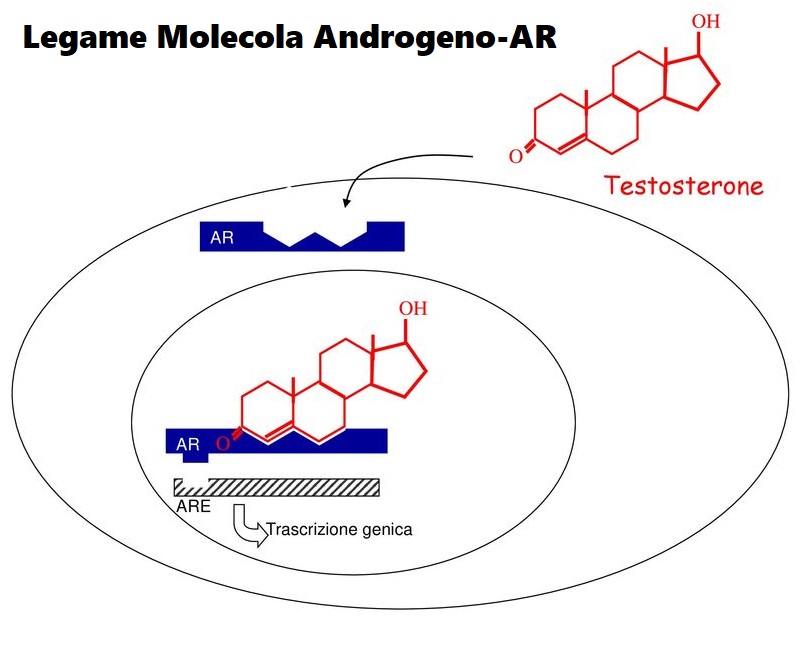
Autore: Gabriel Bellizzi [also known as Ružička, The Biochemist] - CEO BioGenTech -
Negli anni trenta del ventesimo secolo si è verificata una febbre dell’oro scientifica di proporzioni inaudite nel campo della nascente endocrinologia. Questa impresa è stata portata avanti con tanta celerità grazie al pionieristico lavoro di biochimici Adolf Friedrich Johann Butenandt e Lavoslav Stjepan Ružička, entrambi premi Nobel per la chimica nel 1939 grazie proprio alla pubblicazione dell’articolo “Sulla preparazione artificiale dell’ormone testicolare testosterone (androstene-3-one--17-olio)”.
Il potenziale del Testosterone e dei suoi primi derivati che videro la luce nella seconda metà degli anni trenta del 900, arrivo’ all’orecchio degli sportivi d’élite tanto che nel 1938 vi fu una prima pubblicazione che parlava del potenziale uso del Testosterone nel Bodybuilding.
Grazie agli abbattimenti dei costi di produzione delle molecole di sintesi, resi possibili dal genio della chimica Russell Earl Marker e dalla sua “Marker degradation”, nella seconda metà degli anni quaranta l’uso di AAS si è diffuso nelle squadre olimpiche di molti paesi. Successivamente tocco’ al pubblico amatoriale. E' nel 1976 che vi fu una nuova svolta, cioè la nascita della società di biotecnologie “Genetech” nata dall’incontro tra l’imprenditore Robert Swanson e Herbert Boyer, biochimico dell’Università della California. I due decisero di fondare questa società per lo sfruttamento commerciale delle tecniche del DNA ricombinante messe a punto da Boyer. Insulina e hGH divennero parte del corollario di farmaci utilizzati dai bodybuilder, e l’era dei “Freak” venne inaugurata.
Purtroppo, lo “scandalo DOPING” negli anni 80’, e le successive restrizioni di “facciata” hanno smantellato massivamente quella nicchia di ricercatori che lavoravano a stretto contatto con gli atleti e facevano ricerca sul campo. Essi non sono “estinti” ma sono obliati da una certa narrativa di comodo. Da qui il problema presente: l’atleta è in balia di leggende e metodiche partorite da menti non avvezze alla complessità della farmacologia partendo dalle basi della biochimica.
La BioGenTech è un laboratorio di ricerca che opera direttamente sul campo dapprima della sua fondazione grazie al lavoro del CEO Amedeo Gabriel Bellizzi. Nel 2021, ha visto la luce e ha preso concretezza un idea: fornire informazioni valide e affidabili su una scienza multidisciplinare. Nessun circo delle pulci, ma qualcosa che si può vedere e constatare.
Noi alla BioGenTech, la quale è una realtà collaborativa sebbene diretta da una mente, siamo scienziati puri con un atteggiamento snobistico nei confronti dei soli affari. Riteniamo la sola corsa al denaro una cosa da bottegai, poco stimolante dal punto di vista intellettuale. E la ricerca al servizio del commerciale, quindi resa scientismo, può andare bene solo per chi non e’ dotato di etica o è limitato nella materia.
Quindi il nostro atteggiamento nei confronti di chi e’ impegnato nello scientismo speculativo, e’ essenzialmente di critica e avversione. Il Nostro tradizionale antagonismo fa sì che non subiamo la contaminazione del marketing e, ogniqualvolta si scatena un dibattito su questioni biotecnoiogiche, non manchiamo di porci al di sopra delle parti discutendo dei problemi ai massimi livelli.
Contattaci per informazioni su coaching, anti-aging e TRT/HRT sulle piattaforme Instagram, Telegram, Whatsapp o all’indirizzo mail teamympus86@outlook.it
CEO Amedeo Gabriel Bellizzi [Biochimico, esperto in nutrizione sportiva, coach di BodyBuilding, PEDs consulter, esperto in tecniche Anti-Aging, TRT e HRT, ricercatore e divulgatore scientifico indipendente]
Continua la disamina dei principali PEDs utilizzati e del confine che delimita l’uso dall’abuso. Nel primo articolo della serie abbiamo analizzato l’Oxymetholone e come questa molecola sia soggetta a facile abuso da un numero considerevole di atleti di diverse categorie. In questo secondo articolo analizzeremo il Trenbolone, una molecola “must” nel BodyBuilding.
Caratteristiche della molecola di Trenbolone:
Il Trenbolone, noto anche come 19-nor-δ9,11-testosterone o come estra-4,9,11-trien-17β-ol-3-one, è un 19 nor- steroide derivato dal Nandrolone, e condivide con il suo precursore la modifica molecolare in C-19. Nello specifico, il Trenbolone è Nandrolone con due doppi legami aggiuntivi nel nucleo steroideo. Le differenze strutturali tra la molecola di Trenbolone e quella di Nandrolone, quindi, comprendono il doppio legame presente in C9– C10, che inibisce totalmente l’aromatizzazione e aumenta la resistenza al passaggio epatico, e quello in C11-C12 che aumenta l’affinità per il recettore androgeno, rendendo il Trenbolone uno degli anabolizzante con la più forte affinità AR.[1]
Differenze nella struttura molecolare tra Nandrolone e Trenbolone.
Gli esteri del Trenbolone, che presentano un estere in posizione C17β, includono il Trenbolone Acetato, il Trenbolone Enantato, il Trenbolone Hexahydrobenzylcarbonate e il Trenbolone Undecanoato.[2][3][4][5]
Il Trenbolone ha sia effetti anabolizzanti che androgeni.[4] Una volta metabolizzato, il Trenbolone causa un aumento dell’assorbimento di ioni ammonio da parte del tessuto muscolare, effetto che porta ad un aumento del tasso di sintesi proteica. Può anche avere effetti secondari di stimolo dell’appetito (effetto oressizzante) e di diminuzione del tasso catabolico, con quest’ultima caratteristica condivisa in diversa misura con tutti gli AAS.[6] Almeno uno studio svolto sui ratti ha dimostrato che il Trenbolone può indurre l’espressione genica del recettore degli androgeni (AR) almeno allo stesso livello del Diidrotestosterone (DHT). Questa caratteristica sta a indicare che il Trenbolone può causare un aumento delle caratteristiche sessuali secondarie maschili senza la necessità che esso converta in un androgeno più potente.[7]
Gli studi sul metabolismo del Trenbolone sono quanto meno eterogenei, con alcuni studi che mostrano che è metabolizzato dall’enzima Aromatasi o 5α-reduttasi in composti estrogenici e androgenici, nonostante le sue caratteristiche escludano questa potenzialità.[8][9]
Il Trenbolone si lega con alta affinità anche al recettore del Progesterone,[4][10][11][12] e al recettore dei glucocorticoidi.[11]
Per prolungare la sua emivita, il Trenbolone viene somministrato sotto forma di molecola coniugata ad un estere come i precedentemente citati Trenbolone Acetato, Trenbolone Enantato o Trenbolone Hexahydrobenzylcarbonate.[2][3][13][4] Le Lipasi plasmatiche poi scindono il gruppo estere nel flusso sanguigno rendendo libero, e quindi attivo, il Trenbolone.
Dosaggio di Trenbolone per il bestiame Vs dosaggio per esseri umani – :
Il Trenbolone Acetato è stato ed è utilizzato in medicina veterinaria nel bestiame per aumentarne la crescita muscolare e l’appetito, mentre Il Trenbolone Hexahydrobenzylcarbonate in passato è stato utilizzato clinicamente negli esseri umani, ma dalla fine degli anni 90 non è più commercializzato legalmente per tale scopo.[2][3][13][4]
Se si dà un’occhiata alla pagina ufficiale del sito che presenta il prodotto veterinario Finaplix-H, che consiste in pellet da impianto contenenti Trenbolone Acetato e utilizzati ancora oggi nel bestiame, si può notare come il dosaggio somministrato ad una mucca per indurne un aumento di peso è pari a 200mg. Avete capito bene, sono 200mg per capo di bestiame! Ok, sono bovini, hanno caratteristiche diverse tra metabolismo di escrezione, sensibilità recettoriale e risposta genica, ma già questo punto dovrebbe farvi cominciare a riflettere.
E’ altresì utile sottolineare che la somministrazione dei 10 pellet da 200mg di Trenbolone Acetato l’uno avviene per ogni singolo capo di bestiame nel giro di 63 giorni. Ciò significa un totale di 2g di Trenbolone Acetato (1.740mg di Trenbolone effettivo) per capo di bestiame nelle ultime 9 settimane circa prima della macellazione.
E l’uso clinico del Trenbolone negli esseri umani?
Come alcuni di voi già sapranno, nonostante la mancanza di prove cliniche su esseri umani, negli anni 70, in Francia, venne immesso sul mercato farmaceutico il Trenbolone per uso umano, sotto il nome commerciale di Parabolan (Trenbolone Hexahydrobenzylcarbonate), prodotto dalla Negma Laboratories.
Il Parabolan era usato clinicamente come agente anabolizzante per il risparmio proteico in caso di cachessia (deperimento della massa magra) e nella malnutrizione, oltre che per combattere alcune forme di osteoporosi. Le sue linee guida di prescrizione includevano raccomandazioni per il trattamento delle popolazioni androgeno-sensibile, come le donne e gli anziani. Grazie alle sue marcate proprietà androgene, tuttavia, il farmaco è stato controindicato per l’uso nei bambini, e soprattutto nelle giovani donne. Il Parabolan è rimasto sul mercato francese per un tempo molto lungo, anche se è stato interrotto (volontariamente) dalla Negma nel 1997. Da allora, nessun altro preparato a base di Trenbolone è stato approvato per uso umano.
Il Parabolan era generalmente somministrato ad un dosaggio clinico pari a 3 fiale al mese (228mg totali). La terapia veniva avviata il primo mese con tutte e 3 fiale somministrate nel corso dei primi 15 giorni. Durante i successivi 3 mesi, veniva somministrata una iniezione (76 mg) ogni 10 giorni. 76mg di Trenbolone Hexahydrobenzylcarbonate equivalgono a circa 50mg di Trenbolone attivo dopo la scissione del legame molecolare con l’estere.
Il protocollo di dosaggio clinico di Trenbolone negli esseri umani con utilizzo del Parabolan (Negma Laboratories) era il seguente:
114mg Trenbolone Hexahydrobenzylcarbonate (75 mg di ormone attivo) a settimana per le prime due settimane (totale 228mg di Trenbolone Hexahydrobenzylcarbonate);
Successivamente 76mg di Trenbolone Hexahydrobenzylcarbonate (50mg di ormone attivo) ogni 10 giorni.
Si sta ovviamente parlando di dosaggi basati su protocolli clinici, ma sono pienamente considerabili dosaggi più che efficaci specie per un atleta al suo primo utilizzo della suddetta molecola legata al suddetto estere. Parliamo comunque di 75mg di Trenbolone effettivo a settimana. Paragonandolo poi al dosaggio utilizzato per il bestiame (200mg di Trenbolone Acetato equivalente a 174mg di Trenbolone effettivo), si capisce che l’uso di dosaggi nell’ordine di 400-600mg a settimana di Trenbolone Hexahydrobenzylcarbonate (circa 300-400mg di Trenbolone effettivo) o 700mg di Trenbolone Acetato (609mg di Trenbolone effettivo) siano palesemente e a tutti gli effetti un abuso della molecola. Tralasciando certe abitudini yankee che consistono nella somministrazione massiva di Trenbolone a dosaggi fino ad 1g a settimana! Certo, 1g legato all’estere, ma se fate un po’ i conti l’abuso persiste indipendentemente dall’estere, con una variabile discreta tra estere Acetato e estere Enantato o Hexahydrobenzylcarbonate.
Per quanto possa urtare la convinzione di alcuni, usare il Trenbolone a dosaggi più alti di quelli somministrati ad un bovino di 720-1100Kg non è esattamente una pratica sostenibile.
Ad oggi non abbiamo informazioni certe sull’effetto che questo farmaco possa avere negli esseri umani, quindi sarebbe saggio evitarne quantomeno l’abuso. E questo è un dato di fatto.
La questione degli effetti collaterali a carico della sfera cognitiva:
Chi mi segue e legge i miei lavori conoscerà la mia posizioni in merito al Nandrolone e allo sbilanciamento tra “benefici e rischi”, a favore di questi ultimi, dati dal suo utilizzo, in specie a scopi dopanti. Infatti, l’uso del Nandrolone ha un significativo impatto sul SNC, con particolari differenze nel grado di manifestazione rispetto ad altri AAS non progestinici. Questo effetto del Nandrolone sul Sistema Nervoso Centrale è stato osservato scientificamente. Nello studio intitolato “The Impact of Nandrolone Decanoate on the Central Nervous System” vengono descritti chiaramente i numerosi effetti psicologici di questa molecola. Essi comprendono e influenzano:
1- Aggressività 2- Ansia, paura e stress 3- Ricompensa e dipendenza 4- Apprendimento, memoria e capacità di lavoro 5- Locomozione e attività fisica 6- Effetti sulla HPAA (Asse Ipotalamo-Pituitaria-Surrene) 7- Effetto sui neurotrasmettitori: Recettore Acido γ-Aminobutirrico Tipo A (GABAA); Recettori 5-idrossitriptamina (5-HT) e 5-HT; Recettori della Dopamina e Recettori Oppioidi.
Questi effetti sono potenzialmente riconducibili a tutti i progestinici, compreso il Trenbolone. Sebbene vi siano differenze sia nel grado di manifestazione dell’influenza che nel rapporto tra “vantaggi e svantaggi”, senza dubbio a favore del Trenbolone, questi aspetti neurologici non dovrebbero essere comunque tralasciati.
I neuroni nel cervello sono in grado di conservare le informazioni e di elaborarle perché creano continuamente nuove connessioni tra loro. La Proteina precorritrice della beta-amiloide [APP] svolge un ruolo importante in questo processo. Per essere precisi: la APP è una proteina di trans-membrana di tipo 1.
Proteina Precorritrice della Beta-Amiloide [APP]
Gli enzimi scompongono la APP in pezzi e se questo processo si svolge correttamente le cellule cerebrali funzionano in modo normale. Ma se gli enzimi iniziano a non agire come dovrebbero – a causa di geni difettosi o fattori ambientali pericolosi – si formano frammenti proteici tossici. Il più rischioso di questi è l’amiloide-beta-42, che si accumula nel cervello, formando placche e uccidendo infine i neuroni. Il cervello delle persone decedute a causa dell’Alzheimer contengono grandi quantità di amiloidi-beta-42, per cui la maggior parte dei neurologi pensa che l’amiloide-beta-42 sia la causa dell’Alzheimer e di forme legate alla demenza.
Il Testosterone, l’Estradiolo e il DHT offrono protezione contro l’Alzheimer, con influenza incrociata e interdipendente. Ecco perché Ma e Lui si sono chiesti quale effetto avrebbe avuto il Trenbolone sulla formazione di amiloide-beta-42. Così, hanno somministrato a dei topi 5 iniezioni di Trenbolone in un periodo di 48 ore. L’equivalente umano delle dosi utilizzate dai ricercatori sarebbe di circa 0,85mg per kg di peso corporeo.
L’amiloide-beta-42 si è accumulata nel cervello dei ratti maschi trattati con Trenbolone. Il grafico seguente mostra i risultati emersi nel periodo di 48 ore nel quale sono state distribuite le 5 iniezioni di Trenbolone.
Il grafico qui sopra (barre grigie), riporta i dati estrapolati dagli esperimenti in vitro che i ricercatori hanno inoltre svolto con le cellule cerebrali esponendole per 48 ore a 100 nanomoli di Trenbolone [TB]. L’aggiunta di un antiandrogeno come la Flutamina [Flu] ha ridotto l’accumulo di amiloide-beta-42. Così è emerso che i danni cerebrali causati dal Trenbolone sono dovuti agli effetti androgeni di questa molecola.
Una combinazione di Trenbolone e DHT ha mostrato di aumentare l’accumulo di amiloide-beta-42.
In definitiva, per quanto emerso dallo studio, dal momento che i danni ai neuroni possono verificarsi molto prima dei sintomi clinici dei disturbi neurodegenerativi, l’esposizione al Trenbolone dovrebbe essere considerata come un fattore ambientale ad alto rischio per lo sviluppo della malattia di Alzheimer. Ciò nonostante, si parla ancora di ipotesi le quali dovrebbero essere accertate ed avvalorate da ulteriori ricerche.
Siamo più o meno tutti consapevoli del fatto che l’aumento dell’aggressività riscontrabile in diversi bodybuilder “doped” sia una costante con variabili di intensità tra soggetto e soggetto. Ma è piuttosto facile osservare maggiori problemi di instabilità mentale negli utilizzatori di Trenbolone rispetto ad altri AAS.
Generalmente, se un soggetto ha una personalità già di base aggressiva, con l’aumento degli androgeni circolanti tale caratteristica subisce una significativa esacerbazione.
Non stupisce, quindi, che i modelli animali trattati con Trenbolone mostrino effettivamente un degrado cognitivo.
Conclusioni sul Trenbolone:
Grazie ai molti dati enpirici ed aneddotici da me raccolti negli anni riguardo al Trenbolone e al suo utilizzo da parte dei BodyBuilder e PowerLifter, vi esorto innanzi tutto ad essere molto cauti con un suo possibile uso, soprattutto quando si è consapevoli del fatto che i dati clinici a riguardo sono scarsi e basati su modelli animali.
Nonostante alcuni di voi saranno sicuramente sorpresi del fatto che il Trenbolone non è un farmaco che ha avuto studi sugli esseri umani, la verità è questa.
Ci sono molte persone convinte del fatto che, dato l’utilizzo diffuso e annoso nella comunità del Bodybuilding di questa molecola, e dato che è stato precedentemente prescritto clinicamente per il trattamento di malattie degenerative muscolo-scheletriche nell’uomo, il Trenbolone possa essere somministrato con una certa sicurezza anche a dosaggi elevati. Peccato però che di “dati di sicurezza” nell’uso umano non vi sia la minima traccia. Ci si basa solo e soltanto su dati empirici e aneddotici!
Paradossalmente, esistono più dati clinici su esseri umani di molecole relativamente recenti rispetto al Trenbolone che, tanto per delucidazioni temporali, è stato sintetizzato per la prima volta nel 1963.
Qualora una persona decidesse di testare il Trenbolone dovrebbe tenere conto di alcuni punti “conservativi”:
Non superare un dosaggio iniziale pari a 76-100mg di Trenbolone Hexahydrobenzylcarbonato o di 100mg di Trenbolone Enantato a settimana. Una dose massima iniziale può essere pari a 150mg di Trenbolone Acetato a settimana;
Successivamente, calibrare il dosaggio attraverso la formula 2mg/Kg (Trenbolone/Kg di peso corporeo) a settimana (es. 90Kg = 180mg);
Ridurre l’uso della molecola a massimo 1-2 volte l’anno.
ATTENZIONE! Non si tratta di consigli ma di divulgazione preventiva! Non per nulla, questi articoli nascono come mezzo per evitare l’abuso sconsiderato di PEDs!
Continuare ad usare dosaggi molto al di sopra del margine utile e conservativo non porterà a nessun reale vantaggio. E ciò non interessa soltanto gli individui con una genetica nella media, in parte convinti che basti usare un dosaggio maggiore e crescente per essere quello che non si è, ma anche e soprattutto i tendenti “freak”, soggetti che come la mal erba crescono con dosaggi tutt’altro che facenti parte della posologia dei doped “millennial”.
Per il resto, ad ognuno la propria riflessione in merito e il dialogo con la propria coscienza.
Yarrow, Joshua F.; McCoy, Sean C.; Borst, Stephen E. (2010). “Tissue selectivity and potential clinical applications of trenbolone (17β-hydroxyestra-4,9,11-trien-3-one): A potent anabolic steroid with reduced androgenic and estrogenic activity”. Steroids. 75 (6): 377–89. doi:10.1016/j.steroids.2010.01.019. PMID20138077. S2CID205253265.
Gettys, TW; d’Occhio, MJ; Henricks, DM; Schanbacher, BD (1984). “Suppression of LH secretion by oestradiol, dihydrotestosterone and trenbolone acetate in the acutely castrated bull”. The Journal of Endocrinology. 100 (1): 107–12. doi:10.1677/joe.0.1000107. PMID6361192.
È noto ai più che la Creatina porta a ritenzione idrica intracellulare. Ma vi sono alcuni che si pongono una domanda interessante e più completa: come è realmente distribuita quest’acqua? C’è un totale indirizzamento e accumulo intracellulare o vi sono percentuali di accumulo differenti tra l’interno (intracellulare) e l’esterno (extracellulare) del miocita (cellula muscolare)?
La Creatina è prevalentemente immagazzinata nelle cellule muscolari ed è una sostanza osmoticamente attiva. Quindi aumenta l’osmolalità delle cellule. Di conseguenza, le cellule attireranno acqua per osmosi. Logicamente, sembra molto ragionevole supporre che l’acqua sia immagazzinata insieme alla Creatina (depositi di Fosfocreatina e Creatina libera) all’interno delle cellule muscolari.
Alcune ricerche hanno verificato la logicità di tale ragionamento. Dopo tutto, è necessario testare le proprie ipotesi per avvalorarle. Ci sono diverse tecniche che possono essere utilizzate per mettere alla prova questo ragionamento logico. In letteratura troverete le seguenti quattro tecniche utilizzate:
Analisi di bioimpedenza a multifrequenza (MBIA)
Risonanza magnetica (MR)
Analisi di diluizione isotopica (IDA)
Assorbimetria a raggi X a doppia energia (DEXA)
Quindi è utile indagare attraverso la letteratura scientifica e vedere cosa hanno da dire queste tecniche di analisi sulla questione della compartimentalizzazione dell’acqua legata all’assunzione di Creatina.
Analisi di bioimpedenza a multifrequenza (MBIA): la Creatina causa ritenzione idrica intracellulare.
Come ben sappiamo, il corpo umano è composto da vari tipi di tessuti, come il tessuto adiposo, il tessuto osseo, il tessuto muscolare, ecc. Questi tessuti differiscono notevolmente nella quantità di fluido che contengono e nella loro conducibilità elettrica. La MBIA si basa su quest’ultimo principio, le differenze di conduttività.
In breve, piccole scosse elettriche con frequenze variabili sono erogate attraverso il corpo. Si suppone che si possa stimare il volume del fluido extracellulare con la bassa frequenza e il volume totale del fluido con l’alta frequenza. Sottraendo il volume extracellulare dal volume totale si ottiene il volume del fluido intracellulare. Ci sono però molti inconvenienti con questo metodo, ma non li tratterò qui al fine di evitare di discostarmi eccessivamente dai concetti essenziali che questo articolo si pone di esporre.
Il MBIA è stato utilizzato da Ziegenfuss et al. [1]. Ai soggetti sono stati somministrati 0,07g di Creatina per kg di massa grassa per tre giorni. Per un individuo di 70kg con il 15% di grasso corporeo questo ammonterebbe a 21g al giorno. Quindi, in pratica, i ricercatori hanno somministrato ai soggetti dello studio un dosaggio di carico di Creatina consuetudinario. E’ stato quindi osservato un aumento non statisticamente significativo (P=0,07) del fluido corporeo totale del 2%. Il volume del fluido intracellulare è aumentato significativamente del 3% e il volume del fluido extracellulare è rimasto invariato. Questi risultati suggeriscono che la ritenzione idrica legata all’assunzione di Creatina è effettivamente limitata al compartimento del fluido intracellulare.
Risonanza magnetica (MR): la Creatina trattiene l’acqua all’interno delle cellule.
Le MBIA stimano il contenuto di acqua di tutto il corpo. Le tecniche di risonanza magnetica permettono al ricercatore di controllare una particolare area del corpo. La MR sfrutta il fatto che i protoni hanno un momento di dipolo magnetico. Questi momenti di dipolo dei protoni si allineano con un campo magnetico quando questo è presente. Tale allineamento può avvenire nella direzione del campo magnetico (spin up) o contro di esso (spin down). Quando si rilascia un impulso di radiofrequenza su questi protoni, questi momenti di dipolo cambieranno direzione. Quando l’impulso di radiofrequenza non c’è più, ricadono nella loro vecchia direzione (quella del campo magnetico). Questa “ricaduta” richiede un certo tempo e questo varia a seconda del tessuto. Questi tempi sono anche chiamati tempi di rilassamento. Si presume che questi tempi di rilassamento differiscano tra il compartimento extracellulare e intracellulare. Di conseguenza, le misure di questi tempi di rilassamento possono fornire informazioni sui volumi dei compartimenti extracellulare e intracellulare.
Questa tecnica è stata utilizzata da Saab et al. [2]. I ricercatori hanno somministrato ai soggetti 20g di creatina al giorno per 5 giorni e hanno esaminato il muscolo flexor digitorum profundus (un muscolo dell’avambraccio). Gli autori hanno trovato un aumento della concentrazione di protoni che corrisponde al tempo di rilassamento che si adatta al compartimento intracellulare, ma non a quello extracellulare. Come tale, gli autori concludono che l’integrazione di Creatina trattiene l’acqua all’interno delle cellule.
Analisi di diluizione isotopica (IDA): la Creatina causa ritenzione idrica intra ed extracellulare.
Gli isotopi di un particolare elemento hanno lo stesso numero di protoni ma differiscono nel loro numero di neutroni. Di conseguenza, si comportano fisiologicamente allo stesso modo, ma è possibile distinguerli nelle misurazioni a causa del diverso numero di neutroni. Che cosa ha a che fare questo con le misurazioni dei fluidi corporei? Per esempio, prendiamo un isotopo dell’idrogeno: il deuterio. Quando due molecole di deuterio si combinano con l’ossigeno, si ottiene acqua pesante (ossido di deuterio). Se bevi un bicchiere di acqua pesante, sarai in grado di distinguere queste molecole di acqua pesante dalle molecole di acqua normale. Si lascia bere e si aspetta da 2 a 6 ore che si diffonda uniformemente in tutto il corpo. Poi si preleva del sangue e se ne misura la concentrazione. Poi si determina la concentrazione di acqua pesante con un contatore a scintillazione, e voilà. Con un po’ di calcoli si conosce la quantità totale di acqua nel corpo!
Si può fare la stessa cosa con un isotopo diverso che non può entrare nelle cellule, quindi si può calcolare il volume di acqua extracellulare. Uno di questi isotopi è il bromuro di sodio. E poi, sottraendo il volume d’acqua extracellulare dal volume d’acqua totale, conoscerete il volume d’acqua intracellulare.
Questo è un metodo molto affidabile ed è stato applicato da Powers et al. per studiare la ritenzione idrica della creatina [3]. I risultati sono stati in qualche modo inaspettati. Hanno misurato un aumento del fluido corporeo totale 7 e 28 giorni dopo l’assunzione di creatina. Non è una sorpresa. Tuttavia, non hanno misurato alcuna alterazione nella distribuzione dei fluidi. Il che significa che entrambi i compartimenti del fluido extracellulare e intracellulare sono aumentati in proporzione l’uno all’altro.
Questo sembra in contrasto con i due studi precedenti che ho citato sopra. Una notevole differenza con lo studio MR è che quello ha guardato localmente solo un singolo muscolo. IDA misura la distribuzione dei fluidi di tutto il corpo, proprio come MBIA. MBIA d’altra parte è sicuramente meno affidabile della IDA.
Assorbimetria a raggi X a doppia energia (DEXA) e Bioimpedenza bioelettrica spettrale: la Creatina non causa alterazioni della ritenzione idrica extracellulare.
L’assorbimetria a raggi X a doppia energia (DXA, o DEXA) è un mezzo per misurare la densità minerale ossea (BMD) utilizzando l’imaging spettrale. Due fasci di raggi X, con diversi livelli di energia, sono puntati sulle ossa del paziente. Quando l’assorbimento dei tessuti molli viene sottratto, la densità minerale ossea (BMD) può essere determinata dall’assorbimento di ogni raggio da parte dell’osso. L’assorbimetria a raggi X a doppia energia è la tecnologia di misurazione della densità ossea più utilizzata e più studiata.
Le scansioni DEXA possono anche essere utilizzate per misurare la composizione corporea totale e il contenuto di grasso con un alto grado di precisione paragonabile alla pesata idrostatica, con alcune importanti avvertenze.[4] Dalle scansioni DEXA, si può anche generare un’immagine a bassa risoluzione “fat shadow”, che dà un’impressione generale della distribuzione del grasso in tutto il corpo.[5] È stato suggerito che, pur misurando molto accuratamente i minerali e i tessuti molli magri (LST), la DEXA può fornire risultati distorti a causa del suo metodo di calcolo indiretto della massa grassa sottraendola dalla LST e/o dalla massa cellulare corporea (BCM) che la DEXA effettivamente misura.[6]
Tuttavia, le scansioni DEXA sono state suggerite come strumenti utili per diagnosticare condizioni con una distribuzione anomala del grasso, come la lipodistrofia parziale familiare.[7][8][5] Sono anche utilizzate per valutare l’adiposità nei bambini, soprattutto per condurre ricerche cliniche.[9] In definitiva, sufficientemente affidabile per chiarire la questione qui trattata.
Nello studio di Alex S Ribeiro et al.[10]gli autori hanno voluto confrontare gli effetti dell’integrazione di Creatina (Cr) combinata con l’allenamento contro-resistenza sulla massa muscolo-scheletrica (SMM), l’acqua corporea totale, l’acqua intracellulare (ICW) e l’acqua extracellulare (ECW) in uomini allenati contro-resistenza, nonché determinare se il rapporto SMM/ICW cambia in risposta all’uso di questo supplemento ergogenico. Ventisette uomini allenati contro-resistenza hanno ricevuto Cr (n = 14) o placebo (n = 13) per 8 settimane. Durante lo stesso periodo, i soggetti hanno eseguito due routine di allenamento contro-resistenza divise in quattro sedute a settimana. L’SMM è stato stimato dal tessuto molle magro appendicolare valutato mediante assorbimetria a raggi X a doppia energia (DEXA). L’acqua corporea totale, ICW e ECW sono stati determinati dall’impedenza bioelettrica spettrale. Entrambi i gruppi hanno mostrato miglioramenti (p < .05) nella SMM, acqua corporea totale e ICW, con valori maggiori osservati per il gruppo Cr rispetto al placebo. L’ECW è aumentata in modo simile in entrambi i gruppi (p < .05). Il rapporto SMM/ICW non è cambiato in nessuno dei due gruppi (p > .05), mentre il rapporto SMM/ECW è diminuito solo nel gruppo Cr (p < .05). È stata osservata una correlazione positiva (p < .05) tra i cambiamenti SMM e ICW (r = .71). I risultati degli autori suggeriscono che l’aumento della massa muscolare indotto dalla Cr combinato con l’allenamento contro-resistenza avviene senza alterazione del rapporto tra ICW e SMM negli uomini allenati con la resistenza.
Conclusioni:
Che l’assunzione di Creatina porta alla ritenzione di liquidi è stato suggerito dall’intero corpo documentale riportato. La logica impone che il fluido sia contenuto all’interno delle cellule (muscolari) e non all’esterno delle cellule (extracellulare). Le misurazioni MBIA, MR e DEXA supportano questo punto di vista. Tuttavia, l’IDA, che potrebbe essere considerata una misura standard per la distribuzione dei fluidi, suggerisce che la ritenzione di fluidi avviene proporzionalmente sia nel compartimento extracellulare che in quello intracellulare. Non sembra esserci una buona spiegazione per questo, tranne quanto onestamente riportato dagli stessi ricercatori.
Infatti, essi affermano che l’assunzione di calorie e fluidi dei soggetti in esame non è stata registrata , e le differenze in uno di questi fattori potrebbero aver influenzato i cambiamenti nella massa corporea e nel bilancio dei fluidi. Inoltre, ogni soggetto era coinvolto in un programma di allenamento contro-resistenza individualizzato. Questi protocolli di allenamento non sono stati controllati; tuttavia, il volume di ogni sessione di allenamento è stato registrato. A causa della grande variabilità tra i soggetti, non è stato possibile stabilire una relazione tra il volume di allenamento e i cambiamenti della massa corporea e dell’equilibrio dei fluidi. Inoltre, i protocolli di allenamento prima dell’integrazione non sono stati registrati, quindi non è stato possibile fare confronti. Pertanto, è possibile che le differenze nel volume di allenamento abbiano anche influenzato i cambiamenti nella massa corporea e nell’equilibrio dei fluidi.
Ora, anche alla luce dei dati raccolti negli anni e provenienti da preparazioni alla gara di atleti di diverso livello ma sempre nell’ambito culturistico, sono propenso a seguire quanto riportato dai risultati delle misurazioni MBIA, MR e DEXA. Ulteriori ricerche potrebbero sicuramente chiarire questa discrepanza. Ma, ritornando sul discorso empirico/aneddotico, non mi sono mai imbattuto in condizioni di alterata ritenzione extracellulare in atleti supplementati in cronico con dosi pari a 3-5g/die di Creatina quando tutte le variabili di incidenza maggioritaria (vedi, ad esempio, regolare assunzione di liquidi, elettroliti e corretto bilancio acqua:sodio) erano sotto controllo, soprattutto in condizioni di ipocalorica. Alcuni ipotizzano che dosi elevate di Creatina possano causare un certo grado di ritenzione idrica extracellulare dovuta alla saturazione cellulare. Si tratta però di pure supposizioni basate, oltretutto, su possibilità remote e dettate da soli due scenari: 1) Fase di “carico” della Creatina e 2) ignoranza del soggetto (o di chi per lui) sul range di dose efficace del supplemento; in questo caso potrebbero con tutta probabilità aggiungersi altre variabili che influenzano la ritenzione idrica e che sono indipendenti dalla supplementazione di Creatina.
Gabriel Bellizzi
Riferimenti:
Ziegenfuss, Tim N., Lonnie M. Lowery, and Peter WR Lemon. “Acute fluid volume changes in men during three days of creatine supplementation.” J Exerc Physiol 1.3 (1998): 1-9. APA
Saab, George, et al. “Changes in Human Muscle Transverse Relaxation Following Short‐Term Creatine Supplementation.” Experimental physiology 87.3 (2002): 383-389.
Powers, Michael E., et al. “Creatine supplementation increases total body water without altering fluid distribution.” Journal of athletic training 38.1 (2003): 44.
La capacità di riacquisire la condizione della massa muscolare precedente a un periodo di deallenamento o inattività fisica è noto come “memoria muscolare”. Quindi, se un soggetto ha avuto una condizione muscolare ottimale (vedi muscoli più ipertrofici) in passato, ciò lo aiuterà a riportarli nuovamente nelle precedenti condizioni una volta ripreso un regolare stimolo allenante. Il concetto di memoria muscolare si basa in buona parte su qualcosa chiamato permanenza mio-nucleare. Il ‘mio’ in ‘mionucleare’ si riferisce al ‘muscolo’ e il ‘nucleare’ si riferisce alla parola ‘nucleo’: un organello della cellula. Prima di esplorare ulteriormente il concetto di memoria muscolare, e come gli AAS si leghino a questo, cerchiamo prima di rispolverare un po’ di concetti utili sui nuclei muscolari o mionuclei.
Informazioni di base sui nuclei muscolari/mionuclei:
Le cellule muscolo-scheletriche sono le singole cellule contrattili all’interno di un muscolo e sono spesso definite fibre muscolari.[1] Un singolo muscolo come il bicipite in un giovane individuo di sesso maschile adulto contiene circa 253.000 fibre muscolari.[2]
Sezione 3D di una fibra del muscolo-scheletrico
Le fibre muscolo-scheletriche sono le uniche cellule muscolari multinucleate con i nuclei spesso indicati come mionuclei . Ciò si verifica durante la miogenesi con la fusione di mioblasti, ciascuno dei quali contribuisce a un nucleo.[3] La fusione dipende da proteine muscolo-specifiche note come fusogeni chiamate myomaker e myomerger .[4]
Molti nuclei sono necessari alla cellula muscolo-scheletrica per le grandi quantità di proteine ed enzimi necessari per essere prodotti per il normale funzionamento della cellula. Una singola fibra muscolare può contenere da centinaia a migliaia di nuclei.[5] Una fibra muscolare ad esempio nel bicipite umano con una lunghezza di 10cm può avere fino a 3000 nuclei.[5] A differenza di una cellula non muscolare in cui il nucleo è posizionato centralmente, il mionucleo è allungato e si trova vicino al sarcolemma . I mionuclei sono disposti in modo abbastanza uniforme lungo la fibra con ciascun nucleo che ha il proprio dominio mionucleare dove è responsabile del supporto del volume del citoplasma in quella particolare sezione della miofibra.[4,5]
Un gruppo di cellule staminali muscolari conosciute come cellule miosatelliti, anche cellule satelliti che si trovano tra la membrana basale e il sarcolemma delle fibre muscolari, sono normalmente quiescenti ma possono essere attivate dall’esercizio o anche condizioni patologiche per fornire mionuclei aggiuntivi per la crescita o la riparazione muscolare.[6]
Detto più semplicemente, i muscoli sono costituiti da un insieme di fibre muscolari. Ogni fibra muscolare, o cellula muscolare, contiene più nuclei, l’organello di una cellula che contiene il DNA ed è il luogo dove avviene il processo di trascrizione dei geni. La maggior parte degli altri tipi di cellule umane contiene solo un nucleo, o in alcuni casi addirittura nessun nucleo (globuli rossi/Eritrociti). Per dare un’idea di quanti nuclei si stia parlando: le fibre muscolari di ratto contengono da 44 a 116 nuclei per millimetro di lunghezza della fibra, con le fibre muscolari di tipo 1 che contengono più nuclei per millimetro delle fibre muscolari di tipo 2.[7] Il numero sembra più basso negli esseri umani, come riportato da un ricercatore il quale segnala la presenza di circa 30 nuclei per millimetro di lunghezza della fibra nel muscolo del bicipite brachiale.[8] Come tali, le fibre muscolari possono contenere migliaia di mionuclei, dato che possono estendersi per diversi centimetri di lunghezza.
Poiché i nuclei cellulari delle fibre muscolari non sono in grado di dividersi (cioè sono differenziati terminalmente), le fibre muscolari dipendono dalle cellule satelliti circostanti per l’aggiunta di nuovi nuclei. Essenzialmente, le cellule satelliti sono cellule staminali delle fibre muscolari che si trovano schiacciate tra il sarcolemma (la membrana cellulare di una fibra muscolare) e la lamina basale (uno strato di matrice extracellulare che è avvolto intorno al sarcolemma). Sono stati scoperti e descritti per la prima volta da Alexander Mauro nella letteratura scientifica nel 1961.[9] Usando un microscopio elettronico, egli vide delle cellule “incastrate” tra il sarcolemma delle fibre muscolari di rana e la lamina basale. Le descrisse aventi una scarsità di citoplasma, con il nucleo che costituisce quasi l’intero volume della cellula satellite. Ha continuato a speculare sull’origine e sul ruolo delle cellule satelliti, toccando brevemente l’idea che potrebbero essere coinvolte nella risposta al trauma inflitto a una fibra muscolare. Cosa che, in effetti, sono.[10]
L’ipotesi del dominio mionucleare e la permanenza mionucleare
La scoperta delle cellule satelliti e il loro ruolo nella rigenerazione muscolare fanno sorgere la domanda sulla misura in cui le cellule satelliti sono coinvolte nell’ipertrofia. Un’ipotesi chiamata “ipotesi del dominio mionucleare” si è agganciata a questo quesito. Essa postula che un mionucleo controlla una quantità limitata di citoplasma, e quindi, affinché la crescita muscolare abbia luogo, i mionuclei devono essere aggiunti alla fibra muscolare per sostenerla. Tre osservazioni chiave hanno sostenuto questa ipotesi, vale a dire:
L’esposizione alle radiazioni γ rende le cellule satellite incapaci di dividersi e inibisce fortemente l’ipertrofia da sovraccarico nei modelli animali, mantenendo intatto il metabolismo cellulare o la sintesi proteica [11].
I prodotti (organelli, membrane e proteine strutturali) derivati da un nucleo rimangono localizzati nelle sue vicinanze [12].
Il rapporto citoplasma/mionucleo rimane abbastanza costante [13].
Questo implicherebbe un aumento del numero di mionuclei con la crescita di una fibra muscolare (ipertrofia), mentre diminuirebbe con una perdita di dimensioni della stessa (atrofia). Tuttavia, vari studi su animali suggeriscono che i mionuclei non si perdono durante l’atrofia.[14] Così è nato il paradigma della permanenza mionucleare: una volta che i mionuclei sono guadagnati con l’ipertrofia, non vengono persi di nuovo con il deallenamento. Questo potrebbe potenzialmente permettere alle fibre muscolari di ricrescere in modo più efficiente durante il successivo riallenamento e quindi servire come un meccanismo di “memoria muscolare”.
Il concetto di memoria muscolare basato sulla permanenza mionucleare illustrato da Bruusgaard et al.
AAS e permanenza mionucleare:
E gli AAS? Ciò che è chiaro è che l’uso di AAS aumenta il numero di mionuclei. Dosaggi crescenti di Testosterone Enantato portano ad un aumento del numero di mionuclei per mm di fibra muscolare.[15] Questo effetto non è poi così sorprendente: si osserva semplicemente questo effetto con praticamente tutte le modalità di induzione ipertrofica.
Ma che dire della loro permanenza? Questi mionuclei permangono una volta che la massa muscolare diminuisce di nuovo? In un esperimento su animali, da me già riportato anni fa, topi femmina sono stati trattati con Testosterone Propionato per 2 settimane, che ha portato a un aumento del 66% del numero di mionuclei e un aumento del 77% della fibra muscolare CSA [16]. La massa muscolare è tornata alla normalità dopo la successiva interruzione della somministrazione di Testosterone, ma il numero di mionuclei è rimasto elevato per almeno 3 mesi. 3 mesi potrebbe non sembrare molto, ma sulla scala temporale di un topo lo sono: i topi che hanno usato per lo studio vivono per circa 2 anni. Comunque, dopo questi 3 mesi, quando i topi sono stati sottoposti a sovraccarico per induzione ipertrofica, la CSA delle fibre muscolari è aumentata del 30% dopo 6 giorni, mentre quella dei topi di controllo non è aumentata significativamente. Dopo questo, la massa muscolare è aumentata in parallelo tra entrambi i gruppi, ma la CSA era ancora più alta del 20% nel gruppo che era stato precedentemente trattato con Testosterone dopo 14 giorni. Anche se questo non prova un nesso causale tra il numero più alto di mionuclei e l’ipertrofia, è comunque un’osservazione interessante.
Si noti come il gruppo che è stato trattato con Testosterone per 2 settimane, circa 3 mesi prima ha mostrato un forte aumento della massa muscolare rapidamente ottenuto in risposta al sovraccarico.
E negli esseri umani? Due studi hanno valutato questo e sono stati portati all’attenzione da Alexander Kolliari-Turner, uno studente con dottorato di ricerca presso la School of Sport and Health Sciences of the University of Brighton nel Regno Unito. Una è una tesi di master e l’altra è una tesi di dottorato.
Nella tesi di dottorato di Anders Eriksson [17], sono stati reclutati quattro gruppi di soggetti. Un gruppo di soggetti sedentari che fungeva da controllo (gruppo C), un gruppo di PowerLifter natural (gruppo P), un gruppo di powerlifter che usano AAS (gruppo PAS), e un gruppo di PowerLifter che hanno precedentemente usato AAS (gruppo PREV). I mionuclei per fibra muscolare sono stati determinati nei muscoli vasto laterale e trapezio. Il gruppo PREV aveva interrotto l’uso di AAS da almeno un anno (con una media di 8 anni). Infatti, l’area delle fibre muscolari misurata nel gruppo PREV era paragonabile a quella del gruppo P, e notevolmente più piccola di quella del gruppo PAS.
La distribuzione del dominio nucleare (nr. di nuclei per fibra diviso per l’area della fibra) per gruppo si trova nell’immagine qui sotto. Se ci fosse una permanenza dei mioonuclei, ci si aspetterebbe un dominio nucleare più piccolo, cioè più nuclei rispetto all’area delle fibre, nel gruppo PREV rispetto agli altri gruppi.
Chiaramente questo non è il caso del vasto laterale, ma è il caso del trapezio. È difficile dire cosa causa questa apparente discrepanza tra i due muscoli. O qualche proprietà che differisce tra i due muscoli, o il suo modo di utilizzo dopo la cessazione dell’uso di AAS, forse ha portato a apparente permanenza mionucleare nel muscolo trapezio.
Va notato, tuttavia, che questo era uno studio trasversale con un piccolo numero di soggetti (32 in totale). L’ideale sarebbe avere uno studio prospettico che valuti questo, anche se ciò è estremamente difficile su lunghi periodi di tempo, in quanto potrebbe richiedere almeno un anno o più prima che i cambiamenti diventino evidenti. In alternativa, anche uno studio trasversale con un gruppo di soggetti più grande sarebbe piuttosto interessante. Indipendentemente da ciò, questo presta una certa credibilità alla permanenza dei mionuclei negli esseri umani come risultato dell’uso di steroidi anabolizzanti in muscoli selezionati.
In una tesi di laurea di Lindholm et al. sono stati reclutati tre gruppi di soggetti: attuali consumatori di AAS (gruppo CAS), ex consumatori di AAS (gruppo FAS) e controllo allenati alla resistenza (gruppo CON) [18]. Gli ex consumatori di AAS avevano smesso di usarli per una media di 6,5 anni. In questo studio, sono state prese solo biopsie del muscolo vasto laterale. In particolare, non c’erano differenze significative nella CSA delle fibre muscolari tra i tre gruppi. Questo è senza dubbio il risultato delle dimensioni relativamente piccole del gruppo (34 soggetti in totale; un errore di tipo 2).
Una piccola, ma significativa, differenza nel dominio mio-nucleare è stata trovata tra le fibre muscolari di tipo 2 del gruppo FAS rispetto al gruppo CON, come si può vedere nella figura sottostante:
Questo suggerisce una permanenza mionucleare? Forse. La differenza era piccola e può essere facilmente spiegata anche dalla natura trasversale dello studio (e non c’era alcuna differenza rispetto agli attuali utilizzatori di AAS).
Le prove finora sono scarse. In ogni caso, quando si guarda alla permanenza mionucleare in generale, l’evidenza generale indica che questa regge a breve termine, ma mancano prove per il lungo termine [19]. Inoltre, non è chiaro se la permanenza mionucleare possa aiutare o meno il ritorno alla condizione muscolo-scheletrica precedente. E visti i dati di cui sopra, il dibattito sul fatto che l’uso di AAS porti o meno alla manifestazione della memoria muscolare come risultato della permanenza mionucleare, è tutt’altro che risolto.
Conclusione:
Come osservazione conclusiva: c’è anche un concetto di memoria muscolare basato su qualcosa di diverso dalla permanenza mionucleare, vale a dire, la memoria epigenetica.[20] In breve, questa si riferisce a modifiche apportate al DNA senza influenzare la sua sequenza nucleotidica, quindi senza cambiare il codice genetico. Ciò comporta l’aggiunta (o la rimozione) di gruppi metilici ai nucleotidi di Citosina e Adenina o modifiche degli istoni (ad esempio, metilazione o acetilazione di residui di aminoacidi delle proteine istoniche). Il risultato di ciò è che influisce sull’espressione genica. Questo potrebbe forse essere trattato in un futuro articolo, dato che più ricerche vengono gradualmente pubblicate su questa nuova ed interessante strada ipotetica.
A proposito di “memoria epigenetica”: questa figura illustra un modello di sviluppo della persistenza batterica basato sulla presenza di un potenziale effetto di “memoria” epigenetica che include l’eredità stabile di certi modelli di metilazione del DNA. Lo stato di metilazione del DNA cellulare potrebbe portare alla conservazione di alcuni profili di espressione genica che favoriscono la dormienza, conservati in alcune cellule dopo il risveglio dalla dormienza. Cinetica di uccisione bifasica adattata da. (A) Popolazione originale di cellule metabolicamente attive che potrebbero contenere un’intrinseca eterogeneità fenotipica. (B) Quando incontra lo stress, la maggior parte delle cellule metabolicamente attive muore, mentre una piccola frazione di cellule entra nello stato di persistenza. La popolazione di persister può essere in qualche modo eterogenea, cioè formata da diversi percorsi (stocastico contro specifico). (C) Dopo gli stimoli nutrizionali/la rimozione dello stress, alcuni persister si risvegliano. Qui, la maggior parte dei persister inizia rapidamente la crescita e si divide in cellule regolari e metabolicamente attive. Tuttavia, alcune cellule potrebbero sperimentare un effetto di “memoria” epigenetica. Qui, lo stato di metilazione del DNA di alcuni siti che si trovano a monte di regioni codificanti regolate per esprimere tratti che favoriscono la dormienza potrebbe essere mantenuto dopo la replicazione del DNA. (D) A livello di popolazione totale, la popolazione finale dopo il risveglio potrebbe essere ugualmente suscettibile allo stress come la popolazione originale in (A). Tuttavia, a livello di singola cellula, alcune cellule potrebbero contenere un effetto di “memoria” legato alla dormienza, basato sull’eredità di alcuni tratti epigenetici dipendenti dalla metilazione del DNA. (E) L’esistenza di un effetto di “memoria” epigenetica potrebbe potenzialmente aumentare la frequenza dei persister nel tempo durante ripetuti cicli di stress.
Klein, CS; Marsh, GD; Petrella, RJ; Rice, CL (July 2003). “Muscle fiber number in the biceps brachii muscle of young and old men”. Muscle & Nerve. 28 (1): 62–8.
Tseng, Brian S., Christine E. Kasper, and V. Reggie Edgerton. “Cytoplasm-to-myonucleus ratios and succinate dehydrogenase activities in adult rat slow and fast muscle fibers.” Cell and tissue research 275.1 (1994): 39-49.
Schmalbruch H. Skeletal Muscle. Berlin: Springer-Verlag; 1985.
Mauro, Alexander. “Satellite cell of skeletal muscle fibers.” The Journal of Cell Biology 9.2 (1961): 493-495.
Forcina, Laura, et al. “An overview about the biology of skeletal muscle satellite cells.” Current genomics 20.1 (2019): 24-37.
Rosenblatt, J. David, David Yong, and David J. Parry. “Satellite cell activity is required for hypertrophy of overloaded adult rat muscle.” Muscle & nerve 17.6 (1994): 608-613.
Pavlath, Grace K., et al. “Localization of muscle gene products in nuclear domains.” Nature 337.6207 (1989): 570-573.
Allen, David L., Roland R. Roy, and V. Reggie Edgerton. “Myonuclear domains in muscle adaptation and disease.” Muscle & nerve 22.10 (1999): 1350-1360.
Gundersen, Kristian, and Jo C. Bruusgaard. “Nuclear domains during muscle atrophy: nuclei lost or paradigm lost?.” The Journal of physiology 586.11 (2008): 2675-2681.
Sinha-Hikim, Indrani, et al. “Testosterone-induced muscle hypertrophy is associated with an increase in satellite cell number in healthy, young men.” American Journal of Physiology-Endocrinology and Metabolism 285.1 (2003): E197-E205.
Egner, Ingrid M., et al. “A cellular memory mechanism aids overload hypertrophy in muscle long after an episodic exposure to anabolic steroids.” The Journal of physiology 591.24 (2013): 6221-6230.
Eriksson, Anders. Strength training and anabolic steroids: a comparative study of the trapezius, a shoulder muscle and the vastus lateralis, a thigh muscle, of strength trained athletes. PhD Diss. 2006.
Lindholm, Jesper Bøgh, et al. Effects of Long-Term Supplementation of Androgen Anabolic Steroids on Human Skeletal Muscle – Evidence for Muscle Memory? Master’s Thesis, 2019.
Snijders, Tim, et al. “The concept of skeletal muscle memory: Evidence from animal and human studies.” Acta Physiologica 229.3 (2020): e13465.
Seaborne, Robert A., et al. “Human skeletal muscle possesses an epigenetic memory of hypertrophy.” Scientific reports 8.1 (2018): 1-17.
Nonostante decenni di “lotta al doping” esso rimane assai diffuso, e non solo nelle competizioni di alto livello. L’errore alla base di questa campagna mediatico-salutistica è stata la generalizzazione; ossia fornire informazioni imprecise, accentuando i possibili sides senza però premurarsi di una vera e propria informativa preventiva chiara, veritiera ed efficace. In poche parole, quello che non si è fatto è dire: “l’uso di PEDs ha una serie di possibili effetti collaterali di gravità dipendente dal tipo di molecola, dal tempo e dalle modalità di assunzione”. Tutto ciò accompagnato da un manuale scientificamente corretto e di facile comprensione, contenente informazioni utili riguardanti la materia PEDs tale da permettere una migliore comprensione della questione che, a sua volta, renda possibile una più consapevole scelta individuale. Ma ciò non è stato fatto. Con l’unica eccezione di alcuni esperti indipendenti che nel corso degli anni hanno pubblicato libri e scritto articoli di una certa utilità.
Lo scopo di questa serie di articoli sarà quello di arginare il fenomeno dell’abuso dei PEDs, cosa che sta degenerando e che sta mostrando i suoi peggiori effetti su atleti di ambo i sessi.
Per la prima pubblicazione di questa nuova serie iniziamo con l’Oxymetholone…
Una (sempre utile) introduzione alla molecola di Oxymetholone:
L’Oxymetholone, noto anche come 2-idrossimetilene-17α-metil-4,5α-diidrotestosterone (2-idrossimetilene-17α-metil-DHT) o come 2-idrossimetilene-17α-metil-5α-androstan-17β-ol-3-one, è uno steroide androstano sintetico e un derivato 17α-alchilato del DHT.[1][2][3]
Le informazioni disponibili sulla farmacocinetica di questo AAS sono limitate.[4] Sembra essere ben assorbito con la somministrazione orale.[4] L’Oxymetholone ha affinità molto bassa per le globuline leganti gli ormoni sessuali nel siero umano (SHBG), meno del 5% di quella del Testosterone e meno dell’1% di quella del DHT. [5] Il farmaco viene metabolizzato nel fegato tramite ossidazione in posizione C2, riduzione in posizione C3, idrossilazione in posizione C17 e coniugazione. [4][6] Il gruppo C2 idrossimetilene del Oxymetholone può essere scisso per formare il Mestanolone (17α-metil-DHT), che può contribuire agli effetti della molecola precursore.[3] L’emivita del Oxymetholone è sconosciuta sebbene vi siano alcune ipotesi a riguardo.[6] L’Oxymetholone e suoi metaboliti vengono eliminati attraverso le urine.[5][6]
Come altri AAS, l’Oxymetholone è un agonista del recettore degli androgeni (AR).[3] Non è un substrato per la 5α-reduttasi (dal momento che è già 5α-ridotto) ed è uno substrato scarso per il 3α-idrossisteroide deidrogenasi (3α-HSD), e quindi mostra un alto rapporto di attività anabolizzante rispetto all’effetto androgenico.[3]
Data la sua derivanza dal DHT, l’Oxymetholone non è un substrato per l’enzima Aromatasi e quindi non può essere aromatizzato in metaboliti estrogenici.[3] Tuttavia, caratteristica unica tra i derivati del DHT, l’Oxymetholone è comunque associato a un’estrogenicità relativamente elevata ed è noto per avere il potenziale di produrre effetti collaterali estrogenici come ginecomastia (raramente) e ritenzione idrica. [3][7][8][9] È stato suggerito che questo può essere una conseguenza del legame diretto a l’attivazione del recettore degli estrogeni da parte dell’Oxymetholone (estrogenicità intrinseca).[3] L’Oxymetholone non possiede alcuna attività progestinica significativa.[3]
A causa della sua struttura 17α-alchilata, l’Oxymetholone è epatotossico.[3] L’uso a lungo termine del farmaco può causare una varietà di disturbi gravi, tra cui l’epatite, il cancro al fegato e la cirrosi; pertanto si raccomandano test periodici di funzionalità epatica per coloro che assumono l’Oxymetholone a fini terapeutici.[10] Questa molecola ha ottenuto, infatti, la nomea di essere uno tra gli AAS più epatotossici. Ciò deriva da i dosaggi comunemente, ed erroneamente, utilizzati in contesto culturistico. Si parla di dosaggi che facilmente sforano i 100-150mg/die. Ma tali dosaggi sono realmente vantaggiosi in termini di guadagni ipertrofici specie se messi in rapporto con gli effetti collaterali possibilmente verificabili? Questa domanda può ottenere una risposta sufficientemente esaustiva attraverso i risultati di uno studio che ha messo a confronto gli effetti di una dose di Oxymetholone da 50mg/die e una da 100mg/die.[11]
Oxymetholone – 50mg Vs. 100mg:
In questo studio, possiamo vedere i cambiamenti nel peso corporeo, nella massa magra, e la perdita di grasso in risposta a un dosaggio moderato e alto di Oxymetholone (50 mg vs 100 mg).
I cambiamenti nella composizione corporea sono mostrati per i gruppi placebo (barre nere), 50mg di Oxymetholone al giorno (barre bianche) e 100mg al giorno (barre grigie). I numeri sopra le barre rappresentano i cambiamenti assoluti medi e le barre di errore sono ± 1 SE. Per la massa corporea magra totale (LBM) e il grasso totale, le differenze tra i 3 gruppi erano significative (P <0,0001, ANOVA a una via). * Differenze significative rispetto al placebo, P ≤ 0,001.
Come ci si aspetterebbe, il gruppo placebo non ha guadagnato massa magra, né ha perso grasso corporeo.
Il gruppo trattato con 50mg di Oxymetholone ha guadagnato 3,3Kg di massa magra e ha perso 2,6kg di grasso.
Il gruppo trattato con 100mg di Oxymetholone ha guadagnato 4,2Kg di massa magra e ha perso 2,5kg di grasso.
I cambiamenti nella composizione regionale (n = 16) sono mostrati per i gruppi placebo, 50mg/die e 100mg/die. A: i numeri sopra le barre rappresentano i cambiamenti assoluti medi per il grasso del tronco mediante assorbimetria a raggi X a doppia energia (DEXA). B: le barre rappresentano i cambiamenti assoluti medi (kg) per la LBM dell’arto superiore (braccio destro più braccio sinistro) mediante DEXA. C: area della sezione trasversale del muscolo totale prossimale (barre grigie) e posteriore (barre nere) dei muscoli della coscia tramite risonanza magnetica. Le barre di errore sono ± 1 SE. * Differenza significativa rispetto al placebo, P ≤ 0,005. .
Guardando la massa corporea magra, è possibile vedere che quando si confrontano i due gruppi di dosaggio, il gruppo da 100mg ha guadagnato solo 0,9kg di massa corporea magra in più rispetto al gruppo da 50mg.
Questo dopo tre mesi di esposizione al doppio della quantità di farmaco.
Se si confrontano i biomarcatori tra i due gruppi, è possibile vedere che l’effetto di 100mg di Oxymetholone ha avuto sui livelli di ALT e AST era molto più deleterio rispetto al gruppo di 50 mg.
Caratteristiche di base della popolazione dello studio
Come molti di voi già sapranno, l’alanina aminotransferasi (ALT) e l’aspartato aminotransferasi (AST) sono biomarcatori comunemente usati per valutare i danni al fegato.
La somministrazione di un dosaggio di Oxymetholone doppio rispetto al basale di 50mg ha prodotto un ulteriore 27% di crescita muscolare relativa (la massa magra non è composta solo dal muscolo scheletrico!), ma ha provocato un picco 3.4x più alto di ALT e un picco 2.7x più alto nei livelli di AST.
Il calo del HDL è stato simile in entrambi i gruppi 50mg/die e 100mg/die.
Quelli sono solo biomarcatori con valore diagnostico per un eventuale danno epatico ma non sono indicativi di ciò che comporta la variabile del dosaggio sull’ipertrofia ventricolare, o altri fattori comunemente trascurati che dovrebbero essere utilizzati per valutare la salute cardiovascolare.
Anche se è possibile che gli aumenti di massa magra misurati dalla DEXA fossero legati in buona parte alla ritenzione idrica causata dalla terapia con Oxymetholone, i notevoli aumenti di forza muscolare misurati con il metodo 1-RM nei gruppi da 50 e 100mg/die (8,2-18,4%) suggeriscono che gli aumenti di massa magra erano probabilmente dovuti all’accrescimento di proteine miofibrillari oltre che alla semplice massa magra totale, poiché la forza è in una certa misura legata alle dimensioni dei muscoli. Inoltre, i membri del gruppo di ricerca hanno riferito che i cambiamenti nella massa magra appendicolare tramite DEXA sono quantitativamente correlati ai cambiamenti nella forza muscolare scheletrica in risposta a stimoli anabolici. In effetti, nel presente studio, sono stati in grado di corroborare questa relazione dimostrando che gli aumenti significativi del tessuto magro della parte superiore del corpo mediante scansione DEXA appendicolare erano altamente correlati con i cambiamenti nella forza della parte superiore del corpo come valutato da esercizi di Chest Press e Lat Pull-Down. Inoltre, i cambiamenti nella forza muscolare massima volontaria per gli esercizi della parte superiore del corpo hanno mostrato una risposta legata alla dose.
I cambiamenti relativi (%) nella forza sono mostrati per i gruppi placebo (barre nere), 50mg/giorno Oxymetholone (barre bianche) e 100mg/giorno Oxymetholone (barre grigie). I numeri sopra le barre rappresentano il cambiamento relativo (%) dal basale alla settimana 12 per le prove di forza massima a 1 ripetizione. Le barre di errore rappresentano ± 1 SE dalla media. * Differenza significativa rispetto al placebo, P < 0,05; † differenza significativa rispetto al placebo con il test di Wilcoxon, P < 0,02.
Al contrario, c’erano guadagni non significativi tra i tre gruppi di trattamento per la forza degli arti inferiori (3,9-12,0%), coerentemente con la mancanza di un aumento significativo della massa magra degli arti inferiori mediante scansione DEXA. Tuttavia, c’era una differenza quasi significativa (P = 0,052) tra i gruppi per il cambiamento del area della sezione trasversale del muscolo (CSA) dei muscoli della coscia tramite la risonanza magnetica, suggerendo che la terapia dello studio può aver influenzato positivamente i muscoli degli arti inferiori. È possibile che i test di forza di gruppi muscolari multipli e di grandi dimensioni, come quelli utilizzati con l’esercizio Leg Press, siano meno sensibili ai modesti cambiamenti nella massa muscolare, e lo studio potrebbe non aver avuto sufficiente potenza per rilevare piccoli ma significativi guadagni nelle estremità inferiori. Si ipotizza che ciò sia dovuto al fatto che i grandi muscoli delle gambe sono abitualmente utilizzati più frequentemente per sostenere il carico (ad esempio, camminare, alzarsi da una sedia) rispetto ai muscoli dell’estremità superiore negli adulti più anziani. Piccoli ma significativi guadagni nella forza e nella massa muscolare della parte inferiore del corpo possono essere meno dimostrabili che per i muscoli della parte superiore del corpo, che possono essere utilizzati meno per il lavoro ad alto volume e più inclini alla sarcopenia nelle persone anziane. Inoltre, i muscoli degli arti superiori, rispetto ai muscoli degli arti inferiori, hanno proporzioni maggiori di fibre a contrazione rapida di tipo II, che possono essere perse preferibilmente con l’invecchiamento. Inoltre, uno studio longitudinale in uomini anziani ha mostrato che le fibre di tipo I sono state perse principalmente nel vasto laterale della gamba, portando all’ipotesi che ci potrebbe essere una maggiore perdita di fibre di tipo II nelle braccia con l’invecchiamento. Così la risposta agli stimoli anabolici può essere più facilmente dimostrabile nelle estremità superiori di questa popolazione.
C’erano anche significative ma simili diminuzioni del grasso corporeo totale di 2,6 ± 1,2 e 2,5 ± 1,6 kg nei gruppi di 50 e 100mg al giorno, rispettivamente. Una parte importante del miglioramento dell’adiposità riguardava la diminuzione del grasso del tronco (1,7 ± 1,0 e 2,2 ± 0,9 kg nei due rispettivi gruppi di trattamento attivo). Una riduzione significativa del grasso del tronco potrebbe influenzare favorevolmente i fattori di rischio per le malattie cardiovascolari. Anche se ci aspetteremmo che la riduzione del grasso addominale si rifletta in una migliore sensibilità all’insulina, le misure indirette (HOMA-IR e QUICKI) potrebbero non essere state abbastanza sensibili. È anche possibile che ci fossero troppo pochi soggetti in ogni gruppo per rilevare cambiamenti piccoli ma significativi.
Ci sono ragioni teoriche per temere che l’eccesso di androgeni possa provocare o essere associato all’insulino-resistenza, anche se questa relazione è stata dimostrata solo in donne con sindrome dell’ovaio policistico. Non è stata misurata direttamente la sensibilità all’insulina né con il clamp euglicemico iperinsulinemico né con test di tolleranza al glucosio endovena a campionamento frequente. Tuttavia, le misure indirette della sensibilità insulinica (insulina a digiuno, HOMA-IR, QUICKI) non hanno mostrato prove di resistenza insulinica.
Cosa estrapolare?
Questo studio però presenta alcune limitazioni che possono averne influenzato i risultati. In primo luogo, la piccola dimensione del campione di meno di una dozzina di soggetti per gruppo può aver limitato la capacità di rilevare piccoli ma importanti cambiamenti in variabili come la massa magra (LBM) delle estremità inferiori e il CSA della muscolatura della coscia. Allo stesso modo, è possibile che le differenze osservate per i cambiamenti nella LBM totale e nella forza avrebbero potuto essere significative tra i gruppi di trattamento con dimensioni del campione maggiori. Quest’ultimo avrebbe fornito ulteriore supporto alla nostra supposizione di una risposta dose-dipendente con l’Oxymetholone. In secondo luogo, la popolazione rappresentava uomini adulti più anziani, che sono stati caratterizzati come a rischio di sarcopenia legata all’età sulla base dei rapporti che mostrano la perdita di massa e forza muscolare con l’invecchiamento. Tuttavia, i soggetti non sono stati reclutati per la perdita di peso, la fragilità o l’ipogonadismo palese di per sé, dal momento che è stato dimostrato che gli uomini più giovani con concentrazioni di Testosterone normali possono ottenere aumenti apprezzabili della massa muscolare e della forza dopo l’integrazione di androgeni. Inoltre, ci sono prove che la sintesi proteica miofibrillare nelle persone anziane può essere significativamente aumentata a livelli paragonabili a quelli raggiunti nelle persone più giovani in risposta a un potente stimolo anabolico. Infine, poiché l’Oxymetholone è un AAS 17-metilato che provoca un elevato effetto di primo passaggio nel fegato, e che nel presente studio non sono state prese misure di contenimento per l’epatotossicità potenziale, i risultati di AST e ALT ottenuti rappresentano solamente modelli privi di ancillari volti ad una epatoprotezione.
Conclusioni sul dosaggio “ottimale” di Oxymetholone:
Evidenziati i limiti dello studio, pur prendendo i dati ivi riportati universalmente rapportabili al basale d’uso della molecola (es. vedi epatotossicità), possiamo giungere, grazie all’ausilio di dati empirici raccolti negli anni attraverso indagini svolte sulle preparazioni di svariati atleti di medio e alto livello, ad identificare un dosaggio con una ratio “efficacia:rischio (E:R)” favorevole per l’atleta.
Un dato è emerso preponderante nel corso delle indagini svolte: quale fosse il peso dell’atleta e il suo condizionamento atletico, nonché l’utilizzo di una adeguata epatoprotezione e controllo della dislipidemia, il margine della ratio E:R diveniva evidentemente sfavorevole oltre i 150mg/die. Indi per cui, i dosaggi elevati raggiunti da certi atleti, arrivando a picchi di 200-300mg/die, sono risultati inutili al miglioramento delle risposte anabolizzanti complessive e inficianti per il corretto svolgimento della stessa preparazione (vedi, ad esempio, marcata inappetenza e nausea).
Dosaggi standard per un atleta di sesso maschile non dovrebbero discostarsi dal range 50-100mg/die, considerando che la taratura del “dosaggio ideale” si è ottenuta calcolando la dose individuale con la formula 1mg/Kg di peso corporeo. Ovviamente, l’assicurarsi una adeguata protezione epatica e lipidica è il punto parallelo da raggiungere.
Nelle atlete, invece, vista la loro maggiore sensibilità agli aumenti degli androgeni circolanti, la “dose ideale” si è attestata a 25mg/die con punte massime (anche se non necessarie) di 50mg/die. A tal proposito, vorrei ricordare che l’Oxymetholone è risultato essere una molecola più vantaggiosa nel controllo degli effetti collaterali androgenizzanti rispetto a composti quali Methenolone e Boldenone.
La linea tra abuso e uso è spesso molto sottile, ma nel caso del Oxymetholone essa si mostra sufficientemente marcata…
Pavlatos AM, Fultz O, Monberg MJ, Vootkur A (June 2001). “Review of oxymetholone: a 17alpha-alkylated anabolic-androgenic steroid”. Clinical Therapeutics. 23 (6): 789–801, discussion 771.
Saartok T, Dahlberg E, Gustafsson JA (June 1984). “Relative binding affinity of anabolic-androgenic steroids: comparison of the binding to the androgen receptors in skeletal muscle and in prostate, as well as to sex hormone-binding globulin”. Endocrinology. 114 (6): 2100–6.
Hengge UR, Stocks K, Wiehler H, Faulkner S, Esser S, Lorenz C, et al. (March 2003). “Double-blind, randomized, placebo-controlled phase III trial of oxymetholone for the treatment of HIV wasting”. AIDS. 17 (5): 699–710.
Cortesgallegos V, Castaneda G, Alonso R, Perezpasten E, Reyeslugo V, Barron C, Mondragon L, Villalpando S (January 1982). “Spontaneous and Oxymetholone-Induced Gynecomastia”. Journal of Andrology. C/O Allen Press, Inc Po Box 368, Lawrence, Ks 66044: Amer Soc Andrology, Inc. 3 (1): 33.
Villalpando S, Mondragon L, Barron C, Reyeslugo U, Perezpasten E, Alonso R, Castaneda G, Gallegos V (January 1982). “5-Alpha Reductase Blockade May Be Responsible for Spontaneous and Oxymetholone-Induced Gynecomastia”. Archivos de Investigacion Medica. Social Apdo Postal 73-032, Mexico Df 03020, Mexico: Inst Mexicano Seguro. 13 (2): s13.
Il fegato è un organo importante ed è vitale per la sopravvivenza del soggetto. È responsabile di diverse e importanti funzioni nel corpo umano. Produce acidi biliari e proteine plasmatiche, immagazzina glicogeno e produce glucosio attraverso la gluconeogenesi, gioca un ruolo nel sistema immunitario, metabolizza un numero elevato di molecole, ecc. Quindi, si, avete capito bene: è importante. Quando qualcosa risulta dannosa per il fegato, essa si indica come epatotossico (dal greco hêpar-atos, fegato). Un chiaro esempio è l’alcol. Gli alcolisti tendono a sviluppare una malattia del fegato a un certo punto della loro vita. Tuttavia, molti farmaci da prescrizione, o anche over-the-counter, possono essere epatotossici, come l’Acetaminofene. E, come è ben dimostrato, anche gli AAS possono essere epatotossici, anche se specifici. Come sembra, solo quelli con una specifica alterazione chimica sembrano essere maggiormente epatotossici – in particolare, quelli che presentano una metilazione in pozione C-17α.
Modifica della struttura carbossilica del Testosterone (sinistra) in posizione C-17α (destra).
In questo articolo tratterò principalmente ciò che sembra causare questa epatotossicità indotta da AAS. L’effetto epatotossico può essere riscontrato attraverso l’osservazione dei cambiamenti nei marcatori ematici del danno epatico, come Alanina Transaminasi (ALAT), Aspartato Transaminasi (ASAT), γ-glutamiltransferasi (GGT) e la Fosfatasi Alcalina (ALP). Una nota di cautela deve essere presa in considerazione quando si interpretano gli aumenti di ALAT e ASAT, poiché entrambi aumenteranno anche a causa del intyenso lavoro muscolare [1]. È bene sapere che in questi casi, ASAT sarà di solito più alto del ALAT, mantenendo un rapporto ASAT/ALAT superiore a 1. Quindi, quando questi aumentano con un rapporto inferiore a 1, si può essere più sicuri che il danno muscolare non è il colpevole dell’alterazione. Idealmente, nessun esercizio (contro-resistenza) viene svolto 1-2 settimane prima dell’esame del sangue per escludere il danno muscolare muscolare come causa dell’innalzamento, sebbene ciò dipenda anche dall’intensità del allenamento. In rari casi, il danno al fegato potrebbe avanzare clinicamente fino allo sviluppo di ittero colestatico [2]. In questo caso, un prodotto della degradazione dei globuli rossi (bilirubina) si accumula nel corpo. L’ittero può essere osservato visivamente (tono giallo della pelle e della sclera degli occhi), e si possono sviluppare sintomi come nausea, vomito, dolore allo stomaco e prurito. Inoltre, alcuni rari casi di peliosis hepatis (Peliosi Epatica) sono stati segnalati verificarsi come risultato dell’uso di AAS orali ad alte dosi [3]. Questa è una condizione nella quale si vengono a formare cisti piene di sangue nel fegato. La sospensione dell’AAS in questione è solitamente sufficiente e porterà alla scomparsa di queste caratteristiche cliniche entro pochi mesi. In casi più gravi, tuttavia, potrebbero richiedere un intervento chirurgico. Infine, alcuni casi in letteratura hanno riportato un’associazione tra uso di AAS e carcinoma epatico [4] e adenoma [5].
Ho già trattato in passato tale problematica legata all’uso di AAS, ma questa volta voglio trattare la questione più nello specifico, analizzando le due ipotesi che ruotano intorno all’epatotossicità AAS-dipendente: “ipotesi dello stress ossidativo” e “ipotesi di coniugazione dell’anello D”.
L’ipotesi dello stress ossidativo:
L’ipotesi dello stress ossidativo che tratterò qui si basa su un documento che William Llewellyn, Peter Van Mol e Peter Bond hanno pubblicato [6]. Lo stress ossidativo è qualcosa che si pensa possa risultare nell’epatotossicità osservata con l’uso di AAS, e se l’ipotesi è vera, dà qualche opportunità per contrastarla in modo migliore. Quindi, cominciamo con spiegare quello che è lo stress ossidativo. Lo stress ossidativo è descritto da Helmut Sies come un disturbo nell’equilibrio pro-ossidante-antiossidante a favore del primo [7], che si riduce a molecole contenenti ossigeno, che sono altamente reattive (specie reattive dell’ossigeno [ROS]), sopraffacendo il sistema antiossidante. Poiché le ROS sono così altamente reattive, possono reagire con molecole come lipidi, proteine, carboidrati e acidi nucleici (elementi costitutivi del DNA). Quando si dice “reagire con queste molecole”, si intende che danneggia queste molecole (estremamente semplificato, ma è sufficiente per far comprendere il processo). Questi ROS provengono da varie reazioni catalizzate da enzimi come la respirazione cellulare (l’ossidazione dei macronutrienti per fornire energia), altri processi metabolici e radiazioni. La fonte primaria di ROS all’interno di una cellula sono i mitocondri, il che non è sorprendente dato che i mitocondri sono le “centrali energetiche” della cellula. È il posto nella cellula dove i carboidrati alimentari, gli acidi grassi e le proteine (o, meglio, gli amminoacidi che le compongono) finiscono per essere ossidate per produrre energia in un processo chiamato fosforilazione ossidativa. Come suggerisce il nome, la fosforilazione ossidativa ossida e richiede ossigeno per farlo. Questo processo, tuttavia, non è perfetto. Per non complicare troppo le cose al lettore, non mi addentrerò nelle complessità delle reazioni chimiche, ma fondamentalmente, questo processo può produrre ROS come sottoprodotto (superossido in particolare). Le cellule del corpo sono dotate di meccanismi per tenere a bada questi ROS generati (la parte antiossidante dell’equazione). In circostanze normali questo porta ad un sottile equilibrio tra i due. Avere qualche ROS qua e là nelle cellule è normale. Essi giocano un ruolo essenziale nel normale funzionamento di vari processi vitali [8]. Tuttavia, il problema nasce quando questo equilibrio si altera a favore della parte proossidante dell’equazione: lo stress ossidativo. Questo è il momento in cui i ROS prendono il sopravvento, per così dire, e possono iniziare a creare il caos nella cellula. Quanto sopra è un quadro un po’ troppo semplificato. Ci sono diversi tipi di ROS (radicali liberi e non radicali). Ciò che conta è dove si trovano questi ROS nella cellula e come evolvono nel tempo. Inoltre, questo interagisce con il sistema antiossidativo delle cellule, il che complica ulteriormente il quadro. Ma credo che quanto sopra sia sufficiente per dare una buona comprensione di tutto questo. Ciò che conta è che l’epatotossicità indotta da AAS è stata ripetutamente dimostrata essere associata allo stress ossidativo nelle cellule epatiche (fegato) di modelli animali [9]. Questo fa sorgere la domanda: è solo un’associazione, o c’è una relazione causale con l’epatotossicità indotta da AAS? Dopo aver scavato nella letteratura, sono emersi alcuni studi che sembrano sostenere una relazione causale. Uno studio svolto su un carcinoma prostatico umano epiteliale (22Rv1) ha collegato l’attivazione del recettore degli androgeni (AR) a un aumento dei ROS basali [10]. Più tardi, lo stesso gruppo ha pubblicato una ricerca applicando un disegno di studio simile. Questo studio ha confermato i precedenti risultati e ha anche dimostrato che l’aumento dei ROS è dovuto a un aumento indotto dall’AAS nella β-ossidazione mitocondriale degli acidi grassi [11]. Quindi, l’attivazione di l’AR porta a una maggiore ossidazione degli acidi grassi nei mitocondri, con conseguente maggiore produzione di ROS come sottoprodotto. Da notare che questo studio ha anche trovato un aumento dell’mRNA della carnitina palmitoiltransferasi (CPT1). Tutto quello che dovete sapere è che la CPT1 è considerata essere l’enzima che regola la velocità nel processo di ossidazione mitocondriale degli acidi grassi. Quindi, se si aumenta la CPT1, si aumenta l’ossidazione mitocondriale degli acidi grassi. Ora, le cellule del cancro alla prostata non sono cellule del fegato, ovviamente. Ma ciò che è interessante è che l’AAS 17α-alchilato Fluoxymesterone e Metilandrostanolone hanno dimostrato di aumentare l’attività del CPT1 nel fegato di ratto [12]. Inoltre, se si guardano agli epatociti di ratto (cellule epatiche) trattati con AAS 17α-alchilati, si vedrà il gonfiore dei mitocondri e solo cristae leggermente definite [13]. (Le criste sono quelle pieghe caratteristiche della membrana interna dei mitocondri). Infatti, la produzione di ROS è una causa nota di gonfiore mitocondriale, e il gonfiore è un fattore importante che porta alla successiva morte cellulare [14]. Quindi, apparentemente, suggerisce un potenziale ruolo dello stress ossidativo. Questo non vuol dire che qualsiasi aumento nella produzione di energia di una cellula sia negativo. Usando i muscoli aumenta anche la produzione di energia nelle cellule muscolari. Di conseguenza, più ROS vengono prodotti anche in queste cellule. In contrasto con l’aumento di ROS indotto dall’AAS nelle cellule del fegato, questi aumenti sono transitori invece che continui. Inoltre, le cellule muscolari differiscono nei loro meccanismi antiossidanti per gestire questa condizione. Quindi, normalmente, questo non è assolutamente un problema. Tuttavia, l’esercizio intenso e prolungato può anche provocare danni ossidativi alle molecole delle cellule muscolari [15].
L’ipotesi dello stress ossidativo nella epatotossicità indotta da AAS come descritto da Bond et al. [49]. 1 Un androgeno si lega a, e attiva, il recettore degli androgeni (AR) nelle cellule epatiche. Questo porta a 2 la sovra-regolazione della Carnitina Palmitoiltransferasi 1 (CPT1), l’enzima che regola il tasso di β-ossidazione degli acidi grassi (FA). Si pensa che questo porti a 3 un aumento della β-ossidazione degli acidi grassi nei mitocondri. Di conseguenza, 4 la produzione di specie reattive dell’ossigeno (ROS) è aumentata. L’aumento dei ROS poi danneggia i mitocondri, il che sembra essere alla base dell’epatotossicità indotta dall’AAS.
Ora, se si integrassero gli antiossidanti (mitocondriali), si allevierebbe questo danno? Può darsi. Mentre non c’è un trial di buona qualità che valuti questo, uno studio osservazionale su 320 atleti dimostra qualcosa del genere [16]. In breve, gli utilizzatori di AAS che hanno preso un supplemento contenente alcuni composti antiossidanti non ha mostrato alcun aumento dei marcatori di danno epatico dopo il ciclo rispetto a quelli che non hanno assunto quel supplemento. Ancora una volta, questo sarebbe in linea con lo stress ossidativo che gioca un ruolo causale nell’epatotossicità indotta da AAS. Infine, sembra che l’epatotossicità indotta da AAS potrebbe essere legata all’attivazione del AR nelle cellule epatiche. In un vecchio studio del 1964, Marquardt et al. non sono riusciti a dimostrare che l’AAS non 17α-alchilato produce test di funzionalità epatica anormali [17]. Infatti, gli AAS 17α-alchilati mostrano segni di epatotossicità in diversi studi, mentre non si vede questo con AAS non-17αalchilati, nemmeno con un alto dosaggio di 600 mg di Testosterone Enantato settimanale [18]. La 17α-alchilazione sembra quasi necessaria per rendere epatotossico un AAS, probabilmente perché è l’unica alterazione che lo rende sufficientemente biodisponibile per via orale. E, di conseguenza, porta ad alte concentrazioni del composto nel fegato. Ma possiamo individuare le differenze tra i vari AAS 17α-alchilati che riguardano la loro capacità di attivare l’AR? Certamente sembra così. In generale, sembra che sia vero quanto segue:
Epatotossicità = resistenza alla decomposizione epatica×potenza di attivazione del AR
Quindi, facciamo un esempio. Il Methyltrienolone (R1881) ha un’affinità molto alta per l’AR, ha un’alta potenza per la transattivazione dell’AR [19], ed è fortemente resistente al metabolismo epatico. Come tale, è un composto ideale per un saggio dei siti di legame agli androgeni [20]. Infatti, un studio clinico che impiega un basso dosaggio dello steroide (≤1 mg al giorno) ha dimostrato un significativo aumento dei marcatori di danno epatico entro due settimane [21]. Gli autori lo hanno definito “(…) attualmente lo steroide più epatotossico”. Lo steroide 17α-alchilato meno epatotossico è solitamente considerato l’Oxandrolone. Anche con alti dosaggi fino a 80mg al giorno, mostra solo deboli segni di epatotossicità [22]. Mentre lo steroide è abbastanza resistente al metabolismo epatico [23], ha una bassa affinità per il AR [23]. La sua potenza relativa in termini di transattivazione AR è anche quasi 100 volte inferiore a quella del Methyltrienolone [19]. Allo stesso modo, anche l’Oxymetholone ha una bassa affinità per l’AR [23] e la sua potenza in termini di transattivazione AR è molto simile a quella dell’Oxandrolone [19]. Non sorprende che mostri segni di epatotossicità solo in una minoranza di pazienti, nonostante gli alti dosaggi (100-150 mg al giorno) [24].
L’ipotesi di coniugazione dell’anello D:
Avete mai sfogliato il libro Doping in Sports di Thieme e Hemmersbach? [25] In questo libro gli autori notano che non c’è correlazione tra la tossicità epatica e gli effetti farmacologici primari (cioè gli effetti anabolizzanti) – il che è sufficientemente ovvio perché gli AAS non 17α-alchilati sono rapidamente metabolizzati nel fegato, quindi la loro concentrazione in loco non sarebbe come quella dei 17α-alchilati. Naturalmente, non si troverà una correlazione se si guarda solo a questo fattore. Bisogna anche prendere in considerazione la sua resistenza al metabolismo epatico come è stato fatto con l’ipotesi dello stress ossidativo descritta sopra.
In ogni caso, questo ha portato gli autori a formulare un’alternativa ipotesi di ciò che causa l’epatotossicità indotta da AAS. E sembrava essere l’unica. Essi suggeriscono che l’epatotossicità è probabilmente dovuta alla coniugazione dell’anello D con l’acido glucuronico. Questo processo è chiamato glucuronidazione ed è una cosiddetta comune reazione di fase 2 nel metabolismo del farmaco. Rende la molecola madre più solubile in acqua, facilitando così la sua escrezione nelle urine.
Il gruppo 17β-glucuronide (in blu) attaccato al anello D di uno steroide 17α-metilato (gruppo 17α-metilico in rosso).
È semplicemente l’attaccamento (coniugazione) dell’acido glucuronico alla molecola madre (vedi figura sopra). Quando il Testosterone con un gruppo 17β-glucuronide (così come diversi estrogeni con questa modifica) viene iniettato nel ratto, il flusso biliare è inibito [521]. Presumibilmente, perché questi composti condividono somiglianze strutturali con gli acidi biliari, questi composti competono con gli acidi biliari per legarsi a certi recettori. Tuttavia, a parte questo, non c’è molta sostanza per sostenere questa ipotesi come la ragione per l’epatotossicità indotta da AAS, soprattutto perché molti degli AAS non 17α-alchilati, compreso il Testosterone, subiscono la glucuronizzazione del loro gruppo 17β-idrossi. Eppure questi non sono sensibilmente epatotossici. Infatti, la 17βglucuronidazione è stata identificata solo per alcuni AAS 17α-alchilati, e sembra che essi subiscono questo processo solo in piccola misura [26]. Così, ironicamente, se questa ipotesi fosse vera, o significativa, ci si aspetterebbe l’epatotossicità con il Testosterone ma non con gli AAS 17α-alchilati.
Conclusioni sulle ipotesi esposte:
Non è sicuramente una novità per l’utilizzatore medio, ma anche per il semplice soggetto interessato all’argomento PEDs, che gli AAS metilati in C-17 (17α-alchilati) abbiano un effetto epatotossico con lievi variabili tra molecole aventi la stessa modifica strutturale. E non è nemmeno una rivelazione che la supplementazione con antiossidanti (vedi NAC e Silimarina) possa ridurre tale effetto. Di conseguenza, l’ipotesi dello stress ossidativo sembra essere la principale causa del epatotossicità AAS-indotta. Ma non l’unico fattore.
Nell’ultimo decennio si è aggiunto ai classici composti antiossidanti l’uso di acidi biliari come l’Acido Ursodesossicolico e l’Acido Tauroursodesossicolico assunti oralmente.
L’Acido Ursodesossicolico è un acido biliare secondario che deriva dal metabolismo dell’acido colico da parte del microbiota umano intestinale. Il suo nome deriva dal fatto che è il principale acido biliare negli orsi (dal latino ursus). In biologia e biochimica lo si etichetta con l’acronimo UDCA. Il nome completo del UDCA è Acido 3α,7β-diidrossi-5β-colanoico.[27]
Acido Ursodesossicolico (UDCA)
L’Acido Tauroursodesossicolico (TUDCA) è un acido biliare ambifilico. È la forma coniugata di Taurina ed il precedentemente citato Acido Ursodeossicolico (UDCA). Il nome completo del TUDCA è 2-{(4R)-4-[(1R,3aS,3bR,4S,5aS,7R,9aS,9bS,11aR)-4,7-Dihydroxy-9a,11a-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-1-yl]pentanamido} acido etan-1-sulfonico.[28]
Acido Tauroursodesossicolico (TUDCA)
l’UDCA è approvato per il trattamento della cirrosi biliare primaria.[1][2] Di conseguenza, l’Acido Ursodesossicolico (UDCA) ha mostrato effetti epatoprotettivi. Tuttavia, i suoi meccanismi molecolari sottostanti rimangono poco chiari. Per tale motivazione, sono stati condotti alcuni studi come quello di Da Jung Kim et al. nel quale è stato osservato l’effetto epatoprotettivo dell’UDCA e della vitamina E utilizzando la metabolomica e l’analisi metagenomica. In questo studio, sono stati analizzati campioni di sangue e urine di pazienti con obesità e disfunzione epatica. Nove pazienti sono stati assegnati in modo casuale a ricevere UDCA (300 mg due volte al giorno), e 10 soggetti hanno ricevuto la vitamina E (400 UI due volte al giorno) per 8 settimane. L’UDCA ha migliorato significativamente i punteggi della funzionalità epatica dopo 4 settimane di trattamento e ha ridotto efficacemente i livelli epatici di acido Desossicolico e di microRNA-122 nel siero. Per comprendere meglio il suo meccanismo protettivo, è stato condotto uno studio di metabolomica globale ed è stato scoperto che l’UDCA ha regolato le tossine uremiche (acido ippurico, solfato di p-cresolo e metaboliti derivati dall’indolo), gli antiossidanti (solfato di ascorbato e N-acetil-L-cisteina) e il percorso fenilalanina/tirosina. Inoltre, il coinvolgimento del microbioma, in particolare di Lactobacillus e Bifidobacterium, è stato dimostrato attraverso l’analisi metagenomica delle vescicole extracellulari derivate dai batteri. Nel frattempo, il trattamento con vitamina E non ha portato a tali alterazioni, tranne che ha ridotto le tossine uremiche e la disfunzione epatica. I nostri risultati hanno suggerito che entrambi i trattamenti erano efficaci nel migliorare la funzione epatica, anche se attraverso meccanismi diversi.
Schema dei potenziali meccanismi terapeutici del trattamento con UDCA. L’analisi metabolomica ha rivelato che l’UDCA riduce i principali composti nei percorsi fenilalanina/tirosina e triptofano, tra cui fenilalanina, fenilacetato, acetilfenilalanina, aldeide 3,4-idrossifenilacetato, dopamina-3-O-solfato, idrossibenzaldeide, p-cresolo solfato, idrossicynurenamina, idrossindolo e acido ippurico, nel plasma e nelle urine. I metaboliti intermedi degli aminoacidi aromatici come l’idrossimelatonina, l’acido benzoico e l’acido salicilico sono stati aumentati. I forti antiossidanti come l’ascorbato, l’acetiltriptofano e la N-acetil-L-cisteina erano elevati. Inoltre, la disintossicazione delle tossine uremiche tramite glucuronidazione (idrossimetossiindolo glucuronide e p-cresolo glucuronide) è stata osservata dopo il trattamento UDCA. Tuttavia, la vitamina E ha ridotto l’acido indolo-propionico, il solfato di indoxile, la 3-ketosphinganina e la sfingosina, che non sono stati regolati dall’UDCA. Il colore blu indica una diminuzione del livello del metabolita, e il colore rosso indica un aumento del livello del metabolita dopo il trattamento UDCA. I metaboliti che sono cambiati dopo il trattamento con vitamina E sono contrassegnati da un asterisco (*). I metaboliti che sono stati possibilmente regolati da modifiche batteriche sono contrassegnati da un colore viola.
Inoltre, si sa che l’UDCA a livello epatico stimola la secrezione di ATP da parte degli epatociti[29]; sebbene il significato di quest’azione non è ancora noto. Si sa però che interagisce col sistema dei citocromi P450 e che riduce la Glicuronazione degli estrogeni sintetici e non solo.[30] Vi ricorda qualcosa? Esatto! L’ipotesi di coniugazione dell’anello D e la sua potenzialità di essere parte dell’effetto epatotossico AAS-indotto! Se a ciò aggiungiamo che l’UDCA possiede la capacità di attivare direttamente il recettore per i glucocorticoidi, che contribuirebbe ad allargare i meccanismi della sua azione anticolestatica ed antinfiammatoria sul parenchima epatico [31], e che stimola la sintesi del glutatione (GSH), potente antiossidante endogeno, attraverso l’intervento delle chinasi dipendenti dai fosfoinositidi (PI-3K e PKB) [32], ciò fa si che l’UDCA risulti la chiave di volta nella protezione epatica durante l’uso di AAS con marcata resistenza al metabolismo epatico in abbinamento ai largamente utilizzati NAC (precursone ad alta biodisponibilità del Glutatione) e Silimarina.
Quanto detto non rappresenta ne un consiglio medico ne una scusa per abusare di AAS di qualsiasi tipo! Si tratta semplicemente della divulgazione di informazioni che la seria ricerca scientifica ha permesso di estrapolare, per il momento…
Gabriel Bellizzi
Riferimenti:
W. J. Meyer, A. Webb, C. A. Stuart, J. W. Finkelstein, B. Lawrence, and P. A. Walker. Physical and hormonal evaluation of transsexual patients: a longitudinal study. Archives of sexual behavior, 15(2):121–138, 1986.
A. M. Elsharkawy, S. McPherson, S. Masson, A. D. Burt, R. T. Dawson, and M. Hudson. Cholestasis secondary to anabolic steroid use in young men. Bmj, 344, 2012.
J. Nadell and J. Kosek. Peliosis hepatis. twelve cases associated with oral androgen therapy. Archives of pathology & laboratory medicine, 101(8):405–410, 1977.
F. L. Johnson, K. Lerner, M. Siegel, J. Feagler, P. Majerus, J. Hartmann, and E. D. Thomas. Association of androgenic-anabolic steroid therapy with development of hepatocellular carcinoma. The Lancet, 300(7790):1273–1276, 1972.
L. Hernandez-Nieto, M. Bruguera, J. A. Bombi, L. Camacho, and C. Rozman. Benign liver-cell adenom associated with long-term administration of an androgenic-anabolic steroid (methandienone). Cancer,40(4):1761–1764, 1977.
P. Bond, W. Llewellyn, and P. Van Mol. Anabolic androgenic steroid-induced hepatotoxicity. Medical Hypotheses, 93:150–153, 2016.
H. Sies et al. Oxidative stress: introductory remarks. Oxidative stress, 501:1–8, 1985.
K. Brieger, S. Schiavone, F. J. Miller Jr, and K.-H. Krause. Reactive oxygen species: from health to disease. Swiss medical weekly, 142:w13659, 2012.
S. P. Frankenfeld, L. P. Oliveira, V. H. Ortenzi, I. C. Rego-Monteiro, E. A. Chaves, A. C. Ferreira, A. C. Leitáo, D. P. Carvalho, and R. S. Fortunato. The anabolic androgenic steroid nandrolone decanoate disrupts redox homeostasis in liver, heart and kidney of male wistar rats. PloS one, 9(9):e102699, 2014.
J. H. Pinthus, I. Bryskin, J. Trachtenberg, J.-P. Luz, G. Singh, E. Fridman, and B. C. Wilson. Androgen induces adaptation to oxidative stress in prostate cancer: implications for treatment with radiation therapy. Neoplasia, 9(1):68–80, 2007.
H. Lin, J.-P. Lu, P. Laflamme, S. Qiao, B. Shayegan, I. Bryskin, L. Monardo, B. C. Wilson, G. Singh, and J. H. Pinthus. Inter-related in vitro effects of androgens, fatty acids and oxidative stress in prostate cancer: a mechanistic model supporting prevention strategies. International journal of oncology, 37(4):761–766, 2010.
M. Guzmán, A. Saborido, J. Castro, F. Molano, and A. Megias. Treatment with anabolic steroids increases the activity of the mitochondrial outer carnitine palmitoyltransferase in rat liver and fast-twitch muscle. Biochemical pharmacology, 41(5):833–835, 1991.
R. Gragera, A. Saborido, F. Molano, L. Jimenez, E. Muñiz, and A. Megias. Ultrastructural changes induced by anabolic steroids in liver of trained rats. Histology and histopathology, 1993.
X. Chapa-Dubocq, V. Makarov, and S. Javadov. Simple kinetic model of mitochondrial swelling in cardiac cells. Journal of cellular physiology, 233(7):5310–5321, 2018.
S. K. Powers, L. L. Ji, A. N. Kavazis, and M. J. Jackson. Reactive oxygen species: impact on skeletal muscle. Comprehensive Physiology, 1(2):941–969, 2011.
T. A. Pagonis, G. N. Koukoulis, C. S. Hadjichristodoulou, P. N. Toli, and N. V. Angelopoulos. Multivitamins and phospholipids complex protects the hepatic cells from androgenic-anabolic-steroids-induced toxicity. Clinical Toxicology, 46(1):57–66, 2008.
G. H. Marquardt, C. E. Logan, W. G. Tomhave, and R. M. Dowben. Failure of non-17-alkylated anabolic steroids to produce abnormal liver function tests. The Journal of Clinical Endocrinology & Metabolism, 24(12):1334–1336, 1964.
S. Bhasin, L. Woodhouse, R. Casaburi, A. B. Singh, D. Bhasin, N. Berman, X. Chen, K. E. Yarasheski, L. Magliano, C. Dzekov, et al. Testosterone dose-response relationships in healthy young men. American Journal of Physiology-Endocrinology And Metabolism, 281(6):E1172–E1181, 2001.
C. J. Houtman, S. S. Sterk, M. P. Van de Heijning, A. Brouwer, R. W. Stephany, B. Van der Burg, and E. Sonneveld. Detection of anabolic androgenic steroid abuse in doping control using mammalian reporter gene bioassays. Analytica chimica acta, 637(1-2):247–258, 2009.
C. Bonne and J.-P. Raynaud. Assay of androgen binding sites by exchange with methyltrienolone (r 1881). Steroids, 27(4):497–507, 1976.
H. L. Krüskemper and G. Noell. Liver toxicity of a new anabolic agent: methyltrienolone (17α-methyl-4, 9, 11-estratriene-17β-ol-3-one). Steroids, 8(1):13–24, 1966.
C. Grunfeld, D. P. Kotler, A. Dobs, M. Glesby, S. Bhasin, O. S. Group, et al. Oxandrolone in the treatment of hiv-associated weight loss in men: a randomized, double-blind, placebo-controlled study. JAIDS Journal of Acquired Immune Deficiency Syndromes, 41(3):304–314, 2006.
J. A. Kemppainen, E. Langley, C.-i. Wong, K. Bobseine, W. R. Kelce, and E. M. Wilson. Distinguishing androgen receptor agonists and antagonists: distinct mechanisms of activation by medroxyprogesterone acetate and dihydrotestosterone. Molecular Endocrinology, 13(3):440–454, 1999.
U. R. Hengge, K. Stocks, S. Faulkner, H. Wiehler, C. Lorenz, W. Jentzen, D. Hengge, and G. Ringham. Oxymetholone for the treatment of hiv-wasting: a double-blind, randomized, placebo-controlled phase iii trial in eugonadal men and women. HIV clinical trials, 4:150–163, 2003.
A. Sansone, F. Romanelli, M. Sansone, A. Lenzi, and L. Di Luigi. Gynecomastia and hormones. Endocrine, 55(1):37–44, 2017.
W. Schänzer. Metabolism of anabolic androgenic steroids. Clinical chemistry, 42(7):1001–1020, 1996.
Boatright, Jeffrey H.; Nickerson, John M.; Moring, Anisha G.; Pardue, Machelle T. (2009). “Bile acids in treatment of ocular disease”. Journal of Ocular Biology, Diseases, and Informatics. 2 (3): 149–159.
Nathanson MH et al. Stimulation of ATP secretion in the liver by therapeutic bile acids. Biochem J. 2001; 358(Pt 1):1-5.
Weitzel C et al. Ursodeoxycholic acid induced activation of the glucocorticoid receptor in primary rat hepatocytes. Eur J Gastroenterol Hepatol. 2005 Feb; 17(2):169-77.
Sanchez Pozzi EJ et al. Ursodeoxycholate reduces ethinylestradiol glucuronidation in the rat: role in prevention of estrogen-induced cholestasis. J Pharmacol Exp Ther. 2003 Jul; 306(1):279-86.
Arisawa S et al. Ursodeoxycholic acid induces glutathione synthesis through activation of PI3K/Akt pathway in HepG2 cells. Biochem Pharmacol. 2009 Mar 1;77(5):858-66.
Come di mia consuetudine, mi servirò della letteratura scientifica ad oggi disponibile per trattare nel modo più accurato ed esaustivo, rimanendo pur sempre comprensibile da chi non avvezzo alla biochimica e all’endocrinologia, il tema annoso della cadenza di somministrazione del GH.
Iniziamo subito andando ad esaminare la “genesi del dibattito” …
La genesi del dibattito in uno studio:
Il trattamento dei bambini con bassa statura idiopatica mediante iniezioni giornaliere di GH umano (hGH) è seguito, dopo la sua sospensione, da una decelerazione della crescita con livelli sierici normali di GH e IGF-I.
Il studio ivi riportato [1] è stato progettato per capire e prevenire la decelerazione della crescita. I ricercatori hanno ipotizzato che questo fenomeno sia dovuto alla tolleranza a livello dell’organo bersaglio, che la tolleranza si sviluppi in risposta alla farmacocinetica non fisiologica dell’hGH iniettato quotidianamente, e che la terapia con hGH a giorni alterni lo prevenga.
Trentotto bambini prepuberi con bassa statura idiopatica, di età 3.3-9.0 anni, sono stati esaminati. Le loro altezze erano meno di -2 SD score, il tasso di crescita era superiore al 10 ° percentile per l’età, l’età ossea era inferiore al 75% dell’età cronologica, e la concentrazione sierica stimolata di GH era maggiore di 10 μg/litro.
I bambini sono stati abbinati per sesso, altezza e punteggio SD della velocità di crescita per ricevere hGH giornaliero o a giorni alterni alla stessa dose settimanale di 6 mg/m2 per un periodo di 2 anni. Le velocità di crescita medie del 1° e 2° anno erano rispettivamente 3.4 e 2.3 SD score per il gruppo di terapia giornaliera e 3.0 e 2.0 SD score per il gruppo a giorni alterni (P = NS).
Velocità di crescita dei bambini trattati con GH a giorni alterni (▨) o con un regime giornaliero di GH prima, durante e 2 anni dopo l’interruzione della terapia. I valori sono la media ± SD. *, P < 0,05; **, P < 0,01.
Nei 6 mesi iniziali dopo la sospensione della terapia, la velocità di crescita è decelerata fino a un nadir di -3,9 SD score nel gruppo di terapia giornaliera, mentre è decelerata nel gruppo del giorno alternato a solo -0,2 SD score (P < 0,01).
Velocità di crescita pre-trattamento e cumulativa a 4 anni dei bambini trattati con GH a giorni alterni (▨) o con un regime giornaliero di GH . I valori sono la media ± SD. *, P < 0.002.
Durante tutti i 2 anni di interruzione della terapia, quest’ultimo gruppo ha mantenuto tassi di crescita medi da -0,2 a -1,2 SD score, simili alle loro velocità di pretrattamento. Il gruppo giornaliero ha recuperato lentamente per riprendere il loro tasso medio di pretrattamento solo alla quarta valutazione semestrale fuori dalla terapia.
Avanzamento annuale della crescita ossea nei bambini trattati con GH a giorni alterni (▨) o con un regime giornaliero di GH prima, durante e 2 anni dopo l’interruzione della terapia. I valori sono la media ± SD.
La velocità di crescita cumulativa a 4 anni (2 anni con e 2 anni senza terapia) del gruppo a giorni alterni era maggiore di quella del gruppo a terapia giornaliera (media, 0,9 contro 0,3 SD score; P < 0,002). Alla fine del periodo di terapia di 4 anni, la previsione di altezza da adulto del gruppo a giorni alterni era maggiore di quella del gruppo giornaliero di una media di 6,5 cm (P = 0,06).
Punteggio SD dell’altezza dei bambini trattati con GH a giorni alterni (▨) o con un regime giornaliero di GH prima, durante e 2 anni dopo l’interruzione della terapia. I valori sono la media ± SD. *, P < 0,05; **, P < 0,01. Caratteristiche cliniche di 20 pazienti che hanno ricevuto iniezioni giornaliere di hGH, rispetto a 18 pazienti che hanno ricevuto una terapia di GH a giorni alterni a una dose settimanale identica per metro quadrato di superficie corporea. 1= Test di stimolazione dell’Arginina.
Discussione oggettiva sui dati appresi:
Si tratta senza dubbio di uno studio molto approfondito e ben controllato, durato quattro anni e pubblicato sul The Journal of Clinical Endocrinology & Metabolism. Esso mostra chiaramente che le iniezioni di hGH a giorni alterni (EOD) sono molto più vantaggiose a lungo termine delle iniezioni quotidiane.
Le iniezioni quotidiane sembrano abbassare drasticamente la sensibilità del corpo alla propria secrezione di GH, e al GH esogeno. Lo studio comprendeva bambini con bassa statura idiopatica, ma i risultati possono essere estrapolati e trasposti, almeno in buona parte, a soggetti in fisiologia, e cioè non carenti di hGH e che possono utilizzare hGH esogeno periodicamente per Anti-Aging e Bodybuilding, per esempio.
Come abbiamo visto, i 38 bambini sono stati divisi in due gruppi:
Gruppo I: ha ricevuto iniezioni giornaliere di hGH;
Gruppo II: ha ricevuto iniezioni di hGH a giorni alterni.
È importante notare che il dosaggio settimanale totale di hGH era lo stesso per entrambi i gruppi. Entrambi i gruppi hanno ricevuto la terapia di hGH in modo contiguo per due anni. La loro crescita naturale è stata seguita per altri due anni dopo la fine della terapia hGH.
Sono stati tutti misurati a intervalli di tre mesi durante il periodo di quattro anni – due anni con la terapia di hGH e due anni dopo. Il GH sierico è stato misurato con un kit RIA a doppio anticorpo.
Durante la terapia con hGH, entrambi i gruppi hanno accelerato la loro crescita in modo sostanziale:
Gruppo I: ricevendo le iniezioni giornaliere di hGH nel primo e secondo anno la velocità di crescita era di 3.4 e 2.3 SD;
Gruppo II: ricevendo le iniezioni di hGH a giorni alterni aveva un tasso nella velocità di crescita di 3.0 e 2.0 SD per il primo e il secondo anno, rispettivamente.
Nel corso dei sei mesi iniziali dopo il termine della terapia, la velocità di crescita è decelerata ad un basso nadir pari a -3.9 SD di punteggio per il gruppo di terapia a somministrazione giornaliera, mentre è decelerato nel gruppo di terapia a giorni alterni di solo -0.2 SD di punteggio.
Durante i 2 anni seguenti la fine della terapia, quest’ultimo gruppo al quale sono state somministrate iniezioni EOD ha mantenuto tassi di crescita da -0.2 a -1.2 di punteggio SD, che è simile al loro punteggio SD prima del trattamento con hGH esogeno. Il gruppo giornaliero ha anch’esso mostrato un recuperato, seppur molto lentamente, alla quarta valutazione semestrale dopo la conclusione della terapia. La velocità di crescita cumulativa di 4 anni – 2 anni con e 2 anni senza terapia – del gruppo a giorni alterni era maggiore di quella del gruppo con terapia giornaliera: media, 0.9 contro 0.3 SD score.
Alla fine del periodo di terapia di 4 anni, la previsione dell’altezza adulta del gruppo a giorni alterni era maggiore di quella del gruppo giornaliero di una media di 6,5 cm – che è più di 2,5“ in altezza.
Per dirlo il più semplicemente possibile, per tradurre ciò che può significare tutto ciò per un bodybuilder, l’uso giornaliero di hGH darà solo trascurabilmente migliori risultati a breve termine. Tuttavia, l’uso di hGH a giorni alterni darà risultati radicalmente migliori a lungo termine e un recupero molto migliore. Ciò significa che il corpo può tornare all’omeostasi molto più velocemente.
I due gruppi hanno ottenuto lo stesso dosaggio settimanale totale di hGH, così che il gruppo “EOD” è stato trattato con iniezioni che comprendevano il totale del giorno successivo (es. 4UI/die e 8UI/EOD), ovvero il doppio di UI del gruppo trattato ogni giorno, ma con un totale settimanale identico! I ricercatori hanno riportato che la dose era di minore importanza rispetto al programma delle iniezioni. La terapia di hGH quotidiana per 3 anni ha causato una crescita subnormale che persiste per 1,5 anni (molto male).
Può essere che il problema non sia legato tanto ai livelli di secrezione di hGH o IGF-1, ma piuttosto alla diminuita sensibilità del corpo ad esso. La parte interessante è che i livelli sierici di GH e i livelli sierici di IGF-I e IGF-binding protein sono rimasti inalterati, o relativamente mutati.
La secrezione endogena di GH del corpo riprende in pochi giorni, anche dopo una terapia di hGH a lungo termine.
L’ipotesi dei ricercatori è che la tolleranza può essere insita nella trasduzione del segnale del GH in organi bersaglio selettivi in risposta alla scomparsa del modello unico pulsatile di GH sierico durante la terapia con GH esogeno. Ciò è dovuto al fatto che il GH assunto tramite iniezioni SubQ (sottocutanea) non corrisponde alla pulsatilità di rilascio del GH del corpo.
Pulsatilità circadiana del GH negli uomini (in altro) e nelle donne (in basso).
La somministrazione giornaliera SubQ di GH si traduce in un profilo di GH sierico non fisiologico, con livelli di picco a 3-4 ore e un lento declino nel corso delle successive 12-24 ore. Questo modello può essere considerato come una somministrazione continua, piuttosto che i naturali impulsi di GH fisiologici del corpo con una frequenza di circa otto impulsi al giorno.
Farmacocinetica GH esogeno somministrato per via parenterale sottocutanea.
Supponendo che la sindrome da astinenza sia legata alla tolleranza che potrebbe essersi sviluppata verso l’hGH o l’IGF-I, si è cercato di prevenirla con un trattamento a giorni alterni. Inoltre, le dosi di hGH utilizzate in terapia spesso stimolano l’IGF-I a livelli sierici sovrafisiologici, suggerendo che i tessuti bersaglio del IGF-I possono ovviamente essere sovrastimolati rispetto al normale. Il meccanismo sembra, quindi, risiedere nell’azione del hGH e del IGF-I nei confronti di loro tessuti bersaglio. E’ stato dimostrata, fino a prova contraria, quindi, che la terapia a giorni alterni con hGH nei bambini con un asse GH-IGF-I intatto impedisce la sindrome da astinenza.
Legame GH-GHR (Recettore del GH) e seguenti pathways.
I ricercatori collegano l’analogia con un’altra sindrome di tolleranza e astinenza endocrina: “la terapia a giorni alterni con glucocorticosteroidi previene la tolleranza a quell’ormone in misura sostanziale. È interessante notare che la sindrome da astinenza da glucocorticoidi può verificarsi anche mentre l’asse ipotalamo-ipofisi-surrene è intatto, indicando che la tolleranza ai glucocorticoidi si è sviluppata a livello dell’organo bersaglio”.
Conclusioni:
Adesso sappiamo che le iniezioni giornaliere di GH abbassano drasticamente la sensibilità del corpo all’attività dell’ormone a livello dei tessuti bersaglio, sia durante l’uso di GH esogeno sia post utilizzo (bassa risposta ai propri impulsi di GH endogeno).
Come abbiamo potuto constatare, la desensibilizzazione si è verificata, a parità di dosaggio settimanale, in risposta alla somministrazione quotidiana, a differenza del protocollo EOD.
Lo stesso GH ha una breve emivita quando viene iniettato per via endovenosa, la via di somministrazione ottimale, ma l’iniezione IM o subQ porta a un rilascio lento e prolungato e a un’elevazione al di sopra dei livelli basali per 12-24 ore, che comporta una stimolazione cronica dei recettori. Questo porta a una drammatica desensibilizzazione del tessuto bersaglio che può persiste per un lungo periodi di tempo.
Per maggiori benefici, la somministrazione di hGH in ambito Bodybuilding, che sia per la crescita muscolare, la lipolisi e l’antiaging dovrebbe aderire al dosaggio a giorni alterni per massimizzare i risultati e prevenire la tolleranza nei recettori dei tessuti bersaglio. Il dosaggio EOD per ridurre la tolleranza – mantenendo una maggiore sensibilità sia all’HGH esogeno che alla produzione endogena del corpo – ha dimostrato di produrre risultati a lungo termine molto migliori rispetto alla somministrazione quotidiana.
Repetita iuvant: La somministrazione EOD mantiene una maggiore sensibilità sia all’HGH esogeno che alla produzione endogena dell’organismo post utilizzo rispetto alle iniezioni quotidiane, mentre il dosaggio settimanale rimane lo stesso.
Praticamente, il doppio dosaggio di HGH dovrebbe essere somministrato in un giorno con un intervallo di circa 8 ore. Ad esempio al mattino e alla sera e il giorno successivo dovrebbe essere omesso, e così via. Questa somministrazione previene la tolleranza nei recettori del GH e massimizza i risultati a lungo termine. Si prega di notare che il dosaggio settimanale rimane lo stesso.
Un esempio di somministrazione “EOD” potrebbe essere il seguente:
L’hGH assunto per 12-16 settimane o più a 8 UI ogni due giorni, diviso in 4 UI a digiuno subito dopo il risveglio e altre 4 UI prese otto ore dopo. Questo approccio è abbastanza conservativo e può essere ottimale. La dose può essere ulteriormente suddivisa, se lo si desidera, per ridurre il totale delle UI iniettate in qualsiasi momento (es. 2UI appena sveglio – 2UI pre-workout – 2UI 4h dopo – 2UI prima di andare a dormire).
Ovviamente, si può estendere oltre i quattro mesi, e prendere più UI al giorno. L’approccio sopra esposto è di 8UI EOD, quindi è equivalente ad una assunzione giornaliera di 4UI, che è la media utilizzata dalla maggior parte degli utilizzatori di PEDs.
Bisogna però mettere da parte gli assolutismi, dal momento che lo studio in questione ha preso in considerazione l’altezza negli adolescenti, non la massa magra in culturisti adulti, o gli effetti Anti-Aging in adulti di mezza età, quindi è ancora una questione di sperimentazione sul campo ed estrapolazione se i risultati possono essere applicati a questi sottogruppi di utilizzatori. Comunque sia, è vero che i bodybuilder non sono bambini, né carenti di hGH idiopatico, ma la risposta sottoregolativa dei recettori del GH sono una possibilità. Vi ricordo che la “GH resistenza” esiste.
Poiché i dosaggi settimanali rimangono gli stessi, così come la durata dell’uso di hGH, il solo cambiamento del protocollo “die” a quello “EOD” varrebbe la pena di essere testato, dato che sembra statisticamente una pratica migliore rispetto al protocollo ordinario/giornaliero.
Vorrei concludere con il rendere noto che “l’ho usato tutti i giorni per mesi e mi sono tirato!” è una affermazione vuota di significato reale e realmente applicabile al discorso qui trattato: vantaggio di una somministrazione a giorni alterni di GH! Oltretutto, caro il mio bongo, dubito fortemente che tu stessi utilizzando solo GH, e che le altre molecole da te cosomministrate non abbiano avuto, a diverso grado, un impatto sulla massa grassa! Inoltre, dato ciò, non puoi affermare né uno svantaggio né una parità d’effetto delle due metodiche di cadenza nella somministrazione… a meno che tu non abbia testato tale pratica su un numero sufficiente di persone, dividendole in due gruppi trattati con una o l’altra modalità e, con la minore presenza possibile di bias, tu abbia potuto valutare oggettivamente i risultati…
Di AAS/SARM e saturazione recettoriale se ne parla spesso negli ambienti del culturismo “Enhancement“, nei social e nelle community online. Il problema è sempre il medesimo però, il quale colpisce altre argomentazioni le quali richiedono un certo livello culturale per essere trattate: se ne parla in modo confuso e male. Fortunatamente, però, su “Reddit” si tengono discussioni valide, e con letteratura al seguito, riguardo questo argomento, con persone “addette ai lavori”.
Quindi, l’obiettivo di questo breve articolo è principalmente quello di riportare i chiarimenti scientificamente supportati per ciò che concerne l’uso di AAS/SARM e la saturazione dei Recettori degli Androgeni.
Prima di proseguire, è giusto ricordare che ho una vasta conoscenza di biochimica e genetica e faccio ricerca e divulgazione scientifica da anni. Di conseguenza, le mie affermazioni non sono in alcun modo un “punto di vista” dal momento che, ed i miei lavori lo testimoniano già a sufficienza, ho una comprensione alquanto decente di ciò che viene riportato nelle pubblicazioni scientifiche.[1]
Vi espongo di seguito i “punti chiave” necessari per comprendere la questione AAS/SARM e saturazione AR:
I Recettori degli Androgeni nella maggior parte dei tessuti sono saturi all’estremità inferiore del normale intervallo fisiologico di Testosterone.
Nonostante questa saturazione, la crescita muscolare e la diminuzione della massa grassa è ancora legata al Testosterone in modo dipendente dalla dose, anche a livelli sovrafisiologici.
L’aumento della sintesi proteica non è l’unico (e forse non il principale) meccanismo attraverso il quale il Testosterone causa la crescita del muscolo-scheletrico.
Gli Androgeni sembrano causare un aumento delle cellule satelliti e dei mioonuclei nei muscoli. L’aggiunta di mionuclei alle fibre muscolari è uno dei meccanismi principali con cui essi crescono in dimensione. Questo aumento delle cellule satelliti e dei mionuclei avviene attraverso un percorso dipendente dal Recettore degli Androgeni.
In molti tessuti, l’aumento della concentrazione di Androgeni porta a un aumento della densità dei Recettori degli Androgeni. Questo può aiutare a dare una spiegazione alla possibilità di crescita potenziale maggiore “off cycle” attraverso il precedente uso di anabolizzanti. A tal proposito ricordiamoci anche della così detta “Memoria Muscolare”.[2]
L’aumento delle cellule satellite deriva dalla differenziazione delle cellule staminali mesodermiche pluripotenti. Queste sono le stesse cellule che si differenziano in adipociti (cellule del tessuto adiposo, quindi grasso). L’aumento della differenziazione di queste cellule in cellule satellite (che generano mionuclei) spiega il perché dosi più elevate di Androgeni portano a una diminuzione della massa grassa.
L’aumento delle cellule satelliti e dei mionuclei nella fibra muscolare è più che raddoppiato quando si confronta la somministrazione di 300mg vs. 600mg di Testosterone Enantato. Queste, ovviamente, sono già dosi sovrafisiologiche e questo dimostra l’opposto dei rendimenti decrescenti; tuttavia c’è ancora probabilmente un “collo di bottiglia” sconosciuto a questa differenziazione.
Notare le frecce nella figura C, che denotano fibre muscolari divise in un PowerLifter che aveva usato AAS nei precedenti 10 anni. Le fibre più piccole contenevano una isoforma in via di sviluppo della miosina (cioè miosina fetale), suggerendo che erano in realtà fibre di nuova formazione da iperplasia. La teoria qui esposta è che le fibre hanno una certa soglia di crescita, e che una volta raggiunta questa soglia, alla fine si dividono per formare nuove fibre. Con le tradizionali pratiche di allenamento “Natty”, non sembra che i PL raggiungano questa soglia; ma con l’uso di AAS, la crescita può diventare così accentuata che si verifica l’iperplasia (si noti la differenza di dimensioni delle fibre tra il PL “juiced” in Figura A e il sollevatore”Natty” in Figura B). Anche se mancano prove oggettive e inconfutabili, è logico supporre che le fibre aggiunte (e AR sovraespressi) vengano mantenute, anche se il sollevatore interrompe l’uso di AAS. La questione della possibile ipotrofia di queste nuove fibre una volta cessato l’uso di AAS è un altra possibilità.
Conclusioni:
Dosi più elevate di AAS/SARM o abbinamento di questi porteranno a risultati migliori? Ancora non lo sappiamo con certezza, sebbene i dati empirici ci portino ad una parziale conclusione favorevole al quesito posto. Per esempio, sappiamo che il Ki (con tale sigla ci si riferisce al potenziale di legame/saturazione del AR dose-dipendente) del RAD-140 è di 7nM (rispetto a 29nM del Testosterone e i 10nM del DHT).[3] Questo però non ci dà l’efficacia del ligando, ne il tasso di dissociazione (il testosterone si dissocia dal recettore degli androgeni a un tasso 5x rispetto al DHT nonostante abbia un Ki 2,9x maggiore [4]), ma se dovessimo usarlo come unico parametro di misurazione dell’efficacia, sembrerebbe così. Prendendo il tasso di biodisponibilità proposto del 65-75% (vedi riferimento Ki) del RAD nelle scimmie come punto di riferimento per gli esseri umani, un ciclo proposto di 10mg/die (concentrazione stabile intorno ai 25mg), sembrerebbe poter dare ancora dei benefici (e dei danni in termini di effetti collaterali) da dosi più elevate.
Un altro aspetto che non conosciamo è legato agli effetti AR-indipendenti del testosterone. Ci sono state proposte che collegano alcuni degli effetti del testosterone al suo antagonismo degli effetti dei glucocorticoidi attraverso il legame a bassa affinità con il recettore dei glucocorticoidi. Per quanto ne so, non abbiamo alcun indizio circa l’affinità di cui qualsiasi SARMs legano questo recettore.
Non sappiamo in termini assoluti se abbinare AAS/SARM apporti vantaggi superiori alla monoterapia, sebbene, e lo ripeto, i risultati empirici ci portano verso una risposta almeno parzialmente positiva. Ciò che bisogna evitare di fare, è smettere di usare affermazioni semplicistiche e riduttive come “la saturazione dei AR è il fattore principale che determina il tasso soggettivo di ipertrofia muscolare ottenibile”.
Esiste una interessantissima pubblicazione la quale suggerisce che sono le concentrazioni di Recettori degli Androgeni e non i livelli ormonali il fattore limitante della crescita muscolare a livelli fisiologici. Per l’appunto, LIVELLI FISIOLOGICI! Ancora una volta, vi ricordo di tenere a mente che gli androgeni sovraregolano i Recettori degli Androgeni in modo dose dipendente.[5]
Per i più informati, è ormai appurato il fatto che la soppressione dell’attività dell’Asse HPT, per via dell’uso di AAS, è determinata da:
L’origine del AAS
Il tasso di conversione del AAS ad estrogeno, attraverso l’enzima Aromatasi in alcuni tessuti (adiposo, mammario)
Dose e tempo d’uso/abuso del AAS
Attività androgena del AAS.
Di conseguenza, dovrebbe essere chiaro che anche farmaci puramente androgeni o essenzialmente anabolizzanti e con forte potenziale di legame con il AR [vedi SARM non steroidei] possono causare una sotto-regolazione della funzionalità dell’Asse HPT, quindi con meccanismi indipendenti dalla aromatizzazione della molecola.
Infatti, gli AAS [ed i SARM non steroidei] attraversano la barriera ematoencefalica e si legano ai recettori Ipotalamici. Ciò comporterà una marcata soppressione dell’HPTA per via di intermediari quali i peptidi oppioidi endogeni.
Da qualche tempo, e per i risultati ottenuti nel trattamento di soggetti dipendenti da droghe e alcool, è emerso il potenziale degli antagonisti dei recettori oppiacei, come il Naltrexone, di bloccare l’attività dei peptidi oppioidi endogeni sull’Ipotalamo e, consequenzialmente, impedire una riduzione del rilascio di GnRH e, di conseguenza, di LH e FSH.
Questo indubbio potenziale ha attirato l’interesse degli utilizzatori di AAS, sempre in cerca, e a ragione, di un modo per ridurre gli effetti collaterali in largo spetro dato dall’uso di PEDs.
In questo articolo descriverò come i recettori oppioidi situati nel Ipotalamo agiscano causando una riduzione gonadotropica e, di rimando, androgena. Tratterò in dettaglio il farmaco Naltrexone e dove questo potrebbe realmente trovare applicazione.
Ma prima un breve ripasso sul HPTA…
Dettaglia sul HPTA:
Con Asse-Ipotalamo-Ipofisi-Testicoli (HPTA) ci si riferisce alla connessione tra ipotalamo, ghiandola pituitaria e testicoli come se queste singole ghiandole endocrine fossero una singola entità. Poiché queste ghiandole spesso agiscono in concerto, i fisiologi e gli endocrinologi ritengono conveniente e descrittivo parlare di esse come di un unico sistema.
L’asse HPTA svolge una parte critica nello sviluppo e nella regolazione di un certo numero di sistemi del corpo, come i sistemi riproduttivi e immunitari. Le fluttuazioni di questo asse causano variazioni negli ormoni prodotti da ciascuna ghiandola e hanno diversi effetti locali e sistemici nel corpo.
In breve, l’asse HPT rappresenta un sistema di stimolazione/inibizione degli ormoni prodotti dalle rispettiva strutture:
Ipotalamo: GnRH (ormone di rilascio delle gonadotropine; in inglese Gonadotropin-releasing hormone).
Ipofisi (o ghiandola Pituitaria): dalle cellule beta e gamma rispettivamente l’ormone follicolo-stimolante (FSH) e l’ormone luteinizzante (LH).
Come ben sappiamo, diversi AAS sono derivati sintetici del Testosterone, il principale androgeno nei maschi. Il Testosterone sopprime marcatamente l’HPTA, mentre altri derivati lo fanno in misura maggiore o minore.
In questo specifico caso ci concentreremo sulla soppressione/sottoregolazione del HPTA Androgeno-dipendente.
Come accennato pocanzi, l’effetto sotto-regolatore dato dagli androgeni a livello Ipotalamico è mediato dai peptidi oppioidi endogeni. Ma come si verifica la disfunzione endocrina derivata dalla attivazione dei recettori oppioidi?
Oppioidi e disfunzione endocrina:
Gli oppioidi sono stati usati per secoli come metodo principale per alleviare il dolore intenso, in particolare il dolore acuto e il dolore da cancro avanzato. Gli oppioidi somministrati regolarmente 24 ore su 24 continuano a essere il cardine nella gestione del dolore associato alle condizioni di fine vita e sono sanciti nella scala analgesica dell’Organizzazione Mondiale della Sanità. Cinquemila anni fa il Papaver somniferumfu veniva coltivato dai Sumeri. Da allora si è intrecciato con l’esperienza umana di molte culture successive, fungendo da benedizione ma anche da maledizione, come analgesico e ansiolitico, stupefacente e musa. Su di esso sono state combattute guerre; qualcuno potrebbe dire che questo continua ad essere perpetrato. Nel 1804 Friedrich Sertürner isolò la morfina e nel 1843 Alexander Wood utilizzò la prima forma iniettabile di questa.
Le proprietà analgesiche degli oppioidi hanno alleviato la sofferenza di innumerevoli persone e il loro ruolo nella gestione del dolore acuto e del dolore oncologico è ben consolidato. Tuttavia, circa il 50% dei riceventi con dolore non oncologico soffre di effetti avversi e nel 20% di questi ciò porta all’interruzione della terapia. (1) Gli effetti avversi, ben noti sia agli operatori sanitari che ai non addetti, comprendono sonnolenza, costipazione, nausea, prurito e depressione respiratoria. Gli effetti avversi meno noti includono instabilità cardiovascolare, mal di testa e spasmi muscolari sia della muscolatura liscia che striata, che portano a ritenzione urinaria, rallentamento della motilità intestinale e scatti mioclonici. (2) Meno ancora saranno a conoscenza della potenziale compromissione della funzione immunitaria, nota per essere specifica per gli oppioidi.(3) I medici del dolore probabilmente conosceranno l’iperalgesia indotta da oppioidi, anche se raramente viene diagnosticata.
Gli effetti a lungo termine meno conosciuti sono la soppressione della funzione endocrina e l’effetto sulla funzione cognitiva.(3) Con l’invecchiamento della popolazione, l’aumento dei tassi di sopravvivenza al cancro e l’uso crescente di oppioidi per il dolore persistente non oncologico, il numero di prescrizioni di oppioidi nella comunità è aumentato da 6 milioni a 15 milioni nel Regno Unito tra il 1999 e il 2008 (NHS Information Center). Gli Stati Uniti hanno il 5% della popolazione mondiale ma consumano l’80% dell’offerta globale di oppioidi. Il numero di overdose fatali accidentali correlate a oppioidi da prescrizione negli Stati Uniti è ora superiore a quello per l’uso ricreativo di eroina e cocaina combinati. (4)
È per ciò fondamentale che tutti coloro che gestiscono e prescrivono oppioidi per il dolore persistente siano consapevoli degli effetti a lungo termine degli oppioidi sulla funzione endocrina e siano in grado di diagnosticare carenze, monitorare i livelli ormonali, comprendere e spiegare al paziente le possibili conseguenze di queste carenze, sforzarsi di ridurre e sospendere gli oppioidi quando appropriato e collaborare con altri medici per sostituire le carenze ormonali.
Effetti endocrini degli oppioidi:
Nel 1895 il reverendo RH Graves (5) notò come “l’oppio mangiava la virilità dell’individuo” e nel 1925 il chirurgo generale HS Cumming (6) affermò che “l’oppio rende effeminato un uomo”. Katz (7) ha già citato Charles Bruce, che, nel 1839, definì i tossicodipendenti da oppio in Assam “più effeminati delle donne”. (8)
È stato riportato in molte occasioni che gli oppioidi, somministrati per qualsiasi via, sopprimono l’asse ipotalamo-ipofisi-gonadi e hanno un impatto misurabile sulla funzione gonadica. (7)
L’ipotalamo, come sappiamo, è fondamentale per la regolazione degli ormoni sessuali. Esercita il suo controllo attraverso la secrezione dell’ormone di rilascio delle gonadotropine (GnRH) dall’area preottica nella circolazione portale ipofisaria all’eminenza mediana. Il GnRH stimola il rilascio dell’ormone follicolo-stimolante (FSH) e dell’ormone luteinizzante (LH) dall’ipofisi anteriore attivando il proprio recettore del GnRH, che, attraverso l’aumento dei livelli di calcio e proteina chinasi C, porta alla formazione e secrezione di FSH e LH (Figura seguente).
Rappresentazione del normale funzionamento dell’asse ipotalamo-ipofisi-gonadi femminile e maschile (9) . (Riprodotto con il permesso di NIAAA).
Il GnRH viene normalmente rilasciato a impulsi in tutti i vertebrati studiati. Questi impulsi producono picchi circadiani essenziali per il corretto funzionamento del sistema riproduttivo determinando quando ciascun ormone viene rilasciato. La secrezione non pulsatile di GnRH provoca una sottoregolazione dell’ipofisi e porta a una secrezione di LH deficitaria. 7
L’FSH è responsabile della crescita precoce dei follicoli ovarici nelle femmine e del mantenimento dell’epitelio spermatogeno nei maschi. L’LH è responsabile della maturazione finale dei follicoli e della loro secrezione di estrogeni, nonché dell’ovulazione e della formazione iniziale del corpo luteo nella femmina. Nel maschio stimola le cellule di Leydig a secernere testosterone. 10 Questi ormoni sono necessari anche per lo sviluppo appropriato dell’essere umano – fisiologicamente, fisicamente e socialmente.
L’asse ipotalamo-ipofisario è costantemente sotto l’effetto di molteplici sostanze tra cui neurotrasmettitori, ormoni steroidei e oppioidi endogeni. Gli oppioidi esogeni esercitano un effetto sugli stessi recettori degli oppioidi endogeni e hanno dimostrato di interferire con il rilascio (compresa la sua natura pulsatile) di GnRH. 7 , 10 , 11 Il naloxone ha mostrato un aumento dei livelli di GnRH, e quindi un aumento della concentrazione di LH e della frequenza del polso, che ha suggerito un livello basale di inibizione della secrezione di LH a base di oppioidi. È stato suggerito che la morfina inibisca la biosintesi del GnRH. 11 Gli oppioidi riducono anche il feedback negativo degli steroidi sessuali sull’ipofisi anteriore, così come la sua risposta al GnRH. 7Al contrario, la secrezione di FSH non è, o solo in minima parte, influenzata.
Con la riduzione dei livelli di LH, il testosterone e l’estradiolo si abbassano proporzionalmente. Li Shizhen scrisse dell’oppio nel suo Compendium of Materia Medica (1578) che “le persone lo usano per l’arte del sesso, in particolare per “arrestare l’emissione seminale”” 11 . Gli studi sugli animali hanno aggiunto credito all’osservazione di Shizhen, dimostrando una ricettività sessuale inibita in entrambi i sessi di ratti con oppioidi e il contrario con naloxone. 12 Ciò sembra essere dovuto alla diminuzione dell’eccitazione piuttosto che all’impotenza. In modo allarmante, studi correlati hanno mostrato che l’esposizione prepuberale agli oppioidi inibiva la maturazione sessuale. 13 Nell’uomo, in seguito al trattamento con oppioidi per via orale e intratecale, si sono verificate irregolarità mestruali, inclusa l’amenorrea. Daniele14 ha notato che c’era anche una diminuzione degli androgeni surrenali, e quindi spiegando la diminuzione della libido e delle prestazioni sessuali così spesso incontrate in coloro che sono esposti a oppioidi a lungo termine. Sembra che la diminuzione del comportamento sessuale possa anche essere dovuta all’azione diretta degli oppioidi sui recettori µ e ∂ nell’ipotalamo. 15 , 16
Il termine “ormoni sessuali” comprende gli steroidi sessuali (testosterone ed estradiolo) e gli ormoni non steroidei (GnRH, FSH e LH).
Il testosterone è il principale ormone dei testicoli, sintetizzato dal colesterolo nelle cellule di Leydig e dall’androstenedione nella corteccia surrenale. La sua secrezione è controllata da LH a 4–9 mg/die. Piccole quantità sono secrete dalle ovaie e forse dalla corteccia surrenale nelle donne. È legato per il 98% alle proteine (globulina e albumina leganti gli steroidi gonadici) nel plasma. Solo il testosterone libero e debolmente legato all’albumina è disponibile per agire sui recettori degli androgeni. Alcune cellule bersaglio convertono il testosterone in diidrotestosterone, che forma complessi ormone-recettore più stabili. Nei maschi il testosterone ha un ritmo circadiano, con i livelli più alti al mattino. La variazione massima può essere dell’ordine del 35%.
Il testosterone impartisce un feedback negativo sul rilascio di LH dall’ipofisi. Sviluppa e mantiene le caratteristiche sessuali secondarie maschili e incoraggia i comportamenti sessuali maschili. È anabolico, aumenta il tasso di crescita e, insieme all’FSH, promuove la spermatogenesi. Fa tutto questo legandosi ai recettori intracellulari e formando complessi che si legano al DNA, facilitando così una certa espressione genica.
Gli estrogeni (17ß-estradiolo, estrene ed estriolo) sono i principali steroidi sessuali femminili e sono biosintetizzati dal testosterone e dall’androstenedione. Sono secreti dalle cellule della granulosa nei follicoli ovarici. Il novantotto per cento degli estrogeni è legato alle proteine. Sono metabolizzati dal fegato ed escreti nelle urine. Gli estrogeni facilitano lo sviluppo dei follicoli ovarici, aiutano nella regolazione del ciclo mestruale e nei necessari cambiamenti nell’anatomia interna femminile e hanno effetti anabolici sull’utero e sulle tube di Falloppio. Diminuiscono i livelli di FSH e alterano i livelli di LH. Sono responsabili dei comportamenti sessuali femminili e della libido e sono in gran parte responsabili dello sviluppo del seno. Questi effetti, combinati con l’assenza di androgeni, portano a caratteristiche sessuali secondarie femminili.
In sintesi, gli oppioidi portano a una diminuzione della secrezione di GnRH, che a sua volta porta a livelli ridotti di LH. Ciò si traduce in una diminuzione della secrezione di testosterone ed estradiolo, che porta ai segni e sintomi elencati nell’immagine sottostante. È importante notare che questi cambiamenti si sviluppano nel corso di settimane o anni.
Diagnosi di ipogonadismo nel maschio:
La diagnosi di ipogonadismo dipende dall’anamnesi e dall’esame insieme ai test di laboratorio. Nel maschio postpuberale le potenziali cause di ipogonadismo primario menzionate nella tabella sottostante possono causare difficoltà diagnostiche, soprattutto per quanto riguarda la loro graduale insorgenza. Se si sospetta l’ipogonadismo, è quindi importante differenziare l’insufficienza gonadica primaria da quella dell’asse ipotalamo-ipofisario.
Per la seconda consultazione internazionale dell’Organizzazione mondiale della sanità sulla disfunzione erettile, che ha considerato il ritmo circadiano e la natura pulsante della secrezione di Testosterone, dovrebbero essere prelevati due campioni di sangue tra le 8:00 e le 11:00 (quando si presume che i livelli di Testosterone siano al massimo, sebbene il ritmo circadiano può diminuire con l’aumentare dell’età e con variazioni tra sportivi e sedentari). I campioni devono essere inviati per la misurazione del testosterone sierico, della globulina legante gli ormoni sessuali (SHBG), della prolattina, dei livelli di FSH e LH.
Livelli di FSH/LH da normali ad alti potrebbero indicare un ipogonadismo primario (vedi tabella sopra). È importante misurare il livello di FSH poiché ha un’emivita più lunga e dimostra una minore variabilità rispetto all’LH. Ipogonadismo secondario (Tabella seguente) è indicato da bassi livelli di testosterone e livelli di FSH/LH da normali a bassi ( Riquadro seguante).
Con l’invecchiamento si riduce la fluttuazione diurna del testosterone sierico (le variabili vi sono nella popolazione sportivamente attiva). Il livello scende notevolmente durante il giorno, rafforzando l’importanza del campionamento mattutino. Nell’ipogonadismo in questa fascia di età i livelli di testosterone totale possono essere normali se i livelli di globulina legante gli ormoni sessuali (SHBG) sono aumentati. I livelli di SHBG aumentano con l’età e quindi riducono la biodisponibilità del testosterone.
I dati recenti del Massachusetts Male Aging Study (MMAS) forniscono prospettive sui normali intervalli di androgeni, come mostrato nella tabella seguente.
Diagnosi di ipogonadismo nella femmina:
Allo stesso modo, l’ipogonadismo nelle femmine può essere dovuto a un asse ipotalamo-ipofisario o a un difetto gonadico primario. I segni ei sintomi sono strettamente legati al ciclo mestruale negli anni postmenarca e premenopausale. I segni comuni includono oligomenorrea, amenorrea e mancato concepimento. Possono verificarsi sintomi più sottili come vampate di calore e ansia, raramente con cambiamenti nella distribuzione dei peli pubici e nella dimensione del seno 21 . Sebbene gli effetti clinici possano essere gravi per le donne come per gli uomini, i cambiamenti esteriori o visibili non sono così evidenti e non sono stati studiati in dettaglio, specialmente nelle donne in postmenopausa.
Sebbene sia stato dimostrato che la carenza di androgeni è sintomatica nelle donne che utilizzano la terapia con oppioidi intratecali e in quelle in trattamento con metadone di mantenimento, i livelli di testosterone non sono stati misurati di routine. Si ritiene che il diidroepiandrosterone (DHEA) sia abbassato ed è noto che i livelli di LH sono notevolmente ridotti. Il DHEA è un marker della produzione di androgeni surrenali e circa il 50% degli androgeni prodotti nella femmina sono di origine surrenale. Vi è una scarsità di dati per quanto riguarda i livelli ormonali nelle donne e sono necessari ulteriori studi per quantificare il significato clinico di questo aspetto potenzialmente molto interessante della carenza di androgeni indotta da oppioidi (OPIAD).
Effetto degli oppioidi sugli ormoni surrenali:
Il dolore acuto e cronico, come risposte fisiologiche, determina un aumento della secrezione dell’ormone adrenocorticotropo (ACTH) e del cortisolo. Tuttavia, in diversi studi è stato riscontrato che l’uso cronico di oppioidi esogeni riduce i livelli di ACTH e cortisolo e le risposte del cortisolo alle sfide dell’adrenocorticotropina. 19 Gli oppioidi influenzano anche i ritmi circadiani della secrezione di cortisolo, determinando un aumento persistente dei livelli di ACTH e cortisolo e alla fine attenuando la risposta allo stress. 20 Anche i livelli di deidroepiandrosterone solfato (DHEAS), un precursore degli androgeni surrenali, sono stati notevolmente ridotti nei consumatori cronici di oppioidi sia maschi che femmine. 14 , 18Occasionalmente, la somministrazione di oppioidi può causare insufficienza surrenalica franca, ma i fattori di rischio per questo sono attualmente sconosciuti. 15
Prolattina:
La somministrazione acuta di oppioidi stimola il rilascio di prolattina dall’ipofisi anteriore attraverso un effetto a livello dell’ipotalamo. 15 Questo effetto può essere bloccato dalla metoclopramide, suggerendo che sia mediato da meccanismi dopaminergici. L’effetto della somministrazione a lungo termine di oppioidi sulla prolattina è meno chiaro. C’è un aumento occasionale del rilascio di prolattina, probabilmente dipendente dal tipo di oppioide. Il significato clinico di questo è sconosciuto, ma può causare galattorrea.
Ormoni tiroidei:
In generale, gli oppioidi non sembrano alterare in modo significativo la funzione tiroidea, 19 sebbene possano stimolare l’ormone stimolante la tiroide (TSH) attraverso l’ipotalamo. 15 Questo non è importante negli individui con tiroxina libera normale, ma gli individui con ipotiroidismo possono avere risposte prolungate ed esagerate agli oppioidi. 22
Ormone della crescita:
La somministrazione acuta di oppioidi porta ad un aumento della secrezione dell’ormone della crescita (GH), attraverso meccanismi che coinvolgono i recettori degli oppioidi, i livelli di feedback e la trascrizione genica. La dose minima richiesta è di circa 15 mg di morfina. 15 Abs et al. 23 hanno riscontrato una carenza di GH in circa il 15% dei pazienti che ricevevano oppioidi intratecali a lungo termine, ma non in tutti i pazienti. È stato dimostrato che il naloxone inibisce il GH nei soggetti sani, ma lo aumenta nelle donne obese. L’effetto della somministrazione cronica di oppioidi sul GH è complesso e attualmente poco conosciuto, ma sembra essere correlato agli ormoni sessuali, alla composizione corporea e al grado di insulino-resistenza. 15
Vasopressina:
È stato scoperto che il tramadolo causa iponatriemia attraverso il rilascio di vasopressina indotto dalla serotonina. 24 L’effetto degli oppioidi sull’ipofisi posteriore non è chiaro: sono stati riscontrati livelli di vasopressina sia aumentati che diminuiti, a seconda dello stato di idratazione. 22
Ossitocina:
Studi su donne in gravidanza hanno dimostrato che la morfina inibisce la produzione di ossitocina nelle prime fasi del travaglio e durante l’allattamento al seno dopo il parto. 25
Obesità e diabete:
Un numero crescente di dati suggerisce che gli oppioidi svolgono un ruolo nella regolazione dell’assunzione di cibo e della scelta del cibo, e forse la ricompensa associata ai cibi di buon gusto, attraverso meccanismi centrali. 15 L’uso cronico di oppioidi è associato ad aumento di peso, iperglicemia e peggioramento del diabete. Questa può essere un’azione centrale attraverso il sistema nervoso simpatico e una ridotta secrezione di insulina. 26 L’ipogonadismo è associato ad un aumento della resistenza all’insulina e al rischio di diabete mellito, 15 un rischio che è migliorato dalla sostituzione del testosterone. 27
Metabolismo delle catecolamine:
La terapia con oppioidi aumenta la secrezione di catecolamine attraverso l’ipotalamo e il tronco cerebrale. I pazienti che assumono oppioidi a lungo termine devono essere sottoposti a screening per l’ipertensione. 22
Metabolismo osseo:
Esistono molti fattori di rischio per la diminuzione della densità minerale ossea e l’osteoporosi nei pazienti trattati con oppioidi, tra cui possibile scarso stato nutrizionale, ipogonadismo, inibizione degli osteoblasti, ridotta sintesi di osteocalcina, calcio anormale e ormone paratiroideo e aumento del riassorbimento osseo, mediato dall’interleuchina 1. rappresenta un aumento del rischio di frattura ossea nei pazienti che assumono oppioidi. 3
Via di somministrazione:
Gli oppioidi per uso cronico vengono somministrati per via orale o intratecale. Occasionalmente possono essere utilizzate altre vie, ad esempio l’iniezione ripetuta, sebbene questa pratica non sia raccomandata. 28
Cambiamenti ormonali che si verificano dopo la somministrazione intratecale sono stati riportati da diversi autori. 23 , 29 , 30
Si stima che il 90% dei pazienti che assumono oppioidi intratecali svilupperà ipogonadismo. Gli oppioidi orali hanno lo stesso effetto, sebbene l’inizio dell’azione possa essere più lento.
Tipo e dose di oppioide:
Le classi di oppioidi differiscono nel loro effetto sulla soppressione delle gonadi. Tramadolo e buprenorfina non alterano significativamente i livelli di testosterone negli animali e nell’uomo e la buprenorfina non sopprime il cortisolo sierico.
L’incidenza di ipogonadismo era maggiore nei sopravvissuti al cancro che ricevevano una dose equivalente o superiore a 200 mg di morfina al giorno per almeno 1 anno rispetto ai sopravvissuti di pari età non in terapia con oppioidi, suggerendo che gli effetti sono correlati alla dose. 31 Anche la durata della terapia con oppioidi sembra aumentare la possibilità di soppressione ormonale, sebbene ciò debba essere studiato ulteriormente.
Esiste una possibile differenza di genere negli effetti endocrini, tendente a una maggiore sintomatologia nelle donne, ma sono necessari ulteriori studi. 22
Quando e cosa si misura prima di iniziare una terapia con oppioidi:
Prima di iniziare la terapia cronica con oppioidi si raccomanda di misurare quanto segue:
pressione sanguigna;
elettroliti (soprattutto se si utilizza tramadolo);
livelli di glucosio a digiuno;
funzione tiroidea (per escludere l’ipotiroidismo);
livelli di testosterone, globulina legante l’ormone sessuale, LH/FSH ed estradiolo; e
densità ossea (in un gruppo “a rischio”).
Monitoraggio:
Non ci sono standard accettati, ma sembra ragionevole ripetere i test di cui sopra ogni 6 mesi.
Considera i livelli di cortisolo nel sangue, DHEA, ACTH e GH a digiuno mattutino. (Ricorda che un livello di cortisolo nel sangue a digiuno anormalmente alto può rappresentare la perdita della variazione diurna e dovresti chiedere consiglio.)
Ripetere la densità ossea ogni anno nel gruppo “a rischio”.
Misurare i livelli di prolattina se c’è galattorrea.
Terapia sostitutiva:
Non esistono standard accettati per la gestione della disfunzione endocrina indotta da oppioidi.
L’opzione migliore è ridurre gradualmente e ritirare gli oppioidi e monitorare la risposta per un periodo di alcuni mesi, se appropriato. Non è noto se il cambio di oppioidi sia di qualche beneficio. La buprenorfina sembra avere un effetto minore sugli ormoni surrenali ma ha un effetto maggiore sul TSH rispetto alla morfina. La risposta a diversi oppioidi è in gran parte sconosciuta al momento della scrittura.
Se non è possibile ottenere l’astinenza da oppiacei e il paziente presenta sintomi definiti di disfunzione endocrina, si raccomanda la sostituzione ormonale, con il monitoraggio da parte di un endocrinologo.
Il testosterone può essere sostituito, sia negli uomini che nelle donne, come cerotto o gel transdermico o per iniezione. È necessario un attento monitoraggio poiché gli effetti collaterali includono reazioni in sede, policitemia e aumento del rischio di cancro alla prostata negli uomini e irregolarità mestruali e irsutismo nelle donne.
La terapia sostitutiva con estrogeni è meglio monitorata da un ginecologo.
Cosa estrapolare di utile per un utilizzatore di PEDs da quanto detto?
Innanzi tutto bisogna sapere che l’attività di soppressione/sottoregolazione dell’Asse HPT androgeno-dipendente ha come intermediari i peptidi oppioidi endogeni, con attività principale da parte della Beta-Endorfina, delle Encefaline e Dinorfine attraverso il legame con i recettori oppioidi μ.
Quindi, di conseguenza, comprendere i meccanismi e gli effetti dell’uso di oppioidi sulla omeostasi ormonale, soprattutto riguardante l’HPTA, ci permette di capire meglio il legame tra attività AR di una molecola e suoi effetti mediati a livello ipotalamico che causano una sottoregolazione/soppressione della funzione del Asse HPT, anche se la molecola non è aromatizzabile e non possiede attività estrogenica e/o progestinica.
Adesso possiamo approfondire il discorso passando all’analisi del antagonisti degli oppioidi Naltrexone.
Caratteristiche del Naltrexone:
Il Naltrexone, venduto con tra gli altri con i nomi commerciali di ReVia e Vivitrol, è un farmaco utilizzato principalmente per gestire l’abuso di alcol o il disturbo da uso di oppioidi, riducendo le voglie e le sensazioni di euforia associate al disturbo da uso di sostanze “compensative”.[32] È stato anche osservato essere efficace nel trattamento di altre dipendenze e può essere utilizzato per loro in modalità off-label. [33] Una persona dipendente da oppioidi non dovrebbe ricevere il Naltrexone prima della disintossicazione.[4] Viene assunto per bocca o tramite iniezione intra-muscolo.[32] Gli effetti iniziano entro 30 minuti dalla somministrazione.[32] Una diminuzione del desiderio di oppioidi può richiedere alcune settimane per verificarsi.[32]
Il Naltrexone è un antagonista degli oppioidi e funziona bloccando gli effetti degli oppioidi, sia quelli endogeni che esogeni.[32]
Il Naltrexone è stato prodotto per la prima volta nel 1965 ed è stato approvato per uso medico negli Stati Uniti nel 1984.[32][34] Il Naltrexone, come Naltrexone/bupropione (nome commerciale Contrave), è anche usato per trattare l’obesità.[35]
Il Naltrexone, noto anche come N-ciclopropil-metilnorossimorfone, è un derivato dell’Ossimorfone (14-idrossi-diidromorfone). È specificamente il derivato dell’Ossimorfone in cui il sostituto metilico dell’ammina terziaria è sostituito con il Metilciclopropano. Il farmaco strettamente correlato, il Metilnaltrexone (N-metilnaltrexone), è usato per trattare la costipazione indotta dagli oppioidi, ma non tratta la dipendenza in quanto non attraversa la barriera emato-encefalica e, quindi, non è di nostro interesse per l’ipotetico utilizzo in ambito di PEDs. Il Nalmefene (6-desossi-6-metilenaltrexone) è simile al Naltrexone ed è usato per gli stessi scopi. Il Naltrexone non deve essere confuso con il Naloxone (N-allylnoroxymorphone), che è usato in casi di emergenza di overdose da oppioidi. Altri antagonisti degli oppioidi correlati al Naltrexone includono 6β-naltrexol (6β-idrossinaltrexone), Samidorphan (3-carbossamido-4-idrossinaltrexone), β-funaltrexamine (Naltrexone Fumarato Metil Estere), Nalodeina (N-allylnorcodeine), Nalorphine (N-allylnormorphine), e Nalbuphine (N-cyclobutylmethyl-14-hydroxydihydronormorphine).
Farmacocinetica del Naltrexone:
L’assorbimento del Naltrexone con la somministrazione orale è rapido e quasi completo (96%).[36] La biodisponibilità del Naltrexone con la somministrazione orale è dal 5 al 60% a causa di un esteso metabolismo di primo passaggio.[37][38] Le concentrazioni di picco del naltrexone sono da 19 a 44 μg/L dopo una singola dose orale di 100 mg e il tempo per le concentrazioni di picco del naltrexone e del 6β-naltrexol (metabolita) è entro 1 ora. [37][38][36] Aumenti lineari nelle concentrazioni circolanti di naltrexone e 6β-naltrexolo in un intervallo di dosi orali da 50 a 200 mg.[37] Il naltrexone non sembra essere accumulato con la somministrazione orale ripetuta una volta al giorno e non vi è alcun cambiamento nel tempo di picco delle concentrazioni con la somministrazione ripetuta.[37]
6β-naltrexolo
Il legame alle proteine plasmatiche del naltrexone è di circa il 20% in un intervallo di concentrazione di naltrexone da 0,1 a 500 μg/L.[37][36] Il suo volume apparente di distribuzione a 100 mg per via orale è di 16,1 L/kg dopo una singola dose e 14,2 L/kg con dosi ripetute.[37]
Il naltrexone viene metabolizzato nel fegato principalmente dalle diidrodiol deidrogenasi in 6β-naltrexolo (6β-idrossinaltrexone).[37][38] I livelli di 6β-naltrexolo sono da 10 a 30 volte superiori a quelli del naltrexone con la somministrazione orale a causa del vasto metabolismo di primo passaggio. [39] Al contrario, l’esposizione al 6β-naltrexolo è solo circa 2 volte superiore a quella del naltrexone con l’iniezione intramuscolare di naltrexone in microsfere (nome commerciale Vivitrol).[40] Il 6β-Naltrexolo è un antagonista dei recettori degli oppioidi in modo simile al naltrexone e mostra un profilo di legame comparabile ai recettori degli oppioidi. [41] Tuttavia, il 6β-naltrexolo è selettivo a livello periferico e attraversa il cervello molto meno facilmente del naltrexone.[41] In ogni caso, il 6β-naltrexolo mostra ancora una certa attività centrale e può contribuire significativamente alle azioni centrali del naltrexone orale. [41][37] Altri metaboliti del naltrexone includono il 2-idrossi-3-metossi-6β-naltrexolo e il 2-idrossi-3-metossinaltrexone.[37] Dopo la loro formazione, i metaboliti del naltrexone sono ulteriormente metabolizzati per coniugazione con l’acido glucuronico per formare glucuronidi.[37] Il Naltrexone non è metabolizzato dal sistema del citocromo P450 e ha un basso potenziale di interazioni farmacologiche.[42]
L’eliminazione del naltrexone è biesponenziale e rapida nelle prime 24 ore seguita da un terzo declino estremamente lento dopo 24 ore.[37] Le emivite di eliminazione rapida del naltrexone e del suo metabolita 6β-naltrexolo sono rispettivamente di circa 4 ore e 13 ore.[43] Nelle compresse orali Contrave, che contengono anche bupropione e sono descritte come a rilascio prolungato, l’emivita del naltrexone è di 5 ore. [44] L’emivita di eliminazione lenta in fase terminale del naltrexone è di circa 96 ore.[45] Come microsfere di Naltrexone per iniezione intramuscolare (Vivitrol), le emivite di eliminazione del naltrexone e del 6β-naltrexolo sono entrambe di 5-10 giorni.[46] Mentre il naltrexone orale viene somministrato quotidianamente, il naltrexone in microsfere per iniezione intramuscolare è adatto alla somministrazione una volta ogni 4 settimane o una volta al mese.[46]
Livelli ematici di Naltrexone dopo una dose orale di 50mg allo stato stazionario durante il trattamento con 50mg/giorno di Naltrexone.Livelli ematici di Naltrexone dopo una dose di 380mg in microsfere (Vivitrol) per iniezione intramuscolare allo stato stazionario durante il trattamento mensile con 380mg di Naltrexone in microsfere.
Il Naltrexone e i suoi metaboliti sono escreti nelle urine.[36]
Farmacodinamica del Naltrexone:
Il Naltrexone e il suo metabolita attivo 6β-naltrexolo sono antagonisti competitivi dei recettori oppioidi.[47][48] Il naltrexone è specificamente un antagonista preferenzialmente del recettore μ-opioide (MOR), in misura minore del recettore κ-opioide (KOR), e in misura molto minore del recettore δ-opioide (DOR). [47] Tuttavia, il naltrexone non è in realtà un antagonista silenzioso di questi recettori, ma agisce invece come un debole agonista parziale, con valori di Emax dal 14 al 29% al MOR, dal 16 al 39% al KOR, e dal 14 al 25% al DOR in diversi studi.[48][49][50] In accordo con il suo agonismo parziale, sebbene il naltrexone sia descritto come un puro antagonista dei recettori oppioidi, ha mostrato alcune prove di deboli effetti oppioidi in studi clinici e preclinici.[37]
Da solo, il naltrexone agisce come un antagonista o un debole agonista parziale dei recettori oppioidi.[48] In combinazione con agonisti del MOR come la morfina, tuttavia, il naltrexone sembra diventare un agonista inverso del MOR.[48] Al contrario, il naltrexone rimane un antagonista neutro (o un debole agonista parziale) del KOR e DOR. [In contrasto con il naltrexone, il 6β-naltrexolo è puramente un antagonista neutro dei recettori oppioidi.[51] L’agonismo inverso MOR del naltrexone quando è co-presente con agonisti MOR può in parte spiegare la sua capacità di far precipitare l’astinenza in individui dipendenti da oppioidi.[51][48] Ciò può essere dovuto alla soppressione della segnalazione MOR basale attraverso l’agonismo inverso.[51][48]
L’occupazione dei recettori degli oppioidi nel cervello da parte del Naltrexone è stata studiata utilizzando la tomografia a emissione di positroni (PET).[52][53] Il Naltrexone alla dose di 50 mg/giorno è risultato occupare circa il 90-95% dei MOR del cervello e il 20-35% dei DOR del cervello. [52] Il Naltrexone alla dose di 100 mg/giorno è stato osservato raggiungere l’87% e il 92% di occupazione cerebrale del KOR in diversi studi.[54][53][55] Per simulazione, una dose più bassa di Naltrexone di 25 mg/giorno potrebbe essere prevista per raggiungere circa il 60% di occupazione cerebrale del KOR ma ancora vicino al 90% di occupazione del MOR. [53] In uno studio sulla durata del blocco del MOR con il Naltrexone, il farmaco con una singola dose da 50mg ha mostrato un blocco del 91% del legame cerebrale al [11C]carfentanil (un ligando selettivo del MOR) a 48 ore (2 giorni), un blocco dell’80% a 72 ore (3 giorni), un blocco del 46% a 120 ore (5 giorni) e un blocco del 30% a 168 ore (7 giorni). [8][9] Il tempo di dimezzamento del blocco MOR del cervello da parte del Naltrexone in questo studio era da 72 a 108 ore (3. 0 a 4,5 giorni).[56][45] Sulla base di questi risultati, dosi di Naltrexone anche inferiori a 50mg/giorno dovrebbero raggiungere un’occupazione dei MOR cerebrali praticamente completa.[8][9] Il blocco dei MOR cerebrali con Naltrexone è molto più duraturo rispetto ad altri antagonisti oppioidi come il Naloxone (tempo di dimezzamento di ~ 1,7 ore per via intranasale) o il Nalmefene (tempo di dimezzamento di ~ 29 ore).[56][57][58]
Nalmefene
L’emivita di occupazione dei MOR cerebrali e la durata dell’effetto clinico del Naltrexone sono molto più lunghe di quanto suggerito dalla sua emivita di eliminazione plasmatica.[56][59][45] Una singola dose orale di 50mg di Naltrexone ha mostrato di bloccare i MOR cerebrali e gli effetti oppioidi per almeno 48-72 ore. [59][45][60] L’emivita del blocco dei MOR cerebrali da parte del Naltrexone (72-108 ore) è molto più lunga della componente di clearance plasmatica rapida del Naltrexone e del 6β-naltrexolo (~4-12 ore), ma è stato riportato che corrisponde bene alla fase terminale più lunga della clearance plasmatica del Naltrexone (96 ore). [56][45][61] Come possibilità alternativa, la prolungata occupazione cerebrale dei MOR da parte degli antagonisti oppioidi come il Naltrexone e il Nalmefene può essere dovuta alla lenta dissociazione dai MOR conseguente alla loro affinità MOR molto alta (<1,0 nM).[58][62]
Il Naltrexone blocca gli effetti degli agonisti MOR come la Morfina, l’Eroina e l’Idromorfone nell’uomo attraverso il suo antagonismo MOR.[37][42] Dopo una singola dose di 100mg di Naltrexone, gli effetti soggettivi e oggettivi dell’Eroina sono stati bloccati del 90% a 24 ore, con un blocco che è diminuito fino a 72 ore.[1] Allo stesso modo, da 20 a 200mg di Naltrexone hanno antagonizzato in modo dose-dipendente gli effetti dell’Eroina fino a 72 ore. [37] Il Naltrexone blocca anche gli effetti degli agonisti KOR come la Salvinorina A, la Pentazocina e il Butorfanolo negli esseri umani attraverso il suo antagonismo KOR.[63][64][65][66] Oltre agli oppioidi, il Naltrexone è stato osservato bloccare o ridurre gli effetti gratificanti e altri effetti di altre droghe euforizzanti tra cui l’alcol,[67] la Nicotina,[68] e le Anfetamine.[69]
Come visto in precedenza, i recettori degli oppioidi sono coinvolti nella regolazione neuroendocrina.[37] Gli agonisti MOR producono aumenti dei livelli di Prolattina e diminuzioni dei livelli di Ormone Luteinizzante (LH) e Testosterone. [37] Dosi di Naltrexone da 25 a 150mg/giorno sono state osservate produrre aumenti significativi nei livelli di β-endorfina, Cortisolo e LH, cambiamenti equivoci nei livelli di Prolattina e Testosterone, e nessun cambiamento significativo nei livelli di ormone Adrenocorticotropico (ACTH) o Ormone Follicolo-Stimolante (FSH). [37] Il Naltrexone influenza l’Asse Ipotalamo-Ipofisi-Surrene (Asse HPT) probabilmente attraverso l’interferenza con la segnalazione del recettore degli oppioidi da parte delle endorfine.
Si pensa che il blocco dei MOR sia il meccanismo d’azione del Naltrexone nella gestione della dipendenza da oppioidi – blocca o attenua reversibilmente gli effetti degli oppioidi. Si pensa anche che sia coinvolto nell’efficacia del Naltrexone nella dipendenza da alcol riducendo gli effetti euforici di quest’ultimo. Il ruolo della modulazione del KOR da parte del Naltrexone nella sua efficacia per la dipendenza da alcol non è chiaro, ma questa azione potrebbe anche essere coinvolta in base alla teoria e agli studi sugli animali.[70][71]
Oltre ai recettori degli oppioidi, il Naltrexone si lega e agisce come antagonista del recettore del fattore di crescita degli oppioidi (OGFR) e del recettore toll-like 4 (TLR4) e interagisce con siti di legame ad alta e bassa affinità nella filamina-A (FLNA).[72][73][74][75] Si dice che dosi molto basse di Naltrexone (<0. 001-1 mg/die) interagiscono con FLNA, basse dosi (da 1 a 5 mg/die) producono antagonismo di TLR4, e dosi cliniche standard (da 50 a 100 mg/die) esercitano antagonismo del recettore degli oppioidi e OGFR.[72][74] Le interazioni del Naltrexone con FLNA e TLR4 sono ritenute coinvolte negli effetti terapeutici del Naltrexone a basse dosi.[72]
Farmacogenetica del Naltrexone: Prove provvisorie suggeriscono che la storia familiare e la presenza del polimorfismo Asn40Asp predice l’efficacia del Naltrexone.[76][77]
Effetti collaterali del Naltrexone:
Gli effetti collaterali più comuni riportati con il Naltrexone sono disturbi gastrointestinali come diarrea e crampi addominali.[36] Questi effetti avversi sono analoghi ai sintomi dell’astinenza da oppioidi, poiché il blocco del recettore μ-opioide aumenterà la motilità gastrointestinale.
Gli effetti collaterali del naltrexone per incidenza sono i seguenti:[36]
Più del 10%: difficoltà a dormire, ansia, nervosismo, dolori addominali/crampi, nausea e/o vomito, poca energia, dolori articolari/muscolari e mal di testa.[36]
Meno del 10%: perdita di appetito, diarrea, costipazione, sete, aumento di energia, sensazione di depressione, irritabilità, vertigini, eruzione cutanea, eiaculazione ritardata, disfunzione erettile e brividi.[36]
Una varietà di altri eventi avversi sono stati riportati anche con un’incidenza inferiore all’1%.[36] È stato segnalato che il Naltrexone può causare danni al fegato se somministrato a dosi più elevate di quelle raccomandate.[52] È oggetto di un avvertimento della FDA per questo raro effetto collaterale.
Ipotesi d’uso del Naltrexone in contesto PEDs:
Ora, abbiamo tutte le informazioni necessarie per poter comprendere come un antagonista dei recettori oppioidi possa portare a dei possibili vantaggi durante l’uso di PEDs o, meglio, di alcuni farmaci rientranti in tale categoria.
Riassumendo in breve:
Sappiamo che la sottoregolazione/soppressione del Asse HPT avviene anche in modo androgeno-dipendente: quindi, anche nel caso di molecole non aromatizzabili e non aventi attività estrogeniche intrinseche o progestiniche;
Sappiamo che i peptidi oppioidi endogeni agiscono da intermediari dell’attività androgena ipotalamica: attività che vede una maggiore interazione da parte delle Encefaline, Beta-Endorfine e Dinorfine con i recettori oppioidi mu (μ) (MOR OP3(I));
Sappiamo che il Naltrexone può bloccare l’attività dei delle Encefaline, Beta-Endorfine e Dinorfine con i recettori oppioidi mu (μ) (MOR OP3(I));
Sappiamo che tale blocco recettoriale può tradursi in una mancata (o per lo meno marcata e significativa) sottoregolazione/soppressione androgeno-dipendente del Asse HPT.
Se a ciò aggiungiamo che tale potenziale sarebbe al quanto insignificante in un contesto d’uso di AAS aromatizzabili e/o aventi attività estrogenica intrinseca e/o progestinica, anche volendoci abbinare un SERM il gioco non varrebbe la candela, l’unica possibilità d’uso con ritorno “positivo” si verificherebbe senza dubbio con l’uso di soli SARM non steroidei o, tutt’al più, con l’uso di DHT derivati senza la sopramenzionata caratteristica di attività estrogenica intrinseca (quindi niente Oxymetholone).
Per quanto rimanga un idiozia per un atleta di sesso maschile intraprendere protocolli di soli SARM non steroidei o AAS non-aromatizzabili, o progestinici, senza una base per lo meno fisiologica di Testosterone o l’uso di hCG onde garantire almeno un sufficiente “equilibrio” etsrogenico e di DHT, rappresenti il culmine dell’ignoranza e dell’incompetenza (se parliamo di “preparatori”), per i “testofobici” e gli “agofobici” l’uso del Naltrexone potrebbe (e, sottolineo, il condizionale “potrebbe”) garantire una conservazione sufficiente dell’attività del Asse HPT.
Nonostante vi siano scarsi lavori che hanno osservato l’attività cerebrale dei SARM non steroidei, e che uno in particolare abbia affermato che l’Ostarina non passi in modo significativo la barriera ematoencefalica, questa categoria di PEDs è per lo più strutturata su modifiche della molecola antiandrogena Bicalutamide la quale oltrepassa la barriera ematoencefalica.
Molecole di Ostarina (sinistra) e Bicalutamide (destra) a confronto.
Sebbene le modifiche strutturali determinino, per ovvie ragioni, una diversa farmacodinamica, il fatto che la sottoregolazione del HPTA, misurata anche a dosi di 3mg/die di Ostarina, sia con tutta probabilità androgeno-dipendente a livello ipotalamico [androgeno dipendente in senso di legame con i AR non nel senso di effetto androgeno genico] mi spinge ad ipotizzare una potenziale utilità del Naltrexone. Senza considerare che SARM non steroidei come LGD 4033, nonostante abbiano caratteristiche strutturali diverse dall’Ostarina o dalla Andarina, causino sottoregolazioni molto più marcate dei SARM non steroidei con struttura base del Bicalutamide.
LGD4033
Conclusioni:
Questo articolo è un esposizione accurata di una ipotesi e nulla più! Per quanto sensata possa essa essere, non abbiamo attualmente documentazione scientifica che dimostri inconfutabilmente che tale pratica sia funzionale e vantaggiosa o tantomeno sicura.
Il Naltrexone è un farmaco con una serie di potenziali effetti avversi (vedi sopra). Alcuni di essi potrebbero, in alcuni soggetti, vanificare i vantaggi sulla sfera sessuale ottenuti da un ipotetica conservazione del attività del HPTA. Da considerare anche la cura della dose somministrata per possibile danno epatico da sovradosaggio.
Gabriel Bellizzi
Riferimenti:
Moore AR, McQuay HJ. Prevalenza di eventi avversi da oppioidi nel dolore cronico non maligno: revisione sistematica di studi randomizzati di oppioidi orali . Artrite Res Ther 2005; 7 : 1046–1051. [ Articolo gratuito PMC ] [ PubMed ] [ Google Scholar ]2.
Furlan AD, Sandoval JA, Mailis-Gagnon A, et al. Oppioidi per il dolore cronico non oncologico: una meta-analisi di efficacia ed effetti collaterali . Can Med Assoc J 2006; 174 : 1589–1594. [ Articolo gratuito PMC ] [ PubMed ] [ Google Scholar ]3.
Raghavan S, Harvey AD, Humble SR. Nuovi effetti collaterali e implicazioni per gli oppioidi per la terapia a lungo termine . Tendenze Anaesth Crit Care 2011; 1 : 18–21. [ Google Scholar ]4.
Okie S. Un’ondata di oppioidi, una marea crescente di morti . N inglese J Med 2010; 363 ( 21 ): 1981–1983. [ PubMed ] [ Google Scholar ]5.
Graves RH. Quarant’anni in Cina, Baltimora: Woodward , 1895. [ Google Scholar ]6.
Cumming HS, Control of DrugAddiction Mainly a Police Problem, The American City Magazine (novembre 1925, fascicolo 126, riquadro 3, sottoserie 1, serie 3.7.
Katz N, Mazer NA. L’impatto degli oppioidi sul sistema endocrino . Clin J Dolore 2009; 25 ( 2 ): 170–175. [ PubMed ] [ Google Scholar ]8.
Bruce CA. Relazione sulla produzione del tè e sull’estensione e la produzione delle piantagioni di tè nell’Assam . Calcutta: : Bishop’s College Press, 1839. [ Google Scholar ]9.
Hiller-Sturmhofel S, Bartke A. Il sistema endocrino: una panoramica . Alcol Health Res World 1998; 22 ( 3 ): 153–164. [ Articolo gratuito PMC ] [ PubMed ] [ Google Scholar ]10.
Ganong WF. Rassegna di fisiologia medica . USA: Lange/McGraw-Hill, 2005. [ Google Scholar ]11.
Luo Xiwen tr. Bencao Gangmu: Compendio di Materia Medica . Foreign Languages Press, 2003. Pubblicato per la prima volta nel 157812.
Van Vugt DA, Baby N, Stewart M, Reid RL. L’effetto stimolante paradossale della morfina sulla secrezione di LH è dose-dipendente e reversibile al naloxone . Neuroendocrinologia 1989; 50 : 109–116. [ PubMed ] [ Google Scholar ]13.
De la Rosa RE, Hennessey JV. Ipogonadismo e metadone: ipogonadismo ipotalamico dopo l’uso a lungo termine di metadone ad alte dosi . Pratica Endocr 1996; 2 :4–7. [ PubMed ] [ Google Scholar ]14.
Daniel HW. Deficit di DHEAS durante il consumo di oppioidi prescritti ad azione prolungata: evidenza dell’inibizione indotta da oppioidi della produzione di androgeni surrenali . J Dolore 2006; 7 ( 12 ): 901–907. [ PubMed ] [ Google Scholar ]15.
Vuong C, Van Uum SHM, O’Dell L, et al. Gli effetti degli oppioidi e degli analoghi degli oppioidi sui sistemi endocrini animali e umani . Endocr Rev 2010; 31 ( 1 ): 98–132. [ Articolo gratuito PMC ] [ PubMed ] [ Google Scholar ]16.
Van Furth WR, van Emst MG, van Ree JM. Oppioidi e comportamento sessuale dei ratti maschi: coinvolgimento dell’area preottica mediale . Comportamento Neurosci 1995; 109 ; 123–134. [ PubMed ] [ Google Scholar ]17.
Daniell Harry W. Le linee guida cliniche della Endocrine Society sulla terapia con testosterone negli uomini adulti con sindromi da carenza di androgeni . J Clin Metab endocrinolo 2010; 95 ( 6 ): 2536–2559. [ PubMed ] [ Google Scholar ]18.
O’Donne AB, Araujo AB, McKinlay JB. La salute degli uomini che invecchiano normalmente: The Massachusetts Male Aging Study (1987–2004) . Scad. Gerontolo 2004; ( 7 ): 975–984. [ PubMed ] [ Google Scholar ]19.
Katz N. L’impatto degli oppioidi sul sistema endocrino . Round di gestione del dolore 2005; 1 ( 9 ). www.painmanagementrounds.org (visitato il 5 febbraio 2012) [ Google Scholar ]20.
Faccinetti F, Grasso A, Petraglia F, et al. Ridotta ritmicità circadiana di β-lipotropina, β-endorfina e ACTH nei tossicodipendenti . Acta endocrinolo 1984; 105 ( 2 ): 149–155. [ PubMed ] [ Google Scholar ]21.
Daniel HW. Endocrinopatia da oppioidi nelle donne che consumano oppioidi ad azione prolungata prescritti per il controllo del dolore non maligno . J Dolore 2008; 9 ( 1 ): 28–36. [ PubMed ] [ Google Scholar ]22.
Thosani S, Jiminez C. Alterazioni biochimiche dell’asse neuroendocrino indotte da oppioidi . Esperto Rev Endocrinol Metab 2011; 6 ( 5 ): 705–713. [ Google Scholar ]23.
Abs R, Verhelst J, Maeyaert J, et al. Conseguenze endocrine della somministrazione intratecale a lungo termine di oppioidi . J Clin Endo Metab 2000; 85 ( 6 ): 2215–2222. [ PubMed ] [ Google Scholar ]24.
Sarret D, Le Berre JP, Zemraoui N. Iponatriemia indotta da Tramadol . Am J Kidney Dis 2008; 52 ( 5 ): 1026. [ PubMed ] [ Google Scholar ]25.
Lindow SW, Hendricks MS, Nugent FA, et al. La morfina sopprime la risposta all’ossitocina nelle donne che allattano . Gynecol Obstet Invest 1999; 48 ( 1 ): 33–37. [ PubMed ] [ Google Scholar ]26.
Giugliano D. Morfina, peptidi oppioidi e funzione delle isole pancreatiche . Cura del diabete 1984; 7 : 92–98. [ PubMed ] [ Google Scholar ]27.
Kapoor D, Goodwin E, Channer KS, Jones TH. La terapia sostitutiva del testosterone migliora la resistenza all’insulina, il controllo glicemico, l’adiposità viscerale e l’ipercolesterolemia negli uomini ipogonadici con diabete di tipo 2 . Eur J Endocrinolo 2006; 154 : 899–906. [ PubMed ] [ Google Scholar ]28. Oppiacei per il dolore persistente; buona pratica . 2010.
The British Pain Society, Facoltà di Medicina del Dolore del Royal College of Anesthetists, Royal College of General Practitioners e Facoltà di Medicina delle Dipendenze del Royal College of Psychiatrists . Disponibile su: http://www.britishpainsociety.org/book_opioid_main.pdf (visitato il 5 febbraio 2012)
29. Roberts LJ, Finch PM, Pullan PT, et al. Soppressione dell’ormone sessuale da parte degli oppioidi intratecali: uno studio prospettico . Clin J Dolore 2002; 18 : 144–148. [ PubMed ] [ Google Scholar ]
30.Thimineur MA, Kravitz E, Vodapally MS. Trattamento intratecale con oppioidi per il dolore cronico non maligno: uno studio prospettico di 3 anni . Dolore 2004; 109 : 242–249. [ PubMed ] [ Google Scholar ]
31.Rajagopal A, Vasilopoulou-Sellin R, Palmer JL, et al. Ipogonadismo sintomatico nei sopravvissuti maschi al cancro con esposizione cronica agli oppioidi . Cancro 2004; 100 ( 4 ): 851–858. [ PubMed ] [ Google Scholar ]
37. Gonzalez JP, Brogden RN (March 1988). “Naltrexone. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic efficacy in the management of opioid dependence”. Drugs. 35 (3): 192–213.
38. Lee MW, Fujioka K (August 2009). “Naltrexone for the treatment of obesity: review and update”. Expert Opin Pharmacother. 10 (11): 1841–5.
39. Davis MP, Glare PA, Hardy J (28 May 2009). Opioids in Cancer Pain. Oxford University Press. pp. 41–.
40. Dunbar JL, Turncliff RZ, Dong Q, Silverman BL, Ehrich EW, Lasseter KC (March 2006). “Single- and multiple-dose pharmacokinetics of long-acting injectable naltrexone”. Alcohol Clin Exp Res. 30 (3): 480–90.
41.Hipkin, R. William; Dolle, Roland E. (2010). “Opioid Receptor Antagonists for Gastrointestinal Dysfunction”. Annual Reports in Medicinal Chemistry. Vol. 45. Elsevier. pp. 142–155.
42.Sevarino, Kevin A.; Kosten, Thomas R. (2009). “Naltrexone for Initiation and Maintenance of Opiate Abstinence”. Opiate Receptors and Antagonists. Humana Press. pp. 227–245.
48.Wang D, Sun X, Sadee W (May 2007). “Different effects of opioid antagonists on mu-, delta-, and kappa-opioid receptors with and without agonist pretreatment”. J Pharmacol Exp Ther. 321 (2): 544–52.
51.Sadée W, Wang D, Bilsky EJ (February 2005). “Basal opioid receptor activity, neutral antagonists, and therapeutic opportunities”. Life Sci. 76 (13): 1427–37.
52.Ray LA, Chin PF, Miotto K (March 2010). “Naltrexone for the treatment of alcoholism: clinical findings, mechanisms of action, and pharmacogenetics”. CNS Neurol Disord Drug Targets. 9 (1): 13–22.
53.de Laat B, Nabulsi N, Huang Y, O’Malley SS, Froehlich JC, Morris ED, Krishnan-Sarin S (June 2020). “Occupancy of the kappa opioid receptor by naltrexone predicts reduction in drinking and craving”. Mol Psychiatry. 26 (9): 5053–5060.
54.Placzek MS (August 2021). “Imaging Kappa Opioid Receptors in the Living Brain with Positron Emission Tomography”. Handb Exp Pharmacol. Handbook of Experimental Pharmacology. 271: 547–577.
56.Colasanti, Alessandro; Lingford-Hughes, Anne; Nutt, David (2013). “Opioids Neuroimaging”. In Miller, Peter M. (ed.). Biological Research on Addiction. Comprehensive Addictive Behaviors and Disorders. Vol. 2. Elsevier. pp. 675–687.
57.van Waarde, Aren; Absalom, Anthony R.; Visser, Anniek K. D.; Dierckx, Rudi A. J. O. (30 September 2020). “Positron Emission Tomography (PET) Imaging of Opioid Receptors”. PET and SPECT of Neurobiological Systems. Springer International Publishing. pp. 749–807.
58.Soyka M, Rösner S (November 2010). “Nalmefene for treatment of alcohol dependence”. Expert Opin Investig Drugs. 19 (11): 1451–9.
61.Miotto K, McCann M, Basch J, Rawson R, Ling W (2002). “Naltrexone and dysphoria: fact or myth?”. Am J Addict. 11 (2): 151–60.
62.Ingman K, Hagelberg N, Aalto S, Någren K, Juhakoski A, Karhuvaara S, Kallio A, Oikonen V, Hietala J, Scheinin H (December 2005). “Prolonged central mu-opioid receptor occupancy after single and repeated nalmefene dosing”. Neuropsychopharmacology. 30 (12): 2245–53.
65.Preston KL, Bigelow GE (February 1993). “Differential naltrexone antagonism of hydromorphone and pentazocine effects in human volunteers”. J Pharmacol Exp Ther. 264 (2): 813–23.
66.Clark SD, Abi-Dargham A (October 2019). “The Role of Dynorphin and the Kappa Opioid Receptor in the Symptomatology of Schizophrenia: A Review of the Evidence”. Biol Psychiatry. 86 (7): 502–511.
67.Unterwald EM (September 2008). “Naltrexone in the treatment of alcohol dependence”. J Addict Med. 2 (3): 121–7.
68.Modesto-Lowe V, Van Kirk J (August 2002). “Clinical uses of naltrexone: a review of the evidence”. Exp Clin Psychopharmacol. 10 (3): 213–27.
69.Lam L, Anand S, Li X, Tse ML, Zhao JX, Chan EW (April 2019). “Efficacy and safety of naltrexone for amfetamine and methamfetamine use disorder: a systematic review of randomized controlled trials”. Clin Toxicol (Phila). 57 (4): 225–233.
70.Soyka M, Friede M, Schnitker J (March 2016). “Comparing Nalmefene and Naltrexone in Alcohol Dependence: Are there any Differences? Results from an Indirect Meta-Analysis”. Pharmacopsychiatry. 49 (2): 66–75.
74. Lee B, Elston DM (June 2019). “The uses of naltrexone in dermatologic conditions”. J Am Acad Dermatol. 80 (6): 1746–1752.
75.Zagon IS, Verderame MF, McLaughlin PJ (February 2002). “The biology of the opioid growth factor receptor (OGFr)”. Brain Res Brain Res Rev. 38 (3): 351–76.
77.Garbutt JC, Greenblatt AM, West SL, Morgan LC, Kampov-Polevoy A, Jordan HS, Bobashev GV (August 2014). “Clinical and biological moderators of response to naltrexone in alcohol dependence: a systematic review of the evidence”. Addiction. 109 (8): 1274–84.
OPK-88004 è un nuovo SARM non steroideo sviluppato dalla Transition Therapeutics, e che è stato acquistato dalla OKPO nel 2016.
Struttura molecolare del OPK-88004
Uno studio di recente pubblicazione svolto su questo SARM sembra aver mostrato che causa un aumento dose-dipendente della massa muscolare, che diminuisce la massa grassa e aumenta anche la quantità di Testosterone libero. Prima di eccitarvi troppo sull’ultimo punto, vediamo nel dettaglio lo studio. Caratteristiche dello studio:
In questo studio controllato con placebo, randomizzato, in doppio cieco, 114 uomini, di età ≥19 anni, che avevano subito una prostatectomia radicale per un cancro alla prostata di basso grado e localizzato all’organo, PSA non rilevabile (<0,1 ng/mL) per ≥2 anni dopo la prostatectomia radicale e carenza di Testosterone sono stati randomizzati per gradi a placebo [0mg] o 1, 5, o 15mg/die di OPK-88004 per 12 settimane. I risultati includevano la recidiva del PSA, l’attività sessuale, il desiderio sessuale, la funzione erettile, la composizione corporea, la forza muscolare e le misure della funzione fisica, l’umore, la fatica e i marker ossei.
Risultati dello studio:
I partecipanti avevano un’età media di 67,5 anni e una grave disfunzione sessuale (punteggi medi della funzione erettile e del dominio del desiderio sessuale 7,3 e 14,6, rispettivamente). Nessun partecipante ha avuto recidive di PSA o eritrocitosi. OPK-88004 è stato associato a un aumento correlato alla dose della massa magra [non specificatamente muscolare] (P <0,001) e appendicolare (P <0,001) e a una diminuzione significativamente maggiore della percentuale di grasso corporeo (P <0,001) e della fosfatasi alcalina nel siero (P <0,001) rispetto al placebo. I cambiamenti nell’attività sessuale, il desiderio sessuale, la funzione erettile, l’umore, l’affaticamento, le prestazioni fisiche e i marker ossei non differiscono tra i gruppi (P = 0,73).
Risultati dei test per valutare il miglioramento delle prestazioni fisiche.Variabili ormonali riscontrate durante lo studio.
Conclusioni sul OPK-88004:
La somministrazione di OPK-88004 è stata sicura e non è stata associata alla recidiva del PSA in uomini con deficit di androgeni che erano stati sottoposti a prostatectomia radicale per cancro alla prostata confinato all’organo. OPK-88004 ha aumentato la massa corporea magra e diminuito la massa grassa, ma non ha migliorato i sintomi sessuali o le prestazioni fisiche.
In conseguenza dei dati estrapolati dallo studio ivi esposto, e nonostante i dati di sicurezza a breve termine siano rassicuranti, questo SARM non steroideo mostra i difetti dei suoi predecessori:
Nonostante vi sia un aumento del Testosterone libero che potrebbe interessare maggiormente il pubblico rispetto al Testosterone totale, non bisogna dimenticarsi del fatto che gli Androgeni possono interagire con le attività cellulari anche attraverso interazioni non genomiche (non mediate direttamente dal recettore androgeno), le quali avvengono anche con l’ormone legato all’albumina (trasportatore ematico che lega l’ormone sessuale; pari circa al 55-35% del Testosterone). Inoltre, anche SARM non steroidei “datati” come l’Ostarina hanno mostrato le medesime caratteristiche sui livelli di Testosterone.
La diminuzione dell’Estradiolo (E2) può causare, in misura dipendente dall’entità del calo e dalla sensibilità individuale, molteplici problemi come depressione, letargia, affaticabilità, ansia, o disfuzione erettile (o difficolta a raggiungere l’erezione e/o a mantenerla) e riduzione della libido.
Problemi legati ai precedenti sono riscontrabili dalla riduzione del DHT, in maniera sempre dipendente dall’entità del calo e dalla sensibilità individuale. A tal proposito si veda la Testosterone:Estradiolo ratio o la più approfondita DHT:Estradiolo ratio.
Non è un caso che il trattamento con il suddetto SARM non abbia portato ad un aumento della funzione sessuale.
La mancanza di miglioramento nelle prestazioni fisiche e dei marker ossei, non lo rende molto allettante.
L’unico punto interessante rimane la riduzione della massa grassa, essendo l’aumento della massa magra estremamente generico e non inducibile a specifiche miotrofiche. Nonostante ciò, i possibili svantaggi superano di netto il suddetto vantaggio che, oltretutto, è riscontrabile in maniera accentuata in un “vecchio” SARM non steroideo, L’Andarina (S4).
Ora, come si può vedere, analizzando con logica tutti i dati in nostro possesso, possiamo valutare concretamente questo nuovo SARM non steroideo e rilegarlo in una posizione di basso interesse sia per uso terapeutico che “off-label”. Ma la ricerca effettuata non è letteralmente da buttare. I dati raccolti devono spingere la ricerca a migliorare queste molecole, sviluppando nuovi SARM che migliorino non solo il trofismo muscolo-scheletrico ma che portino ad una ottimale funzione sessuale e delle prestazioni psicofisiche.
L’Anamorelina (INN) (nomi in codice di sviluppo ONO-7643, RC-1291, ST-1291), nota anche come Anamorelina Cloridrato (USAN, JAN), è un agonista selettivo non peptidico, attivo per via orale, penetrante-centrale, selettivo del recettore della grelina / un secretagogo dell’Ormone della Crescita (GHSR) con effetti anabolizzanti e di stimolo dell’appetito, sviluppato dalla Helsinn Healthcare SA per il trattamento della cachessia e dell’anoressia da cancro.[2][3][4]
Struttura molecolare della Anamorelina
L’Anamorelina aumenta significativamente i livelli plasmatici dell’Ormone della Crescita (GH), del Fattore di Crescita Insulino-Simile 1 (IGF-1) e della proteina legante il Fattore di Crescita Insulino-Simile 3 (IGFBP-3) nell’uomo, senza influenzare i livelli plasmatici di Prolattina, Cortisolo, Insulina, Glucosio (in acuto), Ormone Adrenocorticotropo (ACTH), Ormone Luteinizzante (LH), Ormone Follicolo-Stimolante (FSH), e dell’Ormone Stimolante la Tiroide (TSH). [3][5] Inoltre, l’Anamorelina aumenta significativamente l’appetito, il peso corporeo complessivo, la massa magra e la forza muscolare,[4][5] con aumenti di peso corporeo direttamente correlati ad aumenti dei livelli plasmatici di IGF-1.[3]
Vie di azione della Anamorelina (e del Ibutamoren).
Un interessante studio sulla Anamorelina del 2018:
Oncologi giapponesi, affiliati all’Institute of Biomedical Research and Innovation, hanno pubblicato uno studio sulla rivista scientifica “Cancer” nel 2018. In questo studio, i ricercatori hanno diviso 174 persone con cancro al polmone avanzato in 2 gruppi.
Un gruppo ha ricevuto un placebo per 12 settimane, l’altro ha assunto giornalmente 2 compresse contenente ognuna 50mg di Anamorelina. I soggetti hanno quindi assunto 100mg di Anamorelina al giorno.
Rispetto al gruppo placebo, la somministrazione di Anamorelina ha portato a un aumento della massa corporea magra di poco meno di 1.5Kg. Anche se la sperimentazione è durata 12 settimane, questo aumento si è verificato completamente durante le prime 3 settimane dello studio. Una classica risposta da adattamento omeostatico.
Si può vedere nella figura sopra il perché l’Anamorelina perde il suo effetto dopo 3 settimane. Durante le prime 3 settimane di prova, le concentrazioni sia di IGF-1 che di IGF-BP3 nel sangue erano aumentate, dopo di che si è verificato un calo. La IGF-BP3 è una proteina legante che, per l’appunto, lega l’IGF-1 proteggendolo dalla degradazione enzimatica, ma non ne riduce l’attività biologica sulla matrice ossea e il muscolo scheletrico.
Un altro studio del 2019 dimostra la riduzione d’efficacia dopo 3 settimane:
Naturalmente bisogna sempre stare attenti a prendere per affidabili le affermazioni riportate in un singolo studio. Ma, seppur non rappresenti un enorme differenza, esiste un altro studio, in qualche misura comparabile con il precedente, ed è sempre uno studio giapponese, condotto tra persone con cancro colorettale, allo stomaco o al pancreas in stadio avanzato, dove la somministrazione di 100mg di Anamorelina ha stimolato il guadagno di massa magra solo per le prime 3 settimane.
Stato legale della Amorelina:
Fino a poco tempo, l’Anamorelina faceva parte della corposa compagine di molecole sperimentali (vedi SARM-non steroidei, SPPARM, ecc…) venduti dal mercato “grigio” dei siti UK.
Infatti, mentre le ricerche sul Ibutamoren sono state interrotte, quelle sulla Anamorelina, d’altra parte, sono proseguite ed essa è stata approvato per uso medico, per lo meno in Giappone.
L’Anamorelina è disponibile nel paese del sol levante sotto il nome di Adlumiz dall’aprile 2021. Il farmaco è destinato al trattamento di persone che stanno perdendo massa muscolare e forza a causa di una condizione di cancro avanzato.
Confezione di Adlumiz venduta in Giappone
A partire dal febbraio del 2016, l’Anamorelina ha completato gli studi clinici di fase III per il trattamento della cachessia e dell’anoressia associate al carcinoma polmonare non a piccole cellule.[6][7]
Il 18 maggio 2017, l’Agenzia europea per i medicinali ha raccomandato il rifiuto dell’autorizzazione all’immissione in commercio del medicinale, destinato al trattamento dell’anoressia, della cachessia o della perdita di peso involontaria in pazienti con carcinoma polmonare non a piccole cellule. La Helsinn ha chiesto un riesame del parere iniziale. Dopo aver considerato le motivazioni di questa richiesta, l’Agenzia europea per i medicinali ha riesaminato il parere, e ha confermato il rifiuto dell’autorizzazione all’immissione in commercio il 14 settembre 2017.[8] L’Agenzia europea per i medicinali ha concluso che gli studi mostrano un effetto marginale dell’Anamorelina sulla massa corporea magra e nessun effetto dimostrato sulla forza della presa delle mani o sulla qualità della vita dei pazienti. Inoltre, a seguito di un’ispezione presso i siti degli studi clinici, l’agenzia ha ritenuto che i dati sulla sicurezza del farmaco non fossero stati registrati adeguatamente. Pertanto, l’agenzia era del parere che i benefici dell’Anamorelina non superassero i suoi rischi.[9]
Conclusioni:
Nonostante la sua autorizzazione in Giappone per l’uso nel trattamento di pazienti umani affetti da cancro in stato avanzato, gli studi non mostrano reali vantaggi terapeutici dal momento che l’effetto del farmaco sembra limitato a tre settimane di uso giornaliero.
Anche se alcuni bodybuilder hanno utilizzato la molecola come sostituto del Ibutamoren, quest’ultimo sembra dimostrare un efficacia prolungata rispetto alla Anamorelina.
Ricordo, inoltre, che secondo legislazione italiana, anche l’acquisto e il possesso di SARM-non steroidei, SPPARM (vedi Cardarina) ed altri è illegale soggetto a sequestro e ammenda dipendenti dalla quantità di materiale in possesso.
Currow DC, Abernethy AP (April 2014). “Anamorelin hydrochloride in the treatment of cancer anorexia-cachexia syndrome”. Future Oncology. 10 (5): 789–802.
Garcia JM, Polvino WJ (June 2009). “Pharmacodynamic hormonal effects of anamorelin, a novel oral ghrelin mimetic and growth hormone secretagogue in healthy volunteers”. Growth Hormone & IGF Research. 19 (3): 267–73.
Garcia JM, Boccia RV, Graham CD, Yan Y, Duus EM, Allen S, Friend J (January 2015). “Anamorelin for patients with cancer cachexia: an integrated analysis of two phase 2, randomised, placebo-controlled, double-blind trials”. The Lancet. Oncology.
Garcia JM, Friend J, Allen S (January 2013). “Therapeutic potential of anamorelin, a novel, oral ghrelin mimetic, in patients with cancer-related cachexia: a multicenter, randomized, double-blind, crossover, pilot study”. Supportive Care in Cancer. 21 (1): 129–37.
Temel JS, Abernethy AP, Currow DC, Friend J, Duus EM, Yan Y, Fearon KC (April 2016). “Anamorelin in patients with non-small-cell lung cancer and cachexia (ROMANA 1 and ROMANA 2): results from two randomised, double-blind, phase 3 trials”. The Lancet. Oncology. 17 (4): 519–531.

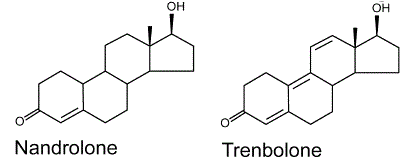
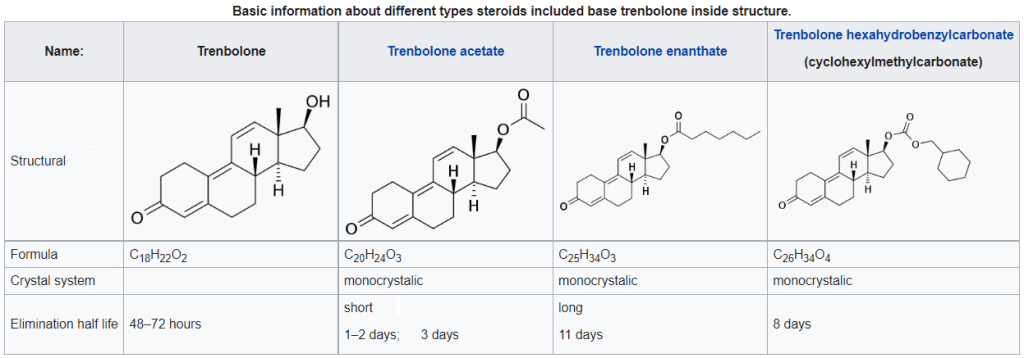













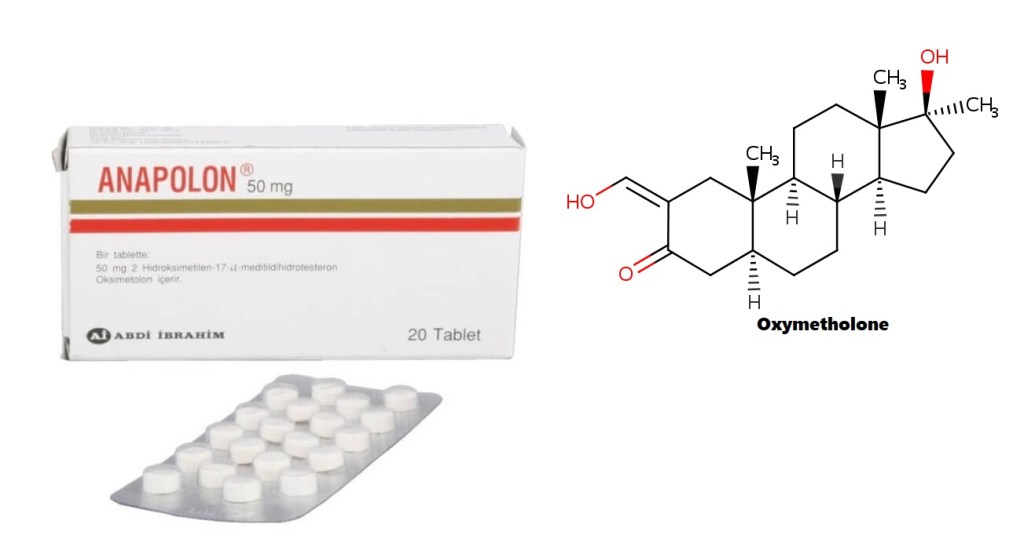

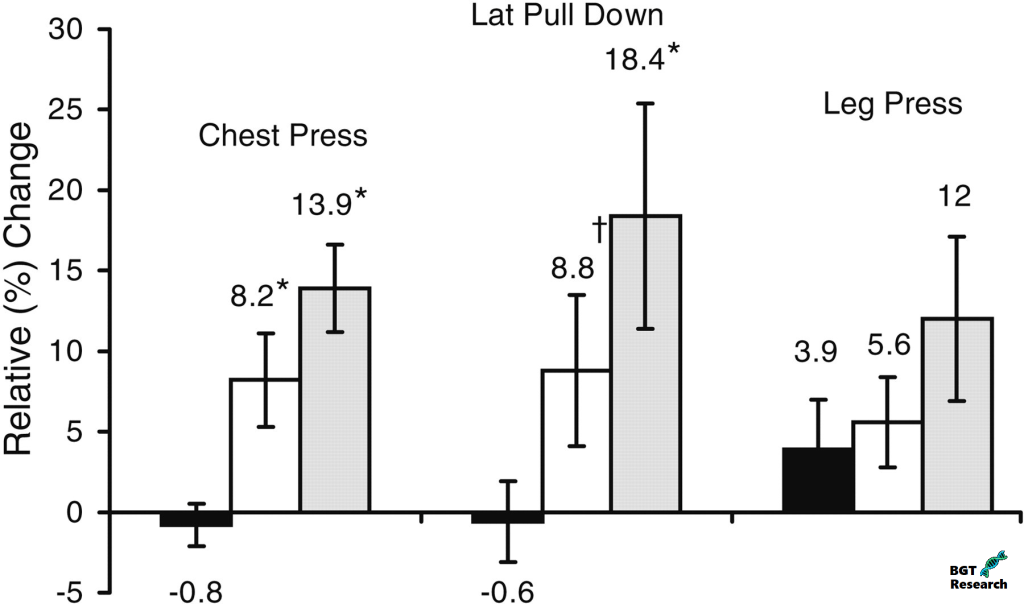

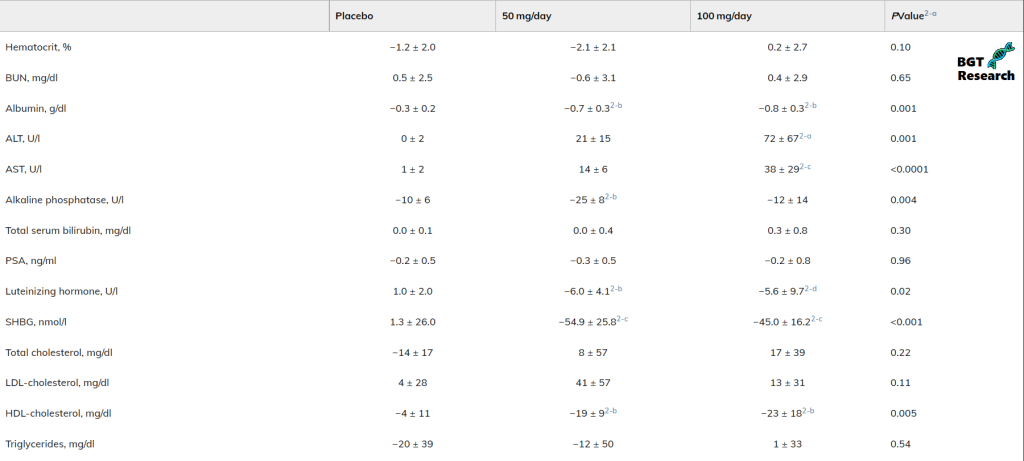

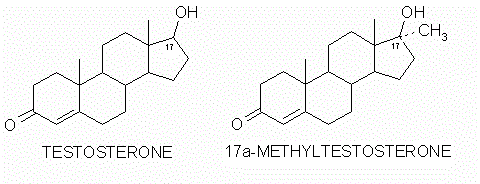
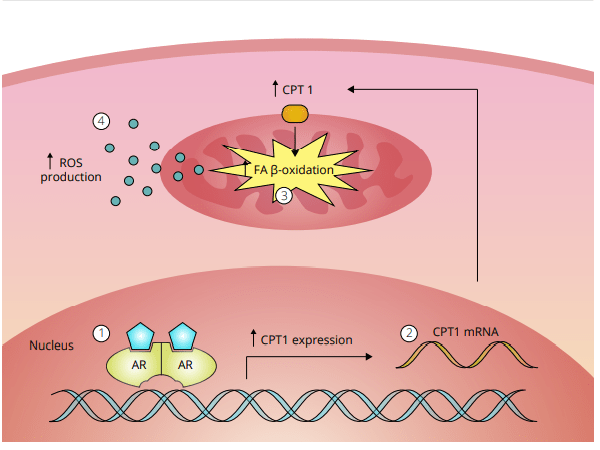



 prima, durante e 2 anni dopo l’interruzione della terapia. I valori sono la media ± SD. *, P < 0,05; **, P < 0,01.
prima, durante e 2 anni dopo l’interruzione della terapia. I valori sono la media ± SD. *, P < 0,05; **, P < 0,01.