*Nota per il lettore: la tesi di seguito esposta si affianca a quanto già ipotizzato dal “web writer”, nonché coach, autore e ricercatore, Type-IIx di MesoRx .
Introduzione:
Abbiamo imparato che il Boldenone, con tutta probabilità, ha una funzione di “ormone esca” per l’enzima Aromatasi. Sappiamo però che, probabilmente, la sua conversione in estrogeno lo vede convertirsi prevalentemente in Estrone [E1] e non in Estradiolo [E2]. Sappiamo che l’Estrone può convertirsi in Estradiolo (e viceversa) ma che il tasso in cui ciò avviene è molto basso. Siamo a conoscenza del fatto che l’E1 è un estrogeno molto meno potente dell’E2 e, come tale, è un estrogeno relativamente debole.[Kuhl H (August 2005), Escande A et al. (May 2006), Ruggiero RJ, Likis FE (2002)] Secondo uno studio, le affinità di legame relative dell’E1 per l’ERα e l’ERβ umani erano rispettivamente il 4,0% e il 3,5% di quelle dell’E2, e le capacità transazionali relative dell’E1 all’ERα e all’ERβ erano rispettivamente il 2,6% e il 4,3% di quelle dell’E2. [ Escande A et al. (May 2006)] In accordo, l’attività estrogenica dell’Estrone è stata riportata a circa il 4% di quella dell’Estradiolo.[Kuhl H (August 2005)] Non sicuramente una caratteristica favorevole per l’uso di una molecola senza la presenza di una base di Testosterone e/o hCG.


Conosciamo molto bene anche il Methenolone che, come derivato del DHT, non è soggetto ad aromatizzazione e quindi non ha la propensione a produrre effetti collaterali estrogenici come la ginecomastia.[William Llewellyn (2011). Anabolics] Come AAS, il Methenolone è antigonadotropo e esercita una soppressione dell’Asse HPT causando ipogonadismo reversibile e infertilità.[van Breda E et al. (Apr 2003)] Essendo un derivato del DHT conserva alcune caratteristiche antiestrogeniche, sebbene esse siano inferiori a quelle osservate con altre molecole simili come il Drostanolone. Queste proprietà, in un ambiente già predisposto a carenza di E2 [vedi mancanza di una base di Testosterone, mancato utilizzo di hCG e/o dosi sufficienti di questa, presenza di una molecola con marcati tassi di conversione in E1] non fanno altro che portare ad effetti avversi tipici dell’ipoestrogenemia [vedi, ad esempio, letargia, debolezza, dolori articolari, bassa libido, difficoltà a raggiungere e mantenere l’erezione ecc…].


Magari avete esperienza nell’uso di Boldenone Undecilenato, di Methenolone Enantato, o forse anche delle due molecole in combinazione ( magari con altri AAS). Forse potrete aver visto riportati i feedback degli utilizzatori in qualche forum in rete, o potreste anche essere a conoscenza di qualcuno che ha avuto effetti completamente diversi dai vostri con l’uso degli stessi farmaci. Nel primo caso (testimonianze su internet), avete, forse, ritenuto che questi utilizzatori si siano probabilmente somministrati prodotti non contenenti le suddette molecole (sperando di non essere voi gli interessati da ciò!). Nel secondo caso, in cui qualcuno che conoscete bene e capite che non ha alcuna motivazione per cui mentire e che sta usando AAS indubbiamente autentici (ad esempio, autenticati da HP/LC) vi riferisce allo stesso modo effetti completamente diversi da quelli da voi riscontrati.
Ma come stanno le cose? – come possono persone diverse sperimentare effetti così marcatamente diversi, persino opposti, dalla stessa molecola (o dalle stesse molecole) a dosi simili?
Non resta che:
- Affrontare questa domanda, in modo rigoroso, per rivelarci ciò che non era immediatamente evidente e, auspicabilmente, imparare alcuni fatti preziosi come risultato.
- Fornire soluzioni a coloro che sperimentano sintomi intollerabili di bassa estrogenicità come conseguenza dell’uso non medico di AAS.
Tesi
Teoria delle potenze estrogeniche dipendenti dalla molecola (per-AAS) e individualizzate (per utilizzatore):
Gli effetti di ogni AAS sull’estrogenicità (effetti associati all’attivazione di ER- α e β) dipendono da fattori dipendenti dalla molecola (per-AAS) e individualizzati (per-utilizzatore) che determinano sia
A. i livelli ematici effettivi che
B. gli effetti a livello tissutale dei prodotti aromatici di ogni AAS.
I prodotti aromatici consequenziali ai processi biochimici degli AAS vanno da quelli nulli (cioè non aromatizzabili), all’E1 (Estrone), un estrogeno debole, all’E2 (Estradiolo), un estrogeno potente (il più potente tra quelli endogeni) di cui tutti i lettori conoscono almeno l’esistenza e che è associato ai classici effetti estrogenici (sia che l’E2 sia “crashato” o meno), fino agli estrogeni non endogeni e altamente potenti come il 7α-metilestradiolo (il prodotto aromatico notevolmente potente del MENT, o anche noto come Trestolone).

Gli effetti di ciascun AAS (alla sua dose e durata) e dei suoi prodotti aromatici (alle loro concentrazioni e durate) determinano l’Androgeno/Estrogeno ratio (A/E), un indicatore degli effetti sistemici generali degli AAS (diretti e collaterali); ad esempio, ginecomastia. Il “braccio” androgeno del rapporto A/E è il prodotto della potenza dell’AAS di attivare l’AR alla sua area sotto la curva (AUC), come nmol×h/L. Il “braccio” estrogenico del rapporto A/E ha due aspetti: effetti estrogenici e antiestrogenici. Per quanto riguarda gli effetti estrogenici, questi sono il prodotto della concentrazione e della durata (AUC come nmol×h/L) dei prodotti aromatici (cioè gli estrogeni) e delle loro capacità di attivare ER- α e β. Reciprocamente, gli effetti antiestrogenici, che sono effetti intrinseci della classe degli AAS ben consolidati nell’uomo e negli animali, derivano dagli effetti ipofisari (cioè antigonadotropi) e tissutali locali (ad esempio, impediscono l’assorbimento degli estrogeni) degli AAS, che si ricollegano al “braccio” degli androgeni.
Gli effetti individualizzati (per utilizzatore) degli AAS sull’estrogenicità dipendono in gran parte da tre (3) fattori ereditabili discreti (cioè, il risultato del proprio fenotipo genetico) che sono soggetti a un’ampia variazione interindividuale (differenze tra utilizzatori): il profilo ormonale legante¹, l’espressione dell’isozima 17β-HSD e l’espressione dell’Aromatasi³. In primo luogo, il profilo ormonale legante dell’utilizzatore (cioè le attività di SHBG, albumina, α₁ glicoproteina acida, globulina legante i corticosteroidi) determina le attività di E1/E2 liberi (estrogeni liberi) e il rapporto E1/E2 liberi:androgeni. In secondo luogo, questo profilo ormonale vincolante¹ interagisce con la velocità di aromatizzazione dell’AAS (Vmax) e la lunghezza della catena di esteri (cioè logP e idrofobicità) quando le concentrazioni del farmaco raggiungono lo stato stazionario, influenzando il gradiente di concentrazione degli estrogeni attivi (E1 ed E2 liberi) poiché l’esterasi libera l’ormone progenitore dal profarmaco mediante idrolisi attiva nel sangue intero [4]. In terzo luogo, l’espressione dell’isoenzima 17β-HSD dell’utilizzatore determina il flusso netto di E1 ( estrogeno debole) rispetto all’E2 (estrogeno potente). Infine, l’espressione dell’Aromatasi dell’utilizzatore – in parte modificabile dall’autoregolazione della massa grassa – determina le concentrazioni assolute di estrogeni (E1 ed E2).
Nota: non lasciatevi dissuadere da questa presentazione così massiccia dei fattori che influenzano le concentrazioni di estrogeni nel sangue e le attività estrogeniche a livello tissutale, poiché non li abbiamo ancora analizzati. Continuate a leggere: questi fattori verranno illustrati man mano che procederemo.
Divergenza negli effetti estrogenici del Boldenone e del Methenolone; e i limiti dei livelli circolanti come indice della regolazione estrogenica tessuto-specifica:
Da referti di casi reali raccolti in rete, i cui soggetti proprietari hanno riferito l’uso di Boldenone e/o Methenolone.
Quattro (4) casi distinti in cui non è stata utilizzata alcuna molecola AI:
1- Innalzamento dell’E2 e dell’E1 sierici con 800mg di Boldenone Undecylenato, 600mg di Trenbolone e 300mg di Testosterone:


Boldenone Undecylenato (800mg) + Trenbolone Enantato (600mg) + Testosterone Enantato (300mg). Analisi del sangue: Estrone (E1): 1.352 pmol/L (Intervallo di riferimento: < 250 pmol/L), cioè 365,6 pg/mL (Molto alto).
2-Elevazioni dell’E2 sierica da 300 mg di Primo, 300 mg di Test:
*Methenolone Enantato + Testosterone Enantato analisi del sangue con E2 basso-moderato

3-Riduzione dell’E2 sotto la norma con 750mg di Testosterone Enantato, 500mg di Boldenone Undecylenato, 400mg di Methenolone Enantato:
*Testosterone Enantato + Boldenone Undecylenato + Methenolone Enantato, analisi del sangue E2

4-Mantenimento dell’E2 nella norma con 300mg di Testosterone Enantato, 180mg di Methenolone Enantato:
- Methenolone Enantato 180mg + Testosterone Enantato 300mg (rosso) vs. Testosterone Cypionato 150mg (blu)

Cosa concludere da questi dati?
Che trarre qualsiasi deduzione (per non parlare delle conclusioni) da questi risultati divergenti è un azzardo. Essi ci indicano una sola cosa: semplicemente che il Boldenone Undecylenato e/o il Methenolone (Enantato) sembrano abbassare l’estrogenicità riflessa dagli esami del sangue in alcuni casi e che per caratteristiche molecolari i meccanismi sono di natura sicuramente diversa.
I risultati di queste analisi del sangue illustrano i rischi di trarre inferenze o conclusioni dalle analisi del sangue di laboratorio postate in rete da diversi utilizzatori.
Dopo che il lettore avrà compreso i limiti dei livelli circolanti come indice della regolazione degli estrogeni specifica per i tessuti, verrà spiegato – nel modo più parsimonioso possibile rispetto alle prove e alla domanda – i fattori che influenzano le concentrazioni di estrogeni nel sangue e le attività estrogeniche a livello tissutale, al fine di “dare un’occhiata sotto il velo” a ciò che potrebbe guidare questa divergenza negli effetti estrogenici del Boldenone e del Methenolone.
Limiti dei livelli circolanti di estrogeni come indice della regolazione estrogenica tessuto-specifica:

AD: Androstenedione

La regolazione della produzione e del metabolismo degli estrogeni nei tessuti periferici è consentita dall’espressione locale dell’Aromatasi (CYP19A1), che converte gli androgeni in estrogeni (T ⇒ E2 e AD ⇒ E1 [l’E2 è l’estrogeno più prevalente nell’uomo; ciò può spiegare la maggiore tollerabilità del Boldenone nelle donne]). Gli estrogeni possono inoltre essere convertiti in solfati di estrogeni e in esteri acilici grassi di estrogeni tramite estrogeno solfotransferasi (EST) e acil-transferasi, rispettivamente. Infine, questi derivati degli estrogeni possono essere riconvertiti in estrogeni progenitori attraverso l’attività della solfatasi steroidea (sulfatasi) e della lipasi [10].
Il tessuto adiposo (AT) è particolarmente ricco di esteri acilici grassi degli estrogeni e, di conseguenza, possiede un ampio sistema di tamponamento che consente la regolazione locale della produzione e del metabolismo degli estrogeni… In particolare, in uno studio condotto su uomini obesi, le concentrazioni di esteri acilici grassi dell’E2 sono risultate correlate nel siero e nel grasso (Wang, et al., 2013) [10], indicando probabilmente che i livelli di estrogeni nel siero influenzano il contenuto di estrogeni immagazzinati nell’AT, ma la conversione in forme bioattive è regolata localmente [10].
Diversi studi clinici hanno dimostrato una dissociazione tra i livelli di estrogeni circolanti e quelli intra-adiposi, anche negli uomini (Blankenstein, et al., 1992; Belanger, et al., 2006; Deslypere, et al., 1985; Wang, et al., 2013) [10].
Fattori confondenti nei dati dell’estrogenicità di Boldenone e/o Methenolone:
In questo articolo si ragionerà sui fattori che determinano un fenomeno di apparenti contraddizioni multiple – per comprendere una realtà (cioè la nostra) in cui praticamente tutti dicono la “verità”, affermando di aver assunto quelli che ritengono essere gli stessi farmaci a dosi comparabili, eppure, sorprendentemente, l’estrogenicità (un fattore coinvolto nella tollerabilità) differisce tra gli individui. I fattori in gioco sono i seguenti:
- Le analisi ematiche di laboratorio possono non riflettere l’estrogenicità perché sono coinvolti meccanismi a livello tissutale (ad esempio, blocco dell’assorbimento degli estrogeni, attività intra- ed endocrina).
- Variazione interindividuale del profilo ormonale legante¹, dell’espressione dell’isoenzima 17β-HSD² e dell’espressione dell’Aromatasi³, per non parlare di fattori come l’espressione del ER (cioè la densità o il numero), ad esempio nel tessuto mammario (fattori che sono coinvolti nella tollerabilità).
- Incompletezza degli esami ematici di laboratorio in cui viene utilizzato il Boldenone (ad esempio, le misure di E2 nel siero sono insufficienti senza le misure di E1).
- Contraffazione o presenza di altra molecola nel prodotto (ad es. Methenolone viene sostituito da Testosterone o Drostanolone).
- Differenze nella lunghezza dell’estere (ad esempio, Boldenone Cypionato vs. Undecylenato) che riflettono il logP: coefficiente di ripartizione e la lipofilia: polarità; profondità di iniezione (ad esempio, nello spazio sottocutaneo vs. intramuscolare profondo) e sito di somministrazione che differiscono nel flusso sanguigno e quindi nell’attività dell’esterasi, influenzando indirettamente il tasso di reazioni dell’Aromatasi.
- Le presunte autodichiarazioni dei professionisti del fitness che traggono un reddito dalla generazione di notizie sui media possono essere motivate da travisamenti e/o frodi al fine di aumentare gli introiti pubblicitari come minimo, se non per integrare le loro scoperte scintillanti e nuove nel loro portafoglio utilizzandole come insegna o segno distintivo, su cui il loro lavoro (ad esempio, video su YouTube, scritti) sarà identificato e distinto.
Fattori che influenzano le concentrazioni di estrogeni nel sangue e le attività estrogeniche a livello tissutale:
Fattori dipendenti dalle molecole (Per-AAS)
- Prodotti aromatici e loro capacità di attivare ER- α e β.
a) Boldenone =[Aromatasi]=> E1 (Estrone, un estrogeno debole, 2% di potenza ER-α rispetto all’E2) ed E2 (Estradiolo, il 17β-OH lo rende 50 volte più potente dell’E2) {aromatizza in E1 ed E2}.
b) Methenolone =X[Aromatasi] {non aromatizza}, quindi non supera:
- Effetti antiestrogenici che sono effetti di classe degli AAS, specie nei DHT derivati:
a) inibizione delle gonadotropine secrete dall’ipofisi (che riducono indirettamente gli estrogeni) e
b) blocco diretto dell’attività degli estrogeni a livello degli organi bersaglio, impedendo l’assorbimento degli estrogeni, ad esempio, nelle cellule sinoviali, causando sintomi di “articolazione secca e dolorante”. È questo l’effetto che rende il Methenolone [1], [2] – e prima che venisse sospeso – Drostanolone [3], così efficace per il cancro al seno metastatico resistente al trattamento.
- Boldenone Undecylenato: a) velocità di aromatizzazione (Vmax) ridotta rispetto al Boldenone libero.
Km: pari alla concentrazione del substrato (ascissa; valori dell’asse delle ascisse) quando la velocità è la metà della velocità massima (1/2Vmax; ordinata; valori dell’asse delle ordinate).
T: Testosterone
L’aromatizzazione è ostacolata (rispetto al T) per gli androsta-1,4-diene-3-oni (come il Boldenone; Undecylenato.), per cui procede lentamente [17].
T =[Aromatasi]=> E2, Κm = 1,83nM, secondo la cinetica di Michaelis-Menten [18].

Non conosciamo il Km per l’attività dell’Aromatasi in vivo rispetto al Boldenone Undecylenato. Sappiamo però che l’enzima Aromatasi è saturabile, per cui al di sopra di una certa dose, che dipende dall’espressione³ o dal numero di proteine dell’Aromatasi (e dal profilo ormonale di legame¹), tale dose non causerà ulteriori aumenti degli estrogeni attivi (E2 ed E1 liberi). Poiché il Boldenone Undecylenato è soggetto a un’aromatizzazione ostacolata, la sua velocità di reazione (Vmax) deve essere relativamente rallentata. Di conseguenza, la sua Km in vivo deve essere spostata verso destra (rispetto a quella di T/E2) e richiede concentrazioni maggiori di T per la saturazione dell’Aromatasi. Questo ci dice che, rispetto al T, sono necessarie dosi più elevate di Boldenone prima che l’Aromatasi si saturi (non è soggetto ad alcun aumento di E2 a dosi superiori al punto di saturazione).
Inoltre sappiamo anche che il 40% in più di Vmax dell’Aromatasi in rapporto al T negli uomini anziani rispetto a quelli giovani è stato praticamente interamente spiegato dalla massa grassa e dalle SHBG (cioè il profilo ormonale legato¹).[18] Poiché l’Aromatasi è espressa anche negli adipociti (cellule grasse), il cui numero è soggetto ad aumentare a causa della lipogenesi di nuove cellule grasse (adipociti), il mantenimento di una bassa percentuale di grasso corporeo per tutta la vita è un fattore importante che può essere controllato dal soggetto. È importante capire che le cellule adipose non vengono distrutte dalla restrizione calorica: l’aspetto visivo di una bassa percentuale di grasso corporeo dopo una dieta ipocalorica non riflette la perdita di numero di adipociti, ma solo la riduzione delle riserve di lipidi all’interno di tali cellule. Solo la lisazione o il congelamento (ad esempio, lisazione chimica come Kybella, CoolSculpting, mesoterapia ecc.) per la successiva rimozione attraverso le feci o la liposuzione (rimozione fisica) delle cellule di grasso distruggono effettivamente queste cellule, in modo tale che si verifichi una riduzione dell’aromatizzazione.

Fattori individuali (per utilizzatore):
- A seconda del profilo ormonale legato di un individuo¹, il rilascio più lento dal deposito per il Boldenone Undecylenato prima di raggiungere lo stato stazionario determinerà quasi certamente una riduzione dell’attività dell’Aromatasi.
- A seconda dell’espressione dell’isozima 17β-HSD di un individuo², il flusso netto di estrogeni potrebbe produrre E1 > E2 dopo la somministrazione di Boldenone Undecylenato, con il risultato che gli estrogeni prevalenti nella circolazione sanguigna sono molto più deboli rispetto all’E2.
- A seconda dell’espressione dell’Aromatasi³ di un individuo, la tollerabilità dell’estrogenicità da parte di androgeni aromatizzabili (ad esempio, il Boldenone) dipende in parte dal numero di Aromatasi.
Figura: Previsione del target molecolare del Methenolone (Primobolan/Rimobolan):

Nota: sebbene vi siano prove (Figura, sopra) che il Methenolone Enantato abbia un’alta probabilità di legare l’Aromatasi (citocromo P450 19A1) (probabilità dell’88%) – la cui inibizione competitiva ridurrebbe l’E2 sierica – e una bassa probabilità di legare la 17β-HSD1, la 17β-HSD2 e la 17β-HSD3 – non farò supposizioni su questi potenziali meccanismi per gli effetti sull’estrogenicità, perché il modello semplicemente non ne ha bisogno. Inoltre, non sappiamo quale modalità di legame utilizzerebbe né la sua rilevanza biologica. È dominio esclusivo della “bro-science” impegnarsi in queste speculazioni sconsiderate.
Fattori individuali per utilizzatore
Fattori individuali (definizioni):
¹: profilo ormonale legato: Le attività di SHBG, albumina, α₁ glicoproteina acida e globulina legante i corticosteroidi influenzano le porzioni inattive legate rispetto a quelle attive libere di androgeni ed estrogeni.
²: Espressione dell’isoenzima 17β-HSD: Il numero relativo di isozimi 17β-HSD di tipo 1 e di tipo 2 determina le proporzioni relative e i livelli assoluti di E2 ed E1 circolanti, rispettivamente.
³: Espressione dell’Aromatasi: Il numero assoluto di proteine Aromatasi determina i livelli di prodotti aromatici (cioè estrogeni).
17β-HSD

La 17β-HSD è un gruppo di enzimi che interconvertono gli steroidi (estrogeni, androgeni) con un gruppo cheto in posizione 17 (ad esempio, E1, AD) e quelli con un gruppo idrossi nella stessa posizione (ad esempio, E2, T).
Tutti gli enzimi 17β-HSD catalizzano l’ossidazione o la riduzione del carbonio in posizione 17 nel substrato steroideo:
preferenze diverse per il substrato (ad esempio, E1, E2, T, 3β-diolo, DHT)
funzioni fisiologiche distinte (Jansson, 2009) [15].
Nell’uomo sono state identificate dodici (12) 17β-HSD… alcune catalizzano reazioni di substrati non steroidei… se il substrato è steroideo, la reazione è di ossidazione o riduzione, a seconda del cofattore e della localizzazione cellulare [16].
Per evitare di sovraccaricare il lettore con informazioni troppo complesse, questo lavoro si concentrerà sulle prime due (2) isoforme principali della 17β-HSD (tipo 1 e tipo 2).
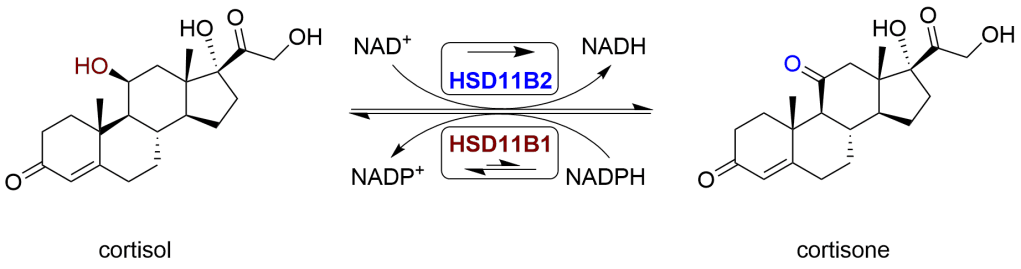
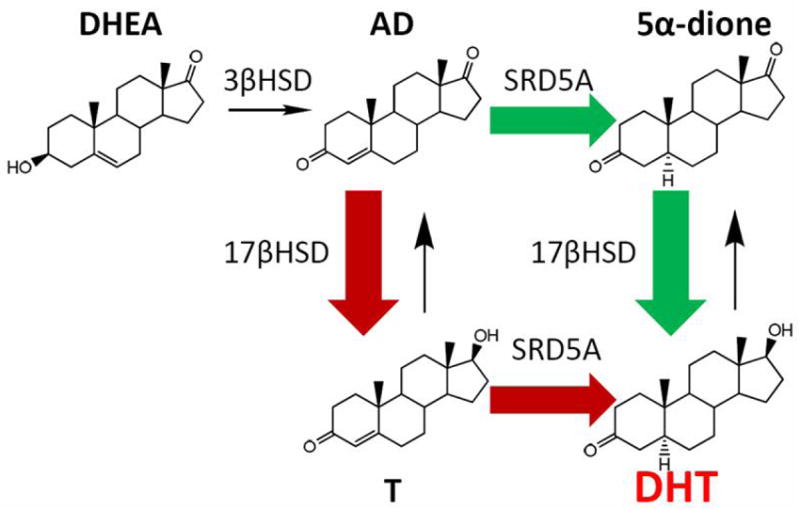
La 17β-HSD1 (tipo 1), sotto il controllo del gene A1-Q327, catalizza la riduzione degli steroidi (estrogeni, androgeni) con un 17-cheto a uno che ha un gruppo idrossi nella stessa posizione. Quindi, da E1 (Estrone) =[17β-HSD1]=> E2 (Estradiolo), e da AD =[17β-HSD1]=> T.
L’espressione della 17β-HSD1 è correlata positivamente all’attivazione dell’E1 e ai livelli di E2 [15] e la sua inibizione li riduce. Inibizione della 17β-HSD1 => ↓E2 [16].
La 17β-HSD2 (tipo 2) inverte le reazioni della 17β-HSD1 (cioè, E2 =[17β-HSD2]=> E1 e E3 =[17β-HSD2]=> 16α-idrossiestrone) e converte il T =[17β-HSD2]=> AD (Androstenedione), ossidando il 17-idrossile per sostituire il C-17 con un gruppo 17-cheto.
La sovraespressione relativa della 17β-HSD2 e la sottoespressione della 17β-HSD1 producono l’effetto netto di un aumento dell’Estrone (E1), soggetto a variazioni interindividuali nel metabolismo.
Aromatasi

L’enzima Aromatasi, sotto il controllo del gene CYP19A1, è presente in vari tessuti dell’uomo… tra cui gonadi, cervello e tessuto adiposo (4) [20].
L’aromatasi è l’unico enzima umano in grado di aromatizzare l’anello A degli steroidi, convertendo così gli androgeni in estrogeni [21].
Questo enzima scinde il 19-metile dall’AAS e riconfigura l’anello A dello steroide in modo da formare tre doppi legami alternati. Questa configurazione dell’anello A è descritta come aromatica (pertanto, questo processo è definito aromatizzazione).
Negli uomini, esiste una variazione della popolazione nell’altezza e nell’espressione del gene dell’Aromatasi [22]. Questo ha senso perché gli estrogeni prodotti dall’aromatizzazione del T endogeno in E2 sono fondamentali per la crescita e il mantenimento delle ossa negli uomini.
Sintomi di bassa estrogenicità
- Articolazioni “secche” e doloranti (artralgia) – Gli estrogeni hanno naturalmente proprietà antinocicettive che potrebbero essere, da una prospettiva teleologica, una caratteristica di design per conferire alle donne la tolleranza al dolore durante il parto, quando i livelli di estrogeni sono naturalmente aumentati [8]. Si ritiene che ciò sia mediato da neuroni del midollo spinale contenenti oppioidi che esprimono ER (24) [8]. I dati sugli animali dimostrano che i topi ovariectomizzati presentano un turnover accelerato della cartilagine (25) che può contribuire alla riduzione dell’ammortizzazione articolare [8]. Gli estrogeni sopprimono la produzione di citochine infiammatorie, mentre una riduzione degli estrogeni aumenta i livelli di citochine infiammatorie come IL-1 e TNF-α (26)… Le cellule sinoviali esprimono l’Aromatasi e, quando questa catalizza la conversione dall’Androstenedione (AD) all’Estrone (E1) e all’Estradiolo (E2), l’espressione di IL-6 si riduce nell’articolazione (28) [8]. Pertanto, un basso livello di estrogeni, e di conseguenza di IA, può provocare un aumento relativo della produzione di IL-6, che notoriamente agisce come citochina pro- e anti-infiammatoria. È anche nota per essere uno dei mediatori chiave dell’aumento della perdita ossea nelle donne in post-menopausa (29) [8].
- Perdita ossea – Gli estrogeni svolgono un ruolo fondamentale nel prevenire la perdita di contenuto/densità minerale ossea. Sebbene gli androgeni abbiano effetti significativi sull’osso maschile, gli estrogeni sono più importanti per la crescita e il mantenimento dell’osso… L’E2 è essenziale per la normale mineralizzazione, massa e turnover dell’osso, ma non per la crescita lineare dell’osso negli uomini (648, 649) [9].
- Resistenza all’Insulina – Il metabolismo del glucosio per kg di muscolo è più alto del 45% nelle donne (756) (probabilmente mediato da ER-α) [9]. Negli uomini, gli effetti metabolici benefici del Testosterone sono mediati più dal suo prodotto aromatico (E2) che dagli androgeni (E2 > T nell’accumulo di ↓AT)… ~15% degli estrogeni circolanti deriva dalla sintesi e dalla secrezione testicolare (cellule di Leydig) e il resto dall’attività dell’Aromatasi periferica… [9].
- Aumento del grasso corporeo (↑AT; AT: tessuto adiposo) – Negli uomini, l’E2 regola le riserve di grasso corporeo > T. I topi maschi ERKO: Estrogen Receptor Knockout (ER null) hanno mostrato depositi di AT superiori del 100% a 9-12 mesi di età (invecchiati)… riflette sia l’iperplasia che l’ipertrofia degli adipociti (281) e si accompagna a intolleranza al glucosio e resistenza all’Insulina (IR) [9]. I topi maschi ERαKO presentano infiammazione del ↑AT, dimensioni degli adipociti e alterata tolleranza al glucosio [9].
- Disfunzioni sessuali – La segnalazione ER-α nell’uomo supporta: i dotti efferenti e le funzioni epididimali; il trasporto di ioni e il riassorbimento di H₂O necessari per sostenere il normale funzionamento degli spermatozoi (riproduzione maschile); il cervello, l’adipe, il muscolo scheletrico, le ossa, i tessuti cardiovascolari e immunitari [9].
Nota: mentre gli estrogeni esogeni causano patologie riproduttive maschili [9], gli estrogeni endogeni (a livelli normali di T) sono fondamentali per il funzionamento sessuale maschile.
- Ridotta reattività del muscolo scheletrico agli stimoli anabolici – Questa affermazione non è attualmente supportata dalle prove relative ai sintomi di bassa estrogenicità indotti dagli AAS. Nonostante sia un luogo comune tra i bodybuilder che l’uso di AI/SERM, attraverso l’azione antiestrogenica nel muscolo scheletrico, riduca l’anabolismo muscolare; o che l’E2 molto alto promuova l’anabolismo muscolare – queste affermazioni non sono supportate da alcuna prova reale (vale a dire, sottoposte a un design di studio rigoroso e a metodi probabilistici e statistici per distinguere causa, effetto e casualità). Ciò che è dimostrato è che la terapia estrogenica sostitutiva (HRT, in letteratura; diversa dalla TRT) aumenta la sintesi proteica muscolare (MPS) indotta dall’allenamento contro-resistenza (RT), ma a scapito della MPS basale (ad es, La sostituzione degli estrogeni nelle donne in post-menopausa riduce la MPS nelle 24 ore) [10]… Mentre le prove nei ruminanti (cioè nei bovini) supportano l’E2 esogeno + androgeni (ad esempio, impianti di Trenbolone Acetato), questo è, come la HRT (sostituzione degli estrogeni) nelle donne in post-menopausa, non analogo agli AAS negli uomini sani.
- Poiché le donne in post-menopausa sono invecchiate e in genere non ricorrono alla terapia ormonale sostitutiva (estrogeni) per periodi di anni dopo la cessazione delle mestruazioni, la semplice associazione tra bassi estrogeni e attenuata reattività agli stimoli anabolici è più probabilmente legata ad altri fattori legati all’età che non alla riduzione degli estrogeni (ad esempio, la diminuzione della capacità rigenerativa delle cellule satelliti e la diminuzione dell’espressione dell’mRNA di IGF-IEc nel muscolo scheletrico).
- Poiché i ruminanti non sperimentano un aumento dell’IGFBP-1 in risposta all’E2 esogeno come gli esseri umani [11], che riduce la disponibilità di IGF-I libero e scatena (endogenamente) la secrezione di GH tramite il ritiro del feedback, qualsiasi connessione estrogeno-anabolismo nel muscolo scheletrico umano è, nella migliore delle ipotesi, tenue e probabilmente un mero fattore terziario, legato invece al T endogeno e al processo di aromatizzazione (che aumenta l’IGF-I) piuttosto che al suo prodotto aromatico. Gli estrogeni (ad esempio, l’E2) aumentano in modo dose-dipendente l’IGFBP-1, motivo per cui le donne hanno livelli di GH endogeno molto più elevati ma livelli di IGF-I proporzionalmente più bassi rispetto agli uomini in base alla superficie corporea (una risposta ridotta al GH) [13], e per cui le donne che assumono contraccettivi ormonali (cioè estrogeni) devono titolare le dosi di rhGH per vedere i benefici sulla crescita e sul metabolismo, ad esempio nella carenza di Ormone della Crescita nell’adulto [14]. Nelle donne in premenopausa, l’Etinilestradiolo orale riduce i livelli di IGF-I fino a una media del 30% (24-27) [13].
I casi di Boldenone Undecylenato e Methenolone Enantato
L’uso di Methenolone Enantato e/o Boldenone Undecylenato può provocare sintomi di bassa estrogenicità, che possono (o meno) essere riflessi da concentrazioni di E2 inferiori alla norma.
Adattamento di Methenolone Enantato e/o Boldenone Undecylenato alla tesi qui esposta
Vedere Teoria delle potenze estrogeniche (modello teorico):
Ogni AAS influisce sul flusso netto di estrogenicità attraverso i suoi particolari effetti sulle concentrazioni di estrogeni nel sangue e sulle attività estrogeniche a livello tissutale nei seguenti modi:
Methenolone:

Il Methenolone, in quanto AAS non aromatizzabile, non converte in estrogeni. Di conseguenza, a dosi moderate/elevate, i suoi effetti sul flusso netto di estrogeni rispetto agli aspetti degli effetti dipendenti dal composto (per-AAS) saranno marcatamente anti-estrogenici – l’inibizione delle gonadotropine secrete dall’ipofisi (che riducono indirettamente gli estrogeni nell’uomo attraverso la soppressione della sintesi e della secrezione di T endogeno [steroidogenesi] da cui dipende la biosintesi dell’Estradiolo [E2] nell’uomo), e il blocco diretto dell’attività degli estrogeni a livello degli organi bersaglio, impedendo l’assorbimento degli estrogeni nelle cellule (ad es. g., cellule sinoviali, causando sintomi di “articolazione secca e dolorante”).
Boldenone:

Il Boldenone, rispetto al Testosterone, aromatizza maggiormente in Estrone (E1) e scarsamente in Estradiolo (E2) [5]. L’E1 è un estrogeno debole perché manca del gruppo 17β-OH dell’E2 e possiede appena il 2% della potenza dell’E2 nel transattivare l’ER-α [6]. Poiché l’espressione dell’isoenzima 17β-HSD² dell’individuo determina il flusso netto dell’equilibrio E1/E2, è particolarmente determinante nel caso degli effetti del Boldenone sul flusso netto di estrogenicità.
Il Boldenone è soggetto a una grande variazione interindividuale rispetto a tutti e tre i fattori enumerati (profilo ormonale legato¹, espressione dell’isozima 17β-HSD² ed espressione dell’Aromatasi³). La sua Vmax relativamente lenta (velocità di reazione dell’Aromatasi), l’aromatizzazione maggiore in E1 (un estrogeno debole) e minore in E2, le sue porzioni libere o legate e il numero assoluto di Aromatasi sono fattori che determinano un’ampia divergenza degli effetti del Boldenone sull’estrogenicità.
Gestione dell’estrogenicità
Per visualizzare il modo in cui l’utente dovrebbe approcciarsi alla gestione dell’estrogenicità si può ricorrere a un semplice modello: la curva a U inversa:

L’asse x è correlato all’attivazione ER a livello tissutale, che potrebbe non essere riflessa dalle concentrazioni di estrogeni nel sangue. L’asse y riflette la tollerabilità. L’area sotto la curva agli estremi (troppo bassa o troppo alta) è caratteristicamente intollerabile. La gestione dell’estrogenicità è quindi un “problema Goldilocks”. L’estrogenicità non può essere troppo bassa o troppo alta, ma deve essere “giusta” rispetto alla tollerabilità individuale.
La sezione che segue è di carattere pratico: un diagramma di flusso decisionale a cui l’utilizzatore può fare riferimento in caso di sospetta bassa estrogenicità (“crash E2”).
Pratica – Un diagramma di flusso del processo decisionale per affrontare la bassa estrogenicità derivante dall’uso di Boldenone e/o Methenolone:

Conclusioni:
L’estrogenicità (sintomi associati all’attivazione dell’ER) degli AAS è soggetta a effetti per-AAS e per utilizzatore. Il Methenolone, in quanto AAS non aromatizzabile e DHT derivato, agisce come antiestrogeno e androgeno. Il Boldenone è un composto interessante proprio per il fatto che è soggetto a effetti divergenti tra gli utilizzatori, che dipendono da fattori quali il profilo ormonale di legame¹, l’espressione dell’isoenzima 17β-HSD² e l’espressione dell’Aromatasi³. Le analisi del sangue di laboratorio spesso non sono sufficientemente precise per gli utilizzatori di AAS che cercano di capire l’estrogenicità a causa di fattori che includono gli effetti locali sui tessuti e le dissociazioni tra intra- ed endocrinologia. È per questo motivo che l’auto interpretazione delle analisi del sangue e il loro utilizzo per dettare il dosaggio e le pratiche dei farmaci ancillari (vedi SERM e/o AI) – che sono più spesso cattiva “bro-science” che medicina – piuttosto che rimanere semplicemente in sintonia con la tollerabilità di questi agenti e lavorare attraverso il diagramma di flusso presentato come necessario, porta il più delle volte a un frustrante gioco di “whack-a-mole” per gli utilizzatori di AAS.
Gabriel Bellizzi [CEO BioGenTech]
Riferimenti e fonti:
- Primo and/or EQ Symptoms of Low vs. High Estrogens, Explained, and a Practical Flowchart of Decisionmaking for Symptoms of Low Estrogens as a Result of Primo and/or EQ Use. By Type-IIx
[1] Suchowsky, GK, Junkmann, K. [Anabolic steroids and their side-effects]. Acta Endocrinol (Copenh). 1962 Jan;39:68-78. German. PMID: 13918121.
[2] Junkmann, K, Suchowsky, G. [Research on anabolic-active steroids]. Arzneimittelforschung. 1962 Mar;12:214-8. German. PMID: 14452833.
[3] Trams, G. (1977). Effect of drostanolone propionate on the binding of oestradiol and dihydrotestosterone by normal and malignant target tissues. European Journal of Cancer (1965), 13(2), 149–153. doi:10.1016/0014-2964(77)90193-1
[4] Kalicharan, R. W., Bout, M. R., Oussoren, C., and Vromans, H. (2016). Where does hydrolysis of nandrolone decanoate occur in the human body after release from an oil depot? International Journal of Pharmaceutics, 515(1-2), 721–728. doi:10.1016/j.ijpharm.2016.10.068
[5] Gual, C., Morato, T., Hayano, M., Gut, M., and Dorfman, R. I. (1962). Biosynthesis of Estrogens. Endocrinology, 71(6), 920–925. doi:10.1210/endo-71-6-920
[6] Houtman, C. J., Sterk, S. S., van de Heijning, M. P. M., Brouwer, A., Stephany, R. W., van der Burg, B., and Sonneveld, E. (2009). Detection of anabolic androgenic steroid abuse in doping control using mammalian reporter gene bioassays. Analytica Chimica Acta, 637(1-2), 247–258. doi:10.1016/j.aca.2008.09.037
[7] Thomas MP, Potter BV. The structural biology of oestrogen metabolism. J Steroid Biochem Mol Biol. 2013;137:27-49. doi:10.1016/j.jsbmb.2012.12.014
[8] P. Niravath, Aromatase inhibitor-induced arthralgia: a review, Annals of Oncology, Volume 24, Issue 6, 2013, Pages 1443-1449, ISSN 0923-7534, doi.org/10.1093/annonc/mdt037.
[9] Cooke PS, Nanjappa MK, Ko C, Prins GS, Hess RA. Estrogens in Male Physiology. Physiol Rev. 2017 Jul 1;97(3):995-1043. doi:10.1152/physrev.00018.2016.
[10] Rubinow KB. Estrogens and Body Weight Regulation in Men. Adv Exp Med Biol. 2017;1043:285-313. doi:10.1007/978-3-319-70178-3_14
[11] Chidi-Ogbolu N, Baar K. Effect of Estrogen on Musculoskeletal Performance and Injury Risk. Front Physiol. 2019;9:1834. Published 2019 Jan 15. doi:10.3389/fphys.2018.01834
[12] Veldhuis, J. D., and Bowers, C. Y. (2003). Human GH pulsatility: An ensemble property regulated by age and gender. Journal of Endocrinological Investigation, 26(9), 799–813. doi:10.1007/bf03345229
[13] Chanson, P., Arnoux, A., Mavromati, M., Brailly-Tabard, S., Massart, C., … Young, J. (2016). Reference Values for IGF-I Serum Concentrations: Comparison of Six Immunoassays. The Journal of Clinical Endocrinology and Metabolism, 101(9), 3450–3458. doi:10.1210/jc.2016-1257
[14] Cook, D. M., Ludlam, W. H., and Cook, M. B. (1999). Route of Estrogen Administration Helps to Determine Growth Hormone (GH) Replacement Dose in GH-Deficient Adults1. The Journal of Clinical Endocrinology and Metabolism, 84(11), 3956–3960. doi:10.1210/jcem.84.11.6113
[15] He, W., Gauri, M., Li, T., Wang, R., and Lin, S.-X. (2016). Current knowledge of the multifunctional 17β-hydroxysteroid dehydrogenase type 1 (HSD17B1). Gene, 588(1), 54–61. doi:10.1016/j.gene.2016.04.031
[16] Thomas MP, Potter BV. The structural biology of oestrogen metabolism. J Steroid Biochem Mol Biol. 2013;137:27-49. doi:10.1016/j.jsbmb.2012.12.014
[17] Gual, C., Morato, T., Hayano, M., Gut, M., and Dorfman, R. I. (1962). Biosynthesis of Estrogens. Endocrinology, 71(6), 920–925. doi:10.1210/endo-71-6-920
[18] Lakshman, K. M., Kaplan, B., Travison, T. G., Basaria, S., Knapp, P. E., Singh, A. B., … Bhasin, S. (2010). The Effects of Injected Testosterone Dose and Age on the Conversion of Testosterone to Estradiol and Dihydrotestosterone in Young and Older Men. The Journal of Clinical Endocrinology and Metabolism, 95(8), 3955–3964. doi:10.1210/jc.2010-0102
[19] Fouad Mansour M, Pelletier M, Boulet MM, Mayrand D, Brochu G, Lebel S, Poirier D, Fradette J, Cianflone K, Luu-The V, Tchernof A. Oxidative activity of 17β-hydroxysteroid dehydrogenase on testosterone in male abdominal adipose tissues and cellular localization of 17β-HSD type 2. Mol Cell Endocrinol. 2015 Oct 15;414:168-76. doi: 10.1016/j.mce.2015.06.016. Epub 2015 Jun 26.
[20] Attardi BJ, Pham TC, Radler LC, Burgenson J, Hild SA, Reel JR. Dimethandrolone (7alpha,11beta-dimethyl-19-nortestosterone) and 11beta-methyl-19-nortestosterone are not converted to aromatic A-ring products in the presence of recombinant human aromatase. J Steroid Biochem Mol Biol. 2008;110(3-5):214-222. doi:10.1016/j.jsbmb.2007.11.009
[21] Thomas MP, Potter BV. The structural biology of oestrogen metabolism. J Steroid Biochem Mol Biol. 2013;137:27-49. doi:10.1016/j.jsbmb.2012.12.014
[22] Ellis, J. A., Stebbing, M., and Harrap, S. B. (2001). Significant Population Variation in Adult Male Height Associated with the Y Chromosome and the Aromatase Gene. The Journal of Clinical Endocrinology and Metabolism, 86(9), 4147–4150. doi:10.1210/jcem.86.9.7875