DISCLAIMER: Il presente articolo è a solo scopo educativo, di intrattenimento e informativo. Non rappresenta in alcun modo una forma di incitamento all’uso/abuso di sostanze dopanti. L’autore ed il sito, per tanto, è esentato da qualsiasi responsabilità dipendente dalla libera scelta individuale.
Introduzione:

L’uso dell’eritropoietina (EPO) per migliorare le prestazioni atletiche, soprattutto nel ciclismo agonistico, è una questione controversa da oltre vent’anni. Nonostante la sua diffusione e le controversie che ne derivano, mancano ancora prove scientifiche solide che ne dimostrino l’efficacia nel migliorare le prestazioni dei ciclisti ben allenati.
Jules Heuberger e il suo team del Centre for Human Drug Research nei Paesi Bassi si sono posti l’obiettivo di affrontare scientificamente proprio questa domanda: L’EPO migliora effettivamente le prestazioni dei ciclisti esperti? Hanno condotto uno studio in doppio cieco, randomizzato e controllato con placebo – considerato il “gold standard” della ricerca scientifica – con dosaggi di EPO che rispecchiano l’uso reale nel ciclismo agonistico. Questo articolo ne condivide i risultati.
Ma partiamo con ordine…
Caratteristiche e azioni dell’Eritropoietina (EPO)

Nel 1905, Paul Carnot propose l’idea che un ormone regolasse la produzione di globuli rossi. Dopo aver condotto esperimenti su conigli sottoposti a salasso, Carnot e la sua studentessa laureata Clotilde-Camille Deflandre[1] attribuirono un aumento dei globuli rossi nei conigli trattati a un fattore emotropico chiamato emopoietina. Eva Bonsdorff e Eeva Jalavisto chiamarono la sostanza emopoietica “eritropoietina”. K.R. Reissman e Allan J. Erslev hanno dimostrato che una certa sostanza, circolante nel sangue, è in grado di stimolare la produzione di globuli rossi e di aumentare l’ematocrito. Questa sostanza è stata purificata e confermata come eritropoietina.[2][3]

Nel 1977, Goldwasser e Kung hanno purificato l’EPO.[4] L’EPO pura ha permesso di identificare parzialmente la sequenza aminoacidica e di isolare il gene.[2] L’EPO sintetica è stata utilizzata per la prima volta con successo per correggere l’anemia nel 1987.[5] Nel 1985, Lin et al. hanno isolato il gene dell’eritropoietina umana da una libreria genomica di fagi e l’hanno utilizzato per produrre l’EPO.[6] Nel 1989, la Food and Drug Administration (FDA) statunitense ha approvato l’ormone Epogen per l’uso in alcune anemie.[7][8]
Gregg L. Semenza e Peter J. Ratcliffe hanno studiato il gene dell’EPO e la sua regolazione ossigeno-dipendente. Insieme a William Kaelin Jr. hanno ricevuto il Premio Nobel 2019 per la Fisiologia o la Medicina per la loro scoperta del fattore inducibile dell’ipossia (HIF), che regola il gene dell’EPO, così come altri geni, in risposta all’ipossia.[9]
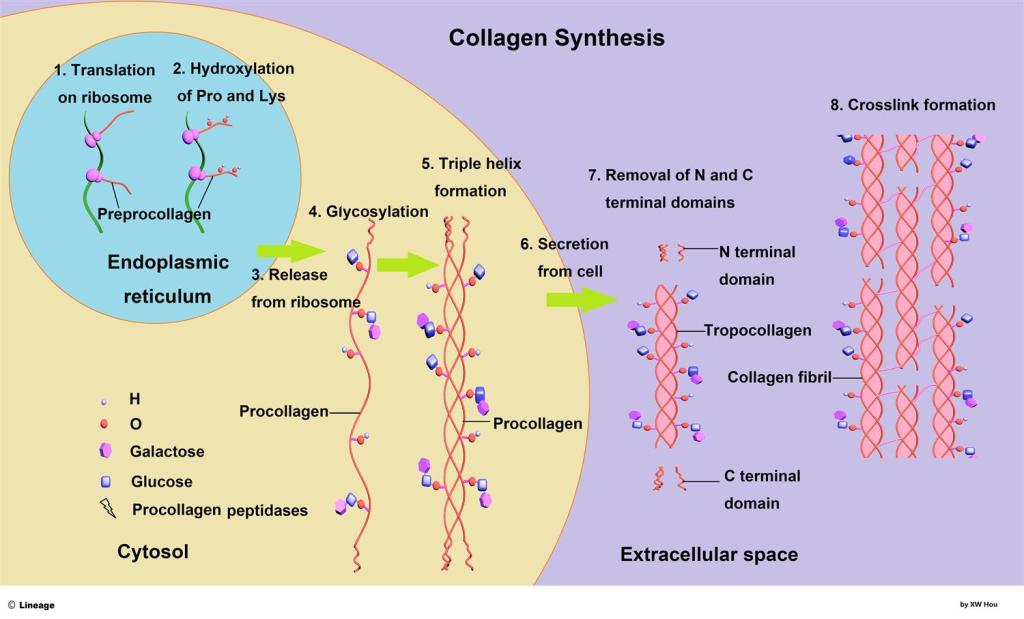
L’Eritropoietina (/ɪˌrɪθroʊˈpɔɪ. ɪtɪn, -rə-, -pɔɪˈɛtɪn, -ˈiːtɪn/; [10][11][12] EPO), nota anche come eritropoetina, ematopoietina o emopoietina, è una citochina glicoproteica secreta principalmente dai reni in risposta all’ipossia cellulare; stimola la produzione di globuli rossi (eritropoiesi) nel midollo osseo. Bassi livelli di EPO (circa 10mU/mL) sono costantemente secreti in quantità sufficiente a compensare il normale ricambio dei globuli rossi. Le cause comuni di ipossia cellulare che determinano livelli elevati di EPO (fino a 10.000mU/mL) comprendono qualsiasi anemia e l’ipossiemia dovuta a malattie polmonari croniche e alla bocca.
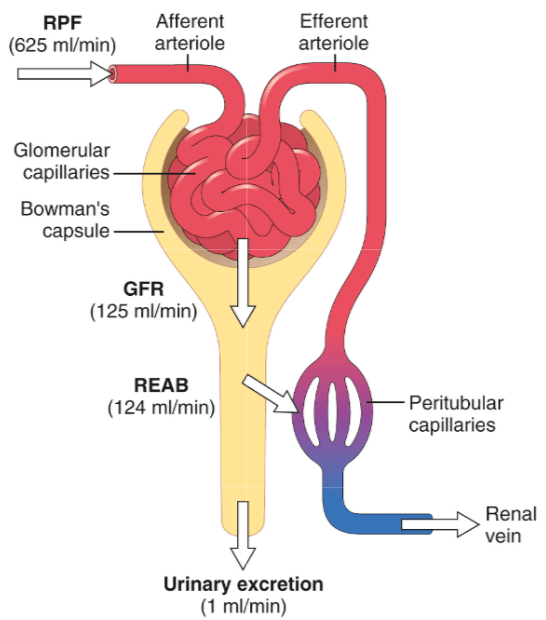
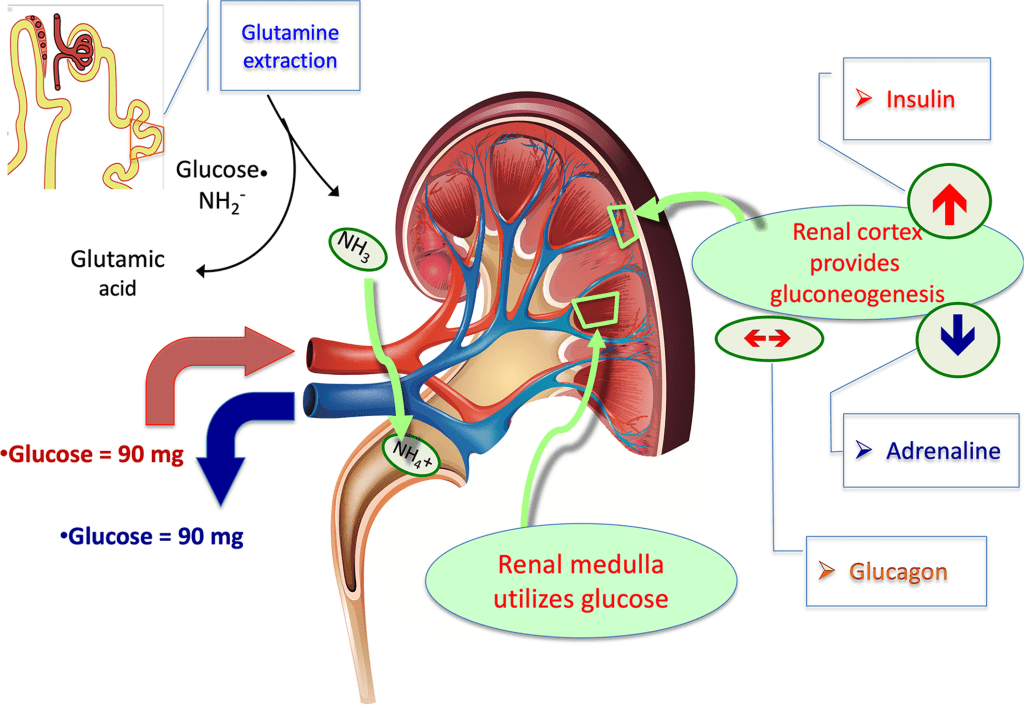
L’Eritropoietina è prodotta dai fibroblasti interstiziali del rene in stretta associazione con il capillare peritubulare e il tubulo contorto prossimale. Viene prodotta anche nelle cellule perisinusoidali del fegato. La produzione epatica predomina nel periodo fetale e perinatale; la produzione renale predomina nell’età adulta. È omologa della trombopoietina.
L’Eritropoietina esogena, l’Eritropoietina umana da DNA ricombinante (rhEPO), viene prodotta con la tecnologia del DNA ricombinante in coltura cellulare e viene chiamata collettivamente agenti stimolanti l’eritropoiesi (ESA): due esempi sono l’epoetina alfa e l’epoetina beta. Gli ESA sono utilizzati nel trattamento dell’anemia nella malattia renale cronica, dell’anemia nella mielodisplasia e dell’anemia da chemioterapia oncologica. I rischi della terapia includono morte, infarto miocardico, ictus, tromboembolismo venoso e recidiva del tumore. Il rischio aumenta quando il trattamento con EPO aumenta i livelli di emoglobina oltre 11g/dL fino a 12g/dL: questo è da evitare.
Come accennato, i livelli di eritropoietina nel sangue sono piuttosto bassi in assenza di anemia, circa 10mU/mL. Tuttavia, in caso di stress ipossico, la produzione di EPO può aumentare fino a 1000 volte, raggiungendo 10.000mU/mL di sangue. Negli adulti, l’EPO è sintetizzata principalmente dalle cellule interstiziali nel letto capillare peritubulare della corteccia renale, con quantità aggiuntive prodotte nel fegato,[13][14][15] e nei periciti del cervello.[16] Si ritiene che la regolazione si basi su un meccanismo di feedback che misura l’ossigenazione del sangue e la disponibilità di ferro.[17] I fattori di trascrizione per l’EPO sintetizzati costitutivamente, noti come fattori inducibili dall’ipossia, sono idrossilati e digeriti proteosomicamente in presenza di ossigeno e ferro. Durante la normossia, GATA2 inibisce la regione promotrice dell’EPO. I livelli di GATA2 diminuiscono durante l’ipossia e permettono di promuovere la produzione di EPO.[18]
La produzione di eritropoietina può essere indotta da HIF-2α e da PGC-1α.[19] L’eritropoietina attiva anche questi fattori, dando luogo a un ciclo di feedback positivo.[19]
È stato dimostrato che l’eritropoietina esercita i suoi effetti legandosi al recettore dell’eritropoietina (EpoR).[20][21] L’EPO si lega al recettore dell’eritropoietina sulla superficie dei progenitori dei globuli rossi e attiva una cascata di segnalazione JAK2. Questo avvia le vie di STAT5, PIK3 e Ras MAPK. Ciò determina la differenziazione, la sopravvivenza e la proliferazione delle cellule eritroidi.[22] Vengono inoltre espressi SOCS1, SOCS3 e CIS, che agiscono come regolatori negativi del segnale delle citochine.[23]
L’espressione del recettore dell’eritropoietina ad alto livello è localizzata nelle cellule progenitrici eritroidi. Sebbene sia stato riferito che i recettori dell’EPO si trovano in una serie di altri tessuti, come il cuore, il muscolo, il rene e il tessuto nervoso periferico/centrale, questi risultati sono confusi dalla non specificità dei reagenti, come gli anticorpi anti-EpoR.[24] In esperimenti controllati, un recettore funzionale dell’EPO non viene rilevato in questi tessuti.[25] Nel flusso sanguigno, gli stessi globuli rossi non esprimono il recettore dell’eritropoietina, quindi non possono rispondere all’EPO. Tuttavia, è stata segnalata una dipendenza indiretta della longevità dei globuli rossi nel sangue dai livelli plasmatici di eritropoietina, un processo definito neocitolisi.[26] Inoltre, vi sono prove inconfutabili che l’espressione del recettore dell’EPO è regolata in modo elevato nelle lesioni cerebrali.[27]
L’eritropoietina è un ormone essenziale per la produzione di globuli rossi. Senza di essa, l’eritropoiesi definitiva non ha luogo. In condizioni di ipossia, il rene produrrà e secernerà eritropoietina per aumentare la produzione di globuli rossi, mirando alle sottopopolazioni di CFU-E, proeritroblasti ed eritroblasti basofili nella differenziazione. L’eritropoietina ha un effetto primario sui progenitori e sui precursori dei globuli rossi (che si trovano nel midollo osseo degli esseri umani), promuovendo la loro sopravvivenza attraverso la protezione di queste cellule dall’apoptosi, o morte cellulare.
L’eritropoietina è il fattore eritropoietico primario che coopera con vari altri fattori di crescita (ad esempio, IL-3, IL-6, glucocorticoidi e SCF) coinvolti nello sviluppo della linea eritroide da progenitori multipotenti. Le cellule eritroidi a formazione di unità di esplosione (BFU-E) iniziano a esprimere il recettore per l’eritropoietina e sono sensibili all’eritropoietina. Lo stadio successivo, l’unità formante colonie eritroidi (CFU-E), esprime la massima densità di recettori per l’eritropoietina ed è completamente dipendente dall’eritropoietina per l’ulteriore differenziazione. Anche i precursori dei globuli rossi, i proeritroblasti e gli eritroblasti basofili, esprimono il recettore dell’eritropoietina e ne sono quindi influenzati.
È stato riferito che l’eritropoietina ha una serie di azioni che vanno oltre la stimolazione dell’eritropoiesi, tra cui l’ipertensione dipendente dalla vasocostrizione, la stimolazione dell’angiogenesi e la promozione della sopravvivenza cellulare attraverso l’attivazione dei recettori dell’EPO, con conseguenti effetti anti-apoptotici sui tessuti ischemici. Questa proposta è tuttavia controversa, in quanto numerosi studi non hanno dimostrato alcun effetto.[28] È inoltre incoerente con i bassi livelli di recettori dell’EPO su queste cellule. Gli studi clinici condotti su esseri umani con tessuti ischemici cardiaci, neurali e renali non hanno dimostrato gli stessi benefici osservati negli animali. Inoltre, alcuni studi hanno dimostrato un effetto neuroprotettivo sulla neuropatia diabetica, ma questi dati non sono stati confermati da studi clinici condotti sui nervi peroneo profondo, peroneo superficiale, tibiale e surale.[29]
Come sappiamo, le eritropoietine disponibili come agenti terapeutici sono prodotte con la tecnologia del DNA ricombinante in coltura cellulare e comprendono Epogen/Procrit (epoetina alfa) e Aranesp (darbepoetina alfa); sono utilizzate per il trattamento dell’anemia derivante da malattie renali croniche,[30] dell’anemia indotta dalla chemioterapia in pazienti affetti da cancro, da malattie infiammatorie intestinali (morbo di Crohn e colite ulcerosa)[31] e da mielodisplasia dovuta al trattamento del cancro (chemioterapia e radiazioni). I foglietti illustrativi includono avvertenze relative all’aumento del rischio di morte, infarto del miocardio, ictus, tromboembolismo venoso e recidiva del tumore, in particolare quando viene utilizzato per aumentare i livelli di emoglobina a più di 11g/dL – 12g/dL.[32]
L’EPO è altamente glicosilata (40% del peso molecolare totale), con un’emivita nel sangue di circa 5 ore. L’emivita dell’EPO può variare tra le versioni endogene e quelle ricombinanti. L’ulteriore glicosilazione o altre alterazioni dell’EPO attraverso la tecnologia ricombinante hanno portato a un aumento della stabilità dell’EPO nel sangue (richiedendo così iniezioni meno frequenti).
EPO come PEDs
Come farmaco per il miglioramento delle prestazioni, l’EPO è stato vietato dall’inizio degli anni ’90, ma un primo test non è stato disponibile fino alle Olimpiadi estive del 2000. Prima che questo test fosse disponibile, alcuni atleti sono stati sanzionati dopo aver confessato di aver fatto uso di EPO, ad esempio nel caso Festina, quando fu trovata un’auto con prodotti dopanti per la squadra ciclistica Festina.
A questo punto, però, è necessario comprendere le questioni di fondo relative all’uso dell’EPO nello sport e, in particolare, il suo impatto sulle prestazioni ciclistiche.
Molti atleti agonisti e osservatori ritengono che l’EPO migliori le prestazioni atletiche aumentando la produzione di globuli rossi e l’apporto di ossigeno ai muscoli. Come abbiamo visto precedentemente, infatti, la rHuEPO clinica viene utilizzata per trattare l’anemia, aumentando il numero di globuli rossi nei pazienti, il che implica che potrebbe fare lo stesso per gli atleti. Questa logica ha contribuito al suo status di sostanza vietata dalla maggior parte delle agenzie antidoping, a partire proprio dal Comitato Olimpico Internazionale nel 1990.
Tuttavia, nonostante questa convinzione diffusa e il suo uso illecito tra gli atleti, è difficile trovare prove scientifiche concrete a sostegno dei suoi effetti di miglioramento delle prestazioni nei ciclisti ben allenati.
Mentre molti si concentrano sul potenziale dell’EPO di aumentare la massa dei globuli rossi e la capacità di trasportare ossigeno, altri, più previdenti, ritengono che il suo uso improprio possa portare a gravi effetti negativi. Sono state rilasciate innumerevoli dichiarazioni relative a complicazioni cardiovascolari come ipertensione, trombosi e aumento del rischio di ictus, nonché a disturbi ematologici come la policitemia.
Inoltre, alcuni avvertono che la somministrazione o il dosaggio improprio dell’EPO possono provocare uno squilibrio nella produzione di globuli rossi, portando a livelli pericolosi di ematocrito e viscosità, che a loro volta possono aumentare il rischio di coaguli di sangue e altri eventi cardiovascolari. Tuttavia, come abbiamo detto in questa serie, i presunti pericoli dell’EPO nel ciclismo di prestazione sono stati oggetto di analisi e studi, che hanno dimostrato che le agenzie sportive si sono spesso basate su affermazioni non comprovate.
Sebbene alcuni considerino l’EPO tra i migliori farmaci per il miglioramento delle prestazioni nel ciclismo, esistono studi che ne confermano la potenziale pericolosità.
Sebbene l’entità dei rischi associati all’uso dell’eritropoietina (EPO) negli atleti rimanga incerta, i dati provenienti da revisioni della letteratura e da studi condotti su soggetti sani e allenati forniscono indicazioni sui potenziali pericoli.
- Pressione sanguigna sistolica
In uno studio, i ricercatori hanno notato un notevole aumento della pressione arteriosa sistolica, sia a riposo che durante l’esercizio submassimale, in seguito alla somministrazione di EPO. L’aumento della pressione arteriosa può predisporre gli atleti a complicazioni cardiovascolari, tra cui l’ipertensione e l’aumento del rischio di eventi trombotici.
- Eventi trombotici
Le evidenze delle revisioni della letteratura evidenziano anche un’elevata incidenza di eventi trombotici nei pazienti trattati con dosi elevate di rHuEPO rispetto a quelli che ricevono un placebo. Tuttavia, è importante notare che questi studi hanno tipicamente utilizzato dosi significativamente superiori a quelle comunemente utilizzate negli studi sulle prestazioni di resistenza. Fattori come l’aumento della viscosità del sangue, l’aumento della coagulazione, l’attivazione endoteliale, la reattività piastrinica e l’infiammazione possono contribuire a questi eventi avversi.
- Impatto con l’esercizio fisico
È inoltre importante considerare i cambiamenti fisiologici indotti dall’esercizio fisico acuto. Questi impatti includono riduzioni del volume plasmatico e del volume sanguigno accompagnate da un aumento dell’ematocrito, potenzialmente in grado di esacerbare il rischio di eventi trombotici negli atleti di resistenza, soprattutto in condizioni di disidratazione e ipertermia.
- Non conclusività delle prove
Sebbene questi risultati suggeriscano potenziali pericoli associati all’uso di EPO negli atleti, mancano prove conclusive. Non è possibile trarre conclusioni definitive senza studi di ricerca ben progettati che analizzino specificamente gli effetti dell’EPO sulle prestazioni e sulla sicurezza dei ciclisti d’élite.
Heuberger et al. [33] hanno progettato uno studio randomizzato, in doppio cieco e controllato con placebo per affrontare le incertezze che circondano gli effetti e la sicurezza dell’uso dell’EPO in ciclisti ben allenati. Questo studio si propone di analizzare in modo rigoroso l’impatto di NeoRecormon, una forma di eritropoietina umana ricombinante sintetica, sulle prestazioni e sui parametri di sicurezza nei ciclisti d’élite.
Lo studio si è concentrato sugli effetti su ciclisti ben allenati, comprendendo 48 soggetti sani e stabili dal punto di vista medico, reclutati tramite pubblicità sui media e associazioni ciclistiche. Questi soggetti sono stati selezionati per rappresentare l’élite del ciclismo, con un alto livello di forma fisica ed esperienza di allenamento. Il protocollo dello studio prevedeva un periodo di 8 settimane durante il quale i partecipanti sarebbero stati assegnati in modo casuale a ricevere NeoRecormon o un placebo.
NeoRecormon è stato somministrato a dosi di 2000, 5000 o tra 6000 e 10.000 UI alla settimana. L’obiettivo era quello di raggiungere l’intervallo prefissato, con aggiustamenti necessari in base ai risultati dell’emoglobina (Hb) o dell’ematocrito (Ht).
Lo studio ha avuto una durata totale di 129 giorni, con un periodo di trattamento di 8 settimane. Questo lasso di tempo ha permesso di valutare in modo completo le prestazioni e la sicurezza dopo la somministrazione di NeoRecormon. Prima e dopo il periodo di intervento sono state condotte valutazioni dettagliate delle metriche di prestazione, tra cui resistenza, potenza e utilizzo dell’ossigeno.
- Obiettivi primari
L’obiettivo dello studio era esplorare gli effetti di NeoRecormon sulle prestazioni ciclistiche in ciclisti ben allenati. L’obiettivo è stato raggiunto con diversi mezzi, tra cui valutazioni separate delle prestazioni in test da sforzo, in condizioni di gara e la misurazione dei marcatori ematologici tramite il Passaporto Biologico dell’Atleta. Sono state effettuate anche misurazioni del flusso sanguigno per valutare le risposte fisiologiche alla somministrazione di EPO.
- Obiettivi secondari
Gli obiettivi secondari comprendevano un’ulteriore esplorazione degli effetti del NeoRecormon in un contesto di gara su strada, per facilitare la determinazione della sua capacità tra gli integratori e i farmaci che migliorano le prestazioni ciclistiche. Altri obiettivi comprendevano una valutazione completa del suo profilo di sicurezza in ciclisti ben allenati e una valutazione dei metodi di rilevamento del doping per l’uso del NeoRecormon.
- I risultati dello studio
La valutazione della sicurezza del trattamento con rHuEPO in ciclisti ben allenati ha rivelato risultati rassicuranti. I segni vitali come il peso, la frequenza cardiaca e la pressione sanguigna erano simili tra i due gruppi di trattamento, mentre gli eventi avversi osservati erano da lievi a moderati e comparabili tra i gruppi. In particolare, non sono stati segnalati eventi avversi gravi (di grado 3 o peggiore) in nessuno dei due gruppi.
Mentre alcuni marcatori della funzione endoteliale hanno mostrato un leggero aumento con il trattamento con rHuEPO, suggerendo un potenziale aumento della trombogenicità, non ci sono stati segni clinici di effetti avversi associati alla somministrazione di rHuEPO.
In termini di miglioramento delle prestazioni, il trattamento con rHuEPO ha portato a miglioramenti nei test di laboratorio di esercizio massimale, con conseguente aumento della resistenza e delle prestazioni. Tuttavia, i suoi effetti sui test di esercizio submassimale e sulle prestazioni nelle corse su strada non sono stati rilevabili. Nel complesso, i risultati dello studio sono stati meno pronunciati rispetto alle affermazioni spesso riportate nella letteratura popolare e nei resoconti aneddotici. Ciò sottolinea l’importanza di una ricerca basata sull’evidenza per valutare l’efficacia e la sicurezza di interventi di miglioramento delle prestazioni come l’EPO.
L’assenza di effetti significativi di miglioramento delle prestazioni osservati nello studio può essere attribuita a diversi fattori:
- Differenze contestuali
Gli effetti dell’EPO possono essere più pronunciati in eventi a più tappe come il Tour de France, in cui la resistenza e il recupero giocano un ruolo critico, rispetto a gare di un solo giorno come quella del Mont Ventoux inclusa nello studio. La durata e l’intensità degli eventi possono influenzare la rilevabilità degli effetti dell’EPO.
- Dimensione dello studio e potenza statistica
La dimensione del campione dello studio potrebbe essere stata insufficiente per rilevare sottili differenze nei risultati delle prestazioni, in particolare nel contesto della corsa su strada. La complessità della misurazione delle prestazioni nelle competizioni ciclistiche reali e la variabilità inerente alle prestazioni dei singoli atleti possono aver limitato la potenza statistica dello studio nel rilevare effetti significativi.
- Entità ridotta dei benefici
È possibile che i benefici della rHuEPO sulle prestazioni, pur essendo presenti, siano minori di quanto si tende a sostenere o a credere, il che li rende difficili da distinguere in assenza di campioni più ampi o di tecniche di misurazione più sensibili.
Conclusioni sulla sicurezza dell’EPO e sul miglioramento delle prestazioni
Sulla base dei risultati dello studio controllato in doppio cieco, la sicurezza dell’uso dell’EPO per migliorare le prestazioni ciclistiche rimane un argomento di dibattito e di cautela. Sebbene non siano state osservate differenze significative negli eventi avversi tra i gruppi EPO e placebo, lo studio ha rivelato un aumento preoccupante dei marcatori endoteliali, in particolare E-selectina e P-selectina, associati a trombogenicità e infiammazione.
Questi risultati suggeriscono un potenziale aumento del rischio cardiovascolare associato al trattamento con rHuEPO, che potrebbe non essere stato adeguatamente colto a causa della bassa incidenza di eventi cardiovascolari negli atleti sani. La limitata potenza dello studio nel rilevare tali rischi sottolinea la necessità di ulteriori ricerche con campioni di dimensioni maggiori e periodi di follow-up più lunghi.
Dato l’uso diffuso e non controllato della rHuEPO tra gli atleti, non si può escludere il rischio potenziale di eventi cardiovascolari. Pertanto, sebbene l’EPO possa offrire benefici per le prestazioni, il suo uso deve essere affrontato con cautela e gli atleti devono essere consapevoli dei potenziali rischi associati alla sua somministrazione.
Per il futuro, è indispensabile condurre studi su larga scala con periodi di follow-up prolungati per valutare in modo completo la sicurezza e l’efficacia dell’uso dell’EPO nei ciclisti ben allenati. È necessario implementare politiche e regolamenti basati sull’evidenza per mitigare i potenziali rischi associati all’abuso di sostanze che migliorano le prestazioni, evitando consapevolmente di usare iperboli o esagerazioni per demonizzare una sostanza.
Comprendere il protocollo di dosaggio dell’EPO
I ricercatori del CHDR hanno progettato e attuato con cura un protocollo di dosaggio dell’EPO [33], con l’obiettivo di replicare le pratiche note nel ciclismo professionistico, garantendo al contempo la sicurezza dei partecipanti e il rispetto degli standard etici. I partecipanti assegnati al gruppo rHuEPO (eritropoietina umana ricombinante) hanno ricevuto otto dosi totali durante il periodo di studio.
Il regime di dosaggio prevedeva la somministrazione di una dose media di rHuEPO, sotto forma di NeoRecormon, di 5000 UI per partecipante a settimana durante le prime 4 settimane dello studio. Successivamente, la dose è stata aumentata a 7000 UI a settimana per le restanti 4 settimane. Per mitigare il rischio di parametri ematologici eccessivi, ai partecipanti che hanno superato un aumento del 15% dell’emoglobina rispetto al basale o che hanno raggiunto una concentrazione di ematocrito superiore al 52% sono state somministrate iniezioni di placebo in cinque occasioni.
La dose media di rHuEPO somministrata per tutto il periodo di studio è stata di 48.000 UI, pari a una media di 6000 UI a settimana. Questa strategia di dosaggio ha determinato un aumento sostanziale della concentrazione di emoglobina, con un incremento medio del 12% fino a 10,2 mmol/L, e un aumento del 16% dei livelli di ematocrito, che ha raggiunto il 50%. Al contrario, i partecipanti al gruppo placebo hanno mostrato concentrazioni di emoglobina ed ematocrito relativamente stabili per tutta la durata dello studio.
I diari dei partecipanti sono stati tenuti con una documentazione meticolosa, contribuendo a confermare l’aderenza al regime di integrazione prescritto per tutto il periodo dello studio, garantendo coerenza e affidabilità nella somministrazione delle dosi di rHuEPO.
In seguito, approfondiremo il razionale di questa strategia di dosaggio, le sue implicazioni per le prestazioni ciclistiche e il contesto più ampio delle linee guida per il dosaggio dell’EPO sotto controllo medico.
- Un esempio di protocollo di 8 settimane
Un esempio di protocollo di 8 settimane per la somministrazione di NeoRecormon potrebbe essere il seguente:
Settimana 1-4: Dosaggio di NeoRecormon da 2000 a 10.000 UI alla settimana (aggiustato in base alle misurazioni di Hb e Ht).
Settimana 5-8: continui aggiustamenti del dosaggio di NeoRecormon secondo le necessità, con regolare monitoraggio dei parametri ematologici.
Integrazione giornaliera: 50 mg di vitamina C e 200 mg di ferro per ottimizzare l’assorbimento e la salute generale.
- I dettagli del dosaggio di NeoRecormon spiegati
Il farmaco sperimentale utilizzato nello studio era l’eritropoietina umana ricombinante (rHuEPO) NeoRecormon, contenente il principio attivo Epoëtine beta. NeoRecormon è stato somministrato per via sottocutanea (nel tessuto adiposo sotto la pelle) ai partecipanti rispettando uno schema di dosaggio attentamente studiato, nella speranza di ottimizzare i parametri ematologici riducendo al minimo i rischi potenziali.
Il protocollo di dosaggio di NeoRecormon si è basato su uno schema decisionale completo, ideato per guidare i ricercatori nell’aggiustamento del dosaggio in base alle caratteristiche e alle risposte dei singoli partecipanti, in particolare alla concentrazione di emoglobina (Hb) e ai livelli di ematocrito (Ht). Questo albero decisionale è stato un aiuto visivo per facilitare il processo decisionale in tempo reale per quanto riguarda gli aggiustamenti durante il periodo di trattamento di 8 settimane.
L’albero decisionale delineava vari scenari basati sulle misurazioni di Hb e Ht prima di ogni somministrazione di NeoRecormon o placebo. Se l’Ht di un partecipante superava il 52%, indicando un alto rischio di complicazioni ematologiche, la somministrazione del dosaggio veniva prontamente interrotta per ridurre i rischi potenziali. Al contrario, se i livelli di Ht erano inferiori al 52%, l’albero decisionale indirizzava i ricercatori a valutare la concentrazione di Hb del partecipante per determinare il dosaggio appropriato di NeoRecormon.
Ecco le fasi dell’albero decisionale in dettaglio:
- Se il livello di Ht raggiungeva un valore superiore o uguale al 52%, il doping veniva interrotto. Se i livelli di Hb scendono al di sotto di circa 1,15x, anche il dosaggio si interrompe.
- Se i livelli di Hb erano superiori o uguali a 1,10x, il dosaggio sarebbe rimasto a 2000IU/settimana.
- Se la situazione non si fosse evoluta prima di 5 settimane, il dosaggio sarebbe arrivato a 5000IU/settimana; se la situazione non si fosse evoluta dopo 5 settimane o più, il dosaggio sarebbe stato aumentato a più o uguale a 6000IU/settimana, con un dosaggio massimo di 10.000IU/settimana.
Il dosaggio di NeoRecormon variava da 2.000 a 10.000 UI alla settimana, con la flessibilità di aggiustare questo intervallo in base alla risposta di ciascun partecipante al trattamento. Questo intervallo di dosaggio è stato concepito per garantire l’efficacia nell’innalzare i livelli di Hb e Ht all’interno dell’intervallo target, riducendo al minimo il rischio di effetti avversi.
Valutazione dei benefici e dei rischi
NeoRecormon è un farmaco registrato con un profilo di sicurezza noto, che lo rende adatto all’uso in contesti di ricerca. Tuttavia, i ricercatori hanno riconosciuto la possibilità di effetti collaterali, tra cui reazioni anafilattoidi, anche se con un basso tasso di incidenza di ≤1 su 10.000 casi. Per questo motivo, tutte le somministrazioni del farmaco in studio sono state condotte in un ambiente clinico sotto stretta supervisione medica, contribuendo a mitigare i rischi.
- Metodologia di monitoraggio
I partecipanti sono stati monitorati attentamente per tutta la durata dello studio e le loro condizioni mediche sono state valutate regolarmente per garantire la sicurezza e il benessere. Gli aggiustamenti del dosaggio sono stati effettuati, se necessario, in base ai parametri ematologici e alle risposte individuali al trattamento, guidati dall’algoritmo dell’albero decisionale.
Rispettando il protocollo di dosaggio prescritto, seguendo accuratamente l’albero decisionale e implementando rigorose misure di sicurezza, i ricercatori miravano a ottimizzare l’efficacia e la sicurezza della somministrazione di NeoRecormon nel migliorare le prestazioni ciclistiche dei partecipanti.
Valutazione della sicurezza e del rischio dell’uso di NeoRecormon
NeoRecormon, un’eritropoietina umana ricombinante (rHuEPO), è comunemente utilizzato in ambito clinico per il trattamento di diverse condizioni mediche, tra cui l’anemia associata a malattie renali croniche e alla chemioterapia del cancro. Il suo profilo di sicurezza è stato ampiamente studiato, con linee guida di dosaggio ben stabilite per garantire l’efficacia (in questi contesti medici) riducendo al minimo i rischi potenziali.
Il rischio più comunemente associato a NeoRecormon riguarda il suo potenziale aumento dei livelli di ematocrito e di emoglobina che, se elevati eccessivamente, possono portare a complicazioni come trombosi, ipertensione ed eventi cardiovascolari. Tuttavia, se somministrato entro gli intervalli di dosaggio raccomandati, NeoRecormon è generalmente considerato sicuro ed efficace per gli scopi medici previsti. La ricerca ha anche dimostrato che l’aumento delle prestazioni dell’EPO probabilmente non è così pericoloso come le organizzazioni antidoping vorrebbero far credere al pubblico.
Confronto con un programma di pre-donazione di sangue autologo
Gli effetti dell’uso di NeoRecormon nei pazienti che partecipano a programmi di pre-donazione di sangue autologo assomigliano molto a quelli dei volontari sani. In entrambi gli scenari, è stato riconosciuto un aumento della produzione di globuli rossi. Il Riassunto delle Caratteristiche del Prodotto (SmPC) per NeoRecormon in un programma di pre-donazione di sangue autologo specifica una dose massima raccomandata di 1200 UI/kg a settimana per somministrazione sottocutanea, equivalente a 90.000 UI per un individuo di 75 kg.
In questo protocollo di dosaggio dell’EPO, che prevede dosi pianificate da 2.000 a 10.000 UI alla settimana, i dosaggi di NeoRecormon sono ben al di sotto del limite massimo raccomandato. Pertanto, il rischio associato alla somministrazione di NeoRecormon nello studio è considerato piccolo e accettabile.
- Integrazione obbligatoria
Per sostenere gli effetti fisiologici della somministrazione di NeoRecormon e ridurre i rischi potenziali, ai partecipanti è stata prescritta un’integrazione giornaliera obbligatoria di 50 mg di vitamina C (acido ascorbico) e 200 mg di ferro (ferrofumarato) per tutto il periodo di trattamento di 8 settimane. Questi integratori contribuiscono a ottimizzare il metabolismo del ferro, l’eritropoiesi e la salute generale durante la terapia con EPO.
Supervisione medica e misure di Harm Reduction
La supervisione medica durante la somministrazione di NeoRecormon controlla da vicino i parametri ematologici, i segni vitali e lo stato di salute generale dei partecipanti, con controlli dettagliati eseguiti prima di ogni dose, il che significa 8 controlli durante tutto il processo. I partecipanti hanno sempre ricevuto le iniezioni in un ambiente clinico sotto la diretta supervisione di un professionista sanitario, garantendo una corretta somministrazione e la gestione immediata di eventuali reazioni avverse.
- Le misure di Harm Reduction includono:
- Valutazione settimanale dei parametri ematologici per individuare e prevenire aumenti eccessivi dei livelli di Hb e Ht.
- Fornitura di un’integrazione obbligatoria di vitamina C e ferro per supportare l’eritropoiesi e minimizzare il rischio di carenza di ferro.
- Interruzione tempestiva del dosaggio di NeoRecormon se i parametri ematologici superano soglie predefinite o se si verificano reazioni avverse.
- Esami del sangue effettuati
Durante lo studio, i partecipanti sono stati inoltre sottoposti a esami del sangue settimanali per monitorare i parametri ematologici, tra cui:
- Concentrazione di emoglobina
- Livelli di ematocrito
- Conteggio dei globuli rossi
- Conteggio delle piastrine
- Profilo di coagulazione
Questi esami del sangue hanno fornito informazioni cruciali per valutare la sicurezza e l’efficacia della somministrazione di NeoRecormon e per guidare gli aggiustamenti del dosaggio, se necessario.
Implicazioni per i medici dello sport e per i pazienti che praticano l’automedicazione
Per i medici sportivi che si trovano di fronte a pazienti che si curano da soli con l’EPO o che ne considerano l’uso per migliorare le prestazioni, questa ricerca fornisce indicazioni preziose su ciò che costituisce un protocollo di dosaggio supervisionato da un medico con le giuste misure di sicurezza. Lo studio sottolinea l’importanza di livelli di dosaggio accurati, di un monitoraggio regolare dei parametri ematologici e dell’aderenza ai regimi di integrazione per ridurre i rischi potenziali e ottimizzare i benefici.
- Criteri per i dosaggi iniziali, per le modifiche o per l’interruzione del trattamento
I criteri che hanno portato alla direzione degli aggiustamenti sono stati:
- Parametri ematologici di base (livelli di Hb e Ht)
- Risposta ai dosaggi iniziali (per esempio, tasso di aumento di Hb e Ht)
- Valori di soglia per le concentrazioni di Hb e Ht per evitare aumenti eccessivi
- Comparsa di reazioni avverse o di sintomi suggestivi di complicazioni ematologiche
Questi criteri hanno guidato i ricercatori nell’individualizzazione dei regimi di trattamento e nel garantire la sicurezza dei partecipanti durante lo studio.
- Rischi ed effetti collaterali monitorati
I ricercatori hanno monitorato attentamente i partecipanti per individuare potenziali rischi ed effetti collaterali, tra cui:
- Aumento eccessivo dei livelli di Hb e Ht con conseguenti complicazioni ematologiche (ad es. trombosi, ipertensione).
- Reazioni anafilattoidi o risposte allergiche alle iniezioni di NeoRecormon.
- Carenza di ferro o disturbi del metabolismo del ferro dovuti all’aumento dell’eritropoiesi.
La valutazione regolare dei parametri ematologici e la valutazione clinica hanno permesso di individuare e gestire precocemente gli eventi avversi, riducendo al minimo il loro impatto sulla sicurezza e sul benessere dei partecipanti.
Conclusioni:
Uno studio del 2007 ha dimostrato che l’EPO ha un effetto significativo sulle prestazioni di esercizio.[chiarisci][https://link.springer.com/article/] Uno studio del 2017 ha dimostrato che allo sforzo submassimale gli effetti dell’EPO non erano distinguibili da quelli del placebo. Si afferma che “[Allo] sforzo [submassimale]…[la potenza media] non differiva tra i gruppi”. Tuttavia, “alla potenza massima [da sforzo] era più alta nel gruppo rHuEPO rispetto al gruppo placebo”. Quindi, anche se non c’erano differenze a livelli inferiori di sforzo, allo sforzo massimale il gruppo EPO ha comunque ottenuto risultati migliori rispetto al gruppo placebo.[https://www.thelancet.com/]
Ma attraverso l’approfondimento di questo articolo abbiamo compreso che il protocollo di dosaggio qui discusso fa luce sull’uso potenziale dell’EPO per migliorare le prestazioni ciclistiche sotto supervisione, e con questo articolo è stato dimostrato che potrebbe essere efficace in determinati contesti. Questo protocollo è un’ottima risorsa per chi sta valutando l’uso dell’EPO e sottolinea l’importanza di aderire a pratiche scientificamente valide per ottimizzare i risultati dando priorità alla sicurezza.
Gabriel Bellizzi [CEO BioGenTech]
Riferimenti:
- Carnot P, Deflandre C (1906). “Sur l’activite hematopoietique du serum au cours de la regeneration du sang”. Compt. Rend. Acad. Sci. 143: 384–386.
- elkmann W (March 2007). “Erythropoietin after a century of research: younger than ever”. European Journal of Haematology. 78 (3): 183–205. doi:10.1111/j.1600-0609.2007.00818.x. PMID 17253966. S2CID 37331032.
- Höke A (2005). Erythropoietin and the Nervous System. Berlin: Springer. ISBN 978-0-387-30010-8. OCLC 64571745.
- Miyake T, Kung CK, Goldwasser E (August 1977). “Purification of human erythropoietin”. The Journal of Biological Chemistry. 252 (15): 5558–64. doi:10.1016/S0021-9258(19)63387-9. PMID 18467.
- Eschbach JW, Egrie JC, Downing MR, Browne JK, Adamson JW (January 1987). “Correction of the anemia of end-stage renal disease with recombinant human erythropoietin. Results of a combined phase I and II clinical trial”. The New England Journal of Medicine. 316 (2): 73–8. doi:10.1056/NEJM198701083160203. PMID 3537801.
- Lin FK, Suggs S, Lin CH, Browne JK, Smalling R, Egrie JC, Chen KK, Fox GM, Martin F, Stabinsky Z (November 1985). “Cloning and expression of the human erythropoietin gene”. Proceedings of the National Academy of Sciences of the United States of America. 82 (22): 7580–4. Bibcode:1985PNAS…82.7580L. doi:10.1073/pnas.82.22.7580. PMC 391376. PMID 3865178.
- “Epogen- epoetin alfa solution”. DailyMed. 25 July 2018. Retrieved 20 April 2022.
- “Epogen: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). 13 January 2017. Retrieved 20 April 2022.
- “The Nobel Prize in Physiology or Medicine 2019”. NobelPrize.org. 7 October 2019. Archived from the original on 31 October 2021. Retrieved 30 October 2019.
- “Erythropoietin”. Merriam-Webster.com Dictionary.
- “Erythropoietin”. Dictionary.com Unabridged (Online). n.d.
- “erythropoietin – definition of erythropoietin in English from the Oxford dictionary”. OxfordDictionaries.com. Archived from the original on 27 September 2012. Retrieved 20 January 2016.
- Jacobson LO, Goldwasser E, Fried W, Plzak L (March 1957). “Role of the kidney in erythropoiesis”. Nature. 179 (4560): 633–4. Bibcode:1957Natur.179..633J. doi:10.1038/179633a0. PMID 13418752. S2CID 4162940.
- Fisher JW, Koury S, Ducey T, Mendel S (October 1996). “Erythropoietin production by interstitial cells of hypoxic monkey kidneys”. British Journal of Haematology. 95 (1): 27–32. doi:10.1046/j.1365-2141.1996.d01-1864.x. PMID 8857934. S2CID 38309595.
- Barrett KE, Barman SM, Boitano S, Brooks H (eds.). Ganong’s review of Medical Physiology (24th ed.). McGraw Hill. p. 709. ISBN 978-1-25-902753-6.
- Ji P (November 2016). “Pericytes: new EPO-producing cells in the brain”. Blood. 128 (21): 2483–2485. doi:10.1182/blood-2016-10-743880. PMID 27884833.
- Jump up to:a b c Jelkmann W (March 2007). “Erythropoietin after a century of research: younger than ever”. European Journal of Haematology. 78 (3): 183–205. doi:10.1111/j.1600-0609.2007.00818.x. PMID 17253966. S2CID 37331032.
- Jelkmann W (March 2011). “Regulation of erythropoietin production”. The Journal of Physiology. 589 (Pt 6): 1251–8. doi:10.1113/jphysiol.2010.195057. PMC 3082088. PMID 21078592.
- Packer M (2020). “Sodium-Glucose Cotransporter-2 Inhibitor (SGLT2i) as a Primary Preventative Agent in the Healthy Individual: A Need of a Future Randomised Clinical Trial?”. Circulation: Heart Failure. 13 (9): e007197. doi:10.1161/CIRCHEARTFAILURE.120.007197. PMID 32894987. S2CID 221540765.
- Middleton SA, Barbone FP, Johnson DL, Thurmond RL, You Y, McMahon FJ, Jin R, Livnah O, Tullai J, Farrell FX, Goldsmith MA, Wilson IA, Jolliffe LK (May 1999). “Shared and unique determinants of the erythropoietin (EPO) receptor are important for binding EPO and EPO mimetic peptide”. The Journal of Biological Chemistry. 274 (20): 14163–9. doi:10.1074/jbc.274.20.14163. PMID 10318834.
- ^ Livnah O, Johnson DL, Stura EA, Farrell FX, Barbone FP, You Y, Liu KD, Goldsmith MA, He W, Krause CD, Pestka S, Jolliffe LK, Wilson IA (November 1998). “An antagonist peptide-EPO receptor complex suggests that receptor dimerization is not sufficient for activation”. Nature Structural Biology. 5 (11): 993–1004. doi:10.1038/2965. PMID 9808045. S2CID 24052881.
- ^ Kasper, C. (2003). “Erythropoietin”. The Cytokine Handbook. pp. 149–166. doi:10.1016/B978-012689663-3/50011-9. ISBN 978-0-12-689663-3.
- ^ Hodges VM, Rainey S, Lappin TR, Maxwell AP (November 2007). “Pathophysiology of anemia and erythrocytosis”. Critical Reviews in Oncology/Hematology. 64 (2): 139–58. doi:10.1016/j.critrevonc.2007.06.006. PMID 17656101.
- ^ Elliott S, Busse L, Bass MB, Lu H, Sarosi I, Sinclair AM, et al. (March 2006). “Anti-Epo receptor antibodies do not predict Epo receptor expression”. Blood. 107 (5): 1892–5. doi:10.1182/blood-2005-10-4066. PMID 16249375.
- ^ Sinclair AM, Coxon A, McCaffery I, Kaufman S, Paweletz K, Liu L, et al. (May 2010). “Functional erythropoietin receptor is undetectable in endothelial, cardiac, neuronal, and renal cells”. Blood. 115 (21): 4264–72. doi:10.1182/blood-2009-10-248666. PMID 20124513.
- ^ Risso A, Ciana A, Achilli C, Antonutto G, Minetti G (2014). “Neocytolysis: none, one or many? A reappraisal and future perspectives”. Frontiers in Physiology. 5: 54. doi:10.3389/fphys.2014.00054. PMC 3924315. PMID 24592241.
- ^ Ott C, Martens H, Hassouna I, Oliveira B, Erck C, Zafeiriou MP, et al. (December 2015). “Widespread Expression of Erythropoietin Receptor in Brain and Its Induction by Injury”. Molecular Medicine. 21 (1): 803–815. doi:10.2119/molmed.2015.00192. PMC 4818269. PMID 26349059.
- Elliott S, Sinclair AM (2012). “The effect of erythropoietin on normal and neoplastic cells”. Biologics: Targets and Therapy. 6: 163–89. doi:10.2147/BTT.S32281. PMC 3402043. PMID 22848149.
- Hosseini-Zare MS, Dashti-Khavidaki S, Mahdavi-Mazdeh M, Ahmadi F, Akrami S (July 2012). “Peripheral neuropathy response to erythropoietin in type 2 diabetic patients with mild to moderate renal failure”. Clinical Neurology and Neurosurgery. 114 (6): 663–7. doi:10.1016/j.clineuro.2012.01.007. PMID 22296650. S2CID 19516031.
- “The Story of Erythropoietin”. http://www.hematology.org. 16 February 2018. Archived from the original on 18 February 2019. Retrieved 18 February 2019.
- Liu S, Ren J, Hong Z, Yan D, Gu G, Han G, Wang G, Ren H, Chen J, Li J (February 2013). “Efficacy of erythropoietin combined with enteral nutrition for the treatment of anemia in Crohn’s disease: a prospective cohort study”. Nutrition in Clinical Practice. 28 (1): 120–7. doi:10.1177/0884533612462744. PMID 23064018.
- “Safety Labeling Changes: Epogen/Procrit (epoetin alfa) and Aranesp (darbepoetin alfa)”. MedWatch: The FDA Safety Information and Adverse Event Reporting Program. United States Food and Drug Administration. 11 August 2011. Archived from the original on 12 January 2017. Retrieved 16 December 2019.
- Heuberger, J. A. A. C., Rotmans, J. I., Gal, P., Stuurman, F. E., van ’t Westende, J., Post, T. E., Daniels, J. M. A., Moerland, M., van Veldhoven, P. L. J., de Kam, M. L., Ram, H., de Hon, O., Posthuma, J. J., Burggraaf, J., & Cohen, A. F. (2017). Effects of erythropoietin on cycling performance of well trained cyclists: A double-blind, randomised, placebo-controlled trial. The Lancet Haematology, 4(8), e374–e386. https://doi.org/10.1016/S2352-3026(17)30105-9