Secondo i manuali e diversi preparatori, il Boldenone non è una molecola dannosa per il fegato. Dopo tutto il Boldenone non ha un gruppo metile in posizione C-17, e gli steroidi che mancano della metilazione in C-17 non influenzano (a diverso grado) negativamente il fegato. Ma questo è quello che dicono i manuali e alcuni preparatori, ma secondo uno studio egiziano svolto su animali, il Boldenone Undecylenato può essere un’eccezione alla regola.(1)
I ricercatori egiziani hanno usato dei conigli per il loro esperimento. Hanno iniettato agli animali del gruppo di controllo del semplice olio di oliva [G1], mentre hanno somministrato ai conigli dei gruppi sperimentali delle iniezioni contenenti una dose di 5mg di Boldenone Undecylenato per kg di peso corporeo. Al gruppo G2 è stata somministrata una singola iniezione; al gruppo G3 ne sono state somministrate due mentre al gruppo G4 sono state somministrate tre iniezioni. Tra ogni iniezione è stato fatto passare un periodo di tempo di tre settimane.
Come ben sappiamo il Boldenone Undecylenato è il principio attivo contenuto nei preparati come l’Equipoise. Il Boldenone è uno steroide con buoni effetti anabolizzanti e relativamente pochi effetti collaterali androgeni ed estrogeni. L’estere Undecylenato rende la molecola attiva per lungo tempo nel corpo. Ecco perché durante i test anti-doping si è in grado di rilevare l’uso di Boldenone Undecylenato per un massimo di diciotto mesi. Questo è anche il motivo per il divario di tre settimane tra le iniezioni nei gruppi G3 e G4.
Alla fine del periodo di somministrazione di Boldenone, i conigli sono stati sezionati dai ricercatori i quali ne hanno studiato il fegato. Quando hanno esaminato i campioni al microscopio hanno visto che, nonostante il fatto che la molecola sia priva di una metilazione in C-17, lo steroide anabolizzante aveva causato danni al fegato. I ricercatori hanno osservato le cellule grasse del fegato e il tessuto connettivo non-funzionante.
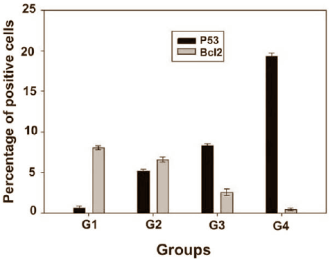
Poiché i ricercatori volevano misurare più oggettivamente quello che era successo nel fegato degli animali, essi hanno misurato la concentrazione della proteina suicida p53 e della proteina anti-suicida Bcl-2 nelle cellule epatiche. Se le cellule epatiche sono sottoposte a eccessivo stress, aumenta la produzione di p53 e quella di Bcl-2 diminuisce. E questo è esattamente quello che è successo nei gruppi G2, G3 e G4.
Le foto di cui sotto mostrano le cellule epatiche nelle quali era attivo il p53 , indicato da una freccia. Un gruppo di controllo =; B = 1 iniezione; C = 2 iniezioni; D = 3 iniezioni. Ancora una volta, si vede che più iniezioni sono state somministrate ai conigli, e più le cellule con proteina p53 sono state trovate nel fegato.
“Non vi è un significativo aumento delle alterazioni istopatologiche e l’incidenza della apoptosi dopo l’iniezione di Boldenone”, concludono i ricercatori. “Così, la gente dovrebbe fare attenzione se vuole utilizzare tale steroide per aumentare la loro forza e resistenza.”
I medici a volte si imbattono in Bodybuilder supplementati chimicamente i quali presentano danni al fegato come conseguenza dell’assunzione di Boldenone, secondo le fonti Ergo-log. Ma i problemi non sono di solito di natura durevole.
Negli anni ‘60 e ’70 del XX secolo i ricercatori hanno testato l’Equipoise più volte sugli esseri umani. Il nome del prodotto allora testato era Parenabol o 29’038-Ba. In uno studio del 1968, iniezioni quindicinali con 50mg di Boldenone Undecylenato non hanno avuto effetti negativi sul fegato.(2)
Tuttavia, le aziende farmaceutiche hanno deciso che sarebbe stato meglio limitare l’uso di Boldenone ai soli preparati per animali. La ricerca sugli esseri umani si è fermata dopo questo, quindi ora sappiamo molto poco circa gli effetti del Boldenone Undecylenato sugli esseri umani.
Comunque sia, affermare che questa molecola sia esente da “sovraccarico” epatico e che addirittura sia benefica per questo organo, è un affermazione eccessiva. Come al solito la regola della “dose che fa il veleno” è pienamente applicabile anche in merito all’uso di Boldenone per il miglioramento delle prestazioni.
Con il termine PCT (Post-Cycle Therapy), come ben sappiamo, ci si riferisce a quella fase che segue la fine di un ciclo di AAS. Dal momento che gli AAS causano una temporanea soppressione della funzione dell’asse HPTA e, quindi della sintesi di Androgeni endogeni , questo è un problema che dovrebbe essere affrontato diligentemente a conclusione di un ciclo. Se gli AAS vengono sospesi bruscamente senza le adeguate procedure di supporto per la rigenerazione dell’HPTA (oltre che un controllo del Cortisolo e degli Estrogeni circolanti in eccesso), il risultato potrebbe essere un prolungato stato di ipogonadismo (bassi livelli di Androgeni), caratterizzato da una sostanziale perdita di massa muscolare, ridotti livelli di energia, depressione, abbassamento della libido, alterata funzionalità sessuale, accumulo di grasso e ginecomastia. Molti culturisti chiamano questa condizione “Crash Post Cycle“.
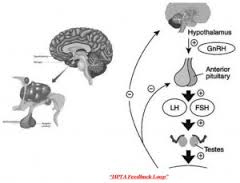
Nel corpo umano, l’asse Ipotalamo-Ipofisi-Gonadi (HPTA) controlla la biosintesi di Testosterone. L’HPTA è un sistema strettamente regolamentato che lavora per assicurare e mantenere il corretto livello di Testosterone. Questo processo di regolazione è diviso in tre livelli:
1. Nella parte superiore v’è la regione ipotalamica del cervello, che rilascia GnRH (Gonadotropin- Releasing Hormone) quando rileva la necessità di aumentare la sintesi di Testosterone.
2. Il GnRH invia un segnale al secondo livello dell’asse, la pituitaria, per produrre Ormone Luteinizzante (LH).
3. LH a sua volta invia un messaggio alle cellule del Leydig nei testicoli che secerneranno Testosterone.
Dato questo ruolo, LH è considerato come il diretto messaggero primario per il controllo della sintesi di Testosterone. Il Testosterone e altri steroidi sessuali che vengono prodotti come risultato della stimolazione di LH servono come contrappeso. Essi forniscono il feedback negativo per abbassare la secrezione di LH e di Testosterone, impedendone una sovrapproduzione. Gli Steroidi Anabolizzanti sintetici, naturalmente, inviano le stesse risposte negative.
Il rilascio da parte dell’Ipotalamo del GnRH (Gonadotropin Releasing Hormone), stimola l’ipofisi a rilasciare Ormone Luteinizzante (LH) e ormone Follicolo-Stimolante (FSH). Questo (LH) promuove il rilascio di Testosterone dai testicoli. Gli Androgeni, nonché Estrogeni e Progestinici, a loro volta causano l’induzione di feedback negativo all’Ipotalamo e all’Ipofisi, abbassando la produzione di Gonadotropine e Testosterone quando troppo ormone è presente.
Recupero dell’HPTA senza supporto
La soppressione della sintesi di Testosterone naturale, da uso di steroidi è tipicamente un fenomeno temporaneo. Anche se non fate niente, la normale sintesi degli androgeni endogeni riprende un paio di mesi dopo la conclusione del ciclo. Il problema è che in questo periodo una produzione adeguata di Testosterone, con un adeguato controllo del Cortisolo, agevola il mantenimento del tessuto muscolare. Gran parte del muscolo ottenuto durante l’assunzione di AAS può essere facilmente perso nelle settimane e nei mesi a seguire, se i livelli di androgeni bassi sono lasciati in declino. La PCT è ampiamente utilizzata dai culturisti e atleti per stimolare l’HPTA, così da normalizzare i livelli di produzione di Androgeni più rapidamente.
Misurazione dei livelli di LH e Testosterone a partire da una settimana dopo l’ultima iniezione da 250mg di Testosterone Enantato. Notare come tra la settimana 1 e 5, i livelli di testosterone sono in calo a causa della cessazione della somministrazione di androgeni esogeni, mentre i livelli di LH cominciano a correggersi. Dalla 5° alla 10° settimana, i livelli di Testosterone cominciano a correggersi. Dalla settimana 5 alla 10, i livelli di Testosterone rimangono molto nei pressi della linea di base, anche se l’LH aumenta da questo punto. Non vi sono correzioni notevoli nei livelli di Testosterone se non dopo la 10° settimana.
Studi sull’aspetto post-utilizzo di steroidi anabolizzanti, in particolare in coloro che abusano di AAS, sono carenti. Nella maggior parte dei casi si deve fare riferimento a studi in monoterapia, di solito di pazienti in sostituzione ormonale . Uno degli studi più dettagliati che tratta della situazione ormonale post-AAS è stato fatto utilizzando Testosterone Enantato. Lo studio comprendeva un gruppo di uomini ai quali era stato somministrato settimanalmente un dosaggio di 250 mg di Testosterone Enantato per 21 settimane, che è una dose certamente superiore a quella normalmente utilizzata in HRT. Diversi ormoni sono stati misurati ogni settimana durante lo studio, e per più di 4 mesi dopo l’interruzione del farmaco. Una revisione dei dati mostra che all’inizio dello studio, i livelli di LH sono stati soppressi in relazione diretta con l’aumento del Testosterone (vedere Figura a lato). Una volta che lo steroide è stato interrotto, tuttavia, c’è stato un ritardo tra il ritorno verso la normale produzione di LH (che ha cominciato a correggersi dalla 3 ° settimana) e di Testosterone (per il quale ci sono volute più di 10 settimane prima della correzione evidente dei livelli). Lo studio sopra suggerisce che una delle prime cose che accadono dopo la cessazione dell’assunzione di AAS è che il cervello riconosce che i livelli di Testosterone sono bassi. Questo farà sì che i livelli di GnRH e LH inizieranno a correggersi abbastanza rapidamente. Il sostanziale ritardo tra questo e un aumento dei livelli di Testosterone è causato in gran parte dalla insensibilità testicolare all’ormone luteinizzante. Dopo mesi dal ricevimento dello stimolo la loro attività è estremamente debole, e le loro dimensioni saranno diminuite (atrofizzazione). Si tratta di un effetto collaterale ben documentata con l’uso di steroidi anabolizzanti, anche se una differenza nelle dimensioni può non essere immediatamente visibile in tutti i casi. Quando i livelli di LH iniziano ad aumentare , i testicoli inizialmente non sono in grado di gestire il carico di lavoro. Ciò è previsto per correggersi nel tempo, ma può richiedere molte settimane affinché i testicoli ripristinino lentamente la loro originale dimensione e attività . Con una buona parte del periodo di recupero post-ciclo effettivamente caratterizzato da livelli normali (anche bassi) di LH, dobbiamo affrontare il recupero, se ci aspettiamo che esso sia efficace.
Programma PCT del dr. Scally
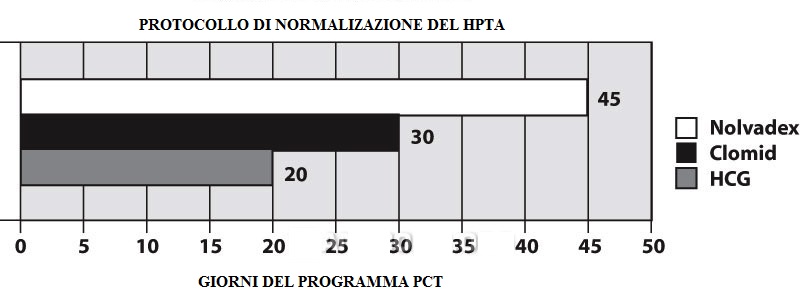
Il seguente programma PCT è stato sviluppato dal Dr. Michael Scally, uno degli individui più noti ed affermati nel campo degli steroidi anabolizzanti e dell’utilizzo medico della terapia ormonale maschile sostitutiva. Scally ha esercitato una pressione particolarmente forte nella comunità medica e verso il governo per il riconoscimento dello squilibrio ormonale seguente all’uso di steroidi, cosa che ha denominato ipogonadismo indotto da steroidi anabolizzanti (ASIH). Ha anche eseguito analisi del sangue su centinaia di pazienti, e così facendo ha sviluppato il seguente programma PCT. Una forma leggermente modificata di questo programma è stata delineata in un rapporto clinico che coinvolge 19 soggetti maschi sani che assumono dosi sovrafisiologiche (altamente soppressive) di Testosterone Cypionato e Nandrolone Decanoato per 12 settimane. Il “HPGA Normalization Protocol” di Scally si concentra sull’uso combinato di hCG, Nolvadex e Clomid, ed è forse il programma di terapia post-ciclo più grande e clinicamente supportato attualmente disponibile.
Questo programma PCT inizia con una dose sostanziale di hCG (2000 UI a giorni alterni per 20 giorni). Sono utilizzati Tamoxifene Citrato (20 mg due volte al giorno) e Clomifene Citrato (50mg due volte al giorno). Il Clomid viene utilizzato per 30 giorni. Mentre nel primo paio di settimane l’uso di questa classe di anti-estrogeni potrebbero non essere altamente efficaci, dovrebbe rivelarsi più critica verso la metà e alla fine del programma. Nella versione pubblicata del programma di Scally (che è leggermente modificato da quanto sopra), la normale funzione ormonale è tornata in tutti i soggetti entro 45 giorni. Questo è un chiaro successo, di gran lunga più favorevole rispetto alla finestra di ripresa protratta riportato nello studio con 250mg a settimana di Testosterone Enantato.
Il rispetto dei giusti tempi per l’inizio di un programma di terapia post-ciclo può essere importante quanto la sua composizione. Se è iniziato troppo tardi, gli AAS circolanti saranno scesi eccessivamente portando alla perdita repentina della massa muscolare ottenuta durante il ciclo. Se si avvia il programma troppo presto, si può perdere la finestra ottimale di efficacia.
Il periodo di 20 giorni di tempo in cui viene utilizzata l’hCG è la più critica. In particolare, si vuol fare in modo che l’hCG sia applicata nel periodo in cui gli steroidi esogeni sono in calo al di sotto della soglia fisiologica di stimolazione degli Androgeni. Nel caso del Testosterone (molecola più facile da capire e spiegare), questo avverrebbe prima che i livelli ematici scendano al di sotto del livello normale (350ng / dL). Ci dovrebbe essere una piccola sovrapposizione con il periodo di attività del ciclo, in modo che l’hCG abbia un breve periodo di tempo per lavorare prima che i livelli di AAS sono drasticamente diminuiti.
Il momento esatto per l’inizio del programma PCT è determinato dall’emivita del farmaco utilizzato. Useremo testosterone Cypionato/Enantato come esempio. Sappiamo che ogni iniezione ha una emivita di circa 8 giorni. Una dose di 200 mg a settimana dovrebbe portare ad una soglia dei livelli ematici a circa 2000-2400 ng/dL dopo diverse settimane di utilizzo. Ci vorrebbero circa 3 settimane (24 giorni) perché i livelli di testosterone scendano a circa 250-300 ng/dL a quel dosaggio. Così, il programma PCT dovrebbe essere iniziato da pochi giorni a una settimana dopo l’ultima iniezione di Testosterone. L’inizio del programma potrebbe essere ritardato con dosi più elevate. Ad esempio, a 500 mg a settimana di T.C./T.E. ci vorrebbero circa 4 settimane (32 giorni) per far si che il Testosterone scenda al di sotto del range di normalità. In questo caso, la PCT verrà avviata circa due settimane dopo l’ultima iniezione di Testosterone. Con un ciclo di soli orali, la PCT dovrebbe essere iniziata 7-10 giorni prima dell’assunzione dell’ultima dose.
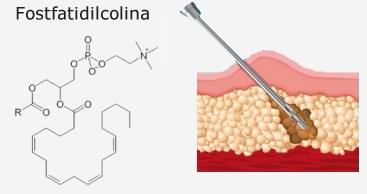
La Fosfatidilcolina (a volte abbreviata come PC) è un tipo di fosfolipide che contiene colina come gruppo di testa. È una molecola anfipatica, ottenuta dalla trimetilazione di una molecola di Fosfatidiletanolammina o sintetizzata ex novo a partire da un digliceride e da CDP-colina.
È stata scoperta nel 1850 da Maurice Gobley, che la estrasse dal tuorlo dell’uovo. La molecola è isolabile anche dai semi di soia attraverso un’estrazione meccanica o chimica utilizzando esano.
Si tratta di uno dei più importanti componenti delle membrane biologiche. In particolare, si tratta del fosfolipide più abbondante sul foglietto esterno della membrana plasmatica.
La Fosfatidilcolina è anche il principale componente della lecitina (dal greco lekithos – λεκιθος, tuorlo d’uovo), il cui estratto consiste appunto in una mistura di Fosfatidilcolina, acido fosforico, colina, acidi grassi, glicerolo, glicolipidi, trigliceridi ed altri fosfolipidi. Spesso, lecitina e Fosfatidilcolina sono utilizzati come sinonimi.
È particolarmente presente nella soia, dalla quale può essere estratta per essere usata nell’industria alimentare come agente emulsionante (sotto il nome di E322). Si trova anche in altri alimenti, tra i quali: uova, caviale e in quantità inferiore in cavolfiori, lenticchie, piselli, riso, fegato di vitello, latte. La lecitinasi è un gruppo di enzimi esterasici in grado di idrolizzare la lecitina. Il colesterolo presente nelle lipoproteine plasmatiche è spesso esterificato con una molecola di lecitina.
La Fosfatidilcolina è un fosfolipide conosciuto e studiato da moltissimo tempo, in quanto , come già eccennato, abbondantemente rappresentato in natura ed introdotto quotidianamente attraverso i vari alimenti.
Il ruolo nutrizionale della Fosfatidilcolina è molto importante, tanto che viene abbondantemente impiegata nell’industria alimentare (come agente emulsionante) e dietetica (come supplemento utile per abbassare il colesterolo e favorire l’efficienza di fegato e cervello).
Nell’organismo umano, la Fosfatidilcolina rappresenta uno dei principali componenti della membrana plasmatica, di cui regola fluidità, integrità e permeabilità.
Le straordinarie virtù della Fosfatidilcolina, sfruttate in campo medico nel trattamento delle iperlipidemie e delle patologie epatiche, derivano dalla sua natura anfifilica, che gli permette di mantenere i grassi in soluzione nel sangue ed in altri fluidi organici (che di per sé sono soluzioni acquose, quindi immiscibili con i lipidi proprio come l’acqua e l’olio). Ed è proprio in questa sua caratteristica che la Fosfatidilcolina ha trovato un suo recente utilizzo nel trattamento delle adiposità localizzate.
E’ piuttosto recente l’impiego della Fosfatidilcolina nel trattamento dell’adiposità localizzata. Fu negli anni ’90, in seguito ad una brillante intuizione, che un medico brasiliano la propose per primo per questo scopo. Infatti, se è vero che la Fosfatidilcolina è in grado di sciogliere il grasso con il quale viene a contatto, ciò può essere fatto da questa al fine di eliminare piccoli depositi adiposi e trattare la pannicolopatia edemato-fibrosclerotica (cellulite) sfruttando tecniche mesoterapiche.
Se la Fosfatidilcolina viene iniettata direttamente nel tessuto adiposo attraverso sottilissimi aghi, essa è in grado di solubilizzare i grassi, riducendo il volume delle cellule che li contengono (“svuota” gli adipociti). La tecnica è chiamata Lipodissolve, ed è scarsamente invasiva e generalmente svolta in regime ambulatoriale. Questa tecnica è particolarmente utile nel trattamento degli accumuli adiposi che, sia per fattori endocrini e metabolici (di base genetici), risultano di difficile e limitata eliminazione con i classici interventi dietetico-comportamentali.
La tecnica “Lipoddisolve” rappresenta quindi una efficace arma contro le adiposità localizzate sia femminili che maschili, anche quando tali inestetismi si localizzano in punti critici, come l’addome, l’interno coscia ed i fianchi: si sono ottenuti buoni risultati anche nel trattamento dei depositi adiposi della palpebra inferiore, del doppio mento e delle guancie.
L’avvento della Fosfatidilcolina ha rappresentato una vera e propria alternativa alla tradizionale liposcultura, la dove i depositi di grasso non sono di grande consistenza: in questo caso la tecnica chirurgica di liposuzione rimane la soluzione più efficace.
Comunque, il trattamento mesoterapico con Fosfatidilcolina consiste principalmente nell’identificazione delle zone di accumulo adiposo, sulle quali viene applicata una dose di crema anestetica. Dopo disinfezione, si infiltra mediante aghi piccoli e sottili la Fosfatidilcolina, direttamente all’interno del pannicolo adiposo distribuendo la sostanza in modo da ottenere una lisi quanto più omogenea e simmetrica.
Il trattamento non è doloroso ed è assolutamente tollerabile. Generalmente il fastidio si limita ad un leggero bruciore nella zona di infiltrazione seguito da un fastidio alla compressione nei giorni successivi, simile ad una contusione.
Può accadere che in alcuni distretti si verifichino dei versamenti, che scompaiono nel giro di una settimana. Ovviamente, nulla in confronto ai postumi di una liposuzione. La letteratura scientifica internazionale non riporta alcun effetto collaterale di rilievo mentre recenti lavori scientifici riportano risultati positivi nel 98% dei pazienti trattati.
Attualmente i protocolli proposti in Medicina Estetica vedono l’esclusione di bambini(direi ovvia), donne in gravidanza o in allattamento, diabetici con vasculopatie, e ovviamente chi è allergico alla Soia. Vi sono altre condizioni a rischio da valutarsi: le insufficienze epatiche e/o renali, l’adiposità con BMI>30, alcune situazioni immuno-endocrine quali la tiroidite o infezioni croniche particolarmente rischiose; alterazioni della coagulazione.
Il trattamento è molto veloce: in genere bastano quindici minuti per due o quattro aree di adiposità. L’iniziale effetto compare a partire dal terzo giorno e raggiungere il suo picco da una a due settimane dopo il trattamento, perdurando in maniera definitiva (dieta permettendo).
Nel corso di una seduta si riescono a trattare da due a quattro cuscinetti, in base all’entità dell’inestetismo. Ipotizzando una situazione tipo, in tre o quattro sedute si possono eliminare accumuli sui fianchi e nel basso addome. Dopo il trattamento si possono riprendere subito le normali attività. Il costo del trattamento (o delle fiale) è ovviamente notevolmente inferiore a quello delle soluzioni chirurgiche classiche, rivelandosi quindi semplice e discretamente accessibile.
Occorre ricordare che le infiltrazioni di Fosfatidilcolina non possono essere considerate sostitutive di strategie dietetiche ed esercizio fisico o di altre soluzioni terapeutiche medico-chirurgiche nel paziente obeso.
Il risultato ottenuto può essere migliorato con l’associazione di Carnitina (amminoacido necessario per veicolare gli acidi grassi all’interno dei mitocondri), di acido desossicolico (un sale biliare) e di altre sostanze capaci di migliorare la salute del microcircolo e limitare le reazioni avverse (antinfiammatori ed antidolorifici).
Ulteriori innovazioni di questo trattamento potrebbero portare alla definitiva sostituzione degli aghi con apparecchiature capaci di indurre la penetrazione delle sostanze tramite l’apertura elettrochimica di specifici canali intracellulari.
Esistono anche cosmetici particolari come gel e patch monouso a base di Fosfatidilcolina e di altri princìpi attivi drenanti, lipolitici e vasoprotettivi. Tra questi ricordiamo la caffeina, la centella, l’equiseto, l’escina, agenti antiossidanti, idratanti, emollienti ed alcune alghe marine. L’efficacia di questa variante non mostra la stessa efficacia mostrata dalle iniziezioni.
L’utilità di questo tipo di trattamento non è da sottovalutare, in specie dai BodyBuilder che cercano di diminuire inestetismi di matrice genetica e difficilmente trattabili con la semplice dieta ed esercizio (o altra supplementazione).
Un altra azione della Carnitina, oltre alle “classiche” riguardanti il metabolismo energetico, è quella di aumentare i recettori androgeni nelle cellule muscolari.
In uno studio condotto dall’Università del Connecticut, i ricercatori hanno testato gli effetti della L-Carnitina (nella sua forma Tartrato) e la sua capacità di migliorare l’utilizzo del Testosterone libero (il 2% prima citato). Durante i 21 giorni di esperimento, è stato verificato come la L-Carnitina abbia aumentato il numero dei recettori androgeni rispetto ai risultati prodotti dal gruppo trattato con placebo.(1)
Questo è davvero molto importante per i BodyBuilder!
Gli effetti della L-Carnitina sui recettori androgeni permettono di aumentare la capacità fisiologica del corpo di interagire con il Testosterone libero a livello recettoriale nelle cellule muscolari.
La L-Carnitina supporta anche la rigenerazione del tessuto muscolare e limita l’accumulo di acido lattico permettendo un miglioramento della prestazione. Inoltre risulta maggiormente utile durante una Dieta Chetogenica dove il minor introito di carboidrati ne riduce la biosintesi endogena. La L-Carnitina facilita la metabolizzazione dei Chetoni. Infatti, questo derivato aminoacidico (prodotto a partire dalla lisina e dalla metionina) è particolarmente attivo durante il digiuno quando la glicemia si abbassa ed i livelli plasmatici di glucagone ed acidi grassi diventano elevati.
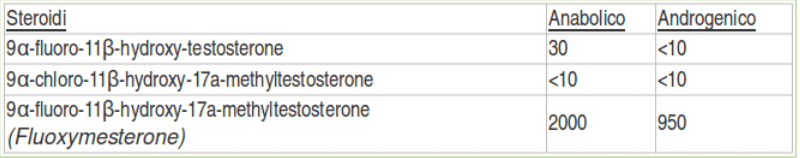
Androgenico: 850 Anabolico: 1,900 Standard: Methyltestosterone (orale) Nome Chimico: 9a-fluoro-11b,17b-dihydroxy-17a-methyl-4-androsten-3-one, 9a-fluoro-11b-hydroxy-17a-methyltestosterone Attività Estrogenica: nessuna Attività Progestinica: nessun dato disponibile (bassa)
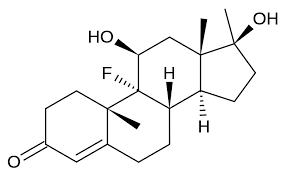
Il Fluoxymesterone (9-fluoro-11,17-dihydroxy-10,13,17-trimethyl-1,2,6,7,8,11,12,14,15,16-decahydrocyclopenta[a]phenanthren-3-one), noto anche con il nome commerciale di Halotestin®, è uno steroide anabolizzante derivato del Testosterone.…
La sua storia è legata alla Upjohn (o The Upjohn Company), una public Company con sede a Kalamazoo nello stato del Michigan in USA, fondata nel 1886 dal Dr. William E. Upjohn e tre suoi fratelli. Essa è stata una delle più grandi produttrici di innovativi farmaci etici degli Stati Uniti.
Il team di ricercatori Upjohn iniziò una serie di studi sul potenziale degli steroidi anabolizzanti in seguito all’osservazione di ampi trial clinici sul “deponortestonate” (prodotto della Upjohn contenente Nandrolone iniettabile) con lo scopo di sviluppare e testare una serie di nuove molecole.
Una delle scoperte più significative del team Upjohn è stata fatta nel 1950, quando scoprirono una tecnica di fermentazione microbiologica per l’introduzione di un atomo di ossigeno nell’11° atomo di carbonio di uno steroide (1). Ciò non permise solo l’introduzione su larga scala di composti corticosteroidei, come il Cortisone, ma aprì la strada anche alla creazione del Fluoxymesterone.
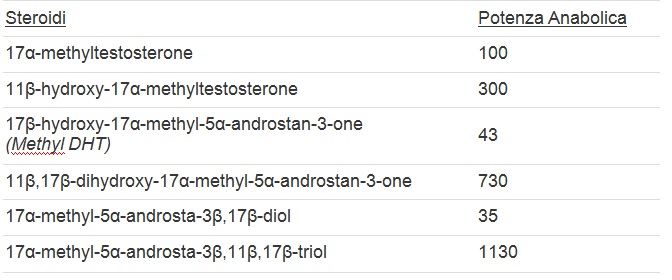
I risultati degli esperimenti sui ratti trattati con alcuni steroidi 11-ossigenati sintetizzati dalla Upjohn si possono osservare nella seguente tabella. (2)
Come si può vedere dalla tabella, tutti gli steroidi 11-ossigenati sono risultati significativamente più potenti rispetto alle loro controparti non-ossigenate.
Derivati corticosteroidei 9α-alogenati sono stati descritti da Fried e Sabo nel 1953 (3) e sono stati osservati possedere una maggiore attività glucocorticoide e anti-infiammatoria. Con questo in mente, Lyster et al alla Upjohn sintetizzarono e testarono una serie di derivati del testosterone 11β-idrossilati e 9α-alogenati (4).
Essi scoprirono che la soppressione dell’attività del gruppo 9-cloro, e la sostituzione 9α-fluoro nello steroide produceva un notevole aumento della attività anabolizzante e androgena orale nei ratti (4):
“l’11β-idrossi-methyltestosterone è 0,9 volte più androgeno e 3 volte più anabolico del Methyltestosterone. Il 9-Fluoro-11β-idrossi-methyltestosterone è 9,5 volte più androgeno e 20 volte più anabolico del Methyltestosterone”
Il composto 17α-methyltestosterone 9α-fluoro 11β-idrossi è stato successivamente denominato Fluoxymesterone.
I ricercatori della Upjohn classificarono il potere Anabolizzante:Androgeno del Fluoxymesterone somministrato oralmente a 2000:950 rispetto al Metiltestosterone.(4)
I dati sono stati successivamente riportati dalla Upjohn nel 1963 dai ricercatori Kincl e Dorfman della Syntex che confermarono rispettivamente i dati sul levator ani, vescicole seminali, e prostata ventrale al 1745%, 757% e 118% (quest’ultimi due come indici androgenici).(5)
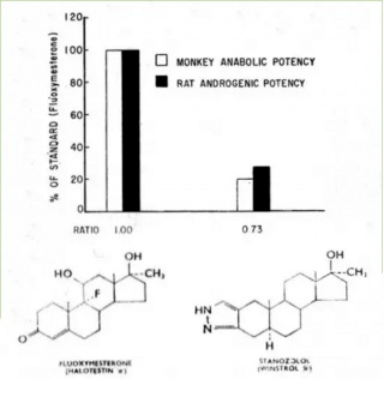
Il rapporto A:A dello Stanozololo non appare così favorevole dalle analisi della Upjhon del 1965.(8)
Quando rivisitato nel 1963 da Beyler et al., il valore anabolizzante 380 (misurato come ritenzione di azoto) e androgeno 140 (misurato con la dimensione della prostata ventrale) sempre rispetto al Metiltestosterone del Bolasterone e del Fluoxymesterone, sono risultati essere “notevolmente inferiore a quelli riportati per alcuni 19-nor steroidi e alcuni steroidi eterociclici di attuale interesse clinico“(6)
Naturalmente le loro conclusioni potrebbero essere state alquanto “edulcorate” dalla loro posizione commerciale; come ricercatori della Sterling-Winthrop erano responsabili della creazione dello steroide Stanozololo (Winstrol).(7)
Struttura molecolare del Fluoxymesterone
Dichloro-Dianabol
Il Fluoxymesterone è strutturalmente classificato come un derivato alogenato del Testosterone. Il gruppo alogeno aggiunto è quello di fluoro in C-9, che ne potenzia l’attività androgena, rendendo la molecola un substrato migliore per la 5α-riduzione, con produzione di metaboliti estremamente androgeni. Mentre la modifica in C-9 è un’anomalia nel mondo degli androgeni, questa è relativamente comune tra i glucocorticoidi sintetici; come nel Desametasone, Betametasone, Fludrocortisone, e Triamcinolone. In realtà ci sono alcuni composti che vengono sostituiti in C-9 con altri alogeni. Ad esempio, il Beclometasone ha una funzione 9α-cloro. La Schering testò alcuni composti 9α, 11β- alogenati, con scarso successo, anche se clorurati derivati dal Dianabol (9α,11β-dichloro-17β-hydroxy-17α- methylandrosta-1,4-dien-3-one) hanno mostrato di essere promettenti come anabolizzanti, con rapporto 300:60 rispetto al Metiltestosterone con somministrazione orale.(9)
Non è del tutto chiaro il motivo per cui la frazione 9α-fluoro conferisce un’elevata attività anabolizzante/androgena al Fluoxymesterone somministrato oralmente, ma l’atomo di fluoro può potenzialmente cambiare la farmacocinetica e la farmacodinamica di un composto in diversi modi:
“1. Il Fluoro, a causa delle sue ridotte dimensioni steriche, assomiglia all’idrogeno bioattivo rispetto all’ambiente sterico in associazione con le regioni proteina recettore;
2. Il Fluoro, l’elemento più elettronegativo, altera gli effetti elettronici;
3. Il Fluoro, quando legato al carbonio ha una energia di legame carbonio-fluoro pari a 107 kcal / mol, aumenta l’ossidabilità, la temperatura e la stabilità metabolica; e
4. Forse ancora più importante, il legami carbonio-fluoro aumenta notevolmente la solubilità dei lipidi e, quindi, migliora i tassi di bio-assorbimento e bio-transporto.”(10)
In conformità con la prima proprietà di cui sopra, l’atomo di fluoro ha un raggio di van der Waals molto simile all’atomo di idrogeno che sostituisce, il raggio standard dell’idrogeno si trova a circa 1.1Å-1.2Å e per il fluoro circa 1.35Å-1.47Å.(11,12,13)
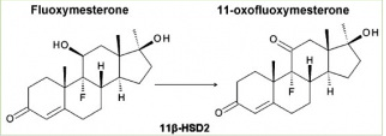
Oltre al suo insolito gruppo 9α-fluoro, il Fluoxymesterone possiede anche un gruppo 11β-idrossile, che è caratteristico di molti corticosteroidi attivi. Un recente studio ha scoperto che a causa della presenza del gruppo 11β-idrossile il Fluoxymesterone interferisce con il metabolismo dei corticosteroidi. Il Cortisolo viene convertito in Cortisone inattivo da parte dell’enzima 11β-idrossisteroide deidrogenasi di tipo 2 (11β- HSD-2). Poiché il recettore mineralcorticoide (MR) può essere attivato sia dai mineralcorticoidi che dai glucocorticoidi, questo enzima è espresso nei tessuti mineralcorticoidi sensibili per impedire l’attivazione del MR da parte del Cortisolo. Il Fluoxymesterone è risultato essere un potente inibitore competitivo della 11β-HSD-2 umana.(14)
Questa inibizione potrebbe causare una attivazione dei MR cortisolo-indotta, che può condurre alla ritenzione di sodio e all’escrezione di potassio, causando ritenzione idrica e ipertensione. Dopo il successivo uso clinico del Fluoxymesterone, sono state apportate alcune variazioni: sono stati sintetizzati composti 16α-metilati da Merck, anche se sono risultati deludenti al confronto.(15)
L’11-keto, analogo del Fluoxymesterone, rispetto a quest’ultimo è leggermente più anabolizzante (22 vs 20) e meno androgeno (8,5 vs 9,5).(16)
Metabolismo del Fluoxymesterone
Nonostante le modifiche 9α-fluoro e 11β-idrossile, il Fluoxymesterone subisce sia la 5α-riduzione che la 5β-riduzione negli esseri umani (17), i 3-cheto metaboliti ridotti sono stati rilevati anche nell’uomo.(17,18)
Nonostante il 9α-fluoro-testosterone aromatizza circa la metà del Testosterone, il gruppo 11β-idrossile del Fluoxymesterone ne impedisce l’aromatizzazione e anche l’affinità di legame con le SHBG.(19,20) ù
“La presenza di sostituenti assiali in posizione C-11 interferisce con l’aromatizzazione dell’anello A”(19)
Come con tutti i 17α-alchil-17b-idrossi steroidi, i metaboliti 17-cheto del Fluoxymesterone sono impossibili. La 17-epimerizzazione del Fluoxymesterone è stata segnalata ed è una comune (anche se minore) via metabolica tra 17α-metil-17β-idrossi steroidi.(21)
La presenza di diversi metaboliti idrossilati, diidrossilati, 11,12 e 13,14 insaturi, e 11-cheto sono stati identificati (post-somministrazione) anche negli esseri umani, secondo un recente studio.(17)
E’ anche interessante notare che il Fluoxymesterone viene metabolizzato al suo corrispondente analogo 11-cheto (11-oxo Fluoxymesterone) dalla 11β-HSD-2 umana (anche se non ampiamente), come dimostrato da un esperimento in vitro contemporaneo.
“Dati biologici hanno mostrato che il Fluoxymesterone è un substrato del 11β-HSD-2, ma con un tasso di conversione inferiore al Cortisolo.”(14)
L’ultima differenza rispetto al Testosterone è la 17-alfa-metilazione, che ovviamente lo rende tossico per il fegato (alcune evidenze mostrano una marcata tossicità).
La sua capacità inibitiva sull’HPTA è più marcata di quella esercitata dal Metilltestosterone, nonostante non sia aromatizzabile, e si manifesta maggiormente a livello testicolare. Nel range dei 20mg/die non sembra mostrare un significativo impatto su FSH e LH ma già sul Testosterone circolante. Il Fluoxymesterone possiede una biodisponibilità del 100%, dovuta alla metilazione in posizione 17α la quale inibisce il metabolismo epatico per ossidazione enzimatica del 17β-idrossile, consentendo l’assorbimento nel flusso sanguigno della molecola. Come molti altri steroidi metilati in C-17, il Fluoxymesterone presenta una scarsa affinità con i recettori AR, ciononostante le sue azioni sono mediate dal recettore degli androgeni, molto probabilmente a causa della sua prolungata emivita plasmatica che è di circa 9,2 ore.(22)
Uso clinico del Fluoxymesterone
Il Fluoxymesterone viene applicato in ambito medico per il trattamento del’ipogonadismo maschile, il ritardo della pubertà nei maschi, e nel trattamento di neoplasie al seno nelle donne. L’attività antitumorale del Fluoxymesterone appare correlata alla riduzione o inibizione competitiva dei recettori della prolattina o dei recettori estrogeni o dell’inibita produzione di questi.
Applicazione nelle preparazioni atletiche
Date le prima citate caratteristiche, il Fluoxymesterone è stato per un lungo periodo di tempo utilizzato in ambito Culturistico e nel Powerlifting. Nel BodyBuilding questo AAS ha visto il suo più largo uso nelle 4-6 settimane precedenti alla gara in abbinamento con una dieta adeguata, grazie alla sua capacità di migliorare la consistenza muscolare e aumentare l’attività mentale (aggressività, resistenza al dolore e alla fatica). Poiché questo farmaco non aromatizza e raramente il suo uso causa ritenzione idrica, funziona piuttosto bene per questo scopo. I powerlifters invece, interessati al forte potenziale androgeno del Fluoxymesterone, usano questa molecola prettamente per aumentare la forza e, di conseguenza, i carichi.
L’avvento degli inibitori dell’aromatase e la maggior disponibilità del Trenbolone hanno tolto parte dei vantaggi pratici derivanti dall’uso di questo prodotto (specie in pre-gara), ma grazie alla sua scarsa affinità per i recettori androgeni (AR) mantiene ancora una certa popolarità in pre-gara e nei cicli di forza in associazione con prodotti spiccatamente affini per i recettori androgeni come il Trenbolone.
Con il dilagare del fenomeno “Crossfit”, il Fluoxymesterone ha trovato una nuova applicazione sportiva oltre alle prima citate categorie (Bodybuilder e Powerlifter). Infatti, questo AAS si presta perfettamente a questa disciplina fornendo all’atleta una maggiore forza, concentrazione e resistenza mentale e fisica. Dal grande potenziale per gli atleti avanzati è lo “stack” Fluoxymesterone, Trenbolone e GW1516.
I dosaggi di Fluoxymesterone mediamente usati dagli atleti di sesso maschile sono tra i 20-40mg/die per un periodo compreso tra le 3 e le 6 settimane; il dosaggio viene diviso in 2-3 assunzioni eguali al giorno. L’emivita del Fluoxymesterone è di circa 6-8 ore. Dati i suoi effetti collaterali a carico del fegato, la sua assunzione non dovrebbe mai superare le 6 settimane totali.
La sua reperibilità nel mercato nero non è così grande, e la presenza di prodotti contenenti altra o nessuna molecola e spacciati per Fluoxymesterone non è una rarità. Gli effetti collaterali che il Fluoxymesterone può portare comprendono cefalea, nervosismo, ansia, oligospermia, iperattività sessuale, atrofia testicolare, carcinoma epatico, alterazione dei lipidi ematici e alopecia.
Conclusioni
Per concludere vorrei sottolineare il fatto che questa molecola non è adatta ai principianti (con meno di 2 cicli ben fatti alle spalle) ed ai soggetti sensibili a livello psicologico. Non abbinare mai il Fluoxymesterone e il Trenbolone prima di aver testato la tollerabilità singola di ognuna di queste due molecole. Il “fai da te” è come al solito caldamente sconsigliato. Per gli agonisti è importante sapere che il tempo di rilevabilità del Fluoxymesterone nei test anti-doping è di 2 mesi.
Gabriel Bellizzi
Riferimenti scientifici:
1. Hogg JA., Steroids, the steroid community, and Upjohn in perspective: a profile of innovation. Steroids 1992 Dec;57(12):593-616.
2. Martini L., Pecile A., Hormonal Steroids Vol. 2, pp. 59-67. New York: Academy Press 1965
3. Fried J., Sabo EF., Synthesis of 17α-hydroxycorticosterone and its 9α-halo derivates from 11-epi-17α- hydroxycorticosterone. J. Am. Chem. Soc., 1953, 75 (9), pp 2273–2274
4. Lyster et al., Androgenic and myotrophic properties of orally administered 9-FLUORO-Il-OXY- METHYLTESTOSTERONES. Endocrinology Vol. 58, pp. 781-785 1956
5. Dorfman RI., Kincl FA., Relative Potency of Various Steroids in an Anabolic-Androgenic Assay Using the Castrated Rat. Endocrinology Vol. 72(2):259-266 1963.
6. Beyler et al., The ratio of anabolic to androgenic activity of 7:17-dimethyltestosterone, oxymesterone, mestanolone and fluoxymesterone. J Endocrinol December 1, 1963 28 87-92.
7. Beyler et al., Influence of molecular unsaturation on hormonal activity pattern ofcertain heterocyclic steroids. Endocrinology Vol. 68(6), pp. 987-995 1961.
8. Proc. 1st Int. Congr. Hormonal Steroids Vol. 2, pp. 119-132. New York: Academic Press, 1965.
9. Robinson et al., New Anabolic Agents: 9α,11β-Dihalogenoandrostane Derivatives. J. Am. Chem. Soc., 1960, 82 (17), pp 4611–4615.
10. Patrick TB., Fluoro-organic biochemistry. Journal of Chemical Education, vol. 56(4) p. 228, 1979.
11. Vida JA., Androgens and anabolic agents; chemistry and pharmacology. New York: Academic Press, 1969, p.63.
12. Bondi A., van der Waals Volumes and Radii. J. Phys. Chem., 1964, 68 (3), pp 441–451
13. Rowland RS., Taylor R., Intermolecular Nonbonded Contact Distances in Organic Crystal Structures: Comparison with Distances Expected from van der Waals Radii. J. Phys. Chem., 1996, 100 (18), pp 7384–7391.
14. Fürstenberger et al., The anabolic androgenic steroid fluoxymesterone inhibits 11β-hydroxysteroid dehydrogenase 2-dependent glucocorticoid inactivation. Toxicol Sci. 2012 Apr;126(2):353-61.
15. Fried et al., 16-Methylated Steroids. III. The Synthesis of 9α – Fluoro -16α,17α- dimethyl -4- androstene-11β,17β- diol-3-one. J. Org. Chem., 1962, 27(2), pp. 682–684.
16. Hogg et al., Synthesis of potent oral anabolic-androgenic steroids. J. Am. Chem. Soc., 1956, 78 (2), pp 500–501
17. Lu et al., Mass spectrometric identification and characterization of new fluoxymesterone metabolites in human urine by liquid chromatography time-of-flight tandem mass spectrometry. Steroids. 2012 Jul; Vol. 77(8-9):871-877. ®
18. Kammerer et al., Testing for fluoxymesterone (Halotestin metabolites by gas chromatography-mass spectrometry. J Steroid Biochem. 1990 Aug 28; Vol. 36(6):659-66.
19. Dorfman et al., Biosynthesis of Estrogens. Endocrinology. 1962 Dec; Vol. 71:920-925.
20. Knudsen JF., Max SR., Aromatization of androgens to estrogens mediates increased activity of glucose 6- phosphate dehydrogenase in rat levator ani muscle. Endocrinology. 1980 Feb; Vol. 106(2):440-443.
21. Schänzer et al., 17-Epimerization of 17 alpha-methyl anabolic steroids in humans: metabolism and synthesis of 17 ) administration to man: Identification of urinary alpha-hydroxy-17 beta-methyl steroids. Steroids. 1992 Nov; Vol. 57(11):537-550.
Il succo di pompelmo è una bevanda conosciuta da tutti e di facile reperibilità. Il suo contenuto di vitamina C, sali minerali, acido citrico e pectine è ciò che lo rende una bevanda salutare e di largo consumo tra gli amanti del Fitness. Ma le propietà del succo di pompelmo non finiscono qui: infatti, esso possiede un altra caratteristica, questa volta utile per gli atleti supplementati chimicamente. Questa caratteristica consiste nel aumentare la biodisponibilità dei farmaci co-somministrati (come gli AAS orali).
Questa capacità emerse già nel 1989, quando casualmente il succo di pompelmo fu utilizzato come componente aggiuntivo di un placebo in un test farmacologico in Canada. (1) Nel gruppo al quale venne somministrato il placebo con succo di pompelmo, la concentrazione plasmatica di felodipina (farmaco vasodilatatore utilizzato per l’ipertensione) risultò più elevata del previsto.
Successivamente furono svolti diversi studi a riguardo grazie ai quali è stato possibile scoprire che il pompelmo interferisce con numerosissimi farmaci tra cui le statine, gli anti-aritmici, gli agenti immunosoppressori ed i bloccanti del canale del calcio .
Ma qual’è il meccanismo d’azione alla base di qiesta caratteristica?
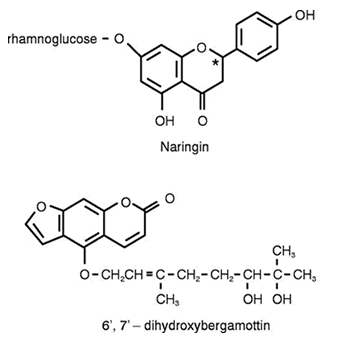
Gli studi condotti sul succo di pompelmo hanno evidenziato che alcune componenti di questo frutto, come il bergamottino (2) e la naringina (3) sono potenti inibitori del citocromo P450 3A4 (CYP3A4).
Questo enzima, espresso a livello intestinale ed epatico, è il principale attore responsabile della detossificazione dell’organismo da circa il 50% dei farmaci attualmente esistenti.
L’interazione del farmaco con il succo di pompelmo avviene principalmente per inibizione del CYP3A4 intestinale, ciò causa un più lento smaltimento del farmaco e una sua prolungata permanenza nell’organismo. Per quesa ragione, a parità di dosaggio, la co-somministrazione di un farmaco orale con succo di pompelmo ne aumenta le concentrazioni ematiche rispetto al farmaco preso singolarmente (con aumento del rischio di incorrere in sovradosaggio).
Struttura del P450 3A4 (CYP3A4)
Dal momento che l’inibizione del CYP3A4 avviene a livello intestinale, le interazioni farmaco-succo del pompelmo si manifestano unicamente con formulazioni orali. Studi effettuati su farmaci metabolizzati dal CYP3A4 epatico, somministrati per via endovenosa, hanno dimostrato che il succo di pompelmo non ne modifica i livelli plasmatici (4).
Il CYP3A4 metabolizza il farmaco fino a ridurne la biodisponibilità del 15%. Il pompelmo, invece, inibendo l’enzima aumenta la biodisponibilità del farmaco del 45%.
I livelli intestinali di CYP3A4 possono essere ridotti del 47% entro un paio d’ore dall’assunzione di pompelmo. (4)
E’stato anche dimostrato che l’interazione farmacologica può verificarsi con l’assunzione di modeste quantità di succo di pompelmo o frutto fresco (250 gr). Questa inattivazione enzimatica può perdurare per un lungo periodo (circa 3 giorni).
Esiste una grossa variabilità biologica d’espressione del CYP3A4 intestinale (polimorfismo genetico), di conseguenza risulta difficile riuscire a predire con certezza, da un soggetto a un altro, una possibile interazione farmacologica che coinvolga il succo di pompelmo. (5,6)Comunque sia, più è ridotta la percentuale del CYP3A4 maggiore sarà la biodisponibilità del farmaco co-somministrato.
E’ giusto sottolineare che l’enzima CYP3A4 è responsabile della deattivazione delle sostanze in un processo che gli scienziati chiamano 6-beta-idrossilazione. Farmaci che sono sensibili a questo processo enzimatico vengono rapidamente eliminati dal corpo.
Il CYP3A4 subisce l’inibizione dovuta dal pompelmo solo nel piccolo intestino. L’enzima si trova anche nel fegato, ma il pompelmo non influisce a quel livello. Meno CYP3A4 nell’intestino tenue significa quindi che un variegato gruppo di sostanze è più facilmente assorbita dal corpo.
Il pompelmo è in grado di inibire un’altra proteina: la P-glicoproteina o P-gp. La P-gp si trova anche nel piccolo intestino e diminuisce anche l’assorbimento di sostanze farmacologiche.
Nella figura si può osservare l’effetto attraverso il grafico. Il grafico mostra la concentrazione del medicinale lovastatina in rapporto al CYP3A4 dopo aver bevuto un quantitativo di acqua o di succo di pompelmo.
I ricercatori hanno affermato che il massimo effetto si raggiunge dopo aver bevuto un bicchiere da 250 ml di succo di pompelmo. Quattro ore dopo l’assunzione, il 47% dell’enzima CYP3A4 era disattivato. Dodici ore dopo aver bevuto il succo, l’effetto era ancora piuttosto ottimale. Ventiquattro ore dopo, l’effetto era ridotto ad un terzo.
L’azione del succo di pompelmo non è ancora del tutto chiara. La vecchia teoria affermava che l’azione era dovuta dalla naringina e dal suo metabolita naringenina. I test di laboratorio non hanno confermato questo però. Un’altra teoria è che il furanocourmarin bergamottina e il suo metabolita 6 ‘, 7’, Dihydroxybergamottin causino l’inibizione, ma in prove di laboratorio l’effetto è stato solo lieve. Ci sono probabilmente diversi composti fitochimici in gioco, e tutti contribuiscono all’effetto.
Una delle conclusioni dei ricercatori è stata che gli utilizzatori di farmaci CYP3A4-sensibili farebbero meglio ad evitare il succo di pompelmo o il frutto intero. I dosaggi di questi farmaci non sono basati su un possibile miglioramento della biodisponibilità dato dal pompelmo, con il rischio che gli utilizzatori possano incorrere in effetti collaterali più severi.
I ricercatori hanno affermato che, in futuro, una volta che l’effetto del pompelmo sarà meglio compreso, potrà essere possibile aggiungere i fitocomposti ai principi attivi dei farmaci in modo che i dosaggi, ed i costi di produzione dei produttori, possano essere abbassati.
La rilevanza di questa scoperta per gli atleti aiutati chimicamente sta nel fatto che alcuni steroidi anabolizzanti orali sono soggetti all’azione del CYP3A4. Nella metà degli anni novanta, Wilhelm Schaenzer pubblicò uno studio sulla beta-idrossilazione del Testosterone, del Boldenone, del Methyltestosterone, del Fluoxymesterone , del Methandrostenolone e del Chlorodehydromethyltestosterone, somministrati oralmente in soggetti di prova.
Schaenzer esaminò i metaboliti nelle urine dei soggetti del suo studio. Scoprì che la 6-beta-idrossilazione del Boldenone, Testosterone e del Methyl Testosterone era stata trascurabile, ma era importante per quanto riguardava l’azione a carico del Chlorodehydromethyltestosterone, del Methandrostenolone e del Fluoxymesterone. Tra il 17 e il 46% di questi ormoni era stato espulso dall’organismo nella forma 6-beta-idrossilata.
Per concludere, il succo di pompelmo può essere utilizzato per aumentare la biodisponibilità degli AAS orali, specie se non metilati in C-17, contribuendo così ad un effetto maggiore con un dosaggio più contenuto.
Gabriel Bellizzi
Riferimenti:
1- Bailey DG, Spence JD, Edgar B, Bayliff CD, Arnold JM. Ethanol enhances the hemodynamic effects of felodipine. Clin Invest Med. 1989;12:357–62.
2- Edwards DJ, Bellevue FH, Woster PM. Identification of 6′,7′-dihydroxybergamottin, a cytochrome P450, in grapefruit juice. Drug Metab Dispos. 1996;24:1287–1290.
3- Kuhnau J. The flavonoids: a class of semi-essential food components; their role in human nutrition. World Rev Nutr Diet. 1976;24:117–191
4- Dahan A, Altman H. Food-drug interaction: grapefruit juice augments drug bioavailabilitymechanism,extent and relevance. Eur J Clin Nutr 2004; 58: 1-9.
5- Dresser GK, Bailey DG. The effects of fruit juices on drug disposition: a new model for druginteractions. Eur J Clin Invest 2003; 33: 10-6.
6- Huang SM, et al. for the Center for Drug Evaluation and Research and Office of Regulatory Affairs,U.S. Food and Drug Administration. Drug interactions with herbal products and grapefruit juice: a conference report. Clin Pharmacol Ther 2004; 75: 1-12.
Androgenico: 50 Anabolico: 100 Standard: Testosterone Nome chimico: 1,4-androstadiene-3-one,17beta-ol, 1-dehydrotestosterone Attività Estrogenica: bassa Attività Progestinica: non disponibile (bassa)
Il Boldenone (INN, BAN) [1,4-androstadiene-3-one,17b-ol],(commercializzato con il nome di Equipoise, Ganabol, Equigan, Ultragan, e Boldane), è uno steroide anabolizzante-androgeno spesso legato all’estere undecylenato. Strutturalmente molto simile al Testosterone, il Boldenone differisce da questo per il raddoppio del legame tra C1 e C2.
La Ciba depositò il brevettato del Boldenone nel 1949. Nel corso degli anni ’50 e ’60, l’azienda ha sviluppato diversi esteri sperimentali di questo farmaco, rilasciando in seguito una versione esterificata della molecola con una lunga durata d’azione: Boldenone undecylenato. Sarebbe stato venduto con il marchio Parenabol, probabilmente in riferimento alle sue caratteristiche come agente anabolizzante parenterale (iniettabile). Il Parenabol vide un certo uso clinico durante la fine degli anni ’60 e primi anni ’70, principalmente come agente anabolizzante in soggetti sotto peso o in pazienti soggetti a malattie cataboliche, e per il mantenimento della massa ossea nell’osteoporosi. Il Boldenone undecylenato come farmaco per uso umano ebbe una breve durata venendo interrotto a livello globale per tale scopo entro la fine del 1970. La Squibb sarebbe stata l’introduttrice di questa molecola nel mercato veterinario, sotto il conosciutissimo marchio Equipoise.
Nel mercato veterinario, il Boldenone undecylenato è più comunemente applicato ai cavalli, anche se in molte regioni è indicato per l’uso su altri animali. Con il suo uso si presenta in genere un marcato effetto sul peso corporeo magro, sull’appetito, e la disposizione generale dell’animale. Il marchio Equipoise è stato venduto dalla Squibb fino al 1985, quando quest’ultima venne acquisita dalla Solvay U.S. . L’Equipoise sarebbe stato venduto sotto l’etichetta Solvay negli anni successivi, fino a quando la Wyeth ha acquisito la divisione salute degli animali della Solvay nel 1995. La divisione è stata formata in Fort Dodge Animal Health, che continua a commercializzare attualmente l’Equipoise negli Stati Uniti e in alcuni mercati esteri. Esistono molti altri nomi generici commerciali per il Boldenone undecylenato in numerosi mercati farmaceutici internazionali, dal momento che i brevetti sul Boldenone undecylenato sono da tempo scaduti.
Come accennato in precedenza, il Boldenone non è altro che Testosterone con un raddoppio del legame tra C1 e C2, caratteristica che:
1- Riduce il tasso di aromatizzazione a circa la metà di quello del Testosterone;
2- rende la molecola un substrato molto meno affine all’enzima 5-alfa reduttasi rispetto al Testosterone; questo aspetto riduce molto la conversione del Boldenone a Diidroboldenone, rendendolo androgeno circa la metà del Testosterone (rapporto androgeno/anabolico del Boldenone è 50/100);
Differenze strutturali tra Boldenone e Testosterone
Nonostante non ci siano dati sperimentali sull’affinità recettoriale del Boldenone, in base alla sua struttura e alla conversione molto limitata a Diidroboldenone, si suppone che l’affinità recettoriale della molecola sia “Mix” a livello muscolare come quella del Testosterone. C’è chi afferma che il Boldenone abbia una affinità AR inferiore a quella del Testosterone, speculando che esso possa essere classificato come “Non-AR”.
Il Boldenone condivide con il Testosterone anche una forte affinità per le SHBG.
Il Boldenone è privo di qualsiasi tipo di metilazione cosa che lo rende sensibilmente meno efficace per via orale, ma meno di quello che ci si potrebbe aspettare: il doppio legame in C1-C2 incrementa leggermente la resistenza al passaggio epatico.
Il Boldenone aromatizza ad estradiolo ad un tasso del 50% rispetto al testosterone. Per questo motivo il Boldenone è considerato uno steroide leggermente estrogenico. A riprova di ciò studi sull’aromatizzazione suggeriscono che il tasso di conversione in estradiolo di questa molecola è di circa la metà di quella del Testosterone.(1) La tendenza a sviluppare effetti collaterali estrogenici evidenti con il Boldenone dovrebbe essere leggermente superiore a quanto accada con il Nandrolone, ma molto inferiore rispetto al Testosterone. Effetti collaterali estrogenici non sono di solito pronunciati a meno che questo farmaco non venga assunto in dosi superiori a 200-400 mg a settimana. Un inibitore dell’aromatasi, come l’Anastrozolo, può essere utile in soggetti con spiccata tendenza all’aromatizzazione.
Anche se il Boldenone è classificato come un androgeno mite, gli effetti collaterali androgeni sono ancora comuni con questa molecola, in particolare con dosi più elevate. Questo può includere pelle grassa, acne, crescita di peli sul corpo e sul viso, e alopecia adrogenetica . Le donne dovrebbero essere messe in guardia sui potenziali effetti virilizzanti. Questi possono includere voce profonda, irregolarità mestruale, cambiamenti nella struttura della pelle, crescita di peli sul viso, e allargamento del clitoride.
Anche se il Boldenone si riduce ad un androgeno più potente (Dihydroboldenone) tramite l’enzima 5-alfa riduttasi nei tessuti bersaglio androgeno-sensibile come la pelle, il cuoio capelluto e la prostata, la sua affinità di farlo nel corpo umano è estremamente bassa.(2) La androgenicità relativa del Boldenone non è, quindi, significativamente influenzata dal finasteride o dal dutasteride.
Il Boldenone non è un metilato in c-17 , e non sono noti effetti epatotossici. Ciò nonostante la possibilità che questa molecola possa causare danni epatici temporanei non è da escludersi.(3)
Come risaputo, gli AAS possono avere effetti deleteri sul colesterolo sierico. Questo include una tendenza alla riduzione delle concentrazioni di colesterolo HDL (buono) e un aumento delle concentrazioni di colesterolo LDL (cattivo), cosa che comporta uno sbilanciamento dell’equilibrio HDL/LDL che si traduce in un rischio maggiore di sviluppare arteriosclerosi. L’impatto relativo all’assunzione di un AAS nei confronti dei lipidi ematici dipende dalla dose, dalla via di somministrazione (per via orale o iniettabile), dal tipo di steroide (aromatizable o non aromatizable), e dal livello di resistenza al metabolismo epatico. Il Boldenone ha un impatto meno drammatico sui fattori di rischio cardiovascolari rispetto agli AAS orali metilati. Ciò è dovuto in parte alla sua blanda resistenza al metabolismo epatico, che permette di avere un effetto minore sulla gestione epatica del colesterolo. L’aromatizzazione del Boldenone in Estradiolo, seppur inferiore a quella del Testosterone, può anche contribuire ad attenuare gli effetti negativi degli androgeni sui lipidi del siero. Gli AAS possono anche influenzare negativamente la pressione del sangue e i livelli dei trigliceridi, riducendo il rilassamento endoteliale, e promuovendo l’ipertrofia ventricolare sinistra, tutti fattori con un potenziale nel aumentare il rischio di malattie cardiovascolari e infarto del miocardio.
Per contribuire a ridurre lo sforzo cardiovascolare si consiglia di mantenere un programma di esercizio cardiovascolare attivo e di ridurre al minimo l’assunzione di grassi saturi, colesterolo e carboidrati semplici in ogni momento durante la somministrazione di AAS.
Con l’uso di Boldenone bisogna prestare particolare attenzione all’ematocrito in quanto questo AAS possiede una spiccata azione sulla eritropoiesi.
La supplementazione con oli di pesce (4 grammi al giorno) e un integratore alimentare di Niacina per il controllo del colesterolo è anche raccomandata.
Tutti gli AAS se assunti in dosi sufficienti per promuovere l’aumento della massa muscolare causano una soppressione del Testosterone endogeno. Senza l’intervento con sostanze Testosterone-stimolante, e una adeguata PCT, i livelli di Testosterone dovrebbero tornare alla normalità entro 1-4 mesi dalla cessione del farmaco. Si noti che un ipogonadismo ipogonadotropo prolungato può svilupparsi secondariamente all’abuso di steroidi, cosa che richiede un intervento medico.
I dosaggi medi di Boldenone utilizzati in ambito maschile si aggirano tra i 250-500 mg (5-10ml, per la versione da 50 mg/ml) a settimana. La posologia può essere ulteriormente divisa per ridurre il volume di ogni iniezione, se necessario, ad esempio somministrando il farmaco due o tre volte alla settimana. Si dovrebbe anche prestare attenzione a ruotare i siti di iniezione regolarmente, in modo da evitare irritazioni o infezioni.
In ambito femminile le dosi medie si aggirano tra i 50-75 mg a settimana. Le donne dovrebbero prestare attenzione alla caratteristica ad azione lenta di questa molecola, che rende i livelli ematici difficili da controllare e lenti a declinare in caso i sintomi virilizzanti si presentino.
Il Boldenone (nella sua forma classica, Undecylenato) è noto per essere una molecola lenta che garantisce un lento ma costante aumento di forza e massa muscolare di qualità. Gli effetti positivi di questo farmaco diventano più evidenti quando viene usato in cicli più lunghi, di solito della durata di 6-8 settimane o più.
Le caratteristiche del Boldenone fanno si che trovi le sue migliori associazioni con AAS di tipo AR e/o in grado di abbassare i livelli di SHBG. Molecole come il Metyldrostanolone (Superdrol) e il Trenbolone rappresentano una scelta ideale. Anche un eventuale associazione con potenti antagonisti delle SHBG come Oral-Turinabol, Winstrol e Oxandrolne rappresentano una buona scelta anche se c’è chi afferma che ciò creerebbe una competizione per l’affinità recettoriale (su questo ho delle riserve). L’uso di un AI con il Boldenone potrebbe essere problematico essendo il Boldenone già poco aromatizzabile e l’Estradiolo noto per la sua mancata proliferazione dei recettori androgeni.
Il Boldenone undecylenato rimane ampiamente disponibile come prodotto medicinale veterinario. Viene prodotto principalmente in America, costantemente alla dose di 25 mg / ml o 50 mg / mL. Un piccolo numero di preparazioni sono fatte ad un dosaggio più alto (in genere 200 mg / ml), soprattutto da parte delle imprese in mercati meno regolamentati dell’Asia dove l’offerta è spesso dettata dalla domanda del mercato nero. Nel mercato delle UGL è emerso un formato di Boldenone a breve durata d’azione, si tratta di Boldenone Propionato.
Il Boldenone acquistato al mercato nero può essere identificato positivamente mediante test di sostanze ROIDTEST ™ B & C. Dopo le recenti tendenze del mercato, troviamo che i preparativi del mercato nero etichettatati come “Boldenone” hanno un alto rischio di non contenente lo stesso o di contenere altri steroidi.
Il test kit ROIDTEST ™ , che può essere utilizzato per confermare la presenza di questo AAS in un prodotto, può essere acquistato qui:
I BodyBuilder più previdenti conoscono l’importanza dell’inserimento degli epatoprotettori nella propria preparazione, la quale contempla l’utilizzo di AAS, specie se metilati in C-17. Tra i composti più utilizzati troviamo la Silimarina e la N-Acetil-cisteina (NAC), facilmente reperibili in quanto prodotti OTC.
Ovviamente, esistono sostanze farmacologiche da prescrizione le quali vengono utilizzate dagli atleti aiutati chimicamente come disintossicanti epatici. Tra queste troviamo il Glutatione, l’Acido Tauroursodesossicolico Biidrato e l’acido ursodesossicolico. Ed è proprio quest’ultimo a dimostrare la sua efficacia nel lavoro scientifico che riporto qui di seguito.
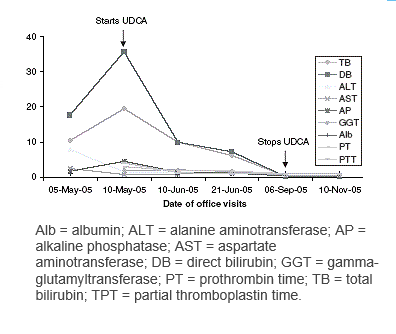
Medici messicani sono riusciti a curare un Bodybuilder con danno epatico utilizzando proprio l’acido ursodesossicolico (UDCA). L’acido ursodesossicolico è un acido biliare secondario che deriva dal metabolismo dell’acido colico da parte del microbiota umano intestinale. Il suo nome deriva dal fatto che è il principale acido biliare negli orsi (dal latino ursus). In biologia e biochimica lo si etichetta con l’acronimo UDCA. Il nome completo del UDCA è Acido 3α,7β-diidrossi-5β-colanoico.
L’atleta in questione era un BodyBuilder di 29 anni che soffriva di mal di stomaco da due mesi. Due settimane prima di andare dal medico aveva inoltre sviluppato ittero e prurito. Aveva perso quattordici chili.
I medici riscontrarono il danno epatico tramite apposite analisi. L’abuso di AAS aveva causato al Bodybuilder la colestasi, una condizione nella quale i condotti nel fegato si infiammano e, pertanto, non è possibile rimuovere la bile e convogliarla al duodeno. La Bile rimuove il colesterolo dal corpo.
Prima di ammalarsi, il Bodybuilder aveva seguito un ciclo di tre mesi composto da 25mg/die di Proviron(Mesterolone), 40mg/die di Andriol (Testosterone Undecanoato orale), 30mg/die di Deca(Nandrolone Decanoato), 50mg/die di Oxymetholone e 800 mg di Testosterone . [Strano ma è riportato “nero su bianco” – NdR.]
I medici, dopo aver fatto interrompere l’assunzione delle suddette molecole al Bodybuilder, diedero al soggetto una dose di 15mg per kg di peso corporeo di acido ursodesossicolico al giorno. L’UDCA rimuove gli acidi biliari tossici dal fegato – e quindi stimola il recupero dei condotti attraverso i quali il fegato invia l’acido biliare al duodeno nell’intestino[http://www.ncbi.nlm.nih.gov/pubmed.]. Ci sono voluti un paio di mesi, ma la funzionalità epatica del culturista è stata recuperata.
Androgenico: non disponibile Anabolico: >100-180 Standard: Methyltestosterone (orale) Nome chimico: 4-chloro-17a-methyl-17b-hydroxyandrosta-1,4-dien-3-one Attività Estrogenica: nessuna Attività Progestinica: non disponibile (bassa)
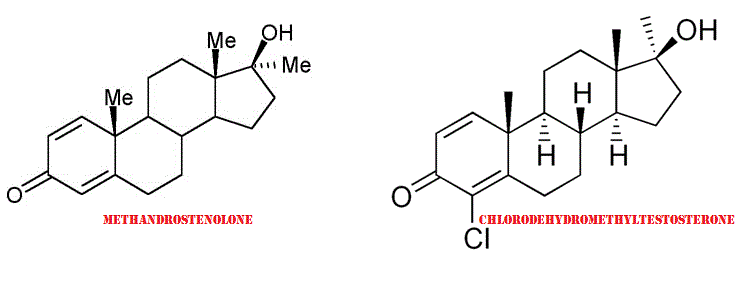
Il Chlorodehydromethyltestosterone (CDMT) [ (8R,9S,10R,13S,14S,17S)-4-chloro-17-hydroxy-10,13,17-trimethyl-7,8,9,11,12,14,15,16-octahydro-6H-cyclopenta[a]phenanthren-3-one ], (nome commerciale Oral Turinabol), è uno steroide androgeno-anabolizzante derivato del Methandrostenolone(Dehydromethyltestosterone) (Dianabol) dal quale differisce per l’aggiunta di un gruppo 4-cloridrico nel nucleo steranico dell’anello A.
Il Chlorodehydromethyltestosterone venne descritto per la prima volta nel 1962.(1) Jenapharm (Jena, Germania) rilasciò questa molecola come farmaco in vendita nella Germania dell’Est, con il marchio Oral Turinabol. Il farmaco è stato favorito dai clinici per la sua natura altamente anabolizzante e blandamente androgena, cosa che rendeva questo farmaco favorevole per l’utilizzo non solo maschile ma anche femminile e nei bambini. Il farmaco venne prodotto in due dosaggi, contenenti 1 mg e 5 mg di principio attivo per compressa, in modo che una versione a basso dosaggio fosse disponibile per i soggetti più sensibili. Il Chlorodehydromethyltestosterone è stato applicato per un certo numero di usi medici; soprattutto quelli incentrati sulla costruzione o il mantenimento del tessuto muscolare e della massa ossea.
L’Oral Turinabol è diventato uno steroide molto discusso nel corso del 1990, quando è stato rivelato che il Clorodeidrometiltestosterone era stato uno dei segreti tenuti gelosamente all’interno della “Macchina doping della Germania dell’Est.” Questo si riferisce al programma doping sponsorizzato dallo stato, chiamato “Staatsplanthema 14.25“, che è stato operativo nella Germania dell’Est tra il 1974 e il 1989. Fu un programma aggressivo di somministrazione di anabolizzante, progettato con un solo obiettivo nella mente: barare al test anti doping olimpico. In molti casi, gli atleti olimpici, sia maschi che femmine, sono stati i protagonisti inconsapevoli di questo “inganno”, semplicemente perché i loro preparatori gli somministravano “vitamine”. Molte di queste “vitamine azzurre” si rivelarono essere Oral Turinabol, un potente e non rilevabile ( all’epoca ) steroide anabolizzanti. Ben 10.000 atleti hanno ricevuto steroidi anabolizzanti durante il periodo di attività del programma, molti di loro assunsero Oral Turinabol. Per uno sguardo più approfondito a questo particolare evento storico, comprese le prove della partecipazione di diversi ex funzionari tedeschi dell’Est, vi consiglio di leggere il libro “Faust’s Gold: Inside the East German Doping Machine” di Steven Ungerleider.
A dispetto di un attività record e di un favorevole margine di sicurezza, la Jenapharm interruppe la produzione di Oral Turinabol nel 1994. Ciò è avvenuto in un momento in cui una grande quantità di attenzione negativa veniva data al doping nello sport, dando credibilità alla speculazione che non collegava questa decisione a problemi finanziari o a preoccupazioni per la salute legate al farmaco. Indipendentemente da ciò, la Jenapharm venne acquisita dalla Schering AG (Germania) nel 1996, una società con nessun interesse a rivivere le controversie del passato (la Schering aveva già interrotto la produzione di molti dei suoi prodotti contenenti steroidi anabolizzanti). Ne prima ne dopo, nessun altro marchio di Clorodeidrometiltestosterone è esistito come farmaco da prescrizione. Oggi, questo agente è ancora disponibile, ma è prodotto solo dal mercato nero delle UGL.
L’aggiunta di un gruppo 4-cloridrico nel nucleo steranico dell’anello A rende questa molecola del tutto non aromatizzabile, mentre, come ben sappiamo, il D-Bol, dal quale deriva, aromatizza a circa il 60% del Testosterone, ma a differenza di quest’ultimo aromatizza al ben più attivo metabolita metil estradiolo. Teoricamente, sebbene non sia 5-alfa ridotto, si suppone che il Clorodeidrometiltestosterone dovrebbe avere una affinità maggiore per i recettori androgeni; non esistono test specifici in proposito, ma si è a conoscenza del fatto che la 5-alfa riduzione non aumenta la stabilità del keto- gruppo in C-3 responsabile dell’affinità per i recettori androgeni, mentre la metilazione in C- 17 aumenta la stabilità generale per l’affinità recettoriale.
Differenze nella struttura molecolare tra Methandrostenolone e Chlorodehydromethyltestosterone.
Esistono altre differenze tra Methandrostenolone e Clorodeidrometiltestosterone, non legate in modo specifico all’aggiunta del gruppo 4-cloridrico. Una differenza di questo tipo è la mancanza di azione soppressiva del Cortisolo, capacità che contraddistingue il D-Bol, per lo meno nelle prime settimane di assunzione, prima che si manifesti l’effetto rebound dell’ACTH.
Molto probabilmente questo è il motivo della minore potenza anabolica/anti-catabolica dell’ Oral Turinabol:
Methandrostenolone 90/210
Clorodeidrometiltestosterone 180
In linea teorica questa differenza dovrebbe diminuire con il passare delle settimane, mentre il rebound dell’ACTH si acutizza.
Come ben sappiamo, tutti gli AAS alchilati mostrano una certa capacità di limitare la produzione epatica di SHBG. L’Oral Turinabol presenta una capacità di decrescita delle SHBG pari al 60% già dopo 2 settimane al dosaggio di 10 mg alla settimana (mentre per il Winstrol si necessita di 30 mg alla settimana per avere lo stesso effetto).
Il valore androgeno del Clorodeidrometiltestosterone è ufficialmente classificato come “0”, anche se ciò sembra legato alla dose; sembra mostrare androgenicità rilevabile a partire da 20 mg.
Il Clorodeidrometiltestosterone presenta una spiccata propensione fibrinolitica, ovvero la capacità di disgregare le piastrine e conseguente aumento del “pro-time” (tempo di coagulazione). Dal momento che tutti gli AAS hanno proprietà fibrinolitiche con andamento dose dipendente (finiscono per inibire la fibrinolisi con il rialzo dei dosaggi) questa caratteristica del Oral Turinabol non rappresenta un problema quando co-somministrato con altri AAS. Con dosaggi consistenti di Clorodeidrometiltestosterone si può verificare un attenuazione della diminuzione del pro-time rispetto ad altre molecole. Si consideri il fatto che una fibrinolisi troppo lenta può essere causa della formazione di coaguli arteriosi e venosi, potenzialmente mortali.(2, 3)
Come già accennato, il Clorodeidrometiltestosterone non è soggetto ad aromatizzazione, e non presenta una attività estrogenica misurabile. Non è quindi necessario l’utilizzo di un anti-estrogeno quando si utilizza questo steroide da solo, ed effetti collaterali come la ginecomastia non dovrebbero essere una preoccupazione anche tra gli individui sensibili. Con l’uso di Oral Turinabol non si verifica ritenzione idrica sottocutanea. Questo lo rende uno steroide favorevole da utilizzare durante i cicli di “Cut”, quando la ritenzione idrica e l’accumulo di grasso sono le maggiori preoccupazioni.
Essendo il Clorodeidrometiltestosterone un composto C17-alfa alchilato, caratteristica che conferisce al farmaco una protezione dalla disattivazione epatica, permettendo ad una percentuale molto elevata del farmaco di entrare nel flusso ematico dopo somministrazione orale, l’esposizione prolungata o ad alte dosi può causare danni al fegato. In rari casi può svilupparsi una disfunzione epatica pericolosa per la vita. Si consiglia quindi di tenere monitorata la funzionalità epatica e la salute generale durante l’uso. L’assunzione di AAS c17-alfa alchilati dovrebbe essere comunque limitata alle 6-8 settimane, nel tentativo di evitare un eccessivo stress epatico. L’uso di un integratore disintossicante epatico come il Liv-52 o l’Essentiale Forte è consigliato durante l’assunzione di AAS epatotossici.
Come risaputo, gli AAS possono avere effetti deleteri sul colesterolo sierico. Questo include una tendenza alla riduzione delle concentrazioni di colesterolo HDL (buono) e un aumento delle concentrazioni di colesterolo LDL (cattivo), cosa che comporta uno sbilanciamento dell’equilibrio HDL/LDL che si traduce in un rischio maggiore di sviluppare arteriosclerosi. L’impatto relativo all’assunzione di un AAS nei confronti dei lipidi ematici dipende dalla dose, dalla via di somministrazione (per via orale o iniettabile), dal tipo di steroide (aromatizable o non aromatizable), e dal livello di resistenza al metabolismo epatico. Il Clorodeidrometiltestosterone ha un forte effetto sulla gestione epatica del colesterolo a causa della sua resistenza strutturale durante il passaggio epatico, dalla sua natura non aromatizable, e per la via di somministrazione. Gli AAS possono anche influenzare negativamente la pressione del sangue e i livelli dei trigliceridi, riducendo il rilassamento endoteliale, e promuovendo l’ipertrofia ventricolare sinistra, tutti fattori con un potenziale nel aumentare il rischio di malattie cardiovascolari e infarto del miocardio.
Per contribuire a ridurre lo sforzo cardiovascolare si consiglia di mantenere un programma di esercizio cardiovascolare attivo e di ridurre al minimo l’assunzione di grassi saturi, colesterolo e carboidrati semplici in ogni momento durante la somministrazione di AAS.
La supplementazione con oli di pesce (4 grammi al giorno) e un integratore alimentare di Niacina per il controllo del colesterolo è anche raccomandata.
Ricerche hanno mostrato una riduzione del Testosterone endogeno del 30% in seguito alla somministrazione di 20 mg al giorno di Chlorodehydromethyltestosterone per 4 settimane. Sembra anche che dopo 5 giorni il livello del Testosterone torni ai livelli iniziali, e dopo 7 giorni il livello del Testosterone risulta superiore ai livelli iniziali del 5-10% mantenendosi a questo livello per altri 8-10 giorni.(4) Ciononostante è bene ricordare che tutti gli AAS se assunti in dosi sufficienti per promuovere l’aumento della massa muscolare causano una soppressione del Testosterone endogeno. Senza l’intervento con sostanze Testosterone-stimolante, e una adeguata PCT, i livelli di Testosterone dovrebbero tornare alla normalità entro 1-4 mesi dalla cessione del farmaco. Si noti che un ipogonadismo ipogonadotropo prolungato può svilupparsi secondariamente all’abuso di steroidi, cosa che richiede un intervento medico.
Anche per il Clorodeidrometiltestosterone se ne consiglia l’assunzione lontano dai pasti in quanto studi hanno dimostrato che l’assunzione di uno steroide anabolizzante orale con cibo può diminuirne la sua biodisponibilità.(5) Questo è causato dalla natura liposolubile degli ormoni steroidei, che può permettere ad una parte del farmaco di sciogliersi con i grassi alimentari non digeriti, riducendone di conseguenza l’assorbimento dal tratto gastrointestinale. Per la massima biodisponibilità , questo steroide deve essere assunto a stomaco vuoto.
Una dose clinica comune di Oral Turinabol per i soggetti di sesso maschile è stimato intorno ai 5 mg al giorno; le linee guida effettive prescrittive non sono disponibili. In ambito sportivo, un efficace dose giornaliera orale rientra nel range dei 15-40 mg, assunto in cicli della durata di non più di 6-8 settimane per minimizzare l’effetto epatotossico. Anche se può essere somministrato con incrementale efficacia anche a dosaggi molto alti, personalmente non andrei oltre i 50mg/die (anche perché è un AAS tipicamente da principiante/intermedio). Comunque sia, il dosaggio prima esposto è sufficiente per aumenti di massa magra e forza verificabili. L’Oral Turinabol trova un forte favore tra gli atleti di sport dove la velocità tende ad essere un obiettivo primario, ottenendo un forte vantaggio anabolizzante senza dover portare in giro acqua e/o grasso addizionale.
Una dose clinica comune di Oral Turinabol per i soggetti di sesso femminile è stimate intorno a 1-2,5 mg al giorno; anche in questo caso le linee guida prescrittive effettive non sono disponibili. In ambito sportivo, le donne avrebbero comunemente bisogno di una sola compressa da 5 mg al giorno, presa in cicli della durata di non più di 4-6 settimane per ridurre al minimo l’epatotossicità. Effetti virilizzanti sono improbabili a questo dosaggio livello. Dosi molto più elevate sono state spesso utilizzate con atleti di sesso femminile nel precedentemente esposto programma di doping della DDR, ma spesso a discapito di forti effetti collaterali virilizzante. Comunque, la dose ideale per una donna si attesta tra i 5mg e non oltre i 20mg/die.
L’emivita del Chlorodehydromethyltestosterone orale è di 8 ore (vita attiva 16 ore); ciò garantisce la possibilità di assumere 2 dosi giornaliere distanziate da un intervallo temporale di 8 ore. Esiste anche una versione iniettabile di Chlorodehydromethyltestosterone del mercato nero con una vita attiva di circa 48-72 ore.
L’ Oral Turinabol trova le sue migliori applicazioni in abbinamento con anabolizzanti aromatizzabili e non particolarmente affini al legame con le SHBG, come il Testoterone (con caratteristiche recettoriali Mix) il Boldenone (con caratteristiche recettoriali Non –ar/Mix) e anche il Primobolan, notoriamente soggetto fortemente al legame con le SHBG.
Come già detto l’Oral Turinabol è stato disponibile come farmaco da prescrizione in Germania (l’unico paese dove è stato prodotto per la maggior parte della sua storia) fino al 1994. Un numero molto piccolo di aziende farmaceutiche hanno commercializzato il farmaco dal momento che, soprattutto nei mercati meno regolamentati dell’Europa orientale e dell’Asia , la domanda del mercato nero influenza ancora la produzione.
Il Chlorodehydromethyltestosterone può essere identificato positivamente mediante test di sostanze ROIDTEST ™ B & C. Dopo le recenti tendenze del mercato, troviamo che i preparativi del mercato nero etichettatati come “Clorodeidrometiltestosterone” hanno un alto rischio di non contenente lo stesso o di contenere altri steroidi. Nonostante le polveri di Chlorodehydromethyltestosterone abbiano un costo contenuto, alcune UGL possono sostituire questo AAS con Methandrostenolone o Metyltestosterone.
ROIDTEST ™ steroidi test kit, che può essere utilizzato per confermare la presenza di questo AAS in un prodotto, può essere acquistato qui:
2- [Activation of the fibrinolytic system with dehydrochlormethyltestosterone] Folia Haematol Int Mag Klin Morphol Blutforsch. 1984;111(4):556-62. German.
3- [Modification of hypofibrinolytic states by dehydrochlormethyltestosterone] Folia Haematol Int Mag Klin Morphol Blutforsch. 1984;111(4):563-6. German.
4- “The pharmacokinetics of Oral-Turinabol in humans” Pharmazie. 1991 Sep;46(9):650-4. German. Department of Urology, Universitaetsklinikum “Carl Gustav Carus,” Technical University of Dresden,Dresden, Germany
5- Anabolic Steroids and Sports Volume II. James E. Wright. Sports Science Consultants, Natick, MA 1982.
A causa della campagna sociale contro il Testosterone e gli steroidi anabolizzanti in generale, risulta difficile anche solo trovare uno studio approvato sull’uso del Testosterone negli uomini. La difficoltà aumenta vertiginosamente quando se ne cerca uno sull’uso del Testosterone nelle donne. Fortunatamente qualcosa c’è. In particolare esiste u…no studio che può aiutare a capire l’importanza di una TRT anche in ambito femminile, dove si sottolinea il potenziale degli androgeni per aiutare le donne a perdere grasso.
Nel 1996, Lovejoy et ali hanno confrontato gli effetti del Nandrolone Decanoato e del farmaco antiandrogeno Spironolattone sulla composizione corporea nelle donne obese postmenopausali (1). La dose data al gruppo Nandrolone era bassa: 30 mg ogni due settimane. Tutte le donne dello studio hanno seguito una dieta con calorie ridotte (500 calorie al di sotto del mantenimento della massa magra) senza modificare le loro abitudini fisiche. Dopo nove mesi le donne che ricevevano il Nandrolone hanno perso in media il 3,6% del loro grasso corporeo mentre il gruppo placebo ha perso solo l’1,8% e il gruppo dello Spironolattone (antiandrogeno) ha perso solo lo 0,5%. Il Nandrolone ha raddoppiato il tasso di riduzione del grasso in confronto al placebo e il gruppo antiandrogeno non ha praticamente perso grasso: il ruolo degli androgeni nella perdita di grasso è chiaramente dimostrato anche nelle donne. Cosa ancora più sorprendente, nonostante la restrizione calorica il gruppo del Nandrolone ha addirittura guadagnato in media 1,8 kg di massa magra, mentre i gruppi placebo e antiandrogeno hanno perso oltre 1 kg di massa magra. Inoltre, il Nandrolone non ha prodotto resistenza all’insulina, cosa che precedentemente gli androgeni erano considerati fare.
Il gruppo di Lovejoy è rimasto sorpreso dalla capacità del Nandrolone di aumentare la massa muscolare nonostante la complessiva perdita di peso. Teniamo sempre a mente che la dose era piuttosto piccola e somministrata solo ogni due settimane e che queste donne seguivano solo una dieta con calorie ridotte senza svolgere un programma di allenamento con i pesi. Immaginatevi il miglioramento della composizione corporea che si sarebbe verificato se queste donne avessero seguito un programma di attività fisica e di dieta ben strutturati con un ricco apporto proteico oltre al Nandrolone!
Nonostante il risultato positivo, gli autori hanno però evidenziato alcuni problemi con l’uso del Nandrolone Decanoato. Nel gruppo del Nandrolone si è osservata una leggera anormalità dei lipidi ematici e un leggero, e paradossale, incremento del grasso addominale. Anche se questi effetti collaterali sono stati contenuti, è probabile che se invece del Nandrolone si opti per il classico Testosterone o Boldenone, questi effetti collaterali sarebbero più bassi o inesistenti grazie alla loro maggiore biocompatibilità.
Anche un uso quotidiano di Testosterone in gel sarebbe più efficace di una dose 3 volte al mese perché il gel terrebbe i livelli di Testosterone a un livello più fisiologico e costante rispetto alle iniezioni (parliamo sempre di ambito TRT). Solitamente le dosi di Testosterone in gel nelle donne si aggirano intorno a 0,25-1 mg ogni due giorni al mattino applicati, per ragioni di assorbimento, in zone nascoste non esposte al sole nell’area del collo dietro la mascella oppure sotto l’ascella. La dose cambia nel corso del tempo.
Dal momento che molte donne sulla trentina già presentano difficoltà a perdere grasso a causa dei bassi livelli di Testostosterone, cosa che notoriamente comincia 10-15 anni prima della menopausa con conseguente aumento di grasso, una semplice TRT ben strutturata garantisce un aiuto fondamentale per il miglioramento della composizione corporea.
Gabriel Bellizzi
Riferimenti scientifici:
(1)Lovejoy, et al, Exogenous androgens influence body composition and regional body fat distribution in obese postmenopausal women-a clinical research center study, J Clin Endocrinol Metab. 1996 Jun;81(6):2198-203