Generalmente, l’abuso di AAS è associato a disturbi percettivi dell’immagine, disturbi dell’alimentazione e dall’esercizio fisico compulsivo. Lo psichiatra Tom Hildebrandt potrebbe aver trovato una spiegazione biologica per quest’ultimo aspetto. Nel 2014, Hildebrandt ha pubblicato uno studio svolto su esseri umani il quale suggerisce che l’uso di AAS aumenta la dipendenza dall’esercizio fisico intenso.[1]
Dettagli dello studio
I ricercatori che hanno partecipato al presente studio, hanno analizzato campioni di sangue di 26 uomini che si allenavano intensamente con i pesi. Dieci uomini erano “natural” [Control; Heavy exercise control], gli altri sedici utilizzavano AAS. L’utilizzatore medio di AAS in questo studio aveva già completato una dozzina di cicli.
Durante questo studio, metà degli utilizzatori di AAS era sotto ciclo, l’altra metà era “off”.
Risultati
Tramite l’analisi del sangue dei soggetti sotto osservazione i ricercatori hanno misurato, attraverso specifici marker, il grado in cui i partecipanti hanno apprezzato l’allenamento fisico utilizzando un test complesso. In breve, i soggetti sotto esame erano sottoposti a sedute di tapis roulant dovendo svolgere compiti sempre più gravosi durante la medesima seduta al fine di poter continuare a correre.
Gli utilizzatori di AAS apprezzavano il loro allenamento più dei non utilizzatori, e coloro i quali erano sotto ciclo hanno ottenuto punteggi notevolmente superiori rispetto agli utilizzatori in “off”.
La tabella seguente è stata semplificata.
Nel sangue analizzato, i ricercatori hanno trovato una possibile spiegazione per la maggiore tendenza di un soggetto utilizzatore di AAS a sottoporsi ad allenamenti intensi, e non è così banale come si potrebbe pensare. La concentrazione di beta-endorfine era più alta in questo gruppo di soggetti.
La beta-endorfina o β-endorfina, è un ormone peptidico, un neuropeptide oppioide endogeno prodotto in alcuni neuroni del sistema nervoso centrale e del sistema nervoso periferico.[2] È una delle tre endorfine prodotte nell’uomo, le altre includono l’α-endorfina e la γ-endorfina.[3]
Le beta-endorfine vengono rilasciate durante l’esercizio fisico intenso. È un fattore importante nelle sensazioni euforiche provate dagli atleti durante la prestazione, che gli atleti di endurance chiamano “lo sballo del corridore”. Secondo gli psicologi dello sport, le beta-endorfine svolgono un ruolo cruciale nella dipendenza dall’attività sportiva.
Le beta-endorfine interagiscono con gli stessi recettori con cui interagiscono gli antidolorifici oppioidi che creano dipendenza come la Morfina. Negli anni ’70, durante la sperimentazione con animali da laboratorio, Horace Loh scoprì che l’effetto analgesico delle beta-endorfine superava quello della Morfina di un fattore 18-33.[4]
Conclusioni
I risultati di questo studio forniscono un supporto continuo per il ruolo dell’esercizio compulsivo nella dipendenza da AAS e la sua possibile incorporazione nel modello di dipendenza da questi ultimi.
Il fatto che gli AAS causino un aumento della massa muscolare e possano anche migliorare l’umore e il valore di rinforzo di comportamenti come l’esercizio tramite effetti sull’asse HPA suggerisce che ciò possa condurre ad una condizione di forte induzione alla cronicizzazione d’uso di AAS.
Sono ormai diversi anni che in nutrizione si discute della questione “dolcificanti artificiali” e se essi siano o meno deleteri nel contesto dell’alimentazione umana. Molti studi hanno “assolto” dalla loro presunta pericolosità dolcificati ipocalorici molto diffusi come l’Aspartame, con le corrette modalità d’uso ovviamente (vedi dosaggio totale giornaliero). Mentre altri dolcificanti artificiali sono decisamente posizionati nella “zona grigia”, come l’Acesulfame-K. Il peggiore, secondo quanto emerso dalle ultime ricerche, sembrerebbe essere il Sucralosio. Il Sucralosio, un dolcificante sintetico mille volte più dolce dello zucchero da cucina (Saccarosio), sembra che possa causare sintomi pre-diabetici nelle persone sane. I ricercatori dell’Università di Yale hanno riportato della comparsa di questi sintomi in un articolo comparso recentemente su “Cell Metabolism”.[1] Sebbene i soggetti dello studio non fossero effettivamente patologici, i risultati sono stati così preoccupanti che l’università ha consigliato ai ricercatori di interrompere lo studio.
Caratteristiche del Sucralosio:
La maggior parte del Sucralosio (E-955) ingerito non viene enzimaticamente scomposto, quindi non apporta calorie. [2] È prodotto dalla clorurazione del saccarosio. Il Sucralosio è da 320 a 1.000 volte più dolce del Saccarosio [3], tre volte più dolce dell’Aspartame e dell’Acesulfame-K, e due volte più dolce della Saccarina Sodica.
Sebbene il Sucralosio è ampiamente considerato stabile e sicuro per l’uso a temperature elevate (come nei prodotti da forno), ci sono alcune prove che mostrano un iniziale degradazione a temperature superiori a 119 gradi Celsius. [4][5] Il successo commerciale dei prodotti a base di Sucralosio deriva semplicemente dal confronto favorevole con altri dolcificanti ipocalorici in termini di gusto, stabilità e sicurezza nelle prima citate circostanze.[6]
Lo studio in questione e risultati emersi…
Per lo svolgimento dello studio che qui andiamo trattando, i ricercatori hanno diviso 45 soggetti sani in tre gruppi. Ogni gruppo si recava al laboratorio di controllo sette volte durante un periodo di due settimane. Li, ai soggetti veniva data una bevanda analcolica da 355ml.
Il contenuto della suddetta bevanda differiva nei tre gruppi esaminati come segue:
Contenuto 1° gruppo [LCS]: 60mg di Sucralosio;
Contenuto 2° gruppo [Sugar]: 30g di Saccarosio [normale zucchero da tavola];
Contenuto 3° gruppo [Combi]: 60mg di Sucralosio + 31g di Maltodestrine.
Come già accennato, la struttura chimica del Sucralosio è molto simile a quella del Saccarosio. In tre punti, tuttavia, il Sucralosio presenta gruppi cloro che mancano nel Saccarosio. A causa di questi gruppi cloro, secondo alcuni studi, il Sucralosio è mille volte più dolce del Saccarosio.
Le bevande analcoliche assunte dai partecipanti dei gruppi 1 e 2 non hanno avuto alcun effetto sulla farmacocinetica del glucosio negli individui esaminati. Quando i ricercatori hanno somministrato a questi soggetti un lotto di glucosio dopo 2 settimane, la glicemia ematica si è ridotta con la stessa velocità osservata prima del periodo di due settimane dello studio. A questo proposito, le bevande analcoliche erano sicure.
Il quadro è cambiato quando i ricercatori hanno esaminato la quantità di insulina che era presente nel sangue dei soggetti dopo la somministrazione del glucosio. Questa quantità era significativamente maggiore nei soggetti che avevano ricevuto bevande analcoliche contenenti Maltodestrine più Sucralosio.
Ciò implica che la combinazione di Sucralosio con un carboidrato ad assorbimento altera il metabolismo glucidico peggiorando, sebbene in acuto, la sensibilità all’Insulina.
I ricercatori hanno anche osservato che in un certo numero di soggetti, la combinazione di Sucralosio e un carboidrato a rapido assorbimento portava ad un aumento dell’Insulina basale, misurata al mattino prima che i soggetti consumassero il loro primo pasto della giornata. Ciò suggerisce anche una possibile ridotta sensibilità all’insulina in cronico.
In bocca, nell’intestino e in altri punti del corpo, i dolcificanti come il Sucralosio interagiscono con i recettori del dolce T1R2 / T1R3. Questi recettori sono in realtà destinati al glucosio e ad altri zuccheri naturali. Regolano l’assorbimento degli zuccheri da parte dell’intestino tenue.
I ricercatori ipotizzano che, tramite questi recettori, il Sucralosio possa indurre il corpo ad assorbire rapidamente i carboidrati assimilandoli ancora più velocemente, interrompendo l’equilibrio tra glucosio e insulina e riducendo la sensibilità all’insulina non solo in acuto ma anche, potenzialmente, in cronico.
I ricercatori hanno scritto che questi risultati suggeriscono che il consumo di Sucralosio altera il metabolismo del glucosio consumato simultaneamente producendo rapidamente effetti deleteri sulla salute metabolica.
Durate di esposizione simili quasi certamente si verificano negli esseri umani nella quotidianità, soprattutto se si considera il consumo di una bevanda dietetica insieme ad un pasto. Ciò solleva la possibilità che l’effetto combinato possa essere un importante contributo all’aumento dell’incidenza del diabete di tipo 2 e l’obesità, in senso indiretto o induttivo.
In tal caso, l’aggiunta di dolcificanti a basso contenuto calorico per aumentare la dolcezza di cibi e bevande già contenenti carboidrati dovrebbe essere scoraggiato e il consumo di bevande dietetiche durante i pasti dovrebbe essere sconsigliato.
Nota:Il Sucralosio risulta particolarmente deleterio anche sul microbiota intestinale. Il primo studio che ha valutato il Sucralosio sul microbiota intestinale è stato eseguito nel 2008 con l’uso di campioni fecali di ratti Sprague-Dawley che hanno ricevuto il dolcificante per 12 settimane. Il consumo di Sucralosio ha ridotto il numero totale di batteri anaerobici e aerobici, bifidobatteri, lattobacilli, Bacteroides e Clostridium.(7) La somministrazione di 15mg di Sucralosio/kg ha influenzato l’abbondanza relativa del Clostridium cluster XIVa nei topi.(8) Più recentemente, la somministrazione di Sucralosio nei topi ha prodotto modifiche nel microbiota intestinale a 14 diversi livelli tassonomici, tra cui Turicibacteraceae, Lachnospiraceae, Ruminococcaceae, Verrucomicrobiaceae, Staphylococcaceae, Streptococcaceae, Dehalobacteriaceae, Dehalobacterium, Lachnospiraceae, Lachnospiraceae ordine Bacillales e cambiamenti nella sintesi e regolazione degli amminoacidi. Queste variazioni erano correlate all’infiammazione nell’ospite.(9)
Nonostante lo studio sia di piccole dimensioni e non sia controllato (non vi è sicurezza nel comportamento alimentare seguito dai soggetti esaminati al di fuori di quanto emergesse durante i controlli), esso rappresenta un forte incentivo verso la ricerca sugli effettivi vantaggi e svantaggi del consumo di dolcificanti in soggetti sani e non.
Nota:Mancano ad oggi prove di un possibile beneficio per la perdita di peso a lungo termine con alcuni dati che supporto il rischio di un aumento di peso e di sviluppo di malattie cardiache con l’uso di questo dolcificante.[10]
Splenda alters gut microflora and increases intestinal p-glycoprotein and cytochrome p-450 in male rats.Abou-Donia MB, El-Masry EM, Abdel-Rahman AA, McLendon RE, Schiffman SSJ Toxicol Environ Health A. 2008; 71(21):1415-29.
Effects of Consuming Xylitol on Gut Microbiota and Lipid Metabolism in Mice.Uebanso T, Kano S, Yoshimoto A, Naito C, Shimohata T, Mawatari K, Takahashi ANutrients. 2017 Jul 14; 9(7):.
Gut Microbiome Response to Sucralose and Its Potential Role in Inducing Liver Inflammation in Mice.Bian X, Chi L, Gao B, Tu P, Ru H, Lu KFront Physiol. 2017; 8():487.
Al principio del mese di giugno di quest’anno ho riportato alcuni casi studio i quali facevano emergere il potenziale effetto epatotossico dato dall’uso dei SARM RAD-140 e LGD-4033. Il caso studio riguardante LGD-4033 non era di per sé convincente, poiché il Bodybuilder in questione era solito consumare discrete quantità di alcol. Di recente, i medici del Baylor College of Medicine negli Stati Uniti hanno segnalato un altro caso di danno epatico legato all’uso di LGD-4033.[1] E in questo nuovo caso studio, non ci sono altri fattori esplicativi del problema.
Il soggetto protagonista del caso studio è un Bodybuilder di 32 anni che ha riferito ai medici di aver usato 10mg/die di LGD-4033 in forma liquida per 15 giorni. Dopo di che aveva cominciato a lamentare malessere e interruppe il suo ciclo con il suddetto SARM. L’uomo aveva dolori di stomaco e nausea, oltre a prurito e ittero. Le sue feci erano grigie, e aveva perso l’appetito. Quando si è rivolto ai medici, l’uomo aveva già perso 18Kg.
Questi sono classici sintomi da danno epatico. Infatti, quando i medici hanno scansionato la cavità addominale del Bodybuilder, hanno notato che il fegato dell’uomo era più grande del normale. Una biopsia ha mostrato che il fegato del aveva cicatrici in alcuni punti. I dotti biliari, che trasportano i sali biliari all’intestino, erano ostruiti.
Nelle settimane successive, i medici hanno monitorato quattro marker del danno epatico nel sangue del Bodybuilder. La figura seguente mostra che le condizioni del fegato dell’uomo sono lentamente migliorate.
Come avevo già riportato nell’articolo di giugno, secondo uno studio del 2012 condotto dai produttori del LGD-4033, questo SARM non è significativamente dannoso per il fegato. Ma in questo studio, i soggetti non hanno ricevuto più di 1mg/die di LGD-4033.[2]
Le aziende che vendono SARM online e alcuni guru del bodybuilding raccomandano dosi significativamente più elevate di 1mg/die. Ad esempio, il paziente del quale si è parlato in questo articolo ha assunto dieci volte la dose più alta testata nello studio del 2012. Con tutta probabilità, un dosaggio di LGD-4033 di tale entità o superiore rappresenta uno stress epatico eccessivo, in specie se l’utilizzatore presenta una marcata sensibilità e manca di una efficace epatoprotezione (comunque non garante di immunità da effetti collaterali a livello epatico).
Alcuni utilizzatori di LGD-4033 hanno pubblicato il loro esame ematico sui forum presenti in rete. Sembra che non mostrino segni di danno epatico, ma l’affidabilità di certi dati è assai scarsa.
Forse il Bodybuilder in questione stava usando un prodotto contaminato o fake. Non tutti i SARM negli store online sono prodotti con le giuste misure di controllo, come riportato da una recente ricerca inglese e americana.[3]
E’ anche possibile che il Bodybuilder del caso studio stava usando altre sostanze oltre al solo LGD-4033 e non ne ha fatto menzione ai medici che lo hanno preso in cura. Le possibilità sono diverse ma ciò che è sufficientemente certo è che l’uso di LGD-4033 ad alte dosi, per vie metaboliche intrinseche, è un potenziale fattore causale per stress e danno epatico.
Nonostante il terrorismo diffuso da alcuni scientisti riguardo alla “malvagità assoluta” del IGF-1 e della sua correlazione con il cancro (esistente ma contestualizzabile), la terapia sostitutiva dell’Ormone della Crescita (GH) sembra non aumentare il rischio di cancro; sembrerebbe che, addirittura, ne riduca il rischio. Ciò è stato evidenziato in una meta-analisi pubblicata da endocrinologi cinesi della Zhejiang University College of Medicine nell’Open Journal of Endocrine and Metabolic Diseases.[1]
I ricercatori hanno esaminato 10 studi pubblicati in precedenza che hanno seguito adulti trattati con Ormone della Crescita per diversi anni. Ai soggetti è stato diagnosticato un deficit dell’ormone della crescita negli adulti. Ovviamente, e mi riferisco al lettore nella media che troppo velocemente trae conclusioni inesatte, gli studi potrebbero non dire molto sugli effetti che gli atleti supplementati farmacologicamente possano incorrere nel contesto dell’incidenza di sviluppo del cancro.
Nella tabella sottostante troverete maggiori informazioni sugli studi utilizzati.
Negli studi esaminati, i soggetti trattati con il GH non hanno sviluppato il cancro in maniera maggiore rispetto ai soggetti dei gruppi di controllo. Da quanto è emerso, per lo meno dagli studi presi in esame, l’Ormone della Crescita ha persino ridotto il rischio di cancro.
I ricercatori hanno però trovato presenza di bias. Ciò significa che sembra che non siano stati pubblicati studi con risultati meno interessanti. Tuttavia, il bias sembrava essere modesto ed i ricercatori sospettano che l’inclusione degli studi potenzialmente mancanti nella loro meta-analisi non ne altererebbe realmente il risultato.
I ricercatori concludono dicendo che la loro analisi corrobora le prove di studi precedenti che dimostrano che la terapia sostitutiva dell’Ormone della Crescita nei pazienti con deficit di questo ormone in età adulta non vedrebbero aumentare il loro rischio di sviluppare il cancro; invece, potrebbe addirittura diminuire il rischio. I risultati hanno suggerito che la terapia sostitutiva dell’Ormone della Crescita nei pazienti con deficit dell’ormone in età adulta era sicura.
Nota: i dosaggi terapici variano nettamente a seconda dell’età e delle finalità della terapia. Sono generalmente dosaggi inferiori a quelli utilizzati a scopo estetico-prestativo. Pertanto, il presente articolo non ha assolutamente lo scopo di far passare un dato che ad oggi non è statisticamente comprovato.
A differenza del trattamento pediatrico con GH, spesso dosato in microgrammi / chilogrammi di peso corporeo / giorno, il dosaggio sostitutivo del GH attualmente raccomandato negli adulti è individualizzato indipendentemente dal peso, tenendo conto dell’età del paziente, del sesso e dello stato degli estrogeni (Johannsson et al., 1997a; Hoffman et al., 2004b). L’inizio della terapia a basse dosi (dose totale 0,2-0,4 mg / die SC) riduce la probabilità di sviluppare effetti collaterali comuni come rigidità articolare, artralgie, mialgie, parestesie ed edema periferico, con ritenzione di liquidi. La dose deve essere titolata a intervalli di 6-8 settimane in base alla risposta clinica, evitando effetti collaterali e monitorando i livelli sierici di IGF-I. Si desidera raggiungere un obiettivo nella metà superiore dell’intervallo normale del IGF-I corretto per l’età del paziente. È ragionevole iniziare con dosi più elevate (0,4-0,5 mg / die) nei pazienti di età inferiore a 30 anni, ma i pazienti più anziani (di età superiore a 60 anni) dovrebbero iniziare con dosi più basse (0,1-0,2 mg / die) e titolato più lentamente per ridurre al minimo il verificarsi di effetti collaterali. Alcuni pazienti con AGHD ad esordio infantile con IGF-I pretrattamento molto basso possono sviluppare effetti collaterali da GH ma non raggiungere livelli medi normali nonostante dosi elevate di quest’ultimo.
Le donne che assumono una terapia sostitutiva degli estrogeni per via orale possono richiedere dosi più elevate per la GHRT, presumibilmente perché l’estrogeno orale inibisce la produzione e la secrezione di IGF-I nel fegato con effetto di primo passaggio (Weissberger et al., 1991). Di solito non è necessario alcun aggiustamento della dose nei pazienti che assumono dosi moderate di estrogeni transdermici.
La Niacina è largamente utilizzata dagli atleti supplementati chimicamente, in special modo da coloro i quali usano molecole con un potenziale negativo marcato sui lipidi ematici. Ma come spesso capita, gli utilizzatori non conoscono a sufficienza le caratteristiche di ciò che assumono, e questa essenziale vitamina del gruppo B (B3) non è da meno. Per la maggior parte degli individui tanto basta sapere che una sua integrazione si traduce in livelli migliorati di Colesterolo e Trigliceridi. Purtroppo, però, si trascurano caratteristiche importanti la cui conoscenza può fare la differenza tra un uso più o meno funzionale per la salute sistemica. Infatti, un effetto collaterale dell’integrazione di Niacina è un peggioramento della resistenza all’insulina, cosa che limita i benefici di tale supplementazione sulla salute cardiovascolare se non vengono prese adeguate precauzioni.
Prima di correre a defenestrare in preda al panico la vostra Niacina, leggete con attenzione (e comprendete) le informazioni che seguono…
Introduzione alla Niacina (vitamina B3)
Niacina
La Niacina, nota anche come Acido Nicotinico, è un composto organico e una forma di vitamina B3, un micronutriente essenziale per l’essere umano. [1] La Niacina ha formula bruta C6H5NO2 e appartiene al gruppo dell’acido piridinecarbossilico.[1] Come precursore di NAD e NADP, la Niacina è coinvolta nella riparazione del DNA.[2] La Niacina viene assunta attraverso la dieta da una varietà di alimenti interi e trasformati, con il più alto contenuto in alimenti confezionati fortificati, carne, pollame, pesce rosso come tonno e salmone, con minori quantità nelle noci, legumi e semi. [1] [3] La Niacina come integratore alimentare viene anche utilizzata per trattare la pellagra, una malattia causata da una sua carenza. Segni e sintomi includono lesioni della pelle e della bocca, anemia, mal di testa e stanchezza.[4] Molti paesi richiedono la sua aggiunta alla farina di grano o ad altri cereali, riducendo così il rischio di pellagra.[1][5] Come vitamina, le raccomandazioni di dosaggio giornaliero indicate in diversi paesi sono 14-18mg/die per gli adulti, quota sufficiente per soddisfare le esigenze delle persone sane. [6] [7] [8]
Sebbene la Niacina e la Nicotinamide (Niacinamide) siano identiche nella loro attività vitaminica, la Nicotinamide non ha gli stessi effetti farmacologici, modificanti i lipidi o gli effetti collaterali della Niacina, cioè quando la Niacina assume il gruppo -amide, non riduce il Colesterolo né causa vampate di calore.[9][10] La Nicotinamide è raccomandata come trattamento per la carenza di Niacina poiché può essere somministrata in quantità correttive senza causare l’effetto negativo del rossore.[11]
La Niacina è anche un farmaco di prescrizione. Quantità molto superiori all’assunzione dietetica raccomandata per le funzioni vitaminiche ridurranno i Trigliceridi nel sangue e le lipoproteine a bassa densità (LDL-C) e aumenteranno le lipoproteine ad alta densità (HDL-C). Ne esistono due forme: Niacina a rilascio immediato e a rilascio prolungato. Le quantità iniziali di prescrizione sono di 500mg/die, con possibilità di essere aumentate nel tempo fino a raggiungere l’effetto terapeutico ricercato. Le dosi a rilascio immediato possono arrivare fino a 3g/die; quelle a rilascio prolungato fino a 2g/die. [12] Nonostante i comprovati cambiamenti lipidici, la Niacina non è stata trovata utile per ridurre il rischio di malattie cardiovascolari nei soggetti già in trattamento con statine. [13] Una review del 2010 aveva concluso che l’efficacia della Niacina si osservava in mono-terapia, [14] ma una review del 2017 che incorporava il doppio del numero degli studi ha concluso che la Niacina su prescrizione, pur influenzando i livelli lipidici, non riduceva la mortalità per tutte le cause, la mortalità cardiovascolare, gli infarti del miocardio, né ictus fatali o non fatali. [15] È stato dimostrato che la Niacina da prescrizione provoca epatotossicità [16] e aumenta il rischio di diabete di tipo 2. [17] [18] Le prescrizioni di Niacina negli Stati Uniti avevano raggiunto il picco nel 2009, a 9,4 milioni, in calo a 1,3 milioni entro il 2017.[19]
Niacina, flusso ematico, pressione e vasodilatazione
Uno studio sulla supplementazione di Niacina che ha valutato il flusso sanguigno dell’avambraccio non è riuscito a trovare un effetto significativo fino a 1g al giorno somministrati nel corso di due settimane in soggetti altrimenti sani, [20] e 1.5g di Niacina a rilascio prolungato negli uomini con sindrome metabolica non sono riusciti a influenzare la dilatazione flusso- mediata (FMD). [21] Un altro studio non è riuscito a trovare un effetto significativo in un intero gruppo di pazienti affetti da afta epizootica, mentre in un gruppo di pazienti con malattia coronarica ha riscontrato un miglioramento in un sottogruppo con bassi livelli HDL-C. [22]
In soggetti con bassi livelli di HDL-C, è stato osservato che 1g di Niacina a rilascio prolungato per una settimana aumenta il flusso sanguigno (via FMD) del 4,5%; questo meccanismo non era correlato alle Prostaglandini, poiché il Laropiprant (un inibitore della Prostaglandine D2) non ha influenzato l’effetto. [23] Questo effetto ha anche coinciso con un aumento della bilirubina indiretta (ma non totale) del 62%. [23] Poiché la bilirubina del acido biliare è un antiossidante endoteliale, [24] e poiché i benefici della niacina sulla funzione endoteliale in questo studio sono stati ritenuti dipendenti dall’ossido nitrico, [23] è stato ipotizzato che un effetto conservativo della bilirubina sulla biodisponibilità dell’ossido nitrico sia alla base della beneficio osservato. Sia l’aumento della bilirubina che il miglioramento del flusso sanguigno si sono dissipati una settimana dopo l’interruzione della Niacina.[23]
Laropiprant
I soggetti che in precedenza avevano subito infarto del miocardio, a seguito del trattamento con Niacina (con Laropiprant) hanno riscontrato un aumento del flusso sanguigno dipendente dall’ossido nitrico (FMD) dopo dodici settimane di terapia insieme a un miglioramento della vasodilatazione indotta da nitroglicerina, entrambe non correlate con alterazioni dei trigliceridi. [25] Miglioramenti simili nel flusso sanguigno sono stati osservati in pazienti con infezione da HIV e con bassi livelli di HDL-C trattati con la sola Niacina. [26]
Prostaglandine D2 (PGD2)
È noto che la Niacina influenza il diametro dei vasi sanguigni, in particolare per via della sua reazione vasodilatativa cutanea (allargamento dei vasi nella pelle), che ha portato a ipotizzare che potrebbe influenzare la pressione sanguigna aumentando il diametro delle arterie e vene. Tuttavia, una review [27] ha notato che un possibile effetto di riduzione della pressione arteriosa della Niacina è indipendente dalla Prostaglandine che media il rossore, nota come PGD2.
È stato osservato che le infusioni di Niacina riducono acutamente la pressione sanguigna negli ipertesi senza alcun effetto nei soggetti con pressione sanguigna normale ed è stata associata ad un aumento della gittata cardiaca e della frequenza cardiaca che era simile in entrambi i gruppi. [28] Un altro studio ha confermato questo risultato, scoprendo che la pressione arteriosa ambulatoriale di 24 ore non sembra essere influenzata da un supplemento di Niacina fino a 1g nell’arco di due settimane in soggetti altrimenti sani. [20]
In termini di effetti della Niacina in cronico sulla pressione sanguigna, una review [27] che ha valutato gli studi che hanno misurato la pressione sanguigna negli ipertesi [29] [30] [31] [32] non ha notato alcun effetto statisticamente significativo nella riduzione della pressione sanguigna associata alla supplementazione di Niacina, sebbene questi studi in quanto a metodologie di misurazione sulle variazioni della pressione sanguigna non fossero ideali secondo gli autori della review. Tuttavia, la review ha osservato che in un ampio studio (il Coronary Drug Project), che inizialmente non è riuscito a trovare alcuna influenza della terapia con Niacina sulla pressione arteriosa, [32] ha osservato variazioni sensibili soltanto sui soggetti con sindrome metabolica. Questi presentavano un lieve riduzione di 2,2mmHg della pressione arteriosa sistolica con una moderata riduzione di 2,9mmHg della pressione diastolica. [33] Un’analisi post-hoc di un altro studio clinico [34] ha rilevato che la pressione arteriosa sistolica è stata abbassata di 2,2mmHg e la pressione sistolica di 2,7 rispetto al placebo nei pazienti dislipidemici trattati per 24 settimane. [35]
Niacina, Trigliceridi, Colesterolo e Aterosclerosi
Apolipoproteina B
La Niacina sembra abbassare i trigliceridi nel sangue inibendo sia la sintesi degli acidi grassi sia la loro esterificazione epatica per formare i trigliceridi, il che aumenta il tasso di degradazione dell’apolipoproteina B riducendo la sua secrezione dalle cellule epatiche. [36] Un meccanismo con cui la Niacina fa questo è attraverso l’inibizione diretta e non competitiva della diacilglicerolo aciltransferasi 2 (DGAT2), l’enzima finale nella sintesi dei trigliceridi nelle cellule epatiche, senza inibizione della DGAT1. [37]
Si è visto che gli effetti della Niacina sulla sintesi dei trigliceridi influenzano i livelli sierici di lipoproteine a densità molto bassa (vLDL-C), dove la terapia con Niacina per 16 settimane in soggetti con malattia del fegato grasso non alcolica (NAFLD) sembra ridurre le vLDL-C nel siero così come i complessi con trigliceridi (vLDL-TG) e apolipoproteina B (vLDL-ApoB) rispetto al placebo e con una potenza paragonabile al fenofibrato. [38] La Niacina lo fa riducendo la secrezione epatica di vLDL-C, sebbene ciò non aumenti la quantità di trigliceridi nel fegato anche nello stato di NAFLD. [38]
Oltre ai suoi effetti sul fegato, la Niacina può anche sopprimere il rilascio di acidi grassi liberi dal tessuto adiposo [39] che normalmente verrebbero reesterificati come trigliceridi nel fegato e quindi secreti via vLDL. [40] Tuttavia, questo meccanismo specifico, che è mediato dal recettore HM74A, [39] non sembra essere rilevante per le proprietà riducenti dei trigliceridi della Niacina. [41]
I benefici sui livelli di trigliceridi possono verificarsi entro una settimana dall’inizio della supplementazione con Niacina a rilascio prolungato (1g), sebbene in misura minore di circa il 4%. [23]
L’integrazione di 1.5-2g di Niacina a rilascio prolungato per due anni con follow-up di un anno nelle persone in terapia con statine caratterizzate da bassi livelli di HDL-C ha mostrato una riduzione dei trigliceridi del 28,6% (statina da sola dell’8,1%). [42]
Esiste un fenomeno noto come “rimbalzo degli acidi grassi” associato alla supplementazione di Niacina, in quanto l’azione iniziale del composto sul suo recettore (HM74A) nel tessuto adiposo può determinare una minore lipolisi e una minore secrezione di acidi grassi non esterificati (NEFA) nel sangue [43] e una migliore conservazione adiposa; [44] si tratta di fenomeni prontamente reversibili in quanto in un giorno di esposizione continua vi è un aumento netto del NEFA piuttosto che la sua soppressione [45] [46] [47] e alterazioni nel NEFA possono non riflette alterazioni dei trigliceridi.
Il primo meccanismo pensato per spiegare il miglioramento del profilo sierico di colesterolo in seguito alla supplementazione di Niacina è stato attraverso la riduzione del rilascio di acidi grassi non esterificati (NEFA) dai tessuti, che non è più considerato un probabile meccanismo in quanto l’integrazione di niacina in cronico è associata ad un aumento, piuttosto che alla soppressione, di NEFA mentre il recettore HM74A appare superfluo in termini di effetti della Niacina nei topi con altri ligandi del HM74A (Acipimox [48] e MK-0354 [49]) che si sono mostrati rispettivamente meno efficaci o inefficaci sul colesterolo. Attualmente si ritiene che l’influenza della Niacina sui NEFA nel siero non sia un fattore determinante nel modo in cui influenza i livelli di colesterolo nel corpo, con le teorie attuali che ipotizzano che il fattore sia determinato dalla sua sintesi e dal suo tasso di catabolismo.
Il primo potenziale meccanismo prevede la sintesi di HDL-C nel fegato attraverso l’aumento della trascrizione del gene ABCA1 (che dipende dal legame LXRα alla regione del promotore DR4 di questo gene). [50] L’attività di ABCA1 promuove la “lipidazione” della principale proteina dell’HDL nota come apolipoproteina AI (ApoAI) aumentando il tasso che associa ai fosfolipidi e al colesterolo, [51] [52] un passaggio obbligatorio nella sintesi dell’HDL-C che è aumentato di 500-1000µM con Niacina in vitro. [50] Questo meccanismo non è stato confermato, poiché mentre l’ApoAI può essere aumentato parallelamente all’aumento dell’HDL-C in soggetti trattati con Niacina e con livelli di HDL-C bassi di base, [53] LXRα sembra richiedere un coattivatore (PPARγ) per esercitare questi effetti, [54] che è attivato dal recettore della Niacina. [55] Tuttavia, l’attività del recettore della Niacina non è stata richiesta per i suoi effetti sui livelli di colesterolo, suggerendo che altri meccanismi potrebbero essere rilevanti.
PPARγ
L’altra teoria relativa alla sintesi di HDL dalla Niacina afferma che ciò dipenda dalla proteina di trasferimento dell’estere del colesterolo (CETP) nonostante la riduzione del colesterolo totale e dei trigliceridi non richieda per entrambe questa proteina. [56] [57] CETP è una proteina che facilita il trasferimento di lipidi tra diverse lipoproteine (generalmente donando un trigliceride da vLDL a HDL e prendendo un estere di colesterolo in un processo noto come trasporto inverso di colesterolo. [58]) La Niacina riduce l’espressione di CETP nel fegato e la sua attività nel sangue dei topi; [56] una riduzione del CETP aumenta la quantità di HDL-C nel sangue poiché i tassi di catabolismo dell’HDL / LDL riflettono l’attività del trasporto inverso del colesterolo e raggiungono rapidamente l’equilibrio, [59] e se il CETP è ridotto allora sarebbe necessario più HDL per normalizzare i tassi di trasporto inverso del colesterolo. Questo meccanismo può anche essere correlato a LXRα, poiché mentre un eteromero di LXRα con il recettore nucleare di vitamina A (RXR) attiva l’elemento DR4 aumenta la CETP [60] la Niacina agevola l’eterodimerizzazione di LXRα e PPARγ che attiva ancora DR4, ma in un modo che promuove l’efflusso di colesterolo. [61-44] Questa eterodimerizzazione competitiva [62] non è stata ancora dimostrata sperimentalmente, e lo studio che ha utilizzato dosi di Niacina da 2g nell’uomo non è riuscito a trovare un’influenza sull’attività del CETP nel siero nonostante un aumento dell’HDL. [63]
L’ultimo potenziale meccanismo per l’aumento dell’HDL non consiste nel suo incremento di sintesi ma piuttosto nel preservare il colesterolo HDL già sintetizzato arricchito con apoAI, riducendo il tasso in cui la lipoproteina viene assunta nelle cellule epatiche nonostante la donazione di colesterolo dall’HDL a queste cellule sia inalterata a causa della riduzione dell’espressione del recettore (catena beta sintasi ATP) che normalmente trasporta l’HDL nella cellula. [64] Questa ipotesi funziona meglio con le osservazioni che suggeriscono che il ridotto catabolismo dell’HDL è il principale fattore determinante dei suoi livelli più elevati, [65] e influenza anche l’apoA1 poiché la sua clearance dal sangue e l’assorbimento da parte dei reni sono ridotti. [66]
Una supplementazione di Niacina a rilascio prolungato (1g) della durata di una settimana in soggetti con bassi livelli di HDL-C non sembra essere sufficiente da aumentare sensibilmente i livelli totali di HDL-C, sebbene sia stata notata una riduzione della dimensione media delle particelle; [23] le variazioni di HDL -C possono mediare un miglioramento della vasodilatazione dipendente dall’ossido nitrico, sebbene sia stato anche osservato un aumento della bilirubina indiretta. [23]
L’integrazione prolungata di Niacina nei diabetici è associata ad un aumento della quantità e delle dimensioni particellari dell’HDL-C (32,7%) mentre le particelle di dimensioni più piccole sono diminuite (8,2%). [67]
È stato osservato che la Niacina conferisce un effetto protettivo sulla mortalità cardiovascolare poiché una metanalisi [68] ha osservato che negli studi su soggetti con malattia coronarica la terapia con Niacina era associata a un minor rischio di rivascolarizzazione dell’arteria coronarica (RR di 0,31; IC al 95% di 0,15-0,63), infarto miocardico non fatale (RR di 0,72; IC al 95% di 0,60-0,86) e attacco ischemico transitorio (RR di 0,76; IC al 95% di 0,61-0,94) mentre la riduzione della mortalità complessiva non è riuscita a raggiungere significatività statistica (RR 0,883; IC 95% di 0,773-1,008). I sette studi inclusi in questa meta-analisi [32] [29] [31] [30] (e un follow-up [69]) hanno totalizzato 5137 pazienti che utilizzavano anche vari prodotti farmaceutici della classe di statine e fibrati .
In uno studio i cui partecipanti erano in terapia con statine e avevano bassi livelli di colesterolo HDL è stato rilevato che 1.5-2g di Niacina a rilascio prolungato sono stati in grado di fornire benefici additivi nel miglioramento dell’HDL-C (20%) e nella riduzione dell’LDL-C (17%) rispetto al placebo, sebbene per quanto riguarda l’endpoint clinico predeterminato (morte o ricovero in ospedale) sia la Niacina che il placebo avevano una uguale quantità di responder. [70] Questo studio ha rilevato un’alta percentuale di pazienti con sindrome metabolica (80%) e commenti [71] hanno suggerito che a causa di una possibile capacità della Niacina a rilascio prolungato di deteriorare l’insulino-resistenza [72] che i suoi benefici potrebbero essere compensati da questo effetto avverso, mentre lo studio stesso ha suggerito che i benefici delle statine hanno sostituito i benefici della Niacina.
Mentre uno studio precedente che utilizzava alte dosi di Niacina a rilascio immediato (3g) ha riscontrato una riduzione della morte del 14% rispetto al placebo insieme a una riduzioni del colesterolo totale, [32] ed è stato osservato che questa riduzione è simile per grandezza agli studi che combinano statine con placebo.
Studi in vitro suggeriscono che la Niacina potrebbe in teoria prevenire la formazione di placche aterosclerotiche riducendo l’infiammazione e il danno alla parete endoteliale attraverso diversi meccanismi. Limitate ricerche su animali hanno mostrato che la Niacina nella dieta, a concentrazioni paragonabili a quelle utilizzate per ridurre il colesterolo, riduce la deposizione della placca sulla parete dell’arteria e ritarda l’aterosclerosi.[73][74][75][76][77][78][79][80]
Niacina e sue interazioni con il metabolismo del glucosio
L’assunzione prolungata di Niacina è stata osservata causare una riduzione della sensibilità all’insulina, causando un aumento compensativo della produzione di insulina da parte delle cellule β del pancreas per mantenere i livelli di glucosio nel sangue. [81] La Niacina non sembra avere effetti diretti sulle cellule β pancreatiche, tuttavia, poiché la perfusione negli isolotti pancreatici (isole di Langerhans) di ratto isolati con Niacina in vitro non ha influenzato la secrezione di insulina. [82] Ciò indica che la Niacina aumenta la produzione di insulina mediante un meccanismo indiretto, secondario a causare insulino-resistenza periferica. È stato osservato che la supplementazione induce resistenza all’insulina a dosi comprese tra 500mg e 1g, che rientrano nell’intervallo di dosaggio che conferisce effetti di riduzione del colesterolo. [83]
In particolare, sembra che sia necessaria una supplementazione cronica di Niacina per aumentare la produzione di Insulina, poiché in uno studio è stato dimostrato che la supplementazione acuta riduce i livelli di questo peptide in soggetti altrimenti sani prima di un picco dopo un giorno, [84] mentre altri studi in acuto hanno notato un effetto minimo o nullo sui livelli di Insulina. [85] [86] [87] [88]
Gli effetti dell’integrazione cronica di Niacina sui livelli di Insulina possono anche dipendere dalla popolazione. È stato osservato che la Niacina provoca iperinsulinemia in soggetti che invecchiano altrimenti sani [83] (1g / die) ed è stato dimostrato che quasi raddoppiano i livelli di Insulina nei soggetti con NAFLD (2g / giorno [89] [90]). Nei pazienti con sindrome metabolica, l’integrazione di Niacina a 6 settimane di somministrazione alla dose di 1.5g / die ha aumentato i livelli di Insulina del 30%. [91]
Nei soggetti obesi con malattia del fegato grasso non alcolico (NAFLD), l’integrazione giornaliera di Niacina a rilascio prolungato (titolata fino a 2g) per 16 settimane sembrava aumentare lo stato di resistenza all’insulina nel fegato, nei muscoli e nel tessuto adiposo [89] con un effetto inibitorio sulle azioni dell’Insulina nel fegato notate negli uomini non diabetici con dislipidemia. [92]
Adiponectina
Negli uomini adulti con sindrome metabolica, è stato osservato che la Niacina a rilascio prolungato alla dose di 1.5g ostacola in modo significativo la sensibilità all’Insulina, valutata dall’HOMA-IR (42%), che è stata associata ad un aumento dell’Insulina sierica nonostante un aumento dell’Adiponectina sierica. [91] Questo è stato notato anche in un altro studio (aumento del 22% dell’HOMA-IR), in cui l’Aspirina assunta insieme alla Niacina non ha impedito la comparsa di una ridotta sensibilità all’Insulina. [93]
Questo effetto può persistere in soggetti altrimenti sani, poiché i soggetti trattati con 1g di Niacina per due settimane a cui veniva somministrato un clamp iperinsulinaemico-euglicemico richiedono meno glucosio per mantenere l’omeostasi, il che è indicativo di una riduzione dell’assorbimento del glucosio (attraverso un aumento dell’Insulino-resistenza). [94]
La resistenza all’Insulina indotta dalla Niacina è stata inizialmente attribuita a un effetto di rebound nel tessuto adiposo in cui un aumento del rilascio di acidi grassi non esterificati (NEFA) da parte della Niacina compromette gli effetti della segnalazione dell’Insulina. [95] [96] Ciò è plausibile, poiché la resistenza all’Insulina può essere indotta con infusione di NEFA in 24 ore nei roditori. [97] Altre fonti suggeriscono che la resistenza all’Insulina non è associata al rebound del NEFA, poiché i soggetti con NAFLD che sperimentano resistenza all’Insulina dalla terapia con Niacina non hanno necessariamente un aumento del NEFA nel siero. [89].
Modello ipotetico per i ruoli intracellulari del DGAT1 e DGAT2.
Un’altra possibile opzione è che la Niacina può inibire in modo non competitivo l’enzima noto come diacilglicerolo aciltransferasi 2 (DGAT2) con un IC50 di 100 µM (potenza simile a circa 300 µM). [98] L’inibizione di questo enzima non causa di per sé resistenza all’insulina con la somministrazione di Niacina, [92] ma poiché il DGAT catalizza il primo stadio della sintesi dei trigliceridi, la sua inibizione può favorire l’accumulo di diacilglicerolo (DAG) che è la molecola che si ritiene spieghi parzialmente la resistenza all’insulina data dalla Niacina. [92] Poiché l’aumento del DAG nelle cellule del fegato sopprime la segnalazione dell’Insulina, [99-162] l’inibizione mediata dalla Niacina del DGAT2 provoca insulino-resistenza, [98] [89] ostacolando così la capacità dell’Insulina di sopprimere la sintesi di glucosio e promuovendo indirettamente uno stato di iperglicemia.
Sebbene l’integrazione cronica di alte dosi di Niacina riduca la sensibilità all’Insulina, ciò non è associato a variazioni dei livelli di glucosio a digiuno. [90] Ciò può essere spiegato da un aumento compensativo della sintesi di Insulina che contrasta la resistenza alla stessa, lasciando sostanzialmente invariati i livelli di glucosio nel sangue. [81]
L’attivazione del recettore della Niacina (HM74A) da parte di alcuni altri agonisti sembra ridurre rapidamente il glucosio sierico nei diabetici migliorando la sensibilità all’Insulina [100] o comunque migliorando i tassi di smaltimento del glucosio. [101] Ciò indica che lo stesso recettore della Niacina può avere effetti benefici sul metabolismo del glucosio e che la resistenza all’Insulina indotta dalla Niacina non si verifica tramite l’attivazione del HM74A.
Quando si osserva il muscolo scheletrico, è stato dimostrato che la terapia con Niacina induce resistenza all’Insulina in questo tessuto in soggetti obesi con NAFLD (2g al giorno nel corso di 16 settimane). Uno studio svolto su ratti a digiuno (il digiuno aumenta la concentrazione plasmatica di acidi grassi non esterificati (NEFA), similmente alla somministrazione di Niacina [102-135] e diminuisce il glicogeno del muscolo scheletrico [103]) in cui sono stati accuratamente somministrati 20mg/kg di Niacina ha mostrato che il glicogeno nel soleo era ridotto mentre il gastrocnemius e il fegato non sono stati influenzati. [103]
Metilgliossale
Quando il processo di glicazione è testato in vitro, la Niacina ha avuto solo effetti inibitori minori sulla glicazione dell’albumina sierica bovina da un noto agente glicante (Metilgliossale [104]) nonostante altri antiossidanti testati come lo Zinco (10-25 µg / mL) avessero più potenti benefici. [105]
È importante sottolineare che qualsiasi effetto della Niacina sulla glicazione in vitro deve essere interpretato con l’avvertenza che la Niacina riduce la sensibilità all’Insulina. Mentre la resistenza all’Insulina indotta dalla Niacina è ben compensata in soggetti sani giovani, lasciando sostanzialmente invariati i livelli di glucosio nel sangue, [81] la compensazione delle cellule β del pancreas negli individui più anziani o in quelli con ridotta tolleranza al glucosio era incompleta in uno studio, [83] causando aumenti nei livelli ematici di glucosio. Pertanto, la misura in cui la Niacina possa influenzare la glicazione in vivo non è chiara e probabilmente dipendente dalla popolazione.
Obesità e massa grassa
L’Adiponectina, un’adipochina nota per migliorare la sensibilità all’Insulina, per essere cardioprotettiva e ritenuta anche antiobesogena, [106] è aumentata in risposta all’attivazione mediata dalla Niacina del recettore HM74A nei topi. [107] La produzione di Adiponectina indotta dalla Niacina è stata rapida in questo studio, aumentando i livelli di questa adipochina del 37% entro 10 minuti da una dose di 30mg / kg per iniezione. I livelli sierici hanno raggiunto il picco dopo 60 minuti e sono rimasti elevati al di sopra del basale fino a 24 ore dopo la somministrazione. [107]
Leptina
È noto anche che la Leptina è aumentata in seguito alla somministrazione di Niacina nell’uomo [91], il che si ritiene si verifichi tramite un meccanismo simile poiché l’agonista farmaceutico HM74A Acipimox induce anch’esso la secrezione di Leptina dal tessuto adiposo in vitro [108] e in vivo. [109]
È stato osservato che la supplementazione di Niacina nel corso di sei settimane negli uomini obesi aumenta l’Adiponectina sierica del 43-56%, con circa metà dell’aumento rappresentato dalla forma ad alto peso molecolare [93] [91] insieme a un aumento del 26,8% della Leptina [91 ] senza cambiamenti osservabili nella Resistina. [91] L’Adiponectina è stata osservata aumentare di circa il 30% in soggetti obesi con NAFLD in risposta alla terapia con Niacina (fino a 2g al giorno), che era correlata con un aumento dell’Insulino-resistenza, [90] portando all’ipotesi che i due meccanismi siano intrecciati, forse come risposta adattativa. [90]
Resistina
Lo “spillover” degli acidi grassi risultante da una conservazione inefficiente del grasso dopo un pasto aumenta i lipidi sierici non esterificati (NEFA), [110] che influenzano negativamente la sensibilità all’Insulina epatica, aumentando la produzione di VLDL e potenzialmente svolgono un ruolo causale nella steatosi epatica. [111] [112] La somministrazione in acuto di Niacina (285 mg per via endovenosa) nell’uomo durante l’alimentazione ha dimostrato di ridurre lo spillover degli acidi grassi, promuovendo l’assorbimento del grasso alimentare nel tessuto adiposo e riducendo i Trigliceridi sierici e i NEFA. [113]
Al contrario, è stato osservato che un trattamento prolungato con Niacina, noto per favorire la resistenza all’Insulina nell’uomo, induce la resistenza all’Insulina adipocitaria, [114] che favorirebbe lo spillover degli acidi grassi, aumentando i livelli sierici di NEFA.[115]
Glucosio-6-fosfato deidrogenasi (G6PD)
È stato osservato che la Nicotinamide sopprime la differenziazione degli adipociti 3T3-L1 in modo dipendente dalla concentrazione con un range superiore a 10mM (il valore ED50), raggiungendo la soppressione completa a 20mM dopo nove giorni. [116] Si ritiene che ciò sia correlato a un effetto inibitorio sulla poli (ADP-ribosio) sintetasi, [116] che la Nicotinamide inibisce a 50µM mentre la Niacina non lo fa. [117] Quando aggiunta dopo differenziazione e in condizioni di glucosio elevato, la Nicotinamide sembra inibire il glucosio-6-fosfato deidrogenasi (G6PD) e prevenire il normale stress ossidativo. [118]
Il recettore dell’Acido Nicotinico è espresso negli dipociti in cui la sua attivazione sopprime l’adenilato ciclasi. [119] Questo effetto sembra essere circa il 30% più efficace negli adipociti rispetto ad altre linee cellulari (milza). [120] Poiché l’attivazione di questo recettore inibisce l’adenilato ciclasi, [119] e i fenolici che agiscono su di esso riducono anch’essi i tassi di lipolisi, [35] l’effetto complessivo dell’Acido Nicotinico sarebbe quello di ridurre la lipolisi negli adipociti, almeno a breve termine.
A lungo termine, tuttavia, il recettore dell’Acido Nicotinico può essere desensibilizzato con esposizione cronica a un agonista [121] e uno studio sui topi ha evidenziato che gli adipociti che sono diventati insulino-resistenti dopo la terapia con Niacina hanno mostrato una maggiore reattività dei recettori adrenergici (β1 e β2) all’aumentare dei livelli di cAMP nella cellula adiposa, [114] (il cAMP viene normalmente soppresso dalla Niacina che agisce sul recettore GRP109A [119]). Ciò potrebbe essere stato correlato alla sottoregolazione dei geni mediata dalla Niacina nella via di segnalazione dell’Insulina incluso il PDE3B, che normalmente degrada il cAMP, [114] una potenziale risposta adattativa nelle cellule adipose che è stata osservata avere la funzione di normalizzare i tassi di lipolisi (nei ratti sotto l’infusione di Niacina) . [97]
Un piccolo studio su sette partecipanti altrimenti sani che assumevano Niacina a 500mg/die, e aumentando la dose a 2g nel corso di due settimane ha mostrato una riduzione dei tassi di ossidazione dei grassi. [94] Tuttavia, a causa di un aumento dei tassi di ossidazione dei carboidrati, non vi era alcuna differenza netta nel tasso metabolico tra Niacina e placebo [94].
I topi privi di PARP-1 sembrano avere tassi metabolici più alti e una minore massa grassa; in assenza di PARP, aumentano le concentrazioni di NAD +, attivando le SIRT1 che quindi lavorano per deacetilare varie proteine (PGC-1α e FOXO1) per promuovere il dispendio energetico attraverso un metabolismo ossidativo aumentato e un incremento dei mitocondri.[122]
La SIRT2 e la SIRT3 non sono influenzate dalla bassa attività del PARP-1, [122] e l’inibizione della ribosilazione dell’ADP con altri mezzi come il knockdown NMNAT-1 sembra conferire anche effetti antiobesità nei roditori. [182] L’alimentazione aumenta acutamente l’attività del PARP-1 nei topi e ostacola transitoriamente l’attività della SIRT1, [122] che si pensa sia correlata al PARP-1 che ha la priorità per l’uso dei donatori di NAD +.
La supplementazione orale di Nicotinamide Riboside a 400mg/kg nel topo sembra aumentare il contenuto di NAD + nel muscolo scheletrico similmente a quanto avviene alla stessa dose di Niacina (Nicotinamide Mononucleotide inefficace in questo organo) [123] e sembra aumentare il dispendio energetico nei topi nutriti con un alto contenuto di grassi insieme all’aumento dell’attività dei geni bersaglio di FOXO1, suggerendo che l’integrazione orale è efficace. [123]
Esistono prove limitate nell’uomo che valutano gli effetti della Niacina sul tasso metabolico, sebbene l’estremità inferiore del dosaggio farmacologico della niacina (1g) in soggetti altrimenti sani non sia riuscito ad aumentare il tasso metabolico rispetto al placebo. [94]
Niacina, muscolo scheletrico e prestazioni fisiche
La somministrazione di Niacina nell’uomo ha dimostrato di aumentare l’espressione dei fattori di trascrizione PPARδ e PPARγ coactivator-1α (PGC-1α) nel muscolo scheletrico. [124] Poiché questi fattori di trascrizione sono importanti regolatori del metabolismo ossidativo e della biogenesi mitocondriale, [125] [126] questo suggerisce che l’integrazione con Niacina può svolgere un ruolo nella resistenza dei muscoli scheletrici.
Gli studi sugli animali hanno supportato questa idea, in cui è stato dimostrato che l’integrazione di Niacina provoca una transizione di fibre muscolari dal tipo II (contrazione rapida) al tipo I (contrazione lenta), aumentando anche il numero complessivo di fibre di tipo I nei muscoli scheletrici nello Zucker (ratto) obeso [127] e suini in crescita [128] (750mg di Niacina/kg di dieta) e pecore (1g di Niacina al giorno). [129] Questo effetto può essere limitato a determinati modelli animali, tuttavia, poiché studi su ratti sani hanno dimostrato che la Niacina ha un effetto trascurabile sulla distribuzione del tipo di fibra muscolare o sul fenotipo metabolico. [130] Inoltre, nonostante la Niacina aumenti l’espressione dei fattori di trascrizione pro-ossidativa nell’uomo, [124] fino ad oggi nessuno studio ha dimostrato che migliora le prestazioni o la capacità di resistenza del muscolo scheletrico.
Tuttavia, come substrato per la sintesi di NAD +, un’adeguata presenza di Niacina può supportare indirettamente il metabolismo ossidativo e la resistenza muscolare. In soggetti altrimenti sani, un lieve esercizio fisico sembra essere associato ad un aumento delle concentrazioni di NAD + nel sangue rispetto a uno stato di riposo (indipendente da qualsiasi integrazione [131]) mentre, quando testato in un esercizio lieve nei roditori, portava anche ad un aumento del NAD + nel sangue prima che diminuisse durante un esercizio ad esaurimento, [131] che è stato notato anche nel muscolo scheletrico. [132] A questo livello di esaurimento c’è un concomitante aumento del contenuto di NADH nel muscolo scheletrico [133] [134] che è stato proposto [135] indicativo di una riduzione del trasferimento di elettroni dal NADH alla sintesi di ATP.
È stato inoltre proposto [135] che da quando l’esercizio aumenta l’ossidazione nei tessuti stimolati e i fattori di stress ossidativo sono noti per alterare l’attività del ciclo di Kreb (TCA) [136] e la catena di trasporto degli elettroni (compresa la NADH deidrogenasi [137]) che forniscono antiossidanti aumenterebbe la resistenza secondaria alla conservazione della cinetica intramuscolare di NAD + / NADH. Quando si forniscono 36mg di picnogenolo [135] come antiossidante durante l’esercizio fisico fino all’esaurimento, sembra che la diminuzione del NAD + nel sangue sia stata invertita in un aumento con gli effetti (sia la diminuzione che l’aumento in attesa di integrazione) più marcati negli atleti allenati. [135]
Impatto organico della Niacina e principali effetti collaterali
Indometacina
In uno studio svolto sui ratti, la Nicotinamide ad un dosaggio di 20mg/kg somministrata un’ora prima di una dose di Indometacina che induceva ulcerazioni allo stomaco ha impedito l’ulcerazione a un livello paragonabile sia al controllo (nessuna induzione di ulcere) sia al farmaco di riferimento di 400mg/kg di sucralfato, che agisce localmente per forma una superficie protettiva per lo stomaco. [138] Questo effetto si è verificato insieme alla conservazione dell’attività del glutatione, alla riduzione della perossidazione lipidica e al miglioramento del muco gastrico. [138] Simili effetti protettivi contro l’ulcerazione indotta da etanolo e stress sono stati notati altrove, con il metabolita primario della Nicotinamide (1-metilnicotinamide; MNA). [139] Questo effetto gastroprotettivo è stato associato con un aumento dell’attività delle prostaglandine, in particolare la PGI2, [139] e nicotinamide, nonché il suo metabolita MNA sono stati implicati nell’aumento del flusso sanguigno gastrico [139] e nella riduzione della permeabilità microvascolare [138] dopo l’ulcerazione.
Interleuchina 10 (IL-10)
Nel colon dei topi, il recettore della Niacina (GPR109A) è necessario per la proliferazione ottimale delle cellule T CD4 + e la produzione di IL-10, che si traduce in un effetto antiinfiammatorio. [140] Questo effetto antinfiammatorio guidato dal GPR109A è mediato dal butirrato, l’acido grasso a catena corta del colon [140], che è un agonista del GPR109A ed è prodotto attraverso la fermentazione della fibra alimentare da parte dei batteri nel colon. [141] [142]
L’effetto riducente dei Trigliceridi dato dalla Niacina sembra da doversi ricondurre al fegato, dove la secrezione di lipoproteine a bassissima densità (vLDL) è ridotta; poiché le vLDL normalmente trasportano i Trigliceridi dal fegato ad altri tessuti, riducendo la secrezione di vLDL ciò si traduce in un livello sierico di Trigliceridi inferiori. [89] La diminuzione della secrezione di vLDL può essere secondaria all’inibizione della lipolisi nel tessuto adiposo, poiché l’aumento cronico di acidi grassi liberi nel siero può regolare negativamente la secrezione di vLDL. [143]
Sembra che l’integrazione di Niacina in acuto (che riduce gli acidi grassi liberi nel siero) sopprime anche la produzione di vLDL e la sua complessazione con i trigliceridi. [144] Ciò suggerisce un altro possibile meccanismo, che può verificarsi attraverso la soppressione acuta del PGC-1β, [145] una proteina nota per promuovere la secrezione di Trigliceridi dal fegato in risposta all’ingestione di grassi nella dieta. [146] In accordo con quest’ultimo meccanismo, la somministrazione di Niacina con un pasto ad alto contenuto di grassi sembra ridurre il picco normale dei trigliceridi postprandiali. [147]
Regioni regolatorie, fattori di trascrizione e molecole di segnalazione (citochine, fattori di crescita, metaboliti, farmaci) che modulano l’espressione del gene ABCA1 nei macrofagi e in altri tessuti. Le frecce e le linee di blocco indicano rispettivamente l’attivazione e la repressione.
Non è confermato come la Niacina riduca le vLDL-C, ma la sua capacità di stimolare l’attività del gene ABCA1 e aumentare il suo contenuto proteico nelle cellule del fegato è alla base dell’aumento dell’HDL-C, [148] che è noto anche per sopprimere la secrezione di vLDL-C. [57] La Niacina (2g per 16 settimane), nonostante riduca le vLDL-C e il complesso con Trigliceridi, non sembra aumentare significativamente il contenuto di trigliceridi intraepatici in soggetti con malattia del fegato grasso non alcolica (NAFLD). [149]
La Niacina sembra anche agire sulle cellule del fegato per promuovere l’accumulo di diacilglicerolo (DAG), che è associato all’insulino-resistenza localizzata. [92] La resistenza all’insulina nelle cellule del fegato riduce l’effetto soppressivo dell’insulina sulla sintesi del glucosio, con conseguente aumento dell’efflusso di glucosio dal fegato nel sangue. [150] Poiché gli stadi iniziali dell’insulino-resistenza indotta dalla Niacina (prima degli aumenti dell’insulina basale e del glucosio) sono stati associati a un fabbisogno ridotto di glucosio per bilanciare un morsetto euglicemico iperinsulinaemico, [109] questo suggerisce che l’inizio dell’insulino-resistenza avviene a livello del fegato. Il ruolo preciso del DAG in questo processo è tuttavia incerto. Mentre il DAG promuove la resistenza all’insulina attraverso l’attivazione di PKCε, [151] l’attivazione di questa proteina non è stata osservata nelle cellule del fegato che sono diventate insulino-resistenti con la Niacina. [92]
TRANSAMINASI. Enzimi intracellulari prodotti principalmente dagli epatociti. normalmente presenti in circolo a bassi livelli nel sangue. Aumentano in caso di danno cellulare. Indici molto sensibili ma moderatamente specifici di danno epatico. ALT è un marker più specifico di danno epatocellulare. (localizzazione citoplasmatica e più lunga emivita)
Nota: La Niacina in dosi terapeutiche può causare aumenti modesti delle transaminasi sieriche e della bilirubina non coniugata, entrambi biomarcatori del danno epatico. Le modifiche vengono invertite se il trattamento farmacologico viene interrotto e di solito si risolvono anche quando si continua l’assunzione. [152] [153] [154] Tuttavia, meno comunemente, la forma di rilascio prolungato del farmaco può portare a gravi epatotossicità, con insorgenza in giorni o settimane. I primi sintomi di gravi danni al fegato includono nausea, vomito e dolore addominale, seguiti da ittero e prurito. Si ritiene che il meccanismo sia una tossicità diretta della Niacina sierica elevata. Abbassare la dose o passare alla forma a rilascio immediato può risolvere i sintomi. In rari casi la lesione è grave e progredisce fino a insufficienza epatica. [152]
È noto che la Niacina rende le cellule β pancreatiche (un tipo di cellula specializzata che secerne insulina in risposta al glucosio) meno sensibile al glucosio sierico. [81] Inoltre, la normale riduzione della sensibilità al glucosio delle cellule β del pancreas associata all’invecchiamento può essere ulteriormente esacerbata dalla supplementazione di Niacina (500mg-1g due volte al giorno). [83] Anche se sembra esserci un aumento compensativo della secrezione di insulina nella risposta alla Niacina [83], in un modello di primati con diabete di tipo I, [155] questo non è stato sufficiente a ridurre la glicemia a livelli normali, con conseguente lieve iperglicemia e iperinsulinemia dopo due settimane di integrazione.[83]
Va notato che una linea cellulare coinvolta nel rossore cutaneo tipico della Niacina, nota come Langerhans, [156] [157] è diversa dall’area del pancreas nota come “Isole di Langerhans”.
Nota: il rossore dato dalla Niacina – una dilatazione a breve termine delle arteriole della pelle, che causa il colore della pelle rossastra – di solito dura circa 15-30 minuti, anche se a volte può persistere per settimane con uso cronico e di forme a lento rilascio. In genere, il viso è maggiormente interessato, ma la reazione può estendersi al collo e alla parte superiore del torace. La causa è la dilatazione dei vasi sanguigni [158] [93] dovuta all’aumento della prostaglandina GD2 (PGD2) e serotonina. [159] [160] [161] [162] Si pensava spesso che il rossore riguardasse l’istamina, ma è stato dimostrato che l’istamina non è coinvolta nella reazione. [159] Il rossore a volte è accompagnato da una sensazione di prurito, in particolare, nelle aree coperte da indumenti. [93]
Acido Acetilsalicilico (Aspirina)
La prevenzione del rossore richiede l’alterazione o il blocco della via mediata dalle prostaglandine. [93] [163] L’Aspirina [165-325mg] assunta mezz’ora prima della Niacina riduce fortemente il rossore, così come l’Ibuprofene [200mg] (una riduzione della frequenza e intensità del rossore fino al 50%). L’assunzione di Niacina ai pasti aiuta anche a ridurre questo effetto collaterale. [93] La tolleranza acquisita aiuterà a ridurre l’effetto; dopo diverse settimane a dosaggio costante, la maggior parte delle persone non ha più esperienza di vampate di calore. [93] Sono state sviluppate forme di Niacina a rilascio lento o “prolungato” per ridurre questi effetti collaterali. [164] [165]
Conclusioni sulla supplementazione di Niacina
Le informazioni che abbiamo a disposizione sulla Niacina e la sua supplementazione, ci presentano un composto senz’altro utile per il controllo o riassesto del quadro lipidico ma allo stesso tempo questa molecola risulta di una complessità d’azione biochimica non trascurabile. Il suo peggiorare l’insulino-resistenza in cronico ma con un maggior picco in acuto, picco che sembra venire controbilanciato da altri fattori come l’aumento della Adiponectina. Lo stesso effetto sulla riduzione della lipolisi può destare preoccupazione nell’atleta, specie se questo si trova in una fase ipocalorica con il principale intento di ridurre la massa grassa. Anche in questo caso sembrerebbe che l’effetto si manifesti in acuto per poi rientrare in condizioni pre-utilizzo. Ciò che è quasi certo, è che le osservazioni sul campo non hanno fatto emergere grosse differenze nell’alterazione della composizione corporea, sia con l’uso della Niacina in regimi ipercalorici che ipocalorici. L’utilizzo di GDA (in specie Berberina) anche alle dosi base efficaci potrebbe essere un “tampone” sufficienti a compensare almeno in parte i possibili peggioramenti dei parametri dell’insulino resistenza. I controlli della glicemia basale e della insulinemia a digiuno sono indicatori da tenere sotto controllo durante l’uso di Niacina. Non è da trascurare la possibilità che la Niacina possa influenzare lo “shift” dalle fibre muscolari di tipo II a quelle di tipo I, cosa non auspicabile per un Bodybuilder o altro atleta di forza.
In definitiva, considerando i dosaggi efficaci e la migliore azione in combinazione con statine (vedi Monacolina K), l’assunzione di Niacina può essere mantenuta con un certo margine di sicurezza tra i 500mg ed 1g/die, ovviamente tarando il dosaggio in risposta agli esami ematici di controllo.
Gabriel Bellizzi
Riferimenti:
“Niacin”. Micronutrient Information Center, Linus Pauling Institute, Oregon State University, Corvallis, OR. 8 October 2018. Retrieved 16 September 2019.
Hegyi J, Schwartz RA, Hegyi V (January 2004). “Pellagra: dermatitis, dementia, and diarrhea”. International Journal of Dermatology. 43 (1): 1–5.
“Why fortify?”. Food Fortification Initiative. 2017. Retrieved 4 April 2017.
Institute of Medicine (1998). “Niacin”. Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline. Washington, DC: The National Academies Press. pp. 123–149.
Bruckert E, Labreuche J, Amarenco P (June 2010). “Meta-analysis of the effect of nicotinic acid alone or in combination on cardiovascular events and atherosclerosis”. Atherosclerosis. 210 (2): 353–61.
“Niacin”. IN: LiverTox: Clinical and Research Information on Drug-Induced Liver Injury (Internet). Bethesda, MD: National Institute of Diabetes and Digestive and Kidney Diseases. February 2014.
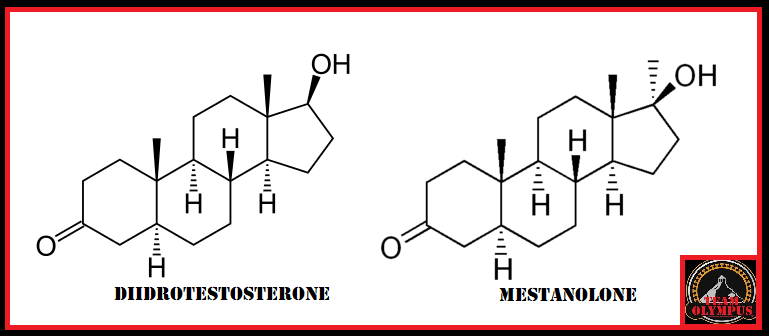
Nel comune pensare dell’uomo (e dell’atleta) medio, il Dihydrotestosterone (DHT) è, al pari degli Estrogeni, visto come un ormone tendenzialmente negativo, da ridurre il più possibile. Ovviamente questa visione è a dir poco ristretta dal momento che valuta l’attività del suddetto metabolita del Testosterone solamente in quelle circostanze dove un suo consistente livello può causare, specie nei soggetti predisposti o in determinate circostanze multifattoriali, acne, perdita accelerata dei capelli e ipertrofia prostatica (ovviamente parliamo di soggetti di sesso maschile). Inoltre, il DHT è considerato un metabolita pressoché insignificante nel miglioramento delle prestazioni, soprattutto per quanto concerne l’ipertrofia muscolare. Ma è veramente così limitato il suo impatto per un atleta? ..
Per rispondere a questo quesito nel presente articolo, in modo simile a quanto già feci nell’articolo dedicato agli Estrogeni, esporrò una panoramica dettagliata di tutto ciò che concerne il Dihydrotestosterone e le sue caratteristiche anche alla luce di recenti ed interessanti studi.
Cos’è il DHT?
Il Dihydrotestosterone (DHT, 5α-dihydrotestosterone, 5α-DHT, Androstanolone o Stanolone) è uno steroide con caratteristiche fortemente androgene, principalmente ottenuto dalla 5α-riduzione del Testosterone. Infatti, l’enzima 5α-reduttasi catalizza la formazione di DHT dal Testosterone in alcuni tessuti tra cui la ghiandola prostatica, le vescicole seminali, le epididimidi, la pelle, i follicoli piliferi, il fegato e il cervello. Questo enzima media la riduzione del doppio legame C4-5 del Testosterone. Rispetto al Testosterone, il DHT è considerevolmente più potente come agonista del recettore degli androgeni (AR), seppure limitato da percorsi enzimatici.
Oltre al suo ruolo di ormone naturale, il DHT è stato usato come farmaco, ad esempio nel trattamento di bassi livelli di Androgeni negli uomini (vedi Androstanolone).
Il DHT nella Storia
Adolf Friedrich Johann Butenandt (24 marzo 1903-18 gennaio 1995) fu un biochimico tedesco. Nel 1939 gli fu assegnato il premio Nobel per la chimica per il suo “lavoro sugli ormoni sessuali”. Inizialmente respinse il premio a causa della politica nazional-socialista, accettandolo solo nel 1949 dopo la seconda guerra mondiale.
Il DHT fu sintetizzato per la prima volta da Adolf Butenandt e dai suoi colleghi nel 1935. [1][2] Venne ottenuto mediante idrogenazione del Testosterone [3], che era stato scoperto all’inizio di quell’anno.[4] Il DHT è stato introdotto per uso medico come AAS nel 1953 ed è stato inizialmente notato per essere più potente del Testosterone ma con maggiore androgenicità.[5][6][7] Ma il suo potenziale androgeno non fu chiaro fino al 1956, quando venne dimostrato che veniva sintetizzato dal Testosterone negli omogenati di fegato di ratto.[2][8] Inoltre, l’importanza biologica del DHT non è stata realizzata fino agli inizi degli anni ’60, quando si è scoperto che era prodotto dalla 5α-riduzione del Testosterone circolante nei tessuti bersaglio come la ghiandola prostatica e le vescicole seminali risultando più potente del Testosterone in test biologici.[9][10][11][12] Le funzioni biologiche del DHT nell’uomo sono state definite in modo molto più chiaro alla scoperta e alla caratterizzazione del deficit di 5α-reduttasi di tipo II nel 1974.[13] Il DHT è stato l’ultimo importante ormone sessuale, gli altri sono Testosterone, Estradiolo e Progesterone, ad essere scoperto, ed è unico in quanto risulta essere il solo ormone sessuale principale che agisce fondamentalmente come ormone intracrino e paracrino piuttosto che come ormone endocrino.[12]
Biosintesi e distribuzione
Il DHT, noto anche come 5α-androstan-17β-ol-3-one, è uno steroide androstano presente in natura con un gruppo chetonico nella posizione C3 e un gruppo idrossile nella posizione C17β. È il derivato del Testosterone in cui il doppio legame tra le posizioni C4 e C5 è stato ridotto o idrogenato.
Differenze strutturali tra Testosterone e DHT
Il DHT è sintetizzato irreversibilmente dal Testosterone dall’enzima 5α-reduttasi. [14] [15] Ciò si verifica in vari tessuti tra cui i genitali (pene, scroto, clitoride, grandi labbra), [16] prostata, pelle, follicoli piliferi, fegato e cervello. [14] Circa il 5-7% del Testosterone subisce una 5α-riduzione in DHT [17] [18], e circa 200-300μg di DHT vengono sintetizzati giornalmente nel corpo. La maggior parte del DHT è prodotta nei tessuti periferici come la pelle e il fegato, mentre la maggior parte del DHT circolante proviene specificamente dal fegato. I testicoli e la ghiandola prostatica contribuiscono relativamente poco alle concentrazioni di DHT nel circolo ematico.[14]
Ghiandola sebacea
Esistono due isoforme principali di 5α-reduttasi, la SRD5A1 (tipo I) e la SRD5A2 (tipo II), quest’ultimo isoenzima ha una maggiore importanza biologica.[14] Esiste anche una terza forma di 5α-reduttasi: SRD5A3. [19] L’SRD5A2 è maggiormente espressa nei genitali, nella ghiandola prostatica, nelle epididimidi, nelle vescicole seminali, nella pelle genitale, nei follicoli piliferi del viso, del torace [20][21] e nel fegato, mentre si osserva un’espressione più bassa in alcune aree del cervello, pelle non genitale / follicoli piliferi, testicoli e reni. L’SRD5A1 è maggiormente espressa nei follicoli non genitali della pelle / dei capelli, nel fegato e in alcune aree del cervello, mentre sono presenti livelli più bassi nella prostata, nelle epididimidi, nelle vescicole seminali, nella pelle genitale, nei testicoli, nelle ghiandole surrenali e nei reni.[14] Nella pelle, la 5α-reduttasi è espressa in ghiandole sebacee, ghiandole sudoripare, cellule epidermiche e follicoli piliferi.[20][21] Entrambi gli isoenzimi sono espressi nei follicoli piliferi del cuoio capelluto [22], sebbene l’SRD5A2 predomina in queste cellule.[21] Il sottotipo SRD5A2 è l’isoforma quasi esclusivamente espressa nella ghiandola prostatica.[23][24]
Globuline leganti gli ormoni sessuali (SHBG)
Il legame del DHT con le proteine plasmatiche è superiore al 99%. Negli uomini, circa lo 0,88% del DHT non è legato e quindi libero, mentre nelle donne in premenopausa, circa lo 0,47-0,48% non è legato. Negli uomini, il DHT è legato per il 49,7% alla globulina legante gli ormoni sessuali (SHBG), il 39,2% per l’albumina e lo 0,22% per la globulina legante i corticosteroidi (CBG), mentre nelle donne in premenopausa il DHT è legato per il 78,1-78,4% alle SHBG, 21,0-21,3% all’albumina e lo 0,12% al CBG. Nella tarda gravidanza, solo lo 0,07% del DHT non è legato nelle donne; Il 97,8% è legato alle SHBG mentre il 2,15% è legato all’albumina e lo 0,04% è legato al CBG. [25][26] Il DHT ha un’affinità maggiore per le SHBG rispetto al Testosterone, all’Estradiolo o qualsiasi altro ormone steroideo.[27][26]
Funzioni e attività biologiche del DHT
Il DHT è biologicamente importante per la differenziazione sessuale dei genitali maschili durante l’embriogenesi, la maturazione del pene e dello scroto durante la pubertà, la crescita dei peli nel viso, nel corpo e dei peli pubici e lo sviluppo e il mantenimento della ghiandola prostatica e delle vescicole seminali. Come già accennato, è principalmente sintetizzato per via della 5α-riduzione del Testosterone in alcuni tessuti ed è il principale androgeno nei genitali, nella ghiandola prostatica, nelle vescicole seminali, nella pelle e nei follicoli piliferi. [28]
3α-Hydroxysteroide dehydrogenasi (3α-HSD)
Il DHT esplica una segnalazione principalmente in maniera intracrina e paracrina nei tessuti in cui viene sintetizzato, svolgendo un ruolo secondario, sebbene non trascurabile, come ormone endocrino circolante.[29][30][31] I livelli circolanti di DHT sono 1/10 e 1/20 di quelli del Testosterone in termini di concentrazioni totali e libere, rispettivamente [32], mentre i livelli locali di DHT possono essere fino a 10 volte quelli del Testosterone nei tessuti con alta espressione del 5α-reduttasi come la prostata.[33] Inoltre, a differenza del Testosterone, il DHT viene inattivato dalla 3α-idrossisteroide deidrogenasi (3α-HSD) nell’androgeno 3α-androstanediolo molto debole in vari tessuti come quello muscolare, adiposo e epatico, tra gli altri [31][34][35], e in relazione a questo, è generalmente stato riportato che il DHT è un agente anabolico molto scarso quando somministrato esogenamente come farmaco. [36] Ma su questo ci torneremo più avanti.
Progressione della alopecia androgenetica
Oltre alle funzioni biologiche di base, il DHT svolge anche un importante ruolo causale in una serie di condizioni dipendenti dagli androgeni, tra cui le condizioni inerenti alla crescita della peluria come l’irsutismo (eccessiva crescita dei peli sul viso / corpo) e anche la perdita di capelli (alopecia androgenetica o calvizie) e malattie della prostata come l’iperplasia prostatica benigna (IPB) e il carcinoma prostatico.[28] Gli inibitori della 5α-reduttasi, che impediscono la sintesi di DHT, sono efficaci nella prevenzione e nel trattamento di queste condizioni, sebbene siano accompagnati da pesanti effetti collaterali.[37][38][39][40] Inoltre, il DHT può svolgere una funzione nel reclutamento e nella funzione del trasportatore di aminoacidi nel muscolo scheletrico.[41] Ed anche su questo punto torneremo tra poco.
Recettore Estrogeno beta (ERβ)
È stato scoperto che i metaboliti del DHT agiscono come neurosteroidi con la propria attività biologica indipendente dall’AR.[42] Il 3α-Androstanediol è un potente modulatore allosterico positivo del recettore GABAA, mentre il 3β-androstanediol è un potente e selettivo agonista del sottotipo ERβ del Recettore degli Estrogeni (ER).[42] Questi metaboliti possono svolgere un ruolo importante negli effetti centrali del DHT e per estensione del Testosterone, inclusi i loro effetti antidepressivi, ansiolitici, gratificanti / edonici, antistress e pro-cognitivi.[42][43] Ed è soprattutto grazie all’azione neurosteroidea dei metaboliti del DHT a conferire a questa molecola i suoi benefici sull’aumento della forza muscolare e del focus mentale, entrambe caratteristiche ricercate negli sport di potenza e propedeutiche ad un migliore stimolo ipertrofico indotto dall’allenamento contro-resistenza.
Il DHT è un potente agonista dell’AR ed è in effetti il ligando endogeno più potente conosciuto per questo recettore. Ha un’affinità (Kd) compresa tra 0,25 e 0,5 nM per la RA umana, che è circa 2-3 volte superiore a quella del Testosterone (Kd = 0,4 a 1,0 nM) [44] e 15-30 volte superiore a quella degli androgeni surrenali.[45] Inoltre, il tasso di dissociazione del DHT dall’AR è 5 volte più lento di quello del Testosterone.[46 L’EC50 del DHT per l’attivazione dell’AR è 0,13 nM, che è circa 5 volte più forte di quello del Testosterone (EC50 = 0,66 nM).[47] Nei biotest, il DHT è risultato essere da 2,5 a 10 volte più potente del Testosterone.[44]
L’emivita di eliminazione del DHT nel corpo (53 minuti) è più lunga di quella del Testosterone (34 minuti), e ciò potrebbe spiegare alcune delle differenze nella loro potenza.[48] Uno studio sul trattamento transdermico con DHT e Testosterone ha riportato emivite terminali rispettivamente di 2,83 ore e 1,29 ore.[49]
A differenza di altri androgeni come il Testosterone, il DHT non può essere convertito dall’enzima aromatasi in estrogeno come l’Estradiolo. Pertanto, viene spesso utilizzato in contesti di ricerca per distinguere tra gli effetti del testosterone causati dal legame con l’AR e quelli causati dalla conversione del Testosterone in Estradiolo e il successivo legame e attivazione del ER.[50] Sebbene il DHT non possa essere aromatizzato, viene comunque trasformato in metaboliti con significativa affinità e attività ER. Questi sono 3α-androstanediolo e 3β-androstanediolo, che sono agonisti predominanti dell’ERβ.[51] Determinano l’effetto anti-estrogenico attribuito al DHT.
I livelli sierici di DHT sono circa il 10% di quelli del Testosterone, ma i livelli nella ghiandola prostatica sono da 5 a 10 volte superiori a quelli del Testosterone a causa di una conversione di oltre il 90% di quest’ultimo in DHT da parte della 5α-reduttasi espressa localmente.[33] Per questo motivo, e oltre al fatto che il DHT è molto più potente come agonista dell’AR rispetto al Testosterone [44], il DHT è considerato il principale androgeno della ghiandola prostatica.[33]
3α-androstanediol
Il DHT è inattivato nel fegato e nei tessuti extraepatici come la pelle in 3α-androstanediol dall’enzima 3α-idrossistoidea deidrogenasi, e in 3β-androstanediol dall’enzimi 3β-idrossisteroidide deidrogenasi.[34][52]Questi metaboliti vengono a loro volta convertiti, rispettivamente, in Androsterone ed Epiandrosterone, quindi coniugati (tramite glucuronidazione e/o solfatazione), rilasciati in circolazione ed escreti nelle urine.[34]
Come già detto, a differenza del Testosterone, il DHT non può essere aromatizzato in estrogeno come l’Estradiolo e, per questo motivo, non ha propensione ad esercitare effetti estrogenici.[53] Quindi, il DHT viene escreto nelle urine sotto forma di metaboliti, come i coniugati di 3α-androstanediol e Androsterone.[54][34]
Uso del DHT in medicina
Il DHT è disponibile in formulazioni farmaceutiche per uso medico come steroide anabolizzante androgeno (AAS) con finalità prettamente androgene.[55] È usato come ancillare principalmente nel trattamento dell’ipogonadismo maschile.[56] Quando usato come farmaco, il DHT viene chiamato Androstanolone (INN) o Stanolone (BAN) [55] [57] [58], e viene venduto sotto nomi commerciali differenti come Andractim. [55] [57] [58] [56] [59] La disponibilità di DHT farmaceutica è limitata; non è disponibile negli Stati Uniti o in Canada, [60] [61] ma è disponibile in alcuni paesi europei. [58] [56] Le formulazioni disponibili di DHT includono compresse orali o sublinguali, gel topici e, come esteri in olio, iniettabili come Androstanolone propionato e Androstanolone Valerato.[55] [56] [59]
L’Androstanolone è disponibile in formulazioni farmaceutiche per uso medico come androgeno.[4] È usato principalmente come forma ancillare nella terapia sostitutiva degli androgeni nel trattamento dell’ipogonadismo maschile ed è specificamente approvato per questa indicazione in alcuni paesi.[62] [13] [63] [64] [65] [66] [67] Non è più raccomandato come solo farmaco nelle terapie sostitutive degli androgeni a causa delle differenze biologiche con il Testosterone come la mancanza di effetti Estrogenici e effetti androgeni parziali.[68] L’Androstanolone topico è utile nel trattamento della ginecomastia.[69] Allo stesso modo, l’Androstanolone Enantato tramite iniezione intramuscolare è risultato efficace nel trattamento della ginecomastia puberale persistente.[70] Il farmaco è stato anche usato come gel topico per il trattamento del pene piccolo nei ragazzi pre e peripubertali con sindrome da insensibilità agli androgeni lieve o parziale.[71] [72] [73]
Drostanolone Propionato
L’Androstanolone è risultato efficace nel trattamento del carcinoma mammario in fase avanzata nelle donne negli anni ’50, sebbene fosse utilizzato in dosi molto elevate e causasse una grave virilizzazione.[74] [75] [76] È stato usato in sospensione acquosa microcristallina mediante iniezione intramuscolare.[77] [78] [79] Poco dopo, il Drostanolone Propionato (2α-Methylandrostanolone Propionato) fu sviluppato per questo uso al fine di sostituire l’Androstanolone a causa della sua superiore farmacodinamica e fu introdotto per questa indicazione negli Stati Uniti e in Europa nei primi anni ’60.[80] [81] [82] [83]
L’Androstanolone è stato usato alla dose di 25mg per via sublinguale da due a tre volte al giorno nella terapia sostitutiva con androgeni per gli uomini.[84] Questo è anche il dosaggio di Androstanolone comunemente utilizzato nel trattamento di individui si sesso maschile.[84]
La questione DHT e ipertrofia muscolare
La sarcopenia, caratterizzata da una perdita di massa muscolare, ossea, forza e resistenza, si verifica con l’invecchiamento e disturbi medici cronici come l’infezione da virus dell’immunodeficienza umana (HIV) e la terapia a lungo termine con glucocorticoidi sistemici (Gcc). D’altra parte, la somministrazione di Testosterone negli uomini più anziani e negli uomini con infezione da HIV con perdita di peso che hanno basse concentrazioni di Testosterone (Bhasin et al. 2001), nonché gli uomini che richiedono un trattamento sistemico a lungo termine di Gcc (Truhan e Ahmed 1989) aumentano il grasso corporeo- massa magra e forza muscolare.
Recettore degli Androgeni
Il muscolo scheletrico è uno dei tessuti bersaglio per l’azione anabolica degli androgeni. Recettori degli Androgeni (AR), localizzati nelle cellule muscolari e adipose, cellule nervose e pluripotenti mesenchimali che risiedono nel tessuto muscolare, probabilmente mediano gli effetti degli androgeni aumentando la massa muscolare, la sintesi proteica, il contenuto ribosomiale, le aree mitocondriali, il numero mioonucleare, il numero di cellule satellite e la miogenesi delle cellule mesenchimali pluripotenti riducendo la degradazione delle proteine e l’adipogenesi delle cellule mesenchimali pluripotenti (Herbst & Bhasin 2004). Sul ligando che si lega all’AR intracellulare, il complesso androgeno-AR viene traslocato nel nucleo e si lega a sequenze specifiche di DNA, elementi di risposta agli androgeni, con conseguente trascrizione di geni specifici (Michel & Baulieu 1980, Simental et al. 1991). Gli androgeni hanno anche azioni rapide non genomiche nel muscolo (Estrada et al. 2000, 2003), tra cui il recettore di membrana accoppiato alla proteina G, il recettore dell’inositolo 1,4,5-trisfosfato (IP3), lo ione calcio (Ca2 +) e la cascata della fosforilazione della proteina chinasi mitogeno-attivata (MAPK) / proteina chinasi regolata da segnali extracellulari (ERK). Oltre al suo ruolo nella contrazione muscolare, si ritiene che Ca2+ intracellulare regola l’espressione genica nel muscolo scheletrico (Estrada et al. 2001, Araya et al. 2003). Pertanto, le azioni genomiche e non genomiche degli androgeni sono responsabili della trascrizione dei geni sensibili agli androgeni (ARG).
Tuttavia, i meccanismi molecolari dell’effetto anabolico degli androgeni nel muscolo scheletrico sono mal compresi. Con l’avvento dell’analisi seriale dell’espressione genica (SAGE) (Velculescu et al. 1995), sono sorte nuove possibilità per l’analisi del trascrittoma su larga scala. Usando questo metodo, si sono precedentemente studiati i meccanismi molecolari responsabili dell’atrofia muscolare causata dall’immobilizzazione nei ratti (St-Amand et al. 2001), nonché il profilo di espressione genica degli uomini allenati per la resistenza (Yoshioka et al. 2003). In un interessante studio del 2006 [85], si sono studiati gli effetti della castrazione (GDX) e del DHT sull’espressione genica globale nel muscolo scheletrico dei topi maschi usando la strategia SAGE. Le trascrizioni modulate DHT sono coinvolte nel rilascio di Ca2 +, nella segnalazione cellulare, nella proliferazione cellulare, nella sintesi di mRNA e proteine e nel metabolismo energetico. Questi risultati costituiscono un primo passo verso una comprensione precisa dei meccanismi molecolari coinvolti negli effetti fisiologici degli androgeni nel muscolo scheletrico.
Topo C57BL6
Nello studio, è stato asportato il muscolo gastrocnemio destro dai topi C57BL6 di età compresa tra 12 e 14 settimane. Gli animali sono stati tenuti con luci accese da 0715 a 1915h, e hanno avuto accesso all’acqua ad libitum. Nessun trattamento è stato eseguito su 26 topi intatti. Il GDX è stato eseguito 7 giorni prima della raccolta di organi in ciascuno dei 14 topi dai gruppi GDX e DHT. I topi del gruppo GDX hanno ricevuto un i.p. della soluzione del veicolo (0,4% (p / v) Methocel A15 LV Premium / 5% etanolo; Dow Chemicals Co, Laval, Quebec, Canada) 24 ore prima della morte, mentre una dose fisiologica di DHT (0,1 mg / topo) è stato iniettato 1, 3, 6 e 24 ore prima della loro uccisione (gruppi DHT 1 h, DHT 3 h, DHT 6 he DHT 24 h). Il muscolo gastrocnemio destro è stato campionato da ciascun topo e messo insieme per l’analisi dello stesso gruppo per eliminare le variazioni inter-individuali ed estrarre quantità sufficienti di mRNA. I tessuti sono stati conservati a -80 ° C fino all’estrazione dell’RNA.
Poliammina Spermina
Gli ormoni anabolizzanti stimolano la crescita muscolare principalmente aumentando la sintesi proteica (Rooyackers & Nair 1997). In questo studio, la condizione GDX ha represso l’espressione del membro della famiglia delle proteine da shock termico 7 (Hspb7), mentre l’iniezione di DHT ha regolato verso l’alto cinque geni che codificano proteine ribosomiali e chaperoni (Mrpl51, Rpl34, Rps20, Cct8 e Cabc1) entro 3 ore e modulando altre tre trascrizioni (Fxr2h, Rps24 e Rps27) a 24 h. Oltre alla sintesi proteica, Rpl34 e Rps20 sono implicati nella biosintesi delle poliammine (Panagiotidis et al. 1995). La proteina Cct8 la cui espressione è fortemente dipendente dalla crescita cellulare (Yokota et al. 1999) piega le proteine appena sintetizzate, compresi i componenti cellulari necessari per la crescita cellulare (Thulasiraman et al. 1999), oltre a comportarsi come proteina associata ai microtubuli (Roobol et al. 1999). Mrpl51 è codificato dal DNA mitocondriale e Cabc1 codifica una proteina mitocondriale essenziale per la corretta conformazione e funzionamento dei complessi proteici nella catena respiratoria (Iiizumi et al. 2002). In effetti, le espressioni di 18 trascrizioni relative alla produzione di OxPhos e ATP sono state sovra-regolate dal DHT in questo studio. Questi dati suggeriscono che il DHT aumenta la sintesi proteica e la stabilizzazione in parallelo con la crescita cellulare entro 3 ore nei topi in vivo.
Le trascrizioni modulate 24h dopo il trattamento con DHT nel presente studio suggeriscono quanto segue:
l’aumento del trasporto di mRNA tra citoplasma e nucleolo da parte delle proteine fragili correlate all’X (FMRP) (Tamanini et al. 1999);
la soppressione della degradazione dell’mRNA da parte della proteina ribosomiale S27 (Revenkova et al. 1999);
la diminuzione della sintesi proteica poiché la proteina ribosomiale S24 è strettamente coinvolta sia nei processi di iniziazione che di allungamento durante la sintesi proteica (Bommer et al. 1988).
Il fatto che il tasso di sintesi proteica diminuisca drasticamente durante la mitosi nelle cellule di mammifero potrebbe spiegare i dati.
La degradazione delle proteine da parte del proteasoma 26S è essenziale per la progressione del ciclo cellulare, il metabolismo delle poliammine e la presentazione della catena pesante di classe I del maggiore complesso di istocompatibilità (MHC) sulla superficie cellulare. Il DHT ha indotto Psmc3 e Psmc5 le cui proteine sono i componenti integrali della subunità normativa 19S del proteasoma 26S, che potrebbe riflettere l’induzione della proliferazione cellulare e la modulazione dell’immunità da DHT.
La regolazione trascrizionale è un punto di controllo essenziale per diverse funzioni cellulari come la proliferazione cellulare, la differenziazione, la trasformazione e l’apoptosi. Il DHT ha sovra-regolato cinque fattori trascrizionali (Ogt, Pttg1, Psmc3, Psmc5 e Smyd2) entro 3 ore dopo il trattamento e il Fxr2h a 24h. L’attivazione della cascata MAPK provoca la traslocazione della proteina citoplasmatica Pttg1 nel nucleo (Pei 2000) dove la proteina Pttg1 transattiva i geni bersaglio che promuovono la proliferazione cellulare (Pei 2001). Le proteine di Psmc3 e Psmc5 suggeriscono ruoli nella transattivazione del recettore dell’ormone tiroideo (Ishizuka et al. 2001). La condizione GDX ha anche ridotto il livello di espressione del Ttr, il cui prodotto è una proteina plasmatica omotetramericana che trasporta tiroxina e retinolo. Sebbene gli ormoni tiroidei siano essenziali durante la crescita, sia un eccesso che una carenza causano un degrado muscolare da meccanismi sconosciuti (Rooyackers & Nair 1997). La proteina Fxr2h mostra una forte attivazione della trascrizione (Hillman & Gecz 2001). La glicosilazione delle proteine nucleari e citoplasmatiche è una modifica post-traduzionale diffusa e reversibile nelle cellule eucariotiche. La glicosilazione intracellulare dei residui di serina e treonina è catalizzata dalla proteina di Ogt, che regola un numero di funzioni cellulari tra cui l’attivazione trascrizionale (geni bersaglio p53) / repressione (RNA polimerasi II) e l’attivazione traslazionale (Wells et al. 2003). La presenza nel gene Smyd2 di domini SET e MYND sarebbe in accordo con gli effetti rispettivamente sulla deacetilazione e metilazione dell’istone (Sims et al. 2002). Pertanto, i risultati suggeriscono che almeno alcune delle azioni del DHT si verificano attraverso l’attivazione o la repressione dei regolatori trascrizionali.
Via di segnalazione MAPK
Nel muscolo scheletrico, il Ca2+ svolge un ruolo chiave nella contrazione e nel rilassamento. Il presente studio ha dimostrato che il trattamento con DHT ha aumentato l’espressione della Pvalb, una proteina di legame Ca2+ ad alta affinità che agisce come fattore di rilassamento muscolare dopo la contrazione, e Trdn che forma un complesso quaternario con il recettore della ryanodina, junctina e calsequestrina nel lume del reticolo sarcoplasmatico ( SR) per il buffering passivo di Ca2+ luminale SR, nonché un rilascio Ca2+ attivo dal processo SR durante l’accoppiamento eccitazione-contrazione. Topi transgenici che sovraesprimono Trdn1 nel cuore mostrano ipertrofia cardiaca con rilassamento alterato e contrattilità attenuata (Kirchhefer et al. 2001). Pertanto, l’induzione di entrambi i fattori di rilassamento muscolare e di contrazione potrebbe contribuire a una generazione di energia generalmente osservata negli atleti che assumono steroidi anabolizzanti.
Fosfolipasi C
La depolarizzazione delle cellule muscolari provoca anche un rilascio transitorio lento di Ca2 +, che è mediato dalla fosfolipasi C (PLC) e IP3 tramite i recettori IP3 (Estrada et al. 2001, Powell et al. 2001) e porta alla fosforilazione di ERK1 / 2 (Powell et al. 2001). Negli osteoblasti, il DHT attiva la proteina Gβ4 accoppiata al PLC-β2 che aumenta la formazione di IP3 e diacilglicerolo (DAG) e innesca il rilascio di Ca2 + intracellulare dal reticolo endoplasmatico (Zagar et al. 2004). Gli aumenti dei livelli di DAG e Ca2 + regolano l’attività della proteina chinasi C (PKC) che stimola ERK1 / 2 attraverso l’attivazione di MAPK chinasi 1/2 (Zagar et al. 2004). I risultati hanno mostrato le induzioni di Camk2g, Dusp1 e Hint1 entro 3 ore dall’iniezione di DHT. Ca2 + multifunzionale / proteinodinasi dipendente dalla calmodulina (CaMKII) media le risposte cellulari alla Ca2 + intracellulare ed è implicato nel controllo di funzioni essenziali quali trasmissione sinaptica, canali ionici, trascrizione genica e progressione del ciclo cellulare (Santella 1998, Anderson 2005). Le cellule proliferanti (Tombes & Krystal 1997) e il cuore ipertrofico con maggiore contrattilità (Colomer et al. 2003) esprimono l’isoforma CaMKIIγ codificata da Camk2g. Dusp1 (chiamato anche CL100 o MAPK fosfatasi 1) è stato originariamente identificato come un gene precoce immediato indotto da mitogeni (Charles et al. 1992, Keyse & Emslie 1992), e il suo livello di trascrizione riflette l’attivazione di ERK1 / 2 (Camps et al. 2000).
Proteina Hint1
La Hint1, una proteina che interagisce con la PKC che originariamente si pensava inibisse quest’ultima, può svolgere un ruolo di soppressore del tumore (Su et al. 2003). Abbiamo osservato la sovra-regolazione di Mpp6, Oaz, Psmc3 e Psmc5 entro 3 ore dal trattamento con DHT. La funzione di Mpp6, un membro della sottofamiglia guanilato chinasi (MAGUK) associata alla membrana p55, non è ancora nota. Tuttavia, il MAGUK interagisce con i recettori del glutammato e vari canali ionici (Godreau et al. 2004). Le poliammine (spermine, spermidina e putrescina) interagiscono anche con alcuni canali ionici e controllano i livelli intracellulari di Ca2 + (Williams 1997). La biosintesi della poliammina nelle cellule di mammifero inizia con una produzione di putrescina da parte dell’ornitina decarbossilasi (ODC). Quando i livelli di poliammina intracellulare aumentano, l’antizima ODC, codificato da Oaz, si lega all’OCD e facilita il suo rapido degrado da parte del proteosoma 26S (Thomas & Thomas 2003). Pertanto, le induzioni di Oaz così come Psmc3 e Psmc5, che sono componenti integranti del proteasoma 26S, possono riflettere il livello aumentato di poliammine intracellulari. Inoltre, le modulazioni di queste trascrizioni e di Mpp6 entro 3 ore, lo stesso decorso di Camk2g, suggeriscono la loro partecipazione al controllo di Ca2 + intracellulare. Inoltre, il percorso Ras / MAPK controlla la trascrizione di Cct8 (Yamazaki et al. 2003) che è stata indotta a 1 ora dopo il trattamento con DHT nel presente studio. Nel loro insieme, il secondo messaggero, ovvero Ca2 + intracellulare, e le sue cascate a valle tra cui PKC e MAPK, che sono essenziali per la regolazione della crescita cellulare, sembrano essere modulati dal DHT.
Le cellule satellite / mioblasti all’interno del tessuto muscolo-scheletrico proliferano in seguito all’esposizione a fattori di crescita e a seguito di lesioni muscolari, ma smettono di dividersi quando si fondono con fibre muscolari preesistenti. La fusione è generalmente accoppiata con l’inizio della proliferazione cellulare. Nel presente studio, la trascrizioni sovra-regolate da parte del DHT e correlate all’entrata in fase S (Pttg1) (Nasmyth et al. 2000), assemblaggio di microtubuli (Cct8) (Roobol et al. 1999), formazione del fuso bipolare (Tctex1) (Vaisberg et al 1993), uguale segregazione cromosomica (Pttg1) (Nasmyth et al. 2000), accatastamento di Golgi cisternae (Gorasp2) (Shorter et al. 1999) e disintossicazione dei metaboliti reattivi prodotti durante la proliferazione cellulare (Akr1a4) (Barski et al. 2004 ), suggeriscono un’induzione della proliferazione cellulare da parte del DHT. D’altra parte, il DHT ha sotto-regola un fattore cistostatico, Lgals1, che mantiene G0 e controlla la traversata G2 (Wells & Mallucci 1991). Il fuso mitotico richiede il montaggio / smontaggio di icrotubuli e l’azione di complessi motori come il dynein (Vaisberg et al. 1993). La proteina Tctex1 è una catena leggera del complesso motorio dynein (Tai et al. 1998). Il Cct8 aumenta durante la transizione G1 / S attraverso la prima fase S (Yokota et al. 1999). Pttg1, protezione umana, si accumula all’inizio della fase S con picchi nelle fasi G2 – M, e previene l’attivazione prematura delle separine durante la mitosi (Nasmyth et al. 2000).
Il DHT sovra-regola Psmc3, Dusp1, Gadd45g e Pttg1 nelle presenti condizioni. La sovraespressione di Psmc3 aumenta le proteine p53 e p21 (Pollice et al. 2004). In risposta al danno al DNA e ad altri stress, il soppressore del tumore p53 induce l’arresto del ciclo cellulare o l’apoptosi a seconda dei contesti cellulari specifici (Yu & Zhang 2005). In risposta al danno al DNA, p53 promuove la riparazione del DNA influenzando il percorso di riparazione dell’escissione del DNA e arrestando le cellule in G1 attraverso l’induzione di p21 che contribuiscono a fornire più tempo per la riparazione (Smith & Seo 2002), mentre anche l’arresto G1 mediato da p53 si verifica per induzione di Dusp1 in assenza di danni al DNA (Li et al. 2003). Le proteine codificate da Gadd45g interagiscono con p21 e sopprimono la crescita cellulare senza alcuna evidenza di apoptosi (Nakayama et al. 1999). L’arresto della crescita mediato dagli inibitori del ciclo cellulare p21 e Gadd45 inibisce la risposta apoptotica indotta da bersagli apoptotici di p53 (Yu & Zhang 2005). Inoltre, la protezione codificata da Pttg1 inibisce la capacità di p53 di indurre la morte cellulare (Bernal et al. 2002). Nel loro insieme, il DHT potrebbe promuovere l’arresto di G1 senza indurre l’apoptosi, almeno secondo ciò che è emerso dal presente studio.
S-adenosilmetionina decarbossilasi (SAMDC)
Il presente studio riporta l’induzione di Amd1 che codifica per la S-adenosilmetionina decarbossilasi (SAMDC), Oaz, Rps20 e Rpl34, nonché Psmc3 e Psmc5 dopo l’iniezione di DHT. D’altra parte, il DHT sotto-regolato Sbp il cui prodotto è correlato all’accumulo di spermatozoi (Moruzzi et al. 1982). I composti policristici sintetizzati dagli enzimi che limitano la velocità, ODC e SAMDC, sono cruciali per la crescita e la proliferazione delle cellule di mammifero. L’antizima ODC codificato da Oaz, così come le proteine ribosomiali L34 e S20, inibiscono le decarbossilasi di arginina e ODC (Panagiotidis et al. 1995). ODC e SAMDC sono degradati dal proteasoma 26S (Yerlikaya e Stanley 2004) che è codificato da Psmc3 e Psmc5. L’antizima ODC inibisce anche l’assorbimento della poliammina e stimola l’escrezione (Sakata et al. 2000). Inoltre, l’assorbimento della poliammina è inibito dal PKC ed è stimolato dalla sua inibizione (Dot et al. 2000). Per coincidenza, Hint1, che inibisce la PKC, aveva un modello di modulazione simile a quello di Oaz con il trattamento a base di DHT. Nel loro insieme, le modulazioni di Pttg1, Cct8, Tctex1, Gorasp2, Akr1a4, Lgals1, Amd1, Oaz, Rpl34, Rps20, Psmc3, Psmc5 e Sbp mediante dal DHT nel presente studio potrebbero riflettere la proliferazione di cellule satelliti / mioblasti nel muscolo scheletrico .
Inoltre, la proteina Pttg1 induce angiogenesi sia in vitro che in vivo (Ishikawa et al. 2001). Nel presente studio, il DHT ha sovra-regolato Pttg1 e Asb5, che è una nuova proteina implicata nell’inizio dell’arteriogenesi (Boengler et al. 2003). La vascolarizzazione è un importante fattore determinante dell’approvvigionamento energetico e della rimozione dei rifiuti durante la contrazione muscolare e la sua stimolazione da parte del DHT in questo studio è quindi in accordo con altri dati presentati.
Lpin1
Nel metabolismo lipidico, la condizione GDX ha spento la Apina2 e il DHT a sovra-regolato la Lpin1. L’apolipoproteina A-II codificata da Apoa2, la seconda proteina più abbondante delle particelle di lipoproteine ad alta densità (HDL), esercita un marcato effetto sul legame HDL e sull’assorbimento selettivo dei lipidi da parte dei recettori scavenger di classe B. Nel topo, l’espressione migliorata di Lpin1 nel muscolo scheletrico promuove l’obesità diminuendo il dispendio energetico dell’intero corpo e l’utilizzo dei grassi, nonché inducendo resistenza all’insulina (Phan & Reue 2005). Contrariamente a quanto accade nel muscolo, la sovraespressione di Lpin1 nel tessuto adiposo provoca obesità senza insulino-resistenza (Phan & Reue 2005). Ho precedentemente riportato che il livello di espressione di Lpin1 nel tessuto adiposo rimane inalterato con il trattamento a base di DHT (Bolduc et al. 2004). L’induzione di Lpin1 solo nel muscolo potrebbe suggerire che il carboidrato fosse usato per aumentare la produzione di OxPhos e ATP. Ulteriori studi sono necessari per chiarire questo intrigante meccanismo.
Le prime risposte all’iniezione di DHT (DHT 1, 3 e 6 h) sono l’induzione sia dei fattori di rilassamento muscolare (Pvalb) che di contrazione (Trdn) che modulano i livelli intracellulari di Ca2+ . Il DHT ha anche indotto le trascrizioni relative alla segnalazione cellulare come Ca2 + (Camk2g), PKC (Hint1) e percorsi MAPK (Dusp1) nonché la biosintesi della poliammina (Amd1, Oaz, Psmc3, Rps20 e Rpl34), proliferazione cellulare (Akr1a4, Cct8 Pttg1 e Tctex1), arresto del ciclo cellulare (Gadd45g), p53 (Cabc1, Dusp1 e Pttg1) e angiogenesi (Asb5 e Pttg1). L’induzione di mRNA correlati alla trascrizione (Ogt, Psmc3 e Psmc5), sintesi proteica (Mrpl51, Ogt, Rps20 e Rpl34), modifica (Cabc1, Cct8 e Ogt) e degradazione (Psmc3 e Psmc5), fosforilazione ossidativa (Cyc1, MtCo1, in questi punti sono stati osservati anche MtCo2, Cyp27a1, Ndufa5 e Ndufb2), produzione di ATP (Atp5j2 e EST Atpaf1), metabolismo lipidico (Lpin1) e immunità (Cd59a e Ga17). Tuttavia, l’induzione di trascrizioni relative alla segnalazione di Ca2+, MAPK, arresto del ciclo cellulare, p53, sintesi proteica e angiogenesi non è più significativa dopo 24 ore dall’iniezione di DHT mentre le trascrizioni relative alla progressione del ciclo cellulare sono ancora sovraregolate. Inoltre, le trascrizioni relative alla sintesi proteica (Rps24 e Rps27) erano sotto-regolate. Questi risultati indicano che l’iniezione di DHT induce la generazione di energia, la sintesi proteica, la funzione mitocondriale e la proliferazione di cellule satellite / mioblasti a livello trascrizionale in vivo, supportando precedenti risultati di un’azione anabolica del composto in questione.
Panoramica degli ARG nel muscolo scheletrico. Le trascrizioni sovra e sotto regolate dal trattamento con DHT sono mostrate rispettivamente in rosa e in blu. Le trascrizioni che mostrano solo una risposta precoce (DHT 1 ora, 3 ore e 6 ore) o tardiva (DHT 24 ore) sono indicate rispettivamente come (E) o (L). Le linee continue e tratteggiate rappresentano rispettivamente attivazione / induzione e inibizione. Akr1a4, aldo-keto reductasi famiglia 1 membro A4; Amd1, S-adenosilmetionina decarbossilasi 1; Asb5, ripetizione di ankyrin e proteina 5 contenente scatola SOCs; Atp5j2, ATP sintasi subunità mitocondriale F0 complessa f isoforma 2; Atp6, ATP sintasi 6; Atpaf1, fattore di assemblaggio complesso mitocondriale F1 sintasi ATP 1; B2m, microglobulina β-2; Cabc1, attività di accompagnatore ABC1 del complesso bc1 simile; Camk2g, protein chinasi IIγ calcio / calmodulina-dipendente; Cct8, subunità chaperonin 8; Cd59a, antigene CD59a; Col1a2, procollagene tipo I α2; Cyc1, citocromo c-1; Cyp27a1, citocromo P450 famiglia 27 sottofamiglia un polipeptide 1; DAG, diacilglicerolo; Dusp1, fosfatasi 1 a doppia specificità; EST, tag di sequenza espressi; Fxr2h, fragile 2 ritardo mentale X gene 2; Ga17, proteina cellulare dendritica GA17; Gadd45g, arresto della crescita e DNA inducibile 45γ; Gorasp2, Golgi riassemblando la proteina 2; Suggerimento 1, proteina 1 di legame alla triade di nucleotidi di istidina; IP3, inositolo 1,4,5-trisfosfato; Lgals1, lectina legante il galattosio solubile 1; Lpin1, lipina 1; MAPK, protein chinasi attivata dal mitogeno; Mrpl51, proteina ribosomiale L51; MtCo1, citocromo c ossidasi 1; MtCo2, citocromo c ossidasi 2; MtCo3, citocromo c ossidasi 3; MtNd2, NADH deidrogenasi 2; MtNd3, NADH deidrogenasi 3; MtNd4, NADH deidrogenasi 4; Ndufa5, NADH deidrogenasi 1α sottocomplex 5; Ndufb2, NADH deidrogenasi 1β sottocomplex 2; Ndufb9, NADH deidrogenasi 1β sottocomplex 9; Oaz, ornitina decarbossilasi antizima; Ogt, N-acetilglucosamina transferasi legata all’O; PKC, proteina chinasi C; PLC, fosfolipasi C; Psmc, subunità ATPase 26S proteasoma; Pvalb, parvalbumina; Rp, proteina ribosomiale; Pttg1, trasformazione del tumore pituitario 1; Sbp, proteina legante lo sperminozoo; Tctex1, testicolo complesso t espresso 1; TPO1, trasportatore di poliammina; Trdn, triadin.
Un altro studio interessante del 2011 e pubblicato sul “The Journal of Physiology” [86] ci offre ulteriori indizzi sul potenziale del DHT nell’ipertrofia muscolare.
Il gruppo di ricerca che ha svolto questo studio ne aveva in precedenza condotto un altro attraverso il quale avevano dimostrato che il Dihydrotestosterone (DHT), ma non il Testosterone, aumenta la produzione di forza nei muscoli a contrazione rapida e la diminuisce in quelli a contrazione lenta. Questi risultati hanno loro suggerito che il DHT poteva essere un androgeno con capacità ben superiori a quelle comunemente attribuiteli. Nel presente studio, i ricercatori hanno esaminato gli effetti di questi ormoni sul trasporto degli aminoacidi nei fasci muscolari dell’apparato muscolo-scheletrico a rapida contrazione del topo. I risultati mostrano che il DHT aumenta la sintesi proteica e l’aumento delle proteine che trasportano aminoacidi essenziali in fasci muscolari a contrazione rapida. Questo non è stato osservato con il Testosterone.
E’ stato osservato che il DHT esercita azioni acute/non genomiche nel muscolo-scheletrico di mammiferi adulti le cui funzioni fisiologiche sono state capite di recente, pertanto l’obiettivo primario di questo studio era di osservare gli effetti acuti / non genomici del DHT sul uptacke di aminoacidi e sugli eventi di trasduzione del segnale cellulare alla base di queste azioni in fasci muscolari scheletrici a contrazione rapida e lenta di topo. Gli aminoacidi marcati con 14C sono stati usati per studiare gli effetti del DHT e del Testosterone (T) sull’assorbimento degli aminoacidi e sono stati usati interventi farmacologici per determinare gli eventi di trasduzione del segnale cellulare che mediano queste azioni. Mentre il T non ha avuto alcun effetto sull’assorbimento di isoleucina (Ile) e acido α-metilamminoisobutirrico (MeAIB) in entrambi i tipi di fibre, il DHT ha aumentato il loro assorbimento nei fasci di fibre a contrazione rapida. Questo effetto è stato invertito dagli inibitori della traslazione proteica, del recettore del prodotto per la crescita epidemica (EGFR), del sistema A, del sistema L, del mTOR e del MEK. Tuttavia, è stato relativamente correlato agli inibitori della trascrizione, dei recettori degli androgeni e dell’IP3K / Akt. In aggiunta, il trattamento con DHT ha aumentato l’espressione della LAT2 e l’insufflazione fosforilata del GRFR, mentre il miscuglio rapido-twitch si mescola e in entrambi i tipi di ERK1 / 2, RSK1 / 2 e ATF2. Inoltre, ha diminuito la fosforilazione di eEF2 e ha aumentato l’incorporazione di proteine di ferro in entrambi i tipi di fibre. La maggior parte di questi effetti è stata superata dagli inibitori di EGFR e MEK. Da questi risultati si ipotizza che un’altra funzione fisiologica delle azioni acute/non genomiche del DHT nelle fibre muscolari isolate dei mammiferi è quella di stimolare l’assorbimento di aminoacidi. Questo effetto è mediato dall’EGFR e comporta l’attivazione della via MAPK e un aumento dell’espressione LAT2.
IGF-1
Un risultato chiave nel presente studio è stato l’osservazione che il trattamento di piccoli fasci di fibre muscolari scheletriche isolati dall’EDL e dal soleo di topi femmine adulti con concentrazioni fisiologiche (630pgml − 1) di DHT, per 1 ora, significativamente (P = 0,001) ha aumentato l’assorbimento di Ile e MeAiB nei fasci di fibre isolati dall’EDL ma non in quelli isolati dal soleo. Sebbene la somministrazione acuta di ormoni come l’insulina (Biolo et al. 1995), il fattore di crescita insulino-simile 1 (IGF-1) (Fryburg et al. 1995) e l’ormone della crescita (Fryburg et al. 1995) hanno dimostrato di aumentare la sintesi proteica e per promuovere l’assorbimento di aminoacidi nel muscolo scheletrico umano, è la prima volta che è stato dimostrato un aumento dell’assorbimento di aminoacidi in risposta alla somministrazione acuta di uno steroide anabolizzante-androgeno nel muscolo scheletrico dei mammiferi adulti. Solo altri due studi hanno precedentemente osservato gli effetti dell’assorbimento di aminoacidi. Entrambi gli studi hanno utilizzato soggetti umani e non sono stati in grado di dimostrare alcun cambiamento nell’assorbimento degli aminoacidi in gruppi muscolari interi (Bhasin et al. 1997; Ferrando et al. 1998). Sebbene nel presente studio siano state utilizzate piccole fibre muscolari e il T sia stata applicato direttamente ai fasci di fibre, è importante notare che questo ormone non ha avuto alcun effetto sull’assorbimento degli aminoacidi in entrambi i tipi di fibre. Al contrario, il trattamento dei fasci di fibre con DHT ha comportato un marcato aumento dell’assorbimento di Ile e MeAIB solo nei fasci di fibre a contrazione rapida (Fig. 1). In precedenza, abbiamo anche dimostrato che il T non ha effetti acuti sulla produzione di forza in fasci muscolari di topo isolati intatti, mentre il DHT ha aumentato la produzione di forza nei fasci di fibre a contrazione rapida, ma l’ha diminuita in quelli a contrazione lenta (Hamdi & Mutungi, 2010). Nel loro insieme, questi risultati suggeriscono che il T potrebbe non avere effetti acuti / non genomici nelle fasce muscolari scheletriche dei mammiferi adulti.
Come già detto, nella maggior parte dei tessuti il T viene convertito in DHT dall’enzima 5 α-reduttasi. Pertanto, è probabile che tutti gli effetti acuti del T osservati in precedenza nei miociti in coltura (Estrada et al. 2003) potrebbero essere stati esercitati dal DHT. In effetti, è stato precedentemente suggerito che gli effetti anabolici del T nei muscoli scheletrici umani possano essere indiretti o secondari al rilascio di un altro ormone come IGF-1 (Ferrando et al. 1998). Con la presente si precisa che oltre all’IGF-1, gli effetti del T nel muscolo scheletrico dei mammiferi possono anche essere esercitati attraverso il rilascio di DHT. I ricercatori suggeriscono anche che il DHT è il principale steroide anabolizzante-androgeno nei muscoli scheletrici dei mammiferi adulti.
Nonostante la maggior parte degli studi è stata effettuata su fasci muscolari di topi femmina, in precedenza era già stato dimostrato che il DHT ha effetti simili sulla produzione di forza nei fasci muscolari scheletrici di topi maschi e femmine (Hamdi & Mutungi, 2010).
I risultati riportati in questo studio (vedi figura seguente) mostrano che sebbene il DHT aumenti l’attività del SNAT2, ciò non influisce sulla sua espressione. Al contrario, aumenta l’attività e l’espressione del LAT2 (vedi Fig. 3). Inoltre, il trattamemto del fascio di fibre muscolari con il classico sistema di inibitori A e L-typeaminoacid ha portato a una marcata riduzione dell’assorbimento basale di L- [U-14C] Ile in entrambi i tipi di fibre e ha completamente soppresso l’aumento indotto da DHT nell’assorbimento di Ile.
Gli effetti del DHT sull’assorbimento di aminoacidi nei fasci di fibre muscolari di topo sono parzialmente mediati attraverso un trasportatore di aminoacidi del sistema A e sono mediati attraverso LAT2.
Effetti simili sono stati osservati anche quando i fasci di fibre sono stati trattati con la soluzione di Ringer contenente anticorpi contro il LAT2, suggerendo che gli effetti del DHT sull’Ile sono mediati attraverso questo trasportatore. Da queste osservazioni si ipotizza che i due trasportatori (LAT2, SNAT2) siano in qualche modo collegati. Pertanto, modulando l’espressione e l’attività di LAT2, anche il DHT sembra regolare indirettamente l’attività di SNAT2.
Recettore del Fattore di Crescita dell’Epidermide (EGFR)
In precedenza, era stato dimostrato che le azioni acute/ non genomiche del DHT sulla produzione di forza nei fasci muscolo-scheletrici di topi adulti sono mediate attraverso il Recettore del Fattore di Crescita dell’Epidermide (EGFR) e comportano l’attivazione di ERK1 / 2 (Hamdi & Mutungi, 2010). È interessante notare che i risultati qui riportati suggeriscono che le azioni acute/non genomiche del DHT sull’assorbimento di aminoacidi nelle fibre muscolari dei mammiferi adulti sono mediate attraverso lo stesso recettore e lo stesso percorso. Inoltre, il pretrattamento dei fasci di fibre con Ciproterone o Flutamide in modo significativo (P <0,05) ha attenuato gli effetti del DHT sull’assorbimento dell’Ile senza sopprimerlo completamente. In precedenza, è stato suggerito che il Testosterone può attivare la via MAPK tramite un recettore degli androgeni accoppiato con proteina G (Estrada et al. 2003). Tuttavia, se questo meccanismo, comunemente indicato come transattivazione, è quello che media gli effetti acuti/non genomici del DHT nelle fibre muscolari dei mammiferi adulti è incerto e sono necessarie ulteriori ricerche per chiarirlo.
Sistema regolatorio di segnalazione ERK1/2.
Dopo l’attivazione, ERK1 / 2 può rimanere nel citosol o traslare nel nucleo dove svolge un ruolo critico nella regolazione dell’espressione genica e della replicazione del DNA (Brunet et al. 1999). Nel nucleo, ERK1 / 2 fosforila una serie di target, inclusi molti fattori di trascrizione e una famiglia di chinasi correlate a RSK, le chinasi proteiche attivate dallo stress e dal mitogeno (MSK) (Deaketal.1998). Anche se nel presente studio è stato esaminato un gran numero di target a valle di ERK1 / 2, il trattamento con DHT ha aumentato soltanto la fosforilazione di RSK1 / 2 e ATF2 . Inoltre, il DHT non ha avuto alcun effetto sulla fosforilazione degli altri modulatori MAPK. Per i ricercatori, questi risultati suggeriscono che le azioni acute / non genomiche del DHT nei muscoli scheletrici dei mammiferi adulti sono esercitate principalmente attraverso l’EGFR e comportano l’attivazione della modulazione ERK1 / 2 del percorso MAPK. L’ipotesi è che il DHT, direttamente o indirettamente, attivi l’EGFR che a sua volta attiva la via MAPK che porta ad un aumento del trasporto di aminoacidi nelle fibre aumentando anche la sintesi proteica in esse. Inoltre, il DHT sembra avere un attività principale sulle fibre a contrazione rapida.
Negli animali adulti, un aumento della massa muscolo scheletrica (ipertrofia) si verifica principalmente a causa di un aumento delle dimensioni piuttosto che del numero di fibre muscolari ed è generalmente ritenuto che sia regolata dal percorso Akt / mTOR (Glass, 2003). Il percorso Akt / mTOR è attivato da molti stimoli tra cui fattori di crescita, altri ormoni e sostanze nutritive. Inoltre, la sua attivazione culmina nel blocco del apoptosi, induzione della sintesi proteica, trascrizione genica e proliferazione cellulare (Dann etal.2007). Pertanto, è stato stimato che il DHT e il T potessero attivare questo percorso. L’assorbimento di amminoacidi indotto dal DHT nelle fibre a contrazione rapida senza la sua completa soppressione, suggerisce che le azioni acute / non genomiche del DHT non sono mediate attraverso Akt. Invece, i risultati che vengono presentati nel presente studio suggeriscono che il DHT aumenta la sintesi proteica regolando la traduzione dell’mRNA già presente nelle cellule. In effetti, il trattamento dei fasci di fibre muscolari con DHT ha portato a una marcata riduzione della fosforilazione di eEF2 e ad un moderato aumento (∼50%) della sintesi proteica nei fasci di fibre a contrazione rapida.
È anche degno di nota il fatto che il pretrattamento dei fasci di fibre con l’inibitore specifico del mTOR non solo ha ridotto l’assorbimento basale di Ile, ma ha anche soppresso completamente l’aumento indotto dal DHT dell’assorbimento di Ile nei gruppi di fibre a contrazione rapida. Per i ricercatori questi risultati suggeriscono che alcuni degli effetti acuti / non genomici del DHT sono probabilmente mediati attraverso mTOR. In effetti, numerosi studi hanno suggerito che mTOR è il legame tra la disponibilità di aminoacidi e una maggiore sintesi proteica (Beugnet et al. 2003; Avruch et al. 2009). Tuttavia, la rapamicina può anche indurre l’autofagia (Ravikumar et al. 2004). Pertanto, un’altra possibilità è che i suoi effetti siano dovuti all’accumulo di aminoacidi nelle fibre muscolari.
Via di segnalazione cellulare che media gli effetti del DHT sul trasporto di aminoacidi nelle fibre muscolo-scheletriche dei mammiferi. Un diagramma schematico che mostra la via di segnalazione cellulare ipotizzata che media gli effetti acuti del DHT sull’amminoacido nei fasci muscolari dei mammiferi. L’ipotesi è che il DHT, attraverso un meccanismo sconosciuto, attiva l’EGFR e questo porta all’attivazione di RSK1 / 2 da parte di ERK1 / 2. L’RSK1 / 2 attivato aumenta quindi l’attività e l’espressione di LAT2 (cerchio blu con una croce), che a sua volta aumenta il trasporto di Ile nei fasci muscolari. Inoltre, RSK1 / 2 migliora la traduzione dell’mRNA in proteine. Si noti che il trasporto di Ile è accoppiato a quello di piccoli aminoacidi neutri come la glutammina (Gln) che vengono trasportati nella cellula dai trasportatori di aminoacidi del sistema A come SNAT2 (cerchio rosso con una croce). Pertanto, un aumento dell’attività di LAT2 aumenta indirettamente l’attività di SNAT2. Gli eventuali effetti di tutti questi processi sono di aumentare la sintesi proteica e quindi la massa muscolare, specialmente nelle fibre muscolari a contrazione rapida.
In conclusione, ci sono state prove che dimostrano che un’altra funzione fisiologica delle azioni acute / non genomiche del DHT nelle fibre muscolari dei mammiferi adulti è quella di aumentare l’assorbimento di aminoacidi essenziali. Sebbene non si possa escludere completamente il coinvolgimento del recettore degli androgeni, i principali risultati suggeriscono che l’aumento dell’assorbimento di aminoacidi e la sintesi proteica sono mediati attraverso l’EGFR e comportano l’attivazione del modulo ERK1 / 2 del pathway MAPK che a sua volta attiva RSK1 / 2 portando ad un aumento dell’espressione del trasportatore di aminoacidi di tipo L LAT2.
Piccola parentesi sul Mesterolone
Arrivati a questo punto, prima di trattare le conclusioni sul DHT, vorrei riportarvi due interessanti studi nei quali si è osservata l’azione di una forma di DHT metilata in C-1, il noto Mesterolone, sulla massa muscolare.
Senza dilungarsi troppo in preamboli, come ben sappiamo, il Mesterolone (nome commerciale Proviron) [1 alpha-methyl-17 beta- hydroxy-5 alpha-androstan-3-one] è un AAS derivato dal Dihydrotestosterone (DHT). A differenza del DHT il Mesterolone presenta l’aggiunta di un gruppo metilico in C1 (similmente al Metenolone) cosa che ne aumenta discretamente la biodisponibilità orale, senza però apportare eccessivo stress epatico. Questa alterazione però non aumenta la stabilità del C3 chetogruppo, essenziale per l’attività anabolizzante a livello muscolare mediata dal legame recettoriale, come il suo precursore.
Ora, sappiamo anche che ci sono frequenti segnalazioni di abuso di Mesterolone negli sport umani ed equini per il miglioramento della prestazione. Tuttavia, sono disponibili informazioni limitate su come questo farmaco esercita i suoi effetti sul muscolo scheletrico.
Le cellule satellite (SC) sono cellule staminali miogeniche mononucleari che contribuiscono alla crescita e alla riparazione dei muscoli postnatali. Poiché l’attivazione delle SC e la successiva differenziazione nei nuovi myonuclei sono un evento importante durante l’ipertrofia muscolare. In uno studio pubblicato nel maggio del 2012 [87], si è osservata l’influenza del Mesterolone sulla distribuzione delle SC all’interno del muscolo pettorale dei polli (Gallus gallus ). Nello specifico, questo studio ha testato le ipotesi secondo cui che il Mesterolone induca l’ipertrofia del muscolo scheletrico aviario e che aumenti anche il numero di SC nel muscolo scheletrico aviario. Sono state utilizzate solide tecniche immunocitochimiche e analisi morfometriche per calcolare il numero di SC e myonuclei. Inoltre, sono stati misurati i livelli di concentrazione di DNA e proteine Pax7 per confermare i risultati immunocitochimici. Il Mesterolone ha aumentato significativamente la massa pettorale e le dimensioni delle fibre. Tutti gli indici delle SC e il numero di myonuclei sono aumentati significativamente con la somministrazione di Mesterolone. Inoltre, negli uccelli trattati con Mesterolone sono state riscontrate una maggiore concentrazione di DNA ed espressione della proteina Pax7. Questo studio indica che il Mesterolone può indurre ipertrofia del muscolo scheletrico aviario e che questo è correlato con un aumento del numero di SC. I ricercatori suggeriscono che le SC siano intermediari cellulari chiave per l’ipertrofia muscolare indotta da Mesterolone. L’ipotesi, che necessità di ulteriori ricerche, è che tale effetto possa riscontrarsi anche in altre specie compreso l’uomo.
Identificazione immunofluorescente di SC nelle sezioni trasversali dei muscoli pettorali ottenuti da polli di controllo e trattati con Mesterolone. (A) e (C) rappresentano una sezione ottenuta dal muscolo pettorale di un esemplare di controllo. (B) e (D) rappresentano una sezione ottenuta dal muscolo pettorale di un esemplare trattato con Mesterolone. (A) e (B) mostrano tutti i nuclei in blu (colorazione DAPI) e le lamine basali delle fibre muscolari in rosso (colorazione anti-laminina). (C) e (D) mostrano SC in verde (etichettatura anti-Pax7) e le lamine basali in rosso. Barre di scala = 50 μm.
In un altro studio pubblicato nel 2010 [88], la microscopia ottica ed elettronica e la morfometria quantitativa sono state utilizzate per determinare gli effetti dell’esercizio e del Mesterolone sul muscolo soleo dei topi. Sia l’esercizio fisico che il Mesterolone hanno causato una significativa ipertrofia delle fibre muscolari extrafusali. L’ipertrofia delle fibre di tipo I era maggiore di quella delle fibre di tipo II. Non c’era iperplasia. I mitocondri erano più numerosi e più grandi rispetto a quanto osservato nei muscoli degli animali sedentari. La capillarità è aumentata e sono comparse piccole fibre muscolari nucleate centralmente, di solito in piccoli gruppi e molto spesso nei muscoli degli animali esposti al Mesterolone. Una piccola percentuale di cellule satelliti mostrava segni di attivazione ma nei muscoli degli animali trattati con Mesterolone c’era una massa muscolare maggiore dopo l’esercizio. I muscoli di animali che erano stati entrambi fatti esercitati e trattati con Mesterolone presentavano i maggiori cambiamenti: la massa muscolare e l’ipertrofia delle fibre muscolari erano maggiori rispetto a tutti gli altri gruppi di animali, la capillarità era maggiore e> il 30% di tutte le cellule satellitari riconosciute mostrava segni di attivazione. Gruppi di piccole fibre muscolari nucleate centralmente sono state comunemente osservate in questi muscoli. Sembravano essere il risultato di lacerazioni rigenerate in fibre muscolari esistenti. Sia con l’esercizio fisico sia con il Mesterolone, da solo o in combinazione, si è verificato un aumento della percentuale di fibre muscolari di tipo I e una diminuzione della proporzione di tipo II.
Entrambi gli studi esposti, sebbene non siano di grande impatto, fanno sicuramente nascere il dubbio in coloro i quali hanno sempre valutato le possibili azioni di una molecola in modo ristretto, sia per mancanza di una conoscenza sufficienza in materia sia per limitatezza nel formulare ipotesi estrapolate dall’osservazione.
Ora, non sto sicuramente dicendo che un culturista trattato con solo Mesterolone possa ambire a grandi cambiamenti nella composizione corporea. Sto semplicemente ipotizzando, che le nozioni in merito alle attività degli AAS sono datate e parziali, anche perchè si basano per lo più, tanto quanto gli studi esposti, sulla capacità di legame degli AAS testati osservata nella prostata e nel levator ani dei topi. Inoltre, il Mesterolone ha visto una riscoperta come farmaco ancillare nella TRT o come anti-depressivo funzionale in alcune tipologie di depressione.
In fondo, il peggior difetto del Mesterolone è dipeso dalla sua consuetudinaria via di somministrazione…
Nota: la biodisponibilità orale del Mesterolone è stata stimata al 3%.
Tiriamo le somme…con possibili critiche…
Già sento i soliti limitati in intelletto e pazienza, sempre pronti a parlare senza cognizione di causa, urlare al “esiste lo studio X che afferma che l’aggiunta di Dutasteride con dosi sovrafisiologiche di Testosterone non cambia il risultato!” … Lo conosco anche io capre!… A proposito, vediamo di cosa si tratta…
Testosterone Enantato
Il principale studio è quello pubblicato su JAMA nel 2012.[89] Si tratta di uno studio in doppio cieco, randomizzato, controllato con placebo. Lo studio è stato diretto dal dott. Bhasin e colleghi, i quali hanno reclutato 139 uomini sani, di età compresa tra 18 e 50 anni, con livelli normali di Testosterone.
Ai pazienti è stata assegnata 1 su 4 dosi di Testosterone Enantato (50, 125, 300 o 600 mg / settimana) più 2,5mg/die di Dutasteride o sono stati assegnati a 1 su 4 gruppi a cui è stato somministrato 1 di 4 dosi di Testosterone Enantato (50, 125, 300 o 600 mg / settimana) più placebo per 20 settimane.
Dutasteride
Il cambiamento nella massa magra rispetto al basale, misurata dall’assorbtiometria a raggi X a doppia energia, è stato il risultato primario. I ricercatori hanno anche misurato i livelli di Testosterone, la forza muscolare, la funzione sessuale, la produzione di sebo e l’acne.
La forza nel Leg Press e del petto sono state misurate con il metodo 1 ripetizione massimale con l’uso di macchine Keizer.
La funzione sessuale è stata valutata con l’indice internazionale della funzione erettile e il questionario sulla salute sessuale maschile per una valutazione più completa del desiderio sessuale.
Il volume della prostata è stato misurato con la risonanza magnetica 1.5-Tesla.
La produzione di sebo è stata misurata con l’uso di nastri di sebo. La scala Palatzi è stata utilizzata per valutare l’acne.
139 uomini furono assegnati in modo casuale; 102 (54 nei gruppi placebo e 48 nei gruppi dutasteride) hanno completato l’intervento di 20 settimane.
Gli uomini assegnati alla Dutasteride erano simili al basale degli uomini assegnati al placebo. Inoltre, i livelli di Testosterone totale e libero non differivano tra i due gruppi.
L’aumento della massa magra ottenuta dai gruppi Dutasteride+Testosterone era di 0,6 kg (IC 95%, da -0,1 a 1,2 kg) quando ricevevano 50 mg/settimana di Testosterone Enantato, 2,6 kg (IC 95%, 0,9 – 4,3 kg) per 125mg/settimana, 5,8 kg (IC 95%, 4,8 – 6,9 kg) per 300mg/settimana e 7,1kg (IC 95%, 6,0 – 8,2 kg) per 600 mg/settimana.
La massa magra ottenuta dai gruppi placebo+Testosterone era di 0,8 kg (IC 95%, da -0,1 a 1,7 kg) quando ricevevano 50mg/settimana di Testosterone Enantato, 3,5 kg (IC 95%, 2,1-4,8 kg) per 125 mg/settimana, 5,7 kg (IC 95%, 4,8 – 6,5 kg) per 300mg/settimana e 8,1 kg (IC 95%, 6,7 – 9,5 kg) per 600mg/settimana.
Le differenze dose-dipendente tra i gruppi Dutasteride e placebo per la massa magra non erano significative (P = .18).
I cambiamenti nella massa grassa, nella forza muscolare, nella funzione sessuale, nel volume della prostata, nella produzione di sebo e nei livelli di ematocrito e lipidi non differivano tra i gruppi.
La frequenza complessiva di eventi avversi, inclusa la frequenza di disfunzione sessuale, acne, vampate di calore e dolorabilità mammaria era simile nei gruppi placebo e dutasteride.
Misure in riferimento alla composizione corporea e alla forza muscolare raccolte nello studio trattato. Nei grafici a sinistra, i marker indicati hanno un margine di errore con intervalli di confidenza al 95%. Nei grafici a destra (variazione dei livelli di Testosterone), le scale dell’asse orizzontale sono state spostate (offset) di 1000ng/dL e quindi è stata applicata una trasformazione della radice quadrata. Il livellamento semiparametrico è stato ottenuto utilizzando modelli di additivi generalizzati. Le aree ombreggiate indicano intervalli di confidenza al 95%.
Bene…possiamo cestinare tutto quanto ipotizzato in precedenza? Visto anche il tipo di studio esposto? Non esattamente …
Ora, da notare la tipologia di soggetti arruolati e il loro grado di “maturità sportiva”, più specificatamente negli allenamenti contro resistenza. Essa non è chiaramente specificata e la misurazione dell’espressione della forza in soggetti poco o per nulla allenati non è un grande parametro sebbene lo scopo dello studio fosse quello di constatare l’impatto nella forza e ipertrofia dato da un protocollo a dosaggio differenziato di Testosterone Enantato con o senza un inibitore della 5α-reduttasi (Dutasteride).
Secondo punto da notare, la mancanza di misurazione specifica della massa muscolare. Infatti, la composizione corporea dei partecipanti è stata valutata per i parametri di massa grassa e massa magra. Come risaputo, quando si parla di massa magra si prende anche in considerazione l’acqua. Sebbene la differenza sia bassa, probabilmente essa è da ricollegare anche al fatto che le dosi di Testosterone utilizzate erano differenti e con limite a 600mg di Testosterone Enantato a settimana (pari a 432mg di Testosterone), e che tale dosaggio non sempre crea iperestrogenemia marcata (considerando anche un lasso di tempo contenuto) tale da causare accumuli sensibili di acqua extracellulare. Considerando poi che uno degli effetti collaterali collegati all’uso di inibitore della 5α-reduttasi sia proprio un aumento degli estrogeni circolanti (per aumento del substrato di sintesi) con ripercussioni a carico di (e non solo limitate a) ritenzione idrica, accumulo di grasso con modello ginoide e ginecomastia, i dubbi sulle metodiche di analisi dei partecipanti allo studio diventano senz’altro dubbie.
Da notare che il Dutasteride è stato osservato ridurre il DHT sierico del >90% e quello intraprostatico del 94% alla dose di 0.5mg/die.[90] Nello studio presentato, i partecipanti assegnati ai gruppi Dutasteride e Dutasteride+Testosterone erano trattati con una dose di Dutasteride pari a 2,5mg/die.
I ricercatori volevano condurre uno studio meticolosamente progettato ed eseguito per rispondere a una domanda vecchia di quasi 5 decenni: il Testosterone, il DHT o entrambi a livelli fisiologici e soprafisiologici sono importanti per il guadagno della massa magra? Purtroppo sembra che non siano stati sufficientemente meticolosi…
Non è di certo mia intenzione far valere di più uno studio svolto in vitro o su animali rispetto ad uno svolto su esseri umani. E’ invece mia intenzione far riflettere il lettore sul design di uno studio, ed in base al livello di questo poter valutare quanto in esso riportato.
Un altra critica riduttiva che potrebbe essere mossa contro le nuove ipotesi di attività del DHT potrebbe essere riferita all’enzima 3α-Hydroxysteroide dehydrogenasi (3α-HSD). Si dimentica però che tale enzima è un “limitatore” dell’attività del DHT a livello di tessuti come quello muscolare e adiposo. Infatti, la sua azione non esclude assolutamente la potenziale attività in acuto e non genomica del DHT nei tessuti dove l’enzima 3α-HSD viene espresso. E’ possibile che la cooperatività del DHT con il Testosterone sia di tipo sequenziale: attività prettamente recettoriale e in cronico (sebbene sotto regolazione organica) data dal Testosterone preceduta e seguita da una attività acuta e non genomica data dal DHT.
Inoltre, ed è una cosa molto importante da tenere in considerazione, la ricerca è tutt’ora in essere. Esistono ipotesi diverse con il loro bagaglio di ricerca. Ciò che il lettore preparato può fare è semplicemente soppesare le informazioni raccolte al fine di farsi un idea il più concreta possibile della questione.
Sebbene la ricerca scientifica abbia fatto enormi passi avanti nell’ultimo secolo, siamo ancora lungi dal conoscere tutto… anche quando si parla di ormoni e la loro attività diretta e indiretta…
Nota conclusiva: è ovvio che non si mette in dubbio l’utilità del controllo terapeutico del DHT in condizioni patologiche (vedi iperplasia prostatica maligna) o preventive in particolari soggetti. Negli individui in fisiologia e senza particolari predisposizioni, l’uso di inibitori della 5α-reduttasi potrebbe risultare una scelta controproducente sotto molteplici aspetti.
Gabriel Bellizzi
Riferimenti:
Schnitzer R (1 January 1967). Experimental Chemotherapy. Elsevier Science. pp. 156–. ISBN 978-0-323-14611-1.
Krüskemper H (22 October 2013). Anabolic Steroids. Elsevier. pp. 12–. ISBN 978-1-4832-6504-9.
Taylor WN (16 January 2002). Anabolic Steroids and the Athlete, 2d ed. McFarland. pp. 178–. ISBN 978-0-7864-1128-3.
William Andrew Publishing (2007). Pharmaceutical Manufacturing Encyclopedia. William Andrew Pub. ISBN 978-0-8155-1526-5.
Newsweek. Newsweek. 1953.
New and Nonofficial Drugs. Lippincott. 1958.
Rubin BL, Dorfman RI (1956). “In vitro conversion of testosterone to 17beta-hydroxyandrostan-3-one”. Proc. Soc. Exp. Biol. Med. 91 (4): 585–6. doi:10.3181/00379727-91-22337. PMID 13323010.
Agmo A (18 April 2011). Functional and Dysfunctional Sexual Behavior: A Synthesis of Neuroscience and Comparative Psychology. Academic Press. pp. 196–. ISBN 978-0-08-054938-5.
Oreopoulos DG, Michelis M, Herschorn S (6 December 2012). Nephrology and Urology in the Aged Patient. Springer Science & Business Media. pp. 495–. ISBN 978-94-011-1822-4.
Webster GF, Rawlings AV (17 May 2007). Acne and Its Therapy. CRC Press. pp. 168–. ISBN 978-1-4200-1841-7.
Smith LB, Mitchell RT, McEwan IJ (1 October 2013). Testosterone: From Basic Research to Clinical Applications. Springer Science & Business Media. pp. 5–. ISBN 978-1-4614-8978-8.
Azzouni F, Mohler J (2012). “Role of 5α-reductase inhibitors in benign prostatic diseases”. Prostate Cancer Prostatic Dis. 15 (3): 222–30.
Melmed S (2016). Williams Textbook of Endocrinology. Elsevier Health Sciences. pp. 621, 711.
Blume-Peytavi U, Whiting DA, Trüeb RM (26 June 2008). Hair Growth and Disorders. Springer Science & Business Media. pp. 161–162.
Rhoades RA, Bell DR (18 January 2012). Medical Phisiology: Principles for Clinical Medicine. Lippincott Williams & Wilkins. pp. 690–.
Rakel D (12 April 2012). Integrative Medicine E-Book. Elsevier Health Sciences. pp. 321–.
Morrison MF (4 May 2000). Hormones, Gender and the Aging Brain: The Endocrine Basis of Geriatric Psychiatry. Cambridge University Press. pp. 17–.
Azzouni F, Godoy A, Li Y, Mohler J (2012). “The 5 alpha-reductase isozyme family: a review of basic biology and their role in human diseases”. Advances in Urology. 2012: 1–18.
Zouboulis CC, Chen WC, Thornton MJ, Qin K, Rosenfield R (2007). “Sexual hormones in human skin”. Horm. Metab. Res. 39 (2): 85–95.
Bolognia JL, Jorizzo JL, Schaffer JV (8 June 2012). Dermatology E-Book. Elsevier Health Sciences. pp. 1094–. ISBN 978-0-7020-5182-1.
Murphy MJ (24 March 2011). Molecular Diagnostics in Dermatology and Dermatopathology. Springer Science & Business Media. pp. 373–.
eam SJ, Scott LJ (2008). “Dutasteride: a review of its use in the management of prostate disorders”. Drugs. 68 (4): 463–85.
Heesakkers J, Chapple C, Ridder DD, Farag F (24 February 2016). Practical Functional Urology. Springer. pp. 280–.
Nieschlag E, Behre HM, Nieschlag S (26 July 2012). Testosterone: Action, Deficiency, Substitution. Cambridge University Press. pp. 61–.
Dunn JF, Nisula BC, Rodbard D (July 1981). “Transport of steroid hormones: binding of 21 endogenous steroids to both testosterone-binding globulin and corticosteroid-binding globulin in human plasma”. J. Clin. Endocrinol. Metab. 53 (1): 58–68.
Williams DA, Foye WO, Lemke TL (2002). Foye’s Principles of Medicinal Chemistry. Lippincott Williams & Wilkins. pp. 707–.
Coutts, S. B.; Kicman, A. T.; Hurst, D. T.; Cowan, D. A. (1997-11-01). “Intramuscular administration of 5α-dihydrotestosterone heptanoate: changes in urinary hormone profile”. Clinical Chemistry. 43 (11): 2091–2098.
Marks LS (2004). “5α-reductase: history and clinical importance”. Rev Urol. 6 Suppl 9: S11–21.
Horton R (1992). “Dihydrotestosterone is a peripheral paracrine hormone”. J. Androl. 13 (1): 23–7. doi:10.1002/j.1939-4640.1992.tb01621.x (inactive 2020-03-09).
Wilson JD (1996). “Role of dihydrotestosterone in androgen action”. Prostate Suppl. 6: 88–92.
Swerdloff RS, Dudley RE, Page ST, Wang C, Salameh WA (2017). “Dihydrotestosterone: Biochemistry, Physiology, and Clinical Implications of Elevated Blood Levels”. Endocr. Rev. 38 (3): 220–254.
Bhasin S (13 February 1996). Pharmacology, Biology, and Clinical Applications of Androgens: Current Status and Future Prospects. John Wiley & Sons. pp. 72–.
Hay ID, Wass JA (26 January 2009). Clinical Endocrine Oncology. John Wiley & Sons. pp. 37–.
Melmed S (2016). Williams Textbook of Endocrinology. Elsevier Health Sciences. pp. 621, 711.
Jin Y, Penning TM (2001). “Steroid 5alpha-reductases and 3alpha-hydroxysteroid dehydrogenases: key enzymes in androgen metabolism”. Best Pract. Res. Clin. Endocrinol. Metab. 15 (1): 79–94.
Llewellyn W (2009). Anabolics. Molecular Nutrition Llc. pp. 19, 163.
Blume-Peytavi U, Whiting DA, Trüeb RM (26 June 2008). Hair Growth and Disorders. Springer Science & Business Media. pp. 161–162.
Azzouni F, Mohler J (2012). “Role of 5α-reductase inhibitors in benign prostatic diseases”. Prostate Cancer Prostatic Dis. 15 (3): 222–30.
Azzouni F, Mohler J (2012). “Role of 5α-reductase inhibitors in prostate cancer prevention and treatment”. Urology. 79 (6): 1197–205.
Lotti F, Maggi M (28 April 2015). “Hormonal Treatment for Skin Androgen-Related Disorders”. In Katsambas A, Lotti T, Dessinioti C, D’Erme AM (eds.). European Handbook of Dermatological Treatments. Springer. pp. 1451–1464.
Wendowski, Oskar; Redshaw, Zoe; Mutungi, Gabriel (February 2017). “Dihydrotestosterone treatment rescues the decline in protein synthesis as a result of sarcopenia in isolated mouse skeletal muscle fibres”. Journal of Cachexia, Sarcopenia and Muscle. 8 (1): 48–56.
Kohtz AS, Frye CA (2012). Dissociating behavioral, autonomic, and neuroendocrine effects of androgen steroids in animal models. Methods Mol. Biol. Methods in Molecular Biology. 829. pp. 397–431.
Mozayani A, Raymon L (18 September 2011). Handbook of Drug Interactions: A Clinical and Forensic Guide. Springer Science & Business Media. pp. 656–.
Hemat RA (2004). Principles Of Orthomolecularism. Urotext. p. 426.
Grino PB, Griffin JE, Wilson JD (February 1990). “Testosterone at high concentrations interacts with the human androgen receptor similarly to dihydrotestosterone”. Endocrinology. 126 (2): 1165–72.
Wilderer PA (1 September 2010). “Bioassays for Estrogenic and Androgenic Effects of Water Constituents”. Treatise on Water Science, Four-Volume Set. Newnes. pp. 1805–.
Diamanti-Kandarakis E (1999). “Current aspects of antiandrogen therapy in women”. Current Pharmaceutical Design. 5 (9): 707–23. PMID 10495361.
von Deutsch DA, Abukhalaf IK, Lapu-Bula R (15 October 2003). “Anabolic Doping Agents”. In Mozayani A, Raymon L (eds.). Handbook of Drug Interactions: A Clinical and Forensic Guide. Springer Science & Business Media. pp. 510–.
Swerdloff RS, Wang C (October 1998). “Dihydrotestosterone: a rationale for its use as a non-aromatizable androgen replacement therapeutic agent”. Baillière’s Clinical Endocrinology and Metabolism. 12 (3): 501–6.
Kohtz AS, Frye CA (2012). Dissociating behavioral, autonomic, and neuroendocrine effects of androgen steroids in animal models. Methods Mol. Biol. Methods in Molecular Biology. 829. pp. 397–431.
Rizner TL, Lin HK, Peehl DM, Steckelbroeck S, Bauman DR, Penning TM (July 2003). “Human type 3 3alpha-hydroxysteroid dehydrogenase (aldo-keto reductase 1C2) and androgen metabolism in prostate cells”. Endocrinology. 144 (7): 2922–32.
Weiner IB, Gallagher M (2003). Handbook of Psychology, Biological Psychology. John Wiley & Sons. pp. 333–.
Schill W, Comhaire FH, Hargreave TB (26 August 2006). Andrology for the Clinician. Springer Science & Business Media. pp. 243–.
Hyde TE, Gengenbach MS (2007). Conservative Management of Sports Injuries. Jones & Bartlett Learning. pp. 1100–.
“Androstanolone Drug Profile”. Adis Insight. 4 December 2006.
Elks J (14 November 2014). The Dictionary of Drugs: Chemical Data: Chemical Data, Structures and Bibliographies. Springer. pp. 640–.
Index Nominum 2000: International Drug Directory. Taylor & Francis. January 2000. pp. 63–.
List PH, Hörhammer L (12 March 2013). Chemikalien und Drogen: Teil B: R, S. Springer-Verlag. pp. 523–.
“Drugs@FDA: FDA Approved Drug Products”. United States Food and Drug Administration. Retrieved 16 November 2016.
“Drug Product Database – Health Canada”. Health Canada. Retrieved 13 November 2016.
Blume-Peytavi U, Whiting DA, Trüeb RM (26 June 2008). Hair Growth and Disorders. Springer Science & Business Media. pp. 161–162.
Azzouni F, Mohler J (2012). “Role of 5α-reductase inhibitors in prostate cancer prevention and treatment”. Urology. 79 (6): 1197–205.
Lotti F, Maggi M (28 April 2015). “Hormonal Treatment for Skin Androgen-Related Disorders”. In Katsambas A, Lotti T, Dessinioti C, D’Erme AM (eds.). European Handbook of Dermatological Treatments. Springer.
Wendowski, Oskar; Redshaw, Zoe; Mutungi, Gabriel (February 2017). “Dihydrotestosterone treatment rescues the decline in protein synthesis as a result of sarcopenia in isolated mouse skeletal muscle fibres”. Journal of Cachexia, Sarcopenia and Muscle. 8 (1): 48–56.
Kohtz AS, Frye CA (2012). Dissociating behavioral, autonomic, and neuroendocrine effects of androgen steroids in animal models. Methods Mol. Biol. Methods in Molecular Biology. 829. pp. 397–431.
Llewellyn W (2009). Anabolics. Molecular Nutrition Llc. pp. 19, 163.
Okeigwe I, Kuohung W (2014). “5-Alpha reductase deficiency: a 40-year retrospective review”. Curr Opin Endocrinol Diabetes Obes. 21 (6): 483–7.
Heesakkers J, Chapple C, Ridder DD, Farag F (24 February 2016). Practical Functional Urology. Springer. pp. 280–.
Imperato-McGinley J, Zhu YS (2002). “Androgens and male physiology the syndrome of 5alpha-reductase-2 deficiency”. Mol. Cell. Endocrinol. 198 (1–2): 51–9.
Coutts, S. B.; Kicman, A. T.; Hurst, D. T.; Cowan, D. A. (1997-11-01). “Intramuscular administration of 5α-dihydrotestosterone heptanoate: changes in urinary hormone profile”. Clinical Chemistry. 43 (11): 2091–2098.
Imperato-McGinley J, Peterson RE, Gautier T, Sturla E (1979). “Androgens and the evolution of male-gender identity among male pseudohermaphrodites with 5alpha-reductase deficiency”. N. Engl. J. Med. 300 (22): 1233–7.
Kang HJ, Imperato-McGinley J, Zhu YS, Rosenwaks Z (2014). “The effect of 5α-reductase-2 deficiency on human fertility”. Fertil. Steril. 101 (2): 310–6.
Katz MD, Cai LQ, Zhu YS, Herrera C, DeFillo-Ricart M, Shackleton CH, Imperato-McGinley J (1995). “The biochemical and phenotypic characterization of females homozygous for 5 alpha-reductase-2 deficiency”. J. Clin. Endocrinol. Metab. 80 (11): 3160–7.
Cilotti A, Danza G, Serio M (2001). “Clinical application of 5alpha-reductase inhibitors”. J. Endocrinol. Invest. 24 (3): 199–203.
Bradbury R (30 January 2007). Cancer. Springer Science & Business Media. pp. 49–.Burchum J, Rosenthal L (2 December 2014). Lehne’s Pharmacology for Nursing Care. Elsevier Health Sciences. pp. 803–.
Bostwick DG, Cheng L (24 January 2014). Urologic Surgical Pathology. Elsevier Health Sciences. pp. 492–.
Sun J, Xiang H, Yang LL, Chen JB (2011). “A review on steroidal 5α-reductase inhibitors for treatment of benign prostatic hyperplasia”. Curr. Med. Chem. 18 (23): 3576–89.
Torres F (2015). Androgenetic, diffuse and senescent alopecia in men: practical evaluation and management. Curr. Probl. Dermatol. Current Problems in Dermatology. 47. pp. 33–44.
Check JH, Cohen R (2015). “An update on the treatment of female alopecia and the introduction of a potential novel therapy”. Clin Exp Obstet Gynecol. 42 (4): 411–5.
Blume-Peytavi U, Whiting DA, Trüeb RM (26 June 2008). Hair Growth and Disorders. Springer Science & Business Media. pp. 182, 369.
Check JH, Cohen R (2015). “An update on the treatment of female alopecia and the introduction of a potential novel therapy”. Clin Exp Obstet Gynecol. 42 (4): 411–5.
Blume-Peytavi U, Whiting DA, Trüeb RM (26 June 2008). Hair Growth and Disorders. Springer Science & Business Media. pp. 182, 369.
Fin dalla loro comparsa nel mercato della supplementazione sportiva “border line” , i SARM hanno goduto fin da subito di una enorme popolarità portata più dalle promesse che i produttori elencavano nelle descrizioni dei loro prodotti che da veri e propri dati di fatto. La “principale promessa” connessa a questa classe di molecole (classe più complessa di quanto comunemente inteso e non del tutto separata dagli AAS) era quella di apportare tutti i benefici degli AAS senza il carico di effetti collaterali ad essi legati. Questa è risultata una affermazione non vera, almeno in buona parte. Personalmente, ho svolto ricerche e studi per molto tempo, un tempo sufficiente per constatare tramite letteratura e sperimentazione che gli effetti collaterali tipici degli AAS continuano a persistere anche nei SARM. Non solo, paradossalmente il loro uso in monoterapia è assai più problematico, per certi versi, di quello che potrebbe essere con l’uso del vecchi Testosterone. Ma il tema di questo articolo è più specifico e si riferisce alla epatotossicità potenziale di due SARM, il RAD-140 ed LGD-4033. Recentemente è stato pubblicato su Epatology Communications un articolo nel quale un equipe australiana ha descritto due casi di danno epatico riscontrati in uomini che avevano usato RAD 140 e LGD4033.[1] Purtroppo, i casi esaminati presentano più variabili che ne impediscono una corretta valutazione del potenziale negativo a livello epatico dei composti in questione.
Ma andiamo per ordine ….
Casi studio e approfondimenti sulla questione
RAD-140
Il primo caso riguarda un uomo di 49 anni che aveva assunto RAD 140 per 5 settimane consecutive. I ricercatori hanno analizzato il contenuto del “supplemento” nel loro laboratorio constatando un effettivo contenuto di RAD 140. Non sono stati trovati contaminanti. L’uomo era sottoposto a terapia antidepressiva a base di anche usato la Venlafaxina, un antidepressivo SSRI che può interferire con la funzionalità epatica in alcune soggetti particolarmente sensibili.[2]
Dopo che l’uomo ha sviluppato ittero e prurito, ha cercato assistenza medica. L’analisi del sangue ha mostrato che la concentrazione dei marker ematici, legati alla valutazione della salute epatica, era sensibilmente aumentata. L’analisi ha suggerito che l’uomo soffriva di colestasi, un classico effetto collaterale da abuso di AAS orali metilati in C-17.
L’esame dei campioni di tessuto epatico ha mostrato che i tubi che drenano i sali biliari nell’intestino erano gravemente compromessi. Il corpo si libera dei prodotti di scarto liposolubili attraverso i sali biliari. Inoltre, i campioni di tessuto epatico erano infiammati e danneggiati.
I dottori hanno impedito all’uomo di assumere le sue medicine e gli hanno somministrato Ursodiolo e Colestiramina. Il suo fegato si riprese nei mesi seguenti. Un anno dopo, tutti i suoi valori erano tornati alla normalità.
LGD-4033
Il secondo caso descritto dagli australiani è quello di un uomo di 24 anni che aveva assunto LGD-4033 per 9 settimane. I ricercatori hanno anche analizzato anche questo “supplemento” trovando effettivamente LGD-4033, e nessun contaminante.
L’uomo si era ammalato una settimana dopo la fine del suo ciclo, ma solo dopo 3 settimane si recò dal medico. Aveva l’ittero, aveva la nausea, mangiava a malapena e aveva già perso 5 chili.
L’uomo non stava usando nessun altro farmaco. Tuttavia, una volta al mese era solito abbandonare lo stile di vita controllato e consumava molto alcol. Nel suo sangue, i ricercatori hanno trovato una maggiore concentrazione di enzimi che indicano danni al fegato. Il fegato dell’uomo aveva dimensioni più grandi della norma, ma secondo una scansione, non c’erano prove di un ridotto drenaggio dei sali biliari.
I ricercatori hanno deciso di aspettare e hanno visto che il fegato si era ripreso 4 mesi dopo.
I ricercatori concludono il loro articolo affermando che la lesione epatica in entrambi i casi rientra nello spettro descritto della lesione epatica associata agli steroidi anabolizzanti. Questi effetti off-target mettono in discussione la cosiddetta selettività tissutale dei SARM, che è stato il loro principale punto di forza.
Resta da vedere se questi casi rappresentino la punta dell’iceberg, ma data la crescente popolarità dell’uso dei SARM, è necessaria una maggiore vigilanza e segnalazione di potenziali casi.
Come abbiamo visto, in entrambi i casi sono presenti delle variabili che non permettono una piena valutazione del potenziale epatotossico delle due molecole. Nel primo caso la variabile era rappresentata dalla Venlafaxina, mentre nel secondo caso era l’abuso di alcool.
Ciò non significa che i due SARM non abbiano un possibile impatto epatico negativo. Studi controllati hanno mostrato infatti una epatotossicità potenziale del RAD-140 dose-dipendente.[3] Mentre con LGD-4033, a dose entro 1mg/die, non hanno osservato alterazioni significative i ALT e AST, sebbene, anche in questo caso, il problema aumenta in modo dose-dipendente.[4] Giova sottolineare che in ambito sportivo, è abbastanza comune per i soggetti di sesso maschile utilizzare dai 10 ai 20mg/die di LGD-4033, mentre le atlete rimangono nel range tra 5 e 10mg/die.
Questi dosaggi sono stati determinati dagli utilizzatori sulla base di informazioni aneddotiche e sperimentazione personale e non rappresentano in alcun modo delle linee guida che determinano un uso corretto o errato della molecola in questione.
Nota: il dosaggio più elevato di LGD-4033 utilizzato negli studi clinici di fase 2 è stato di 2mg al giorno a fini terapeutici.[5]
Come prendere questi dati? In modo costruttivo, ovviamente. Considerate i SARM come un qualsiasi altro farmaco, valutando concretamente il lato “positivo” ed il lato “negativo”, e non come una sorta di “pillola rossa” in grado di darvi la visione e concretizzazione della soluzione definitiva alle vostre smanie estetiche.
Da qualche tempo stanno circolando teorie secondo le quali l’Ibutamoren (comunemente noto come MK-677) può causare danni cerebrali per via dello stimolo cronico dei recettori della Grelina (bersaglio della molecola in questione).
Ma è mai stato osservato un effetto simile sull’uomo negli studi presenti svolti su esseri umani? No, e non risulta nulla di simile nemmeno dai dati aneddotici provenienti dagli utilizzatori, almeno per il momento.
Allora da cosa nasce la teoria? Nasce dai dati provenienti da test di laboratorio svolti su topi. Ed in effetti, i dati provenienti da questi test mostrano collegamenti tra esposizione cronica allo stimolo dei recettori della Grelina e esiti negativi a livello cerebrale.
Ma approfondiamo la questione…
MK-677 e aumento della paura nei ratti.
Uno studio sui ratti ha valutato se l’MK-677 possa causare un aumento dei livelli di paura.[1]
Esistono prove a sostegno del fatto che uno dei modi in cui la Grelina viene modulata è attraverso l’esposizione allo stress.
Ciò che questo studio intendeva valutare specificamente era se i ratti avessero una maggiore probabilità di sperimentare la paura e il disturbo da stress post-traumatico (PTSD) con livelli cronicamente alti di Grelina, situazione “mimata” artificialmente 24/7 con l’utilizzo di un agonista del recettore della Grelina (MK-677).
Ai ratti veniva somministrato in cronico l’MK-677 e nello stesso tempo venivano costantemente spaventati. I ricercatori valutavano quindi la loro differenza di risposta (gruppo trattato con MK-677 Vs. gruppo non trattato).
È stato quindi scoperto che i ratti del gruppo trattato con MK-677 avevano hanno migliorato la memoria della paura rispetto al basale.
Come ho già detto, ci sono prove a sostegno del fatto che uno dei modi in cui la Grelina viene modulata è attraverso l’esposizione allo stress, e questo studio è stato in grado di suscitare un po’ di controversie nella sottocultura del BodyBuilding con i suoi risultati che mostrano un effetto palesemente negativo sull’amigdala in cui gli effetti di una sovrastimolazione del recettore della grelina porta ad un della paura.
La Grelina e l’Ormone della Crescita agiscono insieme nell’amigdala per aumentare la percezione della paura, almeno come osservato in questo modello di roditori.
Si noti che in questo studio, i ratti sono stati trattati con iniezioni intraperitoneale (I.P.) di 1ml/kg.
Nel caso non lo sapeste, una iniezione intraperitoneale consiste nell’iniezione di una sostanza nel peritoneo (cavità corporea).Il peritoneo è la membrana sierosa che forma il rivestimento della cavità addominale. Ed è qui che l’MK-677 è stato somministrato per la prima volta per valutare se una maggiore attivazione del recettore della Grelina è sufficiente per migliorare la memoria della paura.
Dopo aver stabilito che l’attivazione del recettore della Grelina è effettivamente sufficiente per migliorare la memoria della paura, hanno quindi continuato a infondere MK-677 direttamente nel complesso basolaterale dell’amigdala per determinare se l’attivazione ripetuta del recettore della Grelina nel complesso basolaterale dell’amigdala sia sufficiente per migliorare la memoria della paura.
Ripetute infusioni intra-amigdala miglioravano la memoria della paura.[2]
Ora, mentre tutto ciò dipinge un quadro piuttosto negativo dell’MK-677, bisognerebbe tener presente che questo studio ha anche dimostrato che una singola iniezione di un agonista del recettore della Grelina non era sufficiente per aumentare la paura, e persino l’agonismo cronico del recettore della Grelina non alterare la locomozione, l’ansia innata o l’espressione di ricordi di paura acquisiti in precedenza.[2]
Quanto possono interessare l’essere umano i risultati ottenuti?
Negli studi effettuati sull’uomo, l’MK-677 è stato osservato essere ben tollerato per oltre un anno consecutivo di utilizzo di una dose pari a 25mg/die assunta per via orale.
Gli utilizzatori ovviamente non si somministrano cronicamente il composto nella cavità peritoneale o nel cervello.
Questa scoperta non è stata ancora replicata nelle prove umane, e nemmeno nei resoconti aneddotici fino ad oggi.
Detto ciò, la ricerca sugli esseri umani sta indagando su questo particolare effetto?
Probabilmente no, e ovviamente quelli che usano l’MK-677 a livello ricreativo non vengono sottoposti di norma a scansioni cerebrali prima e dopo l’utilizzo del composto.
Molto probabilmente il problema potrebbe emergere se l’uso fosse protratto nel cronico, per anni. Non per nulla esistono meccanismi di autoregolazione nel corpo per una ragione ben specifica.
Anche se non vi fosse alcun rischio neurologico, bisogna ricordare che l’MK-677 causa un peggioramento della sensibilità all’insulina, fattore di certo non da trascurare e da gestire con cicli temporalmente non eccessivi e l’arginamento di tale conseguenza iatrogena con “composti tampone” quali Berberina o Metformina. L’effetto sulla sensibilità all’insulina è certamente una preoccupazione più immediata di qualsiasi altra affermazione estrapolata da studi sui roditori che mostrano i suoi possibili effetti sulla salute del cervello.
Androgenico: 22-55
Anabolico: 100-200
Standard: Methyltestosterone (orale)
Nome Chimico: 17α-Ethylestr-4-en-17β-ol-3-one; 17α-Ethyl-19-norandrost-4-en-17β-ol-3-one.
Attività estrogenica: alta
Attività Progestinica: alta
Aromatizzazione: si
Il Norethandrolone, noto anche come 17α-Ethyl-19-nortestosterone o 17α-Ethylestr-4-en-17β-ol-3-one; 17α-Ethyl-19-norandrost-4-en-17β-ol-3-one, conosciuto con i nomi commerciali di Nilevar e Pronabol, è uno steroide androgene anabolizzante (AAS), disponibile in forma orale, utilizzato in campo clinico come agente per la promozione della crescita muscolare, per curare gravi ustioni, traumi fisici e per il trattamento dell’anemia aplastica, anche se attualmente non è quasi più utilizzato in medicina.[1][2][3] Tuttavia, è ancora disponibile in Francia.[2][3]
Il Norethandrolone è uno steroide estrano sintetico e un derivato metilato in C17 del 19-nortestosterone (Nandrolone).[1][2] È strettamente correlato al Normethandrone (17α-metil-19-nortestosterone) e all’Etilestrenolo (3-deketo-17α-etil-19-nortestosterone).[1][2] In letteratura sono state pubblicate le vie di sintesi chimica del Norethandrolone.[4]
Il Norethandrolone è un AAS con un alto rapporto tra attività anabolica e androgena.[2] Analogamente al caso del nandrolone e del 5α-diidronandrolone, e del 5α-diidronoretandrolone, il metabolita 5α-ridotto del Norethandrolone, mostra una ridotta affinità per il recettore degli androgeni rispetto alla molecola progenitrice.[2][5] Ciò è probabilmente correlato all’alto rapporto tra attività anabolica e androgena osservata con il Norethandrolone.[2][5] Il Norethandrolone ha un’attività estrogenica relativamente elevata per via della sua conversione ad opera dell’enzima Aromatasi in Etilestradiolo.[2] Ha anche una forte attività progestinica.[2] La potenza progestinica del Norethandrolone è simile a quella del Noretisterone in termini di alterazioni dell’endometrio nelle donne.[6-11] Inoltre, il Norethandrolone è epatotossico per via della metilazione in C17.[2]
Il Norethandrolone è stato uno dei primi AAS orali disponibili negli Stati Uniti. Era essenzialmente la risposta della Searles all’AAS di nuova creazione da parte della svizzera Ciba, il Methandrostenolone (Dianabol), che venne rilasciato sul mercato lo stesso anno. In effetti, spesso venne usato il Methandrostenolone come paragone per valutare l’impatto del Norethandrolone sull’aumento di peso, sull’anabolismo e sulla ritenzione idrica.
Sette anni prima del rilascio del Norethandrolone, la Mayo Clinic aveva annunciato l’efficacia drammatica del Cortisone nel trattamento dell’artrite reumatoide. Questo a sua volta ha stimolato un enorme interesse per tutti gli aspetti della chimica degli steroidi, dell’endocrinologia e dei campi correlati. G. D. Searle & Co. ha prontamente avviato un grande sforzo nella ricerca sugli steroidi, con l’obiettivo di scoprire composti steroidei migliori di quelli precedentemente disponibili e che avessero potuto essere utilizzati per condizioni per le quali non erano disponibili altri composti.
Questo sforzo portò all’introduzione del Norethandrolone, commercializzato nel 1956 come Nilevar, il primo agente anabolizzante con una separazione favorevole tra effetti anabolizzanti e androgeni.[7] Per i motivi sopra esposti, negli uomini si riscontrano solo effetti androgeni deboli (probabilmente perché la forma derivante dalla 5-alfa-riduzione ha un ridotto potenziale di legame con i AR), sebbene nelle donne la virilizzazione rimanga una possibilità con l’uso di questa molecola.
Questo AAS è generalmente sconsigliato nelle atlete, non solo per le contenute possibilità di comparsa di effetti androgeni, ma anche per alcuni problemi di infertilità, che sono possibili anche nelle donne.[8][9] L’effetto anabolico di questo farmaco è moderato, e ciò è probabilmente dovuto al suo legame moderatamente forte con il recettore degli androgeni (questo lo rende abbastanza diverso dal Methandrostenolone, che ha uno scarso legame con il recettore degli androgeni agendo per via Non-genomica) e alla sua capacità di stimolare la sintesi proteica e a bloccarne il catabolismo.[10] Il Nilevar fu il primo AAS sviluppato dalla Searles , e fu proprio questo farmaco che alla fine portò alla ricerca e allo sviluppo dell’Oxandrolone (Anavar), molto meno androgeno, molto più anabolizzante e privo di attività estrogenica e progestinica, portando al conseguente declino della popolarità e dell’uso del Nilevar.
Nell’immagine è possibile osservare le due vie attraverso le quali un AAS può svolgere la sua azione a livello cellulare (Azione Genomica e Azione Non-Genomica.
Come risaputo, gli AAS, a diverso grado di impatto, possono avere effetti deleteri sul colesterolo sierico. Il Norethandrolone non è da meno presentando la tendenza a causare una riduzione delle concentrazioni di colesterolo HDL (“buono”) e un aumento delle concentrazioni di colesterolo LDL (“cattivo”), cosa che comporta uno sbilanciamento dell’equilibrio HDL/LDL che si traduce in un rischio maggiore di sviluppare arteriosclerosi. L’impatto relativo all’assunzione di un AAS nei confronti dei lipidi ematici dipende dalla dose, dalla via di somministrazione (per via orale o iniettabile), dal tipo di steroide (aromatizzabile o non aromatizzabile), e dal livello di resistenza al metabolismo epatico.
Essendo il Norethandrolone un AAS con ridotta attività androgenica, la soglia dei possibili forti effetti collaterali androgenici è generalmente bassa ed è paragonabile a quella riscontrata con l’uso di Nandrolone sebbene dosaggi più alti mostrano un aumento della possibilità che tali effetti si manifestino.
Come per gli altri AAS 17α-alchilati, il Norethandrolone presenta un elevato grado di epatotossicità.[1][2]
17-alpha-methyl-estradiolo
Il Norethandrolone è un substrato soggetto all’azione dell’enzima Aromatasi, per la precisione aromatizza in una forma di estradiolo dall’elevato potere biologico, il 17-alpha-methyl-estradiolo e, potenzialmente, 17-alpha-methyl-estrone.[1][2] Questo incrementa le caratteristiche del Norethandrolone come potenziale steroide per una fase “Bulk” grazie alla maggior proliferazione dei recettori degli estrogeni, un miglior utilizzo del glucosio, e un aumentata secrezione di GH e IGF-1. Ovviamente, con il suo uso il fattore estrogenico deve essere attentamente monitorato in quanto effetti collaterali legati all’iperestrogenemia o all’aumentata attività tissutale del Estradiolo (comune con l’uso di progestinici) quali ginecomastia, ritenzione idrica e l’accumulo di grasso con modello femminile sono piuttosto comuni. L’uso di un inibitore dell’aromatasi e di un SERM, dopo comprovata necessità, in soggetti sensibili all’aromatizzazione e/o all’attività estrogenica è risultata una pratica funzionale.
Il Norethandrolone si lega fortemente al recettore del progesterone (PR).[1][2] Dato il noto vincolo del Norethandrolone con il PR, e la tendenza da parte dei farmaci derivati dal Nandrolone di offrire almeno qualche attività progestinica, una significativa attività in tal senso, con gli effetti ad essa legati, non deve essere trascurata per tutto il tempo di assunzione.
Si ricordi inoltre che gli effetti collaterali associati con il Progesterone sono simili a quelli degli estrogeni, compreso il feedback negativo di inibizione della produzione di Testosterone e una maggiore velocità di accumulo di grasso. I progestinici aumentano anche l’effetto stimolante degli estrogeni sulla crescita del tessuto mammario. Sembra che ci sia una forte sinergia tra questi due ormoni, in modo tale che la ginecomastia potrebbe anche verificarsi con l’aiuto dei progestinici, senza eccessivi livelli di estrogeni. L’uso di un AI e/o SERM, che inibisca la componente estrogenica di questa alterazione, come accennato in precedenza, è spesso sufficiente per mitigare la ginecomastia causata da Nandrolone e derivati. Il rialzo della Prolattina è un altra possibile conseguenza da monitorare per via di esami ematici.
L’uso di anti-prolattinici andrebbe presa in considerazione solo quando, attraverso appositi esami ematici, si è appurata una iperprolattinemia.
La soppressione dell’attività dell’Assen HPT con questo composto è piuttosto marcata. Infatti, è risaputo che i progestinici rientrano nella categoria dei composti con attività soppressiva delle gonadotropine.[11]
Tamoxifene
Nota: l’uso del Tamoxifene (Nolvadex) potrebbe risultare controproducente, soprattutto nel lungo periodo, dal momento che potrebbe effettivamente portare ad un aumento dei recettori del progesterone.[12]
Il lato positivo dell’essere un 19-norsteroide si manifesta con la buona affinità di legame con i recettori androgeni, cosa che è positivamente correlata con la lipolisi (per via del legame con i AR sull’adipocita).[13]
I soggetti con dolori articolari che utilizzano il Norethandrolone possono sperimentare una riduzione del sintomo similmente a quanto osservato con l’uso di Nandrolone. Anche per questa ragione, in passato il Nilevar era stato soprannominato “Oral Deca”. Ovviamente, l’uso sul lungo termine del Norethandrolone, per via della sua metilazione in C17, anche per tale fine è caldamente sconsigliato. Comunque, data la sua attività positiva a livello articolare, il suo uso ha visto una certa diffusione anche nel PowerLifting. Nel BodyBuilding, anche per tale caratteristica divenne una scelta inseribile con vantaggio durante cicli di “Bulk”
A differenza del Nandrolone, i cui metaboliti sono rilevabili nelle urine fino a 18 mesi dalla sospensione della somministrazione, il Norethandrolone presenta una rilevabilità d’uso di circa 5 mesi.
Le linee guida di prescrizione per il Norethandrolone (deperimento muscolare [14], pazienti con gravi ustioni, grave trauma e alcune forme di anemia aplastica [2]) indicano un dosaggio giornaliero pari a 20 o 30mg. La somministrazione indicata consiste in uno schema intermittente composto da 12 settimane d’uso seguite da un mese di pausa a seguito del quale può essere riavviato il trattamento. Il Norethandrolone è stato studiato per l’uso come contraccettivo ormonale maschile.[15][16][17] Nelle indicazioni cliniche non vi è menzione di modalità specifiche per l’uso nelle donne. Il dosaggio usuale in ambito sportivo è compreso tra i 20mg e i 40 mg al giorno, anche se il dosaggio massimo generalmente utilizzato risulta essere di 50mg, suddiviso in due assunzioni distanziate da circa 12h, assunte per non più di 6-8 settimane. Le atlete utilizzano dosi pari a 5-10mg/die suddivise sempre in due assunzioni distanziate da circa 12h. Il tempo tra una somministrazione e la successiva è basato sull’emivita della molecola (appunto 12h). Tale timing garantisce il mantenimento di livelli ematici della molecola stabili nelle 24h.
Essendo il Norethandrolone un AAS aromatizzabile e progestinico può causare ritenzione idrica, accumulo di grasso e ginecomastia, in specie se il fattore estrogenico non viene adeguatamente gestito. Queste caratteristiche lo rendono un AAS favorevole da utilizzare durante un ciclo “Bulk” o pre-gara se si è PowerLifter.
Sebbene negli ultimi anni sembra essere tornata di moda tra i culturisti, sempre a causa di quel compulsivo desiderio di provare nuove molecole (sebbene di “nuovo”, in questo caso, non vi sia nulla), la sua reperibilità è molto limitata. Come già detto, è attualmente presente nel mercato farmaceutico francese (venduto in compresse da 10mg). Nel mercato nero, la presenza di questo AAS è minima e spesso prodotti con indicazione in etichetta riportante la presenza di questa molecola in realtà ne contengono un altra, sebbene i costi di produzione siano bassi.
In conclusione, si tratta di una molecola non semplice da gestire, dall’applicabilità limitata e dalla difficile reperibilità. Non rappresenta di certo una molecola dai vantaggi superiori rispetto al Methandrostenolone o all’Oxymetholone, entrambe molecole applicabili in contesti simili a quelli nei quali l’uso del Norethandrolone è più indicato.
Gabriel Bellizzi
Riferimenti:
1- J. Elks (14 November 2014). The Dictionary of Drugs: Chemical Data: Chemical Data, Structures and Bibliographies. Springer. pp. 885–.
2- William Llewellyn (2011). Anabolics. Molecular Nutrition Llc. pp. 575–583.
4- Springer-Verlag. 27 November 2013. pp. 13, 282–283. ISBN 978-3-642-99941-3.
5- Bergink EW, Geelen JA, Turpijn EW (1985). “Metabolism and receptor binding of nandrolone and testosterone under in vitro and in vivo conditions”. Acta Endocrinol Suppl (Copenh). 271: 31–7.
6- Boschann HW (July 1958). “Observations of the role of progestational agents in human gynecologic disorders and pregnancy complications”. Ann. N. Y. Acad. Sci. 71 (5): 727–52.
7- Steroids. 1992 Dec;57(12):624-30.
8- J Reprod Fertil. 1966 Dec;12(3):489-99
9- Contraception. 1975 Feb;11(2):193-207
10- Lancet. 1958 Oct 25;2(7052):885-6
11- Clin Endocrinol (Oxf) 2003 Apr;58(4):506-12
12- Gynecol Oncol. 1999 Mar;72(3):331-6
13- Xu X, et al. “The effects of androgens on the regulation of lipolysis in adipose precursor cells.” Endocrinology 1990 Feb;126(2):1229
14- Walter Sneader (23 June 2005). Drug Discovery: A History. John Wiley & Sons. pp. 206–. ISBN 978-0-471-89979-2.
15- Schearer, S. Bruce; Alvarez-Sanchez, Francisco; Anselmo, Jose; Brenner, Paul; Coutinho, Elsimar; Latham-Faundes, Anibal; Frick, Julian; Heinild, Bent; Johansson, Elof D. B. (1978). “Hormonal Contraception for Men”. International Journal of Andrology. 1 (s2b): 680–712.
16- Neumann F, Diallo FA, Hasan SH, Schenck B, Traore I (1976). “The influence of pharmaceutical compounds on male fertility”. Andrologia. 8 (3): 203–35.
17- Brenner PF, Bernstein GS, Roy S, Jecht EW, Mishell DR (February 1975). “Administration of norethandrolone and testosterone as a contraceptive agent for men”. Contraception. 11 (2): 193–207.
Androgenico: 78-254
Anabolico: 107 (valore sovrastimato)
Standard: Testosterone
Nome Chimico: 17α-Methyl-4,5α-dihydrotestosterone
Attività estrogenica: nessuna
Attività Progestinica: nessuna
Aromatizzazione: no
Il Mestanolone, noto anche come 17α-methyl-4,5α-Diidrotestosterone (17α-methyl-DHT) o 17α-methyl-5α-androstan-17β-ol-3-one, è uno steroide androgeno anabolizzante (AAS), commercializzato in forma orale sotto il nome di Androstalone e Ermalone, ad oggi per lo più in disuso in ambito medico.[1][2][3][4]
Il Mestanolone è uno steroide androstano sintetico e un derivato del Diidrotestosterone (DHT) metilato in posizione C17α. [1][4] Infatti, differisce dal DHT solo per la presenza del gruppo metile nella posizione C17α.[1][4] Stretti parenti sintetici del Mestanolone includono Mesterolone (1α-Methyl-4,5α-dihydrotestosterone), Oxandrolone (2-oxa-17α-methyl-DHT), Oxymetholone (2-idrossimetilene-17α-methyl-DHT) e Stanozololo (un derivato del 17α-methyl-DHT (Mestanolone) con un anello pirazolico fuso con l’anello A.)[1][4]
Differenze nella struttura dello scheletro carbossilico tra Mestanolone (metilazione in C17α) e Mesterolone (metilazione in C1α).
Il Mestanolone fu sintetizzato per la prima volta nel 1935 insieme al Methyltestosterone e al Methandriolo.[5][6] È stato sviluppato dalla Roussel negli anni ’50 ed è stato introdotto per uso medico, con i marchi Androstalone ed Ermalone, intorno al 1960.[4][10][8] Venne inizialmente commercializzato in Germania.[4] Inizialmente si pensava che il farmaco fosse un potente agente anabolizzante, ma le ricerche successive hanno dimostrato che in realtà ha effetti anabolici relativamente deboli ed esplica principalmente azione androgena.[4] Il Mestanolone, insieme al molto più conosciuto 4-Chlorodehydromethyltestosterone (Oral Turinabol) è stato utilizzato come agente dopante negli atleti olimpionici della Germania Orientale all’interno di un programma di doping sponsorizzato dallo stato negli anni ’70 e ’80.[4] Il motivo del suo uso, come precedentemente accennato, era giustificato maggiormente dal suo valore androgeno piuttosto che su quello anabolizzante. Il suo uso garantiva un ottima risposta neuro steroidea con conseguente stimolazione del Sistema Nervoso Centrale e migliorata interazione neuromuscolare, con vantaggi nella velocità, nella forza, nell’aggressività, nella concentrazione, nella resistenza fisica e allo stress mentale.[4] Oggi, l’uso del Mestanolone è stato per lo più sospeso in medicina, sebbene sia ancora disponibile in Giappone.[2][3][4] Il Mestanolone era comunemente disponibile sotto forma di compresse sublinguali da 25mg (marchio Ermalone).[7]
Il Mestanolone è un AAS con effetti molto simili all’Androstanolone (diidrotestosterone; DHT) essendo praticamente una versione orale di quest’ultimo.[4] A causa dell’inattivazione da parte della 3α-idrossisteroide deidrogenasi (3α-HSD) nel muscolo scheletrico, il Mestanolone, sebbene dotato di una metilazione in posizione C-17 la quale ne migliora la stabilità del legame recettoriale, è descritto come un agente anabolizzante molto scarso, analogamente all’Androstanolone e al Mesterolone.[4] Poiché il Mestanolone è un composto 5α ridotto, non è un substrato soggetto all’enzima aromatasi e. quindi, non convertendo in estrogeno oltre a non possedere attività estrogenica intrinseca.[4] Inoltre, il farmaco non ha attività progestinica.[4] Come per gli altri AAS 17α-alchilati, il Mestanolone presenta un certo grado di epatotossicità.[4]
Differenze nella struttura dello scheletro carbossilico tra Diidrotestosterone e Mestanolone (aggiunta metilazione in C17α).
Come risaputo, gli AAS, a diverso grado di impatto, possono avere effetti deleteri sul colesterolo sierico. Il Mestanolone non è da meno presentando la tendenza a causare una riduzione delle concentrazioni di colesterolo HDL (“buono”) e un aumento delle concentrazioni di colesterolo LDL (“cattivo”), cosa che comporta uno sbilanciamento dell’equilibrio HDL/LDL che si traduce in un rischio maggiore di sviluppare arteriosclerosi. L’impatto relativo all’assunzione di un AAS nei confronti dei lipidi ematici dipende dalla dose, dalla via di somministrazione (per via orale o iniettabile), dal tipo di steroide (aromatizzabile o non aromatizzabile), e dal livello di resistenza al metabolismo epatico.
Essendo il Mestanolone un AAS con consistente attività androgenica, la soglia dei possibili forti effetti collaterali androgenici è generalmente alta ed è paragonabile a quella riscontrabile con altri composti come il Mesterolone. Per questa ragione, il suo uso in ambito femminile non è stato molto diffuso dal momento che poteva essere causa di severi effetti virilizzanti.
Quando l’uso del Mestanolone veniva applicato in campo sportivo, i dosaggi comunemente utilizzati erano mediamente tra i 10 ed i 20mg al giorno con un timing di somministrazione tra una dose e la successiva di 12 ore. I tempi di utilizzo rimanevano entro le 6-8 settimane onde evitare di creare un eccessivo stress epatico. Tra i bodybuilder, il Mestanolone era spesso inserito durante la preparazione alla gara vista la sua facile gestibilità non causando ritenzione idrica, non essendo soggetto ad aromatizzazione, esercitando una blanda azione anti-estrogenica (sia recettoriale che come ligando inibitorio dell’enzima Aromatasi) e possedendo una buona azione lipolitica (legame con i AR adipocitari).
Come già detto in precedenza, l’uso del Mestanolone è stato per lo più sospeso in ambito medico anche se rimane disponibile in Giappone.[2][3][4] Nel mercato nero è raramente reperibile per via della sua attuale e pressoché assente richiesta tra gli atleti.
Gabriel Bellizzi
Riferimenti:
1- J. Elks (14 November 2014). The Dictionary of Drugs: Chemical Data: Chemical Data, Structures and Bibliographies. Springer. pp. 775.
2- Index Nominum 2000: International Drug Directory. Taylor & Francis. 2000. pp. 655.
4- William Llewellyn (2017). Anabolics 11th. Molecular Nutrition Llc. p. 284-285-286.
5- Schänzer W (1996). “Metabolism of anabolic androgenic steroids”. Clin. Chem. 42 (7): 1001–20.
6- Ruzicka, L.; Goldberg, M. W.; Rosenberg, H. R. (1935). “Sexualhormone X. Herstellung des 17-Methyl-testosterons und anderer Androsten- und Androstanderivate. Zusammenhänge zwischen chemischer Konstitution und männlicher Hormonwirkung”. Helvetica Chimica Acta. 18 (1): 1487–1498.
7- H.-L. Krüskemper (22 October 2013). Anabolic Steroids. Elsevier. pp. 196.

















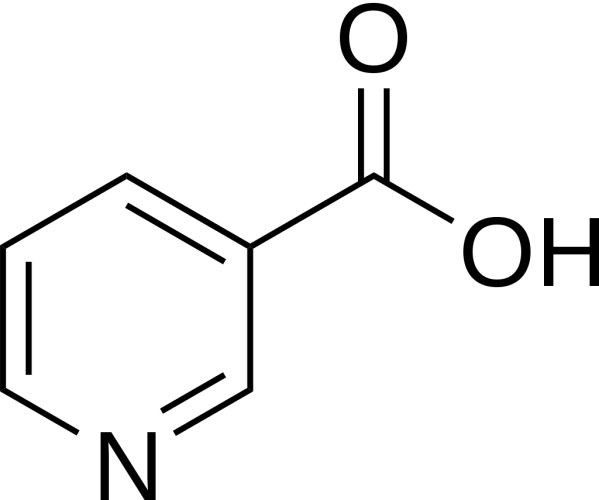
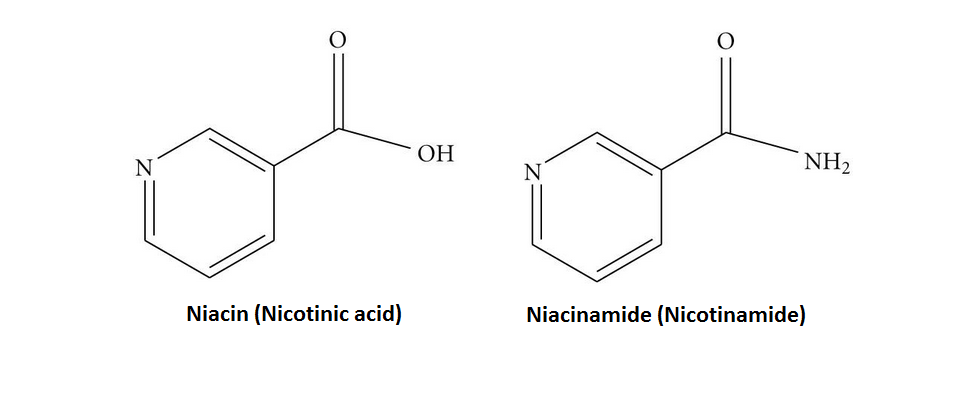


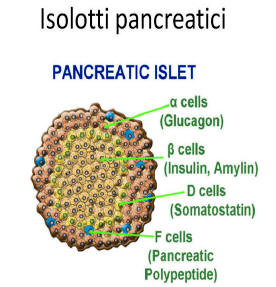
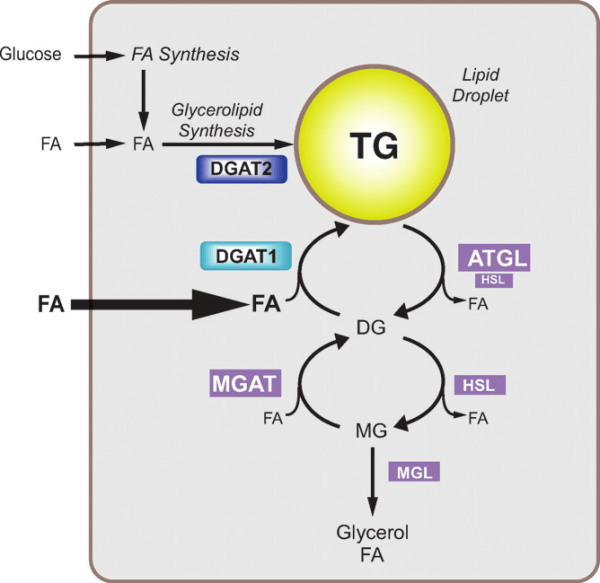
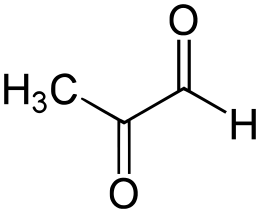
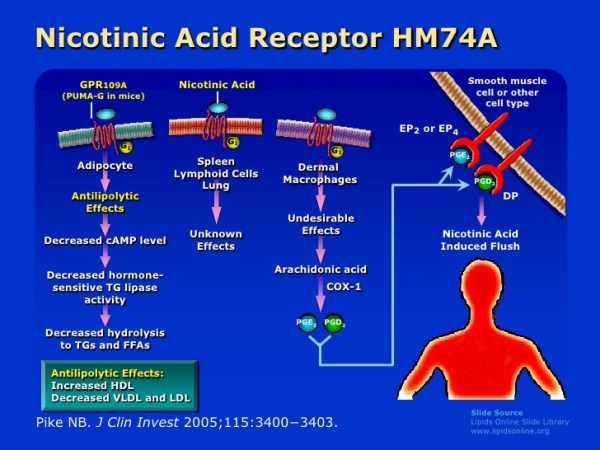
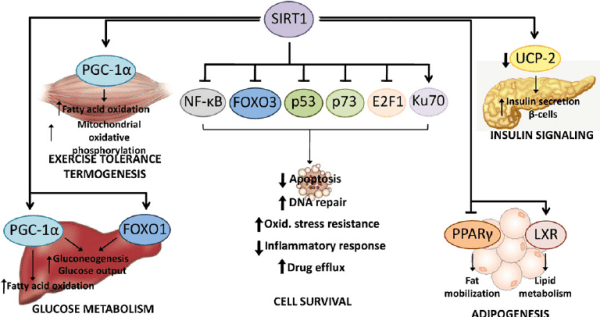
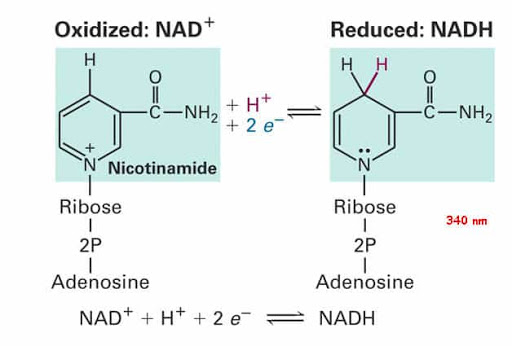
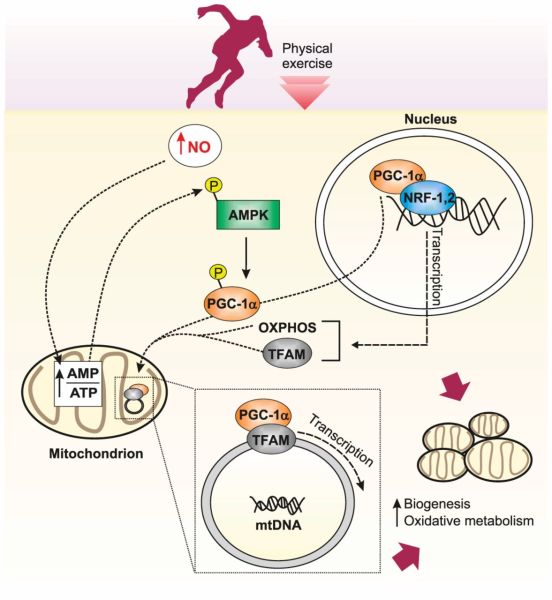
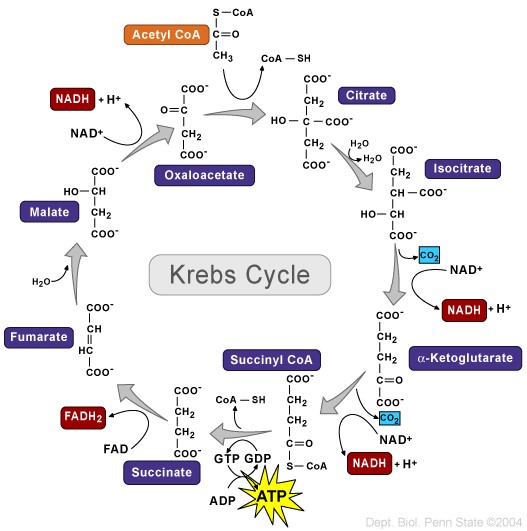

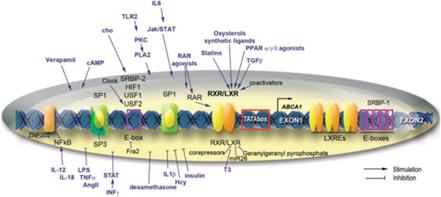



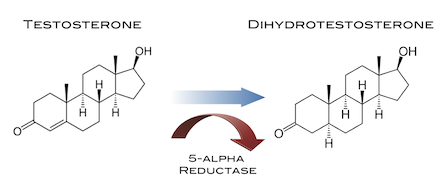
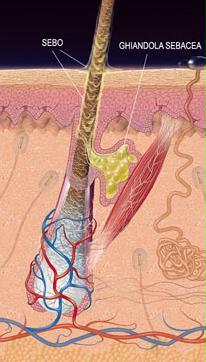

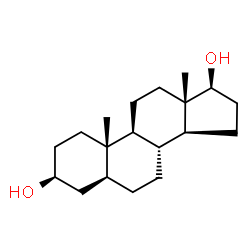


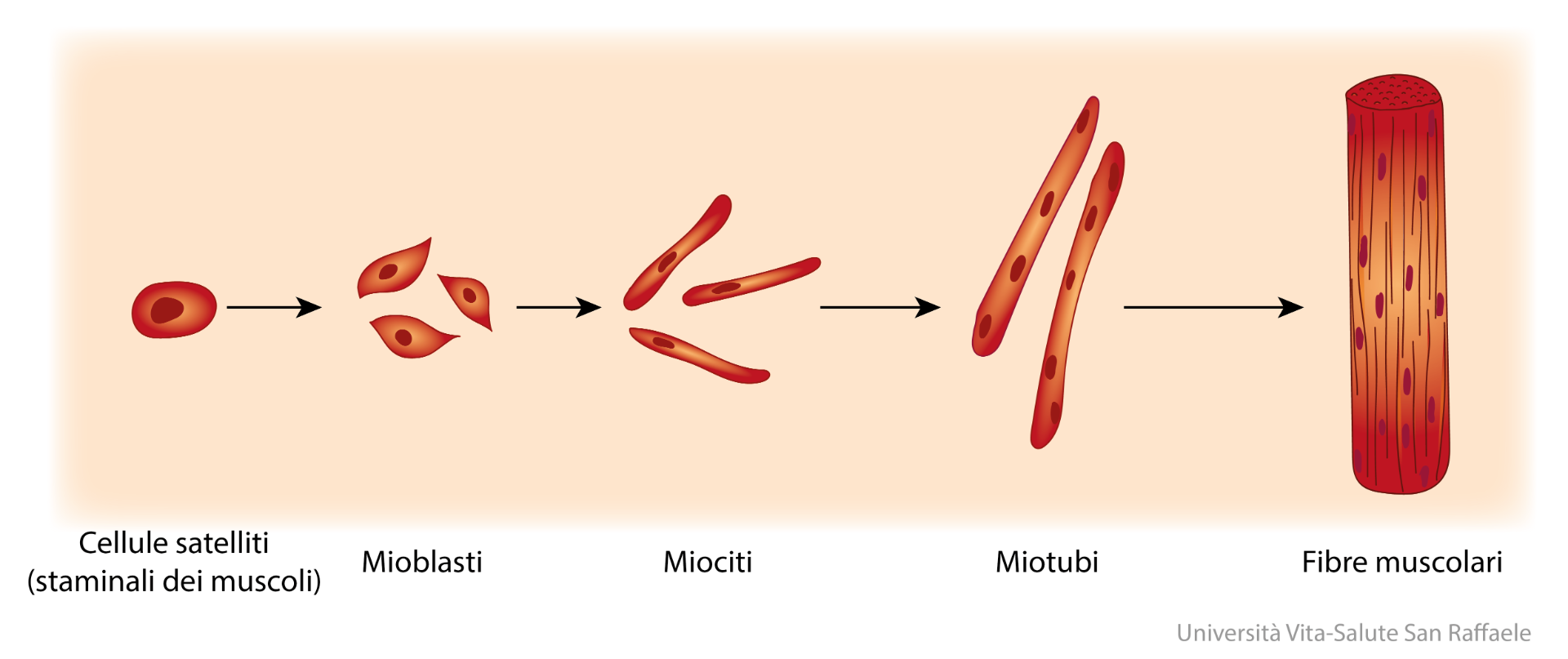

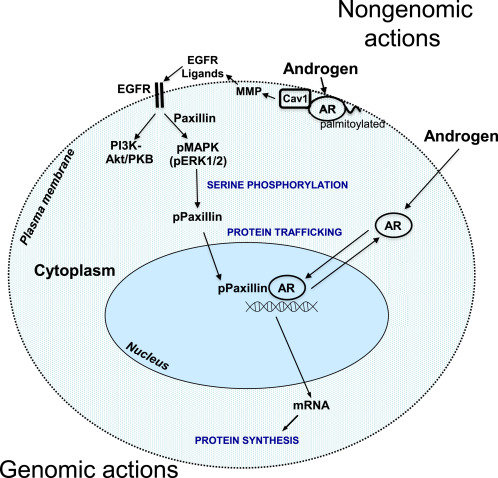





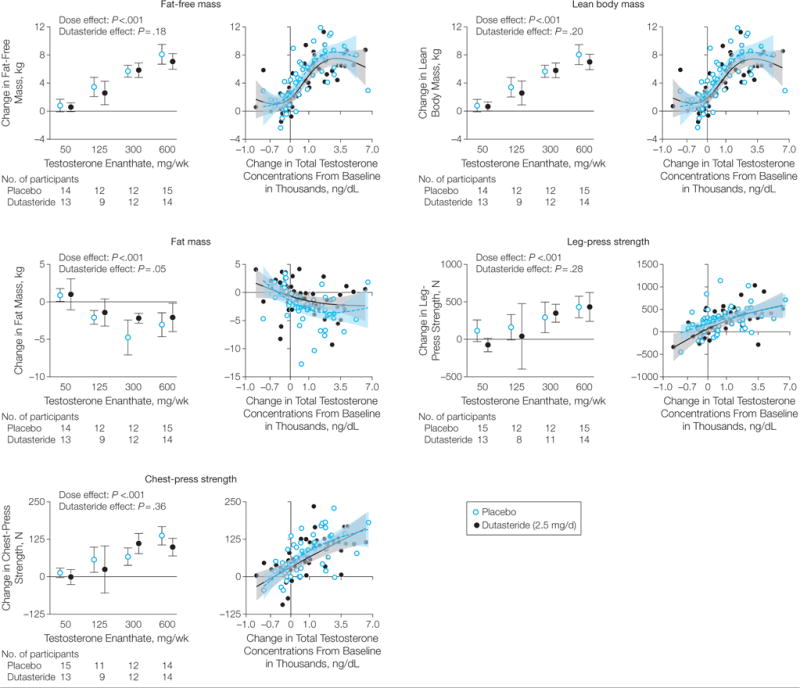
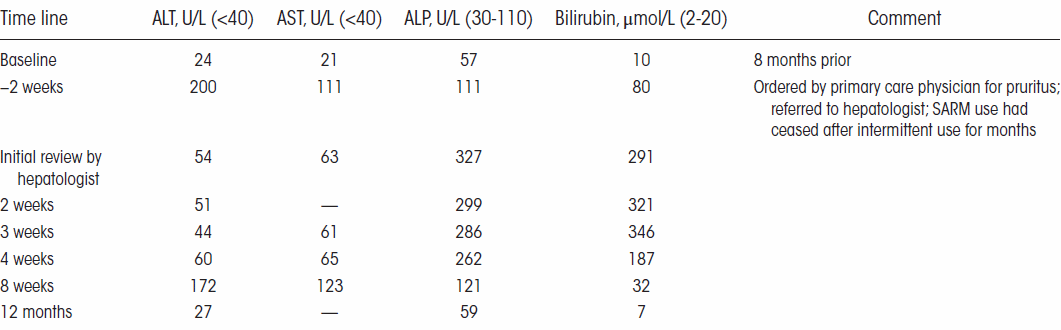

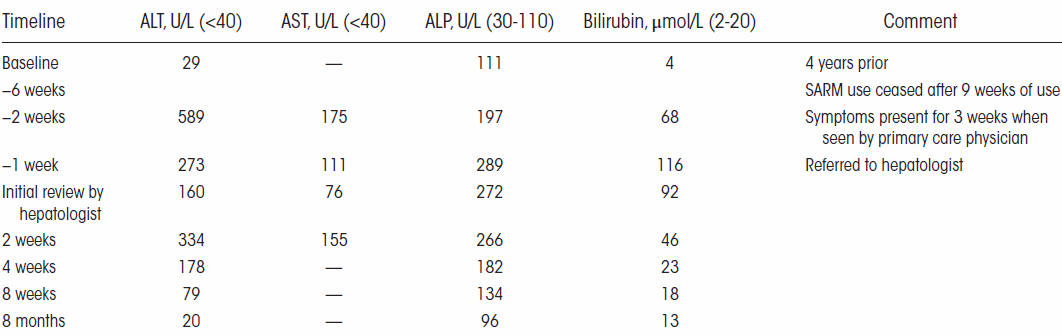
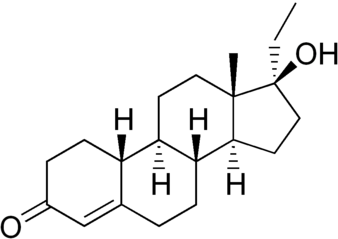



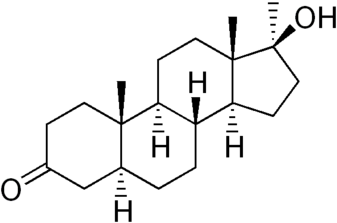

 to come agente dopante negli atleti olimpionici della Germania Orientale all’interno di un programma di doping sponsorizzato dallo stato negli anni ’70 e ’80.[4] Il motivo del suo uso, come precedentemente accennato, era giustificato maggiormente dal suo valore androgeno piuttosto che su quello anabolizzante. Il suo uso garantiva un ottima risposta neuro steroidea con conseguente stimolazione del Sistema Nervoso Centrale e migliorata interazione neuromuscolare, con vantaggi nella velocità, nella forza, nell’aggressività, nella concentrazione, nella resistenza fisica e allo stress mentale.[4] Oggi, l’uso del Mestanolone è stato per lo più sospeso in medicina, sebbene sia ancora disponibile in Giappone.[2][3][4] Il Mestanolone era comunemente disponibile sotto forma di compresse sublinguali da 25mg (marchio Ermalone).[7]
to come agente dopante negli atleti olimpionici della Germania Orientale all’interno di un programma di doping sponsorizzato dallo stato negli anni ’70 e ’80.[4] Il motivo del suo uso, come precedentemente accennato, era giustificato maggiormente dal suo valore androgeno piuttosto che su quello anabolizzante. Il suo uso garantiva un ottima risposta neuro steroidea con conseguente stimolazione del Sistema Nervoso Centrale e migliorata interazione neuromuscolare, con vantaggi nella velocità, nella forza, nell’aggressività, nella concentrazione, nella resistenza fisica e allo stress mentale.[4] Oggi, l’uso del Mestanolone è stato per lo più sospeso in medicina, sebbene sia ancora disponibile in Giappone.[2][3][4] Il Mestanolone era comunemente disponibile sotto forma di compresse sublinguali da 25mg (marchio Ermalone).[7]