Introduzione:
Gli effetti regolatori degli androgeni sull’ematopoiesi sono stati riconosciuti fin dall’inizio del XX secolo. La castrazione dei ratti maschi causa anemia (1) che è reversibile dopo il trattamento con androgeni (2). Un classico studio clinico di Vahlquist ha fornito prove indirette che gli uomini hanno livelli di ematocrito (Hct) intrinsecamente più elevati rispetto alle donne; è stato osservato che le donne in pre-menopausa non hanno livelli di Hct più elevati rispetto alle donne in post-menopausa, mentre hanno un aumento dell’Hct in risposta all’integrazione di ferro (3). Dati aneddotici provenienti da atleti agonisti suggeriscono che l’abuso di androgeni può migliorare le prestazioni, in parte attraverso l’aumento del VO2max (capacità di trasporto di ossigeno nel sangue mediata dall’Hb), anche se a spese di un aumento del rischio di trombosi arteriosa e venosa (4). Allo stesso modo, le donne affette da endocrinopatie iperandrogeniche, come l’iperplasia surrenale congenita e la sindrome di Cushing, possono presentare un’eritrocitosi relativa (5, 6). I risultati storici di cui sopra confermano che gli androgeni stimolano l’eritropoiesi della midollare.
È noto che i parametri ematologici e l’eritropoiesi sono influenzati dall’uso di steroidi androgeni anabolizzanti (AAS), anche se poco si sa in relazione a dosi sovra-fisiologiche di questa classe di farmaci sui dettagli meccanicistici di tale processo.
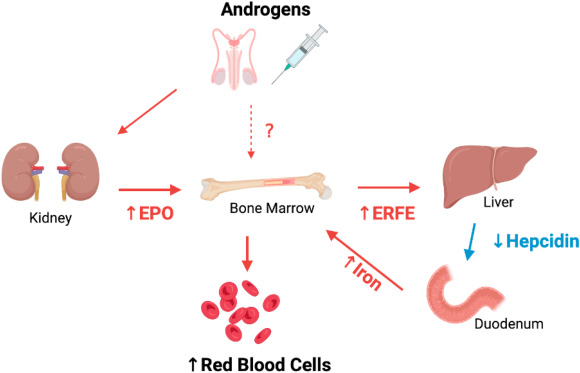
Si è visto che la somministrazione di Testosterone stimola la produzione di EPO, cosa che è stata osservata sia negli uomini ipogonadici che nei volontari sani di sesso maschile, mentre non esiste alcuna correlazione tra le concentrazioni endogene di Testosterone e di EPO durante la pubertà maschile.[7] Il meccanismo alla base dell’aumento dell’EPO indotto dagli androgeni non è stato compreso e, per quanto ne so, i livelli di EPO non sono stati studiati in uomini sani che utilizzano dosi elevate di AAS, in base allo stato ipogonadico. Molti consumatori di AAS presentano ipogonadismo ipogonadotropo (HH), definito ipogonadismo indotto da AAS (ASIH).[8]
Uno studio recente indica che la frazione reticolocitaria ad alta fluorescenza (HFR) è sensibile all’assunzione di Testosterone nelle donne,19 ma l’associazione dell’HFR all’esposizione sovra-fisiologica agli AAS negli uomini non è stata studiata. È possibile che dosi sovra-fisiologiche di AAS esercitino effetti diversi sulle frazioni reticolocitarie.
Sappiamo però che vi è una risposta nell’aumento dei processi eritrocitari in seguito all’assunzione di AAS off-label. Ciò è risultato di grado soggettivo con alcuni utilizzatori che mostrano una risposta minore mentre altri ne mostrano una sensibilmente elevata anche con dosaggi tipici di una TRT [100mg/week di Testosterone Enantato = 72mg di Testosterone effettivo/slegato dall’estere]. La variabile di stimolo eritrocitario sembra variare anche in modo dipendente dal/dagli AAS utilizzato/i con una maggiore risposta a carico del Boldenone e dell’Oxymetholone, ma non solo.
Alcune testimonianze riportate da preparatori e provenienti dai controlli ematici di diversi bodybuilder, suggeriscono che il Methandrostenolone (Dianabol), nonostante sia molto semplicemente una forma metilata in C-17 del Boldenone, al contrario di quest’ultimo eserciti un azione negativa, o sottoregolativa, dell’ematopoiesi e, nel dettaglio, dell’ematopoiesi.
In questo articolo analizzerò quanto attualmente si conosce dei processi di interazione degli androgeni (e AAS esogeni) sulla ematopoiesi e eritropoiesi e quali potrebbero essere le ragioni per le quali è stata osservata la risposta “paradossale” del Methandrostenolone su tali processi (“Dbol Hematocrit Theory”).
Androgeni ed eritropoiesi:
Studi su animali e sull’uomo hanno suggerito un effetto stimolatorio diretto e indiretto degli androgeni sull’eritropoiesi, anche se l’esatto meccanismo di tale relazione rimane vagamente compreso. La somministrazione di androgeni determina un aumento della massa cellulare eritroide, delle unità formanti colonie di eritrociti (CFU-E) e della produzione e secrezione di eritropoietina (EPO) (6), mentre la privazione di androgeni causa una riduzione degli indici di globuli rossi a causa della ridotta proliferazione dei precursori eritroidi del midollo (9).
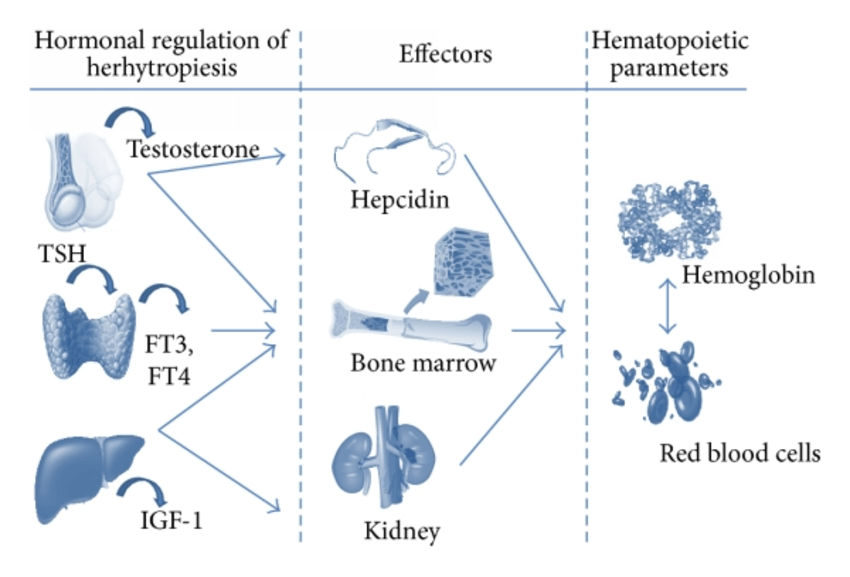
Gli androgeni vengono convertiti in 17-cheto-steroidi in grado di aumentare la sintesi di mRNA nel nucleo, causando la differenziazione delle cellule del midollo osseo da non responsive all’EPO a responsive all’EPO (6). Inoltre, gli androgeni aumentano l’assorbimento di glucosio con conseguente glicolisi e trascrizione genica e sintesi di mRNA negli eritroidi (10-11).
Il Testosterone può aumentare l’Hct inibendo la secrezione di Epcidina, il principale peptide regolatore del ferro, con conseguente aumento del ferro biodisponibile (12), ma può anche aumentare l’incorporazione del ferro nei globuli rossi (13) e migliorare la sopravvivenza di questi ultimi (14). Infine, il riscontro di un aumento dei livelli di IGF-1 nei soggetti che ricevono androgeni ha suggerito un potenziale legame tra gli androgeni e la proliferazione e la differenziazione delle cellule progenitrici eritroidi guidata da IGF-1 (15, 16).
L’effetto del Testosterone sull’eritropoiesi è più pronunciato durante la pubertà, con l’Hb prepuberale che è simile nei ragazzi e nelle ragazze, ma aumenta nei ragazzi dopo i 13 anni in tandem con l’aumento delle concentrazioni di Testosterone(6, 17). I ragazzi con pubertà ritardata hanno livelli di Hb simili a quelli dei ragazzi e delle ragazze prepuberi e il trattamento con Testosterone normalizza i livelli di emoglobina a quelli osservati nella pubertà maschile tardiva (18, 19).
Giova ricordare che prima dello sviluppo della terapia con EPO alla fine degli anni ’80, gli androgeni erano l’unica opzione per il trattamento dell’anemia legata alla Malattia Renale Cronica (CKD) negli uomini. I pazienti con CKD possono presentare una riduzione della densità minerale ossea, della massa muscolare, dei livelli di energia, della qualità della vita e della funzione sessuale, con esiti cardiovascolari avversi, soprattutto in quelli con diabete concomitante (20); sebbene queste caratteristiche abbiano probabilmente un’origine multifattoriale, si verificano anche in caso di ipogonadismo. Il basso livello di Testosterone è prevalente nei pazienti con CKD e può contribuire all’anemia renale (21, 22). Fino a due terzi degli uomini in emodialisi (HD) presentano livelli sierici di Testosterone nell’intervallo ipogonadico, a causa di anomalie a tutti i livelli dell’asse Ipotalamo-Ipofisi-Testicolo (23-24). Dati non pubblicati di una coorte di 113 pazienti in HD e 85 in pre-dialisi (preD) allo stadio 4 e 5 della CKD hanno riportato livelli di Testosterone subnormali nel 76% dei maschi in pre-dialisi e nell’80% dei maschi in HD, con una significativa correlazione inversa della dose di Dα con il T totale (R -0,253; p <0,01) e il T libero (-0,29; <0,01) (25).
Il fatto che l’ipogonadismo sia una causa consolidata di anemia e di ridotta responsività all’EPO negli uomini con CKD può suggerire un possibile ruolo della terapia con Testosterone come aggiunta o alternativa all’EPO in alcuni uomini con anemia correlata alla CKD (26). Ciò è particolarmente rilevante in alcuni sistemi sanitari in cui i pazienti con CKD non possono permettersi la terapia con EPO, che potrebbe causare anemia con necessità di trasfusioni di sangue, soprattutto se manca l’evidenza che l’EPO migliori la morbilità o la mortalità nella CKD (27).
L’anemia negli uomini anziani può aumentare il rischio di morbilità e mortalità. In uno studio retrospettivo su uomini di età superiore ai 65 anni ricoverati con infarto miocardico acuto, livelli di Hct più bassi erano associati a un aumento della mortalità a 30 giorni, mentre il trattamento dell’anemia migliorava i tassi di mortalità (28). Un’osservazione simile di elevata mortalità è stata riportata in una coorte di pazienti anemici che presentavano un’insufficienza cardiaca di nuova insorgenza (29). L’ipogonadismo, o il calo ponderale dei livelli di androgeni legati all’invecchiamento possono entrambi causare anemia, sarcopenia e osteoporosi (30), con studi longitudinali e trasversali che mostrano costantemente un calo dei livelli sierici di Testosterone negli uomini che invecchiano.
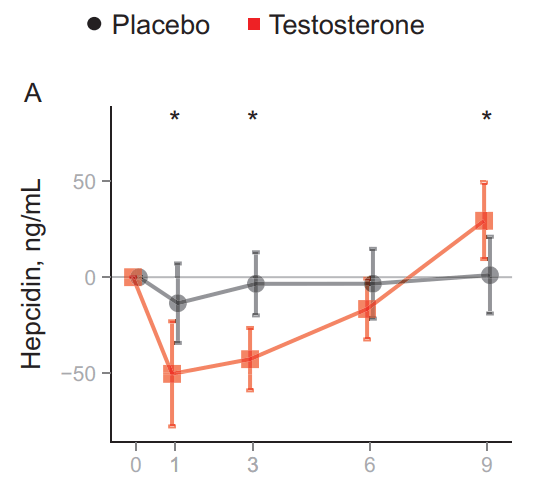
Alcuni studi hanno esaminato i cambiamenti dell’Hb nei pazienti sottoposti a terapia sostitutiva con Testosterone; alcuni hanno sperimentato specificamente la terapia con Testosterone con l’obiettivo primario di migliorare l’anemia, con un accumulo di evidenze. In uno studio randomizzato in doppio cieco controllato con placebo di Dhinsda et al., la terapia con Testosterone ha soppresso l’Epcidina con un marcato aumento dell’Hb, dell’EPO e dell’espressione dei recettori della ferroportina e della transferrina in pazienti ipogonadici con diabete di tipo 2 (31). Gli studi con Testosterone finanziati dal National Institute of Health (NIH) hanno esaminato gli effetti della TRT in uomini ipogonadici di età superiore ai 65 anni (32). Utilizzando un design in doppio cieco, controllato con placebo, 12 mesi di trattamento quotidiano con gel di Testosterone hanno aumentato i livelli di Hb di almeno 1,0g/dL in ~52% degli uomini con ipogonadismo e una causa nota di anemia rispetto al placebo. Inoltre, nei 64 uomini anziani con anemia inspiegabile, l’Hb è migliorata di almeno 1,0g/dL nel 54% degli uomini rispetto al 15% del gruppo placebo. Tuttavia, la carenza di androgeni (AD) è tipicamente trascurata nelle linee guida sull’indagine dell’anemia (33).
È importante notare che la maggior parte delle linee guida cliniche raccomanda di valutare l’Hct e l’antigene prostatico specifico (PSA) prima di iniziare la terapia sostitutiva con Testosterone (34). Dopo tutto l’influenza degli androgeni sulla emopoiesi/eritropoiesi era già piuttosto chiara fin dagli studi della fine degli anni 40′. In definitiva, in ambito terapeutico, il trattamento con androgeni ha anche un potenziale per trattare l’anemia della CKD e negli uomini ipogonadici come aggiunta all’EPO.
Ma per gli utilizzatori off-label lo stimolo eritropoietico dato dagli AAS può diventare un problema non di poca rilevanza.
Dosi sovrafisiologiche di AAS e risposta emopoietica/eritrocitaria:

Come già discusso in un mio precedente articolo trattante l’uso di AAS off-label ed eritrocitosi/policitemia, ho fatto notare che la somministrazione di Testosterone per 20 settimane mostra un aumento dose-dipendente (fino a 600mg di Testosterone Enantato alla settimana; pari a 432mg di Testosterone effettivo) dell’emoglobina e dell’ematocrito, soprattutto negli uomini più anziani, mentre l’EPO non lo fa [35]. E come già esposto, la somministrazione di Testosterone porta alla soppressione dell’Epcidina sierica in uomini giovani e anziani [36]. Il dosaggio del Testosterone (fino a 600mg di Testosterone Enantato a settimana) è altamente correlato con l’ampiezza di questa soppressione. In sintesi, gli androgeni aumentano l’ematocrito/emoglobina attraverso un aumento iniziale dei livelli di EPO e una contemporanea diminuzione dei livelli di Epcidina, che poi scendono gradualmente ai livelli di base di fronte all’aumento dell’ematocrito/emoglobina: un nuovo set point EPO/emoglobina. I meccanismi d’azione responsabili e il loro contributo relativo a questo fenomeno sono ancora da stabilire.
Con dosaggi fino a 600mg di Testosterone Enantato alla settimana, l’emoglobina ha mostrato un aumento di 1,42g/dL nei giovani uomini dopo 20 settimane [37]. Ciò si traduce in un aumento dell’ematocrito di poco superiore al 4%.
Fortunatamente, sembra esserci un limite alla misura in cui gli AAS possono aumentare l’ematocrito. Grazie allo studio HAARLEM, sono stati osservati 100 consumatori di steroidi anabolizzanti seguiti nel tempo mentre si autosomministravano AAS, il cui dosaggio medio, basato sulle informazioni riportate sull’etichetta, era di 898mg a settimana, rendendo così il loro ciclo di AAS abbastanza rappresentativo dell’uso comune da parte dei bodybuilder. Le misurazioni sono state effettuate prima, durante, 3 mesi dopo la fine del ciclo e 1 anno dopo l’inizio del ciclo. I ricercatori hanno riscontrato un aumento del 3% dell’ematocrito dei soggetti dello studio al termine del ciclo. Questo dato è in linea con l’aumento del 4% osservato nello studio nei giovani uomini. L’autore principale ha fatto sapere che l’aumento dell’ematocrito sembra stabilizzarsi a un dosaggio di androgeni di circa 500mg a settimana. Infine, c’è da aggiungere che i soggetti dello studio non hanno effettuato donazioni di sangue, quindi questo non è stato un fattore confondente.
Naturalmente, questi risultati presentano alcune variazioni. Alcuni rispondono agli AAS con un aumento dell’ematocrito maggiore di altri. Tuttavia, livelli molto elevati di ematocrito sembrano essere rari, come si può vedere nei grafici a scatola e baffi dei partecipanti allo studio HAARLEM (T0 = subito prima del ciclo di AAS, T1 = alla fine, T2 = 3 mesi dopo la cessazione dell’uso, T3 = 1 anno dopo l’inizio del ciclo):

Trattamento consuetudinario:
Forse il trattamento migliore consiste nel ridurre notevolmente il dosaggio (ben al di sotto dei 500mg settimanali) o nell’interrompere del tutto l’uso di AAS [tornando in fisiologia controllata]. In questo modo si abbasserà l’ematocrito, con un effetto completo dopo un paio di mesi, e si annullerà il rischio. Tuttavia, questo non è probabilmente il metodo più gradito per contrastare questo problema.

Pratica comunemente diffusa consiste nell’assunzione di un basso dosaggio di CardioAspirina (Acido Acetilsalicilico con gastroprotettore). Sebbene non influisca sui livelli di ematocrito, è ampiamente utilizzata per la prevenzione delle malattie cardiovascolari [38]. Più precisamente, è utilizzata nella prevenzione secondaria delle malattie cardiovascolari. Previene la coagulazione del sangue inibendo un enzima chiamato ciclossigenasi (COX) nei trombociti. Se da un lato riduce il rischio di trombosi, dall’altro aumenta il rischio di emorragie. Parliamo anche di emorragie interne, come l’ictus emorragico. I benefici devono quindi essere attentamente soppesati rispetto ai rischi del suo utilizzo. Attualmente, le linee guida europee sulla prevenzione delle malattie cardiovascolari nella pratica clinica ne sconsigliano l’uso nella prevenzione primaria (anche se questo potrebbe cambiare per alcune popolazioni, come i diabetici) [39]. Oltretutto, nelle settimane successive all’interruzione del farmaco vi è un aumento del rischio di eventi trombotici [40, 41]. Pertanto, anche l’assunzione e la sospensione frequente del farmaco sono sconsigliate.
Nota: prima di un eventuale uso, è caldamente consigliato, oltre il parere medico, un controllo accurato dei fattori che regolano la coagulazione: tempo di tromboplastinaparziale attivata (aPTT), tempo di protrombina (PT) Fibrinogeno, D-Dimero, Antitrombina e Inibitore C1 Esterasi.
Anche la Nattochinasi, un enzima digestivo (una proteasi alcalina) presente nel natto, un alimento tradizionale giapponese fermentato, viene comunemente utilizzata per la prevenzioni di eventi trombotici da parte di utilizzatori di AAS con alterazione del Ematocrito. Ma non ci sono prove sufficienti sull’uomo che utilizzano la Nattochinasi isolatamente e che valutano la formazione di trombi per raccomandarne l’uso come farmaco anti-clottico, anche se sembra esserci qualche promessa. Come per la CardioAspirina, la necessità di utilizzo andrebbe valutata per via esami dei fattori della coagulazione.

Un modo per ridurre efficacemente l’ematocrito è senza dubbio la flebotomia (salasso). Un modo per farlo è la donazione di sangue a una banca del sangue. Tuttavia, molti Paesi (e giustamente) limitano il numero di volte in cui è possibile farlo ogni anno. Vi sono paesi dove è limitato a cinque volte l’anno. Questo potrebbe non essere sufficiente a mantenere i valori nel range desiderato, dato che uno studio ha rilevato livelli di emoglobina persistentemente elevati in occasione di visite ripetute in un numero elevato di pazienti TRT che hanno donato il sangue [42]. Se la donazione di sangue non è sufficiente, si può sempre consultare un medico generico per eseguire una flebotomia terapeutica a intervalli più frequenti.
Occorre ricordare che ad ogni donazione di sangue/salasso si perde Ferro. Di conseguenza, si corre il rischio di esaurire le proprie riserve di Ferro e, consequenzialmente, anche l’emoglobina rimarrà molto bassa e si diventerà temporaneamente anemici. È possibile contrastare questo fenomeno integrando il Ferro, ma questo riduce drasticamente il tempo necessario all’organismo per recuperare i livelli di emoglobina/ematocrito [43]. In uno studio è stato utilizzato un dosaggio di 37,5mg di Ferro elementare al giorno. Pertanto, a intervalli più frequenti, è consigliabile un controllo con analisi del sangue. Inoltre, va ricordato che una flebotomia non monitorata nei tempi di prelievo può portare a rebound dell’ematocrito con peggioramento del quadro clinico ematico.
La “Dbol Hematocrit Theory”:
Come accennato nell’introduzione, alcuni preparatori hanno osservato una “anomalia” con la somministrazione di Methandrostenolone. Questa “anomalia” consisteva in una sensibile riduzione dell’Ematocrico e in una certa misura una alterazione della conta dei globuli bianchi, in special modo dei Linfociti.
La risposta osservata si è verificata in condizioni di monoterapia, quindi priva anche di una base TRT. Ciò è stato fatto per evitare che soggetti sensibili all’aumento del Ematocrito subissero un interazione negativa anche da dosi terapeutiche di Testosterone.

Alcuni di voi diranno “ma come? Senza una base di Testosterone il soggetto andrebbe in uno stato di malessere psicofisico dipendente dalla riduzione marcata di Estradiolo e DHT!”. Questa affermazione è parzialmente vera ma:
- Sebbene il Methandrostenolone sia un derivato 17α-Metilato del Boldenone , sembra soggetto ad una maggiore conversione in 17α-Methylestradiolo. A causa della presenza del suo gruppo metilico C17α, il 17α-Methylestradiolo non può essere disattivato mediante ossidazione del gruppo ossidrile C17β, con conseguente miglioramento della stabilità metabolica e della potenza rispetto al 17β-Estradiolo.[44];
- L’uso di DHT esogeno o di analogo metilato in C-1 sopperisce generalmente alla bassa 5α-riduzione senza interferire con la finalità del protocollo su Ematocrito.
A questo punto la domanda è: “Quali sono i meccanismi attraverso i quali l’uso monoterapico del Methandrostenolone porta ad una riduzione dell’Ematocrito e ad una risposta paradossale del Emopoiesi?”
Al momento possiamo solo ipotizzare utilizzando l’attuale conoscenza in nostro possesso riguardo ai meccanismi androgeni su Emopoiesi e Eritropoiesi. Le ipotesi in merito sono le seguenti:
- Ipotesi della riduzione attività e concentrazioni di 17-Ketosteroidi: Sappiamo che il Methandrostenolone viene metabolizzato nel fegato mediante 6β-idrossilazione, 3α- e 3β-ossidazione, 5β-riduzione, 17-epimerizzazione e coniugazione tra le altre reazioni. Non mi è stato possibile reperire materiale in riferimento riguardante eventuali metaboliti 17-ketosteroidi del Methandrostenolone. Sapendo che, gli androgeni convertendo in 17-Ketosteroidi sono in grado di aumentare la sintesi di mRNA nel nucleo, causando la differenziazione delle cellule del midollo osseo da non responsive all’EPO a responsive all’EPO. Una eventuale riduzione dei metaboliti di conversione ed interconversione dei 17-Ketosteroidi [vedi enzimi 17β-Hydroxysteroide dehydrogenasi;17β-HSD] dati da soppressione/sottoregolazione di sintesi e scarsa affinità enzimatica del Methandrostenolone potrebbero esserne la causa. Mentre per quanto riguarda la riduzione dei Leucociti, essa potrebbe essere riconducibile ad una attività immunosoppressiva osservata anche con l’uso di altri AAS.
- Ipotesi della attività di legame antagonista: si potrebbe ipotizzare che il Methandrostenolone possa agire come una molecola antagonista/agonista dei recettori cellulari del midollo emopoietico portando ad una riduzione dell’attività dei metaboliti 17-ketosteroidi riducendo sia l’Eritropoiesi che la formazione di globuli bianchi e piastrine [derivanti da un unica cellula staminale emopoietica pluripotente]. Il problema è che se ciò fosse vero, la regolazione del segnale indotto dall’attività antagonista/agonista sembra essere maggiore per la risposta eritrocitaria e leucocitaria.
- Ipotesi del metabolita “sintetico” 17-ketosteroideo: si potrebbe ipotizzare che il Methandrostenolone converta in una forma non ancora identificata di 17-ketosteroide avente effetto agonista/antagonista a livello delle cellule staminali emopoietiche multinucleate. Si ricordi, infatti, che di recente (2010) è stato identificato un metabolita del Methandrostenolone nei campioni di urina fino a 19 giorni dopo la sua somministrazione. Il metabolita in questione è il 17beta-idrossimetil-17 alfa-metil-18-norandrosta-1,4,13-trien-3-one (20OH-NorMD), scoperto tramite LC-MS/MS e GC -SM. L’enzima CYP21 e, in misura minore, anche il CYP3A4 possono catalizzare questa idrossilazione dello steroide. Non escluderei in assoluto, quindi, la presenza di un metabolita non ancora identificato[45].
E’ bene ricordare che il Boldenone, precursore strutturale del Methandrostenolone, mostra in corso di trattamento un aumento significativo della conta eritrocitaria totale e dei valori di emoglobina ed ematocrito, mentre gli indici medi di emoglobina corpuscolare e di concentrazione media di emoglobina corpuscolare sembrano diminuire. Il leucogramma, similmente all’effetto notato con il Methandrostenolone, mostra leucopenia, linfopenia e granulocitosi rispetto al controllo negli studi su animali e in alcuni casi studio. Il metabolita androsta–1,4–dien–17 beta-ol-3-one sembra avere un ipotetico ruolo significativo negli effetti emopoietici del Boldenone insieme ad altri steroidi strettamente correlati al 17β-boldenone. E ricordiamoci che le 17β-idrossisteroide deidrogenasi (17β-HSD, HSD17B) (EC 1.1.1.51), anche 17-chetosteroide reduttasi (17-KSR), il gruppo di alcol ossidoreduttasi che catalizzano la riduzione dei 17-Ketosteroidi e la deidrogenazione dei 17β-idrossisteroidi nella steroidogenesi e nel metabolismo degli steroidi, sono implicate nel metabolismo dei 17-ketosteroidi che, a loro volta, sono direttamente correlati alla eritropoiesi/emopoiesi.[46][47]
I dosaggi di Methandrostenolone generalmente utilizzati per tale scopo si attestano nel range dei 10-15mg/die. In questo modo l’atleta, in un discreto numero di casi, non è obbligato a cessare completamente l’utilizzo di AAS sfruttando il forte potenziale anticatabolico del Methandrostenolone con finalità stabilizzative. In corso di terapia il soggetto si sottopone a flebotomia con regolarità in funzione delle risposte al trattamento valutate per via esami ematici.
Nota: durante il periodo di trattamento, l’uso di hCG non è indicato dal momento che l’aumento nella biosintesi testicolare di Testosterone potrebbe alterare la risposta di controllo del Eritropoiesi/Emopoiesi con conseguente non ottenimento della risposta terapeutica ricercata.
Le limitazione di applicazione della monoterapia con Methandrostenolone per il trattamento della eritrocitosi sono le seguenti:
- Stato dislipidemico marcato [alterazione marcata dei livelli di HDL-C (<25mg/dL), LDL-C (>150mg/dL), Trigliceridi (>80mg/dL);
- coesistenza della prima citata dislipidemia ematica con livelli di Omocisteine > 13µmol/L;
- alterazioni marcate delle Transaminasi e/o dei risultati della elettroforesi proteica;
- eGFR < 60, Creatininuria da raccolta urine nelle 24h >2000mg/dL oppure da campione di urine al mattino >300mg/dL, Creatininemia 2mg/dL, Cistatina C >1mg/Lt con valutazione della elettroforesi proteica.
Conclusioni:
L’identificazione del fattore, o dei fattori, implicati nella risposta sottoregolativa sulla Eritropoiesi/Emopoiesi in monoterapia con Methandrostenolone è assai lungi dall’essere con sufficiente sicurezza scoperta. Ad oggi, però, le ipotesi sopra esposte rappresentano le possibilità con la probabilità maggiore di riscontro positivo con eventuali futuri studi specifici.
Si rammenta che ogni cosa esposta concernente la “Dbol Hematocrit Theory” è puramente ipotetico-teorica, non rappresenta quindi una pratica scientificamente avvalorata ne tantomeno un consiglio terapico. Chiunque decida liberamente di utilizzare le informazioni presentate per improvvisarsi ricercatore e/o cavia, lo fa prendendosene la piena responsabilità d’esito.
In conclusione, ringrazio i preparatori (colleghi) che mi hanno fornito esami di confronto e dati raccolti in anni di osservazione attenta degli esami ematici. In particolare modo ringrazio Alberto Prevedi che mi ha fornito gli esami esposti in questo articolo.
Gabriel Bellizzi [CEO BioGenTech]
Riferimenti:
1.Steinglass P, Gordon AS, Charipper HA. Effect of castration and sex hormones on blood of the rat. Proc Soc Exp Biol Med. (1941) 48:169–77. doi: 10.3181/00379727-48-13259
2. Crafts RC. Effects of hypophysectomy, castration and testosterone propionate on hemopoiesis in the adult male rat. Endocrinology. (1946) 39:401–13. doi: 10.1210/endo-39-6-401
3. Vahlquist B. The cause of the sexual differences in erythrocyte, hemoglobin and serum iron levels in human adults. Blood. (1950) 5:874–5. doi: 10.1182/blood.V5.9.874.874
4. Hartgens F, Kuipers S. Effects of androgenic-anabolic steroids in athletes. Sports Med. (2004) 34:513–54. doi: 10.2165/00007256-200434080-00003
5. McCullagh EP, Jones R. A note on the effect of certain androgens upon red blood cell count and upon glucose tolerance. Cleve Clin Q. (1941) 8:79–84. doi: 10.3949/ccjm.8.2.79
6. Shahani S, Braga-Basaria M, Maggio M, Basaria S. Androgens and erythropoiesis: past and present. J Endocrinol Invest. (2009) 32:704–16. doi: 10.1007/BF03345745
7. Shimoda K, Shide K, Kamezaki K, Okamura T, Harada N, Kinukawa N, et al. The effect of anabolic steroids on anemia in myelofibrosis with myeloid metaplasia: retrospective analysis of 39 patients in Japan. Int J Hematol. (2007) 85:338–43. doi: 10.1532/IJH97.06135
8.Jaime-Pérez JC, Colunga-Pedraza PR, Gómez-Ramírez CD, Gutiérrez-Aguirre CH, Cantú-Rodríguez OG, Tarín-Arzaga LC, et al. Danazol as first-line therapy for aplastic anemia. Ann Hematol. (2011) 90:523–7. doi: 10.1007/s00277-011-1163-x
9.Gagliano-Jucá T, Pencina KM, Ganz T, Travison TG, Kantoff PW, Nguyen PL, et al. Mechanisms responsible for reduced erythropoiesis during androgen deprivation therapy in men with prostate cancer. Am J Physiol Endocrinol Metab. (2018) 315:E1185–93. doi: 10.1152/ajpendo.00272.2018
10.Larner J. Intermediary Metabolism and Its Regulation. EnglewoodCliffs, NJ: Prentice Hall Inc. (1971). p. 228.
11.Molinari PF, Neri LL. Effect of a single oral dose of oxymetholone on the metabolism of human erythrocytes. Exp Hematol. (1978) 6:648–54.
12.Bachman E, Feng R, Travison T, Li M, Olbina G, Ostland V, et al. Testosterone suppresses hepcidin in men: a potential mechanism for testosterone-induced erythrocytosis. J Clin Endocrinol Metab. (2010) 95:4743–7. doi: 10.1210/jc.2010-0864
13. Naets JP, Wittek M. The mechanism of action of androgens on erythropoiesis. Ann N Y Acad Sci. (1968) 149:366–76.
14. Rishpon-Meyerstein N, Kilbridge T, Simone J, Fried W. The effect of testosterone on erythropoietin levels in anaemic patients. Blood. (1968) 31:453–60. doi: 10.1182/blood.V31.4.453.453
15. Hagenfeldt Y, Linde K, Sjoberg HE, Zumkeller W, Arver S. Testosterone increases serum 1,25-dihydroxyvitamin D and insulin like growth factor-I (IGF-1) in hypogonadal men. Int J Androl. (1992) 15:93–102. doi: 10.1111/j.1365-2605.1992.tb01118.x
16. Hobbs CJ, Plymate SR, Rosen CJ, Adler RA. Testosterone administration increases insulin-like growth factor-I levels in normal men. J Clin Endocrinol Metab. (1993) 77:776–9. doi: 10.1210/jc.77.3.776
17. Shahidi NT. Androgens and erythropoiesis. N Engl J Med. (1973) 289:72–80. doi: 10.1056/NEJM197307122890205
18. Krabbe S, Christensen T, Worm J, Christiansen C, Transbøl I. Relationship between haemoglobin and serum testosterone in normal children and adolescents and in boys with delayed puberty. Acta Paediatr Scand. (1978) 67:655–8. doi: 10.1111/j.1651-2227.1978.tb17818.x
19. Hero M, Wickman S, Hanhijärvi R, Siimes MA, Dunkel L. Pubertal upregulation of erythropoiesis in boys is determined primarily by androgen. J Pediatr. (2005) 146:245–52. doi: 10.1016/j.jpeds.2004.09.002
20. Tong PC, Kong AP, So WY, Ng MH, Yang X, Ng MC, et al. Hematocrit, independent of chronic kidney disease, predicts adverse cardiovascular outcomes in Chinese patients with type 2 diabetes. Diabetes Care. (2006) 29:2439–44. doi: 10.2337/dc06-0887
21. Karakitsos D, Patrianakos AP, De Groot E, Boletis J, Karabinis A, Kyriazis J, et al. Androgen deficiency and endothelial dysfunction in men with end-stage kidney disease receiving maintenance hemodialysis. Am J Nephrol. (2006) 26:536–43. doi: 10.1159/000097816
22. Carrero JJ, Qureshi AR, Nakashima A, Arver S, Parini P, Lindholm B, et al. Prevalence and clinical implications of testosterone deficiency in men with end-stage renal disease. Nephrol Dial Transplant. (2011) 26:184–90. doi: 10.1093/ndt/gfq397
23. Handelsman DJ. Hypothalamic-pituitary gonadal dysfunction in renal failure, dialysis, and renal transplantation. Endocr Rev. (1985) 6:151–82. doi: 10.1210/edrv-6-2-151
24.National Kidney Foundation. K/DOQI clinical practice guidelines for anemia of chronic kidney disease. Am J Kidney Dis. (2000) 37:182–238. doi: 10.1016/S0272-6386(01)70008-X
25. Kanagasundaram N, Shipley T, Wright R, Playford M, Peaston R, Moochhala S, et al. The Prevalence of Androgen Deficiency in Male Patients With Advanced CKD – An Evaluation in Pre-dialysis and Haemodialysis Population. (2010). Available online at: http://www.renalarchive.org/FetchDoc.aspx?id=6309 (accessed June 26, 2019).
26.DeLong M, Logan JL, Yong KC, Lien YH. Renin angiotensin blockade reduces serum free testosterone in middle-aged men on haemodialysis and correlates with erythropoietin resistance. Nephrol Dial Transplant. (2005) 20:585–90. doi: 10.1093/ndt/gfh638
27. Pfeffer MA, Burdmann EA, Chen CY, Cooper ME, de Zeeuw D, Eckardt KU, et al. A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N Engl J Med. (2009) 361:2019–32. doi: 10.1056/NEJMoa0907845
28.Wu WC, Rathore SS, Wang Y, Radford MJ, Krumholz HM. Blood transfusion in elderly patients with acute myocardial infarction. N Engl J Med. (2001) 345:1230–6. doi: 10.1056/NEJMoa010615
29. Ezekowitz JA, McAlister FA, Armstrong PW. Anemia is common in heart failure and is associated with poor outcomes: insights from a cohort of 12,065 patients with new-onset heart failure. Circulation. (2003) 107:223–5. doi: 10.1161/01.CIR.0000052622.51963.FC
30. Wu FC, Tajar A, Beynon JM, Pye SR, Silman AJ, Finn JD, et al. Identification of late-onset hypogonadism in middle-aged and elderly men. N Engl J Med. (2010) 363:123–35. doi: 10.1056/NEJMoa0911101
31.Dhindsa S, Ghanim H, Batra M, Kuhadiya ND, Abuaysheh S, Green K, et al. Effect of testosterone on hepcidin, ferroportin, ferritin and iron binding capacity in patients with hypogonadotropic hypogonadism and type 2 diabetes. Clin Enocrinol. (2016) 85:772–28. doi: 10.1111/cen.13130
32.Roy CN, Snyder PJ, Stephens-Shields AJ, Artz AS, Bhasin S, Cohen HJ, et al. Association of testosterone levels with anemia in older men: a controlled clinical trial. JAMA Intern Med. (2017) 177:480–90. doi: 10.1001/jamainternmed.2016.9540
33. Al-Sharefi A, Quinton R. Re:male hypogonadism, a treatable yet forgotten cause of unexplained anaemia. REMARQ commentary on Strauder et al. Anemia at older age: etiologies, clinical implications, and management. Blood. (2018) 131:505–14. doi: 10.1182/blood-2017-07-746446
34. Bhasin S, Brito JP, Cunningham GR, Hayes FJ, Hodis HN, Matsumoto AM, et al. Testosterone therapy in men with hypogonadism: an endocrine scoety clinical practice guidelines. J Clin Endocrinol Metab. (2018) 103:715–44. doi: 10.1210/jc.2018-00229
35.Coviello, Andrea D., et al. “Effects of graded doses of testosterone on erythropoiesis in healthy young and older men.” The Journal of Clinical Endocrinology & Metabolism 93.3 (2008): 914-919.
36.Bachman, Eric, et al. “Testosterone suppresses hepcidin in men: a potential mechanism for testosterone-induced erythrocytosis.” The Journal of Clinical Endocrinology & Metabolism 95.10 (2010): 4743-4747.
37.Bhasin, Shalender, et al. “Testosterone dose-response relationships in healthy young men.” American Journal of Physiology-Endocrinology And Metabolism (2001).
38.Baigent, Colin, et al. “Aspirin in the primary and secondary prevention of vascular disease: collaborative meta-analysis of individual participant data from randomised trials.” Lancet 373.9678 (2009): 1849-1860.
39.F. Hobbs, M. Piepoli, A. Hoes, S. Agewall, C. Albus, C. Brotons, A. Catapano, M. Cooney, U. Corra, B. Cosyns, et al. 2016 european guidelines on cardiovascular disease prevention in clinical practice. European Heart Journal, 37(29):2315–2381, 2016.
40.M. Lordkipanidzé, J. G. Diodati, and C. Pharand. Possibility of a rebound phenomenon following antiplatelet therapy withdrawal: a look at the clinical and pharmacological evidence. Pharmacology & therapeutics, 123(2):178–186, 2009.
41.L. A. G. Rodríguez, L. C. Soriano, C. Hill, and S. Johansson. Increased risk of stroke after discontinuation of acetylsalicylic acid a uk primary care study. Neurology, pages WNL–0b013e31820d62b5, 2011
42.B. Chin-Yee, A. Lazo-Langner, T. Butler-Foster, C. Hsia, and I. Chin-Yee. Blood donation and testosterone replacement therapy. Transfusion, 57(3):578–581, 2017
43.Kiss, Joseph E., et al. “Oral iron supplementation after blood donation: a randomized clinical trial.” Jama 313.6 (2015): 575-583.
44.Thieme D, Hemmersbach P (18 December 2009). Doping in Sports. Springer Science & Business Media. pp. 470
45.https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9339561/
46.Labrie F, Luu-The V, Lin SX, Labrie C, Simard J, Breton R, Bélanger A (January 1997). “The key role of 17 beta-hydroxysteroid dehydrogenases in sex steroid biology”. Steroids. 62 (1): 148–58.
47.Brook CG, Truong D, Clayton P, Carroll W, Brown R (2011). Brook’s Clinical Pediatric Endocrinology. John Wiley & Sons. p. 288.