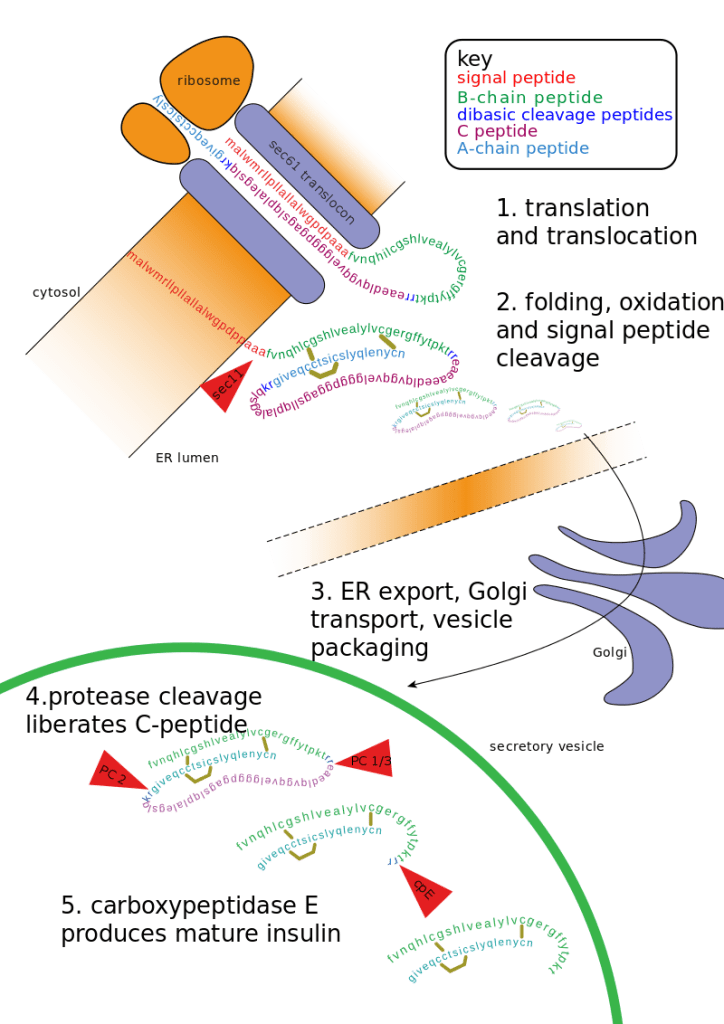
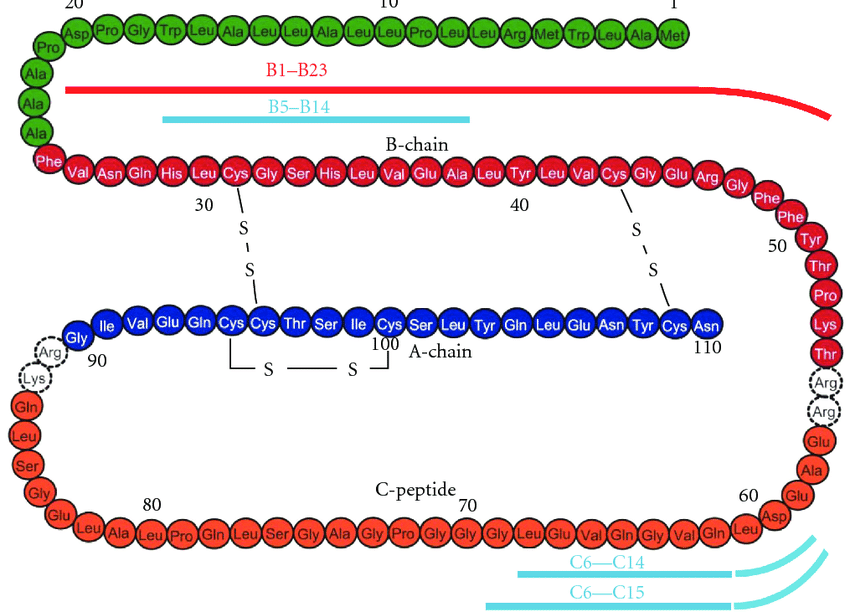
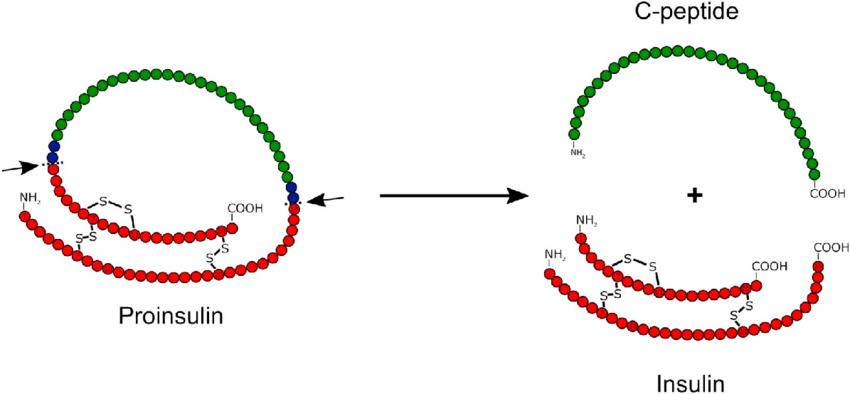
Per accedere alla prima e alla seconda parte clicca qui e qui.
Il ruolo dell’Insulina nella regolazione della sintesi e della degradazione delle proteine del muscolo-scheletrico umano:
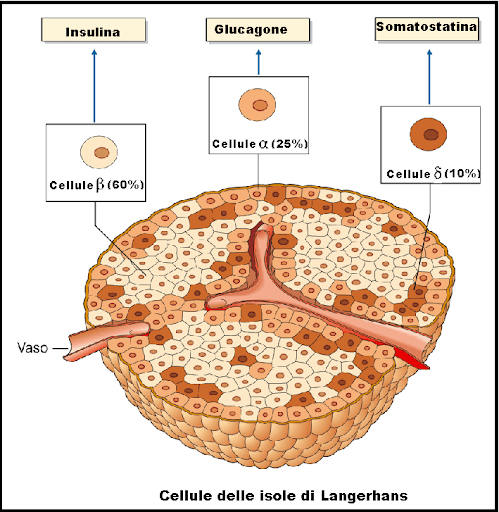
Nell’uomo, le proteine costituiscono circa il 15% del peso corporeo [1]. Sono il principale macronutriente che compone il muscolo-scheletrico, che a sua volta contiene circa il 30-45% delle proteine totali del corpo e contribuisce al 20-35% del turnover proteico dell’intero organismo. È stato dimostrato che sia gli aminoacidi (AA) che l’Insulina svolgono un ruolo cruciale nella regolazione delle variazioni diurne del turnover proteico del muscolo scheletrico [2] e che gli squilibri tra i tassi di sintesi proteica muscolare (MPS) e di degradazione delle proteine muscolari (MPB) hanno importanti conseguenze sulle dimensioni, sulla qualità e sulla funzione del muscolo [3]. La “sarcopenia” descrive la perdita di massa e forza muscolare scheletrica che si verifica con l’avanzare dell’età [4]. Il processo di invecchiamento stesso è caratterizzato dall’incipiente sviluppo della sarcopenia, in cui è stato segnalato un costante declino della massa magra (e della funzione associata) di circa l’1% all’anno oltre i 60 anni di età [5]. A causa della fragilità associata, la sarcopenia porta a una diminuzione della qualità della vita e della salute, caratterizzata da scarsa mobilità, sedentarietà, aumento del rischio di cadute e scarso recupero dalle malattie [6, 7].

I dati provenienti da studi epidemiologici e sperimentali hanno riportato che il diabete di tipo II è correlato a una scarsa forza e funzione muscolare, con un tasso accelerato di declino della qualità e della forza muscolare negli individui anziani fino al 30% [8]. Alla luce della crescente prevalenza del diabete e delle sequele metaboliche della sarcopenia legata all’età, è aumentato l’interesse per i meccanismi con cui il diabete di tipo II esacerba il declino della massa muscolare legato all’età. Inoltre, poiché il muscolo scheletrico è un sito importante per lo smaltimento del glucosio, la riduzione quantitativa del volume del muscolo appendicolare potrebbe potenzialmente influire negativamente sullo smaltimento e sul metabolismo del glucosio [9]. Una maggiore comprensione dei fattori endocrini che regolano la massa muscolare è quindi importante per il controllo glicemico e per contrastare la sarcopenia.
Molti pazienti con diabete di tipo II necessitano di Insulina per raggiungere gli obiettivi ottimali di glucosio, poiché la capacità di produrre Insulina endogena da parte delle cellule beta pancreatiche diminuisce progressivamente [10]. Tuttavia, la terapia insulinica è associata a un aumento di peso [11, 12], soprattutto di massa grassa [13], anche se non correlate all’ormone in se come abbiamo visto nella seconda parte, che aumenta l’insulino-resistenza e rende necessario l’uso di dosi più elevate di Insulina a scapito di un ulteriore aumento di peso. L’esatto ruolo dell’Insulina nel metabolismo del muscolo scheletrico umano, tuttavia, continua a far discutere. Sebbene gli studi sugli animali abbiano riportato che l’Insulina promuove la MPS, questi studi sono stati condotti principalmente su animali in crescita [14, 15]. Il ruolo dell’Insulina nel muscolo scheletrico umano adulto è più complesso e soggetto all’interazione tra altri fattori come la disponibilità di AA, il flusso sanguigno muscolare e il reclutamento microvascolare [16, 17]. Ciò ha portato a diversi studi che riportano conclusioni opposte per quanto riguarda la relazione tra Insulina e turnover proteico del muscolo scheletrico umano [16-22]. Andiamo quindi a tentare di chiarire il ruolo dell’Insulina nella regolazione del metabolismo muscolare nell’uomo.
Per la realizzazione di una interessante review sistematica e meta-analisi riguardante il ruolo dell’Insulina sulla MPS e MPB sono stati cercati studi in lingua inglese pubblicati tra il 1946 e il novembre 2013. Sono stati selezionati gli articoli sottoposti a revisione paritaria che indagavano il ruolo dell’Insulina sulle MPS e/o sulla MPB. Per la review sistematica sono stati selezionati tutti gli studi sperimentali che riportavano cambiamenti nel metabolismo delle proteine muscolari nell’uomo in risposta a interventi con Insulina, indipendentemente dal metodo di valutazione. Sia il metodo a due pool (compartimenti) che quello a tre pool sono stati utilizzati per riportare il metabolismo proteico muscolare, ed entrambi forniscono cambiamenti qualitativamente comparabili nel metabolismo proteico dal sangue e dall’arricchimento intracellulare di fenilalanina [21]. Nel modello a due pool (arteria e vena), la fenilalanina entra ed esce dall’arto rispettivamente attraverso l’arteria e la vena. La velocità di scomparsa della fenilalanina dall’arteria è utilizzata per stimare l’MPS e deriva dalle misurazioni della velocità di MPB e del bilancio netto (NB); MPB è determinato dalla velocità di comparsa della fenilalanina in vena (cioè la diluizione dell’arricchimento del tracciante attraverso l’arto), mentre NB è semplicemente la differenza di concentrazione della fenilalanina attraverso l’arto. Nel modello a tre bacini (arteria, vena e muscolo), la fenilalanina entra ed esce dall’arto come sopra. Il flusso unidirezionale di fenilalanina libera dall’arteria al compartimento intramuscolare è determinato dalla velocità di trasporto verso l’interno. La velocità di comparsa intracellulare della fenilalanina definisce la velocità di rilascio dal MPB. Poiché la fenilalanina non viene ossidata dal muscolo scheletrico, il tasso di utilizzo intracellulare corrisponde al tasso di utilizzo per la MPS [22].
Negli studi esaminati, il modello a due pool è stato il metodo analitico più comunemente utilizzato e la fenilalanina è stata il tracciante AA più comunemente usato. Pertanto, per consentire un’analisi quantitativa comparabile degli studi eleggibili per la review sistematica e per evitare un’eterogeneità significativa, la meta-analisi ha incluso solo gli studi che hanno utilizzato il modello a due pool per analizzare i dati di fenilalanina, rispetto ad altri metodi analitici o ad altri traccianti di AA (ad esempio, la leucina). I dati a tre pool sono stati inclusi nella review sistematica. A causa di un’ampia sovrapposizione tra gli studi che riportavano dati a due e tre pool (n = 10) e del numero significativamente inferiore di studi che riportavano esclusivamente dati a tre pool (n = 5), non è stata eseguita una meta-analisi dei dati a tre pool.
Sono stati esaminati tutti gli studi che rispondevano ai criteri di inclusione. L’esito primario era la variazione di MPS e/o MPB in risposta all’intervento insulinico. I dati pubblicati sono stati estratti dagli studi e sono state calcolate le medie. A causa dei metodi di misurazione simili tra gli studi inclusi, sono stati utilizzati modelli a effetti casuali per calcolare le differenze medie ponderate (WMD), gli CI al 95% e i valori di p corrispondenti. L’eterogeneità tra gli studi è stata valutata utilizzando la statistica I 2, che descrive la percentuale di variazione totale tra gli studi che è il risultato dell’eterogeneità piuttosto che del caso [23]. Poiché gli AA sono il substrato principale per la sintesi proteica, sono state effettuate analisi di sottogruppo in base ai diversi livelli di apporto di AA al muscolo e quindi alla quantità disponibile (aumentata, invariata o diminuita) per il metabolismo proteico. Un’altra analisi di sottogruppo è stata eseguita con studi che coinvolgevano popolazioni con diabete. In questi studi, l’apporto di AA non è cambiato. È stata condotta un’analisi di meta-regressione per verificare le differenze nelle stime in pool tra i sottogruppi e per verificare se le stime in pool differissero in base ad altre covarianti (ad esempio, i livelli di concentrazione di Insulina raggiunti, l’età o la massa corporea magra).
Il bias di pubblicazione è stato valutato esaminando un funnel plot in funzione della dimensione dell’effetto. I test statistici per le meta-analisi sono stati eseguiti utilizzando il pacchetto statistico STATA 13.0 (StataCorp, College Station, TX, USA).
Dopo la rimozione dei duplicati, sono stati recuperati 646 articoli dalla ricerca e dalle liste di riferimento degli articoli selezionati (Fig. seguente). Lo screening del titolo e dell’abstract ha portato all’esclusione di 455 articoli a causa dell’irrilevanza (ad esempio, studi in vitro, studi sul metabolismo delle proteine epatiche) e di altri 87 articoli perché gli studi erano stati condotti su muscolo scheletrico animale. In totale sono stati identificati 104 articoli potenzialmente rilevanti, che sono stati valutati in modo più approfondito. Di questi, altri 60 articoli sono stati esclusi. I principali motivi di esclusione sono stati: (1) gli studi valutavano il ruolo degli interventi nutrizionali e non dell’Insulina in sé; e (2) gli articoli erano revisioni piuttosto che studi di ricerca. Un totale di 44 articoli che comprendevano 75 studi soddisfaceva i criteri per la review sistematica. Di questi 44 articoli, 13 (contenenti 25 studi) sono stati inclusi nella meta-analisi, in quanto rappresentavano il gruppo più numeroso che conteneva dati quantitativamente comparabili. Tutti i 25 studi hanno utilizzato la fenilalanina come tracciante AA, hanno riportato la MPS/MPB in unità di nmol (100ml leg vol.)-1 min-1 e hanno utilizzato l’approccio a due pool (equilibrio arterovenoso) per stimare le variabili di esito.

- MPS e Insulina
Per l’inclusione nella meta-analisi sono stati identificati 13 articoli [16, 19-21, 24-32], contenenti 25 studi sperimentali che hanno utilizzato diverse concentrazioni di insulina (Tabella 1); tutti hanno analizzato l’effetto dell’insulina sia sulla MPS che sulla MPB (Tabella 1). In totale sono stati inclusi 173 individui in questi studi. Un totale di 13 studi ha coinvolto giovani adulti e tre di questi 13 studi hanno coinvolto individui con diabete. Otto studi hanno coinvolto persone anziane e sane, mentre per quattro studi non erano disponibili dati sull’età. L’età media dei partecipanti variava da 18 a 74 anni. La maggior parte degli studi (20 studi) ha utilizzato l’infusione locale di insulina intra-arteriosa per limitare lo sviluppo di ipoglicemia sistemica e un’infusione obbligatoria di glucosio, anche per limitare l’ipoaminoacidemia [33]. Questo aspetto è di fondamentale importanza, soprattutto quando si utilizzano concentrazioni di insulina sovrafisiologiche. Le concentrazioni sistemiche di insulina variavano tra 62,5 e 861,2 pmol/l. L’apporto di AA (concentrazione di AA nell’arteria × flusso sanguigno arterioso) è stato mantenuto in 14 studi e aumentato in otto studi. Tuttavia, come conseguenza diretta della somministrazione sistemica di insulina (vedi discussione sotto), gli AA circolanti sono diminuiti in tre studi.
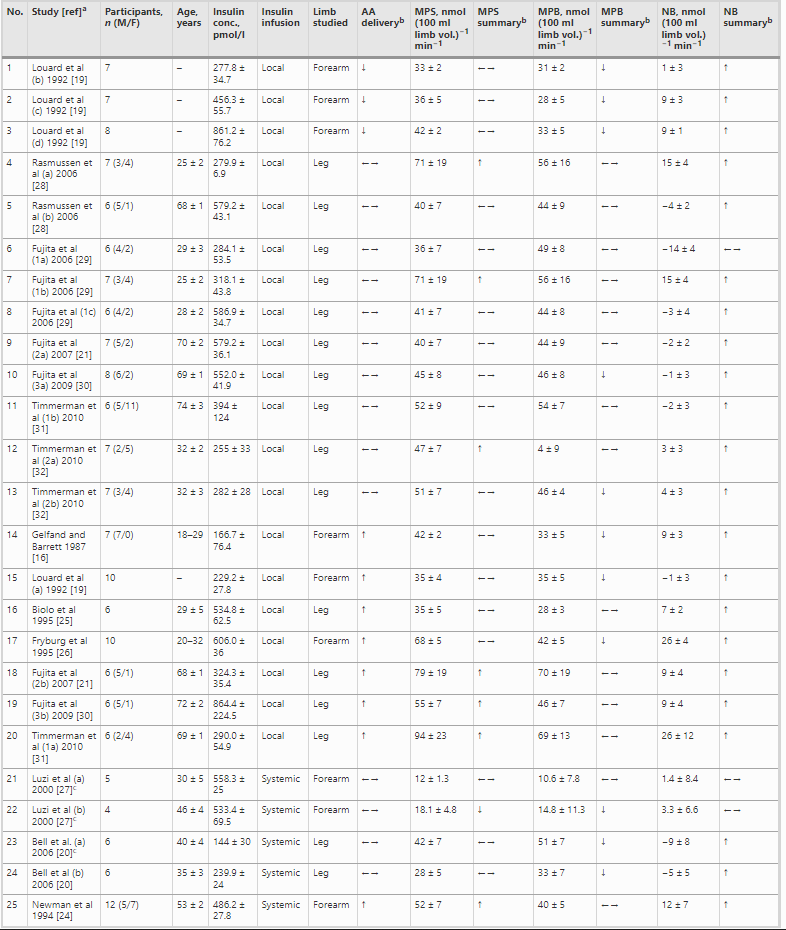
aI numeri accanto al nome dell’autore distinguono studi diversi dello stesso autore; le lettere indicano interventi o caratteristiche dei partecipanti diversi all’interno di uno stesso studio.
bRispetto al basale
cPartecipanti con diabete
I dati della meta-analisi sono stati raggruppati da 13 studi (25 studi o confronti) che hanno coinvolto 173 individui. La WMD per la MPS era 3,90 (95% CI -0,74, 8,55; p = 0,71). L’analisi degli studi basati sulla somministrazione di AA ha rivelato un aumento della MPS (WMD 13,44 [95% CI 4,07, 22,81], p < 0,01) negli studi in cui la somministrazione di AA era aumentata (otto studi, 63 individui). Tuttavia, la MPS non è cambiata significativamente quando la somministrazione di AA è stata ridotta (tre studi; 22 partecipanti; WMD 1,57 [95% CI -3,97, 7,12], p = 0,58) o mantenuta al basale (11 studi; 73 partecipanti; WMD 2,00 [95% CI -5,28, 9,28], p = 0,59). Gli studi che hanno coinvolto individui con diabete (tre studi, 15 individui) hanno mostrato riduzioni significative della MPS in risposta all’Insulina, anche se l’apporto di AA è stato mantenuto (WMD -6,67 [95% CI -12,69, -0,66], p < 0,05).

All’analisi di meta-regressione, la dimensione della stima (WMD) era significativamente diversa tra i sottogruppi basati sulla disponibilità di AA (p = 0,001).
L’I 2 per l’effetto complessivo dell’Insulina sulla MPS era del 49% (p = 0,003). Questa significativa eterogeneità moderata sembrava essere dovuta principalmente all’eterogeneità all’interno del sottogruppo con aumento degli AA (I 2 59%; p = 0,018). Gli altri sottogruppi hanno mostrato valori di p non significativi per l’eterogeneità, suggerendo una maggiore coerenza tra questi studi rispetto ai dati complessivi della MPS (sottogruppi: consegna AA mantenuta: I 2 21%; p = 0,241; diminuzione del rilascio di AA: I 2 0%; p = 0,811; individui con diabete: I 2 0%; p = 0,605).
- MPB e Insulina
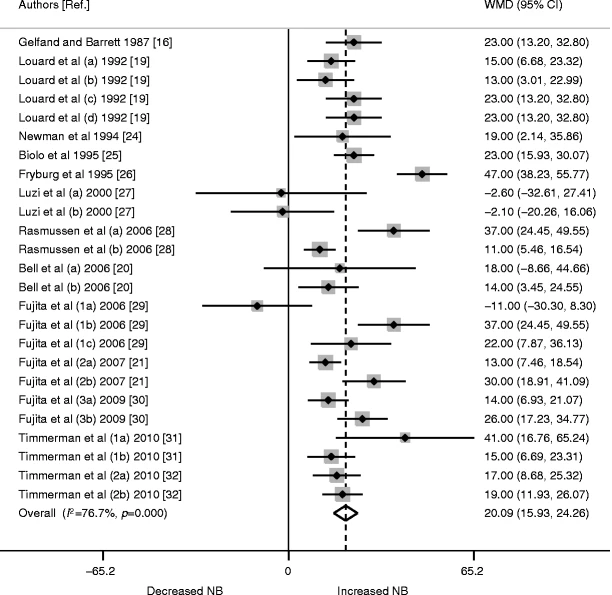
I dati sono stati raggruppati dagli stessi 25 studi come per la MPS. La WMD per la MPB era di -15,46 (95% CI -19,74, -11,18; p < 0,0001; Fig. 3). La disponibilità di AA non ha avuto un impatto significativo sulla dimensione stimata della MPB (p = 0,754). L’I 2 per l’effetto complessivo dell’Insulina sulla MPB era del 13% (p = 0,282), indicando un’eterogeneità non significativa (Fig. 3).

- Insulina e bilancio netto delle proteine [NB]
In un’ulteriore analisi in pool di tutti i 25 studi, è stato riscontrato che l’Insulina aumenta significativamente l’assorbimento di proteine NB (WMD 20,09 [95% CI 15,93, 24,26], p < 0,0001).
- Analisi di meta-regressione di altre variabili
È stata condotta un’analisi di meta-regressione per verificare se altre variabili confondenti di interesse avessero un effetto sulla WMD (ad esempio, la concentrazione di insulina infusa, l’età e la massa corporea magra, se disponibili). Le differenze nelle concentrazioni di Insulina infusa non hanno avuto alcun effetto su MPS (p = 0,955), MPB (p = 0,713) o NB (p = 0,621). Non vi è stato alcun effetto nemmeno per le differenze di età (p = 0,480, p = 0,159 e p = 0,610, rispettivamente) o per le variazioni della massa corporea magra (p = 0,433, p = 0,936 e p = 0,617, rispettivamente).
- Bias di pubblicazione e altri dati
I diagrammi a imbuto dell’effetto dell’insulina su MPS e MPB rispetto a SE non hanno mostrato alcun bias di pubblicazione (vedi materiale supplementare elettronico [ESM]).
Tra gli articoli esaminati, 15 studi hanno utilizzato dati a tre pool [21, 25, 28, 30-35]. I dati a due pool di dieci di questi studi sono stati inclusi nella meta-analisi, che nel complesso ha riportato risultati simili con visualizzazioni quantitativamente diverse. Tutti e cinque gli studi che hanno riportato esclusivamente dati a tre pool hanno dimostrato che l’Insulina ha migliorato l’assorbimento di proteine NB [25, 33-35]. L’aumento dell’NB in questi studi è stato determinato principalmente da una riduzione della MPB [34] o da un aumento della MPS [25, 33-35]. Nessuno di questi studi prevedeva una riduzione della disponibilità di AA. Due degli studi hanno riportato un aumento della MPS in uno stato iperinsulinemico indotto sperimentalmente in soggetti apparentemente insulino-resistenti con gravi ustioni trattati in un’unità di alta dipendenza [33, 35].

aI numeri accanto al nome dell’autore distinguono studi diversi dello stesso autore; le lettere indicano interventi o caratteristiche dei partecipanti diversi all’interno di uno stesso studio.
bRispetto al basale.
- Discussioni conclusive
L’infusione sistemica di Insulina porta a ipoglicemia e a una ridotta disponibilità di AA (ipoaminoacidemia) per la sintesi proteica. Per ovviare a queste conseguenze, glucosio e AA vengono co-infusi per mantenere la glicemia target e la disponibilità di AA. L’infusione locale di Insulina intra-arteriosa sembra limitare l’effetto dell’ipoglicemia sistemica e dell’ipoaminoacidemia, evitando così la necessità di una co-infusione obbligatoria di glucosio (o AA) [33].
La meta-analisi di questi 25 studi non ha mostrato alcun effetto significativo dell’Insulina sulla MPS. L’analisi dei sottogruppi, tuttavia, ha rivelato che negli individui sani l’effetto dell’Insulina sulla MPS diventa significativo solo quando viene aumentata la somministrazione di AA al muscolo scheletrico. Questi risultati sono stati replicati da altri ricercatori in studi in cui la coinfusione di AA e Insulina ha aumentato con successo l’apporto di AA al muscolo [30, 31, 36-42]. In uno studio di Fujita et al, l’esercizio fisico per 45 minuti ha aumentato con successo l’apporto di AA e la MPS rispetto ai controlli non allenati, sebbene l’esercizio fisico di per sé abbia effetti anabolici acuti sulla MPS muscolare [21]. In alcuni studi, tuttavia, l’aumento dell’apporto di AA non ha prodotto un aumento di MPS indotto dall’Insulina [16, 43]. Ciò è probabilmente dovuto al fatto che l’aumento dell’apporto di AA era minimo e ottenuto principalmente attraverso l’aumento del flusso sanguigno, piuttosto che attraverso un aumento della concentrazione di AA. Gli anziani mostrano una resistenza all’effetto anabolico dell’Insulina rispetto alle loro controparti più giovani, probabilmente attraverso meccanismi legati alla disfunzione endoteliale, alla ridotta perfusione tissutale e all’attenuazione della segnalazione anabolica, piuttosto che a una ridotta tolleranza al glucosio [30]. Tuttavia, in presenza di un maggiore apporto di AA, l’Insulina sembra conservare il suo effetto anabolico negli anziani sani. Questo sembra essere il caso sia che l’aumento dell’apporto di AA al muscolo sia ottenuto tramite concentrazioni fisiologiche [24, 38] o sovrafisiologiche [30] di Insulina, farmacologicamente con nitroprussiato di sodio [31] o tramite esercizio fisico [21].
L’aumento delle concentrazioni di Insulina nell’intervallo postprandiale non sembra influire sulla MPS. Uno studio precedente ha riportato che, con aumenti incrementali delle concentrazioni di AA, la MPS ha risposto positivamente a concentrazioni di Insulina di 139,0-194,5 pmol/l, aumentando del 22% rispetto al basale e del 72% quando sono state somministrate concentrazioni di AA più elevate [37]. D’altra parte, un altro studio ha riportato che, in presenza di concentrazioni fisse di AA, l’aumento della concentrazione di Insulina da 34,7 pmol/l a 500,0 pmol/l non ha prodotto ulteriori incrementi significativi della MPS [39]. L’Insulina non ha avuto alcun effetto sulla MPS quando l’apporto di AA è rimasto invariato rispetto al basale. Questi risultati sono supportati anche da altri studi [17, 20, 28-31, 34, 44]. In tutti gli studi sull’uomo sano in cui la disponibilità di AA è stata ridotta, la MPS si è ridotta o è rimasta invariata [18, 19, 37, 45, 46], anche in presenza di concentrazioni sovrafisiologiche di Insulina [18].
La meta-analisi dei 25 studi ha dimostrato che l’Insulina esercita la sua regolazione della massa muscolare magra principalmente attraverso un effetto anticatabolico nella riduzione della MPB. Questo è più evidente se si considerano i dati sulla NB, che hanno mostrato un effetto positivo sulla massa muscolare. Pertanto, le capacità proanaboliche dell’Insulina sono prevalentemente guidate dalla sua capacità di attenuare la MPB scheletrica, piuttosto che da un effetto positivo sulla MPS. Questo risultato è in accordo con le valutazioni di altri ricercatori [16, 29].
La riduzione della MPB indotta dall’Insulina sembra essere più potente quando gli AA sono scarsi. Questi risultati sono coerenti con altri studi che hanno riportato una riduzione della risposta all’Insulina [30, 32, 37, 39, 42, 44], ad eccezione di tre studi che non hanno osservato alcun cambiamento significativo nella MPB [45, 47, 48]. Ciò potrebbe essere dovuto alla resistenza anabolica all’Insulina in una popolazione di studio relativamente anziana e alla presenza di diabete mellito. È interessante notare che l’inibizione massima della MPB da parte dell’Insulina si verifica in risposta a incrementi molto modesti della concentrazione di Insulina (cioè a 104,2 pmol/l) [44].
È stato riferito che il diabete attenua l’effetto positivo dell’Insulina sulle MPB in presenza di una somministrazione prolungata di AA [17, 20, 49]. Tuttavia, in risposta a un trattamento intensivo a lungo termine con Insulina s.c., Halvatsiotis et al. non hanno riscontrato differenze nelle MPS mitocondriali, sarcoplasmatiche o miste rispetto ai controlli sani che non ricevevano Insulina [50]. Non è noto se l’aumento dell’assunzione di AA nei pazienti diabetici trattati con Insulina possa portare a un aumento della massa muscolare. Tuttavia, dato il ruolo facilitante dell’Insulina nel mantenimento della massa muscolare, in particolare in presenza di una maggiore disponibilità di AA, si può ipotizzare la necessità di consigliare ai pazienti con diabete in trattamento insulinico di aumentare l’assunzione di AA per sfruttare gli effetti positivi dell’Insulina sul metabolismo muscolare. Nei pazienti gravemente malati, dove ci si aspetta una significativa resistenza all’Insulina, sono stati osservati aumenti della MPS, ma solo quando sono state utilizzate concentrazioni sovrafisiologiche di Insulina [33, 51]. In uno studio condotto su individui affetti da obesità [40] e insufficienza cardiaca [42], è stato dimostrato che la MPB si riduce in risposta all’Insulina. Questa riduzione, tuttavia, è stata significativamente inferiore a quella osservata nei controlli sani. È evidente che sono necessari ulteriori studi per comprendere appieno il ruolo dell’insulino-resistenza nella regolazione di MPS e MPB nel diabete di tipo II.
Comunque, la review qui esposta presenta diverse limitazioni. I diversi metodi di stima del metabolismo proteico del muscolo scheletrico e la mancanza di dati primari disponibili hanno reso difficile eseguire una valutazione quantitativa completa mediante meta-analisi per tutti gli studi che soddisfacevano i criteri di inclusione della review sistematica. Riconosciamo inoltre che l’uso di più studi per ogni pubblicazione significa che il pooling degli studi non è del tutto indipendente. Inoltre, poiché si tratta di una meta-analisi di studi sperimentali, non è stato possibile effettuare una valutazione completa dei bias, come normalmente si fa nelle meta-analisi di studi controllati randomizzati [52] (ad esempio, generazione della sequenza e occultamento dell’allocazione per controllare i bias di selezione; cecità per controllare eventuali bias di performance, dati di esito incompleti, bias di segnalazione selettiva e altre fonti di bias), poiché nessuno degli studi ha utilizzato questi metodi per l’allocazione dei pazienti. Inoltre, poiché tutti gli studi erano di dimensioni simili, l’analisi del funnel plot potrebbe non essere completamente affidabile nell’informarci di eventuali bias di pubblicazione.
In sintesi, questa review sistematica e meta-analisi suggerisce che il ruolo principale dell’Insulina nell’anabolismo del muscolo scheletrico umano è di tipo facilitativo ed è influenzato dalla velocità di somministrazione degli AA. In situazioni in cui l’apporto di AA è invariato, sono necessarie concentrazioni sovrafisiologiche di Insulina per ottenere l’anabolismo del muscolo scheletrico. Tuttavia, il ruolo dell’Insulina nel ridurre la MPB è chiaramente evidente nella maggior parte degli studi. Questo effetto è attenuato nelle persone anziane e in quelle con resistenza all’Insulina. Questa resistenza è probabilmente legata a un’alterata segnalazione insulinica del metabolismo proteico muscolare e alla disfunzione endoteliale, piuttosto che all’intolleranza al glucosio. Sono necessarie ulteriori prove per tradurre questi risultati in strategie per massimizzare la massa muscolare nei pazienti con diabete insulino-trattato.
Effetto dell’Insulina esogena sull’aumenta del tasso di sintesi proteica muscolare:
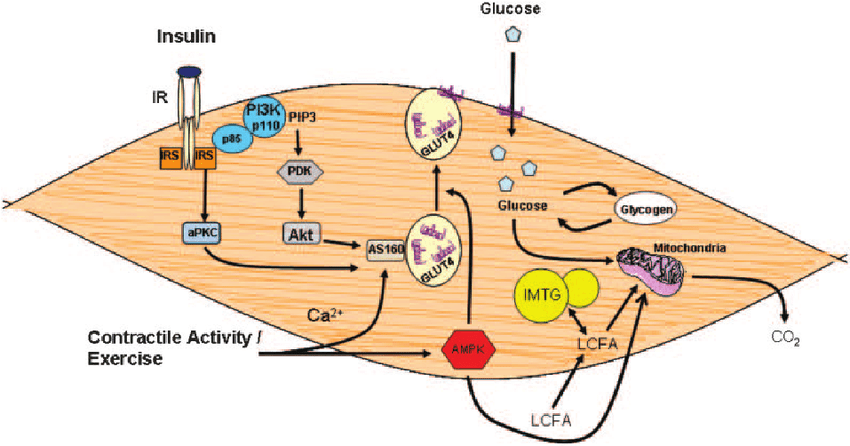
L’Insulina è ben nota come ormone chiave responsabile dell’aumento dell’accumulo endogeno di carboidrati e grassi. Tuttavia, il suo ruolo nel metabolismo delle proteine è più controverso. Studi in vitro hanno dimostrato che l’Insulina stimola la sintesi proteica muscolare mediante l’attivazione diretta del meccanismo di traduzione attraverso la via PI3K→Akt→mTORC1 (53,54,55,56,57,58,59,60,61,62,63). L’Insulina può anche influenzare il metabolismo proteico in vivo grazie alle sue proprietà vasoattive. L’aumento postprandiale dell’Insulina circolante stimola la vasodilatazione endotelio-dipendente in virtù della sua azione sull’ossido nitrico sintasi endoteliale, con conseguente maggiore reclutamento capillare, aumento del volume microvascolare e flusso sanguigno nutritivo al tessuto muscolare scheletrico (64). Si potrebbe ipotizzare che la maggiore perfusione postprandiale aumenti l’esposizione del tessuto muscolare ai nutrienti e ai fattori di crescita e aumenti la sintesi proteica muscolare. Tuttavia, se l’Insulina abbia un effetto stimolante sulla sintesi proteica muscolare postprandiale nell’uomo è stato oggetto di un ampio dibattito come, tra l’altro, abbiamo appena visto (65, 66). Molti ritengono che le concentrazioni di Insulina in circolo siano semplicemente permissive, anziché modulatorie, per consentire un aumento della sintesi proteica muscolare in soggetti giovani e sani (66). In particolare, si ritiene che sia necessaria solo una piccola quantità di Insulina per “innescare” il sistema e che sia il successivo aumento della disponibilità di aminoacidi a guidare la risposta della sintesi proteica muscolare post-prandiale (67). Tuttavia, si ipotizza che gli anziani siano più resistenti all’effetto dell’Insulina sulla sintesi proteica muscolare, un difetto associato alla disfunzione endoteliale (68, 69, 70). Questa nuova review sistematica esamina la letteratura esistente sull’effetto proposto dell’aumento dei livelli di Insulina circolante sulla sintesi proteica muscolare in vivo nell’uomo e cerca di definire se tale effetto differisce tra giovani e anziani.

Insulina e IGF-1: fattore di crescita insulino-simile, PKB/Akt: protein chinasi B, AMPK: adenosina monofasfato protein chinasi, mTOR: mammalian target of rapamycin,
p70S6K: proteina ribosomiale S6 chinasi, 4E-BP1: proteina legante il fattore di iniziazione eucariotica 4E, eIF4G: fattore di iniziazione eucariotica 4G.
È stata eseguita una review sistematica secondo le linee guida PRISMA (71). In breve, nel gennaio 2014 è stata eseguita una ricerca computerizzata della letteratura utilizzando il database PubMed (http://www.ncbi.nlm.gov/pubmed/) e cercando a mano le liste di riferimento degli studi identificati e le principali review della letteratura. Sono stati utilizzati i seguenti termini di ricerca: Insulina; iperinsulinismo; muscolo; gamba; avambraccio; miofibrillare; anabol; sintesi proteica e accrescimento proteico e le funzioni booleane AND e OR. La ricerca elettronica finale è stata effettuata l’8 agosto 2014.
- Tipi di studi
Studi clinici che studiano la somministrazione di Insulina a persone sane. Gli studi sono stati limitati a quelli scritti in lingua inglese. Non sono state imposte restrizioni sulla data di pubblicazione. - Tipi di partecipanti
Sono stati presi in considerazione partecipanti sani di qualsiasi età che ricevevano Insulina esogena e sono stati stratificati in giovani adulti sani (età media del gruppo tra 18 e 65 anni) o adulti anziani (età media del gruppo ≥65 anni). Per studiare l’effetto dell’età sulla sintesi proteica muscolare insulino-mediata di per sé, sono stati esclusi i soggetti con qualsiasi co-morbilità apparente, incluso il diabete. - Tipi di intervento
Questa review è stata limitata agli studi che hanno esaminato la somministrazione di Insulina esogena. - Tipi di misure di esito
La misura di esito primaria è la valutazione qualitativa della sintesi proteica muscolare, ossia un aumento significativo o nessun effetto. Gli studi inclusi hanno valutato la sintesi proteica muscolare come misurata dal tasso di scomparsa dei precursori (leg Rd) nel metodo del bilancio arteriovenoso a due vasche, dall’utilizzo intracellulare dei precursori (Fo,m) nel metodo del bilancio arteriovenoso a tre vasche o dal tasso di sintesi frazionale (FSR) nel modello precursore-prodotto (28). Queste misure tendevano a raggiungere valutazioni qualitative simili della sintesi proteica muscolare (dati non mostrati), pertanto non è stata fatta alcuna distinzione tra i modelli per l’interpretazione dell’effetto riportato dell’Insulina sulla sintesi proteica muscolare.
La valutazione dell’idoneità è stata eseguita individualmente da due autori (J Trommelen e B B L Groen). Le divergenze tra i revisori sono state risolte per consenso. I titoli e gli abstract identificati dalla strategia di ricerca sono stati vagliati per la rilevanza, definita dal rispetto di tutti i seguenti criteri: i) soggetti umani, ii) il disegno dello studio era un trial clinico, iii) l’intervento includeva la somministrazione di insulina esogena in almeno uno dei bracci dello studio, iv) valutava la sintesi proteica muscolare mista (Rd, Fo,m o FSR) e v) l’accessibilità al testo completo.
Due autori (J Trommelen e B B L Groen) hanno estratto individualmente i dati dagli studi inclusi. Le divergenze tra i revisori sono state risolte per consenso. Da ogni studio incluso sono state estratte informazioni su: i) caratteristiche dei soggetti, tra cui età e numero; ii) modello di valutazione della sintesi proteica muscolare (dati non mostrati); iii) tipo di intervento, tra cui dose, co-intervento, compartimento di infusione e gruppi di confronto; e iv) esito dello studio, tra cui effetto dell’insulina esogena sulla sintesi proteica muscolare, raggruppato in aumento significativo o nessun effetto.
Gli studi sono stati esaminati con una tabulazione completa dei risultati di tutti gli studi inclusi. A causa dell’eterogeneità clinica dei disegni sperimentali, non è stato possibile condurre una meta-analisi. Inoltre, è noto che le differenze nei metodi sperimentali introducono variabilità nella sintesi proteica muscolare, complicando l’analisi quantitativa tra gli studi (si rimanda a Smith et al. (72) per una rassegna su questo argomento). Pertanto, per gli studi inclusi è stata determinata una valutazione qualitativa della sintesi proteica muscolare, ossia un aumento significativo o nessun effetto. Sulla base di questi dati, abbiamo costruito diversi modelli in cui i bracci di studio sono stati esclusi sulla base di motivazioni (biologiche). Nel modello 1 sono stati esclusi i bracci di studio con iperamminoacidemia concomitante. Gli aminoacidi possono stimolare in modo indipendente la sintesi proteica muscolare e il rilascio endogeno di insulina, rendendo impossibile distinguere tra l’effetto dell’insulina e quello dell’infusione di aminoacidi (73, 74). Il modello 2 esclude inoltre i bracci di studio con ipoaminoacidemia insulino-mediata. È stato suggerito che l’effetto dell’insulina sulla sintesi proteica muscolare sia mediato da cambiamenti indotti dall’insulina nell’apporto di aminoacidi al muscolo (75, 76, 77, 78). L’infusione sistemica di insulina induce ipoaminoacidemia, che può limitare l’apporto di aminoacidi al muscolo. Pertanto, in questo modello, sono stati esclusi i bracci di studio che hanno permesso ai livelli di aminoacidi di scendere al di sotto del valore basale. Il modello 3 esclude inoltre i bracci di studio che raggiungono concentrazioni di insulina sovrafisiologiche. Ciò è stato fatto per differenziare tra livelli sovrafisiologici che possono essere raggiunti solo con la somministrazione di Insulina (>1200 pmol/l) e livelli fisiologici alla portata della produzione endogena in risposta a un pasto misto (≤1200 pmol/l). Il modello 4 esclude inoltre i bracci di studio in soggetti anziani, perché è stato suggerito che gli anziani sono più resistenti alle proprietà stimolanti proposte dall’insulina sulla sintesi proteica muscolare (79, 80).
La ricerca nel database PubMed ha fornito un totale di 2021 citazioni. Dal totale di 2025 citazioni, sono stati scartati 1980 studi perché, dopo aver esaminato gli abstract, non soddisfacevano i criteri di inclusione. Il testo completo delle restanti 45 citazioni è stato esaminato in modo più approfondito. Dopo un’attenta lettura del testo integrale, altri dieci studi non soddisfacevano i criteri di inclusione descritti. In totale, 40 studi hanno soddisfatto i criteri di ammissibilità e sono stati inclusi nella revisione sistematica.

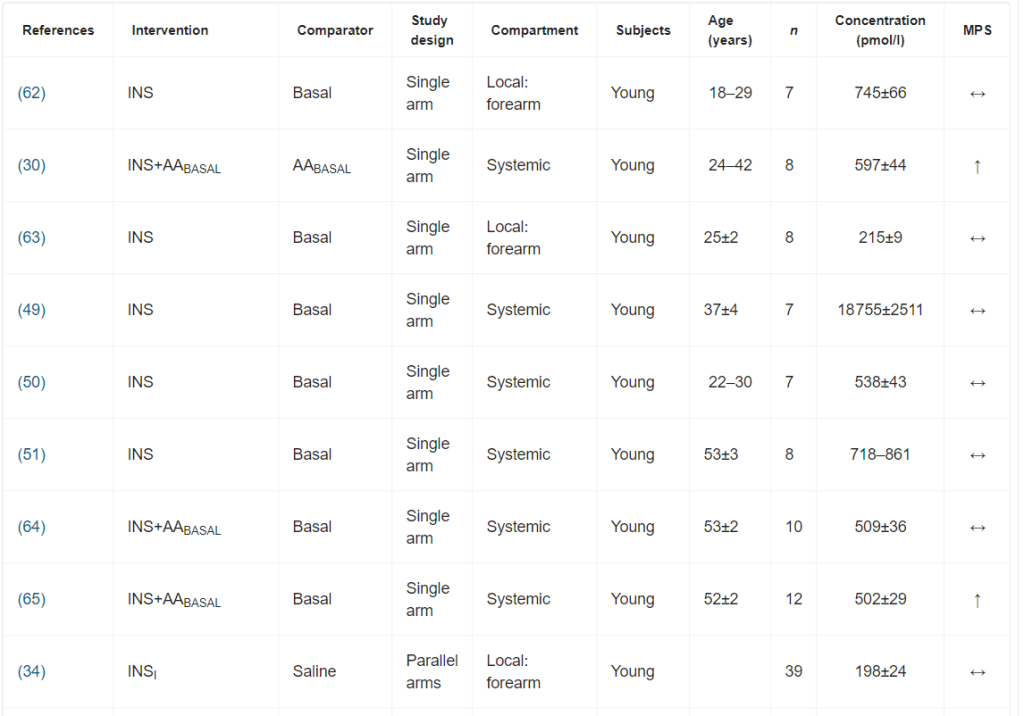


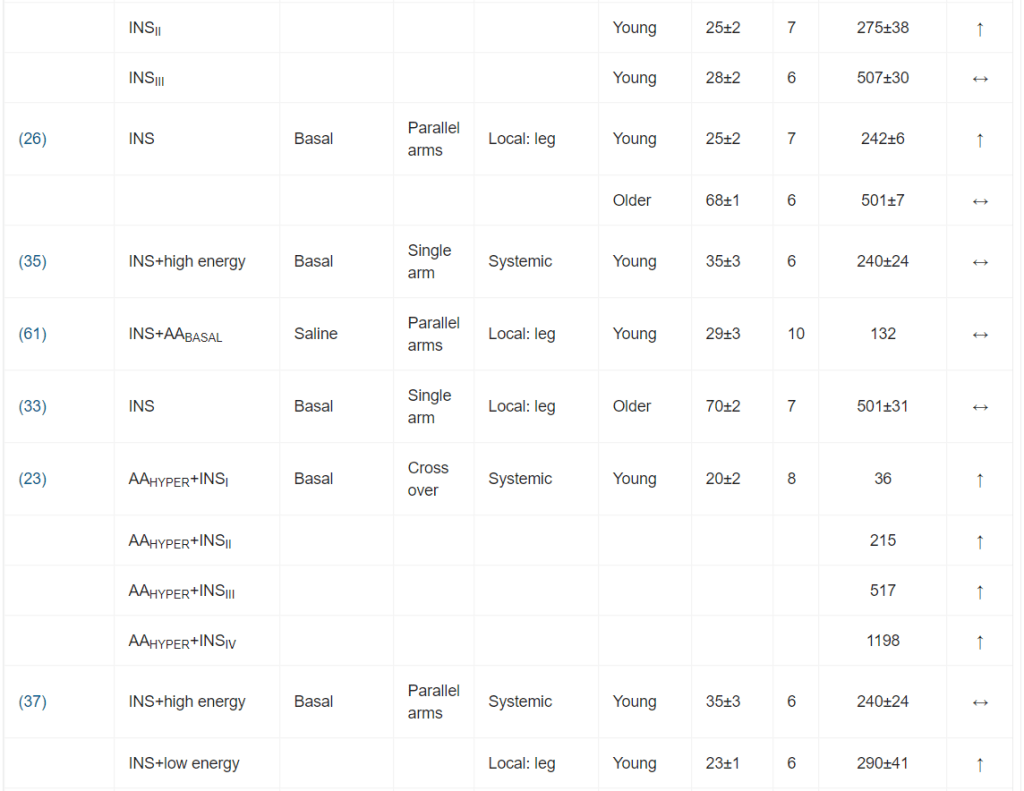
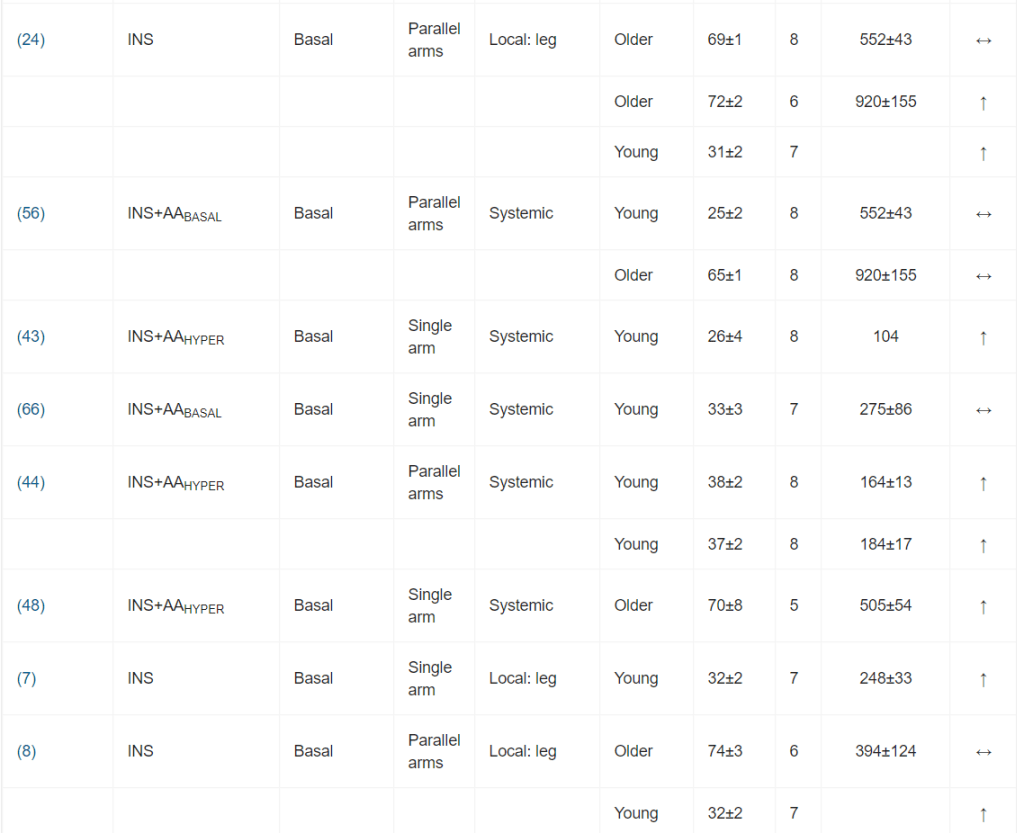

È stata osservata un’elevata eterogeneità metodologica. I design degli studi includevano studi a braccio singolo, a bracci paralleli e crossover. I gruppi di confronto variavano notevolmente, includendo nessun intervento (condizioni basali), soluzione fisiologica, dosaggi insulinici alternativi o protocolli di somministrazione di aminoacidi.
I principali criteri di ammissibilità prevedevano un buono stato di salute. La maggior parte degli studi riportava un esame anamnestico preliminare ed esami del sangue standard (principalmente per la valutazione della tolleranza al glucosio).
I dosaggi di insulina esogena applicati hanno portato a concentrazioni plasmatiche di insulina che variavano da livelli a digiuno (36 pmol/l) a livelli sovrafisiologici (81 078 pmol/l). Sono stati comunemente utilizzati protocolli di infusione sia sistemici che locali. I protocolli di infusione locale di Insulina utilizzavano l’avambraccio o la gamba come compartimento. Il co-intervento variava notevolmente; il più comune era la coinfusione di aminoacidi.
Sono stati identificati 40 studi da includere nella review, che variavano notevolmente nel design sperimentale. Numerosi studi hanno applicato disegni di ricerca che comprendevano più bracci sperimentali con interventi separati e molti dei quali hanno riportato risultati opposti, rendendo difficile trarre conclusioni a livello di studio. Inoltre, molti degli studi identificati includevano co-interventi, come la somministrazione di farmaci o protocolli di esercizio, che influenzano i risultati. Pertanto, la sintesi dei dati è stata effettuata a livello di braccio di studio.
Dai 40 studi selezionati, dopo l’esclusione degli interventi non correlati ai pasti (cioè la co-somministrazione di farmaci), è stato identificato un totale di 66 bracci di studio che includevano il trattamento insulinico. Di questi 66 bracci di studio, 34 hanno riscontrato un effetto insulino-stimolante sulla sintesi proteica muscolare, mentre 32 non hanno riscontrato tale effetto. Dodici bracci di studio consistevano in dati riutilizzati da altri studi inclusi (81, 82, 83, 84, 85, 86, 87, 88). Dopo la deduplicazione, è stato trovato un totale di 54 bracci di studio unici, di cui 28 hanno riportato un aumento dei tassi di sintesi proteica muscolare, mentre 26 non lo hanno fatto.
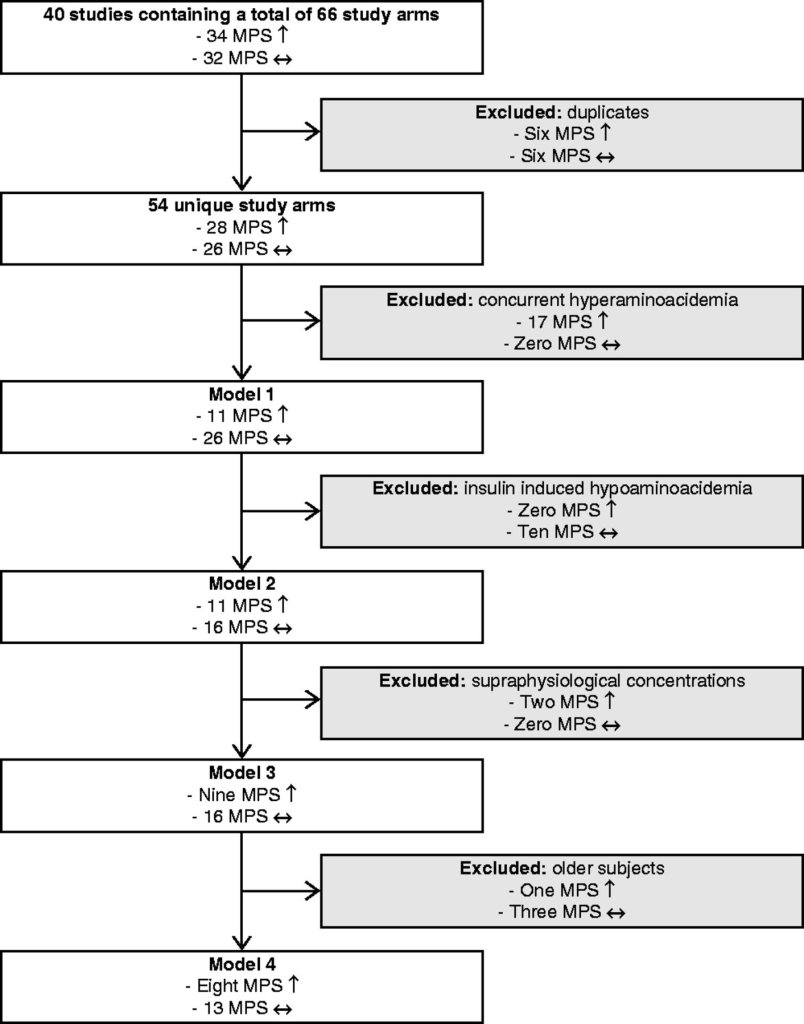
Sulla base di questi dati, sono stati costruiti diversi modelli che escludevano i bracci di studio sulla base di un razionale (biologico), come descritto in precedenza. Nel modello 1, sono stati esclusi i bracci di studio in cui la somministrazione di insulina era combinata con co-interventi di aminoacidi che aumentavano gli aminoacidi plasmatici oltre i livelli basali e non avevano un gruppo di confronto per correggere l’aumento degli aminoacidi plasmatici (89, 90, 91, 92, 93, 94, 95, 96, 97, 98). Questo criterio ha escluso 17 bracci di studio, che hanno tutti riportato un aumento della sintesi proteica muscolare.
Il modello 2 escludeva inoltre i bracci di studio in cui i livelli di aminoacidi potevano scendere al di sotto dei livelli basali (99, 100, 101, 102, 102, 103, 104, 105). Questo criterio ha escluso altri dieci bracci di studio rispetto al modello 1, nessuno dei quali ha riportato un effetto sulla sintesi proteica muscolare.
Il modello 3 ha inoltre escluso i bracci di studio in cui è stata raggiunta una concentrazione sovrafisiologica di insulina (106). Questo criterio ha escluso altri due bracci di studio rispetto al modello 2, che hanno entrambi riportato che l’insulina aumenta la sintesi proteica muscolare.
Il modello 4 ha inoltre escluso i bracci di studio in soggetti anziani (107, 108, 109,110, 111). Questo criterio ha escluso altri quattro interventi rispetto al modello 3, uno dei quali ha riportato un effetto insulino-stimolante sulla sintesi proteica muscolare, mentre gli altri tre non lo hanno fatto. Dopo queste esclusioni finali, il modello 4 includeva un totale di 21 bracci di studio, otto dei quali riportavano un aumento della sintesi proteica muscolare, mentre 13 non lo facevano.
- Discussione finale
Questa review sistematica ha esaminato la letteratura riguardante l’effetto proposto, legato all’età, della somministrazione di Insulina esogena sui tassi di sintesi proteica muscolare in vivo nell’uomo. Sebbene siano stati condotti numerosi studi per valutare l’impatto della somministrazione di Insulina esogena sulla sintesi proteica muscolare, i dati non supportano un ruolo stimolatorio della somministrazione di Insulina esogena sui tassi di sintesi proteica muscolare in vivo nell’uomo.

Gli aminoacidi sono ben noti per la loro capacità indipendente di stimolare la sintesi proteica muscolare (112, 113). Pertanto, è stato costantemente dimostrato che la somministrazione di Insulina e aminoacidi aumenta la sintesi proteica muscolare (114, 115, 116). Senza un adeguato gruppo di controllo con un grado simile di iperamminoacidemia, è impossibile differenziare le proprietà anaboliche proposte dalla somministrazione di Insulina e aminoacidi. Come previsto, tutti i 17 bracci di studio che combinavano la somministrazione di Insulina e aminoacidi, esclusi dalle analisi in base a questo criterio, hanno riportato un aumento della sintesi proteica muscolare. Va notato che uno stato di iperinsulinemia e iperamminoacidemia concomitanti riflette le condizioni fisiologiche successive all’ingestione di un pasto misto. Tre studi hanno esaminato se la somministrazione di Insulina esogena possa aumentare ulteriormente la sintesi proteica muscolare in condizioni di iperamminoacidemia, e tutti non hanno rilevato un effetto incrementale (60, 75, 83). Questi risultati suggeriscono che l’iperinsulinemia e l’iperamminoacidemia concomitanti aumentano la sintesi proteica muscolare ma, almeno nei soggetti giovani e sani, questo effetto sembra interamente attribuito all’iperamminoacidemia.
La somministrazione di Insulina per via endovenosa è seguita da una riduzione dose-dipendente dei livelli plasmatici di aminoacidi, con gli aminoacidi a catena ramificata più sensibili all’aumento dei livelli circolanti di Insulina (90). Questa ipoaminoacidemia indotta dall’Insulina è il riflesso di un aumento dell’assorbimento di aminoacidi dal plasma in combinazione con l’azione inibitoria proposta dall’aumento dei livelli di Insulina sulla proteolisi endogena (80, 70). È stato suggerito che il proposto effetto positivo della somministrazione di Insulina esogena sulla sintesi proteica muscolare sia mediato dall’aumento del flusso sanguigno indotto dall’Insulina e dal conseguente maggiore apporto di aminoacidi al muscolo. Un calo delle concentrazioni circolanti di aminoacidi può limitare l’apporto di aminoacidi al muscolo e di conseguenza limitare la capacità dell’Insulina di stimolare la sintesi proteica muscolare. Per evitare questo calo dei livelli di aminoacidi, diversi studi hanno applicato infusioni i.v. di aminoacidi in combinazione con la somministrazione di Insulina o hanno infuso Insulina esogena localmente nell’arteria femorale o brachiale. Nel modello 2, sono stati esclusi dieci bracci di studio che hanno mostrato un’ipoaminoacidemia indotta dall’Insulina. Nessuno di questi dieci bracci di studio ha riscontrato un effetto insulino-stimolante dell’Insulina sulla sintesi proteica muscolare, sostenendo il razionale che l’ipoaminoacidemia indotta dall’Insulina possa ovviare alle proprietà stimolanti proposte dalla somministrazione di Insulina sull’apporto di aminoacidi al muscolo e sul conseguente aumento della sintesi proteica muscolare.
È stato dimostrato che l’Insulina aumenta la sintesi proteica muscolare in vitro (117, 118, 119). Tuttavia, dagli studi in vivo sull’uomo emergono molte discrepanze sugli effetti positivi proposti dall’Insulina esogena sui tassi di sintesi proteica muscolare. È stato suggerito che l’apparente discrepanza sia attribuita alle concentrazioni di Insulina sovrafisiologiche più che decuplicate (14.000 pmol/l o superiori) applicate nei modelli in vitro rispetto agli aumenti più fisiologici dei livelli di Insulina plasmatica (fino a 1.200 pmol/l) applicati nella maggior parte degli studi umani in vivo (53, 60). Uno studio ha somministrato Insulina esogena localmente nell’avambraccio per ottenere livelli locali di Insulina sovrafisiologica superiori a 50.000pmol/l, mentre si bloccavano gli aminoacidi a livelli arteriosi o venosi basali, e ha riportato un aumento dei tassi di sintesi proteica muscolare (60). I loro risultati suggeriscono che i livelli di Insulina sovrafisiologici possono stimolare efficacemente la sintesi proteica muscolare.

È stato ipotizzato che gli anziani siano più resistenti agli stimoli anabolici, come gli aumenti delle concentrazioni plasmatiche circolanti di Insulina e aminoacidi, rispetto agli adulti più giovani (106). La resistenza anabolica alla somministrazione di aminoacidi e/o Insulina nella popolazione anziana potrebbe essere attribuita a un’alterazione dell’apporto di aminoacidi al muscolo stimolato dall’Insulina (58). Di conseguenza, si è ipotizzato che la somministrazione di Insulina esogena possa aumentare la sintesi proteica muscolare negli adulti più anziani (più resistenti all’Insulina). Dopo aver stratificato i dati in giovani e anziani, è stato identificato uno studio che ha riportato un effetto positivo della somministrazione di Insulina esogena sulla sintesi proteica muscolare nei soggetti anziani (54), mentre tre bracci di studio non hanno osservato tale effetto (112, 113). È interessante notare che l’aumento della sintesi proteica muscolare stimolato dall’Insulina nei soggetti anziani è stato osservato solo in presenza di livelli locali di Insulina relativamente elevati, superiori a 900 pmol/l (54). Dosaggi inferiori di somministrazione locale di Insulina, con livelli plasmatici locali di Insulina di ∼500 pmol, non sembravano aumentare la sintesi proteica muscolare negli anziani. Questi dati suggeriscono che gli anziani potrebbero essere più resistenti alle proprietà anaboliche dell’Insulina, una resistenza che potrebbe essere superata con concentrazioni di Insulina più elevate.
Nel modello 4 sono stati applicati criteri di esclusione rigorosi per escludere i fattori che potrebbero modulare l’effetto stimolante proposto dalla somministrazione di Insulina esogena negli adulti sani, tra cui:
- iperamminoacidemia concomitante;
- ipoaminoacidemia indotta dall’Insulina;
- concentrazioni di insulina sovrafisiologiche e
- soggetti più anziani e più resistenti all’Insulina.
Sono stati identificati otto bracci di studio che hanno riportato un aumento della sintesi proteica muscolare stimolata dall’Insulina, mentre 14 bracci di studio non hanno osservato un aumento dei tassi di sintesi proteica muscolare stimolata dall’Insulina in soggetti giovani e sani. Un sottogruppo del modello 4 comprende otto bracci di studio in cui l’Insulina viene infusa localmente nella gamba, di cui cinque bracci di studio hanno riportato un aumento del tasso di sintesi proteica muscolare, mentre tre non lo hanno fatto. Tre di questi bracci di studio provengono da uno studio dose-risposta, in cui la somministrazione di Insulina a bassa dose (0,05 mU/min×100 ml di gamba) e ad alta dose (0,30 mU/min×100 ml di gamba) non ha aumentato il tasso di sintesi proteica muscolare, mentre la dose intermedia (0,15 mU/min×100 ml di gamba) sì (55). Questa dose intermedia ha aumentato i tassi di sintesi proteica muscolare in tutti e cinque i bracci di studio in cui è stata applicata (7, 24, 28, 54, 55), mentre non è stato osservato alcun aumento dei tassi di sintesi proteica muscolare nei tre bracci di studio che hanno applicato un dosaggio alternato (55, 61). Ciò suggerisce un effetto dose-risposta a forma di U della somministrazione di Insulina esogena sulla sintesi proteica muscolare, dove una dose di ∼0,15mU/min×100 ml di gamba può stimolare la sintesi proteica muscolare. È interessante notare che la somministrazione di Insulina esogena non sembra stimolare la sintesi proteica muscolare quando viene infusa localmente nell’avambraccio. In questo sottogruppo del modello 4, tutti e sei gli interventi non hanno riportato alcun aumento dei tassi di sintesi proteica muscolare, nonostante un’ampia gamma di protocolli di dosaggio studiati (58, 62, 63). Nel tentativo di delineare ulteriormente i risultati, i dati sono stati esaminati per individuare altri potenziali fattori modulanti. La presenza o l’assenza di un effetto stimolante in questi studi non poteva essere attribuita a differenze nei livelli di Insulina circolante o alla scelta dei traccianti aminoacidici.
In tutti i lavori presentati, l’Insulina esogena è stata somministrata con un approccio basato sul clamp. Questo approccio può avere dei limiti, in quanto si potrebbe ipotizzare che il forte aumento postprandiale del livello di Insulina circolante abbia una funzione regolatoria per attivare vari processi fisiologici che facilitano l’aumento postprandiale del tasso di sintesi proteica muscolare. Questi cambiamenti temporali nella secrezione di Insulina, nell’apporto e nell’assorbimento di aminoacidi e nella segnalazione intramiocellulare devono essere strettamente regolati per sostenere la risposta anabolica postprandiale. Inoltre, è stato notato che potrebbero esserci differenze nella rilevanza dei livelli di Insulina circolante sulla modulazione della sintesi di varie (serie di) proteine nel tessuto muscolare scheletrico (60).
Dai dati presentati nell’attuale review sistematica, si può concludere che:
- la somministrazione esogena di Insulina e aminoacidi aumenta efficacemente la sintesi proteica muscolare; tuttavia, questo effetto è attribuito all’iperaminoacidemia;
- l’Insulina esogena somministrata per via sistemica induce ipoaminoacidemia, che ovvia a qualsiasi effetto insulino-stimolatorio sulla sintesi proteica muscolare;
- l’Insulina esogena che determina livelli di Insulina sovrafisiologici superiori a 50.000 pmol/l può stimolare efficacemente la sintesi proteica muscolare anche se i livelli di soglia minima sono di 1.200pmol/l;
- l’Insulina esogena può avere un effetto ridotto sulla sintesi proteica muscolare negli adulti più anziani a causa della resistenza anabolica legata all’età;
- l’Insulina esogena somministrata in range fisiologici per via sistemica non aumenta la sintesi proteica muscolare nei giovani adulti sani.
In definitiva, in base ai dati raccolti dalla letteratura esistente, gli autori della review concludono che la somministrazione di Insulina esogena non aumenta i tassi di sintesi proteica muscolare negli adulti sani, giovani o anziani se non superando la soglia fisiologica.
Di conseguenza, ci troviamo di fronte ad un peptide con funzione prevalentemente anticatabolica e soggetto nella sua efficacia al grado di insulino-sensibilità tissutale?… Così sembrerebbe, specie in contesto fisiologico endogeno e indotto esogenamente. Ma se all’equazione ci aggiungessimo altre variabili che influenzino la risposta di base fisiologica e che non interessino il semplice dosaggio di Insulina utilizzato?…
Continua…
Gabriel Bellizzi
Riferimenti:
- Bier DM (1989) Intrinsically difficult problems: the kinetics of body proteins and amino acids in man. Diabetes Metab Rev 5:111–132Article CAS PubMed Google Scholar
- Prod’homme M, Rieu I, Balage M, Dardevet D, Grizard J (2004) Insulin and amino acids both strongly participate to the regulation of protein metabolism. Curr Opin Clin Nutr Metab Care 7:71–77Article PubMed Google Scholar
- McNurlan MA, Garlick PJ (1989) Influence of nutrient intake on protein turnover. Diabetes Metab Rev 5:165–189Article CAS PubMed Google Scholar
- Cruz-Jentoft AJ, Landi F, Schneider SM et al (2014) Prevalence and intervention for sarcopenia in ageing adults: a systematic review. Report of the International Sarcopenia Initiative (EWGSOP and IWGS). Age Ageing 43:748–759PubMed Central Article PubMed Google Scholar
- Atherton PJ, Etheridge T, Watt PW et al (2010) Muscle full effect after oral protein: time-dependent concordance and discordance between human muscle protein synthesis and mTORC1 signaling. Am J Clin Nutr 92:1080–1088Article CAS PubMed Google Scholar
- Wolfson L, Judge J, Whipple R, King M (1995) Strength is a major factor in balance, gait, and the occurrence of falls. J Gerontol A Biol Sci Med Sci 50:64–67PubMed Google Scholar
- Tinetti ME, Williams CS (1997) Falls, injuries due to falls, and the risk of admission to a nursing home. N Engl J Med 337:1279–1284Article CAS PubMed Google Scholar
- Park SW, Goodpaster BE, Lee JS et al (2009) Excessive loss of skeletal muscle mass in older adults with type 2 diabetes. Diabetes Care 32:1993–1997PubMed Central Article PubMed Google Scholar
- Mavros Y, Kay S, Anderberg KA et al (2013) Changes in insulin resistance and HbA1c are related to exercise-mediated changes in body composition in older adults with type 2 diabetes: interim outcomes from the GREAT2DO trial. Diabetes Care 36:2372–2379PubMed Central Article CAS PubMed Google Scholar
- Prospective UK (1995) Diabetes Study Group. UK Prospective Diabetes Study 16. Overview of 6 years’ therapy of type II diabetes: a progressive disease. Diabetes 44:1249–1258Article Google Scholar
- Koro CE, Bowlin SJ, Bourgeois N, Fedder DO (2004) Glycemic control from 1988 to 2000 among U.S. adults diagnosed with type 2 diabetes: a preliminary report. Diabetes Care 27:17–20Article PubMed Google Scholar
- Biesenbach G, Raml A, Alsaraji N (2006) Weight gain and insulin requirement in type 2 diabetic patients during the first year after initiating insulin therapy dependent on baseline BMI. Diabetes Obes Metab 8:669–673Article CAS PubMed Google Scholar
- Packianathan IC, Fuller NJ, Peterson DB, Wright A, Coward WA, Finer N (2005) Use of a reference four-component model to define the effects of insulin treatment on body composition in type 2 diabetes: the ‘Darwin study’. Diabetologia 48:222–229Article CAS PubMed Google Scholar
- Pain VM, Albertse EC, Garlick PJ (1983) Protein metabolism in skeletal muscle, diaphragm, and heart of diabetic rats. Am J Physiol 245:E604–E610CAS PubMed Google Scholar
- Garlick PJ, Grant I (1988) Amino acid infusion increases the sensitivity of muscle protein synthesis in vivo to insulin. Effect of branched-chain amino acids. Biochem J 254:579–584PubMed Central Article CAS PubMed Google Scholar
- Gelfand RA, Barrett EJ (1987) Effect of physiologic hyperinsulinemia on skeletal muscle protein synthesis and breakdown in man. J Clin Invest 80:1–6PubMed Central Article CAS PubMed Google Scholar
- Pacy PJ, Nair KS, Ford C, Halliday D (1989) Failure of insulin infusion to stimulate fractional muscle protein synthesis in type I diabetic patients; anabolic effect of insulin and decreased proteolysis 38:612–624
- Denne SC, Liechty EA, Liu YM, Brechtel G, Baron AD (1991) Proteolysis in skeletal muscle and whole body in response to euglycemic hyperinsulinemia in normal adults. Am J Physiol 261:E809–E814CAS PubMed Google Scholar
- Louard RJ, Fryburg DA, Gelfand RA, Barrett EJ (1992) Insulin sensitivity of protein and glucose metabolism in human forearm skeletal muscle. J Clin Invest 90:2348–2354PubMed Central Article CAS PubMed Google Scholar
- Bell JA, Volpi E, Fujita S et al (2006) Skeletal muscle protein anabolic response to increased energy and insulin is preserved in poorly controlled type 2 diabetes. J Nutr 136:1249–1255PubMed Central CAS PubMed Google Scholar
- Fujita S, Rasmussen BB, Cadenas JG et al (2007) Aerobic exercise overcomes the age-related insulin resistance of muscle protein metabolism by improving endothelial function and Akt/mammalian target of rapamycin signaling. Diabetes 56:1615–1622PubMed Central Article CAS PubMed Google Scholar
- Biolo G, Fleming RY, Maggi SP, Wolfe RR (1995) Transmembrane transport and intracellular kinetics of amino acids in human skeletal muscle. Am J Physiol 268:E75–E84CAS PubMed Google Scholar
- Higgins JP, Thompson SG, Deeks JJ, Altman DG (2003) Measuring inconsistency in meta-analyses. BMJ 327:557–560PubMed Central Article PubMed Google Scholar
- Newman E, Heslin MJ, Wolf RF, Pisters PW, Brennan MF (1994) The effect of systemic hyperinsulinemia with concomitant amino acid infusion on skeletal muscle protein turnover in the human forearm. Metabolism 43:70–78Article CAS PubMed Google Scholar
- Biolo G, Declan Fleming RY, Wolfe RR (1995) Physiologic hyperinsulinemia stimulates protein synthesis and enhances transport of selected amino acids in human skeletal muscle. J Clin Invest 95:811–819PubMed Central Article CAS PubMed Google Scholar
- Fryburg DA, Jahn LA, Hill SA, Oliveras DM, Barrett EJ (1995) Insulin and insulin-like growth factor-I enhance human skeletal muscle protein anabolism during hyperaminoacidemia by different mechanisms. J Clin Invest 96:1722–1729PubMed Central Article CAS PubMed Google Scholar
- Luzi L, Piceni Sereni L, Spessot M et al (2000) Postabsorptive muscle protein metabolism in type 1 diabetic patients after pancreas transplantation. Acta Diabetol 37:219–224Article CAS PubMed Google Scholar
- Rasmussen BB, Fujita S, Wolfe RR et al (2006) Insulin resistance of muscle protein metabolism in aging. FASEB J 20:768–769PubMed Central CAS PubMed Google Scholar
- Fujita S, Rasmussen BB, Cadenas JG, Grady JJ, Volpi E (2006) Effect of insulin on human skeletal muscle protein synthesis is modulated by insulin-induced changes in muscle blood flow and amino acid availability. Am J Physiol Endocrinol Metab 291:E745–E754PubMed Central Article CAS PubMed Google Scholar
- Fujita S, Glynn EL, Timmerman KL, Rasmussen BB, Volpi E (2009) Supraphysiological hyperinsulinaemia is necessary to stimulate skeletal muscle protein anabolism in older adults: evidence of a true age-related insulin resistance of muscle protein metabolism. Diabetologia 52:1889–1898PubMed Central Article CAS PubMed Google Scholar
- Timmerman KL, Lee JL, Fujita S et al (2010) Pharmacological vasodilation improves insulin-stimulated muscle protein anabolism but not glucose utilization in older adults. Diabetes 59:2764–2771PubMed Central Article CAS PubMed Google Scholar
- Timmerman KL, Lee JL, Dreyer HC et al (2010) Insulin stimulates human skeletal muscle protein synthesis via an indirect mechanism involving endothelial-dependent vasodilation and mammalian target of rapamycin complex 1 signaling. J Clin Endocrinol Metab 95:3848–3857PubMed Central Article CAS PubMed Google Scholar
- Gore DC, Herndon DN, Wolfe RR (2005) Comparison of peripheral metabolic effects of insulin and metformin following severe burn injury. J Trauma Inj Infect Crit Care 59:316–323Article CAS Google Scholar
- Biolo G, Williams BD, Fleming RY, Wolfe RR (1999) Insulin action on muscle protein kinetics and amino acid transport during recovery after resistance exercise. Diabetes 48:949–957Article CAS PubMed Google Scholar
- Ferrando AA, Chinkes DL, Wolf SE, Matin S, Herndon DN, Wolfe RR (1999) A submaximal dose of insulin promotes net skeletal muscle protein synthesis in patients with severe burns. Ann Surg 229:11–18PubMed Central Article CAS PubMed Google Scholar
- Hillier TA, Fryburg DA, Jahn LA, Barrett EJ, Teresa A (1998) Extreme hyperinsulinemia unmasks insulin’s effect to stimulate protein synthesis in the human forearm. Am J Physiol Endocrinol Metab 247:E1067–E1074Google Scholar
- Nygren J, Nair KS (2003) Differential regulation of protein dynamics in splanchnic and skeletal muscle beds by insulin and amino acids in healthy human subjects. Diabetes 52:1377–1385Article CAS PubMed Google Scholar
- Guillet C, Prod’homme M, Balage M et al (2004) Impaired anabolic response of muscle protein synthesis is associated with S6K1 dysregulation in elderly humans. FASEB J 18:1586–1587CAS PubMed Google Scholar
- Greenhaff PL, Karagounis LG, Peirce N et al (2008) Disassociation between the effects of amino acids and insulin on signaling, ubiquitin ligases, and protein turnover in human muscle. Am J Physiol Endocrinol Metab 295:595–604Article Google Scholar
- Guillet C, Delcourt I, Rance M et al (2009) Changes in basal and insulin and amino acid response of whole body and skeletal muscle proteins in obese men. J Clin Endocrinol Metab 94:3044–3050Article CAS PubMed Google Scholar
- Smith GI, Atherton P, Reed DN et al (2009) No major sex differences in muscle protein synthesis rates in the postabsorptive state and during hyperinsulinemia-hyperaminoacidemia in middle-aged adults. J Appl Physiol 107:1308–1315PubMed Central Article CAS PubMed Google Scholar
- Toth MJ, LeWinter MM, Ades PA, Matthews DE (2010) Impaired muscle protein anabolic response to insulin and amino acids in heart failure patients: relationship with markers of immune activation. Clin Sci (Lond) 119:467–476
- Heslin MJ, Newman E, Wolf RF, Pisters PW, Brennan MF (1992) Effect of hyperinsulinemia on whole body and skeletal muscle leucine carbon kinetics in humans. Am J Physiol 262:E911–E918CAS PubMed Google Scholar
- Wilkes EA, Selby AL, Atherton PJ et al (2009) Blunting of insulin inhibition of proteolysis in legs of older subjects may contribute to age-related sarcopenia. Am J Clin Nutr 90:1343–1350Article CAS PubMed Google Scholar
- Arfvidsson B, Zachrisson H, Moller-Loswick AC, Hyltander A, Sandstrom R, Lundholm K (1991) Effect of systemic hyperinsulinemia on amino acid flux across human legs in postabsorptive state. Am J Physiol 260:E46–E52CAS PubMed Google Scholar
- Barazzoni R, Short KR, Asmann Y, Coenen-Schimke JM, Robinson MM, Nair KS (2012) Insulin fails to enhance mTOR phosphorylation, mitochondrial protein synthesis, and ATP production in human skeletal muscle without amino acid replacement. Am J Physiol Endocrinol Metab 303:E1117–E1125PubMed Central Article CAS PubMed Google Scholar
- Wolf RF, Heslin MJ, Newman E, Pearlstone DB, Gonenne A, Brennan MF (1992) Growth hormone and insulin combine to improve whole-body and skeletal muscle protein kinetics. Surgery 112:284–291, discussion 291–292CAS PubMed Google Scholar
- Bell JA, Fujita S, Volpi E, Cadenas JG, Rasmussen BB (2005) Short-term insulin and nutritional energy provision do not stimulate muscle protein synthesis if blood amino acid availability decreases. Am J Physiol Endocrinol Metab 289:E999–E1006PubMed Central Article CAS PubMed Google Scholar
- Charlton MR, Balagopal P, Nair KS (1997) Skeletal muscle myosin heavy chain synthesis in type 1 diabetes. Diabetes 46:1336–1340Article CAS PubMed Google Scholar
- Halvatsiotis P, Short KR, Bigelow M, Nair KS (2002) Synthesis rate of muscle proteins, muscle functions, and amino acid kinetics in type 2 diabetes. Diabetes 51:2395–2404Article CAS PubMed Google Scholar
- Sakurai Y, Aarsland A, Herndon DN et al (1995) Stimulation of muscle protein synthesis by long-term insulin infusion in severely burned patients. Ann Surg 222:283–297PubMed Central Article CAS PubMed Google Scholar
- Wang C, Mamza J, Idris I (2015) Biphasic vs basal bolus insulin regimen in type 2 diabetes: a systematic review and meta-analysis of randomized controlled trials. Diabet Med 32:585–594
- Cuthbertson D, Smith K, Babraj J, Leese G, Waddell T, Atherton P, Wackerhage H, Taylor PM, Rennie MJ. Anabolic signaling deficits underlie amino acid resistance of wasting, aging muscle. FASEB Journal 2005 19 422–424. (doi:10.1096/fj.04-2640fje).
- Abellan Van Kan G. Epidemiology and consequences of sarcopenia. Journal of Nutrition, Health & Aging 2009 13 708–712. (doi:10.1007/s12603-009-0201-z).
- Kinsella K & He W. An aging world: 2008. In International Population Reports, pp P95/09-91. U.S. Department of Health and Human Services, 2009. http://www.census.gov/prod/2009pubs/p95-09-1.pdf.
- Volpi E, Sheffield-Moore M, Rasmussen BB, Wolfe RR. Basal muscle amino acid kinetics and protein synthesis in healthy young and older men. Journal of the American Medical Association 2001 286 1206. (doi:10.1001/jama.286.10.1206).
- Koopman R, van Loon LJC. Aging, exercise, and muscle protein metabolism. Journal of Applied Physiology 2009 106 2040. (doi:10.1152/japplphysiol.91551.2008).
- Katsanos CS, Kobayashi H, Sheffield-Moore M, Aarsland A, Wolfe RR. Aging is associated with diminished accretion of muscle proteins after the ingestion of a small bolus of essential amino acids. American Journal of Clinical Nutrition 2005 82 1065–1073.
- Timmerman KL, Lee JL, Dreyer HC, Dhanani S, Glynn EL, Fry CS, Drummond MJ, Sheffield-Moore M, Rasmussen BB, Volpi E. Insulin stimulates human skeletal muscle protein synthesis via an indirect mechanism involving endothelial-dependent vasodilation and mammalian target of rapamycin complex 1 signaling. Journal of Clinical Endocrinology and Metabolism 2010 95 3848–3857. (doi:10.1210/jc.2009-2696).
- Timmerman KL, Lee JL, Fujita S, Dhanani S, Dreyer HC, Fry CS, Drummond MJ, Sheffield-Moore M, Rasmussen BB, Volpi E. Pharmacological vasodilation improves insulin-stimulated muscle protein anabolism but not glucose utilization in older adults. Diabetes 2010 59 2764–2771. (doi:10.2337/db10-0415).
- Frayn KN, Maycock PF. Regulation of protein metabolism by a physiological concentration of insulin in mouse soleus and extensor digitorum longus muscles. Effects of starvation and scald injury. Biochemical Journal 1979 184 323.
- Fulks RM, Li JB, Goldberg AL. Effects of insulin, glucose, and amino acids on protein turnover in rat diaphragm. Journal of Biological Chemistry 1975 250 290–298.
- Jefferson L, Rannels D, Munger B, Morgan H. Insulin in the regulation of protein turnover in heart and skeletal muscle. Federation Proceedings 1974 33 1098–1104.
- Jefferson L, Li J, Rannels S. Regulation by insulin of amino acid release and protein turnover in the perfused rat hemicorpus. Journal of Biological Chemistry 1977 252 1476.
- Li J, Higgins J, Jefferson L. Changes in protein turnover in skeletal muscle in response to fasting. American Journal of Physiology. Endocrinology and Metabolism 1979 236 E222.
- Lundholm K, Edström S, Ekman L, Karlberg I, Walker P, Schersten T. Protein degradation in human skeletal muscle tissue: the effect of insulin, leucine, amino acids and ions. Clinical Science 1981 60 319.
- Jefferson LS. Lilly Lecture 1979: role of insulin in the regulation of protein synthesis. Diabetes 1980 29 487. (doi:10.2337/diab.29.6.487).
- Jefferson L, Koehler J, Morgan H. Effect of insulin on protein synthesis in skeletal muscle of an isolated perfused preparation of rat hemicorpus. PNAS 1972 69 816. (doi:10.1073/pnas.69.4.816).
- Stirewalt WS, Low R, Slaiby JM. Insulin sensitivity and responsiveness of epitrochlearis and soleus muscles from fed and starved rats. Recognition of differential changes in insulin sensitivities of protein synthesis and glucose incorporation into glycogen. Biochemical Journal 1985 227 355.
- Proud CG, Denton RM. Molecular mechanisms for the control of translation by insulin. Biochemical Journal 1997 328 329.
- Kimball SR, Vary TC, Jefferson LS. Regulation of protein synthesis by insulin. Annual Review of Physiology 1994 56 321–348. (doi:10.1146/annurev.ph.56.030194.001541).
- Muniyappa R, Montagnani M, Koh KK, Quon MJ. Cardiovascular actions of insulin. Endocrine Reviews 2007 28 463–491. (doi:10.1210/er.2007-0006).
- Phillips SM. Insulin and muscle protein turnover in humans: stimulatory, permissive, inhibitory, or all of the above? American Journal of Physiology. Endocrinology and Metabolism 2008 295 E731. (doi:10.1152/ajpendo.90569.2008).
- Timmerman KL, Volpi E. Amino acid metabolism and regulatory effects in aging. Current Opinion in Clinical Nutrition and Metabolic Care 2008 11 45. (doi:10.1097/MCO.0b013e3282f2a592).
- Greenhaff PL, Karagounis L, Peirce N, Simpson EJ, Hazell M, Layfield R, Wackerhage H, Smith K, Atherton P, Selby A. Disassociation between the effects of amino acids and insulin on signaling, ubiquitin ligases, and protein turnover in human muscle. American Journal of Physiology. Endocrinology and Metabolism 2008 295 E595–E604. (doi:10.1152/ajpendo.90411.2008).
- Fujita S, Glynn EL, Timmerman KL, Rasmussen BB, Volpi E. Supraphysiological hyperinsulinaemia is necessary to stimulate skeletal muscle protein anabolism in older adults: evidence of a true age-related insulin resistance of muscle protein metabolism. Diabetologia 2009 52 1889–1898. (doi:10.1007/s00125-009-1430-8).
- Meneilly G, Elliot T, Bryer-Ash M, Floras J. Insulin-mediated increase in blood flow is impaired in the elderly. Journal of Clinical Endocrinology and Metabolism 1995 80 1899.
- Rasmussen BB, Fujita S, Wolfe RR, Mittendorfer B, Roy M, Rowe VL, Volpi E. Insulin resistance of muscle protein metabolism in aging. FASEB Journal 2006 20 768–769. (doi:10.1096/fj.05-4607fje).
- Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Medicine 2009 6 e1000097. (doi:10.1371/journal.pmed.1000097).
- Biolo G, Declan Fleming RY, Wolfe RR. Physiologic hyperinsulinemia stimulates protein synthesis and enhances transport of selected amino acids in human skeletal muscle. Journal of Clinical Investigation 1995 95 811–819. (doi:10.1172/JCI117731).
- Smith GI, Patterson BW, Mittendorfer B. Human muscle protein turnover – why is it so variable? Journal of Applied Physiology 2011 110 480–491. (doi:10.1152/japplphysiol.00125.2010).
- Bennet W, Connacher A, Scrimgeour C, Smith K, Rennie M. Increase in anterior tibialis muscle protein synthesis in healthy man during mixed amino acid infusion: studies of incorporation of [1-13C]leucine. Clinical Science 1989 76 447.
- Kimball S. The role of nutrition in stimulating muscle protein accretion at the molecular level. Biochemical Society Transactions 2007 35 1298–1301. (doi:10.1042/BST0351298).
- DeSouza CA, Shapiro LF, Clevenger CM, Dinenno FA, Monahan KD, Tanaka H, Seals DR. Regular aerobic exercise prevents and restores age-related declines in endothelium-dependent vasodilation in healthy men. Circulation 2000 102 1351–1357. (doi:10.1161/01.CIR.102.12.1351).
- Fujita S, Rasmussen BB, Cadenas JG, Drummond MJ, Glynn EL, Sattler FR, Volpi E. Aerobic exercise overcomes the age-related insulin resistance of muscle protein metabolism by improving endothelial function and Akt/mammalian target of rapamycin signaling. Diabetes 2007 56 1615–1622. (doi:10.2337/db06-1566).
- Louard RJ, Fryburg DA, Gelfand RA, Barrett EJ. Insulin sensitivity of protein and glucose metabolism in human forearm skeletal muscle. Journal of Clinical Investigation 1992 90 2348–2354. (doi:10.1172/JCI116124).
- Bell JA, Volpi E, Fujita S, Cadenas JG, Sheffield-Moore M, Rasmussen BB. Skeletal muscle protein anabolic response to increased energy and insulin is preserved in poorly controlled type 2 diabetes. Journal of Nutrition 2006 136 1249–1255.
- Newman E, Heslin MJ, Wolf RF, Pisters PW, Brennan MF. The effect of systemic hyperinsulinemia with concomitant amino acid infusion on skeletal muscle protein turnover in the human forearm. Metabolism 1994 43 70–78. (doi:10.1016/0026-0495(94)90159-7).
- Drummond MJ, Bell JA, Fujita S, Dreyer HC, Glynn EL, Volpi E, Rasmussen BB. Amino acids are necessary for the insulin-induced activation of mTOR/S6K1 signaling and protein synthesis in healthy and insulin resistant human skeletal muscle. Clinical Nutrition 2008 27 447–456. (doi:10.1016/j.clnu.2008.01.012).
- Biolo G, Williams BD, Fleming R, Wolfe R. Insulin action on muscle protein kinetics and amino acid transport during recovery after resistance exercise. Diabetes 1999 48 949–957. (doi:10.2337/diabetes.48.5.949).
- Zanetti M, Barazzoni R, Kiwanuka E, Tessari P. Effects of branched-chain-enriched amino acids and insulin on forearm leucine kinetics. Clinical Science 1999 97 437–448. (doi:10.1042/CS19990163).
- Hillier TA, Fryburg DA, Jahn LA, Barrett EJ. Extreme hyperinsulinemia unmasks insulin’s effect to stimulate protein synthesis in the human forearm. American Journal of Physiology 1998 274 E1067–E1074.
- Nygren J, Nair KS. Differential regulation of protein dynamics in splanchnic and skeletal muscle beds by insulin and amino acids in healthy human subjects. Diabetes 2003 52 1377–1385. (doi:10.2337/diabetes.52.6.1377).
- Guillet C, Prod’homme M, Balage M, Gachon P, Giraudet C, Morin L, Grizard J, Boirie Y. Impaired anabolic response of muscle protein synthesis is associated with S6K1 dysregulation in elderly humans. FASEB Journal 2004 18 1586–1587.
- Guillet C, Delcourt I, Rance M, Giraudet C, Walrand S, Bedu M, Duche P, Boirie Y. Changes in basal and insulin and amino acid response of whole body and skeletal muscle proteins in obese men. Journal of Clinical Endocrinology and Metabolism 2009 94 3044–3050. (doi:10.1210/jc.2008-2216).
- Smith GI, Atherton P, Reeds DN, Mohammed BS, Jaffery H, Rankin D, Rennie MJ, Mittendorfer B. No major sex differences in muscle protein synthesis rates in the postabsorptive state and during hyperinsulinemia–hyperaminoacidemia in middle-aged adults. Journal of Applied Physiology 2009 107 1308–1315. (doi:10.1152/japplphysiol.00348.2009).
- Chevalier S, Goulet ED, Burgos SA, Wykes LJ, Morais JA. Protein anabolic responses to a fed steady state in healthy aging. Journals of Gerontology. Series A, Biological Sciences and Medical Sciences 66A 2011 681–688. (doi:10.1093/gerona/glr036).
- Smith GI, Atherton P, Reeds DN, Mohammed BS, Rankin D, Rennie MJ, Mittendorfer B. Dietary omega-3 fatty acid supplementation increases the rate of muscle protein synthesis in older adults: a randomized controlled trial. American Journal of Clinical Nutrition 2011 93 402–412. (doi:10.3945/ajcn.110.005611).
- Tessari P, Barazzoni R, Zanetti M. Differences in estimates of forearm protein synthesis between leucine and phenylalanine tracers following unbalanced amino acid infusion. Metabolism 1999 48 1564–1569. (doi:10.1016/S0026-0495(99)90246-9).
- Toth MJ, LeWinter MM, Ades PA, Matthews DE. Impaired muscle protein anabolic response to insulin and amino acids in heart failure patients: relationship to markers of immune activation. Clinical Science 2010 119 467. (doi:10.1042/CS20100110).
- Denne SC, Liechty EA, Liu YM, Brechtel G, Baron AD. Proteolysis in skeletal muscle and whole body in response to euglycemic hyperinsulinemia in normal adults. American Journal of Physiology. Endocrinology and Metabolism 1991 261 E809–E814.
- Tessari P, Inchiostro S, Biolo G, Vincenti E, Sabadin L. Effects of acute systemic hyperinsulinemia on forearm muscle proteolysis in healthy man. Journal of Clinical Investigation 1991 88 27–33. (doi:10.1172/JCI115287).
- Arfvidsson B, Zachrisson H, Moller-Loswick AC, Hyltander A, Sandstrom R, Lundholm K. Effect of systemic hyperinsulinemia on amino acid flux across human legs in postabsorptive state. American Journal of Physiology. Endocrinology and Metabolism 1991 260 E46–E52.
- McNurlan MA, Essen P, Thorell A, Calder AG, Anderson SE, Ljungqvist O, Sandgren A, Grant I, Tjader I, Ballmer PE et al.. Response of protein synthesis in human skeletal muscle to insulin: an investigation with l-[2H5]phenylalanine. American Journal of Physiology 1994 267 E102–E108.
- Meek SE, Persson M, Ford GC, Nair KS. Differential regulation of amino acid exchange and protein dynamics across splanchnic and skeletal muscle beds by insulin in healthy human subjects. Diabetes 1998 47 1824–1835. (doi:10.2337/diabetes.47.12.1824).
- Bell JA, Fujita S, Volpi E, Cadenas JG, Rasmussen BB. Short-term insulin and nutritional energy provision do not stimulate muscle protein synthesis if blood amino acid availability decreases. American Journal of Physiology. Endocrinology and Metabolism 2005 289 E999–E1006. (doi:10.1152/ajpendo.00170.2005).
- Fujita S, Rasmussen BB, Cadenas JG, Grady JJ, Volpi E. Effect of insulin on human skeletal muscle protein synthesis is modulated by insulin-induced changes in muscle blood flow and amino acid availability. American Journal of Physiology. Endocrinology and Metabolism 2006 291 E745–E754. (doi:10.1152/ajpendo.00271.2005).
- Wilkes EA, Selby AL, Atherton PJ, Patel R, Rankin D, Smith K, Rennie MJ. Blunting of insulin inhibition of proteolysis in legs of older subjects may contribute to age-related sarcopenia. American Journal of Clinical Nutrition 2009 90 1343–1350. (doi:10.3945/ajcn.2009.27543).
- Moller-Loswick AC, Zachrisson H, Hyltander A, Korner U, Matthews DE, Lundholm K. Insulin selectively attenuates breakdown of nonmyofibrillar proteins in peripheral tissues of normal men. American Journal of Physiology. Endocrinology and Metabolism 1994 266 E645–E652.
- Fryburg DA, Jahn LA, Hill SA, Oliveras DM, Barrett EJ. Insulin and insulin-like growth factor-I enhance human skeletal muscle protein anabolism during hyperaminoacidemia by different mechanisms. Journal of Clinical Investigation 1995 96 1722–1729. (doi:10.1172/JCI118217).
- Fukagawa N, Minaker K, Young V, Rowe J. Insulin dose-dependent reductions in plasma amino acids in man. American Journal of Physiology. Endocrinology and Metabolism 1986 250 E13–E17.
- Boirie Y, Short KR, Ahlman B, Charlton M, Nair KS. Tissue-specific regulation of mitochondrial and cytoplasmic protein synthesis rates by insulin. Diabetes 2001 50 2652. (doi:10.2337/diabetes.50.12.2652).
- Chow LS, Albright RC, Bigelow ML, Toffolo G, Cobelli C, Nair KS. Mechanism of insulin’s anabolic effect on muscle: measurements of muscle protein synthesis and breakdown using aminoacyl-tRNA and other surrogate measures. American Journal of Physiology. Endocrinology and Metabolism 2006 291 E729–E736. (doi:10.1152/ajpendo.00003.2006).
- Gelfand RA, Barrett EJ. Effect of physiologic hyperinsulinemia on skeletal muscle protein synthesis and breakdown in man. Journal of Clinical Investigation 1987 80 1. (doi:10.1172/JCI113033).
- Fryburg DA, Barrett EJ, Louard RJ, Gelfand RA. Effect of starvation on human muscle protein metabolism and its response to insulin. American Journal of Physiology. Endocrinology and Metabolism 1990 259 E477–E482.
- Heslin MJ, Newman E, Wolf RF, Pisters PW, Brennan MF. Effect of hyperinsulinemia on whole body and skeletal muscle leucine carbon kinetics in humans. American Journal of Physiology 1992 262 E911–E918.
- Newman E, Heslin MJ, Wolf RF, Pisters PW, Brennan MF. The effect of insulin on glucose and protein metabolism in the forearm of cancer patients. Surgical Oncology 1992 1 257–267. (doi:10.1016/0960-7404(92)90086-Z).
- Short KR, Bigelow ML, Nair KS. Short-term prednisone use antagonizes insulin’s anabolic effect on muscle protein and glucose metabolism in young healthy people. American Journal of Physiology. Endocrinology and Metabolism 2009 297 E1260–E1268. (doi:10.1152/ajpendo.00345.2009).
- Barazzoni R, Short KR, Asmann Y, Coenen-Schimke JM, Robinson MM, Nair KS. Insulin fails to enhance mTOR phosphorylation, mitochondrial protein synthesis, and ATP production in human skeletal muscle without amino acid replacement. American Journal of Physiology. Endocrinology and Metabolism 2012 303 E1117–E1125. (doi:10.1152/ajpendo.00067.2012).