Introduzione alla Parte 5:
Nella 4° parte abbiamo analizzato le caratteristiche e funzioni biochimiche dei BCAA. In questa quinta parte, invece, andremo ad analizzare il metabolita della Leucina, l’acido β-idrossi-β-metilbutirrico/HMB.
HMB – storia, ricerca e caratteristiche biochimiche:

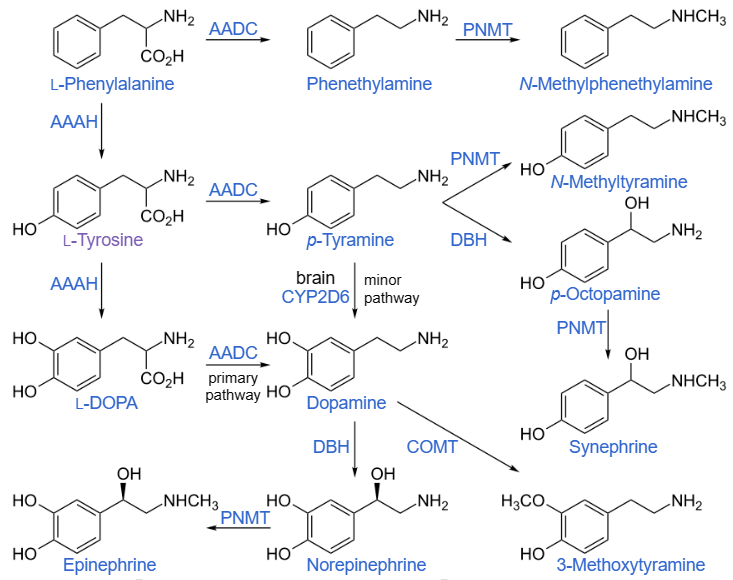
L’HMB, o acido β-idrossi-β-metilbutirrico, è un metabolita naturale dell’aminoacido leucina, dove la leucina si converte nel suo analogo cheto-isocaproato (cheto-isocaproato o KIC) e poi si converte in HMB (attraverso l’enzima citosolico KIC diossigenasi[1]);[2] va notato che la versione mitocondriale della KIC diossigenasi converte il KIC nel derivato CoA dell’acido isovalerico (β-idrossiisovalerato).[1]
Tutto l’HMB endogeno deriva dalla leucina[2] e la produzione di HMB è correlata all’assunzione di leucina con la dieta (sembra una cinetica di primo ordine per la KIC diossigenasi citosolica[3][1]), con circa il 5% di tutta l’ossidazione della leucina in vivo che si traduce nella formazione di HMB.[2] Sebbene l’HMB plasmatico tenda a circolare intorno a 1-4µM, può aumentare di 5-10 volte dopo un pasto ricco di leucina.[3]
L’HMB è un metabolita della leucina alimentare nel corpo umano e media una serie di effetti della leucina. L’assunzione di leucina con la dieta può aumentare la formazione di HMB e circa il 5% della leucina alimentare viene convertita in HMB nell’organismo.

L’HMB è un membro della famiglia dei composti organici dell’acido carbossilico.[4] È un analogo strutturale dell’acido butirrico con un gruppo funzionale idrossile e un sostituente metile situato sul carbonio beta.[4][5] Per estensione, altri analoghi strutturali includono l’acido β-idrossibutirrico e l’acido β-metilbutirrico.[4][5]

La prima sintesi chimica dell’HMB è stata pubblicata nel 1877 dai chimici russi Michael e Alexander Zaytsev.[6] L’HMB è stato isolato dalla corteccia dell’Erythrophleum couminga (un albero del Madagascar) nel 1941 da Leopold Ružička.[7] L’isolamento più precoce dell’HMB come metabolita umano è stato effettuato da Tanaka e collaboratori nel 1968 da un paziente affetto da acidemia isovalerica.[8][9]
Gli effetti dell’HMB sul muscolo scheletrico umano sono stati scoperti per la prima volta da Steven L. Nissen della Iowa State University a metà degli anni ’90.[8][10] Nissen ha fondato un’azienda chiamata Metabolic Technologies, Inc. (MTI) all’epoca della sua scoperta, che in seguito ha acquisito sei brevetti relativi all’HMB che l’azienda ha utilizzato per concedere in licenza il diritto di produrre e incorporare l’HMB negli integratori alimentari. [10][11][12] Quando è stato commercializzato per la prima volta alla fine degli anni ’90, l’HMB è stato commercializzato esclusivamente come integratore per l’esercizio fisico, per aiutare gli atleti e i bodybuilder a costruire i muscoli.[11] MTI ha successivamente sviluppato due prodotti contenenti HMB, Juven e Revigor, di cui Abbott Nutrition ha ottenuto i diritti di commercializzazione rispettivamente nel 2003 e nel 2008.[8][11] Da allora, Abbott ha commercializzato Juven come alimento medico e il marchio Revigor di HMB come ingrediente attivo in prodotti alimentari (ad es, alcune formulazioni di Ensure) e altri alimenti medici (ad esempio, alcune formulazioni di Juven).[8][13][11]
Sono state sviluppate diverse vie sintetiche per l’HMB. Le prime sintesi chimiche riportate hanno avvicinato l’HMB all’ossidazione di precursori alchenici, dioli vicinali e alcol:
- nel 1877, i chimici russi Michael e Alexander Zaytsev riportarono la preparazione dell’HMB per ossidazione del 2-metilpent-4-en-2-olo con acido cromico (H2CrO4);[6]
- nel 1880 e nel 1889, Schirokoff e Reformatsky (rispettivamente) riportarono che la scissione ossidativa del diolo vicinale 4-metilpentano-1,2.,4-triolo con potassio acidificato, 4-triolo con permanganato di potassio acidificato (KMnO4) produce HMB[14][15] – questo risultato è più vicino alla prima sintesi, poiché il KMnO4 diluito a freddo ossida gli alcheni a cis-dioli vicinali che il KMnO4 acido a caldo ossida ulteriormente a composti contenenti carbonile, mentre l’intermedio diolo non si ottiene quando si utilizzano condizioni acide a caldo per l’ossidazione degli alcheni. [In altre parole, il 4-metilpentano-1,2,4-triolo racemico è un derivato del 2-metilpent-4-en-2-olo e l’acido β-idrossi-β-metilbutirrico è un derivato di entrambi,
- nel 1892, Kondakow riportò la preparazione dell’HMB per ossidazione con permanganato del 3-metilbutano-1,3-diolo.
A seconda delle condizioni sperimentali, la cicloaddizione di acetone e chetene produce il β-isovalerolattone o il 4,4-dimetilossetan-2-one,[16][17] che si idrolizzano entrambi in condizioni basiche per produrre la base coniugata dell’HMB. La reazione aloformica fornisce un’altra via per l’HMB che comporta l’alogenazione esaustiva della regione metil-chetonica dell’alcol di diacetone con ipobromito di sodio o ipoclorito di sodio;[5][18][19] l’alcol di diacetone è facilmente disponibile dalla condensazione aldolica dell’acetone. [Un approccio organometallico all’HMB prevede la carbossilazione dell’alcol tert-butilico con monossido di carbonio e reagente di Fenton (perossido di idrogeno e ferro).[5][20] In alternativa, l’HMB può essere preparato attraverso l’ossidazione microbica dell’acido β-metilbutirrico da parte del fungo Galactomyces reessii.[21]
HMB nella supplementazione sportiva:

L’HMB può essere integrato sotto forma di sale di calcio monoidrato (comunemente chiamato HMB di calcio) o come acido libero, ovvero HMB senza il sale di calcio. Il sale di calcio ha una costante di dissociazione simile a quella dell’acetato di calcio[22] e ha un Tmax dell’ordine di 1-2 ore dopo l’ingestione di 1 g di Ca-HMB, con un picco di 487,9+/-19,0nmol/mL (Cmax) e un’emivita di 2,5 ore. Uno studio successivo, condotto con 1 g di HMB calcico, ha rilevato una Cmax di 131+/-10µmol/L e un ritorno al valore basale dopo 12 ore;[23] il motivo di questa discrepanza con la stessa dose non è noto.
Confrontando l’acido libero con il sale di calcio (livelli equivalenti di HMB, quindi 0,8 g di acido libero contro 1 g di HMB di calcio), la Cmax è più alta con l’acido libero del 76-97% e il Tmax più breve (30 minuti), mentre anche l’AUC è aumentata del 91-97%.[23] Quando si tiene la dose di acido libero per via sublinguale per 15 minuti prima di deglutire, non sembrano esserci differenze significative rispetto alla semplice deglutizione.[23]
La forma di acido libero sembra essere assorbita meglio e raggiungere il picco sierico più rapidamente rispetto alla forma di sale di calcio dell’HMB.
Di solito, quando si parla di integrazione alimentare negli atleti, si utilizza una dose di 3 g di HMB. Ciò è dovuto principalmente al fatto che si tratta della dose più comunemente utilizzata, ma le prove limitate che confrontano 3 g con dosi più elevate (di solito 6 g) non trovano alcuna differenza significativa tra le due dosi.[24]
6 g di HMB non sembrano essere significativamente migliori di 3 g di HMB.
- Massa muscolare:
Per quanto riguarda gli studi sugli animali, 460mg/kg di HMB al giorno somministrati a ratti di mezza età sembrano essere efficaci nel ridurre il tasso di declino motorio e l’area della sezione trasversale muscolare durante il successivo processo di invecchiamento, ma non sono riusciti a influenzare la massa magra.[25] Quando questa dose viene somministrata a ratti di sesso femminile di età avanzata, l’aumento della massa muscolare e della produzione di potenza osservato con l’esercizio fisico non viene incrementato.[26]
Gli studi sull’uomo sono in qualche modo simili, con 2 g di HMB (integratore combinato con 5 g di L-arginina e 1,5 g di L-lisina) in grado di migliorare il controllo muscolare e la potenza in uscita per 12 settimane in donne (età media 76 anni. 7) senza influire sulla massa magra[27], anche se il primo studio ha rilevato una tendenza all’aumento della massa magra (e i test in acuto hanno evidenziato un aumento del 20% della sintesi proteica[27]), mentre uno studio successivo ha confermato un aumento della massa magra, ma senza miglioramenti della funzione muscolare.[28] Uno studio con l’aggiunta di vitamina D ha riscontrato benefici sia sulla forza che sulla massa magra nel corso di un anno.[29]

Negli adulti anziani che partecipano all’allenamento con i pesi, l’HMB supplementare è associato a un aumento della massa magra (0,8 kg in 8 settimane) senza influire sulla massa grassa.[30]
È possibile che l’integrazione di HMB nella dieta degli anziani attenui il tasso di perdita muscolare che si verifica durante il processo di invecchiamento.
L’HMB possiede proprietà mitogeniche, valutate da cellule muscolari umane quiscienti stimolate a proliferare con l’incubazione dell’HMB, con un picco di efficacia (aumento della MyoD) a 50ug/mL in questo studio ed effetti negativi a 200ug/mL.[31] Questo effetto mitogenico diretto è stato notato altrove,[32][33] e suggerisce che l’HMB può indurre le cellule muscolari quiscienti (dormienti) alla differenziazione cellulare.
Con l’integrazione di HMB è stata notata una proliferazione cellulare secondaria alla via MAPK/ERK, poiché gli inibitori di MEK aboliscono gli effetti proliferativi dell’HMB in vitro.[31] Questa via è nota per essere un regolatore della proliferazione delle cellule muscolari[34][35] e sembra mediare la proliferazione cellulare indotta dall’HMB.[31]

L’HMB può indurre la proliferazione delle cellule muscolari attraverso la via MAPK/ERK, che è uno dei bersagli molecolari dell’integrazione di HMB.
Esaminando le vie molecolari, è stato riscontrato che l’HMB stimola la sintesi proteica muscolare attraverso la via mTOR[36] a valle di PI3K/Akt[31] e può avvenire indipendentemente dalla leucina.[37][31] Nei ratti (320mg/kg) è stato riscontrato un aumento dell’espressione di mTOR (429,2%) e la successiva fosforilazione di p70S6K.[36]
È stato osservato che gli inibitori di Akt inibiscono la differenziazione muscolare indotta dall’HMB (suggerendo che sia fondamentale per la segnalazione)[31] ed è stato ipotizzato che la via di segnalazione di Akt medi la differenziazione delle cellule muscolari[31].
La sintesi proteica muscolare sembra essere mediata dalla via mTOR (a valle della segnalazione di Akt, il secondo bersaglio molecolare dell’HMB) e dalla successiva fosforilazione di p70S6K.
L’HMB è coinvolto nella riduzione dell’apoptosi (morte cellulare regolata) dei miociti e delle cellule satelliti e, grazie a questi effetti anti-apoptotici, si pensa che l’integrazione di HMB possa svolgere un ruolo in situazioni caratterizzate dall’apoptosi dei miociti (catabolismo associato all’invecchiamento,[38][39] distrofie muscolari,[40][41] e cachessia[42][43]). È stato confermato in vitro che l’HMB riduce l’apoptosi aumentando il rapporto tra Bcl-2/Bcl-X e Bax[31-18] attraverso la segnalazione di Akt[44] che porta le proteine antiapoptotiche Bcl-2 e Bcl-X a sequestrare le proteine pro-apoptotiche Bax.[45]
Analogamente all’induzione della sintesi proteica e della differenziazione muscolare, gli effetti anti-apoptotici dell’HMB sono a valle della segnalazione di Akt.
- Danno Muscolare:

L’integrazione di 3 g di HMB (l’uso del sale di calcio o dell’acido libero non è stato rivelato) prima dell’esercizio fisico in maschi non allenati non ha alterato in modo significativo i livelli di creatinchinasi, sebbene l’integrazione prima dell’esercizio sembrasse ridurre l’LDH sierico.[46] Uno studio successivo, che ha replicato i risultati ma ha utilizzato una forma di sale libero di HMB (assorbito più velocemente[23]), ha osservato che la creatinchinasi indotta dall’esercizio fisico in maschi allenati è stata ridotta (dal 329% al 104%) dopo 3 g di HMB acido libero.[47]
Negli studi che valutano l’indolenzimento muscolare, 3 g di HMB prima dell’esercizio in uomini non allenati non hanno ridotto l’indolenzimento[46], anche se 3 g (di acido libero piuttosto che di sale di calcio) prima dell’esercizio hanno migliorato la capacità percepita degli atleti di eseguire gli allenamenti nei pochi giorni successivi al test.[35] Raddoppiare la dose a 6 g di sale di calcio non ha causato una riduzione dell’indolenzimento acuto.[48]
Sono stati condotti due studi sull’integrazione di HMB e sul recupero. Entrambi hanno utilizzato l’HMB alla dose di 3 g di sale di calcio (con 0,3 g di CCI) e uno ha rilevato che l’integrazione ha favorito il recupero dal sollevamento pesi quando è stata misurata nei tre giorni successivi all’esercizio[49], mentre l’altro studio, che ha utilizzato la stessa dose per favorire il recupero dalla corsa in discesa, non ha riscontrato benefici;[50] quest’ultimo studio, tuttavia, potrebbe aver utilizzato un integratore privo di HMB[51], il che potrebbe spiegare il fallimento.
Non è chiaro se l’integrazione di HMB sia in grado di ridurre l’indolenzimento muscolare, con prove limitate che valutano i tassi di recupero e che suggeriscono che sia l’HMB acido libero sia l’HMB sale di calcio possano avere dei benefici.
- Sintesi Proteica Muscolare:
Uno studio che ha confrontato gli effetti di 3,42 g di HMB con la stessa dose orale di leucina ha rilevato che mentre l’HMB ha aumentato la sintesi proteica muscolare (valutata mediante traccianti di fenilalanina incorporati nei miociti) del 70%, la leucina ha aumentato la sintesi proteica muscolare del 110%.[52]
Sembra essere meno efficace di una pari dose orale di leucina nel promuovere la sintesi proteica muscolare.

È stato osservato che l’aggiunta di 3 g di HMB alla dieta di atleti sottoposti ad allenamento fisico aumenta la massa muscolare dello 0,2+/-2,2% nell’arco di 9 settimane, sebbene questo studio sia confuso con un aumento dell’8% dell’assunzione di cibo (e una riduzione del 10% del placebo)[53] e questo studio si scontra con altri due condotti su persone non allenate, in cui si osserva che l’HMB induce la sintesi proteica muscolare sia nei gruppi ad alto (175 g) che a basso (117 g) contenuto proteico[7] e che non vi sono differenze dovute al sesso o allo stato di allenamento. [54] Anche l’unico studio condotto su giovani atleti ha riportato risultati benefici, ma la composizione della dieta non è stata resa nota (solo una dichiarazione che non presentava differenze).[55]
Al contrario, uno studio comparativo tra 3 g di HMB in formulazione a rilascio ritardato e sale di calcio standard non ha riscontrato un effetto per 6 settimane in nessuno dei due gruppi[56] e il raddoppio della dose a 6 g di calcio-HMB (somministrato tramite frullato proteico) non ha superato il placebo (frullato proteico simile senza HMB) per 28 giorni.[57] Sono stati riportati risultati nulli anche in persone non allenate,[58] a sostegno dell’idea che lo stato di allenamento sia irrilevante.
Le prove a sostegno dell’idea che l’integrazione di HMB promuova la sintesi proteica muscolare negli atleti allenati a 3 g al giorno sono scarse e probabilmente non vi è alcun beneficio.
- Atrofia Muscolare/Catabolismo:

L’HMB possiede un effetto anticatabolico (preserva la massa muscolare) che si ritiene sia in qualche modo nuovo rispetto all’integrazione di leucina, in quanto gli effetti soppressivi della leucina sulla massa muscolare sono massimi a 5-10mM[59] (nettamente superiori ai livelli a digiuno di 0,1mM[60][61] e alle concentrazioni postprandiali che sono state osservate circa raddoppiate dopo infusioni di 162-261mg/kg/h[62]) nonostante le concentrazioni raggiungibili con l’HMB. 1mM[60-51][61] e delle concentrazioni postprandiali che sono state osservate come circa raddoppiate dopo infusioni di 162-261mg/kg/h[62]), nonostante le concentrazioni raggiungibili con la leucina siano sufficienti a promuovere la sintesi proteica muscolare[63] (in misura maggiore rispetto all’HMB[44]), ma la leucina a 0,5mM sembra avere scarsi effetti anticatabolici (6,7% in questo modello animale che ha osservato un aumento della sintesi del 36-38%[64]). È possibile che l’HMB svolga un ruolo di agente anticatabolico nonostante il suo scarso effetto sulla sintesi proteica muscolare, e ciò è in qualche modo supportato dal fatto che gli effetti anticatabolici della leucina sono 10-20 volte superiori alla concentrazione necessaria per promuovere la sintesi proteica muscolare[59] e che circa il 5% della leucina viene convertito in HMB nell’organismo.[6]
È plausibile che l’HMB sia il metabolita anticatabolico della leucina, mentre da solo non è in grado di superare la leucina nella sintesi proteica muscolare (forse perché altri metaboliti della leucina sono più potenti nell’indurre la sintesi proteica), ma può avere un ruolo nella prevenzione della perdita muscolare che non richiede gli altri metaboliti della leucina né la leucina stessa.
A 50μM, si è notato che l’HMB riduce l’atrogina-1 basale in vitro e l’induzione dell’atrogina-1 da parte di stimoli catabolici,[65] che sembra essere una concentrazione raggiungibile di HMB associata a un aumento della sintesi proteica muscolare. [29][18] Ciò suggerisce che gli effetti anticatabolici dell’HMB sono rilevanti (poiché l’atrogin-1 è una proteina che media la disgregazione delle proteine muscolari[66]) e, sebbene siano in parte a valle della segnalazione di mTOR[29], sono completamente dipendenti dall’attivazione di p38/MAPK (p42/44 MAPK sembra non essere coinvolta).[68][65]
Gli effetti anticatabolici (in vitro) sono stati confermati nei confronti dei glucocorticoidi,[65] degli stimoli proinfiammatori LPS[68][24] e TNF-α,[69][24] e dell’angiotensione II.[69][24]
Le ricerche in vitro supportano l’idea che l’HMB sia anticatabolico, e questo effetto anticatabolico sembra estendersi a un’ampia varietà di fattori di stress catabolico e si verifica a una concentrazione raggiungibile dopo l’ingestione orale di integratori di HMB. Ciò avviene attraverso la segnalazione p38/MAPK
Ciò è stato osservato con 3 g di sali di HMB per 10 giorni in adulti anziani sottoposti a riposo a letto, invertendo il declino della massa magra (2,05+/-0,66 kg) a nessun cambiamento significativo (0,17+/-0,19 kg con tendenza all’aumento);[70] che è simile agli aminoacidi a catena ramificata e alla leucina isolata. [71][72] Altri studi hanno osservato che l’integrazione di HMB è efficace nell’attenuare il tasso di perdita di massa magra osservato nella cachessia da cancro[73][74][30] e una combinazione di HMB con L-arginina e L-glutammina ha mostrato efficacia nei pazienti affetti da AIDS[75], anche se in vitro non sembrano avere un effetto anticatabolico sinergico.[29] Attualmente, gli effetti anticatabolici della leucina e dell’HMB non sono stati confrontati direttamente.
Uno studio in acuto che ha utilizzato 3,42 g di HMB rispetto a 3,42 g di leucina ha osservato che mentre la leucina ha superato l’HMB sulla sintesi proteica muscolare, l’HMB è stato in grado di attenuare la disgregazione delle proteine muscolari (57%).[44]
Gli studi sugli atleti volti a valutare la disgregazione delle proteine muscolari sono limitati; uno studio che ha utilizzato 3 g di HMB come sale di calcio per 3 giorni in atlete di judo d’élite durante una grave restrizione calorica (20 kcal/kg e 1,33 g/kg di proteine; per simulare la situazione prima di una gara) non è riuscito a superare il placebo.[36]
Uno studio condotto su atleti di pallavolo d’élite (giovani) non ha rilevato differenze nel cortisolo dopo l’integrazione di 3 g di HMB per un periodo di 7 settimane in concomitanza con l’allenamento.[46]
È stato confermato che l’integrazione di HMB è anticatabolica nei periodi di deperimento muscolare ad alto rischio (cachessia oncologica, AIDS, degenza a letto) a un dosaggio supplementare fattibile, ma non ci sono prove sufficienti per valutare correttamente il suo ruolo negli atleti. Sembra essere migliore della leucina in questo, ma richiede prove più solide per essere confermata.
- Appetito:
Esistono alcuni studi che somministrano HMB a 3 g a maschi allenati alla resistenza che riportano cambiamenti nell’assunzione di cibo, come ad esempio 9 settimane di integrazione che causano una tendenza all’aumento dell’assunzione calorica complessiva e un aumento significativo dell’assunzione di grassi (totali, saturi e monoinsaturi del 44%, 44% e 53% rispetto al basale)[37] e altrove è stato notato che i gruppi integrati con HMB consumano più proteine rispetto al placebo (questo studio ha notato una diminuzione rispetto al basale nel placebo che non era presente nell’HMB); [76] quest’ultimo studio non ha riscontrato differenze nell’assunzione di grassi, ma ha rilevato un aumento relativo dell’apporto calorico. [76]
Altri studi non hanno rilevato differenze significative nella composizione o nella quantità della dieta con 3 g di HMB in gruppi demografici simili[38] e giovani.[77] Alcuni risultati nulli sono stati ottenuti con interventi dietetici (standardizzazione della dieta o introduzione di supplementi calorici, che controllano l’appetito).[78]
Alcuni interventi sull’uomo notano che i gruppi integrati con HMB a 3 g tendono a mangiare di più, anche se questo aumento dell’assunzione di cibo non è affidabile per quanto riguarda la frequenza con cui si verifica e quali macronutrienti vengono consumati in eccesso. Non è certo che l’HMB abbia un ruolo causale in questo caso.
Profilo di sicurezza:
I test tossicologici hanno rilevato che il livello senza effetti avversi osservati (NOAEL; la dose più alta non associata a segni di tossicità) per l’ingestione orale di HMB nei ratti è di 3490mg/kg per i ratti maschi e 4160mg/kg per le femmine;[79] si tratta di un equivalente umano stimato[80-71] di 558mg/kg e 665mg/kg, e ipotizzando un peso corporeo di 150lbs equivale a 38g (maschi) e 45g (femmine). Altri test tossicologici sugli animali includono una dose di circa 5 g/kg nei maiali per 4 giorni, che non ha alterato alcun parametro biochimico o il peso degli organi (Nutritional role of the leucine metabolite B-hydroxy B-methylbutyrate (HMB) 1997; citato tramite una revisione[81]).
Studi tossicologici sull’uomo hanno osservato che circa 6 g di HMB al giorno (78 mg/kg) per un mese in giovani maschi non allenati e sottoposti a esercizio fisico non hanno mostrato effetti tossici sui parametri sierici (metà della dose ha avuto un aumento spontaneo dei basofili, considerato insignificante)[82] e 3 g di HMB al giorno per un massimo di 8 settimane sia in giovani che in anziani non hanno alterato i parametri tossicologici nel siero[83] e questa dose è risultata sicura per un anno di somministrazione (studio confuso con l’ingestione di L-lisina e L-arginina). [Nel complesso, le dosi standard di HMB sembrano essere ben tollerate per lunghi periodi di tempo (meta-analisi).[84]
È stato dimostrato che l’integrazione di HMB fino a 3 g al giorno è molto ben tollerata e si sospetta che dosi maggiori siano altrettanto sicure (ma con meno test sull’uomo). L’integrazione di HMB non desta troppe preoccupazioni in termini di sicurezza.
Conclusioni:

Come abbiamo visto, l’HMB può essere utile in determinate circostanze sebbene il suo margine di efficacia sia tutto sommato sorretto su deboli evidenze scientifiche.
Tralasciando il suo dubbio effetto migliorativo sulla sintesi proteica, sembrerebbe che un suo utilizzo in contesti di ipocalorica e, quindi, tendenzialmente catabolici potrebbe offrire un certo vantaggio. Di conseguenza, il suo uso, se lo si vuole prendere in considerazione, potrebbe essere circoscritto al “Cut” o “Pre-Gara” alla dose di 3-9g/die.
La sua aggiunta in fasi di “Bulk” non ha praticamente mai dimostrato di apportare vantaggi anche minimi rispetto al suo mancato inserimento.
Gabriel Bellizzi [CEO BioGenTech]
Riferimenti:
- Coffman DD, Cramer R, Mochel WE (June 1958). “Syntheses by Free-radical Reactions. V. A New Synthesis of Carboxylic Acids”. Journal of the American Chemical Society. 80 (11): 2882–2887. doi:10.1021/ja01544a072.
- “3-OH-isovaleric acid”. ChemSpider. Royal Society of Chemistry. 2015. Archived from the original on 11 August 2016. Retrieved 10 August 2016.
Experimental Boiling Point: … 128 °C / 7 mm …
Experimental solubility:
Soluble in water - “beta-Hydroxyisovaleric acid”. PubChem Compound. United States National Library of Medicine – National Center for Biotechnology Information. 3 February 2018. Archived from the original on 6 February 2018. Retrieved 6 February 2018.
Chemical Names: Beta-Hydroxyisovaleric acid; 3-Hydroxy-3-methylbutanoic acid; … 3-Hydroxyisovaleric acid; 3-Hydroxy-3-methylbutyric acid
- Wishart, David S.; Guo, An Chi; Oler, Eponine; Wang, Fel; Anjum, Afia; Peters, Harrison; Dizon, Raynard; Sayeeda, Zinat; Tian, Siyang; Lee, Brian L.; Berjanskii, Mark; Mah, Robert; Yamamoto, Mai; Jovel Castillo, Juan; Torres Calzada, Claudia; Hiebert Giesbrecht, Mickel; Lui, Vicki W.; Varshavi, Dorna; Varshavi, Dorsa; Allen, Dana; Arndt, David; Khetarpal, Nitya; Sivakumaran, Aadhavya; Harford, Karxena; Sanford, Selena; Yee, Kristen; Cao, Xuan; Budinsky, Zachary; Liigand, Jaanus; Zhang, Lun; Zheng, Jiamin; Mandal, Rupasri; Karu, Naama; Dambrova, Maija; Schiöth, Helgi B.; Gautam, Vasuk. “Showing metabocard for 3-Hydroxyisovaleric acid (HMDB0000754)”. Human Metabolome Database, HMDB. 5.0.
- “3-hydroxyisovaleric acid”. Chemical Entities of Biological Interest. European Bioinformatics Institute. 23 October 2015. Archived from the original on 12 March 2016. Retrieved 20 August 2016.
- The earliest citation for the synthesis of β-hydroxy β-methylbutyric acid in the Reaxys chemical database as of September 2016 is:
Saytzeff M, Saytzeff A (1877). “Synthese des Allyldimethylcarbinols” [Synthesis of allyldimethylcarbinols]. Justus Liebig’s Annalen der Chemie (in German). 185 (2–3): 151–169. doi:10.1002/jlac.18771850204. - Ružička L, Dalma G, Engel BG, Scott WE (1941). “Zur Kenntnis der Erythrophleum-Alkaloide. (5. Mitteilung). Identifizierung der niedermolekularen Spaltsäure des Coumingins” [Concerning erythrophleum alkaloids. (5th Communication). Identification of the low molecular weight cleavage acids from coumingin]. Helvetica Chimica Acta (in German). 24 (1): 1449–1458. doi:10.1002/hlca.194102401171.
- Tanaka K, Orr JC, Isselbacher KJ (May 1968). “Identification of beta-hydroxyisovaleric acid in the urine of a patient with isovaleric acidemia”. primary source. Biochimica et Biophysica Acta (BBA) – Lipids and Lipid Metabolism. 152 (3): 638–41. doi:10.1016/0005-2760(68)90107-0. PMID 5656832.
- Tanaka K (1975). “Disorders of Organic Acid Metabolism”. In Gaull GE (ed.). Biology of Brain Dysfunction. Boston, MA: Springer US. pp. 145–214. doi:10.1007/978-1-4684-2673-1_3. ISBN 978-1-4684-2675-5.
- Fitzgerald M (May 2014). Diet Cults: The Surprising Fallacy at the Core of Nutrition Fads and a Guide to Healthy Eating for the Rest of Us. Pegasus Books. p. 148. ISBN 978-1-60598-595-4. Retrieved 31 July 2016.
HMB was discovered in the mid-1990s by Steve Nissen, a researcher at Iowa State University
. - “The University of Iowa Economic Development Grow Iowa Values Fund Proposal: Fiscal Year 2011” (PDF). University of Iowa. pp. 13–16. Archived (PDF) from the original on 1 September 2016. Retrieved 1 September 2016.
- “Patents Assigned to Metabolic Technologies, Inc”. Justia Patent. As of March 2018, granted patents include: US8815280, US9259430, US9539224, US9707241, and US9770424.
- “Abbott Nutrition Overview” (PDF). Abbott. Abbott Laboratories. Archived from the original (PDF) on 3 September 2016. Retrieved 3 September 2016.
- Schirokoff A (January 1881). “Ueber die β-Dipropyl- und β-Diäthyläthylenmilchsäure und über die Oxydation des Allyldimethylcarbinols und Diallylcarbinols mit übermangansaurem Kalium” [On the β-dipropyl- and β-diethylenyl-lactic acid, and on the oxidation of the allyl dimethylcarbinol and diallylcarbinol with excess potassium]. Journal für Praktische Chemie (in German). 23 (1): 196–208. doi:10.1002/prac.18810230115.
- Reformatzky B (30 October 1889). “Synthese einiger Glycerine mittelst unterchloriger Säure” [Synthesis of some glycerol by hypochlorous acid]. Journal für Praktische Chemie (in German). 40 (1): 396–419. doi:10.1002/prac.18890400137.
- Gresham TL, Jansen JE, Shaver FW, Beears WL (January 1954). “β-Propiolactone. XIV. β-Isovalerolactone”. Journal of the American Chemical Society. 76 (2): 486–488. doi:10.1021/ja01631a045.
- WO application 2012140276, Noti C, Schmid L, Rittiner B, Hanselmann P, Bierstedt A, “Process for the preparation of 3-hydroxy-3-methylbutyric acid or its calcium salts”, published 10 January 2013, assigned to Lonza Ltd
- Jump up to:a b Kohn M (September 1903). “Zur Kenntnis des Diacetonalkohols und des Mesityloxyds” [Knowledge of diacetone alkohols and mesityl oxide]. Monatshefte für Chemie und Verwandte Teile Anderer Wissenschaften. 24 (9): 765–772. doi:10.1007/BF01526057. S2CID 96317019.
- Doraiswamy LK (February 2001). “Example 5.2”. Organic Synthesis Engineering. New York: Oxford University Press. pp. 102–124. ISBN 978-0-19-509689-7.
- Kochi JK (December 2012). “Homolytic Mechanism in Metal Catalysis”. Organometallic Mechanisms and Catalysis: The Role of Reactive Intermediates in Organic Processes. New York: Elsevier. p. 67. ISBN 978-0-323-14410-0. Archived from the original on 22 March 2018.
- Lee IY, Nissen SL, Rosazza JP (November 1997). “Conversion of beta-methylbutyric acid to beta-hydroxy-beta-methylbutyric acid by Galactomyces reessii“. primary source. Applied and Environmental Microbiology. 63 (11): 4191–4195.
- Calcium β-Hydroxy-β-Methylbutyrate
- Fuller JC Jr, Sharp RL, Angus HF, Baier SM, Rathmacher JAFree acid gel form of β-hydroxy-β-methylbutyrate (HMB) improves HMB clearance from plasma in human subjects compared with the calcium HMB saltBr J Nutr.(2011 Feb)
- Gallagher PM, Carrithers JA, Godard MP, Schulze KE, Trappe SWBeta-hydroxy-beta-methylbutyrate ingestion, Part I: effects on strength and fat free massMed Sci Sports Exerc.(2000 Dec)
- Wilson JM, Grant SC, Lee SR, Masad IS, Park YM, Henning PC, Stout JR, Loenneke JP, Arjmandi BH, Panton LB, Kim JSBeta-hydroxy-beta-methyl-butyrate blunts negative age-related changes in body composition, functionality and myofiber dimensions in ratsJ Int Soc Sports Nutr.(2012 Apr 18)
- Kim JS, Park YM, Lee SR, Masad IS, Khamoui AV, Jo E, Park BS, Arjmandi BH, Panton LB, Lee WJ, Grant SCβ-hydroxy-β-methylbutyrate did not enhance high intensity resistance training-induced improvements in myofiber dimensions and myogenic capacity in aged female ratsMol Cells.(2012 Nov)
- Flakoll P, Sharp R, Baier S, Levenhagen D, Carr C, Nissen SEffect of beta-hydroxy-beta-methylbutyrate, arginine, and lysine supplementation on strength, functionality, body composition, and protein metabolism in elderly womenNutrition.(2004 May)
- Baier S, Johannsen D, Abumrad N, Rathmacher JA, Nissen S, Flakoll PYear-long changes in protein metabolism in elderly men and women supplemented with a nutrition cocktail of beta-hydroxy-beta-methylbutyrate (HMB), L-arginine, and L-lysineJPEN J Parenter Enteral Nutr.(2009 Jan-Feb)
- Fuller JC Jr, Baier S, Flakoll P, Nissen SL, Abumrad NN, Rathmacher JAVitamin D status affects strength gains in older adults supplemented with a combination of β-hydroxy-β-methylbutyrate, arginine, and lysine: a cohort studyJPEN J Parenter Enteral Nutr.(2011 Nov)
- Vukovich MD, Stubbs NB, Bohlken RMBody composition in 70-year-old adults responds to dietary beta-hydroxy-beta-methylbutyrate similarly to that of young adultsJ Nutr.(2001 Jul)
- Kornasio R, Riederer I, Butler-Browne G, Mouly V, Uni Z, Halevy OBeta-hydroxy-beta-methylbutyrate (HMB) stimulates myogenic cell proliferation, differentiation and survival via the MAPK/ERK and PI3K/Akt pathwaysBiochim Biophys Acta.(2009 May)
- Peterson AL, Qureshi MA, Ferket PR, Fuller JC JrIn vitro exposure with beta-hydroxy-beta-methylbutyrate enhances chicken macrophage growth and functionVet Immunol Immunopathol.(1999 Jan 4)
- Siwicki AK, Fuller JC Jr, Nissen S, Ostaszewski P, Studnicka MIn vitro effects of beta-hydroxy-beta-methylbutyrate (HMB) on cell-mediated immunity in fishVet Immunol Immunopathol.(2000 Oct 31)
- The Mitogenic and Myogenic Actions of Insulin-like Growth Factors Utilize Distinct Signaling Pathways
- Elia D, Madhala D, Ardon E, Reshef R, Halevy OSonic hedgehog promotes proliferation and differentiation of adult muscle cells: Involvement of MAPK/ERK and PI3K/Akt pathwaysBiochim Biophys Acta.(2007 Sep)
- Pimentel GD, Rosa JC, Lira FS, Zanchi NE, Ropelle ER, Oyama LM, Oller do Nascimento CM, de Mello MT, Tufik S, Santos RVβ-Hydroxy-β-methylbutyrate (HMβ) supplementation stimulates skeletal muscle hypertrophy in rats via the mTOR pathwayNutr Metab (Lond).(2011 Feb 23)
- Eley HL, Russell ST, Tisdale MJAttenuation of depression of muscle protein synthesis induced by lipopolysaccharide, tumor necrosis factor, and angiotensin II by beta-hydroxy-beta-methylbutyrateAm J Physiol Endocrinol Metab.(2008 Dec)
- Krajnak K, Waugh S, Miller R, Baker B, Geronilla K, Alway SE, Cutlip RGProapoptotic factor Bax is increased in satellite cells in the tibialis anterior muscles of old ratsMuscle Nerve.(2006 Dec)
- Jejurikar SS, Henkelman EA, Cederna PS, Marcelo CL, Urbanchek MG, Kuzon WM JrAging increases the susceptibility of skeletal muscle derived satellite cells to apoptosisExp Gerontol.(2006 Sep)
- Tews DS, Goebel HHDNA-fragmentation and expression of apoptosis-related proteins in muscular dystrophiesNeuropathol Appl Neurobiol.(1997 Aug)
- Tidball JG, Albrecht DE, Lokensgard BE, Spencer MJApoptosis precedes necrosis of dystrophin-deficient muscleJ Cell Sci.(1995 Jun)
- Eley HL, Russell ST, Baxter JH, Mukerji P, Tisdale MJSignaling pathways initiated by beta-hydroxy-beta-methylbutyrate to attenuate the depression of protein synthesis in skeletal muscle in response to cachectic stimuliAm J Physiol Endocrinol Metab.(2007 Oct)
- Aversa Z, Bonetto A, Costelli P, Minero VG, Penna F, Baccino FM, Lucia S, Rossi Fanelli F, Muscaritoli Mβ-hydroxy-β-methylbutyrate (HMB) attenuates muscle and body weight loss in experimental cancer cachexiaInt J Oncol.(2011 Mar)
- Pugazhenthi S, Nesterova A, Sable C, Heidenreich KA, Boxer LM, Heasley LE, Reusch JEAkt/protein kinase B up-regulates Bcl-2 expression through cAMP-response element-binding proteinJ Biol Chem.(2000 Apr 14)
- Green DRApoptotic pathways: ten minutes to deadCell.(2005 Jun 3)
- Wilson JM, Kim JS, Lee SR, Rathmacher JA, Dalmau B, Kingsley JD, Koch H, Manninen AH, Saadat R, Panton LBAcute and timing effects of beta-hydroxy-beta-methylbutyrate (HMB) on indirect markers of skeletal muscle damageNutr Metab (Lond).(2009 Feb 4)
- Wilson JM, Lowery RP, Joy JM, Walters JA, Baier SM, Fuller JC, Stout JR, Norton LE, Sikorski EM, Wilson SM, Duncan NM, Zanchi NE, Rathmacher Jβ-Hydroxy-β-methylbutyrate free acid reduces markers of exercise-induced muscle damage and improves recovery in resistance-trained menBr J Nutr.(2013 Jan 3:1-7)
- Paddon-Jones D, Keech A, Jenkins DShort-term beta-hydroxy-beta-methylbutyrate supplementation does not reduce symptoms of eccentric muscle damageInt J Sport Nutr Exerc Metab.(2001 Dec)
- van Someren KA, Edwards AJ, Howatson GSupplementation with beta-hydroxy-beta-methylbutyrate (HMB) and alpha-ketoisocaproic acid (KIC) reduces signs and symptoms of exercise-induced muscle damage in manInt J Sport Nutr Exerc Metab.(2005 Aug)
- Nunan D, Howatson G, van Someren KAExercise-induced muscle damage is not attenuated by beta-hydroxy-beta-methylbutyrate and alpha-ketoisocaproic acid supplementationJ Strength Cond Res.(2010 Feb)
- Abumrad NN, Rathmacher JAExercise-Induced Muscle Damage is Not Attenuated by Maximuscle β-Hydroxy-β-Methylbutyrate-1000™ SupplementationJ Strength Cond Res.(2011 May 6)
- Wilkinson DJ, et al. Effects of Leucine and its metabolite, β-hydroxy-β-methylbutyrate (HMB) on human skeletal muscle protein metabolism
J Physiol (2013 Apr 8) - Thomson JS, Watson PE, Rowlands DS. Effects of nine weeks of beta-hydroxy-beta- methylbutyrate supplementation on strength and body composition in resistance trained men
J Strength Cond Res (2009 May) - Portal S, Zadik Z, Rabinowitz J, Pilz-Burstein R, Adler-Portal D, Meckel Y, Cooper DM, Eliakim A, Nemet DThe effect of HMB supplementation on body composition, fitness, hormonal and inflammatory mediators in elite adolescent volleyball players: a prospective randomized, double-blind, placebo-controlled studyEur J Appl Physiol.(2011 Sep)
- Slater G, Jenkins D, Logan P, Lee H, Vukovich M, Rathmacher JA, Hahn AGBeta-hydroxy-beta-methylbutyrate (HMB) supplementation does not affect changes in strength or body composition during resistance training in trained menInt J Sport Nutr Exerc Metab.(2001 Sep)
- Kreider RB, Ferreira M, Wilson M, Almada ALEffects of calcium beta-hydroxy-beta-methylbutyrate (HMB) supplementation during resistance-training on markers of catabolism, body composition and strengthInt J Sports Med.(1999 Nov)
- Lamboley CR, Royer D, Dionne IJEffects of beta-hydroxy-beta-methylbutyrate on aerobic-performance components and body composition in college studentsInt J Sport Nutr Exerc Metab.(2007 Feb)
- Zanchi NE, Nicastro H, Lancha AH JrPotential antiproteolytic effects of L-leucine: observations of in vitro and in vivo studiesNutr Metab (Lond).(2008 Jul 17)
- Dardevet D, Sornet C, Balage M, Grizard JStimulation of in vitro rat muscle protein synthesis by leucine decreases with ageJ Nutr.(2000 Nov)
- Filho JC, Bergström J, Stehle P, Fürst PSimultaneous measurements of free amino acid patterns of plasma, muscle and erythrocytes in healthy human subjectsClin Nutr.(1997 Dec)
- Bohé J, Low A, Wolfe RR, Rennie MJHuman muscle protein synthesis is modulated by extracellular, not intramuscular amino acid availability: a dose-response studyJ Physiol.(2003 Oct 1)
- Tischler ME, Desautels M, Goldberg ALDoes leucine, leucyl-tRNA, or some metabolite of leucine regulate protein synthesis and degradation in skeletal and cardiac muscleJ Biol Chem.(1982 Feb 25)
- Buse MG, Weigand DAStudies concerning the specificity of the effect of leucine on the turnover of proteins in muscles of control and diabetic ratsBiochim Biophys Acta.(1977 Mar 2)
- Aversa Z, Alamdari N, Castillero E, Muscaritoli M, Rossi Fanelli F, Hasselgren POβ-Hydroxy-β-methylbutyrate (HMB) prevents dexamethasone-induced myotube atrophyBiochem Biophys Res Commun.(2012 Jul 13)
- Jiang Y, Singh P, Yin H, Zhou YX, Gui Y, Wang DZ, Zheng XLOpposite roles of myocardin and atrogin-1 in L6 myoblast differentiationJ Cell Physiol.(2013 Mar 22)
- Russell ST, Tisdale MJMechanism of attenuation by beta-hydroxy-beta-methylbutyrate of muscle protein degradation induced by lipopolysaccharideMol Cell Biochem.(2009 Oct)
- Eley HL, Russell ST, Tisdale MJMechanism of attenuation of muscle protein degradation induced by tumor necrosis factor-alpha and angiotensin II by beta-hydroxy-beta-methylbutyrateAm J Physiol Endocrinol Metab.(2008 Dec)
- Deutz NE, Pereira SL, Hays NP, Oliver JS, Edens NK, Evans CM, Wolfe RREffect of β-hydroxy-β-methylbutyrate (HMB) on lean body mass during 10 days of bed rest in older adultsClin Nutr.(2013 Mar 4)
- Stein TP, Schluter MD, Leskiw MJ, Boden GAttenuation of the protein wasting associated with bed rest by branched-chain amino acidsNutrition.(1999 Sep)
- Stein TP, Donaldson MR, Leskiw MJ, Schluter MD, Baggett DW, Boden GBranched-chain amino acid supplementation during bed rest: effect on recoveryJ Appl Physiol.(2003 Apr)
- May PE, Barber A, D’Olimpio JT, Hourihane A, Abumrad NNReversal of cancer-related wasting using oral supplementation with a combination of beta-hydroxy-beta-methylbutyrate, arginine, and glutamineAm J Surg.(2002 Apr)
- Smith HJ, Mukerji P, Tisdale MJAttenuation of proteasome-induced proteolysis in skeletal muscle by {beta}-hydroxy-{beta}-methylbutyrate in cancer-induced muscle lossCancer Res.(2005 Jan 1)
- Clark RH, Feleke G, Din M, Yasmin T, Singh G, Khan FA, Rathmacher JANutritional treatment for acquired immunodeficiency virus-associated wasting using beta-hydroxy beta-methylbutyrate, glutamine, and arginine: a randomized, double-blind, placebo-controlled studyJPEN J Parenter Enteral Nutr.(2000 May-Jun)
- EFFECT OF β-HYDROXY-β-METHYLBUTYRATE SUPPLEMENTATION DURING ENERGY RESTRICTION IN FEMALE JUDO ATHLETES
- Jówko E, Ostaszewski P, Jank M, Sacharuk J, Zieniewicz A, Wilczak J, Nissen SCreatine and beta-hydroxy-beta-methylbutyrate (HMB) additively increase lean body mass and muscle strength during a weight-training programNutrition.(2001 Jul-Aug)
- Kreider RB, Ferreira M, Wilson M, Almada ALEffects of calcium beta-hydroxy-beta-methylbutyrate (HMB) supplementation during resistance-training on markers of catabolism, body composition and strengthInt J Sports Med.(1999 Nov)
- Portal S, Zadik Z, Rabinowitz J, Pilz-Burstein R, Adler-Portal D, Meckel Y, Cooper DM, Eliakim A, Nemet DThe effect of HMB supplementation on body composition, fitness, hormonal and inflammatory mediators in elite adolescent volleyball players: a prospective randomized, double-blind, placebo-controlled studyEur J Appl Physiol.(2011 Sep)
- Slater G, Jenkins D, Logan P, Lee H, Vukovich M, Rathmacher JA, Hahn AGBeta-hydroxy-beta-methylbutyrate (HMB) supplementation does not affect changes in strength or body composition during resistance training in trained menInt J Sport Nutr Exerc Metab.(2001 Sep)
- Baxter JH, Carlos JL, Thurmond J, Rehani RN, Bultman J, Frost DDietary toxicity of calcium beta-hydroxy-beta-methyl butyrate (CaHMB)Food Chem Toxicol.(2005 Dec)
- Guidance for Industry: Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers
- International Society of Sports Nutrition Position Stand: beta-hydroxy-beta-methylbutyrate (HMB)
- Gallagher PM, Carrithers JA, Godard MP, Schulze KE, Trappe SWBeta-hydroxy-beta-methylbutyrate ingestion, part II: effects on hematology, hepatic and renal functionMed Sci Sports Exerc.(2000 Dec)
- Nissen S, Sharp RL, Panton L, Vukovich M, Trappe S, Fuller JC Jrbeta-hydroxy-beta-methylbutyrate (HMB) supplementation in humans is safe and may decrease cardiovascular risk factorsJ Nutr.(2000 Aug)
- Rathmacher JA, Nissen S, Panton L, Clark RH, Eubanks May P, Barber AE, D’Olimpio J, Abumrad NNSupplementation with a combination of beta-hydroxy-beta-methylbutyrate (HMB), arginine, and glutamine is safe and could improve hematological parametersJPEN J Parenter Enteral Nutr.(2004 Mar-Apr)