reumatoide. Questo a sua volta ha stimolato un enorme interesse per tutti gli aspetti della chimica degli steroidi, dell’endocrinologia e dei campi correlati. G. D. Searle & Co. ha prontamente avviato un grande sforzo nella ricerca sugli steroidi, con l’obiettivo di scoprire composti steroidei migliori di quelli precedentemente disponibili e che avessero potuto essere utilizzati per condizioni per le quali non erano disponibili altri composti.
Questo sforzo portò all’introduzione del Norethandrolone, commercializzato nel 1956 come Nilevar, il primo agente anabolizzante con una separazione favorevole tra effetti anabolizzanti e androgeni.[7] Per i motivi sopra esposti, negli uomini si riscontrano solo effetti androgeni deboli (probabilmente perché la forma derivante dalla 5-alfa-riduzione ha un ridotto potenziale di legame con i AR), sebbene nelle donne la virilizzazione rimanga una possibilità con l’uso di questa molecola.
Questo AAS è generalmente sconsigliato nelle atlete, non solo per le contenute possibilità di comparsa di effetti androgeni, ma anche per alcuni problemi di infertilità, che sono possibili anche nelle donne.[8][9] L’effetto anabolico di questo farmaco è moderato, e ciò è probabilmente dovuto al suo legame moderatamente forte con il recettore degli androgeni (questo lo rende abbastanza diverso dal Methandrostenolone, che ha uno scarso legame con il recettore degli androgeni agendo per via Non-genomica) e alla sua capacità di stimolare la sintesi proteica e a bloccarne il catabolismo.[10] Il Nilevar fu il primo AAS sviluppato dalla Searles , e fu proprio questo farmaco che alla fine portò alla ricerca e allo sviluppo dell’Oxandrolone (Anavar), molto meno androgeno, molto più anabolizzante e privo di attività estrogenica e progestinica, portando al conseguente declino della popolarità e dell’uso del Nilevar.
Nell’immagine è possibile osservare le due vie attraverso le quali un AAS può svolgere la sua azione a livello cellulare (Azione Genomica e Azione Non-Genomica.
Come risaputo, gli AAS, a diverso grado di impatto, possono avere effetti deleteri sul colesterolo sierico. Il Norethandrolone non è da meno presentando la tendenza a causare una riduzione delle concentrazioni di colesterolo HDL (“buono”) e un aumento delle concentrazioni di colesterolo LDL (“cattivo”), cosa che comporta uno sbilanciamento dell’equilibrio HDL/LDL che si traduce in un rischio maggiore di sviluppare arteriosclerosi. L’impatto relativo all’assunzione di un AAS nei confronti dei lipidi ematici dipende dalla dose, dalla via di somministrazione (per via orale o iniettabile), dal tipo di steroide (aromatizzabile o non aromatizzabile), e dal livello di resistenza al metabolismo epatico.
Essendo il Norethandrolone un AAS con ridotta attività androgenica, la soglia dei possibili forti effetti collaterali androgenici è generalmente bassa ed è paragonabile a quella riscontrata con l’uso di Nandrolone sebbene dosaggi più alti mostrano un aumento della possibilità che tali effetti si manifestino.
Come per gli altri AAS 17α-alchilati, il Norethandrolone presenta un elevato grado di epatotossicità.[1][2]
17-alpha-methyl-estradiolo
Il Norethandrolone è un substrato soggetto all’azione dell’enzima Aromatasi, per la precisione aromatizza in una forma di estradiolo dall’elevato potere biologico, il 17-alpha-methyl-estradiolo e, potenzialmente, 17-alpha-methyl-estrone.[1][2] Questo incrementa le caratteristiche del Norethandrolone come potenziale steroide per una fase “Bulk” grazie alla maggior proliferazione dei recettori degli estrogeni, un miglior utilizzo del glucosio, e un aumentata secrezione di GH e IGF-1. Ovviamente, con il suo uso il fattore estrogenico deve essere attentamente monitorato in quanto effetti collaterali legati all’iperestrogenemia o all’aumentata attività tissutale del Estradiolo (comune con l’uso di progestinici) quali ginecomastia, ritenzione idrica e l’accumulo di grasso con modello femminile sono piuttosto comuni. L’uso di un inibitore dell’aromatasi e di un SERM, dopo comprovata necessità, in soggetti sensibili all’aromatizzazione e/o all’attività estrogenica è risultata una pratica funzionale.
Il Norethandrolone si lega fortemente al recettore del progesterone (PR).[1][2] Dato il noto vincolo del Norethandrolone con il PR, e la tendenza da parte dei farmaci derivati dal Nandrolone di offrire almeno qualche attività progestinica, una significativa attività in tal senso, con gli effetti ad essa legati, non deve essere trascurata per tutto il tempo di assunzione.
Si ricordi inoltre che gli effetti collaterali associati con il Progesterone sono simili a quelli degli estrogeni, compreso il feedback negativo di inibizione della produzione di Testosterone e una maggiore velocità di accumulo di grasso. I progestinici aumentano anche l’effetto stimolante degli estrogeni sulla crescita del tessuto mammario. Sembra che ci sia una forte sinergia tra questi due ormoni, in modo tale che la ginecomastia potrebbe anche verificarsi con l’aiuto dei progestinici, senza eccessivi livelli di estrogeni. L’uso di un AI e/o SERM, che inibisca la componente estrogenica di questa alterazione, come accennato in precedenza, è spesso sufficiente per mitigare la ginecomastia causata da Nandrolone e derivati. Il rialzo della Prolattina è un altra possibile conseguenza da monitorare per via di esami ematici.
L’uso di anti-prolattinici andrebbe presa in considerazione solo quando, attraverso appositi esami ematici, si è appurata una iperprolattinemia.
La soppressione dell’attività dell’Assen HPT con questo composto è piuttosto marcata. Infatti, è risaputo che i progestinici rientrano nella categoria dei composti con attività soppressiva delle gonadotropine.[11]
Tamoxifene
Nota: l’uso del Tamoxifene (Nolvadex) potrebbe risultare controproducente, soprattutto nel lungo periodo, dal momento che potrebbe effettivamente portare ad un aumento dei recettori del progesterone.[12]
Il lato positivo dell’essere un 19-norsteroide si manifesta con la buona affinità di legame con i recettori androgeni, cosa che è positivamente correlata con la lipolisi (per via del legame con i AR sull’adipocita).[13]
I soggetti con dolori articolari che utilizzano il Norethandrolone possono sperimentare una riduzione del sintomo similmente a quanto osservato con l’uso di Nandrolone. Anche per questa ragione, in passato il Nilevar era stato soprannominato “Oral Deca”. Ovviamente, l’uso sul lungo termine del Norethandrolone, per via della sua metilazione in C17, anche per tale fine è caldamente sconsigliato. Comunque, data la sua attività positiva a livello articolare, il suo uso ha visto una certa diffusione anche nel PowerLifting. Nel BodyBuilding, anche per tale caratteristica divenne una scelta inseribile con vantaggio durante cicli di “Bulk”
A differenza del Nandrolone, i cui metaboliti sono rilevabili nelle urine fino a 18 mesi dalla sospensione della somministrazione, il Norethandrolone presenta una rilevabilità d’uso di circa 5 mesi.
Le linee guida di prescrizione per il Norethandrolone (deperimento muscolare [14], pazienti con gravi ustioni, grave trauma e alcune forme di anemia aplastica [2]) indicano un dosaggio giornaliero pari a 20 o 30mg. La somministrazione indicata consiste in uno schema intermittente composto da 12 settimane d’uso seguite da un mese di pausa a seguito del quale può essere riavviato il trattamento. Il Norethandrolone è stato studiato per l’uso come contraccettivo ormonale maschile.[15][16][17] Nelle indicazioni cliniche non vi è menzione di modalità specifiche per l’uso nelle donne. Il dosaggio usuale in ambito sportivo è compreso tra i 20mg e i 40 mg al giorno, anche se il dosaggio massimo generalmente utilizzato risulta essere di 50mg, suddiviso in due assunzioni distanziate da circa 12h, assunte per non più di 6-8 settimane. Le atlete utilizzano dosi pari a 5-10mg/die suddivise sempre in due assunzioni distanziate da circa 12h. Il tempo tra una somministrazione e la successiva è basato sull’emivita della molecola (appunto 12h). Tale timing garantisce il mantenimento di livelli ematici della molecola stabili nelle 24h.
Essendo il Norethandrolone un AAS aromatizzabile e progestinico può causare ritenzione idrica, accumulo di grasso e ginecomastia, in specie se il fattore estrogenico non viene adeguatamente gestito. Queste caratteristiche lo rendono un AAS favorevole da utilizzare durante un ciclo “Bulk” o pre-gara se si è PowerLifter.
Sebbene negli ultimi anni sembra essere tornata di moda tra i culturisti, sempre a causa di quel compulsivo desiderio di provare nuove molecole (sebbene di “nuovo”, in questo caso, non vi sia nulla), la sua reperibilità è molto limitata. Come già detto, è attualmente presente nel mercato farmaceutico francese (venduto in compresse da 10mg). Nel mercato nero, la presenza di questo AAS è minima e spesso prodotti con indicazione in etichetta riportante la presenza di questa molecola in realtà ne contengono un altra, sebbene i costi di produzione siano bassi.
In conclusione, si tratta di una molecola non semplice da gestire, dall’applicabilità limitata e dalla difficile reperibilità. Non rappresenta di certo una molecola dai vantaggi superiori rispetto al Methandrostenolone o all’Oxymetholone, entrambe molecole applicabili in contesti simili a quelli nei quali l’uso del Norethandrolone è più indicato.
Gabriel Bellizzi
Riferimenti:
1- J. Elks (14 November 2014). The Dictionary of Drugs: Chemical Data: Chemical Data, Structures and Bibliographies. Springer. pp. 885–.
2- William Llewellyn (2011). Anabolics. Molecular Nutrition Llc. pp. 575–583.
4- Springer-Verlag. 27 November 2013. pp. 13, 282–283. ISBN 978-3-642-99941-3.
5- Bergink EW, Geelen JA, Turpijn EW (1985). “Metabolism and receptor binding of nandrolone and testosterone under in vitro and in vivo conditions”. Acta Endocrinol Suppl (Copenh). 271: 31–7.
6- Boschann HW (July 1958). “Observations of the role of progestational agents in human gynecologic disorders and pregnancy complications”. Ann. N. Y. Acad. Sci. 71 (5): 727–52.
7- Steroids. 1992 Dec;57(12):624-30.
8- J Reprod Fertil. 1966 Dec;12(3):489-99
9- Contraception. 1975 Feb;11(2):193-207
10- Lancet. 1958 Oct 25;2(7052):885-6
11- Clin Endocrinol (Oxf) 2003 Apr;58(4):506-12
12- Gynecol Oncol. 1999 Mar;72(3):331-6
13- Xu X, et al. “The effects of androgens on the regulation of lipolysis in adipose precursor cells.” Endocrinology 1990 Feb;126(2):1229
14- Walter Sneader (23 June 2005). Drug Discovery: A History. John Wiley & Sons. pp. 206–. ISBN 978-0-471-89979-2.
15- Schearer, S. Bruce; Alvarez-Sanchez, Francisco; Anselmo, Jose; Brenner, Paul; Coutinho, Elsimar; Latham-Faundes, Anibal; Frick, Julian; Heinild, Bent; Johansson, Elof D. B. (1978). “Hormonal Contraception for Men”. International Journal of Andrology. 1 (s2b): 680–712.
16- Neumann F, Diallo FA, Hasan SH, Schenck B, Traore I (1976). “The influence of pharmaceutical compounds on male fertility”. Andrologia. 8 (3): 203–35.
17- Brenner PF, Bernstein GS, Roy S, Jecht EW, Mishell DR (February 1975). “Administration of norethandrolone and testosterone as a contraceptive agent for men”. Contraception. 11 (2): 193–207.
Androgenico: 78-254
Anabolico: 107 (valore sovrastimato)
Standard: Testosterone
Nome Chimico: 17α-Methyl-4,5α-dihydrotestosterone
Attività estrogenica: nessuna
Attività Progestinica: nessuna
Aromatizzazione: no
Il Mestanolone, noto anche come 17α-methyl-4,5α-Diidrotestosterone (17α-methyl-DHT) o 17α-methyl-5α-androstan-17β-ol-3-one, è uno steroide androgeno anabolizzante (AAS), commercializzato in forma orale sotto il nome di Androstalone e Ermalone, ad oggi per lo più in disuso in ambito medico.[1][2][3][4]
Il Mestanolone è uno steroide androstano sintetico e un derivato del Diidrotestosterone (DHT) metilato in posizione C17α. [1][4] Infatti, differisce dal DHT solo per la presenza del gruppo metile nella posizione C17α.[1][4] Stretti parenti sintetici del Mestanolone includono Mesterolone (1α-Methyl-4,5α-dihydrotestosterone), Oxandrolone (2-oxa-17α-methyl-DHT), Oxymetholone (2-idrossimetilene-17α-methyl-DHT) e Stanozololo (un derivato del 17α-methyl-DHT (Mestanolone) con un anello pirazolico fuso con l’anello A.)[1][4]
Differenze nella struttura dello scheletro carbossilico tra Mestanolone (metilazione in C17α) e Mesterolone (metilazione in C1α).
Il Mestanolone fu sintetizzato per la prima volta nel 1935 insieme al Methyltestosterone e al Methandriolo.[5][6] È stato sviluppato dalla Roussel negli anni ’50 ed è stato introdotto per uso medico, con i marchi Androstalone ed Ermalone, intorno al 1960.[4][10][8] Venne inizialmente commercializzato in Germania.[4] Inizialmente si pensava che il farmaco fosse un potente agente anabolizzante, ma le ricerche successive hanno dimostrato che in realtà ha effetti anabolici relativamente deboli ed esplica principalmente azione androgena.[4] Il Mestanolone, insieme al molto più conosciuto 4-Chlorodehydromethyltestosterone (Oral Turinabol) è stato utilizzato come agente dopante negli atleti olimpionici della Germania Orientale all’interno di un programma di doping sponsorizzato dallo stato negli anni ’70 e ’80.[4] Il motivo del suo uso, come precedentemente accennato, era giustificato maggiormente dal suo valore androgeno piuttosto che su quello anabolizzante. Il suo uso garantiva un ottima risposta neuro steroidea con conseguente stimolazione del Sistema Nervoso Centrale e migliorata interazione neuromuscolare, con vantaggi nella velocità, nella forza, nell’aggressività, nella concentrazione, nella resistenza fisica e allo stress mentale.[4] Oggi, l’uso del Mestanolone è stato per lo più sospeso in medicina, sebbene sia ancora disponibile in Giappone.[2][3][4] Il Mestanolone era comunemente disponibile sotto forma di compresse sublinguali da 25mg (marchio Ermalone).[7]
Il Mestanolone è un AAS con effetti molto simili all’Androstanolone (diidrotestosterone; DHT) essendo praticamente una versione orale di quest’ultimo.[4] A causa dell’inattivazione da parte della 3α-idrossisteroide deidrogenasi (3α-HSD) nel muscolo scheletrico, il Mestanolone, sebbene dotato di una metilazione in posizione C-17 la quale ne migliora la stabilità del legame recettoriale, è descritto come un agente anabolizzante molto scarso, analogamente all’Androstanolone e al Mesterolone.[4] Poiché il Mestanolone è un composto 5α ridotto, non è un substrato soggetto all’enzima aromatasi e. quindi, non convertendo in estrogeno oltre a non possedere attività estrogenica intrinseca.[4] Inoltre, il farmaco non ha attività progestinica.[4] Come per gli altri AAS 17α-alchilati, il Mestanolone presenta un certo grado di epatotossicità.[4]
Differenze nella struttura dello scheletro carbossilico tra Diidrotestosterone e Mestanolone (aggiunta metilazione in C17α).
Come risaputo, gli AAS, a diverso grado di impatto, possono avere effetti deleteri sul colesterolo sierico. Il Mestanolone non è da meno presentando la tendenza a causare una riduzione delle concentrazioni di colesterolo HDL (“buono”) e un aumento delle concentrazioni di colesterolo LDL (“cattivo”), cosa che comporta uno sbilanciamento dell’equilibrio HDL/LDL che si traduce in un rischio maggiore di sviluppare arteriosclerosi. L’impatto relativo all’assunzione di un AAS nei confronti dei lipidi ematici dipende dalla dose, dalla via di somministrazione (per via orale o iniettabile), dal tipo di steroide (aromatizzabile o non aromatizzabile), e dal livello di resistenza al metabolismo epatico.
Essendo il Mestanolone un AAS con consistente attività androgenica, la soglia dei possibili forti effetti collaterali androgenici è generalmente alta ed è paragonabile a quella riscontrabile con altri composti come il Mesterolone. Per questa ragione, il suo uso in ambito femminile non è stato molto diffuso dal momento che poteva essere causa di severi effetti virilizzanti.
Quando l’uso del Mestanolone veniva applicato in campo sportivo, i dosaggi comunemente utilizzati erano mediamente tra i 10 ed i 20mg al giorno con un timing di somministrazione tra una dose e la successiva di 12 ore. I tempi di utilizzo rimanevano entro le 6-8 settimane onde evitare di creare un eccessivo stress epatico. Tra i bodybuilder, il Mestanolone era spesso inserito durante la preparazione alla gara vista la sua facile gestibilità non causando ritenzione idrica, non essendo soggetto ad aromatizzazione, esercitando una blanda azione anti-estrogenica (sia recettoriale che come ligando inibitorio dell’enzima Aromatasi) e possedendo una buona azione lipolitica (legame con i AR adipocitari).
Come già detto in precedenza, l’uso del Mestanolone è stato per lo più sospeso in ambito medico anche se rimane disponibile in Giappone.[2][3][4] Nel mercato nero è raramente reperibile per via della sua attuale e pressoché assente richiesta tra gli atleti.
Gabriel Bellizzi
Riferimenti:
1- J. Elks (14 November 2014). The Dictionary of Drugs: Chemical Data: Chemical Data, Structures and Bibliographies. Springer. pp. 775.
2- Index Nominum 2000: International Drug Directory. Taylor & Francis. 2000. pp. 655.
4- William Llewellyn (2017). Anabolics 11th. Molecular Nutrition Llc. p. 284-285-286.
5- Schänzer W (1996). “Metabolism of anabolic androgenic steroids”. Clin. Chem. 42 (7): 1001–20.
6- Ruzicka, L.; Goldberg, M. W.; Rosenberg, H. R. (1935). “Sexualhormone X. Herstellung des 17-Methyl-testosterons und anderer Androsten- und Androstanderivate. Zusammenhänge zwischen chemischer Konstitution und männlicher Hormonwirkung”. Helvetica Chimica Acta. 18 (1): 1487–1498.
7- H.-L. Krüskemper (22 October 2013). Anabolic Steroids. Elsevier. pp. 196.









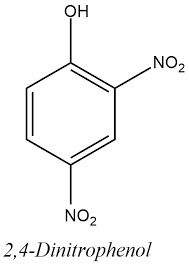
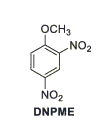
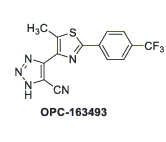

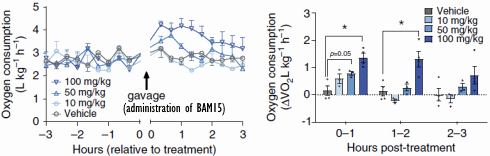
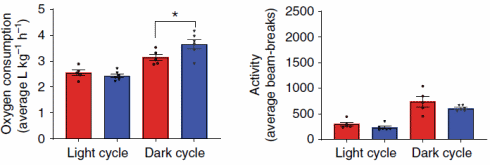
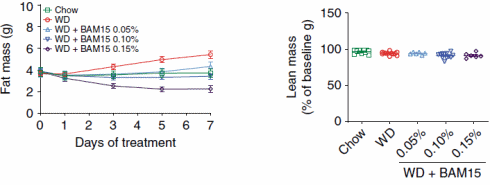

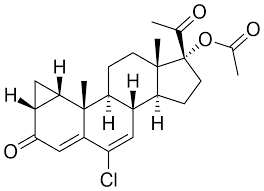

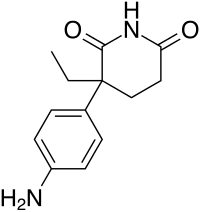

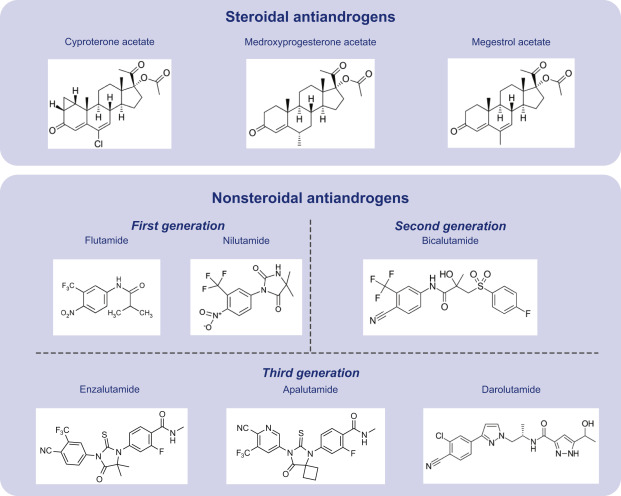
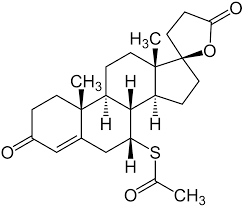
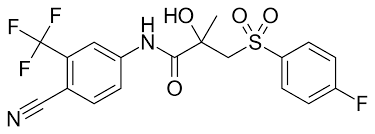
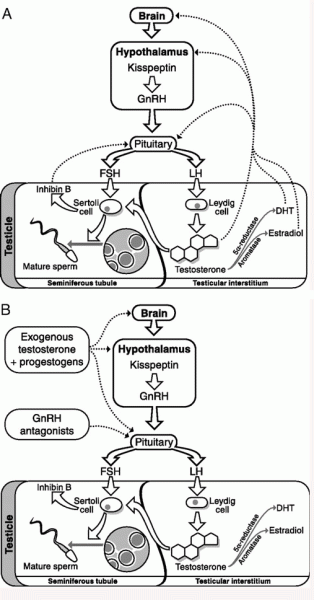



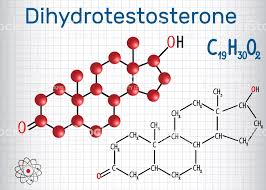

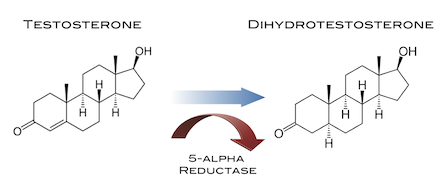
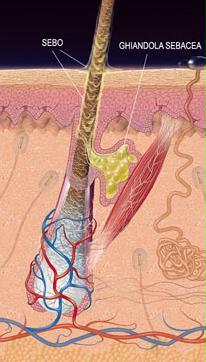

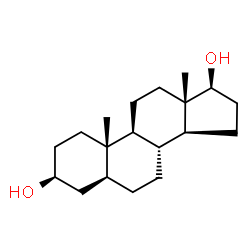

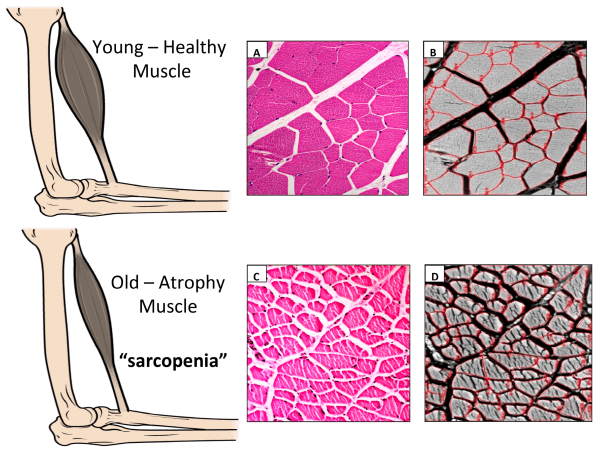

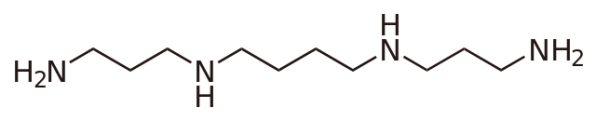
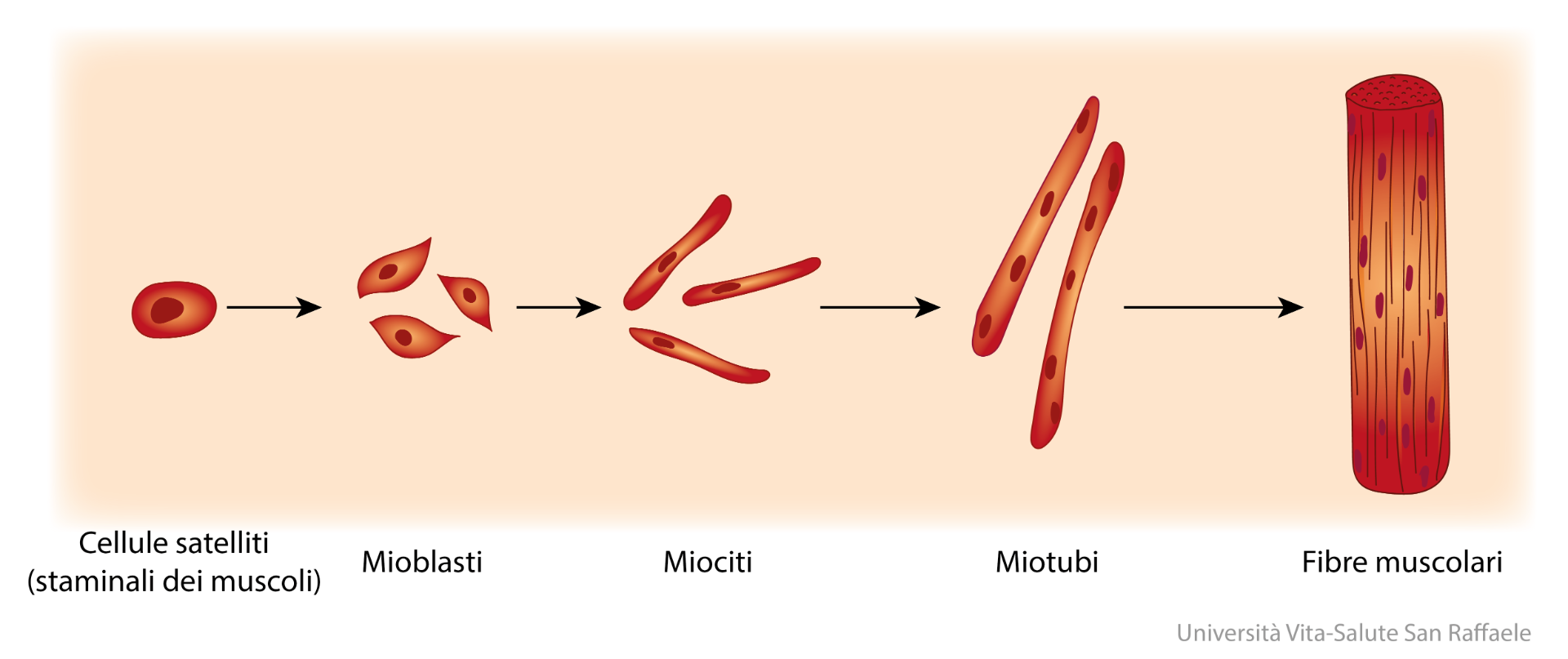

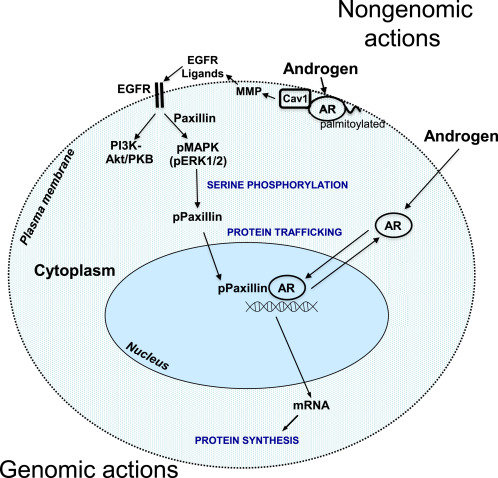





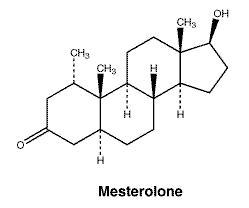
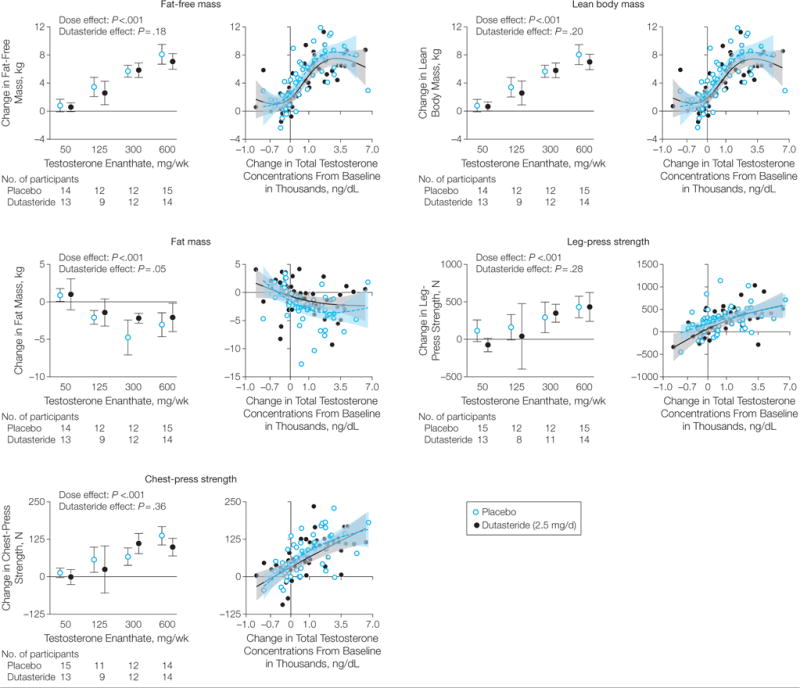



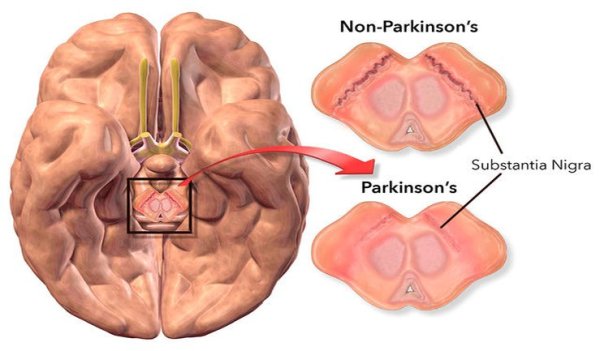





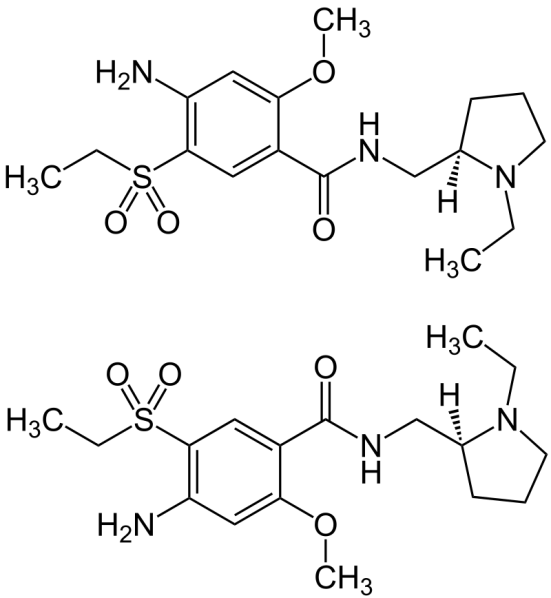

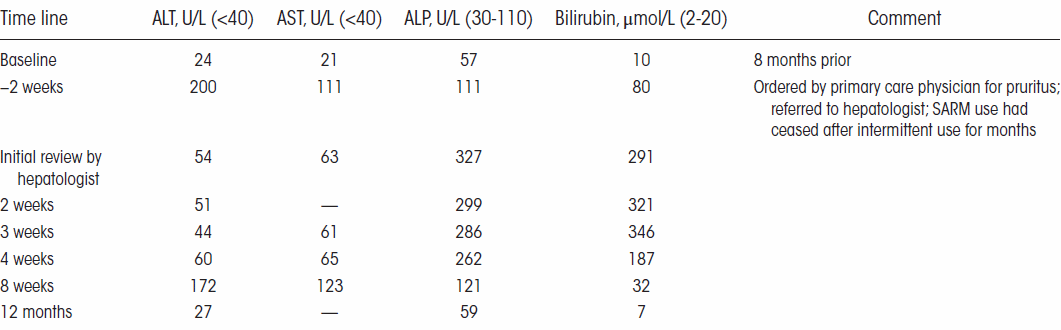

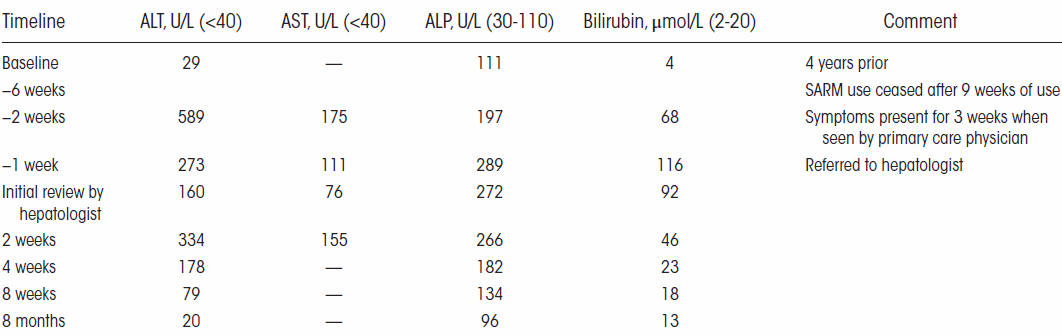
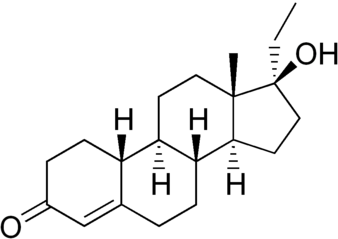
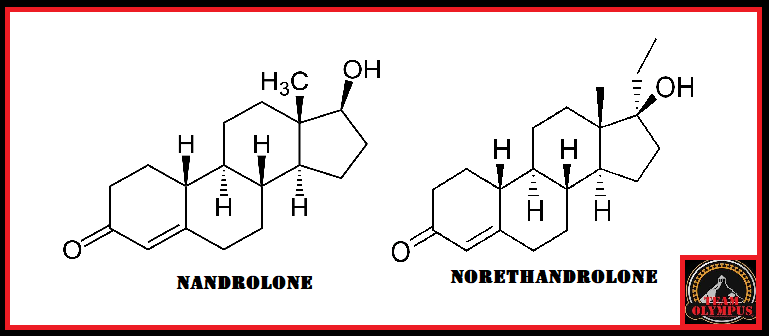


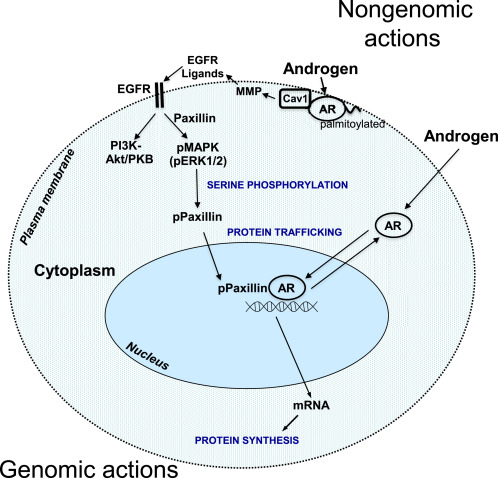

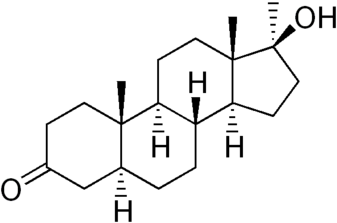

 to come agente dopante negli atleti olimpionici della Germania Orientale all’interno di un programma di doping sponsorizzato dallo stato negli anni ’70 e ’80.[4] Il motivo del suo uso, come precedentemente accennato, era giustificato maggiormente dal suo valore androgeno piuttosto che su quello anabolizzante. Il suo uso garantiva un ottima risposta neuro steroidea con conseguente stimolazione del Sistema Nervoso Centrale e migliorata interazione neuromuscolare, con vantaggi nella velocità, nella forza, nell’aggressività, nella concentrazione, nella resistenza fisica e allo stress mentale.[4] Oggi, l’uso del Mestanolone è stato per lo più sospeso in medicina, sebbene sia ancora disponibile in Giappone.[2][3][4] Il Mestanolone era comunemente disponibile sotto forma di compresse sublinguali da 25mg (marchio Ermalone).[7]
to come agente dopante negli atleti olimpionici della Germania Orientale all’interno di un programma di doping sponsorizzato dallo stato negli anni ’70 e ’80.[4] Il motivo del suo uso, come precedentemente accennato, era giustificato maggiormente dal suo valore androgeno piuttosto che su quello anabolizzante. Il suo uso garantiva un ottima risposta neuro steroidea con conseguente stimolazione del Sistema Nervoso Centrale e migliorata interazione neuromuscolare, con vantaggi nella velocità, nella forza, nell’aggressività, nella concentrazione, nella resistenza fisica e allo stress mentale.[4] Oggi, l’uso del Mestanolone è stato per lo più sospeso in medicina, sebbene sia ancora disponibile in Giappone.[2][3][4] Il Mestanolone era comunemente disponibile sotto forma di compresse sublinguali da 25mg (marchio Ermalone).[7]