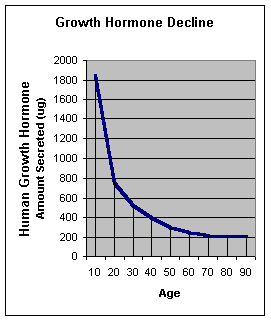
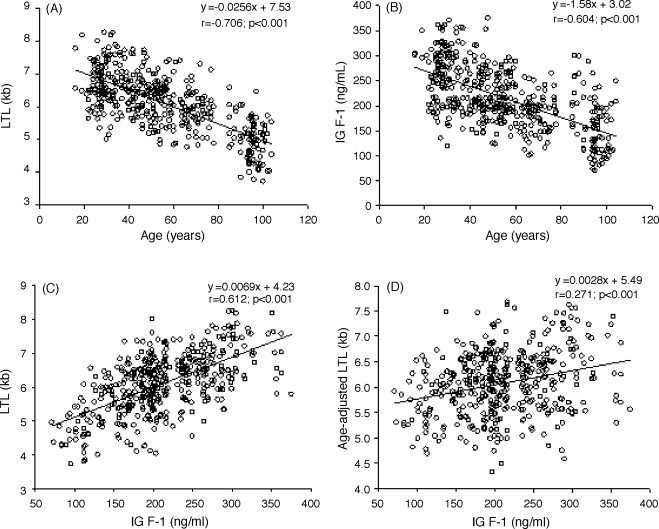
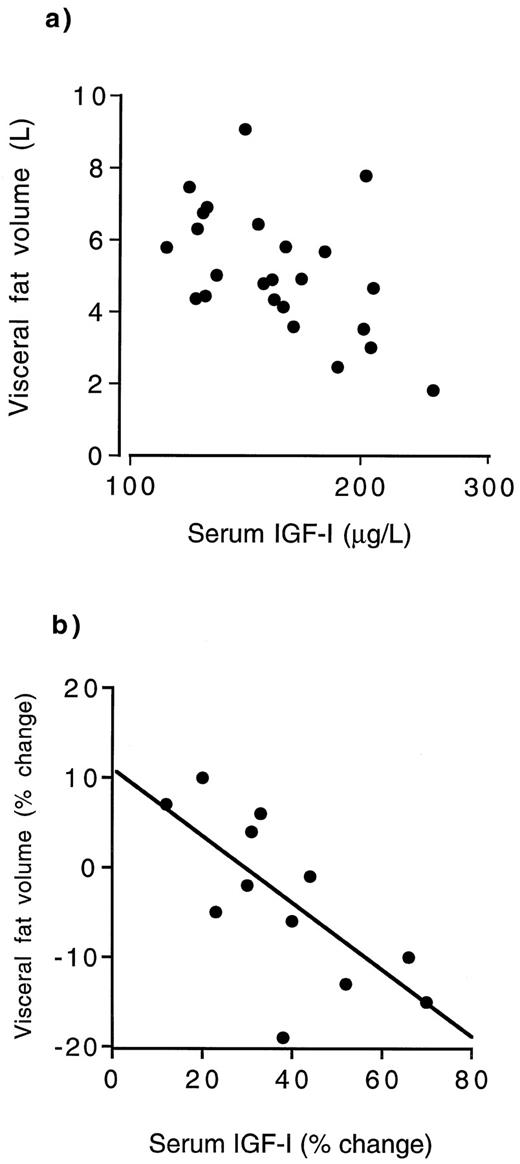
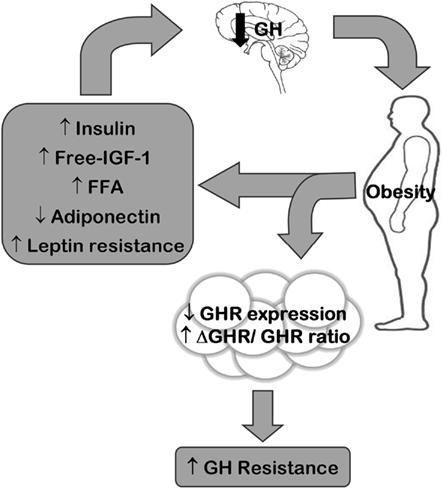
Secondo uno studio su animali svolto da fisiologi rumeni presso l’Università di Medicina e Farmacia Carol Davila e pubblicato su “Medicina”, la supplementazione di Taurina potrebbe offrire protezione cardiovascolare agli utilizzatori di AAS.(1)
Per lo svolgimento del loro studio, i ricercatori hanno diviso dei ratti maschi in 4 gruppi:
Un gruppo di controllo [C] al quale non è stato somministrato alcun farmaco;
Un gruppo al quale veniva somministrata un’iniezione settimanale di Nandrolone Decanoato [A];
Un gruppo al quale veniva somministrata della Taurina miscelata all’acqua [T];
Un gruppo al quale veniva somministrata un’iniezione settimanale di Nandrolone Decanoato e Taurina miscelata all’acqua [AT].
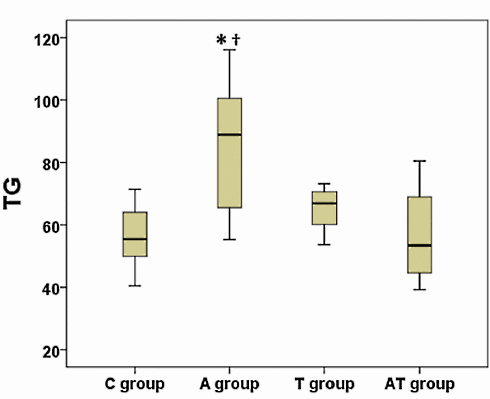
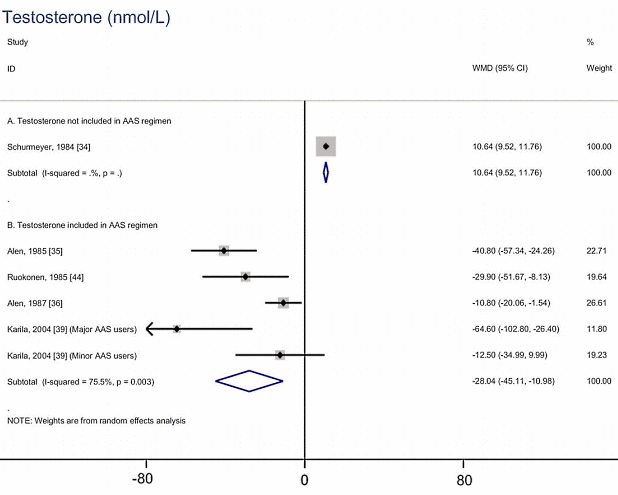
Nei tre mesi di durata dell’esperimento, la concentrazione dei Trigliceridi nel sangue dei ratti trattati con Nandrolone era ovviamente peggiorata. L’ipertrigliceridemia è un noto fattore di rischio per malattie cardiovascolari. La supplementazione di Taurina, tuttavia, ha impedito il verificarsi di questo aumento dei livelli di Trigliceridi, come mostrato nella figura seguente.
La somministrazione di Nandrolone decanoato ha portato anche ad una riduzione delle concentrazioni di HDL, una condizione nota per essere un fattore di rischio cardiovascolare. La figura seguente mostra l’impatto “contenitivo” sulla riduzione del HDL che la Taurina ha esercitato nei topi trattati con Nandrolone Decanoato e la supplementazione dell’aminoacido.
Nel 2016, i ricercatori rumeni avevano pubblicato un altro studio svolto su animali, condotto esattamente nelle stesse modalità, nel Journal of Medical and Biological Research. (2) In quello studio, la somministrazione di Nandrolone Decanoato aveva causato un aumentato della pressione sanguigna dei ratti trattati, ma la co-somministrazione con Taurina aveva ridotto notevolmente questo incremento pressorio.
I ricercatori affermano che, alla luce delle evidenti prove relative alla provata sicurezza della somministrazione di Taurina nell’uomo, il loro studio solleva il presupposto che la Taurina potrebbe essere utile in determinate circostanze associate ad alti livelli di androgeni circolanti in cronico, come nei disturbi endocrini o nell’abuso di AAS da parte degli atleti.
Bisognerebbe comunque prendere i risultati del presente studio con la dovuta cautela. Si tratta pur sempre di uno studio svolto su animali (ratti) e le variabili nella risultante che l’applicazione di questo protocollo integrativo potrebbero avere sull’uomo potrebbero essere assai diverse.
La dose di Nandrolone Decanoato somministrata ai ratti rapportata al dosaggio umano ammonterebbe a 200mg a settimana. Dosaggio decisamente minimale rispetto ai livelli (stupidamente) raggiunti dalla media degli utilizzatori odierni.
Per quanto modesto possa essere il dosaggio di Nandrolone Decanoato utilizzato nel presente studio, la dose di Taurina che i ricercatori somministravano ai ratti era decisamente elevata. Il dosaggio rapportato all’essere umano equivarrebbe a circa 15g di Taurina al giorno. In questo caso la dose è nettamente inferiore agli standard di utilizzo per questo aminoacido (dose massima raccomandata 8g/die). Giova ricordare che vi sono studi in attesa di conferma che indicano come un eccesso di Taurina negli adulti provochi ipertensione (il che andrebbe ad annullare i presunti benefici sulla pressione in co-somministrazione con AAS) e problemi gastrointestinali (diarrea e ulcera peptica). Sembra inoltre che l’assunzione di alti dosaggi di Taurina contribuiscano ad aggravare la psoriasi (comparsa di prurito, squame e diffusione delle lesioni cutanee).
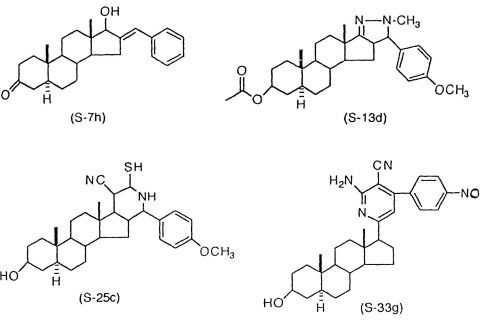
Nel mio quotidiano scartabellare la letteratura scientifica, mi sono imbattuto in un articolo pubblicato da due chimici egiziani nel 2000 su “Scientia Pharmazeutica”. I ricercatori hanno sintetizzato nuovi steroidi anabolizzanti modificando la struttura molecolare del DHT. Uno di questi AAS – l’S7h – risulta essere molto interessante .(1)
Nella pubblicazione, i ricercatori hanno descritto i quattro AAS la cui struttura è riportata di seguito. Tutte e quattro le molecola condividono la struttura simile al DHT con modifiche nell’anello D dello scheletro carbonioso.
La molecola più interessante sintetizzata dai ricercatori – l’S7h – è anche quella con meno modifiche.
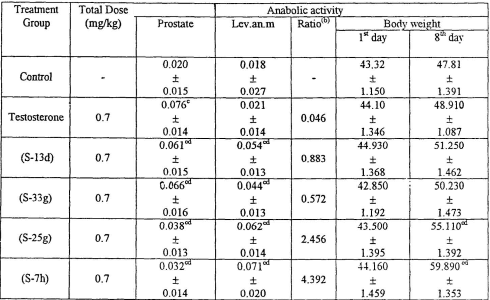
I ricercatori hanno testato l’effetto biologico dei loro nuovi AAS con una variante del test di Hershberger, utilizzando ratti Wistar maschi di 21 giorni di età pre-pubescente. I ricercatori hanno iniettato quotidianamente agli animali Testosterone, S7h, S13d, S25c o S33g alla dose di 0,7mg per Kg al giorno. Un gruppo di controllo è stato trattato con iniezioni senza sostanze attive.
Tutte le molecole hanno mostrato di avere un effetto anabolizzante, ma l’S7h ha mostrato gli effetti migliori nello studio. La somministrazione di S7h ha aumentato il peso corporeo degli animali trattati del 296% rispetto al gruppo di controllo. L’effetto del S7h sul muscolo levator ani [Lev.an.m] è stato del 246% maggiore di quello riscontrato con il Testosterone.
Allo stesso tempo, l’S7h ha avuto un effetto minore sulla dimensione della prostata rispetto a tutte le altre molecole somministrate. Ciò suggerisce che l’S7h ha un potenziale di effetti collaterali androgeni relativamente basso.
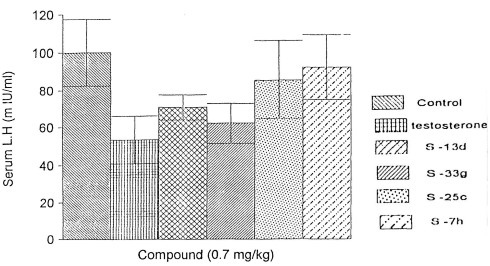
Infine, l’S7h ha esercitato un impatto contenuto sulla soppressione della sintesi di LH e Testosterone, sebbene l’effetto si è presentato e non è trascurabile.
Non ho al momento alcun dato su una sua applicazione sull’uomo, e dato che sono passati diciannove anni dallo studio dubito che ciò possa avvenire nell’ordinario ambiente accademico. E’ più probabile che questa molecola possa comparire nel mercato “grigio” come Designer steroid e/o nel mercato “nero”…
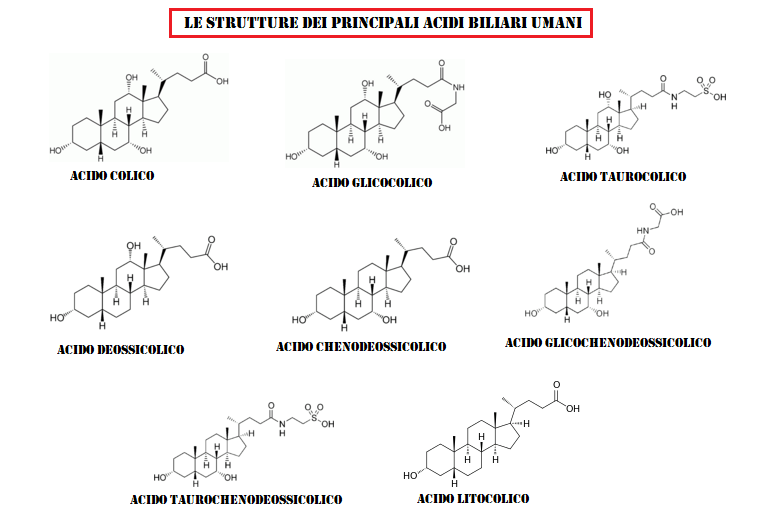
Gli acidi biliari sono acidi steroidei che si trovano principalmente nella bile dei mammiferi e di altri vertebrati. Diverse forme molecolari di acidi biliari possono essere sintetizzate nel fegato da diverse specie.[1] Gli acidi biliari sono coniugati con Taurina o Glicina nel fegato e i sali di Sodio e di Potassio di questi acidi biliari coniugati sono chiamati sali biliari.[2][3][4]
Gli acidi biliari primari sono quelli sintetizzati dal fegato. Gli acidi biliari secondari derivano da azioni batteriche nel colon. Nell’uomo, l’Acido Taurocholico e l’Acido Glicocolico (derivati dell’Acido Colico) e l’Acido Taurochenodesossicolico e l’Acido Glicocchenodesossicolico (derivati dell’Acido Chenodesossicolico) sono i principali sali biliari nella bile e hanno approssimativamente la stessa concentrazione in essa.[5] Si trovano anche i sali coniugati dei loro derivati 7-alfa-deidrossilati, Acido Desossicolico e Acido Litocolico, con derivati degli acidi Colico, Chenodesossicolico e Desossicolico che rappresentano oltre il 90% degli acidi biliari umani.[5]
Enzimi di sintesi dell’acido biliare. Nell’immagine sono mostrati i 16 enzimi e le 17 reazioni che catalizzano la conversione del Colesterolo in un sale biliare coniugato. Sono mostrate la struttura del Colesterolo e un generico sale biliare coniugato. R = OH o H; X = Glicina o Taurina.
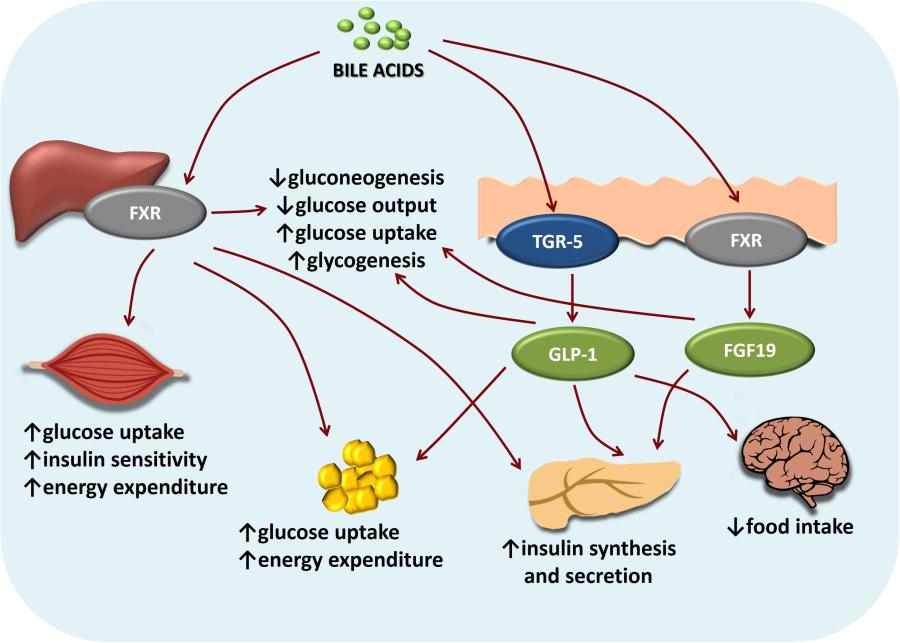
Gli acidi biliari rappresentano circa l’80% dei composti organici presenti nella bile (altri sono Fosfolipidi e Colesterolo).[5] Una maggiore secrezione di acidi biliari produce un aumento del flusso biliare. La funzione principale degli acidi biliari è quella di consentire la digestione di grassi e oli alimentari agendo come un tensioattivo che li emulsiona in micelle, [6] consentendo loro di essere sospesi colloidalmente nel chimo prima dell’ulteriore processazione. Hanno anche azioni ormonali sistemiche (in tutto il corpo), in particolare attraverso il Recettore X farnesoide e GPBAR1 (noto anche come TGR5).[7]
La sintesi degli acidi biliari si verifica nelle cellule epatiche che sintetizzano gli acidi biliari primari (Acido Colico e Acido Chenodesossicolico nell’uomo) mediante ossidazione del Colesterolo citocromo P450-mediata in un processo suddiviso in più fasi. Circa 600 mg di sali biliari vengono sintetizzati quotidianamente per sostituire gli acidi biliari persi nelle feci, anche se, come verrà descritto di seguito, ne vengono secreti quantitativi molto maggiori, riassorbiti nell’intestino e riciclati. La fase di limitazione della velocità in sintesi è costituita dall’aggiunta di un gruppo idrossilico in 7a posizione del nucleo steroideo da parte dell’enzima colesterolo 7 alfa-idrossilasi. Questo enzima è sotto-regolato dall’Acido Colico, sovra-regolato dal Colesterolo ed è inibito dall’azione dell’ormone Ileale FGF15/19.[2][3]
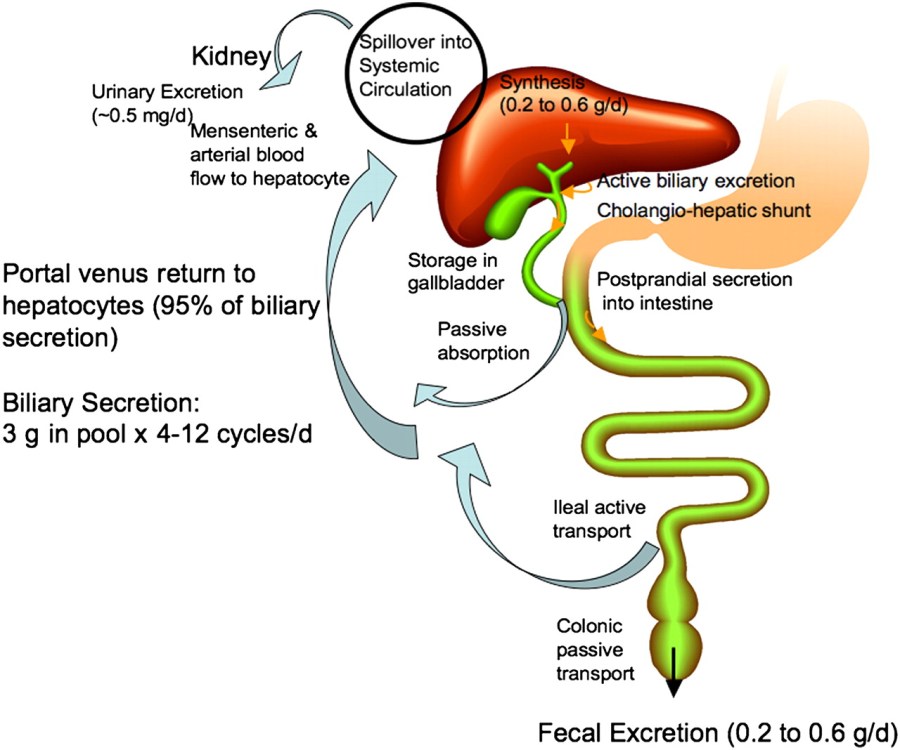
Circolazione enteropatica degli acidi biliari
Prima di secernere uno qualsiasi degli acidi biliari (primario o secondario), le cellule epatiche li coniugano con uno dei due aminoacidi, Glicina o Taurina, per formare un totale di 8 possibili acidi biliari coniugati. Questi acidi biliari coniugati sono spesso indicati come sali biliari a causa delle loro proprietà acido-base fisiologicamente importanti. Il pKa degli acidi biliari non coniugati è compreso tra 5 e 6,5, [4] e il pH del duodeno varia tra 3 e 5, quindi quando gli acidi biliari non coniugati si trovano nel duodeno, sono quasi sempre protonati (forma HA), il che li rende relativamente insolubili in acqua. Gli acidi biliari coniugati con gli aminoacidi riducono la pKa del coniugato acido biliare/aminoacido tra 1 e 4. Pertanto gli acidi biliari coniugati sono quasi sempre nella loro forma deprotonata (A-) nel duodeno, il che li rende molto più idro-solubile e molto più in grado di adempiere alla loro funzione fisiologica di grassi emulsionanti.[8][9]
Quando questi sali biliari vengono secreti nel lume intestinale, la disidrossilazione parziale batterica e la rimozione dei gruppi Glicina e Taurina formano gli acidi biliari secondari, l’Acido Desossicolico e l’Acido Litocolico. L’Acido Colico viene convertito in Acido Desossicolico e l’Acido Chenodesossicolico in Acido Litocolico. Tutti e quattro questi acidi biliari possono essere riportati nel flusso sanguigno, tornare nel fegato ed essere nuovamente secreti in un processo noto come circolazione enteroepatica.[2][3]
Come molecole anfipatiche con regioni idrofobiche e idrofile, i sali biliari coniugati si trovano in una condizione lipidica/acquosa e, alla giusta concentrazione, formano micelle.[9] La solubilità aggiunta dei sali biliari coniugati aiuta nella loro funzione prevenendo il riassorbimento passivo nell’intestino tenue. Di conseguenza, la concentrazione di acidi/sali biliari nell’intestino tenue è abbastanza elevata da formare micelle e solubilizzare i lipidi. Con il termine “concentrazione micellare critica” ci si riferisce sia a una proprietà intrinseca dell’acido biliare stesso sia alla quantità di acido biliare necessaria per adempiere alla funzionalità nella formazione spontanea e dinamica delle micelle.[9] Le micelle contenenti l’acido biliare aiutano le lipasi a digerire i lipidi e li avvicinano alla membrana dell’orletto a spazzola nell’intestino, con conseguente assorbimento dei grassi.[6]
La sintesi degli acidi biliari è una delle principali vie del metabolismo del Colesterolo nella maggior parte delle specie diverse dall’uomo. Il corpo produce circa 800mg di Colesterolo al giorno (con variabili determinate anche dal tipo di alimentazione seguita dall’individuo) e circa la metà di questi viene utilizzata per la sintesi di 400-600mg di acidi biliari al giorno. Gli esseri umani adulti secernono nell’intestino tra i 12 ed i 18g di acidi biliari ogni giorno, principalmente dopo i pasti. La dimensione del pool di acidi biliari è compresa tra i 4 ed i 6g, il che significa che gli acidi biliari vengono riciclati più volte al giorno. Circa il 95% degli acidi biliari viene riassorbito dal trasporto attivo nell’Ileo e riciclato nel fegato per un’ulteriore secrezione nel sistema biliare e nella cistifellea. Questa circolazione enteroepatica degli acidi biliari consente un basso tasso di sintesi ma con grandi quantità che vengono secrete nell’intestino.[5]
Gli acidi biliari hanno altre funzioni, tra cui l’eliminazione del Colesterolo dal corpo, nel flusso biliare per eliminare alcuni cataboliti (compresa la bilirubina), come emulsionante per le vitamine liposolubili e consentirne il loro assorbimento, favorire la motilità e la riduzione della flora batterica presente nell’intestino tenue e il tratto biliare.[5]
Gli acidi biliari hanno azioni metaboliche nel corpo simili a quelle degli ormoni, agendo attraverso due recettori specifici, i prima citati Recettore X farnesoide e il Recettore degli Acidi Biliari accoppiato con Proteine G/TGR5.[7][10] Si legano in modo meno specifico ad alcuni altri recettori ed è stata osservata una loro azione nella regolazione dell’attività di alcuni enzimi [11], canali ionici [12] e la sintesi di diverse sostanze tra cui Etanolamidi di acidi grassi endogeni.
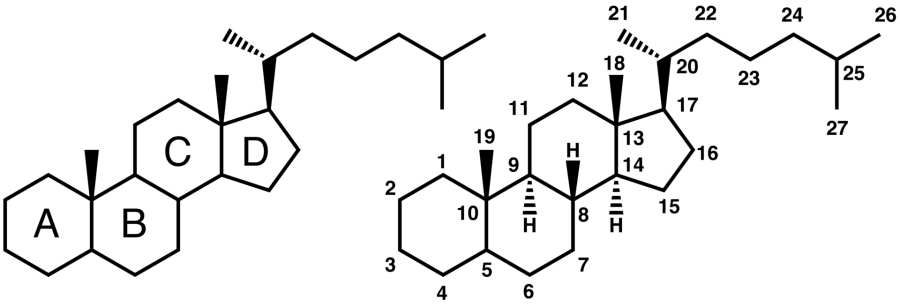
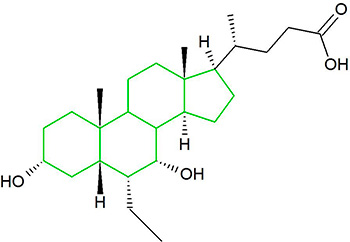
I sali biliari costituiscono una grande famiglia di molecole, ed esse sono formate da una struttura steroidea con quattro anelli, una catena laterale a cinque o otto atomi di Carbonio che termina in un Acido Carbossilico e diversi gruppi idrossilici, il cui numero e orientamento sono diversi tra gli specifici sali biliari.[1] I quattro anelli sono classificati con le lettere A, B, C e D, dal più lontano al più vicino alla catena laterale con il gruppo carbossilico. L’anello D è più piccolo di un carbonio rispetto agli altri tre. La struttura è comunemente rappresentata con A a sinistra e D a destra. I gruppi idrossilici possono essere in una delle due configurazioni: in alto (o fuori), definita beta (β; spesso disegnata per convenzione come linea continua) o in basso, definita alfa (α; visualizzata come linea tratteggiata). Tutti gli acidi biliari hanno un gruppo 3-idrossile, derivato dalla molecola madre, il Colesterolo, in cui il 3-idrossile è beta. [1]
Da sinistra: struttura steroidea del Colesterolo rappresentata con i quattro anelli (A-B-C-D) e la sua struttura con la numerazione degli atomi secondo le indicazione della IUPAC.
Il primo passo nella via classica della sintesi epatica degli acidi biliari è l’aggiunta enzimatica di un gruppo idrossilico in posizione 7α da parte del Colesterolo 7α-idrossilasi (CYP7A1) che forma 7α-idrossicolesterolo. Questo viene quindi metabolizzato in 7α-idrossi-4-colesten-3-one. Ci sono più passaggi nella sintesi di un acido biliare che richiedono in tutto 14 enzimi.[3] Ciò comporta l’alterazione del legame tra i primi due anelli steroidei (A e B), che fa piegare la molecola; in questo processo, il 3-idrossile viene convertito nell’orientamento α. L’acido biliare a 24 atomi di Carbonio più semplice ha due gruppi idrossilici nelle posizioni 3α e 7α. Questo è l’Acido 3α, 7α-diidrossi-5β-colan-24-oico o, come più comunemente noto, Acido Chenodesossicolico. Questo acido biliare fu inizialmente isolato dall’oca domestica, dal cui nome deriva la porzione “cheno” del nome della molecola. Il 5β nel nome indica l’orientamento del legame tra gli anelli A e B del nucleo steroideo (in questo caso, sono piegati). Il termine “colan” indica una particolare struttura steroidea di 24 atomi di Carbonio e “acido 24-oico” indica che l’Acido Carbossilico si trova in posizione 24, alla fine della catena laterale. L’Acido Chenodesossicolico è sintetizzato da molte specie ed è il prototipo dell’acido biliare funzionale.[2][3]
Una via alternativa (acida) della sintesi dell’acido biliare è iniziata dalla sterolo mitocondriale 27-idrossilasi (CYP27A1), espresso nel fegato, nei macrofagi e in altri tessuti. Il CYP27A1 contribuisce in modo significativo alla sintesi totale dell’acido biliare catalizzando l’ossidazione della catena laterale dello sterolo, dopo di che la scissione di un’unità a tre atomi di Carbonio nei perossisomi porta alla formazione di un acido biliare C24. Anche le vie minori iniziate dalla 25-idrossilasi nel fegato e dalla 24-idrossilasi nel cervello possono contribuire alla sintesi dell’acido biliare. La 7α-idrossilasi (CYP7B1) genera ossisteroli, che possono essere ulteriormente convertiti nel fegato in CDCA.[2][3]
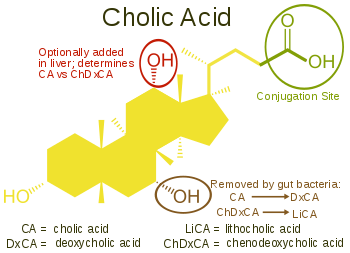
L’Acido Colico, 3α, 7α, 12α-triidrossi-5β-colan-24-oico, l’acido biliare più abbondante nell’uomo e in molte altre specie, è stato scoperto prima dell’Acido Chenodesossicolico. È un acido tri-idrossile-biliare con 3 gruppi idrossilici (3α, 7α e 12α). Nella sua sintesi epatica, l’idrossilazione 12α viene eseguita dall’azione aggiuntiva del CYP8B1. Come già descritto, la scoperta dell’Acido Chenodesossicolico (con 2 gruppi ossidrilici) ha reso la denominazione di questo nuovo acido biliare “Acido Desossicolico” in quanto aveva un gruppo idrossilico in meno rispetto all’Acido Colico.[2][3]
L’Acido Desossicolico è formato dall’Acido Colico mediante 7-deidrossilazione, risultando in 2 gruppi idrossilici (3α e 12α). Questo processo con Acido Chenodesossicolico da come risultato un acido biliare con solo un gruppo idrossilico 3α, chiamato Acido Litocolico (lito = pietra) che è stato isolato e classificato per la prima volta attraverso l’analisi di calcoli prelevati da un polpaccio. È scarsamente solubile in acqua e piuttosto tossico per le cellule.[2][3]
Diverse famiglie di vertebrati sono adatte ad utilizzare le modifiche della maggior parte delle posizioni sul nucleo steroideo e sulla catena laterale della struttura dell’acido biliare. Per evitare i problemi associati alla produzione di Acido Litocolico, la maggior parte delle specie è in grado di aggiunge un terzo gruppo ossidrilico all’Acido Chenodesossicolico. La successiva rimozione del gruppo idrossilico in posizione 7α da parte dei batteri intestinali si tradurrà quindi in un acido biliare diidrossile meno tossico ma ancora funzionale. Si ipotizza che nel corso del tempo, le locazioni per il posizionamento del terzo gruppo ossidrilico nei vertebrati si sia modificato. Sembrerebbe che, inizialmente, la posizione 16α fosse la principale, in particolare negli uccelli. Successivamente, questa posizione avrebbe lasciato il posto in un gran numero di specie alla posizione 12α. I primati (e gli esseri umani) utilizzano il 12α per la loro terza posizione del gruppo ossidrilico, producendo Acido Colico. Nei topi e in altri roditori, l’idrossilazione 6β forma acidi muricolici (α o β a seconda della posizione dell’idrossile 7). I suini hanno una idrossilazione 6α nell’Acido Ecolico (acido 3α, 6α, 7α-triidrossi-5β-cholanoico) e altre specie hanno un gruppo ossidrilico nella posizione 23 della catena laterale.
L’Acido Ursodesossicolico è stato inizialmente isolato dalla bile di orso, la quale è stata usata in medicina per secoli. La sua struttura ricorda l’Acido Chenodesossicolico ma con il gruppo 7-idrossile in posizione β.[1]
L’Acido Obeticolico, l’acido 6α-etil-chenodesossicolico, è un acido biliare semisintetico con maggiore attività come agonista del FXR che è oggetto di ricerche come agente farmacologico.
Acido Obeticolico (OCA). Questo acido biliare semisintetico ha dimostrato di migliorare i livelli ematici di ALT e ALP nei soggetti non responsivi al trattamento con Acido Ursodesossicolico (UDCA), ed è l’unica terapia di seconda linea autorizzata. Negli studi clinici l’OCA ha migliorato i risultati degli esami epatici nell’87% dei soggetti trattati ed è stato di aiuto quasi nel 50% dei pazienti.
Gli acidi biliari fungono anche da ormoni steroidei, secreti dal fegato, assorbiti dall’intestino e con varie azioni metaboliche dirette nel corpo attraverso il recettore nucleare FXR, noto anche con il nome genico NR1H4. [14][15][16] Un altro recettore degli acidi biliari è il recettore della membrana cellulare noto come recettore 1 o TGR5 degli acidi biliari accoppiato con proteine G. Molte delle loro funzioni come molecole di segnalazione nel fegato e nell’intestino sono mediate attivando il FXR, mentre l’interazione con il TGR5 può avviare coinvolgimenti nelle funzioni metaboliche, endocrine e neurologiche.[7]
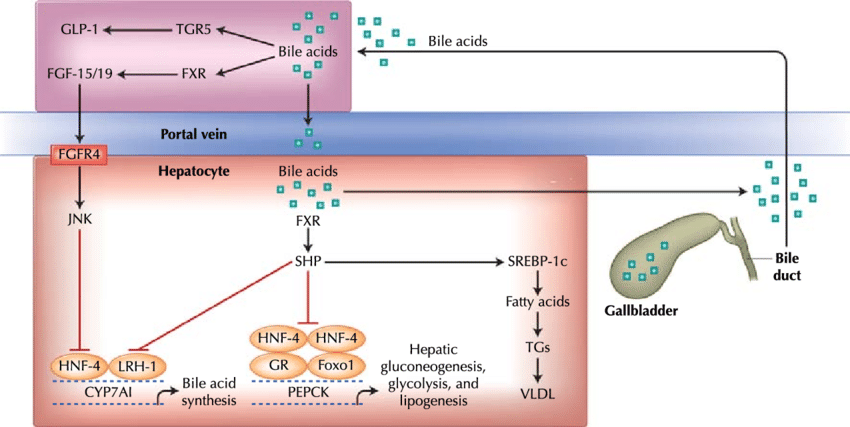
In quanto tensioattivi o detergenti, gli acidi biliari sono potenzialmente tossici per le cellule e quindi le loro concentrazioni sono strettamente regolate. L’attivazione del FXR nel fegato inibisce la sintesi di acidi biliari ed è un meccanismo di controllo a feedback quando i livelli di acidi biliari sono troppo alti. In secondo luogo, l’attivazione del FXR da parte degli acidi biliari durante l’assorbimento nell’intestino aumenta la trascrizione e la sintesi di FGF19, che quindi inibisce la sintesi epatica di acidi biliari.[17]
Regolazione FXR e TGR5-mediata della sintesi degli acidi biliari e del metabolismo dei lipidi e del glucosio nel fegato e nell’intestino.
Prove emergenti associano l’attivazione del FXR con modifiche del metabolismo dei trigliceridi, del metabolismo del glucosio e della crescita del fegato.[7][18]
Gli acidi biliari si legano ad alcune altre proteine oltre ai loro prima citati recettori ormonali (FXR e TGR5) e ai loro trasportatori. Tra questi target proteici, l’enzima N-acil fosfatidiletanolammina specifica fosfolipasi D (NAPE-PLD) genera ammidi lipidici bioattivi (ad esempio l’Anandamide Cannabinoide Endogeno) che svolgono ruoli importanti in diversi percorsi fisiologici tra cui stress e risposte al dolore, appetito e durata della vita. La NAPE-PLD regola un dialogo di segnali incrociati diretti tra ammide lipidica e fisiologia degli acidi biliari.[13]
Poiché gli acidi biliari sono costituiti da Colesterolo endogeno, l’interruzione della circolazione enteroepatica degli acidi biliari abbasserà il Colesterolo. I sequestranti degli acidi biliari si legano a questi ultimi nell’intestino, prevenendone il riassorbimento. Così facendo, il Colesterolo endogeno viene deviato nella produzione di nuovi acidi biliari, riducendo così i livelli di Colesterolo. Gli acidi biliari sequestrati vengono quindi escreti nelle feci.[19]
I test per gli acidi biliari sono utili sia nella medicina umana che in quella veterinaria, poiché aiutano nella diagnosi di una serie di condizioni patologiche, tra cui vi sono i tipi di colestasi come la colestasi intraepatica della gravidanza, shunt portosistemico e displasia microvascolare epatica nei cani.[20] Anomalie strutturali o funzionali del sistema biliare provocano un aumento della bilirubina (ittero) e degli acidi biliari nel sangue. Gli acidi biliari sono correlati alla comparsa di prurito che è comune in condizioni colestatiche come nella Cirrosi Biliare Primaria (PBC), Colangite Sclerosante Primaria o Colestasi Intraepatica della Gravidanza.[21] Il trattamento con Acido Ursodesossicolico è stato usato per molti anni nel trattamento di questi disturbi colestatici.[22][23]
La relazione tra acidi biliari e saturazione del Colesterolo nelle precipitazioni di bile e Colesterolo nella formazione di calcoli biliari è stata ampiamente studiata. I calcoli biliari possono derivare da una maggiore saturazione di Colesterolo o bilirubina o da stasi biliare. Concentrazioni più basse di acidi biliari o fosfolipidi nella bile riducono la solubilità del Colesterolo e portano alla formazione di microcristalli. La terapia orale con acido Chenodesossicolico e/o Acido Ursodesossicolico è stata utilizzata per dissolvere i calcoli biliari di Colesterolo.[24][25][26] I calcoli possono ripresentarsi quando il trattamento viene interrotto. La terapia con un acido biliare può essere utile per prevenire i calcoli in determinate circostanze, come in seguito alla chirurgia bariatrica.[27]
Concentrazioni eccessive di acidi biliari nel colon sono una causa di diarrea cronica. Si manifesta comunemente quando l’Ileo è anormale o è stato rimosso chirurgicamente, come nella malattia di Crohn, o causa una condizione che ricorda la sindrome dell’intestino irritabile predominante nella diarrea (IBS-D). Questa condizione di diarrea da acido biliare/malassorbimento di acido biliare può essere diagnosticata dal test SeHCAT e trattata con sequestranti di acido biliare.[28]
Gli acidi biliari possono avere una certa importanza nello sviluppo del cancro del colon-retto.[29] L’Acido Desossicolico (DCA) risulta aumentato nelle sue concentrazioni nel colon dell’uomo in risposta a una dieta ricca di grassi.[30] Nelle popolazioni con un’alta incidenza di carcinoma del colon-retto, le concentrazioni fecali di acidi biliari sono più elevate, [31] [32] e questa associazione suggerisce che una maggiore esposizione del colon agli acidi biliari potrebbe svolgere un ruolo nello sviluppo del cancro. In un confronto particolare, le concentrazioni fecali di DCA nei nativi africani del Sudafrica (che seguono una dieta povera di grassi) rispetto agli afroamericani (che seguono una dieta ricca di grassi) erano 7,30 contro 37,51 nmol/g di feci umide.[33] I nativi africani del Sud Africa hanno un basso tasso di incidenza di cancro al colon inferiore a 1: 100.000, [34] rispetto all’alto tasso di incidenza per i maschi afroamericani che è di 72: 100.000.[35]
Attraverso studi sperimentali sono stati ipotizzati meccanismi che potrebbero regolare le alte concentrazioni di acidi biliari al carcinoma del colon. L’esposizione delle cellule del colon ad alte concentrazioni di DCA aumenta la formazione di specie reattive dell’ossigeno, causando stress ossidativo e aumentando anche il danno nel DNA.[36] I topi nutriti con una dieta con aggiunta di DCA che mimava i livelli di DCA del colon negli esseri umani che seguono una dieta ricca di grassi hanno sviluppato neoplasie del colon, tra cui adenomi e adenocarcinomi (tumori), a differenza dei topi alimentati con una dieta di controllo che causa un decimo delle concentrazioni di DCA nel colon e non hanno sviluppato neoplasie del colon.[37][38]
Gli effetti dell’Acido Ursodesossicolico (UDCA) nella modifica del rischio di sviluppo del carcinoma del colon-retto sono stati esaminati in diversi studi, in particolare nella colangite sclerosante primitiva e nella malattia infiammatoria intestinale, con risultati variabili in parte correlati al dosaggio.[39][40] La variazione genetica dell’enzima chiave di sintesi dell’acido biliare, CYP7A1, ha influenzato l’efficacia dell’UDCA nella prevenzione dell’adenoma colorettale in un ampio studio.[41]
Gli acidi biliari possono essere utilizzati in soluzione iniettabile da somministrare sottocute per il trattamento delle adiposità localizzate (vedi Mesoterapia). L’Acido Desossicolico in soluzione iniettabile ha ricevuto l’approvazione della FDA per il trattamento del grasso submentale.[42] Gli studi di fase III hanno mostrato risposte significative sebbene molti soggetti abbiano avuto lievi reazioni avverse come lividi, gonfiore, dolore, intorpidimento, eritema e rigidità intorno all’area trattata.[43][44] Della correlazione tra acidi biliari e tessuto adiposo, punto di particolare interesse in questo articolo, ne parlerò a breve.
Acido Ursodesossicolico
Gli atleti che utilizzano PED conoscono ormai da tempo il potenziale “epatoprotettivo” dato dall’uso degli acidi biliari. L’Acido Tauroursodesossicolico (TUDCA) e l’Acido Ursodesossicolico (UDCA) sono generalmente utilizzati come parte integrante dei “mix” epatoprotettivi, in specie durante l’utilizzo di farmaci orali con una forte resistenza al metabolismo epatico (vedi, per esempio, AAS metilati in C17). Infatti, l’UDCA ha mostrato di:
stimolare la secrezione di ATP da parte degli epatociti e di interagire quindi col sistema dei citocromi P450 riducendo la glicuronazione degli estrogeni sintetici (cosa che spiega in parte i suoi effetti benefici sulla colestasi epatica). Gli utilizzatori di AAS potrebbero non essere consapevoli del rischio aumentato di sviluppare calcoli biliari durante l’uso di molecole aromatizzabili e dell’aiuto nell’arginare il fenomeno correlato ad un aumento dei livelli estrogenici sul fattore in questione dato dal UDCA [45];
attivare direttamente il recettore per i glucocorticoidi, il che contribuirebbe ad allargare i meccanismi della sua azione anticolestatica ed antinfiammatoria sul parenchima epatico;
di fungere (come già accennato in precedenza) da agonista parziale del recettore FXRalpha coinvolto nell’espressione di proteine ed enzimi protettivi e/o regolatori del metabolismo intermedio riducendo la colestasi e l’intossicazione epatica stimolando dei meccanismi endogeni di difesa;
stimolare la sintesi del Glutatione (GSH), potente antiossidante endogeno, attraverso l’intervento delle chinasi dipendenti dai Fosfoinositidi (PI-3K e PKB). Questo meccanismo potrebbe giustificarne l’impiego corrente nelle epatiti croniche;
attivare un fattore di trascrizione particolare chiamato Nrf-2, presente allo stato di riposo nel citoplasma di tutte le cellule. Esso si attiva solo quando vi sono variazioni dello stato ossidoriduzione cellulare, ovvero la genesi di eccessive quantità di radicali liberi dell’ossigeno, o delle tossine esterne chiamate (xenobiotici). In risposta a questi stimoli, l’Nrf-2 entra nel nucleo cellulare ed induce una batteria di geni coinvolti nella protezione cellulare dallo stress ossidativo. Si pensa quindi che l’UDCA possa agire da ligando endogeno dell’Nrf-2, anche se non esistono ancora prove dirette o conclusive. Anche questo meccanismo ha azione protettiva sul tessuto epato-biliare.[46]
Come detto pocanzi, un altro uso degli acidi biliari conosciuto dai più in ambito sportivo e, soprattutto, culturistico, è quello, già accennato in precedenza, del trattamento mesoterapico. In particolare, Il Desossicolato di Sodio usato da solo o miscelato con Fosfatidilcolina, viene utilizzato nel trattamento delle adiposità localizzate (soprattutto nella zona submentale e periombellicale) in alternativa all’escissione chirurgica.[47]
Lo sviluppo della ricerca scientifica sugli acidi biliari e metabolismo/composizione corporea
Pratica di utilizzo degli acidi biliari meno conosciuta, ed ancora “embrionale” nella sua applicazione, è quella che vede la loro applicazione per il miglioramento della composizione corporea attraverso assunzione orale.
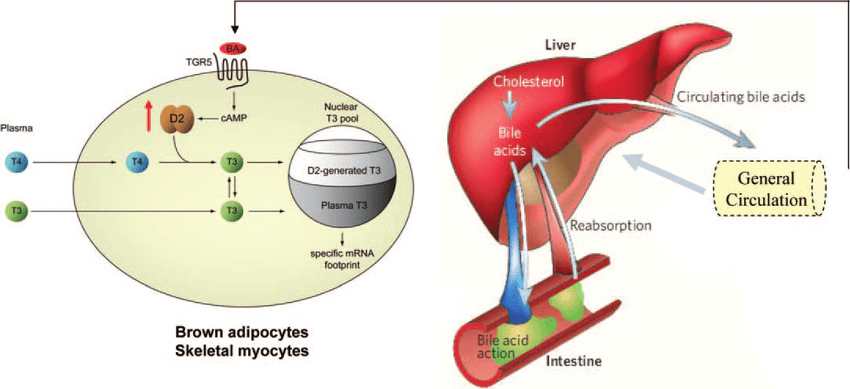
Già nel 2006 uno studio pubblicato sulla rivista Nature aveva rivelato che gli acidi biliari potevano aumentare il tasso metabolico e la perdita di peso nei topi, lasciando intravedere la possibilità che ciò si potesse verificare anche nell’uomo.[48] Mentre gli acidi biliari (AB) erano da tempo noti per essere essenziali nell’assorbimento dei lipidi nella dieta e nel catabolismo del Colesterolo, come abbiamo precedentemente visto, negli anni precedenti a questo studio era emerso un ruolo importante degli acidi biliari come molecole di segnalazione. Gli AB, come già detto, attivano le vie della protein chinasi attivate dal mitogeno, sono ligandi per il recettore TGR5 (GPCR) accoppiato alle proteine G e attivano i recettori dell’ormone nucleare come il recettore alfa X farnesoide (FXR-alfa; NR1H4). Il FXR-alfa regola il riciclo enteroepatico e la biosintesi degli AB controllando l’espressione di geni come il partner eterodimero breve (SHP; NR0B2) che inibisce l’attività di altri recettori nucleari. L’induzione SHP mediata da FXR-alfa è anche alla base della sottoregolazione della biosintesi di acidi grassi e dei Trigliceridi a livello epatico e della produzione di lipoproteine a bassissima densità mediata dalla proteina 1c legante gli elementi regolatori degli steroli. Ciò indicava che l’assunzione di AB avrebbe potuto essere in grado di funzionare al di là del semplice controllo dell’omeostasi degli stessi nelle vie metaboliche generali. Nello studio viene mostrato che la somministrazione di AB nei topi aumenta il dispendio energetico nel tessuto adiposo bruno, prevenendo l’obesità e il peggioramento della resistenza all’insulina. Questo nuovo effetto metabolico degli AB dipende in modo critico dall’induzione della sintesi dell’ormone tiroideo tri-iodotironina (T3) AMP-ciclico-dipendente attraverso l’attivazione l’enzima iodotironina deiodinasi di tipo 2 (D2). Il trattamento degli adipociti bruni e dei miociti muscolo-scheletrici umani con AB mostrò un aumento dell’attività del D2 e del consumo di ossigeno. Questi effetti sono indipendenti dall’FXR-alfa e sono invece mediati dall’aumentata produzione di cAMP che deriva dal legame degli AB con il recettore TGR5 accoppiato con proteine G. Sia nei roditori che nell’uomo, i tessuti più termogenicamente importanti sono specificamente presi di mira da questo meccanismo poiché coesprimono sia l’enzima D2 che il TGR5. Dallo studio emerse quindi che la via di segnalazione AB-TGR5-cAMP-D2 è un meccanismo cruciale per l’ottimizzazione dell’omeostasi energetica che può essere mirata a migliorare il controllo metabolico.
Gli acidi biliari stimolano l’espressione del D2 negli adipociti bruni. Rappresentazione schematica della via dell’acido biliare-TGR5 – D2 negli adipociti bruni Gli acidi biliari nella circolazione generale derivati dalla circolazione enteroepatica possono potenzialmente stimolare l’aumento del TAMP del cAMP, portando così ad un aumento dell’espressione del D2 nei tessuti in cui entrambe le proteine sono coespresse, ad esempio nel BAT e nel muscolo scheletrico. Questo percorso ha dimostrato di aumentare il dispendio energetico e proteggere dall’obesità indotta dalla dieta nei topi. Riprodotto in parte con il permesso di Baxter e Webb: Nature 439: 402–403, 2006 (446).
Dal 2006 ad oggi la ricerca sugli AB ed i loro effetti a livello metabolico-energetico sono proseguiti con ulteriori conferme sulla loro applicabilità per il trattamento dell’obesità negli esseri umani.
Uno studio del 2018 [49] ha evidenziato come il TGR5 sia un mediatore della conversione del tessuto adiposo bianco sottocutaneo (scWAT) in beige (metabolicamente simile al tessuto adiposo marrone) attraverso molteplici stimoli ambientali, tra cui l’esposizione al freddo e una alimentazione ricca di grassi. Inoltre, la somministrazione di mimetici degli acidi biliari TGR5-selettivi nei topi tenuti in ambiente termoneutrale porta alla comparsa di marker adipocitari beige e aumenta il contenuto mitocondriale nel tessuto scWAT dei topi Tgr5+/+ ma non nei loro simili Tgr5 -/- che si trovavano con loro e, quindi, esposti alle medesime condizioni ambientali. Questo fenotipo viene ricapitolato in vitro in adipociti differenziati, in cui l’attivazione del TGR5 aumenta la disponibilità di acidi grassi liberi attraverso la lipolisi, aumentando quindi la β-ossidazione e l’attività termogenica. La segnalazione del TGR5 induce anche la fissione mitocondriale attraverso il percorso ERK/DRP1, migliorando ulteriormente la respirazione mitocondriale. Nel loro insieme, questi dati identificano il TGR5 come un bersaglio farmacologico per promuovere la conversione del tessuto adiposo bianco in beige con potenziali applicazioni nella gestione dei disturbi metabolici.
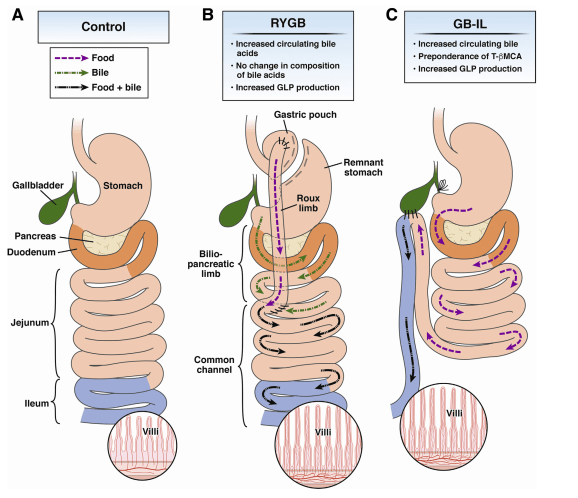
Un altro studio, sempre del 2018, ha approfondito le dinamiche legate allo spiccato effetto metabolico e saziante, molto simile a quello ottenuto tramite bypass gastrico Roux-en-Y (RYGB), correlato alla diversione biliare verso l’Ileo (GB-IL) in modelli animali (roditori) obesi.[50] I ricercatori volevano appurare se i benefici metabolici di queste procedure (bypass gastrico) fossero mediate dall’aumento degli acidi biliari, dal momento che variazioni parallele del peso corporeo e altre variabili confondenti limitavano una possibile interpretazione oggettiva. Per fare ciò hanno utilizzato topi TGR5 -/- o FXRalfa -/- sottoposti ad una alimentazione ricca di grassi, confrontandoli con topi “selvaggi” con un antagonista del recettore per il polipeptide glucagone-simile 1 (Glp-1r) sottoposti ad una dieta chow (ricca in cereali e fibre). Il GB-IL ha indotto la perdita di peso e ha migliorato la tolleranza al glucosio assunto oralmente nei topi Tgr5 – / -, ma non nei topi FxrΔ -/- alimentati con una dieta ricca di grassi, suggerendo un ruolo dell’FXR intestinale. Il GB-IL in topi di tipo “selvaggio” alimentati con cibo convenzionale ha indotto miglioramenti della tolleranza al glucosio e al controllo della glicemia ematica indipendenti dal peso e secondari alla risposta aumentata dell’insulina. I miglioramenti erano concomitanti con un aumento dei livelli di GLP-1 linfatico nello stato a digiuno e un aumento dei livelli del batterio intestinale Akkermansia muciniphila. I miglioramenti nella glicemia a digiuno dopo il GB-IL sono stati mitigati con exendin-9, un antagonista del recettore del GLP-1 o Colestiramina, un sequestrante degli acidi biliari. Gli effetti glucoregolatori del GB-IL sono stati persi nei topi Glp-1r -/- a livello sistemico.
Confronto tra le caratteristiche anatomiche e il flusso di cibo e bile prima (Controllo) e dopo RYGB e GB-IL. (A) In risposta a un pasto, la bile della cistifellea e i succhi pancreatici vengono rilasciati nel duodeno (arancione) dove aiutano la scomposizione e l’assorbimento dei grassi alimentari mentre attraversano l’intestino tenue (digiuno e ileo). Gli acidi biliari vengono riassorbiti nell’ileo terminale (blu) in una circolazione enteroepatica elaborata. (B) Dopo RYGB, il cibo ingerito (punteggiato di viola) e la bile (frecce verdi interrotte) formano un mix (frecce rotte nere) ritardando l’assorbimento dei lipidi al digiuno prossimale / medio. (C) Nella diversione biliare verso l’ileo la miscelazione di nutrienti e bile viene ritardata fino all’ileo terminale.
Per avere una panoramica dettagliata ed estremamente chiara sui potenziali effetti metabolici e, con sequenzialmente, sulla composizione corporea dati dall’uso di acidi biliari ci viene in aiuto una interessantissima review che vaglia tutti gli studi svolti fino al 2018 sulle possibili applicazioni farmacologiche degli acidi biliari e dei loro derivati nel trattamento della sindrome metabolica, pubblicata sulla rivista “Frontiers in Pharmacology” del 3 Dicembre 2018.[51] Anche in questo caso, glia autori sottolineano come, oltre alle classiche funzioni degli acidi biliari nella digestione e nella solubilizzazione dei nutrienti e dei farmaci lipofili nell’intestino tenue (Mikov e Fawcett, 2006; Mircioiu et al., 2012), ci sono prove emergenti che indicano il ruolo degli acidi biliari e il loro derivati come segnalatori, molecole endocrine che esercitano una varietà di effetti metabolici attraverso percorsi complessi e intrecciati, diventando così una classe di moecole molto interessanti per la ricerca volta al vaglio di nuovi trattamenti per la sindrome metabolica (Taoka et al., 2016; Chávez-Talavera et al., 2017; de Boer et al., 2018; Molinaro et al., 2018; Shapiro et al., 2018). Questa review fornisce una panoramica delle attuali conoscenze relative alle implicazioni degli acidi biliari nel metabolismo del glucosio, dei lipidi e delle vie energetiche, nonché la potenziale applicazione degli acidi biliari nel trattamento della sindrome metabolica con raccomandazioni per ulteriori studi.
Nella review è nuovamente riportato come il ricircolo enteroepatico degli acidi biliari amplifica il flusso transepatico degli acidi biliari che attivano il recettore X farnesoide (FXR), che ormai sappiamo essere il fattore di trascrizione più significativo coinvolto nella regolazione della biosintesi e del trasporto degli acidi biliari. Inoltre, il flusso transintestinale di acidi biliari riassorbiti attiva la FXR intestinale, regolando il ricircolo enteroepatico di queste molecole anfifiliche. Questi eventi epatici e intestinali prevengono il sovraccarico e l’accumulo di acidi biliari negli epatociti evitando così la possibilità che si creino concentrazioni tossiche prevenendo così potenziali lesioni epatocellulari, diminuendo la loro stessa sintesi e assorbimento, mantenendo allo stesso tempo quantità sufficienti di acidi biliari nell’albero biliare e nel lume intestinale per l’emulsificazione dei lipidi alimentari . Inoltre, l’assorbimento epatocellulare degli acidi biliari è incompleto e la gamma micromolare delle concentrazioni si riversa dal portale alla circolazione sistemica attraverso anastomosi venose. Queste concentrazioni sistemiche sono sufficienti per interagire con diversi recettori nucleari attualmente identificati; FXR, recettore X della gravidanza (PXR), recettore della vitamina D (VDR) e recettore costitutivo del androstano (CAR), nonché il già più volte citato TGR5 (o GPBAR) che esercita effetti di segnalazione sistemici oltre i tessuti enteroepatici (Gioiello et al., 2014; Comeglio et al., 2017).
E’ interessante notare come la transattivazione ligando-dipendente di geni bersaglio da parte degli acidi biliari è indotta dal legame dell’acido biliare come ligandi endogeni più potenti, con il CDCA come il più potente agonista, mentre l’UDCA, idrofilo, non attiva l’FXR (Kemper, 2011). La potenza degli acidi biliari naturali per il FXR è riassunta nella tabella seguente (secondo Gioiello et al., 2014). Nell’intestino, il FXR attiva la trascrizione del enterokine, fattore di crescita dei fibroblasti-19 (FGF-19 o ortologo FGF-15 roditore) attraverso SHP, governando gli acidi biliari post-prandiale e il metabolismo dei nutrienti (Mertens et al., 2017).
SRC-1
Inoltre, come endobiotici, gli acidi biliari possono attivare la PXR (NR1I2, cromosoma 3q13.33), che possiede la capacità di interagire con una vasta gamma di composti idrofobici strutturalmente diversi tra cui farmaci, integratori alimentari e inquinanti ambientali. La PXR-non attivata più comunemente crea complessi inibitori con il mediatore silenziante per il recettore dell’acido retinoico e il recettore dell’ormone tiroideo (SMRT), SHP e deacetylases dell’istone (Pavlovic et al., 2017). Dopo il legame con il ligando, il recettore subisce cambiamenti conformazionali e il rilascio di fattori corepressori induce l’acetilazione dell’istone o il reclutamento di proteine co-attivanti come il SRC–1 (steroid receptor coactivator–1) (Pavek, 2016; Buchman et al., 2018).
Acidi biliari e metabolismo glucidico
Nell’ultimo decennio, un numero crescente di prove ha fortemente indicato che gli acidi biliari regolano il metabolismo glucidico postprandiale mostrando anche capacità di segnalazione antidiabetica esplicata attraverso i recettori attivati dall’acido biliare, oltre che a migliorare il ripiegamento e la funzione delle proteine, e agli effetti antiapoptotico (Hylemon et al., 2009).
Diagramma schematico del recettore (I) FXR nel fegato, (II) recettore FXR nell’intestino, (IIIA) recettore TGR5 nell’intestino, (IIIB) recettore TGR5 nella cistifellea e (IIIC) recettore TGR5 negli adipociti e nei muscoli.
Vi sono, tuttavia, alcune discrepanze irrisolte in letteratura tra i risultati ottenuti relativi all’implicazione degli acidi biliari nel metabolismo glucidico. Gli studi iniziali che hanno identificato le implicazioni del FXR nel metabolismo del glucosio hanno dimostrato che gli acidi biliari o gli agonisti sintetici specifici del FXR hanno indotto l’espressione del tasso di controllo dell’enzima della gluconeogenesi, fosfoenolpiruvato carbossinasi (PEPCK), aumentando anche la produzione totale di glucosio nell’epatocita umano e di ratto come nei topi in vivo (Stayrook et al., 2005). Al contrario, Yamagata et al. (2004) hanno dimostrato che gli acidi biliari sopprimono l’espressione dei geni della gluconeogenesi PEPCK, glucosio-6-fosfatasi (G6Pase) e fruttosio 1,6-bis-fosfatasi (FBP1) attraverso l’interazione tra SHP con il fattore nucleare degli epatociti 4 (HNF-4) o fattore di trascrizione della fronte Foxo1, sia in vivo che in vitro. La riduzione dell’espressione del PEPCK sembra essere una possibilità interessante per il trattamento del diabete di tipo 2, dato che questa patologia è caratterizzato da un aumento della produzione di glucosio epatico e da iperglicemia (Cariou et al., 2005). Inoltre, l’attivazione epatica del FXR porta ad un aumento dell’attività della glicogeno sintasi mediante fosforilazione e inattivazione della glicogeno sintasi chinasi 3 (GSK3b) (Mencarelli e Fiorucci, 2010).
GW4064
Nello studio del 2006 citato in precedenza (di Zhang et al.) è stato mostrato che la carenza del FXR nei topi è associata a intolleranza al glucosio e resistenza all’insulina manifestate da iperglicemia, ridotta tolleranza al glucosio e grave riduzione della sensibilità insulinica nel fegato, nei muscoli e nel tessuto adiposo. Di conseguenza, l’attivazione del FXR da parte dell’agonista sintetico non steroideo GW4064 nei topi (30mg/kg due volte al giorno) ha significativamente ridotto la produzione di glucosio epatico, abbassato i livelli di glucosio nel sangue, aumentato la glicogenesi, migliorato la sintesi e la secrezione di insulina, migliorato la sensibilità all’Insulina a livello centrale (epatico) e periferico negli animali (Zhang et al., 2006).
L’attivazione indotta del FXR nella trascrizione e nella secrezione di Insulina nelle cellule β del pancreas sono regolate da diversi meccanismi che coinvolgono effetti sia genomici che non genomici. Gli effetti genomici dell’attivazione del FXR si basano sull’induzione del KLF11, che si è dimostrato essere un fattore essenziale per la trascrizione del gene dell’Insulina. Gli effetti non genomici dell’attivazione del FXR nelle cellule βTC6 trasmettono sull’aumento della fosforilazione di Akt e nella traslocazione del trasportatore di glucosio di tipo 2 (GLUT2), un membro delle proteine di membrana che facilita il trasporto del glucosio lungo un gradiente di concentrazione alla membrana plasmatica, aumentando l’assorbimento di glucosio da parte delle cellule β del pancreas (Renga et al., 2010). Inoltre, l’attivazione del FXR induce l’espressione del trasportatore di glucosio GLUT4 nel fegato, la cui espressione risulta ridotta, sia nei soggetti diabetici di tipo 1 che 2 (Garvey et al., 1991, 1992). L’attivazione del FXR è stata in grado di sovraregolare l’espressione dei GLUT4 attraverso il FXRE nel promotore del gene GLUT4. Shen et al. (2008) hanno riferito che l’attivazione del FXR da parte del CDCA in concentrazione di 10 μM potrebbe indurre la trascrizione del GLUT4 nelle linee cellulari 3T3-L1 e HepG2 e aumentare l’espressione della proteina GLUT 4 nei topi C57BL / 6J trattati con CDCA (20 mg / kg / giorno ). Complessivamente, questi cambiamenti hanno comportato una riduzione del livello di glucosio plasmatico, una riduzione della gluconeogenesi epatica e un aumento della sintesi epatica di glicogeno (Mencarelli e Fiorucci, 2010).
Vie di segnalazione intrecciate mediate dagli acidi biliari nel metabolismo del glucosio. L’attivazione da parte degli acidi biliari delle vie di segnalazione FXR e TGR-5 inibisce la gluconeogenesi e promuove la sintesi del glicogeno nel fegato, promuove il rilascio di insulina stimolato dal glucosio nel pancreas, aumenta il dispendio energetico soprattutto nei muscoli scheletrici e nel tessuto adiposo bruno. Nel cervello, la segnalazione degli acidi biliari-TGR5 media la sazietà.
Gli acidi biliari possono anche influenzare l’omeostasi del glucosio in modo indipendente dal FXR (Stanimirov et al., 2012). Oltre al ruolo nel dispendio energetico, è stato dimostrato che l’attivazione del recettore di membrana TGR5 da parte degli acidi biliari aumenta la secrezione intestinale di GLP-1 dalle cellule L endero-endocrine, sia in vitro che in vivo, stimolando la rilascio di Insulina dalle cellule β pancreatiche senza rilascio di Glucagone dalle cellule α e la riduzione della glicemia postprandiale (Katsuma et al., 2005; Kumar et al., 2012; Duboc et al., 2014). Inoltre, Maruyama et al. (2006) hanno mostrato che i topi TGR5-null hanno una riduzione del 25% nelle dimensioni del pool di acido biliare, mentre i topi TGR5-null femmine hanno mostrato un significativo accumulo di grasso con aumento di peso corporeo rispetto a quello dei topi wild-type quando nutriti con una dieta ricca di grassi. La figura seguente mostra chiaramente i percorsi intrecciati delle implicazioni degli acidi biliari nel metabolismo del glucosio mediato dalla segnalazione FXR e TGR5.
L’evidenza genetica in vitro e in vivo indica un forte legame causale tra la capacità funzionale del reticolo endoplasmatico e gli effetti dell’Insulina. Pertanto, la modulazione della funzione del reticolo endoplasmatico potrebbe fornire un nuovo approccio per il trattamento del diabete (Ozcan et al., 2006). Ozcan et al. (2006) hanno mostrato che il tauro-UDCA migliora la resistenza all’Insulina attenuando lo stress del reticolo endoplasmatico negli animali affetti da diabete di tipo 2 con conseguente normalizzazione dell’iperglicemia, miglioramento della sensibilità sistemica all’insulina e dell’azione dell’insulina in vari tessuti.
L’efficacia degli agonisti sintetici del FXR come potenziale terapia per il diabete mellito di tipo 2 è stata dimostrata in uno studio clinico di fase II già menzionato condotto da Mudaliar et al. (2013) nel quale è stato dimostrano che la somministrazione di OCA a pazienti con diabete mellito di tipo 2 e NAFLD per 6 settimane è stata ben tollerata, migliorando la sensibilità all’insulina e riducendo i marker del infiammazione e la fibrosi epatica.
Come già visto, recentemente, la ricerca dei meccanismi alla base dei rapidi miglioramenti glicemici a seguito di procedure chirurgiche bariatriche [bypass gastrico Roux-en-Y (RYGB) e gastrectomia a manica verticale] in pazienti con obesità patologica e diabete di tipo 2 ha confermato i significativi effetti fisiologici degli acidi biliari nella omeostasi del glucosio (Bradley et al., 2012; Pournaras et al., 2012). Queste procedure hanno causato un aumento degli acidi biliari liberi sia nel siero che nell’intestino inferiore (Madsbad et al., 2014; Raghow, 2015). L’aumentata concentrazione di acidi biliari liberi nel lume intestinale ha avuto un effetto diretto sulle Incretine attraverso l’asse TGR5 – GLP-1, migliorando la secrezione di Insulina, nonché attivando il FXR intestinale e il suo target diretto a valle FGF-15/19 – un ormone peptidico postprandiale che migliora la tolleranza al glucosio, una proteina la cui espressione è, altrimenti, ridotta nei pazienti diabetici (Jansen et al., 2011; Schaap, 2012; Madsbad and Holst, 2014). Gli effetti benefici mediati dai recettori degli acidi biliari FXR e TGR5 in seguito a chirurgia bariatrica sono stati ulteriormente confermati poiché in assenza di segnalazione FXR in topi null-FXR o segnalazione GLP-1 antagonizzata dal exendin- (9-39), gli effetti benefici della chirurgia bariatrica sul peso corporeo e il metabolismo del glucosio sono stati annullati (Salehi et al., 2011; Ryan et al., 2014). L’aumento delle concentrazioni di acido biliare libero nelle parti inferiori del tratto intestinale dopo RYGB crea un ambiente adatto per la crescita di batteri tolleranti la bile come il gruppo tassonomico Proteobacter phylum (Osto et al., 2013). La crescita eccessiva dei Proteobacter provoca una diminuzione delle specie secondarie, ceppi batterici che generano attraverso i loro processi metabolici acidi biliari più tossici, con conseguente aumento dei livelli di acido biliare primario (Acido Chenodeossicolico) nel plasma e un pronunciato effetto sulle Incretine (Vrieze et al., 2014).
Acidi biliari e microbiota intestinale
Il tratto intestinale umano è colonizzato da un insieme diversificata di microorganismi, con batteri come membri più numerosi. La composizione del microbiota intestinale è specifica per l’individuo ma rimane relativamente costante nel tempo (Stojančević et al., 2014). Alterazioni del microbioma intestinale umano possono avere un ruolo nello sviluppo di alcune patologie (DeGruttola et al., 2016). Il microbiota intestinale regola il metabolismo dell’ospite producendo numerosi metaboliti che segnalano attraverso i loro recettori affini, influenzando così il peso corporeo, il metabolismo degli acidi biliari, l’attività pro infiammatoria, la resistenza all’Insulina e la modulazione degli ormoni intestinali (Han e Lin, 2014; Wahlstrom et al., 2016). Un’importante classe di metaboliti prodotti dal microbiota sono appunto gli acidi biliari. L’idrolasi batterica dei Sali biliari (BSH), presente in tutte le principali specie batteriche nell’intestino umano, effettua la deconugazione del sale biliare aumentando così la resistenza alla tossicità biliare, mentre la 7α-deidrossilasi converte gli acidi biliari primari CA e CDCA nella bile negli acidi secondaria DCA e LCA, rispettivamente (Wahlstrom et al., 2016). Altre conversioni dei sali biliari eseguite dal microbiota intestinale sono l’ossidazione e l’epimerizzazione dei gruppi idrossilici, 7-disidrossilazione, esterificazione e desolfatazione, contribuendo così alla grande diversità chimica e al pool di acidi biliari più idrofobi (Ridlon et al., 2016). Inoltre, gli acidi biliari possono modulare la composizione del microbiota intestinale sia direttamente, distruggendo le strutture delle membrane batteriche, sia indirettamente, tramite l’attivazione del FXR promuovendo la trascrizione di agenti antimicrobici come iNOS e IL-18 nell’intestino tenue e inibendo così la crescita batterica (Inagaki et al., 2006; Wahlstrom et al., 2016).
Considerando la complessa relazione tra acidi biliari e microbiota intestinale, non sorprende che oltre ai cambiamenti nella composizione degli acidi biliari, i pazienti diabetici abbiano anche una diversa composizione e attività del microbiota intestinale rispetto agli individui sani (Quercia et al., 2014 ). Sia il diabete mellito di tipo 1 che quello di tipo 2 sono associati a una riduzione della diversità microbica complessiva, caratterizzata da riduzione di Firmicutes e batteri produttori di butirrato, nonché da una funzione alterata della barriera intestinale e da una maggiore permeabilità intestinale (DeGruttola et al., 2016; Knip e Siljander, 2016).
La supplementazione con batteri probiotici ha portato a una modulazione benefica del metabolismo dell’ospite in termini di secrezione di GLP-1 stimolata da specifici metaboliti batterici come gli acidi grassi a catena corta (SCFA) attraverso il meccanismo GPR41/43-dipendente (Everard e Cani, 2014). Inoltre, la somministrazione di batteri probiotici ha ridotto l’assunzione di cibo e protetto dall’aumento di peso corporeo e dal peggioramento dell’insulino resistenza nei modelli animali obesi e diabetici (Yadav et al., 2013). Gli studi condotti hanno dimostrato che un approccio multi-terapeutico che utilizza una combinazione di probiotici e acidi biliari come terapia aggiuntiva in un modello di ratto con diabete mellito di tipo 2 ha esercitato una regolazione glicemica ancora migliore e ha portato ad alleviare le complicanze rispetto a ciascun singolo trattamento (Al- Salami et al., 2008, 2012). Gli effetti sinergici degli acidi biliari, probiotici e terapia antidiabetica attuale sono esaminati in dettaglio da Mikov et al. (2017) indicando la potenziale applicazione di questa combinazione nei disturbi metabolici con particolare enfasi sul diabete mellito.
Pertanto, la manipolazione terapeutica del microbiota intestinale mediante integrazione probiotica con effetti secondari sulla composizione del pool di acidi biliari rappresenta una strategia promettente per il trattamento di tali condizioni. Sebbene ulteriori studi siano altamente raccomandati per svelare gli esatti meccanismi responsabili degli effetti benefici della co-somministrazione di acidi biliari-probiotici, l’attivazione di complesse vie di segnalazione FXR e TGR5 è proposta come una delle possibili spiegazioni.
A questo punto è utile precisare che nella composizione corporea il microbiota è un primo “ostacolo” alle calorie assunte. Più esso risulta in equilibrio e in salute e maggiore sembra essere la capacità batterica di esercitare un “blocco” nei confronti dell’eccesso calorico. Il microbiota in condizioni di salute agisce anche sul senso di sazietà, attraverso una segnalazione che parte dagli enterociti (le cellule dell’intestino) per giungere ai centri ipotalamici della fame. Questo avviene perché il microbiota converte le fibre alimentari in acidi grassi a corta catena, i quali sono il nutriente principale degli enterociti. In questo modo le cellule intestinali inviano al cervello il segnale di sazietà.
Acidi biliari e metabolismo lipidico
Come prodotto principale del catabolismo del Colesterolo, gli acidi biliari esercitano effetti profondi, non solo sul metabolismo del Colesterolo, ma anche sul metabolismo dei triacilgliceroli, regolando quindi il metabolismo di varie specie di lipoproteine. L’aumentata sintesi di acidi biliari aumenta l’utilizzo del colesterolo come substrato. Attivando il FXR gli acidi biliari inibiscono il CYP7A1, l’enzima che limita la velocità della sintesi degli acidi biliari e del catabolismo del Colesterolo negli epatociti. Di conseguenza, l’integrazione a lungo termine di 750mg o 375mg/die di CDCA nei pazienti con malattia dei calcoli biliari provoca un modesto aumento del livello di colesterolo lipoproteico a bassa densità (LDL) (Schoenfield e Lachin, 1981). L’aumento del LDL si è verificato nell’85,2% dei pazienti che ricevevano 750mg/die e, nell’82,8% dei pazienti che ricevevano 375mg/die, tuttavia, l’incremento del 67,0% è stato registrato in un gruppo di pazienti che assumevano placebo, probabilmente come conseguenza principale legata alla malattia. D’altra parte, studi in vitro dimostrano che l’agonista del FXR CDCA (250 μM) stabilizza l’mRNA del recettore LDL e aumenta l’attività del recettore LDL nella linea cellulare epatica umana in coltura aumentando così l’assorbimento e la clearance delle particelle di LDL (Nakahara et al., 2002). I cambiamenti nel livello di Colesterolo circolante attraverso l’attivazione del FXR in vivo sono distinti tra modelli animali (roditori) e umani. Vale a dire che, l’espressione del CYP7a1 nei roditori è opposta agli umani nei quali è regolata da due recettori nucleari, il recettore X del fegato-α (LXR-α) e il FXR, entrambi espressi abbondantemente nel fegato. Il LXR-α può essere attivato da derivati del Colesterolo tra cui il 24 (S), 25-epossicolesterolo e il 24 (S) -idrossicolcololo, e in seguito alla sua attivazione LXR interagisce con un elemento di risposta all’interno del promotore del CYP7a1, stimolando in tal modo l’espressione genica. La traduzione degli effetti osservati nei roditori è stata sconcertante poiché in queste specie il Colesterolo circolante è prevalentemente “impacchettato” sotto forma di lipoproteine HDL, contrariamente alle specie LDL dominanti nell’uomo. Nei topi chimerici i cui fegati contengono principalmente epatociti umani e un profilo lipoproteico “umanizzato”, il trattamento con un potente agonista FXR specifico, il derivato dell’acido biliare semisintetico OCA (10mg/kg/giorno), porta all’aumento della riduzione circolante di LDL e HDL , analogamente all’attivazione del FXR nell’uomo. L’aumento del LDL è correlato alla ridotta attività regolante della proteina 2 legante gli elementi sterolici (SREBP-2) e alla sua espressione genica bersaglio, inclusa una significativa sottoregolazione nell’espressione della proteina del recettore LDL (Papazyan et al., 2018). La somministrazione di OCA, 25 o 50mg/die per 2 settimane, durante gli studi clinici ha prodotto effetti simili (Mudaliar et al., 2013; Walters et al., 2015).
La riduzione del livello di HDL mediante attivazione del FXR [utilizzando chow integrato con Acido Taurocolico allo 0,5% p/p (TLCA) per un periodo di 6 giorni] può essere spiegata dalla repressione del gene dell’apolipoproteina A-I, nonché dall’etero-scambio di esteri del colesterolo e triacilgliceroli tra le HDL plasmatiche e le lipoproteine contenenti ApoB inducendo l’espressione della proteina di trasferimento dell’estere del colesterolo (colesteril estere)(Lambert et al., 2003; Gautier et al., 2013). D’altra parte, il targeting FXR può anche rafforzare il trasporto inverso del Colesterolo, un processo nel trasporto del Colesterolo dai tessuti e cellule periferici agli epatociti e al sistema biliare, al fine di eliminare il Colesterolo attraverso la via intestinale. L’analisi Northern blot su campioni di fegato di topi maschi C57BL/6, alimentati con una dieta integrata all’1% per un mese, ha rivelato che gli effetti sopra menzionati sono mediati tramite la proteina di trasferimento dei fosfolipidi, una proteina che media il rilascio di fosfolipidi e Colesterolo dalle lipoproteine LDL a HDL. Gli effetti osservati sono anche una conseguenza dei cambiamenti nell’espressione del recettore scavenger-B1 (SR-B1), che è coinvolto nel riconoscimento delle particelle di HDL e nel loro assorbimento da parte degli epatociti (Urizar et al., 2000). Inoltre, è stato dimostrato che l’attivazione diretta del target FXR, l’enterokina FGF15/19, stimola la consistente secrezione di Colesterolo nel lume intestinale attraverso l’eterodimero di sterolo adenosina trifosfato (ATP)-legante i membri della sottofamiglia G 5/8 (ABCG5 / G8) nei topi (de Boer et al., 2017). Questa scoperta ha potenziali implicazioni nello sviluppo di strategie volte alla riduzione del riassorbimento intestinale del Colesterolo. La somministrazione di 10 o 25mg di OCA al giorno in volontari sani ha indotto un aumento sostenuto della concentrazione sierica di LDL e una riduzione dell’HDL, con un leggero aumento del livello di Colesterolo totale indipendentemente dalla dose (Pencek et al., 2016). Cambiamenti simili sono stati osservati in pazienti con diabete mellito di tipo 2 e in pazienti con steatoepatite non alcolica confermata per biopsia (Mudaliar et al., 2013; Neuschwander-Tetri et al., 2015). Tuttavia, nei pazienti con colangite biliare primitiva trattati con 5-10mg di OCA al giorno per 1 anno, l’aumento di LDL e la riduzione del livello di HDL era minore in ampiezza e transitorio (Nevens et al., 2016). Gli effetti dell’attivazione del FXR mediante somministrazione ripetuta di 5, 10 o 25mg di OCA al giorno per 2 settimane o 20 giorni sono stati stimati nella sperimentazione clinica che recluta volontari sani (Pencek et al., 2016). I risultati osservati in questo studio hanno confermato i risultati di precedenti studi clinici in termini di lievi disturbi dello stato lipidico; vale a dire, il trattamento con OCA ha indotto una riduzione dose-indipendente di HDL, come risultato della riduzione della concentrazione di particelle HDL piccole e medie e un aumento del livello di colesterolo LDL (Pencek et al., 2016). Anche se l’attivazione del FXR da parte degli acidi biliari e dei derivati degli acidi biliari può indurre un fenotipo potenzialmente aterogenico in termini di spostamento delle frazioni di lipoproteine, sono altamente necessari ampi studi clinici multicentrici randomizzati per chiarire l’impatto clinico di tali effetti dislipidemici trattati con acidi biliari o derivati dell’acido biliare.
D’altra parte, è stato dimostrato che l’integrazione di acidi biliari con CA allo 0,5% (w/w) per 8 settimane modula il metabolismo dei triacilgliceroli attraverso diversi meccanismi distinti, principalmente attraverso l’attivazione del FXR. Attivando gli acidi biliari il FXR vanno a reprimere la sintesi di de novo triacilglicerolo attraverso il target downstream FXR, l’inibizione mediata dal SHP della trascrizione dell’elemento regolatore sterolo che lega la proteina-1c (SREBP-1c), un fattore chiave che controlla la trascrizione di diversi geni che regolano la sintesi di acidi grassi e triacilgliceroli, incluso un principale enzima lipogenico nel fegato, acido grasso sintasi (Watanabe et al., 2004). È stato anche dimostrato che l’attivazione del FXR induce l’espressione del recettore-α del perossisoma attivato dal proliferatore (PPAR-α) e dell’isoenzima-4 piruvato deidrogenasi chinasi che promuove l’ossidazione degli acidi grassi, mentre mediante l’inibizione SHP-mediata si verifica l’inibizione dell’espressione proteica microsomiale di trasferimento del triacilglicerolo per ridurre la sintesi di VLDL (Pan et al., 2010). Inoltre, il FXR promuove l’attività dell’enzima ancorato alla superficie luminale delle cellule endoteliali vascolari, una lipoproteina lipasi (LPL) che catalizza quindi l’idrolisi dei triacilgliceroli inducendo l’espressione di apoC-II attivanti LPL in topi C57BL/6J alimentati per 3 settimane con una dieta contenente lo 0,5% di Colato di Sodio. Inoltre, l’attivazione del FXR ha indotto l’espressione del recettore VLDL, facilitando la clearance delle particelle VLDL nelle cellule HepG2 incubate con 50μM di CDCA per 24 ore (Jiao et al., 2015). Oltre a ridurre i sintomi della diarrea da acido biliare nei pazienti, l’integrazione orale di 25mg di OCA al giorno per 2 settimane ha comportato un aumento del livello di FGF-19 e una diminuzione di triacilgliceroli, il che implica il ruolo potenziale dell’FGF-19 sul metabolismo dei triacilgliceroli (Walters et al., 2015).
L’ulteriore complessità della rete di segnalazione tra gli acidi biliari e la rete del triacilglicerolo nasce dalle interazioni con il microbiota intestinale, che è comunemente cambiato nelle persone alimentate con una dieta di tipo occidentale e nell’obesità. In effetti, anche la somministrazione a breve termine di antibiotici non assorbibili orali ha provocato disbiosi intestinale seguita dalla diminuzione delle quantità di acidi biliari secondari DCA e LCA le concentrazioni epatiche e ridotto livello sierico di triacilglicerolo, che non si è ripreso nemmeno dopo l’integrazione con acido biliare (Kuno et al., 2018). I risultati dello studio suggeriscono che gli acidi biliari secondari prodotti dalle specie batteriche intestinali esercitano un ruolo regolatorio significativo nel mantenimento dei livelli sierici di triacilgliceroli e del metabolismo nell’ospite.
Acidi grassi e fegato grasso non alcolico dipendente
Gli effetti positivi dati dall’attivazione del FXR e del TGR5 mediata dagli acidi biliari in una vasta gamma di processi metabolici, tra cui il metabolismo del glucosio e la segnalazione dell’Insulina, il metabolismo dei trigliceridi e del colesterolo, come abbiamo appena visto, nonché sull’infiammazione, focalizzano l’interesse della ricerca sugli acidi biliari per lo sviluppo di un’efficace terapia strategica per trattare la patologia del fegato grasso non alcolica.
Grazie alla sua caratterizzazione e valutazione preclinica, l’OCA è diventato uno dei ligandi più comunemente utilizzati per la decodifica della rete di segnalazione FXR sia in vivo che in vitro. Durante gli studi preclinici la somministrazione di OCA ha avuto effetti benefici significativi in numerosi disturbi del sistema enteroepatico, incluso il miglioramento della colestasi indotta dagli estrogeni, la fibrosi epatica, la NASH, la resistenza all’insulina, l’ipertensione portale, la riduzione dell’infiammazione intestinale e il miglioramento della funzione della barriera ileale durante la colestasi, il miglioramento della diarrea cronica indotta dell’acido biliare (Fiorucci et al., 2004, 2005; Adorini et al., 2012; Verbeke et al., 2014; Walters et al., 2015). È stato dimostrato che la somministrazione per via orale di 25mg di OCA per 72 settimane migliora le caratteristiche istologiche del fegato di pazienti con NASH durante la seconda fase della sperimentazione clinica FLINT (Neuschwander-Tetri et al., 2015). Gli effetti benefici dell’attivazione del FXR nei pazienti con NAFLD comprendono una migliore sensibilità insulinica epatica e di conseguenza un aumento della sintesi di glicogeno, una diminuzione della de novo lipogenesi nel fegato, una migliore sensibilità all’insulina adiposa (riduzione degli effetti lipotossici) e una migliore funzione del recettore attivato dal proliferatore del perossisoma -gamma (PPAR-γ) e PPAR-α, che regolano il metabolismo degli acidi grassi e del glucosio sia nel tessuto adiposo che nel fegato (Fiorucci et al., 2007). L’integrazione dietetica con OCA (10mg/kg/giorno) per il modello animale NASH sottoposto a regime alimentare dieta fast food, che imita la sindrome metabolica, presenta micro RNA-21 ablato (miR21) e PPAR-α attivato che ha portato a una significativa riduzione della steatosi, infiammazione e lipo -apoptosi, svelando il riassetto dell’asse miR21/PPAR-α nel fegato e nei tessuti muscolari mediante attivazione del FXR dato dal OCA (Rodrigues et al., 2017). L’attivazione da parte del OCA (10mg/kg/giorno, somministrato per via orale per 7 settimane) del FXR e del TGR5 mediante un intervento dietetico di 8 settimane contenente 30mg/kg/giorno di INT-777, ha portato ad un miglioramento della steatosi epatica e dell’insulina resistenza nel modello animale (roditori) con NAFLD e obeso (Thomas et al., 2009; Cipriani et al., 2010). La somministrazione di OCA (25, 50mg/die per os per 6 settimane) in pazienti con diabete mellito di tipo 2 e NAFLD ha ridotto significativamente il peso corporeo, ha migliorato la sensibilità all’insulina e ridotto il livello sierico di gamma-glutamiltransferasi, mentre l’aumento sierico dell’FGF-19 enterokine intestinale ha confermato l’attivazione del FXR nei pazienti (Mudaliar et al., 2013). Inoltre, l’OCA (10mg/kg) ha prevenuto l’infiammazione epatica prevenendo l’immunomodulazione e l’infiammazione mediate dal fattore nucleare dannoso κB. Inoltre, attraverso l’inibizione dell’attivazione delle cellule stellate epatiche, l’incubazione con 10μM di OCA ha impedito la progressione verso la fibrosi epatica e lo sviluppo della cirrosi (Goto et al., 2018). Il trattamento con questo agonista del FXR ha comportato un miglioramento delle caratteristiche biochimiche e istologiche del fegato nei pazienti con NASH. Queste funzioni indicano che il FXR è un bersaglio terapeutico interessante per le malattie del fegato (Massafra e van Mil, 2018). Ulteriori studi clinici randomizzati di grandi dimensioni sono altamente desiderabili per confermare gli effetti del “bussare alla porta del FXR” come un potenziale approccio terapeutico in cui il cambiamento nella dimensione e nella composizione del pool di acidi biliari può essere sfruttato per il trattamento dei disturbi metabolici.
Acidi biliari , obesità e patologie cardiometaboliche
È stato riscontrato che un aumento del livello di acidi biliari circolante negli individui obesi è correlato positivamente con l’indice di massa corporea (Prinz et al., 2015). Inoltre, l’insulino-resistenza è associata alla sottoregolazione mediata dal Foxo-1 del CYP8B1 con conseguente deplezione del pool di acido biliare 12α-idrossilato, che può essere spiegato dalla degradazione del Foxo indotta dall’iperglicemia e quindi dalla mancanza di attivazione del CYP8B1, mentre nel diabete di tipo 2 la concentrazione nei pazienti trattati con DCA è risultata elevata (Brufau et al., 2010; Haeusler et al., 2013). Pertanto, gli interventi che manipolano la composizione del pool di acidi biliari potrebbero rappresentare nuove strategie terapeutiche nel trattamento della resistenza all’insulina. I cambiamenti nella dimensione e nella composizione del pool di acidi biliari dopo la chirurgia bariatrica in soggetti obesi si riflettono nel miglioramento dell’omeostasi metabolica (Penney et al., 2015). È quindi ragionevole presumere che l’integrazione con acidi biliari o derivati dell’acido biliare, modificando la segnalazione dell’acido biliare, possa essere considerata come un potenziale intervento cardioprotettivo che migliora il metabolismo e diminuisce il livello infiammatorio. Infatti, l’attivazione del TGR5 nei macrofagi e nelle cellule endoteliali da parte di livelli micromolari di acidi biliari circolanti sia durante il digiuno che nello stato postprandiale quando gli acidi biliari circolanti bilaterali raggiungono il picco di concentrazione, esercitano effetti anti-aterogenici e inibiscono l’aterosclerosi e la malattia coronarica (Steiner et al ., 2011). Il recettore TGR5, espresso negli adipociti, regola il dispendio energetico e il peso corporeo (van Nierop et al., 2017). Il GLP1 indotto dagli acidi biliari esercita anche effetti benefici sulla funzione endoteliale, sulla pressione sanguigna, sul metabolismo miocardico, sulla frazione di eiezione ventricolare sinistra, sull’aterosclerosi e sulla risposta al danno ossidativo indotto dalla riperfusione ischemica (Kang e Jung, 2016). La procedura RYGBP nei pazienti obesi comporta un aumento del livello di GLP1 con effetti cardioprotettivi. Inoltre, un aumento del flusso intestinale di acidi biliari a seguito di RYGBP porta all’attivazione del FXR intestinale e del suo target a valle, Enterokine FGF15/19, che ha dimostrato di reprimere l’espressione genica apo(a) mediante la cascata mediata dalla fosforilazione ERK1/2, e la riduzione del livello circolante di lipoproteine altamente aterogeniche (a) (Chennamsetty et al., 2012). La somministrazione e sovraespressione di FGF19 o FGF19 ricombinante in topi diabetici con deficit di Leptina ha dimostrato di ridurre l’aumento di peso, invertire la condizione diabetica, aumentare il tasso di ossidazione dei lipidi diminuendo anche il contenuto epatico di trigliceridi (Fu et al., 2004). Le procedure di chirurgia bariatrica comportano anche un aumento dei livelli circolanti delle specie di acido biliare, sia in stato di digiuno che postprandiale, nonché cambiamenti qualitativi nella composizione del pool di acidi biliari, a causa di una maggiore sintesi e riassorbimento epatico e riduzione dell’eliminazione intestinale o da cambiamenti nella composizione del microbiota (De Giorgi et al., 2015; Dutia et al., 2016). Di conseguenza, l’aumento della concentrazione di acidi biliari sia nel lume intestinale che nella circolazione sistemica, come potenti ligandi per FXR e TGR5, media il miglioramento del tasso metabolico, del glucosio e del metabolismo lipidico, aumenta la termogenesi con conseguente riduzione del peso corporeo, migliora l’infiammazione sistemica e promuove persino la conversione del tessuto adiposo bianco nel metabolicamente più attivo tessuto adiposo beige, simile al tessuto adiposo marrone (Fang et al., 2015). Questi miglioramenti metabolici implicano che il cambiamento nella composizione e nelle dimensioni del pool degli acidi biliari mediante approccio farmacologico o chirurgia metabolica influisce sul metabolismo sistemico con esito favorevole, suggerendo un nuovo approccio terapeutico nel trattamento dell’obesità e dei componenti della sindrome metabolica. Tuttavia, dato che gli acidi biliari attivano recettori nucleari multipli e possibilmente più di un GPCR, un’accurata dissezione e una valutazione approfondita delle vie di segnalazione mediata dagli acidi biliari su modalità specifiche dei tessuti dovrebbero fornire informazioni utili nel futuro sviluppo di nuovi derivati specifici e selettivi degli acidi biliari come nuovi agenti terapeutici nel trattamento della sindrome metabolica.
Regolazione dipendente dal recettore e indipendente dal recettore delle vie metaboliche e di segnalazione da parte degli acidi biliari nella sindrome metabolica. Gli effetti dei recettori FXR e TGR5 attivati dagli acidi biliari sul glucosio, sui lipidi e sul metabolismo energetico, nonché sugli eventi vascolari associati all’aterosclerosi. FXR e TGR5 hanno un numero significativo di effetti sovrapposti attualmente identificati. D’altro canto, gli effetti dell’UDCA non sono associati all’attivazione di questi recettori degli acidi biliari, mentre l’UDCA esercita vari effetti fisiologici / farmacologici associati alle sue proprietà strutturali specifiche.
Giunti a questo punto abbiamo appurato che le evidenze emergenti dell’ultimo decennio hanno indicato il ruolo degli acidi biliari come molecole di segnalazione endocrine che regolano il glucosio, i lipidi e il metabolismo energetico attraverso percorsi complessi e correlati che includono principalmente la cascata di segnalazione FXR e TGR5. Pertanto, la modulazione di questi percorsi di segnalazione utilizzando gli acidi biliari e i loro derivati e la co-somministrazione con batteri probiotici con effetti secondari sulla composizione del pool di acido biliare è diventata un’area di particolare interesse nella ricerca moderna che offre un nuovo approccio per il trattamento della sindrome metabolica. Tuttavia, molti dei risultati ottenuti derivano da studi condotti su modelli animali che dovrebbero essere presi in considerazione con una attenta interpretazione dei risultati a causa delle principali differenze nel metabolismo degli acidi biliari e nella composizione del microbiota intestinale tra animali e umani. Inoltre, le differenze interindividuali nella composizione del microbiota intestinale, vale a dire l’impronta batterica specifica in alcuni individui, contribuiscono anche a profili di acidi biliari altamente specifici nel singolo soggetto, che influenzano in modo differenziato la patogenesi della malattia e, presumibilmente, la risposta a interventi preventivi e terapeutici correlati agli acidi biliari, che richiedono ulteriori studi e chiarimenti. Da un punto di vista terapeutico, è altamente auspicabile un approfondimento degli effetti metabolici di ciascuna delle specie di acido biliare naturale e sintetico in vivo. I probiotici, come potenziale strategia per modulare la composizione del microbiota intestinale, dovrebbero essere ulteriormente studiati per aiutare il ripristino del metabolismo degli acidi biliari e potenzialmente aiutare nel trattamento dei disturbi metabolici. La ricerca futura dovrebbe basarsi su approcci metabolomici, proteomici e lipidomici in popolazioni sia sane che malate al fine di identificare biomarcatori correlati all’acido biliare che possano essere utili per la previsione della terapia correlata all’acido biliare di diversi disturbi metabolici.
Conclusioni pratiche “sperimentali” per l’uso degli acidi biliari nel miglioramento della composizione corporea
Ma all’atto pratico, l’atleta interessato al miglioramento della composizione corporea come può trarre qualcosa di applicabile in concreto da tutto quello che è stato fino ad ora riportato?
Ovviamente, come spesso è capitato e capita nell’ambiente del BodyBuilding, la “sperimentazione” di nuovi PED non è un comportamento inusuale. E l’applicazione d’uso “sperimentale” degli acidi biliari assunti oralmente al fine di migliorare la composizione corporea ha interessato diversi atleti fin dalle prime pubblicazioni scientifiche.
Alcune pratiche speculari vedono l’uso concomitante della Caffeina durante il periodo di assunzione degli acidi biliari al fine di aumentare l’attività del cAMP (Adenosina Monofosfato Ciclico) stimolato dall’attivazione del TGR5 indotta dall’acido bilare. Pertanto, l’efficacia degli acidi biliari potrebbe essere potenziata assumendo contemporaneamente una quantità ragionevole di Caffeina o altre Xantine. In tessuti come il grasso marrone, nei muscoli scheletrici e nel fegato, l’attivazione del TGR5 aumenta le concentrazioni dell’enzima Deiodinasi 2 (D2) nella cellula.[52] Come ben sappiamo, la D2 converte la forma dell’ormone tiroideo poco attiva T4, che ha per l’appunto un modesto effetto sul metabolismo, nella forma più attiva T3. Questo effetto si tradurrebbe in un vantaggio sia per l’atleta “Natural” in ipocalorica sia nell’atleta che utilizza T4 sintetico nella medesima circostanza. Pertanto, l’utilizzo di acidi biliari in concomitanza con l’uso di T4 porterebbe ad una maggiore conversione di questo in T3. In una fase preparatoria dove l’atleta usa il T4 insieme al GH, il quale ne aumenta già la conversione in T3, l’effetto potrebbe essere additivo. L’effetto in questione è stato osservato con l’uso di Acido Colico e Acido Chenodesossicolico, anche se è potenzialmente riproducibile con l’uso di Acido Ursodesossicolico o Tauroursodesossicolico.
cAMP
Per chi fosse totalmente digiuno di biochimica, il cAMP è il mediatore intracellulare degli effetti dell’ormone. Mentre l’ormone è considerato il primo messaggero che agisce sulle cellule dotate di recettori per l’ormone stesso il cAMP è il secondo messaggero, uguale per tutte le cellule e per tutti gli ormoni che porta il messaggio all’interno della cellula.
Dai risultati di cui sopra, ci sono suggerimenti che indicano i fattori che possono migliorare o compromettere gli effetti sulla composizione corporea legati agli acidi biliari. Come accennato in precedenza, la Caffeina può aumentare l’effetto del segnale del TGR5 agendo sul cAMP nei tessuti. [53] Di conseguenza, per esempio, l’uso di beta-2 agonisti (es. Clenbuterolo) può accentuare ulteriormente l’effetto degli acidi biliari. [53][54] (15-17) Gli Estrogeni e i contraccettivi orali alterano la produzione di acidi biliari, diminuendo in particolare la produzione di Acido Chenodesossicolico; le Statine che abbassano il Colesterolo riducono la produzione totale di acidi biliari fino al 30%.[55][56] Il consumo di alcol riduce la produzione di acidi biliari e gli alcolisti cronici hanno una marcata riduzione della produzione di acidi biliari a causa in parte di un danno epatico avanzato. [57] Gli acidi biliari non raggiungono concentrazioni significative nei soggetti che seguono una dieta povera di grassi. Una dieta ricca di grassi e ricca di proteine induce un aumento del 50% nella produzione di acidi biliari e gli effetti metabolici potrebbero essere ancora maggiori se la fonte proteica è ricca di Taurina e Glicina. [58][59]
Coloro che seguono regimi alimentari ipocalorici “Low-Fat” con uno schema di digiuno intermittente potrebbero notare ulteriori benefici dalla supplementazione con acidi biliari. Questa ipotesi nasce dal fatto che, a digiuno, l’attività della D2 è bassa e le concentrazioni sieriche di acidi biliari sono al minimo, in particolare tra gli obesi. Sebbene non sia stato analizzato in una camera metabolica, è ragionevole ipotizzare che l’integrazione con un acido biliare, insieme a Caffeina o altra molecola con caratteristiche lipolitiche e termogeniche, possa aumentare la beta ossidazione e la termogenesi (dissipazione dell’energia sotto forma di calore), rispetto a un integrazione priva della componente AB durante il digiuno. Ma ciò potrebbe essere vero anche con regimi differenti al digiuno intermittente “Low-Fat”.
Gli acidi biliari hanno una migliore assimilazione se assunti durante un pasto composto da un moderato quantitativo di grassi ed enzimi che ne favoriscono la digestione. In questo modo si verificherà un aumento significativo post-prandiale degli acidi biliari nel flusso ematico. Gli acidi biliari possono anche essere assunti a digiuno e possono aumentare l’effetto beta ossidativo indotto dall’esercizio fisico svolto in tale contesto, sebbene non molto significativo di per se. Le dosi dovrebbero essere contenute , poiché anche i livelli prodotti endogenamente possono provocare ripercussioni negative a livello del colon, dove i batteri convertono gli acidi biliari in metaboliti più tossici. Alcuni acidi biliari sono usati in medicina alla dose di 10 mg/kg/die e sono considerati sicuri – 1g per una persona di 100Kg; gli effetti collaterali legati ad un sovradosaggio includono diarrea ed in cronico portano ad alterazioni degli enzimi epatici. Di conseguenza, la dose di 1g/die è considerabile come il “tetto massimo” di sicurezza. Inoltre, c’è da ricordarsi che l’eccesso delle concentrazioni di acidi biliari dato dalla supplementazione ridurrà marcatamente la loro sintesi endogena, limitandone il beneficio complessivo. In medio stat virtus!
E, ovviamente, evitate tassativamente di darvi al “fai da te” o di affidarvi alla guida di qualche “pitecus gimnicus”…
Jump up to: abcd Fiorucci S, Mencarelli A, Palladino G, Cipriani S (November 2009). “Bile-acid-activated receptors: targeting TGR5 and farnesoid-X-receptor in lipid and glucose disorders”. Trends Pharmacol. Sci. 30 (11): 570–80. doi:1016/j.tips.2009.08.001. PMID19758712.
‘Essentials of Medical Biochemistry, Lieberman, Marks and Smith, eds, p432, 2007’
Wiemuth D, Sahin H, Falkenburger BH, Lefevre CM, Wasmuth HE, Grunder S (2012). “BASIC–a bile acid-sensitive ion channel highly expressed in bile ducts”. FASEB J. 26 (10): 4122–30. doi:1096/fj.12-207043. PMID22735174.
Makishima M, Okamoto AY, Repa JJ, et al. (May 1999). “Identification of a nuclear receptor for bile acids”. Science. 284 (5418): 1362–5. doi:1126/science.284.5418.1362. PMID10334992.
Parks DJ, Blanchard SG, Bledsoe RK, et al. (May 1999). “Bile acids: natural ligands for an orphan nuclear receptor”. Science. 284 (5418): 1365–8. doi:1126/science.284.5418.1365. PMID10334993.
Wang H, Chen J, Hollister K, Sowers LC, Forman BM (May 1999). “Endogenous bile acids are ligands for the nuclear receptor FXR/BAR”. Mol. Cell. 3 (5): 543–53. doi:1016/s1097-2765(00)80348-2. PMID10360171.
Kim, I; Ahn, SH; Inagaki, T; Choi, M; Ito, S; Guo, GL; Kliewer, SA; Gonzalez, FJ (2007). “Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine”. Journal of Lipid Research. 48 (12): 2664–72. doi:1194/jlr.M700330-JLR200. PMID17720959.
Davidson MH (2011). “A systematic review of bile acid sequestrant therapy in children with familial hypercholesterolemia”. J Clin Lipidol. 5 (2): 76–81. doi:1016/j.jacl.2011.01.005. PMID21392720.
Allen L, Stobie D, Mauldin GN, Baer KE (January 1999). “Clinicopathologic features of dogs with hepatic microvascular dysplasia with and without portosystemic shunts: 42 cases (1991-1996)”. J. Am. Vet. Med. Assoc. 214 (2): 218–20. PMID9926012.
Poupon RE, Balkau B, Eschwège E, Poupon R (May 1991). “A multicenter, controlled trial of ursodiol for the treatment of primary biliary cirrhosis. UDCA-PBC Study Group”. N. Engl. J. Med. 324 (22): 1548–54. doi:1056/NEJM199105303242204. PMID1674105.
Glantz A, Marschall HU, Lammert F, Mattsson LA (December 2005). “Intrahepatic cholestasis of pregnancy: a randomized controlled trial comparing dexamethasone and ursodeoxycholic acid”. Hepatology. 42 (6): 1399–405. doi:10.1002/hep.20952. PMID16317669.
Danzinger RG, Hofmann AF, Schoenfield LJ, Thistle JL (January 1972). “Dissolution of cholesterol gallstones by chenodeoxycholic acid”. N. Engl. J. Med. 286 (1): 1–8. doi:10.1056/NEJM197201062860101. PMID5006919.
Thistle JL, Hofmann AF (September 1973). “Efficacy and specificity of chenodeoxycholic acid therapy for dissolving gallstones”. N. Engl. J. Med. 289 (13): 655–9. doi:10.1056/NEJM197309272891303. PMID4580472.
Petroni ML, Jazrawi RP, Pazzi P, et al. (January 2001). “Ursodeoxycholic acid alone or with chenodeoxycholic acid for dissolution of cholesterol gallstones: a randomized multicentre trial. The British-Italian Gallstone Study group”. Aliment. Pharmacol. Ther. 15 (1): 123–8. doi:10.1046/j.1365-2036.2001.00853.x. PMID11136285.
Uy MC, Talingdan-Te MC, Espinosa WZ, Daez ML, Ong JP (December 2008). “Ursodeoxycholic acid in the prevention of gallstone formation after bariatric surgery: a meta-analysis”. Obes Surg. 18 (12): 1532–8. doi:10.1007/s11695-008-9587-7. PMID18574646.
Pattni, S; Walters, JR (2009). “Recent advances in the understanding of bile acid malabsorption”. British Medical Bulletin. 92: 79–93. doi:10.1093/bmb/ldp032. PMID19900947.
Degirolamo C, Modica S, Palasciano G, Moschetta A (2011). “Bile acids and colon cancer: Solving the puzzle with nuclear receptors”. Trends Mol Med. 17 (10): 564–72. doi:10.1016/j.molmed.2011.05.010. PMID21724466.
Reddy BS, Hanson D, Mangat S, et al. (September 1980). “Effect of high-fat, high-beef diet and of mode of cooking of beef in the diet on fecal bacterial enzymes and fecal bile acids and neutral sterols”. J. Nutr. 110 (9): 1880–7. doi:10.1093/jn/110.9.1880. PMID7411244.
Ou J, DeLany JP, Zhang M, Sharma S, O’Keefe SJ (2012). “Association between low colonic short-chain fatty acids and high bile acids in high colon cancer risk populations”. Nutr Cancer. 64 (1): 34–40. doi:10.1080/01635581.2012.630164. PMID22136517.
O’Keefe SJ, Kidd M, Espitalier-Noel G, Owira P (May 1999). “Rarity of colon cancer in Africans is associated with low animal product consumption, not fiber”. Am. J. Gastroenterol. 94 (5): 1373–80. doi:10.1111/j.1572-0241.1999.01089.x. PMID10235221.
Prasad AR, Prasad S, Nguyen H, Facista A, Lewis C, Zaitlin B, Bernstein H, Bernstein C. Novel diet-related mouse model of colon cancer parallels human colon cancer. World J Gastrointest Oncol. 2014 Jul 15;6(7):225-43. doi: 10.4251/wjgo.v6.i7.225. PMID25024814
Singh S, Khanna S, Pardi DS, Loftus EV, Talwalkar JA (2013). “Effect of ursodeoxycholic acid use on the risk of colorectal neoplasia in patients with primary sclerosing cholangitis and inflammatory bowel disease: a systematic review and meta-analysis”. Inflamm. Bowel Dis. 19 (8): 1631–8. doi:10.1097/MIB.0b013e318286fa61. PMID23665966.
Henriksson P, Einarsson K, et al. Estrogen-induced gallstone formation in males. Relation to changes in serum and biliary lipids during hormonal treatment of prostatic carcinoma. J Clin Invest 1989;84:811-6.
Sigma Aldrich; rev. del 12.07.2012
Duncan D, Rotunda AM (2011). “Injectable therapies for localized fat loss: state of the art”. Clin Plast Surg. 38 (3): 489–501, vii. doi:10.1016/j.cps.2011.02.005.
Watanabe M, Houten SM, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 2006;439:484-9.
Teodoro JS, Zouhar P, et al. Enhancement of brown fat thermogenesis using chenodeoxycholic acid in mice. Int J Obes 2013 Dec 6. [E-pub, ahead of print]
Dulloo AG, Seydoux J, et al. Potentiation of the thermogenic antiobesity effects of ephedrine by dietary methylxanthines: adenosine antagonism or phosphodiesterase inhibition? Metabolism 1992;41:1233-41.
van der Lans AA, Hoeks J, et al. Cold acclimation recruits human brown fat and increases nonshivering thermogenesis. J Clin Invest 2013;123:3395-403.
Everson GT, Fennessey P, et al. Contraceptive steroids alter the steady-state kinetics of bile acids. J Lipid Res 1988;29:68-76.
Bertolotti M, Zambianchi L, et al. Influence of newly synthesized cholesterol on bile acid synthesis during chronic inhibition of bile acid absorption. Hepatology 2003;38:939-46.
Monroe P, Vlahcevic ZR, et al. Effects of acute and chronic ethanol intake on bile acid metabolism. Alcohol Clin Exp Res 1981;5:92-100.
Bortolotti M, Kreis R, et al. High protein intake reduces intrahepatocellular lipid deposition in humans. Am J Clin Nutr 2009;90:1002-10.
Liaset B, Hao Q, et al. Nutritional regulation of bile acid metabolism is associated with improved pathological characteristics of the metabolic syndrome. J Biol
L’Adiponectina (denominata anche come GBP-28, apM1, AdipoQ, Acrp30 e Liponectine[1]) è un ormone proteico coinvolto nella regolazione dei livelli di glucosio e nella scomposizione degli acidi grassi. Nell’uomo è codificato dal gene ADIPOQ ed è prodotto nel tessuto adiposo. [2]
Nel 1995, l’Adiponectina è stata inizialmente osservata esercitare una azione di differenziazione degli adipociti 3T3-L1 (Scherer PE et al.).[3] Essa venne infatti scoperta nel 1995 da quattro diversi gruppi di ricerca che lavoravano indipendentemente l’uno dall’altro.[4] Nel 1996 è stato osservato che nei topi l’Adiponectina è la trascrizione dell’mRNA più espressa negli adipociti.[2] Nel 2007, l’Adiponectina è stata osservata essere una trascrizione altamente espressa nei preadipociti [5] (precursori delle cellule adipose) che si differenziano in adipociti.[5][6]
L’omologo umano è stato identificato come la trascrizione più abbondante nel tessuto adiposo. Contrariamente alle aspettative, nonostante venga sintetizzata nel tessuto adiposo, l’Adiponectina risultata essere sottoregolata nei soggetti obesi.[7][8][9] Questa sottoregolazione non è stata completamente spiegata. Il gene è stato localizzato nel cromosoma 3q27, una regione evidenziata per avere una certa influenza nella suscettibilità genetica al diabete di tipo 2 e all’obesità. Il trattamento con diverse forme di Adiponectina è stato in grado di migliorare il controllo dell’Insulina, della glicemia e dei livelli di Trigliceridi nei modelli murini.
Il gene è stato studiato per le variabili che predispongono al diabete di tipo 2.[9][5][10][11][12][13]Diversi polimorfismi a singolo nucleotide nella regione codificante e nella sequenza circostante sono stati identificati in diverse popolazioni, con prevalenze, gradi di associazione e forza di effetto variabili sul diabete di tipo 2. La Berberina, un alcaloide isochinolina, ha dimostrato di aumentare l’espressione del Adiponectina [14], il che spiega, in parte, i suoi effetti benefici sui disturbi metabolici. Topi nutriti con gli acidi grassi Omega-3 Acido Eicosapentaenoico (EPA) e Acido Docosaesaenoico (DHA) hanno mostrato un aumento dell’Adiponectina plasmatica.[15] Anche la Curcumina, la Capsaicina, il Gingerolo e le Catechine hanno mostrato di poter aumentare l’espressione dell’Adiponectina.[16]
La distribuzione filogenetica include l’espressione negli uccelli [17] e nei pesci.[18]
L’Adiponectina è un polipeptide (proteina) composto da una catena di 247 aminoacidi. Esistono quattro regioni distinte nella struttura molecolare della Adiponectina. La prima regione è formata da una breve sequenza di segnali che interessa l’ormone nella secrezione all’esterno della cellula; la seconda regione varia tra le specie; la terza è una regione composta di 65 aminoacidi con somiglianza con le proteine del Collagene; l’ultima è un dominio globale. Nel complesso questa proteina mostra una somiglianza con i fattori del complemento 1Q (C1Q). Tuttavia, quando è stata determinata la struttura tridimensionale della regione globulare, è stata osservata una sorprendente somiglianza con il TNFα (Fattore di Necrosi Tumorale α), nonostante sequenze proteiche non correlate.[19]
Come già accennato, l’Adiponectina è un ormone proteico che modula una serie di processi metabolici, tra i quali la regolazione del glucosio e l’ossidazione degli acidi grassi.[7] L’Adiponectina viene secreta dal tessuto adiposo (e anche dalla placenta in gravidanza [20]) nel flusso ematico ed è molto abbondante nel plasma rispetto a molti altri ormoni. Molti studi hanno scoperto che l’Adiponectina è inversamente correlata all’indice di massa corporea nelle popolazioni di pazienti.[8] Tuttavia, una meta analisi non è stata in grado di confermare questa associazione negli adulti sani.[21] Le concentrazioni circolanti di Adiponectina aumentano durante la restrizione calorica negli animali e nell’uomo, come nei pazienti con anoressia nervosa. Questa osservazione è senza dubbio sorprendente, dato che l’Adiponectina è sintetizzata nel tessuto adiposo. Tuttavia, un recente studio suggerisce che il tessuto adiposo nel midollo osseo, che aumenta durante la restrizione calorica, contribuisce alle elevate concentrazioni ematiche di Adiponectina in tale contesto.[22]
Mitocondrio
Topi transgenici con elevati livelli di Adiponectina mostrano una ridotta differenziazione degli adipociti e un aumento del dispendio energetico associato al disaccoppiamento mitocondriale.[23] L’ormone in questione svolge un ruolo nella soppressione delle alterazioni metaboliche correlate al diabete di tipo 2, [8] obesità, aterosclerosi, [7] epatopatia adiposa non alcolica (NAFLD) e un fattore di rischio indipendente per la sindrome metabolica.[24] L’Adiponectina in associazione con la Leptina ha dimostrato di invertire completamente lo stato di insulino-resistenza nei topi.[25]
L’Adiponectina viene secreta nel flusso sanguigno dove rappresenta circa lo 0,01% di tutte le proteine plasmatiche ad un dosaggio di circa 5-10μg/mL (mg/L). Negli adulti, le concentrazioni plasmatiche sono più elevate nelle femmine rispetto ai maschi e sono ridotte nei diabetici rispetto ai non diabetici. La riduzione del peso ne aumenta significativamente le concentrazioni circolanti.[26]
L’Adiponectina si auto-associa automaticamente in strutture più grandi. Inizialmente, tre molecole di Adiponectina si legano insieme per formare un omotrimero. I trimeri continuano ad auto-associarsi e formano esameri o dodecameri. Come la concentrazione plasmatica, i livelli relativi delle strutture di ordine superiore sono sessualmente dimorfici, dove le femmine hanno livelli maggiori delle forme ad alto peso molecolare. Studi recenti hanno dimostrato che la forma ad alto peso molecolare può essere quella biologicamente più attiva per quanto riguarda l’omeostasi del glucosio.[27] L’Adiponectina ad alto peso molecolare è stata inoltre osservata associarsi ad un minor rischio di diabete con un’entità di associazione simile all’Adiponectina totale.[28] Tuttavia, si è scoperto che la malattia coronarica è associata positivamente con l’Adiponectina ad alto peso molecolare, ma non con l’Adiponectina a basso peso molecolare.[29]
Leptina
L’Adiponectina esercita alcuni dei suoi effetti di riduzione del peso attraverso il cervello. Questa azione è simile a quella esercitata dalla Leptina[9]; l’Adiponectina e la Leptina possono agire sinergicamente.
L’Adiponectina non si lega ad un solo recettore. Finora, sono stati identificati due recettori con l’omologia dei recettori accoppiati a proteine G (GPCRs) e un recettore simile alla famiglia delle Caderine [30][31]:
Recettore 1 dell’adiponectina (AdipoR1)
Recettore 2 dell’adiponectina (AdipoR2)
T-caderina – CDH13
Questi recettori hanno specificità tissutali distinte all’interno del corpo e hanno affinità diverse con le varie forme di Adiponectina. I recettori influenzano a valle l’AMP chinasi, un importante punto di controllo del tasso metabolico cellulare. L’espressione dei recettori è correlata ai livelli di Insulina, cosa osservata nei modelli murini diabetici, in particolare nel muscolo scheletrico e nel tessuto adiposo.[32][33] Nel 2016 l’Università di Tokyo annunciò l’avvio di un’indagine, spinta dalle richieste fatte in modo anonimo, sulla presunta falsificazione dei dati rilasciati sull’identificazione dei recettori AdipoR1 e AdipoR2.[34]
Sia AdipoR1 che AdipoR2 sono orientati in modo opposto ai GPCR nella membrana (cioè N-terminale citoplasmatico, C-terminale extracellulare) e non si legano alle proteine G di memnrana.[35] I recettori dell’Adiponectina, AdipoR1 e AdipoR2, fungono da recettori per l’Adiponectina globulare e integrale e mediano un aumento delle attività dei ligandi AMPK e PPAR-α, nonché l’ossidazione degli acidi grassi e l’assorbimento del glucosio per attività dell’Adiponectina.[35][36]
Gli effetti legati all’attività della Adiponectina sono:
Regolazione del flusso del glucosio ematico
riduzione della gluconeogenesi
Aumento dell’assorbimento cellulare di glucosio [7][9][11]
2. Catabolismo lipidico
β-ossidazione [9]
Liberazione dei trigliceridi [9]
3. Protezione dalla disfunzione endoteliale (aspetto importante della formazione aterosclerotica)
4. Miglioramento della Sensibilità all’Insulina
5. Perdita di peso
6. Controllo del metabolismo energetico [11]
7. Sovraregolazione delle proteine disaccoppianti (UCP) [23]
8. Riduzione del TNF-α.
Regolazione dell’Adiponectina:
L’obesità è associata alla riduzione dell’Adiponectina.
L’esatto meccanismo di regolazione non è noto, ma l’Adiponectina potrebbe essere regolata da meccanismi post-trasduzionali nelle cellule.[37]
Bassi livelli di Adiponectina sono associati all’ADHD negli adulti.[38]
È stato scoperto che i livelli di Adiponectina sono aumentati nei pazienti con artrite reumatoide che rispondono alla terapia con DMARD o inibitori del TNF. [39]
Il rilascio di Adiponectina indotto dall’esercizio fisico ha aumentato la crescita dell’ippocampo e ha portato a risposte antidepressive nei topi.[40]
Un basso livello di Adiponectina è un fattore di rischio indipendente per lo sviluppo di:
Sindrome metabolica [24]
Diabete mellito di tipo II [9] [5] [10] [12] [13]
I livelli circolanti di Adiponectina possono essere indirettamente aumentati attraverso modifiche dello stile di vita e alcuni farmaci come le Statine.[41]
Esistono dei composti sintetici che interagiscono con i recettori dell’Adiponectina come l’AdipoRon, un agonista selettivo, attivo per via orale, del recettore 1 (AdipoR1) e del recettore 2 dell’Adiponectina (AdipoR2) (Kd = 1,8 μM e 3,1 μM, rispettivamente).[42] L’Università di Tokyo che nel 2016 annunciò l’avvio dell’indagine, spinta dalle richieste fatte in modo anonimo, sulla presunta falsificazione dei dati rilasciati sull’identificazione dei recettori AdipoR1 e AdipoR2, si è occupata anche di questo agonista selettivo dei recettori per l’Adiponectina.[34]
È stato riportato che estratti di patate dolci aumentano i livelli di Adiponectina portando, quindi, ad un miglioramento del controllo glicemico nell’uomo.[43] Tuttavia, una review sistematica ha concluso che non vi sono prove sufficienti a supporto del consumo di patate dolci per il trattamento del diabete mellito di tipo 2.[44]
L’Adiponectina è apparentemente in grado di attraversare la barriera emato-encefalica [45] sebbene esistano dati contrastanti a riguardo.[46] L’Adiponectina ha un’emivita di 2,5 ore nell’uomo.[47]
AdipoRon
Ho accennato pocanzi al AdipoRon, agonista sintetico dei recettori per l’Adiponectina AdipoR1 e AdipoR2.[42] Analogamente all’Adiponectina, questa molecola attiva la segnalazione del AMPK e del PPARα causando un miglioramento dell’insulino sensibilità, della dislipidemia e dell’intolleranza al glucosio nei topi db/db (un modello animale per il diabete di tipo II e l’obesità).[42][48] Inoltre, è stato scoperto che AdipoRon estende la durata della vita dei topi db/db alimentati con una dieta ricca di grassi, oltre a migliorarne la resistenza all’esercizio fisico. [42] [48] [49] La molecola è stata scoperta da ricercatori giapponesi nel 2013 attraverso la revisione della letteratura disponibile, ed è il primo agonista dei recettori per l’Adiponectina attivo per via orale ad essere stato identificato.[42][48]
Gli agonisti del recettore dell’Adiponectina come AdipoRon hanno attirato l’interesse come potenziali terapie per il trattamento dell’obesità, del diabete, delle malattie cardiovascolari, della malattia del fegato grasso non alcolico e una panoplia di altre condizioni.[42][48] Inoltre, è stato recentemente chiarito il ruolo di mediatore dell’adiponectina sugli effetti antidepressivi, ansiolitici e neurogenici indottidall’esercizio fisico. [50][51][52] E’ interessante notare che l’aumento dei livelli di Adiponectina dopo una seduta di esercizio fisico moderato perdura per per 24 a 72 ore. La disregolazione dell’espressione dell’Adiponectina è stata anche implicata nella patologia dei disturbi dell’umore, dei disturbi d’ansia, dei disturbi alimentari, dei disturbi neurodegenerativi e di vari altri disturbi neuropsichiatrici.[53] Inoltre, è stato determinato che l’esercizio fisico migliora la resistenza all’insulina attraverso l’attivazione del recettore AdipoR1.[54] Come tale, gli agonisti del recettore dell’Adiponectina sono un target terapeutico molto interessante per il trattamento di una varietà di condizioni diverse.[42][48][52][53] Inoltre, è stato suggerito che potrebbero essere potenzialmente utilizzati come sostituti dell’esercizio fisico per ottenere benefici simili sulla salute fisica e mentale.[42][48][52][55] Questa opzione è da prendere in considerazione solo e soltanto in quei soggetti impossibilitati a svolgere attività fisica “significativa”.
A causa delle limitazioni nella produzione di Adiponectina biologicamente attiva, gli agonisti degli AdipoRs adiponectino-mimetici sono stati suggeriti come possibili alternative per espandere l’opportunità di sviluppare agenti anti-diabetici. Basandosi sulla struttura cristallina del AdipoR1, i ricercatori hanno progettato gli agonisti dei peptidi del AdipoR1 usando la simulazione del docking proteico-peptidico analizzando le loro capacità di legame per i recettori e le funzioni biologiche attraverso la risonanza plasmonica di superficie (SPR) e l’analisi biologica. Sono stati selezionati e confermati tre peptidi candidati, BHD1028, BHD43 e BHD44 per attivare le vie del segnale mediate da AdipoR1. Al fine di migliorare la stabilità e la solubilità degli agonisti peptidici, i peptidi candidati sono stati PEGilati. Il BHD1028 PEGilato ha mostrato la sua attività biologica alla concentrazione nano-molare e potrebbe essere un potenziale agente terapeutico per il trattamento del diabete. Inoltre, l’SPR e tecniche di screening virtuale possono essere potenzialmente applicate ad altri processi di screening di farmaci peptidici per le proteine del recettore di membrana.[56]
Arctiina
Altre forme di agonisti dei recettori per l’Adiponectina sono i peptidi ADP-355 e ADP-399 [57], i non-peptidi (–)-Arctigenina, Arctiina, Gramina, Matairesinol, Deoxyschizandrina, Parthenolide,
Syringing e Taxifoliol.[58] L’ADP-400 è invece un peptide antagonista del recettore per l’Adiponectina. [58]
Date le potenzialità legate all’Adiponectina, l’uso e la diffusione degli agonisti sintetici dei suoi recettori nella sottocultura del BodyBuilding, in particolare, e del Fitness, in generale, non sarà di certo un evento improbabile nel prossimo futuro. In attesa di questo evento, diversi divulgatori d’oltre oceano, più o meno autorevoli, hanno incominciato a cercare soluzioni OTC per incrementare la sintesi endogena di Adipnectina.
Come già detto all’inizio di questo articolo, i ricercatori hanno scoperto che l’assunzione di grassi monoinsaturi presenti nell’olio di pesce (vedi in particolare EPA e DHA), causa un aumento dei livelli di Adiponectina dal 14 al 60%. Anche l’olio di Cartamo ha dimostrato di aumentare la sintesi di Adiponectina. .[15] Per tale ragione alcuni dei prima citati divulgatori consiglia di assumere 4g/die di CLA derivato dall’olio di Cartamo.
I ricercatori hanno scoperto che l’aggiunta di adeguate quantità di fibre alla dieta causa un aumento dei livelli di Adiponectina tra il 60 ed il 115%.[59] Un motivo in più, se mai ce ne fosse stato il bisogno, di consumare la quantità raccomandata di fibre pari a 30g al giorno.[60]
Il consumo regolare di caffè è stato correlato ad un aumento dei livelli di Adiponectina e ad una riduzione delle citochine pro-infiammatorie, che potrebbero aiutare ad aumentare la perdita di peso e ridurre i livelli di infiammazione.[61]
Curcumina
Si è ipotizzato, con ben poche evidenze, che la Curcumina aumenti la sintesi di Adiponectina. La funzione verrebbe esercitata tramite la riduzione della sintesi di sostanza pro-infiammatorie nell’adipocita con un conseguente aumento della sintesi di Adiponectina.[62]
Resveratrolo
Uno studio del 2011, condotto da ricercatori dell’Università del Texas Health Science Center di San Antonio, pubblicato sul Journal of Biological Chemistry, ha riportato che il Resveratrolo stimola anche l’espressione dell’Adiponectina. [63]
Secondo uno studio del 2012, completato all’Università di Gerusalemme, una dieta sperimentale con carboidrati consumata principalmente a cena, piuttosto che durante il giorno, sembra avvantaggiare le persone che soffrono di obesità grave e morbosa. Questa dieta sembra influenzare i modelli di secrezione degli ormoni responsabili della fame e della sazietà, nonché gli ormoni associati alla sindrome metabolica, compreso un aumento della produzione di Adiponectina durante il giorno.[64]
In uno studio pubblicato sull’Iranian Journal of Diabetes and Obesity nel giugno 2012, è stato osservato un aumento significativo dei livelli di Adiponectina nei soggetti che avevano assunto 50mg di Zinco rispetto a al gruppo di controllo.[65] Un altro studio simile, nel quale soggetti diabetici di tipo II sono stati trattati con 30mg/die di Zinco, è stato osservato un aumento significativo della Adiponectina rispetto al basale ma non rispetto al gruppo di controllo (e il 53.3% presentava un insufficienza di Zinco al basale).[66]
Cianidina 3-glucoside
L’antocianina C3G (cianadina 3-glucoside), se assunta in quantità sufficiente in forma di integratore, sembra migliorare la composizione corporea. Si è sempre pensato che questo effetto fosse causato dagli effetti positivi diretti C3G sulla sensibilità all’Insulina. Infatti, è noto da tempo che la C3G possa aumentare la lipolisi stimolando la sintesi di adipocitochine (proteine di segnalazione cellulare) come l’Adiponectina, che regola appunto i livelli di glucosio e l’ossidazione degli acidi grassi. Recentemente sono emerse alcune ricerche secondo cui questo aumento di Adiponectina mediato dal C3G potrebbe anche contribuire alla crescita muscolare.[67]
L’Agaricus blazei Murrill è un fungo medicinale non facente parte della Medicina Tradizionale Cinese (MTC), ma che ha suscitato grande interessa per la sua peculiare capacità di regolare la risposta immunitaria, ha mostrato di poter aumentare la concentrazione plasmatica di Adiponectina del 20%.[68] Favorendo un aumento di concentrazione di Adiponectina, l’AbM risulterebbe molto utile in caso di steatosi epatica non alcolica e insulino-resistenza. C’è da considerare però il fatto che, nello studio citato, l’estratto del AbM era somministrata in concomitanza con Metformina e Gliclazide.
Non fatevi troppe illusioni però, è molto probabile che se vi metteste a provare una o più di queste ipotetiche metodiche per aumentare i livelli di Adiponetina, il risultato che ne ricavereste, nel migliore delle ipotesi, non sarebbe da attribuirsi ad altra cosa se non al effetto placebo. Ma, da ricercatore quale sono, non biasimerò di certo chi vorrà testare e cercare di comprendere, eliminando per quanto possibile le variabili in gioco, se l’effetto ottenuto (se se ne è ottenuto qualcuno) è attribuibile ad un incremento dell’Adiponectina… risultante comunque molto speculativa vista la mancanza per la stragrande maggioranza delle persone di un laboratorio ove sottoporsi a periodiche analisi al fine di quantificare i (se mai ci fossero) incrementi del peptide in questione.
Maeda K, Okubo K, Shimomura I, Funahashi T, Matsuzawa Y, Matsubara K (April 1996). “cDNA cloning and expression of a novel adipose specific collagen-like factor, apM1 (AdiPose Most abundant Gene transcript 1)”. Biochemical and Biophysical Research Communications. 221 (2): 286–9. doi:10.1006/bbrc.1996.0587
Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF (November 1995). “A novel serum protein similar to C1q, produced exclusively in adipocytes”. The Journal of Biological Chemistry. 270 (45): 26746–9. doi:10.1074/jbc.270.45.26746
Lara-Castro C, Fu Y, Chung BH, Garvey WT (June 2007). “Adiponectin and the metabolic syndrome: mechanisms mediating risk for metabolic and cardiovascular disease”. Current Opinion in Lipidology. 18 (3): 263–70.
Matsuzawa Y, Funahashi T, Kihara S, Shimomura I (January 2004). “Adiponectin and metabolic syndrome”. Arteriosclerosis, Thrombosis, and Vascular Biology. 24 (1): 29–33.
Díez JJ, Iglesias P (March 2003). “The role of the novel adipocyte-derived hormone adiponectin in human disease”. European Journal of Endocrinology. 148 (3): 293–300.
Ukkola O, Santaniemi M (November 2002). “Adiponectin: a link between excess adiposity and associated comorbidities?”. Journal of Molecular Medicine. 80 (11): 696–702.
Hara K, Yamauchi T, Kadowaki T (April 2005). “Adiponectin: an adipokine linking adipocytes and type 2 diabetes in humans”. Current Diabetes Reports. 5 (2): 136–40.
Vasseur F, Leprêtre F, Lacquemant C, Froguel P (April 2003). “The genetics of adiponectin”. Current Diabetes Reports. 3 (2): 151–8.
Vasseur F, Meyre D, Froguel P (November 2006). “Adiponectin, type 2 diabetes and the metabolic syndrome: lessons from human genetic studies”. Expert Reviews in Molecular Medicine. 8 (27): 1–12.
Grimshaw CE, Matthews DA, Varughese KI, Skinner M, Xuong NH, Bray T, Hoch J, Whiteley JM (August 1992). “Characterization and nucleotide binding properties of a mutant dihydropteridine reductase containing an aspartate 37-isoleucine replacement”. The Journal of Biological Chemistry. 267 (22): 15334–9.
Yuan J, Liu W, Liu ZL, Li N (2006). “cDNA cloning, genomic structure, chromosomal mapping and expression analysis of ADIPOQ (adiponectin) in chicken”. Cytogenetic and Genome Research. 112 (1–2): 148–51.
Nishio S, Gibert Y, Bernard L, Brunet F, Triqueneaux G, Laudet V (June 2008). “Adiponectin and adiponectin receptor genes are coexpressed during zebrafish embryogenesis and regulated by food deprivation”. Developmental Dynamics. 237 (6): 1682–90.
Shapiro L, Scherer PE (March 1998). “The crystal structure of a complement-1q family protein suggests an evolutionary link to tumor necrosis factor”. Current Biology. 8 (6): 335–8.
Chen J, Tan B, Karteris E, Zervou S, Digby J, Hillhouse EW, Vatish M, Randeva HS (June 2006). “Secretion of adiponectin by human placenta: differential modulation of adiponectin and its receptors by cytokines”. Diabetologia. 49 (6): 1292–302.
Kuo SM, Halpern MM (December 2011). “Lack of association between body mass index and plasma adiponectin levels in healthy adults”. International Journal of Obesity. 35 (12): 1487–94.
Bauche IB, El Mkadem SA, Pottier AM, Senou M, Many MC, Rezsohazy R, Penicaud L, Maeda N, Funahashi T, Brichard SM (April 2007). “Overexpression of adiponectin targeted to adipose tissue in transgenic mice: impaired adipocyte differentiation”. Endocrinology. 148 (4): 1539–49
Renaldi O, Pramono B, Sinorita H, Purnomo LB, Asdie RH, Asdie AH (January 2009). “Hypoadiponectinemia: a risk factor for metabolic syndrome”. Acta Medica Indonesiana. 41 (1): 20–4.
Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, Mori Y, Ide T, Murakami K, Tsuboyama-Kasaoka N, Ezaki O, Akanuma Y, Gavrilova O, Vinson C, Reitman ML, Kagechika H, Shudo K, Yoda M, Nakano Y, Tobe K, Nagai R, Kimura S, Tomita M, Froguel P, Kadowaki T (August 2001). “The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity”. Nature Medicine. 7 (8): 941–6.
Coppola A, Marfella R, Coppola L, Tagliamonte E, Fontana D, Liguori E, Cirillo T, Cafiero M, Natale S, Astarita C (May 2009). “Effect of weight loss on coronary circulation and adiponectin levels in obese women”. International Journal of Cardiology. 134 (3): 414–6.
Oh DK, Ciaraldi T, Henry RR (May 2007). “Adiponectin in health and disease”. Diabetes, Obesity & Metabolism. 9 (3): 282–9.
Rizza S, Gigli F, Galli A, Micchelini B, Lauro D, Lauro R, Federici M (April 2010). “Adiponectin isoforms in elderly patients with or without coronary artery disease”. Journal of the American Geriatrics Society. 58 (4): 702–6.
Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, Sugiyama T, Miyagishi M, Hara K, Tsunoda M, Murakami K, Ohteki T, Uchida S, Takekawa S, Waki H, Tsuno NH, Shibata Y, Terauchi Y, Froguel P, Tobe K, Koyasu S, Taira K, Kitamura T, Shimizu T, Nagai R, Kadowaki T (June 2003). “Cloning of adiponectin receptors that mediate antidiabetic metabolic effects”. Nature. 423 (6941): 762–9.
Fang X, Sweeney G (November 2006). “Mechanisms regulating energy metabolism by adiponectin in obesity and diabetes”. Biochemical Society Transactions. 34 (Pt 5): 798–801.
Bonnard C, Durand A, Vidal H, Rieusset J (February 2008). “Changes in adiponectin, its receptors and AMPK activity in tissues of diet-induced diabetic mice”. Diabetes & Metabolism. 34 (1): 52–61.
Lim S, Quon MJ, Koh KK (April 2014). “Modulation of adiponectin as a potential therapeutic strategy”. Atherosclerosis. 233 (2): 721–8.
Okada-Iwabu M, Yamauchi T, Iwabu M, Honma T, Hamagami K, Matsuda K, Yamaguchi M, Tanabe H, Kimura-Someya T, Shirouzu M, Ogata H, Tokuyama K, Ueki K, Nagano T, Tanaka A, Yokoyama S, Kadowaki T (November 2013). “A small-molecule AdipoR agonist for type 2 diabetes and short life in obesity”. Nature. 503 (7477): 493–9.
Ludvik B, Hanefeld M, Pacini G (July 2008). “Improved metabolic control by Ipomoea batatas (Caiapo) is associated with increased adiponectin and decreased fibrinogen levels in type 2 diabetic subjects”. Diabetes, Obesity & Metabolism. 10 (7): 586–92.
Ooi CP, Loke SC (September 2013). “Sweet potato for type 2 diabetes mellitus”. The Cochrane Database of Systematic Reviews. 9 (9): CD009128.
Spranger J, Verma S, Göhring I, Bobbert T, Seifert J, Sindler AL, Pfeiffer A, Hileman SM, Tschöp M, Banks WA (January 2006). “Adiponectin does not cross the blood-brain barrier but modifies cytokine expression of brain endothelial cells”. Diabetes. 55 (1): 141–7.
Hoffstedt J, Arvidsson E, Sjölin E, Wåhlén K, Arner P (March 2004). “Adipose tissue adiponectin production and adiponectin serum concentration in human obesity and insulin resistance”. The Journal of Clinical Endocrinology and Metabolism. 89 (3): 1391–6.
Nicolas S, Veyssière J, Gandin C, Zsürger N, Pietri M, Heurteaux C, Glaichenhaus N, Petit-Paitel A, Chabry J (2015). “Neurogenesis-independent antidepressant-like effects of enriched environment is dependent on adiponectin”. Psychoneuroendocrinology. 57: 72–83.
Li A, Yau SY, Machado S, Yuan TF, So KF (2015). “Adult neurogenic and antidepressant effects of adiponectin: a potential replacement for exercise?”. CNS Neurol Disord Drug Targets. 14 (9): 1129–1144.
Cho JK, Kim S, Hong HR, Yoon JH, Kang H (2015). “Exercise Training Improves Whole Body Insulin Resistance via Adiponectin Receptor 1”. Int J Sports Med. 36 (13): e24–e30.
Gorman, Katherina, et al, “Adiponectin is necessary for exercise training-induced muscular hypertrophy and vascular adaptation.” The FASEB Journal, April 2016. Vol. 30 no. 1 Supplement 1240.23.
L’Ibutamoren Mesylato (INN) [2-amino-2-methylN-[(2R)-1-(1-methylsulfonylspiro[2H-indole-3,4′-piperidine]-1′-yl)-1-oxo-3-phenylmethoxypropan-2-yl]propanamide] (codice nomi di sviluppo MK-677, MK-0677, L-163.191; tentativo di brand commerciale Oratrope) è un potente agonista selettivo, non peptidico, a lunga durata d’azione del recettore GHS–R1a (Growth Hormone Secretagogue Receptor) della Grelina con azione secretogena dell’Ormone della Crescita (GH) mimando la pulsatilità del rilascio di tale ormone.[1][2][3][4][5] È stato infatti dimostrato che l’Imbutamoren è in grado di aumenta la secrezione di GH e, consequenzialmente, del Fattore di Crescita Insulino-Simile-1 (IGF-1) in modo sostenuto senza influenzare sui livelli di Cortisolo.[6] Infatti, l’MK-677 venne sintetizzato nel 1995 dalla Merck proprio allo scopo terapeutico di ristabilire la pulsatilità del rilascio del GH in anziani e soggetti deficitari, anche in bambini con deficienza idiopatica del GH. [7]
L’Ibutamoren, quindi, non è un SARM (Selective Androgen Receptor Modulators)! Nonostante diverse etichette commerciali tendano a spacciarlo come tale.
Asse GH-IGF-1
L’Ibutamoren ha dimostrato di incrementare l’attività dell’Asse GH – IGF-1 e di aumentare la massa magra senza alterare la massa grassa totale o il grasso viscerale. Questa molecola è sotto osservazione come potenziale trattamento dei pazienti con livelli patologicamente bassi di GH, come nei bambini o negli anziani con deficit dell’Ormone della Crescita. [1] [8] [9] [10] Studi sull’uomo hanno dimostrato che aumenta sia la massa muscolare che la densità minerale ossea, [11] [12] il che lo rende una molecola d’uso potenziale nel trattamento della fragilità ossea negli anziani sebbene, ad oggi, l’Ibutamoren non ha dato riscontri molto significativi nella prevenzione delle fratture senili, nonostante miglioramenti sul turnover osseo con l’aumento dell’osteocalcina sono stati osservati, ed i livelli di IGF-1 e GH risultavano più elevati nei soggetti trattati. [13] [14] A partire da giugno 2017, l’Ibutamoren è nella fase preclinica di sviluppo per il trattamento della carenza dell’Ormone della Crescita. [1]
Grelina
Dal momento che l’MK-677 è tutt’oggi un farmaco sperimentale, non è stata ancora approvata la sua commercializzazione per uso umano negli Stati Uniti ed in altri paesi. [1] Tuttavia, è attualmente utilizzato sperimentalmente da atleti di diverse discipline e in particolare da parte dei BodyBuilder. Poiché questa molecola imita chimicamente l’ormone Grelina, attraversa la barriera emato-encefalica e agisce anche come un neuropeptide a livello del Sistema Nervoso Centrale.[3][4][5][6] Secondo alcune recenti ricerche, vi è la preoccupazione che la sua lunga emivita possa stimolare eccessivamente i recettori della Grelina nel cervello e portare ad alcuni effetti collaterali mentali potenzialmente dannosi. [15] [16]
GHRH
Come già accennato, l’Ibutamoren stimola la secrezione dell’Ormone della Crescita (GH) e aumenta i livelli del Fattore di Crescita Insulino-Simile -1 (IGF-1) legandosi ai recettori GHS-R1a della Grelina, analogamente al GHRP-6 (anche a livello degli effetti riscontrati) dal momento che il rilascio di GH in entrambi i casi è mediato dallo stesso recettore ipofisario e non vi è quindi alcun effetto cumulativo a livello secretorio quando le due molecole vengono co-somministrate (lo stesso vale per tutti i secretagoghi del GH che agiscono per questa via recettoriale); tale effetto si osserva invece quando l’Ibutamoren viene combinato con il GHRH, situazione in cui la secrezione di GH è significativamente più alta rispetto a quella ottenibile dai composti assunti singolarmente, ed è stato inoltre osservato un aumento della concentrazione di cAMP intracellulare. [17][18]
I GHS-R si trovano in regioni del cervello deputate al controllo dell’appetito, del piacere, dell’umore, dei ritmi biologici, della memoria e della capacità cognitiva.[19]
Pertanto, l’utilizzatore potrebbe sperimentare un influenza su queste funzioni da parte dell’Ibutamoren. Tuttavia, almeno finora, gli studi clinici descrivono solo gli effetti dell’ibutamoren sull’appetito e, come previsto, egualmente alla grelina, l’ibutamoren ne causa un aumento.
Come ben sappiamo, GH e IGF-1, a loro volta, hanno un effetto sull’aumento della massa muscolare e della forza (vedi in particolare IGF-1) e sul grasso corporeo (vedi GH).[20][21]
In uno studio dove vennero presi in esame 24 uomini obesi sottoposti a due mesi di trattamento con Ibutamoren, si è osservato un aumento della massa magra e un interessante aumento temporaneo del metabolismo basale (BMR).[22]
In uno studio condotto su otto volontari sani sottoposti ad un forte regime ipocalorico, la somministrazione di Ibutamoren ha causato un inversione della perdita di proteine strutturali del tessuto contrattile, effetto con potenziale preventivo del ipotrofia muscolare.[23]
Su 123 pazienti anziani con frattura dell’anca sottoposti a trattamento con Ibutamoren , si è osservato un miglioramento nell’andatura, nella forza muscolare con una riduzione del numero di cadute.[24]
E’ cosa risaputa che il GH aumenta il turnover osseo e influenza la densità ossea. Tuttavia, a causa dell’aumento del turnover nei soggetti trattati con GH, la densità ossea può inizialmente diminuire prima di subire alcun incremento.[18] [25]
Fondamentalmente, ci vuole un certo periodo di tempo (> 1 anno) per osservare l’aumento della densità ossea.
In 24 volontari maschi obesi “sani” trattati con Ibutamoren si è osservato un aumento del turnover osseo. [26]
In tre studi clinici condotti su 187 adulti anziani (65+ anni) trattati con ibutamoren, si è osservato un aumento della costruzione ossea, misurata attraverso l’analisi dell’Osteocalcina, un indicatore del turnover osseo. [25]
In uno studio condotto su 292 donne in postmenopausa trattate con Ibutamoren, si è osservato un aumento della densità minerale ossea, il che contribuisce ad aumentare la forza ossea e prevenire l’osteoporosi. [27]
Uno studio dove soggetti sia giovani che anziani venivano trattati con Ibutamoren, ha mostrato un miglioramento della qualità del sonno e della durata del sonno REM (rapid eye movement).[28]
Sebbene di marginale importanza, esistono dei dati aneddotici forniti dagli utilizzatori “sperimentali” (vedi atleti) che riportano un miglioramento della qualità del sonno.
Il GH viene secreto soprattutto durante l’infanzia e la gioventù. Dopo i 20 anni, la sua sintesi diminuisce velocemente al punto che di solito la concentrazione di tale ormone in una persona di 50 anni è circa la metà di una di 20. Nell’uomo si osservano picchi secretori a cicli di 3-4 ore con valori massimi durante la notte ed in particolare nella prima fase del sonno profondo. In linea del tutto generale, le donne hanno maggiori livelli di GH e IGF-I rispetto agli uomini di pari età.[29] [30]
In uno studio nel quale 65 adulti anziani sono stati sottoposti a trattamento con Ibutamoren, si è osservato un aumento dei livelli di GH e IGF-1 rispetto a quelli di giovani adulti sani e senza riscontrare gravi effetti avversi.[18]
Un adeguato livello di IGF-1 è anche correlato ad effetti benefici sulla longevità.[31] [32]
In un altro studio nel quale sono stati presi in esami 24 soggetti obesi di sesso maschile (studio randomizzato controllato in doppio cieco) sottoposti a somministrazione di Ibutamoren, si è osservato un miglioramento nei livelli dell’Ormone della Crescita. [24]
Nei soggetti sovrappeso, obesi con diabete di tipo II o con sindrome metabolica si ha un variabile grado di insulino resistenza, che porta ad un’aumentata lipolisi basale nel tessuto adiposo con elevati livelli di FFA nel sangue e talvolta (DM2, sindrome metabolica) alterata glicemia a digiuno. Il GH è un ormone iperglicemizzante e lipolitico, pertanto sia l’iperglicemia che gli acidi grassi liberi (FFA) ne inibiscono la secrezione, infatti questi soggetti hanno ridotti livelli di GH, e la terapia sostitutiva migliora la composizione corporea. Questo avvalora la tesi secondo la quale avere livelli elevati di grasso corporeo deprime la secrezione di GH.
Alcuni soggetti assumono l’Ibutamoren anche per il suo potenziale effetto nootropo. Infatti, tale effetto è legato all’interazione del MK-677 con i recettori della Grelina, la quale ha effetti nootropici. Tuttavia, in ambito scientifico deve ancora essere determinato se e come l’Ibutamoren influenzi le prestazioni cognitive.
Due meccanismi indiretti attraverso i quali l’Ibutamoren potrebbe migliorare le funzioni cerebrale includono:
Aumento del IGF-1, che ha effetti sul miglioramento della memoria e dell’apprendimento .[33] [34]
Aumento della durata del sonno REM e della qualità del sonno, fattore essenziale per una corretta funzionalità cognitiva .[35] [28]
Uno studio ha messo in dubbio l’associazione tra bassi livelli di IGF-1 e morbo di Alzheimer e se l’utilizzo dell’Ibutamoren potesse essere un valido aiuto nel trattamento di questa patologia. Tuttavia, in questo studio, l’Ibutamoren non ha mostrato alcuna efficacia nel rallentamento della progressione dell’Alzheimer negli esseri umani trattati.[36]
L’Ibutamoren ha dimostrato di aumentare i livelli di GH, IGF-1 e IGFBP-3 nei bambini con deficit dell’Ormone della Crescita senza modificare le concentrazioni di Prolattina, Glucosio ematico, Triiodotironina (T3), Tiroxina (T4), Tireotropina, Cortisolo e Insulina.[37]
L’MK-677 ha mostrato effetti simili anche in soggetti adulti con grave carenza di GH, ma in questo caso la glicemia ematica e i livelli di Insulina hanno subito degli incrementi.[38]
L’Ormone della Crescita aumenta la rigenerazione dei tessuti e la guarigione delle ferite, quindi l’Ibutamoren potrebbe avere un potenziale nell’aiutare, per esempio, ad accelerare i tempi di recupero seguenti ad un intervento. [39] [40]
Esistono alcune testimonianze di utilizzatori di Ibutamoren che sembrano avvalorare questa tesi, ma mancano comunque studi scientifici che la avvalorino.
Un altro dato puramente aneddotico sembra attribuire al Ibutamoren un effetto sul trattamento dei postumi di una sbornia. Nulla più che chiacchiere da chat che lasciano spazio a parecchie speculazioni sul perché di questo possibile effetto (vedi, appunto, conseguenza effetto neuro peptidico).
Gli effetti collaterali più frequenti con l’uso dell’Ibutamoren sono aumento dell’appetito, lieve edema negli arti inferiori e dolore muscolare.[18]
Diversi studi riportano un aumento della glicemia a digiuno e una diminuzione della sensibilità all’Insulina. .[18] [38]
Questi effetti collaterali sono simili a quelli osservati negli utilizzatori di dosi sovra fisiologiche di GH o nelle persone con livelli patologicamente elevati di questo ormone, principalmente dolori articolari e insulino-resistenza. [41] [42]
Sebbene la maggior parte degli studi riferisca effetti collaterali minimi o nulli, uno studio è stato interrotto a causa di gravi eventi avversi.
Nei pazienti anziani con frattura dell’anca, il trattamento con Ibutamoren ha aumentato la pressione sanguigna, la glicemia e l’HbA1c e vi sono stati casi di insufficienza cardiaca congestizia. I pazienti che hanno manifestato insufficienza cardiaca congestizia avevano più di 80 anni e con una storia di precedente insufficienza cardiaca.[24]
Sarebbe quindi opportuno sottoporsi a regolari controlli dei marker ematici indicatori dello stato del metabolismo glucidico prima, durante e dopo l’utilizzo di Ibutamoren. Questi comprendono glicemia a digiuno, HbA1c, Insulina a digiuno, indice HOMA-IR e HOMA-B, curva glicemica e insulinica.
Un’altra avvertenza, sebbene possa sembrare ovvia, riguarda il fatto che il potenziale utilizzatore dovrebbe essere certo di non avere alcun tipo di cancro. Sappiamo benissimo che GH e, soprattutto, l’IGF-1 possono favorire la crescita del cancro la dove questo è presente.[43]
Non è raccomandato l’uso di Ibutamoren se si è dominante Th1 poiché il GH presenta proprietà immunostimolatorie.
L’Ibutamoren ha un effetto inibitorio sulla segnalazione della Somatostatina (inibitore della secrezione di GH, TSH, ACTH e Prolattina), ma non si escludono sovra-regolazioni di questo peptide dopo il cessato utilizzo della molecola per lunghi periodi di tempo (sebbene, ad oggi, non sia mai stato documentato). Questo effetto inibitorio, inoltre, è legato allo stimolo indiretto che l’MK-677 può avere (dose dipendente) sulla secrezione di Prolattina e Cortisolo.
Sebbene a dosi contenute (25mg/die) non sono stati registrati casi di sottoregolazione dell’attività tiroidea, dosi elevate (≥50mg/die) potrebbero, e sottolineo potrebbero, causare un feedback negativo a livello tiroideo, simile a quello osservato durante l’uso di GH esogeno. Questa ipotesi, però, potrebbe avere una percentuale di possibile riscontro ridotta per via dell’effetto del Ibutamoren sulla segnalazione della Somatostatina e degli effetti che questo avrebbe sui livelli di TSH, livelli che in cronico, e questa è un altra ipotesi, potrebbero portare ad una sottoregolazione dell’attività tiroidea per desensibilizzazione all’ormone. Si tratta comunque di semplici speculazioni accademiche.
Come detto in precedenza, l’Ibutamoren non presenta effetti stimolatori significativi sui livelli di Cortisolo (aumenti registrati massimi di 2,4 volte il basale [44]), nonostante la Grelina attivi l’Asse HPA causando un aumento del Cortisolo.[45] La risposta potrebbe variare in modo dose dipendente (≥50mg/die).
Come già accennato, attualmente non è stata approvata la vendita e l’uso dell’Ibutamoren negli esseri umani e per questo motivo non esistono linee guida da prescrizione. Nonostante ciò, la molecola è reperibile nel mercato “grigio” degli store online d’oltre Manica e d’oltre oceano e nel mercato nero delle UGL.
In ambito sportivo, in specie nella sottocultura del BodyBuilding e del Fitness, l’Ibutamoren ha mostrato un ottima efficacia alla dose tra i 10 ed i 25mg/die, in un’unica assunzione prima di dormire, sebbene siano stati testati dosaggi giornalieri anche di 0,8mg/Kg [46] o 1mg/Kg [47] con risposte avverse tutto sommato contenute anche se la selettività della molecola si riduceva a seconda dell’entità della dose e con essa aumentavano gli stimoli su Cortisolo e Prolattina, mentre il grado di impatto sulla sensibilità ell’insulina è dose e tempo dipendente. Dosi elevate portano anche ad un significativo aumento del IGFBP-3 (che ricordo essere una delle sei proteine leganti l’IGF; dal IGFBP-1 al IGFBP-6). Usualmente, l’MK-677 viene assunto per periodi di tempo che vanno dai 3 ai 4 mesi (12-16 settimane), sebbene periodi di somministrazione di 6 mesi o più non sono rari sebbene poco intelligenti per via della elevata possibilità di cronicizzare lo stato di insulino resistenza iatrogena e di instaurare di conseguenza una condizione diabetica (diabete di tipo II). Protocolli di assunzione di 8 settimane sono risultati sufficienti al fine di ottenere i benefici sul miglioramento dell’ipertrofia per azione indiretta di questa molecola.
L’Ibutamoren ha mostrato nei beagle di avere un emivita di 4–6 ore [48], mentre negli esseri umani ha causato con una singola dose un aumento dei livelli di IGF-1 fino a 24 ore.[49] Tali risultati, sebbene la questione è dibattuta, portano alla conclusione che negli esseri umani l’emivita sia di circa 12 ore anche se la vita attiva è stimata a circa 24-32 ore. Di conseguenza, sono nate due scuole di pensiero riguardo al timing di assunzione di questa molecola, complice anche la totale mancanza di linee guida cliniche sicure/certe. La prima scuola di pensiero, la più classica, vede la dose giornaliera di Ibutamoren assunta in un’unica somministrazione serale, lontano dall’ultimo pasto prima di dormire. La seconda scuola di pensiero, invece, opta per uno schema di assunzione multiplo, generalmente due assunzioni di pari entità, separate da almeno 12 ore l’una dall’altra. Solitamente, la prima dose viene assunta al mattino appena svegli mentre la seconda la sera prima di dormire. Entrambe le assunzioni sono da preferire lontano dai pasti onde evitare possibili riduzioni e/o alterazioni d’efficacia della molecola.
CJC-1295
Come accennato in precedenza, la combinazione di Ibutamoren con GHRH (growth hormone-releasing hormone) incrementa notevolmente la secrezione di GH rispetto ai singoli composti assunti da soli avendo vie di interazione recettoriale diverse che diventano complementari con la co-somministrazione. Infatti, l’MK-677 è solitamente abbinato a due peptidi secretagoghi del GH analoghi del GHRH, la Sermorelina Acetato e il CJC-1295 (noto anche come DAC:GRF). Entrambe le molecole, data la loro natura peptidica, vengono somministrati per via sottocutanea. Si noti che il CJC-1295 ha un’emivita stimata di circa 6-8 giorni nell’uomo, e ha mostrato dopo una singola iniezione, sempre in soggetti umani, di aumentare i livelli plasmatici di GH da 2 a 10 volte per 6 o più giorni e i livelli plasmatici di IGF-1 da 1,5 a 3 volte da 9 a 11 giorni.[50] Con dosi multiple di CJC-1295, i livelli di IGF-1 sono rimasti elevati negli umani per un massimo di 28 giorni.[50]
Per concludere, l’Ibutamoren è senz’altro una molecola interessante, specie nei periodi “Bulk”, dato il suo impatto sull’appetito che risulterebbe al quanto difficile da gestire durante un deficit calorico (utilizzo di anoressizzanti a parte), in specie in quei casi dove l’acquisto del hGH risulta limitato o impossibilitato. Il potenziale utilizzatore dovrebbe cominciare ad assumere l’MK-677 al dosaggio minimo efficace (10mg/die) per poi testarne la risposta generale e solo dopo riscontro positivo, nel caso, aumentare la dose (al momento non è consigliabile un incremento oltre gli 0,8mg/Kg e per un tempo non più lungo di 12 settimane). L’uso concomitante di GDA (OTC o farmaceutici) potrebbe tamponare almeno in parte l’effetto negativo sulla sensibilità all’Insulina esplicato indirettamente dalla molecola per via dell’aumento in cronico dei livelli di GH e IGF-1.
Evitate assolutamente il “fai da te” o le indicazioni del “palestricolo” privo di qual si voglia nozione utile all’uso corretto di questa o altre molecole.
^ Jump up to: ab Pong SS, Chaung LY, Dean DC, Nargund RP, Patchett AA, Smith RG (January 1996). “Identification of a new G-protein-linked receptor for growth hormone secretagogues”. Molecular Endocrinology. 10 (1): 57–61. doi:1210/me.10.1.57. PMID8838145.
^ Jump up to: ab Cassoni P, Papotti M, Ghè C, Catapano F, Sapino A, Graziani A, Deghenghi R, Reissmann T, Ghigo E, Muccioli G (April 2001). “Identification, characterization, and biological activity of specific receptors for natural (ghrelin) and synthetic growth hormone secretagogues and analogs in human breast carcinomas and cell lines”. The Journal of Clinical Endocrinology and Metabolism. 86 (4): 1738–45. doi:1210/jcem.86.4.7402. PMID11297611.
^ Jump up to: ab Holst B, Frimurer TM, Mokrosinski J, Halkjaer T, Cullberg KB, Underwood CR, Schwartz TW (January 2009). “Overlapping binding site for the endogenous agonist, small-molecule agonists, and ago-allosteric modulators on the ghrelin receptor”. Molecular Pharmacology. 75 (1): 44–59. doi:1124/mol.108.049189.
Copinschi G, Van Onderbergen A, L’Hermite-Balériaux M, Mendel CM, Caufriez A, Leproult R, Bolognese JA, De Smet M, Thorner MO, Van Cauter E (August 1996). “Effects of a 7-day treatment with a novel, orally active, growth hormone (GH) secretagogue, MK-677, on 24-hour GH profiles, insulin-like growth factor I, and adrenocortical function in normal young men”. The Journal of Clinical Endocrinology and Metabolism. 81 (8): 2776–82.
Chapman IM, Bach MA, Van Cauter E, Farmer M, Krupa D, Taylor AM, et al. (December 1996). “Stimulation of the growth hormone (GH)-insulin-like growth factor I axis by daily oral administration of a GH secretogogue (MK-677) in healthy elderly subjects”. The Journal of Clinical Endocrinology and Metabolism. 81 (12): 4249–57.
Thorner MO, Chapman IM, Gaylinn BD, Pezzoli SS, Hartman ML (1997). “Growth hormone-releasing hormone and growth hormone-releasing peptide as therapeutic agents to enhance growth hormone secretion in disease and aging”. Recent Progress in Hormone Research. 52: 215–44, discussion 244-6.
Chapman IM, Pescovitz OH, Murphy G, Treep T, Cerchio KA, Krupa D, Gertz B, Polvino WJ, Skiles EH, Pezzoli SS, Thorner MO (October 1997). “Oral administration of growth hormone (GH) releasing peptide-mimetic MK-677 stimulates the GH/insulin-like growth factor-I axis in selected GH-deficient adults”. The Journal of Clinical Endocrinology and Metabolism. 82 (10): 3455–63.
Murphy MG, Bach MA, Plotkin D, Bolognese J, Ng J, Krupa D, Cerchio K, Gertz BJ (July 1999). “Oral administration of the growth hormone secretagogue MK-677 increases markers of bone turnover in healthy and functionally impaired elderly adults. The MK-677 Study Group”. Journal of Bone and Mineral Research. 14 (7): 1182–8. doi:1359/jbmr.1999.14.7.1182. PMID10404019.
Murphy MG, Weiss S, McClung M, Schnitzer T, Cerchio K, Connor J, Krupa D, Gertz BJ (March 2001). “Effect of alendronate and MK-677 (a growth hormone secretagogue), individually and in combination, on markers of bone turnover and bone mineral density in postmenopausal osteoporotic women”. The Journal of Clinical Endocrinology and Metabolism. 86 (3): 1116–25. doi:1210/jcem.86.3.7294. PMID11238495.
Smith RG, Sun Y, Jiang H, Albarran-Zeckler R, Timchenko N (November 2007). “Ghrelin receptor (GHS-R1A) agonists show potential as interventive agents during aging”. Annals of the New York Academy of Sciences. 1119: 147–64. doi:1196/annals.1404.023. PMID18056963.
Kaklamani VG, Linos A, Kaklamani E, Markaki I, Mantzoros C. Age, sex, and smoking are predictors of circulating insulin-like growth factor 1 and insulin-like growth factor-binding protein 3. J Clin Oncol. 1999; 17:813–817..
L. Willisa, R. F. Wolfb, G. L Whiteb, and D. McFarlane, Age- and gender-associated changes in the concentrations of serum TGF-1β, DHEA-S and IGF-1 in healthy captive baboons (Papio hamadryas anubis), in Gen Comp Endocrinol., 2014 January.
Smith RG, Van der Ploeg LH, Howard AD, Feighner SD, Cheng K, Hickey GJ, Wyvratt MJ, Fisher MH, Nargund RP, Patchett AA (October 1997). “Peptidomimetic regulation of growth hormone secretion”. Endocrine Reviews. The Endocrine Society. 18 (5): 621–45.
Teichman, Sam L.; Neale, Ann; Lawrence, Betty; Gagnon, Catherine; Castaigne, Jean-Paul; Frohman, Lawrence A. (2006). “Prolonged Stimulation of Growth Hormone (GH) and Insulin-Like Growth Factor I Secretion by CJC-1295, a Long-Acting Analog of GH-Releasing Hormone, in Healthy Adults”. The Journal of Clinical Endocrinology & Metabolism. 91 (3): 799–805.
Quattro mesi dopo un ciclo di AAS, i livelli di LH e FSH che, come ben sappiamo, stimolano rispettivamente la biosintesi testicolare di Testosterone e la spermatogenesi, in media si ristabiliscono nei range fisiologici, ma questo parziale assestamento ormonale sembra non riflettersi proporzionalmente sulla sintesi di Testosterone. Ciò è emerso dai risultati di una meta-analisi che endocrinologi greci dell’Università di Ioannina hanno pubblicato nel 2017 su Sports Medicine.(1)
I ricercatori hanno raccolto ed esaminato per la loro meta-analisi 11 studi precedentemente pubblicati nei quali gli autori avevano determinato le concentrazioni di LH, FSH e Testosterone in poche decine di utilizzatori di AAS prima, durante e dopo il ciclo. Gli studi più datati utilizzati risalivano alla metà degli anni ’80.
Negli studi raccolti, i ricercatori hanno esaminato il recupero delle concentrazioni di FSH e LH 13-24 settimane dopo l’interruzione dell’uso di AAS. I ricercatori hanno anche esaminato il recupero dei livelli di Testosterone totale a 16 settimane dall’interruzione d’uso di AAS – non sono stati valutati i livelli di Testosterone libero e biodisponibile (somma del Testosterone libero e del Testosterone legato all’albumina).
I ricercatori hanno osservato un completo recupero delle concentrazioni di LH [vedi la figura seguente] e FSH.
Tuttavia, questo parziale recupero ormonale non era correlato ad un ristabilimento dei livelli di Testosterone [vedi figura seguente]. I livelli di Testosterone totale non erano rientrati nei range fisiologici dopo quattro mesi dall’interruzione d’uso di AAS.
I ricercatori, con una certa banalità espositiva, riportano che la presente meta-analisi ha mostrato che i livelli serici di gonadotropine e di Testosterone endogeno diminuivano durante un periodo di uso attivo di AAS negli atleti di sesso maschile. E continuano scrivendo che i livelli ormonali sono tornati gradualmente alla normalità, sebbene il Testosterone serico sia rimasto inferiore rispetto al basale per diverse settimane dopo l’uso di AAS. Inoltre, riportando altre banalità in modo non prettamente “completo”, i ricercatori hanno affermato che una revisione sistematica della letteratura ha rivelato che gli effetti dell’uso di AAS a lungo termine includono atrofia testicolare, ginecomastia e compromissione delle caratteristiche degli spermatozoi negli uomini, così come la clitoromegalia e l’irregolarità mestruale nelle donne, che possono influire sulla fertilità in entrambi i sessi. L’abuso di AAS ha effetti negativi, potenzialmente gravi sul lungo termine a livello del sistema riproduttivo e sulla salute generale degli utilizzatori; ulteriori azioni sono necessarie per gestire questo problema di salute pubblica globale, insieme all’educazione del pubblico, degli atleti, degli allenatori e degli operatori sanitari.
E’ corretto sottolineare che i casi presi in esame in questa meta-analisi non comprendevano l’applicazione di una corretta PCT al termine d’uso degli AAS. Di conseguenza, le risposte di recupero erano in linea con le capacità medie di riassetto dell’omeostasi ormonale senza alcun sostegno farmacologico iniziale. Si ricorda inoltre che la capacità di recupero è soggetta anche al tipo di molecola/e utilizzata/e durante il ciclo (vedi, per esempio, il Nandrolone).
La bassa biodisponibilità degli AAS orali non metilati in C-17 ha sempre significato una limitazione nella loro applicazione. Dosaggi efficaci e costi correlati hanno di norma spinto atleti e Preparatori verso altre scelte qualora l’uso di un composto orale fosse stato preso in considerazione. Ovviamente, questa limitazione non interessa soltanto questa categoria di AAS orali, e infatti molti altri farmaci potrebbero vedere il loro impiego a dosaggi nettamente più contenuti se somministrati con apposite pratiche. Tuttavia, e dai tempi delle pubblicazioni di Dan Duchaine, scomparso ormai da diciotto anni, che siamo a conoscenza di alcuni metodi per aumentare la biodisponibilità di queste molecole. Queste pratiche di somministrazione vanno ben oltre il semplice, e a volte poco fruttuoso, consiglio di “assumerle a stomaco vuoto”. Senza dubbio, l’utilizzo del DMSO in gel o in soluzione con acqua (50/50) come veicolo per migliorare la biodisponibilità dei farmaci è il più interessante in quanto permette anche una applicazione topica in zone specifiche del corpo, aree nelle quali si trovano accumuli adiposi particolarmente ostici da ridurre e che possono essere trattati in modo più specifico usando determinate molecole veicolate attraverso il DMSO.
Dopo le dovute note introduttive ed aver catturato l’attenzione di alcuni di voi, posso proseguire con la mia disamina riguardante l’argomento chiave di quest’articolo, il DMSO come mezzo per migliorare la biodisponibilità dei farmaci e le loro possibilità applicative.
Ma cos’è il DMSO?
Il Dimetilsolfossido (DMSO), noto anche come Metilsolfossido o Sulfinilbis(metano), è un composto organico appartenente alla categoria dei solfossidi, si tratta di un composto organosulfuro con formula (CH3) 2SO. A temperatura ambiente si presenta come un liquido incolore e inodore particolarmente igroscopico. Sebbene nella sua forma pura sia effettivamente privo di odore, campioni impuri di Dimetilsolfossido odorano fortemente di Dimetilsolfuro. Il DMSO è un solvente aprotico, miscibile con una vasta gamma di solventi, fra cui alcoli, eteri, chetoni, clorurati e aromatici. È inoltre miscibile in tutte le proporzioni con l’acqua. Essendo quindi un solvente aprotico polare, il DMSO scioglie sia i composti polari che quelli non polari. Il DMSO possiede una particolare e insolita proprietà di far percepire un sapore simile all’aglio agli individui la cui pelle entra in contatto con esso.(1)
Il Dimetilsolfossido è un sotto-prodotto della lavorazione della carta, frequentemente usato come solvente in chimica organica: in particolare il Dimetilsolfossido è un solvente particolarmente indicato per alchilazioni SN2. Per esempio è possibile alchilare l’indolo o i fenoli con alte rese utilizzando idrossido di potassio (KOH) come base.
Gli atomi di idrogeno metilici del DMSO presentano un debole carattere acido (pKa= 35) a causa dell’effetto stabilizzante degli anioni del gruppo solfossido.
Il Dimetilsolfossido è stato scoperto nel 1867, tuttavia non venne utilizzato commercialmente fino al termine della Seconda guerra mondiale. In aggiunta ai suoi utilizzi come solvente, sia in chimica organica, sia per applicazioni industriali (chimica dei polimeri, farmaci, prodotti agrochimici), il Dimetilsolfossido è anche un eccellente agente per la rimozione delle vernici da legno e da metallo, caratterizzato da un vantaggio in termini di sicurezza rispetto ad altre sostanze come il Nitrometano e il Diclorometano.
Il DMSO è utilizzato come ossidante nelle reazioni di ossidazione di Swern e nell’ossidazione di Pfitzner-Moffatt.
Il DMSO può anche essere impiegato come agente di lavaggio nell’industria elettronica e, nella sua forma deuterata (DMSO-d6), è un comune solvente utilizzato nelle analisi NMR, grazie alla sua capacità di dissolvere un gran numero di sostanze e alla bassa interferenza dei suoi segnali. (AV)
Il ricorso in campo medico al DMSO risale almeno al 1963 quando una squadra di ricercatori della “Scuola di Medicina dell’Università dell’Oregon” diretta da Stanley Jacob scoprì che tale sostanza era in grado di penetrare in profondità sotto la pelle e altre membrane senza danneggiarle, trasportando altre molecole all’interno del sistema biologico. Il Dimetilsolfossido viene quindi utilizzato per l’applicazione topica di prodotti farmaceutici, accanto ai suoi usi come analgesico locale, anti-infiammatorio ed antiossidante.
Quindi, il compianto Dan Duchaine non scoprì nulla di nuovo ma ebbe quella giusta dose di intuitività in grado di fargli sperimentare combinazioni “artigianali” tra farmaci orali, ridotti in polvere, con bassa biodisponibilità e una soluzione di DMSO e acqua (rapporto 1:1) per applicazioni topiche, soprattutto in quelle zone con depositi adiposi ostici da ridurre.
Adesso abbiamo appurato che il DMSO è un solvente naturale con l’interessante capacità di agire come trasportatore transdermico in modo estremamente efficace. Ma fatta questa constatazione, come dosare correttamente i componenti di questo mix per applicazioni topiche?
DMSO nella pratica
Come accennato precedentemente, per prima cosa il DMSO (al 99,9%) va diluito con acqua in soluzione 50/50 dal momento che il Dimetilsolfossido puro sarebbe troppo irritante per la pelle. È importante utilizzare DMSO farmaceutico (puro) e non di tipo industriale, che avrà una buona dose di impurità. Il quantitativo di DMSO deve essere calcolato in base al quantitativo di principio attivo (addizionato alle compresse) che si vuole utilizzare. Il rapporto tra DMSO e compresse/mg di AAS è di 1ml ogni 10mg. Un semplice e rapido esempio può essere fatto con il Metenolone Acetato: la dose per somministrazione topica efficace risulta essere di 20-25mg. Una volta sbriciolate finemente le compresse contenti tale dosaggio si miscelano con la soluzione DMSO/Acqua composta da 2ml di Dimetilsolfossido e 2ml d’acqua. Una volta miscelata la polvere e la soluzione DMSO/Acqua essa va applicata nella zona desiderata (es. zona ombelicale). Dal momento che si tratta di una soluzione ben poco densa, è utile applicare una pellicola in modo da evitare dispersioni del preparato applicato. In fine, l’applicazione va tenuta per circa quaranta minuti, onde garantire un ottimale assorbimento del principio attivo. Questo trattamento viene generalmente eseguito da una a tre volte al giorno in base anche al numero delle aree da trattare. In alternativa alla soluzione con acqua è possibile utilizzata il DMSO in gel, ma è generalmente più difficile da reperire.
Riassunto pratico della procedura:
Miscelare il DMSO con acqua in rapporto 1:1 (es. 2ml/2ml)
Sbriciolare un quantitativo di compresse mantenendo un rapporto mg/ml di DMSO pari a 1ml ogni 10mg.
Una volta sbriciolate finemente le compresse miscelarle con la soluzione DMSO/Acqua.
Aspirare la miscela ottenuta con una siringa e applicare il contenuto direttamente sulla superfice cutanea della zona da trattare applicando una pellicola per impedire alla soluzione di disperdersi dall’area di applicazione.
Per il trattamento delle adiposità localizzate con AAS miscelati alla soluzione di DMSO, oltre al Metenolone Acetato viene utilizzato il Mesterolone ed il Trenbolone Acetato (in polvere). Alcuni hanno realizzato dei mix topici rassomiglianti al Helios contenenti Clenbuterolo, Chetotifene, Yohimbina e Triiodotironina (T3). Oltre a questo tipo di applicazione, la soluzione con DMSO è stata usata semplicemente per aumentare la biodisponibilità di alcuni AAS non metilati in C-17 come, ad esempio, il Furazabol (noto anche come Androfurazanolo).
Vi sono alcuni effetti avversi derivanti dall’uso del DMSO. Dal momento che il Dimetilsolfossido è un trasportatore molto efficaci capace di veicolare attraverso la pelle qualsiasi struttura con un peso molecolare abbastanza ridotto, gli utilizzatori devono essere cauti e premurarsi di pulire a fondo la l’epidermide che verrà a contatto con la soluzione prima dell’applicazione, e prestare particolare attenzione a ciò che entrerà in contatto con quella specifica area nelle ore immediatamente successive. Alcuni utilizzatori tengono la zona protetta con una pellicola in modo da proteggere la pelle da eventuali contaminanti. Gli utilizzatori di DMSO segnalano frequentemente effetti collaterali minori come prurito o bruciore da eruzione cutanee nel sito di applicazione, spesso dipendenti dalla scorretta diluizione del DMSO con l’acqua. Pat Arnold una volta aveva suggerito la possibilità di diluire il DMSO con circa il 5-10% di isopropile . L’alcol agisce come un carrier similmente al DMSO, ma evapora rapidamente dopo l’applicazione lasciando sulla pelle una lieve patina, dovuta al minor dosaggio necessario, di DMSO. Per quelli molto sensibili al Dimetilsolfossido, questa miscela può consentire un ridotto tasso di irritazione o, comunque, un grado di questa decisamente più contenuto proprio per la minore quantità di DMSO necessaria.
Acido Desossicolico
In conclusione, questa pratica può risultare vantaggiosa nel trattamento delle adiposità localizzate, sebbene esistano soluzioni iniettabili per mesoterapia con un grado di efficacia maggiore (vedi, per esempio, l’Acido Desossicolico iniettabile).
Per il miglioramento della biodisponibilità degli AAS orali, oltre all’assunzione di Naringina o Caffeina, è possibile miscelare le compresse in polvere con dell’olio MCT in modo da veicolarne l’assorbimento attraverso i vasi linfatici intestinali così come avviene per il Testosterone Undecanoato orale (Andriol). E su questo punto è utile aprire una parentesi.
La questione dell’assunzione di AAS orali in concomitanza con pasti a contenuto lipidico significativo e loro biodisponibilità è tanto semplice da trattare quanto soggetta a variabili che definirei di “vantaggio”. In passato citai spesso quanto riportato sul “Anabolic Steroids and Sports Volume II” (2), sul quale veniva riportato come l’assunzione di un AAS per via orale con un pasto poteva determinare una riduzione della su biodisponibilità a causa della natura liposolubile degli ormoni steroidei, che permette ad una parte del farmaco di sciogliersi con i grassi alimentari non digeriti, riducendo il suo assorbimento dal tratto gastrointestinale. Ad una attenta analisi della questione emerge che la possibilità che ciò avvenga in percentuali significative è legata alla quantità totale dei grassi nel pasto e che se questi non sono presenti in quantità elevate la loro presenza può favorire l’assorbimento degli AAS attraverso i vasi linfatici intestinali baipassando la deattivazione epatica di primo passaggio. Ovviamente, la percentuale di AAS che ipoteticamente potrebbe essere soggetta ad una celere espulsione per via dei grassi non digeriti è con tutta probabilità quanto meno discutibile. Esiste un interessante studio del 2016 (3) nel quale neonati operati per cardiopatia congenita e divisi in tre coorti da cinque soggetti sono stati sottoposti a trattamento post-operatorio con Oxandrolone. Ad un gruppo è stato somministrato il farmaco attraverso una soluzione acquosa 0,1 mg / kg / giorno, ad un altro attraverso una soluzione acquosa 0,2 mg / kg / giorno mentre al terzo gruppo è stato somministrato in una soluzione a base di olio di Trigliceride a Catena Media (MCT ) 0,1 mg / kg / giorno. Il gruppo trattato con la soluzione Oxandrolone+MCT ha mostrato il più basso calo di peso per età suggerendo una migliore biodisponibilità di questa preparazione.
Per conclude questa breve parentesi, si potrebbe affermare che gli AAS metilati in C-17 assunti con una quota di lipidi di circa il 20% o più la composizione del pasto potrebbero venire ad una certa loro percentuale assorbiti a livello dei vasi linfatici intestinali sgravando in modo parziale il loro impatto sul fegato, almeno per quanto riguarda i processi di de-attivazione di primo passaggio da parte del parenchima epatico. Ricordatevi, però, che la molecola verrà comunque processata a livello epatico causando stress a quest’organo. Per quanto riguarda gli AAS non metilati, è palese che il vantaggio sul incremento della loro biodisponibilità per via di questa pratica sia di interesse maggiore.
Gabriel Bellizzi
Riferimenti:
Novak, K. M., ed. (2002). Drug Facts and Comparisons (56th ed.). St. Louis, Missouri: Wolters Kluwer Health. p. 619.
Anabolic Steroids and Sports Volume II. James E. Wright. Sports Science Consultants, Natick, MA 1982.
Da quanto emerso attraverso uno studio effettuato dai ricercatori dell’Università di Ahvaz Jundishapur (Iran), l’assunzione giornaliera di Propoli da parte di soggetti con diabete di tipo II causa un miglioramento della loro sensibilità all’Insulina e una consequenziale riduzione dei livelli ematici del peptide. I ricercatori hanno affermato che l’effetto è così forte che l’integrazione con Propoli in questi soggetti può ridurre significativamente il rischio di complicanze legate alla patologia.(1)
Per l’esperimento, della durata di 90 giorni, i ricercatori hanno reclutato un centinaio di soggetti con diabete di tipo II. I soggetti non erano trattati con una terapia ipoglicemizzante a base di Insulina e non erano allergici ai prodotti dell’apicoltura.
A metà dei soggetti presi in esame sono state fatte assumere giornalmente capsule contenenti un placebo. All’altra metà dei soggetti è stata fatta assumere una capsula contenete 500mg di Propoli due volte al giorno (1g di Propoli/die).
I ricercatori hanno usato un prodotto dell’azienda iraniana Shahdine Golha.
La supplementazione con il Propoli ha migliorato la sensibilità all’Insulina dei soggetti trattati.
Nel gruppo sperimentale, i livelli ematici di Insulina sono diminuiti del 45%. Inoltre, dopo che questi soggetti avevano assunto Propoli per 90 giorni, la concentrazione di emoglobina glicata A1c (HbA1c) era diminuita dello 0,98%. Come certamente saprete, emoglobina glicata A1c è una forma di emoglobina usata principalmente per identificare la concentrazione plasmatica media del glucosio per un lungo periodo di tempo. Viene prodotta in una reazione non-enzimatica a seguito dell’esposizione della normale emoglobina al glucosio plasmatico.
Tale diminuzione riduce il rischio di complicanze legate al diabete del 21%.
Nei soggetti del gruppo placebo, l’eGFR (acronimo di “Estimated Glomerular Filtration Rate”; velocità di filtrazione glomerulare) era diminuito del 20,7%. L’eGFR indica il tasso di filtrazione glomerulare, cioè la velocità con cui il sangue viene filtrato (e ripulito) dai reni. Ciò significa che la funzione renale dei soggetti del gruppo placebo era peggiorata. Questa diminuzione era assente nei soggetti che avevano assunto il Propoli.
Secondo i ricercatori, un possibile meccanismo di azione del Propoli è la riduzione della produzione di fattori infiammatori come il TNF-alfa e – in misura minore – del CRP.
E’ interessante notare come molecole che hanno mostrato un certo effetto sulla insulino sensibilità , come l’Acido Clorogenico, posseggano struttura chimica simile a quella Estere Feniletilico dell’Acido Caffeico [CAPE], probabilmente la sostanza bioattiva più importante del Propoli. È molto probabile che queste sostanze condividano il meccanismo d’azione.
Sugli effetti dell’utilizzo di AAS sul lungo termine in soggetti di sesso femminile, ad oggi, sappiamo relativamente poco. Siamo pienamente a conoscenza del fatto che un uso di questa classe di farmaci nelle donne possa portare come conseguenza ad effetti virilizzanti di diverso tipo e entità. Un interessante caso studio realizzato da due medici ORL canadesi, Yael Bensoussan e Jennifer Anderson, dell’Università di Toronto, pubblicato sul Clinical Case Reports, ci offre la possibilità di analizzare la disfonia AAS-indotta e i sui risvolti nel lungo termine in una atleta. (1)
Il soggetto preso in esame nell’articolo di Yael Bensoussan e Jennifer Anderson è una Bodybuilder la quale aveva 27 anni nel 1998. In quell’anno aveva partecipato a una competizione e, per prepararvisi, aveva usato, secondo quanto da lei riportato, 50mg di Nandrolone Decanoato a settimana per 6 settimane.
Due mesi dopo l’inizio della somministrazione di Nandrolone, la voce della bodybuilder era gradualmente divenuta più bassa e roca. Sebbene abbia rinunciato all’uso di altri farmaci nella sua preparazione, l’alterazione del tono vocale risultò permanente. La terapia applicata per trattare l’alterazione della voce, volta quindi a ripristinare una tonalità vocale più alta e più femminile, non risultò efficace.
Il soggetto in questione, come conseguenza di questa alterazione, perse il lavoro perché la gente pensava che fosse un uomo. Contemporaneamente aveva detto addio al Bodybuilding.
Sempre nel 1998 contattò contattato dei medici specialisti, i quali decisero di operare sulla laringe. L’operazione ebbe esito positivo dal momento che la voce della donna tornò ad essere più alta e più femminile. Ma dieci anni dopo la ex Bodybuilder divenne afona. Si era verificata una atrofia delle corde vocali.
Tredici anni dopo la prima operazione, i medici intervenivano chirurgicamente una seconda volta, e di nuovo con successo.
I ricercatori non conoscono il motivo per il quale sia comparsa dell’afonia nella donna. Essa riferì di sentirsi cronicamente stanca e successivi esami ematici mostrarono livelli di Testosterone libero marcatamente bassi. Apparentemente, gli squilibri nella funzionalità dell’Asse HPG dovuti al passato uso di AAS da parte della donna non erano stati trattati e risolti con conseguente mantenimento di livelli bassi di Testosterone cronicizzatisi con il passare degli anni (riduzione ormonale età correlata). Tale circostanza (alterazione a lungo termine del HPTA) si può osservare anche negli ex abusatori di AAS di sesso maschile.
Appurato lo stato ormonale della paziente, i medici le prescrissero una terapia ormonale sostitutiva che risolse lo stato di fatica cronica.
I medici hanno ipotizzato che la possibile causa dell’afonia potrebbe essere ricondotta al livello di Testosterone anormalmente basso della donna il quale ha portato ad una atrofia delle corde vocali. Questa risposta avversa non trova però molto riscontro nell’osservazione di soggetti di sesso maschile con ipogonadismo AAS-dipendente non trattato. Il fatto che non esistano casi studio specifici che abbiano analizzato questo aspetto in tali soggetti, e la presenza di un dimorfismo sessuale di risposta ormonale, fanno si che questa ipotesi rimanga una possibilità plausibile.
Gli autori di questo caso studio affermano che, per quanto ne sanno, il loro è il primo rapporto sugli effetti degli AAS sulla voce di un soggetto di sesso femminile e sulla loro evoluzione in un periodo di 20 anni. Questi cambiamenti sono clinicamente rilevanti dal momento che sono difficili da trattare e pertanto dovrebbe essere comunicato ai soggetti che usano AAS o a coloro che sono sottoposti ad una terapia con androgeni.
Alcuni integratori che dovrebbero aumentare i livelli di Testosterone contengono gli isoflavoni sintetici Methoxyisoflavone e Ipriflavone. Questi due composti potrebbero avere qualche concreto potenziale, o per lo meno questo è quanto emerso da uno studio in vitro svolto da ricercatori italiani, associati alla Federazione Medico Sportiva Italiana, e i cui risultati sono stati pubblicati sul Drug Testing & Analysis.(1) Secondo i ricercatori, sia il Methoxyisoflavone che l’Ipriflavone inibiscono l’Enzima Aromatasi che, come ben sappiamo, converte il Testosterone in Estradiolo.
L’Ipriflavone e il Methoxyisoflavone sono isoflavoni sintetici. Furono sintetizzati nel secolo scorso da parte di chimici dell’Europa dell’Est, i quali erano intenzionati a creare nuovi agenti anabolizzanti mediante la modifica strutturale degli isoflavoni come la Daidzeina, un fitoestrogeno della Soia.
Gli isoflavoni sintetici non ebbero il successo sperato nell’applicazione farmacologica, ma hanno comunque preso piede come integratori alimentari per lo sport.
L’Ipriflavone e il Methoxyisoflavone hanno un azione inibitoria su alcuni enzimi della famiglia del citocromo P450 come il CYP1A2 e il CYP2C9. Questo è il motivo per cui i ricercatori italiani si sono chiesti se i due isoflavoni sintetici avessero potuto inibire l’azione dell’Enzima Aromatasi. L’Enzima Aromatasi, proprio come il CYP1A2 e il CYP2C9, fa parte della famiglia del citocromo P450.
Se così fosse, allora l’Ipriflavone e il Methoxyisoflavone potrebbero essere inseriti nella lista delle sostanze dopanti, insieme ad inibitori dell’Aromatasi di sintesi tra i quali troviamo l’Anastrozolo e il Letrozolo. Come risaputo, gli inibitori dell’Aromatasi possono aumentare i livelli di Testosterone attraverso due vie principali:
la riduzione della conversione del Testosterone in Estradiolo e riduzione delle SHBG con conseguente miglioramento dei livelli complessivi dell’androgeno (sia totale che bioattivo) e della Testosterone:Estradiolo ratio;
attraverso la riduzione dei livelli di Estradiolo e il feedback positivo a livello ipotalamico con aumento del rilascio di GnRH e conseguente aumento di LH e incremento della biosintesi testicolare di Testosterone.
E’ scontato sottolineare che ciò ha come potenziale conseguenza il miglioramento delle prestazioni.
I ricercatori hanno svolto l’esperimento in vitro osservando, in presenza di Testosterone e Aromatasi, se l’Ipriflavone, il Methoxyisoflavone e altri composti simili avessero la capacità di bloccare il processo enzimatico di conversione del Testosterone in Estradiolo.
L’Ipriflavone e il Methoxyisoflavone hanno mostrato di causare l’inibizione della conversione del Testosterone in Estradiolo. Entrambi i composti bloccavano l’azione dell’Enzima Aromatasi. Anche la Daidzeina ha mostrato di poterlo fare, ma in misura molto minore. L’effetto anti-estrogenico dell’Ipriflavone e del Methoxyisoflavone (IN VITRO) è risultato essere simile a quello dell’Anastrozolo.
Quando i ricercatori hanno calcolato il Ki del Methoxyisoflavone e dell’Ipriflavone sulla base dei loro test, hanno visto che era uguale al Ki degli inibitori dell’aromatasi farmacologici.
I ricercatori scrivono che i risultati ottenuti nel loro modello in vitro mostrano, nella gamma di concentrazioni considerate, che il Methoxyisoflavone e l’Ipriflavone hanno un potenziale di inibizione dell’aromatasi simile a quello del Formestano, dell’Anastrozolo e dell’Amminoglutetimmide. Quei farmaci [sono] usati nella terapia antitumorale e banditi dalla WADA per i loro effetti sulla steroidogenesi (l’Amminoglutetimide per particolarità differenti sulla steroidogenesi NdR).
Chiaramente, i risultati presentati non sono sufficienti per esprimere un’opinione finale sulla possibilità di includere gli isoflavoni sintetici nella lista delle sostanze dopanti redatta dalla WADA, dal momento che l’entità effettiva dei loro effetti in vivo sarebbe certamente modulata dalle loro proprietà farmacocinetiche, e specialmente dalla loro bassa biodisponibilità.
Tuttavia, i ricercatori ritengono che il loro monitoraggio possa ancora essere utile nell’analisi del controllo antidoping, considerando anche le alte dosi raccomandate per gli isoflavoni sintetici negli integratori alimentari.

 Secondo uno studio su animali svolto da fisiologi rumeni presso l’Università di Medicina e Farmacia Carol Davila e pubblicato su “Medicina”, la supplementazione di Taurina potrebbe offrire protezione cardiovascolare agli utilizzatori di AAS.(1)
Secondo uno studio su animali svolto da fisiologi rumeni presso l’Università di Medicina e Farmacia Carol Davila e pubblicato su “Medicina”, la supplementazione di Taurina potrebbe offrire protezione cardiovascolare agli utilizzatori di AAS.(1)