Introduzione:
Con il termine Acne Vulgaris (o semplicemente Acne) ci si riferisce ad una malattia cronica della pelle a evoluzione benigna, caratterizzata da un processo infiammatorio del follicolo pilifero e della ghiandola sebacea annessa, chiamata in linguaggio comune “brufolo” o “foruncolo”. Le parti più colpite sono viso, spalle, dorso e regione pettorale del torace.

Nonostante sia connessa maggiormente alla pubertà, anche molte persone in età adulta continuano a soffrirne: si stima che la condizione persista fino ai 20 e ai 30 anni in circa il 64% e il 43% degli individui, rispettivamente [1]. Sebbene la condizione sia facilmente riconoscibile dal punto di vista clinico, la sua patogenesi (il processo attraverso il quale si sviluppa) è ampiamente complessa e i ricercatori di tutto il mondo la stanno ancora decifrando poco a poco.
In generale, si ritiene che l’Acne derivi dai seguenti quattro fattori patogeni (in ordine sparso) [2, 3]:
- Aumento della produzione di sebo
- Cheratinizzazione anomala
- Rilascio di mediatori infiammatori nella pelle
- Ipercolonizzazione batterica del follicolo pilifero da parte di Cutibacterium acnes (C. acnes, precedentemente noto come Propionibacterium acnes [P. acnes])

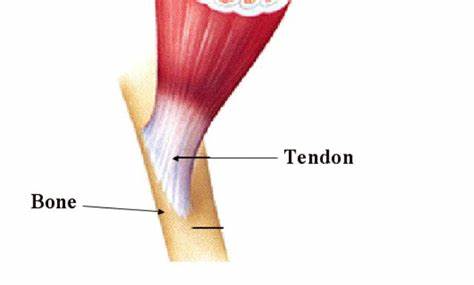
L’aumento della produzione di sebo di per sé non è un problema, nel senso che si limiterebbe a rendere la pelle più grassa. Tuttavia, si ritiene che contribuisca all’Acne fornendo un ambiente più confortevole per il C. acnes e alterando la composizione degli acidi grassi nel sebo. In particolare, una diminuzione del contenuto di Acido Linoleico. L’insieme di questi fattori potrebbe a sua volta disturbare la funzione di barriera delle pareti follicolari dei cheratinociti (cellule che compongono il follicolo pilifero) [4] e portare a una cascata infiammatoria [5].
La cheratinizzazione anomala si riferisce a queste cellule che non si staccano normalmente come dovrebbero. Invece di staccarsi e di essere spinte sulla superficie della pelle, diventano coese e rimangono nel follicolo pilifero, ostruendolo in sostanza. Si ritiene che questo sia un evento precoce nello sviluppo di un comedone, che dà origine a un microcomedone.
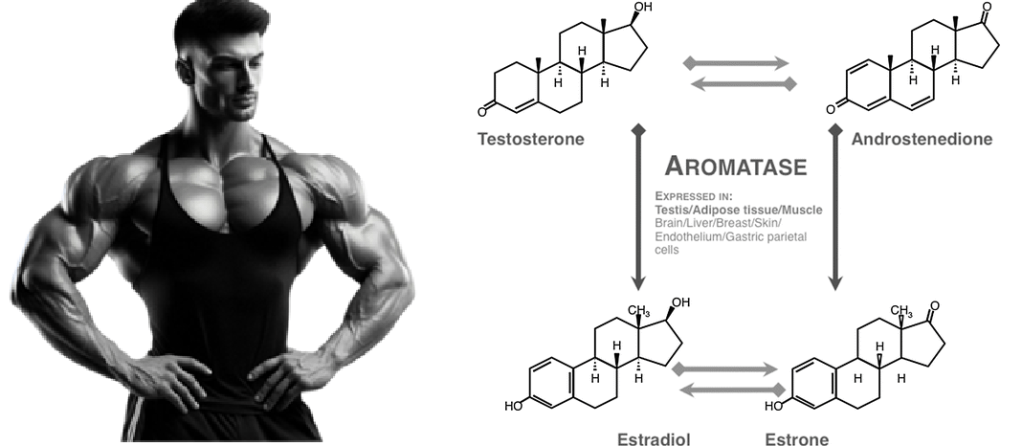
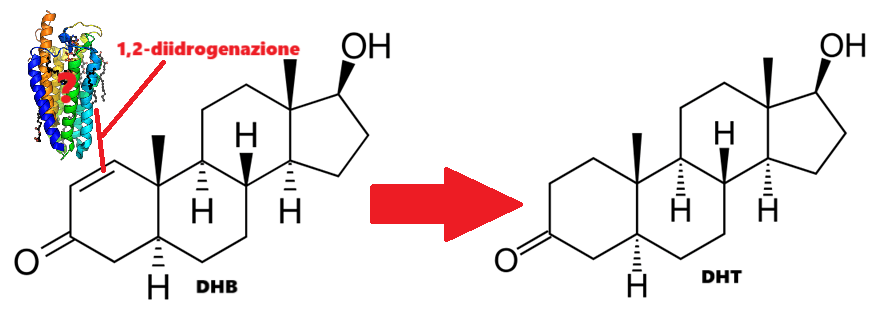
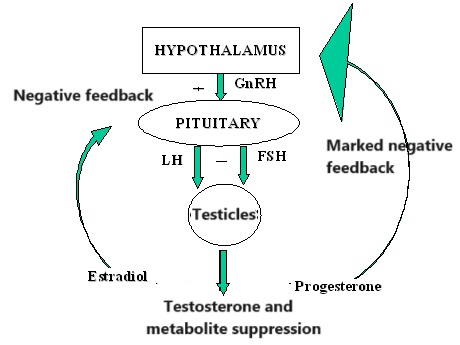
Gli androgeni svolgono un ruolo in entrambi i casi. Ad esempio, gli uomini insensibili agli androgeni non producono livelli dimostrabili di sebo e non sembrano sviluppare l’Acne [6]. Ciò significa che per sviluppare l’Acne è necessaria almeno una certa attività androgenica. Uno studio in cui gli uomini hanno ricevuto dapprima Etinilestradiolo (che sopprime marcatamente la produzione endogena di Testosterone) e successivamente la somministrazione concomitante di Testosterone, ha dimostrato che l’aggiunta di quest’ultimo ha portato a un notevole aumento della produzione di sebo [7]. Infine, il noto studioso di Testosterone Shalender Bhasin e il suo gruppo hanno misurato la produzione di sebo in uomini che ricevevano dosaggi graduali di Testosterone (50, 125, 300 o 600mg settimanali) con o senza l’inibitore della 5α-reduttasi Dutasteride per 20 settimane [8]. Hanno riscontrato che la produzione di sebo nella regione della fronte, ma non sul naso o sulla schiena, era correlata alla dose di Testosterone. Tuttavia, l’associazione era di una settimana e il gruppo da 600mg ha addirittura registrato una diminuzione, anche se non statisticamente significativa, del punteggio di sebo. La produzione di sebo potrebbe apparentemente svolgere un ruolo meno importante del previsto nell’Acne indotta dagli AAS. In effetti, i soggetti hanno riferito raramente di avere la pelle grassa, mentre l’Acne è stata segnalata più frequentemente.
Altri dati interessanti sull’incidenza dell’Acne in seguito all’uso di AAS ad alti dosaggi provengono dallo studio HAARLEM [9]. Come detto più volte, lo studio HAARLEM è uno studio prospettico di coorte in cui 100 consumatori di steroidi anabolizzanti sono stati seguiti nel tempo durante l’autosomministrazione di AAS [9]. Il dosaggio medio, in base alle informazioni riportate sull’etichetta, era di 898mg a settimana, rendendo così il loro ciclo di AAS abbastanza rappresentativo dell’uso comune da parte dei bodybuilder. Le misurazioni sono state effettuate prima, durante, 3 mesi dopo la fine del ciclo e 1 anno dopo l’inizio del ciclo. I ricercatori hanno esaminato visivamente la pelle per verificare la presenza di acne e al basale il 13% degli utenti è risultato affetto da Acne. Questo dato è salito al 29% alla fine del ciclo, per poi scendere al 23% 3 mesi dopo il ciclo e al 10% 1 anno dopo l’inizio del ciclo. L’Acne auto-riferita era notevolmente più alta alla fine del ciclo, con il 10% al basale, il 52% alla fine, il 29% 3 mesi dopo e il 14% 1 anno dopo l’inizio del ciclo.
È chiaro che l’Acne è un effetto collaterale comune dell’uso di AAS ad alti dosaggi. Ma cosa si può fare? In questo articolo illustrerò alcune modalità di trattamento. Tuttavia, occorre prestare attenzione, poiché nessuno di questi studi ha valutato gli effetti di queste modalità di trattamento sull’Acne indotta dagli AAS. Tuttavia, è molto ragionevole supporre che possano funzionare anche in questa situazione.
Integratori orali da banco: Zinco, Vitamina D, Acidi Grassi Omega 3:

Sebbene il suo meccanismo d’azione rimanga piuttosto elusivo, l’integrazione orale di Zinco è risultata efficace in diversi studi clinici [10]. La sua efficacia nel trattamento dell’Acne è stata notata per la prima volta da Fitzherbert [11] e Michaëlsson et al. negli anni ’70 [12]. La somministrazione di Zinco a pazienti con carenza di Zinco ha migliorato l’Acne e Michaëlsson et al. hanno persino avviato uno studio in doppio cieco. In esso hanno confrontato l’integrazione di Zinco con un antibiotico orale (Ossitetraciclina) nel trattamento dell’Acne. Il gruppo Zinco ha ricevuto 45mg di Zinco elementare al giorno sotto forma di Solfato di Zinco. Non sono state riscontrate differenze nei risultati tra i gruppi, con una riduzione media del punteggio dell’Acne del 70% in entrambi. Una review della letteratura descrive i risultati di 8 studi controllati con placebo, di cui la metà ha riscontrato un miglioramento oggettivo significativo dell’Acne nei soggetti trattati con Zinco rispetto a quelli che hanno ricevuto il placebo [10]. Una delle ragioni per cui non tutti gli studi hanno riscontrato un miglioramento potrebbe risiedere nella mancanza di potenza statistica, oltre che nello stato dello Zinco e nella gravità dell’Acne dei soggetti esaminati (è stato suggerito che lo Zinco è più efficace nell’Acne grave rispetto all’Acne lieve-moderata [13]). I dosaggi utilizzati negli studi variano notevolmente e non sembra esserci una chiara relazione tra dosaggio e risultati. Pertanto, raccomanderei di non superare il livello di assunzione superiore tollerabile (UL) di 40mg di Zinco elementare al giorno.
La Vitamina D potrebbe essere un altro integratore da banco con un potenziale di aiuto nella lotta contro l’Acne. Uno studio randomizzato e controllato su soggetti con carenza di Vitamina D (<30nmol/L 25[OH]D) ha dimostrato che un supplemento giornaliero di 1.000UI di Vitamina D per 8 settimane ha portato a una riduzione significativa delle lesioni infiammatorie rispetto al placebo [14]. Dato che molte persone hanno una carenza di Vitamina D, potrebbe essere una buona idea correggerla con un’integrazione. Ad esempio, uno studio olandese ha rilevato che circa due terzi di un campione di 128 atleti altamente allenati erano carenti di Vitamina D (<50nmol/L 25[OH]D) o insufficienti (<75nmol/L 25[OH]D) [15]. Tuttavia, il dosaggio di 1.000 unità al giorno, utilizzato nello studio, è probabilmente troppo basso per correggere una carenza. In effetti, nel corso delle 8 settimane i livelli sono passati da 20nmol/L a 40nmol/L, il che è ancora carente secondo le linee guida della Endocrine Society [16]. I livelli sierici di 25(OH)D superiori a 75nmol/L sono considerati adeguati. La maggior parte dei soggetti richiederebbe probabilmente un dosaggio di 2.000UI al giorno o superiore. L’Autorità Europea per gli Alimenti e la Sicurezza ha fissato una dose giornaliera di 4.000UI come limite superiore tollerabile di assunzione [17].
Infine, uno studio randomizzato controllato con placebo ha rilevato che l’integrazione di acidi grassi Omega 3 (1g di Acido Eicosapentaenoico [EPA] e 1g di Acido Docosaesaenoico [DHA]) al giorno riduce la gravità dell’acne in soggetti con acne da lieve a moderata [18].
Prodotti topici: Perossido di Benzoile e Retinoidi:

Il Perossido di Benzoile è disponibile come crema da banco nella maggior parte dei paesi. È ragionevolmente efficace (anche se la maggior parte degli studi fa schifo dal punto di vista qualitativo) [19] e in particolare contro il C. acnes. Inoltre, sembra aiutare un po’ la cheratinizzazione follicolare [20]. Tuttavia, pur rendendo la pelle secca (marcatamente secca), non sembra effettivamente ridurre la produzione di sebo. Probabilmente rende la pelle secca in virtù della sua capacità ossidativa: ossida i lipidi che altrimenti renderebbero la pelle liscia. Quando si usa questo prodotto, si raccomanda di applicare uno strato sottile di Perossido di Benzoile sulle aree interessate una volta al giorno. Si dovrebbero preferire le preparazioni con una concentrazione del 2,5-5%, invece di quelle più concentrate [21]. Gli effetti collaterali includono secchezza della pelle, arrossamento della pelle, irritazione della pelle, prurito e, in rare occasioni, dermatite da contatto. Assicurarsi inoltre di applicare la protezione solare nei giorni di sole, poiché rende le aree interessate più inclini alle scottature. Infine, ha un forte effetto sbiancante, quindi non andate in giro e non strofinate il viso (o le mani che sono appena entrate in contatto con esso) sui tessuti. È noto per rovinare federe, asciugamani e camicie.

I Retinoidi topici sono disponibili al banco in alcuni Paesi, ma in altri richiedono la prescrizione medica. Sono efficaci soprattutto contro la cheratinizzazione e, in misura minore, contro la cascata infiammatoria. Per questo motivo ritengo che i Retinoidi siano più efficaci nel caso di Acne indotta da AAS rispetto al Perossido di Benzoile. La Tretinoina è un Retinoide comunemente prescritto, mentre altri sono l’Adapalene (Differin) e il Tazarotene (Tazorac). Tutti funzionano relativamente bene, con lievi differenze tra loro. L’Adapalene sembra essere efficace quanto la Tretinoina, ma mostra risultati un po’ più rapidi e viene tollerato meglio [22]. La formulazione più concentrata di Adapelen (0,3% vs 0,1%) sembra funzionare meglio, pur essendo ugualmente ben tollerata [23]. A sua volta, il Tazarotene ha dimostrato di essere leggermente migliore dell’Adapalene in uno studio [24], ma di avere la stessa efficacia in un altro [25], mentre in entrambi i casi l’Adapalene è stato tollerato meglio. Credo che l’Adapalene abbia una certa preferenza a causa della tollerabilità leggermente migliore. Gli effetti collaterali sono simili a quelli del Perossido di Benzoile. Il produttore consiglia di iniziare con una dose giornaliera o a giorni alterni. Tuttavia, questa sostanza è piuttosto impattante per la pelle, quindi due volte a settimana potrebbe essere un punto di partenza migliore, per poi iniziare a lavorare da lì.
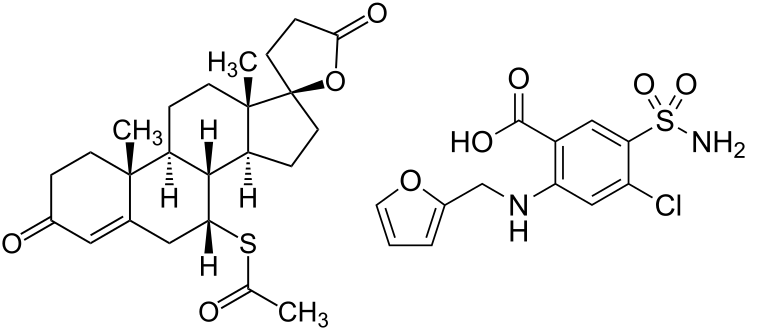
Isotretinoina (Roaccutane/Accutane):

Francamente è il non plus ultra di tutti i trattamenti per l’Acne. Funziona molto bene [26] e agisce su tutti e quattro i fattori coinvolti nella patogenesi dell’acne. È disponibile solo su prescrizione medica (e giustamente). Di solito vengono prescritti dosaggi intorno a 0,5-1,0mg/kg di peso corporeo al giorno. Tuttavia, anche dosaggi inferiori, fino a 5mg al giorno [27], sono abbastanza efficaci e sono molto meglio tollerati. Il che è molto gradito, dato che il trattamento comporta alcuni fastidiosi effetti collaterali, soprattutto di tipo dermatologico. Tra questi, labbra screpolate, pelle secca, prurito, occhi secchi e sanguinamento dal naso. Quando viene prescritto, vengono eseguiti degli esami del sangue (di solito al basale, dopo 1 mese di trattamento e poi ogni 3 mesi). Il motivo è che l’Isotretinoina potrebbe aumentare il Colesterolo, i Trigliceridi e i marker di danno epatico e potrebbe ridurre l’Emoglobina. Tuttavia, una review sistematica ha riportato analisi del sangue anomale nel 4% dei pazienti trattati con Isotretinoina (e solo nello 0,1% dei gruppi di controllo) [26], con solo 1 paziente su 200 che ha dovuto interrompere il trattamento a causa di analisi del sangue anomale (enzimi epatici elevati). In particolare, gli eventi psichiatrici/psicosomatici sono risultati più frequenti di circa il 50% nei soggetti che utilizzano isotretinoina rispetto ai gruppi di controllo. In particolare, nel foglietto illustrativo dell’isotretinoina sono elencati come effetti collaterali “pensieri suicidi” e “suicidio”. Ciò è dovuto più alla prudenza che al fatto che sia stato effettivamente stabilito un nesso causale (a causa della rarità, è molto difficile farlo). Infine, uno strano studio ha descritto che l’integrazione di EPA e DHA (apparentemente 1g in totale, ma lo studio non è riuscito a descriverlo chiaramente) è utile contro alcuni degli effetti collaterali dermatologici [28]. Per l’Acne indotta da AAS, se le altre modalità di trattamento non portano a risultati soddisfacenti, ritengo che un dosaggio basso, compreso tra 5 e 10mg al giorno, sia il più appropriato. Dosaggi più elevati, come quelli comunemente prescritti per l’Acne Vulgaris “normale”, non sembrano giustificati.
Finasteride e Dutasteride? Gli inibitori della 5α-reduttasi non sembrano funzionare :
Forse un po’ a sorpresa, gli inibitori della 5α-reduttasi, che, come ben sappiamo, inibiscono la conversione del Testosterone nel più potente androgeno DHT, non sembrano essere utili contro l’Acne. Il perché? Non è chiaro.

Uno studio ben progettato di 3 mesi, randomizzato e controllato con placebo, condotto su 182 soggetti, ha confrontato l’effetto di un inibitore selettivo della 5α-reduttasi di tipo 1 con quello di un antibiotico (la minociclina, all’epoca un trattamento standard per l’acne, anche se oggi il suo uso è sconsigliato da solo) [30]. La 5α-reduttasi di tipo 1 è fortemente espressa nella ghiandola sebacea [31], e in effetti un inibitore selettivo di tipo 1 mostra una maggiore riduzione del DHT nel sebo rispetto alla finasteride (che non è potente nell’inibire il tipo 1, ma solo i tipi 2 e 3) [32]. Inoltre, hanno anche valutato se la combinazione di minociclina e inibitore di tipo I funzionasse meglio. L’inibitore di tipo I ha funzionato bene quanto il placebo. Anche la terapia combinata non ha migliorato l’efficacia, poiché ha funzionato altrettanto bene della sola minociclina.
Esiste anche un ampio studio (106 partecipanti arruolati) controllato con placebo, registrato dall’azienda farmaceutica Elorac (NCT02502669), in cui i soggetti hanno ricevuto Finasteride (23,5mg al giorno o 33,5mg al giorno) nel trattamento dell’Acne. Lo studio è stato completato nel 2017, ma i risultati non sono mai stati pubblicati. Sospetto che la Finasteride non abbia fatto meglio del placebo. (E questi dosaggi sono elevati).
Bonus – Peptidi sperimentali per il trattamento dell’Acne:
Sono stati proposti alcuni peptidi sperimentali per il trattamento dell’Acne. I principali tra questi sono esposti e descritti di seguito.
- GHK-Cu
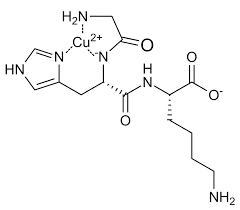
Il Peptide di rame GHK-Cu è un complesso di rame naturale del tripeptide glicil-L-istidil-L-lisina. Il tripeptide ha una forte affinità per il rame(II) ed è stato isolato per la prima volta dal plasma umano. Si trova anche nella saliva e nell’urina. Loren Pickart (1938-2023) ha isolato il peptide di rame GHK-Cu dall’albumina plasmatica umana nel 1973.[33] È stato notato che il tessuto epatico ottenuto da pazienti di età compresa tra i 60 e gli 80 anni presentava un livello maggiore di fibrinogeno. Tuttavia, quando le cellule epatiche dei pazienti anziani venivano incubate nel sangue del gruppo più giovane, le cellule più anziane iniziavano a funzionare quasi allo stesso modo del tessuto epatico più giovane.[34][35] Si scoprì che questo effetto era dovuto a un piccolo fattore peptidico che si comportava in modo simile al peptide sintetico glicil-L-istidil-L-lisina (GHK). Pickart propose che questa attività nell’albumina plasmatica umana fosse un tripeptide glicil-L-istidil-L-lisina e che potesse funzionare chelando gli ioni metallici.[36]
Nel 1977 è stato dimostrato che il peptide che modula la crescita è una glicil-L-istidil-L-lisina.[37] Si propone che il GHK-Cu moduli l’assunzione di rame nelle cellule.[38]
Alla fine degli anni ’80, il peptide di rame GHK-Cu ha iniziato ad attirare l’attenzione come promettente agente di guarigione delle ferite. A concentrazioni da picomolare a nanomolare, il GHK-Cu stimolava la sintesi di collagene nei fibroblasti cutanei, aumentava l’accumulo di proteine totali, glicosaminoglicani (con una curva bifasica) e DNA nelle ferite cutanee dei ratti. Hanno anche scoperto che la sequenza GHK è presente nel collagene e hanno suggerito che il peptide GHK viene rilasciato dopo una lesione tissutale.[39][40] Hanno proposto una classe di molecole di risposta all’emergenza che vengono rilasciate dalla matrice extracellulare nel sito di una lesione.[41] Il GHK-Cu ha anche aumentato la sintesi di decorina, un piccolo proteoglicano coinvolto nella regolazione della sintesi del collagene, nella regolazione della guarigione delle ferite e nella difesa antitumorale.[42]
È stato inoltre stabilito che il GHK-Cu stimola sia la sintesi delle metalloproteinasi, gli enzimi che demoliscono le proteine dermiche, sia i loro inibitori (anti-proteasi). Il fatto che il GHK-Cu non solo stimoli la produzione di componenti dermici, ma ne regoli anche la disgregazione, suggerisce che debba essere usato con cautela.[43]
Una serie di esperimenti sugli animali ha stabilito una marcata attività di guarigione delle ferite del GHK-Cu.[44] Nelle ferite dermiche dei conigli, il GHK-Cu ha facilitato la guarigione delle ferite, causando una migliore contrazione della ferita, uno sviluppo più rapido del tessuto granulare e un miglioramento dell’angiogenesi. Inoltre, ha aumentato il livello degli enzimi antiossidanti.[45][46]
È stato riscontrato che il GHK-Cu induce un miglioramento sistemico della guarigione in ratti, topi e maiali; in altre parole, il peptide GHK-Cu iniettato in un’area del corpo (come i muscoli della coscia) ha migliorato la guarigione in aree del corpo distanti (come le orecchie). Questi trattamenti hanno fortemente aumentato i parametri di guarigione, come la produzione di collagene, l’angiogenesi e la chiusura della ferita sia in camere di ferita che in ferite a tutto spessore.[47] In uno studio, sono state create ferite a tutto spessore di 6 millimetri di diametro in un lembo di pelle ischemica sul dorso dei ratti e per 13 giorni i siti delle ferite sono stati trattati quotidianamente con GHK topico o con un veicolo di idrossipropilmetilcellulosa topico, oppure non sono stati trattati. Alla fine dello studio, le dimensioni della ferita erano diminuite del 64,5% nel gruppo GHK, del 45,6% nel gruppo trattato con il veicolo e del 28,2% nel gruppo di controllo.[48] La differenza tra le ferite del gruppo GHK e quelle del gruppo di controllo era significativa ed era accompagnata da livelli significativamente più bassi di fattore di necrosi tumorale alfa e di metalloproteinasi di matrice che degradano l’elastina.[48]
Il GHK-Cu biotinilato è stato incorporato in una membrana di collagene, utilizzata come medicazione della ferita. Questo materiale arricchito di GHK-Cu ha stimolato la contrazione della ferita e la proliferazione cellulare, oltre ad aumentare l’espressione degli enzimi antiossidanti. Lo stesso materiale è stato testato per la guarigione delle ferite in ratti diabetici. Il trattamento con GHK-Cu ha portato a una contrazione ed epitelizzazione più rapida della ferita, a un livello più elevato di glutatione e acido ascorbico, a una maggiore sintesi di collagene e all’attivazione di fibroblasti e mastociti.[48] Le ferite ischemiche aperte nei ratti trattati con GHK-rame sono guarite più rapidamente e hanno registrato una riduzione della concentrazione delle metalloproteinasi 2 e 9 e del fattore di necrosi tumorale-beta (una delle principali citochine infiammatorie) rispetto al solo veicolo o alle ferite non trattate.[48]
Visti i suoi effetti sulla guarigione delle ferite e la riduzione dell’infiammazione, il GHK-Cu è stato proposto come possibile trattamento per l’Acne. Ad oggi, però, non vi sono dati sufficienti per avvalorarne l’efficacia in questo frangente terapeutico.
- Palmitoyl tetrapeptide-7
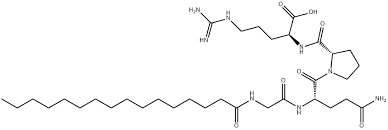
Il Palmitoil tetrapeptide-7 è un peptide sintetico composto dagli amminoacidi glutammina, glicina, arginina e prolina.[49] Agisce come ingrediente rigenerante per la pelle ed è noto per le sue proprietà lenitive, poiché può interrompere i fattori cutanei che causano segni di irritazione (inclusa l’esposizione ai raggi UVB) e perdita di tonicità. Agendo in questo modo, la pelle può ritrovare una sensazione di tonicità e iniziare a ripararsi, riducendo visibilmente le rughe.
Oltre ai quattro amminoacidi, questo peptide contiene anche l’acido grasso palmitico, che ne migliora la stabilità e la penetrazione nella pelle. Il livello di utilizzo tipico è nell’ordine delle parti per milione, che si traduce in percentuali molto basse ma altamente efficaci, comprese tra lo 0,0001% e lo 0,005%, sebbene possano essere utilizzate quantità maggiori o minori a seconda degli obiettivi del formulato.
Il palmitoil tetrapeptide-7 è spesso utilizzato in miscela con altri peptidi, come il palmitoil tripeptide-1. Questo può creare una sinergia ottimale e offrire risultati più mirati su una gamma più ampia di problematiche cutanee.
Da solo, viene fornito in polvere, ma nelle miscele viene combinato con idratanti come glicerina, vari glicoli, trigliceridi o alcoli grassi per facilitarne l’integrazione nelle formule.
Il Cosmetic Ingredient Review Expert Panel ha valutato questo peptide idrosolubile nel 2018 insieme ad altri peptidi e ha concluso che questo ingrediente è sicuro per l’uso nei cosmetici.
- Palmitoyl Tripeptide-1

Il Palmitoyl Tripeptide-1 (sequenza H-Gly-His-Lys-OH), anche noto come Pal-GHK, è un peptide sintetico usato in cosmetica per le sue proprietà anti-aging.[50] Agisce stimolando la produzione di collagene e altri componenti della matrice extracellulare, contribuendo a ridurre rughe e segni dell’invecchiamento, migliorando l’elasticità e la compattezza della pelle. Il Palmitoyl tripeptide-1 è quindi un peptide segnale che invia messaggi alle cellule della pelle per aumentare la produzione di collagene, essenziale per la struttura e l’elasticità della pelle. Stimola anche la produzione di glicosaminoglicani, molecole che contribuiscono all’idratazione e al volume della pelle. Riduce la degradazione del collagene inibendo le metalloproteasi della matrice, enzimi che lo distruggono. Agisce a livello del derma, la parte più profonda della pelle, favorendo la rigenerazione e la riparazione dei tessuti danneggiati.
Di conseguenza, i suoi benefici possono comprendere la riduzione delle rughe e delle linee sottili, il miglioramento dell’elasticità e della compattezza della pelle, la levigatura della superficie cutanea, l’idratazione, l’aumento del volume della pelle e il miglioramento della riparazione dei danni cutanei. Tutti fattori che possono contribuire al miglioramento della condizione legata all’Acne. Ma che in questo caso mancano ancora sufficienti dati specifici.
- LL-37
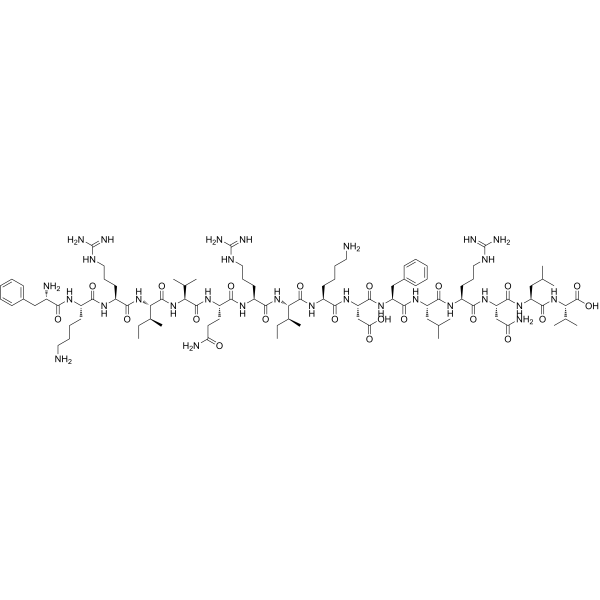
L’LL-37 è la forma attiva del peptide antimicrobico catelicidina (CAMP), un peptide antimicrobico codificato nell’uomo dal gene CAMP.[51-1] Nell’uomo, CAMP codifica il precursore peptidico CAP-18 (18 kDa), che viene elaborato dalla scissione extracellulare mediata dalla proteinasi 3 nella forma attiva LL-37.[52]La famiglia delle catelicidine comprende 30 tipi di cui LL-37 è l’unica catelicidina presente nell’uomo.[53] Le catelicidine sono immagazzinate nei granuli secretori dei neutrofili e dei macrofagi e possono essere rilasciate in seguito all’attivazione da parte dei leucociti.[54] I peptidi delle catelicidine sono molecole a doppia natura chiamate anfifili: un’estremità della molecola è attratta dall’acqua e respinta da grassi e proteine, e l’altra estremità è attratta da grassi e proteine e respinta dall’acqua. I membri di questa famiglia reagiscono ai patogeni disintegrando, danneggiando o perforando le membrane cellulari.
Le catelicidine svolgono quindi un ruolo fondamentale nella difesa immunitaria innata dei mammiferi contro le infezioni batteriche invasive.[55] La famiglia di peptidi delle catelicidine è classificata come peptidi antimicrobici (AMP). La famiglia degli AMP include anche le defensine. Sebbene le defensine condividano caratteristiche strutturali comuni, i peptidi correlati alle catelicidine sono altamente eterogenei.[55] I membri della famiglia di polipeptidi antimicrobici delle catelicidine sono caratterizzati da una regione altamente conservata (dominio della catelina) e da un dominio peptidico della catelicidina altamente variabile.[55]
I peptidi della catelicidina sono stati isolati da molte specie diverse di mammiferi, inclusi i marsupiali.[56] Le catelicidine si trovano principalmente nei neutrofili, nei monociti, nei mastociti, nelle cellule dendritiche e nei macrofagi[57] dopo l’attivazione da parte di batteri, virus, funghi, parassiti o dell’ormone 1,25-D, che è la forma ormonalmente attiva della vitamina D.[58] Sono state trovate in alcune altre cellule, comprese le cellule epiteliali e i cheratinociti umani.[59] Alcuni virus hanno sviluppato meccanismi immunomodulatori per evitare l’esposizione alla catelicidina riducendo il recettore cellulare della vitamina D.[60]
La regola generale del meccanismo che innesca l’azione della catelicidina, come quella di altri peptidi antimicrobici, prevede la disintegrazione (danneggiamento e perforazione) delle membrane cellulari degli organismi verso cui il peptide è attivo.[54]
Le catelicidine distruggono rapidamente le membrane lipoproteiche dei microbi avvolti nei fagosomi dopo la fusione con i lisosomi nei macrofagi. Pertanto, LL-37 può inibire la formazione di biofilm batterici.[61]
I pazienti affetti da rosacea presentano livelli elevati di catelicidina e di enzimi triptici dello strato corneo (SCTE). La catelicidina viene scissa nel peptide antimicrobico LL-37 dalle serin proteasi callicreina-5 e callicreina-7. Si sospetta che l’eccessiva produzione di LL-37 sia una causa concomitante in tutti i sottotipi di rosacea.[62] In passato, gli antibiotici sono stati utilizzati per trattare la rosacea, ma potrebbero essere efficaci solo perché inibiscono alcuni SCTE.[63]
Livelli plasmatici più bassi della proteina antimicrobica umana catelicidina (hCAP18) sembrano aumentare significativamente il rischio di morte per infezione nei pazienti in dialisi.[64] La produzione di catelicidina è sovraregolata dalla vitamina D.[65][66]
SAAP-148 (un peptide sintetico antimicrobico e antibiofilm) è una versione modificata di LL-37 che ha attività antimicrobiche migliorate rispetto a LL-37. In particolare, SAAP-148 è risultato più efficiente nell’uccidere i batteri in condizioni fisiologiche.[67] Inoltre, SAAP-148 agisce in sinergia con l’antibiotico halicina riutilizzato contro batteri e biofilm resistenti agli antibiotici.[68]
Si ritiene che LL-37 svolga un ruolo nella patogenesi della psoriasi (insieme ad altri peptidi antimicrobici). Nella psoriasi, i cheratinociti danneggiati rilasciano LL-37, che forma complessi con materiale autogenetico (DNA o RNA) proveniente da altre cellule. Questi complessi stimolano le cellule dendritiche (un tipo di cellula presentante l’antigene), che a loro volta rilasciano interferone α e β, contribuendo alla differenziazione delle cellule T e al mantenimento dell’infiammazione.[69] LL-37 è stato anche scoperto essere un autoantigene comune nella psoriasi; cellule T specifiche per LL-37 sono state trovate nel sangue e nella pelle in due terzi dei pazienti con psoriasi da moderata a grave.[69]
LL-37 si lega al peptide Ab, associato al morbo di Alzheimer. Uno squilibrio tra LL-37 e Ab potrebbe essere un fattore che influenza le fibrille e le placche associate all’Alzheimer. Le infezioni croniche orali da Porphyromonas gingivalis e dall’herpesvirus (HSV-1) possono contribuire alla progressione della demenza di Alzheimer.[70][71]
LL-37 svolge un ruolo nell’attivazione della proliferazione e della migrazione cellulare, contribuendo al processo di chiusura delle ferite.[72] Tutti questi meccanismi insieme svolgono un ruolo essenziale nell’omeostasi tissutale e nei processi rigenerativi. Inoltre, ha un effetto agonista su vari recettori pleiotropici, ad esempio il recettore del peptide formil-like-1 (FPRL-1),[73] il recettore purinergico P2X7 e il recettore del fattore di crescita epidermico (EGFR).[74]
Inoltre, induce l’angiogenesi[75] e regola l’apoptosi.[76]
Come abbiamo visto, l’LL-37 è un potente peptide antimicrobico che contrasta la proliferazione dei batteri che causano l’Acne. Il suo uso nel trattamento di quest’ultima è con tutta probabilità efficace.
- Acetyl hexapeptide-8
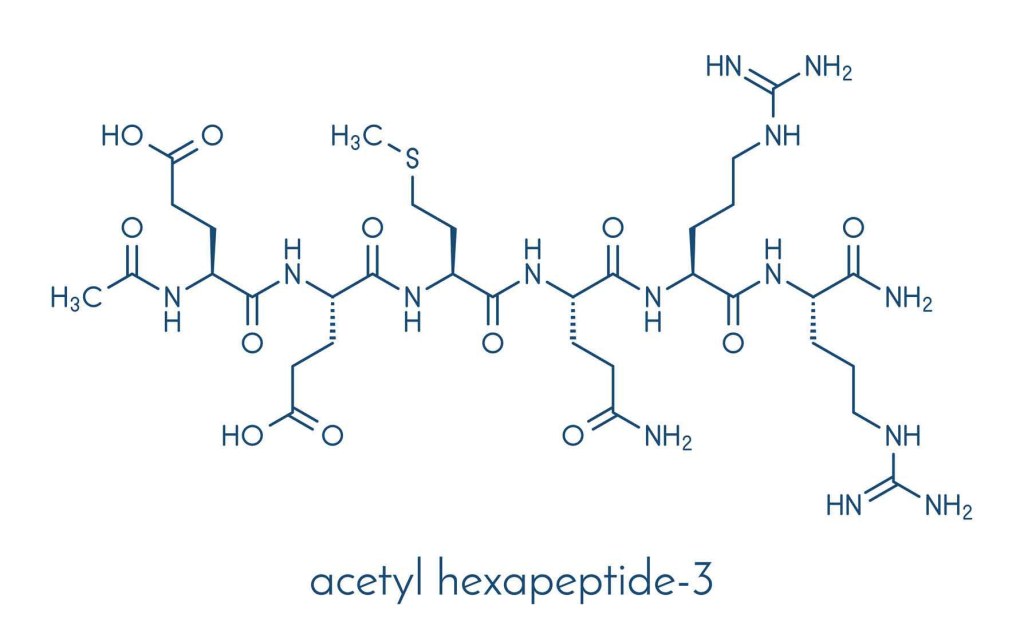
L’Acetyl hexapeptide-8, noto anche come Acetyl hexapeptide-8 ammide (anche erroneamente chiamato Acetyl hexapeptide-3), è un esapeptide sintetico utilizzato come ingrediente cosmetico topico che ha dimostrato di migliorare l’aspetto delle rughe.[77] È un piccolo frammento peptidico di SNAP25, una proteina coinvolta nel rilascio di neurotrasmettitori e uno dei bersagli della tossina botulinica di tipo A (comunemente nota come Botox).
Si propone che l’Acetyl hexapeptide-8 abbia un meccanismo d’azione simile a quello del suo biomimetico, la tossina botulinica, inibendo il complesso SNARE responsabile della fusione delle vescicole sinaptiche, riducendo così le contrazioni dei muscoli facciali. Questo meccanismo proposto ha portato al suo utilizzo nei prodotti anti-aging come potenziale alternativa non invasiva alle neurotossine iniettabili. Nessuno studio clinico ha confrontato direttamente l’efficacia dell’Acetyl hexapeptide-8 con quella della tossina botulinica e la concentrazione necessaria per ottenere effetti simili rimane incerta.[77]
Questo peptide ha un assorbimento cutaneo limitato, probabilmente a causa del suo elevato peso molecolare (889 Da) e dell’idrofilia, che influenzerebbero negativamente i sistemi di somministrazione topici.[77] Uno studio del 2015 ha dimostrato che dopo 24 ore, meno dello 0,2% del peptide applicato penetrava nello strato corneo, lo strato più esterno della pelle, mentre la maggior parte veniva rimossa dopo il lavaggio (99,7%).[78]
L’Acetyl hexapeptide-8 è disponibile dal 2001 ed è commercializzato con il nome commerciale di Argireline da Lubrizol.
La sua funzionale applicabilità per il trattamento dell’Acne è al quanto dubbia.
- LZ1
Un peptide sperimentale, denominato LZ1, con 15 residui amminoacidici, possiede una forte attività antimicrobica contro i batteri patogeni dell’Acne Vulgaris, tra cui Propionibacterium acnes, Staphylococcus epidermidis e S. aureus. In particolare, ha esercitato una forte attività anti-P. acnes. La concentrazione minima inibitoria contro tre ceppi di P. acnes era di soli 0,6 µg/ml, ovvero 4 volte inferiore a quella della Clindamicina. Nel modello sperimentale di colonizzazione cutanea dei topi, LZ1 ha ridotto significativamente il numero di P. acnes colonizzati sull’orecchio, il gonfiore dell’orecchio indotto da P. acnes e l’infiltrazione di cellule infiammatorie. Ha migliorato l’infiammazione indotta da P. acnes inibendo la secrezione di fattori infiammatori, tra cui il fattore di necrosi tumorale-α (TNF-α) e l’interleuchina (IL)-1β. LZ1 ha mostrato scarsa citotossicità sui cheratinociti umani e attività emolitica sui globuli rossi umani. Inoltre, LZ1 è risultato molto stabile nel plasma umano. Grazie alle sue potenziali proprietà battericide e antinfiammatorie, alla struttura semplice e all’elevata stabilità, LZ1 potrebbe essere un candidato ideale per il trattamento dell’acne.
I peptidi antimicrobici (AMP) rappresentano la prima linea dell’immunità innata contro i microrganismi invasori e svolgono un ruolo nel controllo della flora microbica naturale. Le funzioni protettive svolte dagli AMP sulla superficie cutanea esterna erano note fino a poco tempo fa. Ad esempio, la famiglia delle β-defensine umane è stata trovata nelle unità pilosebacee umane, che potrebbero essere coinvolte nella patogenesi dell’acne vulgaris [79]. La capacità di uccidere P. acnes contenuta in hCAP18/LL-37 è stata trovata nelle ghiandole sebacee [80]. Ancora più importante, è stato dimostrato che gli AMP hanno un basso potenziale di indurre resistenza ai farmaci da parte dei microrganismi [80-81,82]. Tra gli AMP, c’è stato un crescente interesse per un sottoinsieme specifico di essi: i peptidi ricchi di triptofano (Trp), lisina (Lys) o arginina (Arg) [83–84]. Questi residui possiedono alcune proprietà chimiche specifiche che li rendono adatti per i peptidi antimicrobici. Studi focalizzati su questi peptidi hanno facilitato la comprensione dei meccanismi molecolari degli AMP. Il Trp idrofobico ha preferenza per la regione interfacciale dei doppi strati lipidici, mentre i residui di Lys e Arg conferiscono ai peptidi cariche cationiche e proprietà di legame idrogeno cruciali per l’interazione con gli abbondanti componenti anionici della membrana batterica [85,86]
L’LZ1 ha una struttura primaria lineare ed è composto da soli 15 residui amminoacidici. Ha mostrato forti capacità antimicrobiche contro i batteri patogeni dell’Acne Vulgaris, come P. acnes, S. epidermidis e S. aureus in vitro. La MIC corrispondente era compresa tra 0,6 e 4,7µg/ml. Ha esercitato le stesse capacità antimicrobiche contro ceppi batterici comuni e resistenti agli antibiotici. La capacità anti-P. acnes di LZ1 in vivo è stata studiata anche in un modello di colonizzazione cutanea di topi. La clearance di P. acnes colonizzato sull’orecchio di topo è stata accelerata da LZ1.
Alcuni studi hanno dimostrato che molti peptidi antimicrobici cationici esercitavano capacità emolitiche sui globuli rossi umani [87,88]. Sono stati testati due possibili effetti collaterali, tra cui emolisi e citotossicità, esercitati da alcuni AMP. LZ1 ha mostrato scarsa attività emolitica e citotossicità anche ad alte concentrazioni (>200µg/ml). Un farmaco può essere modificato o degradato a causa di varie proteasi nel plasma, che rappresenta il problema più importante nell’applicazione del farmaco. Questo peptide sembra essere molto stabile nel plasma umano poiché la sua attività antimicrobica non è stata persa nemmeno dopo l’incubazione di LZ1 con plasma umano per 8 ore a 37°C. Il legame all’Apolipoproteina A-I (apoA-I) e al Glicosaminoglicano inibisce l’attività antibatterica del LL-37 [89,90].
Un’altra caratteristica significativa di LZ1 è che esercita forti effetti antinfiammatori. La colonizzazione follicolare da parte di P. acnes svolge un ruolo importante nella formazione dell’acne. La proliferazione di P. acnes attirerà linfociti CD4+ e macrofagi al microcomedone [91] e quindi indurrà la lesione infiammatoria acneica con rottura della parete follicolare. Come illustrato dalla Figura 4B, l’iniezione di P. acnes ha attratto numerose cellule infiammatorie infiltrate. Dopo la somministrazione epicutanea di LZ1, le cellule infiammatorie infiltrate sono diminuite notevolmente e il gonfiore dell’orecchio indotto da P. acnes è stato inibito significativamente, suggerendo un forte effetto antinfiammatorio. Dopo il trattamento dell’acne con LZ1 per 5 giorni, due importanti citochine infiammatorie, tra cui TNF-α e IL-1β, indotte da P. acnes, sono state significativamente inibite dal peptide, suggerendo che la somministrazione epicutanea di LZ1 sopprimesse l’infiammazione nell’acne, inibendo tuttavia la produzione di citochine infiammatorie.
In conclusione, è stato dimostrato l’effetto antimicrobico di LZ1 contro i batteri della pelle in vitro e il suo potenziale terapeutico per l’Acne Vulgaris infiammatoria indotta da P. acnes in vivo, utilizzando un modello di orecchio di topo. Grazie alla sua semplice struttura primaria con soli 15 residui amminoacidici, che ne facilita la produzione, il trasporto e la conservazione, alla scarsa attività emolitica sui globuli rossi, alla scarsa citotossicità sui cheratinociti umani e all’elevata stabilità nel plasma umano, LZ1 potrebbe essere un eccellente agente terapeutico per il trattamento dell’acne vulgaris, sebbene siano necessari ulteriori studi.
- KPV

Studi di delezione di amminoacidi hanno stabilito che i troncamenti C-terminali di αMSH possiedono anche proprietà antinfiammatorie con la sequenza minima efficace confinata agli ultimi 3 residui, K-P-V [92]. Sono stati testati anche diversi analoghi di questa sequenza, così come omodimeri legati a ponte disolfuro (ad esempio (CKPV)2 [92]) che producono miglioramenti nella capacità del peptide di sopprimere l’attività di NFκB (rivista [93]). Nonostante questi effetti antinfiammatori ben documentati, il meccanismo d’azione di KPV è poco compreso. Il lavoro di Moustafa et al [92] ha dimostrato che l’effetto antinfiammatorio di KPV si estende su un intervallo di concentrazioni che supererebbe la cinetica degli effetti mediati dal recettore e lavori recenti dimostrano un’apparente necessità per il trasporto di membrana di KPV mediato da PEPTL1 [94], sollevando la possibilità che esso medi i suoi effetti interagendo con bersagli intracellulari. Apparentemente, ciò potrebbe verificarsi in due modi. In primo luogo, i residui di KPV possono legare individualmente o collettivamente sequenze amminoacidiche polari o non polari esposte sulla superficie di proteine chiave di segnalazione. La teoria del legame complementare degli amminoacidi prevede che K favorirà le interazioni con i residui L o F, P con W, G e R e V con Y, H, D o N [95], sebbene algoritmi alternativi prevedano alcune varianti più conservative su questo tema [96]. Poiché il residuo di prolina forma un angolo nel peptide KPV, possono verificarsi interazioni complementari tra uno, due o tutti e tre i residui in diversi possibili orientamenti. Questo modello non può, tuttavia, spiegare l’apparente specificità di KPV per la via NFκB poiché la breve sequenza peptidica presumibilmente favorirebbe molteplici bersagli non specifici. Un’ipotesi alternativa prevede che la sequenza di KPV specificherà le sue azioni a una molecola nella via di attivazione di NFκB. KPV mostra le caratteristiche fondamentali di una sequenza di localizzazione nucleare minima (NLS). Sebbene le sequenze NLS siano variabili, le caratteristiche chiave includono un cluster di residui caricati positivamente (ad esempio K-K/R-X-K/R per NLS monopartiti; [97]), spesso preceduti da un residuo di rottura dell’elica come P. Ad esempio, l’NLS monopartito dell’antigene T grande SV40 include residui K, P e V nella sequenza critica di legame al DNA, 126PKKKRKV132 [98] mentre l’NLS del fattore enhancer linfoide-1 (LEF-1) contiene una sequenza simile a KPV in cui la V è sostituita da un altro residuo idrofobico, L, che interagisce con il solco minore del DNA. Una volta libere dal loro inibitore, IκBα, le subunità p50 e p65RelA dell’eterodimero NFκB migrano verso il nucleo legandosi rispettivamente ai terminali N e C dell’importina-α3 (Imp-α3) [99]. L’analisi del dominio Imp-α3 armadillo (arm) 3 che lega p65RelA mostra che la sequenza critica di interazione è ricca di amminoacidi complementari per KPV, suggerendo che in questo sito potrebbe verificarsi un’interazione competitiva che interromperebbe l’importazione nucleare di NFκB.
Per determinare come KPV inibisca la segnalazione infiammatoria indotta da NFκB nell’epitelio delle vie aeree, lo studio attuale ha cercato prove che il peptide potesse funzionare in uno dei seguenti 3 modi: 1) promuovendo la stabilità di IκBα, 2) occupando il solco minore del DNA o 3) interferendo con l’importazione nucleare di p65RelA. Inoltre, è stata studiata l’espressione delle isoforme del recettore della melanocortina nel tessuto epiteliale delle vie aeree per stabilire il potenziale di effetti antinfiammatori mediati dal recettore. I risultati mostrano che KPV trasloca nel nucleo delle cellule epiteliali bronchiali umane e che blocca competitivamente l’interazione tra Imp-α3 e p65RelA di NFκB. Un esame più ampio del ruolo dell’espressione del recettore della melanocortina dimostra che MC3R è il recettore dominante espresso nell’epitelio delle vie aeree e che il suo agonista, γMSH, sopprime l’infiammazione cellulare e sistemica in risposta a stimoli pro-infiammatori. Si conclude che i peptidi della melanocortina possono reprimere la segnalazione infiammatoria nelle cellule epiteliali delle vie aeree sia attraverso la repressione diretta del trasporto nucleare di NFκB sia attraverso vie di segnalazione mediate dal recettore. Pertanto, le melanocortine e i loro derivati rappresentano bersagli robusti per il trattamento delle malattie infiammatorie polmonari.
Questo studio conferma che il tripeptide derivato dalla melanocortina, KPV, sopprime le risposte immunitarie sia locali che sistemiche che comunemente inducono danno alle vie aeree e rimodellamento nelle malattie infiammatorie polmonari. Queste osservazioni sono coerenti con la capacità ampiamente descritta di KPV e dei suoi stereoisomeri (dKPV, KPdV, KdPV, dKPdV e KdPT) di agire come potenti antinfiammatori e antipiretici, rendendoli interessanti bersagli farmacologici per il trattamento di un’ampia gamma di malattie infiammatorie.
Lo scopo di questo studio era determinare come il KPV media i suoi effetti antinfiammatori. Il trasportatore di oligopeptidi accoppiato a H+, PEPT1, media l’assorbimento intracellulare del KPV nell’epitelio e questo è necessario per gli effetti antinfiammatori [94]. Ciò amplia un crescente corpus di prove che suggerisce che l’effetto antinfiammatorio del KPV è indipendente dal sistema di segnalazione del recettore della melanocortina e probabilmente si verifica attraverso un meccanismo intracellulare. Una delle azioni del KPV più costantemente riportate è la riduzione della durata dell’attivazione di NFκB, pertanto l’attenzione si è concentrata sulle sue interazioni con IκBα, p65RelA e Imp-α3 come mediatori critici dell’attivazione e dell’importazione nucleare di NFκB. I risultati corroborano precedenti osservazioni secondo cui KPV promuove la stabilizzazione di IκBα in presenza di citochine pro-infiammatorie; tuttavia, la scoperta principale è che il sito predominante di accumulo di KPV è nel nucleo, dove inibisce competitivamente l’interazione tra p65RelA e Imp-α3. È importante sottolineare che questo effetto si è verificato senza un’interazione significativa con la cromatina.
L’NLS di p65RelA è localizzato nei residui C-terminali 301-304 e contiene la sequenza consenso KRKR, fiancheggiata da due α-eliche, l’elica tre (289-300) e l’elica quattro (305-321). L’elica quattro contiene siti critici necessari per un’interazione stabile con IκBα, che maschera l’NLS e quindi blocca l’interazione con Imp-α3. La fosforilazione e la degradazione proteolitica di IκBα consentono a Imp-α3 di legarsi all’NLS e quindi promuovono la traslocazione nucleare del dimero NFκB [99]. L’osservazione che KPV può interferire con l’interazione tra la subunità p65RelA e Imp-α3 in vitro suggerisce che questo peptide possa legare in modo competitivo sequenze critiche nell’NLS di entrambe le proteine. Sebbene le strutture cristalline di Imp-α3 non siano ancora disponibili, l’analisi del legame con pepsite dell’interazione di KPV con l’isoforma strettamente correlata, Imp-α2 murina, ha dimostrato molteplici possibili interazioni che coinvolgono due o più residui di KPV con gli amminoacidi 360-403 che si estendono sui bracci 7 e 8. L’analisi del pepsite ha rivelato che ciò è piuttosto specifico per questa regione della molecola, senza alcuna interazione prevista in altri siti. Sebbene questo studio non abbia dimostrato questa interazione in vivo, un’interazione tra KPV e proteine della famiglia dell’importina spiegherebbe la tendenza di KPV ad accumularsi nel nucleo in presenza di un’importazione nucleare ostacolata di p65RelA nonostante la fosforilazione di IκBα.
Chiaramente questo solleva la questione se KPV possa interferire con l’importazione di altre proteine nucleari. L’analisi Pepsite suggerisce che l’interazione KPV è specifica per i bracci NLS C-terminali 7 e 8 di Imp-α2 e questo dominio è di fondamentale importanza tra le altre isoforme dell’importina per l’importazione nucleare di HIF-1α e p65relA (da parte di Imp-α3; [100]), STAT1 e proteina nucleare (NP) del virus dell’influenza A (da parte di Imp-α5; [101]). In effetti, questo studio mostra che KPV può migliorare l’inibizione della crescita causata da TNFα oltre al suo effetto antinfiammatorio e induce anche l’attività di mTORC1, un importante regolatore della crescita e della differenziazione cellulare. Pertanto, gli effetti intracellulari di KPV sono pleiotropici e probabilmente coinvolgono una gamma di molecole effettrici che possono spiegare la curva dose-risposta insolitamente prolungata per questa molecola riportata negli studi farmacologici [92].
Questo studio dimostra che il KPV sopprime la segnalazione infiammatoria nelle cellule epiteliali bronchiali polmonari e solleva la questione se il KPV e altri peptidi di melanocortina possano essere utili nel combattere le malattie infiammatorie polmonari. Precedenti lavori sulle cellule polmonari dimostrano che i peptidi di melanocortina possono arrestare varie forme di infiammazione nel polmone. Ad esempio, l’α-MSH ha soppresso la sintesi di PGE nei fibroblasti polmonari umani fetali stimolati con IL-1 [102] e anche l’espressione proteica della mucina indotta da TNFα nell’epitelio nasale umano coltivato [103]. Nei modelli allergici e non allergici di infiammazione polmonare, sia gli agonisti di MC1R che MC3R, αMSH e [D-TRP]-γ-MSH], hanno inibito l’accumulo di leucociti nel polmone e soppresso il rimodellamento polmonare infiammatorio [104]. Lo studio attuale ha scoperto che sia l’α che il γ-MSH possono sopprimere segnali infiammatori intracellulari e sistemici tramite il recettore MC3R nelle cellule epiteliali delle vie aeree sottoposte a stimolazione con RSV o LPS. Questo integra il lavoro di Getting et al [104] che ha dimostrato un ruolo centrale per il recettore MC3R nella soppressione dell’infiammazione delle vie aeree mediata dai leucociti. È importante sottolineare che questo lavoro non ha escluso un ruolo per l’attivazione del recettore MC nell’epitelio delle vie aeree e il presente studio lo conferma dimostrando un’ampia espressione di MC3R nelle cellule epiteliali delle vie aeree, sia in vitro che in vivo, e dimostrando che i peptidi melanocortina sopprimono la secrezione di citochine chemiotattiche dall’epitelio delle vie aeree. Il KPV offre il vantaggio che le sue azioni non sembrano essere mediate dal recettore e la sua struttura può essere potenzialmente modulata per migliorarne il targeting verso una particolare via o un’altra. Inoltre, le sue piccole dimensioni e la sua solubilità in acqua ne consentono la somministrazione in forma nebulizzata nei polmoni, dove i nostri dati dimostrano la sua capacità di mediare effetti antinfiammatori negli epiteli polmonari. La soppressione dell’attività delle MMP è particolarmente degna di nota, poiché questa famiglia di proteasi svolge un ruolo fondamentale nella segnalazione immunitaria e nel rimodellamento polmonare in varie forme di malattie polmonari infiammatorie e cancerose. Nel complesso, questo lavoro suggerisce che gli agonisti di KPV e MC3R rappresentano candidati ideali per la soppressione delle fasi precoci dell’infiammazione negli epiteli delle vie aeree.
In conclusione, il presente studio dimostra che il piccolo tripeptide correlato alla melanocortina, KPV, inibisce la segnalazione infiammatoria nelle cellule epiteliali bronchiali umane attraverso un meccanismo che coinvolge l’interruzione della traslocazione nucleare di p65RelA. I dati mostrano che questa interruzione può verificarsi tramite un’interazione competitiva tra KPV e p65RelA con i domini del braccio di Imp-α3 e l’analisi del Pepsite suggerisce che questa possa essere limitata ai bracci 7 e 8, coinvolti nel traffico nucleare di altri fattori di trascrizione. Pertanto, KPV media il suo principale effetto antinfiammatorio attraverso il sistema di trasporto nucleare e indipendentemente dai recettori della melanocortina. Lo sviluppo farmaceutico di KPV come terapia antinfiammatoria potrebbe quindi dipendere dal grado in cui può essere mirato a specifiche interazioni tra le molecole di importina e il loro carico proteico. Oltre all’effetto KPV, questo studio dimostra che gli agonisti della melanocortina del MC3R possono reprimere le fasi precoci dell’infiammazione cellulare e sistemica, evidenziando i peptidi della melanocortina come uno strumento efficace per il trattamento delle malattie infiammatorie polmonari.
Conclusioni – Esempio di regime di trattamento per l’Acne Vulgaris [con metodi comprovati]:
Un buon punto di partenza sarebbe abbinare ad uno crubs regolare l’utilizzo degli integratori da banco elencati in precedenza: Zinco (fino a 40mg al giorno), Vitamina D (1.000-4.000UI al giorno) e Acidi Grassi Omega 3 (1g di EPA e 1g di DHA al giorno). Se questo non fosse sufficiente, si potrebbe aggiungere un retinoide (come l’Adapalene 0,3%) o il Perossido di Benzoile (2,5-5%) e applicarlo sulle zone interessate. Una volta al giorno per il Perossido di Benzoile e due volte a settimana come punto di partenza per il retinoide. I due possono anche essere combinati, applicando, ad esempio, il Perossido di Benzoile al mattino e il retinoide alla sera. Se dopo diverse settimane i risultati non sono ancora soddisfacenti, si potrebbe optare per l’Isotretinoina a un dosaggio basso, da 5 a 10mg al giorno.
*Si noti che l’acne potrebbe peggiorare inizialmente durante le prime settimane.
Non ho aggiunto alcun protocollo riguardo ai peptidi elencati nell’articolo semplicemente perché si tratta di pratiche ancora non pienamente comprovate. Fatta eccezione per quei peptidi già commercializzati nelle soluzioni topiche anti-aging, quelli maggiormente specifici per l’Acne Vulgaris [LL-37 e IZ1] sono, per l’appunto, in una fase di studio che non permette di esprimere dosaggi univoci per la popolazione nella media.
Gabriel Bellizzi [CEO BioGenTech]
Riferimenti:
- Bhate, K., and H. C. Williams. “Epidemiology of acne vulgaris.” British Journal of Dermatology 168.3 (2013): 474-485.
- D. Thiboutot, H. Gollnick, V. Bettoli, B. Dréno, S. Kang, J. J. Leyden, A. R. Shalita, V. T. Lozada, D. Berson, A. Finlay, et al. New insights into the management of acne: an update from the global alliance to improve outcomes in acne group. Journal of the American Academy of Dermatology, 60(5):S1–S50, 2009
- Williams, Hywel C., Robert P. Dellavalle, and Sarah Garner. “Acne vulgaris.” The Lancet 379.9813 (2012): 361-372.
- P. M. Elias, B. E. Brown, and V. A. Ziboh. The permeability barrier in essential fatty acid deficiency: evidence for a direct role for linoleic acid in barrier function. Journal of Investigative Dermatology, 74(4):230–233, 1980.
- A. H. Jeremy, D. B. Holland, S. G. Roberts, K. F. Thomson, and W. J. Cunliffe. Inflammatory events are involved in acne lesion initiation. Journal of Investigative Dermatology, 121(1):20–27, 2003.
- Imperato-McGinley, Julianne, et al. “The androgen control of sebum production. Studies of subjects with dihydrotestosterone deficiency and complete androgen insensitivity.” The Journal of Clinical Endocrinology & Metabolism 76.2 (1993): 524-528.
- Pochi, Peter E., and John S. Strauss. “Sebaceous gland response in man to the administration of testosterone, D4-androstenedione, and dehydroisoandrosterone.” J Invest Dermatol 52 (1969): 32-36.
- Bhasin, Shalender, et al. “Effect of testosterone supplementation with and without a dual 5α-reductase inhibitor on fat-free mass in men with suppressed testosterone production: a randomized controlled trial.” Jama 307.9 (2012): 931-939.
- Smit, Diederik L., et al. “Positive and negative side effects of androgen abuse. The HAARLEM study: A one‐year prospective cohort study in 100 men.” Scandinavian Journal of Medicine & Science in Sports 31.2 (2021): 427-438.
- Cervantes, Jessica, et al. “The role of zinc in the treatment of acne: A review of the literature.” Dermatologic therapy 31.1 (2018): e12576.
- J. Fitzherbert. Zinc deficiency in acne vulgaris. The Medical journal of Australia, 2(20):685–686, 1977.
- G. Michaëlsson, L. Juhlin, and K. Ljunghall. A double-blind study of the effect of zinc and oxytetracycline in acne vulgaris. British Journal of Dermatology, 97(5):561–566, 1977.
- Y. S. Bae, N. D. Hill, Y. Bibi, J. Dreiher, and A. D. Cohen. Innovative uses for zinc in dermatology. Dermatologic clinics, 28(3):587–597, 2010.
- S.-K. Lim, J.-M. Ha, Y.-H. Lee, Y. Lee, Y.-J. Seo, C.-D. Kim, J.-H. Lee, and M. Im. Comparison of vitamin d levels in patients with and without acne: a case-control study combined with a randomized controlled trial. PloS one, 11(8):e0161162, 2016
- Backx, E. M. P., et al. “The impact of 1-year vitamin D supplementation on vitamin D status in athletes: a dose–response study.” European journal of clinical nutrition 70.9 (2016): 1009-1014.
- M. F. Holick, N. C. Binkley, H. A. Bischoff-Ferrari, C. M. Gordon, D. A. Hanley, R. P. Heaney, M. H.Murad, and C. M. Weaver. Evaluation, treatment, and prevention of vitamin d deficiency: an endocrine society clinical practice guideline. The Journal of Clinical Endocrinology & Metabolism, 96(7):1911–1930, 2011
- N. EFSA Panel on Dietetic Products and A. (NDA). Scientific opinion on the tolerable upper intake level of vitamin d. EFSA Journal, 10(7):2813, 2012.
- J. Y. Jung, H. H. Kwon, J. S. Hong, J. Y. Yoon, M. S. Park, M. Y. Jang, and D. H. Suh. Effect of dietary supplementation with omega-3 fatty acid and gamma-linolenic acid on acne vulgaris: a randomised, double-blind, controlled trial. Acta dermato-venereologica, 94(5):521–526, 2014.
- N. H. Mohd Nor and Z. Aziz. A systematic review of benzoyl peroxide for acne vulgaris. Journal of Dermatological Treatment, 24(5):377–386, 2013.
- J. Waller, F. Dreher, S. Behnam, C. Ford, C. Lee, T. Tiet, G. Weinstein, and H. Maibach. ‘keratolytic’ properties of benzoyl peroxide and retinoic acid resemble salicylic acid in man. Skin pharmacology and physiology, 19(5):283–289, 2006.
- A. J. Brandstetter and H. I. Maibach. Topical dose justification: benzoyl peroxide concentrations. Journal of Dermatological Treatment, 24(4):275–277, 2013
- W. Cunliffe, M. Poncet, C. Loesche, and M. Verschoore. A comparison of the efficacy and tolerability of adapalene 0.1% gel versus tretinoin 0.025% gel in patients with acne vulgaris: a meta-analysis of five randomized trials., 1998
- D. Thiboutot, D. M. Pariser, N. Egan, J. Flores, J. H. Herndon Jr, N. B. Kanof, S. E. Kempers, S. Maddin, Y. P. Poulin, D. C. Wilson, et al. Adapalene gel 0.3% for the treatment of acne vulgaris: a multicenter, randomized, double-blind, controlled, phase iii trial. Journal of the American Academy of Dermatology, 54(2):242–250, 2006.
- E. Tanghetti, S. Dhawan, L. Green, J. R. Del, Z. Draelos, J. Leyden, A. Shalita, D. A. Glaser, P. Grimes, G. Webster, et al. Randomized comparison of the safety and efficacy of tazarotene 0.1% cream and adapalene 0.3% gel in the treatment of patients with at least moderate facial acne vulgaris. Journal of drugs in dermatology: JDD, 9(5):549–558, 2010.
- D. Thiboutot, S. Arsonnaud, and P. Soto. Efficacy and tolerability of adapalene 0.3% gel compared to tazarotene 0.1% gel in the treatment of acne vulgaris. Journal of drugs in dermatology: JDD, 7(6 Suppl):s3–10, 2008
- I. Vallerand, R. Lewinson, M. Farris, C. Sibley, M. Ramien, A. Bulloch, and S. Patten. Efficacy and adverse events of oral isotretinoin for acne: a systematic review. British Journal of Dermatology, 178(1):76–85, 2018.
- M. Rademaker, J. Wishart, and N. Birchall. Isotretinoin 5 mg daily for low-grade adult acne vulgaris–a placebo-controlled, randomized double-blind study. Journal of the European Academy of Dermatology and Venereology, 28(6):747–754, 2014.
- M. Mirnezami and H. Rahimi. Is oral omega-3 effective in reducing mucocutaneous side effects of isotretinoin in patients with acne vulgaris? Dermatology research and practice, 2018.
- J. Leyden, W. Bergfeld, L. Drake, F. Dunlap, M. P. Goldman, A. B. Gottlieb, M. P. Heffernan, J. G. Hickman, M. Hordinsky, M. Jarrett, et al. A systemic type i 5 a-reductase inhibitor is ineffective in the treatment of acne vulgaris. Journal of the American Academy of Dermatology, 50(3):443–447, 2004.
- F. Azzouni, A. Godoy, Y. Li, and J. Mohler. The 5 alpha-reductase isozyme family: a review of basic biology and their role in human diseases. Advances in urology, 2012, 2012.
- F. Azzouni, A. Godoy, Y. Li, and J. Mohler. The 5 alpha-reductase isozyme family: a review of basic biology and their role in human diseases. Advances in urology, 2012, 2012.
- J. I. Schwartz, W. K. Tanaka, D. Z. Wang, D. L. Ebel, L. A. Geissler, A. Dallob, B. Hafkin, and B. J. Gertz. Mk-386, an inhibitor of 5a-reductase type 1, reduces dihydrotestosterone concentrations in serum and sebum without affecting dihydrotestosterone concentrations in semen. The Journal of Clinical Endocrinology & Metabolism, 82(5):1373–1377, 1997.
- Pickart, L; Thaler, MM (1973). “Tripeptide in human serum which prolongs survival of normal liver cells and stimulates growth in neoplastic liver”. Nature New Biology. 243 (124): 85–87. PMID 4349963.
- Pilgeram, L; Pickart, L (1968). “Control of fibrinogen biosynthesis; the role of free fatty acids”. Journal of Atherosclerosis Research. 8 (1): 155–166. doi:10.1016/s0368-1319(68)80089-4. PMID 5642099.
- Pilgeram, L (2010). “Control of fibrinogen biosynthesis; role of FFA/Albumin Ratio”. Cardiovascular Engineering. 10 (2): 78–83. doi:10.1007/s10558-010-9092-1. PMC 2885297. PMID 20383582.
- Pickart, L (1973), A tripeptide in human plasma that increases the survival of hepatocytes and the growth of hepatoma cells, Ph.D. Thesis in Biochemistry: University of California, San Francisco
- Schlesinger, DH; Pickart, L; Thaler, MM (1977). “Growth-modulating serum tripeptide is glycyl-histidyl-lysine”. Cellular and Molecular Life Sciences. 33 (3): 324–325. doi:10.1007/BF02002806. PMID 858356. S2CID 29422959.
- Pickart, L; Freedman, JH; Loker, WJ; et al. (1980). “Growth-modulating plasma tripeptide may function by facilitating copper uptake into cells”. Nature. 288 (5792): 715–717. Bibcode:1980Natur.288..715P. doi:10.1038/288715a0. PMID 7453802. S2CID 4304271.
- Maquart, FX; Pickart, L; Laurent, M; Gillery, P; Monboisse, JC; Borel, JP (1988). “Stimulation of collagen synthesis in fibroblast cultures by the tripeptide-copper complex glycyl-L-histidyl-L-lysine-Cu2+”. FEBS Letters. 238 (2): 343–6. Bibcode:1988FEBSL.238..343M. doi:10.1016/0014-5793(88)80509-x. PMID 3169264. S2CID 19289897.
- Wegrowski, Y.; Maquart, F.X.; Borel, J.P. (1992). “Stimulation of sulfated glycosaminoglycan synthesis by the tripeptide-copper complex Glycyl-L-histidyl-L-lysine-Cu2+”. Life Sciences. 51 (13): 1049–1056. doi:10.1016/0024-3205(92)90504-i. PMID 1522753.
- Maquart, FX; Bellon, G; Pasco, S; Monboisse, JC (2005). “Matrikines in the regulation of extracellular matrix degradation”. Biochimie. 87 (3–4): 353–60. doi:10.1016/j.biochi.2004.10.006. PMID 15781322.
- Siméon, A; Wegrowski, Y; Bontemps, Y; Maquart, FX (2000). “Expression of glycosaminoglycans and small proteoglycans in wounds: modulation by the tripeptide-copper complex glycyl-L-histidyl-L-lysine-Cu(2+)”. The Journal of Investigative Dermatology. 115 (6): 962–8. doi:10.1046/j.1523-1747.2000.00166.x. PMID 11121126.
- Siméon, Alain; Emonard, Hervé; Hornebeck, William; Maquart, François-Xavier (2000). “The tripeptide-copper complex glycyl-L-histidyl-L- lysine-Cu2+ stimulates matrix metalloproteinase-2 expression by fibroblast cultures”. Life Sciences. 67 (18): 2257–2265. doi:10.1016/s0024-3205(00)00803-1. PMID 11045606.
- Pickart, Lorraine; Margolina, Anna (2015). “GHK peptide as a natural modulator of multiple cellular pathways in skin regeneration”. BioMed Research International. 2015 (7): 648108. doi:10.1155/2015/648108. PMC 6073405. PMID 29986520.
- Gul, NY; Topal, A; Cangul, IT; Yanik, K (2008). “The effects of topical tripeptide copper complex and helium-neon laser on wound healing in rabbits”. Veterinary Dermatology. 19 (1): 7–14. doi:10.1111/j.1365-3164.2007.00647.x. PMID 18177285.
- Cangul, IT; Gul, NY; Topal, A; Yilmaz, R (2006). “Evaluation of the effects of topical tripeptide-copper complex and zinc oxide on open-wound healing in rabbits”. Veterinary Dermatology. 17 (6): 417–23. doi:10.1111/j.1365-3164.2006.00551.x. PMID 17083573.
- Pickart L. Compositions for accelerating wound healing in mammals containing cupric salt or complexes with amino acid or peptide. US Patent 5,164,367, 1992.
- Canapp SO Jr, Farese JP, Schultz GS, Gowda S, Ishak AM, Swaim SF, Vangilder J, Lee-Ambrose L, Martin FG (Nov–Dec 2003). “The effect of topical tripeptide-copper complex on healing of ischemic open wounds”. Veterinary Surgery. 32 (6): 515–23. doi:10.1111/j.1532-950x.2003.00515.x. PMID 14648529.
- https://pubchem.ncbi.nlm.nih.gov/compound/Palmitoyl-Tetrapeptide-7
- https://pubchem.ncbi.nlm.nih.gov/compound/Palmitoyl-Tripeptide-1
- “UniProt”. http://www.uniprot.org. Retrieved 8 February 2024.
- “Entrez Gene: CAMP cathelicidin antimicrobial peptide”.
- Dürr U, Sudheendra U, Ramamoorthy, A (September 2006). “LL-37, the only human member of the cathelicidin family of antimicrobial peptides”. Biochimica et Biophysica Acta (BBA) – Biomembranes. 1758 (9): 1408–1425. doi:10.1016/j.bbamem.2006.03.030. PMID 16716248.
- Kościuczuk EM, Lisowski P, Jarczak J, Strzałkowska N, Jóźwik A, Horbańczuk J, et al. (December 2012). “Cathelicidins: family of antimicrobial peptides. A review”. Molecular Biology Reports. 39 (12): 10957–70. doi:10.1007/s11033-012-1997-x. PMC 3487008. PMID 23065264.
- Zanetti M (January 2004). “Cathelicidins, multifunctional peptides of the innate immunity”. Journal of Leukocyte Biology. 75 (1): 39–48. doi:10.1189/jlb.0403147. PMID 12960280. S2CID 14902156.
- Carman R, Simonian MR, Old JM, Jacques NA, Deane EM (2008). Immunohistochemistry using antibodies to the Cathelicidin LL37/hCAP18 in the tammar wallaby (Macropus eugenii). Tissue and Cell. 40(6), 459-466. DOI: 10.1016/j.tice.2008.05.002
- Vandamme D, Landuyt B, Luyten W, Schoofs L (November 2012). “A comprehensive summary of LL-37, the factotum human cathelicidin peptide”. Cellular Immunology. 280 (1): 22–35. doi:10.1016/j.cellimm.2012.11.009. PMID 23246832.
- Liu PT, Stenger S, Li H, Wenzel L, Tan BH, Krutzik SR, et al. (March 2006). “Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response”. Science. 311 (5768): 1770–3. Bibcode:2006Sci…311.1770L. doi:10.1126/science.1123933. PMID 16497887. S2CID 52869005.
- Bals R, Wang X, Zasloff M, Wilson JM (August 1998). “The peptide antibiotic LL-37/hCAP-18 is expressed in epithelia of the human lung where it has broad antimicrobial activity at the airway surface”. Proceedings of the National Academy of Sciences of the United States of America. 95 (16): 9541–6. Bibcode:1998PNAS…95.9541B. doi:10.1073/pnas.95.16.9541. PMC 21374. PMID 9689116.
- Stecher C, Maurer KP, Kastner MT, Steininger C (2022-09-10). “Human Cytomegalovirus Induces Vitamin-D Resistance In Vitro by Dysregulating the Transcriptional Repressor Snail”. Viruses. 14 (9): 2004. doi:10.3390/v14092004. ISSN 1999-4915. PMC 9505537. PMID 36146811.
- Zanetti M, Gennaro R, Romeo D (October 1995). “Cathelicidins: a novel protein family with a common proregion and a variable C-terminal antimicrobial domain”. FEBS Letters. 374 (1): 1–5. Bibcode:1995FEBSL.374….1Z. doi:10.1016/0014-5793(95)01050-o. PMID 7589491. S2CID 34865828.
- Ritonja A, Kopitar M, Jerala R, Turk V (September 1989). “Primary structure of a new cysteine proteinase inhibitor from pig leucocytes”. FEBS Letters. 255 (2): 211–4. Bibcode:1989FEBSL.255..211R. doi:10.1016/0014-5793(89)81093-2. PMID 2792375.
- Dosler S, Karaaslan E (December 2014). “Inhibition and destruction of Pseudomonas aeruginosa biofilms by antibiotics and antimicrobial peptides”. Peptides. 62: 32–7. doi:10.1016/j.peptides.2014.09.021. PMID 25285879. S2CID 207359996.
- Reinholz M, Ruzicka T, Schauber J (May 2012). “Cathelicidin LL-37: an antimicrobial peptide with a role in inflammatory skin disease”. Annals of Dermatology. 24 (2): 126–35. doi:10.5021/ad.2012.24.2.126. PMC 3346901. PMID 22577261.
- Yamasaki K, Di Nardo A, Bardan A, Murakami M, Ohtake T, Coda A, Dorschner RA, Bonnart C, Descargues P, Hovnanian A, Morhenn VB, Gallo RL (August 2007). “Increased serine protease activity and cathelicidin promotes skin inflammation in rosacea”. Nature Medicine. 13 (8): 975–80. doi:10.1038/nm1616. PMID 17676051. S2CID 23470611.
- Gombart AF, Bhan I, Borregaard N, Tamez H, Camargo CA, Koeffler HP, Thadhani R (February 2009). “Low plasma level of cathelicidin antimicrobial peptide (hCAP18) predicts increased infectious disease mortality in patients undergoing hemodialysis”. Clinical Infectious Diseases. 48 (4): 418–24. doi:10.1086/596314. PMC 6944311. PMID 19133797.
- Zasloff M (January 2002). “Antimicrobial peptides of multicellular organisms”. Nature. 415 (6870): 389–95. Bibcode:2002Natur.415..389Z. doi:10.1038/415389a. PMID 11807545. S2CID 205028607.
- Kamen DL, Tangpricha V (May 2010). “Vitamin D and molecular actions on the immune system: modulation of innate and autoimmunity”. Journal of Molecular Medicine. 88 (5): 441–50. doi:10.1007/s00109-010-0590-9. PMC 2861286. PMID 20119827.
- de Breij A, Riool M, Cordfunke RA, Malanovic N, de Boer L, Koning RI, et al. (January 2018). “The antimicrobial peptide SAAP-148 combats drug-resistant bacteria and biofilms”. Science Translational Medicine. 10 (423): eaan4044. doi:10.1126/scitranslmed.aan4044. PMID 29321257.
- van Gent ME, van der Reijden TJ, Lennard PR, de Visser AW, Schonkeren-Ravensbergen B, Dolezal N, et al. (May 2022). “Synergism between the Synthetic Antibacterial and Antibiofilm Peptide (SAAP)-148 and Halicin”. Antibiotics. 11 (5): 673. doi:10.3390/ANTIBIOTICS11050673. PMC 9137631. PMID 35625317.
- Rendon A, Schäkel K (March 2019). “Psoriasis Pathogenesis and Treatment”. International Journal of Molecular Sciences. 20 (6): 1475. doi:10.3390/ijms20061475. PMC 6471628. PMID 30909615.
- Shaykhiev R, Beisswenger C, Kändler K, Senske J, Püchner A, Damm T, et al. (November 2005). “Human endogenous antibiotic LL-37 stimulates airway epithelial cell proliferation and wound closure”. American Journal of Physiology. Lung Cellular and Molecular Physiology. 289 (5): L842-8. doi:10.1152/ajplung.00286.2004. PMID 15964896.
- Chen Q, Schmidt AP, Anderson GM, Wang JM, Wooters J, Oppenheim JJ, Chertov O (October 2000). “LL-37, the neutrophil granule- and epithelial cell-derived cathelicidin, utilizes formyl peptide receptor-like 1 (FPRL1) as a receptor to chemoattract human peripheral blood neutrophils, monocytes, and T cells”. The Journal of Experimental Medicine. 192 (7): 1069–74. doi:10.1084/jem.192.7.1069. PMC 2193321. PMID 11015447.
- von Haussen J, Koczulla R, Shaykhiev R, Herr C, Pinkenburg O, Reimer D, et al. (January 2008). “The host defence peptide LL-37/hCAP-18 is a growth factor for lung cancer cells”. Lung Cancer. 59 (1): 12–23. doi:10.1016/j.lungcan.2007.07.014. PMID 17764778.
- Koczulla R, von Degenfeld G, Kupatt C, Krötz F, Zahler S, Gloe T, et al. (June 2003). “An angiogenic role for the human peptide antibiotic LL-37/hCAP-18”. The Journal of Clinical Investigation. 111 (11): 1665–72. doi:10.1172/JCI17545. PMC 156109. PMID 12782669.
- Ren SX, Shen J, Cheng AS, Lu L, Chan RL, Li ZJ, et al. (2013-05-20). Nie D (ed.). “FK-16 derived from the anticancer peptide LL-37 induces caspase-independent apoptosis and autophagic cell death in colon cancer cells”. PLOS ONE. 8 (5): e63641. Bibcode:2013PLoSO…863641R. doi:10.1371/journal.pone.0063641. PMC 3659029. PMID 23700428.
- Lum, Kalisa; M Hirpara, Milan; Pham, Christine; Nguyen, Megan; Mesinkovska, Natasha (2025-04-01). “Acetyl Hexapeptide-8 as a Topical Alternative to Botulinum Toxin: A Review of the Literature”. Journal of Drugs in Dermatology: JDD. 24 (4): e31 – e32. ISSN 1545-9616. PMID 40196949.
- Zdrada-Nowak, Julita; Surgiel-Gemza, Agnieszka; Szatkowska, Magdalena (2025-06-14). “Acetyl Hexapeptide-8 in Cosmeceuticals-A Review of Skin Permeability and Efficacy”. International Journal of Molecular Sciences. 26 (12): 5722. doi:10.3390/ijms26125722. ISSN 1422-0067. PMC 12193160. PMID 40565185.
- Chronnell CM, Ghali LR, Ali RS, Quinn AG, Holland DB et al. (2001) Human beta defensin-1 and -2 expression in human pilosebaceous units: upregulation in acne vulgaris lesions. J Invest Dermatol 117: 1120–1125. doi:10.1046/j.0022-202x.2001.01569.x. PubMed: 11710922. [DOI] [PubMed] [Google Scholar]
- Lee DY, Yamasaki K, Rudsil J, Zouboulis CC, Park GT et al. (2008) Sebocytes express functional cathelicidin antimicrobial peptides and can act to kill propionibacterium acnes . J Invest Dermatol 128: 1863-1866. doi:10.1038/sj.jid.5701235. PubMed: 18200058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nizet V (2006) Antimicrobial peptide resistance mechanisms of human bacterial pathogens. Curr Issues Mol Biol 8: 11-26. PubMed: 16450883. [PubMed] [Google Scholar]
- Yeaman MR, Yount NY (2003) Mechanisms of antimicrobial peptide action and resistance. Pharmacol Rev 55: 27-55. doi:10.1124/pr.55.1.2. PubMed: 12615953. [DOI] [PubMed] [Google Scholar]
- Schibli DJ, Epand RF, Vogel HJ, Epand RM (2002) Tryptophan-rich antimicrobial peptides: comparative properties and membrane interactions. Biochem Cell Biol 80: 667-677. doi:10.1139/o02-147. PubMed: 12440706. [DOI] [PubMed] [Google Scholar]
- Yang ST, Shin SY, Lee CW, Kim YC, Hahm KS et al. (2003) Selective cytotoxicity following Arg-to-Lys substitution in tritrpticin adopting a unique amphipathic turn structure. FEBS Lett 540: 229-233. doi:10.1016/S0014-5793(03)00266-7. PubMed: 12681513. [DOI] [PubMed] [Google Scholar]
- Strøm MB, Rekdal O, Svendsen JS (2002) Antimicrobial activity of short arginine- and tryptophan-rich peptides. J Pept Sci 8: 431-437. doi:10.1002/psc.398. PubMed: 12212806. [DOI] [PubMed] [Google Scholar]
- Park Y, Lee DG, Jang SH, Woo ER, Jeong HG et al. (2003) A Leu-Lys-rich antimicrobial peptide: activity and mechanism. Biochim Biophys Acta 1645: 172-182. doi:10.1016/S1570-9639(02)00541-1. PubMed: 12573247. [DOI] [PubMed] [Google Scholar]
- Fimland G, Eijsink VG, Nissen-Meyer J (2002) Mutational analysis of the role of tryptophan residues in an antimicrobial peptide. Biochemistry 41: 9508-9515. doi:10.1021/bi025856q. PubMed: 12135373. [DOI] [PubMed] [Google Scholar]
- Yau WM, Wimley WC, Gawrisch K, White SH (1998) The preference of tryptophan for membrane interfaces. Biochemistry 37: 14713-14718. doi:10.1021/bi980809c. PubMed: 9778346. [DOI] [PubMed] [Google Scholar]
- Asthana N, Yadav SP, Ghosh JK (2004) Dissection of antibacterial and toxic activity of melittin: a leucine zipper motif plays a crucial role in determining its hemolytic activity but not antibacterial activity. J Biol Chem 279: 55042-55050. doi:10.1074/jbc.M408881200. PubMed: 15475354. [DOI] [PubMed] [Google Scholar]
- Kondejewski LH, Jelokhani-Niaraki M, Farmer SW, Lix B, Kay CM et al. (1999) Dissociation of antimicrobial and hemolytic activities in cyclic peptide diastereomers by systematic alterations in amphipathicity. J Biol Chem 7: 13181-13192. PubMed: 10224074. [DOI] [PubMed] [Google Scholar]
- Wang Y, Agerberth B, Löthgren A, Almstedt A, Johansson J (1998) Apolipoprotein A-I binds and inhibits the human antibacterial/cytotoxic peptide LL-37. J Biol Chem 273: 33115-33118. doi:10.1074/jbc.273.50.33115. PubMed: 9837875. [DOI] [PubMed] [Google Scholar]
- Moustafa M, Szabo M, Ghanem GE, Morandini R, Kemp EH, MacNeil S, Haycock JW. Inhibition of tumor necrosis factor-α stimulated NFκB/p65 in human keratinocytes by α-melanocyte stimulating hormone and adrenocorticotropic hormone peptides. J Invest Dermatol. 2002;119:1244–1253. doi: 10.1046/j.1523-1747.2002.19602.x. [DOI] [PubMed] [Google Scholar]
- Brzoska T, Luger TA, Maaser C, Abels C, Böhm M. Alpha-melanocyte-stimulating hormone and related tripeptides: biochemistry, antiinflammatory and protective effects in vitro and in vivo, and future perspectives for the treatment of immune-mediated inflammatory diseases. Endocr Rev. 2008;29:581–602. doi: 10.1210/er.2007-0027. [DOI] [PubMed] [Google Scholar]
- Dalmasso G, Charrier-Hisamuddin L, Thu Nguyen HT, Yan Y, Sitaraman S, Merlin D. PepT1-mediated tripeptide KPV uptake reduces intestinal inflammation. Gastroenterology. 2008;134:166–178. doi: 10.1053/j.gastro.2007.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blalock JE. Complementarity of peptides specified by ‘sense’ and ‘antisense’ strands of DNA. TIBTECH. 1991;6:140–144. doi: 10.1016/0167-7799(90)90159-u. [DOI] [PubMed] [Google Scholar]
- Siemion IZ, Cebrat M, Kluczyk A. The Problem of Amino Acid Complementarity and Antisense Peptides. Curr Prot Peptide Sci. 2004;5:507–527. doi: 10.2174/1389203043379413. [DOI] [PubMed] [Google Scholar]
- Chelsky D, Ralph R, Jonak G. Sequence requirements for synthetic peptide-mediated translocation to the nucleus. Mol Cell Biol. 1989;9:2487–2492. doi: 10.1128/mcb.9.6.2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontes MR, Teh T, Kobe B. Structural basis of recognition of monopartite and bipartite nuclear localization sequences by mammalian importin-α. J Mol Biol. 2000;297:1183–1194. doi: 10.1006/jmbi.2000.3642. [DOI] [PubMed] [Google Scholar]
- Fagerlund R, Kinnunen L, Köhler M, Julkunen I, Melén K. NF-κB is transported into the nucleus by importin-α3 and importin-α4. J Biol Chem. 2005;280:15942–15951. doi: 10.1074/jbc.M500814200. [DOI] [PubMed] [Google Scholar]
- Depping R, Steinhoff A, Schindler SG, Friedrich B, Fagerlund R, Metzen E, Hartmann E, Köhler M. Nuclear translocation of hypoxia-inducible factors (HIFs): involvement of the classical importin alpha/beta pathway. Biochim Biophys Acta. 2008;1783:394–404. doi: 10.1016/j.bbamcr.2007.12.006. [DOI] [PubMed] [Google Scholar]
- Melen K, Fagerlund R, Franke J, Kohler M, Kinnunen L, Julkunen I. Importin alpha nuclear localization signal binding sites for STAT1, STAT2, and influenza A virus nucleoprotein. J Biol Chem. 2003;278:28193–28200. doi: 10.1074/jbc.M303571200. [DOI] [PubMed] [Google Scholar]
- Cannon JG, Tatro JB, Reichlin S, Dinarello CA. α-Melanocyte stimulating hormone inhibits immunostimulatory and inflammatory actions of interleukin 1. J Immunol. 1986;137:2232–2236. [PubMed] [Google Scholar]
- Lee SN, Ryu JH, Joo JH, Choi YH, Lee HJ, Kim YJ, Kim KB, Yoon JH. α-Melanocyte-stimulating hormone Inhibits TNFα stimulated MUC5AC expression in human nasal epithelial cells. Am J Respir Cell Mol Biol. 2010 doi: 10.1165/rcmb.2009-0420OC. epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Raap U, Brzoska T, Sohl S, Path G, Emmel J, Herz U, Braun A, Luger T, Renz H. α-Melanocyte -stimulating hormone inhibits allergic airway inflammation. J Immunol. 2003;171:353–359. doi: 10.4049/jimmunol.171.1.353. [DOI] [PubMed] [Google Scholar]