Secondo un recente studio svolto dai ricercatori della Universidade de Sao Paulo, un utilizzatore di AAS su quattro presenta depositi arteriosi. La conclusione dei ricercatori è stata fatta confrontando un gruppo di giovani Bodybuilder utilizzatori di AAS con un gruppo di giovani Bodybuilder non utilizzatori.(1)
I ricercatori hanno preso in esame 20 soggetti di sesso maschile apparentemente sani di età compresa tra i 18 ed i 45 anni i quali si erano allenati con i pesi per almeno due anni e avevano un utilizzo di AAS medio di 2-4 cicli all’anno. Hanno anche esaminato un gruppo altrettanto grande di Bodybuilder “natural”, oltre a un gruppo di 10 soggetti sani che non erano fisicamente attivi.
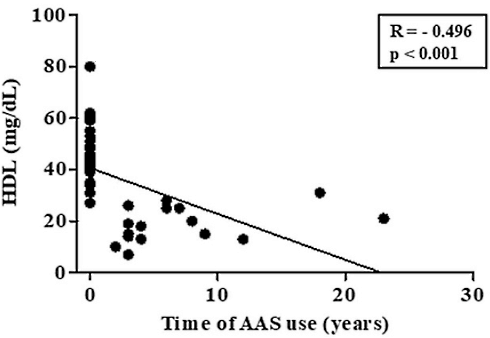
Una prima osservazione dei soggetti esaminati mostrò ovvie caratteristiche di differenziazione nella composizione corporea tra utilizzatori e non utilizzatori. Gli utilizzatori di AAS presentavano una maggiore massa muscolare e una minore massa grassa rispetto agli atleti “natural”. D’altra parte, le ovvie differenze vennero notate anche a livello dei marker ematici base. Gli utilizzatori di AAS avevano livelli inferiori di HDL e livelli maggiori di LDL rispetto ai Bodybuilder “natural”.
I livelli di HDL erano correlati al tempo di utilizzo degli AAS. Maggiore era stato il tempo di esposizione agli AAS e minore risultavano le concentrazioni ematiche di HDL.
Quando i ricercatori hanno determinato le condizioni delle arterie coronarie tramite scansione, non hanno osservato alcuno stato patologico nei soggetti sedentari e nei Bodybuilder “natural”.
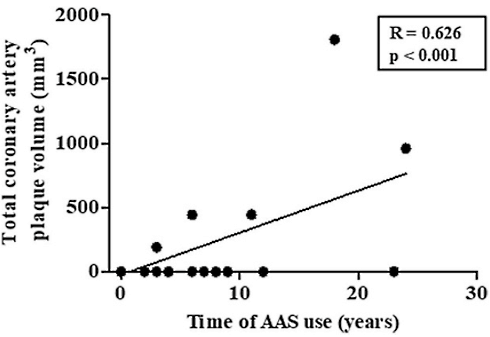
Tuttavia, i ricercatori hanno osservato un deterioramento preclinico delle arterie coronarie tra gli utilizzatori di AAS. In questo gruppo, ¼ dei soggetti aveva arterie coronarie ostruite o calcificate. Le condizioni erano correlate agli anni di utilizzo. Infatti, i soggetti che avevano alle spalle più anni d’uso di AAS presentavano una maggiore quantità di placche coronariche.
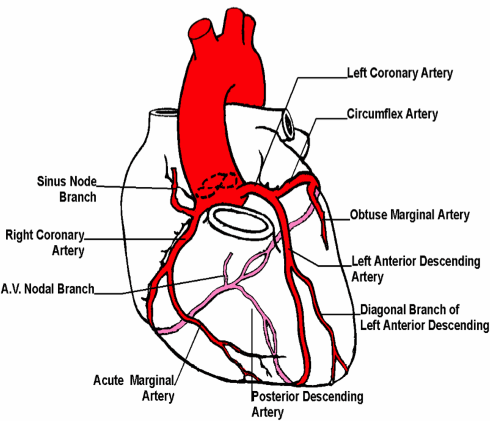
I ricercatori hanno rilevato la maggior parte delle anormalità coronariche degli utilizzatori di AAS nell’arteria discendente anteriore sinistra.
Le scontate conclusioni dei ricercatori sono che l’uso a lungo termine di AAS sembra essere correlato con lo sviluppo di patologie coronariche subclinica precoce in questa popolazione.
Per quanto ad alcuni lettori possa sembrare banale lo studio qui esposto, esso mostra come il peggioramento delle condizioni cardiovascolari possa essere al quanto rapido concretizzandosi in pochi anni. Gli utilizzatori di AAS, oltre ad assumere una adeguata supplementazione volta al controllo dei lipidi ematici, dovrebbero sottoporsi regolarmente a visite cardiologiche comprendenti ecocardiogramma con intervalli massimi tra una visita e la successiva di sei mesi.
Sugli effetti dell’utilizzo di AAS sul lungo termine in soggetti di sesso femminile, ad oggi, sappiamo relativamente poco. Siamo pienamente a conoscenza del fatto che un uso di questa classe di farmaci nelle donne possa portare come conseguenza ad effetti virilizzanti di diverso tipo e entità. Un interessante caso studio realizzato da due medici ORL canadesi, Yael Bensoussan e Jennifer Anderson, dell’Università di Toronto, pubblicato sul Clinical Case Reports, ci offre la possibilità di analizzare la disfonia AAS-indotta e i sui risvolti nel lungo termine in una atleta. (1)
Il soggetto preso in esame nell’articolo di Yael Bensoussan e Jennifer Anderson è una Bodybuilder la quale aveva 27 anni nel 1998. In quell’anno aveva partecipato a una competizione e, per prepararvisi, aveva usato, secondo quanto da lei riportato, 50mg di Nandrolone Decanoato a settimana per 6 settimane.
Due mesi dopo l’inizio della somministrazione di Nandrolone, la voce della bodybuilder era gradualmente divenuta più bassa e roca. Sebbene abbia rinunciato all’uso di altri farmaci nella sua preparazione, l’alterazione del tono vocale risultò permanente. La terapia applicata per trattare l’alterazione della voce, volta quindi a ripristinare una tonalità vocale più alta e più femminile, non risultò efficace.
Il soggetto in questione, come conseguenza di questa alterazione, perse il lavoro perché la gente pensava che fosse un uomo. Contemporaneamente aveva detto addio al Bodybuilding.
Sempre nel 1998 contattò contattato dei medici specialisti, i quali decisero di operare sulla laringe. L’operazione ebbe esito positivo dal momento che la voce della donna tornò ad essere più alta e più femminile. Ma dieci anni dopo la ex Bodybuilder divenne afona. Si era verificata una atrofia delle corde vocali.
Tredici anni dopo la prima operazione, i medici intervenivano chirurgicamente una seconda volta, e di nuovo con successo.
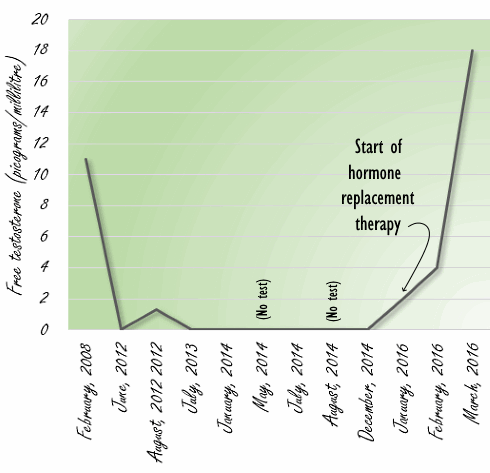
I ricercatori non conoscono il motivo per il quale sia comparsa dell’afonia nella donna. Essa riferì di sentirsi cronicamente stanca e successivi esami ematici mostrarono livelli di Testosterone libero marcatamente bassi. Apparentemente, gli squilibri nella funzionalità dell’Asse HPG dovuti al passato uso di AAS da parte della donna non erano stati trattati e risolti con conseguente mantenimento di livelli bassi di Testosterone cronicizzatisi con il passare degli anni (riduzione ormonale età correlata). Tale circostanza (alterazione a lungo termine del HPTA) si può osservare anche negli ex abusatori di AAS di sesso maschile.
Appurato lo stato ormonale della paziente, i medici le prescrissero una terapia ormonale sostitutiva che risolse lo stato di fatica cronica.
I medici hanno ipotizzato che la possibile causa dell’afonia potrebbe essere ricondotta al livello di Testosterone anormalmente basso della donna il quale ha portato ad una atrofia delle corde vocali. Questa risposta avversa non trova però molto riscontro nell’osservazione di soggetti di sesso maschile con ipogonadismo AAS-dipendente non trattato. Il fatto che non esistano casi studio specifici che abbiano analizzato questo aspetto in tali soggetti, e la presenza di un dimorfismo sessuale di risposta ormonale, fanno si che questa ipotesi rimanga una possibilità plausibile.
Gli autori di questo caso studio affermano che, per quanto ne sanno, il loro è il primo rapporto sugli effetti degli AAS sulla voce di un soggetto di sesso femminile e sulla loro evoluzione in un periodo di 20 anni. Questi cambiamenti sono clinicamente rilevanti dal momento che sono difficili da trattare e pertanto dovrebbe essere comunicato ai soggetti che usano AAS o a coloro che sono sottoposti ad una terapia con androgeni.
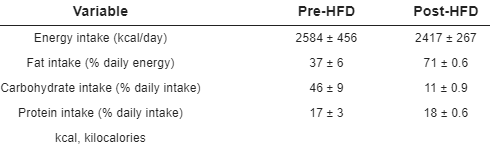
I regimi alimentari “low-carb”, nelle loro varianti, sono largamente utilizzati per la perdita di peso, nonostante non presentino veri e propri vantaggi, a parità di calorie, sulla perdita di massa grassa in rapporto a regimi ipocalorici “low-fat”. Infatti, i loro vantaggi si “limitano” all’impatto psicologico dovuto alla repentina perdita di peso (acqua e glicogeno) delle prime settimane e al senso di sazietà legato all’introito proteico e al livello dei chetoni (dipendente dal tipo di “low carb”). Le varianti di questa tipologia di regime alimentare più applicate sono quelle a schema ciclico, che consistono, come ben sappiamo, nell’alternanza di giorni a ridotta assunzione glucidica e medio/alta assunzione lipidica e proteica con giorni ad alta assunzione glucidica e ridotta/moderata assunzione lipidica e proteica. Questa tipologia di regime “low carb” risulta il più tollerabile sul lungo periodo. Secondo un recente studio di piccole dimensioni svolto da ricercatori canadesi della University of British Columbia un marcato aumento del consumo glucidico (e il creare repentini picchi glicemici) dopo un periodo a ridotto consumo (ma anche in condizioni generali) può causare danni ai vasi sanguinei.(1)
I ricercatori hanno fatto seguire a 9 soggetti sani di sesso maschile una dieta a basso contenuto di carboidrati per 2 settimane. La composizione schematizzata del piano nutrizionale è mostrata qui di seguito.
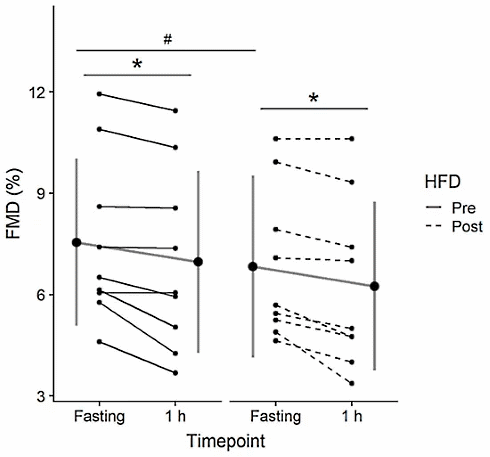
Precedentemente e immediatamente dopo l’applicazione del regime alimentare “low carb”, agli studenti è stato fatto assumere un quantitativo di 75g di glucosio disciolti in acqua. I ricercatori hanno quindi determinato l’effetto dell’assunzione del glucosio sulla flessibilità dei vasi sanguigni. Sono state determinate le concentrazione ematiche di specifici marker indicativi del danno ai vasi sanguigni.
Dopo l’assunzione del glucosio, la flessibilità dei vasi sanguigni era diminuita. Tale condizione risultò in entrambe le assunzioni di glucosio. Il regime a basso contenuto di carboidrati ha incrementato la riduzione dell’elasticità dei vasi sanguigni dopo picco glicemico in misura similare a quanto riscontrato dopo la medesima procedura prima dell’inizio del regime “low carb”.
Questo effetto non era però così allarmante. Nonostante ciò, l’effetto dell’assunzione di glucosio durante la dieta a basso contenuto di carboidrati sulla concentrazione delle microparticelle endoteliali CD31+ CD42b- e CD62E+ è stato comunque motivo di preoccupazione per i ricercatori.
In un comunicato stampa dello scorso mese (2) Cody Durrer, uno dei principali autori dello studio qui in breve esposto, ha affermato che l’intento della ricerca era quello di osservare e valutare le risposte fisiologiche alla reintroduzione di carboidrati dopo un periodo di marcata restrizione di questi ultimi. Poiché la ridotta tolleranza al glucosio e i picchi della glicemia ematica (in cronico) sono noti per essere associati ad un aumentato rischio di sviluppare malattie cardiovascolari, per i ricercatori aveva senso esaminare quello che succede nei vasi sanguigni in risposta ad un picco glicemico.
Cody Durrer continua dicendo che inizialmente lui ed i suoi colleghi cercavano variazioni nella risposta infiammatoria e nella tolleranza glucidica. Ma ciò che hanno scoperto è stato un aumento dei biomarker ematici indicativi di un danno endoteliale vascolare successivo al picco glicemico.
Jonathan Little, un altro dei ricercatori che hanno realizzato lo studio, ha affermato che nonostante la giovane età e il buono stato di salute dei soggetti osservati, quando si esaminava lo stato di salute dei loro vasi sanguigni dopo aver consumato i 75g di glucosio, i risultati sembravano provenire da qualcuno con una salute cardiovascolare compromessa.
La preoccupazione dei ricercatori riguarda il fatto che molte delle persone che seguono una dieta “Keto”, che sia per la perdita di peso, per trattare il diabete di tipo II o qualche altra ragione legata alla salute, potrebbero annullare alcuni degli effetti positivi sul sistema cardiovascolare se assumono improvvisamente (all’interno di un pasto) quantitativi glucidici elevati. Soprattutto se questi soggetti hanno un rischio più elevato per le malattie cardiovascolari.
Il ricercatore conclude dicendo che i dati raccolti suggeriscono che una dieta chetogenica non è un regime da seguire per sei giorni alla settimana e da un giorno alimentarmente “fuori controllo”.
Nonostante lo studio abbia un design piuttosto limitante, i dati che fornisce possono risultare particolarmente interessanti anche in ambito culturistico, in specie nei periodi, come il pre-contest, dove il “carico/scarico” glucidico è un abitudine (o dove l’assunzione glucidica è particolarmente alta, come in “Off Season”) e negli utilizzatori di AAS. Dovrebbe essere infatti risaputo come questi ultimi subiscano un effetto negativo a livello dell’endotelio vascolare dato da livelli sovra fisiologici di AAS (grado dose e tempo dipendente). Una sconsiderata gestione del piano alimentare anche per ciò che concerne il carico glucidico porterebbe con molta probabilità ad un ulteriore peggioramento delle condizioni cardiovascolari in tali soggetti.
Come ben sappiamo, livelli bassi di Testosterone sono un fattore connesso alla riduzione della densità ossea e della sintesi di Collagene. Parimenti, siamo a conoscenza del fatto che dosi sovrafisiologiche di Testosterone possono causare un aumento della sistesi proteica di circa 50 volte, a seconda della dose e del tempo di esposizione, e della densità ossea. Ciò che spesso non si considera, sia per ignoranza che per via di una limitata comprensione del fattore in essere, è il fatto che dosaggi sovra fisiologici di Testosterone (>180mg/week) portano ad una riduzione della sintesi di Collagene ≥ 50% – percentuale variabile in relazione alla dose e alle risposte genotipiche e che può raggiungere l’80%, vale a dire un tasso di sintesi di Collagene di un soggetto anziano. Le fibre dei tendini sono composte per 3 parti da fibre di Collagene e da 1 parte di Elastina. Dato ciò, in condizione di esposizione a dosi sovrafisiologiche di Testosterone, le probabilità di comparsa di lesioni tendinee è maggiore dal momento che si viene a creare una condizione nella quale, facendo un esempio banale ma che rende perfettamente l’idea, l’atleta presenta considerevoli masse muscolari le cui estremità sono connesse a tendini di ridotte dimensioni e resistenza. Questo fattore, quindi, non è dovuto semplicemente ad una variazione del tasso ipertrofico dei due tessuti durante l’uso di AAS o alla marcata soppressione dei livelli estrogenici. Come spesso accade, anche in questo caso ci troviamo di fronte alla presenza di molteplici fattori.
Un AAS che può causare problemi connessi a quanto appena riportato, anche se con dinamica “paradossale”, è lo Stanozololo. Questo AAS aumenta la sintesi del Collagene ma non ne migliora la resistenza, rendendo i tendini soggetti comunque a possibili lesioni. In pratica, lo Stanozololo, aumenta marcatamente la sintesi di Collagene, ma ironicamente riduce l’integrità del cross-linking (formazione di legami incrociati) dello stesso, rendendo così il tendine molto più debole.
Nandrolone, Boldenone, Oxandrolone e Metenolone causano un aumento della sintesi di Collagene senza comprometterne la cross-linking diminuendo la possibilità di infortuni, la comparsa di infiammazioni tendinee e il dolore articolare:
Nandrolone
Nandrolone (privo di esterificazione): la somministrazione di 1,25-2mg/kg a settimana (circa 100-160mg per un uomo di 80Kg) può portare ad un aumento dei livelli di Procollagene III del 270% in un lasso di tempo pari a 2-3 settimane. Il Procollagene III è un indicatore primario utilizzato per determinare il tasso di sintesi di Collagene.
Boldenone
Boldenone (privo di esterificazione): la somministrazione di 2,25mg/Kg a settimana (circa 180mg per un uomo di 80Kg) può portare ad un aumento dei livelli di Procollagene III di circa il 340% in un lasso di tempo pari a 2-3 settimane.
Metenolone
Metenolone (privo di esterificazione): la somministrazione di 3,6mg/kg a settimana (circa 288mg per un uomo di 80Kg) può aumentare la sintesi di Collagene di circa il 180% in un lasso di tempo pari a 2-3 settimane.
Oxandrolone
Oxandrolone: la somministrazione di 0,5mg/Kg al giorno (circa 40mg per un uomo di 80Kg) può aumentare la sintesi di Collagene di circa il ≥50% in un lasso di tempo molto breve (1 settimana circa).
*Esistono oltre un centinaio di studi che documentano l’efficacia del Oxandrolone nel trattamento di pazienti che necessitano di rapidi aumenti nella sintesi di Collagene per migliorare i processi guaritivi.
Altri composti non-steroidei possono ovviamente essere aggiunti al fine di migliorare la risposta preventiva sugli infortuni muscolo-tendinei-articolari:
hGH
hGH: la somministrazione di 6UI/die causa un aumento del tasso di deposizione di Collagene di circa il 250% in strutture di Collagene danneggiate. Questo effetto ha portato all’osservazione di un aumento della forza biomeccanica dei tendini danneggiati trattati con il peptide per via dell’aumento del deposito di Collagene nelle strutture tendinee. L’effetto del hGH sull’aumento della sintesi di Collagene è dose dipendente: ciò significa che la percentuale sopraesposta aumenta in relazione all’aumento della dose e che si può comunque ottenere un ottimo tasso di sintesi (media del +125%) con 3UI/die o 2UI/die (media del +83,3%) con una sensibile riduzione della comparsa di neuropatie (vedi sindrome del Tunnel Carpale).
TB-500
TB-500: in seguito a stime di rapporto con il potenziale di deposito di Collagene dato dall’uso di hGH, la somministrazione di 4-5mg di Timosina beta–4a settimana (per sei settimane seguite da una dose di mantenimento pari a 2-2,5mg ogni quattro settimane) potrebbe causare un aumento del tasso di deposito di Collagene di circa il 350-400%.
In conclusione, da quanto riportato, si può ben capire come l’inserimento dei suddetti composti possa garantire una considerevole prevenzione di infortuni muscolo-tendinei-articolari. Ovviamente, la scelta del composto e dei suoi dosaggi non dovrebbe tenere consto solo del suo potenziale sulla sintesi/deposito di Collagene. L’uso dell’Oxandrolone, per ovvi motivi legati al suo impatto a livello epatico e sui lipidi ematici, è una molecola che dovrebbe comunque essere usata in un lasso di tempo limitato (con una indicazione di massimo 8 settimane). Il Nandrolone, soprattutto a causa dei suoi effetti a livello cerebrale/SNC, sebbene le dosi utili sono tutto sommato contenute, non è ben tollerato da tutti e, consequenzialmente, il suo uso è pienamente funzionale solo su un numero limitato di persone. Il Boldenone, invece, risulta essere superiore al Nandrolone sotto l’aspetto della tollerabilità alla molecola ma, per via della sua azione sulla eritropoiesi di grado maggiore (sebbene non del tutto chiarito nell’uomo) rispetto ad altri AAS, il suo uso potrebbe essere problematico per i soggetti ipersensibili a questo effetto. Il Metenolone, nonostante presenti un tasso d’efficacia inferiore rispetto al Nandrolone e al Boldenone, risulta generalmente il composto con il più alto tasso di tollerabilità rispetto alle precedenti molecole, tollerabilità che ne permette un uso di basse su tempi medio-lunghi. L’uso del hGH, come detto in precedenza, può essere gestito con buoni risultati sotto l’aspetto del deposito di Collagene anche con un dosaggio giornaliero di 2-3UI; i problemi principali con il suo uso sono di natura economica dal momento che il dosaggio contenuto garantisce una limitata possibilità di comparsa di effetti collaterali legati alla molecola. In fine, il TB-500 sembrerebbe avere il potenziale maggiore sul deposito di Collagene sebbene le percentuali esposte siano piuttosto teoriche e la reperibilità della molecola non sia così semplice.
PS:E’ ovvio che i fattori quali livelli di infiammazione sistemica/riduzione delle concentrazioni di Cortisolo e livelli estrogenici devono essere comunque monitorati per far si che la prevenzione di infortuni e/o stati infiammatori tendineo-articolari sia “completa”.
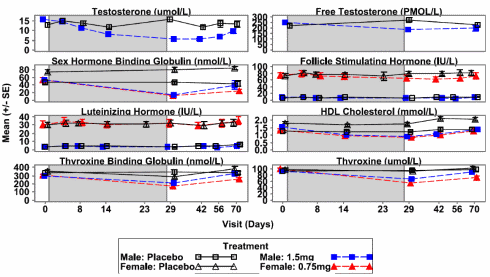
Nel febbraio di quest’anno ho scritto un articolo nel quale riportavo alcuni studi svolti sul SARM GSK20881078. Tra gli studi citati ve ne era uno svolto sull’uomo (studio di fase 1).(1) Attualmente i ricercatori della GlaxoSmithKline stanno proseguendo i test sugli esseri umani. Un recente studio che ha preso in esame gli effetti del GSK20881078 sugli esseri umani, il quale verrà a breve pubblicato sul Journal of Clinical Endocrinology & Metabolism, ha mostrato, per la prima volta, il potenziale anabolizzante di questo SARM nell’uomo.(2) Tuttavia, lo studio suggerisce anche che il GSK20881078 potrebbe avere degli effetti collaterali non di entità non trascurabile.
I ricercatori hanno somministrato il GSK20881078 a 100 persone sane di età superiore ai 50 per 8 settimane. I soggetti presi in esame erano sia maschi che femmine e sono stati trattati con dosaggi differenti.
I soggetti di sesso maschile ai quali è stata somministrata una dose di 4mg/die di GSK20881078 hanno avuto un guadagno di 1,5Kg di massa magra in 8 settimane di trattamento. Le donne alle quali è stata somministrata una dose di 1,5mg/die di GSK2088078, hanno avuto un guadagnato 3Kg di massa magra.
Ovviamente, come per altri SARM o PED in generale, l’uso del GSK20881078 può portare ad alcuni effetti collaterali. Il livello di Testosterone totale negli uomini si è ridotto di due terzi durante il trattamento con il SARM. Due settimane dopo la fine del trattamento, i livelli di Testosterone non erano ancora tornati in soglia basale (nei range della fascia d’età dei soggetti esaminati).
Anche gli effetti sui livelli di HDL non sono da sottovalutare. Infatti, si è verificato un calo del 30-45% delle lipoproteine ad alta densità.
Gli effetti collaterali di cui sopra sono stati rilevati a metà del periodo di somministrazione. Quale entità abbiano gli effetti collaterali dopo 8 settimane di trattamento non è dato saperlo dal momento che i ricercatori, stranamente e con non pochi dubbi, non lo hanno verificato. Per risolvere questo “mistero d’omissione”, o ,per lo meno, per farsi un idea plausibile sulle reali cause di ciò, basta conoscere i finanziatori dello studio (GlaxoSmithKline). Un altro dato mancante è rappresentato dall’impatto della molecola a livello epatico.
I ricercatori concludono affermando che è il trattamento con GSK20881078 può portare ad aumenti potenzialmente significativi a livello clinico della massa magra con una risposta differenziale tra i sessi. I cambiamenti nella chimica clinica sono stati coerenti con quelli precedentemente segnalati per altri SARM ed erano relativamente miti, monitorabili e reversibili. Ulteriori ricerche sono ora in programma per analizzare gli effetti di aumento della massa magra osservati.
Sebbene le risposte nell’aumento della massa magra possano sembrare promettenti ai dosaggi indicati, specie negli individui di sesso femminile, l’entità dei possibili effetti collaterali rende questo composto decisamente meno interessante per gli atleti; specie se paragonato con altri PED attualmente in uso ed il loro rapporto tra possibili benefici ed effetti collaterali. Un calo del HDL del 35-40% con una dose di 4mg/die per 4 settimane non lascia spazio a rosee previsioni sull’effetto riscontrabile con l’uso di dosi più elevate per lo stesso lasso di tempo o per periodi più lunghi.
Una nota interessante, che va oltre lo studio che qui è stato trattato, è data dal fatto che l’antidoping ha già sviluppato un test per la rilevazione del GSK20881078.(3)
A seguito di uno studio in vitro svolto da tossicologi dell’Università di Basilea e pubblicato nel 2012, è emerso che il Fluoxymesterone può causare un aumento significativo dei livelli di Cortisolo. (1) Argomento da me accennato nell’articolo dedicato alla molecola in questione.
Come risaputo, il Fluoxymesterone è un AAS orale metilato in C-17, con un potere androgeno elevato e non soggetto all’enzima aromatasi.
Nonostante quest’ultimo punto, la casa produttrice (Pfizer) riporta nelle avvertenze del prodotto una caratteristica che non ci si aspetta da una molecola priva di attività estrogenica diretta e indiretta: “L’edema, con o senza insufficienza cardiaca congestizia, può essere una seria complicanza in pazienti con preesistente malattia cardiaca, renale o epatica”.(2)
I ricercatori hanno scoperto il meccanismo attraverso il quale il Fluoxymesterone può causare edema e quindi aggravare ulteriormente la sua influenza sulla salute del sistema cardiovascolare. Il Fluoxymesterone si lega all’enzima 11-beta-HSD2, enzima preposto alla conversione del Cortisolo in Cortisone (inattivo). Di conseguenza il gruppo 11-idrossile del Fluoxymesterone viene convertito in un gruppo 11-oxo. Poiché l’attività dell’11-beta-HSD2 subisce una riduzione, si osserva un aumento della concentrazione di Cortisolo.
I ricercatori hanno esteso la loro ricerca ad altri composti al fine di valutarne una possibile azione sui meccanismi di conversione del Cortisolo in Cortisone. L’esito è stata la scoperta della marcata attività inibitoria dell’11-beta-HSD2 da parte del Fluoxymesterone. Durante l’esperimento è stato constatato che il Fluoxymesterone esplica una potenza maggiore sull’alterazione dei livelli di Cortisolo dell’Acido Glicirretico, sostanza presente nella liquirizia che causa un aumentano della produzione endogena di corticosteroidi, attraverso l’inibizione degli enzimi 4 e 5-beta-reduttasi che inattivano gli steroidi.(3)
L’Oxymesterone – o 4-idrossi-17-metil-testosterone – e, in misura minore, l’Oxymetholone inibiscono l’11-beta-HSD2 quasi quanto il Fluoxymesterone.
I ricercatori hanno scoperto anche che il Fluoxymesterone non può interagire direttamente con i Recettori del Cortisolo. Ma, le concentrazioni aumentate di Cortisolo portano, ovviamente, ad un consequenziale aumento dell’attività di quest’ultimo.
Un eccesso di Cortisolo altera l’equilibrio elettrolitico data l’interazione dello steroide con i Recettori Mineralocorticoidi. Induce il corpo a trattenere più sodio e quindi aumenta la ritenzione idrica extracellulare. Ciò significa che la quantità di plasma nel sangue aumenta e di conseguenza aumenta la pressione sanguigna. Oltre a ciò, un livello elevato di Cortisolo ha un effetto restringente sui vasi sanguigni, cosa che, a sua volta, causa un aumento della pressione sanguigna. Infine, un livello elevato di Cortisolo rende i vasi sanguigni più suscettibili ai danni causati dall’accumulo di colesterolo nelle loro pareti.
Pertanto, gli AAS con azione inibitoria nei confronti dell’11-beta-HSD2, come il Fluoxymesterone, possono avere un azione avversa maggiore nel causare effetti cardiovascolari avversi.
Si è ipotizzata anche una differenza nell’impatto sull’11-beta-HSD2 e i livelli di Cortisolo tra assunzione orale e somministrazione per iniezione con maggiore influenza data da quest’ultima. La cosa potrebbe con molta probabilità essere legata alle modifiche che la farmacocinetica subisce con la somministrazione tramite iniezione rispetto alla classica somministrazione orale.
Per avere una visione d’insieme più completa riguardo al Fluoxymesterone vi rimando all’artico ad esso dedicato.
La supplementazione con estratto di semi d’uva sembra poter fornire una protezione a cuore e vasi sanguigni durante l’uso di AAS. Questa possibile azione è emersa da uno studio svolto su animali che i ricercatori dell’Università di Tanta (Egitto) hanno pubblicato sul Oxidative Medicine and Cellular Longevity.(1)
I ricercatori hanno svolto l’esperimento dividendo ratti da laboratorio maschi in 4 gruppi. Durante le otto settimane di durata dell’esperimento, i ricercatori non hanno dato alcuna sostanza attiva al primo gruppo di ratti [Controllo].
Procianidina C1 (membro della famiglia delle Proantocianidine presenti nei semi d’uva)
Il secondo gruppo di ratti ha ricevuto una dose consistente di un estratto di semi d’uva purificato due volte alla settimana tramite un sondino gastrico. Questo estratto, prodotto dalla Merck, conteneva Proantocianidine [GSPE]. Se i ratti fossero stati esseri umani, avrebbero ricevuto una dose media di 700mg di Estratto di semi d’uva due volte a settimana.
Il terzo gruppo di animali esaminati è stato trattato con un iniezione settimanale di Boldenone. Infine, Il quarto gruppo è stato trattato con un iniezione settimanale di Boldenone insieme alla supplementazione con estratto di semi d’uva.
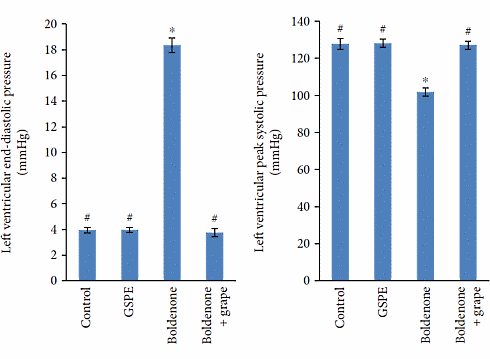
I ratti trattati con Boldenone hanno sviluppato ipertrofia cardiaca. L’aggiunta dell’estratto di semi d’uva al trattamento con Boldenone ha annullato tale effetto.
La figura riportata di seguito mostra come il Boldenone ha indotto l’ipertrofia cardiaca. Se il ventricolo sinistro del cuore – la parte del muscolo cardiaco che pompa sangue ricco di ossigeno nel corpo – si era contratto, il sangue era ancora sotto un elevata pressione nei vasi sanguigni degli animali esaminati a cui era stato somministrato il Boldenone [Sinistra].
Questo indicava che il cuore era sottoposto ad un lavoro maggiore legato alla condizione ipertrofica. Ciò indicava anche che la salute dei vasi sanguigni non era ottimale.
Ancora una volta, l’estratto di semi d’uva ha annullato questo effetto.
Le figure sottostanti mostrano cosa è successo esattamente ai vasi sanguigni degli animali trattati. Il Boldenone aveva attivato gli enzimi NOX. Gli enzimi NOX producono radicali liberi. Le cellule immunitarie, ma anche le cellule che formano i vasi sanguigni, producono enzimi NOX. Questi enzimi sono utili quando il corpo sta combattendo degli agenti patogeni, ma se questi enzimi si attivano senza una buona ragione, possono causare rigidità dei vasi sanguigni e, di conseguenza, danneggiarli.
Il NOX2 è prodotto dalle cellule endoteliali nelle pareti dei vasi sanguigni, il NOX4 nel muscolo cardiaco.
I ricercatori concludono affermando che, queste nuove scoperte sull’attività antiossidante delle Proantocianidine contenute nell’estratto di semi d’uva dovrebbero servire come base per lo sviluppo di migliori strategie chemiopreventive o terapeutiche per la tossicità cardiaca indotta dal Boldenone.
E’ necessario però fare alcune precisazioni…
La dose di Boldenone usata dai ricercatori era tutto sommato contenuta: 5mg/Kg a settimana. Se questo non è un errore di battitura, l’equivalente umano della dose utilizzata dai ricercatori è di circa 70mg a settimana. Gli utilizzatori di AAS assumono dosi settimanali minime di 200-250mg dello steroide preso in esame, e alcuni arrivano anche al grammo. Ovviamente, insieme al Boldenone vengono generalmente somministrati altri AAS. Quest’ultimo punto riduce ulteriormente la validità protettiva del supplemento esaminato nel presente studio in un contesto di utilizzo di AAS a scopo dopante.
In conclusione, è assai improbabile che una supplementazione con estratto di semi d’uva possa fornire una protezione verso l’ipertrofia cardiaca o il danno endoteliale negli utilizzatori di AAS.
Con il termine Antiestrogeni ci si riferisce genericamente ad una classe di farmaci aventi azione diretta o indiretta sull’attività tissutale e/o concentrazione ematica degli estrogeni. Agiscono bloccando il recettore dell’estrogeno (ER) e/o riducendo o sopprimendo la sintesi estrogenica.(1)(2) Una recente categoria di agenti facenti parte di questa classe di farmaci, i SERD (Selective Estrogen Receptor Degrader), esplicano la loro azione antiestrogena degradando/sottoregolando il recettore dell’estrogeno. Gli Antiestrogeni sono una delle tre classi di farmaci antagonisti dell’ormone sessuale, insieme agli Antiandrogeni e agli Antiprogestinici.(3)
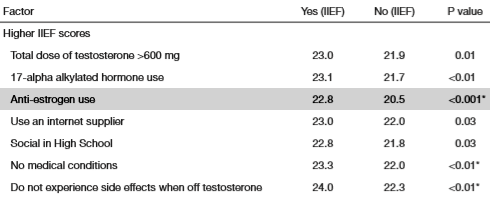
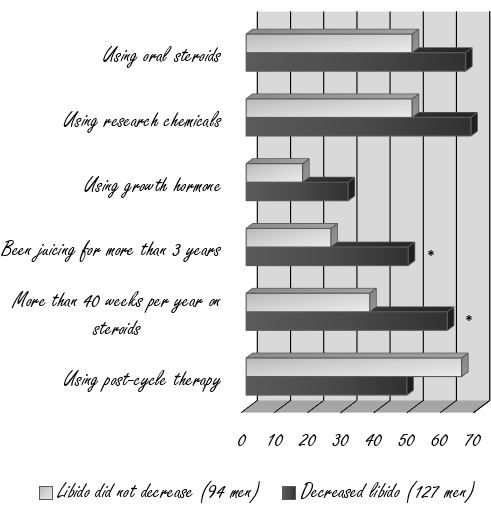
Largamente utilizzati in ambito sportivo, in special modo nell’ambiente culturistico, con il fine di controllare l’attività estrogenica durante l’uso di AAS aromatizzabili, o aventi attività estrogenica intrinseca, e durante la PCT con lo scopo aggiunto di stimolare la ripresa dell’HPTA, questi farmaci hanno un discreto carico di effetti collaterali tra i quali, quelli che destano maggior preoccupazione nell’atleta previdente, vi sono la dislipidemia (aumento dell’LDL, dei Trigliceridi, riduzione del HDL e alterazione delle loro ratio), l’atralgia (dolore articolare), il calo della libido/disfunzione erettile e l’affaticamento/letargia. Non sono di certo da meno le preoccupazioni legate all’alterazione dell’Asse GH/IGF-1 o la riduzione delle potenzialità di induzione ipertrofica di un ciclo in seguito ad una eccessiva soppressione dell’attività e/o delle concentrazioni estrogeniche. Ma esiste un’altra preoccupazione legata all’uso di composti antiestrogeni, ed è la possibilità che si verifichi un rebound estrogenico in seguito al l’oro uso. Purtroppo, la letteratura a disposizione è al quanto scarsa e poco chiara nella specifica del problema. E’ possibile, però, fare maggiore chiarezza sulla questione analizzando le caratteristiche dei composti antiestrogeni e il loro impatto, passando in rassegna tutti i componenti dell’addizione (Recettori Estrogeni e enzima Aromatasi). In questo articolo cercherò di esporre un ragionamento logico grazie al quale, seppur non avendo una risposta definitiva, sarà possibile avere un idea, la più concreta possibile, sul binomio antiestrogeni/rebound estrogenico.
Una analisi della questione…
Recettori dell’Estrogeno, SERM e “rebound estrogenico”
Un dimero della regione legame-ligando del ERa.
I Recettori degli Estrogeni (ER) sono un gruppo di proteine presenti all’interno delle cellule. Sono recettori attivati dall’ormone estrogeno (con maggiore attività del 17β-estradiolo).(4) Esistono due classi di ER: i Recettori degli Estrogeni nucleari (ERα e ERβ), che sono membri della famiglia dei recettori nucleari e dei recettori intracellulari, ed i Recettori degli Estrogeni di Membrana (MR) (GPER (GPR30), ER-X e Gq-mER), che sono per lo più recettori accoppiati alla proteina G. In questa sede ci si riferirà ai primi (ER).
Una volta attivato dall’estrogeno, l’ER è in grado di traslocare nel nucleo e legarsi al DNA per regolare l’attività di diversi geni (ciò significa che è un fattore di trascrizione del DNA). Tuttavia, ha anche funzioni aggiuntive indipendenti dal legame con il DNA.(5)
Poiché l’estrogeno è un ormone steroideo, può passare attraverso le membrane fosfolipidiche della cellula, e pertanto i recettori non hanno bisogno di essere legati alla membrana per potersi legare a loro volta con l’estrogeno.
L’estrogeno esplica la sua attività cellulare attraverso un azione Genomica e Non-Genomica.
• Genomica
In assenza di ormoni, i ER si trovano in gran parte nel citosol. Il legame dell’ormone al recettore innesca un numero di eventi che iniziano con la migrazione del recettore dal citosol nel nucleo, la dimerizzazione del recettore e il successivo legame del dimero del recettore a specifiche sequenze di DNA conosciute come elementi di risposta ormonale. Il complesso DNA / recettore quindi recluta altre proteine che sono responsabili della trascrizione del DNA a valle in mRNA e, infine, in una proteina la quale porta a dei cambiamenti nella funzione cellulare. I recettori degli estrogeni si trovano anche all’interno del nucleo della cellula, ed entrambi i sottotipi del recettore dell’estrogeno hanno un dominio di legame con il DNA e possono funzionare come fattori di trascrizione per regolare la produzione di proteine.
Il recettore interagisce anche con la proteina attivatore 1 e Sp-1 per promuovere la trascrizione, attraverso diversi coattivatori come il PELP-1.(6)
L’acetilazione diretta del recettore alfa dell’estrogeno ai residui della lisina nella regione cerniera mediante il p300 regola la transattivazione e la sensibilità ormonale.(7)
• Non-Genomica
Alcuni recettori per gli estrogeni sono presenti nelle membrana della superficie della cellula e possono essere rapidamente attivati dall’esposizione di questa agli estrogeni.(8)(9)
Inoltre, alcuni ER possono associarsi alle membrane cellulari legandole alla caveolina-1 e formarmando complessi con la proteine G, striatina, tirosina chinasi del recettore (es. EGFR e IGF-1) e tirosina chinasi non recettoriale (es. Src). (6)(8) Attraverso la striatina, alcuni di questi ER legati alla membrana possono portare a livelli aumentati di Ca2 + e ossido nitrico (NO).(10) Attraverso il recettore tirosin chinasi, i segnali vengono inviati al nucleo attraverso la via della proteina chinasi attivata dal mitogeno (MAPK / ERK) e la via del fosfoinositide 3-chinasi (Pl3K / AKT).(11) La glicogeno sintasi chinasi-3 (GSK) -3β inibisce la trascrizione dal ER nucleare inibendo la fosforilazione della serina 118 dell’ERa nucleare. La fosforilazione di GSK-3β rimuove il suo effetto inibitorio, e questo può essere ottenuto tramite il pathway PI3K / AKT e il pathway MAPK / ERK, tramite rsk.
Il 17β-estradiolo ha dimostrato di attivare il recettore GPR30 accoppiato alla proteina G.(12) Tuttavia, la localizzazione subcellulare e il ruolo di questo recettore sono ancora oggetto di controversie.(13)
ERb.
Gli estrogeni e gli ER sono implicati nel cancro al seno, nel carcinoma ovarico, nel cancro del colon, nel cancro alla prostata e nel cancro dell’endometrio. Il carcinoma del colon avanzato è associato a una perdita di ERβ, l’ER predominante nel tessuto del colon, e il tumore del colon è trattato con agonisti specifici per ERβ.(14)
Sappiamo che i recettori degli estrogeni sono sovraespressi in circa il 70% dei casi di cancro al seno, indicati come “ER-positivi”, e possono essere dimostrati in tali tessuti mediante l’immunoistochimica.(15) E’ ipotizzabile ,quindi, che gli atleti più sensibili agli effetti estrogenici presentino un espressione dei ER più elevata del normale, cosa che li porta a sviluppare con maggiore facilità effetti avversi dati da un eccesso dei livelli estrogenici e/o da un aumento della loro attività dato dalla cosomministrazione con progestinici (es. Nandrolone e Trenbolone).
I Modulatori Selettivi del Recettore dell’Estrogeno (SERM) sono composti antiestrogenici che agiscono a livello del ER. (16) Una caratteristica che distingue queste sostanze dagli agonisti e antagonisti ER puri (cioè agonisti completi e antagonisti silenti) è che la loro azione è diversa nei vari tessuti, garantendo in tal modo la possibilità di inibire selettivamente o stimolare l’azione estrogenica in diversi tessuti.
I SERM sono agonisti parziali competitivi del ER.(17) Tessuti diversi presentano differenti gradi di sensibilità all’attività degli estrogeni, pertanto i SERM esplicano effetti estrogenici o antiestrogeni a seconda del tessuto specifico con il quale interagiscono e della percentuale di attività intrinseca (IA) del composto in questione.(18) Un esempio di SERM con alta IA, e quindi di effetti prevalentemente estrogenici, è rappresentato dal Clorotrianisene, mentre un esempio di SERM con bassa IA, e quindi avente per lo più attività antiestrogenica, è rappresentato dall’Ethamoxytrifetolo. SERM come il Clomifene e il Tamoxifene, largamente utilizzati in ambito sportivo, sono considerabili come composti con valore IA intermedio essendo molecole con una azione bilanciata tra effetti estrogenici e antiestrogenici. Il Raloxifene è un SERM che presenta una azione antiestrogenica maggiore del Tamoxifene; entrambi hanno una attività estrogenica (sebbene differente) a livello osseo, ma il Raloxifene presenta una attività antiestrogenica nell’utero mentre il Tamoxifene ha un azione estrogenica nel tessuto dell’utero.(18)
Tamoxifene
Il Tamoxifene è un farmaco di prima linea per il trattamento del carcinoma mammario metastatico ER-positivo. È usato per la riduzione delle possibilità di sviluppo del cancro al seno nelle donne ad alto rischio, come trattamento adiuvante del nodo ascellare negativo e positivo, e nel carcinoma duttale in situ.(19)(20)
Il Tamoxifene è classificabile come un profarmaco, dal momento che la sua affinità per la proteina bersaglio (ER) è limitata. Il Tamoxifene viene metabolizzato nel fegato dall’isoforma del citocromo CYP2D6 e CYP3A4 in metaboliti attivi come l’Afimoxifene (4-idrossitamoxifene; 4-OHT) e l’Endoxifene (N-desmetil-4-idrossitamoxifene) (21) che presentano una affinità da 30 a 100 volte maggiore per il ER rispetto al Tamoxifene. (22) Questi metaboliti attivi competono con gli estrogeni per il legame con il recettore. Nel tessuto mammario, il 4-OHT agisce come un antagonista del ER in modo da inibire la trascrizione dei geni che reagiscono agli estrogeni. (23) Il Tamoxifene ha rispettivamente il 7% e il 6% dell’affinità dell’Estradiolo per il ERα e il ERβ, mentre il 4-OHT ha il 178% e il 338% dell’affinità dell’Estradiolo per il ERα e il ERβ.(24)
Afimoxifene (4-OHT)
Il 4-OHT si lega al ER, il complesso ER/Tamoxifene recluta altre proteine note come co-repressori e quindi si lega al DNA per modulare l’espressione genica. Alcune di queste proteine includono la NCoR e la SMRT. (25) La funzione del Tamoxifene può essere regolata da una serie di variabili diverse, compresi i fattori di crescita.(26) Il Tamoxifene deve bloccare le proteine del fattore di crescita come ErbB2/HER2 (27) perché è stato dimostrato che livelli elevati di ErbB2 si manifestano nei tumori resistenti al Tamoxifene.(28) Il Tamoxifene sembra richiedere una proteina PAX2 affinché possa esplicare il suo pieno effetto antitumorale. (27)(29) In presenza di un elevata espressione della PAX2, il complesso Tamoxifene/ER è in grado di sopprimere l’espressione della proteina pro-proliferativa del ERBB2. Al contrario, quando l’espressione del AIB-1 è superiore alla PAX2, il complesso di Tamoxifene/ER aumenta l’espressione del ERBB2 con conseguente stimolazione della crescita del cancro al seno. (27)(30)
Il 4-OHT si lega al ER in modo competitivo (rispetto all’estrogeno agonista) nelle cellule tumorali e in altri bersagli tissutali, producendo un complesso nucleare che riduce la sintesi del DNA e inibisce gli effetti degli estrogeni. È un agente non steroideo con potenti proprietà antiestrogeniche che competono con gli estrogeni per i siti di legame nel seno e in altri tessuti. Il Tamoxifene fa sì che le cellule rimangano nelle fasi G0 e G1 del ciclo cellulare. Poiché impedisce alle cellule (pre) cancerose di dividersi ma non provoca la morte cellulare, il Tamoxifene è citostatico piuttosto che citocida.
La letteratura scientifica riguardante l’attività del Tamoxifene è a dir poco complessa ed occorre prestare particolare attenzione ai dati disponibili per stabilire se il Tamoxifene, o il suo metabolita 4-idrossi, abbiano il maggiore impatto complessivo.
Norendoxifene
Il Norendoxifene (N, N-didesmetil-4-idrossitamoxifene), un altro metabolita attivo del Tamoxifene, è stato osservato agire come un potente inibitore dell’aromatasi competitivo (IC50 = 90 nM), cosa che a sua volta può amplificare l’attività antiestrogenica complessiva del Tamoxifene.(31)
Come già accennato in precedenza, e come molti sapranno, il Tamoxifene è largamente utilizzato in ambito sportivo, sia da solo che in abbinamento con altri SERM come il Clomifene ( in PCT) o con AI (“on-cycle” e/o in PCT). La sua applicazione all’interno di una preparazione che contempla l’uso di AAS aromatizzabili, alla luce di quanto esposto pocanzi, lo vede come agente preventivo o di trattamento dell’attività estrogenica a livello tissutale, in specie per quanto concerne l’attività estrogenica nel tessuto mammario al fine di evitare (o “tamponare”) la comparsa della ginecomastia. In un contesto PCT tale composto, oltre ad esercitare la funzione di regolazione dell’attività estrogenica appena esposta, agendo a livello ipotalamico stimola il rilascio di GnRH e, consequenzialmente, di LH ed FSH dall’Ipofisi che a loro volta stimoleranno la sintesi di Testosterone e la spermatogenesi.
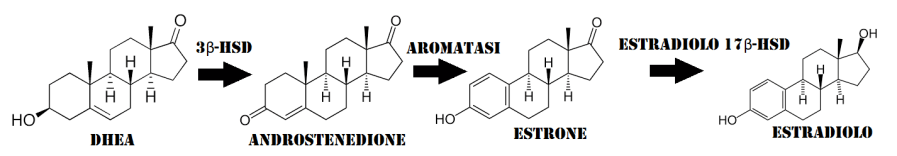
Il suo utilizzo massivo e cronico è stato spesso collegato aneddoticamente a rebound estrogenico. Ora, conoscendo la complessità d’azione che questo composto (ed i suoi metaboliti) ha sul controllo dell’attività estrogenica, si può facilmente ipotizzare che un suo uso protratto (legato anche alla dose e, quindi, al suo impatto sulla attività estrogenica sistemica) possa innescare degli adattamenti reattivi con conseguente aumento dell’attività estrogenica attraverso l’incremento dei livelli serici di Estradiolo e dell’attività non-genomica dello steroide (ipotizzabile anche un aumento del numero dei ER). Nel corso degli anni sono state esposte diverse ipotesi volte a spiegare i meccanismi attraverso i quali un abuso di Tamoxifene possa portare ad un rebound estrogenico. Una di queste ipotesi venne riportata all’inizio del secolo dal compianto A.L. Rea il quale affermava che la causa andasse ricercata nell’aumento del rilascio di DHEA da parte delle ghiandole surrenali e dalla sua successiva (e aumentata) conversione in Androstenedione e, attraverso l’intervento dell’enzima aromatasi che lo converte in Estrone e la successiva azione del estradiolo 17beta-deidrogenasi, Estradiolo.(32) In breve, secondo questa teoria i processi innescati causerebbero l’instaurarsi di livelli di E2 cronicamente alti con conseguente impossibilità del SERM di esplicare la sua azione. Questa teoria seppur, in parte, possa dare una spiegazione logica dei possibili meccanismi implicati manca di alcuni tasselli. Il principale “tallone d’Achille” è rappresentato dai livelli di E2 che, una volta aumentati, diventano dei competitor recettoriali più aggressivi rispetto al 4-OHT (che ricordiamo avere il 178% dell’affinità dell’Estradiolo per il ERα). Ciò potrebbe avvenire in situazioni di calo delle concentrazioni di 4-OHT seguenti alla riduzione del dosaggio del farmaco o alla sua cessazione, quindi, in questo ultimo caso, esplicabili in crescendo nei 7-14 giorni successivi all’interruzione della somministrazione e con una durata indeterminata. Di conseguenza, sembra più plausibile che l’aumento delle concentrazioni di E2, durante l’uso del Tamoxifene, si affianchi ad un consequenziale incremento dell’attività Non-Genomica dell’ormone e da un aumentato numero di ER. Seguendo questa logica, una volta interrotto l’uso del Tamoxifene, queste condizioni tenderanno ad aggravarsi come gli effetti avversi a loro legati.
Consultando la bibliografia scientifica disponibile, non si trovano accenni su un possibile rebound estrogenico in seguito all’uso di Tamoxifene, ma si parla nello specifico di “resistenza al Tamoxifene” o “sottoregolazione degli ER”.(33)(34) Nel caso della “resistenza al Tamoxifene” sembra che l’aumento dell’espressione del gene MACROD2 porti ad una risposta negativa all’azione del SERM con conseguente proliferazione delle cellule cancerose estradiolo-dipendenti. La sovra espressione di tale gene sembra essere di base genetica anche se non si esclude una risposta di adattamento in seguito ad uso cronico del composto in questione.
Raloxifene
Il Raloxifene, un altro SERM discretamente utilizzato nella pratica sportiva, è un agonista-antagonista misto del ER.(35)(36)(37) Ha effetti estrogenici a livello osseo ed epatico con effetti antiestrogenici nei seni e nell’utero. Le azioni biologiche del Raloxifene sono quindi ampiamente mediate dal legame con i ER. Questo legame determina l’attivazione di percorsi estrogenici in alcuni tessuti (agonismo) e il blocco di questi in altri (antagonismo). Le sue caratteristiche d’azione similari a quelle del Tamoxifene, sembrano poter far pensare ad un medesimo e ipotetico meccanismo che possa portare ad un rebound estrogenico. Questa volta la letteratura scientifica sembra dare alcune conferme. In un caso studio (38), una paziente di 66 anni si è presentata con recidiva metastatica acuta estrogeno-positiva e progesterone-positiva, carcinoma mammario Her-2 / neu-negativo, lesioni ossee (colonna lombare, bacino), noduli polmonari, metastasi epatiche, antigene tumorale elevato 15 e enzimi epatici, dispepsia e diarrea. La paziente aveva assunto Raloxifene per circa 8 anni. Dopo la sospensione del farmaco, parametri e sintomi clinici sono migliorati rapidamente senza terapia oncologica o altre forme di trattamento. Tre mesi dopo la sospensione del Raloxifene, l’oncologo ha prescritto alla paziente l’uso della Capecitabina dato che non riteneva plausibile un effetto di rebound estrogenico (anti-estrogen withdrawal effect – AEWE). Tuttavia, la regressione duratura è stata più indicativa di un effetto rebound dato dal Raloxifene rispetto alla chemioterapia o ad altri interventi. In seguito la paziente si è mostrata asintomatica con un buono stato di prestazione. La regressione metastatica epatica è stata confermata, senza alcun trattamento oncologico somministrato negli ultimi 16 mesi e circa 23 mesi dopo il termine d’uso del Raloxifene. Questo caso evidenzia la necessità di esaminare pazienti con carcinoma mammario per la possibilità di un AEWE con l’uso di Raloxifene o con altri SERM . Ovviamente, il caso presentato non è molto comparabile, soprattutto per quanto riguarda i tempi di somministrazione, ad un BodyBuilder supplementato chimicamente nella “media” ma, ciò nonostante, ci offre un indizio sulla probabilità che si possa manifestare un rebound estrogenico con l’uso di SERM.
Enzima Aromatasi, Inibitori della Aromatasi e “rebound estrogenico”
Enzima Aromatasi
L’Enzima Aromatasi, chiamato anche estrogeno sintetasi o estrogeno sintasi, è un enzima responsabile del processo fondamentale della biosintesi degli Estrogeni. Denominato CYP19A1, questo enzima è un membro della superfamiglia del citocromo P450 (EC 1.14.14.1), che sono monoossigenasi che catalizzano molte reazioni coinvolte nella steroidogenesi. In particolare, l’Aromatasi è responsabile dell’aromatizzazione degli Androgeni in Estrogeni. L’enzima Aromatasi è sintetizzato in molti tessuti tra cui le gonadi (cellule della granulosa), cervello, tessuto adiposo, placenta, vasi sanguigni, pelle e ossa, nonché nei tessuti dell’endometriosi, dei fibromi uterini, del cancro al seno e del cancro dell’endometrio.
L’Aromatasi è localizzato nel reticolo endoplasmatico dove è regolato da promotori tissutali che sono a loro volta controllati da ormoni, citochine e altri fattori. Catalizza gli ultimi passaggi della biosintesi degli estrogeni dagli androgeni (in particolare, converte l’Androstenedione in Estrone e il Testosterone in Estradiolo). Queste fasi comprendono tre idrossilazioni successive del gruppo 19-metilico degli androgeni, seguite dall’eliminazione simultanea del gruppo metilico come formiato e aromatizzazione dell’anello A.
Reazioni generali per la conversione del Testosterone in Estradiolo catalizzata dall’Aromatasi. Gli Steroidi sono formati da quattro anelli fusi (A-B-C-D). L’Enzima Aromatasi converte l’anello “A” in uno stato aromatico.
Il gene esprime due varianti di trascrizione. (39) Nell’uomo, il gene CYP19, situato sul cromosoma 15q21.1, codifica per l’Enzima Aromatasi. (40) Il gene ha nove esoni codificanti e un numero di primi esoni non codificanti alternativi che regolano l’espressione specifica del tessuto. (41)
Il CYP19 è presente in un cordato precoce divergente, l’anfiosso cefalocordato (il Florida lancelet, Branchiostoma floridae), ma non nel precedente tunicato divergente Ciona intestinalis. Pertanto, gli evoluzionisti ipotizzano che il gene Aromatasi si sia evoluto precocemente nell’evoluzione dei cordati e non sembra essere presente negli invertebrati non-cordati (ad esempio insetti, molluschi, echinodermi, spugne, coralli). Tuttavia, gli Estrogeni possono essere sintetizzati in alcuni di questi organismi, attraverso altri percorsi sconosciuti.
I fattori noti che aumentano l’attività dell’Aromatasi includono l’età, l’obesità, l’Insulina, le gonadotropine e l’alcol. L’attività dell’Aromatasi risulta diminuita dalla Prolattina, dall’ormone anti-Mülleriano e dal glifosato , un comune erbicida.(42) L’attività dell’Aromatasi sembra essere migliorata in alcuni tessuti estrogeno-dipendenti come il tessuto mammario, nel carcinoma dell’endometrio, nell’endometriosi e nei fibromi uterini.
Gli Inibitori dell’Aromatasi (AI) sono una gruppo di farmaci usati nel trattamento del carcinoma mammario nelle donne in postmenopausa e nella ginecomastia negli uomini. Come i SERM, trovano un largo uso off-label in ambito sportivo durante la somministrazione di AAS aromatizzabili o durante la PCT. Possono anche essere utilizzati per la chemioprevenzione in donne ad alto rischio.
Esistono due tipi di Inibitori dell’Aromatasi approvati per il trattamento del carcinoma mammario e, quindi, diffusi anche per l’uso off-label: (43)
– Gli inibitori steroidei irreversibili, come l’Exemestano (nome commerciale Aromasin), formano un legame permanente e disattivante con l’Enzima Aromatasi.
– Gli inibitori non steroidei, come l’Anastrozolo (nome commerciale Arimidex) e il Letrozolo (nome commerciale Femara), inibiscono la sintesi degli Estrogeni attraverso la competizione reversibile per l’Enzima Aromatasi.
Gli inibitori dell’Aromatasi disponibili (AI) includono:
– Non selettivi:
• L’Aminoglutetimide, il quale però inibisce l’enzima P450scc agendo come inibitore della biosintesi di tutti gli ormoni steroidei (aprirò una nota a riguardo più avanti).
• Testolactone (nome commerciale Teslac) – Selettivi:
• Anastrozolo (Arimidex)
• Letrozolo (Femara)
• Exemestano (Aromasin)
• Vorozolo (Rivizor)
• Formestano (Lentaron)
• Fadrozolo (Afema)
– Non classificati:
• 1,4,6-Androstatrien-3,17-dione (ATD)
• 4-Androstene-3,6,17-trione (“6-OXO”)
Oltre agli AI farmaceutici, alcuni composti naturali hanno mostrato effetti di inibizione dell’Aromatasi, come le foglie di damiana. Il loro impatto non è stato pienamente chiarito sull’uomo.
Gli Inibitori dell’Aromatasi agiscono, proprio come suggerisce il nome, inibendo l’azione dell’enzima Aromatasi, che converte gli Androgeni in Estrogeni mediante un processo chiamato aromatizzazione. Poiché il tessuto mammario è stimolato dagli Estrogeni, diminuirne la produzione è un modo per sopprimere la recidiva del tessuto tumorale del seno. La principale fonte di Estrogeni è rappresentata dalle ovaie nelle donne in premenopausa, mentre nelle donne in post-menopausa la maggior parte degli Estrogeni del corpo viene prodotta nei tessuti periferici (al di fuori del SNC) e anche in alcuni siti del SNC in varie regioni del cervello. L’Estrogeno viene prodotto e agisce localmente in questi tessuti, ma qualsiasi estrogeno circolante, che esercita effetti estrogenici sistemici in uomini e donne, è il risultato dell’Estrogeno che sfugge al metabolismo locale e si diffonde nel sistema circolatorio.(44)
Come già accennato pocanzi, i composti AI sono anch’essi, al pari dei SERM, largamente utilizzati in ambito sportivo, sia come agenti di controllo dei livelli estrogenici durante l’uso di AAS aromatizzabili (uso preventivo della comparsa di effetti estrogenici), in caso di ginecomastia (spesso in combinazione con un SERM, specie se l’AI utilizzato è l’Exemestano) o in combinazione con i SERM in ambito PCT (specie nella fase preliminare dove viene utilizzata l’hCG).
Più che con i SERM, il rebound estrogenico è stato riportato, soprattutto aneddoticamente, con l’uso di AI, specialmente quelli reversibili (vedi Anastrozolo e Letrozolo).
L’Anastrozolo ed il Letrozolo agiscono legandosi in modo reversibile all’Enzima Aromatasi (unità eme del citocromo P450) e, attraverso l’inibizione competitiva, blocca la conversione degli Androgeni in Estrogeni nei tessuti periferici (extragonali).
Il Letrozolo ha dimostrato, attraverso studi clinici, di poter abbassare rapidamente il livello degli estrogeni fino al 65%. Il motivo principale è probabilmente legato alla capacità che la molecola ha di abbassare drasticamente gli estrogeni attraverso un legame competitivo reversibile al gruppo eme della relativa unità del citocromo P450. L’Anastrozolo, il quale agisce similmente al Letrozolo, ha mostrato una riduzione del livello estrogenico in soggetti di sesso maschile del 50%.(45) Il problema di un possibile rebound estrogenico con questi composti nasce proprio dalla loro natura “reversibile”.
Rebound estrogenici sono stati riportati sia con l’uso di Letrozolo che con l’uso di Anastrozolo, sebbene il Letrozolo, avendo un azione inibitoria più marcata, sembra causare rebound di intensità maggiore dopo la sua interruzione. La causa del rebound estrogenico indotto da cessazione d’uso di Letrozolo o di Anastrozolo è proprio legata al comportamento che queste due molecole esplicano nei confronti dell’Enzima aromatasi. Il legame tra la molecola di Letrozolo o di Anastrozolo con l’Enzima Aromatasi è solo temporanea e non decreta la completa de-attivazione dell’enzima responsabile della conversione degli Androgeni in Estrogeni. Una volta interrotta l’assunzione del composto, i livelli di Aromatasi possono salire significativamente con la possibile comparsa di un rebound estrogenico. Una pratica per evitare che ciò si verifichi consiste nell’uso limitato dei due composti e in una loro graduale sospensione. Con questi farmaci, il rebound estrogenico può essere “multifattoriale” derivando non solo dalla cessazione del farmaco in questione ma anche da un incremento dell’espressione dell’Enzima Aromatasi come risposta adattativa all’uso (specie nel lungo termine). Ciò significa che, anche durate un utilizzo cronico, i livelli di E2 possono mostrare degli aumenti, aumenti che diverranno maggiormente significativi una volta cessato l’uso del farmaco. Cessata l’azione del composto non solo viene a mancare un controllo dell’aromatizzazione ma questa risulta anche incrementata rispetto ai tassi pre-utilizzo (l’aumento dell’espressione dell’aromatasi è un comportamento adattativo che si può manifestare anche durante cicli particolarmente lunghi). Prendendo in considerazione la vita attiva del Letrozolo e dell’Anastrozolo, il possibile rebound estrogenico potrebbe manifestarsi in crescendo dopo 64-120h circa dall’ultima assunzione.
Exemestano
L’Exemestano, invece, è un inibitore dell’Aromatasi steroideo irreversibile di tipo I, strutturalmente correlato al substrato naturale 4-androstenedione. Agisce come un falso substrato per l’Enzima Aromatasi e viene trasformato in un intermedio che si lega irreversibilmente al sito attivo dell’enzima causandone l’inattivazione, un effetto noto anche come “inibizione suicida”. Essendo strutturalmente simile agli obiettivi dell’enzima, l’Exemestano si lega in modo permanente a quest’ultimo, impedendo la sua azione di conversione degli Androgeni in Estrogeni. Il tasso di soppressione degli Estrogeni da parte dell’Exemestano varia dal 35% per l’Estradiolo (E2) al 70% per l’Estrone (E1).(46)
Grazie alla sua caratteristica di “inibitore selettivo”, l’Exemestano sembra non causare un rebound estrogenico dopo la sua cessazione. Nonostante ciò, un suo uso temporalmente protratto potrebbe (teoricamente) causare, similmente a quanto accade con l’uso di Letrozolo e Anastrozolo, un aumento dell’espressione dell’Enzima Aromatasi nonché un aumento del numero di ER come risposta adattativa.
Ovviamente, questa possibilità può interessare tutti gli AI con legame irreversibile (es. Formestano).
Queste sono semplici ipotesi nate da una riflessione sulle possibili cause e meccanismi che potrebbero (teoricamente) portare al manifestarsi di un rebound estrogenico con l’uso di tali composti. La letteratura scientifica, purtroppo, non ci aiuta a fare molta chiarezza sulla connessione AI/rebound estrogenico, sebbene esistono alcuni studi nei quali la cosa viene accennata.(47)
Aminoglutetimide
*Nota sull’Aminoglutetimide: inibendo l’enzima P450scc e agendo, di conseguenza, come inibitore della biosintesi di tutti gli ormoni steroidei, l’abuso di Aminoglutetimide può potenzialmente causare non solo un rebound estrogenico ma anche un rebound dei livelli di cortisolo. Lo stesso vale per il farmaco Trylostano.
Conclusioni
Basarsi per la maggior parte sui dati aneddotici è un azzardo, anche perché la maggior parte delle variabili soggettive in gioco rimangono celate. Banalmente, alcuni lamentano rebound estrogenici che alla fine non risultano legati all’uso del SERM o del AI ma alla loro (o del Preparatore) ignoranza, come quando cessano l’utilizzo di AAS, e di SERM e/o AI, senza preoccuparsi di svolgere un adeguata PCT convinti, magari, che un po’ di Mesterolone (Proviron) risolvi tutto. Infatti, la maggior parte dei casi di presunti rebound estrogenici SERM o AI dipendenti sono causati da una repentina cessazione d’uso di questi e di AAS, oppure da una PCT mal pianificata e/o che non ha dato i risultati sperati (vedi anche alterazione della Testosterone:Estradiolo ratio). Queste condizioni sono legate più che altro ad una alterazione del HPTA data dall’uso di AAS e non ad una presunta azione diretta del SERM e/o AI precedentemente utilizzati.
In conclusione, l’uso ponderato e consapevole è l’unica vera arma che l’atleta supplementato chimicamente (o il Preparatore che lo segue) ha per far si che ipotetici rebound non si manifestino.
Gabriel Bellizzi
Riferimenti:
1- “Definition of antiestrogen – NCI Dictionary of Cancer Terms, Definition of antiestrogen – NCI Dictionary of Cancer Terms”.,
2- Jump up ^ “antiestrogen” at Dorland’s Medical Dictionary
3- Jump up ^ Judi Lindsley Nath (2006). Using Medical Terminology: A Practical Approach. Lippincott Williams & Wilkins. pp. 977–. ISBN 978-0-7817-4868-1.
4- Dahlman-Wright K, Cavailles V, Fuqua SA, Jordan VC, Katzenellenbogen JA, Korach KS, Maggi A, Muramatsu M, Parker MG, Gustafsson JA (Dec 2006). “International Union of Pharmacology. LXIV. Estrogen receptors”. Pharmacological Reviews. 58 (4): 773–81. doi:10.1124/pr.58.4.8. PMID 17132854.
5- Levin ER (Aug 2005). “Integration of the extranuclear and nuclear actions of estrogen”. Molecular Endocrinology. 19 (8): 1951–9. doi:10.1210/me.2004-0390. PMC 1249516 . PMID 15705661.
6- Levin ER (Aug 2005). “Integration of the extranuclear and nuclear actions of estrogen”. Molecular Endocrinology. 19 (8): 1951–9. doi:10.1210/me.2004-0390. PMC 1249516 . PMID 15705661.
7- Wang C, Fu M, Angeletti RH, Siconolfi-Baez L, Reutens AT, Albanese C, Lisanti MP, Katzenellenbogen BS, Kato S, Hopp T, Fuqua SA, Lopez GN, Kushner PJ, Pestell RG (May 2001). “Direct acetylation of the estrogen receptor alpha hinge region by p300 regulates transactivation and hormone sensitivity”. The Journal of Biological Chemistry. 276 (21): 18375–83. doi:10.1074/jbc.M100800200. PMID 11279135.
8- Zivadinovic D, Gametchu B, Watson CS (2005). “Membrane estrogen receptor-alpha levels in MCF-7 breast cancer cells predict cAMP and proliferation responses”. Breast Cancer Research. 7 (1): R101–12. doi:10.1186/bcr958. PMC 1064104 . PMID 15642158.
9- Björnström L, Sjöberg M (Jun 2004). “Estrogen receptor-dependent activation of AP-1 via non-genomic signalling”. Nuclear Receptor. 2 (1): 3. doi:10.1186/1478-1336-2-3. PMC 434532 . PMID 15196329.
10- Lu Q, Pallas DC, Surks HK, Baur WE, Mendelsohn ME, Karas RH (Dec 2004). “Striatin assembles a membrane signaling complex necessary for rapid, nongenomic activation of endothelial NO synthase by estrogen receptor alpha”. Proceedings of the National Academy of Sciences of the United States of America. 101 (49): 17126–31. doi:10.1073/pnas.0407492101. PMC 534607 . PMID 15569929.
11- Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, Masushige S, Gotoh Y, Nishida E, Kawashima H, Metzger D, Chambon P (Dec 1995). “Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase”. Science. 270 (5241): 1491–4. doi:10.1126/science.270.5241.1491. PMID 7491495.
12- Prossnitz ER, Arterburn JB, Sklar LA (Feb 2007). “GPR30: A G protein-coupled receptor for estrogen”. Molecular and Cellular Endocrinology. 265-266: 138–42. doi:10.1016/j.mce.2006.12.010. PMC 1847610 . PMID 17222505.
13- Otto C, Rohde-Schulz B, Schwarz G, Fuchs I, Klewer M, Brittain D, Langer G, Bader B, Prelle K, Nubbemeyer R, Fritzemeier KH (Oct 2008). “G protein-coupled receptor 30 localizes to the endoplasmic reticulum and is not activated by estradiol”. Endocrinology. 149 (10): 4846–56. doi:10.1210/en.2008-0269. PMID 18566127.
14- Harris HA, Albert LM, Leathurby Y, Malamas MS, Mewshaw RE, Miller CP, Kharode YP, Marzolf J, Komm BS, Winneker RC, Frail DE, Henderson RA, Zhu Y, Keith JC (Oct 2003). “Evaluation of an estrogen receptor-beta agonist in animal models of human disease”. Endocrinology. 144 (10): 4241–9. doi:10.1210/en.2003-0550. PMID 14500559.
15- Deroo BJ, Korach KS (Mar 2006). “Estrogen receptors and human disease”. The Journal of Clinical Investigation. 116 (3): 561–70. doi:10.1172/JCI27987. PMC 2373424 . PMID 16511588.
16- Riggs BL, Hartmann LC (Feb 2003). “Selective estrogen-receptor modulators — mechanisms of action and application to clinical practice”. The New England Journal of Medicine. 348 (7): 618–29. doi:10.1056/NEJMra022219. PMID 12584371.
17- Cameron JL, Cameron AM (20 November 2013). Current Surgical Therapy. Elsevier Health Sciences. pp. 582–. ISBN 978-0-323-22511-3.
18- Huang X, Aslanian RG (19 April 2012). Case Studies in Modern Drug Discovery and Development. John Wiley & Sons. pp. 392–394. ISBN 978-1-118-21967-6.
19- Pickar JH, Komm BS (Sep 2015). “Selective estrogen receptor modulators and the combination therapy conjugated estrogens/bazedoxifene: A review of effects on the breast”. Post Reproductive Health. 21 (3): 112–21. doi:10.1177/2053369115599090. PMID 26289836.
20- Mirkin S, Pickar JH (Jan 2015). “Selective estrogen receptor modulators (SERMs): a review of clinical data”. Maturitas. 80 (1): 52–7. doi:10.1016/j.maturitas.2014.10.010. PMID 25466304.
21- Desta Z, Ward BA, Soukhova NV, Flockhart DA (Sep 2004). “Comprehensive evaluation of tamoxifen sequential biotransformation by the human cytochrome P450 system in vitro: prominent roles for CYP3A and CYP2D6”. The Journal of Pharmacology and Experimental Therapeutics. 310 (3): 1062–75. doi:10.1124/jpet.104.065607. PMID 15159443.
22- Ahmad A, Shahabuddin S, Sheikh S, Kale P, Krishnappa M, Rane RC, Ahmad I (December 2010). “Endoxifen, a new cornerstone of breast cancer therapy: demonstration of safety, tolerability, and systemic bioavailability in healthy human subjects”. Clinical Pharmacology and Therapeutics. 88 (6): 814–7. doi:10.1038/clpt.2010.196. PMID 20981001.
23- Wang DY, Fulthorpe R, Liss SN, Edwards EA (Feb 2004). “Identification of estrogen-responsive genes by complementary deoxyribonucleic acid microarray and characterization of a novel early estrogen-induced gene: EEIG1”. Molecular Endocrinology. 18 (2): 402–11. doi:10.1210/me.2003-0202. PMID 14605097.
24- Kuhl H (2005). “Pharmacology of estrogens and progestogens: influence of different routes of administration”. Climacteric. 8 Suppl 1: 3–63. doi:10.1080/13697130500148875. PMID 16112947.
25- Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M (Dec 2000). “Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription”. Cell. 103 (6): 843–52. doi:10.1016/S0092-8674(00)00188-4. PMID 11136970.
26- Massarweh S, Osborne CK, Creighton CJ, Qin L, Tsimelzon A, Huang S, Weiss H, Rimawi M, Schiff R (Feb 2008). “Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function”. Cancer Research. 68 (3): 826–33. doi:10.1158/0008-5472.CAN-07-2707. PMID 18245484.
27- Hurtado A, Holmes KA, Geistlinger TR, Hutcheson IR, Nicholson RI, Brown M, Jiang J, Howat WJ, Ali S, Carroll JS (Dec 2008). “Regulation of ERBB2 by oestrogen receptor-PAX2 determines response to tamoxifen”. Nature. 456 (7222): 663–6. Bibcode:2008Natur.456..663H. doi:10.1038/nature07483. PMC 2920208 . PMID 19005469.
28- Osborne CK, Bardou V, Hopp TA, Chamness GC, Hilsenbeck SG, Fuqua SA, Wong J, Allred DC, Clark GM, Schiff R (Mar 2003). “Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer”. Journal of the National Cancer Institute. 95 (5): 353–61. doi:10.1093/jnci/95.5.353. PMID 12618500.
29- “New Mechanism Predicts Tamoxifen Response: PAX2 gene implicated in tamoxifen-induced inhibition of ERBB2/HER2-mediated tumor growth”. http://www.modernmedicine.com. 2008-11-13. Archived from the original on 2011-07-14. Retrieved 2008-11-14.
30- “Study sheds new light on tamoxifen resistance”. News. CORDIS News. Archived from the original on 2009-02-20. Retrieved 2008-11-14.
31- Liu J, Flockhart PJ, Lu D, Lv W, Lu WJ, Han X, Cushman M, Flockhart DA (2013). “Inhibition of cytochrome p450 enzymes by the e- and z-isomers of norendoxifen”. Drug Metab. Dispos. 41 (9): 1715–20. doi:10.1124/dmd.113.052506. PMC 3876808 . PMID 23824607.
32- Chemical muscle enhancement. Report. B.B. desk reference. di Author L. Rea. Pag. 106.
33- https://www.rdmag.com/article/2014/12/journal-watch-tamoxifen-news-again
34- https://www.ncbi.nlm.nih.gov/pubmed/14687597
35- Bryant HU (2001). “Mechanism of action and preclinical profile of raloxifene, a selective estrogen receptor modulation”. Rev Endocr Metab Disord. 2 (1): 129–38. PMID 11704975.
36- Thiebaud D, Secrest RJ (2001). “Selective estrogen receptor modulators: mechanism of action and clinical experience. Focus on raloxifene”. Reprod. Fertil. Dev. 13 (4): 331–6. PMID 11800172.
37- Gizzo S, Saccardi C, Patrelli TS, Berretta R, Capobianco G, Di Gangi S, Vacilotto A, Bertocco A, Noventa M, Ancona E, D’Antona D, Nardelli GB (2013). “Update on raloxifene: mechanism of action, clinical efficacy, adverse effects, and contraindications”. Obstet Gynecol Surv. 68 (6): 467–81. doi:10.1097/OGX.0b013e31828baef9. PMID 23942473.
38- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5739193/
39- “Entrez Gene: CYP19A1 cytochrome P450, family 19, subfamily A, polypeptide 1”.
40- Toda K, Shizuta Y (April 1993). “Molecular cloning of a cDNA showing alternative splicing of the 5′-untranslated sequence of mRNA for human aromatase P-450”. European Journal of Biochemistry. 213 (1): 383–9. doi:10.1111/j.1432-1033.1993.tb17772.x. PMID 8477708.
41- Czajka-Oraniec I, Simpson ER (2010). “Aromatase research and its clinical significance”. Endokrynologia Polska. 61 (1): 126–34. PMID 20205115.
42- Gasnier C, Dumont C, Benachour N, Clair E, Chagnon MC, Séralini GE (August 2009). “Glyphosate-based herbicides are toxic and endocrine disruptors in human cell lines”. Toxicology. 262 (3): 184–91. doi:10.1016/j.tox.2009.06.006. PMID 19539684.
43- Mokbel K (2002). “The evolving role of aromatase inhibitors in breast cancer”. Int. J. Clin. Oncol. 7 (5): 279–83. doi:10.1007/s101470200040 (inactive 2017-01-15). PMID 12402060.
44- Simpson ER (2003). “Sources of estrogen and their importance”. J. Steroid Biochem. Mol. Biol. 86 (3–5): 225–30. doi:10.1016/S0960-0760(03)00360-1. PMID 14623515.
45- Leder BZ, Rohrer JL, Rubin SD, Gallo J, Longcope C (March 2004). “Effects of aromatase inhibition in elderly men with low or borderline-low serum testosterone levels”. J. Clin. Endocrinol. Metab. 89 (3): 1174–80. doi:10.1210/jc.2003-031467. PMID 15001605.
46- Mauras, N; Lima, J; Patel, D; Rini, A; Di Salle, E; Kwok, A; Lippe, B (2003). “Pharmacokinetics and Dose Finding of a Potent Aromatase Inhibitor, Aromasin (Exemestane), in Young Males”. The Journal of Clinical Endocrinology & Metabolism. 88 (12): 5951–6. doi:10.1210/jc.2003-031279. PMID 14671195.
47- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3263690/
L’uso della Gonadotropina Corionica Umana (hCG) è largamente diffuso nell’ambiente culturistico. Usata principalmente per ripristinare la funzionalità gonadale in seguito all’uso di AAS, questo peptide vede la sua applicazione anche durante l’uso di questa classe di farmaci (ciclo, Bridge o TRT), o di altri composti causanti un ciclo di feedback negativo dell’HPTA (vedi SARM), al fine di prevenire l’istaurarsi di una disfunzione testicolare. Fin dai primi anni della sua applicazione su soggetti di sesso maschile, l’hCG è stato oggetto di speculazioni riguardo la possibilità o meno che il suo uso possa portare ad una desensibilizzazione delle cellule di Leydig con conseguente sviluppo di ipogonadismo ipergonadotropo. Il seguente articolo è volto a riportare le caratteristiche del hCG, le sue possibili applicazioni e, in modo approfondito, fare maggiore chiarezza sulla questione legata alla possibile desensibilizzazione hCG-dipendente.
hCG: storia, usi clinici e off-label
Gonadotropina Corionica Umana
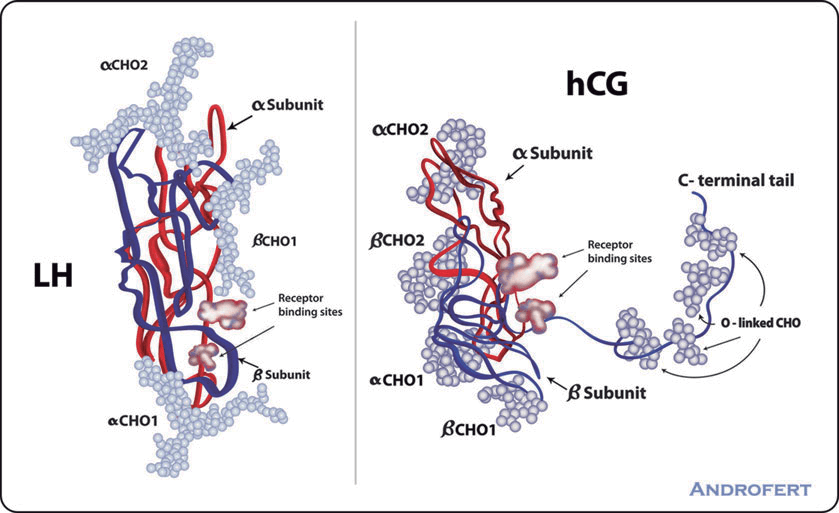
L’hCG (Human chorionic gonadotropin) o Gonadotropina Corionica è un ormone polipeptidico prodotto dall’embrione all’inizio della seconda settimana di sviluppo, in particolare dalle cellule del sinciziotrofoblasto, un tessuto epiteliale monostratificato posto nella porzione profonda del cito-sinciziotrofoblasto, subito dopo l’impianto nell’endometrio. La molecola di hCG è un eterodimero, composto da due subunità (α e β). La subunità α ha struttura identica a quella delle altre gonadotropine (LH e FSH), mentre la subunità β è specifica di ciascun ormone. Per questo motivo, i metodi di dosaggio dell’hCG utilizzano anticorpi diretti contro la subunità β dell’hCG.
Più specificatamente, la Gonadotropina Corionica è una glicoproteina oligosaccaridica composta da 244 aminoacidi. La subunità α è lunga 92 aminoacidi ed è identica a quella dell’Ormone Luteinizzante (LH), dell’Ormone Follicolo-Stimolante (FSH) e dell’Ormone Stimolante la Tiroide (TSH). Come già accennato, la subunità beta è unica per l’hCG.
L’hCG è quindi un analogo del LH, l’ormone prodotto dall’ipofisi che stimola la produzione di ormoni sessuali nei testicoli o nelle ovaie. L’hCG si lega e attiva lo stesso recettore dell’LH ed è ugualmente efficace nello stimolare la produzione di Testosterone negli uomini e di Estrogeni nelle donne.
La Gonadotropina Corionica venne isolata ed identificata per la prima volta nel 1920 (1) venendo in seguito classificata come un ormone della gravidanza circa otto anni dopo.(2) La prima preparazione farmaceutica contenente Gonadotropina Corionica si presentava sotto forma di estratto pituitario animale, il quale venne sviluppato come prodotto commerciale dalla Organon. La Organon introdusse nel mercato l’estratto nel 1931, con il nome commerciale di Pregnon. Una controversia sui marchi obbligò la compagnia a cambiare il nome Pregnyl, che raggiunse il mercato nel 1932. Il Pregnyl è attualmente venduto dalla MSD–Organon, anche se il principio attivo non è più estratto dalla pituitaria animale. Nel 1940 furono introdotte tecniche di produzione che consentivano di ottenere l’ormone filtrando e purificando l’urina delle donne incinta, e alla fine degli anni ’60 questa tecnica di produzione fu adottata da tutti i produttori che avevano usato precedentemente gli estratti animali. Nel corso degli anni i processi di produzione sono stati perfezionati, ma l’hCG è ottenuta essenzialmente nello stesso modo oggi come lo era decenni fa. Nonostante i preparati moderni siano di origine biologica, si afferma che i rischi di contaminanti biologici siano bassi (sebbene non possano essere completamente esclusi).
Al principio della sua applicazione clinica, gli usi indicati per le preparazioni a base di Gonadotropina Corionica erano molto più ampi di quanto non lo siano attualmente. La letteratura inerente al composto degli anni ’50 e ’60 raccomandava l’uso di questo farmaco per, tra le altre cose, il trattamento del sanguinamento uterino e dell’amenorrea, la sindrome di Froehlich, il criptochismo, la sterilità femminile, l’obesità, la depressione e l’impotenza maschile. Un buon esempio degli ampi usi della Gonadotropina Corionica è illustrato nel preparato Glukor, che fu descritto nel 1958 come “Tre volte più efficace del Testosterone. Per i giovani stanchi dal climaterio maschile. Per vecchi stanchi dalla senilità maschile. Benefici nell’impotenza, angina e malattia coronarica, neuropsicosi, prostatite, [e] miocardite.” Tali raccomandazioni, tuttavia, riflettono un’era meno strettamente regolata dall’agenzia governativa e meno dipendente da studi cliniche comprovati. Oggi, le indicazioni approvate dalla FDA per l’uso del hCG sono limitate al trattamento dell’ipogonadotropismo ipogonadico e del criptocridismo negli uomini e alla sterilità anovulatoria nelle donne.
Dr. A.T.W. Simeons
L’hCG non ha alcuna attività significativa di stimolo della tiroide. Questo necessita di essere specificato dato che l’hCG è stata ampiamente usata in passato per il trattamento dell’obesità. Questa applicazione d’uso sembra che sia divenuta popolare nel 1954, dopo la pubblicazione di un articolo del Dr. A.T.W. Simeons nel quale sosteneva che la Gonadotropina Corionica era un’aggiunta efficace alla dieta. Secondo lo studio, i pazienti sono stati in grado di sopprimere efficacemente l’appetito seguendo una dieta con marcata restrizione calorica abbinata alla somministrazione di hCG. Soprannominata la dieta Simeons, le persone in tutti gli Stati Uniti si sottoposero presto a severe restrizioni caloriche (500 Kcal al giorno) e iniezioni di hCG. Poco dopo, l’ormone stesso divenne il coadiuvante principale per la perdita di grasso. Infatti, nel 1957 si diceva che l’hCG era il farmaco più comunemente prescritto per la perdita di peso. Indagini più recenti e complete, tuttavia, confutano l’esistenza di qualsiasi vantaggio anoressizzante o metabolico dato dall’uso di hCG.(3) Nel 1962, il Journal of American Medical Association aveva già avvertito i consumatori circa la dieta Simeons inclusiva di hCG, affermando che la grave restrizione calorica tipica di tale protocollo dimagrante (che si rifletteva in un accentuato catabolismo del tessuto magro) era più pericolosa dell’obesità stessa. Nel 1974, la FDA aveva raccolto abbastanza dichiarazioni sull’uso del hCG per la perdita di grasso che fece inserire una dichiarazione in merito nel bugiardino dei prodotti contenenti l’ormone, nella quale affermava che non vi erano dimostrazioni sulla presunta efficacia nella perdita di peso data dalla somministrazione di hCG in concomitanza con regimi alimentari ipocalorici. Questo avvertimento è tutt’oggi presente su tutti prodotto venduti negli Stati Uniti. Nonostante questo avvertimento e prove che confutano l’efficace di tale pratica, alcune cliniche promuovono ancora l’uso di hCG per la perdita di peso.
La Gonadotropina Corionica Umana è oggi una preparazione farmaceutica molto popolare, poiché rimane una parte indispensabile della terapia di ovulazione per molti casi di infertilità femminile. Sebbene la forma di hCG sintetizzata tramite la tecnica del DNA ricombinante sia stata introdotta sul mercato negli ultimi anni, l’ampia offerta e il basso costo dell’hCG biologico continuano a renderlo un prodotto di base per gli usi clinici e off-label.
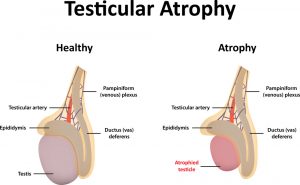
Quando vengono somministrati AAS (o SARM), i livelli di LH diminuiscono rapidamente. Il calo o l’assenza del rilascio ipofisario di LH, e suo consequenziale segnale, induce un calo o interruzione dell’attività testicolare (la quale, ovviamente, si riflette negativamente sulla sintesi di Testosterone) che causa la rapida insorgenza dell’atrofia testicolare. Questa degenerazione testicolare inizia con una riduzione del volume delle cellule di Leydig, seguita da una riduzioni rapida del Testosterone Intra-Testicolare (ITT), dei perossisomi e del fattore insulino-simile 3 (INSL3) – Tutti bio-marcatori e fattori importanti per una corretta funzione testicolare e biosintesi di Testosterone.
Tuttavia, questa degenerazione testicolare viene trattata dai Bodybuilder supplementari chimicamente con la somministrazione di hCG, in special modo all’uscita di un ciclo e per il periodo iniziale della PCT.
Tutte, o quasi tutte, le esperienze pratiche con questo farmaco nel Bodybuilding avvengono con l’uso del hCG biologico (estratto dalle urine di donne gravide), che viene generalmente venduto in vial contenenti polvere liofilizzata da ricombinare con acqua fisiologica o batteriostatica, con un contenuto che va dalle 250 alle 10.000UI per vial.
Il dosaggio clinico di hCG per trattare i casi di ipogonadismo ipogonadotropo è stato tradizionalmente di 5000UI per iniezione. Prima del 1998, la dose tipicamente utilizzata nel bodybuilding per il ripristino della funzione testicolare era la medesima. Di conseguenza, trattandosi di un quantitativo molto elevato, è stato per molto tempo considerato un farmaco di non facile gestione e dagli effetti collaterali, presunti o tali, che destavano non poca preoccupazione (vedi desensibilizzazione delle cellule di Leydig che tratterò più avanti).
Successivamente, venne introdotto l’uso di un dosaggio più basso con un limite di 1500UI per ogni singola iniezione, con una preferenza di dosaggio non superiore alle 1000UI, e con l’uso consigliato di un dosaggio pari a 500UI a somministrazione.
Molti Preparatori danno come raccomandazione quella di non superare le 500UI per ogni somministrazione, poiché non è stato riscontrato alcun vantaggio aggiuntivo nell’utilizzare un dosaggio singolo superiore a questo, a condizione che le iniezioni siano ragionevolmente frequenti (ogni 2-4 giorni).
L’intervallo di dosaggio settimanale comunemente consigliato è compreso tra circa le 700 e le 1750UI. Le dosi di esempio sono 100-250UI al giorno, 250-500 UI a giorni alterni o 250-500UI da tre volte a settimana a somministrazioni distanziate l’una dall’altra da quattro giorni.
Con tali dosaggi sono stati seguiti un numero molto elevato di individui per diversi anni e con eccellenti risultati, e la ricerca scientifica sembra aver convalidato l’utilità del mantenersi all’interno di queste dosi. Come misurato dai livelli intratesticolari di testosterone, questo livello di dosaggio massimizza i risultati. Semplicemente non risulta conveniente la somministrazione di dosi maggiori.
Si raccomandano generalmente iniezioni multiple settimanali dal momento che l’emivita del hCG è di circa 36 ore. Iniezioni meno frequenti comportano uno scarso mantenimento dei livelli ematici.
Prima del 1996, l’uso tradizionale del hCG era quello di inserirla post-ciclo con lo scopo di ripristinare una funzionalità testicolare ottimale. Ma tale pratica non risulta pienamente ottimale dal momento che rallenta comunque i processi di recupero dell’HPTA. Infatti, il tempo medio di recupero della funzionalità testicolare con l’uso del hCG risulta essere in media di 4-8 settimane. Di conseguenza, la scelta migliore, in contesti nei quali i cicli durano più di quattro settimane e/o quando il ciclo viene seguito da un “Bridge” o TRT, l’uso del hCG durante il ciclo permette di conservare una buona attività testicolare permettendo, per esempio, all’atleta in uscita da un ciclo di accelerare i processi di recupero dell’HPTA dal momento che, così facendo, evita quel periodo transitorio (e potenzialmente controproducente) tra la fine del ciclo ed il ripristino di una corretta funzionalità testicolare.
Nei contesti sopra citati, la hCG viene somministrata durante il ciclo con varianti temporali che vanno dalla seconda settimana alla quarta (dipendente dalla durata complessiva del ciclo e da ciò che l’atleta farà nel post ciclo). I dosaggi mediamente utilizzati sono 100 UI al giorno, 200 UI a giorni alterni o 250UI da 3 volte a settimana a ogni 4 giorni.
Un’altra pratica d’uso del hCG è quella di inserirla durante i cicli che non contemplano l’uso di AAS soggetti ad aromatizzazione. Con il solo uso di AAS non aromatizzabili, i livelli di estrogeni diminuiscono in modo anomalo in seguito alla sottoregolazione/soppressione del Testosterone endogeno e la consequenziale diminuzione dei substrati soggetti all’aromatizzazione. Questa condizione interferisce con l’anabolismo, la libido, l’umore, la funzione articolare e, sul lungo termine, la salute cardiovascolare. Un modo ovvio per risolvere questo problema è quello di includere almeno una piccola quantità di uno AAS aromatizzabile (vedi base terapeutica di Testosterone). In questo caso i dosaggi di hCG tipicamente utilizzati sono compresi nella fascia altra d’intervallo del dosaggio efficace suggerito (500UI a giorni alterni). La risultante sarà una sintesi di Testosterone endogeno e Estradiolo.
Terminate le dovute precisazioni sul hCG adesso possiamo trattare l’argomento centrale di questo articolo…
hCG e possibile desensibilizzazione (?)
La questione sulla possibilità secondo cui l’uso prolungato di hCG possa portare ad una condizione di ipogonadismo ipergonadotropo è tutt’ora dibattuta. L’utilizzatore deve comunque tenere a mente che il dosaggio di tale composto deve essere attentamente calibrato in specie con somministrazioni prolungate, poiché alti livelli di hCG possono anche causare un aumento dell’espressione dell’aromatasi testicolare (con conseguente innalzamento dei livelli di estrogeni), (4). Esistono studi piuttosto datati, e svolti per la maggior parte sui ratti, che riportano il verificarsi della desensibilizzazione testicolare al LH in seguito a somministrazione di alti dosaggi e per lunghi periodi di tempo.(5) Il farmaco in questione può effettivamente avere il potenziale di indurre ipogonadismo primario se usato impropriamente, peggiorando notevolmente, non migliorando, la funzionalità testicolare.
I protocolli d’uso di hCG che contemplano la somministrazione di dosi pari a 250UI per via sottocutanea ogni 3 o 4 giorni con una dose massima di 500UI, sviluppati dal Dr. John Crisler, una figura ben nota nel campo dell’Anti-Aging e della terapia ormonale sostitutiva, vengono spesso utilizzati dai soggetti in Terapia Sostitutiva del Testosterone (TRT). L’atrofia testicolare per i pazienti in TRT è un disturbo cosmetico comune. Il programma di somministrazione di hCG del Dr. Crisler è progettato per risolvere questo problema con un uso a lungo termine senza causare l’ipotetica desensibilizzazione. Coloro i quali sono interessati a gestire il timing di somministrazione del hCG con precisione in relazione ad una TRT, il dott. Crisler raccomanda quanto segue: “… i miei pazienti in TRT con Testosterone Cypionato ora somministrano la loro dose di hCG di 250IU nei due giorni precedenti l’iniezione intramuscolare (Testosterone Cypionato NdR.). Tutti i pazienti somministrano la loro dose di hCG per via sottocutanea e il dosaggio può essere aggiustato secondo necessità (devo ancora vedere una necessità di dosaggio superiore alle 350 UI per somministrazione) … Quei pazienti in TRT che preferiscono usare un Testosterone transdermico, o anche Testosterone orale (sebbene io non sia favorevole a ciò) , somministrano la loro dose di hCG ogni tre giorni. ”
Il Dr. John Crisler afferma che è importante non somministrare più di 500UI di hCG in un dato giorno. Egli infatti afferma che vi è solo una quantità massima di stimolazione, e il superamento di questo dosaggio non solo è uno spreco, ma ha conseguenze negative importanti. Dosi più elevate stimolano eccessivamente l’aromatasi testicolare, che aumenta in modo inappropriato i livelli di estrogeni portando alla comparsa di effetti collaterali tipici del iperestrogenemia. Il Dr. Crisler continua dicendo che dosi superiori a quella sopra indicata (500UI) causino anche la desensibilizzazione delle cellule di Leydig verso LH inducendo quindi all’ipogonadismo primario. Egli ribadisce che 250IU ogni 2-4 giorni sia una dose efficace e sicura. Dopotutto, stiamo semplicemente sostituendo ciò che è stato inibito.
Il Dr. Scalley, dal canto suo, critica la posizione del Dr. John Crisler affermando che, la somministrazione dell’hCG per due giorni consecutivi non ha senso, inoltre la dose è omeopatica (inutile). Inoltre, il Dr. Scalley ritiene che, nonostante il Dr. Crisler qualifichi le sue affermazione ricollegandosi a determinati studi, l’errore sta nel considerare come assodato che le dosi più elevate di quelle che consiglia causino la desensibilizzazione. Il Dr. Crisler sembra mancare di una comprensione corretta della letteratura.
Scalley riporta che la desensibilizzazione hCG-dipendente si può potenzialmente verificare in caso di somministrazione prolungata di 5.000UI (cinquemila). Ma, anche in questo caso l’incidenza non è universalmente osservata. C’è anche da aggiungere che il problema della desensibilizzazione non è quasi mai stato osservato nella pratica clinica.
Gli studi solitamente menzionati non danno in realtà alcun supporto a dimostrazione che la desensibilizzazione si verifichi con dosi superiori alle 500UI o che l’uso di 250 UI X2 volte a settimana sia una terapia utile. Se ci si pensaun attimo, qual è lo scopo dell’uso di hCG per due giorni di seguito? Questa pratica risulta completamente bizzarra. Come prima cosa, sfido chiunque a riportare la letteratura (articolo o citazioni) a sostegno del suo trattamento (del Dr. Crisler). Se Crisler è così sicuro di sé, perché non cita alcuna pubblicazione a supporto della sua terapia o, meglio, pubblichi i risultati del trattamento.
Innanzitutto, lo studio che spesso viene citato a sostegno delle tesi del Dr. Crisler (6) valuta il Testosterone Intratesticolare (ITT) e questo, di per se, non è di poca importanza. I partecipanti a questo studio sono stati trattati con Testosterone Enantato (TE), 200 mg alla settimana, per la soppressione rapida della gonadotropina in combinazione con una dose variabile di hCG, somministrata sottocute ogni 2 giorni per 3 settimane: 0 (placebo salino), 125, 250 o 500 UI hCG. Il gruppo placebo è servito come gruppo di controllo. [Nota: la differenza sostanziale è che, anche se lo studio supporta Crisler, il dosaggio è molto diverso da quello da lui raccomandato.]
Quindi, quello che lo studio ci offre sono soggetti di sesso maschile con elevati livelli di Testosterone per via di iniezioni settimanali di 200mg di Testosterone Enantato. La loro produzione endogena di Testosterone è completamente soppressa (teoricamente) come le loro gonadotropine. Il ITT risulta quindi soppresso a causa dell’inibizione delle gonadotropine date dalla somministrazione di Testosterone Enantato. I ricercatori hanno scoperto che ogni dose di hCG (125, 250 e 500 UI) riportava la concentrazione di ITT alla normalità. Si da il caso però che in un maschio normale con un normale livello di Testosterone serico il suo ITT sarà normale. Tutto questo studio è stato semplicemente prendere un maschio normale e sostituire il suo Testosterone con del Testosterone esogeno per poi somministrargli hCG come sostituto del suo LH.
L’unica cosa che può essere salvata di questo studio, è che può essere istruttiva per chi usa hCG a basse dosi in on-cicle, in “Bridge” o in TRT. Più precisamente ci dice qualcosa sulla terapia con hCG mentre si usa un dosaggio “simil-TRT”.
Nello studio risulta interessante esaminare i dati sulle variazioni seriche di Testosterone con ciascuna dose di hCG. I soggetti presi in esame hanno usato una dose contenuta (seppur fisiologicamente alta) di Testosterone Enantato, creando una situazione che per certi versi riproduce quella di un individuo che usa hCG in TRT. Il risultato è stato che la dose di hCG da 125UI a giorni alterni non ha avuto effetti sul Testosterone serico. Le due dosi più elevate (250-500UI) hanno alzato i livelli di Testosterone nel siero al di sopra del normale.
Non ci sono dati individuali (sempre motivo di sospetto quando si esamina la letteratura) e non sono riportati livelli significativi. L’analisi del grafico dello studio riportato di seguito, tuttavia, mostra che il livello di Testosterone del siero non era significativamente diverso dal controllo fino al giorno 21[altra nota a discredito delle affermazioni del Dr. Crisler].
Ci sono quindi molti possibili errori nell’analisi dello studio appena discusso. Dal momento che non ci mostra un analisi sufficientemente accurata tale da permetterci di identificare una soglia di dosaggio che porti alla desensibilizzazione delle cellule di Leydig.
Si può ipotizzare che la modalità di somministrazione dell’hCG nei due giorni precedenti l’iniezione settimanale di Testosterone (come indicato nel protocollo del Dr Crisler) serva da teorico “supporto” al calo della soglia ematica di quest’ultimo. Se si legge la letteratura disponibile sugli effetti dell’hCG, il rialzo dei livelli di Testosterone serico si manifestano in modo significativo a circa 48-72 ore dopo la somministrazione del peptide. Questo dosaggio concentrato in due giorni non da reali vantaggi sulla funzionalità testicolare. Quindi, fino a dimostrazione contraria, le ipotesi del Dr. Crisler sulla somministrazione ottimale di hCG, per effetti e sicurezza riguardo la desensibilizzazione testicolari, non sono altro che opinabili speculazioni.
Per tutti gli scopi pratici, la desensibilizzazione delle cellule di Leydig hCG-dipendente praticamente non sussiste all’interno del quadro clinico, sebbene rimanga una possibilità con l’uso di dosi elevate e per un lungo periodo di tempo (>5000UI)
17 alfa-hydroxyprogesterone
Esiste uno studio che, seppur “isolato”, ritengo sia interessante per farsi un idea delle variabili e della differenza tra possibilità universalmente riscontrate e possibilità di bassa o scarsa incidenza. Si tratta di uno studio nel quale si è osservato che la somministrazione di Tamoxifene in maschi sani ha causato una riduzione dell’accumulo del 17 α-hydroxyprogesterone hCG-indotto.(7)
In questo studio, la somministrazione per via intramuscolare di 1500 UI di hCG al giorno per 3 giorni ha indotto un accumulo transitorio di 17 α-hydroxyprogesterone (17 OHP) rispetto al Testosterone (T) in uomini normali, raggiungendo il massimo nelle 24 ore successive la prima iniezione (rapporto 17 OHP / T, 1,7 +/- 0,3 volte il basale, P <0,01). La somministrazione simultanea di hCG e Tamoxifene (20 mg due volte al giorno) ha quasi completamente soppresso il blocco steroidogenico indotto dal hCG e osservato con il 17 OHP:T ratio (rapporto 17 OHP-T a 24 ore, 1,1 +/- 0,1 volte il basale; 0,01 vs hCG da solo). Questi dati suggeriscono indirettamente che, nell’uomo, la lesione steroidogenica indotta dal hCG potrebbe essere mediata attraverso il suo effetto stimolante sugli estrogeni.
Un altro studio svolto sulla falsariga del precedente, ma ad una distanza di undici anni, ha osservato l’effetto del Tamoxifene sulla risposta testicolare al hCG. (8) Se si legge con attenzione il presente studio, anche alla luce di quanto affermato pocanzi, si riesce ad avere un quadro molto più chiaro sulla questione.
Tamoxifene
In questo studio è stato osservato l’effetto del Tamoxifene (Tx) in concomitanza con la somministrazione acuta e cronica di hCG in pazienti con ipogonadismo ipogonadotropo (HH) e in uomini normali. Un test con hCG (5000 UI hCG) è stato svolto prima e dopo due mesi di somministrazione di hCG (2000 UI di hCG tre volte a settimana) e dopo due mesi di hCG + Tx (2000 UI hCG tre volte a settimana più 20 mg/die di Tamoxifene). I campioni di sangue sono stati prelevati 24 e 72 ore prima e dopo ogni test per determinare i livelli di Testosterone , Estradiolo, 17OHP e SHBG. Il Testosterone è aumentato solo nel gruppo HH con entrambi i trattamenti (X +/- SEM: basale: 97,9 +/- 19,7; hCG: 237,7 +/- 43,2; hCG +/- Tx: 204,7 +/- 10,7 ng / 100 ml). Il 17OHP è aumentato con la somministrazione di hCG da solo, ma non con hCG + Tx in entrambi i gruppi. Il rapporto Estradiolo, SHBG e 17OHP / Testosterone non è cambiato dopo i trattamenti. In risposta al hCG il Testosterone è aumentato 24 ore dopo la somministrazione in ogni test. Il rapporto 17OHP / Testosterone è salito dopo 24 ore nel primo e nel secondo test, ma nel terzo test non è cambiato. Questi risultati supportano il ruolo del Estradiolo nella desensibilizzazione delle cellule di Leydig indotto da una somministrazione acuta di hCG. Tuttavia, l’associazione di Tx non migliora i livelli serici di Testosterone, suggerendo che l’Estradiolo potrebbe non essere il fattore unico coinvolto nei meccanismi di desensibilizzazione testicolare.
Sembrerebbe, quindi, che il fattore determinante legato ad una possibile desensibilizzazione o sottoregolazione della funzionalità testicolare (in particolar modo in riferimento alle cellule di Leydig) sia la dose in acuto e, soprattutto, in cronico. Per ciò che concerne la dose utile questa è invece determinata dal limite fisiologico di stimolo della secrezione di Testosterone che, negli uomini sani con una sensibilità testicolare normale, si è visto corrispondere ad una dose di sole 250UI, con ulteriori aumenti minimi ottenuti con 500UI a 5000UI.
Conclusioni
Sebbene, come già accennato, la questione non sia del tutto chiarita, le informazioni riportate in questo articolo possono senz’altro permettere una pratica d’uso del hCG “sicura” e, soprattutto, intelligente nel contesto di una preparazione farmacologica. Risulta abbastanza chiaro che iniezione intramuscolare o sottocutanee di hCG a dosi di 100-200UI al giorno, 200-250 UI a giorni alterni o 250-500UI da tre volte a settimana a una somministrazione ogni 4 giorni, risultano pienamente efficace per evitare la disfunzione e seguente atrofia testicolare durante l’uso di AAS (o SARM) mantenendone una buona funzionalità in mancanza di stimoli dati dal LH. Dosi superiori a quelle riportate non offrono ulteriori vantaggi. Si tenga inoltre bene a mente che la desensibilizzazione delle cellule di Leydig può manifestarsi con maggiore facilità in una situazione di mancanza di segnale dato dall’LH, condizione che viene spesso osservata in quegli atleti che non usano hCG durante i cicli. Tale desensibilizzazione, però, risulta più semplice da trattare rispetto ad una desensibilizzazione indotta da uno stimolo eccessivo delle cellule di Leydig che, in casi cronici, obbliga il soggetto colpito a doversi sottoporre a trattamento con Testosterone esogeno (TRT).
Quindi, in definitiva, le nozioni base da tenere bene a mente sono:
Uso di dosi e tempi di somministrazione utili allo scopo prefissato (evitare la disfunzione testicolare e avere un ottimale stimolo della biosintesi di Testosterone)
Iniziare la somministrazione di hCG durante il ciclo (tempo variabile dalle 2 alle 4 settimane dall’inizio del ciclo e determinato dalle scelte future al ciclo [PCT, Bridge o TRT]
Non eccedere le 500UI a giorni alterni (principalmente perché dosi più elevate non portano vantaggi considerevoli)
Un’altra nota che mi sento di aggiungere è in riferimento alla ricombinazione del contenuto delle vial e delle procedure per la sterilità del prodotto. Le vial di hCG dovrebbero essere ricostituite con una quantità di soluzione acquosa (sterile o batteriostatica) basata sul quantitativo effettivo in UI della vial. Ad esempio, una vial da 5000UI può essere convenientemente ricombinata con 2,5ml d’acqua. Ciò fornisce una soluzione di 2000IU/ml , che consente un facile calcolo del dosaggio necessario. Ad esempio, una dose di 200 UI richiederebbe quindi l’aspirazione di 0,1mL di soluzione, che sarebbe contrassegnata con “10 UI” su una siringa da insulina.
Se la capacità della vial lo consente, è possibile aggiungere 5,0ml di acqua in una vial da 5000UI. La soluzione risultante sarebbe ovviamente di 1000IU/mL, consentendo un calcolo ancora più semplice del dosaggio necessario.
L’iniezione può essere eseguita intramuscolarmente o sottocute in base alle preferenze personali.
Le vial di hCG non ricostituite devono essere conservate in frigo. Sebbene possano essere spedite a temperatura ambiente. Le vial ricostituite devono sempre essere conservate in frigo; tuttavia, se una vial viene accidentalmente lasciata a temperatura ambiente per un giorno, il principio attivo non subirà alcun deterioramento.
Cosa molto importante quando si manipola l’hCG è quella di impiegare corrette procedure per mantenere la sterilità della vial e della soluzione ivi contenuta. La membrana di gomma deve sempre essere pulita accuratamente con alcool e l’ago deve essere sterile. Il peptide acquoso, o in questo caso le soluzioni glicopeptidiche possono supportare la crescita batterica molto più di quanto possano fare le soluzioni oleose, quindi è raccomandata la massima cura della sterilità del prodotto. Se si nota un intorpidimento della soluzione è consigliabile che il prodotto non venga utilizzato.
Gabriel Bellizzi
Riferimenti:
Exogenous stimulation of corpus luteum formation in the rabbit; influence of extracts of human placenta, decidua, fetus, hydatid mole and corpus luteum on the rabbit gonad. Hirose T 1920 J Jpn Gynecol Sot 16:1055.
Die Schwangerschaftsdiagnose ausdem Ham durch Nachweis des Hypophysenvorderlappen-hormone. II. Pracktishe und theoretische Ergebnisse aus den hamuntersuchungen. Ascheim S, Zondek B 1928 Klin Wochenschr 7:1453-1457.
Un maggior utilizzo di AAS da parte di un atleta si traduce in una maggiore probabilità di avere problemi sessuali durante i periodi di non utilizzo. E no, la PCT non sembra rendere esenti da tali possibili problemi. I ricercatori del American Mayo hospital hanno osservato tale effetto in seguito ad uno studio svolto su diverse centinaia di Bodybuilder supplementati farmacologicamente.(1)
I ricercatori hanno contattato 231 utilizzatori di AAS maschi, attraverso nove forum di bodybuilding, i quali erano pronti a rispondere a domande online sulla loro salute sessuale e sul loro uso di AAS. Per rilevare eventuali disfunzioni erettili, i ricercatori hanno utilizzato una versione abbreviata del questionario IEEF-5. Potete trovarne una copia qui. Gli uomini che totalizzano un punteggio pari o maggiore di 22 non hanno nulla di cui lamentarsi sessualmente parlando, mentre gli uomini che ottengono un punteggio di 17-21 soffrono di un lieve disturbo erettile.
I partecipanti hanno ottenuto un punteggio leggermente superiore sull’IEEF-5 se erano sottoposti ad iniezioni superiori ai 600mg di Testosterone a settimana, se usavano anche un AAS orale o un anti-estrogeno e se erano in buone condizioni di salute. Non molto sorprendente come risultato.
127 utilizzatori hanno affermato che nei loro periodi “off” la libido diminuiva. Questo però non è stato riportato da altri 94 utilizzatori. Quando i ricercatori hanno messo a confronto questi due gruppi, hanno notato che il rischio che si verifichi una riduzione della libido aumentava in modo significativo qualora gli utilizzatori avessero seguito protocolli di AAS per molte settimane all’anno e avessero una lunga storia di utilizzo di questa classe di farmaci.
Lo svolgimento di una PCT era pratica diffusa nel gruppo in cui la libido tra i cicli non diminuiva rispetto al gruppo in cui la libido diminuiva. Tuttavia, questa differenza non era statisticamente significativa.
L’attuale studio rappresenta ad oggi la più grande valutazione sull’impatto dell’utilizzo di AAS ad alto dosaggio e nel lungo periodo sulla funzione sessuale. I risultati dimostrano che l’aumento della durata e della frequenza di utilizzo degli AAS sono associate a più alti tassi di disfunzione erettile de novo e diminuzione della libido dopo l’interruzione d’uso del/i composto/i.
Gli uomini con disfunzione erettile de novo avevano anche maggiori probabilità di riportare altri sintomi legati a bassi livelli di Testosterone, come riduzione della libido, diminuzione dell’energia, depressione, riduzione soggettiva della massa muscolare e aumento soggettivo della massa grassa. Diversamente a ciò, durante l’uso di un dosaggio più alto di Testosterone e l’uso (con tutta probabilità ponderato) di anti-estrogeni si sono osservati punteggi più alti sul questionario IEEF-5.