Androgenico: 78-254
Anabolico: 107 (valore sovrastimato)
Standard: Testosterone
Nome Chimico: 17α-Methyl-4,5α-dihydrotestosterone
Attività estrogenica: nessuna
Attività Progestinica: nessuna
Aromatizzazione: no
Il Mestanolone, noto anche come 17α-methyl-4,5α-Diidrotestosterone (17α-methyl-DHT) o 17α-methyl-5α-androstan-17β-ol-3-one, è uno steroide androgeno anabolizzante (AAS), commercializzato in forma orale sotto il nome di Androstalone e Ermalone, ad oggi per lo più in disuso in ambito medico.[1][2][3][4]
Il Mestanolone è uno steroide androstano sintetico e un derivato del Diidrotestosterone (DHT) metilato in posizione C17α. [1][4] Infatti, differisce dal DHT solo per la presenza del gruppo metile nella posizione C17α.[1][4] Stretti parenti sintetici del Mestanolone includono Mesterolone (1α-Methyl-4,5α-dihydrotestosterone), Oxandrolone (2-oxa-17α-methyl-DHT), Oxymetholone (2-idrossimetilene-17α-methyl-DHT) e Stanozololo (un derivato del 17α-methyl-DHT (Mestanolone) con un anello pirazolico fuso con l’anello A.)[1][4]
Differenze nella struttura dello scheletro carbossilico tra Mestanolone (metilazione in C17α) e Mesterolone (metilazione in C1α).
Il Mestanolone fu sintetizzato per la prima volta nel 1935 insieme al Methyltestosterone e al Methandriolo.[5][6] È stato sviluppato dalla Roussel negli anni ’50 ed è stato introdotto per uso medico, con i marchi Androstalone ed Ermalone, intorno al 1960.[4][10][8] Venne inizialmente commercializzato in Germania.[4] Inizialmente si pensava che il farmaco fosse un potente agente anabolizzante, ma le ricerche successive hanno dimostrato che in realtà ha effetti anabolici relativamente deboli ed esplica principalmente azione androgena.[4] Il Mestanolone, insieme al molto più conosciuto 4-Chlorodehydromethyltestosterone (Oral Turinabol) è stato utilizzato come agente dopante negli atleti olimpionici della Germania Orientale all’interno di un programma di doping sponsorizzato dallo stato negli anni ’70 e ’80.[4] Il motivo del suo uso, come precedentemente accennato, era giustificato maggiormente dal suo valore androgeno piuttosto che su quello anabolizzante. Il suo uso garantiva un ottima risposta neuro steroidea con conseguente stimolazione del Sistema Nervoso Centrale e migliorata interazione neuromuscolare, con vantaggi nella velocità, nella forza, nell’aggressività, nella concentrazione, nella resistenza fisica e allo stress mentale.[4] Oggi, l’uso del Mestanolone è stato per lo più sospeso in medicina, sebbene sia ancora disponibile in Giappone.[2][3][4] Il Mestanolone era comunemente disponibile sotto forma di compresse sublinguali da 25mg (marchio Ermalone).[7]
Il Mestanolone è un AAS con effetti molto simili all’Androstanolone (diidrotestosterone; DHT) essendo praticamente una versione orale di quest’ultimo.[4] A causa dell’inattivazione da parte della 3α-idrossisteroide deidrogenasi (3α-HSD) nel muscolo scheletrico, il Mestanolone, sebbene dotato di una metilazione in posizione C-17 la quale ne migliora la stabilità del legame recettoriale, è descritto come un agente anabolizzante molto scarso, analogamente all’Androstanolone e al Mesterolone.[4] Poiché il Mestanolone è un composto 5α ridotto, non è un substrato soggetto all’enzima aromatasi e. quindi, non convertendo in estrogeno oltre a non possedere attività estrogenica intrinseca.[4] Inoltre, il farmaco non ha attività progestinica.[4] Come per gli altri AAS 17α-alchilati, il Mestanolone presenta un certo grado di epatotossicità.[4]
Differenze nella struttura dello scheletro carbossilico tra Diidrotestosterone e Mestanolone (aggiunta metilazione in C17α).
Come risaputo, gli AAS, a diverso grado di impatto, possono avere effetti deleteri sul colesterolo sierico. Il Mestanolone non è da meno presentando la tendenza a causare una riduzione delle concentrazioni di colesterolo HDL (“buono”) e un aumento delle concentrazioni di colesterolo LDL (“cattivo”), cosa che comporta uno sbilanciamento dell’equilibrio HDL/LDL che si traduce in un rischio maggiore di sviluppare arteriosclerosi. L’impatto relativo all’assunzione di un AAS nei confronti dei lipidi ematici dipende dalla dose, dalla via di somministrazione (per via orale o iniettabile), dal tipo di steroide (aromatizzabile o non aromatizzabile), e dal livello di resistenza al metabolismo epatico.
Essendo il Mestanolone un AAS con consistente attività androgenica, la soglia dei possibili forti effetti collaterali androgenici è generalmente alta ed è paragonabile a quella riscontrabile con altri composti come il Mesterolone. Per questa ragione, il suo uso in ambito femminile non è stato molto diffuso dal momento che poteva essere causa di severi effetti virilizzanti.
Quando l’uso del Mestanolone veniva applicato in campo sportivo, i dosaggi comunemente utilizzati erano mediamente tra i 10 ed i 20mg al giorno con un timing di somministrazione tra una dose e la successiva di 12 ore. I tempi di utilizzo rimanevano entro le 6-8 settimane onde evitare di creare un eccessivo stress epatico. Tra i bodybuilder, il Mestanolone era spesso inserito durante la preparazione alla gara vista la sua facile gestibilità non causando ritenzione idrica, non essendo soggetto ad aromatizzazione, esercitando una blanda azione anti-estrogenica (sia recettoriale che come ligando inibitorio dell’enzima Aromatasi) e possedendo una buona azione lipolitica (legame con i AR adipocitari).
Come già detto in precedenza, l’uso del Mestanolone è stato per lo più sospeso in ambito medico anche se rimane disponibile in Giappone.[2][3][4] Nel mercato nero è raramente reperibile per via della sua attuale e pressoché assente richiesta tra gli atleti.
Gabriel Bellizzi
Riferimenti:
1- J. Elks (14 November 2014). The Dictionary of Drugs: Chemical Data: Chemical Data, Structures and Bibliographies. Springer. pp. 775.
2- Index Nominum 2000: International Drug Directory. Taylor & Francis. 2000. pp. 655.
4- William Llewellyn (2017). Anabolics 11th. Molecular Nutrition Llc. p. 284-285-286.
5- Schänzer W (1996). “Metabolism of anabolic androgenic steroids”. Clin. Chem. 42 (7): 1001–20.
6- Ruzicka, L.; Goldberg, M. W.; Rosenberg, H. R. (1935). “Sexualhormone X. Herstellung des 17-Methyl-testosterons und anderer Androsten- und Androstanderivate. Zusammenhänge zwischen chemischer Konstitution und männlicher Hormonwirkung”. Helvetica Chimica Acta. 18 (1): 1487–1498.
7- H.-L. Krüskemper (22 October 2013). Anabolic Steroids. Elsevier. pp. 196.
L’1-Testosterone ( noto anche come Dihydroboldenone o, più semplicemente, come DHB) ha suscitato molto interesse negli ultimi anni* tra gli appartenenti alla comunità del Bodybuilding. La continua ricerca di nuove molecole dai maggiori potenziali spinge molti atleti facenti parte di questa “sottocultura” a sperimentare nuovi farmaci e/o diversi protocolli di applicazione di questi. Come ovvio che sia, per mancanza di preparazione degli interessati, queste sperimentazioni, e ciò accade molto spesso, si traducono in diffusione di dati non contestualizzati e privi di prove concretamente riconducibili al test su una specifica molecola. Fortunatamente, esiste una piccolissima percentuale di ricercatori indipendenti all’interno di questa comunità ed essi si preoccupano di valutare tutte le variabili in gioco, dalla certezza della molecola testata alla sua contestualizzazione d’uso (es. se essa è usata in mono-terapia o in co-somministrazione ecc…).
*In vero, il DHB è stato venduto legalmente online negli Stati Uniti, in forma prevalentemente orale, forma dalla scarsa biodisponibilità, dal 2002, anno in cui il brevetto del farmaco venne rilasciato per la vendita nel mercato degli integratori alimentari. Anche se tecnicamente la sua legalità era discutibile, la popolarità del nuovo composto è stata rapida e innumerevoli prodotti OTC come “Xtreme” ebbero largo seguito. Ciò durò fino al 2005, quando venne riclassificato come farmaco e inserito all’interno del Controlled Substances Act.
Tornando al punto originario del discorso, il quale ha la finalità di introdurre il lettore all’argomento trattato in questo articolo, il DHB, se attentamente analizzato, risulta decisamente ridimensionato nelle sue potenzialità e valutato sotto un altra luce in riferimento ai suoi possibili effetti avversi rispetto a quanto riportato dai comuni articolisti di settore. Motivo per cui, ora, torno nuovamente sull’argomento DHB, molecola da me già trattata tempo fa su questo sito, in modo maggiormente analitico su alcuni ed essenziali punti.
Ma iniziamo con ordine…
Caratteristiche base del 1-Testosterone
L’1-Testosterone (abbreviato in 1-Testo, 1-T), chiamato anche (nome IUPAC) 4,5α-dihydro-δ1-testosterone (Δ1-DHT) o 5α-androst-1-en-17β-ol-3-one, è uno steroide androgeno-anabolizzante (AAS), uno steroide androstano sintetico, che si differenzia dal Testosterone per l’aggiunta di un doppio legame in posizione C1-C2 e la mancanza del doppio legame in posizione C4-C5 nell’anello A. Il DHB è descrivibile più semplicemente, anche se pur sempre in modo incompleto, come il metabolita 5α-ridotto del Boldenone. [1] Venne sintetizzato negli anni 60 del XX Secolo e descritto per la prima volta nella letteratura medica occidentale nell’anno 1962.[2]
Il doppio legame in C1-C2 presente nello scheletro carbossilico del DHB stabilizza il ketogruppo in C3, essenziale per il legame androgeno.
Due proormoni del 1-Testosterone sono l’1-Androstenediolo e l’1-Androstenedione, l’ultimo dei quali può essere sintetizzato dallo Stanolone Acetato.[3]
Il Mesabolone è un chetale a base di 1-Testosterone.
L’1-Testosterone è anche noto per essere usato per sintetizzare il Metanolone e il Metenolone (comunemente noto come Primoboloan/Rimobolan).
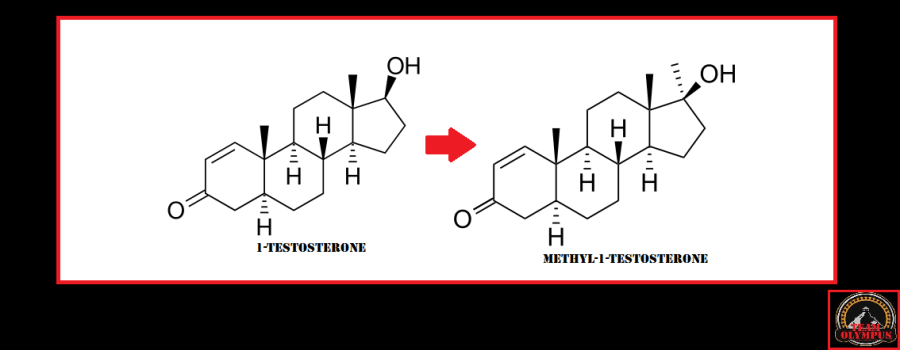
L’aggiunta di un metile in posizione C17 da come risultato la molecola di Methyl-1-Testosterone.
Per via delle sue caratteristiche strutturali, l’1-Testosterone non converte in estrogeno, anche se non ci sono prove che all’interno del corpo (quindi in vivo) possa essere inserito un doppio legame in C4. [4,5] Ciò che sappiamo proviene dai dati aneddotici i quali suggeriscono che l’1-Testosterone sia un AAS con una attività estrogenica irrilevante. Effetti collaterali estrogeno-dipendenti, come ginecomastia, accumuli di grasso con modello femminile e maggiore ritenzione idrica non sono generalmente osservati quando il DHB viene somministrato in mono-terapia.
Essendo un AAS 5α-ridotto, esso non è soggetto all’enzima 5α-reduttasi e, di conseguenza, la sua attività androgena non può essere manipolata dall’uso di inibitori enzimatici con bersaglio l’enzima precedentemente indicato. Quindi, l’androgenicità relativa del 1-Testostosterone non è influenzata dall’uso di Finasteride o Dutasteride).
La Anabolico/Androgeno ratio dell’1-Testosterone riporta un valore pari a 200/100 in riferimento al Testosterone Propionato 100/100. Tale rapporto risulta però dubbio anche per il semplice fatto che il metodo attraverso il quale è stato estrapolato non è chiaro. Ma su questo ritorneremo più avanti.
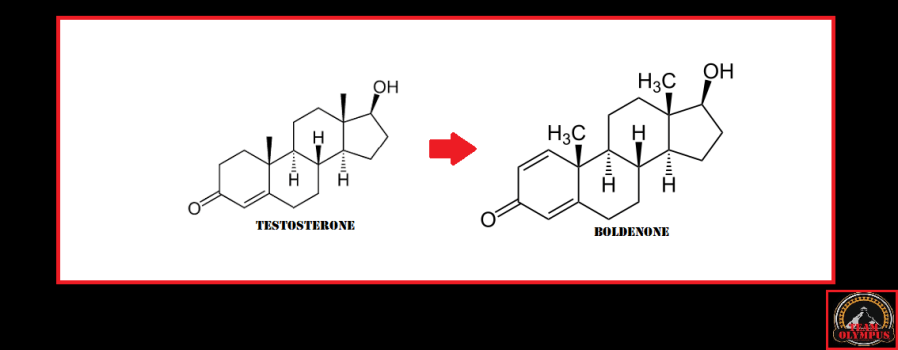
I più sapranno sicuramente che il Boldenone è un derivato del Testosterone il quale è stato sintetizzato modificando la struttura del substrato di derivazione aggiungendo un doppio legame tra gli atomi di carbonio 1 e 2 dell’anello A dello scheletro carbossilico.
Questo legame rallenta drasticamente l’aromatizzazione della molecola rendendola e un substrato scarsamente affina anche per la 5α-reduttasi.
Sebbene il Boldenone rimanga un substrato affine alla 5α-reduttasi potendo essere quindi convertito in DHB, anche con la somministrazione di elevati dosaggi la quantità del metabolita derivante non è sostanziale.
La malsana pratica di utilizzare mega-dosaggi di Boldenone nel tentativo di ottenere concentrazioni sieriche moderate di DHB è stata applicata da molti culturisti in passato.
Poiché l’ormone progenitore (Boldenone) è un substrato così scarsamente affine per la 5α-reduttasi, un numero crescente di culturisti ha iniziato a optare semplicemente per l’uso diretto del DHB.
La scarsa e poco incoraggiante letteratura scientifica sul DHB
Esistono pochissimi dati clinici che valutano l’efficacia o il profilo di sicurezza del DHB.
C’è solo uno studio [6] a cui possiamo fare riferimento il quale mostra in realtà come il DHB sia paragonabile al Testosterone in un ambiente clinico controllato.
Questo studio è stato condotto sul 1-Testosterone (DHB) dopo che esso venne classificato come sostanza sottoposta a controllo e non più commercializzabile negli Stati Uniti.
L’obiettivo dello studio era stabilire che il DHB non è un “proormone”, come era stato affermato da molte società di integratori prima del suo ritiro dal mercato della supplementazione OTC.
Valutando l’effetto del DHB sui tessuti sensibili agli androgeni nel corpo attraverso il Recettore degli Androgeni (AR), lo studio ha stabilito che il DHB è a tutti gli effetti uno Steroide Androgeno Anabolizzante.
Mentre la premessa dello studio aveva lo scopo di esplorare semplicemente la transattivazione dipendente dall’AR mediata dal DHB, l’effetto misurato attraverso l’azione della molecola su vari tessuti del corpo rispetto al Testosterone ha fatto luce su quanto sia selettivo ed efficace il DHB.
Dopo essere stati orchiectomizzati (rimozione chirurgica dei testicoli), i ratti castrati , prima di essere utilizzati per l’esperimento, sono stati lasciati nelle loro gabbie per 7 giorni al fine di consentire un sufficiente declino ormonale.
Questo processo consente ai ricercatori di ridurre le variabili influenti la valutazione della ricerca e di somministrare androgeni esogeni, quindi di pesare diversi organi del corpo e misurare il grado di impatto di un farmaco specifico sul tessuto muscolare, sui tessuti sensibili agli androgeni come la prostata e sugli organi vitali.
Ai ratti dello studio è stato somministrato giornalmente per via sottocutanea un placebo, 1mg/kg/bw/giorno di Testosterone Propionato (TP) o 1mg/kg/bw/giorno di DHB (1-Testo).
Milligrammo per milligrammo, il DHB ha dimostrato di indurre una crescita muscolare leggermente inferiore rispetto al Testosterone, stimolando altrettanto la prostata e la vescicola seminale.
Effetto del Testosterone Propionato (TP) e del 1-Testosterone (1-Testo) sul muscolo Levator Ani dei ratti orchiectomizzati.
Ciò significa che per replicare lo stesso livello di crescita muscolare, il DHB dovrebbe essere somministrato a dosi più elevate del Testosterone.
Tornando alla precedentemente introdotta questione della presunta Anabolico/Androgeno ratio, l’ipotetico valore di 200/100 non si sa bene da dove provenga e come sia stato ricavato. Ciò che risulta piuttosto chiaro è che gli unici dati clinici che ho potuto trovare, e attraverso i quali mi è stato possibile valutare la selettività dei tessuti da parte del DHB, certamente non riflettono tale potenziale.
In effetti sembra essere meno selettivo a livello dei tessuti rispetto al Testosterone e complessivamente un po’ meno anabolico.
Osservando la struttura molecolare del DHB si evince una bassa resistenza alla disattivazione epatica di primo passaggio dovuta alla mancanza di modifiche atte a alterare il metabolismo dell’AAS. Sappiamo che la tossicità epatica si presenta a livelli significativi con l’uso di AAS metilati in C17, anche se molecole come il Trenbolone, aventi per modifiche strutturali non correlate a metilazione, presentano una certa resistenza alla disattivazione epatica causando un certo grado di tossicità potenziale.
I segni chiave della epatotossicità comprendono l’elevazione degli enzimi epatici e, in alcuni casi, persino l’ingrossamento del fegato.
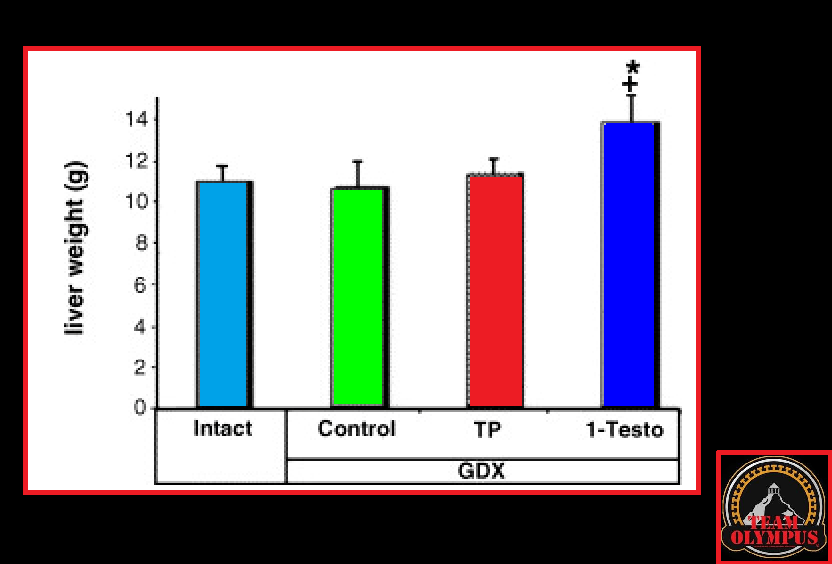
Nello studio qui discusso, il gruppo di topi trattati con Testosterone Propionato non ha mostrato alcun segno di tossicità epatica. Tuttavia, il gruppo trattato con DHB ha avuto un aumento significativo del peso del fegato.
Effetto del Testosterone Propionato (TP) e del 1-Testosterone (1-Testo) sull’aumento del peso del fegato nei ratti orchiectomizzati.
Anche se quanto riscontrato a livello epatico nei topi trattati con DHB non sia una prova assoluta della potenziale tossicità del DHB, l’ipertrofia epatica potrebbe compromettere il funzionamento dell’organo se cronicizzata. Come accennaio tempo a dietro nella scheda tecnica del 1-Testosterone, qualcuno ipotizza (senza documentazione a convalidarlo) che l’ipertrofia epatica DHB-dipendente non è dovuta a fenomeni steatosici (“fegato grasso”) o tumorali, ma semplicemente alla capacità dell’1-Testosterone di legarsi ai recettori androgeni epatici; caratteristica comune ad uno dei prima citati anabolizzanti usatio come substrato di sintesi per il DHB, il Metenolone il quale in passato era ampiamente usato nella cura delle lesioni causate dalla cirrosi epatica.
Valutazione della molecola
Come valutare il DHB alla luce della (scarsa) letteratura disponibile e dei dati aneddotici provenienti dagli atleti (per lo meno quelli affidabili)?
Una cosa che si nota particolarmente nella scelta degli AAS da parte dei bodybuilder è che questi ultimi tendono a optare per molecole non aromatizzabili, terrorizzati dall’ignoranza che aleggia sugli estrogeni e sulla loro corretta gestione. Sebbene un AAS non aromatizzabile si dimostri discretamente più gestibile sotto alcuni aspetti, la mancanza di attività estrogenica indiretta può causare una serie di problemi che vanno dalla sfera psicologica alla disfunzione erettile. Tali effetti sono semplicemente evitabili, insieme a quelli legati ad una condizione di iperestrogenemia, gestendo i fattori estrogenici attraverso dosaggi intelligentemente settati e, nel caso, l’inserimento di anti aromatase e/o SERM. La scelta di una molecola dovrebbe basarsi maggiormente sui potenziali che potrebbero offrire sotto l’aspetto della sicurezza e potenza in rapporto alle altre molecole disponibili. In breve, AAS che sono più selettivi nei tessuti bersaglio e anabolizzanti del Testosterone.
Sebbene il DHB non sia un substrato soggetto all’enzima aromatasi, ciò non è, per esempio, particolarmente vantaggioso per un suo ipotetico uso in monoterapia, poiché richiederebbe la somministrazione esogena di Estradiolo (o di un substrato soggetto alla conversione in esso) al fine di evitare la comparsa di effetti collaterali psicofisici dovuti ad un basso livello estrogenico.
In conclusione, non vi sono dati certi sull’effetto che il DHB può avere sul corpo umano fatta eccezione per le testimonianze raccolta nei forum e, questa volta con qualche valenza in più, dai dati registrati dai ricercatori indipendenti che alternano i libri alla ghisa. Gli unici dati “certi” che abbiamo provengono, come visto, dal modello preclinico di roditori, e non sono nemmeno così promettenti.
Sebbene io non sia contrario alla sperimentazione indipendente, con questa molecola tale pratica può permettersela soltanto chi è in possesso di approfondite conoscenze in campo medico con particolarità nell’Endocrinologia, Andrologia e Farmacologia. I meno esperti, e meno portati, dovrebbero limitarsi alla scelta di molecole con effetti terapeutici riportati basati sul profilo preclinico registrato e anche in sperimentazione umana.
Il DHB nella pratica si è dimostrato una molecola discreta in “Cut” con una capacità miotropica stimabile con quella del Metenolone e del Boldenone.
Ma le sue limitazioni ad oggi persistono e sono:
Nessuno studio svolto su esseri umani e sul/i quale/i poter fare riferimento;
Leggermente meno anabolico e selettivo a livello tissutale rispetto al Testosterone, almeno in base agli studi sui topi;
Potenzialmente deleterio per il fegato sul lungo termine anche in forma iniettabile.
Occorrerebbe svolgere studi più approfonditi, ma dubito che qualcuno con mezzi adegiuati e possibilità maggiori rispetto ad un piccolo ricercatore indipendente si possa muovere in tal senso, anche perché non vi sono necessità che spingerebbero a ciò.
Gabriel Bellizzi
Riferimenti:
1- William Llewellyn (2009). Anabolics (9 ed.). Molecular Nutrition. p. 22,135.
2- Friedel A, Geyer H, Kamber M, Laudenbach-Leschowsky U, Schänzer W, Thevis M, Vollmer G, Zierau O, Diel P (August 2006). “17beta-hydroxy-5alpha-androst-1-en-3-one (1-testosterone) is a potent androgen with anabolic properties”. Toxicol. Lett. 165 (2): 149–55.
3- Zhang H, Qiu Z (December 2006). “An efficient synthesis of 5α-androst-1-ene-3,17-dione”. Steroids. 71 (13–14): 1088–90.
4- 358. K. J. Ryan. Acta Endocrinol. 35, Suppl. 51,697 (1960).
5- C. Gual, T. Morato, M. Gut, R.1. Dorfman, Endocrinology 71,920 (1962). 17beta-hydroxy-5alpha-androst-1-en-3-one (l-testosterone) is a potent androgen with anabolic properties. Friedel A, Geyer H, Kamber M.
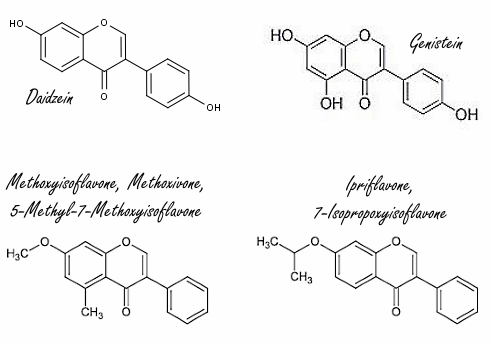
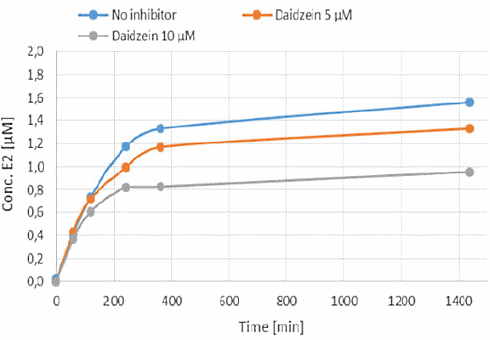
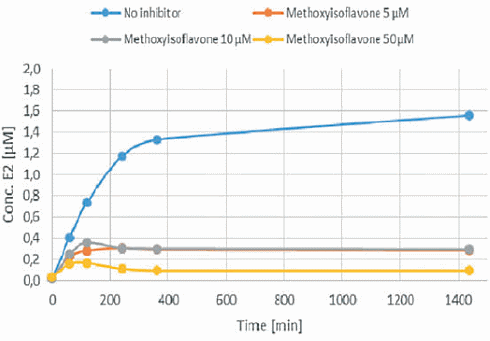
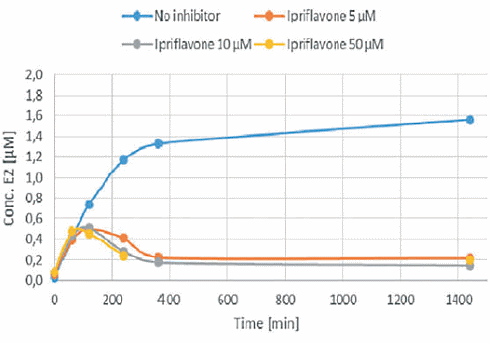
Alcuni integratori che dovrebbero aumentare i livelli di Testosterone contengono gli isoflavoni sintetici Methoxyisoflavone e Ipriflavone. Questi due composti potrebbero avere qualche concreto potenziale, o per lo meno questo è quanto emerso da uno studio in vitro svolto da ricercatori italiani, associati alla Federazione Medico Sportiva Italiana, e i cui risultati sono stati pubblicati sul Drug Testing & Analysis.(1) Secondo i ricercatori, sia il Methoxyisoflavone che l’Ipriflavone inibiscono l’Enzima Aromatasi che, come ben sappiamo, converte il Testosterone in Estradiolo.
L’Ipriflavone e il Methoxyisoflavone sono isoflavoni sintetici. Furono sintetizzati nel secolo scorso da parte di chimici dell’Europa dell’Est, i quali erano intenzionati a creare nuovi agenti anabolizzanti mediante la modifica strutturale degli isoflavoni come la Daidzeina, un fitoestrogeno della Soia.
Gli isoflavoni sintetici non ebbero il successo sperato nell’applicazione farmacologica, ma hanno comunque preso piede come integratori alimentari per lo sport.
L’Ipriflavone e il Methoxyisoflavone hanno un azione inibitoria su alcuni enzimi della famiglia del citocromo P450 come il CYP1A2 e il CYP2C9. Questo è il motivo per cui i ricercatori italiani si sono chiesti se i due isoflavoni sintetici avessero potuto inibire l’azione dell’Enzima Aromatasi. L’Enzima Aromatasi, proprio come il CYP1A2 e il CYP2C9, fa parte della famiglia del citocromo P450.
Se così fosse, allora l’Ipriflavone e il Methoxyisoflavone potrebbero essere inseriti nella lista delle sostanze dopanti, insieme ad inibitori dell’Aromatasi di sintesi tra i quali troviamo l’Anastrozolo e il Letrozolo. Come risaputo, gli inibitori dell’Aromatasi possono aumentare i livelli di Testosterone attraverso due vie principali:
la riduzione della conversione del Testosterone in Estradiolo e riduzione delle SHBG con conseguente miglioramento dei livelli complessivi dell’androgeno (sia totale che bioattivo) e della Testosterone:Estradiolo ratio;
attraverso la riduzione dei livelli di Estradiolo e il feedback positivo a livello ipotalamico con aumento del rilascio di GnRH e conseguente aumento di LH e incremento della biosintesi testicolare di Testosterone.
E’ scontato sottolineare che ciò ha come potenziale conseguenza il miglioramento delle prestazioni.
I ricercatori hanno svolto l’esperimento in vitro osservando, in presenza di Testosterone e Aromatasi, se l’Ipriflavone, il Methoxyisoflavone e altri composti simili avessero la capacità di bloccare il processo enzimatico di conversione del Testosterone in Estradiolo.
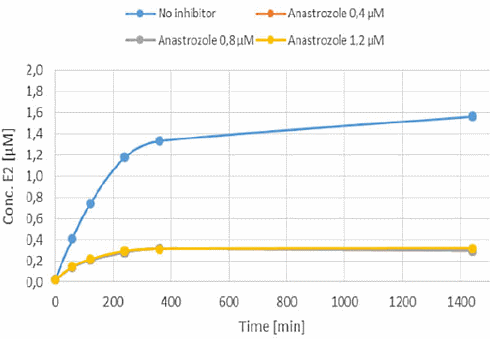
L’Ipriflavone e il Methoxyisoflavone hanno mostrato di causare l’inibizione della conversione del Testosterone in Estradiolo. Entrambi i composti bloccavano l’azione dell’Enzima Aromatasi. Anche la Daidzeina ha mostrato di poterlo fare, ma in misura molto minore. L’effetto anti-estrogenico dell’Ipriflavone e del Methoxyisoflavone (IN VITRO) è risultato essere simile a quello dell’Anastrozolo.
Quando i ricercatori hanno calcolato il Ki del Methoxyisoflavone e dell’Ipriflavone sulla base dei loro test, hanno visto che era uguale al Ki degli inibitori dell’aromatasi farmacologici.
I ricercatori scrivono che i risultati ottenuti nel loro modello in vitro mostrano, nella gamma di concentrazioni considerate, che il Methoxyisoflavone e l’Ipriflavone hanno un potenziale di inibizione dell’aromatasi simile a quello del Formestano, dell’Anastrozolo e dell’Amminoglutetimmide. Quei farmaci [sono] usati nella terapia antitumorale e banditi dalla WADA per i loro effetti sulla steroidogenesi (l’Amminoglutetimide per particolarità differenti sulla steroidogenesi NdR).
Chiaramente, i risultati presentati non sono sufficienti per esprimere un’opinione finale sulla possibilità di includere gli isoflavoni sintetici nella lista delle sostanze dopanti redatta dalla WADA, dal momento che l’entità effettiva dei loro effetti in vivo sarebbe certamente modulata dalle loro proprietà farmacocinetiche, e specialmente dalla loro bassa biodisponibilità.
Tuttavia, i ricercatori ritengono che il loro monitoraggio possa ancora essere utile nell’analisi del controllo antidoping, considerando anche le alte dosi raccomandate per gli isoflavoni sintetici negli integratori alimentari.
Sembra esistere un legame tra, da un lato, il consumo di alimenti trasformati e, dall’altro, l’azione estrogenica tissutale. Ricercatori della Medical University of South Carolina hanno parlato di ciò sul Breast Cancer Research and Treatment. Il loro studio è interessante per le donne con una forma di cancro al seno estradiolo-sensibile e per chiunque, per qualsiasi motivo, voglia ridurre l’azione tissutale estrogenica nel proprio corpo.(1) Gli AGE fanno parte dell’equazione per raggiungere lo scopo.
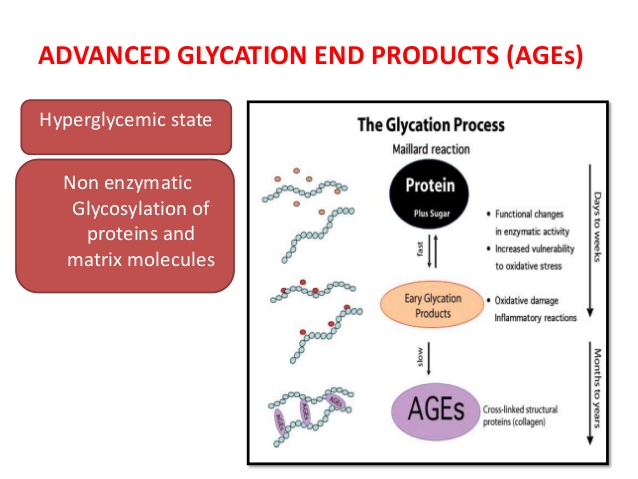
Gli AGE (Advanced Glycation End-product; in italiano prodotto finaledella glicazione avanzata) sono il risultato di una catena di reazioni chimiche successive alla reazione di glicazione iniziale. In breve, si tratta di proteine o lipidi che diventano glicati a seguito dell’esposizione agli zuccheri.(2)
Prodotti intermedi sono conosciuti come il riarrangiamento di Amadori, la base di Schiff e i prodotti di Maillard, chiamati così dai ricercatori che per primi li hanno descritti. La letteratura è ancora indecisa sull’applicare questa terminologia. Per esempio i prodotti della reazione di Maillard sono talvolta considerati prodotti intermedi talvolta prodotti finali. Gli effetti collaterali generati nei passaggi intermedi da agenti ossidanti (come il perossido di idrogeno), e non (come le proteine amiloidi beta).(3) Il termine glicosilazione è talvolta usato per glicazione in letteratura, di solito come glicosilazione non enzimatica.
Gli AGE si trovano sulle superfici dorate o abbrustolite di cibi fritti o grigliati, ed anche sulla crosta del pane. Oltre al colore, gli AGE conferiscono agli alimenti cotti anche il sapore caratteristico, tipico dei prodotti da forno.
Con la dieta moderna e l’utilizzo di prodotti industriali consumiamo più AGE di quanto accadeva anni fa, alcune aziende alimentari trasformano eccessivamente alcuni alimenti o aggiungono AGE artificiali per esaltare il sapore dei propri prodotti.
Furosina
Solo nel latte sono previsti dei limiti, la legge italiana fissa infatti in 8,6 mg/100 g di proteine il limite di Furosina presente nel latte crudo e pastorizzato, ed in 12 mg/100 g di proteine il limite per i formaggi freschi a pasta filata. Nella pasta alimentare secca sono presenti gli AGE (Furosina; ε-furoilmetil-lisina ) da quando è stato introdotto il metodo d’essiccazione ad alta temperatura (HT) ed l’essiccazione ad altissima temperatura (VHT). La legge non è ancora stata aggiornata come per il latte.
I prodotti finali di glicazione avanzata (AGE), noti anche come glicotossine, sono un gruppo eterogeneo di composti altamente ossidanti con un significato patogenetico nel diabete e in molte altre malattie croniche.(4)
La formazione di AGE è una parte del normale metabolismo, ma se si raggiungono livelli eccessivamente alti di AGE nella circolazione ematica e, quindi, nei tessuti possono diventare patogeni. Gli effetti patologici degli AGE sono legati alla loro capacità di promuovere lo stress ossidativo e l’infiammazione legandosi con i recettori della superficie cellulare o reticolazione con proteine del corpo, alterando la loro struttura e funzione.(5)(6) Gli AGE si formano anche endogenamente in condizioni, per esempio, di glicemia cronicamente alta.
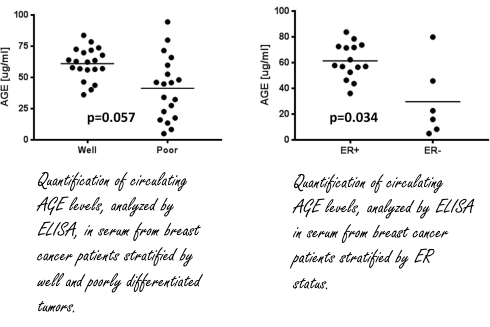
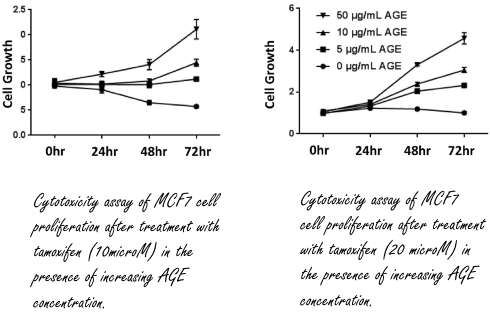
I ricercatori della Medical University of South Carolina volevano scoprire se gli AGE potessero avere un ruolo nello sviluppo del cancro al seno, e per farlo hanno preso in esame un campioni di donne con tale patologia. I ricercatori hanno scoperto che c’erano più AGE nel sangue nelle donne con tumori più sviluppati e quindi più pericolosi rispetto alle donne con tumori meno sviluppati, e quindi meno pericolosi [in basso a sinistra].
I ricercatori hanno anche rilevato livelli più alti di AGE nelle donne con una forma di carcinoma mammario Estradiolo-sensibile rispetto alle donne con un tumore al seno non sensibile all’Estradiolo [in alto a destra]. Questo suggerisce che possa esistere una correlazione tra AGE, cancro della mammella ed Estradiolo.
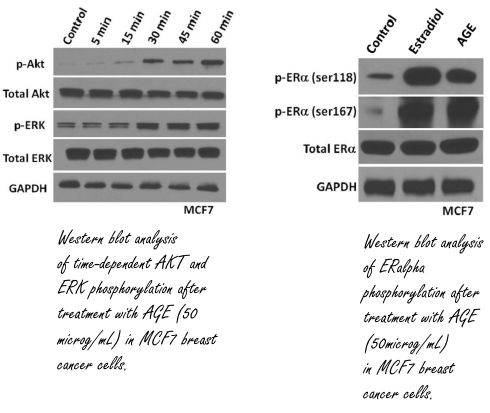
I ricercatori hanno quindi iniziato a svolgere esperimenti in vitro con cellule del cancro al seno estradiolo-sensibile. Se queste cellule venivano esposte agli AGE, il recettore dell’Estradiolo veniva attivato [in basso a destra]. L’Estradiolo attiva molecole di segnale come Akt ed Erk nelle cellule, e gli AGE sembrano fare la stessa cosa. Sembrerebbe quindi che gli AGE mimino l’effetto dell’Estradiolo.
Come risaputo, gli oncologi utilizzano i SERM, che impediscono all’Estradiolo di legarsi ed attivare il recettore bersaglio, nel tentativo di bloccare o rallentare la crescita dei tumori al seno Estradiolo-sensibili. Più AGE circolano nel flusso ematico, e meno efficace risulta questo approccio terapeutico, almeno da quanto emerso dai dati raccolti dai ricercatori che hanno esposto le cellule tumorali in vitro al Tamoxifene e agli AGE.
Quanto scoperto dai ricercatori è ovviamente molto interessante, anche perché le concentrazioni di AGE con le quali sono stati svolti gli esperimenti in vitro sono pressoché uguali alle concentrazioni riscontrabili nel corpo di un soggetto nella media. Appurato ciò, come ridurre i livelli di AGE?
Ridurre fortemente o eliminare gli alimenti processati e mantenere una glicemia basale entro gli 80-90mg/dL sono le pratiche primarie per causare una sensibile riduzione degli AGE. Nello specifico dello studio qui trattato, i ricercatori hanno sottoposto un gruppo di donne obese, trattate per la cura del cancro al seno, ad una alimentazione più salutare e a regolare esercizio fisico per 11 settimane. Il loro approccio – come camminare per 30 minuti al giorno – sebbene blando è risultato efficace come mostrato nella figura seguente.
Con il termine Antiestrogeni ci si riferisce genericamente ad una classe di farmaci aventi azione diretta o indiretta sull’attività tissutale e/o concentrazione ematica degli estrogeni. Agiscono bloccando il recettore dell’estrogeno (ER) e/o riducendo o sopprimendo la sintesi estrogenica.(1)(2) Una recente categoria di agenti facenti parte di questa classe di farmaci, i SERD (Selective Estrogen Receptor Degrader), esplicano la loro azione antiestrogena degradando/sottoregolando il recettore dell’estrogeno. Gli Antiestrogeni sono una delle tre classi di farmaci antagonisti dell’ormone sessuale, insieme agli Antiandrogeni e agli Antiprogestinici.(3)
Largamente utilizzati in ambito sportivo, in special modo nell’ambiente culturistico, con il fine di controllare l’attività estrogenica durante l’uso di AAS aromatizzabili, o aventi attività estrogenica intrinseca, e durante la PCT con lo scopo aggiunto di stimolare la ripresa dell’HPTA, questi farmaci hanno un discreto carico di effetti collaterali tra i quali, quelli che destano maggior preoccupazione nell’atleta previdente, vi sono la dislipidemia (aumento dell’LDL, dei Trigliceridi, riduzione del HDL e alterazione delle loro ratio), l’atralgia (dolore articolare), il calo della libido/disfunzione erettile e l’affaticamento/letargia. Non sono di certo da meno le preoccupazioni legate all’alterazione dell’Asse GH/IGF-1 o la riduzione delle potenzialità di induzione ipertrofica di un ciclo in seguito ad una eccessiva soppressione dell’attività e/o delle concentrazioni estrogeniche. Ma esiste un’altra preoccupazione legata all’uso di composti antiestrogeni, ed è la possibilità che si verifichi un rebound estrogenico in seguito al l’oro uso. Purtroppo, la letteratura a disposizione è al quanto scarsa e poco chiara nella specifica del problema. E’ possibile, però, fare maggiore chiarezza sulla questione analizzando le caratteristiche dei composti antiestrogeni e il loro impatto, passando in rassegna tutti i componenti dell’addizione (Recettori Estrogeni e enzima Aromatasi). In questo articolo cercherò di esporre un ragionamento logico grazie al quale, seppur non avendo una risposta definitiva, sarà possibile avere un idea, la più concreta possibile, sul binomio antiestrogeni/rebound estrogenico.
Una analisi della questione…
Recettori dell’Estrogeno, SERM e “rebound estrogenico”
Un dimero della regione legame-ligando del ERa.
I Recettori degli Estrogeni (ER) sono un gruppo di proteine presenti all’interno delle cellule. Sono recettori attivati dall’ormone estrogeno (con maggiore attività del 17β-estradiolo).(4) Esistono due classi di ER: i Recettori degli Estrogeni nucleari (ERα e ERβ), che sono membri della famiglia dei recettori nucleari e dei recettori intracellulari, ed i Recettori degli Estrogeni di Membrana (MR) (GPER (GPR30), ER-X e Gq-mER), che sono per lo più recettori accoppiati alla proteina G. In questa sede ci si riferirà ai primi (ER).
Una volta attivato dall’estrogeno, l’ER è in grado di traslocare nel nucleo e legarsi al DNA per regolare l’attività di diversi geni (ciò significa che è un fattore di trascrizione del DNA). Tuttavia, ha anche funzioni aggiuntive indipendenti dal legame con il DNA.(5)
Poiché l’estrogeno è un ormone steroideo, può passare attraverso le membrane fosfolipidiche della cellula, e pertanto i recettori non hanno bisogno di essere legati alla membrana per potersi legare a loro volta con l’estrogeno.
L’estrogeno esplica la sua attività cellulare attraverso un azione Genomica e Non-Genomica.
• Genomica
In assenza di ormoni, i ER si trovano in gran parte nel citosol. Il legame dell’ormone al recettore innesca un numero di eventi che iniziano con la migrazione del recettore dal citosol nel nucleo, la dimerizzazione del recettore e il successivo legame del dimero del recettore a specifiche sequenze di DNA conosciute come elementi di risposta ormonale. Il complesso DNA / recettore quindi recluta altre proteine che sono responsabili della trascrizione del DNA a valle in mRNA e, infine, in una proteina la quale porta a dei cambiamenti nella funzione cellulare. I recettori degli estrogeni si trovano anche all’interno del nucleo della cellula, ed entrambi i sottotipi del recettore dell’estrogeno hanno un dominio di legame con il DNA e possono funzionare come fattori di trascrizione per regolare la produzione di proteine.
Il recettore interagisce anche con la proteina attivatore 1 e Sp-1 per promuovere la trascrizione, attraverso diversi coattivatori come il PELP-1.(6)
L’acetilazione diretta del recettore alfa dell’estrogeno ai residui della lisina nella regione cerniera mediante il p300 regola la transattivazione e la sensibilità ormonale.(7)
• Non-Genomica
Alcuni recettori per gli estrogeni sono presenti nelle membrana della superficie della cellula e possono essere rapidamente attivati dall’esposizione di questa agli estrogeni.(8)(9)
Inoltre, alcuni ER possono associarsi alle membrane cellulari legandole alla caveolina-1 e formarmando complessi con la proteine G, striatina, tirosina chinasi del recettore (es. EGFR e IGF-1) e tirosina chinasi non recettoriale (es. Src). (6)(8) Attraverso la striatina, alcuni di questi ER legati alla membrana possono portare a livelli aumentati di Ca2 + e ossido nitrico (NO).(10) Attraverso il recettore tirosin chinasi, i segnali vengono inviati al nucleo attraverso la via della proteina chinasi attivata dal mitogeno (MAPK / ERK) e la via del fosfoinositide 3-chinasi (Pl3K / AKT).(11) La glicogeno sintasi chinasi-3 (GSK) -3β inibisce la trascrizione dal ER nucleare inibendo la fosforilazione della serina 118 dell’ERa nucleare. La fosforilazione di GSK-3β rimuove il suo effetto inibitorio, e questo può essere ottenuto tramite il pathway PI3K / AKT e il pathway MAPK / ERK, tramite rsk.
Il 17β-estradiolo ha dimostrato di attivare il recettore GPR30 accoppiato alla proteina G.(12) Tuttavia, la localizzazione subcellulare e il ruolo di questo recettore sono ancora oggetto di controversie.(13)
ERb.
Gli estrogeni e gli ER sono implicati nel cancro al seno, nel carcinoma ovarico, nel cancro del colon, nel cancro alla prostata e nel cancro dell’endometrio. Il carcinoma del colon avanzato è associato a una perdita di ERβ, l’ER predominante nel tessuto del colon, e il tumore del colon è trattato con agonisti specifici per ERβ.(14)
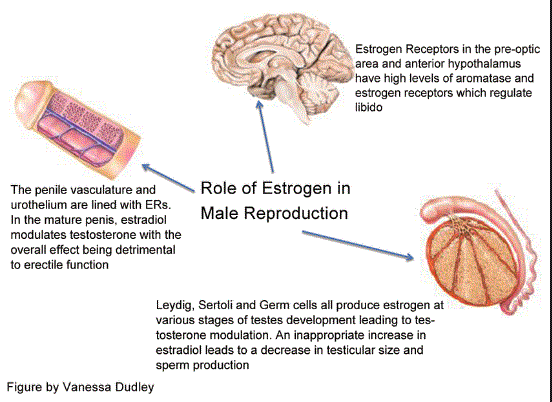
Sappiamo che i recettori degli estrogeni sono sovraespressi in circa il 70% dei casi di cancro al seno, indicati come “ER-positivi”, e possono essere dimostrati in tali tessuti mediante l’immunoistochimica.(15) E’ ipotizzabile ,quindi, che gli atleti più sensibili agli effetti estrogenici presentino un espressione dei ER più elevata del normale, cosa che li porta a sviluppare con maggiore facilità effetti avversi dati da un eccesso dei livelli estrogenici e/o da un aumento della loro attività dato dalla cosomministrazione con progestinici (es. Nandrolone e Trenbolone).
I Modulatori Selettivi del Recettore dell’Estrogeno (SERM) sono composti antiestrogenici che agiscono a livello del ER. (16) Una caratteristica che distingue queste sostanze dagli agonisti e antagonisti ER puri (cioè agonisti completi e antagonisti silenti) è che la loro azione è diversa nei vari tessuti, garantendo in tal modo la possibilità di inibire selettivamente o stimolare l’azione estrogenica in diversi tessuti.
I SERM sono agonisti parziali competitivi del ER.(17) Tessuti diversi presentano differenti gradi di sensibilità all’attività degli estrogeni, pertanto i SERM esplicano effetti estrogenici o antiestrogeni a seconda del tessuto specifico con il quale interagiscono e della percentuale di attività intrinseca (IA) del composto in questione.(18) Un esempio di SERM con alta IA, e quindi di effetti prevalentemente estrogenici, è rappresentato dal Clorotrianisene, mentre un esempio di SERM con bassa IA, e quindi avente per lo più attività antiestrogenica, è rappresentato dall’Ethamoxytrifetolo. SERM come il Clomifene e il Tamoxifene, largamente utilizzati in ambito sportivo, sono considerabili come composti con valore IA intermedio essendo molecole con una azione bilanciata tra effetti estrogenici e antiestrogenici. Il Raloxifene è un SERM che presenta una azione antiestrogenica maggiore del Tamoxifene; entrambi hanno una attività estrogenica (sebbene differente) a livello osseo, ma il Raloxifene presenta una attività antiestrogenica nell’utero mentre il Tamoxifene ha un azione estrogenica nel tessuto dell’utero.(18)
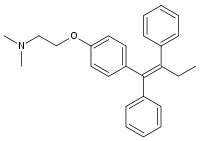
Tamoxifene
Il Tamoxifene è un farmaco di prima linea per il trattamento del carcinoma mammario metastatico ER-positivo. È usato per la riduzione delle possibilità di sviluppo del cancro al seno nelle donne ad alto rischio, come trattamento adiuvante del nodo ascellare negativo e positivo, e nel carcinoma duttale in situ.(19)(20)
Il Tamoxifene è classificabile come un profarmaco, dal momento che la sua affinità per la proteina bersaglio (ER) è limitata. Il Tamoxifene viene metabolizzato nel fegato dall’isoforma del citocromo CYP2D6 e CYP3A4 in metaboliti attivi come l’Afimoxifene (4-idrossitamoxifene; 4-OHT) e l’Endoxifene (N-desmetil-4-idrossitamoxifene) (21) che presentano una affinità da 30 a 100 volte maggiore per il ER rispetto al Tamoxifene. (22) Questi metaboliti attivi competono con gli estrogeni per il legame con il recettore. Nel tessuto mammario, il 4-OHT agisce come un antagonista del ER in modo da inibire la trascrizione dei geni che reagiscono agli estrogeni. (23) Il Tamoxifene ha rispettivamente il 7% e il 6% dell’affinità dell’Estradiolo per il ERα e il ERβ, mentre il 4-OHT ha il 178% e il 338% dell’affinità dell’Estradiolo per il ERα e il ERβ.(24)
Afimoxifene (4-OHT)
Il 4-OHT si lega al ER, il complesso ER/Tamoxifene recluta altre proteine note come co-repressori e quindi si lega al DNA per modulare l’espressione genica. Alcune di queste proteine includono la NCoR e la SMRT. (25) La funzione del Tamoxifene può essere regolata da una serie di variabili diverse, compresi i fattori di crescita.(26) Il Tamoxifene deve bloccare le proteine del fattore di crescita come ErbB2/HER2 (27) perché è stato dimostrato che livelli elevati di ErbB2 si manifestano nei tumori resistenti al Tamoxifene.(28) Il Tamoxifene sembra richiedere una proteina PAX2 affinché possa esplicare il suo pieno effetto antitumorale. (27)(29) In presenza di un elevata espressione della PAX2, il complesso Tamoxifene/ER è in grado di sopprimere l’espressione della proteina pro-proliferativa del ERBB2. Al contrario, quando l’espressione del AIB-1 è superiore alla PAX2, il complesso di Tamoxifene/ER aumenta l’espressione del ERBB2 con conseguente stimolazione della crescita del cancro al seno. (27)(30)
Il 4-OHT si lega al ER in modo competitivo (rispetto all’estrogeno agonista) nelle cellule tumorali e in altri bersagli tissutali, producendo un complesso nucleare che riduce la sintesi del DNA e inibisce gli effetti degli estrogeni. È un agente non steroideo con potenti proprietà antiestrogeniche che competono con gli estrogeni per i siti di legame nel seno e in altri tessuti. Il Tamoxifene fa sì che le cellule rimangano nelle fasi G0 e G1 del ciclo cellulare. Poiché impedisce alle cellule (pre) cancerose di dividersi ma non provoca la morte cellulare, il Tamoxifene è citostatico piuttosto che citocida.
La letteratura scientifica riguardante l’attività del Tamoxifene è a dir poco complessa ed occorre prestare particolare attenzione ai dati disponibili per stabilire se il Tamoxifene, o il suo metabolita 4-idrossi, abbiano il maggiore impatto complessivo.
Norendoxifene
Il Norendoxifene (N, N-didesmetil-4-idrossitamoxifene), un altro metabolita attivo del Tamoxifene, è stato osservato agire come un potente inibitore dell’aromatasi competitivo (IC50 = 90 nM), cosa che a sua volta può amplificare l’attività antiestrogenica complessiva del Tamoxifene.(31)
Come già accennato in precedenza, e come molti sapranno, il Tamoxifene è largamente utilizzato in ambito sportivo, sia da solo che in abbinamento con altri SERM come il Clomifene ( in PCT) o con AI (“on-cycle” e/o in PCT). La sua applicazione all’interno di una preparazione che contempla l’uso di AAS aromatizzabili, alla luce di quanto esposto pocanzi, lo vede come agente preventivo o di trattamento dell’attività estrogenica a livello tissutale, in specie per quanto concerne l’attività estrogenica nel tessuto mammario al fine di evitare (o “tamponare”) la comparsa della ginecomastia. In un contesto PCT tale composto, oltre ad esercitare la funzione di regolazione dell’attività estrogenica appena esposta, agendo a livello ipotalamico stimola il rilascio di GnRH e, consequenzialmente, di LH ed FSH dall’Ipofisi che a loro volta stimoleranno la sintesi di Testosterone e la spermatogenesi.
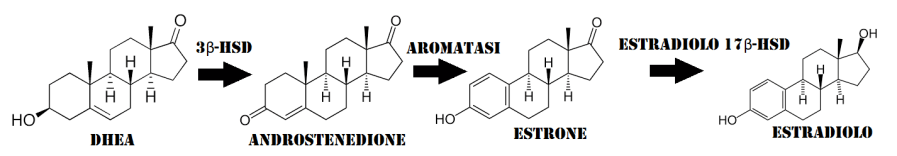
Il suo utilizzo massivo e cronico è stato spesso collegato aneddoticamente a rebound estrogenico. Ora, conoscendo la complessità d’azione che questo composto (ed i suoi metaboliti) ha sul controllo dell’attività estrogenica, si può facilmente ipotizzare che un suo uso protratto (legato anche alla dose e, quindi, al suo impatto sulla attività estrogenica sistemica) possa innescare degli adattamenti reattivi con conseguente aumento dell’attività estrogenica attraverso l’incremento dei livelli serici di Estradiolo e dell’attività non-genomica dello steroide (ipotizzabile anche un aumento del numero dei ER). Nel corso degli anni sono state esposte diverse ipotesi volte a spiegare i meccanismi attraverso i quali un abuso di Tamoxifene possa portare ad un rebound estrogenico. Una di queste ipotesi venne riportata all’inizio del secolo dal compianto A.L. Rea il quale affermava che la causa andasse ricercata nell’aumento del rilascio di DHEA da parte delle ghiandole surrenali e dalla sua successiva (e aumentata) conversione in Androstenedione e, attraverso l’intervento dell’enzima aromatasi che lo converte in Estrone e la successiva azione del estradiolo 17beta-deidrogenasi, Estradiolo.(32) In breve, secondo questa teoria i processi innescati causerebbero l’instaurarsi di livelli di E2 cronicamente alti con conseguente impossibilità del SERM di esplicare la sua azione. Questa teoria seppur, in parte, possa dare una spiegazione logica dei possibili meccanismi implicati manca di alcuni tasselli. Il principale “tallone d’Achille” è rappresentato dai livelli di E2 che, una volta aumentati, diventano dei competitor recettoriali più aggressivi rispetto al 4-OHT (che ricordiamo avere il 178% dell’affinità dell’Estradiolo per il ERα). Ciò potrebbe avvenire in situazioni di calo delle concentrazioni di 4-OHT seguenti alla riduzione del dosaggio del farmaco o alla sua cessazione, quindi, in questo ultimo caso, esplicabili in crescendo nei 7-14 giorni successivi all’interruzione della somministrazione e con una durata indeterminata. Di conseguenza, sembra più plausibile che l’aumento delle concentrazioni di E2, durante l’uso del Tamoxifene, si affianchi ad un consequenziale incremento dell’attività Non-Genomica dell’ormone e da un aumentato numero di ER. Seguendo questa logica, una volta interrotto l’uso del Tamoxifene, queste condizioni tenderanno ad aggravarsi come gli effetti avversi a loro legati.
Consultando la bibliografia scientifica disponibile, non si trovano accenni su un possibile rebound estrogenico in seguito all’uso di Tamoxifene, ma si parla nello specifico di “resistenza al Tamoxifene” o “sottoregolazione degli ER”.(33)(34) Nel caso della “resistenza al Tamoxifene” sembra che l’aumento dell’espressione del gene MACROD2 porti ad una risposta negativa all’azione del SERM con conseguente proliferazione delle cellule cancerose estradiolo-dipendenti. La sovra espressione di tale gene sembra essere di base genetica anche se non si esclude una risposta di adattamento in seguito ad uso cronico del composto in questione.
Raloxifene
Il Raloxifene, un altro SERM discretamente utilizzato nella pratica sportiva, è un agonista-antagonista misto del ER.(35)(36)(37) Ha effetti estrogenici a livello osseo ed epatico con effetti antiestrogenici nei seni e nell’utero. Le azioni biologiche del Raloxifene sono quindi ampiamente mediate dal legame con i ER. Questo legame determina l’attivazione di percorsi estrogenici in alcuni tessuti (agonismo) e il blocco di questi in altri (antagonismo). Le sue caratteristiche d’azione similari a quelle del Tamoxifene, sembrano poter far pensare ad un medesimo e ipotetico meccanismo che possa portare ad un rebound estrogenico. Questa volta la letteratura scientifica sembra dare alcune conferme. In un caso studio (38), una paziente di 66 anni si è presentata con recidiva metastatica acuta estrogeno-positiva e progesterone-positiva, carcinoma mammario Her-2 / neu-negativo, lesioni ossee (colonna lombare, bacino), noduli polmonari, metastasi epatiche, antigene tumorale elevato 15 e enzimi epatici, dispepsia e diarrea. La paziente aveva assunto Raloxifene per circa 8 anni. Dopo la sospensione del farmaco, parametri e sintomi clinici sono migliorati rapidamente senza terapia oncologica o altre forme di trattamento. Tre mesi dopo la sospensione del Raloxifene, l’oncologo ha prescritto alla paziente l’uso della Capecitabina dato che non riteneva plausibile un effetto di rebound estrogenico (anti-estrogen withdrawal effect – AEWE). Tuttavia, la regressione duratura è stata più indicativa di un effetto rebound dato dal Raloxifene rispetto alla chemioterapia o ad altri interventi. In seguito la paziente si è mostrata asintomatica con un buono stato di prestazione. La regressione metastatica epatica è stata confermata, senza alcun trattamento oncologico somministrato negli ultimi 16 mesi e circa 23 mesi dopo il termine d’uso del Raloxifene. Questo caso evidenzia la necessità di esaminare pazienti con carcinoma mammario per la possibilità di un AEWE con l’uso di Raloxifene o con altri SERM . Ovviamente, il caso presentato non è molto comparabile, soprattutto per quanto riguarda i tempi di somministrazione, ad un BodyBuilder supplementato chimicamente nella “media” ma, ciò nonostante, ci offre un indizio sulla probabilità che si possa manifestare un rebound estrogenico con l’uso di SERM.
Enzima Aromatasi, Inibitori della Aromatasi e “rebound estrogenico”
Enzima Aromatasi
L’Enzima Aromatasi, chiamato anche estrogeno sintetasi o estrogeno sintasi, è un enzima responsabile del processo fondamentale della biosintesi degli Estrogeni. Denominato CYP19A1, questo enzima è un membro della superfamiglia del citocromo P450 (EC 1.14.14.1), che sono monoossigenasi che catalizzano molte reazioni coinvolte nella steroidogenesi. In particolare, l’Aromatasi è responsabile dell’aromatizzazione degli Androgeni in Estrogeni. L’enzima Aromatasi è sintetizzato in molti tessuti tra cui le gonadi (cellule della granulosa), cervello, tessuto adiposo, placenta, vasi sanguigni, pelle e ossa, nonché nei tessuti dell’endometriosi, dei fibromi uterini, del cancro al seno e del cancro dell’endometrio.
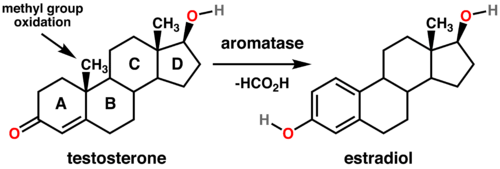
L’Aromatasi è localizzato nel reticolo endoplasmatico dove è regolato da promotori tissutali che sono a loro volta controllati da ormoni, citochine e altri fattori. Catalizza gli ultimi passaggi della biosintesi degli estrogeni dagli androgeni (in particolare, converte l’Androstenedione in Estrone e il Testosterone in Estradiolo). Queste fasi comprendono tre idrossilazioni successive del gruppo 19-metilico degli androgeni, seguite dall’eliminazione simultanea del gruppo metilico come formiato e aromatizzazione dell’anello A.
Reazioni generali per la conversione del Testosterone in Estradiolo catalizzata dall’Aromatasi. Gli Steroidi sono formati da quattro anelli fusi (A-B-C-D). L’Enzima Aromatasi converte l’anello “A” in uno stato aromatico.
Il gene esprime due varianti di trascrizione. (39) Nell’uomo, il gene CYP19, situato sul cromosoma 15q21.1, codifica per l’Enzima Aromatasi. (40) Il gene ha nove esoni codificanti e un numero di primi esoni non codificanti alternativi che regolano l’espressione specifica del tessuto. (41)
Il CYP19 è presente in un cordato precoce divergente, l’anfiosso cefalocordato (il Florida lancelet, Branchiostoma floridae), ma non nel precedente tunicato divergente Ciona intestinalis. Pertanto, gli evoluzionisti ipotizzano che il gene Aromatasi si sia evoluto precocemente nell’evoluzione dei cordati e non sembra essere presente negli invertebrati non-cordati (ad esempio insetti, molluschi, echinodermi, spugne, coralli). Tuttavia, gli Estrogeni possono essere sintetizzati in alcuni di questi organismi, attraverso altri percorsi sconosciuti.
I fattori noti che aumentano l’attività dell’Aromatasi includono l’età, l’obesità, l’Insulina, le gonadotropine e l’alcol. L’attività dell’Aromatasi risulta diminuita dalla Prolattina, dall’ormone anti-Mülleriano e dal glifosato , un comune erbicida.(42) L’attività dell’Aromatasi sembra essere migliorata in alcuni tessuti estrogeno-dipendenti come il tessuto mammario, nel carcinoma dell’endometrio, nell’endometriosi e nei fibromi uterini.
Gli Inibitori dell’Aromatasi (AI) sono una gruppo di farmaci usati nel trattamento del carcinoma mammario nelle donne in postmenopausa e nella ginecomastia negli uomini. Come i SERM, trovano un largo uso off-label in ambito sportivo durante la somministrazione di AAS aromatizzabili o durante la PCT. Possono anche essere utilizzati per la chemioprevenzione in donne ad alto rischio.
Esistono due tipi di Inibitori dell’Aromatasi approvati per il trattamento del carcinoma mammario e, quindi, diffusi anche per l’uso off-label: (43)
– Gli inibitori steroidei irreversibili, come l’Exemestano (nome commerciale Aromasin), formano un legame permanente e disattivante con l’Enzima Aromatasi.
– Gli inibitori non steroidei, come l’Anastrozolo (nome commerciale Arimidex) e il Letrozolo (nome commerciale Femara), inibiscono la sintesi degli Estrogeni attraverso la competizione reversibile per l’Enzima Aromatasi.
Gli inibitori dell’Aromatasi disponibili (AI) includono:
– Non selettivi:
• L’Aminoglutetimide, il quale però inibisce l’enzima P450scc agendo come inibitore della biosintesi di tutti gli ormoni steroidei (aprirò una nota a riguardo più avanti).
• Testolactone (nome commerciale Teslac) – Selettivi:
• Anastrozolo (Arimidex)
• Letrozolo (Femara)
• Exemestano (Aromasin)
• Vorozolo (Rivizor)
• Formestano (Lentaron)
• Fadrozolo (Afema)
– Non classificati:
• 1,4,6-Androstatrien-3,17-dione (ATD)
• 4-Androstene-3,6,17-trione (“6-OXO”)
Oltre agli AI farmaceutici, alcuni composti naturali hanno mostrato effetti di inibizione dell’Aromatasi, come le foglie di damiana. Il loro impatto non è stato pienamente chiarito sull’uomo.
Gli Inibitori dell’Aromatasi agiscono, proprio come suggerisce il nome, inibendo l’azione dell’enzima Aromatasi, che converte gli Androgeni in Estrogeni mediante un processo chiamato aromatizzazione. Poiché il tessuto mammario è stimolato dagli Estrogeni, diminuirne la produzione è un modo per sopprimere la recidiva del tessuto tumorale del seno. La principale fonte di Estrogeni è rappresentata dalle ovaie nelle donne in premenopausa, mentre nelle donne in post-menopausa la maggior parte degli Estrogeni del corpo viene prodotta nei tessuti periferici (al di fuori del SNC) e anche in alcuni siti del SNC in varie regioni del cervello. L’Estrogeno viene prodotto e agisce localmente in questi tessuti, ma qualsiasi estrogeno circolante, che esercita effetti estrogenici sistemici in uomini e donne, è il risultato dell’Estrogeno che sfugge al metabolismo locale e si diffonde nel sistema circolatorio.(44)
Come già accennato pocanzi, i composti AI sono anch’essi, al pari dei SERM, largamente utilizzati in ambito sportivo, sia come agenti di controllo dei livelli estrogenici durante l’uso di AAS aromatizzabili (uso preventivo della comparsa di effetti estrogenici), in caso di ginecomastia (spesso in combinazione con un SERM, specie se l’AI utilizzato è l’Exemestano) o in combinazione con i SERM in ambito PCT (specie nella fase preliminare dove viene utilizzata l’hCG).
Più che con i SERM, il rebound estrogenico è stato riportato, soprattutto aneddoticamente, con l’uso di AI, specialmente quelli reversibili (vedi Anastrozolo e Letrozolo).
L’Anastrozolo ed il Letrozolo agiscono legandosi in modo reversibile all’Enzima Aromatasi (unità eme del citocromo P450) e, attraverso l’inibizione competitiva, blocca la conversione degli Androgeni in Estrogeni nei tessuti periferici (extragonali).
Il Letrozolo ha dimostrato, attraverso studi clinici, di poter abbassare rapidamente il livello degli estrogeni fino al 65%. Il motivo principale è probabilmente legato alla capacità che la molecola ha di abbassare drasticamente gli estrogeni attraverso un legame competitivo reversibile al gruppo eme della relativa unità del citocromo P450. L’Anastrozolo, il quale agisce similmente al Letrozolo, ha mostrato una riduzione del livello estrogenico in soggetti di sesso maschile del 50%.(45) Il problema di un possibile rebound estrogenico con questi composti nasce proprio dalla loro natura “reversibile”.
Rebound estrogenici sono stati riportati sia con l’uso di Letrozolo che con l’uso di Anastrozolo, sebbene il Letrozolo, avendo un azione inibitoria più marcata, sembra causare rebound di intensità maggiore dopo la sua interruzione. La causa del rebound estrogenico indotto da cessazione d’uso di Letrozolo o di Anastrozolo è proprio legata al comportamento che queste due molecole esplicano nei confronti dell’Enzima aromatasi. Il legame tra la molecola di Letrozolo o di Anastrozolo con l’Enzima Aromatasi è solo temporanea e non decreta la completa de-attivazione dell’enzima responsabile della conversione degli Androgeni in Estrogeni. Una volta interrotta l’assunzione del composto, i livelli di Aromatasi possono salire significativamente con la possibile comparsa di un rebound estrogenico. Una pratica per evitare che ciò si verifichi consiste nell’uso limitato dei due composti e in una loro graduale sospensione. Con questi farmaci, il rebound estrogenico può essere “multifattoriale” derivando non solo dalla cessazione del farmaco in questione ma anche da un incremento dell’espressione dell’Enzima Aromatasi come risposta adattativa all’uso (specie nel lungo termine). Ciò significa che, anche durate un utilizzo cronico, i livelli di E2 possono mostrare degli aumenti, aumenti che diverranno maggiormente significativi una volta cessato l’uso del farmaco. Cessata l’azione del composto non solo viene a mancare un controllo dell’aromatizzazione ma questa risulta anche incrementata rispetto ai tassi pre-utilizzo (l’aumento dell’espressione dell’aromatasi è un comportamento adattativo che si può manifestare anche durante cicli particolarmente lunghi). Prendendo in considerazione la vita attiva del Letrozolo e dell’Anastrozolo, il possibile rebound estrogenico potrebbe manifestarsi in crescendo dopo 64-120h circa dall’ultima assunzione.
Exemestano
L’Exemestano, invece, è un inibitore dell’Aromatasi steroideo irreversibile di tipo I, strutturalmente correlato al substrato naturale 4-androstenedione. Agisce come un falso substrato per l’Enzima Aromatasi e viene trasformato in un intermedio che si lega irreversibilmente al sito attivo dell’enzima causandone l’inattivazione, un effetto noto anche come “inibizione suicida”. Essendo strutturalmente simile agli obiettivi dell’enzima, l’Exemestano si lega in modo permanente a quest’ultimo, impedendo la sua azione di conversione degli Androgeni in Estrogeni. Il tasso di soppressione degli Estrogeni da parte dell’Exemestano varia dal 35% per l’Estradiolo (E2) al 70% per l’Estrone (E1).(46)
Grazie alla sua caratteristica di “inibitore selettivo”, l’Exemestano sembra non causare un rebound estrogenico dopo la sua cessazione. Nonostante ciò, un suo uso temporalmente protratto potrebbe (teoricamente) causare, similmente a quanto accade con l’uso di Letrozolo e Anastrozolo, un aumento dell’espressione dell’Enzima Aromatasi nonché un aumento del numero di ER come risposta adattativa.
Ovviamente, questa possibilità può interessare tutti gli AI con legame irreversibile (es. Formestano).
Queste sono semplici ipotesi nate da una riflessione sulle possibili cause e meccanismi che potrebbero (teoricamente) portare al manifestarsi di un rebound estrogenico con l’uso di tali composti. La letteratura scientifica, purtroppo, non ci aiuta a fare molta chiarezza sulla connessione AI/rebound estrogenico, sebbene esistono alcuni studi nei quali la cosa viene accennata.(47)
Aminoglutetimide
*Nota sull’Aminoglutetimide: inibendo l’enzima P450scc e agendo, di conseguenza, come inibitore della biosintesi di tutti gli ormoni steroidei, l’abuso di Aminoglutetimide può potenzialmente causare non solo un rebound estrogenico ma anche un rebound dei livelli di cortisolo. Lo stesso vale per il farmaco Trylostano.
Conclusioni
Basarsi per la maggior parte sui dati aneddotici è un azzardo, anche perché la maggior parte delle variabili soggettive in gioco rimangono celate. Banalmente, alcuni lamentano rebound estrogenici che alla fine non risultano legati all’uso del SERM o del AI ma alla loro (o del Preparatore) ignoranza, come quando cessano l’utilizzo di AAS, e di SERM e/o AI, senza preoccuparsi di svolgere un adeguata PCT convinti, magari, che un po’ di Mesterolone (Proviron) risolvi tutto. Infatti, la maggior parte dei casi di presunti rebound estrogenici SERM o AI dipendenti sono causati da una repentina cessazione d’uso di questi e di AAS, oppure da una PCT mal pianificata e/o che non ha dato i risultati sperati (vedi anche alterazione della Testosterone:Estradiolo ratio). Queste condizioni sono legate più che altro ad una alterazione del HPTA data dall’uso di AAS e non ad una presunta azione diretta del SERM e/o AI precedentemente utilizzati.
In conclusione, l’uso ponderato e consapevole è l’unica vera arma che l’atleta supplementato chimicamente (o il Preparatore che lo segue) ha per far si che ipotetici rebound non si manifestino.
Gabriel Bellizzi
Riferimenti:
1- “Definition of antiestrogen – NCI Dictionary of Cancer Terms, Definition of antiestrogen – NCI Dictionary of Cancer Terms”.,
2- Jump up ^ “antiestrogen” at Dorland’s Medical Dictionary
3- Jump up ^ Judi Lindsley Nath (2006). Using Medical Terminology: A Practical Approach. Lippincott Williams & Wilkins. pp. 977–. ISBN 978-0-7817-4868-1.
4- Dahlman-Wright K, Cavailles V, Fuqua SA, Jordan VC, Katzenellenbogen JA, Korach KS, Maggi A, Muramatsu M, Parker MG, Gustafsson JA (Dec 2006). “International Union of Pharmacology. LXIV. Estrogen receptors”. Pharmacological Reviews. 58 (4): 773–81. doi:10.1124/pr.58.4.8. PMID 17132854.
5- Levin ER (Aug 2005). “Integration of the extranuclear and nuclear actions of estrogen”. Molecular Endocrinology. 19 (8): 1951–9. doi:10.1210/me.2004-0390. PMC 1249516 . PMID 15705661.
6- Levin ER (Aug 2005). “Integration of the extranuclear and nuclear actions of estrogen”. Molecular Endocrinology. 19 (8): 1951–9. doi:10.1210/me.2004-0390. PMC 1249516 . PMID 15705661.
7- Wang C, Fu M, Angeletti RH, Siconolfi-Baez L, Reutens AT, Albanese C, Lisanti MP, Katzenellenbogen BS, Kato S, Hopp T, Fuqua SA, Lopez GN, Kushner PJ, Pestell RG (May 2001). “Direct acetylation of the estrogen receptor alpha hinge region by p300 regulates transactivation and hormone sensitivity”. The Journal of Biological Chemistry. 276 (21): 18375–83. doi:10.1074/jbc.M100800200. PMID 11279135.
8- Zivadinovic D, Gametchu B, Watson CS (2005). “Membrane estrogen receptor-alpha levels in MCF-7 breast cancer cells predict cAMP and proliferation responses”. Breast Cancer Research. 7 (1): R101–12. doi:10.1186/bcr958. PMC 1064104 . PMID 15642158.
9- Björnström L, Sjöberg M (Jun 2004). “Estrogen receptor-dependent activation of AP-1 via non-genomic signalling”. Nuclear Receptor. 2 (1): 3. doi:10.1186/1478-1336-2-3. PMC 434532 . PMID 15196329.
10- Lu Q, Pallas DC, Surks HK, Baur WE, Mendelsohn ME, Karas RH (Dec 2004). “Striatin assembles a membrane signaling complex necessary for rapid, nongenomic activation of endothelial NO synthase by estrogen receptor alpha”. Proceedings of the National Academy of Sciences of the United States of America. 101 (49): 17126–31. doi:10.1073/pnas.0407492101. PMC 534607 . PMID 15569929.
11- Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, Masushige S, Gotoh Y, Nishida E, Kawashima H, Metzger D, Chambon P (Dec 1995). “Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase”. Science. 270 (5241): 1491–4. doi:10.1126/science.270.5241.1491. PMID 7491495.
12- Prossnitz ER, Arterburn JB, Sklar LA (Feb 2007). “GPR30: A G protein-coupled receptor for estrogen”. Molecular and Cellular Endocrinology. 265-266: 138–42. doi:10.1016/j.mce.2006.12.010. PMC 1847610 . PMID 17222505.
13- Otto C, Rohde-Schulz B, Schwarz G, Fuchs I, Klewer M, Brittain D, Langer G, Bader B, Prelle K, Nubbemeyer R, Fritzemeier KH (Oct 2008). “G protein-coupled receptor 30 localizes to the endoplasmic reticulum and is not activated by estradiol”. Endocrinology. 149 (10): 4846–56. doi:10.1210/en.2008-0269. PMID 18566127.
14- Harris HA, Albert LM, Leathurby Y, Malamas MS, Mewshaw RE, Miller CP, Kharode YP, Marzolf J, Komm BS, Winneker RC, Frail DE, Henderson RA, Zhu Y, Keith JC (Oct 2003). “Evaluation of an estrogen receptor-beta agonist in animal models of human disease”. Endocrinology. 144 (10): 4241–9. doi:10.1210/en.2003-0550. PMID 14500559.
15- Deroo BJ, Korach KS (Mar 2006). “Estrogen receptors and human disease”. The Journal of Clinical Investigation. 116 (3): 561–70. doi:10.1172/JCI27987. PMC 2373424 . PMID 16511588.
16- Riggs BL, Hartmann LC (Feb 2003). “Selective estrogen-receptor modulators — mechanisms of action and application to clinical practice”. The New England Journal of Medicine. 348 (7): 618–29. doi:10.1056/NEJMra022219. PMID 12584371.
17- Cameron JL, Cameron AM (20 November 2013). Current Surgical Therapy. Elsevier Health Sciences. pp. 582–. ISBN 978-0-323-22511-3.
18- Huang X, Aslanian RG (19 April 2012). Case Studies in Modern Drug Discovery and Development. John Wiley & Sons. pp. 392–394. ISBN 978-1-118-21967-6.
19- Pickar JH, Komm BS (Sep 2015). “Selective estrogen receptor modulators and the combination therapy conjugated estrogens/bazedoxifene: A review of effects on the breast”. Post Reproductive Health. 21 (3): 112–21. doi:10.1177/2053369115599090. PMID 26289836.
20- Mirkin S, Pickar JH (Jan 2015). “Selective estrogen receptor modulators (SERMs): a review of clinical data”. Maturitas. 80 (1): 52–7. doi:10.1016/j.maturitas.2014.10.010. PMID 25466304.
21- Desta Z, Ward BA, Soukhova NV, Flockhart DA (Sep 2004). “Comprehensive evaluation of tamoxifen sequential biotransformation by the human cytochrome P450 system in vitro: prominent roles for CYP3A and CYP2D6”. The Journal of Pharmacology and Experimental Therapeutics. 310 (3): 1062–75. doi:10.1124/jpet.104.065607. PMID 15159443.
22- Ahmad A, Shahabuddin S, Sheikh S, Kale P, Krishnappa M, Rane RC, Ahmad I (December 2010). “Endoxifen, a new cornerstone of breast cancer therapy: demonstration of safety, tolerability, and systemic bioavailability in healthy human subjects”. Clinical Pharmacology and Therapeutics. 88 (6): 814–7. doi:10.1038/clpt.2010.196. PMID 20981001.
23- Wang DY, Fulthorpe R, Liss SN, Edwards EA (Feb 2004). “Identification of estrogen-responsive genes by complementary deoxyribonucleic acid microarray and characterization of a novel early estrogen-induced gene: EEIG1”. Molecular Endocrinology. 18 (2): 402–11. doi:10.1210/me.2003-0202. PMID 14605097.
24- Kuhl H (2005). “Pharmacology of estrogens and progestogens: influence of different routes of administration”. Climacteric. 8 Suppl 1: 3–63. doi:10.1080/13697130500148875. PMID 16112947.
25- Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M (Dec 2000). “Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription”. Cell. 103 (6): 843–52. doi:10.1016/S0092-8674(00)00188-4. PMID 11136970.
26- Massarweh S, Osborne CK, Creighton CJ, Qin L, Tsimelzon A, Huang S, Weiss H, Rimawi M, Schiff R (Feb 2008). “Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function”. Cancer Research. 68 (3): 826–33. doi:10.1158/0008-5472.CAN-07-2707. PMID 18245484.
27- Hurtado A, Holmes KA, Geistlinger TR, Hutcheson IR, Nicholson RI, Brown M, Jiang J, Howat WJ, Ali S, Carroll JS (Dec 2008). “Regulation of ERBB2 by oestrogen receptor-PAX2 determines response to tamoxifen”. Nature. 456 (7222): 663–6. Bibcode:2008Natur.456..663H. doi:10.1038/nature07483. PMC 2920208 . PMID 19005469.
28- Osborne CK, Bardou V, Hopp TA, Chamness GC, Hilsenbeck SG, Fuqua SA, Wong J, Allred DC, Clark GM, Schiff R (Mar 2003). “Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer”. Journal of the National Cancer Institute. 95 (5): 353–61. doi:10.1093/jnci/95.5.353. PMID 12618500.
29- “New Mechanism Predicts Tamoxifen Response: PAX2 gene implicated in tamoxifen-induced inhibition of ERBB2/HER2-mediated tumor growth”. http://www.modernmedicine.com. 2008-11-13. Archived from the original on 2011-07-14. Retrieved 2008-11-14.
30- “Study sheds new light on tamoxifen resistance”. News. CORDIS News. Archived from the original on 2009-02-20. Retrieved 2008-11-14.
31- Liu J, Flockhart PJ, Lu D, Lv W, Lu WJ, Han X, Cushman M, Flockhart DA (2013). “Inhibition of cytochrome p450 enzymes by the e- and z-isomers of norendoxifen”. Drug Metab. Dispos. 41 (9): 1715–20. doi:10.1124/dmd.113.052506. PMC 3876808 . PMID 23824607.
32- Chemical muscle enhancement. Report. B.B. desk reference. di Author L. Rea. Pag. 106.
33- https://www.rdmag.com/article/2014/12/journal-watch-tamoxifen-news-again
34- https://www.ncbi.nlm.nih.gov/pubmed/14687597
35- Bryant HU (2001). “Mechanism of action and preclinical profile of raloxifene, a selective estrogen receptor modulation”. Rev Endocr Metab Disord. 2 (1): 129–38. PMID 11704975.
36- Thiebaud D, Secrest RJ (2001). “Selective estrogen receptor modulators: mechanism of action and clinical experience. Focus on raloxifene”. Reprod. Fertil. Dev. 13 (4): 331–6. PMID 11800172.
37- Gizzo S, Saccardi C, Patrelli TS, Berretta R, Capobianco G, Di Gangi S, Vacilotto A, Bertocco A, Noventa M, Ancona E, D’Antona D, Nardelli GB (2013). “Update on raloxifene: mechanism of action, clinical efficacy, adverse effects, and contraindications”. Obstet Gynecol Surv. 68 (6): 467–81. doi:10.1097/OGX.0b013e31828baef9. PMID 23942473.
38- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5739193/
39- “Entrez Gene: CYP19A1 cytochrome P450, family 19, subfamily A, polypeptide 1”.
40- Toda K, Shizuta Y (April 1993). “Molecular cloning of a cDNA showing alternative splicing of the 5′-untranslated sequence of mRNA for human aromatase P-450”. European Journal of Biochemistry. 213 (1): 383–9. doi:10.1111/j.1432-1033.1993.tb17772.x. PMID 8477708.
41- Czajka-Oraniec I, Simpson ER (2010). “Aromatase research and its clinical significance”. Endokrynologia Polska. 61 (1): 126–34. PMID 20205115.
42- Gasnier C, Dumont C, Benachour N, Clair E, Chagnon MC, Séralini GE (August 2009). “Glyphosate-based herbicides are toxic and endocrine disruptors in human cell lines”. Toxicology. 262 (3): 184–91. doi:10.1016/j.tox.2009.06.006. PMID 19539684.
43- Mokbel K (2002). “The evolving role of aromatase inhibitors in breast cancer”. Int. J. Clin. Oncol. 7 (5): 279–83. doi:10.1007/s101470200040 (inactive 2017-01-15). PMID 12402060.
44- Simpson ER (2003). “Sources of estrogen and their importance”. J. Steroid Biochem. Mol. Biol. 86 (3–5): 225–30. doi:10.1016/S0960-0760(03)00360-1. PMID 14623515.
45- Leder BZ, Rohrer JL, Rubin SD, Gallo J, Longcope C (March 2004). “Effects of aromatase inhibition in elderly men with low or borderline-low serum testosterone levels”. J. Clin. Endocrinol. Metab. 89 (3): 1174–80. doi:10.1210/jc.2003-031467. PMID 15001605.
46- Mauras, N; Lima, J; Patel, D; Rini, A; Di Salle, E; Kwok, A; Lippe, B (2003). “Pharmacokinetics and Dose Finding of a Potent Aromatase Inhibitor, Aromasin (Exemestane), in Young Males”. The Journal of Clinical Endocrinology & Metabolism. 88 (12): 5951–6. doi:10.1210/jc.2003-031279. PMID 14671195.
47- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3263690/
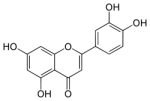
La Luteolina, un flavonoide contenuto nel rosmarino, timo, prezzemolo e negli agrumi, sembra essere un efficace composto anti-aromatase, almeno secondo uno studio svolto su animali e pubblicato da ricercatori del Centro di Scienze Sanitarie dell’Università di Pechino nel Journal of Pharmacology and Experimental Therapeutics.(1) A differenza dei composti anti-aromatasi sintetici, la Luteolina sembra avere il potenziale di migliorare anche l’equilibrio del Colesterolo.
Già nel 2013, in uno studio in vitro svolto da farmacologi dell’Istituto di biologia di Chengdu, dove si erano prese in esame oltre 100 sostanze al fine di valutarne il potenziale antiestrogenico, la Luteolina risultò la più interessante a tal fine.(2)
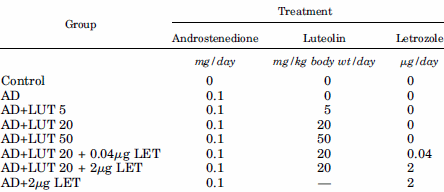
Nello studio del quale ho introdotto brevemente i risultati emersi all’inizio di questo articolo, i ricercatori hanno usato topi di sesso femminile le cui ovaie sono state rimosse chirurgicamente come pratica preliminare per lo svolgimento dell’esperimento. Ad un certo numero di animali sono state somministrate iniezioni giornaliere di Androstenedione [AD]. Come ben sappiamo, l’enzima aromatasi converte l’Androstenedione in Estrone e, attraverso l’azione dell’Estradiolo 17beta-deidrogenasi, in Estradiolo.
Ad alcuni animali è stata anche somministrata per via orale la Luteolina [LUT]. Le dosi utilizzate sono mostrate nella figura riportata di seguito. L’equivalente umano delle dosi utilizzate varia dai 45mg ai 450mg di Luteolina al giorno. Alcuni topi sono stati trattati con iniezioni di Letrozolo [LET], conosciutissimo e potente inibitore dell’aromatasi non steroideo di terza generazione.
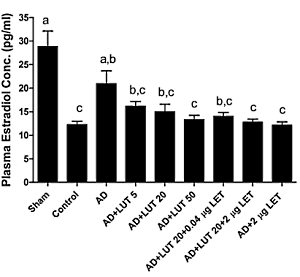
Dopo 12 settimane, i ricercatori hanno misurato le concentrazioni di Estradiolo nel sangue dei topi. La figura seguente mostra che l’effetto anti-aromatasi dato dalla dose più alta di Luteolina era uguale a quello ottenuto con l’uso del Letrozolo.
I ricercatori avevano impiantato nei topi cellule di cancro al seno estradiolo-sensibili. Sia la Luteolina che il Letrozolo hanno inibito la crescita tumorale, ma il Letrozolo ha ottenuto risultati leggermente migliori rispetto alla Luteolina.
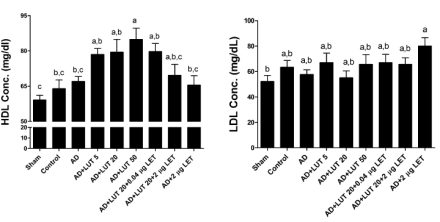
L’uso degli inibitori dell’aromatasi ha tra i suoi effetti collaterali quello di poter causare uno squilibrio delle lipoproteine. Le concentrazioni di LDL aumentano mentre quelle di HDL diminuiscono. La Luteolina, pur agendo attraverso l’inibizione dell’enzima aromatasi, mostra l’effetto opposto sull’equilibrio del Colesterolo. La Luteolina mostra di causare un abbassamento delle concentrazioni di LDL e aumento delle concentrazioni di HDL.
La figura sopra riportata mostra che la Luteolina può anche annullare l’effetto negativo del Letrozolo sui livelli di Colesterolo.
La Luteolina non ha avuto alcun effetto sui livelli di Trigliceridi.
I ricercatori sottolineano il fatto che, sebbene la Luteolina sia ampiamente presenti in diversi alimenti vegetali, i dosaggi utilizzati nello studio qui riportato erano superiori ai livelli normalmente consumati dagli esseri umani. Tuttavia, questo studio potrebbe fornire le basi scientifiche per lo sviluppo nutraceutico o farmacologico di questo flavone.
In fine, i ricercatori concludono che, dato il potenziale di alterazione del rapporto LDL/HDL solitamente associato all’uso a lungo termine degli inibitori dell’aromatasi, la somministrazione di Luteolina può essere un potenziale meccanismo di compensazione senza compromissioni sull’aromatasi.
La questione degli Estrogeni nell’ambito delle preparazioni comprendenti farmaci per il miglioramento delle prestazioni, in special modo nel Bodybuilding, è generalmente mal compresa e, di conseguenza, mal gestita. Complice di questa “mala gestione” è l’ignoranza sia specifica, cioè portata da una scarsa disponibilità di testi di facile accesso (e comprensione) che trattano l’argomento in modo esaustivo, sia indotta dal comportamento della maggior parte dei preparatori o degli atleti “solisti” che ignorano totalmente la necessità di un aggiornamento continuo in tutte le componenti della preparazione (allenamento, nutrizione e supplementazione). Con questa pubblicazione è mia intenzione fornire una buona base di conoscenza sugli Estrogeni (loro biosintesi, azioni ecc…) e sulla migliore gestione di questi nel contesto di una preparazione.
Iniziamo per gradi…
Estrogeni e loro caratteristiche/azioni principali (nella donna e nell’uomo)
Estradiolo
Gli Estrogeni sono i principali ormoni sessuali femminili. Si tratta di ormoni steroidei responsabili dello sviluppo e della regolazione del sistema riproduttivo femminile e delle caratteristiche sessuali secondarie. Con il termine estrogeno ci si può anche riferire a qualsiasi sostanza, naturale o sintetica, che mima gli effetti dell’ormone naturale.(1) L’estrogeno prevalente in quantità e potenza è l’Estradiolo, anche se diversi metaboliti dell’estradiolo hanno anche una marcata attività ormonale estrogenica. Versioni sintetiche degli estrogeni vengono utilizzate come principio attivo di farmaci contraccettivi orali, nella terapia ormonale sostitutiva per le donne in postmenopausa, nelle donne ipogonadiche e nei transgender, oltre che nel trattamento di alcuni tumori ormone-sensibili come il cancro alla prostata e il cancro al seno. Gli Estrogeni sono una delle tre classi di ormoni sessuali, insieme agli Steroidi Androgeni/Anabolizzanti come il Testosterone e i progestinici come il Progesterone.
Gli Estrogeni sono sintetizzati in tutti i vertebrati (2) così come in alcuni insetti.(3) Le tre principali forme di estrogeno presenti naturalmente nelle donne sono l’Estrone (E1), l’Estradiolo (E2) e l’Estriolo (E3). Un altro tipo di Estrogeno chiamato Estetrolo (E4) viene prodotto solo durante la gravidanza. Quantitativamente, gli Estrogeni circolano a livelli inferiori rispetto agli Androgeni sia negli uomini che nelle donne.(4) Mentre i livelli di Estrogeni sono significativamente più bassi nei maschi rispetto alle femmine, gli Estrogeni, tuttavia, hanno anche importanti ruoli fisiologici nei soggetti di sesso maschile.(5)
Come tutti gli ormoni steroidei, gli Estrogeni penetrano facilmente attraverso la membrana cellulare. Una volta all’interno della cellula, si legano e attivano i recettori degli estrogeni (ER) che a loro volta modulano l’espressione di molti geni.(6) Inoltre, gli Estrogeni si legano e attivano rapidamente i recettori degli estrogeni di membrana (mER), (7)(8) come i GPER (GPR30).(9)
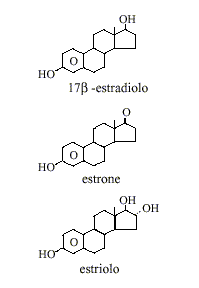
Come già precedentemente accennato, i tre principali Estrogeni presenti naturalmente nelle donne sono l’Estrone (E1), l’Estradiolo (E2) e l’Estriolo (E3). L’Estradiolo è l’estrogeno predominante negli anni riproduttivi sia in termini assoluti nel siero che in termini di attività estrogenica. Durante la menopausa, l’Estrone è l’Estrogeno circolante predominante mentre durante la gravidanza è l’Estriolo ad essere l’Estrogeno circolante predominante in termini di livelli sierici. Anche se l’Estriolo è il più abbondante dei tre estrogeni è anche il più debole, mentre l’Estradiolo è il più forte con una potenza di circa 80 volte quella dell’Estriolo.(10) Quindi, l’Estradiolo è l’Estrogeno più importante nelle donne non in gravidanza che si trovano tra le fasi del menarca e della menopausa.
Estetrolo
Tuttavia, durante la gravidanza questo ruolo passa all’Estriolo e nelle donne in postmenopausa l’Estrone diventa la forma primaria di Estrogeno nel corpo. Come già accennato, un altro tipo di Estrogeno chiamato Estetrolo (E4) viene prodotto solo durante la gravidanza. Oltre che dai follicoli ovarici, tutte le diverse forme di estrogeni sono sintetizzate a partire dagli Androgeni, in particolare dal Testosterone e dal Androstenedione, da parte dell’enzima aromatasi.
Altri substrati utilizzati dal corpo per la sintesi estrogenica, senza il coinvolgimento primario dell’enzima aromatasi, sono il 27-idrossicholesterolo, il Deidroepiandrosterone (DHEA), il 7-oxo-DHEA, il 7α-idrossi-DHEA, il 16α-idrossi-DHEA, il 7β-idrossiepiandrosterone, l’Androstenedione (A4), l’Androstenediolo (A5), il 3α-androstanediolo e il 3β-androstanediolo. Alcuni metaboliti estrogenici, come i catecol-estrogeni 2-idrossiestradiolo, 2-idrossestrone, 4-idrossiestradiolo e 4-idrossestrone, nonché il 16α-idrossiestrone, sono composti estrogenici con vari gradi di attività. L’importanza biologica di questi estrogeni minori non è del tutto chiara. Tutti questi composti possono avere importanti funzioni estrogeniche nel corpo.
Steroidogenesi; gli Estrogeni sono rappresentati in basso a destra.
Recettore degli Estrogeni alfa (ERα)
Le azioni degli Estrogeni sono mediate dal Recettore Estrogeno (ER), una proteina nucleare dimerica che si lega al DNA e controlla l’espressione genica. Come altri ormoni steroidei, l’Estrogeno entra passivamente nella cellula nella quale si lega e attiva il Recettore dell’Estrogeno. Il complesso ER si lega a specifiche sequenze di DNA chiamate “elemento di risposta ormonale” per attivare la trascrizione di geni bersaglio: in uno studio nel quale si è utilizzate una linea di cellule del cancro al seno estrogeno-dipendente come modello, sono stati identificati 89 di questi geni.(11) Poiché l’Estrogeno entra in tutte le cellule, le sue azioni dipendono dalla presenza del ER nella cellula. L’ER è espresso in tessuti specifici tra cui l’ovaio, l’utero e il seno. Gli effetti metabolici degli Estrogeni nelle donne in postmenopausa sono stati legati al polimorfismo genetico dell’ER. (12)
Mentre gli Estrogeni sono presenti sia negli uomini che nelle donne, sono solitamente presenti a livelli significativamente più alti nelle donne in età riproduttiva. Essi promuovono lo sviluppo delle caratteristiche sessuali secondarie femminili, come i seni, e sono anche coinvolti nell’ispessimento dell’endometrio e altri aspetti della regolazione del ciclo mestruale. Nei maschi, l’Estrogeno regola alcune funzioni del sistema riproduttivo importanti per la maturazione dello sperma (13)(14)(15) e sembra essere necessario per una sana libido.(16) Inoltre, vi sono molti altri cambiamenti strutturali indotti dall’Estrogeno oltre ad altre funzioni.
Gli Estrogeni, nelle femmine, sono prodotti principalmente dalle ovaie e, durante la gravidanza, dalla placenta.(17) L’Ormone Follicolo Stimolante (FSH) stimola la produzione ovarica di Estrogeni da parte delle cellule della granulosa dei follicoli ovarici e dei corpi lutei. Alcuni Estrogeni vengono anche prodotti in quantità minori in altri tessuti come il fegato, le ghiandole surrenali e i seni. Queste fonti secondarie di Estrogeni sono particolarmente importanti nelle donne in postmenopausa. Anche nelle cellule adipose vengono sintetizzati Estrogeni (vedi azione dell’Enzima Aromatasi)(18)
Nelle femmine, la sintesi degli Estrogeni inizia nelle cellule della teca follicolare interna nell’ovaio, dalla sintesi del Androstenedione a partire dal Colesterolo. L’Androstenedione è uno steroide con debole attività androgenica il cui scopo è prevalentemente quello di fungere da precursore di più potenti androgeni come il Testosterone così come dell’Estrogeno. Questo steroide attraversa la membrana basale nelle cellule della granulosa circostanti, dove o viene immediatamente convertito in Estrone, o in Testosterone e poi successivamente in Estradiolo. La conversione del Androstenedione in Testosterone è catalizzata dal 17β-idrossisteroide deidrogenasi (17β-HSD), mentre la conversione del Androstenedione e del Testosterone in Estrone ed Estradiolo è rispettivamente catalizzata dal aromatasi, entrambi enzimi che sono espressi nelle cellule della granulosa. Al contrario, le cellule della granulosa non dispongono di 17α-idrossilasi e 17,20-liasi, mentre le cellule della teca follicolare interna esprimono questi enzimi e il 17β-HSD, ma non l’aromatasi. Quindi sia le cellule della granulosa che le cellule della teca follicolare interna sono essenziali per la produzione di Estrogeni nelle ovaie.
I livelli di Estrogeni variano durante il ciclo mestruale, con livelli più alti vicino alla fine della fase follicolare e poco prima dell’ovulazione.
Intervalli di riferimento nelle concentrazioni di Estradiolo, il principale Estrogeno circolante, durante il ciclo mestruale. CYP1A1
Gli Estrogeni sono metabolizzati tramite idrossilazione da enzimi citocromo P450 quali CYP1A1 e CYP3A4 e tramite coniugazione del estrogeno sulfotransferasi (sulfato) e del UDP-glucuroniltransferasi (glucuronidazione). Inoltre, l’Estradiolo viene deidrogenato dal 17β-idrossiesteroide deidrogenasi nel molto meno potente Estrone. Queste reazioni si verificano principalmente nel fegato, ma avvengono anche in altri tessuti.
Gli estrogeni sono implicati nel rilascio di GH e IGF-1 sia nell’uomo che nella donna. Le donne hanno mediamente livelli di GH più elevati sebbene sembri che ne siano meno sensibili (vedi percentuale di grasso corporeo femminile).
Quindi, per riepilogare, gli Estrogeni nelle femmine regolano la maturazione sessuale intervenendo nello sviluppo dell’apparato genitale.
La loro massiccia secrezione in epoca puberale induce la chiusura delle cartilagini di coniugazione delle ossa lunghe, terminando di fatto, la fase di accrescimento staturale.
Gli estrogeni stimolano lo sviluppo stromale della mammella e il mantenimento delle caratteristiche femminili secondarie (crescita delle mammelle, distribuzione dei peli, voce, statura, ossatura, distribuzione del grasso).
Permettono la fecondazione e la gravidanza, intervenendo nella regolazione del ciclo mestruale.
Regolano la distribuzione del grasso corporeo, favorendone il deposito nelle anche, nelle natiche, nelle cosce e nell’addome al di sotto dell’ombelico.
Mantengono il trofismo osseo ed hanno quindi azione protettiva nei confronti dell’osteoporosi
Stimolano la sintesi di trigliceridi e l’aumento delle lipoproteine ad alta densità (HDL) proteggendo le pareti vasali dal danno arteriosclerotico.
Stimolano la lipolisi nel tessuto muscolare ed adiposo. Per questo motivo gli estrogeni migliorano la prestazione degli sport di durata risparmiando il glicogeno muscolare a scapito degli acidi grassi
Regolano molte funzioni cerebrali fra cui l’attenzione e la memoria.
Stimolano la sintesi epatica di numerosi enzimi e proteine (SHBG, angiotensinogeno).
Enzima Aromatasi
Nell’uomo, invece, gli estrogeni circolanti derivano per la maggior parte dall’aromatizzazione degli Androgeni circolanti (19). Il complesso enzimatico conosciuto come aromatasi è responsabile dell’aromatizzazione dell’anello A degli Androgeni che comporta la trasformazione in sostanze ad attività estrogenica. (20) (21) (22)
Recettore degli Estrogeni beta (ERβ)
La trascrizione del gene per l’aromatasi e l’espressione dell’enzima aromatasi avviene in un ampio numero di tessuti, come i testicoli, il tessuto adiposo, il muscolo, il fegato, il cervello, i follicoli piliferi, i fibroblasti etc. (20). L’azione degli Estrogeni, come già accennato, si esplica attraverso il loro legame a recettori specifici, che appartengono alla ‘superfamiglia’ dei recettori nucleari (23). Finora sono stati caratterizzati due tipi di recettori per gli Estrogeni (ER): l’ER classico, ora denominato ERα (24) (25) (26), ed un altro sottotipo denominato ERβ (27). Entrambi sono recettori intranucleari che, una volta attivati, modulano a livello genomico l’attività di trascrizione genica. Di recente è stata documentata anche una via non-genomica dell’azione degli Estrogeni, che prevede un’interazione plasma-membrana del recettore estrogenico (28) (29) (30).
Conversione del Testosterone in Estradiolo
L’Estradiolo (E2) nel maschio è sintetizzato dal Testosterone attraverso l’enzima aromatasi o dall’Estrone attraverso la 17β-idrossisteroido deidrogenasi (17β-HSD) (31). L’E2 prodotto giornalmente nel maschio è stimato essere circa 35-45 μg (0.130- 0.165 μmol), dei quali il 15-20% è direttamente prodotto dal testicolo (32) (19), un 60% è prodotto dall’aromatizzazione periferica del Testosterone circolante ed il restante 20% dalla conversione periferica dell’Estrone. L’Estrone (E1) può anche essere direttamente secreto dai surreni (33) o derivare dall’aromatizzazione periferica dell’Androstenedione, quest’ultimo prodotto in parte dal surrene ed in parte derivante dalla conversione periferica del Testosterone. I testicoli comunque contribuiscono maggiormente, rispetto ai surreni, alla totale produzione di Estradiolo circolante e la via enzimatica di sintesi maggiormente coinvolta nella secrezione di Estrogeni nel maschio è quella aromatasica, mentre la via della 17β-HSD ha una minore rilevanza “in vivo”. Solo il 2-3% dell’Estradiolo circolante è libero, la maggior parte dell’ormone è legato all’albumina o alla globulina legante gli ormoni sessuali (SHBG) (34) e la frazione non legata alla SHBG è considerata bioattiva a livello dei tessuti bersaglio.
SHBG
I progressi negli studi effettuati negli ultimi anni hanno infatti dimostrato in maniera inequivocabile il ruolo fisiologico che gli Estrogeni hanno anche nell’uomo. Gli Estrogeni svolgono un ruolo chiave sul sistema immunitario, a livello del timo, agiscono a livello ipofisario e cardiovascolare, osseo e del metabolismo glucidico e lipidico.
Un eccesso estrogenico nella donna favorisce l’accumulo di tessuto adiposo e la comparsa di ritenzione idrica, oltre ad esporre la donna ad un elevato rischio di sviluppare alcune forme di cancro come quello alla mammella, l’insulinoresistenza, l’infertilità e l’ovaiopolicistico. Nell’uomo l’eccesso estrogenico è legato ad effetti collaterali spiacevoli tra i quali troviamo la ginecomastia, la diminuzione del desiderio sessuale, problemi di erezione (che si verifica anche con un calo estrogenico eccessivo), e diminuzione della fertilità.
Ricordiamoci però che, tanto nella donna come nell’uomo, un livello estremamente basso di Estrogeni comporta diversi problemi tra i quali emergono: elevati livelli sierici di Trigliceridi, LDL e colesterolo totale con ridotti livelli di HDL (35)(36)(37)(38)(39), ridotti livelli di LDL, HDL e colesterolo totale con Trigliceridi normali (40)(41), osteoporosi, dolore osseo, e diabete mellito di tipo II (37).
Si noti che la descrizione di pazienti maschi affetti da deficit congenito di Estrogeni (42) (43) (44) (45) (46) (47) (48) (49) (50) (51) ha fornito un modello in vivo unico che ha permesso di stabilire che la completa ossificazione delle cartilagini epifisarie, anche nel maschio, non può avvenire senza l’azione degli Estrogeni. Inoltre, è stato dimostrato che gli Androgeni da soli non sono sufficienti per garantire una normale mineralizzazione dello scheletro in età adulta, dal momento che un quadro di osteopenia e/o osteoporosi è stato documentato in tutti i pazienti con deficit congenito di Estrogeni (42) (43) (44) (45) (46) (47) (48) (49) (50) (51). Ad oggi è stato chiarito che gli effetti degli steroidi sessuali sul processo di mineralizzazione ossea nel maschio sono solo in parte da ascrivere agli Androgeni di origine testicolare, mentre l’azione degli Estrogeni, che derivano dall’aromatizzazione degli androgeni stessi, sembra giocare un ruolo di maggiore importanza (52) (53). Infatti in letteratura vi sono evidenze riguardo l’associazione tra ipogonadismo e riduzione della massa ossea in entrambi i sessi (54) (55) (56) (57) (58) (59). Inoltre, il concetto generale che gli Androgeni mantengono la massa ossea nell’uomo, così come gli Estrogeni la mantengono nella donna, continua ad essere il punto di vista prevalente tra i medici, sebbene recenti studi suggeriscano il contrario (42) (43) (44) (45) (60) (61). Nel 1997, infatti, è stata descritta l’efficacia della terapia Estrogenica nell’ottenere sia la completa ossificazione delle epifisi che l’aumento di densità minerale ossea (BMD) in un paziente maschio affetto da deficit congenito di aromatasi (44).
Fonti degli estrogeni circolanti nel maschio ed organi e tessuti bersaglio dell’azione degli estrogeni. La figura indica la produzione in µmol/L.(modificata da Baird et al., 1969a e Saez et al., 1972)
Una funzione interessante dell’Estradiolo nel maschio è rappresentata dal suo impatto sulla funzione sessuale. Come già accennato, l‘Estradiolo negli uomini è essenziale per modulare la libido, la funzione erettile e la spermatogenesi. I recettori degli Estrogeni, così come l’aromatasi, sono abbondanti nel cervello, nel pene e nei testicoli, organi importanti per la funzione sessuale. Nel cervello, la sintesi dell’Estradiolo è elevata nelle aree correlate all’auda sessuale. Inoltre, nel pene, i recettori degli Estrogeni si trovano in tutto il corpus cavernosum con elevata concentrazione intorno ai fasci neurovascolari. Un livello basso di Testosterone e elevato di Estrogeni aumenta l’incidenza della disfunzione erettile indipendentemente l’uno dall’altro. Nei testicoli, la spermatogenesi è modulata a tutti i livelli dagli Estrogeni, a partire dall’asse ipotalamo-ipofisi-gonadi, seguita dalle cellule di Leydig, Sertoli e germinali e terminando con l’epitelio ductale, l’epididimo e lo sperma maturo. La regolazione delle cellule testicolari mediante l’Estradiolo mostra sia un’influenza inibitoria che una stimolatoria, indicando un intricata sinfonia di modulazione dose-dipendente e temporalmente sensibile.(62)
Adesso che abbiamo una buona conoscenza di base sugli Estrogeni e le loro azioni sia nell’uomo che nella donna, possiamo passare a trattare la questione della gestione funzionale degli Estrogeni durante la preparazione.
AAS aromatizzabili e gestione estrogenica
Come ormai ben sappiamo, il Testosterone è il substrato primario usato nell’uomo per la sintesi dell’Estradiolo, principale ormone sessuale femminile. Con una leggera alterazione strutturale del Testosterone dovuta all’azione dell’enzima aromatasi, l’Estradiolo viene prodotto nel maschio. L’attività dell’enzima aromatasi nel maschio si verifica in diverse regioni del corpo , compreso il tessuto adiposo, il fegato, le gonadi, il Sistema Nervoso Centrale e il tessuto muscolo scheletrico.(63) (64) (65) (66) (67) In un soggetto sano di sesso maschile , la quantità di Estrogeni prodotta non è generalmente molto significativa ed è basata sulle richieste e caratteristiche fisiologiche divenendo anche un vantaggio, come precedentemente visto, nella qualità e quantità dei valori di colesterolo e nella mineralizzazione ossea. Nei casi in cui le concentrazioni di Estrogeni (in particolare dell’Estradiolo) raggiungono livelli ematici elevati, che ciò avvenga come conseguenza non patologica (vedi obesità e cattiva alimentazione) o patologica (vedi neoplasia al testicolo), o come conseguenza del uso di AAS aromatizzabili e dell’alterazione omeostatica dovuta all’uso di tali composti, il soggetto sperimenta effetti avversi tra i quali vi sono la comparsa di ritenzione idrica, l’accumulo adiposo con modello femminile e la ginecomastia. Per questo motivo, molti atleti si concentrano sulla riduzione dell’incremento e/o dell’attività degli estrogeni nel corpo con l’uso di inibitori dell’aromatasi (es. Anastrozolo, Letrozolo e Exemestane), di Modulatori Selettivi del Recettore degli Estrogeni (SERM) (es. Tamoxifene e Raloxifene) o di inibitori della biosintesi steroidea (es. Aminoglutettimide e Trylostano), in specie quando il ciclo che stanno seguendo è composto da molecole altamente aromatizzabili (es. Testosterone e Methandrostenolone) o quando l’atleta desidera avere un maggior controllo estrogenico in un contesto nel quale si cerca di aumentare la definizione muscolare.
Oxymetholone
Nota:Con l’uso del Oxymetholone, il quale possiede attività estrogenica intrinseca, la gestione del fattore estrogenico (in particolare con la presenza di substrati soggetti all’aromatizzazione) assume caratteristiche leggermente più “complesse”. L’Oxymetholone è infatti un AAS avente azione altamente estrogenica. Tale azione, per l’appunto, non è dipendente dall’aromatizzazione del composto in estrogeno – essendo un derivato del DHT non può essere aromatizzato. L’uso di inibitori dell’aromatasi come l’Anastrozolo o l’Exemestane non influiscone sulla estrogenicità relativa di questo AAS. Alcuni hanno suggerito che l’alto livello di attività estrogenica del Oxymetholone sia in realtà dovuto alla presunta azione progestinica del composto, simile al Nandrolone. Gli effetti collaterali estrogenici e progestinici possono essere molto simili (anche perché l’azione dei due steroidi è co-attiva), il che ha reso plausibile questa ipotesi. Tuttavia, esiste uno studio nel quale è stata esaminata l’attività progestinica del Oxymetholone. Lo studio in questione stabilì che non vi era alcuna attività progestinica derivante dal composto.(68) Con tali risultati, sembra plausibile che l’Oxymetholone possa attivare il recettore degli estrogeni similmente a, ma più profondamente, dell’androgeno estrogenico Methandriolo. In definitiva, l’attività estrogenica di questo composto può essere gestita solamente con l’uso di SERM come il Tamoxifene o il Raloxifene dal momento che, per i motivi sopra detti, composti anti aromatasi e gli inibitori della biosintesi steroidea non hanno alcuna incidenza sull’attività estrogenica di questo AAS.
Anche alla luce di quanto accennato in precedenza, non dobbiamo tuttavia essere indotti a pensare che gli Estrogeni non diano nessun beneficio prestativo. Livelli di Estradiolo mantenuti nei limiti del range di riferimento offrono diversi vantaggi.
Gli atleti sono da anni a conoscenza del fatto che gli AAS aromatizzabili presentano un vantaggio nell’anabolismo muscolare, ma è solo di recente che siamo arrivati finalmente a capire i meccanismi che stanno alla base di tale effetto. Sembra che i motivi vadano oltre l’aumento di peso e di forza che si attribuiscono alla ritenzione idrica data dall’attività estrogenica, comprendendo che questo ormone (Estradiolo) possiede effettivamente un effetto diretto sui processi anabolici. Ciò si manifesta attraverso l’aumento dell’utilizzo del Glucosio, la secrezione dell’Ormone della Crescita (e di IGF-1) e la proliferazione dei recettori degli Androgeni.
Estrogeni e utilizzo del Glucosio
G6PD
Gli Estrogeni possono giocare un ruolo molto importante nella promozione di uno stato anabolico influenzando l’utilizzo del Glucosio nel tessuto muscolare. Ciò avviene attraverso un’alterazione del livello di glucosio 6-fosfato deidrogenasi disponibile, un enzima direttamente legato all’uso del glucosio per la crescita e il recupero del tessuto muscolare.(69) (70) Più specificamente, il G6PD è l’enzima che catalizza la prima reazione della via dei pentoso fosfati (definita anche Shunt dell’Esosomonofosfato [HMP shunt] o PPP da Pentose phosphate pathway), un processo metabolico citoplasmatico, parallelo alla glicolisi, in grado di generare NADPH e zuccheri pentosi (a 5 atomi di carbonio). Durante il periodo di rigenerazione tissutale seguente il danno muscolare, i livelli di G6PD aumentano considerevolmente, il che è ritenuto essere un meccanismo che il corpo attua per migliorare il recupero quando necessario. Sorprendentemente, scopriamo che l’Estrogeno è direttamente legato al livello di G6PD che deve essere messo a disposizione delle cellule in questa fase di recupero. In sintesi, gli Estrogeni svolgono anche una azione metabolica accelerando la sintesi degli acidi nucleici, delle proteine e del glicogeno.
Il legame tra Estrogeno e G6PD è stato stabilito attraverso uno studio nel quale è stato dimostrato che i livelli di questo enzima deidrogenasi aumentano dopo la somministrazione di Testosterone Propionato. Lo studio ha inoltre mostrato che era l’aromatizzazione del Testosterone in Estradiolo ad essere direttamente responsabile di questo aumento e non l’azione Androgena del AAS.(71) In questo studio sono stati presi in esame anche due composti non-aromatizzabili, il Dihydrotestosterone e il Fluoxymesterone, ma l’effetto avuto con l’uso di Testosterone Propionato non è stato replicato. Inoltre, l’effetto positivo del Testosterone Propionato è stato bloccato quando è stato aggiunto l’inibitore dell’aromatasi 4-idrossiandrostenedione (Formestano), mentre la somministrazione di 17-beta Estradiolo da solo ha causato un aumento di G6PD simile a quello osservato con il Testosterone Propionato. Il 17-alfa Estradiolo, isomero ormonalmente inattivo del 17-beta Estradiolo, che non può legarsi al recettore estrogenico, non ha avuto alcun effetto sul G6PD. Ulteriori prove sono state fatte utilizzando il Testosterone Propionato con l’antiandrogeno Flutamide dimostrando che anche quest’ultimo farmaco non ha alterato negativamente le concentrazioni di G6PD, sottolineando l’effetto indipendente dall’azione del Testosterone con il recettore androgeno.
Estrogeni e GH/IGF-1
L’Estrogeno può anche svolgere un ruolo importante nella produzione dell’Ormone della Crescita e del IGF-1. L’IGF-1 (Fattore di Crescita Insulino-Simile) è un ormone anabolizzante rilasciato nel fegato e in vari tessuti periferici attraverso lo stimolo dell’Ormone della Crescita. L’IGF-1 è il principale responsabile dell’attività anabolica dell’Ormone della Crescita, attraverso una maggiore ritenzione di azoto / sintesi proteica e iperplasia cellulare (proliferazione). Uno dei primi studi da portare all’attenzione riguardante la questione qui trattata ha esaminato gli effetti del SERM Tamoxifene sui livelli di IGF-1, dimostrando che il composto esaminato ha un effetto soppressivo.(72) Un secondo studio, forse più degno di nota, svolto nel 1993, ha esaminato gli effetti della terapia sostitutiva del Testosterone sui soli livelli di GH e IGF-1 e li ha confrontati con gli effetti del Testosterone combinato nuovamente con il Tamoxifene. (73) Quando è stato somministrato Tamoxifene, i livelli di GH e IGF-1 hanno subito un notevole calo, mentre entrambi i valori sono stati elevati con la somministrazione di solo Testosterone Enantato. Un altro studio ha mostrato come la somministrazione settimanale di 300mg di Testosterone Enantato causi un lieve aumento del IGF-1 in uomini normali. Questi 300mg di Testosterone Enantato hanno causato un aumento dei livelli di Estradiolo, cosa normale con tale dose. Questo è stato confrontato con l’effetto portato con lo stesso dosaggio di Nandrolone Decanoato; tuttavia, questo AAS non è riuscito a produrre lo stesso aumento nei livelli di IGF-1. Questo risultato è molto interessante, specialmente quando notiamo che i livelli di Estrogeni sono stati effettivamente ridotti quando questo AAS è stato somministrato (ricordiamoci che il Nandrolone aromatizza il 20% del Testosterone). (74) Un altro studio ha dimostrato che la secrezione di GH e IGF-1 è aumentata con la somministrazione di Testosterone in maschi con pubertà ritardata, mentre il Dihydrotestosterone (non aromatizzabile) sembra sopprimere la secrezione di GH e IGF-1. (75)
Estrogeni e Recettori degli Androgeni
Recettore degli Androgeni
È stato anche dimostrato che l’Estrogeno può aumentare la concentrazione dei recettori degli androgeni in alcuni tessuti. Ciò è stato dimostrato in studi svolti su ratti che hanno esaminato gli effetti degli Estrogeni sui Recettori degli Androgeni cellulari in animali sottoposti ad orchiectomia (rimozione dei testicoli, spesso effettuata per diminuire la produzione endogena di Androgeni). Secondo lo studio, la somministrazione di Estrogeni ha determinato un aumento del legame recettoriale nel muscolo levator ani del Metribolone pari al 480%. (76) La spiegazione per tale evento suggerita è che l’Estrogeno stimola direttamente la proliferazione dei Recettori Androgeni, o forse ne diminuisce il tasso di danneggiamento. Anche se la crescita del muscolo levator ani è comunemente usata come riferimento per determinare l’attività anabolica dei composti steroidei, esso è un muscolo dell’organo sessuale ed è diverso dal tessuto muscolo scheletrico in quanto possiede una concentrazione molto più alta di Recettori Androgeni. Questo studio, tuttavia, ha esaminato l’effetto sui AR dato dagli Estrogeni nei tessuti musco scheletrici veloci (tibialis anterior e extensor digitorum longus), ma senza notare lo stesso aumento visto nel levator ani. Anche se scoraggiante come notizia, il fatto che l’Estrogeno possa aumentare il legame del Recettore degli Androgeni in qualsiasi tessuto rimane una scoperta estremamente significativa, soprattutto alla luce del fatto che ora sappiamo che gli Androgeni hanno alcuni effetti positivi sulla crescita muscolare mediati al di fuori del tessuto muscolare.
Estrogeni e fatica/stanchezza
La così detta “Steroid Fatigue” è una frase comunemente utilizzata in riferimento ad un’altra importante funzione dell’Estrogeno sia nel corpo maschile che femminile, vale a dire la sua capacità di promuovere uno stato mentale di vigilanza. Data la larga disponibilità di potenti inibitori dell’aromatasi di terza generazione, i Bodybuilder odierni raggiungono (a volte) un livello di soppressione degli Estrogeni molto più marcata di quanto fosse stato possibile in passato. Spesso associati a questa marcata soppressione si manifestano stati di stanchezza. In tali condizioni, l’atleta, anche se sta seguendo un ciclo correttamente formulato, potrebbe non essere in grado di massimizzare i propri risultati di miglioramento della condizione fisica a causa di un’incapacità di allenarsi con pieno vigore. Questo effetto è talvolta anche soprannominato “letargia steroidea”. La ragione principale per cui ciò accade è legata all’importante azione di supporto all’attività della Serotonina data dall’Estrogeno. La Serotonina è uno dei principali neurotrasmettitori del corpo, di essenziale importanza per un adeguata lucidità mentale e un regolare ciclo sonno / veglia. (77) (78)L’alterazione di questo neurotrasmettitore è associata anche alla sindrome da affaticamento cronico, (79) (80) e ciò ci fa comprendere quanto possa essere incisiva in particolare per la stanchezza. L’abbassamento dei livelli estrogenici nella menopausa è stata associata anche alla stanchezza, (81) così come l’uso clinico di inibitori dell’aromatasi più recenti (e più potenti) come l’Anastrozolo, (82) il Letrozolo, (83) l’Exemestane, (84) e il Fadrozolo (85) in alcuni pazienti. Questo effetto deve essere preso in importante considerazione quando si pianifica un ciclo. Anche se non tutti notano questo problema quando il livello estrogenico è basso, per coloro i quali lo subiscono, l’aggiunta di un po’ di Testosterone o di altro AAS soggetto all’aromatizzazione (oltre che l’abbassamento del composto AI) può aiutare a correggere di molto questo sintomo. È anche da notare che l’uso di AAS non aromatizzabili a volte causa questo effetto, probabilmente dovuto alla soppressione della produzione naturale di Testosterone (abbassando la disponibilità del principale substrato usato dal corpo maschile per sintetizzare Estradiolo).
Methylestradiolo
*Nota:gli AAS metilati in C-17, soggetti ad aromatizzazione (vedi Methyltestosterone e Mathandrostenolone), convertono nel potente Methylestradiolo, o 17α-methylestradiolo (17α-ME). A causa della presenza del gruppo 17α-metile, il Methylestradiolo non può essere disattivato per ossidazione del gruppo 17β-idrossile, con conseguente migliorata stabilità metabolica e potenza di legame recettoriale (e conseguente attività estrogenica) rispetto all’Estradiolo. (86)(87)(88)
Quindi, come gestire il fattore estrogenico durante la preparazione?
Dave Bautista mostra un classico esempio di cattiva gestione estrogenica.
Arrivati a questo punto, quale significato ha quanto è stato trattato per un Bodybuilder che cerca di ottenere una forma ottimale? Fondamentalmente penso che il punto chiave sia quello di mantenere un approccio prudente all’uso di farmaci per il controllo estrogenico, un approccio che deve essere dettato dalle reali esigenze verificate attraverso appositi esami ematici. Ovviamente, è chiaro che l’approccio all’uso dei composti per il controllo estrogenico debba variare a seconda dell’obbiettivo (vale a dire se l’obbiettivo primario è l’aumento della massa muscolare o se è l’aumento della definizione muscolare). Ripeto, è ovvio che l’utilizzo di composti volti al controllo estrogenico è una chiara necessità qualora si manifestassero effetti collaterali estrogenici, come lo è, nel caso, il calo dei composti soggetti all’azione dell’enzima aromatasi, ma gli esami ematici sono sempre di primaria importanza ai fini di una gestione ottimale del problema. La ginecomastia è certamente un problema indesiderato per l’utilizzatore di AAS, come lo è la ritenzione idrica ed un eccessivo accumulo di massa grassa. Ma se tali problemi non emergono, o se dagli esami ematici non emerge la reale necessità, l’aggiunta di composti per il controllo estrogenico (che siano AI, SERM o inibitori della biosintesi steroidea) può inficiare i risultati ottenibili in un contesto “Bulk”.
Quindi, concludendo, in un contesto “Bulk”, al fine di mantenere i benefici estrogenici senza incappare negli effetti collaterali estrogeno-dipendenti, la soglia ematica di Estradiolo dovrebbe mantenersi all’interno del range medio di riferimento; ciò significa che, in teoria, non dovrebbe ne scendere al di sotto dei 30pg/pg ne salire al di sopra dei 60 pg/ml. In un contesto “Cut” o “Pre-Gara”, ovviamente, le cose cambiano. In tali contesti, considerando gli effetti estrogenici anche sullo spessore della pelle, la cosa migliore sarebbe optare per il mantenimento di un livello di estrogeni basso-normale (20-30pg/ml) per tutta la fase “Cut” della preparazione per poi, se si è in preparazione alla gara, calare i livelli a 10pg o meno nell’ultimo paio di settimane prima del contest.
Per le atlete il discorso è simile anche se diverso sotto alcuni aspetti. In ambito femminile il controllo dei livelli e dell’attività estrogenica è di importanza anche maggiore per il raggiungimento di una forma da gara ottimale (vedere ad esempio “Nolvadex (Tamoxifene Citrato)”e “RALOXIFENE E COMPOSIZIONE CORPOREA NELLE DONNE”).
In definitiva, come logica conclusione, il metodo d’approccio per la gestione estrogenica deve basarsi sulle reali necessità, su dati oggettivi, e non su un fallace e molto poco scientifico “passa parola” dettato dall'”esperienza” di qual si voglia agonista o ex tale…
2- Ryan KJ (August 1982). “Biochemistry of aromatase: significance to female reproductive physiology”. Cancer Res. 42 (8 Suppl): 3342s–3344s. PMID7083198.
5- Lombardi G, Zarrilli S, Colao A, Paesano L, Di Somma C, Rossi F, De Rosa M (2001). “Estrogens and health in males”. Molecular and Cellular Endocrinology. 178 (1–2): 51–5. doi:10.1016/S0303-7207(01)00420-8. PMID11403894.
7- Soltysik K, Czekaj P (April 2013). “Membrane estrogen receptors — is it an alternative way of estrogen action?”. J. Physiol. Pharmacol. 64 (2): 129–42. PMID23756388.
12- Darabi M, Ani M, Panjehpour M, Rabbani M, Movahedian A, Zarean E (2011). “Effect of estrogen receptor β A1730G polymorphism on ABCA1 gene expression response to postmenopausal hormone replacement therapy”. Genet Test Mol Biomarkers. 15 (1–2): 11–5. doi:10.1089/gtmb.2010.0106. PMID21117950.
14- Hess RA, Bunick D, Lee KH, Bahr J, Taylor JA, Korach KS, Lubahn DB (1997). “A role for estrogens in the male reproductive system”. Nature. 390 (6659): 447–8. doi:10.1038/37352. PMID9393999.
16- Hill RA, Pompolo S, Jones ME, Simpson ER, Boon WC (2004). “Estrogen deficiency leads to apoptosis in dopaminergic neurons in the medial preoptic area and arcuate nucleus of male mice”. Mol. Cell. Neurosci. 27 (4): 466–76. doi:10.1016/j.mcn.2004.04.012. PMID15555924.
18- Nelson LR, Bulun SE (September 2001). “Estrogen production and action”. J. Am. Acad. Dermatol. 45 (3 Suppl): S116–24. doi:10.1067/mjd.2001.117432. PMID11511861.
19- MacDonald PC, Madden JD, Brenner PF, Wilson JD, Siiteri PK: Origin of estrogen in normal men and in women with testicular feminization. J Clin Endocrinol Metab 49:905-916, 1979
20- Simpson ER, Mahendroo MS, Means GD, Kilgore MW, Hinshelwood MM, Graham-Lorence S, Amarneh B, Ito Y, Fisher CR, Michael MD, et al.: Aromatase cytochrome P450, the enzyme responsible for estrogen biosyn- thesis. Endocr Rev 15:342-355, 1994
21- Simpson ER, Zhao Y, Agarwal VR, Michael MD, Bulun SE, Hinshelwood MM, Graham-Lorence S, Sun T, Fisher CR, Qin K, Mendelson CR: Aromatase expression in health and disease. Recent Prog Horm Res 52:185-213; discus- sion 213-184, 1997
22- Simpson ER: Aromatase: biologic relevance of tissue-specific expression. Semin Reprod Med 22:11-23, 2004
23- Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM: The nuclear recep- tor superfamily: the second decade. Cell 83:835-839, 1995
24- Green S, Walter P, Kumar V, Krust A, Bornert JM, Argos P, Chambon P: Human oestrogen receptor cDNA: sequence, expression and homology to v-erb-A. Nature 320:134-139, 1986
25- Greene GL, Gilna P, Waterfield M, Baker A, Hort Y, Shine J: Sequence and expression of human estrogen receptor complementary DNA. Science 231:1150-1154, 1986
26- Kuiper GG, Carlsson B, Grandien K, Enmark E, Haggblad J, Nilsson S, Gustafsson JA: Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology 138:863-870, 1997
27- Enmark E, Gustafsson JA: Oestrogen receptors – an overview. J Intern Med 246:133-138, 1999
28- Levin ER: Cellular Functions of the Plasma Membrane Estrogen Receptor. Trends Endocrinol Metab 10:374-377, 1999
29- Pedram A, Razandi M, Levin ER: Nature of functional estrogen receptors at the plasma membrane. Mol Endocrinol 20:1996-2009, 2006
30- Filardo E, Quinn J, Pang Y, Graeber C, Shaw S, Dong J, Thomas P: Activation of the novel estrogen receptor G protein-coupled receptor 30 (GPR30) at the plasma membrane. Endocrinology 148:3236-3245, 2007
31- de Ronde W, Pols HA, van Leeuwen JP, de Jong FH: The importance of oestrogens in males. Clin Endocrinol (Oxf) 58:529-542, 2003
32- Baird DT, Horton R, Longcope C, Tait JF: Steroid dynamics under steady- state conditions. Recent Prog Horm Res 25:611-664, 1969
33- Baird DT, Uno A, Melby JC: Adrenal secretion of androgens and oestrogens. J Endocrinol 45:135-136, 1969
34- Dunn JF, Nisula BC, Rodbard D: Transport of steroid hormones: bind- ing of 21 endogenous steroids to both testosterone-binding globulin and corticosteroid-binding globulin in human plasma. J Clin Endocrinol Metab 53:58-68, 1981
35- Carr BR: Disorders of the ovaries and female reproductive tract. Williams Textbook of Endocrinology. 11th ed. W.B. Saunders Company ed. Polonsky KS, Melmed S, Kronenberg HM, Larsen PR, Eds. Philadelphia, 2008
36- Weinbauer GF, Nieschlag E: The role of testosterone in spermatogene- sis. Springer-Verlag ed. Nieschlag E, Behre H, Eds. Berlin Heidelberg, Testosterone action deficiency substitution, 1990, p. 23-50
37- Zondek B: Mass excretion of oestrogenic hormone in the urine of the stallion. Nature 33:209-210, 1934
38- Heard RD, Jellinck PH, O’Donnell VJ: Biogenesis of the estrogens: the con- version of testosterone-4-C14 to estrone in the pregnant mare. Endocrinology 57:200-204, 1955.
39- West CD, Damast BL, Sarro SD, Pearson OH: Conversion of testosterone to estrogens in castrated, adrenalectomized human females. J Biol Chem 218:409-418, 1956.
40- Davis JW, Gut M, Lemon HM, Wotiz HH: Studies in steroid metabolism. V. The conversion of testosterone-4-C14 to estrogens by human ovarian tissue. J Biol Chem 222:487-495, 1956
41- Baggett B, Dorfman RI, Engel LL, Savard K: The conversion of testoster- one-3-C14 to C14-estradiol-17beta by human ovarian tissue. J Biol Chem 221:931-941, 1956 8. Baggett B, Engel LL, Balderas L, Lanman G: Conversion of C14-testosterone to C14-estrogenic steroids by endocrine tissues. Endocrinology 64:600-608, 1959
42- Smith EP, Boyd J, Frank GR, Takahashi H, Cohen RM, Specker B, Williams TC, Lubahn DB, Korach KS: Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. N Engl J Med 331:1056-1061, 1994
43- Morishima A, Grumbach MM, Simpson ER, Fisher C, Qin K: Aromatase deficiency in male and female siblings caused by a novel mutation and the physiological role of estrogens. J Clin Endocrinol Metab 80:3689-3698, 1995
44- Carani C, Qin K, Simoni M, Faustini-Fustini M, Serpente S, Boyd J, Korach KS, Simpson ER: Effect of testosterone and estradiol in a man with aromata- se deficiency. N Engl J Med 337:91-95, 1997
45- Bilezikian JP, Morishima A, Bell J, Grumbach MM: Increased bone mass as a result of estrogen therapy in a man with aromatase deficiency. N Engl J Med 339:599-603, 1998
46- Herrmann BL, Saller B, Janssen OE, Gocke P, Bockisch A, Sperling H, Mann K, Broecker M: Impact of estrogen replacement therapy in a male with con- genital aromatase deficiency caused by a novel mutation in the CYP19 gene. J Clin Endocrinol Metab 87:5476-5484, 2002
47- Pura M, Mittre H, Carreau S, Kottler ML: Clinical findings in an adult man with a novel mutation in the aromatase gene. In Program of the 85th Annual Meeting of the Endocrine Society, Abstract Book Philadelphia, PA, 2003
48- Maffei L, Murata Y, Rochira V, Tubert G, Aranda C, Vazquez M, Clyne CD, Davis S, Simpson ER, Carani C: Dysmetabolic syndrome in a man with a novel mutation of the aromatase gene: effects of testosterone, alendronate, and estradiol treatment. J Clin Endocrinol Metab 89:61-70, 2004
49- Maffei L, Rochira V, Zirilli L, Antunez P, Aranda C, Fabre B, Simone ML, Pignatti E, Simpson ER, Houssami S, Clyne CD, Carani C: A novel com- pound heterozygous mutation of the aromatase gene in an adult man: reinforced evidence on the relationship between congenital oestrogen deficiency, adiposity and the metabolic syndrome. Clin Endocrinol (Oxf) 67:218-224, 2007
50- Luberto A, Rochira V, Zirilli L, Pignatti E, Simpson ER, Maffei L, Carani C: A novel compound heterozygous mutation of the aromatase gene in an adult man: a reinforced estrogen deficincy, adiposity and the metabolic syndrome. J Endocrinol Invest 30 Suppl 4:50, 2007
51- Bouillon R, Bex M, Vanderschueren D, Boonen S: Estrogens are essen- tial for male pubertal periosteal bone expansion. J Clin Endocrinol Metab 89:6025-6029, 2004
52- Vanderschueren D, Boonen S, Bouillon R: Action of androgens versus estrogens in male skeletal homeostasis. Bone 23:391-394, 1998
53- Vanderschueren D, Venken K, Ophoff J, Bouillon R, Boonen S: Clinical Review: Sex steroids and the periosteum–reconsidering the roles of androgens and estrogens in periosteal expansion. J Clin Endocrinol Metab 91:378-382, 2006
54- Horsman A, Gallagher JC, Simpson M, Nordin BE: Prospective trial of oestrogen and calcium in postmenopausal women. Br Med J 2:789-792, 1977
55- Lindsay R, Hart DM, Forrest C, Baird C: Prevention of spinal osteoporosis in oophorectomised women. Lancet 2:1151-1154, 1980
56- Christiansen C, Christensen MS, Transbol I: Bone mass in postmenopausal women after withdrawal of oestrogen/gestagen replacement therapy. Lancet 1:459-461, 1981
57- Finkelstein JS, Neer RM, Biller BM, Crawford JD, Klibanski A: Osteopenia in men with a history of delayed puberty. N Engl J Med 326:600-604, 1992
58- Speroff L, Rowan J, Symons J, Genant H, Wilborn W: The comparative effect on bone density, endometrium, and lipids of continuous hormones as replacement therapy (CHART study). A randomized controlled trial. Jama 276:1397-1403, 1996
59- Wang C, Eyre DR, Clark R, Kleinberg D, Newman C, Iranmanesh A, Veldhuis J, Dudley RE, Berman N, Davidson T, Barstow TJ, Sinow R, Alexander G, Swerdloff RS: Sublingual testosterone replacement improves muscle mass and strength, decreases bone resorption, and increases bone formation markers in hypogonadal men–a clinical research center study. J Clin Endocrinol Metab 81:3654-3662, 1996
60- Faustini-Fustini M, Rochira V, Carani C: Oestrogen deficiency in men: where are we today? Eur J Endocrinol 140:111-129, 1999
61- Vanderschueren D, Venken K, Ophoff J, Bouillon R, Boonen S: Clinical Review: Sex steroids and the periosteum–reconsidering the roles of androgens and estrogens in periosteal expansion. J Clin Endocrinol Metab 91:378-382, 2006
69- Pentose Cycle Activity in Muscle from Fetal, Neonatal and Infant Rhesus Monkeys. Arch Biochem Biophys 117:275-81 1966
70- The pentose phosphate pathway in regenerating skeletal muscle. Biochem J 170: 17 1978
71- Aromatization of androgens to estrogens mediates increased activity of glucose 6-phosphate dehydrogenase in rat levator ani muscle. Endocrinol 106(2):440-43 1980
72- Influence of tamoxifen, aminoglutethimide and goserelin on human plasma IGF-1 levels in breast cancer patients. J steroid Biochem Mol Bio 41:541- 3,1992
73- Activation of the somatotropic axis by testosterone in adult males: Evidence for the role of aromatization. J Clin. Endocrinol Metab 76:1407-12 1993
74- Testosterone administration increases insulin-like growth factor-I levels in normal men. J Clin Endocrinol Metab 77(3):776-9 1993
80- Association between serotonin transporter gene polymorphism and chronic fatigue syndrome. Narita M et al. Biochem Biophys Res Commun 2003 Nov 14;311(2)264-6
81- Premenstrual Syndrome. Dickerson LM et al. Am Fam Physician 2003 Apr 15;67(8):1743-52
82- Phase II trial of anastrozole in women with asymptomatic mullerian cancer. Gynecol Oncol. 2003 Dec;91(3):596-602.
83- Letrozole. A review of its use in postmenopausal women with advanced breast cancer. Drugs. 1998 Dec;56(6):1125-40. Review.
84- Exemestane: a review of its clinical efficacy and safety. Breast. 2001 Jun;10(3):198-208.
85- A study of fadrozole, a new aromatase inhibitor, in postmenopausal women with advanced metastatic breast cancer. J Clin Oncol. 1992 Jan;10(1):111-6.
87- Feenstra A, Vaalburg W, Nolten GM, Reiffers S, Talma AG, Wiegman T, van der Molen HD, Woldring MG (1983). “Estrogen receptor binding radiopharmaceuticals: II. Tissue distribution of 17 alpha-methylestradiol in normal and tumor-bearing rats”. J. Nucl. Med. 24 (6): 522–8. PMID6406650.
E’ possibile che l’uso di SERM risulti maggiormente efficace se il soggetto trattato ha un ritmo del sonno ottimale con maggiore sintesi di Melatonina nelle ore serali e notturne. Questa possibilità è stata riportata da biologi cellulari della Tulane University School of Medicine sul Cancer Research nel 2014. I ricercatori hanno esaminato l’impatto di un sano ritmo del sonno sugli effetti del Tamoxifene in uno studio su animali.(1) I risultati della loro ricerca sono interessanti sia per le donne con una variante del cancro al seno sensibile all’Estradiolo, ma anche per gli atleti supplementati farmacologicamente.
I ricercatori hanno innestato cellule del cancro al seno MCF-7 sensibili all’Estradiolo nei topi. Nelle ore notturne alcuni di questi topi erano tenuti al buio [gruppo LD 12:12], mentre altri erano esposti a luce fioca [gruppo dLEN].
Ad alcuni dei topi del gruppo “dLEN” è stata somministrata Melatonina [dLEN-Mel].
Quando i ricercatori hanno stimato il peso dei tumori a 1,5 grammi, hanno somministrato agli animali presi in esame il Tamoxifene, famoso farmaco antiestrogeno con azione agonista/antagonista nei confronti del recettore dell’Estradiolo.
La somministrazione di Tamoxifene [TAM] non ha avuto alcun effetto sulla crescita dei tumori quando gli animali esaminati erano sottoposti alla luce durante la notte [dLEN], come mostra la figura sottostante. Ma se gli animali presi in esame e che erano stati sottoposti alla luce durante le ore notturne oltre al Tamoxifene avevano ricevuto anche la melatonina [MLT], i loro tumori mostravano di rispondere al trattamento con Tamoxifene.
La supplementazione con Melatonina ha portato le concentrazioni di Melatonina nei ratti esposti alla luce durante le ore notturne ai livelli dei ratti tenuti al nel buio nello stesso lasso di tempo.
La Melatonina, somministrata sotto forma di integratore o rilasciata endogenamente durante le ore di sano sonno, inibisce l’azione del recettore dell’Estradiolo. I ricercatori hanno scoperto questa azione della Melatonina durante l’osservazione delle cellule tumorali degli animali dello studio.
I ricercatori affermano che, in conclusione, il presente studio evidenzia e conferma l’importanza del mantenimento di un ritmo veglia-sonno ottimale così da avere livelli endogeni di Melatonina ottimali i quali agiscono anche sensibilizzando le cellule tumorali del seno umano alla terapia con Tamoxifene.
Inoltre, il presente lavoro dimostra che una comprensione globale della risposta biologico-circadiana cancro/ospite e la natura circadiano-regolata del metabolismo del cancro e la sua segnalazione sono essenziali per ottenere la massima efficacia dal trattamento con Tamoxifene ed eventualmente altre terapie endocrine.
I ricercatori continuano affermando che, è plausibile che molti, se non tutti, i pazienti affetti da cancro al seno siano soggetti a diversi gradi di esposizione alla luce durante la notte e che il ritmo circadiano della Melatonina possa essere alterato a causa della mancanza di sonno e /o di turni di lavoro notturno.
Pertanto, l’esposizione alla luce durante la notte può rappresentare un fattore di rischio unico e precedentemente non apprezzato che potrebbe spiegare alcune forme di resistenza intrinseca e possibilmente acquisita al Tamoxifene e può portare ad un tempo di sopravvivenza ridotto e persino ad un tasso di sopravvivenza ridotto.
Il Phellinus linteus – un fungo che secondo la medicina tradizionale asiatica ha un effetto curativo- contiene il fenolo hispolon. Uno studio coreano in vitro, pubblicato nel 2015 sul Biochemical and Biophysical Research Communications, suggerisce anche che l’hispolon ha un effetto antiestrogeno. (1)
Nella medicina tradizionale asiatica gli estratti di Phellinus linteus – in Corea si chiama Sanghwang – venivano usati come rimedio contro le infiammazioni, i problemi gastrointestinali e vari tipi di cancro. Gli stessi estratti si trovano anche in integratori che vengono venduti come “supporto” del sistema immunitario.
I ricercatori hanno esposto cellule MCF7 e T47D in vitro al hispolon. Le cellule MCF-7 e T47D sono cellule del cancro al seno umano le quali necessitano dell’Estradiolo per potersi sviluppare. L’Estradiolo, per permettere ciò, deve prima legarsi al recettore estrogenico-alfa. Questo recettore svolge un ruolo chiave negli effetti sulla salute più negativi dell’Estradiolo.
L’hispolon ha ridotto il numero di recettori estrogenici-alfa nelle cellule ad esso esposte. Maggiore è la concentrazione di hispolon, maggiore è il suo effetto. L’hispolon ha inibito la produzione di questo tipo di recettore.
Se un composto estrogenico [come l’Estradiolo] si lega al proprio recettore, stimola la cellula attraverso l’elemento di risposta dell’estrogeno nel DNA affinché vengano avviati processi estrogenici di risposta.
I ricercatori hanno scoperto che l’hispolon indebolisce quel segnale. Più alta è la concentrazione e più lunga è l’esposizione al composto, più debole è il segnale.
I ricercatori hanno scritto che, l’hispolon, derivato da Phellinus linteus, è noto per le sue proprietà antitumorali, ma il meccanismo alla base di questa attività non è compreso . Qui abbiamo esaminato gli effetti del hispolon sull’espressione del ER-alfa e sulla crescita dell’ER nelle cellule del cancro al seno estradiolo-sensibili, MCF-7 e T47D.
Coerentemente con l’inibizione data dal hispolon sulla risposta estrogenica nell’attività trascrizionale elemento-dipendente in cellule del cancro al seno umano ER-positive, l’hispolon ha mostrato un attività antitumorale in queste cellule.
I ricercatori affermano di aver scoperto che l’hispolon ha ridotto l’espressione del ER-alfa a livello del mRNA così come i livelli delle proteine specifiche. Le diminuzioni indotte dal hispolon nei livelli di proteina ER-alfa hanno portato alla repressione dell’attività trascrizionale del ER-alfa, che ha provocato una significativa riduzione dell’espressione del gene ER-responsivo pS2d – il gene principale che si presenta nei tessuti del carcinoma mammario che esprimono l’ER ed è un importante indicatore prognostico.
Anche se sono necessari ulteriori studi in vivo per stabilire il potenziale dell’hispolon come farmaco antitumorale per il trattamento del cancro al seno, i risultati dello studio qui esposto suggeriscono che l’hispolon è un efficace inibitore dello sviluppo del cancro al seno nelle cellule del carcinoma mammario umano ER-positive.
L’Oleocantale, una sostanza organica naturalmente presente soprattutto nei frutti dell’ulivo (Olea europea), ed è in pratica la sostanza principalmente responsabile del “bruciore in gola” tipico degli oli extra vergini di oliva, risulta essere un composto che presenta proprietà anti-cancerogene e anti-estrogeniche, almeno secondo quanto riportato da farmacologi della Jordan University of Science and Technology in un articolo sul European Journal of Pharmacology. (1) Forse l’integrazione con Oleocantale può migliorare l’effetto di antiestrogeni come il Tamoxifene.
I ricercatori hanno esposto le cellule tumorali del seno MCF-7, BT-474 e T-47D alla (-) – Oleocantale. Tutti questi tipi di cellule possiedono recettori per l’Estradiolo.
Maggiore è la concentrazione di Oleocantale, maggiori sono gli effetti letali del composto sulle cellule tumorali. La figura sottostante a sinistra mostra che l’Oleocantale uccide le cellule tumorali MCF-7. L’aggiunta di Estradiolo protegge le cellule del cancro al seno contro l’azione del Oleocantale [a destra], ma ad una concentrazione abbastanza elevata l’Oleocantale ha annullato tale effetto protettivo.
Le cellule MCF-7 sono sensibili all’Estradiolo. Nei campioni in vitro contenenti anche Estradiolo, il SERM Tamoxifene ha impedito che l’Estradiolo svolgesse la sua azione, uccidendo di conseguenza le cellule tumorali. L’Oleocantale ha aumentato l’effetto del Tamoxifene.
In uno studio svolto su animali, i ricercatori hanno impiantato cellule tumorali BT-474 nei topi. Nei topi ai quali era stata somministrata una dose di 5 o 10mg di Oleocantale per kg di peso corporeo, le cellule tumorali hanno smesso di crescere.
I ricercatori hanno iniettano l’Oleocantale direttamente nel piccolo intestino. L’equivalente umano orale della dose usata, per un adulto di 80 kg, sarebbe 80-160mg di Oleocantale al giorno.
I ricercatori hanno scoperto che la supplementazione con Oleocantale riduceva la concentrazione del recettore estrogenico alfa nei tumori degli animali da laboratorio del 40%. Utilizzando modelli al computer hanno calcolato che l’Oleocantale si lega al recettore estrogenico, così come l’Estradiolo e il Tamoxifene, dopo di che il recettore si rompe.
Per completare il quadro della situazione: le cellule BT-474 non crescono bene nei ratti e nei topi. I ricercatori hanno dovuto somministrare ai topi dosi extra di Estradiolo per attivare la crescita del tumore. Quindi non è possibile supporre che i risultati promettenti di questo studio sugli animali si possano applicare allo stesso modo negli esseri umani.
In conclusione, l’Oleocantale sopprime la crescita delle cellule tumorali del seno, in parte, riducendo i livelli totali del recettore estrogenico alfa nella coltura cellulare e negli studi sugli animali. La combinazione di Oleocantale con il Tamoxifene ha mostrato un’azione sinergica.
Questi risultati supportano un’ulteriore valutazione del Oleocantale come potenziale opzione terapeutica in combinazione con trattamenti endocrini nel cancro al seno ormone-dipendente.


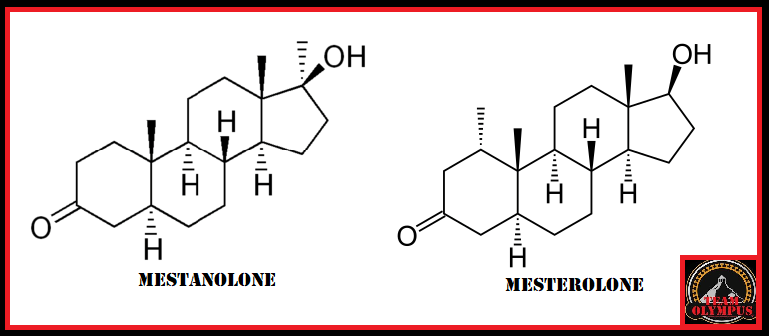
 to come agente dopante negli atleti olimpionici della Germania Orientale all’interno di un programma di doping sponsorizzato dallo stato negli anni ’70 e ’80.[4] Il motivo del suo uso, come precedentemente accennato, era giustificato maggiormente dal suo valore androgeno piuttosto che su quello anabolizzante. Il suo uso garantiva un ottima risposta neuro steroidea con conseguente stimolazione del Sistema Nervoso Centrale e migliorata interazione neuromuscolare, con vantaggi nella velocità, nella forza, nell’aggressività, nella concentrazione, nella resistenza fisica e allo stress mentale.[4] Oggi, l’uso del Mestanolone è stato per lo più sospeso in medicina, sebbene sia ancora disponibile in Giappone.[2][3][4] Il Mestanolone era comunemente disponibile sotto forma di compresse sublinguali da 25mg (marchio Ermalone).[7]
to come agente dopante negli atleti olimpionici della Germania Orientale all’interno di un programma di doping sponsorizzato dallo stato negli anni ’70 e ’80.[4] Il motivo del suo uso, come precedentemente accennato, era giustificato maggiormente dal suo valore androgeno piuttosto che su quello anabolizzante. Il suo uso garantiva un ottima risposta neuro steroidea con conseguente stimolazione del Sistema Nervoso Centrale e migliorata interazione neuromuscolare, con vantaggi nella velocità, nella forza, nell’aggressività, nella concentrazione, nella resistenza fisica e allo stress mentale.[4] Oggi, l’uso del Mestanolone è stato per lo più sospeso in medicina, sebbene sia ancora disponibile in Giappone.[2][3][4] Il Mestanolone era comunemente disponibile sotto forma di compresse sublinguali da 25mg (marchio Ermalone).[7]