Androgenico: 20
Anabolico: 400
Standard: Metyltestosterone
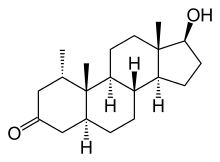
Nome Chimico: 2a,17a-dimethyl-5a-androstane-17b-ol-3-one
Attività Estrogenica: nessuna
Attività Progestinica: nessun dato disponibile (bassa)
Il Methyldrostanolone, detto anche Methasterone, conosciuto al grande pubblico come Superdrol, [2a,17a-dimethyl-5a-androstane-17b-ol-3-one] è uno steroide anabolizzante derivato del Diidrotestosterone (DHT). Per essere precisi il Methyldrostanolone è la forma metilata in C-17 del Drostanolone (da qui l’aggiunta di “Mthyl”).
La molecola presenta le seguenti caratteristiche strutturali:
- L’aggiunta di una metilazione in posizione C-2 (come nel Masteron) che da come risultato un incremento della resistenza alla 3a-HSD da parte del C-3 cheto gruppo, ma senza migliorare la bio-disponibilità orale. Migliora leggermente la bio-attività grazie ad una blanda minor affinità per l’SHBG.
- L’aggiunta di una seconda metilazione in posizione C-17. L’ addizione della metilazione in C-17 comporta, oltre ad un elevata bio-disponbilità orale, anche una diminuzione del legame con le SHBG e un rafforzamento della stabilità del C-3 cheto gruppo, responsabile della potenza della stimolazione del recettore androgeno muscolare. (1)
Il Methyldrostanolone è stato descritto per la prima volta nel 1956 in relazione a una ricerca condotta dalla Syntex Corporation al fine di scoprire un composto con proprietà anti-tumorali. (2) In un articolo pubblicato nel 1959, viene inizialmente menzionato, venendo discusso nel dettaglio il suo metodo di sintesi e le sue proprietà anti-tumorali verificate, descrivendolo come un “potente agente anabolizzante oralmente attivo esibente solo una debole attività androgena.” (3) I risultati dei test successivi alla determinazione dell’attività anabolizzante/androgena del Methyldrostanolone sono stati pubblicati nel Vida’s Androgens and Anabolic Agents, un riferimento datato ma tutt’ora valido, in cui è stato osservato che la molecola possiede la biodisponibilità orale del Metyltestosterone pur essendo il 400% più anabolizzante e androgeno il 20%, ottenendo un Q-ratio (noto anche come rapporto anabolizzante/androgeno) di 20, che è considerato molto elevato. (4)
Rispetto al Testosterone iniettabile il rapporto del Methyldrostanolone diventa circa 20/600.
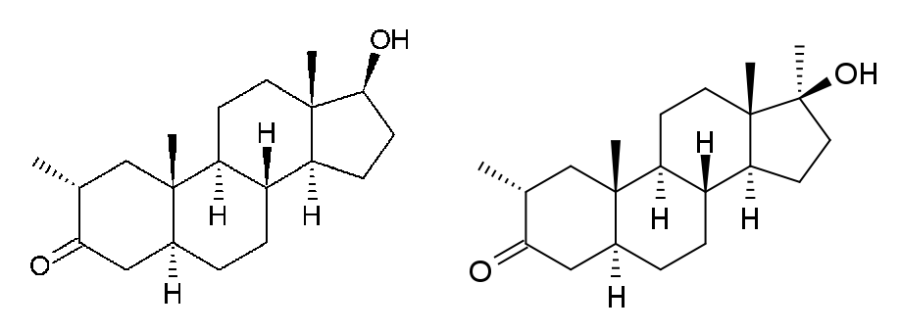
Il Methyldrostanolone non è mai stato commercializzato come farmaco. Come risaputo, la sua controparte non-17α-alchilata, il Drostanolone, è stato commercializzato dalla Syntex Corporation con il marchio Masteron.(5)

Il Methyldrostanolone riemerse nel 2005 nel mercato dei “designer steroid”. (6) E ‘stato portato sul mercato dei designer steroid come ingrediente di un “integratore alimentare chiamato Superdrol. La sua introduzione in commercio può aver rappresentato un tentativo di eludere la U.S. Anabolic Steroids Control Act del 1990 (insieme con la sua revisione del 2004), dal momento che la legge è, in parte, riferita ai farmaci specifici; (7) il Methyldrostanolone, come è accaduto per molti altri designer steroid, non è stato dichiarato nell’atto di controllo delle sostanze anabolizzanti perché non era disponibile in commercio al momento della stesura dell’atto, come non lo era quando fu firmata la successiva revisione. (8) Di conseguenza il Superdrol è stato venduto come un integratore alimentare da banco.
Alla fine del 2005 lo status di steroide anabolizzante del Superdrol, in aggiunta a quello di altri quattro designer steroid, è stato portato a conoscenza del pubblico da un articolo pubblicato sul Washington Post. (9) Don Catlin del Laboratorio Olimpico UCLA, che ha condotto gli studi, ha notato la somiglianza del Superdrol con il Drostanolone. Un avvertimento dalla FDA è stato rilasciato subito dopo al pubblico nonché al distributore, Designer Supplementi LLC, per la commercializzazione di questo composto. (10) Il Methyldrostanolone è stato successivamente aggiunto alla lista World Anti-Doping Agency (WADA) delle sostanze proibite nello sport.(11) Malgrado tutto ciò, il Methyldrostanolone è riemerso nel settore degli integratori in diverse occasioni sin dalla sua messa al bando da parte della WADA. (12)
Come per il Drostanolone, anche il Methyldrostanolone non è soggetto ad aromatizzazione (essendo un composto 5-alfa ridotto) e possiede un certa attività antiestrogena. Questa caratteristica, assieme alla sua forte affinità per i recettori androgeni sia adiposi (che stimolano la liposi) sia muscolari (conservazione marcata della massa magra in regime ipo-calorico), ha reso il Methyldrostanolone un AAS tipicamente usato nel pre-gara.
Il Methyldrostanolone possiede un valore androgeno contenuto (20) ma le possibilità che si verifichino effetti collaterali androgeni non del tutto assenti. Questo può includere pelle oleosa, acne, aumento della peluria, e alopecia androgenetica. Le donne raramente con questo composto possono sperimentare effetti virilizzanti tra i quali un approfondimento della voce, irregolarità mestruale, cambiamenti nella struttura della pelle, crescita di peli sul viso, e allargamento del clitoride.
Si noti che il Methyldrostanolone non è influenzato dal l’enzima 5-alfa reduttasi, quindi la sua androgenicità relativa non è influenzata dall’uso concomitante di finasteride o dutasteride.
Come già accennato, il Methyldrostanolone è un AAS metilato in C-2 ed in C-17. Il Methyldrostanolone presenta una marcata epatotossicità (tossico per il fegato). Molti casi di danni al fegato dovuti all’utilizzo di Methyldrostanolone sono stati citati nella letteratura medica. (13,14,15,16) Molti casi di grave epatotossicità riscontrati con l’uso del Methyldrostanolone sono dovuti ai sovradosaggi utilizzati e non sono esclusivamente attribuibili alla doppia metilazione del composto: anche perché la metilazione in C-2 è praticamente ininfluente a livello epatico.
Gli AAS possono avere effetti deleteri sul colesterolo sierico. Questo include una tendenza alla riduzione del colesterolo HDL (buono) e all’aumento del colesterolo LDL (cattivo), con un alterazione dell’equilibrio tra HDL/LDL favorendo un maggiore rischio di arteriosclerosi. L’impatto relativo di un AAS sui lipidi sierici dipende dalla dose, dalla via di somministrazione (per via orale o iniettabile), dal tipo di steroide (aromatizzabile o non aromatizzabile), e dal livello di resistenza al metabolismo epatico. Il Methyldrostanolone ha un effetto negativo maggiore sulla gestione epatica di colesterolo rispetto al Testosterone o Nandrolone a causa della sua natura non aromatizzabile e alla sua metilazione in C-17. Gli AAS possono anche influenzare negativamente la pressione del sangue e i livelli dei trigliceridi, ridurre il rilassamento endoteliale, e incrementare l’ipertrofia ventricolare sinistra, tutti potenzialmente fattori di rischio per malattie cardiovascolari e infarto del miocardio.
Per contribuire a ridurre lo sforzo cardiovascolare si consiglia di mantenere un programma di esercizio cardiovascolare attivo e di ridurre al minimo l’assunzione di grassi saturi, colesterolo e carboidrati semplici in ogni momento durante la somministrazione di AAS.
La supplementazione con oli di pesce (4 grammi al giorno) e un integratore alimentare di Niacina per il controllo del colesterolo è anche raccomandata.
Tutti gli AAS se assunti in dosi sufficienti per promuovere l’aumento della massa muscolare causano una soppressione del Testosterone endogeno. Senza l’intervento con sostanze Testosterone-stimolante, e una adeguata PCT, i livelli di Testosterone dovrebbero tornare alla normalità entro 1-4 mesi dalla cessione del farmaco. Si noti che un ipogonadismo ipogonadotropo prolungato può svilupparsi secondariamente all’abuso di steroidi, cosa che richiede un intervento medico.
Anche per il Methyldrostanolone se ne consiglia l’assunzione lontano dai pasti in quanto studi hanno dimostrato che l’assunzione di uno steroide anabolizzante orale con cibo può diminuirne la sua biodisponibilità.(17) Questo è causato dalla natura liposolubile degli ormoni steroidei, che può permettere ad una parte del farmaco di sciogliersi con i grassi alimentari non digeriti, riducendo l’assorbimento dal tratto gastrointestinale. Per la massima biodisponibilità , questo steroide deve essere assunto a stomaco vuoto.
Il Methyldrostanolone non è mai stato commercializzato come farmaco da prescrizione. Pertanto non sono disponibili linee guida di prescrizione. Un dosaggio efficace di Methyldrostanolone a fini dopanti in ambito maschile si attesta nel range dei 20-30 mg al giorno, assunti per non più di 4 settimane. A questo dosaggio sembra conferire un effetto assai apprezzabile sulla muscolatura, che di solito è accompagnato da perdita di grasso e maggiore definizione. Con questo composto è possibile ottenere guadagni “puliti” di massa magra (una media di 4kg in 4 settimane) in regime ipocalorico. Nel determinare un dosaggio giornaliero ottimale, è caldamente sconsigliato superare il dosaggio giornaliero di 30mg. La potenziale forte epatotossicità dovrebbe essere tenuta in stretta considerazione con adeguata epatoprotezione ed esami ematici specifici.
Per alleggerire l’impatto epatico della molecola, solitamente si abbina ad un dosaggio giornaliero pari a 20mg di Methyldrostanolone uno steroide iniettabile non tossico, come Testosterone o Boldenone. Essendo una molecola prevalentemente AR, il suo abbinamento con molecole fortemente AR (vedi Trenbolone) non è una buona scelta data la competizione recettoriale che si verrebbe a creare.
In ambito femminile, una dose giornaliera orale efficace si aggira intorno ai 10mg, assunti in cicli della durata non superiore alle 4 settimane per ridurre al minimo le probabilità di virilizzazione (nonostante la bassa androgenicità sono ancora possibili) e lo stress epatico. Il problema però è che il Methyldrostanolone commercializzato è venduto in compresse da 10mg. Avendo un emivita di circa 8 ore, la dose giornaliera andrebbe divisa in due assunzioni per garantire un livello ematico della molecola stabile . Se il prodotto acquistato è contenuto in capsule, è possibile dividere la dose aprendo la capsula e dividendo il contenuto in polvere in 2 dosi eguali.
Anche se rimane la probabilità di incorrere in effetti collaterali virilizzanti, come accade per ogni AAS, il Methyldrostanolone risulta una delle migliori molecole per uso femminile.
Nonostante l’originale Superdrol sia fuori produzione, nel mercato nero e “grigio” è possibile trovare prodotti contenenti Methyldrostanolone.
Gabriel Bellizzi
Riferimenti:
1- William Llewellyn’s ANABOLICS, 9th ed.
2- H. J. Ringold & G. Rosenkranz (1956). “Steroids. LXXXIII. Synthesis of 2-Methyl and 2,2-Dimethyl Hormone Analogs”. Journal of Organic Chemistry. 21: 1333–1335. doi:10.1021/jo01117a625.
3- Jump up ^ Ringold, H. J., E. Batres, O. Halpern, and E. Necoechea (1959). “Steroids. CV.1 2-Methyl and 2-Hydroxymethylene-androstane Derivatives”. Journal of the American Chemical Society. 81 (2): 427–432. doi:10.1021/ja01511a040.
4- Julius A. Vida (1969). Androgens and Anabolic Agents: Chemistry and Pharmacology. New York: Academic Press. pp. 23 & 168.
5-“Superdrol, masteron en oxy komen uit hetzelfde nest” [Superdrol, Masteron, and Oxy come from the same nest]”. Ergogenics.org. Retrieved February 2009. Check date values in: |access-date= (help)
6- Van Enoo, Peter & Frans T. Delbeke (2006). “Metabolism and excretion of anabolic steroids in doping control—New steroids and new insights”. Journal of Steroid Biochemistry & Molecular Biology. 101: 173. doi:10.1016/j.jsbmb.2006.06.024.
7- “Implementation of the Anabolic Steroid Control Act of 2004”. Office of Divesion Control, Drug Enforcement Administration, Department of Justice. Retrieved February 2009. Check date values in: |access-date= (help)
8- Shipley, Amy, Bonnie Berkowitz and Christina Rivero (October 18, 2005). “Designer Steroids Hide and Seek”. The Washington Post. Retrieved February 2009. Check date values in: |access-date= (help) CS1 maint: Multiple names: authors list (link)
9- Amy Shipley (November 30, 2005). “Steroids Detected in Dietary Tablets”. The Washington Post. Retrieved February 2009. Check date values in: |access-date= (help)
10- “FDA Warns Manufacturers About Illegal Steroid Products Sold as Dietary Supplements”. U.S. Food and Drug Administration. March 9, 2006. Retrieved February 2009. Check date values in: |access-date= (help)
11- “The World Anti-Doping Code: The 2009 Prohibited List: International Standard” (PDF). World Anti-Doping Agency. Retrieved February 2009. Check date values in: |access-date= (help)
12- David Epstein; George Dohrmann (May 18, 2009). “What You Don’t Know Might Kill You: Would-be experts and untested products feed a $20 billion obsession with better performance across all levels of sports”. Sports Illustrated.
13- Jasiurkowski, Beata, Jaya Raj, David Wisinger, Richard Carlson, Lixian Zou, and Abdul Nadir (2006). “Cholestatic Jaundice and IgA Nephropathy Induced by OTC Muscle Building Agent Superdrol”. American Journal of Gastroenterology. 101 (11): 2659–2662. doi:10.1111/j.1572-0241.2006.00735.x. CS1 maint: Multiple names: authors list (link)
14- Jump up ^ Nasr, John & Jawad Ahmad (2008). “Severe Cholestasis and Renal Failure Associated with the Use of the Designer Steroid Superdrol (Methasteron): A Case Report and Literature Review”. Digestive Diseases and Sciences. 10: 1007.
15- Jump up ^ L. Shah, Neeral, Isabel Zacharias, Urmila Khettry, Nezam Afdhal, and Fredric D. Gordon (2008). “Methasteron-Associated Cholestatic Liver Injury: Clinicopathologic Findings in 5 Cases”. Clinical Gastroenterology and Hepatology. 6 (2): 255–258. doi:10.1016/j.cgh.2007.11.010. CS1 maint: Multiple names: authors list (link)
16- Jump up ^ Singh V, Rudraraju M, Carey EJ, Byrne TJ, Vargas HE, Williams JE, Balan V, Douglas DD, Rakela J (March 2009). “Severe Hepatotoxicity Caused by a Methasteron-containing, Performance-enhancing Supplement”. Journal of Clinical Gastroenterology. 43 (3). doi:10.1097/mcg.0b013e31815a5796.
17- Anabolic Steroids and Sports Volume II. James E. Wright. Sports Science Consultants, Natick, MA 1982.