
Introduzione:
Chi non conosce, nel 2021, dopo quasi un secolo di ricerca, la Metformina ed i suoi effetti sul miglioramento della sensibilità all’Insulina, con conseguente miglioramento del uptake cellulare di glucosio ? E dei vantaggi che esso può apportare ai Bodybuilder in fase di “Refeed”, magari dopo periodi medio-lunghi a bassi CHO e con una capacità di gestirli non proprio ottimale?
La stessa cosa interessa anche la Berberina, la quale possiede vie farmacodinamiche molto simili alla Metformina. Entrambe le molecole, però, hanno un limite, e questo limite è comune a tutte le Biguanidi oggi in uso clinico o quelle appartenenti ai GDA (come la Berberina): la mancanza di selettività tissutale. Esse, infatti, migliorano sia l’IS del miocita che dell’adipocita, oltre ad attivare l’AMPK con alterazione del mTOR.

Nota: per chi non lo sapesse, le Biguanidi sono una categoria di farmaci ipoglicemizzanti orali di indicazione specifica contro il diabete di tipo II. A differenza di altri farmaci antidiabetici, come ad esempio le sulfaniluree, non determinano un aumento di rilascio di Insulina per cui non causano generalmente ipoglicemia. In questa sede mi riferirò con il termine “Biguanidi” a quelle molecole con tali caratteristiche, sia farmaceutiche (vedi Metformina) che appartenenti al panorama da banco denominato GDA (vedi Berberina).
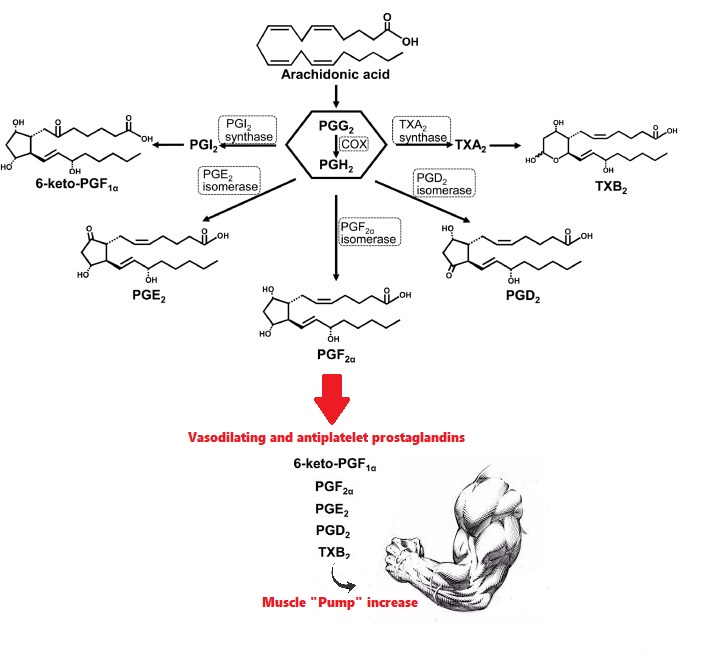
Ora, potremmo anche dire che in un soggetto con una buona massa contrattile e una massa grassa tendenzialmente bassa questo “difetto” non causa particolari problemi nel complesso della preparazione. Ma c’è da considerare che una selettività miocitaria garantirebbe una ripartizione calorica ottimale in un contesto, per esempio, ipercalorico riducendo gli “approvvigionamenti” degli adipociti e prolungando sensibilmente la soglia temporale durante la quale l’atleta in questione potrebbe crescere in modo qualitativamente soddisfacente. Un pò come quando si ipotizzava sulla applicazione di molecole con teorica attività di riduzione dello stoccaggio degli Acidi Grassi. Discorsi ed effetti diversi, ma il fine è uno: aumentare il tempo di durata della “soglia di crescita qualitativa”.
Per “soglia di crescita qualitativa” intendo la possibilità di proseguire con la programmazione in ipercalorica ottenendo maggiori aumenti ipertrofici del muscolo-scheletrico piuttosto che del tessuto adiposo.
“Ma Gabriel! E l’interferenza con l’mTOR osservata con la Metformina ed altre molecole che stimolano l’attività del AMPK non è forse una limitazione ben più importante???!!!” Calma, piccola zecca interattiva, ne parlerò a tempo debito, come parlerò del fatto che è la dose a determinare se l’alterazione risulterà significativa o meno…. Proseguiamo…
In questo articolo tratterò della nuova molecola sperimentale denominata DS20060511, riporterò quanto è a nostra conoscenza ad oggi e quali sono le sue caratteristiche e possibili applicazioni che, tra l’altro, ho già accennato in questa introduzione…
Il principio della scoperta:
La riduzione dell’assorbimento del glucosio nel muscolo scheletrico è un’importante anomalia fisiopatologica nel diabete di tipo II ed è causata dalla alterazione della funzionalità di traslocazione dei GLUT4 sulla superfice cellulare del miocita nel tessuto muscolo-scheletrico.
Il trasportatore del glucosio di tipo 4 (GLUT-4), noto anche come famiglia di trasportatori di soluti 2, membro 4 del trasportatore di glucosio facilitato, è una proteina codificata, nell’uomo, dal gene SLC2A4. Il GLUT4 è il trasportatore del glucosio regolato dall’insulina, ma non solo, che si trova principalmente nei tessuti adiposo e nel muscolo striato (scheletrico e cardiaco). La prima prova di questa distinta proteina di trasporto del glucosio è stata fornita da David James nel 1988. Il gene che codifica per il GLUT4 è stato clonato e mappato nel 1989.

Il GLUT4 è il trasportatore che limita la velocità di assorbimento del glucosio e svolge un ruolo cruciale nel mantenimento dell’omeostasi del glucosio [1, 2]. I soggetti con diabete di tipo II mostrano un ridotto assorbimento di glucosio da parte del muscolo scheletrico a causa della ridotta traslocazione di GLUT4 nella superficie delle cellule del muscolo scheletrico[3]. È stato riportato che i topi diabetici con sovraespressione di GLUT4 mostrano livelli di glucosio plasmatico marcatamente ridotti sia a digiuno che in condizioni postprandiali [4,5,6].
Sebbene il GLUT4 sia immagazzinato in vescicole di stoccaggio intracellulari in condizioni basali, l’Insulina, e l’attività di contrazione del muscolo, induce la traslocazione di GLUT4 sulla superficie cellulare, facilitando l’assorbimento del glucosio [7,8]. L’Insulina attiva Akt tramite il substrato del recettore dell’Insulina (IRS)s-fosfoinositide 3-chinasi (PI3K) [9,10] e l’Akt attivato fosforila e di conseguenza inibisce le proteine Akt substrato di 160 kDa (AS160) e membro della famiglia del dominio TBC1 1 (TBC1D1) , entrambi sono proteine attivanti Rab GTPasi (GAP); ciò si traduce nell’attivazione delle proteine Rab e nella traslocazione di GLUT4 sulla superficie della membrana cellulare [11]. È stato riportato che il substrato 1 (Rac1) della tossina botulinica C3 correlato a RAS, un’altra molecola a valle di PI3K, promuove la traslocazione di GLUT4 indipendentemente dalla via Akt-AS160/TBC1D1-Rab. Rac1 stimola la riorganizzazione della polimerizzazione dell’actina corticale, che consente l’inserimento delle vescicole contenenti GLUT4 nella membrana cellulare[12,13]. È noto che l‘Insulina regola la traslocazione di GLUT4 sia attraverso la via di Akt-AS160-Rab che attraverso la via di polimerizzazione di Rac1-actina[14,15]. Nei soggetti con diabete di tipo II, entrambe le vie di segnalazione dell’Insulina sono compromesse nel muscolo scheletrico, con conseguente riduzione dell’assorbimento del glucosio indotto dall’Insulina in questo tessuto.

Come già accennato, la contrazione durante l’esercizio è un altro importante potenziatore della traslocazione di GLUT4 nel muscolo scheletrico[16]. All’aumentata richiesta di glucosio durante l’esercizio nel muscolo scheletrico, il GLUT4 si trasloca sulla superficie cellulare per promuovere l’apporto di glucosio al muscolo scheletrico[17,18]. L’esercizio aumenta il rapporto AMP/ATP causato dal consumo di ATP, portando all’attivazione della chinasi attivata dall’AMP (AMPK). Nonostante l’evidenza riportata di una contrazione che induce la fosforilazione di TBC1D1 mediante l’attivazione di AMPK[19] o di un aumento dell’assorbimento del glucosio nel muscolo scheletrico mediante attivazione farmacologica di AMPK da parte di AICAR[20], il significato dell’AMPK nell’assorbimento del glucosio stimolato dall’esercizio in vivo rimane controverso [21,22]. Recentemente, l’induzione da parte di Rac1 della produzione NADPH ossidasi 2-dipendente di specie reattive dell’ossigeno è stata implicata nell’assorbimento del glucosio durante l’esercizio, attraverso la regolazione della traslocazione di GLUT4 [23,24]. La contrazione del muscolo scheletrico non ha indotto la fosforilazione di IRS1 o PI3K[25]. La captazione del glucosio indotta dalla contrazione o la traslocazione di GLUT4 nel muscolo scheletrico non è stata inibita dalla Wortmannina, un inibitore di PI3K [26,27]. Inoltre, la combinazione di Insulina e contrazione del muscolo scheletrico ha causato un ulteriore aumento della traslocazione di GLUT4 e dell’assorbimento di glucosio rispetto alla sola Insulina [27]. Questi dati suggeriscono che la contrazione del muscolo scheletrico stimola la traslocazione di GLUT4 indipendentemente dall’Insulina.

Nei soggetti con diabete di tipo II, i campioni bioptici del muscolo scheletrico ottenuti durante un clamp insulinico euglicemico hanno mostrato un’alterata segnalazione dell’Insulina, osservata come riduzione della fosforilazione di IRS1 e dell’attività di PI3K, nel muscolo scheletrico[28], mentre non è stato osservato alcun effetto sulla fosforilazione/attività di Akt [29]. Altri studi hanno dimostrato una riduzione della traslocazione di GLUT4 e dell’assorbimento di glucosio in soggetti con diabete di tipo II [23,28]. Inoltre, è stato riportato che la ridotta traslocazione di GLUT4 nei soggetti con diabete di tipo II è stata migliorata dall’esercizio fisico [30,31]. Questi risultati suggeriscono che l’induzione della traslocazione di GLUT4 nel muscolo scheletrico potrebbe essere un potenziale bersaglio terapeutico nei pazienti con diabete di tipo II.
Recentemente, i ricercatori dell’azienda farmaceutica giapponese Daiichi Sankyo hanno dimostrato che il derivato xantenico DS20060511 induce la traslocazione di GLUT4 specifica del muscolo scheletrico, indipendentemente dall’azioni dell’Insulina. Hanno utilizzato miotubi L6 che esprimono GLUT4 marcato con myc (L6-GLUT4myc) per esaminare la libreria di composti chimici in loro possesso e misurare la traslocazione di GLUT4 sulla superficie cellulare mediante dosaggio immunologico anti-myc quantitativo. Gli effetti del composto sull’assorbimento del glucosio e sul metabolismo del glucosio in tutto il corpo sono stati esaminati in una serie di esperimenti in vitro e in vivo. Il meccanismo d’azione del composto è stato esplorato studiando le vie di segnalazione note coinvolte nella traslocazione di GLUT4 indotta dall’Insulina e dall’esercizio fisico. Infine, abbiamo valutato il potenziale terapeutico del composto in un modello murino obeso e insulino-resistente con diabete di tipo II.

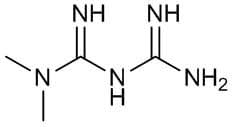
Nota: I derivati xantenici sono modificazioni molecolari dello Xantene (9H-xantene, 10H-9-ossaantracene), un composto organico con la formula CH2[C6H4]2O. È un solido giallo solubile nei comuni solventi organici. Lo stesso xantene è un composto oscuro, ma molti dei suoi derivati sono coloranti utili.
Il DS20060511, è un induttore specifico per la traslocazione di GLUT4 nelle cellule muscolo-scheletriche:
I ricercatori, come detto pocanzi, hanno esaminato la loro libreria chimica, composta da oltre 100.000 composti, utilizzando miotubi L6-GLUT4myc, per identificare i composti che avrebbero indotto la traslocazione di GLUT4 sulla superficie cellulare. Sono stati identificati due composti completamente diversi ed entrambi hanno superato il test per escludere composti che avrebbero esercitato effetti tossici, come l’inibizione della catena respiratoria. Ulteriori test in vitro hanno rivelato che uno dei due composti ha influenzato la via Akt, così che alla fine hanno selezionato l’altro, un composto xantenico originale, come composto con il potenziale effetto di indurre la traslocazione di GLUT4. L’ottimizzazione della struttura molecolare ha infine prodotto il composto xantenico più potente, DS20060511 (vedi immagine seguente). Il trattamento con DS20060511 ha aumentato la traslocazione di GLUT4 nei miotubi differenziati L6-GLUT4myc in modo concentrazione-dipendente, come nel caso del trattamento con Insulina. Tuttavia, mentre il trattamento con Insulina ha anche aumentato la traslocazione di GLUT4 negli adipociti differenziati 3T3-L1-GLUT4myc, il trattamento con DS20060511 non ha avuto quasi alcun effetto sulla traslocazione di GLUT4 in questi adipociti, suggerendo che l’induzione della traslocazione di GLUT4 da parte di DS20060511 è specifica per le cellule del tessuto muscolo-scheletrico. Coerentemente con questi dati, il trattamento con DS20060511 ha aumentato significativamente l’assorbimento di 2-DG in modo concentrazione-dipendente nei miotubi L6-GLUT4myc, come nel caso del trattamento con Insulina. Ancora una volta, mentre è stato dimostrato che l’Insulina aumenta l’assorbimento di 2-DG negli adipociti differenziati 3T3-L1-GLUT4myc, DS20060511 non ha mostrato tale effetto negli adipociti. Questi dati suggeriscono che il composto xantenico DS20060511 promuove l’assorbimento del glucosio mediante l’attivazione specifica della traslocazione di GLUT4 nelle cellule muscolo-scheletriche.

Il trattamento con DS20060511, riduzione dei livelli di glucosio ematico e aumento potenziato dell’assorbimento di glucosio per via della traslocazione di GLUT4 nel muscolo scheletrico in vivo:
Per studiare gli effetti del DS20060511 sulla dinamica del glucosio in vivo, il composto è stato somministrato a topi normali. Nei topi che avevano continuato ad accedere al cibo, la sola somministrazione orale di DS20060511 in modo modesto, ma statisticamente significativo, ha ridotto i livelli di glucosio nel sangue, mentre nei topi che avevano negato l’accesso al cibo durante la notte, il composto non ha esercitato alcun effetto sui livelli di glucosio nel sangue. Quando è stato somministrato prima del carico orale di glucosio nel test di tolleranza al glucosio orale (GTT), il DS20060511 ha prodotto una soppressione dose-dipendente dell’aumento dei livelli di glucosio nel sangue dopo un carico orale di glucosio. La secrezione di Insulina durante il GTT orale è stata ridotta in modo piuttosto significativo in tutti i gruppi trattati con DS20060511, suggerendo che il trattamento con DS20060511 riduce i livelli di glucosio nel sangue indipendentemente dalla secrezione di Insulina. Il trattamento con DS20060511 ha prodotto un aumento significativo dell’assorbimento di [3H]-2-DG nei muscoli soleo e gastrocnemio, ma non nel cuore o nel tessuto adiposo bianco (WAT) durante il GTT intraperitoneale. L’analisi Western blot ha rivelato un aumento dei livelli di espressione della proteina GLUT4 nella frazione della membrana plasmatica dei muscoli scheletrici nel gruppo trattato con DS2006511 come osservato in un gruppo trattato con Insulina. Questi dati suggeriscono che il trattamento con DS20060511 riduce i livelli di glucosio nel sangue aumentando l’assorbimento del glucosio nel muscolo scheletrico inducendo la traslocazione di GLUT4 in vivo.

Valutazione farmacocinetica del DS20060511 nei topi:
In topi normali sono state esaminate le variazioni della concentrazione plasmatica e della distribuzione del DS20060511 in possibili organi/tessuti bersaglio. I livelli di esposizione sistemica al DS20060511 dopo sua somministrazione orale erano dose dipendenti e le concentrazioni massime a 30 min dopo la somministrazione di 1, 10 e 30 mg kg-1 erano rispettivamente di 0,6, 16,5 e 71,4 μM. La misurazione delle concentrazioni di DS20060511 nei tessuti a 75 min dopo la somministrazione orale (30 mg kg-1) ha rivelato concentrazioni quasi comparabili tra il muscolo scheletrico, il WAT e il cuore. Coerentemente con il suo profilo farmacocinetico stabile, la stabilità metabolica del composto nella frazione microsomiale del fegato era elevata (89% e 79% del composto rimanente dopo 1 h di incubazione con la frazione microsomiale del fegato umano e di topo, rispettivamente).
L’effetto ipoglicemizzante del DS20060511 dipende dal GLUT4:
Per confermare che l’effetto ipoglicemizzante del DS20060511 è mediato dal GLUT4, la molecola è stata somministrata a topi GLUT4KO. L’espressione della proteina GLUT4 non era rilevabile nel muscolo scheletrico, nel cuore e nel WAT dei topi GLUT4KO. Mentre il trattamento con DS20060511 ha causato una significativa diminuzione dei livelli di glucosio nel sangue e di Insulina plasmatica nei topi wild-type (WT) durante GTT orale, questi effetti sono stati completamente aboliti nei topi GLUT4KO. Il trattamento con DS20060511 ha aumentato significativamente l’assorbimento di 2-DG da parte dei muscoli soleo ed estensore lungo delle dita (EDL) dei topi WT, mentre non è stato osservato un tale aumento dell’assorbimento muscolare nei muscoli isolati dei topi GLUT4KO trattati con DS20060511 . Questi dati confermano che l’effetto ipoglicemizzante del DS20060511 è mediato da GLUT4 nel muscolo scheletrico.

Il trattamento con DS20060511 induce la traslocazione di GLUT4 senza attivazione delle vie IR-IRS1-PI3K-Akt-AS160 e -PI3K-Rac1:
La traslocazione di GLUT4 indotta dall’Insulina è attivata da (1) la via IR-IRS1-PI3K-Akt-AS16032 e (2) la via IR-IRS1-PI3K-Rac115 nel muscolo scheletrico. L’Insulina lega l’IR, che si traduce nell’attivazione di IRS1, PI3K e Akt. Akt attivato inibisce la proteina di attivazione della Rab GTPasi (GAP) AS160, che si traduce nell’attivazione delle proteine Rab e nella traslocazione di GLUT4 alla membrana plasmatica[33]. D’altra parte, Rac1 è attivato da PI3K e promuove il rimodellamento dell’actina, con conseguente traslocazione di GLUT4[12]. E’ stato esaminato se il trattamento con DS20060511 aumenta la traslocazione di GLUT4 nel muscolo scheletrico attraverso questi percorsi. Sebbene la subunità IRβ e IRS1 siano state fosforilate nei muscoli scheletrici dei topi trattati con Insulina, non è stata osservata tale fosforilazione di queste proteine dopo il trattamento con DS20060511. Allo stesso modo, mentre il trattamento con Insulina ha indotto la fosforilazione di Akt e AS160, il trattamento con DS20060511 non ha avuto tale effetto. Successivamente è stata eseguita la microscopia di immunofluorescenza per indagare se il DS20060511 potesse promuovere la polimerizzazione dell’actina. Sebbene sia stata osservata una forte colorazione di GLUT4 sulla superficie cellulare dopo il trattamento sia con Insulina che con DS20060511, la polimerizzazione dell’actina è stata osservata solo dopo il trattamento con Insulina nei miotubi differenziati L6-GLUT4myc. Inoltre, sebbene la traslocazione di GLUT4 sia stata indotta sia dall’Insulina che dal trattamento con DS20060511, la latrunculina B, un inibitore della polimerizzazione dell’actina, ha soppresso solo la traslocazione di GLUT4 indotta dall’Insulina, ma non quella indotta dal trattamento con DS20060511. Il co-trattamento di DS20060511 e Insulina ha comportato un aumento additivo della traslocazione di GLUT4 nei miotubi L6-GLUT4myc, anche alla concentrazione di Insulina alla quale la traslocazione di GLUT4 da parte della sola Insulina era saturata. Coerentemente con questi dati, anche l’assorbimento di 2-DG indotto dall’Insulina è stato ulteriormente aumentato dal trattamento concomitante con DS20060511 nei muscoli scheletrici isolati. In effetti, i livelli di glucosio nel sangue sono stati ridotti in misura maggiore dopo il trattamento combinato con DS20060511 più Insulina rispetto a quello dopo il solo trattamento con Insulina nei topi trattati con streptozotocina (STZ). Questi dati suggeriscono che l’attivazione né della via IR-IRS1-PI3K-Akt-AS160 né della via IR-IRS1-PI3K-Rac1 è coinvolta nella traslocazione di GLUT 4 indotta dal trattamento con DS20060511.

Il trattamento con DS20060511 aumenta l’ossidazione del glucosio durante l’esercizio fisico:
Poiché l’esercizio fisico, come l’Insulina, è ben noto per migliorare la traslocazione di GLUT4 e aumentare l’assorbimento di glucosio nel muscolo scheletrico[34], i ricercatori hanno successivamente studiato l’effetto del trattamento con DS20060511 sulla capacità di resistenza all’esercizio fisico e l’ossidazione del substrato energetico durante l’esercizio mediante calorimetria. Durante l’esercizio graduale sul tapis roulant, il VO2 è aumentato gradualmente in entrambi i gruppi trattati con il veicolo e DS20060511 e anche la capacità di resistenza all’esercizio era paragonabile tra i due gruppi. Dopo un po’ di tempo dall’inizio della corsa, il gruppo trattato con DS20060511 ha iniziato a mostrare un rapporto di scambio respiratorio (RER) relativamente più elevato rispetto al gruppo trattato con veicolo; inoltre, l’ossidazione stimata del glucosio durante il test era significativamente più alta nei topi trattati con DS20060511 rispetto ai topi trattati con veicolo, mentre l’ossidazione dei grassi era significativamente inferiore. Pertanto, il DS20060511 ha aumentato l’ossidazione del glucosio durante l’esercizio. I livelli di glucosio nel sangue sono diminuiti significativamente dopo l’esercizio nei topi trattati con DS20060511, ma non sono scesi al range di ipoglicemia. I livelli di lattato nel sangue erano comparabili tra i due gruppi.

Mancanza di effetto sulla fosforilazione dell’AMPK con Il trattamento di DS20060511:
Sulla base della scoperta che il DS20060511 ha aumentato l’utilizzo del glucosio nel muscolo scheletrico durante l’esercizio, i suoi effetti combinati con quelli della contrazione muscolare sono stati ulteriormente valutati utilizzando campioni di muscolo scheletrico isolati. L’assorbimento di 2-DG è stato aumentato in misura maggiore dopo l’elettrostimolazione muscolare combinata con il trattamento DS20060511 rispetto a quello dopo l’elettrostimolazione muscolare senza il trattamento DS20060511. Sebbene recenti scoperte suggeriscano che l’AMPK non svolga alcun ruolo nella traslocazione di GLUT4 e nell’assorbimento di glucosio nel muscolo osservato durante l’esercizio[16,22], l’attivazione di AMPK mediante stimolazione elettrica[21], nonché da AICAR[20], potrebbe aumentare l’assorbimento di glucosio nel muscolo scheletrico isolato. E’ stata esaminata la fosforilazione di AMPK dopo il trattamento con DS20060511 mediante western blotting nel muscolo scheletrico isolato. Sebbene il livello di fosforilazione dell’AMPK sia stato elevato dalla stimolazione muscolare elettrica, non è stato osservato alcun cambiamento di questo tipo dopo il trattamento con DS20060511. Il livello di fosforilazione dell’AMPK nel muscolo scheletrico è rimasto invariato dopo il trattamento con DS20060511 rispetto a quello prima del trattamento in vivo, anche in condizioni di non esercizio. Questi dati suggeriscono che l’aumento dell’assorbimento di glucosio indotto da DS20060511 è indipendente dall’attivazione dell’AMPK.

Il trattamento con DS20060511 diminuisce la glicemia in maniera eNOS-indipendente:
È stato dimostrato che il Nitroprussiato di sodio (SNP), un donatore di Ossido Nitrico (NO), aumenta l’assorbimento di glucosio nel muscolo scheletrico e che questo aumento non è inibito dall’inibitore PI3K, Wortmannin[35]. Inoltre, l’assorbimento del glucosio indotto dall’esercizio da parte del muscolo scheletrico non è stato soppresso dall’inibitore di NO NG-monometil-L-arginina (L-NMMA)[35]. Questi dati suggeriscono che il NO induce l’assorbimento del glucosio da parte del muscolo scheletrico attraverso un meccanismo che è distinto sia dall’Insulina che dalle vie di segnalazione dell’esercizio. L’Ossido Nitrico sintasi endoteliale, che è un importante enzima che genera NO, è espresso nel muscolo scheletrico. È stato riportato che l’assorbimento del glucosio è compromesso nei muscoli scheletrici isolati di topi eNOSKO[36]. Per studiare il meccanismo alla base dell’aumento dell’assorbimento di glucosio da parte del muscolo scheletrico indotto da DS20060511, è stato somministrato DS20060511 a topi eNOSKO. Il trattamento con DS20060511 ha ridotto significativamente i livelli di glucosio nel sangue sia nei topi WT che eNOSKO durante GTT orale. Sebbene i livelli di glucosio nel sangue siano stati ridotti dal trattamento con Insulina, i livelli di glucosio nel sangue sono stati ridotti ulteriormente dopo il trattamento con DS20060511, sia nei topi WT che eNOSKO, allo stesso modo. Questi dati suggeriscono che l’effetto ipoglicemizzante di DS20060511 è esercitato in modo eNOS-indipendente.

Il trattamento acuto e cronico con DS20060511 migliora l’intolleranza al glucosio nei topi diabetici obesi:
Per indagare se il trattamento con DS20060511 può attenuare l’intolleranza al glucosio nei topi con obesità indotta dalla dieta e resistenza all’Insulina, i ricercatori hanno condotto GTT orale in topi alimentati con dieta ricca di grassi (HFD) dopo il trattamento con DS20060511. Il trattamento con DS20060511 ha ridotto significativamente i livelli di glucosio nel sangue nei topi nutriti con HFD agli stessi livelli di quelli osservati nei topi alimentati con dieta normale durante il GTT orale. I livelli plasmatici di Insulina erano piuttosto diminuiti nei topi nutriti con HFD trattati con DS20060511. La soppressione dell’assorbimento di 2-DG indotto dall’Insulina nel muscolo scheletrico isolato da topi alimentati con HFD è stata completamente ripristinata dal trattamento con DS20060511. Questi dati suggeriscono che il trattamento acuto con DS20060511 migliora l’intolleranza al glucosio nei topi con obesità indotta dalla dieta e resistenza all’Insulina. Successivamente, è stato studiato l’effetto del trattamento cronico con DS20060511 in topi diabetici geneticamente obesi (db/db). I livelli di glucosio nel sangue sono diminuiti significativamente dal primo al 28° giorno di trattamento con DS20060511 nei topi db/db. Coerentemente con questi dati, anche il valore dell’emoglobina glicata (HbA1c) è stato significativamente ridotto dopo il trattamento cronico con DS20060511. Non ci sono state differenze statisticamente significative nel peso corporeo, nell’assunzione di cibo, nel livello di glucosio nel sangue a digiuno o nei livelli di Insulina plasmatica a digiuno tra i topi db/db trattati con DS20060511 e quelli trattati con il veicolo. Non sono stati inoltre rilevati cambiamenti significativi nei pesi dei tessuti di muscolo, cuore, WAT e fegato, o nel contenuto di glicogeno del muscolo, del cuore e del fegato. Questi dati suggeriscono che il trattamento con DS20060511 sia acuto che cronico migliora il diabete ripristinando l’assorbimento alterato del glucosio da parte del muscolo scheletrico.

Discussioni conclusive:
Come abbiamo visto, è stata passata al vaglio la libreria chimica in possesso dei ricercatori i quali hanno utilizzato miotubi L6-GLUT4myc per lo studio di un nuovo farmaco per il trattamento del diabete di tipo II scoprendo il composto xantenico DS20060511. Il DS20060511 ha aumentato la traslocazione di GLUT4 nei miotubi differenziati L6-GLUT4myc, ma non negli adipociti differenziati 3T3-L1-GLUT4myc, suggerendo che agisca principalmente nei muscoli scheletrici. Coerentemente, in vivo, il DS20060511 ha indotto l’assorbimento di 2-DG nei muscoli soleo e gastrocnemio, ma non nel cuore o nel tessuto adiposo. L’Insulina favorisce l’assorbimento del glucosio nel tessuto adiposo e nel muscolo scheletrico, che inevitabilmente, in condizioni metabolicamente alterate e ipercaloriche, porta all’obesità. Tuttavia, il DS20060511 migliora l’assorbimento del glucosio solo nel muscolo scheletrico e riduce la secrezione di Insulina sopprimendo l’aumento dei livelli di glucosio nel sangue dopo il carico di glucosio, sopprimendo così lo sviluppo dell’obesità; quindi, il composto sembra anche offrire una promessa come farmaco per la prevenzione dell’obesità. Il DS20060511 ha ridotto i livelli di glucosio nel sangue nei topi diabetici obesi, senza causare iperfagia, aumento di peso corporeo o ipoglicemia e senza aumentare la secrezione di Insulina. Inoltre, il DS20060511 non sembra abbassare il livello di glucosio nel sangue a digiuno, indicando il rischio relativamente basso di ipoglicemia associato all’uso di questo composto. Queste caratteristiche potrebbero essere preferibili a un farmaco sicuro ed efficace per il trattamento del diabete di tipo II.

L’effetto ipoglicemizzante del DS20060511 è stato completamente abolito nei topi GLUT4KO, indicando che il DS20060511 aumenta l’assorbimento del glucosio in modo GLUT4-dipendente. È interessante notare che il DS20060511 non è riuscito ad attivare la segnalazione dell’Insulina a monte, inclusa la fosforilazione di AS160 e il rimodellamento dell’actina o il percorso AMPK, che sono anche noti per aumentare la traslocazione di GLUT4 nel muscolo scheletrico. Inoltre, quando somministrato in combinazione con Insulina, il DS20060511 ha ulteriormente migliorato l’assorbimento del glucosio nel muscolo scheletrico sia nei topi normali che in quelli resistenti all’Insulina e ha ulteriormente ridotto i livelli di glucosio nel sangue in un modello murino di diabete di tipo I indotto da STZ. Il DS20060511 ha anche potenziato l’ossidazione del glucosio in tutto il corpo durante l’esercizio fisico, associata a un aumento dell’assorbimento e dell’utilizzo del glucosio nel muscolo scheletrico[16]. Pertanto, il DS20060511 può agire come un agente antidiabetico con un meccanismo d’azione completamente nuovo in pazienti con azioni alterate dell’Insulina nel muscolo scheletrico e in quelli con diabete di tipo I o II che ricevono Insulina e/o terapia fisica.
Alcuni composti sono stati anche segnalati in precedenza per indurre la traslocazione di GLUT4. È stato riportato che nuovi composti della Piridazina inducono fortemente la traslocazione di GLUT4 nei miotubi L6 e mostrano un significativo effetto ipoglicemizzante in un modello murino di diabete grave[37]. È noto che i disaccoppianti protonici, come il 2,4-dinitrofenolo, inducono la traslocazione di GLUT4 in accordo con un rapido calo dei livelli intracellulari di ATP[38]. Tuttavia, a differenza del DS20060511, questi composti promuovono la traslocazione di GLUT4 attraverso la via PI3K o AMPK. È stato riportato che la piccola molecola donatrice di NO NCX 4016 induce la traslocazione di GLUT4 negli adipociti 3T3-L1, ma non nelle cellule del muscolo scheletrico[39]. Questi risultati suggeriscono che un potenziatore della traslocazione di GLUT4 specifico del muscolo scheletrico come il DS20060511 non è mai stato segnalato in precedenza.

Perché il DS2006051 agisce selettivamente sul muscolo scheletrico? La molecola bersaglio del DS2006051 può essere espressa selettivamente nel muscolo scheletrico. La quantità di GLUT4 sulla superficie cellulare è determinata dall’equilibrio tra esocitosi dalle vescicole di stoccaggio intracellulare ed endocitosi dalla membrana cellulare. Il DS2006051 può promuovere l’esocitosi o sopprimere l’endocitosi di GLUT4 tramite l’attivazione della molecola bersaglio. Per studiare il bersaglio selettivo di DS20060511 nel muscolo scheletrico e nei miotubi L6, sono stati adottati tre diversi approcci: legame del composto radiomarcato, purificazione di perline immobilizzate con composto e fotoreticolazione UV di un composto al bersaglio. I composti radiomarcati o modificati chimicamente avevano la capacità di reagire con campioni preparati da tessuto muscolare scheletrico o miotubi L6-GLUT4myc, come lisati, microsomi o cellule viventi. Dopo l’arricchimento e la purificazione abbinati per ciascun approccio, i campioni sono stati analizzati mediante LC-MS/MS. Sfortunatamente, i ricercatori non sono riusciti a identificare nessuna molecola bersaglio specifica che si legasse al DS20060511 con un’alta affinità. Sono necessarie ulteriori indagini per identificare il bersaglio molecolare del DS20060511 e anche la via di segnalazione coinvolta, come la produzione di specie reattive dell’ossigeno associate a Rac1 o NADPH ossidasi 2.
In conclusione, è stato identificato un nuovo composto xantenico, il DS20060511, ed è stato dimostrato che il trattamento con DS20060511 induceva la traslocazione di GLUT4 indipendentemente dalla segnalazione canonica dell’Insulina e dall’attività dell’AMPK, per migliorare l’assorbimento del glucosio da parte del muscolo scheletrico. Inoltre, il trattamento con DS20060511 ha anche migliorato l’intolleranza al glucosio nei topi diabetici obesi. Sebbene non siano stati in grado di identificare la specifica molecola bersaglio del DS20060511 sulla cellula muscolare scheletrica, ulteriori studi con il composto aiuterebbero a sviluppare un nuovo farmaco per il diabete di tipo II.
Le caratteristiche del DS20060511 lo rendono una molecola di particolare interesse per i bodybuilder. La sua selettività per il tessuto muscolo scheletrico e la mancata attivazione dell’AMPK offrono due significativi vantaggi che le molecole con attività di miglioramento del insulino-resistenza (Biguanidi et simili) oggi disponibili non danno:
- Punto 1: la selettività della molecola per il tessuto muscolo-scheletrico e il miglioramento in tale sede dell’uptake del Glucosio da parte del miocita garantisce una ripartizione calorica a sensibile svantaggio del tessuto adiposo (quindi dell’adipocita) in un contesto ipercalorico, prolungando in modo indeterminato (almeno secondo i dati attuali) il periodo di vantaggio che l’atleta può sperimentare in un regime di questo tipo, prima che il peggioramento dei parametri del IR portino ad un aumento significativo della massa grassa e una riduzione dei guadagni muscolari sia in rapporto alla precedente che in termini assoluti;
- Punto 2: la capacità del DS20060511 di bypassare l’attivazione/stimolo del AMPK permette di non sottoregolare/bloccare l’attività del mTOR e della sua attività sull’ipertrofia muscolare. Questo vantaggio è unico nel suo genere dal momento che, per esempio, sia la Metformina che la Berberina, due molecole largamente utilizzate per il miglioramento del IR, interagiscono per via delle PPAR-α nello stimolo dell’attività del AMPK la quale sottoregola/blocca mTOR.

Riguardo all’ultimo punto, c’è da dire che, da quanto osservato empiricamente ed emerso clinicamente, l’interazione negativa di Metformina e Berberina sul mTOR risulta significativa in modo dosaggio-dipendente. Si ipotizza, ma questa è una semplice ipotesi osservazionale, che l’uso di dosaggi non superiori a 500-750mg/die totali di entrambe le molecole non alteri crescita e/o recupero muscolare. Ricordiamoci inoltre che sia la Metformina che la Berberina (compreso anche l’ALA) sembrano avere potenziali inibitori sugli enzimi implicati nella lipogenesi ed esterificazione degli acidi grassi liberi negli adipociti, ma questa è un altra storia.
È interessante notare che alcuni studi dell’ultimo decennio suggeriscono che la Metformina può inibire direttamente l’azione della Leucina sul mTOR. Non solo questo sarebbe, ovviamente, un fattore negativo per la crescita muscolare, ma ipoteticamente l’effetto inibitorio della Metformina sul mTOR potrebbe avere un effetto maggiore in quanto è correlato alla riduzione del rischio di tumori mortali nei diabetici.
E’ a proposito molto interessante quanto postulato dal Dr. Melnik dell’Università di Osnabrück in Germania: “la Metformina può essere un diretto concorrente della Leucina per il legame e l’attività del mTORC1”.
Il medico ha notato nel suo articolo che la dose giornaliera abituale nei diabetici di Metformina (2g) è nell’ordine dei 2g di Leucina derivati dal consumo giornaliero di 100g di carne o formaggio. Poiché le due molecole sono simili per struttura e dimensioni, possono competere per gli stessi siti nell’attivazione del mTOR. Di conseguenza, possiamo affermare, con un buon margine di ragione, che è una questione “dose-risposta dipendente”, come accennato in precedenza, in rapporto all’attività potenziale di alterazione del mTOR sia diretta (legame attivazione leucina-simile) che indiretta (via AMPK).

Per quanto riguarda la questione della potenziale sotto-regolazione sui AR da parte della Metformina, i dati attuali provengono principalmente da studi di linee cellulari in vitro, in donne con PCOS, e da studi sui pazienti con cancro alla Prostata che però non danno comunque dati chiari sul grado di riduzione dei AR a livello del muscolo-scheletrico, di conseguenza si può speculare ancora ampiamente su quali possano essere gli effetti in vivo nell’uomo sulla crescita del tessuto muscolo-scheletrico durante il trattamento con Metformina. Rimango, al momento, dell’idea che sia fondamentalmente una questione di “soglia di efficacia” in rapporto agli “effetti indesiderati”, e la cosa, però, non è così semplice da calibrare come sembra viste le diversità nelle risposte individuali.
Ma, tornando a parlare del DS20060511, potrebbe avere un potenziale anche in un regime ipocalorico? Si, ovviamente, anche se presumibilmente il calo della Leptina sarà più rapido per via della “carestia glucidica adipocitaria indotta”. Sicuramente risulterebbe un vantaggio nei refeed sia pre-contest che quelli di “routine” settimanale. La superiorità rispetto a quanto oggi utilizzato con tali finalità rimane.
Per il momento, non ci resta che attendere nuovi studi sul DS20060511, possibilmente sull’uomo.
Gabriel Bellizzi
Riferimenti:
- Kahn, B. B., Rossetti, L., Lodish, H. F. & Charron, M. J. Decreased in vivo glucose uptake but normal expression of GLUT1 and GLUT4 in skeletal muscle of diabetic rats. J. Clin. Invest. 87, 2197–2206 (1991).
- Wallberg-Henriksson, H. & Zierath, J. R. GLUT4: a key player regulating glucose homeostasis? Insights from transgenic and knockout mice (review). Mol. Membr. Biol. 18, 205–211 (2001).
- Ryder, J. W. et al. Use of a novel impermeable biotinylated photolabeling reagent to assess insulin- and hypoxia-stimulated cell surface GLUT4 content in skeletal muscle from type 2 diabetic patients. Diabetes 49, 647–654 (2000).
- Liu, M. L. et al. Transgenic mice expressing the human GLUT4/muscle-fat facilitative glucose transporter protein exhibit efficient glycemic control. Proc. Natl Acad. Sci. USA 90, 11346–11350 (1993).
- Gibbs, E. M. et al. Glycemic improvement in diabetic db/db mice by overexpression of the human insulin-regulatable glucose transporter (GLUT4). J. Clin. Invest. 95, 1512–1518 (1995).
- Ren, J. M. et al. Overexpression of Glut4 protein in muscle increases basal and insulin-stimulated whole body glucose disposal in conscious mice. J. Clin. Invest. 95, 429–432 (1995).
- Huang, S. & Czech, M. P. The GLUT4 glucose transporter. Cell. Metab. 5, 237–252 (2007).
- Bryant, N. J., Govers, R. & James, D. E. Regulated transport of the glucose transporter GLUT4. Nat. Rev. Mol. Cell. Biol. 3, 267–277 (2002).
- Kubota, T., Kubota, N. & Kadowaki, T. Imbalanced insulin actions in obesity and type 2 diabetes: key mouse models of insulin signaling pathway. Cell. Metab. 25, 797–810 (2017).
- Kubota, N. et al. Dynamic functional relay between insulin receptor substrate 1 and 2 in hepatic insulin signaling during fasting and feeding. Cell. Metab. 8, 49–64 (2008).
- Bhuin, T. & Roy, J. K. Rab proteins: the key regulators of intracellular vesicle transport. Exp. Cell. Res. 328, 1–19 (2014).
- Chiu, T. T., Jensen, T. E., Sylow, L., Richter, E. A. & Klip, A. Rac1 signalling towards GLUT4/glucose uptake in skeletal muscle. Cell. Signal. 23, 1546–1554 (2011).
- Khayat, Z. A., Tong, P., Yaworsky, K., Bloch, R. J. & Klip, A. Insulin-induced actin filament remodeling colocalizes actin with phosphatidylinositol 3-kinase and GLUT4 in L6 myotubes. J. Cell. Sci. 113, 279–290 (2000).
- Sano, H. et al. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J. Biol. Chem. 278, 14599–14602 (2003).
- JeBailey, L. et al. Ceramide- and oxidant-induced insulin resistance involve loss of insulin-dependent Rac-activation and actin remodeling in muscle cells. Diabetes 56, 394–403 (2007).
- Sylow, L., Kleinert, M., Richter, E. A. & Jensen, T. E. Exercise-stimulated glucose uptake—regulation and implications for glycaemic control. Nat. Rev. Endocrinol. 13, 133–148 (2017).
- Hirshman, M. F., Wallberg-Henriksson, H., Wardzala, L. J., Horton, E. D. & Horton, E. S. Acute exercise increases the number of plasma membrane glucose transporters in rat skeletal muscle. FEBS Lett. 238, 235–239 (1988).
- Goodyear, L. J., Hirshman, M. F. & Horton, E. S. Exercise-induced translocation of skeletal muscle glucose transporters. Am. J. Physiol. 261, E795–E799 (1991).
- Vichaiwong, K. et al. Contraction regulates site-specific phosphorylation of TBC1D1 in skeletal muscle. Biochem. J. 431, 311–320 (2010).
- Merrill, G. F., Kurth, E. J., Hardie, D. G. & Winder, W. W. AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. Am. J. Physiol. 273, E1107–E1112 (1997).
- Sylow, L. et al. Rac1 and AMPK account for the majority of muscle glucose uptake stimulated by ex vivo contraction but not in vivo exercise. Diabetes 66, 1548–1559 (2017).
- McConell, G. K. It’s well and truly time to stop stating that AMPK regulates glucose uptake and fat oxidation during exercise. Am. J. Physiol. Endocrinol. Metab. 318, E564–E567 (2020).
- Henríquez-Olguin, C. et al. Cytosolic ROS production by NADPH oxidase 2 regulates muscle glucose uptake during exercise. Nat. Commun. 10, 4623 (2019).
- Sylow, L. et al. Rac1 governs exercise-stimulated glucose uptake in skeletal muscle through regulation of GLUT4 translocation in mice. J. Physiol. 594, 4997–5008 (2016).
- Goodyear, L. J., Giorgino, F., Balon, T. W., Condorelli, G. & Smith, R. J. Effects of contractile activity on tyrosine phosphoproteins and PI 3-kinase activity in rat skeletal muscle. Am. J. Physiol. 268, E987–E995 (1995).
- Yeh, J. I., Gulve, E. A., Rameh, L. & Birnbaum, M. J. The effects of wortmannin on rat skeletal muscle. Dissociation of signaling pathways for insulin- and contraction-activated hexose transport. J. Biol. Chem. 270, 2107–2111 (1995).
- Lund, S., Holman, G. D., Schmitz, O. & Pedersen, O. Contraction stimulates translocation of glucose transporter GLUT4 in skeletal muscle through a mechanism distinct from that of insulin. Proc. Natl Acad. Sci. USA 92, 5817–5821 (1995).
- Krook, A. et al. Characterization of signal transduction and glucose transport in skeletal muscle from type 2 diabetic patients. Diabetes 49, 284–292 (2000).
- Kim, Y. B., Nikoulina, S. E., Ciaraldi, T. P., Henry, R. R. & Kahn, B. B. Normal insulin-dependent activation of Akt/protein kinase B, with diminished activation of phosphoinositide 3-kinase, in muscle in type 2 diabetes. J. Clin. Invest. 104, 733–741 (1999).
- Kennedy, J. W. et al. Acute exercise induces GLUT4 translocation in skeletal muscle of normal human subjects and subjects with type 2 diabetes. Diabetes 48, 1192–1197 (1999).
- Martin, I. K., Katz, A. & Wahren, J. Splanchnic and muscle metabolism during exercise in NIDDM patients. Am. J. Physiol. 269, E583–E590 (1995).
- Kramer, H. F. et al. Distinct signals regulate AS160 phosphorylation in response to insulin, AICAR, and contraction in mouse skeletal muscle. Diabetes 55, 2067–2076 (2006).
- Jaldin-Fincati, J. R., Pavarotti, M., Frendo-Cumbo, S., Bilan, P. J. & Klip, A. Update on GLUT4 vesicle traffic: a cornerstone of insulin action. Trends Endocrinol. Metab. 28, 597–611 (2017).
- Richter, E. A. & Hargreaves, M. Exercise, GLUT4, and skeletal muscle glucose uptake. Physiol. Rev. 93, 993–1017 (2013).
- Higaki, Y., Hirshman, M. F., Fujii, N. & Goodyear, L. J. Nitric oxide increases glucose uptake through a mechanism that is distinct from the insulin and contraction pathways in rat skeletal muscle. Diabetes 50, 241–247 (2001).
- Duplain, H. et al. Insulin resistance, hyperlipidemia, and hypertension in mice lacking endothelial nitric oxide synthase. Circulation 104, 342–345 (2001).
- 37.Tsuji, T. et al. Discovery of novel pyridazine derivatives as glucose transporter type 4 (GLUT4) translocation activators. Bioorg. Med. Chem. Lett. 29, 1785–1790 (2019).
- 38.Klip, A., Schertzer, J. D., Bilan, P. J., Thong, F. & Antonescu, C. Regulation of glucose transporter 4 traffic by energy deprivation from mitochondrial compromise. Acta Physiol. (Oxf.). 196, 27–35 (2009).
- Kaddai, V. et al. The nitric oxide-donating derivative of acetylsalicylic acid, NCX 4016, stimulates glucose transport and glucose transporters translocation in 3T3-L1 adipocytes. Am. J. Physiol. Endocrinol. Metab. 295, E162–E169 (2008).










































































{kind=link}